Phasenumwandlungsanalyse von Cu-Au-Legierungen mittels Röntgendiffraktion
Werbung

Institut für Materialphysik
Charakterisierung der Phasenumwandlung
in Kupfer-Gold Legierungen mittels
Röntgendiffraktometrie
Versuchsanleitung
Im fortgeschrittenen Praktikum am Institut für Materialphysik
Stand: April 2015
Inhaltsverzeichnis
1. Einleitung
2. Theoretische Grundlagen
2.1. Das Phasendiagramm von Kupfer-Gold Legierungen . . . . . . . . . . . . . . . . . . .
2.1.1. Phasenumwandlung von CuAu . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.2. Röntgenbeugung am Kristall . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.2.1. Die Millerschen Indizes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.2.2. Braggsche Reflexionsbedingung . . . . . . . . . . . . . . . . . . . . . . . . . .
2.3. Einfluss der Wärmebewegung auf die Interferenz von Röntgenstrahlen . . . . . . . . .
2.3.1. Die Kernaussagen von Debye . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.3.2. Experimentelle Bestimmung des Debye-Waller Faktors . . . . . . . . . . . . .
2.3.3. Temperaturabhängigkeit der Gitterkonstanten . . . . . . . . . . . . . . . . . .
2.3.4. Zusammenhang zwischen der mittleren Auslenkung der Atome und der Gitterkonstanten des Kristalls . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.4. Theorie der Fernordnung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.4.1. Experimentelle Bestimmung der Fernordnung . . . . . . . . . . . . . . . . . .
2.5. Kalorimetrie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
2.5.1. Messung thermodynamischer Größen . . . . . . . . . . . . . . . . . . . . . . .
2.6. Keimbildung und Wachstum . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
4
5
5
6
9
9
11
13
13
14
16
17
19
19
24
26
26
3. Experimentelle Vorbereitung und Versuchsdurchführung
29
3.1. Proben . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
3.2. Die Kombination von Röntgendiffraktometer und Heizkammer . . . . . . . . . . . . . 30
3.3. Durchzuführende Messreihen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
4. Auswertung und Protokoll
33
A. Charakterisierung von Phasenübergängen
34
A.1. Phasenübergang 1.Ordnung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34
A.2. Phasenübergang 2.Ordnung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35
Inhaltsverzeichnis
B. Debeye-Waller-Faktor: Mathematische Modellierung der Problematik
36
B.1. Theoretische Ergebnisse des Einflusses der Wärmebewegung für CuAu . . . . . . . . . 38
C. Näherung des Integrals im Parameter M
40
D. Temperaturabhängigkeit der Gitterkonstanten
41
E. Keimbildung und Wachstum
45
F. Charakterisierung der Kristallstruktur
48
Literaturverzeichnis
51
© SoSe 2015
3
1. Einleitung
Der Begriff der Phasenumwandlung beschreibt in der Thermodynamik die Umwandlung ein oder
mehrerer Phasen in andere. Phasenübergänge können zwischen festen, flüssigen und gasförmigen
Phasen auftreten und nach Ehrenfest klassifiziert werden. Die Legierung Kupfer-Gold mit einem
Mischungsverhältnis von 75 : 25 Atomprozent ist einerseits geeignet um Grundlagen von Phasenübergängen zu erläutern, andererseits bietet sie interessante Möglichkeiten um das Verständnis zu
vertiefen. Abhängig von der Temperatur der Legierung ordnen sich die Kupfer- und Goldatome mehr
oder weniger geordnet an und Veränderungen in der Kristallstruktur können beobachtet werden.
Die röntgenographische Methode ist in der Materialphysik ein gängiges Verfahren um die Gitterstruktur zu analysieren. Mit Hilfe eines Röntgendiffraktometers in Kombination mit einer Heizkammer
ist es möglich, eine Röntgenuntersuchung in unterschiedlichen Temperaturbereichen vorzunehmen.
Röntgendiffraktometrie der CuAu-Probe bei gleichzeitigem Aufheizen in die Nähe der entsprechenden Phasenübergangstemperatur ermöglicht eine Untersuchung der Phasenumwandlung.
Kalorimetrische Methoden werden in der Materialphysik üblicherweise genutzt um Phasenumwandlungen näher zu charakterisiern. Eine vollständige Beschreibung der Kinetik ist durch Kombination
beider Methoden möglich, in dem die Phasenumwandlung im Kalorimeter beobachtet wird und die
Änderung der Gitterstruktur mittels der Röntgendiffraktometrie nachvollzogen werden kann.
2. Theoretische Grundlagen
Die röntgenographische Analyse von Materialien ist für sich alleine schon ein komplexes und technisch aufwendiges Verfahren zur Merkmalsbeschreibung von Kristallstrukturen. Die Funktionsweise
eines Röntgendiffraktometers und der theoretische Hintergrund auf dessen Basis die Auswertung der
Interferenzspektren vorgenommen wird, stellt neben der Ordnungsproblematik der CuAu-Legierung
den ersten Schritt in der theoretischen Betrachtung dar. Eine parallele Temperaturbehandlung der
Proben erfordert zudem, dass die Theorie eine Wärmebewegung der Atome im Kristallgitter berücksichtigt wird
2.1. Das Phasendiagramm von Kupfer-Gold Legierungen
Um eine Gesamtübersicht über die möglichen Zustände von Kupfer-Gold Legierungen in Abhängigkeit von Temperatur und Mischungsverhältnis zu erhalten, empfiehlt sich ein Blick auf das entsprechende Phasendiagramm (s. Abb. 2.1). Das System Kupfer-Gold ist oberhalb von 390 °C lückenlos
mischbar und abhängig von der Zusammensetzung der Legierung ergeben sich unterschiedliche Temperaturen für Schmelze, 2-Phasensysteme und Umwandlung in die jeweiligen Mischkristalle.
Bei einer theoretischen Zusammensetzung von 25 Atom-% Gold und 75 Atom-% Kupfer lassen sich
aus dem Phasendiagramm folgende Aussagen ableiten:
• CuAu schmilzt bei einer Temperatur von etwa 950 °C. Dies muss bei der Wärmebehandlung
der Proben berücksichtigt werden.
• In einem Temperaturbereich zwischen 390 °C und 900 °C erstarrt CuAu aus der Schmelze und
liegt anschließend in einem kubisch-flächenzentriertem (kfz) Gitter vor, wobei aber die Kupferund Goldatome einen beliebigen Platz im Gitter einnehmen. Sie sind ungeordnet.
• Unterhalb einer Temperatur von 390 °C stellt sich eine Ordnung der Atome ein und es bilden
sich Überstrukturen in der Legierung aus.
• Kühlt man CuAu langsam auf Raumtemperatur herunter, so bildet sich die Ordnungsphase
Cu3 AuI aus, die auch stabil bestehen bleibt.
2.1 Das Phasendiagramm von Kupfer-Gold Legierungen
Abbildung 2.1.: Phasendiagramm von Kupfer-Gold [nach IFW11].
2.1.1. Phasenumwandlung von CuAu
Die für die Versuche verwendeten Proben weisen ein Mischungsverhältnis von 25 Atom-% Gold und
75 Atom-% Kupfer auf (CuAu), sodass sich der für die Beobachtungen relevante Teil des Phasendiagramms auf den mittleren Ausschnitt reduziert. Oberhalb von 390 °C besitzt CuAu eine kubischflächenzentrierte Kristallstruktur, wobei die Gitteratome statistisch verteilt sind, d.h. die Positionen
an denen sich Gold- oder Kupferatome befinden, sind zufällig. Ursächlich hierfür ist der mit steigender Temperatur T zunehmende Einfluss der Entropie S und das daraus resultierende Absenken der
Gibbsschen freien Energie G:
G = H − TS ,
(2.1)
wobei H die Enthalpie der thermodynamischen Phase ist und der Energie des Kristallgitters entspricht. Diese Energie berücksichtigt die Mischungstendenz der Komponenten Cu und Au und bein
haltet als wesentlichen Parameter die Konfigurationsenergie εAB − 12 (εAA + εBB ) zwischen den einzelnen Gitteratomen. Sie ist die wesentliche treibende Kraft die eine Ausbildung der Ordnung voran
treibt. Welche Phase sich bei welchen Temperaturen als stabil herausbildet, entscheidet sich über die
Minimierung der freien Energie. Oberhalb von 390 °C nimmt der Einfluss der Entropie (Unordnung)
zu und wirkt der Konfigurationsenergie entgegen. Dadurch bildet sich eine regellose Atomverteilung
aus (s. Abb. 2.2 links).
Unterhalb einer Temperatur von 390 °C (vgl. Abb. 2.1) ist der Einfluss der Entropie gering genug,
sodass eine Minimierung der Vertauschungsenergie zu einem Minimum der freien Energie führt. Unter Berücksichtigung der chemischen Ähnlichkeit, des Atomradius und der Gitterstruktur durchläuft
© SoSe 2015
6
2.1 Das Phasendiagramm von Kupfer-Gold Legierungen
Abbildung 2.2.: Statische Verteilung der Atome auf die Gitterplätze:
(a) links: Ohne eine Ausbildung von Überstrukturen.
(b) rechts: Mit ausgeprägter Ordnungsstruktur.
die Legierung beim Abkühlen eine Phasenumwandlung und es stellt sich eine geordnete Struktur ein,
in der die Kupfer- und Goldatome wie in Abbildung 2.2 (rechts) gezeigt, angeordnet sind.
Phasenübergänge können sich unter verschiedenen thermodynamischen Randbedingungen ausbilden
und weisen merkliche Unterschiede auf. Zwei grundlegende Klassen sind zusätzlich im Anhang A
vorgestellt.
In der geordneten Struktur sitzen die Goldatome auf den Kanten und die Kupferatome auf den Flächen. Festzustellen ist, dass in Summation die Elementarzelle wie gefordert insgesamt aus 3 Kupferatomen (6 · 12 ) und 1 Goldatom (8 · 18 ) besteht. Diese geordnete Kristallstruktur trägt die Bezeichnung
Cu3 AuI. Sie besteht aus 4 sc-Gittern (einfach kubisch) die ineinander liegen. Dabei befinden sich die
Gold-Atome auf einem sc-Gitter, welches mit den bisherigen Ecken des fcc-Gitters übereinstimmt. Die
flächenzentrierten Plätze des ungeordneten fcc-Gitters kann man nun als drei sc-Gitter beschreiben,
die von Kupferatomen besetzt sind. Allgemein wird dieser Typ als L12 bezeichnet.
Die Ausbildung der Ordnung in dem Kristallgitter hängt allerdings wesentlich davon ab, wie schnell
der Abkühlprozess verläuft, da das “Suchen“ nach dem richtigen Gitterplatz, bzw. das Minimieren
der Vertauschungsenergie durch Diffusionsprozesse stattfindet und damit an die thermische Aktivität
der Atome gekoppelt ist. Im Idealfall wird die Legierung also unendlich langsam auf Umgebungstemperatur abgekühlt, sodass sie sich ständig im thermodynamischen Gleichgewicht befindet. Damit
haben die Atome ausreichend Zeit um sich ideal, also im energetisch günstigsten Zustand, im Gitter anzuordnen. Je höher die Abkühlrate, desto unwahrscheinlicher wird es, dass alle Atome ihren
energetisch günstigsten Gitterplatz einnehmen können. Im Extremfall wird die Legierung durch Abschrecken (etwa durch Eiswasser) so schnell abgekühlt, dass die Gitteratome nun praktisch keine
Möglichkeit haben sich anzuordnen. Sie verharren gewissermaßen in der zufälligen Position die sie
aufgrund der thermischen Energie temporär eingenommen haben.
Wie lässt sich nun die Ordnungsstruktur der Legierung im Hinblick auf ihre Ausprägung beschreiben?
Wie groß ist der Anteil zum Beispiel der Goldatome die auf dem für sie vorgesehenen Gitterplatz
sitzen. Diese Fragestellung bedarf noch einer ausführlichen Behandlung.
© SoSe 2015
7
2.1 Das Phasendiagramm von Kupfer-Gold Legierungen
Der vorangegangene Abschnitt lässt sich wie folgt zusammenfassen:
Bei der Kinetik der Ordnungsbildung spielen Temperatur und Vertauschungsenergie die wesentlichen
Rollen. Die Temperatur beeinflusst die nötigen Diffusionsvorgänge damit die Gitteratome den energetisch günstigsten Gitterplatz erreichen können und die Vertauschungsenergie bestimmt in Konkurrenz
zur Entropie, ab welcher Temperatur eine Ordnung energetisch günstiger ist. In welcher Ausprägung
sich eine Ordnungsstruktur nach einem Abkühlvorgang einstellt, lässt sich durch den Ordnungsparameter η ausdrücken. Wie dieser theoretisch beschrieben und experimentell bestimmt werden kann
wird in Abschnitt 2.4 diskutiert.
