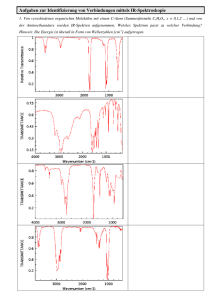
Grundlagen der Spektroskopie
Werbung

PC III: Grundlagen der Spektroskopie Peter Hamm • Elektronische Spektroskopie (UV-VIS) • Schwingungsspektroskopie (FTIR und Raman) • (Rotationsspektroskopie) • NMR-Spektroskopie Bücher: - Atkins: Physikalischer Chemie - Atkins und Friedman: Molecular Quantum Mechanics NMR Spektroskopie Rotations- Schwingung- elektronische X-ray elektronische Spek. Schwingungsspek. vibronische Spek. RotationsSchwingungsspek. rovibronische Spek. Rotationsspek. p* n p N-Methylthioacetamid nm Propenal in CCl4 elektronische Spek. Schwingungsspek. vibronische Spek. RotationsSchwingungsspek. rovibronische Spek. Rotationsspek. elektronische Spek. Schwingungsspek. vibronische Spek. RotationsSchwingungsspek. rovibronische Spek. Rotationsspek. Absorbance [norm.] N-Methylthioacetamid C N cis C-C-N-C trans 1400 1500 1600 -1 Frequency [cm ] Absorbance [norm.] CO2 in Wasser 2300 2320 2340 2360 -1 Frequency [cm ] 2380 2400 Absorbance [norm.] CO2 in Luft 2300 2320 2340 2360 -1 Frequency [cm ] 2380 2400 Absorbance [norm.] CO2 in Edelgasmatrix (10 K) 2300 2320 2340 2360 -1 Frequency [cm ] 2380 2400 elektronische Spek. Schwingungsspek. vibronische Spek. RotationsSchwingungsspek. rovibronische Spek. Rotationsspek. Rotationsspektrum von HCl Absorbance [norm.] Rotations-Schwingungsspektrum von CO2 2300 2320 2340 2360 -1 Frequency [cm ] 2380 2400 elektronische Spek. Schwingungsspek. vibronische Spek. RotationsSchwingungsspek. rovibronische Spek. Rotationsspek. S2 S1 DEel DErot S0 Hˆ tot Hˆ el Hˆ vib Hˆ rot DErot DEvib DEel DEvib Wechselwirkung von Licht mit Materie Absorption |1 hn |0 hn E1 E0 Gitterspektrometer (UV/VIS) Gitter Lampe Linse Probe Linse Spektrometer Intensity I0(n) I(n) n Lambert-Beersche Gesetz I (n ) A (n )cl 10 10 I 0 (n ) Integrierter Extinktionskoeffizient (n )dn ˆ 0 1 2 ˆ e 0 x 1 2 2 01 01: Übergangsdipolmoment n Verknüpfung Spektroskopie ↔ Quantenmechanik E1, 1 Frequenz: hn E1 E0 hn E0, 0 Integrierter Extinktionskoeffizient: (n )dn 01 2 ˆ 0 1 2 ˆ e 0 x 1 2 x x 01 y e y z z Crystalline Acetanilid b-Axis a-Axis (5) c-Axis (5) x x 01 y e y z z elektronische Spek. Schwingungsspek. vibronische Spek. RotationsSchwingungsspek. rovibronische Spek. Rotationsspek. Kurze Wiederholung PCII ˆ H( x) E( x) Zeitunabhängige Schrödingergleichung Beispiel: H2-Molekül 2 2 ˆ p e 1 e 1 1 ˆ H (1) 2m 4p 0 ra 4p 0 rb (1) 2 e- ra(1) p+ rb R ra (1) r12 p+ rb(2) (2) e- 2 2 ˆp2 2 e 1 e 1 ( 2) 2m 4p 0 ra 4p 0 rb ( 2 ) e 2 1 4p 0 R e2 1 4p 0 r12 Konzepte zur näherungsweise Lösung der SGL: LCAO Ansatz (Variationsprinzip) Schrödingergleichung Operatorgleichung/Differentialgleichung: 2 2 ˆ V ( x) ( x) E ( x) 2 2m dx Atomorbitale als Basis: cii i Entwickeln in dieser Basis Matrizen-Eigenwertgleichung Hc S c mit H ij dxdydz i* Hˆ j Sij dxdydz i* j