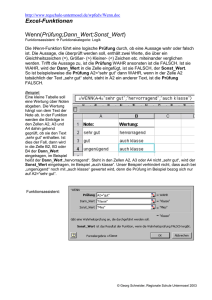
Netzwerke von Nervenzellen auf strukturierten Oberflächen
Werbung

Netzwerke von Nervenzellen auf strukturierten Oberflächen charakterisiert mit optischen und elektrophysiologischen Methoden Dissertation zur Erlangung des Grades "Doktor der Naturwissenschaften" am Fachbereich Chemie und Pharmazie der Johannes Gutenberg-Universität Mainz Lars Lauer geboren in Bonn Mainz im Mai 2001 Tag der Prüfung 31.05.2001 Die vorliegende Arbeit wurde angefertigt unter Betreuung von Herrn Prof. Dr. W. Knoll und Herrn PD Dr. A. Offenhäusser in der Zeit von April 1999 bis Mai 2001 am Max-Planck-Institut für Polymerforschung in Mainz. I INHALT 1 Einleitung 1-1 1.1 Aufgabenstellung............................................................................... 1-4 1.2 Aufbau der vorliegenden Arbeit.....................................................1-5 2 Neuronale Zellen 2.1 2-1 Charakteristika neuronaler Zellen..................................................2-2 2.1.1 Morphologie .................................................................................................. 2-2 2.1.2 Membranpotential ........................................................................................ 2-4 2.1.3 Ionenkanäle.................................................................................................... 2-8 2.1.4 Aktionspotential ........................................................................................... 2-9 2.2 Signalübertragung zwischen Neuronen........................................2-12 2.2.1 Aktive Signalleitung im Axon..................................................................... 2-12 2.2.2 Synaptische Kopplung ................................................................................. 2-16 2.2.2.1 2.2.2.2 Gap Junctions ............................................................................................................ 2-16 Funktionale Synapsen.............................................................................................. 2-18 2.3 Neuronale Zellkultur......................................................................... 2-22 2.4 Eingesetzte Zellsysteme und Kulturprotokolle ...........................2-25 2.4.1 2.4.1.1 2.4.1.2 2.4.2 2.4.2.1 2.4.2.2 2.4.2.3 2.5 2.5.1 2.5.1.1 2.5.1.2 2.5.1.3 2.5.2 3 Zellinien ......................................................................................................... 2-26 Mz1 und MzN........................................................................................................... 2-26 P19 .............................................................................................................................. 2-29 Primärkulturen.............................................................................................. 2-30 Organotypische Hirnschnitte des Hirnstamms.................................................... 2-30 Dissoziierte hippocampale Neuronen ................................................................... 2-35 Kardiomyozyten ....................................................................................................... 2-36 Protokolle zur Fluoreszenzfärbung ................................................2-39 Funktionale Antikörperfärbung ................................................................. 2-40 MAP 2......................................................................................................................... 2-40 Tau.............................................................................................................................. 2-41 GFAP .......................................................................................................................... 2-42 Färbung durch Mikroinjektion ................................................................... 2-42 Die Patch-Clamp Technik 3-1 3.1 Whole-Cell Konfiguration................................................................3-3 3.2 Voltage-Clamp und Current-Clamp Messverfahren...................3-8 3.2.1 Signalverarbeitung und Kompensation - Voltage-Clamp ...................... 3-10 II 3.2.2 Signalkorrektur ............................................................................................. 3-13 3.2.3 Signalverarbeitung und Kompensation - Current-Clamp ...................... 3-14 3.3 Versuchsaufbau ..................................................................................3-18 3.3.1 Dreifach Patch-Clamp Aufbau.................................................................... 3-18 3.3.2 Fertigung von Mikropipetten...................................................................... 3-21 4 Kontrolliertes Zellwachstum 4-1 4.1 Extrazelluläre Matrix und Laminin ................................................4-3 4.2 Strukturierung mit Mikrostempeln................................................4-7 4.2.1 4.2.1.1 4.2.1.2 4.2.1.3 4.2.2 4.3 4.3.1 4.3.2 4.3.2.1 4.3.2.2 4.3.2.3 4.4 4.4.1 4.4.1.1 4.4.1.2 4.4.1.3 4.4.1.4 4.4.2 4.4.2.1 4.4.2.2 4.4.3 5 Stempelfertigung .......................................................................................... 4-8 Strukturentwurf........................................................................................................ 4-9 Photolithographie..................................................................................................... 4-10 Abformung ................................................................................................................ 4-12 Stempelübertrag............................................................................................ 4-16 Oberflächenchemie für maximalen Kontrast ...............................4-19 Beschichtung mit Polystyrol........................................................................ 4-20 Kovalente Bindung von Laminin an Aktivester-Bürsten........................ 4-20 Mechanismus von Polymerisation und Lamininbindung .................................. 4-22 Kinetik der Reaktion mit Laminin und Schichtdickenmessung ........................ 4-26 Copolymerzusammensetzung und Zellwachstum.............................................. 4-33 Mikrostrukturen für Lokalisation und Vernetzung....................4-36 Charakterisierung mit der Zellinie MzN................................................... 4-36 Eingesetzte Strukturen............................................................................................. 4-37 Vermeiden von Zellklumpen.................................................................................. 4-40 Positionierung der Zellkörper ................................................................................ 4-43 Induzierte neuronale Polarisation.......................................................................... 4-46 Strukturierung von Hirnschnittkulturen .................................................. 4-48 Proliferation und Überwuchs ................................................................................. 4-48 Quantitative Wachstumsanalyse............................................................................ 4-51 Komplexe neuronale Schaltkreise .............................................................. 4-54 Elektrophysiologische Messungen 5-1 5.1 Zellinien Mz1, MzN und P19...........................................................5-2 5.2 Primäre Neuronen aus Hirnschnitten ............................................5-6 5.3 Zell-Zell Kopplung ............................................................................5-16 5.3.1 Kopplungstypen ........................................................................................... 5-17 5.3.2 Prinzip Ohm'scher Kopplung ..................................................................... 5-18 5.3.3 Prinzip kapazitiver Kopplung .................................................................... 5-19 5.3.4 Prinzip funktionaler Kopplung .................................................................. 5-19 5.3.5 Behandlung der Kopplungstypen .............................................................. 5-20 Inhalt 5.4 5.4.1 5.4.1.1 5.4.1.2 III Grundlagen der Kopplung...............................................................5-22 Zell-Zell Kopplung als lineares System..................................................... 5-22 Theorie linearer Systeme ......................................................................................... 5-23 Analyse linearer Systeme ........................................................................................ 5-25 5.4.2 Messkonfiguration........................................................................................ 5-26 5.4.3 Messverfahren ............................................................................................... 5-29 5.4.4 Physikalisches Kopplungsmodell .............................................................. 5-33 5.4.4.1 5.4.4.2 5.4.4.3 5.4.5 5.4.5.1 5.4.5.2 5.4.6 5.4.6.1 5.4.6.2 5.5 5.5.1 5.5.1.1 5.5.1.2 5.5.2 5.5.2.1 5.6 Beschreibung der Zellen .......................................................................................... 5-35 Beschreibung der Kopplung ................................................................................... 5-35 Beschreibung des Kabels ......................................................................................... 5-36 Vereinfachtes Ohm'sches Kopplungsmodell ............................................ 5-40 Kopplung von Gleichspannungen ......................................................................... 5-42 Kopplung von Wechselspannungen...................................................................... 5-43 Betrachtung systematischer Fehlerquellen ............................................... 5-44 Frequenzgang des Patch-Clamp Verstärkers ....................................................... 5-45 Zuleitungswiderstand.............................................................................................. 5-48 Messergebnisse aus Ohm'scher Kopplung ...................................5-51 Gleichspannungsanalyse ............................................................................. 5-51 Validierung des Kopplungsmodells ...................................................................... 5-51 Auswertung der Kopplungsdaten ......................................................................... 5-58 Wechselspannungsanalyse.......................................................................... 5-66 Auswertung der Kopplungsdaten ......................................................................... 5-66 Messergebnisse aus funktionaler Kopplung................................5-72 5.6.1 Messverfahren ............................................................................................... 5-72 5.6.2 Auswertung der Kopplungsdaten ............................................................. 5-73 5.7 6 Ergebnis der Kopplungsexperimente ............................................5-76 Ausrichtung von Struktur und Substrat 6-1 6.1 Sensorchips zur extrazellulären Signalerfassung........................ 6-3 6.2 Ausgerichtete Mikrostrukturierung ...............................................6-5 6.2.1 Hybrid-Stempelsystem aus Glas und PDMS............................................ 6-5 6.2.2 Optische Ausrichtung auf Chip Sensorpunkte......................................... 6-8 6.2.3 Kontrolle des Anpressdrucks...................................................................... 6-10 6.2.4 Spezialwerkzeuge für ausgerichtetes Mikrostempeln ............................ 6-11 6.2.5 Bewertung der ausgerichteten Mikrostrukturierung .............................. 6-13 6.3 In vitro Positionierung ......................................................................6-15 6.3.1 Substrate für in vitro Positionierung.......................................................... 6-15 6.3.2 Versuchsablauf.............................................................................................. 6-16 6.3.3 Messung an Kardiomyozyten ..................................................................... 6-17 6.3.4 Bewertung der in vitro Positionierung...................................................... 6-20 IV 7 Zusammenfassung und Ausblick 7-1 8 Anhang 8-1 8.1 Mikrostrukturen.................................................................................8-1 8.2 Glossar.................................................................................................. 8-7 8.3 Geräteverzeichnis............................................................................... 8-9 8.3.1 Laborgeräte.................................................................................................... 8-9 8.3.2 Mikrostempel-Ausrichtung......................................................................... 8-9 8.3.3 Dreifach Patch Clamp Messaufbau ............................................................ 8-9 8.4 9 Software, Materialien und Rezepturen..........................................8-11 8.4.1 Software ......................................................................................................... 8-11 8.4.2 Technische Materialien ................................................................................ 8-11 8.4.3 Chemikalien allgemein ................................................................................ 8-13 8.4.4 Chemikalien zur Zellkultur......................................................................... 8-13 8.4.5 Antikörper und Fluoreszenzfarbstoffe ...................................................... 8-16 8.4.6 Proteine zur Mikrostrukturierung.............................................................. 8-18 8.4.7 Versuchstiere ................................................................................................. 8-19 8.4.8 Rezepturen..................................................................................................... 8-19 Literaturverzeichnis 9-1 1-1 1 Einleitung Das Gehirn ist das zentrale Steuerorgan jedes höheren Lebewesens. Es empfängt die wahrgenommenen Sinneseindrücke, verarbeitet diese, und kontrolliert in Reaktion darauf alle Bewegungsabläufe des Körpers. Jede Form von Verhalten eines Lebewesens in seiner Umwelt hat seinen Ursprung im Gehirn. Auch Gedächtnis und Lernen sowie die Entstehung von Bewusstsein entspringen diesem zentralen Organ. Vom Körper wird das Gehirn besonders gut geschützt durch massive Schädelknochen und die Cerebrospinalflüssigkeit. Das Organ selbst erscheint mit bloßem Auge betrachtet jedoch eher bedeutungslos. Man erkennt lediglich eine mit vielen Blutgefäßen überzogene gräulich weiße Masse mit der Oberfläche einer Das Gehirn im Wandel der Zeit • (A) Früher Beobachtung mit bloßem Auge: – Oberfläche einer gigantischen Walnuss – Konsistenz einer reifen Avocado – Sehr gut geschützt durch Schädelknochen und Cerebrospinalflüssigkeit Aristoteles: „Kühlorgan für erhitztes Blut“ • (B) Heute Mikroskopie, Färbetechniken, ... : – Komplexe zelluläre Strukturen im Gehirn (Neurone mit Zellkörper und Nervenfasern) – 3% der Körpermasse, aber 20 % des Energieverbrauchs - zentrales Steuerorgan des Körpers - Sitz von Bewusstsein, Gedächtnis und Lernen Abbildung 1-1 Ohne Mikroskopie und Färbetechniken können einzelne Nervenzellen nicht in der Gehirnmasse nachgewiesen werden. Abbildungen entnommen aus [www_1, 2001; www_2, 2001] (A) Abbildung des Gehirns bei Betrachtung mit bloßem Auge. (B) Silberfärbung von Neuronen in einem Gehirnschnitt nach Golgi. 1-2 1 Einleitung riesigen Walnuss und der Konsistenz einer reifen Avokado (siehe Abbildung 1-1 A). Die enorme Bedeutung des Gehirns wurde daher lange Zeit vollkommen unterschätzt. Der griechische Philosoph und Gelehrte Aristoteles schrieb dem Gehirn noch die Funktion eines "Kühlorgans für überhitztes Blut" zu. Erst nach Entwicklung der Lichtmikroskopie und Entdeckung der selektiven Silberfärbung einzelner Neurone durch Golgi gegen Ende des 19. Jahrhunderts, wurde es möglich, einzelne Nervenzellen von ~15 µm Durchmesser und mit langen, dünnen Fortsätzen in der ansonsten diffusen Gehirnmasse zu identifizieren (Abbildung 1-1 B). Dadurch wurde schlagartig die ungeheure Komplexität des Organs offenbar. Das Gehirn eines Menschen besteht aus insgesamt 1011 einzelnen Nervenzellen, die über 1014 Synapsen zu einem gigantischen Netzwerk verbunden sind. Heute ist die Funktion einzelner Nervenzellen und deren Kommunikation mit Nachbarzellen über synaptische Kontakte, bis hinunter auf molekulare Ebene weitgehend wissenschaftlich erklärt. Einen entscheidenden Beitrag zu diesem Verständnis haben die elektrophysiologischen Experimente von Hodgkin, Huxley und Katz [Hodgkin, 1949] am Riesenaxon des Tintenfischs geliefert sowie die Entwicklung der Patch-Clamp Technik durch Sakmann und Neher [Neher, 1976]. Die Funktion des Gehirns als ganzes, bestimmt durch das Zusammenspiel aller Nervenzellen, ist jedoch immer noch weitgehend unverstanden. So gibt es zwar verschiedene Modelle für Gedächtnis und Lernen, diese sind aber in ihrer Gesamtheit nicht experimentell bestätigt. Für das Entstehen von Bewusstsein im Gehirn gibt es noch keinen Erklärungsansatz. Durch die Entwicklung innovativer medizinischer Bildgebungsverfahren wie der Kernspintomographie (MRT) und der Positronen-Computertomographie (PCT), ist es in den letzten Jahren gelungen, bestimmte Gehirnfunktionen spezifischen Gehirnregionen zuzuordnen [Brownell, 1982]. Funktionale Bildgebung mit MRT und PCT sind insbesondere deshalb vielversprechend für die weitere Aufklärung der Gehirnfunktion, da sie Analysen der Gehirnaktivität am lebenden Probanden ermöglichen. Detektierte neuronale Aktivität kann so in direkten Kontext zu 1-3 bewusst gesteuerten Denkprozessen gesetzt werden. Das Auflösungsvermögen dieser Bildgebungsverfahren geht allerdings nicht bis auf die Ebene einzelner Nervenzellen herab. Für die Analyse neuronaler Interaktion auf Einzelzellebene, ist daher auch heute die direkte elektrophysiologische Messung an jeder einzelnen Zelle eines neuronalen Netzwerks die einzige Möglichkeit. Mit neuen elektrophysiologischen Messverfahren auf Basis von extrazellulären Halbleitersensoren [Fromherz, 1991] und Mikroelektroden [Gross, 1979; Pine, 1980] ist es inzwischen möglich, die Signale von vielen hundert Zellen gleichzeitig aufzunehmen. Die entscheidende Schwierigkeit bei der Interpretation der so gewonnenen Daten ist jedoch die ungeheure Komplexität der Verschaltung neuronaler Zellen. Selbst wenn man nur eine Monolage von Nervenzellen auf einer Abbildung 1-2 Die Komplexität neuronaler Verschaltung in Kulturen von Nervenzellen kann mit strukturierten Substraten reduziert werden. (A) Neuronales Wachstum auf unstrukturierten Kultursubstraten. (B) Neuronales Wachstum auf mit Laminin strukturierten Substraten aus Polystyrol. 1-4 1 Einleitung flachen Substratoberfläche kultiviert, entstehen nach kurzer Zeit unüberschaubar viele Verbindungen zwischen den Neuronen (Abbildung 1-2 A). Die Aufstellung eines exakten "Schaltplans" zur Interpretation und Deutung der elektrophysiologischen Messergebnisse in einem solchen System ist quasi unmöglich. Mit bestimmten Proteinen der extrazellulären Matrix wie z.B. Laminin, kann das Wachstum von Neuronen und Nervenfasern jedoch gezielt gesteuert werden [Sanes, 1989]. Bringt man Gitter oder Linien von Laminin auf eine geeignete Substratoberfläche auf, so wachsen die Zellen entlang der vorgegebenen Strukturen (Abbildung 1-2 B). Basierend auf diesem Ansatz soll in dieser Arbeit versucht werden, einfache zweidimensionale Netzwerke von neuronalen Zellen mit definierter Verschaltung herzustellen. Die elektrophysiologische Stimulation und Messung von Neuronen in solchen geordneten Netzwerken könnte in naher Zukunft dazu beitragen, Modelle für Lernen, Gedächtnis und Bewusstsein im Experiment zu überprüfen. 1.1 Aufgabenstellung In dieser Arbeit sollen durch Stempelübertrag des Proteins Laminin strukturierte Substratoberflächen erzeugt werden, auf welchen neuronale Zellen zu geordneten Netzwerken wachsen. Dazu sind im einzelnen folgende Schritte erforderlich: 1. Geeignet erscheinende Serien von Mikrostrukturen mit systematisch variierten Abmessungen müssen entworfen werden. Für die Festlegung der Größenordnung der Abmessungen sollen Ergebnisse früherer Arbeiten verwendet werden. 2. Mikrostempel mit den entsprechenden Strukturen müssen in einem geeigneten Verfahren produziert werden. 3. Die individuelle Eignung jeder Struktur für eine gezielte Steuerung neuronalen Wachstums, muss in Experimenten mit neuronalen Zellen ermittelt werden. Nur die am besten geeigneten Strukturen werden für Folgeexperimente eingesetzt. 1-5 Die einzelnen Neurone in den erzeugten Netzwerken sollen mit Hilfe der Patch-Clamp Technik elektrophysiologisch untersucht werden. Durch gleichzeitige Verwendung mehrerer Patch-Clamp Systeme, sollen synaptische Verbindungen zwischen verschiedenen Nervenzellen in einem Netzwerk detektiert und charakterisiert werden. Hierzu ist es erforderlich, einen elektrophysiologischen Messplatz aufzubauen, geeignete Stimulationssequenzen für die geplanten Untersuchungen zu entwerfen sowie ein Analyseprogramm für die Auswertung der Messdaten zu programmieren. Für die theoretische Beschreibung der Signalübertragung zwischen Nervenzellen in den realisierten Netzwerken, soll ein geeignetes elektrisches Kopplungsmodell aufgestellt werden. Dieses Modell ist anhand der experimentell erzielten Messergebnisse zu validieren. Mit Hilfe von Modell und Messergebnissen soll anschließend versucht werden, Rückschlüsse auf einen Zusammenhang zwischen Strukur des Netzwerks und Kopplung der Zellen zu ziehen. Mit der klassischen Patch-Clamp Technik können mit vertretbarem messtechnischem Aufwand nur die Signale von zwei bis maximal drei Nervenzellen gleichzeitig gemessen werden. Zukünftige Messungen an sehr vielen Neuronen eines Netzwerks, müssen daher auf Basis von extrazellulären Halbleitersensoren erfolgen. Um diese Messungen vorzubereiten, soll eine Methode entwickelt werden die es ermöglicht, kontrolliertes neuronales Wachstum auch auf den Oberflächen solcher Sensoren zu erzeugen. Dazu soll eine geeignete Oberflächenmodifikation sowie ein Verfahren zur Ausrichtung des Stempelübertrags auf die Messpunkte der Halbleitersensoren entwickelt werden. 1.2 Aufbau der vorliegenden Arbeit Die vorliegende Arbeit umfasst einschließlich der Einleitung sieben Kapitel sowie einen Anhang und ein Literaturverzeichnis. In Kapitel 2 wird grundlegendes Wissen über neuronale Zellen als theoretische Basis für das Verständnis aller folgenden Kapitel vermittelt. 1-6 1 Einleitung Im Einzelnen werden neuronale Zellen mit ihren charakteristischen Eigenschaften beschrieben, Formen neuronaler Zellkultur vorgestellt und verglichen sowie die in dieser Arbeit verwendeten Kultur- und Färbeprotokolle dokumentiert. Kapitel 3 enthält eine Einführung in die Patch-Clamp Technik. Es beschreibt den experimentellen Ablauf der durchgeführten elektrophysiologischen Messungen, die Funktion des Patch-Clamp Verstärkers sowie den elektrophysiologischen Messplatz. In Kapitel 4 wird untersucht wie Mikrostrukturen auf Substratoberflächen neuronales Wachstum steuern können. Die angewandte Methode zur Mikrostrukturierung, auf Basis von speziell hierfür entwickelten Mikrostempeln und chemisch modifizierten Substratoberflächen, wird beschrieben. Anhand von Experimenten mit neuronalen Zellen wird gezeigt, wie eine gezielte Steuerung neuronalen Wachstums durch Auswahl geeigneter Abmessungen der Mikrostrukturen möglich ist. Die Durchführung und die Ergebnisse elektrophysiologischer Messungen an strukturiert gewachsenen Nervenzellen werden in Kapitel 5 diskutiert. Insbesondere werden in diesem Kapiel unterschiedliche Formen synaptischer Kopplung zwischen Neuronen in künstlich strukturierten Netzwerken analysiert. In Kapitel 6 wird beschrieben, wie eine Ausrichtung der erzeugten Netzwerke auf Strukturen an der Substratoberfläche erfolgen kann. Eine Untersuchung durch extrazelluläre Halbleitersensoren mit Messpunkten in regelmäßigen Abständen an der Sensoroberfläche wird dadurch möglich. Kapitel 7 fasst alle Ergebnisse zusammen und gibt einen Ausblick auf zukünftige Arbeiten und Perspektiven, basierend auf den in dieser Arbeit entwickelten Methoden und gewonnenen Erkenntnissen. Der Anhang enthält Entwurfszeichnungen der verwendeten Mikrostrukturen, ein Glossar aller verwendeter Fachbegriffe und Abkürzungen, Listen mit den eingesetzten Geräten, Materialien und Chemikalien sowie eine Sammlung von Rezepturen. 2-1 2 Neuronale Zellen Neurone im Gehirn und im peripheren Nervensystem bilden die Grundlage für jegliche Form von Verhalten. Ob einfache Reflexreaktionen bei Fischen, oder abstraktes Lernen bei Affe oder Mensch, immer sind es Nervenzellen, die mit ihren spezifischen Eigenschaften die Aufnahme, Verarbeitung und Weiterleitung von Informationen erst ermöglichen. Neurone sind damit die Basis jeder schnellen Reaktion eines Organismus auf seine Umwelt. Der grundlegende Ablauf ist dabei immer ähnlich: Informationen aus der Umwelt werden über neuronale Sinneszellen wahrgenommen und in elektrische Signale umgesetzt. Diese Signale werden entlang von Nervenbahnen in eine ebenfalls mit Neuronen aufgebaute Schaltzentrale weitergeleitet. Dort wird die Information verarbeitet und als Reaktion ein elektrisches Signal erzeugt, welches über Motorneurone zu Muskeln geleitet wird, wo es schließlich eine bestimmte physische Reaktion auslöst. Viele Prozesse in den Nervenzellen des zentralen (ZNS) und peripheren Nervensystem (PNS) sind heute bis hinunter auf molekulare Ebene wissenschaftlich erklärt. Für das Verständnis dieser Arbeit ist eine grundlegende Kenntnis dieser Zusammenhänge und ihrer Bedeutung unbedingt erforderlich. In den folgenden Abschnitten werden daher die wichtigsten Eigenschaften einzelner neuronaler Zellen sowie die Mechanismen der Signalübertragung zwischen neuronalen Zellen dargestellt. 2-2 2 Neuronale Zellen 2.1 Charakteristika neuronaler Zellen Gegenüber allen anderen Zellen im Organismus zeichnen sich Neurone durch eine ganze Reihe einzigartiger Eigenschaften aus: − Sie haben eine komplexere Morphologie, − sie sind elektrisch aktiv, d.h. sie können elektrische Impulse erzeugen, − und sie haben die Fähigkeit zur Kommunikation mit anderen Neuronen sowie einigen nicht-neuronalen Zelltypen. Dies ist möglich durch ihre Fähigkeit zur gerichteten und schnellen Weiterleitung elektrischer Impulse entlang von langen Ausläufern, den sog. Axonen. Insgesamt besteht das Nervensystem des erwachsenen menschlichen Körpers aus ca. 1011 Nervenzellen und einer noch größeren Zahl von ca. 1012 Gliazellen. Gliazellen findet man immer in direkter Nachbarschaft zu Nervenzellen. Dort unterstützen sie die Funktion von Neuronen1. Eine gesicherte, umfassende Erklärung der Funktion von Gliazellen, insbesondere ihrer Funktion im ZNS und an Synapsen, gibt es jedoch noch nicht. 2.1.1 Morphologie Neuronale Zellen weisen in ihrer Morphologie eine ausgesprochene Vielfalt auf. Dies erscheint zuerst verwirrend, bei einer genaueren Betrachtung werden jedoch Zusammenhänge zwischen Morphologie und Funktion deutlich. Man teilt Neurone daher entsprechend ihrer Funktion in verschiedene Klassen ein. In Abbildung 2-1 A sind Zeichnungen von vier Neuronen aus unterschiedlichen Bereichen des Nervensystems abgebildet: Ein Motorneuron aus dem Rückenmark, eine Purkinjezelle aus dem Kleinhirn, eine Mitralzelle aus dem Bulbus olfactorius sowie eine Pyramidenzelle aus dem Cortex. Man erkennt deutlich die Unterschiede in Form und Gestalt. 1 Beispielsweise hüllen sie Axone in eine Myelinschicht. Dadurch wird die Signalausbreitung beschleunigt. 2.1 Charakteristika neuronaler Zellen 2-3 Morphologie neuronaler Zellen (A) Formen und Größen von Neuronen (B) Schema eines Neurons Abbildung 2-1 Neuronale Zellen zeigen eine sehr vielfältige Morphologie, der zugrundeliegende Aufbau ist jedoch immer gleich. Abbildungen entnommen aus [Nicholls, 1995]. (A) Zeichnungen verschiedener neuronaler Zellen: Pyramidales Neuron aus dem Großhirn einer Maus, Motorneuron aus dem Rückenmark eines Säugers, Purkinjezellen aus dem menschlichen Kleinhirn, Mitralzellen aus dem Bulbus olfaktorius einer Ratte. (B) Aufbau eines Neurons, dargestellt am Beispiel eines Motorneurons. Allerdings variiert nur die äußere Form. Der funktionale Aufbau aller Neurone ist identisch. Jedes Neuron setzt sich aus vier Kompartimenten zusammen: Dendriten, Zellkörper, Axon sowie Synapsen am Ende des Axons. In Abbildung 2-1 B ist der Aufbau exemplarisch an einem Motorneuron dargestellt. Die einzelnen Kompartimente erfüllen spezifische Funktionen bei der Verarbeitung und Weiterleitung elektrischer Signale: 1. Am oberen Ende des Motorneurons ist ein verästelter Baum von Dendriten zu erkennen. Ihre Aufgabe ist der Empfang von elektrischen Signalen. Die Signale werden entlang der Verästelungen passiv zum Zellkörper weitergeleitet. 2-4 2 Neuronale Zellen 2. Im Zellkörper werden alle Signale integriert. Übersteigt die resultierende Signalamplitude einen Schwellenwert, so kommt es zur Ausbildung eines neuen elektrischen Impulses, eines sog. Aktionspotentials. 3. Das Aktionspotential wird entlang des Axons aktiv von der Nervenzelle weg zum axonalen Ende weitergeleitet. Myelinscheiden um das Axon isolieren dieses von der Umgebung und beschleunigen dadurch die Signalausbreitung. An Ranvier'schen Schnürringen erfolgt eine aktive Verstärkung des Signals, so dass die Signalamplitude entlang des Axons nicht abnimmt. 4. Am Ende des Axons trifft das Aktionspotential auf synaptische Kontakte zu einer Muskelzelle (Kontakte sind auch zu Dendriten oder zu Zellkörpern anderer Neuronen möglich). Das Signal bewirkt hier präsynaptisch eine Ausschüttung von Neurotransmittermolekülen. Diese werden postsynaptisch an Rezeptoren gebunden, wodurch es zur aktiven Erzeugung eines neuen Signals in der postsynaptischen Zelle kommt. Die komplexe Morphologie von vollständig ausdifferenzierten, erwachsenen Neuronen hat Konsequenzen: Im Unterschied zu allen anderen Zellen eines Organismus teilen sich Neurone nach Abschluss der embryonalen Entwicklung nicht mehr. Einzig die Verschaltung der Neurone untereinander ist plastisch und ändert sich mit der Zeit. Die Versorgung der weit vom Zellkern entfernten axonalen und dendritischen Verästelungen mit Proteinen und anderen Stoffwechselprodukten, erfolgt über spezielle Transportmechanismen. Man unterscheidet anterograden (vom Kern weg) und retrograden (zum Kern hin) sowie schnellen und langsamen Transport2. 2.1.2 Membranpotential Das Cytoplasma jeder Zelle ist über die Zellmembran vom extrazellulären Raum getrennt. Die Zellmembran besteht aus einer Doppellage von Phospholipiden, welche sich mit ihren hydrophoben Enden zu einer Doppelschicht zusammenlagern und die hydrophilen Kopfgruppen nach aussen strecken. Eine reine 2 Der Transport erfolgt mit einer Geschwindigkeit zwischen 0,1-40 cm pro Tag, je nach Transportmechanismus. 2.1 Charakteristika neuronaler Zellen 2-5 Zellmembran und Membranpotential (A) Aufbau der Zellmembran (B) Entstehung des Membranpotentials Intrazelluläre Seite Extrazelluläre Seite Konzentrationsgradient Spannungsgradient UMem Abbildung 2-2 Aufbau einer Zellmambran und Entstehung des Membranpotentials. Abbildungen entnommen aus [Nicholls, 1995]. (A) In die Doppellipidschicht einer Zellmembran sind Transmembranproteine eingelagert, welche den Transport von Ionen und Molekülen durch die Zellmembran ermöglichen. (B) Ein Konzentrationsgradient einzelner Ionenarten über die Zellmembran hinweg ist verantwortlich für die Ausbildung des Membranpotentials. Phospholipidmembran ist undurchlässig für Wasser und wasserlösliche Ionen. Transmembranproteine, welche in die Membran eingebunden sind, ermöglichen der Zelle einen kontrollierten Austausch von Ionen und Molekülen über diese Barriere hinweg. Für die Entstehung des Membranpotentials einer Zelle sind zwei Typen von Transmembranproteinen verantwortlich, Ionenkanäle und die Natrium-Kalium Pumpe. Ionenkanäle sind wassergefüllte Poren, welche mit hoher Selektivität spezifische Ionen passieren lassen. Die Natrium-Kalium Pumpe ist ein spezielles Transportprotein, welches in einem elektrogenen Prozess unter ATP-Verbrauch Ionen durch die Lipidmembran schleust. In einem Zyklus von + Konformationsänderungen transportiert die Na-K-ATPase drei Na -Ionen aus der Zelle hinaus und zwei K+-Ionen in die Zelle hinein. 2-6 2 Neuronale Zellen In Abbildung 2-2 A ist eine Zellmembran mit eingelagerten Transmembranproteinen sowie ein Schnitt durch die Pore eines Ionenkanals schematisch dargestellt. Durch das Zusammenspiel von Na-K-ATPase und Ionenkanälen kommt es, wie in Abbildung 2-2 B dargestellt, zu einer Trennung von positiven und negativen Ladungen an der Zellmembran: − Die Na-K-ATPase erzeugt einen Konzentrationsgradienten der Ionenkonzentrationen über die Zellmembran. Daraus resultiert ein osmotischer Druck. − Kalium-Ionenkanäle in der Zellmembran ermöglichen einen Ausstrom von positiv geladenen Kaliumionen entlang dieses Konzentrationsgradienten aus der Zelle heraus. Dies führt zu einem Überschuss von negativen Ladungen im Zellinneren. − Die resultierende Potentialdifferenz über der Zellmembran wirkt dem Kaliumausstrom entgegen. Es stellt sich ein Gleichgewichtszustand ein, in welchem die Kräfte aufgrund von osmotischem Druck und elektrischer Potentialdifferenz gleiche Beträge haben. Für den Spezialfall nur eines Ionen-Konzentrationsgradienten an einer ideal permeablen Membran, wird der Gleichgewichtszustand durch die Nernst Gleichung beschrieben. Die resultierende Potentialdifferenz über der Membran UMem bezeichnet man als Nernst Potential: U Mem = UMem R T z F ci,a = = = = = = RT ca ln zF ci Gleichung 2-1 Membranpotential [mV] Gaskonstante [J/K/mol] Temperatur [K] Wertigkeit des Ions [1] Faradaykonstante [C/mol] Innen / Aussenkonzentration [mol/l] Das Membranpotential einer Nervenzelle im Ruhezustand, das sog. Ruhepotential, setzt sich aus einer Überlagerung mehrerer Membranpotentiale verschiedener Ionentypen zusammen. Beteiligt sind Na+, K+, Ca2+, und Cl- Ionen. In Tabelle 2-1 sind 2.1 Charakteristika neuronaler Zellen 2-7 die typischen Konzentrationen dieser Ionen in Neuronen von Mensch und 3 Tintenfisch aufgelistet . Die Nernst Potentiale jedes einzelnen Ionentyps wurden berechnet. Ionentyp cinnen [mM] Mensch caussen [mM] UMem [mV] cinnen [mM] Tintenfisch caussen [mM] UMem [mV] 10 120 63 50 440 55 Na+ + 140 3 -97 400 20 -75 K 2+ 0,0001 1 116 0,0001 10 145 Ca Cl4 121 -86 60 560 -56 Tabelle 2-1 Ionenkonzentrationen in Neuronen von Mensch und Tintenfisch mit den resultierenden Nernst Potentialen UMem . Konzentrationen entnommen aus [Nicholls, 1995; Kleinig, 1999]. Um das Ruhepotential URuhe einer Nervenzelle zu bestimmen, müssen die Ionenkonzentrationen c aller Ionen sowie spezifische Permeabilitäten P der Membran für jeden einzelnen Ionentyp berücksichtigt werden. Die Goldmann Gleichung beschreibt den Zusammenhang quantitativ: U Ruhe URuhe R T F P ci,a a a i RT PK + c K + + PNa + c Na + + PCl − cCl − = ln i a F PK + c Ki + + PNa + c Na + + P −c Cl Cl − = = = = = = Gleichung 2-2 Ruhepotential [mV] Gaskonstante [J/K/mol] Temperatur [K] Faradaykonstante [C/mol] Membranpermeabilität für das jeweilige Ion [cm/s] Innen / Aussenkonzentration [mol/l] des jeweiligen Ions Der Beitrag der Ca2+ Ionen fehlt in dieser Gleichung, da er aufgrund der geringen 2+ -9 Membranpermeabilität für Ca -Ionen (PCa << 10 ) vernachlässigt werden kann. Die Membranpermeabilitäten eines menschlichen Neurons für die anderen beteiligten Ionen sind PK = 10-7, PNa = 10–9 und PCl = 10-8. Anhand dieser Permeabilitäten und den Konzentrationen aus Tabelle 2-1 erhält man rechnerisch ein Ruhepotential von -89 mV. Bei elektrophysiologischen Messungen findet man gewöhnlich Ruhepotentiale zwischen -60 und -90 mV. 3 Für beide Spezies erhält man trotz deutlich variierender Absolutkonzentrationen jeweils ähnliche Konzentrationsquotienten und Nernst Potentiale. Dies gilt allgemein für alle neuronalen Zellen in unterschiedlichsten Spezies. 2-8 2 Neuronale Zellen Die Anzahl Ionen, welche insgesamt an der Ausbildung des Membranpotentials beteiligt sind, ist verschwindend gering verglichen mit der Gesamtzahl aller Ionen in der Zelle. Dadurch ist eine schnelle Änderung der Membranspannung, induziert nur durch Änderungen der spezifischen Leitfähigkeit der Membran für bestimmte Ionen möglich. Der Konzentrationsgradient jedes Ionentyps bleibt dabei weitgehend stabil. 2.1.3 Ionenkanäle Alle Zellen haben Ionenkanäle, bei Neuronen ist die Dichte und Vielfalt an Kanälen jedoch besonders groß. Neben passiven, permanent geöffneten Ionenkanälen, welche wie zuvor beschrieben für die Entstehung des Ruhepotentials verantwortlich sind, exprimieren Neurone aktive, schaltbare Ionenkanäle. Diese ermöglichen Schaltbare Ionenkanäle (A) Spannungsgesteuerter Ionenkanal (C) G-Protein gekoppelter Rezeptor (B) Ligandengesteuerter Ionenkanal Abbildung 2-3 Mechanismen zum aktiven Schalten von Ionenkanälen. Abbildungen A und B entnommen aus [Stryer, 1996]. Abbildung C entnommen aus [Munk, 2000]. (A) Prinzip des spannungsgesteuerten Ionenkanals. Die Membranpore öffnet und schließt mit geänderter Membranspannung. (B) Prinzip des ligandengesteuerten Ionenkanals. Binden eines Liganden an den Kanal bewirkt eine Konformationsänderung. (C) Indirekte Kanalöffnung durch Ligandenbindung an einen örtlich separiertes Rezeptorprotein. Kopplung von Rezeptor erfolgt auf dem Umweg über ein G-Protein. 2.1 Charakteristika neuronaler Zellen 2-9 sowohl die Erzeugung eines Aktionspotentials, dessen aktive Weiterleitung sowie die Signalübertragung an Synapsen. Das aktive Öffnen und Schließen der Pore eines Ionenkanals erfolgt durch eine Konformationsänderung der beteiligten Transmembrandomänen des Kanals. Man unterscheidet drei Mechanismen: 1. Spannungsgesteuerte Ionenkanäle ändern ihre Konformation mit der Membranspannung an der Zellmembran (Abbildung 2-3 A). Geladene Domänen des Proteins werden dabei durch elektrostatische Wechselwirkung verschoben + (Na -Kanal: S4 Region). Dabei ändert sich die Konformation des Proteins, wodurch sich die Pore im Kanal öffnet oder schließt. 2. Bei ligandengesteuerten Ionenkanälen wird die Konformationsänderung verursacht durch Anbindung eines Liganden an einen spezifischen Rezeptor. Entweder bindet der Ligand direkt an eine Rezeptordomäne des Kanals (schnelles Öffnen im ms-Bereich, Abbildung 2-3 B) oder an ein anderes Rezeptorprotein in der Zellmembran, welches das Signal zur Öffnung über eine G-Protein gekoppelte Signalkaskade an den Kanal weitergibt (langsames Öffnen > 100 ms, Abbildung 2-3 C). 3. Mechanisch gesteuerte Ionenkanäle werden rein mechanisch durch Kräfte entlang der Zellmembran aufgezogen oder zugedrückt. Alle kationischen Ionenkanäle haben eine ausgesprochen hohe Selektivität (bis zu 1:100) für bestimmte Ionen. Über die Größe der Transmembranpore selektieren die Kanäle die hindurchtretenden Ionen nach dem Ionenradius. Na+ ist mit 0,095 nm Ionenradius deutlich kleiner als K+ (0,133 nm). Behält das Na+-Ion jedoch seine Hydrathülle, so beträgt der Radius 0,5 nm. Zur Selektion von K+ Ionen streift der Kaliumkanal daher die Hydrathülle ausschließlich von den K+ Ionen. 2.1.4 Aktionspotential Die Fähigkeit Aktionspotentiale zu bilden ist eine ganz charakteristische Eigenschaft neuronaler Zellen. Ein Aktionspotential entsteht aus dem Wechselspiel von spannungsgesteuerten Natrium- und Kaliumkanälen in der Zellmembran von 2-10 2 Neuronale Zellen Neuronen. Durch kurzzeitige Änderung der Membranleitfähigkeit für Na+ und K+ Ionen wird eine schnelle Depolarisation und Repolarisation der Zelle ausgelöst, und so ein kurzer Spannungsimpuls (100 mV, 1 ms) erzeugt. Das zugrundeliegende Prinzip ist bei allen Neuronen und in allen Spezies gleich4: − Wird ein Neuron über eine spezifische Schwellenspannung (ca. –40 mV) hinweg depolarisiert (z.B. durch Na+-Einstrom an einer Synapse), so öffnen sich + spannungsgesteuerte Na -Kanäle in der Zellmembran. Es kommt zu einem explosionsartigen Spannungsanstieg aufgrund von Natriumeinstrom, da mit steigender Depolarisation immer mehr Natriumkanäle öffnen. Der Anstieg endet erst beim Na+-Gleichgewichtspotential (+63 mV, Tabelle 2-1). − Jetzt öffnen langsamere, spannungsgesteuerte Kaliumkanäle. Gleichzeitig werden die Natriumkanäle durch eine weitere Konformationsänderung wieder undurchlässig und werden gleichzeitig inaktiviert. In der Folge kommt es zu einer Repolarisation der Zelle durch Kaliumausstrom in Richtung des K+Gleichgewichtspotentials (-97 mV, Tabelle 2-1). − Mit wachsender Repolarisation schließen die K+-Kanäle wieder. Das Membranpotential schwingt kurz über das Ruhepotential hinaus und pendelt sich dann wieder auf dieses ein. − Nach einer kurzen Erholungsphase fallen die Natriumkanäle wieder in die aktivierbare Ausgangskonformation zurück. Ein neues Aktionspotential kann ausgelöst werden. In Abbildung 2-4 A ist der zeitliche Verlauf der am Aktionspotential beteiligten + + Ionenströme durch die Na - und K - Kanäle dargestellt. Die Signale wurden an Neuronen aufgenommen, bei denen jeweils ein Kanaltyp durch ein spezifisches Toxin blockiert war. Blockade des Na+-Kanals erfolgte durch TTX (Tetrodotoxin), des Kaliumkanals durch TEA (Tetraethylammonium). Beginnend zum Zeitpunkt 0 ms und für die gesamte abgebildte Dauer, wurde die Zellmembran mit einer im Cytoplasma angelegten, positiven Spannung depolarisiert. 4 Lediglich Auslöseschwelle und Form eines Aktionspotentials an verschiedenen Neuronen oder verschiedenen + + Stellen eines Neurons variieren durch unterschiedliche Konzentrationen von Na - und K -Kanälen. 2.1 Charakteristika neuronaler Zellen 2-11 Das Aktionspotential (A) Beteiligte Ionenströme Natriumstrom (B) Zeitlicher Verlauf eines AP Membranpotential [mV] Kaliumstrom 0 1 2 3 4 5 Zeit [ms] Abbildung 2-4 Zeitlicher Verlauf eines Aktionspotentials und beteiligte Ionenströme. Abbildungen entnommen aus [Nicholls, 1995; Kleinig, 1999]. + + (A) Getrennte Aufnahme von Na und K Strom eines Neurons durch Blockade des jeweils anderen Ionenstroms durch die Toxine TTX und TEA. (B) Signalverlauf eines Aktionspotentials. Man erkennt deutlich die unterschiedliche Geschwindigkeit des Anstiegs von Na+ + + und K Strom. Das Öffnen der Na -Kanäle erfolgt deutlich schneller als das der K+-Kanäle. Weiterhin ist die Inhibition der Na+-Kanäle sichtbar. Nach wenigen Millisekunden nimmt der Na+-Strom wieder bis auf Null ab, wohingegen der K+-Strom für die gesamte Dauer der Depolarisation anhält. Wird die Membranspannung nicht festgehalten sondern kann frei schwingen, so führt die Überlagerung von Na+- und K+-Strom zur Änderung des Membranpotentials der Zelle in der charakteristischen Form des Aktionspotentials (Abbildung 2-4 B). Depolarisation der Zellmembran über die Auslöseschwelle hinaus (gestrichelt eingezeichnet), löst ein Aktionspotential aus. 2-12 2 Neuronale Zellen 2.2 Signalübertragung zwischen Neuronen Anders als bei Signalen in Computern ist im Nervensystem außer der Signalintensität (zeitliche Dichte der Aktionspotentialpulse) keine Information im Signal selber verschüsselt. Die Information über die Bedeutung eines Aktionspotentials ergibt sich vielmehr aus der Bahn, auf welcher das Signal das Gehirn erreicht. Insgesamt hat das enge Geflecht von Axonen und Dendriten im Gehirn eine Gesamtlänge von über 500.000 km. Die Mechanismen, mit welchen eine schnelle Signalleitung entlang dieser Pfade erfolgt sowie die Funktionsweise der Signalübertragung von einem Neuron zum nächsten an elektrischen und funktionalen Synapsen sind im Folgenden beschrieben. 2.2.1 Aktive Signalleitung im Axon Entlang myelinisierter Axone im peripheren Nervensystem werden Aktionspotentiale verlustfrei über Distanzen von bis zu 2 m mit einer Geschwindigkeit von 100 m/s weitergeleitet. Dies ist nur möglich durch aktive Signalverstärkung mittels spannungsgesteuerter Ionenkanäle sowie Isolation des Axons durch Myelin. Misst man die passive Signalausbreitung in einem nicht myelinisierten Axon (Abbildung 2-5A1) im Experiment, so beobachtet man einen exponentiellen Abfall der Signalamplitude über die zurückgelegte Weglänge (Abbildung 2-5 A2). Theoretisch beschreiben kann man diese Beobachtung mit dem in Abbildung 2-5 A3 dargestellten elektrischen Modell. Das Axon wird darin als eine Kaskade von in Reihe und parallel geschalteten Widerständen beschrieben. Längswiderstände Ri längs des Axons repräsentieren den Widerstand des Axoplasmas entlang der Signalausbreitung. Querwiderstände Rm berücksichtigen die nur unvollständige Isolation des Axoplasmas von der Umgebung durch die Axonmembran. 2.2 Signalübertragung zwischen Neuronen 2-13 Signalleitung im Axon (A) Passive Signalleitung (B) Aktive Signalleitung (A1) I U [mV] Schwellenwert Ruhepotential (A2) U [mV] U U0 Schwellenwert Ruhepotential U [mV] (A3) I Rm Ri Rm Ri Rm Rm Rm Ri Ri Ri Schwellenwert Ruhepotential Ri X Abbildung 2-5 Passive und aktive Signalleitung im Axon. Abbildungen entnommen aus [Nicholls, 1995]. (A) Passive Signalleitung im Axon, gemessen durch Stimulation unter der Auslöseschwelle eines Aktionspotentials. (A1) Messanordnung, (A2) Messergebnis, (A3) Elektrischer Ersatzschaltkreis zur Beschreibung passiver Signalleitung im Axon. (B) Aktive Leitung eines Aktionspotentials. Die Amplitude ist an allen Punkten gleich groß. Für einen Grenzübergang zu infinitesimal kleinen Axonabschnitten führt man die spezifischen Widerstände R'i [Ohm/cm] (Der Längswiderstand eines Axonstücks ist proportional zu dessen Länge) und R'm [Ohm*cm] (Die Leitfähigkeit der Membran eines Axonstücks ist proportional zur Membranfläche und damit zur Länge des Axonstücks) ein. Mit dem Ohm'schen Gesetz ergibt sich folgende homogene Differentialgleichung (DGL) zweiter Ordnung: R ' d 2U ( x) U ( x) = m' dx 2 Ri U(x) R'i R'm x = = = = Spannung über der Axonmembran am Ort x [mV] Spezifischer Widerstand des Axoplasmas [Ohm/cm] Spezifischer Widerstand der Axonmembran [Ohm*cm] Abstand vom Punkt der Stimulation [cm] Gleichung 2-3 2-14 2 Neuronale Zellen Als Lösung dieser DGL erhält man eine einfache Exponentialfunktion: U ( x) = U 0 e Uo λ = = − x λ ,λ = Rm' Ri' Gleichung 2-4 Spannung über der Axonmembran am Ort der x=0 [mV] Abklinglänge [cm] Der gemessene Potentialverlauf entlang des Axons lässt sich mit dieser Gleichung exakt beschreiben. 9 Setzt man die spezifischen Leitfähigkeiten R'i = 6,4 * 10 Ohm/cm und 6 R'm = 3,2 * 10 Ohm*cm eines isolierten Axons mit einem Radius von 1 µm ein [Katz, 1985], so erhält man eine Abklingkonstante von λ = 0,02 cm. Das Ausgangssignal fällt also bereits nach 0,2 mm Signalweg auf 37 % seiner Ausgangsgröße ab. Eine Weiterleitung von Aktionspotentialen über größere Distanzen von mehreren Metern ist mit einem solchen Axon ausgeschlossen. Eine mögliche Lösung des Problems besteht in der Vergrößerung des Axondurchmessers. Da der Innenwiderstand des Axons mit dem Quadrat des Axonradius abnimmt, der Membranwiderstand aber nur linear mit dem Radius sinkt, resultiert eine Netto-Zunahme der Abklinglänge λ mit wachsendem Axonradius: λ (r ) = α r λ(r) = α = r = Gleichung 2-5 Räumliche Abklinglänge der Signalamplitude [cm] Proportionalitätskonstante [1] Radius des Axons [µm] Erweitert man das elektrische Modell um die Membrankapazität und betrachtet auch die zeitliche Komponente der Signalleitung im Axon (siehe Kapitel 5), so findet man für die Ausbreitungsgeschwindigkeit von Impulsen die gleiche Abhängigkeit von der Wurzel des Axondurchmessers. Im Riesenaxon des Tintenfischs ist dieses Prinzip ausgenutzt. Dieses Axon ermöglicht aufgrund seiner besonders großen Dicke eine schnelle Signalausbreitung mit geringen Verlusten. 2.2 Signalübertragung zwischen Neuronen 2-15 Bei allen vertebraten Lebewesen wird der gleiche Effekt jedoch wesentlich effizienter durch die Ummantelung des Axons mit einer isolierenden Myelinschicht erzielt. Dadurch erhöht sich der Widerstand R'm in den myelinisierten Bereichen, und Abklinglänge sowie Ausbreitungsgeschwindigkeit nehmen zu. Die extrem schlechte Leitfähigkeit des Axoplasmas kann dadurch aber immer noch nicht vollständig ausgeglichen werden. Ohne aktive Signalverstärkung würde man immer noch einen deutlichen Signalabfall über längere Strecken beobachten. Misst man die Signalausbreitung an einem Axon, so ist die Signalamplitude jedoch an allen Stellen gleich groß (Abbildung 2-5 B). Dies wird möglich durch eine aktive Verstärkung des Signals entlang des Signalwegs. In regelmäßigen Abständen entlang eines Axons beobachtet man Einschnürungen der Myelinisierung an sog. Ranvier'schen Schnürringen (siehe Abbildung 2-1 B). An diesen Punkten sorgen Ionenkanäle für die Ausbildung eines neuen lokalen Aktionspotentials. Ein Prinzip der aktiven Signalleitung im Axon Zeit Punkt 1 Punkt 2 Punkt 3 Stimulation (A) Stimulation an Punkt 1 (t=0) (B) Aktionspot. an Punkt 2 (t=1) (C) Aktionspot. an Punkt 3 (t=2) Abbildung 2-6 Aktive Ausbreitung eines Aktionspotentials entlang eines Axons. (A) Durch Stimulation an Punkt 1 wird ein Aktionspotential ausgelöst. + (B) Passive Ströme entlang des Axoplasmas bewirken das Öffnen von Na -Kanälen an Punkt 2. Ein neues Aktionspotential wird an Punkt 2 gebildet. (C) Passive Ströme des Aktionspotentials an Punkt 2 lösen an Punkt 3 ein Aktionspotential aus. 2-16 2 Neuronale Zellen Aktionspotential springt bei seiner Ausbreitung im myelinisierten Axon also förmlich von Schnürring zu Schnürring. In Abbildung 2-6 A bis C ist am Beispiel eines nicht-myelinisierten Axons dargestellt wie durch das Zusammenwirken von passiven Strömen entlang des Axoplasmas und + + aktiven Strömen durch Na -und K -Ionenkanäle in der Axonmembran eine verlustfreie Signalübertragung erfolgt. Das zugrundeliegende Prinzip ist auch bei Myelinisierung das gleiche. Für die gerichtete Ausbreitung des Aktionspotentials ist die nach Öffnung der Na++ Kanäle einsetzende, zeitlich begrenzte Deaktivierung der Na -Kanäle von entscheidender Bedeutung. Ohne diesen Mechanismus würde ein Aktionspotential ohne Unterlass im Axon hin- und herlaufen. 2.2.2 Synaptische Kopplung Die Übertragung elektrischer Signale zwischen zwei Nervenzellen, oder einer Nervenzelle und einer Muskelzelle kann über zwei grundsätzlich verschiedene Mechanismen erfolgen: Entweder fliesst direkt der elektrische Strom eines Pulses von einer Zelle zur nächsten. Dies geschieht über eine elektrische Synapse, welche das Cytoplasma beider Zellen miteinander verbindet. Oder das Signal wird indirekt durch Ausschüttung von Botenstoffen (Neurotransmittern) an die andere Zelle übertragen. Dies ist das Prinzip einer funktionalen Synapse. 2.2.2.1 Gap Junctions Elektrische Synapsen verbinden das Cytoplasma benachbarter Zellen durch Poren in den Membranen, sog. Gap Junctions, leitend miteinander. Der Abstand der Zellmembranen im Bereich der Poren beträgt dabei nur 3,5 nm. Die Poren haben einen Durchmesser von 1,5 nm und gestatten den Austausch beliebiger Ionen zwischen den Zellen. Es resultiert eine ungerichtete Leitfähigkeit für elektrische Signale, wobei die Signalamplitude von einer Zelle zur nächsten je nach Anzahl der Poren und deren Leitfähigkeit mehr oder weniger stark abgeschwächt wird. 2.2 Signalübertragung zwischen Neuronen 2-17 Obwohl bei Säugetieren nur wenige Zellen nicht durch Gap Junctions zu Zellverbänden zusammengeschlossen sind, spielen die elektrischen Eigenschaften von Gap-Junctions nur vereinzelt eine Rolle im Organismus. Eine Schlüsselrolle kommt ihnen jedoch bei der Synchronisation der elektrischen Aktivität von Muskelzellen im Herzmuskel zu. Im Nervensystem finden elektrische Synapsen Elektrische Synapsen - Gap Junctions (A) Schematischer Aufbau (B) Anordnung in der Zellmembran Abbildung 2-7 Gap Junctions bilden eine Pore zwischen zwei Zellen, eine elektrische Synapse entsteht. Abbildungen entnommen aus [Nicholls, 1995; Kleinig, 1999]. (A) Modell einer Gap Junction. Jeweils ein hexameres Connexon bildet einen Kanal durch eine Plasmamembran. Connexine (farbig dargestellt) verbinden die beiden Poren über den Interzellularraum (2-3 nm) hinweg. An den intrazellulär liegenden N- und C- Termini der 2+ Connexine können Regulationsmechanismen (Ca , pH) angreifen. (B) Schematische Darstellung der Anordnung von Gap Junctions in den Zellmembranen zweier verbundener Zellen. insbesondere dort Anwendung, wo es auf eine besonders schnelle Signalübertragung ankommt. Anders als bei funktionalen Synapsen erfolgt die Signalübertragung an elektrischen Synapsen nämlich ohne jede Zeitverzögerung. So findet man elektrische Synapsen beispielsweise im Schaltkreis zur Steuerung der Fluchtreaktion von Fischen. 2-18 2 Neuronale Zellen Gap Junctions setzen sich durch Aneinanderlagerung zweier Transmembranproteine, den sog. hexameren Connexonen, aus den Membranen benachbarter Zellen zusammen. Zwei Connexone verbinden sich dabei im Interzellularraum, so dass eine lange Pore entsteht, welche die Phospholipidmembranen beider Zellen durchspannt (Abbildung 2-7 A). Der Außendurchmesser einer solchen Pore beträgt 6,5 nm, ihre Länge 18 nm. Jedes Connexon der Pore besteht aus sechs Untereinheiten, den Connexinen. In Zellmembranen treten Gap Junctions regelmäßig nebeneinander in Feldern angeordnet auf (Abbildung 2-7 B). Eine Verringerung der Leitfähigkeit von Gap Junctions wird beobachtet bei 2+ intrazellulärem pH-Abfall oder Erhöhung der interzellulären Ca -Konzentration. Dies ist ein Schutzmechanismus um gekoppelte Zellverbände im Fall der Lyse einzelner Zellen von den zerstörten Zellen zu entkoppeln. 2.2.2.2 Funktionale Synapsen Bei funktionalen Synapsen ist das Cytoplasma der Zellen vollständig voneinander getrennt. Auch der Abstand der Zellmembranen an der synaptischen Kopplungsstelle ist mit 10 bis 50 nm deutlich größer als bei Gap Junctions. Die Signalübertragung erfolgt aktiv (unter ATP Verbrauch) durch Freisetzung von Neurotransmitter aus der Präsynapse in den synaptischen Spalt zwischen beiden Zellmembranen. Der Neurotransmitter bindet an Rezeptoren der Postsynapse, worauf sich Ionenkanäle in der postsynaptischen Membran öffnen und eine Depolarisation der postsynaptischen Zelle bewirken. Die Signalausbreitung erfolgt also gerichtet von der Präsynapse zur Postsynapse. Aufgrund des aktiven Öffnens von Ionenkanälen durch den Neurotransmitter, ist je nach Typ des geöffneten Ionenkanals eine Signalverstärkung in positiver (Depolarisation der Postsynapse z.B. durch Na+-Einstrom) und negativer Richtung (Hyperpolarisation der Postsynapse z.B. durch Cl--Einstrom) möglich. Funktionale Synapsen sind an allen Formen neuronaler Signalverarbeitung im Gehirn und im peripheren Nervensystem beteiligt. Jedes Neuron im Gehirn hat zwischen 1000 und 100000 funktionale Synapsen zu anderen Neuronen. Im 2.2 Signalübertragung zwischen Neuronen 2-19 Funktionale Synapsen (A) Ablauf der synaptischenTransmission (B) Synapsen im Elektronenmikroskop 1 µm Abbildung 2-8 Prinzip und Morphologie einer funktionalen Synapse. Abbildungen entnommen aus [Kleinig, 1999]. (A) Mechanismus der synaptischen Übertragung am Beispiel einer neuromuskulären Synapse. (B) Elektronenmikroskopische Aufnahme eines Schnitts durch fünf Synapsen an einem Dendriten. Präparat aus dem Gehirn eines Affen. Der Dendrit enthält im Zentrum des Bildes ein dunkel erscheinendes Mitochondrion. Unterschied zu Nervenzellen werden funktionale Synapsen auch im adulten Organismus weiter gebildet. Bis zu 250.000 Stück entstehen im menschlichen Gehirn in jeder einzelnen Minute. Aber auch bestehende Synapsen sind keine starren Verbindungen. Sie haben eine gewisse Plastizität, wodurch ihre Kopplungsstärke kurzfristig und langfristig modulieren kann. Man nimmt an, dass synaptische Modulation auch für komplexe Vorgänge im Gehirn wie Lernen und Gedächtnis verantwortlich ist. In Abbildung 2-8 A ist das Prinzip der synaptischen Übertragung an einer funktionalen neuromuskulären Synapse dargestellt. Die einzelnen Schritte des Übertragungsprozesses (entsprechend der Numerierung in der Abbildung) sind im folgenden kurz erläutert: 1. Ein Aktionspotential erreicht über das Axon die Präsynapse. 2-20 2 Neuronale Zellen 2. Nach Depolarisation der präsynaptischen Membran öffnen spannungssensitive 2+ 2+ Ca -Kanäle. Ca strömt ein. 3. Die erhöhte Ca2+-Konzentration bewirkt die Exocytose von synaptischen Vesikeln. Der Neurotransmitter Acetylcholin (ACh) wird aus den Vesikeln in den synaptischen Spalt ausgeschüttet (Dauer ca. 1 ms). 4. ACh diffundiert durch den Spalt zur postsynaptischen Membran (Dauer ca. 2 µs) und bindet dort an ACh-Rezeptoren. 5. Die Transmitterbindung löst das Öffnen von Na+-Kanälen in der postsynaptischen Membran aus (Dauer ca. 1,5 µs). + 6. Na - Einstrom durch die ACh-Rezeptoren depolarisiert die postsynaptische Membran. Ein neues Aktionspotential wird postsynaptisch ausgelöst. 7. Der Neurotransmitter ACh wird wieder freigesetzt und durch ACh-Esterase abgebaut. 8. Die Abbauprodukte des Neurotransmitters werden wieder in die Präsynapse aufgenommen. Neues ACh wird generiert und in Vesikel eingelagert. Funktionale synaptische Übertragung im PNS erfolgt ausschließlich auf Basis des Neurotransmitters ACh nach dem beschriebenen Mechanismus. Im ZNS können hingegen zahlreiche Neurotransmitter an der Übertragung beteiligt sein. Man unterscheidet niedermolekulare Neurotransmitter von Neuropeptiden. Niedermolekulare Neurotransmitter (ACh, Amine, Aminosäurederivate, …) bewirken eine schnelle Signalübertragung im ms-Bereich. Neuropeptide (Endorphine, Enkephaline, Opiode, …) hingegen wirken eher langsam modulierend innerhalb von Minuten bis Stunden. Weitere Unterschiede zwischen Synapsen des ZNS und PNS sind die Form des Transmitterabbaus sowie die Morphologie am synaptischen Spalt. Im ZNS werden Neurotransmitter nicht im synaptischen Spalt abgebaut, sondern diffundieren in die Umgebung und werden anschließend wieder in Präsynapsen aufgenommen. Im PNS ist der synaptische Spalt (50 nm) mit einer Basalmembran ausgefüllt, welche auch die gesamte Präsynapse umhüllt. Im ZNS findet man keine Basalmembran und 2.2 Signalübertragung zwischen Neuronen 2-21 der synaptische Spalt ist mit 10 nm deutlich enger. Insgesamt ist die Synapse im PNS mit durchschnittlich 10 µm Ausdehnung deutlich größer als im ZNS (0,5 µm). Abbildung 2-8 B zeigt einen Schnitt durch fünf funktionale Synapsen an einem Dendriten im ZNS. Deutlich sind die synaptischen Vesikel in der Nähe der Zellmembran zu erkennen (Pfeile). 