1 1 GRUNDLAGEN DER THERMODYNAMIK _____ 3 2
Werbung

1
GRUNDLAGEN DER THERMODYNAMIK _____ 3
1.1
Einleitung 1,2,3 ______________________________________3
1.2
Das thermodynamische System 1,3 ______________________3
1.3
Ideale Gase 1,2,3 _____________________________________6
1.3.1
Ideale Gasgleichung________________________________6
1.3.2
Zustandsänderungen eines idealen Gases _______________9
1.3.3
Ideale Gemische von Gasen _________________________11
1.4
Reale Gase 1 _______________________________________15
1.4.1
Van-der-Waalssche Gleichung und Kondensation und
kritische Größen _________________________________________15
1.4.2
Gesetz der korrespondierenden Größen (dimensionslose
VDW-Gleichung) ________________________________________19
1.4.3
Virialkoeffizienten ________________________________20
1.5
Aufbau von Diagrammen in der Thermodynamik 3 ______21
1.6
Wärmedehnung____________________________________22
1.6.1
Wärmedehnung fester Körper _______________________22
1.6.2
Wärmedehnung bei Flüssigkeiten ____________________24
1.7
Übungen zum ersten Kapitel _________________________25
1. Aufgabe _____________________________________________25
2. Aufgabe _____________________________________________25
3. Aufgabe _____________________________________________25
4. Aufgabe _____________________________________________26
5. Aufgabe _____________________________________________26
6. Aufgabe _____________________________________________26
7. Aufgabe _____________________________________________27
8. Aufgabe _____________________________________________27
2
HAUPTSÄTZE DER THERMODYNAMIK1,3 ___ 28
2.1
Arbeits- und Energieformen3 _________________________28
2.1.1
Wärme _________________________________________28
2.1.2
Arbeit __________________________________________29
2.1.3
Innere Energie und Enthalpie________________________33
2.2
Der Erste Hauptsatz der Thermodynamik______________34
2.2.1
Formulierungen des ersten Hauptsatzes________________34
2.2.2
Zustandsgleichung der inneren Energie ________________35
2.2.3
Zustandsgleichung der Enthalpie _____________________37
2.2.4
Zusammenhang zwischen CV und Cp bei idealen Gasen ___40
2.2.5
Der erste Hauptsatz für offene Systeme________________41
2.3
Der Zweite Hauptsatz der Thermodynamik1,3 ___________44
2.3.1
Formulierungen des Zweiten Hauptsatzes ______________44
2.3.2
Umkehrbare und nicht umkehrbare Prozesse____________45
2.3.3
Entropie ________________________________________46
2.3.4
Mathematische Definition der Entropie ________________47
2.3.5
Hauptgleichungen der Thermodynamik________________50
2.4
Einfache Zustandsänderungen idealer Gase 3 ___________50
2.4.1
Isochore Zustandsänderung _________________________51
2.4.2
Isobare Zustandsänderungen ________________________52
2.4.3
Isotherme Zustandsänderung ________________________54
1
2.4.4
2.4.5
Adiabate Zustandsänderung _________________________56
Polytrope Zustandsänderung ________________________59
2.5
Prozesse in der Thermodynamik1,3 ____________________60
2.5.1
Thermodynamischer Prozess und Kreisprozess __________60
2.5.2
Definition von Kreisprozessen_______________________60
2.5.3
Einfache Kreisprozesse ____________________________61
2.5.4
Darstellung von Kreisprozessen im p,v-Diagramm _______62
2.5.5
Carnot-Prozess für ideale Gase ______________________64
2.6
Übungsaufgaben ___________________________________67
9. Aufgabe _____________________________________________67
10. Aufgabe ____________________________________________68
11. Aufgabe ____________________________________________68
12. Aufgabe ____________________________________________68
13. Aufgabe ____________________________________________69
14. Aufgabe ____________________________________________69
15. Aufgabe ____________________________________________70
3
FEUCHTE LUFT³________________________ 70
3.1
Grundlagen _______________________________________70
3.2
Kalorische Zustandsgleichungen für feuchte Luft________73
3.3
Das h, x-Diagramm nach Molier ______________________78
3.4
Anwendungen des h, x-Diagramms____________________81
3.4.1
Erwärmung und Kühlung im Einphasengebiet bei konstantem
Dampfgehalt ____________________________________________81
3.4.2
Trocknung feuchter Luft ___________________________82
3.4.3
Adiabate Vermischung zweier Luftströme _____________83
2
1 Grundlagen der Thermodynamik
1.1 Einleitung 1,2,3
Die Thermodynamik beschäftigt sich mit der Untersuchung
des Verhaltens von Systemen bei Temperaturänderung mit
der Ab- bzw. Zufuhr von Wärme bzw. Arbeit. Ein
wesentlicher Bestandteil dieser Vorlesung ist die
Beschreibung der Zustandsänderung von Gasen und der
Zustand von Gasmischungen. Aber auch die Änderung des
Energieinhalts (1. Hauptsatz der Thermodynamik) und der
Qualität der Energie (2. Hauptsatz der Thermodynamik) im
Zuge einer Zustandsänderung.
1.2 Das thermodynamische System 1,3
In der Thermodynamik ist es zweckmäßig, Betrachtungen auf
eine definierte und abgegrenzte Menge oder einen Bereich zu
konzentrieren. Diese begrenzte Quantität bezeichnet man als
„System“. Dieses System wird durch eine Systemgrenze
festgelegt. Prinzipiell ist diese Abgrenzung willkürlich, aber
in den meisten Fällen ist es sinnvoll, sie an der Fragestellung
auszurichten, da sich dadurch die Lösung eines Problems
vereinfacht. Alles, was sich außerhalb des Systems befindet,
wird normalerweise als Umgebung bezeichnet.
Nach dem inneren Aufbau eines Systems unterscheidet man:
• Homogene einfache Systeme: Sie bestehen aus einer
einzigen, homogenen, isotropen Phase eines Stoffes.
(Homogen: Die Eigenschaften innerhalb des Systems sind
überall gleich, das heißt zum Beispiel keine Dichte- oder
Temperaturunterschiede in dem System. Isotrop: alle
Eigenschaften der Phase sind unabhängig von der
Richtung, d. h. z. B. leitet ein isotroper Stoff den
elektrischen Strom in alle Richtungen gleich gut.)
• Heterogene einfache Systeme: Bestehen aus mehreren
Phasen oder mehreren Apparaten innerhalb der
Systemgrenzen.
Durch geschickte Umordnung der Systemgrenzen kann ein
heterogenes System in homogene Systeme umbaut werden.
Häufig ist dies jedoch nicht zweckmäßig.
3
Man unterscheidet je nach ihren Eigenschaften verschiedene
Systemgrenzen.
• In einem isolierten oder vollständig abgeschlossenen
System ist weder Transport von Energie als Wärme Q
bzw. Arbeit W, noch von Masse m über die Grenzen des
Systems hinweg möglich (Abb. 1).
Abb. 1: Vollständig abgeschlossene Systeme 3
• Bei wärmeisolierten oder adiabaten Systemen ist kein
Wärmeaustausch mit der Umgebung möglich. Sie können
offen (Massenaustausch möglich) oder geschlossen sein,
auch die Übertragung von Arbeit ist möglich (Abb. 2).
Abb. 2: Adiabate Systeme 3
• Bei arbeitsisolierten Systemen wird keine Arbeit mit der
Umgebung ausgetauscht. Sie können offen oder
geschlossen, auch die Übertragung von Wärme ist
möglich (Abb. 3). Beispiele: Rohrströmungen,
Destillation, Rektifikationskolonne
Abb. 3: Arbeitsisolierte Systeme 3
•
Bei geschlossenen Systemen ist kein Massetransport an
die Umgebung, jedoch Wärme- und Arbeitsübertragung
möglich. (Abb. 4)
4
Abb. 4: Geschlossene Systeme 3
•
Offene Systeme können Wärme, Arbeit und Masse
übertragen (Abb. 5). Beispiele: WkA, Kolonnen mit
Rührwerk, Kessel, usw.
Abb. 5: Offene Systeme 3
Zustandsgrößen
Ein thermodynamisches System wird durch sogenannte
Zustandsgrößen beschrieben. Zustandsgrößen sind nutzbar
und kennzeichnen Zustand eines Systems wie zum Beispiel
Druck, Temperatur, Volumen und Masse.
Man unterscheidet diese Zustandsgrößen nach ihrer
Massenabhängigkeit.
• Intensive Größen sind von der Masse unabhängige
Größen, sie ändern ihren Wert durch Teilung des
Systems nicht. Druck und Temperatur sind intensive
Größen. Sie werden in der Regel mit kleinen Buchstaben
abgekürzt. (Ausnahme: Temperatur T) Beispiel: 1 Liter
Wasser besitzt die Temperatur von 25°C. Ist der
Wärmetransport mit der Umgebung zu vernachlässigen
und entnimmt man schnell 0,2 l Wasser, so haben beide
Teilmengen immer noch die gleiche Temperatur von
25°C.
• Extensive Größen sind von der Masse abhängig und
lassen sich additiv aus den entsprechenden
Zustandsgrößen der Teilsysteme zusammensetzen. Sie
werden in der Regel mit großen Buchstaben abgekürzt
(Ausnahme: Masse m). Masse, Volumen und Stoffmenge
sind zum Beispiel extensive Größen. Beispiel für die
5
Verwendung
von
m = m1 + m 2 , V = V1 + V2
extensiven
Größen:
In manchen Fällen ist es sinnvoll, die Massenabhängigkeit
auszuschalten und damit eine extensive Zustandsgröße in
eine intensive Größe umzuwandeln. Dies erreicht man durch
Division einer extensiven Größe mit der Masse oder
Stoffmenge. Wird die Größe auf die Masse bezogen,
bezeichnet man sie als spezifisch, wird sie auf die Stoffmenge
bezogen erhält sie die Bezeichnung molar.
spezifische Zustandsgröße =
Extensive Zustandsgröße
Systemmasse
Beispiel: Spezifisches Volumen v =
molare Zustandsgröße =
V
m³
in
m
kg
Extensive Zustandsgröße
Stoffmenge
Beispiel: molares Volumen v =
V
m³
in
n
mol
Spezifische Zustandsgrößen werden in der Regel mit dem
kleinen Buchstaben der jeweiligen extensiven Größe
dargestellt. Molare Größen erhalten zusätzlich einen
Überstrich (Beispiel: v für molares Volumen, h für molare
Enthalpie, usw.).
Hinweis: Die sogenannte molare Masse oder das Molgewicht,
ebenso, wie das Atomgewicht sind „molare“ Größen, deren
Einheiten jedoch nicht kohärent sind, d. h. deren Einheit nicht
identisch beziehungsweise zusammenhängend sind.
1.3 Ideale Gase 1,2,3
1.3.1 Ideale Gasgleichung
Das ideale Gas stellt in der Thermodynamik eine
zweckmäßige Idealisierung dar. Sie geht auf die Erfahrung
zurück, dass alle Gase bei „niedrigem“ Druck einfache und
übereinstimmende Zusammenhänge zwischen Druck,
Temperatur und Volumen aufweisen. Beim idealen Gas trifft
man folgende Annahmen:
6
•
Das ideale Gas besteht aus einer großen Anzahl von
Atomen oder Molekülen, deren
Abmessungen
vernachlässigbar klein gegenüber ihrer mittleren
Entfernung
voneinander
und
gegenüber
der
Behälterdimension sind.
•
Die Moleküle befinden sich in völlig regelloser,
translatorischer Bewegung.
•
Es gibt keine weiteren Wechselwirkung zwischen den
Molekülen außer elastischen Stößen. Die Zusammenstöße
zwischen den Molekülen und der Behälterwand sind
streng elastisch, d. h. Energie- und Impulsänderungen
unterliegen den Erhaltungssätzen der klassischen
Mechanik.
Beispiel für ein ideales Gas ist Luft in einem bestimmten
Druckbereich.
Abb.
6:
Verhalten
Zustandsänderungen.1
idealer
Gase
bei
verschiedenen
Die von Boyle-Mariotte und Gay-Lussac gefundenen Ansätze
ermöglichen die Erstellung der Zustandgleichung idealer
Gase oder kurz die ideale Gasgleichung.
p⋅V = n ⋅R ⋅T
p⋅ v = R ⋅T
oder
(1)
p⋅V = m⋅R ⋅T : m
p⋅ v = R ⋅T
mit
7
p = Druck des Systems in Pa
V = Systemvolumen in m³
n = Stoffmenge in mol
R = Universelle (oder molare) Gaskonstante,
häufig auch R.
J
mol ⋅ K
T = Absolute Temperatur in K
R = 8,314
m = Masse in kg
R = spezifische Gaskonstante in
J
kg ⋅ K
Hinweis: Die universelle Gaskonstante gilt für alle idealen
Gase, während die spezifische Gaskonstante stoffspezifisch
ist. Sie kann mit Hilfe der molaren Masse berechnet werden.
(2)
R = M ⋅R
Ein Maß für die Abweichung vom idealen Verhalten ist der
Realgasfaktor Z. Abbildung 7 zeigt durch Erfahrung
gewonnene Werte von Z als Funktion des Drucks.
Es gilt:
Z = f(p, T) =
p⋅v
R ⋅T
(3)
Bei kleinen Drücken verhalten sich die Gase nahezu ideal
( Z ≈ 1 ). Für höhere Drücke ( Z > 1 ) sind, im Gegensatz zu
idealen, reale Gase schwerer zu komprimieren, die
Teilchenabstoßung dominiert. Bei mäßigen Drücken ( Z < 1 )
überwiegen bei den meisten Gasen die anziehenden Kräfte
der Teilchen, das Gas ist vergleichsweise leicht
komprimierbar.
8
Abb. 7: Verhalten des Kompressionsfaktors Z für verschiedene Gase
bei 273,15 K. 1
1.3.2 Zustandsänderungen eines idealen Gases
Eine wichtige Eigenschaft von Zustandsgleichungen ist ihre
Wegunabhängigkeit, dass heißt es ist beliebig, auf welchem
Weg ein Zustand erreicht wird. In diesem Kapitel wird mit
Hilfe des Satzes von Schwarz ein einfaches Kriterium
vorgestellt, mit dessen Hilfe überprüft werden kann, ob eine
Beziehung eine Zustandsgleichung ist.
Die ideale Gasgleichung ist eine Funktion von zwei variablen
intensiven Größen, falls die dritte intensive Größe als
Funktionswert fungiert und die Stoffmenge beziehungsweise
die Masse konstant ist, so können sich folgende funktionale
Zusammenhänge ergeben:
p =p(v, T)
v = v(p, T)
T = T(v, p)
(4)
Betrachtet man nun die Änderung einer gewählten intensiven
Größe unter idealen Bedingungen, so ändern sich beide
Variablen ebenfalls. Ist die Wegunabhängigkeit gegeben, so
kann man ein totales Differential bilden.
9
Am Beispiel der Änderung des spezifischen Volumens ergibt
sich folgende Gleichung für das totale Differential.
(5)
v = v(p, T)
dv =
∂v
∂T
⋅ dT +
p
∂v
∂p
⋅ dp
(6)
T
Dabei wird (∂v ∂T )p als die partielle Änderung des
spezifischen Volumens mit der Temperatur bei konstantem
Druck und (∂v ∂p )T als die partielle Änderung des
spezifischen Volumens mit dem Druck bei konstanter
Temperatur bezeichnet. Die partiellen Differentialquotienten
(∂v ∂T )p und (∂v ∂p )T werden auch mit A und B benannt.
Abbildung 8 beschreibt graphisch das vollständige oder totale
Differential für die Änderung des Druckes.
Abb. 8: Darstellung des totalen Differenzials (Zustandsfläche) im p, v,
T – Diagramm.³
Das Differential kann allerdings nur als vollständig
angesehen werden, falls der Satz von Schwarz die
Vertauschung der einzelnen Differentiationsschritte zulässt
und damit die Unabhängigkeit vom Weg garantiert ist.
