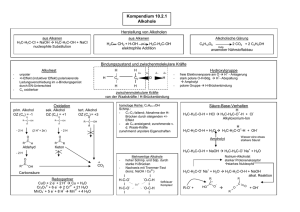
1. Bor - Chempage
Werbung

1. Bor 1.1 Vorkommen: Wegen seiner Reaktionsfähigkeit kommt Bor nicht elementar vor. Natürliche Borverbindungen sind die Borate Die wichtigsten Mineralien sind: - Kernit Na2B4O7 · 4H2O - Borax Na2B4O7 · 10H2O - Borocalcit CaB4O7 · 4 H2O - Colemanit Ca2B6O11 · 5H2O (Borverbindungen lassen sich in den USA und der Türkei finden.) 1.2 Darstellung: Kristallines hochreines Bor lässt sich durch Reduktion aus seinen Halogeniden mit Wasserstoff bei 1000-1400°C gewinnen: 2 BCl3 + 3 H2 → 2 B + 6 HCl ∆H = + 262kJ/mol Außerdem lässt es sich durch thermische Zersetzung von BI3 an Wolframdrähten gewinnen. Welche Modifikation des Bors vorliegt hängt von der Reaktionstemperatur ab. Amorphes Bor lässt sich durch Reduktion von B2O3 mit Natrium oder Magnesium gewinnen: B2O3 + 3 Mg → 2B + 3MgO ∆H = -533kJ/mol Technisch wird Bor heutzutage meist per Schmelzflusselektrolyse aus einem Gemisch von KBF4, KCl und B2O3 bei 800°C hergestellt. Bor wird benötigt als Desoxidationsmittel und zum vergüten von Stahl (Erhöhung der Härtbarkeit). 1.3 physikalische und chemische Eigenschaften: Normalerweise kristallisieren Hauptgruppenelemente mit weniger als vier Valenzelektronen in Metallgittern aus. Eine Ausnahme stellt Bor dar. Da es eine hohe Ionisierungsenergie sowie eine relativ hohe Elektronegativität besitzt, bildet es kovalente Bindungen aus. Die besonderen Bindungseigenschaften des Bors resultiert aus dem Elektronenmangel am Boratom. Bor besitzt zwar 4 Valenzorbitale aber nur 3 Valenzelektronen. Somit können Raumnetzstrukturen nur durch die Ausbildung von Mehrzentren Bindungen gebildet werden. Von Bor sind verschiedene Modifikationen bekannt wobei immer B12-Ikosaeder Einheiten auftauchen. Im Bor-Ikosaeder sind alle 12 Boratome äquivalent und jedes besitzt 5 Boratomnachbarn und liegen auf einer fünfzähligen Achse. α-rhomboedrisches Bor besitzt die einfachste Struktur. Die B12-Ikosaeder sind in einer annähernd kubisch-dichtesten-Packung angeordnet. 6 B-Atome besitzen die Koordinationszahl 7. Sie sind über eine Dreizentrenbindung an zwei B-Atome zweier Nachbar-Ikosaeder innerhalb einer Schicht gebunden. Die anderen 6B-Atome haben die Koordinationszahl 6. Sie sind durch zweizentren-Bindungen an je drei Ikosaeder der darüber und darunter liegenden Ikosaederschicht gebunden. Abb.: B12-Ikosaeder Abb.: a) Die Struktureinheit sind B12-Iokosaeder. Sie haben die Anordnung einer kubisch-dichten Packung. b) 6 B-Atome eines Ikosaeder sind durch eine geschlossene 3 Zentren-BBB-Bindung gebunden β-rhomboedrsisches Bor ist die thermodynammisch stabile Modifikation. Man erhält es durch Erhitzen von α-rhomboedrischem Bor auf 1200°C. Die Struktur ist kompliziert. Die Elementarzelle enthält 105 Boratome. Ein Teil der Struktureinheiten stellen Ikosaeder dar. α-tetragonales Bor enthält B12-Ikosaeder und einzelne Boratome. Die B12-Ikosaeder sind in einer kexagonal-dichten Packung angeordnet. ¼ der vorhandenen Teetraederlücken sind mit B-Atomen besetzt. Die einzelnen B-Atome sind tetraedrisch koordiniert, sie verbinden 4 Ikosaeder. Die B-Atome der Ikosaeder besitzen die KZ von 6. Jedes Ikosaeder ist mit 10 weitern Ikosaedern durch B-B-Einfachbindungen verbunden. Die Verbindung zum 11. und 12. Ikosaeder erfolgt über das EINFACHE Boratom. Außerdem gibt es noch eine β-tetragonale Modifikation mit 190 Boratomen pro Elementarzelle. Bormodifikationen sind sehr hart und halbleitend. Die thermodynamisch stabile ist das βrhomboedrische Bor. Es ist mit einer Mohs-Härt von 9,3 nach dem Diamanten das zweit härteste Element. Bindungseigenschaften des Bors Bor bildet in Verbindungen aufgrund seiner Elektronenkonfiguration sp²-hybridisierte BAtome. Es entstehen somit Verbindungen mit trigonal-planarer Koordination. Verbindungen mit Halogenen und Sauerstoff sind stark polar. Wasserstoff ist in Boratomen negativ polarisiert und somit ähneln die Borane eher den Silane als den Alkanen in iherer chemischen Eigenschaft. Die Verbindungen des Typs BX3 stellen Elektronenmangel Verbindungen dar und besitzen nur ein Elektronensextett. Sie können sich auf drei verschiedene Weisen unter Ausbildung eines Oktetts stabilisieren. I. Ausbildung von π-Bindungen Am Beispiel BF3 lässt sich zeigen, dass Bor mit nichtbindenden Elektronenpaare des Fluors wechselwirkt und somit sich die Bindungslänge aufgrund von delokalisierten π-Elektronen auf 130pm (B-F: 145pm; B=F: 125pm) verkürzt. Dies führt zu einer geringeren Lewisacidität. Ebenso wie beim BF3 existieren auch beim BCl3 und BBr3 (auch beim BN) πBindungsanteile. II. Mehrzentrenbindungen In Verbindungen, in denen keine freien Elektronenpaare für die Ausbildung von π-Bindungen zur Verfügung stehen, kann das Bor durch Ausbildung von Mehrzentrenbindungen stabilisiert werden. Am Beispiel des BH3 lässt sich zeigen, dass BH3 als Monomer nicht beständig ist, es reagiert sofort mit einem weiteren BH-Molekül zum Diboran (B2H6). Mehrzentrenbindungen treten auch bei anderen Boranen, den Modifaktionen und den Metallboriden auf. III. Anlagerung von Donormolekülen Die Elektronenlücke des Bors kann durch Elektronenpaare eines Donormoleküls geschlossen werden. Dabei erfolgt beim B-Atom eine Änderung der Hybridisierung von sp² zu sp³. Beispiele: BF4-, BH4-, BF3OR2 Mit stärker werdender π-Bindung nimmt die Akzeptorstärke ab: BH3 > BBr3 > BCl3 > BF3. Die Bindungslängen im BF4- entsprechen Einfachbindungen. Im Gegensatz zu den BX3Verbindungen ist BX4- hydrolyseunempfindlich. Aufgrund der Eigenschaft Mehrzentrenbindungen ausbilden zu können, kann Bor neben den Koordinationszahlen 3 und 4 auch noch Koordinationszahlen zwischen 5 und 9 erreichen. Diese Eigenschaft besitzt kein anderes Element. Bor bildet äußerst selten Doppelbindungen aus, Einfachbindungen des Bors (B-B) treten bei den Halogeniden auf. 1.4 wichtige Verbindungen des Bors: Bortrifluorid BF3 ist einfarbloses, stechend riechendes Gas. Es entsteht durch Erhitzen von B2O3 und CaF2 mit konzentrierter Schwefelsäure: CaF2 + H2SO4 → CaSO4 + 2HF B2O3 + 6HF → 2BF3 + 3H2O Eigenschaften: Bortrifluorid wird als Druckgas oder als Diethylether-Addukt BF3 · OEt2 gehandelt. BF3 wirkt als starke Lewissäure und vereinigt sich mit Donatoren D wie Wasser, Alkoholen, Ethern und Ammoniak leicht zu Addukten: D + BF3 D→BF3 Mit Wasser hydrolysiert es gemäß: BF3 + 3 H2O → B(OH)3 + 3 HF. Zusammen mit Flusssäure bildet sich aus Borsäure eine starke Säure, welche nur in wässriger Lösung bekannt ist: B(OH)3 + 4HF → HBF4 + 3H2O Bortrichlorid BCl3 ist einfarbloses an der Luft rauchendes Gas, welches mit Wasser zu B(OH)3 hydrolysiert. Bortrichlorid wird durch die reduktive Chlorierung von B2O3 gewonnen: B2O3 + 3C + 3Cl2 → 2BCl3 + 3 CO (bei 550°C) Diboran B2H6 entsteht durch Hydridolyse bei der Umsetzung von BCl3 mit etherischer LiAlH4-Lösung sowie beim Eintropfen von BF3·OEt2 in eine Lösung von NaBH4 in Diglym: 4BCl3 + 3LiAlH4 → 2(BH3)2 + 3LiAlCl4 4BF3 + 3NaBH4 → 2(BH3)2 + 3NaBF4 technisch wird Diboran durch die Hydridolyse von Bortrifluorid mit Natriumhydrid gewonnen: 2BF3 + 6 NaH → (BH3)2 + 6NaF Im Labor gewinnt man es durch Protolyse von Boranaten mit nichtoxidierenden Säuren: 2BH4- + 2H+ → (BH3)2 + H2 2BH4- + I2 → (BH3)2 + 2I- + H2 Bindungsverhältnisse: Beständigkeit: Bei Normalbedingungen ist Diboran bis zu einer Temperatur von 50°C metastabil und zersetzt sich oberhalb im wesentlichen zu: H2, B4H10, B5H9, B5H11, B10H14, sowie höhermolekularen, festen, gelben Boranen. Gasförmiges Diboran bildet bei höheren Drücken B4H10, welches sich in Anwesenheit von B2H6 zu B5H11 umwandelt. Durch Einwirken von starken Lewisbasen entstehen Addukte des Borans: B2H6 + 2D → 2D-BH3 D = CO, NH3, PH3, PF3 etc. In Wasser hydrolisiert es zu H2 und B(OH)3 B2H6 + 6H2O → 2 B(OH)3 + 6 H2 ∆H = -467kJ/mol B2H6 brennt unter hoher Wärmeentwicklung: B2H6 + 3O2 → B2O3 + 3H2O ∆H =-2066kJ/mol Enthält Diboran Spuren von höheren Boranen, so ist es schon bei Raumtemperatur selbstentzündlich. Borsäure B(OH)3 kristallisiert in einer Schichtstruktur, in der planare B(OH)3-Moleküle über Wasserstoffbrückenbindungen zu zweidimensionalen Schichten verbunden sind. Zwischen den Schichten wirken nur van der Waals-Kräfte. B(OH)3 + 2 H2O H3O+ + [B(OH)4]3H3BO3 → [B3O3(OH)4]- + H3O+ + H2O Bornitrit: Die technische Darstellung des Bornitrits erfolt in einer Ca3(PO4)2-Matrix durch umsetzen von B2O3 mit NH3 bei 800-1200°C: B2O3 + 2NH3 → 2BN + 3H2O reines Bornitrit: B2O3 + 3C + N2 → 2BN + 3CO (bei 1800-1900°C) Im Labor kann BN durch Schmelzen von Borax (Na2B4O7) mit Ammoniumchlorid dargestellt werden. Eigenschaften: Bornitrit existiert in vier Modifikationen. Die thermodynamisch stabilste Form ist das hexagonale α-BN welches eine dem Graphit entsprechende Schichtstruktur aufweist. Wegen der großen EN-Differenz zwischen Bor und Stickstoff sind jedoch die π-Elektronen nicht delokalisiert, sondern weitgehend am N lokalisiert. abb. riedel s. 581 BN ist ist thermisch sehr beständig und chemisch ziemlich inert. β-BN (kubisch) besitzt eine dem Diamanten entsprechende Struktur. 2. Kohlenstoff 2.1 Vorkommen: Kohlenstoff kommt in der Natur elementar als Diamant und als Graphit vor. Außerdem lässt es sich in Form von Carbonaten in großen Mengen finden. 2.2 chemische und physikalische Eigenschaften: Es existieren drei Modifikationen des Kohlenstoffs: Graphit, Diamant und Buckminster Fullerene. Der Diamant Im Diamanten sind alle Kohlenstoffatome tetraedrisch von 4 weiteren Kohlenstoffatomen umgeben (Kohlenstoff ist in diesem Falls sp3 hybridisiert). Aufgrund der hohen C-CBindungsenergie (348kJ/mol) ist der Diamant sehr hart und chemisch inert. Da alle Elektronen der sp3-Hybridorbitale lokalisiert sind, ist der Diamant farblos und elektrisch nicht leitend. Der Diamant ist metastabil und wandelt sich bei Temperaturen über 1500°C in Graphit um. In Gegenwart von Luftsauerstoff verbrennt er bei 800°C zu CO2. Graphit kristallisiert in einer Schichtstruktur. Die C-Atome sind dabei anders als im Diamanten sp2 hybridisiert und bilden mit ihren Nachbarn σ-Bindungen aus. Das vierte Elektron befindet sich in einem zur Ebene senkrechtstehenden p-Orbital. Diese p-Orbitale sind in der Lage delokalisierte (p-p)-π-Bindungen auszubilden, welche sich über die gesamte Ebene erstrecken. Daher beträgt der Abstand der C-C-Atome nur noch 142pm statt 154pm im Diamanten. Die gut beweglichen π-Elektronen sind für den metallischen Glanz, die schwarze Färbung sowie der guten elektrischen Leitfähigkeit (innerhalb einer Schicht) des Graphit verantwortlich. Zwischen den Schichten ist die Leitfähigkeit sehr viel geringer und die einzelnen Schichten werden lediglich durch van der Waals-Kräfte zusammengehalten (Abstand der Schichte 355pm). Dies erklärt die leichte Verschiebbarkeit der einzelnen Schichten. Man setzt Graphit daher als Schmiermittel und als Elektrodenmaterial. Graphit ist chemisch reaktionsfähiger als Diamant und verbrennt schon an Luft bei 700°C zu CO2. Bei hohen Drücken ist der Diamant thermodynamisch stabiler als Graphit. Kohlenwasserstoffe: Kohlenwasserstoffe bilden zahlreiche kettenförmige und ringförmige Verbindungen, wobei die Alkane die einfachsten Vertreter sind. Sie bilden kettenförmige Verbindungen der allgemeinen Form CnH2n+2. Die Alkane bilden eine homologe Reihe. Der einfachste Vertreter ist das Methan (CH4), darauf folgt das Ethan (C2H6) und das Propan (C2H8). Ab dem Butan gibt es eine Reihe von Strukturisomeren. In allen Alkanen ist das zentrale Kohlenstoffatom tetraedrisch verbunden mit je zwei Wasserstoffatomen und zwei Kohlenstoffatome. Alle Kohlenstoffatome sind in Alkanen sp³ hybridisiert. Kohlenwasserstoffe welche eine C-CDoppelbindungen aufweisen werden als Alkene bezeichnet. Die Kohlenstoffatome die an einer Doppelbindung beteiligt sind, sind sp² hybridisiert und bilden eine trigonal planare Geometrie. Die Verbindungen dreifach gebundener Kohlenstoffatome werden als Alkine bezeichnet. Hierbei sind die Kohlenstoffatome sp hybridisiert und bilden eine lineare Geometrie. 