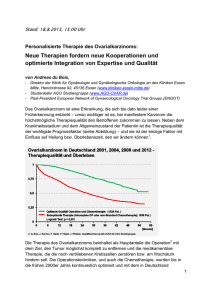
Titel der Arbeit
Werbung

I Aus der Klinik und Poliklinik für Frauenheilkunde und Geburtshilfe der Universität zu Köln Direktor: Universitätsprofessor Dr. med. Peter Mallmann Quantitative Untersuchung der Proteinkinase AKT am Ovarialkarzinom Inaugural- Dissertation zur Erlangung der Doktorwürde der Hohen Medizinischen Fakultät der Universität zu Köln Vorgelegt von Ines Kristin Middel aus Mainz promoviert am 16. Dezember 2009 II Dekan: Universitätsprofessor Dr. med. J. Klosterkötter 1. Berichterstatter: Universitätsprofessor Dr. med. P. Mallmann 2. Berichterstatter: Universitätsprofessor Dr. med. A. Engert Erklärung Ich erkläre hiermit, dass ich die vorliegende Dissertationsschrift ohne unzulässige Hilfe Dritter und ohne Benutzung anderer als der angegebenen Hilfsmittel angefertigt habe; die aus fremden Quellen direkt oder indirekt übernommenen Gedanken sind als solche kenntlich gemacht. Bei der Auswahl und Auswertung des Materials sowie bei der Herstellung des Manuskripts habe ich Unterstützungsleistungen von folgenden Personen erhalten: - Dr. Markus Hoopmann Priv.- Doz. Dr. Dr. Thomas Schöndorf Frau Martina Becker Weitere Personen waren an der geistigen Herstellung der vorliegenden Arbeit nicht beteiligt. Insbesondere habe ich nicht die Hilfe eines Promotionsberaters in Anspruch genommen. Dritte haben von mir weder unmittelbar noch mittelbar geldwerte Leistungen für Arbeiten erhalten, die im Zusammenhang mit dem Inhalt der vorgelegten Dissertation stehen. Die Dissertationsschrift wurde von mir bisher weder im Inland noch im Ausland in gleicher oder ähnlicher Form einer anderen Prüfungsbehörde vorgelegt. Köln, den 17.03.2009 III Die dieser Arbeit zugrunde liegenden Experimente sind von mir mit Unterstützung von Herrn Privatdozent Priv.- Doz. Dr. Dr. Th. Schöndorf und der medizinischtechnischen Assistentin Frau M. Becker durchgeführt worden. Die Proben wurden durch Frau Dr. Gunhild Rot vorbehandelt und eingefroren. Die Krankengeschichten wurden von mir selbst ausgewertet. IV Danksagung Ich danke Herrn Prof. Dr. med. Peter Mallmann für die Möglichkeit, die Arbeit an seiner Klinik zu erstellen. Mein besonderer Dank geht an Dr. med. Markus Hoopmann, der mit viel Geduld alle Korrekturen der Arbeit vornahm und immer für Fragen ansprechbar war. Frau Martina Becker als MTA war eine große Hilfe zur Erstellung der Daten, ebenfalls Priv.- Doz. Dr. Dr. Thomas Schöndorf als damaliger Leiter des Labors. Vielen Dank für die Zusammenarbeit. Mein Dank gilt dem Institut für medizinische Statistik, Informatik und Epidemiologie der Universität zu Köln, welches Hilfestellung bei der Auswertung der Daten leistete. Und zuletzt danke ich meiner Familie, welche immer die größte Unterstützung für mich war. V Inhaltsverzeichnis 0. 1. 1.1. 1.2. 1.3. Abkürzungsverzeichnis .............................................................................. Einleitung .................................................................................................... Ovarialkarzinom ......................................................................................... Das Protein AKT/PKB ............................................................................... Fragestellung .............................................................................................. 1 2 2 13 22 2. 2.1. 2.1.1. 2.1.2. 2.2. 2.3. 2.4. 2.5. Material und Methoden .............................................................................. Geräte und Chemikalien ............................................................................. Verwendete Geräte ..................................................................................... Verwendete Reagenzien ............................................................................. Isolation der Tumorzellen und Protein-Extraktion ..................................... Proteingehaltbestimmung ........................................................................... Western Blots ............................................................................................. Statistische Methoden ................................................................................ 23 23 23 23 24 25 26 27 3. 3.1. Ergebnisse ................................................................................................... Beschreibung des Patientinnenkollektivs und der untersuchten Gewebeproben ......................................................................................................... Tumorhistologie, Grading, FIGO-Stadium der Gewebeproben ................. AKT- und P-AKT-Konzentration ............................................................... Korrelation zwischen den Proteinkonzentrationen von AKT und P-AKT mit verschiedenen Krankheitsparametern .................................................. AKT- und P-AKT-Konzentration in Relation zum FIGO-Stadium ........... AKT- und P-AKT-Konzentration in Relation zum Grading ...................... AKT- und P-AKT-Konzentration in Relation zur Platinsensibilität .......... Korrelation der AKT-Konzentration mit der prä- und postoperativen Ca-125-Konzentration ................................................................................ Korrelation der P-AKT-Konzentration mit der prä- und postoperativen Ca-125-Konzentration ................................................................................ Korrelation der AKT- und der PAKT-Konzentration mit der rezidivfreien postoperativen Überlebenszeit ......................................................... Korrelation der AKT- und der P-AKT-Konzentration mit dem postoperativen progessionsfreien Überleben .................................................... 29 4. Diskussion .................................................................................................. 48 5. Zusammenfassung ...................................................................................... 59 6. Literaturverzeichnis .................................................................................... 61 3.2. 3.3. 3.4. 3.4.1. 3.4.2. 3.4.3. 3.4.4. 3.4.5. 3.4.6. 3.4.7. 29 31 35 36 36 38 40 42 44 45 46 1 0. Abkürzungsverzeichnis Abbildung Aktivierte Proteinkinase Area under the curve Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften B-Zell-Lymphom/Leukämie-2(-Gen) BCL2 BReast CAncer BRCA Computertumographie CT Desoxyribonucleic Acid DNA Enhanced Chemoluminescence ECL und Mitarbeiter (Mitautoren) et al. Firma Fa. Fédération Internationale de Gynécologie et d‘Obstétrique FIGO HER/NEU Human Epidermal Growth Factor Receptor HNPCC Hereditary Nonpolyposis Colon Cancer (Syndrom) HMFG Human Milk Fat Globulin ICD-10 International Classification of Diseases, 10th Revision kDa kiloDalton K-RAS GTP-bindende Protein K-RAS mM milliMolar mg Milligramm ml Milliliter MRT Magnetresonanztomographie MYC Myelocytomatosis viral oncogene homologue µg Mikrogramm nm Nanometer PET Positronen-Emissions-Tomographie PTEN Protein- und Phosphoinositid-Phosphatase rpm rounds per minute; Umdrehungen pro Minute TGFb Transforming Growth Factor beta TNM Tumorklassifikation (T = lokales Wachstum, N = noduläres Wachstum; M = Metastasenbildung) v.a. vor allem Abb. AKT AUC AWMF 2 1. Einleitung 1.1. Ovarialkarzinom Das Ovarialkarzinom ist eine der häufigsten Ursachen für Frauen in der westlichen Welt (v.a. West-Europa und die weiße Bevölkerung der USA) an Krebs zu sterben und rangiert auf Platz fünf der häufigsten weiblichen Tumore. Es erkranken etwa 15 - 20 von 100000 Frauen jährlich neu an einem Ovarialkarzinom. In Deutschland erkranken jährlich etwa 8.000 Frauen daran (Robert Koch Institut 2006) - siehe Abb. 1 und 2. Abbildung 1: Altersspezifische Inzidenz des Ovarialkarzinoms in Deutschland (www.rki.de) Im Jahr 2007 lag die absolute Inzidenz des Ovarialkarzinoms bei 9.950 Personen, was 23,5 Erkrankungsfällen pro 100.000 Einwohnern entspricht (Beckmann et al. 2007). In den Vereinigten Staaten von Amerika wurden im Jahr 2006 durch das Ovarialkarzinom 3 15.310 Todesfälle verursacht (Birrer 2006). In westlichen Industrienationen erkranken jährlich 15 von 100.000 Frauen neu an Ovarialkarzinom, weltweit geht man von einer jährlichen Zahl von Neuerkrankungen in einer Höhe von 205.000 Fällen aus, wobei weltweit jährlich 125.000 Frauen an Ovarialkarzinomen sterben (Tropé und Kaern 2007, IARC 2002). Das Risiko einer Frau im Laufe ihres Lebens an einem Ovarialkarzinom zu erkranken, beträgt 1,5% und das Risiko, an diesem Karzinom zu sterben, erreicht 1%. Die größte Inzidenz bösartiger Ovarialkarzinome findet man bei Frauen im 6. und 7. Lebensjahrzehnt (Robert Koch Institut 2005, Beckmann et al. 2007) (s. Abb. 1 und 2). Abbildung 2: Altersstandardisierte Inzidenz und Mortalität des Ovarialkarzinoms in Deutschland (www.rki.de) Das Ovarialkarzinom gehört zu den gefürchtetsten Krebserkrankungen. 33% aller Malignome der Adnexe und des Uterus sind Ovarialkarzinome. Sein Anteil an Todesfällen durch Genitalkarcinome beträgt fast 75%. Das Ovarialkarzinom stellt damit nach dem Mammakarzinom die Haupttodesursache unter den gynäkologischen Tumoren in Europa und den Vereinigten Staaten dar (Auersbergs et al. 2001, Arbeitsgemeinschaft bevölkerungsbezogene Krebsregister in Deutschland 1997, Chaudhuri et al. 2007, Hashiguchi et al. 2006, Pfleiderer et al. 2001, Sanseverino et al. 2005, Weir et al. 2007, 4 Xia et al. 2006, Yan et al. 2006). Rassenzugehörigkeit, unterschiedliche Lebensgewohnheiten und genetische Determination sind ätiologisch relevant (Martin 2006, Kolasa et al. 2006, Sanseverino et al. 2005). Als protektive Faktoren gelten Schwangerschaften, lange Stillperioden, orale Kontrazeptiva, Hysterektomie und Sterilisation (Chaudhuri et al. 2007). Man geht davon aus, dass jede Ovulation und anschließende Proliferation im Keimepithels des Ovars Bedeutung für das spätere Auftreten eines Ovarialkarzinoms haben kann (Brinton 2007). Als Risikofaktoren gelten zunehmendes Lebensalter, familiäres Vorkommen, Nulliparität/Infertilität, frühe Menarche, späte Menopause und häufiger Wohnortwechsel innerhalb Nordeuropa, Westeuropa und Nordamerika (Chaudhuri et al. 2007, Brinton 2007, Schulz et al. 2007). 90% der Ovarialkarzinome gelten als „sporadisch“, im Gegensatz zu den genetisch fixierten familiär-erblichen Ovarialkarzinomen, die eher jüngere Frauen betreffen. Die meisten dieser letztgenannten Patientinnen tragen das BRCA 1-Gen (Mutation auf Chromosom 17q21) bzw. BRCA 2-Gen (auf Chromosom 13q12-13). Dabei handelt es sich um Tumorsuppressor-Gene, welche mit dem BreastOvarian-Cancer-Syndrom assoziiert sind. Die Häufigkeit dieser Mutation beträgt zwischen 1:8000 bis 1:16000. Bei Trägerinnen dieser Mutation beträgt die Wahrscheinlichkeit, an einem Ovarialkarzinom zu erkranken, 87% bzw. 63%. Seltener ist das Ovarialkarzinom mit einem HNPCC-Syndrom (Hereditary Nonpolyposis Colon Cancer Syndrom) assoziiert (Holinski-Feder et al. 1998, Meden 1996, Pfleiderer et al. 2001, Stratton et al. 1997, Wang-Y et al. 2005, Woenckhaus et al. 2007). Abhängig von Typ und Malignitätsgrad werden auch Veränderungen der Tumorsupressor- bzw. Onkogene KRAS, PTEN, BCL2/HER/NEU, p53, MYC und TGFβ beobachtet (Bell 2005, Wang-HQ et al. 2005b, Woenckhaus et al. 2007). So kann man beobachten, dass 5-7% der Patientinnen mit einem Ovarialkarzinom von einem weiteren Fall in der Familie berichten können. Sind zwei oder mehrere Familienmitglieder betroffen, steigt das persönliche Risiko auch ein Ovarialkarzinom zu entwickeln von 1% auf 40% an. 85-90% der bösartigen Neoplasien des Ovars leiten sich von der undifferenzierten Oberfläche des Ovars ab, dem sogenannten Keimepithel. Man unterscheidet papillär-seröse, muzinöse, endometrioide, hellzellige und undifferenzierte Ovarialkarzinome (Staebler und Diebold 2007) - siehe auch Abbildung 3). 5 Abbildung 3: Histologische Beispiele für Ovarialkarzinome (Nunez et al. 2005). A-C: seröse Ovarialkarzinome E-F: endometroide Ovarialkarzinome J-L: mucinöse Ovarialkarzinome 3 Außerdem gibt es Neoplasien, die aus dem sexuell differenzierten Gonadenmesenchym hervorgehen (z.B. Granulosazelltumore) und solche, die aus den Keimzellen hervor- 6 gehen (z.B. Dysgerminom, Dottersacktumor). Man kann Ovarialkarzinome auch aufgrund ihrer Lokalisation einteilen in Karzinome, die innerhalb des Ovarialstromas unter Einstülpung des Oberflächenepithels entstehen und erst nach Durchbruch durch die Tumorkapsel zur peritonealen Aussaat führen sowie Karzinomen, die an der Oberfläche des Ovars entstehen und so primär zu einer peritonealen Streuung führen, auch wenn nur ein mikroskopischer Tumornachweis gelingt. Schließlich kann man noch Ovarialkarzinome abgrenzen, die mit primärer Lokalisation außerhalb des Ovars auftreten, histologisch aber einem Ovarialkarzinom entsprechen und eine ausgedehnte Peritonealkarzinose bewirken (Pfleiderer et al. 2001). Die Früherkennung des Ovarialkarzinoms ist eines der Hauptprobleme. Im Gegensatz zu anderen humanen epithelialen Tumoren ist das Ovarialkarzinom dadurch charakterisiert, dass die lokale Tumormasse ohne typische Symptome wächst und bei Diagnosestellung bereits in das Peritoneum metastasiert hat (Aletti et al. 2007, Reddy et al. 2004). Wenn Symptome auftreten, sind diese meistens unspezifisch und treten häufig erst dann auf, wenn bereits eine Metastasierung eingesetzt hat. Solche Symptome sind abdominelle Schmerzen, Völlegefühl, Veränderungen der Darmtätigkeit, frühes Sättigungsgefühl und Dyspepsie. In weit fortgeschrittenem Stadium kann es zu Aszites und tastbaren abdominellen Tumoren kommen. Auch eine Beteiligung der Pleura ist dann möglich. Eventuell können bei der Palpation auch vergrößerte inguinale, supraclaviculäre und axilläre Lymphknoten getastet werden. Paraneoplastische Symptome sind selten, z.B. zerebelläre Degeneration, oberflächliche Thrombophlebitis, Dermatomyositis oder Polyarthritis ( Alletti et al. 2007, Martin 2006). Durch den symptomarmen Verlauf und die sehr frühe peritoneale Metastasierung werden 70% der Fälle erst im FIGO Stadium III oder IV erkannt. Hier beträgt die 5Jahres-Überlebensrate bei unter 60jährigen Patientinnen 42% und bei älteren Patientinnen 13%. Es besteht deshalb dringender Bedarf an Methoden der Früherkennung bzw. eines Screenings, um die Prognose verbessern zu können. Hier auf molekularer Ebene einen Ansatz zu finden, wäre ein Schritt von größter Bedeutung. Zur Früherkennung stehen zur Zeit die bimanuelle gynäkologische Untersuchung und der transvaginale Ultraschall zur Verfügung. Beide tragen zur Erkennung eines fortgeschrittenen Ovariakkarzinoms bei, führen aber zu keiner signifikanten Senkung 7 von Mortalität oder Morbidität. Bei prämenopausalen Patientinnen ist eine Aussage zur Dignität vor allem durch funktionelle gutartige Veränderungen am Ovar erschwert. Tumormanifestationen im kleinen Becken, im Bereich des Diaphragmas und der Leberoberfläche sind im MRT sowie im modernen Mehrzeilen-CT teilweise erkennbar. Allerdings kann eine Peritonealkarzinose, die oft den Erstbefund darstellt, häufig erst ab einer Tumorgröße von einigen Zentimetern dargestellt werden. Ein CT sollte bei Verdacht auf ein FIGO IV-Karzinom im Bereich des Thorax und Abdomens erfolgen. Der Tumormarker CA-125 ist bei nur 50% der Patientinnen im Stadium I und II erhöht, bei 80% der Patientinnen im Stadium III und IV. Ab einem Wert von 35 U/ml gilt der CA-125 als erhöht und ein Wert ab 65 U/ml gilt als Hinweis auf ein Ovarialkarzinom. Allerdings besitzt der Tumormarker eine zu geringe Sensitivität, um im Rahmen eines Tumorscreening eingesetzt zu werden. Die Tumormarker CA 72-4 und CA 19-9 sind beim muzinösen Ovarialkarzinom dem CA 125 an Sensitivität überlegen. Bei Patientinnen mit familiärer Belastung ist eine jährliche rektovaginale Untersuchung und ein transvaginaler Ultraschall anzuraten sowie eine nachfolgende CA125 Bestimmung bei diagnostischen Auffälligkeiten (AWMF online 2002, Deutsche Krebsgesellschaft 2000, Pfleiderer et al. 2001). Die Stadieneinteilung gemäß FIGO/TNM-Klassifikation erfolgt ausschließlich nach pathologisch-anatomischen Gesichtspunkten (siehe Tabelle 1). 