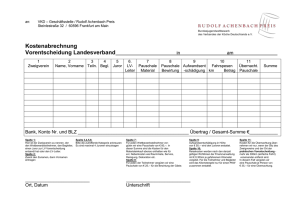
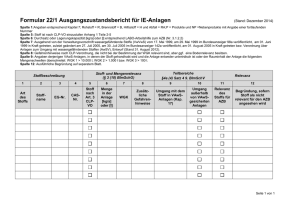
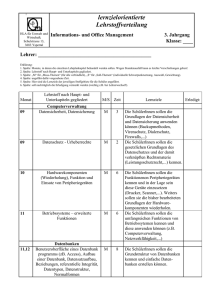
Ergänzungen 2002 – Mitte 2005 zu
Werbung

Ernst Mutschler, Gerd Geisslinger, Heyo K. Kroemer, Monika Schäfer-Korting Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 Sehr geehrte Leserinnen und Leser der „Arzneimittelwirkungen“, in Übereinstimmung mit unserem Verleger, der Wissenschaftlichen Verlagsgesellschaft Stuttgart, hatten wir uns 2003 entschlossen, Ihnen über das Internet in regelmäßigen Abständen wichtige Ergänzungen zu unserem Lehrbuch zukommen zu lassen. Nachstehend erhalten Sie die vereinigten Beiträge (2002 – Mitte 2005), von denen wir hoffen, dass sie für Sie nützlich sind und dazu beitragen, die Aktualität der „Arzneimittelwirkungen“ auch in der Zeit zwischen zwei Auflagen zu gewährleisten. Bitte tragen Sie auf den angegebenen Seiten die entsprechenden Ergänzungen ein. Nicht zuletzt möchten wir Ihnen weiterhin viel Erfolg und Freude beim Arbeiten mit unserem Lehrbuch und Nachschlagewerk wünschen. 2 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 87 am Ende der rechten Spalte einfügen: Zur Häufigkeitsangabe von unerwünschten Arzneimittelwirkungen werden die europäischen Richtlinien (Häufigkeitsangaben von Nebenwirkungen zur Zusammenfassung von Produktmerkmalen, Summary of product characteristics, SPC) verwendet, die auch vom Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) übernommen wurden (Tab. A 4–1a). Tab. A 4–1a. Angaben zur Häufigkeit unerwünschter Arzneimittelwirkungen nach der SPC-Guideline Englisch Deutsch %-Angabe very common sehr häufig >10% common häufig >1% bis <10% uncommon gelegentlich >0,1% bis <1% rare selten >0,01% bis <0,1% very rare sehr selten <0,01%* *Darin sind Einzelfälle enthalten, die nicht mehr gesondert aufgeführt werden auf S. 170 vor 1.2.2.4 Reserpin einfügen: 1.2.2.3.4 Aripiprazol ® Aripiprazol (Abilify ) unterscheidet sich von den bisher beschriebenen atypischen Neuroleptika dadurch, dass es an D2-Rezeptoren als Partialagonist/-antagonist wirkt. Außerdem ist es ein potenter 5-HT2AAntagonist. Ferner hat es keine anticholinergen und nur geringe H1- und ·1-antagonistische Wirkungen. Dieses Wirkprofil erklärt, warum Aripiprazol bei Schizophrenen im mesolimbischen System infolge des hohen dopaminergen Tonus als D2-Antagonist, im mesokortikalen System dagegen wegen der verminderten dopaminergen Aktivität als partieller Agonist wirkt, und warum es nur selten Gewichtszunahme und auch nur gelegentlich orthostatische Dysregulationen hervorruft. auf S. 176 Aripiprazol wird rasch und gut resorbiert (Bioverfügbarkeit ca. 85%), die Proteinbindung beträgt über 99%, die Plasmahalbwertszeit 75 Stunden. Hauptmetabolit ist das noch wirksame Dehydro-Aripiprazol. Die Dosierung beträgt 15–30 mg/Tag. rechte Spalte nach (s. Tab. B 1–15) einfügen: Citalopram und dessen S-Enantiomer Escitalopram, auf S. 186 rechte Spalte, und S. 186, linke Spalte: ganzes Unterkapitel 1.2.5.4 Kavapyrone streichen (auf Anordnung des BfArM wegen Leberschäden aus dem Handel genommen) (Abilify®) 3 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 189 rechte Spalte, am unteren Ende einfügen: 1.2.6.3a Atomoxetin Ein weiterer Wirkstoff zur Behandlung des Aufmerksamkeitsdefizit-/Hyperaktivitäts-Syndroms ist Atomoxetin (Strattera®). Sein Wirkungsmechanismus besteht in der selektiven Hemmung des (präsynaptischen) Noradrenalin-Transporters. Atomoxetin wird rasch und vollständig resorbiert, die Halbwertszeit beträgt bei extensiven Metabolisierern 3,6 und bei langsamen Metabolisieren 21 Stunden. Die Ausscheidung erfolgt vor allem renal als 4Hydroxy-Atomoxetin-Glucuronid. Die Erhaltungsdosis für Kinder und Jugendliche mit einem Körpergewicht > 70 kg liegt nach einer Initialdosis von 40 mg/Tag bei 80 mg täglich. Als Nebenwirkungen treten – oft vorübergehend – insbesondere gastrointestinale Störungen und verminderter Appetit auf. Weitere unerwünschte Wirkungen auf S. 211 sind Schlafstörungen, Mydriasis, Sinustachykardie, Ejakulationsstörungen u.a. Atmoxetin darf nicht zusammen mit MAO-Hemmern angewandt werden. Vorsicht ist angezeigt bei der gleichzeitigen Verordnung von ‚2-AdrenozeptorAgonisten und CYP2D6-lnhibitoren. (Strattera®) rechte Spalte, vor 1.5.8.1 „Schmerzgedächtnis“ einfügen: 1.5.1.7.1 Dronabinol Die Gabe von Dronabinol (vgl. auch S. 193), einem Stereoisomer von 9-Tetrahydrocannabinol (THC), wird als vielversprechende additive Maßnahme in der Schmerztherapie diskutiert. In Deutschland ist bislang zwar kein Dronabinol-Fertigarzneimittel im Handel, doch steht es als verschreibungsfähige Rezeptursubstanz (Betäubungsmittelrezept!) zur Verfügung. Da vom Gesetzgeber keine Indikationsgebiete für Dronabinol angegeben sind, darf der Arzt die Substanz bis zu einer Verordnungshöchstmenge von 500 mg in 30 Tagen verschreiben und zwar für jeden Krankheitszustand, bei dem er sich einen Therapieerfolg verspricht. Positive Effekte des adjuvanten Einsatzes von Dronabinol werden vor allem bei spastisch bedingten Schmerzen (z. B. bei Multipler Sklerose), neuropathischen und amputationsbedingten sowie Arthrose- und Osteoporose-Schmerzen berichtet. Als Wirkungsmechanismus nimmt man eine Wechselwirkung mit zahlreichen Neurotransmittern und Neuromodulatoren über Cannabinoid-Rezeptoren (CB1, CB2, s. Abb. B 1–26 und Tab. B 1–24) an. Neben den Cannabinoid-Rezeptoren wurden auch endogene Cannabinoide entdeckt (s. Abb. B 1-26), die vermutlich eine wichtige Rolle bei der Regulation immunologischer, gastrointestinaler, reproduktiver und kardiovaskulärer Körperfunktionen haben und darüber hinaus an Gedächtnisbildung, neuronaler Plastizität, nozizeptiver Übertragung und Schmerzhemmung beteiligt sind. Nach peroraler Applikation ist die Bioverfügbarkeit von Dronabinol wegen eines ausgeprägten First-PassEffektes mit 5-10 % sehr gering. Andere Applikations- formen werden daher derzeit untersucht. Dronabinol passiert die Blut-Hirn- und die Plazentaschranke und tritt in die Muttermilch über. Außerdem verteilt es sich in fettreiches Gewebe, aus dem es nur relativ langsam wieder in die Blutbahn und andere Gewebe umverteilt wird. In der Leber wird Dronabinol zu 95 % metabolisiert. Der Hauptmetabolit 11-Hydroxy-9-THC ist ebenfalls pharmakologisch aktiv. Die terminale Plasmahalbwertszeit liegt bei 20-30 Stunden. Die Elimination der Dronabinol-Metabolite erfolgt zu einem Drittel renal, zu etwa zwei Dritteln fäkal. Da die interindividuelle Ansprechbarkeit auf Dronabinol sehr variabel ist, wird eine einschleichende Dosierung mit zweimal 2,5 bzw. zweimal 5 mg oral /Tag mit einer Steigerung bis zu fünfmal 10 mg täglich als Tageshöchstdosis empfohlen. Als Nebenwirkungen werden Tachykardie und Mundtrockenheit als Folge einer Wirkung auf die Acetylcholin-Freisetzung und -Biotransformation beobachtet. Außerdem werden ZNS-Effekte, u.a. eine Beeinträchtigung der Reaktionsfähigkeit und des Gedächtnisses, berichtet. Obwohl das Suchtpotential von Dronabinol relativ gering sein soll, können schon nach therapeutischen Dosen Entzugssymptome (Angst, Unruhe, Schlaflosigkeit, Speichelfluss, Durchfall, Erhöhung des Augeninnendrucks) auftreten. Außerdem ist eine physische Abhängigkeit möglich. Bislang wurden bei der Kombination von Dronabinol mit einer Vielzahl von Medikamenten keine klinisch relevanten Wechselwirkungen beschrieben. Allerdings sind diesbezüglich weitere Untersuchungen notwendig. 4 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 221 rechte Spalte, vor dem Petitabschnitt: ganzen Abschnitt Levacetylmethadol entfernen. [Das Präparat wurde wegen nicht vorhersehbarer schwerer Herzrhythmusstörungen bis hin zu Torsades de pointes (paroxysmalem Kammerflattern vom Spit- auf Seite 222 zenumkehrtyp) auf Empfehlung der Europäischen Arzneimittelbehörde (EMEA) vom Markt genommen.] linke Spalte, vor 1.5.2.6 Opioid-Agonisten/Antagonisten einfügen: Zur Behandlung von Durchbruchschmerzen bei Patienten, deren Tumorschmerzen bereits mit Opioiden behandelt werden, wurde Fentanyl als oral-transmukosales therapeutisches System (o-TTS) zugelassen (ACTIQ®). Vorteil dieser Arzneiform ist der schnelle Wirkungseintritt, der ähnlich schnell erfolgt wie nach i.v. verabreichtem Morphin. Das Fentanyl-o-TTS ist in 6 Wirkstärken (200 µg, 400 µg, 600 µg, 800 µg, 1200 µg, 1600 µg) erhältlich. auf S. 232 linke Spalte, vor Vergiftungen einfügen: Wechselwirkungen mit anderen nichtsteroidalen Antiphlogistika. Bei gleichzeitiger Einnahme von ASS (zur Herzinfarktprophylaxe) und Ibuprofen kann dieses vor ASS an das katalytische Zentrum der Cyclooxygenase-1 binden, so dass je nach Einnahmemodus eine Aufhebung der kardioprotektiven Wirkung von ASS möglich ist. Patienten, die ASS in niedriger Dosis zur Herzinfarktprophylaxe einnehmen, sollten daher bei entsprechender Indikation nicht mehr als eine Einzeldosis Ibuprofen pro Tag erhalten und diese mindestens 2 Stunden nach der Applikation von ASS anwenden. Die gleichzeitige Zufuhr von Paracetamol, Diclofenac oder Rofecoxib beeinflusste die thrombozytenaggregationshemmende Wirkung der Acetylsalicylsäure dagegen nicht. Abb. B 1–36a. Mechanismus der Cyclooxygenase -1-Hemmung durch Acetylsalicylsäure (ASS). Irreversible Acetylierung von Serin 529 und dadurch sterische Hinderung des Zugangs von Arachidonsäure zum katalytischen Zentrum. Cyclooxygenase 1 liegt physiologisch als Dimer vor. Aus Med. Mo. Ph. Jahrgang 25, Heft 4, April 2002, Seite 111. auf S. 233 Abb. B 1–36b. Vorherige reversible Belegung des katalytischen Zentrums durch Ibuprofen verhindert den Zugang von Acetylsalicylsäure zur SerinBindestelle an Pos. 529 rechte Spalte, vor 1.5.2.1.4 Oxicame einfügen: Dexibuprofen [S(+)-Ibuprofen, Deltaran®] ist das rechtsdrehende, hinsichtlich der Prostaglandinsynthese-Hemmung aktive Enantiomer von IbuprofenRazemat. Dexibuprofen ist wie das Razemat zur symptomatischen Therapie von Schmerzen und Entzündungen indiziert, allerdings unterliegt es auch bei niedriger Dosis noch der Verschreibungspflicht. Die Tagesdosis beträgt bei leichten bis mäßigen Schmerzen dreimal 200 mg, bei Dysmenorrhö bis zu dreimal 300 mg und in der antirheumatischen Therapie bis zu dreimal 400 mg. Neben- und Wechselwirkungen sowie Kontraindikationen entsprechen denen von Ibuprofen-Razemat. 5 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf Seite 233 unter 1.5.4.2.1.3 Arylpropionsäurederivate (am Ende des Abschnitts) einfügen: Mit Oxaprozin (Danoprox®, Dayrun®) wurde ein weiteres Propionsäurederivat als nichtsteroidales Antiphlogistikum, das in den USA bereits seit ca. 10 Jahren zur Verfügung steht, nun auch in Deutschland zugelassen. Wie die anderen Substanzen dieser Gruppe ist Oxaprozin ein nicht-selektiver CyclooxygenasenHemmer. Erwähnenswert ist seine lange Eliminationshalbwertszeit von 50–60 h. Die Tagesdosis beträgt 600 mg bis maximal 1800 mg. Indikationen, Neben- und Wechselwirkungen sowie Kontraindikationen entsprechen denen anderer nichtselektiver COX-Hemmer. auf S. 238 (Danoprox®, Dayrun®) linke und rechte Spalte, gesamten Text 1.5.4.2.1 und Tab. B 1-32 ersetzen: 1.5.4.2.1.7 COX-2-selektive nichtsteroidale Antiphlogistika („Coxibe“) Für die antiphlogistischen, analgetischen und antipyretischen Wirkungen klassischer NSAIDs wird die Hemmung der COX-2, für viele Nebenwirkungen dagegen die der COX-1 verantwortlich gemacht. Folglich war es das Ziel, mit selektiven Hemmern der COX-2 nebenwirkungsärmere NSAIDs zu entwickeln. Solche Substanzen, die in therapeutisch eingesetzten Dosen nur die COX-2 hemmen, sind (z.T. noch nicht oder nicht mehr zugelassen, vgl. Tab. B 1.32): Celexoxib (Celebrex®), Etoricoxib (Arcoxia®), Lumaricoxib, Parecoxib (Dynastat®), wirkungen belastet als herkömmliche NSAIDs, aber nicht nebenwirkungsfrei, da auch die COX-2 physiologische Funktionen ausübt. Nebenwirkungen, die am häufigsten genannt werden, sind Infektionen der oberen Atemwege, Durchfall, Dyspepsie, Oberbauchbeschwerden und Kopfschmerzen. Periphere Ödeme und eine Erhöhung des Blutdrucks treten ebenso häufig auf, gastrointestinale Komplikationen dagegen seltener als bei herkömmlichen NSAIDs. Rofecoxib und Valdecoxib wurden aufgrund einer erhöhten Rate für kardiovaskuläre Ereignisse (Schlaganfall, Herzinfarkt) wieder vom Markt genommen. Die zugrunde liegenden Mechanismen sind noch unklar. In jüngster Zeit wurden Studien publiziert, dass auch einige nicht selektive NSAIDs (z.B. Naproxen) vermehrt kardiovaskuläre Ereignisse verursachen, so dass unter Umständen alle NSAIDs, selektive wie nicht selektive COX-Hemmer, mit diesem Risiko belastet sind. Rofecoxib und Valdecoxib. Parecoxib ist ein wasserlösliches, parenteral applizierbares Prodrug von Valdecoxib. Die Pharmakokinetik der Substanzen ist in Tab. B 1–32 zusammengefasst. Die Indikationen der „Coxibe“ sind prinzipiell mit denen der nicht-selektiven NSAIDs identisch (s. S. 247), auch wenn bisher nicht alle Coxibe für das ganze Anwendungsspektrum klassischer NSAIDs zugelassen sind. Infolge der fehlenden COX-1-Inhibition sind COX2-selektive Hemmer zwar mit weniger Neben- (Arcoxia®) 6 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 238 linke und rechte Spalte, gesamten Text 1.5.4.2.1 und Tab. B 1-32 ersetzen: (Dynastat®) Tabelle B 1–32. Pharmakokinetische Kenndaten selektiver COX-2-Hemmer Verteilungsvolumen (l) tmax (h) t1/2 (h) Bemerkungen Celecoxib ~ 400 2-4 6-12 Sulfonamidallergie Valdecoxib ~ 50-90 2-4 6-11 nicht mehr im Handel Etoricoxib ~ 120 ~ 1-2 20-26 lange Halbwertszeit Rofecoxib ~ 90 2-4 12-18 nicht mehr im Handel Lumiracoxib ~ 13 1-3 2-6 noch nicht im Handel tmax: Zeit bis zum Erreichen der maximalen Plasmakonzentration; t1/2, Eliminationshalbwertszeit; auf S. 250 rechte Spalte, ganz unten einfügen: Nach den neuesten Daten des deutschen Spontanerfassungssystems kann es unter der Therapie mit Leflunomid häufig, das heißt bei 1–10% der Patienten, zu einer Blutdruckerhöhung kommen. Deshalb muss der Blutdruck vor Beginn und während einer Leflunomid-Therapie in regelmäßigen Abständen kontrolliert werden. Ebenso wurde über zwar seltene, aber schwere Leberschäden berichtet, so dass Leflunomid nicht bei leberinsuffizienten Patienten eingesetzt werden soll. Der aktive Metabolit von Leflunomid ist ein CYP2C9Hemmstoff. Folglich werden ebenfalls über CYP2C9 metabolisierte Substanzen (z.B. Phenprocoumon) bei gleichzeitiger Gabe mit Leflunomid langsamer abgebaut. 