Gilt für ein Differential der Form dz = A ⋅ dx + B ⋅ dy , das
Kriterium
⌈ ∂A ⌉
⌈ ∂B ⌉
=
,
⌊ ∂y ⌋ x ⌊ ∂x ⌋ y
(7)
10
so ist dieses ein vollständiges Differential. Die damit
verbundene Wegunabhängigkeit macht das vollständige
Differential zu einer Zustandsgleichung. In den folgenden
Kapiteln werden wir auch Größen kennenlernen (sogenannte
Prozessgrößen), die wegabhängig sind und von denen sich
kein vollständiges Differential bilden lässt.
In manchen Fällen ist es hilfreich, einen unbekannten
partiellen Differentialquotient aus zwei anderen zu
berechnen. Aus Gleichung 6 und 7 lässt sich nun ein
Zusammenhang zwischen den einzelnen partiellen
Differentialen herstellen.
∂T
∂p
⋅
v
∂p
∂v
⋅
T
∂v
∂T
= -1
(8)
p
1.3.3 Ideale Gemische von Gasen
Ein Gasgemisch besteht aus zwei oder mehreren reinen
Gasen, die auch Komponenten genannt werden. Diese
Komponenten dürfen für unsere Betrachtungen nicht
miteinander
reagieren
und
keine
physikalischen
Veränderungen
ihrer
Eigenschaften
durch
Volumenveränderungen
oder
sogenannten
Wärmeerscheinungen.
Allgemein gelten die folgenden Definitionen, wobei zu
beachten ist, dass folgende Einheiten verwendet werden.
[m] = kg
[n] = mol
[M] = g/mol
Die Massenkonzentration (Massenanteil) eines beliebigen
Stoffes i
wi=
mi
mG
(9)
ist das Verhältnis der Teilmasse mi zur Gesamtmasse mG. Die
Gesamtmasse des Gemisches setzt sich aus der Summe der
Teilmassen zusammen.
j=1
∑m
j
= m1 + m 2 + m 3 + ........ + m n = m G
n
Da nach (10)
11
(10)
∑m
j
mG
=1
(11)
= w 1 + w 2 + w 3 + ........ + w n = 1
(12)
folgt
j=1
∑w
j
n
Analog
kann
man
(Stoffmengenanteil) gemäß
xi =
die
ni
nG
Stoffkonzentration
(13)
definieren. Sie ist das Verhältnis aus Teilstoffmenge zur
Gesamtstoffmenge.
Die Teilstoffmenge einer Komponente i ist über die Masse
und die molare Masse der Komponente i gegeben.
ni =
mi
Masse
=
M i molare Masse
(14)
Die relative Atom- bzw. Molekülmasse ist mit der molaren
Masse verknüpft. Für Atome entspricht
A r (i) ≡ M i
(15)
die relative Atommasse Ar der Komponente i der molaren
Masse der Komponente i. Der Index r steht für „relativ“ oder
im korrekten Fall für die relativen Atom- und
Molekülmassen.
Die relative Molekülmasse Mr(i) ist die Summe der relativen
Atommassen, aus denen das Molekül besteht und entspricht
der molaren Masse des Moleküls.
∑ A (i) = M
r
r
(k ) ≡ M k
(16)
Beispiel:
M r (CO 2 ) = A r (C) + 2 ⋅ A r (O) ≡ M CO2
Gleichung 16 gilt lediglich für Moleküle. Der geklammerte
Buchstabe i steht als Platzhalter für die Bezeichnung des
jeweiligen Atoms mit dieser relativen Atommasse. Analog
hierzu steht k als Platzhalter für die Bezeichnung des
12
Moleküls, z. Bsp. die chemische Formel oder der chemische
Name.
Die Gesamtstoffmenge ist die Summe der Teilstoffmengen.
nG = ∑ n j
(17)
Wie in Gleichung (12) erhält man auch hier
∑x
=1
j
(18)
Die Volumenkonzentration (Volumenanteil)
ri =
Vi
VG
(19)
ist das Verhältnis aus Teilvolumen und Gesamtvolumen.
VG = ∑ Vj
(20)
Gleichung 20 gilt nur für ideale Gase.
∑r
j
=1
(21)
Für ideale Gase gilt,
x i = ri
(22)
Das Verhältnis der Komponente i zur Komponente k, wird
wie folgt ausgedrückt.
ψi =
Vi
Vk
(23)
Die Stoffmengenkonzentration (veraltert: Molarität) einer
Komponente i erhält man aus dem Verhältnis der
Teilstoffmenge der Komponente i zum Gesamtvolumen.
ci =
ni
VG
(24)
Das Dalton-Gesetz besagt, dass die Summe der Partialdrücke
einer Gasmischung den Gesamtdruck ergibt (im
Gesamtvolumen).
13
p G = p1 + p 2 + p 3 + ....... + p K = ∑ p j
(25)
Zusammenhänge der Größen
Für die molare Masse eines Gemisches MG ergibt sich aus
m
; m i = n i ⋅ M i und ∑ m j = m G
n
M=
MG =
mj
nj
mG
=∑
= (∑
⋅ M j)
nG
nG
nG
M G = ∑ (x j ⋅ M j ) .
(26)
Mit
wi =
mi
=
mG
MG =
n i ⋅ Mi
n ⋅ Mi
M
= i
= xj ⋅ i
MG
∑ (n j ⋅ M j ) n G ⋅ M G
xi
⋅ Mi
wi
(27)
Oder mit
∑x
j
= 1 und w i = x i ⋅
Mi
MG
folgt
1 = ∑ (w j ⋅
wj
MG
) = MG ⋅ ∑ ( )
Mj
Mj
1
w
= ∑( i )
MG
Mi
(28)
Die individuelle Gaskonstante des Gemisches RG erhält man
aus dem idealen Gasgesetz und dem Dalton-Gesetz bei
konstantem Gesamtvolumen VG .
Partialdr
ücke
6Daltonsche
44
47
44
4
8
( p1 + p2 + .... + p n ) ⋅ VG
pG ⋅ VG
=
( m1 + m 2 + .... + m n ) ⋅ R G ⋅ T m G ⋅ R G ⋅ T
144424443
Summe der Massen der Teilsystem
14
(29)
RG =
m1 ⋅ R1 + m2 ⋅ R2
=
m1 + m2
∑ (m ⋅ R ) = ( w
∑
∑m
j
j
j
⋅ Rj )
(30)
j
Der Zusammenhang zwischen der universellen und der
spezifischen Gaskonstante des Gemisches wird über die
molare Masse des Gemisches hergestellt (s. Gleichung 2).
(31)
RG ⋅ M G = R
1.4 Reale Gase 1
Der Hauptgrund der Abweichung realer Gase vom idealen
Verhalten sind die zwischenmolekularen Wechselwirkungen,
das heißt Abstoßungs- oder Anziehungskräfte wirken. Wie
bereits in Kapitel 1.3 diskutiert, verhalten sich alle Gase bei
niedrigen Drücken nahezu ideal und ändern ihr Verhalten mit
dem Druck. Ein Maß für das ideale Verhalten war der
Kompressionsfaktor Z.
1.4.1 Van-der-Waalssche
Gleichung
Kondensation und kritische Größen
und
Die von Johannes van der Waals 1873 entwickelte Gleichung
stellt eine gute Nährung für das Verhalten mancher realen
Gase dar. Als Grundlage für seine Nährungsgleichung
benutzte er experimentelle und theoretische Befunde. Die
van-der-Waalssche Gleichung berücksichtigt sowohl das
Eigenvolumen der Moleküle als auch die Anziehungs- und
Abstoßungskräfte, die zwischen ihnen wirken.
Eine Formulierung der Van-der-Waals-Gleichung lautet:
p=
R ⋅T
a
−
( v − b) ( v)²
(32)
mit
R = spezifische Gaskonstante in J/mol K
p = Druck in Pa
v = V/m = spezifisches Volumen in m³/kg
a, b = temperaturunabhängige, stoffspezifische Van-derWaals-Koeffizienten
T = Absolute Temperatur in K
Gültigkeit der Gleichung
15
Abbildung 9 stellt den funktionalen Verlauf der Van-derWaals-Gleichung dar. Zwischen den gedachten Punkten C
und D durchläuft die Funktion eine Schwingung.
Abb. 9: Van-der-Waals-Isothermen bei verschiedenen Werten von
T/Tkrit. 1
Diese Schwingungen, auch Van-der-Waals-Schleifen
genannt, werden von der Erfahrung nicht bestätigt.
Abb. 10: Nähere Betrachtung der Van-der-Waals-Schleife.
Es wurde nie beobachtet, dass sich das Volumen bei
sinkendem Druck verringert, wie dies in Abbildung 10
zwischen dem Maximum und dem Minimum geschieht. Dies
ist ein Fehler in der Gültigkeit der Van-der-Waals-Gleichung.
Die Zustandsänderung von Punkt E zum Minimum ist
möglich, sie tritt bei einem überhitzten Fluid auf. Ebenso
kann die Zustandsänderung von Punkt C bis zum Maximum
durchgeführt werden. Das dabei entstehende unterkühlte
Fluid führt zu Gasimplosionen, d. h. große Mengen an Fluid
kondensieren schlagartig, es kommt zu Kavitation. Die
Strecke zwischen Minimum und Maximum besitzt keine
Gültigkeit.
Kondensation und kritische Größen
Das Volumen einer Gasprobe, welches sich zunächst im
Zustand A (vgl. Abb. 11) befand, soll bei konstanter
Temperatur verringert werden.
16
Zunächst erfolgt eine Zunahme des Druckes nach dem
Boyleschen Gesetz nahezu ideal, erst bei Punkt B kann man
die erste Abweichung vom idealen Verhalten erkennen. Am
Punkt C ist das Verhalten vollständig nichtideal. Versucht
man nun das Volumen weiter zu verringern, stellt man fest,
dass eine Flüssigkeit vom Zustand E auftritt.
Das Gas kondensiert auf der gedachten Linie CDE, wobei der
Druck konstant bleibt und der Anteil der flüssigen Phase
zunimmt (Die Linie CDE existiert in der Natur nicht.).
Diesen konstanten Druck zwischen dem Punkt C und E, bei
dem sich ein genau definiertes Gleichgewicht zwischen
flüssiger und gasförmiger Phase einstellt, bezeichnet man als
Dampfdruck.
Abb. 11: Experimentelle Isothermen von Kohlenstoffdioxid bei
verschiedenen Temperaturen. 1
Eine wichtige Rolle für die Beschreibung des Zustands eines
Stoffes spielen die kritischen Größen. Der auf der 31,04°CIsothermen mit einem Stern versehene Punkt in Abbildung 11
wird als kritischer Punkt, die Temperatur der dazugehörigen
Isotherme als kritische Temperatur TK bezeichnet. Alle
Isothermen unterhalb dieses Punktes verhalten sich wie oben
beschrieben, es findet eine Verflüssigung mit verschiedenen
Gleichgewichten zwischen Punkt C und E statt. Genau am
kritischen Punkt allerdings fallen der Punkt E und C direkt
zusammen, das heißt es kann sich keine Phasentrennung
ergeben. Bei TK findet man nur eine Phase, die man
definitionsgemäß Gas nennt. Oberhalb der kritischen
Temperatur (des kritischen Punktes) kann man eine Substanz
nicht verflüssigen.
17
Die einzige Phase, die bei T > TK das gesamte
Probenvolumen ausfüllt, weist in der Regel eine viel größere
Dichte auf, als man normalerweise für ein Gas erwartet, man
nennt sie überkritisches Fluid.
Am kritischen Punkt heißen die Zustandsgrößen kritische
Temperatur TK, kritischer Druck pK und kritisches
spezifisches Volumen vK oder zusammengefasst kritische
Größen.
Kritischer Punkt der Van-der-Waals-Gleichung
Am kritischen Punkt muss gelten:
a) VK' = VK''
(33)
Die Isotherme
Sattelpunkt:
b)
c)
∂p
∂v
der
kritischen
Temperatur
= 0 (Steigung, notwendig)
hat
einen
(34)
T, K
∂ ²p
∂ (v)²
= 0 (Wendepunkt, hinreichend)
(35)
T, K
Für die van-der-Waals-Gleichung gilt:
∂p
∂v
=-
RT
2a
+
=0
(v - b)² (v)³
(36)
=
2RT
6a
−
=0
(v - b)³ ( v) 4
(37)
T, K
∂ ²p
∂ (v)²
T, K
Die Lösungen aus den Bedingungen und den Gleichungen:
v K = 3b
TK ⋅ R =
(38)
8a
27b
(39)
18
a
27b²
PK =
(40)
Zur Überprüfung der Richtigkeit dieser Ausdrücke, wird der
Realgasfaktor ZK für den kritischen Punkt bestimmt.
ZK =
pK ⋅ vK
=
R ⋅ TK
3
8
{
oder
1{
(41)
Für ideale Gase
Für VDW -Gase
Im Vergleich mit realen Gasen, bei denen die Werte für ZK in
den meisten Fällen um ca. 0,3 schwanken, ist die
Abweichung des Van-der-Waals-Gases zum realen Verhalten
mit einem ZK von 3/8 relativ klein.
Fazit: Eindeutiger Zusammenhang zwischen VDWKoeffizienten a, b und den kritischen Größen existiert, man
kann die kritischen Größen in guter Nährung berechnen.
1.4.2 Gesetz der korrespondierenden Größen
(dimensionslose VDW-Gleichung)
Mit Hilfe der kritischen Größen kann man dimensionslose
Zustandsgrößen definieren.
pr =
p
pK
(42)
mit
pr = reduzierte Variable oder reduzierter Druck
Analog dazu erhält man die reduzierte Temperatur und das
reduzierte Volumen.
Tr =
T
TK
(43)
Vr =
v
vK
(44)
Man erhält so die dimensionslose Van-der-Waals-Gleichung,
die Lösungen für alle VDW-Gase und für einige reale Gase,
für die nicht die VDW-Gleichung gültig ist, liefert.
19
pr =
8Tr
3
−
3Vr − 1 Vr
(45)
Die Bedeutung des Prinzips dieser dimensionslosen
Gleichung beruht nicht so sehr auf seiner theoretischen
Interpretation als auf der Möglichkeit, die Eigenschaften
einer ganzen Reihe von Gasen in einem Diagramm
gemeinsam wiederzugeben (siehe Abb. 11 im Vergleich zu
Abb. 7).
Abb. 12: Auftragung des Kompressionsfaktors von vier
verschiedenen Gasen unter Verwendung reduzierter Variablen. 1
1.4.3 Virialkoeffizienten
Die Virialgleichung stellt eine vollständige Beschreibung des
Zustands eines realen Gases dar. Sie ist eine Potenzreihe, die
aus
sogenannten
Virialkoeffizienten
besteht.
Eine
Formulierung lautet:
p⋅v
= 1 + B(T) ⋅ p + C(T) ⋅ p² + ........
R ⋅T
(46)
Die Virialkoeffizienten A ( A ≡ 1 und A ⋅ p0 ) , B, ... sind
temperaturabhängig, stoffspezifisch und werden in der Regel
durch Erfahrung bestimmt. In der Praxis werden solche
Probleme numerisch bzw. rechnergestützt gelöst oder, falls es
die Abweichung zulässt, einige Glieder vernachlässigt, um
einen lösbaren Zusammenhang zu erhalten.
Es existiert keine Viralgleichung, die für den gesamten
Zustandsraum (p, v, T) gültig ist. So muss für jede
20
Viralgleichung exakt der Gültigkeitsbereich angegeben
werden mit Übergangslösungen zwischen benachbarten
Gleichungen. Aus der Wasserdampftafel liegen in
Abhängigkeit ihrer „Raumgröße“ 9 bis 13 Virialgleichungen
zugrunde.
1.5 Aufbau von Diagrammen
Thermodynamik 3
in
der
Um Zustandänderungen in der Thermodynamik einfach und
anschaulich darzustellen, verwendet man in der
Thermodynamik Diagramme.
Abb. 13: Darstellung einer Zustandsänderung im p, v-Diagramm mit
Isothermen als Parameter. 3
Aus der idealen Gasgleichung erkennt man, dass eine
Funktion Z(p, v, T) = 0 existiert. Man benötigen also
entweder drei Zustandsgrößen oder bei bekannter Funktion
zwei um die dritte berechnen zu können. Die dritte
Zustandsgröße ist in Darstellung 13 die Temperatur T als
Parameter.
