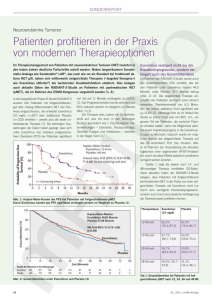
DESIREE Everolimus dose-escalation
Werbung

2.2 Protocol Synopsis German Studientitel DESIREE - Eine multizentrische, randomisierte doppelblinde, Phase-IIStudie zum Vergleich der Verträglichkeit bei einschleichender Dosierung von Everolimus bei Patientinnen mit metastasiertem Brustkrebs Studiennummer GBG 86 EudraCT Nummer 2014-005126-35 Sponsor GBG Forschungs GmbH, Neu-Isenburg Phase Randomisierte, doppelblinde Phase II Studie Hintergrund und Die BOLERO-2 Studie zeigte einen Vorteil für Patienten, die Everolimus Rationale zusammen mit Exemestan erhielten nachdem sie während oder nach der Gabe eines nicht steroidalen Aromatase-Inhibitors (NSAI) einen Progress erlitten hatten (Baselga NEJM 2012) und führte schließlich zur Zulassung von Everolimus in dieser Indikation. Beim Routineeinsatz wird eine hohe Rate der Unerträglichkeit dieses innovativen Behandlungsansatzes besonders während der ersten 12 Wochen der Behandlung beobachtet. Häufigste Nebenwirkung und Hauptursache für die Beendigung der Therapie vor einem Progress ist die orale Mukositis / Stomatitis. Diese Beobachtungen im Routineeinsatz sind gegensätzlich zu Erkenntnissen aus BOLERO-2, in dem die Zahl der Patienten noch die volle Dosis (10 mg) von Everolimus nach 4, 8 und 12 Wochen 77,8%, 75,6% und 75,6% beträgt. Diese Ergebnisse sind in Übereinstimmung mit nicht-interventionellen Studien (NIS). Allerdings könnten die Ergebnisse durch eine günstige Vorauswahl der Patienten herrühren. Im Non-Responder Teil (Setting III) der neoadjuvanten GeparQuinto Studie (Therapieresistente) wurde Everolimus als Salvage-Therapie (zweite und Alternativtherapie) in Kombination mit Paclitaxel bei Patienten ohne Reaktion auf 4 Zyklen Epirubicin / Cyclophosphamid +/Bevacizumab gegeben. Ein Dosis-Eskalationsschema wurde erfolgreich eingesetzt, um die Verträglichkeit von Everolimus zusammen mit dem zytotoxischen Mittel zu verbessern. Primäres Studienziel Vergleich der kumulativen Rate von Mukositis Grad 2-4 (WHO Skala OTS), 12 Wochen nach Beginn der Behandlung jeweils mit dem Standardschema GBG 86 - Desiree Protocol Version 2 – 01.04.2015-Confidential 14 oder einer eskalierenden Dosierung von Everolimus in Kombination mit Exemestan bei Patienten mit metastasierendem Brustkrebs und Progress oder Rezidiv nach nicht-steroidalen Aromatase--Inhibitor Behandlung. Weitere Studienziele Sekundäre Studienziele x Vergleich der kumulativen Rate an Mukositis/Stomatitis Grad 2-4 ((WHO‘s oral toxicity scale (OTS); die erste Episode wird bei jedem Patienten in der Zählung miteinbezogen) 24 Wochen nach x Behandlung mit Everolimus. Vergleich der kumulativen Rate an Mukositis/Stomatitis Grade 1 und jegliches Grads (WHO‘s oral toxicity scale (OTS)) 12 und 24 x Wochen nach Behandlung mit Everolimus. Vergleich des klinischen Benefits (CR, PR und SD >=16 Wochen) zwischen den Behandlungsarmen nach mindestens 24 Wochen x x x x x x Everolimustherapie Ermittlung der Sicherheit hinsichtlich anderer Gesundheitsparameter