2-22 2 Neuronale Zellen 2.3 Neuronale Zellkultur Die Anfänge neuronaler Zellkultur gehen auf Ross Granville Harrison zurück. 1907 berichtete dieser erstmals von Beobachtungen an in Kultur gewachsenen Nervenfasern [Harrison, 1907]. Dieser Nachweis, dass neuronales Gewebe auch außerhalb des Körpers überleben und wachsen kann, gab den Anstoß für die Begründung einer neuen wissenschaftlichen Disziplin, der neuronalen Zellkultur. 1955 gelang Peterson und Muray der Nachweis, dass auch komplexe Vorgänge wie Synapsenbildung und Myelinisierung von Axonen unter geeigneten Bedingungen in Kultur ablaufen [Peterson, 1955]. Mit der Verfügbarkeit von sterilen Arbeitsmaterialien auf Polymerbasis sowie von Filtersystemen für die Entkeimung der Luft im Zellabor seit ca. 30 Jahren, bekam das Feld der neuronalen Zellkultur schließlich breite Bedeutung in der Neurobiologie. Sterile Kulturbedingungen waren durch diese Entwicklungen mit einem Mal ohne größere Schwierigkeiten in jedem Labor zu realisieren. Heute sind neuronale Zellkulturen Gegenstand umfangreicher Forschung zur weiteren Ergründung der Funktion des Nervensystems. Im Gegensatz zu allen anderen Zellen im Organismus haben Nervenzellen die Eigenschaft, dass sie nach Abschluss einer Phase schneller Zellteilung im embryonalen Entwicklungsstadium, ihre Zellteilung vollständig einstellen und zu erwachsenen Neuronen ausdifferenzieren. Trotz wissenschaftlicher Fortschritte in den letzten Jahren [Gage, 1995] ist es bisher noch nicht gelungen, ausdifferenzierte Neurone wieder zurück in eine Phase des Wachstums zu überführen und die Schranke zur Zellteilung zu überwinden [Banker, 1998]. Experimente an Nervenzellen in Kultur erfordern daher das Opfer eines Versuchstieres für jedes einzelne Experiment. Auch ist die Anwendung genetischer Techniken aufgrund der ohne Zellteilung fehlenden Vererbung ausgeschlossen. Weiterhin werden die Experimente dadurch erschwert, dass die Kulturen, die bei 2.3 Neuronale Zellkultur 2-23 verschiedenen Experimenten folglich aus verschiedenen Tieren stammen, eine signifikante Streuung in den Eigenschaften der enthaltenen Nervenzellen aufweisen. Da die Ausbeute an Neuronen jeder Präparation begrenzt ist, kann diese Streuung auch nicht durch Einfrieren eines für mehrere Experimente ausreichenden Zellvorrats eliminiert werden. Aus den oben genannten Gründen gibt es seit langer Zeit Bestrebungen Zellsysteme zu entwickeln, welche die Fähigkeit zur Zellteilung besitzen und anstelle von primär isolierten Neuronen für wissenschaftliche Experimente eingesetzt werden können. Mit Zellen aus neuronalem Tumorgewebe ist dies inzwischen auch gelungen. Man spricht in diesem Zusmmenhang von neuronalen Tumorzellinien. Tumorzellinien basieren meist auf Gewebe, welches aus Hirntumoren von Ratten oder Mäusen gewonnen wurde. Einzelne Zellen des Tumorgewebes werden daraus isoliert und zu einer Zellinie kloniert. Die hierzu verwendeten Tumore sind entweder natürlichen Ursprungs oder werden durch Bestrahlung und Verabreichung andere karzinogener Substanzen gezielt induziert. Die Vorteile von solchen neuronalen Zellinien sind offensichtlich: − Sie sind einfach zu Handhaben. Für Experimente ist keine aufwendige Primärisolation von Nervenzellen aus dem tierischen Organismus erforderlich sondern es können Zellen direkt aus einer kontinuierlich geführten Kultur entnommen werden. − Alle Zellen einer Zellinie sind genetisch homogen. Zwar kommt es im Laufe der Kultur zu spontanen Mutationen, jedoch kann bei regelmäßiger Erneuerung der Kultur durch frische Stammzellen eine gute Homogenität erzielt werden. − Die Anwendung genetischer Methoden ist möglich. Neuronale Zellinien eignen sich aber leider nur begrenzt als Modell für Nervenzellen. Abgeleitet von entarteten, karzinogenen Zellen weist bisher keine bekannte Zellinie das volle Spektrum neuronaler Eigenschaften auf, das an primär isolierten Neuronen beobachtet wird. So wird i.d.R. keine spezifische Polarisation 2-24 2 Neuronale Zellen der Neuriten in Axone und Dendriten beobachtet. Auch eine Myelinisierung von 5 Axonen oder die Bildung synaptischer Verknüpfungen erfolgt nicht [Banker, 1998]. Trotz dieser Einschränkungen sind viele aufschlussreiche Experimente mit neuronalen Zellinien möglich. Auch in dieser Arbeit wurden neben Primärkulturen verschiedene Tumorzellinien eingesetzt. 5 Die Zellinie P19 stellt eine hiervon abweichende vielversprechende Ausnahme dar. Es konnten funktionale Synapsen, verbunden mit axonaler und dendritischer Polarisation der beteiligten Neurone, an diesem Zellsystem nachgewiesen werden. 2.4 Eingesetzte Zellsysteme und Kulturprotokolle 2-25 2.4 Eingesetzte Zellsysteme und Kulturprotokolle Für die Durchführung der Experimente an lebenden neuronalen Zellen wurden verschiedene Zellsysteme eingesetzt. Je nach experimenteller Aufgabenstellung kamen dissoziierte Neurone aus Zellinien oder neuronale Primärkulturen in dissoziierter und organotypischer Form zum Einsatz. Bei dissoziierten Kulturen wurden die neuronalen Zellen zunächst durch Enzyme aus dem Zellverband gelöst und anschließend in Form einer Suspension auf Substratoberflächen ausgesät. Bei organotypischen Kulturen wurden Gewebsschnitte angefertigt und diese direkt auf die Substratoberfläche plaziert. Allgemeine Vorversuche und eine systematische Charakterisierung von strukturierten Kultursubstraten erfolgten auf Basis von Zellinien. Dies geschah einerseits im Hinblick auf die größere Stabilität dieser Systeme über mehrere Versuche hinweg, andererseits aber auch zur Minimierung der Zahl der benötigten Versuchstiere. Elektrophysiologische Experimente wurden vor allem an proliferierten Neuronen aus Hirnschnitten durchgeführt. Der Schwerpunkt bei diesen Experimenten lag auf der Detektion synaptischer Verbindungen innerhalb der Kultur. Die minimale Schädigung des Gewebes bei den organotypischen Schnitten sollte die Wahrscheinlichkeit für die Ausbildung funktionaler Synapsen in vitro erhöhen. Neben neuronalen Zellsystemen wurde noch ein weiteres elektrisch aktives primäres Zellsystem auf Basis von Herzmuskelzellen (Kardiomyozyten) verwendet. Kardiomyozyten sind neben neuronalen Zellen der einzige Zelltyp, welcher in der Lage ist Aktionspotentiale zu bilden. Zwar fehlen Kardiomyozyten alle anderen für Neurone charakteristischen Eigenschaften, ihre Aktionspotentiale sind für experimentelle Untersuchungen jedoch besonders interessant: Die elektrischen Signale sind, verglichen zu Neuronen, besonders stark und die Aktionspotentiale bilden sich spontan ohne externe Stimulation. Außerdem ist es möglich aktive Zellen an ihrer rhytmischen Kontraktion im Mikroskop zu identifizieren. 2-26 2 Neuronale Zellen Im Folgenden werden die verwendeten Zellsysteme beschrieben und alle Schritte zur Präparation und Kultivierung erläutert. 2.4.1 Zellinien 2.4.1.1 Mz1 und MzN Die Zellinien PCC7-Mz1 und PCC7-MzN (kurz Mz1 und MzN) sind beides Subklone eines Azaguanin-resistenten Klons von PCC7-S-Zellen (PCC7-S- AzaR1) [Fiellous, 1978; Pfeiffer, 1981; Lang, 1989]. PCC7-S-Zellen stammen von einem spontan entwickelten testikulären Tumor des rekombinierten Inzuchtmäusestammes 129xC57B1/6J. Mz1 differenziert sowohl in neuronale Zellen, als auch in andere Zelltypen wie beispielsweise Glia-Zellen. Bei MzN ist die Differenzierung zu nicht-neuronalen Zellen hingegen weitestgehend unterdrückt. Nach Ausdifferenzierung mit RA (All-Trans-Retinsäure) und cAMP (zyklisches Adenosin Monophosphat) entwickeln sich aus den Zellinien neuronale Zellen mit deutlich ausgeprägten Ionenkanälen und der Fähigkeit zur Ausprägung von Aktionspotentialen. Insbesondere ist es auf diesem Wege in früheren Arbeiten gelungen neuronale Polarität zu induzieren und durch Antikörperfärbungen nachzuweisen [Berger, 1997]. Beide Zellinien wurden entsprechend den Angaben in früheren Veröffentlichungen kultiviert [Lang, 1989; Berger, 1997; Herget, 1998; Jostock, 1998]. Als Kulturmedium wurde DMEM/Ham's F12 (DMEM/F12, 21331-012, Life Technologies GmbH) eingesetzt, zusätzlich versetzt mit 16 % FCS (F-7524, Sigma), 1 % P/S (P11-010, PAA ) sowie 200 mM Glutamax I (35050-038, Life Technologies GmbH). Die Inkubation der Zellen erfolgte bei 37 °C, 5 % CO2 und 95 % Luftfeuchtigkeit. Als Kulturgefäße wurden Falcon Zellkulturflaschen (Falcon 3108, Becton Dickinson) verwendet. Die Zellinien wurden für die gesamte Dauer der Experimente in Kultur gehalten. Je nach Bedarf wurden daraus Zell-Suspensionen gewonnen und diese auf Substraten ausgesät. Die dazu erforderlichen Schritte sind im Folgenden beschrieben: 2.4 Eingesetzte Zellsysteme und Kulturprotokolle 2-27 1. Abziehen des Kulturmediums. 2. Abwaschen der Serumreste durch Spülen mit PBS (14040-083, Life Technologies GmbH). 3. Ablösen der Zellen vom Kulturgefäß durch Zugabe eines Gemisches aus 0,25 % Trypsin (T-8128, Sigma) und 0,25 % EDTA (G-7020, Sigma) in PBS. 4. Auswaschen des Trypsins mit frischem Kulturmedium und Neutralisation des Trypsins durch zugesetztes fötales Kälberserum. 5. Zählen der Zelldichte im Hämocytometer und Berechnen des Verdünnungsverhältnisses für die Aussaat mit einer Zelldichte von 3500 Zellen je cm² . Verdünnen der Suspension mit Kulturmedium im errechneten Verhältnis. 6. Aussaat der Zellen durch Auftropfen einer definierten Menge der fertigen Suspension auf die Substrate. 7. Auffüllen der Substrate mit Kulturmedium nach ca. 1 bis 2 Stunden Sedimentationszeit, nachdem die Zellen adhärent sind. 8. Nach 48 Stunden Kulturzeit: Auslösen des neuronalen Differenzierungsprozesses auf den Substraten durch Zugabe von RA (R-2625, Sigma) in das Kulturmedium in einer Endkonzentration von 0,1 µM. 9. Nach 72 Stunden Kulturzeit: Vervollständigung der Stimulation durch Zugabe von cAMP (104 396, Roche Diagnostics GmbH) in einer Endkonzentration von 1 mM. 10. Von jetzt an, regelmäßiger Wechsel des Kulturmediums supplementiert mit RA und cAMP in den oben angegebenen Konzentrationen. Die genauen Zeitpunkte können aus Abbildung 2-9 B entnommen werden. Nach Beginn der neuronalen Ausdifferenzierung ist die Überlebenszeit neuronaler Zellinien begrenzt. Mit den Zellinien Mz1 und MzN ist es in den durchgeführten Versuchsreihen gelungen, die Zellen maximal zwei Wochen in Kultur zu halten. Der zeitliche Verlauf der Kultur ist in Abbildung 2-9, gemeinsam mit Beobachtungen zu Zellwachstum und Elektrophysiologie, im Überblick dargestellt. 2-28 2 Neuronale Zellen Kulturprotokoll für MzN- und Mz1- Zelllinien Kulturtag -1 0 1 2 3 4 5 6 7 8 9 10 11 12 13 (A) Vorbereitung der Substrate – Oberflächenstrukturierung (B) Zellkultur – Aussaat – Mediumwechsel – Differenzierung • RA • C-AMP • DMEM / HAM's F12 (Gibco), 12.5% FCS, Glutamax I, 1% PS • ..., 16% FCS, ... (C) Beobeachtung des Wachstums – Schnelle Zellteilung – Neuritenwachstum – Bildung von Zellklustern – Zelltod (D) Elektrophysiologische Messung – Membranpotential [mV] – Na-Kanäle – K-Kanäle -20 -30 -40 -20 Abbildung 2-9 Die neuronalen Zellinien wurden nach einem exakten Kulturprotokoll bis zu zwei Wochen auf den Substraten kultiviert. (A) Zeitpunkt der Substratvorbereitung (B) Schritte der Zellkultur (C) Beobachtungen am Mikroskop (D) Elektrophysiologische Untersuchungsergebnisse Eine erste Ausprägung neuronaler Eigenschaften nach Beginn der Differenzierung an Tag 2 wurde ab Tag 6 beobachtet. Zu diesem Zeitpunkt begann das Wachstum von Neuriten (Abbildung 2-9 C). Das in elektrophysiologischen Untersuchungen gemessene Membranpotential stieg ab Tag 6 von -20 mV auf -40 mV an Tag 10 an. Natrium und Kalium Ionenkanäle waren ab Tag 8 nachzuweisen. Diese Ergebnisse sind in guter Übereinstimmung mit früheren Untersuchungen der zeitlichen Entwicklung von Mz1 und MzN Zellen nach neuronaler Ausdifferenzierung [Berger, 1997]. 2.4.1.2 P19 Neuronal ausdifferenzierte Zellen der Zellinie P19 gelten derzeit als einziges nicht primäres Zellsystem, in welchem funktionale Synapsen nachgewiesen werden 2.4 Eingesetzte Zellsysteme und Kulturprotokolle 2-29 können [Banker, 1998]. P19 Neurone zeigen spezifische axonale und dendritische Polarität, und bilden elektrophysiologisch nachweisbare Synapsen [Parnas, 1995; Finley, 1996]. Die Zellinie P19 wurde 1982 aus einem künstlich induzierten Tumor gewonnen, gebildet durch Implantation eines 7 Tage alten Mäuseembryos in die Testikularkapsel einer erwachsenen Maus [McBurney, 1982]. Der Phenotyp der Zellen ist vergleichbar zu Zellen des primitiven Ectoderms. Sie zeigen ein breites Entwicklungspotential in unterschiedliche Zelltypen wie Herzmuskelzellen, Skelettmuskelzellen und neuronale Zellen [Rossant, 1982]. Mit All Trans Retinalsäure und einem speziellen Protokoll ist es möglich, eine P19 Kultur in neuronale Zellen auszudifferenzieren [Bain, 1994]. Das in dieser Arbeit eingesetzte Kulturprotokoll basiert auf diesen Angaben und ist im Folgenden beschrieben: − Kultivierung der Stammzellen: 1. In Abständen von 4 Wochen wird die bestehende Stammzellkultur verworfen und eine Kultur von neuen Stammzellen aus einem tiefgefrorenen Zellstamm begonnen, um eine Veränderung der Zellinie durch spontane Mutationen auszuschließen. 2. Kultur der Stammzellen erfolgt in MEM Alpha Medium (10888-022, Life Technologies GmbH), versetzt mit 10 % FCS (F-7524, Sigma) und 1 % P/S, in Falcon Zellkulturflaschen (Falcon 3108, Becton Dickinson). 3. In Abständen von 2 Tagen werden die Zellen im Verhältnis 1:10 – 1:20 passagiert. − Neuronale Ausdifferenzierung: 1. Abziehen des Kulturmediums. 2. Abwaschen der Serumreste durch Spülen mit PBS 2-30 2 Neuronale Zellen 3. Konfluent gewachsene Stammzellen 3 Minuten mit einer Lösung aus 0,25 % Trypsin (T-8253, Sigma) und 0,025 % EDTA (E-6758, Sigma) in PBS von der Kulturflasche ablösen. 4. Trypsin mit Kulturmedium auswaschen. 5. Zelldiche im Hämocytometer bestimmen, und die Zellen in einer Dichte von 12.800 Zellen/cm² in einer Non-TC-Schale mit 10 cm Durchmesser (663102, Greiner) aussäen. Kultumedium: 20ml MEM Alpha Medium mit 5 % FCS und 0,5 µM RA (R-2625, Sigma). 6. Nach 2 Tagen Mediumwechsel: Überführen der Zellen in ein 50 ml Röhrchen; Abwarten der Sedimentation der Zellen (ca. 10 Min.); Abziehen des Mediums; Aussaat des Sediments in neuem RA-haltigem Medium analog zu Schritt 2. 7. Nach 4 Tagen erfolgt die Übertragung auf normale Kultursubstrate: Auswaschen des RA-haltigen Mediums in normalem MEM Alpha Medium ohne Serum; Vereinzeln der gebildeten Zellaggregate in 2ml 0,25 % TrypsinEDTA Lösung in PBS mit 50 µg/ml DNAse (D-4527, Sigma); Abzentrifugieren mit 2 ml Serumhaltigem MEM Alpha Medium mit DNAse. 8. Zellen in 10 ml MEM Alpha Medium (10888-022, Life Technologies GmbH) mit B-27 Supplement (17504-044, Life Technologies GmbH) und 50 ng/ml NGF (13290-010, Life Technologies GmbH) aufnehmen, zählen und in einer Dichte von 20.000 Zellen/cm² auf Kultursubstrate aussäen. 2.4.2 Primärkulturen 2.4.2.1 Organotypische Hirnschnitte des Hirnstamms Schnitte des Hirnstamms von E18 Rattenembryos wurden für Primärkulturen als Reservoir für proliferierende neuronale Zellen genutzt. Neurone des Hirnstamms sind im Organismus verantwortlich für eine Reihe unterbewusst ablaufender Prozesse wie z.B. Atmung und Kontrolle des Blutdrucks. Sie zeigen daher gewöhnlich auch in Kultur spontane Aktivität sowie besonders stark ausgeprägte Natrium- und Kaliumkanäle. 2.4 Eingesetzte Zellsysteme und Kulturprotokolle 2-31 Aufbau des Gehirns Abbildung 2-10 Sagitalschnitt durch das Gehirn. Abbildung entnommen aus [Nicholls, 1995]. In Abbildung 2-10 ist ein Mediosagittalschnitt durch das Gehirn abgebildet. Darin ist der Hirnstamm mit seinen drei Unterbereichen Mittelhirn, Pons und Medulla gut zu erkennen. Die Methode der Präparation lehnt sich an ein bekanntes Kulturprotokoll von B. H. Gähwiler an. Dieses wurde erstmals 1981 veröffentlicht [Gähwiler, 1981], und in der Folge in weiteren Details dokumentiert [Gähwiler, 1984b; Gähwiler, 1990]. Um den Auswuchs von Neuronen aus dem Hirnschnitt auf die Substratoberfläche hinaus zu steigern, wurde das Protokoll bezüglich des verwendeten Kulturmediums, der Dicke der Schnitte sowie des Verfahrens der Plazierung der Schnitte auf den Substraten modifiziert. Insbesondere wurde auf eine Beschichtung der Substrate mit Zellplasma verzichtet. Anstelle dessen wurden die verwendeten Substrate entweder homogen mit Laminin beschichtet oder mit einer Lamininstruktur bestempelt (siehe Kapitel 4). Die einzelnen Schritte der Präparation laufen wie folgt ab: 2-32 2 Neuronale Zellen 1. Vollständige Extraktion des Gehirns aus 15-18 Tage alten, embryonalen CD®6 Sprague-Dawley Ratten . Die für die Experimente verwendeten Ratten stammten aus einer kontinuierlichen Verpaarung des Unternehmens Charles River Wiga Deutschland GmbH. 2. Abtrennen von Rückenmark und Pons mittels transversalem Schnitt durch den Schnabelförmigen Pons. 3. Entfernen des Kleinhirns durch Sektion des Hirnstiels. 4. Anfertigen kranzförmiger Hirnschnitte (6-12 Schnitte je Ratte) in einer Dicke von je 250 µm mit einer automatisierten Schnittmaschine (Tissue Chopper, McIlwain, England). 5. Einlegen der fertigen Schnitte in kaltem Hirnschnitt-Kulturmedium: Ham's F10 (N-1387, Sigma-Aldrich Chemie GmbH) ergänzt mit 20-25% FCS (F-7524, SigmaAldrich Chemie GmbH) und 4 mM Glutamin (G-1573, Sigma-Aldrich Chemie GmbH). 6. Lagerung für 4-5 Stunden im Inkubator (BB 6060, Heraeus Instruments GmbH) bei 37 °C, 5 % CO2 und 95 % Luftfeuchtigkeit. Während dieser Zeit lösen sich geschädigte Zellen vom Hirnschnitt ab und bleiben bei der späteren Entnahme der Schnitte im Medium zurück. 7. Vorbereitung der Kultursubstrate: Petrischalen aus Polystyrol (Greiner Labortechnik) homogen mit Laminin (1243217, Boehringer Mannheim GmbH) beschichten7, oder strukturiert bestempeln. Petrischalen mit HirnschnittKulturmedium füllen8. 8. Umbetten der Schnitte auf die Kultursubstrate: Die Schnitte einzeln mit einem kleinen, oberflächenpolierten Spatel anheben und nach Eintauchen in die mit 6 Der Stamm dieser Versuchstiere geht aus der Kreuzung einer weiblichen Wistar-Ratte mit einer männlichen Hybridratte (Hooded) hervor. Er wurde 1925 von Robert W. Dawley entwickelt. 7 Beschichtung erfolgt durch Aufbringen eines Tropfens einer PBS-Laminin-Lösung mit 25 µg/ml Laminin für die Dauer einer Stunde. 8 Kritisch ist hierbei die Wahl der richtigen Füllhöhe. Bei einer zu großen Menge an Medium schwimmt der Hirnschnitt von der Oberfläche weg. Gibt man hingegen zu wenig Medium zu, so kann der Hirnschnitt über Nacht austrocknen. Bei Petrischalen mit 3 cm Durchmesser wurden die besten Ergebnisse mit 1 ml Medium erzielt. 2.4 Eingesetzte Zellsysteme und Kulturprotokolle 2-33 Medium gefüllten Kultursubstrate durch Wegziehen des Spatels zu Boden sinken lassen. 9. Auffüllen der Kultursubstrate mit mehr Medium erst ab Tag 4-5 in Kultur. Nach dieser Zeit liegt gewöhnlich eine ausreichende Anhaftung der Hirnschnitte vor. Bis zu diesem Zeitpunkt muss die Menge an Kulturmedium täglich kontrolliert werden, um einem Austrocknen oder Aufschwimmen des Schnitts entgegenzuwirken. 10. Unterdrücken übermäßigen Wachstums nicht neuronaler Zellen, z.B. Gliazellen, an Tag 5 in Kultur: Austausch des Mediums gegen ein mit 1 µM ARA-C (C-6645, Sigma-Aldrich Chemie GmbH) angereichertes Hirnschnitt-Kulturmedium für ca. 12 Stunden9. Dieses blockiert die Zellteilung und stoppt damit die Ausbreitung von Gliazellen über das Substrat. 11. Rücktausch des Mediums gegen normales Hirnschnitt-Kulturmedium. 12. Ohne weiteren Mediumwechsel können die Hirnschnitte nun mehrere Wochen inkubiert werden. Alle Arbeitsschritte der Präparation müssen unter sterilen Bedingungen ausgeführt werden, um eine Infektion der Kultur mit Bakterien oder Pilzsporen unter allen Umständen zu vermeiden. Erfolgsentscheidend für die Präparation sind daneben insbesondere die Geschwindigkeit (maximal 45 Minuten vom Töten des Versuchstiers bis zum Einlegen der fertig präparierten Schnitte in Kulturmedium) sowie eine ausreichende Kühlung der Arbeitsfläche und der verwendeten Medien während der Präparation. In Abbildung 2-11 ist in Mikroskopieaufnahmen der typische zeitliche Verlauf dieser Hirnschnittkultur auf einem unstrukturierten (Abbildung 2-11 A) und einem mit Linien strukturierten Substrat (Abbildung 2-11 B) dargestellt. Zunächst haften die Hirnschnitte nur an der Substratoberfläche an und bleiben ca. 1-2 Tage unverändert als kompakter Zellverband erhalten (Abbildung 2-11 A1, 9 Bei sehr starkem Glia- und Fibroplastenwachstum muss die Konzentration von ARA-C auf 2 µM erhöht werden, und für mehrere Tage im Medium verbleiben. 2-34 2 Neuronale Zellen Abbildung 2-11 Die Hirnschnitte beginnen nach Tag 1-2 in Kultur mit einer starken laterale Ausbreitung. (A) Isotrope Ausbreitung des Zellverbands auf einem unstrukturierten Kontrollsubstrat (B) Geordnete Zellmigration entlang vorgegebener Wachstumspfade B1). Nach 2-3 Tagen in Kultur beginnen erste Neuronen aus dem Hirnschnitt herauszuwachsen und breiten sich in der direkten Umgebung des Gewebes aus. Dies geschieht auf dem unstrukturierten Substrat A isotrop und ungeordnet in allen Richtungen (Abbildung 2-11 A2), auf dem mit Linien strukturierten Substrat hingegen geordnet in Richtung der vorgegebenen Wachstumspfade (Abbildung 2-11 B2). Zwischen Tag 8-14 in Kultur wird die maximale Ausbreitung von Neuronen über die Substrate erreicht (Abbildung 2-11 A3, B3). Eine Reichweite zwischen ein und zwei Millimeter Radius um den Hirnschnitt wurde regelmäßig beobachtet. Nach Abschluss dieser Phase raschen Wachstums kommt die Ausbreitung der Neuronen langsam wieder zum Stillstand. Nach Tag 14 in Kultur ist in i.d.R. keine deutliche Vergrößerung der lateralen Ausbreitung mehr zu beobachten. Die bis dahin mit Neuronen besiedelten Areale 2.4 Eingesetzte Zellsysteme und Kulturprotokolle 2-35 werden durch Wachstum von Neuriten und Gliazellen langsam verdichtet, bis im ungünstigsten Fall alle Neuronen von Gliazellen und Neuriten eingehüllt sind. Auf den unstrukturierten Substraten sind in Abbildung 2-11 A2 und A3 eine große Anzahl von ungeordnet um den Hirnschnitt herum verteilten Neuronen in Form von hellen Punkten zu erkennen10. Auf dem strukturierten Substrat liegen die Neurone entlang der Linien wie an einer Perlenschnur aufgereiht (Abbildung 2-11 B2, B3). Als bester Zeitpunkt für elektrophysiologische Messungen an der beschriebenen Kultur hat sich der Zeitraum zwischen Tag 12 und Tag 16 erwiesen. In dieser Periode sind alle Ionenkanäle des Zellsystems bereits hinreichend stark ausgeprägt und die Neuronen liegen für Patch-Clamp Messungen weitestgehend frei zugänglich an der Oberfläche des Substrats. Erfolgt eine Messung zu spät, so ist ein verstärkter Überwuchs der zuvor freiliegenden Neurone zu beobachten. Elektrophysiologische Messungen können in diesem Fall nur noch mit sehr geringer Erfolgsquote durchgeführt werden. 2.4.2.2 Dissoziierte hippocampale Neuronen Eine dissoziierte Primärkultur hippocampaler Neuronen wurde verwendet, um strukturiertes Wachstum auf den Oberflächen von extrazellulären Halbleitersensoren nachzuweisen. Für diese Versuche wurde ein aus früheren Versuchen [Scholl, 2000] bekanntes Kulturprotokoll benutzt. Die einzelnen Schritte der Präparation sind: 1. Herauspräparieren der Hippocampi aus Embryos (E16-E18) einer trächtigen Sprague Dawley Ratte. 2. Inkubation des Gewebes in 0,25 % Trypsin (T-8253, Sigma-Aldrich GmbH), gelöst in Ca2+-und Mg2+-freiem HBSS (14185-045, Life Technologies GmbH) für 8 Minuten bei 37 °C. 10 Die Zellkörper der Neuronen heben sich in der mit Phasenkontrast aufgenommenen Mikroskopieaufnahme hell vom Hintergrund ab. 2-36 2 Neuronale Zellen 3. Fünffaches Abspülen der Hippocampi während 5 Minuten in 2 ml Kulturmedium, bestehend aus Neurobasalmedium (10888-022, Life Technologies GmbH) mit zusätzlichem B-27 Supplement (17504-044, Life Technologies GmbH). 4. Dissoziieren der Zellaggregate durch abwechselndes Einsaugen und Auspressen mit einer feuerpolierten Glaspipette. 5. Aussaat der dissoziierten Zellen in einer Dichte von 5000 bis 15000 Zellen/cm² auf die Oberfläche der Halbleitersensoren. 6. Inkubation bei 37 °C, 5 % CO2 und 95 % Luftfeuchtigkeit. 7. Nach 60 Minuten Kulturzeit, Waschen der Substratoberfläche mit frischem Kulturmedium, um die Oberfläche von nicht-haftenden Zellen zu säubern. 2.4.2.3 Kardiomyozyten Kardiomyozyten sind ein sehr gut untersuchtes elektrisch aktives Zellsystem. Nach einigen Tagen in Kultur bilden die Zellen in regelmäßigen Zeitabständen spontane Aktionspotentiale aus. Als Folge des durch jedes Aktionspotential ausgelösten starken Calciumeinstroms in die Zelle, kommt es zur Kontraktion von Myofibrillen in der Zelle. Das Ergebnis ist eine unter dem Mikroskop direkt zu beobachtende rhytmische Kontraktion. Kardiomyozyten bilden auf homogen mit Fibronektin (F 3667, Sigma) beschichteten Substraten eine konfluente Schicht aus. Das Cytoplasma aneinanderliegender Zellen ist über Gap-Junctions (gebildet von Transmembranproteinen des Typs Connexin Cx 43 ) leitend verbunden. Dadurch kommt es bei hinreichend dichter Aussaat der Zellen zu einer synchronen Kontraktion der gesamten Zellschicht. Insbesondere die spontane Aktivität von Herzmuskelzellen sowie die Möglichkeit zur direkten mikroskopischen Identifikation aktiver Zellen anhand ihrer Kontraktion macht das Zellsystem besonders interessant für die Erprobung neuer Messverfahren. Im Rahmen dieser Arbeit wurden Kardiomyozyten für die Erprobung einer neuen Technik zur extrazellulären Signalableitung eingesetzt. 2.4 Eingesetzte Zellsysteme und Kulturprotokolle 2-37 Die für diese Versuche verwendeten Zellen wurden aus Herzen von embryonalen Ratten (E16-E18) wie folgt präpariert [Bhatti, 1989]: 1. Herzen von 10-12 Embryonen steril entnehmen. Sammeln der Herzen in HBSS (14185-045, Life technologies GmbH). 2. Entfernen des Blutes durch mehrfaches manuelles Pumpen der Herzen mit einer Pinzette. 3. Zerkleinern des Gewebes mit einem Skalpell. 4. Sammeln der Gewebsstücke in einem Zentrifugenröhrchen und dreimaliges Waschen mit eiskaltem HBSS. 5. Ersetzen des HBSS durch 0,05 % Trypsin (T-8253, Sigma) gelöst in Fixierungspuffer (Rezeptur siehe Anhang). 6. Inkubation für 20 Minuten bei 37 °C, 5 % CO2 und 95 % Luftfeuchtigkeit. 7. Verwerfen des Überstandes. 8. Zugabe von 100 µl DNAse Typ II (D 4527, Sigma) zur weiteren enzymatischen Dissoziation des Gewebes. Nach 2 Minuten Wartezeit werden weitere 2,5 ml Trypsinlösung (0,025 % in Fixierungspuffer) hinzugefügt. 9. Inkubation für 20 Minuten. 10. Sammeln des Überstandes, welcher nun vereinzelte Zellen enthält und überführen in ein Medium aus HEPES Puffer mit 0,5 % Insulin-, Transferrin-, Selenit-Lösung (1884, Sigma) und 25 % FCS (F-7524, Sigma). 11. Zentrifugieren der Zellsuspension bei 1500 min-1 für 5 Minuten. Anschließend Überstand verwerfen. 12. Resuspension des Sediments aus noch nicht dissoziiertem Gewebe in eiskaltem Ham's F10 (21955-018, Life Technologies GmbH) mit 0,5 % ITS und 10% FCS. 13. Wiederholung der Schritte 8 -12 bis alles Gewebe dissoziiert ist. 14. Sammeln der endgültigen Zellsuspension (aus Schitt 10) und Inkubation in Zellkulturflaschen für 30 Minuten. Da Fibroblasten schneller adhärieren als 2-38 2 Neuronale Zellen Kardiomyozyten, erhält man anschließend beim Ausgießen eine Suspension mit einer erhöhten Anzahl Kardiomyozyten. 15. Einstellen der gewünschten Zellkonzentration durch Zählen in einem Hämycytometer und entsprechende Verdünnung mit weiterem Medium. Mit Zelldichten von 20.000 bis 40.000 Zellen/cm² wurden die besten Ergebnisse erzielt. 2.5 Protokolle zur Fluoreszenzfärbung 2-39 2.5 Protokolle zur Fluoreszenzfärbung Eine exakte morphologische Identifikation von neuronalen Zellen sowie ihrer Axone und Dendriten ist auch mit modernen Phasenkontrastoptiken im Lichtmikroskop sehr schwierig und häufig ungenau. Anhand einiger Merkmale11 ist es mit etwas Übung zwar möglich neuronale Zellen von Gliazellen zu unterscheiden, jedoch ist insbesondere in Kulturen mit hoher Zelldichte eine Unterscheidung von Axonen und Dendriten sowie deren Zugehörigkeit zu einzelnen Neuronen im Phasenkontrastbild nicht mehr möglich. Hat man ein Fluoreszenzmikroskop zur Verfügung, so können bestimmte Zelltypen, einzelne Zellen, oder funktionaler Einheiten wie Axone oder Dendriten gezielt mit einem Fluoreszenzmarker angefärbt werden. Unter dem Fluoreszenzmikroskop heben sich die markierten Bereiche dann kontraststark vom Hintergrund ab. Für Experimente dieser Art wurden zwei verschiedene Verfahren der Floreszenzfärbung eingesetzt: 1. Zur Identifikation von neuronalen Zellen sowie deren Axone und Dendriten, wurden Zellsubstrate mit fluoreszenzmarkierten Antikörpern gefärbt. Dieses Verfahren wird als funktionale Antikörperfärbung bezeichnet, da die eingesetzten Antikörper gegen spezifische, funktionale Proteine der Zellen gerichtet sind. 2. Parallel zu elektrophysiologischen Messungen an synaptisch verschalteten Zellen, wurden die exakten Pfade der elektrischen Verbindung durch Mikroinjektion von Farbstoffen in das Zell Cytoplasma sichtbar gemacht. Im Folgenden sind beide Methoden mit den exakten Protokollen beschrieben. Die erzielten Ergebnisse werden in späteren Kapiteln der Arbeit diskutiert. 11 Neurone: Großer Zellkern, erhabener Zellkörper, lange Fortsätze in zylindrischer Form; Unterscheidung eines langen Axons von vielen stark verzweigten Dendriten. Gliazellen: Polygonale Form, flacher Zellkörper, Auswüchse radialsymmetrisch und stark verzweigt. 2-40 2 Neuronale Zellen 2.5.1 Funktionale Antikörperfärbung Antikörper wurden eingesetzt, um in den verwendeten Zellsystemen neuronale Zellen und Gliazellen zu identifizieren. Neuronale Zellen wurden dabei mit einem Antikörper gegen das Mikrotubuli assoziierte Protein 2 (kurz MAP 2) markiert [Caceres, 1984], Gliazellen mit einem Antikörper gegen das Glial Fibrillary Acidic Protein (kurz GFAP) [Debus, 1983]. Weiterhin wurde versucht, neuronale Polarität durch spezifische Anfärbung von Axonen und Dendriten auf strukturierten Substraten nachzuweisen. Dendriten wurden dabei mit anti-MAP 2 markiert, Axone mit einem Antikörper gegen das Mikrotubuli assoziierte Protein Tau (kurz Tau) [Mandell, 1996]. Alle eingesetzten Antikörper gelten als weitreichend erprobt und sind wissenschaftlich anerkannt [Banker, 1998]. Zur Absicherung der Ergebnisse wurden bei jeder Färbung jeweils zwei Isotypkontrollen mitgeführt, um eine unspezifische Färbung ausschließen zu können. Die verwendeten Protokolle stützen sich auf Angaben in früheren Arbeiten [Berger, 1994; Berger, 1997; Jostock, 1998]. 2.5.1.1 MAP 2 Die selektive Anfärbung von neuronalen Zellen und ihrer Dendriten erfolgte in drei Schritten: Zuerst wurden die Zellen fixiert und permeabilisiert. Dann wurde das spezifisch in Neuronen exprimierte Protein MAP 2 mit einem primären MausAntikörper markiert. Im letzten Schritt wurde ein Cy-3 fluoreszenzmarkierter Sekundärantikörper an den Primärantikörper gebunden. − Aufbereiten der Zellen in CS-Puffer12 (siehe Rezeptur im Anhang): 1. Zweimaliges Waschen der Zellsubstrate mit vorgewärmten CS-Puffer. 2. Fixieren bei Raumtemperatur in 3,7 % Paraformaldehyd (PFA) in CS-Puffer während 15 Minuten. 3. Dreimaliges Waschen mit CS-Puffer. Insgesamt 10 Minuten. 4. Permeabilisieren mit 0,5 % Triton-X-100 (T-9284, Sigma) in PBS für 5 Minuten. 12 Alternativ kann auch in allen Schritten PBS anstelle von CS-Puffer verwendet werden. 2.5 Protokolle zur Fluoreszenzfärbung 2-41 5. Falls Substrate aus mehreren Versuchen für eine gemeinsame Färbung gesammelt werden sollen, Substrate jetzt in CS-Puffer bei 4 °C einlagern. − Markierung mit dem Primärantikörper Maus anti-MAP 2 (MAB 3418, Chemicon) mit einer Konzentration des Antikörpers von 10 µg/ml in Verdünnungspuffer (Rezept siehe Anhang): 1. Aufpipettieren von 100 µl Antikörperlösung auf die Substrate. 2. Abdecken der Substrate mit einem Deckglas (22x22 mm). 3. Inkubation für 40 Minuten bei 37 °C, 5 % CO2 und 95 % Luftfeuchtigkeit. 4. Dreimaliges Auswaschen der Antikörperlösung mit PBS. Insgesamt 10 Minuten. − Applikation des Cy-3 markierten Sekundärantikörpers F(ab)2-Esel anti Maus (715096-150, Dianova) analog zum Vorgehen bei dem Primärantikörper. Auf die fertig gefärbten Präparate wurden 100 µl PBS aufpipettiert, und der Tropfen mit einem Deckglas abgedeckt13. Zur Auswertung wurden die Substrate unter einem Fluoreszenzmikroskop betrachtet. 2.5.1.2 Tau Axonale Färbungen mit dem Marker Kaninchen anti-Tau (T 6402, Sigma) sowie dem FITC markierten Sekundärantikörper F(ab)2-Esel anti Kaninchen (711-166-152, Dianova) wurden nach exakt dem gleichen Protokoll wie MAP 2 Färbungen durchgeführt. Bei Einzelfärbung ohne zusätzliche MAP 2 Markierung kann allerdings der Triton-X-100 Permeabilisierungsschritt entfallen. Die Verdünnungen der verwendeten Antikörper waren: − anti-Tau: 1:100 − F(ab)2-Esel anti Kaninchen: 1:100 13 Alternativ kann auch eine Langzeitversiegelung in Eindeckmittel (Rezept siehe Anhang) erfolgen. 2-42 2.5.1.3 2 Neuronale Zellen GFAP Auch die Markierung von Gliazellen mit dem Marker Maus anti-GFAP (814 369, Roche Diagnostics GmbH) erfolgte nach dem Protokoll der MAP 2 Färbung. Anstelle der Triton-X-Permeabilisierung erfolgte ein zusätzlicher Fixierschritt in Eisessig. Dazu wurden die bereits mit PFA fixierten Proben für 5 Minuten bei –20 °C mit 100 µl Eisessig überschichtet und anschließend mit PBS gewaschen. Der primäre Antikörper der Färbung, anti-GFAP wurde in einer Konzentration von 2 µg/ml eingesetzt. Als sekundärer Antikörper wurde analog zur anti-MAP 2 Färbung der Sekundärantikörper F(ab)2-Esel anti Maus (715-096-150, Dianova) verwendet. 2.5.2 Färbung durch Mikroinjektion Bei intrazellulären Patch-Clamp Messungen erfolgt die Messung von Ionenströmen über eine mit einer Elektrolytlösung gefüllten Mikropipette. Diese Pipette wird an die Membran der untersuchten Zelle geführt und die Zellmembran durch leichten Unterdruck in der Pipette geöffnet. Während der Messung besteht also eine Verbindung des Zellcytoplasmas zur Elektrolytlösung in der Patch-Pipette. Durch Einbringen eines Fluoreszenzfarbstoffs in die Elektrolytlösung kann folglich das Zellcytoplasma angefärbt werden. Verwendet man geeignete polare Farbstoffe, die aufgrund ihrer Polarität nicht in der Lage sind aus der Zelle durch die Zellmembran hinauszudiffundieren, so kann man auf diese Weise eine kontrastreiche Anfärbung des Zellcytoplasmas erreichen. Der Farbstoff gelangt dabei rein diffusiv in die Zelle. Die Anfärbung kann aber auch entlang eines Spannungsgradienten beschleunigt werden. Dazu werden mit dem Patch-Clamp Verstärker Hyperpolarisationspulse an die Zelle angelegt. Eine ganze Reihe polarer Fluoreszenzfarbstoffe für diese Anwendung sind kommerziell erhältlich. Der wohl populärste Farbstoff unter ihnen ist Lucifer Yellow. 2.5 Protokolle zur Fluoreszenzfärbung 2-43 Abbildung 2-12 Verwendete Fluoreszenzfarbstoffe für die Anfärbung von Zellen durch Mikroinjektion, zusammen mit jeweils einer charakteristischen Färbung eines Neurons. Die gefärbten Neurone stammen aus der oben beschriebenen Kultur organotypischer Hirnschnitte des Hirnstamms von E18 Rattenembryos. (A) Sulforhodamine (B) Lucifer Yellow (C) Cascade Blue Um gleichzeitig bis zu drei Zellen getrennt färben zu können, wurden in dieser Arbeit neben Lucifer Yellow noch zwei weitere polare Farbstoffe, Sulforhodamine und Cascade Blue verwendet. In Abbildung 2-12 sind alle drei Farbstoffe mit den zugehörigen Strukturformeln sowie einer exemplarischen Fluoreszenzaufnahme dargestellt. Die abgebildeten Färbungen stammen von einer neuronalen Kultur aus organotypischen Hirnschnitten des Hirnstamms von E18 Rattenembryos. In allen drei Aufnahmen ist die mit Fluoreszenzfarbstoff gefüllte Mikropipette als hell leuchtendes spitz auf die Zelle zulaufende Spitze deutlich zu erkennen. Über die Visualisierung einzelner Zellen hinaus, können durch Mikroinjektion auch Verbindungen des Cytoplasmas zu benachbarten Zellen (gebildet durch GapJunctions) detektiert werden [Steward, 1978; Eckert, 1993]. 2-44 2 Neuronale Zellen Ziel der in dieser Arbeit durchgeführten Färbungen durch Mikroinjektion war eine Visualisierung und Vermessung der Pfade von Axonen und Dendriten an elektrophysiologisch untersuchten Zellen. Bei einer elektrophysiologisch detektierten elektrischen Verbindung zwischen zwei benachbarten Neuronen kann auf diesem Weg der Pfad der Signalübertragung genau identifiziert und seine Länge bestimmt werden. Das in den Versuchen eingesetzte Protokoll zur Färbung durch Mikroinjektion lehnt sich an Angaben aus früheren Veröffentlichungen an [Traub, 1994; Eckert, 1999] und umfasst folgende Schritte: − Herstellung angefärbter intrazellulärer Lösungen: 1. Jeweils 5 mg der Farbstoffe Lucifer Yellow (L-435, Molecular Probes), Sulforhodamine (S-359, Molecular Probes) und Cascade Blue (C-687, Molecular Probes) in 20 ml intrazellulärer Patch-Clamp Lösung (Rezept siehe Anhang) auflösen. 2. Ungelöste Schwebstoffe durch Filtern der Lösungen mit einem Mikrofilter (1,25 µm Porenweite) entfernen. − Füllen der Mikropipetten mit Farbstoff: 1. Mikropipetten durch Injektion von ungefärbter, intrazellulärer Patch-Clamp Lösung wie bei einem gewöhnlichen elektrophysiologischen Experiment füllen. 2. In einem zweiten Schritt wenige µl der gefärbten Lösung mit einer dünnen Kapillare von hinten ca. 1mm hinter die Spitze der Pipette injezieren. Wichtig hierbei ist, dass die gefärbte Lösung nicht ganz in die Spitze gelangt. Nur so kann eine Verteilung von Farbstoff in das Kulturmedium um die untersuchten Zellen herum vermieden werden, da der Farbstoff erst nach einiger Zeit an die Öffnung der Pipette diffundiert. − Einfärben der untersuchten Zellen: 2.5 Protokolle zur Fluoreszenzfärbung 2-45 1. Nach Eröffnung einer Zelle für die elektrophysiologische Messung (siehe hierzu Kapitel 3) diffundiert der Fluoreszenzfarbstoff durch die Pipettenspitze in die Zelle. 2. Ca. 5-10 Minuten nach Zellöffnung ist ausreichend Farbstoff in der Zelle vorhanden, um den Zellkörper und die Neuriten im Fluoreszenzmikroskop zu beobachten. Eine Beschleunigung der Anfärbung ist durch Anlegen von Spannungsgradienten seitens des Patch-Clamp Verstärkers möglich. Dies ist jedoch in der Regel nicht erforderlich, da sich während der Durchführung elektrophysiologischer Messungen eine ausreichende Wartezeit ergibt. 3. Der Beleuchtungsstrahlengang des Fluoreszenzmikroskops darf nur so kurz wie möglich und erst unmittelbar zum Zeitpunkt der Fluoreszenzmessung geöffnet werden. Durch photochemische Reaktion des angeregten Farbstoffs mit Sauerstoff entstehen in der gefärbten Zelle freie Radikale, die zytotoxisch wirken und die Zelle von innen zerstören können. Aus diesem Grund wurden elektrophysiologische Messung ausschließlich vor Beginn der Fluoreszenzmikroskopie durchgeführt. 2-46 2 Neuronale Zellen 3-1 3 Die Patch-Clamp Technik Seit Entwicklung der Patch-Clamp Technik durch Erwin Neher und Bert Sakmann im Jahre 1976 [Neher, 1976], steht der Wissenschaft ein präzises Messverfahren für die Untersuchung der elektrophysiologischen Eigenschaften von elektrisch aktiven Zellen und deren Ionenkanälen zur Verfügung. Im Jahre 1991 erhielten die beiden Wissenschaftler dafür den Nobelpreis. Je nach gewählter Konfiguration, ist es mit der Patch-Clamp Technik möglich, die Elektrophysiologie ganzer Zellen (Whole-Cell Konfiguration) oder sogar einzelner Ionenkanäle in der Zellmembran (Single-Channel Konfiguration) zu messen. In der Whole-Cell Konfiguration wird mittels einer Glaselektrode eine leitende Verbindung zum Cytoplasma der untersuchten Zelle hergestellt. Die elektrischen Eigenschaften der gesamten Zellmembran werden dadurch messtechnisch zugänglich. Bei der Single-Channel Konfiguration wird die Glaselektrode nur auf die Zellmembran aufgesetzt (Cell-attached Konfiguration), oder es wird mit der Pipette ein Membranstück aus der Zellmembran herausgelöst (Patch Konfiguration). In beiden Fällen wird die Pipettenöffnung von einem intakten Membranfragment überspannt und elektrisch von der Umgebung isoliert. Somit tragen nur einzelne Ionenkanäle zum Messignal bei, welche sich in dem Membranfragment über der Pipettenöffnung befinden. In dieser Arbeit wurde die Patch-Clamp Technik auschließlich in der Whole-Cell Konfiguration verwendet. Mit dem Verfahren wurden sowohl Ionenströme und Aktionspotentiale einzelner neuronaler Zellen gemessen als auch synaptische Kopplungen zwischen zwei Neuronen elektrisch charakterisiert. Insbesondere für die Interpretation der Messignale der untersuchten synaptischen Kopplungen ist ein genaues Verständnis des Messverfahrens selbst sowie darüber hinaus der Funktionsweise aller elektronischen Verstärkerkomponenten des verwendeten Messaufbaus erforderlich. Im Folgenden wird die Whole-Cell 3-2 3 Die Patch-Clamp Technik Konfiguration im Detail beschrieben und die Signalverarbeitung im Patch-Clamp Verstärker anhand von Blockschaltbildern erläutert. 3.1 Whole-Cell Konfiguration 3-3 3.1 Whole-Cell Konfiguration Für die Durchführung einer Patch-Clamp Messung in Whole-Cell Konfiguration, wird das Cytoplasma der untersuchten Zelle mit einer Glaselektrode leitend kontaktiert. Die Leitfähigkeit der Glaselektrode wird durch vorherige Füllung mit einer ionenleitenden, intrazellulären Elektrolytlösung hergestellt. Die untersuchte Zelle selbst befindet sich in einem Bad mit extrazellulärer Elektrolytlösung. So entsteht ein geschlossener Stromkreis zwischen Glaselektrode und Bad über die Zellmembran der kontaktierten Zelle hinweg. In das Bad und in die Glaselektrode sind chlorierte Siberdrähte eingetaucht, an welche schließlich der Patch-Clamp Verstärker angeschlossen ist. Whole-Cell Konfiguration 2 Zellberührung mit Gigaseal 3 Zellöffnung und Messung Pipette Pipette Zelle Zelle Zelle RPipette RPipette 1 Annäherung an die Zelle (A) Schematische Zeichnung Pipette RKanal RMembran bran CMem- UMembran Abbildung 3-1 Arbeitsschritte zur Durchführung einer Patch-Clamp Messung in Whole-Cell Konfiguration. (A) Schematische Darstellung des Systems aus Pipette und Zelle. (B) Typische Messignale vor und nach erfolgter Kompensation. (C) Elektrische Ersatzschaltbilder der einzelnen Konfigurationen. RKanal RAbdicht RAbdicht UOffset CPipette RPipette RAbdicht CPipette - Kompensiert RPipette + Schritt 1 Schritt 2 - Kompensation UMembran - - Unkompensiert + - RPipette RZugang + (C) Elektrisches Ersatzschaltbild CPipette (B) Messignal - Testpuls - Signal - Signal, kompensiert 3-4 3 Die Patch-Clamp Technik Um das Innere einer Zelle zu kontaktieren und zu der oben beschriebenen Messkonfiguration zu gelangen, sind mehrere vorbereitende Schritte erforderlich. Parallel zur mechanischen Herstellung der Konfiguration, erfolgt dabei auch eine elektrische Kompensation der Messchaltung um störende Artefakte. In Abbildung 3-1 sind die einzelnen Schritte zusammen mit den typischen Messignalen sowie einem elektrischen Ersatzschaltbild der jeweiligen Konfiguration dargestellt: 1. Im ersten Schritt wird die Glaselektrode (ab hier kurz Pipette genannt) mit 1 intrazellulärer Lösung gefüllt, mit einem leichten Überdruck versehen, in die extrazelluläre Badlösung eingetaucht und schließlich in unmittelbare Nähe der Zelle gebracht (Abbildung 3-1 A1). Ein elektrischer Stromkreis zwischen Pipettenöffnung und Badlösung entsteht. Bei Anlegen eines schwachen Spannungspulses von wenigen mV zwischen Pipette und Bad wird ein zur Pulshöhe proportionaler Strom (Abbildung 3-1 B1) über den Innenwiderstand der Pipette RPipette (Abbildung 3-1 C1) beobachtet. Der Stromfluss über die Pipettenkapazität CPipette ist dagegen vernachlässigbar klein. Das gemessene Stromsignal ist allerdings um einen Offsetanteil gegenüber der Nullinie verschoben. Die Spannung UOffset resultiert im wesentlichen aus der elektrochemischen Potentialdifferenz zwischen den chlorierten Siberdrähten und den extrazellulären und intrazellulären Elektrolytlösungen sowie aus unkompensierten Offsetkomponenten des Messverstärkers. Beide Effekte können in guter Näherung über die gesamte Messzeit als konstant angenommen 2 werden. Sie werden daher einmalig elektronisch kompensiert . Nach erfolgreicher Kompensation verläuft der gemessene Strom entlang der Nullinie (Abbildung 3-1 B1). 1 So wird durch ausströmende intarzelluläre Lösung eine Verschmutzung der Pipettenöffnung beim Eintauchen in die extrazelluläre Badlösung verhindert. 2 Je nach Zusammensetzung der intarzellulären und extrazellulären Lösungen, muss nach Eröffnen der Zellmembran manuell eine weitere Offsetkorrektur vorgenommen werden, um die in der automatischen Kompensation enthaltene Kontaktspannung zwischen intra- und extrazellulärer Lösung wieder zu entfernen [Neher, 1992]. Der Kontakt der beiden Lösungen wird in der endgültigen Messkonfiguration nämlich durch die Zellmembran unterbrochen. Entsprechende Tabellenwerte für die Korrektur können der Literatur entnommen werden [Schulze, 1999]. Bei den durchgeführten Messungen wurde entsprechend der verwendeten Lösungen ein Korrekturwert von -10 mV berücksichtigt. 3.1 Whole-Cell Konfiguration 3-5 2. Im zweiten Schritt wird die Pipette bis an die Zellmembran herangeführt (Abbildung 3-1 A2), und es wird der zuvor angelegte Überdruck aus der Pipette abgelassen. Es bildet sich ein dichter Kontakt zwischen Pipettenöffnung und Zellmembran. War die Abdichtung erfolgreich, so bildet sich ein Abdichtwiderstand RAbdicht von mehreren Gigaohm (Gigaseal) und der Stromfluss (Abbildung 3-1 B2) verschwindet fast vollständig. Lediglich der kapazitive Ladeund Entladestrom über die Pipettenkapazität CPipette wird weiterhin beobachtet (Abbildung 3-1 B3). Dieser wird nun ebenfalls elektronisch kompensiert3, bis der gemessene Strom nicht mehr von der Nullinie abweicht. 3. Im letzten Schritt wird durch kontrolliertes Anlegen von Unterdruck4 an die Pipette die Zellmembran geöffnet. Eine leitende Verbindung zum Zellinneren ist hergestellt (Abbildung 3-1 A3). Der Ersatzschaltkreis dieser Konfiguration (Abbildung 3-1 C3) umfasst nun auch den in Serie zum Pipettenwiderstand liegenden Zugangswiderstand RZugang und alle Membraneigenschaften der untersuchten Zelle: Die Kapazität der Zellmembran CMembran , das Ruhepotential der Zelle UMembran , den festen Leckwiderstand der Membran RMembran sowie eine variable Widerstandskomponente RKanal , welche sich aus der Leitfähigkeit der schaltbaren Ionenkanäle in der Zellmembran zusammensetzt. Der Abdichtwiderstand RAbdicht ist gestrichelt gezeichnet, da er aufgrund seiner Große gegenüber den anderen Komponenten vernachlässigt werden kann. Betrachtet man wieder den gemessenen Strom als Antwort auf einen schwachen SpannungsTestpuls (Abbildung 3-1 B3), so erkennt man den Stromfluss durch die hinzugekommenen Ohm'schen und kapazitiven Komponenten im Messignal. In zwei weiteren Kompensationsschritten ist es möglich, zuerst den Stromfluss durch das RC-Glied aus den Zugangswiderständen RPipette , RZugang und der Membrankapazität CMembran aus dem Messignal zu eliminieren, und anschießend auch den Einfluss des Leckwiderstandes RMembran zu kompensieren. Stellt man die Messpannung des Patch-Clamp Verstärkers nun auch noch exakt auf die 3 Siehe hierzu Punkt 3.2.1. 4 Dies geschieht durch leichtes Saugen an einem mit der Pipette verbundenem Silikonschlauch. 3-6 3 Die Patch-Clamp Technik Membranspannung ein, so verschwindet der Stromfluss wieder vollständig (Abbildung 3-1 B3). Die Whole-Cell Konfiguration ist nun vollständig hergestellt. Durch Anlegen gezielter Stimulationspulse, ist es jetzt möglich, das elektrische Verhalten der Ionenkanäle in der Zellmembran als Änderung der Leitfähigkeit der Komponente RKanal zu messen. Um die aufwendigen Kompensationsschritte des Messverfahrens überprüfen zu können, wurde ein Testschaltkreis mit bekannten elektronischen Komponenten verwendet. Der Testschaltkreis simuliert die in Abbildung 3-2 A dargestellte Konfiguration einer Zelle in Badlösung, welche in Whole-Cell Konfiguration mit einer Pipette kontaktiert wird. Abbildung 3-2 B zeigt ein Photo des Bauteils. Es wird über einen BNC-Stecker an Testschaltkreis - einzelne Zelle (A) Beschriebenes System • passive Zelle – keine Ionenkanäle – kein Membranpotential (B) Bauteil Pipette Zelle (C) Schaltplan – – 1: Pipette im Bad 2: Pipette an Zelle (Gigaseal) 3: Zelle geöffnet 10 MΩ Ω 123 0.5 GΩ Ω – 6 pF • Modi 22 pF 5.1 MΩ Ω Abbildung 3-2 Testschaltkreis zur Überprüfung des Kompensationsverfahrens des Patch-Clamp Verstärkers. (A) Schematische Darstellung des beschriebenen Systems aus Pipette, Zelle und Badlösung. (B) Photographie des Bauteils. (C) Schaltplan. 3.1 Whole-Cell Konfiguration 3-7 den Eingang des Patch-Clamp Verstärkers angeschlossen. Eine Verbindung zur Masse des Verstärkers wird mit einer zusätzlichen Erdungsleitung hergestellt. Mit dem Schalter auf der Gehäuseoberseite kann der in Abbildung 3-2 C gezeigte Schaltplan zwischen drei Konfigurationen umgeschaltet werden: 1. "Pipette im Bad" (CPipette = 6 pF, RPipette = 10 MΩ) 2. "Gigaseal" (CPipette = 6 pF) 3. "Zelle geöffnet" (CPipette = 6 pF, RZugang = 5,6 MΩ, CMembran = 22 pF, RMembran = 0,5 GΩ) Referenzmessungen haben ergeben, dass bei der Signalkompensation des Patch-Clamp Verstärkers alle Komponenten des Testschaltkreises mit einer Genauigkeit von besser als 1 % bestimmt werden. 3-8 3 Die Patch-Clamp Technik 3.2 Voltage-Clamp und Current-Clamp Messverfahren Mit einem Patch-Clamp Verstärker lassen sich zwei unterschiedliche Messschaltungen zur Untersuchung der elektrischen Membraneigenschaften von Zellen realisieren. Im Voltage-Clamp Modus (kurz VC-Modus) wird die Spannung über der Zellmembran vom Patch-Clamp Verstärker fest vorgegeben und gleichzeitig der Strom durch die Membran gemessen (Abbildung 3-3 A). Im CurrentClamp Modus (kurz CC-Modus) ist die Membranspannung die Messgröße und der Strom wird gezielt kontrolliert (Abbildung 3-3 B). Beide Messverfahren sind für die Charakterisierung der elektrophysiologischen Eigenschaften neuronaler Zellen von Nutzen: − Der VC-Modus wird verwendet, um gezielt die Stärke von Ionenströmen über die Zellmembran bei fest vorgegebenen Membranspannungen zu untersuchen. Anhand des zeitlichen Verlaufs des gemessenen Stromsignals, können Beiträge spezifischer Ionenkanäle im Signal identifiziert werden. Im dargestellten Beispiel in Abbildung 3-3 A wurde ein primär isoliertes Neuron aus dem Hirnstamm eines Rattenembryos, ausgehend von einer Haltespannung von -50 mV, mit Spannungspulsen von 1: +20 mV, 2: +50 mV und 3: +100 mV stimuliert. Wie in Kapitel 2 bereits theoretisch beschrieben, tritt bei schwacher Depolarisation mit +20 mV zunächst nur ein negativer, in die Zelle gerichteter Stromfluss von Na+-Ionen auf, welcher schnell abklingt. Bei starker Depolarisation mit +100 mV dominiert hingegen ein positiver, auswärts gerichteter Strom von K+-Ionen. Dieser klingt, anders als der Na+-Strom, nicht durch Inhibition der Kanäle ab sondern bleibt über die gesamte Dauer des Depolarisationspulses bestehen. Anhand des gemessenen Stromsignals ist es folglich möglich, Rückschlusse auf Typ und Häufigkeit bestimmter Ionenkanäle in der Zellmembran zu ziehen. 3.2 Voltage-Clamp und Current-Clamp Messverfahren 3-9 Voltage-Clamp und Current-Clamp Modus (A) Voltage-Clamp (VC) • Kontrolle der Spannung • Messen des Stroms – Messsignal Strom [pA] 20 0 2 -20 1 -40 -60 3 2 3 1 2 0 1 -1 -2 0 5 10 15 20 25 30 35 40 I • Kontrolle des Stroms • Messen der Spannung 3 40 Strom [nA] – Stimulationspuls Spannung [mV] 60 Beispiele I (B) Current-Clamp (CC) U 120 100 80 60 40 20 0 -20 Spannung [mV] Messverfahren U 40 20 Aktionspotential 0 -20 Schwellenwert -40 -60 -80 Zeit [ms] 0 5 10 Zeit [ms] 15 20 Abbildung 3-3 Patch-Clamp Messungen im Voltage-Clamp und Current-Clamp Modus. (A) Voltage-Clamp (VC). (B) Current-Clamp (CC). − Der CC-Modus greift idealerweise garnicht in die elektrophysiologische Eigendynamik der Zelle ein. Das Membranpotential der Zelle kann sich frei bewegen. In dieser Konfiguration werden spontane Aktionspotentiale oder postsynaptisch induzierte Potentialänderungen detektiert. Depolarisiert man im CC-Modus die Zelle von aussen durch einen einwärts gerichteten, positiven Strompuls, so wird ein Aktionspotential ausgelöst. Die Aufnahme des Aktionspotentials in Abbildung 3-3 B, erfolgte nach Umschalten aus dem VC-Modus in den CC-Modus am gleichen Neuron wie die Messung im VC-Modus in Abbildung 3-3 A. Der angelegte Strompuls von +100 pA depolarisiert zunächst langsam die Zellmembran durch Aufladen der Membrankapazität, bis bei Erreichen eines Schwellenwerts von -25 mV die + Na -Kanäle öffnen und sich ein Aktionspotential ausbildet. Im VC-Modus arbeitet der Patch-Clamp Verstärker als Spannungsquelle, im CCModus als Stromquelle. Für eine möglichst genaue Messung muss das Verhalten 3-10 3 Die Patch-Clamp Technik a b c Voltage-Clamp Schaltung x Addierer: x = a+b+c 2 Kompensation 1 Stimulation (A) Eingang + UStim a + b - x Komparator: x = (a-b)*∞ 4 Signalbereinigung 3 UOffset 1 1 + R 2 3 + 2 + 6 (C) Fehlerkompensation (D) Ausgang Subtrahierer: x = a-b Zelle Haltespannung UHalte x 3 Messung / U-I-Wandlung Offsetspannung Stimulationsspannung (B) Signalverarbeitung a + b - 4 + -4 - 5 7 5 Korrekturschaltkreis Spannungsmonitor Strommonitor Abbildung 3-4 Blockschaltbild des Patch-Clamp Verstärkers im Voltage-Clamp Modus. (A) Signaleingang. (B) Signalverstärkung und -verarbeitung. (C) Fehlerkompensation des Messignals um kapazitive und Ohm'sche Störgrößen. (D) Ausgabe der Signale. des Messverstärkers dabei dem einer idealen Spannungsquelle (mit Innenwiderstand Null), bzw. einer idealen Stromquelle (mit Innenwiderstand unendlich) entsprechen. Beide Anforderungen in einer einzigen Verstärkerschaltung zu realisieren ist nicht trivial und nur durch den Einsatz spezieller Operationsverstärker hoher Güte5 möglich. 3.2.1 Signalverarbeitung und Kompensation - Voltage-Clamp In Abbildung 3-4 ist der Schaltplan des in dieser Arbeit verwendeten Patch-Clamp Verstärkers (EPC-9/3, HEKA Elektronik) für den Betrieb im VC-Modus als Blockschaltbild dargestellt. In der Beschreibung werden im Folgenden vier Ebenen 5 7 Möglichst große Differenzverstärkung (≥ 10 ) bei gleichzeitig maximaler Unterdrückung unerwünschter -7 Gleichtaktverstärkung (≤ 10 ). 3.2 Voltage-Clamp und Current-Clamp Messverfahren 3-11 A bis D des Signalflusses, und vier Schritte 1 bis 4 der Signalverarbeitung unterschieden. Die Eingangssignale des Verstärkers aus Ebene A (Abbildung 3-4 A) werden in Ebene B (Abbildung 3-4 B) verarbeitet. Die Fehlerkompensation, beispielsweise um die kapazitive Störgröße der Zellmembran, erfolgt in Ebene C (Abbildung 3-4 C) und wird in die Ebene B der Signalverarbeitung zurückgekoppelt. In Ebene D (Abbildung 3-4 D) werden die Messignale Membranspannung und Membranstrom über die Anschlüsse von Spannungs- und Strommonitor ausgegeben. Das Prinzip der Signalverarbeitung wird wie folgt entlang der Schritte 1 bis 4 unter Zuhilfenahme der eingezeichneten Messpunkte beschrieben. Mit Ui wird dabei die Spannung am Messpunkt i bezeichnet. Ii bezeichne den in Pfeilrichtung fließenden Strom: 1. Das Stimulationssignal UStim und die Haltespannung UHalte aus Ebene A werden in Ebene B im Addierer 1 addiert. Die Summe der Signale U1 = UStim + UHalte wird an den Spannungsmonitor in Ebene D ausgegeben und gleichzeitig an den zweiten Addierverstärker als Vorgabewert der einzustellenden Membranspannung weitergegeben. 2. Durch Addition von Korrekturspannungen zum Vorgabewert U1 , berücksichtigt der Addierer 2 Korrekturen für messtechnisch bedingte Signalfehler. Er bildet die Summe aus der in Ebene A eingestellten Offsetspannung UOffset , dem Vorgabewert für die Membranspannung U1 sowie einer Korrekturspannung U6 (siehe Punkt 3.2.2): U2 = UOffset + U1 + U6 . 3. Herzstück der gesamten Verstärkerschaltung ist der als Strom-SpannungsWandler betriebene Operationsverstärker 3. Sein Arbeitspunkt wird über die Spannung U2 an seinem nicht-invertierenden Eingang kontrolliert. Durch die Rückkopplung des Ausgangs über Widerstand R an den invertierten Signaleingang, liegen beide Eingänge des Verstärkers immer auf dem gleichem Potential. Es gilt daher U3 = U2 . Somit folgt mit U3 das Potential der PatchClamp Pipette in Ebene A immer der vorgegebenen Spannung U2 . Wenn Spannungsverluste in der Pipette durch in Schritt 2 addierte 3-12 3 Die Patch-Clamp Technik Kompensationsignale ausgeglichen werden, folgt das Potential in der Zelle exakt dem in Schritt 1 vorgegebenen Stimulationssignal U1 . Fließt bei einer vorgegebenen Spannung U1 nun ein Strom I3 aus oder in die Zelle, so wird dieser über den Referenzwiderstand R zu Punkt 4 geleitet. Dadurch kommt es zu einem dem Strom proportionalen Sannungsabfall über R (∆U = R * I3). Damit das durch die Schaltung vorgegebene Gleichgewicht beider Eingänge des Operationsverstärkers nicht verletzt wird, regelt dieser seine Ausgangsspannung U4 soweit nach, bis beide Eingänge wieder auf gleichem Potential liegen. Der volle Spannungsabfall an R schlägt sich dadurch als Potentialänderung U4 an Punkt 4 nieder. Der Stom I3 wird mit der beschriebenen Schaltung also in ein zu I3 proportionales Spannungssignal U4 verwandelt. Proportionalitätskonstante ist der Widerstandswert von R . 