7 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 253 Anstelle der ersten beiden Abschnitte im Petit-Teil einfügen: Adalimumab (Humira) ist ein völlig humanisierter monoklonaler Antikörper gegen TNF-· mit im Vergleich zu Infliximab niedrigerer Inzidenz zur Antikörperbildung. Er wurde in Kombination mit Methotrexat oder als Monotherapeutikum zur Behandlung der rheumatoiden Arthritis bei Patienten zugelassen, die auf andere DMARDs, einschließlich Methotrexat, nicht angesprochen haben. Die Dosierung beträgt 40mg alle 2 Wochen subkutan. Anakinra. Anakinra (Kineret®), ein rekombinant hergestellter humaner Interleukin-1-Rezeptorantagonist, ist nach Etanercept, Infliximab und Adalimumab der vierte im Handel befindliche „biologische Zytokinhemmstoff“. Anakinra hemmt kompetitiv die Bindung von Interleukin-1 (IL-1) an den Interleukin1-Rezeptor. IL-1 (s. S. 927) ist ein wichtiges proinflammatorisches Zytokin, dessen Konzentration bei Patienten mit rheumatoider Arthritis in aktiven Krankheitsphasen sowohl im Plasma als auch in der Synovialflüssigkeit erhöht ist. Darüber hinaus ist bei diesen Patienten die Konzentration des endogenen Interleukin-1-Rezeptorantagonisten erniedrigt. auf S. 263 Nach subkutaner Bolusinjektion beträgt die Bioverfügbarkeit von Anakinra 95%. Der Absorptionsprozess verläuft allerdings langsam. Maximale Plasmakonzentrationen werden nach 3–7 h erreicht. Die Eliminationshalbwertszeit liegt bei 4–6 h. Anakinra ist zur Behandlung der rheumatoiden Arthritis in Kombination mit Methotrexat indiziert. Die Dosierung beträgt einmal täglich 100 mg subkutan. Als häufigste Nebenwirkung (ca. 70% der Fälle) wird eine leichte bis mäßige Reaktion (Erythem, Entzündung, Schmerzen) an der Einstichstelle beschrieben. Sehr häufig treten Kopfschmerzen auf. Ferner wurden Blutbildveränderungen (Neutropenien) und Infektionen, möglicherweise als Folge der Immunsuppression beobachtet. Wechselwirkungen mit NAIDs, Gluccocorticoiden oder anderen Basis-Therapeutika wurden bisher nicht beschrieben. Vor der gleichzeitigen Anwendung von Etanercept und Anakinra muss dagegen aufgrund eines erhöhten Risikos von Neutropenien und schwerwiegenden Infektionen gewarnt werden, zumal die Kombination keinen zusätzlichen klinischen Nutzen im Vergleich zur Etanercept-Monotherapie gezeigt hat. Ergänzung der Tab. B 1–36: Tab. B 1–36. 5–HT1B/1D-Rezeptoragonisten (Triptane) im Vergleich Sumatriptan (Imigran®) Zolmitriptan Naratriptan Rizatriptan Eletriptan (AscoTop®) (Naramig®) (Maxalt®) (Relpax®) Almotriptan Frovatriptan (Almogran®) (Allegro®) Einzeldosis 50–100 mg oral, 2,5 mg oral 25 mg rektal, 20 mg nasal, 6 mg s.c. 2,5 mg oral 5–10 mg oral 40–80 mg oral 12,5 mg oral 2,5 mg oral Lipophilie (+) + ++ + +++ +++ + Orale Bioverfügbarkeit (%) 14 40 60–70 40–45 ~50 ~70 20–30 tmax (h) 1,5–2,5 2–3 2–3 1–1,5 1–1,5 2,2–2,5 2–4 t1/2 (h) 2 ~3 6 2–3 4–5 3–4 ~25 Ansprechquote (%) 50–70 oral 80 s.c. 60–80 60–68 62–71 64 (40 mg) 71 (80 mg) 57–70 37–46 Wiederauftreten 30–40 oral von Schmerzen (%) 50 s.c. 30 27 30–35 21 (80 mg) ~25 20 (Almogran®) (Allegro®) 8 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 273 rechte Spalte oben, ab 4. Zeile ändern: ...als die S-Enantiomere, wurde von Bupivacain auch das reine S-Enantiomer in der Therapie eingeführt (Levobupivacin; Chirocain). Von Ropivacain ist nur das S-Enantiomer im Handel. auf S. 306 Tab. B 1-45: Tiagabin-Formel ändern: auf S. 308 linke Spalte, unteres Drittel vor Als Nebenwirkungen einfügen: Vor einiger Zeit erhielt Gabapentin auch die Zulassung für alle Indikationen neuropathischer Schmerzen im Erwachsenenalter. Die Dosierung erfolgt individuell nach Verträglichkeit und Wirkung. Die erste Aufdosierung auf 900 mg/Tag sollte ca. 3 Tage dauern (1. Tag 300 mg, 2. Tag 600 mg, 3. Tag 900 mg). Die anzustrebende auf S. 308 Tageszieldosis beträgt ab dem 6. Tag 1800 mg. Bei Bedarf ist eine Steigerung auf 3600 mg/Tag möglich. Bei niereninsuffizienten Patienten muss die Dosis reduziert werden. linke Spalte, ganz unten einfügen: Ein Analogpräparat mit denselben Indikationen ist Pregabalin (Lyrica®), das an eine Untereinheit von spannungsabhängigen Calciumkanälen bindet und dort [3H-]-Gabapentin zu verdrängen vermag. (Lyrica®) Die Dosierung beträgt initial 150/Tag und kann bei Bedarf bis auf 600 mg gesteigert werden. Bei eingeschränkter Nierenfunktion ist eine Dosisanpassung erforderlich. 9 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 310 linke Spalte, vor 1.9.2.3.6. Hormone einfügen: 1.9.2.3.6 Levetiracetam Das Piracetam-Derivat (s. S. 195) Levetiracetam (Keppra®) ist das S-Enantiomer von Etiracetam. Es wird nicht wie Piracetam als Nootropikum, sondern als antiepileptisches Zusatzmedikament bei therapierefraktären partiellen Anfällen Erwachsener eingesetzt. Der Wirkungsmechanismus ist (noch) unbekannt. Nach peroraler Einnahme wird Levetiracetam rasch und vollständig resorbiert, die Bioverfügbarkeit liegt bei nahezu 100%. Maximale Plasmakonzentrationen werden nach etwas mehr als einer Stunde erreicht. Die Substanz wird nur wenig metabolisiert, der Hauptmetabolit ist die freie Säure (Hydrolyse der Amidgruppe). Die Ausscheidung erfolgt fast ausschließlich renal. Die Dosierung beträgt initial zweimal täglich auf S. 312 500 mg und kann bis auf zweimal täglich 1 500 mg gesteigert werden. Als Nebenwirkungen werden häufig Asthenie und Somnolenz, ferner – deutlich seltener – andere zentralnervöse Störungen (u. a. Kopfschmerzen, Schlafstörungen, depressive Verstimmung, Ängstlichkeit), ferner gastrointestinale Beschwerden sowie Hautreaktionen beschrieben. Relevante Wechselwirkungen wurden nicht beobachtet. (Keppra®) linke Spalte oben, letzter Satz im Petit-Abschnitt: „Der Tremor verstärkt sich bei willkürlicher Muskelanspannung (Intentionstremor)“ streichen. auf S. 318 rechts oben, Pramipexol-Formel: in der Seitenkette NH anstelle N N H (Sifrol®) auf S. 318 linke Spalte, anstelle des bisherigen Petit-Abschnitts einfügen: Apomorphin (vgl. auch S. 596) und S. 961), das in Deutschland nicht als Antiparkinsonmittel im Handel ist, eignet sich besonders zur Behandlung schmerzhafter Dystonien. 10 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 322 rechte Spalte, ganz unten einfügen: Neurokinin-1-(NK1-)Rezeptorantagonist. Aprepitant (EMEND®) hemmt die Stimulation des NK1-Rezeptors durch Substanz P und verringert dadurch die Symptome des Früherbrechens und vor allem die des verzögerten Erbrechens bei einer hoch emetogenen Chemotherapie. Es wird in Kombination mit 5-HT3-Antagonisten und Dexamethason eingesetzt. Nach oraler Applikation wird es gut resorbiert. Seine fast vollständige Metabolisierung erfolgt hauptsächlich über CYP3A4. Aprepitant induziert die Expression von CYP3A4 und hemmt gleichzeitig dieses Enzym moderat. Dies führt zu potentiellen Wechselwirkungen mit Pharmaka, die CYP3A4 blockieren, und mit solchen, die über CYP3A4 abgebaut werden. Die Dosierung beträgt 125 mg p.o. 1 Stunde vor Beginn der Chemotherapie und je 80 mg an den Tagen zwei und drei morgens. auf S. 332 Häufige Nebenwirkungen sind Schluckauf, Müdigkeit, Obstipation und Kopfschmerzen. (EMEND®) rechte Spalte, 6. Zeile von unten: Wirkstoffe aus dieser Gruppe sind Etilefrin (Cardanat®, Effortil®, Etilefrin-ratiopharm®, Thomasin® u.a.) und Oxilofrin (Carnigen®). (Effortil® u.a.) (Carnigen®) auf S. 359 Tab. B 1–69: Tiotropium-Formel ändern: 11 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 361 rechte Spalte, vor Pirenzepin einfügen: Bei derselben Indikation werden Darifenacin (Emselex®) und Solifenacin (Vesicur®), selektive M3-Rezeptorantagonisten, in einer Dosierung von 7,5 (–15) bzw. 5 (–10) mg täglich eingesetzt. (Vesicur®) (Emselex®) auf S. 362 Tab. B 1–70 auf S. 374 linke Spalte, vor Somatostatin und Octreotid einfügen: Ein Analogpräparat ist Ganirelix (Orgalutran®). auf S. 376 linke Spalte, ab drittletzter Zeile ändern: tropin und Lutropin) gewonnen. Alle drei Gonadotropine werden außerdem gentechnologisch hergestellt. auf S. 376 rechte Spalte, anstelle der 10. und 11. Zeile von oben: Bei der zuletzt genannten Indikation wird neben Menotropin auch rekombinant gewonnenes Lutropin eingesetzt. Abschnitt Handelspräparate: Handelspräparate: Choragon®, Ovitrelle® und Predalon® enthalten Choriongonadotropin, Menogon® HP enthält Urogonadotropin; in Puregon® und GONAL® ist Follitropin und in Luveris® Lutropin enthalten. 12 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 379 rechte Spalte, vor 2.2.2 Hypophysenhinterlappenhormone einfügen: Außerdem kann bei Akromegalen, bei denen eine Operation oder eine Strahlentherapie sowie eine Behandlung mit Somatostatin-Analogen keinen Erfolg gebracht hat, Pegvisomant (Somavert®) eingesetzt werden. Pegvisoman ist ein gentechnologisch gewonnener, pegylierter selektiver Wachtumshor- monrezeptor-Antagonist, der subkutan appliziert wird. Die Dosierung der gut verträglichen Substanz beträgt initial einmalig 80 mg, danach täglich 10 bis maximal 30 mg, der IGF-I-Serumkonzentration entsprechend. auf S. 390 2.3.4.3.1.3 Iodid-Ionen und Iod anstelle Iodid-Ionen und Kaliumiodid auf S. 393 linke Spalte, vor 2.4.3 Störungen der Nebenschilddrüsenfunktion einfügen: Teriparatid (FORSTEO®), ein gentechnologisch hergestelltes, aus den Aminosäuren 1–34 bestehendes Parathormon-Fragment, dient zur Osteoporose-Behandlung (s. S. 395 ff.). Außer zur Behandlung von Frauen mit postmenopausaler manifester Osteoporose wird es bei Männern zur Therapie einer primären oder hypogonadalen Osteoporose mit hohem Frakturrisiko auf S. 394 eingesetzt. Die Plasmahalbwertszeit liegt bei ca. 1 Stunde. Als Peptid wird Teriparatid zu zahlreichen Bruchstücken biotransformiert. Die Dosierung beträgt 20 µg/Tag subkutan. Als Nebenwirkungen wurden vor allem Übelkeit, Glieder- und Kopfschmerzen sowie Schwindel beobachtet. linke Spalte,2.4.4 Calcitonin (Thyreocalcitonin) einfügen: Zur Behandlung eines sekundären Hyperparathyreoidismus bei dialysepflichtigen Patienten mit terminaler Niereninsuffizienz wird außerdem als Teil des therapeutischen Regimes Cinacalcet (Mimpara®) eingesetzt. Die calcimimetische Substanz erniedrigt den Parathormonspiegel durch Steigerung der Empfindlichkeit des Calcium-sensiblen Rezeptors auf extrazelluläres Calcium. Die Dosierung beträgt initial 30 mg täglich und kann bis zu einer Maximaldosis von 180 mg/Tag gesteigert werden. auf S. 394 rechte Spalte: Zeile 12, streichen: und die Umwandlung von Osteoklasten in Osteoblasten fördert. auf S. 394 linke Spalte, ab 9. Zeile ergänzen: Bisphosphonate, Calcitonin, Teriparatid (s. S. 393) und Strontiumranelat. (Mimpara®) 13 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 397. in Tabelle B 2–4 unten einfügen: Tab. B 2–4. Bisphosphonate R1 R2 N OH CH2 Internationaler Freiname Handelspräparat (Eingetragenes Warenzeichen) Zoledronsäure Zometa N auf S. 397 linke Spalte einfügen: nach Bisphosphonate mit einem basischen Heterocyclus: (Risedronat, Zoledronsäure) auf S. 398 linke Spalte: anstelle der beiden letzten Zeilen ganz unten: auf S. 398 Osteolysetherapie Clodronat, Ibandronat, Pamidronat und Zoledronsäure zugelassen. rechte Spalte: vor dem Petit-Abschnitt einfügen: auf S. 398 Zoledronat einmal 4 mg als Infusion. rechte Spalte, ganz unten einfügen: 2.4.5.2.3 Strontiumranelat Der neueste Wirkstoff zur Osteoporosetherapie ist Strontiumranelat (Protelos®).Der Arzneistoff besteht aus zwei Strontium-Ionen und einem Molekül Ranelinsäure, einer Tetracarbonsäure. Er steigert den Knochenaufbau und hemmt gleichzeitig die Knochenresorption. auf S. 411 in Tab. B 2–8 unter II. Insulinglulisin Apidra unter III.1 Insulindetemir Levemir Die Bioverfügbarkeit von Strontium beträgt ca 20%, die Halbwertszeit ca. 60 Stunden. Die Ausscheidung erfolgt renal und mit den Fäzes. Die tägliche Dosis liegt bei 2 g peroral. Häufige Nebenwirkungen sind Übelkeit und Diarrhö. Nahrung, insbesondere Milch und Milchprodukte, verringern die Bioverfügbarkeit. 