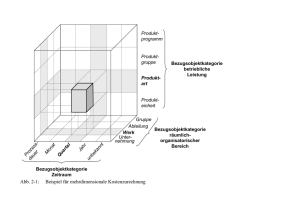
Strategien zur Charakterisierung von ausgesuchten Streptomyces
Werbung

was du nicht willst das man dir tu das füg auch keinem anderen zu Strategien zur Charakterisierung von ausgesuchten Streptomyces lividans Genen und deren Funktionen Dissertation zur Erlangung des akademischen Grades Doktor der Naturwissenschaften (Dr. rer. nat.) des Fachbereiches Biologie/Chemie der Universität Osnabrück vorgelegt von Jens Overbeck September 2006 Dekan: 1. Berichterstatter: 2. Berichterstatter: Prof. Dr. Karl Altendorf Prof. Dr. Hildgund Schrempf PD Dr. Ullrich Keller Inhaltsverzeichnis 1. EINLEITUNG 2. MATERIALIEN UND METHODEN I 1-9 11-46 2.1. Chemikalien und Enzyme 2.2. Bakterienstämme, Vektoren, Konstrukte, Phagen und Oligos 2.2.1. Bakterienstämme 2.2.2. Vektorplasmide 2.2.3. Rekombinante Konstrukte 2.2.4. Bakteriophagen (Lambda-Derivate) 2.2.5. Im Rahmen der Arbeit erwähnte Oligos 11 13 13 14 14 19 20 2.3. Protein- und DNA-Standards 2.4. Medien 2.5. Mikrobiologische Methoden 2.5.1. Kulturbedingungen und Stammhaltung von Streptomyceten 2.5.2. Kulturbedingungen und Stammhaltung von Escherichia coli 24 25 30 30 30 2.6. Biochemische Methoden 2.6.1. Gewinnung von His-Tag Proteinen 2.6.1.1. Gewinnung von His-Tag Proteinen (denaturierend) 2.6.1.2. „Native“ Gewinnung von His-Tag Proteinen 2.6.2. Konzentration von Proteinen und Wechsel des Puffers 2.6.3. Gelfiltration 2.6.4. Massenspektrometischer Nachweis von FAD 2.6.5. Bestimmung von Proteinkonzentrationen 2.6.6. Spektrophotometrische Messungen 2.6.7. Bestimmung des FAD/KsuF-Verhältnisses 31 31 31 31 33 33 33 33 33 34 2.7. Elektrophoretische Techniken 2.7.1. Agarose-Gelelektrophorese 2.7.2. SDS Polyacrylamid-Gelelektrophorese (PAGE) 2.7.3. Denaturierende Gelelektrophorese in Schägger/v.Yagow Gelen 2.7.4. Färbung mit Coomassie-Brilliant-Blau 2.7.5.Silbernitratfärbung 2.7.6. Proteintransfer auf Membranen („Semidry-Electro-Blotting“) 34 34 35 35 35 35 36 Inhaltsverzeichnis II 2.8. Immunologische Nachweismethoden 2.8.1. Immunologischer Nachweis von Proteinen auf Nylonmembranen („Western Blot“) 36 2.9. Molekulargenetische Methoden 2.9.1. Aufreinigung von DNA 2.9.1.1. Plasmidisolierung aus E.coli (alkalische Lyse) 2.9.1.2. Plasmidaufreinigung aus E.coli („Quick and dirty“) 2.9.1.3. Plasmidaufreinigung aus E.coli (Ionenaustauscher) 2.9.1.4. Schnelltest zum Nachweis von Plasmiden in E.coli (SCFSB) 2.9.1.5. Aufreinigung von einzelsträngiger Phagen-DNA (M13) 2.9.1.6. Isolierung von Plasmiden aus Streptomyceten 2.9.1.7. Isolierung von chromosomaler DNA aus Streptomyceten 2.9.1.8. Aufreinigung von DNA-Fragmenten aus Agarosegelen 2.9.2. Transformation von Plasmiden 2.9.2.1. Transformation von E.coli (CaCl2-Methode) 2.9.2.2. Transformation von E.coli (Elektroporation) 2.9.2.3. Transformation von Streptomyceten (PEG-Methode) 2.9.2.4. Integration von Genen in das Chromosom von S.lividans 2.9.3. Enzymatische in vitro Reaktionen von DNA 2.9.3.1. Restriktion von DNA 2.9.3.2. Ligation 2.9.3.3. Klenow-Behandlung 2.9.3.4. Polymerasekettenreaktion (PCR) 2.9.3.5. Herstellung von „Sonden“-DNA 2.9.4. DNA-DNA Hybridisierung 2.9.5. Sequenzierung 37 37 37 37 37 38 38 38 39 39 39 39 40 40 41 42 42 42 42 43 43 44 44 2.10. Fluoreszensmikroskopie 2.11. Verwendete biologische Softwareprogramme 45 45 3. ERGEBNISSE 36 47-137 3.1. Analyse der Sequenzen 47 3.2. Klonierung der Gene des untersuchten Bereiches und Produktion der kodierenden Genprodukte in E.coli 49 3.2.1. Strategie zur Gewinnung des C-Terminus von KcsA und der Antikörper 49 3.2.2. Produktion von KsuB und von gegen KsuB gerichteten Antikörpern 53 3.2.3. Herstellung von Konstrukten für die Synthese und Reinigung von KsuL 54 3.2.4. Gewinnung von KsuC und von gegen KsuC gerichteten Antikörpern 56 Inhaltsverzeichnis III 3.3. Physiologische Studien von S.lividans und ausgewählten Mutanten 3.3.1. Immunologischer Nachweis der Proteine in S.lividans WT 3.3.2. Synthese der Proteine in S.lividans TK21 pJO1 3.3.3. Analyse von S.lividans NI Transformanten 3.3.3.1 Herstellung der Konstrukte 3.3.3.2. Nachweis der Protein-Produktion 3.3.4. Produktion der Proteine in einer sigH-Mutante 3.3.5. Abhängigkeit der Synthese von KsuF vom Vorhandensein des Thiostrepton Resistenzproteins 58 58 60 62 62 64 66 3.4. Gewinnung von His-Tag Proteinen aus S.lividans 3.4.1. Konstruktion der Plasmide pOVV1, pOVV2 und pOVE3 3.4.2. Konstruktion der bifunktionellen Plasmide pOVE4 und pOVE5 3.4.3. Herstellung kcsA-haltiger Konstrukte und Analyse der S.lividansTransformanten 3.4.4. Produktion von KsuB in S.lividans-Transformanten 3.4.5. Produktion von KsuC in S.lividans-Transformanten 3.4.6. Produktion von KsuF in S.lividans-Transformanten 68 69 70 3.5. Studien von S.lividans-Transformanten mit ausgewählten Konstrukten 3.5.1. Herstellen der Konstrukte 3.5.2. „In vivo“ Nachweis der Synthese von EGFP (enhanced green fluorescent protein) 67 72 74 75 77 80 80 85 3.6. Charakteristika der Proteine 3.6.1. Klassifizierung des abgeleiteten Flavo-Proteins (KsuF) 3.6.2. Reinigung und Charakterisierung des Flavoproteins 3.6.3. Analyse des extrazellulären Proteins KsuL 3.6.4. Analyse der Aldo-Keto Reduktasen KsuC und KsuB 3.6.5. Analyse des abgeleiteten ORFI-Proteins 88 88 93 100 103 107 3.7. Disruption verschiedener Gene in S.lividans 3.7.1. Auswahl der Strategien zur Disruption von Genen 3.7.2. Disruption der Gene in mit Hilfe des Vektors pGM160 3.7.3. Disruption von kcsA 3.7.4. Disruption von ksuF 3.7.5. Disruption von ksuL 3.7.6. Disruption von orfI 3.7.7. Disruption von ksuB 3.7.8. Disruption von ksuC 108 108 109 110 111 113 114 116 118 3.8. Charakteristika der Mutanten 3.8.1. Wachstumseigenschaften 3.8.2. Immunologischer Nachweis der relevanten Proteine 3.8.3. Analyse der Pigment-Produktion 120 120 120 123 IV Inhaltsverzeichnis 3.9. Konstruktion von chromosomalen Fusionsgenen in S.lividans 3.9.1. Herstellung von Konstrukten und Analyse der Transformanten 3.9.2. Herstellen von Hygromycin-resistenten Mutanten mit chromosomalen Fusionsgen 4. DISKUSSION 4.1. Sequenzanalyse und Charakteristika der abgeleiteten Proteine 4.2. Synthese von drei Proteinen unter verschiedenen physiologischen Bedingungen 4.3. Undecylprodigiosin-Überproduktion in Mutanten 5. ZUSAMMENFASSUNG 6. LITERATURVERZEICHNIS ANHANG Anhang A: Der sequenzierte 13920 Bp lange Abschnitt aus S.lividans 66 Anhang B: Terminatorstruktur zwischen ksuF und ksuL Anhang C: Konstruktion von pACYC001 bis pACYC010 Anhang Tabelle D: Messwerte der FAD/KsuF Verhältnisbestimmung Anhang E: Sequenzvergleich von KsuL mit den homologen Proteinen 128 128 130 139-167 139 157 161 169 171-194 i-xi ii iv xvi xx xi Abkürzungsverzeichnis Abb. Act Bp BSA CAPS Casa. CDA DIG DMSO DNA dNTP DTT ECF EDTA EGFP EtOH FAD FPLC GuHCl HEPES HPLC IPTG kBp kDa Lsg. MBp MeOH MW MWCO NAD(P) Ni-NTA OD Oligo Orf PAA PAGE PCR PEG PHBH pI RED RNA RT SARP SDS seradest H2O SSAO TES Tir Tris ü.N. UpM. UV WT V Abbildung Actinorhodin Basenpaare bovine serum albumine 3-Cyclohexylamino-1-propansulfonsäure casaaminoacids, Casaaminosäuren calcium-dependend antibiotic Digoxygenin Dimethylsulfoxid deoxyribonucleic acid, Desoxyribonukleinsäure Desoxynucleotidphosphat Dithiothreitol extra-cytoplasmic function Ethyldiamintetraessigsäure enhanced green fluorescent protein Ethanol Flavin-adenin-dinucleotid fast protein liquid chromatography Guanidin-Hydrochlorid N-(2-Hydroxyethyl)piperazin-N`-ethansulfonsäure high pressure liquid chromatography Isopropyl-β-D-Thiogalactopyranosid Kilo-Basenpaare Kilo-Dalton Lösung Mega-Basenpaare Methanol molecular weight, Molekulargewicht molecular weight cut-off Nicotinamid-adenin-dinucleotid(-phosphat) nickel-nitrilotriacetic acid, Nickel Nitrilotriessigsäure optische Dichte Oligonukleotid open reading frame, offenes Leseraster Polyacrylamid Polyacrylamid-Gelelektrophorese polymerase chain reaction, Polymerase-Ketten-Reaktion Polyethylenglycol p-Hydroxybenzoat Hydroxylase isoelektrischer Punkt Undecylprodigiosin/Butyl-meta-Cycloheptylprodiginin ribonucleic acid, Ribonukleinsäure Raumtemperatur Streptomyces antibiotic regulatory proteins Laurylsulfat, Natrium Salz über Ionenaustauscher gereinigtes H2O Semicarbizide sensitive Amin-Oxidasen n-Tris(hydroxymethyl)methyl-2-aminoethansulfonsäure terminal inverted repeats Tris-(hydroxymethyl)aminomethan über Nacht Umdrehungen pro Minute Ultraviolett Wildtyp Einleitung -1- 1. EINLEITUNG Streptomyceten sind Gram-positive, aerobe, saprophytische, frei lebende Bodenbakterien, die sich durch einen komplexen Lebenszyklus mit morphologischer Differenzierung in Substrat-, Luftmycel, und Sporen, auszeichnen. Sie sind mit 104 bis 107 Kolonie-formenden Einheiten pro Gramm Boden zu finden und bilden 1-20 % oder mehr der gesamten Lebend-Zellmasse. Sie sind auch in Sedimenten des Seebodens, in marinen Sedimenten, als vermutliche Symbionten in der Rhizosphäre von Pflanzen und in Pflanzengeweben oder im Darmtrakt von Würmern und Insekten auffindbar. Streptomyceten gehören zur Ordnung der Actinomycetales (im Weiteren mit dem gebräuchlicheren Synonym „Actinomyceten“ bezeichnet). Neben den Streptomyceten, die zu den Streptomycetaceae gehören, sind auch weitere, wie die Actinomycetaceae, Arthrobacteriaceae, Bifidobacteriaceae, Cellulomonadaceae, Corynebacteriaceae, Dermathopylaceae, Frankiaceae, Microbacteriaceae, Mycobacteriaceae, Propionobacteriaceae, Nocardiaceae, Nocardioidaceae, Pseudonocardiaceae und Streptosporangiaceae, Mitglieder dieser Ordnung. Alle haben sie einen sehr hohen GC Gehalt gemeinsam, Streptomyceten einen mit mehr als 70 %. Wie zuerst für Streptomyces lividans nachgewiesen, besitzen Streptomyceten, anders als die meisten anderen Prokaryoten, ein lineares Chromosom (Lin, Y.-S., et al.; 1993). Weitere Beispiele linearer Chromosomen bei Bakterien, außerhalb der Familie der Actinomyceten, finden sich auch bei Borrelia burgdorferi und Agrobacterium tumefaciens (Fraser, C.M. et al.; 1997, Goodner, B., et al.; 2001). An den Enden des Chromosoms von Streptomyceten finden sich häufig „terminal inverted repeats“ (TIRs), die, je nach Stamm, 20-550 kBp umspannen. Die von Streptomyces coelicolor A3(2) sind ca. 20 kBp lang, die des 356 kBp langen linearen Plasmides SCP1 jeweils 75 kBp (Bentley, S.D., et al.; 2004; Bentley, S.D., et al.; 2002). TIRs wurden jedoch im Rahmen des Sequenzierungsprojektes von Streptomyces avermitilis nicht gefunden (Hopwood, D.A.; 2003; Ikeda, H., et al.; 2003). Für die äußersten ca. 200 Bp langen 3’ Enden der linearen Chromosomen und Plasmide wird eine für die Replikation bedeutsame Sekundärstruktur postuliert (Bentley, S.D., et al.; 2004). Nicht alle Mitglieder der Actinomyceten besitzen lineare Chromosomen, z.B. die der Mitglieder der Unter-Ordnung Einleitung -2- der Corynebacterineae, zu denen die Coryne- und Mycobakterien gehören, sind circulär (Li, L., et al.; 2005; Kalinowski, J., et al.; 2003; Cerdeňo-Tárranga, A. M., et al.; 2003, Cole, S.T., et al.; 1998). Des Weiteren finden sich bei Streptomyceten lineare aber auch circuläre und ins Chromosom integrierte Plasmide. So trägt S.coelicolor A3(2) das lineare 356 kBp große Plasmid SCP1 (Bentley, S.D., et al.; 2004, Vivian, A. and Hopwood, D.A.; 1970), daneben noch das 31 kBp lange circuläre Plasmid SCP2 (Haug, I., et al.; 2003, Schrempf, H., et al.; 1975), ein ins Chromosom integriertes Plasmid mit einer Länge von 17,2 kBp (SLP1INT; Lee, S.C., et al.; 1988) und vermutlich SLP4, ein Plasmid unbekannter Struktur (Hopwood, D.A., et al.; 1983). SCP1 und SCP2 sind sequenziert (Bentley, S.D., et al.; 2004; Haug, I., et al.; 2003). Wie lange bekannt und durch die Sequenzierungsprojekte von S.coelicolor A3(2), S.avermitilis und Streptomyces scabies gezeigt, besitzen Streptomyceten ein außergewöhnlich großes Genom. So ist das von S.avermitilis 9 MBp lang. Es wurden 7574 potentielle offene Leseraster identifiziert. Das von S.coelicolor besitzt eine Länge von 8,7 MBp, mit 7825 potentiellen offenen Leserastern (Ikeda, H., et al.; 2003; Bentley, S.D., et al.; 2002). Eigene Erkenntnisse zeigen, dass die hohe Zahl der potentiellen offenen Leseraster für S.coelicolor etwas zu optimistisch postuliert ist. Die Sequenz von S.scabies ist veröffentlicht, jedoch noch nicht ausgewertet. Das Genom besitzt eine Länge von 10,15 MBp (http://www.sanger.ac.uk/Projekts/S_scabies). Wie vergleichende Sequenzanalysen des S.coelicolor A3(2) (Bentley, S.D., et al.; 2002) und S.avermitils Genoms (Ikeda, H., et al.; 2003), jeweils untereinander und teilweise zu vier weiteren Actinomyceten Genomen, zweier Mycobacterium tubercolosis Stämmen (H37Rv und CDC1551; Cole, S.T., et al.; 1998; Fleischmannn, R.D., et al.; 2002), Mycobacterium leprae TN (Cole, S.T., et al.; 2001) sowie Corynebacterium diphteriae (http://www.sanger.ac.uk/Projects/C_diphtheriae/) zeigten, besitzen Streptomyceten einen zentralen, im Rahmen der Zusammensetzung und der Anordnung der Gene hoch konservierten Abschnitt, der alle bekannten essentiellen Gene enthält (Hopwood, D.A.; 2003; Ikeda, H., et al.; 2003; Bentley, S.D., et al.; 2002) und den vollständigen Genombereich von M.tuberculosis und C.diphteriae umfasst (Bentley, S.D., et al.; 2002). Dieser betrifft den ca. 6,5 MBp zentralen Abschnitt des S.avermitilis und den ca. 5 MBp zentralen Bereich des S.coelicolor Genoms (Ikeda, H., et al.; 2003; Bentley, S.D., et al.; 2002). Die äußeren Armbereiche, diese umfassen die äußeren 1,5 MBp (linker Arm) und 2,3 MBp (rechter Arm) von S.coelicolor A3(2) und 0,5 MBp (rechter Arm) und 2,0 MBp (linker Arm) von S.avermitilis, tragen nicht essentielle Gene, hiervon einen Großteil der für die Sekundärmetabolit Synthese determinierenden. Die Anordnung und Zusammensetzung der Gene ist deutlich geringer konserviert und nimmt zu den Telomeren hin ab (Ikeda, H., et Einleitung -3- al.; 2003). 44,5 % aller Gene von S.avermitilis und 42,1 % von S.coelicolor sind in den Armregionen lokalisiert (Ikeda, H., et al.; 2003). Seit im Labor von S.Waksman im Jahre 1940 das Antibiotikum Actinomycin und im Jahre 1944 Streptomycin, das erste effektive Medikament zur Behandlung der Tuberkulose, entdeckt wurden, sind die die Actinomyceten und hier besonders die Streptomyceten als Produzenten von Antibiotika und anderer biologisch wirksamer Sekundärmetaboliten bekannt. Von Mitgliedern der Actinomyceten produzierte Sekundärmetabolite finden als Antibiotika, antifungale-, antiparasitäre-, cytostatische-/Antitumorund immunsupprimierende- Medikamente und Substanzen mit weiteren biologischen Bedeutungen, wie z.B. Acarbose (Pseudomaltotetraose) zur Behandlung von Diabetes und agro-aktiven Substanzen, Verwendung. Des Weiteren besitzen sie ihre Bedeutung als Produzenten von Enzymen, Aminosäuren und bei der Degradation von Polymeren. Entsprechend ist die großtechnische Fermentation von Actinomyceten, vor allem von Streptomyceten, gut etabliert. Heutzutage sind Tausende von Antibiotika und Sekundärmetabolite mit biologisch wirksamer Aktivität bekannt. Aufbauend auf den Daten des Jahres 2002 nennt Berdy eine Zahl von 33485 entdeckten antibiotisch wirksamer Substanzen. Von diesen werden ca. 25 % (8700) von Actinomyceten produziert und unter diesen ca. 70-80 % von Mitgliedern der Streptomyceten (Berdy, J.; 2005). Als Folge wurden vor allem die der Streptomycyten in den letzten Jahrzehnten systematisch und bevorzugt nach neuen Antibiotika und biologisch aktiven Substanzen untersucht. Neuere Erkenntnisse zeigen, dass noch viele zu finden sein werden und es werden Fragen nach den bisher verwendeten Methoden aufgeworfen. Vor Beginn des Sequenzierungsprojektes des am Besten untersuchten Streptomycetenstammes, S.coelicolor A3(2), waren 4 (Hopwood, D.A.; 2003) bzw. 6 (Challis, G.L., Hopwood, D.A.; 2003) chromosomal determinierte Sekundärmetabolite bekannt. Im Rahmen des Projektes wurden 24 für Sekundärmetabolite determinierende Cluster gefunden, die ca. 4,5 % des Genoms einnehmen (Hopwood, D.A.; 2003, Bentley, S.D., et al.; 2002), in S.avermitilis, dem zweiten Streptomycetenstamm, dessen Genom sequenziert und ausgewertet ist, sogar 30 (Hopwood, D.A.; 2003; Ikeda, H., et al.; 2003). Acht (16 %) von 50 willkürlich ausgesuchten Actinomycetenstämmen tragen eine Kassette von 5 Genen für die Synthese von Antitumor„Antibiotika“ der Enediyne-Klasse. Für keinen der untersuchten 50 Stämme war die Produktion von Antibiotika dieser Klasse zuvor bekannt, noch ist es möglich, sie unter den üblichen Testbedingungen zu synthetisieren. Es ist anzunehmen, dass diese Stämme sehr wohl zur Produktion dieser Antibiotika in der Lage sind (Zazopoulos, E., et al.; 2003). Die Synthese der Sekundärmetabolite ist zumeist abhängig von der Wachstumsphase und Nährstofflimitationen. Es wird postuliert, dass viele der Sekundärmetabolite die Funktion eines alternativen chemischen Verteidigungsmechanismus besitzen und so die Überlebenschance der Organismen erhöhen (Maplestone, R.A., et al.; 1992). Aber warum sind Streptomyceten so potente Sekundärmetabolit-Produzenten, auch im Vergleich zu -4- Einleitung anderen Actinomyceten? Streptomyceten sind (außer als Sporen) als Hyphen-bildende Bodenbakterien immobil. Ihr Lebensraum ist komplex, mit unzähligen chemischen, physikalischen und biologischen Stressfaktoren und unterliegt mit der Zeit großen Veränderungen. Sie sind in der Lage, mit Hilfe ihrer zahlreichen Abbauwege verschiedenste Substrate als Nährstoffquellen zu verwenden, um in diesem Lebensraum zu bestehen; sie sind Generalisten. Sie sind nicht in der Lage, den Stressbedingungen auszuweichen, optimale Lebensbedingungen zu suchen oder der räumlichen Veränderung der optimalen Bedingungen zu folgen, anders als optimal für bestimmte Bedingungen angepasste mobile Spezialisten. Die große Zahl der in den S.coelicolor A3(2) und S.avermitilis Sequenzierungsprojekten gefundenen Gene, die für Regulatorproteine, Transporter und Nährstoffverwertung determinieren, liegt hierin vermutlich begründet (Bentley, S.D., et al.; 2002). So besitzt S.coelicolor z.B. 84 für Sensor-Kinasen determinierende Gene. Hiervon liegen 67 als SensorKinase/Antwort-Regulator Paare vor (Hutchings, M.I., et al.; 2004). S.avermitilis besitzt 60 für Sigma-Faktoren determinierende Gene, hiervon 45 zur Familie der ECF-Sigma Faktor (extra-cytoplasmic function) gehörend, (Ikeda, H., et al.; 2003), S.coelicolor 65, hievon 45 der ECF-Familie (Bentley, S.D., et al.; 2002). Weitere 3 Sigmafaktoren der ECF-Gruppe fanden sich auf dem linearen Plasmid SCP1 (Bentley, S.D., et al.; 2004). Dies ist eine deutlich höhere Zahl als bei allen anderen Bakterienstämmen. Die nächst höhere Zahl findet sich mit 23 bei Mesorhizobium loti (Kaneko, T., et al.; 2000). ECF-Sigma Faktoren sind in S.coelicolor an der Reaktion auf externen Stimuli, wie Osmo- oder Salz-Stress, beteiligt und aktivieren Gene, die in der Stressantwort, Sekundärmetabolitsynthese, Zell-Wand Homeostasis und in der Zelldifferenzierung involviert sind (Helmann, J.D.; 2002). Es wird postuliert, dass Streptomyceten zur Verteidigung ihres Lebensraumes und der Nährstoffquellen Antibiotika und andere Sekundärmetabolite, wie Siderophore, produzieren. Zur Abwehr möglicher resistenter Konkurrenten werden von einigen Stämmen auch synergistisch sowohl Antibiotika als auch Antibiotikaresistenzprotein-Hemmer synthetisiert. So produzieren Streptomyces clavigulus, Streptomyces jumonjinensis, Streptomyces katsurahamanus und ein nicht klassifizierter Streptomyces spec. zugleich sowohl das β-Lactam Cephamycin C als auch den β-Lactamase Inhibitor Clavulansäure (Challis, G.L. and Hopwood, D.A.; 2003; Jensen, S.E. and Paradkar, A.S.; 1999). Nach Ausbildung des Substratmycels (vegetative Phase) folgt, nach einer Übergangsphase (transiente Phase), die Ausbildung des Luftmycels und der Sporen (Vermehrungsphase). Hierbei kommt es zur teilweisen Autolyse des Substratmycels, um mit Hilfe des Recyclings von Makromolekülen das Wachstum des Luftmycels und der Sporen zu verbessern (Miguélez, E.M., et al.; 1999). Es wird vermutet, dass die durch die Autolyse freigesetzten Aminosäuren, Zucker und weitere Nährstoffe, Konkurrenten anlocken, die durch Antibiotika abgetötet und von den Streptomyceten-Kolonien wiederverwertet werden, als „fatal attraction“ bekannt (Challis, G.L. and Hopwood, D.A.; 2003, Shi, W. and Zusman, D.R.; 1993). Streptomyces violaceus-ruber, auch S.violaceoruber genannt, S.coelicolor A3(2) und der in dieser Arbeit verwendete Stamm S.lividans 66, sind Mitglieder der S.violaceoruber Familie. Einleitung -5- Sie sind sehr nahe miteinander verwandt, S.coelicolor A3(2) und S.lividans 66 sind genetisch nahezu identisch. Sie unterscheiden sich, wie auch von S.violaceoruber, vorwiegend durch den Besitz unterschiedlicher Plasmide. Wie das Sequenzierungsprojekt gezeigt hat, besitzt S.coelicolor A3(2) 24 für Sekundärmetabolite determinierende „Cluster“ und Gene, plasmiddeterminiert ein weiteres „Cluster“ (Bentley, S.D., et al.; 2004; Bentley, S.D., et al.; 2002). Hierunter finden sich neben für Antibiotika, Siderophore, Pigmente, Carotine, Geosmin, γ-Butyrollactone und z.B. Hopanoid und Eicosapentaenoidsäuren (mit postulierter Funktion der Membranstabilisierung unter niedrigen Temperaturen und zum Schutz vor Wasserverlust der Sporen) determinierenden „Clustern“ und Genen auch weitere mit unbekannten Funktionen. S.coelicolor A3(2) produziert vier Antibiotika, die, wie für die Synthese von Sekundärmetaboliten typisch, von in „Clustern“ zusammengefassten Genen determiniert werden. Die Mischung aus Actinorhodin, γ-Actinorhodin (Lactonform von Actinorhodin) und weiteren Actinorhodin-Derivaten (Bystrykh, L.v., et al.; 1996) wird kurz als Act bezeichnet. Sie sind aromatische Polyketid-Antibiotika, determiniert vom TypII Polyketidsynthesecluster SCO5071-5092, mit einer Länge von 22 kBp. OligopyrrolProdiginine, die eine Mischung des linearen Tripyrrols Undecylprodigiosin und des cyklischen Derivates Butyl-meta-Cycloheptylprodiginin sind und in einem 2 zu 1 Verhältnis vorliegen (Cerdeno, A.M., et al.; 2001; Tsao, S.W., et al.; 1985), werden von SCO5877-5898 (33 kBp) determiniert. Sie werden in „Kurzform“ als RED bezeichnet. Mitglieder der Prodiginine und Prodigiosine besitzen anti-fungale, anti-bakterielle, anti-Protozoen/antiMalaria, immunsuppressive und cytostatische Wirkungen. Sie sind von großem potentiellen pharmakologischen Interesse, jedoch auf Grund starker Zytotoxidität bisher nicht in pharmakologischer Anwendung (Williamson, N.R., et al.; 2005). Das „nicht-ribosomale“ cyclische Lipo-Peptidantibiotika CDA (calcium-dependend antibiotic, determiniert vom 80 kBp langen „Cluster“ SCO3210-3249), besitzt eine anti-fungale Wirkung. Methylenomycin A ist ein Cyclopentanon Antibiotikum, mit antibakterieller Wirkung gegen Gram-positive, wie auch Gram-negative Bakterien. Methylenomycin A, vor allem ein Derivat hiervon, Methylenomycin B, unterliegen wachsendem pharmakologischem Interesse. Die Methylenomycin-Synthese ist in S.coelicolor (SCP1) wie auch S.violaceoruber (pSV1), wo es ursprünglich identifiziert wurde, plasmiddeterminiert. Zusammen mit zwei pSAL2-L determinierten Polyketiden aus S.rochei (Mochizuki, S., et al.; 2003; Suwa, M., et al.; 2000) sind dies bisher die einzigen Beispiele einer plasmiddeterminierten Antibiotikasynthese. S.lividans 66 besitzt keine für Methylomycin determinierenden Gene. Somit ist S.lividans 66 nur in der Lage, Act, RED und CDA zu produzieren. Anders als S.coelicolor zeigt S.lividans im Verhältnis eine deutlich geringere Antibiotika-Produktion, die einer unterschiedlichen Regulation unterliegt; sie wird teilweise als „silent“ beschrieben, jedoch ist die Bezeichnung „strikt reguliert“ eher zutreffend. Sowohl Act, als auch RED sind farbig. Act wird als lösliches rot gefärbtes Molekül ausgeschieden, das unter basischem pH einen Farbumschlag zu blau zeigt. RED ist rotfarbig und mycelassoziiert. Die pH-Indikator Eigenschaft von Act wurde schon 1947 beschrieben (Brockmann, H. und Pini, H.; 1947). Auf Grund der einfachen -6- Einleitung Nachweisbarkeit und der Möglichkeit einer relativ einfachen Konzentrationsbestimmung, mit Hilfe spektrophotometrischer Messmethoden, ist die Regulation der Act- und REDBiosynthese am Besten von allen Antibiotikasynthesen untersucht. Die Gene der Sekundärmetabolitsynthese werden zumeist direkt von Biosynthesewegspezifischen Transkriptionsaktivatoren reguliert. Die der Antibiotikasynthese sind häufig Mitglieder der SARP Regulator Familie (Streptomyces antibiotic regulatory proteins, Wietzorrek, A. and Bibb, M.; 1997). Die Biosyntheseweg-spezifischen Regulatoren der Act-, CDA- und RED-Synthese sind das von actII-orf4 determinierte ActII-ORF4 (Act), das von cdaR determinierte Regulatorprotein CdaR (CDA) und das Regulatorprotein RedD, von redD determiniert und der dazugehörige Transkriptionsaktivator von RedD, RedZ, von redZ determiniert (RED). ActII-ORF4, CdaR und RedD sind Mitglieder der SARP Familie. Neueste Erkenntnisse zeigen für diese Biosyntheseweg-spezifischen Regulatoren eine pleiotrope, regulatorische Funktion untereinander, wie auch auf eine Reihe weiterer von der Biosynthese unabhängiger Gene, als auch für RedZ auf das höherrangige Regulatorpaar afsR2/afsS (Huang, J., et al.; 2005). Wie die der meisten anderen Sekundärmetabolite ist die Produktion von Act und RED abhängig von der Wachstumsphase. In der vegetativen Phase erfolgt keine Produktion. Sie beginnt in der Zwischenphase, auch transiente Phase genannt und ist in der Vermehrungsphase mit Beginn der Ausbildung des Luftmycels deutlich erhöht. Die Differenzierung wird hierbei vor allem durch Verminderung der vorhandenen Nährstoffquellen ausgelöst. Mutationen in bestimmten Genen (bld), die die Ausbildung des Luftmycels verhindern und eine glatte „bald“ Kolonieausbildung zur Folge haben, korellieren zum Teil mit dem Fehlen der Antibiotikasynthese. Hierzu gehört z.B. bldC, determinierend für ein DNA-Bindeprotein der MerR Familie (Hunt, A.C., et al.; 2005) und der globale Regulator bldA, das für die t-RNA des raren Leucin Codons UUA determiniert (Takano, E., et al.; 2003; White, J. and Bibb, M.; 1997). Die Reduktion der Wachstumsgeschwindigkeit in der transienten Phase zeichnet sich durch eine Erhöhung der Konzentration des intrazellulären Signalmoleküls ppGpp, synthetisiert von der Synthetase RelA und Reduzierung der GTPKonzentration aus. Hohe Konzentrationen an ppGpp induzieren die Act- und REDProduktion; die Transkription von actII-orf4 wird erhöht, jedoch nicht die von redD. Die RED-Produktion ist von weiteren regulatorischen Faktoren abhängig (Hesketh, A., et al.; 2001; Martinez-Costa, O.H., et al.; 1996). Eine Reihe weiterer Substrate, Nährstoffe, Stickstoffquellen, Signalmoleküle, pleiotrope und globale Regulatoren, Transportproteine und Zweikomponentensysteme haben Einfluss sowohl auf die Act- als auch die RED-Produktion. Hierzu gehören das extrazelluläre Signalmolekül γ-Butyrollacton SCB1 und die regulatorischen SCB1-Bindeproteine ScbR, ScbR2, CrpA und CprB (Butler, M.J., et al.; 2003; Takano, E., et al.; 2001; Onaka, H., et al.; 1998). Des Weiteren das DNA-Bindeprotein AfsS, das vom globalen Regulator AfsR (auch als AfsR2 bezeichnet) reguliert wird, der wiederum von den Sensor-Kinasen PkaG, AfsL und AfsK phosphoryliert wird, dessen Kinase Aktivität von KbpA moduliert wird (Horinouchi, S.; 2003; Bibb, M.; 1996). Ebenso Einfluss Einleitung -7- haben die Antibiotika Thiostrepton und Apramycin (Beeinflussung der Transkription von absB, Huang, J., et al.; 2005), hohe Salzkonzentrationen (Sevcikova, B. and Kormanec, J.; 2004) und die Phosphatkonzentration, reguliert durch das Sensor Protein PhoR und das DNABindeproteins PhoP der OmpR-Familie (Sola-Landa, A., et al.; 2003). Der an der Stressregulation involvierte Sigma-Faktor σB und sein Anti-Sigmafaktor RbsA sind ebenfalls an der Regulation der Antibiotikasynthese beteiligt (Lee, E.-J., et al.; 2004a; Cho, Y.-H., et al.; 2001), direkt über die Regulation der Transkription von redZ und actIV (Lee, E.-J., et al.; 2005), wie auch indirekt über σL und σM (Lee, E.-J., et al.; 2005) durch die Katalase CatB (Cho, Y.-H., et al.; 2001; Cho, Y.-H., et al.; 2000). Beteiligt sowohl an der Act- als auch RED-Synthese sind das ribosomale Protein S12 (Okamoto-Hosoya, Y., et al.; 2003a und b) und die S−Adenosylmethionin Synthetase wie auch die Gabe von S−Adenosylmethionin im Medium (Okamoto, S., et al.; 2003). Ebenso beteiligt sind Prolin-Transportmutanten (Hood, D.W, et al.; 1992) und die Rifampicin resistente Mutante rif-10 der RNA Polymerase βUntereinheit des S.lividans 66 DV-1 Stammes, determiniert von rpoB (Hu, H., et al.; 2002). Auch die Polyphosphat Kinase (Chouayekh, H and Virolle, M.-J.; 2002) und das an der Sporulation beteiligte amfC (Yonekawa, T. et al.; 1999) sind für die Antibiotikasynthese bedeutsam. Der Promoterbereich des zur LysR Familie gehörenden Transkriptionsregulators AbaB aus S.antibioticus ATCC11891 zeigt in hoher Kopienzahl in S.lividans Einfluss auf die Act- und RED-Produktion (Scheu, A.-K., et al.; 1997). Ebenso Einfluß haben die Phosphotyrosin Protein-Phosphatase (Umeyama, T., et al.; 1996) und der pleiotrope Regulator AbsB, ein Endoribonuklease RNaseIII-Homolog (Price, B., et al.; 1999; Champness, W., et al.; 1992). Die für „Zwei-Komponenten Systeme“ determinierenden absA1/A2 (Anderson, T.B., et al.; 2001; Adamidis, T. and Champness, W.; 1992), cutR/S (Chang, H.M., et al.; 1996) und afsQ1/Q2 (Ishizuka, H., et al.; 1992) sind an der Antibiotikaproduktion beteiligt. Die Disruption von afsB inhibiert die Act- und RED-Produktion (Hara, O., et al.; 1983), durch eine erhöhte Expression des Hauptsigmafaktors, determiniert von hrdB, kann diese supprimiert werden (Aigle, B., et al.; 2000). Des Weiteren scheint die Poly-3-Hydroxybutyrat Konzentration der Zellen für Antibiotika-Produktion bedeutsam zu sein (Verma, S., et al.; 2002). Eine Reihe von Nährstoffquellen haben einen Einfluss auf die Antibiotikaproduktion, wie Stickstofflimitation, Zucker oder Aminosäuren (Hobbs, G., et al.; 1990), auch Mutanten, die in den Pentose Phosphat Weg eingreifen und so vermutlich die Effektivität der Glycolyse erhöhen (Butler, M.J., et al.; 2002). Auch fördert „…die Farbstoffbildung...“ „…ganz wesentlich…“ der „…Zusatz eines wässrigen Maisauszugs“ (Plotho, O.v., 1947). Auswirkungen ausschließlich auf die Act- jedoch nicht auf die RED-Produktion haben, neben Mutanten des Act-Biosyntheseweges, folgende Faktoren und Proteine. Die Deletion des Biosyntheseweg-spezifischen Regulators ActII-Orf4 inhibiert, die Überproduktion erhöht die Produktion von Act (Gramajo, H.C., et al.; 1993; Strauch, E., et al.; 1991). Durch die Deletion von atrA, das für den Transkriptionsfaktor AtrA determiniert, ist die Act-Produktion reduziert (Uguru, G.C., et al.; 2005). Zwei ATP-abhängige Proteasen, determiniert von clp1 und clp2, sind ebenfalls indirekt an der Act-Produktion beteiligt. Überexpression des dazugehörigen -8- Einleitung von clpX determinierten Transkriptionsregulators führt zur erhöhten Act-Produktion (de Crécy-Lagard, V., et al.; 1999). Die rif-2, rif-15 und rif-17 Mutanten des für die RNA Polymerase β-Untereinheit determinierenden rpoB Gens von S.lividans 66 zeigen eine deutliche Erhöhung der Act-Produktion (Hu, H., et al.; 2002, Lai, C., et al.; 2002) Die ActSynthese ist abhängig von der Ammoniumkonzentration. Schon geringe Konzentrationen hemmen vollständig die Act-Produktion in S.coelicolor (Hobbs, G., et al.; 1990; Doul, J.L. and Vining, L.C.; 1990). Eine Reihe von Proteinen hat ausschließlich Einfluss auf die RED-Biosynthese, aber beeinflusst nicht die Act-Produktion. Mutationen und Deletionen von den an der REDBiosynthese involvierten Genen unterbrechen die Synthese von RED. Die Deletion der Biosyntheseweg-spezifischen Regulatoren RedD und RedZ, determiniert von redD und redZ, inhibiert die RED-Biosynthese (Feitelson, J.S., et al.; 1985; Narva, K.E. and Feitelson, J.S.; 1990; White, J. and Bibb, M.; 1997). Durch hohe Kopienzahlen von redD und redZ wird die RED-Produktion erhöht (Malpartida, F., et al.; 1990; Narva, K.E. and Feitelson, J.S.; 1990; White, J. and Bibb, M.; 1997). Auch ein „Zwei Komponenten System“ mit unbekannter Funktion, determiniert von ercA1/A2, der Antwort-Regulator wird hierbei von ercA1determiniert, die Sensor-Kinase von ercA2, zeigt eine positive regulatorische Funktion für die RED-Biosynthese (Li, Y.-q., et al.; 2004). Des Weiteren ist in der rif-9 Mutante der RNA Polymerase β−Untereinheit, determiniert von rpoB, die RED-Produktion deutlich erhöht (Hu, H., et al.; 2002). Die Regulation der Act- und RED-Synthese in S.coelicolor und S.lividans ist äußerst komplex und, wenn auch am Besten untersucht, bei weitem noch lange nicht vollständig verstanden. Es ist anzunehmen, dass die Regulation der Antibiotikasynthese anderer Streptomyceten-Stämme vergleichbar komplex ist. Wie beschrieben, tragen die äußeren Armbereiche vorwiegend nicht essentielle Gene und zeichnen sich durch eine hohe Variabilität aus. In unserer Arbeitsgruppe konnte aus den äußeren Armbereichen von S.lividans WT ein Genomabschnitt isoliert werden. Dieser zeichnet sich durch die Eigenschaft aus, unter hohen Konzentrationen an Chloramphenicol amplifiziert zu werden und den so entstandenen Mutanten eine deutlich erhöhte Resistenz gegenüber dem Antibiotikum zu verleihen. Auf diesem Genomabschnitt wurden neben dem für das Chloramphenicol Efflux-Protein determinierenden Gen, ein offenes Leseraster mit unbekannter Funktion (ORFI) und das für den Kaliumkanal KcsA determinierende Gen gefunden. Des Weiteren wurde das 3’ Ende eines weiteren Gens identifiziert, das den Namen ksuF erhielt. Nach Vervollständigung von ksuF, der Überproduktion und Aufreinigung des Genprodukts aus E.coli im Rahmen der Diplomarbeit, wurde KsuF zur Herstellung von Antikörpern verwendet und die Synthese in einer das Gen auf einem Konstrukt tragenden S.lividans Transformante nachgewiesen. Einleitung -9- Ziel der Arbeit war, die Rolle dieser und benachbarter Gene und deren Genprodukte aufzuschlüsseln. Deshalb sollten Strategien entwickelt werden, um größere Mengen dieser Produkte zu gewinnen. Die gezielte Erstellung von Promoterfusionen und Disruptionsmutanten, sowie deren Analyse, sollten diese Studien ergänzen. Materialien und Methoden - 11 - 2. MATERIALIEN UND METHODEN 2.1. Chemikalien und Enzyme AppliChem GmbH, Darmstadt Dodecyl-β-D-maltosid, Harnstoff Amersham Biosciences Europe GmbH, Freiburg Superdex 75 ARK Scientific GmbH, Darmstadt (jetzt Sigma-Genosys) Oligonukleotide BioWhittaker Molecular Applications, Rockland, USA LE-Agarose Carl Roth GmbH & Co., Karlsruhe Rotiphorese Gel30, Guadinin HCl, Phenol-Chloroform, Laurylsulfat Natrium-Salz (SDS, Ultra pur) Difco Laboratories, Detroit, USA Agar, Aminosäuren aus Casein (Casaminoacids), Hefeextrakt, Trypton, Pepton Eurogentec Deutschland GmbH, Köln DNA-Größenstandard (SmartLadder), Low-meltig Agarose Fermentas GmbH, St. Leon-Rot Restriktionsenzyme Gibco/BRL Ltd. Eggenstein T4-Ligase, Restriktionsenzyme, Taq-Polymerase Henselwerk GmbH, Magstadt Sojamehl - 12 - Materialien und Methoden Millipore Corp., Bedford, USA Ultrafiltrationsmembranen und -einheiten, Filtereinheiten MWG-Biotech AG, Ebersberg Oligonukleotide New England Biolabs GmbH, Frankfurt/M. Restriktionsenzyme, Protein-Größenstandard 8S Pall Europe Ltd., Portsmouth, Großbritannien FluoroTrans®-Membran für Proteintransfer, Biodyne B für DNA-Transfer Promega GmbH, Mannheim Restriktionsenzyme, Pfu-Polymerase Qiagen GmbH, Hilden Taq PCR Core Kit, Qiafilter Plasmid Midi Kit, Qiagen Plasmid Midi Kit, QIAprep Spin M13 Kit, QIAquick PCR Purification Kit, QIAquick Gel Extraction Kit, QIAEX II Gel Extraction Kit, QIAquick Nucleotide Removal Kit, Ni-NTA-Agarose Roche Diagnostics GmbH, Mannheim Restriktionsenzyme, Dig-DNA Labeling and Detection Kit, Blocking-Reagenz Schleicher & Schuell, Dassel Faltenfilter, Papierfilter, Membranfilter, Filtereinheiten Serva Feinbiochemika GmbH & Co., Heidelberg Agar, Ammoniumpersulfat, Aminosäuren, Coomassie Brilliant Blue R-250, Coomassie Brilliant Blue G-250, Bromphenolblau, n-Dodecylsulfat-Natrium-Salz (Laurylsulfat, SDS), Isopropyl-β-D-Thiogalactopyranosid (IPTG) Squibb E. R. & Sons, Princeton, USA Thiostrepton Stratagene, La Jolla, USA SanDI Whatman International Ltd., Maidstone, Großbritannien Filterpapiere Alle nicht gesondert aufgeführten Chemikalien wurden von den Firmen Sigma Aldrich Chemie (Steinheim), F. Merck (Darmstadt), Fluka/Riedel-de-Haën (Seelze) und Serva (Heidelberg) bezogen. Materialien und Methoden - 13 - 2.2. Bakterienstämme, Vektoren, Konstrukte, Phagen und Oligos 2.2.1. Bakterienstämme Stamm E.coli DH5α S.lividans 66 Phänotyp/Genotyp F , φ80d/lacZ∆M15, recA1, endA1, gyrA96, thi−, hsdR17 (rK-,mK+), supE44, relA1, deoR, ∆ (lacZYA-argF) U169 recA1, endA1, gyrA96, thi-1, hsdR17,supE44, relA1, lac, [F proAB, lacIqZ∆M15, Tn10 (tetR)] hsdR-, hsdM+, gal-, met-, supE, recAnalS, strS, rifS, thi-, lac-, ara+, gal+, mtlrecA+, uvr+, lon+ [pREP4: lacIq, neoR] rpsL (Strr), thr, leu, thi-1, lacY, galK, galT, ara, tonA, tsx, dam, dcm, supE44 ∆(lacproAB), [F´ traD36 proAB lacIqZ∆M15]. Wildtyp, SLP2+, SLP3+ S.lividans TK21 SLP2-, SLP3+ S.lividans TK24 SLP2-, SLP3- S.lividans NI arg, Ntr, cmlS, tetS S.coelicolor M145 SCP1-, SCP2- S.coelicolor M145 sigH:tsr S.coelicolor J1984 SCP1-, SCP2-, sigH:tsr S.lividans 66 ∆kcsA (∆kcsA9, 13 und 14) S.lividans 66 ∆ksuF (∆ksuF2, 6 und 7) S.lividans 66 ∆ksuL (∆ksuL17, 20, 21 und 24) S.lividans 66 ∆orfI (∆orfI35, 36, 37 und 39) S.lividans 66 ∆ksuB (∆ksuB21, 22, 24, 25, 26, 28, 29, 31 und 32) S.lividans 66 ∆ksuC (∆ksuC1, 3, 4, 5, 6, 7, 8, 11, 12, 15, 19 und 20) kcsA:Ωhyg, Disruption von kcsA, hygR, Integration des Ωhyg-Fragmentes, ksuF:Ωhyg, Disruption von ksuF, hygR, Integration des Ωhyg-Fragmentes ksuL:Ωhyg, Disruption von ksuL, hygR, Integration des Ωhyg-Fragmentes E.coli XL1-blue E.coli K802 recAE.coli M15 [pREP4] E.coli JM110 - [M145] SCP1-, SCP2-, sigF:tsr Referenz Woodcock, D.M., et al.; 1989 Bullock, W.O., et al.; 1987 Wood, W.B.; 1966 Villarejo M.I. and Zabin, I.; 1974 Yanisch-Perron, C., et al.; 1985 Lomovskaya et al., 1972 Hopwood, D.A., et al.; 1985 Hopwood, D.A., et al.; 1985 Dyson und Schrempf, 1987 Hopwood, D.A., et al.; 1985 Sevcikova, B., et al.; 2001 Keleman, G.M.; et al.; 1998 Diese Arbeit Diese Arbeit Diese Arbeit orfI:Ωhyg, Disruption von orfI, hygR, Integration des Ωhyg-Fragmentes Diese Arbeit ksuB:Ωhyg, Disruption von ksuB, hygR, Integration des Ωhyg-Fragmentes Diese Arbeit ksuC:Ωhyg, Disruption von ksuC, hygR, Integration des Ωhyg-Fragmentes Diese Arbeit Materialien und Methoden - 14 S.lividans 66 KEH5 (K ,L und M) S.lividans 66 FEH11 (A, B, C, D, E, F, G, H, I, und N) S.lividans 66 FEH4 (P, Q, R, S, T, U, V, W, Y und Z) kcsA::EGFP-GenΩhyg, hygR, Integration von EGFP-Gen „in Frame“ hinter kcsA ksuF::EGFP-GenΩhyg, hygR, Integration von EGFP-Gen „in Frame“ hinter ksuF ksuF::EGFP-GenΩhyg, hygR, Integration von EGFP-Gen „out of Frame“ hinter ksuF Diese Arbeit Diese Arbeit Diese Arbeit 2.2.2. Vektorplasmide Vektor pBR322 pUC18 pUC19 pUS18 pBSK(+) Grösse 4,4 kBp 2,7 kBp 2,7 kBp 2,7 kBp 2,96 kBp Selektion bla, tet bla, lacZ bla, lacZ bla, lacZ bla, lacZ pACYC184 M13mp18 pHP45Ωhyg 4,25 kBp cat, tetZ 7,1 kBp lacZ unbekannt hygR, cat, trägt Ωhyg-Fragment pQE32 pQE70 pEGFP1 3,5 kBp 3,4 kBp 4,2 kBp pIJ702 pGM160 5,7 kBp 7,8 kBp pWHM3be 7,1 kBp bla bla kanR/neoR, trägt EGFP-Gen tsr, melC1, melC2 bla, tsr, aacC1, Temps bla, lacZα, tsr Referenz Balbas, P., et al.; 1988 Yanisch-Perron, C., et al.; 1985 Yanisch-Perron, C., et al.; 1985 Yanisch-Perron, C., et al.; 1985 Alting-Mees, M. A. and Short, J.M.: 1989 Rose, R.E.; 1988 Yanisch-Perron, C., et al.; 1985 Saito, A. Blondelet-Rouault, M.H., et al.; 1997 Qiagen, Hilden Qiagen, Hilden Clontech Laboratories inc. Katz, E., et al.; 1983 Muth, G., et al.; 1989 Vara, J., et al.; 1989 2.2.3. Rekombinante Konstrukte Konstrukte pOBH23 pOSP13 pK8 pBRS1 pBRS2 Beschreibung Rekombinante E.coli Konstrukte 4,5 kBp BglII/HindIII Fragment aus CCM1 ligiert mit HindIII/BamHI geschnittenen pUC18 1,05 kBp PvuII/SstI Fragment aus pOBH23, mit SmaI/SstI geschnittenen pUC18 ligiert 4,2 kBp SphI/HindIII-Fragment aus CCM1, ligiert mit pBSK(+) 2,1 kBp NotI/KpnI-Fragment aus CCM3, mit pBSK(+) ligiert 4,3 kBp PstI/NotI-Fragment aus CCM3, mit pBSK(+) ligiert Referenz Diplomarbeit Diplomarbeit Diese Arbeit Ralf Steger Ralf Steger Materialien und Methoden - 15 - Rekombinante E.coli/S.lividans-Konstrukte (pACYC/pOVE-Reihe) pACYC001 Aus den Oligos Pr184I und Pr184II fusionierte 55 BpDiese Arbeit Fragment, ligiert mit AvaI/XbaI-geschnittenen pACYC184 pACYC002 1,9 kBp SstI/HindIII-Fragment aus pJO1, mit pACYC001 Diese Arbeit ligiert pACYC003 3,2 kBp SstI/SgfI-Fragment aus pOBH23, mit pACYC002 Diese Arbeit ligiert pACYC004 4,2 kBp SgfI/PstI-Fragment aus pBRS2, mit pACYC003 Diese Arbeit ligiert pACYC005 4,6 kBp BglII/MluI-Fragment aus pACYC006 mit Diese Arbeit pACYC004 ligiert pACYC006 3,5 kBp Bpu1102I/MluI-Fragment aus pK8 mit pACYC002 Diese Arbeit ligiert pACYC007 4,6 kBp BglII/MluI- Fragment aus pACYC006 mit Diese Arbeit pACYC003 ligiert pACYC008 3,2 kBp SstI/SgfI-Fragment aus pOBH23 mit pACYC001 Diese Arbeit ligiert pACYC009 4,2 kBp SgfI/PstI-Fragment aus pBRS2 mit pACYC001 Diese Arbeit ligiert pACYC010 4,2 kBp SgfI/PstI-Fragment aus pBRS2 mit pACYC008 Diese Arbeit ligiert pOV001 50 Bp PstI/XbaI-Fragment aus pACYC001 mit pWHM3be Diese Arbeit ligiert pOV002 2 kBp PstI/XbaI-Fragment aus pACYC002 mit pWHM3be Diese Arbeit ligiert pOV003 5,1 kBp PstI/XbaI-Fragment aus pACYC003 mit pWHM3be Diese Arbeit ligiert pOV004 9,3 kBp PstI/XbaI-Fragment aus pACYC004 mit pWHM3be Diese Arbeit ligiert pOV005 12,1 kBp PstI/XbaI-Fragment aus pACYC005 mit Diese Arbeit pWHM3be ligiert pOV006 4,7 kBp PstI/XbaI-Fragment aus pACYC006 mit pWHM3be Diese Arbeit ligiert pOV007 7,9 kBp PstI/XbaI-Fragment aus pACYC007 mit pWHM3be Diese Arbeit ligiert pOV008 3,2 kBp PstI/XbaI-Fragment aus pACYC008 mit pWHM3be Diese Arbeit ligiert pOV009 4,2 kBp PstI/XbaI-Fragment aus pACYC009 mit pWHM3be Diese Arbeit ligiert pOV010 7,4 kBp PstI/XbaI-Fragment aus pACYC010 mit pWHM3be Diese Arbeit ligiert Sonstige rekombinante E.coli/S.lividans-Konstrukte pJO1 2,1 kBp StuI/Bpu1102 Fragment aus CCM1 in Diplomarbeit Ecl136/Bpu1102 Schnittstelle von pWH115 (2,8 kBp Insert) pWH115 pWHM3be-Derivat, trägt ein 1,2 kBp HindIII/EcoRII. Borovok, Fragment, mit 5’-Teil von ksuF, kcsA und 5’-Teil von orfI H. Schrempf, nicht veröffentlicht - 16 pWH297 pOVH101 Materialien und Methoden pWHM3be-Derivat, trägt ein 3,05 kBp SstI/XbaI-Fragment, mit 5’-Teil von ksuF, kcsA, orfI und 5’-Teil von cmlR I. Borovok, H. Schrempf, nicht veröffentlicht Diese Arbeit Ωhyg-Fragment, BamHI-geschnitten, mit pOV001 ligiert, Leserichtung von hygR entgegegesetzt von tsr pOVH201 Ωhyg-Fragment, BamHI-geschnitten, mit pOV001 ligiert, Diese Arbeit Leserichtung von hygR in Richtung von tsr Überproduktion von Proteinen in E.coli M15 [pREP4] oder XL1blue Heiner Splitt pQE837 pQE32-Derivat, trägt ein 0,57 kBp Fragment (kcsA), für die Produktion von His-KcsA in E.coli M15 [pREP4] oder XL1blue pORFZ 0,9 kBp SphI/HindIII PCR-Fragment ligiert mit pUC18 Diese Arbeit pQE309 0,9 kBp SphI/HindIII PCR-Fragment aus pORFZ, ligiert mit Diese Arbeit pQE32, für Produktion von His-KsuB in E.coli M15 [pREP4] pQE709 0,9 kBp SphI/HindIII PCR-Fragment aus pORFZ, ligiert mit Diese Arbeit pQE70, für Produktion von KsuB in E.coli M15 [pREP4] pQE314 1,38 kBp SphI/MscI Fragment aus pJO1 in SmaI/SphI Diplomarbeit geschnittenen pQE32, für Produktion von His-KsuF in E.coli M15 [pREP4] pQE714 1,4 kBp SphI/HindIII Fragment aus pQE314 in pQE70, für Diplomarbeit Produktion von KsuF in E.coli M15 [pREP4] pORFX1 1,76 kBp SphI/HindIII PCR-Fragment, ligiert mit pUC18 Diese Arbeit pORFX2 1,7 kBp SphI/HindIII PCR-Fragment, ligiert mit pUC18 Diese Arbeit Diese Arbeit pQE315 1,76 kBp SphI/HindIII Fragment aus pORFX1, ligiert mit pQE32, ermöglicht die Produktion von His-KsuL mit Signalpeptid in E.coli M15 [pREP4] Diese Arbeit pQE715 1,76 kBp SphI/HindIII Fragment aus pORFX1, ligiert mit pQE70, ermöglicht die Produktion von KsuL mit Signalpeptid in E.coli M15 [pREP4] Diese Arbeit pQE316 1,76 kBp SphI/HindIII Fragment aus pORFX2, ligiert mit pQE32, ermöglicht die Produktion von His-KsuL ohne Signalpeptid in E.coli M15 [pREP4] Diese Arbeit pQE716 1,76 kBp SphI/HindIII Fragment aus pORFX2, ligiert mit pQE32, ermöglicht die Produktion von KsuL ohne Signalpeptid in E.coli M15 [pREP4] Diese Arbeit pQE317 0,2 kBp SphI/HindIII PCR-Fragment, ligiert mit pQE32, für Produktion des His-C-terminalen-KcsA in E.coli M15 [pREP4] pORS1 0,9 kBp SphI/HindIII PCR-Fragment ligiert mit pWHM3be Ralf Steger Ralf Steger pQE321 0,9 kBp SphI/HindIII PCR-Fragment aus pORS1 ligiert mit pQE32, für Produktion von His-KsuC in E.coli M15 [pREP4] pQE721 0,9 kBp SphI/HindIII PCR-Fragment aus pORS1 ligiert mit Ralf Steger pQE70, für Produktion von KsuC in E.coli M15 [pREP4] Materialien und Methoden pOVV1 pOVV2 pOVE3 pOVE4 pOVE5 pOVE31 pOVE41 pOVE51 pOVE32 pOVE42 pOVE52 pWRS1 pOVE54 pOVE33 pOVE43 pOVE53 pOVG1 pOVG11 pOVG12 pOVG13 pOVG14 pOVG15 pOVG17 Protein-Produktionskonstrukte in S.lividans TK21 80 Bp XbaI/HindIII PCR-Fragment aus pQE32, ligiert mit pWHM3be 356 Bp EcoRI/Mph1103I PCR-Fragment (Bereich vor ksuF), ligiert mit pOVV1 158 Bp HindIII/MluI PCR-Fragment (Terminator-Bereich), ligiert mit pOVV2 336 Bp EcoRI/SphI PCR-Fragment (Bereich vor ksuF), ligiert mit pOVE3 Aus den Oligos PrPoly3I und PrPoly3II fusionierte 34 Bp Fragment, ligiert mit dem Mph1103I/BamHI geschnittenen pOVE3 560 Bp SphI/HindIII-Fragment aus pQE837, ligiert mit pOVE3 560 Bp SphI/HindIII-Fragment aus pQE837, ligiert mit pOVE4 560 Bp SphI/HindIII-Fragment aus pQE837, ligiert mit pOVE5 929 Bp SphI/HindIII-Fragment aus pORFZ, ligiert mit pOVE3 929 Bp SphI/HindIII-Fragment aus pORFZ, ligiert mit pOVE4 929 Bp SphI/HindIII-Fragment aus pORFZ, ligiert mit pOVE5 897 Bp SphI/HindIII Fragment aus pQE321, ligiert mit pOVE3 897 Bp SphI/HindIII Fragment aus pWRS1, ligiert mit pOVE5 1,4 kBp SphI/HindIII Fragment aus pQE314, ligiert mit pOVE3 1,4 kBp SphI/HindIII Fragment aus pQE314, ligiert mit pOVE4 1,4 kBp SphI/HindIII Fragment aus pOVE33, ligiert mit pOVE5 EGFP-Promoter Test-Konstrukte in S.lividans TK21 0,73 kBp Mph1103I/XbaI PCR-Fragment aus pEGFP1 mit pOVE3 ligiert 1 kBp BglII/Mph1103I PCR-Fragment (Bereich vor kcsA), Ligation mit pOVG1 0,35 kBp BglII/Mph1103I PCR-Fragment (Bereich vor cmlR), Ligation mit pOVG1 1 kBp BglII/Mph1103I PCR-Fragment (Bereich vor ksuF), Ligation mit pOVG1 0,98 kBp BglII/Mph1103I-Fragment (Bereich vor ksuL), Ligation mit pOVG1 0,53 kBp Mph1103I/BglII-Fragment (Bereich vor ksuC), Ligation mit pOVG1 0,5 kBp Mph1103I/BglII PCR-Fragment (Kontrolle), Ligation mit pOVG1 - 17 - Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Ralf Steger Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit - 18 - pTSK pUTSK1H pGMK pTSF pUTSF1H pGMF pTSL pUTSL1H pGML pTSO pUTSO1H pGMO pTSB pUTSB1H pGMB pTSC pUTSC1H pGMC Materialien und Methoden Disruption von Genen in S.lividans 66 725 Bp HindIII/ SnaBI PCR-Fragment (Bereich vor kcsA) und 696 Bp KpnI/SnaBI PCR-Fragment (hinter kcsA), Ligation mit dem HindIII/KpnI geschnittenen pUC18 2,3 kBp DraI-Ωhyg Fragment aus pHP45Ωhyg, Ligation mit SnaBI geschnittenen pTSK 3,6 kBa HindIII/NcoI Fragment aus pUTSK1H, Ligation mit pGM160 758 Bp HindIII/ SnaBI PCR-Fragment (Bereich vor ksuF) und 672 Bp KpnI/SnaBI PCR-Fragment (hinter ksuF), Ligation mit dem HindIII/KpnI geschnittenen pUC18 2,3 kBp DraI-Ωhyg Fragment aus pHP45Ωhyg, Ligation mit SnaBI geschnittenen pTSF 3,7 kBa HindIII/NcoI Fragment aus pUTSF1H, Ligation mit pGM160 764 Bp HindIII/ SnaBI PCR-Fragment (Bereich vor ksuL) und 723 Bp KpnI/SnaBI PCR-Fragment (hinter ksuL), Ligation mit dem HindIII/KpnI geschnittenen pUC18 2,3 kBp DraI-Ωhyg Fragment aus pHP45Ωhyg, Ligation mit SnaBI geschnittenen pTSL 3,7 kBa HindIII/NcoI Fragment aus pUTSL1H, Ligation mit pGM160 731 Bp HindIII/ SnaBI PCR-Fragment (Bereich vor orfI) und 539 Bp KpnI/SnaBI PCR-Fragment (hinter orfI), Ligation mit dem HindIII/KpnI geschnittenen pUC18 2,3 kBp DraI-Ωhyg Fragment aus pHP45Ωhyg, Ligation mit SnaBI geschnittenen pTSO 3,5 kBa HindIII/NcoI Fragment aus pUTSO1H, Ligation mit pGM160 755 Bp HindIII/ SnaBI PCR-Fragment (Bereich vor ksuB) und 642 Bp KpnI/SnaBI PCR-Fragment (hinter ksuB), Ligation mit dem HindIII/KpnI geschnittenen pUC18 2,3 kBp DraI-Ωhyg Fragment aus pHP45Ωhyg, Ligation mit SnaBI geschnittenen pTSB 3,7 kBa HindIII/NcoI Fragment aus pUTSB1H, Ligation mit pGM160 731 Bp HindIII/ SnaBI PCR-Fragment (Bereich vor ksuC) und 593 Bp KpnI/SnaBI PCR-Fragment (hinter ksuC), Ligation mit dem HindIII/KpnI geschnittenen pUC18 2,3 kBp DraI-Ωhyg Fragment aus pHP45Ωhyg, Ligation mit SnaBI geschnittenen pTSC 3,6 kBa HindIII/NcoI Fragment aus pUTSC1H, Ligation mit pGM160 Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Diese Arbeit Materialien und Methoden - 19 - Integration vom EGFP-Gen hinter kcsA in S.lividans 66 Diese Arbeit pBSK2 2 kBp PvuII/PstI Fragment (melC1/melC2) aus pIJ702 und 2,2 kBp DraI Fragment aus pHP45Ωhyg (Ωhyg), zugleich ligiert mit SmaI/PstI geschnittenen pBSK(+) Diese Arbeit pBSK3 740 Bp SstI/KpnI PCR-Fragment (Bereich vor kcsA und kcsA), ein 529 Bp NotI/XbaI PCR-Fragment (Bereich hinter kcsA) und ein 757 Bp KpnI/NotI Fragment aus pEGFP1 (EGFP-Gen) wurden zugleich mit SstI/XbaI geschnittenen pBSK(+) ligiert Diese Arbeit pGM4 2 kBp XbaI/partiell NcoI-Fragment aus pBSK3, 4,2 kBp XbaI/HindIII-Fragment, ligiert zugleich mit NcoI/HindIII geschnittenen pGM160 Integration von EGFP-Gen und hygR hinter Genen in S.lividans 66 Diese Arbeit pUCK1K2 762 Bp BamHI/XbaI PCR-Fragment (Bereich vor kcsA und kcsA) und 721 Bp BamHI/HindIII PCR-Fragment (Bereich nach kcsA), zugleich ligiert mit XbaI/HindIII pUC18 Diese Arbeit pUCKEH 1,56 kBp SanDI/NotI Fragment aus pHP45Ωhyg, 757 Bp KpnI/NotI Fragment aus pEGFP1, zugleich ligiert mit SanDI/partiell KpnI geschnittenen pUCK1K2 pGMKEH 3,8 kBp HindIII/XbaI Fragment aus pUCKEH, ligiert mit Diese Arbeit pGM160 Diese Arbeit pUCF1F2 706 Bp BamHI/XbaI PCR-Fragment (3’ Bereich von ksuF) und 576 Bp BamHI/HindIII PCR-Fragment (Bereich nach ksuF), zugleich ligiert mit XbaI/HindIII pUC18 Diese Arbeit pUCFEH 1,56 kBp SanDI/NotI Fragment aus pHP45Ωhyg, 757 Bp KpnI/NotI Fragment aus pEGFP1, zugleich ligiert mit SanDI/partiell KpnI geschnittenen pUCF1F2 pGMFEH 3,6 kBp HindIII/XbaI Fragment aus pUCFEH, ligiert mit Diese Arbeit pGM160 pUCFEH2 pUCFEH ApaI geschnitten, 3’ Ende mit T4 Polymerase Diese Arbeit abverdaut und wieder aufgefüllt, religiert Diese Arbeit pUCFEH4 pUCFEH2 NcoI geschnitten, 5’ Ende mit „Klenow“ aufgefüllt, religiert pGMFEH4 3,6 kBp HindIII/XbaI Fragment aus pUCFEH4, ligiert mit Diese Arbeit pGM160 2.2.4. Bakteriophagen (Lambda-Derivate) Phage Charon 35 CCM1 CCM3 (RS1) Beschreibung λsbhI1°, lac5, srI, lacZ, srIλ3, ∆WL113, KH54, nin5, shndIII, λ6°, srIλ5° 9,65 kBp partiell verdautes Sau3A Fragment aus S.lividans 66 Chromosom ligiert mit BamHI geschnittenen Charon 35 Ca. 9kBp partiell verdautes Sau3A Fragment aus S.lividans 66 Chromosom ligiert mit BamHI geschnittenen Charon 35 Referenz Loenen, W.A. and Blattner, F.R.; 1983 Michael Betzler, unsere Arbeitsgruppe, 1986 Ralf Steger, unsere Arbeitsgruppe, nicht veröffentlicht Materialien und Methoden - 20 2.2.5. Im Rahmen der Arbeit erwähnte Oligos Oligo Sequenz Beschreibung Konstruktion von pOVV1, pOVV2, pOVE3 und pOVE4 PrPCR1HI GAGCGTTCTGAACAAATCC Konstruktion von pOVV1 2RPCR1XN GAGGAGATCTAGATGCATAGAG Konstruktion von pOVV1, trägt zusätzlich eine Mph1103I und GATCTCAC XhoI- Schnittstelle Pr70 GTTGTAGTCGTGGATGTCC pOVV2 und pOVE4, Amplification Promoterbereich von ksuF Pr71 GCAGTTTGACCAATCTGG pOVV2, Amplification Promoterbereich von ksuF PRPCR2E GACAGGAATTCAGATCTGCTCC pOVV2 und pOVE4, Amplification Promoterbereich GCCACCGG von ksuF, zusätzlich EcoRI und BglII