In den meisten Fällen werden Zustandsänderungen so
durchgeführt, dass sie über Parameterkurven (Parameter, ist
eine neben den eigentlichen Variablen auftretende konstante
Hilfsvariable) erfolgen. Für das ideale Gasgesetz würde man
zum Beispiel Temperatur, Druck oder das Volumen konstant
halten.
In einem Diagramm werden solche Parameterkurven für V =
konstant als Isochore, für T = konstant als Isotherme und für
p = konstant als Isobare bezeichnet.
In unserem Beispiel (Abbildung 12) wird über die T1Isotherme der Zustand 1’ und über eine isochore der Zustand
2 erreicht. Die Zustandsänderung 1 -> 2 kann auf unendlich
vielen verschiedenen Wegen geschehen.
21
1.6 Wärmedehnung
Wie aus der Erfahrung bekannt, ändern sich bei Zufuhr von
Wärme die Temperatur und das Volumen eines Stoffes (Bsp.:
Metallstab
bei
Wärmezufuhr).
Charakteristische
Proportionalitätsfaktoren, die Volumenänderung und
Temperatur
mathematisch
verbinden,
sind
die
Ausdehnungskoeffizienten.
1.6.1 Wärmedehnung fester Körper
Man definiert als Längenausdehnungskoeffizient,
˺ :=
1 ∂L
⋅
L ∂T
(47)
P
und als kubischer Volumenausdehnungskoeffizient,
˼ :=
1 ∂V
⋅
V ∂T
(48)
P
Zwischen den
Zusammenhang,
Koeffizienten
˼ ≈ 3⋅ ˺ .
besteht
der
folgende
(49)
der wie folgt hergeleitet wird.
∂V = ∂L³
∂V
= 3 ⋅ L² → ∂V = 3 ⋅ L² ⋅ ∂L
∂L
einsetzen in Gleichung (25)
˼ :=
1 3L/ ²/ ⋅ ∂L
⋅
L/³
∂T
= 3⋅
p
⌈1
∂L ⌉
∂T p ⌋
⌊ L42
1
43
˺
⋅
˼ ≈ 3⋅ ˺
Die Ausdehnungskoeffizienten sind häufig temperaturabhängig, deshalb ist eine integrale Mittelwertbildung
22
notwendig, falls man die Wärmedehnung über einen
bestimmten Temperaturbereich bestimmen will. Man nehme
an, α ist im Temperaturbereich zwischen T1 und T2 (T1< T2)
nicht konstant, wobei die integralen Mittelwerte von einer
Bezugszugstemperatur T0 zu T1 beziehungsweise T2 gegeben
sind.
T2
T2
T1
T1
T0
T0
∫ ˺ ⋅ dT ≡ ∫ ˺ ⋅ dT − ∫ ˺ ⋅ dT
Durch Integration erhält man
˺ |TT12 ⋅ (T2 − T1 ) ≡ ˺ |TT20 ⋅(T2 − T0 ) − ˺ |TT10 ⋅(T1 − T0 )
und
˺ |TT12 ≡
1
˺ |TT02 ⋅(T2 − T0 ) − ˺ |TT10 ⋅(T1 − T0 )
(T2 − T1 )
(
)
(50)
Diese Methode der Mittelwertbildung gilt auch für andere
Anwendung, z. Bsp. bei cP über T.
Abb.
14:
Lineare
Ausdehnungskoeffizienten.
Mittelwertbildung
23
bei
dem
Tabelle 1 zeigt einige Längenausdehnungskoeffizienten für
verschiedene Stoffe.
Tab. 1: Längenausdehnungskoeffizienten von verschiedenen
festen Stoffen und in verschiedenen Temperaturbereichen
Stoffe
̅m
̅m
100° C
200° C
˺ 0°C in
˺ 0°C in
K⋅m
K⋅m
Al (99,5 %)
23,8
24,5
Gusseisen
10,4
11,1
Glas (techn.)
3,5 – 8,1
3,6 – 84
Quarzglas
0,5
0,6
Kupfer
16,5
16,9
Messing (62% Cu) 18,4
19,3
Stahl (0,2 – 0,6 %) 11
12
Formeln der Dehnung
Zur Berechnung der Volumen- und Längendehnung gilt:
T
(51)
∆L = α T2 ⋅ L1 ⋅ (T2 − T1 )
1
T
˝V = ˼ T2 ⋅ V1 ⋅ (T2 − T1 )
(52)
1
1.6.2 Wärmedehnung bei Flüssigkeiten
Die Wärmedehnung muss auch bei inkompressiblen
Flüssigkeiten (̊(p)=const) berücksichtigt werden, weil V(T)
≠ const, wie die überschlägige Betrachtung
γ H 2 0 = (3 − 6) ⋅ γ Stahl
zeigt, aus der die Notwendigkeit von Überlastarmaturen
direkt ablesbar ist.
Tab. 2: Längenausdehnungskoeffizienten von verschiedenen
flüssigen Stoffen und in verschiedenen Temperaturbereichen
Stoffe
cm³
cm³
50° C
100° C
˼ 0°C in
˼ 0°C in
K ⋅ m³
K ⋅ m³
Hg
182,2
182,6
Glycerin
520
Benzol
1270
Für H 2 0 :
˼ (0°C) = - 0,085 dm³/K ⋅ m ˼ (50°C) = 0,462 dm³/K ⋅ m
24
1.7 Übungen zum ersten Kapitel
Zur Behandlung einiger Aufgaben benötigen Sie folgende
relative Atommassen:
1,008H
12,011C
14,007N
15,999O
32,064S
126,9I
1. Aufgabe
Beweisen Sie, dass die
a) ideale Gasgleichung
b) Van-der-Waals-Gleichung
eine Zustandsgleichung ist. Welches Kriterium verwenden
Sie dazu?
2. Aufgabe
In einem starren Gefäß mit einem Volumen von 10 dm3
befinden sich 1 mol Stickstoff und 3 mol Wasserstoff bei
einer Temperatur von 25 oC.
a) Wie groß sind die Partialdrücke und der Gesamtdruck?
Welche Annahmen müssen Sie treffen und welche Gesetze
wenden Sie an?
p N 2 /1 = 2,5 bar, p H 2 = 7,4 bar und pGes/1 = 9,9 bar
(
)
b) In das Gefäß werden isotherm zusätzlich 1 mol Stickstoff
und 1 mol Sauerstoff gebracht. Berechnen Sie die
Partialdrücke und den Gesamtdruck der neuen Mischung
sowie deren mittlere relative molare Masse.
pN 2 /2 = 5 bar, pH 2/2 = 2,5 bar und pGes21 = 14,9 bar
M = 15,679 g/mol
3. Aufgabe
Berechnen Sie die spezielle Gaskonstante, die Masse und die
Stoffmenge der Luft von 20 °C und 8 bar, die sich in einem
starren Speicher von 14 m3 Inhalt befindet und sich wie ein
25
ideales Gas mit M = 28,96 g/mol verhält.
(RLuft = 287,1 J/(kg*K), m=133,12 kg, n = 4,6 kmol)
4. Aufgabe
a) Bestimmen Sie die Masse und die Stoffmenge der Luft
von 20° C und 1 bar, die sich in einem starren Speicher von
70 m3 Inhalt befindet; sie kann im betrachteten
Zustandsbereich als ein ideales Gas mit der Gaskonstanten R
= 287,1 J/(kgK) angesehen werden. (m = 83,17 kg, n = 2,87
kmol)
b) Welche Menge und Masse müssen zugeführt werden, dass
der Druck bei gleicher Temperatur auf 8 bar steigt?
(∆m = 582,2 kg, ∆n = 20,1 kmol)
5. Aufgabe
Die Analyse eines Hochofen-Gichtgases ergab folgende
Zusammensetzung in Massenanteilen (Angabe in Prozent):
2,07 % H2; 25,1 % CO; 16,7 % CO2 und 56,13 % N2.
Bestimmen Sie unter der Annahme idealen Gasverhaltens:
a) Seine Raumanteile und seine molare Masse
(r (H2) = 23,85 %, r (CO) = 20,81, r (CO2)=8,81%, r(N2)=
46,53%, M=23,22 g/mol)
b) sowie seine Dichte unter Normbedingungen (1,01325
bar; 0° C).(ρ = 1,036 kg/m³)
6. Aufgabe
Leuchtgas besteht aus den Volumenanteilen 50 % H2, 30 %
CH4, 15 % CO, 3 % CO2 und 2 % N2. Bestimmen Sie
a) seine Gaskonstante, seine mittlere molare Masse und die
jeweiligen Massenanteile, (R=698,7 J/(kg*K), M=11,9
g/mol, w(H2)= 8,47%, w(CH4)= 40,44%, w(CO)= 35,30%,
w(CO2)= 11,09 %, w(N2)= 4,70%)
b) die Dichte des Leuchtgases bei 25 °C und 1 bar, (ρ=0,48
kg/m³)
c) die
prozentuale
Drucksteigerung,
wenn
die
Leuchtgastemperatur in einem starren Behälter unter
Sonneneinstrahlung von 20 °C auf 80 °C steigt. (A=20,4%)
26
d) Zeigen Sie, dass für ideale Gasmischungen Raum- und
Stoffmengenanteile gleich sind.
7. Aufgabe
Methan wird mit Luft vollständig verbrannt. Es entstehen 292
kg Verbrennungsgas, das bei idealer Reaktion aus 15,05 %
Kohlendioxid, 12,32 % Wasserdampf (jeweils in
Massenprozent) und aus Stickstoff besteht. Bestimmen Sie:
a) die Stoffmengen und die Stoffmengenanteile der
Komponenten, ( n(H2O)=1,997 mol, n(N2)= 7,571 mol,
n(CO2)= 0,999 kmol, x(H2O)=18,898%, x(N2)=71,648%,
x(CO2)= 9,454% )
b) die Gaskonstante und die molare Masse des
Verbrennungsgases und (R= 300,8 J/(kg*K), M=27,64
g/mol)
c) seine Dichte bei 25 °C und 101325 Pa. (ρ = 1,13 kg/m³)
8. Aufgabe
Die Abgasanalyse einer Steinkohle-Feuerung ergab bei
Normbedingungen (101325 Pa, 0 °C) folgende
Volumenanteile: 11,2 % CO2, 3 % H2O, 0,8 % SO2, 7 % O2
und 78 % N2.
Wie groß sind die Massenanteile, die Dichte im Normzustand
und die Gaskonstante des Abgases, wenn die Einzelgase und
ihre Mischung als ideale Gase zu betrachten sind?
(w(H20)= 1,8%, w(SO2)= 1,7%, w(O2)= 7,44%, w(CO2)=
16,39 %, w(N2)= 72,67%, R= 276,6 J/(kg*K),
ρ=1,342 kg/m³)
27
2 Hauptsätze der Thermodynamik1,3
2.1 Arbeits- und Energieformen3
2.1.1 Wärme
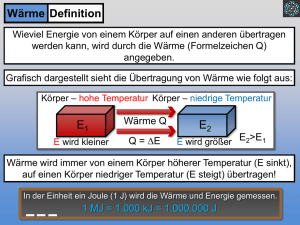
Wärme ist eine Größe, die bei der Wechselwirkung zwischen
einem System und seiner Umgebung auf Grund eines
Temperaturunterschieds zwischen dem System und der
Umgebung auftritt. Man sagt, es wird eine gewisse Menge an
Wärme übertragen und damit auch eine Änderung der
Temperatur hervorgerufen. Wärme ist eine Form von
Energie, welche beim Transport über die Systemgrenze
hinweg auftritt. Sie wird mit Q bezeichnet und hat die Einheit
der Energie Joule (1J = 1 Nm).
Die Temperatur spielt für die Wärme eine wesentliche Rolle,
da sie ein Maß für die Fähigkeit eines Systems ist, Wärme
aufzunehmen oder abzugeben. Da die Wärme nur bei
überschreiten der Systemgrenze in Erscheinung tritt, kann sie
natürlich nicht im System festgestellt werden. In dem System
kann lediglich die Temperatur gemessen und damit der
Zustand des Systems bestimmt werden. Die über die
Systemgrenzen hinweg übertragene Wärme hängt von dem
gewählten Prozess ab (wegabhängig), deshalb ist die Wärme
keine Zustandsgröße, sondern eine Prozessgröße.
Die Wärme ist festgelegt, durch
(53)
Q = m ⋅ c ⋅ ∆T
mit
Q = Wärme in J
m = Masse in kg
c = spezifische Wärmekapazität in J/(kg K)
T= Absolute Temperatur in K.
In der Regel wird die Änderung der Wärme betrachtet (Gl.
54). In einigen Fällen, wie bei offenen Systemen, ist es
zweckmäßig, die Änderung der Wärme mit der Zeit zu
verfolgen (Gl. 55). Man nennt diese neue Größe
Wärmestrom.
δQ = d(m ⋅ c ⋅ T)
mit
m = const , c = const. → δQ = m ⋅ c ⋅ dT
28
(54)
δQ & d
= Q = (m ⋅ c ⋅ T)
dt
dt
(55)
2.1.2 Arbeit
Der Begriff Arbeit ist aus der Mechanik vertraut. Das
Verrichten von Arbeit ist ebenfalls eine Wechselwirkung
zwischen System und Umgebung. Drückt man zum Beispiel
einen Kolben in ein zylindrisches Gefäß ein, so verändert sich
das Volumen, sowie die Grenze des Systems (vgl. Abb. 15).
Die verrichtete Arbeit erhöht die Energie des Systems. Arbeit
ist also wie Wärme eine Energieform, die in Erscheinung
tritt, wenn sie die Systemgrenze überschreitet. Ein System
kann keine Arbeit, sondern lediglich Energie enthalten. Somit
beschreibt die Arbeit keinen Zustand und ist damit auch eine
Prozessgröße.
Volumenänderungsarbeit
Volumenänderungsarbeit tritt bei der Veränderung des
Volumens eines Systems auf.
1
2
Abb. 15: Kolben
Volumenarbeit.
im
Zylinder
zur
Veranschaulichung
der
Abbildung 15 zeigt einen Zylinder, dessen beweglicher
Kolben Gas durch die Kraft F vom Zustand 1 zum Zustand 2
komprimiert. Auf der Strecke dx wird bei diesem Vorgang
Arbeit geleistet. Betrachtet man das System allgemein, so
setzt sich die Arbeit W12 = ∫ F ⋅ dx zusammen, aus der
Volumenänderungsarbeit WV12 und der Reibungsarbeit,
zwischen den Gas- beziehungsweise Fluidteilchen. Die
Reibungsarbeit zwischen Koben und Zylinderwand, wird hier
nicht betrachtet, da sie außerhalb der Systemgrenze geleistet
wird. Die wirkliche an der Kolbenstange angreifende Kraft
muss größer sein.
29
Unter der Annahme, dass der Vorgang quasistatisch verläuft,
d. h. der Druck im gesamten Volumen gleich groß ist, gilt:
2
WV12 = − ∫ F ⋅ dx = − ∫ p ⋅ A ⋅ dx = − ∫ p ⋅ dV
(56)
1
mit
WV12 = Volumenänderungsarbeit in J (von Zustand 1 nach
Zustand 2)
A = Kolbenfläche in m²
p = absoluter Druck in Pa
V = Volumen des Systems
Da bei Kompression des Systems „Gas“ sein Volumen
verringert wird, wird der Term dV negativ. Da allerdings bei
der Kompression dem System Energie zugeführt wird, wird
ein negatives Vorzeichen verwendet, so dass der
Energieeintrag positiv ist.
Abb. 16: Zustandsänderung von 1 nach 2 entlang der Kurve p(V) im
p, v-Diagramm. Die Volumenarbeit stellt sich als Fläche unterhalb
der Kurve p(V) dar.
Nutzarbeit
30
Im Allgemeinen befindet sich ein Zylinder in einer
Umgebung mit einem charakteristischen Umgebungsdruck.