Hückeltheorie: - nur p-Elektronen - vollständig parametrisiert - Überlappintegrale vernachlässigt Extended Hückeltheorie: - nur Valenzelektronen - Ionisierungsenergien und Zeff parametrisiert. - aber: Überlappintegrale ‚exakt‘ - semi-empirisch H2+: - ‚ab initio‘: startet von Schrödingergleichung, keine Parameter - Näherung, da Basis endlich - aber: nur ein Elektron ab initio Behandlung von Mehrelektronensystemen: - Hartree Fock, Dichtefunktionaltheorie, Configuration Interaction Hückeltheorie: Benzol LUMO HOMO N2 4s* 2p* 2px,2py, 2pz 3s 1p 2px,2py, 2pz 2s* 2s 2s 1s Heteronukleare Bindung: CO 4s* C2p 2p* 3snb C2s O2p 1p 2snb 1s O2s N2 4s* S0 S1 HOMOLUMO s p* 2p* 2px,2py, 2pz 3s 1p 2px,2py, 2pz 2s* 2s 2s 1s s p* x e s x p * e s x p *dxdydz ·x· · dxdydz dxdydz= 0 s p* y e s y p *dxdydz ·y· · dxdydz dxdydz= 0 s p* z e s z p *dxdydz ·z· · dxdydz dxdydz= 0 N2 4s* S0 S2 HOMO-1LUMO p p* 2p* 2px,2py, 2pz 3s 1p 2px,2py, 2pz 2s* 2s 2s 1s p p* x e p x p *dxdydz ·x· · dxdydz dxdydz 0 p p* y e p y p *dxdydz ·y· · dxdydz dxdydz= 0 p p* z e p z p *dxdydz ·z· · dxdydz dxdydz= 0 CO n p* 4s* C2p 2p* 3snb C2s O2p 1p 2snb 1s O2s n p* x e n x p *dxdydz ·x· dxdydz · dxdydz= 0 n p* z e n z p *dxdydz ·z· dxdydz · dxdydz 0 p* n p N-Methylthioacetamid Verknüpfung Spektroskopie ↔ Quantenmechanik E1, 1 Frequenz: hn E1 E0 hn E0, 0 Integrierter Extinktionskoeffizient: (n )dn 2 01 ˆ 0 1 2 ˆ e 0 x 1 2 x 01 y z elektronische Spek. Schwingungsspek. vibronische Spek. RotationsSchwingungsspek. rovibronische Spek. Rotationsspek. Lampe Linse Probe Linse Spektrometer I (n ) 10 A 10 (n )cl I 0 (n ) Intensity I0(n) I(n) n Gitterspektrometer (UV/VIS) Gitter FTIR Spektrometer 50% Strahlteiler Dx Detektor FTIR Spektrometer I (n , Dx) I (n )1 cos(2pnDx / c) 0 0 I (Dx) I (n )1 cos(2pnDx / c) dn const I (n ) cos(2pnDx / c)dn Inverse Fourier Transformation → I (n ) Lampe Linse Probe Linse Spektrometer I (n ) 10 A 10 (n )cl I 0 (n ) Intensity I0(n) I(n) n elektronische Spek. Schwingungsspek. vibronische Spek. RotationsSchwingungsspek. rovibronische Spek. Rotationsspek. H2+-Molekül: LCAO Ansatz Basis: Atomorbitale (a) 1s , , (b ) 1s 2s u 1s (a) 1s (b ) 1(sa ) 1(sb) 1s g 1s (a) 1s (b) H2+-Molekül: LCAO Ansatz Basis: Atomorbitale (a) 1s eV 4 , , (b ) 1s 2s u 1s 3 1s (a) (b ) 2 Antibindendes Orbital 1 1 -1 -2 2 3 1s g 1s (a) 4 R/Å 1s (b) Bindendes Orbital HONO: Schwingungsgrundzustand HONO: Streckschwingung HONO: Torsionsschwingung HONO: Biegeschwingung elektronische Spek. Schwingungsspek. vibronische Spek. RotationsSchwingungsspek. rovibronische Spek. Rotationsspek. H2+-Molekül: Harmonische Näherung E/H 0.02 2 4 6 8 R/a0 -0.02 -0.04 -0.06 Taylorentwicklung: V V ( R) V ( Re ) R 1 2V R Re 2 2 R R Re R Re 2 R Re Harmonischer Oszillator m1 k m2 V(x) x Harmonischer Oszillator k V(x) mit: m1 m2 m1 m2 x Harmonischer Oszillator k k 2 V ( x) x 2 H E m2 V(x) k 2 x E 2 2 2 x 2 2 x Lösungen: Harmonischer Oszillator n ( y) H n ( y)e mit k 2 y x 2 2 1 y2 2 1 En n n 0,1,2,... 