und Symptome Vergleich der Zeiten bis zum Auftreten ein Mucositis >= 2 Vergleich der kumulativen Dosis nach 4 Wochen Vergleich der relativen Dosisintensität von Everolimus Vergleich der Lebensqualität anhand des FACT-B Fragebogens und des QSDQ Vergleich der Dauer des Ansprechens zwischen den Behandlungsarmen Weitere Studienziele: Potentielle prädiktive Biomarker hinsichtlich Verträglichkeit und Patientenkooperation werden nach Abschluss der Behandlung und der Studie festgelegt Studiendesign und Dies ist eine multizentrische, randomisierte, doppelblinde Phase-II- Behandlung Studie zur Untersuchung einer möglichen Verbesserung der Mukositis Grad 2-4 (WHO Skala OTS) und anderer Compliance und Verträglichkeitsendpunkte 12 Wochen nach Therapiebeginn bei Vergleich einer herkömmlichen Dosierung beginnend mit 10 mg bei erster Gabe versus einer Dosiseskalation über 21 Tage von Everolimus beginnend mit 2,5mg in Kombination mit Exemestan bei Patienten mit metastasierendem Brustkrebs und Progress oder Rezidiv nach nichtGBG 86 - Desiree Protocol Version 2 – 01.04.2015-Confidential 15 steroidalem AI. Alle Patienten werden im Rahmen des Zulassungslabels von Everolimus in Kombination mit Exemestan behandelt. Die Patienten werden im Verhältnis 1:1 dazu randomisiert und erhalten entweder: x Everolimus 10 mg/Tag, oral, Woche 1-3: 4X2,5mg Tag (verblinded); Woche 4-24: 10mg/Tag (open-label entsprechend Fachinformation) x oder Eine vorgeschaltete Dosiseskalation von Everolimus-Plazebo wie folgt: Woche 1 1x2,5mg Verum + 3x Plazebo/Tag; Woche 2: 2x 2,5mg Verum +2x Plazebo/Tag; Woche 3: 3x 2,5 mg Verum + 1x Plazebo/Tag; Woche 4-24: 10mg/Tag (open-label entsprechend Fachinformation) Die Behandlung wird gegeben bis sich ein Fortschreiten der Krankheit einstellt, inakzeptable Toxizitäten der Studienmedikation auftreten, das Einverständnis des Patienten zurückgezogen wird. Die Behandlung ab Woche 24 sollte nach den allgemeinen Richtlinie erfolgen. Einschlusskriterien Patienten dürfen nur dann in die Studien aufgenommen werden, wenn sie folgende Einschlusskriterien erfüllen: 1. Schriftliches Einverständnis studienspezifischer vor Untersuchungen; Kooperationsbereitschaft der Beginn (dies Patientin jeglicher schließt die hinsichtlich der protokollgemäßen Verfahren mit ein) nach lokal gültigen regulatorischen Richtlinien. 2. Die komplette Baselinedokumentation muss mittels des webbasierten Dokumentationssystems MedCODES® an die GBG Forschungs GmbH übermittelt werden. 3. Histologisch gesicherter Hormonrezeptor-positiver (HR+), HER2negativer Brustkrebs.ER/PR-positiv ist definiert als >1% gefärbter Zellen und HER2-positiv ist definiert als HercepTest ICH 3+ oder GBG 86 - Desiree Protocol Version 2 – 01.04.2015-Confidential 16 in-situ hybridisation (ISH) Ratio >2.0. Es muss alles vertretbar mögliche unternommen werden, um das paraffineingebettete Gewebe oder Schnitte Metastasengewebe für des die Primärtumors zentrale und/oder Überprüfung und weiterführende translationale Untersuchungen zur Verfügung zu stellen. 