8 Tabelle 1: Klinische Stadieneinteilung (FIGO) (AWMF online 2002, Deutsche Krebsgesellschaft 2000) FIGO-Stadium Ausbreitung FIGO I Tumor begrenzt auf Ovarien Ia Tumor auf ein Ovar begrenzt; Kapsel intakt; kein Tumor auf der Oberfläche des Ovars Ib Tumor auf beide Ovarien begrenzt; Kapsel intakt, kein Tumor auf der Oberfläche beider Ovarien Ic Tumor begrenzt auf ein oder beide Ovarien mit Kapselruptur, Tumor an Ovaroberfläche oder maligne Zellen im Aszites oder bei Peritonealspülung FIGO II Tumor befällt ein/beide Ovarien und breitet sich im Becken aus IIa Ausbreitung auf und/oder Implantate an Uterus und/oder Tube(n) IIb Ausbreitung auf andere Beckengewebe IIc Ausbreitung im Becken (2a oder 2b) und maligne Zellen im Aszites oder bei Peritonealspülung FIGO III Tumor befällt ein oder beide Ovarien, oder N1 mit histologisch nachgewiesenen Peritonealmetastasen außerhalb des Beckens und/oder regionären Lymphknotenmetastasen IIIa mikroskopische Peritonealmetastasen jenseits des Beckens IIIb makroskopische Peritonealmetastasen jenseits des Beckens, größte Ausdehnung < 2cm IIIc Peritonealmetastasen jenseits des Beckens, oder N1 größte Ausdehnung > 2 cm, und/oder regionäre Lymphknotenmetastasen FIGO IV Fernmetastasen; ein Pleuraerguss muss zytologisch positiv sein, parenchymale Lebermetastasen. N-Stadien Nx regionäre Lymphknoten können nicht beurteilt werden N0 keine regionären Lymphknotenmetastasen N1 regionäre Lymphknotenmetastasen Die Prognose des Ovarialkarzinoms hat sich in den letzten Jahrzehnten nur unwesentlich verändert. Sie ist abhängig von der Ausdehnung/Staging, dem Differenzierungsgrad/Grading, dem postoperativen Tumorrest, dem Lymphknotenstatus, einer Aszitesbildung und dem histologischen Typ (Noske et al. 2007). Auch das Alter und der Allgemeinzustand (z.B. Karnofsky-Index) der Patientinnen muss vor allem unter dem Aspekt der Operabilität berücksichtigt werden, da bei jüngeren Patientinnen in gutem Allgemeinzustand die Möglichkeit eines aggressiveren therapeutischen Vor- 9 gehens besteht (AWMF online 2002, Deutsche Krebsgesellschaft 2000, Stratton et al. 2001). Die Bedeutung des Östrogen/Progesteronrezeptorgehaltes ist nicht sicher einzustufen. In einigen Arbeiten wurde eine prognostische Bedeutung der palliativen endokrinen Therapie beschrieben. Insgesamt ist dieser Effekt eher von geringer Bedeutung (AWMF online 2002, Kommoss et al. 1991). Prognosefaktoren wie DNA-Euploidie, Wachstumsfaktoren, Onkogene, Tumorsuppressorgene und -Rezeptoren (beispielsweise KRAS, PTEN, BCL2/HER/NEU, p53, MYC und DGFβ) sind noch nicht hinreichend geklärt und Gegenstand zahlreicher Studien. Die Therapie des Ovarialkarzinoms besteht zunächst in der operativen Entfernung und sollte in spezialisierten Zentren erfolgen (Fader und Rose 2007, Cadron et al. 2007, Gershenson 2007, Colombo et al. 2007, Martin 2006). Die Primäroperation sollte als „Staging-Laparotomie“ vorgenommen werden. Sie umfasst Abdominallängsschnitt, Hysterektomie, Adnexektomie beidseits (mit hoher Resektion der Ovarialgefäßbündel), infragastrische Omentektomie, Appendektomie, Entnahme von Peritonealbiopsien (ggf. mit Entnahme von Douglas- und Blasenperitoneum), Entnahme von zytologischen subdiaphragmatischen Abstrichen und peritonealer Spülzytologie sowie die Entfernung allen suspekten Tumorgewebes. Bei Befall des Mittel- und Oberbauchs können zusätzliche Darmresektionen sowie oberbauchchirurgische Eingriffe die Rate der postoperativen Tumorfreiheit und somit die Prognose verbessern. Da der postoperativ verbleibende Tumorrest bei Patientinnen mit fortgeschrittenem Ovarialkarzinom den entscheidenden Prognosefaktor darstellt, ist das Ziel des operativen Vorgehens die komplette Tumorresektion, oder zumindest eine Tumorreduktion auf eine Größe unter 1 cm. Hierbei ist für die Größenangabe bei mehreren verbleibenden Tumorherden der größte Durchmesser des Residualtumors maßgeblich (Cadron et al. 2007). Auch bei ausgedehnten Tumormassen und bedeutsamen Stenosen bei inoperablen Tumoren sollte eine Reduktion des Tumors erfolgen, das sogenannte „Debulking“. Es 10 dient palliativen Zwecken und der Verbesserung des Ansprechens auf die Chemotherapie. Bei Patientinnen mit Tumoren im Stadium IV wird eine Prognoseverbesserung diskutiert, die bis heute aber noch fraglich erscheint. Im Stadium FIGO Ia GI ist bei jungen Patientinnen nach diagnostischer Laparotomie eine fertilitätserhaltende Operation unter Erhaltung des Uterus und der kontralateralen Adnexe möglich (Fader und Rose 2007, Colombo et al. 2007). Die pelvine und paraaortale Lymphadenektomie ist bisher eher von diagnostischem als von therapeutischem Nutzen. Sie gilt als umstritten, wird allerdings bei fortgeschrittenen Karzinomen aufgrund des häufigen Lymphknotenbefalls gelegentlich durchgeführt, da dieser Eingriff möglicherweise einen positiven Einfluss auf die Überlebensrate hat (de Poncheville et al. 2001). Die Lymphadenektomie wird in drei Gruppen unterteilt, nämlich die Biopsie palpabler Knoten, die radikale Lymphadenektomie und das selektive Sammeln von Lymphknoten. Das Hauptproblem der Biopsie ist, dass 55% der LymphknotenMetastasen kleiner als 2 mm sind (Petru et al. 1994, Stratton et al. 2001, Wu et al. 1986). Die radikale Lymphadenektomie birgt hohe Komplikationsraten, und sollte nicht durchgeführt werden, solange nicht von einem eindeutigen Benefit dieser Maßnahme auszugehen ist (Stratton et al. 2001, 43). Das selektive Sammeln der Lymphknoten ist für das Staging wichtig, z.B. um die Indikation zur Chemotherapie in Frühstadien abzuwägen. (Burghardt et al. 1991, Stratton et al. 2001). Die „Second-Look“ Operation wird nur noch innerhalb von Studien durchgeführt. Sie dient dem Nachweis des Chemotherapieerfolges und hat keinen Einfluß auf die Prognose. Bei Spätrezidiven (rezidivfreies Intervall nach Beendigung der Primärtherapie > 12 Monate) kann eine Überlebenszeitverlängerung durch eine Rezidivoperation mit Verringerung der Tumormasse erreicht werden. Allerdings bringt dies nur bei Patientinnen mit initial frühem Tumorstadium, makroskopischer Entfernung des Primärtumors und gutem Ansprechen auf die Chemotherapie bei möglichst langem Abstand zur Primäroperation einen Vorteil. Die Sekundäroperation sollte nur durchgeführt werden, wenn man auf eine makroskopische Tumorentfernung hoffen kann. Bei Frührezidiven oder Fernmetastasen wird keine sekundäre Operation empfohlen (Allen et al. 1995, AWMF online 2002, Deutsche Krebsgesellschaft 2000, Le et al. 1998, Onda et al. 1998, Spirtos et al. 1995, Stratton et al. 2001, Vergote et al. 1993). 11 Die Festlegung der postoperativen systemischen Therapie wird durch das Ergebnis der Laparatomie bestimmt. Die Chemotherapie erfolgt nach therapeutischen Standards: bei Patientinnen im FIGO Stadium I profitieren `Low Risk´-Patientinnen (Stadium Ia/Ib bei G1) nicht von einer Chemotherapie. Ihre 5-Jahres-Überlebensrate beträgt bei adäquatem Staging und chirurgischer Primärtherapie mehr als 90%. Eine adjuvante Chemotherapie wird allerdings bei High Risk Patientinnen (Stadium Ia/Ib bei G3, Ic, ausgeprägter Aszites, klarzelliger Subtyp) empfohlen. Hier ist eine Absenkung des Rezidivrisikos zu erwarten, eine Verlängerung der Überlebenszeit ist möglich. Die Chemotherapie in den Stadien II bis IV erfolgt nach Primäroperation mit einer Chemotherapie bestehend aus Carboplatin/Paclitaxel (Trapé und Kaern 2007, Kim et al. 2007, Weir et al. 2007, Yang et al. 2006). Die Standarddosis beträgt bei Cisplatin 5075 mg/m² und bei Carboplatin AUC 4-5 mg/m². Sie wird jeweils im Abstand von drei Wochen über 5-6 Zyklen verabreicht. Eine weitere Dosissteigerung oder längere Behandlungsdauer konnte keine Ergebnisverbesserung erzielen. Ein Ansprechen auf die Chemotherapie erfolgt in 90% der Fälle (Verschwinden des Aszites), Komplettremissionen sind in 30% der Fälle zu beobachten. Neben den allgemeinen Nebenwirkungen (Alopezie, Nausea, Knochenmarkdepression) ist bei Cisplatin auf die Neurotoxizität sowie Nephrotoxizität (ab einer kumulativen Dosis von 300 mg/m²) zu achten. Bei Chemotherapie zur Tumorreduktion erfolgt ein Staging nach 3 Zyklen mit Sonographie, CT, Zysto- und Rektoskopie. Bei Chemotherapie nach kompletter Operation mit einem Tumorrest < 2cm intraperitoneal erfolgt das apparative Staging und Tumormarkerkontrolle nach 6 Zyklen (AWMF online 2002, Bella et al. 1994, Bolis et al. 1997, Decker et al. 1982, De Oliveira et al. 1990, Gadducci et al. 1996, Goerke et al. 2003, Trope et al. 1997, Young 1995, Young 1998). Liegt ein Tumorrezidiv vor, so muss der Einsatz einer weiteren Chemotherapie im Hinblick auf die Sicherung der Lebensqualität der Patientin abgewogen werden, da in diesem Fall keine Aussicht auf einen kurativen Erfolg mehr besteht. Patientinnen mit Platin-sensiblen Tumoren (Ansprechen auf platinhaltige Substanzen in der Primärtherapie, nachfolgend rezidivfreies Intervall von mehr als 12 Monaten Dauer) werden allerdings auch in einer Rezidivsituation auf eine platinhaltige Reduk- 12 tionstherapie ansprechen und zwar desto besser, je länger das rezidivfreie Intervall andauerte. Ein großes Problem der Therapie des Ovarialkarzinoms ist die Resistenz auf die Chemotherapeutika (Sabbatini und Odunsi 2007, Schöndorf et al. 2003, Selvendiran et al. 2007, Shaw und van der Hyden 2007, Yan et al. 2006, Yang et al. 2006). Es kommt häufig relativ früh zu Rezidiven (Delord et al. 2005). Bei platin-resistenten Patientinnen (rezidivfreies Intervall < 6 Monate oder Tumorprogression unter der Chemotherapie) wird keine kombinierte Chemotherapie mehr empfohlen, da sie keine Verbesserung der Protnose verspricht. Nur etwa 20% der Patientinnen sprechen auf eine Monotherapie z.B. mit Etoposid, Topotectan, Paclitaxel und einigen andere Substanzen an (AWMF online 2002). Die Strahlentherapie ist heute weitgehend durch die Chemotherapie verdrängt worden. Eine Indikation besteht lediglich für einzelne inoperable Herde, Tumorrezidive sowie palliativ zur Analgesie von Knochenmetastasen. Eine Strahlentherapie kann außerdem eine Option für Patientinnen sein, die aus internistischen Gründen (z.B. Nierenschäden, Kardiomyopathie) keine Chemotherapie erhalten können. Primär sind lediglich Dysgerminome und Granulosazelltumore strahlensensibel (Goerke et al. 2003). Alternative Therapiemodelle sind beim Ovarialkarzinom limitiert. Zu nennen wäre vor allem die Hormontherapie etwa mit Progesteron, Tamoxifen oder GnRH-Analoga. Diese gewinnt unter anderem bei platin-resistenten Tumoren an Bedeutung, insbesondere wenn wegen der hohen Toxizität auf eine weitere Chemotherapie verzichtet werden muss. Auch bei den genannten Hormonen liegen die Ansprechraten bei unter 20%, sie haben jedoch einen positiven Effekt vor allem auf die tumorinduzierte Anorexie und Kachexie (AWMF online 2002, Emons und Kavanagh 1999, Williams 1998). Weitere Möglichkeiten, die sich im Moment noch in klinischer Erprobung befinden, sind die Gentherapie und die Immuntherapie. Das Prinzip der Immuntherapie umfasst die Erzeugung einer Interaktion zwischen Tumorzellen und Abwehrsystem. Derzeit steht der Einsatz monoklonaler Antikörper aufgrund ihrer Effektivität und der weniger komplizierten Applikationsweise (CA 125, Her2-neu, HMFG-1, Anti-Idiotypen-Antikörper) im Vordergrund. Obgleich die beschriebenen imunologischen Ansätze Erfolg versprechend erscheinen, stehen Ergeb- 13 nisse größerer randomisierter Studien zur Überführung in die klinische Routine zur Zeit noch aus. Ähnlich verhält es sich mit Studien der Gentherapie. Sie umschreibt das Einbringen eines Fremdgens in somatische oder Keimzellbahnen, um die Funktion eines mutierten oder fehlenden Gens zu rekonstruieren oder eine neue genetische Eigenschaft hervorzurufen. Die Hauptzielsetzung konzentriert sich auf eine erhöhte Zellempfindlichkeit gegenüber Zytostatika durch Integration von Fremdgenen (u.a. p53), bzw. die Toleranz der hämatopoetischen Stammzelle gegenüber Zytostatika durch Transfer der MultiDrug-Resistance-Genen zu erhöhen oder auf Prodrug-Aktivierung (AWMF online 2002, Deutsche Krebsgesellschaft 2000). Die Nachsorge des Ovarialkarzinoms erfolgt individuell. Das Ziel ist die Erkennung der Rezidiverkrankung, die Erkennung und Behandlung therapieassoziierter Nebenwirkungen (gastrointestinale Morbidität, Parästhesien, sekundäre Malignome, Hormonausfallserscheinungen), die psychosoziale Betreuung sowie die Verbesserung der Lebensqualität. Während einer Langzeitchemotherapie ergeben sich zwangsläufig kurze Intervalle von einer bis vier Wochen. Bei ausgedehnten Tumoren sollte bis zum zweiten Jahr post operationem alle drei Monate, danach alle fünf Monate eine Kontrolluntersuchung erfolgen. Bei jeder Untersuchung erfolgt eine Spiegeleinstellung (Scheidenstumpfrezidiv), Kolposkopie, Zytologie-Entnahme, vaginale und rektale Palpation, Palpation des gesamten Bauchraumes, inklusive der Leber, Inspektion und Palpation der Mammae. Einmal jährlich wird eine Mammographie empfohlen. Bei Rezidivverdacht ist die Kolposkopie, Zystoskopie, i.v.-Pyelographie, evtl. CT, MRT, PET indiziert (AWMF online 2002, Goerke et al. 2003). 1.2. Das Protein AKT/PKB Da aufgrund der schlechten Prognose des Ovarialkarzinoms auf der Basis von Chemotherapieresistenz und später Diagnose erheblicher Forschungsbedarf besteht, versucht man, die Chemosensilbilität vo Ovarialkarzinomen zu erhöhen. Einen möglichen Ansatz stellen Signaltransduktionswege der Proteinkinase AKT dar. Während der Entwicklung eines Tumors durchläuft die Zelle eine Reihe phenotypischer 14 Charakteristika, welche ihr vor allem ermöglicht schnell und unlimitiert zu proliferieren, in die Umgebung einzuwandern, ohne ihr natürliches Umfeld zu überleben und in andere Gewebe zu metastasieren. Diese Entwicklung ist meist das Resultat wachsender genomischer Instabilität, welche zu einer Hochregulierung (Up-Regulation) der Onkogene, bzw. Hinunterregulierung (Down-Regulation) der Tumor Suppressor Gene führt. In den letzten Jahren sind wichtige Entwicklungsstufen der Tumorgenese mit ihren vielfältigen Signaltransduktionswegen, welche zu unregulierten Prozessen wie Proliferation und Überleben führen, entschlüsselt worden. Die Serin/Threonin Protein Kinase AKT/PKB (Protein Kinase B) hat sich als wichtiger Regulator von unterschiedlichen Prozessen wie Apoptose, Proliferation, Differen- zierung und Metabolisation in der Tumorzelle herausgestellt. Die Unterbrechung der normalen AKT Regulation ist eine in einigen menschlichen Tumoren übliche Erscheinung. Das Enzym spielt anscheinend eine erhebliche Rolle in der Tumorentstehung und der Tumorprogression. Zuerst wurden 1977 zwei Gene (AKT1 und AKT2) isoliert, welche Homologe des viralen Onkogens v-AKT sind. Sie befinden sich am Genlokus 19q13.1-q13.2 und erzeugen eine Form von Leukämie bei Mäusen (Brasil und Hemmings 2001, Staal 1987). V-AKT und seine Homologen kodieren die Proteinkinase B (PKB). Die drei Mitglieder die aus dieser Familie 1991 isoliert werden konnten werden PKBa (AKT1), PKBb (AKT2) und PKBg (AKT3) genannt. Jede Isoform exprimiert eine N-terminale Pleckstrin homologe Domäne (PH) mit ca. 100 Aminosäuren, welche ahnlich wie andere signalisierende Moleküle 3-Phosphoinositid binden. Dies weist darauf hin, dass diese PH Domäne die Bindung von AKT mit 3-Phosphoinositiden ermöglicht. Außerdem befindet sich in dieser Region ein Threonin Rest (Thr308) dessen Phosphorylierung für die Aktivierung von AKT maßgeblich ist. Dieser Region folgt ein hydrophobes C-terminales Ende, ein Serin Rest (Ser473), welcher den zweiten phosphorylierbaren Regulator des Proteins darstellt. Thr308 und Ser473 werden durch Wachstumsfaktoren und andere extrazelluläre Stimuli indirekt phosphoryliert Dies ist der essentielle Schritt, um AKT zu aktivieren (Andelkovic et al. 1999, Alessi et al. 1996, Nicholson und Anderson 2002). 