14 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 413: Miglitol-Formel ändern: auf S. 415 linke Spalte: schaft und Stillzeit ist Metformin kontraindiziert. Außerdem darf es zwei Tage vor und zwei Tage nach intravenöser Anwendung eines Röntgenkontrastmittels wegen der Gefahr eines akuten Nierenversagens nicht gegeben werden. anstelle von Zeile 4: auf S. 417 linke Spalte, anstelle von 2.6.7.3.2 Repaglinid einfügen: 2.6.7.2.3.2 Glinide Am Ende des Kapitels: Ein Analogpräparat ist Nateglinid (Starlix®). Das stark eiweißgebundene D-Phenylalanin-Derivat wird vorwiegend durch CYP2C9 metabolisiert. Die Dosierung, jeweils zu den drei Hauptmahlzeiten, beträgt initial 60 mg und kann auf 120 mg gesteigert werden. Repaglinid-Formel ändern: (NovoNovum®) auf S. 433 (starlix®) rechte Spalte, Zeile 7: drittels (anstelle monats) auf S. 437 in der rechten Spalte, 5. Zeile von unten streichen: Progesteron wird vor allem in der Leber hydroxyliert. 15 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 438 Tab. B 2-13: In der Norethisteron-Formel die anguläre Methylgruppe an C-19 durch H ersetzen. auf S. 439 Tab. B 2–13 (Fortsetzung): Norgestimat-Formel-Formel ändern: auf S. 440 linke Spalte, vor Indikationen einfügen: Das 17·-Spironolacton-Derivat Drospirenon ist ein neues Gestagen mit schwacher antiandrogener sowie antimineralocorticoider Wirkung. Es wird in einer Dosierung von 3 mg in Kombination mit Ethinylestradiol (30 µg) in Einphasenpräparaten zur Kontrazeption eingesetzt (Petibelle®, Yasmin®). auf S. 441 rechte Spalte, nach 8. Zeile von oben: Etonogestrel ist der Wirkstoff in einem Implantat (Implanon®), das – am Oberam angewandt – einen Konzeptionsschutz über 3 Jahre vermittelt. rechte Spalte, anstelle der 14. Zeile von unten: wiegend antiovulatorisch sowie nidationshemmend. 16 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 445 Tab. B 2–15 durch folgende Tabelle ersetzen: Tab. B 2–15. Präparate zur Hormonersatztherapie Estrogen Gestagen Handelspräparat (Eingetragenes Warenzeichen) Estrogen/Gestagen-Kombinationen zur oralen Therapie Einphasenpräparate Estradiol Dydrogesteron Femoston conti Estradiol Norethisteronacetat Activelle, Kliogest N Estradiolvalerat Dienogest Climodien Estradiol Dydrogesteron Femoston Estradiol Medroxyprogesteronacetat Osmil Estradiol Norethisteronacetat Gynamon, Novofem Estradiolvalerat Cyproteronacetat Climen Estradiolvalerat Levonorgestrel Klimonorm, Östronara Estradiolvalerat Medroxyprogesteronacetat Estrafemol, Gianda, Procyclo, Sisare Estradiolvalerat Norethisteron Mericomb Estradiolvalerat Norgestrel Cyclo-Progynova Estradiol Norethisteronacetat Trisequens Estradiolvalerat Medroxyprogesteronacetat Sisare 28 Tage Zweiphasenpräparate Dreiphasenpräparate Reine Estrogenpräparate zur oralen Gabe Estradiol - Estrifam, Estronorm, Femoston mono Estradiolvalerat - Estradiol JENAPHARM, Gynokadin, Merimono, Progynova konjugierte Estrogene - Climopax mono Estriol - Estriol JENAPHARM, OeKolp; Ovestin, Synapause E Präparate zur transdermalen Applikation Estradiol - Cutanum, Dermestril, Estrabeta, Estraderm TTS, Estradot, Estramon, Evorel, Fem 7, Gynokadin Gel, GynPolar Gel, Sandrena, Sisare Gel, Tradelia Estradiol Norethisteronacetat Estalis sequi, Estragest TTS Estradiol Levonorgestrel Fem7 Combi Selektive Estrogenrezeptor-Modulatoren Raloxifen auf S. 448 - EVISTA, Optruma linke Spalte, vor 2.8.2. Testes einfügen: Ein neuartiges Tokolytikum ist der Oxytocin-Antagonist Atosiban (Tractocile®). Er unterscheidet sich von der physiologischen Substanz in vier Aminosäuren. Die Substanz wird i.v. verabreicht, auf eine Bolusinjektion von 6,75 mg folgen Infusionen von 300 µg/min über 3 Stunden und 100 µg/min über insgesamt maximal 48 h. 17 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 451 Strukturformel Tab. B 2-16. Androgene durch nachfolgende Tabelle ersetzen: R Internationaler Handelspräparat (Einge- Dosierung Freiname tragenes Warenzeichen) Testosteron Androderm Androtop Gel, Testim Gel auf S. 459 2,5–5 mg transdermal/Tag 50 mg/Tag Testosteronundecanoat Andriol 40-160 mg/Tag p. o. Testosteronenantat Testosteron Depot 250 mg 250-750 (-1000) mg/Woche i. m. Eifelfango Testosteron-Depot 250-750(-1000) mg/Woche i.m JENAPHARM Testosteron Depot250 mg i.m. alle 2-3 Wochen. Rotexmedica linke Spalte, vor Wirkungen einfügen: Ebastin (Ebastel®), Desloratadin (Aerius®) und Levocetirizin (Xusal®) sind neue H1-Antihistaminika der 2. Generation ohne prinzipielle Unterschiede zu den bislang verfügbaren Vertretern dieser Wirkstoffgruppe. Ebastin wird bei oraler Gabe gut resorbiert und bei der ersten Leberpassage über CYP3A4 fast vollständig in den aktiven Metaboliten Carebastin umgewandelt. Konjugate von Carebastin werden mit einer Halbwertszeit von 17 Stunden renal ausgeschieden. Die Indikationen entsprechen denen der anderen H1-Antihistaminika der 2. Generation, die Tagesdosis beträgt 10 mg. Auch hinsichtlich der Nebenwirkungen lässt sich Ebastin in diese Wirkstoffgruppe einordnen. Bei gleichzeitiger Gabe von CYP3A4-Inhibitoren ist ein Anstieg der Ebastin-Konzentrationen zu erwarten. CYP3A4 (Ebastel®) Desloratadin ist der wirksame Hauptmetabolit von Loratadin. Aufgrund der höheren Rezeptoraffinität und einer längeren Verweildauer beträgt die Desloratidin-Dosis (5 mg) nur die Hälfte von der der Muttersubstanz, die vom Originalanbieter vom Markt genommen wurde. Levocetirizin ist das aktive R-Enantiomer von Cetirizin. Da das S-Enantiomer inaktiv ist, kann die Dosierung gegenüber dem Razemat auf die Hälfte (5 mg) reduziert werden. (Aerius®) 18 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 468 Abb. B 3–3: Die kurzen Pfeile zwischen LTC4 und LTD4 umdrehen. COOH 15-LOX 12-LOX 15-HPETE 12-HPETE Arachidonsäure 15-HETE 12-HETE 5-LOX 5-LOX OOH COOH Lipoxine A und B 5-HETE 5-HPETE OH O H2O COOH COOH Hydrolase LTA4 LTB4 OH Glutathion-Stransferase Gly γ−Glu Cys COOH OH LTC4 γ−Glutamyltranspeptidase Glu Gly Cys Cys Dipeptidase COOH COOH OH OH LTD4 auf S. 470 Gly LTE4 Abb. B 3-6: in der Dinoproston-Formel –COOH (anstelle –COOCH) auf S. 484 rechte Spalte, vor Kombinationspräparate einfügen: Eisen(III)-hydroxid-Dextran-Komplex (CosmoFer®) ist ein dem Ferritin nachempfundener Komplex, der nach intravenöser Gabe in Leber und Milz in seine Komponenten gespalten wird und dort langsam Eisen freisetzt. Die Substanz ist indiziert zur Behandlung eines Eisenmangels bei nachgewiesener Intoleranz oder mangelnder Wirksamkeit von oralen Darreichungs- formen sowie bei klinischer Erfordernis zur raschen Auffüllung der Eisenspeicher. Die Dosierung beträgt zwei- bis dreimal wöchentlich 100 bis 200 mg. Wie bei anderen parenteral verabreichten Eisenpräparaten können nach der Gabe schwere, möglicherweise letale, anaphylaktoide Reaktionen auftreten. 19 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 486 linke Spalte, nach dem Petit-Abschnitt einfügen: Darbopoetin alfa (Aranesp®) ist ein rekombinantes Erythropoetin mit einer Molmasse von 38500 Dalton, das aufgrund seines höheren Kohlenhydratanteils (51% im Vergleich zu 40% bei Erythropoetin) eine längere terminale Halbwertszeit als rekombinantes humanes Erythropoetin aufweist (25,3 Stunden im Vergleich zu 8,5 Stunden). Die Dosierung liegt bei 0,45 µg/kg Körpergewicht/Woche. auf S. 502 linke Spalte, vor 4.1.7.2 Thrombozytenfunktionshemmer einfügen: 4.1.7.2a Anagrelid Anagrelid (Xagrid®) erniedrigt die Thrombozytenzahl bei Risikopatienten mit essentieller Thrombozytämie. In der postmitotischen Phase der Megakaryozyten-Entwicklung verringert es die Ploidie, die Reifung und die Größe der Megakaryozyten. Zumindest ein Teil des Wirkungsmechanismus beruht auf einer Hemmung der cAMP-Phosphodiesterase. Die empfohlene Anfangsdosis beträgt zweimal täglich 0,5 mg für mindestens eine Woche. Danach erfolgt eine individuelle Dosiseinstellung anhand der Thrombozytenzahl (maximale Einzeldosis 2,5 mg). auf S. 503 Die häufigsten Nebenwirkungen waren Kopfschmerzen, Palpitationen, Flüssigkeitsretention und Übelkeit. (Xagrid®) linke Spalte, zweite Zeile unter 4.1.7.2.2 ändern : Adenosindiphosphat (anstelle Adenosinphosphat) auf S. 509 rechte Spalte, nach Zeile 9 einfügen: Das sulfatierte Pentasaccharid Fondaparinux (Arixta®) ist ein synthetisch hergestellter, selektiver Inhibitor des aktivierten Faktors X (Xa). Fondaparinux verstärkt die durch Antithrombin vermittelte Hemmung des Faktors Xa, indem es selektiv an Antithrombin bindet, um das ca. 300fache. Auf die Thrombozytenfunktion hat es keinen Einfluss. Fondaparinux ist indiziert zur Prophylaxe venöser thromboembolischer Ereignisse bei Patienten, die sich größeren orthopädischen Eingriffen an den unteren Extremitäten unterziehen müssen. Studien zur Erweiterung dieser Indikation laufen. Die Dosierung beträgt einmal täglich 2,5 mg subkutan. Als Nebenwirkungen können neben einer erhöhten Blutungsneigung auch Thrombozytopenien auftreten, daher sollte die Substanz bei Patienten mit Heparininduzierter Thrombozytopenie nicht eingesetzt werden. (Arixta®) 20 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 510 linke Spalte, vor 4.1.7.3.4 Vitamin-K-Antagonisten einfügen: Ein weiteres Hirudin-Analoges ist Bivalirudin (Angiox®). Es wird zur Reduktion des Thromboserisikos bei Patienten mit koronarer Herzkrankheit nach einer perkutanen Koronarintervention eingesetzt. auf S. 512 Initial wird ein Bolus (0,75 mg/kg) intravenös appliziert, gefolgt von einer i.v. Infusion (1,75 mg/kg/Stunde mindestens während der Dauer des Eingriffs). rechte Spalte, vor Streptokinase: Tenecteplase (Metalyse®) ist ebenfalls ein rekombinanter fibrinspezifischer Plasminogenaktivator, der im Vergleich zu t-PA an drei Stellen (Position 103, 117 sowie 296 bis 299; s. Abb. B 4–9a) modifiziert wurde.). Diese Modifikation führt zu einer längeren Plasmahalbwertzeit, einer höheren Fibrinspezifität und einer geringeren Inaktivierung durch PlasminAktivator-Inhibitor-1. Die Substanz wird körpergewichtsbezogen in einer maximalen Dosis von 10 000 U ( = 50 mg Tenecteplase) verabreicht. G A S C L E S P W L P N S L V H C D S S O K W A N M P W H N I 117 I K N R T R L A L T G R K S P I D W D C G R L R C G G D E L Y T P D Y K G N R A A W P N V V C N S Y N H G E F Y R S G S E F Y K A S Y T C G K C E N 184 C A S A Y G D T 103 T O N T F V S W P P N H N G R Y S I G Y A S OD P A S H C K C C P S I E D G S E S D W Y LG G N S OA A A L T C O F C T A L L C G G A A V P G K L A KG C I RO T R P C Y S O P O F R I F K F R A A R E I DT A R R G D V A 296 E P C A OC L V S A G P V O S L I G G 299 L F RE F WL RP I C S L D S RC F V S O P S P N L E R F P P H S H L H F OE D F E O R G C HC T O A S W Y G A S V W I L S A O L I T C C T N L S R E A Y C O R K O G P V S I C V N D D I S DN L V S R A Y L G M E D T S K L O L H E C T L O R D Y T C S Y Y D M K L G O R C D E Y T A P G W V D F L K R G H C R V V E E A G 478 G N S G C L P S P F S K I Y S I I G D H E S G V E R W O V G O L C E L I A Y K G E D S Y C H E O K G L A E V E F K O N H K A O D V P V R O P G L G S Y V R P Y T S T D S K G R 527 V A C P T C R T N L M S Y M L O N N DR WD H I D 448 LL T V T RN N S A O K P Y S L A GR R T Kringle 1 Kringle 2 T Finger K Epidermal Growth Factor Protease Abb. B 4–9a: Tenecteplase (Metalyse®) 21 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 520 Abb. B 4–11: In der Cholesterol-Formel Methylgruppe an C–19 ergänzen, Seitenkette an C–17 um eine CH2-Gruppe verkürzen. HMG-CoAReduktaseBlockade durch CSE-Hemmer Isopentenylpyrophosphat (C5) HO CH3 COOH OH Geranylpyrophosphat (C10) Mevalonat HO Farnesylpyrophosphat (C15) CH3 COOH O Squalen (C30) 3-Hydroxy-3-methylSCoA glutaryl-CoA Squalenepoxid Acetoacetyl-CoA (C4) Acetyl-CoA Lanosterol (C2) Cholesterol HO (C30) (C27) auf S. 520–522: Cerivastatin (LIPOBAY®, Zenas®) streichen. auf S. 522 Tab. 4–12: Atorvastatin-Formel ändern Das Präparat wurde aufgrund einer im Vergleich mit anderen Statinen höheren Rate schwerer Nebenwirkungen (Rhabdomyolysen, insbesondere in Kombination mit Gemfibrozil) im August 2001 vom Markt genommen. 