2 mit k Harmonischer Oszillator Harmonischer Oszillator: Übergangsdipolmoment mn m n d m R n dR Auswahlregeln: • Zweiatomiges Molekül: Permanentes Dipolmoment • Dn=±1 • Hochfrequente Moden bei nicht zu hohen Temperaturen ( kBT): 01 Born Oppenheimer Approximation Hˆ Tˆel Tˆnuc Vˆel,nuc solve electronic SEQ Hˆ el el (r; R) Eel (R)el (r; R) Eel (R) is potential energy surface on which nuclei move solve vibrational SEQ by reintroducing: Hˆ nucnuc (R) nuc (R) with Tˆnuc Hˆ nuc Tˆnuc E(R) total wavefunction: tot el (r; R)nuc ( R) Transition dipole for vibration S0 S0 el (r ; R)i ( R) er el (r ; R ) j ( R) integrate over r: i ( R) el ( R) j ( R) Taylor expansion: el ( R) el ( Req ) del ( Req ) dR R Req R Req i ( R) el ( Req ) j ( R ) el ( Req ) ij dˆ el ( R ) i ( R) R j ( R) dR static dipole transition dipole Dn 1 CO2: Normalmoden Symmetrische Streckschwingung 1388 cm-1 Asymmetrische Streckschwingung 2349 cm-1 Biegeschwingung, 2-fach entartet 667 cm-1 Anzahl Normalmoden: • allgemein: 3N-6 • lineares Molekül: 3N-5 d 01 0 x 1 dx CO2: Normalmoden n1 ? nas(OCO) n3 s,deg OCO n 2 mi 2 1 ˆ H xi xi K ij x j 2 2 ij i Massegewichtete Koordinaten: 2 qi 1 ˆ H qi Fij q j 2 2 ij i qi mi xi mit: Fij K ij mi m j Normalmodentransformation (i. e. Eigenwerte und Vektoren von F): 2 1 Q 2 Hˆ i i Qi 2 2 i i N entkoppelte Oszillatoren mit: Ei i ni 1/ 2 mit: i i CO2: Normalmoden k m1 x1 k m2 x2 m1 x3 1 1 1 2 2 V k x1 x2 k x3 x2 K ij xi x j 2 2 2 i, j k mit K k 0 k 2k k k m1 0 k F k oder m1m2 k 0 k m1m2 2k m2 k m1m2 0 k m1m2 k m1 CO2: Normalmoden 1 0 k 2 m1 2km1 km2 3 m1m2 1 m2 A mm 1 2 1 1 0 1 1 2 m1m2 m2 1 k Translation: k 0 cm-1 m1 Symmetrische Streckschwingung: 1388 cm-1 Asymmetrische Streckschwingung: 2349 cm-1 m2 m1 CO2: Normalmoden 1 0 2 k m1 2km1 km2 3 m1m2 1 1 1 A' 1 0 2m1 m2 1 1 1 k Translation: k 0 cm-1 m1 Symmetrische Streckschwingung: 1388 cm-1 Asymmetrische Streckschwingung: 2349 cm-1 m2 m1 CO2: Normalmoden Symmetrische Streckschwingung 1388 cm-1 Asymmetrische Streckschwingung 2349 cm-1 Biegeschwingung, 2-fach entartet 667 cm-1 Anzahl Normalmoden: • allgemein: 3N-6 • lineares Molekül: 3N-5 d 01 0 x 1 dx CO2: Normalmoden n1 ? nas(OCO) n3 s,deg OCO n 2 Hessian Matrix 2 ˆ p 1 i ˆ H xi Kij x j 2 ij i 2mi Taylorentwicklung: V V ( x ) V (0) i xi 2V K ij xi x j 1 2V xi 2 i , j xi x j x 0 i xi x j 0 xi x j xi x j 0 Normal Modes Benzene Formaldehyd: Normal Modes 2783 cm-1 1746 cm-1 1500 cm-1 1167 cm-1 2843 cm-1 1249 cm-1 Anharmonizität: CO2 Four vibrational modes, two of them degenerate Two bands in IR nas(OCO), n3 = 2349 cm-1 and s,deg OCO, n2 = 667 cm-1 One band in Raman ns(OCO), n1= 1388 cm-1, not seen in IR n1+ n3 Combination bands (NIR) 2n2+ n3 nas(OCO) n3 