PRPCR2N CAGGGGTATGCATGGTGGCTCC pOVV2, Amplification Promoterbereich von ksuF, TTCGGG zusätzlich Mph1103I (Startkodon) pRPCR3MH CACACGAACGCGTTAACGCGAA pOVE3, Amplifikation des Terminators, zusätzlich MluI CTGACCC und HpaI pRPCR3SX GATCAAGCTTCTAGACTAGTGAG pOVE3, Amplifikation des Terminators, zusätzlich HindIII, CAGGGAGGGC XbaI und SpeI Konstruktion von pOVE5 PrPoly3I TCGCGGCTCCCACCACCACCACC Für N-terminalen His-Tag, Verschmelzung mit PrPoly3II ACCACGG PrPoly3II GATCCCGTGGTGGTGGTGGTGGT Für N-terminalen His-Tag, Verschmelzung mit PrPoly3I GGGAGCCGCGATGCA Konstruktion von pOVG1 pREGFPmp CGCCACCATGCATAGCAAGGGCG Amplifikation des EGFP-Gens, AGGAGCTG zusätzlich Mph1103I (Startkodon) pREGFPxb CACTCAACCCTATCTCGG Amplifikation des EGFP-Gens, zusätzlich XbaI Konstruktion von pOVG11, pOVG12, pOVG13, pOVG14, pOVG15 und pOVG17 pkcsam1 GTTTGACATGCATGGCCAGAAGA pOVG11, Amplifikation Promoterbereich von kcsA, CCGGAC zusätzlich Mph1103 im Startkodon Pr23 GCCCAGCCGTCGAATTCC pOVG11, Sequenzierungsoligo, Amplifikation Promoterbereich von kcsA PcmlRm1 CATGGCGCAATGCATCACGGCGA pOVG12, Amplifikation Promoterbereich von cmlR, GCAGGTAC zusätzlich Mph1103 im Startkodon Materialien und Methoden PcmlRb2 pksuFm1 pksuFb2 Pksulm1 Pksulb2 PorfYm1 PorfYb2 pkontm1 pkontb1 pkcsas1 pkcsah2 orfZstart orfZende orfX1Start orfX2start orfXende CCGGTCAAGATCTCGGTGACGAA GGG - 21 - pOVG12, Amplifikation Promoterbereich von cmlR, zusätzlich BglII GTCCGCCATGCATGGCGCCGATG pOVG13, Amplifikation Promoterbereich von ksuF, ATCGTG zusätzlich Mph1103 im Startkodon GTCCACCGAGATCTGCCAGCCGC pOVG13, Amplifikation Promoterbereich von ksuF, CGTG zusätzlich BglII CAGTGGATGCATCAGCCGGGTGG pOVG14, Amplifikation Promoterbereich von ksuL, TGCGCG zusätzlich Mph1103 im Startkodon GCCGCGCAGATCTCGGACCTCCC pOVG14, Amplifikation Promoterbereich von ksuL, GCTC zusätzlich BglII TCGCGGAATGCATCCGGCCGCCG pOVG15, Amplifikation Promoterbereich von ksuC, ATGGTG zusätzlich Mph1103 im Startkodon GCCCGCTAGATCTGCGGGGACGC pOVG15, Amplifikation Promoterbereich von ksuC, GCTC zusätzlich BglII GACTGCTATGCATCCACGACTAC pOVG17, Amplifikation eines internen Abschnittes von ksuF, AAC zusätzlich Mph1103 CACCCGCAGATCTCGGTGATCAG pOVG17, Amplifikation eines internen Abschnittes von ksuF, GTC zusätzlich BglII Konstruktion von pQE317 CCGCGCGCATGCCCTGGTTCGTC Amplifikation des 3’ Endes von kcsA, Mph1103 im Startkodon GGCCG CGCACAAGCTTCGCAGACTCATC Amplifikation des 3’ Endes von kcsA, HindIII GGTGC Konstruktion von pQE309 und pQE709 GCCGCATGCAGACCTTCACCCTG Amplifikation von ksuB, zusätzlich SphI im Startkodon CC GCGCCGAAGCTTACGGCACCGTG Amplifikation von ksuB, zusätzlich HindIII TAGC Konstruktion von pQE315, pQE715, pQE316 und pQE716 GGGATGCATGCCCAGAACGCGC Amplifikation von ksuL, 5’ Ende mit Signalpeptid, ACC zusätzlich SphI CGCCGGCATGCCCGTGACCGCC Amplifikation von ksuL, 5’ Ende ohne Signalpeptid, GGACTC zusätzlich SphI CCGAGGAAGCTTCAGTTCGCGTT Amplifikation von ksuL, 3’ Ende, zusätzlich HindIII CACC - 22 - Pr184I Pr184II PrkcsAIHin PrkcsAISna PrkcsAIISna PrkcsAIIKpNc PrksuFIHin PrksuFISna PrksuFIISna PrksuFKpNc PrksuLIHin PrksuLISna PrksuLIISna PrksuLIIKpNc ProrfIHin Materialien und Methoden Konstruktion von pACYC001 CCGACTGCAGCGGCCGCGCGATC Verschmelzung mit Pr184II, GCGCATGCGAGCTCATGCATAAG intern PstI, NotI, SgfI, SphI, CTTACGCGT Mph1103I, HindIII und MluI, überhängende Enden: XbaI und AvaI CTAGACGCGTAAGCTTATGCATG Verschmelzung mit Pr184I, AGCTCGCATGCGCGATCGCGCGG intern PstI, NotI, SgfI, SphI, CCGCTGCAG Mph1103I, HindIII und MluI, überhängende Enden: XbaI und AvaI Disruption von kcsA, ksuF, ksuL, orfI, ksuB und ksuC ACGGCTAAGCTTGTGACCCCACC ∆kcsA, Amplifikation Bereich fluss-aufwärts von kcsA, GCAC +HindIII ATCTGGTACGTAGACCGGACAGC ∆kcsA, Amplifikation Bereich flussATGGG aufwärts von kcsA, +SnaBI CCACTCTACGTAGGCCGCCGAGG ∆kcsA, Amplifikation Bereich fluss-abwärts von kcsA, +SnaBI AGGCG GGCTCCGGTACCATGGGAATCGC ∆kcsA, Amplifikation Bereich fluss-abwärts von kcsA, +KpnI CCCGTCGG +NcoI CAGCGCAAGCTTCACCAGACCGA ∆ksuF, Amplifikation Bereich fluss-aufwärts von ksuF, AGGAGG +HindIII GCGCGCTACGTAGGTGACGAGTC ∆ksuF, Amplifikation Bereich fluss-aufwärts von ksuF, GGCC +SnaBI GACGGGTACGTACTGGGTACGGC ∆ksuF, Amplifikation Bereich fluss-abwärts von ksuF, +SnaBI CCTCG CCGACCGGTACCATGGAATGGGA ∆ksuF, Amplifikation Bereich fluss-abwärts von ksuF, +KpnI CC +NcoI GCTGGCAAGCTTGCCGGACGCCG ∆ksuL, Amplifikation Bereich fluss-aufwärts von ksuL, AACCGC +HindIII CGACCGTACGTATGCCTGTGAGC ∆ksuL, Amplifikation Bereich fluss- aufwärts von ksuL, AGTGGAC +SnaBI CTGCCTTACGTACACCGACGCGA ∆ksuL, Amplifikation Bereich fluss-abwärts von ksuL, +SnaBI TCGAC GGGGTGGGTACCATGGGCGACGC ∆ksuL, Amplifikation Bereich fluss-abwärts von ksuL, +KpnI GGCCCACC +NcoI GTGAAGAAAGCTTACGGACCGTA ∆orfI, Amplifikation Bereich fluss-aufwärts von orfI, AAGG +HindIII Materialien und Methoden ProrfISna GCCGTGTACGTAGCCGCTCTCGG CGTCCTC - 23 - ∆orfI, Amplifikation Bereich fluss- aufwärts von orfI, +SnaBI ProrfIISna GACGTCTACGTACTGGCAGAACT ∆orfI, Amplifikation Bereich fluss- abwärts von orfI, +SnaBI CGACCC ProrfIIKpNc TCCGAGGGTACCATGGCGCAGAC ∆orfI, Amplifikation Bereich fluss-abwärts von orfI, +KpnI GGCCAC +NcoI PrksuBIHin CGCGAGAAGCTTGCGGCCGGCGC ∆ksuB, Amplifikation Bereich fluss-aufwärts von ksuB, CGACGTC +HindIII PrksuBISna GGATGCTACGTAGGCCGAGGAC ∆ksuB, Amplifikation Bereich fluss- aufwärts von ksuB, GATGTTG +SnaBI PrksuBIISna GAGCGATACGTACGGCGTGCCG ∆ksuB, Amplifikation Bereich fluss-abwärts von ksuB, +SnaBI GCCGAG PrksuBIIKpNc CTCGCCGGTACCATGGCTGAGCG ∆ksuB, Amplifikation Bereich fluss-abwärts von ksuB, +KpnI GCAGTTCCG +NcoI PrksucIHin ACCCGGAAGCTTCAGGGGGCAG ∆ksuC, Amplifikation Bereich fluss-aufwärts von ksuC, GACCGCG +HindIII PrksuCISna CACGCTTTACGTATGCGGGGTCC ∆ksuC, Amplifikation Bereich fluss-aufwärts von ksuC, CCGTTG +SnaBI PrksuCIISna CGCTCGTACGTATCGCCAACGCC ∆ksuC, Amplifikation Bereich fluss-abwärts von ksuC, +SnaBI AAGG PrksuCIIKpNc CTGGCGGGTACCATGGTTCGAGA ∆ksuC, Amplifikation Bereich fluss-abwärts von ksuC, +KpnI CGCCGAAG +NcoI Konstruktion von pBSK3 (pGM4), Integration des EGFP-Gens hinter kcsA PrIEK GACGGCGAGCTCCATGGAGCCAC Bereich von kcsA und flussaufwärts liegender CGACATCCGAC Abschnitt, +SstI +NcoI PrIIEK CGGCGGGGTACCCGGCGGTTGTC Bereich von kcsA und flussaufwärts liegender GTCGAG Abschnitt, +KpnI PrIIIEK ACGACAGCGGCCGCTGACTCCGC Bereich des flussabwärts liegenden Abschnittes von CGGTGACC kcsA, +NotI PrIVEK GGTACTTCTAGACGAGGCCGTGC Bereich des flussabwärts liegenden Abschnittes von CGCATG kcsA, +XbaI pUCK1K2 und pUCF1F2, Integration des EGFP-Gens hinter kcsA und ksuF PrkcsAK CGGTCAGGATCCGGGTACCCGG pUCK1K2, Amplifikation Bereich von kcsA und Bereich CGGTTGTCGTCG flussaufwärts, +KpnI, BamHI PrkcsAX GCCACCTCTAGAAGGTGACGGC pUCK1K2, Amplifikation Bereich von kcsA und TGTC flussaufwärts liegenden Abschnitt, +XbaI Materialien und Methoden - 24 PrkcsAS AATGCTGGATCCCGGGACCCGG TGACTCCGCCGGTGACC PrkcsAH GGCACCAAGCTTCCTCCGGCGTC CTAG PrksuFK GAAGCCGGATCCGGGTACCCCG CCGTTCTCGGTG PrksuFX GACGTTTCTAGACGGCGGCACG GTCGTCAC TCGACCGGATCCCGGGACCCGG CGGGTGAGCAGGGAG PrksuFS PrksuFH GCACTGAAGCTTCACCGACTTCG CCAG pUCK1K2, Amplifikation Bereich flussabwärts von kcsA, +SanDI +BamHI pUCK1K2, Amplifikation Bereich flussabwärts von kcsA, +HindIII pUCF1F2, Amplifikation Bereich von ksuF, +KpnI, BamHI pUCF1F2, Amplifikation Bereich von ksuF, +XbaI pUCF1F2, Amplifikation Bereich flussabwärts von ksuF, +SanDI +BamHI pUCF1F2, Amplifikation Bereich flussabwärts von ksuF, +HindIII 2.3. Protein- und DNA-Standards Protein-Standards: 7B (prestained, Sigma Aldrich Chemie, Steinheim) Apparente Moleculargewichte: 172,5 kDa, 112,5 kDa, 86 kDa, 62,5 kDa, 53 kDa, 33,5 kDa und 31,5 kDa 8S (prestained, New England Biolabs GmbH, Frankfurt/M.) Apparente Molekulargewichte: 175 kDa, 83 kDa, 62 kDa, 47,5 kDa, 32,5 kDa, 25 kDa, 16,5 kDa und 6,5 kDa DNA-Standards: Digoxygenin markierter Molekulgewichtsmarker II (Roche Diagnostics GmbH, Mannheim) 23130 Bp, 9416 Bp, 6557 Bp, 4361 Bp, 2322 Bp, 2027 Bp und 564 Bp Digoxygenin markierter Molekulgewichtsmarker III (Roche Diagnostics GmbH, Mannheim) 21226 Bp, 5148 Bp, 4973 Bp, 4268 Bp, 3530 Bp, 2027 Bp 1904 Bp, 1584 Bp, 1375 Bp, 947 Bp, 831 Bp und 564 Bp Materialien und Methoden - 25 - 2.4. Medien R-S Medium (nach Hopwood, D.A., et al.; 1985) -zur Stammhaltung von Streptomyceten K2SO4 MgCl2 x 6 H2O Glucose Casaminoacids Hefeextrakt Tris Serva-Agar (für festes Medium) 0,25 g 10,12 g 10 g 0,10 g 5g 3,03 g 14 g auf 1 l mit seradest H2O auffüllen, den pH-Wert auf 7,6 einstellen und autoklavieren. Anschließend erfolgt Zugabe von: Spurenelementlsg. KH2PO4 (0,5 %) CaCl2 x 2 H2O (5 M) 2 ml 1 ml 0,4 ml R+S Medium (Hopwood, D.A., et al.; 1985) - zur Regeneration von Protoplasten Zusammensetzung wie R-S Medium mit zusätzlich 103 g Sucrose, jedoch Verwendung von Difco-Agar und 5,73 g Tes statt Tris, nach Autoklavieren Zusatz von: Spurenelementlsg. KH2PO4 (0,5 %) CaCl2 x 2 H2O (5 M) L-Prolin 20 % 2 ml 10 ml 4 ml 15 ml Materialien und Methoden - 26 Vollmedium (nach Jasenka Pigac, pers. Mitteilung) - zur Anzucht von Streptomyceten in Flüssigkultur Sucrose Tryptic Soy Broth Hefeextrakt 100 g 20 g 5g auf 990 ml mit seradest H2O auffüllen; nach Autoklavieren, Zugabe von: MgCl2 x 6 H2O (5 M) 10 ml Yeme Medium (ohne Sucrose) - zur Anzucht von Streptomyceten in Flüssigkultur Hefeextrakt Pepton (Oxoid) 10 g 5g mit seradest H2O auf 1 l auffüllen, pH auf 7,5 einstellen und nach Autoklavieren erfolgt Zugabe von: MgCl2 x 6 H2O (2,5 M) Glucose (50 %) 2 ml 20 ml Sojamehl-Mannit Medium (SM Medium) - zur Stammhaltung und zur Sporenbildungsförderung von Streptomyceten Sojamehl Mannit Agar 10 g 10 g 7,5 g mit seradest H2O auf 500 ml auffüllen, in eine 1 l Flasche geben, pH-Wert auf 7,6 einstellen und autoklavieren. Achtung! Kocht in der Mikrowelle sehr schnell über, Flaschen immer nur maximal zur Hälfte füllen. Spitznamen: „Saubermach-Medium“! Materialien und Methoden - 27 - RSM-Medium - zur Stammhaltung und zur Sporenbildungsförderung von Streptomyceten Nach Autoklavieren 2 Anteile (500 ml) Sojamehl-Mannit Medium mit einem Anteil (250 ml) R-S Festmedium mischen. Anschließend Zugabe von: Spurenelementlsg. KH2PO4 (0,5 %) CaCl2 x 2 H2O (5 M) 0,5 ml 0,25 ml 0,1 ml MM40 Medium - zur Anzucht von Streptomyceten. KH2PO4 (NH4)2SO4 Asparagin Agar (für festes Medium) 1g 3,5 g 1,5 g 14 g mit seradest H2O auf 1 l auffüllen, den pH-Wert auf 7,2 einstellen; für Festmedium auf 500 ml auffüllen, Agar (14 g) auf 500 ml auffüllen, jeweils getrennt autoklavieren und anschließend zusammengeben. Nach Autoklavieren und Erkalten, Zusatz von: NaCl (10 %) 1 ml CaCl2 (10 %) 1 ml MgSO4 (5 %) 10 ml Spurenelementlsg. 1 ml Glycerin (80 %) 12,5 ml (Zugabe von 35 µg/l Arginin für S.lividans N1-Stämme) Spurenelementlsg. (10x) - benötigt für R-S, R+S und MM40 Medium ZnCl2 FeCl3 x 6 H2O CuSO4 x 2 H2O MnCl2 x 4 H2O Na2B4O7 x 10 H2O (NH4)6Mo7O24 x 4 H2O 40 mg 200 mg 10 mg 10 mg 10 mg 10 mg auf 100 ml mit seradest H2O auffüllen, sterilfiltrieren. Materialien und Methoden - 28 - LB (Luria-Bertani) Medium (Maniatis, T., et al., 1982) - Für die Stammhaltung, Plasmidisolierung und Proteinüberproduktion (E.coli) Trypton Hefeextrakt NaCl Agar (für festes Medium) 10 g 5g 10 g 14 g auf 1 l mit seradest H2O auffüllen, den pH-Wert auf 7,5 einstellen; anschließend autoklavieren. NZY Medium - zur Propagierung von Phagen in E.coli NZ-Amin Hefeextrakt NaCl MgCl2 x 6 H2O CaCl2 Agar (für feste Medien) 10 g 5g 5g 2,5 g 0,37 g 14 g mit seradest H2O auf 990 ml auffüllen, den pH-Wert auf 7,0 einstellen und autoklavieren; anschließend Zugabe von: Maltose (20 %) 10 ml SOB/SOC Medium (Maniatis, T., et al.; 1982) - zur Transformation von E.coli - SOB Medium Bacto Trypton Hefeextrakt NaCl KCl 20 g 5g 0,584 g 0,186 g auf 1 l mit seradest H2O auffüllen und autoklavieren. Materialien und Methoden - 29 - - SOC Medium zu 98 ml SOB Medium werden nach Autoklavierung hinzugefügt: Mg2+ Stocklsg. (2 M) Glucose (2 M) 1 ml 1 ml Mg2+ Stocklsg. (2 M) - benötigt für SOC Medium MgCl2 x 6 H2O MgSO4 x 7 H2O 20,33 g 24,65 g auf 100 ml seradest H2O auffüllen, anschließend autoklavieren. Protoplastenpuffer (P-Puffer, Hopwood, D.A. and Wright, H.M.; 1978) -Zur Transformation von Streptomyceten Sucrose K2SO4 MgCl2 x 6 H2O 103 g 0,25 g 2,02 g auf 800 ml auffüllen, autoklavieren in 10 Portionen (20 Min. bei 121 °C), für P-Puffer fest vorher Zugabe von 0,4 % Agarose pro Portion. Anschließend je 80 ml Zugabe von: KH2PO4 (0,5 %) 1 ml CaCl2 x 2 H2O (3,68 %) 10 ml TES-Puffer pH 7,2 (5,73 %)10 ml Spurenelementlösung 2 m - 30 - Materialien und Methoden 2.5. Mikrobiologische Methoden 2.5.1. Kulturbedingungen und Stammhaltung von Streptomyceten Die Anzucht von Streptomyceten in Flüssigkultur erfolgte durch Zugabe von Sporen im Erlenmeyerkolben mit grober Schikane in Jasenka-Medium (mit Sucrose). Nach Wachstum ü.N. bei 30°C als Standkultur erfolgte die Inkubation der Kulturen für ca. 6-8 h bei 140 UpM. als Schüttelkulturen. Zur Stammhaltung wurden RSM-Agarplatten mit Sporen oder Mycelien beimpft, bis zur Sporulation bei 30°C inkubiert und anschließend bei 4°C gelagert. Für eine längerfristige Lagerung wurden Mycelien aus Flüssigkulturen nach Abzentrifugation in 50 % Glycerin oder mit Wasser abgeschwemmte Sporen von RSM-Agarplatten im identischen Volumen 100 % Glycerin oder 1,2 Volumen 80 % Glycerin gelöst und bei -20°C aufbewahrt. Bei Stämmen, die Plasmidderivate von pWHM3 enthielten, wurde dem Medium bei Flüssigkultur 12,5 µg/ml und bei Agarplatten 25 µg/ml Thiostrepton zugesetzt, Stämmen, die Derivate von pGM160 mit einer integrierten Ωhyg-Kassette trugen, wurde zusätzlich zu Thiostrepton auch 25 µg/ml Hygromycin zugesetzt. 2.5.2. Kulturbedingungen und Stammhaltung von Escherichia coli E.coli XL1-Blue, DH5α und M15[pREP4] wurden ü.N. bei 37°C mit LB Medium auf Platten oder in Flüssigkultur (120 UpM.) angezogen. E.coli K802 recA- wurde zur Phagenpropagierung in NZY Medium bei 37°C ü.N. auf Platten oder in Flüssigkultur (150 UpM.) kultiviert. Zur Lagerung wurden die Bakterien bis zu einer OD600 von 0,8 angezogen, mit dem identischen Volumen an 100 % Glycerin oder mit 1,2 Volumen 80 % Glycerin versetzt und bei -20°C aufbewahrt. Auf Platten ausgestrichene Stämme konnten für einige Wochen bei 4°C gelagert werden. Zur Überprüfung der fehlenden Rekombinationsfähigkeit von E.coli K802 recA- wurde dieser auf einer NZY-Platte ausgestrichen und mit UV-Licht bis zu 10“ bestrahlt. Anschließend wurde die Platte mit Alufolie umwickelt und bei 37°C ü.N. inkubiert. (Die Zellen wurden bereits nach 4-6“ UVBestrahlung vollständig abgetötet). Materialien und Methoden - 31 - 2.6. Biochemische Methoden 2.6.1. Gewinnung von His-Tag Proteinen 2.6.1.1. Gewinnung von His-Tag Proteinen aus E.coli (denaturierend) Anzucht, Induktion und Aufschluß: 50 ml LB-Medium mit 100 mg/l Ampicillin und 25 mg/l Kanamycin wurden angeimpft und ü.N. inkubiert (37°C, 150 UpM.). 1 l LB-Medium mit den entsprechenden Antibiotika in einem 2 Kolben mit grober Schikane wurden 1/50 von der ü.N. Kultur angeimpft und bei 37°C bis zu einer OD600 von ca. 0,6-0,7 (140-150 UpM.) angezogen, dann erfolgte die Induktion mit 1 mM IPTG. Nach weiteren 3-4 h Inkubation (37°C, 140-150 UpM) wurden die Zellen abzentrifugiert, in 3,5-5 Volumen/g Nassgewicht Ultraschallpuffer (50 mM NaPO4-Puffer, 300 mM NaCl, pH 7,5) aufgenommen und mit Ultraschall aufgeschlossen (Sonifier 250, Branson, ca. 40 Watt, ca. 2’ Beschallungszeit). Reinigung der Inclusionbodies: Inclusionbodies sedimentieren deutlich schneller als Zelltrümmer. Die aufgeschlossenen Zellen wurden bei ca. 1000 UpM. abzentrifugiert, der Überstand und die Zelltrümmerfraktion wurde vorsichtig von dem weißlichen oder leicht gräulichen Inclusionbodie-Niederschlag abgenommen und verworfen. Der Niederschlag wurde erneut in 3,5-5 Volumen/g Nassgewicht aufgenommen. Dieser Waschschritt wurde mehrfach wiederholt. Teilweise erfolgten die Waschschritte mit Zusatz von 0,01 % Triton X100. Solubilisierung der Inclusionbodies und Aufreinigung über Ni-NTA: Der gereinigte Inclusionbodie-Niederschlag wurde in 3,5-5 Volumen „Lysis“-Puffer (50 mM NaPO4, 6 M GuHCl, pH 7,5) aufgenommen, unlösliche Reste wurden anschließend bei 35000 g sedimentiert, der Überstand mit ca. 0,75 ml Ni-NTA versetzt und auf 50 ml mit dem oben erwähnten Puffer aufgefüllt. Nach mehrstündigem Anbinden oder Anbinden ü.N., jeweils bei RT, wurde das Material auf eine entleerte Qiagen Plasmid Midi Säule aufgetragen, mit ca. 30−50 Volumen 50 mM NaPO4-Puffer, 8 M Harnstoff, 25 mM Imidazol pH 7,5 gewaschen und anschließend erfolgte die mehrfache Elution mit 50 mM NaPO4, 8 M Harnstoff, 500 mM Imidazol, pH 7,5. Die eluierten Fraktionen wurden vereinigt, 0,75 oder 0,5 ml Ni-NTA hinzugegeben und mit 8 M harnstoffhaltigen Puffer ohne Imidazol auf ca. 50ml aufgefüllt, so dass eine Imidazol-Endkonzentration von 20-30 mM erreicht wurde. Nach Anbinden über mehrere Stunden und Auftragen auf eine Säule erfolgten erneute Waschschritte und Elution, wie oben beschrieben. 2.6.1.2. „Native“ Gewinnung von His-Tag Proteinen Anzucht, Induktion und Aufschluß von E.coli Überexpressionsstämmen: 50 ml LB-Medium mit 100 mg/l Ampicillin und 25 mg/l Kanamycin wurden angeimpft und ü.N. inkubiert (30°C, 150 UpM.). Je 1 l LB-Medium mit den entsprechenden Antibiotika in 2 l Kolben mit grober Schikane wurden 1/50 von der ü.N. Kultur angeimpft und bei 30°C bis zu einer OD600 von - 32 - Materialien und Methoden ca. 0,3 (140-150 UpM.), dann bis zu einer OD600 von 0,5 bei 25°C inkubiert (140150 UpM.), anschließend bei 16°C im Wasserbad (ca. 120 UpM.). Bei Erreichen einer OD600 von ca. 0,8-1,2 (je nach Versuch) erfolgte die Induktion mit 0,1 mM IPTG. Nach Wachstum ü.N. bei 16°C und Zentrifugation wurde das Nassgewicht der Zellen bestimmt und diese in 3,5-5 Volumen Ultraschallpuffer (50 mM NaPO4-Puffer mit 300 mM NaCl, pH 7,5) aufgenommen (teilweise wurden bei diesem Schritt Protease-Inhibitoren hinzugefügt) und die Zellen mit Hilfe von Ultraschall aufgeschlossen. Zur Trennung der Cytoplasmafraktion von den Zelltrümmern und anderen unlöslichen Produkten, erfolgten zwei Zentrifugationsschritte bei 5000 g und anschließend bei ca. 35000 g. Expression von His-Tag-Proteinen aus S.lividans Stämmen und Aufschließen der Zellen: 20 ml Jasenka-Medium mit Sucrose und 12,5 mg/l Thiostrepton in 100 ml Kolben wurden mit Sporen von Platte oder aus Glycerinkultur „dick“ angeimpft und ü.N. bei 30°C als Standkultur inkubiert. Diese wurde in 250 ml Kolben mit grober Schikane übertragen, auf 50 ml mit dem oben erwähnten Medium mit Thiostrepton aufgefüllt und nach Inkubation von ca. 4-6 h bei 30°C als Schüttelkultur, abzentrifugiert und die Zellen in 1 l MM40 Medium mit 300 mM KCl oder NaCl in einen 2 l Kolben mit grober Schikane übertragen. Nach Inkubation ü.N. als Schüttelkultur (30°C, ca. 150 UpM) wurden die Zellen abzentrifugiert. Diese wurden in 3,5-5 Volumen Ultraschallpuffer (50 mM NaPO4-Puffer mit 300 mM NaCl) pro Gramm Nassgewicht mit 5 mg Lysozym/ml und einer Tablette Proteaseinhibitor (C@mplete Mini, EDTA-free, Roche) aufgenommen und ca. 30’ -1h bei 30°C inkubiert, anschließend durch Ultraschall aufgeschlossen (ca. 40 Watt, ca. 2’). Die Zelltrümmer wurden in zwei Zentrifugationsschritten (ca. 10’ bei 4000 UpM und 15’ bei 35000 g) von der Cytoplasmafraktion getrennt. Zur Aufreinigung von His-Tag KcsA wurde die Membranfraktion durch Ultrazentrifugation (ca. 1 h bei 300000 g) von der Cytoplasmafraktion getrennt und in Ultraschallpuffer (50 mM NaPO4-Puffer mit 300 mM NaCl) und 1 mg/ml Dodecylmaltosid (DDM) aufgenommen. Native Aufreinigung von Proteinen über Ni-NTA: Die Cytoplasmafraktion wurde mit 0,51 ml reinem Ni-NTA versetzt und mit Ultraschallpuffer und Ultraschallpuffer mit 500 mM Imidazol so auf 50 ml aufgefüllt, dass sich eine Endkonzentration von 20 mM ergab. Nach Anbindung der Proteine an Ni-NTA (3-4 h bei RT oder ü.N. bei 4°C, Roller), wurde die Probe auf eine entleerte Säule (Qiagen Plasmid Midi) gegeben, eluiert und mit 25-50 Volumen des Ni-NTA-Volumens mit Ultraschallpuffer mit 20 mM Imidazol gewaschen. Anschließend erfolgte die Elution entweder mit Ultraschallpuffer mit 500 mM Imidazol oder mit Ultraschallpuffer mit 100-500 mM Imidazol in 100 mM Imidazolschritten. Zur erneuten Aufreinigung wurden die eluierten Fraktionen mit dem höchsten Anteil an His-Tag Protein vereinigt und nach Zugabe von 0,5-1 ml Ni-NTA mit Hilfe von Ultraschallpuffer so auf 50 ml verdünnt, dass eine Imidazol-Endkonzentration von 25-30 mM Imidazol erreicht wurde. Nach Anbindung (3-4 h bei RT oder ü.N. bei 4°C, Roller), Auftragung auf eine Säule und Waschung mit Ultraschallpuffer mit 20 mM Imidazol (25-50 Volumen Ultraschallpuffer mit 20 mM Imidazol) erfolgte die Elution mit Ultraschallpuffer mit 500 mM Imidazol. Materialien und Methoden - 33 - 2.6.2. Konzentration von Proteinen und Wechsel des Puffers Die Ankonzentration und Pufferwechsel von Proteinen erfolgte mit Hilfe von Zentrifugationsfiltersystemen. Verwendet wurden, je nach aufzureinigendem Protein, Filtrationssysteme mit einem Ausschlussvolumen von 3000, 10000 und 30000 Dalton (Centricon YM3, YM10 und YM30, Millipore), jeweils nach Vorschrift des Herstellers. 2.6.3. Gelfiltration Fraktionen von His-Tag KsuF, nach Aufreinigung aus S.lividans pOVE33 und E.coli M15[pREP4] pQE314 über Ni-NTA, wurden über Centricon YM30 Zentrifugationskonzentratoren ankonzentriert und auf 50 mM Hepespuffer mit 200 oder 300 mM NaCl (pH 7,5) umgepuffert. Anschließend erfolgte eine Gelfiltration. Verwendet wurden SUPERDEX 75 HR 10/30 und SUPERDEX 200 HR 10/30 Säulen (Pharmacia Biotech), als Laufpuffer (Flussrate 0,4 ml/min) wurde jeweils der oben erwähnte, für die Umpufferung verwendete Hepes-Puffer verwendet. 2.6.4. Massenspektrometischer Nachweis von FAD Über Gelfiltration aufgereinigtes His-Tag KsuF der gelbgefärbten monomeren Fraktionen (aus S.lividans TK21 pOVE33) und käuflich erworbenes FAD wurde jeweils mit 30 % Acetonitril versetzt. Nach Zentrifugation der proteinhaltigen Proben, um ausgefallenes Protein zu sedimentieren (15’, 30000 g), wurden die Proben über HPLC aufgetrennt (Agilent 1100 Series, ThermoQuest 250 x 4,6 mm, 5 µm HyPURITY Elite C18) und anschließend massenspektrometrisch analysiert (esquire HCT, Bruker Daltonics). 2.6.5. Bestimmung von Proteinkonzentrationen Die Proteinkonzentrationsbestimmungen erfolgten mit Hilfe von Lowry- (Dulley, J. R. and Grieve, P. A.; 1975), Bradford- (Bradford, M. M.; 1976) und BCA-Tests von „Novagen“ (Smith, P.K, et al.; 1985; Kessler, R.J. and Fanestil, D.D.; 1986), jeweils nach Vorschrift, mit BSA als Referenz. 2.6.6. Spektrophotometrische Messungen Die spektrophotometrischen Messungen von FAD und His-Tag KsuF erfolgte an einem Spektrophotometer in der Biophysik. Gemessen wurde jeweils das Spektrum von 200/230 nm bis 650/700 nm. Alle Messungen erfolgten in 50 mM Hepespuffer mit 300 mM NaCl (pH 7,5), FAD wurde in einer 0,01 mM Konzentration eingesetzt, His-Tag KsuF wurde nach Aufreinigung aus E.coli M15[pREP4] pQE314 und aus S.lividans pOVE33 über Gelfiltration - 34 - Materialien und Methoden weiter aufgereinigt und die Fraktionen, die dimeres und die monomeres His-Tag KsuF enthielten, wurden unverdünnt eingesetzt. Die weitere Bearbeitung der Daten erfolgte mit Hilfe des Computerprogramms „EXEL“. 2.6.7. Bestimmung des FAD/KsuF-Verhältnisses In Versuchen wurde gezeigt, dass, anders als die beiden Absorptionsmaxima von FAD bei 265 nm und 375 nm, das bei 450 nm nur geringfügig durch die Bindung an das Protein und die durch Protein hervorgerufenen Absorption in der Stärke beeinflusst wurde (um ca. 8,8 % reduziert; Palfey, B.A., et al, 2003). Dieses wurde im Vergleich mit 0,01 mM FAD zur Konzentrationsbestimmung verwendet, der bestimmte Wert wurde mit dem Korrekturfaktor 1,097 multipliziert, um die FAD-Konzentration zu erhalten. Die Proteinkonzentration wurde mit Hilfe des BCA-Tests im Vergleich zu BSA (geeicht, Novagen) bestimmt. Der BCA-Test zeigt eine im Verhältnis zur Reaktion mit Protein sehr schnell abgeschlossene und schwache Farbreaktionen mit FAD, zur Bestimmung der Proteinkonzentration wurde die hierdurch hervorgerufene Absorption jeweils durch Vergleich mit FAD und der spektophotometrisch bei 450 nm bestimmten FAD-Konzentration der Fraktionen berechnet und von der Absorption subtrahiert. Nach Bestimmung der jeweiligen molaren Konzentrationen wurde die FAD durch die Proteinkonzentration geteilt. 2.7. Elektrophoretische Techniken 2.7.1. Agarose-Gelelektrophorese Zur Auftrennung wurden 0,7-1,2 % Agarosegele, für die Auftrennung von sehr kleinen Fragmenten (Fragmentgrößen von ca. 100 Bp und kleiner) wurden „Low-melting“ Agarosegele mit einer Konzentration von bis zu 2,5 % verwendet, als Laufpuffer 1 x TBEPuffer (0.89 M Tris/HCl, 0,89 M Borsäure, 25 mM Na2EDTA, pH 8,0). Die DNA-Proben wurden 1 zu 4 mit Gelladepuffer III (0,25 % Bromphenol Blau, 0,25 % Xylene Cyanol, 30 % Glycerin in H2O) versetzt und bei 60-80 V in ca. 2-3 h oder ü.N. bei 12 V in horizontalen Elektrophoresekammern (Pharmacia, Owl oder Institutseigenbau) aufgetrennt. Anschließend konnte die DNA mit Ethidiumbromid-Lösung (4 µg/ml) oder (seltener) in SYBRE Green Lösung (1/10000 der Stammlösung) in jeweils 1x TBE-Puffer angefärbt und durch UV-Licht bei 302 nm sichtbar gemacht und fotografisch ausgewertet werden. Materialien und Methoden - 35 - 2.7.2. SDS Polyacrylamid-Gelelektrophorese (PAGE) Die SDS-Gelelektrophorese erfolgte nach Laemmli (Laemmli, U.K.; 1970). Verwendet wurden 12,5 % und 15 % Gele. Als Ladepuffer wurde ein 4x SDS-Probenpuffer (200 mM Tris/HCl, pH 6,8; 4 % SDS (Laurylsulfat); 20 % Glycerin), reduzierende Agenzien (Mercapthoethanol und Dithiothreitol(DTT)) wurden bei Bedarf frisch zugesetzt. Um unerwünschte Signale nach immunologischem Nachweis so weit wie möglich zu reduzieren, wurden das Tris in Ultra- und SDS in Elektrophorese- oder in Ultra-Qualität eingesetzt. 2.7.3. Denaturierende Gelelektrophorese in Schägger/v.Yagow Gelen Die Gelelektrophorese erfolgte nach Schägger und von Jagow (Schägger, H. and Jagow, v.G.; 1987). Verwendet wurden 15 % denaturierende Gele mit 8 M Harnstoff. Tris wurde in Ultra-, SDS (Laurylsulfat) in Elektrophorese-Qualität eingesetzt. 2.7.4. Färbung mit Coomassie-Brilliant-Blau Nach der Elektrophorese wurden die SDS und Nativen PAA-Gele mit 0,1 %iger CoomassieBrillant-Blau R 250 Lösung in 10 % Essigsäure und 50 % Methanol gefärbt. Nach 10-30’ erfolgte eine Entfärbung in 10 % Essigsäure und 50 % Methanol. 2.7.5.Silbernitratfärbung Nach der SDS Polyacrylamid-Gelelektrophorese wurden die Gele für 20’ in Lösung I (10 % Trichloressigsäure) fixiert. Nach 2 x 10’ Waschen mit seradest H2O erfolgte eine weitere 10minütige Fixierung in Lösung II (1 g Na2B4O7 x 10 H2O, 2 ml Glutaraldehyd (50 %), 98 ml H2O). Nach anschließendem dreimaligem Waschen mit seradest H2O für jeweils 10’ und Wechseln der Wanne, wurden die Gele für 15’ in einem kurz zuvor angesetztem Gemisch aus Lösung III (0,8 g Silbernitrat, 4 ml seradest H2O) und Lösung IV (70 ml H2O, 1,84 ml NaOH (1 M), 1,96 ml NH4 konzentriert) inkubiert. Nach 7’ Waschen in seradest H2O, erfolgte eine sehr kurze Inkubation in Lösung V (100 ml H2O, 6 mg Citronensäure, 50 µl Formaldehyd), bis die ersten Proteinbanden zu erkennen waren. Um eine zu starke „Nachfärbung“ zu verhindern, wurde die Färbereaktion durch mehrmaliges Waschen in seradest H2O beendet. - 36 - Materialien und Methoden 2.7.6. Proteintransfer auf Membranen („Semidry-Electro-Blotting“) Die Übertragung von Proteinen aus einem SDS-Polyacryamid-Gel auf eine PVDF-Membran (Fluorotrans-Membran, Pall, Pourtsmouth, England) erfolgte mit einer GrafitplattenTransferkammer (CTI, Idstein) durch „Semidry-Electro-Blotting“ nach der von Burnette, W.N. modifizierten Methode (Towbin, H., et al.; 1979; Burnette, W.N.; 1981). Als Transferpuffer wurde 10 mM Caps-Puffer pH 11 mit 10 % Methanol verwendet. Die Transferzeit betrug ca. 1h bei 1 mA/cm2. Zum Transfer von KcsA, His-Tag KcsA und dem His-Tag C-terminalen Ende von KcsA wurde 25 mM Tris/Glycin-Puffer mit 7 % MeOH und 0,07 % SDS als Transferpuffer verwendet, die Transferzeit betrug ca. 3 h bei 1 mA/cm2 Membranfläche. 2.8. Immunologische Nachweismethoden 2.8.1. Immunologischer Nachweis von Proteinen auf Nylonmembranen („Western Blot“) Nach Transfer wurde die Membran von der Gelmatrix getrennt und für 1 h in einer Schale mit 100 ml „Blocking-Puffer“ (1 % Blocking-Reagenz (Boehringer) und 0,5 % BSA oder 5 % Milchpulver) in 1x PBS-Puffer (8,18 g NaCl, 0,2 g KCl, 1,798 g Na2HPO4, 0,245 g KH2PO4, auf 1 l mit seradest H2O auffüllen) inkubiert. Danach erfolgte eine Inkubation der Membran ü.N. bei 4°C oder für 4 h bei RT mit dem primären, in 1 x PBS-Puffer verdünnten Antikörper. Die Membran wurde dreimal für 10-15’ mit 1 x PBS-Puffer gewaschen und anschließend für 2-3 h bei RT mit dem entsprechenden sekundären, in 1 x PBS-Puffer verdünnten Antikörper (1/30000) inkubiert. Nach Entfernung des ungebundenen sekundären Antikörpers durch zweimaliges Waschen mit 1 x PBS-Puffer für je 10’ wurde die Membran kurz in Substratpuffer (25 mM Tris/HCl, pH 8,3) äquilibriert. Die Farbreaktion erfolgte durch Inkubation mit der Entwicklungslösung, bis die Farbsignale erschienen. Entwicklungslösung: Lösung A: 2 mg Naphtol-AS-E-Phosphat in 200 µl DMSO Lösung B: 10 mg Fast Violett B in 10 ml Substratpuffer - Lösung A und B erst kurz vor Gebrauch zusammengeben. Nach Erscheinen der Farbsignale wurde die Membran mehrfach mit H2O gewaschen und getrocknet. Materialien und Methoden - 37 - 2.9. Molekulargenetische Methoden 2.9.1. Aufreinigung von DNA 2.9.1.1. Plasmidisolierung aus E.coli (alkalische Lyse) 3 ml LB Medium mit einem entsprechenden Antibiotikum wurden mit einer E.coliEinzelkolonie angeimpft und ü.N. bei 37°C inkubiert. 2 x 1,5 ml der Kultur wurden für eine Min. bei 14000 UpM. in einem Eppendorfröhrchen abzentrifugiert und der entstandene Niederschlag in 100 µl Lösung 1 (50 mM Glucose, 25 mM Tris/HCl pH 8, 25 mM Na2EDTA) resuspendiert. Nach 2’ bei RT erfolgte eine Zugabe von 200 µl Lösung 2 (1 % SDS, 0,2 M NaOH). Nach einer 5minütigen Inkubation bei RT wurden 150 µl Lösung 3 (3 M Kalium- oder Natriumacetat, 11,5 % Essigsäure, pH 5,5) hinzugegeben und bei 14000 UpM. (15’) zentrifugiert. Zum Überstand wurde ein Volumen Phenol/Chloroform gegeben. Nach kräftigem Mischen und Zentrifugation, wurde die wässrige Phase entnommen, ein Volumen Chloroform zu dieser hinzugegeben, wiederum gut gemischt und nach Zentrifugation die Plasmid-DNA mit Hilfe von 0,7 Volumen Isopropanol aus dieser gefällt. Der nach Zentrifugation und Abnahme des Überstandes zurückbleibende Niederschlag wurde anschließend einmal mit 70 % Ethanol gewaschen und nach Trocknen in 50 µl seradest H2O oder in einmal TE-Puffer (10 mM Tris/HCl, 1 mM EDTA, pH 7,4) aufgenommen. 2.9.1.2. Plasmidaufreinigung aus E.coli („Quick and dirty“) Diese Methode entspricht im Prinzip der oben beschriebenen Plasmidaufreinigung (Alkalische Lyse), jedoch wurden jeweils 420 µl der Lösungen I, II und III eingesetzt, nach Zentrifugation erfolgte kein Phenolisierungsschritt, sondern es wurde 0,7 Volumen Isopropanol dem Überstand hinzugesetzt, der nach Zentrifugation zurückbleibende Niederschlag wurde zweimal mit 70 % EtOH gewaschen, getrocknet und in 50 µl seradest H2O aufgenommen. Die über diese Methode gereinigte DNA zeigte teilweise deutlich bessere Ergebnisse bei enzymatischen Reaktionen, wie z.B. Sequenzierung und Restriktionen als die über die klassische alkalische Lyse aufgereinigte DNA. 2.9.1.3. Plasmidaufreinigung aus E.coli (Ionenaustauscher) Für Aufreinigungen von Plasmiden aus E.coli, für die größere Mengen an DNA und besonders saubere DNA benötigt wurde (z.B. Sequenzierung), wurde das Qiagen Plasmid Midi Kit und das Qiafilter Plasmid Midi Kit (Qiagen, Hilden) nach Vorschrift der Hersteller verwendet, mit folgenden Änderungen: Statt einer 25 ml Kultur in der logarithmischen Phase wurden 30-50 ml einer ü.N.-Kultur verwendet, das Volumen der Puffer P1, P2 und P3 wurde auf 6 ml erhöht, die DNA-Pellets wurde in je 25 µl seradet H2O aufgenommen, da Tris-Puffer die Sequenzierung störten. - 38 - Materialien und Methoden 2.9.1.4. Schnelltest zum Nachweis von Plasmiden in E.coli (SCFSB) Single colony final sample buffer (SCFSB-Puffer): 2,5 % Ficoll 400 (w/v) 1,25 % SDS (w/v) 0,015 % Bromphenolblau (w/v) In 1 x TBE-Puffer (0.89 M Tris/HCl, 0,89 M Borsäure, 25 mM Na2EDTA, pH 8,0) aufnehmen. Mit Hilfe dieser Methode können relativ schnell Anhaltspunkte gewonnen werden, welche E.coli-Kolonien nach Ligation und Transformation Plasmide tragen, bei denen ein DNAFragment korrekt inseriert sein könnte. Hierzu wurden die Zellen mit Hilfe des SCFSBPuffers lysiert und die Plasmide durch Agarose-Gelelektrophorese der Größe nach aufgetrennt. Durch zusätzliches Auftragen von E.coli Zellen mit Plasmiden bekannter Größe als Kontrolle und/oder Plasmiden bekannter Größe in SCFSB-Puffer, wurde eine ungefähre Bestimmung der Plasmidgrößen ermöglicht. Mit einem Zahnstocher wurde eine ungefähr stecknadelkopfgroße Menge der zu testenden E.coli-Zellen von einer LB-Agarplatte mit dem entsprechendem Antibiotika „abgekratzt“ und in 100 µl SCFSB-Puffer aufgenommmen. Nach einer 10minütigen Inkubation bei 37°C erfolgte eine Zentrifugation von 15’ bei 14000 g, der wässerige Überstand wurde anschließend auf ein 0,7 % Agarosegel aufgetragen. Hierbei war zu beachten, dass der 1 x TBE-Pufferstand in der Gelelektrophoresekammer knapp unter der Agarosegeloberfläche gehalten werden musste, da sonst die detergenzhaltige Probe herausgespült würde. Nach 510minütigen Einlaufen ins Gel (60 V), konnte der TBE-Pufferstand auf normale Werte erhöht werden. Die weitere Arbeitsmethode erfolgte wie unter Agarose-Gelelektrophorese beschrieben. 2.9.1.5. Aufreinigung von einzelsträngiger Phagen-DNA (M13) Zur Aufreinigung von einzelsträngiger M13 Phagen-DNA aus E.coli wurde das „QIAprep Spin M13 Kit“ der Firma „Qiagen“, Hilden nach Vorschrift der Hersteller eingesetzt. 2.9.1.6. Isolierung von Plasmiden aus Streptomyceten 5 ml Vollmedium mit einem entsprechenden Antibiotikum wurden mit Mycel angeimpft, ü.N. als Standkultur und weitere 24 h als Schüttelkultur bei 30°C inkubiert. 1,5 ml der Kultur wurden in ein Eppendorfgefäß überführt und das Mycel (ca. 70 mg) abzentrifugiert. Anschließend wurde es in 500 µl Lysozym- und RNAse- haltiger Lösung I (15 mM Glucose, 25 mM Tris/HCl, 10 mM EDTA, pH 8,0; 5 mg/ml Lysozym, 1 mg/ml RNAse) resuspendiert. Nach 30’ bei 37°C, erfolgte die Zugabe von 300 µl alkalischem SDS (2 % SDS in 0,3 M NaOH), nach 5’ auf Eis erfolgte die Zugabe von 270 µl 3 M Kaliumacetat, pH 4,8. Nach weiteren 5’ auf Eis wurde die Mischung zentrifugiert (14000 UpM., 4°C) und der Überstand mit einem ungefähr identischen Volumen an saurem Phenol versetzt und erneut zentrifugiert Materialien und Methoden - 39 - (14000 UpM., 4°C). Der Überstand wurde abgenommen und der Phenolisierungsschritt wurde so oft wiederholt, bis sich keine Interphase mehr bildete, anschließend erfolgte ein Chloroformschritt. Die Plasmid-DNA wurde mit 0,7 Volumen Isopropanol gefällt, mit 70 % EtOH gewaschen und in 50 µl TE-Puffer aufgenommen. 2.9.1.7. Isolierung von chromosomaler DNA aus Streptomyceten Die S.lividans Stämme, aus denen die chromosomale DNA aufgereinigt werden sollte, wurden für ca. 24 h in Jasenka-Medium + Sucrose als Standkultur inkubiert (30°C), anschließend ü.N. als Schüttelkultur (30°C, 150 UpM.). 50-70 µg Zellen wurden in einem 1,5 ml Reaktionsgefäß abzentrifugiert (4000 g), in 700 µl Lösung I (50 mM Glucose, 25 mM Tris/HCl pH 8, 25 mM Na2EDTA) mit 2 mg/ml Lysozym resuspendiert und 30’ bei 37°C protoplastiert. Nach Zugabe von 35 µl 20 % SDS erfolgte ein 5’ Schritt bei RT, anschließend wurde die Suspension mehrfach mit 500 µl Phenol/Chloroform phenolisiert, bis keine Interphase mehr sichtbar war. Nach einem Chloroform-Schritt (500 µl) wurde der Überstand mit 80 µl 3 M NaAc und 600 µl Isopropanol versetzt und das Reaktionsgefäß mehrfach gewendet, bis die chromosomale DNA ausgefallen war. Die DNA wurde vorsichtig mit einer Pipette angesaugt, in 1 ml 70 % EtOH gewaschen, anschließend getrocknet und in 50 µl TEPuffer aufgenommen. 2.9.1.8. Aufreinigung von DNA-Fragmenten aus Agarosegelen Hierzu wurden, nach Anfärbung mit Ethidiumbromid, die über Agarose-Gelelektrophorese aufgetrennten DNA-Fragmente mit Hilfe eines Skalpells ausgeschnitten und weiterverarbeitet. Aufreinigung mittels Glasmilch: Die Elution der DNA-Fragmente aus den Agaroseblöckchen erfolgte mit dem „QIAEX-Kit“ der Firma Qiagen, Hilden, nach Vorschrift des Herstellers. Aufreinigung mittels Säulen: Die Elution der DNA-Fragmente aus den Agaroseblöcken erfolgte mit Hilfe des „QIAquick Gel Extraction Kit“ der Firma Qiagen, Hilden, nach Vorschrift des Herstellers. 2.9.2. Transformation von Plasmiden 2.9.2.1. Transformation von E.coli (CaCl2-Methode) Die E.coli-Stämme JM109 und JM110 konnten mit Hilfe von Elektroporation nicht transformiert werden. Zur Herstellung CaCl2 kompetenter Zellen wurden 50 ml LB-Medium mit 0,5 ml einer ü.N. Kultur angeimpft und bei 37°C (Schüttler, 120 Upm.) inkubiert, bis eine OD600 von 0,6-0,7 erreicht war. Nach 10’ auf Eis wurden die Zellen abzentrifugiert (6000 Upm., 4°C), in 0,5 ml 0,1 M CaCl2 (eiskalt) aufgenommen und 20 % Glycerin (eiskalt) wurden hinzugegeben. Nach >20’ auf Eis wurden je 100 µl Portionen bei -70°C eingefroren. - 40 - Materialien und Methoden Für die Transformation wurde zu einer 100 µl Portion Zellen die zu transformierende DNA gegeben, nach 30’ auf Eis erfolgte ein Hitzeschock (2’ bei 42°C oder 5’ bei 37°C), die Zellen wurden anschließend in 100 µl SOC-Medium aufgenommen und nach 45’ bei 37°C auf den entsprechenden Selektionsmedien ausgestrichen. 2.9.2.2. Transformation von E.coli (Elektroporation) Die Herstellung elektrokompetenter Zellen erfolgte, wie im Electroporator II Manual, Version 3.0 (Invitrogen) beschrieben. Die Elektroporation erfolgte in Elektroporationsküvetten mit einem Spaltmaß von 0,1 cm (Invitrogen), verwendet wurde ein Invitrogen Elektroporator II, nach dem offiziellen Rückruf des Gerätes auf Grund von Sicherheitsmängeln, ein Gene PulserTM und Pulse Controller (BioRad). Je 80 µl der elektrokompetenten Zellen wurden mit der DNA gemischt und in eine eisgekühlte Küvette gegeben. Nach Elektroporation (Einstellungen: 10 W, 100 mA, 1500 V für die Stromquelle, 150 Ω, 50 µF für den Elektroporator (Elektroporator II, Invitrogen), 1200 V, 200 Ω, 25 µF für den Gen Pulser/Pulse Controller (BioRad)) wurden die Zellen in 1 ml SOC-Medium aufgenommen und nach ca. 45’ bei 37°C auf den entsprechenden Seleltionsmedien ausgestrichen. 2.9.2.3. Transformation von Streptomyceten (PEG-Methode) Herstellung von Protoplasten: 10-20 ml Jasenka-Medium mit Sucrose wurden von Platte mit Sporen angeimpft und ü.N. bei 30°C als Standkultur inkubiert, anschließend erfolgte eine Inkubation als Schüttelkultur (30°C, 150 Upm.) für ca. 24 h. Die Kultur wurde abzentrifugiert (10’, 4500 Upm.), zweimal mit je 5 ml 10,3 % Sucroselösung gewaschen und anschließend das Mycel in 3,5 ml P-Puffer/g Mycel mit 7 mg Lysozym/g Mycel resuspendiert und für 3060’ bei 30°C (oder 37°C) inkubiert, bis etwa 90 % des Mycels protoplastiert waren. Nach dreimaligem vorsichtigen Aufziehen der Suspension in einer 5 ml Pipette, zum Lösen der Protoplasten voneinander, wurde die Suspension zentrifugiert (1’, 1000 Upm.), der Überstand, der die Protoplasten enthielt, abgenommen und erneut zentrifugiert (7’, 3000 Upm.). Das Protoplastenpellet wurde in 1 ml P-Puffer resuspendiert und nach Bestimmung der Protoplastenkonzentration (Auszählen in einer Thomakammer) und mit PPuffer auf eine Endkonzentration von 3-6x109 Protoplasten/ml verdünnt. Nach Aliquotierung (100-300 µl jeweils) und 30’ auf Eis wurden die Protoplasten bei -70°C gelagert. Materialien und Methoden - 41 - Transformation: Lösung A: 25 ml 10,3 % Sucrose 0,2 ml Spurenelementlösung 1 ml 2,5 % K2SO4 0,2 ml 5 M CaCl2 0,5 ml 1 M Tris/Maleinsäure Puffer pH 8,0 Transformationspuffer (T-Puffer) modifiziert: 2 Teile Lösung A, 1 Teil P-Puffer und 1 Teil PEG1000 (autoklaviert, vor Gebrauch aufschmelzen und etwas abkühlen lassen) Zu 50 µl der Protoplastensuspension wurden 1-10 µl DNA-Lösung pipettiert, vorsichtig gemischt und nach mehren Minuten auf Eis erfolgte die Zugabe von 200 µl T-Puffer. Nach ca. 20’’ wurde der Ansatz von 10-1 bis 10-2 mit P-Puffer verdünnt und je 100 µl auf R+S Platten mit 2 ml Weichagar (P-Puffer mit 0,4 % Agarose, 40°C) ausplattiert. Nach ca. 24 h Inkubation bei 30°C waren die Protoplasten regeneriert (Ausbildung von Mycel, mikroskopische Kontrolle) und konnten mit 2,5-3 ml Weichagar, der das zur Selektion geeignete Antibiotikum enthielt, überschichtet werden. 2.9.2.4. Integration von Genen in das Chromosom von S.lividans Nach Austreichen und Vereinzeln der Transformanden auf RSM-Medium Platten mit 25 µg/ml Thiostrepton und 25 µg/ml Hygromycin, wurden die Transformanden, entweder über Stempeln oder Ausstreichen als Rasen, auf RSM Medium Platten mit 25 µg/ml Hygromycin übertragen und für mehrere Tage bei 37°C inkubiert, bis Kolonien gewachsen waren. Diese wurden auf RSM Hygromycin- und RSM Hygromycin/Thiostrepton-Platten ausgestrichen und bei 30°C inkubiert. Die Kolonien, die auf RSM Hygromycin-Platten wuchsen, jedoch nicht auf RSM Hygromycin/Thiostrepton Platten, hatten das hygRResistenzgen über eine Doppelrekombination ins Chromosom integriert, die auch auf Hygromycin/Thiostrepton Platten wuchsen, hatten das temperatursensitive pGM160-Derivat Konstrukt nicht komplett verloren. Es wurden nie „Einzel-Rekombinationen“ gefunden, die das vollständige Konstrukt integriert hatten. Der Nachweis der korrekten Integration erfolge nach Aufreinigung und Restriktion der chromosomalen DNA mit Hilfe von Hybridisierung gegen verschiedene Proben. - 42 - Materialien und Methoden 2.9.3. Enzymatische in vitro Reaktionen von DNA 2.9.3.1. Restriktion von DNA Plasmid- oder Phagen-DNA wurde in den passenden Reaktionspuffern verschiedener Hersteller (Gibco, Roche, Invitrogen, Promega, MBI, Stratagene und New England Biolabs) nach Vorschrift der Hersteller gespalten, eingesetzt wurden 5-10 U der entsprechenden Restriktionsenzyme, die Inkubationszeit betrug ca. 4 h. Sollte mit mehr als einem Restriktionsenzym gleichzeitig geschnitten werden, wurde ein Restriktionspuffer verwendet, der die optimale Restriktionbedingungen der Enzyme ermöglichte. Im Falle, dass kein optimaler Reaktionspuffer für mehrfach-Restriktionen vorhanden war, erfolgte eine sequentielle Reaktion, hierfür wurde der Restriktionsansatz nach der ersten Reaktion gefällt und in dem für das zweite Enzym geeigneten Puffer aufgenommen. 2.9.3.2. Ligation Vektor und Fragmente wurden im Verhältnis 1 zu 4 für überhängende Enden, 1 zu 1 bei glatten Enden, gemischt, nach Zugabe von 4 µl 5 x T4-Ligasepuffer (250 mM Tris/HCl pH 7,6, 50 mM MgCl2, 5 mM ATP, 5 mM DTT, 25 % PEG8000) auf ein Volumen von 20 µl mit seradest H2O aufgefüllt. Anschließend wurde 1 U T4 DNA-Ligase hinzugegeben. Die Ligation erfolgte bei cohesiven Enden der Fragmente für 1h bei RT, bei glatten Enden über Nacht bei 16 °C. Desweiteren wurde das T4-Quick-Ligase Kit (NEB-Biolabs) nach Vorschrift des Herstellers verwendet. Da die hohe Ionenstärke von Ligationsansätzen sowie die darin enthaltenen Konzentrationen an PEG auf Transformationen einen störenden Einfluß ausüben, wurden diese wie folgt aufgereinigt: Der Reaktionsansatzes wurde mit seradest H2O auf 50 µl aufgefüllt, 500 µl nButanol zugegeben und der Ansatz kurz gemischt. Nach Zentrifugation bei 14000 UpM. für 10’ wurde der Rückstand getrocknet und in 5-10 µl seradest H2O aufgenommen. Diese Lösung wurde dann direkt für die Transformation eingesetzt. 2.9.3.3. Klenow-Behandlung Um 3’ überstehende Enden von Vektor oder Fragment aufzufüllen oder 5’ überstehende Enden zu hydrolisieren, wurde die DNA nach der Restriktion gefällt und in Klenow-Puffer (10 mM Tris/HCl, pH 7,5, 10 mM MgCl2, 50 mM NaCl) aufgenommen. Nach Zugabe von dNTP's in einer Endkonzentration von je 0,125 mM und 1 U Klenow-Enzym/µg DNA wurde der Ansatz 30’ bei RT inkubiert. Materialien und Methoden - 43 - 2.9.3.4. Polymerasekettenreaktion (PCR) Die Amplifikation von DNA-Fragmenten mittels PCR wurde in einem Trio-Termocycler oder Mono-Termocycler (jeweils Biometra, Göttingen) durchgeführt (nach: Mullis, K.B. and Faloona, F.A.; 1987). Verwendet wurden Taq-Polymerase (Qiagen, Hilden oder Eigenaufreinigung der Arbeitsgruppe) und die „proof-reading“ Polymerasen Vent (New England Biolabs) und Pfu (Promega). Kontrollsequenzierung mit Vektoren ligierter Fragmente ergab bei der Vent-Polymerase eine unerwartet hohe Fehlerrate von einem Fehler auf ungefähr 1500-1600 Bp (sequenzierte Gesamtlänge ca. 10000 Bp), auf Grund dessen wurde in späteren Versuchen nur die Pfu-Polymerase für „proof-reading“-Reaktionen verwendet. Für eine Standardreaktion wurde die DNA für 3’ bei 96°C geschmolzen, anschließend erfolgten 25-30 Zyklen mit Aufschmelzen bei 96-97°C für 40-45’’, Anbindung der Oligos für 40’’ bei von den Oligos abhängiger Temperatur und Verlängerung der Fragmente bei 72°C mit einer Zeit von 1 min/1000 Bp. Für die Pfu-Polymerase wurde eine Verlängerungszeit von 2 min/1000 Bp gewählt. Um die Amplifikation der erwarteten Fragmente zu optimieren und die Bildung nicht erwünschter Fragmente zu verringern/verhindern, wurde in seltenen Fälle eine so genannte „Touch-Down“ PCR durchgeführt, hierzu wurde eine ca. 6°C höhere Temperatur als die theoretische, für die Anbindung verwendet und nach 3 Zyklen auf eine 3°C höhere, nach weiteren 3 Zyklen auf die theoretische Anbindungstemperatur gesenkt. Zur Auflösung von Sekundärstrukturen wurde DMSO in Konzentrationen von 5-10 % und „Q-Solution“ (Qiagen, Hilden) in Konzentrationen bis maximal 20 % eingesetzt. Zur Aufreinigung der Fragmente nach PCR wurde, wenn benötigt, das „QIAquick PCR Purification Kit“ der Firma „Qiagen“, Hilden, verwendet, nach Vorschrift des Herstellers. 2.9.3.5. Herstellung von „Sonden“-DNA Es wurden zwei Methoden zur Herstellung von Proben-DNA verwendet. „random primed“ DNA-Markierungsmethode: Die Methode wurde, wie in der Vorschrift: „DNA Labeling and Detection Kit, Nonradioactive“ (Roche/Boehringer Mannheim) beschrieben, durchgeführt. Linearisierte DNA wurde durch Hitze denaturiert, markierte Proben DNA wurde mit Hilfe des Klenow-Enzyms, Hexanukleotid-Mix (als Oligos) und dNTP-Markierungsgemisch (mit Digoxigenin markierten dUTP) hergestellt. Herstellung von Proben-DNA mittels PCR: Die Proben-DNA wurde mit Hilfe von PCR hergestellt, als Polymerase wurde die Taq-Polymerase verwendet, jedoch wurden statt 1 µl dNTP’s, 3 µl dNTP-Markierungsgemisch (mit DIG-dUTP) eingesetzt. Der Erfolg der Markierung konnte durch Agarose-Gelelektrophorese kontrolliert werden, die markierte Proben-DNA läuft deutlich langsamer als die vergleichbaren unmarkierten Fragmente. - 44 - Materialien und Methoden 2.9.4. DNA-DNA Hybridisierung Transfer: Nach Auftrennung der DNA in Agarosegelen wurde der Teil, der große DNAFragmente enthielt (>2 kBp), für 10’ mit UV-Licht (260 nm) bestrahlt, um Einzelstrangbrüche zu erzeugen. Eine Denaturierung der DNA erfolgte durch leichtes Schwenken in Denaturierungspuffer (0,5 M NaOH, 1,5 M NaCl) für zweimal 20’. Der Transfer der einzelsträngigen DNA auf Nylonmembranen erfolgte mit 20 x SSC (3 M NaCl, 0,3 M Na2Citrat; pH 7,0). Die Membran wurde vor dem Auflegen auf das Gel kurz in 20 x SSC gespült. Das Übertragen der DNA erfolgte ü.N. bei RT durch Kapillarkräfte. Anschließend wurde die Nylonmembran kurz in 2 x SSC geschwenkt, zwischen zwei Whatman-Filtern luftgetrocknet und zur Anbindung der DNA entweder bei 80°C für 1 h gebacken oder für 10’ mit UV-Licht (260 nm) bestrahlt. Hybridisierung und immunologischer Nachweis: Die DNA-DNA Hybridisierung und der immunologischer Nachweis erfolgte wie im Protokoll „DNA Labeling and Detection Kit Nonradioactive“ (Boehringer/Roche) beschrieben, verwendet wurden Dig-dUTP markierte Proben-DNAs und Hybridisierungs- und Waschtemperaturen zwischen 72°C und 68°C. 72°C wurde für über PCR hergestelle Sonden verwendet, nach mehrmaligem Gebrauch wurde die Temperatur sukzessive auf bis zu 68°C gesenkt, für über „random primed“ DNAMartkierungsmethode hergestellt Proben-DNA wurde eine Temperatur von 68°C verwendet. Alkalische Phophatase Antikörper-Konjugat wurde für den immunologischen Nachweis verwendet, für die Farbreaktion NBT und X-Phosphat, die Farbreaktion wurde, anders als beschrieben, mit seradest H2O gestoppt, sobald die Signale erschienen waren. Sollte die Membran wieder verwendet werden, durfte sie nicht getrocknet werden. Entfärbung: Die Entfärbung erfolgte, wie im Protokoll „DNA Labeling and Detection Kit Nonradioactive“ (Boehringer/Roche) beschrieben, verwendet wurde DMF bei 50-60°C. Die Probe wurde, wie in der Methode I beschrieben mit Hilfe von NaOH abgewaschen. Nach Trocknung konnte die Membran wieder verwendet werden. 2.9.5. Sequenzierung Für die erste Methode wurden die Reaktionen mit dem „GATC-BioCycle Sequencing Kit“ der GATC GmbH, Konstanz und der Thermo SequenaseTM der Firma Amersham LIFE SCIENCE, Cleveland Ohio, USA, durchgeführt. Für die Sequenzierung wurden unmarkierte Primer verwendet, die Reaktion über „Cycle Sequencing“ mit Digoxigenin-markierten ddNTP`s durchgeführt, hierbei wurden 10 % DMSO zugesetzt. Die Reaktions-Produkte wurden mit Hilfe des GATC 1500-Systems aufgetrennt und mittels Farbreaktion nachgewiesen (jeweils nach Vorschrift des Herstellers). Die zweite Methode mit dem „Cy5TM-dATP Labelling Mix“ der Firma Pharmacia (Uppsala, Schweden), ist im Prinzip eine Sequenzreaktion mit radioaktiv markierten alpha S35 dATP, nur das statt der radioaktiven eine Markierung mit dem Fluoreszensfarbstoff Cy5TM an den Materialien und Methoden - 45 - dATP`s verwendet wird. Die Sequenzreaktion erfolgte nach Vorschrift des Herstellers, alle weiteren Arbeiten wurden auf einem „ALFexpressTM“ DNA Sequenzierer (Fa. Pharmacia) durch Doris Hörnschemeyer, Arbeitsgruppe Genetik durchgeführt. Bei der dritten Methode erfolgte die Rektion wie bei der ersten Methode über „Cycle Sequencing“ allerdings mit Fluoreszens-markierten ddNTP`s (ABI Big Dye Cycle Sequencing Kit). Die Sequenzreaktion erfolgte nach Vorschrift des Herstellers. Der Sequenzlauf wurde von Ulrike Coja (Spezielle Botanik) auf einem ABI-Sequenzer durchgeführt. Des Weiteren wurden auch zeitweilig als Auftragsarbeit Konstrukte zur Sequenzierung an einen kommerziellen Anbieter (MWG-Biotech AG, Ebersberg) verschickt. 2.10. Fluoreszensmikroskopie Die Fluoreszensmikroskopie erfolgte an einem Zeiss Axiovert 10 Fluoreszensmikroskop, für alle dargestellten Abbildungen wurde ein Achroplan 40x/0,65 Objektiv verwendet. Ein HQFilter-Satz der Firma „AHFanalysentechnik“ (Tübingen) für den Nachweis von FITC fand Verwendung (F41-001, Anregungsfilter HQ480/40, Strahlungsteiler Q505LP, Sperrfilter HQ535/50), die Aufnahmen wurden mit Hilfe einer Phothometrix Sensys 1400 Kamera aufgenommen und mit Hilfe der Softwareprogramme IPLap 5.32 und IPLap 3.00 der Firma „Scananalytics“ verarbeitet. 