15 Die zelluläre Aktivierung von AKT erfolgt durch eine phosphorylierende Kaskade über das Enzym Phosphoinositide 3-Kinase (PI-3K) welches durch verschiedene Stimuli (insbesonders Insulin, IGF-I, IGF-II, TNFa, PDGF), die an der Plasmamembran binden, selber aktiviert wird. Es phosphoryliert direkt Phosohatidylinositol-4,5biphosphate (PIP2) an dessen 3-OH Gruppe und wandelt es dadurch in die aktive Form PIP3 um, welches AKT an die Plasmamembran bindet und dessen Konfiguration ändert. Dadurch wird indirekt eine Phosphorylierung von AKT ermöglicht. PIP3 wird nicht nur durch die Proteinkinase PI-3K reguliert, sondern auch durch andere Phosphatasen wie PTEN, SHIP oder den Inhibitor LY294002, welche PIP3 durch Entfernung der OH Gruppe inaktivieren (AWMF online 2002, Fraser et al. 2003). Ein „latent membran Protein“(LMP1) des Eppstein-Barr-Virus kann durch Phosphorylierung an p85 zu einer PI-3K Aktivierung führen und somit als Oncogen wirken (Dawson et al. 2003). Die Phosphorylierung von AKT erfolgt durch die Phosphoinositide-Dependent-Kinase1 (PDK-1) am Thr308 und Ser473-Rest und durch PDK-2 am Ser473-Rest. Beide sind Serine/Threonin-Kinasen mit hoher Affinität zu 3-Phosphoinositiden. Meist werden Ser473 und Thr308 simultan stimuliert, es bestehen aber auch unabhängige Regulationsmechanismen, wie eine direkte Aktivierung durch Insulin oder ILK sowie auch Autophosphorylierungsmechanismen (Alessi et al. 1997, Kroner et al. 2000, Hill et al. 2001, Nicholson und Anderso 2002, Stokoe et al. 1997, Toker und Newton 2000). Theoretisch hängt das Aktivitätsniveau von AKT in den Zellen von der Balance zwischen „on“-Signalen (hervorgerufen durch erhöhte PIP3 Werte) und dem Einfluss von „off“-Signalen ab, welche zu AKT-Dephosphorylierung führen. Dies können abgeschwächte PI-3K/PIP3-Konzentrationen sein sowie osmotischer Stress und Ceramide, die durch Dephosphorylierung über Okadaicsäure-sensitive Phosphatasen entstehen (Chen et al. 1999, Nicholson und Anderson 2002, Schubert et al. 2000). Die Proteinphosphatase PTEN, ursprünglich identifiziert als Tumor Suppressor Gen, wird oft als Gegenspieler von AKT angesehen, da es ein AKT direkt vorgeschaltetes Upstream Target (PIP3) inhibiert (Lee et al. 2005a). Mutationen werden regelmäßig in fortgeschrittenen Stadien einiger Karzinome, wie zum Beispiel beim Ovarial-, Mamma-, Endometrium- und Prostatakarzinom, sowie 16 beim Glioblastom gefunden. Beim Ovarialkarzinom findet man in 30-50% der Fälle eine PTEN Verminderung. Seine Funktion besteht vor allem darin, das Zellwachstum zu inhibieren und eine zunehmende Anfälligkeit der Zelle für Apoptose und Anoikisis zu induzieren. Als Substrat gilt vor allem PIP3, welches direkt von PTEN dephosphoryliert wird und somit eine Hemmung der PI-3K Kaskade bewirkt. Es scheinen weitere unabhängige Mechanismen zu bestehen, sowie ein dephosphorylierender Einfluss auf ILK und das AKT Substrat BAD. Ein PTEN Mangel, untersucht an PTEN-null embryonalen Fibroblasten, bewirkt eine PIP3 Zunahme und nachfolgende unregulierte AKT Aktivierung mit unkontrollierter Zell Proliferation. Umgekehrt bedeutet eine PTEN Reexpression eine AKT Down-Regulierung (Brasil und Hemmings 2001, Haas-Kogan 1998, Kurose et al. 2001, Nicholson und Anderson 2002, Nieh et al. 2005, Stambolic et al. 1998, Tang et al. 2006, Whang 1998). AKT ist essentiell für das Überleben der Zelle. So findet man AKT1 und AKT3 ubiquitär in humanen Zellen. AKT2 findet man eher in Insulin abhängigem Gewebe wie braunem Fettgewebe, Skelettmuskel und Leber (Bellacosa et al. 2004). Jede der drei Isoformen der Proteinkinase AKT wird aber auch in speziellen Tumoren exprimiert.. So findet man AKT1 vor allem bei Ovarial-, Magen-, Prostata- und Mammakarzinomen. AKT2 wird vermehrt in Ovarial-, Pankreas-, Magen- und Mammakarzinomen gefunden. Eine Überexpression von AKT3, mit selektiver Aktivierung durch verschiedene Wachstumsfaktoren findet man bei hormonunabhängigen Mamma- und Prostatakarzinomen. Man geht davon aus, dass vor allem eine Erhöhung der AKT2 Konzentration, regelmäßig in zahlreichen Tumoren gefunden werden kann. Ab welchem Wert man allerdings von einer erhöhten Konzentration ausgehen kann und ob es Grenzwerte und Vorhersagewerte gibt, ist kaum dokumentiert (Nicholson und Anderson 2002, Ruggeri et al. 1998). Um die Wirkmechanismen von AKT zu verstehen, muss man die zahlreichen Substrate und deren Funktion kennen. In ruhenden, nicht stimulierten Zellen befindet sich AKT im Zytoplasma, erst nach Aktivierung findet man es an der Plasmamembran, sowie im Zytosol und im Nucleus. Im Nucleus befinden sich zahlreiche Substrate von AKT. Es wird vor allem das Zellwachstum und Zellüberleben gefördert. Dies kann direkt über Hemmung der Apoptose geschehen, wie auch über eine veränderte Regulierung der 17 Transkription von pro- oder anti- apoptotisch wirkenden Genen. Zum Beispiel wirkt die Phosphorylierung von Mitgliedern der Forkhead Familie (FKHR) positiv regulierend auf die Induktion von zellerhaltenden Genen, da diese nicht mehr inhibiert werden können, nachdem FKHR durch Phosphorylierung vom Nucleus in das Zytoplasma transferiert wurde. Dieser Mechanismus scheint beim Mamma Karcinom eine besondere Bedeutung zu haben, da auch das Östrogen abhängige Wachstum durch FKHR Verschiebung ins Zytoplasma verstärkt werden kann (Brunet et al. 1999, Dijkers et al. 2000, Nicholson und Anderson 2002). Bestimmte Mitglieder der Forkhead Familie sind in der Lage die Zelle in der G0 oder G1 Phase zu halten. Dieser Zellzyklus Arrest hindert die Zelle an einer Proliferation und kann durch AKT induzierte Phosphorylierung aufgehoben werden (Burgering und Kops 2002). Viele Substrate werden von AKT stimuliert, um die Proliferation, Anti-Apoptose und das Überleben der Zellen zu förden. Hierzu gehört auch die Tumorgenesis und vermehrte Einsprossung von Gefäßen, um das Wachstum durch gute Versorgung zu fördern. Zu nennen ist hier zum Beispiel NF-κB, welches umgekehrt zu FKHR in den Nucleus transloziert wird, um dort spezielle Gene der Entwicklung, Entzündung, Abwehr und Zell Überleben zu verändern (Chen et al. 1999, Nicholson und Anderson 2002, Romashkova und Makarov 1999, Schubert et al. 2000). AKT phosphoryliert auch direkt Schlüsselproteine der Apoptose-Regulation. Zum Beispiel die Kinase BAD aus der Bcl-2 Familie, welche durch AKT Phosphorylierung gehindert wird ihre üblichen Mechanismen wie Apoptose Anregung und Antagonisierung von zellüberlebensfördernden Mitgliedern der eigenen Familie, auszuführen (Del Peso et al. 2001, Nicholson und Anderson 2002). Dieser Mechanismus fällt zum Beispiel bei Patienten mit Zustand nach Leberteilresektion und anderen Ursachen einer Leber-regeneration auf, bei denen Insulin, IL-6, EGF und HGF die AKT-Kaskade aktivieren und somit unter anderem BAD-vermittelt einer Apoptose entgegenwirken und somit die Regeneration fördern (Hong et al. 2000). Interessant ist auch die Verbindung von AKT zur stressaktivierten Proteinkinase (SAPKs). Diese ist involviert in eine durch ionisierende Strahlen, Hitzeeinwirkung und osmotischen Stress induzierte Apoptose. Nach AKT-Intervention kann die SAPKsinduzierte Apoptose unterdrückt werden (Nicholson und Anderson 2002). 18 Bei direkter Bindung an ein Retinoblasten-Protein (pRB) im Nucleus, welches verschiedene Gene des Zellzyklus inhibiert, kann AKT eine Progression der G1/S Phase induzieren und somit den Zellzyklus beschleunigen (Assoian und Schwartz 2001, Nicholson und Anderson 2002). Ein weiteres Substrat, welches den Zellzyklus verkürzt, ist CyclinD1. Es findet sich in einigen Karzinomen, vor allem im Mammakarzinom und es ist in 50% der Fälle hochreguliert. Durch Verkürzung des Zellzyklus wirkt es direkt auf die Tumorprogression. Außerdem wirkt es auf die Transkription, mRNA-Translation und auf die Proteinstabilität. Es scheint, dass AKT in jedem Schritt regulativ auf CyclinD1 einwirken kann (Gille und Downward 1999, Nicholson und Anderson 2002, Takuwa et al. 1999). Dies erfolgt über eine Hemmung durch Phosphorylierung von GSK-3, wodurch CyclinD1 stabilisiert wird. Außerdem fallen durch abnehmende GSK-3Konzentrationen hemmende Mechanismen auf das β-Catenin weg, was eine Zunahme von CyclinD1 bewirkt. GSK-3 wird außerdem als Substrat in der Insulin-AKT-Kaskade genutzt. Durch dessen Inhibierung wird die Glycogensynthese via Glycogen-Synthase (GS) reguliert. Auch Prozesse der Differenzierung, Proliferation und Trans-formation werden unterstützt (Cohen 1998, Cross et al. 1995, Nicholson und Anderson 2002). Von besonderer Bedeutung ist der Einfluss von AKT`s auf die Wirkungsweise verschiedener Chemotherapeutika. In soliden Tumoren ist die Effiktivität der Chemotherapeutika abhängig von der Vaskularisation, der Proliferationsaktivität und von Veränderungen in der Tumorumgebung wie der Abstand zu versorgenden Gefäßen. Viele Tumore entwickeln eine Hypoxietoleranz, um einer Apoptose zu widerstehen und proliferieren zu können. AKT scheint Einfluss auf diese Adaptionsvorgänge zu haben. Außerdem besitzt AKT eine angiogenetische Fähigkeit. Es reagiert auf proangiogenetische Stimuli wie Angiopoetin und Vascular Endothelial Growth Factor (VEGF). AKT phosphoryliert in aktiviertem Zustand Endothelial Nitric Oxide Synthase (eNOS), welche durch NO Freisetzung eine Neovaskularisation und VEGF induzierte Endothelzellmiigration, Gefäßtonusveränderung und Differenzierung erlaubt (Bellacosa et al. 2004, Dimmeler et al. 1999, Fulton et al. 1999). Weitere Schlüsselregulatoren, welche auf Hypoxie reagieren und durch Insulin und Wachstumsfaktoren verstärkt werden, können AKT-abhängig die Transkription und Translation verschiedener Gene der Angiogenese und des Glukose-Transports 19 induzieren und eine verstärkte VEGF Produktion auslösen (Cheng et al. 2002, Gerber et al. 1998, Nicholson und Anderson 2002, Zhong et al. 2000, Zundel et al. 2000). Im Glukose-Metabolismus wirkt AKT stimulierend auf die Glycogen-Synthese durch GSK-3-Phosphorylierung. Außerdem unterstützt AKT die Translokation von GLUT4 an die Plasmamembran und induziert die GLUT1 Gen Transkription. Desweiteren soll AKT die Toleranz der Zellen gegenüber Minderernährung fördern. Dies kann es über das Substrat mTOR, welches oft in Tumoren aktiviert ist und Ernährungsmechanismen im Tumor verändert und die Zellproliferation fördert, sowie eine chromosomale Instabilität hervorruft (Bellacosa et al. 2004). All diese Mechanismen erlauben es Tumoren, unter hypoxischen Bedingungen zu proliferieren (Nicholson und Anderson 2002, Izuishi et al. 2000). Speziell beim Ovarialkarzinom ist das Ansprechen auf die Chemotherapie, vor allem mit platin- und taxolhaltigen Substanzen, von essentieller Bedeutung für die Progression des Tumors und somit für das Überleben der Patientinnen. Der Wirkmechanismus von Platin beruht auf dessen Fähigkeit der Vernetzung von DNASträngen und Induzierung der Apoptose. Es scheinen auch Zusammenhänge zwischen der Expression von spezifischen Genen für den Zelltod und der Down-Regulierung der für das Zellüberleben wichtigen Gegenspieler von Platin zu existieren (Cheng et al. 2002, Li et al. 2001). Bei vielen Patientinnen besteht eine Resistenz gegen Platin. Diese ist in 75% der Fälle mit einer Resistenz gegen Taxol verbunden und stellt einen limitierenden Faktor in der adjuvanten und palliativen Therapie dar. Die Mechanismen dieser Chemoresistenz sind multifaktoriell. Es muss unterschieden werden zwischen primär bestehender und im Laufe der Therapie entstandener Resistenz. Man geht von verändertem Transport der Substanzen in die Zellen aus und einer speziellen Gen Expression (Multi-Drug- Resistance-Gen). Die erhöhte Rate von DNA-Reparaturmechanismen und eine erniedrigte Rate von chemotherapie-induzierten DNA und makromolekularen Schäden spielen eine Rolle (Cheng et al. 2002, Reed et al. 1996). Also eine Vielzahl von intraund extrazellulären Resistenz-Faktoren gegen zytotoxische Substanzen. Die empfindliche Balance zwischen Tumorsuppressorgenen und die für ein verlängertes zelluläres Überleben wichtigen Faktoren, wie Inhibitoren der Apoptose und Bestandteile der gesamten PI-3K/AKT-Kaskade, scheint im Zusammenhang mit der 20 Chemoresistenz gestört zu sein. Man findet veränderte Proteinzusammensetzungen (unter anderem eine Hochregulierung von AKT) in verschiedenen menschlichen Karzinomen. Dies lässt teilweise Rückschlüsse auf eine schlechtere Prognose zu (Cheng et al. 2002, Sun et al. 2001). Eine veränderte Platinsensitivität wird durch viele molekularbiologische Mechanismen hervorgerufen. Man findet in Platin resistenten Tumoren zum Beispiel häufig eine erhöhte Konzentration von X-linked Inhibitor of Apoptosis Protein (XIAP). Daraus folgt eine herabgesetzte Apoptoserate der Tumorzellen. Der Mechanismus beruht auf einer Hemmung der Spaltung von AKT und einer erhöhten AKT-Phosphorylierung und somit einer AKT-Aktivierung. XIAP vermittelt das von AKT verwendete Substrat. Daraus resultiert eine indirekte Platinresistenz. Eine Reduzierung von XIAP sensibilisiert die Zelle für Platin. Platin bewirkt aber auch direkt eine XIAP-Reduzierung, eine AKT-Spaltung und somit eine Induktion der Apoptose. In platinssensiblen Zellen beobachtet man eine hohe Rate von AKT-Spaltung, nicht dagegen in platinrresistenten Zellen, in denen die AKT-Spiegel erhöht sind. Viele andere AKT-induzierte Wege wirken hier ähnlich und meist hemmend auf die Apoptose. Man kann festhalten, dass man bei Tumoren, die eine erhöhte Resistenz gegen Platin und eventuell auch gegen Taxane aufweisen, erhöhte AKT-Spiegel findet. Diese Tumorzellen gehen nicht in eine platininduzierte Apoptose über (Cheng et al. 2002, Fraser et al. 2003, Li et al. 2001, Page et al. 2000, Sasaki et al. 2000). Weitere Substratbindungsstellen, welche die Tumorgenese potentiell verändern können, sind Östrogen- und Androgen-Rezeptoren. In steroidabhängigen Tumoren lassen sich wegen herabgesetzter Stimulierbarkeit oft nach einiger Zeit Resistenzen gegen die antihormonale Therapie feststellen mit konsekutiver hormonunabhängiger Proliferation. AKT kann hier als Mediator fungieren, indem es eine Stimulierung der Rezeptoren durch EGF und IGF-1 fördert. Außerdem phosphoryliert AKT direkt Steroidrezeptoren, die dadurch hormonunabhängig aktiviert werden und eine Resistenz für antihormonelle Substrate entwickeln (Campbell et al. 2001, Nicholson und Anderson 2002). Bei speziellen Mäusearten, welche phänotypisch einen AKT-Mangel besitzen, kann man eine kürzere Zellüberlebenszeit beobachten mit beeinträchtigter Insulin Wirkung in der Leber und im Skelett Muskel (Brasil und Hemmings 2001, Cho 2001). 