22 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf Seite 522 rechte Spalte, vor 4.1.8.2.2 Anionenaustauscherharze ergänzen: 4.1.8.2.1.a Cholesterolresorptionshemmer Mit Ezetimib (Ezetrol®) wurde der erste Vertreter der neuen Substanzklasse der Cholesterolresorptionshemmer zugelassen. Die Substanz hemmt selektiv die intestinale Resorption von Cholesterol und verwandten Phytosterolen. Ezetimib ist indiziert (allein oder in Kombination mit Statinen) zur Behandlung von Hypercholesterolämien sowie einer Sitosterolämie. Nach oraler Gabe wird Ezetimib nicht über das P450-Enzymsystem abgebaut, sondern durch Glucuronosyltransferasen (s. S. 28 f.) zum Glucuronid verstoffwechselt, das ebenfalls die Cholesterolresorption hemmt. Die Plasmaproteinbindung beträgt 99,7 %, die Halbwertszeit der Ausgangssubstanz bzw. die des Metaboliten 22 Stunden. Ezetimib kann daher einmal täglich gegeben werden (Standarddosis 10 mg). Als Nebenwirkung von Ezetimib wurde in Einzelfällen eine Erhöhung der Serumtransaminasen beobachtet, die im Allgemeinen asymptomatisch und reversibel ist. Außerdem wurde über gastrointestinale Störungen berichtet. Kombination mit anderen Lipidsenkern. Da die Hemmung der Cholesterolresorption zu einer gesteigerten endogenen Cholesterol-Synthese führt, ist die Kombination von Ezetimib mit einem Cholesterolsynthese-Enzym-Hemmstoff (CSE-Hemmer; Statin; s. S. 520 ff.) sinnvoll. Aufgrund der unterschiedlichen Metabolisierungswege – CSE-Hemmer werden überwiegend über P-450-Enzyme abgebaut – treten bei der Kombination keine pharmakokinetischen Interaktionen auf. Die Kombination mit Fibraten erscheint aufgrund der unterschiedlichen Angriffspunkte aus pharmakologischer Sicht ebenfalls geeignet. Da die Verträglichkeit der Kombination bisher aber nicht untersucht wurde, wird die gleichzeitige Anwendung von Ezetimib und Fibraten (derzeit) nicht empfohlen. (Ezetrol®) auf Seite 523 in der rechter Spalte am Ende einfügen: 4.1.8.2.4 Sonstige Omega-3-Säurenethylester (Omacor®) enthält Ethylester semisynthetisch hergestellter Omega-3-Fettsäuren. Wesentliche Inhaltsstoffe sind die Ethylester von Eicosa- (EPA; 46%) und Docosahexaensäure (DHE; 38%), die als „falsche“ Substrate der triglycerid-synthetisierenden Enzyme eine Senkung der VLDL (s. S. 514 f.) bewirken. Das Präparat wird zur Behandlung von Hypertriglyzeridämien, bei Typ IIb/III-Hyperlipoproteinämien auch in Kombinationen mit Statinen (s. S. 520) eingesetzt, wenn eine auf S. 533 Monotherapie mit diesen nicht ausreicht. Besonders ist es bei Hypertriglyzeridämie nach Myokardinfarkt indiziert. Als Nebenwirkungen treten insbesondere gastrointestinale Störungen auf. Bei gleichzeitiger Gabe von Antikoagulantien kann die Blutungszeit etwas verlängert sein. In diesem Fall ist eine sorgfältige Überwachung der Gerinnungsparameter indiziert. Aufgrund der Möglichkeit einer mäßigen Erhöhung der LeberTransaminasen ist bei Patienten mit Leberfunktionsstörungen Vorsicht geboten. rechte Spalte, bei 4.2.2.4 Aldosteronantagonisten ergänzen: (Spironolacton, Eplerenon) auf S. 533 rechte Spalte, ab Zeile 8 ändern: ..... Aldosteronantagonisten (Spironolacton, Eplerenon, s.S. 692 f.) bei der Therapie ..... 23 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf Seite 570 linke Spalte vor Kapillaren, anstelle der zwei letzten Sätze im Petit einfügen: Mit Bosentan (Tracleer®) wurde nunmehr der erste Vertreter dieser Stoffgruppe zugelassen. Bosentan ist ein nicht-selektiver, dualer Antagonist an ETA- und ETB-Rezeptoren. Da Studien bei Patienten mit chronischer Herzinsuffizienz bisher keine deutlich positiven Ergebnisse erbrachten, ist das Pharmakon vorerst nur zur Behandlung der pulmonalen Hypertonie indiziert. Die Bioverfügbarkeit von Bosentan beträgt ca. 50 %, die Plasmaproteinbindung mehr als 98% und die terminale Halbwertszeit etwa 5,5 Stunden. Die Clearance von 8,2 l/Stunde erfolgt im Wesentlichen durch hepatischen Metabolismus über CYP3A4 und CYP2C9 (s. S. 22 f.). Durch Autoinduktion der verstoffwechselnden Enzyme nimmt unter Dauertherapie die Clearance von Bosentan zu (Rückgang der Plasmakonzentrationen auf 50 bis 65% der ursprünglichen Werte). In-vitro-Befunde zeigen ferner eine ausgeprägte Hemmung eines hepatischen Transportproteins, der Gallensalzexport-Pumpe (bile salt export pump; BSEP). Die Therapie wird mit einer Dosierung von 62,5 mg pro Tag über vier Wochen begonnen, anschließend wird eine Erhaltungsdosis von zweimal 125 mg pro Tag angestrebt. Um einen möglichen, bisher aber nicht nachgewiesenen Reboundeffekt zu vermeiden, sollte die Substanz ausschleichend abgesetzt werden. Die meisten Nebenwirkungen von Bosentan sind mit der Hemmung der Endothelinwirkung erklärbar (z.B. Kopfschmerzen und Flush). Eine besondere Nebenwirkung von Bosentan besteht in einem dosisabhängigen Anstieg der hepatischen Aminotransferasen (Aspartat- und Alaninaminotransferase), die möglicherweise im Zusammenhang mit der Hemmung der BSEP (s.o.) steht. Diese Enzymerhöhung verläuft meist asymptomatisch und ist nach Dosisreduktion reversibel. Bei einigen Patienten wurde eine Normalisierung der Leberenzyme trotz Weiterführung der Therapie beobachtet. Aufgrund der Induktion von CYP3A4 und CYP2C9 (s. S. 31 f.) werden gleichzeitig gegebene Arzneistoffe, die über diese Enzyme metabolisiert werden, schneller abgebaut und wirken somit möglicherweise abgeschwächt. Eine Dosisanpassung sollte in diesen Fällen in Betracht gezogen werden. Bei gleichzeitiger Gabe von Warfarin (s. S. 510) oder damit verwandten Antikoagulantien, die durch CYP2C9 verstoffwechselt werden, sollte die INR (International Normalized Ratio) engmaschig kontrolliert werden. Bei hormonalen Kontrazeptiva besteht die Möglichkeit eines Verlusts der kontrazeptiven Wirkung durch die Enzyminduktion. Die gleichzeitige Gabe von CYP3A4und CYP2C9-Inhibitoren (z.B. Fluconazol), die zu einer starken Erhöhung der Konzentrationen von Bosentan führen können, sollte vermieden werden. Aus demselben Grund ist die Kombination von Bosentan und Ciclosporin kontraindiziert. Wegen des hemmenden Einflusses von Glibenclamid auf die BSEP sollte auch diese Substanz nicht gleichzeitig mit Bosentan verwendet werden. (Tracleer®) 24 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 576 Abb. B 4–28: im rechten, grünumrandeten Teil der Abbildung Valin durch Leucin ersetzen (eine zusätzliche CH2Gruppe einfügen). Angiotensin I Angiotensin II Arg Tyr Val ✂ Asp lle His Pro Phe Leu His Zn2+ Zn2+ N CH3 HS H2N – C NH N H C O O + N O N O O + NH2 C O O H2N NH – C NH2 NH Angiotensin-Konversions-Enzym blockiert durch Captopril auf S. 594 ACE Angiotensin-Konversions-Enzym spaltet Angiotensin I Abb. B 4–32: in der Flunaricin-Formel beide Fluoratome jeweils in p-Stellung (nicht in m-Stellung) auf Seite 581 am Ende Tab. B 4-30 (Fortsetzung) einfügen: Tab. B. 4–30. AT1-Blocker (Fortsetzung) Strukturformel Internationaler Freiname Handelsname (Eingetragenes Warenzeichen) Dosis (mg) Halbwertszeit (h) Olmesartanmedoxomil Olmetec, Votum 10 10 – 15 25 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf Seite 596 am Ende der rechten Spalte einfügen: Als weitere Phosphodiesterase-5-Hemmer – und damit als Sildenafil-Analoge – wurden Tadalafil (Cialis®) und Vardenafil (Levitra®) zugelassen. Den schnellsten Wirkungseintritt (ca. 20 Minuten nach Einnahme) der drei Substanzen besitzt Vardenafil, die mit 17,5 Stunden längste Plasmahalbwertszeit Tadalafil. (Levitra®) (Cialis®) auf S. 596 am Seitenende: Apomorphin. Ein zusätzlicher Wirkstoff, der zur Behandlung einer erektilen Dysfunktion nicht injiziert werden muss, sondern sublingual angewandt werden kann, ist der bisher als Antiparkinsonmittel (s. S. 318) und Emetikum (s. S. 961) eingesetzte Dopaminagonist Apomorphin (Ixense®, Uprima®). Die vorwiegend an D2-Rezeptoren angreifende Substanz wird in einer Dosierung von 2–3 mg etwa 20 Minuten vor dem beabsichtigten Geschlechtsverkehr unter die Zunge gelegt. (Wegen des hohen First-pass-Effekts von Apomorphin bei peroraler Gabe ist diese Applikationsform erforderlich.) Wie Sildenafil ist Apomorphin kein Aphrodisiakum, zum Erreichen der erwünschten Wirkung ist daher eine sexuelle Stimulation erforderlich. Bezogen auf die subkutane Verabreichung beträgt die Bioverfügbarkeit bei Verwendung einer Sublingualtablette etwas weniger als 20%. Hauptsächliche Biotransformationsprodukte sind Konjugate (Glucuronide, Sulfate) der Muttersubstanz sowie des N-Desmethylprodukts. Die Plasmahalbwertszeit wird mit ca. 3 Stunden angegeben. Die Ausscheidung erfolgt sowohl renal als auch mit den Fäzes. Bei den Nebenwirkungen, die bei mehrfacher Applikation vielfach abnehmen, stehen Übelkeit sowie Kopfschmerzen, Schwindel und Blutdruck- abfall im Vordergrund. Als weitere – meist nur schwach ausgeprägte – unerwünschte Wirkungen wurde über Rhinitis, Pharyngitis, Benommenheit, Schmerzen, verstärkten Husten, Hitzewallungen, Schwitzen u.a. berichtet. Bei Patienten mit instabiler Angina pectoris, vor kurzem erlittenem Myokardinfarkt, schwerer Herzinsuffizienz oder Hypotonie ist Apomorphin kontraindiziert. Doch kann es im Gegensatz zu Sildenafil zusammen mit Nitraten angewandt werden, falls die Dosis von 3 mg nicht überschritten wird. Gleichzeitige Einnahme von Alkohol verstärkt die Gefahr einer Hypotonie. Zu vermeiden ist auch die Kombination mit anderen zentral wirkenden Dopaminagonisten oder Dopaminantagonisten. (Ixense®, Uprima®) 26 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 607 rechte Spalte unten, vor Lungenemphysem einfügen: Ähnlich wie bei Asthma bronchiale hat die Deutsche Atemwegsliga ein Stufenschema erarbeitet, nach dem die Therapie der COPD je nach Schweregrad erfolgen soll. Tab. B 1–5a. Schweregradeinteilung der COPD. Der Schweregrad der COPD wird nach der 1-Sekunden-Kapazität (FEV1), dem Verhältnis FEV1/VC (VC: inspiratorische Vitalkapazität) und der Symptomatik beurteilt (nach Deutsche Atemwegsliga) Schweregrad Kriterien 0 (Risikogruppe) • normale Spirometrie • chronische Symptomatik (Husten, Auswurf) I (leichtgradig) • FEV1>80%, FEV1/VC<70% • mit/ohne Symptomatik (Husten, Auswurf) II (mittelgradig) • 30%<FEV1<80%, FEV1/VC<70% • mit/ohne chronische Symptome (Husten, Auswurf, Dyspnoe) III (schwer) • FEV1<30%, FEV1/VC<70% • FEV1<50% und respiratorische Insuffizienz oder Zeichen der Rechtsherzinsuffizienz Tab. B 1–5b. Stufenschema für die Langzeittherapie bei COPD. Die Behandlung von Patienten mit COPD sollte sich vor allem nach dem Schweregrad der Erkrankung richten (nach Deutsche Atemwegsliga). Schweregrad Medikamentöse Therapie Nicht-medikamentöse Therapie 0 keine Risikofaktoren meiden (Raucherentwöhnung) I Bei Bedarf Beta-2-Sympathomimetika und/oder Anticholinergika Schutzimpfungen Beta-2-Sympathomimetika und/oder Anticholinergika, zusätzlich Theophyllin Zusätzlich Rehabilitation: Körperliches Training, Physiotherapie, adäquate Ernährung, Patientenschulung bei fehlender Besserung II Wie oben; ferner Therapieversuch mit inhalativen Glucocorticoiden über 3 Monate, Weiterverordnung bei nachgewiesenem Therapieeffekt III Zusätzlich prüfen, ob eine Langzeit-O2-Therapie angezeigt ist auf S. 616 rechte Spalte oben: anstelle des letzten Satzes vor 5.3.4. Stufenplan der Asthmatherapie: Viani® und atmadisc® enthalten die Kombination Salmeterol und Fluticason-17-propionat, Symbicort® ist eine Kombination aus Formoterol und Budesonid. auf S. 653 linke Spalte oben: Diphenoxylat (Bestandteil von Reasec®) einschließlich Formel streichen. Weitere Möglichkeiten: Heimbeatmung, Emphysemchirurgie, Lungentransplantation 27 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf Seite 657 linke Spalte, am Ende des dritten Absatzes ergänzen: Vor einiger Zeit wurde Adefovir-dipivoxil (Hepsera®) zur Behandlung der chronischen Hepatitis B bei Erwachsenen zugelassen. Nach oraler Applikation entsteht im Körper Adefovir, ein Adenosin-Monophosphat- und damit ein Nucleotid-Analogon, das in der Zelle nach Umwandlung in das Diphosphat die virale DNA-Polymerase hemmt. Da die humane DNAPolymerase erst bei vergleichsweise