Betrachten wir die Atmosphäre der Erde als Umgebung, so ist
der Umgebungsdruck der Atmosphärendruck pu. In Abb. 16
ist pu < p1 eingetragen. Es ist offenkundig, dass pu *A einen
Anteil zur Kraft liefert, die an der Kolbenstange angreift und
diesen verschiebt.
Expandiert ein Gas in diesem Zylinder, so muss es gegen den
Druck der Umgebung arbeiten, um sein Volumen um dV zu
vergrößern. Mit anderen Worten muss das Gas im Zylinder
die Umgebungsluft wegschieben, um zu expandieren.
Abb. 17: Das sich im Zylinder befindliche Gas expandiert gegen den
Atmosphärendruck und leistet dabei Umgebungsarbeit.
Bei der Expansion eines Gases ist also nicht die gesamte
Volumenänderungsarbeit nutzbar (außer der Zylinder wäre
von einem Vakuum umgeben). Die sogenannte Nutzarbeit ist
eine Differenz (Gl. 57 und 58).
Expansion:
2
WNE12 = − ∫ p ⋅ dV + p U ⋅ (V2 − V1 )
(57)
1
Bei einer einmaligen Kompression des Gases liefert der
Umgebungsdruck zusätzliche Umgebungsarbeit, man
benötigt weniger Kompressionsarbeit.
1
WNK12 = − ∫ p ⋅ dV + p U ⋅ (V1 − V2 )
(58)
2
Arbeit zur Veränderung äußerer Energien
Die obengenannte Volumenänderungsarbeit führt zu einer
Änderung der Inneren Energie des Systems. Einem System,
31
welches sich nicht in Ruhe befindet oder eine Höhendifferenz
überbrücken muss, muss Arbeit in Form von potentieller oder
kinetischer Energie ab- oder zugeführt werden.
Wa = E a , 2 − E a ,1 =
m
⋅ w 22 − w12 + m ⋅ g ⋅ (z 2 − z1 )
1
424
3
2
(
)
(59)
h
Technische Arbeit
Technische Arbeit (oder Druckänderungsarbeit) wird von
einem stetig fließenden Stoffstrom eines Fluids verrichtet. Sie
tritt
deshalb
nur
in
offenen
Systemen
auf.
Abb. 18: Darstellung der technischen Arbeit. Das strömende Fluid
verrichtet über die Welle Arbeit.
Technische Arbeit kann kontinuierlich an der Welle einer
Strömungsmaschine zu- oder abgeführt werden.
2
Wt12 = ∫ V ⋅ dp
(60)
1
Reibungsverluste
Neben den erwähnten Arbeitsformen treten bei irreversiblen
(nicht umkehrbaren) Prozessen Verluste als Reibung auf.
Dadurch wird die Qualität der Energie verringert, sie ist nicht
im gleichen Maße nutzbar. Der gewählte Prozess kann nicht
mehr reversibel, d. h. umkehrbar sein, da der entsprechende
Teil an Energie fehlt.
Wegabhängigkeit der Arbeit und der Wärme
Abb. 19 zeigt eine Zustandsänderung auf den Wegen a, b und
c im p, v-Diagramm. Die Arbeit stellt sich dabei als Fläche
zwischen der jeweiligen Zustandsänderungskurve (a oder b)
und der x-Achse (v-Achse) dar.
Da die Flächen unterhalb der jeweiligen Kurven verschieden
sind, in unserem Fall ist die Fläche unterhalb der Kurve (oder
32
Zustandsänderung) b größer als die unter Kurve a, ist der
Verlauf der Zustandsänderung für die Größe der Arbeit
entscheidend.
Abb. 19: Verschiedene Zustandsänderungen (a, b, c) im p,vDiagramm die zum selben Endzustand führen und dabei
unterschiedliche Arbeit verrichten.
2.1.3 Innere Energie und Enthalpie
Die innere Energie U ist eine extensive Größe mit der Einheit
Joule. Sie nimmt mit der Temperatur eines Systems streng
monoton zu und ist die Summe der Translations-, Rotationsund Schwingungsenergien (Innere Energien Ei aller Moleküle
eines Systems). Neben der inneren Energie kann ein System
weitere Energien enthalten. Bewegt sich das System, so
besitzt es kinetische Energie Ekin, wenn es auf einer Höhe
über einen Nullniveau liegt, potentielle Energie Epot.
∑E
i
= U + E kin + E pot
14243
(61)
Ea
Generell kann die absolute innere Energie nicht bestimmt
werden, in der Technik gibt man in der Regel, die Änderung
der inneren Energie ∆U an.
∑ ∆E
i
=∆U + ∑ ∆E a
(62)
Enthalpie
33
Die Enthalpie ist eine extensive Zustandsgröße mit dem
Zeichen H und der Einheit Joule. Sie ist eine definierte
Größe, die bei einigen Anwendungen eine praktische
Abkürzung darstellt. (Beispiel: bei offenen System wird
durch
die
Enthalpie
die
Volumenänderungsarbeit
berücksichtigt, siehe 2.2 Erster Hauptsatz)
H := U + p ⋅ V
(63)
2.2 Der
Erste
Thermodynamik
Hauptsatz
der
2.2.1 Formulierungen des ersten Hauptsatzes
Um den Zusammenhang zwischen Wärme und Arbeit zu
finden, führte Joule ein Experiment (Abb. 20) durch. Ein mit
Luft gefüllter Zylinder wird mit einer Wärmequelle beheizt.
Der Kolben im Zylinder sei ideal und reibungsfrei, sowie
freibeweglich.
Abb. 20: Zufuhr von Wärme mit gleichzeitiger Abfuhr von Arbeit.
Infolge der Wärmedehnung des Gases bewegt sich der
Kolben nach außen und das Gewicht wird gehoben, dadurch
wird die Arbeit W12 vom System geleistet. Das Ergebnis
dieses Versuchs zeigt, dass Wärme und Arbeit gleichwertig
sind. Wärme kann in Innere Energie und diese wiederum in
Arbeit umgewandelt werden.
Mathematische Form des 1. Hauptsatzes
Die Erkenntnis, dass Wärme und Arbeit austauschbare
Energieformen sind, führte zur mathematischen Formulierung
des Ersten Hauptsatzes, in dessen Mittelpunkt die innere
Energie U eines Systems steht. U ist eine Zustandsgröße und
damit vom Weg der Zustandsänderung unabhängig. Der erste
Hauptsatz
in
seiner
allgemeinen
mathematischen
34
Formulierung für geschlossene
Energiebilanz (Gl. 64).
Systeme
ist
Q12 + W12 = U 2 − U1 + E a 2 − E a1
eine
(64)
Die Energiebilanz in der differenziellen Formulierung zeigt
Gleichung 65.
δQ + δW = dU + dE a
(65)
Eine Formulierung des Ersten Hauptsatzes haben wir bereits
kennen gelernt:
„Wärme und Arbeit sind gleichwertig.“
Über die Systemgrenzen eines abgeschlossenen Systems kann
weder Arbeit, äußere Energie noch Wärme transportiert
werden. Aus dieser Erkenntnis folgt eine weitere
Formulierung des ersten Hauptsatzes:
„Die innere Energie eines abgeschlossenen Systems ist
konstant.“
2.2.2 Zustandsgleichung der inneren Energie
Betrachtet man die innere Energie U makroskopisch, so ist
sie grundsätzlich eine Funktion von Druck, Temperatur und
Volumen. Da die drei Größen untereinander verknüpft sind,
wählt man als Variablen lediglich das Volumen und die
Temperatur aus (Ist T und V gegeben, so ist p bei m=const.
auch bestimmt, man könnte als Variablen ebenso p und T
oder p und V verwenden. Dies ist eine reine Festlegung.). Da
U = f (T, V) (mit m=const.) und die innere Energie eine
Zustandsgröße ist, kann man für die Änderung der inneren
Energie das totale Differential bilden.
dU =
∂U
∂U
⋅ dT +
⋅ dV
∂T V
∂V T
123
123
CV
(66)
πT
Die Wärmekapazität bei konstantem Volumen Cv ist eine
extensive Größe die einen Zusammenhang zwischen der
Änderung der Temperatur und der Wärme herstellt. Der
Innendruck πT (oder auch Binnendruck von der VDWGleichung genannt) ist die Änderung der Inneren Energie
mit dem Volumen bei konstanter Temperatur.
Joul´sches Experiment
35
Joule wollte mit Hilfe der in Abbildung 21 dargestellten
Versuchanordnung den Innendruck eines Systems
bestimmen, indem er ein Gas in ein Vakuum expandieren ließ
und versuchte die Temperaturänderung des Wasserbads mit
Hilfe eines Thermometers zu bestimmen. Nachdem Joule den
Gashahn zwischen den zwei Metallgefäßen (eines evakuiert,
das andere mit dem Gas gefüllt) öffnete, stellte er fest, dass
sich die Temperatur nicht änderte.
Nach dem ersten Hauptsatz bedeutet dies, dass keine Arbeit
bei der Expansion verrichtet wurde (W12=0). Wäre Arbeit
geleistet worden, so hätte die Arbeit, die Temperatur (Innere
Energie) des geschlossenen Systems der metallenen Gefäße
steigen lassen. Die Temperaturdifferenz zwischen Wasserbad
und Gefäßinnenraum würde zur Wärmeübertragung führen,
dass Wasserbad müsste sich erwärmen.
Abb. 21: Versuchanordnung von Joule zur Bestimmung des
Binnendrucks π.1
Aus thermodynamischer Sicht bedeutet dies, dass bei
isothermer, freier Expansion, sich die innere Energie eines
Gases sich nicht ändert. Im Sinne des ersten Hauptsatzes hat
dies zu Folge, dass die Änderung der Inneren Energie nicht
vom Volumen abhängig ist, sondern lediglich von der
Temperatur
Allerdings ließ die Versuchanordnung keine genauen
Messungen zu, insbesondere war die Wärmekapazität der
Gefäße so hoch, dass die bei dem Experiment tatsächlich
auftretende Temperaturdifferenz viel zu klein war, als dass
man sie hätte messen können. Deshalb gilt:
Treten geringe oder keine Wechselwirkungen zwischen den
Atomen oder Molekülen eines Gases auf, wie dies bei idealen
Gasen der Fall ist, so ist die Innere Energie unabhängig von
der Änderung des Volumens.
36
In der mathematischen Konsequenz bedeutet dies, dass für
die differentielle Änderung der Inneren Energie eines
idealen Gases Gleichung 70 gilt.
dU =
∂U
∂U
⋅ dT +
⋅ dV
∂T V
∂V T
123
123
CV
=0
Aus diesem totalen Differential folgt der Zusammenhang:
dU = C V ⋅ dT
(67)
2.2.3 Zustandsgleichung der Enthalpie
Analog zu Kapitel 2.2.2 kann man die Enthalpie, da ihre
Definitionsgleichung ( H := U + p ⋅ V ) nur aus Zustandsgrößen
besteht, ebenfalls als Zustandsgröße bezeichnen.
Mit einer analogen Begründung wie im vorangestellten
Kapitel, nämlich eine Folge des folgenden Joule-ThomsonExperiments definiert man die Enthalpie als Funktion der
variablen Temperatur T und Druck p und erhält das
vollständig Differential für die Änderung der Enthalpie:
dH =
∂H
∂H
⋅ dT +
∂T
∂p
123p
CP
⋅ dp
Der erste Koeffizient entspricht der
Wärmekapazität bei konstantem Druck.
CP =
∂H
∂T
(68)
T
Definition
der
(69)
p
Für ideale Gase gilt analog:
dH = C P ⋅ dT
(70)
Joule-Thomson-Effekt
Von James Joule und William Thomson stammt die Idee zu
einer Messanordnung (Abb. 22), in der eine
Zustandsänderung bei konstanter Enthalpie ablaufen kann.
37
In einem Zylinder, in dessen Mitte ein Drosselventil
eingebaut ist und der mit freibeweglichen und nahezu
reibungsfreien Kolben verschlossen ist, wird ein Gas des
Volumens VA so eingesperrt, dass es sich zwischen linkem
Kolben und Drossel befindet. Der konstante Druck pA auf den
linken Kolben sei größer als der konstante Druck pB auf dem
rechten Kolben. Beide Systeme befinden sich in Ruhe und
liegen horizontal, so dass keine äußeren Kräfte wirken. Durch
den Druckunterschied geht System A in System B über, d.h.
der Kolben auf der rechten Seite wird weggeschoben. Dieser
Vorgang verläuft quasistatisch, also durch endlich viele
Gleichgewichte, dass die Drücke konstant bleiben.
Die gesamte Anordnung ist thermisch isoliert, so dass der
Prozess adiabatisch verläuft. Meist beobachtet man nun auf
der Niederdruckseite eine niedrigere Temperatur TB als die
Temperatur TA auf der Hochdruckseite. Die Temperatur- und
Druckdifferenz sind zueinander proportional. Diese
Änderung der Temperatur bei einer adiabatischen Expansion
nennen wir heute Joule-Thomson-Effekt.
Abb. 22: Messanordnung für den Ablauf einer isenthalpischen
Zustandsänderung.1
Um das Experiment aus thermodynamischer Sicht näher zu
betrachten, verwendet man den ersten Hauptsatz.
Q
/ aB − W
/ aA
/ AB + WAB = U B − U A + W
{
14
243
= 0,
adiabates
System
= 0,
das System befindet sich in Ruhe
und besitzt keine
Geschwindi gkeit
Während des Experiments wird zweimal Volumenarbeit
geleistet. Zunächst wird mit dem konstanten Druck pA das
Gas durch das Drosselventil gedrückt, um sich danach gegen
den konstanten Gegendruck pB auszudehnen.
38
V ( ∆t ) = VB
V ( ∆t ) = 0
WAB = WV ,AB = −
∫
p A ⋅ dV −
V(t = 0) =VA
∫p
B
V(t = 0) = 0
⋅ dV
Da alle Drücke konstant sind kann man relativ einfach
integrieren.
WAE = −p E ⋅ VE − p A ⋅ (−VA )
Durch Einsetzen in den angepassten ersten HS erhält man:
WAE = p A ⋅ VA − p E ⋅ VE = U E − U A
p A ⋅ VA + U A = U E + p E ⋅ VE
142
4 43
4 142
4 43
4
HA
HE
HA = HE
Daraus kann man erkennen, dass es sich bei diesem Vorrang
um eine Volumenausdehnung ohne Änderung der Enthalpie
handelt, man spricht von einem isenthalpen Prozess.
Linde-Verfahren
Den Joule-Thomson-Effekt verwendete Linde um ein
Verfahren (Abb. 23) zur großtechnischen Kühlung und
Verflüssigung von Gasen unterhalb der Inversionstemperatur
beim Entspannen zuentwickeln. Ein Gas wird im Kreislauf
geführt und kühlt, falls es sich unterhalb der
Interventionstemperatur befindet, ab.
Abb. 23: Prinzipieller Aufbau einer Linde-Maschine. 1
39
Er verwendete dazu den sogenannten Joule-ThomsonKoeffizient, der die Änderung der Temperatur mit der
Änderung des Drucks bei konstanter Enthalpie verfolgt.
µ :=
∂T
∂p
H
∂H
∂p
=−
cp
T
=−
ε
cp
(71)
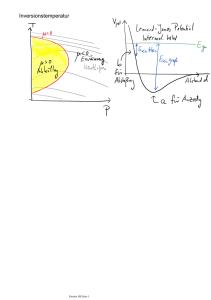
Abbildung 24 zeigt aus der Erfahrung gewonnene Werte von
Druck und Temperatur bei Durchströmung einer Drossel,
wobei die konstante Enthalpie als Parameter dient. Aus dem
Diagramm ist zu erkennen, dass für µ > 0 eine Abkühlung
des Gases (d.h. dp negativ und dT ebenfalls negativ) eintritt.
Die Funktionswerte T der Grenzkurve zwischen dem
Erwärmungs- und Abkühlungsgebiet, entsprechen der
Inversionstemperatur bei dem jeweiligen Druck. So erhalten
wir auch eine Definition der Inversionstemperatur. Strömt ein
Gas durch eine Drosslung mit einem Druckunterschied ∆p,
so erwärmt es sich oberhalb der Inversionstemperatur
(kühlt unterhalb der Inversionstemperatur ab).
