Singulett (gepaarte Spins): 2 +1/2 – 1/2 + 1 = 1
Werbung
: 2 +1/2 – 1/2 + 1 = 1")
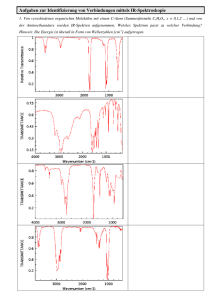
PHILIPPS-UNIVERSITÄT MARBURG FACHBEREICH CHEMIE _____________________________________________________________________________________________________________________________ ______________________ Spektroskopischer Kurs – Teil IR-Spektroskopie Prof. Dr. Thomas Schrader Fachbereich Chemie, Universität Marburg Hans-Meerwein-Straße Phone: int. + 6421 / 28-25544 fax: int. + 6421 / 28-28917 e-mail: [email protected] ___________________________________________________________________________________________________________________________________________________ 1. Einleitung: Moleküle und elektromagnetische Strahlung ~ E = h = hc/ = hcν h = 6.626.10-34 Js c = 3 x 108 ms-1 = 3 x 1010cms-1 NA = 6.022 x 1023 mol-1 Ionisierungsenergien... Energie Energie ...von Elektronen in inneren Schalen (Elektronenspektrosk. f. d. chem. Anal.) ...von Valenzelektronen (Photoelektronenspektroskopie) Bsp.: Ethen - Formaldehyd - n - >100 eV >103 kJmol-1 ESCA 10-20eV ~103 kJmol-1 PES 10.5 eV 10.9 eV 14.0 eV [1 eV = 100 kJmol-1] Elektronische Übergänge 200-800 nm 293 nm Bsp.: n* - Acetaldehyd n* - CH3-N=N-CH3 n* - Indigo Achtung: Bindungsstärken von 347 nm 610 nm Spektroskopie 600-150 kJmol-1 UV/VIS Verdopplung der Wellenlänge = Halbierung der Energie Kovalenzbindungen: Photochemie! Schwingungen 2500 – 50-5 kJmol-1 25000 nm (2.5-25m) 4000 cm1 – [1 cm –1 12 400 cm-1 Jmol-1] IR Rotationen 100 cm-1 – ~1000 – 10 1 cm-1 Jmol-1 (0.1 mm – 10 mm) 19.9 cm-1 MW Bsp.: NH3 (J=0 J=1) Energiedifferenzen zwischen Spinniveaus E = geB x B (e-) bei 0.3 T ESR E = giN x B = x ђ B (Kernspin) bei 2.35 T 10 GHz 1010 Hz 4 Jmol [ = 3 cm] 10 MHz 107 Hz -2 -1 4x10 Jmol [ = 3 m] -1 NMR (Radiowellen) 2. Grundlagen der IR-Spektroskopie 2.1. Valenzschwingung Zweiatomiges Molekül: harmonischer, besser anharmonischer Oszillator; Abb. 2.1. (Feder) und 2.2. (Harmon.) und 2.3. (Anharmon. Oszillator) Morse-Potential: n = Schwingungsquantenzahl; r0 = Atomabstand = Bindungslänge bei RT: n = 0 hauptsächlich populiert (E ist nicht Null). Aus Schrödingergleichung folgt: Evib (n = 0) = ½ h osc = ђ k/osc = ½ k/harmonischer Oszillator). Evib (n = 1) = 3/2 h osc; E = h osc = hc x ~ ν = m1 x m2 / m1 + m2 (reduzierte Masse). osc. k = Kraftkonstante (~Bindungsstärke). osc 1013 – 1014 Hz. 2 Konsequenzen: a) je größer die Kraftkonstante k, desto größer ist osc ( ~ ν). Bsp.: ~ ν [cm -1 CC-Bindungen CH-Bindungen ~ ν[cm -1 ] ~ ν [cm -1 ] C≡C C=C C-C 2220 1640 1000 ≡C-H Car-H Calk-H 3300 3050 2950 b) je kleiner die reduzierte Masse , desto größer ist osc ( vgl. H-X vs. D-X: ] ~ ν). HX = mH x mX / mH + mX ; DX = mD x mX / mD + mX also ist DX ≈ 2 HX und damit DX = 1/2 HX = 0.7 HX Bsp.: Calk-H: 2950 cm-1; Calk-D: 2150 cm-1 (!) , auch bei C-X ( X = C,O,F,Cl) Bsp.: LAH vs. LAD-Reduktion eines Oxetans... OH O LAH OH H OH OH OH Ph OH O Ph 2.2. Auswahlregeln Ph LAD OH 2950 cm-1 OH D OH OH Ph 2150 cm-1 a) n = +/- 1, d.h. Oberschwingungen haben sehr geringe Intensität; i.a. beobachtet