4. Ohne weitere Bearbeitung ist dem Stromsignal R * I3 in der Spannung U4 noch die Spannung U2 überlagert. Es gilt U4 = U2 + R * I3 . Um die eigentliche Messgröße R * I3 zu isolieren, wird in Subtrahierer 4 U2 von U4 abgezogen. Das bereinigte Signal U5 = R * I3 ist dann dem gesuchten Messwert I3 direkt proportional und wird am Stommonitor in Ebene D ausgegeben. Weitere Signalkorrekturen (siehe Punkt 3.2.2) fließen über U7 am dritten Eingang des Subtrahierers 4 in das Ergebnis ein. Wie zuvor bereits angemerkt, arbeitet der Operationsverstärker in Schritt 3 aus Sicht der Zelle im VC-Modus wie eine Spannungsquelle. Nur wenn diese Spannungsquelle einen idealen Innenwiderstand von Null Ohm aufweist weicht die Spannung an der Pipette auch bei starken Strömen I3 durch die Zellmembran nicht vom vorgegebenen Wert U1 ab. Dieses ideale Verhalten ist mit realen Bauteilen nur annähernd zu erreichen. Die effektive Impedanz Z der Spannungsquelle kann man anhand der Größe des Referenzwiderstandes sowie der Spannungsverstärkung gU des verwendeten Operationsverstärkers 3 berechnen. Per Definition gilt für die Impedanz Z eines elektronischen Bauteils: Z= ∂U ∂I Gleichung 3-1 3.2 Voltage-Clamp und Current-Clamp Messverfahren 3-13 Angewendet auf die hier verwendete Schaltung folgt daraus: Z = ∂U 3 ∂I 3 U 2 = const . Gleichung 3-2 Durch Anwendung grundlegender Rechenregeln für Schaltungen mit Operationsverstärkern [Tietze, 1993] sowie der Kirchhoff'schen Gesetze kann man weiterhin folgende Beziehungen aufstellen: gU = − ∂I 3 = − ∂U 4 ∂U 3 U 2 = const . ∂U 4 R U 2 = const . Gleichung 3-3 Gleichung 3-4 Einsetzen von Gleichung 3-3 und Gleichung 3-4 in Gleichung 3-2 führt zum gewünschen Ergebnis: Z= Z R gU R gU = = = Gleichung 3-5 Impedanz der Spannungsquelle [Ohm]. Referenzwiderstand im Rückkopplungszweig [Ohm]. Spannungsverstärkung des Operationsverstärkers [1]. Nimmt man für den Referenzwiderstand6 der Schaltung R = 500 MΩ an [Beckers, 1993] sowie für die Spannungsverstärkung7 einen typischen Wert von gU = 107 [Tietze, 1993], so resultiert eine Impedanz von Z = 50 Ω. Berücksichtigt man, dass der Zugangswiderstand von Patch-Pipette zu Zell-Cytoplasma im Bereich einiger MΩ liegt, so kann man den Messverstärker als eine ideale Spannungsquelle betrachten. 3.2.2 Signalkorrektur Der Korrekturschaltkreis 5 in Ebene C dient dazu, einerseits Verluste in der Zuleitung zur Zelle durch entsprechende Signalüberhöhung auszugleichen, andererseits das Messignal von unerwünschten Störeinflüssen zu befreien. Wie in Punkt 3.1 beschrieben, werden bei den einzelnen Schritten zur Whole-Cell 6 Eine exakte Angabe des Widerstandswertes ist in den Herstellerangaben nicht enthalten. 7 Der Typ des verwendeten Operationsverstärkers ist in den Herstellerangaben nicht offengelegt. 3-14 3 Die Patch-Clamp Technik Konfiguration mehrere Kompensationsschritte durchgeführt. Diese Kompensationsschritte legen das elektrische Verhalten des Kompensationsschaltkreises fest. So wird beispielsweise ein Spannungsabfall über dem Zugangswiderstand der Pipette zur Zelle durch entsprechende Überhöhung des Stimulationspulses mit einem Faktor α (Korrektursignal U6 = U1 * α) ausgeglichen. Die Kompensation der kapazitiven Verluste in Pipette und Zellmembran erfolgt auf Basis des gemessenen Stromsignals U5 auf ähnlichem Wege. Allerdings ist für diese Effekte nur eine Kompensation bis zu ~95 % möglich, da es sonst wegen der Phasenverschiebung an den kapazitiven Schaltungskomponenten zu einer Oszillation des Verstärkers kommt. Eine Übersteuerung des Operationsverstärkers 3 kann die Korrektur allerdings auch schon deutlich unterhalb von 95 % begrenzen. Die Entfernung der kapazitiven Stromspitzen sowie des Leckstroms aus dem Messignal erfolgt über Spannung U7 . Die entsprechenden, erwarteten Störsignale werden auf Basis der Stimulationsspannung U2 im Korrekturschaltkreis gebildet und als U7 in Subtrahierer 4 vom Messignal abgezogen. 3.2.3 Signalverarbeitung und Kompensation - Current-Clamp Der Schaltplan des Patch-Clamp Verstärkers für den Betrieb im CC-Modus ist in Abbildung 3-5 als Blockschaltbild dargestellt. Gegenüber der Schaltung zur VoltageClamp Messung in Abbildung 3-4 gibt es in diesem Modus fünf statt vier Schritte der Signalverarbeitung. Es ist ein zusätzlicher Operationsverstärker 1 (dunkelgrau hervorgehoben) in den Schaltplan hinzugekommen. Dafür ist der Stimulationsschaltkreis um eine Stelle nach links an Position 0 gerückt. Alle Schaltungskomponenten der Schritte 2 bis 4 sind unverändert geblieben und haben weiterhin die gleiche Funktion. Schritt 0 entspricht dem vorigen Schritt 1. Diese Schritte werden daher nicht weiter erläutert. 3.2 Voltage-Clamp und Current-Clamp Messverfahren a b c Current-Clamp Schaltung 0 Stimulation (A) Eingang + x Addierer: x = a+b+c a + b - 2 Kompensation 1 Stromvergleich 3-15 x Subtrahierer: x = a-b IHalte IStim 0 + 3 0 Gegenkopplung 1 + R UOffset 2 1 3 + 2 + 6 (C) Fehlerkompensation (D) Ausgang Komparator: x = (a-b)*∞ Zelle Haltestrom (B) Signalverarbeitung x 3 Messung / 4 SignalbeU-I-Wandlung reinigung Offsetspannung Stimulationsstrom a + b - 4 5 + - 4 -- 7 5 Korrekturschaltkreis Spannungsmonitor Strommonitor Abbildung 3-5 Blockschaltbild des Patch-Clamp Verstärkers im Current-Clamp Modus. (A) Signaleingang. (B) Signalverstärkung und -verarbeitung. (C) Fehlerkompensation des Messignals um kapazitive und Ohm'sche Störgrößen. (D) Ausgabe der Signale. Die grundsätzlich verschiedene Funktionsweise des Schaltkreises beruht einzig auf dem hinzugekommenen Operationsverstärker 1 . An den invertierenden Eingang dieses Bauteils wurde das Ergebnis der Strommessung U5 zurückgekoppelt. Der Operationsverstärker 1 arbeitet als Komparator. Er vergleicht U5 mit dem an seinem nicht-invertierenden Eingang anliegenden Signal U0 . Das Signal U0 erhält damit die Bedeutung eines Stroms. Die Stimulationssignale der Eingangsebene A bekommen damit die Bedeutung eines Stimulations- und eines Haltestroms. Die Rückkopplungsschleife bewirkt, dass sich am Ausgang des Komparators 1 gerade eine solche Spannung U1 einstellt, dass der mit U5 gemessene Strom I3 in die Zelle exakt dem vorgegebenen Stromsignal U0 entspricht. Ändert sich das Potential U3 der Zelle, so bewirkt die erzwungene Konstanz des Stroms I3 , dass diese Potentialänderung im Verhältnis 1:1 auch von der Spannung U1 beschrieben wird. 3-16 3 Die Patch-Clamp Technik Das zum Spannungsmonitor geführte Messignal U1 entspricht folglich wie gewünscht dem Potential der Zellmembran U3 . Die Schaltung aus den Operationsverstärkern8 1 und 3 bildet eine kontrollierte Stromquelle für die Zelle an Punkt 3. Wie zuvor im VC-Modus für die Spannungsquelle durchgeführt, macht es Sinn auch an dieser Stelle die Impedanz der Stromquelle zu berechnen. Nur wenn die Stromquelle einen unendlich großen Innenwiderstand aufweist, entspricht der Strom in die Zelle I3 auch tatsächlich dem eingestellten Wert. Die effektive Impedanz Z der Stromquelle kann man auch für den CC-Modus anhand der Größe des Referenzwiderstandes sowie der Spannungsverstärkung gU der verwendeten Operationsverstärker 1 und 3 berechnen. Innerhalb der Schaltung gelten die Beziehungen9: ∂U 1 = ∂U 2 = − gU ∂U ∂U 4 − ∂U 2 = ∂U 5 5 U = const / U = const 0 6 U 7 = const Gleichung 3-6 Gleichung 3-7 ∂U 4 = g U (∂U 2 − ∂U 3 ) Gleichung 3-8 ∂U 3 = ∂U 4 + R∂I 3 Gleichung 3-9 Kombination von Gleichung 3-6 mit Gleichung 3-7 ergibt: ∂U 1 = − gU ∂U 4 gU − 1 Gleichung 3-10 U 0 = const / U 6 = const / U 7 = const Einsetzen dieses Ergebnisses in Gleichung 3-8 liefert: ∂U 4 = − g U2 − g U ∂U 3 g U2 − g U + 1 U Gleichung 3-11 0 = const / U 6 = const / U 7 = const 8 Die Addierer und Subtrahierer 3 und 5 schleifen das Messignal unverändert mit dem Skalierungsfaktor 1 durch und können daher bei der Analyse der Schaltungsfunktion vernachlässigt werden. 9 Der Einfluss des Korrekturschaltkreises 5 wird für die Betrachtung vernachlässigt. U6 und U7 werden als konstant angenommen. 3.2 Voltage-Clamp und Current-Clamp Messverfahren 3-17 Ersetzt man ∂U4 in Gleichung 3-9 durch Gleichung 3-11, so kann man nach der gesuchtem Impedanz auflösen: Z= Z R gU ∂U 3 = R g U2 − g U + 1 ∂I 3 ( = = = ) Gleichung 3-12 Impedanz der Spannungsquelle [Ohm]. Refrenzwiderstand im Rückkopplungszweig [Ohm]. Spannungsverstärkung des Operationsverstärkers [1]. Wegen gU >> 1 kann man schließlich vereinfacht schreiben: Z = g U2 R Gleichung 3-13 Die Impedanz der Stromquelle entspricht also dem Referenzwiderstand R der Schaltung, skaliert mit dem Quradrat der Spannungsverstärkung gU . Bei R = 500 MΩ [Beckers, 1993] und gU = 107 [Tietze, 1993], liegt die Impedanz um den Faktor g2U = 1014 über dem typischen Leckwiderstand einer Zellmembran (~500 MΩ). Man kann daher in sehr guter Näherung von einer idealen Stromquelle ausgehen. Ein Vergleich der Impedanzen von VC-Modus (Gleichung 3-5) und CC-Modus (Gleichung 3-13) ergibt, dass diese über den Widerstandswert R miteinander gekoppelt sind. Vergrößert man den Referenzwiderstand R, um für den Betrieb im CC-Modus eine "idealere" Stromquelle mit größerem Innenwiderstand zu erhalten, so resultiert gleichzeitig ein schlechteres Verhalten der Schaltung für den Betrieb im VC-Modus; der Innenwiderstand der Spannungsquelle im VC-Modus vergrößert sich. Eine Steigerung der Verstärkerqualität in beiden Messmodi ist nur durch Verwendung von Operationsverstärkern mit größerer Spannungsverstärkung gU zu erzielen. Die maximale Größe des Parameters gU eines Operationsverstärkers 10 unterliegt jedoch technischen und physikalischen Grenzen . Entscheidend für die messtechnische Qualität des Patch-Clamp Verstärkers ist eine ausgewogene, exakt abgestimmte Dimensionierung der Operationsverstärker 1 und 3 sowie des Referenzwiderstands R. 10 8 Kommerziell erhältlich sind Operationsverstärker mit einer Differenzspannungsverstärkung bis gU ~ 10 . 3-18 3 Die Patch-Clamp Technik 3.3 Versuchsaufbau Für die praktische Durchführung der elektrophysiologischen Messungen sowie für Mikroskopieaufnahmen im Phasenkontrast- und Fluoreszenzmodus, wurde im Rahmen dieser Arbeit ein Patch-Clamp Messplatz aufgebaut. Das System setzt sich zusammen aus einem Fluoreszenzmikroskop, Mikromanipulatoren zur Steuerung der Patch-Pipetten, einem Videosystem zur Bilderfassung sowie einem computergesteuerten dreifachen Patch-Clamp Verstärker. Die Fertigung von Mikropipetten für die Messungen erfolgte in einem separat aufgestellten Pipettenpuller. 3.3.1 Dreifach Patch-Clamp Aufbau Der Messaufbau gliedert sich in zwei Zweige: − einen elektrischen Zweig (Abbildung 3-6 A, linke Seite) und − einen optischen Zweig (Abbildung 3-6 A, rechte Seite). Im elektrischen Messzweig befinden sich alle Komponenten für die Durchführung der Patch-Clamp Messungen. Dieser Zweig besteht aus dem Patch-Clamp Verstärker 2 (EPC-9/3, Heka Elektronik), drei Messköpfen 3, und den zugehörigen Mikromanipultoren 4 (Mini 25, Luigs&Neumann GmbH) inklusive Steuereinheit 5. Der optische Messzweig setzt sich zusammen aus einer Framegrabber-Karte zur Bilderfassung 6 (Meteor II, Matrox Electronic Systems GmbH), einer hochsensitiven Farb-Videokamera 7 (Sony XC-333 P, Sony Deutschland GmbH) sowie dem Mikroskop 8 (Olympus IX-50). Die Messdaten beider Messzweige werden in einem gemeinsamen Messcomputer 1 (500 MHz Pentium IV mit 30 GB SCSI Festplatte und 256 MB RAM) verarbeitet, gespeichert und ausgewertet. Abbildung 3-6 B zeigt eine Photographie des Messaufbaus. Die einzelnen Komponenten sind darin mit den Komponentennummern aus Abbildung 3-6 A identifiziert. Zur Isolation der vibrationsempfindlichen Anordnung aus 3.3 Versuchsaufbau 3-19 Messaufbau zur Patch-Clamp Messung (A) Schematische Darstellung Datenerfassung (B) Technische Realisierung Bildverarbeitung (1) Computer (1) Computer (2) Patch-Clamp Verstärker 3 4 7 (6) VideobildDigitalisierkarte (7) Videokamera 10 8 5 9 (3) Messkopf z (4) Mikromanipulator y x (5) Manipulatorsteuerung (8) Mikroskop (9) Basisplatte (10) Marmorplatte Abbildung 3-6 Gesamtansicht des Messaufbaus zur dreifach Patch-Clamp Messung. (A) Schematische Darstellung. (B) Photographie des Messaufbaus. Patch-Pipetten und Zellsubstrat von Gebäudeschwingungen, sind Mikroskop und Mikromanipulatoren auf einer luftgefederten Basisplatte (9) festgeschraubt. Zusätzlich steht der Aufbau auf einer schwingungsisolierten Marmorplatte (10) mit einem Eigengewicht von ca. 200 kg. Die elektrische und optische Vermessung der Zellsubstrate erfolgt auf dem optischen Verschiebetisch des Mikroskops im Zentrum des Aufbaus. Abbildung 3-7 zeigt eine Detailansicht dieses Bereichs. Auf dem Verschiebetisch ist ein speziell angefertigter, kontrolliert beheizbarer Substrathalter 7 montiert. Dieser ermöglicht eine passgenaue Fixierung von Zellkultur-Petrischalen mit 3 cm Durchmesser 6 (627 102, Greiner Labortechnik). Vor Einsetzen eines Zellsubstrats in den Halter wird dieser mit einem Tropfen Wasser (ca. 0,1 ml) angefeuchtet, um die Wärmeleitung zwischen Halter und Substrat zu erhöhen. Die Temperatur des extrazellulären Mediums in der Petrischale kann dann 3-20 3 Die Patch-Clamp Technik Patch-Clamp Messkopf und Substrathalter (B) Technische Realisierung (A) Schematische Darstellung Zum Patch-Clamp Verstärker or r ) V ke (1 stär r ve (2) Druckschlauch 2 1 (3) Pipettenhalter (4) Chlorierter Silberdraht zur Kontaktierung (5) Glaspipette gefüllt mit Salzlösung 2 5 3 (6) Petrischale mit extrazellulärem Patch-Medium 6 Zelle (7) Substrathalter mit Heizplatte 4 7 Abbildung 3-7 Patch-Clamp Messaufbau in Substratnähe. (A) Schematische Darstellung der Komponenten. (B) Photos von der technischen Realisierung. über einen elektronischen Regelkreis auf ± 0,25 °C konstant gehalten werden (gemessen bei einer Raumtemperatur im Bereich von 18 °C bis 24 °C). Damit optische Messungen am Zellsubstrat möglich sind, wurde in die Mitte des Substrathalters eine Öffnung mit 1 cm Durchmesser eingefräst und ein spezielles, ringförmiges Heizelement darum herum angeordnet. Der Substrathalter 7 ist aus Metall gefertigt, um eine Abschirmung des Substrats gegenüber der Einstreuung elektrischer Störsignale zu erzielen. Die Patch-Clamp Messungen erfolgen über drei unabhängige Messköpfe, jeweils bestehend aus Vorverstärker 1, Pipettenhalter 3 und Glaspipette 5. Der elektrische Kontakt zwischen der Verstärkerschaltung und den Elektrolytlösungen in Glaspipette 5 und Petrischale 6 wird über chlorierte Silberdrähte 4 hergestellt. Die Kontrolle des Pipetten-Innendrucks während der Messung wie unter Punkt 3.1 3.3 Versuchsaufbau 3-21 beschrieben, erfolgt durch den Druckschlauch 2, welcher von einem Stutzen am 11 Pipettenhalter 3 bis zum Mund des Experimentators führt. 3.3.2 Fertigung von Mikropipetten Da ein Gigaseal zwischen Messpipette und Zellmembran nur mit einer vollkommen sauberen Glaspipette möglich ist, muss für jede einzelne Patch-Clamp Messung jeweils eine neue, unbenutzte Mikropipette angefertigt werden. Die Herstellung von Mikropipetten ist daher ein zentraler Bestandteil der durchgeführten elektrophysiologischen Messungen. Zur Herstellung der Pipetten wurde ein Mikroprozessor gesteuerter Pipettenpuller verwendet. Das Prinzip der Pipettenfertigung ist in Abbildung 3-8 A dargestellt: 1. Zuerst wird eine frische Glaskapillare 1 (1810016, Hilgenberg) in den Pipettenpuller 2 (P-97, Sutter Instrument Company) eingespannt. 2. Erhitzen der Glaskapillare durch einen glühenden Wolframdraht 3 bringt das Glas zum Schmelzen. Gleichzeitig wird die Kapillare mit kontrollierter Kraft an beiden Enden symmetrisch auseinandergezogen. Der Querschnitt der Kapillare verengt sich. 3. Nach mehreren Schmelzschritten reisst die Kapillare schließlich in zwei Hälften auseinander. Zwei Mikropipetten 4 entstehen. Die Form und der Enddurchmesser der fertigen Mikropipetten wird über die Parameter: Temperatur12, Zugkraft und Zuggeschwindigkeit kontrolliert. Der resultierende Pipettenwiderstand hängt zusätzlich von der Zusammensetzung der intrazellulären und extrazellulären Patch-Clamp Lösung ab. Auch der Typ des verwendeten Glases beeinflusst die Eigenschaften der Mikropipetten. Harte Gläser mit Schmelzpunkten über 900 °C haben geringere Dielektrizitätskonstanten und dadurch eine geringere Leitfähigkeit im 11 Mit Mund und Lunge ist der Mensch in der Lage sehr geringe Druckunterschiede im Bereich einiger mbar gegenüber der Umgebung aufzubauen und zu kontrollieren. 12 Entscheidend ist die Temperaturdifferenz zum Schmelzpunkt des Glases. Die Bestimmung des Schmelzpunktes ist durch Fahren einer Temperaturrampe möglich. 3-22 3 Die Patch-Clamp Technik Abbildung 3-8 Darstellung des Verfahrens zur Anfertigung und Befüllung von Mikropipetten. (A) Pipetten mit einer Öffnung < 1 µm werden durch kontrolliertes Erhitzen und Ziehen in einem Pipettenpuller aus Glaskapillaren gefertigt. (B) Unmittelbar vor der Patch-Clamp Messung erfolgt die Befüllung der Pipetten mit intrazellulärer Lösung. Hochfrequenzbereich. Sie zeichnen sich durch ein besonders geringes thermisches Rauschen aus und werden daher insbesondere für Patch-Clamp Messungen an einzelnen Ionenkanälen verwendet [Sakmann, 1995]. Bei Whole-Cell Messungen ist die Signalstärke jedoch auch bei weichen Gläsern hinreichend groß gegenüber dem thermischen Rauschen. Man verwendet für Whole-Cell Messungen daher vorzugsweise Gläser mit Schmelzpunkten unter 800 °C. Der geringere Schmelzpunkt bewirkt rundere Abrisskanten an der Pipettenspitze, dies wiederum verbessert die Ausbildung des Gigaseals bei der Messung. Weiterhin wird durch die geringere Arbeitstemperatur der Wolframdraht des Pipettenpullers geschont und bleibt über eine größere Zahl gefertigter Pipetten in seinem thermischen Verhalten konstant. Für die Versuche in dieser Arbeit wurden Mikropipetten bauchiger Form mit einem Innendurchmesser der Spitze von ~1 µm aus weichem, bei ca. 785 °C schmelzendem 3.3 Versuchsaufbau 3-23 Borosilikatglas angefertigt. Der Innenwiderstand der gefüllten Pipetten lag im Bereich zwischen 5 und 10 MΩ . Die Fertigung der Pipetten kann schon einige Stunden vor der Messung erfolgen. Eine längere Lagerung ist aufgrund der Gefahr der Verschmutzung durch Staubpartikel nicht ratsam. Ca. ¼ Stunde vor Beginn einer elektrophysiologischen Messung wurde das Kulturmedium des untersuchten Substrats durch extrazelluläre Patch-Clamp Lösung ersetzt. Die Mikropipetten wurden einzeln, jeweils unmittelbar vor der Montage im Pipettenhalter, mit intrazellulärer Patch-Clamp Lösung gefüllt. Dies geschah wie in Abbildung 3-8 B dargestellt, durch Füllen der Pipette von hinten mit einer dünnen 13 Füllkapillare 7 aus Kunstoff . Die intrazelluläre Lösung wurde mit Hilfe einer Spritze 5 durch einen Mikrofilter 6 in die Füllkapillare gepresst. Aufgabe des Mikrofilters war es, größere Partikel14 zurückzuhalten, welche die Pipette verstopfen können. Die genauen Rezepturen der verwendeten intrazellulären und extrazellulären PatchClamp Lösungen sind im Anhang angegeben. 13 Füllkapillaren wurden manuell durch Ausziehen einer Eppendorf Pipettenspitze in der Flamme eines Bunsenbrenners gefertigt. 14 Partikel können sich insbesondere durch unvollständig gelöste Farbstoffe in der intrazellulären Lösung befinden. 3-24 3 Die Patch-Clamp Technik 4-1 4 Kontrolliertes Zellwachstum Das Wachstum von Zellen auf künstlichen Oberflächen in vitro kann durch Aufbringen von haftungsvermittelnden Proteinen gezielt beeinflusst werden. Dies gelingt umso besser, je größer der Haftungs-Kontrast zwischen proteinbeschichteten und freien Bereichen einer Oberfläche ist. Wird beispielsweise eine Schicht aus Laminin (Protein der extrazellulären Matrix im Gehirn) auf einer Oberfläche aus Polystyrol physisorbiert, so wird ein Anhaften von neuronalen Zellen nahezu ausschließlich in den mit Laminin bedeckten Bereichen beobachtet (Abbildung 4-1 A). Zellwachstum auf unterschiedlich strukturierten Oberflächen Oberfläche Grad der Strukturierung (A) Flächen (B) Linien Dreicksflächen mit 500 µm Kantenlänge aus Laminin1 Linien aus 2 µm breit (C) Linien mit Knoten Laminin1, Linien mit Knoten aus Laminin1, 4 µm / 20 µm breit Zellwachstum 250 µm Ergebnis 1 2 <V10_1_s35_ b1> Zellen2 wachsen ungeordnet auf den mit Laminin bedeckten Flächen. 100 µm <V10_1_s1_ b2> Zellen2 wachsen entlang der vorgegebenen Linien. 100 µm <V10_1_s11_ b4> Zellen2 liegen in regelmäßigen Abständen auf den Knoten entlang der vorgegebenen Linien. Struktur hergestellt durch Stempelübertrag von Laminin auf Polystyrol Zellsystem: MzN Zellinie an Tag 4 nach neuronaler Ausdifferenzierung; Zelldichte jeweils 3500 Zellen/cm² Abbildung 4-1 Mit dem Grad der Oberflächenstrukturierung steigt die Ordnung des beobacheten Zellwachstums. (A) Dreiecksflächen makroskopischer Ausdehnung: ungeordnetes Wachstum auf den mit Laminin beschichteten Flächen. (B) Linienstruktur: Zellen ordnen sich entlang der 2µm breiten Linien an. (C) Linien mit Knoten: Zellen setzen sich auf die 20 µm große Knotenpunkte, Neuriten wachsen entlang der 4 µm breiten Linien. 4-2 4 Kontrolliertes Zellwachstum Geht man bei der Oberflächenstrukturierung zu mikroskopischen Strukturen in der Größenordnung neuronaler Zellen von einigen µm über, so bilden die anhaftenden Neuronen die Strukturen nach. Dabei siedeln sich die Zellkörper bevorzugt in Bereichen größerer Strukturbreite (≥ 10 µm) an. Neuriten wachsen auch entlang engerer Pfade. (Abbildung 4-1 B, C). Physisorbierte Mikrostrukturen von Proteinen wie Laminin auf zellabweisenden Oberflächen wie Polystyrol eignen sich daher dazu, neuronale Zellen definiert auf einer Oberfläche anzuordnen und mit Neuriten zu verbinden. Ein geeignetes Verfahren für den Übertrag von Proteinen auf eine künstliche Substratoberfläche, basiert auf der Verwendung von strukturierten Stempeln [Blawas, 1998]. Diese Technik wurde auch in dieser Arbeit eingesetzt und ist in Punkt 4.2 beschrieben. Andere Verfahren zur Herstellung strukturierter Substrate für die Steuerung neuronalen Wachstums bedienen sich photolithographischer Methoden [Kleinfeld, 1988; Curtis, 1997], Laserabtragungstechniken [Corey, 1991; Dulcey, 1991; Stenger, 1992; Matsuzawa, 1996] sowie der Photoimmobilisierung [Clémence, 1995]. Neben einer rein chemisch induzierten Steuerung neuronalen Wachstums durch Wechselwirkung der Zellen mit Proteinen auf der Substratoberfläche, können Neuronen auch durch topographische Strukturen gezielt beeinflusst werden [Kleinfeld, 1988; Curtis, 1997]. 4.1 Extrazelluläre Matrix und Laminin 4-3 4.1 Extrazelluläre Matrix und Laminin Während der embryonalen Entwicklung des Nervensystems werden unzählige Verbindungen von Nervenzellen untereinander, zu Muskelzellen, und zu Sinneszellen geknüpft. Im Gehirn selbst baut jedes einzelne Neuron Verbindungen zu fast allen Nachbarzellen im Umkreis einiger Millimeter auf [Hebb, 1949]. Ein komplexes Netzwerk verschalteter Nervenzellen entsteht. er Seit Ende der 80 Jahre ist klar, dass die Organisation bei der Erstellung dieses komplexen Schaltplans nicht allein von intrinsischen, in den einzelnen Nervenzellen selbst gespeicherten Informationen ausgeht sondern auch von aussen durch Vorgabe von axonalen und dendritischen Wachstumspfaden erfolgt. Ein Gerüst aus Molekülen in der Umgebung der wachsenden Nervenzellen, die sog. extrazelluläre Matrix (ECM), wurde als zentraler Faktor bei dieser Steuerung des neuronalen Wachstums identifiziert [Sanes, 1983]. Der Kontakt von Nervenzellen mit Proteinen der extrazellulären Matrix beeinflusst die neuronale Entwicklung, axonales und dendritisches Wachstum sowie die Entstehung synaptischer Verbindungen. Erste Beobachtungen einer Präferenz von axonalem Wachstum entlang bevorzugter Pfade wurden bereits 1928 gemacht [Ramon y Cajal, 1928]. In den Experimenten von P. Letourneau im Jahre 1975 [Letourneau, 1975] zeigte sich eine selektive axonale und dendritische Beeinflussung neuronalen Wachstums: Axone bevorzugten bei Angebot von Pfaden aus verschiedenen Substanzen den Untergrund mit der stärkeren Haftung. Diese Beobachtung lenkte den Fokus von einer rein mechanischen, undifferenzierten Steuerung neuronalen Wachstums auf eine selektive chemische Wechselwirkungen zwischen Zelle und Umgebung. Die Produktion der chemisch aktiven Substanzen, der sog. Wachstumsfaktoren, erfolgt in Zellen in unmittelbarer Nachbarschaft zu den Neuronen [Collins, 1980; Berg, 1984; Collins, 1984]. Folgt man den Ergebnissen dieser Veröffentlichungen, so wird das Wachstum von Neuriten in der embryonalen Entwicklung des Nervensystems von extrazellulären Substanzen gelenkt, welche von benachbarten Zellen bereitgestellt werden. Die 4-4 4 Kontrolliertes Zellwachstum spezifischen, für die Wachstumssteuerung verantwortlichen Proteine in diesen Substanzen wurden von zahlreichen Autoren mit drei Analysemethoden identifiziert: Bekannte chemische Bestandteile der ECM wurden auf ihre Fähigkeit, Neuritenwachstum zu stimulieren getestet; wachstumsfördernde Kulturmedien wurden auf ihre chemischen Wirkkomponenten hin untersucht; und die örtliche Verteilung entsprechender Substanzen im Organismus wurde dokumentiert. Die Ergebnisse der durchgeführten Untersuchungen führen insgesamt zu dem Schluss, dass das Protein Laminin eine Schlüsselfunktion in der Stimulation neuronalen Wachstums hat. Im Detail liegen diesem Schluss folgende Einzelergebnisse zugrunde: 1. Neurone einer Vielzahl von Tierarten zeigten Neuritenwachstum auf Substraten welche mit reinem Laminin beschichtet sind [Baron-Van Evercooren, 1982; Lander, 1983; Manthorpe, 1983; Rogers, 1983; Wewer, 1983; Edgar, 1984; Faivre-Bauman, 1984; Liesi, 1984; Adler, 1985; Davis, 1985; Hammarback, 1985; Hopkins, 1985; Unsicker, 1985; Unsicker, 1985b; Engvall, 1986; Hammarback, 1986]. Bei einem direkten Vergleich von Laminin mit anderen ECM Komponenten wie Kollagenen, Proteoglykanen und Fibronektin erwies sich Laminin jeweils als überlegene Wachstumsgrundlage [Gundersen, 1987; Gundersen, 1987b]. Ausserdem zeigen bestimmte Neuronentypen nur auf Laminin Neuritenwachstum, nicht aber auf Fibronektin oder Kollagen [Rogers, 1983]. Weiterhin ist einzig Laminin in der Lage konditionierte Kulturmedien (zumindest bedingt und zeitweise) zu ersetzen: Zellen die gewöhnlich auf neuronale Wachstumsfaktoren (NGF) im Kulturmedium angewiesen sind, überleben kurze Zeit auf lamininbehandelten Oberflächen auch in NGF-freiem Medium [Baron-Van Evercooren, 1982; Edgar, 1984]. 2. Laminin wurde als zentrales, aktives Molekül in den ECM Substanzen zahlreicher ECM erzeugender Zellen identifiziert. Laminin kommt dabei in einem engen aber nicht kovalenten HeparansulfatProteoglykankomplex und manchmal in einem ChondroitinsulfatProteoglykankomplex sowie in einem nicht kollagenen Glykoprotein der ECM, Entarkin, vor. Eingehende Untersuchungen legen nahe, dass nahezu 4.1 Extrazelluläre Matrix und Laminin 4-5 ausschließlich Laminin für die wachstumsfördernde Wirkung des Komplexes auf Neuriten verantwortlich ist [Lander, 1982; Davis, 1985; Lander, 1985; Lander, 1985b; Davis, 1987]. 3. Mittels Antikörperfärbungen in vivo wurde Laminin entlang zentraler axonaler Wachstumspfade identifiziert: An den Innervationspunkten neuro-muskulärer Verbindungen [Chiu, 1984] sowie entlang spinaler Axone [Rogers, 1986; Riggot, 1987]. Bei einer Verletzung der Nervenbahn degenerieren die durchtrennten Axone. Schwann'sche Zellen, welche die Axone zuvor ummantelt haben überleben jedoch. Nachwachsende Axone tasten sich entlang der mit Laminin bedeckten Innenseite der Schwann'schen Zellen [Scherer, 1984]. Aber selbst wenn die Schwann Zellen durch Kälteeinwirkung abgetötet wurden, findet ein Nachwachsen von Axonen entlang des immer noch an der Zelloberfläche vorhandenen Laminins statt [Ide, 1983]. Auch die ECM Komponenten Fibronektin, Kollagen I und Kollagen IV fördern nachweislich das Neuritenwachstums. Jedoch ist der bei Laminin beobachtete Einfluss auf neuronales Wachstum deutlicher und klar reproduzierbar. Im Rahmen dieser Arbeit wurde daher ausschließlich Laminin zur Strukturierung von Zellsubstraten eingesetzt. Es wurde primär isoliertes Laminin (1243217, Roche Diagnostics GmbH) mit einem Molekulargewicht von 600-900 kD verwendet. Die Tertiärstruktur dieser natürlichen Form des Proteins Laminin besteht aus 3 Polypeptidketten, einer α und zwei β Ketten, mit jeweils ca. 1000 Aminosäuren. Die drei Untereinheiten sind miteinander über Cystein-basierte Disulfidbrücken verknüpft [Stetefeld, 1996]. Laminin zeigt eine sehr gute Löslichkeit in Wasser. Dies ist auf die große Anzahl an Lysinen (ca. 52 Stück je Kette) innerhalb des Proteins zurückzuführen. In Abbildung 4-2 A ist die Tertiärstruktur einer der drei Lamininketten abgebildet. 4-6 4 Kontrolliertes Zellwachstum Laminin und extrazelluläre Matrix (A) Tertiärstruktur einer einzelnen Kette des Proteins Laminin (B) Anbindung von Zellen an die ECM Abbildung 4-2 Das ECM Protein Laminin als Ankerpunkt für Integrine der Zellmembran. (A) Tertiärstruktur einer der drei Ketten des Laminins. Domänen mit α-Helix (Zylinder in der Darstellung) sowie β-Faltblätter (Pfeile in der Darstellung) sind in der Struktur enthalten. Abbildung entnommen aus [Stetefeld, 1996]. (B) Schematische Darstellung der Verankerung einer Zelle an Proteinen der extrazellulären Matrix (hier Fibronektin). Integrine durchspannen die Phospholipidmembran der Zelle und stellen so eine Verbindung zwischen Actinfilamenten des Zell-Cytoskeletts und Proteinen der ECM her. Abbildung entnommen aus [Munk, 2000]. Die Wechselwirkung von Laminin mit neuronalen Zellen erfolgt über Ankerproteine, die sog. Integrine. Diese durchspannen als Transmembranproteine die Phospholipidmembran der Zelle. Auf der Cytoplasmischen Seite sind sie über ihre β-Untereinheit fest mit einzelnen Actinfilamenten des Cytoskeletts der Zelle verbunden. Extrazellulär binden sie mit α- und β-Untereinheit an Laminin, Fibronektin, oder andere Proteine der extrazellulären Matrix an (siehe Abbildung 4-2 B). 4.2 Strukturierung mit Mikrostempeln 4-7 4.2 Strukturierung mit Mikrostempeln Eine Strukturierung von Oberflächen im µm Bereich mit Proteinen der ECM ist grundsätzlich über verschiedene technische Verfahren zu realisieren. Besonders geeignet sind photolithographische Methoden, Laser-Abtragung sowie Strukturierung mit Mikrostempeln. Bei photolithographischen Methoden wird durch strukturierte Abdeckung der Substratoberfläche mit Photolack und chemischer Modifikation der freien Bereiche eine örtlich lokalisierte Anbindung des ECM Moleküls erzielt [Corey, 1996]. Bei der Anwendung von Laserabtragungstechniken wird eine zunächst homogen aufgebrachte Schicht aus reaktiven chemischen Ankergruppen für die spätere Anbindung des ECM Moleküls aus ungewünschten Bereichen entfernt [Corey, 1996; Stenger, 1998]. Bei Strukturierung mit Mikrostempeln überträgt man das ECM Molekül, physisorbiert an der Oberfläche eines Polymerstempels, direkt aus wässriger Lösung auf das Substrat [Bernard, 1998; Chen, 1998; James, 1998; Branch, 2000]. Diese Methode bietet drei wesentliche Vorteile gegenüber den ersten zwei Verfahren: 1. Einfachheit: Nach Fertigung des Mikrostempels ist für die Herstellung von Substraten kein aufwendiges technisches Gerät mehr erforderlich. 2. Keine Störfaktoren: Die Substratoberfläche wird weder chemisch noch physikalisch beeinflusst. 3. Übertragbarkeit: Die Strukturierung kann ohne Änderung des Versuchsablaufs mit unterschiedlichen ECM Molekülen und auf beliebigen, ebenen Substratoberflächen erfolgen. Defizite der Methode liegen in der begrenzten Präzision, bedingt durch mögliche Fehlstellen im Stempel und Deformation der Struktur sowie in der örtlich unvollständigen Übertragung bei Kontaktinsuffizienzen. Bei ausreichend großer Dimensionierung der Strukturausdehnung (1 cm² Strukturierte Fläche gegenüber Elementarstrukturen von 50 bis 100 µm Länge) sind Fehlstellen im Stempelübertrag aufgrund der hohen lateralen Redundanz jedoch vernachlässigbar. Es können in 4-8 4 Kontrolliertes Zellwachstum diesem Fall innerhalb eines Stempelabdrucks immer Bereiche mit intaktem Stempelübertrag für Experimente gefunden werden. Zur Herstellung strukturierter ECM Substrate wurde in dieser Arbeit daher ausschließlich mit Mikrostempeln gearbeitet. 4.2.1 Stempelfertigung Die Herstellung eines Mikrostempels umfasst drei Prozessschritte: Zuerst werden die gewünschten Strukturen in einem Grafiksystem (Autocad 2.0, Autodesk, Inc.) im Maßstab 1:1 gezeichnet (Strukturentwurf: Abbildung 4-3 A). Im zweiten Schritt wird eine photolithographische Maske gefertigt und diese in Photolack belichtet. Nach der Entwicklung sind die Strukturen als Topographie in den Photolack eingeprägt (Photolithographie: Abbildung 4-3 B). Im letzten Schritt wird durch Ausgießen der Photolackstrukturen mit flüssigem Polymer und anschließendem Aushärten des Herstellung von Mikrostempeln Prozessschritt Arbeitsablauf (B) (C) (A) Strukturentwurf (B) Photolithographie (C) Abformung • Design geeigneter • Fertigung einer Chrom • Ausgießen der Strukturen Elementarstrukturen • Vervielfältigung der Elementarstrukturen zu 1 cm² großen Flächen • Regelmäßige Anordnung auf einer 7" Scheibe (Waferformat) Darstellung (A) CAD-Design Maske mit dem Elektronenstrahlschreiber • Belacken von mit unvernetztem Polymer • Härten im Ofen • Ablösen strukturierter Glaswafern Polymerstempel • Photostrukturierung Strukturen im Photolack (Lichtmikroskopie mit DIC) Strukturiertes Polymer (REM Aufnahme) <991103_No27.dib> <R0574b-01.tif> <991103_No3.dib> 3 cm <Maske_Gesamt.tif> 200 µm Abbildung 4-3 Ein Mikrostempel wird in drei Prozessschritten hergestellt. (A) Strukturentwurf im Grafiksystem (B) Photolithographische Übertragung der Strukturen in Photolack (C) Abformung von Polymerstempeln an den Photolackstrukturen 100 µm 4.2 Strukturierung mit Mikrostempeln 4-9 Kunstoffs im Ofen ein Stempel mit erhabenen Strukturen abgeformt (Abformung: Abbildung 4-3 C). Je nach Anwendung wurden Stempel dreier unterschiedlicher Bauformen eingesetzt. Im Folgenden ist der Herstellungsprozess für den Grundtyp eines Polymerstempels aus 100 % PDMS (Polydimethylsiloxan) beschrieben. Die Herstellung zweier weiterer Bauformen in Glas-PDMS-Hybridbauweise, speziell zur Strukturierung von extrazellulären Sensorchips entwickelt, weicht hiervon im letzten Schritt ab. Dies wird später diskutiert. 4.2.1.1 Strukturentwurf Insgesamt 37 verschiedene Einzelstrukturen1 wurden entworfen, um den Einfluss von Größe und Form einer Mikrostruktur auf das neuronale Zellwachstum zu untersuchen. Dabei sind grundlegende Ergebnisse bereits veröffentlichter Arbeiten berücksichtigt worden. Der Entwurf der Stempelstrukturen in Prozessschritt A, Abbildung 4-3 A, erfolgte in drei Teilschritten A1, A2 und A3: (A1) Zeichnen der Elementarstruktur2: Durch Angabe einer Serie von Ortskoordinaten (Xi/Yi) wurden alle Elemente der Elementarstruktur als geschlossene Kurvenzüge erzeugt; z.B. eine 2 µm dicke und 100 µm lange Linie als Kurvenzug mit vier Eckpunkten (0/0)(2/0), (2/0)(2/100), (2/100)(0/100), (0/100)(0/0). Alle Elementarstrukturen wurden auf einer Grundfläche von 1000 x 1000 µm² erstellt. (A2) Vervielfältigung der Elementarstruktur: Durch 10-fache Vervielfältigung der Elementarstruktur in X- und Y-Richtung wurden 1 cm² große, strukturierte Quadrate erzeugt. (A3) Anordnung auf dem Wafer: Im letzten Schritt wurden die fertigen Quadrate mit jeweils 5 mm Zwischenraum regelmäßig auf der Oberfläche einer 5" 1 In Kapitel 4.4 sind die eingesetzten Strukturen und deren Wirkung im Detail beschrieben. 2 Es wurde jeweils die kleinste, sich regelmäßig wiederholende Einheit der zu erstellenden Struktur als Elementarstruktur gewählt. Also beispielsweise für ein Gitter aus 2 µm dicken Linien im Abstand von 50 µm ein Quadrat mit 48 µm Kantenlänge, welches sich alle 50 µm in X- und Y-Richtung wiederholt. 4-10 4 Kontrolliertes Zellwachstum Kreisfläche angeordnet und in den Zwischenräumen mit Markierungskreuzen versehen. Eine abschließende Überprüfung des Layouts erfolgte lokal durch Ausdruck vergrößerter Ausschnitte aus den einzelnen strukturierten Bereichen sowie global in 3 der Gesamtansicht. Diese ist in Abbildung 4-3 A dargestellt . 4.2.1.2 Photolithographie Um aus den fertig gezeichneten Strukturen ein reales topographisches Relief zur Stempelabformung herzustellen (im Folgenden als Strukturmaske bezeichnet), wurde die Technik der Photolithographie eingesetzt. Hierbei wird durch partielle Belichtung einer photosensitiven Schicht (Photolack) eine chemische Reaktion in den belichteten Bereichen ausgelöst. Diese hat zur Folge, dass sich nach Entwicklung in einem Entwicklungsbad die Lackschicht an den belichteten Stellen löst und an den unbelichteten auf der Oberfläche haften bleibt4. Aufgrund der physikalischen Beugung des Lichts muss für die Abbildung von Strukturen in der Größenordnung weniger Mikrometer eine Lichtquelle im ultravioletten Wellenlängenbereich eingesetzt werden. Ein geeigneter, in diesem Wellenlängenbereich hinreichend intransparenter Werkstoff für die Fertigung der Maske ist Chrom. Das Trägermaterial der Maske besteht aus Quarzglas um eine ausreichende Transmission der UV-Strahlung zu gewährleisten. Die Herstellung von Chrommasken erfolgt mit einem in der Halbleiter- und Mikrostrukturtechnik etablierten Verfahren, dem Elektronenstrahlschreiben. Dabei wird mit Hilfe eines Elektronenstrahls Chrom von der Oberfläche einer zuvor homogen mit Chrom bedampften Quarzglasscheibe verdampft. Der Elektronenstrahl wird dazu, von einem Computer gesteuert, durch elektromagnetische Ablenkung gezielt über die Oberfläche geführt und zeichnet so die gewünschte Struktur in die Maske. Es entsteht ein Transmissionsmuster an der Maskenoberfläche. Zur Herstellung einer solchen Chrom-Quarzglas-Maske wurde der im CAD-System erstellte 3 Die in der Abbildung nur unvollständige Darstellung der meisten strukturierten Quadrate als Kreuze ist auf den für diese Darstellung nicht ausreichenden Speicherplatz des verwendten Rechners zurückzuführen. 4 In diesem Fall spricht man von einem Negativ-Lack; bleiben hingegen die belichteten Bereiche erhalten, so wird der Photolack als Positiv-Lack bezeichet. 4.2 Strukturierung mit Mikrostempeln 4-11 Strukturentwurf mit einem Konvertierungsprogramm in Steuercodes für den Elektronenstrahlschreiber übersetzt und anschließend mit diesem in die Chromschicht der Maske übertragen. Die Strukturen der Maske wurden nun in drei Teilschritten B1, B2 und B3 in Photolack übertragen: (B1) Im ersten Schritt wurde ein Glaswafer (0,6 mm dicke Glasscheibe im Durchmesser von 5", MEMC Electronic Materials, Deutschland) im sog. SpinCoating Verfahren mit einer 12,5 µm dicken Photolackschicht belackt. Dazu wurde ein definiert großer Tropfen von AZ 4562 (Positiv Photolack, Clariant GmbH, Deutschland) auf das Zentrum des Glaswafers aufgebracht, der Glaswafer in schnelle Rotation versetzt und dabei der Photolack durch die radial wirkende Zentrifugalkraft gleichmäßig zu einer Schicht homogener Dicke auseinandergetrieben (Belacken: Abbildung 4-4 B1). Durch anschließendes Ausheizen des belackten Glaswafers für 60 s bei 110 °C auf einer Heizplatte, "Soft bake", wurde die Lackschicht getrocknet. (B2) Im nächsten Schritt erfolgte die eigentliche Photostrukturierung. Die ChromQuarzglas-Maske wurde in einer UV-Belichtungsmaschine (MA-25, Karl Süss GmbH) auf die belackte Oberfläche des Wafers gepresst (Kontaktbelichtung). In dieser Anordnung erfolgte die Belichtung mit einer Energie von 30 mJ/cm² (Abbildung 4-4 B2). Nach Entwicklung des Photolacks für 30 s im Entwicklerbad (AZ 726 MIS, Clariant) und Abspülen aller löslichen Lackreste mit Milli-Q erschien die belichtete Struktur als Topographie in den Lack eingeprägt. Zur Festigung des Lacks und zur Glättung der Strukturkanten erfolgte eine abschließende Wärmebehandlung ,"Hard bake", bei 110 °C für 90 Minuten. 4-12 4 Kontrolliertes Zellwachstum Photolithographische Herstellung von Strukturmasken Teilschritt Arbeitsablauf (B) (C) (B1) Belacken (B2) Belichtung/ Entwicklung (B3) Lagerung • Glaswafer auf • Einspannen von Wafer • Zersägen des Wafers in Rotationsplatte ansaugen • Applikation eines Photolacktropfens (B1a) • Verteilen des Tropfens durch Rotation (B1b) • Trocknen des Photolacks Darstellung (A) B1a B1b und Chrommaske in ein Belichtungssystem • Belichten (B2a) • Entwickeln und Spülen einzelne Strukturmasken (B3a) • Lagerung in Kunstoffbehältern (B3b) • Thermisches Nachhärten auf einer Heizplatte B3a B2a B3b 3 cm Abbildung 4-4 Durch photolithographische Strukturierung einer Schicht aus Photolack wird in drei Schritten ein topographisches Relief (Strukturmaske) für die Abformung von Mikrostempeln hergestellt. (B1) Belacken des Glaswafers. (B2) Belichten mit UV Strahlung. (B3) Zuschneiden und Lagern der fertigen Strukturmasken. (B3) Im letzten Schritt wurden die einzelnen strukturierten Bereiche auf dem Wafer entlang der zu diesem Zweck im CAD-Entwurf angebrachten Markierungskreuze mit einer Wafersäge auseinandergesägt und in 24-WellPlatten (Falcon 3047, Becton Dickinson & Company) eingeordnet (Abbildung 4-4 B3). 4.2.1.3 Abformung Zur Abformung gebrauchsfertiger Stempel von den Strukturmasken wurde der Polymerwerkstoff Polydimethylsiloxan (PDMS; Sylgard 182, Dow Corning) verwendet. Dieser Werkstoff ist transparent, während der Verarbeitung flüssig und nach dem Aushärten gummiartig fest. Das Aushärten des PDMS erfolgt durch 4.2 Strukturierung mit Mikrostempeln 4-13 Beimischung eines Härters im Gewichtsverhältnis 1:10 und anschließendem Erhitzen 5 im Ofen . Schwierig bei der Abformung ist insbesondere das Ablösen des gehärteten PDMS von der Strukturmaske. Sind PDMS und Strukturmaske verklebt, so wird die Strukturmaske beim Ablösevorgang beschädigt. Dies kann wirksam verhindert werden, wenn die Strukturmaske vor Aufbringen des flüssigen PDMS einige Minuten mit einem Detergens benetzt wird. Hierzu wurde eine Lösung von 200 mM Natriumdodecylsulfat (SDS; SDS 71729, Fluka) in destilliertem Wasser verwendet. Die an der Oberfläche von Stempel und Strukturmaske verbleibenden Seifenreste können durch Spülen mit destilliertem Wasser und Reinigen im Ultraschallbad vollständig entfernt werden. Eine weitere Schwierigkeit bei der Abformung liegt in der geringen Hitzebeständigkeit des Photolacks. Es ist daher erforderlich das Aushärten auf der Strukturmaske bei einer Temperatur unter 70 °C durchzuführen. Um eine vollständige Härtung des PDMS dennoch zu gewährleisten, wurden alle abgeformten Stempel nach Ablösen der Strukturmaske einem zweiten Härteschritt bei 110 °C unterworfen. Werden diese beiden Vorsichtsmassnahmen zum Schutz der Strukturmasken beachtet, so können diese mehrfach wiederverwendet werden. Von einzelnen Strukturmasken wurden bis zu 6 Abformungen ohne erkennbare Beschädigung durchgeführt. Im Folgenden sind die einzelnen Schritte der Abformung als Anleitung beschrieben: 1. Anmischen von gebrauchsfertigem PDMS aus Rohmasse und Härter im Gewichtsverhältnis 10:1. Entgasen des fertigen Gemischs unter Vakuum bis optisch keine Blasenbildung mehr erkennbar ist6, zur Vermeidung späterer Blasenbildung durch im Werkstoff eingeschlossene Luft. 5 24 Stunden bei 60°C, 2 Stunden bei 100°C 6 Erfahrungsgemäß ist Evakuieren für ca. 1 Stunde ausreichend. 4-14 4 Kontrolliertes Zellwachstum Stempelabformung Teilschritt Arbeitsablauf (A) (C1) Aufbau Gusswerkzeug (C2) PDMS Ausguss • Auswahl einer • Aufsetzen eines PDMS (C2a) • Aushärten des PDMS bei Kunstoffzylinders auf die Strukturmaske (C1b) 60°C für 24h (C2b)2 • Entfernen der Metallscheibe und Ablösen der Strukturmaske (C3a) • Aufschneiden des Kunstoffzylinders und Herauslösen des fertigen Stempels (C3b) C2b • Beschweren des (C) (C3) Stempelentnahme • Ausgießen der Form mit Strukturmaske (C1a)1 (B) Kunstoffzylinders mit einer Metallscheibe (C1c) Darstellung C1a C1b C3a C3b C1c/C2a 1 cm 1 Die Strukturmaske wird vor der Abformung mit einem Detergens (SDS) benetzt, dies erleichtert das spätere Ablösen 2 Durch Ausgießen und Aushärten in zwei Stufen kann eine Beschriftung in den Stempel eingegossen werden Abbildung 4-5 Die Abformung eines Mikrostempels von einer Strukturmaske geschieht mit Hilfe eines speziellen Gusswerkzeugs. (C1) Aufbau des Gusswerkzeugs. (C2) Ausgießen der Gussform mit PDMS. (C3) Entnahme des fertigen Stempels. 2. Auswahl der gewünschten Stempelstruktur. Entnehmen der entsprechenden Strukturmaske aus dem staubdichten Vorratsbehälter. Strukturmaske in eine Petrischale legen und für ca. 15 Minuten an der strukturierten Oberfläche mit einem Tropfen SDS benetzen. Mit Druckluft Tropfen wegblasen und die Oberfläche vollständig trocknen. 3. Kunstoffzylinder als Gussform für den Ausguss mit PDMS auf der strukturierten Oberfläche der Strukturmaske anordnen: 1,5 ml Eppendorf Röhrchen (0030.121.589, Eppendorf) am Boden aufschneiden und auf den Kopf stellen. Zur erhöhten Abdichtung zwischen Gussform und Strukturmaske die Eppendorf Röhrchen mit Gewicht beschweren. Dazu eine 12 g schwere, im Innendurchmesser exakt auf das Röhrchen passende Beilagscheibe aus Metall von 4.2 Strukturierung mit Mikrostempeln 4-15 oben auf das Röhrchen stecken. Die gesamte Anordnung jetzt mittig auf der Strukturmaske plazieren (Abbildung 4-5 C1). 4. Die fertige Abgussform zur Hälfte mit PDMS auffüllen. Mit Hilfe einer 1 ml Einweg-Spritze 0,5 ml des zähflüssigen Werkstoffs von oben in die Gussform tropfen (Abbildung 4-5 C2). 5. Aushärten des PDMS in der Gussform während 24 h bei 60 °C auf einer 7 Heizplatte . 6. Beschriftung des Stempels zur späteren einfachen Identifizierung: Transparentfolie mit der Nummer der abgeformten Strukturmaske beschriften. Transparentfolie zuschneiden, auf die ausgehärtete erste PDMS-Schicht legen und durch Zugabe von weiterem PDMS eingießen. 7. Aushärten der hinzugegebenen PDMS Masse; diesmal während 12 h bei 60 °C im Ofen. 8. Herauslösen des fertigen Stempels aus der Gussform: Strukturmaske mit der Hand vorsichtig an den Ecken anheben und vom Stempel lösen. Die Beilagscheibe von der Gussform abziehen. Die Kunstoffhülse mit einem Skalpell der Länge nach vom strukturierten Ende aus aufschneiden und den fertigen Stempel herauspellen (Abbildung 4-5 C3). 9. Zur vollständigen Aushärtung des PDMS, den fertigen Stempel noch einmal bei 110 °C für eine Stunde in den Ofen legen. Um das Verfahren zu beschleunigen, wurden jeweils sechs oder zwölf Stempel gleichzeitig abgeformt. Dazu wurden je sechs Strukturmasken gleichmäßig im Deckel einer Petrischale aus Glas verteilt, darauf die Gussformen gestellt und die gesamte Anordnung wie ab Schritt 4 beschrieben behandelt. 7 Die Verwendung einer Heizplatte in diesem Schritt bewirkt durch den entstehenden Temperaturgradienten im Stempel eine Erwärmung des PDMS zuerst am Boden der Gussform, also an der Grenzfläche zur Strukturmaske. Dadurch entweichen etwaige, beim Ausgießen eingebrachte Luftblasen nach oben. 4-16 4 Kontrolliertes Zellwachstum 4.2.2 Stempelübertrag Die fertigen Mikrostempel wurden bis zum Gebrauch trocken in staubdichten Kunstoffgefäßen gelagert. 24 Stunden vor jedem Experiment wurden die benötigten Stempel aus den Gefäßen entnommen, 15 Minuten mit destilliertem Wasser bei 60 °C im Ultraschallbad gereinigt und bis zum Experiment in destilliertem Wasser Stempelübertrag Teilschritt Arbeitsablauf (A) Benetzen (B) Trocknen (C) Stempelübertrag • Stempel kopfüber • Stempeloberfläche • Stempel auf das Substrat in eine wässrige Proteinlösung tauchen in einem Stickstoffstrahl abtrocknen • Stempel wieder abheben > 30 Sekunden Stempel • Stempel 10 Sekunden andrücken • Benetzungszeit Darstellung aufsetzen Stempel Stempel Stempel Stickstoff Stempel ProteinLösung Stempel Substrat Substrat Substrat Abbildung 4-6 Der eigentliche Stempelübertrag von Proteinen auf ein Substrat erfolgt in drei Schritten. (A) Benetzen des Stempels mit Proteinen. (B) Trocknen der Stempeloberfläche. (C) Aufbringen des Stempelabdrucks auf das Substrat. aufbewahrt. Unmittelbar vor Beginn des Experiments erfolgte eine Desinfektion der Stempeloberfläche durch einminütiges Eintauchen in 70 % iges Ethanol und abschließendes Trocknen im Stickstoffstrahl. Der eigentliche Stempelübertrag erfolgte wie in Abbildung 4-6 dargestellt, in drei Schritten: 4.2 Strukturierung mit Mikrostempeln 4-17 1. Zunächst wurde die Stempeloberfläche mit dem zu übertragenden Protein benetzt (Abbildung 4-6 A). Dazu wurden die Stempel jeweils für mindestens 30 8 9 Sekunden in eine wässrige Laminin-Lösung getaucht . 2. Die angefeuchteten Stempel wurden dann mit einem 1,5 bar Stickstoffstrahl vorsichtig getrocknet (Abbildung 4-6 B). Insbesondere wurde auf ein langsames Eintrocknen an der Oberfläche geachtet, ein Wegblasen der aufgenommenen Substanz von der Oberfläche wurde vermieden10. 3. Jetzt erfolgte der eigentliche Stempelübertrag durch Aufsetzen und Andrücken des Stempels auf die Substratoberfläche für ca. 10 Sekunden (Abbildung 4-6 C). Mit einer Andruckkraft von 15 g/cm² wurden die besten Ergebnisse erzielt (Abbildung 4-7 B). Wesentlich kleinere Kräfte führten zu Fehlstellen im Stempelübertrag (Abbildung 4-7 A). Größere Kräfte resultierten in einer Deformation des Stempels, verbunden mit einer Veränderung der übertragenen Struktur (Abbildung 4-7 C). Dieses Ergebnis für die optimale Größe der Andruckkraft ist in Übereinstimmung mit früheren Veröffentlichungen [Branch, 2000]. Zwischen den Stempelüberträgen einer Versuchsreihe wurden die Stempel nicht gereinigt oder desinfiziert. Alle Arbeiten wurden im gefilterten Luftstrom einer Sterilbank (HB 2436, Heraeus Instruments GmbH) ausgeführt. Dadurch war eine Kontamination der Stempel und der Substrate mit Keimen ausgeschlossen. 8 Bei Benetzungszeiten unter 30 Sekunden war ein deutlich schlechterer Stempelübertrag zu beobachten. 9 25 µg/ml (~0,25 µM) Laminin (1243217, Roche Diagnostics GmbH) in PBS Pufferlösung. 10 Die Stempeloberfläche muss in jedem Fall vollständig getrocknet sein. Bei unvollständiger Trocknung zurückgebliebene Tropfen verteilen sich bei Aufsetzen des Stempels auf das Substrat durch Kapillarkräfte über weite Bereiche der Struktur und verschmieren den Stempelübertrag. 4-18 4 Kontrolliertes Zellwachstum Einfluss der Andruckkraft auf den Stempelübertrag Andruckkraft FA (A) FA < 5 g/mm² (B) 10 g/mm² < FA < 20 g/mm² (C) FA > 35 g/mm² Strukturübertrag einer Gitterstruktur1 50 µm Bewertung <Abb5_ucp_20g.tif> • Ungleichmäßiger und unvollständiger Kontakt. • Nur Strukturfragmente werden übertragen. <Abb5_ucp_50g.tif> • Gleichmäßiger Strukturübertrag. • Präzise Abbildung der <Abb5_ucp_100g.tif> • Starke Deformation des Stempels. • Strukturen sind verzerrt2 . Stempelstruktur. 1 Die effektive Oberfläche dieser Struktur beträgt 588 µm² je 5000 µm² Stempelfläche. Für Stempel mit wesentlich kleineren oder größeren effektiven Oberflächen muss die Andruckkraft im entsprechenden Verhältnis angepasst werden. 2 Der Stempel wölbt sich aufgrund des hohen Drucks zwischen den einzelnen Strukturelementen zum Substrat durch. In Bereichen großer Strukturzwischenräume kommt es zum Kontakt dieser Wölbung mit dem Substrat. Abbildung 4-7 Die Andruckkraft beim Stempelübertrag beeinflusst ganz wesentlich die Qualität des Strukturübertrags auf das Substrat. (A) Bei unzureichendem Stempelandruck werden nur Fragmente der Struktur übertragen. (B) Ein vollständiger, präziser Strukturübertrag wird mit Kräften zwischen 10 und 15 g/cm² erzielt. (C) Zu große Kräfte führen, bedingt durch Verformung des Stempels, zu einer Verzerrung der Struktur sowie zu Artefakten in den Strukturzwischenräumen. Nach Abschluss der Stempelarbeiten wurden die benutzten Stempel erneut wie oben beschrieben im Ultraschallbad gereinigt, mit Stickstoff getrocknet und zurück in die Kunstoffgefäße gelegt. Die strukturierten Substrate wurden bis zur Aussaat der Zellen bei +10 °C im Kühlschrank gelagert. 4.3 Oberflächenchemie für maximalen Kontrast 4-19 4.3 Oberflächenchemie für maximalen Kontrast Eine Steuerung neuronalen Wachstums mit Strukturen aus Laminin ist nur dann erfolgreich, wenn die Substratoberfläche in den nicht mit Laminin beschichteten Bereichen ausreichend zellabweisend ist. Bietet die unmodifizierte Oberfläche den Zellen zuviel Halt, so siedeln sich Nervenzellen auch neben den Lamininstrukturen 11 an . Die für die Haftung erforderlichen Proteine werden dabei von bestimmten Zellen selbständig gebildet [Collins, 1980]. Entscheidend für den zellabweisenden oder zellfreundlichen Charakter einer Oberfläche ist, wie gut bestimmte Proteine an der Oberfläche anhaften. Hydrophobe Oberflächen wirken im allgemeinen zellabweisend. Hydrophile Oberflächen hingegen begünstigen die Zellhaftung [Kleinfeld, 1988], ebenso wie Oberflächen mit einer hohen Dichte an Aminogruppen [Stenger, 1992]. Für elektrophysiologische Messungen an Nervenzellen gibt es keine technische Einschränkung für das Material des Zellsubstrats. Es ist folglich ohne Schwierigkeit möglich, direkt ein Basissubstrat aus einem geeignet hydrophoben Material, wie beispielsweise Polystyrol, einzusetzen. Petrischalen aus Polystyrol sind kommerziell erhältlich. Die optische Transparenz von Polystyrol gestattet zudem eine Beobachtung der Probe mit Durchsicht-Phasenkontrastmikroskopie. Elektrophysiologische Untersuchungen wurden daher ausschließlich an NervenzellNetzwerken auf Substraten aus Polystyrol durchgeführt. Andere Untersuchungsverfahren als die Patch-Clamp Technik hingegen, sind unmittelbar an ein bestimmtes Substrat-Material gekoppelt. Um strukturierte Netzwerke von Nervenzellen auch in diesem Fall an der Substratoberfläche erzeugen zu können, ist eine chemische Modifikation der Oberfläche erforderlich. Sensorchips für die extrazelluläre Messung der Signale von Nervenzellen beispielsweise, haben eine Oberfläche aus Siliziumoxid [Sprössler, 1997; Krause, 11 Wie schwierig es im allgemeinen ist, den Bewuchs von Oberflächen wirksam zu unterbinden, wird anhand eines ganz anderen Beispiels deutlich: Im Schiffbau werden auch heute noch hochgiftige, zinn- und bleihaltige Farben für den Außenanstrich von Schiffsrümpfen verwendet. Nur so kann ein übermäßiger Bewuchs mit Muscheln, etc. zumindest für einige Jahre vermieden werden. 4-20 4 Kontrolliertes Zellwachstum 2000]. Eine Zielsetzung dieser Arbeit war es, Netzwerke von Nervenzellen auch einer Messung mit solchen Sensoren zugänglich zu machen. Im Folgenden werden zwei chemische Verfahren beschrieben, welche eine geeignete Modifikation von Siliziumoberflächen gestatten. 4.3.1 Beschichtung mit Polystyrol In verschiedenen Experimenten wurden die Oberflächen von Siliziumwafern mit Polystyrol beschichtet. Die Beschichtung erfolgte im "Grafting-to-Verfahren" [Prucker, 1995]. Dazu wurde zunächst eine Monolage von Benzophenonsilan auf Siliziumoxid immobilisiert. Die Kopplung des Polystyrol erfolgte anschließend in einer Lösung von Polystyrol (2,38 kg/mmol), gelöst in Toluol, durch Photoaktivierung für eine Dauer von 30 Minuten bei Raumtemperatur. Nach Ablauf der Reaktion wurde die Schichtdicke12 des kovalent gebundenen Polymers zu ~10 nm bestimmt. Wurden diese Substrate mit Laminin strukturiert, so war ein ebenso hoher Haftungskontrast der Zellen13 zu beobachten wie auf Petri-Schalen aus reinem Polystyrol. Die auf reinen Polystyrolsubstraten erzielten Ergebnisse strukturierten neuronalen Wachstums können daher auf Siliziumoberflächen übertragen werden, wenn eine Beschichtung der Oberfläche mit Polystyrol erfolgt. 4.3.2 Kovalente Bindung von Laminin an Aktivester-Bürsten Durch die Beschichtung von Siliziumoxid mit Polystyrol ändern sich die physikalischen Eigenschaften der Oberfläche dahingehend, dass sich die Oberfläche ebenso hydrophob wie ein massives Substrat aus Polystyrol verhält. Der Übertrag von Laminin auf diese Oberfläche erfolgt weiterhin durch Physisorption. Man kann bei der Oberflächenmodifikation aber auch noch einen Schritt weitergehen und reaktive Gruppen für eine kovalente Anbindung des übertragenen Laminins an der Oberfläche immobilisieren. Ein solches reaktives System hat gegenüber der Strukturierung durch physisorbiertes Laminin vor allem zwei Vorteile: 12 Ellipsometrische Messung. 13 Die Versuche wurden mit der Zellinie MzN durchgeführt. 4.3 Oberflächenchemie für maximalen Kontrast 4-21 1. Die Menge des übertragenen Laminins kann kontrolliert werden und ist über die gesamte Fläche des Stempelabdrucks weitgehend homogen. Spült man das Substrat nach Abschluss der Reaktion zwischen Laminin und den reaktiven Gruppen gründlich ab, so bleibt nur noch kovalent gebundenes Laminin an der Oberfläche zurück. Dessen Menge ist korreliert mit der Anzahl freier Bindungsstellen der ursprünglichen Oberflächenbeschichtung [Golze, 2001]. Bei einem Stempelübertrag durch Physisorption hingegen ist eine solche Kontrolle nicht möglich. Mehrere Faktoren bestimmen in diesem Fall die Menge des übertragenen Laminins: Die Lamininkonzentration im "Tintenfass" beeinflusst die Menge adsorbierten Laminins auf der Stempeloberfläche. Die tatsächlich übertragene Menge variiert mit den Oberflächeneigenschaften von Substrat und Stempel. Weiterhin kommt es zu Inhomogenitäten im Stempelübertrag und Abweichungen der übertragenen Menge zwischen verschiedenen Experimenten. 2. Der zweite Vorteil liegt in der größeren Stabilität der strukturierten Oberfläche. Diese bleibt über längere Zeit stabil und das Substrat ist insgesamt einfacher zu handhaben. Kovalent angebundenes Laminin geht auch bei unkontrolliertem Spülen der Substratoberfläche mit Kulturmedium nicht in Lösung. Spült man hingegen ein mit Laminin strukturiertes Polystyrolsubstrat vor der Zellaussaat, so lösen sich im ungünstigsten Fall weite Bereiche der Struktur von der Oberfläche ab. Diese Vorteile rechtfertigen den mit der Herstellung einer reaktiven Oberfläche verbundenen erhöhten Aufwand. Auch für Patch-Clamp Messungen ist eine solche Substratoberfläche attraktiv. Verwendet man in diesem Fall als Basissubstrat Quarzglas, so ist weiterhin Durchlicht-Phasenkontrastmikroskopie möglich. In den folgenden Abschnitten wird ein System mit einer speziellen reaktiven Oberfläche, synthetisiert auf Basis von Copolymerbürsten aus N,NDimethylacrylamid (DMAA) und einem C5Aktivester (C5AE) beschrieben. Dieses System wurde ausgewählt, da es in anderem Kontext bereits erfolgreich für die Immobilisierung von DNA an DNA-Chips erprobt wurde [Golze, 2001]. Es zeichnete sich dabei gegenüber Systemen mit nur einer Monolage aktiver Gruppen vor allem 4-22 4 Kontrolliertes Zellwachstum durch die erzielbare höhere Menge an gebundener DNA aus. Im Hinblick auf neuronale Zellkulturen bietet das System insofern eine weitere interessante Eigenschaft: Aufgrund der Dreidimensionalität der Copolymerbürsten entsteht nach Anbindung von Laminin an diese Struktur eine ebenfalls dreidimensionale extrazelluläre Matrix. Das resultierende System ist den natürlichen Gegebenheiten im Organismus in dieser Beziehung also ähnlicher als andere Substrate auf Basis von reaktiven Monolagen oder einfacher Physisorption. 