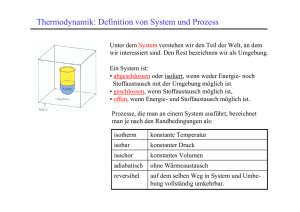
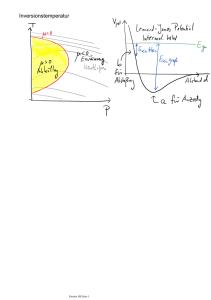
Vorlesung Physikalische Chemie
Werbung

Vorlesung Physikalische Chemie Im Studiengang Energy Science (Christian Mayer) Vorlesung: Übungen: Mittwoch, 08:00 bis 10:00, LF 035 Mittwoch, 10:00 bis 11:00, LF 035 Beginn der Vorlesung: Ende der Vorlesung: Mittwoch, den 9. April 2014 Mittwoch, den 16. Juli 2014 Inhalt: 1 2 3 4 5 6 7 Einführung Grundlagen der Thermodynamik 2.1 Grundlegende Begriffe und Parameter 2.2 Der Nullte Hauptsatz der Thermodynamik und die Temperaturskala 2.3 Zustandsfunktionen Beschreibung gasförmiger Systeme 3.1 Die Gasgesetze und das ideale Gas 3.2 Das Kinetische Gasmodell 3.3 Reale Gase Der Erste Hauptsatz der Thermodynamik 4.1 Weiterführende Begriffe: Arbeit, Wärme und Energie 4.2 Die Innere Energie und der Erste Hauptsatz 4.3 Das Perpetuum mobile: ein Gedankenexperiment 4.4 Messung von Änderungen der Inneren Energie 4.5 Die Enthalpie 4.6 Messung von Änderungen der Enthalpie 4.7 Relation zwischen Innerer Energie und Enthalpie 4.8 Die totalen Differentiale von Innerer Energie und Enthalpie 4.9 Die Temperaturabhängigkeit der Inneren Energie und der Enthalpie 4.10 Die Volumenabhängigkeit der Inneren Energie 4.11 Die Druckabhängigkeit der Enthalpie 4.12 Reaktionsenergie und Reaktionsenthalpie Der Zweite Hauptsatz der Thermodynamik 5.1 Die Entropie 5.2 Entropieänderungen bei ausgewählten Prozessen 5.3 Der Carnotsche Kreisprozess 5.4 Wirkungsgrade von Wärmekraftmaschinen Der Dritte Hauptsatz der Thermodynamik 6.1 Formulierung des Dritten Hauptsatzes 6.2 Erzeugung tiefer Temperaturen Freie Energie und Freie Enthalpie 7.1 Definition und Bedeutung 7.2 Die Fundamentalgleichung 7.3 Die Maxwellschen Gleichungen 7.4 Die Abhängigkeit der Freien Enthalpie von Temperatur und Druck 7.5 Das chemische Potential 7.6 Das chemische Gleichgewicht 7.7 Die Elektromotorische Kraft 2 1 Einführung Die Wissenschaft der Thermodynamik beschäftigt sich, wie der Wortstamm verrät, mit dem Begriff der Temperatur und den damit verbundenen dynamischen Vorgängen. Historisch betrachtet verdankt sie ihre Entstehung in erster Linie einer Vielzahl an Beobachtungen, die im praktischen Umgang mit dem Phänomen Temperatur gesammelt wurden. In der langen Geschichte der Anwendungen thermischer Prozesse standen insbesondere folgende Zielsetzungen im Vordergrund: - Das Erreichen einer bestimmten vorgegebenen Temperatur zum Zweck des menschlichen Wohlbefindens, der Konservierung, etc. (z.B. Heizung, Klimaanlage, Kühlschrank) - Die gezielte Änderung des Zustands eines vorgegebenen Werkstoffs durch Temperaturänderung (z.B. Konservieren oder Garen von Lebensmitteln, Erweichen und Schmelzen von Metallen, Glas oder Kunststoff, thermische Ausdehnung) - Die Gewinnung von Arbeit unter Nutzung thermischer Energie (z.B. Dampfmaschine, Stirlingmotor, Peltier-Element) Insbesondere der letztgenannte Punkt, die Herausforderung, Wärme möglichst effektiv in Arbeit zu verwandeln, führte dazu, dass sich bereits vor etwa 200 Jahren eine Anzahl von Wissenschaftlern intensiv mit grundlegenden Fragestellungen der Thermodynamik beschäftigte. Er gab schließlich auch den Anlass zur Formulierung wichtiger Naturgesetze, der Hauptsätze der Thermodynamik. Aus heutiger Sicht, unter Einbeziehung aller historischen Entwicklungen, welche die Thermodynamik als Wissenschaft vollzogen hat, lässt sich zusammenfassend vielleicht folgende Definition festlegen: „Die Thermodynamik ist die Lehre von der Nutzung und Umwandlung von Energie.