2.3 Verbindungen des Kohlenstoffs: 2.3.1 Calciumcarbid Calciumcarbid CaC2 ist von großtechnischer Bedeutung es wird durch die Umsetzung von Calciumoxid mit Koks im elektrischen Ofen erzeugt: CaO + 3C → CaC2 + CO ∆H= + 465kJ/mol Calciumcarbid kristallisiert in einer verzerrten Natriumchloridstruktur. Der größte Teil des Calciumcarbids wird zu Herstellung von Acetylen verwendet: CaC2 + H2O → CaO + C2H2 2.3.2 Calciumcyanid 2.3.3 Kohlenstoffmonoxid und Kohlenstoffdioxid CO und CO2 sind die wichtigsten Kohlenstoffoxide. Kohlenstoffmonoxid ist ein farbloses, geruchloses und sehr giftiges Gas. Die Moleküle CO und N2 sind isoelektronisch, in beiden Verbindungen sind die Atome durch eine σ-Bindung und durch 2 π-Bindungen. CO entsteht bei der unvollständigen Verbrennung von Kohlenstoff: C + ½ O2 → CO ∆H = -111kJ/mol Technisch wird CO fällt es bei der Wassergasreaktion an und ist Bestandteil des Leuchtgases. Im Labor kann man es durch Eintropfen von Ameisensäure in warme konz. H2SO4 darstellen: H 2SO 4 → H2O + CO HCOOH An der Luft verbrennt CO mit einer charakteristisch blauen Flamme zu CO2. CO wird häufig als Reduktionsmittel verwendet (beispielsweise bei der Eisendarstellung). Zusammen mit Übergangsmetallen bildet CO sehr stabile Carbonylkomplexe. Die Komplexbildung mit Übergangsmetallen ist auch der Grund für die giftige Eigenschaft des CO. Es komplexiert das Eisen des Hämoglobins und verhindert somit die O2-Aufnahme. Bedeutend ist CO für die Darstellung von Methanol und Ethanol von Bedeutung. Dabei reagiert CO zusammen mit H2 und es entstehen Alkanole oder gesättigte und ungesättigte Kohlenwasserstoffe: nCO + (2n+1) H2 → CnH2n+2 + nH2O nCO + 2nH2 → CnH2n + nH2O Diese Form der Umsetzung wird als Fischer-Tropf-Synthese bezeichnet. Setzt man Kohlenstoff mit Wasser bei 800-1000°C um, so erhält man das Synthesegas: H2O + C H2 + CO Hieran lässt sich das Wassergasgleichgewicht anschließen. Kohlenstoffdioxid CO2 ist ein farbloses, geruchloses Gas, das nicht brennt und die Verbrennung nicht unterhält. Es ist anderthalbmal dichter als Luft und sammelt sich somit am Boden geschlossener Räume. In Wasser lässt es sich gut lösen (0,9l CO2 in 1l Wasser). CO2 entsteht beim vollständigen Verbrennen von Kohlenstoff: C + O2 → CO2 ∆H = -394kJ/mol Es fällt außerdem beim Kalkbrennen an: CaCO3 ∆T → CO2 + CaO CO2 ist sehr beständig und lässt sich erst bei sehr hohen Temperaturen dissoziieren (bei 1200°C zu 0,03%; bei 2600°C zu 52%). CO2 CO + ½ O2 ∆H = +283kJ/mol Diese Reaktion wird als Boudouard-Gleichgewicht bezeichnet. CO2 kann nur schwer durch sehr starke Reduktionsmittel zu CO reduziert werden (zB. durch H2, C, Na oder Mg). Auch durch H2 wird CO2 nur bei sehr hohen Temperaturen zu reduzieren. Beim Wassergasgleichgewicht liegt das Gleichgewicht erst bei Temperaturen von über 1000°C auf der rechten Seite: CO2 + H2 CO + H2O ∆H = +41 kJ/mol Von technischer Bedeutung ist diese Reaktion bei der technischen Erzeugung von Wasserstoff. Setzt man CH4 und H2O miteinander um, so erhält man CO und H2: CO + 3H2 CH4 + H2O Das Gasgemisch wird als Spaltgas bezeichnet. 3. Stickstoff 3.1 Vorkommen: Stickstoff ist mit 78,1% (O2: 20,95%; CO2: 0,03%; Edelgase: 0,935%) Hauptbestandteil der Luft. Außerdem ist es im Chilesalpeter enthalten NaNO3. 3.2 Darstellung: Stickstoff wird ebenso wie Sauerstoff durch fraktionierte Destillation von Luft nach dem Linde-Verfahren gewonnen. Die angesaugte Luft wird im Verdichter bei 200bar kompromiert und dann im Kühler vorgekühlt und mittels Drosselventils wieder entspannt und dabei abgekühlt. Mit dieser abgekühlten Luft wird im Gegenstrom-Wärmetauscher die nachkommende verdichtete Luft vorgekühlt. Die Temperatur sinkt immer mehr, bis schließlich bei der Entspannung flüssige Luft entsteht. Bei Druckerniedrigung um 1bar sinkt die Temperatur um etwa ¼ °C. Die Abkühlung beim Linde-Verfahren beruht auf dem Joule-Thomson-Effekt. Demnach kühlt sich ein Gas beim wieder Ausdehnen ab. Bei der Ausdehnung muss Arbeit geleistet werden um die Anziehungskräfte zwischen den Gasteilchen zu überwinden. Die Energie dazu wird der inneren Energie des Gases entnommen, die kinetische Energie und damit die Temperatur nehmen ab. Im Labor kann man Stickstoff aus der Luft gewinnen. Dazu wird der Luftsauerstoff mittels Kupfer gebunden und man erhält im wesentlichen Stickstoff: 2Cu + O2 + 4N2 → 4N2 + 2CuO Außerdem lässt sich N2 durch Oxidation aus NH3 gewinnen. Dazu versetzt man NH3-Lösung mit salpetriger Säure: NH3 + HNO2 → N2 + 2H2O 3.3 wichtige Stickstoffverbindungen Ammoniak NH3: Ammoniak ist ein farbloses, stechend riechendes Gas, das sich leicht verflüssigen lässt. NH3 ist pyramidal gebaut, die Bindungswinkel betragen 107°. Flüssiges Ammoniak ist ein gutes Lösungsmittel für viele Salze. es bildet außerdem im flüssigen Zustand Wasserstoffbrücken aus. Wie Wasser findet in flüssigem Ammoniak die Autoprotolyse statt: 2NH3 NH4+ + NH2Alkalimetalle und Erdalkalimetalle lassen sich in flüssigem NH3 unter Bildung von solvatisierten Elektronen lösen: Me + NH3 Me+am. + e-am. Diese Lösungen sind sehr gute elektrische Leiter. Sie sind blau gefärbt und paramagnetisch. Solvatisierte Elektronen sind sehr starke Reduktionsmittel. Sie reduzieren viele Schwermetallkationen zum elementaren Zustand und die meisten Nichtmetalle zu Anionen. Die Lösungen sind metastabil und zersetzen sich beim Erwärmen oder bei Zusatz von Katalysatoren: NH3 + e-am. NH2- + ½ H2 ∆H= -67kJ/mol NH3 lässt sich gut in Wasser lösen und stellt wegen seines freien Elektronenpaars eine Lewisbase dar. Wässrige NH3-Lösungen reagieren schwach basisch: NH3 + H2O NH4+ + OHpKB= 4,75 Das Gleichgewicht liegt weit auf der linken Seite. Zusammen mit OH--Ionen reagieren Ammoniumsalze zu NH3 und Wasser: NH4Cl + OH- → NH3 + H2O + ClMit Protonendonatoren reagiert NH3 quantitativ zu den entsprechenden Ammoniumsalzen: NH3 + HCl → NH4Cl Großtechnisch wird Ammoniak mit dem Haber-Bosch-Verfahren aus den Elementen erzeugt: 3/2 H2 + ½ N2 NH3 ∆H=-46kJ/mol Darstellung von Amiden, Nitriden und Imiden: Beim Erhitzen reagiert gasförmiges Ammoniak mit Alkali- und Erdalkalimetallen zu Amiden: 2Na + 2NH3 → 2NaNH2 + H2 Aus Amiden der Erdalkalimetalle erhält man bei weiterem Erhitzen Imide: Ca(NH2) → CaNH + NH3 und schließlich Nitride: 3CaNH → Ca3N2 + NH3 In Wasser entsteht aus den Ionen sofort NH3. Hydrazin N2H4: N2H4 ist eine farblose Flüssigkeit, die an der Luft raucht. Im N2H4-Molekül ist eine N-NEinfachbindung vorhanden, die Bindungswinkel entsprechen etwa einer sp³-Hybridisierung. Die NH2-Gruppen sind um die N-N-Achse des Moleküls um ca. 100° verdrillt. Dadurch ist die Abstoßung der freien Elektronenpaare am geringsten. Im Gleichgewicht besteht N2H4 zu gleichen Teilen aus zwei spiegelbildlichen Isomeren, die sich mit hoher Frequenz ineinander umwandeln. Beim Erhitzen zerfällt Hydrazin explosionsartig: 3N2H4 → 4NH3 + N2 In Wasser ist es unbegrenzt löslich und kann gefahrlos gehandhabt werden. Wässrige Lösungen besitzen reduzierende und basische Eigesnchaften. Cu(II)-Salze werden zu Cu2O, Ag- und Hg-Salze zu den Metallen reduziert. Dabei entsteht N2. N2H4 + O2 → N2 + 2H2O ∆H =-623kJ/mol N2H4 ist eine schwächere Base als NH3 N2H4 + H2O N2H5+ + H2O N2H5+ + OHN2H62+ + OH- KB1 = 8·10-7mol/l KB2 = 8·10-16mol/l Es gibt zwei Reihen von Hydraziniumsalzen. N2H5+-Salze sind in Wasser beständig. N2H62+Salze wie N2H6Cl2 und N2H6SO4 hydrolysieren, da KB2 sehr klein ist: N2H62+ + H2O N2H5+ + H3O+ Hydrazin wird durch die Raschig-Synthese erzeugt. Dabei wird NH3 mit NaOCl oxidiert, wobei als Zwischenprodukt Chloramin NH2Cl auftritt: NH3 + NaOCl → NaOH + NH2Cl NH2Cl + NH3 + NaOH → N2H4 + NaCl + H2O Gesamtreaktion: 2NH3 + NaOCl → N2H4 + NaCl + H2O Da schon Spuren von Schwermetallen die Konkurrenzreaktion katalysieren wird EDTA zur Komplexierung zugesetzt: 2NH2Cl + N2H4 → 2NH4Cl + N2 Heutzutage wird Hydrazin über den Bayer-Prozeß gewonnen. Dabei wird NH3 in Anwesenheit von Aceton mit Natriumhypochlorid zu Hydrazin oxidiert: 2 NH3 + NaOCl + CH3COCH3 → (CH3)2C=N-N=C(CH3)2 + NaCl + 3H2O (CH3)2C=N-N=C(CH3)2 + 2H2O → CH3COCH3 + N2H4 Derrivate des Hydrazins sind als Polymerisationsinitiatoren, als Herbizide und Pharmaka von Bedeutung. Stickstoffwasserstoffsäure HN3 Wasserfreies HN3 ist eine farblose explosive Flüssigkeit: 2 HN3 + H2O → 3N2 + H2 ∆H = -538kJ/mol Wässrige Lösungen bis zu einem Massenanteil von 20% HN3 sind gefahrlos zu handhaben, sie reagieren schwach sauer. HN3 + H2O H3O+ + N3pKs= 4,9 Die Salze der Stickstoffwasserstoffsäure heißen Azide. Das N3--Ion ist ein Pseudohalogenidion. Schwermetallazide wie AgN3 und Pb(N3)2 sind schwerlösliche und explodieren beim Erwärmen. Pb(N3)2 wird als Initialzünder verwendet. Die Alkalimetall- und Erdalaklimetallazide lassen sich bei höheren Temperaturen kontrolliert zersetzten. 2NaN3 → 2Na + 3N2 bei 300°C HN3 ist ein starkes Oxidationsmittel. Metalle (Zn, Fe, Mn, Cu) lösen sich unter Stickstoffentwicklung. Me + 3HN3 → Me(N3)2 + N2 + NH3 Das Azidion ist linear und symmetrisch gebaut. Im Gegensatz dazu enthält das HN3-Molekül zwei unterschiedliche N-N-Bindungen. NaN3 stellt man durch Überleiten von N2O über NaNH2 her. NaNH2 + N2O → NaN3 + H2O bei 190°C Distickstoffmonoxid N2O N2O ist ein farbloses, reaktionsträges Gas. Es ist metastabil zerfällt aber erst bei Temperaturen oberhalb von 600°C in die Elemente. Es wird als Anästetikum verwendet, unterhält aber die Atmung nicht. Da es eingeatmet zu Halluzinationen und Lachlust hervorruft wird es als Lachgas bezeichnet. N2O wird durch thermische Zersetzung von Ammoniumnitrat gewonnen: NH4NO3 → N2O + 2 H2O (bei 200°C) ∆H =-124kJ/mol Oberhalb von 300°C kann explosionsartiger Zerfall von NH4NO3 erfolgen. Das N2O Molekül ist linear gebaut, isoelektrisch mit CO2, N3- und NO2+ und kann durch die folgende Grenzstruktur beschrieben werden: Stickstoffmonoxid NO NO ist ein farbloses, giftiges Gas, das aus N2 und O2 in endothermer Reaktion entsteht: ½ N2 + ½ O2 NO ∆H = +90kJ/mol Bei Raumtemperatur liegt das Gleichgewicht vollständig auf der linken Seite. NO ist ein Zwischenprodukt bei der Salpetersäureherstellung. 