sehr viel höheren Konzentrationen inhibiert wird, war erwartungsgemäß die Substanz bei den klinischen Prüfungen relativ gut verträglich. Dosisabhängige Nebenwirkungen sind Kopfschmerzen, Übelkeit, Durchfall und Nierenfunktionsstörungen. In bisherigen Untersuchungen (maximale auf S. 658 Adefovir-dipivoxil (Hepsera®) linke Spalte, anstelle des Petit-Abschnitts einfügen: Für die Therapie der chronischen Hepatitis C sind ferner 2 pegylierte Interferone zugelassen, Peginterferon ·-2a (Pegasys®) und Peginterferon ·-2b (PegIntron®). Bei der Pegylierung wird Polyethylenglycol kovalent an das entsprechende Interferon gebunden mit dem Ziel, dass sowohl der enzymatische Interferon-Abbau als auch die renale Ausscheidung deutlich verlangsamt werden. Im Vergleich zu auf S. 692 Dauer 48 Wochen) wurden keine Resistenzentwicklungen beobachtet. Peginterferon ·-2b enthält Peginterferon ·-2a eine verzweigte Polyethylenglycol-Kette, wodurch die Eliminationshalbwertszeit von ca. 30 Stunden bei Peginterferon ·-2b auf ca. 80 Stunden erhöht werden konnte (Halbwertszeit des Standard-Interferons 3–4 Stunden). Pegylierte Interferone müssen deshalb nur noch einmal wöchentlich injiziert werden. linke Spalte bei 7.4.3.1 Aldosteronantagonisten ab Zeile 5 ändern: ....verospiron®), dessen Metabolit Canrenon (bzw. Kaliumcanrenoat, s.u.) und die Analogsubstanz Eplerenon (Inspra®) Eingang in die Therapie gefunden haben. auf S. 693 linke Spalte, vor 7.4.3.2 Cycloamidin-Derivate einfügen: Eplerenon. Die Analogsubstanz Eplerenon zeichnet sich durch geringere Nebenwirkungen (u.a. keine Gynäkomastie) und Interaktionen im Vergleich mit Spironolacton aus. Sie wird zusätzlich zur üblichen Standardtherapie zur Verringerung des kardiovaskulären Mortalitätsrisikos bei Patienten mit linksventrikulärer Dysfunktion nach Herzinfarkt angewendet. Die Behandlung wird mit 25 mg einmal täglich peroral begonnen, die Zieldosis beträgt, ebenfalls einmal täglich, 50 mg. (Inspra®) 28 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf Seite 696 rechte Spalte, vor Phytopharmaka einfügen: (Proscar®) (Avodart®) Dutasterid (Avodart®) ist ein stärker wirkendes Analogpräparat von Finasterid, das daher niedriger (0,5 mg täglich) als dieses dosiert werden kann. Während Finasterid nur die 5·-Reduktase vom Typ 1 hemmt, blockiert Dutasterid sowohl die Typ-1- als auch die Typ-2-5·-Reduktase. auf S. 696 rechte Spalte, am Ende des vorletzten Abschnitts einfügen: Eine weiterer, zur Behandlumg der mittelschweren bis schweren Belastungsinkontinenz von Frauen zugelassener Wirkstoff ist Duloxetin (Yentreve®), der sowohl im ZNS als auch im sakralen Rückenmark die Wiederaufnahme von Serotonin und Noradrenalin hemmt. Dadurch wird der Harnröhrenschließmuskeltonus während der Speicherphase des Miktionsyklus erhöht. Duloxetin wird nach oraler Gabe gut resorbiert, die Bioverfügbarkeit schwankt von 30–80%. Die Eiweißbindung ist mit 96% hoch. Dulexetin wird in hohem Maße durch CYP2D6 und CYP1A2 metabolisiert und vor allem in Form der Metabolite renal ausgeschieden. Die Dosierung beträgt zweimal täglich 20–40 mg. Als Nebenwirkungen können Schlaflosigkeit, Müdigkeit, Übelkeit, trockener Mund, Obstipation oder auch Diarrhö sowie Libidoverringerung auftreten. Duloxetin darf nicht mit nichtselektiven MAOHemmern oder CYPlA2-Hemmstoffen angewandt werden. auf S. 706, Dipivefrin-Formel: Substituent in p-Stellung am Aromaten enthält wie der Substituent in m-Stellung eine Ester- und keine Keto-Gruppe. auf S. 707 rechte Spalte, anstelle von 8.5.1.5 Latanoprost: 8.5.1.5 Prostaglandine (Yentreve®) 29 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 708 linke Spalte, vor 8.5.1.6 Osmodiuretika einfügen: Eine chemisch mit Latanoprost nahe verwandte Analogsubstanz ist Travoprost (Travatan®), ebenfalls ein Ester-Prodrug. Bimatoprost (Lumigan®) ist zwar ebenfalls mit Prostaglandin F2· chemisch verwandt, jedoch ist es kein Ester, sondern ein Amid und wirkt es solches. Auch interagiert es nicht mit Prostaglandin-, sondern mit Prostamid-Rezeptoren. (Prostamide sind erst vor kurzem entdeckte körpereigene Substanzen, die an der Regulation des Augeninnendrucks beteiligt sind.) Bimatoprost verbessert sowohl den trabekulären als auch den uveoskleralen Kammerwasserabfluss und senkt daher den intraokularen Druck etwas stärker als Latanoprost. Die Nebenwirkungen entsprechen weitgehend denen der anderen Prostaglandin-Derivate. (Travatan®) auf S. 710 (Lumigan®) rechte Spalte, am Seitenende einfügen: 8.5.10 Verteporfin Verteporfin (Visudyne®) ist ein lichtaktivierbares Benzoporphyrin-Derivat (Photosensibilisator), das zur Therapie der sog. feuchten Form der altersbedingten Makuladegeneration (AMD) eingesetzt wird. Bei dieser Erkrankung kommt es zu einer Exsudation aus den Kapillaren der Choriodea (daher „feuchte“ Form) sowie zu einer subfovealen chorioidalen Neovaskularisation (Einsprossung von chorioidalen Blutgefäßen unter die Netzhaut). Verteporfin wird intravenös infundiert, im Blut bindet es an Lipoproteine (LDL) und lagert sich als Komplex mit LDL vor allem an schnell proliferierende Zellen, damit auch intraokular an das Endothel der neugebildeten Gefäßen, an. Ca. 15 Minuten nach Begin der Infusion wird der Farbstoff gezielt im Auge mit rotem Laserlicht aktiviert. Die bei seiner Rückkehr in den Grundzustand freiwerdende Energie führt zur Bildung von Singulettsauerstoff, der durch Zellschä- digung und anschließende lokale Thrombosierung einen Verschluss der krankhaften Gefäße – ohne signifikante Beeinflussung des umliegenden Gewebes – bewirkt. Verteprofin wird zu einem geringen Prozentsatz zur Dicarbonsäure hydrolysiert, die Plasmahalbwertszeit liegt bei 5–6 Stunden, die Ausscheidung erfolgt vorwiegend mit den Fäzes. Die Dosierung beträgt 6 mg/m2 Körperoberfläche. Als Nebenwirkungen wurden Sehstörungen (u.a. unklares, verschwommenes Sehen, Gesichtsfelddefekte), Übelkeit, Pruritus, Rückenschmerzen während der Infusion sowie allergische Reaktionen beschrieben. Bei Porphyrie und schweren Leberfunktionsstörungen ist Verteporfin kontraindiziert. Die Patienten sind darauf hinzuweisen, dass sie nach der Behandlung zwei Tage lang Augen und Haut vor starker Lichteinstrahlung schützen müssen. 30 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 Abb. B 9–3: PPA durch PEP ersetzen in der Legende zur Abbildung % 50 II-1·-Suppression in Keratinozyten auf S. 723 PC 40 PEC BMV DCE 30 PD PEP BM DM 20 10 0 auf S. 724 10 20 30 IL-1·-Suppression in Fibroblasten rechte Spalte, oben einfügen: 9.4.3.2a Immunsuppressiva In jüngster Zeit wurde mit dem zur Behandlung der Transplantatabstoßung dienenden Tacrolimus (s. S. 932f.; Protopic®) ein neues Therapieprinzip für das atopische Ekzem etabliert. In gleicher Weise wirkt auch ein weiteres Immunsuppressivum (Pimecrolimus; Elidel®). Die beiden 1%igen Cremezubereitungen sollen gut wirksam und verträglich sein. Als Nebenwirkungen dominieren lokale Reizerscheinungen; eine Hautatrophie ist nicht zu befürchten. (Elidel®) % 40 31 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf Seite 727 in der rechten Spalte am Ende des Abschnittes 9.4.5.5 Immunsuppressiva einfügen: Ein weiteres Immunsuppressivum für die Behandlung der Psoriasis ist Alefacept (in den USA AMEVIVE®), ein Fusionsprotein des humanen Lymphozytenfunktion-assoziierten Antigens 3 (LFA-3) und des Fc-Teils von Immunglobulin G-1 (IgG1). Durch Bindung an einen speziellen Rezeptor, den CD2-Rezeptor, blockiert der LFA-3-Teil die Aktivierung von T-Lymphozyten. Die Bindung von IgG1 an den Fc-Rezeptor natürlicher Killerzellen führt dann zum Tod von Gedächtnis-T-Lymphozyten, die in der Pathogenese der Psoriasis eine große Bedeutung besitzen (s. nachstehende Abb. B 9–5a). Ergebnisse der bisherigen kli- nischen Prüfung lassen eine lang anhaltende Besserung der Symptome erwarten. Die Plasmahalbwertszeit beträgt mehrere Tage. Alefacept ist bei chronischer mittelschwerer bis schwerer Psoriasis indiziert, sofern diese auf eine andere Behandlung nicht anspricht. Die Verträglichkeit wird als gut bezeichnet; über das vermehrte Auftreten von Infektionen bzw. Tumoren ist noch keine abschließende Aussage möglich. Bei allergischen Reaktionen gegen den Wirkstoff ist Alefacept kontraindiziert. Antigen-präsentierende Zelle zerstörte T-Gedächtniszelle LFA-3 Alefacept Antigen Alefacept CD2 T-Gedächtniszelle Natürliche Killerzelle Abb. B 9–5a. Zytotoxische Wirkung von Alefacept infolge einer Reaktion mit T-Zellen und natürlichen Killerzellen. auf S. 730 rechte Spalte, vor 9.4.5.7 Benzoylperoxid und Azelainsäure einfügen: 9.4.5.6a Efalizumab Efalizumab (Raptiva®) ist ein humanisierter monoklonaler Antikörper, der an die CD11a-Untereinheit von LFA-1 (lymphocyte function-associated antigen1), ein auf der Oberfläche von Leukozyten lokalisiertes Protein, bindet. Dadurch wird die Bindung von LFA-1 an das Adhäsionsmolekül ICAM-1, das in den Keratinozyten der Psoriasisplaques hochreguliert ist, verhindert. Als Folge davon werden Psoriasis-Symptome abgeschwächt. Efalizumab ist indiziert zur Behandlumg erwachsener Patienten mit mittelschwerer bis schwerer Psoriasis, die auf andere Therapiemaßnahmen nicht angesprochen oder diese nicht vertragen haben. Die Dosierung beträgt initial einmal 0,7 mg/kg, danach wöchentlich 1,0 mg/kg subkutan. Als Nebenwirkungen sind u.a. Leuko- und Lymphozytose, Arthralgien, grippeähnliche Symptome sowie Anstiege der alkalischen Phosphatase und AlaninAminotransferase beschrieben. 32 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 731 rechte Spalte vor 9.4.9 Lichtschutzsubstanzen: 9.4.9a Wachstumsfaktoren Ergänzend zur chirurgischen und enzymatischen Wundreinigung kann eine Förderung der Wundheilung durch eine Lokaltherapie mit dem platelet deri- auf Seite 733 ved growth factor (PDGF) erfolgen, der gentechnisch gewonnen als Becaplermin (REGRANEX®) zur Behandlung chronischer Ulzera zur Verfügung steht. nach Tab. B 9-7 einfügen: 9.4.10 Photodynamische Therapie Zur photodynamischen Therapie von Hauterkrankungen vgl. S. 906 in dieser Ergänzung. auf S. 744 linke Spalte oben, Strukturformeln: R1 = H; R2 = R3 = CH3: Á-Tocopherol auf S. 793 rechte Spalte oben, Cephalosporin-C-Formel ändern: auf Seite 798 linke Spalte, unter 11.3.1.1.3 Carbapeneme ergänzen: Imipenem [in fixer Kombination mit Cilastatin (= ZIENAM®)], Meropenem (Meronem®) und Ertapenem (Invanz®). Ertapenem-Natrium (Invanz®) Rechte Spalte, am Ende des 2. Abschnitts ergänzen: Ertapenem wird mit einer Halbwertszeit von 4 Stunden renal eliminiert. Rechte Spalte, am Ende des 3. Abschnitts einfügen: Ertapenem wird einmal täglich in einer Dosis von 1 g als i.v. Kurzinfusion gegeben. 33 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 809, linke Spalte, vor 11.3.2.4 Chloramphenicol einfügen: 11.3.2.3.1 Ketolide Auch die Ketolide, deren erster Vertreter Telithromycin (Ketek®) ist, können zu den Makroliden gerechnet werden. Telithromycin wirkt gegen die häufigsten Erreger von Atemwegsinfektionen. Es unterscheidet sich von Makroliden durch eine Ketogruppe an C–3 anstelle der Cladinose, wodurch eine hohe Säurestabilität und ein geringeres Risiko einer Resistenzsteigerung erreicht wird. Durch Eingriff in die Translation an zwei Punkten – Blockade der Translokation der an tRNA-gebundenen Aminosäurenkette von der Akzeptor- zur Donorposition der 50S-Untereinheit der Ribosomen und der Domänen II und V der 23S-Untereinheit der ribosomalen RNA – wirkt das Ketolid vielfach noch bei Resistenz gegen Makrolide. Telithromycin wird bei oraler Gabe im nüchternen Zustand sowie auch nach einer Mahlzeit gut resorbiert, die Bioverfügbarkeit wird mit 57% angegeben. Die Elimination erfolgt durch Metabolisierung über CYP3A4 und andere Enzyme, die terminale Halbwertszeit beträgt 10 h. Telithromycin dient zur Behandlung von Pneumonien, akuten Exazerbationen einer chronischen Bronchitis und anderen Atemwegserkrankungen. Die Dosierung beträgt einmal täglich 800 mg. Die Nebenwirkungen entsprechen denen der Makrolide. Wie diese ist auch Telithromycin ein starker CYP3A4- und CYP2D6-Inhibitor und darf daher nicht mit Arzneistoffen kombiniert werden, bei denen dadurch eine Kumulationsgefahr resultiert. (Ketek®) auf S. 813 rechte Spalte, Zeile 2: an N-1 (anstelle an C-1) auf S. 814 Haemophilus influenzae streichen linke Spalte unter Gruppe 2; Die Standardchinolone: 34 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 831 Abb. B 11–22 ersetzen: Squalen Allylamine 2,3-Oxidosqualen (Squalenepoxid) O Lanosterol HO Azole HO Morpholine HO Fecosterol HO Morpholine Episterol HO Ergosterol HO 35 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf Seite 839 rechte Spalte, vor 11.4.5 Ciclopirox einfügen: 11.4.5a Glucansynthesehemmer (Candine) Vor dem Hintergrund der zunehmend häufigeren systemischen Pilzinfektionen sowie von Resistenzen gegen Azol-Antimykotika (s. S. 775) infolge der Bildung einer strukturell abweichenden Lanosteroldemethylase und/oder von Effluxpumpen kommt der Entwicklung einer völlig neuen Gruppe von Antimykotika, den sog. Candinen, große Bedeutung zu. Diese blockieren die Synthese des essentiellen Pilzzellwandbestandteils 1,3-‚-D-Glucan durch irreversible Hemmung der ‚-1,3-D-Glucansynthase. Die Pilze sterben durch Aufbrechen der Zellwand. Candine wirken daher ähnlich wie die antibakteriell wirksamen ‚-Lactam-Antibiotika. Erster Vertreter dieser Substanzklasse ist Caspofungin (CANCIDAS®). Dabei handelt es sich um ein partial- synthetisches Derivat des Lipopeptids Pneumocandin Bo. Das Wirkungsspektrum umfasst Candida- und Aspergillusarten, nicht aber Cryptococcus neoformans, dessen Zellwand nur wenig 1,3-‚-D-Glucan enthält. In-vitro-Versuche weisen auf eine synergistische Wirkung mit Fluconazol und Amphotericin B hin. Caspofungin ist bei oraler Gabe nur zu <1% bioverfügbar und muss daher parenteral gegeben werden. Die Plamaeiweißbindung beträgt 80–96%, die Halbwertszeit ca. 9 Stunden. Die Substanz wird nahezu vollständig biotransformiert. Caspofungin ist indiziert bei invasiver Aspergillose, die Dosierung beträgt am ersten Tag 70 mg, an den Folgetagen 50 mg. Als Nebenwirkung der – nach dem gegenwärtigen Kenntnisstand – gut verträglichen Substanz wird ein Anstieg des Serumkreatinins angegeben. (CANCIDAS®) auf S. 848 rechte Spalte, vor Idoxuridin einfügen: Ein oral applizierbares Prodrug von Ganciclovir ist Valganciclovir (Valcyte®). (Valcyte®) 36 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 850 auf S. 850 rechte Spalte,nach 2. Zeile ergänzen: Fusionshemmern, nucleosidischen Reverse-TranskriptaseInhibitoren (Nucleosiden, NRTI), nucleosidischen Reverse-TranskriptaseInhibitoren (Nucleotid-Analoga, NTRTI), nicht-nucleosidischen Reverse-TranskriptaseInhibitoren (Nichtnucleosiden, NNRTI) und HIV-Proteasehemmern. 11.5.5.0 Enfuvirtid Enfuvirtid (Fuzeon®), ein synthetisches, aus 36 Aminosäuren bestehendes Peptid, verhindert als Fusionshemmer die Übertragung des genetischen Virusmaterials in das Zytoplasma der Wirtszelle. Dadurch werden gesunde Zellen geschützt. Es ist indiziert in Kombination mit anderen retroviralen Substanzen bei AIDS-Patienten, bei denen es zu einer Resistenz gegen die Standardtherapie gekommen ist oder bei denen eine Unverträglichkeit gegen das vorausgegangene Therapieregime vorliegt. Nach subkutaner Injektion beträgt die Bioverfügbarkeit ca. 65%, die Eliminationshalbwertszeit etwa 4 Stunden. Die Dosierung beträgt zweimal täglich 90 mg subkutan. Als Nebenwirkungen wurden neben Lokalreaktionen an der Injektionsstelle u.a. gehäuftes Auftreten von bakteriellen Pneumonien, Schlaflosigkeit, Kopfschmerzen, Störungen der Geschmacksempfindung, verminderter Appetit, periphere Neuropathien, Myalgien und Lymphadenopathien beschrieben. 11.5.5.1 Nucleosidische und nucleotidische Reverse-Transkriptase-Inhibitoren (NRTI und NTRTI) Als Nucleoside zur HIV-Therapie dienen Zidovudin (Azidothymidin, AZT), ein bereits 1964 synthetisiertes Thymidin-Analogon, Abacavir, Didanosin (Dideoxyinosin, DDI) Lamivudin (3TC), Zalcitabin (Dideoxycytidin) und Emtricitabin. (Emtriva®) 37 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 855 linke Spalte, vor 12.1.2 Antagonisten von Purin- und Pyrimidinbasen einfügen: Amprenavir (Agenerase®), Atazanavir (REYATAZ®), Fosamprenavir (Amprenavir-Calciumphosphat-Ester; Telzir®) und Lopinavir sind neuere HIV-Proteasehemmer. Das Handelspräparat von Lopinavir (Kaletra®) enthält eine kleine Menge Ritonavir (33,3 mg/Weichkapsel bzw. 100 mg/5 ml Lösung), das den First-pass-Metabolismus von Lopinavir (durch CYP3A) hemmt und dadurch dessen Bioverfügbarkeit und Wirksamkeit erhöht. Die Halbwertszeiten liegen von Amprenavir und Fosamprenavir bei 7–10 Stunden, von Atazanavir bei 7–9 Stunden und von Lopinavir bei 12 Stunden. Die oralen Tagesdosen betragen von Amprenavir 2400 mg, von Atazanavir 300 mg, von Fosamprenavir 1400 mg und von Lopinavir 800 mg. Die Nebenwirkungen entsprechen denen der anderen HIV-Proteasehemmer, zusätzlich kann Lopinavir Lipidstoffwechselstörungen auslösen. (Agenerase®) (REYATAZ®) (in Kaletra®) 38 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf Seite 861 linke Spalte, anstelle des zweiten Petit-Abschnittes einfügen: Ein neues Malariamittel ist die Kombination von Artemether und Lumefantrin. Das Handelspräparat Riamet® enthält die beiden Komponenten in einer Menge von 20 bzw. 120 mg. Das Kombinationspräparat soll selbst in Gebieten mit Multiresistenz von P. falciparum gut gegen Blutschizonten wirken. Das Sesquiterpenlacton Artemether leitet sich von Artemisinin, einem Inhaltsstoff von Artemisia annua, ab. Über seine Endoperoxidstruktur tritt Artemether in Wechselwirkung mit Häm-Eisen und gehört damit ebenso wie Lumefantrin, einem Strukturanalogon von Halofantrin, zu den Hämpolymerase-Inhibitoren. Beide Substanzen wirken synergistisch, die Wirkung tritt sehr rasch ein. Artemether wird schnell, Lumefantrin dagegen langsamer resorbiert. Einnahme mit einer Mahlzeit fördert die Resorption beider Komponenten. Die Plasmaproteinbindung ist bei Artemether mit 95,4% auf Seite 885 hoch, bei Lumefantrin mit 99,9% sehr hoch. Über CYP3A4/5 der Leber wird Artemether zum biologisch aktiven Metaboliten Dihydroartemisinin demethyliert, Lumefantrin N-debutyliert. Die Plasmahalbwertzeit von Artemether beträgt nur 2 Stunden, die von Lumefantrin dagegen mehrere Tage. Die Kombination dient zur Behandlung der akuten unkomplizierten Infektion mit P. falciparum. Innerhalb von 48 Stunden sind 24 Tabletten einzunehmen. Die Nebenwirkungen entsprechen denen von Chloroquin, doch können auch – wie bei Halofantrin – lebensbedrohliche Arrhythmien auftreten. Riamet® ist bei Patienten mit Herzerkrankungen (auch in der Familienanamnese; s. Halofantrin, S. 863), bei einer Behandlung mit CYP3A4-Inhibitoren sowie mit Substraten von CYP2D6, das durch Lumefantrin gehemmt wird, kontraindiziert. linke Spalte, vor 12.1.2 Antagonisten von Purin- und Pyrimidinbasen einfügen: Pemetresed (Alimta®) ist ein weiterer Folsäureantagonist, der für die Rezidivtherapie bei Patienten mit nichtkleinzelligem Bronchialkarzinom und in Kombination mit Cisplatin für die Primärtherapie des Pleuramesothelioms zugelassen ist. (Alimta®) 39 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 887 rechte Spalte, anstelle des Petit-Abschnitts: Tegafur/Uracil (UFT®) wird im Verhältnis 1:4 in Kombination mit Calciumfolinat zur Primärtherapie des metastasierten kolorektalen Karzinoms eingesetzt. Tegafur ist eine Vorstufe von 5-Fluorouracil. Nach seiner Bioaktivierung zu 5-FU mittels CYP2A6 wird der von der Dihydropyrimidindehydrogenase (DPD) katalysierte 5-FU-Abbau dadurch gehemmt, dass Uracil dieses Enzyms reversibel blockiert. Als Folge dieser kompetitiven Hemmung steigt die 5-FUPlasmakonzentration. Außerdem wird die Zytotoxizität von 5-FU durch gleichzeitig gegebenes Calciumfolinat gesteigert. Die Dosierung beträgt täglich 300 mg/m2 Tegafur und 672 mg/m2 Uracil in Kombination mit 90 mg Calciumfolinat oral. Die Nebenwirkungen sind qualitativ denen von 5-FU vergleichbar, treten jedoch signifikant seltener auf. Wie bei 5-FU besteht ein erhöhtes Toxizitätsrisiko bei solchen Patienten, bei denen ein genetisch bedingter DPD-Mangel besteht. Capecitabin (Xeloda®) ist ein ebenfalls ein Prodrug von 5-Fluorouracil, das nach enzymatischer Abspaltung der Seitenkette in der Leber durch die im Tumor vermehrt vorkommende Thymidin-Phosphorylase in die Wirkform 5-Fluorouracil überführt wird. Die Substanz wird wie UFT® bei metastasierendem Dickdarmkrebs eingesetzt, wobei wiederum ein besseres Ansprechen im Vergleich zu 5-Fluorouracil beobachtet wurde. Die Dosierung beträgt 1250 mg/m2 zweimal täglich über 14 Tage, gefolgt von einer 7-tägigen Therapiepause. auf S. 903 linke Spalte, vor Aromatasehemmer einfügen: Fulvestrant (Faslodex®) ist ein Estrogen-Rezeptorantagonist, der die Estrogen-Rezeptoren vollständig blockiert und keine partielle agonistische Wirkung aufweist. Seine Rezeptor-Affinität ist höher als die von Tamoxifen (s.o.). Darüber hinaus verhindert die lange Seitenkette von Fulvestrant Konformationsänderungen des Rezeptors, so dass Transkriptionsfaktoren nicht aktiviert werden können. Da auch die Dimerisierung des Rezeptors durch Fulvestrant blockiert wird, kommt es zu einer vollständigen Rezeptor-Deaktivierung. Der Rezeptor-AntagonistKomplex wird außerdem beschleunigt abgebaut, die Folge ist eine Rezeptor-Downregulation. Fulvestrant ist indiziert zur Behandlung von postmenopausalen Frauen mit Estrogenrezeptor-positivem, lokal fortgeschrittenem oder metastasiertem Mammakarzinom, bei Mammakarzinom-Rezidiven während oder nach adjuvanter Antiestrogen-Therapie sowie bei Progression der Erkrankung unter der Behandlung mit einem Antiestrogen. Die Substanz wird einmal monatlich intramuskulär in einer Dosis von 250 mg verab- reicht. Maximale Plasmakonzentrationen werden aufgrund langsamer Resorption aus der Injektionsstelle erst nach sieben Tagen erreicht. Die Substanz wird über CYP3A4 verstoffwechselt, ohne den Abbau anderer Substanzen zu beeinflussen. Die häufigsten Nebenwirkungen sind Hitzewallungen und Störungen im Gastrointestinalbereich (Übelkeit, Erbrechen, Diarrhö). Fulvestrant ist kontraindiziert bei schwerer Einschränkung der Leberfunktion. (Faslodex®) 40 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf Seite 905 linke Spalte, vor Asparaginase einfügen: Arsentrioxid. Arsentrioxid (Trisenox®) wird bei Patienten mit promyeloischer Leukämie (akuter Promyelozytenleukämie; APL), bei denen eine Therapie mit Zytostatika bzw. Alltrans-Retinoiden keinen Behandlungserfolg gebracht hat, als letzte Behandlungsoption eingesetzt. Bei der Promyelozytenleukämie, die durch eine chromosomale Translokation gekennzeichnet ist, wird ein Protein gebildet, das die Reifung der Vorläuferzellen der weißen Blutzellen beeinträchtigt. Arsentrioxid schädigt dieses Protein und induziert Apoptose (s. S. 876 ff.) von Leukämiezellen. Im Rahmen einer Induktionstherapie wird Arsentrioxid täglich zunächst in einer Dosis von 0,15 mg/kg intravenös gegeben, bis eine Knochenmarkremission erreicht ist. Drei bis vier Wochen nach Ende der Induktionstherapie wird eine Konsolidierungstherapie eingeleitet, bei der ebenfalls 0,15 mg/kg an fünf aufeinanderfolgenden Tagen – gefolgt von zwei Tagen Pause – verabreicht werden. Die Konsolidierungsphase wird über fünf Wochen durchgeführt. Die Nebenwirkungen von Arsentrioxid umfassen Hyperglykämie, Hypokaliämie, Neutropenie und erhöhte Alaninaminotransferase-Plasmaspiegel. Zu beachten ist außerdem die Verlängerung der QT-Zeit, die zu gefährlichen Herzrhythmusstörungen führen kann und insbesondere in der Kombination von Arsentrioxid mit solchen Medikamenten relevant ist, die ebenfalls die kardiale Repolarisation verlängern (z. B. Makrolide, Neuroleptika). Als spezifische Nebenwirkung kann ferner das so genannte Leukozyten-Aktivierungssyndrom (APL-Differenzierungssyndrom) auftreten, das durch Fieber, Dyspnoe, Gewichtszunahme, Lungeninfiltrate und Pleura- bzw. Perikardergüsse gekennzeichnet ist. Die Behandlung dieser Nebenwirkung erfolgt mittels hoher GlucocorticoidGaben. Wegen der hauptsächlichen Ausscheidung über die Niere ist bei Patienten mit eingeschränkter Nierenfunktion besondere Vorsicht geboten. 