Abb. 24: Interversionskurve und Isenthalphen zur Kennzeichnung
des Verhaltens eines Gases beim Durchströmen einer Drossel.1
Für alle idealen Gase ist der Joule-Thomson-Koeffizient
gleich null.
2.2.4 Zusammenhang zwischen CV und Cp bei
idealen Gasen
40
Wir können mit Hilfe der Differentialquotienten bei
konstantem Druck beziehungsweise Volumen beide
Wärmekapazitäten bestimmen.
Da gilt
Cp − C V =
∂H
∂T
−
p
∂U
∂T
V
mit
H = U + pV = U + n ⋅ R ⋅ T
folgt für die Ableitung
∂H ∂U
=
+ n⋅R
∂T ∂T
und erhalten für das ideale Gas
Cp − C V =
∂U
∂T
+ n⋅R −
p
∂U
∂T
= n⋅R = m⋅R
(72)
p
2.2.5 Der erste Hauptsatz für offene Systeme
Kompressoren,
Turbinen,
kontinuierlich
betriebene
Reaktoren oder Wärmeübertrager, alle diese Apparate und
Maschinen arbeiten nach dem Prinzip eines offenen Systems.
Über die Grenzen des offenen Systems wird sowohl Arbeit,
Wärme und Masse übertragen.
Um den ersten Hauptsatz für offene Systeme zu finden,
betrachten wir ein allgemein übertragbares Modell.
Ein Zylinderraum sei so als geschlossenes System definiert,
dass es zwei massendurchlässige Grenzen an der Stelle 1 und
der Stelle 2 besitzt, dieses definierte System wird
Kontrollraum genannt. Das System sei stationär, das heißt
während des gesamten Vorgangs ändert sich im Kontrollraum
Temperatur, Druck, usw. nicht oder mit anderen Worten alle
Stoff- und Energieströme sind zeitlich konstant. Die
mathematische Formulierung wird in Gleichung 73
wiedergegeben.
41
∂
=0
∂t
(73)
Abb. 25: Das geschlossene System als Modell für das offene System. 3
Wie in Abbildung 25 gezeigt, wird ein Volumen der Masse
˝m1 zum Zeitpunkt t = 0 in den Kontrollraum eingeschoben.
Beim Eintreten wird Verschiebe- beziehungsweise
Volumenarbeit geleistet. Nach einer Zeit ˝t ist die Masse
˝m1 vollständig eingeschoben. Am Austritt hat nach der
gleichen Zeit ˝t eine Masse der Bezeichnung ˝m2 den
Kontrollraum
verlassen.
Herrschen
im
festen
Kontrollvolumen stationäre Bedingungen so gilt ˝m1 = ˝m2
= ˝m.
Anhand des Ersten Hauptsatzes kann man den Vorgang näher
beschreiben.
Q12 + W12 = U 2 − U1 + ∆E a
Betrachten wir uns die einzelnen Bestandteile des Ersten
Hauptsatzes näher.
U 2 − U1 = ∆U = (U 0 + ∆m ⋅ u 2 ) − (U 0 + ∆m ⋅ u1 )
U0 ist die Innere Energie des Kontrollraums. Da wir einen
stationären Prozess betrachten, bleibt die Energie im
Systeminneren konstant. u2 und u1 sind die spezifischen
inneren Energien der ein- und austretenden Stoffportionen,
sie seien verschieden.
∆E a = E a 2 − E a1 = (E a 0, 2 + ∆m ⋅ e a , 2 ) − (E a 0,1 + ∆m ⋅ e a ,1 )
Für den ruhenden und auf der Höhennulllinie stehenden
Kontrollraum kann man sagen, das die äußeren Energien im
Kontrollraum bei Zustand 1 (t = 0) und Zustand 2 (t = ˝t)
gleich sind, d.h. E a 0, 2 − E a 0,1 = 0 . Die spezifischen äußeren
42
Energien der ein- und austretenden Stoffportionen ea,2 und ea,1
sind ungleich.
Bei diesem offenen System wirken zwei Formen von Arbeit
zum einen die Volumenarbeit oder Verschiebearbeit, zum
anderen die von der Welle verrichtete technische Arbeit.
Da das Volumen im Kontrollraum konstant ist, berechnet
man lediglich Volumenänderung der ein- und austretenden
Stoffportion. Die Drücke p1 und p2 seien konstant und ebenso
alle übrigen Zustandsgrößen im Eintrittsquerschnitt 1 und im
Austrittsquerschnitt 2.
2
2
W12 = WV12 + Wt12 = − ∫ p1 ⋅ dV − ∫ p 2 ⋅ dV + Wt12
1
= −
∫
1
∆V ( ∆t ) = ∆V2
∆V ( ∆t ) = 0
p1 ⋅ dV −
∆V (0 ) = ∆V1
∫p
2
∆V (0 ) = 0
⋅ dV + Wt12
= (p1 ⋅ ∆V1 − p 2 ⋅ ∆V2 ) + Wt12
Einsetzen in den Ersten Hauptsatz:
Q12 + (p1 ⋅ ∆V1 − p 2 ⋅ ∆V2 ) + Wt12 = ∆m ⋅ (u 2 − u 1 + e a , 2 − e a ,1 )
Mit ∆Vi = ∆m ⋅ v i erhält man
Q12 + ∆m ⋅ (p1 ⋅ v1 − p 2 ⋅ v 2 ) + Wt12 = ∆m ⋅ (u 2 − u 1 + e a , 2 − e a ,1 )
oder durch umstellen
Q12 + Wt12 = ∆m ⋅ (u 2 + p 2 ⋅ v 2 − u 1 − p1 ⋅ v1 + e a , 2 − e a ,1 ) .
Mit Hilfe der spezifischen Form von Gleichung 63
( h := u + p ⋅ v ), lässt sich der Hauptsatz für offene Systeme
formulieren.
Q12 + Wt12 = ∆m ⋅ (u 2 + p 2 ⋅ v 2 − u 1 − p1 ⋅ v1 + e a , 2 − e a ,1 )
14243 14243
h2
h1
Q12 + Wt12 = ∆m ⋅ (h 2 − h 1 + e a , 2 − e a ,1 )
(74)
Grundvoraussetzung für die obenstehenden Gleichungen ist,
dass der Grenzwertübergang ∆T->0 für alle Gleichungen
durchgeführt werden kann. Ist dies möglich wird Gleichung
74 mit ∆t dividiert und der Grenzwert gebildet.
43
dm1
∆m1
∆m1
& 1 , ebenso für m2
=
=m
= lim
∆t − >0
dt
∆t
∆t
W12
W12
= lim
= P12
∆t ∆t − > 0 ∆t
∆V1
∆V1
&
= lim
=V
∆t ∆t − > 0 ∆t
∆U1
∆U1
&
= lim
=U
∆t − > 0
∆t
∆t
Man erhält
Hauptsatzes:
die
folgende
Formulierung
des
& +P =m
& ⋅ (h 2 − h1 + e a , 2 − e a ,1 )
Q
12
12
2.3 Der
Zweite
Thermodynamik1,3
Ersten
(75)
Hauptsatz
der
2.3.1 Formulierungen des Zweiten Hauptsatzes
Der Zweite Hauptsatz liefert die empirische Beschränkung
für den ersten Hauptsatz der feststellt, dass Arbeit und
Wärme gleichwertig sind. Nach dem Zweiten Hauptsatz kann
nur ein Teil der Wärme in Arbeit beziehungsweise nur am
absoluten Nullpunkt lässt sich Wärme vollständig in Arbeit
umwandeln.
Nach Definition von Thomson und Plank:
Eine dauernd oder zyklisch funktionierende Maschine, die
einem Behälter nur Wärme entnimmt und daraus
ausschließlich Arbeit erzeugt, ist unmöglich.
Was dies in der Praxis bedeutet macht uns folgendes Beispiel
klar. Nach dem ersten Hauptsatz ist es möglich, dass ein
Schiff seine Maschinen dadurch betreibt, dass es
kontinuierlich Wärme aus dem Wasser entzieht und es in
Arbeit umwandelt. Das Schiff würde eine Spur kalten
Wassers hinter sich herziehen. Der erste Hauptsatz stellt
lediglich fest, dass eine zyklisch-betriebene Maschine nicht
44
kontinuierlich Arbeit liefert ohne Wärme oder eine andere
Art von Energie zu erzeugen. Wärme und Arbeit sind
gleichartige Energieformen.
Die Erfahrung und der zweite Hauptsatz bestätigen, dass ein
solches Schiff aus unserem Beispiel nicht existieren kann.
Somit ist dies ein Beispiel für die Gültigkeit und
Notwendigkeit des zweiten Hauptsatzes.
Aus der abstrakten Formulierung von Thomson und Plank
ergeben sich folgende Schlussfolgerungen:
•
•
•
Soll in einem Kreisprozess Arbeit gewonnen
werden, so muss Wärme mit mindestens zwei
Behältern unterschiedlicher Temperatur ausgetauscht
werden.
Soll Wärme vom kalten zum warmen Behälter
gebracht werden, so muss Arbeit zugeführt werden.
Wärme kann nicht dauernd und vollständig in Arbeit
umgewandelt werden.
2.3.2 Umkehrbare
Prozesse
und
nicht
umkehrbare
Ein Prozess, der auf keine Weise vollständig rückgängig
gemacht werden kann heißt irreversibler Prozess.
Aus der Mechanik kennen wir bereits Prozesse, die
annähernd reversibel sind. Schwingungen, wie zum Beispiel
das Schwingen eines Pendels, gehören dazu.
Abb. 26: Schwingung eines Pendels, ein nahezu reversibler Prozess.
In der Thermodynamik kann von reversibelen Vorgängen
nur gesprochen werden, wenn es sich um einen adiabaten,
quasistatischen und reibungsfreien Prozess handelt.
45
Eine quasistatische Zustandsänderung kann man am besten
anhand der Bewegung eines Kolbens nachvollziehen.
Nehmen wir an, dass sich ein Kolben quasistatisch bewegt, so
bewegt er sich so langsam im Vergleich zu allen anderen in
der
Umgebung
ablaufenden
Stoffund
Energieaustauschprozessen, dass sich Temperatur und Druck
Abb. 27: Reversible, d. h. adiabate, quasistatische und reibungsfreie
Volumenarbeit.3
des Systems während des Vorgangs stetig und überall
gleichmäßig
ändern.
Anders
ausgedrückt,
Zustandsänderungen,
die
durch
unendliche
viele
Gleichgewichtszustände verlaufen und bei denen es zu
keinem Energie- oder Stoffaustausch mit der Umgebung und
insbesondere im System kommt, bezeichnen wir als
quasistatisch.
Die meisten in der Natur vorkommenden Prozesse sind
allerdings irreversibel, das heißt sie sind nicht umkehrbar.
Abb. 28: Einströmen von Luft in einen evakuierten Behälter;
irreversible Zustandsänderung.³
Weitere Beispiele für irreversible Zustandsänderungen:
•
•
•
•
Temperaturausgleich
Konzentrationsausgleich
Druckausgleich
Bestimmte chemische Reaktionen (Verbrennungen)
2.3.3 Entropie
46
Die Entropie S ist eine extensive Zustandsgröße mit der
Einheit J/K (Joule pro Kelvin). Sie ist ein Maß für die
Umkehrbarkeit von Prozessen und ergibt sich aus den
Zweiten Hauptsatz. Sind alle Vorgänge während eines
gesamten Prozesses umkehrbar (d.h. reversibel), so bleibt die
Entropie im Prozess und in der Umgebung konstant. Ist der
Prozess im Gegensatz dazu nicht umkehrbar (irreversibel), so
nimmt die Entropie in der Umgebung zu. Eine von
Boltzmann gefundene Definition der Entropie setzt sie als
Maß für den Ordnungszustand eines Systems. Dabei ist das
ungeordnete System am wahrscheinlichsten.
Die Entropie ist außerdem ein Maß für die Qualität der
Energie. Steigt die Entropie, so wird die Einsatzfähigkeit der
Energie vermindert. Ein Beispiel wird in Abbildung 29
sichtbar. Ein Stein fällt ohne größeres zu tun zur Erde. Die
potentielle Energie, die er besitzt, wird beim Aufschlag auf
den Boden zum Teil als Wärme an die Umgebung abgegeben.
Die Entropie steigt.
Abb. 29: Nach dem ersten Hauptsatz möglich, ein Stein sammelt
Energie und fliegt in Richtung Tisch.
Noch nie wurde beobachtet, dass derselbe Stein dem Boden
Wärme entzieht, diese in Energie umwandelt wird und dann
in die Luft fliegt. Die Qualität der Energie hat sich bei dem
vorausgegangen Aufschlag des Stein so verringert, dass eine
Umkehrung des Prozesses unmöglich ist.
2.3.4 Mathematische Definition der Entropie
Die Wärme Q ist eine Prozessgröße, die sich aus dem ersten
Hauptsatz gemäß
δQ = dU + p ⋅ dV
oder in der spezifischen Form zu
δq = du + pdv
47
ergibt. Da Prozessgrößen wegabhängig sind, kann kein
vollständiges Differential gebildet werden.
Um ein vollständiges Differential zu bilden, wird ein Faktor
eingeführt, den man als „Eulerscher Multiplikator“ oder
integrierenden Faktor bezeichnet. Im Falle der Wärme wird
die reziproke Temperatur 1/T verwendet und man erhält
Gleichung 80.
δq dU p
=
+ ⋅ dV
T
T
T
(76)
δq
als neue Zustandsgröße, bei der
T
eine Integration möglich ist. Diese Tatsache macht man sich
zu nutze, um die Entropie quantitativ erfassen zukönnen.
Man erhält den Term
Durchläuft ein System eine Zustandsänderung von Zustand 1
zu Zustand 2 auf dem Weg a und wieder auf dem Weg b
zurück, so hat es einen Kreisprozess durchlaufen (vgl.
Abbildung 30).
Abb. 30: Zustandsänderungen auf verschiedenen Wegen a und b.
War der gesamte Vorgang reversibel, so gilt das Theorem
von Clausius.
∫
δQ rev
δQ
=∫
=0
T
T
(77)
Dieses Integral lässt sich für die beiden Wege aufteilen in
2
δQ
∫1 T
1
a
+
∫
2
δQ
T
b
=0
(78)
Ein Folgesatz des 2. Hauptsatzes stellt fest, dass bei
reversiblen (Kreis-)Prozessen, wie dem idealem Carnot48
Prozess (vgl. Kap. 2.5.3) das Kreisintegral aller Verhältnisse
δQ
gleich null ist.
T rev
Reversible Kreisprozesse
∫
δQ rev
=0
T
(79)
Irreversible Kreisprozesse
∫
δq irr
<0
T
(80)
Allgemeine Kreisprozesse
δQ
(81)
≤0
T
Gleichung 78 liefert den Zusammenhang zwischen den
Integralen der beiden Wege (vgl. Abb. 30).
∫
2
δQ
∫1 T
2
a
=∫
1
δQ
T
b
Beide Integrationswerte sind gleich, das bedeutet das
Verhältnis von Wärme und Temperatur ist unabhängig vom
Weg und damit eine Zustandsgröße, die nach Clausius
Entropie genannt wird.
dS =
δQ rev
T
(82)
Damit ist nachgewiesen, dass der integrierende Faktor die
Prozessgröße Wärme in die Zustandsgröße S umwandelt.
Für einen irreversiblen Prozess gilt
dS >
δQ irr
T
(83)
Aus den Gleichungen 82 und 83 erhält man eine Gleichung
(Gl. 84). Wenn man die Änderung der Entropie dS zerlegt in
einen Anteil dSrev, der von der reversiblen Übertragung der
Wärme von oder nach außen herrührt und in einen Anteil
dSirr, der von einer irreversiblen (Index irr) EntropieErzeugung im Innern des System stammt.