4 NH3 + 5O2 → 4 NO + 6H2O (bei 800-950°C; Pt) ∆H = -960kJ/mol Ein NH3-Luft-Gemisch wird über einen Platinnetz-Katalysator geleitet. Die Kontaktzeit am Katalysator beträgt nur etwa 1/1000 s. Dadurch wird NO sofort aus der heißen Reaktionszone entfernt und auf Temperaturen abgeschreckt, bei denen das metastabile NO nicht mehr in die Elemente zerfällt. Im Labor wird NO durch Reduktion von Salpetersäure mit Kupfer hergestellt: 8 H3O+ + 2 NO3- + 3 Cu → 3 Cu2+ + 2 NO + 12 H2O Da NO 11 Valenzelektronen besitzt und daher das π*-Orbital nur einfach besetzt ist, ist das Molekül paramagnetisch. Wie auch CO kann NO Komplexe mit Übergangsmetallen bilden. Ein Beispiel dafür ist die Ringprobe: NO + [Fe(H2O)6]2+ → [Fe(H2O)5NO]2+ + H2O Für das Radikal sollte man eine Dimerisierung erwarten. Dies ist allerdings erst im kondensierten Zustand der Fall. Mit Sauerstoff reagiert NO spontan zu NO2: 2 NO + O2 2 NO2 ∆H = -114kJ/mol Distickstofftrioxid N2O3 N2O3 entsteht als blaue Flüssigkeit beim Abkühlen einer Mischung aus gleichen Stoffmengen der beiden Radikalmolekülen NO2 und NO. NO + NO2 N2O3 ∆H = -40kJ/mol Bereits oberhalb -10°C zerfällt N2O3 in Umkehrung der Bildungsgleichung, bei 25°C enthält der Dampf nur noch 10% undissoziiertes N2O3. N2O3 ist das Anhydrid der salpetrigen Säure. Mit Laugen reagiert N2O3 daher zu Nitriten: N2O3 + 2 OH- → 2 NO2- + H2O Das N2O3-Molekül ist planar gebaut und enthält eine schwache N-N-Bindung, es kann als Nitrosylnitrit beschrieben werden. Stickstoffdioxid NO2; Distickstofftetraoxid N2O4 NO2 ist ein braunes, giftiges, paramagnetisches Gas, das zu farblosem diamagnetischen N2O4 dimerisiert. 2 NO2 N2O4 ∆H = -57kJ/mol Bei 27°C sind 20%, bei 100°C 90% N2O4 dissoziiert. Bei -11°C erhält man farblose Kristalle von N2O4. NO2 ist ein Zwischenprodukt bei der Salpetersäureherstellung. Im Labor erhält man es durch thermische Zersetzung von Schwermetallnitraten im Sauerstoffstrom. Pb(NO3)2→ PbO + 2 NO2 + 1/2 O2 bei 250-600°C Oberhalb von 150°C beginnt NO2 sich in NO und O2 zu zersetzen, bei 600°C ist der Zerfall vollständig. NO2 → 2NO + O2 NO2 und N2O4 sind starke Oxidationsmittel. NO2 ist das gemischte Anhydrid der Salpetersäure und der salpetrigen Säure. Mit Lauge reagiert NO2 bzw. N2O4 nach: N2O4 + 2 OH- → NO3- + NO2- + H2O NO2 ist gewinkelt und kann mit den folgenden mesomeren Grenzstrukturen beschrieben werden: N2O4 besteht in der Gasphase und auch im festen Zustand aus planaren Molekülen mit einer schwachen N-N-Bindung. Distickstoffpentaoxid N2O5 N2O5 ist das Anhydrid der Salpetersäure und kann aus dieser durch Entwässern mit P4O10 erhalten werden. 2 HNO3 → N2O5 + H2O N2O5 bildet farblose Kristalle, die bei 32°C sublimieren, mit Wasser zu HNO3 reagieren und sich bereits bei Raumtemperatur zu NO2 und O2 zersetzen. Festes N2O5 besitzt ionogene Struktur [NO2+][NO3-] und ist also ein Nitrylnitrat. Im gasförmigen Zustand liegt die folgende Struktur vor. Salpetersäure HNO3 HNO3 wird großtechnisch durch Einleiten von N2O4 in Wasser hergestellt, wobei zur Oxidation noch Sauerstoff erforderlich ist. N2O4 + H2O + ½ O2 → 2 HNO3 (bei 20-35°C; 3-10bar) Im einzelnen laufen folgende Reaktionen ab: Aus N2O4 entsteht mit Wasser durch Disproportionierung Salpetersäure und Salpetrige Säure. N2O4 + H2O → HNO3 + HNO2 HNO2 → HNO3 + 2 NO + H2O NO reagiert mit Luftsauerstoff zu NO2, das überwiegend dimerisiert. 2 NO + O2 → N2O4 Letztlich wird Salpetersäure durch mehrere großtechnische Reaktionen aus dem Stickstoff der Luft hergestellt: Haber − Bosch − Verfahren / + H 2 → NH3 Ostwald − Verfahren / + O 2 +O2 + O , H 2O → NO → NO2 2 → HNO3 N2 Wasserfreie HNO3 ist eine farblose Flüssigkeit. Beim Sieden erfolgt eine teilweise Zersetzung, die schon durch Lichteinwirkung bei Raumtemperatur einsetzt. 4 HNO3 → 4NO2 + 2 H2O + O2 HNO3 wird daher in braunen Flaschen aufbewahrt. HNO3 ist ein starkes Oxidationsmittel. NO + 6 H2O NO3- + 4 H3O+ + 3eE0= +0,96V Die konzentrierter Säure löst Kupfer, Quecksilber und Silber, nicht aber Gold und Platin. 3 Cu + 2 NO3- + 8 H3O+ → 3 Cu2+ + 2 NO + 12 H2O Einige unedle Metalle (Cr, Al, Fe) werden von konz. HNO3 nicht gelöst, da sich auf ihnen eine dichte Oxidhaut bildet, die das Metall vor weiterer Säureeinwirkung schützt (Passivierung). Diese Metalle lösen sich nur in verdünnter HNO3. Die Mischung von konz. HNO3 und konz HCl im Volumenverhältnis von 1:3 heißt Königswasser. Es löst fast alle Metalle, auch Gold und Platin, da aktives Chlor entsteht und mit dem Metallionen Chlorokomplexe gebildet werden, die das Redoxpotential beeinflussen. HNO3 + 3 HCl → NOCl + 2 Cl + 2 H2O Wie beim HNO3-Molekül ist das N-Atom sp² hybridisiert, die völlige Delokalisierung des πElektronenpaares führt zu einer Stabilisierung des NO3--Ions, daher ist das NO3--Ion stabiler als das HNO3-Molekül. Nitrate sind in Wasser leicht löslich. Alkalimetallnitrat zersetzen sich beim Erhitzen in Nitrite, während aus Schwermetallnitraten NO2 und Metalloxide entstehen. KNO3 → KNO2 + ½ O2 Hg(NO3)2 → HgO + 2 NO2 + ½ O2 Nitrate sind, besonders bei höheren Temperaturen, Oxidationsmittel. Durch starke Reduktionsmittel wird das NO3--Ion zu NH3 reduziert. NaNO3 (Chilesalpeter), KNO3 (Salpeter) ist im ältesten Explosivstoff Schwarzpulver enthalten, der aus einer Mischung von Schwefel, Holzkohle und Kaliumnitrat besteht. Salpetrige Säure HNO2 HNO2 ist in reinem Zustand nicht darstellbar, sondern nur in verdünnter Lösung einige Zeit haltbar. HNO2 ist eine mittelstarke Säure, sie zersetzt sich unter Disproportionierung. 3 HNO2 → HNO3 + 2 NO + H2O HNO2 kann je nach Reaktionspartner reduzierend oder oxidierend wirken. Als Reduktionsmittel NO2- + 3 H2O NO3- + 2 H3O+ + 2eE = +0,94V fungiert sie gegenüber MnO4-, PbO2 und H2O2, als Oxidationsmittel gegenüber I- und Fe2+. NO2- + 2 H3O+ + eNO + 3 H2O E = +0,996V Mit NH3 reagiert HNO2 zu N2: NH3 + HNO2 N2 + 2 H2O HNO2 besteht aus planaren cis- und trans-Isomeren. Darstellung von Nitriten: NO + NO2 (bzw. N2O4) + 2 NaOH → 2 NaNO2 + H2O Erhitzen KNO3 → KNO2 + ½ O2 4. Phosphor 4.1 Vorkommen: Da Phosphor sehr reaktionsfähig ist, kommt er in der Natur nur in Verbindungen vor. Die wichtigsten Mineralien sind die Phosphate. Häufig ist Apatit Ca5(PO4)3(OH, F, Cl). 4.2 Darstellung: Phosphor wird aus Calciumphosphat durch Reduktion mit Koks bei 1400°C im Lichtbogenofen hergestellt, wobei der Phosphor als Dampf entweicht und als weißer Phosphor gewonnen wird. Quarzsand wird als Schlackenbildner zugesetzt. 2 Ca(PO4)2 + 6 SiO2 + 10 C → 6 CaSiO3 + 10 CO + P4 4.3 physikalische und chemische Eigenschaften Modifikationen Weißer Phosphor: Weißer Phosphor entsteht bei der Kondensation von Phosphordampf. Er ist wachsweich, weiß bis gelblich, schmilzt bei 44°C und löst sich in CS2, nicht in H2O. Er ist sehr reaktionsfähig und sehr giftig. Er verbrennt zu P4O10 in feinverteilter Form entzündet er sich an der Luft von selbst. Er wird daher unter Wasser aufbewahrt. Im Dunklen leuchtet weißer Phosphor. Er bildet P4 Tetraeder. Roter Phosphor: Erhitz man weißen Phosphor unter Luftabschluß auf 180-400°C, so wandelt er sich in den polymeren, amorphen roten Phosphor um. Iod beschleunigt die Umwandlung katalytisch. Er besteht aus einem unregelmäßigen, dreidimensionalen Netzwerk, dessen Ordnungszustand von der Temperatur und Temperzeit abhängig. Roter Phosphor ist ungiftig und luftstabil und entzündet sich erst oberhalb von 300°C. Violetter Phosphor (Hittorfscher Phosphor) entsteht beim Erhitzen von rotem Phosphor auf 550°C, er kristallisiert in einer komplizierten Schichtstruktur. Schwarzer Phosphor ist bei Standarddruck bis 550°C thermodynamisch stabile Modifikation. Er entsteht aus weißem Phosphor bei 200°C und 12bar oder bei 380°C in Gegenwart von Hg als Katalysator. Schwarzer Phosphor zeigt Metallglanz, ist ein elektrischer Halbleiter und reaktionsträge. Es kristallisiert in einer rhombischen Schichtstruktur, die aus Doppelschichten besteht. Oberhalb von 550°C erfolgt Umwandlung in violetten Phosphor, der bis 620°C die stabile Modifikation ist. Bei 620°C sublimiert er bei Normaldruck, bei einem Druck von 49bar schmilzt er. Gas und Schmelze bestehen aus P4-Molekülen. 4.4 Phosphorverbindungen Phosphan PH3 Das PH3-Molekül ist pyramidal gebaut. PH3 kann in wässriger Lösung aus Phosphoniumsalzen gewonnen werden. PH4+ + H2O → PH3 + H3O+ Hydrolyse von Phosphiden: Mg2P2 + 6 HCl → 2 PH3 + 3 MgCl2 PH3 Darstellung aus weißem Phosphor und Kalilauge unter Erwärmen: P4 + 3 KOH + 3 H2O → PH3 + 3 KH2PO2 Phosphan ist ein farbloses, knoblauchartig riechendes , sehr giftiges Gas. Mit Hydrogenhalogeniden bilden sich Phosphoniumsalze, die in wässriger Lösung hydrolytisch zersetzt werden. PH4I + H2O → PH3 + H3O+ + IPhosphortrichlorid PCl3 Die technische Darstellung von PCl3 erfolgt durch direkt Umsetzung von trockenem Chlorgas mit gasförmigem weißem Phosphor in einem Brenner: ¼ P4 + 1½Cl2 → PCl3 Der Phosphor entzündet sich dabei von selbst und verbrennt mit fahler Flamme, während in die gekühlte Vorlage ein Gemisch von Phosphortrichlorid PCl3 und etwas Phosphorpentachlorid PCl5 destilliert. Um letzteres zu entfernen, fügt man zum Destillat etwas weißen Phosphor hinzu und destilliert erneut (6 PCl5 + P4 → 10 PCl3). Die technische Gewinnung erfolgt auch durch Einleiten von Chlor in eine P4-haltige PCl3-Lösung. Hierbei wird P4 kontinuierlich zugegeben, gebildetes PCl3 kontinuierlich abdestilliert. Eigenschaften: Phosphortrichlorid ist eine farblose, stechend riechende, bei 75°C siedende und bei -93,6°C erstarrende Flüssigkeit. Von Wasser wird PCl3 sehr leicht unter Bildung von Phosphonsäure und Salzsäure zersetzt und raucht daher an feuchter Luft stark: PCl3 + 3 HOH → P(OH)3 + 3 HCl Phosphorpentachlorid PCl5 Die technische Darstellung von PCl5 erfolgt in mit Blei ausgekleideten Türmen, in welchen man PCl3 (von oben) und Cl2 (von unten) einander entgegenführt: PCl3 + Cl2 PCl5 PCl5 sammelt sich am Boden an und wird dort ausgetragen. Eigenschaften: PCl5 stellt im reinen Zustand eine weiße, gewöhnlich aber wegen teilweiser Spaltung in PCl3 und Cl2 grünlich weiße Masse dar. Beim Erhitzen unter Normaldruck sublimiert PCl5 bei 159°C, ohne zu schmelzen. PCl5 ist im gasförmigen Zustand trigonal-bipyramidal gebaut. Im festen Zustand liegt es in ionischer Form vor [PCl4]+[PCl6]- . Wegen der leichten Abspaltbarkeit von Chlor wird Phosphorpentachlorid vielfach als Chlorierungsmittel benutzt. An der Luft zieht Phosphorpentachlorid Wasser an und geht in Phosphorylchlorid bzw. Phosphorsäure sowie Chlorwassertoff über und raucht daher an feuchter Luft: PCl5 + H2O − 2 HCl −3HCl → POCl3 + 3 H2O → PO(OH)3 PCl5 stellt eine Lewissäure dar