“ Der Bezug zur Chemie konnte erst wesentlich später geschaffen werden, als klar wurde, dass die Triebkraft aller chemischen Reaktionen auf den Grundlagen der Thermodynamik beruht. Darüber hinaus wurde gefunden, dass auch die Lage eines chemischen Gleichgewichts und die Geschwindigkeit einer chemischen Reaktion eng mit thermodynamischen Gegebenheiten verknüpft sind. Die so genannte chemische Thermodynamik beantwortet damit sowohl die Frage nach dem „warum“, als auch die Frage nach dem „wie“ einer chemischen Umsetzung. 3 2 Physikalische Beschreibung eines Systems 2.1 Grundlegende Begriffe und Parameter Aus der Sicht der Thermodynamik betrachtet man bei allen anstehenden Messungen, Versuchen oder theoretischen Betrachtungen stets eine mehr oder weniger scharf umrissene Gesamtheit von Stoffen. Diesen momentan interessierenden Ausschnitt des Universums nennt man ein System. Das kann der Inhalt eines Kolbens oder Reaktors sein, oder auch eine lebende Zelle, die Atmosphäre der Erde, ein Teich oder einige wenige Moleküle im Raum. Hat man sich einmal für ein System entschieden, so betrachtet man den Rest des Universums als Umgebung. Je nachdem, wie das System von der Umgebung abgetrennt ist, unterscheidet man drei Arten von Systemen: a) Offene Systeme: Hier werden sowohl Stoffe als auch mindestens eine Form der Energie zwischen System und Umgebung ausgetauscht. b) Geschlossene Systeme: Hier findet zwar ein Austausch von Energie statt, nicht aber ein Austausch von Stoffen. c) Abgeschlossene Systeme: Hier werden weder Stoffe noch Energie mit der Umgebung ausgetauscht. Darüber hinaus gibt es noch weitere Charakteristika eines Systems, die thermodynamisch von Bedeutung sind. Eines der wichtigsten ist das thermodynamische Gleichgewicht: ein System kann sich in Bezug auf verschiedene Systemvariablen (chemische Umsetzungen, Temperatur, Druck etc.) im Gleichgewicht oder im Ungleichgewicht befinden. Weiterhin unterscheidet man zwischen homogenen und inhomogenen Systemen, je nachdem, ob im System Phasengrenzen vorliegen oder nicht. So betrachtet man eine Emulsion oder einen Nebel als inhomogenes System, eine Kochsalzlösung oder ein Gasgemisch dagegen als homogenes System. Bei der Beobachtung von Materie bei unterschiedlichen Vorgängen wurde früh erkannt, dass äußerliche Veränderungen bei jeder Art von Materie stets mit Änderungen gewisser Zustandsparameter verbunden sind. Von den drei wichtigsten Phasenzuständen fest, flüssig und gasförmig ist der gasförmige mit Abstand am leichtesten physikalisch zu beschreiben, daher wurden Gase in diesem Zusammenhang besonders intensiv untersucht. Um den Zustand eines Gases in Abhängigkeit von der Temperatur vollständig festzulegen, benötigt man zunächst die Begriffe Stoffmenge, Volumen, Druck und Temperatur: n Stoffmenge: Die Stoffmenge wird über die Teilchenzahl definiert: Einheit der Teilchenzahl: 1 Mol Definition: Ein Mol eines Stoffes enthält dieselbe Anzahl an Teilchen wie 0,012 kg reiner Kohlenstoff des Isotops 12 C (1 Mol ≈ 6.022.1023 Teilchen) Dabei muss eindeutig festgelegt sein, was unter einem Teilchen des Stoffes jeweils zu verstehen ist. Ist die Stoffmenge konstant, so spricht man von einem geschlossenen System. 