41 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf Seite 906 linke Spalte, vor Tretinoin einfügen: Photodynamische Therapie. Unter einer photodynamischen Therapie versteht man die Behandlung mit einem Wirkstoff, der durch Licht lokal aktiviert wird. Temoporfin (Foscan®) ist eine aus der Gruppe der Porphyrine stammende Substanz, die im Rahmen einer solchen Therapie zur palliativen Behandlung von Plattenepithelkarzinomen im Kopf- und Halsbereich verwendet werden kann. Die Substanz wird in einer Dosierung von 0,15 mg/kg Körpergewicht infundiert. Nach einer Wartezeit von 96 Stunden wird der Tumorbereich mit einem Laser der Wellenlänge 652 nm bestrahlt. Die dabei gebildeten hochreaktiven Sauerstoffspezies sollen zu einer lokalen Apoptoseinduktion (s. S. 876 ff.) und damit zu einer Reduktion des Tumors führen. Während der Behandlung kann eine unerwünschte Aktivierung von Temoporfin auch durch Tageslicht erfolgen. Die Patienten müssen daher während der ersten 15 Tage nach einer TemoporfinInjektion Haut und Augen vor direktem Son-nenlicht und heller Raumbeleuchtung schützen. Temoporfin und seine konjugierten Metabolite werden über die Galle ausgeschieden. Die terminale Halbwertszeit der Substanz beträgt 65 Stunden, die Plasmaeiweißbindung >85 %. Temoporfin darf bei Patienten mit Porphyrie (s. S. 108) nicht angewendet werden. Die gleichzeitige Gabe weiterer photosensibilisierender Substanzen ist zu vermeiden. An Nebenwirkungen treten neben der generellen Photosensibilisierung Schmerzen am Injektionsort sowie Dysphagie und Gesichtsödeme auf. Eine weitere, zur photodynamischen Therapie geeignete Substanz ist Methyl-5-amino-4-oxopentanoat (Metvix® 160 mg/g Creme), der Methylester der 5Aminolävulinsäure, der zur Therapie aktinischer Keratosen und von Basalzellkarzinomen eingesetzt wird. Bei aktinischen Keratosen handelt es sich um Verhornungsstörungen der Keratinozyten, die auf eine langjährige, chronische Lichtexposition zurückzuführen sind. Keratosen gelten als Präkanzerosen, die sich zu invasiven, epithelialen Tumoren entwickeln können und somit behandlungsbedürftig sind. Die relativ selektive Wirkung an Tumor- im Vergleich zu normalen Zellen ergibt sich aus der stärkeren Anreicherung von 5-Aminolävulinsäure-Methylester im entarteten Gewebe und der geringen Eindringtiefe von Rotlicht. Das Prodrug wird in der Haut zu 5-Aminolävu-linsäure, einem frühen Baustein der Porphyrinsynthe-se (s. S. 108), hydrolysiert. Im Rahmen der Hämsynthese entsteht der sehr starke Photosensibilisator Protoporphyrin IX. Seine Licht-induzierte Anregung führt zur Übertragung der Anregungsenergie auf freien Sauerstoff in der Umgebung der Substanz und damit wie bei Temoporfin zur Bildung von reaktiven Sauerstoffspezies, insbesondere von Singulettsauerstoff. Dieses hochwirksame Zellgift spaltet oxidativ C=C-Doppelbindungen. Schäden, die zum Zelltod durch Nekrose und/oder Apoptose führen, betreffen essentielle Zellstrukturen, z.B. Membranen und Mikrotubuli. Aufgrund der kurzen Lebensdauer des Singulettsauerstoffs soll die DNA dagegen kaum angegriffen werden. Zweimalige okklusive Applikation der Creme auf die Läsionen und Bestrahlung mit Licht der Wellenlänge 570–670 nm (75 J/cm2) im Abstand von einer Woche reduziert die Größe des veränderten Areals. Als unerwünschte Wirkungen treten Zeichen einer vorübergehenden lokalen phototoxischen Reaktion (u.a. lokale Ödeme, Erytheme) auf. Photochemische Reaktionen liegen auch der Photosensibilisierung von Dantrolen (s. S. 297) und Tetracyclinen (s. S. 806) sowie der PUVA-Therapie (s. S. 726) zugrunde. (Foscan®) (Metvix®) 42 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 906 rechte Spalte, vor 12.8. Radioaktive Isotope: Bexaroten (Targretin®), ein Retinoid der 3. Generation ( s. S. 727), wird zur Behandlung von Hautmanifestationen beim kutanen T-Zelllymphom eingesetzt. Die Substanz bindet an RX-Rezeptoren (s. S. 728) und aktiviert diese, wodurch die Expression zahlreicher Transkriptionsfaktoren sowie nachgeschaltete Prozesse der Zelldifferenzierung und Apoptose beeinflusst werden. Der genaue Wirkmechanismus beim T-Zelllymphom ist unbekannt. Die Dosierung beträgt 300 mg/m2. Nebenwirkungen bestehen in erster Linie in ausgeprägten Hyperlipidämien (Triglycerid- und Cholesterol-Erhöhung) sowie Hypothyreosen. Die Substanz wird durch CYP3A4 abgebaut und induziert dieses Enzym möglicherweise selbst. Dementsprechend wurde eine ausgeprägte Interaktion mit Gemfibrozil beobachtet. Bortezomib. Bortezomib (VELCADE®) ist der erste zugelassene Proteasomen-Inhibitor und damit Vertreter einer neuen Substanzklasse. Es hemmt eine definierte Untereinheit des Proteasoms (Chymotrypsin-artige Aktivität des 26S-Proteasoms), das für den Abbau von zellulären Proteinen von zentraler Bedeutung ist. Für einen solchen Abbau bestimmte Proteine werden von der Zelle mit Ubiquitin markiert und anhand dieser Markierung von dem 26S-Proteasom erkannt und abgebaut. Auf die Hemmung dieses ubiquitären Abbauwegs reagieren insbesondere Tumorzellen sehr empfindlich. Es resultiert eine Veränderung der Faktoren, die den Zellzyklus kontrollieren, und damit ein eingeschränktes zelluläres Wachstum. In der Folge sind die Fähigkeiten der Tumorzellen zur Metastasierung reduziert, und es kommt zur Apoptose. Bortezomib ist zugelassen zur Behandlung von Patienten mit multiplem Myelom, weitere Indikationsgebiete werden derzeit untersucht. Als Peptid wird Bortezomib intravenös verabreicht. Die Dosierung beträgt 1,3 mg/m2 Körperoberfläche zweimal wöchentlich über einen Zeitraum von 2 Wochen, gefolgt von einer zehntägigen Therapiepause. Die häufigsten Nebenwirkungen sind gastrointestinale Symptome (Erbrechen, Diarrhö, Obstipation), Blutbildveränderungen (Thrombozytopenie) sowie (Targretin®) Neuropathien. Bei Diabetikern kann es zu Hyper- bzw. Hypoglykämien kommen, so dass eine häufige Blutzuckerkontrolle indiziert ist. Da Bortezomib ein schwacher Hemmstoff verschiedener Cytochrom-P-450-Enzyme (CYP1A2, 2C9, 2C19, 2D6 und 3A4) und damit ein mögliches Substrat ist, sollte bei gleichzeitiger Gabe von Hemmstoffen dieser Enzyme (z.B. Ketoconazol) oder Induktoren (z.B. Rifampicin) eine engmaschige Überwachung der Patienten erfolgen. (VELCADE®) 43 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 907 linke Spalte, vor Zytokine einfügen: Alemtuzumab (MabCampath®) ist ein gentechnisch hergestellter, humanisierter IgG1-Antikörper, der auf der Lymphozytenoberfläche an CD52 bindet und dort zu einer zellvermittelten Zytotoxizität führt. Als Folge davon kommt es zu einer Lyse der Lymphozyten. Indiziert ist Alemtuzumab bei Patienten mit chronischer lymphatischer Leukämie. Die (steigende) Dosierung beträgt 3 mg am 1. Tag, 10 mg am 2. Tag und 30 mg am 3.Tag, gefolgt von 30 mg dreimal pro Woche. Als Nebenwirkungen treten insbesondere Symptome auf, die auf eine Zytokinfreisetzung zurückzuführen sind (Hypotonie, Rigor, Fieber, Dyspnoe). Eine Prämedikation mit einem Antihistaminikum (Diphenhydramin) und einem Analgetikum (Paracetamol) wird daher empfohlen. Als weitere Nebenwirkungen werden opportunistische Infektionen beobachtet. Außerdem können schwere Blutungen auftreten. Cetuximab (Erbitux®) ist ein chimärer monoklonaler IgG1-Antikörper gegen den epidermalen Wachstumsfaktor-Rezeptor (Epidermal Growth Factor Receptor, EGRF). EGFR-Signalwege sind an der Steuerung der Überlebensfähigkeit von Zellen, des Zellzyklus sowie der zellulären Invasion bzw. Metastasierung beteiligt. Cetuximab, das an EGFR mit etwa 5- bis 10fach höherer Affinität als endogene Liganden bindet, hemmt die Funktion des Rezeptors und induziert dessen Internalisierung sowie die Apoptose von EGFRexprimierenden Tumorzellen. Außerdem wird die Expression von Angiogenesefaktoren durch Tumorgewebe und dadurch die Neovaskularisierung von Tumoren unterdrückt. Cetuximab ist zur Therapie des fortgeschrittenen kolorektalen Karzinoms nach Versagen einer Behandlung mit Irinotecan (s. S. 894 f.) indiziert. Für weitere Indikationen befindet es sich in klinischer Prüfung. Die Dosierung beträgt initial 400 mg/m2 Körperoberfläche, danach einmal wöchentlich 250 mg/m2 Körperoberfläche. Als Nebenwirkungen wurden u.a. allergische Reaktionen, Konjunktivitis und Atemnot beschrieben. Bevacicumab (Avastin®), ein humanisierter monoklonaler Antikörper, bindet an den Gefäßwachstumsfaktor VEGF (Vascular Endothelial Growth Factor) und hemmt dadurch die Wechselwirkung von VEGF mit seinen Rezeptoren. Als Folge davon kommt es zu einer Reduzierung der Tumorvaskularisation und damit des Tumorwachstums. Die Behandlung mit Bevacizumab führte dementsprechend zu einer ausgeprägten antitumoralen Aktivität bei einer Reihe bösartiger Tumore. Die Dosierung beträgt 5 mg/kg alle 14 Tage als i.v. Infusion. Als Nebenwirkungen wurden u.a. Stomatitis, Hypertonie, Blutungen, Obstipation, Asthenie, Fieber, Dyspnoe, intraabdominale Entzündungen und Hautveränderungen beschrieben. Mit Ibritumomab-Tiuxetan (Zevalin®) wird erstmalig ein Radiotherapeutikum mit einem Immuntherapeutikum bei der Behandlung von Tumorerkrankungen kombiniert. Dabei wird der Betastrahler 90Yttrium mittels eines Chelators (Tiuxetan) stabil an einen antiCD20-Antikörper (Ibritumomab) gebunden. Dieser Antikörper wird bei CD20-positiven Non-HodgkinLymphomen eingesetzt, wenn die Behandlung mit dem gegen die gleiche Zielstruktur gerichteten Antikörper Rituximab (s.o.) nicht erfolgreich war. Nach Gabe von Ibritumomab-Tiuxetan bindet der Antikörper an die Zielstruktur auf dem Lymphom. Die Reichweite der Strahlung von 90Yttrium (ca. 5 mm) reicht bei entsprechender lokaler Strahlungsstärke, die durch die Kombination mit dem Antikörper erreicht wird, aus, um Zielzellen und benachbarte Zellen abzutöten. Zur Verhinderung, dass CD20-Strukturen auf normalen B-Zellen durch Ibritumomab-Tiuxetan angegriffen werden und damit der tumorspezifische Effekt vermindert wird, gibt man vor Behandlungsbeginn eine geringe Dosis von Rituximab, die genügt, um die Bindungsstellen der nicht-tumoralen B-Zellen abzusättigen. Ibritumomab-Tiuxetan ist zugelassen zur Behandung eines nach Rituximab-Gabe rezidivierenden oder Therapie-refraktären CD20-positiven follikulären Non-Hodgkin-Lymphoms vom B-Zell-Typ. An Nebenwirkungen treten insbesondere Fieber, Infektionen sowie Blutbildveränderungen auf. 44 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 907 rechte Spalte, vor dem Petit-Abschnitt: Imatinib. Imatinib (Glivec®) ist ein Hemmstoff der Tyrosinkinase Bcr-Abl, deren Aktivität bei Patienten mit chronisch myeloischer Leukämie (CML) deutlich erhöht ist, wodurch die Proliferation weißer Blutzellen stark zunimmt. Bcr-Abl entsteht durch reziproke GenTranslokation zwischen Chromosom 9 und 22 (wodurch es zur Bildung des so genannten PhiladelphiaChromosoms kommt). Imatinib hemmt die Bindung von ATP an die Tyrosinkinase und verhindert so deren Aktivierung. Aufgrund der sehr positiven Studienergebnisse wurde die Substanz in einem beschleunigten Verfahren von der FDA zur Behandlung von Erwachsenen mit Bcr-Abl-positiver CML zugelassen. Derzeit läuft eine Reihe von weiteren Untersuchungen bei verschiedenen Tumorarten. Die Dosierung beträgt 400 mg/Tag bei der chronischen und 600 mg bei der akzelerierten Phase einer CML. auf S. 917 Häufige Nebenwirkungen sind Übelkeit, Erbrechen und Durchfälle sowie Störungen des Blutbildes. Nach oraler Gabe wird eine hohe interindividuelle Variabilität der Plasmakonzentrationen beobachtet, die zum Teil auf die Biotransformation durch CYP3A4 zurückgeführt werden kann. Bei gleichzeitiger Gabe von CYP3A4-Hemmstoffen oder -Induk-toren ist daher Vorsicht geboten. (Glivec®) Abb. B 13–6 durch die folgende Abbildung ersetzen:. (Die Ständige Impfkommission (STIKO) am RobertKoch-Institut hat ihre Impfempfehlungen neu gefasst. Der Impfkalender enthält nun auch generelle Empfehlungen für Erwachsene. Außerdem rät die Impfkommission bei mehr Erkrankungen als bisher zur Expositionsprophylaxe.) Abb. B 13–6. Impfkalender für Säuglinge, Kinder, Jugendliche und Erwachsene. Empfohlenes Impfalter und Mindestabstände zwischen Impfungen aus: http://www.rki.de/GESUND/IMPFEN/IMPFEN.HTM?/GESUND/IMPFEN/STI_NEU.HTM&1 Impfstoff/ Antigen-Kombinationen Alter in vollendeten Monaten Geburt 2 3 1. DTaP * DT/Td 2. 4 3. 11-14 15-23 s.a) Alter in vollendeten Jahren 4-5 9-17 ab 18 > s.a.) s.a.) 60 4. b) A A A aP Hib* 1. s. c) 2. 3. IPV* 1. s. c) 2. 3. A 1. s. c) 2. 3. G HB* MMR** A*** s. d) 1. 