und bidet z.B. mit Chloriddonatoren Chlorokomplexe PCl6-. Außerdem wirkt PCl5 als Lewisbase und verbindet sich mit vielen lewissauren Chloridakzeptoren zu Phosphonium-Salzen gemäß: PCl5 + MCln → [PCl4]+[MCln+1]Phosphor(III)-oxid P4O6 P4O6 entsteht bei der Oxidation von Phosphor mit der stöchiometrischen Menge Sauerstoff als sublimierbare, wachsartige, giftige Masse: P4 + 3 O2 → P4O6 ∆H = 1641kJ/mol Die Struktur lässt sich aus dem P4-Molekül ableiten; die P-P-Bindungen sind durch P-O-PBindungen ersetzt. Phosphor(III)-oxid besteht in allen Phasen und auch in Lösung aus P4O6Molekülen. Bei 25°C ist P4O6 an der Luft beständig, bei 70°C verbrennt es zu P4O10. Nur mit kaltem Wasser erfolgt Reaktion zu Phosphonsäure, dessen Anhydrid P4O6 ist: P4O6 + 6 H2O → 4 H2PHO3 Mit heißem Wasser außerdem P, PH3 und H3PO4. Phosphor(V)-oxid P4O10 P4O10 entsteht bei der Verbrennung von Phosphor in überschüssigem Sauerstoff als weißes, geruchloses Pulver, das bei 358°C sublimiert. P4 + 5 O2 → P4O10 ∆H = -2986kJ/mol Die Struktur leitet sich ebenfalls vom P4-Tetraeder ab. Jedes P-Atom ist tetraedrisch von Sauerstoff umgeben. Das P-Atom ist sp³ hybridisiert, es bildet vier tetraedrische σ-Bindungen und eine π-Bindung: P4O10 reagiert mit Wasser äußerst heftig über Zwischenstufen zu Orthophosphorsäure. P4O10 + 6 H2O → 4 H3PO4 ∆H = -378kJ/mol P4O10 ist eine der wirksamsten wasserentziehenden Substanzen und dient als Trockenmittel und zur Darstellung von Säureanhydriden. An der Luft zerfließt P4O10 zu einem sirupösen Gemisch von Phosphorsäuren. Im Gegensatz zu N2O5 ist P4O10 kein Oxidationsmittel. Durch Erhitzen im abgeschlossenen System auf 450°C wandelt sich das aus P4O10-Molekülen aufgebaute Phosphor(V)-oxid nacheinander in zwei polymere Formen mit einer Schichtstruktur und einer Raumnetzstruktur um: H2PHO3 Die Darstellung erfolgt durch Umsetzen von PCl3 mit Wasser: PCl3 + 3 H2O → H3PO3 + 3 HCl bzw. P2O3 + 3 H2O → 2 H3PO3 Eigenschaften: Die reine Phosphonsäure bildet farblose, in Wasser sehr leicht lösliche Kristalle vom Schmelzpunkt 73,8°C. Als zweibasige Säure dissoziiert sie in zwei Stufen und bildet zwei Reihen von Salzen: primäre Phosphonate MH[PHO3] (Hydrogenphosphonate) und sekundäre Phosphonate M2[PHO3] (Phosphonate). Von diesen sind die Alkaliphosphonate in Wasser leicht, die anderen schwer löslich. Die primären Phosphonate gehen beim Erwärmen unter vermindertem Druck in Diphosphonate über: 2 HPHO3- → HO2P-O-P-PO2H2- + H2O Charakteristisch für die Phosphonsäure ist ihr starkes Reduktionsvermögen, da sie das Bestreben hat, in die höhere Oxidationsstufe der Phosphorsäure überzugehen: H3PO3 + H2O H3PO4 + 2 H+ + 2eSo reduziert es sich beim Erhitzen selbst zu PH3: H3PO3 + 3 H3PO3 → H3P + 3 H3PO4 H3PO4 Als Ausgangsmaterial dient Apatit Ca5(PO4)3(F, OH, Cl). Ihre Überführung in Phosphorsäure erfolgt durch nassen Aufschluss mit Schwefelsäure und durch trockenen Aufschluss mit Koks und Quarz im elektrischen Ofen auf dem Wege über weißen Phosphor. Der Phosphat-anteil der gemahlenen Apatits reagiert gemäß: Ca3(PO4)2 + 3 H2SO4 → 3 CaSO4 + 2 H3PO4 Eigenschaften: Phosphorsäure ist eine dreibasige mittelstarke Säure und bildet dementsprechend drei Reihen von Salzen: primäre Phosphate (Dihydrogenphophate) MH2PO4, sekundäre Phosphate (Hydrogenphosphate) M2HPO4 und tertiäre Phosphate (Phosphate) M3PO4. Die Dissoziation der Säure erfolgt in drei Stufen H3PO4 H2PO4- + H+ HPO42- + H+ PO43- + H+ Im geschmolzenen Zustand leitet die wasserfreie Phosphorsäure gut den elektrischen Strom, was auf die Bildung von Phosphatacidium-Ionen (P(OH)4+) hinweist. 2 H3PO4 H4PO4+ + H2PO4Anders als die homologe Salpetersäure ist Phosphorsäure kein Oxidationsmittel, da die Affinität des Phosphors zu Sauerstoff wesentlich größer ist als die des Stickstoffs. Phosphorsäure ist daher ein gutes Reduktionsmittel. Die primären Phosphate lassen sich alle gut in Wasser lösen. Bei den sekundären und tertiären Phosphate lassen sich nur die Alkalisalze gut lösen. Diphosphate sind die Salze der Diphosphorsäure Sie entsteht beim Erhitzen von H3PO4 auf Temperaturen über 200°C. Dabei erfolgt eine intermolekulare Wasserabspaltung: abb. riedel s.492 Diphosphate erhält man durch erhitzen von Hydrogenphosphaten: 2 Me2HPO4 → Me4P2O7 + H2O Na2H2P2O7 wird als Treibmittel im Backpulver verwendet. 5. Sauerstoff 5.1 Vorkommen: Sauerstoff ist zu ca. 20% in der Luft enthalten und außerdem in einer Reihe von Metalloxiden. 5.2 Darstellung: Sauerstoff wird großtechnisch durch fraktionierte Destillation von verflüssigter Luft (nach dem Linde-Verfahren) hergestellt. Die angesaugte Luft wird im Verdichter bei 200bar komprimiert und dann im Kühler vorgekühlt und mittels Drosselventils wieder entspannt und dabei abgekühlt. Mit dieser abgekühlten Luft wird im Gegenstrom-Wärmetauscher die nachkommende verdichtete Luft vorgekühlt. Die Temperatur sinkt immer mehr, bis schließlich bei der Entspannung flüssige Luft entsteht. Bei Druckerniedrigung um 1bar sinkt die Temperatur um etwa ¼ °C. Die Abkühlung beim Linde-Verfahren beruht auf dem Joule-Thomson-Effekt. Demnach kühlt sich ein Gas beim wieder Ausdehnen ab. Bei der Ausdehnung muss Arbeit geleistet werden um die Anziehungskräfte zwischen den Gasteilchen zu überwinden. Die Energie dazu wird der inneren Energie des Gases entnommen, die kinetische Energie und damit die Temperatur nehmen ab. Chemisch kann man Sauerstoff durch Erhitzen von BaO an Luft isolieren 2 BaO + O2 500°C 2 BaO2 700°C Im Labor lässt sich Sauerstoff durch katalytische Zersetzung von H2O2 darstellen: 2 H2O2 → 2 H2O + O2 Durch Elektrolyse von Kalilauge Kathode: H3O+ + 2e- → H2 + 2 H2O Anode: 2 OH- → H2O + ½ O2 + 2e5.3 chemische und physikalische Eigenschaften Unter Normalbedingungen ist Sauerstoff (ein Dimer O2) ein farbloses, geruch- und geschmackloses Gas, das verflüssigt und im festen Zustand hellblau ist. In Wasser ist O2 (0,049l/l bei 0°C und 1bar) etwas besser löslich als N2. Im O2-Molekül sind die Sauerstoffatome durch eine σ-Bindung und eine π –Bindung miteinander verbunden. Die Bindungslänge beträgt 121pm. Die Lewisformel beschreibt das Molekül unzureichend, da Sauerstoff paramagnetisch ist und zwei ungepaarte Elektronen besitzt. Mit der MO-Theorie ist sowohl die Bindungsordnung als auch der Paramagnetismus zu verstehen. O O Das O2- Molekül ist ziemlich stabil und es dissoziiert erst bei hohen Temperaturen. Bei 3000°C beträgt der Dissoziationsgrad 6%. O2 → 2 O ∆H = 498kJ/mol Die Umsetzung mit Sauerstoff erfolgt meist erst bei hohen Temperaturen. Mit vielen Stoffen erfolgen langsame Oxidationen, z.B. das Rosten und das Anlaufen von Metallen. In reinem Sauerstoff laufen Oxidationen viel schneller ab. Ein glimmender Holzspan brennt in Sauerstoff mit heller Flamme, Schwefel verbrennt mit intensiv blauem Licht zu SO2. S + O2 → SO2 Noch stärker laufen Oxidation in flüssigem Sauerstoff. Singulettsauerstoff Mit Triplettsauerstoff bezeichnet man den normalen Sauerstoff, bei dem sich im antibbindenden π*-MO zwei Elektronen mit parallelem Spin befinden. Der Singulettsauerstoff ist energetisch angeregter Sauerstoff bei dem die Spinrichtung kurzzeitig antiparallel sind. Somit ist Singulettsauerstoff nicht mehr paramagnetisch, sondern diamagnetisch: Ozon O3 Sauerstoff kommt in einer weiteren Modifikation vor, dem Ozon. Ozon ist ein charakteristisch riechendes, blassblaues Gas, das sich bei -111°C verflüssigen lässt und bei -193°C in den festen Zustand über geht. Die kondensierte Phasen sind schwarzblau und diamagnetisch. Ozon besteht aus gewinkelten O3-Molekülen (Bindungswinkel 117°C), die beiden O-OAbstände sind gleich lang (128pm), es ist daher eine delokalisierte π-Bindung vorhanden. (+) (+) O O O O O (-) (-) O Ozon ist eine endotherme Verbindung: 3/2 O2 → O3 ∆H = 143kJ/mol Reines Ozon, besonders in kondensiertem Zustand, ist explosiv. In verdünntem Zustand erfolgt bei Normaltemperatur nur allmählicher Zerfall, der sich beim Erwärmen und in Gegegenwart von Katalysatoren beschleunigt. O3 ist ein starkes Oxidationsmittel. PbS wird zu PBSO4 oxidiert, S zu SO3. Das Standardpotential zeigt, dass das Oxidationsvermögen von O3 fast das des atomaren Sauerstoffs erreicht und nur von wenigen Stoffen übertroffen wird. O2 + 3 H2O O3 + H3O+ + 2e∆H = 2,07V + 3 H2O O + 2 H3O + 2e ∆H= 2,42V Beim Einleiten von O3 in eine KI-Lösung entsteht I2. O3 + 2 I- + H2O → I2 + O2 + 2 OHDurch Titration des Iods kann O3 quantitativ bestimmt werden. Darstellung ½ O2 O + O2 O O3 1½ O2 O3 Um die einzelnen O-Atome zu gewinnen muss Energie zugeführt werden. Dies kann in Form von Wärme, Lichtenergie oder elektrischen Strom erfolgen. Es ist jedoch zweckmäßig die Gase nicht zu hoch zu temperieren, da somit die Zersetzung des Ozons bevorzugt abläuft. Man erhält aufgrund der Abbaureaktion O3 + O → 2 O2 auch nach dem Siemensschen Oszillator nur ein Gemisch mit 10% O3. Durch fraktionierte Destillation lässt sich aber reines O3 gewinnen. Beim Einwirken von Fluor auf Wasser entsteht elementarer Sauerstoff, welcher mit O2 zu Ozon reagieren kann: F2 + H2O → 2 HF + O Wasserstoffperoxid H2O2 ist eine sipuröse, fast farblose Flüssigkeit. In den Handel kommt sie als 30%ige Lösung. Wasserstoffperoxid besitzt die Strukturformel: aber es liegt in verdrillter Ketten von 4 Atomen vor. Die O-O-Bindung ist schwach, die Bindungsenergie ist klein. H2O2 ist daher eine metastabile Verbindung, die sich bei höherer Temperatur – eventuell auch explosionsartig- zersetzt. H2O2 → H2O + ½ O2 ∆H = -98kJ/mol Die Zersetzung wird durch Spuren von Schwermetallkationen wie Cu2+ oder Pt und alkalisch reagierenden Stoffen katalysiert. Stabilisierend wirkt Phosphorsäure. H2O2 ist eine sehr schwache Säure (Ks= 10-12). Gegenüber vielen Verbindungen wirkt H2O2 sowohl in saurer als auch in alkalischer Lösung oxidierend. H2O2 + 2 H3O+ + 2e4 H2O E = +1,78V H2O2 oxidiert SO2 zu SO42-, NO2- zu NO3-, Fe(II) zu Fe(III), Cr(III) zu Chromat. Gegenüber starken Oxidationsmitteln wirkt es reduzierend. H2O2 + 2 H2O O2 + 2 H3O+ + 2e- E= +0,68V Dies ist gegenüber MnO4-, Cl2, Ce(IV), PbO2 und O3 der Fall. Die Reaktion 2 MnO4- + 6 H3O+ + 5 H2O2 → 2 Mn2+ + 14 H2O + 5 O2 wird titrimetrisch zur Bestimmung von H2O2 benutzt. H2O2 bildet ein tiefblaues Chromperoxid CrO5 und ein gelbes Peroxotitanylion [TiO2]2+, die zum H2O2-Nachweis geeignet sind. Darstellung: H2O2 lässt sich durch elektrolytische Oxidation von H2SO4-SO42--Lösung zu Peroxodisulfat und daraus per Hydrolyse zu H2O2 synthetisieren: Heute wird es zum größten Teil nach dem Anthrachinon-Verfahren hergestellt. Dabei wird Anthrachinon zu Anthrahydrochinon hydriert. Durch Oxidation mit Luftsauerstoff entsteht H2O2 und Anthrachinon, das wieder hydriert werden kann. O22-: Peroxide enthalten den Sauerstoff mit der Oxidationsstufe -1. Ionische Peroxide enthalten das Anion O22- und reagieren unter Kühlung in Wasser zu alkalischen H2O2 Lösungen: Na2O2 + 2 H2O → H2O2 + NaOH Ohne Kühlung zersetzt sich wegen der Temperaturerhöhung und der katalytischen Wirkung des OH- das gebildete H2O2 sofort unter O2-Entwicklung. Na2O2 + H2O → 2 NaOH + ½ O2 O2-: Hyperoxid entstehen beim Verbrennen von Rb, Cs und K in Sauerstoff. Sie besitzen Sauerstoffatome mit der formalen Oxidationsstufe -½. Sie kristallisieren in einer tetragonalverzerrten NaCl-Struktur. Hyperoxide sind paramagnetisch und sind starke Oxidationsmittel. Mit Wasser reagieren sie heftigst unter Disproportionierung: 2 O2- + 2 H2O → O2 + H2O2 + 2OHDioxygenylverbindungen Sie enthalten das Kation O2+ mit der Oxidationsstufe +½. Das O2+-Kation ist paramagnetisch. Die Entferung eines Elektrons aus einem der π*-Orbitale erfordert eine hohe Ionisierungsenergie. O2 → O2+ + e- ∆H = 1168kJ/mol Der Reaktionspartner muß daher eine große Elektronenaffinität haben: Beispiel O2 + PtF6 → O2+[PtF6]Außerdem sind bekannt: O2BF4, O2PF6, O2SbF6, O2AuF6… Die O-O-Bindung wird in der Reihe vom O2+ zum O22- geschwächt. Dies ist nach den MOEnergieniveau-Diagramme auch zu erwarten. 