2.11. Verwendete biologische Softwareprogramme Die Sequenzauswertung erfolgte mit Hilfe des Programms „Chromas“, Version 1.42 von „Conor McCarty“ und „DNA-Manager“ in verschiedenen Versionen, die weitere Bearbeitung wurde in „Clone Manager“ in verschiedenen Versionen durchgeführt, wie auch die Konstruktion von Plasmiden und z.B. der Disruptionsmutanten. Die Bestimmung offener Leseraster erfolgte mit Hilfe des Programms „FramePlot“ in unterschiedlichen Versionen von „Jun Ishikawa“. Zur Suche nach verwandten Proteinen oder DNA-Sequenzen wurden primär verschiedene „Blast“-Versionen der NCBI- und EMBL-EBIServer verwendet, des weiteren „Blitz“ und „Fasta“ (EMBL-EBI-Server). Alignments verschiedener Proteine erfolgten primär mit Hilfe der Programme „ClustalW“ in verschiedenen Versionen und auf unterschiedlichen Servern und „MultAlin“ von „Florence Corpet“, das sich besonders geeignet bei Untersuchungen von KsuF erwies. Die graphische Darstellung von Alignments wurde mit Hilfe des Programms „Boxshad“ durchgeführt, mit anschließender Bearbeitung in CorelDraw. Die Bestimmung von Signalpeptiden erfolgte mit - 46 - Materialien und Methoden einem Programm auf Basis des „Neural Networks-“ und des „Hidden Markov-Modells“ auf dem SignalP 3.0 Server. Die theoretische Berechnung von Kristallstrukturen auf Basis von Homologien erfolgte mit Hilfe des „First Approach Mode“ und „Alignement Interfaces“ des „Swiss-Modell Servers“ des „SIB Biozentrums Basel“. Ergebnisse - 47 - 3. ERGEBNISSE 3.1. Analyse der Sequenzen Vor Beginn meiner Diplomarbeit war der in Abb. 3.1 mit schwarz markierte Bereich sequenziert. Im Rahmen der Diplomarbeit wurde die Sequenz von ksuF vervollständigt, des Weiteren wurde die 3’ Hälfte von KsuL sequenziert (Abb. 3.1., in grün dargestellt). In dieser Arbeit erfolgte die Sequenzierung des restlichen Teils des auf CCM1 lokalisierten Fragmentes (in rot dargestellt), des Weiteren wurde der in früheren Arbeiten sequenzierte Abschnitt (in schwarz dargestellt) zur Korrektur der wenigen vorhandenen Fehler erneut einzelsträngig sequenziert. Zur Vervollständigung von ksuC erfolgten, in Zusammenarbeit im Rahmen der Diplomarbeit von Ralf Steger, die Isolierung und Sequenzierung des entsprechenden Abschnitts (in blau) aus einer Charon 35 S.lividans 66 Phagenbank (CCM3). Mit einer Länge von 13920 Bp unterscheidet sich die S.lividans 66 Sequenz von der entsprechenden aus S.coelicolor A3(2) nur an 66 Stellen, ein Zeichen der nahen Verwandtschaft dieser beiden zur S.violacoeruber-Familie gehörenden Stämme. Es findet sich ein offenes Leseraster, orfP benannt, mit einer Länge von 1472 Bp, das für ein putatives extrazelluläres Protein mit einer Länge von 490 Aminosäuren determiniert, ein N-terminales putatives Signalpeptid ist zu identifizieren. OrfP zeigt Homologien zu extrazellulären Hydrolasen, vor allem zu Triaminopeptidasen. Flussabwärts und in entgegengesetzter Flussrichtung von orfP endet, nur durch 1 Base getrennt, orfT (612 Bp), das für einen 204 Aminosäuren langen Transkriptionsregulator der TetR-Familie determiniert. orfT folgt in entgegengesetzter Richtung, mit einem intergenischen Abstand von 51 Bp, ein 549 Bp langes offenes Leseraster mit dem Namen orf?ace. Dieses determiniert für eine 183 Aminosäuren lange Acetyltransferase der GNAT Acetyltransferase Familie. orfP, orfT und orf?ace wurden im Rahmen dieser Arbeit nicht weiter betrachtet. Alle weiteren Gene und hiervon determinierenden Proteine werden in folgenden Kapiteln besprochen. Die vollständige Sequenz ist in Anhang A dargestellt. - 48 - Ergebnisse Abb. 3.1: Der 13920 Bp lange Abschnitt des S.lividans 66 Chromosoms. Die in den beiden Charon 35 Phagen CCM1 und CCM3 (RS1) und dem Konstrukt pJO1 integrierten Abschnitte sind im unteren Bereich als schwarze Linien dargestellt, die gefundenen Gene und offenen Leseraster als rote Pfeile. Der in früheren Arbeiten sequenzierte Bereich ist als schwarzer, der im Rahmen der Diplomarbeit sequenzierte als grüner, im Rahmen dieser Arbeit sequenzierte als roter und in Zusammenarbeit mit Ralf Steger sequenzierte als blauer Balken dargestellt. Anders als in den potentiellen Promoterbereichen von ksuC/ksuB, ksuL und cmlR, finden sich im intergenischen 325 Bp langen Abschnitt zwischen den Startkodons von ksuF und kcsA eine auffällig hohe Anzahl von Palindromen, „direct-“ und „invertet repeats“ (siehe auch Anhang A und Abb. 3.2). Es können zwei identische palindromische Sequenzen (schließen den DNA-Bereich, der die restlichen repetitiven Sequenzen enthält, ein), acht „inverted repeats“, mit einer Mindestlänge von 7 Nukleotiden und vier verschiedene „direct repeat“Sequenzen, identifiziert werden. Repetitive Strukturen im intergenischen Bereich vor Genen besitzen oft eine Funktion als Operator-Bindestellen von regulatorischen Proteinen. Auf Grund des zumeist verhältnismäßig großen Abstandes zwischen den repetitiven Sequenzen von 50 und mehr Basenpaaren ist die Funktion als Operatorsequenzen für die „inverted repeats“ und die palindromische Sequenzwiederholung unwahrscheinlich. Eine Ausnahme bildet „inverted repeat 11“ mit einem Abstand von 22 Bp. Die „direct repeats“ sind auffällig. Das Hexamer „direct repeat 1“ mit der Consensus-Sequenz CCG/tACA/g findet sich siebenmal, davon fünfmal mit der Sequenz CCGACA. Die „repeats“ sind fünfmal durch 5 Nukleotide getrennt, einmal durch 4. Das Hexamer CGACGG „direct repeat 2“ ist viermal, durch 10, 9 und 10 Nukleotide, das Hexamer AGGAGT „direct repeat 3“ dreimal, durch 6 und 22 Bp und das Nonamer „direct repeat 4“ ACCGCCGCC zweimal, durch 9 Basen voneinander getrennt, auszumachen. Ergebnisse - 49 - Abb. 3.2: Repetitive Sequenzen im intergenischen Abschnitt zwischen ksuF und kcsA. Dargestellt sind die repetitiven „direct-“, „inverted repeat“ und „palindromischen“ Sequenzen. Die Positionen sind jeweils den Sequenzen vorangestellt (siehe auch Anhang A). Die jeweils identischen Basen sind in Grossbuchstaben dargestellt und unterstrichen. Der Abstand zwischen der letzen Base des vorherigen und der ersten des nachfolgenden „repeats“ ist jeweils zwischen den „repeats“ eingetragen. Der Abstand zwischen der jeweiligen ersten Base ist vorangestellt. 3.2. Klonierung der Gene des untersuchten Bereiches und Produktion der kodierenden Genprodukte in E.coli 3.2.1. Strategie zur Gewinnung des C-Terminus von KcsA und der Antikörper Die in früheren Arbeiten hergestellten Antikörper gegen KcsA (H. Splitt) zeigten eine relativ geringe Antigenität gegen KcsA, jedoch relativ starke Kreuzreaktionen mit den Proteinen der Membranfraktionen aus E.coli und S.lividans (nicht dargestellt). Die membrandurchspannenden Teile eines Membranproteins sind häufig wenig antigen. Deshalb sollte als Antigen der aufgereinigte cytoplasmatische C-Terminus von KcsA verwendet werden. Hierzu wurde ein 217 Bp Fragment mit Hilfe von PCR (Vent-Polymerase) amplifiziert und nach SphI/HindIII-Restriktion das entstandene 199 Bp Frg. mit dem ebenso geschnittenen Vektor pQE32 (Qiagen) ligiert. Unter Induktion mit IPTG sollte ein 63 Aminosäuren langes Peptid hergestellt werden, das N-terminal einen 6x His-Tag und dahinter den 48 Aminosäuren langen vorwiegend cytoplasmatischen C-terminalen Teil von KcsA trägt. Anschließend wurde - 50 - Ergebnisse das entstandene Konstrukt (erhielten den Namen pQE317, Abb. 3.3) in E.coli M15[pREP4] transformiert. Abb. 3.3: Produktionsvektor pQE317. Für die Amplifikaton des für den C-terminalen Teil von KcsA kodierenden Bereiches wurden die Oligos pkcsas1 (enthält zusätzlich eine SphI-Schnittstelle) und pkcsah2 (enthält zusätzlich eine HindIII-Schnittstelle) verwendet. Nach Überproduktion (1 mM IPTG, 4 h, 37°C) und Aufreinigung über Ni-NTA unter nativen oder denaturierenden Bedingungen zeigte sich, dass neben dem erwarteten Protein mit einer Größe von ca. 6,3 kDa, auch zwei weitere Proteine bei ca. 9 kDa und ca. 30 kDa aufgereinigt wurden (Abb. 3.4 a). Verschiedene Kolonien zeigten ein ähnliches Produktionsverhalten. Das bei 9 kDa migrierende Protein verhält sich wie das erwartete bei 6,3 kDa. Beide eluieren z.B. unter identischen Bedingungen und sind schwer aus einem Gel auf Membranen transferierbar (siehe Abb. 3.4 b). Weder durch Erhitzen (wie bei KcsA), noch durch reduzierende Agenzien (DTT/ Mercaptoethanol) konnte die 9 kDa Bande aufgetrennt werden (Abb. 3.4 a und b). Sowohl das 6,3 kDa Protein, als auch das bei 9 kDa laufende Protein zeigten Kreuzreaktionen gegen Anti-His-Tag-Antikörper, nicht jedoch das bei 30 kDa (nicht dargestellt). Unter „nativen“ und stringenteren (Erhöhung der Imidazolkonzentration beim Anbinden) Bedingungen konnte die Co-Aufreinigung des Proteins bei ca. 30 kDa verhindert werden, jedoch nicht die des bei 9 kDa migrierenden Proteins (Abb. 3.4 c). Ergebnisse - 51 - Abb. 3.4 a-d): Aufreinigung des C-terminalen Bereiches von KcsA. In allen 4 Abbildungen sind mit Coomassie gefärbte Gele dargestellt, links jeweils die Mobilität der Proteine. a) und b) Aufgetragen wurden jeweils 4 Proben von unter denaturierenden Bedingungen (8 M Harnstoff) aufgereinigtem Protein. In b) ist das Gel nach Transfer der Proteine auf eine Membran gezeigt. Zu sehen sind die nicht transferierten Proteine. Spur 1: Probe nicht aufgekocht, Spur 2: Probe aufgekocht, Spur 3: Probe nicht aufgekocht mit Mercaptoethanol, Spur 4: Probe aufgekocht mit Mercaptoethanol c) Spur 1: aufgetragen wurde unter „nativen“ und stringenten Bedingungen aufgereinigtes Protein und in d), Spur 1: nach Aufreinigung des 6,3 kDa Peptid über ein Biorad Modell 422 Electro-Eluter-System. 8S: Proteinstandard Die Cytoplasmafraktion von E.coli M15[pREP4] pQE317 (4 Liter Kultur) wurde nach Überproduktion (Induktion mit 1 mM IPTG, ca. 4 h bei 37°C) unter „nativen“ Bedingungen zweimal über Ni-NTA aufgereinigt (siehe Material und Methoden). Nach Auftrennung über ein denaturierendes Gel (Schägger-von Yagow) wurde die bei ca. 6,3 kDa migrierende Proteinbande ausgeschnitten, über ein Electro-Eluter-System eluiert (Abb. 3.4 d) und ca. 100 µg für die Immunisierung in Meerschweinchen eingesetzt. Um die Menge der Antikörper zu erhöhen, erfolgte, nach dem Test der zweiten gelieferten Antikörper-Charge („grand bleeding“), eine weitere Immunisierungsrunde mit 100 µg Peptid. Spätere Analysen zeigten, dass die nach dieser weiteren Immunisierung ausgelieferten Antikörperchargen eine deutlich geringere Kreuzreaktion gegen KcsA besaßen, als die der zweiten Charge (Ergebnisse nicht dargestellt). Sowohl gegen KcsA (Abb. 3.5 a), als auch - 52 - Ergebnisse gegen den C-terminalen Teil (Abb. 3.5 c) werden gute Kreuzreaktionen gefunden. Gegen E.coli M15[pREP4] pQE317-Zellextrakt finden sich keine unspezifischen Signale (Abb. 3.5. c), gegen S.lividans NI-Zellextrakt erscheinen einige unspezifische Kreuzreaktionen (Abb. 3.5 b). Abb. 3.5 a-c): Test der gegen den C-terminalen Teil von KcsA hergestellten Antikörper. Getestet wurden jeweils drei verschiedene Antikörperkonzentrationen. Aufgetragen wurden: Membran a) Spuren 1-3: His-Tag KcsA aufgereinigt, nicht aufgekocht; Spuren 5-7: His-Tag KcsA aufgereingt, aufgekocht (nicht vollständig monomerisiert). Membran b) Spuren 1-3: S.lividans NI Membranfraktion. c) Spuren 1, 3 und 5: Aufgereinigtes His-Tag C-term KcsA; Spuren 2, 4 und 6: Zellextrakt von E.coli M15[pRep4] pQE317. 8S: Proteinstandard. Die Laufhöhe der Tetramerform von His-Tag KcsA ist in a) durch ein X markiert, die der Monomer-Form mit einem Punkt. Um die Erkennbarkeit des 8S-Proteinstandards zu verbessern, wurde der Kontrast des entsprechenden Abschnittes in b) verstärkt. Wie in zahlreichen Versuchen gezeigt wurde, kann KcsA unter Standard-Bedingungen („Semi-dry-Transfer“, Puffer: 10 mM Caps/NaOH, 10 % MeOH, pH 11, ca. 1 h bei 1 mAmp/cm2) nur sehr schlecht auf Membranen übertragen werden, wobei das His-Tag Protein schlechter übertragen wird als das native, die Tetramer-Formen schlechter als die Monomere (siehe Abb.3.5 a). Auch das His-Tag C-terminale Ende von KcsA, das bei ca. 6,3 kDa migriert und die Bande bei 9 kDa, können unter diesen Bedingungen nur sehr schlecht transferiert werden (Abb. 3.4 b). Vermutlich verantwortlich sind die ungewöhnlich hohen pK-Werte der Proteine (theoretische pI für KcsA: 10,3; für His-Tag KcsA: 10,76; für His-Tag C-term KcsA: 11) und die Eigenschaft, dass KcsA ohne Detergenz offensichtlich aggregiert und nicht transferierbare Komplexe bildet. Dies hat zur Folge, dass geringere Mengen an KcsA-Protein besser zu transferieren sind als größere. Höhere Konzentrationen an Protein führen dazu, dass bei Transfer nur die Randbereiche der Monomer und TetramerBanden auf die Membran übertragen wird; der Zentralbereich ist maskiert, hier ist kein Transfer zu erkennen (Abb. 3.5. a). Ergebnisse - 53 - Zur Optimierung des Transfers wurden verschiedene Bedingungen getestet. Tris/Glycin/Methanol-Puffer zeigte eine leichte Verbesserung im Transfer gegenüber dem CAPS/Methanol Standardpuffer. Bei Gegenwart der Detergenzien Dodecylmaltosid (DDM, 5 mM) und Triton X100 (0,4 %) wurde die Bindung von Proteinen an die Membran verhindert und die Elution von KcsA aus dem Gel nicht verbessert. Steigende SDSKonzentrationen im Transferpuffer verbessern den Transfer aus dem Gel in Tris/GlycinPuffer. Die Bindung an die Membran wurde bei höheren Konzentrationen reduziert, eine Konzentration von 0,07 % SDS erwies sich als optimal. Methanol wird für die Aktivierung der Membran benötigt, Konzentrationen ab ca. 10 % führten zur Reduzierung des Transfers aus dem Gel. Bei 20 % war der Transfer sehr stark eingeschränkt. Bei einer Konzentration von 7 % konnte keine Einschränkung des Transfers beobachtet werden. Die Methode des „Semi-Dry“-Transfers erwies sich gegenüber der des „Nass“-Transfers bei allen getesteten Bedingungen als effektiver. Die optimalen Bedingungen sind: „Semi-Dry“-Transfer, Transferpuffer: 25 mM Tris/GlycinPuffer pH 7,5 mit 7 % MeOH und 0,07 % SDS, 1 mAmp/cm2. Durch eine Verlängerung der Transferzeit auf ≥3 h gelang es, sowohl die Monomere, als auch die Tetramer-Form fast vollständig zu transferieren. 3.2.2. Produktion von KsuB und von gegen KsuB gerichteten Antikörpern Für die Synthese von KsuB in E.coli wurden das Gen jeweils in die Überproduktionsvektoren pQE32 und pQE70 integriert. Nach Induktion mit IPTG sollte die Produktion einer Nterminalen His-Tag-Form der Proteine (pQE32-Derivat) und einer Form in Originallänge ohne zusätzlichen His-Tag erfolgen (pQE70-Derivat). Das Gen ksuB wurde vom Plasmid pOBH23 amplifiziert (Vent-Polymerase), als HindIII/SphI Fragment mit den ebenso geschnittenen Vektor pUC18 ligiert. Nach Kontrollsequenzierung wurde aus diesem pORFZ benannten Konstrukt wieder über HindIII/SphI-Restriktion das Fragment ausgeschnitten, mit den ebenso geschnittenen Vektoren pQE32 und pQE70 ligiert und in E.coli M15[pREP4] transformiert. Die Konstrukte erhielten den Namen pQE309 (pQE32-Derivat, Abb. 3.6) und pQE709 (pQE70-Derivat, Abb. 3.6). Nach Induktion durch IPTG sollte der pQE309 Transformant ein 323 Aminosäuren langes Protein synthetisieren mit einem N-terminalen His-Tag, der pQE709 Transformant das 310 Aminosäuren lange dem aus dem Wildtypstamm entsprechende Protein. - 54 - Ergebnisse Abb. 3.6: Konstrukte pQE309 und pQE709. Das in die Vektoren pQE32 und pQE70 integrierte Fragment ist in gelb dargestellt. Für die Amplifikation des Fragmentes wurden die Oligos orfZstart und orfZende verwendet, das HindIII/SphI- Fragment besitzt eine Größe von 929 Bp. Bei der Produktion von KsuB und His-Tag KsuB aus den Stämmen E.coli M15[pREP4] pQE709 und pQE309, zeigte sich, dass bei Induktion mit 1 mM IPTG und Inkubation bei 37°C die gesuchten Proteine mehr oder weniger vollständig in Form von „Inclusion-Bodies“ vorlagen. Durch Senkung der Inkubationstemperatur und der IPTG-Konzentration verbesserte sich die Löslichkeit, bei Inkubation bei 16°C, Induktion mit 0,1 mM IPTG und weitere Inkubation bei 16°C, konnte erreicht werden, dass die Proteine vollständig löslich vorlagen. Zur Antikörperherstellung wurden die „Inclusion-Bodies“ aus E.coli M15[pREP4] pQE309 mit Triton X100 gewaschen und unter denaturierenden Bedingungen zweimal über Ni-NTA aufgereinigt (siehe Abb. 3.8 a). Nach Konzentration wurde das Protein über SDS-Page aufgetrennt, über das „Biorad“ Modell 422 Electro-Eluter-System aufgereinigt und zur Herstellung von Antikörpern in Meerschweinchen versendet (siehe Abb. 3.8 b). 3.2.3. Herstellung von Konstrukten für die Synthese und Reinigung von KsuL Vom Konstrukt pOBH23 wurde ksuL, sowohl mit, als auch ohne den für die Signalsequenz kodierenden Bereich, amplifiziert (Vent-Polymerase) und nach HindIII/SphI-Restriktion jeweils mit dem ebenso geschnittenen Vektor pUC18 ligiert; die Konstrukte erhielten die Namen pORFX1 (Originallänge, mit dem für die Signalsequenz kodierenden Bereich) und pORFX2 (ohne den für die Signalsequenz kodierenden Bereich). Nach Verifizierung des in pORFX1 und in pORFX2 integrierten Bereiches durch Sequenzierung, wurde das Insert durch eine HindIII/SphI Restriktion ausgeschnitten, mit den ebenso geschnittenen Vektoren pQE32 und pQE70 ligiert und in E.coli M15[pREP4] transformiert. Die Konstrukte erhielten die Namen pQE315 (mit Signalsequenz und His-Tag, 598 Aminosäuren), pQE715 (Mit Signalsequenz, 585 Aminosäuren), pQE316 (Ohne Signalsequenz, mit His-Tag, 576 Ergebnisse - 55 - Aminosäuren) und pQE716 (Ohne Signalsequenz, 563 Aminosäuren) und sind in Abb. 3.7 dargestellt. Abb. 3.7: Konstrukte pQE315, pQE715, pQE316 und pQE716. Die in die Vektoren pQE32 und pQE70 integrierten Fragmente sind in gelb dargestellt. Für die Amplifikation von ksuL mit Signalsequenz wurden die Oligos orfX1Start und orfXende verwendet (pQE315 und pQE715), für ksuL ohne Signalsequenz orfX2start und orfXende (pQE316 und pQE716). Bei der Synthese von KsuL in den verschiedenen Formen (KsuL mit und ohne Signalsequenz, jeweils mit oder ohne His-Tag) zeigte sich, das nur bei den Stämmen, die KsuL mit His-Tag synthetisieren sollten, eine Produktion nachgewiesen werden konnte (E.coli M15[pREP4] pQE315 und pQE316). Eine Kontrollsequenzierung der Konstrukte pQE715 und pQE716 zeigte weder im integrierten Bereich, noch im Promoter/Operator-Abschnitt des Vektors Fehler, es konnte nicht geklärt werden, warum keine Synthese nachweisbar war. Bei Inkubation bei 37°C und Induktion mit 1 mM IPTG wurden His-Tag-KsuL mit und ohne Signalsequenz wie bei KsuB vollständig in Form von „Inclusionbodies“ gefunden. Anders als bei KsuB konnte jedoch die Löslichkeit der Proteine, z.B. durch Inkubation bei 16°C und Induktion mit geringeren Konzentrationen an IPTG, nicht erhöht werden. Für die Immunisierung wurde His-Tag KsuL mit Signalsequenz (E.coli M15[pREP4] pQE315) aus „Inklusionbodies“, wie schon für KsuB beschrieben, aufgereinigt und zur Herstellung von Antikörpern versendet; als Tierart wurde Ratte gewählt (siehe Abb. 3.8 c und d). - 56 - Ergebnisse Abb. 3.8 a bis d): Aufreinigung von His-Tag KsuB und His-Tag KsuL aus E.coli und Test der gegen KsuB und KsuL hergestellten Antikörper. Dargestellt sind in a) und c) mit Commassie gefärbte Gele. Aufgetragen wurden, nach Aufreinigung über Ni-NTA, in a) vier verschiedene His-Tag KsuB- und in c) vier verschieden HisTag KsuL- (mit Signalsequenz) Elutionen. Jeweils Spur 1: Elution mit 200 mM NaCl, Spur 2: 300 mM NaCl, Spur 3: 400 mM NaCl und Spur 4: 500 mM NaCl. In b) und d) ist der Nachweis der Antikörperaktivität der Antikörper gegen KsuB in b) und gegen KsuL in d) gezeigt. Verwendet wurden 1/10000 Verdünnungen der jeweiligen Antikörper. Aufgetragen wurden: Aus E.coli aufgereinigtes His-Tag KsuB- in b), Spur 1 und His-Tag KsuL-Protein mit Signalsequenz in c), Spur 1. 7B und 8S: Proteinstandards. Zur Verbesserung der Erkennbarkeit wurde jeweils der den 8S-Standard tragende Abschnitt bildlich bearbeitet. 3.2.4. Gewinnung von KsuC und von gegen KsuC gerichteten Antikörpern Die Herstellung der Konstrukte, die Überproduktion und Aufreinigung der Proteine und die Aktivitätskontrolle der erhaltenen Antikörper erfolgte unter direkter Betreuung im Rahmen der Diplomarbeit von Ralf Steger. Vom Konstrukt pBRS1 wurde ksuC als 0,9 kBp Fragment amplifiziert, mit einer eingefügten SphI- und HindIII-Schnittstelle im Start- bzw hinter dem Stopcodon. Nach entsprechender Restriktion erfolgte die Ligation mit dem Vektor pWHM3be, das Konstrukt wurde pORS1 benannt. Nach Verifizierung der Sequenz wurde das Insert nach SphI/HindIII-Restriktion mit den ebenso geschnittenen Vektoren pQE32 und pQE70 ligiert und in E.coli M15[pREP4] transformiert. Die Konstrukte erhielten die Namen pQE321 und pQE721 (Abb. 3.9). Ergebnisse - 57 - Abb. 3.9: KsuC-Überproduktionskonstrukte pQE321 und pQE721 Das in die beiden Vektoren pQE32 und pQE70 integrierte ca. 900 Bp lange ksuC- tragende Fragment ist in Gelb dargestellt. Von pQE321 sollte unter Induktion ein 298 Aminosäuren langes His-Tag KsuC synthetisiert werden, von pQE721 ein 285 Aminosäuren langes Protein, das in der Sequenz und Länge dem Wildtyp KsuC entspricht. Nach Induktion mit IPTG erfolgte von E.coli M15[pREP4] pQE321 die Produktion eines HisTag KsuC-Proteins, von pQE721 eines KsuC in Originallänge (nicht dargestellt). Nach Synthese fanden sich His-Tag KsuC und KsuC ohne His-Tag anscheinend vollständig in „Inklusionbodies“ wieder; ähnlich wie für KsuL gelang es nicht, die Solubilisierung durch Reduzierung der IPTG-Induktionsmenge und Senkung der Kultivierungstemperatur zu erreichen. Die Aufreinigung von His-Tag KsuC für die Immunisierung erfolgte ähnlich, wie für die Aufreinigung von His-Tag KsuB und His-Tag KsuL beschrieben. Nach Waschen der Inklusionbodies erfolgte die Aufreinigung unter denaturierenden Bedingungen über Ni-NTAAffinitätschromatographie (nicht dargestellt), anschließend über ein „Mini-Prep-CellSystem“. Die Immunisierung wurde in Meerschweinchen und in Huhn durchgeführt. Während aus Meerschweinchen spezifische Antikörper erhalten werden konnten, waren die Antikörper aus Huhn auf Grund einer zu großen Unspezifität unbrauchbar (nicht dargestellt). - 58 - Ergebnisse 3.3. Physiologische Studien von S.lividans und ausgewählten Mutanten 3.3.1. Immunologischer Nachweis der Proteine in S.lividans WT S.lividans 66 WT wurde in Minimalmedium ohne zusätzlich Salz, 100 mM, 200 mM, 300 mM, 400 mM, 500 mM und 700 mM NaCl angezogen und die Produktion von KsuB, KsuC und KsuF immunologisch untersucht (Abb. 3.10 b bis d). Für KsuB und KsuC zeigt sich jeweils eine sehr hohe Synthese der Proteine bei NaCl-Konzentrationen über 300 mM NaCl, wobei die Produktion bei 700 mM NaCl vermutlich auf Grund geringer Wachstumsraten reduziert ist. Bei der Synthese von KsuB ist eine leichte Steigerung bei Erhöhung der NaCl-Konzentration auf 200 mM zu erkennen und eine deutlich stärkere ab 300 mM NaCl. Für KsuC findet sich erst ein Nachweis bei 200 mM NaCl und eine deutliche Steigerung der Synthese bei 300 mM NaCl. Eine Produktion von KsuF kann im Wildtyp nicht nachgewiesen werden (Abb. 3.10 d). Sie findet sich nur in der Kontrolle S.lividans TK21 pJO1 (Abb. 3.10 d, Spuren 8 und 9, siehe auch nächstes Kapitel). Vergleichbare Ergebnisse finden sich bei Anzucht in Minimalmedium mit identischen KCl-Konzentrationen statt NaCl (nicht dargestellt). Ergebnisse - 59 - Abb. 3.10 a-d): Nachweis der Synthese von KsuB, KsuC und KsuF in S.lividans 66 WT unter verschiedenen Salzbedingungen. Dargestellt ist in a) ein Coomassie gefärbtes Gel. In den weiteren Abbildungen ist der Nachweis der Synthese in verschiedenen Cytoplasmafraktionen nach Transfer auf Membranen mit Antikörpern gegen KsuB in b), gegen KsuC in c) und gegen KsuF in d), dargestellt. Aufgetragen wurden auf Spur 1: S.lividans 66 WT nach Wachstum in MM40-Medium ohne Salz, auf Spur 2: mit 100 mM NaCl, auf Spur 3: mit 200 mM NaCl, auf Spur 4: mit 300 mM NaCl, auf Spur 5: mit 400 mM NaCl, auf Spur 6: mit 500 mM NaCl und auf Spur 7: mit 700 mM NaCl. 7B Proteinstandard. Auf Spur 8 und 9 sind Cytoplasmafraktionen von S.lividans TK21 pJO1 nach Wachstum in MM40-Medium ohne Salz auf Spur 8 und 500 mM KCl auf Spur 9, dargestellt. Aus einer Jasenka Medium mit Sucrose ü.N. Standkultur und nach Schütteln für ca. 5 h, wurden die Zellen in die unterschiedlichen MM40 Kulturen mit unterschiedlichen Salzkonzentrationen überführt und für eine weitere Nacht als Schüttelkultur inkubiert. - 60 - Ergebnisse 3.3.2. Synthese der Proteine in S.lividans TK21 pJO1 Das Konstrukt pJO1 ist ein pWHM3be Derivat, das ein 2,75 kBp Insert mit dem 5’ Teil von orfI, vollständig kcsA und ksuF und dem 3’ Ende von ksuL trägt. Die Synthese des Plasmidund Chromosomal- determinierten KsuF und KcsA und des nur Chromosmal- determinierten KsuB und KsuC wurde in der S.lividans TK21 pJO1 Transformante bei Wachstum in Minimalmedium mit verschiedenen Salzkonzentrationen zwischen 0 mM und 700 mM KCl oder 300 mM Sucrose untersucht (Abb. 3.11 a-d). Vergleichbare Ergebnisse finden sich bei Synthesen in Minimalmedium mit NaCl (nicht dargestellt). Für KsuB, KsuC und KsuF ist eine von der KCl-Konzentration abhängige Produktion nachweisbar. Die höchste wird jeweils bei Wachstum bei KCl Konzentrationen zwischen 200 und 500 mM KCl erreicht. Sie reduziert sich jeweils bei 700 mM KCl; KCl-Konzentrationen von mehr als 500-600 mM KCl reduzieren die Wachstumsrate der Zellen, bei 700 mM KCl ist das Wachstum deutlich reduziert. Für KsuB zeigt sich bei steigenden Konzentrationen bis 300 mM KCl eine deutliche Steigerung der Synthese (Abb. 3.11 b). Für KsuC ist in diesem Fall bei Wachstum in Minimalmedium ohne zusätzlich Salz oder mit 100 mM Salz nur eine gleich bleibend sehr geringe Produktion nachweisbar (in Abb. 3.11 c, Spuren 1 und 2). KsuF wird bei Plasmid determinierter Synthese in so großen Mengen produziert, dass die Produktion auf einem mit Coomassie gefärbten Gel deutlich zu erkennen ist (Abb. 3.11 a, Laufhöhe mit Pfeilen markiert). Auch hier steigt die Synthese von KsuF mit Erhöhung der KCl-Konzentration im Medium an, was aufgrund der sehr hohen Syntheserate am deutlichsten auf dem mit Coomassie gefärbten Gel zu erkennen ist. KsuF findet sich vorwiegend in drei Formen, jeweils mit gering unterschiedlicher Migration. Bei Wachstum in 200-700 mM KCl sind primär zwei Signale mit Laufhöhen von ca. 40 und 36 kDa nachzuweisen (siehe Abb. 3.11 a und d, Spuren 3-7). Bei Wachstum, in Medium ohne Zusatz von Salz, in 100 mM KCl oder in 300 mM Sucrose, ist vor allem ein Signal mit einer Migration von 38 kDa zu finden. Die bei 40 und 36 kDa migrierenden Formen des KsuF sind nur bei Synthese in Minimalmedien mit hohen Salzkonzentrationen nachweisbar. Jedoch finden sich auch Versuche, bei denen nur die bei 38 kDa migrierende Form nachweisbar ist (nicht dargestellt). Die Zugabe von 300 mM Sucrose hat keinen Effekt auf die Synthese von KsuB, KsuC und KsuF (Abb. 3.11 a-d, Spur 8). Sie entspricht jeweils der gefundenen Produktionsmenge in Minimalmedium ohne Zusatz (Abb. 3.11 a-d, Spur 1). KcsA konnte in der Membranfraktion nicht nachgewiesen werden (nicht dargestellt). Ergebnisse - 61 - Abb. 3.11 a-d): Nachweis der Synthese von KsuB, KsuC und KsuF in S.lividans TK21 pJO1 unter verschiedenen Salzbedingungen und Sucrose. Dargestellt ist in a) ein mit Coomassie gefärbtes Gel. In den Abbildungen b-d) wurden verschiedenen Cytoplasmafraktionen auf Membranen transferiert und mit Antikörpern gegen KsuB in b), gegen KsuC in c) und gegen KsuF in d), untersucht. Aufgetragen wurden jeweils Cytoplasmafraktionen von S.lividans TK21 pJO1 nach Wachstum in MM40 Medium mit verschiedenen Salzkonzentrationen und Sucrose. Spur 1: nach Wachstum in MM40-Medium ohne Salz. Auf Spur 2: mit 100 mM KCl. Auf Spur 3: mit 200 mM KCl. Auf Spur 4: mit 300 mM KCl. Auf Spur 5: mit 400 mM KCl. Auf Spur 6: mit 500 mM KCl. Auf Spur 7: mit 700 mM KCl und auf Spur 8: mit 10,3 % Sucrose (300 mM Sucrose). 8S Proteinmarker. Das mit drei leicht unterschiedlichen Migrationen laufende KsuF ist in a) und d) mit drei Pfeilen markiert. Die Anzucht erfolgte jeweils, wie in der Legende zu Abb. 3.10 beschrieben. - 62 - Ergebnisse 3.3.3. Analyse von S.lividans NI Transformanten 3.3.3.1 Herstellung der Konstrukte S.lividans N1 ist ein Stamm, in dem, neben dem um kcsA liegenden Bereich, nach Puls- Feld Gelelektrophorese- Ergebnissen, mindestens 300 kBp des Chromosoms deletiert sind (pers. Mitteilung von H. Schrempf). Neuere Erkenntnisse deuten auf großräumige Deletionen beider äußerer Armbereiche des Chromosoms hin. In diesen Stamm sollte ein Großteil des um kcsA liegenden sequenzierten Bereiches auf Plasmiden als Subfragmente und als Gesamtfragment transformiert werden, um die Produktion verschiedener, auf den jeweiligen Abschnitten liegenden Genen, in S.lividans NI zu untersuchen. Der für die Propagierung in S.lividans NI vorgesehene Vektor pWHM3be konnte, aufgrund unpassender Schnittstellen, nicht direkt für die verschiedenen Konstruktionen verwendet werden. Hierfür wurde das den p15A ori tragende „low-copy“-Plasmid pACYC184 verwendet, das dafür bekannt ist, auch nach Integration größerer Fragmente stabil in E.coli propagiert werden zu können. Das Tetracyclin-Resistenzgen wurde durch einen neu konstruierten Polylinker ersetzt, in den die unterschiedlichen Fragmente integriert werden konnten. Die verschiedenen „Inserts“ wurden anschließend mit dem E.coli/StreptomycesTransfer-Vektor pWHM3be ligiert und in S.lividans NI transformiert. Zur Konstruktion des benötigten Polylinkers wurden 2 Oligos mit einer Länge von jeweils 55 Bp verwendet. Nach 5’ Phosphorylierung und Verschmelzung der beiden Oligos, wurde ein doppelsträngiges Fragment erhalten, mit den überhängenden Enden einer XbaI und einer AvaI-Schnittstelle an den Flanken und den internen Schnittstellen PstI, NotI, SgfI, SphI, Mph1103I, HindIII und MluI. Dieses Fragment wurde mit dem AvaI/XbaI geschnittenen Vektor pACYC184 ligiert und anschließend in E.coli transformiert. Erhalten wurde der Vektor pACYC001 (siehe Abb. 3.12). Ergebnisse - 63 - Abb. 3.12: Vektor pACYC001 Im Vektor pACYC184 wurde das Tetracyklin-Resistenzgen durch einen neu konstruierten Polylinker ersetzt, hergestellt aus den beiden Oligos Pr184I und Pr184II. Der eingesetzte Bereich ist in gelb dargestellt. In den Vektor pACYC001 sind nacheinander verschiedene Fragmente integriert worden, so dass 9 verschiede Konstrukte erhalten wurden, die unterschiedliche Bereiche des um kcsA liegenden Bereiches enthalten (siehe Abb. 3.13 und „Klonierungsstrategie“ im Anhang C). Die Namen der Konstrukte setzen sich jeweils aus dem Namen pACYC und einer fortlaufenden Nummer von 002 bis 010 zusammen. Aus dem Vektor pACYC001 und den 9 Konstrukten wurde jeweils der Linker (pACYC001), bzw. das Insert mit Hilfe einer PstI/XbaI-Restriktion ausgeschnitten und mit dem ebenso geschnittenen E.coli/StreptomycesShuttle-Vektor pWHM3be ligiert und in E.coli transformiert. Die Konstrukte erhielten jeweils die Namen pOV001 bis pOV010. Nach Aufreinigung erfolgte die Transformation in S.lividans N1. Versuche, die größeren pOV-Konstrukte (pOV003, pOV004, pOV005, pOV006, pOV007 und pOV010) in S.lividans 66 Wildtyp oder TK21 Wildtyp zu transformieren, scheiterten, da die inserierten Fragmente innerhalb kurzer Zeit offensichtlich mit den homologen Bereichen des Chromosoms rekombinierten. Es waren nur noch Konstrukte mit ungefähr der Größe des „Leervektors“ pOV001 nachweisbar. - 64 - Ergebnisse Abb. 3.13: Konstrukte pACYC/pOV002 bis pACYC/pOV010. Dargestellt ist im oberen Teil der vollständige bisher sequenzierte um kcsA gelegene Bereich, mit einer Länge von ca. 14 kBp, mit den als Pfeile dargestellten Genen und den für die Konstruktionen verwendeten Restiktionsschnittstellen. Im unteren Teil ist die Lage, der in die pACYC002 bis pACYC010 und pOV002 bis pOV010 integrierten Fragmente, durch Striche dargestellt. Senkrechte Striche ( | ) an den Flanken zeigen in der Sequenz vorkommende Restriktionsschnittstellen, schräge Striche( / ) Restriktionsschnittstellen aus dem Vektorteil. pACYC002: Ein 1,9 kBp langes SstI/HindIII-Fragment aus pJO1 wurde mit dem ebenso geschnittenen Vektor pACYC001 ligiert. pACYC003: In pACYC002 ist ein 3,2 kBp langes SstI/SgfI-Fragment aus pOBH23 eingefügt worden. pACYC004: Aus pBRS2 wurde ein 4,2 kBp langes SgfI/PstI-Fragment in pACYC003 integriert. pACYC006: Aus dem Konstrukt pK8 ist ein 3,5 kBp langes Bpu1102I/MluI-Fragment mit dem ebenso geschnittenen Konstrukt pACYC002 ligiert. pACYC005: Aus pACYC006 wurde mit BglII/MluI ein 4,6 kBp-Fragment ausgeschnitten und mit pACYC004 ligiert. pACYC007: Ligation des 4,6 kBp BglII/MluIFragmentes aus pACYC006 mit pACYC003. pACYC008: Ligation des auch zur Konstruktion von pACYC003 verwendeten SgfI/SstI-Fragmentes aus pOBH23 mit pACYC001. pACYC009: Ligation des zur Konstruktion von pACYC004 verwendeten SgfI/PstI-Fragmentes aus pBRS2 in pACYC001. pACYC010: Das für die Konstruktion von pACYC004 und pACYC009 verwendete SgfI/PstI-Fragment aus pBRS2 ist mit dem ebenso geschnittenen pACYC008 ligiert worden. 3.3.3.2. Nachweis der Protein-Produktion Die Produktion der Proteine KsuB, KsuC und KsuF wurde in den Transformanten nach Kultivierung in Minimalmedium mit Hochsalz oder ohne getestet (siehe Abb. 3.14 a-c). Bei der Negativ-Kontrolle S.lividans NI pOV001 kann, wie zu erwarten war, kein Signal gegen KsuF, KsuB und KsuC gefunden werden (Abb. 3.14 a-c, Spur 1 und 2). KsuF findet sich bei allen Stämmen, die das vollständige Gen für KsuF tragen (Abb. 3.14 c, Spuren 5-10, 13 und 14, pOV003-, pOV004- und pOV005-Transformanten). pOV006 trägt nur den 5’-Teil von ksuF, es kann kein Signal nachgewiesen werden (die schwachen Signale werden durch einen Überladungseffekt der Nachbarspuren hervorgerufen; Abb. 3.14 c, Spuren 11 und 12). Die Konstrukte pOV004, pOV005 und pOV010 tragen ksuC und ksuB. Hier können auch jeweils deutliche Signale der erwarteten Größe gefunden werden (Abb. 3.14 a und b). Das Konstrukt pOV009 trägt vollständig ksuC, jedoch nur einen Teil von ksuB. Die Produktion von KsuC kann nachgewiesen werden (Abb. 3.14 b, Spuren 17 und 18), KsuB ist nicht Ergebnisse - 65 - nachweisbar (Abb. 3.14 a, Spuren 17 und 18). Ein vergleichbares Ergebnis konnte in Minimalmedium mit 300 mM NaCl statt 300 mM KCl gefunden werden (Ergebnisse nicht dargestellt). Die Produktion von KsuB, KsuC und KsuF ist bei Wachstum in Minimalmedium mit 300 mM KCl (Abb. 3.14 a-c, gerade Nummern) oder 300 mM NaCl, (nicht dargestellt) gegenüber der Synthese in Minimalmedium ohne Zusatz von Salz (Abb. 3.14 a-c, ungerade Nummern), deutlich um ca. das fünf- bis zehnfache erhöht. Die Zugabe von 10,3 % (300 mM) Sucrose statt Salz erbringt eine, der Produktion in Minimalmedium ohne zusätzlich Salz, vergleichbares Ergebnis (nicht dargestellt). Abb. 3.14 a-c) Test der Synthese von KsuB in a), KsuC in b) und KsuF in c). Aufgetragen wurde auf den jeweiligen Spuren die Cytoplasmafraktionen der S.lividans NI Stämme mit folgendem Plasmid, jeweils angezogen in MM40 (Spuren mit ungerade Nummern) und in MM40 + 300 mM KCl (Spuren mit gerade Nummern): 1-2: pOV001, 3-4: pOV002, 5-6: pOV003, 7-8: pOV004, 9-10: pOV005, 11-12: pOV006, 13-14: pOV007, 15-16:pOV008, 17-18:pOV009, 19-20: pOV010. Getestet wurde mit Antikörpern gegen KsuB (Membran a), KsuC (Membran b) und KsuF (Membran c). Es sind jeweils nur die Proben aufgetragen worden, deren Stämme auch jeweils ein Plasmid mit den entsprechenden Genen trugen (siehe Abb. 3.13). Als NegativKontrolle wurde der den Leervektor tragende S.lividans NI pOV001-Stamm verwendet. - 66 - Ergebnisse 3.3.4. Produktion der Proteine in einer sigH-Mutante Der Sigma-Faktor SigH (σH) ist an der osmotischen Regulation in S.coelicolor beteiligt, vor sigH findet sich ein Promoter (P2), der unter hohen Salz-, jedoch nicht Sucrosekonzentrationen die erhöhte Transkription von sigH bewirkt (Kormanec, J., et al.; 2000). Von der SigH-Regulation abhängige Proteine zeigen ein zu KsuB, KsuC und KsuF vergleichbares Produktionsverhalten, unter hohen KCl und NaCl Konzentrationen (Jan Kormanec, pers. Mitteilung). Zum Nachweis, ob eine SigH abhängige Regulation der drei Proteine vorliegt, wurde die sigH-Mutante S.coelicolor M145 sigH:tsr (Sevcikova, B., et al.; 2001) und zur Kontrolle S.coelicolor J1984 (M145 sigF:tsr, Kelemen, G.M.; et al.; 1998), S.coelicolor M145 WT, S.lividans 66 WT (nicht dargestellt), S.lividans 66 pOV001 und als „Positiv“ Kontrolle die S.lividans NI pOV005 Transformante (trägt ksuC, ksuB und ksuF auf dem Konstrukt) in Minimalmedium ohne zusätzlich Salz (nicht dargestellt) und in Minimalmedium mit 300 mM KCl angezogen (siehe 3.15 a-c). Das Produktionsverhalten von KsuB, KsuC und KsuF der S.coelicolor-Stämme und des S.lividans Wildtyps war jeweils vergleichbar. Die S.lividans 66 pOV001 Transformande, die einen „Leervektor“ ohne ein integriertes Fragment trägt, zeigt eine deutliche, chromosomal determinierte KsuF-Produktion (siehe Abb. 3.15 c, Spur 5), alle anderen Stämme nicht (Abb. 3.15 c, Spuren 1-4). Eine von SigH regulierte Synthese von KsuB, KsuC und KsuF konnte nicht nachgewiesen werden. Abb. 3.15 a-c): Test der SigH-Abhängigkeit der KsuB, KsuC und KsuF Synthese. Alle Stämme wurden jeweils nach Vorkultur in Jasenka-Medium mit Sucrose ü.N., in MM40 Minimalmedium mit 300 mM KCl angezogen und die Cytoplasmafraktionen mit Antikörpern gegen KsuB in a), KsuC in b) oder gegen KsuF in c), getestet. Aufgetragen wurden: Spur 1: S.coelicolor M145 Wildtyp, Spur 2: S.coelicolor M145 sigH:tsr, Spur 3: S.colicolor 1984 (M145 sigF:tsr), Spur 4: S.lividans 66 pOV001 und Spur 5: S.lividans NI pOV005. 8S: Proteinstandard. Die Anzucht der S.coelicolor-Stämme erfolgte in Medium ohne Thiostrepton, die der S.lividans Transformanten mit 12,5 mg/l Thiostrepton. Zur besseren Erkennbarkeit des Proteinstandards und der Signale, wurden die Abbildungen jeweils bearbeitet (Tonwertekorrektur). Das von KsuB stammende Signal der S.coelicolor Stämme ist schwer zu erkennen, die Laufhöhe ist durch einen Pfeil markiert. Ergebnisse - 67 - 3.3.5. Abhängigkeit der Synthese von KsuF vom Vorhandensein des Thiostrepton Resistenzproteins Eine Produktion von KsuF in Wildtypstämmen (z.B. S.lividans 66 oder S.coelicolor M145) konnte mit Hilfe von Antikörpern nach Transfer auf Membranen nicht nachgewiesen werden, anders als bei Synthese in S.lividans WT Stämmen, die pWHM3be und pIJ702 oder Derivate derselben, ohne ein ksuF tragendes Fragment, enthielten (z.B. siehe vorheriges Kapitel, Abb. 3.15 c, Spur 5). Die Synthese des aus Streptomyces azureus stammenden Thiostrepton Resistenzproteins Tsr ist durch Thiostrepton induzierbar. Geringe Konzentrationen sind ausreichend, um durch Thiostrepton induzierbare Proteine zu induzieren (Ali, N., et al., 2002, Holmes, D.J., et al.; 1993). Es wurde 1 µg/ml Thiostrepton verwendet, da diese Konzentration keine Auswirkung auf das Wachstum der Wildtypstämme zeigte. In unserer Arbeitsgruppe sind keine S.lividans Stämme vorhanden, die tsr im Chromosom integriert haben. Um auszuschließen, dass weitere, plasmiddeterminierte Proteine von pIJ702 oder pWHM3be für die Synthese von KsuF verantwortlich sind, wurden zwei S.coelicolorDisruptionsmutanten von Sigma-Faktoren verwendet, die nur tsr im Chromosom integriert haben. Dies sind die Stämme S.coelicolor M145 sigH:tsr (Sevcikova, B., et al.; 2001) und das M145 Derivat S.coelicolor J1984 (sigF:tsr, Keleman, G.M.; et al.; 1998). Die Gabe von Thiostrepton hat keine Auswirkung auf die Synthese von KsuF in Wildtypstämmen (Abb. 3.16 a, Spuren 1-3). Eine KsuF-Produktion konnte auch im Stamm S.lividans 66 FEH11 nicht nachgewiesen werden; dieser Stamm trägt ein EGFP-Gen „in frame“ integriert hinter ksuF (Abb. 3.16 a, Spur 6). Die tsr im Chromosom integriert tragenden Stämme S.coelicolor M145 sigH:tsr und S.coelicolor J1984 (sigF:tsr) zeigen bei Gabe von 1 µg/ml Thiostrepton eine hohe Synthese des chromosomal determinierten KsuF (Abb. 3.16 a, Spuren 4 und 5); im Vergleich hierzu der S.coelicolor M145 Wildtyp-Stamm keine (Abb. 3.16 a, Spur 3). Bei Wachstum in Medium ohne Thiostrepton zeigen beide Deletionsmutanten keine Synthese von KsuF, bzw. eine, die unterhalb der Nachweisgrenze liegt (siehe vorheriges Kapitel, Abb. 3.15 c, Spuren 2 und 3). Alle Stämme, die die Vektoren pIJ702, pWHM3be oder Derivate derselben tragen, zeigen bei Wachstum in Medium mit 12,5 µg/ml Thiostrepton eine Produktion von chromosomal determiniertem KsuF, die in der Synthesemenge vergleichbar ist mit der der beiden sigH:tsr und sigF:tsr S.coelicolor Stämme (Abb. 3.16. a, Spur 7 und b, Spuren 1-3, 6 und 7). Die Synthesesrate von KsuF ist somit abhängig vom Vorhandensein des ThiostreptonResistenzproteins und gleichzeitiger Gabe von Thiostrepton zur Induktion der Produktion desselben. Sie ist nicht alleine direkt abhängig von Thiostrepton und von weiteren pIJ702 oder pWHM3be determinierten Proteinen. Das Vorhandensein des potentiellen Promoterbereiches oder eines Teils von ksuF auf einem Konstrukt reduziert die Synthese des chromosomal determinierten KsuF (Abb. 3.16 b, Spur 4). Dies konnte auch bei anderen, die Konstrukte pOVE31, pOVE32, pOVE34 und pOVE 43 - 68 - Ergebnisse tragenden S.lividans Stämmen, beobachtet werden (nicht dargestellt). Die geringere Migrierung des von pOVE33 determinierten His-Tag KsuF ist zu erkennen (Abb. 3.16 b, Spur 5). Abb. 3.16 a und b): Nachweis der KsuF-Synthese in unterschiedlichen Stämmen unter Thiostrepton. Die Stämme wurden jeweils in Minimalmedium mit Salz angezogen (MM40 Medium mit 300 mM KCl), Stämme, die das Plasmid pWHM3be trugen oder unterschiedliche Konstrukte desselben, wurden mit 12,5 µg/ml Thiostrepton angezogen, Stämme ohne Plasmide oder Konstrukte, mit 1 µg/ml Thiostrepton oder ohne. Der Nachweis erfolgte nach Transfer auf Membran mit Hilfe von Antikörpern gegen KsuF. Auf a) wurde aufgetragen: Spur 1: S.lividans 66 WT, angezogen ohne Thiostrepton und in Spur 2 angezogen mit 1 µg/ml Thiostrepton, Spur 3: S.coelicolor M145 WT, angezogen mit 1 µg/ml Thiostrepton, Spur 4: S.coelicolor M145 sigH:tsr (1 µg/ml Thio), Spur 5: S.colicolor J1984 (M145 sigF:tsr) (1 µg/ml Thio), Spur 6: S.lividans 66 FEH11 (1 µg/ml Thio), Spur 7: S.lividans 66 pIJ702 (12,5 µg/ml Thio) und in Spur 8: S.lividans NI pOV005 (12,5 µg/ml Thio). In b) wurde aufgetragen auf: Spur 1: S.lividans TK21 pOV001 (12,5 µg/ml Thio), Spur 2: S.lividans 66 pOV001, Spur 3: S.lividans 66 pWHM3be (12,5 µg/ml Thio), Spur 4: S.lividans TK21 pOVE3 (12,5 µg/ml Thiostrepton), Spur 5: S.lividans TK21 pOVE33 (12,5 µg/ml Thio, His-Tag KsuF-Produktionsstamm), Spur 6: S.lividans 66 pOV005 (12,5 µg/ml Thio), Spur 7: S.lividans pOV201 (12,5 µg/ml Thio) und 8: S.lividans NI pOV005. 8S: ist jeweils der Proteinstandard. Es wurden jeweils ca. 0,45 µl Cytoplasmafraktion aufgetragen, für S.lividans NI pOV005 ca. 1/10 der Menge. Spätere Tests des Stammes S.lividans 66 pOV005 zeigten, dass das in pOV005 integrierte homologe 13 kBp Fragment vollständig deletiert war, das Konstrukt besaß ungefähr die Größe des „Leervektors“ pOV001. Die gefundene Produktion von KsuF ist chromosomal determiniert. 3.4. Gewinnung von His-Tag Proteinen aus S.lividans Um verschiedene Proteine in S.lividans in größeren Mengen zu produzieren und über Ni-NTA aufreinigen zu können, wurde der Vektor pOVE3 konstruiert, da in unserer Arbeitsgruppe kein entsprechender Vektor vorhanden war. In den Streptomyces/E.coli-Vektor pWHM3be wurde der den potentiellen Promotor von ksuF tragende intergenische Bereich zwischen ksuF und kcsA, der für den His-Tag kodierende- und Polylinker-Bereich aus pQE32 und der zwischen ksuF und ksuL liegende Terminator integriert. Der Promotor von ksuF ermöglichte Ergebnisse - 69 - auf einem Konstrukt (pJO1 oder z.B. pOV005) in S.lividans eine sehr hohe Produktion von KsuF (Abb. 3.11 a und d und Abb. 3.14 c). Das Startkodon liegt innerhalb der Erkennungssequenz des SphI-Restriktionsenzyms (GCATGC). Zur Inaktivierung der Schnittstelle wurde die zweite Base (G) vor dem A des ATG durch ein C ausgetauscht. 3.4.1. Konstruktion der Plasmide pOVV1, pOVV2 und pOVE3 Das bifunktionelle Produktionsplasmid pOVE3 wurde über zwei Zwischenkonstrukte hergestellt, pOVV1 und pOVV2. In einem ersten Schritt wurde der für den 6fach His-Tag kodierende- und der Polylinker-Bereich aus pQE32 (Qiagen) amplifiziert und nach XbaI/HindIII-Restriktion das entstandene 80 Bp lange Fragment mit dem ebenso geschnittenen Vektor pWHM3be ligiert. Hierbei wurde eine Mph1103IRestriktionsschnittstelle in das Startkodon des His-Tag’s eingefügt. Der entstandene Vektor erhielt den Namen pOVV1 (Abb. 3.17) Im nächsten Schritt wurde der vor ksuF liegende potentielle Promotor-Bereich in den Vektor pOVV1 eingefügt. Nach Amplifikation mit Hilfe von „nested-PCR“ wurde, nach Restriktion mit EcoRI/Mph1103I, das entstandene 356 Bp lange Fragment mit dem ebenso geschnittenen Vektor pOVV1 ligiert. Das Konstrukt erhielt den Namen pOVV2 (Abb. 3.17). Im letzten Schritt wurde der zwischen ksuF und ksuL gelegene Terminator amplifiziert, mit HindIII und MluI geschnitten und als 158 Bp-Fragment mit dem ebenso geschnittenen Vektor pOVV2 ligiert. Der entstandene Vektor trägt den Namen pOVE3 (Abb. 3.17). Abb. 3.17: Vektoren pOVV1, pOVV2 und pOVE3. Gelb markiert ist das den 6x His- kodierenden TagBereich tragende Fragment aus pQE32. In grau, der den potentiellen Promoter tragende Bereich und blau markiert, der Terminator-Bereich mit dem Terminator selbst, in rot. Zur Amplifikation des Fragmentes (in gelb), das die sechs Histidin-Kodons enthält, wurden die Oligos 2PrCR1XN und PrPCR1H1 verwendet. Für die Amplifikation des vermutlichen Promoterbereiches wurden in einer ersten Amplifikation die Oligos Pr70 und Pr71 und pJO1 als Matrize verwendet. Von dem so entstandenen 530 Bp Fragment erfolgte eine weitere Amplifikation mit Hilfe der Oligos PRPCR2E und PRPCR2N, wobei die entsprechenden Schnitttstellen eingefügt wurden. Der Terminatorbereich wurde mit Hilfe der Primer PRPCR3MH und PRPCR3SX vom Konstrukt pOSP13 amplifiziert. - 70 - Ergebnisse 3.4.2. Konstruktion der bifunktionellen Plasmide pOVE4 und pOVE5 pOVE4 unterscheidet sich von pOVE3 durch eine SphI-Restriktionsschnittstelle im Startkodon und besitzt keinen für die 6 Histidine determinierenden Abschnitt. Die 2. Base (G) vor dem ATG des Startkodons wurde, anders als bei pOVE3, nicht ausgetauscht. Mit diesem Plasmid sollte die Überproduktion von nativen Proteinen ermöglicht werden. Hierzu wurde das für die Konstruktion von pOVV2 verwendete 530 Bp PCR-Fragment als Matrize für eine PCR-Reaktion verwendet. Das entstandene Fragment wurde nach EcoRI- und SphIRestriktion als 336 Bp-Fragment mit dem entsprechend geschnittenen Vektor pOVE3 ligiert. Der resultierende Vektor erhielt den Namen pOVE4 (Abb. 3.18). Abb. 3.18: Überproduktionsvektor pOVE4. Grün makiert ist der neu eingefügte Promoterbereich. Das ATGStartkodon des ksuF-Promoters liegt in der SphI-Schnittstelle. Gelb makiert ist der Polylinker-Bereich aus pQE32, blau der Terminatorbereich, rot der Terminator selbst. Für die Amplifikation des potentiellen Promotorbereiches wurden die beiden Oligos PR70 und PRPCR2E verwendet. Die Produktion der His-Tag Proteine bei den pOVE3-Derivat-Transformanten in S.lividans fiel deutlich geringer aus, als erwartet. Es wurde vermutet, dass der aus pQE32 stammenden Abschnitt verantwortlich sein könnte, aufgrund des Vorhandenseins von für E.coli typischen jedoch für Streptomyceten untypischen Kodons. Dieser Abschnitt umfasst die für die ersten 11 Aminosäuren kodierenden Basentripletts, unter anderem die für das 6x His-Tag (MHRGSHHHHHH). Einige dieser sind selten in Streptomyceten zu finden (AGA-> 0,8 von 1000; 4x CAT->1,7; GGA->7,2 und TCT ->0,6 von 1000 Basentripletts bei S.coelicolor A3(2), NCBI-Genbank Flat File Release 144.0). Durch Austausch seltener Basentripletts im 5’ Bereich konnte die Produktionsrate bei anderen Stämmen deutlich beeinflusst werden (Liljenstrom H. and von Heijne, G.; 1987); sechs der ersten 11 Kodons sollten gegen Ergebnisse - 71 - Streptomyceten gängigere ersetzt werden. Um diesen Bereich auszutauschen, wurden zwei komplementäre Oligos phosphoryliert und anschließend zum Doppelstrang assoziiert. Die Oligos waren so gewählt, dass das assoziierte Fragment an einer Seite das überhängende Ende einer Mph1103I- und auf der anderen einer BamHI-Schnittstelle besaß. Dieses wurde mit dem Mph1103I/BamHI geschnittenen Vektor pOVE3 ligiert und in E.coli transformiert. Erhalten wurde pOVE5 (Abb. 3.19). Nach Sequenzierung fanden sich zwei korrekt inserierte Konstrukte, die verwendet wurden. In den drei weiteren getesteten fanden sich Basenaustausche im integrierten Bereich, hervorgerufen durch minderwertige Oligos. Abb. 3.19: Überproduktionsvektor pOVE5. Grün markiert ist der ausgetauschte Bereich, gelb der aus pQE32 stammende Linkerbereich, blau der Terminatorbereich und rot der Terminator selbst. Die beiden Oligos PrPoly3I und PrPoly3II wurden für die Konstruktion des ausgetauschten Bereiches verwendet. Von den für den Nterminalen His-Tag kodierenden ersten 11 Kodons (MHRGSHHHHHH) wurden folgende ausgetauscht: drei der vier CAT-Kodons (die drei im His-Tag vorhandenen) gegen CAC (kodierend für Histidin), AGA zu CGC (Arginin), GGA zu GGC (Glycin) und TCT zu TCC (Serin). - 72 - Ergebnisse 3.4.3. Herstellung kcsA-haltiger Konstrukte und Analyse der S.lividans-Transformanten Zur Klonierung von kcsA in pOVE3 wurde das entsprechende Gen aus pQE837 (H. Splitt) mit Hilfe einer SphI/HindIII-Doppelrestriktion ausgeschnitten und in die entsprechenden Restriktionsschnittstellen von pOVE3 eingefügt (pOVE31, Abb. 3.20). Dasselbe Fragment wurde auch mit den ebenso geschnittenen Vektoren pOVE4 und pOVE5 ligiert. Erhalten wurden die Konstrukte pOVE41 und pOVE51 (Abb. 3.20). Nach Aufreinigung aus E.coli erfolgte jeweils die Transformation in S.lividans TK21. Abb. 3.20: Konstrukte pOVE31, pOVE41 und pOVE51 In dunkelgrau und grün sind die potentiellen Promoter-Bereiche, in hellgrau das inserierte kcsA, in blau der Terminatorbereich mit dem Terminator in rot, dargestellt. Mit den Konstrukten pOVE3, pOVE4 und pOVE5 wurde jeweils das kcsA tragende 560 Bp große SphI/HindIII-Fragment aus pQE837 ligiert. Die pOVE31 und pOVE51 S.lividans-Transformanten sollten die Produktion eines 174 Aminosäuren langen N-terminalen His-Tag KcsA ermöglichen, die der pOVE41 Transformanten eines mit 160 Aminosäuren in der Länge und Sequenz dem originalem KcsA entsprechenden Proteins. Nach Anzucht der Transformanten und Aufreinigung des His-Tag KcsA über Ni-NTA aus den Membranfraktionen, wurde die Produktion immunologisch getestet („Western Blot“, Abb. 3.21 a-d). Die pOVE31- und die pOVE51- Transformanten produzieren His-Tag KcsA. Die Produktion ist bei den pOVE51-Transformanten deutlich geringer. Sowohl die tetramere (theoretisches Molekulargewicht von 77,1 kDa), als auch monomere Form (theoretisches MW von 19,3 kDa), haben im Vergleich zum Standard eine etwas geringere Mobilität als erwartet. Dies wurde auch schon früher beobachtet und ist typisch für Membranproteine. Nach Aufkochen in Probenpuffer ohne reduzierende Agenzien findet sich KcsA als Monomer. Eine Produktion von KcsA in den pOVE41-Transformanten konnte nie nachgewiesen werden (Abb. 3.20 a, Spuren 5 und 6). Da pOVE41 über Restriktionskontrolle und Sequenzierung verifiziert wurde, ist keine Ursache hierfür zu erkennen. Ergebnisse - 73 - Abb. 3.21 a-d): Test der Produktion von KcsA und His-Tag KcsA in S.lividans TK21 pOVE3, pOVE31-, pOVE41- und pOVE51-Transformanten. Getestet wurden die Membranfraktionen (Membran a und b) und die über Ni-NTA aufgereinigten Proteine (Membran c und d), jeweils mit Antikörpern gegen KcsA (Membran a und c) und gegen den His-Tag (Membran b und d). Es wurden jeweils Fraktionen nicht gekocht (ungerade Zahlen) und gekocht (gerade Zahlen) aufgetragen. Die Mobilität der tetramären und der monomeren Form von His-Tag KcsA ist jeweils mit Pfeilen markiert. a) und b): Aufgetragen wurden die Membranfraktionen von: Spuren 1 und 2 pOVE3-, Spuren 3 und 4 pOVE31-, Spuren 5 und 6 pOVE41- und Spuren 7 und 8 pOVE51-Transformanten. Spur 9: 8S-Proteinstandard. c) und d): Aufgetragen sind die Elutionsfraktionen von: Spuren 1 und 2 pOVE3-, Spuren 3 und 4 pOVE31- und Spuren 5 und 6 pOVE51-Transformanten. Spur 7: 8S-Proteinstandard. Um die Erkennbarkeit des Proteinstandards zu erhöhen, wurde der Kontrast der Spur 9 in a) und der Spur 7 in c) verstärkt. Alle Kulturen wurden in MM40 Medium mit 300 mM KCl angezogen. - 74 - Ergebnisse 3.4.4. Produktion von KsuB in S.lividans-Transformanten Aus dem für die Konstruktion des His-Tag KsuB Überproduktionsvektor pQE309 verwendeten Konstrukt pORFZ wurde ksuB mit Hilfe einer SphI/HindIII-Restriktion ausgeschnitten und mit den ebenso geschnittenen Vektoren pOVE3, pOVE4 und pOVE5 ligiert und in E.coli transformiert. Die Konstrukte erhielten die Namen pOVE32, pOVE42 und pOVE52 (siehe Abb. 3.22). Nach Aufreinigung aus E.coli erfolgte die Transformation in S.lividans TK21. Abb. 3.22: Konstrukte pOVE32, pOVE42 und pOVE52 Die potentiellen Promoter-Bereiche sind in dunkelgrau und grün dargestellt. Das inserierte ksuB tragende Fragment ist in hellgrau, in blau der Terminatorbereich mit dem Terminator in rot, dargestellt. Mit den Konstrukten pOVE3, pOVE4 und pOVE5 wurde jeweils das ksuB tragende, 929 Bp lange SphI/HindIII-Fragment aus pORFZ ligiert. Die Produktion eines 323 Aminosäuren langen N-terminalen His-Tag KsuB sollten die pOVE32 und pOVE52 S.lividansTransformanten ermöglichen, die der pOVE42 Transformanten eines dem Original entsprechenden Proteins (310 Aminosäuren lang). Der Nachweis der Produktion erfolgte immunologisch. Getestet wurden die cytoplasmatischen Fraktionen und die in Folge über Ni-NTA aufgereinigten Proteine (Abb. 3.23 a und b). Neben den in allen fünf Cytoplasma-Fraktionen gefundenen, vom Chromosom determinierten KsuB (Abb. 3.23 a, Spuren 1-5, theoretisches MW von 34,3 kDa), findet sich bei den pOVE32 und pOVE52 Transformanten ein weiteres Protein, das His-Tag KsuB (theoretisches MW von 36 kDa) repräsentiert (Abb. 3.23 a und b, Spuren 3 und 5). Die Produktion von His-Tag KsuB ist bei den pOVE32-Transformanten größer, was auch an den über Ni-NTA aufgereinigten Fraktionen zu erkennen ist (siehe Abb. 3.23 a und b, Spuren 7 und 8). Es konnte keine erhöhte Produktion von KsuB in den pOVE42-Transformanten gegenüber den einen „Leervektor“ tragenden pOVE3 oder pOVE4 Transformanten identifiziert werden (Abb. 3.23 a, Spur 4 und Spuren 1 und 2). Ergebnisse - 75 - Abb. 3.23 a und b): Test der Produktion von KsuB und His-Tag KsuB in S.lividans TK21 pOVE3-, pOVE4- pOVE32-, pOVE42- und pOVE52-Transformanten. Die Anzucht der Kulturen erfolgte in MM40 Medium mit 300 mM KCl. Aufgetragen und mit Antikörpern gegen KsuB (Membran a) und gegen den His-Tag (Membran b) getestet, wurden Cytoplasma-Fraktionen von pOVE3- (jeweils Spur 1), pOVE4- (Spur 2), pOVE32- (Spur 3), pOVE42- (Spur 4) und von pOVE52-Transformanten (Spur 5). Auf den Spuren 6 bis 8 wurden jeweils über Ni-NTA aufgereinigte Protein-Fraktionen aufgetragen: Spur 6, von pOVE3-, Spur 7 von pOVE32-, Spur 8 pOVE52-Transformanten. Spur 9: 8S-Proteinstandard. 