man nur Schwingungen zwischen n = 0 → n = 1 (Grundschwingung). b) Dipolmoment-Änderung: Wechselwirkung mit elektromagnetischer Strahlung Bsp.: ←H-H→ H-H-Valenzschwingung: IR-inaktiv ←C=O→ C=O-Valenzschwingung: IR-aktiv Dipolmoment: = Q x l (Ladung x Abstand); die Absorptionsintensität ~ Änderung des Dipolmoments, denn großes Q (~Polarisation)→ großes Merken: Schwingungen symmetrisch zum Symmetriezentrum sind IR-inaktiv, aber Raman-aktiv (mißt Streulicht)! c) Rotationsquantelung: Aufspalten der Schwingungslinie in mehrere Linien (Rotations-Schwingungsspektren); jedoch scharf nur in der Gasphase. Realfall: Linienverbreiterung (später mehr im UV-Teil). 2.3. Schwingungsformen Mehratomige Moleküle mit N Atomen: 3N-5 Schwingungs-Freiheitsgrade (lineare) bzw. 3N-6 (nicht lineare Moleküle). Bsp. Wasser (9 - 6 = 3 Normalschwingungen): =H =O Sym. Valenz Asym. Valenz Sym. Deformation Valenzschwingungen: Bindungslängen ändern sich. Deformationsschwingungen: Bindungswinkel ändern sich. In Molekülverbänden sind viele Schwingungssysteme voneinander unabhängig: lokalisierte Schwingungen. Abb.: Lokalisierte Schwingungen! Probleme: a) gekoppelte Schwingungen – energetisch und räumlich benachbarte Schwingungsszustände beeinflussen sich gegenseitig. Bsp. C-O und C-C –Valenzschwingungen koppeln in C-C-O-Gruppen: H3C-OH: H3C-CH2-OH: (H3C)2HC-OH: (H3C)3C-OH: ~ ν (CO) = 1034 cm ~ ν (CO) = 1054 cm ~ ν (CO) = 1110 cm ~ ν (CO) = 1200 cm -1 -1 -1 -1 Kopplungen zwischen Normal- und Oberschwingungen: Fermi-Resonanz (Aldehydgruppe, später). b) Gerüstschwingungen, z. B. in C-Ketten oder –Ringen: „Fingerprint“-Bereich ab < 1500 cm-1 Allgemein: Kopplungen werden vemieden, wenn die Eigenfrequenzen benachbarter Schwingungen sich stark unterscheiden (d.h. unterschiedliche Kraftkonstanten bzw. reduzierte Massen) 2.4. Probenaufnahme und Absorptionsbereiche 2.4.1. Probenbereitung: a) Feststoffe: KBr-Preßling oder Nujol-Verreibung (= Paraffinöl); in Lösung (CH2Cl2, CCl4, CS2). Vorsicht: Viele polare Lösungsmittel absorbieren zu stark! Als Pulver (ATR-Aufnahme) b) Flüssigkeiten: Film auf KCl, Lösung 2.4.2. Spektrometer a) Gitterspektrometer: klassische Meßmethode (zeitaufwendige Zweikanalgeräte) b) FT-Geräte: Simultane Erfassung aller Frequenzen des IR-Spektrums im Detektor →kein Wellenlängen-Scan mehr nötig. Trick: Zerlegung der polyfrequenten IR-Strahlung in ein, das keine Funktion der Frequenz, sondern der Zeit ist Interferogramm (Michelson-Interferometer: Ein beweglicher und ein fester Spiegel überlagern die Strahlung konstruktiv oder destruktiv). Nach Probendurchgang Umwandlung der optischen Signale in elektrische durch Detektor und Fourier-Transformation ins das gewohhnte Bandenspektrum. Vorteile: schnell (wenige Sekunden), präzise Wellenlängen (Laser als interne Eichung), günstiges Signal-Rauschverhältnis (→ GC-IR). Abb. 2.6. Spektrometer! 