6. Schwefel 6.1 Vorkommen: Verbindungen des Schwefels, vor allem die Schwermetallsulfide besitzen größte Bedeutung als Erzlagerstätten. Einige Mineralien sind: FeS2 (Zinkblende), ZnS, PbS(Bleiglanz) 6.2 Darstellung Ein kleiner Teil des Weltverbrauchs an Schwefel wird durch elementaren Schwefel gedeckt. Meist wird Schwefel aus H2S-haltigem Gas nach dem Claus-Prozeß gewonnen. Zuerst wird dazu in einer Brennerkammer H2S zu SO2 oxidiert: H2S + 3/2 O2 → SO2 + H2O ∆H = -581kJ/mol Die Sauerstoffzufuhr muss so geregelt werden, dass sich ein Verhältnis H2S/SO2 = 2 einstellt. Dieses Gemisch reagiert in hintereinander geschalteten Reaktoren katalytisch zu Schwefel. 2 H2S + SO2 → 3 S + 2 H2O bei 200 bis 300°C Diese Reaktion findet auch in der Brennerkammer statt, so dass dort bereits 60% des H2S in Schwefel umgewandelt werden. Schwefel wird vor allem in großen Mengen zur Herstellung von Schwefelsäure gebraucht. 6.3 chemische und physikalische Eigenschaften Modifikationen des Schwefels Der Schwefel besitzt eine ausgeprägte Tendenz, Ringe oder Ketten auszubilden. Dabei ist die S8-Konfiguration am stabilsten. Die S-Atome sind durch Einfachbindungen verbunden. Bei Normalbedingungen ist der rhombische α-Schwefel thermodynamisch stabil. Dabei liegen 16 S8-Moleküle in einer Elementarzelle. Die Kristalle sind hellgelb und spröde. Sie lassen sich nicht in Wasser lösen, aber sehr gut in CS2. Erhitzt man den α-Scwefel auf 95,6°C, so erfolgt eine reversible Umwandlung in den monoklinen β-Schwefel, der ebenfalls aus S8-Molekülen besteht. Bei Raumtemperatur wandelt sich der β-Schwefel langsam wieder in den rhombischen α-Schwefel ab. Der Dampfdruck des Schwefels ist bei 100°C so groß, dass der Schwefel sublimiert. Der βSchwefel schmilzt bei 119,6°C. Die Schmelze besteht aus S8-Ringen (γ-Schwefel), und bei sofortigem Abkühlen erstarrt sie wieder bei 119,6°C. Nach längerem Stehen erstarrt die Schmelze bei 114,5°C (natürlicher Schmelzpunkt). Die Schmelzpunkterniedrigung ist auf die Bildung von etwa 5% an Fremdmolekülen in der Schmelze zurückzuführen (2,8% S7; 0,5% S6; 1,5% > S8). In der Nähe des Schmelzpunktes ist der Schwefel hellgelb und dünnflüssig. Mit steigender Temperatur wächst der Anteil an niedermolekularen Schwefelringen Sn (πSchwefel; n = 6-26; hauptsächlich 6, 7, 9, 12) sowie hochmolekularen Schwefelketten Sx (µSchwefel; x = 103-106). Bei 159°C nimmt die Viskosität sprunghaft zu, die Schmelze wird dunkelrot, das Gleichgewicht verschiebt sich drastisch in Richtung µ-Schwefel. Durch Abschrecken dieser Schmelze erhält man plastischen Schwefel, der hochmolekulare Schwefelketten enthält. Er ist instabil und wandelt sich nach kurzer Zeit in kristallinen Schwefel um. Bei 187°C erreicht die Viskosität ein Maximum, bei höheren Temperaturen nimmt die Molekülgröße infolge thermischer Crackung ab und beim Siedepunkt ist die Schmelze dunkel-rotbraun und wieder dünnflüssig. In der Gasphase existiert ein temperaturabhängiges Gleichgewicht von Molekülen Sn mit n = 1-8. S-Atome überwiegen erst bei 2200°C. S8, S7, S6, S5 sind ringförmig gebaut. S4 ist kettenförmig und von roter Farbe. S3 ist blau und wie O3 gewinkelt gebaut. S2 ist blauviolett, paramagnetisch und enthält eine Doppelbindung (die Elektronenkonfiguration ist analog der von O2). Die Reaktion 4 S2 (g) → S8 (g) ∆H = -412kJ/mol ist exotherm, während die Berechnung für die analoge hypothetische Reaktion von O2Molekülen zu einem O8-Molekül eine Reaktionsenthalpie ∆H = +888kJ/mol ergibt. Cyclooctaschwefel S8 ist bei Normaltemperatur nicht sehr reaktionsfähig. Bei Raumtemperatur reagiert Schwefel nur mit Fluor und Quecksilber. Bei erhöhter Temperatur verbindet er sich direkt mit vielen Metallen und Nichtmetallen (nicht mit Au, Pt, Ir, N2, Te, I, Edelgasen). Beispiele: Cu + S → CuS H2 + S → H2S Gegen Wasser und nichtoxidierende Säuren wie HC list S8 inert, von oxidierenden Säuren und Alkalien wird er angegriffen. Synthetisch lassen sich Schwefelmodifikationen mit den Ringmolekülen Sn mit n = 6, 7, 9, 10, 11, 12, 13, 15, 18, 20 herstellen. Nach thermischer Stabilität und Reaktionsfähigkeit können 4 Gruppen unterschieden werden. S8 S12S18S20 S6S9S10S11S13S15 thermische Stabilität ← Reaktionsfähigkeit → S7 S6 entsteht z.B. bei der Zersetzung von Thiosulfat mit Säuren: Na2S2O3 + 2 HCl → 1/6 S6 + SO2 + 2 NaCl + H2O Gasförmiger Schwefel Der Dampf über flüssigem Schwefel, der bei 44,6°C 1,013bar erreicht, besteht zu mindestens 90% aus S8, S7 sowie S3 und nur untergeordnet aus den kleinen Molekülen S5, S4, S3 und S2. Letztere zeichnen sich durch charakteristische Farben aus: S5 orangerot S4 rot S3 blau S2 violett Mit steigender Temperatur bilden sich S2-5 aus S>5 in temperatur- und druckabhängigen Gleichgewichten in zunehmendem Maße. Bei 700°C und 1mbar besteht der Schwefeldampf überwiegend aus S2-Molekülen. Oberhalb 1800°C beginnen auch die S2-Moleküle in SAtome zu dissoziieren, die dann oberhalb von 2200°C bei Drücken < 10-5mbar dominieren. 6.4 Schwefelverbindungen Schwefelwasserstoff H2S H2S kann aus den Elementen dargestellt werden: H2 + S → H2S (bei 600°C) ∆H = -20kJ/mol Im Labor stellt man H2S durch folgende Reaktion dar (aus Al2S3 oder aus FeS): FeS + 2HCl → H2S + FeCl2 H2S ist ein farbloses, sehr giftiges, und unangenehm riechendes Gas. H2S zerfällt bei hoher Temperatur in die Elemente. Bei 1000°C sind 25% zerfallen. An der Luft verbrennt H2S mit blauer Flamme. H2S + 1,5 O2 → H2O + SO2 In 1l Wasser lösen sich bei 20°C 2,6l H2S. H2S wirkt reduzierend, z.B. aus Cl2 und konz. H2SO4. H2S + Cl2 → 2 HCl + S H2S + H2SO4 → SO2 + S + 2 H2O H2S ist eine schwache zweibasige Säure: H2S + H2O H3O+ + HSH3O+ + S2HS- + H2O Ks = 1,0·10-7 Ks = 1,3·10-13 Die Säurestärke nimmt innerhalb der Gruppe der Chalkogene von H2O zu H2Te zu. S2- ist eine starke Anionenbase. Dies zeigt sich bei der Fällung der Sulfide in sauren Lösungen (geringe Konzentration an S2--Ionen ). Sulfide sind die Salze des Schwefelwasserstoff. Dabei gibt es zwei Reihen von Salzen und zwar die Hydrogensulfide und die Sulfide. Die Sulfide stark elektropositiver Metalle sind ionisch. (Beispiele: Na2S, K2S, Al2S3) Technisch erhält man Na2S durch Reduktion von Na2SO4. Na2SO4 + 4C → Na2S + 4CO (bei 700-100°C) Die aus NH3 und H2S im Stoffmengenverhältnis 2 : 1 hergestellte “farblose Ammoniumsulfidlösung” enthält keine S2--Ionen, sondern HS--Ionen. Von Übergangsmetallen sind zahlreiche Sulfide bekannt, die in Strukturen mit überwiegend kovalenten Bindung kristallisieren: Natriumchlorid-Struktur, Zinkblende-Struktur, WurzitStruktur, Nickelarsenid-Struktur, Cadmiumiodid-Struktur, Pyrit-Struktur. Die Schwerlöslichkeit der Metallsulfide benutzt man in der analytischen Chemie zur Trennung von Metallen. Bei pH = 0 beträgt in einer gesättigten H2S-Lösung die Konzentration c(S2-) = 10-21mol/l. Schwerlösliche Sulfide fallen daher mit H2S schon aus saurer Lösung aus (Schwefelwasserstoffgruppe): As2S3 gelb Sb2S3 orange SnS braun HgS schwarz PbS schwarz Bi2S3 CuS dunkelbraun schwarz CdS gelb Weniger schwerlösliche Sulfide fallen erst in ammoniakalischer Lösung aus, in der die S2-Konzentration wesentlich größer ist (Ammoniumsulfidgruppe): NiS CoS FeS MnS ZnS schwarz schwarz schwarz fleischfarben weiß Als Reagenz eignet sich Thioacetamid, das mit Wasser zu H2S reagiert. Schwefeldioxid SO2 SO2 ist ein farbloses, stechend riechendes, korrodierendes Gas. Es löst sich gut in Wasser, die Lösung reagiert schwach sauer und wirkt reduzierend. Es wird als Desinfektionsmittel (Ausschwefeln von Weinfässern) verwendet. Das SO2-Molekül ist gewinkelt (119°), der Bindungsgrad beträgt 2. Die beiden σ-Bindungen werden sp²-Hybridorbitalen des S-Atoms gebildet. Nach der VBTheorie stehen für die beiden π-Bindungen ein p- und ein d-Elektron zur Verfügung, die zwei der drei für π-Bindungen geeigneten pd²-Hybridorbitale besetzen. Technisch wird SO2 durch Verbrennen von Schwefel hergestellt: S + O2 → SO2 ∆H = -297kJ/mol Außerdem lässt es sich durch Rösten von Sulfiden gewinnen: 4 FeS2 + 11 O2 → 2 Fe2O3 + 8 SO2 SO2 wird zu Herstellung von H2SO4 benötigt. Schwefeltrioxid SO3 SO3 kommt in mehreren Modifikationen vor. Monomer existiert es nur im Gaszustand in Gleichgewicht mit S3O9-Molekülen. 3 SO3 S3O9 ∆H = -126kJ/mol Das SO3-Molekül ist trigonal planar gebaut und enthält drei gleichstarke S-ODoppelbindungen. Die Atomorbitale pz, dxy und dyz bilden nach der VB-Theorie pd²-Hybridorbitale, die für πBindungen geeignet sind. Kühlt man gasförmiges SO3 auf -80°C ab, entsteht kristallines, eisartiges γ-SO3, das bei 17°C schmilzt und bei 44°C siedet. γ-SO3 ist aus S3O9-Molekülen aufgebaut. Es sind gewellte Ringe, in denen die S-Atome verzerrt tetraedrisch von Sauerstoff umgeben sind. O O S O O O S O S O O O Unterhalb der Raumtemperatur wandelt sich γ-SO3 in stabilere, asbestartige Modifikation (βSO3, α-SO3) um, die weiße, seidigglänzende Nadeln bilden. β-SO3 besteht aus kettenförmigen Molekülen und ist eigentlich eine Polyschwefelsäure. Die genaue Struktur von α-SO3 ist nicht bekannt, aber wohl ähnlich der β-SO3-Struktur. SO3 ist eine sehr reaktive Verbindung, ein starkes Oxidationsmittel und das Anhydrid der Schwefelsäure. Darstellung SO3 siehe Schwefelsäure. Schwefelsäure H2SO4 Schwefelsäure wird heutzutage ausschließlich durch das Kontaktverfahren gewonnen. Dazu wird zunächst Schwefeldioxid durch Verbrennen von Schwefel oder durch Rösten von sulfidischen Erzen gewonnen und weiter oxidiert (mit Luftsauerstoff): 1/8 S8 + O2 → SO2 ∆H = -297,03kJ/mol SO2 + ½ O2 SO3 ∆H = -98,98kJ/mol Beim Kontaktverfahren dienen dabei Vanadiumverbindungen als Sauerstoffüberträger. Bei zunehmender Temperatur verschiebt sich das Gleichgewicht in Richtung SO2, da die Reaktion exotherm ist. Bei Raumtemperatur reagieren SO2 und O2 praktisch nicht miteinander. Bei höheren Temperaturen stellt sich zwar das Gleichgewicht schnell ein, es liegt aber dann auf der Seite von SO2. Damit die Reaktion bei günstiger Gleichgewichtslage mit gleichzeitig ausreichender Reaktionsgeschwindigkeit abläuft, müssen Katalysatoren verwendet werden. Beim Kontaktverfahren benutzt man V2O5 auf SiO2 als Trägermaterial und arbeitet bei 420-440°C. Bei der Sauerstoffübertragung durch den Katalysator laufen schematisch folgende Reaktionen ab. V2O5 + SO2 → V2O4 + SO3 V2O4 + ½ O2 → V2O5 SO3 löst sich schneller in H2SO4 als in Wasser. Dabei bildet sich Dischwefelsäure. Diese wird dann mit Wasser zu H2SO4 umgesetzt. SO3 + H2SO4 → H2S2O7 H2S2O7 + H2O → 2 H2SO4 Reine Schwefelsäure ist eine farblose, ölige Flüssigkeit. Die konzentrierte Säure des Handels ist 98%ig, sie siedet azeotrop bei 338°C. Schwefelsäure mit einem Überschuß an SO3 heißt rauchende Schwefelsäure. Konzentrierte Schwefelsäure wirkt wasserentziehend und wird deshalb als Trocknungsmittel verwendet (Gaswaschflaschen, Exsikatoren). Auf viele organische Stoffe wirkt konz. H2SO4 verkohlend. Beim Vermischen mit Wasser tritt eine hohe Lösungsenthalpie auf. Konz. H2SO4 wirkt oxidierend, heiße Säure löst Kupfer, Silber und Quecksilber: 2 H2SO4 + Cu → CuSO4 + SO2 + 2 H2O Schwefelsäure ist aufgrund ihrer Autoprotolyse elektrisch leitend. 