© SoSe 2015
8
2.2 Röntgenbeugung am Kristall
2.2. Röntgenbeugung am Kristall
Im vorangegangenen Abschnitt wurde die Existenz verschiedener Phasen in der CuAu-Legierung
ausführlich beschrieben. Unordnung, Ordnung mit tetragonaler und mit rhombischer Struktur stellen
sich unter bestimmten thermodynamischen Randbedingungen ein. Jetzt geht es um die Frage, wie
sich diese unterschiedlichen Strukturen erkennen lassen. Dazu gibt es in der Materialphysik drei
gängige Verfahren:
• Laue-Verfahren: Ein Einkristall wird unter festen Winkel mit Röntgenstrahlung kontinuierlicher Wellenlänge bestrahlt, um dessen Struktur zu bestimmen.
• Debye-Scherrer-Verfahren: Es wird eine polykristalline Probe mit monochromatischer Röntgenstrahlung bestrahlt, dabei wird der Reflexionswinkel variiert.
• Drehkristallverfahren: Hierbei ist der Betrachtungswinkel nicht konstant und der Einkristall
wird mit monochromatischer Röntgenstrahlung bestrahlt.
Da es sich bei Kupfer-Gold um einen Polykristall handelt ist das Debye-Scherer-Verfahren das Mittel
der Wahl. Die Theorie hinter dieser technisch aufwendigen Methode soll im Folgenden vorgestellt
werden. Dabei werden in diesem Abschnitt die allgemeinen Grundsätze diskutiert die die Grundlage
der Röntgendiffraktometrie bilden. Im darauf folgendem Abschnitt geht es um die spezielle Theorie
einer Röntgenanalyse unter hoher thermischer Aktivität.
2.2.1. Die Millerschen Indizes
Die Theorie der röntgenographischen Methode und die Auswertung der gewonnen Spektren erfolgt
unter einem speziellen Terminus, welcher in dieser Form explizit nur in der Kristallographie angewandt wird. Die Millerschen Indizes dienen der eindeutigen Identifizierung von Kristallebenen und
sind eine wichtige Hilfe bei der Identifizierung der gewonnenen Röntgenpeaks.
Betrachtet wird ein kubischer Kristall, dem sich ein orthogonales Koordinatensystem zuordnet lässt
→
− −
−
und dessen Basis durch die Vektoren →
a , b ,→
c gebildet wird (s. Abb. 2.3). Beliebige Richtungen im
Kristallgitter können somit aus den Elementen dieser Basis linear kombiniert werden. Beispielsweise
→
−
−
−
−
−c .
ist die Richtung →
r1 eine Linearkombination aus →
r1 = →
a + b +0·→
Die Millerschen Indizes stellen ein Zahlentripel hkl (mit h,k,l ∈ Z) dar, welches den Vorfaktoren der
Linearkombination entspricht. Diese Vorfaktoren müssen allerdings ganzzahlig sein. Negative h,k,l
werden mit h̄, k̄, ¯l bezeichnet. Zusätzlich wird, um deutlich zu machen das es sich um eine Richtung
→
−
−
−
−c = [110]. Die
im Kristall handelt, die Schreibweise [hkl] gewählt. Beispiel: →
r1 = →
a + b +0·→
−
Richtung [1̄1̄0] würde der entgegengesetzten Richtung von →
r1 entsprechen.
Kristallebenen können ebenfalls durch die Millerschen Indizes dargestellt werden, allerdings in der
→
− −
−
Schreibweise (hkl). Diese beschreiben diejenige Ebene, welche die Achsen →
a , b ,→
c an den Punkten
© SoSe 2015
9
2.2 Röntgenbeugung am Kristall
Abbildung 2.3.: Kristall im kartesischen Koordinatensystem (KOS) und verschiedene Netzebenen
zu jeweiligen hkl-Indizes.
→
− −
−
1→
a , k1 b , 1l →
c
h
schneidet. Da sich das Raumgitter durch Symmetrieoperationen über den gesamten
Kristall erstreckt, beschreibt ein Tripel von Indizes direkt die vollständige Ebenenschar (vgl. Abb.
2.3). Die Netzebene (100) beschreibt somit alle Ebenen die den rechten Würfelflächen entsprechen.
Zusätzlich gilt in einem kubischen Kristall, dass die Ebenen (hkl) immer senkrecht auf den jeweiligen
Richtungen [hkl] stehen. Dies veranschaulichen die Ebenen (110) und (220) aus Abbildung 2.3. Die
→
− −
→
− −
−
−
Ebene (220) schneidet die Vektoren →
a , b ,→
c an den Punkten 12 →
a , 21 b , 10 →
c wobei 0 bedeutet, dass
→
−
die Ebene keinen Schnittpunkt mit der Achse c besitzt sondern parallel dazu verläuft. Abbildung
2.4 stellt einige Beispiele verschiedener (hkl)-Ebenen in einem kubischen Kristall dar2 .
Abbildung 2.4.: Beispiele von Netzebenen in einem kubischen Kristall.
2
Die Netzebene (111) erreicht durch eine Translation ~t = ~a die dargestellte Position.
© SoSe 2015
10
2.2 Röntgenbeugung am Kristall
2.2.2. Braggsche Reflexionsbedingung
In einem Röntgendiffraktometer wird ein Kristall kontinuierlich einer monochromatischen Röntgenstrahlung ausgesetzt, welche mit den Gitteratomen wechselwirkt. Dabei ist die Beugung der Röntgenstrahlung an den Gitteratomen der zu berücksichtigende physikalische Sachverhalt. Das Beugungsbild
hängt signifikant davon ab, ob die Streuung an einem Gold- oder Kupferatom stattfindet. Daher kann
aus dem gewonnenen Röntgenspektrum auf die vorliegende Gitterstruktur rückgeschlossen werden.
In Abbildung 2.5 ist schemenhaft dargestellt, wie der physikalische Hintergrund zu diskutieren ist.
Dabei wird eine Ebene von Gitteratomen als Ebene bzw. Netzebene des Kristallgitters betrachtet.
Somit wird die Problematik der Wechselwirkung mit den einzelnen Gitteratomen überführt in eine
Wechselwirkung mit der entsprechenden Netzebene.
Abbildung 2.5.: Konstruktive Interferenz von Röntgenstrahlen an Kristallebenen nach William Lawrence Bragg.
Trifft monochromatische Röntgenstrahlung auf einen Kristall, so wird ein Teil der Strahlung an jeder
Netzebene transmittiert und reflektiert. Eine konstruktive Interferenz der reflektierten Strahlung
tritt bekanntermaßen nur dann auf, wenn der Gangunterschied einem Vielfachen der Wellenlänge
entspricht. Dieser Gangunterschied berechnet sich gemäß Abbildung 2.5 und einfachen geometrischen
Überlegungen zu:
2d · sin ϑ = nλ n ∈ N,
(2.2)
mit dem Netzebenen Abstand d, der Ordnung n des betrachteten Maximums und dem Winkel ϑ der
einfallenden Strahlung. Diese Bedingung ist sehr scharf und führt daher zu sehr genauen Interferenzpeaks im Beugungsbild. Der Netzebenenabstand d einer bestimmten (hkl)-Ebene (kurz: dhkl ) hängt
von der Symmetrie der Kristallstruktur und damit von den für eine vollständige Beschreibung nötigen Gitterkonstanten ab (s. Abb. 2.2). Für die in dieser Arbeit behandelten CuAu-Proben werden
© SoSe 2015
11
2.2 Röntgenbeugung am Kristall
folgende Zusammenhänge benötigt, welche sich direkt mit Hilfe der Hesseschen Normalform und der
sich daraus ergebenden Abstandberechnung zum Ursprung des R3 herleiten lassen:
Bei einem einfachen kubischen Kristall mit der Gitterkonstanten a:
dhkl = √
h2
a
+ k 2 + l2
(2.3)
Bei einem tetragonal verzerrtem Kristall mit den Gitterkonstanten a und c:
1
d2hkl
=
h2 + k 2
l2
+
a2
c2
(2.4)
Bei einem rhombischen Kristallsystem mit den Gitterkonstanten a,b und c:
1
d2hkl
=
h2 k 2
l2
+
+
a2
b2
c2
(2.5)
Wenn sich in einem Kristall eine ausgeprägte Ordnungsstruktur zwischen den Legierungspartnern
einstellt, dann bilden sich Überstrukturen aus, für die weitere Netzebenen verantwortlich sind. Im
einfachen 2-dimensionalen Fall zeigt Abbildung 2.6 anschaulich wie durch Ordnungsbildung ein weiteres Untergitter entsteht, welches einen anderen Netzebenenabstand besitzt. Die Gitterkonstante a
ist logischerweise in beiden Fällen immer noch identisch.
Abbildung 2.6.: Zusammenhang zwischen Ordnung und Netzebenenabstand [Got07].
Wie gelingt es nun diese zusätzlichen Netzebenen mit Hilfe eines Röntgendiffraktometers zu identifizieren? Die Antwort auf diese Frage liegt in dem oben schon erwähnten unterschiedlichen Streuvermögen der beteiligten Kupfer- und Goldatome. Sie haben verschiedenen Einfluss auf die Beugung
der Röntgenstrahlen und daher treten in dem entsprechendem Beugungsbild zusätzliche Reflexe (sog.
Überstrukturreflexe) auf welche sich daraufhin einer bestimmten Netzebene zuordnen lassen.
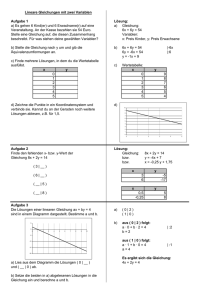
⇒ Aufgabe zur theoretischen Vorbereitung: Überlegen Sie wie Sie vorgehen müssen, um die ge-
© SoSe 2015
12
2.3 Einfluss der Wärmebewegung auf die Interferenz von Röntgenstrahlen
wonnenen Röntgenspektren mit den geeigneten Netzebenen (hkl) zu identifizieren. Anders ausgedrückt, wie ist es möglich jedem Peak in einem Röntgenspektrum unter Kenntnis der Kristallstruktur (kubisch, tetragonal o. rhombisch) die ihm zugehörige Netzebene zuzuordnen.
2.3. Einfluss der Wärmebewegung auf die Interferenz von
Röntgenstrahlen
Die in den vorangegangenen Abschnitten erwähnten Zusammenhänge und theoretischen Modelle zwischen der Interferenz von Röntgenstrahlen und der Bestimmung der Kristallstruktur müssen nunmehr
um eine wichtige Sachlage ergänzt werden. In diesem Praktikumsversuch soll die Kristallstruktur bei
bestimmten Temperaturen analysiert werden. Es ist nunmehr überhaupt nicht klar, ob das Verhalten der Röntgenpeaks bzw. des Röntgenspektrums unabhängig von einer solchen Wärmebehandlung
der Probe ist. Peter Debye hat Anfang des 20. Jahrhunderts eine Abhandlung veröffentlicht, in der
die theoretische Beschreibung des Einflusses der Wärmebewegung auf die Interferenz von Röntgenstrahlung mathematisch hergeleitet und diskutiert wird [Deb14]. Die Kernaussagen werden hier kurz
aufgeführt und ein experimenteller Zugang vorgestellt. Für näheres Verständnis der mathematischen
Zusammenhänge kann bei Verlangen der Anhang B konsultiert werden.
2.3.1. Die Kernaussagen von Debye
Zusammenfassend seien die zentralen Aussagen von Debye hier festgehalten. Sie resultieren zum
einen direkt aus der strengen mathematischen Modellierung und zum anderen aus den experimentellen Vorhersagen welche in Anhang B dargestellt sind. Ob es gelingen wird diese Aussagen in den
Experimenten auch quantitativ genau wiederzufinden wird sich zeigen, sollte aber mit Skepsis betrachtet werden. Der anschließende Abschnitt greift die Problematik der Quantifizierung nochmals
auf. Hauptziel sollte in erster Linie sein, die richtigen Tendenzen in den Röntgenspektren wiederzufinden, nämlich die Kernaussagen von Debye und Waller:
1. Die Schärfe der Interferenzmaxima wird durch die Wärmebewegung nicht beeinflusst.
2. Durch die Wärmebewegung ändert sich die räumliche Intensitätsverteilung.
3. Aufgrund der Wärmebewegung nimmt die Interferenzintensität
a) mit zunehmender Temperatur
b) mit zunehmendem Winkelabstand zwischen Einfalls- und Beobachtungsrichtung
c) mit abnehmender Wellenlänge
exponentiell ab.