2.4.3. Absorptionsbereiche und diagnostische Schwingungen (4000-600 cm-1) Abb. 2.9. Absorptionsbereiche! a) 4000 – 3000 cm-1 3000 – 2000 cm-1 Kumulierte Doppelbdg.: OH: 3600-3200 (stark,breit) – variierend NH: 3500-3300 (mittel) CH: C≡C-H 3300 (s) =C-H: 3100-3000 (m oder v) CH: CH2, CH3 3000 – 2800 cm-1 (s) SH: 2600-2550 (w-m) PH: 2440-2450 (m-s) SiH: 2160-2110 (m-s) C≡C: R‘C≡CR 2260-2150 (m-w) RC≡CH 2140-2100 (w) C≡N: 2260-2200 (m) CD: 2300-2200 (m-w) N=C=O: 2275-250 (vs) N3: 2160-2120 (s) 2000 cm-1 – 600 cm-1 C=O: 1800-1650 (s-vs) NO2: 1560 (s) und 1350 (s) (as. u. sym. V.-S.) Deformation von Aromaten mit typischem Substitutionsmuster: C-H: 900-600 (m-s) Allgemein: Der diagnostische Wert solcher IR-Banden steigt, wenn Ausgangsmaterial und Reagentien einer Reaktion bekannt sind. So können Strukturdaten kontrolliert werden: Sind die Zuordnungen vernünftig? Wichtig: IR- und NMR- sowie MS-Spektren ergänzen sich hervorragend: Im IR sind die funktionellen Gruppen direkt zu sehen, im NMR die Protonen und Gerüst-C-Atome, im MS das Molekulargewicht sowie typische Fragmente. Bsp.: NMR- und IR-Spektrum von p-Nitrobenzonitril. 3. Funktionelle Gruppen und ihre Schwingungen Zur besseren Übung behandeln wir zunächst zu jeder Gruppe die typischen Schwingungen und demonstrieren sie an Beispielspektren. Danach üben wir das Gelernte sofort an Aufgaben A-M im Skript! 3.1. Alkane: Einfache Spektren: Viele Banden fallen zusammen und sind intensitätsschwach (vor allem C-Ketten!). Keine H-Brücken: sehr verläßliche Bandenlagen. Gut zu unterscheiden: gesättigte CH-Valenz immer bei ≤ 3000 cm-1 (≠ unges. =CH-Valenz immer bei > 3000 cm-1 und schwächer. Genauer: 2960-2850: CH-Valenz; 1470-1370: CH-Deformation. Aldehyde: O=C-H-Valenz typische Doppelbande bei 2900-2700 cm-1 (FermiResonanz), Beispiel später bei Carbonylen. CH3-C=O-: 1380-1350 cm-1 (starke CH-Def.) Ether: 1250-1060 cm1 (starke C-O-Val.) Bsp.: Dia 7: Dipropylether – CH-Valenz und C-O-Valenz! 3.2. Alkene / Alkine typische Valenzen: C≡C-H: 3300 cm-1 (s); =C-H: 3100-3000 cm-1 (oft verdeckt); R2C=CR-H: typische Deformationsschwingungsmuster! (um 800-1000 cm-1). Auch unterscheidbar: cis/trans-Alkene; Vinyliden (CH2=CH-). C=C-Valenz: ~1650 cm-1. Konjugation verringert DB-Charakter (~k)→kleinere Werte! C≡C-Valenz: ~2200-2100 cm-1. Aromaten: charakteristische, aber schwache Obertöne bei 2000-1650 cm-1; typisch: Trio bei 1600, 1500 und 1450 cm-1 (C=C-Valenzen, m) Typische Substitutionsmuster erkennbar im Fingerprint bei 700-900 cm-1 (CHout of plane-Deformationsschw.) Bsp.: Dia 9: cis- HC≡C-CH=CH-OCH3 : man sieht ≡C-H, C≡C, C=C, =C-O-C (1270), CH3-O (1120), HC=CH (cis, 730). + Dia 10: Ph-C≡C-CH(O-CH2-CH3)2: Trio und Fingerprint des monosubst. Aromaten (760, 700). --------------------------------------------- Aufgaben A und B ! ---------------------------------------------- 3.3. Alkohole, Phenole, Amine: Alkohole, Phenole - Wichtig: OH-Valenz (frei, Gasphase): 3650-3600 (scharf). Meist aber in H-Brücken: O-H...O, dabei ist die O-H-Bindung geschwächt und liefert kleinere Wellenzahlen; H-Brücken zu sp3-O-/N- (Alkohole, Aminoalkohole): 3600-3200 (s); ... zu sp2-O=/N- (Carbonsäuren): 3200-2500 cm-1 (v). Kristallwasser: 3600 – 3100 cm-1 (oft in Preßlingen). Intramolekulare H-Brücken (1,2-Diole): 3570-3450 (scharf) bei Verdünnung unverändert! ≠ : Intermolekulare H-Brücken (Carbonsäuredimere): 3500-3200 (breit, bes. bei Polymeren) verschwinden bei Verdünnung! Typisch auch: C-OH-Valenz: 1150-1040 cm1 (vgl. Ether), dazu oft in planeDeformation bei 1420-1260 cm-1 (s). Amine - NH-Valenz: 3500-3300 cm-1 (primäre Amine 2 Banden!): schwächer, aber schärfer als OH (weniger H-Brücken). H-Brücken zu Carbonylen erniedrigen deren C=O-Valenz (Peptide)! Amid-NH: 2 Banden (Amid I, monomer, 3500 und II, dimer, 3400), auch für die C=O-Valenz (1690, 1600) NH3+ (Aminos.): 3130-3030 cm-1: breit; dazu oft 2 Buckel bei 2500 / 2100 cm-1. NH2+, NH+: 2700-2250 cm-1. Amide, monosubst.: 3460-3400 cm-1 (bei H-Brücken erniedrigt). NH-Deformation: NH2 1650 cm-1; NH3+: 1600/1500 cm-1 (s!); auch bei 800. Bsp.: Dia 13: 1-Ethinylcyclohexanol: O-Hfrei (sterisch abgeschirmt! 3600); in HBrücken: 3470, C≡C: 3310. Dia 15: 2-Hydroxyacetophenon: intramol. H-Brücke im Chelat: breite O-HValenz: 3000, unverändert bei Verdünnung, 1640: konjugierte C=O (später), C=Carom.: darunter. Dia 28: n-Butylamin: NH-Valenz 3350; NH2-Def. in plane 1600; NH2-Def. out of plane 830 cm-1. 3.4. Carbonsäuren Intensität der C=O-Bande nimmt ab von Säuren > Ester > Ketone ~ Aldehyde ~ Amide (Dipolmoment). OHfrei -Val.: 3600-3500 cm1, aber nur sehr verdünnt. OHgeb. 3300-2500 cm-1 (sehr breit, oft typisch strukturiert!). C=O-Val.: 1725-1700 (s, meist Dimer); CO2-: 1610-1550 cm-1 (Aminosäuren). Bsp.: Dia 26: CH3-CH=CH-CH=CH-CO2H: OH-Val 3000, bei Verd. auch 3530, konj. C=O: 1690 cm-1. --------------------------------------------- Aufgaben L (Amine) und E + F (Phenole, Alkohole, Säuren) ---------------------------------------------- 3.5. Ester: Gesättigte Ester bei 1750-1735 cm-1; O-Vinylester 1800-1750 cm-1 (–I-Effekt am Ester-O → höherer DB-Charakter bei C=O). Lactone: 1840-1730 cm-1(Ringspannung!); stark elektronegative Subst. in -Pos. (-Chloro-, -Ketoester): 1770-1740 cm-1; ungesättigt (auch bei -Ketoestern in Enolform: ~1650 cm-1 (Konjugation OH-C=C-CO2R), Schwächung des DBCharakters C=O durch Mesomerie. Alle Klassen: C-O-Valenz: 1300-1050 (2 starke Banden, sym. + as.) Anhydride: 1850-1750 cm-1; Säurechloride: 1820-1750 cm-1 (stark elektronenziehende Subst.). Bsp.: Dia 31: Essigsäure-(dimethylaminoethyl)ester: C=O 1750; C-O 1240 (Acetyl), ebenso 1380 (s). 3.5. Ketone, Aldehyde: Merken: Die Lage der Carbonylbande hängt ab von: a) physikalischem Zustand (fest etwas niedriger); b) induktiven Effekten von Nachbarsubstituenten (je stärker elektronenziehend, desto höheres k); c) Konjugation (15-40 cm-1 niedriger wg. partiellem Einfachbindungscharakter O=C-C=C ↔O--C=C-C+); d) Ringspannung (bis zu 100 cm-1 höher; typisch für Ringgröße der Ketone (kleien Ringe: endocyclische Orbitale am Carbonyl-C mehr p-Charakter, exocyclische mehr s-Charakter →stärkere C=O-Bindung); e) H-Brücken: C=O...H-O; 40-60 cm-1 niedriger). Ketone: gesättigte 1725-1700 cm-1; aromatische 1700-1680 cm-1; Enone: 16801660 cm-1. 6-Ringe u. größer: 1720 cm-1; 5-Ringe: 1750 cm-1; 4-Ringe 1780 cm-1. -Halogenketone: 1745-1725 cm-1; -Dihalogen: 1765-45 cm-1; -Fluor: 1770 cm-1. 