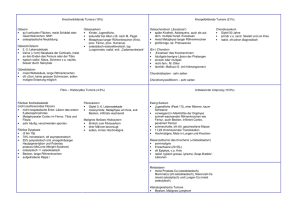
Tumore des Endolymphatischen Sacks und
Werbung

Aus der Neurochirurgischen Universitätsklinik Sektion Neuroradiologie der Albert-Ludwigs-Universität Freiburg i.Br. Tumore des Endolymphatischen Sacks und Hämangioblastome bei von Hippel-Lindau Erkrankung: Prävalenz und natürlicher Verlauf INAUGURAL-DISSERTATION zur Erlangung des Medizinischen Doktorgrades der Medizinischen Fakultät der Albert-Ludwigs-Universität Freiburg i.Br. Vorgelegt 2007 von Claudia Hader geboren in Augsburg Dekan Prof. Dr. Ch. Peters 1. Gutachter Prof. Dr. J. Spreer 2. Gutachter Prof. Dr. H.P.H. Neumann Jahr der Promotion 2007 Meinen Eltern Anneliese und Walter Hader 1 EINLEITUNG 1 1.1 Die von Hippel-Lindau Erkrankung 1 1.1.1 Historischer Überblick 1 1.1.2 Klinische Manifestation und Diagnostische Kriterien 2 1.1.3 Screening 3 1.1.4 Genetische Grundlagen der von Hippel-Lindau Erkrankung 4 1.2 Tumore des Endolymphatischen Sacks 5 1.2.1 Geschichte der ELS-Tumore 5 1.2.2 ELS-Tumore als Manifestation der von Hippel-Lindau Erkrankung 6 1.2.3 Klinische Symptomatik und Therapie 7 1.2.4 Radiologische Diagnostik 8 1.2.5 Histopathologie der ELS-Tumore 9 1.3 Zerebrale Hämangioblastome 10 1.3.1 Klinische Symptomatik und Therapie 11 1.3.2 Radiologische Diagnostik 11 1.3.3 Histopathologie 12 2 FRAGESTELLUNGEN 13 3 PATIENTEN, MATERIAL UND METHODEN 14 3.1 Patienten 14 3.1.1 Patientenkollektiv 14 3.1.2 Einschlusskriterien 14 3.1.3 Ausschluss von Patienten 14 3.2 MR-Technik 15 3.2.1 Bildmaterial 15 3.2.2 Technische Parameter 16 3.3 Bildbefundung 17 3.3.1 Analyse der ELS-Tumore 18 3.3.2 Analyse der zerebralen Hämangioblastome 18 3.3.3 Analyse der spinalen Hämangioblastome 20 3.4 Genetik 20 3.5 Statistik 20 4 ERGEBNISSE 21 4.1 Patientenkollektiv 21 4.1.1 Alters- und Geschlechtsverteilung 21 4.1.2 Familienzugehörigkeit 22 4.1.3 Nachweis der von Hippel-Lindau Erkrankung 22 4.1.4 Einteilung in Genotypen 22 4.2 MR-Tomographien 24 4.3 ELS-Tumore 25 4.3.1 Retrospektive Analyse der 6 mm Standardsequenzen des Zerebrums 25 4.3.2 Analyse der 3 mm Felsenbeinsequenzen in Fettsättigungstechnik 26 4.3.3 Vergleich der 6 mm Standardsequenzen mit den 3 mm Felsenbeinsequenzen 28 4.3.4 Fallberichte der Patienten mit ELS-Tumoren 29 4.3.5 Mutationen der Patienten mit ELS-Tumoren 34 4.4 Zentralnervöse Hämangioblastome 35 4.4.1 Häufigkeit zerebraler Hämangioblastome 35 4.4.2 Größe zerebraler Hämangioblastome und Operationshäufigkeit 37 4.4.3 Einfluss der Tumorlokalisation 40 4.4.4 Einfluss des Tumortyps 41 4.4.5 Einfluss der Mutation 42 4.4.6 Natürlicher Verlauf zerebraler Hämangioblastome 43 4.4.7 Einfluss von Lokalisation, Tumorgröße und Tumortyp auf das Wachstum 44 4.4.8 Wachstumsgeschwindigkeit zerebraler Hämangioblastome 46 4.4.9 Neuauftreten zerebraler Hämangioblastome 50 4.4.10 Spinale Hämangioblastome 50 5 DISKUSSION 51 5.1 ELS-Tumore 51 5.1.1 Prävalenzrate der ELS-Tumore 51 5.1.1.1 Einfluss der Bildqualität 52 5.1.1.2 MR-Tomographie der Felsenbeine 54 5.1.1.3 Zusammensetzung des Patientenkollektivs 54 5.1.2 Aquaeductus vestibuli und Saccus/Ductus endolymphaticus 56 5.1.3 Mutationen der Patienten mit ELS-Tumoren 57 5.1.4 Therapie der ELS-Tumore 58 5.2 Zentralnervöse Hämangioblastome 59 5.2.1 Häufigkeit zentralnervöser Hämangioblastome 59 5.2.2 Operationsfrequenz zerebraler Hämangioblastome 61 5.2.3 Bedeutung tumorassoziierter Zysten 62 5.2.4 Bedeutung des Mutationstyps des VHL-Gens 63 5.2.5 Wachstum zerebraler Hämangioblastome 63 5.2.6 Neuauftreten zerebraler Hämangioblastome 64 6 ZUSAMMENFASSUNG 65 7 LITERATUR 66 8 DANKSAGUNG 74 9 LEBENSLAUF 75 1 1 Einleitung 1 Einleitung 1.1 Die von Hippel-Lindau Erkrankung Die von Hippel-Lindau Erkrankung ist eine hereditäre Multisystemerkrankung. Betroffene Patienten können benigne und maligne Tumore des zentralen Nervensystem und des Abdomens entwickeln. 1.1.1 Historischer Überblick 1904 beschrieb Eugen von Hippel seltene Veränderungen des Augenhintergrunds, die er zunächst als entzündliche Komplikation einer Knochentuberkulose interpretierte. Nachdem ein Auge eines Patienten aufgrund eines Sekundärglaukoms entfernt werden musste hatte von Hippel Zugang zu histologischen Präparaten, erkannte den vaskulären Charakter der Veränderungen und interpretierte sie als Angiomatosis retinae. 1911 veröffentlichte er die Arbeit „Die anatomische Grundlage der von mir beschriebenen „sehr seltenen Erkrankung der Netzhaut““ (von Hippel 1904 und 1911). Der schwedische Pathologe Arvid Lindau diskutierte 1927 ausführlich den Zusammenhang zwischen zentralnervösen Hämangioblastomen und der Angiomatosis retinae, welche mittlerweile in der Literatur auch als „von Hippelsche Krankheit“ bezeichnet wurde (Lindau 1927). In Lindaus Sammlung von 19 Fällen wurden bereits die familiäre Disposition und das Auftreten von Nierentumoren, Pankreaszysten, Nierenzysten, Nebennierentumoren und Nebenhodentumoren erwähnt. 2 1 Einleitung 1.1.2 Klinische Manifestation und Diagnostische Kriterien Als Hauptläsionen gelten retinale Angiome und zentralnervöse Hämangioblastome. Häufige Läsionen sind Nierenzysten, Nierenzellkarzinome, Pankreaszysten, Phäochromozytome und Zystadenome des Nebenhodens. Seltene Läsionen sind Tumore des Endolymphatischen Sacks, Inselzelltumore des Pankreas und Zystadenome der breiten Mutterbänder (Neumann et al. 1998). Klinische Kriterien, die die Diagnose einer von Hippel-Lindau Erkrankung belegen, wurden erstmals von Melmon et al. zusammengestellt und später von Neumann modifiziert (Melmon et al. 1964, Neumann 1987). Demnach wird die Diagnose gestellt, wenn bei positiver Familienanamnese (retinales Angiom oder zerebrales Hämangioblastom bei einem Blutsverwandten) ein zerebraler oder viszeraler Tumor aus Tab 1.1 nachgewiesen wurde. Ohne Familienanamnese müssen zwei zerebrale Hämangioblastome oder ein zerebrales Hämangioblastom oder retinales Angiom und ein viszeraler Tumor vorliegen. Zysten der Nieren und der Nebenhoden werden wegen ihres allgemein häufigen Auftretens als viszerale Manifestationen nicht miteinbezogen (Lonser et al. 2003). War man bis 1993 zur Diagnosestellung allein auf Anamnese, klinische Untersuchung, bildgebende und laborchemische Diagnostik angewiesen, so ist seit der Identifikation des von Hippel-Lindau Gens (VHL-Gen) auch der Nachweis der Mutation möglich (Latif et al. 1993). Vor der Einführung multidisziplinärer Vorsorgeuntersuchungen und regelmäßiger Magnet-Resonanz-Tomographien (MRT) lag die geschätzte mittlere Lebenserwartung der Patienten bei 49 Jahren. Haupttodesursachen waren dabei das Nierenzellkarzinom gefolgt von zerebellären Hämangioblastomen (Maher et al. 1990). Die in der Literatur angegebene Häufigkeit der einzelnen Tumormanifestationen gibt Tabelle 1.1 wieder. 1 Einleitung 3 Tumormanifestation Häufigkeit bei VHL-Patienten Alter bei Erstmanifestation Retinale Angiome 25-60 % 25 (1-67) Tumore des Endolymphatischen 10-16 % 22 (12-50) Zerebelläre Hämangioblastome 44-72 % 33 (9-78) Hirnstamm Hämangioblastome 10-25 % 32 (12-46) Spinale Hämangioblastome 13-50 % 33 (12-66) <1% Unbekannt Nierenzellkarzinome 24-45 % 39 (16-67) Phäochromozytome 10-20 % 30 (5-58) Pankreastumore oder –zysten 35-70 % 36 (5-70) Zystadenome des Nebenhodens 25-60 % Unbekannt Unbekannt Unbekannt Sacks Supratentorielle Hämangioblastome Zystadenome der breiten Mutterbänder Tab 1.1: Häufigkeit der Tumormanifestationen und durchschnittliches Alter bei Erstmanifestation (in Jahren) bei von Hippel-Lindau Erkrankung (Lonser et al. 2003, Maher et al. 1990, Choo et al. 2004) 1.1.3 Screening Betroffene Patienten sollten in einer Spezialambulanz vorgestellt werden und einem regelmäßigen Screening unterzogen werden (Choyke et al. 1995). Potentiellen Mutationsträgern (Verwandte 1. Grades) wird die genetische Untersuchung empfohlen. Kann die Mutation ausgeschlossen werden, sind keine weiteren Untersuchungen nötig und der Betroffene wird keine von Hippel-Lindau assoziierten Tumore entwickeln. Wird die Mutation nachgewiesen, werden die Patienten in das Screening aufgenommen und lebenslang betreut. Das Screening umfasst jährliche Ophthalmoskopien ab dem Kindesalter, ein- bis zweijährliche Untersuchungen des 24-Stunden-Sammelurins auf Katecholamine ab dem Kindesalter oder bei erhöhten Blutdruckwerten, jährliche Kontrastmittel (KM) verstärkte MR-Tomographien des Schädels und des Spinalkanals ab dem Jugendalter, jährliche Abdomensonographien ab dem Jugendalter sowie jährliche Computertomographien (CT) des Abdomens ab 18 Jahren. Heute sollte angesichts der Strahlenbelastung alternativ eine MR-Tomographie des Abdomens empfohlen werden. 4 1 Einleitung 1.1.4 Genetische Grundlagen der von Hippel-Lindau Erkrankung Die Erkrankung wird autosomal dominant vererbt mit einer Penetranz von 97 % bei über 60-jährigen Patienten (Maher et al. 1990). Die geschätzte Inzidenz liegt bei 1:36000 Lebendgeburten (Maher et al. 1991). Das VHL-Gen wurde 1993 isoliert (Latif et al. 1993). Es liegt auf dem kurzen Arm des Chromosoms 3 (3p25-26) und besteht aus 3 Exons. Das Genprodukt, das VHL-Protein, ist ein Tumorsuppressorprotein, das an der ischämieinduzierten Zellantwort partizipiert. Betroffene Familienmitglieder tragen in allen Zellen ein mutiertes Allel und ein normales Allel (Wildtyp). Nach der „2-Treffer-Theorie“ nach Knudson muss zur Entstehung eines Tumors das Wildtypallel mutieren (Knudson 1986). Die Mutation muss in einem empfänglichen Organ (ZNS, Niere, Nebenniere, Pankreas) stattfinden. Der Nachweis der Mutation bei einer betroffenen Familie ist in 75-100 % der Fälle möglich (Chen et al. 1995, Stolle et al. 1998). Zum Nachweis einer Deletion des gesamten VHL-Gens wird ein quantitativer Southern Blot, zum Nachweis einer partiellen Deletion ein Southern Blot und zum Nachweis einer Punktmutation eine Sequenzierung des VHL-Gens mittels Fluoreszenz-in-situ-Hybridisierung (FISH) empfohlen (Stolle et al. 1998). Bis 1998 waren 146 verschiedene Mutationen bekannt, diese Zahl nimmt noch zu (Neumann et al. 1998). Gefunden wurden missense und nonsense Punktmutationen, Mikrodeletionen, Insertionen, Splice-siteMutationen, Deletionen eines, zweier oder aller Exons. Die Mutation kann zu einer inkompletten Translation des VHL-Proteins (Truncation) führen. Hierzu zählen schwerwiegende Mutationen wie große Deletionen, Nonsense-Punktmutationen, Mikrodeletionen/Mikroinsertionen mit Verschiebung des Leserasters und Splice-siteMutationen. Alternativ kommt es zu einer kompletten Translation des VHL-Proteins (Full-length), wobei eine Aminosäure ausgetauscht wird. Dazu zählen MissensePunktmutationen und Mikrodeletionen/Mikroinsertionen ohne Verschiebung des Leserasters. Clusteranalysen zeigten eine Korrelation zwischen Genotyp und Phänotyp. Bei Familien ohne Phäochromozytome, aber allen anderen Tumormanifestationen (Typ 1) findet man überwiegend schwerwiegende Mutationen, welche wahrscheinlich zu einem Funktionsverlust des VHL-Proteins führen. Bei Familien mit Phäochromozytomen findet man in 96 % Missense-Mutationen (Typ 2) (Chen et al. 1995, Neumann et al. 1998). Letztere werden in 3 Subgruppen eingeteilt (siehe 5 1 Einleitung Tabelle 1.2). Identische Mutationen führen bei Kaukasiern und Japanern zu gleichen Phänotypen (Zbar et al. 1996). Typ 1 Geringes Risiko für Phäochromozytom Typ 2 Hohes Risiko für Phäochromozytom 2A Niedriges Risiko für Nierenzellkarzinom 2B Hohes Risiko für Nierenzellkarzinom 2C Nur Phäochromozytom Tab 1.2: Einteilung der Phänotypen der von Hippel-Lindau Krankheit (Brauch et al. 1995, Lonser et al. 2003). 1.2 Tumore des Endolymphatischen Sacks 1.2.1 Geschichte der ELS-Tumore 1984 beschrieben Hassard et al. erstmals eine Neoplasie im Endolymphatischen Sack (ELS) (Hassard et al. 1984). Eine 34-jährige Patientin litt über mehrere Jahre an einer als Morbus Ménière eingeschätzten Symptomatik, verbunden mit einer einseitigen progredienten Hörminderung. Während der operativen Exploration des Endolymphatischen Sacks wurde eine stark vaskularisierte Raumforderung gefunden, welche die anteriore Schicht des Endolymphatischen Sacks arrodierte, jedoch weder den Knochen des Felsenbeins, noch die Dura mater infiltrierte. Histologisch wurde der Tumor als Adenom mit papillärer Wachstumsform interpretiert und entsprach wahrscheinlich einem auf den Saccus endolymphaticus begrenzten ELS-Tumor. Gaffey et al. veröffentlichten 1988 den Fall einer 29-jährigen Patientin, welche ebenfalls eine einseitige Hörminderung erlitt, zudem jedoch eine Fazialisparese entwickelte (Gaffey et al. 1988). Die CT-Diagnostik zeigte einen aggressiv wachsenden Tumor, welcher sowohl die Hinterwand des Os petrosum als auch Teile des Mittelohrs destruierte. Die histologische Untersuchung zeigte ebenfalls einen infiltrativ wachsenden papillären Tumor. Gaffey nahm an, dass der Tumor primär im Mittelohr entstand und versuchte diesen als eigenständige Entität, bezeichnet als APMET (aggressiv papillary middle ear tumor), von nicht infiltrativen Mittelohrtumoren, welche er als MEA (middle ear adenoma) bezeichnete, abzugrenzen. Die für ELS-Tumore typische Destruktion der Hinterwand des Os 6 1 Einleitung petrosum unter Einbeziehung des Endolymphatischen Sacks ist in der veröffentlichten Abbildung klar zu erkennen, so dass es sich wohl auch hier um eine frühe Beschreibung eines ELS-Tumors handelt. 1989 veröffentlichte Heffner eine Serie von 20 Patienten mit einseitig destruierend wachsenden Felsenbeintumoren (Heffner 1989). Die Tumore zeigten ein einheitliches makropathologisches Bild mit Destruktion der Felsenbeinrückfläche zwischen Meatus acusticus internus und Sinus sigmoideus, Infiltration des Felsenbeines und Tumorausdehnung in die hintere Schädelgrube. Histopathologisch waren den Tumoren der epitheliale Ursprung, das papillär-zystische Wachstumsverhalten, die teilweise sehr starke Vaskularisation und intratumorale, ossäre Anteile gemeinsam. Heffner folgerte daraus, dass es sich um eine eigenständige Tumorentität handeln müsse. Als mögliches epitheliales Ursprungsgewebe kamen der Endolymphatische Sack oder die epitheliale Auskleidung der Mastoidzellen in Betracht. Unter den untersuchten Fällen war auch der von Hassard bereits publizierte Fall, bei welchem der Ursprung im Endolymphatischen Sack operativ belegt war. Zudem war Heffner der Meinung, dass bei mastoidalem Ursprung die Lokalisation der Tumoren weitaus variabler sein müsse. Er nannte sie „Low-grade Adenocarcinoma of Probable Endolymphatic Sac Origin“. Interessanterweise zeigte der erste, von Eugen von Hippel beschriebene Patient in der Sektion, die nach 22-jährigem Krankheitsverlauf durchgeführt und von Rudolf Brandt 1921 diskutiert wurde, neben retinalen Angiomen zerebelläre und spinale Hämangioblastome, ossär metastasierte Nierenzellkarzinome, Nieren- und Pankreaszysten und „An der Spitze der linken Felsenbeinpyramide … ein walnussgroßer Tumor, der den Knochen arrodiert hat, und in dessen Bereich der 3.6. Hirnnerv liegen.“ (Brandt 1921). Ohne genaue Kenntnis des histologischen Befundes kann jedoch nicht geklärt werden, ob es sich um einen Tumor des Endolymphatischen Sacks oder eine Metastase des Nierenzellkarzinoms handelte. 