A: Organische Analyse – Teil 1 1. Theoretische Grundlagen Extraktion
Werbung

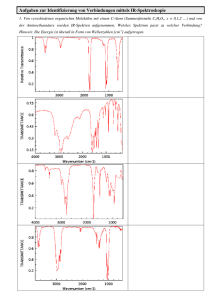
LU Synthesechemie Extraktion A: Organische Analyse – Teil 1 Prinzipielle Aufgabenstellung und Extraktion In der vorliegenden Übung wird ein 3-Komponentengemisch bestehend aus zwei organischen Neutralstoffen und einer organischen Verbindung mit sauren oder basischen Eigenschaften ausgehändigt. Durch geeignete Wahl des wässrigen Extraktionsmittels kann der Stoff mit sauren oder basischen Eigenschaften von den beiden Neutralstoffen abgetrennt werden. Die beiden Neutralstoffe sollen anschließend mittels Säulenchromatographie getrennt werden. Alle drei Stoffe werden nun isoliert und getrocknet und anschließend mittels Schmelzpunkt, IR, MS und NMR Messung charakterisiert und identifiziert. 1 Falls für eine eindeutige Charakterisierung erforderlich sind die kristallinen Produkte ggf. noch umzukristallisieren. 1. Theoretische Grundlagen Extraktion 1.1. Extraktion einer einzelnen Komponente Unter Extraktion versteht man die Überführung eines Stoffs aus einer Phase, in der er gelöst oder suspendiert ist, in eine andere flüssige Phase. Diese Überführung ist möglich, weil sich der Stoff in einem bestimmten Verhältnis auf die beiden Phasen verteilt. In einem System, welches aus zwei praktisch nicht mischbaren Flüssigkeiten und einem dritten, in beiden Flüssigkeiten löslichen Stoff besteht, stellt sich bei konstanter Temperatur ein Verteilungsgleichgewicht ein. In diesem Verteilungsgleichgewicht ist das Verhältnis der Konzentration des gelösten Stoffes der einen Flüssigkeit cA (mol/L) zur Konzentration des gelösten Stoffes der anderen Flüssigkeit cB (mol/L) konstant und wird durch den Nernst´schen Verteilungssatz (NVS) bestimmt: cA ── = K cB Die Konstante K nennt man den Nernst’schen Verteilungskoeffizienten. Die angeführte Beziehung gilt aber nur dann, wenn der gelöste Stoff in beiden Lösungsmitteln die gleiche Molekülform besitzt. Zusätzlich ist K temperaturabhängig. Der NVS gilt in der gegebenen Form für geringe Konzentration (ideale Verhältnisse) und unter der Annahme, dass der gelöste Stoff in beiden Phasen identischen Assoziationszustand besitzt. Daraus folgt, die angeführte Gleichung gilt in dieser Form nicht, wenn z.B. der Stoff in einem Lösungsmittel dissoziiert und im anderen nicht. Dieser zweite Fall wird in Kapitel 3 besprochen. Die unterschiedliche Löslichkeit wird in der Chemie zur Trennung und Reindarstellung von Stoffen aus Gemischen herangezogen. Das entsprechende Laborverfahren nennt man „Ausschütteln“. Der entsprechende technische Prozess wird als Flüssig-Flüssig-Extraktion bezeichnet. Wie aus dem NVS leicht abzuleiten ist, ist die Extraktion eines Stoffes leicht möglich wenn er im Extraktionsmittel signifikant besser löslich ist als in der anderen Phase, d.h. K >> 1 ist. Schon bei K < 100 reicht eine einfache Extraktion nicht aus. In diesen Fällen muss die Extraktion mit frischem Lösungsmittel mehrmals wiederholt werden. Allgemein ist es ist beim Ausschütteln sinnvoller, mehrere Male mit geringeren Flüssigkeitsmengen auszuschütteln als nur einmal mit einer größeren Menge Flüssigkeit. 1 Die Messdaten sollen mit Vergleichsdaten aus der Literatur verglichen werden. Vergleichsdaten können z.B. aus Datenbanken wie SciFinder oder Reaxys bezogen werden. Siehe hierfür das Literatursuche Seminar. LU Synthesechemie Extraktion 1.2. Trennung eines Zweikomponentengemisches Das bisher gesagte gilt für den einfachen Fall der Extraktion einer einzelnen Komponente aus einer flüssigen Phase. Für eine Mischung von zwei Substanzen ergibt sich ein etwas anderes Bild. Zuallererst ist festzuhalten, dass sich die beiden Substanzen im Idealfall unabhängig voneinander auf die beiden flüssigen Phasen verteilen. Bei genügend großem Unterschied ihrer Verteilungskoeffizienten, ist eine Trennung der beiden Komponenten durch einfache Extraktion möglich. Die Schwierigkeit des Trennproblems wird durch den Trennfaktor ß (ß > 1; d. h. man dividiert den größeren Verteilungskoeffizienten durch den kleineren) bestimmt: K1 β = ── K2 Die beiden Substanzen lassen sich nur dann befriedigend durch einfache Extraktion trennen, wenn ß > 100 ist. Zur Trennung von Gemischen mit ß < 100 müssen multiplikative Verteilungsverfahren angewendet werden. 2 Der Stoffaustausch ist wie bei allen Verteilungsverfahren nur an der Phasengrenzfläche möglich und die Phasengrenzfläche sollte daher möglichst groß sein um die Einstellung des Gleichgewichts zu beschleunigen. Bei der Flüssig-Flüssig Extraktion werden die Flüssigkeiten daher geschüttelt (beim Ausschütteln) oder durch Fritten fein verteilt (bei der Perforation). 2. Praktische Durchführung Die auszuschüttelnde wässrige Lösung oder seltener die Suspension wird in einem Scheidetrichter (siehe Abbildung rechts) mit etwa einem Fünftel bis einem Drittel ihres Volumens an Extraktionsmittel versetzt. Dabei ist zu beachten, dass der Scheidetrichter maximal zu zwei Drittel gefüllt sein darf. Die Größe des Scheidetrichters ist also den entsprechenden Flüssigkeitsmengen anzupassen! Der Scheidetrichter wird mit einem Plastikstopfen (keine Glasstopfen verwenden!) verschlossen und umgedreht, wobei man sowohl das Hahnküken als auch den Stopfen festhält (siehe Abbildung). Bevor noch mit dem Schütteln begonnen wird, wird ein erstes Mal belüftet. Dabei ist der Auslauf stets nach oben und weg von einem selbst und anderen Personen gerichtet! Nach Schließen des Hahns schüttelt man zunächst vorsichtig und belüftet (langsames Öffnen des Hahns!) anschließend wieder. Durch das Belüften soll ein sich eventuell entwickelnder Überdruck abgeführt werden. Schütteln und Lüften müssen so lange wiederholt werden, bis der Gasraum im Scheidetrichter mit dem Lösungsmitteldampf gesättigt ist. Das ist der Fall wenn kein zischendes Entweichen von Überdruck mehr wahrgenommen wird. Erst jetzt wird etwa l Minute kräftig geschüttelt. Anschließend lässt man stehen, wobei sich die Phasentrennung einstellt. Die untere Phase wird dann nach Abnehmen des Stoppels durch den Hahn des Scheidetrichters abgelassen, die obere Phase durch die obere Öffnung ausgegossen. 3 Es ist festzuhalten dass beim einfachen einmaligen 2 Bei der multiplikativen Verteilung handelt es sich um eine Vielstufenextraktion, bei der die beiden flüssigen Phasen im Gegenstrom zueinander bewegt und ständig ins Gleichgewicht gebracht werden. Das heißt, teilweise mit gelöstem Stoff angereicherter Extrakt kommt mit frischer Substanzlösung in Berührung und teilweise extrahierte Lösung mit frischem Extraktionsmittel. 3 In Zweifelsfällen prüft man, welches die wässrige Phase ist, indem man einer Phase einen Tropfen entnimmt und diesen in etwas Wasser gibt LU Synthesechemie Ausschütteln im günstigsten Fall einer vollständigen Gleichgewichtseinstellung jeweils nur die durch den Nernst´schen Verteilungssatz und die angewandte Menge Extraktionsmittel festgelegte Menge der zu extrahierenden Substanz in das Extraktionsmittel übergehen kann. Aus diesem Grunde muss man im Allgemeinen wiederholt ausschütteln. Typischerweise wird 3-4mal extrahiert. Es ist allerdings ratsam mittels DC auf vollständige Extraktion zu prüfen. Dafür genügt im Normalfall ein Tüpfel DC der letzten Extraktionsphase. 