3.4.5. Produktion von KsuC in S.lividans-Transformanten Die Konstruktion von pWRS1 erfolgte unter direkter Anleitung durch Ralf Steger. Aus dem für die Produktion von His-Tag KsuC in E.coli verwendeten Konstrukt pQE321 wurde über eine HindIII/SphI- Restriktion ksuC ausgeschnitten und mit dem ebenso geschnittenen Vektor pOVE3 ligiert und in E.coli transformiert. Erhalten wurde das Konstrukt pWRS1 (Abb. 3.24). Aus pWRS1 wurde über eine HindIII/SphI-Restriktion ksuC wieder ausgeschnitten und mit dem ebenso geschnittenen Vektor pOVE5 ligiert. Das Konstrukt erhielt nach Transformation in E.coli den Namen pOVE54 (Abb. 3.24). Beide Konstrukte wurden in S.lividans TK21 transformiert. Der Nachweis der Produktion von His-Tag KsuC erfolgte mit Hilfe von Antikörpern durch Test der Cytoplasma-Fraktionen und des über Ni-NTA aufgereinigten Proteins. In den beiden His-Tag KsuC-Überproduktionstransformanten (pWRS1 und pOVE54, Abb. 3.25 a und b, Spuren 3, 4, 6 und 7) können gegenüber den Kontrollen (pOVE3 und pOVE5Transformanten, Membranen a und b, Spuren 1 und 2) zusätzlich Kreuzreaktionen gefunden werden. Die Mobilitäten sind geringer, als das theoretische Molekulargewicht von 29,9 kDa für KsuC und 31,6 kDa für His-Tag KsuC erwarten lassen würde. Die Produktion von HisTag KsuC ist bei der pOVE5-Derivat- (pOVE54; Abb. 3.25 a und b, Spuren 4 und 7) geringer als bei der pOVE3-Derivat-Transformante (pWRS1; Abb. 3.25 a und b, Spuren 3 und 6). - 76 - Ergebnisse Abb. 3.24: Konstrukte pWRS1 und pOVE54 Dargestellt sind: die potentiellen Promoter-Bereiche in dunkelgrau, das inserierte, 897 Bp lange ksuC tragende SphI/HindIII Fragment in hellgrau, in blau der Terminatorbereich mit dem Terminator in rot. S.lividans-Transformanten, die einen der beiden Konstrukte trugen, sollten jeweils die Produktion eines 299 Aminosäuren langen His-Tag KsuC ermöglichen. Abb. 3.25 a und b): Test der Produktion von KsuC und His-Tag KsuC in S.lividans TK21 pOVE3-, pOVE5- pWRS1-, und pOVE54-Transformanten. Die Anzucht erfolgte in MM40 Medium mit 300 mM KCl. Mit Antikörpern gegen KsuC (Membran a) und gegen den His-Tag (Membran b) wurden die CytoplasmaFraktionen von pOVE3- (jeweils Spur 1), pOVE5- (Spur 2), pWRS1- (Spur 3) und von pOVE54Transformanten (Spur 4) getestet. Über Ni-NTA aufgereinigte Protein-Fraktionen wurden jeweils auf den drei nachfolgenden Spuren aufgetragen: Spur 5, von pOVE3-, Spur 6 von pOVE34-, Spur 7 pOVE54Transformanten. Spur 8: 8S-Proteinstandard. Der Kontrast des den Standard tragenden Abschnittes wurde jeweils erhöht, um die Erkennbarkeit zu verbessern. Ergebnisse - 77 - 3.4.6. Produktion von KsuF in S.lividans-Transformanten Aus pQE314 (His-Tag KsuF-Produktionsvektor) wurde das ksuF-tragende Fragment mit Hilfe der Restriktionsenzyme SphI/HindIII ausgeschnitten und mit den ebenso geschnittenen Vektoren pOVE3 und pOVE4 ligiert. Es wurden die Konstrukte pOVE33 und pOVE43 erhalten (siehe Abb. 3.26). Anschließend wurde aus pOVE33 dasselbe Fragment wieder ausgeschnitten und mit dem Vektor pOVE5 ligiert. Nach Transformation in E.coli wurde das Konstrukt pOVE53 erhalten (Abb. 3.26). Nach Aufreinigung erfolgte die Transformation in S.lividans TK21. Abb. 3.26: Konstrukte pOVE33, pOVE43 und pOVE53. Die potentiellen Promoter-Bereiche sind in dunkelgrau und grün dargestellt. Das inserierte, ksuF tragende Fragment in hellgrau, in blau ist der Terminatorbereich mit dem Terminator selbst in rot, dargestellt. Das in hellgrau dargestellte, aus pQE314 stammende SphI/HindIII-Fragment, besitzt jeweils eine Größe von 1399 Bp. Von den pOVE33 und pOVE53 Transformanten sollte eine Produktion eines 387 Aminosäuren langen His-Tag KsuF erfolgen, von pOVE43 eines dem Original entsprechenden 373 langen KsuF-Proteins. Die Synthesen von His-Tag KsuF konnte sowohl in den Cytoplasma-Fraktionen der pOVE33 und pOVE53-Transformante, als auch in den über Ni-NTA aufgereinigten Fraktionen, nachgewiesen werden (Abb. 3.27 a und b, Spuren 3, 4, 6 und 7). Die Mobilität entspricht den erwarteten 40,8 kDa und ist deutlich höher, als das chromosomal determinierte KsuF mit einem theoretischen MW von 39,1 kDa (Abb. 3.27 a, Spuren 1 und 2). Die His-Tag KsuFund KsuF-Lauffront ist jeweils mit einem Pfeil markiert. Auch hier ist die Produktion vom pOVE5- deutlich geringer als vom pOVE3-Derivat. Die Produktion von KsuF der pOVE43Transformanten erwies sich als vergleichbar stark, wie die der pOVE4-Transformanten (nicht dargestellt). Sie war geringer als die chromosomal determinierte Produktion von Transformanten, die pWHM3be-Derivate, ohne ksuF auf einem Fragment inseriert, trugen. - 78 - Ergebnisse Abb. 3.27 a und b): Test der Produktion von KsuF und His-Tag KsuF in verschiedenen S.lividans Transformanten. Die Anzucht erfolgte in MM40 Medium mit 300 mM KCl. Aufgetragen wurden die Cytoplasma-Fraktionen (Spuren 1 bis 4) und über Ni-NTA aufgereinigte Fraktionen (jeweils Spuren 5-7), der Nachweis erfolgte mit Antikörpern gegen KsuF (Membran a) und den His-Tag (Membran b). Spur 1: pOVE3-, Spur 2: pOVE5-, Spur 3: pOVE33-, Spur 4: pOVE53-, Spur 5: pOVE3-, Spur 6: pOVE33- und Spur 7: pOVE53Transformanten. Auf der Spur 8 wurde jeweils 8S-Proteinstandard aufgetragen. In S.lividans-Transformanten, die entweder eines der pOVE3- oder der pOVE5-DerivatKonstrukte trugen, konnte die Produktion eines His-Tag-Proteins nachgewiesen werden. Die Produktionsrate der jeweiligen His-Tag Proteine war bei den pOVE5-Derivat-S.lividans Transformanten deutlich geringer, als bei den vergleichbaren pOVE3-Derivat-Transformanten (ca. um das Fünf- bis Zehnfache). Beide Derivatfamilien unterscheiden sich nur durch eine alternative Kodonverwendung im N-terminalen Bereich des His-Tag’s. Da die Kodonverwendung in den pOVE5-Derivaten für Streptomyceten optimiert war, sollte hier eine mit den pOVE3-Derivaten zumindest vergleichbare Produktion erfolgen. Die Ergebnisse zeigten jedoch, dass die für E.coli optimierten Kodons für den His-Tag in den pOVE3-Derivaten, auch in S.lividans eine höhere Produktion ermöglichen, als die für S.lividans optimierten Kodons in den pOVE5-Derivaten. Dass die Verwendung von den in Streptomyceten häufiger vorkommenden Kodons zu einer geringeren Synthese führte, war unerwartet. Ob dieses Ergebnis auf eine veränderte Termination der Transkription oder auf eine verringerte Stabilität der mRNA zurückzuführen war, wurde nicht näher analysiert. Die Produktionsrate war bei Verwendung eines Vollmedium (Jasenka-Medium) deutlich geringer (ca. um das Fünffache pro Gramm Nassgewicht), als bei Verwendung von Minimalmedium mit Hochsalz (Ergebnisse nicht dargestellt). Es konnte nicht gezeigt werden, dass von den pOVE4-Derivat Transformanten (pOVE41, pOVE42 und pOVE43) die Produktion eines Proteins, determiniert vom Plasmid, erfolgt. Bei den pOVE42- und pOVE43-Transformanten entspricht die Produktion von KsuB bzw. KsuF jeweils der chromosomal determinierten. Bei einer Reihe von Tests unterschiedlicher pOVE41-Transformanten konnte KcsA nicht nachgewiesen werden. Da alle notwendigen Kontrollen erfolgten ist keine Ursache hierfür zu erkennen. Ergebnisse - 79 - Die von den pOVE3 Konstrukten determinierte Proteinsynthese ist deutlich geringer als erwartet. Sie ist geringer, als die chromosomal determinierte Produktion von KsuF von S.lividans-Stämmen, die pWHM3be Derivate oder pIJ702 trugen, ohne in ihnen inseriertes ksuF (siehe z.B. Abb. 3.16 a und b) und deutlich geringer, als z.B. in S.lividans NI-Stämmen mit ksuF tragenden pOVE-Derivaten (siehe Abb. 3.14 c) oder dem S.lividans TK21 pJO1 Stamm (Abb. 3.11 a und d). Vergleichbare Ergebnisse zeigen auch die Fluoreszensmikroskopie-Ergebnisse der Stämme S.lividans TK21 pOVG1 und S.lividans TK21 pOVG13, die den für pOVE3 verwendeten und einen deutlich längeren potentielle Promoterbereiche von ksuF trugen (siehe unten, Abb. 3.34 und Abb. 3.35, jeweils b und h). - 80 - Ergebnisse 3.5. Studien von S.lividans-Transformanten mit ausgewählten Konstrukten 3.5.1. Herstellen der Konstrukte Um die Induktion und die Aktivierung von Streptomycetenpromotoren zu untersuchen und die Produktion verschiedener Proteine zu testen, wurde der Vektor pOVG1 (Abb. 3.28) aus dem Überproduktionsvektor pOVE3 konstruiert. Der für den His-Tag kodierende Bereich wurde ausgeschnitten und durch ein EGFP-Gen ersetzt. Hierzu wurde das EGFP-Gen aus dem Vektor pEGFP1 (Clontech) amplifiziert und nach Restriktion mit dem ebenso geschnittenen Vektor pOVE3 ligiert. Nach Mph1103I/EcoRI- oder Mph1103I/BglII-Restriktion von pOVG1 kann ein DNA-Fragment, mit einem mutmaßlichen Promotor-Bereich, vor das EGFP-Gen eingesetzt werden. Das Konstrukt pOVG1 trägt den vermutlich nicht voll funktionellen Promoter von ksuF, der, nach den Ergebnissen mit verschiedenen pOVE3-Konstrukten, in S.lividans nur eine geringe Synthese von EGFP erlauben sollte. Abb. 3.28: Bifunktioneller Produktionstestvektor pOVG1. In grün dargestellt ist das EGFP-Gen tragende Fragment. Dieses wurde nach Amplifikation mit pEGFP1 (Clontech) als Template und den Primer pREGFPmp und pREGFPxb (Fragmentgröße 1109 Bp, Vent-Polymerase) und Mph1103I/XbaI Restriktion als 728 Bp Fragment mit pOVE3 ligiert. In grau dargestellt ist der jeweils zu ersetzende Bereich, in blau der TerminatorBereich, der Terminator selbst ist in rot dargestellt. Zum Test der von den verschiedenen putativen Promotoren abhängigen Proteinproduktion, wurde versucht, die flussauwärts liegenden Bereiche verschiedener Gene mit Hilfe von PCR (Vent-Polymerase) zu amplifizieren und nach Mph1103I/BglII Restriktion in die entsprechenden Schnittstellen von pOVG1 zu klonieren. Die Oligos für die Amplifikation wurden so gewählt, dass jeweils ein Fusionsprotein produziert werden sollte, bestehend aus den ersten ca. 10 Aminosäuren des entsprechenden Proteins und nachfolgend dem EGFP- Ergebnisse - 81 - Protein. Als „Negativ“-Kontrolle wurde ein interner Sequenzbereich aus ksuF entgegen der Leserichtung in pOVG1 eingesetzt (pOVG17). Zur Integration des potentiellen Promoters von kcsA wurde aus dem Vektor pJO1 ein 1135 Bp langes Fragment mit Hilfe der Oligos Pr23 und pkcsam1 amplifiziert (Vent-Polymerase) und nach BglII/Mph1103I Restriktion das erhaltene 987 Bp Fragment mit dem ebenso geschnittenen Vektor pOVG1 ligiert. Das erhaltene Konstrukt erhielt den Namen pOVG11. Das Insert enthält neben dem vor kcsA liegenden Bereich die für den Anfangsbereich kodierenden Nukleotide, nachfolgend im selben Leseraster den EGFP-Gen-Teil (Abb. 3.29). Als Kontrollplasmid wurde der Vektor pOVG17 verwendet (Abb. 3.29). Hier wurde mit Hilfe der Oligos pkontm1 und pkontb1 ein 991 Bp Fragment vom Konstrukt pJO1 amplifiziert und nach Mph1103I/BglII Restriktion das entstandene 492 Bp Fragment mit dem ebenso geschnittenen Vektor pOVG1 ligiert. Von diesem Konstrukt sollte in S.lividans keine Synthese von EGFP erfolgen. Abb. 3.29: Konstrukte pOVG11 und pOVG17. Das in den Vektor pOVG1 jeweils integrierte Fragment ist in gelb dargestellt, das EGFP-Gen tragende Fragment in grün. Das den Terminator tragende Fragment ist in blau, der Terminator selbst ist in rot dargestellt. In S.lividans sollte von pOVG17 keine Synthese erfolgen, während von pOVG11 die Produktion eines 249 Aminosäuren langen KcsA’-EGFP-Fusionsproteins erfolgen sollte, bestehend aus den ersten 10 Aminosäuren von KcsA und nachfolgend den 239 von EGFP. - 82 - Ergebnisse Um die vom potentiellen Promoterbereich von ksuL determinierte Synthese zu testen, wurde der entsprechende Bereich amplifiziert und nach Mph1103I/BglII-Restriktion mit dem ebenso geschnittenen Vektor pOVG1 ligiert. Nach Aufreinigung aus E.coli wurde das Konstrukt, das den Namen pOVG14 erhielt (Abb. 3.30), in S.lividans TK21 transformiert. Abb. 3.30: Konstrukt pOVG14. Das potentielle Promoter- ist in gelb, das EGFP-tragende Fragment in grün und der Terminatorbereich in blau, der Terminator selbst in rot dargestellt. Für die Amplifikation des potentiellen Promoter-Bereiches wurden die Oligos Pksulm1 und Pksulb2, als Template das Konstrukt pOBH23, verwendet. Das entstandene 893 Bp Fragment wurde nach BglII/Mph1103I-Restriktion als 878 Bp großes Fragment in pOVG1 eingefügt. Das ksuL-EGFP-Gen kodiert für ein Protein von 248 Aminosäuren, bestehend aus den ersten 9 Aminosäuren von KsuL und den nachfolgenden 239 Aminosäuren von EGFP. Die beiden Gene ksuC und ksuB bilden vermutlich eine einzige Transkriptionseinheit; es wurde nur ein Konstrukt entwickelt mit dem vor ksuC liegenden potentiellen PromoterBereich. Zum Test der Aktivität wurde dieser potentielle Promotorbereich amplifiziert und nach Mph1103I/BglII-Restriktion mit dem ebenso geschnittenen Vektor pOVG1 ligiert. Das so entstandene Konstrukt pOVG15 wurde nach Aufreinigung aus E.coli in S.lividans TK21 transformiert (Abb. 3.31). Ergebnisse - 83 - Abb. 3.31: Konstrukt pOVG15. Dargestellt sind: Das den potentiellen Promoter tragende Fragment in gelb, das EGFP tragende in grün, der Terminatorbereich in blau und der Terminator selbst in rot. Für die Amplifikation des potentiellen gemeinsamen Promoter-Bereiches von ksuC/ksuB wurden die Oligos PorfYm1 und PorfYb2, als Template das Konstrukt pBRS2, verwendet. Das entstandene 527 Bp Fragment wurde nach BglII/Mph1103IRestriktion als 511 Bp großes Fragment mit dem ebenso geschnittenen Vektor pOVG1 ligiert. Vom entstandenen Konstrukt sollte die Synthese eines KsuC’-EGFP-Proteins bestehend aus den ersten 9 Aminosäuren von KsuC und nachfolgend dem EGFP-Protein erfolgen. Der Abstand zwischen dem Ende von orfI und dem Start von cmlR beträgt 236 Bp. Daher wurde für den Test der Synthese von CmlR nur ein relativ kurzer Abschnitt des intergenischen Bereiches und einem Teil von orfI amplifiziert und in pOVG1 eingesetzt. Das erhaltene Konstrukt erhielt den Namen pOVG12 und wurde nach Aufreinigung aus E.coli in S.lividans TK21 transformiert (Abb. 3.32). - 84 - Ergebnisse Abb. 3.32: Konstrukt pOVG12. Das den potentiellen Promoter von cmlR tragende Fragment ist in gelb dargestellt. Für die Amplifikation wurden die beiden Oligos PcmlRm1 und PcmlRb2 und als Template pWH297 gewählt. Das erhaltene Fragment, mit einer Größe von 366 Bp, wurde nach BglII/Mph1103I-Restriktion als 348 Bp langes Fragment mit pOVG1 ligiert. Das EGFP-Gen tragende Fragment ist in grün dargestellt, der Terminatorbereich in blau und der Terminator selbst in rot. Vom Konstrukt sollte die Produktion eines CmlR’EGFP-Proteins, bestehend aus den ersten neun Aminosäuren von CmlR und dem nachfolgenden EGFP-ProteinTeil, erfolgen. Der Vektor pOVG1 besitzt den verhältnismäßig kurzen, nur gering exprimierenden, potentiellen Promoter von ksuF aus pOVE3. Zum Test der Expression von ksuF wurde ein mit ca. 1 kBp deutlich größerer Abschnitt des vor ksuF liegenden Abschnittes amplifiziert und nach BglII/Mph1103I-Restriktion mit dem ebenso geschnittenen Vektor pOVG1 ligiert. Das Konstrukt, welches den Namen pOVG13 erhielt (Abb. 3.33), wurde aus E.coli aufgereinigt und in S.lividans TK21 transformiert. Abb. 3.33: Konstrukt pOVG13 Dargestellt ist in grün das EGFP-Gen tragende Fragment, in blau der den Terminator (in rot dargestellt) tragende Abschnitt und in gelb der potentielle Promoterbereich von ksuF. Der potentielle Promotorbereich wurde mit Hilfe der Oligos pksuFm1 und pksuFb2 vom Konstrukt pJO1 Ergebnisse - 85 - amplifiziert, das entstandene 1057 Bp große Fragment nach BglII/Mph1103I Restriktion als 1032 Bp großes Fragment mit pOVG1 ligiert. Das Konstrukt sollte die Synthese eines 249 Aminosäuren langen KsuF’-EGFPProteins ermöglichen, bestehend aus den ersten 10 Aminosäuren von KsuF und nachfolgend dem EGFPProteinteil. 3.5.2. „In vivo“ Nachweis der Synthese von EGFP (enhanced green fluorescent protein) Unter den getesteten Bedingungen (Minimal-Medium mit 300 mM KCl und Vollmedium mit Sucrose, jeweils mit 12,5 mg/l Thiostrepton) konnte bei 4 der 7 Transformanten eine Produktion von EGFP nachgewiesen werden (siehe Abb. 3.34 und Abb. 3.35). In Minimalmedium mit 300 mM KCl (Abb. 3.34) zeigt S.lividans pOVG13, das den PromoterBereich von ksuF trägt, die stärkste Fluoreszens. Es folgen, mit einer vergleichbaren Fluoreszenz, die Transformanten S.lividans pOVG14, das den Promoterbereich von ksuL und S.lividans pOVG15, welche den gemeinsamen Promoterbereich von ksuC und ksuB trägt; die schwächste Fluoreszens zeigt die pOVG1 Transformante. Für die S.lividans-Transformanten pOVG11, welches den Promoterbereich von kcsA und pOVG12, das den Promoterbereich des für das Chloramphenicol-Resistenz determinierende cmlR trägt, konnte, wie auch für die Negativ-Kontrolle, die pOVG17-Transformante, keine Synthese von EGFP gezeigt werden. Bei Wachstum in Vollmedium mit Sucrose (siehe Abb. 3.35) ist ebenfalls bei den pOVG13-, pOVG14-, pOVG15- und den pOVG1-Transformanten Fluoreszens nachzuweisen. Sie ist jedoch jeweils deutlich geringer, als bei Wachstum in Minimalmedium mit 300 mM KCl. Die stärkste Fluoreszens zeigt die pOVG14 Transformante, gefolgt von der pOVG15- und der pOVG13-Transformante. Auch hier ist die Fluoreszens der pOVG1 Transformante sehr gering. Das Konstrukt pOVG1 trägt wie pOVG13, den flussaufwärts gelegenen Bereich von ksuF, ist jedoch um ca. 660 Bp kürzer. Die pOVG11- und die pOVG17-Transformante, als Negativ-Kontrolle, zeigen keine Fluoreszens. Bei der pOVG12-Transformante ist bei Wachstum auf Jasenka-Medium mit Sucrose unter Umständen eine sehr schwache Fluoreszens zu erkennen, die jedoch, wenn tatsächlich vorhanden, nur unwesentlich stärker ist, als die der pOVG11- und pOVG17-Transformanten. Die Produktion von EGFP kann von pOVG12 (trägt den potentiellen Promoterbereich des für die Chloramphenicol-Resistenz determinierenden Gens cmlR) mit Hilfe von Chloramphenicol induziert werden (Abb. 3.36 a bis l). Die EGFP Produktion ist in den hier dargestellten Abbildungen bei Wachstum in Minimalmedium und Induktion mit Chloramphenicol (Abb. 3.36 d) höher, als bei Wachstum in Vollmedium (Abb. 3.36 j). Es finden jedoch auch Beispiele, bei denen die Synthese jeweils vergleichbar ist (nicht dargestellt). Die pOVG11 Transformante (trägt den potentiellen Promoter von kcsA) zeigt keine nachweisbare EGFPProduktion (nicht dargestellt). Das Wachstum der pOVG12 Transformante ist nach Induktion mit 5 mg/l Chloramphenicol gegenüber der Kontrolle und den weiteren pOVGTransformanten deutlich verlangsamt (nicht dargestellt). - 86 - Ergebnisse Abb. 3.34 a-n): Fluoreszensmikroskopie der S.lividans pOVG Transformanten bei Wachstum in MM40 Medium mit 300 mM KCl. Dargestellt ist jeweils links eine Aufnahme in Hellfeld, rechts jeweils die dazugehörige Aufnahme als Fluoreszenzbild. a) und b) pOVG1 Transformante, c) und d) pOVG11 Transformante, e) und f) pOVG12 Transformante, g) und h) pOVG13 Transformante, i) und j) pOVG14 Transformante, k) und l) pOVG15-Transformante und in m) und n) die pOVG17-Transformante als NegativKontrolle. Die Stämme wurden jeweils als Sporen in Jasenka-Medium mit Sucrose ca. 20 h inkubiert, 5 h geschüttelt und anschließend in MM40 Medium mit 300 mM KCl aufgenommen und für 3 h geschüttelt Belichtungszeit: 7000 ms, 40x Objektiv, Binning 1x1, der Schwarzwert wurde jeweils auf 67 gesetzt, der Weißwert auf 600. Ergebnisse - 87 - Abb. 3.35 a-n): Fluoreszensmikroskopie der S.lividans pOVG Transformanten bei Wachstum in JasenkaMedium mit Sucrose. Dargestellt ist jeweils links eine Aufnahme in Hellfeld, rechts jeweils die dazugehörige Aufnahme als Fluoreszenzbild. a) und b) pOVG1 Transformante, c) und d) pOVG11 Transformante, e) und f) pOVG12 Transformante, g) und h) pOVG13 Transformante, i) und j) pOVG14 Transformante, k) und l) pOVG15-Transformante und in m) und n) die pOVG17-Transformante als Negativ-Kontrolle. Die Stämme wurden jeweils als Sporen in Jasenka-Medium mit Sucrose ca. 20 h inkubiert und anschließend für 9 h geschüttelt. Belichtungszeit: 7000 ms, 40x Objektiv, Binning 1x1, der Schwarzwert wurde jeweils auf 67 gesetzt, der Weißwert auf 400. - 88 - Ergebnisse Abb. 3.36 a-l): EGFP Synthese der S.lividans TK21 pOVG12 Transformante unter CloramphenicolInduktion. Dargestellt ist jeweils links die Hellfeldaufnahme, rechts das dazugehörige Fluoreszensbild. Die Stämme wurden jeweils in MM40 Medium mit 300 mM KCl (in a bis h) oder in Jasenka Medium mit 10,3 % Sucrose angezogen. Die Induktion erfolgte jeweils mit 5 mg/l Chloramphenicol (in c, d, g, h und i bis l). a bis d, i und j): pOVG12 Transformante, e) bis h), k) und l): pOVG17 Transformante (Negativ-Kontrolle). Belichtungszeit: 7000 ms, 40x Objektiv, Binning 2x2, der Schwarzwert wurde jeweils auf 70 gesetzt, der Weißwert auf 700. 3.6. Charakteristika der Proteine 3.6.1. Klassifizierung des abgeleiteten Flavo-Proteins (KsuF) 327 Basenpaare stromaufwärts von kcsA findet sich in entgegengesetzter Flussrichtung ein offenes Leseraster, mit einer Länge von 1119 Bp. Es determiniert für ein Protein mit einer Länge von 373 Aminosäuren (siehe Anhang A) und erhielt den Namen ksuF bzw. KsuF für das hiefür determinierte Protein. Vor dem Startkodon findet sich eine potentielle ShineDalgarno-Sequenz. Hinter ksuF liegt ein Terminator, nachfolgend ksuL, das in entgegengesetzter Flußrichtung von ksuF lokalisiert ist (siehe Anhang A und B). KsuF gehört zur Familie der „Aromatic-ring hydroxylase“ (RNGMNOXGNASE, Acc.Nr.: IPR003042, auch als „Flavoprotein Monooxygenase“-Famile bezeichnet; Harayama, S., et al.; Ergebnisse - 89 - 1992), mit 1896 Mitgliedern, die sich sowohl bei Bakterien (1298 identifizierte Mitglieder), als auch bei Archaen (102 Mitglieder) und Eukaryoten (einschließlich von 136 Plastiddeterminierten: 496 Mitglieder) finden (Stand 2003, InterPro). Die Signatur dieser Familie wurde aus 30 Proteinsequenzen gebildet und besteht aus sechs kurzen Motiven (PR00420, Search PRINTS-S der Print Datenbank, 1999). Die meisten Mitglieder dieser Familie besitzen jedoch nur einen Teil der Motive, die häufigste Anzahl sind 2 Motive (PR00420, Search PRINTS-S der Print Datenbank, 1999). Bei KsuF finden sich 3 Motive (Motiv 1, 2 und 3), die jeweils an der Bindung des FAD-Cofaktors beteiligt (Abb. 3.37 a) und hoch konserviert sind. Die außerhalb dieser 6 Motive liegenden Bereiche sind jeweils hoch variabel. Die größte Untergruppe (Unterfamilie) der „Aromatic-ring hydroxylase“ zeichnet sich durch den Besitz der „Monooxygenase, FAD-binding“ Domäne („FAD_binding_3“, auch als „mOase_FAD_bd“ bezeichnet, Acc.Nr.: IPR002938, Harayama, S., et al.; 1992) aus. In diese ist die „Monooxygenase“ (single domain) Familie (pfam01360.11) aufgegangen. „FAD_binding_3“ ist in KsuF vollständig enthalten und erstreckt sich, bis auf einen kurzen Cterminalen Bereich, über die komplette Länge von KsuF (siehe Abb. 3.37 b). Von den 835 Mitgliedern, bei denen diese Domäne identifiziert wurde, finden sich 550 unter den Bakterien (davon 15 bei den Cyanobakterien), 8 bei den Archaeen und 277 bei den Eukaryoten (einschließlich 51 plastiddeterminierten) (ebenfalls Stand 2003, InterPro). Der Archetyp dieser Untergruppe ist die p-Hydroxybenzoat Hydroxylase (PHBH, EC 1.14.13.2), aus Pseudomonas aeruginosa und Pseudomonas fluorescens, das im Lignin-Degradationsweg involviert, durch Röntgenstrukturanalysen analysiert und am besten untersucht ist. Abb. 3.37 a-c): Homologe Bereiche von KsuF zu Erkennungssequenzen einer Familie und einer Domäne. Durch Flavin hervorgerufene Gelbfärbung des aufgereinigten Proteins. Dargestellt sind in a) die 3 gefundenen Erkennungsmotive der Familie der „Aromatic-ring hydroxylase“ (RNGMNOXGNASE) als grüne Kästchen ( ), die für die Bindung von FAD am Protein verantwortlich und hoch konserviert sind. Die Positionen sind: Aminosäure 4-26 für die erste konservierte Struktur, 147-162 für die zweite und 287-302 für die dritte. In b) ist der zu der „FAD_binding_3“-Domäne (Monooxygenase, FAD binding) homologe Bereich als blauer Kasten ( ) dargestellt, der sich von Aminosäure 2-348 von KsuF erstreckt. Das 373 Aminosäuren lange KsuF ist jeweils als schwarzer Balken dargestellt. In c) ist über Ni-NTA aufgereinigtes His-KsuF aus E.coli M15[pREP4] pQE314 (linkes Reaktionsgefäß) im Vergleich zum verwendeten Puffer (rechtes Reaktionsgefäß) gezeigt. Die Gelbfärbung, durch das nicht kovalent gebundene Flavin hervorgerufen, ist deutlich zu erkennen. - 90 - Ergebnisse Mitglieder der „FAD_binding_3“ Familie (im Weiteren als Flavoprotein Monooxygenasen bezeichnet) verwenden aromatische Verbindungen als Substrat und hydroxylieren diese. Hierbei wird NAD(P)H oxidiert, O2 reduziert, ein Sauerstoffatom wird in die Hydroxylgruppe eingeführt, das zweite als H2O freigesetzt (siehe Abb. 3.38). Abb. 3.38: Reaktionsschema der Flavoprotein Monooxygenasen Typische Substrate sind z.B. 4-Hydroxybenzoat (PobA, p-Hydroxybenzoat Hydroxylase (PHBH, EC 1.14.13.2) aus P.aeruginosa und P.fluorescens), 2-Hydroxybenzoat (Salicylat Hydroxylase, EC 1.14.13.1, z.B. aus P.fluorescens (NahG), und Pseudomonas stutzeri AN10 (NahW)) und Phenol (Phenol Hydroxylase (PHHY, EC 1.14.13.7) aus Trichosporon cutaneum). Sie sind auch beteiligt an der Inaktivierung von Tetracyklinen (TetX aus Bacteroides fragilis, zeigt bei Synthese in E.coli eine Tetracycline Resistenz), an der Synthese von Tetracyklinen (TcsC, Anhydrotetracyclin Oxygenase, z.B. aus Streptomyces aureofaciens) oder der Synthese von Ubichinon (UbiH, 2-Octaprenyl-6-methoxyphenol Hydroxylase aus E.coli). Bei den meisten Mitgliedern dieser Unterfamilie ist das Substrat jedoch unbekannt. Als Kofaktoren besitzen sie NADPH oder NADH, des Weiteren FAD, das nicht kovalent am Protein gebunden ist. Nukleotid-Kofaktoren binden an Proteine gewöhnlich über eine „Rossman fold“ Sekundärstruktur (Rao, S. T. and Rossman, M.G.; 1973). Sie findet sich im N-terminalen Teil und ist vorwiegend für die Bindung des Adenosin-Teils des FADs verantwortlich (β1α1β2α2β3-Sekundärstruktur, siehe Abb. 3.39). Flavoprotein Monooxygenasen sind somit Mitglieder des „Clans“ „FAD/NAD(P)-binding Rossmann fold Superfamily“. Die Bindung von NAD(P)H erfolgt bei Flavoprotein Monooxygenasen nicht über diese Sekundärstruktur. His-Tag KsuF zeigt nach Aufreinigung aus E.coli (Abb. 3.37 c) wie auch aus S.lividans (nicht dargestellt) eine starke Gelbfärbung, hervorgerufen durch das nicht kovalent gebundenen FAD. Drei Proteine der Flavoprotein Monooxygenasen wurden durch Röntgenstrukturanalysen aufgeklärt. Die Phenol Hydroxylase (EC 1.14.13.7) aus Trichosporon Cutaneum (1foh.pdb, Enroth, C., et al.; 1998), die Cholesterol Oxidase (EC 1.1.3.6) aus Brevibacterium sterolicum (1coy.pdb, Li, J., et al.; 1993) und die p-Hydroxybenzoat Hydroxylase (PHBH, EC 1.14.13.2) aus P.aeruginosa und P.fluorescens. KsuF zeigt zur Phenol Hydroxylase aus Trichosporon Ergebnisse - 91 - Cutaneum zu geringe Homologien, um eine Strukturaufklärung zu betreiben. Vor allem, da sich bei 1foh.pdb die Abschnitte zwischen den homologen Bereichen in der Größe zu stark unterscheiden. Die Cholesterol Oxidase zeigt (mit Ausnahme eines Motivs, „Rossman fold“) keine Ähnlichkeiten zu KsuF oder weiteren Mitgliedern der„FAD_binding_3“-Familie. Die Kristallisation der p-Hydroxybenzoat Hydroxylase ist jeweils mit dem Cofaktor FAD und dem Substrat p-Hydroxybenzoat (pOHB) gelungen. Der zweite Cofaktor, NADPH, konnte nicht zusammen mit dem Enzym kristallisiert werden; die Cokristallisation von NAD(P)H gelang auch nicht bei der Phenol Hydroxylase und Cholesterol Oxidase. Drei unterschiedliche Konformationen von PHBH sind kristallisiert, eine „in“ Konformation, in der das FAD in der Nähe des pOBH lokalisiert ist. Hierfür ist 1pbe.pdb (Schreuder, H.A., et al; 1989) aus P.fluoreszens der Archetyp. Eine „out“ Konformation, in der das FAD an der Oberfläche des Proteins und der Flavinteil des FAD gegenüber der „in“ Konformation vom Substrat entfernt, lokalisiert ist, hierfür wird 1dob.pdb aus P.aeruginosa (Gatti, D.L., et al; 1994) als Archetyp verwendet. Eine „open“ Konformation, in der das Substrat aufgenommen und das Produkt freigesetzt wird, 1k0j.pdb (Wang, J. et al; 2002) aus P.aeruginosa ist hierfür der Archetyp. In 1dob.pdb wurde Tyr 222 gegen Phe 222 (Y222F) ausgetauscht, so dass die „out“ Konformation erhalten wurde. In 1k0j.pdb wurde Arg 220 gegen Gln220 ausgetauscht, so dass die „open“ Konformation erhalten wurde, des Weiteren unterscheidet sich PHBH aus P.aeruginosa an 2 zusätzlichen Positionen von der aus P.fluoreszens (siehe Abb. 3.39). Trotz relativ geringer Homologien von 30,6 % (davon 19,6 % identische Aminosäuren zu Sequenz von 1dob und 1k0j und 20,1 % identische zu 1pbe) gelang es, für KsuF zu den pHydroxybenzoat Hydroxylasen ein Alignment zu erstellen (Abb. 3.39). Hieraus konnte sowohl für die „in“ als auch die „out“ Konfiguration jeweils eine putative Kristallstruktur generiert werden (Abb. 3.40 a-d). Zu der „open“ Konfiguration gelang dies nicht. Die höchsten Homologien finden sich in den drei primär an der Bindung von FAD beteiligten Abschnitten. Diese entsprechen den drei bei KsuF gefundenen Erkennungsmotiven für die „Aromatic-ring hydroxylase“ Familie (Abb. 3.37 a und Abb. 3.39). Die in PHBH an der Bindung von FAD, des Substrates und NADPH beteiligten Aminosäuren sind identifiziert und im Alignement eingetragen (Abb. 3.39). - 92 - Ergebnisse Abb. 3.39: Sequenzvergleich von KsuF mit der p-Hydroxybenzoat Hydroxylase. Dargestellt ist ein Alignment von KsuF mit der Sequenz von 1dob.pdb („out“-Konfiguration) und 1pbe.pdb („in“-Konfiguration). Die mit KsuF identischen Aminosäuren sind schwarz hinterlegt, die homologen grau. 1dob und 1pbe unterscheiden sich an drei Positionen, die in 1dob mutierte Aminosäure Y222F und die Aminosäuren an den Positionen 228 und 249. Diese sind rot umrandet. Die Sekundärstrukturen von 1dob und 1pbe und die im Vergleich hierzu ermittelten von KsuF sind unter dem Alignment dargestellt, s steht für β-sheet, h für α-helix. Die Lücken in den jeweiligen Sequenzen sind durch – markiert. Die Sekundärstrukturen der „out“ Konformation sind rot dargestellt, die der „in“-Konformation in grün. In PHBH wurden die für die Bindung des Substrates (pOBH), FAD und NADPH beteiligten Aminosäuren ermittelt. Die Positionen sind oberhalb des Alignments markiert, mit einem * markierte Aminosäuren sind beteiligt an der Bindung von FAD, die mit einem + markierten an der Bindung von NADPH und die mit einem ^ markierten an der Bindung des Substrates und dem Wasserstoffbrückennetzwerk. Die für die Bindung des Adenosin-Teils des FAD beteiligten β-sheet und α-helix Sekundärstrukturen (Rossman fold) sind eingetragen (β1 α1 β2 α2 β3). Ergebnisse - 93 - Abb. 3.40 a-d): Putative Kristallstrukturen von KsuF in der „in“ und „out“ Konformation. Dargestellt sind in a) und b) jeweils die potentiellen monomeren, in c) und d) die potentiellen dimeren Strukturen. In a) und c) ist KsuF in der „in“ Konfiguration, berechnet aus der Kristallstruktur der p-Hydroxybenzoat Hydroxylase aus P.fluoreszens (1pbe.pbd), in b) und d) in der „out“ Konfiguration, berechnet aus der Kristallstruktur der Y222F Mutante aus P.aeruginosa (1dob.pdb), abgebildet. Die zwei Untereinheiten des Dimers sind in grün und gelb, FAD ist allen vier Abbildungen in schwarz dargestellt. Die Sichtweise der in a) und b) dargestellten monomeren Strukturen entsprechen der Aufsicht der in c) und d) in grün dargestellten Untereinheit, das Molekül ist um 180 Grad gedreht. Berechnet wurden die Kristallstrukturen jeweils im „Alignement Interface“-Modus des SwissModel Servers, mit Hilfe des in Abb. 3.39 vorgestellten Alignements. Da keine Kristalldaten des potentiellen Dimers veröffentlicht worden sind, wurde die potentielle Dimer-Struktur jeweils von Hand modelliert. 3.6.2. Reinigung und Charakterisierung des Flavoproteins Nativ aus E.coli M15[pREP4] pQE314 und aus S.lividans TK21 pOVE33 über Ni-NTA aufgereinigtes His-Tag KsuF wurde über Gelfiltration zusammen mit GrößenstandardProteinen aufgetrennt (Abb. 3.41 a-d und Abb. 3.42 a-f). His-Tag KsuF kann jeweils in zwei Fraktionen nachgewiesen werden, die im Vergleich zu den Standard-Markerproteinen in der Größe dem Monomer und einem Multimer, mit einer Größe zwischen Dimer und Trimer, von His-Tag KsuF entsprechen. Auf Grund von fehlenden und unpassenden GrößenstandardProteinen war eine genauere Bestimmung, auch bei Verwendung einer anderen Gelmatrix - 94 - Ergebnisse (Superose 6), nicht möglich. Der His-Tag KsuF Flavin-Komplex kann für das, aus E.coli M15[pREP4] pQE314 aufgereinigte Protein, spektrophotometrisch nur in den gelbfarbigen, der Monomergröße entsprechenden Fraktionen, nachgewiesen werden (Abb. 3.41 d). In den Multimerfraktionen ist FAD nicht nachweisbar (Abb. 3.41 c). Der spektrophotometrische Nachweis des Flavins nach Aufreinigung aus S.lividans ist trotz geringer Synthese und der Verluste bei den nachfolgenden Aufreinigungsschritten gelungen. FAD findet sich ebenfalls nur in den Monomerfraktionen. Die für FAD typischen Absorptionsmaxima bei 375 nm und 450 nm sind, wenn auch nicht deutlich, zu erkennen (Abb. 3.42 f). In den Multimerfraktionen von His-Tag KsuF aus S.lividans TK21 pOVE33 sind zwei Proteine, mit der erwarteten Mobilität für His-Tag KsuF von 40,8 kDa und mit einer um ca. 2,5 kDa geringeren Mobilität, zu finden (Abb. 3.42 b, Spuren 1-3). Die Vermutung, dass neben His-Tag KsuF das chromosomal determinierte KsuF (theoretische Größe von 39,1 kDa) als Dimer coaufgereinigt worden ist, bestätigte sich nicht. Antikörper gegen den His-Tag zeigen Kreuzreaktionen gegen beide Proteine (Abb. 3.42 d, Spur 1). Das mit einer Mobilität von ca. 38-39 kDa laufende Protein ist wahrscheinlich ein C-terminales Abbauprodukt von His-Tag KsuF. In den das Multimer enthaltenen Fraktionen findet sich eine gleichmäßig exponentiell abfallende Absorption in Richtung der längerwelligen Bereiche. Dies ist ein Zeichen für Streuung, hervorgerufen durch unlösliches, ausgefallenes Protein (Abb. 3.41 c und Abb. 3.42 f). Durch Zentrifugation der Probe vor der Gelfiltration findet sich ein durch His-Tag KsuF gebildetes Proteinpellet, welches keine durch FAD hervorgerufene Gelbfärbung zeigt. Der Anteil des Multimers ist bei einer nachfolgenden Gelfiltration deutlich reduziert (nicht dargestellt). Bei Gelfiltration von His-Tag KsuF sind im Lauf-Bereich des monomeren Proteins zwei sich überlagernde Elutionsspitzen zu erkennen. In den Fraktionen derjenigen, die früher eluiert wird, kann Streuung nachgewiesen werden. Das FAD/His-Tag KsuF Verhältnis ist deutlich in Richtung der Proteinkonzentration verschoben (nicht dargestellt). Die nachgewiesene FAD-Konzentration stammt vermutlich von dem bei der Gelfiltration überlagernd laufenden monomeren FAD-tragendem His-Tag KsuF. Nach Zentrifugation der Streuung hervorrufenden Fraktionen des Multimers findet sich wieder ein von His-Tag KsuF gebildeter nicht gelber Proteinniederschlag. In den monomeren FAD-enthaltenden Fraktionen ist keine Streuung zu erkennen. Es findet sich nach Zentrifugation kein Niederschlag, His-Tag KsuF ist nur in der monomeren FAD-tragenden Form löslich. Ergebnisse - 95 - Abb. 3.41 a-d): Nachweis des Multimerisierungsgrades von His-Tag KsuF aus E.coli M15[pREP4] pQE314 und spektrophotometrischer Nachweis von FAD. His-Tag KsuF wurde zusammen mit Markerproteinen über Gelfiltration (Superdex75) aufgetrennt. In a) ist der Plot des Laufes dargestellt, in b) das mit Coomassie gefärbte Gel, auf dem Proben von 5 Fraktionen (sind entsprechend in a) mit der entsprechenden Zahl und mit für den entsprechenden Fraktionsbereich markiert) aufgetragen worden sind. Auf der rechten Seite der Abbildung sind die jeweiligen Proteine markiert (BSA, Albumin Bovine Serum, apparentes Molekulargewicht von 66 kDa; His-Tag KsuF mit einem theoretischen Molekulargewicht von 40,8 kDa; CA, Carbonic-Anhydrase, apparentes Molekulargewicht von 29 kDa; CyC, CytochromC mit apparentem Molekulargewicht von 12,4 kDa). In c) und d) ist das Spektrum von 2 Fraktionsbereichen dargestellt, die der Fraktion 1 (dargestellt in c) und 3 (dargestellt in d) aus Abb. a) entsprechen, allerdings aus einer Aufreinigung stammen, in der ca. die 30fache Menge eingesetzt wurde. Auf der X-Achse ist die Wellenlänge in nm aufgetragen, auf der Y-Achse die Absorption. c) Dargestellt ist in rot das Spektrum der potentiellen MultimerFraktion, in gelb FAD als Kontrolle. d) Dargestellt ist in rot das Spektrum der FAD tragenden Monomerfraktion, in blau das des errechneten FAD-Anteils (FAD tragende Monomeranteil minus Spektrum von BSA) und in gelb die FAD-Kontrolle. - 96 - Ergebnisse Abb. 3.42 a-f): Nachweis des Multimerisierungsgrades von His-Tag KsuF aus S.lividans TK21 pOVE33 und spektrophotometrischer Nachweis von FAD. His-Tag KsuF wurde zusammen mit Markerproteinen über Gelfiltration (Superdex75) aufgetrennt, in a) ist der Plot des Laufes, in b) und c) sind die über SDS-Page aufgetrennten Fraktionen dargestellt (Coomassie gefärbt). Die Laufhöhen von BSA (Albumin Bovine Serum, apparentes Molekulargewicht von 66 kDa), His-Tag KsuF (theoretisches Molekulargewicht von 40,8 kDa) und von CA (Carbonic Anhydrase, apparentes Molekulargewicht von 29 kDa) sind markiert. Die in den „Multimer“Fraktionen zu erkennenden 2 KsuF-Derivate sind mit ( ) markiert. Die Nummerierung der Spuren entspricht den mit ( ) markierten Fraktionsbereichen in a). Die im Plot a) gezeigte rechte Absorptionspitze wird von CytochromC (apparentes Molekulargewicht von 12,4 kDa) gebildet, wurde jedoch nicht in b) und c) aufgetragen. In d) und e) erfolgte der Nachweis von His-Tag KsuF nach Transfer auf Membranen mit Hilfe von Antikörpern gegen den His-Tag (jeweils Spur 1) und gegen KsuF (jeweils Spur 2). In d) wurde die Fraktion 2 aufgetragen, in e) die Fraktion 5. 8S ist jeweils der Protein-Größenstandard. Das Absorptionsspektrum der „Multimer“- (in rot) und der „Monomer“- (in blau) Fraktion ist in f) dargestellt, stammen allerdings aus einer Aufreinigung über Gelfiltration, in der eine größere Menge an His-Tag KsuF eingesetzt wurde. Die „Multimer“-Fraktion entspricht den Fraktionen 1-3, die „Monomer“-Fraktion der Fraktion 5. Auf der X-Achse ist die Wellenlänge in nm dargestellt, auf der Y-Achse die Absorption. Ergebnisse - 97 - Mitglieder der Flavoprotein Monooxygenasen weisen pro Untereinheit jeweils ein Flavin/FAD auf. Zur Untersuchung des FAD/KsuF-Verhältnisses des monomeren FADtragenden KsuF wurde die FAD-Konzentration spektrophotometrisch bei 450 nm bestimmt, die Proteinkonzentration mit Hilfe eines Proteintests (Abb. 3.43). Errechnet wurde ein Verhältnis von 0,9744 FAD pro KsuF-Untereinheit mit einer Standardabweichung σx von 0,0741. Die für die Berechnung verwendeten Werte sind im Anhang D dargestellt. Abb. 3.43: Darstellung des FAD/KsuF-Verhältnisses. Aus drei unterschiedlichen Anzuchten und Aufreinigungen von His-Tag KsuF aus von E.coli M15[pREP4] pQE314, wurde das FAD/KsuF-Verhältnis jeweils der zusammen 14 monomeren FAD tragenden Fraktionen bestimmt. In A, B und C ist jeweils das FAD/Proteinverhältnis der drei unterschiedlichen Aufreinigungen mit je 4, 5 und 5 gemessenen Fraktionen dargestellt, in D der Mittelwert der 14 Proben, jeweils als Balkengrafik (genaue Werte siehe Anhang D). Der maximale Streuungsbereich der Werte ist jeweils durch ein schwarzes (I) dargestellt, in D zusätzlich die Standardabweichung σx durch ein rotes (I). Die Proteinkonzentration wurde jeweils mit Hilfe des BCAProteintests bestimmt, die FAD-Konzentration durch Messung der Absorptionsspitze bei 450 nm im Vergleich zu FAD (zur Bestimmung der FAD-Konzentration wurde der erhaltene Wert mit dem Korrekturfaktor 1,097 (Palfey, B.A., et al, 2003) multipliziert, siehe auch: Materialen und Methoden, 2.6.7.). - 98 - Ergebnisse Zur Bestimmung des Kofaktors in KsuF wurde aufgereinigtes His-Tag KsuF Protein aus S.lividans pOVE33 und käuflich erworbenes FAD über HPLC aufgetrennt und anschließend massenspektrometrisch untersucht. Nach ca. 12,3 Minuten Elutionsdauer erschien jeweils in der Absorptionsmessung (nicht dargestellt) der HPLC eine Spitze, die in der massenspektrometrischen Analyse von einem Molekül mit einer Größe von 786,3 m/z bzw. 786,2 m/z gebildet wird (Abb. 3.44 a und e). Dies entspricht der erwarteten Größe von FAD mit einem Molekulargewicht von 785 MW + 1 Proton. Das FAD ist jeweils einfach positiv geladen. Dieses Molekül wurde jeweils massenspektrometrisch in einem MS2-Spektrum untersucht. Es wurden präferentiell zwei einfach geladene Moleküle mit 348 m/z (347 MW + 1 Proton) und 439 m/z bzw. 439,1 m/z (438 MW + 1 Proton) erhalten (Abb. 3.44 b und f). Zusammen ergeben die Molekulargewichte jeweils eine Größe von 787 m/z (785 MW + 2 Protonen), was der von FAD entspricht. Das 347 MW + 1 Proton Fragment von FAD besteht aus dem Adenosin Teil und einer Phosphatgruppe (AMP), das 437 MW + 1 Proton aus dem Flavin- und dem Ribitol-Teil und PO3H. Von beiden Molekülen wurde ein MS3 Spektrum erstellt. Die Analyse des Moleküls mit einer Größe von 348 m/z ergab ein Molekül mit 136 m/z (135 MW + 1 Proton, siehe Abb. 3.44 c und g), das dem einfach positiv geladenen Adenin entspricht. Die Analyse des 439 m/z Flavin-Ribitol-PO3H Teils des FAD ergab jeweils 5 identische Molkül-Bruchstücke, mit 297 m/z, 341,1 m/z, 359/359,1 m/z, 396/395,9 m/z und 421,1 m/z. Bei dem aus dem gekauften FAD fand sich zusätzlich noch ein Fragment von 295 m/z, bei dem aus dem gereinigten His-Tag KsuF aus S.lividans pOVE33 fanden sich drei weitere mit 242,9 m/z, 323.1 m/z und 403 m/z (Abb. 3.44 d und h). Das 242,9 m/z Fragment wird vermutlich vom Flavinteil gebildet, das 395 m/z Fragment vom Flavin-Ribitolteil. In dem aus E.coli M15[pREP4] pQE314 aufgereinigtem His-Tag KsuF konnte FAD, mit Hilfe von HPLC, durch ein identisches Laufverhalten mit der Kontrolle (käuflich erworbenen FAD), nachgewiesen werden (nicht dargestellt). Ergebnisse - 99 - Abb. 3.44 a-h): Massenspektrometrische Analyse von FAD. Dargestellt sind die MS- in a) und e), MS2- in b) und f) und je zwei MS3-Spektren in c), d), g) und h) von käuflich erworbenen FAD in a-d) und von FAD aus S.lividans aufgereinigtem His-Tag KsuF in e-h). Auf der Y-Achse ist jeweils die Intensität der Signale, auf der X-Achse der m/z-Wert (Molekulargewicht + Anzahl positiver Ladungen/Anzahl positiver Ladungen) dargestellt. Mit einem roten Quadrat sind die Moleküle jeweils markiert, die in den nachfolgenden MS-Spektren untersucht wurden. Mit einem grünen Quadrat ist in den MS2 und MS3 Spektren auf der X-Achse jeweils die Ursprungsgröße des untersuchten Moleküls abgebildet. In den beiden MS2-Spektren in b) und f) wurde jeweils das im MS-Spektrum gefundene Molekül mit einer m/z von 786 untersucht, das 348 m/z große Molekül wurde in den MS3-Spektren c) und g), das Molekül mit einer m/z von 439 in den MS3-Spektren in d) und h). Die Proben wurden vor den MS-Messungen über HPLC aufgetrennt (ThermoQuest 250 x 4,6 mm, 5 µm HyPURITY Elite C18). - 100 - Ergebnisse Für die Kristallisation von KsuF wurde der Stamm E.coli M15[pREP4] pQE314, wie im Methodenteil beschrieben, angezogen und His-Tag KsuF zweimal unter „nativen“ Bedingungen über Ni-NTA aufgereinigt. Nach Umpufferung erfolgte die Aufreinigung über FPLC (Superose 6 Gelfiltration). Nach Ankonzentration und Umpufferung der gelb gefärbten, Flavin tragenden monomeren Fraktion von His-Tag KsuF auf 25 mM Hepespuffer pH 7,5 wurde je 1 µl mit einer Proteinkonzentration von 15 µg/µl als Kristallisations-Ansatz für die 240 unterschiedlichen Puffer eines Kristallisations Screening Kits verwendet. Die erste Kontrolle nach 48-72 h zeigte, dass es in 238 zur Ausbildung eines Präzipitats gekommen war, zwei waren auf Grund von Pipettierfehlern misslungen. Auch bei weiteren Kontrollen nach ca. 3 und 9 Monaten konnte keine Kristallbildung festgestellt werden. Es gilt die Grundregel, dass, wenn in keinem oder in allen Testpuffern eines Kristallisationsansatzes das Protein präzipitiert, es aussichtslos sein wird, das Protein zu kristallisieren (A.Saito, persöhnliche Mitteilung). Auf weitere Versuche wurden verzichtet. 3.6.3. Analyse des extrazellulären Proteins KsuL 776 Bp hinter ksuB findet sich in identischer Leserichtung ein offenes Leseraster mit einer Länge von 1755 Bp, das für ein Protein mit einer Länge von 585 Aminosäuren determiniert, eine potentielle Shine-Dalgarno Sequenz ist zu identifizieren (siehe Anhang A). In der Zwischenzeit wurde dieses offene Leseraster in molS und MolS für das hierfür determinierte Protein umbenannt. In dieser Arbeit wird weiterhin der vorherige Name ksuL für das Gen und KsuL für das hierfür determinierende Protein verwendet. Hinter ksuL folgt in 133 Bp Abstand in entgegengesetzter Leserichtung ksuF. In dem intergenischen Bereich zwischen ksuL und KsuF findet sich ein Terminator (siehe Anhang A und B). KsuL besitzt im N-terminalen Bereich eine Signalsequenz von 39 Aminosäuren Länge. Dies konnte sowohl theoretisch durch Berechnung (SignalP 3.0 Server) mit Hilfe des „Neural Networks-“ (Bendtsen, J.D., et al; 2004; Nielsen, H., et al; 1997) und des „Hidden Markov-Modells“ (Nielsen, H. and Krogh, A.; 1998) gezeigt, als auch experimentell durch Ansequenzierung des aufgereinigten Proteins, nachgewiesen werden (pers. Mitteilung H.Schrempf). KsuL zeigt im C-terminalen Bereich Ähnlichkeiten zu der C-terminalen katalytisch aktiven Domäne von Mitgliedern der Lysyl-Oxidasen, auch als Protein Lysin 6-Oxidasen bezeichnet und zur katalytisch aktiven Domäne der Lysyl-Oxidase ähnlichen Proteinen (siehe Abb. 3.45). Diese gehören zur Amine Oxidase (AO) Familie, die sich in die FAD enthaltenden Amine Oxidase und in die Kupfer enthaltenden Semicarbizide sensitiven Amin Oxidasen (SSAO) aufteilen (Jalkanen, S. and Salmi, M.; 2001). Die Lysyl Oxidasen (EC 1.4.3.6) sind Mitglieder der SSAO-Familie und sind extrazellulär. SSAO’s besitzen gewöhnlich Topa-Quinone (TPQ) als Kofaktor (Klinman, J.P. and Mu,D.; 1994; Klinman, J.P.; 1996; Lyles, G.A.; 1996), Lysyl Oxidasen allerdings eher Lysin-Tyrosylquinon (Wang S.X., et al.; 1997). Die beiden hierfür benötigten Aminosäuren (Tyrosin und Lysin) sind auch in KsuL zu finden (siehe Abb. 3.45). Ergebnisse - 101 - SSAO’s sind dimere Glycoproteine mit einer durchschnittlichen Größe von 140-160 kDa (Klinman, J.P. and Mu, D.; 1994; Lyles, G.A.; 1996), die zwei Kupferatome pro Dimer enthalten; diese sind über Histidine an die SSAO’s gebunden (siehe Abb. 3.45). Lysyl Oxidasen sind an der Verknüpfung von Collagen- oder Elastin-Ketten untereinander involviert und führen folgende Reaktion durch: Peptidyl-L-Lysyl-Peptide + O2 + H2O = Peptidyl-Allysyl-Peptide + NH3 + H2O2 Im Folgenden kommt es zur Autokondensation der Peptidyl-Allysyl-Peptide, mit Ausbildung einer Matrix. In den letzten Jahren wurden weitere Funktionen der Lysyl Oxidasen und der Lysyl Oxidase ähnlichen Proteine vermutet. KsuL zeigt nur zu zwei Proteinen über die volle Länge Homologien, diese sind das vermutliche Pedant in S.avermitilis (BAC72171), mit einer Länge von 563 Aminosäuren und aus Nocardia sp. JS614 (ZP_00656873), mit einer Länge von 527 Aminosäuren, beide sind nicht weiter untersucht. Des Weiteren findet sich in der kürzlich veröffentlichten S.scabiesSequenz ein KsuL-Homolog mit einer vermutlichen Länge von 586 Aminosäuren (Basen: 4945557-4943764, http://www.sanger.ac.uk/Projects/S_scabies/). Ein Sequenzvergleich dieser drei Proteine mit KsuL ist im Anhang E dargestellt. Abb. 3.45: Sequenzvergleich des C-terminalen Teils von KsuL mit den entsprechenden Bereichen von 3 Lysyl Oxidasen und einem Lysyl Oxidase ähnlichen Protein. Dargestellt ist ein Mitglied der Lysyl Oxidase ähnlichen Proteine (Plo1 aus Perca flavescens, AAC79085, 819 Aminosäuren) und 3 Lysyl Oxidasen (LYOX_CHICK aus Gallus gallus, Q05063, 420 Aminosäuren; LYOX_MOUSE aus Mus musculus, NP_034858, 411 Aminosäuren und LYOX_HUMAN aus Homo sapiens, P283000, 417 Aminosäuren) im Vergleich zu KsuL (rot markiert). Die jeweils identischen Aminosäuren sind schwarz hinterlegt, die homologen mit grau. Die an der Ausbildung des Lysin-Tyrosylquinon beteiligten Lysin (K467) und Tyrosin (Y518) sind jeweils durch einen markiert, die drei an der Koordination des Cu-Ions beteiligten Histidine durch . - 102 - Ergebnisse KsuL kann in den Überständen der Medien in drei Formen nachgewiesen werden: Als Protein mit einer Laufhöhe von ca. 60 kDa (Abb. 3.46 Spur 5) und/oder in 2 Fragmenten von ca. 31 und 26 kDa (siehe Abb. 3.46 Spuren 2 und 3). In Stämmen, die nicht KsuL tragen, wie S.lividans NI pOV001 (Abb. 3.46 Spur 1) oder der KsuL Deletionsmutante S.lividans ∆L17 (Abb. 3.46 Spur 4), ist KsuL nicht nachweisbar. Das Protein mit der Laufhöhe von ca. 60 kDa entspricht in der Größe dem „reifen“ KsuL ohne Signalsequenz von 57,8 kDa, die beiden kleineren Signale dem N-terminalen- (ohne Signalsequenz) und C-terminalen-Fragment von 32,1 kDa und 25,7 kDa. Der Nachweis erfolgte durch Aufreinigung und Ansequenzierung der jeweiligen Fragmente in unserer Arbeitsgruppe (pers. Mitteilung H.Schrempf). Bei Synthese in S.lividans NI, die ksuL tragende Konstrukte besitzen, finden sich nur die N-terminalen oder C-terminalen Fragmente. In S.lividans WT kann nur das vollständige Protein (ohne Signalsequenz) nachgewiesen werden (wie hier dargestellt), es finden sich aber auch Synthesen, in denen sich das „reife“ Protein und die N- und C-terminalen Fragmente, nur die N- und C-terminalen Fragmente oder nur das N-terminale Fragment (siehe Abb. 3.64 Spuren 1-7) nachweisen lassen. Eigene Versuche zeigten, dass vor allem das C-terminale Fragment von KsuL sehr schnell abgebaut wurde. Abb. 3.46: Synthese von KsuL. Aufgetragen wurden jeweils mit Amoniumsulfat gefällte Überstände nach Kultivierung in MM40 Medium mit 300 mM KCl. Spur 1: S.lividans NI pOV001, Spur 2: S.lividans NI pOV004, Spur 3: S.lividans NI pOV005, Spur 4: KsuL Deletionsmutante S.lividans ∆ksuL20 und Spur 5: S.lividans WT. 8S: Proteinstandard. Die drei Pfeile markieren jeweils die Laufhöhen der Proteine. Diese liegen bei ungefähr 60 kDa für den Wildtyp und bei 30 kDa und 26 kDa für die beiden S.lividans NI-Stämme mit den Konstrukten pOV004 und pOV005. Die aufgetragenen Mengen entsprechen jeweils ca. 20 ml mit 90 % Ammoniumsulfat gefälltem Kulturüberstand. Das mit einer Laufhöhe von ca. 23 kDa zu erkennende Signal ist unspezifisch. Die Herstellung der ksuL-Deletionsmutante wird weiter unten besprochen. Ergebnisse - 103 - 3.6.4. Analyse der Aldo-Keto Reduktasen KsuC und KsuB Nach einem 1082 Bp langen intergenischen Bereich finden sich 2 offenen Leseraster mit einer Länge von 855 Bp und 930 Bp, getrennt durch einen intergenischen Bereich von 55 Bp. Diese erhielten die Namen ksuC und ksuB bzw. KsuC und KsuB für die hierfür determinierenden 285 und 310 Aminosäuren lange Proteine. Hinter ksuB folgt in derselben Leserichtung, in einem Abstand von 744 Bp, ksuL. Sowohl vor ksuC, als auch ksuB, kann jeweils eine potentielle Shine-Dalgarno Sequenz identifiziert werden (siehe Anhang A). In Sequenzvergleichen zeigt sich, dass sowohl KsuC, als auch KsuB Mitglieder der Aldo-Keto Reduktase Familie sind (IPR001395, Aldo/ket_red). Zu dieser werden mehr als 1900 Mitglieder gezählt (Stand 2003, InterPro, EMBL-EBI). Sie werden jeweils von verschiedenen Servern und Datenbanken hierin eingeteilt (Abb. 3.47 a und b). Abb. 3.47 a und b): Homologien von KsuB und KsuC zur Familie der Aldo-Keto Reduktasen. In a) sind jeweils die homologen Bereiche von KsuB, in b) von KsuC, zur Familie der Aldo Keto Reduktase Superfamilie, identifiziert jeweils mit unterschiedlichen Programmen verschiedener Server, dargestellt. In rot sind die identifizierten Bereiche mit Hilfe des Prodom-Servers (Aldo/Keto_red, PD000288), in grün die kurzen Signalabschnitte mit Hilfe der Print S Datenbank (ALDKETRDTASE, PR00069) und in gelb die Bereiche mit Hilfe der Pfam-Datenbank (aldo_ket_red, PF00248), zu erkennen. Das 310 Aminosäuren lange KsuB in a) und das 285 Aminosäuren lange KsuC in c) ist jeweils als schwarzer Strich in der entsprechenden Länge dargestellt. Eine Unterfamilie der Aldo-Keto Reduktasen sind die tetrameren β-Untereinheiten der Kaliumkanäle, die im N-terminalen Teil eine Tetramerisierungsdomäne (T1) besitzen. Diese wird jedoch weder bei KsuC noch bei KsuB gefunden. Die Mitglieder der Aldo-Keto Reduktase Familie sind vorwiegend monomere Proteine (Jez, J.M. and Penning, T.M.; 2001), die überwiegend NADP weniger NAD als Cofaktor verwenden, dieser bindet an eine Rossman-Fold Struktur. Es finden sich aber auch dimere Mitglieder der Familie, wie die - 104 - Ergebnisse Ratten Leber Aflatoxin Dialdehyde Reductase (AKR7A1, Pdb: 1Gve Chain a und b). AldoKeto Reduktasen sind Oxidoreductasen. Zu diesen gehören z.B. Aldehyd Reduktasen, Aldose Reduktasen, Prostaglandin F Synthasen, Xylose Reduktasen und Rho Crystallin. Sie besitzen eine Länge von ca. 320 Aminosäuren und ein durchschnittliches Molekulargewicht von ca. 36 kDa (El-Kabbani, O., et al, 1998), KsuB besitzt ein theoretisches MW von 34,3 kDa, KsuC von 30 kDa. Mitglieder der Aldo-Keto Reduktase Familie zeichnen sich durch den Besitz eines parallelen β-8/α-8-Fasses (TIM Barrel; Farber, G.K. and Petsko, G.A.; 1990) als Tertiärstruktur aus (Abb. 3.48 und 3.49). Die Strukturen verschiedener Mitglieder der Familie konnten durch Röntgen-Strukturanalysen aufgeklärt werden. Zu Mitgliedern mit bekannter Struktur dieser Familie, zeigen sowohl KsuB als auch KsuC die höchsten Ähnlichkeiten zu einer potentiellen Aldo-Keto Reduktase aus E.coli (YDHF, Pdb:1og6 Chain a-c und 1ur3 Chain M, unterscheidet sich von 1og6 durch den Besitz eines N-Terminalen His-Tag, keine Cokristallisation von NADP). 