© SoSe 2015
13
2.3 Einfluss der Wärmebewegung auf die Interferenz von Röntgenstrahlen
Die Änderung der räumlichen Intensitätsverteilung, gegeben durch
(2.6)
I = I0 · exp (2M )
lässt sich in der Praxis sehr deutlich erkennen und wird im nachfolgendem Abschnitt näher behandelt.
2.3.2. Experimentelle Bestimmung des Debye-Waller Faktors
Der grundlegende Einfluss der Temperatur auf die Interferenzen der Röntgenpeaks ist aus den obigen Aussagen deutlich geworden. Experimentelle Ergebnisse haben allerdings gezeigt, dass die tatsächliche Abnahme der Intensität von der Debyeschen Theorie deutlich unterschätzt wird. Folgende
kritischen Punkte müssen angeführt werden:
• Es ist unklar ob für den Mischkristall CuAu nur ein einzelner Debye-Waller Faktor zu berücksichtigen ist.
• In seiner Arbeit [Deb14] nähert Debye die Atommasse von Sylvin aus den Mittelwerten der
beteiligten Komponenten Kalium und Chlorid. Dies ist zulässig, da sich die Atommassen der
beiden Elemente sehr ähnlich sind (K = 39 u, Cl = 35 u). Für CuAu ist diese Methode unter
Umständen nicht geeignet weil die Atommassen in einem deutlich ungünstigeren Verhältnis
stehen (1:3).
• Die Debye-Temperatur wurde als konstant angenommen und aus den Werten für Kupfer und
Gold gemittelt. Dies ist als wesentliche Kritik an dieser Methodik festzuhalten.
Unter Berücksichtigung dieser Aspekte erscheint es verständlich, dass die theoretische Modellierung
den realen Verlauf der Intensitätsabnahme nicht exakt darstellen kann. Aus diesem Grund wurde der
Debye-Waller Faktor auf Basis experimenteller Ergebnisse korrigiert. Im folgenden wird, wie unten
beschrieben, der DWF M durch den leicht abgewandelten Faktor B ersetzt. Die experimentellen
Ergebnisse ergaben folgende Korrektur:
25 °C
200 °C 300 °C
380 °C 400 °C 500 °C
700 °C
Bkorr
0,1749
0,8487
1,2337
1,523
1,6187
2,0037
2,7737
BTheo
Debye mit NPE
0,4120
0,6475
0,7825
0,8538
0,9141
1,0526
1,3262
Tabelle 2.1.: Experimentell bestimmte und theoretisch berechnete Werte für den Debye-Waller Fak2
tor im Vergleich (alle Werte in Å ).
Dieser korrigierte Verlauf soll in der Auswertung dazu dienen den Einfluss der Wärmebewegung zu
berücksichtigen und somit den Fernordnungsparameter sinnvoll um diesen Einfluss zu korrigieren.
© SoSe 2015
14
2.3 Einfluss der Wärmebewegung auf die Interferenz von Röntgenstrahlen
Abbildung 2.7.: Abnahme der Intensität gemäß der Debyeschen Theorie (vgl. Abb. B.1) und nach
2
einer Korrektur auf Basis experimenteller Ergebnisse. M = B · sinλ2(ϑ) (s. unten).
Ausführlich wird die Bestimmung des Fernordnungsparameters in Abschnitt 2.4.1 behandelt.
Folgende Überlegung zeigt wie der Parameter M in den Parameter B überführt wurde:
Gemäß der Debyeschen Theorie lässt sich der Abfall der Intensität nach Gleichung B.2 (im Anhang)
darstellen mit M gemäß B.6 (Anhang), wobei der Faktor P zu 0, 073 berechnet wurde und die Werte
für Φ(x)
für einige Temperaturen in Tabelle B.1 (Anhang) aufgeführt sind. Wird die Winkelabhängigx
keit und die Wellenlänge aus dem Ausdruck isoliert und werden zusätzlich die restlichen konstanten
Faktoren zusammenfasst, so lässt sich M auch darstellen als:
1
M = Pe 2 (1 − cos ϕ)
λ
1
= Pe 2 (1 − cos(2ϑ))
λ
1
= Pe 2 (1 − (cos2 (ϑ) − sin2 (ϑ)))
λ
1
= Pe 2 · 2 sin2 (ϑ)
λ
sin2 (ϑ)
⇒M =B·
,
λ2
© SoSe 2015
(2.7)
15
2.3 Einfluss der Wärmebewegung auf die Interferenz von Röntgenstrahlen
2
wobei jetzt der Braggsche Winkel ϑ die unabhängige Variable darstellt und B die Einheit Å besitzt.
implizit enthalten
Die Temperatur lässt sich nicht direkt isoliert darstellen, weil sie im Ausdruck Φ(x)
x
ist. Die Transformation in den Faktor B ist inhaltlich eigentlich unbedeutend, ermöglicht aber noch
einen weiteren wesentlichen Schritt. James hat in seiner Arbeit ([Jam62]) hergeleitet, wie aus dem
Faktor B ein Maß für die mittlere quadratische Auslenkung jedes einzelnen Atoms gewonnen werden
kann:
hu2 i =
3
B
8π 2
(2.8)
2
hu2 i trägt die Einheit Å . Es ist somit möglich aus den gewonnenen Debye-Waller Faktoren darauf
zu schließen welchen Einfluss Temperaturschwankungen auf die Gitterschwingungen haben.
2.3.3. Temperaturabhängigkeit der Gitterkonstanten
Nach Debye ändert sich die räumliche Intensitätsverteilung der Reflexe mit einer von der Temperatur
abhängigen Wärmebewegung der Atome. In den Röntgenspektren lässt sich dies deutlich erkennen.
Mit steigender Temperatur verschieben sich die Röntgenpeaks um wenige zehntel Grad, hin zu kleineren Winkeln was einer Vergrößerung der Gitterkonstanten entspricht. Es gilt jetzt die Frage zu
klären, in welchem Zusammenhang diese Vergrößerung mit einem Anstieg der Temperatur steht.
Eine Lösung dieser Fragestellung wird nur gewonnen, wenn das Wechselwirkungspotential Ψ über
eine anharmonische Näherung modelliert wird. Im Fall einer harmonischen Näherung ist der mittlere Abstand der Atome temperaturunabhängig. Er bleibt auch mit steigender Temperatur immer
mittig im Potential positioniert. Bei einer anharmonischen Näherung des Wechselwirkungspotential
verschiebt sich der mittlere Abstand der Atome mit steigender Temperatur hin zu größeren Werten
(s. Abb. 2.8 [GM11]).
Um die Gitterenergie zu beschreiben ist es nötig das Wechselwirkungspotential in eine Taylor-Reihe
zu entwickeln und die übliche Anzahl der Termine dieser Entwicklung in einer Näherung zu berücksichtigen. Die ausführliche Berechnung findet sich im Anhang D. An dieser Stelle sei nur das Ergebnis
dargestellt:
hr − r0 i =
3 b
kB · T + const.
4 a2
(2.9)
Es wird somit deutlich, dass zwischen einer Temperaturänderung und dem mittleren Abstand der
Gitteratome ein linearer Zusammenhang besteht. In der Auswertung soll mit diesem Wissen überprüft
werden ob die Gitterkonstante, welche den mittleren Abstand der Atome repräsentiert, tatsächlich
linear mit höheren Temperaturen ansteigt.
© SoSe 2015
16
2.3 Einfluss der Wärmebewegung auf die Interferenz von Röntgenstrahlen
Abbildung 2.8.: Verschiebung des mittleren Abstands im Kristallgitter mit steigender Temperatur. Nur mit dem Ansatz einer anharmonischen Näherung für das interatomare
Wechselwirkungspotential lässt sich die thermische Ausdehnung beschreiben. Im
Fall einer harmonischen Näherung ändert sich der mittlere Abstand mit der Temperatur nicht.
2.3.4. Zusammenhang zwischen der mittleren Auslenkung der Atome und
der Gitterkonstanten des Kristalls
Aus experimentellen Messungen ist es mit obigen Überlegungen möglich die mittlere Auslenkung der
Gitteratome für verschiedene Temperaturen zu bestimmen. Parallel zu den experimentellen Ergebnissen hat sich auf Basis der Debyeschen Theorie ergeben, dass der Faktor B ungefähr linear mit der
Temperatur zusammenhängt (s. Anhang C,
qAbb. C.1). Resultiert allerdings die mittlere Auslenkung
p
2
direkt über den Zusammenhang hu i = 8π3 2 B aus diesem Faktor, so zeigt sich offensichtlich eine
√
Abhängigkeit von der Temperatur mit T .
p
√
hu2 i ∝ T
(2.10)
Folgender interessanter Vergleich kann nun gezogen werden: Wie ändert sich das Verhältnis zwischen der mittleren quadratischen Auslenkung der Gitteratome hu2 i und der Gitterkonstanten a mit
steigender Temperatur? Nach Lindemann gilt folgender Zusammenhang:
Lindemann Kriterium: Ein Kristall schmilzt, wenn die Amplitude der Gitterschwingungen einen
kritischen Wert erreicht, der ungefähr einem Zehntel des interatomaren Abstands entspricht.
Ist ã = √12 a der Abstand nächster Nachbarn im Kristallgitter, dann muss am Schmelzpunkt
© SoSe 2015
17
2.3 Einfluss der Wärmebewegung auf die Interferenz von Röntgenstrahlen
ungefähr gelten:
p
hu2 i
· 100 > 10%
ã
(2.11)
√
hu2 i
Durch Kenntnis dieses Verhältnisses für niedrige Temperaturen kann eine Extrapolation ( ã gegen
√
T ) für den Schmelzpunkt gewonnen und ein Vergleich mit diesem Kriterium angestrebt werden.
© SoSe 2015
18
2.4 Theorie der Fernordnung
2.4. Theorie der Fernordnung
In Abschnitt 2.1.1 wurde die Fragestellung eröffnet, wie sich nun die ausgeprägte Ordnung in einem Kristallgitter qualitativ beschreiben lässt. Im Folgenden wird eine theoretische Modellierung
der Fernordnung hergeleitet und anschließend eine Möglichkeit vorgestellt diese auch aus den experimentellen Ergebnissen zu bestimmen.
Die nachfolgende thermodynamische Modellierung basiert auf Überlegungen der statistischen Physik und behandelt als Kernproblematik die Wahrscheinlichkeit, dass ein bestimmtes Atom am für
ihm vorgesehenen “richtigen“ 5 Platz sitzt. Der Grad der Fernordnung ist definiert als der Bruchteil
der A-Atome die sich auf dem richtigen Gitterplatz befinden im Verhältnis zu denjenigen, die falsch
positioniert sind.
1+η
PeA
=
,
(2.12)
PA
1−η
wobei PA die Wahrscheinlichkeit beschreibt, dass ein A-Atom auf dem richtigen Platz sitzt und PeA
dementsprechend über 1−PA die Wahrscheinlichkeit für eine Fehlbesetzung ausdrückt. Aus Gründen
der Nullteilerfreiheit wird der Ordnungsparameter η eingeschränkt auf 0 < η ≤ 1, wobei 1 vollständige
Ordnung und 0 vollständige Unordnung beschreibt.
2.4.1. Experimentelle Bestimmung der Fernordnung
Die Intensität Ihkl eines Röntgenreflexs zu einer bestimmten Netzebene kann über den komplexen
Strukturfaktor Fhkl , den Flächenhäufigkeitsfaktor p, den Lorentz-Polarisationsfaktor Lp und den
Debye-Waller-Faktor DT über den Zusammenhang
Ihkl ∝ |Fhkl |2 · p · Lp · DT
(2.13)
beschrieben werden. Die Intensität ist also bis auf eine Proportionalitätskonstante theoretisch zu
bestimmen.
• Der Strukturfaktor Fhkl berücksichtigt das Streuvermögen einer Elementarzelle. Abhängig vom
Ordnungsgrad η steigt oder sinkt der Beitrag des Streuvermögens zur Intensität. Da der Strukturfaktor eine komplexe Größe ist geht er mit seinem Betragsquadrat in die Intensität der
Röntgenpeaks ein. Er wird im Anschluss ausführlich beschrieben.