1.2.2 ELS-Tumore als Manifestation der von Hippel-Lindau Erkrankung Seit der Beschreibung der ELS-Tumore als eigenständige Entität sind in der Literatur Fallberichte von ELS-Tumoren bei von Hippel-Lindau Patienten veröffentlicht worden (Megerian et al. 1995). Bilaterale ELS-Tumoren sind bisher nur bei von Hippel-Lindau 1 Einleitung 7 Patienten beschrieben worden. Eine ausführliche Literaturrecherche von Gaffey ergibt 47 Felsenbeintumore, die nach den Beschreibungen einem ELS-Tumor entsprechen. 7 der Patienten hatten eine gesicherte von Hippel-Lindau Erkrankung. Dies ergibt einen hochsignifikanten Zusammenhang (p = 1,4 x 10-24) zwischen ELSTumor und von Hippel-Lindau Erkrankung (Gaffey et al. 1994). Manski et al. untersuchten erstmals systematisch ein großes Kollektiv von 121 VHL-Patienten retrospektiv anhand von MR-Tomographien auf das Vorliegen von ELS-Tumoren und fanden bei 13 Betroffenen 15 ELS-Tumore, einer Prävalenzrate von 11 % entsprechend. Im Kontrollkollektiv mit 253 Patienten fand sich kein ELS-Tumor (Manski et al. 1997). 1.2.3 Klinische Symptomatik und Therapie Die meisten Patienten mit einem ELS-Tumor erleiden einen sensoneuralen Hörverlust. Dabei bestehen die Symptome bei Diagnosestellung oft bis zu 10 Jahren. Weitere häufige Symptome sind Tinnitus und Schwindel. Bei einem Wachstum in den Kleinhirnbrückenwinkel kann es zu einer Fazialisparese kommen. Selten wird durch den ELS-Tumor eine Ataxie verursacht. Die Häufigkeit der klinischen Beschwerden zeigt Tab. 1.3. Symptom Schwerhörigkeit Häufigkeit Manski Mukherji Choo n = 13 n = 20 n = 21 100 % 100 % 95 % Tinnitus 92 % 50 % 81 % Schwindel 62 % 20 % 67 % 8% 60 % Keine Angaben Fazialisparese Tab 1.3: Häufigkeit der Symptome von Patienten mit Tumoren des Endolymphatischen Sacks. (Manski et al. 1997, Mukherji et al. 1997, Choo et al. 2003). Die Therapie besteht aus der radikalen chirurgischen Resektion der Tumore. Eine Metastasierung ist bisher nicht beobachtet worden. Über Ergebnisse einer Strahlentherapie liegen keine Daten vor. 8 1 Einleitung 1.2.4 Radiologische Diagnostik In der Bildgebung zeigen ELS-Tumore typischerweise eine Destruktion der Felsenbeinhinterwand. Der Tumor liegt zwischen dem Meatus acusticus internus und dem Sinus sigmoideus. In der Computertomographie sind am Tumorrand glatt begrenzte Osteolysen zu erkennen. Typisch sind intratumorale Knochenfragmente (Lo 1993) und eine dünne periphere Verkalkung. Die Knochenfragmente entsprechen am ehesten Resten des Felsenbeines und nicht einer Knochenneubildung. Der verkalkte Tumorrand könnte einer stark ausgedünnten Kortikalis des Felsenbeins entsprechen (Mukherji et al. 1997). Kleine Tumore (< 3 cm) sind auf die Region des Aquaeductus vestibuli beschränkt, größere Tumore können den Kleinhirnbrückenwinkel, den Sinus cavernosus, das Foramen jugulare und die Paukenhöhle infiltrieren (Mukherji et al. 1997). In der MR-Tomographie sind in 80 % der Fälle T1-gewichtet (T1-w) nativ ein hyperintenser Rand, oder, bei größeren Tumoren, fokale Hyperintensitäten zu sehen. Diese stellen zystische Tumoranteile mit proteinreichem Inhalt oder Einblutungen dar. Die soliden Tumoranteile sind T1-w nativ signalarm und reichern nach Kontrastmittelgabe an (Patel et al. 2006). T2-w sind die Tumore entweder homogen oder inhomogen hyperintens im Vergleich zum Kleinhirnmarklager (Mukherji et al. 1997). Die Blutversorgung erfolgt aus Ästen der Arteria carotis externa, größere Tumore können zusätzlich über Äste der Arteria carotis interna und der Arteria vertebralis versorgt werden (Mukherji et al. 1997). 9 1 Einleitung Abb. 1.1: Tumor des Endolymphatischen Sacks des linken Felsenbeins bei einem von Hippel-Lindau Patienten (schwarzer Pfeil). Hämangioblastom der linken Kleinhirnhemisphäre (weißer Pfeil) und postoperative Veränderungen nach subokzipitarer Kraniektomie und Hämangioblastomresektion. (a) – (e) Invasiv wachsende Raumforderung mit Destruktion der Felsenbeinhinterkante. Die KMverstärkte CT zeigt im Weichteilfenster (a) den typischen verkalkten Tumorrand, der Tumor ist isodens zum Hirnparenchym. Im Knochenfenster (b) kommen die intratumoralen Knochenfragmente gut zur Darstellung (Pfeil). Der Tumor zeigt im T2-w (c) eine inhomogene Signalintensität, im T1-w nativ (d) hyperintense Foci und reichert nach KM-Applikation (e) an. (a) CT mit KM, (b) CT im Knochenfenster, (c) MRT, T2-w, (d) MRT, T1-w nativ, (e) MRT, T1-w mit Gadolinium-Chelat (Gad-Chelat). 1.2.5 Histopathologie der ELS-Tumore Makropathologisch werden ELS-Tumore in der Literatur einheitlich als rötlich-blau und hypervaskularisiert beschrieben (Kempermann et al. 1998). Mikroskopisch zeigen die Tumore unterschiedliche Komponenten. Zum einen sind kolloidale Zysten mit schmalem Stroma, welche an Schilddrüsenmetastasen erinnern, beschrieben, zum zweiten sind papilläre Anteile mit teilweise klarzelligem Stroma vorhanden, wodurch die Differenzierung zur Nierenzellkarzinommetastase erschwert wird. Zum dritten sind intratumorale Knochenfragmente, Blutungen und regressive 10 1 Einleitung Veränderungen möglich. Da ELS-Tumore nicht alle Komponenten aufweisen müssen, ist verständlich, dass die histologische Zuordnung schwierig sein kann (Heffner et al. 1989). Elektronenmikroskopisch zeigen die Tumore Ähnlichkeiten mit normalem Epithel des Endolymphatischen Sacks (Kempermann et al. 1998). Immunhistochemisch zeigen sie die stärkste Expression für den epithelialen Marker Cytokeratin (CK). Die Expression der neuronen spezifischen Enolase (NSE) unterstreicht den neuroektodermalen Ursprung (Gaffey et al. 1994, Kempermann et al. 1998). Die Tumore zeigen ein langsames Wachstum und metastasieren nicht. Abb. 1.2: Tumor des Endolymphatischen Sacks mit papillären und zystischen Anteilen. H-E-Färbung, 10-fache Vergrößerung. Mit freundlicher Genehmigung von Dr. K. Müller, Neuropathologie Universitätsklinikum Freiburg. 1.3 Zerebrale Hämangioblastome Hämangioblastome sind histologisch gutartige, gefäßreiche Tumore. Sie können sporadisch oder in Zusammenhang mit der von Hippel-Lindau Erkrankung auftreten. Bevorzugt liegen sie in der hinteren Schädelgrube oder am Rückenmark. 34 - 38 % der Patienten mit zentralnervösen Hämangioblastomen sind VHL-Patienten (Richard et al. 1998, Conway et al. 2001). Patienten mit Hämangioblastomen im Rahmen der 11 1 Einleitung von Hippel-Lindau Erkrankung sind bei Erstdiagnose signifikant jünger als Patienten mit sporadischen Tumoren (Richard et al. 1998). Liegen multiple Tumore vor, gilt die Diagnose VHL als gesichert. Lindau beschrieb bereits 1927 die bevorzugte Lokalisation in der hinteren Schädelgrube mit Affinität zum Rautenhirn und der Neigung zur Zystenbildung. Als mögliche Komplikation der Tumore erwähnte er Transsudation und Erweichung und empfahl als Therapie die Resektion des angioplastischen Tumors, da ihm die alleinige Punktion der Zysten langfristig sinnlos erschien (Lindau 1927). 1.3.1 Klinische Symptomatik und Therapie Kleine Tumore sind häufig asymptomatisch und werden oft im Rahmen des Screenings entdeckt. Größere Tumore können raumfordernd wirken und durch Schwindel oder Gangstörungen auffällig werden oder Symptome erhöhten Hirndrucks wie Kopfschmerzen, Übelkeit und Erbrechen verursachen. Tumore am Hirnstamm führen früher zu Beschwerden als Tumore der Kleinhirnhemisphären (Wanebo et al. 2003). Tumore mit Zysten sollen rascher zu einem Tumorprogress mit neurologischer Symptomatik führen als solide Tumore (Slater et al. 2003). Therapie der Wahl ist die mikrochirurgische Resektion des soliden Tumorknotens. Bei großen und häufig sehr gut vaskularisierten Tumoren kann eine prächirurgische Tumorembolisation indiziert sein. Therapieversuche mit stereotaktischer Bestrahlung (Gammaknife) wurden unternommen, hierzu liegen noch keine Langzeitergebnisse vor. 1.3.2 Radiologische Diagnostik Hämangioblastome können, je nach Vorliegen von zystischen und soliden Anteilen in 4 Typen eingeteilt werden (Richard et al. 1998). Die Übersicht zeigt Tab. 1.4. Die MRT ist der CT überlegen, da insbesondere in der hinteren Schädelgrube Aufhärtungsartefakte den Nachweis kleiner Tumore in der CT erschweren (Lee et al. 1989). Tumorzysten sind in der MR-Tomographie liquorisointens, solide Tumore zeigen eine homogene KM-Aufnahme. 12 Typ 1 Einfache Zyste ohne Tumorknoten Typ 2 Zyste mit wandständigem Tumorknoten Typ 3 Solider Tumor ohne Zyste Typ 4 Solider Tumor mit intratumoralen Zysten 1 Einleitung Tab. 1.4: Einteilung der Hämangioblastome anhand des Vorliegens von soliden und zystischen Tumoranteilen (Richard et al. 1998). Abb. 1.3: Typische Beispiele von Hämangioblastomen nach der oben genannten Einteilung. (a) Typ 2: Zyste mit wandständigem Tumorknoten der rechten Kleinhirnhemisphäre, (b) Typ 3: zwei solide Tumore beider Kleinhirnhemisphären , (c) Typ 4: solider Tumor der rechten Kleinhirnhemisphäre mit intratumoralen Zysten. (a) – (c) MRT, T1-w mit Gad-Chelat, transversal. 1.3.3 Histopathologie Hämangioblastome bestehen aus einem dichten Kapillargeflecht und zwischengelagerten Stromazellen. Das Stroma besteht in der Regel aus großen Zellen mit Vakuolen (zellulärer Typ) oder, seltener, aus dicht gepackten Zellen (retikulärer Typ) (Hasselblatt et al. 2005). Die Zellkerne liegen sehr dicht beieinander, Mitosen sind sehr selten (Richard 2000). Stromazellen stellen den neoplastischen Anteil des Tumors dar, Kapillaren entstehen reaktiv (Vortmeyer 1997). Tumorassoziierte Zysten haben keine Wand, sondern zeigen einen gliotischen Rand. In der Zyste ist oft eine gelbliche Flüssigkeit vorhanden, Zysteneinblutungen sind möglich (Lee et al. 1989). Vortmeyer et al. konnten zeigen, dass zentralnervöse Hämangioblastome aus Angioblasten hervorgehen, die Erythropoetin (Epo) und EpoRezeptoren koexprimieren (Vortmeyer et al. 2003). 13 2 Fragestellungen 2 Fragestellungen 1. Wie hoch ist die Prävalenzrate von Tumoren des Endolymphatischen Sacks im Freiburger von Hippel-Lindau Patientenkollektiv? 2. Lässt sich der Aquaeductus vestibuli durch MR-Tomographien der Felsenbeine zuverlässig nachweisen? 3. Können durch MRT-Dünnschichtaufnahmen der Felsenbeine bei von HippelLindau Patienten häufiger Tumore des Endolymphatischen Sacks nachgewiesen werden als in der Standarduntersuchung? 4. Wie häufig und in welcher Lokalisation sind zerebrale Hämangioblastome bei von Hippel-Lindau Patienten nachweisbar? 5. Besteht ein Zusammenhang zwischen Größe, Lokalisation, Mutationstyp und Operationshäufigkeit zerebraler Hämangioblastome? 6. Unterscheidet sich der Verlauf zystischer und solider Hämangioblastome? 7. Wie ist der natürliche Verlauf zerebraler Hämangioblastome? 8. Besteht ein Zusammenhang zwischen Größe, Lokalisation, Tumortyp, Mutationstyp und Wachstum zerebraler Hämangioblastome? 14 3 Patienten, Material und Methoden 3 Patienten, Material und Methoden 3.1 Patienten 3.1.1 Patientenkollektiv Von 607 Patienten der Freiburger von Hippel-Lindau Datenbank wurden 251 zwischen Mai 1988 und März 2006 in Freiburg MR-tomographisch untersucht. Von 17 Patienten waren die Aufnahmen nicht mehr zugänglich. MR-Tomographien von 234 Patienten wurden reevaluiert. Davon mussten nachträglich 6 Patienten ausgeschlossen werden: eine Patientin war doppelt aufgeführt, bei einer war die Untersuchung inkomplett und nicht auswertbar, bei vier Patienten war die von HippelLindau Erkrankung nicht gesichert. Somit wurden 228 Patienten in diese Arbeit aufgenommen und ausgewertet. 3.1.2 Einschlusskriterien In diese Studie wurden nur Patienten mit gesicherter von Hippel-Lindau Erkrankung aufgenommen. Dies ist der Fall, wenn entweder die Mutation auf Chromosom 3 nachgewiesen werden konnte und/oder die in Kapitel 1.1.2 aufgeführten klinischen Kriterien erfüllt waren. Radiologisches Einschlusskriterium war das Vorliegen einer zerebralen, in der Regel biplanaren, kontrastmittelgestützten MRT in T1-Wichtung. Das Vorliegen einer Sequenz in transversaler Orientierung war obligat. MRT, von denen lediglich der schriftliche Befund vorlag, oder welche ohne Kontrastmittel angefertigt worden waren, wurden nicht in die Untersuchung aufgenommen. 3.1.3 Ausschluss von Patienten Bei allen 228 eingeschlossenen Patienten war die Beurteilung des Saccus endolymphaticus möglich. 15 3 Patienten, Material und Methoden Zwei Patienten wurden von der Auswertung der Hämangioblastome ausgeschlossen, da sowohl zerebrale Metastasen eines Nierenzellkarzinoms als auch Hämangioblastome vorlagen und die Tumore nicht sicher zu unterscheiden waren. 3.2 MR-Technik 3.2.1 Bildmaterial Zerebrale MR-Tomographien von 228 Patienten wurden retrospektiv analysiert, wobei zunächst jeweils die aktuellste Untersuchung herangezogen wurde. 147 der Patienten hatten mindestens zwei MR-Tomographien. Die Anzahl der vorliegenden MRT zeigt Tabelle 3.1. Lagen zerebrale Hämangioblastome oder ELS-Tumore vor, wurden auch alle Voraufnahmen zur Verlaufsuntersuchung ausgewertet. Die Bilder standen entweder in digitaler Form via PACS (Tiani, Agfa-GWI, Version 3.3.16) oder als Laserprint-Film zur Verfügung. Bei 220 Patienten lag zusätzlich eine MR-Tomographie der Wirbelsäule vor. Anzahl MRT im Verlauf Anzahl Patienten 1 85 2 53 3 27 4 25 5 12 6 11 7 6 8 3 9 1 10 0 11 1 12 1 13 1 14 1 15 0 16 1 Tab. 3.1: Anzahl der vorliegenden MRT des untersuchten Patientenkollektivs. Von 228 Patienten hatten 85 jeweils ein MRT, 143 hatten mindestens 2 MRT. 16 3 Patienten, Material und Methoden 3.2.2 Technische Parameter Die vorliegende Arbeit umfasst MRT aus dem Zeitraum 1988 bis 2006. Die MRTomographien zwischen 1988 und 1995 wurden an unterschiedlichen Geräten von 1,0 und 1,5 T Feldstärke angefertigt und beinhalteten eine kontrastmittelgestützte biplanare Darstellung des Schädels in T1-Wichtung mit einer maximalen Schichtdicke von 7,5 mm. Die MR-Tomographien von 1996 bis März 2006 wurden an einem 1,5 T Kernspintomographen (Magnetom Vision, Siemens, Erlangen) angefertigt. Das Untersuchungsprotokoll bestand während dieses Zeitraums unverändert aus einer biplanaren, in der Regel transversalen und sagittalen, in seltenen Fällen transversalen und coronaren Darstellung des Schädels in T1-Wichtung sowie sagittaler Darstellung der gesamten Wirbelsäule in drei aufeinander folgenden Untersuchungsblöcken (Schädel, HWS bis Mitte BWS und Mitte BWS bis LWS). Die Akquisitionen erfolgten nach intravenöser Gabe von 0,5 molarem Gadolinium-Chelat mit einer Dosierung von 0,1 mMol Gad-Chelat pro kg Körpergewicht. Von Juli 2005 bis März 2006 wurden bei 60 Patienten zusätzlich zum Standardprotokoll kontrastmittelgestützte dünnschichtige Sequenzen der Felsenbeine angefertigt. Um Verfettungen in den Ossa petrosa von Kontrastmittelanreichernden Tumoren zu differenzieren, wurde dabei eine spektrale Fettsättigung angewandt. Die Untersuchungsparameter im Einzelnen zeigt Tabelle 3.2. 