4 2.1. Praktische Tipps Bei in Wasser verhältnismäßig leicht löslichen Substanzen kann man die wässrige Schicht mit Ammoniumsulfat oder Kochsalz sättigen. Manche Systeme neigen zur Bildung von Emulsionen. In solchen Fällen schüttelt man den Scheidetrichter nicht, sondern schwenkt ihn nur. Entstandene Emulsionen lassen sich brechen, wenn man etwas Antischaummittel oder Pentylalkohol zugibt, die wässrige Phase mit Kochsalz sättigt (bei spezifisch leichteren Lösungsmitteln) oder die gesamte Lösung filtriert. Das sicherste Mittel ist stets, längere Zeit stehenzulassen. Substanzen, die in Wasser schwer löslich sind, schüttelt man drei- bis viermal aus, während die Operation bei gut wasserlöslichen Stoffen u. U. viele Male wiederholt werden muss. Es ist effizienter, mit wenig Lösungsmittel mehrere Male auszuschütteln, als die ganze Menge Extraktionsmittel auf einmal einzusetzen. 3. Trennung aufgrund unterschiedlicher Säure/Basen Eigenschaften Das bis jetzt Gesagte gilt für Verbindungen, die in beiden flüssigen Phasen im selben Assoziationsgrad vorliegen. Allerdings haben viele organische Verbindungen saure oder basische Eigenschaften und können daher, abhängig vom pH Wert des Extraktionsmediums, in den beiden Phasen unterschiedlich assoziiert vorliegen. Das kann zur Trennung von Mehrkomponentengemischen ausgenützt werden, wie anhand einfacher Beispiele erläutert werden soll. Fall 1: Ein Zweikomponentengemisch aus einer organischen Säure (z.B. Benzoesäure) und einem organischen Neutralstoff. In einem typischen organischen Lösungsmittel wie Diethylether oder Ethylacetat werden beide Verbindungen gut löslich sein. Extraktion mit Wasser (pH 7) wird auch zu keiner befriedigenden Trennung führen aufgrund eines K <<100. In wässrig basischer Lösung dissoziiert allerdings die Säure zum analogen Carboxylatanion und wird dadurch sehr gut wasserlöslich, wohingegen der Neutralstoff im organischen Lösungsmittel verbleibt. Fall 2: Ein Zweikomponentengemisch aus einer organischen Base (z.B. Anilin) und einem organischen Neutralstoff. Analog zum oberen Fall wird eine wässrige neutrale Extraktion keinen Trennerfolg bringen. Extraktion im wässrig sauren Milieu allerdings führt die organische Base in ein Salz über, welches wiederum gut wasserlöslich sein wird. 4. Praktische Übung In der vorliegenden Übung wird ein 3 Komponentengemisch bestehend aus 2 organischen Neutralstoffen und einer organischen Verbindung mit sauren oder basischen Eigenschaften ausgehändigt. Durch geeignete Wahl des wässrigen Extraktionsmittels kann der Stoff mit sauren oder basischen Eigenschaften von den beiden Neutralstoffen abgetrennt werden. Die beiden Neutralstoffe sollen anschließend mittels Säulenchromatographie getrennt werden. Alle drei Stoffe werden nun isoliert und getrocknet und anschließend mittels Schmelzpunkt, IR, MS und NMR Messung charakterisiert und identifiziert. Eine Gesamtübersicht der Übung ist im folgenden Schema dargestellt. 4 siehe Kapitel DC Extraktion LU Synthesechemie 4.1. Eprouvettenvorversuche Am Beginn der Übung wissen Sie nicht, ob der dritte Stoff saure oder basische Eigenschaften hat, d.h. ob er sich durch Extraktion mit 2N aq. HCl (basischer Stoff) oder 2N aq. NaOH bzw. gesättigter aq. NaHCO3 Lösung abtrennen lässt. Um das festzustellen sollen Vorversuche in Eprouvetten durchgeführt werden. Im Folgenden wird ein solcher Vorversuch für eine hypothetische Probe bestehend aus zwei Neutralstoffen N1 und N2 und einer basischen Verbindung B1 beschrieben (siehe auch Grafik). Am Beginn der Übung soll ein geeignetes DC Laufmittel gefunden werden. Bei dem 3-Komponentengemisch haben die beiden Neutralstoffe in jedem Fall unterschiedlichen Rf Wert (bei geeigneter Wahl des Laufmittels), die dritte Komponente kann unter Umständen den identen Rf Wert eines der Neutralstoffe haben. Es sollten also mindestens zwei Spots auf dem DC zu erkennen sein, idealerweise drei. Zur Optimierung des DC-Laufmittels siehe Skriptum Grundlagen der Chemie und Kapitel B2.1. Extraktion LU Synthesechemie Extraktion Nun können die eigentlichen Eprouvettenversuche durchgeführt werden. Hierfür lösen Sie eine kleine Menge (ca. eine Spatelspitze) der homogenisierten Probe in einer Eprouvette in ca. 3mL Diethylether (bei schlechter Löslichkeit kann ein polareres Lösungsmittel verwendet werden). Nun extrahiert man diese Lösung zum Beispiel mit 2mL 2N aq. HCl. Für den Fall einer Mischung aus N1, N2, und B1 wird B1 in die wässrige Phase übergehen (Dieser Fall ist in der Grafik schematisch dargestellt). Neutralstoffe und Verbindungen mit sauren Eigenschaften verbleiben in der organischen Phase (d.h. im Falle einer Mischung bestehend aus N1, N2, und einer Säure S1, bleiben alle drei Verbindungen in der organischen Phase!). Nachdem die organische Phase spezifisch leichter als die wässrige ist, kann man direkt aus der oberen Phase eine DC-Probe nehmen und mit dem ursprünglichen 3-Komponentengemisch vergleichen. Um zu überprüfen ob eine Verbindung in die wässrige Phase extrahiert wurde, trennt man zuerst die wässrige Phase von der organischen. Die wässrige Phase wird basisch gestellt (z.B. mit 2N aq. NaOH; pH Kontrolle!) und wieder mit Diethylether extrahiert. Man kann nun mittels DCs aus der organischen Phase feststellen, ob sich Substanz in dieser befindet. Im Falle einer Mischung N1, N2, und S1, wird sich zeigen dass keine Extraktion stattgefunden hat. In diesem Fall wiederholt man nun dieselbe Prozedur, extrahiert allerdings mit einer wässrig basischen Lösung anstelle von 2N aq. HCl. Dadurch kann S1 abgetrennt werden. Dabei ist zu beachten, dass durch die Wahl der Base auch schon Information über die Struktur der Verbindung gewonnen werden kann. In der vorliegenden Übung ist S1 entweder eine Carbonsäure, oder eine phenolische Verbindung. Carbonsäuren sind natürlich deutlich acider als Phenole, was sich aus den pKA Werten für Phenol und Benzoesäure (siehe Tabelle) ablesen lässt. Die höhere Acidität einer Carbonsäure, wie z.B. Benzoesäure (pKA 4.2) ergibt sich aus der größeren Stabilität der konjugierten Base (in diesem Fall des entsprechenden Carboxylat Anions), da hier eine Verteilung der Ladung über beide Sauerstoffe der Säuregruppe möglich ist. Beim Phenol (pKA 9.95) hingegen ist die negative Ladung des Phenolats nur an einem Sauerstoff lokalisiert. 5 Anilin, als Beispiel einer basischen Verbindung hat natürlich nochmals einen deutlich höheren pKA Wert von 25. Verbindung pKA Phenol 9.95 Benzoesäure 4.20 Anilin 25 5 Der M-Effekt des Phenylrings hat natürlich ebenfalls einen Einfluss und bewirkt z.B. die höhere Acidität von Phenolen im Vergleich zu aliphatischen Alkoholen. LU Synthesechemie Dadurch kann eine Carbonsäure bereits durch eine schwache Base extrahiert werden, ein Phenol allerdings nicht. Wenn man nun im Eprouvettenversuch zuerst mit gesättigter wässriger NaHCO3 Lösung extrahiert, dann wird eine Carbonsäure in die wässrige Phase übergehen, ein Phenol nicht. Hierfür wird 2N aq. NaOH nötig sein. Diese Tatsache kann man sich zu nutzen machen, um bereits zu diesem Zeitpunkt eine Information über die Art der Verbindung zu bekommen, noch bevor spektroskopische Methoden zum Einsatz kommen. Erst wenn zweifelsfrei festgestellt wurde, welcher Art die vorliegenden Verbindungen sind, wird mit der Gesamtprobe im Scheidetrichter die Trennübung durchgeführt. Das Prinzip folgt hierbei dem bei den Eprouvettenversuchen beschriebenen. 