s,deg OCO n Fundamental bands 2 Anharmonizität: CO Overtone band (NIR) 2n(CO) n(CO) Expanded Morsepotential V V De 1 e a ( R Re ) 2 1 1 En n x n 2 2 2 mit: a und x 4 De De 2De Dn=±1 Auswahlregel aufgeweicht Re R Anharmonizität: CO2 Four vibrational modes, two of them degenerate Two bands in IR nas(OCO), n3 = 2349 cm-1 and s,deg OCO, n2 = 667 cm-1 One band in Raman ns(OCO), n1= 1388 cm-1, not seen in IR n1+ n3 Combination bands (NIR) 2n2+ n3 nas(OCO) n3 s,deg OCO n Fundamental bands 2 Elektrische und mechanische Anharmonizität Elektrische Anharmonizität: Höhere-Ordnung Terme in der Taylorentwicklung des Dipoloperators: m n d m x n ... dx Mechanische Anharmonizität: Höhere-Ordnung Terme in der Taylorentwicklung der Potentialfläche: 1 2V V ( R) V ( Re ) 2 R 2 R Re 2 R Re Leiteroperatoren: p 2 k 2 p 2 2 m 2 H xˆ xˆ 2m 2 2m 2 mit m x' xˆ p' 1 p m 2 p' xˆ '2 H 2 2 Leiteroperatoren: definiere: 1 xˆ ipˆ a 2 1 a a† oder: xˆ 2 1 xˆ ipˆ a 2 † [a, a† ] aa† a†a 1 mit: a a n Nˆ n n n † 1 pˆ i a a † 2 (folgt aus: [ x , pˆ ] i ) 1 † H a a 2 † a n n 1 n 1 Aufsteigeoperator a n n n 1 Absteigeoperator Besetzungszahloperator Elektrische Anharmonizität:Oberton d 1 d 2 2 (Q) 0 Q Q ... 2 dQ 2 dQ Oberton: d 1 d 2 2 2 ˆ 0 2Q0 2 Q 0 ... 2 dQ 2 dQ Elektrische Anharmonizität: Kombinationsmode 2 0 Qi QiQ j ... i Qi i , j Qi Q j Kombinationsmode: 1i1 j ˆ 0i 0 j 1i Qi 0i 1 j 0 j 1i 0i 1 j Q j 0 j Qi Q j 2 1i Qi 0i 1 j Q j 0 j ... Qi Q j Anharmonizität: CO2 Four vibrational modes, two of them degenerate Two bands in IR nas(OCO), n3 = 2349 cm-1 and s,deg OCO, n2 = 667 cm-1 One band in Raman ns(OCO), n1= 1388 cm-1, not seen in IR n1+ n3 Combination bands (NIR) 2n2+ n3 nas(OCO) n3 s,deg OCO n Fundamental bands 2 Mechanische Anharmonizität: Fermiresonanz CO2 Mehr symmetrische Streckschwingung n1 Mehr Oberton Biegeschwingung n2 Mechanische Anharmonizität: Fermiresonanz Absorbance Benzoylchloride 1720 1760 1800 -1 Wavenumber [cm ] Mechanische Anharmonizität: Fermiresonanz mit einem Oberton 1 2V 2 1 3V 2 V V0 Qi Qi Q j ... 2 2 2 i Qi 2 i , j Qi Q j Vanh Kopplung: 1i 0 j Vanh 0i 2 j wenn: 1 3V 2 1i Qi 0i 0 j Q j 2 j 2 2 Qi Q j i 2 j 1i 2 j 1i 1i 2 j i 0i 2j j 1j j 0j Mechanische Anharmonizität: Fermiresonanz mit einem Oberton 1 0i 0 j ˆ 1i 0 j 0i 2 j 2 1 0i 0 j ˆ 1i 0 j 0i 0 j ˆ 0i 2 j 2 1 0i ˆ 1i 2 wenn: i 2 j 1i 2 j 1i 1i 2 j i 0i 2j j 1j j 0j Mechanische Anharmonizität: Fermiresonanz mit einer Kombinationsmode 1 V 2 2 3V V V0 Qi Qi Q j Qk ... 