2 H2SO4 H3SO4+ + HSO4- Bei 25°C beträgt das Löslichkeitsprodukt 2,7·10-4mol²/l² In wässriger Lösung ist H2SO4 eine starke, zweibasige Säure und ist praktisch vollständig in H3O+ und HSO4- protolysiert. H2SO4 + H2O H3O+ + HSO4pKs = -3,0 + 2HSO4 + H2O H3O + SO4 pKs = +1,96 Von H2SO4 leiten sich Hydrogensulfate mit dem Anion HSO4- und Sulfate mit dem Anion SO42- ab. Schwerlöslich sind BaSO4, SrSO4 und PbSO4. Die wasserfreie Dischwefelsäure bildet auch eine durchsichtige, kristalline Masse. Disulfate entstehen beim Erhitzen von Hydrogensulfaten. 2 NaHSO4 → Na2S2O7 + H2O H2SO4, HSO4-, SO42- und H2S2O7 können mit den folgenden Strukturformeln beschrieben werden. Die SO-Bindungslänge im tetraedrisch gebauten SO42--Ion sind gleich (151pm), die πBindungen delokalisiert. Im HSO4--Ion sind die π-Bindungen über drei Bindungen delokalisiert (Bindungslänge 147pm). Im H2SO4-Molekül sind beide π-Bindungen lokalisiert (Bindungslänge 143pm). Schweflige Säure H2SO3 Löst man SO2 in Wasser, so erhält man eine ausgesprochen sauer reagierende, den elektrischen Strom leitende Lösung: SO2 + H2O H2SO3 Das Gleichgewicht liegt weit auf der linken Seite. Die zweibasige Säure dissoziiert in zwei Stufen: H2SO3 + H2O H3O+ + HSO3K1 = 1,54·10-2 HSO3- + H2O H3O+ + SO32K2 = 1,02·10-7 Es gibt zwei Reihen von Salzen. Man erhält die Salze durch Einleiten von SO2 in wässrige Lösungen von Alkalien: 2 KOH + SO2 → K2SO3 + H2O Na2CO3 + SO2 → Na2SO3 + CO2 Das Sulfit-Ion besitzt eine pyramidale Gestalt, wobei der Schwefel an der Spitze der Pyramide steht. Die wichtigste Eigenschaft der schweflige Säure und ihrer Salze ist ihre reduzierende Eigenschaft. Sie beruht auf dem Bestreben der schwefligen Säure, in die höhere Oxidationsstufe der Schwefelsäure überzugehen: SO2 + O → SO3 bzw. SO32-→ SO3 + 2e- Diese Eigenschaft ist in alkalischer Lösung ausgeprägter als in saurer: SO32- + 2 OHSO42- + H2O + 2e- E = -0,936V SO2 + 2 H2O SO42- + 4 H+ + 2eE = +0,158V 7. Halogene Fluor F Ordnungszahl Z 9 Elektronenkonfiguration [He]2s22p5 EN 4,1 Elektronenaffinität in -3,4 eV Ionisierungsenergie in 17,5 eV Nichtmetallcharakter Reaktionsfähigkeit Affinität zu elektropositiveren Elementen Affinität zu elektronegativeren Elementen Chlor Cl 17 [Ne]3s23p5 2,8 -3,6 Brom Br 35 [Ar]3d104s24p5 2,7 -3,4 Iod I 53 [Kr]4d105s25p5 2,2 -3,1 13,0 11,8 10,4 → nimmt ab → nimmt ab → nimmt ab → nimmt zu 7.1 Darstellung Fluor. Wegen seines hohen Standardpotentials kann Fluor aus seinen Verbindungen nicht durch chemische Oxidationsmittel freigesetzt werden. F2 wird daher durch anodische Oxidationsmittel von F--Ionen in wasserfreien Elektrolyten hergestellt. In Gegenwart von Wasser erfolgt Entladung von OH-Ionen zu O2. Da wasserfreies HF ein schlechter Leiter ist, verwendet man zu Elektrolyse wasserfreie Schmelzen der Zusammensetzung KF·xHF. Die Schmelzpunkte sinken mit wachsendem HF-Gehalt: KF·HF 217°C, KF·3HF 66°C. Im technisch verwendeten Mitteltemperaturverfahren elektrolysiert man Schmelzen mit x = 2-2,2 bei Temperaturen von 70 bis 130°C. Für die Herstellung im Laboratorium benutzt man Hochtemperaturzellen mit KF·HF-Schmelzen, die Temperaturen von 250°C erfordern. Da KHF2 praktisch nicht hygroskopisch ist, enthält das damit erzeugte F2 nur sehr wenig O2 bzw. OF2. Die Elektrolysezellen bestehen aus Stahl oder Monel. Die verwendeten Metalle überziehen sich bei Betriebsbedingungen mit einer vor weiterem Fluorangriff schützende Fluoridschicht (Passivierung). Die Darstellung von Fluor auf chemischen Wege gelingt mit dem Trick, ein instabiles Fluorid herzustellen, das sich unter Entwicklung von elementarem Fluor zersetzen lässt. Aus K2MnF6 wird mit SbF5 das instabile Fluorid MnF4 freigesetzt, das spontan in MnF3 und F2 zerfällt. K2Mn6 + 2 SbF5 → 2 KSbF6 + MnF3 + ½ F2 K2MnF6 und SbF5 werden nach den folgenden Reaktionen hergestellt: 2 KMnO4 + 2 KF + 10 HF + 3 H2O2 → 2 K2MnF6 + 8 H2O + 3 O2 SbCl5 + 5 HF → SbF5 + 5 HCl F2 wird gebraucht um Kampfstoffe wie ClF3 und für Atombomben benötigtes UF6 herzustellen. Chlor. Technisch wird Chlor fast ausschließlich durch Elektrolyse wässriger NaCl-Lösungen hergestellt. 2 Na+ + 2 Cl- + 2 H2O → 2 Na+ + 2OH- + H2 + Cl2 Früher wurde es nach dem Deacon-Verfahren produziert: 2 HCl + ½ O2 → Cl2 + H2O Das Verfahren wurde bei 430°C mit Luftsauerstoff und CuCl2 als Katalysator durchgeführt wurde. Im Labor lässt es sich durch die Reaktion von MnO2 oder KMnO4 mit HCl hergestellt: 4 HCl + MnO2 → Cl2 + MnCl2 + 2 H2O 16 HCl + 2 KMnO4 → 5 Cl2 + 2 MnCl2 + 2 KCl + 8 H2O Chlor wird in großen Mengen in der organischen Chemie gebraucht. Außerdem wird es benötigt um Brom herzustellen. Brom. Bei der Aufarbeitung von Kalisalzen entstehen Br--haltige Lösungen. In die schwach sauren Lösungen wird Cl2 eingeleitet und das entstandene Br2 mit einem Luftstrom ausgetrieben: 2 Br- + Cl2 → Br2 + 2 ClIm Labor kann Br2 durch Oxidation von KBr mit konz. H2SO4 hergestellt werden. 2 HBr + H2SO4 → Br2 + SO2 + 2 H2O Iod. Die Hauptmenge des Iods wird aus iodhaltigen Lösungen gewonnen, die bei der Kristallisation von Chilesalpeter zurückbleibt. Zunächst wird ein Teil der Iodsäure HIO3 mit SO2 reduziert. HIO3 + 3 SO2 + 3 H2O → HI + 3 H2SO4 HI wird durch noch vorhandene Iodsäure oxidiert. HIO3 + 5 HI → 3 I2 + 3 H2O Gesamtreaktion: 2 HIO3 + 5 SO2 + 4 H2O → 5 H2SO4 + I2 Außerdem wird Iod aus Salzsolen gewonnen, die oft bei der Erdöl- und Erdgasförderung anfallen. Aus Iodiden (z.B. in der Asche der Meeresalgen) kann I2 durch Oxidation (z.B. mit MnO2 oder H2SO4) hergestellt werden. Technisch ist die Gewinnung aus Algen oder Tang heute ohne Bedeutung. Iod und Iodverbindungen werden für Katalysatoren, pharmazeutische Zwecke, Futtermittelzusätze und Farbstoffe verwendet. 7.2 chemische und physikalische Eigenschaften physikalische Eigenschaften Aufgrund der Valenzelektronenkonfiguration s2p5 bestehen die elementaren Halogene in allen Aggregatzuständen aus zweiatomigen Molekülen. Zwischen den Molekülen sind schwache van der Waals-Kräfte wirksam, die Schmelz- und Siedetemperaturen sind daher z.T. sehr niedrig. Innerhalb der Gruppe steigen die als Folge der zunehmenden van der Waals-Kräfte regelmäßig an. Fluor ist bei Raumtemperatur ein gelbliches Gas. Es ist stark ätzend und extrem giftig. Es kann noch in sehr kleinen Konzentrationen am Geruch erkannt werden, der dem Geruch eines Gemisches aus O3 und Cl2 ähnelt. Chlor ist bei Raumtemperatur ein gelbgrünes, giftiges, die Schleimhäute angreifendes Gas. Es ist 2,5mal so schwer wie Luft und durch Kompression leicht zu verflüssigen. Die kritische Temperatur beträgt 144°C, der Dampfdruck bei 20°C 6,7bar. Brom ist bei Raumtemperatur eine dunkelbraune Flüssigkeit, die schon bei -7°C dunkelbraunrot kristallisiert. Bromdampf reizt die Schleimhäute, flüssiges Brom erzeugt auf der Haut schmerzhafte Wunden. In Wasser ist Brom weniger gut löslich als Chlor. Es ist aber mit unpolaren Lösungsmitteln (z.B. CCl4, CS2) gut mischbar. Iod bildet bei Raumtemperatur grauschwarze, metallisch glänzende, halbleitende Kristalle. Es schmilzt bei 114°C zu einer braunen Flüssigkeit und siedet bei 185°C unter Bildung eines violetten Dampfes. Alle Phasen bestehen aus I2-Molekülen. Schon bei Raumtemperatur ist Iod flüchtig, beim Schmelzen beträgt der Dampfdruck 0,13bar. Man kann daher Iod sublimieren und durch Sublimation reinigen. I2 kristallisiert wie Br2 und Cl2 in einer Schichtstruktur mit ausgeprägter Spaltbarkeit der Kristalle parallel zu den Schichten. Zwischen den Schichten sind die Moleküle durch van der Waals-Kräfte aneinander gebunden. Die Abstände betragen 435-450pm (van der WaalsAbstand 430pm). Innerhalb der Schichten sind die Abstände zwischen den I2-Molekülen kürzer, so dass auch schwache kovalente Teilbindungen auftreten. Es liegen Mehrzentrenbindungen vom σ-Typ vor, die sich über die ganze Schicht erstrecken. Die damit verbundene Elektronendelokalisierung erklärt Farbe, Glanz und elektrische Leitfähigkeit des Iods (parallel zu den Schichten). Bei Normaldruck ist Iod ein Halbleiter mit einem gefüllten Valenzband und einem leeren Leitungsband. Bei etwa 170kbar wird Iod ein metallischer Leiter, die Packung der Moleküle ist so dicht geworden, dass Valenzband und Leitungsband überlappen. Bei 210kbar erfolgt eine Strukturänderung, alle Iodabstände werden gleich groß, es entsteht ein aus Atomen aufgebauter metallischer Kristall. Iod löst sich in unpolaren Lösungsmitteln (CCl4, CHCl3) mit violetter Farbe. Die Lösungen enthalten wie der Dampf I2-Moleküle. In anderen Lösungsmitteln, wie H2O, Ether, löst sich Iod mit brauner Farbe, in aromatischen Kohlenwasserstoffen mit roter Farbe. Die Farbänderung ist auf die Bildung von Charge-Transfer-Komplexen zurückzuführen. Sie kommen durch den teilweisen Übergang eines Elektronenpaares des Lösungsmittelmoleküls auf ein I2-Molekül zustande. Der Grundzustand der Charge-Transfer-Komplexe kann mit mesomeren Grenzstrukturen beschrieben werden, wobei die Grenzstruktur I überwiegt. I2 --- D ↔ I2-D+ I II Donoreigenschaften besitzen z.B. π-Elektronensysteme und die einsamen Elektronenpaare des O-Atoms. Charge-Transfer-Komplexe zeichnen sich meist durch eine intensive Lichtabsorption aus. Dabei erfolgt ein Elektronenübergang in einen angeregten Zustand des Komplexes, bei dem die Grenzstruktur II überwiegt. Die Charge-Transfer-Absorptionen der I2-Komplexe liegen in nahen Ultraviolett. Durch die Bildung der Charge-Transfer-Komplexe wird die I-I-Bindung geschwächt und damit auch die Energie der Elektronenanregung, die im ungestörten I2-Molekül die violette Farbe verursacht, beeinflusst. Eine Farbänderung in Abhängigkeit von den Donoreigenschaften ist die Folge. Chemische Eigenschaften Fluor ist das reaktionsfähigste Element. Es reagiert direkt mit allen Elementen außer He, Ne, Ar, N2. In Verbindungen mit Fluor erreichen die Elemente hohe und höchste Oxidationszahlen: IF7, SF6, XeF6, ClF5, BiF5, AgF2, AuF5, UF6. Nickel, Kupfer, Stahl sowie Legierungen Monel (Cu-Ni) und Elektron (Al-Mg) werden von Fluor nur oberflächlich angegriffen. Es bildet sich eine dichte, fest haftende Fluoridschicht, die den weiteren Angriff von Fluor verhindert (Passivierung). Cu kann bis 500°C, Ni und Monel bis 800°C für Arbeiten mit Fluor verwendet werden. Fluor ist in Stahlflaschen mit Drücken bis 30bar im Handel. In Quarz- und Glasgefäßen kann nur gearbeitet werden, wenn weder H2O noch F zugegen ist, da sonst ein ständiger Angriff erfolgen würde. 