• Der Flächenhäufigkeitsfaktor p berücksichtigt, dass in einem Kristall die verschiedenen Netzebenen unterschiedlich häufig vorkommen aber bezüglich der Bragg-Bedingung identisch sind.
Es gibt zum Beispiel sechs symmetrisch äquivalente Ebenen6 zu (001) aber acht zu (111). Er
5
“Richtig“ in Bezug auf die geordnete Struktur.
© SoSe 2015
19
2.4 Theorie der Fernordnung
ist in Tabelle 2.2 für einen kubischen Kristall und mögliche Netzebenen aufgeführt. Dabei bezeichnet die Schreibweise {hkl} alle entsprechenden Symmetrieebenen.
(z.B.: {h00} ∼ (100) ∼ (200) ∼ (050) ∼ (00l))
Netzebene
p - kubisch
{h00}
6
{00h}
6
{hhh}
8
{0kk}
12
{0kl}
24
{hhl}
24
{hkl}
48
Tabelle 2.2.: Flächenhäufigkeitsfaktor p für ein kubisches Kristallsystem.
• Mit dem Lorentz-Polarisationsfaktor Lp werden üblicherweise verschiedene geometrische Ef2 (2ϑ)
fekte der Röntgenbeugung korrigiert. Er lässt sich über den Zusammenhang Lp = sin1+cos
2 (ϑ)·cos(ϑ)
bestimmen, wenn ϑ der Reflektionswinkel ist.
• Der Debye-Waller-Faktor DT berücksichtigt wie beschrieben den Einfluss der Wärmebewegung
auf die Intensität, abhängig vom Winkel7 ϕ und der Temperatur. Er lässt sich wie oben schon
einmal erläutert anhand von Gleichung B.7 beschreiben.
Der komplexe Strukturfaktor Fhkl berücksichtigt die Lage der Atome in der Elementarzelle und die
Atomformfaktoren fi der beteiligten Legierungspartner um das Streuvermögen jeder Netzebene in
Abhängigkeit vom Ordnungsparameter η beschreiben zu können.
Fhkl ∝
n
X
fi e2πi(hxi +kyi +lzi )
(2.14)
i=1
wenn n die Anzahl der Basisatome beschreibt und sich die Atomformfaktoren über die Gleichung
fAtom (s) =
n
X
2
ai e−bi s + c ,
(2.15)
i=1
mit s = sin(ϑ)
([λ] = Å), für verschiedenen Streuwinkel annähernd bestimmen lassen. Die Parameter
λ
ai , bi , c sind in Tabelle 2.3 für Kupfer und Gold aufgeführt.
6
Zur Äquivalenzklasse von (001) gehören (100), (010), (001) und deren Gegenüberliegende.
7
Vorsicht: Dies ist ein anderen Winkel als ϑ (s. 2.3.1).
© SoSe 2015
20
2.4 Theorie der Fernordnung
i=1
i=2
i=3
i=4
i=5
6,932996
0,265666
Cu
ai
bi
c
14,014192
3,738280
-3,254477
4,784577
0,003744
5,056806
13,034982
1,457971
72,554794
Au
ai
bi
c
16,777390
0,122737
-6,279078
19,317156
8,621570
32,979683
1,256902
5,595453 10,576854
38,008820 0,000601
2
Tabelle 2.3.: Die Parameter ai , bi und c (in Å ) zur näherungsweisen Bestimmung der Atomformfaktoren für Kupfer und Gold [WK95].
Die Elementarzele von Cu3 Au hat eine vieratomige Basis (s. Abb. 2.9) an den Positionen
a
a
a a
a a
(0, 0, 0) ; ( , , 0) ; ( , 0, ) und (0, , ) ,
2 2
2
2
2 2
im Falle eines kubischen Gitters mit der Kantenlänge a.
Abbildung 2.9.: Die Elementarzelle eines kfz-Gitters im Fall CuAu.
Damit lässt sich die Summation des Strukturfaktors ausführen:
Fhkl = f1 + f2 eπi(h+k) + f3 eπi(h+l) + f4 eπi(k+l)
(2.16)
Dieser Ausdruck liefert interessante Ergebnisse wenn er hinsichtlich eines Ordnungsparameters untersucht wird.
© SoSe 2015
21
2.4 Theorie der Fernordnung
Für vollständige Ordnung (η = 1) ist jedes Atom an der richtigen Stelle positioniert. Somit ist
auch die Basis der Elementarzelle wohlgeordnet. Wie in Abbildung 2.9 ersichtlich besteht die
Basis an der Atompositionen (0, 0, 0) aus einem Goldatom und und bei (0, a2 , a2 ), ( a2 , a2 , 0) und
( a2 , 0, a2 ) aus Kupferatomen. Somit reduziert sich der Strukturfaktor aus Gleichung 2.16 zu:
η=1
= fAu + fCu · (eπi(h+l) + eπi(k+l) + eπi(h+k) )
Fhkl
Diskutiert man diese Summe im Hinblick auf gerade oder ungerade Kombinationen der Millerschen Indizes, so gilt für den Strukturfaktor:
η=1
Fhkl
fAu + 3 fCu ,
f − f ,
Au
Cu
=
f − f ,
Au
Cu
f − f ,
Au
Cu
für nur gerade oder ungerade h,k,l
für h ∼ k und h, k l
8
für h ∼ l und h, l k
(2.17)
für k ∼ l und k, l h
Für vollständige Unordnung (η = 0) ist jedes Atom mit einer Wahrscheinlichkeit 14 ein Goldatom.
Somit setzt sich auch die Basis der Elementarzelle zufällig aus den Kupfer- bzw. Goldatomen
zusammen. Alle Atomformfaktoren f1 · · · f4 entsprechen daher dem Ausdruck ( 34 fCu + 14 fAu ),
wodurch sich der Strukturfaktor insgesamt reduzieren lässt auf die Form:
1
3
η=0
Fhkl
= ( fCu + fAu ) · (1 + eπi(h+k) + eπi(h+l) + eπi(k+l) )
4
4
Diskutiert man diese Summe nochmals im Hinblick auf gerade oder ungerade Kombinationen
der Millerschen Indizes, so gilt für den Strukturfaktor in diesem Fall:
η=0
Fhkl
=
f
Au
0,
+ 3 fCu , für nur gerade oder ungerade h,k,l
für gemischte h,k,l
(2.18)
Die Kombination dieser Fallunterscheidungen macht deutlich, dass an bestimmten Netzebenen immer Reflexe zu sehen sein werden, wohingegen es auch Röntgenpeaks gibt, welche nur bei einer
entsprechender Ordnungsstruktur auftreten dürfen. Eine formale Kombination des Strukturfaktors bindet den Ordnungsparameter mit in die Auswahlregeln ein, wenn als Ansatz gewählt
8
∼ bedeutet, dass die Grad- oder Ungeradzahligkeit identisch ist (h und k beide gerade oder beide ungerade) und
sagt, dass l eine entgegengesetzte Grad- oder Ungeradzahligkeit hat wie h und k. Hinweis: 0 ist gerade.
© SoSe 2015
22
2.4 Theorie der Fernordnung
wird das der Strukturfaktor linear mit dem Ordnungsparameter η zusammenhängt.
Fhkl
fAu + 3 fCu ,
η · ( f − f ),
Au
Cu
=
η · ( fAu − fCu ),
η · ( f − f ),
Au
Cu
für nur gerade oder ungerade h,k,l
für h ∼ k und h, k l
für h ∼ l und h, l k
(2.19)
für k ∼ l und k, l h
Röntgenreflexe die immer, unabhängig vom Ordnungsparameter auftreten werden als Fundamentalreflexe (F) bezeichnet, wohingegen diejenigen die nur bei ausgeprägter Ordnungsstruktur ausgebildet werden die Bezeichnung Überstrukturreflex (Ü) tragen.
Sei der Ansatz aufgegriffen der den Ordnungsparameter mit dem Strukturfaktor verbindet, so hängt
die Intensität der Überstrukturreflexe quadratisch von der Ordnung ab, da der Strukturfaktor wie
oben beschrieben (vgl. Gl. 2.13) quadratisch auf die Intensität Einfluss nimmt. Anders gesagt, ein
Überstrukturreflex bei beliebiger Ordnung η lässt sich beschreiben als Produkt des perfekten Überstrukturreflexes (η = 1) multipliziert mit η 2 .
I Ü (η) = η 2 · I Ü (1)
⇔ η2 =
I Ü (η)
I Ü (1)
Wie in Gleichung 2.13 erwähnt ist I Ü (η) bis auf eine Konstante theoretisch beschrieben. Diese Konstante representiert allerdings auch das Verhältnis zwischen den theoretisch beschriebenen Fundamentalreflexen und ihren experimentellen Pendants.
⇔ η2 =
Ü
(η) · I F
Iexp
F · I Ü (1)
Iexp
Jetzt wird für die theoretischen Intensitäten die Gleichung 2.13 eingesetzt:
Ü
Iexp
(η) ((fAu + 3fCu )F )2 pF LFp DTF
·
·
·
· Ü
⇔η =
F
Iexp
((fAu − fCu )Ü )2 pÜ LÜ
DT
p
2
Da klar ist, dass der experimentelle Überstrukturreflex von der Ordnung abhängt kann dieser Zusatz
weggelassen werden. Kürzen und verschönern liefert die nutzbare Formel um den Ordnungsparameter
η experimentell bestimmen zu können.
Ü
Iexp
η = F ·
Iexp
2
© SoSe 2015
(fAu + 3fCu )F
(fAu − fCu )Ü
2
·
pF LFp DTF
·
· Ü
pÜ LÜ
DT
p
(2.20)
23
2.5 Kalorimetrie
2.5. Kalorimetrie
Mit dem dynamischen Differenzkalorimeter, kurz DSC (Differential Scanning Calorimetry), lassen
sich Wärmetönungen bei einer bestimmten Temperatur oder einer Temperaturänderung messen. Mit
dem DSC kann so unter anderem eine Phasenübergangstemperatur ermittelt und die Enthalpie oder
die Wärmekapazität gemessen werden. Im dem DSC befinden sich eine Probe und eine Referenz;
diese Paarstruktur ist ein Hauptmerkmal des DSC [hoehne1996].
Mit Hilfe des DSCs wird nun untersucht, wie sich die Probe und die Referenz zueinander verhalten.
Beide werden in Aluminiumtiegeln verkapselt, wobei die Referenz meist ein leerer Tiegel ist.
Abbildung 2.10.: Aufbau eines ’disk-type’ Wärmestrom-DSC. P: Probe, R: Referenz, 1: Scheibe,
2: Thermoelemente, 3: Temperaturregelung [hoehne1996]
Man unterscheidet zwei Methoden, wie das DSC betrieben werden kann. Beim dynamischen Leistungsdifferenzkalorimeter (˝power compensating DSC) wird die Energie gemessen und beim dynamischen Wärmestromdifferenzkalorimeter (˝heat flux DSC) wird die Temperaturdifferenz gemessen.
Das auch für diese Arbeit verwendete Wärmestrom-DSC existiert in zwei verschiedenen Ausführungen, die sich im Aufbau unterscheiden. Beim relevanten ˝disk-typebefinden sich beide Tiegel in einer
Kammer, in der sie symmetrisch zur Mitte auf einer Scheibe angeordnet werden (siehe Abbildung
2.10).