21 Betrachtet man all diese Mechanismen wird klar, dass AKT eine wichtige Rolle in weiten Bereichen der Tumorgenese spielt. AKT wirkt direkt auf Tumorprogression, Hyperplasie und Zellüberleben. Aber vor allem als Glied langer Enzymketten und in Kooperation mit anderen onkogenen Wegen wirkt es auch zum Anstoß von Dysplasie und Neoplasie. Cheng et al. (2002) fand erhöhte AKT Spiegel bei fortgeschrittenen Tumoren mit hohem Grading, allerdings nicht in frühen Tumorstadien. Dies scheint die These zu untermauern, dass AKT eher die Tumor Progression unterstützt als die Initiation (Tang et al. 2006, Nieh et al. 2005). Es gibt verschiedene experimentelle Ansätze, um alle diese Erkenntnisse therapeutisch zu nutzen. Ein wichtiger Ansatz ist die Sensitivität gegenüber den Chemotherapeutika Platin und Taxol zu erhöhen, da sie die Chemotherapeutika der ersten Wahl beim Ovarialkarzinom sind und die Chemoresistenz eines der Hauptprobleme in der Therapie darstellt. Der Versuch der Genmanipulation, vor allem über adenovirale Vektoren, scheint schon geglückt zu sein. Man ersetzt zum Beispiel Tumor Suppressorgene oder initiiert einen Transfer von Genen der Multi-Drug-Resistance. Auch eine direkte XIAP Reduzierung ist auf diesem Weg möglich, was eine bedeutende Rolle in der Überwindung der Platinresistenz spielt (Cheng et al. 2002, Collinet et al. 2000). Eine Reduzierung von AKT sensibilisiert die Zelle direkt für eine Chemotherapie. Außerdem bewirkt man durch die Reduzierung eine höhere Apoptoserate und eine Verminderung der Proliferation mit verlangsamtem Zellzyklus. Außerdem würde eine vermehrte Anfälligkeit der Zelle für Hypoxie und die Reduktion von einsprossenden Gefäße mit Unterversorgung an nährenden Substanzen, den Metabolismus der Tumorzellen herabsetzen (Cheng et al. 2002, Datta et al. 1999, Sun et al. 2001). Versuche zur AKT-Reduktion beinhalten die Verabreichung von AKT-Antikörpern und anderen Peptiden, welche die Aktivierung von AKT verhindern und in die Downstream Kaskade eingreifen. Es ist ein pharmakologischer AKT-Inhibitor auf dem Markt (Calbiochem), genaue Studien zur Wirkung stehen allerdings noch aus (Cheng et al. 2002). 22 1.3. Fragestellung Die zahlreichen Signaltransduktionswege der Proteinkinase AKT sind vielfach untersucht und erscheinen uns mittlerweile als gut bekannt. Durch ihre maßgebliche Beteiligung am Tumorgeschehen, vor allem an der Tumorinduktion, Entwicklung und Progression, muss ein vermehrtes Augenmerk auf die klinische Relevanz gelegt werden. Wir isolierten das Protein AKT und dessen phosphorylierte Form P-AKT aus 99 Tumorproben von Ovarialkarzinompatientinnen. Die Konzentrationen wurden mit verschiedenen biologischen Parametern und der Rezidivfreiheit in Bezug gesetzt, um somit einen Zusammenhang Krankheitsverlauf zu erfassen. zwischen Konzentration von AKT und dem 23 2. Material und Methoden 2.1. Geräte und Chemikalien 2.1.1. Verwendete Geräte Es wurden die nachfolgend aufgeführten Geräte eingesetzt: - Zentrifuge Biofuge 15 (Fa. Heraeus, Hanau) - Spannungsgeräte Power Pack 300 und 200, Imaging Densitometer GS 690, Criterion Elektrophoresekammer, Criterion Blotter und Microplate Reader Model 550 (Fa. Bio--Rad, München) - Filmentwicklung: Curix 60 (Fa. AGFA, Leverkusen) - Eindampfen: Concentrator 5301 (Fa. Eppendorf, Köln) - Ultraschallbad Electric Sono Gerätetyp TK 52 (Fa. Bandelin, Berlin) - Mikro-Dismembrator (Fa. Braun, Melsungen) 2.1.2. Verwendete Reagenzien Es wurden folgende Reagenzien benutzt. - TPER™ Tissue Protein Extraction Reagent (Fa. Pierce, Rockfort, IL, USA) - 12,5% Tris-HCL Criterion Precast Gel, Criterion Blotting Sandwiches, Protein Standard Marker, DC Protein Assay, Protein Assay Standard 1, Precision Protein Standards (sämtlich Fa. Bio Rad, München) - ECL-Marker, ECL-System, Chemilumineszenz-Filme (Fa. Amersham, Braunschweig) - Antikörper anti-AKT (Ser 473), polyclonal anti rabbit (Fa. Cell Signaling, Taufkirchen) - Antikörper anti-P-AKT (Ser 473), polyclonal anti rabbit (Fa. Bio Source, Solingen) - zweiter Antikörper anti rabbit und HRP (Fa. Amersham, Braunschweig) - Entwickler, Fixierer, Algezid II special biocide (Fa. AGFA, Leverkusen) 24 Alle weiteren Chemikalien der Qualitätsstufen "zur Analyse" oder "reinst" wurden von der Fa. Merck (Darmstadt), der Fa. Sigma (Taufkirchen), der Fa. Serva (Heidelberg) und der Fa. Fluka (Neu-Ulm) bezogen. 2.2. Isolation der Tumorzellen und Protein-Extraktion Die Tumorzellen wurden aus frischem Tumorgewebe, malignem Aszites bzw. Pleuraerguß oder metastatisch befallenen Lymphknoten gewonnen. Eine vorläufige Tumorzellsuspension wurde direkt nach Gewinnung des Materials weiterverarbeitet oder, wenn eine direkte Verarbeitung nicht erfolgen konnte, bei -70°C gelagert. Bei der Aufbearbeitung wurden die Zellen "geerntet", d.h. das Tumorgewebe wurde mechanisch im Dismembrator homogenisiert und daraufhin bei 5.000 rpm für fünf Minuten dichtezentrifugiert. Der Überstand wurde verworfen. Es folgte ein Waschschritt in sterilem PBS (pH-Wert 7,2-7,4; 0,9% NaCl, 8 mM KH2PO4, 1 mM KH2PO4) und eine erneute Zentrifugation bei 5.000 rpm für fünf Minuten. Auch hier wurde der Überstand verworfen. Die auf diese Weise gewonnenen Pellets könnten bei -70°C gelagert werden. Bei endgültiger Verwendung der Proben zur Proteinisolation, wurden diese mit Extraktionspuffer (100 ml TPER; 0,3 µ/ml Proteinaseinhibitor Aprotinin und 1 mM ß-Mercaptoethanol) versetzt und für 15 Minuten bei 4°C inkubiert. Pro 100 mg Tumorgewebe wurde 1 ml Extraktionspuffer zugegeben. Bei Aszites bzw. Pleuraerguß wurde 1 ml tumoröse Flüssigkeit mit 1 ml Extraktionspuffer versetzt. Auch diese Proben konnten dann bei -20°C gelagert werden. Die Zellen wurden im Ultraschallbad für 5 Minuten lysiert und anschließend bei 1400 rpm fünf Minuten dichtezentrifugiert. Der gewonnene Überstand wurde in ein frisches Eppendorf-Cup überführt und bis zur Messung der Proteinkonzentration, bzw. Weiterverarbeitung in der Western Blot Analyse bei -20°C gelagert. 25 2.3. Proteingehaltbestimmung Da bei jeder Probe eine festgesetzte Menge von 50 µg Gesamtprotein eingesetzt werden sollte, musste die Proteinkonzentration jeder einzelnen Probe bestimmt werden. Dies erfolgte unter Verwendung des Folin-Phenol-Reagenz. Dazu wurde das Protein mit Kupfer vorbehandelt, welches nun eine Reduktion des Folin-Reagenz bewirkte. Geschieht diese Reaktion der Proteine in alkalischem Milieu, kann man eine Farbentwicklung beobachten. Eine Farbentwicklung in Abwesenheit von Kupfer entsteht durch den Tyrosin- und Tryptophangehalt des Proteingemisches. Durch die Zugabe von Kupfer kommt es unter alkalischen Bedingungen zu einer 3-15fachen Erhöhung der Farbentwicklung, welche den generellen Proteingehalt des Gemisches widerspiegelt. Kupfer hat jedoch nur eine geringe Wirkung auf die Farbentstehung durch freies Tyrosin und Tryptophan. Die Farbentwicklung ist nach 5-10 Minuten maximal und verändert sich innerhalb einer Stunde nur unwesentlich (Lowry et al. 1951). Die Messung der Proteinkonzentration erfolgte unter Verwendung eines Standards. Die drei Komponenten des verwendeten Protein Assay sind folgende: - Reagenz A: alkalische Kupfertartrat-Lösung, - Reagenz S: Natriumdodecylsulfat, - Reagenz B: Folin Reagenz. Aus den Reagenzien A und S wurde kurz vor Gebrauch im Verhältnis S zu A von 1:50 das Reagenz A´ hergestellt. Die Verdünnungsreihe des Standards und der Proben wurden jeweils in Doppel- bestimmung in einer Mikrotiterplatte angelegt. Zur Kontrolle wurde der Standard ein zweites Mal aufgetragen und wie eine Probe behandelt. Die Verdünnung erfolgte bis 1:16 beim Standard und bis 1:128 bei den Proben. Dazu mussten 10 µl Probe bzw. Standard in Reihe A pipettiert werden. Darauf wurden 50 µl A`-Reagenz pipettiert. In alle restlichen Vertiefungen wurden 30 µl A`-Reagenz vorgelegt. Es folgte die Verdünnung durch Entnahme von 30 µl aus Reihe A, die in Reihe B pipettiert wurde. Nach kurzem umrühren wurden nun aus Reihe B 30µl entnommen und Reihe C zugefügt usw. Zum Schluß wurde 200 µl Reagenz B in alle Vertiefungen eingefügt. 26 Die charakteristische blaue Färbung entwickelte sich maximal nach einer Stunde. In Dunkelheit wurde der Proteingehalt anhand der Extinktion bei 655 nm quantifiziert 2.4. Western Blots Die Auftrennung der Proteine aus dem Lysat erfolgte in einer diskontinuierlich denaturierenden 12,5%igen Polyacrylamid-Gelelektophorese. Dafür wurden jeweils 50 µg Gesamtprotein pro Probe aufgetragen, aufgenommen in 10 µl Aqua destillata. Bei eingedampften Proben und bei Proben, dessen Volumen unter 10 µl bei 50 µg Gesamtprotein lag, wurde mit Aqua dest. auf 10 µl aufgefüllt. Zusätzlich wurden 10 µl SDS Page-Probenpuffer (6% ß-MCE; 6% SDS; 20% Glycerin; 0,6% Bromphenolblau; 0,2M DTT) hinzupipettiert. Es entstand ein Gesamtvolumen von 20 µl für jede Geltasche. Ein Molekulargewichtsmarker wurde mitpipettiert, sowie ein ECL-Marker und Positiv-Kontrollen. Die Proben wurden anschließend im Thermobad für 5 Minuten bei 95°C denaturiert und 10 Sekunden zentrifugiert, um das genaue pipettieren der Volumina zu gewährleisten. Es folgte die Pipettierung der Proben in das Gel (0,5 M Tris Criterion Precast Gel) und Einfüllung des SDS-Gel-Laufpuffers (25 mM Tris-Base; 0,19 M Glycine; 0,1% SDS bei pH 8,3). Angeschlossen an das Spanungsgerät Power Pack 300 wurden die Proben für 10 Minuten bei 50 Volt auf einer Ebene gesammelt und danach bei konstant 120 Volt für 110 Minuten getrennt. Im Anschluss wurden die aufgetrennten Proteine auf eine Nitrozellulose-Membran transferiert. Dies erfolgte in der Criterion-Blotting Kammer in Transferpuffer (48 mM Tris-Base; 39 mM Glycine; 20% Methanol; 0,1% SDS) bei 100 Volt am Spannungsgerät Power-Pack 400 für 50 Minuten. Die Expression des Proteins, welches bei 65 kDa nachweisbar ist, erfolgte durch Inkubation mit einem Primär- und Sekundär-Antikörper. Zunächst aber wurde die Nitrozellulose-Membran mit den übertragenen Proteinen in TBS-Puffer (10mM TrisBase; 0,9% NaCl; 0,1% TWEEN 20 bei pH7,4) vorblockiert und 60 Minuten in 6%iger TBS-Milk blockiert, um unspeziefischen Reaktionen vorzubeugen. 27 Der verwendete AKT-Antikörper wurde im Verhältnis 1:4000 in TBS-Puffer gelöst (der P-AKT-Antikörper hingegen in einem Verhältnis von 1:5000) und pro Membran mit 4 ml aufgetragen. Über Nacht wurde bei 4°C inkubiert. Anschließend wurde der Blot dreimal für fünf Minuten in TBS gewaschen. Um die Bindung des primären Antikörpers an das zu bestimmende Protein nachzuweisen, inkubierte man die Membran für 60 Minuten mit einem sekundären Peroxidase-gekoppelten-Antikörper (Verdünnung des zweiten Antikörpers im Verhälsnis 1:10000, Verdünnung des HRP 1:1000), gelöst in 30 ml TBS-Puffer bei Raumtemperatur. Erneut wurde der Blot dreimal für fünf Minuten in TBS-Puffer gewaschen. Es folgte die Inkubation des Blots mit der ECL-Detektionslösung (100 mM TRIS/HCl bei pH 8,5; 0,2% p-Comuarinsäure; 0,5% 3-Aminophtalhydrazid (Luminel); 0,03% v/v H2O2) für 30 Sekunden. Die entstehende Chemilumineszenz ist direkt proportional zur Menge des Epitops, da die Lichtentstehung spezifische Antigene nachweist, die an die Rettich-Peroxidase-gekoppelten Antikörper gebunden sind. Dies wurde auf jeweils zwei Röntgenfilmen dargestellt, um die autoradiographische Reproduzierbarkeit des quantitativen Ergebnisses zu gewährleisten. Die Autoradiogramme wurden densitometrisch mit einem Grünfilter aufgenommen und die entstandenen Banden bei 65 kDa mit der Multi-Analyst Software quantifiziert. Die Menge des AKT/P-AKT Proteins, phosphoryliert an Serin 473, konnte somit berechnet und ausgewertet werden. 2.5. Statistische Methoden Die gewonnenen Untersuchungsdaten wurden in das Statistikprogramm Statistical Package for Social Sciences (Fa. SPSS GmbH, München) eingegeben und ausgewertet. Für die beschreibende Statistik wurden absolute (n) und relative (%) Häufigkeit sowie Mittelwert (Med), Standardfehler des Mittelwertes (SEM), Median (Med), Minimum (Min) und Maximum (Max) errechnet. Die intervall- und rationalskalierten Daten der Studie (Alter der Patientinnen, Gesamtproteingehalt, AKT, P-AKT, Ca-125 prä- und postoperativ, Rezidiv- und Progressionsfreiheit postoperativ) wurden zunächst einer Prüfung auf Normalverteilung 28 mittels Kolmogorov-Smirnov-Test unterzogen. Mit Ausnahme des Patientinnenalters und der Progressionsfreiheit musste jedoch eine Gauß-Verteilung der Daten abgelehnt werden. Deshalb wurden generell streuungsunempfindliche, nicht-parametrische Prüfverfahren verwendet (z.B. Kruskal-Wallis-Test, Korrelation nach Spearman). Für die Kalkulation der 5-Jahres-Überlebensraten wurden Überlebenstabellen nach KaplanMeier verwendet. Das Signifikanzniveau wurde auf p < 0,05 festgelegt. Die für die Signifikanzprüfung verwendeten Testverfahren sind im Ergebnisteil benannt. 29 3. Ergebnisse 3.1. Beschreibung des Patientinnenkollektivs und der untersuchten Gewebeproben Das Patientinnenkollektiv bestand aus 85 Frauen mit Ovarialkarzinom, deren mittleres Alter zum Zeitpunkt des ersten operativen Eingriffs 58,4 + 1,3 Jahre (Median 59,5 Jahre) betrug. Die jüngste Patientin war zu diesem Zeitpunkt 27 Jahre und die älteste Patientin 85 Jahre alt. Wie die nachfolgende Abbildung zeigt, stellten die 61-70jährigen Patientinnen die mit Abstand größte Gruppe im Untersuchungskollektiv (33,7%), gefolgt von den 41-50jährigen (23,3%) und den 51-60jährigen (20,9%). Die über 70jährigen Patientinnen stellten 15,1% des Kollektivs. Jüngere Patientinnen im Alter bis zum 40. Lebensjahr waren mit 7% selten vertreten. Abbildung 4: Altersverteilung der 86 Patientinnen in Jahresklassen 30 Im Untersuchungszeitraum vom 18.02.1998 bis 19.07.2001 wurden von den Tumorpatientinnen insgesamt 99 Gewebeproben im Rahmen von 43 primären (43,4%) und 56 sekundären (56,6%) Tumoroperationen gewonnen - vergleiche Abbildung 5. Abbildung 5: Untersuchte Patienten und gewonnene Gewebeproben (GP) Die nachfolgenden Ausführungen beziehen sich nicht mehr auf die ursprüngliche Patientinnenzahl (n=86) sondern auf die Anzahl insgesamt untersuchter Gewebeproben (n=99). 