4.3.2.1 Mechanismus von Polymerisation und Lamininbindung In Abbildung 4-8 ist der verwendete Prozess zur Herstellung von Siliziumsubstraten mit kovalent gebundenem Laminin beschrieben. Er umfasst vier Schritte A bis D. Im ersten Schritt A erfolgt eine Aktivierung der Oberfläche mit 25 % iger Schwefelsäure bei 80 °C für 20 Minuten. In Schritt B wird der Initiator für die spätere Polymerisation während einer 15 stündigen Reaktionszeit bei 20 °C unter Schutzgas an die aktivierte Oberfläche angebunden. Die Reaktion erfolgt in Toluol und wird durch Triethylamin katalysiert. Schritt C umfasst die eigentliche Polymerisation; die Substrate werden hierzu in ein Gemisch der Monomere DMAA und C5AE gelegt. Die Polymerisation erfolgt bei 60 °C während 16 h in DMF. Durch anschließende Extraktion in CH2Cl2 wird freies, ungebundenes Polymer von der Oberfläche gelöst. Im letzten Schritt D erfolgt die Strukturierung des Substrats mit Laminin. Nach Übertrag der Lamininstruktur muss das Substrat vor der Verwendung noch mindestens 10 h bei 37 °C und gesättigter Luftfeuchte gelagert werden, um eine vollständige Reaktion aller aktiven Gruppen mit dem Laminin sicherzustellen (siehe hierzu Punkt 4.3.2.2). 4.3 Oberflächenchemie für maximalen Kontrast 4-23 Herstellung von Substraten mit kovalent gebundenem Laminin (A) Oberflächenaktivierung (B) Initiatorreaktion (C) Polymerisation Chemikalien • H2SO4, 25 % • Initiator (I) • Katalysator Lösungsmittel • H2O • Toluol Reaktionsbedingungen • 80 °C, 20 Min. • 20 °C, Argon, 15 h • 60 °C, 16 h • C5AE:DMAA = 1:4, Reaktionsschema Resultierende Substratoberfläche 1 Triethylamin (K) • Aktivester (C5AE) • Dimethylacrylamid --> Si-OH + H2O • Si-OH + I --> Si-O-I' + HCl • HCl + K --> Cl- + KH+ Initiator (I) Aktiviertes Siliziumoxid (Si-OH) Siliziumoxid (SiO2) Silizium (Si) • Laminin (L) (DMAA) • DMF • Extraktion in CH2Cl2 1,5 mol/l (1) • Si-O + H3O+ (D) Bestempeln mit Laminin • PBS • Mikrostempeln • 37 °C, 100 % Luftfeuchte, 10 h • Si-O-I' -- 60 °C--> • …-C5AE + L --> Si-O-I'' + I''frei …-C5AE'-L • Si-O-I'' + C5AE + DMAA --> Si-O-I''-[C5AE]n-[DMAA]m Laminin (L) Aktivester-DMAA-Copolymer (CoPo) Höhere Konzentrationen (z.B. 3.5 Mol/l) ergeben eine dickere AE-Schicht. Die Oberflächenrauhigkeit steigt aber ebenfalls. Abbildung 4-8 Substrate zur kovalenten Bindung von Lamininstrukturen wurden auf Basis eines reaktiven Copolymers hergestellt. Der Herstellungsprozess umfasst vier Schritte. (A) Aktivierung der Siliziumoberfläche (B) Anbinden eines Initiators für die Polymerisation (C) Polymerisation (D) Kovalente Anbindung von Laminin Das fertig strukturierte Substrat hat wie in Abbildung 4-8 unten dargestellt, am Ende des Prozesses an seiner Oberfläche eine Schichtstruktur von 6 unterschiedlichen Materialien. Auf dem Basismaterial Silizium (Schicht 1: Si) sitzt eine Oxidschicht (Schicht 2: SiO2). An der obersten Lage dieser Schicht wurden im Aktivierungsschritt A freie OH-Gruppen erzeugt (Schicht 3: Si-OH). Darauf folgt der Initiator (Schicht 4: I), das Copolymer (Schicht 5: CoPo) und schließlich das Laminin (Schicht 6: L). Abbildung 4-9 und Abbildung 4-10 zeigen die Reaktionsschritte B, C und D auf molekularer Ebene: Die Anbindung von symmetrisch aufgebauten Initiatormolekülen an die OH-Gruppen der aktivierten Oberfläche (Abbildung 4-9 B1) erfolgt unter Freisetzung von HCl. Der Katalysator Triethylamin ist notwendig um dieses abzufangen und dadurch das Reaktionsgleichgewicht 4-24 4 Kontrolliertes Zellwachstum Reaktionsablauf - Oberflächenmodifikation (B1) Freier Initiator Cl H3C Initiator gebunden an Siliziumoberfläche CH3 Si (C) Nach thermischer Spaltung des Initiators wächst ein statistisches Copolymer (C1) m (B) Der Initiator bindet an die aktivierte Siliziumoberfläche O Cl H3C CH3 Si H C5AE N H3C O O O O O n O O CH3 CN H3C N DMAA N N NC H3C CH3 CN N NC CH3 NC CH3 O Startradikal N O N O CH3 CH3 NC O H3C Si Cl Si OH Si O Si (B2) N [ CH2 O + + OH O O O O H3C OH O O CH3 Si OH Si CH3 O O H3C Si CH3 OH Si O Si CH3 ]3 Triethylamin H N + Si Si CH3 N [ CH2 CH3 ]3 Triethylammoniumchlorid Si O (C2) H2C Cl- H3C O O DMAA CH3 H2C O CH3 CH3 O O N H C AE 5 O N O Abbildung 4-9 Details der Oberflächenmodifikation auf Molekularer Ebene. (B) Anbindung des Initiators. (C) Aufwachsen des statistischen Copolymers aus C5AE und DMAA zugunsten der Produkte zu verschieben (Abbildung 4-9 B2). Durch die Erwärmung auf 60 °C in Reaktionsschritt C kommt es zur Spaltung des Initiators unter Freisetzung von Stickstoff. Das Fragment des Initiators, welches nicht an die Siliziumoberfläche gebunden ist, geht dabei in Lösung und bildet freies, 14 ungebundenes Polymer . Das an die Siliziumoberfläche gebundene Fragment des Initiators hingegen bildet den Ankerpunkt für das Aufwachsen einer statistischen Copolymerkette auf die Siliziumoberfläche. Die beiden Komponenten DMAA und C5AE der Copolymerkette, dargestellt in Abbildung 4-9 C2, lagern sich dabei beginnend beim freien radikalischen Ende des gebundenen Initiators in zufälliger Reihenfolge zu einem statistischen Copolymer zusammen (Abbildung 4-9 C1). 14 Aus diesem Grund wird die Substratoberfläche nach abgeschlossener Polymerisation mittels Extraktion in CH2Cl2 von freiem, ungebundenem Polymer gesäubert. 4.3 Oberflächenchemie für maximalen Kontrast 4-25 Reaktionsablauf - Kovalente Bindung von Laminin m m (D) Laminin bindet durch Reaktion mit dem Aktivester kovalent an die Oberfläche an H O H C5AE O N H3C O N H3 C O O O DMAA n n O C5AE N N H CH3 CH3 N N DMAA CH3 O CH3 NC CH3 O NC CH3 + O + O O O H O O O H H3C Si O Abgangsgruppe N CH3 H N Laminin Si Copolymeroberfläche auf Silizium H3C Si CH3 O Si Laminin kovalent an Copolymer gebunden Abbildung 4-10 Das auf das Copolymer übertragene Laminin wird durch Reaktion mit dem C5AE kovalent an die Oberfläche angebunden. Beide Komponenten DMAA und C5AE haben spezielle Eigenschaften, die in Kombination gemeinsam das Verhalten des Copolymers bestimmen: − Das DMAA verleiht der Copolymerbürste die Eigenschaft der Quellbarkeit in Wasser. Nur aufgrund dieser Eigenschaft sind tiefer unter der Oberfläche liegende C5AE-Gruppen später für in Wasser gelöste Reaktionspartner wie z.B. Laminin zugänglich [Golze, 2001]. − Die reaktiven Komponenten der Copolymerbürste werden durch die Aktivestergruppen C5AE gebildet. In Abbildung 4-10 ist ihre Rolle bei der kovalenten Anbindung von Laminin an das Substrat in Schritt D dargestellt. Eine C5AE-Gruppe der Copolymerbürste reagiert dabei mit einer Aminogruppe des Laminins. Es wird eine kovalente Bindung zu einem Stickstoffatom des Laminins gebildet. Bei dieser Reaktion spaltet sich eine Abgangsgruppe vom C5AE ab und geht in Lösung. 4-26 4 Kontrolliertes Zellwachstum Der relative Anteil von DMAA zu C5AE hat wesentlichen Einfluss auf die Eigenschaften der Copolymeroberfläche. In den durchgeführten Versuchen haben verschiedene Verhältnisse von DMAA zu C5AE zu unterschiedlichen Ergebnissen in der Zellkultur geführt. Die besten Resultate wurden mit einem molaren Mischungsverhältnis der Monomere von C5AE : DMAA = 1 : 3 erzielt (siehe Punkt 4.3.2.3). 4.3.2.2 Kinetik der Reaktion mit Laminin und Schichtdickenmessung Für eine quantitative Untersuchung des Systems ist es notwendig, nicht nur den Reaktionsmechanismus zu kennen sondern auch die Länge und die quantitative Zusammensetzung der an der Oberfläche verankerten Copolymerbürsten kontrollieren zu können. Hierzu sind geeignete Messverfahren zur Bestimmung der entsprechenden Parameter erforderlich. Als Maß für die Länge kann die Schichtdicke der resultierenden Copolymerschicht herangezogen werden. Diese läßt sich z.B. durch Ellipsometrie ermitteln. Das Verhältnis von DMAA zu C5AE im Copolymer kann über IR-Spektroskopie bestimmt werden. Eine Bestimmung über NMR-Methoden ist nicht möglich, da die charakteristischen Signale von C5AE und DMAA sich überlagern [Golze, 2001]. In Abbildung 4-11 A ist ein charakteristischer Ausschnitt des IRAbsorptionsspektrums einer DMAA-C5AE-Copolymerschicht aufgetragen. Zwischen den Wellenzahlen 1825 cm-1 und 1575 cm-1 erkennt man deutlich vier Absorptionsbanden. Die Banden 1, 2 und 3 zwischen 1825 cm-1 und 1700 cm–1 resultieren dabei aus Schwingungsmoden der C5AE-Komponente des Copolymers. Die vierte Bande zwischen 1700 cm-1 und 1575 cm-1 kann der DMAA-Komponente des Copolymers zugeordnet werden. 4.3 Oberflächenchemie für maximalen Kontrast 4-27 Reaktionsablauf - Nachweis der Bindung von Laminin (B) IR-Spektren im Verlauf der Reaktion mit Laminin1 (A) IR-Spektrum einer Copolymerschicht 0 2 4 6 8 10 10 DMAA (4) 2,0 8 C AE 5 1,5 6 (3) 1,0 0,5 4 2 (1) (2) 2,5 IR-Absorption [%] IR-Absorption [%] 2,5 0 2 4 1,5 8 10 10 DMAA (4) 0h 0,5 h 17 h 2,0 8 6 C AE (3) 5 1,0 0,5 6 4 2 (1) (2) 0 1800 1750 1700 1650 0 1600 1800 Wellenzahl [cm-1] 1700 1650 1600 Wellenzahl [cm-1] • C5AE und DMAA haben charakteristische • Nach Reaktion mit Laminin verschwindet die • Integration über die Fläche der Banden ermöglicht • Die Bande des DMAA bleibt unverändert. Absorptionsbanden. Bande des C5AE. einen quantitativen Vergleich. 1 1750 Reaktion im Inkubator bei 37 °C und gesättigter Luftfeuchte Abbildung 4-11 Die Zusammensetzung des Copolymers und die Reaktion des C5AE mit Laminin kann anhand von IR-Absorptionsspektren untersucht werden. (A) Typisches IR-Spektrum einer Copolymerschicht. (B) Veränderung des IR-Spektrums bei Reaktion mit Laminin. Integriert man über die zwischen x-Achse und Absorptionsspektrum eingeschlossenen Flächen der einzelnen Banden (grau schattiert in Abbildung 4-11 A), so kann man aus einem Vergleich des Flächeninhalts der C5AE-Banden mit dem Flächeninhalt der DMAA-Bande das Mengenverhältnis von C5AE : DMAA im Copolymer bestimmen. X (C5 AE : DMAA) = X 0 * X X0 = = A(n) = 1700 1575 1825 1700 ∫ A(n)dn ∫ A(n)dn Gleichung 4-1 Molenbruch von C5AE zu DMAA im Copolymer. Eichkonstante zur Berücksichtigung der abweichenden Absorptionsstärke von C5AE und DMAA; wird ermittelt aus den Spektren der Homopolymere. Absorptionsgrad [%]. Basierend auf diesem Messverfahren wurden quantitative Untersuchungen mit systematisch variierten Copolymerzusammensetzungen durchgeführt [Golze, 2001]. Das Ergebnis ist folgende Wertetabelle. Die Zusammensetzung der Copolymerbürste 4-28 4 Kontrolliertes Zellwachstum kann damit aus dem Mischungsverhältnis von C5AE- zu DMAA-Monomer zum Zeitpunkt der Polymerisation bestimmt werden: C5AE - Anteil am Monomergemisch 0% 25% 33% 47% 64% 100% C5AE - Anteil im Copolymer 0% 45% 48% 55% 62% 100% Tabelle 4-1 Zusammenhang zwischen der Zusammensetzung von Monomergemisch vor der Polymerisation und Copolymer nach der Polymerisation. Anhand des IR-Absorptionsspektrums kann jedoch nicht nur die Zusammensetzung des Copolymers bestimmt werden sondern auch der zeitliche Verlauf der Reaktion von C5AE mit Laminin analysiert werden. Überträgt man wie in Punkt 4.2.2 beschrieben, mit einem unstrukturierten Stempel aus PDMS eine Schicht Laminin auf die Copolymerschicht und nimmt erneut ein IR-Spektrum auf, so erkennt man zunächst keinen Unterschied zu vorher (Spektrum aufgenommen bei 0 h Reaktionszeit in Abbildung 4-11 B). Das Protein Laminin zeigt folglich keine Absorptionsbanden im betrachteten Bereich des Spektrums15. Läßt man das Laminin jedoch eine ½ Stunde in einem Inkubationsschrank mit der Copolymeroberfläche reagieren und nimmt erneut ein Spektrum auf, so tritt eine deutliche Veränderung des Spektrums auf (Spektrum bei 0,5 h Reaktionszeit in Abbildung 4-11 B). Zumindest die dritte Absorptionsbande des Aktivesters hat gegenüber dem ersten Spektrum deutlich abgenommen. Der Absorptionspeak des DMAA (Bande 4 im Spektrum) hingegen ist nicht geschrumpft. Nach weiteren 16 ½ Stunden Reaktionszeit sind alle drei Banden des Aktivesters nahezu vollständig verschwunden (Spektrum bei 17 h Reaktionszeit in Abbildung 4-11 B). Die reaktiven C5AE-Gruppen der Copolymeroberfläche sind zu diesem Zeitpunkt durch Reaktion mit Laminin umgesetzt und haben ihr charakteristisches Absorptionsspektrum verloren. 15 Eine messbare Photoabsorption tritt bei Proteinen erst im UV-Bereich auf. Diese geht aus von den Resonanzen der Elektronenverteilung in den aromatischen Ringen der Aminoäuren Phenylalanin, Tyrosin und Tryptophan. 4.3 Oberflächenchemie für maximalen Kontrast 4-29 Reaktionsablauf - Geschwindigkeit der Bindung von Laminin (A) Zeitlicher Verlauf der Reaktion mit Laminin Luftfeuchte: Reines Copolymer Copolymer mit Laminin 100% Luftfeuchte: 0% 100% 100 80 τ = 28,4 ± 5,5 h C AE-Anteil [%] 60 40 τ = 8,1 ± 1,6 h 80 60 40 5 C5AE-Anteil [%] 100 0%, (B) Zeitliche Stabilität des Copolymers 20 0 0 12 24 36 48 60 72 84 96 20 0 Tag 0 Tag 30 Reaktionszeit [h] • Der Reaktionsverlauf wird durch eine Exponentialfunktion beschrieben. • Die Reaktionsgeschwindigkeit ist abhängig von der umgebenden Luftfeuchtigkeit. Tag 30 + 12 h • Unter Schutzgas (0 % Luftfeuchte) reagiert auch nach 30 Tagen nur 20 % des AE. • Wird Feuchtigkeit zugeführt, setzt eine schnelle Reaktion ein wenn Laminin an der Oberfläche ist. Abbildung 4-12 Durch Auswertung der IR-Spektren kann der zeitliche Verlauf der Reaktion mit Laminin ermittelt werden. (A) Reaktion bei bei 0 % und 100 % Luftfeuchte. (B) Vergleich der Stabilität von mit Laminin bestempeltem und unbestempeltem Copolymer. Der Anteil an noch nicht umgesetztem Aktivester, XAE ,läßt sich aus dem C5AE : DMAA-Verhältnis X nach Gleichung 4-1 zu den unterschiedlichen Zeitpunkten tn wie folgt bestimmen: X AE (t n ) = X (tn ) X (t0 ) XAE(t)= X(t) = t0 = = tn Gleichung 4-2 Anteil des noch nicht umgesetzten C5AE der Copolymerbürste zum Zeitpunkt t [%]. Verhältnis von C5AE zu DMAA zum Zeitpunkt t [%]. Zeitpunkt des Bestempelns mit Laminin [h]. Zeitpunkt der Messung [h]. Trägt man die Werte für XAE gegen die Zeit t auf (Abbildung 4-12 A), so erkennt man einen deutlichen Abfall des Aktivesteranteils über die Reaktionszeit. 4-30 4 Kontrolliertes Zellwachstum Der Reaktionsverlauf läßt sich als Reaktion erster Ordnung16 mit einer Exponentialfunktion beschreiben: X AE (t ) = A + (1 − A) e XAE(t)= A t τ = = = − t τ Gleichung 4-3 Anteil des noch nicht umgesetzten C5AE der Copolymerbürste zum Zeitpunkt t [%]. Verbleibender C5AE-Anteil zum Ende der Reaktion [%]. Zeitpunkt der Reaktion [h]. Zeitkonstante der Reaktion [h]. Mit diesem Fomalismus wurden die Reaktionsbedingungen für die kovalente Anbindung von Laminin näher untersucht. In Abbildung 4-12 A ist der zeitliche Verlauf der Reaktion für zwei unterschiedliche Reaktionsbedingungen, 0 % Luftfeuchte unter Schutzgas und gesättigte Luftfeuchte im Inkubationsschrank, aufgetragen. Beide Verläufe lassen sich gut mit Gleichung 4-3 beschreiben. Obwohl beide Reaktionen bei gleicher Temperatur T = 37 °C und Druck p = 1 bar abgelaufen sind, verläuft die Reaktion unter Schutzgas mit einer Halbwertszeit von τ = 28,4 ± 5,5 h wesentlich langsamer als bei gesättigter Luftfeuchte mit τ = 8,1 ± 1,6 h. Neben der langsameren Reaktionsgeschwindigkeit ist auch der insgesamt umgesetzte Anteil an C5AE deutlich geringer. Bei 100 % Luftfeuchte reagieren annähernd 80 % des Aktivesters. Bei 0 % Luftfeuchte auch nach sehr langen Reaktionszeiten von bis zu 30 Tagen (siehe auch Abbildung 4-12 B) weniger als 20 %. Der Grad der Luftfeuchte bestimmt also ganz wesentlich den Ablauf der Reaktion. Dies wird auch in folgendem Experiment deutlich: Überführt man ein zuvor bei 0 % Luftfeuchte gelagertes Substrat in eine Umgebung mit gesättigter Luftfeuchte, so kommt es wie bei dem unmittelbar in gesättigter Luftfeuchte gelagertem Substrat auch hier zu einer schnellen Reaktion des Aktivesters mit Laminin. Dies geschieht unabhängig von der vorherigen Dauer der Lagerung unter Schutzgas (vergleiche Abbildung 4-12 A und B). 16 Man kann davon ausgehen, dass Laminin im Überangebot vorliegt. Die Reaktionsgeschwindigkeit wird dann dominant vom Reaktionspartner C5AE bestimmt. In diesem Fall ist die Annahme einer Reaktion erster Ordnung gerechtfertigt. 4.3 Oberflächenchemie für maximalen Kontrast 4-31 Dieses Verhalten kann man sich aus der Quellbarkeit der Copolymerschicht erklären. Nur wenn das Copolymer durch Einlagerung von Wasser aufquillt, kann das aufgebrachte Protein an die tiefer unter der Oberfläche liegenden C5AE-Gruppen gelangen und daran kovalent gebunden werden. Dass eine Reaktion der C5AE-Gruppen tatsächlich mit dem aufgebrachten Laminin erfolgt, wird mit den Messergebnissen in Abbildung 4-12 B belegt: Darin ist der Aktivesteranteil eines Substrats mit übertragenem Laminin dem eines Substrats ohne Laminin gegenübergestellt. Wurde kein Laminin auf das Substrat übertragen, so misst man nach 30 Tagen Lagerung unter Schutzgas noch immer einen unveränderten Anteil von 100 % Aktivester. War hingegen Laminin auf der Oberfläche, so hat dieser Anteil auf 80 % abgenommen. Unter Schutzgas ist die reine Copolymeroberfläche also über mehrere Wochen absolut stabil. Bringt man beide Substrate anschließend für 12 h in eine Umgebung mit gesättigter Luftfeuchte, so nimmt der Aktivesteranteil auf dem mit Laminin beschichteten Substrat auf 43 % ab. Dies ist in Übereinstimmung mit der zuvor beschriebenen Reaktionsgeschwindigkeit. Der Aktivesteranteil des nicht mit Laminin bestempelten Substrats schrumpft bei 100 % Luftfeuchte jedoch ebenfalls. Zwar ist der absolute Effekt mit einer Abnahme auf 84 % in diesem Fall wesentlich geringer, aber deutlich nachweisbar. Diese Abnahme geht auf eine Hydrolyse der reaktiven Aktivestergruppen zurück. Um möglichst alle Aktivestergruppen für die kovalente Bindung von Laminin zu erhalten, wurden die mit Copolymer beschichteten Substrate daher bis zur Strukturierung mit Laminin unter Schutzgas aufbewahrt. Basierend auf diesen Ergebnissen wurden die Substrate in allen weiteren Experimenten wie folgt behandelt: 1. Polymerisation, 2. Lagerung unter Schutzgas, 3. Bestempeln mit Laminin, 4. Reaktion im Inkubator bei 37 °C und 100 % Luftfeuchte für die Dauer der doppelten Halbwertszeit (16 h), 5. Verwendung für Folgeexperimente (Zellkultur, etc.). 4-32 4 Kontrolliertes Zellwachstum Messung der Schichtdicken zu verschiedenen Zeitpunkten (A) Arbeitsschritt 1. Polymerisation 3. Spülen 4. Extraktion Stempel Stempel t tra bs Su Substrat Substrat Substrat Substrat Substrat • Eine 7,4 nm dicke 60 Schichtdicke [nm] (B) Gemessene Schichtdicke 2. Stempelübertrag SiO2 50 Copolymer 40 29,9 29,9 4,1 4,1 26,0 26,6 10 0 2,8 1 Schicht aus Laminin wird beim Stempeln übertragen. • Spülen mit Wasser 7,4 30 20 Laminin 2,8 2 3 Arbeitsschritt 2,8 2,8 4 entfernt einen Teil des Laminins. Gleichzeitig nimmt die Dicke der Copolymerschicht ab. • Extraktion führt zu keiner weiteren Abnahme der Schichtdicke. Abbildung 4-13 Kovalent gebundenes Laminin löst sich auch bei Extraktion nicht mehr von der Oberfläche. (A) Ablauf der Experimente. (B) Ellipsometrisch bestimmte Schichtdicken. Für ein derart behandeltes System wurden durch Ellipsometrische Messungen die Schichtdicken der SiO2-Schicht, des Copolymers sowie der darauf aufgestempelten Lamininschicht bestimmt. Hierzu wurden die folgenden Brechungsindizes angenommen [Braun, 1998; Golze, 2001]: nSiO2 = 1,451 nCopolymer = 1,497 nLaminin = 1,497 (optisch äquivalent zum Copolymer) Abbildung 4-13 fasst die Ergebnisse dieser Experimente zusammen. Die darin angegebenen Schichtdicken der einzelnen Teilschichten, wurden durch Differenzbildung aus Einzelmessungen an unmittelbar vergleichbaren Substraten bestimmt. In Abbildung 4-13 A sind die einzelnen Schritte der Substratbehandlung dargestellt. Eine Schichdickenmessung erfolgte zu vier Zeitpunkten: Nach Polymerisation, nach 4.3 Oberflächenchemie für maximalen Kontrast 4-33 Stempelübertrag und Inkubation, nach Spülen des Substrats mit Wasser und nach Extraktion in Wasser. Abbildung 4-13 B zeigt die Ergebnisse: Zu allen Zeitpunkten befindet sich eine unverändert 2,8 nm dicke Siliziumoxidschicht auf den Siliziumchips. Nach abgeschlossener Polymerisation gemäß dem in Punkt 4.3.2.1 beschriebenen Verfahren, findet man darauf ein Copolymer mit einer Schichtdicke 17 von 29,9 nm. Stempelt man im zweiten Schritt Laminin auf die Substrate, so wächst die gesamte Schichtdicke weitere 7,4 nm um die Schichtdicke des Laminins an. Spült man die bestempelten Substrate im Anschluss mit Wasser ab, so wird ungebundenes Laminin von der Oberfläche gewaschen, die Schichtdicke des Laminins beträgt nur noch 4,1 nm. Gleichzeitig nimmt aber auch die Schichtdicke des Copolymers auf 26,0 nm ab. Eine weitere Behandlung der Substrate durch Extraktion mit Wasser führte zu keiner weiteren signifikanten Veränderung der gemessenen Schichtdicken. Die wichtigsten Ergebnisse dieses Experiments sind die Quantifizierung der Schichtdicken sowie die damit verbundene Beobachtung, dass sich nur ein Teil des Laminins von der Oberfläche entfernen lässt. Führt man den gleichen Versuch mit einem Siliziumwafer ohne Copolymerschicht durch, so ist nahezu die gesamte gemessene Schichtdicke des Laminins nach dem Spülen verschwunden (Messwerte nicht abgebildet). Der nicht zu entfernende Teil des Laminins ist also derjenige Teil, welcher durch Reaktion mit den C5AE-Gruppen kovalent am Copolymer verankert wurde. 4.3.2.3 Copolymerzusammensetzung und Zellwachstum Das in den Punkten 4.3.2.1 und 4.3.2.2 beschriebene System wurde nun noch hinsichtlich des erzielbaren Wachstumskontrastes in Experimenten mit lebenden Zellen optimiert. Wie zuvor beschrieben, lässt sich die Zusammensetzung des Copolymers über die Zusammensetzung des Monomergemischs bei der Polymerisation kontrollieren. Auf diesem Wege wurden Substrate mit unterschiedlichem Verhältnis von C5AE : DMAA hergestellt und gitterförmig mit Laminin bestempelt. Anschließend wurde das Wachstum von neuronalen Zellen des Typs MzN auf den Substratoberflächen an 17 Der Stempelübertrag erfolgte gemäß den in Punkt 4.2.2 beschriebenen Bedingungen. 4-34 4 Kontrolliertes Zellwachstum Abbildung 4-14 Wachstum von neuronalen Zellen des Zelltyps MzN auf unterschiedlichen Copolymeroberflächen an Tag 4 in Kultur. Nur mit einem ausgewogenen Gemisch aus C5AE und DMAA werden befriedigende Ergebnisse erzielt. (A) Zellwachstum auf einer Oberfläche aus reinem DMAA-Polymer. (B) Zellwachstum auf einer Oberfläche aus C5AE-DMAA-Copolymer. (C) Zellwachstum auf einer Oberfläche aus reinem C5AE-Polymer. Tag 4 in Kultur verglichen. Die Zellen wurden wie in Kapitel 2 beschrieben kultiviert. Ihre Aussaat auf der Substratoberfläche erfolgte jeweils in einer Dichte von 10.000 Zellen je cm². In Abbildung 4-14 ist für drei unterschiedliche Polymerzusammensetzungen jeweils eine exemplarische Mikroskopieaufnahme des Zellwachstums auf der strukturierten Fläche sowie auf dem unbestempelten Hintergrund abgebildet. Man erkennt deutlich, dass nur bei einem Aktivesteranteil > 0 % ein klar strukturiertes Wachstum erfolgt. Ist kein Aktivester im Copolymer enthalten (Abbildung 4-14 A), so bindet das Laminin offenbar nur unvollständig an die Oberfläche und die Zellen wachsen weitgehend ungeordnet. Wird hingegen der DMAA-Anteil vollständig weggelassen (Abbildung 4-14 C), so kommt es zu einem unerwünschten starken Bewuchs des unstrukturierten Hintergrunds. Der hydrophobe Charakter der beiden 4.3 Oberflächenchemie für maximalen Kontrast 4-35 (A) Anteil Zellen auf Lamininstruktur1 Zellen auf Struktur [% ] Zusammensetzung des Copolymers - Statistische Auswertung 100 90 80 70 60 50 40 30 20 10 0 • C5AE ist für guten Haftungskontrast notwendig. • Bester Haftungskontrast (94%) bei einem Mischungsverhältnis C5AE : DMAA = 1 : 3. 0:1 1:5 1:3 1:2 1:0 Verhältnis C 5AE : DMAA 70 • Ohne DMAA wachsen 60 Anzahl Zellen [1] (B) Anzahl Zellen auf Hintergrund1 viele Zellen auf dem Hintergrund. 50 40 • Sauberster Hintergrund 30 (< 5 Zellen) bei einem Mischungsverhältnis C5AE : DMAA = 1 : 3. 20 10 0 0:1 1:5 1:3 1:2 1:0 Verhältnis C5AE : DMAA 1 Zellen wurden jeweils in einem Bildausschnitt von 550 x 700 µm² gezählt; Zellaussaat mit 10.000 Zellen / cm² Abbildung 4-15 Die besten Ergebnisse wurden mit einer Zusammensetzung von C5AE : DMAA = 1:3 erzielt. (A) Prozentualer Anteil der Zellen auf der Lamininstruktur. (B) Bewuchs des unstrukturierten Hintergrunds. CH3 Endgruppen des DMAA ist für einen ausreichend großen Haftungskontrast der Oberfläche also offenbar unbedingt erforderlich [Kleinfeld, 1988; Stenger, 1992]. Das Ergebnis einer statistischen Auswertung der Versuche zeigt, dass mit einer Zusammensetzung von C5AE : DMAA = 1 : 3 die besten Resultate erzielt werden können (Abbildung 4-15). Bei diesem Verhältnis ist sowohl der relative Kontrast innerhalb der strukturierten Bereiche mit 94 % am größten als auch der absolute Bewuchs des unstrukturierten Hintergrundes mit weniger als 10 Zellen auf 550 x 700 µm² minimal. Weicht die Copolymerzusammensetzung von diesem Idealwert ab, so verringert sich entweder das Wachstum auf den Lamininstrukturen (Abbildung 4-15 A) oder es erhöht sich der unerwünschte Bewuchs auf unstrukturiertem Hintergrund (Abbildung 4-15 B). 4-36 4 Kontrolliertes Zellwachstum 4.4 Mikrostrukturen für Lokalisation und Vernetzung Ein wesentliches Ziel dieser Arbeit war es, Mikrostrukturen zu finden welche neuronales Wachstum gezielt steuern. Mit derartigen Strukturen und dem in Punkt 4.2 beschriebenen Verfahren des Mikrostempelns sollten einfache und reproduzierbare neuronale Netzwerke erzeugt und einer elektrophysiologischen Untersuchung zugänglich gemacht werden. Die maximale Kontrolle über ein Neuron auf einer künstlichen Oberfläche ist erreicht, wenn sowohl die Position des Zellkörpers, die Pfade der Neuriten und auch die Polarisation der einzelnen Neuriten in Axone oder Dendriten von aussen vorgegeben werden kann. Dass eine Kontrolle neuronalen Wachstums mittels Mikrostrukturierung grundsätzlich möglich ist, wurde bereits in früheren Arbeiten gezeigt [Corey, 1996; Wheeler, 1999; Scholl, 2000]. Auch eine mögliche Beeinflussung neuronaler Polarisation wurde beschrieben [Stenger, 1998]. Die Ergebnisse dieser Arbeiten hinsichtlich der Abmessungen der verwendeten Mikrostrukturen sind als Rahmenbedingungen in den Entwurf der in dieser Arbeit eingesetzten Mikrostrukturen eingeflossen. Um dem Ziel einer systematischen Kontrolle neuronalen Wachstums näher zu kommen, wurden basierend auf diesen Rahmenbedingungen Serien von Mikrostrukturen mit gezielt veränderten Strukturparametern gefertigt und das Wachstum von neuronalen Zellen auf diesen Strukturen untersucht. In Vorversuchen waren mit Substraten aus Polystryrol und physisorbiertem Laminin regelmäßig vergleichbar gute Ergebnisse erzielt worden, wie mit Substraten auf Copolymerbasis und kovalent gebundenem Laminin. Substrate aus Polystyrol sind im Gegensatz zu Copolymersubstraten kommerziell erhältlich und einfacher zu handhaben. Für die im Folgenden beschriebenen Experimente wurden aus diesem Grund ausschließlich Substrate aus Polystyrol verwendet. 4.4.1 Charakterisierung mit der Zellinie MzN Um eine einheitliche Beurteilung des neuronalen Wachstums auf den verschiedenen Mikrostrukturen zu gewährleisten, wurden unter immer gleichen Bedingungen 4.4 Mikrostrukturen für Lokalisation und Vernetzung 4-37 Stempelabdrücke aus Laminin unterschiedlicher Struktur auf Substrate aus Polystyrol (3 cm Polystyrol Petrischalen Nr. 627 102, Greiner Labortechnik) übertragen. Der exakte Ablauf des Stempelübertrags erfolgte wie in Punkt 4.2.2 beschrieben. Als Zellmodell für die Untersuchungen wurde die neuronale Zellinie MzN unter Anwendung des in Kapitel 2 beschriebenen Kulturprotokolls verwendet. Entscheidendes Argument für die Verwendung einer Zellinie und keiner Primärkultur war deren hohe Stabilität und Reproduzierbarkeit über mehrere Versuchsreihen hinweg. Jeweils 3500 Zellen wurden am ersten Tag des Versuchs auf jedes strukturierte Substrat ausgesät. Insgesamt drei Substrate je Struktur wurden verwendet. An den darauffolgenden Tagen wurde das Zellwachstum in einem Phasenkontrastmikroskop verfolgt. Jeweils an den Tagen 1, 4, 7 und 10 nach der Zellaussaat wurden mit einer Videokamera Mikroskopieaufnahmen des Zellsystems auf der strukturierten Oberfläche angefertigt. Anhand der Bilder von Tag 1 und 4 erfolgte im Anschluss eine statistische Analyse der Qualität des Zellwachstums auf der Struktur. Dazu wurden in zufällig gewählten Bildausschnitten von 200 x 300 µm² folgende fünf Größen durch Abzählen einzelner Zellen bestimmt: 1. Anzahl der Zellen insgesamt, 2. Anzahl der Zellen auf der Struktur, 3. Anzahl der Zellen an spezifischen Punkten der übertragenen Struktur, 4. Anzahl der Zellklumpen auf der Struktur sowie 5. Anzahl der Zellen mit verschobenem Zellkern auf der Struktur. Diese Daten bildeten die Grundlage für die Bewertung der Strukturen. 4.4.1.1 Eingesetzte Strukturen Eine Tabelle mit allen verwendeten Strukturen befindet sich im Anhang. Darin ist jeder Struktur eine spezielle Identifikationsnummer (ID) zugeordnet. Im Folgenden wird diese ID benutzt um eine bestimmte Struktur eindeutig zu identifizieren. 4-38 4 Kontrolliertes Zellwachstum Stempelstrukturen - Beispiele (A) Kategorie 1: Linien 100 µm (C) Kategorie 3: Polarisationsstrukturen (B) Kategorie 2: Gitter 100 µm (D) Kategorie 4: Komplexe Schaltungen 100 µm 100 µm Abbildung 4-16 Beispiele von Mikrostrukturen aus jeder Strukturkategorie. (A) Linienstruktur (ID 10). 2 µm breite Linien mit 20 µm weiten Knotenpunkten. (B) Gitterstruktur (ID 9). 6 µm breite Gitterlinien mit 14 µm weiten Knotenpunkten. (C) Polarisationsstruktur (ID 25). 4 µm breite Gitterlinien mit 22 µm weiten Knotenpunkten und 5 µm weiten Lücken in drei von vier abgehenden Linien eines Knotenpunktes. (D) Komplexe Schaltung (ID 33). Rückkopplungskreis für bis zu drei Neuronen. 2 µm breite Linien, 20 µm weite Knotenpunkte und 5 µm weite Lücken. Insgesamt wurden vier Kategorien von Strukturen verwendet (siehe Abbildung 4-16): Kategorie 1: Linien im Abstand von 100 µm oder 50 µm (ID: 1-3 und 10-12). Kategorie 2: Gitter mit einer Maschenweite von 50 x 100 µm² (ID: 4-9 und 13-15). Kategorie 3: Polarisationsstrukturen (ID: 16-27). Kategorie 4: Komplexe Schaltungen (ID: 28-37). Gemeinsam ist allen Kategorien die Verwendung von Linien mit 2-6 µm Breite zur Kontrolle des Neuritenwachstums und von Knoten bis 24 µm Durchmesser als Inseln für die Anhaftung der Zellkörper. In den Kategorien 3 und 4 wurden zusätzlich definierte Lücken in einzelne Linien eingefügt um eine Vorzugsrichtung für die Ausbildung des Axons vorzugeben [Stenger, 1998]. Innerhalb dieser Kategorien 4.4 Mikrostrukturen für Lokalisation und Vernetzung 4-39 wurden bestimmte Parameter systematisch variiert um Aussagen über deren Einfluss auf neuronales Wachstum treffen zu können. In Kategorie 1 (siehe Abbildung 4-16 A) ist in den Strukturen 1-3 die Linienbreite von 2 - 6 µm variiert, die Strukturen 10 und 12 ermöglichen einen Vergleich von 10 µm Knoten mit 20 µm Knoten bei 2 µm Linien; die Strukturen 10 und 11 einen Vergleich zwischen Linienbreite 2 und 4 µm bei 20 µm Knoten. Kategorie 2 (siehe Abbildung 4-16 B) umfasst drei Serien a: 4-6, b: 7-9 sowie c: 13-15 mit Knotendurchmesser von a: Linienbreite + 0 µm, b: Linienbreite + 8 µm und c: Linienbreite + 18 µm. Die Linienbreite steigt in jeder Serie von 2 µm über 4 µm auf 6 µm an. Mit den Kategorien 1 und 2 ist folglich eine Charakterisierung des Einflusses von Linien- und Knotenweite auf neuronales Wachstum entlang von Linien- und Gitterstrukturen 18 möglich . In Kategorie 3 (siehe Abbildung 4-16 C) wird in zwei Serien a: 16-21 und b: 22-27 mit fester Linien- und Knotenweite von a: 2 µm Linien und 10 µm Knoten sowie b: 4 µm Linien und 22 µm Knoten, die Größe von einzelnen Lücken (a1: 16-18, b1: 22-24) und mehreren Unterbrechungen (a2: 19-21, b2: 25-27) in den Verbindungslinien zwischen den Knoten variiert. Jeweils drei der vier von jedem Knoten abgehenden Verbindungslinien sind dabei entweder durch eine einzelne Lücke vom Knoten getrennt (a1, b1) oder mehrfach unterbrochen (a2, b2). Die Größe der Lücken beträgt dabei in den Serien a1 und b1 jeweils 5, 10 und 20 µm, und in den Serien a2 und b2 jeweils 5, 2 und 1 µm. Kategorie 3 ermöglicht damit eine gezielte Untersuchung des Einflusses von Unterbrechungen entlang neuritischer Pfade auf Neuritenwachstum und Differenzierung in axonale oder dendritische Spezifität. Kategorie 4 (siehe Abbildung 4-16 D) schließlich umfasst einige ausgewählte komplexe Schaltungen. Die Wahl der Strukturparameter für diese Strukturen wurde basierend auf Ergebnissen von Vorversuchen und den oben genannten Veröffentlichungen getroffen. Insgesamt sind die Strukturen aus Kategorie 4 weniger auf systematische Vergleiche und Analysen ausgerichtet. Sie stellen einen ersten Versuch dar, Neuronen über einfache regelmäßige Anordnungen in Linien18 Zur vereinfachten Benennung werden an einigen Stellen dieser Arbeit die Knotenweiten von 10, 12 und 14 µm als 10 µm Knoten bezeichnet; Knotenweiten 20, 22 und 24 µm als 20 µm Knoten. 4-40 4 Kontrolliertes Zellwachstum und Gitterform hinaus, gezielt zu funktionalen Netzwerken komplexer Struktur zusammenzufügen. Die Strukturen 28 und 34 zielen darauf, eine Reihenschaltung von bis zu vier Neuronen zu erzeugen. Die Strukturen 29 und 35 beschreiben eine Parallelschaltung von bis zu 3 Neuronen. Mit den Strukturen 32 und 33 soll eine Rückkopplung in einem Verbund von bis zu 3 Neuronen möglich werden. Die Strukturen 30, 31, 36 und 37 schließlich beschreiben Kaskaden zur Signaladdition bzw. Signalausdünnung. Die Strukturen aller Kategorien wurden kompatibel zu einem einheitlichen Raster von 50 x 100 µm² angelegt. Alle Knoten der Strukturen befinden sich auf Punkten dieses Rasters. Dies geschah, um die Übertragbarkeit der erzeugten neuronalen Netze auf extrazelluläre Sensoren zu gewährleisten, welche in vorangegangenen Arbeiten am Max-Planck Institut für Polymerforschung entwickelt worden sind [Offenhäusser, 1997; Sprössler, 1997; Krause, 2000]. Auf der Oberfläche dieser Sensoren sind Messpunkte zur extrazellulären Erfassung neuronaler Signale gitterförmig in regelmäßigen Abständen von 50, 100 oder 200 µm angeordnet. 4.4.1.2 Vermeiden von Zellklumpen In Abbildung 4-1 A wurde bereits dargestellt wie sich neuronale Zellen auf einer großflächig strukturierten Oberfläche anordnen. Ist die mit Laminin beschichtete Fläche groß gegenüber dem Zelldurchmesser, so bilden die Zellen einen dichten Rasen auf der Fläche aus. Dieser Effekt stellt sich auch auf einer Linienstruktur ein, wenn die Linienbreite eine kritische Grenze überschreitet. In diesem Fall bilden sich Klumpen von Zellen entlang der Linien (Abbildung 4-17A). Für eine gezielte Beeinflussung des neuronalen Wachstums einzelner Zellen muß eine derartige Verklumpung durch Wahl ausreichend enger Linien vermieden werden (Abbildung 4-17 B). Andererseits beeinträchtigen zu enge Linien die Wanderung von Neuronen 19 entlang der Linien auf die Knotenpunkte . Angestrebt wird die maximal mögliche Linienbreite bei der es noch zu keiner Klumpenbildung kommt. Mit den Strukturen 1-3, bzw. 4-6 der Kategorien 1 und 2 wurde systematisch untersucht, ab welcher 19 Dies ist insbesondere bei Verwendung nicht-dissoziierter Kulturen von Bedeutung, bei welchen eine Proliferation der Neuronen über weite Strecken und über mehrere Knotenpunkte hinweg aus einem Gewebeschnitt auf die strukturierte Oberfläche erfolgen soll. 4.4 Mikrostrukturen für Lokalisation und Vernetzung 4-41 Linenstruktur - Einfluss der Linienbreite auf das Zellwachstum (A), (B) Beispiele Struktur Linienbreite (A) 6 µm Linien (B) 2 µm Linien Zellwachstum1 <V10_1_s3> (C) Statistische Auswertung Einzelne Zellen [%] 100 µm <V10_1_s1> Tag 1 Tag 4 100% 80% • Ab einer Linienbreite 60% <= 4µm dominieren einzelne Zellen über Zellhaufen. 40% 20% 0% 1 100 µm 6 4 2 Linienbreite [µm] Zellsystem: MzN Zellinie an Tag 4 nach neuronaler Ausdifferenzierung; Zelldichte jeweils 3500 Zellen/cm² Abbildung 4-17 Bei Wahl einer Linienbreite <= 4 µm dominieren einzelne Zellen über Zellhaufen. (A) Phasenkontrastbild von Zellhaufen auf 6 µm breiten Linien an Tag 4. (B) Phasenkontrastbild einer Perlenschnur von Zellen auf 2 µm breiten Linien an Tag 4. (C) Histogramm des Anteils einzelner Zellen bei Linienweiten von 2, 4 und 6 µm an Tag 1 und 4. Linienstärke es zur Bildung von Zellklumpen auf Linien bzw. Gittern aus Laminin kommt. In Abbildung 4-17 C ist das Ergebnis der Untersuchungen für Linienstrukturen in Form eines Histogramms dargestellt. Betrachtet man die Ergebnisse für Tag 1, so erkennt man deutlich eine Abhängigkeit zwischen der Linienbreite und dem prozentualen Anteil einzelner Zellen auf den Linien: Bei 6 µm Linien sind weniger als 20 % aller Zellen vereinzelt auf den Linien zu finden, bei 4 µm Linien sind es bereits über 70 % und bei 2 µm Linien annähernd 100 %. Eine statistische Analyse der Rohdaten auf Korrelation bestätigt diese Abhängigkeit (p = 0,0001). Auch an Tag 4 ist eine Korellation deutlich nachweisbar (p = 0,0095), allerdings hat sich gegenüber Tag 1 der Anteil einzelner Zellen auf 6 µm und 4 µm Linien erhöht, also gerade auf jenen Linienbreiten mit der stärksten Bildung von Zellhaufen. Offenbar haben sich beim Wechsel des Kulturmediums zwischen Tag 1 und Tag 4 verstärkt 4-42 4 Kontrolliertes Zellwachstum Gitterstruktur - Einfluss von Linien, Knoten und Lücken (A) Linienbreite Einzelne Zellen [%] Statistische Auswertung • Auch bei dicken Linien 60% von 6 µm bilden sich keine Zellklumpen. 40% 20% Zellen auf Knoten [%] 100% 6 4 2 Linienbreite [µm] Tag 1 Tag 4 • Der Fokus auf Knoten steigt nur schwach mit der Knotenweite. 80% 60% • Wird das Gitter durch 40% Lücken aufgebrochen, so sinkt der Fokus wieder. 20% 0% Knoten [µm]: keine Lücken [µm]: keine 1 Tag 4 80% 0% (B) Knotenweite Tag 1 100% 12 keine 22 keine 20 5 Antikörperfärbung an Tag 10 nach neuronaler Ausdifferenzierung des MzN Zellsystems Abbildung 4-18 Einfluss von Linienbreite, Knotenweite und Lücken auf Zellen in einer Gitterstruktur. (A) Histogramm des Anteils einzelner Zellen bei Linienweiten von 2, 4 und 6 µm an Tag 1 und 4. (B) Histogramm des Anteils an Zellen auf Knotenpunkten an Tag 1 und 4 bei Knotenweiten von 0, 10 und 20 µm sowie bei zusätzlichen Lücken von 5 µm in der Struktur. Zellhaufen und weniger einzelne Zellen von der Substratoberfläche gelöst. Dies erscheint logisch, da ein Zellhaufen bezogen auf die gleiche Haftungsfläche am Substratboden eine größere Angriffsfläche im strömenden Medium bietet. Zellhaufen werden folglich leichter weggespült als einzelne Zellen20, wodurch sich im Laufe der Kultur das Verhältnis zugunsten einzelner Zellen verschiebt. Diese Verschiebung erfolgt jedoch auf Kosten der insgesamt mit Zellen besetzten strukturierten Fläche und aufgrund eines vollkommen anderen Prinzips als die ursprüngliche Strukturierung. Für den direkten Vergleich der Strukturen kommt daher Tag 1 das stärkste Gewicht zu. Betrachtet man im Vergleich zur Linienstruktur die Ergebnisse für Gitterstrukturen in Abbildung 4-18 A, so kann man zwar immer noch einen signifikanten Anstieg des 20 In Untersuchungen mit definierter Strömungsgeschwindigkeit [Thiébaud, 2001] konnte dies direkt beobachtet werden. 4.4 Mikrostrukturen für Lokalisation und Vernetzung 4-43 Anteils einzelner Zellen mit sinkender Linienbreite des Gitters beobachten (p = 0,0007 an Tag 1), die beobachteteVariation ist jedoch sehr viel kleiner als bei den Linienstrukturen. Es dominieren bei allen verwendeten Linienbreiten von 2, 4 und 6 µm einzelne Zellen mit einem Anteil größer 80 % über Zellhaufen. Bei den verwendeten Gitterstrukturen von 50 x 100 µm² Maschenweite kommt es also offenbar erst bei größeren Linienbreiten als 6 µm zur vermehrten Bildung von Zellhaufen. Interpretiert man die Anordnung der Zellen entlang der Strukturen als Resultat von Kräften zwischen den Zellen sowie zwischen den Zellen und dem gestempelten Laminin, so ist einsichtig, dass die gitterartige Struktur aufgrund der Verbindungsstege zwischen den Linien zu zusätzlichen Kräften führt. Diese zusätzlichen Kräfte wirken auf jede einzelne Zelle entlang der Gitterlinien gleichermaßen. Liegt nun ein ungeordneter Haufen von Zellen auf einer Linie, so werden diejenigen Zellen des Zellhaufens die stärksten Kräfte spüren, die der Gitterlinie am nächsten sind. Die erhöhte Spannung der Oberfläche in diesen Zellen resultiert in einer Schwächung der Bindung an den Rest des Zellhaufens. In diesem Modell ist die verminderte Häufigkeit von Zellhaufen einer Gitterstruktur also erklärbar. Für beide untersuchte Strukturtypen (Kategorien 1 und 2) konnte innerhalb des Parameterbereichs von 2 - 6 µm Linienstärke eine klare Dominanz von mehr als 80 % einzelner Zellen über Zellhaufen erzielt werden. Zur Erzeugung linienartiger neuronalen Strukturen sind Linienbreiten zwischen 2 und 4 µm am besten geeignet . Gitterartige Strukturen mit 50 x 100 µm² Maschenweite können auch noch mit 6 µm breiten Linien erzeugt werden. 4.4.1.3 Positionierung der Zellkörper Die genaue Kontrolle über die Position einzelner Nervenzellen war das nächste Ziel 21 der Untersuchungen. Anhand der Strukturen 10-12 aus Kategorie 1 sowie 8 und 14 aus Kategorie 2 wurde die fokussierende Wirkung unterschiedlich großer quadratischer Knotenpunkte auf Neuronen in Linien- oder Gitteranordnung untersucht. Zusätzlich wurde anhand der Gitterstrukturen 22-24 aus Kategorie 3 der 21 Mit Ausnahme von Struktur 12 wurden alle Vergleiche auf der Basis von 4 µm breiten Linien durchgeführt. 4-44 4 Kontrolliertes Zellwachstum Abbildung 4-19 Die Lokalisation einzelner Zellen auf Knotenpunkte in der Struktur wächst mit der Knotenweite. (A) Phasenkontrastbild von ungeordneten Zellen auf 2 µm Linien mit 10 µm Knoten an Tag 4. (B) Phasenkontrastbild von geordneten Zellen auf 4 µm Linien mit 20 µm Knoten an Tag 4. (C) Histogramm des Anteils an Zellen auf Knotenpunkten an Tag 1 und 4 bei Knotenweiten von 10 und 20 µm sowie bei zusätzlichen Lücken von 10 und 20 µm in der Struktur. Einfluss von Lücken in der Struktur auf den Fokus studiert. Lücken größer als 5 µm resultierten dabei in einem Aufbruch der Gitterstruktur in linienartige Struktur. Die Strukturen 23 und 24 (10 und 20 µm Lücken) wurden daher für Vergleiche zu den Linienstrukturen aus Kategorie 1 verwendet. Wie bereits in Abbildung 4-1 C exemplarisch gezeigt, ist es möglich neuronale Zellen gezielt an bestimmte Positionen auf einer Linie zu bringen, wenn die Linie an den entsprechenden Stellen zu einem Knotenpunkt aufgeweitet ist. Eine geringfügige Aufweitung der Linie genügt hierzu jedoch nicht. Bei 10 µm Knotenweite ist auf den ersten Blick noch keine regelmäßige Anordnung zu erkennen (Abbildung 4-19 A), bei einer Knotenweite von 20 µm hingegen wird ganz deutlich die Regelmäßigkeit der Zellpositionen sichtbar (Abbildung 4-19 B). Die Größe der Knotenpunkte spielt also eine entscheidende Rolle. 4.4 Mikrostrukturen für Lokalisation und Vernetzung 4-45 Werden die Linien zusätzlich durch Lücken hinter den Knotenpunkten unterbrochen, so ergibt sich ein noch geordneteres Bild (Abbildung 4-19 C). Die statistische Auswertung dieser Beobachtungen ist in Abbildung 4-19 D dargestellt: Bei 10 µm Knoten sind nur 40 % aller Zellen auf Knotenpunkten lokalisiert, bei 20 µm Knoten sind es bereits mehr als 70 % und bei Einführung von 10 µm Lücken hinter 20 µm Knoten sogar über 85 %. Eine Vergrößerung der Lücken auf 20 µm führt zu keiner weiteren Steigerung. Folglich hat nicht die Größe der Lücke einen Einfluss auf den Fokus, sondern die unabhängig von der Größe der Lücke resultierende Unsymmetrie in der Linienstruktur. Zwischen Tag 1 und Tag 4 ändern sich diese Ergebnisse nur unwesentlich. Ein Tag reicht für die Wanderung der Zellen entlang der Linien auf die Knotenpunkte also offenbar aus. Auf die Messergebnisse der beiden Strukturpaare 10 µm und 20 µm Knoten sowie 0 µm Lücke und 10 µm Lücke bei 20 µm Knoten wurde der Student t-Test angewendet. Die beobachteten Unterschiede sind danach statistisch signifikant. Für Tag 1 ergibt sich p1 = 0,0114 sowie p2 = 0,0005. Für Tag 4 ist p1 = 0,0036 sowie p2 = 0,086. Die gleichen Untersuchungen wurden mit Gitterstrukturen wiederholt. In Abbildung 4-18 B sind die entsprechenden Ergebnisse dargestellt. Wie bei der Linienstruktur steigt der Anteil an fokussierten Zellen mit dem Knotendurchmesser an: 40 % der Zellen sind auf dem Gitter ohne Knoten an den Gitterschnittpunkten lokalisiert, 50 % der Zellen auf 12 µm Knoten und 60 % der Zellen auf 22 µm Knoten. Dieser Anstieg ist statistisch signifikant (p = 0,0027 an Tag 1 und p = 0,0014 an Tag 4). Im Vergleich zur Linienstruktur ist die Absolute Änderung deutlich geringer, andererseits erfolgt aber bereits ohne Knoten eine deutliche Fokussierung. Bricht man das Gitter durch 5 µm Lücken hinter den Knoten auf, so kommt es im Gegensatz zur Linienstruktur nicht zu einer weiteren Steigerung des Fokus, sondern zu einem leichten Rückgang. Die gitterartige Struktur bewirkt also auch ohne Knoten einen Fokus auf die Kreuzungspunkte des Gitters. Aufbrechen des Gitters durch Lücken stört diesen Effekt und hat daher einen Rückgang des Fokus zur Folge. Eine klare Lokalisation von einzelnen neuronalen Zellen auf spezifische Punkte wurde sowohl für linienartige, als auch für gitterartige Strukturen erreicht. Die besten Ergebnisse wurden jeweils mit 20 µm großen Knoten erzielt. Durch 4-46 4 Kontrolliertes Zellwachstum zusätzliche Lücken konnte der Fokus bei linienartigen Strukturen weiter gesteigert werden. Mit Struktur 23 (4 µm Linien, 20 µm Knoten und 10 µm Lücken) wurde mit einem Anteil von 85 % aller Zellen auf Knotenpunkten das beste Ergebnis erzielt. 4.4.1.4 Induzierte neuronale Polarisation In Abbildung 4-20 A und B sind Fluoreszenzaufnahmen von neuronal ausdifferenzierten MzN Zellen an Tag 10 nach RA-Differenzierung abgebildet. Die Zellen wurden mit anti-MAP 2 und anti-Tau rot und grün angefärbt. Zellkörper und Dendriten der Zellen erscheinen bei dieser Färbung rot (anti-MAP 2), Axone grün (anti-Tau) [Berger, 1994]. Die erste Aufnahme (A) zeigt zwei Zellen auf einer Induzierte axonale und dendritische Polarisation (A), (B) Färbung Struktur (A) Durchgehende Struktur (B) 10 µm Lücken • Spezifische Anfärbung von Axonen (grün) und Dendriten (rot). Zellwachstum1 • Gerichtete (C) Statistische Auswertung 1 Zellen mit verschobenem Kern [%] 100 µm <V10_2_Fluor_s10> 100% 80% 100 µm <V10_2_ Fluor_s23> Tag 1 Tag 4 Polarisation auf unterbrochener Struktur • Der Anteil von Zellen mit verschobenem Zellkern steigt mit der Weite der Lücken. 60% 40% 20% 0% 10 5 20 Weite der Lücken [µm] Antikörperfärbung an Tag 10 nach neuronaler Ausdifferenzierung des MzN Zellsystems Abbildung 4-20 Lücken in der Struktur bewirken eine gezielte Polarisation der neuronalen Zellen. (A) Fluoreszenzbild von MzN Zellen an Tag 10 auf einer ununterbrochenen Linienstruktur. Die Zellen liegen auf den Knotenpunkten der Struktur, aber es ist keine spezifische Anfärbung zu erkennen. (B) Fluoreszenzbild von MzN Zellen an Tag 10 auf einer Polarisationsstruktur. Die Zellen liegen auf den Knotenpunkten der Struktur. Eine charakteristische Färbung von Axonen (grün) sowie Dendriten und Zellkörpern (rot) ist zu beobachten. (C) Histogramm des Anteils von Zellen mit verschobenem Zellkern an Tag 1 und 4 auf Strukturen mit Lücken von 5, 10, und 15 µm. 4.4 Mikrostrukturen für Lokalisation und Vernetzung 4-47 durchgehenden Linienstruktur. Wie ein Vergleich mit der zugrundeliegenden Stempelstruktur zeigt, sind die Zellkörper auf den Knotenpunkten der Linienstruktur lokalisiert. Die Färbung ist jedoch unspezifisch. Es ist keine eindeutige rote oder grüne Farbdominanz zu erkennen. Auf der zweiten Aufnahme (B) hingegen sind ganz deutlich rot und grün eingefärbte Bereiche erkennbar. Diese Aufnahme zeigt zwei Zellen auf einer unterbrochenen Polarisationsstruktur der Kategorie 3. Beide Zellkörper sitzen jeweils auf einem Knotenpunkt und sind rot angefärbt. Der neuritische Fortsatz der oberen Zelle folgt der ununterbrochenen Struktur entlang nach links und erscheint in der Färbung deutlich grün. Es handelt sich also um einen axonalen Fortsatz. Der Fortsatz der unteren Zelle wächst nach rechts über die 10 µm breite Lücke in der Struktur hinweg. Dieser Fortsatz erscheint rot in der Fluoreszenzaufnahme. Hier handelt es sich folglich um einen dendritischen Fortsatz. Die Beobachtung, dass sich der dominante axonale Fortsatz einer neuronalen Zelle vornehmlich entlang des Weges des geringsten Widerstands und damit entlang ununterbrochener Pfade ausbildet wurde bei den Strukturen 22, 23 und 24 mehrfach beobachtet und ist in Übereinstimmung zu anderen Beschreibungen [Stenger, 1998]. Dies kann ausgenutzt werden um eine spezifische Polarität einzelner Neuronen in einem neuronalen Netzwerk gezielt zu induzieren. Da die für eine Erkennung mit anti-MAP 2 und anti-Tau notwendigen spezifischen axonalen und dendritischen Proteine erst zum Ende des neuronalen Differenzierungspfades der verwendeten Zellinie voll exprimiert werden [Lang, 1989] ist eine erfolgreiche Antikörperfärbung erst nach ca. 10-12 Tagen ab Beginn der Ausdifferenzierung mit RA möglich. Zu diesem Zeitpunkt war die Zahl der auf dem Substrat verfügbaren Zellen in den meisten Versuchsreihen jedoch bereits stark durch Apoptose reduziert. Für einen quantitativen Vergleich der unterschiedlichen Strukturen im Hinblick auf neuronale Polarisation wurde daher ein anderer Weg gewählt: Die Ausbildung von neuronaler Polarisation beim Zellsystem MzN ist verbunden mit einer Verschiebung des Zellkerns aus dem Zentrum der Zelle heraus. Dabei verschiebt sich der Zellkern zum neuritischen Fortsatz der später die Funktion des Axons übernimmt [Berger, 1994]. An den Tagen 1 und 4 nach RA-induzierter 4-48 4 Kontrolliertes Zellwachstum Differenzierung wurde nun der Prozentsatz der Zellen auf den Knotenpunkten der jeweiligen Struktur ermittelt, bei welchen unter dem Mikroskop ein verschobener Zellkern beobachtet werden konnte. In Abbildung 4-20 C ist das Ergebnis dieser Untersuchung für die Strukturen 22, 23 und 24 in einem Histogramm aufgetragen. Bei 5 µm Lücken in der Struktur werden erst ab Tag 4 Zellen mit verschobenem Zellkern gezählt. Bei 10 µm Lücken weisen an Tag 1 und 4 ca. 40 % der Zellen einen verschobenen Kern auf und bei 20 µm Lücken sogar annähernd 60 %. Dieses Ergebnis zeigt, dass offenbar ein direkter Zusammenhang zwischen der Größe der Hindernisse in den dendritisches Wachstumspfaden, den Lücken in der Struktur, und der Ausbildung neuronaler Differenzierung besteht. 4.4.2 Strukturierung von Hirnschnittkulturen Für elektrophysiologische Experimente mit primär isolierten Neuronen (siehe Kapitel 5) wurde eine neuronale Primärkultur auf Basis organotypischer Hirnschnitte verwendet. Im Gegensatz zu anderen dissoziierten neuronalen Zellsystemen22, wurden bei organotypischen Hirnschnitten signifikante Unterschiede zum Zellsystem MzN bezüglich des Wachstums auf mit Laminin strukturierten Oberflächen beobachtet. Aus diesem Grund wurde mit diesem Zellsystem erneut eine Untersuchung des Wachstums auf systematisch variierten Strukturen durchgeführt [Yeung, 2001]. Die Ergebnisse sind im Folgenden zsammengefasst. 4.4.2.1 Proliferation und Überwuchs Zwei grundsätzlich neue Aspekte von organotypischen Hirnschnitten sind die neuronale Proliferation aus dem Hirnschnitt in die Struktur hinein sowie die Ausbreitung von nicht neuronalem Überwuchs über die strukturierte Fläche. Bei der Aussaat dissoziierter neuronaler Zellen, sinken die Neurone homogen verteilt auf die gesamte Substratoberfläche herab. Die gesamte Substratoberfläche wird also unmittelbar nach der Zellaussaat gleichmäßig mit Zellen besetzt. Treffen Zellen dabei auf Lamininstrukturen, so können sie anhaften und auf den nächstgelegenen Knotenpunkt wandern. Zellen hingegen welche auf nicht 22 Weitere Experimente mit einzelnen ausgewählten Strukturen wurden sowohl an der neuronalen Zellinien P19 als auch an einer Primärkultur von dissoziierten hippocampalen Neuronen durchgeführt. 4.4 Mikrostrukturen für Lokalisation und Vernetzung 4-49 Abbildung 4-21 Bei neuronaler Proliferation aus Hirnschnitten kommt Linienbreite und Knotenweite der verwendeten Lamininstrukturen eine neue Bedeutung zu. (A) Ausbreitung nur von Neuriten auf eine Gitterstruktur mit 2 µm Linien und 20 µm Knoten. (B) Ungeordneter Überwuchs einer Gitterstruktur mit 4 µm Linien und 22 µm Knoten. (C) Klar strukturiertes Wachstum auf einem Gitter mit 4 µm Linien und 12 µm Knoten. strukturierte Flächen des Substrats fallen, haben keine ausreichende Substrathaftung und werden in der Regel beim nächsten Mediumwechsel weggespült. Fallen mehrere Zellen eng nebeneinander auf die Struktur, so bilden sich Zellhaufen. Plaziert man hingegen einen Hirnschnitt auf ein strukturiertes Substrat, so erfolgt die Besetzung der Struktur mit neuronalen Zellen auf einem vollkommen anderen Weg. Nicht simultan sondern nacheinander proliferieren einzelne neuronale Zellen aus dem Hirnschnitt heraus und wandern die aufgeprägten Lamininpfade entlang. Der Hirnschnitt kann dabei als ein Reservoir von neuronalen Zellen angesehen werden. Eine Bildung von Zellhaufen bei zu großer Linienweite, wie bei dissoziierten Kulturen beobachtet, ist daher sehr unwahrscheinlich. Im Gegensatz zu dissoziierten Kulturen ist bei Hirnschnitten also auch die Verwendung von Strukturen mit einer Linienbreite > 4 µm möglich. Verwendet man zu enge Linien, z.B. 2 µm Linienbreite 4-50 4 Kontrolliertes Zellwachstum in Abbildung 4-21 A, so wird die Proliferation der Neurone in die Struktur sogar behindert. In diesem Fall beobachtet man ein reines Neuritenwachstum entlang der vorgegebenen Pfade, Neurone hingegen wandern nicht in die Struktur. Als weitere Implikation der neuronalen Ausbreitung durch Proliferation ergibt sich, dass die Ausbreitung an Unterbrechungen der Führungsstrukturen stoppt. Experimente mit unterbrochenen Strukturen aus Kategorie 3 haben ergeben, dass sich unterbrochene Strukturen nicht für die Anwendung mit Hirnschnitten eignen. Ein wesentlicher Vorteil von Hirnschnitten ist die erhaltene natürliche Heterogenität des Zellverbandes. Bis auf die Schnittkanten ist das Gewebe unversehrt. Im Gegensatz zu Zellinien sind daher auch alle anderen, nichtneuronalen Zellen der jeweiligen Hirnregion im Schnitt vorhanden. Dies ist für die Vitalität der Neurone aufgrund der in diesem natürlichen Zellverband synthetisierten Wachstumsfaktoren von Vorteil. Andererseits wird die kontrollierte Erzeugung von Netztwerkstrukturen durch die Anwesenheit nichtneuronaler Zellen erschwert. Verwendet man keine antimitotischen Agentien, so kommt es in der Regel nach wenigen Tagen in Kultur zur Ausbreitung nichtneuronalen Überwuchses über die strukturierten Bereiche. Das Ausmaß des störenden Wachstums hängt jedoch entscheidend von einem Strukturparameter, der Größe der Knotenpunkte in der Struktur, ab. Große Knotenpunkte wirken wie Inseln über die sich nichtneuronale Zellen in die Struktur ausbreiten. In Abbildung 4-21 B ist deutlich zu sehen wie sich ein Teppich von Zellen enlang der Knotenpunkte (hier 22 µm) ausbreitet und die Gitterstruktur überwuchert. Verwendet man hingegen kleine Knotenpunkte, so ist es möglich auch ohne Zugabe von antimitotischen Agentien große Bereiche der Strukturen über mehrere Wochen frei von Überwuchs zu halten. In Abbildung 4-21 C ist ein klar strukturiertes Netzwerk ohne Überwuchs, gebildet von Neuronen eines Hirnschnittes an Tag 12 in Kultur, abgebildet. 4.4.2.2 Quantitative Wachstumsanalyse Wie bereits qualitativ beschrieben, haben Linienbreite und Knotenweite der Lamininstruktur einen entscheidenden Einfluss auf die Qualität des aus einem Hirnschnitt auswachsenden neuronalen Netzwerks. Beeinflusst durch diese Parameter werden insbesondere die Proliferation von Neuronen, der Grad nicht 4.4 Mikrostrukturen für Lokalisation und Vernetzung 4-51 neuronalen Überwuchses sowie die Fokussierung der neuronalen Zellen auf die Knotenpunkte der Struktur. Der quantitative Zusammenhang zwischen den Parametern der Struktur und der Güte des resultierenden neuronalen Netzwerks wurde für Linien- und Gitterstrukturen durch statistische Auswertung von Mikroskopieaufnahmen entsprechender Kulturen ermittelt. Bei jeweils gleichen Kulturbedingungen (siehe Kulturprotokoll für Hirnschnitte in Kapitel 2) wurden Hirnschnitte des Hirnstamms von embryonalen (E15 bis E18) Sprague-Dawley Ratten angefertigt und auf zuvor mit Laminin strukturierte Substrate aufgebracht. Im Zeitraum zwischen ein und zwei Wochen in Kultur wurden im Anschluss Mikroskopieaufnahmen von zufällig gewählten Ausschnitten der Substratoberfläche (Bildausschnitt jeweils 300 x 400 µm²) angefertigt. Insgesamt 54 Aufnahmen von 24 Substraten wurden wie folgt ausgewertet: − Die Gesamtzahl der Zellen auf den Strukturen sowie die Zahl der Zellen auf Knotenpunkten wurde gezählt. Die Gesamtzahl der Zellen ist dabei ein Maß für die Proliferation von neuronalen Zellen in die Struktur. Das Verhältnis von Zellen auf Knoten zu der Gesamtzahl von Zellen auf der Struktur gibt die Fokussierung auf Knotenpunkte wieder. − Der Grad des Überwuchses wurde anhand der Fläche bewertet, welche in der jeweiligen Mikroskopieaufnahme von Überwuchs bedeckt war. Das Verhältnis von Überwachsener zu nicht überwachsener Fläche ist dabei als Grad des Überwuchses definiert. In Abbildung 4-22 A bis C sind die Ergebnisse der Untersuchung zusammengestellt. Folgende Schlussfolgerungen können daraus gezogen werden: 1. Die Proliferation von neuronalen Zellen in die Struktur nimmt unabhängig vom Strukturtyp (Linen oder Gitter) mit steigender Linienbreite zu. Aus Abbildung 4-22 A geht dies deutlich hervor. So steigt die Anzahl Zellen im Bildausschnitt bei Linienstrukturen von 10 ± 6 Zellen bei 2 µm Linien auf 17 ± 8 Zellen bei 4 µm Linien an23. Bei Gitterstrukturen 23 6 µm Linien wurden in dieser Versuchsreihe nicht eingesetzt. 4-52 4 Kontrolliertes Zellwachstum Struktureinfluss auf Hirnschnittkultur - Statistische Auswertung (A) Linienweite - Proliferation entlang der Struktur Zellen auf Struktur [1] 30 25 Linien Gitter • Proliferation in die Struktur 20 steigt mit der Linienweite an. 15 • Stärkere Proliferation entlang 10 von Linienstrukturen als entlang von Gitterstrukturen. 