2 2 i Qi i , j , k Qi Q j Qk Vanh Kopplung: 1i 0 j 0k Vanh wenn: i 3V 0i1 j1k 1i Qi 0i 0 j Q j 1 j 0k Qk 1k Qi Q j Qk j k 1i 1 j1k 1i 1 j1k i 0i 1i 1 j1k k j 1 j 0k 0 j 0k Mechanische Anharmonizität: Fermiresonanz Absorbance Benzoylchloride 1720 1760 1800 -1 Wavenumber [cm ] Raman Spektroskopie Probe Spektrometer Linse GaAs cw-Laser Raman Spektroskopie Polarisierbarkeit: ind d R Req ... ( R) Req dR Durch eine Schwingung wird R zeitabhängig: R Req DR cos(vibt ) (t ) eq D cos(vibt ) mit d D DR dR Wenn wir Licht einstrahlen (nicht-resonant mit dem Schwingungsübergang, und nicht-resonant mit elektronischen Übergang ): (t ) 0 cosopt t ind (t ) (t ) (t ) 0 D cosvibt 0 cosopt t 0 0 cosopt t D 0 cosopt vib t cosopt vib t Raleigh Anti-Stokes Stokes Raman Spektroskopie mn d m R n dR Auswahlregeln: • Zweiatomiges Molekül: immer • Mehratomiges Molekül mit Inversionssymmetrie: IR-aktiv und Raman aktiv schliessen sich gegenseitig aus • Dn=±1 Schwingungsspektroskopie CO2 Symmetrische Streckschwingung 1388 cm-1 Raman-aktiv Asymmetrische Streckschwingung 2349 cm-1 IR-aktiv Biegeschwingung, 2-fach entartet 667 cm-1 IR-aktiv CO2: Normalmoden Raman Spektroskopie: CO2 Fermiresonanz: Streckschwingung + Oberton Biegeschwingung Raman Spektroskopie: Quantenmechanische Beschreibung GaAs Raman Spektroskopie Polarisierbarkeit: ind d R Req ... ( R) Req dR Durch eine Schwingung wird R zeitabhängig: R Req DR cos(vibt ) (t ) eq D cos(vibt ) mit d D DR dR Wenn wir Licht einstrahlen (nicht-resonant mit dem Schwingungsübergang, und nicht-resonant mit elektronischen Übergang ): (t ) 0 cosopt t ind (t ) (t ) (t ) 0 D cosvibt 0 cosopt t 0 0 cosopt t D 0 cosopt vib t cosopt vib t Raleigh Anti-Stokes Stokes Raman Spektroskopie Polarisierbarkeit: ind d R Req ... ( R) Req dR Durch eine Schwingung wird R zeitabhängig: R Req DR cos(vibt ) (t ) eq D cos(vibt ) mit d D DR dR Klassisch: thermische Anregung Quantum-mechanisch: „Nullpunkts-Schwingung“ + thermische Anregung Raman Spektroskopie: Quantenmechanische Beschreibung virtuelles Niveau virtuelles Niveau opt vib opt Stokes opt opt vib n=1 n=1 n=0 n=0 Anti-Stokes Raman Spektroskopie: Quantenmechanische Beschreibung I AntiStokes e I Stokes GaAs vib k BT Resonanz-Raman Spektroskopie Elektronischer Übergang opt vib opt Stokes opt opt vib n=1 n=1 n=0 n=0 Anti-Stokes Resonanz-Raman Spektroskopie: Bacteriorhodopsin 3 ps 0.5 ps J K 2 μs L 50 μs ms O ms M N ms Mathies, 1987 Resonanz-Raman Spektroskopie: Bacteriorhodopsin 3 ps 0.5 ps J K 2 μs L 50 μs ms O ms M N ms Mathies, 1987 IR Spektroskopie: Bacteriorhodopsin 2.0 D2O Absorbance 1.5 1.0 Amide 2 0.5 0.0 1000 1100 1200 1300 1400 1500 -1 Frequency [cm ] Amide I 1600 1700 Zusammenfassung: Schwingungsspektroskopie • Zwei komplementäre Messmethoden: IR-Absorptionsspektroskopie und Ramanspektroskopie • 3N-6(5) Normalmoden • in harmonischer Näherung und Dipolnäherung: maximal 3N-6(5) Banden: Auswahlregel Dn=±1 IR- aktiv: d/dR ≠0 Raman-aktiv: d/dR ≠0 • Mehratomiges Molekül mit Inversionssymmetrie: IR-aktiv und Raman aktiv schliessen sich gegenseitig aus • Anharmonizität Obertöne, Kombinationsmoden, Fermisresonanz (meist klein) elektronische Spek. Schwingungsspek. vibronische