31 3.2. Tumorhistologie, Grading, FIGO-Stadium der Gewebeproben Die Tumorhistologie konnte bei 98 der 99 entnommenen Gewebeproben untersucht werden. Die Mehrzahl der Gewebeproben waren aus einem papillär-serösem Adenom (Zyste) entnommen worden (57,1%). Bei acht Gewebeproben handelte es sich um ein endometroides Karzinom (8,2%) und bei weiteren fünf Gewebeproben um ein muzinöses Karzinom. Jeweils zwei Gewebeproben stammten aus poly-pleomorphen Karzinomen bzw. einem Granulosazelltumor. Eine Gewebeprobe war einem Borderline-Tumor entnommen worden. Bei den verbleibenden 24 Gewebeproben lagen nicht näher klassifizierte Ovarialkarzinome vor (vgl. Abbildung 6). Abbildung 6: Tumorhistologie von 98 untersuchten Gewebeproben (Ca = Karzinom; Tu = Tumor) 32 Das Grading konnte bei 88 Gewebeproben aus den Akten entnommen werden. Bei mehr als der Hälfte der Ovarialkarzinome lag ein G3-Tumor vor, bei knapp einem weiteren Drittel der Fälle handelte es sich um G2-Tumoren. G4-Ovarialkarzinome stellten knapp 10% der untersuchten Gewebeproben, während G1-Tumoren selten vorhanden waren (siehe Abbildung 7). Abbildung 7: Tumor-Grading bei 88 Gewebeproben von Ovarialkarzinomen 33 Aus den Unterlagen von 96 Gewebeproben waren Angaben zum FIGO-Stadium des Ovarialkarzinoms eruierbar gewesen. Dabei waren bei 12,5% dieser Fälle die Tumoren im Stadium FIGO I bis II erkannt worden. Zwei Drittel der Tumoren war erst im Stadium FIGO III und ein weiteres Fünftel erst im Stadium FIGO IV erkannt worden (siehe auch Abbildung 8). Abbildung 8: FIGO-Stadium der auswertbaren 96 Gewebeproben 34 Bei 39 Gewebeproben von insgesamt 21 Patientinnen war aus den Krankenunterlagen ein Zusammenhang zur Platinsensibilität des Tumors herstellbar. Die Voraussetzung für diesen Zusammenhang war die Durchführung einer Platintherapie sowie einer R0Resektion des Ovariakarzinoms. Deshalb wurden aus der Betrachtung die 25 Gewebeproben, bei denen keine Platintherapie vorgenommen worden war, sowie weitere 35 Gewebeproben, bei denen das Ovarialkarzinom nicht R0-reseziert worden war, ausgeschlossen. Von den verbleibenden 39 Gewebeproben zeigte sich die Mehrzahl (87,2%) als platinsensibel, lediglich 7,7% waren platinresistent (siehe Abbildung 9). Abbildung 9: Platinsensibilität bei 39 untersuchten Gewebeproben 35 3.3. AKT- und P-AKT-Konzentration Die chirurgische Therapie der Ovarialkarzinompatientinnen erfolgte bei 43 Gewebeproben als Primäreingriff (43,4%). Bei weiteren 56 Fällen (56,6%) war die Probenahme im Rahmen eines Rezidiveingriffs vorgenommen worden. Es konnten allerdings nicht von allen oben genannten Gewebeproben erfolgreich Untersuchungen hinsichtlich der AKT- und P-AKT-Bande erfolgen. Bei insgesamt 76 Gewebeproben war eine AKT-Bestimmung möglich. Der durchschnittliche AKT-Wert lag bei Geweben, die im Rahmen von Primäreingriffen gewonnen worden waren, etwas höher als bei Proben aus Rezidiveingriffen, jedoch zeigte sich kein signifikanter Unterschied zwischen Gewebeproben aus einer Primäroder Sekundär-operation (Mann-Whitney: p = 0,8746). Demgegenüber war der durchschnittliche P-AKT-Wert von Proben aus Sekundäreingriffen deutlich höher als jener von Gewebeproben aus Primäreingriffen, aber auch hier ergab sich kein signifikanter Unterschied der P-AKT-Bande zwischen den Gewebeproben, die bei einem Primärbzw. Sekundäreingriff gewonnen wurden (Mann-Whitney: p = 0,3408) - siehe auch Tabelle 2. Tabelle 2: AKT- und P-AKT-Konzentration bei primär und sekundär operierten Fällen Parameter AKT P-AKT Primäroperation Min-Max n n mean + SEM Med 32 648,6 + 120,6 524,2 0,14 - 3224,5 44 38 662,7 + 122,8 371,9 17,8 - 3673,6 46 Sekundäroperation mean + SEM Med Min-Max 626,0 + 85,2 445,3 13,6 - 2670,3 604,3 18,2 - 4439,7 752,2 + 115,8 36 3.4. Korrelation zwischen den Proteinkonzentrationen von AKT und P-AKT mit verschiedenen Krankheitsparametern 3.4.1. AKT- und P-AKT-Konzentration in Relation zum FIGO-Stadium Ein Zusammenhang zwischen dem FIGO-Stadium der Gewebeproben und der AKTKonzentration war nicht festzustellen. Es fand sich diesbezüglich kein signifikanter Unterschied (vgl. Abbildung 10 und Tabelle 3). Abbildung 10: AKT-Konzentration in Abhängigkeit vom FIGO-Stadium (73 Gewebeproben); Boxplot-Graphik 37 Es fand sich ebenfalls kein signifikanter Unterschied der P-AKT-Konzentration zwischen den unterschiedlichen FIGO-Stadien der auswertbaren Gewebeproben (siehe Abbildung 11 und Tabelle 3). Abbildung 11: P-AKT-Konzentration in Abhängigkeit vom FIGO-Stadium (81 Gewebeproben); Boxplot-Graphik Tabelle 3: AKT- und P-AKT-Konzentration in Abhängigkeit vom FIGO-Stadium FIGOAKT-Konzentration P-AKT-Konzentration Stadium Min-Max n mean + SEM Med Min-Max n mean + SEM Med FIGO I 906,3 3 18,2 - 1793,5 381,5 + 198,8 427,4 16,4 - 700,6 3 906,0 + 512,5 FIGO II 454,8 + 177,8 324,4 77,8 - 1061,5 7 397,8 + 139,3 304,5 5 17,9 - 1035,8 FIGO III 49 713,4 + 94,8 535,7 0,1 - 3224,5 55 781,7 + 111,8 567,9 22,3 - 4439,7 FIGO IV 16 421,7 + 76,5 317,7 14,5 - 1066,9 16 422,7 + 93,4 302,1 80,8 - 1329,9 p-Wert1) 0,4371 0,2239 1) Kruskal-Wallis-Test 38 3.4.2. AKT- und P-AKT-Konzentration in Relation zum Grading Zwischen den unterschiedlichen Grading-Stufen der auswertbaren OvarialkarzinomGewebeproben fand sich kein statistisch signifikanter Unterschied der AKT-Konzentration (vgl. Abbildung 12 und Tabelle 4). Abbildung 12: AKT-Konzentration in Abhängigkeit vom Grading (68 Gewebeproben); Boxplot-Graphik 39 Es fand sich ebenfalls kein signifikanter Unterschied der P-AKT-Konzentration zwischen den vier Grading-Stufen bei den Ovarialkarzinom-Gewebeproben (siehe Abbildung 13 und Tabelle 4). Abbildung 13: P-AKT-Konzentration in Abhängigkeit vom FIGO-Stadium (73 Gewebeproben); Boxplot-Graphik Tabelle 4: AKT- und P-AKT-Konzentration in Abhängigkeit vom Grading Grading AKT-Konzentration Min-Max n mean + SEM Med G1 3 238,6 + 195,5 68,9 18,4 -628,6 G2 20 511,1 + 142,1 373,2 0,1 - 2670,3 G3 41 679,3 + 95,0 535,7 14,4 - 3224,5 G4 4 1143,9 + 479,6 986,2 248,7 - 2354,8 p-Wert1) 0,1498 1) Kruskal-Wallis-Test P-AKT-Konzentration n mean + SEM Med Min-Max 245,5 62,8 - 330,5 4 221,1 + 61,3 466,5 17,8 - 4439,7 23 699,5 + 197,0 570,6 35,7 - 3673,6 41 729,6 + 105,9 527,9 205,5 - 2039,2 5 792,6 + 330,8 0,2478 40 3.4.3. AKT- und P-AKT-Konzentration in Relation zur Platinsensibilität Die AKT-Konzentration unterschied sich nicht statistisch signifikant zwischen Ovarialkarzinom-Gewebeproben von Tumoren mit unterschiedlicher Platinsensibilität (vgl. Abbildung 14 und Tabelle 5). Abbildung 14: AKT-Konzentration in Abhängigkeit von der Platinsensibilität des Ovarialkarzinoms (30 Gewebeproben); Boxplot-Graphik 41 Auch die P-AKT-Konzentration zeigte keinen signifikanten Unterschied zwischen Gewebeproben aus unterschiedlich platinsensiblen Ovarialkarzinomen (vgl. Abbildung 15 und Tabelle 5). Abbildung 15: P-AKT-Konzentration in Abhängigkeit von der Platinsensibilität des Ovarialkarzinoms (34 Gewebeproben); Boxplot-Graphik Tabelle 5: AKT- und P-AKT-Konzentration in Abhängigkeit von der Platinsensibilität PlatinAKT-Konzentration sensibilität n Min-Max mean + SEM Med resistent 3 599,0 + 341,4 568,0 23,9 - 1205,2 intermediär 2 740,3 + 306,4 740,3 433,9 - 1046,7 sensibel 25 708,5 + 127,5 535,7 0,1 - 2670,3 p-Wert1) 0,9661 1) Kruskal-Wallis-Test P-AKT-Konzentration n mean + SEM Med Min-Max 2 1218,7 + 51,6 1218,7 1167,1 - 1270,4 771,9 872,7 - 1543,7 2 771,9 + 100,8 544,1 22,3 - 4439,7 30 782,5 + 158,7 0,2265 42 3.4.4. Korrelation der AKT-Konzentration mit der prä- und postoperativen Ca-125Konzentration Eine Korrelation zwischen der AKT-Konzentration und der präoperativ gemessenen Konzentration des Tumormarkers Ca-125 lag nicht vor (siehe Abbildung 16). Abbildung 16: Korrelation (Spearman) zwischen AKT-Konzentration und präoperativer Ca-125-Konzentration (57 auswertbare Gewebeproben) Präoperativ war der Ca-125-Serumspiegel bei 10 Proben unter der Norm von 35 U/ml gelegen, bei 66 Gewebeproben jedoch über der Norm. Daten sowohl zum CA-125Spiegel als auch zur AKT-Konzentration lagen präoperativ aber nur bei 57 Proben vor. Bei fünf dieser 57 Proben war der präoperative Tumormarker unauffällig und bei 52 über der Norm gewesen, die korrespondierenden AKT-Konzentrationen betrugen im Durchschnitt 242,4 + 76,4 bzw. 596,3 + 83,8, unterschieden sich aber nicht signifikant (Mann-Whitney: p = 0,2713). 43 Eine Korrelation zwischen der AKT-Konzentration und der postoperativ gemessenen Konzentration des Tumormarkers Ca-125 war ebenfalls nicht feststellbar (siehe Abbildung 17). Abbildung 17: Korrelation (Spearman) zwischen AKT-Konzentration und postoperativer Ca-125-Konzentration (39 auswertbare Gewebeproben) Postoperativ war der Ca-125-Serumspiegel bei 5 Proben unter der Norm von 35 U/ml gelegen, bei 47 Gewebeproben jedoch über der Norm. Daten sowohl zum CA-125Spiegel als auch zur AKT-Konzentration lagen präoperativ aber nur bei 43 Proben vor. Bei drei dieser 43 Proben war der postoperative Tumormarker unauffällig bzw. bei 40 über der Norm gewesen. Die entsprechenden AKT-Konzentrationen betrugen im Durchschnitt 1546,4 + 928,6 bzw. 556,5 + 84,9, unterschieden sich aber nicht signifikant (Mann-Whitney: p = 0,3908). 44 3.4.5. Korrelation der P-AKT-Konzentration mit der prä- und postoperativen Ca-125Konzentration Eine Korrelation zwischen der P-AKT-Konzentration und der präoperativ gemessenen Konzentration des Tumormarkers Ca-125 lag nicht vor (siehe Abbildung 18). Abbildung 18: Korrelation (Spearman) zwischen P-AKT-Konzentration und präoperativer Ca-125-Konzentration (62 auswertbare Gewebeproben) Präoperativ war der Ca-125-Serumspiegel bei 10 Proben unter der Norm von 35 U/ml gelegen, bei 66 Gewebeproben jedoch über der Norm. Daten sowohl zum CA-125Spiegel als auch zur P-AKT-Konzentration lagen präoperativ aber nur bei 62 Proben vor. Bei neun dieser 62 Proben war der präoperative Tumormarker unauffällig und bei 53 über der Norm gewesen, die korrespondierenden P-AKT-Konzentrationen lagen im Mittel bei 341,1 + 89,3 bzw. 653,3 + 92,4, unterschieden sich aber nicht signifikant (Mann-Whitney: p = 0,2424). 45 Es war ebenso keinerlei Korrelation zwischen der P-AKT-Konzentration und der postoperativ gemessenen Konzentration des Tumormarkers Ca-125 ermittelbar (siehe Abbildung 19). Abbildung 19: Korrelation (Spearman) zwischen P-AKT-Konzentration und postoperativer Ca-125-Konzentration (39 auswertbare Gewebeoproben) Postoperativ war der Ca-125-Serumspiegel bei 5 Proben unter der Norm von 35 U/ml gelegen, bei 47 Gewebeproben jedoch über der Norm. Daten sowohl zum CA-125Spiegel als auch zur AKT-Konzentration lagen präoperativ aber nur bei 43 Proben vor. Bei drei dieser 43 Proben war der postoperative Tumormarker unauffällig bzw. bei 40 über der Norm gewesen. Die entsprechenden AKT-Konzentrationen betrugen im Durchschnitt 1614,3 + 1072,8 bzw. 639,0 + 127,1, unterschieden sich aber nicht signifikant (Mann-Whitney: p = 0,3809). 46 3.4.6. Korrelation der AKT- und der PAKT-Konzentration mit der rezidivfreien postoperativen Überlebenszeit Bei 59 Gewebeproben war das postoperative rezidivfreie Überleben der Patientin und der AKT-Gehalt bekannt. Bei 68 Gewebeproben lagen Daten zum P-AKT-Gehalt und gleichzeitig zum postoperative rezidivfreien Überleben vor. Diese Überlebenszeitspanne wurde mit dem AKT- und dem PAKT-Gehalt korreliert. Es zeigte sich jedoch bei keinem der beiden Proteingehalte eine signifikante Korrelation mit der rezidivfreien Überlebenszeit (vgl. Abb. 20). Abbildung 20: Korrelation der AKT- und der P-AKT-Konzentration mit dem rezidivfreien postoperativen Überleben (Monate) 47 3.4.7. Korrelation der AKT- und der P-AKT-Konzentration mit dem postoperativen progessionsfreien Überleben Bei 16 Gewebeproben war das postoperative progressionsfreien Überleben der Patientin und der AKT-Gehalt bekannt. Bei weiteren 19 Gewebeproben lagen Daten zum P-AKT-Gehalt und gleichzeitig zum postoperativen progressionsfreien Überleben vor. Das progressionsfreie Überlebens wurde mit dem AKT- und dem PAKT-Gehalt korreliert, zeigte aber in beiden Fällen keinen signifikanten Zusammenhang (vgl. Abb. 21). Abbildung 21: Korrelation der AKT- und der P-AKT-Konzentration mit dem postoperativen progressionsfreien Überleben (Monate) 4. Diskussion 48 Die Prognose des Ovarialkarzinoms hat sich in den letzten 20 Jahren nicht wesentlich verändert. Trotz optimalem chirurgischem Management und extensiver Chemotherapie mit standardisiertem Behandlungsplan, stellt die späte Tumorerkennung und die Hämoresistenz nach wie vor ein erhebliches Behandlungsproblem dar. Viele Studien der letzten Jahre haben gezeigt, dass der AKT-Signaltransduktionsweg essentiell ist für ein ungehemmtes Zellwachstum und ein verlängertes Überleben der Zellen. Die genannten Wirkmechanismen, die Hemmung der Apoptose und der erhebliche Einfluss auf die Chemoresistenz veranlassen, die Therapiemöglichkeiten einiger Karzinomarten zu überdenken und neue Wege, vor allem auf molekularer Ebene, zu begehen. Während die klinischen und histologischen Prognosefaktoren des Ovarialkarzinoms vor allem nach chirurgischem Staging und Grading gut bekannt sind, muss der biologische Prozess, welcher zu ungehemmtem Wachstum führt, genauer beleuchtet werden. Die Imbalance zwischen Zellwachstum und Apoptose spielt eine wesentliche Rolle und nicht nur die die Wachstumsrate der Tumorzellen. Das Ansprechen auf die Chemotherapie ist im ersten Moment erfreulich, da die Mehrzahl der Patientinnen eine Remission erfährt. Leider entwickeln die meisten Patientinnen im Verlauf eine Resistenz gegen die jeweiligen Chemotherapeutika. Der Mechanismus beruht auf einem verändertem Transport der Medikamente zur den entsprechenden Zellen, der Expression von Multi-Drug Resistance Genen, einer erhöhten DNA-Reparaturrate und einer verminderten DNA- oder makromolekularen Schädigung durch die Chemotherapeutika. Die Studien belegen eine Schädigung der intra- und extrazellulären apoptotischen Wege als einen der Hauptgründe der Chemoresistenz. Eine gestörte Balance zwischen Expression oder Aktivierung und Inaktivierung von Zellüberlebensfaktoren und Tumorsuppressoren, wie zum Beispiel AKT und dessen Up- und DownstreamSubstraten sowie deren Gegenspieler, zeigt die regelmäßige Veränderung der Konzentration dieser Proteine in vielen menschlichen Karzinomen, vor allem auch beim Ovarialkarzinom. 49 Um die Chemotherapie, den Hauptpfeiler der Therapie des Ovarialkarzinoms, wirksamer eizusetzen und deren langfristige Erfolgsrate zu erhöhen, sind deshalb GenManipulationen, zum Beispiel via Adenoviren, oder eine direkte Hemmung von Onkogenen durch Antikörper denkbar. Diese Therapieansätze sind in klinischer Erprobung, vor allem sind Antikörper gegen AKT und Peptide, welche an dessen PHterminales Ende binden, anstatt zum Beispiel PI3K, vorhanden und im klinischen Alltag auf dessen Wirkung zu überprüfen. Durch diesen Therapiezusatz würde die AKT Konzentration und die Aggressivität der Tumorzellen verringert. Außerdem wurde eine vermehrte Apoptoserate in verschiedenen Zelllinien beobachtet, sowie vermindertes Zellwachstum und Überleben der Tumorzellen (Cheng et al. 2002, Liu et al. 2001, Sato et al. 2000). Die meisten Forschungsgruppen, die sich vor allem in den letzten 15 Jahren mit der Proteinkinase AKT und deren Substraten befassten, legten ihr Augenmerk auf die Erforschung der Bedeutung von AKT in vivo und in vitro auf molekularer Ebene. Es wurde dann das Ziel verfolgt, die Signaltransduktionswege langer Enzymketten, AKT vor- und nachgeschaltet, zu erkennen und im Ganzen zu verstehen. Diese Erkenntnisse wurden genutzt, um die Enzyme den verschiedenen Zellstrukturen zuzuordnen, die Gegenspieler zu identifizieren, die Lokalisation innerhalb der Zelle zu bestimmen und die genaue Wirkung auf die verschiedenen Systeme, vor allem in humanen Zellen darzustellen. Nur mit diesen Vorarbeiten lässt sich nun die klinische Bedeutung erarbeiten und es lassen sich die therapeutischen Möglichkeiten für die Zukunft nutzen. Der Weg für neue Optionen in der Therapie bzw. Therapieverbesserungen und unterstützende Maßnahmen sind durch diese Studien möglich. Die wenigen Arbeiten, die sich nun mit der klinischen Relevanz für das Ovarialkarzinom befassen, gehen bevorzugt auf die quantitative Bestimmung von AKT in Tumorzellen ein, im besonderen auch in Ovarialkarzinomzellen im Gegensatz zu normalem Stroma. Die qualitative Bewertung ist jedoch nur im Ansatz erfasst. Das Patientinnenkollektiv der vorliegenden Studie bestand aus 85 Frauen mit Ovarialkarzinom, deren mittleres Alter zum Zeitpunkt des ersten operativen Eingriffs 58,4 Jahre betrug. Insgesamt wurden von diesen Patientinnen 99 Proben gewonnen. Die Mehrzahl der Gewebeproben waren aus einem papillär-serösem Adenom (Zyste) 50 entnommen worden (57,1%). Bei acht Gewebeproben handelte es sich um ein endometroides Karzinom (8,2%) und bei weiteren fünf Gewebeproben um ein muzinöses Karzinom. Jeweils zwei Gewebeproben stammten aus poly-pleomorphen Karzinomen bzw. einem Granulosazelltumor. Eine Gewebeprobe war einem Borderline-Tumor entnommen worden. Bei den verbleibenden 24 Gewebeproben lagen nicht näher klassifizierte Ovarialkarzinome vor. Es wird von einigen Autoren beobachtet, dass vor allem in späten Stadien wie in den FIGO Stadien III und IV erhöhte AKT Konzentrationen zu finden sind (Noske et al. 2007). Bei den Proben der eigenen Patientinnen war das FIGO-Stadium nur bei einem Teil der Proben eruierbar. 