17 Orientierung 3 Patienten, Material und Methoden Tra T1-w KM Sag T1-w KM Tra T1-w KM Sag T1-w KM Sag T1-w KM Schädel Schädel Fettsättigung Wirbelsäule Wirbelsäule Felsenbein HWS/BWS BWS/LWS Transversal Sagittal Transversal Sagittal Sagittal TR 532 ms 532 ms 705 ms 697 ms 642 ms TE 17,0 ms 17,0 ms 17 ms 12 ms 12 ms 90° 90° 90° 150° 150° 202 x 256 230 x 256 222 x 256 198 x 512 306 x 512 220 mm 230 mm 230 mm 280 mm 290 mm Rechteck-FOV 7/8 8/8 7/8 6/8 6/8 Schichtanzahl 19 19 15 11 11 6 mm 6 mm 3 mm 3 mm 4 mm 0,6 mm 0,6 mm 0,3 mm 0,3 mm 0,4 mm 2 2 3 3 4 1:59 min 2:14 min 4:15 min 4:40 min 4:26 min Fettsättigung Nein Nein Ja Nein Nein Gadolinium- Ja Ja Ja Ja Ja Standard Standard 07/2005 - Standard Standard Flipwinkel Matrix FOV Schichtdicke Distanz Akquisitionen Messzeit Chelat Anwendung 03/2006 Tabelle 3.2: Untersuchungsparameter des Standardprotokolls des Screenings von Hippel-Lindau Erkrankter. Die Dünnschichtung der Felsenbeine erfolgte ab Juli 2005 zusätzlich zur Standarddiagnostik. (TR: Relaxationszeit, TE: Echozeit, FOV: Field of view, tra: transversal, sag: sagittal) 3.3 Bildbefundung Alle vor Juli 2005 angefertigten MR-Tomographien wurden von der Verfasserin retrospektiv analysiert. Die Aufnahmen wurden in Hinblick auf das Vorliegen von ELS-Tumoren und Hämangioblastomen beurteilt. Die zusätzlich angefertigten Dünnschichtaufnahmen der Felsenbeine wurden nur zur Diagnostik von ELSTumoren herangezogen. Auf einem Erhebungsbogen wurden die relevanten Daten dokumentiert. 18 3 Patienten, Material und Methoden 3.3.1 Analyse der ELS-Tumore Die 6 mm Standardsequenzen wurden auf das Vorliegen von ELS-Tumoren begutachtet. Als ELS-Tumore wurden hyperintense Läsionen im Verlauf des Aquaeductus vestibuli oder Saccus endolymphaticus gewertet, welche über die normale Ausdehnung des Aquädukts hinausgingen und/oder eine Destruktion der Felsenbeinhinterwand und/oder eine Tumorausdehnung in die hintere Schädelgrube zeigten. Lag eine komplette oder inkomplette Petrosektomie vor, wurde der histologische Befund herangezogen. Die Läsionen mussten in zwei Ebenen abgrenzbar sein und sicher vom Sinus sigmoideus und mastoidalen Fetteinlagerungen zu differenzieren sein. Eine alleinige Hyperintensität im anatomisch normal konfigurierten Ductus oder Saccus endolymphaticus wurde nicht als ELS-Tumor gewertet. In Zweifelsfällen wurde als Zweitbefunder ein erfahrener Neuroradiologe hinzugezogen. Die Beurteilung erfolgte anhand der genannten Kriterien im Konsens. Lag ein Tumor vor, wurden der maximale Längsdurchmesser und die senkrecht dazu stehenden maximalen Querdurchmesser am transversalen und sagittalen Bild bestimmt. Lagen MR-Tomographien im Verlauf vor, wurde die Größe der Tumore im Verlauf ermittelt. Sequenzen der Felsenbeine in 3 mm Schichtdicke wurden nach Kontrastmittelgabe in Fettsättigungstechnik akquiriert. Die Auswertung erfolgte durch die Verfasserin und einen erfahrenen Neuroradiologen. Dabei wurde unabhängig voneinander beurteilt, ob der Aquaeductus vestibuli je Felsenbein abgrenzbar ist, ob der Aquädukt im Vergleich zu den Strukturen des inneren Gehörgangs hyperintens erscheint und ob ein Tumor vorliegt. Die Ergebnisse wurden auf den Grad der Übereinstimmung geprüft. 3.3.2 Analyse der zerebralen Hämangioblastome Die Untersuchung der zerebralen Hämangioblastome wurde von der Verfasserin an den 6 mm Standardsequenzen durchgeführt. Als Hämangioblastome wurden scharf berandete, hyperintense, intradural gelegene Raumforderungen gewertet. Sie mussten in zwei Ebenen abgrenzbar sein. Die Lokalisation der Tumore wurde in 19 3 Patienten, Material und Methoden folgende Bereiche eingeteilt: supratentoriell, Kleinhirnhemisphären, Vermis cerebelli oder Medulla oblongata / Hirnstamm. Der Tumortyp wurde nach Richard et al. in vier Kategorien eingeteilt (Typ 1 einfache Zyste, Typ 2 Tumorknoten mit assoziierter Zyste, Typ 3 solider Tumor und Typ 4 Tumor mit intratumoralen Zysten) (Richard et al. 1998). Die Tumore wurden von apikal nach basal in der Reihenfolge ihres Erscheinens nummeriert, um bei Verlaufsuntersuchungen die korrekte Zuordnung zu gewährleisten. Die aufeinander senkrecht stehenden Längsdurchmesser in drei Raumebenen wurden für den soliden und zystischen Anteil und für die Gesamtgröße gemessen und in eine Datenbank eingetragen. Ein Beispiel zeigt Abb. 3.1. Das Volumen eines Ellipsoids berechnet sich nach der Formel V = 4 / 3 * π * (x / 2 * y / 2 * z / 2); x, y und z entsprechen den aufeinander senkrecht stehenden Durchmessern. Durch Kürzung ergibt sich V = π / 3 * (x * y * z) / 2. Setzt man π / 3 = 1, erhält man die Näherung V = (x * y * z) / 2. Da Hämangioblastome in der Regel kugelig bis ellipsoid sind, wurde das Tumorvolumen V nach der Näherung V = (x * y * z) / 2 ermittelt. Wurde im Verlauf ein Tumor reseziert, wurde das OP-Datum notiert, postoperative Defekte wurden als reseziertes Hämangioblastom ohne Größenangabe gewertet. Abb. 3.1: Messung der aufeinander senkrecht stehenden Durchmesser eines Hämangioblastoms mit exzentrischer Zyste. Der solide Anteil hat ein Volumen von 10*6*10/2 = 300 mm3, die Zyste ein Volumen von 41*33*24/2 = 16236 mm3. 20 3 Patienten, Material und Methoden 3.3.3 Analyse der spinalen Hämangioblastome Bei 215 Patienten lag zusätzlich zur zerebralen Bildgebung eine sagittale Darstellung der gesamten Wirbelsäule vor. Es wurde registriert, ob Hämangioblastome vorlagen oder nicht. Eine Quantifizierung der Tumore in Hinblick auf Anzahl und Größe erfolgte nicht. 3.4. Genetik Die Ergebnisse der genetischen Untersuchungen aller Patienten wurden freundlicherweise von Prof. Neumann, Abteilung für Nephrologie und Hypertension, Universitätsklinikum Freiburg zur Verfügung gestellt. DNA wurde aus peripheren Blutleukozyten extrahiert. Als Screening wurde anfangs eine Single-Stranded Conformational Polymorphism (SSCP) Analyse durchgeführt. Später wurde diese Technik durch eine Denaturation High Performance Liquid Chromatography (DHPLC) ersetzt. Im Falle aberrierender Banden wurde die DNA amplifiziert und zur weiteren Sequenzierung in ein Speziallabor gesandt. Deletionen wurden initial mittels Southern Blot (Eco RI) nachgewiesen. In letzter Zeit wurde zum Nachweis großer Deletionen eine MLPA (multiplex ligation-dependent probe amplification) angewandt. 3.5. Statistik Ein Ergebnis wurde als signifikant eingestuft, wenn die Wahrscheinlichkeit mindestens 95 % betrug (p ≤ 0,05). Lag der Erwartungswert über 5, wurde der ChiQuadrat Test angewandt, bestand diese Voraussetzung nicht, der exakte FischerTest. Mittelwerte wurden mit dem unpaaren t-Test auf Signifikanz überprüft. 21 4 Ergebnisse 4 Ergebnisse 4.1 Patientenkollektiv 4.1.1 Alters- und Geschlechtsverteilung In die vorliegende Arbeit wurden 228 Patienten eingeschlossen, davon 106 Männer und 122 Frauen. Dies ergibt ein Verhältnis m:w von 1:1,2. Der Altersdurchschnitt lag zum Zeitpunkt der aktuellsten MR-Tomographie im Mittel bei 39,5 Jahren (Median = 39 Jahre). Der jüngste Patient war vier, der älteste 82 Jahre alt. Abbildung 4.1 zeigt die Altersverteilung. Abb. 4.1: Altersverteilung 228 untersuchter Patienten zum Zeitpunkt der jeweils aktuellsten MRTomographie. 22 4 Ergebnisse 4.1.2 Familienzugehörigkeit Die Zugehörigkeit zur jeweiligen von Hippel-Lindau Familie war bei 219 Patienten (96% der Fälle) bekannt. Es handelte sich um 106 verschiedene Familien mit einem bis maximal zwölf betroffenen Mitgliedern. 4.1.3 Nachweis der von Hippel-Lindau Erkrankung Der Nachweis der von Hippel-Lindau Erkrankung gilt als sicher, wenn die in Kapitel 1.1.2 aufgeführten klinischen Kriterien erfüllt sind oder die Mutation nachgewiesen wurde. Die Diagnose VHL war bei allen 228 eingeschlossenen Patienten gesichert. In 96 % der Fälle (218 Patienten) war die Mutation bekannt, bei 10 Patienten war der Mutationsnachweis nicht möglich. Bei diesen Patienten waren jeweils die klinischen Kriterien eindeutig erfüllt. Ein Großteil der Patienten (200 Fälle) konnte allein anhand der klinischen Kriterien sicher als betroffen eingruppiert werden. Bei 28 Patienten stützte sich die Diagnose allein auf den Nachweis der Mutation (18 Patienten mit positiver Familienanamnese ohne nachweisbare Manifestation, 4 Patienten ohne Familienanamnese und einer Manifestation, 6 Patienten ohne Kenntnis der Familienzugehörigkeit, davon 5 mit einer Manifestation und einer ohne Manifestation). 4.1.4 Einteilung in Genotypen Weltweit sind ca. 500 verschieden Mutationen des VHL-Gens bekannt. Es werden zwei Mutationstypen unterschieden. Bei einer Missense-Mutation (Punktmutation, Mikroinsertion / -deletion ohne Verschiebung des Leserasters) wird im VHL-Protein eine Aminosäure verändert und das Protein wahrscheinlich in voller Länge translatiert (Full-length, F-Typ). Bei einer Nonsense-Mutation (Einfügen eines StopCodons, Mikroinsertion / -deletion mit Verschiebung des Leserasters, große Deletion) und Slice-site-Defekten bricht die Translation des VHL-Proteins wahrscheinlich ab (Truncation, T-Typ). Im Patientenkollektiv dieser Arbeit kamen 52 unterschiedliche Mutationen vor. Die Ergebnisse der Mutationsanalysen wurden freundlicherweise von Prof. Neumann zu Verfügung gestellt. Eine Mutation, der so genannte 4 Ergebnisse 23 Schwarzwaldtyp (c.505 T>C) war mit 102 Patienten deutlich überrepräsentiert. Die Mutationen sind im Einzelnen in Tabelle 4.1 aufgeführt. Nukleotid-Austausch Mutationsname Mutationstyp T-/F-Typ Anzahl Patienten Ala56Gly Missense F 1 Frameshift T 1 Exon 1 c.380 C>G c.393 ins C c.407 C>A Ser65X Nonsense T 1 c.407 C>G Ser65Trp Missense F 2 Frameshift T 1 c.417 dup G c.430 C>T Gln73X Nonsense T 1 c.434 T>G Val74Gly Missense F 2 c.437-9 del TCT Ile75-Phe76delinsIle In-frame Deletion F 9 c.443 ins TCT 443ins3 Frameshift T 1 c.446 A>G Asn78Ser Missense F 1 c.452 G>T Ser80Ile Missense F 3 c.469 C>T Pro86Ser Missense F 1 c.475 T>A Trp88Arg Missense F 2 c.479 T>C Leu89Pro Missense F 6 c.490 G>A Gly93Ser Missense F 2 c.505 T>C Tyr98His Missense F 102 Frameshift T 1 In-frame Insertion F 1 Splice-site-Defekt T 2 c.508-518 dup11 c.529 ins GCC 529ins3 c.553+1 G>T Exon 2 c.557 A>G His115Arg Missense F 2 c.557/558 AC/GT His115Arg Missense F 1 c.595 C>T Leu128Phe Missense F 1 c.601 G>T Val130Phe Missense F 3 c.605 A>C Asn131Thr Missense F 1 c.610 A>C Thr133Pro Missense F 2 c.617 T>A Leu135Stop Nonsense T 1 c.620 T>C Phe136Ser Missense F 1 c.658-671 del 14 658del14 Frameshift T 2 Splice-site-Defekt T 3 c.677-1 G>C Splice-site-Defekt T 2 c.677-2 A>G Splice-site-Defekt T 4 Missense F 4 c.676+2 T>C Exon 3 c.680 A>G Tyr156Cys 4 Ergebnisse 24 c.694 C>T Arg161Stop Nonsense T 5 c.695 G>A Arg161Gln Missense F 1 c.695 G>C Arg161Pro Missense F 2 c.699 C>G Cys162Trp Missense F 2 c.701 T>A Leu163His Missense F 1 c.712 C>T Arg167Trp Missense F 3 c.713 G>A Arg167Gln Missense F 2 c.738 C>A Tyr175Stop Nonsense T 1 c.742 A>T Arg177Stop Nonsense T 1 c.746 T>A Leu178Gln Missense F 1 c.761 C>A Ser183Stop Nonsense T 2 c.768 C>A Tyr185Stop Nonsense T 1 c.775 C>G und 454 Leu188Val und Missense F 2 C>T Pro81Ser c.806 T>A Leu198Gln Missense F 1 Deletion Exon 1 Deletion T 2 Deletion Exon 2 Deletion T 5 Deletion Exon 3 Deletion T 5 Deletion Exon 1-2 Deletion T 2 Deletion Exon 2-3 Deletion T 1 Deletion Exon 1-3 Deletion T 11 Tab. 4.1: Auflistung der 52 Mutationen. In Spalte eins ist die Veränderung des jeweiligen Nukleotids aufgeführt. Der Mutationsname gibt an, an welcher Position im VHL-Protein eine Aminosäure ausgetauscht wird. 4.2 MR-Tomographien Zunächst wurde die jeweils aktuellste MR-Tomographie der Patienten ausgewertet. 194 der 228 untersuchten Patienten (85 %) waren nach MRT-Standardprotokoll untersucht worden. In 13 Fällen (6 %) lag statt einer sagittalen eine coronare Ebene, in 4 Fällen (2 %) ausschließlich eine transversale Ebene vor. In 3 Fällen (1 %) waren die Schichten dicker (bis 7,5 mm) und in 14 Fällen (6 %) dünner als 6 mm (1 – 5 mm). Der Zeitraum der Untersuchungen erstreckte sich von März 1989 bis März 2006. 4 MRT (2 %) stammten aus den Jahren 1989 bis 1995, 44 MRT (19 %) von 1996 bis 2000 und 180 MRT (79 %) aus den Jahren 2001 bis 2006. Bei 143 Patienten waren Voraufnahmen aus den Jahren 1988 bis 2005 vorhanden. 4 Ergebnisse 25 4.3 ELS-Tumore 4.3.1 Retrospektive Analyse der 6 mm Standardsequenzen des Zerebrums Es wurden 228 MR-Tomographien von 228 von Hippel-Lindau Patienten analysiert. 6 der 228 Patienten zeigten 8 Läsionen, welche die in Kapitel 3.3.1 genannten Kriterien eines ELS-Tumors erfüllten. Die Prävalenzrate, also die Anzahl der Erkrankten mit ELS-Tumoren im Verhältnis zur Gesamtzahl der untersuchten von Hippel-Lindau Patienten beträgt 6 aus 228 oder 2,6 %. 2 der 6 Patienten waren bilateral betroffen. Bei 3 Patienten waren 4 Tumore komplett oder partiell reseziert, die Diagnose war jeweils histologisch gesichert, bei 3 Patienten waren die radiologischen Kriterien erfüllt. Eine Auflistung der 6 Patienten zeigt Tabelle 4.2. Initialen Alter Geschlecht Tumor Seite Größe [mm³] H., F. 37 m Histologisch gesicherter Links 4784 Radiologische Evidenz für Links 1320 ELST, Rezidiv- oder Resttumor K., H-G. 56 m ELST N., D. 29 m Histologisch gesicherter Rechts 1176 ELST, Rezidiv N., S. 26 w Bds. radiologische Beidseits Li: 8830 Evidenz für ELST P., L. 13 w Re: 6804 Radiologische Evidenz für Rechts 270 ELST S., J. 49 w Bds. histologisch gesicherter ELST, links Rezidiv Tab. 4.2: Auflistung der Patienten mit ELS-Tumoren. Beidseits Li: 6630 26 4 Ergebnisse ELS-Tumore waren in der MR-Tomographie nach primärer KM-Gabe T1-w inhomogen hyperintens, teils glatt, teils lobuliert abgrenzbar. Das mittlere Tumorvolumen betrug 4259 mm3. Bei 3 Patienten war eine Verlaufsbeobachtung der ELS-Tumore möglich, der Beobachtungszeitraum lag zwischen 7 und 91 Monaten. Abb. 4.2 zeigt das Tumorvolumen in Relation zum Beobachtungszeitraum. Patient H.F. wurde nach 66 Monaten operiert. Eine Auswertung der Wachstumskinetik der Tumore ist aufgrund der geringen Anzahl nicht möglich. Geringere Schwankungen der Tumorvolumina sind am ehesten auf Messfehler zurückzuführen, da die Tumore oft eine sehr irreguläre Begrenzung zeigen. Abb. 4.2 Größenänderung der ELS-Tumore bei 3 Patienten im zeitlichen Verlauf. 4.3.2 Analyse der 3 mm Felsenbeinsequenzen in Fettsättigungstechnik Die 3 mm dicken Sequenzen der Felsenbeine wurden in T1-Wichtung, nach Kontrastmittelgabe in Fettsättigungstechnik akquiriert. Die Auswertung erfolgte durch zwei unabhängige Befunder. Es wurden 60 MRT von 60 Patienten untersucht. Festgehalten wurde, ob der Saccus endolymphaticus je Felsenbein abgrenzbar ist, ob im Vergleich zum Labyrinth ein hyperintenses Signal zu erkennen ist und ob zusätzlich ein Tumor vorliegt. Das Ergebnis der Befunder A und B zeigt Tabelle 4.3. 4 Ergebnisse 27 Der Aquaeductus vestibuli war in 74 % bzw. 58 % (Befunder A / Befunder B) abgrenzbar und zeigte in 23 % bzw. 28 % ein hyperintenses Signal. Analyse von 60 Befunder A Befunder B Beidseits 40 27 Rechts 4 13 Links 5 3 Nicht abgrenzbar 11 17 Beidseits 9 11 Rechts 4 7 Links 6 5 Keine KM-Anreicherung 41 37 Beidseits 0 0 Rechts 0 0 Links 2 2 Keine Raumforderung 58 58 3mm-Felsenbein-MRT Abgrenzbarkeit des Saccus endolymphaticus KM-Anreicherung des Saccus endolymphaticus Tumor im Saccus endolymphaticus Tabelle 4.3: Zweifachauswertung von 60 MRT der Felsenbeine mit 3 mm Schichtdicke, T1-w mit KM und Fettsättigung durch zwei unabhängige Befunder. Vergleicht man die unabhängig voneinander erhobenen Ergebnisse der einzelnen Patienten direkt miteinander, so ergibt sich für die Abgrenzbarkeit des Aquaeductus vestibuli eine Übereinstimmung von 73 %, für das Vorliegen eines hyperintensen Signals eine Übereinstimmung von 88 % und für das Vorliegens eines Tumors eine Übereinstimmung von 100 %. Tabelle 4.4 fasst die Daten zusammen. Abbildung 4.2 zeigt jeweils ein Beispiel eines gut abgrenzbaren Aquaeductus vestibuli mit normaler und hoher Signalintensität. 