Spek. RotationsSchwingungsspek. rovibronische Spek. Rotationsspek. Benzol 925 cm-1 995 cm-1 Heteronukleare Bindung: CO 4s* C2p 2p* 3snb C2s O2p 1p 2snb 1s O2s Übergang zwischen zwei elektronischen Potentialflächen Energy S1 S0 R Franck-Condon Prinzip: klassische Beschreibung S1 10fs „<1fs“ S0 R „senkrechter Übergang“ Franck-Condon Prinzip: quantenmechanisch S1 el vib ˆ el 'el 'vib el ˆ el 'el vib 'vib S0 R elektronisches Dipolmatrixelement Franck-Condon Faktor nicht Franck-Condon aktiv n=2 FC 0 ( S0 ) n ( S1 ) 2 n=1 n=0 n=0 R (0,0) (0,1) (0,2) (0,3) (0,4) Frequenz schwach Franck-Condon aktiv n=2 FC 0 ( S0 ) n ( S1 ) 2 n=1 n=0 n=0 R (0,0) (0,1) (0,2) (0,3) (0,4) Frequenz stark Franck-Condon aktiv n=2 FC 0 ( S0 ) n ( S1 ) 2 n=1 n=0 n=0 R (0,0) (0,1) (0,2) (0,3) (0,4) Frequenz Franck-Condon Prinzip: klassisch QM n=2 n=1 n=0 n=0 R R Der senkrechte Übergang ist der intensivste Benzol 925 cm-1 995 cm-1 0-14 n=0 0-15 0-16 Vibronisches Spektrum I2 Aus. B. Snadden J. Chem Education 64 (1987) 919 1-14 n=1 1-15 n=2 Vibronische Übergänge in mehratomigen Molekülen R2 R2 R1 Vibronische Übergänge in mehratomigen Molekülen R2 R1 R1 Eine stark Franck-Condon aktive Mode, hohe Frequenz 00 01 02 03 04 Eine stark Franck-Condon aktive Mode, hohe Frequenz + eine schwach Franck-Condon aktive Mode, niedrige Frequenz 0,01,0 0,00,0 0,02,0 0,03,0 0,01,1 0,00,1 0,2 0,04,0 1,2 0,3 1,3 el1,vib2,vib ˆ el 'el '1,vib '2,vib el ˆ el 'el 1,vib '1,vib 2,vib '2,vib Evib E1,vib E2,vib 0,0 0,0 gleichzeitige Anregung R2 R1 0,0 1,0 gleichzeitige Anregung R2 R1 0,0 0,1 gleichzeitige Anregung R2 R1 0,0 1,1 gleichzeitige Anregung R2 R1 Eine stark Franck-Condon aktive Mode, hohe Frequenz + eine schwach Franck-Condon aktive Mode, niedrige Frequenz FC 1,vib '1,vib 2,vib '2,vib Evib E1,vib E2,vib 0,01,0 0,00,0 0,02,0 0,03,0 0,01,1 0,00,1 0,2 0,04,0 1,2 0,3 1,3 Franck-Condon Übergang CO2 asymmetrische Streckschwingung: nicht Franck-Condon aktiv symmetrische Streckschwingung: Franck Condon aktiv Benzol 925 cm-1 995 cm-1 Totally symmetric C-C Stretch Vibration: 995 cm-1 asymmetric C-C Stretch Vibration: 1010 cm-1 Franck-Condon Übergang Benzol asymmetrische Streckschwingung: nicht Franck-Condon aktiv symmetrische Streckschwingung: Franck Condon aktiv Resonanz-Raman Spektroskopie opt vib opt n=1 n=0 Stokes „1fs“ n=1 n=0 Raman-aktiv Franck-Condon aktiv Auswahlregeln: Franck-Condon Übergang • Symmetrie: Raman-aktiv Franck-Condon aktiv • Molekül mit Inversionssymmetrie: IR-aktiv und FranckCondon aktiv schliessen sich gegenseitig aus • Moden die stark resonanz-Raman aktiv sind, sind auch stark Franck-Condon-aktiv • Im Gegensatz zur Ramanspektroskopie: keine Dn=±1 Auswahlregel Benzol 925 cm-1 995 cm-1 Totally symmetric C-C Stretch Vibration: 995 cm-1 Elektronenkonfiguration Benzol LUMO HOMO Absorption und Floureszenz: Benzol 925 cm-1 995 cm-1 Einstein