4 V Volumen: Die Definition des Volumens erfolgt über die festgelegte Längeneinheit und den geometrischen Volumenbegriff: Einheit des Volumens: 1 m³ Definition: Ein m³ ist das Volumen eines würfelförmigen Raums mit einer Kantenlänge von einem Meter. Ist das Volumen konstant, so spricht man von einem isochoren Vorgang. P Druck: Die Definition erfolgt über die Kraft, die ein Stoff auf jede Flächeneinheit eines ihn einschließenden Behälters ausübt. Einheit des Drucks: = 10-5 bar 1 Pascal = 1 Pa = 1 N/m² Definition: Ein Pascal ist der Druck, bei dem auf jeden Quadratmeter der Behälterwände eine Kraft von einem Newton ausgeübt wird. Ist der Druck konstant, so spricht man von einem isobaren Vorgang. Die vielleicht wichtigste Zustandsgröße der Thermodynamik ist gleichzeitig diejenige, die sich am schwierigsten definieren lässt, die Temperatur. Die menschliche Wahrnehmung liefert zwar einen subjektiven Temperaturmaßstab („warm“, „kalt“), ermöglicht jedoch keine objektive Festlegung einer Temperaturskala. Man kann, abgeleitet aus dem allgemeinen Erfahrungsschatz, zunächst nur eine Methode zum Vergleich von Temperaturen festlegen. Entscheidend hierfür ist die Beobachtung, dass zwischen zwei Systemen mit unterschiedlicher Temperatur Wärme fließt. Dies äußert sich darin, dass die Temperatur des Mediums, aus dem Wärme abfließt, abnimmt, während gleichzeitig die Temperatur des Mediums, dem Wärme zufließt, zunimmt. T Temperatur: Der Temperaturvergleich erfolgt über die Bestimmung der Richtung des Wärmeflusses. Definition: Stehen zwei Systeme S1 und S2 miteinander in thermischem Kontakt, so bestimmt die Temperaturdifferenz den Wärmefluss. Ist T1 größer als T2, so fließt Wärme von S1 nach S2. Ist T1 kleiner als T2, so fließt Wärme von S2 nach S1. Ist T1 gleich T2, so kommt der Wärmefluss zum Stillstand. Man nennt diesen Zustand das thermische Gleichgewicht. hohe Temperatur niedrige Temperatur Wärme niedrige Temperatur hohe Temperatur gleiche Temperaturen Wärme Ist die Temperatur konstant, so spricht man von einem isothermen Vorgang. 5 2.2 Der „Nullte Hauptsatz“ der Thermodynamik und die Temperaturskala Die so festgelegte Form des Temperaturvergleichs führte zu grundsätzlichen Überlegungen zur Natur eines thermischen Gleichgewichts und schließlich zur Formulierung eines grundlegenden Naturgesetzes, des „Nullten Hauptsatzes“ der Thermodynamik: Nullter Hauptsatz der Thermodynamik: Sind zwei Systeme im thermischen Gleichgewicht mit einem dritten System, dann sind sie auch untereinander im thermischen Gleichgewicht. Auf den ersten Blick scheint diese Aussage banal und ohne größere Konsequenz zu sein. Sie erst ermöglicht allerdings, bei genauerer Betrachtung, das Einbringen eines „fremden“ Systems, beispielsweise eines Thermometers, in ein zu untersuchendes System zum Zweck der Temperaturbestimmung. Nur unter der obengenannten Voraussetzung erhält man dann, nach Einstellung des thermischen Gleichgewichts zwischen dem Thermometer, der Umgebung des Thermometers und dem übrigen zu untersuchenden System, schließlich eine Situation, bei der die Temperaturen aller beteiligten Komponenten identisch sind: Umgebung des Thermometer Thermometers Übriges System 1) Gleichgewicht zwischen Thermometer und Umgebung des Thermometers 2) Gleichgewicht zwischen Umgebung des Thermometers und dem übrigen System Aus 1) und 2) folgt gemäß dem Nullten Hauptsatz: 3) Gleichgewicht zwischen dem Thermometer und dem übrigen System Aus 1) , 2) und 3) folgt gemäß der Definition des Temperaturvergleichs: 4) Die Temperatur des Thermometers ist gleich der Temperatur des übrigen Systems Wie alle Hauptsätze der Thermodynamik ist auch der Nullte Hauptsatz nicht bewiesen, sondern beruht auf einer Vielzahl von Beobachtungen. Seine elementare Aussage bildet die Ausgangsbasis für das Verständnis fast aller Systemparameter, insbesondere der Temperatur. Erst auf dieser Grundlage kann man sich der nächsten Aufgabe zuwenden, der Messung der Temperatur und der Festlegung von Temperaturskalen. 