Beim Erhitzen der Kammer können über die leitfähige Scheibe, auf der sich die Tiegel befinden,
die Temperaturen mit Thermoelementen gemessen werden. Es kann beispielsweise bei einem Phasenübergang die Temperaturdifferenz zwischen der Probe und der Referenz ermittelt werden. Die
© SoSe 2015
24
2.5 Kalorimetrie
Kammer, in der sich die Tiegel befinden, kann mit einer konstanten Rate geheizt oder abgekühlt
werden. Auch eine isotherme Temperaturhaltung kann eingestellt werden. Für den Wärmefluss gilt
dann:
ΦF S − ΦF R ∼ −∆T
(2.21)
∆T = TP − TR
(2.22)
mit
Hierbei ist ΦF S der Wärmefluss von der Kammer zur Probe, ΦF R der Wärmefluss von der Kammer
zur Referenz, TP die Temperatur der Probe und TR die Temperatur der Referenz. Für den gesamten
Wärmefluss ΦM gilt dann:
ΦM = −k · ∆T
(2.23)
Der Faktor k hängt vom Messgerät ab und dient zur Kalibrierung. Für die Wärmekapazität CP ergibt
sich mit der Heizrate β und einem Messgerät bezogenen Faktor l [hoehne1996]:
CP = −l ·
∆T
β
(2.24)
Das dynamische Leistungsdifferenzkalorimeter zeichnet sich durch zwei getrennte kleine Kammern
bzw. Öfen aus. Die Probe und die Referenz kommen jeweils in eine Kammer identischer Bauart, die
sich in einem Metallblock mit konstanter Temperatur befinden. In den beiden Kammern sind jeweils
zwei Heizelemente und ein Temperaturfühler. Beim Aufheizen werden beide Öfen mit derselben Energie gespeist. Kommt es im Verlauf der Messung zu einem Temperaturunterschied, wird nicht dieser
gemessen, sondern die Energiedifferenz, die benötigt wird, um beide Tiegel mit Hilfe eines zweiten
Heizkreislaufes auf derselben Temperatur zu halten. Es wird also die elektrische Leistungsdifferenz
∆P und nicht die Temperaturdifferenz ∆T gemessen.
Für den Wärmefluss gilt also:
ΦM = −k2 · ∆T
(2.25)
∆P = −k1 · ∆T
(2.26)
mit
© SoSe 2015
25
2.6 Keimbildung und Wachstum
Die Faktoren k1 und k2 ergeben sich durch das Messgerät [hoehne1996].
2.5.1. Messung thermodynamischer Größen
Der Wärmefluss ΦM ist gleichbedeutend mit einer zeitlichen Änderung der Wärme
ΦM =
dQ
.
dt
(2.27)
Über die Heizrate β lässt sich, wie in Gleichung 2.24 eine direkte Verbindung zwischen Wärmekapazität und Messsignal zeigen
dQ cP =
.
dT p
(2.28)
Da bei einer konstatanten Heizrate während der Messung der Temperaturverlauf linear in der Zeit
ist, verienfacht sich der Ausdruck zu:
1
ΦM .
β
cP =
(2.29)
Da auch gilt
dH cp =
T ,
d p
(2.30)
lässt sich die Umwandlungenthalpie schreiben als
1
∆H =
β
Z
φ(T )dT,
(2.31)
was der Fläche unter dem Peak entspricht [hoehne1996].
2.6. Keimbildung und Wachstum
Eine Frage welche in den vorherigen Abschnitten noch nicht gestellt wurde ist die Klärung des Verlaufs der Entwicklung einer Ordnungsstruktur. Die Ursache einer Ordnungsausbildung in einer
Legierung wurde hinlänglich, mit Hinweis auf die thermodynamischen Zusammenhänge, geklärt (vgl.
2.1.1). Wie sich der Prozess allerdings kinetisch vollzieht, kann bisher noch nicht beantwortet werden.
Als Ansatz sei ein mathematisches Konzept gewählt, welches die Wachstumskinetik von Körner beschreibt. Das Johnson-Mehl-Avrami-Kolmogorov Model (JMAK) nimmt an, dass sich eine isotherme
Phasenumwandlung in einem Material durch Keimbildungsprozesse und Kornwachstum beschreiben
© SoSe 2015
26
2.6 Keimbildung und Wachstum
lässt. M.a.W.: Wenn verstanden werden soll, wie schnell ein Material zum Beispiel von der Schmelze aus kristalliert, dann beschreibt das JMAK-Modell dies als Zusammenspiel von Nukleationen
(Keimbildungsprozesse) und dem anschließenden Wachstum dieser Körner.
Abbildung 2.11.: Schemenhafte Darstellung des Kornwachstums mit der Zeit t.
Die Ordnungsausbildung von CuAu hängt direkt mit der Phasenumwandlung zwischen CuAu und
CuAu I zusammen und geschieht ebenfalls über Bildung und Wachstum von Körnern. Ein Korn ist
ein kleiner Bereich des Kristalls, welcher durch eine Korngrenze von weiteren Körnern abgegrenzt
ist. Polykristalline Materialien bestehen aus einer vielzahl einzelner Körner mit individueller Orientierung.
Wenn der Fernordnungsparameter η ein Maß für die Ausprägung der Ordnungsstruktur darstellt und
zeitlich aufgelöst in seiner Entstehung ermittelt werden kann, wobei zu berücksichtigen ist, dass dies
nur unter isothermen Randbedingungen geschehen darf, dann müsste dies mit einer Modellierung
auf Basis der JMAK-Gleichung vergleichbar sein. Dieses würde ein Maß für die Kinetik des Phasenübergangs beschreiben.
Im Anhang wird nach [WBS97] ein solches Modell vorgestellt, anhand dessen versucht werden soll
diese Kinetik verständlich zu beschreiben. Es lässt sich zeigen, dass sich das umgewandelte Volumen
X(t) mit der Zeit t über folgenden Zusammenhang beschreiben lässt:
1
⇒ X(t) = 1 − e− 3 πN g
3 tn
n
= 1 − e−K·t
(2.32)
Wenn der Fernordnungsparameter stellvertretend als ein Maß für die Entwicklung der Phasenumwandlung anzusehen ist und er im Experiment unter isothermen Bedingungen zeitlich aufgelöst
werden kann, dann ist ein Vergleich mit dieser Gleichung anzustreben. In der Auswertung soll insbesondere darauf eingegangen werden, ob der Exponent 4 den Verlauf des Parameters gut beschreibt,
oder ob er möglicherweise anders gewählt werden sollte.
In isothermen Kalorimeterexperimenten ist der Wärmefluss (˝heatflow ) ein Maß für den umgewandelten Volumenbruchteil im Falle eines Phasenübergangs. Dabei ist der Zusammenhang als zeitliche
Ableitung gegeben.
dX(t)
= H · K · n · exp (−K · (t)n ) · tn−1
dt
© SoSe 2015
(2.33)
27
2.6 Keimbildung und Wachstum
Die Enthalpie H hängt dabei von der Aktivierungsenergie des Prozess ab und wird als Paramter
betrachtet.
© SoSe 2015
28
3. Experimentelle Vorbereitung und
Versuchsdurchführung
Da die Vorbereitungen zu den Messungen einen zeitlichen Umfang erfordern der nicht im Rahmen
des Praktikums sichergestellt werden kann, wird das Versuchssetting schon vom Betreuer vorbereitet.
Daher soll an dieser Stelle etwas ausführlicher vorgestellt werden wie man sich den Aufbau bzw. die
Kombination aus Röntgendiffraktometer und Heizkammer vorstellen kann.
3.1. Proben
Die Kupfer-Gold Proben in einer Zusammensetzung von 75:25 (Atom-%) wurden aus 5N Kupfer
und 5N Gold hergestellt (5N =
b 99, 999%). Insgesamt dreimal hintereinander wurden die Gold- und
Kupferanteile im Lichtbogenofen zusammengeschmolzen um eine möglichst vollständige Durchmischung zu erzielen. Die entstandenen Halbkugeln wurden nun in mehrere Scheiben geschnitten und
jede wurde mit einer Bohrung von 0, 6 mm Durchmesser versehen (s. Abb. 3.1). Diese Bohrung dient
dazu den Thermoelement (Typ K) in der Heizkammer aufzunehmen um eine möglichst exakte Probentemperatur zu erhalten.
Abbildung 3.1.: Geschnittene CuAu-Probe mit Bohrung.
3.2 Die Kombination von Röntgendiffraktometer und Heizkammer
3.2. Die Kombination von Röntgendiffraktometer und
Heizkammer
Schon mehrfach wurde die Besonderheit angesprochen, dass in diesem Setting eine Röntgenuntersuchung parallel zu einer Wärmebehandlung durchgeführt werden kann. Realisiert wurde dies indem ein
Kristalloflex D5000 der Firma Siemens mit einer Heizkammer kombiniert wurde. Die Heizkammer
besteht aus einer runden Edelstahlplatte (mit Loch) worauf ein Heizband montiert ist und einem
soliden, abnehmbaren Deckel welcher auf diese Platte aufgeschraubt wird. Der Deckel besitzt auf
der oberen Hälfte einen breiten Schlitz welcher mit einer Folie abgedeckt ist. Diese Folie hat die
Eigenschaft für die Röntgenstrahlung durchlässig zu sein und gleichzeitig eine gute Wärmeisolation
zu bieten. Alle Anschlüsse, Befestigungen und Messinstrumente werden vakuumdicht durch die Metallplatte hindurch in den Kristalloflex geführt und von dort aus mit der Steuerelektronik verbunden.
Abbildung 3.2.: Heizband mit Probe und Temperaturfühler.
Bevor die Proben in der Heizkammer getempert werden muss ein Vakuum vorhanden sein um eine
Oxidation der Proben zu verhindern. Dazu wird auf die Kammer der Deckel aufgeschraubt und eine
Vakuumpumpe an das Loch der Edelstahlplatte angeschlossen. Die Messungen in der Heizkammer
wurden bei Drücken um 10−5 mbar durchgeführt. Es wurde überprüft wie hoch der Einfluss des
Vakuums und der Folie auf die Intensität der Röntgenstrahlen ist. Durch die Folie wird die Intensität
der Reflexe leicht verringert, steigt aber unter Vakuum wiederum an. Dies hat seine Ursache darin,
dass die Röntgenstrahlung nunmehr weniger stark an den Luftmolekülen gestreut wird.
Wird ein Kristall monochromatischer Röntgenstrahlung mit einer Wellenlänge von (λ = 1, 54 Å)
ausgesetzt, so entsteht konstruktive Interferenz unter Berücksichtigung der Braggschen Bedingung
aus Abschnitt 2.2.2. Die Röntgenanalyse wird im gekoppelten ϑ/ϑ-Scan zwischen Quelle und Detektor
durchgeführt (s. Abb. 3.4). Der genau im entsprechenden Winkel mitdrehende Detektor registriert
die Intensität des reflektierten Röntgenstrahls.
⇒ Aufgabe zur theoretischen Vorbereitung: Machen Sie sich mit dem Aufbau und der Funktionsweise einer Röntgenröhre vertraut.
© SoSe 2015
30
3.2 Die Kombination von Röntgendiffraktometer und Heizkammer
Abbildung 3.3.: Deckel der Heizkammer mit röntgendurchlässiger Folie.
Abbildung 3.4.: 2ϑ Geometrie des Kristalloflex D5000.
Die Einstellungen des Diffraktometers mit welchen die Röntgenanalyse durchgeführt werden soll muss
den Anforderungen an den jeweiligen Versuchsteil (s. nächster Abschnitt) genügen. Die Analyse wird
mit einem ortssensitiven Detektor durchgeführt der einen Winkelbereich von 4° gleichzeitig erfassen
kann.
Versuchsteil 1: Winkelbereich zwischen 22° und 90° bei einem Increment (Auflösung/Step Size) von
ca. 2ϑ = 0, 04° (Kanal 3 von 4). Messdauer pro Step: 1 sec.
Versuchsteil 2: Winkelbereich geeignet für eine Analyse des (001)- bzw. (111)-Peak. Increment: ca.
2ϑ = 0, 04°; Messdauer: Was wäre ein geeignetes Konzept?
© SoSe 2015
31
3.3 Durchzuführende Messreihen
3.3. Durchzuführende Messreihen
Mit Blick auf die in den theoretischen Vorbereitungen behandelten Fragestellungen sollen folgende
folgende Messreihen am Röntgendiffraktometer durchgeführt und ausgewertet werden werden:
1: Röntgenanalyse des vollständigen Spektrums eines ungeordneten CuAu-Kristalls bei verschiedenen Temperaturen und anschließend Analyse des Spektrums eines geordneten Kristalls nach
gemächlichem Abkühlen (ca. 10 °C pro Minute). Die Reihenfolge ist dabei beliebig, aber folgendes Schema wird als sinnvoll erachtet. Reihenfolge.
a) Bei 400 °C
b) Bei 500 °C
c) Bei 700 °C
d) Übergang in den geordneten Zustand: Langsames und möglichst zweimaliges (nochmal
auf max. 420 °C aufheizen) Abkühlen auf Raumtemperatur (T < 100 °C).
2: Von einer hohen Temperaturen ausgehend (ca. 400 °C) den Kristall auf eine Temperatur von
375 °C abkühlen und die Entwicklung der Ordnungsausbildung zeitaufgelöst analysieren.
a) Die Entwicklung der Fernordnung wird über die Ausbildung des (001)-Peaks im Vergleich
zum (111)-Reflex registriert.
b) In den ersten Minuten und nach ca. 1 Stunden wird zudem die Intensität des Fundamentalreflex (111) registriert.