4. Postmenopausale Frauen. 5. Lokal fortgeschritten oder metastasiert und nicht kurativ durch Operation oder Radiotherapie alleine behandelbar. 6. Keine Indikation für eine Chemotherapie. 7. Die Patienten haben ein komplettes Staging innerhalb von 4 Wochen vor der Randomisierung inklusive entsprechend der Routine wie eines CT der Brust und des Abdomens oder ein MRI und einen Knochenscan (Ausnahme Knochenscan bis zu 8 Wochen). Weitere Untersuchungen müssen nach klinischer Notwendigkeit durchgeführt werden. 8. Fortschreiten der Erkrankung während oder nach vorheriger Letrozol oder Anastrozol. Behandlung wie folgt: Wiederauftreten während oder nach Abschluss einer adjuvanten Behandlung mit Letrozol oder Anastrozol. Wiederauftreten während oder nach Abschluss einer Behandlung mit Letrozole oder Anastrozole für metastasierten Brustkrebs Hinweis: nicht-steroidale Aromatase Inhibitorsen (z.B. Letrozol oder Anastrozol) sind nicht notwendigerweise die letzte Therapie vor Einschluss. Andere Therapien sind auch vor Einschluss möglich, z.B. Tamoxifen, Fulvestrant, Exemestan. Die Patienten müssen sich vor Studieneinschluss auf Grad1 oder besser von unerwünschten Ereignissen erholt haben (Außer Alopezie und Nagelveränderungen). 9. Mindestens 4 Wochen seit Radiotherapie mit vollständiger Erholung. Die messbare Zielläsion muss vollständig außerhalb des bestrahlten Bereichs liegen oder es muss ein pathologischer Nachweis eines erneuten Progresses vorliegen. 10. Alter von mindestens 18 Jahren. 11. ECOG-Status 0-2. GBG 86 - Desiree Protocol Version 2 – 01.04.2015-Confidential 17 12. Laborwerte: x x x x x x x x Absolute Neutrophile (ANC) t1500 Zellen/Pl, ,ćŵŽŐůŽďŝŶшϵ͘ϬŐͬĚ>;,ćŵŽŐůŽďŝŶфϵ͘ϬŐͬĚ>ŝƐƚ akzeptabel falls durch Wachstumsfaktoren oder Transfusion korrigiert wird), Thrombozyten t100.000 Zellen/Pl, Bilirubin d1.5x die Obergrenze des Normalwertes der Institution (ULN), Erhöhung der Transaminasen und alkalische PŚŽƐƉŚĂƚĂƐĞфϯdžh>E oĚĞƌфϱdžh>E bei Patienten mit Lebermetastasen, BHS (Blutharnstoffstickstoff) чULN, Nüchtern Blutzucker;&W'ͿчϭϲϬŵŐͬĚ>ŽĚĞƌчϴ͘ϵŵŵŽůͬ>, EƺĐŚƚĞƌŶ ^ĞƌƵŵĐŚŽůĞƐƚĞƌŝŶ ч ϯϬϬ mg/dl oder 7.75 mmol/L (LDL CŚŽůĞƐƚĞƌŝŶ фϭϵϬŵŐͬĚůͿ Ƶnd Nüchtern TƌŝŐůLJĐĞƌŝĚĞ чϮ͘ϱdžh>E ;фϯϬϬ mg/dl). Falls ein oder beide Limits überschritten werden, kann der Patient nur nach Beginn einer Statintherapie und nach x x dem Erreichen obiger Grenzwerte eingeschlossen werden, Kreatinin d2.0 x ULN oder Kreatininclearance >40 ml/min (nach Cockcroft-Gault), Urin-dĞƐƚƐƚƌĞŝĨĞŶ Ĩƺƌ WƌŽƚĞŝŶƵƌŝĞ фϮн͘ Bei Patienten, bei denen ĞŝŶĞшϮнProteinurie bei der Teststreifenanalyse festgestellt wird, sollte eine 24-Stunden-Urinsammlung durchgeführt werden, und ĞƐŵƵƐƐƐŝĐŚчϭŐWƌŽƚĞŝŶŝŶϮϰ^ƚƵŶĚĞŶnjĞŝŐĞŶ, 13. Die Patienten müssen hinsichtlich der Behandlung und der Nachbeobachtung zur Verfügung stehen und kooperativ sein. Patienten, die in diese Studie eingeschlossen werden, müssen in einem teilnehmenden oder mit diesem kooperierenden Zentrum behandelt und nachverfolgt werden. Ausschlusskriterien