5 0 2 4 6 Linienbreite [µm] (B) Knotenweite - Fokussierung Linien 80% 60% 40% 20% 0% 100% Gitter Überwuchs [%] Zellen auf Knoten [%] 100% (C) Knotenweite - Überwuchs 10 20 Knotenweite [µm] Linien Gitter 60% 40% • Sehr starker 20% 0% • Besserer Fokus durch große Knoten nur bei Linien. 80% 10 20 Knotenweite [µm] Überwuchs durch große Knoten bei Gittern. Abbildung 4-22 Linienbreite und Knotenweite beeinflussen erheblich das Wachstum von Hirnschnitten auf Strukturen. (A) Histogramm der Anzahl an in die Struktur proliferierten Zellen bei 2, 4, und 6 µm Linienbreite. (B) Histogramm des Anteils an Zellen auf Knotenpunkten bei Knotenweiten von 10 und 20 µm. (C) Histogramm des Prozentualen Überwuchses der Struktur bei 10 und 20 µm Knotenweite. wurde gleichfalls ein Anstieg von 1 ± 1 Zellen bei 2 µm Linen, über 9 ± 5 Zellen bei 4 µm Linien, auf 13 ± 6 Zellen festgestellt. Die im Vergleich höhere Absolutzahl von proliferierten Zellen auf Linienstrukturen verglichen zu Gitterstrukturen kann man sich über die stärkere Ausrichtung der Proliferation auf Linien erklären. Auf einer Linie ist die Richtung der Proliferation vom Hirnschnitt weg klar vorgegeben. Auf einer Gitterstruktur hingegen, gibt es in regelmäßigen Abständen Verzweigungen, wodurch die radiale Ausbreitung gebremst wird. 2. Wie bei dissoziierten neuronalen Zellen beeinflusst die Knotenweite den Grad der Fokussierung auf die Knotenpunkte. Abbildung 4-22 B zeigt den Grad der Fokussierung in Abhängigkeit von der Knotenweite vergleichend für Linien und Gitterstrukturen. Man erhält insgesamt ein ähnliches Ergebnis wie bei dissoziierten Zellen: Bei Linienstrukturen steigt der Fokus stark mit der 4.4 Mikrostrukturen für Lokalisation und Vernetzung 4-53 Knotenweite an. Nur 30 ± 10 % der Zellen liegen auf 10 µm Knoten, aber 70 ± 9 % auf 20 µm Knoten. Die Gitterstruktur hingegen fokussiert die Zellen unabhängig von der Knotenweite auf die Kreuzungspunkte des Gitters. Der beobachtete Fokus steigt nur um 1 % von 75 ± 8 % bei 10 µm Knoten auf 76 ± 5 % bei 20 µm Knoten. 3. Die Betrachtung des Überwuchses ergibt (Abbildung 4-22 C), dass Linien und Gitterstrukturen unterschiedlich anfällig für nicht neuronalen Überwuchs sind. Absolut betrachtet ist der Überwuchs bei Gitterstrukturen wesentlich stärker als bei Linienstrukturen. Und auch die relative Steigerung des Überwuchses mit wachsender Knotenweite ist bei Gitterstrukturen stärker ausgeprägt. Bei den untersuchten Linienstrukturen war bei 10 µm Knoten durchschnittlich nur eine Fläche von 10 ± 20 % des Beobachtungsfensters von Überwuchs bedeckt. Auch bei 20 µm Knoten war die Fläche nicht größer als 18 ± 25 %. Gitterstrukturen hingegen zeigten bei 10 µm Knoten mit 30 ± 25 % einen deutlich stärkeren Überwuchs, der bei 20 µm Knoten mit 58 ± 25 % auf mehr als die Hälfte der Fläche anstieg. Betrachtet man die Ergebnisse aus Punkt 2 und 3 gemeinsam, so kommt man zu dem Schluss, dass sich für Linienstrukturen die besten Ergebnisse mit großen (20 µm) Knoten erzielen lassen. Gitternetze hingegen werden am effizientesten mit kleinen (10 µm) Knoten erstellt. Aus Punkt 1 geht hervor, dass Linienbreiten von 4 µm und 6 µm am besten geeignet sind um eine ausreichende Proliferation von neuronalen Zellen in die Struktur zu gewährleisten. Für elektrophysiologische Experimente (siehe Kapitel 5) wurden insbesondere Strukturen mit diesen Parametern eingesetzt. 4-54 4 Kontrolliertes Zellwachstum 4.4.3 Komplexe neuronale Schaltkreise Fasst man die erzielten Ergebnisse aus Punkt 4.4.1 und 4.4.2 zusammen, so erscheint es möglich, neuronale Zellen auf einer künstlichen Substratoberfläche definiert zu einem funktionalen Schaltkreis zusammenzusetzen. Erste Versuche hierzu wurden mit der Zellinie MzN und einigen ausgewählten Strukturen aus Stempelkategorie 4 durchgeführt. Knotenpunkte mit einem Durchmesser von 20 µm definieren dabei die Position der Zellen, 2 µm dünne Linien legen die Pfade der Neuriten fest, und durch gezielte einseitige Unterbrechung der Linien wird die Polarität der auswachsenden Neuriten als Axon oder Dendrit gesteuert. In Abbildung 4-23 sind drei Beispiele dargestellt: Eine Paralellschaltung von bis zu 3 Zellen (A), eine Reihenschaltung von bis zu 3 Zellen mit Rückkopplungszweig (B) sowie eine baumartige Kaskade von Zellen (C). Wie die Mikroskopieaufnahmen in Abbildung Abbildung 4-23 Exemplarische Beispiele neuronalen Wachstums in komplexen Netzwerken. (A) Phasenkontrastbild einer Paralellschaltung von Zellen an Tag 1. (B) Phasenkontrastbild von Zellen in einer Reihenschaltung mit Rückkopplungsschleife an Tag 1. (C) Phasenkontrastbild einer Zellkaskade an Tag 1. 4.4 Mikrostrukturen für Lokalisation und Vernetzung 4-55 4-23 zeigen, folgen die Zellen mit ihrem Wachstum auch diesen komplexen Strukturen. Verglichen mit den regelmäßigen, sich vielfach in x- und y- Richtung wiederholenden Strukturen in Linien- oder Gitterform ist die Redundanz komplexer Strukturen auf der Substratoberfläche viel kleiner. Je mehr Zellen in einem Netzwerk definiert verschaltet werden sollen, desto geringer ist die Fehlertoleranz. Bedingt durch unvollständigen Stempelübertrag oder ungleichmäßige Zellaussaat kommt es aber bei fast jedem Substrat zu Fehlstellen im resultierenden Netzwerk. Ist in einer einfachen Linienstruktur eine Lücke, so kann man die Bereiche rechts und links neben der Lücke immer noch als Linienstruktur betrachten. Ist hingegen eine Zellkaskade in der Mitte unterbrochen, so sind bestenfalls noch Fragmente der Struktur zu gebrauchen. Aus diesem Grund ist die Ausbeute bei komplizierten Strukturen deutlich reduziert gegenüber einfachen Linien- oder Gitterstrukturen. Ziel dieser Arbeit war insbesondere auch eine grundlegende elektrophysiologische Charakterisierung strukturierter Netzwerke. Um hierzu mit akzeptabler Ausbeute eine ausreichende Datenmenge zu erzielen wurden elektrophysiologische Messungen im Rahmen dieser Arbeit ausschließlich an einfachen Linien- und Gitterstrukturen durchgeführt. Auf diesen Strukturen ist es gelungen synaptische Kontakte nachzuweisen (siehe Kapitel 5). In zukünftigen Arbeiten sollten auf Basis dieser Ergebnisse elektrophysiologische Messungen unbedingt auch auf funktionale Schaltkreise wie in Abbildung 4-23 ausgeweitet werden. 4-56 4 Kontrolliertes Zellwachstum 5-1 5 Elektrophysiologische Messungen Die Patch-Clamp Technik gestattet eine präzise Analyse der elektrophysiologischen Eigenschaften von neuronalen Zellen. Wie in Kapitel 3 beschrieben, wird dazu das Cytoplasma einer Zelle mit einer Mikropipette kontaktiert. In dieser Konfiguration ist eine direkte Messung des Membranpotentials sowie der Ionenströme durch die Zellmembran möglich. Die in dieser Arbeit erzeugten Netzwerke von Nervenzellen wurden mit Hilfe der Patch-Clamp Technik auf zwei Wegen untersucht: 1. Die Ionenströme einzelner Zellen wurden gemessen. Durch einen Vergleich der Messdaten von Zellen auf Lamininstrukturen mit Daten von Zellen auf unstrukturierten Kultursubstraten sind Rückschlüsse auf eine Wechselwirkung zwischen Substratstruktur und elektrophysiologischen Eigenschaften möglich. 2. Mit mehreren Patch-Clamp Verstärkern wurden bis zu drei Zellen gleichzeitig kontaktiert. Durch die Stimulation einer Zelle bei simultaner Beobachtung der anderen Zellen kann auf diesem Wege eine Kopplung im Netzwerk detektiert und analysiert werden. Im Folgenden sind die durchgeführten elektrophysiologischen Versuche und Messergebnisse beschrieben. Zunächst werden vorbereitende Einzelmessungen an den Zellinien Mz1, MzN und P19 diskutiert. Daran schließen sich Messergebnisse an, die in Einzelmessungen an primären Neuronen aus Hirnschnitten gewonnen wurden. Den Schwerpunkt des Kapitels bildet schließlich die Detektion, Analyse und Interpretation von Zell-Zell Kopplungen zwischen benachbarten Zellen in den erzeugten Netzwerken. 5-2 5 Elektrophysiologische Messungen 5.1 Zellinien Mz1, MzN und P19 Neuronale Zellinien zeichnen sich gegenüber Primärkulturen vor allem durch eine große Stabilität der Kultur über zahlreiche Versuche aus (siehe auch Kapitel 2). In dieser Arbeit wurden Zellinien aus diesem Grund für Vorversuche zur systematischen Optimierung der Strukturen zur Netzwerk-Erzeugung verwendet. Wie in Kapitel 4 beschrieben, wurden dabei in Versuchsreihen mit den neuronalen Zellinien Mz1, MzN und P19 geeignete Stempelstrukturen identifiziert. Um zu überprüfen, dass die beobachteten Zellen tatsächlich neuronaler Natur waren, erfolgten parallel zur Wachstumsbeobachtung Patch-Clamp Messungen an einzelnen Stichproben der Substrate. Der elektrophysiologische Nachweis von spannungssensitiven Na+- und K+Ionenkanälen in Verbindung mit der Stimulation von Aktionspotentialen erlaubt es, Zellen sicher als Neuronen zu identifizieren. Insondere ist auf diesem Weg auch eine Abgrenzung von morphologisch ähnlichen Gliazellen möglich. Diese weisen zwar ebenfalls K+-, und vereinzelt auch sehr viel schwächere Na+- Kanäle auf, Aktionspotentiale können bei Gliazellen jedoch in keinem Fall ausgelöst werden [Banker, 1998]. In Abbildung 5-1 sind die Ergebnisse einer Stichprobe bei Mz1 Zellen an Tag 3 nach neuronaler Differenzierung dargestellt. Abbildung 5-1 A zeigt zwei Bilder der Zellen auf einer mit Laminin strukturierten Substratoberfläche. Man erkennt deutlich, dass die Zellen wie auf einer Perlenschnur aufgereiht, entlang der aufgestempelten Linien aus Laminin wachsen. Das zweite Bild zeigt einen vergrößerten Bildausschnitt. Die untersuchte Zelle ist in einen Kreis eingerahmt. Die zugehörige Patch-Clamp Messung im VC-Modus (Abbildung 5-1 B), ausgehend von einer Membran-Haltespannung von -50 mV, zeigt deutliche Anteile von Na+und K+- Strömen, mit ~ 200 pA Na+-Anteil bei einer Depolarisation der Zellmembran um +40 mV und ~ 550 pA K+- Anteil bei einer Depolarisation um +100 mV. Im CC-Modus konnten Aktionspotentiale stimuliert werden. Bei der untersuchten Zelle handelt es sich also eindeutig um eine neuronale Zelle. 5.1 Zellinien Mz1, MzN und P19 5-3 Abbildung 5-1 Beispiel einer Patch-Clamp Messung an Mz1 Zellen am dritten Tag nach neuronaler Differenzierung. (A) Wachstum der Zellen auf Linien aus Laminin. (B) Elektrophysiologische Messung im VC-Modus. In Analogie zu früher durchgeführten Untersuchungen an der Zellinie Mz1 [Berger, 1997], wurde auch bei den elektrophysiologischen Stichproben an Mz1-Neuronen in dieser Arbeit eine deutliche Steigerung der Na+- und K+- Ströme mit der Kulturdauer beobachtet. Maximal wurden Na+-Ströme von 1 nA und K+-Ströme von 1,5 nA gemessen. Die Signale erreichten jedoch in keinem Fall eine Stärke mehrerer nA welche regelmäßig bei primär isolierten Neuronen aus Hirnschnitten beobachtet werden konnte. Abbildung 5-2 zeigt das Ergebnis einer Stichprobe an neuronal differenzierten P19 Zellen. In dem dargestellten Beispiel wurden P19 Zellen auf einem homogen mit Laminin beschichteten Kontrollsubstrat an Tag 14 nach neuronaler Differenzierung untersucht. Die zugehörige Patch-Clamp Messung im VC-Modus (Abbildung 5-2 B), ausgehend von einer Membran-Haltespannung von -70 mV, zeigt wie bei der zuvor betrachteten 5-4 5 Elektrophysiologische Messungen Abbildung 5-2 Beispiel einer Patch-Clamp Messung an P19 Zellen am zehnten Tag nach Aussaat auf dem Kultursubstrat (14 Tage nach neuronaler Differenzierung). (A) Wachstum auf einer homogen mit Laminin beschichteten Substratoberfläche. (B) Elektrophysiologische Messung im VC-Modus. + + Messung an einem Mz1-Neuron, deutliche Anteile von Na - und K - Strömen. Die Ionenströme betrugen ~ 600 pA Na+-Anteil bei einer Depolarisation der + Zellmembran um +60 mV und ~ 800 pA K - Anteil bei einer Depolarisation um +100 mV. Im CC-Modus konnten auch bei dieser Zelle Aktionspotentiale stimuliert werden. Die untersuchte Zelle zeigt also wiederum eindeutig neuronale Eigenschaften. Eine statistische Auswertung der Messergebnisse aller untersuchter P19-Neuronen an Tag 7 und Tag 14 nach neuronaler Differenzierung ergibt auch für das P19Zellsystem eine deutliche Steigerung der neuronalen Eigenschaften mit der Kulturdauer: 5.1 Zellinien Mz1, MzN und P19 5-5 − Am 7. Tag nach neuronaler Ausdifferenzierung wurden nur schwache Natriumkanäle mit INa = 180 ± 150 pA und IK = 260 ± 140 pA gemessen. Aktionspotentiale konnten nur in ~ 30 % aller Stichproben stimuliert werden. − Nach 14 Tagen hatten ~ 90 % der untersuchten Zellen die Fähigkeit zur Ausbildung von Aktionspotentialen. Die durchschnittlichen Ionenströme betrugen INa = 760 ± 570 pA und IK = 610 ± 350 pA. Die bereits nachgewiesene Fähigkeit von P19-Neuronen zur Bildung von echten physiologischen Synapsen [Finley, 1996] macht eine Verwendung der Zellinie für Experimente mit künstlichen Netzwerken sehr interessant. Simultane elektrophysiologische Messungen an mehreren Neuronen zum Nachweis synaptischer Kontakte wurden in dieser Arbeit jedoch auf Kulturen mit primär isolierten Neuronen beschränkt. Daher ist an dieser Stelle keine Aussage über die Fähigkeit von P19-Neuronen zur Synapsenbildung auf strukturierten Substraten möglich. Eine Ausdehnung zukünftiger Versuche auch auf P19-Neuronen erscheint aber vielversprechend. 5-6 5 Elektrophysiologische Messungen 5.2 Primäre Neuronen aus Hirnschnitten Systematische elektrophysiologische Messreihen an Neuronen auf strukturierten Substratoberflächen wurden auf Basis des in Kapitel 2 beschriebenen neuronalen Zellsystems aus Hirnschnitten embryonaler Ratten durchgeführt. Ziel der Untersuchungen war der Nachweis und die Charakterisierung synaptischer Kopplung in künstlich strukturierten Netzwerken. Die Verwendung von Hirnschnitten für die neuronale Primärkultur stützt sich auf folgende Überlegungen und Fakten: 1. Neuronale Zellen in Hirnschnitten wachsen und differenzieren unter Kulturbedingungen weiter, vergleichbar zu in vivo Bedingungen [Annis, 1993]. Alle wesentlichen Typen von Neuronen und Gliazellen können in den Kulturen nachgewiesen werden [Gähwiler, 1984; Caeser, 1989; Streit, 1989]. 2. Gewebeschnitte zeichnen sich gegenüber dissoziierten Kulturen allgemein durch eine größere Ähnlichkeit zu in vivo Bedingungen aus. So wird beispielsweise bei hippocampalen Kulturen von einem natürlicheren Verhältnis pyramidaler Neuronen zu Granule Zellen berichtet [Hoch, 1986]. Für Kulturen aus dem Hirnstamm von Ratten wurde eine besondere Ähnlichkeit der elektrischen Membraneigenschaften zu in vivo Bedingungen beschrieben [Lohrke, 1998]. 3. Die Schädigung der Zellen bei der Präparation ist gegenüber dissoziierten Kulturen deutlich geringer. Es werden keine die extrazelluläre Matrix und die Zellmembranen schädigenden Verdauungsenzyme wie Trypsin zur Vereinzelung der Zellen eingesetzt. Einzig die Zellen, welche unmittelbar an der Schnittkante liegen, werden beim Schnitt des Gewebes verletzt. Insgesamt kann bei Hirnschnitten von einer weitgehenden Unversehrtheit aller Zellen sowie von einer Konservierung des intakten Zellverbandes ausgegangen werden. 4. Die genauen Voraussetzungen für die Bildung von Synapsen im ZNS sind bis heute noch weitgehend unverstanden [Kandel, 1991]. An Synapsen im PNS konnte zwar inzwischen das Protein "Agrin" als zentraler Botenstoff der Synaptogenese identifiziert werden [McMahan, 1992; Bowe, 1995], die 5.2 Primäre Neuronen aus Hirnschnitten 5-7 Mechanismen der Synaptogenese im ZNS sind aber weitaus komplexer und lassen sich nach neueren Erkenntnissen nicht alleine auf Basis von Agrin erklären [Kröger, 2001]. Es ist daher bis heute nicht möglich, Synaptogenese in Kultur durch Zugabe entsprechender Botenstoffe zu stimulieren. Innerhalb des intakten neuronalen Zellverbandes eines Hirnschnittes jedoch, sollte die Synthese etwaiger für die Synaptogenese erforderlicher Botenstoffe auf natürliche Weise auch in Kultur erfolgen. 5. Die Auswahl der Region des Hirnstamms als Ursprungsgewebe für die Schnitte, wurde vor allem im Hinblick auf eine Übertragung des Zellmodells auf Sensorchips (siehe hierzu auch Kapitel 6) gewählt [Ingebrandt, 2001]. Im Hirnstamm sind Motorneuronen mit einem großen Anteil vertreten. Motorneuronen zeigen spontane Aktivität [Banker, 1998]. Dadurch ist wie bei spontan schlagenden Herzmuskelzellen, eine extrazelluläre Ableitung der Signale ohne Fremdstimulation möglich. In Abbildung 5-3 sind die elektrophysiologischen Signale einer Messung an drei Neuronen aus einer 16 Tage alten Kultur dargestellt. Wie erwartet zeigen die Neuronen starke spontane Aktivität. In der Langzeitaufnahme (Abbildung 5-3 A) erkennt man bei allen drei Zellen eine unregelmäßige Folge von Aktionspotentialen im Abstand von Sekundenbruchteilen bis Sekunden. Betrachtet man den Signalverlauf in einem kleinen Zeitfenster von 100 ms (Abbildung 5-3 B), so erkennt man neben voll ausgeprägten Aktionspotentialen bei Zelle 2 in den Signalen von Zelle 1 und Zelle 3 auch schwächere Depolarisationen unterhalb der Auslöseschwelle für ein Aktionspotential (sog. Sub-ThresholdStimulationen). Diese Signale sind ein Indiz für vorhandene chemische Synapsen in der Kultur. 5-8 5 Elektrophysiologische Messungen Spontane Aktionspotentiale an Tag 16 Zelle 1 Membranpotential [mV] (A) Langzeitaufnahme -20 -40 • Spontane Aktivität -60 -80 0 2 4 6 8 10 Zeit [s] Zelle 1 Membranpotential [mV] <Spontane_APs_Detail.OPJ> Zelle 3 0 <Spontane_APs_Langzeit.OPJ> (B) Einzelsignale Zelle 2 20 Zelle 2 Zelle 3 20 0 nach 2 Wochen: – Aktionspotentiale in Abständen von 100-500 ms. • Indiz für synaptische Kopplung: – Depolarisation unterhalb der Auslöseschwelle. -20 -40 -60 -80 60 80 100 120 140 Zeit [ms] Abbildung 5-3 Elektrophysiologischer Nachweis spontaner Aktionspotentiale an drei simultan vermessenen Neuronen eines embryonalen Hirnschnitts an Kulturtag 16. Die Zellen wurden auf einem homogen mit Laminin beschichteten Kontrollsubstrat kultiviert. (A) Langzeitaufnahme über 10 Sekunden. (B) Einzelne Signale in einem Zeitfenster von 100 ms. Die verwendete Hirnschnittkultur erfüllt also alle in sie gesetzten Erwartungen: − Die untersuchten Zellen bilden Aktionspotentiale und sind nach ca. zwei Wochen in Kultur spontan aktiv. Es handelt sich folglich um Neuronen mit voll + + exprimierten Na - und K - Ionenkanälen. Aufgrund der spontanen Aktivität kann man auf die Anwesenheit von Motorneuronen in der Kultur schließen. − Der Nachweis von Sub-Threshold-Stimulationen ist ein wichtiges Indiz für physiologisch aktive Synapsen. Es sollte daher möglich sein, funktionale synaptische Kontakte in der Kultur nachzuweisen. Auf Basis dieser positiven Ergebnisse wurden die Versuche auf strukturierte Substrate ausgedehnt. In einer Serie von Experimenten wurden Hirnschnitte wie in Kapitel 2 beschrieben, unter immer gleichen Bedingungen auf homogen mit Laminin beschichteten 5.2 Primäre Neuronen aus Hirnschnitten 5-9 Zeitliche Entwicklung der Kultur und des Membranpotentials (A) Wachstum Tag 3: Tag 4: Tag 7: • Nach ca. 1 Woche Überwuchs mit Gliazellen. (B) Membran potential 50 µm • Messungen an Kontrolle Membranpotential [mV] <Growth_series_V20-1.tif> Lamininstruktur -60 strukturierten Substraten daher nur bis Tag 16 möglich. • In diesem Zeitraum keine signifikanten Unterschiede im Membranpotential. -40 -20 <Membranpotential_normal_struktur.opj> 0 8 12 16 20 Tag in Kultur [1] 24 Abbildung 5-4 Zeitliche Entwicklung von Hirnschnitt-Neuronen auf strukturierten Kultursubstraten. (A) Neuronales Wachstum auf einem Laminingitter mit 6 µm Linien und 14 µm Knoten. (B) Größe des Membranpotentials im Vergleich zu unstrukturierten Substraten. Kontrollsubstraten sowie auf mit Mikrostempeln strukturierten Substratoberflächen kultivert. Zu verschiedenen Zeitpunkten wurden im Phasenkontrastmikroskop Aufnahmen der Substrate angefertigt (Abbildung 5-4 A) sowie einzelne, aus dem Schnitt auf die Substratoberfläche proliferierte Neuronen elektrophysiologisch vermessen (Abbildung 5-4 B und Abbildung 5-5). Insgesamt wurden auf Kontrollsubstraten 90 und auf Lamininstrukturen 63 Einzelmessungen durchgeführt. Zum Nachweis von Gliazellen in der Kultur (Abbildung 5-6), wurden außerdem Antikörperfärbungen auf Basis von anti-GFAP vorgenommen (siehe Kapitel 2). In Abbildung 5-4 A kann man anhand von drei Momentaufnahmen an Tag 3, 4 und 7 nach Kulturbeginn, exemplarisch den zeitlichen Verlauf neuronalen Wachstums auf einem strukturierten Substrat beobachten. Bereits an Tag 3 sind die Neuronen auf den Knotenpunkten der Gitterstruktur plaziert. In den folgenden Tagen wird verstärktes Neuritenwachstum zwischen den Zellen entlang der Lamininpfade beobachtet. Zwischen der zweiten und dritten Zelle von unten ist an Tag 3 noch 5-10 5 Elektrophysiologische Messungen keine Verbindung erkennbar, an Tag 4 wächst ein Fortsatz zwischen beiden Zellen, 1 und an Tag 7 ist schließlich eine durchgehende Verbindung vorhanden. Dieser zeitliche Ablauf ist mit kleineren Abweichungen repräsentativ für alle beobachteten Substrate. Nach einer frühen Phase (Tag 2 - Tag 5) schneller Proliferation aus dem Schnitt heraus auf Knotenpunkte der Lamininstruktur begann eine Phase starken Neuritenwachstums (Tag 3 - Tag 8). Danach blieb die neuronale Struktur weitgehend unverändert. In Abbildung 5-4 A ist aber auch noch eine weitere Veränderung der Kultur zu beobachten: An Tag 3 erscheinen die Zellkörper noch deutlich hell im Phasenkontrastbild, wohingegen die Zellen an Tag 7 dunkel erscheinen. Patch-Clamp Messungen an solchen dunkel erscheinenden Neuronen waren nur sehr eingeschränkt möglich, da sich bei diesen Zellen meist kein Gigaseal ausbildete oder die Zellmembran nach Ausbildung des Gigaseals nicht geöffnet werden konnte. Eine für statistische Auswertungen ausreichende Anzahl elektrophysiologischer Messungen war an strukturierten Substraten daher nur bis Tag 16 möglich. Auf homogen beschichteten Kontrollsubstraten, wurde dieser Effekt zwar auch beobachtet, jedoch in deutlich geringerem Ausmaß. Das Ergebnis der Messung des Membranpotentials an Neuronen auf strukturierten Substraten und auf Kontrollsubstraten (Abbildung 5-4 B) zeigt, dass die Zellen nach einer Woche, unabhängig von der Substratoberfläche, gleich große elektrische Potentiale von -40 bis -50 mV aufbauen. Diese Größe des Ruhepotentials ist in guter Übereinstimmung mit früher veröffentlichten Messwerten an vergleichbaren Kulturen [Lohrke, 1998]. Die Struktur der Oberfläche hat also offenbar keinen Einfluss das Ruhepotential. Die Ionenkonzentrationen innerhalb und außerhalb der untersuchten Zellen waren in allen Experimenten gleich, determiniert durch die Zusammensetzung der PatchClamp Lösungen. Anhand der Goldmann Gleichung (siehe Kapitel 2) kann man daher im Ruhezustand auf eine unveränderte Gewichtung der 1 Der optische Nachweis einer Verbindung lässt keinerlei direkten Schluss auf die Präsenz einer synaptischen Verbindung zu. Er ist nur eine notwendige Voraussetzung für die Ausbildung einer Synapse. 5.2 Primäre Neuronen aus Hirnschnitten 5-11 Membranpermeabilitäten P für die einzelnen Ionentypen Na+, K+ und Cl- relativ zueinander schließen. + + Betrachtet man die Stärke der Ionenströme durch spannungsgesteuerte Na - und K - Ionenkanäle in beiden Zellsystemen (Abbildung 5-5), so erkennt man signifikante Unterschiede zwischen Neuronen auf mit Laminin strukturierten Substraten und Neuronen auf homogen mit Laminin beschichteten Kontrollsubstraten. Zwar ist die Ionenströme auf Strukturen und Kontrollen (A) Natriumstrom (Na+) Natriumstrom [nA] Kontrolle (B) Kaliumstrom (K+) Kaliumstrom [nA] <Natriumstrom_normal_struktur.opj> <Kaliumstrom_normal_struktur.opj> Lamininstruktur 5 4 3 2 • Na+- und K+- Ströme sind auf strukturierten Substraten vermindert. 1 0 • Die relative zeitliche 5 Entwicklung der Ionenströme stimmt überein. 4 3 2 1 0 8 12 16 20 24 Tag in Kultur [1] Abbildung 5-5 + + Vergleich der Na - und K - Ionenströme von Neuronen auf strukturierten und unstrukturierten Substraten. (A) Zeitliche Entwicklung der Natriumströme. (B) Zeitliche Entwicklung der Kaliumströme. relative zeitliche Entwicklung der gemessenen Na+- und K+- Ströme gleich, die absolute Größe der Ströme ist auf strukturierten Substraten gegenüber Kontrollen jedoch um einen Faktor 2 bis 3 reduziert. Die Verhältnisse der Verringerung des + + Na - und K - Stroms stimmen dabei im Rahmen der Fehlerbalken überein. 5-12 5 Elektrophysiologische Messungen Durch das Wachstum auf einer Lamininstruktur wird gegenüber unstrukturiertem + Wachstum folglich eine gleichmäßige Abnahme von spannungsgesteuerter Na - und K+- Permeabilität der Membran bewirkt. Alle vermessenen Kulturen basierten auf dem gleichen Zellsystem und die Substrate waren jeweils mit dem gleichen ECM-Protein Laminin beschichtet. Die Typen der + + von den Zellen exprimierten Na - und K - Kanäle sollten sich daher nicht grundsätzlich unterscheiden. Für die Reduzierung der Ionenströme auf strukturierten Substraten gibt es folglich nur zwei mögliche Erklärungen: − Entweder haben die Zellen, aufgrund der durch die Struktur festgelegten räumlichen Ausdehnung, eine insgesamt kleinere Oberfläche, und daher in der Summe weniger Ionenkanäle bei unveränderter Oberflächendichte der Kanäle, − oder aber, die Dichte der aktiven Ionenkanäle in der Membran ist reduziert; möglicherweise durch eine geringere Anzahl von Ionenkanälen in der Membran insgesamt, oder durch Blockade vorhandener der Ionenkanäle. Beide Erklärungen stellen keinen Widerspuch zu der weiter oben gefundenen Konservierung des Ruhepotentials dar. Das Ruhepotential wird nur von der relativen Zahl von Kanälen zueinander beeinflusst, nicht aber von deren Absolutanzahl. Die erste Möglichkeit kann man anhand eines Vergleichs der Membrankapazitäten2 ausschließen. Die Kapazität einer Zellmembran CMem lässt sich mit dem Modell eines Plattenkondensators berechnen. Dies wird sofort anschaulich klar, wenn man sich die Zellmenbran aufgetrennt und flach zwischen zwei Metallplatten ausgebreitet vorstellt. Die Metallplatten ersetzen in diesem Bild die elektrisch leitfähige intrazelluläre und extrazelluläre Lösung. Der Abstand der Platten ist dann durch die Dicke d der Zellmembran festgelegt, die Plattenausdehnung durch die Fläche A der Zellmembran. Die elektrischen Eigenschaften des Plattenzwischenraums ergeben sich aus den Eigenschaften der Zellmembran. 2 Vor jeder Patch-Clamp Messung wird bei der Kompensation des Patch-Clamp Verstärkers auch die Membrankapazität ermittelt. 5.2 Primäre Neuronen aus Hirnschnitten 5-13 Für die Kapazität CMem dieser Anordnung gilt in Analogie zu einem physikalischen Plattenkondensator: C Mem = εε 0 C ε ε0 A d = = = = = A d Gleichung 5-1 Kapazität der Zellmembran [F] Relative Dielektrizitätskonstante der Zellmembran [1] Vakuum-Dielektrizitätskonstante [F/m] Oberfläche der Zellmembran [m²] Dicke der Zellmembran [m] Die Membrankapazität CMem ist also direkt proportional zur Membranoberfläche A. Bei unveränderten Werten für ε und d (beide Parameter sind durch die spezifischen Eigenschaften des Systems "Zell-Lipidmembran" in engen Grenzen festgelegt), kann man folglich von der Membrankapazität unmittelbar auf die Membranfläche schließen. Die durchschnittliche Membrankapazität der gemessenen Zellen auf Kontrollsubstraten war mit CMem = 21 ± 13 pF gleich dem Messwert CMem = 22 ± 15 pF auf strukturierten Substraten. Die Größe der Membranoberflächen stimmt daher ebenfalls überein. Betrachtet man dieses Ergebnis im Kontext zu der oben anhand von Abbildung 5-4 A diskutierten morphologischen Entwicklung, so liegt es nahe die Abnahme der Ionenströme durch eine Form von nicht-neuronalem Überwuchs zu erklären, welcher die Zellen dunkel erscheinen lässt und Ionenkanäle verschließt. Als möglicher Ursprung eines derartigen Überwuchses kommen insbesondere Gliazellen in Frage, welche auf strukturierten Substraten Neuriten und ganze Neuronen mit einer Hülle aus Myelin umgeben könnten. Glia-Überwuchs wird auch in [Banker, 1998] als eine mögliche Ursache für erschwerte Patch-Clamp Bedingungen angeführt. Das Ergebnis einer Glia-spezifischen Antikörperfärbung mit dem Antikörper antiGFAP stützt diese Theorie. Abbildung 5-6 zeigt Mikroskopieaufnahmen der gefärbten Substrate im Phasenkontrast- und Fluoreszenzmodus. Gliazellen erscheinen in den Aufnahmen mit grüner Fluoreszenz. Es wurden wieder Kontrollsubstrate (Abbildung 5-6 A) und strukturierte Substrate (Abbildung 5-6 B) miteinander verglichen. In beiden Fällen kann man Gliazellen 5-14 5 Elektrophysiologische Messungen Abbildung 5-6 Antikörperfärbung mit anti-GFAP identifiziert Gliazellen in der Kultur. (A) Nachweis von Gliazellen (grün) auf einem homogen mit Laminin beschichteten Kontrollsubstrat. (B) Nachweis von Gliazellen (grün) auf einer Lamininstruktur mit 6 µm Linien und 14 µm Knoten. Zellkerne wurden blau mit einem DNA-Marker angefärbt. erkennen, welche aus dem Hirnschnitt heraus auf die Substratoberfläche gewachsen sind. Auf dem strukturierten Substrat folgt der Glia-Bewuchs der gestempelten Lamininstruktur. Entscheidend ist die Beobachtung, dass auf dem Kontrollsubstrat nicht alle Neuronen (dunkle Punkte) von Gliazellen überwachsen sind, wohingegen das strukturierte Substrat entlang der Lamininstrukturen vollständig von Gliazellen bedeckt ist. Dies liegt möglicherweise an den begrenzten Ausbreitungsmöglichkeiten der Gliazellen auf der Lamininstruktur. Die beobachtete starke Abschwächung der Ionenströme bei strukturierten Substraten ist also offenbar tatsächlich auf eine Überdeckung der Neuronen mit Gliazellen zurückzuführen. Auch durch die Zugabe anti-mitotischer Agentien, wie z.B. ARA-C, in das Kulturmedium ist es nicht gelungen, den Überwuchs der Strukturen mit Gliazellen 5.2 Primäre Neuronen aus Hirnschnitten 5-15 vollständig zu verhindern. Allerdings konnte der Zeitpunkt vollständigen Überwuchses von ~ 1 Woche ohne Zugabe von ARA-C auf maximal 2 ½ Wochen mit ARA-C herausgezögert werden. 5-16 5 Elektrophysiologische Messungen 5.3 Zell-Zell Kopplung Eine Kopplung zwischen neuronalen Zellen wurde unter gleichzeitiger Verwendung von zwei oder drei Messkanälen des Patch-Clamp Verstärkers detektiert. Der Ablauf der Messungen war dabei wie folgt: 1. Zunächst wurde mit Hilfe des Mikroskops eine geeignet erscheinende Anordnung von zwei oder drei neuronalen Zellen gesucht. Kriterium für die Auswahl war die Anordnung mehrerer Neuronen in unmittelbarer Nachbarschaft und mit deutlich erkennbaren Verbindungen. Auf strukturierten Substraten wurde außerdem die Qualität der Zell-Anordnung auf der Struktur bewertet. 2. Die ausgewählten Zellen wurden dann gleichzeitig, unter Verwendung mehrerer Patch-Clamp Messköpfe, jeweils mit einer Patch-Clamp Pipette in Whole-Cell Stimulation Beobachtung Stimulationsfolge zum Test auf Kopplung Wiederholung (A) Stimulationspuls an Zelle 1 Beobachtung von Zelle 2 (B) Stimulationspuls an Zelle 2 Beobachtung von Zelle 1 ? + ? + - Zelle 1 Zelle 1 Zelle 2 ? Zelle 2 Zelle 1 ? Zelle 2 t t Abbildung 5-7 Eine mögliche Kopplung zweier Zellen wird durch alternierende Stimulation unter Beobachtung der jeweils unstimulierten Zelle detektiert. (A) Test auf Kopplung von Zelle 1 nach Zelle 2. (B) Test auf Kopplung in die entgegengesetzte Richtung von Zelle 2 zu Zelle 1. 5.3 Zell-Zell Kopplung 5-17 Konfiguration kontaktiert. Die einzelnen Kanäle des Patch-Clamp Verstärkers 3 wurden dabei wie in Kapitel 3 beschrieben kompensiert . 3. Schließlich erfolgte der eigentliche Test auf Kopplung durch abwechselnde Stimulation der kontaktierten Zellen und gleichzeitige Beobachtung der Messignale aus den nicht stimulierten Zellen. Abbildung 5-7 zeigt das Messprinzip und den zeitlichen Ablauf der programmierten Stimulationssequenz. 5.3.1 Kopplungstypen Im Verlauf der Experimente wurden mehrere hundert Messungen an Zellen auf strukturierten Substraten sowie auf homogen mit Laminin beschichteten Kontrollsubstraten durchgeführt. Drei unterschiedliche Typen der Kopplung wurden in diesen Messungen beobachtet. Abbildung 5-8 zeigt für jeden Kopplungstyp eine typische Zell-Konfiguration mit den zugehörigen Messignalen: 1. Ohm'sche Kopplung (Abbildung 5-8 A). In diesem Fall bestand eine elektrisch leitende Verbindung zwischen zwei Zellen. Eine an Zelle 1 angelegte Gleichspannung U1 hatte eine um den Faktor XU12 proportionale Spannungsänderung U2 in Zelle 2 zur Folge. Kopplungskoeffizienten bis zu XU12 > 50 % waren möglich. Wurde eine Ohm'sche Kopplung von Zelle 1 nach Zelle 2 gemessen, so war stets auch eine Kopplung in der Gegenrichtung von Zelle 2 zu Zelle 1 nachweisbar. 2. Kapazitive Kopplung (Abbildung 5-8 B). Waren zwei Zellen kapazitiv miteinander gekoppelt, so hatte ein in Zelle 1 induzierter Spannungssprung zeitgleich eine kapazitive Spannungsspitze in Zelle 2 zur Folge. Die Stärke der kapazitiven Kopplung war in den meisten Fällen sehr schwach (XU12 < 1 %, bei einem Spannungssprung mit 2 µs Anstiegszeit in Zelle 1). Kapazitive Kopplung war wie Ohm'sche Kopplung immer auch in der Gegenrichtung möglich. 3 Um Störungen der Kompensation durch Kopplung an die anderern Messkanäle in den benachbarten Zellen zu vermeiden, wurde jeweils nur der aktuell kompensierte Kanal im VC-Modus betrieben. Die anderen Kanäle wurden für die Dauer der Kompensation mit 0 pA Haltestrom in den CC-Modus geschaltet. 5-18 5 Elektrophysiologische Messungen Kopplungstypen und Signalformen 2 (B) Kapazitive Kopplung 50 µm <V20_1__s11-3-1 p13> C 1 2 2 Streukapazität an Berührungspunkten der Zellmembranen (C) Funktionale Kopplung 2 1 Leitende Verbindung des Cytoplasmas durch Transmembranporen Membranspannung [mV] R 1 1 <V30-3__s2-1 p5> 50 µm N 1 1 2 Chemische Signalübertragung durch Neurotransmitter 2 50 µm Zelle 1 Zelle 2 Messignale* <V20_1__s11-3-4 p18> 20 0 -20 -40 Zeit [ms] 100 125 150 175 200 Membranspannung [mV] (A) Ohm'sche Kopplung Konfiguration 60 40 20 0 -20 -40 -60 Zeit [ms] Membranspannung [mV] Prinzip ** 25 50 75 100 125 20 0 -20 -40 -60 Zeit [ms] 100 125 150 175 200 * Jeweils Zelle 1 stimuliert. ** Signal x10 verstärkt. Abbildung 5-8 Drei grundsätzlich verschiedene Kopplungstypen wurden identifiziert. (A) Ohm'sche Kopplung über eine elektrisch leitfähige Verbindung des Zell-Cytoplasmas beider Zellen. (B) Kapazitive Kopplung über die Kontaktkapazität eng aneinanderliegender Bereiche der Zellmembranen. (C) Funktionale Kopplung an einer physiologisch aktiven Synapse. Zeitverzögerte Signalübertragung auf Basis von Neurotransmittern. 3. Funktionale Kopplung (Abbildung 5-8 C). In diesem Fall waren beide Zellen bei schwacher präsynaptischer Stimulation elektrisch voneinander isoliert, die Stimulation eines Aktionspotentials in Zelle 1 hatte jedoch mit einem charakteristischen Zeitversatz weniger Millisekunden eine elektrische Depolarisation der postsynaptischen Zelle zur Folge. Die Kopplung war stets gerichtet, d.h. Signalübertragung war nur in einer bestimmten Richtung möglich. 5.3.2 Prinzip Ohm'scher Kopplung Das Prinzip der Ohm'schen Kopplung basiert auf einer leitenden Verbindung des Cytoplasmas der gekoppelten Zellen. Dabei sind jedoch nicht die Zellmembranen mehrerer Zellen fusioniert, sondern die elektrische Verbindung wird über permanent 5.3 Zell-Zell Kopplung 5-19 geöffnete, wassergefüllte und ionendurchlässige Transmembranporen, sog. Gap Junctions, hergestellt (siehe auch Kapitel 2). Diese Verhalten sich elektrisch wie ein Ohm'scher Widerstand RKopplung zwischen den Zellen [Kandel, 1991]. Ohm'sche Kopplung wurde als wichtiges Schaltungselement in funktionalen Netzwerken von Nervenzellen nachgewiesen [Carew, 1976]. Die Leitfähigkeit einzelner Gap Junctions liegt in der Größenordnung von ~100 pS [Neyton, 1985; Traub, 1994]. Je nach Anzahl der an der Kopplung beteiligten Gap Junctions, ist eine Gesamtleitfähigkeit in der Größenordung von einigen nS messbar [Bennett, 1967; Neyton, 1986]. 5.3.3 Prinzip kapazitiver Kopplung Bei kapazitiver Kopplung ist keine direkte elektrische Leitfähigkeit zwischen den untersuchten Zellen nachweisbar. Das Cytoplasma kapazitiv gekoppelter Zellen ist nicht leitend verbunden, sondern wird auch im Kontaktbereich durch die Zellmembranen isoliert. Die Messignale kapazitiver Kopplung können über ein kapazitives Leck zwischen beiden Zellen erklärt werden. Liegen die Zellmembranen zweier Zellen, bzw. des Axons einer Zelle und eines Dendriten der anderen Zelle auf einer hinreichend großen Fläche dicht genug aneinander, so wird zwischen beiden Zellen eine signifikante Kontaktkapazität CKontakt gebildet. Ändert sich die Membranspannung in Zelle 1, so lädt diese Spannungsänderung die Kontaktkapazität über den Membranwiderstand RMem2 der zweiten Zelle auf. Die Folge ist eine messbare Spannungsänderung in Zelle 2. Das Zeitverhalten des Ladevorgangs wird durch die Zeitkonstante τ = RMem2 * CKontakt bestimmt. Man kann daraus CKontakt ermitteln. Für die Messung in Abbildung 5-8 B resultiert eine Kontaktkapazität von CKontakt ~ 1,4 pF. 5.3.4 Prinzip funktionaler Kopplung Die Signalübertragung an funktionalen Synapsen erfolgt, wie in Kapitel 2 beschrieben, nicht elektrisch sondern auf chemischem Weg. Neurotransmittermoleküle werden präsynaptisch in den synaptischen Spalt 5-20 5 Elektrophysiologische Messungen ausgeschüttet, diffundieren zur postsynaptischen Membran, binden dort an Rezeptoren und triggern so das Öffnen ligandengesteuerter Ionenkanäle. Insbesondere die Ausschüttung des Neurotransmitters verzögert dabei die Signalübertragung um Bruchteile von Millisekunden. Der Nachweis einer konstanten Zeitverzögerung zwischen präsynaptischem Aktionspotential und postsynaptischer Depolarisation ist daher ein sicheres Zeichen für funktionale synaptische Kopplung. Jede funktionale Kopplung hat auch eine kapazitive Komponente. Im Bereich des synaptischen Spalts sind die Zellmembranen auf einer Fläche von ~ 0,25 µm² bis auf einen Abstand von ~ 10 nm aneinander angenähert. Daraus resultiert analog zum Fall rein kapazitiver Kopplung eine Kontaktkapazität. In einem vereinfachten Modell kann man die Größe dieser Kapazität leicht abschätzen: Die spezifische Membrankapazität einer typischen Nervenzelle beträgt ungefähr CMem = 1 µF/cm² [Katz, 1985]. Vernachlässigt man den Einfluss des synaptischen Spalts4, so ist die Kontaktkapazität gleich der Reihenschaltung der Membrankapazitäten beider sich berührender Zellmembranen, also CKontakt = CMembrankontakt/2 . Die Kapazität der Zellmembran im Kontaktbereich kann man über die Größe des Kontaktbereichs direkt berechnen. Man erhält als Endergebnis CKontakt ~ 1 fF (1 Femtoferat = 10-15 F). Die kapazitive Kopplung an einer einzelnen funktionalen Synapse ist folglich mindestens um einen Faktor 1000 schwächer als im oben betrachteten Messbeispiel rein kapazitiver Kopplung in Abbildung 5-8 B. 5.3.5 Behandlung der Kopplungstypen Der Beitrag kapazitiver Kopplung zur Signalübertragung ist im Vergleich zu funktionaler und Ohm'scher Kopplung sehr gering. In der Literatur wird von keinem physiologisch relevantem Beispiel kapazitiver Kopplung berichtet. Von einer Analyse der Messdaten rein kapazitiver Kopplung wird aus diesem Grund abgesehen. Ohm'sche und funktionale Kopplung hingegen haben eine zentrale Bedeutung für die Funktion des Nervensystems [Kandel, 1991]. Sie bilden nachweislich die 4 Innerhalb des synaptischen Spalts wird die Kopplung durch Ohm'sche Leckströme abgeschwächt. 5.3 Zell-Zell Kopplung 5-21 Grundlage der Signalübertragung zwischen Neuronen und ermöglichen so erst die Entstehung funktionaler neuronaler Schaltkreise im ZNS. Im Rahmen dieser Arbeit gelang der Nachweis Ohm'scher Kopplung in einer Vielzahl von Fällen sowohl auf homogenen als auch auf strukturierten Substraten. Die Ergebnisse dieser Messungen konnten in Bezug zur verwendete Substratstruktur gezielt analysiert werden. Funktionale Kopplung wurde in den durchgeführten Experimenten nur in wenigen Einzelfällen beobachtet. Die begrenzte Menge der Messdaten reicht für eine statistische Auswertung nicht aus, eine Betrachtung ausgewählter Einzelmessungen ist aber dennoch sehr aufschlussreich. Im Folgenden wird ein physikalisches Modell eingeführt, um die elektrischen Eigenschaften eines Systems aus zwei gekoppelten Zellen zu beschreiben. Auf Basis dieses Modells werden im Anschluss die Messergebnisse der detektierten Ohm'schen und funktionalen Kopplungen analysiert und diskutiert. 5-22 5 Elektrophysiologische Messungen 5.4 Grundlagen der Kopplung Bei der Diskussion der Kopplungstypen in Punkt 5.3 wurde bereits deutlich, dass sich die Signalübertragung bei Ohm'scher und kapazitiver Kopplung mit den elektronischen Bauteilen Widerstand und Kondensator erklären lässt. Es liegt daher nahe anzunehmen, dass das elektrische Verhalten eines Systems aus zwei gekoppelten Zellen vollständig mit einer geeigneten Schaltung passiver elektronischer Bauelemente beschrieben werden kann. In Punkt 5.4.1 wird die Theorie linearer Systeme als allgemeiner Formalismus zur Beschreibung passiver elektronischer Schaltungen eingeführt. Im Anschluss wird in Punkt 5.4.2 und 5.4.3 das verwendete Messverfahren zur elektrischen Charakterisierung des Kopplungssystems beschrieben. Ein konkretes physikalisches Kopplungsmodell wird in den Punkten 5.4.4 und 5.4.5 entwickelt. Die Grenzen der Messgenauigkeit der Parameter dieses Modells werden in Punkt 5.4.6 diskutiert. 5.4.1 Zell-Zell Kopplung als lineares System Besteht eine elektronische Schaltung ausschließlich aus passiven Bauelementen des Typs Widerstand R, Kapazität C und Induktivität L, so bildet sie ein lineares System. Definiert man zwei beliebige Punkte 1 und 2 als Eingang X der Schaltung und zwei weitere Punkte 3 und 4 als Ausgang Y, dann kann die Übertragung eines Signals vom Eingang X zum Ausgang Y in einem einfachen Formalismus beschrieben werden. Dieser ist unabhängig von der konkreten Form des Netzwerks. Wie in Abbildung 5-9 A dargestellt, kann man ein lineares System daher als eine "Black-Box" auffassen. Linear ist ein beliebiges System mit einem Eingang X und einem Ausgang Y genau dann wenn gilt: Y (α X 1 + β X 2 ) = α Y ( X 1 ) + β Y ( X 2 ) Y(X) = X1, 2 = α, β = Gleichung 5-2 Spannung am Ausgang der Black-Box in Abhängigkeit von der Eingangsspannung X [V] Am Eingang der Black-Box angelegte Spannungen beliebiger Größe [V] Dimensionslose Faktoren beliebiger Größe [1] 5.4 Grundlagen der Kopplung 5-23 Dies ist die Linearitätsbedingung. Sie besagt, dass die Antwort Y der Black-Box auf eine zusammengesetzte Eingangsgroße X gleich der Summe aller Teilantworten auf die einzelnen Teileingangsgrößen ist. Für elektronische Schaltungen aus passiven Komponenten ist diese Bedingung erfüllt. Widerstände, Kapazitäten und Induktivitäten bezeichnet man daher auch als lineare Bauelemente. Für sie gilt das Ohm'sche Gesetz. Ihr Widerstand ist eine ausschließlich von der Frequenz abhängige Konstante, unabhängig von der absoluten Größe des Stroms und der Spannung. Enthält eine Schaltung nichtlineare Bauelemente wie Dioden und Transistoren, so können die Gesetze der Theorie linearer Systeme hingegen nicht angewendet werden. In Bezug auf die Beschreibung des Systems "gekoppelte Zellen" heißt dies, dass ausschließlich Ohm'sche und kapazitive Kopplung als lineares System beschrieben werden können. Bei funktionaler Kopplung funktioniert dies aufgrund der Nichtlinearität an der funktionalen Synapse nicht. 5.4.1.1 Theorie linearer Systeme Die Theorie linearer Systeme, auch bekannt als Vierpoltheorie, beschreibt die Signalverarbeitung in linearen Systemen allgemein. Definiert wird eine Übertragungsfunktion h(t) so dass gilt: Y (t ) = h (t ) ∗ X ( t ) , wobei h (t ) ∗ X (t ) ≡ +∞ ∫ h(t − τ ) X (τ )dτ Gleichung 5-3 −∞ Y(t) = X(t) = ∗ h(t) = Spannung am Ausgang der Black-Box zum Zeitpunkt t [V] Spannung am Eingang der Black-Box zum Zeitpunkt t [V] Faltungsprodukt Übertragungsfunktion im Zeitraum [1] Die Antwort Y(t) des Systems zu einem beliebigen Zeitpunkt t wird berechnet als Faltung der Übertragungsfunktion h(t) mit dem Eingangssignal X(t). Transformiert man das System mit einer Fouriertransformation vom Zeitraum in den Frequenzraum, so transformiert das Faltungsprodukt in ein gewöhnliches skalares Produkt: 5-24 5 Elektrophysiologische Messungen Y (ω ) = h(ω ) X (ω ) Y(ω) = X(ω) = h(ω) = Gleichung 5-4 Spannung am Ausgang der Black-Box mit Frequenz ω [V] Spannung am Eingang der Black-Box mit Frequenz ω [V] Übertragungsfunktion im Frequenzraum [1] Im Frequenzraum ist die Beschreibung der Signalübertragung deutlich einfacher. Die Übertragungsfunktion h(ω) kann unmittelbar aus dem Quotienten von Y(ω) und X(ω) gewonnen werden. Nach Rücktransformation von h(ω) in den Zeitraum ist im Anschluss die Beschreibung des Zeitverhaltens möglich. Ein besonderer Vorzug des Frequenzraums liegt darin, dass Kapazitäten und Induktivitäten mit den gleichen Rechenregeln wie Ohm'sche Widerstände durch komplexe Widerstände, den Impedanzen Z, beschrieben werden können. Wie in Abbildung 5-9 B dargestellt gilt für die einzelnen Bauteile: Z C (ω ) = ZC(ω) ZL(ω) ZR −i ωC = = = , Z L (ω ) = iωL und ZR = R Gleichung 5-5 Komplexer Widerstand einer Kapazität C bei Frequenz ω [Ω] Komplexer Widerstand einer Induktivität L bei Frequenz ω [Ω] Ohm'scher Widerstand; frequenzunabhängig [Ω] Der Phasenverschiebung von Strom und Spannung an den einzelnen Bauteilen wird durch die komplexe Zahl i Rechnung getragen. Kennt man den Schaltplan eines linearen Systems, so kann man anhand der komplexen Widerstände und mit Hilfe der Kirchhoff'schen Gesetze unmittelbar die Übertragungsfunktion h(ω) ausrechnen. Für einen Tiefpass (siehe Abbildung 5-9 C) erhält man beispielsweise: h (ω ) = 1 mit iωRC + 1 h(ω ) = 1 ω 2 R 2C 2 + 1 ϕ (ω ) = − arctan(ωRC ) h(ω) h(ω ) = = Übertragungsfunktion im Frequenzraum [1] Betrag der Übertragungsfunktion im Frequenzraum [1] ϕ (ω) = Phase der Übertragungsfunktion [°] Gleichung 5-6 5.4 Grundlagen der Kopplung 5-25 Signalverarbeitung in linearen Systemen (A) Lineares System als "Black-Box" 1 (B) Schaltelemente im Frequenzraum 3 BlackBox X ZR = R Y 2 ZC = ZL = i ω L 4 0 (C) Beispiel: Tiefpass ϕ (ω ) [-45 °] R X 3 Y C 2 ∧ ∧ Y (ω ) = h(ω ) X (ω ) Y (α X 1 + β X 2 ) = α Y ( X 1 ) + β Y ( X 2 ) 1 −i ωC Bode-Plot -90 1 h(ω ) 4 0,1 0,1 1 10 ω ω0 Abbildung 5-9 Ein lineares System wird als "Black-Box" mit charakteristischer Übertragungsfunktion h(ω) beschrieben. (A) Allgemeine Form eines linearen Systems als "Black-Box". (B) Komplexe Widerstände und Übertragungsfunktion h(ω) im Frequenzraum. (C) Das Frequenzverhalten eines linearen Systems am Beispiel des Tiefpass. 5.4.1.2 Analyse linearer Systeme Physikalisch messbar sind nur der Betrag und die Phase der Übertragungsfunktion. h(ω ) erhält man aus den Amplitudenwerten von Eingangs- und Ausgangssignal: ∧ h(ω ) = Y (ω ) ∧ X (ω ) ∧ Y (ω ) Gleichung 5-7 ∧ X (ω ) = Amplitude des Eingangssignals X [V] = Amplitude des Ausgangssignals Y [V] Den Phasenwinkel ϕ(ω) bestimmt man aus der Phasenverschiebung zwischen beiden Signalen. 5-26 5 Elektrophysiologische Messungen Weit verbreitet ist die graphische Darstellung von h(ω ) und ϕ(ω) in Form des Bode-Plots. Dabei werden die beiden Größen gegen den Logarithmus der Frequenz aufgetragen. Beim Tiefpass in Abbildung 5-9 C knickt der Frequenzgang bei ω =ω0 nach unten ab. Gleichzeitig erreicht der Phasenwinkel einen Wert von -45 °. ω0 ist die Grenzfrequenz des Systems: ω0 = 1 RC Gleichung 5-8 Im Folgenden wird für h(ω ) kurz h(ω) geschrieben; gemeint ist immer der Betrag der Übertragungsfunktion im Frequenzraum. Mit h wird die Übertragungsfunktion des Systems für den Spezialfall h = h(ω=0) einer Übertragung von Gleichspannungen bezeichnet. Um den Schaltplan eines unbekannten linearen Systems zu ermitteln, kann man den Frequenzgang von h(ω) und ϕ(ω) durch direkte Messung bestimmen und diesen mit simulierten Werten, berechnet aus Modellannahmen für die Schaltung, vergleichen. Die freien Parameter des Modells können auf diesem Weg Schritt für Schritt eliminiert werden. Die Phaseninformation ϕ(ω) ist dazu nicht in jedem Fall erforderlich. Enthält eine Schaltung, wie im Fall der Zell-Zell Kopplung, mehrere unbekannte Größen, so ist es hilfreich in einem ersten Schritt den Spezialfall ω = 0 zu analysieren. Gemäß Gleichung 5-5 können alle Kapazitäten in diesem Fall vernachlässigt werden. Mögliche Induktivitäten werden durch eine feste Verbindung ersetzt. Die Schaltung vereinfacht sich. Diese Form der Analyse wird im Folgenden als Gleichspannungsanalyse bezeichnet. Im zweiten Schritt können die Messungen dann auf Wechselspannungen ausgedehnt werden. Hierfür wird der Begriff Wechselspannungsanalyse eingeführt. 5.4.2 Messkonfiguration Sowohl der initiale Test auf Kopplung sowie ausführliche Messreihen nach Detektion einer Kopplung wurden durch Umschalten der einzelnen Messkanäle zwischen VC- und CC- Modus in verschiedenen elektrischen Konfigurationen 5.4 Grundlagen der Kopplung 5-27 Patch-Clamp Konfigurationen zur Kopplungsanalyse Modus Zelle 1 * Zelle 2 ** (A) VC-VC Voltageclamp (VC) Voltageclamp (VC) U1 (B) VC-CC 1 Currentclamp (CC) 1 • Messung des postsynaptischen • Leitfähigkeit der Stroms als Funktion der präsynaptisch angelegten Spannung • Messung der postsynaptischen Spannung als Funktion der präsynaptischen Spannung Kopplung G12 [S] • SpannungsKopplungskoeffizient XU12 [%] • XU12 entspricht h12 Voltageclamp (VC) • Messung des postsynaptischen Stroms als Funktion des präsynaptischen Stroms • StromKopplungskoeffizient XI12 [%] 2 Currentclamp (CC) U2 1 Ergebnis 2 I2 Currentclamp (CC) I1 2 U2 Currentclamp (CC) I1 (D) CC-CC 1 Voltageclamp (VC) U1 (C) CC-VC I2 Art der Messung • Zeitanalyse der Übertragung von Aktionspotentialen bei funktional gekoppelten Zellen 2 • Übertragungszeit t12[ms] • Postsynaptisch induziertes Potential U2 [mV] * Es wird jeweis Zelle 1 Stimuliert. ** Bei dreifach Messungen bezieht sich diese Spalte auch auf Zelle 3. Abbildung 5-10 Unter Einbeziehung von VC- und CC-Modus beider Patch-Clamp Verstärker werden für die Untersuchung eines gekoppelten Zellpaares vier elektrische Messkonfigurationen ausgenutzt. (A) VC-VC-Modus. (B) VC-CC-Modus. (C) CC-VC-Modus. (D) CC-CC-Modus. durchgeführt. In Abbildung 5-10 sind alle möglichen Messkombinationen in einer Tabelle zusammengefasst. Bei Messungen an drei Zellen wurde jeweils nur eine Zelle stimuliert und beide anderen Zellen beobachtet. Alle beobachteten Zellen wurden immer im gleichen Messmodus betrieben. Der VC-VC-Modus (Abbildung 5-10 A) ermöglicht die Messung des postsynaptischen Stroms I2 als Funktion der präsynaptisch angelegten Spannung U1 . Das Membranpotential beider Zellen wird von außen durch die Messanordnung vorgegeben. In dieser Konfiguration kann man die elektrische Leitfähigkeit G12 der Kopplung von Zelle 1 zu Zelle 2 bestimmen [Neyton, 1985]. Es gilt: 5-28 5 Elektrophysiologische Messungen G12 = G12 I2 U1 Gleichung 5-9 I2 U1 = = = Leitfähigkeit von Zelle 1 zu Zelle 2 [nS] Gemessener Strom in Zelle 2 [pA] Angelegte Spannung an Zelle 1 [mV] Weiterhin ist in dieser Konfiguration eine gezielte Messung postsynaptisch induzierter stimulierender und inhibierender Ströme möglich [Bi, 1998]. Der VC-CC-Modus (Abbildung 5-10 B) dient zur Messung der postsynaptisch induzierten Spannung U2 als Funktion der präsynaptisch angelegten Spannung U1 . Das Membranpotential der stimulierten Zelle 1 wird von außen durch die Messanordnung vorgegeben. Die Membranspannung von Zelle 2 wird als Messgröße ermittelt [Neyton, 1986]. Aus dem Quotient von U2 und U1 kann man einen Kopplungskoeffizient XU12 der Spannungsübertragung von Zelle 1 zu Zelle 2 bilden. Dieser entspricht im Bild der linearen Systemtheorie der Übertragungsfunktion h12 des Systems: X U 12 = h12 = XU12 h12 U2 U1 = = = = U2 U1 Gleichung 5-10 Spannungs-Kopplungskoeffizient von Zelle 1 zu Zelle 2 [1] Übertragungsfunktion des linearen Systems "gekoppelte Zellen" [1] Gemessene Spannung in Zelle 2 [mV] Angelegte Spannung an Zelle 1 [mV] Im CC-VC-Modus (Abbildung 5-10 C) wird die Kopplung der Ströme untersucht. Es wird der postsynaptisch in Zelle 2 induzierte Strom I2 als Funktion des präsynaptisch in Zelle 1 geleiteten Stroms I1 gemessen [Carew, 1976]. Der Quotient von I2 und I1 ergibt den Kopplungskoeffizienten XI12 der Stromübertragung von Zelle 1 zu Zelle 2: X I 12 = XI12 = = I2 I1 = I2 I1 Gleichung 5-11 Strom-Kopplungskoeffizient von Zelle 1 zu Zelle 2 [1] Gemessener Strom in Zelle 2 [mV] Angelegter Strom an Zelle 1 [mV] 5.4 Grundlagen der Kopplung 5-29 In der vierten und letzten Messkonfiguration, dem CC-CC-Modus (Abbildung 5-10 D), können beide Zellen ihr Membranpotential frei ändern und sind daher in der Lage Aktionspotentiale auszubilden. Dieser Modus eignet sich zur Bestimmung der synaptischen Übertragungszeit t12 sowie der Amplitude von postsynaptisch induzierten Potentialen U2 bei funktional gekoppelten Zellen. In allen Messmodi wurden jeweils nur relative Änderungen der postsynaptisch gemessenen Spannungen und Ströme als Reaktion auf präsynaptisch angelegte Signale betrachtet. 5.4.3 Messverfahren Die Vermessung der elektrischen Eigenschaften jedes einzelnen untersuchten Systems gekoppelter Zellen erfolgte nach einem einheitlichem Ablauf. Dadurch sollte die Vergleichbarkeit der Messdaten für statistische Auswertungen sichergestellt werden. Nur wenn eine funktionale Kopplung vorlag wurde von diesem Standard abgewichen (siehe hierzu Punkt 5.6). Der gesamte Messprozess umfasst drei Teilschritte, welche im Folgenden beschrieben sind: Im ersten Schritt werden nach Herstellung der Whole-Cell Konfiguration Messdaten im VC-VC-Modus und im VC-CC-Modus erfasst (Abbildung 5-11). Dabei werden durch abwechselnde Stimulation beider5 untersuchter Zellen insgesamt vier Datensätze aufgenommen. 5 Werden gleichzeitig 3 Zellen untersucht, so werden drei anstelle von zwei Einzelmessungen in jeder Konfiguration durchgeführt. Auch in alle späteren Berechnungen ist dann immer der dritte Messkanal einbezogen. 5-30 5 Elektrophysiologische Messungen Messverfahren - Teil 1: Datenerfassung 1.1 Messung in mehreren Konfigurationen VC-VC VC-VC VC-CC VC-VC VC-CC Membranspannung [mV] (A) DC Messung CC-VC VC-VC VC-VC VC-CC CC-VC DC - Datentabelle Stimulierte Zelle Gemessene Zelle 0 -20 -40 Schlüsseldaten Zelle 1 Zelle 2 Experiment Modus Zeit [ms] U1 [mV] I1 [pA] U2 [mV] I2 [pA] V21-2_s2_1 DC_VCVC V21-2_s2_1 DC_VCVC V21-2_s2_1 DC_VCVC ... 0 0,1 0,2 -50,2 -50,1 -50,4 4,2 4,7 3,8 -49,6 -49,4 -50,3 2,2 3,7 1,9 V21-2_s2_1 DC_VCCC ... 0 -49,3 3,2 -27,2 0,1 -60 -80 Zusatzinformationen 0 VC-VC VC-CC Membranspannung [mV] (B) AC Messung 1.2 Datenexport in relationale Datenbank 1 2 Messzeit [s] Stimulierte Zelle Schlüsseldaten 3 DB Gemessene Zelle (MSAccess) Mikroskopie PC-Parameter KabelCMem1 CMem2 länge Experiment Struktur [pF] ... [µm] Bild [pF] V21-2_s2_1 9 30 X 20,7 22,3 ... V21-2_s5_2 7 120 Y 33,4 28,5 ... V22-1_s3_4 10 70 Z 24,6 38,5 ... ... -20 AC - Datentabelle -40 Schlüsseldaten Zelle 1 Zelle 2 Experiment Modus Zeit [ms] U1 [mV] I1 [pA] U2 [mV] I2 [pA] -60 -80 0,0 0,5 1,0 Messzeit [s] 1,5 V21-2_s2_1 AC_VCVC V21-2_s2_1 AC_VCVC V21-2_s2_1 AC_VCVC ... 0 0,1 0,2 -50,1 -49,2 -49,3 3,4 3,2 2,9 -51,2 -51,3 -49,1 1,1 0,5 0,9 V21-2_s2_1 AC_VCCC ... 0 -50,8 2,1 -32,8 -0,7 Abbildung 5-11 Messverfahren zur Charakterisierung der Kopplung - Datenerfassung. (A) Gleichspannungsmessung (B) Wechselspannungsmessung Es erfolgt sowohl eine Untersuchung des Kopplungsverhaltens von Gleichspannungen (Abbildung 5-11 A), als auch von Wechselspannungen zwischen 0 Hz und 8000 Hz (Abbildung 5-11 B). Die Gleichspannungskopplung wird analysiert durch stufenweise Variation der Membranspannung der stimulierten Zelle zwischen -70 und +50 mV in Schritten von +10 mV (Abbildung 5-11 A1.1). Für die Messung der Wechselspannungskopplung werden jeweils 5 Wellenzüge von Wechselspannungen steigender Frequenz und fester Amplitude (30 mV) angelegt (Abbildung 5-11 B1.1). Die nacheinander eingestellten Frequenzen des Stimulationssignals sind: 0, 10, 14, 20, 29, 42, 60, 86, 122, 175, 250, 358, 511, 731, 1046, 1495, 2138, 3058, 4372,6252 und 8941 Hz. Während der Stimulation, wird neben Spannung U1 und Strom I1 der stimulierten Zelle 1, gleichzeitig Spannung U2 und Strom I2 der gekoppelten Zelle 2 gemessen. Diese Messdaten werden zusammen mit dem jeweiligen Messzeitpunkt gespeichert. Die Abtastung der Signale erfolgt dabei mit einer Abtastrate von ~ 10 kHz. Nach 5.4 Grundlagen der Kopplung 5-31 Messverfahren - Teil 2: Datenverarbeitung 2.1 Amplitudenbestimmung in ausgewählten Zeitfenstern VC-VC VC-VC DB VC-CC VC-VC VC-CC Membranspannung [mV] (A) DC Messung CC-VC VC-VC VC-VC DB VC-CC CC-VC Messintervall Gemessene Zelle DC - Ergebnistabelle Schlüsseldaten Experiment Modus -20 -40 -60 2,00 2,25 2,50 2,75 Messzeit [s] VC-VC VC-CC Stimulierte Zelle Membranspannung [mV] (B) AC Messung 0 Stimulierte Zelle 2.2 Speichern der Ergebnisse in neuen Tabellen Messintervall Gemessene Zelle V21-2_s2_1 V21-2_s2_1 V21-2_s2_1 V21-2_s2_1 V21-2_s2_1 ... V21-2_s2_1 ... DC_VCVC DC_VCVC DC_VCVC DC_VCVC DC_VCVC 0 0 0 0 0 -20 -10 0 10 20 -2 0 1 2 2 0 0 0 0 0 -10 -5 0 5 10 DC_VCCC 0 -20 0 -12 0 AC - Ergebnistabelle Schlüsseldaten -20 Experiment Modus -40 -60 -80 1,000 1,125 Messzeit [s] 1,250 Zelle 1 Zelle 2 Frequenz [Hz] Û1 [mV] Î1 [pA] Û2 [mV] Î2 [pA] V21-2_s2_1 V21-2_s2_1 V21-2_s2_1 V21-2_s2_1 V21-2_s2_1 ... V21-2_s2_1 ... Zelle 1 Zelle 2 Frequenz [Hz] Û1 [mV] Î1 [pA] Û2 [mV] Î2 [pA] DC_VCVC DC_VCVC DC_VCVC DC_VCVC DC_VCVC 0 10 14 20 29 20 20 20 20 20 1 0 2 1 0 9 10 9 8 8 1 0 0 2 1 DC_VCCC 0 20 1 5 2 Abbildung 5-12 Messverfahren zur Charakterisierung der Kopplung - Datenverarbeitung. (A) Gleichspannungsmessung (B) Wechselspannungsmessung Abschluss der Messung werden alle erfassten Daten in Tabellen einer relationalen Datenbank (Microsoft Access, Version Office '97) exportiert und innerhalb der Datenbank zusammen mit Zusatzinformationen wie Mikroskopieaufnahmen und Parametern des Patch-Clamp Verstärkers gespeichert. Im nächsten Schritt werden die Messdaten innerhalb der Datenbank verarbeitet (Abbildung 5-12). Ein fest vorgegebenes, automatisch ablaufendes Analyseprogramm bestimmt die Amplituden Û1 , Û2 , Î1 und Î2 der Messignale in fest vorgegebenen Zeitintervallen. Bei Gleichspannungsmessung (Abbildung 5-12 A2.1) entspricht die Amplitude dem Sprung des Messwertes aus einem Zeitintervall unmittelbar vor Anlegen des Stimulationspulses auf den asymptotischen Endwert kurz vor Ende des angelegten Pulses. Bei Wechselspannungsmessung (Abbildung 5-12 B2.1) ist die Amplitude gleich dem Abstand zwischen Maximum und Minimum der Messwerte im betrachteten Zeitintervall. 