4 Ergebnisse 28 Übereinstimmung Abgrenzbarkeit Diskrepanz 87 (120) 73 % 33 (120) 27 % 106 (120) 88 % 14 (120) 12 % 120 (120) 100 % 0 (120) 0% Aquaeductus vestibuli KM-Anreicherung Aquaeductus vestibuli Raumforderung im Aquaeductus vestibuli Tabelle 4.4: Rate der Übereinstimmung bzw. der Diskrepanz der beiden unabhängigen Untersucher A und B bei der Beurteilung des Aquaeductus vestibuli. Abb. 4.2: Aquaeductus vestibuli in (a) bds. isointens zum Labyrinth gut abgrenzbar, in (b) rechts hyperintens. (a) und (b) MRT, T1-w mit Gad-Chelat, Fettsättigung, transversal. 4.3.3 Vergleich der 6 mm Standardsequenzen mit den 3 mm Felsenbeinsequenzen Alle drei ELS-Tumore, welche auf den 3 mm Dünnschichtsequenzen der Felsenbeine detektiert wurden, waren auch auf den 6 mm Standardsequenzen eindeutig abgrenzbar. Keiner der ELS-Tumoren war nur auf den Dünnschichtaufnahmen zu erkennen. Die Prävalenzrate für ELS-Tumore beträgt für 6 mm Sequenzen 6 von 228 oder 2,6 %, für 3 mm Sequenzen 2 von 60 oder 3,3 %. Der Unterschied ist erwartungsgemäß statistisch nicht signifikant (p = 0,65). 29 4 Ergebnisse 4.3.4 Fallberichte der Patienten mit ELS-Tumoren Patient H., F., 37 Jahre Die VHL-Erkrankung wurde 1994 mit drei zerebellären Hämangioblastomen manifest. Im Verlauf wurden multiple zerebelläre und spinale Hämangioblastome, beidseitige Nierenzellkarzinome und retinale Angiome diagnostiziert und therapiert. Seit 1998 war eine Raumforderung im linken Felsenbein bekannt, welche im Mai 2003 aufgrund der Größenzunahme mittels gehörerhaltender partieller Petrosektomie subtotal reseziert wurde. Die histologische Untersuchung ergab einen ELS-Tumor. MRT Verlaufskontrollen zeigten postoperativ eine langsam progrediente, T1-w hyperintense Raumforderung in der Hinterwand des linken Os petrosum, einem Rezidiv- oder Resttumor entsprechend. Zum Zeitpunkt der aktuellsten MRT (August 2005) bestand eine Anakusis links. Abb. 4.3: ELS-Tumor des linken Felsenbeins und Hämangioblastom der linken Kleinhirnhemisphäre mit angrenzendem erweitertem Gefäß. (a) bis (c) präoperative Darstellung des ELS-Tumors (schwarzer Pfeil) im Januar 2003 mit Destruktion der Felsenbeinhinterkante. Gut zu erkennen sind die intratumoralen knöchernen Spiculae (c). Kalottendefekt und Metallartefakte aufgrund vorangegangener Operationen. Benachbartes Hämangioblastom der linken Kleinhirnhemisphäre (weißer Pfeil). (d) Der ELS-Tumor ist im Januar 2004 subtotal reseziert (Pfeilspitzen). (e) Geringe Größenzunahme des Resttumors im August 2005, das Hämangioblastom wurde zwischenzeitlich entfernt. (a) MRT, T1-w mit Gad-Chelat, transversal, (b) CT mit Kontrastmittel, transversal, (c) CT im Knochenfenster, transversal, (d) und (e) MRT, T1-w mit Gad-Chelat, transversal 30 4 Ergebnisse Patient K., H-G., 56 Jahre Die VHL-Erkrankung wurde erkannt, nachdem 1999 ein zerebelläres Hämangioblastom reseziert wurde und im darauf folgenden Jahr eine Nephrektomie bei Nierenzellkarzinom durchgeführt wurde. Im Januar 2002 erfolgte eine Resektion einer thorakalen intramedullären Metastase und im Juli 2002 eine Resektion einer pulmonalen Filia. Der Patient stellte sich im August 2002 zur Beratung und MRDiagnostik erstmals in Freiburg vor. In der Bildgebung sah man zerebelläre und spinale Tumore, wobei nicht sicher zwischen Hämangioblastomen und Metastasen des Nierenzellkarzinoms differenziert werden konnte. Eine 20 x 12 x 11 mm große, T1-w hyperintense Raumforderung im Aquaeductus vestibuli des linken Felsenbeins wurde nach radiologischen Kriterien als ELS-Tumor eingestuft, da eine aktuelle Skelettszintigraphie keine Speicherung zeigte, und somit eine ossäre Metastasierung unwahrscheinlich war. Bei fortschreitendem Tumorprogress verstarb der Patient Januar 2003. Abb. 4.4: Raumforderung im linken Felsenbein, welche nach radiologischen Kriterien als ELS-Tumor eingestuft wurde. (a) ELS-Tumor mit typischem hyperintensem Signal (schwarzer Pfeil). Die hyperintense Struktur im rechten Os petrosum entspricht einem Anschnitt des Sinus sigmoideus (weißer Pfeil). (b) ELS-Tumor isointens zum Hirnparenchym, beidseitige Ödeme der Kleinhirnhemisphären bei multiplen zerebellären Raumforderungen (nicht abgebildet). (a) MRT, T1-w mit Gad-Chelat, transversal (b) MRT, T2-w, transversal 31 4 Ergebnisse Patient N., D., 29 Jahre Der Patient N., D. ist der Bruder der Patientin N., S.. Es liegen nicht behandlungsbedürftige zerebelläre und spinale Hämangioblastome vor. Retinale Angiome wurden beidseits laserkoaguliert, Pankreaszysten und beidseitige Nierenzysten sind bekannt. 1997 wurde rechts ein ELS-Tumor mittels Mastektomie und Labyrinthektomie entfernt. Zu diesem Zeitpunkt war der Patient seit ca. sieben Jahren auf der betroffenen Seite ertaubt. Die postoperativen MRT Verlaufskontrollen zeigten rechts ein größenprogredientes ELS-Tumorrezidiv, welches im September 2002 reseziert wurde. Histologisch konnte das Rezidiv bestätigt werden. Abb. 4.5: Histologisch gesichertes Rezidiv eines ELS-Tumors des rechten Felsenbeins. (a) bis (c) zeigen den hyperintensen ELS-Tumor an der rechten Felsenbeinhinterkante (schwarzer Pfeil), anterolateral davon eine Zyste (weißer Pfeil) und lateral fettäquivalente postoperative Veränderungen nach subtotaler Petrosektomie (Pfeilspitze). (a) MRT, T1-w mit Gad-Chelat, transversal, (b) MRT, T1-w mit Gad-Chelat, coronar, (c) MRT, T2-w, transversal. 32 4 Ergebnisse Patientin N., S., 26 Jahre Die Patientin (Schwester von Patient N., D.) wurde bereits im Alter von 13 Jahren mit morgendlichem Erbrechen, Schwindel und einer Netzhautablösung des rechten Auges symptomatisch. 1982 erfolgte im Alter von 14 Jahren die Resektion eines 3 cm großen Hämangioblastoms der Medulla oblongata. Viszeral wurden Pankreasund Nierenzysten diagnostiziert. In den nachfolgenden Jahren wurden mehrfach Hämangioblastome des Kleinhirns, des kraniozervikalen Übergangs und des Rückenmarks operiert. Es kam zu zunehmenden neurologischen Ausfällen mit spastischer Hemiparese und bilateralen Hirnnervenausfällen. Am linken Auge wurden mehrfach retinale Angiome laserkoaguliert. 1989 wurden erstmals Raumforderungen beider Felsenbeine beschrieben. Zu diesem Zeitpunkt bestand rechts eine Taubheit und links eine Schwerhörigkeit von 20 dB. Die Tumore zeigten bis 1993 eine Größenzunahme. Mittlerweile war die Patientin beidseits ertaubt, klagte jedoch über Ohrgeräusche. Aufgrund des schlechten Allgemeinzustandes wurde auf weitere Operationen verzichtet, die Patientin verstarb 1994 im Alter von 26 Jahren. Abb. 4.6: Beidseitige Raumforderungen der Felsenbeine und zystisches Hämangioblastom der linken Kleinhirnhemisphäre. (a) MRT aus dem Jahr 1989 mit nativ hyperintensen Arealen der ELS-Tumore (schwarze Pfeile), Hämangioblastomzyste (weißer Pfeil). (b) Nach Kontrastmittelgabe Anreicherung der ELS-Tumore (schwarze Pfeile). (c) Deutliche Größenzunahme der Tumore bis 1993, jetzt mit Hirnstammkompression. Der linke Tumor wächst in die mittlere Schädelgrube ein und durchbricht die Schädelkalotte (Pfeilspitzen). Das Hämangioblastom wurde zwischenzeitlich reseziert. (a) MRT, T1-w nativ, transversal, (b) MRT, T1-w mit Gad-Chelat, transversal, (c) CT mit Kontrastmittel, transversal. 33 4 Ergebnisse Patientin P., L., 13 Jahre Die beschwerdefreie Patientin kam im Rahmen eines Familien-Screenings zur MRDiagnostik. Abdominal war eine große Pankreaszyste bekannt, zentralnervöse Hämangioblastome konnten nicht nachgewiesen werden. Im rechten Felsenbein fand sich eine 9 x 6 x 10 mm große, T1-w hyperintense Raumforderung im Aquaeductus vestibuli, welche den radiologischen Kriterien eines ELS-Tumors entsprach. Eine operative Therapie wurde bisher nicht durchgeführt. Abb. 4.7: Tumor an der rechten Felsenbeinhinterkante mit Ausdehnung in den Aquaeductus vestibuli und Saccus endolymphaticus. (a) und (b) MRT, T1-w mit Gad-Chelat, transversal, (c) MRT, T1-w mit Gad-Chelat, sagittal. Patientin S., J., 49 Jahre Als Manifestation der von Hippel-Lindau Erkrankung sind zerebelläre und spinale Hämangioblastome, retinale Angiome und beidseitige Nierentumore bekannt. Bereits 1984 wurden beidseits invasiv wachsende Tumore der Felsenbeine reseziert. Initial wurden sie histologisch als Metastasen eines Schilddrüsenkarzinoms diagnostiziert. Die darauf folgende Thyroidektomie erbrachte keinen Primärtumor. Eine Reevaluierung der histologischen Ergebnisse ergab beidseits Tumore des Endolymphatischen Sacks. Bei beidseitiger Ertaubung wurde 1994 rechts ein Cochlear-Implant eigesetzt, welches im Verlauf gegen eine Hirnstammelektrode (auditory brainstem implant, ABI) ausgetauscht wurde. Letztere wurde bei Funktionsverlust aufgrund einer aufsteigenden neuronalen Degeneration entfernt. Die vorliegenden MR-Tomographien zeigen rechts einen postoperativen Defekt nach Petrosektomie. Links ist ein seit 2001 langsam progredientes Rezidiv des ELSTumors zu erkennen. 34 4 Ergebnisse Abb. 4.8: Defektzustand nach beidseitiger ELS-Tumor Operation. (a) Das CT der Schädelbasis von April 2000 zeigt eine rechtsseitige Mastoidektomie und eine linksseitige Petrosektomie. Rechts sind Kabel der Hirnstammelektrode zu erkennen. (b) und (c) ELS-Tumorrezidiv des linken Felsenbeins im Juni 2002 (Pfeile). Rechts besteht kein Hinweis auf ein Tumorrezidiv. (a) CT im Knochenfenster, transversal, (b) MRT, T1-w mit Gad-Chelat, transversal, (c) MRT, T2-w, transversal. 4.3.5 Mutationen der Patienten mit ELS-Tumoren Bei allen sechs Patienten mit ELS-Tumoren ist die Mutation bekannt. Zwei Patienten stammen aus einer Familie, die restlichen sind nicht miteinander verwandt. Die Mutationen im Einzelnen zeigt Tabelle 4.5. Es handelt sich überwiegend um T-Typ Mutationen (zwei große Deletionen, zwei Spilce-site-Defekte) und zwei F-Typ Mutationen (eine Missense-Mutation und eine In-frame-Deletion). Nukleotid-Austausch Mutationstyp Deletion Exon 3 Deletion c.605A>C Missense c.676+2T>C Splice-site-Defekt * c.676+2T>C Splice-site-Defekt * c.437-9delTCT In-frame-Deletion Deletion Exon 3 Deletion Tab 4.5: Mutationen der 6 Patienten mit ELS-Tumor. Die mit * gekennzeichneten Patienten stammen aus einer Familie. 35 4 Ergebnisse 4.4 Zentralnervöse Hämangioblastome 4.4.1 Häufigkeit zerebraler Hämangioblastome Es wurden MR-Tomographien von 228 gesicherten von Hippel-Lindau Patienten retrospektiv auf das Vorliegen von zerebralen Hämangioblastomen untersucht. Zwei Patienten wurden ausgeschlossen, da neben Hämangioblastomen auch Metastasen eines Nierenzellkarzinoms vorlagen und die Differenzierung der Tumore anhand der MR-Tomographien nicht sicher möglich war. Es wurden bei 103 von 226 Patienten zerebrale Hämangioblastome oder Parenchymdefekte nach stattgehabter Tumorresektion gefunden. Das entspricht 46 % der untersuchten Patienten. 41 % der Patienten hatten Tumore des Kleinhirns, 17 % Tumore des Hirnstamms und 5 % Tumore des Großhirns. Von den 103 Patienten mussten sich 70 Patienten im Laufe ihrer Erkrankung einer zerebralen Tumorresektion unterziehen (67 %). Es lagen 455 zerebrale Hämangioblastome vor. Die Anzahl der Tumore pro Patient reicht von 1 bis 24 Tumoren. Die Verteilung der Tumorhäufigkeit zeigt Abbildung 4.9. Abb. 4.9: Häufigkeit zerebraler Hämangioblastome. 103 von Hippel-Lindau Patienten hatten 455 zerebrale Tumore. 4 Ergebnisse 36 Im arithmetischen Mittel hatten die Patienten 4,4 Tumore, der Median lag bei 3 Tumoren. 86 der 103 Patienten (84 %) hatten multiple Tumore, 97 Patienten (94 %) hatten 1 bis 5 Tumore. Bei 402 der 455 Hämangioblastome war die Lokalisation bekannt. Bei den restlichen Patienten lag ein postoperativer Defekt vor, der keinen sicheren Rückschluss auf die Tumorlokalisation zuließ. 97 % der Tumore lagen infratentoriell. Die supratentoriellen Tumore lagen jeweils oberflächlich pial. Die genaue Lokalisation der Tumore zeigt Tab. 4.6. Es bestand keine Seitenpräferenz (169 Tumore rechts, 169 Tumore links, 49 Tumore median, 15 Defekte ohne sichere Seitenzuordnung). Lokalisation Anzahl der Tumore Infratentoriell Relativer Anteil 389 97 % 300 75 % Vermis cerebelli 41 10 % Hirnstamm 48 12 % Supratentoriell 13 3% Frontal 3 <1% Parietal 3 <1% Temporal 2 <1% Thalamus 1 <1% Colliculus superior 1 <1% Sella 1 <1% Hypophysenstiel 1 <1% Chiasma opticum 1 <1% Kleinhirnhemisphäre Tab. 4.6: Lokalisation zerebraler Hämangioblastome. Ausgewertet wurden 402 Tumore. 402 Tumore konnten nach der in Kapitel 1.3.2 genannten Einteilung nach Richard zugeordnet werden (Richard et al. 1998). Am häufigsten zeigte sich ein rein solider Tumor (Typ 3), gefolgt von Tumorknoten mit angrenzender Zyste (Typ 2) und Tumoren mit intratumoralen Zysten (Typ 4). Eine einfache Zyste ohne Tumorknoten (Typ 1) kam nicht vor. Tabelle 4.7 zeigt die Häufigkeit der einzelnen Tumortypen. 12 % aller Tumore waren zystisch (Typ 2 und 4). 4 Ergebnisse 37 Tumortyp Zahl der Tumore Relativer Anteil Typ 1 0 0% Typ 2 42 10,5 % Typ 3 354 88,1 % Typ 4 6 1,5 % 402 100 % Gesamt Tab. 4.7: Häufigkeit solider und zystischer Hämangioblastome. Typ 1: einfache Zyste, Typ 2: Tumor mit angrenzender Zyste, Typ 3: solider Tumor, Typ 4: Tumor mit intratumoralen Zysten. 4.4.2 Größe zerebraler Hämangioblastome und Operationshäufigkeit Zerebrale Hämangioblastome können winzige Knötchen oder große raumfordernde Tumore mit Kompression benachbarter Strukturen darstellen. Die Tumorgröße konnte bei 386 Tumoren bestimmt werden. Bei Tumoren mit Zysten wurde der Gesamttumordurchmesser gemessen. Der minimale messbare Tumordurchmesser lag bei 2 mm, somit betrug das kleinste Tumorvolumen nach der Näherungsformel für Ellipsoide 4 mm3. Im arithmetischen Mittel lag das Tumorvolumen bei 1807 mm3, der Median jedoch bei nur 40 mm3. Dieser Unterschied beruht darauf, dass einzelne Tumore sehr groß sind. Zur exakteren Vergleichbarkeit der Tumoren wurde in der Auswertung jeweils das Tumorvolumen herangezogen. Um eine bessere Vorstellung von der Größe der Tumore zu erhalten, zeigt Tabelle 4.8 den Zusammenhang zwischen Durchmesser und Volumen einer kugelförmigen Struktur. So entspricht ein Tumorvolumen von 40 mm3 dem Volumen einer Kugel mit 4,3 mm Durchmesser. Kugelvolumen Kugeldurchmesser 3 63 mm 5 mm 500 mm3 10 mm 1688 mm3 15 mm 3 20 mm 3 25 mm 3 30 mm 3 35 mm 3 40 mm 3 45 mm 4000 mm 7813 mm 13500 mm 21438 mm 32000 mm 45563 mm Tabelle 4.8: Gegenüberstellung von Kugelvolumen und Kugeldurchmesser nach der Näherungsformel (a * b * c) / 2. 38 4 Ergebnisse Die meisten Tumore waren relativ klein. 79 % der zerebralen Hämangioblastome hatten ein Tumorvolumen von maximal 500 mm3 (Kugeldurchmesser 10 mm). Der größte Tumor hatte ein Volumen von 58212 mm3, einem Kugeldurchmesser von 49 mm entsprechend. Die Verteilung der Tumorgrößen veranschaulicht Abbildung 4.10. Abb. 4.10: Größenverteilung zerebraler Hämangioblastome. 