49
dS = dS rev + dSirr
(84)
mit
dSrev =
δQ
T
dS irr ≥ 0
Bsp. :
dS irr =
δWR12
T
2.3.5 Hauptgleichungen der Thermodynamik
Kombiniert man den 1.Hauptsatz und 2. Hauptsatz der
Thermodynamik, so erhält man eine sogenannte
Hauptgleichung der Thermodynamik:
dU = T ⋅ dS + W12
(85)
Unter der Annahme, dass lediglich Volumenarbeit geleistet
wird erhält man mit W12 = −p ⋅ dV folgenden Ausdruck.
(86)
dU = T ⋅ dS − p ⋅ dV
oder für die Enthalpie gilt
(87)
dH = T ⋅ dS + V ⋅ dp
2.4 Einfache Zustandsänderungen idealer
Gase 3
Einfache Zustandsänderungen sind solche, bei denen eine
Zustandsgröße konstant bleibt oder keine Wärme übertragen
wird.
Betrachten wir nun einige Zustandsänderungen, verwenden
wir die folgenden bekannten Gleichungen:
δq = c v ⋅ dT + p ⋅ dv
oder
δq = c p ⋅ dT − v ⋅ dp
c V = konst.
c p = konst.
p⋅v = R ⋅T
δq = T ⋅ ds
50
2.4.1 Isochore Zustandsänderung
v = konst.
a) Geschlossene Systeme
Abb. 31: Darstellung der isochoren Zustandsänderung in
Diagrammen. 3
Zunächst betrachten wir die Zustandsgrößen bei der
isochoren Zustandsänderung.
Aus der idealen Gasgleichung folgt
p1 p 2
=
= konst.
T1 T2
(88)
Aus dem Ersten Hauptsatz und der Definition der Entropie:
T ⋅ ds = c V ⋅ dT + p ⋅ dv : T
2
cv
p
∫ ds = T ⋅ dT + T ⋅ dv
1
nach der idealen Gasgleichung gilt
p R
=
T v
und durch Einsetzen erhält man
2
2
cv
R
⋅ dT + ⋅ dv
T
v
1
∫ ds = ∫
1
durch Integration und mit dv = 0, da v = konst., erhält man
s 2 − s1 = c v ⋅ ln
T2
T1
(89)
Für
die
Prozessgrößen
Zusammenhänge.
erhalten
Volumenarbeit:
51
wir
ähnliche
Da die Änderung des Volumens null ist, ist auch die
Volumenarbeit null.
Wärme:
Da die Volumenarbeit null ist, erhalten wir den Ersten
Hauptsatz in der folgenden Form:
2
q12 = u 2 − u1 = ∫ c V ⋅ dT
1
da c V = konst. gilt
2
2
1
1
q12 = u 2 − u1 = ∫ c V (T) ⋅ dT = c V ⋅ (T2 − T1 ) = ∫ T ⋅ ds
(90)
b) Offene Systeme
Nur die technische Arbeit unterscheidet sich im Gegensatz
zum geschlossenen System. Alle anderen Zusammenhänge
bleiben gleich.
Technische Arbeit:
2
Wt12 = ∫ v ⋅ dp = v ⋅ (p 2 − p1 )
(91)
1
Abb. 32: Technische Arbeit bei der isochoren Zustandsänderung.3
2.4.2 Isobare Zustandsänderungen
p = konst.
a) Geschlossene Systeme
52
Zustandsgrößen:
Aus dem idealen Gasgesetz folgt:
v1 T1
=
= konst.
v 2 T2
(92)
und
v
1
=
R ⋅T p
Aus
T2
v
+ R ⋅ ln 2 und c p − c V = R
T1
v1
s 2 − s1 = c v ⋅ ln
folgt
s 2 − s1 = c v ⋅ ln
T2
v
T
v
+ R ⋅ ln 2 = c p ⋅ ln 2 = c p ⋅ ln 2 (92)
T1
v1
T1
v1
Abb. 33: Darstellung der isobaren Zustandsänderung in
Diagrammen. 3
Wärme:
2
q12 = u 2 − u 1 + ∫ p ⋅ dv = (u 2 + p ⋅ v 2 ) − (u1 + p ⋅ v1 )
1
(97)
q12 = h 2 − h 1 = c p ⋅ (T2 − T1 )
Volumenarbeit:
2
w V12 = − ∫ p ⋅ dv = −p ⋅ ( v 2 − v1 ) = −R ⋅ (T2 − T1 )
1
b) Offenes System
53
(98)
Nur die technische Arbeit unterscheidet sich in der
Volumenarbeit; alle anderen Zusammenhänge bleiben gleich.
Da sich der Druck nicht ändert, ist die technische Arbeit
gleich null.
Abb. 34: Technische Arbeit bei der isobaren Zustandsänderung ist
null.3
2.4.3 Isotherme Zustandsänderung
p = konst.
Abb. 35: Darstellungen der isothermen Zustandsänderung.³
Zustandsgrößen:
Aus dem Gesetz von Bolye-Mariott und dem idealen
Gasgesetz erhält man
p ⋅ v = R ⋅ T = const
und
p1 v 2
=
p 2 v1
(95)
Aus
54
T2
v
+ R ⋅ ln 2
T1
v1
s 2 − s1 = c v ⋅ ln
folgt für T=konst.
v2
p
= R ⋅ ln 1
v1
p2
s 2 − s1 = R ⋅ ln
(96)
Wärme:
2
Aus u 2 − u 1 = ∫ c V ⋅ dT = 0 folgt
1
2
q12 = ∫ p ⋅ dV = − w 12
(97)
1
Mit Gleichung 100 folgt:
2
q12 = ∫ T ⋅ ds = T1 ⋅ (s 2 − s1 ) = R ⋅ T1 ⋅ ln
1
v2
p
= R ⋅ T ⋅ ln 1
v1
p2
(98)
Volumenarbeit:
2
2
1
1
w V12 = − ∫ p ⋅ dv = − ∫ R ⋅ T ⋅ dv = − p1 ⋅ v1 ⋅ ln
w V12
p
= − p1 ⋅ v1 ⋅ ln 1
p2
v2
v1
(99)
b) Offenes System
Alle Funktionen sind gleich. Die technische Arbeit entspricht
der Volumenarbeit.
w t12 = w V12 = − p1 ⋅ v1 ⋅ ln
v2
V1
(100)
55
Abb. 36: Technische Arbeit bei der isothermen Zustandsänderung.3
2.4.4 Adiabate Zustandsänderung
q12 = 0
a) Geschlossenes System
Zustandsgrößen:
Jede der Zustandsgrößen kann sich ändern. Die folgenden
Betrachtungen gelten für reibungsfreie und quasistatische
Zustandsänderungen.
Abb. 37: Darstellungen der adiabaten Zustandsänderung.³
Logarithmiert man das ideale Gasgesetz
p⋅v = R ⋅T
folgt
ln p + ln v = ln R + ln T
Mit
56
d(ln p) =
dp
p
und d(ln(R )) = 0
ergibt sich
dp dv dT
+
=
p
v
T
multipliziert mit p ⋅ v folgt
v ⋅ dp + p ⋅ dv =
p⋅v
dT = R + dT
T
(101)
mit dem sogenannten Isentropenexponenten
κ=
cp
(102)
cv
und
cp − c v = R
cv =
R
und
(κ − 1)
δq + δw = du
folgt
δw = − p ⋅ dv = du = c v ⋅ dT =
R
⋅ dT
κ −1
(103)
Aus
v ⋅ dp + p ⋅ dv = − κ ⋅ p ⋅ dv + p ⋅ dv : (p ⋅ v )
folgt
dp
dv
+ κ⋅
= 0 bzw. ln p + κ ⋅ ln v = const.
p
v
oder
Error! Objects cannot be created from editing field codes.
(104)
Kombiniert man Gleichung 104, mit der idealen
Gasgleichung, so erhält man folgende Zusammenhänge der
Zustandsgrößen für adiabate Zustandsänderung.
57
T1 ⌈ v 2 ⌉
=
T2 ⌊ v1 ⌋
κ −1
⌈p ⌉
= 1
⌊ p2 ⌋
κ −1
κ
(105)
Für die Entropie gilt bei reversibler Zustandsänderung
2
q12 = ∫ T ⋅ ds = 0
1
und
ds = 0 oder s1 = s 2
(106)
Die Adiabate bei reversibler Zustandsänderung ist zugleich
Isentrope (Kurve bei konstanter Entropie).
Wärme: Adiabate Zustandsänderung, kein Wärmetransfer.
Volumenarbeit:
w V12 = ∆u = c V ⋅ (T2 − T1 )
(107)
Die innere Energie des Systems wird in Arbeit umgewandelt.
Bezieht man die Volumenarbeit auf den Adiabatenexponent
erhält man
2
2
w V12 = − ∫ p ⋅ dv = − ∫ p ⋅ v κ ⋅
1
1
2
dv
= − p1 ⋅ v1κ ⋅ ∫ v −κ ⋅ dv
κ
v
1
(108)
Aus Gleichung 105 und 107 werden neue Zusammenhänge
gefunden
⌉ R ⋅T ⌈T
⌉
p ⋅ v ⌈ ⌈ v1 ⌉
1
= 1 1⋅
−1 =
⋅ 2 −1
κ −1 ⌊ v2 ⌋
κ − 1 ⌊ T1 ⌋
⌊
⌋
κ −1
⌈
⌉
R ⋅ T1 ⌈ p 2 ⌉ κ
=
⋅
−1
κ − 1 ⌊ p1 ⌋
⌊
⌋
κ −1
w V12
w V12
(109)
b) Offenes System
Nur die technische Arbeit unterscheidet sich von der
Volumenarbeit.
Technische Arbeit:
58
2
2
1
1
w t12 = ∫ v ⋅ dp = ∫ c p ⋅ dT = c p ⋅ (T2 − T1 )
(110)
oder unter Verwendung von κ
w t12 = κ ⋅ w V12
(111)
Abb. 38: Technische Arbeit bei der adiabaten Zustandsänderung.3
2.4.5 Polytrope Zustandsänderung
Die einfachen Zustandsänderungen in den Unterkapiteln 2.4.1
bis 2.4.4 kann man als Spezialfälle der Gleichung
p ⋅ v n = const.
sehen.
Es gilt:
•
Isochore Zustandsänderung:
n=∞
p ⋅ v ∞ = konst. oder p1 / ∞ ⋅ v = konst.
oder p 0 ⋅ v = konst. oder
•
v = konst.
Isobare Zustandsänderung:
p ⋅ v n = konst.
•
p = konst.
Isotherme Zustandsänderung:
p ⋅ v n = konst. = R ⋅ T
•
n=0
n =1
T = konst.
Adiabate Zustandsänderung:
59
n=κ
p ⋅ v κ = konst.
Für die reversible Zustandsänderung des idealen Gases kann
man die Kurven der Zustandsänderungen im p, v- und T, sDiagramm darstellen. Abbildung 39 zeigt diese
Zustandsänderungen.
Abb. 39: Polytrope Zustandsänderungen im p, v- und T, s-Diagramm
2.5 Prozesse in der Thermodynamik1,3
2.5.1 Thermodynamischer
Kreisprozess
Prozess
und
Ändert sich durch äußere Einwirkung der Zustand eines
Systems, so bezeichnet man diesen Vorgang als
thermodynamischen Prozess. Ein thermodynamischer Prozess
kann aus einer Zustandsänderungen oder aus mehreren
Zustandsänderungen hintereinander bestehen.
Eine wichtige Feststellung bei der Einordnung von Prozessen
ist die Frage, ob die Prozesse stationär, quasistationär oder
nichtstationär.
Wenn Stoff- und Energieströme zeitlich konstant sind, so
spricht man von stationären Prozessen. Ist dies nahezu der
Fall, spricht man von quasistationären Prozessen. Ändern
sich die Größen mit der Zeit, so spricht man von
nichtstationären Prozessen.
Eine weitere wichtige Unterscheidung trifft man zwischen
reversiblen und irreversiblen Prozessen. Diese wurde im
Zuge des Zweiten Hauptsatzes behandelt.
2.5.2 Definition von Kreisprozessen
60
Aufeinanderfolgende Zustandsänderungen, welche so
verlaufen, dass sie nach einer Anzahl von Teilschritten den
Anfangszustand des Systems wieder erreichen, bezeichnet
man als Kreisprozesse (Anfangszustand = Endzustand).
Deshalb
müssen sich, während eines Kreisprozesses,
sämtliche Änderungen von Zustandsgrößen ZG jeweils
aufheben. Für ein Kreisintegral über alle Zustandsgröße ZG
muss gelten:
∫ dZ
G
=0
2.5.3 Einfache Kreisprozesse
Ein Beispiel für einen einfachen Prozess ist der Satz von
Hess, der eine spezielle Anwendung des Ersten Hauptsatzes
darstellt.
Der Heß’sche Satz besagt, dass die Reaktionsenthalpie (die
Wärme, die bei konstantem Druck während einer chemischen
Umsetzung mit der Umgebung in Wechselwirkung tritt)
gleich der Summe einer Folge von Reaktionen, in die die
betreffende Reaktion formal zerlegt werden kann, ist.
Dabei müssen die jeweiligen Teilschritte nicht realisierbar
sein, sie können rein hypothetisch sein.
Abb. 40: Nach dem Satz von Hess ist die Reaktionenthalpie eine
Zustandsgröße, die nicht vom Reaktionsweg abhängig ist. Eine
Reaktion kann auf verschiedenem Weg beschrieben, egal wir viele
Teilschritte sie beinhaltet.
Born-Haber-Kreisprozeß
Die Bildungsenthalpie eines Feststoffes hat je nach Art der
Bildung des Stoffes einen unterschiedlichen Wert. So kann
man zum Beispiel die Herstellung von Natriumchlorid und
seiner Elemente auf verschiedenstem Wege erreichen.
61
Durch die Umkehrung der Reaktion erhalten wir aus
NaCl(S)ջNa(S)+1/2Cl(g)
einen Kreisprozess (vgl. Abb. 41).
Abb. 41: Born-Haber-Kreisprozess1
Mit Hilfe des Born-Haber-Kreisprozesses kann man die
Gitterenthalpie von Stoffen bestimmen. Er ist ein Beispiel für
einen einfachen Kreisprozess in der Thermodynamik.
2.5.4 Darstellung von Kreisprozessen im p,vDiagramm
62
Abb. 42: Rechtsgängiger Kreisprozess im p, V-Diagramm.³
In Abbildung 42 ist ein Kreisprozess dargestellt, der aus einer
Isothermen (T = konst.), einer Isochoren (V = konst.), einer
Isobaren (p = konst.) und einer Isentropen (S = konst.)
reversiblen Zustandsänderung zusammengesetzt ist.
Der in Abbildung 42 gezeigte Prozess, wird als
rechtsgängiger Prozess bezeichnet, da die einzelnen
Zustandsänderungen im Uhrzeigersinn verlaufen. Weil bei
diesem Prozess auch Arbeit gewonnen wird, bezeichnet man
ihn als Wärmekraftprozess.
Bei der isothermen Zustandsänderung, von Zustand 1 nach 2
wird Volumenarbeit zu und Wärme abgeführt.
Die Fläche unterhalb der Isothermen, spiegelt die zugeführte
Arbeit W12 Fläche [1,2,a,b,1], während die Fläche [3,4,c,a3]
die abgeführte Arbeit W34 und die Fläche [4,1,b,c,4] die
abgeführte Arbeit W41 darstellt.
Anhand der Flächen kann man erkennen, dass die Summe aus
zu- und abgeführter Arbeit, einen Gewinn an Nettoarbeit WN
zur Folge hat, die der schraffierten Fläche in Abbildung 42
entspricht.
WN =W34 +W41+W12
4
1
2
3
4
1
WNE12 = − ∫ p ⋅ dV − ∫ p ⋅ dV − ∫ p ⋅ dV
(112)
Dabei ergibt sich aus der Integration das richtige Vorzeichen.
Linksgängige Prozesse
63
Neben den rechtsgängigen Kreisprozessen gibt es auch die
entsprechende Umkehrung, also Prozesse die gegen den
Uhrzeigersinn verlaufen und linksgängige Prozesse genannt
werden.
Abb. 43: Linksgängiger Prozess im p, V-Diagramm.³
Bei diesen Prozessen kehrt sich die Richtung des
Energieumsatzes um, d.h. zum Beispiel aus W12 wird -W12.