3: Für das heatflow DSC wird eine Probe von ca. 20 mg eingewogen. Nach anfänglichem Aufheizen
über die Umwandlungstemperatur soll die Probe nach einem kurzen Abkühlschritt isotherm gehalten und die Phasenumwandlung beobachtet werden. Dazu wird die Probe auf einer Temperatur unterhalb der Umwandlungstemperatur für 120 Minuten isotherm gehalten. Aufgenommen
wird der Heatflow gegen Zeit.
© SoSe 2015
32
4. Auswertung und Protokoll
Eine ausführliche und sorgfältige Auswertung ist für ein erfolgreiches Praktikum unerlässlich. Damit
den Studierenden allerdings unnötiger Arbeitsaufwand erspart wird, werden die konkret erwarteten
Punkte welche das Protokoll beinhalten muss explizit erwähnt.
Zu Versuchsteil 1: Auszuwerten ist die Vergrößerung der Gitterkonstanten des CuAu-Kristalls mit
steigender Temperatur (a-c) gemäß Abschnitt 2.3.3 (hierbei soll auch auf die Gültigkeit des
Lindemannschen Kriteriums eingegangen werden (Abschnitt 2.3.4)), sowie die Gitterkonstanten des geordneten Zustands (d).
Hinweise: Die Bestimmung des Fernordnungsparameters erfolgt über die sinnvollen Reflexpaare gem. Abschnitt 2.4.1. Die zur Abschätzung des Lindemann Kriterium notwendigen DebyeWaller Faktoren können Tabelle 2.1 im Anhang (B) entnommen werden.
Zu Versuchsteil 2: Auszuwerten ist die Entwicklung der Fernordnung gemäß des JMAK-Modells.
Dabei ist darauf einzugehen, wie der Exponent in der Zeit am besten zu wählen ist. Die Fernordnung berechnet sich aus dem Verhältnis des Überstrukturreflexes (001) zum Fundamentalreflex
(111) (a). Um zu überprüfen in welchem Maß die Intensität des (111)-Reflex mit steigender
Ordnungsausprägung variiert werden zwei Referenzwerte erhoben (b).
Zu Versuchsteil 3: Auch hier soll die Umwandlung mit Blick auf das JMAK-Modell ausgewertet
werden. Dazu sei gesagt, dass die Betrachtung von Heatflow gegen Zeit als Ableitung der
umgwandelten Stoffmenge im Sinne des JMAK-Modells betrachtet werden kann. Daher sollte
der Avrami Koeffizient gemäß
dX(t)
= H · K · n · exp (−K · (t)n ) · tn−1
dt
gefittet werden.
(4.1)
A. Charakterisierung von
Phasenübergängen
A.1. Phasenübergang 1.Ordnung
Die Charakterisierung eines Phasenübergang hängt wesentlich mit dem Verhalten der Gibbschen
freien Energie (auch: freie Enthalpie) zusammen. Notwendige Bedingung: G1 = G2 .
Definition: Ein Phasenübergang 1. Ordnung ist dadurch gekennzeichnet,dass die ersten Ableitungen
der freien Enthalpie G nicht stetig sind.
Abbildung A.1.: Verlauf der thermodynamischen Eigenschaften bei einem Phasenübergang 1.Ordnung. Die Unstetigkeit von H beim Phasenübergang führt zu einer Singularität von Cp .
= −S und
Die natürlichen Variablen von G sind die Temperatur T und der Druck p. Da gilt ∂G
∂T p
∂G
= V , weisen die Verläufe von Volumen und Entropie eine Unstetigkeit (Sprung) auf. Betrach∂p
T
tet man die Enthalpie bei einem Phasenübergang 1. Ordnung, so ändert sich diese im Moment des
Phasenübergangs. Es erfolgt somit eine Wärmeaufnahme- oder abgabe ohne eine Temperaturänderung. Man spricht auch vom Auftreten latenter Wärme. Da die Wärmekapazität Cp als Ableitung der
Enthalpie nach der Temperatur definiert ist, weist sie bei einem Phasenübergang 1. Ordnung als Folge der momentanen Änderung von H, was einer unendlichen Steigung entspricht, eine Singularität
auf. Die Wärmekapazität ist bei einem solchen Phasenübergang bei der Umwandlungstemperatur
nicht definiert. Ein hierfür typisches Beispiel ist kochendes Wasser beim Übergang von flüssig zu
gasförmig.
A.2 Phasenübergang 2.Ordnung
A.2. Phasenübergang 2.Ordnung
Im Unterschied zum Phasenübergang 1. Ordnung tritt in diesem Fall bei der Umwandlungstemperatur keine sprunghafte Änderung von Volumen und Entropie auf. Die notwendige Bedingung für
einen Phasenübergang (G1 = G2 ) gilt natürlich weiterhin.
Definition: Ein Phasenübergang 2. Ordnung ist dadurch gekennzeichnet, dass zwar die ersten Ableitungen der freien Enthalpie G stetig, jedoch die zweiten Ableitungen unstetig sind.
Abbildung A.2.: Verlauf der thermodynamischen Eigenschaften bei einem Phasenübergang 2.Ordnung. Die 2. Ableitungen von G nach T und p sind unstetig. Die Wärmekapazität besitzt zwar
immer noch einen Sprung, aber keine Singularität mehr.
Die Unstetigkeit der 2. Ableitungen von G ist an der sprunghaften Änderung der Steigung (Knick)
von V und S zu erkennen. Die Enthalpie ändert sich bei einem Phasenübergang 2. Ordnung zwar
ebenfalls, aber in diesem Fall erfolgt diese Änderung stetig. Dadurch gibt es keine Singularität
der Wärmekapazität mehr. Ein hierfür klassischen Beispiel stellt der Phasenübergang Normal/Supraleitend dar.
© SoSe 2015
35
B. Debeye-Waller-Faktor: Mathematische
Modellierung der Problematik
Debye betrachtet als Ausgangspunkt ein rechtwinkliges Koordinatensystem, eine ebene, monochromatische Röntgenwelle die den Kristall trifft und die Koordinaten x = x0 + u, y = y0 + v, z = z0 + w
eines beliebigen Atoms. Die Koordinate setzt sich somit aus der absoluten Ruhelage und einer Verschiebungskomponente zusammen, welche bei Wärmebewegung großer Null ist. Dies charakterisiert
das Auftreten von Gitterschwingungen die einer Verrückung jedes Atoms um seine Ruhelage ursächlich sind. Diese Gitterschwingungen werden in der Literatur auch durch das Quasiteilchen Phonon
vertreten. Debye beschrieb mathematisch wie groß der Einfluss der Temperatur und damit der Einfluss dieser Gitterschwingungen auf ein Röntgenspektrum ist. Im ersten Schritt fand Debye, nach
verschiedenen Mittelungen, für die Amplitude der von dem beliebigen Atom gebeugten Strahlung
den Ausdruck
Jm =
A2 X X M ix[(α−α0 )(x0 −x0 0 )+(β−β0 )(y0 −y0 0 )+(γ−γ0 )(z0 −z0 0 )]
e e
.
r2
(B.1)
In dieser Gleichung ist eM der entscheidene Faktor. Es ist ein Mittelwert für den Einfluss der Verschiebung u, v, w auf die Intensität. Ivar Waller bestätigte einige Jahre später diesen Faktor, berechnete
ihn allerdings zu e2M [Wal27]. Seither trägt er die Bezeichnung Debye-Waller-Faktor (DWF).
Die Amplitude I der rückgestreuten Strahlung lässt sich durch Gleichung B.2 ausdrücken und hängt
somit wesentlich von dem exponentiellen Ausdruck e2M ab.
(B.2)
I = I0 · e2M ,
wobei I0 der theoretischen Intensität am absoluten Nullpunkt, also ohne Einfluss von Gitterschwingungen, entsprechen soll. Für M gilt:
κ 2 h2
3
−M = 2 (1 − cos ϕ)
4π
µkΘ
1
1
+ 2
4 x
Z
0
x
ξ
dξ
ξ
e −1
,
(B.3)
hωk
wobei gilt ξ = 2πkT
, κ = 2π
,x = Θ
, Θ die charakteristische Temperatur3 , µ die Masse eines Atoms
λ
T
der betreffenden Substanz und ϕ der Winkel zwischen Beobachtungsrichtung und Einfallsrichtung.
3
Die charakteristische Temperatur wird für CuAu gemittelt zu Θ = 255 K (vgl. B.1).
A.2 Phasenübergang 2.Ordnung
Dieser entspricht gerade dem Doppelten des Winkels zwischen Probenoberfläche und einfallendem
Röntgenstrahl (ϕ = 2ϑ, vgl. Abb. 2.5). µ lässt sich auch ausdrücken durch den Quotient NAA , wobei
NA die Avogadro-Konstante ist und A für das Atomgewicht steht. Der Summand 14 berücksichtigt in
Gleichung B.3 eine Nullpunktsenergie (NPE) über deren Existenz sich Debye nicht 100%ig im klaren
war, sie dennoch berücksichtigte um umfangreichere theoretische Vorhersagen treffen zu können.
Rx
Der Temperaturverlauf von M wird wesentlich durch das Integral x12 0 eξξ−1 dξ bestimmt. Mit der
Substitution
Z
Φ(x)
1 x ξ
dξ
=
x 2 0 eξ − 1
x
lässt sich für Φ(x) mit der Reihenentwicklung4
Entwicklung finden:
Φ(x) =
1 −
π2
6x
x
4
2
4
x
ex −1
6
= 1−
x
2
+
B 1 x2
2!
8
+ B1 x3! − B2 x5! + B3 x7! − B4 x9! + · · · ,
1
− e−3x 13 +
− e−x 1 + x1 − e−2x 12 + 4x
−
B2 x4
4!
+
B3 x6
6!
− · · · folgende
für kleine x
1
9x
− · · · , für große x
(B.4)
Berechnet man die Werte für Φ(x) und zwar indem x ab einer Größe von 2 als großer Wert betrachtet
wird und man zudem beide Reihenentwicklungen bis zum ausreichenden dritten Gliede führt, dann
zeigt Tabelle C.1 die ermittelten Werte für Φ(x) und Abbildung C.1 den Verlauf der dominierenden
Funktion Φ(x)
bzw. 41 + Φ(x)
ohne bzw. mit Berücksichtigung einer Nullpunktsenergie (s. Anhang B).
x
x
Sei an dieser Stelle einmal vorgegriffen, dass die Simulation in B.1 für fünf verschiedene Temperaturen
durchgeführt wird, so muss berechnet werden welchen Werten für x bzw. Φ(x) dies entsprechen würde
(s. Tab. B.1).
4
Bn sind die Bernoulli Zahlen. Bn =
© SoSe 2015
(2n)!
22n−1 π 2n
·
P∞
1
k=0 k2n
37
B.1 Theoretische Ergebnisse des Einflusses der Wärmebewegung für CuAu
T0
T1
T2
T3
T4
T5
T in °C
T in K
x
Φ(x)
Φ(x)
x
25
100
300
400
500
700
298
373
573
673
773
973
0,86
0,68
0,44
0,38
0,33
0,26
0,81
0,84
0,89
0,91
0,92
0,94
0,94
1,23
2,01
2,39
2,79
3,58
Tabelle B.1.: Berechnung des Quotient
Φ(x)
x
für verschiedene Temperaturen über x =
Θ
.
T
Soll Gleichung B.3 vollständig beschrieben werden so fehlt noch eine Betrachtung des gesamten
Vorfaktors. Sei
3 κ 2 h2
4 π2 µ k Θ
h2 NA
=3 2
kλ AΘ
P =
so lässt sich dieser mit den bekannten Konstanten h (Plancksches Wirkungsquantum) und k (BoltzmannKonstante) der Vorfaktor bestimmen zu:
P =
0, 574 · 10−16
A Θ λ2
(B.5)
Somit ergibt sich für den Faktor M aus Gleichung B.1:
1 Φ(x)
M = −P (1 − cos ϕ)
+
4
x
,
(B.6)
wobei der Summand 14 für die Berücksichtigung einer Nullpunktsenergie steht und bei Bedarf weggelassen werden kann.