12,5% der Proben betrafen Karzinome des FIGO-Stadiums I-II, 2/3tel betrafen FIGO-Stadium III und lediglich 1/5tel das FIGO-Stadium IV. Mehr als die Hälfte der Tumoren betraf G3-Tumoren, knapp ein Drittel G2-Tumoren, knapp 10% G4-Tumoren. G1-Tumoren waren nur selten vorhanden. Allerdings hatte in den eigenen Proben die AKT- und die P-AKT-Aktivität keinen signifikanten Zusammenhang zum FIGO-Stadium oder zu den Gradingstufen gezeigt. Die AKT-Werte waren bei den eigenen Proben aus Primäreingriffen leicht, jedoch nicht statistisch signifikant, höher als bei den Proben aus Rezidiveingriffen. Umgekehrt verhielt es sich mit der P-AKT-Aktivität. Diese war bei den Primäreingriff-Proben, aber ebenfalls nicht signifikant, niedriger als bei den Proben aus Rezidiveingriffen. Damit werden die Befunde von Noske et al. (2007) durch die eigenen Ergebnisse nicht gestützt. Die mittlere AKT-Konzentrationen lagen bei den Proben aus Primäreingriffen bei 648,6 und jenen aus Sekundäreingriffen bei 626,0. Die P-AKT-Konzentrationen betrugen im Mittel bei den Proben aus Primäreingriffen 662,7 und jenen aus Sekundäreingriffen 752,2. Die eigenen Ergebnisse lassen also nicht den Schluss zu, dass Ovarialkarzinome in späteren Tumorstadien höhere AKT- oder P-AKT Konzentrationen aufweisen. Eine erhöhte AKT-Expression bei fortgeschrittenen Ovarialkarzinomen wird von einigen Autoren allerdings beschrieben (Noske et al. 2007, Cristiano et al. 2006). Cristiano et al. (2006) fanden im Rahmen einer Studie an Proben von 92 primären Ovarialtumoren bei 93% der Proben eine AKT-3-Erhöhung. Noske et al. (2007) beschrieben bei 58% ihrer 45 primär invasiven Ovarialkarzinom-Proben eine AKT-Expression, wobei die Werte umso höher ausfielen, je fortgeschrittener der Tumor war. 51 Dies impliziert eine Aufrechterhaltung und Progression des Tumors durch AKT, sowie eine Überführung in aggressivere Phänotypen, mehr denn die Annahme einer Initiation. Hier besteht sicherlich noch Bedarf an Studien mit höheren Fallzahlen, um zu eindeutigen Ergebnissen zu kommen. Eine der ersten Arbeiten, welche sich mit der quantitativen Erhöhung von AKT in malignem Tumorgewebe befasste, stammt von Bellacosa et al. (1995). Sie bestimmten AKT2-Gen-Konzentrationen durch Southern Blot- und Northern Blot-Analyse in Ovarial- und Mammakarzinomgewebe. Bei einem Patientenkollektiv von 132 Patientinnen mit Ovarialkarzinom beobachteten sie eine AKT2-Erhöhung bei 16 Patientinnen (12,1%) im Southern Blot Verfahren. Ein signifikanter Anstieg wurde bei undifferenzierten Tumoren festgestellt (4 von 8 Fällen). In einer gesonderten Gruppe wurden keine AKT-Erhöhungen bei benignen- oder Borderline-Tumoren gefunden (0 von 24 Fällen). In der Northern Blot Analyse fielen nur drei Patientinnen mit AKTErhöhung bei 25 Untersuchten auf. Die Erhöhung wurde im Vergleich zu plazentarer DNA/RNA gemessen und galt bei densitometrischer Messung ab einer Verdopplung der Dichte als erhöht. Die meisten Erhöhungen lagen bei 3-5facher Dichte. Eine leichte AKT Erhöhung ließ sich auch bei Patientinnen über dem 50. Lebensjahr messen. Ebenso fiel bei der Gruppe mit erhöhten Werten eine erhöhte Mortalität im Gegensatz zu der Gruppe mit normalen Werten auf (keine signifikante Erhöhung). Dies lässt auf eine schlechtere Prognose bei AKT-Erhöhung schließen sowie auf eine aggressivere Tumorform, wenn man die AKT-Erhöhung bei undifferenzierten Tumoren bedenkt. Im Vergleich zur oben beschriebenen Messung der DNA und RNA Konzentrationen von AKT2, wurde in der vorliegenden Studie die AKT-Proteinkonzentration durch Western Blot Technik, vor allem aber die aktivierte Form als P-AKT bewertet. Man kann nicht von erhöhten Werten sprechen, da kein „normales“ Gewebe zum Vergleich herangezogen wurde. In dieser Studie beziehen sich die Daten auf Vergleiche innerhalb des Kollektivs, welches ausschließlich maligne Ovarialkarzinome beinhaltete. Innerhalb des Patientenkollektivs wurde die AKT/P-AKT-Konzentration bei einer Fallzahl von insgesamt 84 Patientinnen miteinander verglichen. Zu bedenken ist, dass sich 52 einige Patientinnen im Untersuchungszeitraum mehrfach Operationen unterzogen, so dass die Probenanzahl auf 99 stieg. Die Dichte wurde am Densitometer gemessen und die somit errechnete Konzentration in Beziehung zu Überleben, FIGO Stadium, Grading, Platin-Sensibilität und Erst- und Zweitoperation gesetzt. Es konnte kein signifikanter Unterschied zwischen den AKT-Konzentrationen der verschiedenen Parameter festgestellt werden. Die Cox-Überlebensanalyse, in die nur die 42 Proben einbezogen wurden, bei denen der genaue Zeitraum der Progressionsund Rezidivfreiheit aus den Akten zu ermitteln war, zeigte keine signifikante AKTVeränderung bei kürzeren Intervallen zu Progression und Rezidiv. Es konnten auch keine signifikant höheren Werte bei Patientinnen mit hohem Grading (n=88) oder hohen FIGO Stadien (n=82) ermittelt werden, ebenso wenig bei Patientinnen mit Platinresistenz im Gegensatz zu Patientinnen mit intermediärem Verhalten gegenüber Platin oder gegenüber Platin-sensiblen Patientinnen (n=39). Ein Trend ließ sich nur in der Gruppe der sekundär operierten Patientinnen mit im Durchschnitt höheren P-AKTWerten feststellen. Die eigenen Ergebnisse zeigen, dass man innerhalb eines erkrankten Kollektivs keine signifikanten Konzentrationsveränderungen von AKT/P-AKT beobachten kann, die auf ein vorangeschrittenes Krankheitsstadium oder eine schlechtere Prognose schließen helfen lassen würden. Es lässt sich hier allerdings keine Aussage über eventuelle AKT-Erhöhungen insgesamt im Kollektiv treffen. Da hiervon aber auszugehen ist, dürfen die eigenen Ergebnisse nicht zur Unterschätzung der Bedeutung von AKT in der Tumorgenese beitragen. Es bleibt die Frage, ob man wie bei Bellacosa et al. (1995) überhaupt direkt gesundes mit krankem Gewebe vergleichen kann. Ebenso stellt sich die Frage, ob eine Grenze bei der Verdopplung der Dichte zur Erhöhung zu ziehen ist. Deshalb war ein Vergleich innerhalb eines kranken Patientenkollektivs wichtig, um hier eventuelle Prognoseunterschiede und Unterschiede in der Ausprägung des Tumors herauszuarbeiten. Vergleichbar aufgebaute Studien zu Bellacosa et al. (1995) führten Yuan et al. (2000) und Sun et al. (2001) in Western Blot Technik zur AKT-Proteingehaltbestimmung durch. Wie auch die eigene Arbeit, so benutzten auch sie einen Phospho-Ser-473 AKTAntiköper, durch welchen sich die aktivierte Form von AKT (P-AKT) darstellen lässt. 53 Yuan et al. (2000) wiesen eine regelmäßige P-AKT2 Erhöhung bei Patientinnen mit Ovarialkarzinom nach. Hohe P-AKT Werte fanden sie in 42 von 91 Fällen (46%). Es wurde von ihnen aber nicht definiert, ab wann die P-AKT Werte als erhöht gelten können. Zusätzlich untersuchten Yuan et al. (2000) die AKT2-Kinase-Aktivität durch in vitro Kinase Assay und wiesen eine erhöhte AKT2 Kinase-Aktivität nach. Hier wurde die Grenze zu normalen Werten bei einer dreifachen Erhöhung festgelegt. Diese erhöhten Werte fanden sich in 33 von 91 Proben (36,3%) - vor allem bei Tumoren mit hohem Grading (52,9%) und Staging (FIGO III und IV mit 48,5%). Allerdings fanden sich keine erhöhten Werte bei Endometrium-, Borderline-, Muzinösen- oder Granulosazelltumoren. Die Ergebnisse der Studie sprechen für eine Erhöhung der AKT-Werte bei Tumorprogression und aggressiverem Phänotyp und decken sich mit den Ergebnissen von Bellacosa et al. (1995). Auch in dieser Studie erfolgt allerdings kein direkter Vergleich zwischen den einzelnen Patientinnen bzw. keine Beurteilung der Quantität der Erhöhung. Ein ähnlicher Studienaufbau findet sich bei Sun et al. (2001). Mit geringeren Fallzahlen wiesen sie regelmäßig erhöhte P-AKT1-Werte bei Patientinnen mit Ovarialkarzinom nach. Nach der Aktivitätsuntersuchung der AKT1-Kinase mit einer Erhöhung der Werte bei 11 von 23 Patientinnen mit serösen Adenokarzinomen erfolgte die Bestätigung der Werte durch die Proteinbestimmung von P-AKT durch Western Blot Technik, jedoch ohne Angabe einer Grenze der Erhöhung zu normalem Gewebe. Regelmäßig wurden bei Patientinnen mit erhöhter AKT1-Kinaseaktivität erhöhte P-AKT-Werte gemessen. In „normalem“ Gewebe (Gewebe nicht benannt) sowie in Gewebe ohne AKT1-Kinase-Aktivität waren keine P-AKT-Werte messbar. Es fand sich keine Erhöhung der AKT1-Kinase-Aktivität bei den zwei untersuchten Borderlinekarzinomen, aber die Mehrheit der erhöhten Werte war in der Gruppe der Patientinnen mit hohem Staging und Grading zu beobachten. Auch diese Befunde decken sich mit den vorangegangenen Studienergebnissen bzw. belegen, dass nicht nur AKT2, sondern auch AKT1 regelmäßig im Gewebe maligner Ovarialtumoren erhöht vorzufinden ist. Eine neuere Studie von Altomare et al. (2004) bestätigt diese Vorarbeiten. Es zeigten 21 von 31 Patientinnen (68%) mit Ovarialkarzinom in immunhistochemischen Untersu- 54 chungen mit Antikörpern gegen die phosphorylierte (aktive) Form aller drei AKT Isoformen an Ser473 erhöhte Werte im Vergleich zu normalem ovariellem Gewebe. Diese Erhöhungen waren aber von den Autoren nicht eindeutig definiert worden, sondern lediglich mit „++“ und „+++“ beschrieben worden. Erklärt wird der relativ hohe Anteil der Patientinnen mit erhöhten Werten durch die Berücksichtigung aller drei AKT Isoformen. Dies ist zulässig, da vor allem für AKT1 und AKT2 eine Rolle in der Tumorgenese des Ovarialkarzinoms als erwiesen gelten kann. Bei 39 Gewebeproben von insgesamt 21 eigenen Patientinnen war aus den Krankenunterlagen ein Zusammenhang zur Platinsensibilität des Tumors herstellbar. Dabei zeigte sich die Mehrzahl (87,2%) als Platin-sensibel, lediglich 7,7% waren platinresistent. Die Ergebnisse der eigenen Studie zeigten allerdings keinerlei signifikante Zusammenhänge der AKT- und P-AKT-Aktivität zur Platinsensibilität der Ovarialkarzinome. Die eigenen Ergebnisse flossen jedoch in eine weitere Studie unserer Forschungsgruppe ein. Untersucht wurde hier zusätzlich die Korrelation zwischen dem Tumorsuppressionsprotein PTEN und der Platinsensitivität von Patientinnen mit malignem Ovarialkarzinom sowie die Korrelation zwischen der PTEN und P-AKT Expression in malignen Ovarialkarzinomen (Schöndorf et al. 2003). Im Kollektiv befanden sich 20 Patientinnen mit fortgeschrittenem Tumorstadium, welche einem standardisierten Behandlungsverfahren mit radikaler Operation und platinhaltiger Chemotherapie unterzogen worden waren. Sieben Patientinnen galten als platinsensibel mit einer Tumorprogressionsfreiheit von über 12 Monaten. Erfolgte die Progression des Tumors innerhalb der ersten 12 Monate, galten die Patientinnen als platinresistent, dies war bei 13 Patientinnen des Kollektivs der Fall. Zum einen zeigte sich in der platinresistenten Gruppe eine signifikante PTENVerminderung sowie in der platinsensiblen Gruppe ein Anstieg der PTENKonzentration. Dieses Ergebnis ist im Zusammenhang mit einigen Arbeiten aus der Literatur interessant. Yan et al. (2006) verglichen chemosensitive und -resistente Ovarialkarzinomzelllinien miteinander und stellten fest, dass die PTEN-Überexpression chemoresistente Ovarialkarzinomzelllinien sensibilisieren konnte. In diesen Prozess ist eine p53-mediierte apoptotische Kaskade involviert. Zu ähnlichen Ergebnissen kamen Selvendiran et al. (2007). Eine Suppression der PTEN-Expression verminderte 55 signifikant die p53- und p21-Spiegel und aktivierte die AKT-Phosphorylisierung, was zu einer verminderten Apoptoserate und einer erhöhten Zellüberlebensrate führte. Die Überexpression von PTEN induzierte andererseits aber auch eine Apoptose. Die Ergebnisse von Selvendiran et al. (2007) bestätigten, dass eine Hochregulation von PTEN das AKT inhibiert, was wiederum p53-Spiegel stimuliert und damit die Apoptose fördert. Eine Studie von Lee et al. (2005b) an verschiedenen Ovarialkarzinomzelllinien führte zu dem Ergebnis, das ein niedriger Spiegel einer PTEN-Expression mit einer erhöhten AKT-Aktivierung einhergeht. Eine verminderte PTEN-Expression ging mit einer Resistenzentwicklung gegen Cisplatin einher. Nach Yang et al. (2006) ist die AKTmediierte Cisplatinresistenz bei Ovarialzellkarzinomen abhängig von der p53Regulation. Ein möglicher Mechanismus der Beeinflussung der Apoptose bei cisplatinresistenten Ovarialzelllinien beschrieben Weir et al. (2007). Sie stellten fest, dass Curcumin die Proliferation von cisplatinresistenten und -sensitiven menschlichen Ovarialkarzinomzellen hemmte. Curcumin hemmte auch die Phosphorylierung von AKT, während die Phosphorylierung von p38 MAPK verstärkt wurde. Zusammengefasst zeigten die Ergebnisse von Weir et al. (2007), dass Curcumin in der Lage ist, die Proliferation von cisplatinresistenten Ovarialkarzinomzellen durch eine Induktion von Superoxidbildung und Apoptose zu hemmen. Kim et al. (2007) beschrieben einen weiteren interessanten Befund, wonach auch Paclitaxel die Anzahl phosphorylierter AKT-Moleküle bei Ovarialkarzinomzelllinien in einer zeitabhängigen Weise hemmt. Die Inhibition von AKT durch spezifische Phosphatidylinositol-Kinase-Inhibitoren erhöhte die Effizienz der Paclitaxel-induzierten Apoptose bei diesen Zelllinien. Dies lässt den Rückschluss zu, dass die Applikation eines AKT-Inhibitors die therapeutische Effizienz von Paclitaxel bei Patienten mit Ovarialkarzinom eventuell steigern könnte. Die AKT- und P-AKT-Bestimmung der eigenen Proben zeigte keinen signifikanten Zusammenhang zu den prä- oder postoperativen Ca-125-Konzentrationen und auch keinerlei signifikante Zusammenhänge zum rezidivfreien Überleben oder zum postoperativen progressionsfreien Überleben. Dieses Ergebnis wird von einer Studie von Wang et al. (2005) bestätigt. An 118 fortgeschrittenen Ovarialkarzinom-Patientinnen 56 zeigte sich, dass 51,6% der Proben positiv für P-AKT waren, wobei allerdings die PAKT-Werte kein statistisch unabhängiger prognostischer Marker waren. Nakayama et al. (2006) berichteten bei 65 Ovarialtumoren im Fish-Test über eine 18,2%ige Amplifikation von AKT2 bei Karzinomen in höheren Tumorstadien. Daraus zogen die Autoren den Schluss, dass das AKT2 eine wichtige Rolle in der Entwicklung von Ovarialkarzinomen spielt. Nach Meng et al. (2006) ist AKT1 bei Ovarialzellkarzinomzelllinien in die Zellbewegung, Invasion und Proliferation involviert. Auch Woenckhaus et al. (2007) konnten keinen signifikanten Zusammenhang zwischen den P-AKT-Spiegeln und dem Outcome von 74 Patientinnen mit Ovarialkarzinomen feststellen. Die Autoren fanden lediglich, dass bei denjenigen Patientinnen, die eine PAKT-Immunoreaktivität zeigten, ein leichter Trend zu einer besseren Überlebensrate auszumachen war. Es fanden sich aber keine statistischen Signifikanzen. Die Autoren postulierten eine wichtige Rolle der AKT-Phosphorylierung bei Ovarialkarzinomen. Verglich man die PTEN-Entwicklung mit den P-AKT-Werten in den vorliegenden Operationspräparaten, zeigte sich bei 14 der 20 Patientinnen eine inverse Entwicklung der PTEN- und der P-AKT-Werte. Dies bedeutet eine P-AKT-Erhöhung bei rückläufigen PTEN-Werten. Zu ähnlichen Ergebnissen auch andere Forschungsgruppen. Sun et al. (2001) beobachtet eine fehlende PTEN-Expression bei 2 von 11 Patientinnen mit erhöhten AKT-Werten und zusätzlicher PI 3-Kinase-Aktivierung. Yuan et al. (2000) zeigten diese Entwicklung bei 3 von 18 Patientinnen