97 % der Tumore waren maximal 500 mm3 groß. Von Hippel-Lindau Patienten entwickeln oft multiple zerebrale Hämangioblastome. Tumore werden in der Regel erst reseziert, wenn sie Symptome verursachen. Gelegentlich werden kleine Tumore, welche im Zugangsweg von größeren Tumoren liegen, ebenfalls entfernt. Von 455 Tumoren sind 126 operativ entfernt worden (28 %). Die restlichen wurden durch MR-Tomographien beobachtet. Die präoperative Größe des Tumors war bei 76 Tumoren messbar. Bei 50 Tumoren war nur der postoperative Defekt dokumentiert. Die präoperative Tumorgröße lag im arithmetischen Mittel bei 7756 mm3 (4 – 58212 mm3), im Median bei 1624 mm3. 310 Tumore, die nicht operiert wurden, konnten ausgewertet werden. Das mittlere Tumorvolumen betrug in dieser Gruppe 348 mm3 (Median 23 mm3), die Spanne reichte von 4 – 21285 mm3. Tumore, welche operativ entfernt wurden, waren im Durchschnitt mehr als zehnmal größer als Tumore, welche beobachtet wurden. 4 Ergebnisse 39 Abbildung 4.11 veranschaulicht das Verhältnis von operierten zu nicht operierten Tumoren in Relation zu ihrer Größe. Abb. 4.11: Anteil resezierter zerebraler Hämangioblastome an der Gesamtzahl der Tumore in Abhängigkeit von der Tumorgröße. Zwischen 4 mm3 und 500 mm3 wurden 22 Tumore operativ entfernt und 283 Tumore beobachtet (7,2 % OP), über 500 mm3 wurden 54 Tumore reseziert und 27 beobachtet (66,7 % OP). Tumore > 500 mm3 wurden signifikant häufiger operiert, p ≤ 0.000001. Die Zahlen im Einzelnen zeigt Tabelle 4.9. Tumorgröße [mm3] 4 – 63 Zahl der operierten Zahl der beobachteten Tumore Tumore Relativer Anteil OP 7 216 3% 64 – 500 15 67 18 % 501 – 1688 17 15 53 % 1689 – 4000 10 8 56 % 4001 – 7813 4 1 80 % 7814 – 13500 5 0 100 % 18 3 86 % > 13500 Tab. 4.9: Auflistung der operierten und unter Beobachtung stehenden Tumore in Relation zu ihrer Gesamtgröße. 4 Ergebnisse 40 4.4.3 Einfluss der Tumorlokalisation Das Ausmaß raumfordernder Effekte hängt nicht nur von der Tumorgröße ab, sondern auch von der Lokalisation. Es ist anzunehmen, dass Tumore des Hirnstamms durch Kompression früher zu Symptomen führen als Tumore der Kleinhirnhemisphären oder des Großhirns. Um zu analysieren, ob Tumore an unterschiedlichen Lokalisationen unterschiedliche Größen zeigen, wurde die mittlere Tumorgröße in Abhängigkeit von der Lokalisation in Tabelle 4.10 aufgelistet. Tumore des Kleinhirns waren im Mittel am größten, gefolgt von Tumoren des Hirnstamms und des Großhirns. Lokalisation Kleinhirn Anzahl Mittleres Tumore Tumorvolumen 325 Standard- Median Minimum Maximum abweichung 3 6910 mm3 32 mm3 4 mm3 58212 mm3 1987 mm Hirnstamm 48 903 mm3 2077 mm3 67 mm3 4 mm3 10962 mm3 Großhirn 13 634 mm3 1224 mm3 108 mm3 4 mm3 4554 mm3 Tab 4.10: Durchschnittliches Tumorvolumen in Bezug zur Tumorlokalisation. Tabelle 4.11 zeigt den Einfluss der Tumorlokalisation auf die Operationshäufigkeit. Tumore des Hirnstamms wurden signifikant häufiger operiert als Tumore des Kleinhirns (p ≤ 0,05). Die mittlere Tumorgröße bei OP war kleiner als bei Tumoren des Kleinhirns. Das deutet darauf hin, dass Hämangioblastome des Hirnstamms häufiger und früher reseziert werden. Die Zahl der Tumore des Großhirns ist zu gering für eine statistische Auswertung. Lokalisation Kleinhirn Hirnstamm Großhirn operierte Tumore beobachtete Tumore Relativer Anzahl Anzahl Anteil OP Größe 59 9317 mm3 15 3 2 2501 mm 3 1143 mm Größe 266 361 mm3 18,4 % 33 3 31,3 % 3 15,4 % 11 176 mm 542 mm Tab. 4.11: Anzahl der operativ entfernten Tumore im Verhältnis zur Anzahl der unter Beobachtung stehenden Tumore in Abhängigkeit von der Lokalisation der Tumore. Angabe der Tumorgröße vor OP oder zum Zeitpunkt der aktuellsten MR-Tomographie (Arithmetisches Mittel). 4 Ergebnisse 41 4.4.4 Einfluss des Tumortyps 12 % der zerebralen Hämangioblastome haben tumorassoziierte Zysten (Typ 2 und 4). Zystische Tumore sind an allen Lokalisationen in etwa gleich häufig (siehe Tabelle 4.12). Es besteht kein signifikanter Unterschied zwischen Kleinhirn und Hirnstamm (p = 0,57) oder Hirnstamm und Großhirn (p = 0,51). Zystisch (Typ 2 und 4) Kleinhirn Solide (Typ 3) gesamt 40 11,7 % 301 88,3 % 341 Hirnstamm 7 14,6 % 41 85,4 % 48 Großhirn 1 7,8 % 12 92,3 % 13 48 11,9 % 354 88,1 % 402 gesamt Tab. 4.12: Häufigkeit zystischer Tumore in Relation zu ihrer Lokalisation. Zystische Tumoren (Typ 2 und 4) waren im Mittel 13021 mm3 groß, solide Tumoren (Typ 1) 289 mm3. Der Median betrug 8922 mm3, respektive 24 mm3. Der Unterschied ist hochsignifikant (p ≤ 0,000001). Eine Auflistung der Häufigkeit des Tumortyps in Abhängigkeit von der Tumorgröße zeigt Tabelle 4.13. Kugeldurchmesser Tumorvolumen 3 [mm ] [mm] Alle Tumore Zystische Tumore Solide Tumore (Typ 1 bis 4) (Typ 2 und 4) (Typ 3) 2–5 4 – 63 224 58,0 % 0 0% 224 58,0 % 6 – 10 64 – 500 81 21,0 % 3 0,8 % 78 20,2 % 11 – 15 501 – 1688 32 8,3 % 8 2,1 % 24 6,2 % 16 – 20 1689 – 4000 18 4,7 % 8 2,1 % 10 2,6 % 21 – 25 4001 – 7813 5 1,3 % 4 1,0 % 1 0,3 % 26 – 30 7814 – 13500 5 1,3 % 2 0,5 % 3 0,9 % > 30 > 13500 21 5,4 % 21 5,4 % 0 0% 386 100 % 46 11,9 % 340 88,1 % Gesamtzahl der Tumore Tab. 4.13: Häufigkeit zerebraler Hämangioblastome in Abhängigkeit von Größe und Tumortyp. Große Tumore wurden signifikant häufiger operiert als kleine Tumore. Zystische Tumore sind signifikant größer als solide Tumore. Erwartungsgemäß wurden zystische Tumore häufiger operiert als solide. Von 48 zystischen Tumoren wurden 35 4 Ergebnisse 42 Tumore reseziert (73 %), von 354 soliden Tumoren wurden nur 42 operativ entfernt (12 %). Der Unterschied ist hoch signifikant (p ≤ 0.00001). 4.4.5 Einfluss der Mutation Von 226 Patienten, die in die Analyse der zerebralen Hämangioblastome aufgenommen wurden, war bei 216 die Mutation bekannt. Die Mutationen wurden in zwei Gruppen aufgeteilt: F-Typ, wenn die Translation des Proteins in voller Länge zu erwarten ist, und T-Typ, wenn die Translation wahrscheinlich abbricht. Bei 161 Patienten lag der F-Typ vor, bei 55 Patienten der T-Typ. Betrachtet man das Auftreten zerebraler Hämangioblastome in Abhängigkeit vom Mutations-Typ, so ergibt sich ein hochsignifikanter Unterschied (p ≤ 0,00001) (siehe Tabelle 4.14). Mutation Patienten mit zerebralen Patienten ohne zerebrale Hämangioblastomen Hämangioblastome gesamt F-Typ 49 30,4 % 112 69,6 % 161 T-Typ 50 90,9 % 5 9,1 % 55 Tab 4.14: Anzahl der Patienten mit oder ohne zerebralen Hämangioblastomen in Abhängigkeit vom Mutations-Typ. Patienten mit zerebralen Hämangioblastomen und einer Mutation vom F-Typ hatten im Mittel 3,8 Tumore, Patienten mit einer Mutation vom T-Typ hatten im Mittel 5,2 Tumore. Von 455 Tumoren war bei 444 die Mutation bekannt. 184 Tumore hatten eine Mutation vom F-Typ und 260 Tumore eine Mutation vom T-Typ. Beide Mutationstypen kamen im Kleinhirn und im Hirnstamm in etwa gleich verteilt vor. Mutationen vom F-Typ waren im Großhirn signifikant häufiger als im Kleinhirn (p < 0,05) (siehe Tabelle 4.15). 4 Ergebnisse 43 Kleinhirn Hirnstamm Großhirn Anzahl TU Anzahl TU Anzahl TU gesamt F-Typ 133 81,1 % 22 13,4 % 9 5,5 % 164 T-Typ 201 87,0 % 26 11,3 % 4 1,7 % 231 Tab. 4.15: Anzahl zerebraler Hämangioblastome Tumore in Relation zu Mutations-Typ und Lokalisation. Hinsichtlich der Tumorgröße unterschieden sich die beiden Mutationstypen nicht. Tumore vom F-Typ maßen im Mittel 1739 mm3, Tumore vom T-Typ 1911 mm3. Der Median betrug 30 mm3 bzw. 50 mm3. Zystische Tumore waren bei beiden Mutationstypen gleich häufig. Beim F-Typ waren 18 von 164 Tumoren zystisch. Dies entspricht 11 %. Beim T-Typ waren 30 von 231 Tumoren zystisch, 13 % entsprechend. Der Unterschied war nicht signifikant (p = 0,55). Auch bezüglich der Operationshäufigkeit bestand kein Unterschied. 46 von 184 Tumoren wurden beim F-Typ entfernt (25 %), 76 von 260 Tumoren des T-Typs wurden reseziert (29 %). Das Signifikanzniveau lag bei p = 0,33. 4.4.6 Natürlicher Verlauf zerebraler Hämangioblastome Bei 257 Tumoren lag eine Verlaufsuntersuchung vor. Die Nachbeobachtungszeit lag zwischen einem und 168 Monaten, im Mittel bei 44,5 Monaten. Um Messfehler nicht fehl zu deuten, wurde als Progress mindestens eine Verdoppelung des Tumorvolumens definiert. Unter dieser Voraussetzung zeigten 112 der 257 Tumore einen Progress (44 %). Tabelle 4.16 veranschaulicht, wie das Tumorvolumen zunimmt, wenn die Durchmesser in allen drei Raumebenen um jeweils einen, zwei oder drei Millimeter zunehmen. Abbildung 4.12 zeigt die Anzahl der progredienten und nicht progredienten Tumore in Anhängigkeit von der Nachbeobachtungszeit. Bis zu einer Nachbeobachtungszeit von 20 Monaten zeigen 11 von 80 Tumoren (14 %) einen Progress. Über 20 Monaten Beobachtungszeit zeigen 101 von 177 Tumoren einen Progress (57 %). Dies erklärt sich am ehesten dadurch, dass die Tumore in der Regel langsam wachsen und erst nach längerer Beobachtung ein Progress nachweisbar ist. Der Anteil progredienter Tumore bleibt ab 20 Monaten Beobachtungszeit relativ konstant. 4 Ergebnisse 44 Zunahme der Durchmesser x, y und z AusgangsDurchmesser +1 mm +2 mm +3 mm 5 mm 73 % 174 % 310 % 10 mm 33 % 73 % 120 % 15 mm 21 % 46 % 52 % 20 mm 16 % 33 % 52 % 30 mm 10 % 21 % 33 % Tab. 4.16: Relative Zunahme des Tumorvolumens in Abhängigkeit von den Ausgangsdurchmessern. Abb. 4.12: Anzahl der Tumore mit und ohne Progress in Abhängigkeit vom Beobachtungszeitraum. 4.4.7 Einfluss von Lokalisation, Tumorgröße und Tumortyp auf das Wachstum Um den Einfluss von Lokalisation, Größe und Tumortyp auf den Progress zu prüfen, wurden jeweils nur Tumore mit einer Nachbeobachtungszeit von mindestens 20 Monaten berücksichtigt. Diese Voraussetzung traf auf 186 Tumore zu. 104 Tumore zeigten einen Progress, 82 waren stabil. 4 Ergebnisse 45 Den Anteil progredienter Tumore in Abhängigkeit von der Lokalisation zeigt Tabelle 4.17. Es bestand kein signifikanter Unterschied zwischen Tumoren des Kleinhirns und des Hirnstamms (p = 0,6), des Kleinhirns und des Großhirns (p = 0,5) und Tumoren des Hirnstamms und des Großhirns (p = 0,4). Progress Kein Progress gesamt Kleinhirn 92 56 % 73 44 % 165 Hirnstamm 10 63 % 6 37 % 16 2 40 % 3 60 % 5 Großhirn Tab. 4.17: Anzahl und relativer Anteil progredienter und stabiler Tumore in Abhängigkeit von der Tumorlokalisation. Um den Einfluss der Tumorgröße auf das Wachstum zu analysieren, wurde die Ausgangsgröße der Tumore herangezogen. Das mittlere Tumorvolumen betrug bei den progredienten Tumoren 71 mm3 (4 – 910 mm3) und bei den stabilen Tumoren 68 mm3 (4 – 1274 mm3). Beide Gruppen unterscheiden sich nicht. Tabelle 4.18 zeigt die Abhängigkeit des Wachstums zerebraler Hämangioblastome vom Tumortyp. Alle zystischen Tumore (Typ 2 und 4) zeigten bei einer Nachbeobachtungszeit von mindestens 20 Monaten einen Tumorprogress, während nur 52 % der soliden Tumore ein Wachstum zeigten. Der Unterschied ist signifikant (p < 0,0001). Progress Kein Progress gesamt Typ 2 14 0 14 Typ 3 88 82 170 Typ 4 2 0 2 Tab. 4.18: Anzahl der progredienten Tumore in Relation zum Tumortyp. Die minimale Nachbeobachtungszeit betrug 20 Monate. Um die Bedeutung des Tumorprogresses für den Patienten zu beurteilen, wurde analysiert, ob wachsende Tumore häufiger operiert wurden als stabile Tumore. Tab. 4.19 zeigt die Auflistung. Progrediente Tumore wurden signifikant häufiger operiert (p < 0,005). 4 Ergebnisse 46 OP TU-Progress Stabiler Tumor gesamt Keine OP gesamt 23 (22 %) 81 104 5 (6 %) 77 82 28 (15 %) 158 186 Tab. 4.19: Anteil operierter zerebraler Hämangioblastome in Abhängigkeit von der Wachstumstendenz. Progrediente Tumore wurden signifikant häufiger operiert als stabile Tumore. 4.4.8 Wachstumsgeschwindigkeit zerebraler Hämangioblastome Zerebrale Hämangioblastome können sich in ihrer Wachstumsgeschwindigkeit stark unterscheiden. Um zu analysieren, ob Faktoren wie Tumorlokalisation, Tumortyp oder Mutationstyp einen Einfluss auf das Wachstum haben, wurde die absolute Zunahme des Tumorvolumens aus der Differenz des maximalen und minimalen Tumorvolumens gebildet. Diese Werte wurden jeweils über die Nachbeobachtungszeit in einem Diagramm aufgetragen. Zur übersichtlicheren Darstellung wurde für „Zunahme des Tumorvolumens“ ein logarithmischer Maßstab gewählt. In den Abbildungen 4.13, 4.14 und 4.15 sind jeweils 112 zerebrale Hämangioblastome eingetragen, welche einen Progress zeigten (Siehe 4.4.4). Jeder Punkt steht für einen Tumor. Die Wachstumsrate in mm³/Monat errechnet sich aus dem Verhältnis von „Zunahme des Tumorvolumens“ zu „Nachbeobachtungszeit“. 4 Ergebnisse 47 Der Einfluss der Tumorlokalisation ist in Abbildung 4.13 dargestellt. Tumore des Kleinhirns, des Hirnstamms und des Großhirns sind in unterschiedlichen Farben abgebildet. Die durchschnittlichen Wachstumsraten zeigt Tabelle 4.20. Statistisch besteht kein Zusammenhang zwischen Tumorlokalisation und Wachstumsrate (p > 0,09, unpaarer t-Test). Abb. 4.13: Einfluss der Lokalisation zerebraler Hämangioblastome auf die Zunahme des Tumorvolumens in Relation zur Beobachtungszeit. Blaue Rauten symbolisieren Tumore des Kleinhirns, rote Quadrate Tumore des Hirnstamms und gelbe Dreiecke Tumore des Großhirns. Arith. Mittel Kleinhirn (n = 97) Standardabweichung Median Spanne 111,7 566,2 2,3 0,1 – 5310,2 Hirnstamm (n = 13) 13,9 27,3 1,1 0,4 – 103,3 Großhirn (n = 2) 14,8 4,6 14,8 10,2 – 19,3 Tab. 4.20: Durchschnittliche Wachstumsrate zerebraler Hämangioblastome in mm³/Monat an unterschiedlichen Lokalisationen. 4 Ergebnisse 48 Den Einfluss des Tumortyps veranschaulicht Abbildung 4.14. Zystische Tumore (Typ 2 und 4) finden sich eher im oberen Anteil des Diagramms und scheinen somit deutlicher an Volumen zuzunehmen als solide Tumore. Der Unterschied der durchschnittlichen Wachstumsraten ist jedoch statistisch nicht signifikant (p = 0,07, unpaarer t-Test) (siehe Tabelle 4.21). Abb. 4.14: Einfluss des Tumortyps zerebraler Hämangioblastome auf die Zunahme des Tumorvolumens in Relation zur Beobachtungszeit. Blaue Rauten symbolisieren solide Tumore, rote Quadrate zystische Tumore (Typ 2) und gelbe Dreiecke zystische Tumore (Typ 4). Arith. Mittel Standardabweichung Median Spanne Typ 2 (n = 17) 595,4 1242,3 151,9 2,9 – 5310,2 Typ 3 (n = 93) 8,3 20,3 1,2 0,1 – 123,9 Typ 4 (n = 2) 76,0 76,0 32,3 – 119,7 Tab. 4.21: Durchschnittliche Wachstumsrate zerebraler Hämangioblastome unterschiedlicher Tumortypen. 4 Ergebnisse 49 Den Einfluss des Mutationstyps auf das Wachstum veranschaulichen Abbildung 4.15 und Tabelle 4.22. Die Wachstumsraten zerebraler Hämangioblastome mit Mutationen vom F-Typ und T-Typ scheinen gleich verteilt zu sein. Es besteht kein signifikanter Unterschied der durchschnittlichen Wachstumsraten beider Gruppen (p = 0,6). Abb. 4.15: Einfluss des Mutationstyps zerebraler Hämangioblastome auf die Zunahme des Tumorvolumens in Relation zur Beobachtungszeit. Blaue Rauten symbolisieren Tumore mit Mutationen vom F-Typ, rote Quadrate Tumore mit Mutationen vom T-Typ. Arith. Mittel Standardabweichung Median Spanne F-Typ (n = 41) 146,9 817,9 0,9 0,1 – 5310,2 T-Typ (n = 67) 74,9 233,7 4,1 0,1 – 1296,3 Tab. 4.22: Durchschnittliche Wachstumsrate zerebraler Hämangioblastome mit Mutationen vom F-Typ oder T-Typ. 4 Ergebnisse 50 4.4.9 Neuauftreten zerebraler Hämangioblastome Von 455 zerebralen Hämangioblastomen sind 69 Tumore im Verlauf neu aufgetreten. Es wurden 66 Patienten mit zerebralen Tumoren verlaufsbeobachtet. Die gesamte Beobachtungszeit dieser Patienten betrug 3470 Monate. Daraus lässt sich errechnen, dass durchschnittlich 0,24 Tumore pro Jahr neu aufgetreten sind. Zum Zeitpunkt des Nachweises waren neu aufgetretene Tumore im Median 9 mm3 groß. Die Spanne reichte von 4 mm3 bis 12896 mm3. 