4.2. Isolation der sauren/basischen Verbindung Wenn die beiden Neutralstoffe abgetrennt sind, geht es daran den Stoff mit den sauren/basischen Eigenschaften zu isolieren. Dazu wird der pH-Wert der wässrigen Lösung, die den entsprechenden Stoff enthält invertiert, 6 um ihn wieder in ein organisches Lösungsmittel extrahieren zu können. Man extrahiert mehrmals mit z.B. Diethylether und überprüft die Vollständigkeit der Extraktion mittels Tüpfel-DC. Zur Vortrocknung werden die vereinigten organischen Phasen mit gesättigter aq. NaCl Lösung einmal rückgeschüttelt und anschließend über Na2SO4 getrocknet. Das Na2SO4 wird abfiltriert und der Filterkuchen mehrmals mit frischem Lösungsmittel gewaschen. Anschließend wird am Rotavapor das Lösungsmittel abgezogen 7 (Kolben tarieren; es ist über alle präparativen Arbeitsschritte eine Mengenbilanz zu führen) und die Verbindung noch im Vakuum (Einhalsrundkolben mit Absaugstück am Wasserbad) getrocknet. Charakterisierung erfolgt über den Schmelzpunkt, IR, NMR und MS Spektren. Sobald der Schmelzpunkt konstant ist (d.h. die Verbindung trocken ist), kann eine NMR Probe abgefüllt werden. 4.2.1. Umkristallisieren Sollte der Schmelzpunkt ein großes Intervall aufweisen kann das auf eine Verunreinigung in der Probe hindeuten. Diese ist durch Umkristallisieren zu beseitigen. Das Umkristallisieren ist eine der wichtigsten Reinigungsoperationen für Feststoffe. Dabei wird im Allgemeinen das Rohprodukt in einem geeigneten Lösungsmittel in der Siedehitze gelöst, wobei eine heiß gesättigte Lösung hergestellt wird. Beim nachfolgenden, langsamen Auskühlen kristallisiert das Produkt in reinerer Form aus, da dessen Löslichkeit in der Kälte herabgesetzt wird, die in geringerer Menge vorhandenen Verunreinigungen aber immer noch gelöst bleiben. Es kann aber auch vorkommen, dass sich die Verunreinigungen schlechter lösen und daher als unlöslicher Rückstand in der heißen Lösung überbleiben. In diesem Fall müssen diese durch Abgießen oder Heiß-Filtrieren abgetrennt werden und erst dann wird das Filtrat langsam abgekühlt. Nach Erreichen der Raumtemperatur wird das Gemisch zur Vervollständigung der Kristallisation noch in den Eiskasten gestellt. Die abgeschiedenen Kristalle werden dann durch Saugfiltration von der „Mutterlauge“ abgetrennt, mit wenig vorgekühltem Lösungsmittel nachgewaschen und nach Überführen in eine Kristallisierschale im Exsikkator getrocknet. 6 d.h. im Falle der Extraktion einer Base mit HCl wird die wässrige Phase jetzt basisch gestellt, im Falle der Extraktion einer sauren Verbindung mit NaOH oder NHCO3 wird die wässrige Phase sauer gestellt. 7 Für das korrekte Vorgehen beim Einrotieren wenden Sie sich im Zweifelsfall an einen Saalassistenten Extraktion LU Synthesechemie 4.2.2. Praktische Durchführung der Umkristallisation Zuerst muss ein geeignetes Lösungsmittel zur Umkristallisation gefunden werden. Dies geschieht wieder in Eprouvettenversuchen. Eine kleine Menge der Verbindung wird mit Lösungsmittel versetzt. Bei Raumtemperatur soll die Verbindung im gewählten Lösungsmittel nur wenig löslich sein, ansonsten ist es für eine Umkristallisation ungeeignet. Anschließend wird zum Sieden erhitzt. In einem geeigneten Lösungsmittel sollte sich die Probe in der Siedehitze lösen. Wichtig ist vor allem aber auch, dass die Substanz beim Wiederabkühlen auch wieder kristallisiert. Häufig verwendete Lösungsmittel zur Umkristallisation sind z.B. Diisopropylether, Ethanol, Ethylacetat, Toluol, Ligroin etc. Ist ein geeignetes Lösungsmittel gefunden kann mit der Umkristallisation der Gesamtprobe begonnen werden. Die rohe, trockene Substanz wird in einem Rundkolben geeigneter Größe mit Lösungsmittel beschichtet und das Gemisch zum Sieden erhitzt. Das siedende Gemisch wird durch den Dimrothkühler portionsweise mit weiterem Lösungsmittel versetzt, bis sich der Feststoff vollständig gelöst hat (in kleinen Portionen zugeben). Anschließend lässt man die Umkristallisationslösung wieder abkühlen. Wenn die abkühlende Lösung Raumtemperatur erreicht hat, wird der Kolben noch für 30 min in den Eiskasten gestellt. Die gebildeten Kristalle werden abgesaugt, mit wenig gekühltem Lösungsmittel nachgewaschen und in der Glassinternutsche trocken gesaugt. Um das Wegsaugen des Lösungsmittels in die Membranpumpe zu vermeiden, soll das Filtrat vorher aus der Saugflasche entfernt werden). Die Kristalle werden in eine gewogene Kristallisierschale übergeführt, ausgewogen und der Schmelzpunkt bestimmt. 4.3. Isolation des Neutralstoffgemisches Die Etherphase, die nunmehr nur noch die beiden Neutralstoffe enthalten sollte, wird zur Vortrocknung mit gesättigter aq. NaCl Lösung einmal rückgeschüttelt und anschließend über Na2SO4 getrocknet. Das Na2SO4 wird abfiltriert (Papierfilter und Glastrichter) und der Filterkuchen mehrmals mit frischem Lösungsmittel gewaschen. Anschließend wird am Rotavapor das Lösungsmittel abgezogen (Kolben tarieren zur Bestimmung der Gemischausbeute). Zur Trennung der beiden Neutralstoffe mittels Säulenchromatographie siehe Teil 2 dieses Skriptums. Extraktion LU Synthesechemie Säulenchromatographie B: Chromatographie Dünnschicht- und Säulenchromatographie (DC und SC) 1. Grundlagen Unter Chromatographie versteht man physikalische Methoden bei denen eine Stofftrennung durch Verteilung von Substanzen zwischen einer ruhenden (stationären) und einer sich bewegenden (mobilen) Phase erfolgt. Dabei kann die Verteilung zwischen den Phasen durch verschiedene Effekte hervorgerufen werden, z.B. Adsorption, Absorption, Ionengleichgewichte, Diffusionseffekte, etc. Für die formale Beschreibung der chromatographischen Trennung ist i.a. die Art der Verteilung unerheblich. Im Folgenden sollen nur für das Verständnis der Übung mindestens notwendigen Grundlagen erläutert werden, für eine umfassendere Betrachtung sei auf einschlägige Literatur verwiesen. Die in der Übung angewandte Chromatographie ist eine Adsorptionschromatographie. Dabei ist die stationäre Phase fest, die mobile Phase flüssig. Man spricht daher auch von einer FestFlüssig Chromatographie (im Englischen Liquid-Solid Chromatography, LSC). Da es sich bei den auftretenden Wechselwirkungen um adsorptive Vorgänge handelt, ist die entscheidende Stoffeigenschaft, die es zu betrachten gilt, die Polarität der beteiligten Stoffe. Dies umfasst stationäre Phase, mobile Phase und Analyt(en). Naturgemäß ist die adsorptive Wechselwirkung zwischen zwei Stoffen umso größer, je höher die Polarität beider Stoffe ist. 1.1. Die stationäre Phase Kieselgel ist die meistverwendete stationäre Phase. Das in diesen Übungen verwendete Material hat eine Korngröße von 0.04 mm - 0.063 mm. An der Oberfläche des Kieselgels befinden sich freie OH-Gruppen: Si O Si O O Si O O Si O O H H Freie OH-Gruppen an der Oberfläche Abb. 1. Struktur von Kieselgel Diesen freien OH-Gruppen weisen eine hohe Polarität auf, treten also leicht mit polaren Anteilen der mobilen Phase sowie mit polaren Analyten in Wechselwirkung. Dabei ist besonders die Bildung von Wasserstoffbrücken von Bedeutung. 