Linksgänge Prozesse werden bei der Verdichtung von Gasen
in Kompressoren und zur Kälteerzeugung verwendet, deshalb
nennt man sie auch Kälteprozesse.
Abb. 43 stellt einen linksgängigen Kreisprozess da, bei dem
die Arbeit W21 abgeführt (zur Expansion) und die Arbeit W43
zugeführt (zur Kompression) wird.
Die Nettoarbeit erkennt man als schraffierte Fläche, die
aufzuwendende Arbeit (also Nettoarbeit) ergibt sich in
Gleichung 113.
WN = W43 + W21
(113)
Dabei darf nicht einfach die Arbeit W21 positiv gesetzt
werden, vielmehr muss sich das Vorzeichen aus der
Gleichung ergeben.
2.5.5 Carnot-Prozess für ideale Gase
Der Carnot-Prozess ist einer der wichtigsten Kreisprozesse
der Thermodynamik. Er gibt an, wie Wärme am Besten in
Arbeit umgewandelt werden kann.
Der Carnot-Prozess besteht aus zwei reversiblen Isothermen,
sowie zwei reversiblen Adiabaten. Dabei ist der Prozess ein
idealisierter Vorgang. (vgl. Abb. 44)
64
Abb. 44: Der Carnot -Prozess im p, v- und T, s-Diagramm.³
Dabei gliedern sich die einzelnen Teilprozesse in:
1ջ2 Isotherme Expansion
T1 = T2 = Tmax
2
q12 = ∫ T ⋅ dS = T1 ⋅ (s 2 − s1 )
1
q12 = R ⋅ T1 ⋅ ln
v2
v1
q12 = R ⋅ T1 ⋅ ln
v2
v1
w 12 = w V12 = −q12 = w t12
2ջ3 Reversible adiabate Expansion
q 23 = 0
v2
T
= 3
v3
T2
1
κ −1
w 23 = w V 23 = c v ⋅ (T3 − T2 ) =
w t 23
κ
3ջ4 Isotherme Kompression
65
T3 = T4 = Tmin
4
q 34 = ∫ T ⋅ ds = T3 ⋅ (s1 − s 2 )
3
q 34 = R ⋅ T3 ⋅ ln
v4
v3
w 34 = w V 34 = −q 34 = w t 34
4ջ1 Reversible adiabate Kompression
q 41 = 0
v4
T
= 1
v1
T4
1
κ −1
w 41 = c v ⋅ (T1 − T4 ) = c v ⋅ (T2 − T3 ) =
w t 41
κ
w 41 = − w 23
Der Carnot-Prozess hat nicht nur Gültigkeit für geschlossene
Systeme, sondern auch für offene Systeme. Abbildung 44
zeigt einen solchen „offenen“ Carnot-Prozess.
Im vorausgegangenen Kapitel haben wir bereits die
Nettoarbeit kennen gelernt. In Abbildung 44 erscheint sie uns
als schraffierte Fläche im Kreisprozess. Da wir dem CarnotProzess als rechtsgängigen Prozess darstellen, ergibt sich:
4
WN = ∑ w = w12 + w 23 + w34 + w 41
1
w N = −R ⋅ T1 ⋅ ln
v2
v
+ cv ⋅ (T3 − T2 ) − R ⋅ T3 ⋅ ln 4 + cv ⋅ (T2 − T3 )
v1
v3
Mit
v 2 ⌈ T3 ⌉
=
v 3 ⌊ T2 ⌋
1
Κ −1
und
v1 ⌈ T4 ⌉
=
v 2 ⌊ T1 ⌋
sowie T1=T2 und T3=T4
v1 v 2
=
v4 v3
oder
v 2 v3
=
v1 v 4
w gew = − R ⋅ (T1 − T3 ) ⋅ ln
v2
v1
66
1
Κ −1
Die Wärme kann relativ einfach beschrieben werden, da für
alle Kreisprozesse gilt, dass die innere Energie als
Zustandsgröße konstant bleibt. (Anfangs- und Endenergie
gleich).
∑ q = −∑ w
(114)
Für die zugeführte Wärme kann man setzen:
2
q12 = q zu = R ⋅ T1 ⋅ ln
v2
= ∫ T ⋅ ds = T1 ⋅ (s 2 − s1 )
v1
1
− w N = q12 + q 34 = q12 − q 34 = q zu − q ab
Die Nettowärme, also q zu − q ab ist als schraffierte Fläche
[1,2,3,4] im T,s-Diagramm erkennbar.
Der thermische Wirkungsgrad ist definiert als
ηth =
w gew
q zu
=1−
Qab
Qzu
=
Wgew
(115)
Q
Für den Carnot-Prozess erhält man
R ⋅ (T1 − T3 ) ⋅ ln
ηth =
v
R ⋅ T1 ⋅ ln 2
v1
v2
v1
=
T1 −T3
T1
(116)
und kann damit den Carnot-Wirkungsgrad, wie folgt
definieren:
ηcarnot = ηc = 1 −
Q
Tmin
= 1 − ab
Tmax
Qzu
(117)
Der Wirkungsgrad des Carnot-Prozesses kann von keinem
anderen Kreisprozess übertroffen werden.
2.6 Übungsaufgaben
9. Aufgabe
In einem 50 m hohen Wasserfall wird nahezu die gesamte
Fallenergie (g = 9,81 m/s2) des Wassers in innere Energie
67
umgewandelt.
a) Um wieviel Grad erwärmt sich das Wasser, wenn durch
Wärmeabgabe an die Umgebung (infolge Konvektion und
insbesondere
Verdunstung)
2
%
und
durch
Verformungsarbeit 3 % der Fallenergie aufgebraucht werden
und seine spezifische Wärmekapazität den Wert 4187 J/(kgK)
besitzt? (0,111 K)
b) Beschreiben und definieren Sie das betrachtete System.
10. Aufgabe
Eine 10 kg schwere Bleikugel fällt aus 50 m Höhe auf eine
harte Unterlage (z. B. Straßenpflaster) wobei sich ihre
Fallenergie (g = 9,81 m/s2) zum Teil in innere Energie
umwandelt und zum Teil als Verformungsarbeit aufgebraucht
wird.
a) Um wieviel Grad erwärmt sich das Blei, wenn seine
spezifische Wärmekapazität 146,5 J/(kgK) und die
Verformungsarbeit 1635 J beträgt? (4,4K)
b) Beschreiben und definieren Sie das betrachtete System.
11. Aufgabe
Welche Bleimenge kann von 15 °C bis zur
Schmelztemperatur von 327 °C durch den Aufschlag eines
200 kg schweren Hammers erwärmt werden, wenn dieser aus
einer Höhe von 2 m fällt und die gesamte am Blei verrichtete
Arbeit zu einer Erhöhung von dessen innerer Energie führt?
Die spezifische Wärmekapazität von Blei beträgt konstant
146,5 J/(kgK). (85,8 g)
Formulieren Sie den 1. Hauptsatz sowohl für das System
„Hammer“ als auch für das System „Blei“ und erläutern Sie
die aufgeführten Terme an Hand von „System-Skizzen“.
12. Aufgabe
In der Turbine einer Kälteanlage werden 156,88 m3/h Luft
adiabat-reversibel von 42oC und 2,6 bar (Zustand 1) auf 1 bar
entspannt (Zustand 2) und anschließend von der Luft eines
Kühlraumes isobar auf 5oC erwärmt (Zustand 3). Für die
folgenden Berechnungen und Darstellungen kann Luft als
ideales Gas mit R = 287,1 J/(kg K) und der kalorischen
68
Zustandsgleichung h = cpT + const mit cp = 1007 J/(kg K)
betrachtet werden.
a) Skizzieren Sie beide Prozesse und sämtliche Isothermen in
je einem p, v- und T, s-Diagramm.
b) Welchen Wärmestrom nimmt die Luft im Kühlraum auf?
(17,33 MJ/h)
c) Welche technische Arbeit gibt die Luft an die Turbine ab?
Erläutern Sie durch Abschätzung, dass die Änderungen der
kinetischen und potentiellen Energie vernachlässigbar sind.
(w=75,71 kJ/kg)
13. Aufgabe
Im Kompressor einer Kälteanlage werden 360 m3/h Luft
eines Kühlraumes adiabat-reversibel von ϑ1 = 5 °C und p1 =
1 bar auf p2 = 2,6 bar verdichtet und anschließend in einem
Kühler isobar auf ϑ3 = 42 °C gekühlt. Für Ihre Berechnungen
können Sie Luft als ideales Gas konstanter spezifischer
Wärmekapazitäten mit R = 287,1 J/(kg K) und ̃ = 1,4
annehmen.
a) Stellen Sie beide Zustandsänderungen in jeweils einem p,
v- und T, s-Diagramm mit Isothermen, Isentropen und
Isobaren als Parameter dar. Skizzieren und vervollständigen
Sie die Kaltluftanlage.
b) Wie groß ist der Luft-Massenstrom? (450 kg/h)
c) Welche Leistung wird der Luft im Kompressor zugeführt?
(10,97 kW). Erläutern Sie durch eine Abschätzung, dass
kinetische
und
potentielle
Energieänderungen
vernachlässigbar sind.
d) Welcher Wärmestrom wird der Luft im Kühler entzogen?
(6,318 kW)
14. Aufgabe
Durch die adiabat isolierte Turbine eines Wasserkraftwerkes
strömen 8 m3/s Wasser. Der Höhenunterschied zwischen
Unterund
Oberwasserspiegel
beträgt
100
m;
Turbineneinlauf und Unterwasserspiegel liegen auf gleicher
Höhe; der Austrittsstutzen ist unter dem Unterwasserspiegel
angeordnet und als Diffusor mit großem Endquerschnitt
ausgebildet. Die Turbinenleistung beträgt 7 MW. Das Wasser
sei inkompressibel und seine spezifische Wärmekapazität
69
konstant 4,19 J/(gK). ρ W = 1000
kg
m³
a) Wie groß wäre die Leistung (7,848 MW), wenn je nach
Systemgrenze die kinetische bzw. potenzielle Energie des
Wassers verlustfrei in mechanische Arbeit umgewandelt
werden könnte? Skizzieren Sie die Anlage und tragen Sie
beide Systemgrenzen ein.
b) Erläutern Sie Ihre Annahme: „vernachlässigbar kleine
Wasseraustrittsgeschwindigkeit“ und zeigen Sie, dass die
Tiefenlage des Austrittsstutzens „unter Unterwasserspiegel“
ohne Einfluss auf die Rechnung ist.
c) Wie groß ist der isentrope Wirkungsgrad (das Verhältnis
der isentropen zur tatsächlichen Leistung) der Turbine (89,2
%) und um wieviel Grad erwärmt sich das Wasser im
System? (0,025 K)
15. Aufgabe
1 kg/s Luft von 105 Pa und 298 K wird auf 106 Pa und 616 K
adiabat verdichtet. Berechnen Sie unter Vernachlässigung
kinetischer und potentieller Energieänderungen und der
Annahme Luft sei ein ideales Gas mit R = 287,1 J/(kg K) und
cv = 720 J/(kg K) = konst.:
a) Die spezifischen Entropieänderungen der Luft: ∆sges,
∆srev und ∆sirr (∆sges = ∆sirr = 68,6 (J/(kgK)),
b) Austrittstemperatur (575 K) und Leistung (278,8 kW),
wenn die Kompression adiabat-reversibel erfolgte und den
isentropen Verdichter-Wirkungsgrad: das Verhältnis der
isentropen zur tatsächlichen Leistung (87,2 %).
c) Stellen Sie beide Zustandsänderungen im p, v- und im T,
s-Diagramm dar.
3 Feuchte Luft³
3.1 Grundlagen
Feuchte Luft ist ein Gasgemisch, welches in unserer
Atmosphäre vorkommt. Das Gemisch aus Wasser und Luft
wird in der Klimatechnik, bei der Trocknung oder bei der
70
Entneblung eingesetzt. Das in der feuchten Luft vorhandene
Wasser kann als Sattdampf, Flüssigkeit, Feststoff (Eismantel)
oder überhitzter Dampf vorliegen.
Für Luft-Dampf-Gemische kann das Dalton-Gesetz
verwendet werden, wonach sich der Gesamtdruck einer
Gasmischung aus den Partialdrücken der Bestandteile
zusammensetzt.
pG = ∑ pi
Um ein Maß für das in der Luft vorhandene Wasser zu
erhalten, definiert man eine neue Größe, die wir Wassergehalt
nennen.
x=
Masse Wasser m W
=
Masse Luft
mL
(118)
Vorsicht Verwechselungsgefahr: x ist hier nicht die
Stoffmengenkonzentration.
Man kann den Wassergehalt auch mit Hilfe der molaren
Massen aus den Stoffmengen berechnen.
x=
nW ⋅ MW
MW nW
=
⋅
nL ⋅ ML
ML
n
123 L
≈ 0,622
(119)
Die gesamte Masse des Gemisches aus Wasser und Luft mG
kann über den Massenerhaltungssatz und den Wassergehalt
ausgedrückt werden.
(120)
m G = m L + m W = m L ⋅ (1 + x )
Aus dem idealen Gasgesetz folgt
p L ⋅ VG = m L ⋅ R L ⋅ T
p D ⋅ VG = m D ⋅ R D ⋅ T
Bedingungen für diese Gesetze:
•
•
•
Luft und Wasserdampf als ideales Gas.
Beide Gase nehmen z. B. in einem Behältervolumen
VG den gleichen Raum ein.
Die Temperatur T der Luft sei gleich der des
Wasserdampfes.
71
Setzt man die Partialdrücke an Dampf und Luft ins
Verhältnis, so erhält man:
pD mD ⋅ R D
R
=
= x⋅ D
pL mL ⋅ R L
RL
(121)
oder
pD ⋅ R L
pD
R
pD
MD
=
⋅ L =
⋅
=x
p L ⋅ R D pG − pD R D pG − pD M L
{
≈ 0,622
x = 0,622 ⋅
pD
n
= 0,622 ⋅ D
pG − pD
nL
(122)
(123)
Ist der größte Wasser- oder Dampfgehalt in der Luft erreicht,
so wird bei gegebener Temperatur ein maximaler Druck
gemessen. Dieser Druck pS wird auch als Sättigungsdruck
bezeichnet, da die Luft mit Wasser vollständig gesättigt ist.
Mit Hilfe von Gleichung 136 erhält man für den größten
Dampfgehalt xS folgenden Zusammenhang:
x S = 0,622 ⋅
pS
p G − pS
(124)
In der Regel sind Luft- und Wasserdampf Gemische nicht
gesättigt. Ein Maß für die Sättigung eines Gemisches ist der
Sättigungsgrad ψ (oder relative Sättigung), er wird durch den
jeweiligen Wassergehalt der Luft x und dem Sättigungsgehalt
xS gebildet.
ψ=
x
xS
(125)
Ein weiteres Maß ist die sogenannte relative Feuchte ϕ
verwendet
ϕ=
pD
x
p
=
⋅ G
pS x + 0,622 pS
(126)
Setzt man die beiden Maße ins Verhältnis erhält man
ψ p G − pS
=
ϕ pG − p D
(127)
Die Gemischdichte ρ G ist allgemeingültig definiert als
72
mW + mL mG
=
V
V
ρ=
(128)
Bei folgenden Bedingungen,
•
•
•
Luft und Wasserdampf als ideales Gas,
Beide Gase nehmen zum Beispiel in einem
Behältervolumen VG den gleichen Raum ein,
Die Temperatur T der Luft sei gleich der des
Wasserdampfes,
kann die Dichte aus dem idealen Gasgesetz gewonnen
werden.