4.4.10 Spinale Hämangioblastome Bei 214 Patienten lag neben der zerebralen auch eine MR-Tomographie des Spinalkanals vor. Von diesen 214 Patienten hatten 97 Patienten zerebrale Hämangioblastome (45 %), entsprechend der im vorangehenden Kapitel ermittelten Rate. 132 Patienten hatten spinale Hämangioblastome (62 %), 142 Patienten hatten zerebrale oder spinale Tumore (66 %). 87 Patienten hatten zerebrale und spinale Hämangioblastome (41 %), 45 Patienten nur spinale Tumore (21 %), 10 Patienten nur zerebrale Tumore (5 %). 72 Patienten hatten weder zerebrale noch spinale Tumore (34 %). Eine Übersicht zeigt Tabelle 4.23. Spinale Zerebrale Keine zerebralen Gesamtzahl Hämangioblastome Hämangioblastome Patienten 87 40,7 % 45 21,0 % 132 10 4,7 % 72 33,6 % 82 Hämangioblastome Keine spinalen Hämangioblastome Gesamtzahl 97 117 214 Patienten Tab. 4.23: Häufigkeit zerebraler und spinaler Hämangioblastome bei 214 von Hippel-Lindau Patienten. 51 5 Diskussion 5 Diskussion 5.1 ELS-Tumore ELS-Tumore führen durch ihr destruktives Wachstum oder lokale Einblutungen zu einer Hörminderung oder einem vollständigen Hörverlust des betroffenen Ohres. Im Falle bilateraler Tumore kann es zur Ertaubung kommen. Die Frage, wie häufig diese Tumore bei von Hippel-Lindau Patienten vorkommen ist für die Betroffenen von enormer Bedeutung, insbesondere, da durch retinale Angiome das Sehvermögen ebenfalls gefährdet ist. 5.1.1 Prävalenzrate der ELS-Tumore In der Literatur gibt es zwei Arbeiten, die jeweils Gruppen von über 100 von HippelLindau Patienten in Hinblick auf ELS-Tumore untersucht haben (Manski et al. 1997, Choo et al. 2004). Dabei fanden Manski et al. bei 13 von 121 Patienten Hinweise auf einen ELS-Tumor und Choo et al. bei 21 von 129 Patienten. Dies ergibt eine Prävalenzrate von 11% beziehungsweise 16%. Die Prävalenzrate von ELS-Tumoren ist in der vorliegenden Arbeit mit 2,6% signifikant niedriger (p ≤ 0,001). In der Studie von Choo et al. wurden von Hippel-Lindau Patienten mit und ohne ELSTumore hinsichtlich ihrer subjektiven und objektiven Symptome und deren natürlicher Verlauf untersucht. Eingeschlossen wurden Patienten, welche im Rahmen eines multidisziplinären Screenings prospektiv untersucht wurden. Eine Angabe des Zeitrahmens erfolgte nicht, auch wurde nicht erwähnt, ob bereits publizierte Fälle in der Arbeit enthalten sind. Patienten, bei denen die von Hippel-Lindau Erkrankung nicht bestätigt werden konnte, wurden als Kontrollgruppe herangezogen. Die Prävalenz von ELS-Tumoren war nicht primäres Studienziel und wurde von den Autoren nicht direkt aus den Ergebnissen abgeleitet. Da ein systematischer Fehler zugunsten otologisch symptomatischer Patienten nicht ausgeschlossen werden kann, darf aus den publizierten Zahlen keine Prävalenzrate abgeleitet werden. Die Arbeit von Manski et al. hatte die Prävalenz der ELS-Tumore als primäres Studienziel, sodass im Folgenden diskutiert wird, in wieweit systematische Unterschiede wie Bildqualität, Patientenselektion oder genetische Unterschiede der 52 5 Diskussion Patientenkollektive die Diskrepanz mit den Ergebnissen der vorliegenden Arbeit erklären können. 5.1.1.1 Einfluss der Bildqualität Zum Nachweis von ELS-Tumoren sind Schnittbildverfahren wie MRT oder CT geeignet. Bei beiden Verfahren hängt der Tumornachweis entscheidend von der Bildqualität ab. Diese wird in erster Linie von Akquisitionsparametern wie Schichtdicke und Bildmatrix bestimmt. Zudem sind für die MR-Tomographie Kontrast beeinflussende Faktoren wie Echozeit (TE), Repetitionszeit (TR), Feldstärke, „Numbers of excitations“ (NEX), Fettsuppression, Kontrastmittelgabe und in der Computer-Tomographie der Rekonstruktionsalgorithmus und die applizierte Dosis wichtig. In der vorliegenden Arbeit lagen zu 85 % standardisierte T1-w MR-Tomographien, durchgeführt an einem 1,5 T Gerät in 6 mm Schichtdicke biplanar nach Kontrastmittelgabe vor. In 15 % bestanden, wie in Kapitel 4.2 aufgeführt, geringe Abweichungen vom Standardprotokoll. In der Arbeit von Manski et al. wird angegeben, dass MR-Tomographien aus den Jahren 1988 bis 1994 retrospektiv analysiert wurden. Die Aufnahmen stammten von einem 0,5 T Gerät und lagen in 5 mm Schichtdicke vor und nach Kontrastmittelgabe vor. Mit dieser Technik wurden 10 der 13 ELS-Tumor Patienten identifiziert. Als ELS-Tumor wurde dabei eine Läsion gewertet, die an der Hinterwand des Os petrosum lokalisiert war und ein hohes oder intermediäres Signal vor oder nach KM-Gabe zeigte. Patienten, bei denen aus der Krankengeschichte eine Hörminderung oder Schwindel bekannt waren, wurden nachträglich gezielt mittels 3 mm Felsenbein-MRT und 1,5 mm Felsenbein-CT nachuntersucht. So konnten drei weitere Patienten identifiziert werden. Eine Auflistung der Untersuchungsparameter im Einzelnen zeigt Tabelle 5.1. 5 Diskussion 53 Manski et al. Manski et al. Freiburg Freiburg Screening Nachuntersuchung Screening Felsenbein MR T1-w MR T1-w FS Modalität MR T1-w MT T1-w CT Zeitraum 1988 – 1984 k. A. 1989 - 2006 2005 - 2006 Patientenzahl 121 30 228 60 Patient mit 10 3 6 2 ELS-Tumor Untersuchte Schädel Felsenbein Felsenbein Schädel Felsenbein Feldstärke /mA 0,5 T 0,5 oder 1,5 T 170 mA 1,5 T 1,5 T FOV k. A. 200 mm 130 mm 220 mm 230 mm Matrix k. A. 192 x 256 k. A. 202 x 256 222 x 256 Schichtdicke 5 mm 3 mm 1,5 mm 6 mm 3 mm ohne KM Ja Ja Ja Nein Nein mit KM Ja Ja Nein Ja Ja Region Tab. 5.1: Auflistung der technischen Parameter des in dieser Arbeit und von Manski et al. durchgeführten ELS-Tumor Screenings. T = Tesla, k. A. = keine Angaben. Vergleicht man die Screening-Protokolle, so sind die Aufnahmen von Manski et al. mit geringerer Feldstärke akquiriert. Dies ergibt weniger Signal und führt zu einer schlechteren Bildqualität. Andererseits wird dieser Effekt durch eine etwas geringere Schichtdicke teilweise ausgeglichen. Man darf daher annehmen, dass die Bildqualität der Screening-Protokolle annähernd gleich ist, bzw. dass die Bildqualität der retrospektiven Analyse von Manski et al. der Bildqualität dieser Arbeit nicht überlegen ist. Der Unterschied der Prävalenzrate von ELS-Tumoren (6/228 versus 13/121) ist statistisch signifikant (p ≤ 0,011) und nicht durch eine unterschiedliche Bildqualität zu erklären. Die Tatsache, dass bei Manski et al. von Hippel-Lindau Patienten mit Hörminderung oder vestibulären Symptomen gezielt nachuntersucht wurden, und dadurch 3 weitere ELS-Tumoren entdeckt wurden, kann zu einem systematischen Fehler zugunsten symptomatischer Patienten führen. Der Unterschied der Prävalenzrate bleibt jedoch statistisch signifikant, wenn man nur die Patienten des Screenings vergleicht (6/228 versus 10/121, p ≤ 0,017). 54 5 Diskussion 5.1.1.2 MR-Tomographie der Felsenbeine Im Standard-Screening wurden 6 mm Schichten gemessen. Der Saccus/Ductus endolymphaticus misst im Verlauf durch den Aquaeductus vestibuli, dort wo die Tumorentstehung vermutet wird, durchschnittlich 1,5 mm. Obwohl das umgebende Gewebe aus Knochen und Luft besteht und folglich kein Signal gibt, ist es denkbar, dass die kleine Struktur des Saccus/Ductus endolymphaticus bei dicken Schichten durch den Partialvolumeneffekt nicht zur Darstellung kommt. Um zu klären, ob mit dünneren Schichten häufiger ELS-Tumoren nachweisbar sind, wurden ab Juli 2005 konsekutiv alle von Hippel-Lindau Patienten zusätzlich zum Standard-Screening mit einer Dünnschicht-Felsenbeinsequenz untersucht. Es wurden Daten von 60 Patienten gewonnen und zwei ELS-Tumoren diagnostiziert. Beide Tumore waren sowohl auf den 6 mm Standardsequenzen, als auch auf den Felsenbeinaufnahmen zu erkennen und aus der Krankengeschichte bereits bekannt. Kein Tumor war nur auf den Felsenbein-MRT zu sehen. Natürlich ist anzunehmen, dass kleinere Tumore auf dünneren Schichten eher zu erkennen sind, andererseits zeigt der Vergleich, dass ELS-Tumore im Standard-Screening nicht in größerem Maße übersehen wurden. Daraus lässt sich folgern, dass die Prävalenzrate der 6 mm Sequenzen nicht technisch bedingt fälschlich niedrig ist. 6 mm dicke Schichten sind für das Screening ausreichend. Bei klinischem Verdacht auf einen ELS-Tumor sollte ergänzend eine Felsenbein- Dünnschicht-MRT mit Fettsättigung und eine Dünnschicht-CT des Felsenbeins durchgeführt werden. Durch die hohe Auflösung der CT ist der knöcherne Aquaeductus vestibuli besser nachweisbar als in der MR-Tomographie (Lo et al. 1997) und es ist anzunehmen, dass die knöcherne Destruktion als Zeichen eines ELS-Tumors gut darstellbar ist. Diese Hypothese wäre in einer gezielten Studie zu überprüfen. 5.1.1.3 Zusammensetzung des Patientenkollektivs Vergleicht man Daten zur Häufigkeit eines Tumors, muss darauf geachtet werden, ob die verglichenen Patientenkollektive systematische Unterschiede aufweisen. Bezüglich Alters- und Geschlechtsverteilung sind in der Arbeit von Manski keine Angaben gemacht. 55 5 Diskussion Die Sicherung der von Hippel-Lindau Erkrankung ist anhand klinischer Kriterien und seit 1993 auch durch den Nachweis der Mutation möglich. Das hat zur Folge, dass ein Teil der Patienten allein durch den Mutationsnachweis in das Screening aufgenommen wurde. In unserem Patientenkollektiv war der Nachweis der Mutation bei 218 von 228 Patienten möglich (96 %). Der Anteil unserer Patienten ohne nachweisbare Tumormanifestationen liegt bei 28/228 (12 %). Ob bei Manski et al. Patienten ohne Tumormanifestationen eingeschlossen wurden, geht aus der Arbeit nicht hervor. Untersucht waren Patienten aus den Jahren 1988 bis 1994, sodass möglicherweise keine Patienten ohne Tumormanifestation eingeschlossen wurden, da die Diagnosestellung allein durch den Mutationsnachweis erst seit 1993 möglich ist. Bereinigt man die Daten der vorliegenden Arbeit um die tumorfreien Mutationsträger, um einen systematischen Unterschied zu vermeiden, ergibt sich eine Prävalenzrate von ELS-Tumoren von 6/200 oder 3 %. Unsere Prävalenzrate ist trotz dieser Korrektur signifikant niedriger als in der Arbeit von Manski et al. (p ≤ 0,01). Betrachtet man die Häufigkeit der einzelnen Mutationen, so fällt auf, dass im untersuchten Kollektiv die Mutation c.505 T>C mit 102 Patienten deutlich überrepräsentiert ist. Diese Patienten gehören zum VHL-Typ 2A und haben einen relativ milden Krankheitsverlauf und eine normale Lebenserwartung (Bender et al. 2001). Keiner der Patienten zeigte einen ELS-Tumor. Da die Mutation im Schwarzwald und in einer Region in Pennsylvania (USA) am häufigsten vorkommt, kann ein systematischer Fehler zugunsten wenig symptomatischer Patienten in unserem Kollektiv vorliegen. Nimmt man aus der Gesamtzahl der Patienten jene mit der Mutation c.505 T>C heraus, ergibt sich eine Prävalenzrate von ELS-Tumoren von 6/126 oder 4,8 %. Korrigiert man diese Zahl wiederum um die Patienten ohne Tumormanifestation, so bleiben 6/116 oder 5,2 %. Der Unterschied zu den Daten von Manski et al. ist nun mit p ≤ 0,15 nicht mehr signifikant. Nach Ausschluss der überprüfbaren systematischen Fehlerquellen ist die Prävalenzrate von ELS-Tumoren im Freiburger VHL-Kollektiv signifikant niedriger ist als im Amerikanischen Patientenkollektiv. Dies ist auf das häufige Vorkommen der Mutation c.505 T>C im Freiburger Kollektiv zurückzuführen. 56 5 Diskussion 5.1.2 Aquaeductus vestibuli und Saccus/Ductus endolymphaticus Der Aquaeductus vestibuli ist ein knöcherner Kanal im Os petrosum, der von der anteromedialen Wand des Vestibulums zur Felseneinrückfläche zieht und dort in einer flachen Mulde zwischen Meatus acusticus internus und Sinus sigmoideus in der Apertura externa endet. Der Aquädukt verläuft retrolabyrinthär, parallel zum posterioren Bogengang und enthält den Ductus und Saccus endolymphaticus, welche zum membranösen Labyrinth gehören. Der Ductus endolymphaticus ist nur sehr kurz und geht bereits im distalen Anteil des Aquaeductus vestibuli in den Saccus endolymphaticus über. Dieser Teil des Saccus endolymphaticus wird auch Pars rugosa oder Pars canalicularis genannt, da hier der Sack nicht aus einer einzigen Röhre, sondern aus zahlreichen miteinander kommunizierenden Kanälen besteht (Lo et al. 1997). Hier sollen die Tumore des endolymphatischen Sacks entstehen (Kempermann et al. 1998, Gläsker et al. 2005). Der distale Teil des Saccus endolymphaticus liegt in einer Mulde des Felsenbeins zwischen den Durablättern. Der Aquaeductus vestibuli misst im mittleren Abschnitt maximal 1,5 mm und ist am besten auf hochauflösenden CT der Felsenbeine zu erkennen (Lo et al.). Der Saccus und Ductus endolymphaticus sind dagegen besser mit der MR-Tomographie darstellbar. Im Vergleich zum restlichen Labyrinth zeigt der Saccus endolymphaticus sowohl eine T1-, als auch eine T2-Verkürzung. Das bedeutet, dass er sich T1-w hyperintens, T2-w hypointens im Vergleich zum Labyrinth darstellt. Der Inhalt des Aquaeductus vestibuli ist T1-w besser zu erkennen als T2-w (Schmalbrock et al. 1996). Spin-Echo Sequenzen gelten aufgrund der geringeren Suszeptibilitätsartefakte als geeigneter als Gradienten-Echo Sequenzen (Dahlen et al. 1997). In der vorliegenden Arbeit war der Saccus endolymphaticus in 74 % (Befunder A) bzw. in 58 % (Befunder B) der Felsenbeine mit der Felsenbein-MRT abgrenzbar. Dahlen konnte mit einer 2mm T2-w Turbo-Spin-Echo bei 75 % gesunder Probanden den Endolymphatischen Sack erkennen (Dahlen et al. 1997). Schmalbrock et al. erreichten mit einer Kombination aus T2-w und T1-w MRT im Submilimeterbereich eine Nachweisbarkeit von 95% (Schmalbrock et al. 1996). Durch eine dünnere Schichtdicke ließe sich die Nachweisbarkeit wahrscheinlich erhöhen, die dadurch bedingte längere Messzeit kann jedoch zu Bewegungsartefakten führen und die Bildqualität reduzieren. Die Spin-Echo Sequenz 57 5 Diskussion dieser Arbeit hat sich als sehr stabil erwiesen. Durch die zusätzlich gewählte Fettsättigungstechnik konnten Fetteinlagerungen im Mastoid unterdrückt werden, sodass diese bei der Befundung nicht zu Schwierigkeiten führten. Saccus und Ductus endolymphaticus sind von lockerem, vaskularisiertem Bindegewebe umgeben (Hultgård-Eckwall et al. 2003). Die Vena aquaeducti vestibuli verläuft parallel zum Aquädukt in einem winzigen knöchernen Kanal und erreicht die Felsenbeinhinterkante in unmittelbarer Nachbarschaft zum Endolymphatischen Sack. Sie drainiert Blut aus dem Labyrinth zum Sinus sigmoideus, Sinus petrosus inferior und Bulbus venae jugularis (Mazzoni 1979). In der vorliegenden Arbeit war der Aquaeductus vestibuli in 23 % (Untersucher A) bzw. in 28 % (Untersucher B) der Felsenbeine hyperintens im Vergleich zum restlichen Labyrinth. Diese T1Verkürzung ist am ehesten durch eine Anreicherung der Vena aquaeducti vestibuli oder des vaskularisierten periduktalen Bindegewebe zu erklären. Eine ähnliche Beobachtung machten Naganawa et al.; mit einer T1-3D-SPGR (spoiled gradientecho sequence) mit Kontrastmittel zeigten 22,5 % gesunder Probanden eine Anreicherung des Saccus endolymphaticus (Naganawa et al. 2002). In einer Folgestudie konnte gezeigt werden, dass eine 3D-Fast-Spin-Echo Sequenz einer 3DSPGR Sequenz beim Nachweis der KM-Anreicherung des Aquaeductus vestibuli leicht überlegen ist (Naganawa et al. 2003). 