1og6 wurde als Trimer kristallisiert. Mit Hilfe eines Sequenzvergleiches (Abb. 3.48) gelang es, sowohl für KsuB, als auch für KsuC, eine potentielle Tertiär- und Quartärstruktur gegen YDHF (Pdb:1og6 Chain a-c) über die komplette Proteinlänge zu errechnen (Abb.3.49 a-d). Die Kristallisation von 1og6 Chain a-c erfolgte mit dem Cofaktor NADP (siehe Abb. 3.49 a-d). Die an der Bindung von NADPH beteiligten Aminosäuren sind unter den Mitgliedern der Aldo-Keto Reduktase Familie konserviert. In Sequenzvergleichen mit der Aldehyd Reductase ALR1 (Pdb: 1ae4), Aldose Reduktase ALR2 (Pdb: 1mar) und einem durch einen Fibroblast Wachstums Faktor induzierten Protein FR-1 (Pdb:1frb), konnten die entsprechenden Aminosäuren identifiziert werden (Abb. 3.48). KsuB zeigt mit 50 % ähnlichen und identischen Aminosäuren, davon 36 % identischen, eine relativ hohe Ähnlichkeit zu YDHF; KsuC zeigt eine geringere Ähnlichkeit zu YDHF mit 33 %, davon 23 % identischen. Vergleichbar hoch ist die Ähnlichkeit von KsuB zu KsuC mit 33 %, davon 21 % identischen Aminosäuren (Abb. 3.48). KsuB unterscheidet sich von KsuC und YDHF durch das Vorhandensein einer postulierten zusätzlichen antiparallelen β/βFaltblattstruktur (Aminosäuren 186-202 von KsuB). Diese liegt zwischen der sechsten Faltblatt- und Helixstruktur des Tim-Barrels (Abb. 3.48 und Abb. 3.49 a und c). Ergebnisse - 105 - Abb. 3.48: Sequenzvergleicht von YDHF gegen KsuB und KsuC und Sekundärstrukturen der Proteine. Das Alignment wurde mit Hilfe des Programmes „Multalin“ (Corpet, F.; 1988) erstellt und anschließend von Hand optimiert. Verglichen wurde YDHF (putative Aldo-Keto Reductase aus E.coli, Pdb: 1og6 Chain a) mit KsuB und KsuC aus S.lividans. Identische Aminosäuren sind schwarz hinterlegt, homologe in grau. Unterhalb des Sequenzvergleiches ist die bekannte Sekundärstruktur für YDHF (Pdb: 1og6 Chain a) in rot, die aus dem Sequenzvergleich berechnete für KsuB in grün und für KsuC in blau, dargestellt. α-helicale Sekundärstrukturen sind als h dargestellt, β-Faltblattstrukturen als s, Lückenbereiche jeweils durch einen Bindestrich -. Die an der Bindung von NADPH beteiligten Aminosäuren sind unter den Mitgliedern der Aldo-Keto Reduktase Familie konserviert. Oberhalb des Sequenzvergleiches sind in gelb die an der Bindung von NADPH beteiligten und identifizierten Aminosäuren aus ALR1, ALR2 und FR-1 eingetragen. ALR1 unterscheidet sich von ALR2 und FR-1 an Position 272 (bei YDHF, als Z für Glx dargestellt). Bei ALR1 findet sich Glutamin, bei ALR2 und FR-1 ein Glutamat an der entsprechenden Position. - 106 - Ergebnisse Abb. 3.49 a-d): Putative Kristallstrukturen von KsuB und KsuC. Die Berechnung der putativen Kristallstrukturen erfolgte jeweils gegen YDHF, eine putative Aldo-Keto Reduktase aus E.coli (PDB: 1og6). Verwendet wurde das in Abb. 3.9 dargestellte Alignement von KsuB, KsuC und YDHF. Die Berechnung erfolgte mit Hilfe des Swiss Model Server des SIB Biozentrums Basel im „Alignment Interface“ (Schwede, T., et al; 2003). Gezeigt sind jeweils die Tertiärstrukturen in der „Backbone“ Konfiguration. a) Dreidimensionale Struktur von KsuB, in b) von KsuC, jeweils errechnet gegen 1og6 „Chain a“. c) und d) Putative Quartärstruktur von KsuB in c) und KsuC in d). Die aus 1og6 „Chain a“ errechnete Struktur ist in gelb dargestellt, die von „Chain b“ in grün und von „Chain c“ in blau. Strukturen, die nicht mit 1og6 homolog waren und errechnet wurden, sind rot markiert. NADPH ist jeweils in braun dargestellt. Ergebnisse - 107 - 3.6.5. Analyse des abgeleiteten ORFI-Proteins Hinter kcsA folgt in einem 86 Bp Abstand ein offenes Leseraster, mit einer Länge von 603 Bp. Es erhielt den Namen orfI und ORFI, für das hierfür determinierte Protein, mit einer Länge von 201 Aminosäuren. Hinter orfI folgt in gleicher Leserichtung das für die Cloramphenicol Resistenz CmlR determinierende cmlR (Dittrich, W., et al; 1991), in einem Abstand von 238 Bp. Vor orfI ist eine potentielle Shine-Dalgarno Region gelegen (siehe Anhang A). OrfI zeigt Homologien zu bakteriellen Deaminasen/Reduktasen, die am zweiten und dritten Schritt der Riboflavinsynthese aus GTP beteiligt sind (Richter, G., et al.; 1997). Beispiele für Mitglieder dieser Familie sind RibD aus E.coli und RibG aus B.subtilis, die in einem ersten Schritt den Pyrimidinring von 2,5-Deamino-6-ribosylamino-4(3H)-pyrimidone 5’-Phosphat (Substrat II) zu 5-Amino-6-ribosylamino-2,4(1H,3H)-pyriminedione 5’-Phosphat (Substrat III) deaminieren und anschließend in einer Reductase-Reaktion an der RibosylSeitenkette Substrat III zu 5-Amino-6-ribitylamino-2,4(1H,3H)-pyriminedione 5’-Phosphat (Substrat V) reduzieren. Bakterielle Deaminasen/Reduktasen bestehen aus 2 Domänen, einer N-terminalen Deaminase-Domäne (CMP_dCMP_Zn_bd, IPR002125), die eine ZincBindedomäne besitzen und einer C-terminalen Reductase-Domäne (RibDG_C, IPR002734), die NAD(P)H als Cofaktor verwenden. OrfI besteht nur aus der C-terminalen ReductaseDomäne (Abb. 3.50 a), die N-terminale Deaminase-Domäne ist nicht vorhanden. Nur eine RibDG_C-Domäne findet sich z.B. auch bei Hefen. Hier führt RIB7 in einem ersten Schritt eine Reductase-Reaktion des Substrates II zu 2,5-Diamino-6-ribitylamino-4(3H)-pyrimidone 5’Phosphat (Substrat IV) durch, anschließend RIB2 die Deaminierung zu Substrat V. Des Weiteren zeigt OrfI über fast die komplette Länge (siehe Abb. 3.50 b) Ähnlichkeiten zur Dihydrofolat Reductase Superfamilie (DHFR, SSF53597). Dihydrofolat Reductasen katalysieren die Reduktion des Kofaktors Dihydrofolat zu Tetrahydrofolat, das an der de novo Synthese von Glycin, Purinen und Deoxythymidin Phosphat beteiligt ist. Die Ähnlichkeiten zu Proteinen, mit nachgewiesener Funktion der bakteriellen Deaminasen/Reduktasen und Dihydrofolat Reductasen, sind relativ gering. Abb. 3.50 a und b): Homologien von OrfI zu Erkennungssequenzen einer Domäne und einer Superfamilie. Das 201 Aminosäuren lange Protein OrfI ist jeweils als schwarzer Strich dargestellt. - 108 - Ergebnisse 3.7. Disruption verschiedener Gene in S.lividans 3.7.1. Auswahl der Strategien zur Disruption von Genen Zur Etablierung der Disruption (Unterbrechung) von Genen in S.lividans sollte ein System neu in unserer Arbeitsgruppe eingeführt werden, um eine Reihe von Genen (kcsA, ksuF, orfI, ksuL, ksuB und ksuC), in der Nähe von kcsA gelegen, jeweils durch eine AntibiotikaResistenzkassette (bis auf einem kurzen 5’ und 3’ Bereich zu ersetzen). Die Inaktivierung der Gene sollte über eine „Doppel-Rekombination“ erfolgen. Hierbei musste die Antibiotikakassette, flankiert von den kurzen 5’ und 3’ Bereichen der entsprechenden Gene, über homologe Rekombination integriert werden; die homologen Bereiche besaßen jeweils eine Länge zwischen 700 Bp und 1 kBp. Vier grundsätzliche Methoden, das für die Rekombination benötigte Fragment zuzuführen, werden bei Arbeiten mit Streptomyceten-Stämmen angewendet. Dies sind die Verwendung von nicht replikativen Plasmiden oder Fragmenten, Plasmide mit dem von Natur aus Temperatur sensitiven Replikon von pSG5 aus Streptomyces ghanaensis (Muth, G., et al.; 1995), Derivate des Phagen-Vektors ΦC31 (Malpartida, F. and Hopwood D.A.; 1986) oder von Vektoren mit instabilen Replikons. In einer ersten Versuchsreihe wurde versucht, mit Hilfe von nicht replikativen Plasmiden und Fragmenten, die Disruptionen zu erreichen. Verschiedene, jeweils die Antibiotika Resistenz mit den flankierenden homologen Bereichen tragende Konstrukte wurden verwendet. Sie wurden über Transformation oder Konjugation in S.lividans transferiert. Unter Selektionsdruck sollten nur die Transformanten überleben, die das Gen für die Antibiotikaresistenz ins Chromosom integriert hatten. So wurde ein konjugative E.coli/Streptomyceten Vektorsystem verwendet. Die vollständigen Kassetten wurden auch als pUC-, pBR322- und M13-Derivate in S.lividans 66 WT oder den Plasmid-reduzierten Mutanten TK21 oder TK24 transformiert, sowohl als doppelsträngige Derivate, als auch in einzelsträngiger Form (nach Denaturierung oder bei M13-Derivaten nach Aufreinigung), da einzelsträngige DNA eine 10 bis 100fach höhere Transformationseffizens in Streptomyceten bieten soll (Hillemann D., et al.; 1993). Da einige Streptomyceten-Stämme methylierungsspezifische Restriktionsenzyme besitzen, hierzu zählt z.B. auch Streptomyces coelicolor ATCC 10147 (Mueller) und Streptomyces avermitilis, kann die Transformationshäufigkeit der Konstrukte durch vorherige Passage über einen methylierungsdefizienten E.coli-Stamm deutlich erhöht werden (MacNeil, D.J.; 1988). Ein relativ schwaches methylierungsspezifisches Restriktionssystem besitzt S.lividans TK21. Dieser unterscheidet sich von S.lividans 66 durch das Fehlen der beiden Plasmide SLP2 und SLP3. Die Transformationshäufigkeit kann hier nur geringfügig erhöht werden (MacNeil, D.J.; 1988). Zur Aufreinigung von nicht methylierter Plasmid-DNA wurde der Dam- und Dcm- Stamm E.coli JM110 verwendet. Ergebnisse - 109 - Des Weiteren wurde versucht, die Resistenzkassetten mit den flankierenden homologen Bereichen sowohl als doppelsträngiges als auch als einzelsträngiges Fragment zu transformieren, sowohl als PCR-Fragment, als auch nach Restriktion und Aufreinigung. Bei den verschiedenen Versuchen wurden viele potentielle Disruptionsmutanten erhalten, die sich als resistent gegen Thiostrepton oder Kanamycin erwiesen. Von mehr als 350 wurde die chromosomale DNA aufgereinigt und mit Hilfe von Hybridisierungen auf mögliche Integration der Resistenzgene ins Chromosom untersucht, was jedoch in keinem Fall gelang. Es lag jeweils eine spontan entstandene Resistenz vor. Sowohl die Methoden als auch die gewählten Resistenzen erwiesen sich als ungeeignet. 3.7.2. Disruption der Gene in mit Hilfe des Vektors pGM160 Da alle Versuche, mit Hilfe von in Streptomyceten nicht replizierenden Plasmiden („suicide“Vektoren) und isolierten Fragmenten Disruptionen einzuführen, erfolglos blieben, wurde versucht, diese mit Hilfe von Konstrukten, die Temperatur-sensitive Replikons besitzen, zu generieren. Verwendet wurde der 7,78 kBp große Streptomyces/E.coli bifunktionelle („shuttle“) Vektor pGM160, der einen Temperatur-sensitiven Streptomyceten pSG5-Replicon und einen E.coli pUC Plasmid-Teil besitzt. Als Selektionsgene in Streptomyceten trägt er Thiostrepton- (tsr) und Gentamicin- (aacC1) Resistenzgene, für E.coli eine Ampicillin- (bla)-Resistenz. Um die entsprechenden chromosomalen Gene durch ein Hygromycin- (hygR) Resistenzgen zu ersetzen, wurde aus pGM160 das aacC1-Gen ausgeschnitten und hygR, beidseitig flankiert von einem ca. 700 Bp großen DNA-Abschnitt aus dem jeweiligen Anfangs- und Endbereich der zu ersetzenden Gene, eingesetzt. Nach Transformation in S.lividans und Wachstum auf 37°C sollten nur die Kolonien wachsen können, die die Hygromycin-Resistenz chromosomal integriert hatten (siehe Abb. 3.51). - 110 - Ergebnisse Abb. 3.51: . Rekombinationsschema. Die jeweils homologen Bereiche des Plasmides und des Chromosoms sind in grau dargestellt, das Ω-hyg-Fragment ist in weiß dargestellt. Die Leserichtung des Resistenzgens ist immer identisch mit dem des zu unterbrechenden Gens. Ebenfalls in weiß dargestellt ist das ThiostreptonResistenzgen, welches auf dem Vektorteil liegt. Die Ω-hyg-Kassette wurde so zwischen den homologen Bereichen eingesetzt, dass nach Rekombination ins Chromosom das zu unterbrechende Gen bis auf einen kurzen 5’- und 3’-Rest vollständig ersetzt wurde. Die Integration ins Chromosom kann in einem einzigen „doublecrossing-over“-Fall eintreten oder über 2 „single-crossing-over“-Fälle. Dargestellt ist nur das „Double-crossover“ Endprodukt. Der jeweilige Phänotyp ist auf der linken Seite dargestellt. 3.7.3. Disruption von kcsA Mit Hilfe der PCR wurden vom flußaufwärts und flussabwärts gelegenen Bereich von kcsA ca. 700 Bp lange Fragmente synthetisiert. Nach HindIII/SnaBI-Restriktion des N-terminal gelegenen und SnaBI/KpnI Restriktion des C-terminal gelegenen Fragmentes, wurden beide zugleich mit einem HindIII/KpnI geschnittenen Vektor pUC18 ligiert (pTSK, Abb. 3.52). In diesen wurde nach SnaBI-Restriktion („blunt“) die DraI („blunt“) geschnittene HygromycinResistenzkassette aus pHP45Ωhyg eingefügt. Von den beiden möglichen Endkonstrukten wurde nur das weiterverwendet, das hygR in Flussrichtung von kcsA integriert hatte (pUTSK1H, Abb. 3.52). Die komplette Kassette mit den N- und C-terminalen Fragmenten und der dazwischen liegenden Resistenzkassete wurde mit HindIII/NcoI ausgeschnitten und in den ebenso geschnittenen Vektor pGM160 eingefügt. Das Plasmid erhielt den Namen pGMK (Abb. 3.52). Dieses wurde in S.lividans 66 WT transformiert und anschließend die Transformanten auf potentielle Disruptionsmutanten selektiert. Ergebnisse - 111 - Abb. 3.52: pTSK, pUTSK1H und pGMK. Der flußaufwärts gelegene Bereich ist in rot dargestellt (verwendete Oligos: PrkcsAIHin und PrkcsAISna, Fragmentlänge: 745 Bp, nach Restriktion mit HindIII und SnaBI 725 Bp), der flussabwärts gelegene Bereich in gelb (PrkcsAIISna und PrkcsAIIKpNc, Fragmentgröße 716 Bp, nach KpnI/SnaBI- Restriktion 696 Bp), das hygR tragende Ω-hyg-Fragment in grün. Für die Amplifikationen wurde pOV005 als Template und die Pfu-Polymerase verwendet. Die chromosomalen DNA’s der potentiellen Mutanten wurden mit Hilfe von Hybridisierung getestet (Abb. 3.54 a und b). 3.7.4. Disruption von ksuF Die Disruption von ksuF wurde, wie im Abschnitt 3.7.3 für die Disruption von kcsA beschrieben, durchgeführt. Die flussaufwärts und flussabwärts liegenden Bereich von ksuF wurden gleichzeitig mit dem Vektor pUC18 ligiert und in E.coli transformiert. In dieses pTSF benannte Konstrukt (Abb. 3.53) wurde das Ω-hyg-Fragment eingefügt und in E.coli transformiert. Weiter Verwendung fand nur das der beiden möglichen Konstrukte, bei dem das Ω-hyg-Fragment in Flussrichtung von ksuF integriert war (pUTS1H, Abb. 3.53). Das komplette Insert wurde nach Restriktion mit dem Vektor pGM160 ligiert und erhielt den Namen pGMF (Abb. 3.53). Nach Aufreinigung aus E.coli und Transformation in S.lividans 66 WT erfolgte die Selektion auf mögliche Disruptionsmutanten. - 112 - Ergebnisse Abb. 3.53 a-c): Konstrukte pTSF, pUTSF1H und pGMF. In rot dargestellt ist der amplifizierte flussaufwärts gelegene Bereich von ksuF (Oligos: PrksuFIHin und PrksuFISna, Fragmentgröße: 778 Bp, nach Restriktion 758 Bp), in gelb jeweils der flussabwärts gelegene Bereich (Oligos PrksuFIISna und PrksuFKpNc, Fragmentgröße 792 Bp, nach Restriktion 772 Bp). Das Ω-hyg-tragende Fragment ist in grün dargestellt. Die korrekte Integration des Hygromycin-Resistenzgens in die chromosomale DNA wurde durch Hybridisierungen untersucht (Abb. 3.54 a und b). Abb. 3.54 a und b): Nachweis der Disruption von ksuF und kcsA. Die Tests erfolgten mit Sonden gegen das ksuF und kcsA tragende Fragment (Membran a) und gegen das Hygromycin-Resistenzgen (Membran b). Alle DNA’s wurden mit NcoI/PvuI geschnitten. Die Membran aus a) wurde nach Abwaschen der Sonden („Dehybridisierung“) für die zweite Hybridisierung in b) wieder verwendet. Aufgetragen wurden auf den ersten 4 Spuren potentielle ksuF-Mutanten (Spur 1: ∆ksuF2, Spur 2: ∆ksuF5, Spur 3: ∆ksuF6 und Spur 4: ∆ksuF7), auf den weiteren 5 potentielle kcsA-Mutanten (Spur 5: ∆kcsA9, Spur 6: ∆kcsA10, Spur 7: ∆kcsA11, Spur 8: ∆kcsA13 und Spur 9: ∆kcsA14). Auf Spur 10 wurde S.lividans WT-DNA, auf Spur 11 Dig-markierter MolekularGewichts-Marker III, Plasmid pHP45Ωhyg auf Spur 12 und auf Spur 13 unmarkierter λ-PstI-Marker aufgetragen. Ergebnisse - 113 - Bei drei der potentiellen ksuF- (∆ksuF2, ∆ksuF6 und ∆ksuF7) und drei der kcsA-Mutanten (∆kcsA9, ∆kcsA13 und ∆kcsA14) wurden Hybridisierungen der erwarteten Mobilität (Größe von 4354 Bp für die ksuF- und 5187 Bp für die kcsA-Mutanten) gefunden, mit einer deutlich geringeren, gegenüber der Hybridisierung für den Wildtyp (Größe von 3357 Bp, Abb. 3.54, Membran a, Spur 10). Diese wurden weiterverwendet. 3.7.5. Disruption von ksuL Auch die Disruption von ksuL wurde, wie für die Disruption von kcsA (Abschnitt 3.7.3) beschrieben, durchgeführt. Das pUC18-Derivat, in das die flussabwärts und flussaufwärts gelegenen Bereiche von ksuL integriert wurde, erhielt den Namen pTSL (Abb. 3.55). Das nachfolgende Derivat, mit der in Flussrichtung integrierten Ω-hyg-Kassette, erhielt den Namen pUTSL1H (Abb. 3.55) und das pGM160 Konstrukt, in welches anschließend das vollständige Insert eingefügt wurde, den Namen pGML (Abb. 3.55). Die Selektion auf potentielle Disruptionsmutanten erfolgte nach Transformation von pGML in S.lividans WT, die Kontrolle der korrekten Integration durch Hybridisierung (Abb. 3.56 a und b). Abb. 3.55: Konstrukte pTSL, pUTSL1H und pGML Der flußaufwärts gelegene amplifizierte Bereich von ksuL (verwendete Oligos: PrksuLIHin und PrksuLISna, Fragmentgröße 762 Bp nach HindIII/SnaBI-Restriktion 746 Bp) ist in rot dargestellt, in gelb, der flussabwärts gelegene Bereich (Oligos PrksuLIISna und PrksuLIIKpNc, 739 Bp, 723 Bp nach SnaBI/KpnI-Restriktion) und in grün das Ω-hyg-Fragment. Die Hybridisierungen gegen die gespaltenen DNAs der 5 ∆ksuL-Mutanten erschienen in Mobilität der erwarteten Größe von 4145 Bp (Abb. 3.56 a, Spur 1-5), mit einer deutlich geringeren Mobilität als die WT-DNA mit einer erwarteten Größe von 3315 Bp (Spur 6). Die Hybridisierung von ∆ksuL23 scheint bei einer geringfügig geringeren Größe zu laufen als die vier anderen Mutanten. Diese wurde nicht weiterverwendet. - 114 - Ergebnisse Abb. 3.56 a und b): Nachweis der Disruption von ksuL. Getestet wurde mit Sonden gegen a): das ksuL tragende DNA-Fragment und b): das Hygromycin-Resistenzgen. Die DNA’s wurden mit BglII/SgfI geschnitten. Die in a) verwendete Membran wurde „dehybridisiert“ und für die Hybridisierung gegen das Hygromycinresistenzgen in b) verwendet. Aufgetragen wurden 5 verschiedene potentielle ∆ksuL-Mutanten: Spur 1: ∆ksuL17, 2: ∆ksuL20, 3: ∆ksuL21, 4: ∆ksuL23 und 5: ∆ksuL24, S.lividans WT-DNA (Spur: 6), Dig-markierter Molekular-Gewichts-Marker III (Spur 7) und Plasmid pHP45Ωhyg (Spur 8). Auf Spur 9 ist unmarkierter λ-PstI Marker aufgetragen. 3.7.6. Disruption von orfI Die Disruption von orfI erfolgte wie für kcsA (siehe Abschnitt 3.7.3) beschrieben; das pUC18-Konstrukt, das die beiden flußaufwärts und flussabwärts gelegenen Bereich von orfI trägt, erhielt den Namen pTSO (Abb. 3.57). In dieses wurde anschließend die Ω-hyg-Kassette eingefügt. Aus dem Konstrukt, das das Resistenzgen in Leserichtung integriert hatte (pUTSO1H, Abb. 3.57), wurde das komplette Insert ausgeschnitten, in pGM160 eingefügt und in E.coli transformiert. Ergebnisse - 115 - Abb. 3.57: Konstrukte pTSO, pUTSO1H und pGMO Der amplifizierte flussaufwärts liegende Bereich ist in rot dargestellt (Oligos: ProrfIHin und ProrfIISna, Fragment-Größe 748 Bp, nach HindIII/SnaBI-Restriktion 731 Bp), der flussabwärts liegende amplifizierte Bereich in gelb (Oligos: ProrfIISna und ProrfIIKpNc; Größe des Fragmentes 609 Bp, nach Restriktion 593 Bp) und der des Ω-hyg-Fragmentes in grün. Dieses pGMO benannte Konstrukt (Abb. 3.57) wurde in S.lividans WT transformiert und nachfolgend auf Mutanten selektiert. Der Nachweis der Integration erfolgte durch Hybridisierungen gegen die chromosomalen DNA’s (Abb. 3.58 a-c). Abb. 3.58 a-c): Hybridisierungen, Nachweis der Disruption von orfI. Getestet wurde mit Sonden gegen das Hygromycin-Resistenzgen in a), das orfI tragende DNA-Fragment in b) und gegen das ThiostreptonResistenzgen in c). Die DNA’s wurden jeweils mit BamHI/BglII restringiert. Es wurde immer dieselbe Membran verwendet, die jeweils „dehybridisiert“ und neu hybridiesiert wurde. Aufgetragen wurden 6 verschiedene potentielle ∆orfIMutanten: Spur 1: ∆orfI33, 2: ∆orfI34, 3: ∆orfI35, 4: ∆orfI36, 5: ∆orfI37 und 6: ∆orfI39. Des Weiteren wurde S.lividans WT-DNA (Spur: 7), Plasmid pOV005 (Spur 8), Plasmid pGMO (Spur 9), ein unmarkierter λPstIMarker (Spur 10) und Dig-markierter Molekular-Gewichts-Marker III aufgetragen (Spur 11). - 116 - Ergebnisse Bei den Disruptionsmutanten ∆orfI35, ∆orfI36, ∆orfI37 und ∆orfI39 (Abb. 3.58 a und b, Spuren 3-6) finden sich die Hybridisierungen in Höhe der erwarteten Mobilität (Größe von 6981 Bp). Die Hybridisierung gegen WT-DNA zeigt die erwartet größere Mobilität (Größe von 4995 Bp, Abb. 3.58 b, Spur 7). Die beiden potentiellen Mutanten ∆orfI33 und ∆orfI34 sind nicht korrekt. Bei ∆orfI34 ist es zu keiner Rekombination des Plasmids pGMO mit dem Chromosom gekommen, es findet sich sowohl eine Hybridisierung in Mobilität des Plasmides (Abb. 3.58 a und c, Spur 2) als auch auf der Mobilität der chromosomalen WT-DNA (Abb. 3.58 b, Spur 2). Weiter verwendet wurden die 4 korrekten Mutanten. 3.7.7. Disruption von ksuB Das ksuB Gen wurde wie für kcsA beschrieben, unterbrochen (Abschnitt 3.7.3). Die amplifizierten flussaufwärts und flussabwärts gelegenen Fragmente wurden nach Restriktion mit pUC18 ligiert und in E.coli transformiert. In dieses pTSB benannte Konstrukt wurde die Ω-hyg-Kassette eingefügt und in E.coli transformiert (Abb. 3.59). Aus dem, das Hygromycin Resistenz-Gen in Flussrichtung tragende Konstrukt pUTSB1H (Abb.3.59), wurde das Insert ausgeschnitten und mit pGM160 ligiert. Nach Transformation und Aufreinigung aus E.coli erfolgte die Transformation des Konstruktes pGMB (Abb. 3.59) in S.lividans WT, nachfolgend die Selektion auf mögliche Disruptanten. Abb. 3.59: Konstrukte pTSB, pUTSB1H und pGMB In rot dargestellt ist der flußaufwärts gelegene amplifizierte Bereich von ksuB (verwendete Oligos: PrksuBIHin und PrksuBISna, Fragmentgröße 755 Bp nach HindIII/SnaBI-Restriktion), in gelb der flussabwärts gelegene (Oligos PrksuBIISna und PrksuBIIKpNc, 742 BP nach SnaBI/KpnI-Restriktion) und in grün das Ω-hyg-Fragment. Der Nachweis der korrekten Integration ins Chromosom erfolgte durch Hybridisierungen (Abb. 3.60 a-c). Ergebnisse - 117 - Abb. 3.60 a-c): Hybridisierungen, Nachweis der Disruption von ksuB. Getestet wurde mit Sonden gegen das Hygromycin-Resistenzgen a), das ksuB tragende DNA-Fragment b) und gegen das Thiostrepton-Resistenzgen c). Die DNA’s wurden jeweils mit BglII/KpnI restringiert. Jeweils dieselbe Membran wurde verwendet, die „dehybridisiert“ und neu hybridisiert wurde. Es wurden 12 verschiedene potentielle ∆ksuB-Mutanten getestet: Spur 1: ∆ksuB21, Spur 2: ∆ksuB22, Spur 3: ∆ksuB23, Spur 4 ∆ksuB24, Spur 5: ∆ksuB25, Spur 6: ∆ksuB26, Spur 7: ∆ksuB27, Spur 8: ∆ksuB28, Spur 9: ∆ksuB29, Spur 10: ∆ksuB30, Spur 11: ∆ksuB31 und ∆ksuB32 auf Spur 12. Zur Kontrolle wurden auf Spur 13 S.lividans WT-DNA, Plasmid pGMB auf Spur 14, ksuL-Disruptionsmutante ∆ksuL21 auf Spur 15 und auf Spur 16 Dig-markierter Molekular-Gewichts-Marker III aufgetragen. Bei neun der zwölf potentiellen Disruptionsmutanten finden sich Hybridisierungen in Mobilität der erwarteten Größe von 6383 Bp, bei einer deutlich geringeren Mobilität als die Hybridisierung für den Wildtyp bei einer erwarteten Größe von 5393 Bp zeigt (Abb. 3.60 b, Spur 13) und geringfügig geringer als die Hybridisierung der ksuL-Disruptionsmutante (6222 Bp, Abb. 3.60 a und b, Spur 15). Drei Mutanten sind nicht korrekt, ∆ksuB23 (Spur 3), ∆ksuB27 (Spur 7) und ∆ksuB30 (Spur 10). Bei ∆ksuB30 wurde die Hygromycin-Kassette vermutlich korrekt ins Chromosom integriert, jedoch eine weitere Hybridisierung in Mobilität des Plasmides lässt auf eine heterogene Population schließen (Abb. 3.60 a und c, Spur 10). Diese drei Mutanten wurden nicht weiterverwendet. - 118 - Ergebnisse 3.7.8. Disruption von ksuC Die Disruption von ksuC erfolgte, wie in Abschnitt 3.7.3 für kcsA beschrieben. Das pUC18Derivat, in das die flussabwärts und flussaufwärts gelegenen Bereiche von ksuB integriert wurden, erhielt den Namen pTSC, das nachfolgende Derivat mit der in Flussrichtung integrierten Ω-hyg-Kassette den Namen pUTSC1H, das pGM160 Konstrukt, in das das vollständige Insert eingefügt wurde, den Namen pGMC (Abb. 3.61). Nach Transformation von pGMC in S.lividans WT erfolgte die Selektion auf potentielle Disruptionsmutanten. Zwanzig potentielle Mutanten wurden anschließend mit Hilfe von Hybridisierungen getestet (Abb. 3.62 a-f). Abb. 3.61 a-c): Konstrukte pTSC, pUTSC1H und pGMC Der amplifizierte flussaufwärts liegende Bereich ist in rot dargestellt (Oligos: PrksucIHin und PrksuCISna, Fragment Größe 748 Bp, nach HindIII/SnaBI-Restriktion 731 Bp), der flussabwärts liegende amplifizierte Bereich in gelb (Oligos: PrksuCIISna und PrksuCIIKpNc, Größe des Fragmentes 609 Bp, nach SnaBI/KpnI-Restriktion 593 Bp) und der des Ω-hyg-Fragmentes in grün. Zwölf der zwanzig potentiellen Mutanten scheinen das Hygromycin-Resistenzgen korrekt integriert zu haben (∆ksuC1, ∆ksuC3, ∆ksuC4, ∆ksuC5, ∆ksuC6, ∆ksuC7, ∆ksuC8, ∆ksuC11, ∆ksuC12, ∆ksuC15, ∆ksuC19 und ∆ksuC20, Abb. 3.62 a bis f) Hier finden sich Hybridisierungen im Mobilitätsbereich der erwarteten 8746 Bp Größe, bei einer deutlich geringeren Mobilität als bei der Hybridisierung gegen die Wildtyp-DNA (5393 Bp, Abb. 3.62 b und e, Spur 11). Ergebnisse - 119 - Abb. 3.62 a-f): Nachweis der Disruption von ksuC: Getestet wurde mit Sonden gegen das HygromycinResistenzgen (Membranen a und d), gegen das ksuC tragende Fragment (Membranen b und e) und gegen das Thiostrepton Resistenzgen (Membranen c und f). Insgesamt 20 verschiedene potentielle Mutanten wurden gestestet. Alle chromosomalen DNA’s wurden mit BglII/KpnI restringiert. Für die Hybridisierungen in a-c) und in d-f) wurde jeweils eine Membran verwendet, die „dehybridisiert“ und neu hybridisiert wurde. Aufgetragen wurde auf den Membranen a-c): 10 potentielle ksuC-Mutanten, Spur 1: ∆ksuC1, Spur 2: ∆ksuC2, Spur 3: ∆ksuC3, Spur 4: ∆ksuC4, Spur 5: ∆ksuC5, Spur 6: ∆ksuC6, Spur 7: ∆ksuC7, Spur 8: ∆ksuC8, Spur 9: ∆ksuC9 und Spur 10: ∆ksuC10. Auf Spur 11 wurde S.lividans WT-DNA aufgetragen, auf 12 die ksuLDisruptionsmutante ∆ksuL21, auf 13 das Plasmid pGMB und auf 14 Dig-markierter Molekular-Gewichts-Marker III. Auch auf den Membranen d-f) wurden auf den 10 ersten Spuren potentielle ksuC-Disruptionsmutanten aufgetragen: Spur 1: ∆ksuC11, Spur 2: ∆ksuC12, Spur 3: ∆ksuC13, Spur 4: ∆ksuC14, Spur 5: ∆ksuC15, Spur 6: ∆ksuC16, Spur 7: ∆ksuC17, Spur 8: ∆ksuC18, Spur 9: ∆ksuC19 und Spur 10: ∆ksuC20. Auf Spur 11 wurde S.lividans WT-DNA, auf Spur 12 Plasmid pGMC, auf 13 die ksuL-Disruptionsmutante ∆ksuL21 und auf 14 Digmarkierter Molekular-Gewichts-Marker III aufgetragen. Versuche, die Mutanten und den S.lividans WT, mit Hilfe entsprechender Konstrukte (pOV004, pOV005, pOV006 und pOV010) zu komplementieren, waren nicht erfolgreich. Die inserierten Fragmente rekombinierten jeweils mit den homologen Bereichen des Chromosoms (nicht dargestellt). Nach mehrmaligem Ausstreichen konnten jeweils nur Konstrukte der Größe des „Leervektors“ pOV001 nachgewiesen werden. Ein Transformationsversuch mit dem Konstrukt pOV008 zur Komplementierung der ∆ksuL Mutante ist anscheinend vor kurzem gelungen (pers. Mitteilung H.Schrempf); pOV008 trägt nach pOV002 das zweitkürzeste Fragment (3,2 kBp) aller pOV Konstrukte. - 120 - Ergebnisse 3.8. Charakteristika der Mutanten 3.8.1. Wachstumseigenschaften Auf Sporulationsmedium (RSM-Agarose Platten) zeigen die Disruptionsmutanten keinen Unterschied im Wachstum und der Differenzierung zum Wildtyp (nicht dargestellt). Auxotrophien konnten nicht gefunden werden (nicht dargestellt). Hierzu wurden jeweils die Deletionsmutanten von ksuB, ksuC, ksuL, ksuF, kcsA und orfI auf Minimalmedium (MM40) ausgestrichen und im Wachstum mit dem Wildtyp (S.lividans 66) verglichen. 3.8.2. Immunologischer Nachweis der relevanten Proteine Zum Test, ob die Deletion einzelner Gene Auswirkungen auf die Protein-Synthese, determiniert von benachbarten Genen, zeigt (KsuB, KsuC, KsuF und KcsA), wurden die Disruptionsstämme, S.lividans 66 WT und der auf dem Konstrukt pOV005 ein 13 kBp Fragment mit den zu untersuchenden Genen tragende Stamm S.lividans NI, in Minimalmedium und in Minimalmedium mit 400 mM KCl angezogen. Die Produktion der Proteine wurde mit Hilfe von Antikörpern, nach Transfer auf Membranen, nachgewiesen. KsuB kann in allen Stämmen, außer in den Deletionsstämmen S.lividans ∆ksuB und ∆ksuC (Abb. 3.63 a, Spuren 1-4) nachgewiesen werden. Das durch einen grünen Pfeil markierte Doppel-Signal mit einer Migration bei ca. 50 kDa ist unspezifisch und kann auch bei S.lividans ∆ksuB identifiziert werden. Es ist jedoch zu schwach um hier erkennbar zu sein (Abb. 3.63 a, Spur 1 und 2). Die Synthese von KsuC erfolgt in allen Stämmen außer der ksuC Deletionsmutante (siehe Abb. 3.63 c und d). Die Deletion von ksuC zeigt einen polaren Effekt auf die Produktion von KsuB. Dieses erklärt sich dadurch, dass eine Co-Transkription beider Gene abgeleitet werden kann. Die Synthese von KsuF kann außer beim Stamm S.lividans NI, der das Konstrukt pOV005 trägt, nicht nachgewiesen werden (siehe Abb. 3.63 e und f). KcsA konnte weder in den Membranfraktionen der Disruptionsstämme noch bei S.lividans NI pOV005 nachgewiesen werden (nicht dargestellt). Ergebnisse - 121 - Abb. 3.63 a-f): Nachweis von polaren Effekten der Disruptionsmutanten. Getestet wurden jeweils die Cytoplasma-Fraktionen von sechs verschiedenen Disruptionsmutanten, von S.lividans WT und von S.lividans NI pOV005, jeweils nach Anzucht in MM40 Medium ohne zusätzlich Salz (ungrade Nummern) und MM40 Medium mit 400 mM KCl (grade Nummern). Getestet wurde jeweils mit Antikörpern gegen KsuB (a und b), KsuC (c und d) und gegen KsuF (e und f). Aufgetragen wurden jeweils die Cytoplasma-Fraktionen auf den Membranen a), c) und e) von: Spuren 1 und 2: S.lividans ∆ksuB, Spuren 3 und 4: S.lividans ∆ksuC, Spuren 5 und 6: S.lividans ∆ksuF, Spuren 7 und 8: S.lividans WT. Auf den Membranen b), d) und f) wurden jeweils die Cytoplasmafraktionen von: Spuren 1 und 2: S.lividans ∆kcsA, Spuren 3 und 4: S.lividans ∆ksuL, Spuren 5 und 6: S.lividans ∆orfI und Spuren 7 und 8: S.lividans NI pOV005 aufgetragen. 8S ist ein Proteinstandard. Die Laufhöhe von KsuB, KsuC und KsuF ist jeweils auf der rechten Seite durch einen schwarzen Pfeil markiert. - 122 - Ergebnisse Der Test der KsuL Produktion erfolgte nach Ammoniumsulfatfällung der Zellkultur nach Wachstum in Minimalmedium mit 300 mM KCl (siehe Abb. 3.64). Die Synthese von KsuL kann in allen Disruptionsmutanten und dem Wildtyp nachgewiesen werden, außer der ksuL Deletionsmutante (Abb. 3.64 Spur 3). Die KsuL-Produktion ist in den ∆ksuC und ∆ksuB Deletionsmutante (Abb. 3.64 Spuren 1 und 2) gegenüber der ∆ksuF, ∆kcsA und ∆orfI Deletionsmutante und dem Wildtyp (Spuren 4-7), reduziert. ksuL liegt, durch einen 776 Bp langen intergenischen Bereich getrennt, hinter ksuB und ksuC in gleicher Leserichtung, vermutlich auf einem gemeinsamen Operon. Vor ksuL findet sich ein weiterer Transkriptionsstart. Abb. 3.64: Nachweis polarer Effekte auf die Synthese von KsuL. Aufgetragen wurden die mit Ammoniumsulfat gefällten Kulturüberstände der 6 Deletionsmutanten, vom Wildtyp und von S.lividans NI pOV005, nach Wachstum in M40 Medium mit 300 mM KCl. Nach Transfer auf eine Membran erfolgte der Nachweis mit Hilfe von Antikörpern gegen KsuL. Spur 1: ksuB-, Spur 2: ksuC-, Spur 3: ksuL-, Spur 4: ksuF-, Spur 5: kcsA- und Spur 6: orfI-Deletionsmutante. Auf Spur 7 wurde S.lividans WT aufgetragen und auf Spur 8: S.lividans NI pOV005. 8S: Proteinstandard. Der Kontrast des Proteinstandards wurde zur besseren Erkennbarkeit erhöht. Die auf Spur 8 aufgetragene Probe ist dieselbe Probe, die auch für Abb. 3.46, Spur 3 verwendet wurde. Auf den Spuren 1, 2, 4-7 ist jeweils nur das mit einem Pfeil markierte 32,1 kDa große Nterminale Fragment zu identifizieren, das C-terminale Fragment ist vermutlich abgebaut (ich bitte die schlechte Bildqualität zu entschuldigen). Polare Auswirkungen der Disruptionsmutanten (im Vergleich zum Wildtyp) auf die Synthese der durch cmlR determinierten Cloramphenicol-Resistenz wurde durch Wachstum bei drei verschiedenen Chloramphenicol-Konzentrationen (ohne, 5 und 10 µg/ml) durch Bestimmung des Nassgewichtes untersucht. Es konnten jeweils keine Unterschiede im Wachstum festgestellt werden (nicht dargestellt). Ergebnisse - 123 - 3.8.3. Analyse der Pigment-Produktion Undecylprodigiosin ist ein Mycel assoziiertes rotfarbiges Antibiotikum (siehe Einleitung), das von S.lividans WT unter bestimmten Bedingungen produziert wird. Getestet wurden die 6 Disruptionsmutanten S.lividans ∆kcsA, ∆ksuB, ∆ksuC, ∆orfI, ∆ksuL und ∆ksuF im Vergleich zu S.lividans WT bei Wachstum auf einer Minimalmedium-Agaroseplatte (siehe Abb. 3.65). Die Disruptionsmutanten S.lividans ∆ksuB und ∆ksuC zeigen im Vergleich zu den restlichen Mutanten und dem Wildtyp eine verstärkte Produktion an Undecylprodigiosin, erkennbar an der Rotfärbung. Undecylprodigiosin-Produktion findet sich bei allen anderen Stämmen, anders als bei den beiden überproduzierenden Mutanten, vor allem im Randbereich der Austriche, zu einem späteren Zeitpunkt auch auf der gesamten Ausstrichsfläche, jedoch mit einer deutlich geringeren Konzentration. Auch bei Wachstum in MM40 Flüssig-Medium ist eine verstärkte RED-Produktion der ∆ksuB und ∆ksuC Mutanten im Vergleich zum Wildtyp und den weiteren Mutanten zu beobachten. Die Synthese der ∆ksuB Mutante ist deutlich höher als die der ∆ksuC Mutante (nicht dargestellt). Abb. 3.65: Undecylprodigiosin-Produktion der Disruptionsstämme und S.lividans 66. Die Stämme wurden auf einer Minimalmedium-Platte (MM40) ausgestrichen und nach ca. 4,5 Tagen fotografisch protokolliert. Zur Verdeutlichung der Unterschiede ist der Kontrast erhöht worden. 1: S.lividans Wildttyp, 2: S.lividans ∆kcsA, 3: S.lividans ∆ksuB, 4: S.lividans ∆ksuC, 5: S.lividans ∆orfI, 6: S.lividans ∆ksuL und 7: S.lividans ∆ksuF. Abgebildet ist die Rückseite der Platte. Streptomyceten produzieren, „A-factor“ genannte, extrazelluläre γ-Butyrollactone als Signalstoffe, die, wie zuerst für S.griseus nachgewiesen (Khokhlov, A.S., et al.; 1967, Mori, K.; 1983), Einfluss auf die Antibiotika-Produktion und teilweise auf die Differenzierung der Zellen besitzen. S.coelicolor A3(2) produziert mehrere γ-Butyrolactone wovon SCB1 genauer - 124 - Ergebnisse charakterisiert ist (Kawabuchi, M., et al.; 1997), SCB1 hat eine negative regulatorische Funktion auf die Antibiotika-Produktion. Zum Test, ob SCB1, weitere diffundierende Signalmoleküle oder ausgeschiedenen Zwischenprodukte einen Einfluss auf die verstärkte Undecylprodigiosin-Produktion der ∆ksuC und ∆ksuB Mutanten besitzen, wurden diese zusammen mit dem S.lividans Wildtyp ausgestrichen (siehe Abb. 3.66). Es konnte kein Einfluss nachgewiesen werden. Abb. 3.66: Einfluss möglicher Signalmoleküle auf die Undecylprodigiosin Produktion. Ausgestrichen auf MM40 Minimalmedium wurden die beiden S.lividans ∆ksuC- (oben links) und ∆ksuB-Mutanten (unten links) und auf der rechten Seite zweimal der Wildtyp S.lividans 66. Dargestellt ist die Rückseite der Platte. Die beiden Gene ksuC und ksuB werden vermutlich von einer polycistronischen mRNA transkribiert, ksuC ist hierbei vor ksuB lokalisiert. Die Disruption von ksuC verhindert auch die Produktion von KsuB (siehe Abb. 3.63 a, Spuren 3 und 4), während die Disruption von ksuB keinen Effekt auf die Synthese von KsuC zeigt (siehe Abb. 3.63 c, Spuren 1 und 2). Die Disruption der Gene erfolgte jeweils durch die Insertion von hygR einschließlich eines Terminators in derselben Leserichtung wie ksuC und ksuB. Die Transkription von hygR ist durch die Gabe von Hygromycin induzierbar. Um zu identifizieren, ob nur das Fehlen von KsuB oder KsuC/KsuB zusammen für die erhöhte Undecylprodigiosin-Produktion verantwortlich ist, wurden die beiden Disruptionsmutanten, zusammen mit den ∆orfI und ∆ksuF Disruptionsmutanten, die keinen Unterschied zum Wildtyp zeigen, auf Minimalmedium mit Hygromycin ausgestrichen, in der Hoffnung, dass hinter hygR liegende Terminator der ∆ksuC Mutante teilweise überlesen und die Produktion von KsuB ermöglichen würde (siehe Abb. 3.67). Die ∆ksuC Mutante zeigt eine, gegenüber der ∆orfI und ∆ksuF Mutante, leicht erhöhte Undecylprodigiosin (RED)-Produktion. Die RED-Produktion der ∆ksuB-Mutante ist gegenüber der Produktion der ∆ksuC Mutante jedoch deutlich erhöht, Ergebnisse - 125 - auch ist die RED-Produktion deutlich früher nachweisbar (ca. 4-6 h). Dies deutet auf das erhoffte teilweise „durchlesen“ des Terminators der ∆ksuC Mutante hin. Somit kann angenommen werden, dass der beobachtete Phänotyp der Mutanten nur durch die nichtProduktion von KsuB verursacht wird und unabhängig von KsuC ist. Abb. 3.67: Undecylprodigiosin-Produktion der ∆ksuC und ∆ksuB Mutanten auf Minimalmedium mit Hygromycin. Ausgestrichen wurden die ∆ksuB (oben links), ∆ksuC (unten links), ∆orfI (oben rechts) und ∆ksuF (unten rechts) Mutanten auf Minimalmedium (MM40) mit 10 mg/l Hygromycin. Abgebildet ist der Zustand nach ca. 44 h Wachstum bei 30°C, dargestellt ist die Rückseite Platte. Verschiedene Supplemente beeinflussen die Undecylprodigiosinproduktion. So ist bekannt, dass Prolin, das ein Grundbaustein der Synthese ist, die Phosphat-Konzentration und hohe NaCl-Konzentrationen Auswirkungen auf die Synthese zeigen. Des Weiteren wurden die für die Synthese, neben dem Grundbaustein Prolin, benötigten Substrate Serin und Glycin und die Auswirkung hoher KCl- und Sucrose-Konzentrationen getestet, ebenso das Wachstum auf Mannitol und Casaaminosäuren. Die Ergebnisse sind in Abb. 3.68 a bis l dargestellt und in der Tabelle 3.1 zusammengefasst. Die Produktion von Undecylprodigiosin (RED) ist abhängig von der Wachstumsphase. Unter Bedingungen, unter denen keine Ausbildung von Luftmycel erfolgt (z.B. Minimalmedium mit 20 % Sucrose oder mit 0,1 g/l KH2PO4, nicht bildlich dargestellt und Minimalmedium mit Casaaminosäuren und Casaaminosäuren + Mannitol, siehe Abb. 3.68 i und j), ist keine Antibiotikaproduktion erkennbar. Die Ausbildung des Luftmycels begann jeweils nach ca. 40 h. Bei Zugabe von 300 mM NaCl oder 10 % Sucrose ist die Antibiotikaproduktion anscheinend schon während der transienten - 126 - Ergebnisse Phase, zwischen der vegetativen und der Vermehrungsphase, nachweisbar (Abb. 3.68 e und g). Ansonsten ist sie frühestens mit Beginn der Luftmycelbildung erkennbar. Bei Wachstum auf Minimalmedium mit 10 g/l KH2PO4 statt 1 g/l erscheint nach ca. 48-51 h das deutlich erkennbare, diffundierende, violette Antibiotika Actinorhodin (Abb. 3.68 l). Die Actinorhodin Produktion ist zu einem späteren Zeitpunkt (nach ca. 6 Tagen) auch bei Wachstum auf Minimalmedium, Minimalmedium mit nur 0,2 g/l KH2PO4 und auf Minimalmedium mit Mannitol oder Prolin nachweisbar. Es ist jeweils kein Unterschied der Actinorhodin Produktion der Mutanten zum Wildtyp feststellbar. Abb. 3.68 a-l): Undecylprodigiosinproduktion der ∆ksuB und ∆ksuC Mutanten im Vergleich zum Wildtyp. Getestet wurde jeweils die ∆ksuB- (jeweils links oben) und die ∆ksuC-Mutante (jeweils links unten), des Weiteren zweimal S.lividans 66 Wildtyp (jeweils rechte Seite). Gezeigt ist der Zustand nach ca. 65 h Wachstum bei 30°C. Ausgestrichen wurde jeweils auf MM40 Minimalmedium in a), mit zusätzlich 1 g/l Prolin (8,69 mM) in b), mit zusätzlich 1 g/l Glycin (13,3 mM) in c), zusätzlich 1 g/l Serin (9,5 mM) in d), mit zusätzlich 300 mM NaCl in e), mit zusätzlich 300 mM KCl in f), zusätzlich 10 % Sucrose (300 mM) in g), mit 10 g/l Mannitol in h), mit zusätzlich 2 g/l Casaaminosäuren in i), mit 2 g/l Casaaminosäuren und 10 g/l Mannitol in j), mit nur 0,2 g/l KH2PO4 (1,47 mM) statt 1 g/l (7,3 mM), wie in MM40 Medium, in k) und mit 10 g/l KH2PO4 (73,5 mM) in l). Die deutlich erkennbare starke Violettfärbung in l) wird durch Actinorhodin hervorgerufen. Ergebnisse - 127 - Medium Stamm Beginn REDProduktion RED-Produktion nach 52 h Erscheinungsbild nach 72 h/RED Produkt Act Produktion nach 6 Tagen MM40 WT Nach ca. 48-50 h + ∆ksuB/ksuC Nach ca. 41-42 h ++ WT Nach ca. 48-50 h + ∆ksuB/ksuC Nach ca. 41-43 h ++ +1 g/l Glycin WT Keine Produktion +1 g/l Serin ∆ksuB/ksuC WT Nicht genau bestimmbar Nach ca. 48-50 h Nach ca.48-50 h + + Actinorhodinbildung (Act, wie Mutanten) ++ + Act (wie WT) + + Act (wie Mutanten) ++ + Act (wie WT) + geringer als Mutanten + + WT ∆ksuB/ksuC WT ∆ksuB/ksuC WT Nach ca. 46-48 h Nach ca. 38 h Nach ca. 38 h Nicht genau bestimmbar Nicht genau bestimmbar Nach ca. 36 h Nach ca. 36 h Keine Produktion Keine Produktion Nach ca. 41 h + +++ +++ Randbereich? +/Randbereich? +/++++ ++++ Kein Luftmycel Kein Luftmycel ++ ∆ksuB/ksuC Nach ca. 41 h +++ WT ∆ksuB/ksuC WT Keine Produktion Keine Produktion Keine Produktion Kein Luftmycel Kein Luftmycel Kein Luftmycel ∆ksuB/ksuC WT Keine Produktion Keine Produktion Kein Luftmycel Kein Luftmycel ∆ksuB/ksuC KeineProduktion Kein Luftmycel WT Nach ca. 42-43 h ++ ∆ksuB/ksuC Nach ca. 39 h +++ WT Nach ca. 44 h + ∆ksuB/ksuC Nach ca. 40 h + +1 g/l Prolin 300 mM NaCl 300 mM KCl ∆ksuB/ksuC WT ∆ksuB/ksuC WT ∆ksuB/ksuC 10 % Sucrose 20 % Sucrose 10 g/l Mannitol 2 g/l Casa. 2 g/l Casa +Mannitol 0,1 g/l KH2PO4 0,2 g/l KH2PO4 10 g/l KH2PO4 +/+ + ++++ (wie Mutanten) ++++ (wie WT) sehr gering sehr gering gering stärker als WT +++ (wie Mutanten) +++ (wie WT) Keine Luftmycelbildung (n.a.) Keine Luftmycelbildung (n.a.) ++ + Act (wie Mutanten) +++ + Act (wie WT) Keine Luftmycelbildung Keine Luftmycelbildung Keine Luftmycelbildung Keine Luftmycelbildung Zu wenig Phosphat Wachstum eingestellt (n.a.) Zu wenig Phosphat Wachstum eingestellt (n.a.) +++ (wie Mutanten) ++ Act (wie Mutante) +++ (wie WT) ++ Act (wie WT) ++++ (wie Mutanten) +++ Act (wie Mutante) ++++ (wie WT) +++ Act (wie WT) Tabelle 3.1: Antibiotikaproduktion unter verschiedenen Mediumbedingungen. Die Anzucht erfolgte jeweils auf MM40 Minimalmedium mit unterschiedlichen Supplementen. Das verwendete Medium ist jeweils in der ersten Spalte dargestellt, in der zweiten der jeweils verwendete Stamm. WT steht für den S.lividans 66 Wildtypstamm, die beiden S.lividans ∆ksuB und ∆ksuC Deletionsmutanten sind gemeinsam dargestellt, da beide Mutanten jeweils einen identischen Phänotyp zeigen. In der dritten Spalte ist der Zeitpunkt des Beginns der Undecylprodigiosin (RED) Produktion aufgeführt. In den Spalten 4 und 5 ist jeweils die Stärke der Undecylprodigiosin-Produktion dargestellt, +/- steht für eine kaum identifizierbare Antibiotika Produktion, + für eine schwache, ++ für eine stärkere, +++ für eine starke und ++++ für eine sehr starke Produktion, zum jeweiligen Zeitpunkt, wobei die Antibiotikaproduktion der Deletionsmutanten bei Wachstum auf Minimalmedium ohne Supplemente bei RED mit zwei ++ und bei Act mit + als Standard verwendet wurde. (n.a.) steht für: nicht in Abb. 3.68 a-l) dargestellt. - 128 - Ergebnisse 3.9. Konstruktion von chromosomalen Fusionsgenen in S.lividans 3.9.1. Herstellung von Konstrukten und Analyse der Transformanten Um mit Hilfe der Fluoreszenz-Mikroskopie die Synthese von KcsA nachweisen zu können, wurde in einem ersten Experiment versucht, das Gen für das „enhanced green fluorescent protein“ EGFP „in Frame“ am 3’ Ende von kcsA ins Chromosom von S.lividans 66 zu integrieren, so dass ein KcsA-EGFP-Fusionsprotein produziert werden sollte. Um polare Effekte möglichst auszuschließen oder so weit wie möglich zu reduzieren, sollten hierbei keine weiteren Selektionsgene mit integriert werden (Rekombinationsschema in Abb. 3.69). Für die Integration wurde ein Temperatur-sensitives Vektor-Derivat von pGM160 verwendet, in dem über die HindIII/NcoI-Schnittstellen ein 6,2 kBp Insert eingefügt wurde, aacC wurde hierbei ersetzt. Dieses sollte in S.lividans 66 transformiert werden. Nach Temperaturerhöhung (37°C) erfolgte eine Selektion auf „single crossing over“ (Selektion auf Phänotyp: ThioR, HygR und Mel+), anschließend auf „double crossing over“ (ThioS, HygS und Mel-). Abb. 3.69: Rekombinationsschema. Die jeweils homologen Bereiche auf dem Plasmid und Chromosom sind in grauen Boxen dargestellt; die in der Darstellung linke Box trägt das vollständige kcsA, ohne Stoppcodon. Das EGFP-Gen tragende Fragment ist in grün dargestellt, das Gen liegt im gleichen Leseraster direkt hinter kcsA. Das Ω-hyg-Fragment ist in weiß dargestellt, die Leserichtung des Hygromycin-Resistenzgens ist ebenso, wie von melC1 und melC2, die für die Melanin-Synthese verantwortlich sind (in hellgrau dargestellt), entgegen der Leserichtung des kcsA-EGFP-Fusionsgens. Dargestellt sind, der das Plasmid tragende Transformant (in A), die beiden möglichen „Single-crossing over“ Zwischenstufen (B1 und B2) und die „Double-Crossing over“ Endform (in C), bei der das Gen für EGFP „in Frame“ hinter kcsA integriert ist. Da das kcsA tragende homologe Fragment mit ca. 700 Bp um ca. 200 Bp größer ist, als das zweite, dem kcsA nachfolgende homologe Fragment, sollte die „Single-Crossing-over“Rekombination über das größere Fragment bevorzugt werden (durch die 1 im Kreis dargestellt). Der jeweilige Phänotyp ist rechts dargestellt. Ergebnisse - 129 - Es wurden zwei Vorkonstrukte geschaffen. Die für die Melanin-Synthese verantwortlichen Gene melC1 und melC2 wurden mit PvuII/PstI aus pIJ702, das Ω-hyg-Frg aus dem Vektor pHP45Ωhyg mit DraI ausgeschnitten. Beide Fragmente wurden gleichzeitig mit dem SmaI/PstI-geschnittenen Vektor pBSK(+) ligiert. Es wurden 2 verschiedene Konstrukte erhalten. Weiter verwendet wurde das Konstrukt, bei dem die Leserichtung von hygR der, der Melanin-Synthese Gene, entsprach. Der Vektor erhielt den Namen pBSK2 (Abb. 3.70). Vom Konstrukt pOV006 wurde ein ca. 700 Bp-Fragment amplifiziert, welches den flussaufwärts liegenden Bereich von kcsA und kcsA bis auf das Stopcodon enthielt. Dieses Fragment wurde mit SstI/KpnI restringiert. Der flussabwärts liegende Bereich von kcsA (einschließlich des Stopkodons) wurden von demselben Konstrukt amplifiziert, das erhaltene ca. 500 BpFragment mit NotI/XbaI restringiert. Aus pEGFP1 wurde mit Hilfe von KpnI/NotI das EGFPGen ausgeschnitten. Alle drei restringierten Fragmente wurden gleichzeitig in einer einzigen Ligationsreaktion mit dem SstI/XbaI geschnittenen Vektor pBSK(+) ligiert. Das Konstrukt erhielt den Namen pBSK3 (Abb. 3.70). Nach NcoI/XbaI-Restriktion von pBSK3 (partieller Verdau, da eine weitere NcoI-Schnittstelle im EGFP-Teil zu finden ist), XbaI/HindIII-Restriktion von pBSK2, wurden die beiden erhaltenen Fragmente zugleich mit dem NcoI/HindIII-geschnittenen Vektor pGM160 ligiert. Das Konstrukt erhielt den Namen pGM4 (Abb. 3.70). Abb. 3.70: Konstrukte pBSK2, pBSK3 und pGM4. Mit grün markiert ist der, das EGFP-Gen tragende Abschnitt, mit braun markiert, der die für die Melanin-Synthese zuständigen Gene tragende Abschnitt und in gelb das Ω-hyg-Fragment. Mit blau markiert sind die die amplifizierten Bereiche von kcsA mit dem flussaufwärts liegenden Bereich (verwendete Oligos PrIEK und PrIIEK, Fragmentgröße 758 Bp, nach Restriktion 740 Bp) und dem flussabwärts liegenden Bereich von kcsA (Oligos PrIIIEK und PrIVEK, Fragmentgröße 547 Bp, nach Restriktion 528 Bp). pGM4 wurde in S.lividans 66 transformiert. Nach 37°C Temperaturerhöhung erfolgte die Selektion auf „single crossing over“ (ThioR, HygR, Mel+), anschließend auf „double crossing over“ (ThioS, HygS, Mel-) durch mehrmaliges Ausstreichen auf unselektivem Medium. Es konnten eine Reihe potentieller Mutanten erhalten werden. Nach Restriktion und Transfer der - 130 - Ergebnisse chromosomalen DNA zeigte sich, dass keine Hybridisierung mit Sonden, spezifisch gegen hygR und tsr, erhalten werden konnten, jedoch auch nicht gegen das DNA-Fragment der erwarteten Größe oder ein anderes. Die Kontrollen waren positiv. (Ergebnisse nicht dargestellt). Anscheinend waren kcsA und die umliegenden Bereiche deletiert. 