2 F2 + 2 H2O → 4 HF + O2 SiO2 + 4 HF → SiF4 + 2 H2O Wie mit H2O reagiert F2 auch mit Wasserstoffverbindungen unter Bildung von HF. Chlor gehört zu den reaktionsfähigsten Elementen, es reagiert außer mit den Edelgasen, O2, N2 und C mit allen Elementen, meist schon bei niedrigen Temperaturen. Mit vielen Metallen reagiert es beim Erwärmen oder bei großer Metalloberfläche unter Feuererscheinung, z.B. mit Alkalimetallen, Erdalkalimetallen, Cu, Fe, As, Sb, Bi. Die Reaktion mit W zu WCl6 und dessen thermische Zersetzung dient zur Reinigung des Metalls. W + 3 Cl2 WCl6 (Siedepunkt 346°C) Nichtmetalle wie Phosphor und Schwefel werden je nach Reaktionsbedingungen in die kovalente Chloride PCl3, PCl5, S2Cl2, SCl2, SCl4 überführt. Die Reaktion mit H2 verläuft nach Zündung explosionsartig (Chlorknallgas) in einer Kettenreaktion. H2 + Cl2 2 HCl ∆H = -185kJ/mol Cl2 löst sich gut in Wasser, dabei bildet sich in einer Disproportionierungsreaktion HCl und Hypochlorige Säure HOCl. Cl2 + H2O HCl + HOCl HOCl wirkt stark oxidierend, daher wird feuchtes Chlor zum oxidativen Bleichen (Papier), sowie zum Desinfizieren (Trinkwasser, Abwässer) verwendet. Brom reagiert analog zum Chlor, die Reaktionsfähigkeit ist jedoch geringer. Iod ist weniger reaktiv, verbindet sich aber immer noch mit einigen Elementen, z.B. P, S, Al, Fe, Hg. Charakteristisch für Iod und als Nachweisreaktion für kleine Iodmengen geeignet ist die intensive Blaufärbung mit wässrigen Stärkelösungen. Bei dieser Iodstärkereaktion erfolgt ein Einschluß von Iod (Einschlußverbindung). Fluor und Chlor sind starke Oxidationsmittel. Fluor ist – abgesehen von KrF2 – das stärkste Oxidationsmittel überhaupt. Innerhalb der Gruppe nimmt das Oxidationsvermögen mit zunehmender Ordnungszahl ab. Fluor kann deshalb alle anderen Halogene aus ihren Verbindungen verdrängen. F2 + 2 Cl- → 2 F- + Cl2 F2 + 2 Br- → 2 F- + Br2 Chlor kann Brom und Iod, Brom nur Iod freisetzen. Cl2 + 2 Br- → 2 Cl- + Br2 Br2 + 2 I- → 2 Br- + I2 Das Oxidationsvermögen eines Halogens ist um so größer, je mehr Energie bei der Reaktion X2(g) + 2e- → 2 X-(aq) freigesetzt wird. Die Gesamtenergie ist durch die Energiebeträge der folgenden Teilschritte bestimmt. ½ X2 (g) → ½ D (Dissoziationsenergie) → X (g) → E (Elektronenaffinität) → X (g) → ∆H (Hydratationsenthalpie) → X (aq) Obwohl die Elektronenaffinität von Chlor größer als die von Fluor ist, ist Fluor das wesentlich stärkere Oxidationsmittel. Dies liegt an der kleinen Dissoziationsenergie von F2 und der großen Hydrationsenergie der kleinen F--Ionen. Die viel größere Dissoziationsenergie von Cl2 entspricht im Vergleich mit Br2 und I2 der Erwartung. F2 hat eine kürzere Bindungslänge, daher ist eine starke Abstoßung nichtbindender Elektronenpaare wirksam. 7.3 Verbindungen des Halogene 7.3.1 Interhalogenverbindungen Von Verbindungen der Halogene untereinander sind die Zypen XY, XY3, XY5 und XY7 bekannt, in denen das elektropositivere Halogen X in der Oxidationsstufe +1, +3, +5 oder +7 vorliegt. Die Interhalogenverbindungen sind typische kovalente Verbindungen. Sie lassen sich aus den Elementen synthetisieren uns sind sehr reaktionsfähig. Von den VERBINDUNGEN DER Zusammensetzung XY sind alle Kombinationen bekannt. Die Interhalogenverbindungen XY sind wie die Halogene sehr reaktive Substanzen. Sie sind Oxidationsmittel und Halogenüberträger. Die Reaktionsfähigkeit und die Disproportionierungsenergie ist um so größer je weiter die Halogene im PSE voneinander entfernt sind. Beispiele: ClF ist disproportionierungsstabil, es wird als Fluorierungsmittel benutzt. BrF disproportioniert nach 3 BrF → Br2 + BrF3. IF ist nur bei tiefen Temperaturen beständig, oberhalb von -14°C zerfällt es nach 5 IF → 2 I2 + IF5. Die Zerfallsneigung der Interhalogenverbindungen XY wächst in der Reihe ClF < ICl < BrF < IBr < BrCl. Mit Wasser findet die Reaktion XY + H2O → Hy + HOX statt, X ist das elektropositivere Atom. Mit Ausnahme von ICl3 sind alle anderen Interhalogenverbindungen Fluoride Die Halogenide XY3 sind T-förmig gebaut, die Pentahalogenide XY5 haben die Geometrie einer quadratischen Pyramide, IF7 bildet eine pentagonale Bipyramide. Nur Br, Cl und I sind Zentralatome und hauptsächlich F als Substituent geeignet. Eine Erklärung liefert die VB-Theorie. Die Promotionsenergie des s- und p-Elektronen in die dOrbitale nimmt von Cl zu I ab. Daher ist verständlich, dass die thermodynamische Stabilität der Verbindungen XY3 und XY5 von Cl zu I zunimmt und nur I ein Heptafluorid bildet. Von allen Elemente besitzt Fluor die größte Fähigkeit zur Stabilisierung hoher positiver Oxidationsstufen. Chlor kann nur noch mit Iod zu Iodtrichlorid reagieren. 7.3.2 Halogenide Hydrogenfluorid HF, Hydrogenchlorid HCl, Hydrogenbromid HBr und Hydrogeniodid HI sind farblose, stechend riechende Gase. In den Hydrogenhalogeniden liegen polare Einfachbindungen vor. Die Polarität der Bindung wächst entsprechend der zunehmender Elektronennegativitätsdifferenz von HI nach HF. Zwischen HX-Molekülen wirken nur schwache van der Waals-Kräfte, daher sind alle Verbindungen flüchtig. Erwartungsgemäß nehmen die Schmelzpunkte, Siedepunkte und die Verdampfungsenthalpien von HI zu HCl ab, HF zeigt aber anomal hohe Werte. Die Ursache sind zusätzliche Bindungskräfte, die durch Wasserstoffbrücken zustande kommen. Im festen Hydrogenfluorid sind die HF-Moleküle über unsymmetrische Wasserstoffbrücken F-H---F- zu Zick-Zack-Ketten verknüpft. Ähnliche Brückenbindungendürften im flüssigen HF vorliegen. Es ist eine farblose, bewegliche, hygroskopische Flüssigkeit. Im Dampf sind gewellte (HF)6Ringe im Gleichgewicht mit HF-Molekülen, erst oberhalb von 90°C ist Hydrogenfluorid monomolekular. Alle Hydrogenhalogenide lösen sich gut in Wasser. Bei 0°C lösen sich in 1l Wasser 507l HCl-Gas und 712l HBr-Gas. Da sie dabei Protonen abgeben, fungieren sie als Säuren. HX + H2O H3O+ + XDie Säurestärke nimmt von HF nach HI zu. Die Ursache dafür ist die von HF nach HI abnehmende Bindungsenergie. Alle Hydrogenhalogenide bilden sich in direkter Reaktion aus den Elementen. H2 + X2 2 HX Die Reaktionen von F2 und Cl2 verlaufen explosionsartig. Br2 reagiert auch in Gegenwart von Pt-Katalysatoren erst bei 200°C. HI zersetzt sich bereits bei mäßig hohen Temperaturen zum Teil in die Elemente (19% bei 300°C). Die Bildungsreaktionen der Hydrogenhalogenide verlaufen nach einem Radikalkettenmechanismus, bei I2 allerdings erst bei Temperaturen oberhalb 500°C. Hydrogenfluorid HF. Die übliche technische Darstellung von HF ist die Umsetzung von CaF2 mit konz. H2SO4 bei 270°C. CaF2 + H2SO4 → 2 HF + CaSO4 Reinstes, wasserfreies HF gewinnt man durch thermische Zersetzung von KHF2. KHF2 → KF + HF Mit den im Flussspat als Nebenprodukt vorhandenen Silicaten entsteht SiF4, das mit HF zu Hexafluorokieselsäure umgesetzt wird. 2 CaF2 + SiO2 + 2 H2SO4 → SiF4 + 2 CaSO4 + 2H2O SiF4 + 2 HF → H2SiF6 Die Hauptmenge HF wird zur Herstellung von AlF3, Kryolith und Fluorhalogenkohlenwasserstoffen verwendet. In der Glasindustrie dient es zum Ätzen und Polieren von Glas. Aus H2SiF6 gewinnt man AlF3 und Kryolith. Wässrige Lösungen von HF heißen Flusssäure. Flusssäure ist eine mittelstarke Säure; sie ätzt Glas und kann daher nicht in Glasflaschen aufbewahrt werden (aber in Polyethylenflaschen). SiO2 + 4HF → SiF4 + 2H2O Hydrogenchlorid HCl. Bei der technischen Darstellung von HCl aus den Elementen benutzt man einen nach dem Prinzip des Daniellschen Hahns arbeitenden Quarzbrenner. Beim Chlorid-Schwefelsäure-Verfahren wird NaCl mit konz. H2SO4 umgesetzt. NaCl + H2SO4 → NaHSO4 + HCl bei 20°C NaCl + NaHSO4 → Na2SO4 + HCl bei 80°C Das meiste HCl entsteht als Zwangsanfall bei der technisch wichtigen Chlorierung organischer Verbindungen. Technisch nicht verwendbares HCl wird durch Elektrolyse in Cl2 und H2 umgesetzt. Wässrige Lösungen von HCl heißen Salzsäure. In konzentrierter Salzsäure ist ein Massenanteil von ca. 38% HCl-Gas gelöst. Salzsäure ist eine starke, nichtoxidierende Säure, sie löst daher nur unedle Metalle wie Zn, Al, Fe, nicht aber Cu, Hg, Ag, Au, Pt und Ta. Zn + 2 HCl → H2 + ZnCl2 Hydrogenbromid HBr. Hydrogeniodid HI. HBr und HI können nicht aus ihren Salzen mit konz. H2SO4 hergestellt werden, da teilweise Oxidation zu Br2 und I2 erfolgt. Sie werden durch Hydrolyse von PBr3 bzw. PI3 hergestellt. PBr3 + 3H2O → 3HBr + H3PO3 PI3 + 3H2O → 3HI + H3PO3 Dazu kann roter Phosphor und das Halogen direkt in Gegenwart von Wasser umgesetzt werden, intermediär bildet sich das Phosphortrihalogenid. Hydrogeniodid ist eine sehr starke Säure, sie ist oxidationsempfindlich. Bei Einwirken von Luftsauerstoff wird Iod frei. 4HI + O2 → 2I2 + 2H2O Die Halogenide der Alkalimetalle und der Erdalkalimetalle sind typische Salze, die überwiegend in Ionengittern kristallisieren. Typisch für Fluor ist die Existenz von Hydrogenfluoriden, so z.B. der Alkalimetallhydrogenfluoride Me+HF2-. Me+H2F3- und Me+H3F4-. Mit Nichtmetallen bilden die Halogenide flüchtige, kovalente Halogenide, die in Molekülgittern kristallisieren. Beispiele: BF3, SiF4, SF4, PF5, CF4 SCl2, PCl2, CCl4, SiBr4 Gase bei 25°C Flüssigkeiten bei 25°C 7.3.3 Sauerstoffsäuren der Halogene Beim gleichen Halogen steigt die Stabiltät der Sauerstoffsäure mit wachsender Oxidationszahl. In reiner Form lassen sich nur HClO4, HIO3, H5IO6, H7I3O14 und (HIO4)n isolieren. Die anderen Oxosäuren existieren nur in wässrigen Lösungen. BrO- und IO2- treten nur als instabile Reaktionszwischenprodukte auf. Die Nomenklatur der Sauerstoffsäuren des Chlors und ihrer Salze, sowie die Bindungsverhältnisse nach VB-Theorie stehen in der Tabelle 4.10.. Durch Anregung von Elektronen aus dem Grundzustand in die leeren d-Orbitale stehen den Chloratomen maximal sieben ungepaarte Elektronen für Bindungen zur Verfügung. Die dOrbitale des Chlors überlappen mit p-Orbitalen des Sauerstoffs unter Ausbildung von (p-d)-πBindungen. Ergeben die rein kovalenten Grenzstrukturen zu hohe (p-d)-π-Bindungsanteile, so kann dies durch ionogene Grenzstrukturen entsprechenden Gewichts berücksichtigt werden. Mit zunehmender Zahl der π-Bindungen wächst die Anzahl mesomerer Grenzstrukturen, die Anionen werden dadurch stabilisiert, die negative Ladung an den O-Atomen wird gerringer, und die Protonen werden weniger stark angezogen. Die Säurestärke wächst daher mit zunehmender Oxidationszahl. Dies ist auch die Ursache für den Anstieg der Säurestärke in der Reihe H4SiO4, H3PO4, H2SO4, HClO4. Mit zunehmender Koordinationszahl nimmt mit Anzahl freier, reaktiver Elektronenpaare am Cl-Atom ab, die Stabilität erhöht sich. Dies erklärt die typischen Disproportionierungsreaktionen, bei denen aus sauerstoffärmeren Ionen Cl- und sauerstoffreichere Anionen entstehen. 