Koeffizienten Absorption Fluoreszenz |1 |1 hn 2 |1 hn hn |0 B01 01 Stimulierte Emission |0 A10 B01n 3 |0 B10 B01 Absorption R Schwingungsrelaxation 1-10 ps R Fluoreszenz 10 ns R n=1 Absorption: FC 0 Emission: FC 0 ( S0 ) ( S1 ) 1 n=0 n=1 n=0 R ( S1 ) ( S0 ) 1 Benzol 925 cm-1 995 cm-1 Jablonski Diagram schnell (Kasha‘s Rule) kIVR 1-10 ps 2.Periode: langsam Schwere Elemente: schnell 100ns 10ns (dipolerlaubt) 200fs –100ns Harmonische Potentialflächen R2 R1 Photochemie: H2+-Molekül eV 4 3 2 1 1 -1 -2 2 3 4 R/Å Femtochemistry: Ahmed Zewail, Nobel Prize 1999 Photodissociation of ICN M. Dantus, M. J. Rosker, A. H. Zewail, J. Chem. Phys. 87 (1987) 2395 A. H. Zewail, J. Phys. Chem. A 104 (2000) 5660 Femtosecond Lasers CPM Laser 1980 Pump-Probe Spectroscopy Pump-Probe Spectroscopy Pump-Probe Spectroscopy Pump-Probe Spectroscopy Prototype Photochemical Reaction: ICN M. Dantus, M. J. Rosker, A. H. Zewail, J. Chem. Phys. 87 (1987) 2395 A. H. Zewail, J. Phys. Chem. A 104 (2000) 5660 probe Photochemie: H2+-Molekül eV 4 1 pump 2 1 -1 -2 probe 3 2 3 4 R/Å Prototype Photochemical Reaction: ICN M. Dantus, M. J. Rosker, A. H. Zewail, J. Chem. Phys. 87 (1987) 2395 A. H. Zewail, J. Phys. Chem. A 104 (2000) 5660 Prototype Photochemical Reaction: NaI T. S. Rose, M. J. Rosker, A. H. Zewail, J. Chem. Phys. 91 (1989) 7415 A. H. Zewail, J. Phys. Chem. A 104 (2000) 5660 Electronic Photochemistry of HONO hn HONO HO· + · NO rN=O S10 S R2 R1 Cotting, Huber, JCP 195 (1996) 6208 rO-N Photochemical Reaction: Conical Intersection S1 and S2 in pyrazine in the Q6a and Q10a subspace Photochemical Reactions in Nature: Isomerization of Rhodopsin T. P. Sakmar, Curr. Opinion in Cell Biology 14 (2002) 189 Photochemical Reactions in Nature: Isomerization of Rhodopsin Q. Wang, R. W. Schoenlein, L. A. Peteanu, R. A. Mathies, C. V. Shank, Science 266 (1994) 422 Photochemical Reaction: Conical Intersection S. Hahn, G. Stock, J. Phys. Chem. B, Vol. 104, 2000, 1146 elektronische Spek. Schwingungsspek. vibronische Spek. RotationsSchwingungsspek. rovibronische Spek. Rotationsspek. Rotationsspektrum von HCl Rotationsspektroskopie Trägheitsmoment: I mi xi2 i xi : Abstand von der Drehachse, welche durch Schwerpunkt geht m1 m2 2-atomiges Molekül I R2 R mit: m1 m2 m1 m2 Rotationsspektroskopie Drehimpuls l I Energie m1 m2 Erot 1 2 l2 I 2 2I Korrespondenzprinzip R ˆ l l i Schrödingergleichung (in 2D) E 2 2I 2 2 Korrespondenzprinzip klassisch Ort x Impuls p Drehimpuls l Energie E quantenmechanisch xˆ x pˆ i x ˆl i ˆ E i t Rotationsspektroskopie Energiezustände Entartung: J=5: E=30B 11 J=4: E=20B 9 J=3: E=12B 7 J=2: E=6B J=1: E=2B J=0: E=0 5 3 1 Erot 2 J ( J 1) BJ ( J 1) 2I mit J 0,1,2, mJ J ,..., J Auswahlregeln: Permanentes Dipolmoment DJ 1 DmJ 0,1 Rotationsspektrum von HCl EJ BJ ( J 1) EJ EJ 1 2 BJ Rotationsspektroskopie