5.1.3 Mutationen der Patienten mit ELS-Tumor Mutationen des VHL-Gens können entweder zu einer inkompletten Translation des VHL-Proteins (Truncation) führen, dazu zählen schwerwiegende Mutationen wie große Deletionen, Nonsense-Punktmutationen, Mikrodeletionen/Mikroinsertionen mit Verschiebung des Leserasters und Splice-site-Mutationen, oder zu einer kompletten Translation des VHL-Proteins (Full-length), bei der eine Aminosäure ausgetauscht wird. Dazu zählen Missense-Punktmutationen und Mikrodeletionen/Mikroinsertionen ohne Verschiebung des Leserasters. Diese Mutationen werden als weniger schwerwiegend eingestuft. In der vorliegenden Arbeit waren bei sechs Patienten mit ELS-Tumoren 4 schwerwiegende Mutationen nachweisbar (2 große Deletionen, 2 Splice-siteDefekte) und 2 weniger gravierende Mutationen (1 Missense-Mutation, 1 Mikrodeletion ohne Verschiebung des Leserasters). Letztere gehörten zu Familien 58 5 Diskussion mit Nierenzellkarzinomen. Die beiden Patienten mit Splice-site-Defekten waren miteinander verwandt. Eine sichere Zuordnung zu Phänotyp 1 oder 2 war, bei oft kleiner Patientenanzahl, nicht zuverlässig möglich. Manski et al. konnte bei 7 von 13 Patienten die Mutation nachweisen, zwei Patienten stammten aus einer Familie. Er fand 6 schwerwiegende Mutationen (4 große Deletionen, 2 Nonsense-Mutationen) und eine weniger schwerwiegende Missense-Mutation (Manski et al. 1997). Choo et al. konnten bei 19 von 21 Patienten die Mutation angeben. Da die Autoren zur gleichen Arbeitsgruppe gehören wie Manski, kann sich das Patientenkollektiv teilweise überschneiden. Choo fand 13 schwerwiegende Mutationen (10 Rearrangements, 2 Nonsense-Mutationen, 1 Splice-site-Defekt) und 5 MissenseMutationen, eine Mutation war nicht zuzuordnen. Alle 5 Patienten mit MissenseMutationen hatten Nierenzellkarzinome. Aus dieser Übersicht lässt sich folgern, dass ELS-Tumore häufiger bei schwerwiegenden Mutationen mit unvollständiger Kodierung des VHL-Proteins vorkommen. Bei Missensmutationen mit ELS-Tumoren hatten die Patienten immer auch Nierenzellkarzinome. Im Umkehrschluss ist bei Missense-Mutationen ohne Nierenzellkarzinome (Typ 2A) das Risiko für einen ELSTumor extrem gering. 5.1.4 Therapie der ELS-Tumoren ELS-Tumore sind lokal invasiv wachsende Tumore, welche zu einer Destruktion des Felsenbeins samt Labyrinth und einer Raumforderung in der hinteren Schädelgrube führen können. Sie können zu einer irreversiblen Ertaubung des betroffenen Ohres führen. Nach Literaturangaben wird die radikale Resektion des Tumors empfohlen. Für kleine Tumore ist die gehörerhaltende retrolabyrinthäre posteriore Petrosektomie beschrieben worden (Kim et al. 2005). Nicht bei allen von Hippel-Lindau Patienten mit Hörminderung und Schwindel lassen sich ELS-Tumore nachweisen. Lonser et al. beschrieb einen Patienten, dessen Beschwerden dem Tumornachweis 3 Jahre vorausgingen. In einer initialen MRT war kein Tumor abgrenzbar, drei Jahre später zeigte sich eine 3 mm große Raumforderung, die reseziert wurde und einem ELSTumor entsprach (Lonser et al. 2004). Lonser geht davon aus, dass kleine, bildgebend nicht nachweisbare Tumore zu Blutungen führen können und somit symptomatisch werden können. Gläsker et al. konnten an vier Felsenbeinen asymptomatischer von Hippel-Lindau Patienten im endolymphatischen Sack 59 5 Diskussion atypisches Epithel mit „loss of heterozygozity“ nachweisen. Dies wurde als Vorläufer von ELS-Tumoren interpretiert. Allerdings wurde keine gesunde Kontrollgruppe von nicht von Hippel-Lindau Erkrankten untersucht (Gläsker et al. 2005). Es ließe sich folgern, dass von Hippel-Lindau Patienten mit plötzlicher Hörminderung, Schwindel und Tinnitus frühzeitig gehörerhaltend petrosektomiert werden sollten. Auf der anderen Seite ist die Komplikationsrate dieser OP-Technik bisher nicht an größeren Fallzahlen untersucht. Auch liegen über das Rezidivrisiko bisher keine ausreichenden Daten vor. Ein Nachbeobachtungszeitraum von maximal 18 Monaten, wie in der Verlaufsbeobachtung von Kim et al. ist bei den langsam wachsenden Tumoren nicht ausreichend (Kim et al. 2005). In der vorliegenden Arbeit zeigten drei der vier operierten ELS-Tumore ein Rezidiv oder Tumorreste mit Größenprogredienz. Ein Felsenbein ist seit 16 Jahren rezidivfrei. Ob die frühzeitige Resektion das Hörvermögen langfristig günstig beeinflusst, werden Verlaufsbeobachtungen zeigen. Bei ertaubten Patienten mit ELS-Tumor ohne Kompression neuraler Strukturen besteht keine OP-Indikation, da eine Metastasierung des Tumors bisher nicht beobachtet wurde, und die klinische Situation durch die Resektion nicht verbessert werden kann. Besteht eine Kompression des Hirnparenchyms mit entsprechender Symptomatik, so ist eine operative Dekompression angezeigt. Ob eine radikale Resektion die Gesamtprognose günstig beeinflusst, ist nicht untersucht. 5.2 Zentralnervöse Hämangioblastome Hämangioblastome des Zentralen Nervensystems können sporadisch oder im Rahmen der von Hippel-Lindau Erkrankung auftreten. Es handelt sich um benigne, stark vaskularisierte Tumore, welche bevorzugt zerebellär und spinal auftreten und Zysten ausbilden können. Sie zählen zu den Hauptmanifestationen der von HippelLindau Erkrankung und werden bei bis zu 72 % der VHL-Patienten beobachtet (Richard et al. 2000). Nach Nierenzellkarzinomen stellen sie die zweit häufigste Todesursache bei VHL-Patienten dar (Maher et al. 1990). 5 Diskussion 60 5.2.1 Häufigkeit zentralnervöser Hämangioblastome In der vorliegenden Arbeit zeigten 66 % der untersuchten von Hippel-Lindau Patienten zentralnervöse Hämangioblastome. 46 % der Patienten hatten zerebrale und 62 % hatten spinale Tumore. Einen Vergleich mit der Literatur zeigt Tabelle 5.2. Übereinstimmend kommen zerebrale Hämangioblastome im Kleinhirn am häufigsten vor. Der relativ niedrigere Anteil von Patienten mit zerebellären Tumoren in den neueren Studien ist am ehesten dadurch bedingt, dass in der Arbeit von Maher et al. vorwiegend symptomatische Patienten untersucht wurden. Der geringe Anteil von Patienten mit spinalen Tumoren in der Arbeit von Maher et al. ist am ehesten dadurch zu erklären, dass die diagnostischen Möglichkeiten vor 1990 den heutigen Methoden deutlich unterlegen waren. Insbesondere ist heutzutage der Nachweis kleinster Tumore des Rückenmarks mittels MR-Tomographie möglich. Der Anteil der Patienten mit multiplen Hämangioblastomen lag in Freiburg bei 84 %. Das deckt sich mit den Erfahrungen von Wanebo et al., die in 79 % der Patienten multiple Tumore fanden. Autor Maher et Jahr Patienten- Kleinhirn Hirnstamm Großhirn anzahl HB HB HB 1990 152 1998 215 59 % 4% Spinale HB 1% 13 % al. Richard Zerebrale und spinale HB 72 % et al. Lonser et 2003 44 – 72 % 10 – 25 % <1% 13 – 50 % al. Wanebo 2003 231 47 % 22 % 4% 53 % 2006 226 41 % 17 % 5% 62 % et al. Freiburg Tab. 5.2: Relativer Anteil von VHL-Patienten mit Hämangioblastomen aufgegliedert nach der Tumorlokalisation. (Maher et al. 1990, Richard et al. 1998, Lonser et al. 2003, Wanebo et al. 2003) Betrachtet man die Gesamtheit der zerebralen Hämangioblastome, so lagen im Freiburger Kollektiv 97 % der Tumore infratentoriell. Das entspricht exakt dem Ergebnis von Wanebo et al. Die Autoren fanden bei 324 zerebralen Tumoren 250 Tumore in den Kleinhirnhemisphären, 64 Tumore am Hirnstamm (97 % infratentoriell) und 10 Tumore im Großhirn (Wanebo et al. 2003). 61 5 Diskussion 5.2.2 Operationsfrequenz zerebraler Hämangioblastome Ein Großteil der Patienten (67 %) mit zerebralen Hämangioblastomen wurde im Laufe der Erkrankung mindestens einmal am Kopf operiert. In der Literatur sind genaue Angaben zur Operationsfrequenz nicht vorhanden. Richard berichtet lediglich, dass sich die Mehrheit der Patienten mit zentralnervösen Hämangioblastomen irgendwann einer Operation unterziehen musste (Richard et al. 1998). Betrachtet man die Gesamtzahl der Tumore, so wurden in Freiburg nur 26 % der zerebralen Hämangioblastome operativ entfernt. Folglich haben viele Patienten mit einem operationspflichtigen Tumor auch asymptomatische Tumore, welche keiner Therapie bedürfen. Größere Tumore (über 500 mm³) wurden erwartungsgemäß signifikant häufiger operiert als kleine Tumore. Hierbei spielt auch die Lokalisation der Tumore eine wichtige Rolle. Hämangioblastome des Hirnstamms wurden signifikant häufiger operiert als Tumore des Kleinhirns, obwohl sie im Mittel geringere Tumorvolumina aufwiesen. Der raumfordernde Effekt hängt also vom jeweiligen Kompartiment ab. Dies deckt sich mit den Erfahrungen anderer Arbeitsgruppen (Wanebo et al. 2003, Richards et al. 1998). Nicht alle großen Tumore wurden operiert. Das ist am ehesten darauf zurückzuführen, dass einzelne Tumore inoperabel sind, oder andere Probleme des Patienten vordringlicher erscheinen. Ein Beispiel eines inoperablen Hämangioblastoms im Verlauf zeigt Abbildung 5.1. Abb. 5.1: Hämangioblastom des linken Kleinhirnbrückenwinkels. Eine radikale Resektion des Tumors war aufgrund der Infiltration des Cavum Meckeli nicht möglich. (a) 2002 solider Tumor, (b) 2004 deutliche Größenzunahme des soliden Tumors mit Kompression des Hirnstamms, (c) 2006 Ausbildung eine tumorassoziierten Zyste mit erheblicher Zunahme der Hirnstammkompression. (a) – (c) MRT, T1-w mit Gad-Chelat, transversal. 62 5 Diskussion 5.2.3 Bedeutung tumorassoziierter Zysten In der vorliegenden Arbeit waren 12 % der zerebralen Hämangioblastome zystisch (Typ 2 und 4). Die Rate liegt etwas unterhalb der von Wanebo et al., die bei 62 von 314 zerebralen Tumoren assoziierte Zysten (20 %) fanden (Wanebo et al. 2003) und deutlich unterhalb der Rate der französischen Arbeitsgruppe, welche bei 74 % der Tumore assoziierte Zysten fand (Richard et al. 1998). Eine mögliche Erklärung der Diskrepanz kann die Verbesserung der MR-Tomographie im Laufe der Jahre darstellen. So ist heute der Nachweis 2 mm großer Tumore möglich. Wie in der Arbeit gezeigt wurde, kommen kleine Tumore häufig vor und sind überwiegend solide. Es ist anzunehmen, dass diese meist asymptomatischen Tumore der Diagnostik früher entgangen sind. Typ 1 der Hämangioblastome (einfache Zyste) wurde bei Richard mit einer Häufigkeit von 5 % angegeben, kam jedoch im Freiburger Kollektiv nicht vor (Richard et al. 1998). Auch dies ist am ehesten auf die verfeinerte Diagnostik zurückzuführen, da heute auch kleinste Tumorknötchen nachweisbar sind. Es ist zu bezweifeln, ob Typ 1 überhaupt existiert. Hämangioblastomzysten haben keine Wand und man nimmt an, dass ihr Inhalt einem Transsudat entspricht. Die Entstehung einer Zyste ohne verursachenden Tumor ist pathophysiologisch schwer zu erklären. In einer kleinen Sammlung von 17 Hämangioblastomen, welche mittels MR-Tomographie untersucht wurden, kam Typ 1 ebenfalls nicht vor (Lee et al. 1989). Es besteht keine Präferenz zystischer Tumore für bestimmte Lokalisationen. Auch konnte kein Zusammenhang zwischen der Häufigkeit zystischer Tumore und dem Mutationstyp des VHL-Gens nachgewiesen werden. Hingegen besteht ein Zusammenhang zwischen Tumorgröße und dem Vorkommen assoziierter Zysten. Zystische Tumore waren in der vorliegenden Arbeit signifikant größer als solide Tumore und wurden entsprechend signifikant häufiger operiert. Das passt zu der Feststellung von Wanebo et al., dass Hämangioblastome mit assoziierten Zysten häufiger symptomatisch werden als solide Tumore (Wanebo et al. 2003). Die Frage, ob große Tumore besonders häufig Zysten bilden, oder ob zystische Tumore besonders groß werden ist schwer zu beantworten. Betrachtet man den Anteil progredienter Tumore, so zeigten in der vorliegenden Arbeit zystische Tumore signifikant häufiger einen Progress, wohingegen die Tumorgröße keinen Einfluss auf den Progress zeigte. Dies legt nahe, dass das Vorliegen einer 63 5 Diskussion Tumorzyste die Größenzunahme bedingt. Diese Annahme unterstützt die Feststellung von Wanebo et al., dass Tumorzysten um ein vielfaches größer waren als die verursachenden Tumore (Wanebo et al. 2003). Patienten, die bei der Screening-Untersuchung zystische Tumore zeigen, sollten auf die Möglichkeit der raschen Entwicklung klinischer Beschwerden hingewiesen und eventuell engmaschiger untersucht werden. 5.2.4 Bedeutung des Mutationstyps des VHL-Gens Die Mutationen des VHL-Gens wurden entsprechend der zu erwartenden Translation des VHL-Proteins in zwei Typen eingeteilt: F-Typ (Full-length) und T-Typ (Truncation). Zu 75 % lag ein F-Typ vor. Patienten mit einer Mutation vom T-Typ entwickelten signifikant häufiger zerebrale Hämangioblastome (91 %) als Patienten mit Mutationen vom F-Typ (30 %). Patienten mit zerebralen Hämangioblastomen und einer T-Typ Mutation hatten tendenziell mehr Tumore als Patienten mit F-Typ Mutation. Wenn Patienten zerebrale Hämangioblastome entwickelten, zeigten die Tumore des F-Typs und des T-Typs keinen Unterschied bezüglich durchschnittlicher Tumorgröße, Anteil assoziierter Zysten, Häufigkeit von Operationen oder Wachstumsrate der Tumoren. 5.2.5 Wachstum zerebraler Hämangioblastome Nach den RECIST-Kriterien (Response Evaluation Criteria in Solid Tumors) muss eine Zunahme des Tumorvolumens um 73 % vorliegen, um einen Progress zu diagnostizieren (Wormans 2005). In der vorliegenden Arbeit haben Messfehler in seltenen Fällen bei kleinen Tumoren zu einer scheinbaren Abnahme des Tumorvolumens bis 50 % geführt. Deshalb wurde in der vorliegenden Arbeit eine Verdoppelung des Tumorvolumens als Progress definiert. Durch diese Definition wurden 8 von 455 Tumoren als stabil definiert, die nach RECIST-Kriterien progredient gewesen wären. Nicht alle zerebralen Hämangioblastome zeigten einen Tumorprogress. Bei kurzer Nachbeobachtungszeit (bis 20 Monate) war der Anteil progredienter Tumore gering (14 %). Erst ab einer Nachbeobachtungszeit von 20 Monaten war dieser Anteil relativ 64 5 Diskussion stabil (ca. 50 %). Das unterstreicht die Notwendigkeit langer Nachbeobachtungszeiten, da die Tumore oft sehr langsam wachsen. Bei der Analyse, welche Faktoren einen Einfluss auf den Tumorprogress haben, wurden daher nur Tumore berücksichtigt, die mindestens 20 Monate beobachtet wurden. Unter dieser Voraussetzung zeigten 57 % der Tumore einen Progress. Die Tumorlokalisation und die Tumorgröße schienen keinen Einfluss auf das Tumorwachstum zu haben, zystische Tumore waren dagegen signifikant häufiger progredient als solide Tumore (100 % versus 52 %). Auch Slater et al. konnten in einer kleinen Studie von 28 Tumoren zeigen, dass zystische Tumore häufiger eine Größenzunahme zeigen als solide Tumore (57 % versus 7 %) (Slater et al. 2003). Die Größenzunahme der Tumore hat eine Konsequenz für die betroffenen Patienten, denn es konnte ebenfalls gezeigt werden, dass progrediente Tumore signifikant häufiger operiert wurden als stabile Tumore. Diese Ergebnisse unterstreichen die in Kapitel 5.2.3 diskutierte Bedeutung tumorassoziierter Zysten. 