1,2-Diketone: 1720–1 (s-trans); 1750 cm-1 (s-cis, z.B. 1,2-Dioxocyclohexan), s.o. 1,3-Diketone: 1650-1540 (breite Bande wg. Konjugation und intramol. HBrücken [O=C-C=C ↔O--C=C-C]. Aldehyde: O=C-H-Valenz: 2 typische Banden bei 2880-2650 cm-1 (FermiResonanz mit CH-Deformations-Oberton). Gesättigte: 1740-1720 cm-1; aromatische: 1715-1695 cm-1; Enale: 1700-1680 cm-1. Bsp.: Dia 18: 2-Pentanon: C=O 1720, CH 2900, unverzweigte Kette. Dia 19: 4-Methyl-2-pentanon: C=O 1720; CH 2900; Isomer im Fingerprint! Dia 21: 4-Oxo-2-pentanon: C=O 1620; OH 3000 (br); Enol-Tautomerie! --------------------------------------------- Aufgaben G (Ester) und C/D (Aldehyde, Ketone) ---------------------------------------------- Anhang: Zu Amiden, Urethanen, Thioestern, Iminen, Ebolethern, Enaminen, Nitrogruppen sowie Fingerprint-Banden (Thioketon, Sulfoxid, Sulfonate, Phosphonsäurealkyl- und arylester, Alkylhalogenide) sowie zu anorganischen IonenAmmonium, Phosphat, Nitrat, Cyanid, Cyanat, Sulfat, Carbonat) siehe Hesse-Meier-Zeeh, Kap. 9-11: Tab. 2.10-2.21! ------------------4. Moderne Aspekte der IR-Spektroskopie a) Peak-Erkennung, -Überlagerung, Subtraktion von ganzen Spektren durch spezielle Software heute Standard. b) Online-Spektrenkataloge (SpecInfo etc.) erlauben automatischen Vergleich und Identifizierung unbekannter Verbindungen. c) Spektreninterpretation: Simulation ganzer Spektren; pattern recognition etc. d) Verknüpfung von IR-Spektrum mit zugehörigen NMR-Signalen, Fragmentierungen im MS oder UV-Banden → koordinierte Spektreninterpretation (artificial intelligence). Bsp.: Komplexbildung von Noradrenalin mit macrozyklischem Wirt (O. Molt). 5. Quantitative IR-Spektroskopie: Lambert-Beer-Gesetz: lg Io/I = x c x d = E = Extinktion E = Extinktion wird gemessen; = Stoffkonstante; d = Schichtdicke (Küvette): → c = Konzentration bestimmbar! Achtung: Lambert-Beer gilt nur bei niedrigen Konzentrationen (→Eichkurve). Anwendungen: Kunststoffanalytik und Qualitätskontrolle von Pharmaka und Pflanzenschutzmitteln. Spektroskopischer Kurs – Teil 2: UV/VIS-Spektroskopie 1. UV/VIS-Sektor des elektromagnetischen Spektrums: [nm] X-ray fernes UV nahes UV VIS IR < 10 10-280 B: 320-280 A: 400-320 Sunglasses! 400-750 > 750 Folie: Ultraviolett / Sichtbar Folie: Atomspektren 2. Wieviele Elektronen machen den Übergang? Klassisch: Übergangswahrscheinlichkeit = Oszillatorstärke f01 Gegenstück in der Quantenmechanik: Dipolstärke (Veränderung des Dipolmoments) Zusammenhang: Oszillatorstärke F01 Dipolstärke D01 = Übergangsmoment M01 2 Verbotener Übergang: f01 « 1 Erlaubter Übergang: f01 1 Folie: Polarisationsrichtung der Absorption Folie: *-Absorption von Ethylen 3. Auswahlregeln: a) Spinverbot: Gesamtspin S bzw. Multiplizität M = 2 S + 1 darf sich nicht ändern! b) Symmetrieverbot: z.B. in zentrosymmetrischen Molekülen: verboten: gg bzw. uu. c) Überlappungsverbot: Orbitale müssen sich überlappen ( Charge-Transfer) Aber: Alle Verbote könen umgangen werden, z.B. durch Spin-Bahn-Kopplung (vgl. n*-Übergang). Dabei ist lediglich f01 oft erniedrigt. 