2. Influenza**** S Pneumo-***** S kokken 45 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 A G S a) b) c) d) Auffrischimpfung: Diese sollte möglichst nicht früher als 5 Jahre nach der vorhergehenden letzten Dosis erfolgen. Grundimmunisierung aller noch nicht geimpften Jugendlichen bzw. Komplettierung eines unvollständigen Impfschutzes Standardimpfungen mit allgemeiner Anwendung = Regelimpfungen Zu diesen Zeitpunkten soll der Impfstatus unbedingt überprüft und gegebenenfalls vervollständigt werden. Ab einem Alter von 5 bzw. 6 Jahren wird zur Auffrischimpfung ein Impfstoff mit reduziertem Diphtherietoxoid-Gehalt (d) verwendet. Antigenkombinationen, die eine Pertussiskomponente (aP) enthalten, werden nach dem für DTaP angegebenen Schema benutzt. Siehe Anmerkungen ›Postexpositionelle HepatitisB-Immunprophylaxe bei Neugeborenen‹. Abstände zwischen den Impfungen mindestens 4 Wochen; Abstand zwischen vorletzter und letzter Impfung mindestens 6 Monate ** Mindestabstand zwischen den Impfungen 4 Wochen *** jeweils 10 Jahre nach der letzten vorangegangenen Dosis **** jährlich mit dem von der WHO empfohlenen aktuellen Impfstoff ***** Impfung mit Polysaccharid-Impfstoff; Wiederimpfung im Abstand von 6 Jahren * Um die Zahl der Injektionen möglichst gering zu halten, sollten vorzugsweise Kombinationsimpfstoffe verwendet werden. Impfstoffe mit unterschiedlichen Antigenkombinationen von D/d, T, aP, HB, Hib, IPV sind bereits verfügbar. Bei Verwendung von Kombinationsimpfstoffen sind die Angaben des Herstellers zu den Impfabständen zu beachten. Anmerkungen zu den im Impfkalender aufgeführten Impfungen Diphtherie. Ab einem Alter von 5 bzw. 6 Jahren (je nach Angaben des Herstellers) wird bei Auffrischimpfungen und zur Grundimmunisierung ein Impfstoff mit reduziertem Diphtherietoxoid-Gehalt (d) verwendet, in der Regel kombiniert mit Tetanustoxoid oder weiteren Antigenen. Haemophilus influenzae Typ b (Hib). Nach dem 12. bzw. 15. Lebensmonat (Packungsbeilage beachten) ist eine einmalige Hib-Impfung ausreichend. Ab einem Alter von 5 Jahren ist eine Hib-Impfung nur in Ausnahmefällen indiziert (z.B. bei funktioneller oder anatomischer Asplenie). Für die einzelnen Impfungen bei der Grundimmunisierung sollte – wenn möglich – ein Impfstoff mit gleichem Trägerprotein verwendet werden. Wenn jedoch nicht bekannt ist, mit welchem Impfstoff zuvor geimpft wurde, weil der Handelsname nicht – wie erforderlich – dokumentiert wurde, muss die Grundim- munisierung nicht erneut begonnen werden, sondern kann mit jedem Hib-Impfstoff fortgesetzt werden. Postexpositionelle Hepatitis-B-Prophylaxe bei Neugeborenen von HBsAg-positiven Müttern bzw. von Müttern mit unbekanntem HBs-Ag-Status. Entsprechend den Mutterschafts-Richtlinien ist bei allen Schwangeren nach der 32. Schwangerschaftswoche, möglichst nahe am Geburtstermin, das Serum auf HBsAg zu untersuchen. Ist das Ergebnis positiv, dann ist bei dem Neugeborenen unmittelbar post partum, d.h. innerhalb von 12 Stunden nach der Geburt, mit der Immunisierung gegen Hepatitis B zu beginnen. Dabei werden simultan die erste Dosis HBImpfstoff und HB-Immunglobulin verabreicht. Die begonnene HB-Grundimmunisierung wird einen Monat nach der 1. Impfung durch eine 2. und sechs Monate nach der 1. Impfung durch eine 3. Impfung vervollständigt. Bei Neugeborenen inklusive Frühgeborenen von Müttern, deren HBsAg-Status nicht bekannt und bei denen noch vor bzw. sofort nach der Geburt die serologische Kontrolle nicht möglich ist, wird unabhängig vom Geburtsgewicht ebenfalls unmittelbar post partum die Grundimmunisierung mit HB-Impfstoff begonnen. Bei nachträglicher Feststellung einer HBsAg-Positivität der Mutter kann beim Neugeborenen innerhalb von 7 Tagen postnatal die passive Immunisierung nachgeholt werden. Nach Abschluss der Grundimmunisierung von Neugeborenen ist eine serologische Kontrolle erforderlich (s. a. Epid. Bull. 10/2000 und 8/2001). Masern, Mumps, Röteln (MMR). Die Impfung gegen Masern, Mumps und Röteln sollte mit einem Kombinationsimpfstoff (MMR-Impfstoff) durchgeführt werden, in der Regel im Alter von 11 bis 14 Monaten. Bis zum Ende des 2. Lebensjahres soll auch die zweite MMR-Impfung erfolgt sein, um den frühestmöglichen Impfschutz zu erreichen. Steht bei einem Kind die Aufnahme in eine Kindereinrichtung an, kann die MMR-Impfung auch vor dem 12. Lebensmonat, jedoch nicht vor dem 9. Lebensmonat, erfolgen. Sofern die Erstimpfung vor dem 12. Lebensmonat vorgenommen wurde, muss die MMR-Impfung bereits zu Beginn des 2. Lebensjahres wiederholt werden, da persistierende maternale Antikörper im 1. Lebensjahr die Impfviren neutralisieren können. Die Eliminierung der Masern ist ein erklärtes Ziel der deutschen Gesundheitspolitik. Masern können eliminiert werden, wenn die Durchimpfungsrate gegen Masern bei Kindern mehr als 95% erreicht. Diesem Ziel sind bisher die Länder nahe gekommen, die eine zweimalige Impfung im Kindesalter empfehlen und dabei hohe Durchimpfungsraten realisieren. Hierzu gehören die skandinavischen Länder, Großbritannien, die Niederlande und die USA. Die STIKO empfiehlt eine zweite MMR-Impfung seit 1991. Mit dieser sollen Immunitätslücken geschlossen werden. Die zweite Impfung kann bereits 4 Wochen nach der ersten erfolgen. Bei Mädchen wird mit der zweimaligen MMRImpfung auch der unverzichtbare Schutz vor einer 46 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 Röteln-Embryopathie weitgehend gesichert. Selbst bei anamnestisch angegebener Masern-, Mumps- oder Rötelnerkrankung sollte die zweite MMR-Impfung durchgeführt werden, da anamnestische Angaben über eine Masern- oder Rötelnerkrankung ohne mikrobiologisch-serologische Dokumentation der Erkrankungen in der Regel unzuverlässig und nicht verwertbar sind, und es in der Fachliteratur keine Hinweise auf vermehrte Nebenwirkungen nach mehrmaligen Masern-, Mumps- oder Rötelnimpfungen gibt. Eine Altersbegrenzung für die MMR-Impfung besteht ebenfalls nicht, sie kann daher in jedem Alter erfolgen. Empfohlen wird die MMR-Impfung auch für alle ungeimpften bzw. empfänglichen Personen in Einrichtungen der Pädiatrie sowie in Gemeinschaftseinrichtungen für das Vorschulalter und in Kinderheimen. Eine zusätzliche monovalente Rötelnimpfung für Mädchen ist nicht erforderlich, wenn bereits zwei Impfungen mit MMR-Impfstoff dokumentiert sind. Ist nur eine MMR-Impfung vorausgegangen, muss die zweite möglichst frühzeitig bei allen Kindern und Jugendlichen nachgeholt werden. Bei der Jugendgesundheitsuntersuchung ist sicherzustellen, dass alle Jugendlichen zwei MMR-Impfungen erhalten haben. Pertussis. In Anbetracht der Pertussis-Situation in Deutschland und der Schwere des klinischen Verlaufs Auf S. 929 eines Keuchhustens im Säuglingsalter ist es dringend geboten, die Grundimmunisierung der Säuglinge und Kleinkinder zum frühestmöglichen Zeitpunkt, d.h. unmittelbar nach Vollendung des 2. Lebensmonats, zu beginnen und zeitgerecht fortzuführen. Empfohlen wird je eine Impfung mit einem Impfstoff, der Pertussis-Antigene (aP) enthält, im Alter von 2, 3 und 4 Monaten und eine weitere Impfung im Alter zwischen 11 und 14 Monaten. Das Nachholen oder die Vervollständigung der Pertussis-Immunisierung wird im Kindes- und Jugendalter empfohlen (Fachinformation beachten). Für bereits viermal gegen Pertussis geimpfte Kinder bzw. Jugendliche ist im Alter von 9 bis 17 Jahren eine weitere Dosis (aP) angezeigt (s. a. Epid. Bull. 17/2000). Poliomyelitis. Der Polio-Lebendimpfstoff, die orale Polio-Vakzine (OPV), wird wegen des – wenn auch sehr geringen – Risikos einer Vakzine-assoziierten paralytischen Poliomyelitis (VAPP) nicht mehr eingesetzt. Zum Schutz vor Poliomyelitis wird nur noch ein zu injizierender Impfstoff, die inaktivierte Polio-Vakzine (IPV), mit gleicher Wirksamkeit empfohlen. Im Alter von 9 bis 17 Jahren soll damit eine Auffrischimpfung erfolgen. Eine mit OPV begonnene Grundimmunisierung wird ebenfalls mit IPV komplettiert. Zeile Interferon-· 2a in Tab. B 13–8 einfügen: Tab. B 13–8. Indikationen von Interferonen Internationaler Freiname Handelspräparat (Eingetragenes Warenzeichen) Indikation Interferon ·-2a Roferon-A Haarzell-Leukämie, Hepatitis B und C, malignes Melanom, chronischmyeloische Leukämie, kutanes T-Zell-Lymphom, Nierenzellkarzinom, follikuläres Non-Hodgkin-Lymphom Interferon ·-2a Pegasys Chronische Hepatitis C mit oder ohne Ribavirin Interferon ·-2b Intron A Chron. Hepatitis B und C, chronisch-myeloische Leukämie, follikuläre Lymphome, Karzinoid, malignes Melanom, multiples Myelom, Kaposi-Sarkom Peginterferon-· 2b PegIntron Chronische Hepatitis C im Fall einer Ribavirin-Unverträglichkeit Interferon-‚ 1a Avonex, Rebif Schubförmige Multiple Sklerose Interferon-‚ 1b Betaferon Schubförmige Multiple Sklerose Interferon-Á 1b Imukin Chronische Granulomatose Interferon-‚, human Fiblaferon Fiblaferon Gel Virusenzephalitis, Herpes zoster generalisatus, virale Innenohrinfekte mit Gehörverlust, undifferenziertes Nasopharynxkarzinom Feigwarzen (Condylomata accuminata bis 3 mm) Inferax Chronische Hepatitis C Interferon-alfacon-1, (sog. ConsensusInterferon) Mit Ausnahme von Interferon-β, human werden alle Interferone rekombinant hergestellt 47 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf Seite 929 rechte Spalte, am Ende des ersten Absatzes einfügen: Mittlerweile steht mit Pegfilgrastim (Neulasta®) auch eine pegylierte Form des Wachstumsfaktors Filgrastim zur Verfügung. Bei der Pegylierung wird Polyethylenglycol kovalent an Filgrastim gebunden, wodurch die renale Ausscheidung des Wachstumsfaktors verzögert wird. Durch die Pegylierung verlän- auf S. 931 rechte Spalte, vor 13.4 Immunsuppressiva einfügen: Glatirameracetat (Copolymer-1, Copaxone®) ist ein Immunmodulator zur Behandlung der Multiplen Sklerose (MS). Es besteht aus einem polymerisierten Gemisch der vier wichtigsten Aminosäuren des basischen Myelinproteins im selben Verhältnis wie im Myelin. Es ist indiziert zur Reduktion der Schubfrequenz bei Patienten mit MS, die ohne Hilfe gehfähig sind und bei denen mindestens zwei Schübe mit neurologischen Funktionsstörungen während der letzten beiden Jahre auftraten. Glatirameracetat eignet sich wie auch die ß-Interferone hauptsächlich für Patienten mit relativ wenig Behinderungen in einem frühen Stadium der Erkrankung. Das Gemisch der verschiedenen Polypeptide moduliert Immunprozesse, die für die Pathogenese der MS verantwortlich gemacht werden. Als Wirkungsmechanismus vermutet man, dass die Substanz das Autoantigen aus seiner Bindung an den MHC-Komplex und an spezifischen Effektorzellen verdrängt und die Zahl von Suppressorzellen, die die auf S. 931 gert sich dementsprechend die Filgrastim-Halbwertszeit von ca. 4–6 Stunden auf 15–80 Stunden, ohne dass sich die pharmakologische Aktivität ändert. Pegfilgrastim kann daher weniger häufig als Filgrastim injiziert werden. Entzündung im ZNS unterdrücken können, erhöht. Mit dem Wirkungseintritt der Substanz ist in frühestens 3 Monate nach Therapiebeginn zu rechnen. Zur Pharmakokinetik am Menschen liegen nur relativ wenig Erfahrungen vor. Subkutan appliziertes Glatirameracetat wird gut absorbiert, der überwiegende Teil der Dosis wird bereits im Gewebe abgebaut. Die Dosierung beim Erwachsenen beträgt einmal täglich 20 mg subkutan. Als Nebenwirkungen sind vor allem Reaktionen an der Injektionsstelle beschrieben. Als „sofortige PostInjektionsreaktion” traten subjektive Luftnot mit Engegefühl im Thorax, Angst, Gefäßerweiterung, Herzjagen und Brustschmerzen auf. Ferner wird häufig über Herzklopfen, Vasodilatation, Synkopen und Tachykardie berichtet. Auch Übelkeit, Angst und Schwitzen werden häufig beobachtet. Über Wechselwirkungen ist noch wenig bekannt. rechte Spalte, vor 13.4.1 ergänzen: 13.4.1 Ciclosporin, Tacrolimus, Sirolimus, Everolimus auf S. 933 rechte Spalte, vor 13.4.2 Glucocorticoide letzte Satz im Abschnitt Sirolimus streichen. Danach einfügen: Everolimus. Everolimus (Certican®) ist ein synthetisches Derivat von Sirolimus und nach diesem der zweite TOR-Inhibitor. Wirkungen, Wirkungsmechanismus und Nebenwirkungen von Everolimus sind daher mit Sirolimus vergleichbar. Die orale Bioverfügbarkeit von Everolimus ist größer und seine Halbwertszeit geringer als die von Sirolimus. Wie dieses wird Everolimus hauptsächlich durch CYP3A4 metabolisiert. Es kann dehalb zu Interaktionen mit CYP3A4-Induktoren oder -Hemmern kommen. (Certican®) 48 Ergänzungen 2002 – Mitte 2005 zu „Mutschler Arzneimittelwirkungen“, 8. Auflage, 2001 auf S. 944 Spalte Ferucarbotran in Tab. B 14–3 aufnehmen: Tab. B 14–3. Eisen-(II/III)-Oxid- und Mangan-haltige MRT-Kontrastmittel Internationaler Freiname Handelspräparat (Eingetragenes Warenzeichen) Applikation Anwendung Ferristen (=Eisenferrit) Abdoscan oral zur Abgrenzung des Darms Eisen-(II/III)-Oxid Endorem i. v. zur Darstellung von Lebertumoren Eisen-(II/III)-Oxid Lumirem oral und rektal zur Markierung des Verdauungstrakts Mangafodipir Teslascan i. v. zur Darstellung von Leberzellkarzinomen und Lebermetastasen Ferucarbotran (Eisen(II/III)-Oxid) Resovist i. v. zur Darstellung fokaler Leberläsionen auf S. 945 rechte Spalte, vor 14.3 Kontrastmittel für die Sonographie: Ferucarbotran (Resovist®) besteht ebenfalls aus paramagnetischen Eisen-(II)- und Eisen-(III)-Oxidpartikeln, die mit einer Carboxydextranschicht ummantelt sind. Dadurch kann eine Aggregation der Eisen-Oxidpartikel verhindert und die Stabilität der kolloidalen wässrigen Suspension garantiert werden. Als Nebenwirkungen wurden hauptsächlich Schmerzen, Vasodilatation (Wärmegefühl) und Parästhesie (Kältegefühl) beschrieben. Bei Patienten mit bekannter Überempfindlichkeit gegenüber Dextran ist Ferucarbotran kontraindiziert.