B.1. Theoretische Ergebnisse des Einflusses der
Wärmebewegung für CuAu
Gleichung B.6 hat zusammenfassend beschrieben, wie der Einfluss der Wärmebewegung auf die Intensität der Röntgenpeaks theoretisch zu modellieren ist. Dies sei im Folgenden für die verwendeten
CuAu-Proben ausgeführt um die experimentellen Ergebnisse mit den Vorhersagen vergleichen zu
−16
können. Der Faktor P hat nach Gleichung B.5 die Gestalt P = 0,574·10
. Diese Berechnung scheint
A Θ λ2
trivial, erfordert aber Umsichtigkeit beim Einsetzen der Faktoren.
Beispiel muss schon bei der
h 2 Zum
i
s
Berechnung von Gleichung B.5 eine Umrechnung von [eV ] zu m2 kg2 durchgeführt werden, wobei zu
© SoSe 2015
38
B.1 Theoretische Ergebnisse des Einflusses der Wärmebewegung für CuAu
beachten ist das die Atommasse [u], in Kombination mit der Teilchenanzahl pro Mol, in Gramm auftritt und nicht in Kilogramm und daher noch ein Faktor 1000 berücksichtigt werden muss. Kostbare
Zeit, die schon umfangreich investiert worden ist, kann dem geneigten Leser an dieser Stelle erspart
werden.
Die Röntgenversuche werden mit einer Kupferanode durchgeführt deren charakteristische Strahlung (Kα ) eine Wellenlänge von λ = 1, 54 · 10−10 m besitzt. Die Atommasse A von CuAu wird
aus den jeweiligen Werten (AAu = 197 u, ACu = 63 u) gemittelt zu A = 130 u. Mit der charakteristischen Temperatur Θ wurde identisch Verfahren. Somit ergibt sich ein Wert von Θ = 255 K
(ΘAu = 165 K, ΘCu = 345 K). Insgesamt bestimmt sich der gesamte Vorfaktor P zu: P = 0, 073.
1 Φ(x)
+
⇒ M = −0, 073 · (1 − cos ϕ)
4
x
(B.7)
Die Abbildungen B.1 stellen die Ergebnisse der theoretischen Modellierung dar. Auf der y-Achse
ist der Debye-Waller-Faktor e2M für verschiedene Temperaturen mit zunehmendem Winkel zwischen
Beobachtungs- und Einfallsrichtung aufgetragen.
Abbildung B.1.: (a) links: Der Debye-Waller Faktor e2M für T0 , . . . , T5 ohne Berücksichtigung und,
(b) rechts: mit Berücksichtigung einer Nullpunktsenergie.
Deutlich wird der zunehmende Abfall der Intensität bei steigender Temperatur und zunehmendem
Winkelabstand zwischen Einfalls- und Beobachtungsrichtung ebenso, wie die leichte Verstärkung
des DWF durch Existenz einer Nullpunktenergie. Ob diese theoretische Modellierung allerdings die
experimentellen Ergebnisse ausreichend genau wiederspiegelt oder ob es nicht Anhaltspunkte zur
kritischen Diskussion gibt, wird in Abschnitt 2.3.2 aufgegriffen.
© SoSe 2015
39
C. Näherung des Integrals im Parameter
M
x
Φ(x)
x
Φ(x)
x
Φ(x)
x
Φ(x)
x
Φ(x)
0
0,1
0,2
0,3
0,4
0,5
0,6
1,000
0,975
0,951
0,927
0,904
0,882
0,860
0,7
0,8
0,9
1
1,1
1,2
1,3
0,839
0,818
0,797
0,778
0,758
0,739
0,721
1,4
1,5
1,6
1,7
1,8
1,9
2
0,703
0,686
0,669
0,653
0,637
0,622
0,607
2,5
3
3,5
4
4,5
5
5,5
0,578
0,500
0,441
0,393
0,355
0,322
0,295
6
6,5
7
7,5
8
9
10
0,272
0,252
0,234
0,219
0,205
0,183
0,164
Tabelle C.1.: Näherung des Integralausdrucks Φ(x) für verschiedenen x.
Abbildung C.1.: Verlauf von
Φ(x)
x
aufgetragen gegen x1 .
D. Temperaturabhängigkeit der
Gitterkonstanten
Um die Gitterenergie zu beschreiben ist es nötig das Wechselwirkungspotential in eine Taylor-Reihe
zu entwickeln und zu befinden wie viele Glieder dieser Entwicklung in einer Näherung berücksichtigt
werden sollen. Bei einer harmonischen Näherung wird die Taylor-Entwicklung nach dem zweiten
Glied abgebrochen. Therme höherer Ordnung als eine quadratische bleiben somit unberücksichtigt.
Die gesamte Gitterenergie U lässt sich als Summation des Wechselwirkungspotentials Ψ über alle
Gitteratome beschreiben:
U=
1 X
Ψ((r0m + um ) − (r0n + un )) ,
2 n,m; n6=m
(D.1)
wenn r0m,n für die Ruhelage und un,m für die Auslenkung des m-ten, oder n-ten Atoms steht. Der Faktor 21 resultiert aus der Tatsache, dass in der obigen Summe über jedes Atompaar zweimal summiert
wird. Dieser Ausdruck ist nun um die Ruhelage der Gitteratome in eine Taylor-Reihe zu entwickeln:
Allgemein gilt für das Taylorpolynom vom Grade n zu einer Funktion f (x) an der Entwicklungsstelle
x0 folgende Gleichung:
Txn0 f (x)
=
n
X
f (k) (x0 )
k=0
k!
(x − x0 )k
angewandt auf die Gleichung für die Gitterenergie folgt:
Trn0m −r0n (U )
=
n
X
U (k) (rm − rn )
0
k=0
n
X
1
=
k!
k=0
k!
0
((r0m + um ) − (r0n + un ) − (r0m − r0n ))k
!(k)
1 X
Ψ(r0m − r0n )
(um − un )k
2 n,m; n6=m
B.1 Theoretische Ergebnisse des Einflusses der Wärmebewegung für CuAu
Im Fall einer anharmonischen Näherung werden nun auch Glieder höherer Ordnung als 2 berücksichtigt. Sei die Reihe daher bis zum Grade n = 4 entwickelt:
⇒U =
1 X
1 X
Ψ(r0m − r0n ) +
∇Ψ(r0m − r0n )(um − un )
2 n,m; n6=m
2 n,m; n6=m
|
{z
}
U0
1
1 X
∇2 Ψ(r0m − r0n )(um − un )2 +
∇3 Ψ(r0m − r0n )(um − un )3
4 n,m; n6=m
12 n,m; n6=m
1 X
∇4 Ψ(r0m − r0n )(um − un )4
+
48 n,m; n6=m
X
+
Es fällt auf das der lineare Anteil verschwindet, da gilt:
X
∇Ψ(r0m − r0n )(um − un ) =
X
um ∇Ψ(r0m − r0n ) −
n,m; n6=m
n,m; n6=m
=
X
m
u
m
X
un ∇Ψ(r0m − r0n )
n,m; n6=m
X
∇Ψ(r0m
−
r0n )
n
−
X
n
un
X
∇Ψ(r0m − r0n )
m
Zusätzlich ist festzustellen, dass:
X
n
∇Ψ(r0m − r0n ) −
X
∇Ψ(r0m − r0n ) = 0 ,
m
weil in der Ruhelage eine Summation über alle Kräfte, die von den Atomen an den Positionen r0m
bzw. r0n auf das Atom an der Position r0n bzw. r0m ausgeübt wird, verschwindet. Somit fällt der lineare
Anteil der Taylor-Entwicklung weg. Beschreibt man zur Vereinfachung die einzelnen Summanden in
der Entwicklung durch allgemeine Konstanten9 a, b, c ∈ R≥0 und einem potenziertem Ausdruck uk
und verschiebt man das Energieniveau um U0 , so lässt sich die Gitterenergie U in ihrer grundlegenen
Gestallt kompakt zusammenfassen:
⇒ U = au2 − bu3 − cu4
(D.2)
Das negative Vorzeichen vor dem dritten und vierten Glied berücksichtigt, dass die Energie für kleine
Abstände zunimmt und mit wachsendem Abstand verschwindet. Wenn die Gitterenergie durch den
obigen Ausdruck beschrieben ist, wie verhält sich dann der mittlere Abstand der Atome mit variabler
Temperatur? Um dies zu analysieren sei der mittlere Abstand der Atome hr − r0 iim thermodynamischen Mittelwert [GM11] betrachtet. Dies ist eine übliche Methode aus der statistischen Physik in
der im allgemeinen große Systeme mit vielen Teilchen betrachtet werden. Es findet eine Integration
über alle möglichen Abstände statt, wobei jeder mit einem von der Temperatur abhängigen Wahr9
Dies ist erlaubt, da alle Summanden im Falle eines bekannten Potentials Ψ vollständig determiniert sind.
© SoSe 2015
42
B.1 Theoretische Ergebnisse des Einflusses der Wärmebewegung für CuAu
scheinlichkeitsfaktor gewichtet wird. Dies entspricht der Idee der Boltzmann Verteilung wonach jeder
Zustand abhängig von seinem Energieniveau mit unterschiedlicher Anzahl von Atomen besetzt ist:
Uε
nε = e kB T ,
wobei nε = der Anzahl der Atome ε mit der Energie Uε entspricht. Mit dieser Gewichtung ergibt sich
der thermodynamische Mittelwert des mittleren Abstandes der Gitteratome:
R∞
−U (r−r0 )
(r − r0 ) e kB T d(r − r0 )
hr − r0 i = −∞R
−U (r−r0 )
∞
kB T
e
d(r − r0 )
−∞
(D.3)
In diesem Ausdruck wird jetzt auch der Einfluss der Temperatur auf den mittleren Abstand berücksichtigt. Die Lösung dieser Integrale liefert somit einen Zusammenhang zwischen einer Änderung der Temperatur und einer Änderung des mittleren Atomabstands bzw. einer Änderung der
Gitterkonstanten. Die Energie U (r − r0 ) lässt sich mit obigen Überlegungen über den Ausdruck
U (r − r0 ) = a(r − r0 )2 + b(r − r0 )3 + c(r − r0 )4 beschreiben. Für kleine Auslenkungen sind die Terme
(r − r0 )3 und (r − r0 )4 sehr viel kleiner als (r − r0 )2 . Daher kann die Exponentialfunktion für diese
Therme in eine Reihe entwickelt und nach niedrigem Gliede (n=1) abgebrochen werden.
e
−U (r−r0 )
kB T
=e
−a(r−r0 )2
kB T
=e
−a(r−r0 )2
kB T
=e
−a(r−r0 )2
kB T
b(r−r0 )3 +c(r−r0 )4
kB T
·e
"∞
#
X 1 b(r − r0 )3 + c(r − r0 )4 k
k!
kB T
k=0
b(r − r0 )3 + c(r − r0 )4
+ Rest
· 1+
kB T
Zur Vereinfachung sei (r − r0 ) durch r̂ und kB1 T im Folgendem durch β substituiert. Die Integrale
aus Gleichung D.3 sind somit übersichtlicher zu lösen.
Z
∞
−βU (r̂)
r̂ e
−∞
Z
∞
2
e−βa r̂ (r0 + βb r̂4 + βc r̂5 ) dr̂
Z−∞
Z ∞
Z ∞
∞
2
−βa r̂2
4 −βa r̂2
=
r̂ e
dr̂ +
βb r̂ e
dr̂ +
βc r̂5 e−βa r̂ dr̂
−∞
| −∞ {z
}
| −∞
{z
}
=0 da ungerade
=0 da ungerade
Z ∞
2
=
βb r̂3 · r̂ e−βa r̂ dr̂
dr̂ =
−∞
© SoSe 2015
43
B.1 Theoretische Ergebnisse des Einflusses der Wärmebewegung für CuAu
Eine doppelte partielle Integration liefert mit Kenntnis des Grenzwertes für das Gaußsche Fehlerintegral
1
lim erf(z) = lim √
z→∞
z→∞
2π
Z
z
1 2
e− 2 t dt = 1
−∞
und geeigneter Substitution 12 t2 = βar̂2 den Wert:
Z
3
∞
2
βb r̂4 e−βa r̂ dr̂ =
−∞
3 √ (kB T ) 2
πb
5
4
a2
(D.4)
Das zweite Integral aus Gleichung D.3 ergibt sich sehr analog zu:
Z
∞
−βa r̂2
e
dr̂ =
−∞
√
√
kB T
π √
a
(D.5)
Daraus lässt sich der thermodynamische Mittelwert abschließend einfach berechnen:
2
3√
πb (kB 5T )
4
a2
√ √k B T
√
π
a
3
hr − r0 i =
⇒ hr − r0 i =
3 b
kB · T
4 a2
(D.6)
Es wird somit deutlich, dass zwischen einer Temperaturänderung und dem mittleren Abstand der
Gitteratome ein linearer Zusammenhang besteht. Dabei handelt es sich jedoch nicht um eine Proportionalität (Kein Nulldurchgang).