auf. Die höhere positive Korrelation in der vorliegenden Arbeit lässt sich mit der Beschreibung einer Entwicklung in beide Richtungen erklären. Es wird hier von inverser Korrelation gesprochen, es liegt keine beschränkte Betrachtungsweise auf nur ansteigende P-AKT-Werte bzw. abfallende PTEN-Werte vor. Es zeigt sich, dass die ovarielle Karzinogenese mit einer Verminderung der PTEN Expression einhergeht, ausgelöst durch verschiedene Faktoren, darunter die Phosphorylierung (Aktivierung) der Proteinkinase AKT. Es ist bekannt, dass das AKTabhängige Zellüberleben negativ durch PTEN beeinflusst wird. Zellen des Ovarialkarzinoms mit erhöhten AKT-Spiegeln zeigen niedrigere PTEN-Werte mit intensiviertem Transduktionsweg für das Zellüberleben. Es wurde auch demonstriert, dass 57 Patientinnen bei längerem progressionsfreiem Intervall erhöhte PTEN-Werte zeigen, also die klinische Entwicklung und Ausprägung tatsächlich abhängig zu sein scheint von Tumorsuppressoren und Aktivatoren. Gomes und Andrade (2006) berichteten, dass die PTEN-Spiegel weder mit dem Tumorvolumen noch mit dem Tumorstadium in einem signifikanten Zusammenhang standen. Sie hatten in ihrer Studie 70 Patientinnen mit Ovarialkarzinomen aufgenommen. Die PTEN-Inaktivierung ist laut Gomes und Andrade (2006) ein sehr früher Schritt in der Karzinogenese des Ovarialkarzinoms. In einer Untersuchung von Lee et al. (2005b) an 54 Adenokarzinomen und 23 Borderline-Karzinomen des Ovars ging eine reduzierte PTEN-Expression mit signifikant höheren Adenokarzinomstadien im Vergleich zu den Borderline-Tumoren einher. Eine verminderte PTEN-Expression bei den Adenokarzinomen korrelierte jedoch nicht mit dem FIGO-Stadium. Auch Lee et al. (2005b) hielten Veränderungen des PTEN-Gens für einen wichtigen, tumorauslösenden Faktor. Willner et al. (2007) berichteten bei 12 Clearzell-, 26 endometroiden und 51 serösen Ovarialkarzinomen über den Einfluss verschiedener Marker, u.a. PTEN. PTEN-Mutation waren bei low-grade-Karzinomen häufiger. In einer Studie von Sui et al. (2006) war die PTEN-Expression von benignen zu malignen Stadien des Ovarialkarzinoms vermindert und zwar statistisch signifikant. Ferner zeigte sich eine inverse Korrelation zwischen der PTEN-Expression und des Survivins. Die PTENExpression korrelierte signifikant mit dem Tumorgrading, des histologischen Subtypes, dem Vorliegen von Aszites. Ferner zeigte sich eine signifikante Korrelation zwischen einer verminderten PTEN-Expression und einem schlechteren Gesamtüberleben. Der jeweilige Einfluss der Proteine der an der Tumorgenese beteiligten Enzymketten bei verschiedenen Individuen erweist sich als sehr unterschiedlich. Dies zeigen alle angeführten Studien durch die fehlende Ausschließlichkeit der Ergebnisse in verschiedenen Untersuchungen. Natürlich existieren zahlreiche unbekannte und wenig erforschte Einflüsse, sowie persönliche Ausprägungen und Determinationen. Große Anstrengungen werden deshalb unternommen, das chemotherapeutische Regime zur Behandlung des Ovarialkarzinoms zu intensivieren und zu verbessern. Ein Erfolg wird sicher auf molekularer Ebene zu erzielen sein, da nun zahlreiche Wege bekannt sind, um in einige der Transduktionswege einzugreifen. Dies wäre zum Beispiel durch 58 eine Hemmung des AKT-Signaltransduktionsweges denkbar, vor allem durch Antikörper als antineoplastische Therapie oder durch Unterstützung der PTEN regulierten Wege, zum Beispiel über Hemmung von PI3K, den direkten Antagonisten von PTEN. All diese Ansätze beschränken ihre Wirkung auf proliferierende Tumorzelllinien in fortgeschrittenen Karzinomen, was bedeutet, dass gesundes Gewebe geschont würde. Außerdem würde durch eine solche additive Therapie der Effekt der platinhaltigen Chemotherapeutika auf die Tumorzellen intensiviert werden. Die Verlängerung des Intervalls zu Progression des Tumors oder zur Entwicklung eines Rezidivs ist für jede Patientin von essentieller Bedeutung und speziell beim Ovarialkarzinom ein lohnenswertes Ziel, um die Überlebenszeit zu verlängern und Lebensqualität zu verbessern. 59 5. Zusammenfassung Die Prognose des Ovarialkarzinoms hat sich in den letzten Jahrzehnten nur unwesentlich verändert. Ein großes Problem der Therapie des Ovarialkarzinoms ist die Resistenz auf platinhaltige Chemotherapeutika. Da aufgrund der schlechten Prognose des Ovarialkarzinoms auf der Basis von Chemotherapieresistenz und später Diagnose erheblicher Forschungsbedarf besteht, versucht man, die Chemosensilbilität vo Ovarialkarzinomen zu erhöhen. Einen möglichen Ansatz stellen Signaltransduktionswege der Proteinkinase AKT dar. In der vorliegenden Arbeit wurde das Protein AKT und dessen phosphorylierte Form P-AKT aus 99 Tumorproben von Ovarialkarzinompatientinnen isoliert. Die Konzentrationen wurden mit verschiedenen biologischen Parametern und der Rezidivfreiheit in Bezug gesetzt, um somit einen eventuellen Zusammenhang zwischen Konzentration von AKT und dem Krankheitsverlauf zu erfassen. Es wurde von einigen Autoren beobachtet, dass vor allem in späten Stadien erhöhte AKT Konzentrationen zu finden waren. Allerdings hatte in den eigenen Proben die AKT- und die P-AKT-Aktivität keinen signifikanten Zusammenhang zum FIGOStadium oder zu den Gradingstufen gezeigt. Die AKT-Werte waren bei den eigenen Proben aus Primäreingriffen leicht, jedoch nicht statistisch signifikant, höher als bei den Proben aus Rezidiveingriffen. Umgekehrt verhielt es sich mit der P-AKT-Aktivität. Diese war bei den Primäreingriff-Proben, aber ebenfalls nicht signifikant, niedriger als bei den Proben aus Rezidiveingriffen. Es konnte auch kein signifikanter Unterschied zwischen den AKT-Konzentrationen der übrigen untersuchten Parameter festgestellt werden. Die Cox-Überlebensanalyse, in die nur die 42 Proben einbezogen wurden, bei denen der genaue Zeitraum der Progressions- und Rezidivfreiheit aus den Akten zu ermitteln war, zeigte keine signifikante AKT- Veränderung bei kürzeren Intervallen zu Progression und Rezidiv. Es konnten auch keine signifikant höheren Werte bei Patientinnen mit Platinresistenz im Gegensatz zu Platin-sensiblen Patientinnen festgestellt werden. Ein Trend ließ sich nur in der Gruppe der sekundär operierten Patientinnen mit im Durchschnitt höheren P-AKT- Werten feststellen. Die eigenen Ergebnisse zeigen, dass man innerhalb eines erkrankten Kollektivs keine signifikanten Konzentrationsveränderungen von AKT/P-AKT beobachten kann, die auf 60 ein vorangeschrittenes Krankheitsstadium oder eine schlechtere Prognose schließen helfen lassen würden. Es lässt sich hier allerdings keine Aussage über eventuelle AKT-Erhöhungen insgesamt im Kollektiv der Ovarialkarzinompatientinnen treffen. Da hiervon aber auszugehen ist, dürfen die eigenen Ergebnisse nicht zur Unterschätzung der Bedeutung von AKT in der Tumorgenese beitragen. 61 6. Literaturverzeichnis 1. Aletti GD, Gallenberg MM, Cliby WA, Jatoi M, Harmann LC (2007): Current management strategies for ovarian cancer. Mayo Clin Proc 82: 751-770 2. Allen DG, Heintz AP, Touw FW (1995): A meta-analysis of residual disease and survival in stage III and IV carcinoma of the ovary. Eur J Gyn Oncol 16: 349-356 3. Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA (1996): Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15: 4066-4075 4. Alessi DR, Deak M, Casamayor A, Caudwell FB, Morrice N, Normann DG, Gaffney P, Reese CB, MacDougall CN, Harbison D, Ashworth A, Bownes M (1997): 3-Phosphoinositide-dependent protein kinase-1 (PDK-1): structural and functional homology with the Drosophila DSTP K61 Kinase. Curr Biol 7: 776-89 5. Altomare DA, Wang HQ, Skele KL, De Rienzo A, Klein-Szanto AJ, Godwin AK, Testa JR (2004): AKT and mTor phosphorylation is frequently detected in ovarian cancer and can be targeted to disrupt ovarian tumor cell growth. Oncogene 23: 5853-5857 6. Andjelkovic M, Maira SM, Cron P, Parker PJ, Hemmings BA (1999): Domain swapping used to investigate the mechanism of protein kinase B regulation by 3phosphoinositide-dependent kinase1 and Ser 473 kinase. Mol Cell Biol 19: 50615072 7. Assoian RK, Schwartz MA (2001): Coordinate signaling by integrins and receptor tyrosine kinase in the regulation of G1 phase cell-cycle progression. Curr Opin Genet Dev 11: 48-53 8. Auersbergs N , Wong AS, Choi KC, Kang SK, Leung PC (2001): Ovarian surface epithelium: biologie, endocrinologie, and pathologie. Endocr Rev 22: 255-288 9. Arbeitsgemeinschaft bevölkerungsbezogene Krebsregister in Deutschland (1997): Krebs in Deutschland - Häufigkeiten und Trends. Saarbrücken, 12. Auflage 10. Beckmann MW, Kimmig R, Gissmann L, Schneider A, Friese K, Hillemanns P (2007): Gynäkologische Tumore. Vortrag auf der Nationalen onkologischen Präventionskonferenz. Congress Cencer Essen, 16.6.2007 11. Bell DA (2005): Origins and molecular pathology of ovarian cancer. Mod Pathol 18 (Suppl.): 29-32 62 12. Bella M, Cocconi G, Lottici R (1994): Mature results of a prospective randomized trial comparing two different dose- intensity regimes of cisplatin in advanced ovarian carcinoma. Ann Oncol 5 (Suppl. 8): 2-5 13. Bellacosa A, De Feo D, Godwin AK, Bell DW, Cheng JQ, Altomare DA, Wan MM, Dubeau L, Scambia G, Masciullo V, Ferrandina G, Mancuso S, Neri G, Testa JR (1995): Molecular alterations of the AKT2 oncogene in ovarian and brest carcinomas. Int J Cancer 64: 280-285 14. Birrer MJ (2006): Ovarian cancer. Not a borderline issue. Cancer Biol Ther 5: 779-785 15. Bolis G, Favelli G, Danese S (1997): Weekly cisplatin given for 2 month versus cisplatin plus cyclophosphamide given for 5 month after cytoreductive surgery for advanced ovarian cancer. J Clin Oncol 15: 1939-1944 16. Brasil D, Hemmings B (2001): Ten years of protein kinase B signalling: a hard AKT to follow. Trends Biochem Sci 26: 657-664 17. Brinton L (2007): Long-term effects of ovulation-stimulating drugs on cancer risk. Reprod Med Online 15: 38-44 18. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME (1999): AKT promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96: 857-868 19. Burgering BM, Kops GJ (2002): Cell cycle and death control: long live Forkheads. Trends Biochem Sci 27: 352-359 20. Burghardt E, Girardi F, Lahousen M (1991): Patterns of pelvic and paraaortic lymph node involvement in ovarian cancer. Gynecol Oncol 40: 103-106 21. Cadron I, van Gorp T, Amant F, Neven P, Vergote I (2007): Management of borderline ovarian neoplasms. J Clin Oncol 25: 2928-2937 22. Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S, Nukshatri H (2001): Phosphatidyl-inositol 3Kinase/AKT-mediated activation of estrogen receptor α: a new model for anti-estrogen resistance. J Biol Chem 276: 9817-9824 23. Chaudhuri D, Orsulic S, Ashok BT (2007): Antiproliferative activity of sulforaphane in Akt-overexpressing ovarian cancer cells. Mol Cancer Ther 6: 334-345 24. Chen D, Fucini RV, Olson AL, Hemmings BA, Pessin JE (1999): Osmotic shock inhibits insulin signaling by maintaining AKT/protein kinase B in an inactive dephosphorylated state. Mol Cell Biol 19: 4684-4694 63 25. Cheng JQ, Jiang X, Fraser M, Li M, Dan HC, Sun M, Tsang BK (2002): Role of Xlinked inhibitor of apoptosis protein in chemoresistance in ovarian cancer: possible involvement of the phosphoinositide-3 kinase/AKT pathway. Drug Resist Updat 5: 131-146 26. Cho H (2001): Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase AKT2(PKBβ). Science 292: 1728-1731 27. Cohen P (1998): The central roles of PI3-Kinase and AKT in Insulin Signaling. Upstate News and Views 28. Collinet P, Lanvin D, Vereecque R, Quesnel B, Querleu D (2000): Gene therapy and ovarian cancer: update of clinical trials. J Gynecol Obstet Biol Reprod 29: 532537 29. Colombo N, Parma G, Zanagnolo V, Insinga A (2007): Management of ovarian stromal cell tumors. J Clin Oncol 25: 2944-2951 30. Cristiano BE, Chan JC, Hannan KM, Lundie NA, Marmy-Conus NJ, Campbell IG, Phillips WA, Robbie M, Hannan RD, Perason RB (2006): A specific role for AKT3 in the genesis of ovarian cancer through modulation of G2-M phase transition. Cancer Res 66: 11718-11725 31. Cross DA, Alessi DR, Cohen P, Andjelkovic M, Hemmings BA (1995): Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378: 785-789 32. Dawson CW, Tramountanis G, Eliopoulos AG, Young LS (2003): Epstein-Barr virus latent membrane protein1(LMP1) activates the phosphatidylinositol 3-kinase/ AKT pathway to promote cell survival and induce actin filament remodeling. J Biol Chem 278: 3694-3704 33. Datta SR, Brunet A, Greenberg ME (1999): Cellular survival: a play in three AKTs. Genes Dev 13: 2905-2927 34. Decker DG, Thomas MD, Fleming R (1982): Cyclophosphamide plus cisplatinum in combination: Treatment program for stage III or IV ovarian carcinoma. Obstet Gynecol 60: 418-427 35 Delord JP, Allal C, Canal M, Mery E, Rochaix P, Hennebelle I, Pradines A, Chatelut E, Bugat R, Guichard S, Canal P (2005): Selective inhibition of HER2 inhibits AKT signal transduction and prolongs disease-free survival in a micrometastasis model of ovarian carcinoma. Ann Oncol 16: 1889-1897 36. Del Peso L, Gonzalez GM, Page C, Herrera R, Nunez G (1997): Interleukin-3induced phosphorylation of BAD through the protein kinase AKT. Science 278: 687-689 64 37. De Poncheville L, Perrotin F, Lefranc T, Lansac J, Body G (2001): Does paraaortic lymphadenectomy have a benefit in the treatment of ovarian cancer that is apparently confined to the ovaries? Eur J Cancer 37: 210-215 38. De Oliveira CF, Lacave AJ, Villani C (1990): Randomized comparison of cyclophosphamide, doxorubicin and cisplatin for the treatment of advanced ovarian cancer. Eur J Gynecol Oncol 11: 323-330 39 Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ (2000): Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol 10: 1201-1204 40. Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM (1999): Activation of nitric oxide synthase in endothelial cells by AKT-dependent phosporylation. Nature 399: 601-605 41. Emons G, Kavanagh JJ (1999): Hormonal interactions in ovarian cancer. Hematol Oncol Clin North Am 13: 145-161 42. Fader N, Rose PG (2007): Role of surgery in ovarian carcinoma. J Clin Oncol 25: 2873-2883 43. Fraser M, Leung BM, Yan X, Dan HC, Cheng JQ, Tsang BK (2003): p53 is a determinant of X-linked inhibitor of apoptosis protein/AKT-mediated chemoresistance in human ovarian cancer cells. Cancer Res 63: 7081-7088 44. Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K (1999): Regulation of endothelium-derived nitric oxide production by the protein kinase AKT. Nature 399: 597-601 45. Gadducci A, Bruzzone M, Carnino F (1996): Twelf-year follow-up of a randomized trial comparing cisplatin and cyclophosphamide, with cisplatin, doxorubicin and cyclophosphamide in patients with advanced epithelial ovarian cancer. Int J Gynecol Cancer 6: 286-290 46. Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, Ferrara N (1998): Vascular endothelial cell survival through the phosphatidylinositol 3kinase/AKT signal transduktion pathway. Requirement for Flk-1/KDR activation. J Biol Chem 273: 30336-30343 47. Gershenson DM (2007): Management of ovarian cell tumors. J Clin Oncol 25: 2938-2943 48. Gille H, Downward J (1999): Multiple ras effector pathways contribute to G(1)cell cycle progression. J Biol Chem 274: 22033-22040 65 49. Goerke G, Steller J, Valet A (2003): Gynäkologie und Geburtshilfe. Urban und Fischer Verlag, München, 6. Auflage 50. Gomes CP, Andrade LA (2006): PTEN and p53 expression in primary ovarian carcinomas: immunohistochemical study and discussion of pathogenetic mechanisms. Int J Gynecol Cancer 16 (Suppl.1): 254-258 51. Haas-Kogan D (1998): Protein kinase B (PKB/AKT) activity is elevated in glioblastoma cells due to mutation of the tumor suppressor PTEN/MMAC. Curr Biol 8: 1195-1198 52. Hashiguchi Y, Tsuda H, Inoue T, Berkowitz RS, Mok SC (2006): PTEN expression in clear cell adenocarcinoma of the ovary. Gynecol Oncol 101: 71-75 53. Hill MM, Andjelkovic M, Brazil DP, Ferrari S, Fabbro D, Hemmings BA (2001): Insulin-stimulated protein kinase B phosphorylation on Ser-473 is independent of its activity and occurs through a staurosporine-insensitive kinase. J Biol Chem 23: 23-27 54. Holinski-Feder E, Brandau O, Nestle-Krämling C (1998): Genetik des erblichen Mammacarcinoms - Grundlagen, Forschung, Diagnostik. Dt. Ärzteblatt 95: 600-605 55. Hong F, Nguyen VA, Shen X, Kunos G, Gao B (2000): Rapid activation of protein kinase B/AKT has a key role in antiapoptotic signalling during liver regeneration. Biochem Biophys Res Com 279: 974-979 56. IARC (2002): Globocan 2002: Cancer incidence, mortality and prevalence worldwide (2002 estimates). http://www.dep.iarc.fr 57. Izuishi K, Kato K, Ogura T, Kinoshita T, Esumi H (2000): Remarcable tolerance of tumor cells to nutrient deprivation: possible new biochemical target for cancer therapy. Cancer Res 60: 6201-6207 58. Kim SH, Juhnn YS, Song YS (2007): Akt involvement in paclitaxel chemoresistance of human ovarian cancer cells. Ann NY Acad Sci 1095: 82-89 59. Kolasa IK, Rembiszewska A, Janiec-Jankowska A, Dansonka-Mieszckowska A, Lewandowska AM, Konopka B, Kupryjanczyk (2006): PTEN mutation, expression and LOH at its locus in ovarian carcinomas. Relaton to TP53, K-RAS and BRCA1 mutations. Gynegol Oncol 103: 692-697 60. Kommoss F, Pfisterer J, Thome M (1992): Steroid receptors in ovarian carcinoma: Immunhistochemical determination may lead to new aspects. Gynecol Oncol 47: 317-322 61. Kroner C, Eybrechts K, Akkerman J-WN (2000) Dual regulation of platelet protein kinase B. J Biol Chem 275: 27790-27822 66 62. Kurose K, Zhou XP, Araki T, Cannistra SA, Mahrer ER, Eng C (2001): Frequent loss of PTEN expression is linked to elevated phosphorylated AKT levels, but not associated with p27 and Cyclin D1 Expression, primary epithelial ovarian carcinomas. AJP 158: 2097-2104 63. Le T, Krepart GV, Lotocki RJ (1998): Does Debulking surgery improve survival in biologically aggressive cancer. Gynecol Oncol 67: 208-214 64. Lee JS, Choi YD, Choi C, Lee MC, Park CS, Min KW (2005a): Expression of PTEN in ovarian epithelial tumors and its relation to tumor behavior and growth. Anal Quant Cytol Histol 27: 202-210 65. Lee S, Choi EJ, Jin C, Kim DH (2005b): Activation of PI3K/Akt pathway by PTEN reduction and PIK3CA mRNA amplification contributes to cisplatin resistance in an ovarian cancer cell line. Gynecol Oncol 97: 26-34 66. Li J, Feng Q, Kim JM, Schneiderman D, Liston P, Li M, Vanderhyden B, Faught W, Fung MF, Senterman M, Korneluk RG, Tsang BK (2001): Human ovarian cancer and cisplatin resistance: possible role of inhibitor of apoptosis proteins. Endocrinology 142: 370-380 67. Liu X, Shi Y, Han EK, Chen Z, Rosenberg SH, Giranda VL, Luo Y (2001): Downregulation of AKT1 inhibits anchorage-independent cell growth and induces apoptosis in cancer cells. Neoplasia 3: 278-286 68. Lowry ON, Rosenbrough NJ, Farr A, Randall RL (1951): Protein measurement with the Folin phenol reagent. J Biol Chem 193: 265-275 69. Martin VR (2006): Ovarian cancer. An overview of treatment options. Clin J Oncol Nurs 11: 201-207 70. Meden H (1996): Ovarialcarcinom: Aktuelle Aspekte in Diagnostik und Therapie in Klinik und Praxis. De Gruyter Verlag, Berlin, New York 71. Meng Q, Xia C, Fang J, Rojanasakul Y, Jiang BH (2006): Role of PI3K and AKT specific isoforms in ovarian cancer cell migration, invasion and proliferation through the p70S6K1 pathway. Cell Signal 18: 2262-2271 72. Nakayama K, Nakayama N, Kurman RJ, Cope L, Pohl G, Samuels Y, Velculescu VE, Wang TL, Shih IM (2006): Sequence mutations and amplification of PIK3CA and AKT2 genes in purified ovarian serous neoplasms. Cancer Biol Ther 5: 779785 67 73. Nieh S, Chen SF, Chu TY, Lai HC, Lin YS, Fu E, Gau CH (2005): Is p16INK4A expression more useful than human papillomavirus test to determine the outcome of atypical squamous cells of undetermined significance-categorized Pap smear? A comparative analysis using abnormal smears with follow-up biopsies. Gynecol Oncol 97: 35-40 74. Nicholson KM, Anderson N (2002): The protein kinase B/AKT signalling pathway in human malignancy. Cell Signal 14: 381-395 75. Noske A, Kaszubiak A, Weichert W, Sers C, Niesporek S, Koch I, Schaefer B, Sehouli J, Dietel M, Lage H, Denkert C (2007): Specific inhibition of AKT2 by RNA interference results in reduction of ovarian cancer cell proliferation: increased expression of AKT in advanced ovarian cancer. Cancer Lett 246: 190-200 76. Nunez MI, Rosen DG, Ludes-Meyers JH, Abba MC, Kil H, Page R, Klein-Szanto AJ, Godwin AK, Liu J, Mills GB, Aldaz CM (2005): WWOX protein expression varies among ovarian carcinoma histotypes and correlates with less favorable outcome. BMC Cancer 5: 64 77. Onda T, Yoshikawa H, Yasugi T (1998): Patients with ovarian carcinoma upstaged to stage III after systemic lymphadenectomie have similar survival to stage II patients and superior survival to other stage III patients. Cancer 83:1555-1560 78. Page C, Lin HJ, Jin Y, Castle VP, Nunez G, Huang M, Lin J (2000): Overexpression of AKT can modulate chemotherapy-induced apoptosis. Anticancer Res 20: 407-416 79. Petru E, Lahousen M, Tamussino K (1994): Lymphadenektomy in stage I ovarian cancer. Am J Obstet Gynecol 170: 656-662 80. Pfleiderer A, Breckwoldt M, Martius G (2001): Gynäkologie und Geburtshilfe, Sicher durch Studium und Praxis, G. Thieme Verlag, New York, 4. Aufl. 81. Reed JC, Toshiuki M, TakayamaS, Wang H-G, Sato T, Krajewski S, Aime-Sempe C, Bodrug S, Kitada S, Hanada M (1996): Bcl-2 family proteins:regulators of cell death involved in the pathogenesis of Cancer and resistance to therapy. J Cell Biochem 60: 23-32 82. Reddy P, Liu L, Ren C, Lindgren P, Boman K, Shen Y, Lundin E, Ottander U, Rytinki M, Liu K (2004): Formation of E-cadherin-mediated cell-cell adhesion activates akt and mitogan activated protein kinase via phosphatidlinositol 3 kinase and ligand-independent activation of epidermal growth factor receptor in ovarian cancer cells. Mol Endocrinol 19: 2564-2578 83. Robert Koch Institut (2005): Krebs in Deutschland. Häufigkeiten und Trends. 5. Aufl., Saarbrücken 68 84. Romashkova JA, Makarov SS (1999): NF-κB is a target of AKT in anti-apoptotic PDGF signalling. Nature 401: 86-90 85. Ruggeri BA, Huang L, Wood M, Cheng JQ, Testa JR (1998): Amplification and over expresion of the AKT2 oncogene in a subset of human pancreatic ductal adenocarcinomas. Mol Carcinog 21: 81-86 86. Sabbatini P, Odunsi K (2007): Immunologic approaches to ovacian cancer treatment. J Clin Oncol 25: 2894-2893 87. Sanseverino F, D'Andrilli G, Petraglia F, Giordano A (2005): Molecular pathology of ovarian cancer. Anal Quant Cytol Histol 27: 121-124 88. Sasaki H, Sheng Y, Kotsuji F, Tsang BK (2000): Down-regulation of x-linked inhibitor of apoptosis protein induces apoptosis in chemoresistance human ovarian cancer cells. Cancer Res 60: 5659-5666 89. Sato S, Fujita N, Tsuruo T (2000): Modulation of AKT kinase activity by binding to Hsp9O. Proc Natl Acad Sci USA 97: 10832-10837 90. Schöndorf T, Göhring UJ, Roth G, Middel I, Becker M, Moser N, Valter MM, Hoopmann M (2003): Time to progression is dependent on the expression of the tumour suppressor PTEN in ovarian cancer patients. Eur J Clin Invest 33: 256360 91. Schubert KM, Scheid MP, Duronio V (2000): Ceramide inhibits protein kinase B/AKT by promoting dephosphorylation of Serine 473. J Biol Chem 275: 1333013335 92. Schulz M, Nöthlings U, Allen N, Onland-Moret C, Agnoli C, Engeset D, Galasso R, Wirfält E, Tjönneland A, Olsen A, Overvad K, Boutron-Ruault MC, Chajes V Clavel-Chapelon F, Ray J, Hoffmann K, Chang-Claude J, Kaaks R, Trichopoulos D, Georgila C, Zourna P, Palli D, Berrino F, Tumino R, Vineis P, Panico S, Buenode-Mesquita HB, Ocke MC, Peeters PHM, Gonzalez CA, Quiros JR, Dorronson M, Martinez C, Tormo MJ, Barricate A, Bingham S, Khaw KT, Key TJA, Rinaldi S, Slimani N, Riboli E (2007): No association of consumption of animal foods with risk of ovarian cancer. Cancer Epidemiol Biomarkers Prev 16: 852-855 93. Selvendiran K, Tong L, Vishwanath S, Bratasz A, Trigg NJ, Kutala VK, Hideg K, Kuppusamy P (2007): EF24 induces G2/M arrest and apoptosis in cisplatin-resistant human ovarian cancer cells by increasing PTEN expression. J Biol Chem, http://www.jbc.org/cgi/doi/10.1074/jbc.M703796200 94. Shaw TJ, van der Hyden BC (2007): AKT mediates the pro-survival effects of KIT in ovarian cancer cells and is a determinant of sensitivity to imatinib mesylate. Gynecol Oncol 105: 122-131 69 95. Spirtos NM, Gross GM, Freddo JL (1995): Cytoreductive surgery in advanced epithelial cancer of the ovary.The impact of aortic and pelvic lymphadenectomie. Gynecol Oncol 56: 345-52 96. Staal SP (1987): Molekular cloning of the akt oncogene and its human homologues AKT1 and AKT2: Amplification of AKT1 in a primary human gastric adenocarcinoma. Proc Natl Acad Sci USA 84: 5034-5037 97. Staebler A, Diebold J (2007): Molekularpathologie der epithelialen Ovarialneoplasien. Von der Phänotyp-Genotyp-Korrelation zu neuen Ansatzpunkten in Diagnostik und Therapie. Pathologe 28: 180-186 98. Stambolic V, Suzuki A, de la Pompa JL, Brothers GM; Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW (1998): Negative regulation of PKB/AKT-dependent cell survival by the tumor suppressor PTEN. Cell 95: 29-39 99. Stokoe D, Stephens LR, Copeland T, Gaffney PR, Reese CB, Painter GF, Holmes AB, McCormick F, Hawkins PT (1997): Dual role of phosphoinositol-3,4,5-triphosphate in the activation of protein kinase B. Science 277: 567-570 100. Stratton JF, Gayther SA, Russel P (1997): Contribution of BRCA1 mutation to ovarian cancer. N Engl J Med 336: 1123-30 101. Stratton JF, Tidy JA, Paterson MEL (2001): The surgical management of ovarian cancer. Cancer Treat Rev 27: 111-118 102. Sui L, Dong Y, Watanabe Y, Yamaguchi F, Sugimoto K, Tokuda M (2006): Alteration and clinical relevance of PTEN expression and its correlation with survivin expression in epithelial ovarian tumors. Oncol Rep 15: 773-778 103. Sun M, Wang G, Paciga JE, Feldmann RI, Yuan Z, Ma X, Shelley SA, Jove R, Tsichlis PN, Nicosia SV, Cheng JQ (2001): AKT1/PBKα kinase is frequently elevated in human cancers and its constitutive activation is required for oncogenic transformation in NIH3T3 cells. Am J Pathol 159: 431-437 104. Takuwa N, Fukui Y, Takuwa Y (1999): CyclinD1 expression mediated by phosphatidylinositol 3-kinase through mTor-p70(S6K)-independent signalling in growth factor-stimulated NIH 3T3 fibroblasts. J Biol Chem 19: 1346-1358 105. Tang HJ, Jin X, Wang S, Yang D, Cao Y, Chen J, Gossett DR, Lin J (2006): A small molecule compound inhibits AKT pathway in ovarian cancer cell lines. Gynecol Oncol 100: 308-317 106. Toker A, Newton AC (2000): AKT/ protein kinase B is regulated by autophosphorylation at the hypothetical PDK-2 site. J Biol Chem 275: 8271-8274 70 107. Tropé C, Kaern J (2007): Adjuvant chemotherapy for early-stage ovarian cancer: review of the literature. J Clin Oncol 25: 2909-2920 108. Trope C, Kaern J, Vergote I (1997): Randomized trial of adjuvant carboplatin versus no treatment in stage I high risk ovarian cancer by the Nordic Ovarian Cancer Study Group (NOCOVA). Proc Am Soc Clin Oncol 16: 352-355 109. Vergote IB, Kaern J, Abeler VM (1993): Analysis of prognostic factors in stage I epithelial ovarian carcinoma; importance of degree of differentiation and DNA ploidy in predicting relapse. Am J Obstet Gynecol 169: 40-52 110. Wang HQ, Altomare DA, SKele KL, Poulikakos PI, Kuhajda FP, Di Cristofano A, Testa JR (2005): Positive feedback regulation between AKT activation and fatty acid synthase expression in ovarian carcinoma cells. Oncogene 24: 3574-3582 111. Wang Y, Kristensen GB, Helland A, Nesland JM, Borresen-Dale AL, Holm R (2005): Protein expression and prognostic value of genes in the erb-b signaling pathway in advanced ovarian carcinoma. Am J Clin Pathol 124: 392-401 112. Weir NM, Selvendrian K, Kutala VK, Tong L, Vishwanath S, Rajaram M, Tridandapani S, Anant S (2007): Curcumin induces G2/M arrest and apoptosis in cisplatin-resistant human ovarian cancer cells by modulating Akt and p38 MAPK. Cancer Biol Ther 6: 178-184 113. Whang YE (1998): Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc natl acad sci USA 95: 5246-5250 114. Williams CJ (1998): Tamoxifen in relapsed ovarian cancer: A systemic review. Int J Gynecol Cancer 8: 89-94 115. Willner J, Wurz K, Allison KH, Galic V, Garcia RL, Goff BA, Swisher EM (2007): Alternate molecular genetic pathways in ovarian carcinomas of common histological types. Hum Pathol 38: 607-613 116. Woenckhaus J, Steger K, Sturm K, Münstedt K, Franke FE, Fenic I (2007): Prognostic value of PIK3CA and phosphorylated AKT expression in ovarian cancer. Virchows Arch 450: 387-395 117. Wu PC; Qu JY, Lang JH (1986): Lymph node metastases of ovarian cancer. A preliminary survey of 74 cases of lymphadenectomy. Am J Obstet Gynecol 155: 1103 118. Xia C, Meng Q, Cao Z, Shi X, Jiang BH (2006): Regulation of angiogenesis and tumor growth by p110 alpha and AKT1 via VEGF expression. J Cell Physiol 209: 56-66 71 119. Yan X, Fraser M, Qui Q, Tsang BK (2006): Over-expression of PTEN sensitizes ovarian cancer cells to cisplatin-induced apoptosis in a p53-dependent manner. Gynecol Oncol 102: 348-355 120. Yang X, Fraser M, Moll UM, Basak A, Tsang BK (2006): Akt-mediated cisplatin resistance in ovarian cancer: modulation of p53 action on paspase-dependent mitochondrial death pathway. Cancer Res 66: 3126-3136 121. Young RC (1995): The treatment of early stage ovarian cancer. Semin Oncol 22: 76-79 122. Young RC (1998): Management of early ovarian cancer. Semin Oncol 25: 335339 123. Yuan ZQ, Sun M, Feldman RI, Wang G, Ma X-L, Jiang C, Coppola D, Nicosia SV, Cheng JQ (2000): Frequent activation of AKT2 and induction of apoptosis by inhibition of phospho-Inositide-3-OH kinase/AKT pathway in human ovarian cancer. Oncogene 19: 2324-2330 124. Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, GeorgescuMM, Simons JW, Semenza GL (2000): Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/ Phosphatidylinositol 3-kinase/PTEN/AKT/ FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res 60: 1541-1545 125. Zundel W, Schindler C, Haas.Kogan D, Koong A, Kaper F, Chen E, Gottschalk AR, Ryan HE, Johnson RS, Jefferson AB, Stokoe D, Giaccia AJ (2000): Loss of PTEN faciliates HIF-1-mediated gene expression. Genes Dev 14: 391-396 1 Lebenslauf PERSÖNLICHE DATEN: Name: Geburtsdatum: Geburtsort: Staatsangehörigkeit: Familienstand: Adresse: Ines Kristin Middel 15.03.1977 Mainz deutsch ledig Siedlungsstr. 9, 65719 Hofheim SCHULBILDUNG: 1983 – 1987 1987 – 1996 1996 Taunusblick- Grundschule, Wallau Elly- Heuss- Gymnasium, Wiesbaden Abitur HOCHSCHULBILDUNG: WS 1998/ 99WS 2000/ 01 5 Semester des Medizinstudiums an der Freien Universität Berlin SS 2001- 2005 Beendigung des Medizinstudiums an der Universität zu Köln BERUFSTÄTIGKEIT: 2005 Assistenzärztin im chirurgischen Zentrum der Asklepios Paulinen Klinik Wiesbaden, bis heute SPRACHKENNTNISSE: Englisch: sehr gut in Wort und Schrift Französisch: gut in Wort und Schrift Hofheim, den 12.03.2009