5-32 5 Elektrophysiologische Messungen Messverfahren - Teil 3: Datenanalyse und Ausgabe VC-VC VC-VC DB G12 = I2 / U1 G21 = I1 / U2 h12 = U2 / U1 VC-CC G12 (ω) = Î2 (ω) / Û1 (ω) VC-VC VC-VC DB G21 (ω) = Î1 (ω) / Û2 (ω) h12 (ω) = Û2 (ω) / Û1 (ω) VC-CC CC-VC h12 h21 50 0 -20 0 h21 (ω) = Û1 (ω) / Û2 (ω) 20 40 60 80 100 Stimulation [mV] G [nS] (B) AC Messung G12 G21 1 h21 = U1 / U2 <V21_5_12-5-3> 2 G12 G21 1 0 100 h [%] CC-VC 2 0 100 h [%] (A) DC Messung 3.2 Ausgabe der Ergebnisse G [nS] 3.1 Berechnen der Kopplungsparameter h12 h21 50 0 10 100 1000 10000 Frequenz [Hz] <V21_5_12-5-3> Abbildung 5-13 Messverfahren zur Charakterisierung Ohm'scher Kopplung - Datenanalyse und Ausgabe. (A) Gleichspannungsmessung (B) Wechselspannungsmessung Die Ergebnisse der Amplitudenbestimmung werden innerhalb der Datenbank als neue Datensätze in einer DC- und AC-Ergebnistabelle gespeichert. Der letzte Schritt umfasst die Analyse und Ausgabe der erhaltenen Messergebnisse. Innerhalb der relationalen Datenbank werden dabei nach festen Rechenvorschriften spezifische Kopplungsparameter ermittelt und diese graphisch dargestellt (Abbildung 5-13). Aus den Messdaten des VC-VC-Modus wird eine Leitfähigkeit G12 der Kopplung von Zelle 1 nach Zelle 2 in nS berechnet. Aus den Messdaten des VC-CC-Modus erhält man einen dimensionslosen Kopplungsfaktor XU12 . Dieser ist gleichzusetzen mit der Übertragungsfunktion h(ω) des Systems. Die Darstellung dieser Parameter erfolgt für die Gleichspannungsanalyse als Funktion der jeweils angelegten Stimulationsamplitude in Zelle 1 (Abbildung 5-13 A3.2). Die Kopplungsparameter der Wechselspannungsanalyse werden als 5.4 Grundlagen der Kopplung 5-33 Funktion der jeweils verwendeten Stimulationsfrequenz dargestellt (Abbildung 5-13 B3.2). In den Graphen in Abbildung 5-13 sind die berechneten Parameter einer typischen Ohm'schen Kopplung dargestellt. Man erkennt, dass im Fall der Gleichspannungsnanalyse (Abbildung 5-13 A3.2) beide Kopplungsparameter bis zu einer kritischen Spannung von ca. +40 mV Stimulationsamplitude nahezu konstant sind. Das Absinken der Kopplung oberhalb von +40 mV Stimulationsspannung kann durch das Einsetzen nichtlinearer Effekte aufgrund öffnender Ionenkanäle erklärt werden. Die Graphen der Wechselspannungsanalyse (Abbildung 5-13 B3.2) zeigen einen deutlichen Abfall der Kopplung mit steigender Frequenz. Dies ist auf die Membrankapazität der Zellen und der Verbindung zwischen den Zellen zurückzuführen. Der scheinbare erneute Anstieg der Kopplung bei hohen Frequenzen (VC-VC-Modus) ist nicht real sondern ein Artefakt des Messverstärkers, welcher aufgrund der großen Ströme bei hohen Frequenzen auftritt (siehe hierzu Punkt 5.4.6). 5.4.4 Physikalisches Kopplungsmodell Für die physikalische Beschreibung der elektrischen Signalübertragung zwischen zwei Nervenzellen erscheint es sinnvoll sich an der Morphologie des realen Systems zu orientieren. Das System gliedert sich dann wie in Abbildung 5-14 A schematisch dargestellt, in vier Kompartimente: 1. Eine Zelle 1, von welcher die Signalübertragung ausgeht. Physikalisch lässt sich dieses Kompartiment vereinfacht als eine Sphäre mit der Zellmenbran als Wand und gefüllt mit Cytoplasma beschreiben. 2. Ein Kabel von Zelle 1 zu einer zweiten Zelle 2. Entlang dieses Kabels breitet sich das elektrische Signal aus. Dieses Kompartiment kann man als einen zylindrischen Leiter ansehen, umgeben mit einer Wand als Isolationsschicht. In seinem Inneren ist dieser mit Cytoplasma gefüllt und sein Innenraum ist unmittelbar mit Zelle 1 verbunden. Die Wand des Leiters ist morphologisch eine Ausstülpung der Zellmembran von Zelle 1 und hat 5-34 5 Elektrophysiologische Messungen Physikalisches Modell der Zellkopplung (B) Kabelmodell l (A) Kompartimente des Systems Zelle 1 Kabel X r Zelle 2 ∆l ∆Ri (C) Elektrisches Ersatzschaltbild Kabel ∆Cm Kopplung X Zelle 2 X1: RPore ∆Ri ∆ Cm ∆ Rm ∆ Cm ∆ Rm X2: CKontakt ∆ Cm Um RMem2 ∆Ri ∆ Rm CMem1 RMem1 ∆Ri ∆Rm CMem2 Zelle 1 K X3: NFunktional Abbildung 5-14 Die elektrischen Eigenschaften der Kopplung zweier Nervenzellen werden in einem auf vier Kompartimenten basierenden physikalischen Modell beschrieben. (A) Kompartimente des Systems. (B) Beschreibung des Kabels zwischen Zelle 1 und Zelle 2 als zylindrischer Hohlleiter. (C) Elektrisches Ersatzschaltbild des Gesamtsystems. daher in erster Näherung die gleichen elektrischen Eigenschaften wie die Zellmembran. 3. Die eigentliche synaptische Verbindung X zwischen beiden Zellen. Je nach Art der Kopplung (siehe Abbildung 5-14 C) erfolgt an dieser Stelle eine Signalweiterleitung zu Zelle 2 entweder über eine leitende Pore mit Widerstand RPore (Gap-Junctions bei Ohm'scher Kopplung), über eine Kontaktkapazität CKontakt (kapazitive Kopplung), oder aber auf chemischem Weg durch Ausschüttung von Neurotransmitter NFunktional in den synaptischen Spalt einer funktionalen Synapse. Die im Modell getroffene Annahme, dass die synaptische Verbindung X am Ende des Kabels liegt und nicht in der Mitte ist willkürlich. Für die elektrische Beschreibung hat dies aufgrund der Symmetrie des Systems aber keine Relevanz. Siehe hierzu auch Punkt 5.5.2.1. 5.4 Grundlagen der Kopplung 5-35 4. Eine Zelle 2, welche das übertragene Signal empfängt. Die physikalische Beschreibung ist analog zu Zelle 1. Die elektrischen Eigenschaften dieser vier Kompartimente können nun einzeln, wie in Abbildung 5-14 C dargestellt, mit elektrischen Ersatzschaltbildern modelliert werden. Durch Verbinden aller Kompartimente entsteht ein Gesamtschaltplan des Systems. 5.4.4.1 Beschreibung der Zellen Die Beschreibung beider Zellen auf Basis von Membranwiderstand RMem1,2 und Membrankapazität CMem1,2 folgt unmittelbar aus elementaren Eigenschaften der Zellmenbran (siehe auch Kapitel 2). 5.4.4.2 Beschreibung der Kopplung Die Modellierung von Ohm'scher Kopplung durch einen Widerstand RPore sowie von kapazitiver Kopplung durch eine Kontaktkapazität CKontakt ergibt sich aus den in Punkt 5.3 angestellten Überlegungen. In beiden Fällen entsteht ein geschlossener elektrischer Schaltkreis zwischen Zelle 1 und Zelle 2. Bei funktionaler Kopplung hingegen besteht keine direkte elektrische Verbindung zwischen beiden Zellen. An der Kopplungsstelle X ist der Schaltkreis bei Kopplungstyp X3 unterbrochen. Die Signalübertragung geschieht in diesem Fall nicht elektrisch sondern, wie in Kapitel 2 beschrieben, auf chemischem Weg. Im Gegensatz zu Ohm'scher und kapazitiver Kopplung ist funktionale Kopplung ein aktiver Prozess, bei dem auch eine Signalverstärkung erfolgt. Aufgrund der damit verbundenen Nichtlinearitäten können die Gesetze der linearen Systemtheorie auf funktionale Synapsen nicht angewendet werden. Für die funktionale Kopplung zweier Zellen kann man daher alleine auf Basis der linearen Systemtheorie kein geschlossenes Modell aufstellen. Die Auswertung der Messdaten funktionaler Kopplung erfolgt aus diesem Grund nicht über ein geschlossenes Kopplungsmodell sondern anhand ausgewählter Parameter wie der Übertragungszeit und der Kopplungsstärke. Die genaue Vorgehensweise wird in Punkt 5.6 beschrieben. 5-36 5 Elektrophysiologische Messungen 5.4.4.3 Beschreibung des Kabels Neben der Kopplungsstelle X, hat auch das Kabel einen ganz entscheidenden Einfluss auf die Signalübertragung. Im Folgenden werden die elektrischen Eigenschaften dieses zentralen Schaltungselements ausführlich diskutiert. Wie in Abbildung 5-14 B dargestellt, hat das Axon eines Neurons in erster Näherung die Form eines zylindrischen Rohrs. Die isolierende Wand des Rohrs besteht aus einer Membran ähnlich der Zellmenbran. Das innere des Rohrs ist mit leitfähigem Cytoplasma gefüllt. Die Umgebung des Axons besteht aus ebenfalls leitfähiger, extrazellulärer Lösung. Beachtet man die Größenordnung aller physikalischen Effekte, so kann man ohne relevante Fehler den Widerstand der Umgebung des Axons als identisch Null annehmen sowie induktive Komponenten des Kabels vernachlässigen [Katz, 1985]. Für ein kleines Stück des Kabels der Länge ∆l kann man dann im Grenzfall ∆l 0 das Ersatzschaltbild in Abbildung 5-14 B aufstellen. Dieses besteht aus dem Innenwiderstand ∆Ri längs des Kabelstücks (Widerstand der Cytoplasmasäule) sowie einer Parallelschaltung von Membranwiderstand ∆Rm und Membrankapazität ∆Cm (Oberfläche des zylindrischen Membransegments). Durch Aneinanderreihen unendlich vieler solcher Kabelsegmente erhält man das Schaltbild eines Kabels mit realer Länge l (siehe Abbildung 5-14 C). Die physikalische Größe der Bauteile ∆Ri , ∆Rm und ∆Cm folgt aus den Abmessungen des Kabels mit Hilfe von Literaturwerten für die spezifische Leitfähigkeit des Cytoplasmas ρi und der Zellmenbran ρm sowie der spezifischen Kapazität der Zellmenbran ζm : ∆Ri = ρ i ∆Ri ∆Rm ∆Cm ρi ρm ζm ∆l r = = = = = = = = ∆l πr 2 ∆R m = ρ m 1 2πr∆l ∆C m = ζ m 2πr∆l Gleichung 5-12 Längswiderstand eines Kabelsegments der Länge ∆l [Ohm] Oberflächenwiderstand eines Zylinderstücks der Länge ∆l [Ohm] Oberflächenkapazität eines Zylinderstücks der Länge ∆l [µF] Spezifischer Widerstand des Cytoplasmas [Ohm*cm] (ρi ~ 200 Ω*cm) Spezifischer Widerstand der Zellmenbran [Ohm*cm²] (ρm ~ 2000 Ω*cm²) Spezifische Kapazität der Zellmenbran [µF/cm²] (ζm ~ 1 µF/cm²) Länge des Kabelsegments [cm] Innenradius des Kabels [cm] 5.4 Grundlagen der Kopplung 5-37 Anwendung der Kirchoff'schen Knotenregel auf ein einzelnes Kabelsegment (Knotenpunkt K in Abbildung 5-14 B) liefert eine Beziehung für die Ströme IRi , IRm und ICm durch die einzelnen Bauteile: I Ri = I Rm + I Cm Gleichung 5-13 Aus Rechenregeln für kapazitive und Ohm'sche Bauteile erhält man unter Einbeziehung der Membranspannung Um am betrachteten Knotenpunkt K sowie des Spannungsabfalls ∆Um über dem betrachteten Membransegment die Gleichungen: I Ri = ∆U m ∆Ri I Rm = Um ∆R m I Cm = ∆ C m ∂U m ∂t Gleichung 5-14 Im Grenzübergang zu infinitesimal kleinen Kabelsegmenten ∆l ∂l und nach Einsetzen von Gleichung 5-14 in Gleichung 5-13 sowie Ersetzen von ∆Ri , ∆Rm und ∆Cm durch die Beziehungen aus Gleichung 5-12 resultiert als Endergebnis eine inhomogene Differentialgleichung (DGL) zweiter Ordnung: U m (l , t ) = λ 2 Um(l,t) = ρi = ρm = ζm = r = ∂ 2U m ( l , t ) ∂U m (l , t ) −τ 2 ∂t ∂l mit λ2 = rρ m τ = ρ mζ m 2 ρ i und Gleichung 5-15 Membranspannung zum Zeitpunkt t an Punkt l des Kabels [mV] Spezifischer Widerstand des Cytoplasmas [Ohm*cm] (ρi ~ 200 Ω*cm) Spezifischer Widerstand der Zellmenbran [Ohm*cm²] (ρm ~ 2000 Ω*cm²) Spezifische Kapazität der Zellmenbran [µF/cm²] (ζm ~ 1 µF/cm²) Innenradius des Kabels [cm] Diese beschreibt die räumliche und zeitliche Ausbreitung einer am Ort l = 0 und zur Zeit t = 0 an das Axon angelegten Spannung Um0 = Um(l = 0, t = 0). Für den stationären Fall (System ist im eingeschwungenen Zustand, die Ableitung von Um nach der Zeit ist Null) wird diese DGL durch eine ortsabhängige Exponentialfunktion gelöst: U m (l ) = U e 0 m − l λ Gleichung 5-16 5-38 5 Elektrophysiologische Messungen Die Signalamplitude Um fällt entlang des Axons exponentiell mit dem Abstand l vom Ursprungsort der Erregung ab. Diese Beziehung wurde bereits in Kapitel 2 verwendet. Die Lösung der nicht-stationären DGL erhält man aus einem Vergleich von Gleichung 5-15 mit der Diffusionsgleichung [Feynman, 1991]. Gleichung 5-15 stimmt formal mit einer inhomogenen, eindimensionalen Diffusionsgleichung überein. Für die Geschwindigkeit der Signalausbreitung kann man in Anlehnung an die Diffusionsgeschwindigkeit daher unmittelbar schreiben: 2λ2 τ2 v= v = Gleichung 5-17 Ausbreitungsgeschwindigkeit eines Spannungspulses entlang des Axons [cm/µs] Für ein Axon mit einem Radius von r = 1 µm = 10-4 cm erhält man daraus unter Verwendung der angegebenen Literaturwerte für ρi , ρm und ζm aus Gleichung 5-12 sowie den Definitionen von λ und τ in Gleichung 5-15 eine Signalausbreitungsgeschwindigkeit von v = 16 cm/s. Die Abklinglänge λ, innerhalb derer die Signalamplitude gemäß Gleichung 5-16 auf den Faktor 1/e abgeklungen ist, beträgt für ein solches Axon λ = 200 µm. Beide Ergebnisse sind für eine schnelle Weiterleitung von Signalen über größere Strecken im Körper zu gering. Zwar können Ausbreitungsgeschwindigkeit und Abklinglänge durch Ausdehnung des Axonradius gesteigert werden (λ und v wachsen proportional zu r ), dies hätte jedoch eine enorme Vergrößerung des gesamten PNS und ZNS zur Folge. Bei vertebraten Organismen hat die Natur dieses Problem elegant umgangen indem die Axone der Nervenzellen durch eine Isolationsschicht, den sog. Myelinmantel, umhüllt werden (siehe Kapitel 2). Dieser vergrößert ρm und verringert gleichzeitig ζm . So werden Signalgeschwindigkeiten von bis zu v = 100 m/s möglich. Der trotz Isolation verbleibende Abfall der Signalamplitude entlang des Axons wird vollständig durch eine aktive Verstärkung des Signals mit spannungssensitiven 5.4 Grundlagen der Kopplung 5-39 Ionenkanälen entlang des Kabels kompensiert. In allen vertebraten Organismen ist daher eine verlustfreie Übertragung von Signalen vom Gehirn bis in die äußesten Extremitäten möglich. Im Hinblick auf die später beschriebene Analyse der Kopplung von Nervenzellen, erscheint es an dieser Stelle sinnvoll, auch die Impedanz Z des Kabels zu berechnen. Man gelangt wieder zum Ziel indem man das Kabel als eine Kaskade von kleinen Teilstücken betrachtet wie in Abbildung 5-14 C dargestellt. Feynman beschreibt für eine solche Schaltung [Feynman, 1991] einen geeigneten Rechenansatz: Man betrachtet die kleinste Einheit der Kaskade, in diesem Fall die Schaltung in Abbildung 5-14 B, und hängt diese als neues Glied an ein bereits unendlich langes Netzwerk an. Das unendlich lange Netzwerk habe dabei bereits die gesuchte Impedanz Z . Im Grenzfall ∆l 0 ändert sich die Impedanz des Gesamtsystems nicht durch Hinzuschalten eines weiteren Gliedes. Die Impedanz, welche man hinter dem neu angehängten Glied misst, entspricht also wieder der Impedanz Z. Auf Basis dieser Überlegungen kann man eine Gleichung aufstellen, welche nach der Unbekannten Z aufgelöst werden kann. Anwendung dieses Ansatzes auf die Schaltung in Abbildung 5-14 C ergibt: Z = ∆ Ri + Z = ∆Ri = ∆Zm = Z∆Z m Z + ∆Z m Gleichung 5-18 Gesuchte Impedanz des Kabels [Ohm] Längswiderstand eines Kabelsegments der Länge ∆l [Ohm] Impedanz der Parallelschaltung aus Membranwiderstand ∆Rm und Membrankapazität ∆Cm in der Membran des Kabelsegments [Ohm] Man erhält eine quadratische Gleichung in Z . Da der Realteil von Z größer Null sein muss, besitzt diese eine einzige, eindeutige Lösung: ∆Ri Z= + 2 ∆R i2 + ∆ R i ∆Z m 4 Gleichung 5-19 5-40 5 Elektrophysiologische Messungen Für ∆Zm kann man auf Basis der Parallelschaltung von Membranwiderstand ∆Rm und Membrankapazität ∆Cm schreiben: ∆Z m = ω = ∆ Rm 1 + i ω ∆ Rm ∆ C m Gleichung 5-20 Kreisfrequenz des angelegten Signals [Hz] Durch Einsetzen von ∆Zm aus Gleichung 5-20 und der Formeln für ∆Ri , ∆Rm und ∆Cm aus Gleichung 5-12 in Gleichung 5-19 sowie unter Beachtung von ∆l 0, erhält man als Ergebnis die komplexe Impedanz Z des zylindrischen Kabels: ρi ρ m 1 2 3 2π r 1 + iωρ mζ m Z= Z ρi ρm ζm r = = = = = Gleichung 5-21 Impedanz des Kabels [Ohm] Spezifischer Widerstand des Cytoplasmas [Ohm*cm] (ρi ~ 200 Ω*cm) Spezifischer Widerstand der Zellmenbran [Ohm*cm²] (ρm ~ 2000 Ω*cm²) Spezifische Kapazität der Zellmenbran [µF/cm²] (ζm ~ 1 µF/cm²) Innenradius des Kabels [cm] Der Betrag der Impedanz ist: Z = ρi ρ m 1 2 3 2π r 1 + ω 2 ρ 2ζ 2 m m Gleichung 5-22 Für ein Axon mit Radius r = 1 µm resultiert daraus eine Impedanz von Z = 140 MΩ bei Anlegen einer Gleichspannung. Für eine Wechselspannung mit ω = 2*π*250 Hz (Grenzfrequenz eines normalen Aktionspotentials) verringert sich die Impedanz auf Z = 78 MΩ. Für physiologisch relevante Signale kann man folglich eine axonale Impedanz Z zwischen 78 und 140 MΩ annehmen. 5.4.5 Vereinfachtes Ohm'sches Kopplungsmodell Mit einigen Nebenannahmen kann man für die Beschreibung Ohm'scher Kopplung eine vereinfachte Form des zuvor vorgestellten physikalischen Kopplungsmodells verwenden. In Abbildung 5-15 ist dieses vereinfachte Modell dargestellt. 5.4 Grundlagen der Kopplung 5-41 Die unbekannte Kopplungsgröße X des allgemeinen Modells aus Abbildung 5-14 wurde in Abbildung 5-15 A ersetzt durch eine Verengung des Kabelquerschnitts vor Zelle 2. Diese Verengung repräsentiert elektrisch die Funktion der Gap-Junctions an der Synapse. Die Vereinfachten elektrischen Ersatzschaltbilder der Kopplung in Abbildung 5-15 B und C basieren auf folgender Überlegung: Die typische Leitfähigkeit Ohm'scher Synapsen liegt in der Größenordnung einiger nS [Neyton, 1985]. In den durchgeführten Experimenten wurden meist schwache Ohm'sche Kopplungen mit einer Leitfähigkeit kleiner als 2 nS detektiert. Daraus resultiert ein Ohm'scher Widerstand der synaptischen Verbindungsstelle von mehr als 500 MΩ. Dieser Wert liegt deutlich über der Impedanz Z ~ 100 MΩ des axonalen Kabels vor der Synapse (siehe Punkt 5.4.4.3). Vereinfachtes Modell der Ohm'schen Kopplung Kopplung über Transmembranpore mit Ionenleitfähigkeit (Gap Junction) (A) Kopplungsmodell Zelle 2 Kabel (B) Ersatzschaltbild für Gleichspannungen 0 3 0 Abbildung 5-15 Die Beschreibung Ohm'scher Kopplung erfolgt in einem vereinfachten elektrischen Modell. (A) Kopplungsprinzip. (B) Elektrisches Ersatzschaltbild für die Kopplung von Gleichspannungen. (C) Elektrisches Ersatzschaltbild für die Kopplung von Gleich- und Wechselspannungen. RMem2 RPore 2 Zelle 2 CMem2 RKabel 1 Kopplung CMemKabel 3 Kabel RMemKabel RPore Zelle 1 CMem1 RMemKabel RKabel 2 RMem1 1 Kopplung Zelle 2 RMem2 Zelle 1 Kabel (C) Ersatzschaltbild für Wechselspannungen RMem1 Zelle 1 5-42 5 Elektrophysiologische Messungen In einem vereinfachten Modell kann man für die elektrische Beschreibung des Systems aus diesem Grund annehmen, dass das Kabel durch Ströme vom Kabelende über die Kopplungsstelle zu Zelle 2 nicht belastet wird. Das System aus Zelle 1 und Kabel kann als vollständig entkoppelt von Zelle 2 angesehen werden. Das Kabel wirkt in diesem Fall als Spannungsteiler, welcher die Spannung aus Zelle 1 verringert bevor diese den Kopplungswiderstand zu Zelle 2 erreicht. 5.4.5.1 Kopplung von Gleichspannungen Betrachtet man den Spezialfall einer Übertragung reiner Gleichspannungen, so entfallen alle kapazitiven Elemente aus der Schaltung in Abbildung 5-14. Das Ersatzschaltbild kann wie in Abbildung 5-15 B dargestellt, als reines Widerstandsnetz beschrieben werden. Zelle 1 und Zelle 2 treten darin mit ihrem jeweiligen Membranwiderstand RMem1,2 auf. Die Ohm'sche Kopplung wird repräsentiert durch den Widerstand der Pore RPore , und das Kabel wird durch einen einfachen Spannungsteiler aus Kabelserienwiderstand RKabel und Kabelmembranwiderstand RMemKabel ersetzt. Auf Basis der oben begründeten Entkopplung von Kabel und Zelle 2, kann man für den Fall schwacher Kopplung die Spannung U2 am Ende des Kabels (Punkt 2) als Funktion der Spannung U1 in Zelle 1 (Punkt 1) direkt berechnen. Es gilt: U2 R MemKabel = = hKabel U 1 R MemKabel + R Kabel U1 = U2 = RMemKabel = RKabel = hKabel = U 2 = hKabel U 1 Gleichung 5-23 Spannung in Zelle 1 [mV] Spannung am Ende des Kabels [mV] Querwiderstand in vereinfachtem Kabelmodell [MOhm] Längswiderstand in vereinfachtem Kabelmodell [MOhm] Übertragungsfunktion des Kabels [1] Die Spannung U2 am Ende des Kabels ist also proportional zur Spannung U1 in Zelle 1, mit dem Wert der Übertragungsfunktion des Kabels hKabel als Proportionaliätskonstante. Zelle 2 ist über den Kopplungswiderstand RPore und das Kabel an die Spannungsquelle aus Zelle 1 angeschlossen. Für die Spannung U3 in Zelle 2 gilt: 5.4 Grundlagen der Kopplung U3 R Mem 2 = = hPore U 2 R Mem 2 + R Pore U1 = U2 = U3 = RMem2 = RPore = hPore = hKabel = 5-43 U 3 = h PoreU 2 = hKabel hPoreU 1 Gleichung 5-24 Spannung in Zelle 1 [mV] Spannung am Ende des Kabels [mV] Spannung in Zelle 2 [mV] Widerstand der Zellmenbran von Zelle 2 [MOhm] Widerstand der Ohm'schen Kopplung [MOhm] Übertragungsfunktion der Ohm'schen Synapse [1] Übertragungsfunktion des Kabels [1] Werden die Spannungen in beiden Zellen bei einer Messung im VC-VC-Modus von aussen kontrolliert, so gilt für die Änderung des über die Verbindung in Zelle 2 fließenden Stroms IPore bei einer Spannungsänderung ∆U1 in Zelle 1: ∆I Pore = ∆U 2 R Pore = U 3 = const ∆U 1 hKabel R Pore U 3 = const Gleichung 5-25 Der Widerstand der synaptischen Verbindung RPore , skaliert um die Übertragungsfunktion des Kabels hKabel , kann im VC-VC-Modus also direkt gemessen werden. 5.4.5.2 Kopplung von Wechselspannungen Für die Kopplung von Wechselspannungen gelten grundsätzlich die gleichen Überlegungen wie für die Kopplung von Gleichspannungen. Die in diesem Fall hinzukommenden kapazitiven Elemente der Schaltung (siehe Abbildung 5-15 C) werden in den zuvor beschriebenen Gleichungen durch Einführen komplexer Impedanzen berücksichtigt. RPore und RKabel bleiben unverändert, RMem1, RMemKabel und RMem2 werden durch die Impedanzen ZMem1, ZMemKabel und ZMem2 ersetzt. Die Impedanzen Z gehen aus der Parallelschaltung jeweils eines Ohm'schen Widerstandes und einer Kapazität wie folgt hervor: 5-44 5 Elektrophysiologische Messungen Z Mem1, 2 = ZMem1,2 ZMemKabel RMem1,2 RMemKabel CMem1,2 CMemKabel ω = = = = = = = R Mem1, 2 1 + iωR Mem1, 2 C Mem1, 2 Z MemKabel = R MemKabel 1 + iωR MemKabel C MemKabel Gleichung 5-26 Impedanz der Zellmenbranen [MOhm] Impedanz des Kabelquerwiderstands [MOhm] Widerstand der Zellmenbranen [MOhm] Widerstand des Kabelquerwiderstands [MOhm] Kapazität der Zellmenbranen [pF] Kapazität über die Kabelwand [pF] Kreisfrequenz des übertragenen Wechselspannungssignals [Hz] 5.4.6 Betrachtung systematischer Fehlerquellen Impedanzmessungen an gekoppelten Nervenzellen sind mit einem Patch-Clamp Messaufbau grundsätzlich nur eingeschränkt möglich. Der große Zugangswiderstand durch die Patch-Pipette in die Zelle bildet einen Tiefpass mit der Membrankapazität und beschneidet den Frequenzgang [Fromherz, 1993]. Weiterhin kann es durch ein nicht 100 % lineares Verhalten des Patch-Clamp Verstärkers zu einer zusätzlichen Verzerrung des Frequenzgangs kommen. Um den Einfluss dieser Störfaktoren auf die durchgeführten Messungen zu bestimmen, wurden entsprechend dem Kopplungsmodell in Abbildung 5-15 C mehrere elektrische Testschaltkreise aus Präzisionswiderständen und Kondensatoren bekannter Wertigkeit aufgebaut. Aus einem Vergleich des mit dem Patch-Clamp Verstärker real gemessenen Frequenzgangs dieser Schaltungen mit einer Simulation in PSpice (PSpice Mixed Mode Simulator V6.0, MicroSim Corporation) können systematische Fehlerquellen ermittelt werden. Ein Vergleich des Frequenzgangs simulierter Testschaltkreise mit realen Messdaten gekoppelter Zellen ermöglicht darüber hinaus auch eine Prüfung des Kopplungsmodells. 5.4 Grundlagen der Kopplung 5.4.6.1 5-45 Frequenzgang des Patch-Clamp Verstärkers Abbildung 5-16 B zeigt eine Photographie eines fertig aufgebauten Testschaltkreises. Alle Bauteile sind in einem HF-dichten Metallgehäuse untergebracht, um Einstreuungen von Störsignalen zu minimieren. In Abbildung 5-16 B ist ein allgemeiner Schaltplan abgebildet. Über das Testschaltkreis - Ohm'sche Kopplung (A) Beschriebenes System (B) Bauteil • zwei passive Zellen, elektrisch gekoppelt – Ohm'sche Kopplung – Kabel mit kapazitiven und Ohm'schen Verlusten Pipetten 1 & 2 Zelle 1 Zelle 2 (C) Schaltplan Pipette 1 R1 1 C1 Zelle 1 R2 Kabel / Kopplung R3 2 C2 R4 Zelle 2 R5 3 C4 Pipette 2 R7 5 4 C6 R6 C7 0 Abbildung 5-16 Testschaltkreise aus Präzisionsbauteilen wurden für die Überprüfung von Kopplungsmodell und Frequenzgang des Patch-Clamp Verstärkers verwendet. (A) Der Testschaltkreis beschreibt alle elektrischen Komponenten eines Systems aus zwei gekoppelten Zellen einschließlich der Patch-Pipetten. (B) Photographie eines fertig aufgebauten (geöffneten) Testschaltkreises. (C) Allgemeiner Schaltplan der Testschaltkreise. Ersatzschaltbild der Ohm'schen Kopplung in Abbildung 5-15 C hinaus, sind darin zusätzliche Komponenten (R1 und C1 sowie R2 und C2) zur elektrischen Beschreibung der beiden Patch-Pipetten enthalten. Insgesamt beschreibt der Testschaltkreis also ein System aus zwei Patch-Pipetten, zwei Zellen, einem Kabel sowie einer Ohm'schen Kopplung (Abbildung 5-16 A). 5-46 5 Elektrophysiologische Messungen Für die Untersuchungen wurde ein Testschaltkreis mit Bauteilen folgender Größe6 verwendet: R1 = R 7 = 10 MΩ R2 = R 6 = 3300 MΩ C2 = 22,0 pF R3 = R 5 = 330 MΩ C4 = 4,7 pF 33000 MΩ C6 = 33,0 pF R4 = C1 = C 7 = 6,8 pF Messungen an diesem Schaltkreis wurden mit Simulationen in PSpice verglichen. In Abbildung 5-17 A und Abbildung 5-17 B sind die experimentell ermittelten Messkurven des Frequenzgangs (einzelne Messpunkte) dem Ergebnis einer Simulation in PSpice (durchgezogene Linie) überlagert dargestellt. Abbildung 5-17 A beschreibt die Messung im VC-VC-Modus. Aufgetragen ist die Leitfähigkeit der Kopplung G12 gegenüber der Frequenz: − Die Schaltung verhält sich wie ein Tiefpass. Nur bei tiefen Frequenzen wird eine signifikante Kopplung detektiert, und diese nimmt ab ca. 50 Hz mit steigender Frequenz stark ab. − Bis zu einer Frequenz von ca. 400 Hz sind nur geringe Abweichungen der Messung von der Simulation zu erkennen. Der Patch-Clamp Verstärker arbeitet für Frequenzen zwischen 0 und 400 Hz annähernd linear. Abweichungen oberhalb von 400 Hz sind für die Auswertung der Messdaten unerheblich, da aufgrund anderer Überlegungen (siehe unten) nur Messwerte bis ~ 100 Hz verwendet werden können. 6 Die Wertigkeit der Bauelemente von Pipette 1, Pipette 2, Zelle 1, Zelle 2 und Kopplungspore wurde in Anlehnung an reale Messdaten gewählt. Der Wert der Kabelkapazität ergibt sich aus der Annahme, dass ein typisches Kabel von 100 µm Länge etwa die Oberfläche von 1/10 der Zelloberfläche hat. 5.4 Grundlagen der Kopplung 5-47 Frequenzgang der Ohm'schen Kopplung (B) VC-CC-Messmodus (A) VC-VC-Messmodus U1 Testschaltkreis U1 I2 G12 (ω ) = Î2 (ω ) / Û1 (ω) Kopplung h12 [%] Kopplung G12 [nS] h12-gemessen G12-simuliert 1,5 1,0 0,5 10 100 1000 Frequenz [Hz] Simulation_VCVC_normal.OPJ U2 h12 (ω ) = Û2 (ω) / Û1(ω) G12-gemessen 0,0 Testschaltkreis 10000 h12-simuliert 80 60 40 20 0 10 100 1000 10000 Frequenz [Hz] Simulation_VCCC_normal.OPJ Abbildung 5-17 Messung und Simulation des Frequenzgangs der Ohm'schen Kopplung. Simulation und Messergebnis stimmen in weiten Bereichen überein. (A) Frequenzgang im VC-VC-Messmodus. (B) Frequenzgang im VC-CC-Messmodus. Abbildung 5-17 B beschreibt die Messung im VC-CC-Modus. Aufgetragen ist hier die Übertragungsfunktion h12(ω) gegenüber der Frequenz: − Auch in dieser Messkonfiguration verhält sich die Schaltung wie ein Tiefpass, allerdings mit wesentlich kleinerer Grenzfrequenz. Im VC-CC-Modus kann die Spannung an Punkt 4 der Schaltung aus Abbildung 5-16 C frei schwingen. Im Gegensatz zum VC-VC-Modus hat der Kondensator C6 darum erheblichen Einfluss auf den Frequenzgang. Das RC-Glied aus R3, R5 und C6 bestimmt mit der Zeitkonstante τ = 22 ms dominant den Frequenzgang und schneidet die Übertragungsfunktion h12(ω) bereits bei einer Grenzfrequenz unter 50 Hz ab. Für eine Charakterisierung der Kopplung ist der VC-CC-Modus deshalb ungeeignet. Das Frequenzverhalten von Kabel und Gap-Junctions wird durch das Zeitverhalten dieses RC-Gliedes überlagert. 5-48 5 Elektrophysiologische Messungen − Der gemessene Frequenzgang im VC-CC-Modus folgt exakt der Simulation. Bei den durchgeführten Messungen wurde keine signifikante Abweichung detektiert. Das Ergebnis des Tests ist, dass sich für eine Analyse des Frequenzverhaltens Ohm'scher Kopplung der VC-VC-Modus am besten eignet. Bis zu einer Grenzfrequenz von ~ 400 Hz sind dabei keine Korrekturen des Frequenzgangs zu berücksichtigen. 5.4.6.2 Zuleitungswiderstand Ist das gewählte Kopplungsmodell korrekt, so muss es möglich sein durch gezielte Parametervariation der einzelnen Bauteile im Schaltkreises aus Abbildung 5-16 C den Verlauf des Frequenzgangs real gemessener Ohm'scher Kopplungen nachzubilden. Entsprechende Simulationen in PSpice wurden an ausgewählten Einzelmessungen durchgeführt. Für Frequenzen kleiner als 100 Hz war eine exakte Modellierung des Frequenzgangs in jedem Fall möglich. Für höhere Frequenzen allerdings beobachtet man in realen Messungen einen erneuten Anstieg der Kopplung (siehe Abbildung 5-13 B). Dies kann mit keiner Parameterkonstellation des Schaltkreises aus Abbildung 5-16 C nachvollzogen werden. Als Ursache dieser Abweichung wurde ein zusätzlicher Widerstand in der Erdungsleitung des Messaufbaus identifiziert: Im realen Experiment sind die einzelnen elektrischen Komponenten der Schaltung nicht, wie in Abbildung 5-16 C angenommen, direkt an den gemeinsamen Erdungspunkt 0 angeschlossen sondern werden über die extrazelluläre Patch-Clamp Lösung im Kulturgefäß mit der Badelektrode verbunden. Diese Verbindung hat einen nicht zu vernachlässigenden 7 Widerstand zwischen 50 und 400 kΩ, je nach Füllhöhe der Badlösung im Kulturgefäß. Fügt man einen Widerstand entsprechender Größe in die Erdungsleitung des Testschaltkreises ein, so kann auch der Anstieg der Kopplung bei hohen Frequenzen mit dem Modell beschrieben werden. 7 Gemessen an der in allen Versuchen verwendeten extrazellulären Patch-Clamp Lösung, durch Eintauchen von zwei chlorierten Silberdrähten im Abstand von 1 cm in ein gefülltes Zellkulturgefäß. 5.4 Grundlagen der Kopplung 5-49 Scheinkopplung bei hohen Frequenzen durch Massewiderstand (B) VC-CC-Messmodus (A) VC-VC-Messmodus Testschaltkreis U1 Testschaltkreis I2 U1 RMasse G'12 (ω ) = Î'2 (ω ) / Û1 (ω) h'12 (ω ) = Û'2 (ω) / Û1(ω) h'12-gemessen h'12-simuliert mit RMasse = 400K h'12-simuliert mit RMasse = 0K Kopplung h'12 [%] Kopplung G'12 [nS] G'12-gemessen G'12-simuliert mit RMasse = 400K G'12-simuliert mit RMasse = 0K 8 6 4 2 0 10 100 1000 60 40 20 0 10 Frequenz [Hz] Simulation_VCVC_sweepground.OPJ U2 RMasse 100 1000 Frequenz [Hz] Simulation_VCCC_sweepground.OPJ Abbildung 5-18 Der scheinbare Wiederanstieg Ohm'scher Kopplung bei hohen Frequenzen kann über den Widerstand des Badmediums erklärt werden. (A) Modell und Frequenzgang im VC-VC-Modus (B) Modell und Frequenzgang im VC-CC-Modus In Abbildung 5-18 A und Abbildung 5-18 B ist das Ergebnis entsprechender Simulationen in PSpice für den VC-VC-Modus und den VC-CC-Modus dargestellt. An den Verlauf des Frequenzgangs einer realen Messung (einzelne Datenpunkte) konnte dabei unter Berücksichtigung eines Massewiderstands von RMasse = 400 kΩ ein simulierter Frequenzgang (durchgezogene Linie) angeglichen werden. Zum Vergleich ist in den Graphen zusätzlich der simulierte Frequenzgang ohne Massewiderstand (gestrichelte Linie) dargestellt. Mit dem in Punkt 5.4.5 beschriebenen Modell der Ohm'schen Kopplung kann man folglich unter Beachtung des Massewiderstandes der Badlösung den Frequenzgang der Kopplung im gesamten betrachteten Messbereich vollständig erklären. Der beobachtete Wiederanstieg der gemessenen Kopplungparameter bei hohen Frequenzen ist kein Effekt der Ohm'schen Kopplung. Eine Kopplung in diesem 5-50 5 Elektrophysiologische Messungen Frequenzbereich erfolgt nicht entlang des Kabels sondern über die Badlösung. Informationen über Kabel und Gap-Junctions sind daher im Frequenzbereich oberhalb von 100 Hz nicht mehr aus dem Messignal zu isolieren. Für die Auswertung der AC-Messdaten wird aus diesem Grund nur ein eingeschränkter Frequenzbereich von 0 Hz bis 100 Hz betrachtet. In diesem Bereich kann der Einfluss des Badwiderstandes RMasse auf die Kopplung noch vollständig vernachlässigt werden (siehe Abbildung 5-18). Die Auswertung der Messdaten kann in diesem Fall ohne einen zusätzlichen Massewiderstand auf Basis des normalen Kopplungsmodells aus Punkt 5.4.5. erfolgen. 5.5 Messergebnisse aus Ohm'scher Kopplung 5-51 5.5 Messergebnisse aus Ohm'scher Kopplung Aufgrund der unterschiedlichen Modelle für die Kopplung von Gleich- und Wechselspannungen (siehe Abbildung 5-15 B und C in Punkt 5.4.5), werden auch die Ergebnisse der durchgeführten Messungen getrennt diskutiert. Unter dem Oberbegriff Gleichspannungsanalyse sind alle Ergebnisse aus der Analyse von Messdaten zusammengefasst, welche die Kopplung von Gleichspannungen betreffen. Die Wechselspannungsanalyse in Punkt 5.5.2 behandelt alle Ergebnisse aus der Kopplung von Wechselspannungen. 5.5.1 Gleichspannungsanalyse Die Gleichspannungsanalyse umfasst den Datenbestand aus der DC Messung aller untersuchter Zellpaare. Anhand der Messdaten war es möglich das in Punkt 5.4.5 vorgestellte Kopplungsmodell im Experiment zu überprüfen. Darüber hinaus konnten anhand der in Punkt 5.4.4 beschriebenen Kabelgleichungen Rückschlüsse auf die Kabeleigenschaften der Kopplung erfolgen. 5.5.1.1 Validierung des Kopplungsmodells Der Interpretation aller erzielter Messergebnisse liegt das allgemeine Kopplungsmodell aus Punkt 5.4.4 sowie die daraus abgeleitete elektrische Beschreibung Ohm'scher Kopplung in Punkt 5.4.5 (speziell Abbildung 5-15 B) zugrunde. Um die Anwendbarkeit dieses Modells auf das untersuchte biologische System gekoppelter neuronaler Zellen nachzuweisen, wurden anhand experimenteller Messdaten die wesentlichen Grundannahmen des Modells überprüft. Die elektrische Beschreibung von Zelle 1 und Zelle 2 durch Ohm'sche Membranwiderstände RMem1 und RMem2 ist für eine schwache GleichspannungsDepolarisation unterhalb der Auslöseschwelle von spannungsgesteuerten Ionenkanälen sicher korrekt. Die Richtigkeit dieser Annahme ist allgemein bekannt [Katz, 1985] und konnte bei Messungen an einzelnen, ungekoppelten Zellen auch regelmäßig im Experiment beobachtet werden. 5-52 5 Elektrophysiologische Messungen Das grundlegende Prinzip Ohm'scher Kopplung basiert auf ionenpermeablen Poren, den Gap-Junction's. Diese werden in dem verwendeten Modell aus Punkt 5.4.5 mit einem Ohm'schen Widerstand beschrieben. Eine solche Beschreibung ist nur dann richtig, wenn die Poren tatsächlich vollkommen passiv sind und in beiden Richtungen einen spannungsunabhängigen Widerstand gleicher Größe aufweisen. In der Literatur werden neben passiv ungerichteten Ohm'schen Kopplungen [Hagiwara, 1958] aber auch Ohm'sche Kopplungen mit gleichrichtender Wirkung beschrieben [Furshpan, 1959 #3]. Außerdem ist bekannt, dass der spezifische Widerstand einer Gap-Junction nicht unter allen Umständen konstant ist, sondern im Einzelfall auch einer Modulation durch intra- und extrazelluläre Botenstoffe, wie z.B. 2+ + Ca , H , oder cAMP, unterliegen kann [Neyton, 1986]. Auch eine Abhängigkeit des Widerstands von der Membranspannung ist dadurch möglich. Bezogen auf das hier verwendete Zellsystem und die daran detektierten Ohm'schen Kopplungen ist die Beschreibung des Kopplungsmechanismus durch einen einfachen passiven Widerstand jedoch vollkommen korrekt. Dies zeigen die Ergebnisse von zwei Analysen: 1. Eine gleichrichtende Funktion der Kopplung konnte in einem direkten Vergleich der Kopplungsstärke in beiden Richtungen nicht nachgewiesen werden. 2. Eine mögliche spannungsabhängige Modulation des Kopplungswiderstands wurde durch den Nachweis strenger Linearität des an der Kopplung beteiligten elektrischen Netzwerks ausgeschlossen. Abbildung 5-19 A zeigt das Prinzip der im VC-VC-Modus durchgeführten Messungen zum Ausschluss der Gleichrichterfunktion. Wie in Punkt 5.4.3 beschrieben, wurde in jedem Experiment abwechselnd Zelle 1 und Zelle 2 als präbzw. postsynaptische Zelle aufgefasst und so nacheinander die Kopplung in beide Richtungen gemessen. In der ersten Messung erfolgte dabei eine Stimulation von Zelle 1 mit einer Spannung U1 = +20 mV, in der zweiten Messung wurde Zelle 2 mit U2 = +20 mV stimuliert. Die jeweils andere Zelle wurde konstant auf einer Spannung von 0 mV gehalten, um den Kopplungsstrom I2 bzw. I1 direkt messen zu können. Aus dem Quotienten des Kopplungsstroms an der Postsynapse und der 5.5 Messergebnisse aus Ohm'scher Kopplung 5-53 Bidirektionalität der Ohm'schen Kopplung Messung 1: (A) Messprinzip Messung 2: U2 U1 – Jeweils zwei Messungen je Zellpaar I2 I1 • Die Kopplung war bei – U1 = U2 = -20 mV G12 = I2 / U1 Kopplung vorwärts G12 [nS] (B) Ergebnis Messpunkte 8 7 allen Messungen in beiden Richtungen gleich groß. G21 = I1 / U2 – Ungerichtete Kopplung. Fitfunktion – Keine sinnvolle Unterteilung in Präund Postsynapse möglich. G12 = (0,96 ± 0,03) * G21 6 5 4 3 2 1 0 0 1 2 3 4 5 6 7 8 Kopplung rückwärts G21 [nS] DC_VCVC_U-20_Bidirektionalität.OPJ Abbildung 5-19 Die gemessenen Ohm'schen Kopplungen sind bidirektional und haben in beiden Kopplungsrichtungen die gleiche Kopplungsstärke. (A) Prinzip der Messung: Im VC-VC-Modus wird die Leitfähigkeit der Kopplung bei jedem untersuchten Zellpaar in beiden Richtungen ermittelt. (B) Messergebnis: Die Leitfähigkeiten in beiden Richtungen stimmen überein. Stimulationsspannnung an der Präsynapse erhält man so die Leitfähigkeit der Kopplung G12 bzw. G21 als Kopplungsparameter. In Abbildung 5-19 B sind die gemessenen Leitfähigkeiten G12 und G21 als x- und yWert jedes einzelnen Experiments in einem Punktdiagramm gegeneinander aufgetragen. Wie man deutlich sieht, besteht eine enge Korellation zwischen x- und y- Wert jedes Datenpunktes. Legt man eine Ausgleichsgerade durch die Messpunkte, so verläuft diese durch den Ursprung mit einer Steigung von g = 0,96 ± 0,03 ≈ 1 . Die beobachteten Kopplungen haben folglich eine identische Leitfähigkeit in beiden Kopplungsrichtungen. Sie zeigen keine Gleichrichterfunktion. Eine Unterteilung in Prä- und Postsynapse ist damit nicht möglich; beide Zellen sind gleichermaßen prä- und postsynaptisch und die Beschreibung der Kopplung durch einen passiven Widerstand ist korrekt. 5-54 5 Elektrophysiologische Messungen Linearität der Ohm'schen Kopplung – Mehrfache Messung von I2 bei verschiedenen Spannungen U2 Anordnung: I2 G12 (U1) = I2 (U1 ) / U1 Kopplungsstrom I12 [pA] (B) Ergebnis DC_VCVC_Linearität.OPJ Ablauf: U1 Spannung U1 [mV] (A) Messprinzip 4 -40 -50 3 -60 -70 0,0 2 • Linearer 1 0,5 1,0 1,5 2,0 Zeit [s] Messung 1 Messung 2 150 5 -30 Messung 3 100 50 Zusammenhang zwischen angelegter Spannung und Kopplungsstrom. – Verhalten mit einem Ohm'schen Widerstand zu beschreiben. 0 -50 -100 -150 -20 -10 0 10 20 Spannungsgradient U12 [mV] Abbildung 5-20 Das elektrische Verhalten Ohm'scher Kopplung ist streng linear. (A) Prinzip der Messung: Im VC-VC-Modus werden an Zelle 1 verschiedene Gleichspannungen angelegt und der Stromfluss nach Zelle 2 wird gemessen. (B) Messergebnis: Der Stromfluss nach Zelle 2 wächst linear mit der Spannung in Zelle 1 an. Mit dieser Analyse ist allerdings noch nicht gezeigt, dass der Kopplungswiderstand spannungsunabhängig ist. Dies kann man wie folgt überprüfen: Nimmt man einen rein Ohm'schen, spannungsunabhängigen Widerstand zur Beschreibung von Kabel und Kopplung an (siehe Punkt 5.5), so entsteht ein Netzwerk aus rein Ohm'schen Widerständen. Für ein solches Netzwerk aus Ohm'schen Widerständen folgt aus der Theorie linearer Systeme zwingend ein linearer Zusammenhang zwischen einer, an einem beliebigen Punkt des Netzwerks angelegten Spannnung und dem Stromfluss durch jedes einzelne Bauteil des Netzwerks [Tietze, 1993]. Insbesondere muss daher auch der Stromfluss von Zelle 1 zu Zelle 2 über den Kopplungswiderstand RPore linear mit der Potentialdifferenz zwischen beiden Zellen anwachsen. Diese Linearität des Kopplungsstroms wurde durch Messungen im VC-VC-Modus gezielt überprüft. 5.5 Messergebnisse aus Ohm'scher Kopplung 5-55 Abbildung 5-20 A zeigt die gewählte Messanordnung und den Ablauf der Messung: An Zelle 1 wurden im VC-Modus verschiedene Spannungen U1 (-20, -10, 0, +10 und +20 mV) angelegt. Gleichzeitig wurde der zu Zelle 2 fließende Strom I2 gemessen. Während der gesamten Messung war Zelle 2 konstant auf eine Spannung von U2 = 0 mV geklemmt. In Abbildung 5-20 B ist exemplarisch das Ergebnis von drei Messungen dargestellt. Die ausgewählten Messungen sind repräsentativ für alle Versuche. Man erkennt einen streng linearen Zusammenhang zwischen dem angelegten Spannungsgradienten U12 (U12 = U1 - U2 = U1) und dem gemessenen Kopplungsstrom I12 (Wegen U2 = 0 mV während der Messung fließt kein Strom über die Zellmenbran von Zelle 2 ab und der in Zelle 2 gemessene Strom I2 entspricht dem Kopplungsstrom I12). Die gefundene strenge Linearität zeigt, dass die Wahl eines Netzwerks aus Widerständen für die Beschreibung der Ohm'schen Kopplung von Gleichspannungen korrekt ist. Einzelne Ohm'sche Kopplungen unterscheiden sich je nach Stärke der Kopplung nur in der Steigung der Geraden des Spannungs-StromGraphen in Abbildung 5-20 B. Die Gap-Junctions der nachgewiesenen Ohm'schen Kopplungen verhalten sich also tatsächlich wie ein einfacher Ohm'scher Widerstand. Das unter Punkt 5.4.5 eingeführte Modell in Abbildung 5-15 B ist folglich korrekt. Für praktische Berechnungen ist es nun noch erforderlich zu überprüfen, unter welchen Bedingungen es möglich ist, wie in Punkt 5.4.5 beschrieben, die Signalübertragung von Zelle 1 bis zum Kabelende getrennt von der Kopplung zu Zelle 2 zu betrachten. Nur wenn der Kopplungswiderstand RPore hinreichend groß ist gegenüber der Kabelimpedanz, kann man die beiden Systeme rechnerisch separieren. Abbildung 5-21 zeigt den Versuchsaufbau, das verwendete Rechenmodell und die Ergebnisse der zur Überprüfung dieser Annahme durchgeführten Experimente. Das Prinzip der Analyse ist ein Vergleich der experimentell gemessenen 5-56 5 Elektrophysiologische Messungen Übertragungsfunktion h12 des Netzwerks mit einem auf Basis des Kopplungsmodells theoretisch berechneten Wert h'12 . Die Übertragungsfunktion h12 beschreibt, welcher Bruchteil einer an Zelle 1 angelegten Spannung U1 als Messwert U2 zu Zelle 2 gekoppelt wird. Bei Stimulation von Zelle 1 im VC-Modus und Messung von Zelle 2 im CC-Modus sind die Spannungen U1 und U2 messtechnisch unmittelbar zugänglich. Abbildung 5-21 A zeigt die Messkonfiguration. Andererseits gestattet eine Messung mit beiden Zellen im VC-Modus die direkte Bestimmung der Leitfähigkeit G12 der Kopplung (siehe oben). In beiden Messkonfigurationen wird die Spannung U1 in Zelle 1 durch den Patch-Clamp Verstärker vorgegeben. Der Membranwiderstand RMem1 hat für die Beschreibung daher keine Bedeutung. Geht man weiterhin davon aus, dass die oben angeführte rechnerische Separation des Systems korrekt ist, so kann man den Überprüfung des Modells - Überführen von G in h (A) Direkte Messung von h12 U1 h12 = U2 / U1 (B) Berechnung von h'12 aus G12 K 0 Zelle 1 Kopplung h' U12 = R Mem2 R'Pore + R Mem2 2 RMem2 Kabel RMem U1 RKabel R'Pore RMem1 1 U2 Zelle 2 h12' = h12-Berechnet [%] (C) Ergebnis des Vergleichs U2 Messpunkte 80 Fitfunktion h12' = (0,96 ± 0,07) * h12 60 40 20 0 0 20 40 60 80 h12-Gemessen [%] ; R'Pore = 1 / G12 Abbildung 5-21 Ist das Kopplungsmodell korrekt, so müssen G und h ineinander umrechnenbar sein. (A) Prinzip der Messung des Kopplungskoeffizienten h12 im VC-CC-Modus. (B) Berechnung des Kopplungskoeffizienten h'12 auf Basis der Kopplungs-Leitfähigkeit G'12. (C) Vergleich der Messgröße h12 mit dem berechneten Wert h'12 . 5.5 Messergebnisse aus Ohm'scher Kopplung 5-57 Einfluss des Spannungsteilers aus RKabel und RMemKabel auf die Kopplung alternativ auch durch eine Skalierung des Kopplungswiderstands RPore um einen Faktor hKabel zu R'Pore berücksichtigen. Es entsteht das stark vereinfachte Schaltbild der Kopplung in Abbildung 5-21 B. Aus diesem kann man leicht den theoretischen Wert der Übertragungsfunktion h'12 ableiten. Es gilt: h '12 = h'12 RMem2 RPore R'Pore hKabel G12 = = = = = = RMem 2 R ' Pore + R Mem 2 mit R ' Pore = R Pore 1 = hKabel G12 Gleichung 5-27 Berechnete Übertragungsfunktion für die Kopplung von Zelle 1 nach Zelle 2 [1] Experimentell bestimmter Widerstand der Zellmembran von Zelle 2 [MΩ] Widerstand der Ohm'schen Kopplung [MΩ] Widerstand der Ohm'schen Kopplung, skaliert um Kabeleffekte [MΩ] Übertragungsfunktion des Kabels [1] Experimentell bestimmte Leitfähigkeit der Kopplung [µS] Den Widerstand R'Pore der Ohm'schen Kopplung in dieser Gleichung, erhält man unmittelbar aus der gemessenen Leitfähigkeit der Kopplung G12 . Den Widerstand RMem2 der Zellmembran von Zelle 2 aus der Kompensation des Patch-Clamp Verstärkers zu Beginn der Messung (siehe hierzu auch Kapitel 3). Gleichung 5-27 ist aber nur korrekt, wenn die Kopplung tatsächlich "schwach" ist. Schwach bedeutet dabei in Bezug auf das verwendete Modell, dass gilt: R Pore >> R Kabel und R Pore >> R Mem 2 Gleichung 5-28 Nur in diesem Fall kann man die Kopplung tatsächlich in zwei getrennte Kopplungsschritte separieren. Im ersten Schritt wird dabei die Spannung U1 durch den Spannungsteiler des Kabels aus RKabel und RMemKabel mit einem Faktor hKabel auf die Spannung UK vermindert: U K = h Kabel U 1 mit h Kabel = R MemKabel R MemKabel + R Kabel Gleichung 5-29 Im zweiten Schritt bewirkt diese Spannung UK im VC-VC-Modus einen Kopplungsstrom I2 in Zelle 2, wobei für die gemessene Leitfähigkeit G12 der Kopplung gilt: 5-58 G12 = 5 Elektrophysiologische Messungen h I2 = Kabel U 1 R Pore Gleichung 5-30 Im VC-CC-Modus resultiert ein Spannungsabfall U2 über dem Membranwiderstand der zweiten Zelle. Die gemessene Übertragungsfunktion h12 ist hierbei: h12 = R Mem 2 U2 U = hKabel 2 = hKabel U1 UK R Mem 2 + R Pore Gleichung 5-31 Unter Beachtung der Bedingungen aus Gleichung 5-28 kann man durch Einsetzen des Messwertes von RPore aus Gleichung 5-30 in Gleichung 5-31 die zuvor aufgestellte Gleichung 5-27 ableiten. Der Skalierungsfaktor hKabel der Kabeleigenschaften fällt dabei aus den Gleichungen heraus. Ist die gewählte Beschreibung korrekt, so muss der gemessene Wert h12 mit dem anhand von G12 berechneten Wert h'12 übereinstimmen. Das in Abbildung 5-21 C dargestellten Ergebnis der durchgeführten Analyse zeigt für Messungen bis zu einem Betrag von h12 < 25 % eine Übereinstimmung von h12 und h'12. Legt man durch die zugehörigen Messpunkte eine Ausgleichsgerade, so verläuft diese durch den Ursprung und hat eine Steigung von 0,96 ± 0,07 . Für Kopplungen mit h12 >> 25 % ist das Modell aber offenbar nicht korrekt. Die Messwerte h12 dieser Kopplungen weichen deutlich von den aus G12 berechneten Werten h'12 ab. Bei "starken" Kopplungen wird h12 in der Berechnung unterbewertet. Aus den Messdaten geht weiterhin hervor, dass die Bedingung h12 < 25 % für alle detektierten Kopplungen bis zu einer Leitfähigkeit von G12 ≤ 2,5 nS erfüllt ist. In die nun folgende Ergebnisauswertung der Messdaten wurden daher nur Kopplungen bis zu diesem Grenzwert einbezogen. 5.5.1.2 Auswertung der Kopplungsdaten Die erzielte Steuerung neuronalen Wachstums durch Mikrostrukturen aus Laminin hat entscheidende Bedeutung für die Auswertung der Kopplungsdaten: − Die Zahl möglicher Verbindungen einer Zelle ist auf die Pfade der vorgegebenen Lamininstruktur begrenzt. Eine Zelle auf einer Linienstruktur hat deshalb maximal Verbindungen zu zwei direkten Nachbarn, eine Zelle auf einem Gitter 5.5 Messergebnisse aus Ohm'scher Kopplung 5-59 zu maximal vier. Aufgrund dieser starken geometrischen Einschränkung möglicher Verbindungswege, konnte jeder detektierten Kopplung exakt ein Kopplungspfad zugeordnet und dessen Länge bestimmt werden. Eine Analyse des Zusammenhangs zwischen Kabellänge und Kopplungsstärke war dadurch möglich. − Anhand eines Vergleichs der Kopplungen auf Lamininstrukturen mit unterschiedlichen Abmessungen kann der geometrische Einfluss der Lamininstrukturen auf die Kopplung betrachtet werden. Mit den verwendeten Strukturen war ein Vergleich von Kopplungsstärke und Pfadbreite möglich. Die erforderliche Bestimmung der Kabellänge zwischen den untersuchten Zellen erfolgte anhand von Phasenkontrast- und Fluoreszenzaufnahmen. Unmittelbar vor Beginn jeder Patch-Clamp Messung wurde mit Hilfe des Phasenkontrast-Mikroskops die jeweilige Zellkonfiguration photographiert. Konnte in den anschließend durchgeführten elektrophysiologischen Messungen eine Kopplung detektiert werden, so wurde nach Abschluss aller elektrophysiologischen Experimente die untersuchte Zellkonfiguration zusätzlich mittels Fluoreszenzmikroskopie aufgenommen und exakt vermessen. Während der Durchführung der elektrophysiologischen Messungen, war die Fluoreszenzlampe des Mikroskops grundsätzlich abgeschaltet und die Phasenkontrastbeleuchtung auf ein Minimum reduziert. Diese Abdunkelung ist notwendig, um eine vorzeitige Schädigung der Zellen durch photoinduzierte, freie Radikale zu vermeiden [Banker, 1998]. In Abbildung 5-22 ist an zwei Beispielen der optische Messablauf dargestellt: 1. Zwei Phasenkontrastbilder der Zellanordnung wurden vor Beginn der elektrophysiologischen Messungen aufgenommen. Eines von der Anordnung der untersuchten Zellen (Abbildung 5-22 A1) ohne Patch-Pipetten, und ein zweites nach Annäherung der Patch-Pipetten an die Zellen und erfolgreicher Herstellung von Gigaseals (Abbildung 5-22 B1). 5-60 5 Elektrophysiologische Messungen Abbildung 5-22 Die Kabellänge zwischen zwei gekoppelten Zellen wird anhand von Phasenkontrast- und Fluoreszenzaufnahmen ausgemessen. (A) Beispiel 1: Zwei verbundene Zellen auf einer Linienstruktur (ID 11) aus Laminin. (B) Beispiel 2: Mehrere Zellen auf einer Gitterstruktur (ID 9) aus Laminin. Das Kabel zwischen den beiden mit Mikropipetten kontaktierten Zellen ist deutlich zu erkennen. 2. Nach Abschluss der elektrophysiologischen Messungen wurde die Fluoreszenzlampe zugeschaltet und durch Wahl geeigneter Anregungs- und Emissionsfilter die Fluoreszenz des Zell-Cytoplasmas aufgenommen. Die hierfür erforderliche Fluoreszenzmarkierung der Zellen war zuvor wie in Kapitel 2 beschrieben, durch Mikroinjektion polarer Fluoreszenzfarbstoffe über die Patch-Pipetten hergestellt worden. Nach Überlagerung der Fluoreszenzbilder mit den zuvor angefertigten Phasenkontrastbildern entsteht ein klares, plastisches Bild der Zellanordnung (Abbildung 5-22 A2 und Abbildung 5-22 B2). Insbesondere der exakte Weg der Verbindung zwischen beiden Zellen kann anhand dieser Aufnahmen eindeutig bestimmt werden. 3. Auf Grundlage dieser Bilder erfolgte eine quantitative Vermessung der Zellanordnung. Hierbei war die relative Lage der gekoppelten Zellen zueinander 5.5 Messergebnisse aus Ohm'scher Kopplung 5-61 sowie die Länge des Kabels zwischen beiden Zellen Gegenstand der Untersuchung. In Abbildung 5-22 A3 und Abbildung 5-22 B3 ist das Vorgehen zur Bestimmung der Kabellänge für die beiden Messbeispiele dargestellt. Die Länge l des Kabels wird dabei ermittelt, als die Länge des Kabels zwischen den Ansatzpunkten Punkt 1 und Punkt 2 des Kabels an die Zellkörper. Eine interessante zusätzliche Beobachtung ist auf den Fluoreszenzaufnahmen in Abbildung 5-22 B zu sehen. Nicht nur das Kabel zwischen den beiden, mit PatchPipetten kontaktieren Zellen ist zu erkennen, sondern auch eine weitere, dritte Zelle erscheint mit gelber Fluoreszenz. Offenbar besteht also auch von dieser Zelle eine Verbindung zu der kontaktierten, gelb gefärbten Zelle. Über diese Verbindung ist der verwendete Fluoreszenzmarker in einer für den optischen Nachweis ausreichenden Menge in die Nachbarzelle diffundiert. Eine solche Ausbreitung von Fluoreszenzfarbstoff in Nachbarzellen wurde in den durchgeführten Experimenten häufig beobachtet, wenn zwischen Kontaktierung der Zellen und Fluoreszenzmessung eine Zeit von mehr als 10 - 15 Minuten lag.. Dies stützt die Annahme von Gap-Junctions als Ursache der Ohm'schen Kopplung. Gap-Junctions sind mit einem Innendurchmesser von 1,5 nm durchlässig für die verwendeten Fluoreszenzfarbstoffe, die Diffusion ist jedoch gegenüber der Diffusion im freien Cytoplasma deutlich gebremst [Eckert, 1999]. Die Untersuchung des Zusammenhangs zwischen Zellabstand und Kopplungsstärke führt auf das Ergebnis in Abbildung 5-23. Abbildung 5-23 A zeigt das Prinzip der Analyse: Von allen detektierten Kopplungen wurde im VC-VC-Modus die Leitfähigkeit der Kopplung G12 ermittelt. Zusätzlich wurde, wie oben beschrieben, jeweils die Länge l des Kabels zwischen beiden Zellen bestimmt. Anschließend wurde jede gemessene Leitfähigkeit G12 entsprechend der zugehörigen Kabellänge l in ein bestimmtes Abstandsintervall eingeordnet (10 µm breite Intervalle im Bereich von 10 bis 120 µm). Für jedes Abstandsintervall wurde dann auf Basis der darin enthaltenen Messdaten G12 der Mittelwert und die Streuung berechnet. In Abbildung 5-23 B ist das Ergebnis der Analyse dargestellt. Für jedes 5-62 5 Elektrophysiologische Messungen Abnahme der Kopplung mit dem Zellabstand (A) Messprinzip G12 (U1) = I2 (U1 ) / U1 U1 – Messung vieler Zellpaare, verbunden mit verschiedenen Kabellängen l – U1 = -20 mV I2 I2 l1 • Zusammenhang l2 zwischen Kopplungsstärke und Länge des Kabels. (B) Ergebnis Mittelwerte Kopplung G12 [nS] 3 Fitfunktion – Die Kopplungsstärke nimmt mit der Länge des Kabels ab. 2 3 4 1 14 λ = 67 ± 29 µm 12 8 5 3 5 100 120 4 0 0 20 40 60 80 Kabellänge l [µm] DC_VCVC_U-20_Kopplung-Abstand.OPJ Abbildung 5-23 Die Kopplungsstärke nimmt mit dem Abstand der Zellen ab. (A) Prinzip der Messungen. (B) Messergebnis. Abstandsintervall wurde die mittlere Leitfähigkeit als Datenpunkt mit Fehlerbalken und Angabe der Anzahl zugrundeliegender Messungen dargestellt. Man erkennt deutlich einen Abfall der mittleren Leitfähigkeit G12 mit zunehmender Länge des Kabels. Allerdings wird dieser Effekt im Bereich kurzer Abstände bis ca. 50 µm von einer sehr großen Streuung der Einzelmessungen verwischt. Für Kabellängen > 50 µm ist der Abfall jedoch sehr deutlich und verläuft wie eine abklingende Exponentialfunktion. Mit dem Kopplungsmodell aus Punkt 5.4.5 kann man das beobachtete Verhalten klar interpretieren: Die Leitfähigkeit G12 der Kopplung setzt sich nach Gleichung 5-25 aus zwei Komponenten zusammen, der Übertragungsfunktion des Kabels hKabel und dem Widerstand der Pore RPore . Führt man mit GPore die Leitfähigekit der Pore ein, so kann man G12 als Produkt zweier Komponenten schreiben: 5.5 Messergebnisse aus Ohm'scher Kopplung G12 = h Kabel G Pore G12 hKabel GPore RPore = = = = G Pore = mit 1 5-63 Gleichung 5-32 R Pore Leitfähigkeit der Kopplung [µS] Übertragungsfunktion des Kabels [1] Leitfähigkeit der Ohm'schen Kopplung [µS] Widerstand der Ohm'schen Kopplung [MΩ] Die Leitfähigkeit GPore wird dabei nur von der Anzahl Gap-Junctions an der jeweiligen Ohm'schen Synapse bestimmt und konnte in den durchgeführten Experimenten von aussen nicht kontrolliert werden. Die Übertragungsfunktion hKabel des Kabels hingegen, unterliegt gemäß Punkt 5.4.4 den von außen zugänglichen geometrischen Rahmenbedingungen eines zylindrischen Kabels. Aus Gleichung 5-15 und Gleichung 5-16 folgt für die Übertragungsfunktion hKabel : h Kabel = e l λ r ρm ρi = = = = = − l λ mit λ= rρ m 2ρi Gleichung 5-33 Länge des Kabels [cm] Charakteristische Abklinglänge des Kabels [cm] Innenradius des Kabels [cm] Spezifischer Widerstand der Zellmenbran [Ohm*cm²] (ρm ~ 2000 Ω*cm²) Spezifischer Widerstand des Cytoplasmas [Ohm*cm] (ρi ~ 200 Ω*cm) Auf Basis dieser theoretischen Überlegungen, wird das Messergebnis in Abbildung 5-23 B verständlich: − Für kleine Kabellängen < 50 µm dominiert die zufällige Streuung der Porenleitfähigkeit GPore über die Gesamtleitfähigkeit G12 der Kopplung. Die Messwerte streuen in diesem Bereich daher sehr stark. − Bei größeren Kabellängen wird der Signalverlust im Kabel zunehmend dominant. Die Gesamtleitfähigkeit der Kopplung G12 wird in diesem Fall von der Übertragungsfunktion hKabel bestimmt. In Übereinstimmung mit Gleichung 5-33 beobachtet man einen Abfall von G12 mit der Kabellänge. Gleichzeitig, skaliert um denselben Faktor hKabel , verringert sich mit wachsender Kabellänge auch die von GPore ausgehende Streuung der Messwerte. Der beobachtete Abfall der 5-64 5 Elektrophysiologische Messungen Kopplung geht also offenbar unmittelbar auf die Verlängerung des Kabels zurück. Legt man anhand von Gleichung 5-33 eine exponentiell abfallende Fitfunktion durch die Messwerte, so erhält man eine Abklinglänge λ = 67 ± 29 µm. Daraus resultiert ein Kabelinnenradius von r = 0,1 ± 0,07 µm. Dieses Ergebnis liegt in der erwarteten Größenordnung. In einer zweiten Analyse wurde die Stärke der Kopplung auf Strukturen mit unterschiedlich breiten Pfaden aus Laminin verglichen. Dadurch sollte geklärt werden, ob eine Beeinflussung der Kabeleigenschaften durch die geometrische Struktur der Pfade möglich ist. Es wurden Kopplungen entlang von Pfaden mit 2 µm, 4 µm und 6 µm Linienbreite verglichen. Abbildung 5-24 A zeigt das Prinzip der Analyse. Nur Kopplungen mit einer Kabellänge zwischen 30 und 80 µm wurden einbezogen. Dieser Bereich wurde nach drei Kriterien ausgewählt: 1. Der Einfluss der Kabeleigenschaften auf die Kopplung sollte hinreichend stark sein gegenüber dem Einfluss der Poren. In der vorherigen Analyse wurde ab einer Kabellänge von 40-50 µm eine klare Dominanz der Kabeleigenschaften nachgewiesen. 2. Die Streuung aufgrund unterschiedlicher Kabellängen sollte weitgehend unterdrückt werden. Nur Messwerte aus einem begrenzten Kabellängenintervall sollten daher verwendet werden. 3. Insgesamt sollte eine ausreichende Anzahl Messwerte für eine statistische Auswertung berücksichtigt werden. Der Kabellängenbereich zwischen 30 und 80 µm ist ein Kompromiss aus diesen Überlegungen. Die Bereichsgrenze 30 µm wurde ausgewählt, da Kopplungen auf 2 µm breiten Pfaden nur bis zu einem Abstand von 30 µm gefunden wurden. Schränkt man den Bereich auf 50 - 80 µm ein, so bleibt das Ergebnis für 4 µm und 5.5 Messergebnisse aus Ohm'scher Kopplung 5-65 Zunahme der Kopplung mit der Linienbreite des Verbindungspfades (A) Messprinzip G12 (U1) = I2 (U1 ) / U1 U1 – Messung vieler Zellpaare, verbunden über Pfade verschieder Breite b – U1 = -20 mV I2 2 1 • Zusammenhang l = 30 - 80 µm (B) Ergebnis Mittelwerte für 30-80 µm Abstand Kopplung G12 [nS] 3 zwischen Kopplungsstärke und Dicke des Verbindungspfads. – Die Kopplungsstärke nimmt mit der Dicke des Pfades zu. 2 10 1 3 0 DC_VCVC_U-20_Kopplung-Linienweite.OPJ 2 2 4 6 Linienbreite b [µm] Abbildung 5-24 Kopplungen entlang dicker Lamininpfade sind stärker als Kopplungen entlang dünner Pfade. (A) Prinzip der Messungen. (B) Messergebnis. 