4. Welche elektronischen Vorgänge gibt es? Jablonski-Termschema! Abb.1.4. a) Absorption A b) Internal Conversion IC c) Intersystem Crossing ISC d) Fluoreszenz F e) Phosphoreszenz P f) Relaxation R Singulett (gepaarte Spins): 2 +1/2 – 1/2 + 1 = 1 Triplett ( 2 ungepaarte Spins): 2 +1/2 + 1/2 + 1 = 3 Allgemein: Bandenverbreiterung durch überlagerte Schwingungen und Rotationen! Folie: Molekülspektren (Bandenspektren) Folie: Ein typisches ... Folie: Bandenlage Folie: Absorptionsintensität ~Bandenfläche, Näherungsmethode 5. Lambert-Beer-Gesetz: A = E = Extinktion = log I/Io = c d A = Absorption = Extinktion (dimensionslos) I = Intensität des eingestrahlten Lichts Io = Intensität des durchgelassenen Lichts (Transmission) = molarer Extinktionskoeffizient [cm2 mmol-1] c = Konzentration [mol / l) d = Schichtdicke in der Küvette [cm] UV-Spektrum (Werte bis zu 105) 6. Übergänge zwischen MO’s: Folie: Arten der Elektronenanregung Folie: Absorptionsbereiche Folie: *-Übergänge Folie: n*-Übergänge Substituenteneffekte bewirken bathochrome (längerwellige) und hypsochrome (kürzerwellige) Verschiebungen. Ausgedehntere -Systeme wirken hyperchrom (Intensitätsverstärkend) und hypochrom (Intensitätsschwächend). Abstufung der Intensität: verboten schwach erlaubt stark Franck-Condon-Prinzip: erst bei Versuch mit Tetracen – später! 7. Apparatives: Zweistrahl-Spektrometer: Abb. 1.2. Gute Lösungsmittel ohne Eigenabsorption: a) Unschlagbar: Perfluoralkane b) Bis 180-190 nm: Pentan-Heptan, Cyclohexan, Wasser, Acetonitril c) Bis 210 nm: Methanol, Ethanol, Diethylether d) Bis 249nm: Methylenchlorid, Chloroform, Tetrachlorkohlenstoff UV-Spektren zeichnen die Absorption A oder besser den molaren ~ Extinktionskoeffizienten (substanzspezifisch) gegen oder besser auf (~ Energie). 8. Chromophore und Auxochrome: a) Konjugation (=Vinylogie) von Chromophoren (Farbträger) bathochrome Verschiebung durch Erhöhung der MO-Zahl auf energetisch engem Raum (*-Übergänge werden längerwellig und intensiver). Folie: *-Übergänge b) Auxochrome (Farbvertiefer) haben + oder –M-Effekt-Heteroatome mit freien Elektronenpaaren (Vergrößerung des delokalisierten Systems) Abb. 1.15. Ethylen mit Auxochrom X. Folie: Polymethinfarbstoffe 1 und 2. Auf Versuch hinweisen! c) Inkrementsysteme – später bei der Nachbesprechung der Versuche! 9. Benzol und Aromaten: Abb. 1.19. Elektronenanregungen beim Benzol Im Grundzustand zwei Paare von entarteten MO’s (2 / 3 und 4* / 5*). Nach Anregung von 1-2 Elektronen in höhere Niveaus ergeben sich ein Grundniveau und 4 angeregte Niveaus (die beiden obersten sind entartet). Also gibt es drei Übergäne, die man -, p- und -Bande nennt. Abb. 1.20 oder Folie: Aromaten Die längstwellige -Bande ist stark strukturiert durch aufmodulierte Schwingungsübergänge. Dabei ist die symmetrische Gerüstschwingung (923 cm1 ) IR-verboten, erst die unsymmetrische nächsthöhere (520 cm-1) wird beobachtet. + und –M-Substituenten verändern die Lage der p- und -Bande durch Vergrößerung des Chromophors durch Mesomerie. Besonders effektiv: pushpull-Systeme! Bsp.: p-Nitrophenol (max (): 310 nm vs. Benzol: 256 nm). Kondensierte Aromaten: auf Versuch hinweisen – später. 