3.9.2. Herstellen von Hygromycin-resistenten Mutanten mit chromosomalen Fusionsgen Da der erste Ansatz, das EGFP-Gen ins Chromosom zu integrieren, nicht gelang, wurde ein neuer Ansatz gewählt. Anders als bei ersten Versuch wurde kein „single-cross over“Zwischenschritt benötigt, die Selektion erfolgte auf das „Double cross over“-Endprodukt. Zur positiven Selektion wurde flussabwärts des EGFP-Gens das Hygromycin-Resistenzgen hygR, entgegen der Flussrichtung von EGFP, eingesetzt; um polare Effekte so weit wie möglich zu reduzieren, wurde der Terminator von hygR deletiert. Nach Ligation der Inserts mit dem Temperatur-sensitiven Plasmid pGM160, Transformation in S.lividans 66 WT und Selektion auf Thiostrepton und Hygromycin, wurden die Transformanten auf Selektionsmedien mit Hygromycin transferiert und bei 37°C angezogen. Unter diesen Bedingungen sollte pGM160 nicht replizieren, so dass nur die Zellen, die die Hygromycin-Resistenz- (und somit auch das EGFP-Gen) integriert haben, wachsen (Rekombinationsschema siehe Abb. 3.71). Abb. 3.71: Rekombinationsschema. In grau markiert sind die jeweils homologen Bereiche des Plasmides und des Chromosoms. Das EGFP-Gen ist so integriert, dass jeweils ein KcsA- oder KsuF-EGFP-Fusionsprotein synthetisiert werden kann; das EGFP-Gen tragende Fragment ist in grün dargestellt. In der zum Fusionsgen umgekehrten Leserichtung ist das Ω-hyg-Fragment für die positive Selektion inseriert (in weiß); das Fragment ist um den hinter dem Resistenzgen liegenden Terminator verkürzt. Ebenfalls in weiß dargestellt ist das Thiostrepton-Resistenzgen, welches auf dem Vektorteil liegt. Bei der Kontrolle wurde das Gen für EGFP durch eine Leserasterverschiebung so integriert, dass statt eines KsuF-EGFP-Fusionsproteins nur ein geringfügig verlängertes KsuF-Protein synthetisiert werden kann. Die Rekombination ins Chromosom kann direkt in einem Schritt über eine Doppel-Rekombination oder über den Zwischenschritt einer Einzelrekombination erfolgen. Dargestellt ist nur das Endergebnis (Doppel-Rekombination). Der jeweilige Phänotyp ist links dargestellt. Ergebnisse - 131 - Vom Konstrukt pACYC007 wurde kcsA bis auf das Stopkodon und der flussaufwärts liegende Bereich amplifiziert und das erhaltene ca. 750 Bp Fragment anschließend mit BamHI/XbaI geschnitten. Vom selben Konstrukt wurde der flussabwärts liegende Bereich einschließlich des Stopkodons von kcsA amplifiziert und das ca. 750 Bp Fragment mit BamHI/HindIII restringiert. Beide Fragmente wurden zugleich mit dem XbaI/HindIII geschnittenen Vektor pUC18 ligiert. Es entstand pUCK1K2 (Abb. 3.72). Das Hygromycin-Resistenz-Gen enthaltene SanDI/NotI Fragment aus pHP45Ωhyg und das KpnI/NotI EGFP-Fragment aus pEGFP1 wurden gleichzeitig mit dem SanDI/partiell KpnI geschnittenen Konstrukt pUCK1K2 ligiert. Das erhaltene Konstrukt bekam den Namen pUCKEH (Abb. 3.72). Aus diesem wurde das Insert als HindIII/XbaI Fragment mit dem ebenso geschnittenen Vektor pGM160 ligiert. Es entstand pGMKEH (Abb. 3.72). Abb. 3.72: Konstrukte pUCK1K2, pUCKEH und pGMKEH. Die beiden homologen Bereiche sind in blau dargestellt, das EGFP-Gen tragende Fragment in grün und das Ω-hyg-Fragment in gelb. Für die Amplifikation von kcsA und dem flussaufwärts gelegenen Bereiches wurden die Oligos PrkcsAK und PrkcsAX (781 Bp, 763 Bp nach Restriktion) verwendet, für den flussabwärts gelegenen PrkcsAS und PrkcsAH (739 Bp, 721 Bp nach Restriktion). Nach Transformation in S.lividans 66 und Selektion auf Rekombinanten, wurden von den Stämmen, die HygR und ThioS sensitiv waren, die chromosomalen DNA’s aufgereinigt. Die Analyse der Rekombinanten erfolgte mittels DNA-Hybridisierung (Sonden gegen das EGFPGen, Ωhyg, tsr und das AhdI-Frg; Abb. 3.73). Die Signale finden sich jeweils bei der erwarteten Größe von 9399 Bp für die drei Mutanten (Membranen a-c, Spur 1-3) und den 7074 Bp beim Wildtyp (Membran b, Spur 4). Zusätzlich wurden zur Kontrolle der korrekten Insertion jeweils der homologe 5’-Bereich der drei Mutante vom Chromosom mit Hilfe von PCR amplifiziert und durch doppelsträngige Sequenzierung verifiziert. Alle drei getesteten Mutanten scheinen in Ordnung zu sein (siehe auch Tabelle 3.2). Ergebnisse - 132 - a) c) b) d) Abb. 3.73 a-d): Hybridisierung, Nachweis der Integration in kcsA. Hybridisierung mit Sonden gegen a) Ωhyg, b) AhdI-Frg c) EGFP-Gen d) tsrR. Die chromosomalen DNA’s der Rekombinanten und S.lividans TK21 wurden mit AhdI geschnitten. Jeweils in den Spuren 1-3: EGFP-Insertionsmutanten K, L und M. Spur 4: S.lividans TK21 WT. Spur 5: Dig-markierter Molekular-Gewichts-Marker II. BuchstabenCode K L M Stamm 66 66 66 CodeInsertion KEH56 KEH56 KEH56 kcsA-EGFP-Insertionsmutanten, Nr. ThioR Morphologie 11 16 18 OK OK OK OK OK OK Sequenzierung OK OK OK Besonderheiten - Tabelle 3.2: Auswertung der 3 kcsA-EGFP-Insertionsmutanten (KEH). Die Oligos PrKcsAvor und PrEGFPback wurden für die Amplifikation verwendet, die Oligos PrEGFPseq und PrKcsAseq für die doppelsträngige Kontrollsequenzierung des entstandenen 1029Bp-Fragmentes. Ergebnisse - 133 - Wie schon für die Integration des EGFP-Gens „in frame“ hinter kcsA beschrieben, wurde auch für die Integration hinter ksuF der „hintere“ Teil von ksuF bis auf das Stopkodon und vom selben Konstrukt, der flussabwärts gelegene Abschnitt einschließlich des Stopkodons amplifiziert und nach Restriktion mit pUC18 ligiert. Nach Transformation in E.coli erhielt das Konstrukt den Namen pUCF1F2 (Abb. 3.74). Nach Integration der beiden hygR und EGFPGen tragenden Fragmente in pUCF1F2 und Transformation, erhielt das Konstrukt den Namen pUCFEH (Abb. 3.74). Aus diesem wurde das Insert als HindIII/XbaI-Fragment mit dem ebenso geschnittenen Vektor pGM160 ligiert, in E.coli transformiert und erhielt nach Kontrolle den Namen pGMFEH (Abb. 3.74). Abb. 3.74: Konstrukte pUCF1F2, pUCFEH und pGMFEH. Die beiden homologen Bereiche sind in blau dargestellt, das EGFP-Gen tragende Fragment in grün und das Ω-hyg-Fragment in gelb. Für die Amplifikation des ksuF-Teils wurden die Oligos PrksuFK und PrksuFX (724 Bp, 706 Bp nach Restriktion), für den flussabwärts gelegenen Bereich PrksuFS und PrksuFH verwendet (594 Bp, 576 Bp nach Restriktion). Als „Template“ fand das Konstrukt pACYC007 Verwendung. Nach Transformation in S.lividans 66 WT und Selektion auf Rekombinanten, wurden von 12 Stämmen, die HygR und ThioS sensitiv waren, die chromosomalen DNA’s aufgereinigt und mittels DNA-Hybridisierung auf korrekte Rekombination getestet (Sonden gegen das EGFPGen, Ωhyg, tsr und das AhdI-Frg; Abb. 3.75). Bei allen fand sich ein Signal in Höhe der erwarteten Größe von 9403 Bp (Membranen a-c, Spuren 1-12), deutlich höher, als das Signal beim Wildtyp (Membran c, Spur 13, erwartete Größe von 7074 Bp). - 134 - Ergebnisse Abb. 3.75 a-d): Hybridisierung, Nachweis der Integration vom EGFP-Gen hinter ksuF. Hybridisierung mit Sonden gegen a) Ω-hyg, b) EGFP-Gen, c) AhdI-Frg d) tsrR. Die chromosomalen DNA’s der potentiellen Insertionsmutanten und S.lividans TK21 wurden mit AhdI geschnitten. Jeweils aufgetragen wurden auf den ersten 12 Spuren die potentiellen Mutanten: Spur 1: Mutante A, Spur 2: B, Spur 3: C, Spur 4: D, Spur 5: E, Spur 6: F, Spur 7: G, Spur 8: H, Spur 9: I, Spur 10: J, Spur 11: N, und Spur 12: O. Auf Spur 13 wurde zur Kontrolle S.lividans TK21 WT-DNA aufgetragen, auf Spur 14: Dig-markierter Molekular-Gewichts-Marker II. Zusätzlich wurde, zur Kontrolle der korrekten Insertion, jeweils der homologe 5’-Bereich der 12 Mutante vom Chromosom mit Hilfe von PCR amplifiziert und durch doppelsträngige Sequenzierung verifiziert. Nur bei der Mutante mit dem Buchstabenkode O findet sich, ca. 50 Bp vor Ende des ksuF-Teils des Fusionsgens, ein Basenaustauch (siehe auch Tabelle 3.3). Ergebnisse - 135 - BuchstabenCode A B C D E F G Stamm 66 66 66 66 66 66 66 CodeInsertion FEH111 FEH111 FEH111 FEH111 FEH111 FEH111 FEH111 H I J 66 66 66 N O 66 66 KsuF-EGFP-Insertionsmutanten Nr. ThioR Morphologie 1 2 3 4 6 8 9 OK OK OK OK OK OK OK OK OK OK OK OK OK OK Sequenzierung OK OK OK OK OK OK OK FEH112 FEH112 FEH112 15 16 24 OK OK OK OK OK OK OK OK OK FEH1111 FEH1111 27 39 OK OK OK OK OK Basenaustausch Besonderheiten Kolonien unterschiedlicher Größe Tabelle 3.3: Auswertung der 12 ksuF-EGFP-Insertionsmutanten (FEH). Mutante J zeigt leichte Auffälligkeiten in der Morphologie. Für die Amplifikation wurden die Oligos PrksuFvor und PrEGFPback, für die doppelsträngige Sequenzierung die Oligos PrksuFseq und PrEGFPseq, verwendet. Bei Mutante O ist die Base 1070 des KsuF-EGFP-Fusionsgens von G zu C ausgetauscht. Dies hat keine Auswirkungen auf die Sequenz des Proteins. Um auszuschließen, dass die Integration eines EGFP- und des Hygromycin-Resistenz-Gens ins Chromosom Auswirkungen auf die Expression des zu testenden Gens hat, sollte eine Leserasterverschiebung so eingefügt werden, dass statt eines KsuF-EGFP-Fusionsprotein ein gegenüber KsuF um 11 Aminosäuren verlängertes Protein produziert würde. Das für die ksuF-EGFP-Fusionsmutante benötigte Zwischenkonstrukt pUCFEH (Abb. 3.74) wurde mit ApaI geschnitten, die 3’ überhängenden Basen mit Hilfe der T4-Polymerase „abverdaut“, wieder aufgefüllt und nach Religation transformiert. Bei Kontrollsequenzierung verschiedener Klone des nun pUCFEH2 genannten Konstruktes zeigte sich, dass jeweils statt der erwarteten Verkürzung um 4 Nukleotide eine von 5 Nukleotiden vorlag. Das Konstrukt pUCFEH2 wurde mit NcoI geschnitten, nach „auffüllen“ (Klenow) des 5’ Überhanges religiert und in E.coli transformiert. Aus diesem (pUCFEH4 benannt) wurde nach Kontrollsequenzierung das Insert als HindIII/XbaI-Fragment mit dem ebenso geschnittenen Vektor pGM160 ligiert, in E.coli transformiert und erhielt nach der Kontrolle den Namen pGMFEH4 (Abb. 3.76). - 136 - Ergebnisse Abb. 3.76: Konstrukt pGMFEH4. Die mit dem Chromosom homologen Bereiche sind in blau dargestellt, das EGFP-Gen tragende Fragment in grün und das Ω-hyg-Fragment in gelb. Nach Transformation in S.lividans 66 WT und Selektion auf Rekombinanten, wurden die chromosomalen DNA’s von 11 potentiellen Insertionsmutanten aufgereinigt und nach Restriktion gegen verschiedene Sonden hybridisiert (Abb. 3.77 a-c). Abb. 3.77 a-c): Hybridisierung, Nachweis der Integration vom EGFP-Gen hinter ksuF außerhalb des Leserasters. Hybridisierung mit Sonden gegen a) Ω-hyg, b) AhdI-Frg und c) tsrR. Die chromosomalen DNA’s der potentiellen Insertionsmutanten und S.lividans TK21 wurden mit AhdI geschnitten. Jeweils aufgetragen wurden auf den ersten 12 Spuren die potentiellen Mutanten: Spur 1: Mutante P, Spur 2: Q, Spur 3: R, Spur 4: S, Spur 5: T, Spur 6: U, Spur 7: V, Spur 8: W, Spur 9: X, Spur 10: Y und Spur 11: Z. Auf Spur 12 wurde zur Kontrolle S.lividans TK21 WT-DNA aufgetragen, auf Spur 13 (mit M markiert): Dig-markierter MolekularGewichts-Marker II. Ergebnisse - 137 - Zehn der elf getesteten potentiellen Mutanten scheinen das EGFP-Gen korrekt ins Chromosom integriert zu haben, Mutante Nr.9 (Mutante X) ist nicht korrekt inseriert. Von diesen Mutanten sollte die Synthese eines C-terminal um 12 Aminosäuren verlängerten KsuFProteins erfolgen. BuchstabenCode P Q R S T U V W X Y Z Stamm 66 66 66 66 66 66 66 66 66 66 66 CodeInsertion FEH4 FEH4 FEH4 FEH4 FEH4 FEH4 FEH4 FEH4 FEH4 FEH4 FEH4 Nr. ThioR Morphologie 1 2 3 4 5 6 7 8 9 10 11 OK OK OK OK OK OK OK OK OK OK OK OK OK OK OK OK OK OK OK OK OK OK Besonderheiten fehlerhaft inseriert Tabelle 3.4: Auswertung der 11 ksuF/EGFP-Insertionsmutanten (FEH4) die zur Kontrolle dienen. Zehn der 11 Mutanten sind korrekt inseriert und wurden weiter verwendet. Eine Synthese des chromosomal determinierten KcsA-EGFP (S.lividans 66 KEH5) und KsuFEGFP (S.lividans 66 FEH11), im Vergleich zum leicht verlängerten KsuF der NegativKontrolle (S.lividans 66 FEH4), konnte weder durch Fluoreszensmikroskopie (nicht dargestellt), noch mit Hilfe von Antikörpern nach Transfer auf Membranen, nachgewiesen werden (siehe Abb. 3.16 a, Spur 6 für S.lividans 66 FEH11). Eine Produktion der Proteine war auch auf Grund der vorher dargestellten Ergebnisse nicht zu erwarten (siehe 3.3.5.). Eine Synthese des KsuF-EGFP Fusionsproteins und des leicht verlängerten KsuF der NegativKontrolle sollte nach Transformation eines für die Thiostrepton Resistenz determinierenden tsr tragenden Plasmides, wie z.B. pWHM3be oder pIJ702 erfolgen, aufgrund von Zeitmangel konnte dies nicht mehr durchgeführt werden. Diskussion - 139 - 4. DISKUSSION 4.1. Sequenzanalyse und Charakteristika der abgeleiteten Proteine Wie vergleichende Sequenzanalysen des S.coelicolor A3(2) (Bentley, S.D., et al.; 2002) und S.avermitils Genoms (Ikeda, H., et al.; 2003) gezeigt haben, ist die Anordnung und Zusammensetzung der Gene in den äußeren Armbereichen deutlich geringer konserviert und nimmt zu den Telomeren hin ab (Ikeda, H., et al.; 2003, siehe auch Einleitung). Der im Rahmen dieser Arbeit untersuchte Abschnitt findet sich im äußersten Bereich des rechten Arms des S.coelicolor Genoms, nur 163 kBp-177 kBp entfernt vom telomeren Ende. Die äußersten 20 kBp werden hierbei von den „Terminal inverted repeats“ (TIR’s) gebildet. Somit verwundert es nicht, dass eine vergleichbare Zusammensetzung der Gene dieses Abschnittes mit einer Ausnahme, weder bei S.avermitilis noch bei einem anderen Bakterienstamm, außer bei den Mitgliedern der S.violacoeruber Familie, S.violaceoruber (Puls-Feld Gelexperimente im Rahmen der Diplomarbeit), S.lividans 66 und S.coelicolor A3(2) zu finden ist. Kineococcus radiotolerans ist ein aus einem radioaktiven Absetzteich isolierter, aerober, kockoider, Gram-positiver Actinomycetenstamm mit hohem G+C-Gehalt, der in symetrischen Multizell Gruppen wächst und sich durch eine, wie der Name schon sagt, sehr hohe Toleranz gegenüber radioaktiver Strahlung auszeichnet (Phillips, R.W., et al.; 2002). Auf dem Kineococcus radiotoleranz-Genom findet sich ein vermutliches Operon, bestehend aus drei Genen (EAM73830, EAM73831 und EAM73832), die jeweils für Proteine der Aldo-Keto Reduktase-Familie kodieren. EAM73830 zeigt hierbei mit 73 % identischen und 85 % homologen Aminosäuren eine sehr große Ähnlichkeit zu KsuC, EAM73831 mit 77 % identischen und 85 % homologen eine sehr große zu KsuB, die entsprechenden DNA Sequenzen sind jeweils 81 % identisch zu den für KsuC und KsuB determinierenden. Des Weiteren findet sich, beginnend 80 Bp flussaufwärts vor EAM73830 und 130 Bp vor ksuC, ein jeweils 70 Bp langer AT reicher homologer Abschnitt (siehe Anhang A, rot markiert). Es ist zu vermuten, dass dieser Abschnitt jeweils mögliche Operator-Binderegionen, die -35 und -10 Region und den Transkriptionsstart enthält. In S.lividans sind ksuC und ksuB durch einen 35 Bp langen intergenischen Abschnitt getrennt, EAM73830 und EAM73831 sind um 4 Nukleotide überlappend. Hier unterscheiden sich die beiden Stämme. Eine Funktion für dieses, für drei Aldo-Keto Reduktasen determinierenden putativen Operon aus K.radiotolerans, ist weder bekannt noch postuliert. - 140 - Diskussion Das aus der Sequenz abgeleitete Protein KsuF ist ein Mitglied der FlavoproteinMonooxygenasen. Diese führen in aromatischen Substrate eine neue Hydroxylgruppe an der ortho Position der vorhandenen Hydroxylgruppe ein (Harayama, S., et al.; 1992). Es sind jedoch auch Enzyme, wie die 3-Hydroxybenzoat-6-Hydroxylase aus Pseudomonas cepacia und Klebsiella pneumoniae M5a1, bekannt, die eine para-Hydroxylierung durchführen (Harayama, S.M., et al.; 1992; Wang, L.-H., et al.; 1987; Liu, D.Q., et al.; 2005). Wie am Beispiel der p-Hydroxybenzoat-Hydroxylase (PHBH), die p-Hydroxybenzoat (4Hydroxybenzoat) zu 3,4-Hydroxybenzoat hydroxyliert, explizit gezeigt wurde, führen Flavoprotein-Monooyxygenasen die enzymatische Reaktion in zwei Schritten durch: (1) Reduktion des FAD durch NAD(P)H als Antwort auf die Bindung des Substrates am Enzym und (2) Oxidation des reduzierten FAD mit O2 in einer wasserfreien Umgebung mit Bildung eines Hyperoxids, welches anschließend das Substrat zum Endprodukt oxygeniert (Cole, L.J., et al.; 2005; Entsch, B., et al.; 2005, Entsch, B. and van Berkel, W.J., et al.; 1995). In Gelfiltrationsexperimenten wurde His-KsuF als Monomer und in der Größe eines Dimers/Trimers aufgereinigt (siehe Abb. 3.41 a-d und Abb. 3.42 a-f). Anders als das Monomer, ist das Dimer/Trimer „FAD frei“ und neigt zur Aggregation. Es ist anzunehmen, dass die Dimer/Trimer Form artifiziell ist. Aus den S.lividans TK21 pOVE33 oder pOVE53 Transformanten konnte das chromosomal determinierte KsuF nicht als Multimer mit dem Plasmid-kodierten His-KsuF über Ni-NTA co-aufgereinigt werden, was ebenfalls dafür spricht, dass KsuF als Monomer vorliegt. Alle Untersuchungen wurden unter hohen Salzkonzentrationen durchgeführt (200 mM oder 300 mM NaCl), so dass nicht auszuschließen ist, das KsuF unter anderen Bedingungen als Multimer vorliegt. Die Mitglieder der Flavoprotein-Monooxygenasen werden als monomere oder homodimere Proteine beschrieben (Harayama, S., et al.; 1992), jedoch finden sich auch Beispiele als Homotetramer. So liegen z.B. die Salicylat-5-Hydroxylase (SAL5H), m-Hydroxybenzoat-6Hydroxylase (MHB6H) und die p-Hydroxybenzoat-3-Hydroxylase (PHB3H) aus Rhodococcus erythopolis als Homotetramer vor (Suemori, A., et al.; 1995). Die PhenolHydroxylase aus Trichosporon cutaneum wurde als Homodimer charakterisiert (Sejlitz, T. and Neujahr, H.Y.; 1987), jedoch als Tetramer aus 2 Homodimeren kristallisiert (1Foh, Chain A-D; Enroth, C., et al.; 1998). Die 3-Hydroxybenzoat-6-Hydroxylase aus P.cepacia ist ein Monomer von 44 kDa (Wang, L.-H., et al.; 1987), während sie aus Micrococcus spec. als Dimer identifiziert wurde (Rajasekharan, S., et al.; 1990). KsuF wie auch TetX bzw. TetX2 aus Bacteroides Stämmen (Whittle, G., et al.; 2001; Park, B.H. and Levy, S.B.; 1988; Speer, B.S. and Salyers, A.A.; 1988) und eine Reihe weiterer nicht genauer charakterisierte Flavoprotein-Monooxygenasen zeichnen sich durch den Besitz eines relativ kurzen C-terminalen Endes aus. TetX und TetX2 zeigen nach Produktion in E.coli eine kryptische, sauerstoffabhängige, durch Monohydroxylierung an Position 11a hervorgerufenen Resistenz gegen Tetrazykline. Wie an TetX2 gezeigt, liegen sie jeweils als Monomer vor (Yang, W., et al.; 2004). KsuF zeigt unter den Flavoprotein-Monooxygenasen mit bekannter Funktion die höchsten Ähnlichkeiten zu TetX und TetX2 einschließlich des C- Diskussion - 141 - terminalen Endbereiches, mit jeweils 46 % homologen, hiervon 30 % identischen Aminosäuren. Die p-Hydroxybenzoat-Hydroxylase (PHBH) aus Pseudomonas fluoreszens und Pseudomonas aeruginosa, wie auch weiteren Pseudomonaden, ist ein Homodimer (Seibold, B., et al.; et al.; 1996; Müller, F., et al.; 1979). Wie aus den 3D Strukturdaten ersichtlich, ist das C-terminale Ende der PHBH primär an der Dimerisierung beteiligt (Abb. 4.1 b und d). Die hier dargestellten dimeren Strukturen sind aus Kristalldaten des Monomers postuliert. Vor kurzem gelang zum ersten Mal die Kristallisation als Dimer. Die Daten zeigen ebenfalls die Bedeutung des C-terminalen Endes für die Dimerisierung (Entsch, B., et al.; 2005). Abb. 4.1: Postulierte Kristallstruktur des C-terminalen Endes von KsuF und der PHBH („in“ Konformation). Dargestellt sind in a) und c) die putative Kristallstrukturen der C-terminalen 26 Aminosäuren von KsuF und in b) und d) die Kristallstruktur der um 3 Aminosäuren gekürzten C-terminalen 52 Aminosäuren der PHBH (1pbe.pdb), jeweils in dimerer Form. Die 2 Untereinheiten sind jeweils in grün und gelb dargestellt. Die Ansicht in a) und b) entspricht der Seitenansicht in Abb. 3.40 c, in c) und d) wurden die Peptide um die Hochachse um 90° in „Uhrlauf“-Richtung rotiert. Wie aus dem „Alignment“ ersichtlich, ist das C-terminale Ende von KsuF um 29 Aminosäuren gegenüber der PHBH verkürzt. Nach Ser356 von KsuF (PHBH: Ser348) sind keine korrespondierenden Aminosäuren zu finden (Abb. 3.39). Ob die im Vergleich zu den Kristallstrukturen der PHBH postulierte C-terminale α-helicale Struktur (Leu367 bis Gly373 von KsuF) vorhanden ist, ist zu bezweifeln (siehe Abb. 4.1 a und c). KsuF liegt vermutlich als Monomer vor. Flavoprotein-Monooxygenasen besitzen FAD als Cofaktor, das, wie z.B. bei der pHydroxybenzoat-Hydroxylase (PHBH), nicht kovalent gebunden ist (Eppink, M.H.M, et al.; - 142 - Diskussion 1998b). Jeweils ein FAD-Molekül ist pro Untereinheit gebunden (Entsch, B., et al.; 2005; Yang, W., et al.; 2004; You, I.S., et al.; 1990; Tu, S.C., et al.; 1981). KsuF kann sowohl nach Überproduktion aus E.coli als auch nach Aufreinigung aus Streptomyceten (nicht dargestellt) als gelb gefärbtes Protein aufgereinigt werden (siehe Abb. 3.37 c). In Streptomyceten finden sich neben FMN und FAD auch FO (7,8-Dedimetyl8-hydroxy-5-deazariboflavin), z.B. bei der Tetrazyclin-Dehydrogenase aus Streptomyces aureofaciens (Nakano, T. et al.; 2004) als Flavin-Cofaktoren. Weitere FAD-Derivate sind bekannt, wie z.B. Arabino-FAD, gefunden in der Alkohol-Oxidase methylothropher Hefen (Bystrykh, L.V., et al.; 1991). In massenspektrometrischen Untersuchungen wurde FAD als Cofaktor nachgewiesen (siehe Ergebnis 3.6.2., Abb. 3.44 a-h). Die Ergebnisse zeigten, dass jeweils ein FAD pro Monomer vorhanden ist (siehe Anhang D und Abb. 3.43). FAD ist an KsuF nicht kovalent gebunden. Eine geringfügige Dissoziation des FAD vom KsuF konnte immer während dessen Aufreinigung und Konzentrierung beobachtet werden. Die Ablösung von FAD führt zur Bildung von oligomeren Formen (Dimeren/Trimeren, siehe Abb. 3.41 a-d und 3.42 a-f), die leicht präzipitieren (ab ca. 14000 g). Anders als für andere Flavoprotein-Monooxygenasen ist die Ablösung von FAD anscheinend irreversibel. Es war nicht möglich, wie z.B. bei der PHBH und Salicylat-Hydroxylase gezeigt (Bhushan, B., et al.; 2004; Palfey, B.A., et al.; 2003), unter vergleichbaren Bedingungen (Red-A Methode, Gatti, D.L., et al.; 1994) den KsuF-FAD-Komplex zu regenerieren. Die Stabilität des KsuF-FADKomplexes ist unabhängig vom verwendeten Puffer. Die Untersuchungen wurden mit 15 verschiedenen anorganischen oder organischen Puffern bei pH 7,5 durchgeführt (nicht dargestellt). Auch haben (getestete) Salzkonzentrationen von 0-300 mM NaCl oder KCl auf die Stabilität des FAD-KsuF Komplexes keinen Einfluß. Anders als z.B. bei der PHBH (Palfey, B.A., et al.; 2003) besitzt KsuF nur einen sehr geringen pH-Bereich in dem es stabil ist; pH-Werte unter pH 7-6,5 oder über pH 8-8,5 führen zur sofortigen, vollständigen, irreversiblen und sehr starken Aggregation und Präzipitation von KsuF, mit Ablösung von FAD. KsuF fällt hierbei als braun gefärbter „Klumpen“ aus, die unter diesen Bedingungen beobachtete Aggregation unterscheidet sich hierin von der oben beschriebenen. Überraschender Weise ist KsuF empfindlich gegenüber Metall, so führt z.B. auch das „Aufziehen“ der Proteinlösung von KsuF-FAD durch eine Metallnadel zur sofortigen und vollständigen Verklumpung des Proteins unter Freisetzung des FAD, vergleichbar der beobachteten bei niedrigen oder hohen pH-Werten. Wie an der PHBH gezeigt wurde, sind vier Bereiche der Flavoprotein-Monooxygenasen für die Bindung von FAD bedeutsam (zusammenfassend dargestellt in: Wang. J,. et al.; 2002). Die an der Bindung von FAD beteiligten Aminosäuren sind in der Abb. 3.39 markiert und auch in KsuF zu einem großen Teil konserviert, bzw. liegen in Bereichen, die „hoch“ konserviert sind. Flavoprotein-Monooxygenasen sind Mitglieder der „FAD/NAD(P)-binding Rossmann fold Superfamily“. Im N-terminalen Teil (Aminosäuren 1-48) von KsuF findet sich die typische „Rossman fold“ Struktur (Rao, S.T. and Rossman, M.G.; 1973), bestehend aus alternierenden β-Faltblatt und α-helikalen Sekundärstrukturen (β1α1β2α2β3, siehe Abb. 3.39). Diskussion - 143 - Der Bereich der Aminosäuren 9-14 von KsuF bindet hierbei am Adenosin des FAD, vor allem an der Ribosegruppe, die Aminosäuren 31-47 am Ribityl und dem Isoalloxazin von FAD. Der Bereich der Aminosäuren 154-162 von KsuF ist an der Bindung der Pyrophosphate involviert, 293-310 von KsuF an der Bindung der Pyrophosphate, der Ribosyl- und der Isoalloxazingruppe. Als zweiten Cofaktor neben FAD besitzen Flavoprotein-Monooxygenasen NAD(P)H. TetX bzw. TetX2 verschiedener Bacteroides-Stämme (Yang, W., et al.; 2004) und die PHBH aus P.fluorescens und aeruginosa sind NADPH abhängig. Die Salicylat-1-Monooxygenase des Pseudomonas spec. Stammes ATCC 29352 bevorzugt NADH gegenüber NADPH (Bhushan, B., et al.; 2004) und die Salicylat-Hydroxylase aus Pseudomonas stutzeri AN10 benötigt NADH (Bosch, R.; et al.; 1999). Eine Bindung von NADH oder NADPH an gereinigtem HisTag KsuF oder an KsuF-haltigen Zellextrakten (aus S.lividans 66 pJO1 bzw. S.lividans NI pOV005 Transformanten) konnte nicht nachgewiesen werden (nicht dargestellt). Ursächlich verantwortlich ist vermutlich, dass es nicht gelungen ist, ein Substrat für KsuF zu finden (siehe unten). Wie für die die Phenol-Hydroxylase aus Trichosporon cutaneum und die PHBH gezeigt wurde, erfolgt die Bindung des NAD(P)H Cofaktors erst nach der Bindung des Substrates (Entsch, B., et al.; 2005; Sejlitz, T. and Neujahr, H.Y.; 1987). Die Cokristallisation der PHBH mit NADPH ist bisher nicht gelungen, auch wenn NADPH in der Zwischenzeit sekundär in die Kristallstruktur modelliert wurde (Entsch, B., et al.; 2005; Wang, J, et al.; 2002). Wie für die PHBH gezeigt, ist an der Bindung des Nukleotid-Cofaktors NADPH keine „Rossman“-fold Struktur beteiligt. Dies unterscheidet die FlavoproteinMonooxygenasen von allen anderen Proteinen (Entsch, B. and van Berkel, W.J.H., 1995). Durch Aminosäureaustausche konnte die Beteiligung einer Reihe von Aminosäuren an der Bindung von NADPH nachgewiesen werden (Eppink, M.H.M., et al.; 1999; Eppink, M.H.M., et al.; 1998a; Eppink, M.H.M., et al.; 1998b; Eppink, M.H.M., et al.; 1997; Eppink, M.H., et al.; 1995). Für einen Großteil wird eine duale Rolle in der Bindung sowohl von FAD als auch von NADPH angenommen, hierunter Arg166 (Eppink, M.H.M., et al.; 1997). Für His162 und Arg269 wird postuliert, dass sie mit dem Pyrophophatteil von NADPH interagieren (Eppink, M.H.M., et al.; 1998a). Nur Arg166, für das eine duale Rolle in der Bindung sowohl von FAD als auch von NADPH angenommen wird (Eppink, M.H.M., et al.; 1997), ist in KsuF konserviert (KsuF: Arg162). Arg269 ist durch ein konservatives Histidin ersetzt (KsuF: His276); für Arg269 wird postuliert, dass es mit dem Pyrophophatteil von NADPH interagiert (Eppink, M.H.M., et al.; 1998a). Wie verschiedene eigene Sequenzvergleiche zeigten, sind die in der PHBH an der NADPH Bindung beteiligten Aminosäuren auch in anderen Flavoprotein-Monooxygenasen kaum konserviert. Eine Ausnahme bildet das hochkonservierte Arg166 (KsuF: Arg162) (Eppink, M.H.M., et al.; 1997). Die typischen Substrate der Flavoprotein-Monooxygenasen, p-Hydroxybenzoat, Salicylat, Phenol, 2,4-Dichlorophenol, Tetrazyclin und Anhydrotetrazyclin, des Weiteren auch Chloramphenicol werden von KsuF nicht umgesetzt (nicht dargestellt). - 144 - Diskussion Flavoprotein-Monooxygenasen zeichnen sich durch eine sehr hohe Substratspezifität aus (Seibold, B., et al.; 1996), dies ist vermutlich der Grund, dass es nicht gelang, ein Substrat für KsuF oder die Bindung eines Substrates an KsuF nachzuweisen. So ist für die PHBH nur bekannt, dass sie, außer 4-Hydroxybenzoat, nur 2-Fluoro-4-hydroxybenzoat, 2-Chloro-4hydroxybenzoat und 2,4-Hydroxybenzoat als Substrat umsetzt (Seibold, B., et al.; 1996). Ursächlich verantwortlich ist die „Passwort“ Erkennung des Substrates, die gegenüber dem „Lock and Key“ Modell (Fischer, E.; 1894) und dem „Induced Fit“ Modell (Koshland, D.E.; 1958), bei denen die Substraterkennung nur durch die Bindung des Substrates an der strukturell passenden Substratbindestelle erfolgt, einen weiteren Schritt der Substraterkennung besitzt. Nach Bindung des Substrates und des NADPH Cofaktors in der „open“ erfolgt der Wechsel in die „out“ Konformation. Hier ist die „Ionisierbarkeit“ (Deprotonierung, siehe unten) des Substrates notwendig. Erst anschließend erfolgt die Reduzierung des FAD durch NADPH und der Wechsel in die „in“ Konformation mit Oxidation des Substrates (Palfey, B.A., et al.; 1999). Tyr201, Ser212, Arg214, Tyr222 und Pro293 sind direkt in der p-HydroxybenzoatHydroxylase (PHBH) über Wasserstoffbrücken an der Bindung des Subtrates pHydroxybenzoat (4-Hydroxybenzoat) beteiligt (Ortiz-Maldonado, M., et al.; 2003). Ser212, Arg214 und Tyr222 binden an der −COO− Gruppe des Substrates; sie sind nicht an der katalytischen Reaktion beteiligt. Ihre Aufgabe ist die Erkennung und die Stabilisierung des Substrates. In KsuF ist nur Tyr223 (PHBH: Tyr222) konserviert. An den Positionen Ser212 und Arg214 finden sich His213 und Glu215, was auf eine unterschiedliche Substratspezifität hinweist. Aus der errechneten 3D Struktur von KsuF kann vermutet werden, dass auf Grund der räumlichen Position, His213, His221 und Tyr223 eine entsprechende Aufgabe übernehmen könnten. Tyr201 und Pro293 binden über Wasserstoffbrücken an der 4-OH Gruppe des p-Hydroxybenzoat. Im Gegensatz zu Tyr201 findet sich in KsuF ein entsprechend positionierter Prolin-Rest (Pro302, siehe Abb. 3.39). Pro293 ist in FlavoproteinMonooxygenasen hoch konserviert (Eppink, M.H.M., et al.; 1997). Während der Reaktion wird ein Proton über ein Wasserstoffbrücken-Netzwerk von 4-OH-Tyr201-Pro293 über Tyr201, Tyr385 und zwei Wassermolekülen auf His72 auf der Außenseite des Proteins übertragen (Ortiz-Maldonado, M., et al.; 2004; Palfey, B.A., et al.; 1999; Gatti, D.L., et al.; 1996). An Position 72 ist in KsuF Glu72, Tyr385 befindet sich im C-terminalen Ende der PHBH, der in KsuF nicht vorhanden ist. Die Deprotonierung der 4-OH Gruppe mit Bildung einer „Phenolat“ Form führt zur Abstoßung von Pro293. Dies bewirkt die Konformationsänderung des Proteins und ist der Schlüsselschritt von der „out“ zur „in“ Konformation (Palfey, B.A., et al.; 2002). KsuF führt vermutlich eine ortho- Hydroxylierung durch. Eigene Sequenzvergleiche zeigen, dass Flavoprotein-Monooxygenasen, die eine paraHydroxylierung durchführen, keinen dem Pro293 entsprechenden Prolin-Rest tragen. Die Produktion von KsuF ist in S.lividans wie auch in S.coelicolor abhängig von der Synthese des Thiostreptonresistenzproteins. Dies wird in einem der folgenden Kapitel besprochen (4.2. Synthese von drei Proteinen unter verschiedenen physiologischen Bedingungen). Des Diskussion - 145 - Weiteren ist sie abhängig von der Salzkonzentration im Medium, vergleichbar der Produktion von KsuC und KsuB. Hohe Salzkonzentrationen an NaCl und KCl erhöhen die Synthese bei Vorhandensein des Thiostreptonresistenzproteins deutlich; dies wird ebenfalls in diesem folgenden Kapitel besprochen. Die Startkodons von ksuF und kcsA sind durch einen 325 Bp langen intergenischen Abschnitt getrennt, kcsA liegt in entgegengesetzter Leserichtung von ksuF. Im 132 Bp langen intergenischen Abschnitt, jeweils flussabwärts von ksuF und ksuL, findet sich ein Terminator (cgtggagccgacggccaccacctcgcggtggccgtcggctccacg), mit 19 Bp langen perfekten „inverted repeats“ und einem 7 Bp umspannenden „Loop“- Bereich (siehe Anhang A und B). In Transformanten, die ksuF zusammen mit tsr auf einem Konstrukt trugen, konnte in Transkriptionsstudien gezeigt werden, dass ksuF von einer monocistronischen mRNA determiniert wird (J.Ritz, pers. Mitteilung). Der Terminator erwies sich hierbei als besonders effizient. Es gelang nicht, ein mRNA-Transkript in S.lividans WT nachzuweisen. Weder mit Hilfe entsprechender Programme, noch beim Durchsuchen „von Hand“, konnte vor ksuF oder einem der in dieser Arbeit untersuchten Gene, eine σ70 RNA-Polymerasen typische -35 -10 Erkennungssequenz gefunden werden (Strohl, W.R.; 1992). Dies lässt schließen, dass die Transkription jeweils von einem der 41 im Rahmen des S.coelicolor A3(2) Genomprojektes gefundenen ECF-Sigma Faktoren abhängig ist; auf Grund von variablen und untypischen Erkennungs-Sequenzen sind diese schwer zu identifizieren (Bentley, S.D., et al.; 2002; Bourn, W.R. and Babb, B.; 1995; Strohl, W.R.; 1992). Es ist durch Abgleich mit dem 3’ Ende der 16S rRNA von S.lividans gelungen, sowohl vor ksuF als auch vor allen anderen im Rahmen dieser Arbeit untersuchten Genen jeweils eine putative Shine-Dalgarno Sequenz zu identifizieren (siehe Anhang A, Strohl, W.R.; 1992; Bibb, M.J., et al.; 1985; Bibb, M.J. and Cohen, S.N.; 1982). Im 325 Bp langen intergenischen Abschnitt finden sich eine überraschend hohe Zahl von palindromischen Sequenzen, „direct“ und „inverted repeats“; hierbei sind besonders die vier identifizierten „direct repeats“ auf Grund ihrer jeweils geringen und regelmäßigen Abstände auffällig (siehe Abb. 3.2). Repetitive Sequenzen in intergenischen Bereichen besitzen häufig eine Funktion als Bindestelle von regulatorischen Proteinen. Experimentell bekannte oder durch Sequenzanalysen identifizierte Operator-Sequenzen zeigen keine Sequenzidentitäten zu den repetitiven Sequenzen. Die Anordnung des dreimal gefundenen Hexamers „direct repeats 3“, mit einem Abstand von sechs und 22 Nukleotiden, erinnert an die Operatorsequenzen der SARP Regulator-Familie (Streptomyces antibiotic regulatory proteins, Wietzorrek, A., Bibb, M.; 1997). Wie für neun von ActII-ORF4 und DnrI kontrollierten Promoterregionen gezeigt, binden diese an „direct repeats“ als Operatorsequenzen (Sheldon, P.J., et al.; 2002). Die Promoter-Bereiche von actI-ORF1, actIII, dnrC und dnrD tragen jeweils drei Heptamere „direct-repeats“, die durch vier und 15 Nukleotide getrennt sind. Die hohe Anzahl der repetitiven Sequenzen mit einer vermutlich regulatorischen Funktion deutet auf eine sehr komplexe Regulation von ksuF und kcsA hin. - 146 - Diskussion Die Überproduktionskonstrukte (pOVE3-Serie) und das EGFP-Translations-Test Konstrukt pOVG1 tragen nur den 325 Bp langen intergenischen Abschnitt zwischen ksuF und kcsA als Promoterbereich, das KsuF-EGFP-Translations-Test Konstrukt (pOVG13) besitzt einen deutlich längeren (1057 Bp), flussaufwärts von ksuF liegenden Abschnitt. Die EGFP Produktion in der pOVG1-Transformante entspricht derjenigen von Transformanten mit einem pOVE3-Derivat, deutlich geringer als erwartet (siehe Abb. 3.34 und 3.35, a und b; Abb. 3.23 a, Spur 3). Die pOVG13 Transformante zeigt die erwartete hohe Produktion, wie sie auch in Transformanten, die ksuF auf einem Konstrukt trugen, gefunden wurde (siehe z.B. Abb. 3.34 und 3.35, g und h; Abb. 3.11 d). In Bakterien findet sich nahezu immer der Transkriptionsstart weniger als 300 Bp vor dem Translationsstart-Kodon (Studholme, D.J., et al.; 2004), bei Streptomyceten zumeist innerhalb von 100 Bp (Strohl, W.R.; 1992). Unter 139 Streptomyceten Promotoren haben 3 (nicht genauer benannt) einen weiten Abstand von 298, 335 und 345 Nukleotiden zum jeweiligen Translations Startkodon (Strohl, W.R.; 1992). In der 5’ untranslatierten Sequenz dieser drei finden sich repetitive Sequenzen und Sekundärstruktur-bildende Abschnitte, vergleichbar denen im intergenischen Bereich zwischen ksuF und kcsA (Strohl, W.R.; 1992). Somit erscheinen zwei mögliche Transkriptionsstartpunkte bzw. Operator/Promoter-Bereiche vor ksuF wahrscheinlich zu sein, einer im intergenischen 325 Bp Bereich gelegen und ein weiterer zumindest zum Teil außerhalb dieses Bereiches im für kcsA determinierenden Abschnitt. Die C-terminale Hälfte von KsuL zeigt in mehreren kurzen Motiven Ähnlichkeiten zur Cterminalen katalytischen Domäne eukaryotischer extrazellulärer Protein-Lysin-6-Oxidasen (bezeichnet als Lysyl-Oxidasen: LO; EC1.4.3.13) sowie zur katalytischen Domäne von „lysyl-oxidase-like“ Proteinen (LOXL, Csiszar, K.; 2001; Kagan, H.M. and Wande, L.; 2003). Diese gehören zur Familie der Amine-Oxidasen (AOs) und deren Unterfamilie der Kupfer enthaltenden „semicarbazide-sensitive amine oxidases“ (SSAOs; Jalkanen, S. and Salmi, M; 2001). Bestimmte Prototypen der Lysyl-Oxidasen sind an der Quervernetzung von Collagen bzw. Elastin beteiligt. Die dabei durchgeführte Reaktion erfolgt in zwei Teilschritten. Im ersten Schritt wird Lysyl, bzw. Hydroxylysyl zum Aldehyd oxidiert, die Aminogruppe bleibt hierbei am Lysyl-Tyrosyl Quinon Cofaktor gebunden. Im zweiten Schritt wird die Aminogruppe unter Wasser- und O2-Verbrauch zum Ammonium reduziert und freigesetzt, zusätzlich entsteht H2O2 (Kagan, H.M. and Wande, L.; 2003; Williamson, M.A. and Kagan, H.M.; 1985; Williamson; P.R. and Kagan, H.M.; 1986). Die Teilreaktionen können wie folgt dargestellt werden: (1) LOox + RCH2NH2 → LOred(-NH2) + RHC=O (2) LOred(-NH2) + O2 +H2O → LOox + H2O2 + NH3 Die Aldehydgruppe kann im Folgenden spontan mit benachbarten Aldehyden (Aldol Kondensation) oder mit anderen Lysyl-Gruppen (über eine „Schiffsche“-Base) kondensieren unter Bildung von „intra“- und „inter“-molekularen Quervernetzungen der Collagen- und Elastinketten (Rucker, R.B., et al.; 1998; Reiser, K., et al.; 1992). Diskussion - 147 - KsuL kann immunologisch als extrazelluläres Protein nachgewiesen werden (siehe Abb. 3.46, Spur 5). Ein 39 Aminosäuren langes Signalpeptid lässt sich vorhersagen und wurde in weiterführenden Arbeiten durch Ansequenzieren des „reifen“ extrazellulären Proteins nachgewiesen (pers. Mitteilung I.Koebsch und M.Kotasińska). In Ergänzung werden zwei prozessierte Subformen gefunden (Abb. 3.46 Spuren 2 und 3). Wie durch Ansequenzierung gezeigt wurde, besteht die größere Subform (ca. 30 kDa) aus den N-terminalen 312 Aminosäuren (ohne Signalsequenz), die kleinere (ca. 26 kDa) aus den C-terminalen 234 Aminosäuren (pers. Mitteilung I.Koebsch und M.Kotasińska). Vergleichbares findet sich bei eukaryotischen Lysyl-Oxidasen. Sie werden als katalytisch inaktive Proteine nach Abspalten des Signalpeptids ausgeschleust (Trackman, P.C., et al.; 1992). Die C-terminale (ca. 28-32 kDa) Domäne des Proteins wird anschließend durch die Metalloendoproteinase Procollagen C-Proteinase proteolytisch abgetrennt (Uzell, M.I., et al.; 2001) und ist dann katalytisch aktiv. Für die N-terminale Domäne wird eine Schutzfunktion durch Blockierung angenommen, zum Schutz vor unerwünschter Aktivität innerhalb der Zelle. Ob die N-terminale Domäne von KsuL eine vergleichbare Blockierungsfunktion besitzt, wird zu untersuchen sein. Eine mögliche Funktion als Bindedomäne und/oder eine Chaperon-Funktion für die C-terminale Domäne wird vermutet. Ob die proteolytische Trennung der N-terminalen und C-terminalen Domäne durch eine Metalloendoprotease erfolgt, wird durch die Verwendung „nicht toxischer“ Proteaseinhibitoren im Medium zu untersuchen sein. Bei verschiedenen extrazellulären Enzymen von Streptomyceten (z.B. Cellulasen, Xylanasen und Chitinasen) wurden jeweils spezifische Proteasen identifiziert (Fernández-Abalos, J.M., et al.; 2003; Schrempf, H. and Walter, S.; 1995; Blaak, H. and Schrempf, H.; 1995). KsuL ist ein Orphan-Protein, das sich nur in den bisher sequenzierten Streptomycetenstämmen und einem Nocardia-Stamm findet; in allen weiteren Actinomycetenstämmen ist kein KsuL-Homolog zu entdecken. Die im Rahmen von Sequenzierungsprojekten identifizierten KsuL-Homologen aus S.avermitilis (BAC72171) und Nocardia spec. JS614 (ZP_00656873) sind bisher nicht weiter untersucht worden (siehe Anhang E). Vor kurzem wurden die Sequenzdaten von S.scabies veröffentlicht (http://www.sanger.ac.uk/Projects/S_scabies/). Auch hier findet sich ein KsuL-Homolog mit einer vermutlichen Länge von 586 Aminosäuren (Basen: 4945557-4943764). Diese drei und KsuL zeigen untereinander besonders in der C-terminalen Hälfte sehr hohe Aminosäureidentitäten, die der N-terminalen Hälfte sind geringer, im Bereich des Signalpeptids sind kaum Ähnlichkeiten zu finden (siehe Anhang E). Der Translationsstart des Homologs aus S.avermitilis ist falsch postuliert, der vermutlich richtige Translationsstart beginnt am Methionin an der Position 9. Die genannten abgeleiteten Proteine unterscheiden sich untereinander und zu KsuL vor allem durch Verkürzungen und eine hohe Divergenz im Bereich vor der experimentell nachgewiesenen proteolytischen Schnittstelle von KsuL (KsuL: ca. 245-334, siehe Anhang E). Ob dieser Abschnitt als Verbindungs-Domäne dient, die die Beweglichkeit der N-terminalen zur C-terminalen Domäne bedingt, wird zu untersuchen sein. - 148 - Diskussion Die im Bereich der proteolytischen Schnittstelle zwischen der N- und C-terminalen Domäne liegenden Aminosäuren sind konserviert, die Konsensussequenz lautet Pro−X−Gly−Lys/Arg−Ala−║−X−Val/Glu. In „reifen“ eukaryotischen Lysyl-Oxidasen sind zehn Cysteine zu finden, die fünf DisulfidBrücken bilden und für die Stabilität der Konformation bedeutend sind (Williams, M.A. and Kagan, H.M.; 1985). In KsuL finden sich vier Cysteine, jeweils zwei in der N-terminalen (Cys 145 und Cys167), wie auch in der C-terminalen Domäne (Cys471 und Cys498). Diese sind auch in den oben genannten KsuL-Homologen konserviert (siehe Anhang E). Durch Verwendung reduzierender Agenzien konnte sowohl für die N- als auch für die C-terminale Domäne jeweils eine Disulfid-Brücke nachgewiesen werden (eigene Ergebnisse, nicht dargestellt, pers. Mitteilung H.Schrempf). Anders als bei den eukaryotischen Lysyl-Oxidasen ist der Anteil der Lysin-Reste in KsuL sehr hoch. In KsuL sind 40 Lysine zu finden, im Vergleich zu nur fünf bis sechs bei eukaryotischen Lysyl-Oxidasen (Kagan, M.H. and Li, W.; 2003). Die Kupfer enthaltenden „semicarbazide-sensitive amine oxidases“ (SSAOs) besitzen Topaquinon (2,4,5-Trihydroxyphenylalanin Quinon; TPQ) als Cofaktor (Janes, S.M., et al.; 1990); als Ausnahme konnte experimentell für die Lysyl-Oxidase aus der Ratte LysylTyrosyl-Quinon (LTQ) als Cofaktor identifiziert werden (Wang, S.C., et al.; 1996). Auf Grund von Sequenzvergleichen wird auf das Vorhandensein von LTQ auch in allen weiteren eukaryotischen Lysyl-Oxidasen und LOXL Proteinen geschlossen. Die an der Lysyl Tyrosylquinon Cofaktorbildung beteiligten Tyrosin und Lysin sind an den Positionen Tyr518 und Lys468 in KsuL konserviert (siehe Abb. 3.45), ebenso an den entsprechenden Positionen in den oben genannten KsuL-Homologen. Somit ist anzunehmen, dass KsuL wie die eukaryotischen Lysyl-Oxidasen ein Lysyl-Tyrosyl-Quinon als Cofaktor trägt. In unserer Arbeitsgruppe erfolgen zurzeit Untersuchungen zum Nachweis desselben (M.Kotasińska, pers. Mitteilung). Lysyl-Oxidasen gehören zur Gruppe der Kupfer enthaltenden Semicarbazid-sensitiven Amin Oxidasen (SSAO, Jalkanen, S. and Salmi, M.; 2001). Pro Lysyl-Oxidase Molekül ist ein Cu(II) Atom als Cofaktor relativ fest gebunden (Gacheru, S.N., et al.; 1990). Es wird vermutet, dass die Bindung an je drei Histidine erfolgt. Das His-X-His-X-His Motiv ist in KsuL wie auch den oben genannten Homologen konserviert und findet sich in KsuL an der Position 441-445 (siehe Abb. 3.45). Cu(II) ist am Elektronentransfer vom und zum Sauerstoff beteiligt, zur oxidativen Deaminierung der Peptidyl-Lysyl Gruppe und an der Quinon Cofaktor Bildung (Rucker, R.B., et al.; 1998). Zurzeit laufen weiterführende Versuche zum Nachweis des Cu(II) Cofaktors in unserer Arbeitsgruppe (I.Koebsch, pers. Mitteilung). Weiterführende Studien zeigten auf, dass aus S.lividans isoliertes KsuL mit der Zeit zur Aggregation neigt und als Niederschlag ausfällt; es bilden sich hierbei kovalent verknüpfte Multimere, vermutlich zurückzuführen auf autokatalytische Quervernetzung der Monomere. Vergleichbares wurde auch bei eukaryotischen Lysyl-Oxidasen gefunden (Romero-Chapman, N, et al.; 1997; Bedell-Hogan, D., et al.; 1993; Kagan, H.M., et al.; 1979). Diskussion - 149 - Wie eigene Versuche z.B. mit Hilfe des EGFP Expressionstestkonstruktes (S.lividans TK21 pOVG14) gezeigt haben (siehe Abb. 3.34 und Abb. 3.35, jeweils i und j) und in weiterführenden Arbeiten in unserer Arbeitsgruppe bestätigt wurde (pers. Mitteilung H.Schrempf), wird KsuL unabhängig von der Wachstumsphase und des verwendeten Mediums produziert. Flussaufwärts von ksuL, getrennt durch einen 776 Bp langen intergenischen Bereich, finden sich in derselben Leserichtung die Gene ksuC und ksuB. Im intergenischen Bereich sind keine für einen Terminator typischen „inverted repeats“ zu finden, wie flussabwärts von ksuL (siehe Anhang A und B). Die KsuL Synthese ist in den ∆ksuC und ∆ksuB Disruptionsmutanten im Vergleich zum Wildtyp und weiteren Disruptionsmutanten reduziert (siehe Abb. 3.64). Somit ist zu vermuten, dass ksuL von einer polycistronischen mRNA, beginnend vor ksuC, wie auch von einer monocistronischen mRNA, mit einem Startpunkt im intergenischen Bereich zwischen ksuB und ksuL, transkribiert wird. Die Medium- und Wachstumsphasen- unabhängige Produktion von KsuL wird hierbei wahrscheinlich von der monocistronischen mRNA determiniert. Die Produktion von KsuC und KsuB ist unter hohen Salzkonzentrationen deutlich erhöht, eine erhöhte Transkription von KsuL unter Hochsalzbedingungen ist anzunehmen. Dies wurde in Vorversuchen beobachtet (nicht dargestellt). In Bakterien stehen häufig die von einem Operon determinierten Proteine in einem funktionellen Zusammenhang. Dieser ist jedoch für die zwei cytoplasmatischen Aldo-Keto Reduktasen und der extrazellulären Lysyl-Oxidase KsuL nicht zu erkennen. Das von einem flussabwärts von kcsA gelegene offenen Leseraster orfI determinierte OrfI wurde im Rahmen dieser Arbeit nur am Rande untersucht. Es sind weder Antikörper gegen OrfI vorhanden, noch konnte eine Synthese des Proteins gezeigt werden. Die im Rahmen dieser Arbeit hergestellte Diruptionsmutante ∆orfI zeigt keine Unterschiede im Phänotyp zum Wildtyp. Eine Auswirkung auf die Resistenz gegen Chloramphenicol, determiniert vom flussabwärts von orfI in gleicher Leserichtung gelegenen cmlR, ist nicht zu erkennen. Es ist zu vermuten, dass kcsA und orfI auf einem Operon liegen und von einer polycistronischen mRNA mit einem Transkriptionsstart vor kcsA determiniert werden. Jürgen Ritz konnte in Transkriptionsstudien weder für kcsA noch für orfI ein Transkriptionsprodukt nachweisen (J.Ritz, pers. Mitteilung). Im S.coelicolor A3(2) Genomprojekt wurde ein um 11 Aminosäuren früherer Translationsstart postuliert (SCO7661), als der vermutlich richtige (siehe Anhang A). Analysen des GC Gehaltes lassen diesen jedoch sehr unwahrscheinlich erscheinen, auch ist flussaufwärts von diesem keine potentielle Shine Delgarno Region zu finden. OrfI ist ein vermutlich cytoplasmatisches Protein, das über die vollständige Länge in kurzen Motiven Ähnlichkeiten zur Dihydrofolat-Reduktase Superfamilie (DHFR, SSF53597) zeigt. Dihydrofolat-Reduktasen katalysieren die Reduktion von Dihydrofolat zu Tetrahydrofolat, das als Cofaktor z.B. an der de novo Synthese von Glycin, Purinen und Deoxythymidin-Phosphat beteiligt ist. OrfI enthält einige Motive der C-terminalen Domäne bakterieller bifunktionaler Deaminasen-Reduktasen, die am zweiten Schritt der - 150 - Diskussion Riboflavinsynthese aus GTP beteiligt sind (Richter, G., et al.; 1997), der Vorstufe von FAD. Es ist deshalb vermutet worden, dass OrfI eine Modifikation des Flavin Cofaktors von KsuF durchführen könnte. Da nur FAD als Cofaktor gefunden wurde, ist dies auszuschließen. Die Funktion von OrfI bleibt unter den getesteten Bedingungen obskur, auch ist kein funktioneller Zusammenhang mit KcsA oder einem weiteren der in dieser Arbeit untersuchten Proteine zu erkennen. Im Rahmen dieser Arbeit wurde ksuB und der für die C-terminale Hälfte determinierende Abschnitt von ksuC sequenziert. KsuB wurde in E.coli als His-Tag Protein überproduziert, aufgereinigt und zur Herstellung von Antikörpern verwendet. In Zusammenarbeit wurde von Ralf Steger ksuC mit Hilfe einer S.lividans Charon 35 Genom-Phagenbank vervollständigt, KsuC als His-Tag Protein aus E.coli aufgereinigt und zur Herstellung von Antikörpern verwendet. Sowohl KsuC als auch KsuB sind Mitglieder der Aldo-Keto Reduktase (AKR) Superfamilie, deren Mitglieder reduzieren als Oxidoreduktasen Aldehyd- oder Ketogruppen zu primären oder sekundären Alkoholgruppen. Als Cofaktoren verwenden sie NADPH, seltener NADH, wie z.B. AKR1C19 aus der Maus, das „in vitro“ sowohl NADPH als auch NADH gleichwertig als Cofaktor verwenden kann aber „in vivo“ vermutlich ausschließlich NADH verwendet (Ishikura, S., et al.; 2005). Im Rahmen dieser Arbeit erfolgten keine Arbeiten zur Enzymaktivität oder zur Identifizierung des Cofaktors von KsuC und KsuB. Als Substrate der Aldo-Keto Reduktasen dienen aliphatische und aromatische Aldehyde und Ketone wie Zucker, Steroide, Prostaglandine und Xenobiotika (Jez, J.M., et al.; 1997). Die Substratspezifität ist gering. So ist die Ratten-Leber Aflatoxin-Dialdehyd Reduktase (AKR7A1) in der Lage, neben Aflatoxin B1 Dialdehyd auch eine Reihe weiterer Aldehyde und Diketone zu reduzieren. Hierbei zeigt sich eine sehr hohe Affinität zu den strukturell sehr unterschiedlichen Succinat-Semialdehyd, 2-CarboxyBenzalaldehyd und 9,10-Phenanthrenquinon (Kozma, E. et al.; 2002; Kelly, V.P., et al.; 2000; Ellis, E.M. and Hayes, J.D.; 1995). Wie für „konjugierte Polyketon-Reduktasen“ aus verschiedenen Pilzstämmen gezeigt wurde, besitzen auch diese eine sehr große Substratdiversität, mit strikter Stereospezifität (Hidalgo, A.