1.2. Die mobile Phase – die Eluotrope Reihe Die eluotrope Reihe stellt eine Anordnung der als mobile Phasen üblichen Lösungsmittel dar. Dabei werden die Lösungsmittel in aufsteigender Reihenfolge nach ihrer Elutionskraft bei Kieselgel (oder Aluminiumoxid) als stationäre Phase angeordnet. Die in Tab. 1 angegeben Zahlenwerte charakterisieren die jeweiligen Eluotionsstärken, diese ist proportional zur Polarität des Lösungsmittels. LU Synthesechemie Säulenchromatographie Tab. 1 Elutionsstärkeparameter ( εo-Werte) für einige Lösungsmittel Lösungsmittel εo εo Pentan Diisopropylether 0.00 0.28 0.00 0.21 Dichlormethan 0.42 0.32 Essigsäureethylester Acetonitril 0.58 0.65 0.38 0.50 Methanol 0.95 0.73 Al2O3 SiO2 Man kann in erster Näherung davon ausgehen, dass die Elutionskraft bei gleichem εo -Wert ungefähr gleich bleibt, auch dann wenn eine unterschiedliche Zusammensetzung (d.h. anderes Lösungsmittel) der mobilen Phase vorliegt. 1.3. Der Trennvorgang Betrachten wir nun den einfachsten Fall der Verteilung, das System aus stationärer Phase, mobiler Phase und einem Analyten. Formal lässt sich dieser Verteilungsvorgang mit dem schon eingeführten Nernst’schen Verteilungskoeffizienten K beschreiben, es gilt bei konstanter Temperatur die Gleichung der Adsorptionsisothermen: 𝐾𝐾 = 𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾 𝑣𝑣𝑣𝑣𝑣𝑣 𝑥𝑥 𝑖𝑖𝑖𝑖 𝑑𝑑𝑑𝑑𝑑𝑑 𝑠𝑠𝑠𝑠𝑠𝑠𝑠𝑠𝑠𝑠𝑠𝑠𝑠𝑠ä𝑟𝑟𝑟𝑟𝑟𝑟 𝑃𝑃ℎ𝑎𝑎𝑎𝑎𝑎𝑎 [𝑥𝑥]𝑠𝑠𝑠𝑠𝑠𝑠𝑠𝑠𝑠𝑠𝑠𝑠𝑠𝑠ä𝑟𝑟 = [𝑥𝑥]𝑚𝑚𝑚𝑚𝑚𝑚𝑚𝑚𝑚𝑚 𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾𝐾 𝑣𝑣𝑣𝑣𝑣𝑣 𝑥𝑥 𝑖𝑖𝑖𝑖 𝑑𝑑𝑑𝑑𝑑𝑑 𝑚𝑚𝑚𝑚𝑚𝑚𝑚𝑚𝑚𝑚𝑚𝑚𝑚𝑚 𝑃𝑃ℎ𝑎𝑎𝑎𝑎𝑎𝑎 Die polaren Zentren der stationären Phase werden nun einerseits vom Analyten und andererseits (quasi in einer Konkurrenzreaktion) von der mobilen Phase belegt. Die Belegung mit der mobilen Phase ist entsprechend den obigen Ausführungen umso größer, je höher deren Polarität ist, was aber andererseits heißt, dass der Anteil des Analyten auf der stationären Phase sinken muss, der K-Wert wird kleiner. Phänomenologisch bedeutet das, dass eine Erhöhung der Polarität der mobilen Phase eine Erhöhung der Analyten in derselben erzeugt (unabhängig davon ob der Analyt nun polar oder unpolar ist!). Wenn nun ein Stoffgemisch aus zwei unterschiedlich polaren Analyten vorliegt, so kann mit einigermaßen hoher Wahrscheinlichkeit davon ausgegangen werden, dass die resultierenden K-Werte bei gegebenem Trennsystem (Kombination aus stationärer und mobiler Phase) unterschiedlich sind. Graphisch dargestellt ergibt sich eine Kurvenschar, welche im idealen Bereich (verdünnte Lösungen, keine Wechselwirkungen zwischen den Analyten und keine anderen Wechselwirkungen zwischen Analyten und stationärer bzw. mobiler Phase als die bisher betrachteten) als Geraden mit verschiedenen Steigungen erscheint: Cstat Substanz A Cstat A Substanz B Cstat B Substanz C Cstat C Noch linearer Bereich Cmob N Cmob Abb. 2, Adsorptionsisothermen dreier Stoffe A, B, C LU Synthesechemie Säulenchromatographie Aus diesen Ausführungen ergibt sich zwingend, dass ein Substanzpaar nur dann chromatographisch trennbar ist, wenn es sich in den K-Werten unterscheidet (wenngleich angemerkt werden sollte, dass auch ein sehr geringer Unterschied für eine erfolgreiche Trennung ausreicht). 2. Durchführung 2.1. Dünnschichtchromatographie Die Dünnschichtchromatographie ist eine (i.a.) analytische Trennmethode. Die verwendeten DC-Platten werden vom Institut bereitgestellt und bestehen aus einem Trägermaterial aus dünnem Aluminium mit einer Schichtdicke der stationären Phase von ca. 0.25 mm. Die Größe der Platten beträgt für Standarddünnschichtchromatogramme ca. 2.5 cm x 5 cm für das Auftragen von 1 bis 3 Startflecken. Für mehr als 3 Startflecke werden DC-Platten verwendet, welche die Breite der DC-Kammer ausnutzen. Vor Anfertigen eines Dünnschichtchromatogramms (DC) wird eine DC-Kammer mit dem Laufmittelgemisch (z.B. PE/EE Mischung) beschickt und bei geschlossenem Deckel ca.5-10 min stehen gelassen, um eine Konditionierung zu ermöglichen. Vor Auftragen der Probe wird in 7-10 mm Entfernung vom unteren Rand der DC-Platte eine Startlinie mit Bleistift (!) gezogen. Punkte auf der Startlinie kennzeichnen den genauen Ort der Probenaufgabe. Die Aufgabe des Substanzgemisches erfolgt mittels Mikropipette, sie ist mit Aceton sauber zu halten (Die Mikropipette wird befüllt, indem man sie in die Probelösung taucht; die Flüssigkeit wird durch die Kapillarwirkung in die Pipette gesaugt. Sie wird entleert, indem man sie senkrecht auf eine saugfähige Oberfläche tüpfelt; das kann die DC Platte für die Probenaufgabe oder ein Filterpapier bei den Reinigungsvorgängen sein). Feststoffe wie auch Flüssigkeiten werden immer in Form von Lösungen auf die DC-Platte aufgebracht. Bevor eine Platte zur Entwicklung in die DC-Kammer gestellt wird, wird das Lösungsmittel entweder abgeföhnt (kühl) oder es wird das Abtrocknen abgewartet (bei leicht flüchtigen Lösungsmitteln). Die so vorbereitete DCPlatte wird senkrecht in die konditionierte DC-Kammer gestellt, der Deckel der Kammer wird geschlossen und das Aufsteigen der mobilen Phase bis etwa 5-7 mm unter den oberen Rand der Platte abgewartet. Nach Entnehmen der DC-Platte aus der Kammer wird sofort die Front des Laufmittels mit Bleistift markiert und anschließend das Laufmittel abgedampft. Nicht färbige Substanzflecken werden unter der UV-Lampe sichtbar gemacht und mit Bleistift markiert. Eine weitere Möglichkeit, farblose Substanzen auf der DC Platte sichtbar zu machen ist, die Platte nach dem Entwickeln mit Anfärbereagenzien zu behandeln. Ein für organische Verbindungen recht universell einsetzbares Reagenz ist Cer-Phophormolybdänsäure, welches auch im Praktikum verfügbar ist. Die DC Platte wird in die gelbe Flüssigkeit getaucht, gleich wieder herausgezogen und ein wenig abgestreift. Anschließend wird mit der Heißluftpistole vorsichtig (fächelnd) bis zum Maximalkontrast erwärmt. Anfärbbare Substanzen verfärben sich blau. Zur zahlenmäßigen Beschreibung eines Dünnschichtchromatogramms dient der Rf-Wert (Abb 3). Er ist charakteristisch für einen Analyten im jeweiligen Trennsystem, d.h. die Angabe eines Rf-Wertes ohne Angabe des Trennsystems (stationäre und mobile Phase) ist sinnlos! Der Rf berechnet sich wie folgt: C B R fC = c f Entfernung des Verbindungsfleckes vom Start c = f Entfernung der Laufmittelfront vom Start Abb. 3 - Definition des Rf-Wertes A 1 cm LU Synthesechemie Säulenchromatographie Zur Messung von c wird der optische Mittelpunkt des Fleckes (die Stelle der höchsten Intensität) angenommen. Für die erfolgreiche Trennung des Dreikomponentengemisches ist es zunächst nötig, eine DC Methode zu entwickeln, die die Analyse des Gemisches bzw. dessen Komponenten ermöglicht. Dazu löst man eine Spatelspitze des (homogenen!) Gemisches in etwa 1 mL Ethylacetat. Von dieser Probelösung bringt man mittels Mikrokapillare eine Menge von 1-3 µL durch wiederholtes kurzes Tüpfeln auf eine DC Platte auf (es soll ein möglichst kleiner Startfleck entstehen) und entwickelt in einem Gemisch aus Petrolether / Ethylacetat mit mittlerer Polarität (z.B. PE/EE=5/1). Die Trennung ist erfolgreich und als Analysenmethode gut geeignet, wenn 3 klar getrennte Substanzflecken sichtbar sind (UV Kammer bzw. Anfärben). Sollte die Trennung nicht erfolgreich sein (Substanzen werden zu viel/zu wenig eluiert), so wird durch Änderung des Mischungsverhältnisses die Polarität der mobilen Phase solange angepasst, bis eine brauchbare Trennung erzielt wird. Mit dieser mobilen Phase kann nun das Ergebnis der jeweiligen Extraktions-Eprouvetten Versuche überprüft werden. Dazu wird wie folgt vorgegangen: Es wird ein Vergleichs-DC erstellt, d.h. man bringt auf eine DC Platte sowohl die Referenzprobe (die Lösung des Dreikomponentengemisches) als auch die organische (!) Phase des jeweiligen Extraktionsversuches auf. Zusätzlich wird ein dritter Punkt getüpfelt (vorzugsweise zwischen den beiden anderen Punkten), der sogenannte Co-Spot; wie aus dem Namen hervorgeht wird hier sowohl die Referenz als auch die Probe getüpfelt, und zwar ca. 1/1. Diese Vorgehensweise ist zwingend nötig, wenn Referenz und Probe nicht in demselben Lösungsmittel gelöst sind oder die Probe aus einer Aufarbeitung (z.B. Extraktion einer wässerigen Lösung!) hervorgegangen ist. Das DC wird entwickelt und nach Visualisierung der Substanzen entschieden, ob der Extraktionsversuch erfolgreich war. Bei sauren oder basischen Verbindungen kommt es oft zu einem die ideale Punktform verzerrenden Effekt, dem sogenannten Tailing. Dabei werden die Flecken kometenschweifartig verformt. Das kommt dadurch zustande, dass die Substanzen neben den adsorptiven auch noch Säure-Basen Wechselwirkungen mit der stationären Phase eingehen. Dem Effekt kann begegnet werden, indem man bei sauren Analyten dem Laufmittel eine kleine Menge Säure (z.B. Essigsäure, ca. 3 Tropfen/5mL Laufmittel) zusetzt, bei basischen Analyten eine Base (z.B. Triethylamin). Die Flecken werden dadurch i.a. wieder rund indem der Dissoziationsgrad des Analyten herabgesetzt und somit hauptsächlich die nicht-ionisierte (und somit apolarere) Form vorliegt, was eine schärfere Trennung ermöglicht. 2.2. Säulenchromatographie Die Säulenchromatographie kann sowohl als analytische als auch als präparative Methode ausgeführt sein. Im Falle dieses Übungsbeispiels wird sie zur präparativen Trennung des Zweiphasengemisches der Neutralstoffe verwendet. 2.2.1. Aufgabe Säulenchromatographie (Flashchromatographie) In Abb. 4 ist die verwendete Apparatur zur Flashchromatographie abgebildet. Zwei Faktoren beeinflussen maßgeblich die Güte der Trennung. Dies ist einerseits die Wahl eines adäquaten Trennsystems (im Normalfall ist die stationäre Phase vorgegeben, d.h. die Frage reduziert sich auf die Wahl des passenden Elutionsmittels; für organisch-chemische Fragestellungen im Labormaßstab stellt Kieselgel die hauptsächlich verwendete stationäre Phase dar) und andererseits das Verhältnis stationäre Phase zu Probe. Die besten Ergebnisse werden mit mobilen Phasen erzielt, die in einem als Vergleich herangezogenen DC für den obersten Substanzfleck einen Rf-Wert von ca. 0.2-0.3 ergeben. Als Faustregel gilt ein Mengenverhältnis Probe:Kieselgel = 1:30-1:100, typische Werte sind 1:50 – 1:70. LU Synthesechemie Säulenchromatographie Belüftung PVC Schläuche Schraubquetschhahn zur Druckregulation Absaugstück mit Klammer Druckluft ein Vorratsbehälter für mobile Phase T-Stück mobile Phase 1 cm Seesand Schliff mit Klammer Kieselgel Ca. 1 cm Glaswolle Seesand bis zum Ende der Verengung Hahn Eluat Fraktionen, werden in Eprouvetten aufgefangen Abb. 4 Schema einer Flashchromatographiesäule Der zeitliche Verlauf einer typischen säulenchromatographischen Trennung ist wie folgt: LU Synthesechemie Säulenchromatographie Signalstärke am Detektor tbrutto t0 tnetto t0 = Durchbruchszeit tbrutto = Gesamtretentionszeit tnetto = Nettoretentionszeit Peaks Start des Chromatogramms k ´= Retentionszeit Nettoretentionszeit t netto = Durchbruch szeit t0 Abb. 5 - Ermittlung des k´-Wertes aus einem Säulenchromatogramm Die Lage eines Signales im Säulenchromatogramm wird durch den Kapazitätsfaktor (k´-Wert) beschrieben. Dieser ist definiert nach Abb. 5. Für jeden getrennten Peak gibt es einen bestimmten k´-Wert. Diese Definition gilt genau nur für symmetrische Peaks. Die Durchbruchszeit t0 entspricht der Aufenthaltszeit einer Substanz in der Säule, die von der stationären Phase nicht zurückgehalten wird. Die Gesamtretentionszeit tbrutto entspricht der Gesamtzeit, die sich die Substanz in der Säule befindet (bis zum Maximum des Detektorsignals, das kann sein UV-Absorption, Brechungsindex der mobilen Phase, Intensität eines getüpfelten DC-Flecks,…). Daher ist die Nettoretentionszeit tnetto jene Zeit. die sich die Substanz nur in der stationären Phase aufhält. Diese Nettoretentionszeit ist für die Substanz in einem bestimmten Trennsystem charakteristisch. 3. Vorgehensweise zur Trennung des Neutralstoffgemisches (2 Komponenten) 3.1. Bestimmung der optimalen mobilen Phase Eine geringe Menge des Substanzgemisches wird in einem organischem Lösungsmittel (vorzugsweise Ethylacetat) gelöst und mittels DC eine geeignete mobile Phase bestehend aus PE/EE gesucht. Dies ist dann der Fall, wenn der oberste Substanzfleck einen Rf-Wert von ca. 0.5 nicht überschreitet und ein Unterschied des Rf-Wertes von 0.2 - 0.3 zum nächsten Fleck vorahnden ist. Das DC, das mit der optimalen mobilen Phase erstellt wurde, muss einem Saalassistenten gezeigt werden. Dieser bestätigt das verwendete LM-Gemisch im Journal und auf der DC Platte (diese wird dann ins Journal geklebt; mit Klebestreifen oder Buchbindefolie LU Synthesechemie Säulenchromatographie überkleben, Kleber auf der Rückseite hält nicht dauerhaft). Aus ökologischen und ökonomischen Gründen wird zur eigentlichen Trennung ein Elutionsmittel aus PE/EE Redestillaten verwendet. Diese Gemische (ungefähre Zusammensetzung wird angegeben) sind im Praktikum verfügbar; durch Mischen zweier dieser Destillate wird ein Elutionsmittel hergestellt (zunächst kleine Mengen, ca. 10mL), das am DC die gleichen Elutionseigenschaften zeigt wie das zuvor bestimmte Lösungsmittel. Auch dieses DC wird vom Saalassistenten abgezeichnet und im Journal eingeklebt. 3.2. Befüllen der Säule mit stationärer Phase Beim Umfüllen des Kieselgels ist unnötige Staubentwicklung zu vermeiden und die Säule im Abzug zu befüllen, da Kieselgel lungengängig und gesundheitsschädlich ist. Vor Beginn der Arbeiten muss kontrolliert werden, ob alle Teile vorhanden und in einwandfreiem Zustand und trocken sind. Der Ablaufhahn muss gut geschmiert und geschlossen sein. Zuerst wird ein Stück Glaswolle oder Watte mittels Glasstab in die Verengung der Säule oberhalb des Hahnes gedrückt, um so den Austrag von Kieselgel und Seesand zu verhindern (Achtung: Bruchgefahr des Glasstabes und somit Verletzungsrisiko!). Anschließend wird über einen Feststofftrichter Seesand bis zum Ende der Verengung der Säule zugegeben. Eine ebene, waagrechte Oberfläche des Seesandes ist durch vorsichtiges Rütteln der Säule erreichbar. Die Säule wird nun senkrecht am Gestänge befestigt, darunter platziert man für die Dauer des Befüllens einen Erlenmeyerkolben. Dann wird das Kieselgel (ca. 70g) und langsam so viel von der mobilen Phase in ein 400ml Becherglas gegeben, dass eine umfüllbare Aufschlämmung entsteht. Durch vorsichtiges Schwenken und Rühren mit einem Glasstab können