10.Carbonyle: Am günstigsten ist der n*-Übergang von Ketonen bei 275-300 nm. Konjugation an die C=O-Gruppe in Enonen verschiebt die -Niveaus stark, nicht aber das n-Niveau. Konsequenz: der längstwellige *-Übergang verschiebt sich in den sichtbaren Bereich und verdeckt den schwächeren n*Übergang. Abschätzung der Absorptionsmaxima mit den Woodward-Regeln: evtl. nach den Versuchen auf Folien – später! Solvatochromie: Solvatation stabilisiert einseitig entweder das LUMO (bathochrome Verschiebung des Übergangs) oder das HOMO (hypsochrome Verschiebung des Übergangs) eines Farbstoffs – auf Versuch hinweisen! Neue Folie? 11. Anwendungen: A) Quantitative Analyse: Kolorimetrie und Photometrie (Lambert-Beer: E ~ c) B) Potentiometrische Titration C) Bestimmung von Gleichgewichten und Dissoziationskonstanten D) Spurenanalyse – Bsp. Blutalkoholbestimmung: Ethanol wird im Test durch eine Dehydrogenase zu Acetaldehyd oxidiert, dabei entsteht H2. Dies wird anschließend sofort von NAD+ aufgenommen und reduziert dieses zu NADH. Der stark veränderte Chromophor von NADH wird durch quantitative Absorptionsmessungen verfolgt. Formeln? E) UV-Detektoren für die HPLC und GC. Elegant: Photodiodenarray-Detektor – er liefert zu jedem Meßpunkt (~ Retentionszeit) ein vollständiges UVSpektrum. Abbildung Beckmann-Diodenarray F) Kinetikmessungen direkt im Reaktionsgefäß über Lichtleiter. LaserBlitzapparaturen: Anregungsblitz startet Reaktion, Meßblitze erfassen nach kurzer Zeit den Fortgang der Reaktion (Lebensdauern im ns- und psBereich). Nobelpreis 1999: Achmed Zewail: Femtosekunden-Spektroskopie – man kann heute Übergangszustände anschauen! Abb.: Titelblatt Angewandte Chemie – Nobelvortrag Bsp.: N O h N O 365 nm O + N2 + CO Nach Einstrahlung ins langwellige Absorptionsmaximum nimmt die Transmission bei 2 Banden des Edukts zu, d.h. das Edukt wird verbraucht. Zwischen den beiden isosbestischen Punkten nimmt die C=O-Absorption (n*)des Cyclopentanons ständig zu. Isosbestische Punkte (alle Kurven schneiden sich in solchen Punkten) zeigen das Fehlen von Zwischenstufen an! 12. Derivativ-Spektroskopie: Junge Entwicklung: Überführung der UV-Absorptionskurven ind die erste (dA() / d) bzw. zweite Ableitung (d2A() / d2) führt zu einer ausgeprägteren Struktur, die kleine Veränderungen im Originalspektrum (z.B. eine Schulter) gut herausarbeitet. Abb. 1.29. Erste Ableitung: Maximum 0; Wendepunkt Maximum. 13.UV-Titration: Ein Wirtmolekül bindet ein farbiges Gastmolekül unter Veränderung seines UV-Spektrums (Bsp.: Anslyn Tartrat-Receptor über Bisguanidiniumboronat). Bei der Zugabe des besser bindenden Tartrats verdrängt dieses den Farbstoff aus dem Komplex und das UV-Spektrum geht wieder auf das des freien Farbstoffs zurück. Isosbestische Punkte zeigen die direkte Komplexierung und Verdrängung des Farbstoff-Moleküls vom Receptor an. Die Extinktion im Absorptionsmaximum gibt direkt die Konzentration an Tartrat in der Weinprobe an (Eichgerade nötig).