5.2.6 Neuauftreten zerebraler Hämangioblastome Conway et al. berechneten in einer Studie mit 15 von Hippel-Lindau Patienten mit zentralnervösen Hämangioblastomen, dass im Durchschnitt 1 Tumor pro 1,5 Jahren neu auftritt (Conway et al. 2001). Das entspricht 0,67 Tumoren pro Jahr. In der vorliegenden Studie lag die Rate neu aufgetretener Hämangioblastome bei 0,24 Tumoren pro Jahr. Diese Berechnung beruht auf 66 Patienten mit zerebralen Hämangioblastomen und einer mittleren Nachbeobachtungszeit von 52 Monaten. Im Umkehrschluss bedeutet das, dass Patienten mit zerebralen Tumoren im Durchschnitt alle 4,2 Jahre einen neuen Tumor entwickeln. Im Vergleich zu Conway et al. ist somit das Neuauftreten von Hämangioblastomen im Freiburger VHLKollektiv seltener, jedoch nicht unerheblich für die Patienten. 65 6 Zusammenfassung 6 Zusammenfassung Die von Hippel-Lindau Erkrankung ist eine hereditäre Multisystemerkrankung. Betroffene Patienten können intrakraniell Hämangioblastome und Tumore des Endolymphatischen Sacks entwickeln. Ziel der Arbeit war es die Häufigkeit der Tumore und deren natürlichen Verlauf am Freiburger VHL-Patientenkollektiv zu bestimmen. Hierzu wurden MRT-Verlaufsaufnahmen von 228 VHL-Patienten retrospektiv analysiert. Zusätzlich wurden prospektiv bei 60 VHL-Patienten Felsenbein-Dünnschicht-MRT akquiriert und ausgewertet. Tumore des Endolymphatischen Sacks waren mit einer Prävalenzrate von 3 % deutlich seltener als bisher in der Literatur angegeben wurde. Der Aquaeductus vestibuli war bei einer Doppelbefundung in 74 % bzw. 58 % der Felsenbeine nachweisbar. Die MRT-Diagnostik des Screening-Programms hat sich zum Nachweis der Tumore als ausreichend dargestellt. Es konnte gezeigt werden, dass die niedrigere Prävalenzrate durch die Überrepresentation der „Schwarzwald-Mutation“ im Freiburger Patientenkollektiv bedingt ist. Patienten mit dieser Mutation zeigen einen relativ milden Krankheitsverlauf und ELS-Tumore wurden bei Patienten mit dieser Mutation bisher nicht nachgewiesen. Die Prävalenzrate zerebraler Hämangioblastome war mit 46 % vergleichbar mit den Angaben der Literatur. 97 % der Tumore lagen infratentoriell. Große Tumore und Tumore des Hirnstamms wurden häufiger operiert als kleine Tumore oder Hämangioblastome der Kleinhirnhemisphären oder des Großhirns. 12 % der Hämangioblastome zeigten tumorassoziierte Zysten. Zystische Tumore waren im Durchschnitt größer als solide Tumore und wurden folglich häufiger operiert. Im Verlauf konnte bei zystischen Hämangioblastomen signifikant häufiger ein Progress nachgewiesen werden als bei soliden Tumoren. Es konnte gezeigt werden, dass zerebrale Hämangioblastome im Verlauf neu auftreten können. Aus den vorliegenden Daten kann gefolgert werden, dass Patienten mit zerebralen Tumoren ca. alle 4,2 Jahre einen neuen Tumor entwickeln. Mutationen des VHL-Gens kann man in zwei Gruppen einteilen. Patienten mit Mutationen vom T-Typ entwickelten signifikant häufiger zerebrale Hämangioblastome als Patienten mit Mutationen vom F-Typ. Die Tumore selbst unterschieden sie sich jedoch nicht hinsichtlich ihrer mittleren Größe, ihres Typs oder ihrer Wachstumsrate. 66 7 Literatur 7 Literatur Bender B.U., Eng C., Olschewski M., Berger D.P., Laubenberger J., Altehöfer C., Kirste G., Orszagh M., van Velthoven V., Miosczka H., Schmidt D., Neumann H.P.H. (2001) VHL c.505 T>C mutation confers a high age related penetrance but no increased overall mortality. J Med Genet 38:508-514 Brandt R. (1921) Zur Frage der Angiomatosis retinae. Graefes Arch Ophthalmol 106:127-165 Brauch H., Kishida T., Glavac D., Chen F., Pausch F., Höfler H., Latif F., Lerman M.I., Zbar B., Neumann H.P.H. (1995) Von Hippel-Lindau (VHL) disease with pheochromocytoma in the Black Forest region of Germany: evidence for a founder effect. Hum Genet 95:551-556 Chen F., Kishida T., Yao M., Hustad T., Glavac D., Dean M., Gnarra J.R., Orcutt M.L., Duh F.M., Glenn G., Green J., Hsia Y.E., Lamiell J., Li H., Wei M.H., Schmidt L., Tory K., Kuzmin I., Stackhouse T., Latif F., Linehan W.M., Lerman M., Zbar B. (1995) Germline mutations in the von Hippel-Lindau disease tumor suppressor gene: correlations with phenotype. Hum Mutat 5:66-75 Choo D., Shotland L., Mastroianni M., Glenn G., van Waes C., Linehan W.M., Oldfield E.H. (2004) Endolymphatic sac tumors in von Hippel-Lindau disease. J Neurosurg 100:480-487 67 7 Literatur Choyke P.L., Glenn G.M., Walther M.M., Patronas N.J., Linehan W.M., Zbar B. (1995) von Hippel-Lindau disease: genetic, clinical, and imaging features. Radiology 194:629-642 Conway J.E., Chou D., Clatterbuck R.E., Brem H., Long D.M., Rigamonti D. (2001) Hemangioblastomas of the central nervous system in von Hippel-Lindau syndrome and sporadic disease. Neurosurgery 48:55-62 Dahlen R.T., Harnsberger H.R., Gray S.D., Shelton C., Allan R., Parkin J.L., Scalzo D. (1997) Overlapping thin-section fast spin-echo MR of the large vestibular aqueduct syndrome. AJNR 18:67-75 Gaffey M.J., Mills S.E., Fechner R.E., Intemann S.R., Wick M.R. (1988) Aggressive papillary middle-ear tumor: a clinicopathologic entity distinct from middleear adenoma. Am J Surg Pathol 12:790-797 Gaffey M.J., Mills S.E., Boyd J.C. (1994) Aggressive papillary tumor of middle ear/temporal bone and adnexal papillary cystadenoma: manifestations of von Hippel-Lindau disease. Am J Surg Pathol 18:1254-1260 Gläsker S., Lonser R.R., Tran M.G.B., Ikejiri B., Butman J.A., Zeng W., Maxwell P.H., Zhuang Z., Oldfield E.H., Vortmeyer A.O. (2005) Effects of VHL deficiency on endolymphatic duct and sac. Cancer Res 65:10847-10853 68 7 Literatur Hassard A.D., Boudreau S.F., Cron C.C. (1984) Adenoma of the endolymphatic sac. J Otolaryngol 13:213-216 Hasselblatt M., Jeibmann A., Gerß J., Behrens C., Rama B., Wassmann H., Paulus W. (2005) Cellular and reticular variants of haemangioblastoma revisited:a clinicopathologic study of 88 cases. Neuropathol Appl Neurobiol 31:618-622 Heffner D.K. (1989) Low-grade adenocarcinoma of probable endolymphatic sac origin: a clinicopathologic study of 20 cases. Cancer 64:2292-2302 Hultgård-Ekwall A.K.H., Couloigner V., Rubin K., Rask-Andersen H. (2003) Network organisation of interstitial connective tissue cells in the human endolymphatic duct. J Histochem Cytochem 51:1491-1500 Kim H.J., Butman J.A., Brewer C., Zalewski C. Vortmeyer A.O., Glenn G., Oldfield E.H., Lonser R.R. (2005) Tumors of the endolymphatic sac in patients with von Hippel-Lindau disease: implications for their natural history, diagnosis, and treatment. J Neurosurg 102:503-512 Kempermann G., Neumann H.P.H., Volk B. (1998) Endolymphatic sac tumours. Histopathology 33:2-10 Knudson A.G. (1986) Genetics of human cancer. Ann Rev Genet 20:231-251 69 7 Literatur Lindau A. (1927) Zur Frage der Angiomatosis retinae und ihrer Hirnkomplikationen. Acta Ophthalmol (Copenh) 4:193-226 Latif F., Tory K., Gnarra J., Yao M., Duh F.M., Orcutt M.L., Stackhouse T., Kuzmin I., Modi W., Geil L., Schmidt L., Zhou F., Li H., Wei M.H., Chen F., Glenn G., Choyke P., Walther M.M., Weng Y., Duan D.R., Dean M., Glavac D., Richards F.M., Crossey P.A., Ferguson-Smith M.A., Le Paslier D., Chumakov I., Cohen D., Chinault A.C., Maher E.R., Linehan W.M., Zbar B., Lerman M.I. (1993) Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 260:1317-20 Lee S.R., Sanches J., Mark A.S., Dillon W.P., Norman D., Newton T.H. (1989) Posterior fossa hemangioblastomas: MR Imaging. Radiology 171:463-468 Lo W.W.M., Applegate L.J., Carberry J.N., Solti-Bohman L.G., House J.W., Brackmann D.E., Waluch V., Li J.C. (1993) Endolymphatic sac tumors: radiologic appearance. Radiology 189:199-204 Lo W.W.M., Daniels D.L., Chakeres D.W., Linthicum F.H., Ulmer J.L., Mark L.P., Swartz J.D. (1997) The endolymphatic duct and sac. AJNR 18:881-887 Lonser R.R., Glenn G.M., Walther M., Chew E.Y., Libutti S.K., Linehan W.M., Oldfield E.H. (2003) Von Hippel-Lindau disease. Lancet 361:2059-2067 70 7 Literatur Lonser R.R., Kim H.J., Butman J.A., Vortmeyer A.O., Choo D.I., Oldfield E.H. (2004) Tumors of the endolymphatic sac in von Hippel-Lindau disease. N Engl J Med 350:2481-2486 Maher E.R., Yates J.R., Harries R., Benjamin C., Harris R., Moore A.T., Ferguson-Smith M.A. (1990) Clinical features and natural history of von Hippel-Lindau disease. Q J Med 77:1151-63 Maher E.R., Iselius L., Yates J.R., Littler M., Benjamin C., Harris R., Sampson J., Williams A., Ferguson-Smith M.A. Morton N. (1991) Von Hippel-Lindau disease: a genetic study. J Med Genet 28: 443-447 Manski T.J., Heffner D.K., Glenn G.M., Patronas N.J., Pikus A.T., Katz D., Lebovics R., Sledjeski K., Choyke P.L., Zbar B., Linehan W.M., Oldfield E.H. (1997) Endolymphatic sac tumors: a source of morbid hearing loss in von Hippel-Lindau disease. JAMA 277:1461-1466 Mazzoni A. (1979) Vein of the vestibular aqueduct. Ann Otol 88:759-767 Megerian C.A., McKenna M.J., Nuss R.C., Maniglia A.J., Ojemann R.G., Pilch B.Z., Nadol J.B. (1995) Endolymphatic sac tumors: histopathologic confirmation, clinical characterisation, and implication in von Hippel-Lindau disease. Laryngoscope 105: 801-808 71 7 Literatur Melmon K.L., Rosen S.W. (1964) Lindau’s disease: rewiew of the literature and study of a large kindred. Am J Med 36:595-617 Mukherji S.K., Albernaz V.S., Lo W.W.M., Gaffey M.J., Megerian C.A., Feghali J.G., Brook A., Lewin J.S., Lanzieri C.F., Talbot J.M., Meyer J.R., Carmody R.F., Weissman J.L., Smirniotopoulos J.G., Rao V.M., Jinkins J.R., Castillo M. (1997) Papillary endolymphatic sac tumors: CT, MR Imaging, and angiographic findings in 20 patients. Radiology 202:801-808 Naganawa S., Koshikawa T., Fukatsu H., Ishigaki T., Nakashima T., Ichinose N. (2002) Contrast-enhanced MR imaging of the endolymphaic sac in patients with sudden hearing loss. Eur Radiol 12:1121-1126 Naganawa S., Koshikawa T., Nakamura T., Fukatsu H., Ishigaki T., Aoki I. (2003) High-resolution T1-weighted 3D real IR imaging of the temporal bone using tripledose contrast material. Eur Radiol 13:2650-2658 Neumann H.P.H. (1987) Basic criteria for clinical diagnosis and genetic counselling in von Hippel-Lindau syndrome. VASA 16:220-226 Neumann H.P.H., Bender B.U. (1998) Genotype-phenotype correlations in von Hippel-Lindau disease. J Intern Med 243:541-545 Patel N.P., Wiggins R.H., Shelton C. (2006) The radiologic Diagnosis of endolymphatic sac tumors. Laryngoscope 116:40-46 72 7 Literatur Richard S., Campello C., Taillandier L., Parker F., Resche F. (1998) Haemangioblastoma of the central nervous sytem in von Hippel-Lindau disease. J Intern Med 243:547-553 Richard S., David P., Marsot-Dupuch K., Giraud S., Beroud C., Resche F. (2000) Central nervous sytem hemangioblastomas, endolymphatic sac tumors, and von Hippel-Lindau disease. Neurosurg Rev 23:1-22 Schmalbrock P., Dailiana T., Chakers D.W., Oehler M.C., Welling D.B., Williams P.M., Roth L. (1996) Submillimeter-resolution MR of the endolymphatic sac in healthy subjects and patients with Meniere disease. AJNR 17:1707-1716 Slater A., Moore N.R., Huson S.M. (2003) The natural history of cerebellar hemangioblastomas in von Hippel-Lindau disease. AJNR 24:1570-1574 Stolle C., Glenn G., Zbar B., Humphrey J.S., Choyke P., Walther M., Pack S., Hurley K., Andrey C., Klausner R., Linehan W.M. (1998) Improved detection of germline mutations in the von Hippel-Lindau disease tumor suppressor gene. Hum Mutat 12:417-423 von Hippel E. (1904) Über eine sehr seltene Erkrankung der Netzhaut. Graefes Arch Ophthalmol. 59:83-106 von Hippel E. (1911) Die anatomische Grundlage der von mir beschriebenen „sehr seltenen Erkrankung der Netzhaut“. Graefes Arch Ophthalmol. 79:350-377 73 7 Literatur Vortmeyer A.O., Gnarra J.R., Emmert-Buck M.R., Katz D., Linehan W.M., Oldfield E.H., Zhuang Z. (1997) Von Hippel-Lindau gene deletion detected in the stromal cell component of a cerebellar hemangioblastoma associated with von Hippel-Lindau disease. Hum Pathol 28:540-543 Vortmeyer A.O., Frank S., Jeong S.Y., Yuan K., Ikejiri B., Lee Y.S., Bhowmick D., Lonser R.R., Smith R., Rodgers G., Oldfield E.H., Zhuang Z. (2003) Developmental arrest of angioblastic lineage initiates tumorigenesis in von HippelLindau disease. Cancer Res 63:7051-7055 Wormanns D. (2005) Radiologische Messverfahren zur Beurteilung des Therapieansprechens solider Tumoren. Radiologie up2date 3:261-271 Zbar B., Kishida T., Chen F., Schmidt L., Maher E:R:, Richards F.M., Crossey P.A., Webster A.R., Affara N.A., Ferguson-Smith M.A., Brauch H., Glavac D., Neumann H.P.H., Tisherman S., Mulvihill J.J., Gross D.J., Shuin T., Whaley J., Seizinger B., Kley N., Olschwang S., Boisson C., Richard S., Lips C.H.M., Linehan W.M., Lerman M. (1996) Germline mutations in the von Hippel-Lindau disease (VHL) Gene in families from North America, Europe, and Japan. Hum Mutat 8:348-357 74 8 Danksagung 8 Danksagung Herrn Prof. Spreer danke ich für die Überlassung des Themas und die Betreuung während der Zusammenstellung dieser Arbeit. Mein besonderer Dank gilt Herrn Prof. Neumann. Ohne die jahrelange konsequente Dokumentation klinischer und genetischer Daten zahlreicher von Hippel-Lindau Patienten wäre diese Arbeit nicht möglich gewesen. Bei den stets freundlichen Damen der von Hippel-Lindau Studienzentrale, Frau Schluh, Frau Schonhardt und Frau Nabulsi möchte ich mich für die Hilfsbereitschaft und prompte Beantwortung all meiner Fragen bedanken. Schließlich danke ich allen medizinisch technischen Assistenten und Assistentinnen der Sektion Neuroradiologie für ihre Unterstützung bei der Durchführung der MRTomographien. 75 9 Lebenslauf 9 Lebenslauf Name Claudia Hader Geboren 09.11.1967 in Augsburg Familienstand ledig Schulbildung 1974 - 1978 Grundschule, Augsburg 1978 - 1987 Peutinger Gymnasium, Augsburg Hochschulausbildung 1987 – 1994 Studium der Humanmedizin, Ludwig-Maximilians-Universität München Okt 1989 Physikum Sep 1990 1. Staatsexamen Apr 1993 2. Staatsexamen Apr 1994 3. Staatsexamen Praktisches Jahr Apr – Aug 1993 Chirurgie/Neurochirurgie, Klinikum Ingolstadt Aug – Dez 1993 Neurologie, Klinikum Großhadern, München Jan – Mär 1994 Innere Medizin, Kreisspital Pfäfffikon ZH, Schweiz AiP Mai 1994 – Okt 1995 Neurochirurgische Klinik, Klinikum Ingolstadt, Approbation 01.11.1995 76 9 Lebenslauf Weiterbildungszeiten Nov 1995 – Dez 1995 Neurochirurgische Klinik, Klinikum Ingolstadt Apr 1996 – Jul 2000 Neurochirurgische Klinik, Medizinische Hochschule Hannover, davon 6 Monate: Abteilung Neuroradiologie, Medizinische Hochschule Hannover Aug 2000 – Dez 2004 Institut für diagnostische und interventionelle Radiologie, Klinikum Ingolstadt Seit Jan 2005 Abteilung für Neuroradiologie, Universitätsklinikum Freiburg Facharztanerkennung Neurochirurgie 16.05.2001 Diagnostische Radiologie 08.09.2004