-R. G. D., et al.; 2001). Eine strikte Stereospezifität des verwendeten Substrates ist typisch für NAD(P)H abhängigen Enzymen, wie schon 1953 für die Alkohol-Dehydrogenase gezeigt wurde (Fisher, H.F., et al.; 1953). Auf Grund der sehr geringen Substratspezifität der Aldo-Keto Reduktasen und der Möglichkeit, sowohl KsuC als auch KsuB als His-Tag Proteine in S.lividans-Transformanten zu produzieren und als lösliches Protein aufzureinigen, sollte ein Nachweis und Untersuchung einer enzymatischen Aktivität und des Cofaktors möglich sein. Es konnten keine Hinweise gefunden werden, dass KsuC und KsuB als Oligomer vorliegen. Die meisten Mitglieder der Aldo-Keto Reduktase Superfamilie sind monomere Proteine. Es finden sich jedoch auch Dimere, wie AKR7A1 und AKR7A4 und vermutlich die gesamte AKR7-Familie der Aldo-Keto Reduktasen (Kozma, E., et al.; 2002) und Tetramere, wie AKR6A2, der β-Untereinheit der spannungsabhängigen Kalium-Kanäle (Gulbis, J.M., et al.; Diskussion - 151 - 2000). YDHF, eine Aldo-Keto Reduktase aus E.coli unbekannter Funktion wurde als Trimer kristallisiert (Pdb:1og6 Chain a-c, keine Publikation). Bisher ist die strukturelle Aufklärung von einer Reihe von Aldo-Keto Reduktasen gelungen. Sowohl KsuC als auch KsuB zeigen die größten Übereinstimmungen zu Proteinen dieser Familie mit bekannter Struktur zu einer Aldo-Keto Reduktase aus E.coli (YDHF, Pdb:1og6 Chain a-c). Aus einem Sequenzvergleich zu YDHF (siehe Abb. 3.48) wurden sowohl für KsuC wie auch KsuB, einschließlich des Cofaktors NADP, die Kristallstrukturen errechnet und in Abb. 3.49 a-d) dargestellt. Eine enzymatische Aktivität für dieses Protein ist bisher nicht publiziert. YDHF wurde als Trimer kristallisiert und die abgeleitete Struktur in die Datenbank gestellt. Da trotz Ankündigung (2003) bisher keine Publikation zu YDHF veröffentlicht wurde (Jeudy, S., et al.; to be published), erscheint die Korrektheit der für Mitglieder der Aldo-Keto Reduktasen einzigartigen trimeren Struktur fraglich. Wie zuerst für die menschliche Aldose-Reduktase nachgewiesen (AKR1B1; Rondeau, J.M., et al.; 1992; Wilson, D.K., et al.; 1992), besitzen die Mitglieder der Aldo-Keto Reduktase Superfamilie ein β-8/α-8-Fass (TIM Barrel) als Tertiärstruktur (Farber, G.K. and Petsko, G.A.; 1990), das sich auch in KsuC und KsuB findet (siehe Abb. 3.48 und 3.49). KsuB zeichnet sich im Vergleich zu YDHF durch den Besitz einer 17 Aminosäuren langen Insertion (Aminosäuren 186-202 von KsuB) im Anfangsbereich der α-6-Helix aus, für die eine antiparallele β/β-Faltblattstruktur postuliert wird (siehe Abb. 3.48 und Abb. 3.49 a). Diese liegt im Kontaktbereich der Untereinheiten des postulierten Trimers und lässt die Wahrscheinlichkeit einer Oligomerisierung von KsuB gering erscheinen (siehe Abb. 3.49 c). Die putative Ausbildung von neun Wasserstoffbrücken, hiervon sieben zwischen den antiparallelen β-Faltblattstrukturen, spricht für die Korrektheit der postulierten antiparallelen β/β-Faltblattstruktur (erstellt mit Deep View/Swiss PDB Viewer). Die an der Cofaktorbindung beteiligten Aminosäuren sind aus einer Reihe von Röntgenstrukturanalysen von Mitgliedern der Aldo-Keto Reduktase Familie bekannt. In Abb. 3.48 sind die über Salzbrücken und Wasserstoffbrückenbindungen beteiligten, der Aldehyd-Reduktase (ALR1), der Aldose-Reduktase (ALR2) und des „Fibroblast Wachstumsfaktor induzierten Proteins“ (FR-1), eingetragen (El-Kabbani, O., et al.; 1998, ElKabbani, O., et al.; 1997; Wilson, D.K., et al.; 1993; Wilson, D.K., et al.; 1995). Von den 17 an der Cofaktor-Bindung beteiligten Aminosäuren sind acht in YDHF, sieben in KsuB und sechs in KsuC konserviert. Alle Aminosäuren, die an der Bindung des katalytisch aktiven Nicotinamid Teils von NADP beteiligt sind, sind sowohl in KsuB, als auch in KsuC vollständig konserviert (KsuB: Asp47, Ser151, Asn152 und Gln175; KsuC: Asp48, Ser158, Asn159 und Gln182). Auf Grund der Kristalldaten wird postuliert, dass der katalytische Mechanismus der Aldo-Keto Reduktasen auf einem stereospezifischen Transfer des 4-pro-R Wasserstoff des C-4 des Nicotinamids von NADPH zum C-Atom der Carbonylgruppe des Substrates basiert, gefolgt von der Protonierung des Sauerstoffes der Carbonylgruppe des Substrates durch eine Protonen Donor Gruppe. Tyr50 und Lys80 bilden eine Wasserstoffbrücke mit der Ammonium Seiten-Kette des Lys80 in ALR1 aus, welches eine - 152 - Diskussion Salz-Brücke mit Asp45 ausbildet. Diese sind am Protonen-Transfer beteiligt (El-Kabbani, O., et al.; 1998) und sowohl in KsuC wie auch KsuB konserviert (KsuB: Tyr52, Lys82 und Asp47; KsuC: Tyr53, vermutlich Lys86 und Asp48). Für beide Proteine ist somit ein vergleichbarer katalytischer Mechanismus anzunehmen (siehe Abb. 3.48.). Eigene Auswertung der Kristalldaten von YDHF ergaben, das Gly264 eine Wasserstoffbrücke zur Phosphatgruppe und Gly213 eine zum Pyrophosphat von NADP ausbildet (Deep View/Swiss PDB Viewer). Gly264 ist in KsuC (Gly257) und KsuB (Gly275) konserviert, wie auch Gly213 in KsuC (Gly211); in KsuB findet sich an dieser Position ein Glutamin (Gln223). Mit Hilfe der putativen Kristalldaten kann nicht bestimmt werden, ob NAD oder NADP als Cofaktor für KsuC und KsuB verwendet wird. Die Synthese von KsuC und KsuB wird vermutlich von einer polycistronischen mRNA determiniert, mit einem Transkriptionsstart vor ksuC; ksuC und ksuB sind durch einen intergenischen Bereich von 55 Bp Länge getrennt. Es konnten keine Hinweise gefunden werden, dass in diesem Bereich ein Promoter zu finden ist. Dies konnte auch mit Hilfe der ∆ksuC und ∆ksuB Disruptionsmutanten gezeigt werden. Weder KsuC noch KsuB konnten in der ∆ksuC Disruptionsmutante nachgewiesen werden. In der ∆ksuB Disruptionsmutante ist KsuC vergleichbar dem Wildtyp nachweisbar (siehe Abb. 3.63 a-d). Beide Disruptionsmutanten zeigen bei Wachstum auf Minimalmediumplatten eine erhöhte Produktion von Undecylprodigiosin (RED), jedoch keine Veränderung bezüglich der Produktion von Actinorhodin (Act) (siehe folgendes Kapitel: UndecylprodigiosinÜberproduktion in Mutanten). Wie auch bei KsuF gefunden, ist die Synthese von KsuC und KsuB bei Wachstum in Minimalmedium mit hohen Salzkonzentrationen deutlich erhöht. Die Synthese ist jeweils unabhängig davon, ob NaCl oder KCl verwendet wurden (siehe Abb. 3.10 und Abb. 3.11, jeweils b und c) und wird in einem der folgenden Kapitel besprochen (siehe: Synthese von drei Proteinen unter verschiedenen physiologischen Bedingungen). Diskussion - 153 - Das in unser Artbeitsgruppe entdeckte Kalium Kanalprotein KcsA (Schrempf, H. et al.; 1995) ist, seit der gelungenen Kristallisation und Aufklärung der Struktur, der Archetyp aller Kanalproteine (Doyle, D.A., et al.; 1998). Vorwiegend für die Strukturaufklärung von KcsA wurde im Jahre 2003 der Nobelpreis an R. MacKinnon im Bereich Chemie vergeben. Wie Sequenzvergleiche zeigen, ist KcsA mit seiner geringen Länge und dem im Verhältnis zu anderen Kanalproteinen sehr kurzen cytoplasmatischen C-terminalen Teil einzigartig. Es konnte im Rahmen der Diplomarbeit in Hybridisierungsexperimenten an chromosomalen DNA’s verschiedener Streptomycetenstämme nach „Puls Feld“ Gelelektrophorese gezeigt werden, dass kcsA nur in Mitgliedern der S.violaceoruber Familie (S.violaceoruber, S.lividans und dem getesteten S.coelicolor A3(2) Derivat M145) vorhanden ist. Die gegen KcsA vorhandenen Antikörper (H.Splitt) waren, da sie nur eine verhältnismäßig geringe Spezifität gegen KcsA besaßen und sowohl gegen die Proteine der Membranfraktionen aus E.coli als auch S.lividans WT sehr starke unspezifische Kreuzreaktionen zeigten, nur bedingt zu gebrauchen. Da die membrandurchspannenden Teile eines Membranproteins häufig nur eine geringe Antigenität besitzen, erfolgte die Antikörperherstellung gegen das cytoplasmatische C-terminale Ende von KcsA. Hierbei zeigte sich, dass die vorletzte „Charge“ eine deutlich größere Reaktion gegen KcsA besaß als die zuletzt gelieferte. Mit nur 6,3 kDa ist das zur Immunisierung eingesetzte Peptid verhältnismäßig klein. Wie in zahlreichen Versuchen gezeigt wurde, besitzen kurze Peptidfragmente eine geringere Antigenität als große Proteine (The QIAexpressionistTM, Qiagen, Hilden). Die vorgefundenen Probleme sind vermutlich hierauf zurückzuführen. Es ist nicht gelungen, KcsA im Wildtyp oder in S.lividans Transformanten, die kcsA auf einem Konstrukt trugen, in der Membranfraktion mit Hilfe von Antikörpern („Western Blot“) nachzuweisen. Auch in Transkriptionsstudien war weder im Wildtyp noch in kcsA auf einem Konstrukt tragenden Transformanten ein entsprechendes Transkriptionsprodukt zu finden (J. Ritz, pers. Mitteilung). In den EGFP Insertionsmutanten, die die Synthese eines KcsAEGFP Fusionsproteins ermöglichen sollten, ist weder mit Hilfe von „Western Blot“ noch durch Fluoreszensmessungen eine Produktion von KcsA-EGFP nachweisbar. Auch in den KcsA-EGFP Produktions-Test Transformante S.lividans TK21 pOVG11 war unter keiner der getesteten Bedingungen eine EGFP-Produktion zu finden (siehe Abb. 3.34. und Abb. 3.35, jeweils c und d). Ein Nachweis im „Western Blot“ ist nur in den S.lividans pOVE31 und pOVE51 Produktions-Transformanten, die die Synthese eines N-terminalen His-Tag KcsA ermöglichen, erfolgreich (siehe Abb. 3.21). Es ist unklar, ob die Synthesesrate von KcsA zu gering ist, um KcsA mit Hilfe von Antikörpern nach Transfer auf Membranen nachzuweisen oder ob der KcsA-Transfer auf Grund der Bildung nicht auflösbarer hochmolekularer Aggregate („Cluster“) verhindert wird. Ein Nachweis von KcsA mit Hilfe der in dieser Arbeit hergestellten Antikörper im Wildtyp ist bisher ausschließlich elektronenmikroskopisch möglich (Hegermann, J., et al.; 2006, im Druck). KcsA liegt hierbei in „Clustern“ vor und ist im S.lividans WT nur in ca. einer von fünfzig angeschnittenen Hyphen nachweisbar, jedoch nicht in den in dieser Arbeit - 154 - Diskussion hergestellten Deletionsmutanten. In Zusammenarbeit mit Jan Heggemann konnte KcsA in den S.lividans pOVE31 Transformanten ebenfalls in „Clustern“ nachgewiesen werden. Sie sind jedoch anscheinend größer und in nahezu jedem Schnitt einer Hyphe nachweisbar. Die S.lividans pOVE3 Transformante als Kontrolle zeigt eine dem S.lividans Wildtyp vergleichbar geringere Anzahl (nicht dargestellt). Jan Hegermann konnte in weiterführenden Arbeiten mit Hilfe von „Energy-filtering-TEM“ (EFTEM) für die ∆kcsA Mutante, gegenüber dem Widtyp, eine ca. 30 % geringere Akkumulation von Cäsium-Ionen in der Zelle nachweisen (Hegermann, J., et al.; 2006, im Druck). Der S.lividans Wildtyp und die ∆kcsA-Disruptionsmutante unterscheiden sich weder bezüglich des Wachtums und der Differenzierung, noch finden sich Unterschiede im Wachstumsverhalten bei Verwendung von unterschiedlichen NaCl- und KCl-Konzentrationen im Medium. Auch Stämme, die zusätzlich zum chromosomalen kcsA weitere Kopien auf einem Konstrukt tragen und die His-KcsA Produktionstransformanten S.lividans TK21 pOVE31 und pOVE51, zeigen keine Unterschiede zu den entsprechenden Kontrollen. Im S.coelicolor A3(2) Genom finden sich neben kcsA eine Reihe weiterer Gene für putative Kalium-Transportproteine. So findet sich ein weiteres Kanalprotein (SCO7196), ein vollständiges Kalium-Transport ATPase A Operon (SCO3719-3716), Gluthathion regulierte Kalium-Efflux-System Proteine der KefB- (SCO3279 und SCO5782) und der KefC-Familie (SCO3602 und SCO3826), zwei Kupfer/Kalium-Transport ATPase A Proteine (SCO0860 und SCO6460), zwei auf einem Operon liegende TrkA Kalium-Aufnahme Proteine (SCO5876 und SCO5875) und ein Kalium-Efflux-System Protein (SCO6958), das hohe Identitäten zu zwei Proteinen aus Rhizobium meliloti zeigt. Des Weiteren ist eine große Anzahl von weiteren abgeleiteten Kation-Efflux Proteinen und ATPasen zu finden. Es ist zu vermuten, dass diese in der Lage sind, den Verlust von KcsA durch Disruption vollständig zu kompensieren. KcsA findet sich nach Produktion in E.coli als Tetramer, das sich durch eine außergewöhnliche Stabilität gegenüber Detergenzien und Hitze auszeichnet. Längeres Erhitzen von KcsA über 80°C in SDS-haltigem Probenpuffer führt zum Zerfall des Tetramers in Monomere (Meuser, D., et al.; 2001, siehe auch Abb. 3.5 a, Spuren 1-3 und 4-6). Mutationen im Bereich des Selektivitätsfilters (GYG) von KcsA haben einen Einfluß auf die Stabilität des Tetramers gegenüber Hitze (Meuser, D., et al.; 2001). KcsA ist frei von Cysteinresten. Die Gabe von reduzierenden Agenzien hat somit keinen Einfluss auf die Stabilität des Tetramers. Wie im Rahmen dieser Arbeit gezeigt wurde, zeigt KcsA nach Produktion als His-Tag Protein in S.lividans (siehe z.B. Abb. 3.21 a, Spuren 3 und 4) und nach Synthese mit Hilfe des Baculovirus-Systems in Insektenzellen als His-Tag Protein oder als Protein „nativer“ Länge ein vergleichbares Verhalten. KcsA inseriert sich von selbst innerhalb der kürzesten messbaren Zeit (<30’’) nach Synthese vollständig als Monomer in die Membran (van Dalen, A., et al.; 2000). Die elektrische Komponente des „proton motive force“ (pmf) wird für die korrekte Tetramerisierung in der Membran benötigt. Die Integration des Monomers in die Membran ist unabhängig von aktiven ATP verbrauchenden Systemen wie z.B. SecA (van Dalen, A., et al.; 2000). Dies Diskussion - 155 - konnte auch im Rahmen dieser Arbeit gezeigt werden. Es wurde versucht, KcsA, His-KcsA, und KcsA und His-KcsA als EGFP-Fusionsprotein (mit und ohne N-terminalen Signalpeptid) mit Hilfe des Baculovirussystems in Insektenzellen zu produzieren und die Kanalaktivität mit der „Patch Clamp“ Methode zu messen (in Zusammenarbeit mit K.Hill, nicht im Ergebnisteil dargestellt). Nach Herstellung der Konstrukte und Transfektion zeigte sich, das diese Versuche nicht erfolgreich waren. Die Translokation von KcsA und der entsprechenden Derivate in die äußere Membran gelang nicht. KcsA (und Derivate) konnte sowohl nach Produktion mit als auch ohne Signalpeptid ausschließlich in den inneren Zellmembranen der Insektenzellen nachgewiesen werden, das Signalpeptid war (wenn vorhanden) nicht abgeschnitten. Des Weiteren wurde vermutlich im C-terminalen Endbereich von KcsA eine Proteaseschnittstelle gefunden, KcsA und die Derivate lagen jeweils C-terminal verkürzt vor; mit Hilfe von Floreszenzmikroskopie wurde gezeigt, dass EGFP im Cytoplasma lokalisiert und nicht Membran-assoziiert vorlag (nicht dargestellt). Eine Kanalaktivität konnte jeweils nicht nachgewiesen werden (pers. Mitteilung K.Hill). Weitere Arbeiten wurden aufgegeben. Bis zum heutigen Zeitpunkt finden sich keine Veröffentlichungungen über Versuche, KcsA mit Hilfe des Baculovirus-Systems in Insektenzellen produzieren und die Kanalaktivität mit Hilfe der „Patch Clamp“-Methode oder nach Aufreinigung und Rekonstitution mit Hilfe „planarer Bilyer-Systeme“ zu messen. Es ist zu vermuten, dass KcsA nach Synthese mit Hilfe des Baculovirussystems nicht funktionell ist. Das von cmlR determinierte Cloramphenicol-Resistenzprotein CmlR wurde in dieser Arbeit nur marginal untersucht. CmlR wurde in früheren Arbeiten in unserer Arbeitsgruppe entdeckt und als Chloramphenicol-Resistenzprotein charakterisiert. Es schleust das Antibiotikum Chloramphenicol als membranständiges Transportprotein aus (Dittrich, W., et al.; 1991). Antikörper gegen CmlR sind nicht vorhanden. Wie im Rahmen dieser Arbeit mit Hilfe der S.lividans pOVG12 Transformante, die das EGFP-CmlR-Produktionstestkonstrukt trägt, gezeigt wurde, wird die CmlR-Synthese durch die Gabe von Chloamphenicol induziert. In Medien ohne Chloramphenicol ist keine oder nur eine marginal über der Nachweisgrenze liegende Produktion von EGFP nachweisbar. Nach Gabe von Chloramphenicol ist eine starke Fluoreszenz zu erkennen (Abb. 3.34 und 3.35, jeweils e und f und Abb. 3.36 a-d, i und j). Die Synthese von CmlR wird somit, wie für andere Resistenzproteine beschrieben, durch das Antibiotikum induziert (Wright, G.D.; 2005). Die S.lividans pOVG12 Transformante zeigt, bei Gabe von Chloramphenicol im Medium, eine deutlich verringerte Wachstumsrate im Vergleich zu anderen Stämmen; nach ca. vier Tagen Wachstum als Schüttelkultur wird ein vergleichbares Zellvolumen erreicht. Es ist zu vermuten, dass der in hoher Kopienzahl vorliegende konstruktdeterminierte Promoter-Bereich von cmlR, das für die Transkription von cmlR verantwortliche Regulatorprotein abfängt. Dies hat eine verringerte Synthese des chromosomal determinierten Resistenzproteins zur Folge. Flussaufwärts von cmlR finden sich orfI und kcsA, die jeweils in identischer Leserichtung zu cmlR vorliegen. Es ist in diesem Abschnitt kein potentieller Terminator zu erkennen. Zwischen orfI und cmlR liegt ein 238 Bp langer intergenischer Bereich. Es wurde postuliert, das CmlR sowohl von einer - 156 - Diskussion polycistronischen mRNA mit einem Transkriptionsstartpunkt vor kcsA, als auch einer monocistronischen mRNA mit einem Startpunkt im intergenischen Bereich zwischen orfI und cmlR, determiniert würde. Jürgen Ritz führte in seinen Arbeiten zu kcsA auch Transkriptionsstudien zu cmlR durch. Er konnte bei Arbeiten mit S.lividans WT und den in dieser Arbeit hergestellten S.lividans NI pOV001/005/006/007 Transformanten unter Chloramphenicol-Induktion ein monocistronisches cmlR-Transkript nachweisen. Für die postulierte polycistronische mRNA gelang dies nicht (Jürgen Ritz, pers. Mitteilung). Dies entspricht den zu den ∆kcsA und ∆orfI-Disruptionsmutanten erhaltenen Ergebnissen. Beide Mutanten zeigten im Vergleich zum Wildtyp keinen Unterschied in der Resistenz gegen Chloramphenicol. Die cmlR Transkription wird durch die Anwesenheit von Chloramphenicol induziert und CmlR wird unter den getesteten Bedingungen primär von einer monocistronischen mRNA determiniert. Diskussion - 157 - 4.2. Synthese von drei Proteinen unter verschiedenen physiologischen Bedingungen Wie in dieser Arbeit gezeigt wurde, ist die Synthese von KsuF abhängig vom Vorhandensein des tsr-Gens und der Synthese des hiervon determinierenden Thiostreptonresistenzproteins Tsr aus Streptomyces azureus (siehe Ergebnisse 3.3.5). In Stämme, die kein tsr-Gen enthalten (S.lividans 66 WT und S.coelicolor M145), ist die Synthese von KsuF auch unter Thiostrepton-Gabe nicht nachweisbar. Eine Beteiligung von Konstrukt-determinierten Genen konnte durch die Verwendung von Stämmen, die das tsr-Gen ins Chromosom integriert hatten (S.coelicolor M145 sigH:tsr und S.colicolor J1984 sigF:tsr) ausgeschlossen werden. Thiostrepton wird für die Transkription des tsr-Gens und die nachfolgende Synthese des Resistenzproteins benötigt. Es kann nicht vollkommen ausgeschlossen werden, dass das Thiostreptonresistenzprotein zusammen mit Thiostrepton für die Induktion der KsuFSynthese benötigt wird. Dies erscheint jedoch unwahrscheinlich, da Thiostrepton nicht in der Lage ist, an A1067 O2methylierte 23S mRNA zu binden (Thompson, C.J., et al.;1982b). In Zusammenarbeit führte Jürgen Ritz in seinen mRNA Arbeiten zu kcsA auch Arbeiten zu ksuF durch. Während im S.lividans Wildtyp kein ksuF-Transkript nachzuweisen war, konnte in S.lividans 66 pJO1 und S.lividans NI pOV005 Transformanten, beides Konstrukte, die ksuF tragen, eine extrem hohe Menge eines monocistronischen ksuF-Transkriptes gefunden werden. Es wurden keine Versuche mit einer der S.lividans Wildtyp Transformanten durchgeführt, die ein tsr haltiges Konstrukt ohne ksuF tragen. Nach den Produktionsdaten zu schließen, wäre hier ein ksuF-Transkript gefunden worden. Die Transkriptions- und Produktionsstudien zeigen, dass das Vorhandensein des Tsr-Proteins die Transkription von ksuF bedingt. Es konnten in der Literatur keine Beispiele gefunden werden, dass das Thiostreptonresistenzprotein zur Transkription von Genen führt. Dieses Phänomen wurde auch für andere Resistenzproteine, die eine Modifikation der 16S bzw. 23S rRNA durchführen, nicht beschrieben. Die in dieser Arbeit gefundenen Erkenntnisse sind neu. Das von Streptomyces azureus synthetisierte Thiostrepton (multizyklisches Thiazol PeptidAntibiotikum) hemmt die Peptid Synthese, indem es am L11-23S rRNA Komplex bindet, die Bindung von EF-G am Ribosom verhindert (Lentzen, G., et al.; 2003; Cameron, D.M., et al.; 2002) und so die Termination der Translation bewirkt (übersichtlich dargestellt in: Bowen, W.S., et al.; 2005). Des Weiteren inhibiert es die prokaryotischen Terminationsfaktoren RF1, RF2 sowie RelA (Brot, N., et al.; 1974; Haseltine, W.A., et al.; 1972). Trotz jahrzehntelanger Forschung ist die genaue Ursache der Peptidsynthese-Inhibition durch Thiostrepton noch nicht verstanden. Das von tsr determinierte Thiostreptonresistenzprotein von S.azureus (Bibb, M.J., et al.; 1985; Thompson, C.J., et al.; 1982a) ist eine O2Methylase und methyliert das Adenosin 1067 der 23S rRNA bevor es in die 50S ribosomale Untereinheit eingebaut wird (Bechthold, A. and Floss, H.G.; 1994; Cundliffe, E.; 1989). Thiostrepton induziert die Synthese des Thiostreptonresistenzproteins (Janssen, G.R. and Bibb, M.J.; 1990) und tsr wird seit langem als Resistenz zur Selektion in Streptomyceten Vektoren eingesetzt; es ist zurzeit der gebräuchlichste Marker. - 158 - Diskussion Im S.coelicolor A3(2) Genom-Sequenzierungsprojekt (Bentley, S.D., et al.; 2002) konnte kein tsr Homolog gefunden werden. Somit ist anzunehmen, dass die durch das Resistenzprotein induzierte Synthese von KsuF artifiziell ist. Es ist zu vermuten, dass die durch das Thiostreptonresistenzgen durchgeführte Methylierung des Adenin 1067 der 23S rRNA und u.U. hierdurch variierte Konformation des Ribosoms oder des ribosomalen Proteins L11, die Translation regulatorischer Proteine oder Sigma-Faktoren beeinflusst, die nachfolgend die Transkription von ksuF ermöglichen. Außer den präsentierten eigenen Daten sind keine Studien zu diesem Effekt bekannt. Neben der spezifischen Methylierung in der 23S rRNA wurden bisher keine weiteren Stellen gefunden, die durch das Tsr-Protein methyliert werden und so die Transkription von ksuF bewirken könnten. Durch weiterführende Studien wie 2D Gelelektrophoresen und Microarraystudien, mit S.lividans Wildtyp, tsr und tsr unter z.B. einem konstitutiven Regulator tragenden S.lividans Transformanten als Vergleich, sollte es gelingen, die für die Synthese von KsuF beteiligten Proteine zu identifizieren. Es wird für Thiostrepton und weiteren komplexen Thiopeptiden die Funktion als Hormon vermutet (Chiu, M.L., et al.; 1999). Durch die Gabe von „sublethalen“ Konzentrationen von Thiostrepton (<10-9 M) wird in S.lividans die Synthese von 17 Proteinen induziert und von 4 reprimiert (Chiu, M.L., et al.; 1999, Holmes, D.J., et al.; 1993, Murakami, T. et al.; 1989). Nur die von tipA determinierten zwei „in-frame“ Translationsprodukte TipAL und TipAS (werden hierbei als ein Protein gezählt) wurden identifiziert und im Folgenden genauer charakterisiert. Die N-terminale Domäne von TipAL ist ein Transkriptions-Aktivator der MerR Familie. TipAS ist um die N-terminale Domäne verkürzt und besteht nur aus der Cterminalen Domäne von TipAL, die in der Lage ist, Thiostrepton kovalent zu binden (Chiu, M.L., et al.; 1996). TipAL bindet als Dimer an einem Operatorbereich des eigenen Promoters ptipA (Kahmann, J.D., et al.; 2003), induziert unter Thiostrepton die Produktion des eigenen Proteins und weiteren sieben, zwei werden reprimiert (Chiu, M.L., et al.; 1999). Die Regulation der 11 weiteren durch Thiostrepton induzierten (9) oder reprimierten (2) Proteine ist unklar und nicht untersucht (Chiu, M.L., et al.; 1999). Alle oben genannten Arbeiten zur Thiostrepton abhängigen Induktion erfolgten an S.lividans Transformanten, die tsr tragende Konstrukte enthielten. Es ist nicht auszuschließen, dass deren Translation, wie bei KsuF gezeigt, alle oder teilweise vom Vorhandensein des Thiostreptonresistenzproteins abhängig sind. Dies wird in weiteren Studien zu untersuchen sein auch ob eines dieser 11 Proteine die beobachtete Transkription von ksuF bedingt. In den S.lividans ksuF-EGFP Fusions-Insertionsmutanten konnte keine Synthese des KsuFEGFP nachgewiesen werden. Dies ist mit diesen Erkenntnissen auch nicht zu erwarten, da die Produktion von KsuF abhängig von der Synthese des Thiostreptonresistenzproteins ist. Auf Grund von Zeitmangel unterblieb die Transformation eines Thiostreptonresistenzgen tragenden Vektors. Diskussion - 159 - Wie dargelegt wurde, ist die Synthese von KsuC, KsuB und KsuF abhängig von der erhöhten NaCl- bzw. KCl-Konzentration, für die Produktion von KsuF ist zusätzlich das Vorhandensein des Thiostreptonresistenzproteins notwendig. In Gegenwart hoher Konzentrationen von NaCl oder KCl erfolgt eine deutlich erhöhte Synthese dieser Proteine, mit einer starken Erhöhung der Produktion ab einer Salzkonzentration von ca. 150-200 mM und Erreichen der maximalen um ca. 200-250 mM. Transkriptionsstudien zu ksuF untermauern dieses Ergebnis mit einer deutlich erhöhten mRNA-Produktion unter hohen NaCl- und KCl-Konzentrationen (J. Ritz, pers. Mitteilung). Die Gabe von 10 % (300 mM), 15 % (450 mM) Sucrose hat keinen Einfluss auf die Synthese von KsuC, KsuB oder KsuF, durch 20 % (600 mM) Sucrose wird das Wachstum inhibiert. Da sich die Synthese von KsuC, KsuB und KsuF unter hohen Salzkonzentrationen jeweils identisch verhält, ist anzunehmen, dass die Produktion der gleichen Regulation unterliegt und die erhöhte Synthese von KsuC und KsuB unter Hochsalz ebenfalls Transkriptions-abhängig ist. Die Synthese von KsuC, KsuB und KsuF unterscheidet sich in S.coelicolor M145-Mutanten, bei denen jeweils das chromosomale Gen für einen Sigma-Faktor (σH bzw. σF) zerstört worden war, nicht von denen des S.coelicolor M145 und des S.lividans Wildtyps. Der von sigF determinierte Sigma-Faktor σF ist in einem der letzten Schritte der Sporenreifung und der Sporen-Pigmentbildung involviert und nicht an der Osmoregulation beteiligt (Kormanec, J., pers. Mitteilung; Potuckova, L., et al.; 1995). Die erhöhte Produktion einiger SigH (σH) regulierter Proteine erfolgt bei Anwesenheit von NaCl/KCl (200-300 mM), wird jedoch nicht durch Sucrose beeinflusst (J. Kormanec, pers. Mitteilung; Kormanec, J. and Sevcikova, B.; 2003; Kormanec, J. and Sevcikova, B.; 2002a; Viollier, P.H., et al.; 2003a; Kelemen, G.H., et al.; 2001; Sevcikova, B., et al.; 2001; Kormanec. J., et al.; 2000). Diese Beobachtung entspricht derjenigen, die für KsuC/B und KsuF gemacht wurde. Neben der Hochsalzabhängigen Regulation ist σH über dem von ssgB determinierten Sporulations-spezifischen Zell-Teilungs Proteins SsgB an der Differenzierung beteiligt (Kormanec, J., Sevcikova; 2002b). Die Hochsalz-abhängige Transkription von sigH und nachgeschaltete σH-abhängige Transkription von weiteren Genen erfolgt vom sigH-P2 Promoter (Kormanec, J., et al.; 2000). Eine putative sigH-P2 ähnliche Promotersequenz konnte nicht vor ksuC/ksuB und ksuF gefunden werden. S.coelicolor A3(2) verwendet ein osmotisches Sensor System, welches die Aktivität verschiedener Sigma-Faktoren koordiniert und somit die Aktivität verschiedener Promotoren unter ähnlichen Stress-Bedingungen kontrolliert (Viollier, P.H., et al.; 2003b). Sowohl σH als auch das in der Kontrolle getestete σF gehören zu einer in S.coelicolor A3(2) identifizierten Gruppe von zehn Sigma-Faktoren, die zum Stress-„Master-Regulator“ σB aus Bacillus subtilis (Price, C.W., et al.; 2001; Völker, U. et al.; 1999; Hecker, M. and Völker, U.; 1998) ähnlich sind. Dies sind σWhiG, σB, σF, σG, σH, σI, σK, σL, σM und σN (Lee, E.-J., et al.; 2005; Lee, E.-J., et al.; 2004a), in weiteren Arbeiten wird mit σJ zusätzlich ein elfter hinzugezählt (Viollier, P.H. et al.; 2003a und b). Für einige dieser konnte bisher eine Beteiligung an der Osmoregulation in gezeigt werden, es muss allerdings kritisch angemerkt werden, dass außer - 160 - Diskussion für σB und σH anscheinend ausschließlich hohe NaCl- bzw. KCl-, jedoch nicht z.B. hohe Sucrose-Konzentrationen getestet wurden (Cho, Y.-H., et al.; 2001, J. Kormanec, pers. Mitteilung). σB ist in S.coelicolor A3(2) an der allgemeinen Osmoregulation, Reaktion auf oxidativen Stress und Differentierung involviert (Cho, Y.-H., et al.; 2001) und ist hauptverantwortlich für die Osmoregulation (Lee, E.-J., et al.; 2005), sowohl unter hohen Salz- wie auch SucroseKonzentrationen (Cho, Y.-H., et al.; 2001). Die Gene für σB, σL, σM und σHrdD werden unter hohen KCl-Konzentrationen in σB abhängiger Weise induziert (Lee, E.-J., et al.; 2005). Drei weitere der elf oben erwähnten Sigma-Faktoren werden unter „osmotischem“ Stress induziert. Zwei hiervon konnten σI und σJ zugerechnet werden, die unabhängig von σB und σH reguliert werden (Lee, E.-J., et al.; 2005; Viollier, P.H., et al.; 2003b). Ein weiteres, als SigX bezeichnet, konnte nicht identifiziert werden (Viollier, P.H., et al.; 2003b). σB-Gruppe typischen Regulator-Bindestellen (Sevcikova, B., et al.; 2005; Lee, E.-J., et al.; 2004a und b; Kormanec, J., and Sevcikova, B.; 2002b) sind weder vor ksuC/ksuB und ksuF noch vor allen anderen Genen des im Rahmen dieser Arbeit untersuchten Genomabschnittes des S.lividans Chromosoms zu finden. Die Transkription des für TipAL/AS determinierenden Gens tipA wird durch die Bindung von Thiostrepton bzw. weiteren komplexen Thiopeptiden an den Transkriptionsregulator TipAL autoreguliert und ist bei Vorhandensein von Thiostrepton deutlich erhöht. Des Weiteren wird die Transkription von tipA auch unter hohen osmotischen Bedingungen deutlich erhöht (Ali, N., et al.; 2002). Unter diesen Bedingungen erfolgt eine reduzierte negative Überwindung der DNA. Ähnliche Beobachtungen gibt es für verschiedene Promoterbereiche, wie z.B. beim merT Promoter pmerT unter hohen Hg-Konzentrationen in E.coli (Ansari, A.Z., et al.; 1992; Parkhill, J., et al.; 1993). Eine osmotische Regulation der Transkription von ksuC/B und ksuF durch TipAL ist auszuschließen. Die Synthese von KsuC, KsuB und KsuF ist unabhängig von der Gegenwart von Thiostrepton (für KsuC und KsuB nicht dargestellt), Thiostrepton ist nur indirekt über die Induktion der Transkription von tsr an der Produktion von KsuF beteiligt (siehe oben). Im Rahmen des S.coelicolor A3(2) Genom Projektes wurden 965 Gene (12,3 %) identifiziert, für dessen Genprodukte eine putative Beteiligung an regulatorischen Funktionen angenommen wird (Bentley, S.D., et al.; 2002). Ob eines dieser Regulatorproteine einen ptipA vergleichbaren osmotisch regulierten Promoter besitzt, ist nicht untersucht aber anzunehmen. Diskussion - 161 - 4.3. Undecylprodigiosin-Überproduktion in Mutanten Die ∆ksuC und ∆ksuB Mutanten zeigen bei Wachstum auf Minimal-Festmedium eine, im Verhältnis zum Wildtyp und der anderen, in dieser Arbeit hergestellten Mutanten, erhöhte und verfrühte Produktion eines roten, Mycel-assoziierten Farbstoffes. Dieser wird aus einer Mischung der Antibiotika Undecylprodigiosin und des cyclischen Derivats Butyl-metacycloheptylprodiginin gebildet (zusammen als RED bezeichnet) und vom RED-„Cluster“ (SCO5877-5898) determiniert (Butner, S.D., et al.; 2002). Die Produktion von RED erfolgt z.B. bei Wachstum auf Minimalmedium um ca. sechs Stunden früher als beim Wildtyp (siehe Tabelle 3.1). Vergleichbares findet sich bei einer Reihe von Mutanten, die Einfluss auf die Antibiotikaproduktion von S.coelicolor A3(2) haben (Bibb, M.; 1996). Die Actinorhodin (Act)-Produktion ist in den Mutanten unverändert; sowohl der Produktionsbeginn als auch die Menge des produzierten Antibiotikums ist unbeeinflusst. Die Produktion von RED und Act der Mutanten ist weiterhin abhängig von der Wachstumsphase und der Fähigkeit, ein Luftmycel auszubilden; sie ist frühestens mit Beginn der Zwischenphase zwischen der vegetativen und der Vermehrungsphase nachweisbar. Unter den meisten getesteten Wachstumsbedingungen erfolgt sie erst nach Start der Ausbildung des Luftmycels (ca. 40 h nach Ausstreichen auf Minimalmedium, siehe Tabelle 3.1). Der Beginn der Luftmycelbildung findet deutlich früher als bei S.coelicolor statt (nach ca. 72 h, Huang, J., et al.; 2005 und eigene Versuche, nicht dargestellt). Unter Kulturbedingungen bei denen keine Luftmycelbildung beobachtet werden konnte (Standard-Minimalmedien mit Zusatz von Casaaminosäuren oder 20 % Sucrose oder reduziertem Phosphatgehalt von 0,1 g/l KH2PO4, siehe Tabelle 3.1), ist auch bei verlängertem Wachstum keine Antibiotikaproduktion nachweisbar. Eine Wachstumsphasen-abhängige Synthese der Sekundärmetabolit- vor allem der Antibiotikasynthese ist typisch für viele Streptomyceten, einschließlich S.coelicolor A3(2) und S.lividans (Challis, G.L. and Hopwood, D.A.; 2003, Demain, A.L. and Fang, A.; 1995; Martin, J.F. and Demain, A.L.; 1980). Bei Wachstum auf Minimal-Festmedium mit Hygromycin zeigt die ∆ksuC Mutante eine gegenüber der ∆ksuB Mutante deutlich erhöhte und früher einsetzende RED-Produktion (siehe Abb. 3.67). Die geringere RED-Produktion der ∆ksuB Mutante wird vermutlich durch das Durchlesen des hinter hygR liegenden Terminators und der Synthese von KsuB hervorgerufen. Vergleichbares ist bei Wachstum in Minimalmedium-Flüssigkulturen ohne zusätzlich Salz zu beobachten (nicht dargestellt). Die RED-Produktion der ∆ksuB Mutante ist sehr viel stärker als die der ∆ksuC Mutante, die wiederum intensiver ist als die des Wildtyps. Dies muss als guter Hinweis gewertet werden, dass ausschließlich das Fehlen von KsuB für die beobachtete erhöhte und verfrühte RED-Produktion verantwortlich ist, KsuC jedoch keinen Einfluss hat. KsuC und KsuB sind, als Mitglieder der Aldo-Keto Reduktase-Familie, Oxidoreduktasen. Mit Ausnahme der Strukturgene der jeweiligen Biosynthesewege betreffen alle bisher gefundenen Mutanten und Überexpressionsstämme, die Einfluss auf die Act- und RED-Biosynthese - 162 - Diskussion zeigen, Gene, die für Proteine mit regulatorischer Funktion, für Transportproteine oder die RNA Polymerase β-Untereinheit determinieren. Die in dieser Arbeit gefundenen Erkenntnisse sind einzigartig. Der Phänotyp der erhöhten RED-Produktion der ∆ksuC- und ∆ksuBMutanten wurde zu einem verhältnismäßig späten Zeitpunkt der Arbeit gefunden. Im Folgenden werden die ersten Versuche diskutiert, die zur Aufklärung dieses Phänotyps durchgeführt wurden. Die Ergebnisse eines Ausstrichtests zeigten, dass die erhöhte RED-Produktion der ∆ksuCund ∆ksuB-Mutanten unabhängig von möglichen ausgeschiedenen Zwischenprodukten und extrazellulären Hormonen ist (Abb. 3.66). S.lividans und S.coelicolor A3(2) produzieren während der transienten Phase in hohen Konzentrationen das extrazelluläre γ-ButyrolactonHormon SCB1. Die Produktion ist in der vegetativen- und Vermehrungs-Phase reduziert (Takano, E., et al.; 2001; Onaka, H., et al.; 1998). Die Deletion des entsprechenden Gens scbA in S.coelicolor A3(2) oder S.lividans WT erhöht unter anderem die Produktion von Act und RED (Takano, E., et al.; 2001; Butler, M.J., et al. 2003). Niedrige extrazelluläre Phospatkonzentrationen führen zu einer erhöhten Produktion von RED, Act sowie den zwei weiteren Antibiotika in S.coelicolor A3(2), während hohe Konzentrationen deren Synthese inhibieren (Doull, J. and Vining, C.L.; 1990; Hobbs, G., et al.; 1990; Hobbs, G., et al.; 1992). Dasselbe wird auch für die Antibiotika-Produktion in S.lividans 1326 WT (entspricht dem in dieser Arbeit verwendete Stamm S.lividans 66 WT, der von D. A. Hopwood umbenannt wurde) und der Plasmid-reduzierten S.lividans-Variante TK24 postuliert (Ghorbel, S, et al.; 2006; Sola-Landa, A., et al.; 2003; Martín, J.F.; 2004). Wie an diesen Stämmen untersucht, erfolgt die Phospat-Regulation über das „ZweiKomponenten System“ PhoR-PhoP, wobei PhoR das Membran-gebundene Sensorprotein ist (es liegt keine Namensverwechslung vor!). Dieses Zwei-Komponenten System besitzt eine negative regulatorische Funktion auf die Antibiotikasynthese von RED und Act (SolanaLanda, A., et al.; 2003). Sowohl die ∆ksuC und ∆ksuB Mutanten als auch der S.lividans WT zeigen bei Wachstum auf niedrigen Phosphatkonzentrationen (1,47 mM) die erwartete hohe Produktion von Act und RED (siehe Abb. 3.68 k und Tabelle 3.1). Die RED-Produktion erfolgt in den Disruptionsmutanten um ca. 3-4 h früher als im Wildtyp (Beginn nach ca. 39 h). Überraschender Weise zeigt sich bei Wachstum in Gegenwart sehr hoher Phosphatkonzentrationen (73,5 mM) nach 72 h die stärkste RED- wie auch Act-Produktion; nach 48 h ist diese nicht erhöht und entspricht ca. der bei mittleren (7,35 mM) Phosphatkonzentrationen (Abb. 3.68 a und l, Tabelle 3.1). S.coelicolor M145 ist im Gegensatz zu S.lividans in der Lage, bei 0,735 mM Phosphat zu wachsen, hier findet sich die erwartete hohe Produktion an Act und RED (nicht dargestellt). Bei Wachstum auf sehr hohen Phosphatkonzentrationen (73,5 mM) ist im Vergleich zu S.lividans die Auskeimungsrate deutlich reduziert, die Kolonien sind deutlich kleiner und die Act- und RED-Synthese ist nicht Diskussion - 163 - erhöht (nicht dargestellt). S.coelicolor A3(2) und S.lividans könnten in einem Ökosystem unterschiedliche Nischen besetzen. Eine Abhängigkeit niedriger Phospatkonzentration auf die Synthese von Sekundärmetaboliten konnte schon seit den 70er Jahren (Aussage von Martín, J.F.; 2004) für eine Reihe von Streptomyceten produzierten Antibiotika und weiteren Sekundärmetaboliten nachgewiesen werden. Hohe Phosphatkonzentrationen inhibieren diese (Martin, J.F.; 2004; McDowall, K.A. et al.; 1999; Liras, P., et al.; 1990; Asturias, J.A., et al.; 1990; Rebollo, A., et al.; 1989; Masuma, R., et al.; 1986; Martin, J.F. and Demain, A.L.; 1980). Es wird postuliert, dass die erhöhte Antibiotikaund Sekundärmetabolit-Produktion unter niedrigen Phosphatkonzentrationen induziert wird, um die Nährstoffquelle gegen andere Konkurrenten zu verteidigen (reviewed in: Martin, J.F.; 2004). Die gefundene deutlich erhöhte Antibiotikasynthese in S.lividans unter sehr hohen Phosphatkonzentrationen widerspricht allen bisherigen Erkenntnissen. Vergleichbares wurde bisher nicht beschrieben. Dies überrascht, da ein Großteil der Arbeiten zur Phosphat-abhängigen Regulation der Antibiotikasynthese an S.lividans WT und Mutanten dieses Stammes erfolgten, wenn auch nicht unter vergleichbar hohen Phosphatkonzentrationen (Ghorbel, S, et al.; 2006; Martin, J.F., 2004; Sola-Landa, A., et al.; 2003). Im S.coelicolor A3(2) Genom können durch Sequenzvergleiche zwei weitere PhoR-PhoP ähnliche putative Regulator Paare identifiziert werden (SCO3741/3740 und SCO3013/3012). Eines dieser könnte die Regulation der Act und RED-Produktion gegenüber PhoR-PhoP unter sehr hohen Phosphatkonzentrationen (73,5 mM) und zu einem späten Zeitpunkt durchführen, jedoch anscheinend nicht im genetisch nah verwandten S.coelicolor A3(2). Das Verhältnis der Act- zur RED-Produktion zueinander scheint sowohl unter niedrigen, wie auch sehr hohen Phosphatkonzentrationen, identisch zu sein. Der in den Mutanten gefundene Phänotyp der erhöhten RED-Biosynthese ist vermutlich unabhängig von der Phosphatkonzentration. Diese hat sowohl Einfluss auf die RED-, als auch auf die Act-Biosynthese. Die Deletion von ∆ksuC und ∆ksuB beeinflusst jedoch ausschließlich die RED-Biosynthese. Hohe Salzkonzentrationen im Medium führen in S.coelicolor M145 zu einer stark erhöhten RED-Produktion; die Act-Produktion ist unter diesen Bedingungen jedoch deutlich reduziert. Die Transkription der jeweiligen Regulatorgene redD und actII-orf4 korreliert unter diesen Bedingungen mit der Antibiotikaproduktion (Sevkikova, B. and Kormanec, J.; 2004; Sevkikova, B., et al.; 2001). Die Synthese von RED und Act ist vermutlich ausschließlich abhängig von der Salzkonzentration und unabhängig von der Osmolarität; Wachstum auf Festmedium mit Zusatz von 10,3 % Sucrose hatte keinen Effekt auf die RED- und ActBiosynthese; Transkriptionsstudien wurden hierzu jedoch nicht durchgefüht (Sevcikova, B. and Kormanec, J.; 2004). Der Effekt ist unabhängig des vom sigH determinierten StressAntwort Sigma-Faktor (σH) auf hohe Salzkonzentrationen (Sevcikova, B. and Kormanec, J.; 2004). - 164 - Diskussion Hohe Konzentrationen von NaCl und KCl steigern die Produktion von KsuC und KsuB deutlich. Auf Grund dessen erfolgten der Ausstrich auf Minimal-Medium mit Zusatz von NaCl, KCl und Sucrose. Bei Wachstum auf 300 mM NaCl zeigt sich der gleiche Phänotyp, wie für S.coelicolor M145 bei Wachstum auf hohen Salzkonzentrationen beschrieben (Sevcikova, B. and Kormanec, J.; 2004). Die RED-Produktion erfolgt verfrüht und ist deutlich erhöht, eine Act-Produktion ist nicht zu erkennen (siehe Abb. 3.68 e, Tabelle 3.1). Die ∆ksuC und ∆ksuB Mutanten verhalten sich hierbei identisch mit dem Wildtyp. Überraschender Weise zeigt sich bei Wachstum auf hohen KCl Konzentrationen eine sehr stark reduzierte RED-Produktion, die in den Mutanten geringfügig stärker ausfällt als im WT. Eine Act-Produktion ist, wie auch bei Wachstum auf 300 mM NaCl, nicht zu erkennen (siehe Abb. 3.68 f und Tabelle 3.1). Somit wird durch hohe NaCl-Konzentrationen die REDBiosynthese gesteigert und durch hohe KCl-Konzentrationen reduziert. Diese Beobachtungen, erstmals im Rahmen dieser Arbeit dargestellt, widersprechen der Aussage einer Arbeit anderer Autoren, dass die RED-Produktion unter hohen Salz-Konzentrationen gesteigert ist. Alle Versuche mit hohen Salz-Konzentrationen in dieser Veröffentlichung erfolgten ausschließlich mit Zusatz von 2,5 % NaCl im Medium, jedoch nicht mit KCl (Sevcikova, B. and Kormanec, J.; 2004). Flussaufwärts von redD (SCO5877) finden sich in entgegengesetzter Flussrichtung, durch einen 268 Bp langen intergenischen Bereich getrennt, ein Operon aus zwei offenen Leserastern (SCO5876 und SCO5875), die für zwei TrkA Kalium-Transportprotein Homologe aus E.coli determinieren. Die Transkription von SCO5876/SCO5875 könnte ausschließlich KCl-abhängig und nicht osmotisch reguliert sein. Die bei Wachstum auf hohen KCl Konzentrationen beobachtete reduzierte RED-Synthese wäre somit durch Überlappung der jeweiligen Promoter- oder Operatorbereiche und Blockierung der redD Transkription hervorgerufen worden. Dies hätte eine deutlich verringerte Konzentration des REDBiosyntheseweg spezifischen Regulators RedD zur Folge. Untersuchungen zu SCO5876 und SCO5875 und der hiervon determinierenden putativen Kalium-Transportproteine wurden bisher nicht veröffentlicht; in einem Nebensatz wird erwähnt, jedoch nicht genauer beschrieben, dass das trkA-Operon den RED-Titer bei Wachstum in Flüssigmedium „beeinflussen“ kann (Cerdeno, A. M., et al.; 2001). Bei Wachstum auf 10 % Sucrose ist zwischen den Mutanten und dem S.lividans WT kein Unterschied zu erkennen; die RED-Produktion ist deutlich erhöht, eine Act-Produktion ist nicht zu erkennen. Der Produktionsbeginn von RED in S.lividans erfolgt hierbei am Ende der vegetativen Wachstumsphase oder zu Beginn der Zwischenphase, ca. 4-6 h vor Beginn der Luftmycelbildung, deutlich früher als unter allen anderen getesteten Bedingungen (siehe Abb. 3.68 g und Tabelle 3.1). Die hier gefundenen Ergebnisse stehen im direkten Widerspruch zu den für S.coelicolor M145 gewonnen, dass die Zugabe von 10,3 % Sucrose die RED- und Act-Synthese nicht beeinflusst (Sevcikova, B. and Kormanec, J.; 2004). Dies kann als ein Beispiel für die unterschiedliche Regulation der Antibiotikasynthese der nahe verwandten S.coelicolor M145 und S.lividans angesehen werden; auf Grund einiger Unstimmigkeiten Diskussion - 165 - erscheint jedoch auch ein Fehler in der oben erwähnten Veröffentlichung nicht unwahrscheinlich zu sein. Da unter hohen NaCl-, KCl- oder Sucrose-Konzentrationen in S.lividans eine vergleichbare (vermutlich vollständige) Inhibierung der Act- und deutlich erhöhte RED-Produktion (mit Ausnahme von hohen KCl-Konzentrationen, vermutlich aus den oben beschriebenen Gründen) gefunden wurde, kann eine jeweils von einer hohen Osmolarität abhängige komplexe Regulation der Act- und RED-Synthese angenommen werden. Vermutlich unterliegt auch die Act- und RED-Biosynthese in S.coelicolor M145 einer von einer hohen Osmolarität und nicht wie behauptet, einer Hochsalz abhängigen komplexen Regulation (Sevcikova, B. and Kormanec, J.; 2004). Hauptverantwortlich für die osmotische Regulation (getestet wurden bisher NaCl, KCl und Sucrose) in S.coelicolor A3(2) sind der Sigmafaktor σB und die niederrangigen von σB regulierten σL und σM (Lee, E.-J., et al.; 2005). Die beobachtete erhöhte RED- und inhibierte Act-Synthese unter hoher Osmolarität könnte durch eine gesteigerte Transkription von catB oder eine gesteigerte intrazelluläre ProlinKonzentration verursacht sein. Die Transkription von catB, das für die Katalase CatB determiniert, ist (direkt oder indirekt) σB-abhängig (Lee, E.-J., et al.; 2005; Cho, Y.-H.; et al.; 2001) und osmotisch reguliert (Cho, Y.-H.; et al.; 2001). Die Deletion von catB führt zu einer erhöhten und verfrühten Act-Produktion, die von RED ist deutlich reduziert, die Zellen sind osmosensitiv und zeigen einen „bald“-Phänotyp; hohe Kopienzahlen von catB führen zu einer reduzierten Act- und erhöhten RED-Produktion (Lee, E.-J., et al.; 2005; Cho, Y.-H.; et al.; 2001; Cho, Y.-H., et al.; 2000). Die intrazelluläre Prolin-Konzentration in Streptomyceten wird zur Osmotolerans unter hohen osmotischen extrazellulären Konzentrationen um bis zu 50-80fach erhöht (Kilham, K. and Firestone, M.K.; 1984). Prolin Transportmutanten, die gleichzeitig auch eine geringere Fähigkeit zur Degradation von Prolin besitzen und eine gesteigerte intrazelluläre Prolinkonzentration aufweisen, überproduzieren RED, die ActProduktion ist deutlich reduziert (Hood, D.W., et al.; 1992). Da eine hohe Osmolarität sowohl die RED- wie auch die Act-Synthese beeinflusst, scheint sie nicht verantwortlich für die in den ∆ksuC und ∆ksuB Mutanten beobachtete erhöhte Produktion von RED zu sein. Es ist zu vermuten, dass dieser in den Mutanten gefundene Phänotyp durch die durch Sucrose und NaCl induzierte verfrühte und sehr starke REDProduktion überdeckt wird. Hierfür spricht auch die Beobachtung, der gegenüber dem Wildtyp leicht erhöhten RED-Produktion der Mutanten bei Wachstum auf KCl. Der Biosyntheseweg von RED wurde in der Veröffentlichung von Cerdeno (Cerdeno, A.M., et al.; 2001) aus der Sequenz des dazugehörigen „Clusters“ postuliert und in der Veröffentlichung von Williamson (Williamson, N.R., et al.; 2005) sehr gut grafisch zusammenfassend dargestellt. Prolin, Serin und Glycin, des Weiteren Acetyl- und MalonylCoA, sind die Grundbausteine der RED-Biosynthese. Durch den Zusatz von Prolin (8,96 mM) wird die Antibiotikaproduktion sowohl der Mutanten als auch des Wildtyps im Vergleich zum Wachstum auf Minimalmedium nicht verändert - 166 - Diskussion (siehe Abb. 3.68 a und b; Tabelle 3.1). Die externe Gabe von Prolin ist nicht in der Lage, die intrazelluläre Prolinkonzentration zu steigern, so die erhöhte Produktion von RED zu induzieren und die Act-Produktion zu inhibieren, wie in Prolin-Transportmutanten gefunden (siehe oben, Hood, D.W., et al.; 1992). Durch die Zugabe von Glycin (13,3 mM) und Serin (9,5 mM) wird die RED-Überproduktion der Mutanten teilweise (bei Glycin) oder vollständig (bei Serin) komplementiert. Nach längerem Wachstum von 6 Tagen entspricht die REDProduktion jeweils ungefähr der des Wildtyps auf Minimalmedium. Die SerinHydroxymethyltransferase katalysiert die Umwandlung von Serin in Glycin, unter Freisetzung von Tetrahydrofolsäure~CH2OH. Die Reaktion ist bidirektional. Da die Mutanten bei Zugabe von Glycin, trotz einer höher eingesetzten Konzentration als bei Serin, eine leicht höhere RED-Produktion zeigen, ist anzunehmen, dass die Komplementation Serin-abhängig ist. Im Rahmen des S.coelicolor A3(2) Genomprojektes wurden drei potentielle SerinHydroxymethyltransferasen identifiziert, determiniert von glyA1 (SCO5470), glyA2 (SCO5470) und glyA3 (SCO5364). Serin ist der Grundbaustoff der Cysteinbiosynthese. Ob Cystein oder das Zwischenprodukt O-Acetylserin für die Komplementation verantwortlich sind, bzw. ebenfalls eine Komplementation bewirken, wurde nicht untersucht. Da Gabe von externen Glycin, vor allem von Serin, die RED-Überproduktion der Mutanten auf die Produktionsmenge des Wildtyps reduziert (ca. in der Stärke des Wildtyps bei Wachstum auf Minimalmedium), wurde eine Beteiligung am gefundenen Phänotyp der REDÜberproduktion angenommen. Dies ist jedoch auszuschließen, da die Act-Produktion inhibiert wird. Vergleichbares wurde auch in früheren Versuchen zu S.coelicolor A3(2) gefunden, Glycin, Glutamin und Ammoniumnitrat beeinflussen die RED-Biosynthese nur unwesentlich, die Act-Produktion wird inhibiert (Hobbs, G., et al.; 1990). KsuC und KsuB sind Mitglieder der Aldo-Keto Reduktase-Familie, Aminosäuren sind keine typischen Substrate dieser Familie. Die Regulation der Antibiotika-Synthese ist, wie explizit für S.coelicolor A3(2) und S.lividans gezeigt, äußerst komplex und nur zum Teil verstanden, viele unterschiedliche Faktoren sind hieran beteiligt (siehe Einleitung). Ein vergleichbarer Phänotyp einer veränderten RED- und einer gering oder unbeeinflussten Act-Synthese zeigen Mutanten von an der REDBiosynthese involvierten Genen (Cerdeno, A.M., et al.; 2001), Mutanten und hohe Kopienzahlen von redZ oder redD, die für die „Biosyntheseweg spezifischen Regulatoren“ determinieren (White, J. and Bibb, M.; 1997, Malpartida, F., et al.; 1990; Narva, K.E. and Feitelson, J.S.; 1990; Feitelson, J.S., et al.; 1985), Mutanten von ercA1/A2, determinierend für ein Zwei-Komponenten System der NarL Familie (Li, Y.-q., et al.; 2004) und eine der spontan entstandenen Rifampicin resistenten Mutanten der RNA Polymerase β-Untereinheit, determiniert von rpoB (S.lividans EN1 rif-9, trägt eine Deletion des für Lys430 determinierenden Kodons; Hu, H., et al.; 2002). Nicht ausgeschlossen werden kann, dass KsuC/B, auf Grund der großen Substratdiversität der Aldo-Keto Reduktasen, Zwischenprodukte der RED-Biosynthese modifizieren. Eine Reihe Diskussion - 167 - von Zwischenprodukten der RED-Biosynthese sind Ketone, Diketone und Aldehyde und somit potentielle Substrate der Aldo-Keto Reduktasen (Williamson, N.R., et al.; 2005; Cerdeňo, A.M., et al.; 2001). Für das von ercA1/A2 determinierte „Zwei Komponenten System“ wird eine positive regulatorische Funktion für den RED-Biosynthese „Cluster“ postuliert. Jedoch ist weder bekannt noch postuliert, auf welche Art und Weise dieses an der Regulation der RED-Biosynthese beteiligt ist (Li, Y.-q., et al.; 2004). Es kann vermutet werden, dass der Antwort-Regulator (determiniert von ercA1) über eine Kaskade weiterer Regulatorproteine die Transkription von redD und redZ reguliert. In der oben erwähnten Rifampicin resistenten Mutante der RNA Polymerase β-Untereinheit könnte hierbei die Bindung des niederrangigsten Regulatorproteins dieser Kaskade vor redD oder redZ mimikrieren sein. Es ist möglich, dass ein Endprodukt von KsuC/KsuB an eines der Regulatorproteine der postulierten Kaskade bindet und so die RED-Produktion beeinflusst. Des Weiteren könnte KsuB (auf Grund der großen Substratdiversität der Aldo-Keto Reduktasen) ein Molekül, das durch Bindung an eines dieser Regulatorproteine an der Kaskade beteiligt ist, als Substrat modifizieren. Eine Koordinierung der Synthese von KsuC/B mit der RED-Biosynthese wurde in dieser Arbeit nicht beobachtet. Wie in Microarray-Studien zu 1500 Genen aus S.coelicolor A3(2) gezeigt wurde, werden ecrA1/ecrA2 koordiniert mit den Genen des RED spezifischen Biosyntheseweges transkribiert, jedoch nicht ksuC und ksuB (Zusatzmaterial Fig.S3 und Fig.S1 zu: Huang, J., et al.; 2005, http://www.blackwellsynery.com). Zusammenfassung - 169 - 5. ZUSAMMENFASSUNG Das lineare Chromosom von S.lividans zeichnet sich durch eine hohe Variabilität insbesondere der chromosomalen Endbereiche aus. Hier finden sich unter anderen auch verschiedene Gene, die bisher einzigartig sind. Um die Funktion dieser zu untersuchen wurden verschiedene Strategien angewandt. Nach Klonierung der Gene in E.coli wurden die entsprechenden Genprodukte als His-Tag Fusionsproteine überproduziert, aufgereinigt und zur Herstellung von Antikörpern verwendet. Der untersuchte Abschnitt, als Ganzes und in Unterabschnitten, wurde auf einem „Hoch Kopien Vektor“ in S.lividans transformiert. In extra hierfür konstruierten Vektorsystemen erfolgte die Produktion von His-Tag Proteinen in S.lividans. Nach Fusion von potentiellen Promoterbereichen mit dem promoterlosen EGFP-Gen, gelang deren Identifizierung in „enhanced green fluorescent protein“ (EGFP) produzierenden S.lividans Transformanten. Mit Hilfe eines Vektors, der ein Thiostrepton-Resistenzgen trägt und ein Temperatur sensitives Replikon besitzt, wurden Gene durch die Integration eines Hygromycin-Resistenzgenes ersetzt, bzw. als Fusionsgen mit dem EGFP-Gen erstellt. Mit weiteren pysiologischen, biochemischen und immunologischen Untersuchungen, zum Teil in Kooperation mit Mitgliedern der Arbeitsgruppe, wurde erstmals ein Gen identifiziert, dessen Genprodukt für die Interaktion zwischen Streptomyceten-Hyphen wichtig ist. Ein Flavoprotein wurde zur Homogenität gereinigt. Es wurde nachgewiesen, dass in S.lividans pro Monomer ein FAD-Molekül interagiert. Physiologische Studien zeigen, dass die Synthese des chromosomal determinierten Proteins in S.lividans nur erfolgt, wenn dieser Stamm ein Plasmid- oder chromosomal- kodiertes Thiostrepton Resistenzprotein (23S rRNA Methylase) enthält. Es muss geschlussfolgert werden, dass die Methylierung der 23S rRNA die Translation verschiedener mRNAs beeinflusst. Die Synthese dieses Proteins ist des Weiteren abhängig von hohen Konzentrationen an NaCl und KCl im Medium, wie auch die zweier Aldo-Keto Reduktasen. Disruptionsmutanten eines dieser zwei Aldo-Keto Reduktase-Gene zeigen jeweils eine erhöhte und verfrühte Produktion eines rot gefärbten Mycel-assoziierten Antibiotikums (Undecylprodigiosin), während die eines weiteren (Actinorhodin) unbeeinflusst blieb. Die Beteiligung einiger Faktoren an diesem Phänotyp konnte ausgeschlossen werden. Als Nebenaspekt zeigten sich eine hier erstmals beschriebene, zu einem späten Zeitpunkt erfolgende, deutlich erhöhte Produktion der beiden Antibiotika bei Wachstum auf sehr hohen Phosphat-Konzentrationen und zusätzlich eine vermutlich osmotische Regulation der Synthese dieser beiden Antibiotika. Literaturverzeichnis - 171 - 6. LITERATURVERZEICHNIS Adamidis T, Champness W. Genetic analysis of absB, a Streptomyces coelicolor locus involved in global antibiotic regulation. J Bacteriol. 1992 Jul;174(14):4622-8. Aigle B, Wietzorrek A, Takano E, Bibb MJ. A single amino acid substitution in region 1.2 of the principal sigma factor of Streptomyces coelicolor A3(2) results in pleiotropic loss of antibiotic production. Mol Microbiol. 2000 Sep;37(5):995-1004. Ali N, Herron PR, Evans MC, Dyson PJ. Osmotic regulation of the Streptomyces lividans thiostrepton-inducible promoter, ptipA. Microbiology. 2002 Feb;148(Pt 2):381-90. Alting-Mees MA, Short JM. pBluescript II: gene mapping vectors. Nucleic Acids Res. 1989 Nov 25;17(22):9494. Anderson TB, Brian P, Champness WC. Genetic and transcriptional analysis of absA, an antibiotic gene cluster-linked two-component system that regulates multiple antibiotics in Streptomyces coelicolor. Mol Microbiol. 2001 Feb;39(3):553-66. Ansari AZ, Chael ML, O'Halloran TV. Allosteric underwinding of DNA is a critical step in positive control of transcription by Hg-MerR. Nature. 1992 Jan 2;355(6355):87-9. Asturias JA, Liras P, Martin JF. Phosphate control of pabS gene transcription during candicidin biosynthesis. Gene. 1990 Sep 1;93(1):79-84. Balbas P, Soberon X, Bolivar F, Rodriguez RL. The plasmid, pBR322. Biotechnology. 