Patienten, die mindestens eines der folgenden Ausschlusskriterien erfüllen, dürfen nicht in die Studie eingeschlossen werden: 1. Kein dokumentierter Progress unter nicht steroidalen Aromatase Inhibitoren 2. Bekannte Überempfindlichkeit gegen Wirk- oder Zusatzstoffe 3. Gleichzeitige Immuntherapie (empfängnisverhütend oder oder Hormontherapie Hormonersatztherapie). Bisphosphonate oder Denosumab dürfen weiter eingenommen GBG 86 - Desiree Protocol Version 2 – 01.04.2015-Confidential 18 werden. 4. Lebenserwartung von weniger als 3 Monaten. 5. Parenchymale Hirnmetastasen, es sei denn diese werden adäquat durch OP oder Radiotherapie kontrolliert. 6. Jede noch bestehende Toxizität einer früheren Antikrebstherapie die Grad 2-4 ist und/oder sich verschlimmert, außer Alopezie und Nagelveränderungen. 7. Bekannte oder vermutete kongestive Herzinsuffizienz (>NYHA I) und/oder koronare Herzerkrankung, Angina pectoris mit Notwendigkeit einer antianginösen Medikation, Myocardinfarkt in der Krankengeschichte in den letzten 6 Monaten, Hinweis auf einen transmuralen Infarkt im EKG, un- oder unzureichend kontrollierter Bluthochdruck (z.B. BP >150/100 mmHg unter Behandlung mit Rhythmusanomalitäten, zwei die einer Blutdrucksenkern), dauerhaften Therapie bedürfen, klinisch signifikante Herzklappenerkrankung. 8. Gegenwärtig aktive Infektion. 9. Andere maligne Erkrankung in der Krankengeschichte innerhalb der letzten 5 Jahre, die signifikant die Diagnose, Untersuchung oder Prognose des metastasierten Brustkrebses beeinflussen. 10. Malabsorptionssyndrom oder unzureichende gastrointestinale Funktion, vorherige Diagnose einer ulzerierenden Kolitis. 11. Gleichzeitige Behandlung mit anderen experimentellen Substanzen; Teilnahme an anderen klinischen Studien mit Substanzen ohne Marktzulassung innerhalb von 30 Tagen vor Studieneintritt. 12. Unzureichend kontrollierter Diabetes, bekannte HIV-Infektion oder chronische Hepatitis B oder C oder eine Hepatitis B oder C in der Krankengeschichte, ernsthaft beeinträchtigte Leberfunktion (Child-Pugh, Klasse A, B oder C). 13. Jegliches schwerwiegendes und/oder instabiles bereits existierendes medizinisches, psychiatrisches oder anderweitiges Leiden, welches mit der Sicherheit des Studienteilnehmers, der Einholung GBG 86 - Desiree des Patienteneinverständnisses Protocol Version 2 – 01.04.2015-Confidential oder der 19 Kooperationsbereitschaft (inklusive zur schwerwiegende Studienbehandlung Lungenerkrankung, kollidiert AIDS und schwerwiegende aktive Infektion sowie Diabetes mellitus). 14. Männliche Patienten. 