Luftblasen entfernt werden. Die Aufschlämmung wird solange stehengelassen bis keine Wärmeentwicklung mehr stattfindet. Es sind die Vorratsgefäße der mobilen Phase verschlossen zu halten, da die leichter flüchtige Komponente bei längerem Stehen verdampft und sich damit die Zusammensetzung der mobilen Phase mit der Zeit ändert. Danach befüllt man die Säule vorsichtig bei geschlossenem Hahn ca. 10 cm hoch mit mobiler Phase. Luftblasen werden durch Klopfen und sachtes Schütteln entfernt. Nun wird das aufgeschlämmte Kieselgel möglichst in einem Guss in die Säule eingebracht. Dann wird der Ablaufhahn geöffnet. Kieselgelreste an der Glaswand werden mittels Pasteurpipette mit Laufmittel von der Glaswand gespült. Es sollte über dem sich setzenden Kieselgel nun ein deutlicher Lösungsmittelüberstand (mind. 5 cm) zu erkennen sein, sollte das nicht der Fall sein wird mittels Pasteurpipette vorsichtig (um die stationäre Phase nicht aufzuwirbeln) Lösungsmittel zugegeben. Achtung: Von nun an darf die Säule nicht mehr „Trockenlaufen“, d.h. das Kieselgel muss immer von Laufmittel bedeckt sein! Nun wird durch Anlegen von Druckluft (Vordruck wird auf max. 0.5 bar eingestellt, bei vollständig geöffnetem Schlauchquetschhahn wird das Ausgangsventil der Pressluft vorsichtig geöffnet, die Regulierung des Drucks erfolgt über den Schlauchquetschhahn, s. Abb.4) das Kieselgel zusammengepackt. Sollten sich nun noch Luftblasen in der Säule befinden müssen diese mit Laufmittel aus der Säule gedrückt werden. Dazu wird die Säule vorsichtig bis 10 cm unter den Rand mit Solvens beschickt, das Vorratsgefäß aufgesetzt (Klemme!), mit weiterem Solvens befüllt und die mobile Phase durch Anlegen von Druckluft durch die Säule gepresst. Der Druck sollte nun so reguliert werden, dass die Tropfgeschwindigkeit bei ca. 2 Tropfen/s liegt. (Das Eluat, das dabei im Erlenmeyerkolben gesammelt wird, kann und soll wieder als Laufmittel eingesetzt werden). Die Säule ist fertig gepackt, wenn sie keine (sichtbaren) Luftblasen und Risse enthält. Abschließend wird ein ca. 1 cm hohes Seesandbett aufgebracht, um das Aufwirbeln von Kieselgel während späterer Arbeitsschritte zu verhindern. Dazu wird der Lösungsmittelspiegel bis etwa 10cm über die Kieselgeloberfläche abgesenkt. Dann wird Seesand vorsichtig zugegeben (das Kieselgel darf nicht aufgewirbelt werden), bis eine ca. 5-10 mm hohe Schicht entstanden ist. LU Synthesechemie Säulenchromatographie 3.3. Probeaufgabe Die Probe des Zweikomponentengemisches (sollte ca. 1 g sein, abwägen und im Journal dokumentieren!) wird in möglichst wenig mobiler Phase gelöst (5 bis max. 10 mL); sollte dies nicht möglich sein, kann die Polarität des Lösemittels erhöht werden. Die mobile Phase in der Säule wird bis auf Höhe des oberen Seesandbettes abgelassen, und der Hahn geschlossen. Die gelöste Probe wird mittels Pasteurpipette auf den Seesand gleichmäßig aufgebracht (bevorzugt über die innere Glaswand der Säule, um Aufwirbeln zu vermeiden). Man öffnet den Hahn und wartet bis die Probelösung wieder das Niveau des Seesandbettes erreicht hat. Mit einer weiteren kleinen Menge mobiler Phase (Pasteurpipette!) wird das zu trennende Gemisch vollständig ins Kieselgel eingetragen (mindestens 2x wiederholen, auch um die Säuleninnenwand von der Aufgabelösung abzuspülen). Dann wird die Säule vorsichtig (zuerst mit der Pasteurpipette, dann unter zu Hilfenahme eines Glastrichters) mit Laufmittel befüllt (bis ca. 5 cm unter den Rand), das Vorratsgefäß aufgesetzt, die mobile Phase bis zum Vorratsgefäß aufgefüllt und mit dem Sammeln der Fraktionen begonnen (in Eprouvetten, ca. bis 2cm unter den Rand anfüllen). 3.4. Eluieren Um festzustellen, ab welcher Fraktionen Produkt(e) enthalten sind, wird auf einem Standard DC Plättchen (2.5x5cm) mit Bleistift ein Raster aus Quadraten (Seitenlänge ca. 5mm) eingezeichnet. Man beginnt nun startend mit der 1. Fraktion fortlaugend jede Fraktion (=Eprouvette) auf ein eigenes Quadrat zu tüpfeln (entwickelt wird nicht, man nennt diese Vorgehensweise Tüpfel DC); durch Kontrolle im UV Licht erkennt man, ab welcher Fraktion Produkt eluiert wurde. Beginnend mit 2 Eprouvetten vor der ersten Produktfraktion (diese enthalten noch kein Produkt) wird nun ein DC auf der breiten Platte (es sollten mind. 10 Startpunkte Platz finden; es bietet sich an, in der Mitte als zusätzlichen Punkt das Ursprungsgemisch aufzutragen, um einen Vergleichswert zu haben) getüpfelt und entwickelt. Wird festgestellt, dass das erste Produkt vollständig eluiert wurde, kann man eine Gradientenelution durchführen, d.h. man ändert die Polarität der mobilen Phase in Richtung höherer Polarität zu mehr Ethylacetat und eluiert die zweite Substanz vollständig. Der Erfolg der Trennung ist durch Beurteilung dieser Chromatogramme zu bewerten. Es ist das Ziel, möglichst keine Mischfraktionen zu erhalten, sondern beide Substanzen in chromatographisch reiner Form. Die Chromatogramme werden einem Saalassistenten gezeigt (abzeichnen lassen und ins Journal einkleben) und mit diesem entschieden, welche Fraktionen vereinigt werden können. Nach dem vereinigen wird über Natriumsulfat getrocknet, abfiltriert und in einem tarierten Kolben einrotiert. Von den getrennten Substanzen (und von etwaigen Mischfraktionen) bestimmt man die Auswaage (eine Mengenbilanz ist zu erstellen!) und fertigt noch ein DC an (abzeichnen lassen und ins Journal einkleben). Danach kann mit den Substanzen weiter gearbeitet werden (ggf. umkristallisieren, spektroskopische Proben, Schmelzpunkt, etc). 3.5. Entsorgung Die Entsorgung des gebrauchten Kieselgels erfolgt nach „Trockenblasen“ der Säule in der Feststofftonne. Alle PE/EE Gemische werden in den dafür vorgesehenen Behältern für die Recyclierung gesammelt (hier darf kein Aceton hineingeleert werden!!!) LU Synthesechemie IR Spektroskopie C: IR Spektroskopie Probenvorbereitung, Einreichen für die Messung, Interpretation der Messdaten 1. Probenvorbereitung und Einreichen für die Messung Substanzen müssen in gereinigter und trockener Form als Feststoff vorliegen, um ein unverfälschtes IR Spektrum aufnehmen zu können. Auch geringe Spuren von Restfeuchte oder Lösungsmitteln liefern mehr oder weniger intensive IRBanden, die eine Substanzidentifizierung erschweren oder verfälschen. Die trockene Probe bitte etikettiert mit Platznummer und Beschriftung Substanz A, B oder C in ein Eppendorfer Vial abfüllen. 