Energiezustände Entartung: J=5: E=30B 11 J=4: E=20B 9 J=3: E=12B 7 J=2: E=6B J=1: E=2B J=0: E=0 5 3 1 Erot 2 J ( J 1) BJ ( J 1) 2I mit J 0,1,2, mJ J ,..., J Auswahlregeln: permanentes Dipolmoment DJ 1 DmJ 0,1 Rotationsspektroskopie: mehratomige Moleküle Molekül Trägheitsellipsoid: Linearer Kreisel: m1 m2 m3 I m x HCN 2 i i i Erot BJ ( J 1) Auswahlregeln: Dipolmoment DJ 1 DmJ 0,1 mit J 0,1,2, mJ J ,..., J Rotationsspektroskopie: mehratomige Moleküle Molekül Trägheitsellipsoid: Symmetrischer Kreisel: oblate: I|| I prolate: I|| I m1 m2 m3 m3 m3 Benzol Erot BJ ( J 1) ( A B) K 2 mit 2 2 B , A 2I 2I|| Auswahlregeln: Dipolmoment DJ 1 DK 0 DmJ 0,1 CH3Cl J 0,1,2, K J ,..., J mJ J ,..., J Rotationsspektroskopie: mehratomige Moleküle Molekül Trägheitsellipsoid: Sphärischer Kreisel: m1 m2 m1 m1 CH4 m1 Erot BJ ( J 1) mit 2 B 2I J 0,1,2, K J ,..., J mJ J ,..., J Auswahlregeln: Dipolmoment immer 0 DJ 1 DK 0 DmJ 0,1 elektronische Spek. Schwingungsspek. vibronische Spek. RotationsSchwingungsspek. rovibronische Spek. Rotationsspek. Rotations-Schwingungsspektrum von CO J=4 n=1 J=3 J=2 J=1 J=0 Etot Evib Erot 1 n BJ ( J 1) 2 Auswahlregeln: P-Zweig R-Zweig J=4 J=3 n=0 J=2 J=1 J=0 Übergangsdipolmoment bezüglich Schwingung D n 1 D J 1 CO2: Normalmoden elektronische Spek. Schwingungsspek. vibronische Spek. RotationsSchwingungsspek. rovibronische Spek. Rotationsspek. S2 S1 DEel DErot Hˆ tot Hˆ el Hˆ vib Hˆ rot Etot Eel Evib Erot tot el vib rot S0 DEvib S2 S1 DEel DErot S0 Hˆ tot Hˆ el Hˆ vib Hˆ rot Eel Evib Erot DEvib Ende Zeit: Ort: jeweils Dienstag 8-10, Mittwoch 10-12 34-K-01 Übungen: • Mittwoch 10.15-12.00 (jede 2. Woche, erstmals 3.Okt, 34K01 und 13K05) • Abgabe jeweils am Dienstag davor, 12.00 • Übung obligatorisch, 10 min Quiz, zählt 20% zur Endnote Betreuer: Steven Waldauer Halina Strzalka 34 K 92 34 K 90 Raman Spektroskopie: Polarisierbarkeit Polarisierbarkeit: Hˆ Hˆ (0) Hˆ (1) Hˆ (0) exˆ Hˆ (0) Vergleich Störungstheorie 1. und 2. Ordnung mit Taylorentwicklung dEn (0) E n En d En (0) 1 d 2 En 2 2 d 0 1 2 ... 2 Störungstheorie 1.Ordnung: En (1) n (0) n (0) 2 ... 0 Raman Spektroskopie: Polarisierbarkeit Polarisierbarkeit: Hˆ Hˆ (0) Hˆ (1) Hˆ (0) exˆ Hˆ (0) Vergleich Störungstheorie 1. und 2. Ordnung mit Taylorentwicklung dEn (0) E n En d En (0) 1 d 2 En 2 2 d 0 2 ... 0 1 2 ... 2 Störungstheorie 2.Ordnung: En ( 2) m n n ( 0) m ( 0) n E ( 0) E ( 0) m 2 2 n (1) mn n (0) m(0) E (0) n E (0) m Beispiel: Polarisierbarkeit atomarer Wasserstoff 2 px 1s 1s c2 px + Störungstheorie 2.Ordnung: En ( 2) m n n ( 0) m ( 0) n E ( 0) E ( 0) m 2 2 n (1) mn n (0) m(0) E (0) n E (0) m Raman Spektroskopie: Polarisierbarkeit Hˆ Hˆ (0) Hˆ (1) Hˆ (0) exˆ Hˆ (0) Vergleich Störungstheorie 1. und 2. Ordnung mit Taylorentwicklung dEn (0) E n En d En (0) 1 d 2 En 2 2 d 0 2 ... 0 1 2 ... 2 Dipolmoment: dE 0 d Wasserstoff: 9 2 a3