15. Prüfsubstanz Siehe Referenzdokument für Everolimus hinsichtlich Information zu physikalischen und chemischen Eigenschaften von Everolimus und eine Liste der sonstigen Bestandteile und Trägerstoffe. Nach Abschluss der vorgeschalteten Dosiseskalation (3 Wochen), ist der interventionelle Teil der Studie abgeschlossen und die Patienten führen Ihre Behandlung von Everolimus entsprechend der Fachinformation und Grundversorgung fort. Nicht-Prüfsubstanzen Die endokrine Behandlung mit Exemesthane erfolgt gemäß Herstellerbeschreibung in der vorgesehenen Weise und nach den deutschen Richtlinien der AGO Breast Kommission. Primärer Endpunkt Die Inzidenz der ersten Episode einer Mukositis WHO´s OTS Grad 2-4 vor jedem Zeitpunkt der 12 Wochen nach Behandlungsbeginn mit Everolimus. Sekundäre Inzidenz der ersten Episoden von Mukositis/Stomatitis WHO’s OTS Grad Endpunkte 2-4 während der gesamten 24 Behandlungswochen. Inzidenz der ersten Episoden von Mukositis/Stomatitis WHO’s OTS Grad 1 und jedes Grades während der gesamten 24 Behandlungswochen. Die gemittelte Behandlungsdosis an Woche 12 und Woche 24 Klinische Benefitrate ist definiert als die Anzahl der Patienten mit keinerlei Anzeichen eines Progresses 24 Wochen nach Behandlungsbeginn mit Everolimus. Die Sicherheit ist nach Toxizitätsgraden durch die Kriterien nach NCICTCAE Version 4.03 , die orale Mukositis nach den WHO Kriterien(OTS) definiert. Die kumulative Behandlungsdosis wird berechnet durch Addition der täglichen Everolimusdosen bis zum Behandlungsende oder Absetzen der Medikation . Die relative Dosisintensität für Everolimus ist das Verhältnis in Prozent der tatsächlichen Gesamt Dosis (ATDI) zur geplanten Gesamtdosis (PTDI). Vergleich der Rate an Mucositis/ Stomatitis zwischen den Behandlungsarmen bei 12 und 24 Wochen nach Behandlungsbeginn mit Everolimus GBG 86 - Desiree Protocol Version 2 – 01.04.2015-Confidential 20 Die Lebensqualität wird mit Hilfe folgender Patientenfragebögen ermittelt: FACT-B und QSDQ. Biomaterial x Vollblut für SNP Analyse bei Studienbeginn x Serum und Plasma bei Studienbeginn und bei Ende der Studientherapie x Optional Paraffin-eingebettetes Tumorgewebe der Primärerkrankung und/oder der Metastase Exploratorische Potentielle Biomarkers, die auf Sicherheit und Verträglichkeit hinweisen Endpunkte werden nach Studienabschluss bestimmt, wie SNPs, CTC, pi3 Kinase Marker und andere Biomarker für Brustkrebs. Statistische 156 auswertbare Patientinnen (78 in jedem Arm) sind erforderlich, um Methoden / Anzahl eine klinisch relevante Differenz von 20% in der Mukositis Rate (Grad 2-4 an Patienten gemäß WHO Skala OTS) zwischen den Behandlungsarmen zu entdecken, wenn ein kontinuitätskorrigierter ʖ2-test mit einem Signifikanzniveau alpha von 0.2 und einem beta von 0.9 verwendet wird. Im Kontrollarm wird eine Rate von 40% und im Behandlungsarm von 20% angenommen. Das Hauptzielkriterium wird gemäß einem modifizierten intention-totreat Prinzip ausgewertet, d.h. alle randomisierten Patientinnen mit mindestens einer Dosis Studienmedikation werden eingeschlossen. Anzahl der Zentren Bis zu 60 Zentren die sich aus den BRAWO Zentren rekrutieren und Erfahrung mit Everolimus haben. Ungefähre Fristen Rekrutierungszeitraum: 24 Monate (Q-II 2015 – Q-II 2017) Studienstart (FPFV): 01.04.2015 Rekrutierungsende (LPFV): 01.04.2017 Finale Analyse: 30.04.2018 Studienende (LPLV): 30.10.2017 GBG 86 - Desiree Protocol Version 2 – 01.04.2015-Confidential 21