2.1. Interpretation der Messdaten - allgemeines Sämtliche Proben werden auf dem Perkin-Elmer Spectrum Two FTIR in ATR-Technik gemessen. Wie im PS Strukturaufklärung erwähnt, bedeutet dies generell, dass die Intensitäten der Banden oberhalb von ca. 2000 cm-1 schwächer ausfallen, als würde man in Transmission messen. Zuordnung der Banden: Anhand von einigen typischen Gruppenfrequenzen für organische Moleküle sind funktionelle Gruppen bzw. Informationen über die Stoffklasse (aromatische/aliphatische Kohlenwasserstoffverbindung) zu erhalten. Weiters ist etwaig vorhandenes Wasser (Restfeuchte oder Kristallwasser) identifizierbar. Tabelle einiger wichtiger Funktionalitäten und deren typische Absorptionsbereiche im IR C≡C ca. 2200 – 1900 cm-1 CH4 2917 cm-1 CCl4 459 cm-1 C=C ca. 1900 – 1500 cm-1 CD4 2109 cm-1 CBr4 267 cm-1 C–C ca. 1500 – 1300 cm-1 CF4 909 cm-1 CJ4 178 cm-1 C≡N ca. 2200 – 1900 cm-1 O–H ca. 3600 – 3500 cm-1 (ohne H-Brücken) O–H aus COOH 3000 – 2500 cm-1 O–H ca. 3500 – 3200 cm-1 (mit H-Brücken) C=N ca. 1900 – 1500 cm-1 N–H ca. 3500 – 3300 cm-1 C=O ca. 1900 – 1500 cm-1 C–H ca. 3150 – 3000 cm-1 (aromatisch) N=O ca. 1900 – 1500 cm-1 C–H ca. 3000 – 2850 cm-1 (aliphatisch) C–O ca. 1300 – 1030 cm-1 C–H Deformation an aromatischen Ringen: 730-780 (monosubstituierter Ring) C–H Deformation an aromatischen Ringen: 740-780 (disubstituierter Ring) C–H Deformation an aromatischen Ringen: 750-810 (trisubstituierter Ring) C–H Deformation an aromatischen Ringen: 850-800 (tetrasubstituierter Ring) C–H Deformation an aromatischen Ringen: 900-800, schwach (pentasubstituierter Ring) LU Synthesechemie 3600 O-H ohne HBrücken 3500 IR Spektroskopie 3400 3300 3200 3100 3000 2900 2800 2700 1900 1800 1700 800 700 450 400 O-H mit H-Brücken N-H C-H aromatisch O-H aus -COOH C-H aliphatisch 2600 2500 O-H aus -COOH 2400 2300 2200 2100 2000 C≡C C≡N C-D C=C C=N C=O N=O 1600 C=C C=N C=O N=O 1500 1400 1300 1200 1100 1000 900 C-C C-O C-F C-H Def. an pentasubst. Aromaten 850 800 750 700 650 600 550 500 LU Synthesechemie IR Spektroskopie C-H Def. an tetrasubst. Aromaten C-H Def. an trisubst. Aromaten C-H Def. an disubstituierten Aromaten C-H Def. an monosubstituierten Aromaten C-Cl LU Synthesechemie IR Spektroskopie 2.2 Interpretation der Messdaten – ein Beispiel 100 90 1587 1587 3071 3071 1497 1497 80 3031 3031 2885 2885 2624 2624 1003 1003 1452 1452 1376 1376 70 3389 3389 767 767 1029 1029 2964,5 2964,5 1227 1227 60 856 856 937 937 885 885 1191 1191 %T 1284 1284 606 606 50 730 730 677 677 521 521 491 491 40 695 695 1059 1059 1706 1706 30 20 10 0 4000 3500 3000 2500 2000 1500 1000 500 450 cm-1 Beginnend mit den Banden der hohen Wellenzahlen ist prominent die Bande bei 3359 cm-1 zu erkennen, die nur durch eine sekundäre Amin-Gruppe -NH- oder OH-Gruppe (nicht isoliertes, sondern H-Brücken bildendes OH) erklärbar ist. Da die (schwache) –NHDeformationsschwingung zwischen 1580 cm-1 und 1490 cm-1 fehlt, kann ein sekundäres Amin ausgeschlossen werden. Die alkoholischen oder phenolischen O-H-Deformationsschwingungen sind jedoch prominent erkennbar (siehe 1376 cm-1 und 1284 cm-1). Die beiden (schwachen) Banden bei 3071 cm-1 und 3031 cm-1 weisen auf C-H-Streckschwingungen eines aromatischen Ringsystems hin. Die beiden (schwachen) Banden bei 2965 cm-1 und 2855 cm-1 weisen auf zumindest eine aliphatische C-H-Streckschwingung hin. Die schwache breite Bandenstruktur um 2624 cm-1 liegt im Bereich der O-H-Valenzschwingungen von Carbonsäuren (oft mir kleineren Nebenbanden wegen intramolkularen HBrücken) Die intensive scharfe Bande bei 1706 cm-1 ist typisch für eine Carbonyl-Streckschwingung einer Carbonsäure. Die mittelstarke scharfe Bande um 1500 (hier 1497 cm-1) gemeinsam mit einer eher schwachen Bande um die 1600 cm-1 ist typisch für einen aromatischen Ring, könnte also auf einen Benzolring hinweisen. Da es im Bereich um 1580 cm-1 keine intensivere Bande gibt als die 1500er Bande, schließt das eine weitere Konjugation zum Arylring aus. Die weiteren Banden zwischen 1225 und 950 cm-1 stellen unspezifische Kombinationsschwingungen von aromatischen Ringen dar. Deren Existenz untermauert aber das Vorhandensein zumindest eines aromatischen Ringsystems. Die intensiven Banden bei 1376 cm-1 und 1284 cm-1 liegen im Bereich der alkoholischen oder phenolischen O-H-Deformationsschwingungen (1410 – 1260 cm-1) und deutet somit auf zumindest eine O-H-Gruppe neben einer Carbonsäure hin. Die intensive Bande bei 730 cm-1 ist eine C-H Deformationsschwingung typisch bei 5 benachbarten aromatischen C-H (= monosubstituierter Benzolring). LU Synthesechemie IR Spektroskopie Die intensive Bande bei 695 cm-1 ist eine Ring-Deformationsschwingung typisch bei 5 benachbarten aromatischen C-H (= monosubstituierter Benzolring). Die weiteren Banden zwischen 950 und 750 cm-1 sowie unterhalb von 650 cm-1 stellen Gerüstschwingungsmoden des gesamten Moleküls von geringem diagnostischem Wert dar. 3. Zusammenfassung • • • • Es muss sich um ein aromatisches Ringsystem (vermutlich Benzolring) handeln, der nur monosubstituiert ist. Es gibt zumindest ein aliphatisches C-H (Seitenkette des Ringes). Es gibt zumindest ein alkoholisches oder phenolisches O-H. Da ein phenolisches O-H bei gleichzeitiger Monosubstitution keine C-H-Seitenkette mehr erlaubt, muss also ein alkoholisches O-H vorliegen. Es muss eine Carbonsäure sein. Eine mögliche Lösung, auf die obige Bedingungen passen, wäre die Mandelsäure. Ein endgültiger Beweis ist nur mit den korrespondierenden 1H-NMR Daten zu erbringen. OH OH O LU Synthesechemie NMR Spektroskopie D: NMR Spektroskopie Probenvorbereitung, Einreichen für die Messung, Interpretation der Messdaten 1. Probenvorbereitung Zunächst wird für alle Proben die Löslichkeit in Eprouvettenversuchen überprüft: ca. 15 mg Substanz werden mit 0.5 mL CHCl3 versetzt; ist die Substanz problemlos löslich, ist CDCl3 das für die Messung geeignete Lösungsmittel, andernfalls ist DMSO-d6 zu verwenden (Ausnahme: für Carbonsäuren wird jedenfalls DMSO-d6 verwendet). Die NMR-Rohre werden mit Aceton gewaschen und über Nacht im Trockenschrank getrocknet. In diese sauberen und trockenen NMR-Rohre werden je ca. 15 mg Substanz abgefüllt (kein Lösungsmittel einfüllen! Die deuterierten Lösungsmittel werden vom Laboranten/der Laborantin zugegeben). Die NMR-Zettel sind jeweils mit folgenden Angaben auszufüllen: • • • • • Name und Platznummer des/der Studierenden H als zu messender Kern Präparatcode: xxx_ANA_1 bis xxx_ANA_3 (xxx ist die Platznummer; 1 bis 3 ist die Substanznummerierung, z.B. aus der Reihenfolge der Spots im DC von oben nach unten) Das zu verwendende Lösungsmittel (CDCl3 oder DMSO-d6, wie im Vorversuch ermittelt) Als Kommentar wird Neutralstoff/Säure/Base angegeben. 1 Die befüllten NMR-Rohre werden gemeinsam mit den vollständig ausgefüllten NMR-Zetteln bei der Probeneinreichung (einem eigens dafür vorgesehenen Probengestell im Bereich der Chemikalieneinreichung) deponiert. Nach der Messung sind die NMR-Rohre an derselben Stelle wieder abzuholen. 2. Auswertung 1H-NMR 2.1. Chemische Verschiebung Die chemische Verschiebung der einzelnen Signale gibt Auskunft über die unmittelbare chemische Umgebung der jeweiligen Protonen: Der Bereich der rein aliphatischen Protonen ohne Einfluss von Funktionalitäten ist etwa 0.5 1.5 ppm. Induktive Effekte benachbarter Gruppen verschieben solche Signale entweder zu niedrigeren ppm-Werten (+I - Substituenten wie Si oder Metalle) oder zu höheren Werten (-I Gruppen; zunächst Hs benachbart zu Mehrfachbindungen, dann zu N-, Halogen- und OAtomen); der Bereich geht dabei bis zu ca. 5 ppm bei Substitution mit einer ziehenden Gruppe (z.B. -O-CO-R) bzw. bis 6 ppm bei zwei ziehenden Gruppen (z.B. Acetale). Der olefinische Bereich umfasst etwa 5 - 6.5 ppm (diese Signalseparation von aliphatischen Signalen ist eine Konsequenz von Anisotropieeffekten, wie in Vor-LVAs dargelegt), der aromatische Bereich wird üblicherweise mit 6.5 - 8 ppm angegeben. In beiden Fällen haben mesomere Einflüsse stärkere Wirkung als induktive; wiederum führt eine Erhöhung der Elektronendichte durch die Substituenten zu niedrigeren ppm-Werten, eine Erniedrigung der Elektronendichte zu höheren Werten. Auch bei Heteroaromaten gelten analoge Regeln. Einen Sonderfall stellen an Heteroatome gebundene Protonen dar: stark saure Protonen (Säuren, Enole, …) finden sich meist im Bereich 10 - 15 ppm, Phenol-OHs von 6 bis 9 ppm, Alkoholund Amin-Protonen sind meist im "aliphatischen" Bereich zwischen 0 und 5 ppm zu finden. Allen diesen Signalen ist gemeinsam, dass sie meistens auffällig breit sind; unter ungünstigen Umständen können derartige Protonen auch gar nicht im Spektrum sichtbar sein. LU Synthesechemie NMR Spektroskopie Die folgende Grafik zeigt eine Übersicht über 1H-Verschiebungsbereiche; weitere Unterlagen zur Analyse der chemischen Verschiebungen (einschließlich Inkrement-Tabellen) werden im IChemLab zur Verfügung gestellt. 