1988;10:5-41. Review. Bechthold A, Floss HG. Overexpression of the thiostrepton-resistance gene from Streptomyces azureus in Escherichia coli and characterization of recognition sites of the 23S rRNA A1067 2'-methyltransferase in the guanosine triphosphatase center of 23S ribosomal RNA. Eur J Biochem. 1994 Sep 1;224(2):431-7. - 172 - Literaturverzeichnis Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004 Jul 16;340(4):783-95. Bentley SD, Brown S, Murphy LD, Harris DE, Quail MA, Parkhill J, Barrell BG, McCormick JR, Santamaria RI, Losick R, Yamasaki M, Kinashi H, Chen CW, Chandra G, Jakimowicz D, Kieser HM, Kieser T, Chater KF. SCP1, a 356,023 bp linear plasmid adapted to the ecology and developmental biology of its host, Streptomyces coelicolor A3(2). Mol Microbiol. 2004 Mar;51(6):1615-28. Bentley SD, Chater KF, Cerdeno-Tarraga AM, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, Bateman A, Brown S, Chandra G, Chen CW, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Hornsby T, Howarth S, Huang CH, Kieser T, Larke L, Murphy L, Oliver K, O'Neil S, Rabbinowitsch E, Rajandream MA, Rutherford K, Rutter S, Seeger K, Saunders D, Sharp S, Squares R, Squares S, Taylor K, Warren T, Wietzorrek A, Woodward J, Barrell BG, Parkhill J, Hopwood DA. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature. 2002 May 9;417(6885):141-7. Berdy J. Bioactive microbial metabolites. J Antibiot (Tokyo). 2005 Jan;58(1):1-26. Review. Erratum in: J Antibiot (Tokyo). 2005 Apr;58(4):C-1. Bhushan B, Halasz A, Spain JC, Hawari J. Initial reaction(s) in biotransformation of CL20 is catalyzed by salicylate 1-monooxygenase from Pseudomonas sp. strain ATCC 29352. Appl Environ Microbiol. 2004 Jul;70(7):4040-7. Bibb M. 1995 Colworth Prize Lecture. The regulation of antibiotic production in Streptomyces coelicolor A3(2). Microbiology. 1996 Jun;142 ( Pt 6):1335-44. Review. Bibb MJ, Bibb MJ, Ward JM, Cohen SN. Nucleotide sequences encoding and promoting expression of three antibiotic resistance genes indigenous to Streptomyces. Mol Gen Genet. 1985;199(1):26-36. Bibb MJ, Cohen SN. Gene expression in Streptomyces: construction and application of promoter-probe plasmid vectors in Streptomyces lividans. Mol Gen Genet. 1982;187(2):265-77. Blanco J, Coque JJ, Martin JF. The folate branch of the methionine biosynthesis pathway in Streptomyces lividans: disruption of the 5,10-methylenetetrahydrofolate reductase gene leads to methionine auxotrophy. J Bacteriol. 1998 Mar;180(6):1586-91. Literaturverzeichnis - 173 - Blondelet-Rouault MH, Weiser J, Lebrihi A, Branny P, Pernodet JL. Antibiotic resistance gene cassettes derived from the omega interposon for use in E. coli and Streptomyces. Gene. 1997 May 6;190(2):315-7. Bosch R, Garcia-Valdes E, Moore ER. Genetic characterization and evolutionary implications of a chromosomally encoded naphthalene-degradation upper pathway from Pseudomonas stutzeri AN10. Gene. 1999 Aug 5;236(1):149-57. Bourn WR, Babb B. Computer assisted identification and classification of streptomycete promoters. Nucleic Acids Res. 1995 Sep 25;23(18):3696-703. Bowen WS, Van Dyke N, Murgola EJ, Lodmell JS, Hill WE. Interaction of thiostrepton and elongation factor-G with the ribosomal protein L11-binding domain. J Biol Chem. 2005 Jan 28;280(4):2934-43. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976 May 7;72:248-54. Brockmann H, Pini H. Actinorhodin, ein roter Farbstoff aus Actinomyceten. Die Naturwissenschaften. 1947; 34:190. Brot N, Tate WP, Caskey CT, Weissbach H. The requirement for ribosomal proteins L7 and L12 in peptide-chain termination. Proc Natl Acad Sci U S A. 1974 Jan;71(1):89-92. Bullock WO, Fernandez JM, Short JM. XL1-Blue: A high efficiency plasmid transforming recA Escherichia coli strain with beta-galactosidase selection. BioTechniques 1987 5:367 Burnette WN. "Western blotting": electrophoretic transfer of proteins from sodium dodecyl sulfate--polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal Biochem. 1981 Apr;112(2):195-203. Butler MJ, Takano E, Bruheim P, Jovetic S, Marinelli F, Bibb MJ. Deletion of scbA enhances antibiotic production in Streptomyces lividans. Appl Microbiol Biotechnol. 2003 Jun;61(5-6):512-6. Butler MJ, Bruheim P, Jovetic S, Marinelli F, Postma PW, Bibb MJ. Engineering of primary carbon metabolism for improved antibiotic production in Streptomyces lividans. Appl Environ Microbiol. 2002 Oct;68(10):4731-9. - 174 - Literaturverzeichnis Bystrykh LV, Fernandez-Moreno MA, Herrema JK, Malpartida F, Hopwood DA, Dijkhuizen L. Production of actinorhodin-related "blue pigments" by Streptomyces coelicolor A3(2). J Bacteriol. 1996 Apr;178(8):2238-44. Bystrykh LV, Dijkhuizen L, Harder W. Modification of flavin adenine dinucleotide in alcohol oxidase of the yeast Hansenula polymorpha. J Gen Microbiol. 1991 Oct;137(10):2381-6. Cameron DM, Thompson J, March PE, Dahlberg AE. Initiation factor IF2, thiostrepton and micrococcin prevent the binding of elongation factor G to the Escherichia coli ribosome. J Mol Biol. 2002 May 24;319(1):27-35. Cerdeno AM, Bibb MJ, Challis GL. Analysis of the prodiginine biosynthesis gene cluster of Streptomyces coelicolor A3(2): new mechanisms for chain initiation and termination in modular multienzymes. Chem Biol. 2001 Aug;8(8):817-29. Cerdeno-Tarraga AM, Efstratiou A, Dover LG, Holden MT, Pallen M, Bentley SD, Besra GS, Churcher C, James KD, De Zoysa A, Chillingworth T, Cronin A, Dowd L, Feltwell T, Hamlin N, Holroyd S, Jagels K, Moule S, Quail MA, Rabbinowitsch E, Rutherford KM, Thomson NR, Unwin L, Whitehead S, Barrell BG, Parkhill J. The complete genome sequence and analysis of Corynebacterium diphtheriae NCTC13129. Nucleic Acids Res. 2003 Nov 15;31(22):6516-23. Challis GL, Hopwood DA. Synergy and contingency as driving forces for the evolution of multiple secondary metabolite production by Streptomyces species. Proc Natl Acad Sci U S A. 2003 Nov 25;100 Suppl 2:14555-61. Review. Champness W, Riggle P, Adamidis T, Vandervere P. Identification of Streptomyces coelicolor genes involved in regulation of antibiotic synthesis. Gene. 1992 Jun 15;115(12):55-60. Chang HM, Chen MY, Shieh YT, Bibb MJ, Chen CW. The cutRS signal transduction system of Streptomyces lividans represses the biosynthesis of the polyketide antibiotic actinorhodin. Mol Microbiol. 1996 Sep;21(5):1075-85. Chiu ML, Folcher M, Katoh T, Puglia AM, Vohradsky J, Yun BS, Seto H, Thompson CJ. Broad spectrum thiopeptide recognition specificity of the Streptomyces lividans TipAL protein and its role in regulating gene expression. J Biol Chem. 1999 Jul 16;274(29):20578-86. Literaturverzeichnis - 175 - Chiu ML, Folcher M, Griffin P, Holt T, Klatt T, Thompson CJ. Characterization of the covalent binding of thiostrepton to a thiostrepton-induced protein from Streptomyces lividans. Biochemistry. 1996 Feb 20;35(7):2332-41. Cho YH, Lee EJ, Ahn BE, Roe JH. SigB, an RNA polymerase sigma factor required for osmoprotection and proper differentiation of Streptomyces coelicolor. Mol Microbiol. 2001 Oct;42(1):205-14. Cho YH, Lee EJ, Roe JH. A developmentally regulated catalase required for proper differentiation and osmoprotection of Streptomyces coelicolor. Mol Microbiol. 2000 Jan;35(1):150-60. Chouayekh H, Virolle MJ. The polyphosphate kinase plays a negative role in the control of antibiotic production in Streptomyces lividans. Mol Microbiol. 2002 Feb;43(4):919-30. Cole LJ, Entsch B, Ortiz-Maldonado M, Ballou DP. Properties of p-hydroxybenzoate hydroxylase when stabilized in its open conformation. Biochemistry. 2005 Nov 15;44(45):14807-17. Cole ST, Eiglmeier K, Parkhill J, James KD, Thomson NR, Wheeler PR, Honore N, Garnier T, Churcher C, Harris D, Mungall K, Basham D, Brown D, Chillingworth T, Connor R, Davies RM, Devlin K, Duthoy S, Feltwell T, Fraser A, Hamlin N, Holroyd S, Hornsby T, Jagels K, Lacroix C, Maclean J, Moule S, Murphy L, Oliver K, Quail MA, Rajandream MA, Rutherford KM, Rutter S, Seeger K, Simon S, Simmonds M, Skelton J, Squares R, Squares S, Stevens K, Taylor K, Whitehead S, Woodward JR, Barrell BG. Massive gene decay in the leprosy bacillus. Nature. 2001 Feb 22;409(6823):1007-11. Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998 Jun 11;393(6685):537-44. Erratum in: Nature 1998 Nov 12;396(6707):190. Corpet F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988 Nov 25;16(22):10881-90. - 176 - Literaturverzeichnis de Crecy-Lagard V, Servant-Moisson P, Viala J, Grandvalet C, Mazodier P. Alteration of the synthesis of the Clp ATP-dependent protease affects morphological and physiological differentiation in Streptomyces. Mol Microbiol. 1999 May;32(3):505-17. Dittrich W, Betzler M, Schrempf H. An amplifiable and deletable chloramphenicolresistance determinant of Streptomyces lividans 1326 encodes transmembrane protein. Mol Microbiol. 1991 Nov;5(11):2789-97. a putative Doull JL, Vining LC. Nutritional control of actinorhodin production by Streptomyces coelicolor A3(2): suppressive effects of nitrogen and phosphate. Appl Microbiol Biotechnol. 1990 Jan;32(4):449-54. Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998 Apr 3;280(5360):69-77. Dulley JR, Grieve PA. A simple technique for eliminating interference by detergents in the Lowry method of protein determination. Anal Biochem. 1975 Mar;64(1):136-41. Ellis EM, Hayes JD. Substrate specificity of an aflatoxin-metabolizing aldehyde reductase. Biochem J. 1995 Dec 1;312 ( Pt 2):535-41. El-Kabbani O, Wilson DK, Petrash M, Quiocho FA. Structural features of the aldose reductase and aldehyde reductase inhibitor-binding sites. Mol Vis. 1998 Sep 29;4:19. Review. el-Kabbani O, Carper DA, McGowan MH, Devedjiev Y, Rees-Milton KJ, Flynn TG. Studies on the inhibitor-binding site of porcine aldehyde reductase: crystal structure of the holoenzyme-inhibitor ternary complex. Proteins. 1997 Oct;29(2):186-92. Enroth C, Neujahr H, Schneider G, Lindqvist Y. The crystal structure of phenol hydroxylase in complex with FAD and phenol provides evidence for a concerted conformational change in the enzyme and its cofactor during catalysis. Structure. 1998 May 15;6(5):605-17. Entsch B, Cole LJ, Ballou DP. Protein dynamics and electrostatics in the function of phydroxybenzoate hydroxylase. Arch Biochem Biophys. 2005 Jan 1;433(1):297-311. Review. Literaturverzeichnis - 177 - Entsch B, van Berkel WJ. Structure and mechanism of para-hydroxybenzoate hydroxylase. FASEB J. 1995 Apr;9(7):476-83. Review. Eppink MH, Bunthol C, Schreuder HA, van Berkel WJ. Phe161 and Arg166 variants of phydroxybenzoate hydroxylase. Implications for NADPH recognition and structural stability. FEBS Lett. 1999 Jan 29;443(3):251-5. Eppink MH, Schreuder HA, van Berkel WJ. Interdomain binding of NADPH in phydroxybenzoate hydroxylase as suggested by kinetic, crystallographic and modeling studies of histidine 162 and arginine 269 variants. J Biol Chem. 1998a Aug 14;273(33):21031-9. Eppink MH, Schreuder HA, van Berkel WJ. Lys42 and Ser42 variants of phydroxybenzoate hydroxylase from Pseudomonas fluorescens reveal that Arg42 is essential for NADPH binding. Eur J Biochem. 1998b Apr 1;253(1):194-201. Eppink MH, Schreuder HA, Van Berkel WJ. Identification of a novel conserved sequence motif in flavoprotein hydroxylases with a putative dual function in FAD/NAD(P)H binding. Protein Sci. 1997 Nov;6(11):2454-8. Eppink MH, Schreuder HA, Van Berkel WJ. Structure and function of mutant Arg44Lys of 4-hydroxybenzoate hydroxylase implications for NADPH binding. Eur J Biochem. 1995 Jul 1;231(1):157-65. Farber GK, Petsko GA. The evolution of alpha/beta barrel enzymes. Trends Biochem Sci. 1990 Jun;15(6):228-34. Review. Feitelson JS, Malpartida F, Hopwood DA. Genetic and biochemical characterization of the red gene cluster of Streptomyces coelicolor A3(2). J Gen Microbiol. 1985 Sep;131(9):243141. Fisher HF, Conn EE, Vennesland B, Westheimer FH. The enzymatic transfer of hydrogen. I. The reaction catalyzed by alcohol dehydrogenase. J Biol Chem. 1953 Jun;202(2):687-97. Fischer, E. Effekt der Zuckerkonfiguration auf die Enzymwirkung. Berichte der Deutschen Chemischer Gesellschaft 1894; 27: 2984-2993. - 178 - Literaturverzeichnis Fleischmann RD, Alland D, Eisen JA, Carpenter L, White O, Peterson J, DeBoy R, Dodson R, Gwinn M, Haft D, Hickey E, Kolonay JF, Nelson WC, Umayam LA, Ermolaeva M, Salzberg SL, Delcher A, Utterback T, Weidman J, Khouri H, Gill J, Mikula A, Bishai W, Jacobs Jr WR Jr, Venter JC, Fraser CM. Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains. J Bacteriol. 2002 Oct;184(19):5479-90. Fraser CM, Casjens S, Huang WM, Sutton GG, Clayton R, Lathigra R, White O, Ketchum KA, Dodson R, Hickey EK, Gwinn M, Dougherty B, Tomb JF, Fleischmann RD, Richardson D, Peterson J, Kerlavage AR, Quackenbush J, Salzberg S, Hanson M, van Vugt R, Palmer N, Adams MD, Gocayne J, Weidman J, Utterback T, Watthey L, McDonald L, Artiach P, Bowman C, Garland S, Fuji C, Cotton MD, Horst K, Roberts K, Hatch B, Smith HO, Venter JC. Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature. 1997 Dec 11;390(6660):580-6. Gatti DL, Entsch B, Ballou DP, Ludwig ML. pH-dependent structural changes in the active site of p-hydroxybenzoate hydroxylase point to the importance of proton and water movements during catalysis. Biochemistry. 1996 Jan 16;35(2):567-78. Gatti DL, Palfey BA, Lah MS, Entsch B, Massey V, Ballou DP, Ludwig ML. The mobile flavin of 4-OH benzoate hydroxylase. Science. 1994 Oct 7;266(5182):110-4. Ghorbel S, Kormanec J, Artus A, Virolle MJ. Transcriptional studies and regulatory interactions between the phoR-phoP operon and the phoU, mtpA, and ppk genes of Streptomyces lividans TK24. J Bacteriol. 2006 Jan;188(2):677-86. Goodner B, Hinkle G, Gattung S, Miller N, Blanchard M, Qurollo B, Goldman BS, Cao Y, Askenazi M, Halling C, Mullin L, Houmiel K, Gordon J, Vaudin M, Iartchouk O, Epp A, Liu F, Wollam C, Allinger M, Doughty D, Scott C, Lappas C, Markelz B, Flanagan C, Crowell C, Gurson J, Lomo C, Sear C, Strub G, Cielo C, Slater S. Genome sequence of the plant pathogen and biotechnology agent Agrobacterium tumefaciens C58. Science. 2001 Dec 14;294(5550):2323-8. Gramajo HC, Takano E, Bibb MJ. Stationary-phase production of the antibiotic actinorhodin in Streptomyces coelicolor A3(2) is transcriptionally regulated. Mol Microbiol. 1993 Mar;7(6):837-45. Gulbis JM, Zhou M, Mann S, MacKinnon R. Structure of the cytoplasmic beta subunit-T1 assembly of voltage-dependent K+ channels. Science. 2000 Jul 7;289(5476):123-7. Literaturverzeichnis - 179 - Haug I, Weissenborn A, Brolle D, Bentley S, Kieser T, Altenbuchner J. Streptomyces coelicolor A3(2) plasmid SCP2*: deductions from the complete sequence. Microbiology. 2003 Feb;149(Pt 2):505-13. Hara O, Horinouchi S, Uozumi T, Beppu T. Genetic analysis of A-factor synthesis in Streptomyces coelicolor A3(2) and Streptomyces griseus. J Gen Microbiol. 1983 Sep;129(9):2939-44. Harayama S, Kok M, Neidle EL. Functional and evolutionary relationships among diverse oxygenases. Annu Rev Microbiol. 1992;46:565-601. Review. Hecker M, Volker U. Non-specific, general and multiple stress resistance of growthrestricted Bacillus subtilis cells by the expression of the sigmaB regulon. Mol Microbiol. 1998 Sep;29(5):1129-36. Review. Hegermann J, Overbeck J, Schrempf H. In vivo monitoring of the potassium channel KcsA in Streptomyces lividans hyphae using immuno-electron microscopy and energyfiltering transmission electron microscopy. Microbiology. 2006 Sep;152(Pt 9):2831-41. Helmann JD. The extracytoplasmic function (ECF) sigma factors. Adv Microb Physiol. 2002;46:47-110. Review. Hesketh A, Sun J, Bibb M. Induction of ppGpp synthesis in Streptomyces coelicolor A3(2) grown under conditions of nutritional sufficiency elicits actII-ORF4 transcription and actinorhodin biosynthesis. Mol Microbiol. 2001 Jan;39(1):136-44. Hidalgo AR, Akond MA, Kita K, Kataoka M, Shimizu S. Isolation and primary structural analysis of two conjugated polyketone reductases from Candida parapsilosis. Biosci Biotechnol Biochem. 2001 Dec;65(12):2785-8. Hillemann D, Dammann T, Hillemann A, Wohlleben W. Genetic and biochemical characterization of the two glutamine synthetases GSI and GSII of the phosphinothricyl-alanyl-alanine producer, streptomyces viridochromogenes Tu494. J Gen Microbiol. 1993 Aug;139(8):1773-83. Hobbs G, Obanye AI, Petty J, Mason JC, Barratt E, Gardner DC, Flett F, Smith CP, Broda P, Oliver SG. An integrated approach to studying regulation of production of the antibiotic methylenomycin by Streptomyces coelicolor A3(2). J Bacteriol. 1992 Mar;174(5):1487-94 - 180 - Literaturverzeichnis Hobbs G, Frazer CM, Gardner DCJ, Flett F, Oliver SG. Pigmented antibiotic production by Streptomyces coelicolor A3(2): kinetics and the influence of nutrients. J Gen Microbiol. 1990;136:2291-2296. Holmes DJ, Caso JL, Thompson CJ. Autogenous transcriptional activation of a thiostrepton-induced gene in Streptomyces lividans. EMBO J. 1993 Aug;12(8):3183-91. Hood DW, Heidstra R, Swoboda UK, Hodgson DA. Molecular genetic analysis of proline and tryptophan biosynthesis in Streptomyces coelicolor A3(2): interaction between primary and secondary metabolism--a review. Gene. 1992 Jun 15;115(1-2):5-12. Review. Hopwood DA. The Streptomyces genome--be prepared! Nat Biotechnol. 2003 May;21(5):505-6. Hopwood DA, Bibb MJ, Chater KF, Kieser T, Burton CJ,Kieser HM, Lydrate DJ, Smith CP, Ward JM and Schrempf H. Genetic manipulation of Streptomyces. A laboratory manual (Norwich: The John Innes Foundation) 1985 Hopwood DA, Kieser T, Wright HM, Bibb MJ. Plasmids, recombination and chromosome mapping in Streptomyces lividans 66. J Gen Microbiol. 1983 Jul;129(7):2257-69. Hopwood DA, Wright HM. Bacterial protoplast fusion: recombination in fused protoplasts of Streptomyces coelicolor. Mol Gen Genet. 1978 Jul 4;162(3):307-17. Horinouchi S. AfsR as an integrator of signals that are sensed by multiple serine/threonine kinases in Streptomyces coelicolor A3(2). J Ind Microbiol Biotechnol. 2003 Aug;30(8):462-7. Review. Hu H, Zhang Q, Ochi K. Activation of antibiotic biosynthesis by specified mutations in the rpoB gene (encoding the RNA polymerase beta subunit) of Streptomyces lividans. J Bacteriol. 2002 Jul;184(14):3984-91. Huang J, Shi J, Molle V, Sohlberg B, Weaver D, Bibb MJ, Karoonuthaisiri N, Lih CJ, Kao CM, Buttner MJ, Cohen SN. Cross-regulation among disparate antibiotic biosynthetic pathways of Streptomyces coelicolor. Mol Microbiol. 2005 Dec;58(5):1276-87. Huang J, Lih CJ, Pan KH, Cohen SN. Global analysis of growth phase responsive gene expression and regulation of antibiotic biosynthetic pathways in Streptomyces coelicolor using DNA microarrays. Genes Dev. 2001 Dec 1;15(23):3183-92. Literaturverzeichnis - 181 - Hunt AC, Servin-Gonzalez L, Kelemen GH, Buttner MJ. The bldC developmental locus of Streptomyces coelicolor encodes a member of a family of small DNA-binding proteins related to the DNA-binding domains of the MerR family. J Bacteriol. 2005 Jan;187(2):716-28. Hutchings MI, Hoskisson PA, Chandra G, Buttner MJ. Sensing and responding to diverse extracellular signals? Analysis of the sensor kinases and response regulators of Streptomyces coelicolor A3(2). Microbiology. 2004 Sep;150(Pt 9):2795-806. Review. Ikeda H, Ishikawa J, Hanamoto A, Shinose M, Kikuchi H, Shiba T, Sakaki Y, Hattori M, Omura S. Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nat Biotechnol. 2003 May;21(5):526-31. Ishikura S, Horie K, Sanai M, Matsumoto K, Hara A. Enzymatic properties of a member (AKR1C19) of the aldo-keto reductase family. Biol Pharm Bull. 2005 Jun;28(6):1075-8. Ishizuka H, Horinouchi S, Kieser HM, Hopwood DA, Beppu T. A putative two-component regulatory system involved in secondary metabolism in Streptomyces spp. J Bacteriol. 1992 Dec;174(23):7585-94. Jalkanen S, Salmi M. Cell surface monoamine oxidases: enzymes in search of a function. EMBO J. 2001 Aug 1;20(15):3893-901. Review. Janssen GR, Bibb MJ. Tandem promoters, tsrp1 and tsrp2, direct transcription of the thiostrepton resistance gene (tsr) of Streptomyces azureus: transcriptional initiation from tsrp2 occurs after deletion of the -35 region. Mol Gen Genet. 1990 May;221(3):33946. Jensen SE, Paradkar AS. Biosynthesis and molecular genetics of clavulanic acid. Antonie Van Leeuwenhoek. 1999 Jan-Feb;75(1-2):125-33. Review. Jeudy, S, Abergel, C, Claverie, JM. Crystal Structure of Ydhf, an Aldo-Keto Reductase from Escherichia Coli. To be Published Jez JM, Penning TM. The aldo-keto reductase (AKR) superfamily: an update. Chem Biol Interact. 2001 Jan 30;130-132(1-3):499-525. Jez JM, Bennett MJ, Schlegel BP, Lewis M, Penning TM. Comparative anatomy of the aldo-keto reductase superfamily. Biochem J. 1997 Sep 15;326 ( Pt 3):625-36. Review. - 182 - Literaturverzeichnis Kahmann JD, Sass HJ, Allan MG, Seto H, Thompson CJ, Grzesiek S. Structural basis for antibiotic recognition by the TipA class of multidrug-resistance transcriptional regulators. EMBO J. 2003 Apr 15;22(8):1824-34. Kalinowski J, Bathe B, Bartels D, Bischoff N, Bott M, Burkovski A, Dusch N, Eggeling L, Eikmanns BJ, Gaigalat L, Goesmann A, Hartmann M, Huthmacher K, Kramer R, Linke B, McHardy AC, Meyer F, Mockel B, Pfefferle W, Puhler A, Rey DA, Ruckert C, Rupp O, Sahm H, Wendisch VF, Wiegrabe I, Tauch A. The complete Corynebacterium glutamicum ATCC 13032 genome sequence and its impact on the production of L-aspartate-derived amino acids and vitamins. J Biotechnol. 2003 Sep 4;104(1-3):5-25. Review. Kaneko T, Nakamura Y, Sato S, Asamizu E, Kato T, Sasamoto S, Watanabe A, Idesawa K, Ishikawa A, Kawashima K, Kimura T, Kishida Y, Kiyokawa C, Kohara M, Matsumoto M, Matsuno A, Mochizuki Y, Nakayama S, Nakazaki N, Shimpo S, Sugimoto M, Takeuchi C, Yamada M, Tabata S. Complete genome structure of the nitrogen-fixing symbiotic bacterium Mesorhizobium loti. DNA Res. 2000 Dec 31;7(6):331-8. Katz E, Thompson CJ, Hopwood DA. Cloning and expression of the tyrosinase gene from Streptomyces antibioticus in Streptomyces lividans. J Gen Microbiol. 1983 Sep;129(9):2703-14. Kawabuchi M, Hara Y, Nihira T, Yamada Y. Production of butyrolactone autoregulators by Streptomyces coelicolor A3(2). FEMS Microbiology Letters 1997 157:1 81 Kelemen GH, Viollier PH, Tenor J, Marri L, Buttner MJ, Thompson CJ. A connection between stress and development in the multicellular prokaryote Streptomyces coelicolor A3(2). Mol Microbiol. 2001 May;40(4):804-14. Kelemen GH, Brian P, Flardh K, Chamberlin L, Chater KF, Buttner MJ. Developmental regulation of transcription of whiE, a locus specifying the polyketide spore pigment in Streptomyces coelicolor A3 (2). J Bacteriol. 1998 May;180(9):2515-21. Kelly VP, Ireland LS, Ellis EM, Hayes JD. Purification from rat liver of a novel constitutively expressed member of the aldo-keto reductase 7 family that is widely distributed in extrahepatic tissues. Biochem J. 2000 Jun 1;348 Pt 2:389-400. Kessler RJ, Fanestil DD. Interference by lipids in the determination of protein using bicinchoninic acid. Anal Biochem. 1986 Nov 15;159(1):138-42. Literaturverzeichnis - 183 - Khokhlov AS, Tovarova II, Borisova LN, Pliner SA, Shevchenko LN, Kornitskaia EIa, Ivkina NS, Rapoport IA. [The A-factor, responsible for streptomycin biosynthesis by mutant strains of Actinomyces streptomycini]. Dokl Akad Nauk SSSR. 1967 Nov-Dec;177(1):2325. Russian. Killham K, Firestone MK. Salt Stress Control of Intracellular Solutes in Streptomycetes Indigenous to Saline Soils. Appl Environ Microbiol. 1984 Feb;47(2):301-306. Klinman JP. New quinocofactors in eukaryotes. J Biol Chem. 1996 Nov 1;271(44):2718992. Review. Klinman JP, Mu D. Quinoenzymes in biology. Annu Rev Biochem. 1994;63:299-344. Review. Kormanec J, Sevcikova B. A relation between stress and differentiation in Streptomyces. Meeting: Biology of streptomycetes and related actinomycetes. Talk 10. 2003 Febr. 27. Kormanec J, Sevcikova B. Stress-response sigma factor sigma(H) directs expression of the gltB gene encoding glutamate synthase in Streptomyces coelicolor A3(2). Biochim Biophys Acta. 2002a Aug 19;1577(1):149-54. Kormanec J, Sevcikova B. The stress-response sigma factor sigma(H) controls the expression of ssgB, a homologue of the sporulation-specific cell division gene ssgA, in Streptomyces coelicolor A3(2). Mol Genet Genomics. 2002b Jun;267(4):536-43. Kormanec J, Sevcikova B, Halgasova N, Knirschova R, Rezuchova B. Identification and transcriptional characterization of the gene encoding the stress-response sigma factor sigma(H) in streptomyces coelicolor A3(2). FEMS Microbiol Lett. 2000 Aug 1;189(1):31-8. Koshland DE. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc Natl Acad Sci U S A. 1958 Feb;44(2):98-104. Kozma E, Brown E, Ellis EM, Lapthorn AJ. The crystal structure of rat liver AKR7A1. A dimeric member of the aldo-keto reductase superfamily. J Biol Chem. 2002 May 3;277(18):16285-93. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970 Aug 15;227(5259):680-5. - 184 - Literaturverzeichnis Lai C, Xu J, Tozawa Y, Okamoto-Hosoya Y, Yao X, Ochi K. Genetic and physiological characterization of rpoB mutations that activate antibiotic production in Streptomyces lividans. Microbiology. 2002 Nov;148(Pt 11):3365-73. Lee EJ, Karoonuthaisiri N, Kim HS, Park JH, Cha CJ, Kao CM, Roe JH. A master regulator sigmaB governs osmotic and oxidative response as well as differentiation via a network of sigma factors in Streptomyces coelicolor. Mol Microbiol. 2005 Sep;57(5):1252-64. Lee EJ, Cho YH, Kim HS, Ahn BE, Roe JH. Regulation of sigmaB by an anti- and an antianti-sigma factor in Streptomyces coelicolor in response to osmotic stress. J Bacteriol. 2004a Dec;186(24):8490-8. Lee EJ, Cho YH, Kim HS, Roe JH. Identification of sigmaB-dependent promoters using consensus-directed search of Streptomyces coelicolor genome. J Microbiol. 2004b Jun;42(2):147-51. Lee SC, Omer CA, Brasch MA, Cohen SN. Analysis of recombination occurring at SLP1 att sites. J Bacteriol. 1988 Dec;170(12):5806-13. Lentzen G, Klinck R, Matassova N, Aboul-ela F, Murchie AI. Structural basis for contrasting activities of ribosome binding thiazole antibiotics. Chem Biol. 2003 Aug;10(8):769-78. Li J, Vrielink A, Brick P, Blow DM. Crystal structure of cholesterol oxidase complexed with a steroid substrate: implications for flavin adenine dinucleotide dependent alcohol oxidases. Biochemistry. 1993 Nov 2;32(43):11507-15. Li L, Bannantine JP, Zhang Q, Amonsin A, May BJ, Alt D, Banerji N, Kanjilal S, Kapur V. The complete genome sequence of Mycobacterium avium subspecies paratuberculosis. Proc Natl Acad Sci U S A. 2005 Aug 30;102(35):12344-9. Li YQ, Chen PL, Chen SF, Wu D, Zheng J. A pair of two-component regulatory genes ecrA1/A2 in S. coelicolor. J Zhejiang Univ Sci. 2004 Feb;5(2):173-9. Liljenstrom H, von Heijne G. Translation rate modification by preferential codon usage: intragenic position effects. J Theor Biol. 1987 Jan 7;124(1):43-55. Lin YS, Kieser HM, Hopwood DA, Chen CW. The chromosomal DNA of Streptomyces lividans 66 is linear. Mol Microbiol. 1993 Dec;10(5):923-33 Literaturverzeichnis - 185 - Liras P, Asturias JA, Martin JF. Phosphate control sequences involved in transcriptional regulation of antibiotic biosynthesis. Trends Biotechnol. 1990 Jul;8(7):184-9. Review. Liu DQ, Liu H, Gao XL, Leak DJ, Zhou NY. Arg169 is essential for catalytic activity of 3hydroxybenzoate 6-hydroxylase from Klebsiella pneumoniae M5a1. Microbiol Res. 2005;160(1):53-9. Loenen WA, Blattner FR. Lambda Charon vectors (Ch32, 33, 34 and 35) adapted for DNA cloning in recombination-deficient hosts. Gene. 1983 Dec;26(2-3):171-9. Lyles GA. Mammalian plasma and tissue-bound semicarbazide-sensitive amine oxidases: biochemical, pharmacological and toxicological aspects. Int J Biochem Cell Biol. 1996 Mar;28(3):259-74. Review. MacNeil DJ. Characterization of a unique methyl-specific restriction system in Streptomyces avermitilis. J Bacteriol. 1988 Dec;170(12):5607-12. Malpartida F, Niemi J, Navarrete R, Hopwood DA. Cloning and expression in a heterologous host of the complete set of genes for biosynthesis of the Streptomyces coelicolor antibiotic undecylprodigiosin. Gene. 1990 Sep 1;93(1):91-9. Malpartida F, Hopwood DA. Physical and genetic characterisation of the gene cluster for the antibiotic actinorhodin in Streptomyces coelicolor A3(2). Mol Gen Genet. 1986 Oct;205(1):66-73. Maniatis T, Fritsch EF, Sambrook J. Molecular Cloning: A LABORATORY MANUAL. 1982. (Cold Spring Harbor Laboratory) Maplestone RA, Stone MJ, Williams DH. The evolutionary role of secondary metabolites-a review. Gene. 1992 Jun 15;115(1-2):151-7. Review. Martin JF. Phosphate control of the biosynthesis of antibiotics and other secondary metabolites is mediated by the PhoR-PhoP system: an unfinished story. J Bacteriol. 2004 Aug;186(16):5197-201. Review. Martin JF, Demain AL. Control of antibiotic biosynthesis. Microbiol Rev. 1980 Jun;44(2):230-51. - 186 - Literaturverzeichnis Martinez-Costa OH, Arias P, Romero NM, Parro V, Mellado RP, Malpartida F. A relA/spoT homologous gene from Streptomyces coelicolor A3(2) controls antibiotic biosynthetic genes. J Biol Chem. 1996 May 3;271(18):10627-34. Masuma R, Tanaka Y, Tanaka H, Omura S. Production of nanaomycin and other antibiotics by phosphate-depressed fermentation using phosphate-trapping agents. J Antibiot (Tokyo). 1986 Nov;39(11):1557-64. McDowall KJ, Thamchaipenet A, Hunter IS. Phosphate control of oxytetracycline production by Streptomyces rimosus is at the level of transcription from promoters overlapped by tandem repeats similar to those of the DNA-binding sites of the OmpR family. J Bacteriol. 1999 May;181(10):3025-32. Meuser D, Splitt H, Wagner R, Schrempf H. Mutations stabilizing an open conformation within the external region of the permeation pathway of the potassium channel KcsA. Eur Biophys J. 2001 Sep;30(5):385-91. Miguelez EM, Hardisson C, Manzanal MB. Hyphal death during colony development in Streptomyces antibioticus: morphological evidence for the existence of a process of cell deletion in a multicellular prokaryote. J Cell Biol. 1999 May 3;145(3):515-25. Mochizuki S, Hiratsu K, Suwa M, Ishii T, Sugino F, Yamada K, Kinashi H. The large linear plasmid pSLA2-L of Streptomyces rochei has an unusually condensed gene organization for secondary metabolism. Mol Microbiol. 2003 Jun;48(6):1501-10. Mori K. Revision of the absolute configuration of A-factor, the inducer of streptomycin biosynthesis, basing on the reconfirmed (R)-configuration of (+)-paraconic acid. Tetrahedon. 1983; 38:2919-2921 Muller F, Voordouw G, Van Berkel WJ, Steennis PJ, Visser S, Van Rooijen PJ. A study of phydroxybenzoate hydroxylase from Pseudomonas fluorescens. Improved purification, relative molecular mass, and amino acid composition. Eur J Biochem. 1979 Nov 1;101(1):235-44. Mullis KB, Faloona FA. Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol. 1987;155:335-50. Murakami T, Holt TG, Thompson CJ. Thiostrepton-induced gene expression in Streptomyces lividans. J Bacteriol. 1989 Mar;171(3):1459-66. Literaturverzeichnis - 187 - Muth G, Farr M, Hartmann V, Wohlleben W. Streptomyces ghanaensis plasmid pSG5: nucleotide sequence analysis of the self-transmissible minimal replicon and characterization of the replication mode. Plasmid. 1995 Mar;33(2):113-26 Muth G, Nussbaumer B, Wohlleben W, Puehler A. A vector system with temperaturesensitive replication for gene disruption and mutational cloning in streptomycetes. Mol Gen Genet. 1989, 219:341-348 Nakano T, Miyake K, Endo H, Dairi T, Mizukami T, Katsumata R. Identification and cloning of the gene involved in the final step of chlortetracycline biosynthesis in Streptomyces aureofaciens. Biosci Biotechnol Biochem. 2004 Jun;68(6):1345-52. Narva KE, Feitelson JS. Nucleotide sequence and transcriptional analysis of the redD locus of Streptomyces coelicolor A3(2). J Bacteriol. 1990 Jan;172(1):326-33. Nielsen H, Krogh A. Prediction of signal peptides and signal anchors by a hidden Markov model. Proc Int Conf Intell Syst Mol Biol. 1998;6:122-30. Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997 Jan;10(1):1-6. Okamoto-Hosoya Y, Hosaka T, Ochi K. An aberrant protein synthesis activity is linked with antibiotic overproduction in rpsL mutants of Streptomyces coelicolor A3(2). Microbiology. 2003a Nov;149(Pt 11):3299-309. Okamoto-Hosoya Y, Okamoto S, Ochi K. Development of antibiotic-overproducing strains by site-directed mutagenesis of the rpsL gene in Streptomyces lividans. Appl Environ Microbiol. 2003b Jul;69(7):4256-9. Okamoto S, Lezhava A, Hosaka T, Okamoto-Hosoya Y, Ochi K. Enhanced expression of Sadenosylmethionine synthetase causes overproduction of actinorhodin in Streptomyces coelicolor A3(2). J Bacteriol. 2003 Jan;185(2):601-9. Onaka H, Nakagawa T, Horinouchi S. Involvement of two A-factor receptor homologues in Streptomyces coelicolor A3(2) in the regulation of secondary metabolism and morphogenesis. Mol Microbiol. 1998 May;28(4):743-53. - 188 - Literaturverzeichnis Ortiz-Maldonado M, Entsch B, Ballou DP. Oxygen reactions in p-hydroxybenzoate hydroxylase utilize the H-bond network during catalysis. Biochemistry. 2004 Dec 7;43(48):15246-57. Ortiz-Maldonado M, Entsch B, Ballou DP. Conformational changes combined with charge-transfer interactions are essential for reduction in catalysis hydroxybenzoate hydroxylase. Biochemistry. 2003 Sep 30;42(38):11234-42. by p- Palfey BA, Murthy YV, Massey V. Altered balance of half-reactions in phydroxybenzoate hydroxylase caused by substituting the 2'-carbon of FAD with fluorine. J Biol Chem. 2003 Jun 20;278(25):22210-6. Palfey BA, Basu R, Frederick KK, Entsch B, Ballou DP. Role of protein flexibility in the catalytic cycle of p-hydroxybenzoate hydroxylase elucidated by the Pro293Ser mutant. Biochemistry. 2002 Jul 2;41(26):8438-46. Palfey BA, Moran GR, Entsch B, Ballou DP, Massey V. Substrate recognition by "password" in p-hydroxybenzoate hydroxylase. Biochemistry. 1999 Jan 26;38(4):1153-8. Park BH, Levy SB. The cryptic tetracycline resistance determinant on Tn4400 mediates tetracycline degradation as well as tetracycline efflux. Antimicrob Agents Chemother. 1988 Dec;32(12):1797-800. Parkhill J, Ansari AZ, Wright JG, Brown NL, O'Halloran TV. Construction and characterization of a mercury-independent MerR activator (MerRAC): transcriptional activation in the absence of Hg(II) is accompanied by DNA distortion. EMBO J. 1993 Feb;12(2):413-21. Phillips RW, Wiegel J, Berry CJ, Fliermans C, Peacock AD, White DC, Shimkets LJ. Kineococcus radiotolerans sp. nov., a radiation-resistant, gram-positive bacterium. Int J Syst Evol Microbiol. 2002 May;52(Pt 3):933-8. Plotho Ov. Farbstoffbildung bei Actinomyceten. Die Naturwissenschaften. 1947; 34:190. Potuckova L, Kelemen GH, Findlay KC, Lonetto MA, Buttner MJ, Kormanec J. A new RNA polymerase sigma factor, sigma F, is required for the late stages of morphological differentiation in Streptomyces spp. Mol Microbiol. 1995 Jul;17(1):37-48. Literaturverzeichnis - 189 - Price B, Adamidis T, Kong R, Champness W. A Streptomyces coelicolor antibiotic regulatory gene, absB, encodes an RNase III homolog. J Bacteriol. 1999 Oct;181(19):6142-51. Price CW, Fawcett P, Ceremonie H, Su N, Murphy CK, Youngman P. Genome-wide analysis of the general stress response in Bacillus subtilis. Mol Microbiol. 2001 Aug;41(4):757-74. Rajasekharan S, Rajasekharan R, Vaidyanathan CS. Substrate-mediated purification and characterization of a 3-hydroxybenzoic acid-6-hydroxylase from Micrococcus. Arch Biochem Biophys. 1990 Apr;278(1):21-5. Rao ST, Rossmann MG. Comparison of super-secondary structures in proteins. J Mol Biol. 1973 May 15;76(2):241-56. Rebollo A, Gil JA, Liras P, Asturias JA, Martin JF. Cloning and characterization of a phosphate-regulated promoter involved in phosphate control of candicidin biosynthesis. Gene. 1989 Jun 30;79(1):47-58. Richter G, Fischer M, Krieger C, Eberhardt S, Luttgen H, Gerstenschlager I, Bacher A. Biosynthesis of riboflavin: characterization of the bifunctional deaminase-reductase of Escherichia coli and Bacillus subtilis. J Bacteriol. 1997 Mar;179(6):2022-8. Rondeau JM, Tete-Favier F, Podjarny A, Reymann JM, Barth P, Biellmann JF, Moras D. Novel NADPH-binding domain revealed by the crystal structure of aldose reductase. Nature. 1992 Jan 30;355(6359):469-72. Rose RE. The nucleotide sequence of pACYC184. Nucleic Acids Res. 1988 Jan 11;16(1):355. Schagger H, von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem. 1987 Nov 1;166(2):368-79. Scheu AK, Martinez E, Soliveri J, Malpartida F. abaB, a putative regulator for secondary metabolism in Streptomyces. FEMS Microbiol Lett. 1997 Feb 1;147(1):29-36. Schrempf H, Bujard H, Hopwood DA, Goebel W. Isolation of covalently closed circular deoxyribonucleic acid from Streptomyces coelicolor A3(2). J Bacteriol. 1975 Feb;121(2):416-21. - 190 - Literaturverzeichnis Schreuder HA, Prick PA, Wierenga RK, Vriend G, Wilson KS, Hol WG, Drenth J. Crystal structure of the p-hydroxybenzoate hydroxylase-substrate complex refined at 1.9 A resolution. Analysis of the enzyme-substrate and enzyme-product complexes. J Mol Biol. 1989 Aug 20;208(4):679-96. Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003 Jul 1;31(13):3381-5. Seibold B, Matthes M, Eppink MH, Lingens F, Van Berkel WJ, Muller R. 4Hydroxybenzoate hydroxylase from Pseudomonas sp. CBS3. Purification, characterization, gene cloning, sequence analysis and assignment of structural features determining the coenzyme specificity. Eur J Biochem. 1996 Jul 15;239(2):469-78. Sejlitz T, Neujahr HY. Phenol hydroxylase from yeast. A model for phenol binding and an improved purification procedure. Eur J Biochem. 1987 Dec 30;170(1-2):343-9. Sevcikova B, Mazurakova V, Kormanec J. Characterization of the alternative sigma factor sigmaG in Streptomyces coelicolor A3(2). Folia Microbiol (Praha). 2005;50(1):47-58. Sevcikova B, Kormanec J. Differential production of two antibiotics of Streptomyces coelicolor A3(2), actinorhodin and undecylprodigiosin, upon salt stress conditions. Arch Microbiol. 2004 May;181(5):384-9. Sevcikova B, Benada O, Kofronova O, Kormanec J. Stress-response sigma factor sigma(H) is essential for morphological differentiation of Streptomyces coelicolor A3(2). Arch Microbiol. 2001 Dec;177(1):98-106. Sheldon PJ, Busarow SB, Hutchinson CR. Mapping the DNA-binding domain and target sequences of the Streptomyces peucetius daunorubicin biosynthesis regulatory protein, DnrI. Mol Microbiol. 2002 Apr;44(2):449-60. Shi W, Zusman DR. Fatal attraction. Nature. 1993 Dec 2;366(6454):414-5. Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985 Oct;150(1):76-85. Erratum in: Anal Biochem 1987 May 15;163(1):279. Speer BS, Salyers AA. Characterization of a novel tetracycline resistance that functions only in aerobically grown Escherichia coli. J Bacteriol. 1988 Apr;170(4):1423-9. Literaturverzeichnis - 191 - Sola-Landa A, Moura RS, Martin JF. The two-component PhoR-PhoP system controls both primary metabolism and secondary metabolite biosynthesis in Streptomyces lividans. Proc Natl Acad Sci U S A. 2003 May 13;100(10):6133-8. Strauch E, Takano E, Baylis HA, Bibb MJ. The stringent response in Streptomyces coelicolor A3(2). Mol Microbiol. 1991 Feb;5(2):289-98. Strohl WR. Compilation and analysis of DNA sequences associated with apparent streptomycete promoters. Nucleic Acids Res. 1992 Mar 11;20(5):961-74. Review. Studholme DJ, Bentley SD, Kormanec J. Bioinformatic identification of novel regulatory DNA sequence motifs in Streptomyces coelicolor. BMC Microbiol. 2004 Apr 8;4:14. Suemori A, Nakajima K, Kurane R, Nakamura Y. o-, m- and p-hydroxybenzoate degradative pathways in Rhodococcus erythropolis. FEMS Microbiol Lett. 1995 Jan 1;125(1):31-5. Suwa M, Sugino H, Sasaoka A, Mori E, Fujii S, Shinkawa H, Nimi O, Kinashi H. Identification of two polyketide synthase gene clusters on the linear plasmid pSLA2-L in Streptomyces rochei. Gene. 2000 Apr 4;246(1-2):123-31. Takano E, Tao M, Long F, Bibb MJ, Wang L, Li W, Buttner MJ, Bibb MJ, Deng ZX, Chater KF. A rare leucine codon in adpA is implicated in the morphological defect of bldA mutants of Streptomyces coelicolor. Mol Microbiol. 2003 Oct;50(2):475-86. Takano E, Chakraburtty R, Nihira T, Yamada Y, Bibb MJ. A complex role for the gammabutyrolactone SCB1 in regulating antibiotic production in Streptomyces coelicolor A3(2). Mol Microbiol. 2001 Sep;41(5):1015-28. Thompson CJ, Kieser T, Ward JM, Hopwood DA. Physical analysis of antibiotic-resistance genes from Streptomyces and their use in vector construction. Gene. 1982a Nov;20(1):5162. Thompson CJ, Skinner RH, Thompson J, Ward JM, Hopwood DA, Cundliffe E. Biochemical characterization of resistance determinants cloned streptomycetes. J Bacteriol. 1982b Aug;151(2):678-85. from antibiotic-producing Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979 Sep;76(9):4350-4. - 192 - Literaturverzeichnis Tsao SW, Rudd BA, He XG, Chang CJ, Floss HG. Identification of a red pigment from Streptomyces coelicolor A3(2) as a mixture of prodigiosin derivatives. J Antibiot (Tokyo). 1985 Jan;38(1):128-31. Tu SC, Romero FA, Wang LH. Uncoupling of the substrate monooxygenation and reduced pyridine nucleotide oxidation activities of salicylate hydroxylase by flavins. Arch Biochem Biophys. 1981 Jul;209(2):423-32. Uguru GC, Stephens KE, Stead JA, Towle JE, Baumberg S, McDowall KJ. Transcriptional activation of the pathway-specific regulator of the actinorhodin biosynthetic genes in Streptomyces coelicolor. Mol Microbiol. 2005 Oct;58(1):131-50. Umeyama T, Tanabe Y, Aigle BD, Horinouchi S. Expression of the Streptomyces coelicolor A3(2) ptpA gene encoding a phosphotyrosine protein phosphatase leads to overproduction of secondary metabolites in S. lividans. FEMS Microbiol Lett. 1996 Nov 1;144(2-3):177-84. van Dalen A, Schrempf H, Killian JA, de Kruijff B. Efficient membrane assembly of the KcsA potassium channel in Escherichia coli requires the protonmotive force. EMBO Rep. 2000 Oct;1(4):340-6. Vara J, Lewandowska-Skarbek M, Wang YG, Donadio S, Hutchinson CR. Cloning of genes governing the deoxysugar portion of the erythromycin biosynthesis pathway in Saccharopolyspora erythraea (Streptomyces erythreus). J Bacteriol. 1989 Nov;171(11):5872-81. Verma S, Bhatia Y, Valappil SP, Roy I. A possible role of poly-3-hydroxybutyric acid in antibiotic production in Streptomyces. Arch Microbiol. 2002 Dec;179(1):66-9. Villarejo MR, Zabin I. Beta-galactosidase from termination and deletion mutant strains. J Bacteriol. 1974 Oct;120(1):466-74. Viollier PH, Weihofen A, Folcher M, Thompson CJ. Post-transcriptional regulation of the Streptomyces coelicolor stress responsive sigma factor, SigH, involves translational control, proteolytic processing, and an anti-sigma factor homolog. J Mol Biol. 2003a Jan 24;325(4):637-49. Viollier PH, Kelemen GH, Dale GE, Nguyen KT, Buttner MJ, Thompson CJ. Specialized osmotic stress response systems involve multiple SigB-like sigma factors in Streptomyces coelicolor. Mol Microbiol. 2003b Feb;47(3):699-714. Literaturverzeichnis - 193 - Vivian A, Hopwood DA. Genetic control of fertility in Streptomyces coelicolor A3(2): the IF fertility type. J Gen Microbiol. 1970 Nov;64(1):101-17. Volker U, Maul B, Hecker M. Expression of the sigmaB-dependent general stress regulon confers multiple stress resistance in Bacillus subtilis. J Bacteriol. 1999 Jul;181(13):3942-8. Wang J, Ortiz-Maldonado M, Entsch B, Massey V, Ballou D, Gatti DL. Protein and ligand dynamics in 4-hydroxybenzoate hydroxylase. Proc Natl Acad Sci U S A. 2002 Jan 22;99(2):608-13. Wang LH, Hamzah RY, Yu YM, Tu SC. Pseudomonas cepacia 3-hydroxybenzoate 6hydroxylase: induction, purification, and characterization. Biochemistry. 1987 Feb 24;26(4):1099-104. Wang SX, Nakamura N, Mure M, Klinman JP, Sanders-Loehr J. Characterization of the native lysine tyrosylquinone cofactor in lysyl oxidase by Raman spectroscopy. J Biol Chem. 1997 Nov 14;272(46):28841-4. White J, Bibb M. bldA dependence of undecylprodigiosin production in Streptomyces coelicolor A3(2) involves a pathway-specific regulatory cascade. J Bacteriol. 1997 Feb;179(3):627-33. Whittle G, Hund BD, Shoemaker NB, Salyers AA. Characterization of the 13-kilobase ermF region of the Bacteroides conjugative transposon CTnDOT. Appl Environ Microbiol. 2001 Aug;67(8):3488-95. Wietzorrek A, Bibb M. A novel family of proteins that regulates antibiotic production in streptomycetes appears to contain an OmpR-like DNA-binding fold. Mol Microbiol. 1997 Sep;25(6):1181-4. Williamson NR, Simonsen HT, Ahmed RA, Goldet G, Slater H, Woodley L, Leeper FJ, Salmond GP. Biosynthesis of the red antibiotic, prodigiosin, in Serratia: identification of a novel 2-methyl-3-n-amyl-pyrrole (MAP) assembly pathway, definition of the terminal condensing enzyme, and implications for undecylprodigiosin biosynthesis in Streptomyces. Mol Microbiol. 2005 May;56(4):971-89. Wilson DK, Nakano T, Petrash JM, Quiocho FA. 1.7 A structure of FR-1, a fibroblast growth factor-induced member of the aldo-keto reductase family, complexed with coenzyme and inhibitor. Biochemistry. 1995 Nov 7;34(44):14323-30. - 194 - Literaturverzeichnis Wilson DK, Tarle I, Petrash JM, Quiocho FA. Refined 1.8 A structure of human aldose reductase complexed with the potent inhibitor zopolrestat. Proc Natl Acad Sci U S A. 1993 Nov 1;90(21):9847-51. Wilson DK, Bohren KM, Gabbay KH, Quiocho FA. An unlikely sugar substrate site in the 1.65 A structure of the human aldose reductase holoenzyme implicated in diabetic complications. Science. 1992 Jul 3;257(5066):81-4. Wood WB. Host specificity of DNA produced by Escherichia coli: bacterial mutations affecting the restriction and modification of DNA. J Mol Biol. 1966 Mar;16(1):118-33. Woodcock DM, Crowther PJ, Doherty J, Jefferson S, DeCruz E, Noyer-Weidner M, Smith SS, Michael MZ, Graham MW. Quantitative evaluation of Escherichia coli host strains for tolerance to cytosine methylation in plasmid and phage recombinants. Nucleic Acids Res. 1989 May 11;17(9):3469-78. Wright GD. Allostery trumps antibiotic resistance. Structure. 2005 Aug;13(8):1089-90. Yang W, Moore IF, Koteva KP, Bareich DC, Hughes DW, Wright GD. TetX is a flavindependent monooxygenase conferring resistance to tetracycline antibiotics. J Biol Chem. 2004 Dec 10;279(50):52346-52. Yanisch-Perron C, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene. 1985;33(1):10319. Erratum in: Gene. 1992 May 1;114(1):81-3. Yonekawa T, Ohnishi Y, Horinouchi S. Involvement of amfC in physiological and morphological development in Streptomyces coelicolor A3(2). Microbiology. 1999 Sep;145 ( Pt 9):2273-80. You IS, Murray RI, Jollie D, Gunsalus IC. Purification and characterization of salicylate hydroxylase from Pseudomonas putida PpG7. Biochem Biophys Res Commun. 1990 Jun 29;169(3):1049-54. Zazopoulos E, Huang K, Staffa A, Liu W, Bachmann BO, Nonaka K, Ahlert J, Thorson JS, Shen B, Farnet CM. A genomics-guided approach for discovering and expressing cryptic metabolic pathways. Nat Biotechnol. 2003 Feb;21(2):187-90. Anhang A-E ANHANG i ii Anhang A Anhang A iii iv Anhang A Anhang A v vi Anhang A Anhang A vii viii Anhang A Anhang A ix x Anhang A Anhang A xi xii Anhang A Anhang A xiii xiv Anhang A Anhang A: Der sequenzierte 13920 Bp lange Abschnitt aus S.lividans 66. Die Sequenz ist doppelsträngig dargestellt, die jeweils determinierte Aminosäuresequenz der Proteine in grün. Die jeweils 19 Bp langen „inverted repeats“ des zwischen ksuL und ksuF gelegenen Terminators sind durch rote Pfeile markiert. Die potentiellen Ribosomen-Bindestellen (Shine-Delgarno) sind jeweils in Fett dargestellt, eine zweite mögliche Bindestelle vor ksuC ist unterstrichen. Die möglichen Bindestellen wurden aus der Sequenz des 3’ Endes der 16S rRNA von S.lividans erstellt (3’-OH-UCUUUCCUCCACUAG-5’; Bibb, M.J., et al; 1985). Alle liegen im beschriebenen Bereich zwischen 5-12 Nukleotiden (Durchschnitt von 8,5) vor dem Startkodon, hierbei wird der Abstand vom A, bzw. des entsprechenden Nukleotids der GGAGG Konsensussequenz zum ersten Nukleotid des Starkodons berechnet (Strohl, W. R.; 1992). Die zwischen ksuF und kcsA gefundenen repetetiven Sequenzen sind in schwarz (Palyndrome), in rot („direct repeats“) und in grün („invertet repeats“) durch Linien oder Pfeile markiert. Es wurden nur „direct repeats ab einer Länge von 6 und „invertet repeats“ ab 7 Nukleotiden eingetragen. Die Signalpeptidase-Schnittstelle in KsuL ist durch einen roten Pfeil markiert, die Schnittstelle zwischen der N-terminalen und C-terminalen Domäne durch einen blauen Pfeil. Anhang B xv Anhang B: Terminatorstruktur zwischen ksuF und ksuL. Im intergenischen Bereich zwischen ksuL und ksuF findet sich eine 45 Bp lange Terminator-Struktur, die aus zwei 19 Bp „invertet repeats“ und einem 7 Bp langen Loop-Bereich gebildet wird. Dargestellt ist die Terminator-Struktur in Leserichtung von ksuF. xvi Anhang C Anhang C xvii xviii Anhang C Anhang C xix Anhang C: Konstruktion von pACYC001 bis pACYC010. Die in pACYC001 bis pACYC010 inserierten Fragmente wurden zur Konstruktion der pOV001-pOV010 Konstrukte mit XbaI/PstI ausgeschnitten und mit pWHM3be ligiert. Anhang Tabelle D xx Probe A1 A2 A3 A4 Durchschnitt A1-4 σx A1-4 B1 B2 B3 B4 B5 Durchschnitt B1-5 σx B1-5 C1 C2 C3 C4 C5 Durchschnitt C1-5 σx C1-5 Gesamt D σx D Konz. FAD (in mM) 0,01534 0,01967 0,01592 0,00923 0,015037 Konz. Protein (in mM) 0,0165 0,0183 0,0158 0,0095 0,015025 0,00642 0,01320 0,01145 0,01086 0,00641 0,01028 0,007 0,0146 0,016 0,0123 0,0072 0,01142 0,01506 0,01642 0,01377 0,007942 0,003521 0,0115361 0,0142 0,0163 0,0123 0,008 0,0036 0,01088 Verhältnis FAD/Protein 0,92945818 1,07482022 1,00743481 0,97113368 0,99571172 0,06161493 0,91677857 0,90389795 0,90776750 0,88295122 0,88978889 0,90023683 0,01370853 1,06038 1,00749 1,119297 0,992786 0,978158 1,03162242 0,0580149 0,9744388 0,074055 Anhang Tabelle D: Messwerte der FAD/KsuF Verhältnisbestimmung. Dargestellt sind die drei unterschiedlichen Messreihen (A1-A4, B1-B5 und C1-C5), die jeweiligen FAD- und Proteinkonzentrationen und das FAD/Protein-Verhältnis, des Weiteren die Mittelwerte der einzelnen Messreihen, das Gesamtdurchschnittverhältnis (D) und jeweils die Standartabweichung (mittlerer Fehler) σx. Anhang E xxi Anhang E: Sequenzvergleich von KsuL mit den homologen Proteinen. Dargestellt sind, neben KsuL, die homologen Proteine aus S.avermitilis (BAC72171), S.scabies (4945557-4943764) und Nocardia spec. JS614 (ZP_00656873). Die mit KsuL identischen Aminosäuren sind schwarz hinterlegt, die homologen grau. Die vier jeweils konservierten Cysteine sind rot umrandet, die Position der Signalpeptidase Schnittstelle in KsuL ist durch einen roten Pfeil, die proteolytische Schnittstelle zwischen der N- und C-terminalen Domäne von KsuL durch einen blauen Pfeil markiert. Hiermit versichere ich, dass die vorliegende Dissertation selbständig und ohne unerlaubte Hilfe verfasst und keine anderen Quellen und Hilfsmittel als die angegebenen verwendet wurden. Ich versichere, dass ich weder an der Universität Osnabrück noch anderweitig versucht habe, eine Dissertation einzureichen oder mich einer Promotionsprüfung zu unterziehen. ______________________ Ort, Datum ______________________ Unterschrift Ich danke Frau Prof. Dr. H. Schrempf für die Überlassung des interessanten Themas, für die Betreuung und die Korrekturen. Herrn PD Dr. U. Keller danke ich für die Übernahme des Koreferats. Bei Herrn Jan Kormanec bedanke ich mich für die Überlassung der sigH- und sigFDeletionsstämme. Und meiner gesamten Arbeitsgruppe, der „Angewandten Genetik der Mikroorganismen“, danke ich für das gute Arbeitsklima. Und natürlich bedanke ich mich bei allen Personen, die mich in irgendeiner Weise unterstützt haben, wie auch für die Unterstützung durch meine Familie. >>…Das ist das gleiche<< Karl Edward Wagner, Lynortis’ Reprise