© SoSe 2015
44
E. Keimbildung und Wachstum
Abbildung E.1.: Schemenhafte Darstellung des Kornwachstums mit der Zeit t.
Angenommen die Keimbildung findet in der Legierung rein zufällig statt und das Wachstum der
Körner vollzieht sich in alle Raumrichtungen identisch, dann sei die Modellierung fokussiert um einen
Punkt x der mit der Zeit t nicht umgewandelt wird. Eine Umwandlung kann in diesem Sinne nur
geschehen, wenn eine Nukleation in der Nähe von x stattfindet und das daraufhin wachsende Korn
den Punkt x erreicht (s. Abb. E.1). Die Keime bilden sich im Zeitintervall i = τi , . . . , τi + ∆τi und
wachsen radial mit der Zeit t in alle Richtungen, sodass sich der Radius ri und damit das Volumen
der Körner beschreiben lässt als
Z
t
ri =
g(t0 ) dt0
τi
4
⇒ V = π(ri )3 ,
3
wobei g(t0 ) die Wachstumsrate ist. Wenn N (τi ) die Nukleationsrate zum Zeitpunkt τi beschreibt,
dann ist der Punkt x mit einer Wahrscheinlichkeit
N (τi ) ∆τi
4
π ri3
3
bis zum Zeitpunkt t umgewandelt worden. Also ist er mit einer Wahrscheinlichkeit
Pi = 1 − N (τi ) ∆τi
4
π ri3
3
B.1 Theoretische Ergebnisse des Einflusses der Wärmebewegung für CuAu
noch nicht von der Umwandlung erfasst worden. Dabei ist zu beachten das dies eine Wahrscheinlichkeit für ein einzelnes Zeitintervall i ist. Um die gesamte Wahrscheinlichkeit zu berechnen müssen
somit alle Zeitintervalle berücksichtigt werden.
⇒ P = Πi
|
4
3
1 − N (τi ) ∆τi π ri
3
{z
}
Z(t)
Wenn X(t) der Anteil des umgewandelten Volumens in der Probe ist und die Rechenregeln des
Logarithmus und der Exponentialfunktion ausgenutzt werden, dann gilt für X(t):
4
3
X(t) = 1 − Z(t) = 1 − Πi 1 − N (τi ) ∆τi π ri
3
4
X
⇔ X(t) = 1 − exp
ln 1 − N (τi ) ∆τi π ri3
|
{z 3
}
i
a
P
ak
da gilt ln(1−a) = ∞
k=1 (−1) k und a im Grenzwert für ∆τi → 0 sehr klein wird, kann die Summation
nach dem ersten Glied abgebrochen werden.
k #
1
4
⇒ X(t) = 1 − exp
(−1) · N (τi ) ∆τi π ri3
3
k
i k=1
!
X
4
lim (X(t)) = 1 − exp −
N (τi ) ∆τi π ri3
∆τi →0
3
i
#
"
Z t
3
Z t
4
g(t0 ) dt0
N (τ )
dτ
= 1 − exp − π
3 0
τ
"
∞
XX
(E.1)
In diesem Ausdruck ist die Nukleations- und Wachstumsrate der Dreh- und Angelpunkt und leider
nicht ohne weiteres zu konkretisieren. Wie sich diese in der Realität wirklich verhalten und durch
welche Parameter sie vollständig beschrieben werden können ist nicht trivial. Daher sei in erster
Näherung die Phasenumwandlung von CuAu bei konstantem N (τ ) und g(t) beschrieben.
Die Integrale aus Gleichung E.1 lassen sich somit sehr einfach lösen:
"
#
Z t
3
Z t
4
0
0
X(t) = 1 − exp − π
N (τ )
g(t ) dt
dτ
3 0
τ
Z t
4
3
N (τ ) (gt − gτ ) dτ
= 1 − exp − π
3 0
"
t #
4
1
= 1 − exp − π(−1)N g 3 (t − τ )4 3
4
0
© SoSe 2015
46
B.1 Theoretische Ergebnisse des Einflusses der Wärmebewegung für CuAu
1
⇒ X(t) = 1 − e− 3 πN g
© SoSe 2015
3 t4
4
= 1 − e−C·t
(E.2)
47
F. Charakterisierung der Kristallstruktur
Nach der Braggschen Bedingung entsteht ein Intensitätspeak genau dann, wenn zwischen dem einfallenden Strahl und den an verschiedenen Netzebenen reflektiertem Strahl konstruktive Interferenz
vorliegt. Wenn nun im Spektrum der reflektierten Röntgenstrahlung für verschiedene 2ϑ einen Intensitätspeak identifiziert werden kann ist es möglich über die Braggsche Bedingung auf den Gangunterschied und damit auf den Netzebenenabstand der beiden Strahlen rückzuschließen. Wie in
a
Abschnitt 2.2.2 erläutert, gilt für kubisch primitive Gitter dhkl = √h2 +k
2 +l2 . Um jetzt die Werte für
die h, k, l verschiedener Netzebenen zu gewinnen ist es sinnvoll folgende Überlegung anzustellen: Der
Abstand zweier Netzebenen di und dj ist selbstverständlich konstant. Daraus folgt mit ddji = z der
Zusammenhang zwischen den jeweiligen hkl:
a
a
=
z
·
d
·z
di = p 2
j = 2
h2 + k22 + l22
h1 + k12 + l12
1
1
⇒p 2
=p 2
·z
2
2
h1 + k1 + l1
h2 + k22 + l22
(F.1)
(F.2)
Aus dem bekannten Verhältnis z können somit durch ausprobieren geeignete h, k, l bestimmt werden
a
die in der Berechnung über dhkl = √h2 +k
2 +l2 zu einem identischen dhkl führen wie nach der Braggschen
Bedingung. Diese Berechnung kann wiederum nur durchgeführt werden wenn die Gitterkonstante
a schon bekannt ist. Das verdeutlicht, dass eine streng analytische Vorgehensweise nicht möglich
ist, sondern nur das wiederholte Einsetzen unterschiedlicher Kombinationen zum Ziel führt. Eine
√
Tabelle der Verhältnisse √hi ki li zu den verschiedenen Netzebenen i, j führt schnell zum Ziel, weil
hj kj lj
die passende Kombination, also die zutreffende Konstante z, einfach heraus gesucht werden kann.
Da diese Überlegungen wie oben schon erwähnt nur für kubisch primitive Gitter sinnvoll sind, gilt
folgender Ansatz im Fall einer tetragonal verzerrten Kristallstruktur:
1. Identifikation zweier Reflexe die aufgrund ihres Winkels und ihrer Intensität aus dem Spektrum
des ungeordneten Kristalls bekannt zu sein scheinen.
2. Zu den identifizierten Winkeln werden die entsprechenden Netzebenenabstände dhkl ermittelt.
B.1 Theoretische Ergebnisse des Einflusses der Wärmebewegung für CuAu
3. Bei geeigneter Wahl lässt sich Gleichung 2.4 der Art vereinfachen, dass Werte für a und c
gewonnen werden können.
4. Daraufhin ist zu prüfen, ob mit diesen Achsenmaßen auch bei den anderen Reflexen eine Übereinstimmung zwischen den zwei verschiedenen Möglichkeiten der Berechnung des Netzebenenabstands dhkl auftritt oder nicht.
Für ein tetragonal verzerrtes Gitter gilt nach 2.2.2 folgender Zusammenhang zwischen den Gitterkonstanten a und c, den Millerschen hkl-Indizes und dem Netzebenenabstand d:
1
d2hkl
=
h2 + k 2
l2
+
a2
c2
(F.3)
wobei c die tetragonal verzerrte Kantenlänge beschreibt. Wie gelingt es jetzt diese Größen aus dem
aufgenommenem Spektrum zu berechnen? Folgende Überlegungen im Sinne der oben schon angesprochenen Vorgehensweise führen zum Ziel:
Ein Vergleich der Spektren im Falle des geordneten und des ungeordneten Kristalls lässt vermuten,
dass eine Übereinstimmung in gewissen Fundamentalreflexen vorliegt. Nach wie vor gibt es einen
Winkel im Bereich von 40, 5°und einen in der Umgebung um 46, 5°die auch eine ausgeprägte Intensität aufweisen. Daher liegt es nahe, dass diesen Reflexen auch die jeweils identische Netzebene wie
im Falle des ungeordneten Kristalls zugewiesen werden kann. Somit kann begründet angenommen
werden, dass der Reflex bei 40, 5°der Netzebene (111) und der Reflex bei 46, 5°der Netzebene (200)
zugesprochen werden kann. Durch diese Annahmen reduziert sich die obige Gleichung auf die leicht
händelbaren Ausdrücke
1
d2200
=
4
+0
a2
(F.4)
=
2
l
+ 2
2
a
c
(F.5)
und
1
d2111
Die Netzebenenabstände d200 und d111 können über die Braggsche Bedingung bestimmt werden,
wodurch es gelingt auf die Achsenmaßen a und c zu schließen.
© SoSe 2015
49
B.1 Theoretische Ergebnisse des Einflusses der Wärmebewegung für CuAu
2d200 · sin ϑ = λ
⇒ d200 = 1, 98Å
⇒ a = 3, 958Å
(F.6)
(F.7)
(F.8)
(F.9)
und somit bestimmt sich c über den Ausdruck
a · d111
c= p
a2 − 2d2111
(F.10)
mit d111 = 2, 23Å analog wie oben und dem berechneten a zu c = 3, 676Å. Wenn diese Werte Gültigkeit besitzen sollen, müssten sich den anderen Reflexen hkl-Indizes der Art zuordnen lassen, sodass die
experimentellen Ergebnisse der entsprechenden Netzebenenabständen mit den theoretischen Werten
gemäß Gleichung F.3 übereinstimmen.
© SoSe 2015
50
Literaturverzeichnis
[Deb14] Peter Debye, Interferenz von Röntgenstrahlen und Wärmebewegung,
Annalen der Physik Nr. 348, S. 49-95, 1914.
[FT98] M. Fanfoni & M. Tomellini, The Johnson-Mehl-Avrami-Kolmogorov model: A brief review,
IL NUOVO CIMENTO, Vol. 20D, N.7-8, Luglio-Agosto, 1998.
[Got07] Günter Gottstein, Physikalische Grundlagen der Materialkunde,
3. Auflage, Heidelberg, 2007.
[GM11] Rudolf Gross & Achim Marx, Festkörperphysik Kapitel 6,
Vorlesungsskript zur Vorlesung im SS 2011, Walther-Meissner-Institut, Lehrstuhl für Technische Physik (E23), S. 219,
http://www.wmi.badw.de/teaching/Lecturenotes/FKP/FKP_Kapitel6.pdf;
Stand: November 2011.
[IFW11] Internetauftritt des Leipniz-Instituts für Festkörper- und Werkstoffforschung Dresden,
Kapitel 15: Ordnung-Unordnung,
http://www.ifw-dresden.de/institutes/imw/lectures/lectures/pwe; Stand: Oktober 2011.
[Jam62] R.W. JAMES, The Optical Principles of the Diffraction of X-Rays,
The Crystalline State Vol. II, London, 1962.
[Wal27] Ivar Waller, Die Einwirkung der Wärmebewegung der Kristallatome auf die Intensität, Lage
und Schärfe der Röntgenspektrallinien,
Annalen der Physik, Band 83, 4. Folge, 1927.
[WBS97] Michael C. Weinberg, Dunbar P. Birnie III & Vitaly A. Shneidman,
Crystallization kinetics and the JMAK equation,
Journal of Non-Crystalline Solids, 219, S. 89-99, 1997.
[WK95] D. Waasmaier & A. Kirfel,
New Analytical Scattering-Factor Functions for Free Atoms and Ions,
Acta Crystallographica, A51, S. 416-431, 1995.
Literaturverzeichnis
[hoehne1996] Höhne, G. and Hemminger, W. and Flammerheim, H.-J.,
Differential Scanning Calorimetry: An Introduction for Practitioners,
Springer-Verlag, 1996
© SoSe 2015
52