2.2. Integrale Die Integrale über die einzelnen Signale spiegeln die relativen Intensitäten der Peaks wider. Üblicherweise wird das kleinste zur Substanz gehörige Integral auf 1 gesetzt; sollten dann (unter Vernachlässigung von geringfügigen Ungenauigkeiten) einige Signale nicht-ganzzahlige Werte aufweisen, wird so mit dem kleinst-möglichen konstanten Faktor multipliziert, dass alle Werte ganzzahlig werden. Die erhaltenen Werte geben dann die Verhältnisse der im Molekül vorhandenen Protonenzahlen wieder - Achtung: die tatsächlichen Protonenzahlen können auch Vielfache dieser Werte sein. 2.3. Kopplungen Aus den Kopplungsaufspaltungen der einzelnen Signale können Informationen über benachbarte NMR-aktive Kerne erhalten werden, wobei sich "benachbart" auf den Weg über Bindungen bezieht und in der Regel einen Abstand von einer (kommt für Protonenspektren selten in Betracht), zwei, drei (der Normalfall) oder vier (in Falle ungesättigter Strukturelemente) Bindungen bedeutet. Jede einzelne dieser Kopplungs-Wechselwirkungen zweier Kerne wird durch eine spezifische Kopplungskonstante charakterisiert, deren Größe von der Anzahl der Bindungen zwischen den betrachteten Kernen und der jeweiligen lokalen Struktur bestimmt wird. Typische Werte sind z.B.: • 12-18 Hz für Kopplungen über zwei Bindungen sowie über drei Bindungen, wenn der Diederwinkel etwa 180° beträgt (trans-ständige olefinische Hs, aliphatische Elemente mit fixierter Konformation) • 6-10 Hz für die meisten aliphatischen Kopplungen über drei Bindungen und cis-ständige olefinische sowie ortho-ständige aromatische Protonen • 1-2 Hz für Kopplungen über mehr als drei Bindungen. Im allgemeinsten Fall müssen für die Berechnung des Kopplungsmusters auch Kerne mit Spinquantenzahl > ½ betrachtet werden; die Anzahl der Linien ergibt sich dann als Produkt über alle Ausdrucke (2nI+1), wobei n die Anzahl äquivalenter Kopplungspartner und I die jeweilige LU Synthesechemie NMR Spektroskopie Spinquantenzahl bedeuten. Betrachtet man vereinfachend nur Kopplungen zwischen Protonen, ergeben sich zwei relevante Möglichkeiten: 1. alle Kopplungspartner eines betrachteten Kerns sind äquivalent, d.h. haben mit diesem gleiche Kopplungskonstanten: in diesem einfachen Fall ist die Anzahl der Linien gleich (n+1), und die Linienintensitäten lassen sich aus dem Pascal'schen Dreieck ableiten; z.B. also ein Duplett (Intensität 1:1) bei einem Kopplungspartner, ein Triplett (1:2:1) bei zwei Partnern, und ein Quartett (1:3:3:1) bei drei Partnern. Diese Form der Aufspaltung findet man meist in aliphatischen Ketten bei freier Drehbarkeit um die C-C - Bindungen (z.B. n-Alkylgruppen). 2. alle Kopplungspartner zeigen unterschiedliche Kopplungskonstanten: in diesem Fall ist die Anzahl der Linien gleich 2n, und alle Linien haben im Idealfall gleiche Intensität. Solche Muster (häufig auch als Kopplungsbäume aufgelöst) finden sich oft in starren (polycyclischen) aliphatischen Systemen sowie in ungesättigten Strukturen (z.B. mehrfach substituierte aromatische Verbindungen). In der Praxis sind natürlich auch Mischformen dieser beiden Spezialfälle möglich, die dann durch geeignete Kombinationen der beiden Regeln erklärbar sind. Auch Kopplungen mit anderen Spin-½-Kernen (z.B. 19F, 31P) folgen diesen Gesetzmäßigkeiten. 3. Auswertung 13C-NMR In der 13C-NMR-Spektroskopie wird im Routinefall nur die chemische Verschiebung zur Analyse herangezogen. Wie im Fall von 1H-NMR ist auch bei 13C-Spektren die Hybridisierung des Kohlenstoffatoms der wichtigste Faktor für die chemische Verschiebung. • sp3-hybridisierte C-Atome geben Signale im Bereich von 5 bis etwa 80 ppm. Wie beim 1H bewirken elektronenspendende Substituenten eine Verschiebung zu niedrigeren ppmWerten (im Falle von sehr elektropositiven Resten sind Werte <0 ppm möglich); elektronenziehende Reste führen zu einer Erhöhung der Verschiebung (im Fall von mehreren stark ziehenden Gruppen auch bis zu 100 ppm und darüber, z.B. Acetale/Ketale oder CF3-Gruppen). • sp-hybridisierte C-Atome (Alkine) erscheinen im relativ engen Bereich zwischen 70 und 90 ppm. • bei sp2-hybridisierten C-Atomen gibt es keinen Unterschied zwischen olefinischen und (hetero-)aromatischen Bereichen, diese Signale erscheinen im Bereich 100-150 ppm. Wie im 1H ist auch hier der mesomere Einfluss der Substituenten (wenn vorhanden) stärker wirksam als induktiver Einfluss; letzterer kann allerdings das Signal des direkt substituierten C-Atoms stark verschieben (z.B. bis ca. 160 ppm im Fall eines CH3OSubstituenten). Deutlich abgesetzt vom olefinisch/aromatischen Bereich erscheinen Signale von C=O-Gruppen; hier ist gut zu unterscheiden zwischen Aldehyden/Ketonen einerseits (185 bis 220 ppm) und Carbonsäuren und deren Derivaten andererseits (165 bis 180 ppm). Die im IChemLab bereit gestellten Unterlagen enthalten auch Tabellen zu 13C-NMR. Die für die vorliegende Übung zur Verfügung gestellten 13C-Spektren wurden mit der APT-Die für die vorliegende Übung zur Verfügung gestellten 13C-Spektren wurden mit der APT-Technik ("Attached Proton Test") aufgenommen, die eine direkte Ermittlung der Multiplizitäten der einzelnen Signale ermöglicht. Die Differenzierung basiert dabei auf dem unterschiedlichen Kopplungsverhalten der C-Atome in Abhängigkeit von der Anzahl der direkt gebundenen Protonen; die Unterscheidung zwischen direkt gebundenen und weiter entfernten Protonen ist durch die deutlich unterschiedliche Kopplungskonstante möglich. Typischerweise werden im fertig prozessierten Spektrum Peaks von CH3- und CH-Gruppen in positiver Richtung (oberhalb der Grundlinie) und Peaks von CH2-Gruppen und quartären Kohlenstoffatomen in entgegengesetzter Richtung dargestellt. Im Gegensatz zu 1H-Spektren sind im 13C die Signalintensitäten der routinemäßig aufgenommenen Spektren nicht zur Anzahl der jeweiligen Atome proportional; es steht also keine Integral-Information zur Verfügung. Standard-13C-Spektren werden so aufgenommen, dass keine Kopplungen zu Protonen sichtbar sind (andernfalls wären die Spektren sehr komplex und würden wesentlich längere LU Synthesechemie NMR Spektroskopie Aufnahmedauer benötigen). Sind im Molekül allerdings andere NMR-aktive Kerne vorhanden (z.B. 19F, 31P), dann sind die entsprechenden Kopplungen sehr wohl sichtbar; die entsprechenden Aufspaltungen sind nach den für 1H beschriebenen Gesetzmäßigkeiten erklärbar.