Aus
(p L + p D ) ⋅ VG = (m L ⋅ R L + m D ⋅ R D ) ⋅ T = p G ⋅ VG
folgt
ρG =
p
p
1+ x
1+ x
⋅ G =
⋅ G
R L + x ⋅ R D T 1+ x ⋅ R D / R L R L ⋅ T
(129)
oder mit Zahlenwerten ausgedrückt
p
1+ x
⋅ G
(130)
1 + 1,61⋅ x R L ⋅ T
Für das spezifische Volumen eines Gemisches vG gilt analog
für die gleichen Bedingungen
ρG =
vG =
VG
1 1 + 1,61⋅ x R L ⋅ T
=
=
⋅
m G ρG
1+ x
pG
(131)
Bezieht man das Volumen VG nicht auf die Masse des
Gemisches, sondern auf die Masse der trockenen Luft, so gilt
für dieses spezifische Volumen
vG / L = v =
VG
R ⋅T
= ( x + 0,622) ⋅ D
mL
pG
3.2 Kalorische Zustandsgleichungen
feuchte Luft
73
(132)
für
Für die in diesem Kapitel verwendeten Formeln müssen
folgende Bedingungen erfüllt sein:
•
Wasserdampf und Luft seien ideale Gase
•
cp sei keine Funktion der Temperatur: cp = const.
•
Luft- und Wassertemperatur sind gleich groß.
•
Alle Gase und Flüssigkeiten seien inkompressibel.
( ρ = konst. )
•
Die Bezugstemperatur sei 0°C, bei der die Enthalpie
der Luft und des flüssigen Wassers zu 0 kJ/kg
definiert ist.
•
Die Enthalpie ist Funktion des Wassergehalt und der
Temperatur. H = f(T,x))
Um den Energiezustand eines strömenden Gases, wie feuchte
Luft zu beschreiben, verwendet man zweckmäßigerweise die
Enthalpie.
dh =
∂h
∂h
⋅ dT +
⋅ dp
∂T p
∂p T
123
123
c p =konst .
=o ,ideales Gas
dh = c p ⋅ dT
Integriert man den Term erhält man
h
T
h0
T0
∫ dh = c p ⋅ ∫ dT
(133)
h = c p (T − T0 ) + h 0
Die Enthalpie der feuchten Luft HG setzt sich zusammen aus,
der Summe der Enthalpie der Luft HL und der Enthalpie des
Wassers Hw.
HG = HL + HW
(134)
Für die spezifische Form gilt
HW = mW ⋅ hW
(135)
HL = mL ⋅ h L
(136)
74
Es ist zweckmäßig die gesamte Enthalpie auf die Masse an
trockener Luft zu beziehen. In diesem Kapitel verwenden wir
die spezifische Enthalpie h (ohne Index) als
h G / l := h :=
HG
mL
(137)
Achtung: Für alle folgende Formeln in diesem Kapitel wird
die spezifische Enthalpie auf die Masse an trockener Luft
bezogen. In manchen Literaturquellen wird diese spezifische
Enthalpie auch mit dem Index 1+x, also h1+x bezeichnet.
Die spezifische Enthalpie bezogen auf die trockene Luft setzt
sich dann zusammen aus:
h = hL + x ⋅ hW
(138)
Bezugstemperatur
Die absolute spezifische Enthalpie h (nicht die Änderung der
spezifischen Enthalpie) kann über eine Bezugstemperatur T0
bestimmt werden. Dabei spielt die spezifische Enthalpie bei
der Bezugstemperatur eine Rolle.
Für
h L = c p,L ⋅ (T − T0 ) + h 0 (T0 )
(139)
und der Bezugstemperatur T0=273,15 K oder ϑ =0°C (mit
Theta ( ϑ ) wird die Temperatur in °C bezeichnet.) gilt
∆T = T − T0 = ϑ
(140)
So kann man die Temperatur in °C verwenden, die weitaus
umgänglicher ist.
h L = c p ,L ⋅ ϑ + h 0 (T0 )
Legt man für Luft und flüssiges Wasser bei ϑ = 0°C die
Enthalpie fest zu h0=0 kJ/kg, so gilt
(141)
h L = c p ,L ⋅ ϑ
Zustände des Wassers und kalorische Zustandsgleichung
75
Wasser kann in feuchter Luft, je nach Temperatur und
Wassergehalt verschiedene Aggregatzustände
Phasenzusammensetzungen haben. Man unterscheidet hierbei
vier verschiedene Zusammensetzungen und Phasengebiete.
A) Einphasengebiet (ungesättigte oder gerade gesättigte
feuchte Luft)
Die feuchte Luft besteht im Fall A aus (Wasser-) Dampf
und Luft. Es gilt x ≤ x S , ϑ > 0°C und da wir den
Wasserdampf als ideales Gas ansehen, kann man
annehmen
(142)
h D ≈ c P ,D ⋅ ϑ + x ⋅ ∆h V ,0
˝hv,0 ist die Verdampfungsenthalpie, die aufgrund des
Verdampfungsvorgangs von Wasser zu Dampf bei 0 °C
benötigt wird.
Berücksichtigt man nun noch die Enthalpie der Luft, so
erhält man
h = (c P ,L + x ⋅ c P ,D ) ⋅ ϑ + x ⋅ ∆h V ,0
(143)
Mit den Stoffdaten,
kJ
kg ⋅ K
kJ
≈1
kg ⋅ K
c P ,D = 1,86
c P ,L
∆h V , 0 = 2501
kJ
kg
h
ϑ
= (1 + 1,86 ⋅ x ) ⋅ + 2501⋅ x
kJ / kg
°C
(144)
Hinweis: Für [h]=kJ/kg und [ ϑ ]=°C entspricht die
Ordinateneinteilung {h}={ ϑ }
B) Zweiphasengebiet (übergesättigte feuchte Luft als
Nebel)
Die feuchte Luft besteht im Fall B aus (Wasser-) Dampf,
flüssigem Wasser (Nebel) und Luft. Es gilt x > x S ,
ϑ > 0°C und da wir den Wasserdampf als ideales Gas
76
ansehen, kann man annehmen (Achtung: Die feuchte Luft
ist mit Wasserdampf gesättigt!),
H = H L + H D + H W ,f
h=
m L ⋅ h L + m D ⋅ h D + m w ,f ⋅ h w ,f
mL
h = (c P ,L − x S (c W − c P ,D ) + x ⋅ c W ) ⋅ ϑ + x S ⋅ ∆h V , 0
(145)
mit
c W = 4,19
kJ
kg ⋅ K
h
ϑ
= (1 − 2,33 ⋅ x S + 4,19 ⋅ x ) ⋅ + 2501 ⋅ x S
kJ / kg
°C
(146)
Der Wassergehalt an Nebel (flüssiges Wasser) in der
feuchten Luft xf, kann über den Sättigungsgehalt
bestimmt werden.
x f = x − xS
(147)
C) Zweiphasengebiet (übersättigte Luft als Eisnebel)
Feuchte Luft besteht im Fall C aus Dampf, Eis und Luft.
Es gilt x > x S , ϑ > 0°C (Feuchte Luft ist mit
Wasserdampf gesättigt),
H = HL + HD + HE
h = h L + h D ⋅ x S + (x − x S ) ⋅ h E
mit
h E = −∆h S − c E ⋅ ϑ
˝hs
ist
die
Schmelzenthalpie,
bei
dem
Verdampfungsvorgang von Eis zu Wasserdampf frei
wird.
77
kJ
kg
kJ
c E = 2,09
kg ⋅ K
∆h S = 334
h = (c P ,L − x S ⋅ (c E − c P ,D ) + x ⋅ c E ) ⋅ ϑ + x S (∆h V , 0 + ∆h S ) − x ⋅ ∆h S
h
ϑ
= (1 − 0,23x S + 2,09x ) ⋅ + 2835x S − 334x
kJ / kg
°C
(148)
Analog zu Fall kann man nun den Gehalt an Eis bestimmen.
xe =
mE
= x − xS
mL
(149)
Das Dreiphasengebiet in dem Luft, Dampf, Eis und flüssiges
Wasser gemeinsam existieren, wird aufgrund seiner
Seltenheit hier nicht behandelt.
Man kann allerdings alle vier Gebiete mit Hilfe eines h, xDiagramms darstellen.
Abb. 45: Phasengebiete feuchter Luft im h,x-Diagramm.³
3.3 Das h, x-Diagramm nach Molier
Würde man ein h,x-Diagramm in gewohnter Weise
aufzeichnen, so würde man die Auftragung, wie in Abb. 45
erhalten.
78
Die Neigungen der Isothermen, ergeben sich je nach
∂h
Phasengebiet aus dem Differenzialquotienten
. Da
∂x ϑ
bei feuchter Luft die Enthalpie eine Funktion von x und
ϑ ist, kann man die Steigung aus der Änderung der Enthalpie
mit dem Wassergehalt bestimmen.
Zu den in Kapitel 3.2 zugeordneten Fälle A-C werden hier
die Steigungen der Isothermen in den jeweiligen
Phasengebieten bestimmt.
A)
B)
C)
∂h
∂x
ϑ
∂h
∂x
ϑ
∂h
∂x
ϑ
= 1,86 ⋅ ϑ + 2500
(150)
= 4,19ϑ
(151)
= 2,05ϑ − 333
(152)
Das interessanteste Gebiet, der schraffierte und ungesättigte
Bereich in Abbildung 46, kann nicht genau abgelesen
werden. Deshalb musste eine Lösung gefunden werden, die
eine bessere Ablesung gewährleistet.
Abb. 46: h, x-Diagramm bei rechtwinkliger Auftragung.³
Nach dem Vorschlag von Molier kann die Isotherme ϑ = 0°C
so gedreht, dass sie parallel zur x-Achse ist. (die Pfeile in
Abbildung 46 zeigen diese Bewegung) Alle Isothermen im
ungesättigten Gebiet (mit ϑ >0°C) verlaufen dann leicht
schräg nach oben. Im Zweiphasengebiet verlaufen sie rechts
nach unten. Eine Darstellung des h, x-Diagramms nach
Molier zeigt Abbildung 45.
79
Abb. 47: Schematische Darstellung eines h, x-Diagramms für feuchte
Luft (nach Mollier).³
Auf der x-Achse wird der Wassergehalt, auf der y-Achse die
spezifische Enthalpie (bezogen auf die Masse an trockener
Luft) aufgetragen.
Die gestrichelten Linien kann man als Geraden konstanter
Enthalpie (Isenthalpen) erkennen.
Von zentraler Bedeutung sind die Kurven konstanter Feuchte
(̏-Linien). An der ϕ = p D p' = 1 -Linie (Sättigungslinie)
knicken die Isothermen ab, während die Isenthalpen weiter
verlaufen.
Die Linien konstanter Feuchte sind abhängig vom
Gesamtdruck des Systems. Sie sind durch Erfahrung
gewonnen und verschieben sich, wie in Abb. 47 dargestellt.
80
Abb. 48: Linien
Gesamtdrücken pG.³
konstanter
Feuchte
bei
verschiedenen
3.4 Anwendungen des h, x-Diagramms
3.4.1 Erwärmung und Kühlung im
Einphasengebiet bei konstantem
Dampfgehalt
Strömt feuchte Luft über eine Heiz- oder Kühlplatte, so
verändert sich neben der Temperatur auch sich seine
Enthalpie.
Feuchte Luft vom Zustand 1 (vgl. Abb. 49) ist
gekennzeichnet durch den Dampfgehalt x1 , sowie die
Temperatur ϑ1 . Damit sind auch die relative Feuchte ϕ1 und
die spezifische Enthalpie h1 festgelegt.
Da sich der Dampfgehalt nicht ändert liegen Zustand 2
(Erwärmung) und Zustand 3(Abkühlung) senkrecht zu
Zustand 1. Mit der Abkühlung nimmt die relative Feuchte ̏
zu, da sich mit der Temperatur auch die maximale Sättigung
ändert.
81
Abb. 49: Erwärmung und Kühlung feuchter Luft bei konstantem
Dampfgehalt.³
Aus dem 1.Hauptsatz können wir mit
Q12 = m 2 ⋅ (h 2 − h1 )
die aufgenommene oder abgegebene Wärmemenge
bestimmen. Findet dies kontinuierlich statt, gilt es den
Wärmestrom zu beachten.
& =m
& 2 ⋅ (h 2 − h1 )
Q
12
3.4.2 Trocknung feuchter Luft
Abbildung 50 zeigt idealisiert, die Zustandsänderungen,
welche Luft durchlaufen muss, um dem Dampfgehalt zu
verändern.
Abb. 50: Trocknung feuchter Luft als idealisierter Vorgang.³
82
1. Zunächst findet eine Abkühlung von Zustand 1 auf
Zustand 2 statt. Nach Überschreitung der
Sättigungslinie bei Zustand 1s („s“ für Sättigung, im
Diagramm mit 1’ benannt) bildet sich Nebel, da die
Mischung übersättigt ist.
2. Entfernt man nun das flüssige Wasser (Nebel) bis zur
Sättigungskonzentration (Isotherm), so führt dies zu
Zustand 2s (2’). Da sich die Temperatur nicht ändert,
findet die Zustandsänderung auf der ϑ2 -Isothermen
statt.
3. Erwärmt man nun die Luft von Zustand 2s (2’) auf
Zustand 3, wobei der Dampfgehalt konstant bleibt
(senkrecht),
trifft
man
erneut
auf
die
Ausgangsisotherme ϑ1
Dieser Vorgang ist eine Idealisierung, für dessen Teilschritte
die Wärmemengen und Wärmeströme aus dem 1. Hauptsatz
der Thermodynamik errechnet werden können.
Als Beispiel die Wärmemengenänderung bei der Entnahme
des Wassers:
Q 22 ' = m l ⋅ (h 2s − h 2 )
analog der Wärmestrom
& =m
& 2 ⋅ (h 2s − h 2 )
Q
3.4.3 Adiabate Vermischung zweier Luftströme
Abb. 51: Adiabate Vermischung zweier Luftströme im h, xDiagramm.³
Man stelle sich folgenden Fall vor: Zwei Luftströme
& L ,1 ⋅ (1 + x 1 ) und m
& L , 2 ⋅ (1 + x 2 ) werden gemischt. Die beiden
m
83
Zustände sind uns in Abb. 51 als Punkt 1 und Punkt 2
gegeben.
Vermischen sich die beiden Luftströme, so wird der Zustand
3 erreicht. Dieser Zustand 3 muss bei adiabater Vermischung
irgendwo auf der Mischungsgeraden (die Gerade ist durch
Punkt 1 und 2 bestimmt und schneidet diese) liegen.
Es gibt verschiedene Möglichkeiten um den Zustand 3 (Punkt
3) zu errechnen.
Dazu sei der Gesamtmassenstrom der Luft gegeben durch
& L ,3 = m
& L, 2 + m
& L ,3
m
sowie der Gesamtmassenstrom des Wasserdampfes durch
& L ,3 = x 1 ⋅ m
& L ,1 + x 2 ⋅ m
& L, 2
x3 ⋅ m
x3 =
& L ,1 + x 2 ⋅ m
& L,2
x1 ⋅ m
=
& L ,1 + m
& L, 2
m
x1 + x 2 ⋅
& L, 2
m
& L,1
m
&
m
1 + L, 2
& L ,1
m
&
x1 − x 3 m
c
= L, 2 =
& L,1 d
x3 − x2 m
(153)
(154)
Mit der Mischungsgeraden und dem Wassergehalt x3 ist
Punkt 3 gegeben. Man kann nun auch für die und mit der
Enthalpie eine entsprechende Aussage finden. Mit
h3 =
& L ,1 + h 2 ⋅ m
& L, 2
h1 ⋅ m
& L ,1 + m
& L, 2
m
und
&
h1 − h 3 m
c a
= L, 2 = =
& L ,1 d b
h3 − h2 m
(155)
Liegen ähnliche Dreiecke vor, das heißt, die Zustände 1,3 und
2 bilden eine Gerade, so gilt auch:
&
Strecke 1 − 3 m
c a
= L, 2 = =
& L ,1 d b
Strecke 3 − 2 m
(156)
84
Bei bekannten Zuständen 1 und 2, sowie den Massenströmen
& L ,1 , m
& L, 2 kann man Punkt 3 auf der Mischungsgeraden
m
bestimmen. (Gesetz der abgewandten Hebelarme)
Liegen die Zustände der beiden Luftmengen nahe am
Sättigungsgebiet, so erhält man Nebel (vgl. Abb. 52).
Abb. 52: Auftreten von Nebel bei der Vermischung zweier feuchter
Luftmengen.³
85