6 µm breite Pfade das gleiche, die Kopplungen auf 2 µm breiten Pfaden fallen jedoch vollständig heraus. Abbildung 5-24 B zeigt das Ergebnis der Analyse. Man erkennt einen deutlichen Anstieg der Leitfähigkeit G12 mit der Linienbreite b. Offenbar wird die Geometrie des Kabels von der Breite des Lamininpfades beeinflusst. Für eine quantitative Analyse des zugrundeliegenden funktionalen Zusammenhangs ist mit nur drei Stützpunkten (2, 4 und 6 µm) leider eine zu geringe Datenbasis vorhanden. Geht man aber hypothetisch davon aus, dass die Breite der Linien mit der Dicke des Kabels korreliert ist, so kann man das beobachtete Kopplungsverhalten über die Radiusabhängigkeit der Abklinglänge λ erklären. Für eine feste Kabellänge l resultiert aus Gleichung 5-33 die Übertragungsfunktion: 5-66 hKabel = e 5 Elektrophysiologische Messungen − 2 ρi l ρm r Gleichung 5-34 Der Betrag der Übertragungsfunktion hKabel wächst also mit steigendem Kabelradius r an. Dies deckt sich mit dem Ergebnis der Experimente. 5.5.2 Wechselspannungsanalyse Die Wechselspannungsanalyse Ohm'scher Kopplung umfasst die Auswertung aller in AC Messungen (siehe hierzu auch Punkt 5.4.3) gewonnenen Messdaten. Die Beschreibung der Kopplung von Zelle 1 zu Zelle 2 erfolgt wie bei der Gleichspannungsanalyse im vereinfachten Ohm'schen Kopplungsmodell aus Punkt 5.4.5 . Aufgrund der Frequenz der angelegten Wechselspannungssignale können jetzt allerdings die Kapazitäten CMem1 und CMem2 der Zellmembranen sowie der Kabelmembran CMemKabel nicht vernachlässigt werden sondern das vollständige Ersatzschaltbild aus Abbildung 5-15 C muss betrachtet werden. 5.5.2.1 Auswertung der Kopplungsdaten Um ein geeignetes Verfahren für die Auswertung der gewonnenen Messdaten zu entwickeln, wurde in Simulationen mit PSpice analysiert, welchen Einfluss die einzelnen Parameter des Kopplungsmodells auf den Frequenzgang der Kopplung haben. Zugrundeliegendes Schaltbild für alle folgenden Analysen ist das bekannte Kopplungsmodell aus Abbildung 5-15 C, erweitert um die elektrischen Eigenschaften der Patch-Pipetten (analog zu Abbildung 5-16). Folgende Vorüberlegungen zur Eingrenzung der Anzahl an erforderlichen Simulationen können angestellt werden: 1. Es werden ausschließlich Messungen im VC-VC-Modus ausgewertet. Die Bauteile CMem2 und RMem2 haben daher keinen Einfluss auf den Frequenzgang. Dies gilt, da die Membranspannung der zweiten Zelle vom Patch-Clamp Verstärker konstant auf einer festen Spannung gehalten wird. 2. Der Membranwiderstand RMem1 der ersten Zelle kann vernachlässigt werden, da er groß ist gegenüber dem Zugangswiderstand der Patch-Pipette. Er beeinflusst das 5.5 Messergebnisse aus Ohm'scher Kopplung 5-67 Zeitverhalten des RC-Gliedes aus Pipettenzugangswiderstand und Membrankapazität CMem1 der ersten Zelle nicht. 3. Aufgrund der Symmetrie der Schaltung haben die Widerstände RKabel und RPore beide den gleichen Einfluss auf die Leitfähigkeit G12 der Kopplung8. Es genügt daher die Variation eines der beiden Widerstände RKabel und RPore zu simulieren. Die freien Parameter für die Simulation können folglich auf die Bauelemente CMem1 , RPore , RMemKabel und CMemKabel eingeschränkt werden. Abbildung 5-25 A bis D zeigt das Simulationsergebnis der Variation jedes dieser vier Parameter. Für alle Simulationen wurde jeweils der gleiche Satz an Grundparametern gewählt. Dies sind die Parameter des in Punkt 5.4.6 beschriebenen Testschaltkreises. Ausgehend von dieser Grundkonfiguration wurde dann der jeweils betrachtete Parameter in sinnvollen Grenzen variiert. Jede Simulation in Abbildung 5-25 zeigt drei Szenarien. Szenario 1 beschreibt das Frequenzverhalten der Kopplung G12 bei einem minimalen Wert des variierten Parameters, Szenario 2 bei unverändertem Standardwert, und Szenario 3 bei einem Maximalwert. Simuliert wurde über den gesamten Frequenzbereich der durchgeführten Messungen. Interessant für die Auswertung der experimentellen Daten ist aufgrund von Messartefakten bei hohen Frequenzen allerdings nur der Frequenzbereich zwischen 0 und 100 Hz (vergleiche hierzu auch Punkt 5.4.6). Die Variation der Membrankapazität CMem1 der ersten Zelle (Abbildung 5-25 A) und des Kabelquerwiderstandes RMemKabel (Abbildung 5-25 C) bewirken im Frequenzbereich von 0 bis 100 Hz keine signifikante Änderung des Frequenzgangs. Der Einfluss dieser Bauteile kann bei der Messdatenanalyse also vernachlässigt werden. Eine Änderung des Kopplungswiderstandes RPore oder der Kabelkapazität CMemKabel hingegen, verändert den Frequenzgang in diesem Bereich stark. 8 Bei der vorliegenden Schaltung handelt es sich um ein sog. T-Glied, eine klassische Anordnung von Widerständen und Kondensatoren [Tietze, 1993]. 5-68 5 Elektrophysiologische Messungen Einfluss der freien Parameter auf die Übertragungsfunktion G12 (B) Widerstand der Pore 1,5 G12 bei CMem1 = 10pF G12 bei CMem1 = 33pF G12 bei CMem1 = 50pF Kopplung G12 [nS] Kopplung G12 [nS] (A) Membrankapazität der 1. Zelle 1,0 0,5 0,0 10 100 1000 10000 3,0 2,5 2,0 1,5 1,0 0,5 0,0 10 Frequenz [Hz] Kopplung G12 [nS] Kopplung G12 [nS] G12 bei RMemKabel = 10G G12 bei RMemKabel = 33G G12 bei RMemKabel = 50G 0,5 100 1000 10000 (D) Kabelkapazität 1,0 0,0 10 100 Frequenz [Hz] (C) Kabelquerwiderstand 1,5 G12 bei RPore = 200M G12 bei RPore = 330M G12 bei RPore = 600M 1000 Frequenz [Hz] 10000 1,5 G12 bei CMemKabel = 1pF G12 bei CMemKabel = 4.7pF G12 bei CMemKabel = 10pF 1,0 0,5 0,0 10 100 1000 10000 Frequenz [Hz] Abbildung 5-25 Nur der Kopplungswiderstand und die Kapazität des Kabels haben einen signifikanten Einfluss auf den Fre quenzgang der Leitfähigkeit G12 der Ohm'schen Kopplung. (A) Frequenzgang von G12 je nach Membrankapazität CMem1.der 1. Zelle. (B) Frequenzgang von G12 je nach Kopplungswiderstand RPore . (C) Frequenzgang von G12 je nach Kabelquerwiderstand RMemKabel . (D) Frequenzgang von G12 je nach Kabelkapazität CMemKabel . Anhand der Messdaten sollte folglich eine direkte Bestimmung des Zeitverhaltens des RC-Gliedes aus RPore bzw. RKabel und CMemKabel möglich sein. Eine Überprüfung des Frequenzverhaltens dieser Komponenten des Kabelmodells ist deshalb möglich. Aus den Gleichungen der Beschreibung von Gleichspannungs- und Wechselspannungskopplung (siehe Punkt 5.4.5.1 und 5.4.5.2) erhält man das Frequenzverhalten der Kopplungsleitfähigkeit G12 (Einsetzen von Gleichung 5-23 und Gleichung 5-26 in Gleichung 5-25). Bei Annahme eines unendlich großen Kabelquerwiderstandes9 RMemKabel resultiert folgende einfache Form für den Betrag von G12 : 9 Dies ist aufgrund des vernachlässigbaren Einfusses dieses Parameters auf den Frequenzgang möglich. 5.5 Messergebnisse aus Ohm'scher Kopplung G12 = RPore ω τ RKabel CMemKabel 1 1 R Pore 1+ ω 2 τ 2 = = = = = 5-69 τ = R Kabel C MemKabel mit Gleichung 5-35 Widerstand der Pore [MOhm] Kreisfrequenz des übertragenen Wechselspannungssignals [Hz] Zeitkonstante des Kabels [s] Längswiderstand des Kabels [MOhm] Kapazität über die Kabelwand [pF] Eine Bestimmung des Kabelparameters τ aus den Messdaten ist mit Gleichung 5-35 unmittelbar möglich. Den Widerstand RPore erhält man aus dem Messwert bei ω0 = 0 Hz. Zusammen mit einer zweiten Messung von G12 bei ω1 > 0 Hz, folgt daraus: τ= 1 ω1 (G 12 1 (ω1 ) R Pore ) 2 −1 mit R Pore = 1 G12 (ω 0 ) Gleichung 5-36 Insgesamt gilt: G12 (ω 0 ) G (ω ) − 1 12 1 2 1 τ= ω1 Gleichung 5-37 Mit dieser Rechenvorschrift wurde im Frequenzbereich von 0 Hz bis 100 Hz aus den Messergebnissen der Kabelparameter τ ermittelt. Das Modell für ein zylindrisches Kabel aus Punkt 5.4.4, beschreibt die Abhängigkeit des Kabelwiderstandes RKabel und der Kapazität der Kabelwand CMemKabel von den Abmessungen des Kabels. Danach gilt für die Kabelkonstante τ : τ= τ ρi ζm l r 2 l 2 0,2 ms 2 l ρ ζ = i m cm r r 10 6 = = = = = Gleichung 5-38 Zeitkonstante des Kabels [s] Spezifischer Widerstand des Cytoplasmas [Ohm*cm] (ρi ~ 200 Ω*cm) Spezifische Kapazität der Zellmenbran [µF/cm²] (ζm ~ 1 µF/cm²) Länge des Kabels [cm] Innenradius des Kabels [cm] Für ein Kabel mit einem Radius von 0,1 µm und einer Länge von 100 µm beispielsweise resultiert eine Kabelkonstante von τ = 2 ms, bzw. eine Grenzfrequenz von 500 Hz. 5-70 5 Elektrophysiologische Messungen Nach dem Modell besteht eine direkte Abhängigkeit der Zeitkonstante von der Kabellänge und dem Kabelradius. Die Messergebnisse für die Kabelkonstante τ wurden daher gegenüber der Kabellänge (Abbildung 5-26 A) und gegenüber der Breite des Lamininpfades (Abbildung 5-26 B) aufgetragen. Ist das Modell korrekt, so sollten die Messdaten einen Anstieg von τ zum Quadrat der Kabellänge l zeigen. Diese Beziehung kann unmittelbar im Experiment überprüft werden. Eine exakte experimentelle Bestimmung auch der Abhängigkeit vom Kabelradius r ist hingegen nicht möglich, da zwischen der Breite des Lamininpfades und dem Kabelradius kein unmittelbarer Zusammenhang besteht. Ein Ergebnis der Gleichspannungsanalyse in Punkt 5.5.1.2 war jedoch, dass der Kabeldurchmesser mit der Breite des Lamininpfades zunimmt. Insofern ist zumindest qualitativ eine Abnahme der Kabelkonstante τ mit der Breite der Linien zu erwarten. Die Messergebnisse in Abbildung 5-26 zeigen tatsächlich die erwarteten Abhängigkeiten: − Die Zunahme von τ mit der Kabellänge l in Abbildung 5-26 A kann mit einer Parabel angefittet werden. Aus den Fitparametern erhält man ∆τ/∆l² = 2,6*10-4 ± 2*10-5 ms/µm². Für ein 100 µm langes Kabel ergibt sich damit ein Wert von τ = 2,6 ± 0,2 ms. Ausgehend von Gleichung 5-38 hätte ein solches Kabel einen Radius von r = 0,08 ± 0,01 µm. Dies ist in Übereinstimmung mit dem Ergebnis r = 0,1± 0,07 µm aus der Gleichspannungsanalyse (siehe Punkt 5.5.1.2). Das Modell ist also konsistent. − In Abbildung 5-26 B ist die Auswertung der Messungen mit unterschiedlichen Pfadbreiten dargestellt. In die Analyse wurden Zellpaare mit Kabellängen zwischen 60 und 80 µm einbezogen. Auf Linien mit einer Breite b = 4 µm und b = 6 µm wird eine größere Zeitkonstante τ beobachtet als auf Linien mit nur 2 µm Breite. 5.5 Messergebnisse aus Ohm'scher Kopplung 5-71 Abhängigkeit des Kabelparameters τ von Kabellänge und Pfadbreite (A) τ gegenüber Kabellänge (B) τ gegenüber Pfadbreite Mittelwerte Fitfunktion Mittelwerte für 60-80 µm Abstand 1,2 2,0 1,5 -5 τ = (2,6*10 ± 2*10 ) * l 1,0 2 9 τ [ms] τ [ms] -4 1,0 0,5 0,0 0 20 1 2 0,2 40 60 Kabellänge l [µm] τ nimmt zu mit dem Quadrat der Kabellänge l. AC_VCVC_Teta-Kabellänge.OPJ 0,6 0,4 2 2 3 4 0,8 80 0,0 2 4 Linienbreite b [µm] 6 τ nimmt ab mit der Pfadbreite. AC_VCVC_Teta-Pfadbreite.OPJ Abbildung 5-26 Das Frequenzverhalten des Kabels wird beeinflusst von Länge und Durchmesser des Kabels. Die erzielten Messergebnisse können mit dem Kopplungsmodell erklärt werden. (A) Gemittelte Messwerte des Kabelparameter τ aufgetragen gegenüber der Kabellänge. (B) Gemittelte Messwerte des Kabelparameters τ von Kabeln mit 60 bis 80 µm Länge entlang von Lamininstrukturen verschiedener Breite. Wie diese Ergebnisse zeigen, ist es mit dem Kopplungsmodell aus Punkt 5.4.5. und dem Kabelmodell aus Punkt 5.4.4 möglich, auch die Kopplung von Wechselspannungen zwischen zwei Zellen vollständig zu erklären. 5-72 5 Elektrophysiologische Messungen 5.6 Messergebnisse aus funktionaler Kopplung Bei der Kopplung an funktionalen Synapsen treten nichtlineare Effekte auf. Man kann daher im Gegensatz zu Ohm'scher und kapazitiver Kopplung kein geschlossenes elektrisches Modell auf Basis der Theorie linearer Systeme aufstellen. Die Messergebnisse funktionaler Kopplung wurden aus diesem Grund separat anhand eines geeigneten Messverfahrens analysiert. 5.6.1 Messverfahren Die Charakterisierung funktionaler synaptischer Kopplung erfolgte im CC-CCModus. Abbildung 5-27 zeigt die beiden Schritte des Messverfahrens. Durch repetitive Stimulation der präsynaptischen Zelle mit Strompulsen einer Stärke von 100 pA und einer Dauer von 100 ms wurden Aktionspotentiale stimuliert (Abbildung Funktionale Kopplung - Messverfahren Stimulations-Strompuls [pA] Präsynaptische Zelle [mV] Postsynaptische Zelle [mV] 80 60 40 20 0 -20 -40 -60 200 400 600 800 1000 Zeit [ms] • Funktionale Kopplung: – postsynaptisches Signal folgt auf präsynaptisches AP. – fester Zeitversatz zwischen AP und postsynaptischem Signal. (B) Bestimmen der mittleren Übertragungszeit Membranpotential [mV] Membranpotential [mV] Stimulationspuls [pA] (A) Präsynaptische Stimulation von Aktionspotentialen Präsynaptische Zelle Postsynaptische Zelle 20 0 -20 -40 ∆t -60 540 ∆UPIP 545 550 555 Zeit [ms] 560 • Übertragungszeit: – Zeitintervall ∆t zwischen präsynaptischem AP-Maximum und postsynaptischem Signalanstieg. • Postsynaptisch induziertes Potential: – Amplitude ∆UPIP der postsynaptisch induzierten Potentialänderung. Abbildung 5-27 Übertragungszeit und Kopplungsstärke einer funktionalen Synapse werden durch präsynaptische Stimulation einer Serie von Aktionspotentialen ermittelt. (A) Stimulationspulse und Messignale. (B) Bestimmung von Übertragungszeit und postsynaptischem Potential. 5.6 Messergebnisse aus funktionaler Kopplung 5-73 5-27 A). Gleichzeitig wurde der zeitliche Verlauf der Membranspannung der postsynaptischen Zelle aufgezeichnet. Nach Abschluss der Messungen wurden die aufgezeichneten Daten am Bildschirm des Patch-Clamp Verstärkers ausgewertet. Dazu wurde für jedes detektierte postsynaptische Depolarisationssignal der Zeitversatz ∆t zum Aktionspotential der präsynaptischen Zelle sowie die Amplitude ∆UPIP des postsynaptisch induzierten Depolarisationspotentials bestimmt. Abbildung 5-27 B zeigt wie die beiden Parameter anhand des überlagerten Signalverlaufs der Membranspannungen von prä- und postsynaptischer Zelle ermittelt wurden. 5.6.2 Auswertung der Kopplungsdaten Eine Ausbildung funktionaler Synapsen in vitro ist selten und wird zwischen vereinzelt wachsenden Neuronen nur bei optimalen Kulturbedingungen erreicht [Banker, 1998]. Im Rahmen dieser Arbeit ist es dennoch gelungen, eindeutig synaptische Kontakte nicht nur auf homogen mit Laminin beschichteten Kontrollsubstraten (Abbildung 5-28 A), sondern auch auf strukturierten Substraten eindeutig nachzuweisen (Abbildung 5-28 B). Die Anzahl an insgesamt detektierten Kopplungen war für eine statistische Auswertung der gewonnenen Daten leider zu gering. Eine grundsätzliche, qualitative Diskussion der Einzelmessungen in Abbildung 5-28 ist jedoch ebenfalls sehr aufschlussreich und wird im Folgenden ausgeführt. In beiden Messungen waren die gekoppelten Zellen um ein Vielfaches ihres Zelldurchmessers voneinander entfernt. 47 µm bei Messung A auf dem Kontrollsubstrat und 96 µm bei Messung B auf der Lamininstruktur. Nimmt man an, dass die Axone in beiden Fällen nicht myelinisiert10 sind, so erfolgt die Ausbreitung eines Aktionspotentials entlang des Axons mit einer Geschwindigkeit von einigen Zentimetern pro Sekunde (v = 16 cm/s , berechnet unter Punkt 5.4.4 für ein Axon mit einem Radius von 1 µm). Auf einer Strecke von 50 - 100 µm erwartet man daher theoretisch eine Signallaufzeit von 0,3 - 0,6 ms. Eine weitere Zeitverzögerung 10 Anhand der Mikroskopieaufnahmen kann eine Myelinisierung nicht eindeutig ausgeschlossen werden. 5-74 5 Elektrophysiologische Messungen Abbildung 5-28 Funktionale Synapsen zwischen Neuronen auf einem Kontrollsubstrat und einer Struktur aus Laminin. (A) Funktionale Synapse zwischen zwei unkontrolliert verbundenen Neuronen. Messung durchgeführt an Tag 16, in einer Hirnschnitt-Kultur auf einem homogen mit Laminin beschichteten Kontrollsubstrat. (B) Funktionale Synapse entlang einer künstlich aufgeprägten Verbindungslinie aus Laminin. Messung durchgeführt an Tag 15, in einer Hirnschnitt-Kultur auf einem mit Linien aus Laminin strukturierten Substrat. Stempelstruktur Nr. 11: 4 µm Linen, 20 µm Knoten. entsteht an der Synapse. Hier kann man von einem Zeitbedarf zwischen 1 und 2 ms für die aktive synaptische Übertragung ausgehen [Kandel, 1991]. In Summe erwartet man eine Zeitverzögerung von 1,3 bis 2,6 ms, wobei die Signallaufzeit im Axon davon nur einen kleinen Teil ausmacht. Die experimentell gemessenen Laufzeiten sind 5,25 ± 0,49 ms in Fall A und 2,42 ± 0,23 ms in Fall B . Sie liegen in der erwarteten Größenordnung. Auch ein Vergleich mit Literaturwerten bestätigt die Plausibilität der gemessenenen Übertragungszeiten: In einer dissoziierten Kultur hippocampaler Neuronen wird für monosynaptische Verbindungen eine mittlere Übertragungszeit zwischen 1,5 und 2,6 ms angegeben [Fitzsimonds, 1997]. Die gekoppelten Neuronen waren in diesen Messungen maximal 950 µm voneinander entfernt. 5.6 Messergebnisse aus funktionaler Kopplung 5-75 In der letzten Zeile von Abbildung 5-28 wurde unter Annahme einer festen Signalverzögerung von 1 ms an der Synapse die Übertragungsgeschwindigkeit im Axon berechnet. Auf dem Kontrollsubstrat (Abbildung 5-28 A) ist die berechnete Geschwindigkeit mit 1,1 ± 0,5 cm/s deutlich kleiner als 6,5 ± 1,1 cm/s entlang der durch den Lamininpfad vorgegebenen Verbindungslinie in Abbildung 5-28 B. Man könnte diesen Unterschied auf einen "Umweg" der Axonalen Verbindung in Messung A zurückführen, derart, dass die tatsächliche Verbindungsstrecke zwischen beiden Neuronen auf der unstrukturierten Substratoberfläche deutlich größer ist, als die direkte Distanz zwischen beiden Zellen. Die dieser Betrachtung zugrundeliegende Annahme exakt gleicher synaptischer Übertragungszeiten ist jedoch rein hypothetisch. Ohne die Möglichkeit zur statistischen Auswertung vieler Messungen mit unterschiedlichen Zellabständen, ist eine Interpretation der Ergebnisse spekulativ. 5-76 5 Elektrophysiologische Messungen 5.7 Ergebnis der Kopplungsexperimente Mit den durchgeführten Analysen zur Kopplung von Gleich- und Wechselspannungen konnte nachgewiesen werden, dass für das untersuchte Zellmodell eine Beschreibung der passiven elektrischen Eigenschaften von axonalen Fortsätzen mit dem physikalischen Modell eines zylindrischen Kabels korrekt ist. Für makroskopische Nervenfasern des PNS ist die Anwendbarkeit dieses Modells seit langem bekannt [Hodgkin, 1946]. Die Übertragbarkeit auch auf mikroskopische Axone im ZNS, war jedoch bislang mit elektrophysiologischen Methoden nicht detailliert untersucht worden. Die Ergebnisse dieser Arbeit legen den Schluss nahe, dass jedes Axon im ZNS das elektrische Verhalten eines zylindrischen Kabels aufweist. Erkenntnisse aus Untersuchungen der Signalübertragung im PNS können demnach nach entsprechender Skalierung der Kabelparameter unmittelbar auf das ZNS übertragen werden. Aus den Messergebnissen funktionaler Kopplung kann man folgende Schlussfolgerungen ziehen: 1. Neuronen der verwendeten neuronalen Primärkultur bilden funktionale Synapsen in vitro. 2. Auch auf mit Laminin strukturierten Substratoberflächen entstehen funktionale Synapsen. Die Ausbeute ist jedoch sehr gering. 3. Eine gezielte Anordnung von Nervenzellen zu künstlichen Netzwerken, miteinander verknüpft durch funktionale Synapsen entlang vorgegebener Pfade ist experimentell möglich. Im Rahmen dieser Arbeit ist damit der Nachweis gelungen, dass sich mit Mikrostrukturen aus Laminin Nervenzellen nicht nur definiert auf einer Oberfläche anordnen lassen sondern dass diese untereinander auch über Ohm'sche und funktionale Synapsen kommunizieren. 5.7 Ergebnis der Kopplungsexperimente 5-77 Ein zentrales Ziel zukünftiger Arbeiten muss es jetzt sein, die Ausbeute an funktionalen synaptischen Kontakten deutlich zu steigern. Gelingt dies, so können an synaptisch verschalteten Modellnetzwerken weitreichende Fragestellungen der Gehirnforschung experimentell untersucht werden. Beispielsweise könnten mit entsprechenden Mikrostrukturen ringförmige Schaltungen von Nervenzellen erzeugt werden. Solche Netzwerke wären für eine Überprüfung des Modells assoziativen Lernens geeignet. 5-78 5 Elektrophysiologische Messungen 6-1 6 Ausrichtung von Struktur und Substrat Wie in Kapitel 4 beschrieben, können Netzwerke aus Nervenzellen durch gezielte Strukturierung einer Substratoberfläche mit Laminin erzeugt werden. Das Protein Laminin wird dazu mit Hilfe eines Mikrostempels auf die Substratoberfläche übertragen. Ohne weitere technische Maßnahmen ist die Orientierung der Stempelstruktur gegenüber dem Substrat dabei nicht zu kontrollieren. Für eine Anwendung der Methode auf homogenen Substratoberflächen ist dies auch nicht erforderlich. Möchte man das Verfahren jedoch für inhomogene Substratoberflächen verwenden und die Stempelstruktur mit einer bereits an der Subsratoberfläche vorhandenen Struktur gezielt überlagern, so ist es notwendig die Position und die Orientierung des Stempels vor dem Stempelübertrag exakt justieren zu können. Ein hierfür geeigneter experimenteller Aufbau wurde im Rahmen dieser Arbeit entwickelt. Eine Ausrichtung des Stempels ist z.B. erforderlich, um Strukturen aus unterschiedlichen Proteinen der ECM zusammenzusetzen. In Kapitel 4 wurde beschrieben, wie neuronale Polarität durch bestimmte Strukturelemente auf einer Substratoberfläche beeinflusst werden kann. Eine weitere Möglichkeit selektiv axonales und dendritisches Wachstum zu fördern besteht darin, zwei verschiedene ECM-Proteine zu einer heterogenen Struktur zusammenzusetzen [Wheeler, 1999]. Nur wenn ein zweiter Stempelabdruck einem bereits vorhandenen exakt überlagert werden kann, ist die Herstellung solcher Substrate mit Mikrostempeln möglich. Eine weitere wichtige Anwendung ist die Verwendung von Sensorchips zur extrazellulären Erfassung der Signale von Nervenzellen. Die Oberfläche eines solchen Sensors hat eine vorgegebene Struktur von regelmäßig angeordneten Messpunkten. Ein Stempelübertrag von Laminin auf die Sensoroberfläche muss auf diese Messpunkte ausgerichtet werden, damit später die elektrischen Signale der einzelnen Neuronen an den Messpunkten detektiert werden können. Sensorchips sind insbesondere für eine Analyse größerer Netzwerke aus Nervenzellen erforderlich. Kleine Netzwerke aus bis zu drei Neuronen können, wie 6-2 6 Ausrichtung von Struktur und Substrat in Kapitel 5 beschrieben, ohne Schwierigkeiten mit der Patch-Clamp Technik untersucht werden. Für Netzwerke aus mehr als drei bis vier Nervenzellen stößt man aber an technische Grenzen. Einerseits wird die Anordnung der einzelnen Messköpfe um die Probe herum sehr eng und unübersichtlich. Andererseits benötigt man für eine einzelne Messung, bedingt durch die aufwendige Positionierung jeder einzelnen Mikroelektrode, sehr viel Zeit. Die maximal verfügbare Messzeit ist jedoch aufgrund der Schädigung der Zellen durch die Mikroelektroden begrenzt. Mit einem geeigneten Sensorchip können die Signale von vielen Nervenzellen gleichzeitig aufgenommen werden. Die Signalerfassung kann unmittelbar nach Anschluss des Chips an einen Messrechner beginnen. Eine Kontaktierung einzelner Zellen wie bei der Patch-Clamp Technik ist nicht notwendig. Darüber hinaus wird die Zellmembran der Nervenzellen nicht durch Mikropipetten verletzt. Es sind daher auch Langzeitstudien möglich. 6.1 Sensorchips zur extrazellulären Signalerfassung 6-3 6.1 Sensorchips zur extrazellulären Signalerfassung Mit speziellen Sensorchips auf Halbleiterbasis ist eine extrazelluläre Messung der elektrischen Signale von Nervenzellen möglich. Die Signalerfassung erfolgt dabei ohne leitende Verbindung zum Cytoplasma ausschließlich über rein kapazitive Kopplung [Fromherz, 1991]. Verschiedene Sensoren für dieses Messverfahren sind in den letzten Jahren entwickelt worden. Grundsätzlich zu unterscheiden sind Sensoren auf Basis von Feldeffekttransitoren (Typ1) und Mikroelektroden (Typ 2). In Abbildung 6-1 sind zwei Sensoren von Typ 1 (A1) und Typ 2 (B1) zusammen mit Mikroskopieaufnahmen der Sensoroberflächen (A2, B2) abgebildet. Bei Sensoren vom Typ 1 erfolgt die Signalverstärkung unmittelbar an der Oberfläche Verwendete Sensorchips Sensortyp (A) Typ 1: Sensorchip auf FET1-Basis (B) Typ 2: Sensorchip auf MEA2-Basis Gesamtansicht A1 B1 Mikroskopieaufnahme der SensorB1a oberfläche A2 B2 400 µm 200 µm 1 FET: Feld-Effekt-Transitor (Feldeffekttransitoren an der Oberfläche eines Siliziumchips arbeiten als aktive Sensoren) 2 MEA: Mehrfach-Elektroden-Anordnung (Goldelektroden auf einer Quarzglasoberfläche leiten passiv die Zellsignale ab) Abbildung 6-1 Zwei unterschiedliche Typen von Sensorchips wurden für die Experimente verwendet. (A) Sensorchip mit Feldeffekttransitoren. (A1) Gesamtansicht, (A2) Mikroskopische Vergrößerung der sensitiven Oberfläche. (B) Sensorchip mit Goldelektroden. (B1) Gesamtansicht, (B2) Mikroskopische Vergrößerung der sensitiven Oberfläche 6-4 6 Ausrichtung von Struktur und Substrat des Sensors, so nah wie nur irgend möglich an der zu vermessenden Zelle. Verstärkt wird mit speziell für diese Anwendung entwickelten Feldeffekttransistoren in der Chipoberfläche [Fromherz, 1991; Offenhäusser, 1997; Sprössler, 1998]. Bei Typ 2 werden die Signale von der Zelle durch Mikroelektroden abgeleitet. Verstärkt werden die Signale einige Zentimeter entfernt mit kommerziell erhältlichen Feldeffekttransistoren [Krause, 2000] oder Operationsverstärkern [Gross, 1979; Pine, 1980; Connolly, 1990]. Gemeinsam ist beiden Systemen die regelmäßige Anordnung einer Vielzahl sensitiver Messpunkte auf der Oberfläche des Sensors. Nach gegenwärtigem Stand der Technik ist es mit solchen Systemen möglich, simultan Signale von bis zu 64 Messkanälen aufzunehmen. Entsprechende Messplätze werden inzwischen auch kommerziell vertrieben (ME System, Multi Channel Systems MCS GmbH). Aufgrund der großen Anzahl von simultan zur Verfügung stehenden Messkanälen, werden solche Sensorchips in Zukunft mit großer Wahrscheinlichkeit auch eine entscheidende Rolle bei der Aufklärung der Prozesse in neuronalen Netzwerken spielen. Anders als die Mikropipetten bei der Patch Clamp Technik, können die Messpunkte an der Oberfläche extrazellulärer Senoren jedoch nicht bewegt werden. Will man daher Neuronen in einem durch Stempelübertrag von Laminin erzeugten neuronalen Netzwerkverbund mit einem Sensorchip analysieren, müssen für die Messung die einzelnen Nervenzellen exakt über den Messpunkten an der Oberfläche des Sensors liegen. Alle in dieser Arbeit realisierten Stempelstrukturen basieren auf einem Grundraster von 100 x 50 µm. Dieses Raster ist kompatibel zu Sensorchips mit Messpunkten im Abstand von 50 µm, 100 µm und 200 µm. Wachsen Nervenzellen auf den Knotenpunkten dieses Rasters, so können sie mit den Sensorpunkten zur Deckung gebracht werden. Die Ausrichtung der Netzwerke auf die Sensorpunkte der Chips kann auf zwei verschiedenen Wegen erfolgen: Entweder wird die Lamininstruktur ausgerichtet auf den Sensor aufgeprägt, so dass Neuronen aus einer später aufgebrachten Neuronenkultur sich direkt in der gewünschten Form auf der Oberfläche des Sensors anordnen [James, 2000; Lauer, 2001b]. Oder aber, die Netzwerke werden auf 6.2 Ausgerichtete Mikrostrukturierung 6-5 strukturierten Substraten außerhalb des Sensors erzeugt, und zur Messung kopfüber auf den Sensor gelegt. Im ersten Fall muss der Mikrostempel beim Stempelvorgang auf die Messpunkte des Sensorchips ausgerichtet werden (ausgerichtete Mikrostrukturierung). Im zweiten Fall erfolgt die Positionierung des Zellnetzes auf die Sensorpunkte unmittelbar vor der Messung (in vitro Positionierung). Für beide Verfahren ist ein spezieller apparativer Aufbau und eine besondere Technik erforderlich. Beides wurde im Rahmen dieser Arbeit entwickelt und ist im Folgenden beschrieben. 6.2 Ausgerichtete Mikrostrukturierung Aufgabenstellung für den Entwurf eines experimentellen Aufbaus zur ausgerichteten Mikrostrukturierung war es, eine möglichst präzise Ausrichtung von Mikrostempeln auf die Messpunkte eines Sensorchips zu erzielen (∆x, ∆y < 5 µm), bei gleichzeitig schnellstmöglichem Ablauf des Stempelprozesses. Weiterhin sollte die Strukturierung extrem kleiner Oberflächen (FET < 2,5 mm ∅, MEA < 5 mm ∅) möglich sein. Diese Anforderungen ergeben sich aus der Größe der zu übertragenden Mikrostrukturen (10-20 µm an Zelladhäsionspunkten), der erfahrungsgemäß erforderlichen großen Substratanzahl je Experiment sowie den Abmessungen der verwendeten Sensoren. 6.2.1 Hybrid-Stempelsystem aus Glas und PDMS Die Anforderung, für die Strukturierung der FET-Sensoren Mikrostempel mit einem maximalen Durchmesser von nur 2,5 mm zu verwenden, erforderte ein vollkommen neues Design der Stempel. Anstelle eines massiven Stempelkörpers aus PDMS (siehe oben) trat ein Hybridaufbau aus einem starren Glaszylinder und einer mikrostrukturierten dünnen Schicht aus PDMS auf der einen Stirnseite des Glaszylinders. Dieses Design ist erforderlich, um trotz des geringen Durchmessers der Stempel eine ausreichende Stabilität zu gewährleisten. Ultradünne Mikrostempel auf Glasträgern sind zwar bereits in der Literatur beschrieben [James, 1998], jedoch basierten diese Systeme bislang immer auf dünnen Glasträgern mit größerer lateraler Ausdehnung als 2,5 mm. Solche Stempel sind in 6-6 6 Ausrichtung von Struktur und Substrat der Praxis nur sehr schwer zu handhaben. Ein µm-genauer Strukturübertrag auf Sensorchip-Oberflächen ist mit einem solchen Design nur mit erheblichem technischem Aufwand möglich. Beschrieben werden Systeme mit einem Aufbau ähnlich zu einem Maskenausrichter (Mask-Aligner) aus der Photolithographie [James, 2000; Wheeler, 2001]. Das hier vorgestellte Design mit einem Glaszylinder als Stempelrückrat hingegen, kann mit bloßen Händen und einer einfachen Pinzette gehandhabt werden und es lässt sich mit einem kommerziell erhältlichen Positioniersystem verbinden. Dadurch ist eine sekundenschnelle Justage möglich. Gleichzeitig sind alle in der Literatur beschriebenen Vorteile von ultradünnen Mikrostempeln [James, 1998] vorhanden, 1 insbesondere auch die erhöhte Druck-Belastbarkeit der Stempeltopographie . In Abbildung 6-2 sind die wesentlichen drei Schritte des Herstellungsprozesses von Hybridstempeln dargestellt. Die einzelnen Fertigungsschritte sind im Folgenden stichpunktartig in Form einer Anleitung beschrieben: 1. Bereitlegen von Fertigungswerkzeug W (Abbildung 6-2 A), Glassockeln2 S und Strukturmaske M. 2. Reinigen von Fertigungswerkzeug und Glassockeln mit Aceton. Ohne eine gründliche Entfettung der Glassockel in diesem Schritt ist die Haftung des später aufgebrachten PDMS-Tropfens an der Sockeloberfläche unzureichend. 3. Vorbehandlung der Oberfläche der Strukturmaske mit SDS. 4. Auftropfen von ca. 0,5 ml blasenfreiem PDMS (zuvor entgast im Excitator) auf einen Glas-Objektträger. 1 Aufgrund des steifen Glasrückrats wird eine Verformung der Stempelstruktur beim Stempelandruck erst bei deutlich größeren Andruckkräften beobachtet, als bei PDMS Stempeln aus Vollmaterial. Aufgrund des bei gleicher Andruckkraft mit sinkender Stempelfläche reziprok steigenden Drucks ist dies von entscheidender Bedeutung. 2 Die Glassockel werden zuvor aus Duran-Glasstäben mit 2 mm Durchmesser geschnitten und an den Enden poliert. Die Länge der Schnittstücke wird so präzise wie möglich auf 2 cm eingestellt. Zur Herstellung von Hybridstempeln mit größerem Stempelquerschnitt als 2 mm werden die Glasstäbe vor dem Zuschnitt im Bunsenbrenner tropfenförmig verdickt. Der Schnitt erfolgt dann durch die Mitte des Tropfens. Solche, an einem Ende auf 4 mm verdickte Hybridstempel wurden für die Strukturierung von MEASensoren mit einem Substratdurchmesser von 5 mm verwendet. 6.2 Ausgerichtete Mikrostrukturierung 6-7 Herstellung von Hybridstempeln Teilschritt Arbeitsablauf (A) Vorbereitung (B) Justage • Abformwerkzeug W, • PDMS-Tropfen mit Glassockel S und Strukturmaske M bereitlegen • Entfetten der Glassockel in Aceton • Benetzen der Strukturmaske mit SDS Glassockel aufnehmen • Glassockel S in das Abformwerkzeug W schieben • Strukturmaske M aufsetzen • Justage mit Feintrieb F bis die PDMS-Tropfen aller Glassockel die Maske berühren Darstellung (C) Abformung • Aushärten des PDMS bei 60 °C für 24 h im Ofen • Abheben der Strukturmaske • Entnahme der Stempel M M W S W S W S F M F 1 cm Abbildung 6-2 Die Herstellung von Hybridstempeln erfolgt mit Hilfe eines Abformungswerkzeugs. In diesem können bis zu fünf Glassockel senkrecht zur Strukturmaske und in definiertem Abstand zu dieser gehaltert werden. (A) Abformwerkzeug W, Glassockel S und Strukturmaske M. (B) Glassockel S und Strukturmaske M gehaltert im Abformwerkzeug W. (C) Mit Feintrieb F vollständig justierter Aufbau. 5. Senkrechtes Eintauchen des Kopfendes jedes einzelnen Glassockels in den PDMSFilm auf dem Objektträger. Beim Anheben der Glassockel bleibt jeweils ein Tropfen PDMS an der Spitze haften. 6. Einschieben des Fußendes aller fünf Glassockel S in die Bohrungen des Fertigungswerkzeugs W (Abbildung 6-2 B). 7. Aufsetzen der Strukturmaske M auf das Fertigungswerkzeug W, mit der strukturierten Seite nach unten. (Abbildung 6-2 B) 8. Eindrehen der Schrauben des Feintriebs F auf der Unterseite des Fertigungswerkzeuges W, bis der PDMS-Tropfen auf dem dadurch nach oben geschobenen Glassockel S die Strukturmaske M berührt, und die 6-8 6 Ausrichtung von Struktur und Substrat Berührungsfläche dem Durchmesser des Glassockels entspricht (Abbildung 6-2 C). Das Fertigungswerkzeug muss dabei aufrecht gehalten werden. 9. Erhitzen der gesamten Anordnung im Ofen. Aushärten des PDMS bei 60 °C für 24 h. 10. Abheben der Strukturmaske. Gewöhnlich kleben die Stempel nun an der Strukturmaske und werden mit dieser aus dem Fertigungswerkzeug gezogen. 11. Ablösen der Stempel. Dazu vorsichtig an den Glassockeln ziehen, bis sich der gehärtete PDMS-Tropfen von der Oberfläche der Strukturmaske zu lösen beginnt. Beim Ablösen den Glassockel leicht verdrehen. Etwaige PDMS-Fäden werden dadurch aufgewickelt ohne die strukturierte Oberfläche des neuen Stempels zu verkleben. 12. Die abgelösten Stempel erneut in das Abformwerkzeug stellen und im Ofen erhitzen. Zum endgültigen Aushärten diesmal bei 110 °C für 1 h. 13. Entnahme der Stempel. Die staubgeschützte Lagerung der fertigen Hybridstempel erfolgt in mit Milli-Q gefüllten Glasgefäßen. 6.2.2 Optische Ausrichtung auf Chip Sensorpunkte Für die exakte Positionierung der Hybridstempel beim Stempelvorgang wird der Glaszylinder des Stempels in einer speziell entwickelten Halterung formschlüssig fixiert. Die Halterung selbst ist Teil eines Justiersystems in welchem durch Überlagerung zweier optischer Strahlengänge die Aufsicht auf Stempeloberfläche und Substratoberfläche zur Deckung gebracht wird. In Abbildung 6-3 ist das vollständige Justiersystem schematisch (A) und als Photographie (B, C) dargestellt. Kernelement des Systems ist ein kommerziell erhältliches Positioniersystem aus der Halbleiterindustrie (FINEPLACER-145 "PICO", FINETECH electronics). Der 6.2 Ausgerichtete Mikrostrukturierung 6-9 Abbildung 6-3 Die Ausrichtung des Mikrostempels auf die Oberfläche des Sensorchips erfolgt unter optischer Kontrolle mit einer speziellen Positioniereinheit. (A) Schemische Darstellung des Aufbaus sowie der Arbeitsschritte. (B) Photo des Aufbaus in Justagestellung (C) Photo des auf das Substrat abgesenkten Stempels beim Stempelabdruck. Positionierarm A dieses Systems kann punktgenau mit einer Genauigkeit3 von 1 µm um 90° zum Boden geschwenkt werden. In angehobener Position des Hebelarms beobachtet der Experimentator durch eine Stereolupe L (Stemi 2000, Carl Zeiss GmbH) gleichzeitig die in Prisma P überlagerten Bilder von Stempel S und Sensorchip C. Durch Justage an Verschiebetisch4 T (MVT 40B & WVT 40, OWIS) bringt er diese zur Deckung. Ist die Justage abgeschlossen, ein Vorgang der nach etwas Übung nur wenige Sekunden in Anspruch nimmt, wird der Arm A aus der Justageposition in Abbildung 6-3 B abgesenkt bis der Stempel wie in Abbildung 6-3 C gezeigt, die Chipoberfläche berührt. 3 Die vom Hersteller zertifizierte Genauigkeit des Systems beträgt 5 µm. In den durchgeführten Experimenten wurde bei konstanter Umgebungstemperatur jedoch eine Positioniergenauigkeit von besser als 1µm erzielt. 4 Alle Freiheitsgrade x,y,z und Rotation in der x-y-Ebene sind mit Mikrometerschrauben einstellbar. 6-10 6 Ausrichtung von Struktur und Substrat Nach erfolgtem Stempelübertrag wird der Arm wieder angehoben. Nun werden Stempel und Chip rasch ausgetauscht um die nächste Strukturierung vornehmen zu können. Der Austausch des Mikrochips erfolgt dabei durch Lösen eines Schnellspannhebels am Null-Kraft-Sockel der Chiphalterung. Der Stempel wird mit einer Pinzette am Glassockel aus seiner Halterung gezogen und gegen einen frischen, mit Laminin benetzten Stempel ersetzt. 6.2.3 Kontrolle des Anpressdrucks Wie in Kapitel 4 beschrieben, ist der Anpressdruck beim Stempelübertrag ein kitischer Parameter des Verfahrens. Zu kleiner Druck führt zu unvollständigem Stempelübertrag, zu großer Druck verformt die übertragene Struktur. Je kleiner die effektive Stempeloberfläche, umso genauer muss die Dosierung der Andruckkraft erfolgen. Erzielt man bei einem gewöhnlichen Mikrostempel5 (Oberfläche A1 = 78,5 mm²) mit Kräften zwischen 7,85 kN < F1 < 15,7 kN gute Ergebnisse, so muss die Kraft für einen verkleinerten Hybridstempel der gleichen Struktur6 (Oberfläche A2 = 3,1 mm²) um einen Faktor 25 geringer sein7. Die Toleranz von 7,85 kN schrumpft damit ebenfalls um diesen Faktor auf 0,31 kN. Bei einem Toleranzfenster der Andruckkraft von 7,5 kN (~ 750 g) kann der Stempelandruck durchaus mit bloßen Händen erfolgen. Eine Kontrolle der Kraft auf 0,31 kN (~31g) genau ist jedoch nur mit technischen Hilfsmitteln möglich. Der Hebelarm der Positioniereinheit wurde dazu mit einem verschiebbaren Gegengewicht G ausgestattet (siehe Abbildung 6-3 A und B). Mit der Position des Gewichts entlang einer geeichten Skala kann damit die Andruckkraft des Stempels zwischen 50 N und 600 N auf 25 N genau eingestellt werden. Bei entsprechender Anpassung der Andruckkraft ist es mit dem beschriebenen Positioniersystem möglich, präzise ausgerichtete und nahezu fehlerfreie Stempelabdrücke auf die Oberfläche der eingesetzten Sensoren zu übertragen. In 5 Annahme: Zylinderförmiger Stempel mit 1 cm Durchmesser der strukturierten Oberfläche 6 Annahme: Zylinderförmiger Stempel mit 2 mm Durchmesser der strukturierten Oberfläche 7 Der Effekt der größeren Stabilität durch das Glasrückrat des Mikrostempels ist bei dieser Abschätzung nicht berücksichtigt. 6.2 Ausgerichtete Mikrostrukturierung 6-11 Ausgerichteter Strukturübertrag auf Sensorchips Sensortyp (A) Typ 1: Sensorchip auf FET-Basis (B) Typ 2: Sensorchip auf MEA-Basis Technische Zeichnung der Stempelstruktur A1 B1 100 µm 100 µm Mikroskopieaufnahme der Sensoroberfläche B1a A2 B2 100 µm <V1_1_s9> 100 µm <V12_5_s13> Abbildung 6-4 Mit dem entwickelten Positioniersystem ist es möglich, die Oberfläche von Sensorchips mit unterschiedlichen Strukturen gezielt zu modifizieren. Die jeweilige Struktur kann dabei exakt auf die Messpunkte des Sensors ausgerichtet werden. (A) Ausgerichtete Lamininstruktur auf einem FET-Sensor. (B) Ausgerichtete Lamininstruktur auf einem MEA-Sensor. Abbildung 6-4 sind entsprechende Mikroskopieaufnahmen von Lamininstrukturen auf der Oberfläche eines FET-Sensors (A) sowie eines MEA-Sensors (B) dargestellt. 6.2.4 Spezialwerkzeuge für ausgerichtetes Mikrostempeln Sowohl die Herstellung von Hybridstempeln, als auch deren Halterung und Positionierung erfordern spezielle Hilfswerkzeuge. Diese sind in Abbildung 6-5 in Detailaufnahmen abgebildet. Wie in Punkt 6.2.1 beschrieben, ist für die Herstellung von Hybridstempeln ein spezielles Fertigungswerkzeug zur Halterung der Glassockel bei der Abformung erforderlich. Abbildung 6-5 A zeigt dieses Fertigungswerkzeug von allen Seiten. In Abbildung 6-5 A3 sind die Schrauben des Feintriebs auf der Unterseite des Fertigungswerkzeugs deutlich zu erkennen. Diese können für die Justage der Glassockel mit einem Miniaturschraubenzieher verdreht werden. 6-12 6 Ausrichtung von Struktur und Substrat Spezialwerkzeuge für ausgerichtetes Mikrostempeln (A) Werkzeug zur Stempelabformung A1 A2 A3 1 cm (B) Stempelhalterung B1 B2 (C) Justagewerkzeug C1 B1a B1b C2 B1c 1 cm 1 cm Abbildung 6-5 Für Herstellung und Handhabung von Hybrid-Mikrostempeln sind Spezialwerkzeuge und – bauteile erforderlich. (A) Werkzeug zur Stempelfertigung. Bis zu fünf Glaszylinder (mit einem Tropfen PDMS versehen) werden zur Strukturabformung in die Bohrlöcher (A1) gesteckt. Mit den Stellschrauben des Feintriebs an der Unterseite (A3) wird der Abstand zur Strukturmaske justiert. (B) Halterung zur formschlüssigen Aufnahme der Hybridstempel. (B1) zeigt die zerlegte Halterung: Ein Sockel (B1a) nimmt den Glaszylinder des Mikrostempels (B1b) in einer Bohrung auf. Im vorderen Teil des Sockels ist die Bohrung auf das Aussenmaß zweier kleiner O-Ringe aufgeweitet. Diese gleiten im Abstand von 0,5 mm (Beilagscheiben) auf dem Glaszylinder des Stempels. Durch Aufschrauben der Abschlusskappe (B1c) auf den Sockel werden die O-Ringe auf den Glassockel des Stempels gepresst (B2). (C) Justagewerkzeug. Für die Justage der Optik wird der Justierstift (C1) anstelle eines Stempels in die Stempelhalterung (B) eingespannt. Durch Absenken des Stempelarms der Stempelapparatur wird die Justierkappe (C2) auf den Justierstift (C1) mittig zentriert. Der Stempelarm wird wieder angehoben und das im Mikroskop sichtbare Überlagerungsbild von (C1) und (C2) wird durch Justage der optischen Komponenten zur Deckung gebracht. Um die Präzision von 1 µm des in Punkt 6.2.2 beschriebenen Positioniersystems auch tatsächlich nutzen zu können, muss der Hybridstempel ohne jedes Spiel mit dem Arm des Positioniersystems verbunden werden können. Gleichzeitig soll aber auch ein schneller und einfacher Austausch des Stempels möglich sein, um in rascher Folge frisch mit Laminin benetzte Stempel zum Stempelübertrag in das System einsetzen zu können. Beides ist mit der speziell entwickelten Halterung in 6.2 Ausgerichtete Mikrostrukturierung 6-13 Abbildung 6-5 B möglich. Der Glaszylinder eines Hybridstempels wird darin formschlüssig von O-Ringen fixiert. Zum Stempeltausch muss lediglich die Verschlusskappe der Halterung leicht gelockert werden. Das dritte in Abbildung 6-5 C dargestellte Spezialwerkzeug dient zur Justage des Positioniersystems. Um zu gewährleisten, dass die überlagerten Bilder von Stempel und Chip auch tatsächlich einen exakt ausgerichteten Stempelübertrag zur Folge haben, muss die Optik des Positioniersystems gelegentlich nachjustiert werden. Insbesondere Temperaturschwankungen können zu einer Dejustage des Systems führen. Die Justage der Optik erfolgt unter Beobachtung der überlagerten Bilder zweier auf mechanischem Wege zu 100 % korrekt ausgerichteter Bauteile. Diese sind in Abbildung 6-5 C dargestellt. Durch die konische Passform der Bauteile werden diese bei abgesenktem Stempelarm mechanisch zentriert. Hebt man anschließend den Stempelarm an und beobachtet die überlagerten Bilder, so ist die Optik genau dann richtig justiert, wenn sich die Passformen beider Bauteile optisch decken. 6.2.5 Bewertung der ausgerichteten Mikrostrukturierung Es konnte gezeigt werden, dass mit dem Verfahren der ausgerichteten Mikrostrukturierung ein exakt positionierter Übertrag von Wachstumsstrukturen auf die inhomogene Oberfläche der extrazellulären Sensorchips möglich ist. Abbildung 6-6 zeigt ausgerichtet wachsende hippocampale Nervenzellen an Tag 3 auf einem MEA-Sensorchip. Die Oberflächen mehrerer Chips wurden in diesem Versuch mit einer Linienstruktur (4 µm Linien und 20 µm Knoten, Abbildung 6-6 A) aus Laminin bestempelt. Entweder wurden die Linien zwischen die Elektrodenreihen eines Chips positioniert (Abbildung 6-6 B1) oder exakt über die Elektroden (Abbildung 6-6 B2). In beiden Fällen waren die Knotenpunkte der Linienstruktur auf gleicher Höhe zu den Elektroden des Chips. Anhand der Mikroskopieaufnahmen des Zellwachstums in Abbildung 6-6 B1 und B2 ist deutlich zu erkennen, dass sich die Nervenzellen entlang der gestempelten Linien anordnen und auf die Knotenpunkte der Linien wandern. Viele Nervenzellen in Abbildung 6-6 B liegen somit wie gewünscht direkt über einer Elektrode des Sensorchips. 6-14 6 Ausrichtung von Struktur und Substrat Ausgerichtetes Wachstum von Neuronen auf einem Sensorchip (A) Verwendete Stempelstruktur 100 µm (B) Wachstum von Nervenzellen auf der strukturierten Chip-Oberfläche (B1) Wachstum parallel zu den Elektroden des Chips (B2) Wachstum ausgerichtet auf die Elektroden des Chips B1a 100 µm <V12_2_s11> 100 µm <V12_2_s11> Abbildung 6-6 Die Position von Nervenzellen auf einem MEA-Sensorchip kann durch ausgerichtete Mikrostrukturierung exakt auf die Position der Elektroden an der Chipoberfläche festgelegt werden. (A) Verwendete Stempelstruktur: 4 µm breite Linien mit 20 µm Knotenpunkten. (B) Wachstum auf der strukturierten Chip-Oberfläche: (B1) Zellwachstum parallel zu den Elektroden des Chips. (B2) Zellwachstum exakt ausgerichtet auf die Elektroden des Chips. Ohne eine geeignete Oberflächenmodifikation der Sensorchips, entsprechend der Verfahren aus Kapitel 4, war die Qualität der erzeugten Wachstumsstrukturen jedoch nur bis wenige Tage nach Zellaussaat stabil. Wurden die Chipoberflächen chemisch behandelt, so ergab sich eine andere Komplikation: Sollten die Siliziumchips nicht nur als inhomogenes, vorstrukturiertes Kultursubstrat dienen, sondern zu elektrisch funktionsfähigen Sensoren zusammengesetzt werden, so war es erforderlich die elektrischen Anschlüsse jedes Siliziumchips durch BondingDrähte mit makroskopischen Kontakten in einem Chip-Carrier-Gehäuse zu verbinden. Ist die Chipoberfläche aber mit Polystyrol oder Aktivester beschichtet, so wird diese Kontaktierung sehr schwierig und schlägt häufig sogar ganz fehl. Eine nachträgliche Beschichtung nach erfolgter Montage des Chip aber war ebenfalls nicht möglich, da die zur Versiegelung des Bauteils eingesetzte biokompatible Vergussmasse PDMS (Sylgard 182, Dow Corning) in DMF (DMF als Lösungsmittel 6.3 In vitro Positionierung 6-15 ist erforderlich für den Beschichtungsprozess; siehe Kapitel 4) stark quillt. Aus diesem Grund konnten nur sehr wenige Experimente mit beschichteten und gleichzeitig voll funktionsfähigen Sensoren durchgeführt werden. Kurz vor Abschluss dieser Arbeit wurde in unserer Arbeitsgruppe die Entwicklung eines neuen Chiptyps abgeschlossen. Bei diesem erfolgt die Kontaktierung zum Gehäuse nicht mehr über Bonding Drähte, sondern über Kontaktpunkte auf der Rückseite des Chips [Ingebrandt, 2001]. Bei diesem Chiptyp sollte eine Montage auch nach vorheriger Oberflächenmodifikation problemlos möglich sein, sofern die Rückseite des Chips während des chemischen Prozesses geschützt wird. 6.3 In vitro Positionierung Wie in 6.2 beschrieben, ist es durch ausgerichtetes Mikrostempeln von Laminin auf einer geeignet chemisch modifizierten Chipoberfläche möglich, neuronale Zellen gezielt auf den Sensorpunkten von planaren Sensorchips wachsen zu lassen. Es wurde jedoch auch deutlich, dass es sich hierbei um ein aufwendiges und kompliziertes, mehrstufiges Verfahren handelt. Der Ausschuss an Substraten bei jedem einzelnen der drei Schritte Oberflächenmodifikation, Strukturierung und Zellkultur ist signifikant. Im Rahmen dieser Arbeit wurde daher noch ein weiterer, davon grundsätzlich verschiedener Ansatz erprobt, die in vitro Positionierung. Dabei erfolgte die Ausrichtung von Wachstumsstruktur und Chipoberfläche erst nach dem Zellwachstum auf der Struktur, also "in vitro". Die Substrate wurden dabei kopfüber, mit der bewachsenen Seite nach unten, auf die Oberfläche von Sensorchips gelegt und mit einer an einem Mikromanipulator befestigten Nadel ausgerichtet. 6.3.1 Substrate für in vitro Positionierung Als Substrate für diese Versuche wurden kleine Plättchen (2 mm x 2 mm x 0,5 mm) aus Polystyrol verwendet (Abbildung 6-7 B1). Diese waren zuvor mit einer Wafersäge aus dem Boden von großen Petrischalen aus Polystyrol mit 10 cm Durchmesser (663 102, Greiner) geschnitten worden. 6-16 6 Ausrichtung von Struktur und Substrat In Vitro Positionierung (A) Ablauf 1. Strukturierung 2. Zellkultur 3. Positionierung Nadeln an Mikromanipulatoren Medium Stempel Stempel Substrat Substrat Zellen Substrat Substrat (B) Beispiele Sensorchip (B2) Justage unter dem Mikroskop (B1) Laminin auf Polystyrolplättchen 200 µm <V17_2> 100 µm <V17_2> Abbildung 6-7 Bei in vitro Positionierung wird ein bereits strukturiertes und mit Zellen bewachsenes Substrat mittels zweier Mikromanipulatoren auf einen Sensorchip ausgerichtet. (A) Ablauf der in vitro Positionierung. (B1) Mit Laminin strukturiertes Polystyrolplättchen. (B2) Bewegung des Substrats über der Sensoroberfläche. Das Basismaterial Polystyrol wurde gewählt, um ohne zusätzliche Oberflächenmodifikation einen ausreichenden Wachstumskontrast zu erzielen. Die kleinen Abmessungen der Plättchen von 2 mm x 2 mm waren erforderlich um das Substrat in der im Durchmesser maximal 5 mm großen Öffnung von EGESensorchips plazieren und justieren zu können. 6.3.2 Versuchsablauf In Abbildung 6-7 A ist der Ablauf der in vitro Positionierung dargestellt: Im ersten Schritt werden die Polystyrolplättchen mit Laminin strukturiert. Dies geschieht analog zum Vorgehen bei der Strukturierung von normalen Substraten8. In 8 Um die Strukturierung mit den leichter zu handhabenden Makrostempeln durchführen zu können, wurden jeweils 9 Substrate zu einem Quadrat zusammengelegt. 6.3 In vitro Positionierung 6-17 Abbildung 6-7 B1 ist eine Mikroskopieaufnahme eines strukturierten Plättchens abgebildet. In Schritt 2 erfolgt die Aussaat von neuronalen Zellen. Hierzu werden die Plättchen mit der strukturierten Seite nach oben in eine mit Medium gefüllte, große Petrischale gelegt. Die Zellen sinken auf die Plättchen hinab und wachsen entlang der Lamininstrukturen am Substrat fest. In Schritt 3 erfolgt die Positionierung auf den Sensorchip. Dazu wird ein einzelnes Plättchen mit einer spitzen Pinzette aus dem Zellmedium gehoben und kopfüber in die ebenfalls mit Zellmedium gefüllte Petrischale des Sensorchips getaucht. Die Pinzette wird geöffnet und das Plättchen sinkt auf die Oberfläche des Sensorchips ab. Die eigentliche Positionierung erfolgt jetzt unter optischer Kontrolle im Mikroskop mit Hilfe von zwei, mit Nadeln (Glaspipetten) ausgestatteten Mikromanipulatoren. Der erste Mikromanipulator drückt das Plättchen von hinten auf die Sensoroberfläche. Mit der x- und y- Achse dieses Manipulators wird die laterale Position des Plättchens eingestellt. Die Nadel des zweiten Mikromanipulators wird neben dem Plättchen plaziert. Durch Randrücken dieser Nadel an die Seite des Plättchens ist es möglich dieses um die erste Nadel und damit um seine z-Achse zu drehen. In Abbildung 6-7 B2 ist in zwei Aufnahmen dargestellt wie das Plättchen mit den darauf angewachsenen Zellen bei der Positionierung über die Substratoberfläche geschoben wird. 6.3.3 Messung an Kardiomyozyten Bei der in vitro Positionierung sind die untersuchten Zellen zwischen Sensorchip und Substrat eingeschlossen. Es ist daher nicht möglich, simultan zur extrazellulären Messung die Zellen mit Patch-Clamp Pipetten zu stimulieren. Will man dennoch Aktionspotentiale messen, so muss entweder eine extrazelluläre Stimulation der Zellen seitens des Sensorchips erfolgen oder aber ein Zellsystem verwendet werden, welches auch ohne Stimulation spontane Aktivität zeigt. Für die Erprobung des Messverfahrens wurden aus diesem Grund keine Nervenzellen sondern Herzmuskelzellen (Kardiomyozyten) eingesetzt. 6-18 6 Ausrichtung von Struktur und Substrat Kardiomyozyten zeigen in Kultur spontane Aktivität. In regelmäßigen 2+ Zeitabständen kontrahieren sie sich: Ca Einstrom in die Zelle bewirkt eine langsame Depolarisation bis zum Ansprechen spannungssensitiver Na+ Kanäle. Ein Aktionspotential mit schneller Depolarisation der Zelle wird ausgelöst. Dadurch öffnen weitere, spannungssensitive Ca2+ Kanäle. Die Folge ist eine deutliche 2+ Erhöhung der Ca Konzentration im Cytoplasma, wodurch das Actomyosinsystem der Zelle aktiviert wird, die Zelle kontrahiert sich. Schließlich erfolgt die Repolarisation der Zelle durch K+ Ausstrom über ebenfalls spannungsgesteuert + geöffnete K Kanäle. Das vor jeder Kontraktion ausgebildete Aktionspotential kann extrazellulär gemessen werden [Sprössler, 1999]. Für die durchgeführten Versuche wurden Kardiomyozyten aus den Herzen von neonatalen Ratten (N1 bis N3) entsprechend dem Protokoll in Kapitel 2 präpariert. Die Zellen wurden auf homogen mit Laminin9 beschichteten Substraten (2 x 2 mm², geeignet zur in vitro Positionierung) ausgesät und inkubiert. Nach zwei bis drei Tagen in Kultur konnten unter dem Mikroskop erste rhytmische Kontraktionen der Zellschicht beobachtet werden. Messungen an den Zellen erfolgten zwischen Tag 4 und Tag 6 in Kultur. Als extrazellulärer Sensor wurde ein Chip mit 16 n-Kanal Feldeffekttransistoren eingesetzt [Sprössler, 1998; Ingebrandt, 2001]. In Abbildung 6-8 sind untereinander versetzt die Messignale abgebildet, welche von 3 Feldeffekttransistoren des Chips simultan aufgenommen wurden. In Abbildung 6-8 A erfolgte die Messung nach der Positionierung eines schlagenden Bereichs der Zellschicht über die Sensorpunkte des Chips. In Abbildung 6-8 B wurde zusätzlich Druck ausgeübt. Dies geschah, indem die zur Positionierung auf der Rückseite des Zellsubstrats aufgesetzten Glaspipetten mit den Mikromanipulatoren weiter abgesenkt wurden. In beiden Fällen erkennt man Aktionspotentiale der Herzmuskelzellen als deutliche Spitzen im Messignal. 9 Die Beschichtung wurde hergestellt durch Lagerung der Substrate in einer Lamininlösung (50 µg/ml ) für 1 Stunde. 6.3 In vitro Positionierung 6-19 Extrazelluläre Messung an in vitro positionierten Kardiomyozyten Messung Andruckkraft 4 Signalamplitude [mV] • Langzeit 2 0 -2 -4 Signalamplitude [mV] 2 • Detail (B) Starker Druck 4 6 Zeit[ms] [s] Zeit 2 0 -2 -4 2,80 Zeit[ms] [s] Zeit 2,85 2 0 -2 -4 2 4 2,75 4 8 Signalamplitude [mV] Messignale Signalamplitude [mV] (A) Schwacher Druck 2,90 4 6 Zeit[ms] [s] Zeit 8 4 2 0 -2 -4 2,85 2,90 Zeit[ms] [s] Zeit 2,95 3,00 Abbildung 6-8 Extrazellulär gemessene Signale der Aktionspotentiale von spontan schlagenden Kardiomyozyten. Messung an Tag 6 in Kultur bei 35 °C. (A) Zellsubstrat mit kleinsmöglichster Kraft auf den Sensorchip gepresst. (B) Messung bei größerer Andruckkraft. Ein Vergleich der Messignale bei schwachem Andruck (Abbildung 6-8 A) und bei starkem Andruck (Abbildung 6-8 B) ergibt folgende interessante Beobachtungen: 1. Die Periodendauer der Kontraktionen ändert sich nicht. In beiden Fällen wird ein zeitlicher Abstand zwischen zwei Aktionspotentialen von ca. 4 s gemessen. Der erhöhte Druck auf die Zellen beeinflusst also offenbar nicht deren Aktivität. 2. Die Amplitude der gemessenen Signale erhöht sich mit zunehmendem Druck drastisch. Vergleicht man die Gesamtamplitude USS vom Minimalwert des Signals zum Maximalwert, so ergibt sich eine Erhöhung um den Faktor 6. Da mit dem verfügbaren Messaufbau keine Möglichkeit zur direkten Messung der ausgeübten Kräfte bestand, ist diese Beobachtung rein qualitativer Natur. Eine Druckverstärkung der Signale konnte in mehreren Experimenten in ähnlicher Größenordnung reproduziert werden. In der Beobachtung der Zellen mit dem Mikroskop war bei den ausgeübten Kräften optisch keine Deformation der Zellen 6-20 6 Ausrichtung von Struktur und Substrat erkennbar. Insgesamt waren die gemessenen Signalamplituden auch bei geringstmöglichem Druck größer als bei Messungen an fest auf dem Sensorchip aufgewachsenen Zellen. 3. Die Form der detektierten Signale ändert sich mit dem Anpressdruck. Vergleicht man die Form der Signale in der Detaildarstellung, so ist bei geringem Druck zuerst ein kleiner positiver und dann ein größerer negativer Ausschlag zu erkennen. Bei hohem Druck hingegen beobachtet man Ausschläge mit annähernd gleicher Amplitude in beide Richtungen. Diese Beobachtung ist besonders interessant, da sich hieraus eine Möglichkeit ergibt die elektrische Kopplung zwischen Sensor und Zelle genauer zu untersuchen [Ingebrandt, 2001]. 6.3.4 Bewertung der in vitro Positionierung Es konnte gezeigt werden, dass eine in vitro Positionierung wie oben beschrieben möglich ist. Die Zellen werden nicht zwischen dem Polystyrolplättchen und der Sensoroberfläche zerquetscht, sondern können unbeschädigt auf die Messpunkte eines Chips ausgerichtet werden. Es muss allerdings darauf geachtet werden, eine Verschiebung des Plättchens über größere Strecken immer nur mit der für die Bewegung gerade noch ausreichenden Andruckkraft auszuführen. Ist die Andruckkraft bei einer Verschiebung zu groß, so werden einzelne Zellen von der Substratoberfläche geschert. Sind die Zellen spontan elektrisch aktiv, so sind ihre extrazellulären elektrischen Signale mit Sensorchips messbar. Die erzielbaren Signalamplituden eingekoppelter Aktionspotentiale sind dabei deutlich größer als bei Zellen, die direkt auf der Oberfläche eines Sensorchips kultiviert werden. Das Verfahren eignet sich daher insbesondere auch für extrazelluläre Messungen an Netzwerken aus Nervenzellen. Die Aktionspotentiale von Nervenzellen sind gewöhnlich deutlich schwächer als bei Kardiomyozyten. Die hinzugewonnene Sensitivität des Sensors bei in vitro Positionierung ist daher von entscheidender Bedeutung. Weitere Vorteile der in vitro Positionierung sind: 6.3 In vitro Positionierung 6-21 − Es besteht die Möglichkeit über die Andruckkraft die Kopplung zwischen Zelle und Sensor zu beeinflussen. Dadurch wird es möglich, das gegenwärtig allgemein anerkannte Modell der Kopplung zwischen Zelle und extrazellulärem Sensor, das Punktkontaktmodell [Fromherz, 1999], bei kontinuierlich variierter Kopplung experimentell zu untersuchen. − Es ist keine Oberflächenmodifikation des Sensorchips erforderlich. − Die Strukturierung der Kultursubstrate kann mit gewöhnlichen Mikrostempeln erfolgen, ohne einen speziellen Aufbau zur Ausrichtung. − Viele Substrate können mit einem einzigen Sensorchip gemessen werden. − Es ist ohne großen Aufwand möglich eine sehr große Zahl strukturiert bewachsener Substrate herzustellen. Aus diesen können dann unmittelbar vor der Messung die besten ausgewählt werden. Es gibt jedoch aber auch zwei entscheidende Nachteile: − Durch den Einschluss der Zellen zwischen Polystyrolsubstrat und Chipoberfläche ist kein direkter Zugang zu den Zellen möglich. Eine gezielte Stimulation oder Signalableitung der Zellen durch Patch-Clamp Messung mit Mikropipetten ist daher ausgeschlossen. Aus diesem Grund wurden elektrische Messungen im Rahmen dieser Arbeit ausschließlich an spontan aktiven Kardiomyozyten durchgeführt. − Für die Messung sind immer zwei Mikromanipulatoren und ein Mikroskop zur Justage erforderlich, ein sehr aufwendiger und kompliziert zu bedienender Messaufbau. Wird der Sensorchip hingegen vor der Zellaussaat ausgerichtet strukturiert, so können alle weiteren Schritte einschließlich der Messung auch in einem technisch einfacher ausgestatteten Zellabor durchgeführt werden. Die in vitro Positionierung ist eine sehr vielversprechende neue Technik zur extrazellulären Messung der Signale von elektrisch aktiven Zellen. Insbesondere in Kombination mit extrazellulärer Stimulation ergeben sich interessante neue Möglichkeiten zur Untersuchung neuronaler Netzwerke. 