3 ClO- → ClO3- + 2 ClIn saurer Lösung sind alle Chlorsauerstoffsäuren starke Oxidationsmittel. Ein besonders starkes Oxidationsvermögen besitztt HClO. Mit wachsendem pH-Wert nimmt das Oxidationvermögen stark ab. Die Potentiale zeigen auch, dass z.B. die Disprportionierung von Cl2 in Cl- und ClO- nur in alkalischen Lösungen möglich ist. In sauren Lösungen ist die Komproportionierung von HClO und Cl- zu Cl2 energetisch begünstigt. Hypochlorige Säure HOCl in einer Disproportionierungsreaktion beim Einleiten von Cl2 in Wasser. Cl2 + H2O HCl + HOCl Das Gleichgewicht der Reaktion liegt aber ganz auf der linken Seite (Chlorwasser). Eine Verschiebung des Gleichgewichts nach rechts erreicht man durch Abfangen von HCl mit einer HgO-Suspension als unlösliches HgO·HgCl. Es entsteht 20%ige HOCL, die sich aber schon bei 0°C langsam zersetzt. 2 HOCl → 2HCl + O2 HOCl ist eine schwache Säure und ein starkes Oxidationsmittel (Desinfektion von Wasser). Sie ist im wasserfreien Zustand nicht bekannt, beim Entwässern entsteht ihr Anhydrid Cl2O. In Lösungen ist Cl2O im Gleichgewicht mit HOCl so dass nebeneinander Cl2, HOCl und Cl2O vorliegen. 2 HOCl Cl2O + H2O Die Salze der Hypochlorigen Säure, die Hypochlorite, erhält man durch Einleiten von Chlor in kalte alkalische Lösungen. Cl2 + 2 NaOH → NaCl + NaOCl + H2O Brom und Iod reagieren analog zu Hypobromiten bzw. zu Hypoioditen. Technisch kann man die Darstellung von NaOCl an die Chloralkalieelektrolyse anschließen, indem man das anodisch entwickelte Chlor in die kathodisch gebildete Natronlauge einleitet. Chlorkalk erhält man aus Cl2 und Ca(OH)2. Cl2 + Ca(OH)2 → CaCl(OCl) + H2O Mit Salzsäure entsteht aus Chlorkalk Chlor. CaCl(OCl) + 2HCl → CaCl2 + Cl2 + H2O Hypochlorite sind schwächere Oxidationsmittel als HOCl, sie werden als Bleich- und Desinfektionsmittel verwendet. Wässrige Lösungen reagieren basisch, da ClO- eine Anionenbase ist. Chlorige Säure HClO2 ist bedeutungslos, da sie sich schnell zersetzt. 5 HClO2 → 4 ClO2 + HCl + 2 H2O Beständiger sind ihre Salze, die Chlorite. Sie werden technisch durch Einleiten von ClO2 in NaOH-H2O2-Lösungen hergestellt. 2ClO2 + H2O2 + 2 NaOH → 2 NaClO2 + O2 + 2 H2O Verwendet werden sie als Bleichmittel für Textilien, da das beim Ansäuern freiwerdende ClO2 faserschonend bleicht. Chlorsäure HClO3 erhält man aus ihren Salzen, den Chloraten. Ba(ClO3)2 + H2SO4 → 2 HClO3 + BaSO4 Lösungen mit mehr als 40% HClO3 zersetzen sich. HClO3 ist eine starke Säure (pKs = -2,7) und ein starkes Oxidationsmittel. Chlorate entstehen durch Disproportionierung von Hypochloriten in erwärmten Lösungen. 3ClO- → ClO3- + 2ClWahrscheinlich wird dabei das Anion ClO- durch die freie Säure HClO oxidiert. 2HClO + ClO- → ClO3- + 2HCl Da Cl2 in NaOH zu ClO- und Cl- disproportioniert, erhält man ClO3- durch Einleiten von Cl2 in heiße Laugen. 3 Cl2 + 6 OH- → 5Cl- + ClO3- + 3H2O Technisch elektrolysiert man heiße NaCl-Lösung ohne Trennung des Kathoden- und Anodenraums. Chlorate sind kräftige Oxidationsmittel. Gemische von Chloraten mit oxidierbaren Substanzen (Phosphor, Schwefel, organische Substanzen) sind explosiv. Perchlorsäure HClO4 ist die beständigste und die einzige in reiner Form herstellbare Chlorsauerstoffsäure. HClO4 ist eine farblose Flüssigkeit, die bei 120°C siedet und bei -101°C erstarrt. Beim Erwärmen zersetzt sie sich, manchmal explosionsartig. Mit brennbaren Substanzen erfolgt Explosion. In wässriger Lösung ist HClO4 stabil, sie ist eine der stärksten Säuren. Trotz des hohen Redoxpotentials wirkt sie aus kinetischen weit weniger oxidierend als HClO3. Von HClO3 wird z.B. HCl zu Cl2 und S zu H2SO4 oxidiert, aber nicht von HClO4. HClO4 kann aus Perchloraten dargestellt werden. KClO4 + H2SO4 → HClO4 + KHSO4 Die entstandene Perchlorsäure wird im Vakuum abdestilliert. Perchlorate werden technisch durch anodische Oxidation von Chloraten hergestellt. ClO3- + H2O → ClO4- + 2H+ + 2ePerchlorate entstehen auch bei der thermischen Disproportionierung von Chloraten. 4 KClO3 → 3 KClO4 + KCl bei 400°C Bei noch stärkerem Erhitzen zersetzt sich KClO4. KClO4 → KCl + 2 O2 Die Perchlorate sind die beständigsten Salze von Oxosäuren des Chlors. Schwerlöslich sind die Perchlorate von K, Rb, Cs. NH4ClO4 wird als Raketentreibstoff verwendet. Iodsäure HIO3 kristallisiert in farblosen Kristallen. Sie ist ein starkes Oxidationsmittel, durch Entwässern erhält man aus ihr I2O5. HIO3 kann durch Oxidation von I2 mit HNO3, Cl2 oder H2O2 hergestellt werden. I2 + 6 H2O + 5 Cl2 → 2 HIO3 + 10 HCl HCl muss aus dem Gleichgewicht entfernt werden, da es HIO3 reduziert. 7.3.4 Oxide der Halogene In den Oxiden von Chlor, Brom, Iod kommen die Halogene in positiveren Oxidationszahlen vor. Gesichert ist die Existenz der in der folgenden Tabelle aufgelisteten Halogenoxide. Mit Ausnahme von I2O5 sind die Halogenoxide endotherme Verbindungen, die beim Erwärmen teilweise explosionsartig zerfallen. Sie sind sehr reaktionsfähig und starke Oxidationsmittel. Die Strukturen sind zum Teil noch ungeklärt. Technische Bedeutung hat ClO2. Dichloroxid Cl2O ist ein gelbrotes Gas, das beim Erwärmen explosionsartig in Cl2 und O2 zerfällt. Es entsteht durch Reaktion von Cl2 mit HgO. 2 Cl2 + 2 HgO → Cl2O + HgO·HgCl2 In analoger Reaktion entsteht Br2O. Cl2O ist da Anhydrid von HOCl, es bildet sich in Alkalilaugen Hypochlorit. Cl2O und Br2O sind gewinkelte Moleküle mit schwachen Einfachbindungen. Chlordioxid ClO2 ist ein gelbes, sehr explosives Gas. Mit CO2 verdünnt, wird es als Oxidationsmittel zum Bleichen und als Desinfektionsmittel verwendet. Es wird durch Reduktion von NaClO3 mit SO2 oder Salzsäure hergestellt. 2 NaClO3 + SO2 + H2SO4 → 2 ClO2 + 2 NaHSO4 Im Labor entsteht es aus KClO3 und konz. H2SO4 durch Disproportionierung der Chlorsäure. KClO3 + H2SO4 → HClO3 + KHSO4 3 HClO3 → 2 ClO2 + HClO4 + H2O In alkalischen Lösungen disproportioniert ClO2. 2 ClO2 + 2 OH- → ClO2- + ClO3- + H2O ClO2 ist ein gewinkeltes Molekül, es enthält ein ungepaartes Elektron. Wahrscheinlich ist das ungepaarte Elektron über das ganze Molekül delokalisiert. Bei tiefen Temperaturen existiert im festen Zustand Dimere mit kompensierten Spinmomenten. BrO2-Dimere entstehen bei elektrischen Entladung aus Br2/O2-Gemischen. Die Struktur ist die eines Bromperborats, Br – O – BrO3. Räumlicher Bau von HalOF3, HalOF5, HalO2F3, HalO3F 8. Edelgase Heliu m He Neon Ne Argon Ar Krypto n Kr 36 [Ar]3d 10 2 4s 4p Ordnungszahl Z 2 Elektronenkonfigurati 1s2 on 10 [He]2s22p 18 [Ne]3s23p 54 86 10 [Kr]4d 5 [Xe]4f145d106s26p6 s25p6 Ionisierungsenergie in eV Promotionsenergie np → (n+1) in eV Schmelzpunkt in °C Siedepunkt in °C Kritische Temperatur in °C Van der WaalsRadien in pm Farbe des in Gasentladungsröhren ausgestrahlen Lichts 24,6 21,6 15,8 14,0 12,1 10,7 - 16,6 11,5 9,9 8,3 6,8 -272 -269 -268 -249 -246 -229 -189 -186 -122 -157 -153 -64 -112 -108 17 -71 -62 105 120 160 190 200 220 - gelb rot rot gelbgr ün violett weiß 6 6 6 Xenon Xe Radon Rn 8.1 Darstellung Edelgase sind Bestandteile der Luft. Ihr Volumenanteil in der Luft beträgt 0,935%. Im einzelnen ist die Zusammensetzung der Luft in der Tabelle aufgelistet: N2 O2 Ar CO2 78,09% 20,95% 0,93% 0,03% Ne He Kr Xe 1,6·10-3% 5·10-4% 1·10-4% 9·10-6% He ist in Erdgasen enthalten. Ergiebige Erdgasquellen in den USA enthalten Volumenanteile He bis 8%. Die He-Reserven in Erdgasen werden auf 5·109m³ geschätzt. Die technische Gewinnung erfolgt aus der Luft durch fraktionierende Destillation verflüssigter Luft. He erhält man aus Erdgasen. Bei der Abkühlung auf -205°C bleibt nur He gasförmig zurück. Ar wird auch aus Industrieabgasen gewonnen. Bei der NH3-Synthese reichert sich in dem im Kreislauf gefahrenen Gasgemisch Ar an (ca. 10%). Die wichtigsten Verwendungsgebiete sind die Lichttechnik und die Schweißtechnik. Argon wird als Schutzgas, z.B. beim Umschmelzen von Metallen und bei der Lichtbogenschweißung verwendet. Gasentladungsröhren mit Edelgasfüllung dienen als Lichtreklame. He-O2Gemische werden als Atemgas für Taucher eingesetzt, da sich unter Druck weniger He im Blut löst als N2. In der Kerntechnik wird es als Kühlmittel eingesetzt. 8.2 Halogenverbindungen Xenonfluoride lassen sich durch direkte Reaktion der Elemente gewinnen. Das Fluor muss jedoch durch Erhitzen, Licht oder elektrischer Entladung aktiviert werden. Xenon reagiert mit Fluor nach den folgenden Gleichgewichtsreaktionen schrittweise und exotherm: Xe + F2 XeF2 XeF2 + F2 XeF4 XeF4 + F2 XeF6 Darstellungsbedingungen der Xenonfluoride XeF2 Stoffmengenverhältnis Xe/F2 2:1 1:5 400°C XeF4 1:20 400°C, 6bar XeF6 300°C, 60bar Xe(II)-fluorid XeF2 und Xenon(IV)-fluorid XeF4 sind in allen Phasen monomer. Xenon(VI)fluorid XeF6 ist nur in der Gasphase monomer, im festen Zustand sind quadratisch-pyramidale XeF5+-Ionen durch F--Ionen zu tetrameren oder hexameren Ringen verbunden. Die Xenonfluoride sind bei Raumtemperatur beständig, zersetzen sich aber beim Erhitzen in die Elemente. Sie sind flüchtig und sublimieren bereits bei Raumtemperatur. Sie sind starke Oxidations- und Fluorierungsmittel. Bei Redoxreaktionen entsteht Xe. Mit der Oxidation einer Verbindung ist vielfach eine Fluorierung verbunden. Beispiele: XeF2 + H2 → Xe + 2HF bei 300°C XeF4 + 4I- → Xe + 2I2 + 4FXeF4 + SF4 → Xe + 2 SF6 XeF6 + 6HCl → Xe + 3 Cl2 + 6HF Die Xenonfluoride können sowohl F--Donatoren als auch F--Akzeptoren reagieren. XeF6 reagiert z.B. mit PtF5 zu einem gelben Salz [XeF5+][PtF6-]. Die XeF5+-Ionen sind quadratisch-pyramidal gebaut. Mit Alkalimetallfluoriden (außer LiF) entstehen Fluoroxenate(VI) CsF + XeF6 → CaXeF7 → ½ Cs2XeF8 + ½ XeF6 Xenon(VI)-oxid XeO3 entsteht bei der Hydrolyse von XeF6 und XeF4. XeF6 + 3H2O → XeO3 + 6HF 3 XeF4 + 6H2O → Xe + 2 XeO3 + 12HF Die farblosen Kristalle bestehen aus einem Molekülgitter mit isolierten XeO3-Einheiten, sie sind hochexplosiv. XeO3 → Xe + 1,5 O2 ∆H = -402kJ/mol Betsändig sind wäßrige Lösungen von XeO3, in denen es überwiegend molekular gelöst ist. Außerdem entsteht etwas Xenonsäure H2XeO4, deren Anhydrid XeO3 ist. XeO3 + H2O H2XeO4 H2XeO4 + H2O H3O+ + HXeO4Die Lösungen ragieren schwach sauer und wirken stark oxidierend. Bei Zusatz von Lauge bildet sich Xenat(VI). H2XeO4- + 4 Na+ + 2 OH- → HXeO4- + H2O Auf diese Weise können thermisch ziemlich stabile Perxenate(VIII) Na4XeO6·nH2O und Ba2XeO6·1,5H2O erhalten werden. Perxenate(VIII) sind sehr starke Oxidationsmittel. H4XeO6 + 2H3O+ + 2e- XeO3 + 5 H2O E = +2,36V