6-22 6 Ausrichtung von Struktur und Substrat 7-1 7 Zusammenfassung und Ausblick Das Wachstum von Nervenzellen und deren Verbindungen im zentralen und peripheren Nervensystem wird durch Proteine der extrazellulären Matrix kontrolliert [Sanes, 1989]. Artifizielle Strukturen dieser Proteine auf künstlichen Substratoberflächen beeinflussen daher auch neuronales Wachstum in vitro [Letourneau, 1975]. In dieser Arbeit wurde das Protein Laminin verwendet, um damit Netzwerke von Nervenzellen auf strukturierten Oberflächen zu erzeugen. Entsprechende Proteinstrukturen wurden dazu mit Polymerstempeln aus Polydimethylsiloxan auf Zellkultursubstrate übertragen. Die Fertigung der Stempel erfolgte in einem mehrstufigen Verfahren durch Abformung von photolithographisch hergestellten Masken. Als Standardsubstrat wurde Polystyrol verwendet. Dieses ist ausreichend hydrophob, um ohne Oberflächenmodifikation ein Anhaften von neuronalen Zellen zu verhindern. Übertragenes Laminin haftet durch Physisorption an der Oberfläche dieser Substrate. In Experimenten mit chemisch reaktiven Oberflächen aus Aktivesterbürsten konnte darüber hinaus eine kovalente Anbindung von Laminin an die Oberfläche erzielt werden. In systematischen Wachstumsversuchen mit neuronalen Zellen der Zellinien MzN und P19 wurden geeignete Strukturen für die gezielte Steuerung neuronalen Wachstums identifiziert. Mit einer Geometrie von 20 µm breiten Adhäsionspunkten und 4 µm breiten Linien ist es gelungen, mehr als 85 % der Zellen in einer Kultur auf vorgegebene Positionen an der Substratoberfläche zu fokussieren und entlang definierter Pfade zu verbinden. Auf Basis dieser Ergebnisse wurden Linien- und Gitternetzwerke sowie komplexere Schaltungen von Nervenzellen realisiert. Für systematische elektrophysiologische Messreihen wurden Nervenzellen aus Hirnschnitten verwendet. In dieser Form von Primärkultur sollten durch Erhalt des intakten Zellverbundes im Schnitt Kulturbedingungen möglichst nahe zur Situation 7-2 7 Zusammenfassung und Ausblick in vivo geschaffen werden, um die Bildung von Synapsen zu begünstigen. Bei geeigneter Plazierung der Schnitte auf zuvor strukturierten Substratoberflächen, konnte ein Auswachsen einzelner Nervenzellen entlang der Proteinstrukturen erzielt werden. Mit denselben Strukturen, die bei Versuchen mit dissoziierten Neuronen (MzN und P19) eine Kontrolle des Wachstums ermöglicht hatten, gelang es auch in diesem Fall geordnete Netzwerke herzustellen. Einzig Strukturen mit unterbrochenen Linien erwiesen sich als ungeeignet, da die Lücken in den Linien ein schnelles Auswachsen von Nervenzellen aus dem Hirnschnitt behinderten. Netzwerke aus Nervenzellen dieser Hirnschnitt-Kulturen wurden mit der PatchClamp Technik elektrophysiologisch untersucht und mit Nervenzellen auf unstrukturierten Kontrollsubstraten verglichen. Eine deutliche Verringerung der + + Dichte an aktiven Na - und K - Ionenkanälen um einen Faktor 2-3 wurde dabei auf strukturierten Substraten festgestellt. Antikörperfärbungen mit anti-GFAP zeigten einen verstärkten Überwuchs der Nervenzellen mit Gliazellen. Dies ist eine mögliche Ursache für die abgeschwächten Ionenströme. In simultanen Patch-Clamp Messungen, an bis zu drei Neuronen gleichzeitig, gelang der eindeutige elektrophysiologische Nachweis synaptischer Kopplung in den erzeugten Netzwerken. Sowohl funktionale synaptische Kopplung über chemische Synapsen, als auch Ohm'sche Kopplung über Gap-Junctions wurde beobachtet. Funktionale Kopplung wurde allerdings nur sehr selten detektiert, Ohm'sche Kopplung hingegen in einer für statistische Analysen ausreichenden Zahl. Aus den Messergebnissen Ohm'scher Kopplung wurde ein elektrisches Kopplungsmodell abgeleitet. Die Analyse der Messdaten ergab, dass die Signalleitung in den Nervenfasern der erzeugten Netzwerke wie in einem zylindrischen, durch die Zellmembran von der Umgebung isolierten Kabel erfolgt. Dieses Kabelmodell entspricht dem von Axonen mit größerem Durchmesser [Hodgkin, 1946]. Für dieses Kabelmodell ist ein exponentielles Abklingen der Übertragungsfunktion mit der Länge des Kabels charakteristisch. Dieses Verhalten konnte auch in den durchgeführten Versuchen nachgewiesen werden. Zur Bestimmung des exakten Verbindungswegs und dessen Länge, wurde dabei das Cytoplasma der untersuchten Nervenzellen durch Mikroinjektion von 7-3 Fluoreszenzfarbstoff angefärbt. Auch ein Zusammenhang zwischen Breite der vorgegebenen Verbindungspfade aus Laminin und den Eigenschaften des Kabels konnte nachgewiesen werden. Die Signalübertragung durch Kabel entlang von dicken, 6 µm breiten Pfaden war signifikant stärker als entlang von dünneren Pfaden mit 2 µm oder 4 µm Breite. Die Breite des Lamininpfades beeinflusst offenbar die Dicke des darauf wachsenden Axons. Für eine quantitative Interpretation des Zusammenhangs waren jedoch nicht genügend Messdaten verfügbar. Insgesamt konnte im Rahmen dieser Arbeit gezeigt werden, dass mit Hilfe von Mikrostrukturen aus Laminin synaptisch verschaltete Netzwerke von Nervenzellen in vitro erzeugt werden können. Die Neuronen bilden darin Ohm'sche und funktionale Synapsen aus. Strukturierte neuronale Zellkultur eignet sich daher insbesondere auch für das Studium synaptischer Plastizität in einem vorgegebenen Netzwerk. Interessant erscheint eine Kombination dieses Ansatzes mit extrazellulären elektrophysiologischen Messverfahren, realisiert durch Sensorchips auf Basis von Halbleitersensoren [Fromherz, 1991; Sprössler, 1999], oder Elektrodenfeldern [Gross, 1977; Krause, 2000]. Beide Systeme ermöglichen eine nicht-invasive Erfassung der Signale vieler hundert Nervenzellen gleichzeitig. Langzeitstudien synaptischer Plastizität in definierten Netzwerken von Nervenzellen sind auf diesem Wege denkbar. Dies eröffnet die Möglichkeit zu einer gezielten experimentellen Überprüfung von Modellen für Lernen und Gedächtnis. Innerhalb dieser Arbeit wurden bereits wesentliche Voraussetzungen für derartige Versuche geschaffen. Ein Positioniersystem zur exakten Ausrichtung von Mikrostempeln auf die Oberflächenstruktur der Sensorchips wurde entwickelt. Lamininstrukturen können dadurch mit 1 µm Positioniergenauigkeit auf die Messpunkte an der Chipoberfläche übertragen werden. 7-4 7 Zusammenfassung und Ausblick Entscheidende Hürden für gezielte quantitative Experimente sind derzeit jedoch noch die geringe Sensitivität der extrazellulären Sensoren sowie die extrem geringe Ausbeute an funktionalen Synapsen in den erzeugten Netzwerken. Die erste Hürde wird durch weitere Optimierung des physikalischen Chipdesigns in naher Zukunft voraussichtlich deutlich kleiner werden. Wissenschaftliche Arbeiten mit diesem Ziel sind Gegenstand aktueller Forschung [Ingebrandt, 2001]. Eine Steigerung der Anzahl funktionaler Synapsen ist mit großer Wahrscheinlichkeit durch weitere Optimierung des Zellkulturprotokolls möglich. Ein Wechsel von Hirnschnitten zu einer dissoziierten Primärkultur in zukünftigen Experimenten ist ein möglicher Ansatzpunkt. Beispielsweise könnten dissoziierte Neurone in Kokultur zu einem Hirnschnitt auf dem gleichen Substrat ausgesät werden, um so die Vorteile beider Zellsysteme zu kombinieren. Ein wesentlicher Vorzug von dissoziierten Nervenzellen gegenüber Hirnschnitten in diesem Zusammenhang ist, dass für die Substratstrukturierung auch komplexe Strukturen mit Lücken verwendet werden können. Mit derartigen Strukturen ist es bereits gelungen, gezielt neuronale Polarisation zu induzieren [Stenger, 1998; Lauer, 2001]. Nur Nervenzellen mit definierter axonaler und dendritischer Polarität können funktionale Synapsen ausbilden. Die Voraussetzungen zur Synapsenbildung könnten mit entsprechenden Strukturen also gezielt gefördert werden. Geeignete Strukturen wurden in dieser Arbeit bereits anhand von axonalen und dendritischen Antikörperfärbungen an Neuronen der Zellinie MzN identifiziert. 8-1 8 Anhang 8.1 Mikrostrukturen Insgesamt 37 verschiedene Mikrostrukturen wurden zur Strukturierung von Zellkultursubstraten in dieser Arbeit eingesetzt. Eine vollständige Übersicht aller Strukturen ist in Tabelle 8-1 wiedergegeben. In den Abbildungen in Spalte 4 der Tabelle ist dazu jeweils ein charakteristischer Auschnitt der zugrundeliegenden technischen Zeichnung dargestellt. Durch periodische Vervielfältigung des darin abgebildeten Strukturelements in x- und y- Richtung wird die gesamte Strukturfläche von jeweils 1 cm² ausgefüllt. Alle Strukturen basieren auf einem Raster von 100 x 50 µm. Tabelle 8-1 Auflistung aller für die Herstellung von Mikrostempeln eingesetzten Strukturen. Die jeweilige Strukturnummer in Spalte "ID" wird an verschiedenen Stellen dieser Arbeit zur eindeutigen Identifizierung der Strukturen verwendet. ID Struktur Abmessungen Technische Zeichnung 1 Linien Linien: 2 µm Abstand: 100 µm 2 Linien Linien: 4 µm Abstand: 100 µm 3 Linien Linien: 6 µm Abstand: 100 µm 4 Gitter Linien: 2 µm Maschen: 100 x 50 µm 8-2 8 Anhang 5 Gitter Linien: 4 µm Maschen: 100 x 50 µm 6 Gitter Linien: 6 µm Maschen: 100 x 50 µm 7 Gitter mit Knoten Linien: 2 µm Knoten: 10 µm Maschen: 100 x 50 µm 8 Gitter mit Knoten Linien: 4 µm Knoten: 12 µm Maschen: 100 x 50 µm 9 Gitter mit Knoten Linien: 6 µm Knoten: 14 µm Maschen: 100 x 50 µm 10 Linien mit Knoten Linien: 2 µm Knoten: 20 µm Linienabstand: 50 µm Knotenabstand: 100 µm 11 Linien mit Knoten Linien: 4 µm Knoten: 22 µm Linienabstand: 50 µm Knotenabstand: 100 µm 12 Linien mit Knoten Linien: 2 µm Knoten: 10 µm Linienabstand: 50 µm Knotenabstand: 100 µm 8.1 Mikrostrukturen 13 Gitter mit Knoten 8-3 Linien: 2 µm Knoten: 20 µm Maschen: 100 x 50 µm 14 Gitter mit Knoten Linien: 4 µm Knoten: 22 µm Maschen: 100 x 50 µm 15 Gitter mit Knoten Linien: 6 µm Knoten: 24 µm Maschen: 100 x 50 µm 16 Gitter mit Knoten und Linien: 2 µm Lücken Knoten: 10 µm Lücken: 5 µm Maschen: 100 x 50 µm 17 Gitter mit Knoten und Linien: 2 µm Lücken Knoten: 10 µm Lücken: 10 µm Maschen: 100 x 50 µm 18 Gitter mit Knoten und Linien: 2 µm Lücken Knoten: 10 µm Lücken: 20 µm Maschen: 100 x 50 µm 19 Gitter mit Knoten und Linien: 2 µm unterbrochenen Knoten: 10 µm Linien Unterbrechungen: 5 µm Maschen: 100 x 50 µm 8-4 8 Anhang 20 Gitter mit Knoten und Linien: 2 µm unterbrochenen Knoten: 10 µm Linien Unterbrechungen: 2 µm Maschen: 100 x 50 µm 21 Gitter mit Knoten und Linien: 2 µm unterbrochenen Knoten: 10 µm Linien Unterbrechungen: 1 µm Maschen: 100 x 50 µm 22 Gitter mit Knoten und Linien: 4 µm Lücken Knoten: 22 µm Lücken: 5 µm Maschen: 100 x 50 µm 23 Gitter mit Knoten und Linien: 4 µm Lücken Knoten: 22 µm Lücken: 10 µm Maschen: 100 x 50 µm 24 Gitter mit Knoten und Linien: 4 µm Lücken Knoten: 22 µm Lücken: 20 µm Maschen: 100 x 50 µm 25 Gitter mit Knoten und Linien: 4 µm unterbrochenen Knoten: 22 µm Linien Unterbrechungen: 5 µm Maschen: 100 x 50 µm 26 Gitter mit Knoten und Linien: 4 µm unterbrochenen Knoten: 22 µm Linien Unterbrechungen: 2 µm 8.1 Mikrostrukturen 8-5 Maschen: 100 x 50 µm 27 Gitter mit Knoten und Linien: 4 µm unterbrochenen Knoten: 22 µm Linien Unterbrechungen: 1 µm Maschen: 100 x 50 µm 28 Reihenschaltung mit Linien: 2 µm unterbrochenen Knoten: 20 µm Linien Unterbrechungen: 5 µm Linenabstand: 100 µm Knotenabstand: 100 µm 29 Paralellschaltung mit Linien: 2 µm unterbrochenen Knoten: 20 µm Linien Unterbrechungen: 5 µm Linenabstand: 100 µm Knotenabstand: 100 µm 30 Kaskade Linien: 2 µm (zunehmend) mit Knoten: 20 µm unterbrochenen Unterbrechungen: 5 µm Linien Linenabstand: 50 µm Knotenabstand: 100 µm 31 Kaskade Linien: 2 µm (abnehmend) mit Knoten: 20 µm unterbrochenen Unterbrechungen: 5 µm Linien Linenabstand: 50 µm Knotenabstand: 100 µm 8-6 8 Anhang 32 Rückkopplung Linien: 2 µm (enge Schleife) mit Knoten: 20 µm unterbrochenen Unterbrechungen: 5 µm Linien Linenabstand: 50 µm Knotenabstand: 100 µm 33 Rückkopplung (weite Linien: 2 µm Schleife) mit Knoten: 20 µm unterbrochenen Unterbrechungen: 5 µm Linien Linenabstand: 100 µm Knotenabstand: 100 µm 34 Reihenschaltung Linien: 2 µm Knoten: 20 µm Linenabstand: 100 µm Knotenabstand: 100 µm 35 Paralellschaltung Linien: 2 µm Knoten: 20 µm Linenabstand: 100 µm Knotenabstand: 100 µm 36 Kaskade Linien: 2 µm (abnehmend) Knoten: 20 µm - Teil 1: Knoten Linenabstand: 50 µm Knotenabstand: 100 µm 37 Kaskade Linien: 2 µm (abnehmend) Unterbrechungen: 5 µm – Teil 2: Verbindungen Linenabstand: 50 µm 8.2 Glossar 8-7 8.2 Glossar Ach Acetylcholin. AchR Acetylcholinrezeptor. AP Aktionspotential. Elektrischer Puls eines Neurons, ausgelöst durch Erregung des Neurons über einen Schwellenwert. Axon Einzelner, langer und dünner Fortsatz eines Neuron mit der Fähigkeit zur aktiven, verlustfreien Weiterleitung eines Aktionspotentials. BNC Normierte Anschlussnorm für Hochfrequenzverbindungen. CAD Computer Aided Design; Grafiksystem zur Erstellung technischer Zeichnungen, wie z.B. Autocad. Cell-Attached Patch-Clamp Konfiguration zur Messung einzelner Ionenkanäle. Clipping Übersteuerung eines Verstärkers. Kann ein elektronisches Verstärkerbauteil ein Signal innerhalb seines begrenzten Dynamikumfangs nicht mit voller Amplitude übertragen, so werden die Spitzen des Signals gekappt. Man spricht in diesem Zusammenhang von Clipping. Dendriten Stark verzweigte Fortsätze eines Neurons. Empfangen Signale und leiten diese passiv zur Zelle weiter. DIC Tag in Kultur (engl.: Day in Culture). ECM Extrazelluläre Matrix (engl.: Extra Cellular Matrix). FET Sensorchip zur extrazellulären Signalmessung basierend auf Feldeffekttransistoren. Gigaseal Abdichtwiderstand im Bereich einiger Gigaohm. 8-8 8 Anhang In vitro Am lebenden Präparat außerhalb des Organismus. In vivo Am lebenden Organismus. MEA Multi-Elektroden-Array. Sensorchip zur extrazellulären Signalmessung basierend auf Goldelektroden. NGF Nerve Growth Factor. Wachstumsfaktor welcher das Überleben neuronaler Zellen unterstützt. Passagieren Ablösen von Zellen von Kulturgefäßen und verdünnte Neuaussaat zur weiteren Kultivierung. Patch Membranfragment einer Zelle. PNS Periphäres Nervensystem. SAC Stretch Activated Cation Channels. Durch Druck oder Zug geöffnete Ionenkanäle. Sub-Thresehold Stimulation einer Nervenzelle unterhalb der Auslöseschwelle (engl. thresehold) für ein Aktionspotential. UV Ultraviolette Strahlung. Whole-Cell Patch-Clamp Konfiguration zur Messung aller Ionenkanäle der gesamten Zellmembran. ZNS Zentrales Nervensystem. 8.3 Geräteverzeichnis 8-9 8.3 Geräteverzeichnis 8.3.1 Laborgeräte Glaselektroden-Puller Horizontaler Pipettenpuller. P-97, Sutter Instrument Company, USA. Inkubator Inkubator, CO2-Begasung. BB 6060, Heraeus Instruments GmbH, D-63450 Hanau. Oszilloskop Speicheroszilloskop. 9400 A, LeCroy, Schweiz. Sterilbank Sterilarbeitsbank. Lamin Air HB 2436, Heraeus Instruments GmbH, D-63450 Hanau. Gewebsschnittmaschine 8.3.2 Tissue Chopper, McIlwain, England. Mikrostempel-Ausrichtung Positioniereinheit FINEPLACER-145 "PICO", FINETECH electronics, D10247 Berlin. Stereolupe Stemi 2000, 6.5 – 45x, Carl Zeiss GmbH, D-60335 Frankfurt. Verschiebetisch 8.3.3 MVT 40B & WVT 40, OWIS, D-79219 Staufen i. Br. Dreifach Patch Clamp Messaufbau Framegrabber Meteor II, Matrox Electronic Systems GmbH, D-82008 Unterhaching. Manipulatoren Mikromanipulatoren, Schrittmotor gesteuert in XYZRichtung für alle drei Patch-Clamp Verstärkerköpfe, sowie für den Mikroskop-Fokustrieb. Mini 25, Luigs&Neumann GmbH, D-40880 Ratingen. Mikroskop 1 Olympus IX-50, Olympus, D-20097 Hamburg. 8-10 Mikroskop 2 8 Anhang Zoom 100 D, EHD Physikalische Technik GbR, D-49401 Damme. Patch Clamp Verstärker Whole Cell / Patch-Clamp Amplifier. Modell: EPC-9/3, Steuersoftware: TIDA 4.11. HEKA Elektronik, Dr. Schulze GmbH, D-67466 Lambrecht/Pfalz. Videokamera 1 Sony XC-333 P, Sony Deutschland GmbH, D-50829 Köln. Videokamera 2 Sony XC-003 P, Sony Deutschland GmbH, D-50829 Köln 8.4 Software, Materialien und Rezepturen 8-11 8.4 Software, Materialien und Rezepturen 8.4.1 Software Autocad CAD System zur Arfertigung technischer Zeichnungen. Autocad LT V2.0, Autodesk, Deutschland. MIL Treiberpaket zur Ansteuerung der MatroxFramegrabberkarten über Visual Basic. MIL Lite, Matrox Electronic Systems GmbH, D-82008 Unterhaching. MSAccess Relationale Datenbank. MSAccess '97, Microsoft, USA. PSpice Softwarepaket zur Simulation elektronischer Schaltungen. PSpice Mixed Mode Simulator, Version 6.0, MicroSim Corporation, USA. Tida Steuer- und Messprogramm für den Betrieb des PatchClamp Verstärkers. Tida 4.11, HEKA Elektronik, Dr. Schulze GmbH, D-67466 Lambrecht/Pfalz. Visual Basic Graphische Entwicklungsumgebung auf Basis der Programmiersprache Basic. Visual Basic, Microsoft, USA. 8.4.2 Technische Materialien 24-Well Platten MultiwellTM Gewebekulturplatte. Falcon 3047, Becton Dickinson & Company, USA. 75 ml Flaschen zur Zellkultur. Falcon 3111, Becton Dickinson, D-69126 Heidelberg. 8-12 Eppendorf Röhrchen 8 Anhang 1,5 ml Eppendorf Röhrchen zur Stempelfertigung. 0030 121.589, Eppendorf – Netheler – Hinz GmbH, D-22331 Hamburg. Glaselektroden Borosilicatglaskapilare mit Filament. Länge L = 100 mm, Aussendurchmesser DA = 1,5 mm Innendurchmesser DI = 0,87 mm, Wandstärke s = 315 µm, Filamentdurchmesser DF = 315 µm. 1810016, Hilgenberg, D-34323 Maisfeld. Glaswafer 0,6 mm dicke Glasscheiben im Durchmesser von 5". MEMC Electronic Materials, USA. Hellmanex Reinigungsmittel für Ultraschallreinigung. 320001 Hellmanex II, Hellma GmbH & Co, D-79379 Müllheim. Milli-Q Wasser -1 Reinstwasser, 18.2 MQ cm . Millipore GmbH, D-65760 Eschborn. PDMS Polydimethylsiloxan. Transparenter Polymerwerkstoff auf zwei-Komponenten-Basis. Sylgard 182, Dow Corning, bezogen über Sasco Semiconductor, D-85640 Putzbrunn. Photolack Positiv Photolack. AZ 4562, Clariant GmbH, D-65843 Sulzbach. Polystyrol Substrate Petrischalen aus Polystyrol mit 3 cm Durchmesser. 627 102, Greiner Labortechnik, D-72636 Frickenhausen. Petrischalen aus Polystyrol mit 10 cm Durchmesser. 663 102, Greiner Labortechnik, D-72636 Frickenhausen. SDS Sodium Dodecyl Sulfate. SDS 71729, Fluka (Sigma-Aldrich GmbH), D-82041 Deisenhofen. 8.4 Software, Materialien und Rezepturen Zellkulturflaschen 8-13 25 ml Flaschen zur Zellkultur. Falcon 3108, Becton Dickinson, D-69126 Heidelberg. 8.4.3 Chemikalien allgemein Benzophenonsilan (4-β'-Chlorodimethylsilyl)propyloxybenzophenon C5AE Aktivester angebunden als Kopfgruppe an eine Kette von 5 Kohlenstoffatomen. Monomer synthetisiert nach einem Protokoll von S. Golze. CH2Cl2 Dichlormethan. DMAA Dimethylacrylamid. Monomer synthetisiert nach einem Protokoll von S. Golze. DMF Organisches Lösungsmittel. ETOH Ethanol. H2SO4 Schwefelsäure. HCl Salzsäure. Initiator Initiatoren für die Anbindung von Polystyrol und C5AEDMAA-Copolymerbürsten an Siliziumoberflächen. Synthetisiert nach Protokollen von S. Golze. NaOH Natronlauge KOH Kalilauge Toluol Organisches Lösungsmittel. Triethylamin Katalysator zur Bindung freier Salzsäure. 8.4.4 Accutase Chemikalien zur Zellkultur Enzym zur Ablösung und Vereinzelung von Zellen. L11-007, PAA, D-35091 Cölbe. (Verwendet anstelle von Trypsin für besonders empfindliche Zellen) 8-14 ARA-C 8 Anhang Cytosine ß-D-Arabinofuranoside. C-6645, Sigma-Aldrich GmbH, D-82041 Deisenhofen. ATP Adenosin 5'-Triphosphate. A 8937, Sigma-Aldrich GmbH, D-82041 Deisenhofen. B-27 B-27 Supplement. 17504-044, Life Technologies GmbH, D-76131 Karlsruhe. cAMP Cyclic Adenine Mono Phosphate. 104 396, Roche Diagnostics GmbH, D-68305 Mannheim. (100 mM Stammlösung, gelagert bei -20 °C) DMEM/F12 Dulbecco's Modifiziertes Eagle Medium / Nutrient Mix F12 (1:1). 21331-012, Life Technologies GmbH, D-76131 Karlsruhe. DNAse Typ II DNAse Typ II. Geliefert in 40.000 Gebrauchseinheiten. D 4527, Sigma-Aldrich GmbH, D-82041 Deisenhofen. EDTA Ethylendiamine Tetra-Acetic Acid. E 6758, Sigma-Aldrich GmbH, D-82041 Deisenhofen. EGTA Ethyleneglycol-bis(β-Aminoethyl Ether)-Tetra-Acetic Acid. E 4378, Sigma-Aldrich GmbH, D-82041 Deisenhofen. FCS Fetal Calf Serum. F-7524, Sigma-Aldrich GmbH, D-82041 Deisenhofen. Glutamax I 200 mM L-Alanyl-L-Glutamine. 35050-038, Life Technologies GmbH, D-76131 Karlsruhe. Gluconsäure Gluconic Acid. G 4500, Sigma-Aldrich Chemie GmbH, D-82039 Deisenhofen. 8.4 Software, Materialien und Rezepturen Glucose 8-15 D-(+)-Glucose. G 7021, Sigma-Aldrich Chemie GmbH, D-82039 Deisenhofen. Glutaminsäure Glutaminsäure. 12419-016, Life Technologies GmbH, D-76131 Karlsruhe. HAMS F10 Zellkulturmedium. 21955-018, Life Technologies GmbH, D-76131 Karlsruhe. HAMS F12 Zellkulturmedium. N-1387, Sigma-Aldrich GmbH, D-82041 Deisenhofen. HAT HAT Supplement; Hypoxanthin 5mM, Aminopterin 20 µM, Thymidin 0,8 mM. 21060-017, Life Technologies GmbH, D-76131 Karlsruhe. HBSS Hanks Balanced Salt Solution; Ca- und Mg- frei. 14185-045, Life Technologies GmbH, D-76131 Karlsruhe. ITS Insulin-, Transferrin-, Selenit-Lösung 1884, Sigma-Aldrich GmbH, D-82041 Deisenhofen. L-Glutamin L-Glutamin. G-1573, Sigma-Aldrich GmbH, D-82041 Deisenhofen. MEM Minimal Essential Medium. Neurobasal Alpha Medium 10888-022, Life Technologies GmbH, D-76131 Karlsruhe. (enthält Ribonukleoside, Desoxiribonukleoside und Glutamax I) Na-Pyruvat 100mM Natriumpyruvat 11360-039, Life Technologies GmbH, D-76131 Karlsruhe. NGF Nerve Growth Factor 7S. 13290-010, Life Technologies GmbH, D-76131 Karlsruhe. 8-16 8 Anhang PBS Mit Phosphat gepufferte Salzlösung (engl.: Phosphate Buffered Saline). Die mit deionisiertem Wasser hergestellte Lösung enthält 2,7 mM Kaliumchlorid und 138 mM Natriumchlorid bei pH 7,4 . 14040-083, Life Technologies GmbH, D-76131 Karlsruhe. P/S Penicillin / Streptomycin Lösung. P11-010, PAA, D-35091 Cölbe. (Einsatz in Verdünnung 1:100) PFA Paraformaldehyd, reinst. 1.04005.1000, Merck, D-64293 Darmstadt. Pipes Pipes. P 8658, Sigma-Aldrich GmbH, D-82041 Deisenhofen. RA All-Trans Retinoic Acid. R-2625, Sigma-Aldrich GmbH, D-82041 Deisenhofen. (1 mM Stammlösung hergestellt aus 50 mg RA, gelöst in 156 ml 100% ETOH und 8 ml Milli-Q. Bei 4°C mehrere Monate haltbar.) Triton-X-100 Permeabilisierungsmittel für Antikörperfärbungen. T-9284, Sigma-Aldrich GmbH, D-82041 Deisenhofen. Trypsin Trypsin Typ III. T-8253, Sigma-Aldrich GmbH, D-82041 Deisenhofen. 8.4.5 Antikörper und Fluoreszenzfarbstoffe anti-MAP-2 Antikörper gegen das Mikrotubuli assoziierte Protein 2. Geeignet zur selektiven Markierung neuronaler Zellen und deren Dendriten von Mensch, Ratte, Rind, Maus und Huhn. MsIgG1, MAB 3418, Chemicon, D-65719 Hofheim. (Gelagert bei –20 °C in einer Konzentration von 200 µg/ml. Zur Anwendung 1:20 auf 10 µg/ml verdünnt.) 8.4 Software, Materialien und Rezepturen anti-Tau 8-17 Antikörper gegen das Mikrotubuli assoziierte Protein Tau. Geeignet zur selektiven Anfärbung von Axonen bei Zellen von vielen Säugetieren und Vögeln. IgG vom Kaninchen, T 6402, Sigma-Aldrich GmbH, D82041 Deisenhofen. (Gelagert bei –20 °C. Zur Anwendung 1:100 verdünnt.) anti-GFAP Antikörper gegen Glial Fibrillary Acidic Protein. Geeignet zur selektiven Anfärbung von Astrozyten und Bergman-Gliazellen aus Mensch, Schwein, Huhn und Ratte. MsIgG1, MAB 3402, Chemicon, D-65719 Hofheim. (Gelagert bei 4°C in einer Konzentration von 20 µg/ml. Zur Anwendung 1:10 auf 2 µg/ml verdünnt.) F(ab)2-Esel anti Maus Cy-3 (rot) fluoreszenzmarkierter Sekundärantikörper gegen Primärantikörper der Maus. 715-096-150, Dianova, D-20148 Hamburg. (Gelagert bei +4 °C. Zur Anwendung 1:200 verdünnt.) F(ab)2-Esel anti Kaninchen FITC (grün) fluoreszenzmarkierter Sekundärantikörper gegen Primärantikörper des Kaninchens. 711-166-152, Dianova, D-20148 Hamburg. (Gelagert bei +4°C. Zur Anwendung 1:100 verdünnt.) MsIgG1 MsIgG1 Purified Immunoglobulin. Verwendet zur Isotypkontrolle bei MsIgG1 Primärantikörpern. CBL600, Dianova, D-20148 Hamburg. (Gelagert bei 4 °C in einer Konzentration von 100 µg/ml. Zur Anwendung jeweils passend verdünnt.) Kaninchen Gamma-Globulin Verwendet zur Isotypkontrolle bei IgG Primärantikörpern des Kaninchens. 011-000-002, Dianova, D-20148 Hamburg. 8-18 8 Anhang (Gelagert bei 4 °C in einer Konzentration von 100 µg/ml. Zur Anwendung jeweils passend verdünnt.) Lucifer Yellow Gelbes Lithiumsalz (Absorption: 375 nm, Emission: 525 nm). Polarer Farbstoff zur Anfärbung des Cytoplasmas durch Mikroinjektion. L-453, Molecular Probes, bezogen über Mo Bi Tec GmbH, D-37083 Göttingen. Sulforhodamine Sulforhodamine (Emission: 610 nm). Polarer Farbstoff zur Anfärbung des Cytoplasmas durch Mikroinjektion. S-359, Molecular Probes, bezogen über Mo Bi Tec GmbH, D-37083 Göttingen. Cascade Blue Blaues Natriumsalz (Absorption: 370 nm, Emission: 420 nm). Polarer Farbstoff zur Anfärbung des Cytoplasmas durch Mikroinjektion. C-687, Molecular Probes, bezogen über Mo Bi Tec GmbH, D-37083 Göttingen. 8.4.6 Proteine zur Mikrostrukturierung Laminin 1243217, Roche Diagnostics GmbH, D-68305 Mannheim. (1 ml Stammlösung mit 500 µg/ml. Lagerung bei –20 °C über mehrere Monate möglich. Zum Gebrauch bis max. 8 Wochen bei 4 °C gelagert.) Fibronektin Fibronectin from bovine plasma. (lat.: fibra=Faser, nectere=verbinden) F 1141, Sigma-Aldrich GmbH, D-82041 Deisenhofen. Fibronectin Adhesion promoting Peptide. F 3667, Sigma-Aldrich GmbH, D-82041 Deisenhofen. (Stammlösung mit 100 µg/ml hergestellt durch Lösen von 5 mg in 50 ml PBS. Lagerung in 2 ml Portionen bei -20 °C.) 8.4 Software, Materialien und Rezepturen Poly-L-lysine 8-19 Poly-L-lysine (70-150 kDa). P 6282, Sigma-Aldrich GmbH, D-82041 Deisenhofen. (Stammlösung mit 100 µg/ml hergestellt durch Lösen von 10 mg in 100 ml PBS. Lagerung in 2 ml Portionen bei -20 °C.) 8.4.7 Versuchstiere ® CD -Ratte E15-E18 trächtiges weibliches Versuchstier für die Präparation embryonaler Neurone und Herzmuskelzellen. CD®-Ratte, Charles River Wiga (Deutschland GmbH, D97633 Sulzfeld. 8.4.8 Rezepturen Laminin Lösung 50 µl Laminin (0,5 mg/ml) (25 µg/ml) 1 ml PBS Fibronektin Lösung 1 ml Fibronektin Stamnmlösung (100 µg/ml) (25 µg/ml) 3 ml PBS Poly-L-lysine Lösung 1 ml Poly-L-lysine Stammlösung (100 µg/ml) (25 µg/ml) 3 ml PBS Intrazelluläre 125,0 mM 2,928 g Kaliumgluconat Patchelektrodenlösung 20,0 mM 0,149 g Kaliumchlorid (Mengen in g für 100 ml) 0,5 mM 0,006 g Kalziumdichlorid 2,0 mM 0,019 g Magnesiumdichlorid 5,0 mM 0,190 g EGTA 10,0 mM 0,238 g HEPES 4,0 mM 2,332 g ATP pH-Wert eingestellt auf pH 7,3 mit KaOH; Osmolarität: 300 ± 10 mOs/kg. 8-20 Extrazelluläre 8 Anhang 150,0 mM 8,77 g Natriumchlorid Patchelektrodenlösung 5,0 mM 0,37 g Kaliumchlorid (Mengen in g für 1 l) 1,0 mM 0,10 g Magnesiumdichlorid 2,5 mM 0,28 g Kalziumdichlorid 10,0 mM 2,38 g HEPES 10,0 mM 1,80 g Glucose pH-Wert eingestellt auf pH 7,3 mit NaOH; Osmolarität: 300 ± 10 mOs/kg. CS-Puffer zur Fixierung 100,0 mM (Mengen in g für 2l) 60,50 g Pipes 1,0 mM 0,76 g EGTA 4,0 % 8,00 g PEG 100,0 mM 8,00 g NaOH pH-Wert eingestellt auf pH 6,9. Verdünnungspuffer 89,98 % PBS 10,00 % FCS 0,02 % Eindeckmittel 1 mg/ml 70 % Natriumazid (NaN3) p-Phenylendiamin in PBS Glycerin pH-Wert eingestellt auf pH 8,5. Lagerung bei –20 °C. Fixierungspuffer 140,0 mM NaCl 5,0 mM KCl 5,0 mM Glucose 10,0 mM HEPES 0,5 mM EDTA pH-Wert eingestellt auf pH 5 mit 5 M NaOH. 9-1 9 Literaturverzeichnis [Adler, 1985] R. Adler, J. Jerden und A.T. Hewitt, 1985, Responses of cultured neural retinal cells to substratum-bound laminin and other extracellular matrix molecules, Dev Biol, 112, S. 100-14. [Annis, 1993] C.M. Annis, R.T. Robertson und D.K. O'Dowd, 1993, Aspects of early postnatal development of cortical neurons that proceed independently of normally present intrinsic influences, J Neurobiol, 24, S. 1460-1480. [Bain, 1994] G. Bain, W.J. Ray, M. Yao und D.I. Gottlieb, 1994, From embryonal carcinoma cells to neurons: The P19 Pathway, Bioessays, 16, S. 343-348. [Banker, 1998] G. Banker und K. Goslin, 1998, Culturing Nerve Cells, Auflage 2, MIT Press, Cambridge, Massachusetts. [Baron-Van Evercooren, 1982] A. Baron-Van Evercooren, H.K. Kleinman, S. Ohno, P. Marangos, J.P. Schwartz und M.E. Dubois-Dalcq, 1982, Nerve growth factor, laminin and fibronectin promote neurite growth in human fetal sensory ganglia cultures, J Neurosci Res, 8, S. 179-93. [Beckers, 1993] M. Beckers und e. al., 1993, The Axon Guide, Axon Instruments, Inc. [Bennett, 1967] M.V. Bennett, Y. Nakajima und G.D. Pappas, 1967, Physiology and ultrastructure of electrotonic junctions. I. Supramedullary neurons, Journal of Neurophysiology, 30, S. 161-79. [Berg, 1984] D.K. Berg, 1984, New neuronal growth factors, Ann Rev Neurosci, 7, S. 14970. [Berger, 1994] C. Berger, 1994, Untersuchungen zur Polarität neuronaler Derivate einer ECZellinie, Diplomarbeit, Universität Johannes Gutenberg-Universität Mainz. [Berger, 1997] C. Berger, S. Reinhard, M. Rentrop, M. Bachmann, T. Weiser, E. Link, M. Wienrich, R. Jahn und A. Maelicke, 1997, De novo acquisition of neuronal polarity in retinoic acid-induced embryonal carcinoma cells, Eur J of Cell Biol, 74, S. 230-245. [Bernard, 1998] A. Bernard, E. Delamarche, H. Schmidt, B. Michel, H.R. Bossard und H. Biebuyck, 1998, Printing Patterns of Proteins, Langmuir, 14 (9), S. 2225-2229. [Bhatti, 1989] S. Bhatti, G. Zimmer und J. Bereiter-Hahn, 1989, Enzyme release from chick myocytes during hypoxia and reoxygenation: dependence on pH, J Mol Cell Cardiol, 10, S. 995-1008. [Bi, 1998] G.Q. Bi und M.M. Poo, 1998, Synaptic Modifications in Cultured Hippocampal Neurons: Dependence on Spike Timing, Synaptic Strength, and Postsynaptic Cell Type, The J of Neuroscience, 18(24), S. 10464-10472. [Blawas, 1998] A.S. Blawas und W.M. Reichert, 1998, Protein patterning, Biomaterials, 19, S. 595-609. [Bowe, 1995] M.A. Bowe und J.R. Fallon, 1995, The role of agrin in synapse formation, Annu Rev Neurosci, 18, S. 443-462. [Branch, 2000] D.W. Branch, B.C. Wheeler, G.J. Brewer und D.E. Leckband, 2000, Long-term maintenance of patterns of hippocampal pyramidal cells on substrates of polyethylene glycol and microstamped polylysine, IEEE Transactions on biomedical engineering, 47, S. 290-300. 9-2 9 Literaturverzeichnis [Braun, 1998] D. Braun und P. Fromherz, 1998, Fluorescence Interferometry of Neuronal Cell Adhesion on Microstructured Silicon, Physical Review Letters, 81 (23), S. 5241-5244. [Brownell, 1982] G.L. Brownell, T.F. Budinger, P.C. Lauterbur und P.L. McGeer, 1982, Positron tomography and nuclear magnetic resonance imaging, Science, 215, S. 619-626. [Caceres, 1984] A. Caceres, G. Banker, O. Steward, L. Binder und M. Payne, 1984, MAP 2 is localized to the dendrites of hippocampal neurons which develop in culture, Brain Res, 315, S. 314-318. [Caeser, 1989] M. Caeser, T. Bonhoeffer und J. Bolz, 1989, Cellular organization and development of slice cultures from rat visual cortex, Exp Brain Res, 77, S. 234-244. [Carew, 1976] T.J. Carew und E.R. Kandel, 1976, Two functional effects of decreased conductance EPSP's: synaptic augmentation and increased electrotonic coupling, Science, 192, S. 150-3. [Chen, 1998] C.S. Chen, M. Mrksich, S. Huang, G.M. Whitesides und D.E. Ingber, 1998, Micropatterned Surfaces for Control of Cell Shape, Position, and Function, Biotechnol Prog, 14, S. 356-363. [Chiu, 1984] A.Y. Chiu und J.R. Sanes, 1984, Differentiation of basal lamina in synaptic and extrasynaptic portions of embryonic rat muscle, Dev Biol, 103, S. 456-67. [Clémence, 1995] J.F. Clémence, J.P. Ranieri, P. Aebischer und H. Sigrist, 1995, Photoimmobilization of a bioactive laminin fragment and pattern-guided selective neuronal cell attachment, Bioconj Chem, 6, S. 411-417. [Collins, 1980] F. Collins und J.E.J. Garreth, 1980, Elongated nerve fibers are guided by a pathway of material released from embryonic nonneuronal cells, Proc Natl Acad Sci USA, 77, S. 6226-28. [Collins, 1984] F. Collins und M.R. Lee, 1984, The spatial control of ganglionic neurite growth by the substrate-associated material from conditioned medium: An experiemntal model of haptotaxis, J Neurosci, 4, S. 2823-29. [Connolly, 1990] P. Connolly, P. Clark, A.S.G. Curtis, J.A.T. Dow und C.D.W. Wilkinson, 1990, An extracellular microelectrode array for monitoring electrogenic cells in culture, Biosensors & Bioelectronics, 5, S. 223-234. [Corey, 1991] J.M. Corey, B.C. Wheeler und G.J. Brewer, 1991, Compliance of Hippocampal Neurons to Patterned Substrate Networks, Journal of Neuroscience Research, 30, S. 300-307. [Corey, 1996] J.M. Corey und B.C. Wheeler, 1996, Micrometer Resolution Silane-Based Patterning of Hippocampal Neurons: Critical Variables in Photoresist and Laser Ablation Processes for Substarte Fabrication, Transactions on Biomedical Engineering, 43 (9). [Curtis, 1997] A. Curtis und C. Wilkinson, 1997, Topographical control of cells, Biomaterials, 18, S. 1573-83. [Davis, 1985] G.E. Davis, M. Manthorpe und S. Varon, 1985, Parameters of neuritic growth from ciliary ganglion neurons in vitro: Influence of laminin, Schwannoma polyornithine-binding neurite promoting factor and ciliary neurotrophic factor, Dev Brain Res, 17, S. 75-84. [Davis, 1985] G.E. Davis, M. Manthorpe, E. Engvall und S. Varon, 1985, Isolation and characterization of rat Schwannoma neurite-promoting factor: Evidence that the factor contains laminin, J Neurosci, 5, S. 2662-71. 9-3 [Davis, 1987] G.E. Davis, G. Klier, E. Engvall, C. Cornbrooks, S. Varon und M. Manthorpe, 1987, Association of Laminin with heparan and chrondroitin sulfate-bearing proteoglycans in neurite-promoting factor complexes from rat Schwannoma cells, Neurochem Res, 12, S. 909-21. [Debus, 1983] E. Debus, K. Weber und M. Osborn, 1983, Differentiation, 25, S. 193-203. [Dulcey, 1991] C.S. Dulcey, J.H. Georger, V. Krauthamer, D.A. Stenger, T.L. Fare und J.M. Calvert, 1991, Deep UV photochemistry of chemisorbed monolayers patterned coplanar molecular assemblies., Science, 252, S. 551-554. [Eckert, 1993] R. Eckert, A. Dunina-Barkovskaya und D.F. Hulser, 1993, Biophysical characterization of gap-junction channels in HeLa cells, Pflugers Archiv European Journal of Physiology, 424, S. 335-42. [Eckert, 1999] R. Eckert, B. Adams, J. Kistler und P. Donaldson, 1999, Quantitative determination of gap junctional permeability in the lens cortex, Journal of Membrane Biology, 169, S. 91-102. [Edgar, 1984] D. Edgar, R. Timpl und H. Thoenen, 1984, The heparin-binding domain of laminin is responsible for its effects on neurite outgrowth and neuronal survival, EMBO J, 3, S. 1463-68. [Engvall, 1986] E. Engvall, G.E. Davis, K. Dickerson, E. Ruoslathi, S. Varon und M. Manthorpe, 1986, Mapping of domains in human laminin using monoclonal antibodies: Localization of the neurite-promoting site, J Cell Biol, 103, S. 2457-65. [Faivre-Bauman, 1984] A. Faivre-Bauman, J. Puymirat, C. Loudes, A. Barret und A. Tixier-Vidal, 1984, Laminin promotes attachment and neurite elongation of fetal hypothalamic neurons grown in serum-free medium, Neurosci Lett, 44, S. 83-89. [Feynman, 1991] R.P. Feynman, R.B. Leighton und M. Sands, 1991, Feynman Vorlesungen über Physik, 2, Auflage 2, R. Oldenburg Verlag München Wien, ISBN 3-48622058-6. [Fiellous, 1978] M. Fiellous, E. Günther, R. Kemler, J. Wiels, R. Berger, J.L. Guenet und H. Jakob, 1978, Association of the H-Y male antigen with b2-microglobulin on human lymphoid and differentiated mouse teratocarcinoma cell lines, J Exp Med, 148, S. 58-70. [Finley, 1996] M.F. Finley, N. Kulkarni und J.E. Huettner, 1996, Synapse formation and establishment of neuronal polarity by P19 embryonic carcinoma cells and embryonic stem cells, J Neurosci, 16, S. 1056-1065. [Fitzsimonds, 1997] R.M. Fitzsimonds, H.-J. Song und M.-m. Poo, 1997, Propagation of activitydependent synaptic depression in simple neural networks, Nature, 388, S. 439-448. [Fromherz, 1991] P. Fromherz, A. Offenhäusser, T. Vetter und J. Weis, 1991, A Neuron-Silicon Junction: A Retzius Cell of the Leech on an Insulated-Gate Field-Effect Transistor, Science, 252, S. 1290-1293. [Fromherz, 1993] P. Fromherz, C.O. Müller und R. Weis, 1993, Neuron Transistor: Electrical Transfer Function Measured by the Patch-Clamp Technique, Physical Review Letters, 71 (24), S. 4079-4082. [Fromherz, 1999] P. Fromherz, 1999, Extracellular recording with transistors and the distribution of ionic conductances in a cell membrane, European Biophysics Journal, 28(3), S. 254-258. 9-4 9 Literaturverzeichnis [Gage, 1995] F.H. Gage, P.W. Coates, T.D. Palmer, H.G. Kuhn, L.J. Fisher, J.O. Suhonen, D.A. Peterson, S.T. Suhr und J. Ray, 1995, Survival and differentiation of adult neuronal progenitor cells transplanted to the adult brain, Proc Natl Acad Sci USA, 92, S. 11879-11883. [Gähwiler, 1981] B.H. Gähwiler, 1981, Organotypic Monolayer Cultures Of Nervous Tissue, Journal of Neuroscience Methods, 4, S. 329-342. [Gähwiler, 1984] B.H. Gähwiler, 1984, Development of the hippocampus in vitro: cell types, synapses, and receptors., Neuroscience, 11, S. 751-760. [Gähwiler, 1984b] B.H. Gähwiler, 1984, Slice cultures of cerebellar, hippocampal and hypothalamic tissue, Expertina, 40, S. 235-243. [Gähwiler, 1990] B.H. Gähwiler und T. Knöpfel, 1990, Preparations of Vertebrate Central Nervous System In Vitro, John Wiley & Sons Ltd. [Golze, 2001] S. Golze, 2001, Oberflächengebundene Polymermonolagen für die Herstellung von DNA-Chips, Dissertation, Universität Mainz. [Gross, 1977] G.W. Gross, E. Rieske, G.W. Kreutzberg und A. Meyer, 1977, A new fixedarray multi-microelectrode system designed for long-term monitoring of extracellular single unit neuronal activity in vitro, Neurosci Lett, 6, S. 101105. [Gross, 1979] G.W. Gross, 1979, Simultaneous single unit recording in vitro with a photoetched laser deinsulated gold multimicroelectrode surface, IEEE trans on biomed engineering, 26, S. 273-279. [Gundersen, 1987] R.W. Gundersen, 1987, Response of chick sensory growth cones to laminin and fibronectin: External morphology, Soc Neurosci Abstr, 13, S. 1484. [Gundersen, 1987b] R.W. Gundersen, 1987b, Response of sensory neurites and growth cones to patterned substrata of laminin and fibronectin in vitro, Dev Biol, 121, S. 42332. [Hagiwara, 1958] S. Hagiwara und I. Tasaki, 1958, A study of the mechanism of impulse transmission across the giant synapse of the squid, J Physiol, 143, S. 114-137. [Hammarback, 1985] J.A. Hammarback, S.L. Palm, L.T. Furcht und P.C. Letourneau, 1985, Guidance of neurite outgrowth by pathways of substratum adsorbed laminin, J Neurosci Res, 13, S. 213-20. [Hammarback, 1986] J.A. Hammarback und P.C. Letourneau, 1986, Neurite extension across regions of low cell-substratum adhhesivity: Implications for the guidepost hypothesis of axonal pathfinding, Dev Biol, 118, S. 129-40. [Harrison, 1907] R.G. Harrison, 1907, Observations on the living developing nerve fiber, Anat Rec, 1, S. 116-118. [Hebb, 1949] D.O. Hebb, 1949, The organization of behaviour, Auflage 8, Wiley, New York. [Herget, 1998] T. Herget, H. Specht, C. Esdar, S.A. Oehrlein und A. Maelicke, 1998, Retinoic Acid Induces Apoptosis-Associated Neural Differentiation of a Murine Teratocarcinoma Cell Line, J of Neurochemistry, 70 Nr. 1, S. 47-58. [Hoch, 1986] D.B. Hoch und R. Dingledine, 1986, GABAergic neurons in rat hippocampal culture, Brain Res, 390, S. 53-64. [Hodgkin, 1946] A.L. Hodgkin und R.D. Keynes, 1946, The electrical constants of a crustacean nerve fibre, Proc Roy Soc Ser B, 133, S. 444-479. [Hodgkin, 1949] A.L. Hodgkin, A.F. Huxley und B. Katz, 1949, Ionic currents underlying activity in the giant axon of the squid, Arch Sci Physiol, 3, S. 129-150. 9-5 [Hopkins, 1985] J.M. Hopkins, T.S. Ford-Holevinski, J.P. McCoy und B.W. Agranoff, 1985, Laminin and optic nerve regeneration in the goldfish, J Neurosci, 5, S. 303038. [Ide, 1983] C. Ide, K. Tohyama, R. Yokota, T. Nitatori und S. Onodera, 1983, Schwann cell basal lamina and nerve regeneration, Brain Res, 288, S. 61-75. [Ingebrandt, 2001] S. Ingebrandt, 2001, Charakterisierung der Zell-Transistor Kopplung, Dissertation, Universität Mainz. [James, 1998] C.D. James, R.C. Davis, L. Kam, H.G. Craighead, M. Isaacson, J.N. Turner und W. Shain, 1998, Patterned Protein Layers on Solid Substrates by Thin Stamp Microcontact Printing, Langmuir, 14, S. 741-744. [James, 2000] C.D. James, R. Davis, M. Meyer, A. Turner, S. Turner, G. Withers, L. Kam, G. Banker, H. Craighead, M. Isaacson, J. Turner und W. Shain, 2000, Aligned microcontact printing of micrometer-scale poly-L-lysine structures for controlled growth of cultured neurons on planar microelectrode arrays, IEEE Trans on Biomed Eng, 47 (1), S. 17-21. [Jostock, 1998] R. Jostock, M. Rentrop und A. Maelicke, 1998, Cell fate specification in an in vitro model of neuronal development, Eur J of Cell Biol, 76, S. 63-76. [Kandel, 1991] E.R. Kandel, J.H. Schwartz und T.M. Jessel, 1991, Principles of neural science, Auflage 3, Appleton & Lange. [Katz, 1985] B. Katz, 1985, Nerv, Muskel und Synapse, Einführung in die Elektrophysiologie, Auflage 4, Georg Thieme Verlag, ISBN 3-13-465304-4. [Kleinfeld, 1988] D. Kleinfeld, K.H. Kahler und P.E. Hockberger, 1988, Controlled outgrowth of dissociated neurons on patterned substrates, J Neurosci, 8, S. 4098-4120. [Kleinig, 1999] H. Kleinig und U. Maier, 1999, Zellbiologie, Auflage 4, Fischer, ISBN 3-82740806-7. [Krause, 2000] M. Krause, 2000, Untersuchungen zur Zell-Transistor Kopplung mittels der Voltage-Clamp Technik, Dissertation, Universität Mainz. [Kröger, 2001] S. Kröger, 2001, Private Communication. [Lander, 1982] A.D. Lander, D.K. Fujii, D. Gospodarowitcz und L.F. Reichardt, 1982, Characterization of a factor that promotes neurite outgrowth: Evidence linking activity to a herpan sulfate proteoglycan, J Cell Biol, 94, S. 574-85. [Lander, 1983] A.D. Lander, K. Tomaselli, A.L. Calof und L.F. Reichardt, 1983, Studies on extracellular matrix components that promote neurite outgrowth, Cold Spring Harbor Symp Quant Biol, 48, S. 611-24. [Lander, 1985] A.D. Lander, D.K. Fujii und L.F. Reichardt, 1985, Purification of a factor that promotes neurite outgrowth: Isolation of laminin and associated molecules, J Cell Biol, 101, S. 898-913. [Lander, 1985b] A.D. Lander, D.K. Fujii und L.F. Reichardt, 1985b, Laminin is associated with the "neurite outgrowth promoting factors" found in conditioned media, Proc Natl Acad Sci USA, 82, S. 2183-87. [Lang, 1989] E. Lang, M.L. Marizauric-Stüker und A. Maelicke, 1989, States of developmental commitment of a mouse embryonal carcinomal cell line differentiating along a neuronal pathway, J Cell Biol, 109, S. 2481-2493. [Lauer, 2001] L. Lauer, K. Klein und A. Offenhäusser, 2001, Spot compliant neuronal networks by structure optimized micro contact printing, Biomaterials, in press. 9-6 9 Literaturverzeichnis [Lauer, 2001b] L. Lauer, S. Ingebrandt und A. Offenhäusser, 2001b, Aligned Microcontact Printing of Biomolecules on Microelectronic Device Surfaces, IEEE Transactions on Biomedical Engineering, in press. [Letourneau, 1975] P.C. Letourneau, 1975, Cell-to-substratum adhesion and guidance of axonal elongation, Dev Biol, 44, S. 92-101. [Liesi, 1984] P. Liesi, D. Dahl und A. Vaheri, 1984, Neurons cultured from developing rat brain attach and spread preferentially to laminin, J Neurosci Res, 11, S. 24151. [Lohrke, 1998] S. Lohrke, M. Kungel und E. Friauf, 1998, Electrical membrane properties of trapezoid body neurons in the rat auditory brain stem are preserved in organotypic slice cultures, Journal of Neurobiology, 36, S. 395-409. [Mandell, 1996] J.W. Mandell und G.A. Banker, 1996, A spatial gradient of tau protein phosphorylation in nascent axons, J Neurosci, 16, S. 5727-5740. [Manthorpe, 1983] M. Manthorpe, E. Engvall, E. Ruoslahti, F.M. Longo, G.E. Davis und S. Varon, 1983, Laminin promotes neuritic regeneration from cultured peripheral and central neurons, J Cell Biol, 97, S. 1882-90. [Matsuzawa, 1996] M. Matsuzawa, P. Liesi und W. Knoll, 1996, Chemically modifying glass surfaces to study substratum-guided neurite outgrowth in culture, J of Neuroscience Methods, 69, S. 189-196. [McBurney, 1982] M.W. McBurney und B.J. Rogers, 1982, Isolation of male embryonal carcinoma cells and their chromosome replication patterns, Dev Biol, 82, S. 503-508. [McMahan, 1992] U.J. McMahan, S.E. Horton, M.J. Werle, L.S. Honig, S. Kroger, M.A. Ruegg und G. Escher, 1992, Agrin isoforms and their role in synaptogenesis, Curr Opin Cell Biol, 4, S. 869-874. [Munk, 2000] K. Munk, 2000, Biochemie, Zellbiologie, Ökologie, Evolution, Grundstudium Biologie, Spektrum Akademischer Verlag GmbH, ISBN 3-8274-0910-1. [Neher, 1976] E. Neher und B. Sakman, 1976, Single channel currents recorded from membrane of denervated frog muscle fibres, Nature, 260, S. 799-802. [Neher, 1992] E. Neher, 1992, Correction for liquid junction potentials in patch-clamp experiments, Methods Enzymol, 207, S. 123-131. [Neyton, 1985] J. Neyton und A. Trautmann, 1985, Single-channel currents of an intercellular junction, Nature, 317, S. 331-5. [Neyton, 1986] J. Neyton und A. Trautmann, 1986, Physiological modulation of gap junction permeability, Journal of Experimental Biology, 124, S. 93-114. [Nicholls, 1995] Nicholls, Martin und Wallace, 1995, Vom Neuron zum Gehirn, ISBN 3-43720517-X. [Offenhäusser, 1997] A. Offenhäusser, C. Sprössler, M. Matsuzawa und W. Knoll, 1997, FieldEffect transistor array for monitoring electrical activity from mammalian neurons in culture, Biosensors & Bioelectronics, 12 (8), S. 819-826. [Parnas, 1995] D. Parnas und M. Linial, 1995, Cholinergic Properties of Neurons Differentiated from an Embryonal Carcinoma Cell-Line (P19), Int J Devl Neuroscience, 13 (7), S. 767-781. [Peterson, 1955] E.R. Peterson und M.R. Murray, 1955, Myelin sheat formation of avian spinal ganglia, Am J Anat, 96, S. 319. 9-7 [Pfeiffer, 1981] S.E. Pfeiffer, H. Jakob, K. Mikoshiba, P. Dubois, J.L. Guenet, J.-F. Nicolas, J. Gaillard, G. Chevance und F. Jakob, 1981, Differentiation of a teratocarcinoma line: Preferential development of cholinergic neurons, J Cell Biol, 88, S. 57-66. [Pine, 1980] J. Pine, 1980, Recording action potentials from cultured neurons with extracellular microcircuit electrodes, J of neurosci methods, 2, S. 19-31. [Prucker, 1995] O. Prucker, 1995, Molekular dünne Schichten aus kovalent gebundenem Polystyrol über immobilisierte Azoinitiatoren, Dissertation, Universität Bayreuth. [Ramon y Cajal, 1928] S. Ramon y Cajal, 1928, Degeneration and Regenration of the Nervous System, neue Auflage aus dem Jahr 1968, Hafner. [Riggot, 1987] M.J. Riggot und S.A. Moody, 1987, Distribution of laminin and fibronectin along peripheral trigeminal axon pathways in the developing chick, J Comp Neurol, 258, S. 580-96. [Rogers, 1983] S.L. Rogers, P.C. Letourneau, S.L. Palm, J. McCarthy und L.T. Furcht, 1983, Neurite extension by peripheral and central nervous system neurons in response to substratum-bound fibronectin and laminin, Dev Biol, 98, S. 21220. [Rogers, 1986] S.L. Rogers, K.J. Edson, P.C. Letourneau und S.C. McLoon, 1986, Distribution of laminin in the developing peripheral nervous system of the chick, Dev Biol, 113, S. 429-35. [Rossant, 1982] J. Rossant und M. McBurney, 1982, The developmental potential of a euploid male teratocarcinoma cell line after blastocyst injection, J Embryol Exp Morphol, 67, S. 167. [Sakmann, 1995] B. Sakmann, 1995, Single-channel recording, Auflage 2, Plenum Press. [Sanes, 1983] J.R. Sanes und A.Y. Chiu, 1983, The basal lamina of the neuromuscular junction, Cold Spring Harbor Symp Quant Biol, 48, S. 667-678. [Sanes, 1989] J.R. Sanes, 1989, Extracellular matrix molecules that influence neural development, Annu Rev Neurosci, 12, S. 491-516. [Scherer, 1984] S.S. Scherer und S.J. Easter, 1984, Degenerative and regenerative changes in the trochlear nerve of goldfish, J Neurocytol, 13, S. 519-65. [Scholl, 2000] M. Scholl, C. Sprössler, M. Denyer, M. Krause, K. Nakajima, A. Maelicke, W. Knoll und A. Offenhäusser, 2000, Ordered networks of rat hippocampal neurons attached to silicon oxide surfaces, Journal of Neuroscience Methods, 104, S. 65-75. [Schulze, 1999] P. Schulze, 1999, EPC-9 Manual, Heka elektronik GmbH. [Sprössler, 1997] C. Sprössler, 1997, Extrazelluläre Signalableitung durch ein Sensorfeld mit Feldeffekttransistoren, Dissertation, Universität Darmstadt. [Sprössler, 1998] C. Sprössler, D. Richter, M. Denyer und A. Offenhäusser, 1998, Long-term recording system based on field-effect transistor arrays for monitoring electrogenic cells in culture, Biosensors & Bioelectronics, 13, S. 613-618. [Sprössler, 1999] C. Sprössler, M. Denyer, S. Britland, W. Knoll und A. Offenhäusser, 1999, Electrical recordings from rat cardiac muscle cells using field-effect transistors, Physical Review E, 60, S. 2171-2176. [Stenger, 1992] D.A. Stenger, J.H. Georger, C.S. Dulcey, J.J. Hickman, A.S. Rudolph, T.B. Nielsen, S.M. McCort und J.M. Calvert, 1992, Coplanar molecular assemblies of amino- and perfluorinated alkylsilanes: characterization and geometric definition of mammalian cell adhesion and growth., J American Chemical Society, 114, S. 8435-8442. 9-8 9 Literaturverzeichnis [Stenger, 1998] D.A. Stenger, J.J. Hickman, K.E. Bateman, M.S. Ravenscroft, W. Ma, J.J. Pancrazio, K. Shaffer, A. Schaffner, D.H. Cribbs und C.W. Cotman, 1998, Microlithographic determination of axonal/dendritic polarity in cultured hippocampal neurons, Journal of Neuroscience Methods, 82, S. 167-173. [Stetefeld, 1996] J. Stetefeld, U. Mayer, R. Timpl und R. Huber, 1996, Crystal Structure of Three Consecutive Laminin-type Epidermal Growth Faktor-like (LE) Modules of Laminin gamma1 Chain Harboring the Nidogen Binding Site, J Mol Biol, 257, S. 644-657. [Steward, 1978] W.W. Steward, 1978, Functional Connections between cells as revealed by dye-coupling with highly fluorescent naphthalamide tracer, Cell, 14, S. 741759. [Streit, 1989] P. Streit, S.M. Thompson und B.H. Gähwiler, 1989, Anatomical and physiological properties of GABAergic neurotransmission in organotypic slice cultures of rat hippocampus, Eur J Neurosci, 1, S. 603-615. [Stryer, 1996] L. Stryer, 1996, Biochemie, Auflage 4, Spektrum Akademischer Verlag GmbH, ISBN 3-86025-346-8. [Thiébaud, 2001] P. Thiébaud, L. Lauer, W. Knoll und A. Offenhäusser, 2001, PDMS device for patterned application of microfluids to neuronal cells arranged by microcontact printing, Biosensors Bioelectronics, accepted. [Tietze, 1993] U. Tietze und C. Schenk, 1993, Halbleiter-Schaltungstechnik, Auflage 10, Springer, ISBN 3-540-56184-6. [Traub, 1994] O. Traub, R. Eckert, H. Lichtenberg-Frate, C. Elfgang, B. Bastide, K.H. Scheidtmann, D.F. Hulser und K. Willecke, 1994, Immunochemical and electrophysiological characterization of murine connexin40 and -43 in mouse tissues and transfected human cells, European Journal of Cell Biology, 64, S. 101-12. [Unsicker, 1985] K. Unsicker, S.D. Skaper und S. Varon, 1985, Developmental changes in the responses of rat chromaffin cells to neuronotrophic and neurite-promoting factors, Dev Biol, 111, S. 425-33. [Unsicker, 1985b] K. Unsicker, S.D. Skaper, G.E. Davis, M. Manthorpe und S. Varon, 1985, Comparison of the effects of laminin and the polyornithine-binding neuritepromoting factor from RN22 Schwannoma cells on neurite regeneration from cultured newborn and adult rat dorsal root ganglion neurons, Dev Brain Res, 17, S. 304-8. [Wewer, 1983] U. Wewer, R. Albrechtsen, M. Manthorpe, S. Varon, E. Engvall und E. Ruoslathi, 1983, Human laminin isolated in a nearly intact, biologically active form from placenta by limited proteolysis, J Biol Chem, 258, S. 1265460. [Wheeler, 1999] B.C. Wheeler, J.M. Corey, G.J. Brewer und D.W. Branch, 1999, Microcontact Printing for Precise Control of Nerve Cell Growth in Culture, Journal of Biomechanical Engineering-Transactions of the Asme, 121, S. 73-78. [Wheeler, 2001] B.C. Wheeler, 2001, Private communication. [www_1, 2001] www_1, 2001, http://rpiwww.mdacc.tmc.edu/se/anatomy/brain/gross_brain_left.jpg. [www_2, 2001] www_2, 2001, http://www.csic.es/hispano/patrimo/imagenes/cajal6.jpg. [Yeung, 2001] C.-K. Yeung, L. Lauer, A. Offenhäusser und W. Knoll, 2001, Modulation of the growth and guidance of rat brain stem neurons using patterned extracellular matrix proteins, Neuroscience Letters, 301 (2), S. 147-150. Lebenslauf Lars Oliver Lauer geboren 24. Mai 1972 in Bonn wohnhaft Dr.-Karl-Schramm-Str. 2 D-55129 Mainz 1978 – 1982 Martinus-Grundschule Mainz-Gonsenheim 1982 – 1991 Gymnasium Theresianum Mainz, Gymnasium des Johannesbundes zu Leutesdorf 17.06.1991 - 15.09.1992 Zivildienst in der ev. Kirchengemeinde Mainz-Finthen/Drais WS 1991/92 Teizeitstudium Elektrotechnik an der Fernuniversität Hagen WS 1992/93 – SS 1996 Studium der Physik an der Johannes Gutenberg-Universität Mainz 22.09.1996 – 09.10.1997 Diplomarbeit bei Prof. Dr. E. Otten am FB Physik der Johannes Gutenberg-Universität Mainz seit 01.12.1997 Unternehmensberater bei McKinsey & Company, Inc. in Frankfurt seit 01.04.1999 Doktorand im Arbeitskreis Materialwissenschaften am Max-Planck-Institut für Polymerforschung Mainz Veröffentlichungen L. Lauer, S. Ingebrandt und A. Offenhäusser, 2001, Aligned Microcontact Printing of Biomolecules on Microelectronic Device Surfaces, IEEE Transactions on Biomedical Engineering, in press. L. Lauer, K. Klein und A. Offenhäusser, 2001, Spot compliant neuronal networks by structure optimized micro contact printing, Biomaterials, in press. P. Thiébaud, L. Lauer, W. Knoll und A. Offenhäusser, 2001, PDMS device for patterned application of microfluids to neuronal cells arranged by microcontact printing, Biosensors Bioelectronics, accepted. C.K. Yeung, L. Lauer, A. Offenhäusser und W. Knoll, 2001, Modulation of the growth and guidance of rat brain stem neurons using patterned extracellular matrix proteins, Neuroscience Letters, 301 (2), S. 147-150. A.J. Deninger, B. Eberle, M. Ebert, T. Großmann, W. Heil, H.-U. Kauczor, L. Lauer, K. Markstaller, E. Otten, J. Schmiedeskamp, W. Schreiber, R. Surkau, M. Thelen und N. Weiler, 1999, Quantification of regional intrapulmonary oxygen partial pressure evolution during apnea by ³He MRI, Journal of Magnetic Resonance, 141 (2), S. 207-216. Konferenzbeiträge L. Lauer, C.-K. Yeung, M. Scholl, A. Offenhäusser, 2000, Structure aligned micro contact printing controls nerve cell growth and network formation, European Biophysics Journal, 29 (4-5), S. 353. Patente G. Nelles, A. Yasuda, W. Knoll, A. Offenhäusser, C.-K. Yeung und L. Lauer, 2000, A method of forming a cell pattern on a surface, cellular networks and tissues based thereon, EP 00 122915.2 A. Deninger, B. Eberle, M. Ebert, T. Großmann, W. Heil, H.-U. Kauczor, L. Lauer, K. Markstaller, T. Roberts, W. Schreiber, R. Surkau und N. Weiler, 1998, Use of a hyperpolarized gas for MRI detection of regional variations in oxygen uptake from the lungs, US 1998 005 7979, GB 1998 000 7879, PCT/GB99/01095, WO 9953332. N. Weiler, B. Eberle, M. Ebert, T. Grossmann, W. Heil, H.-U. Kauczor, L. Lauer, K. Markstaller, E. Otten und R. Surkau, 1997, 1998, Aparatus for administering fluids, DE 1997 1050 467, DE 1998 1013 790, PCT/EP98/07515-07516, PL 340503. Danksagung Abschließend möchte ich mich bei allen bedanken, die mir diese Promotion ermöglicht und zu ihrem Gelingen beigetragen haben. Mein ganz besonderer Dank gilt … … Prof. Dr. W. Knoll für die herzliche Aufnahme in seinen Arbeitskreis und die spannende interdisziplinäre Themenstellung. Die breitgefächerten wissenschaftlichen Möglichkeiten in seinem Labor und das sehr angenehme Arbeitsklima waren ideale Rahmenbedingungen für meine Promotion. … PD Dr. A. Offenhäusser für die freundschaftliche Betreuung. Sein anspornendes Interesse an meiner Arbeit sowie seine fachliche und praktische Unterstützung waren von ganz entscheidender Bedeutung. … Prof. Dr. H.-J. Butt für die Vertretung meiner Arbeit vor dem Fachbereich Chemie und Pharmazie der Universität Mainz. … Prof. Dr. A. Maelicke und Dr. C. Klein vom Institut für Physiologische Chemie und Pathobiochemie der Universtät Mainz für die fruchtbare Kooperation und praktische Unterstützung in allen Fragen der Zellkultur. Weiterhin für die Möglichkeit zur Präsentation und Diskussion meiner Ergebnisse im Gruppenseminar des Instituts. … Dr. A. Yasuda und Dr. G. Nelles von der Firma Sony International Europe GmbH in Stuttgart für die finanzielle Unterstützung des Projektes. … W. Staab vom Institut für Mikrotechnik Mainz (IMM) für die gute Zusammenarbeit bei der Herstellung von Stempelstrukturen. … Dr. M. Scholl, Dr. C.-K. Yeung und C. Kauff für die Präparation von primären dissoziierten Neuronen und Hirnschnitten. M. Hemmerlein und A. Kosan für die Kultivierung neuronaler Zellinien und viele nützliche Hinweise für den Umgang mit neuronaler Zellkultur. … S. Golze für die Beschichtung von Siliziumsubstraten mit Polymeren und ihre Unterstützung bei gemeinsamen Experimenten. … S. Ingebrandt für seine Hilfsbereitschaft bei gemeinsamen Messungen mit von ihm aufgebauten Sensorchips. … A. Vogt für ihre Unterstützung bei Antikörperfärbungen und biologischen Fragen. … T. Baumgart und P. Thiébaud für fachkundige Hilfe bei Fragen zu Chemie und Mikrotechologie. … Dr. S. Schmitgen und Dr. G. Strube, Partner der Unternehmensberatung McKinsey & Company, Inc., für die wohlwollende Unterstützung der zeitlichen Planung meines Promotions-Leaves im Rahmen des McKinsey Fellow-Programms. Und nicht zuletzt meinen Eltern für ihre beständige Unterstützung zu allen Zeiten meines Studiums. Hiermit erkläre ich an Eides statt, dass ich die vorliegende Arbeit selbständig und ausschließlich unter Verwendung der angegebenen Hilfsmittel angefertigt habe. Ich erkläre außerdem, dass ich noch keinen vorherigen Promotionsversuch unternommen habe