6 Thermometer Zur Verwendung für Thermometer eignet sich prinzipiell jeder physikalische oder chemische Vorgang, der reproduzierbar mit einer Temperaturänderung verknüpft ist. Klassisch sind dies insbesondere die Ausdehnungsvorgänge von Gasen, Flüssigkeiten und Festkörpern: Festkörperthermometer werden gewöhnlich nach dem Prinzip des Bimetall-Thermometers ausgelegt (ganz links). Dabei werden zwei verschiedene Festkörper (z.B. zwei Bleche aus verschiedenen Metallen) flächig miteinander in Kontakt gebracht. Bedingt durch die unterschiedliche thermische Ausdehnung der Materialien krümmt sich das Bimetall-Blech abhängig von der Temperatur mehr oder weniger stark zu einer Spirale. Gas Hg Flüssigkeitsthermometer (Mitte) und Gasthermometer (rechts) nutzen die Volumenänderung eines fluiden Mediums mit der Temperatur. Die Genauigkeit kann erhöht werden, indem einem großvolumigen Vorratsbehälter ein relativ kleinvolumiger Ausdehnungs- und Ablesebereich gegenübergestellt wird. In der Praxis kommen mehr und mehr die elektronischen Varianten der Temperaturmessung zum Zug, die zumeist auf der Messung der Thermospannung basieren. Neben der Messmethode ist die Festlegung einer Temperaturskala wichtig. Dazu dienten zunächst einige Fixpunkte, die heute teilweise noch historische Bedeutung haben: 1) Die tiefste Temperatur des Winters 1708/1709 in Danzig: - 17,8°C 2) Die Temperatur von schmelzendem Eis bei 760 Torr (760 Torr = 1 atm = 101 325 Pa): 0°C 3) Koexistenztemperatur von Eis, Wasser und Wasserdampf: 0,01°C (exakt) 4) Die durchschnittliche Körpertemperatur eines gesunden Menschen: 37,8°C 5) Die Siedetemperatur des Wassers bei 760 Torr (760 Torr = 1 atm = 101 325 Pa): 100°C Die Punkte 1 und 4 bildeten die erste vorläufige Grundlage des FahrenheitSystems, die Punkte 2 und 5 die der Celsius-Skala. Bei beiden Systemen wurde der definierte Bereich zunächst in 100 gleiche Teile (Grade) aufgeteilt, dann extrapoliert. Beide Definitionen wurden später verfeinert (Celsius: 99,99 Grade C zwischen den Fixpunkten 3 und 5, Fahrenheit: 180 Grade F zwischen den Fixpunkten 1 und 5). Trotzdem mangelt es außer Punkt 3 allen genannten Fixpunkten an Genauigkeit und Reproduzierbarkeit. Der Punkt 3, der so genannte Tripelpunkt des Wassers, bei dem Wasserdampf, flüssiges Wasser und Eis miteinander im Gleichgewicht stehen, ist als einziger exakt einstellbar. Das zweite Problem, nach der Unvollkommenheit der meisten Fixpunkte, besteht in der Festlegung einer systemunabhängigen linearen Teilung. 7 Gewöhnlich ist der Verlauf der Skala vom gewählten Medium abhängig: Eine lineare Teilung auf der Skala eines Quecksilberthermometers entspricht daher nicht einer linearen Teilung auf der Skala eines Alkoholthermometers, da die Ausdehnung bei jedem Medium in unterschiedlicher Weise von der Temperatur abhängt. Beide Probleme, sowohl die Wahl der passenden Fixpunkte als auch die Festlegung einer sinnvollen linearen Teilung werden heute durch die Festlegung der so genannten absoluten Temperaturskala gelöst. Grundlage hierfür sind übereinstimmende Beobachtungen an Gasthermometern: V -273,15°C -300 -200 -100 0 100 200 T Bei wiederholten Messungen mit verschiedenen Gasthermometern, verschiedenen Gasen und Gasvolumina und bei verschiedenen Drucken stellt man fest, dass sich die Verlängerungen aller in den jeweiligen Diagrammen erhaltenen Linien in einem Punkt schneiden. Dieser Punkt entspricht auf der Volumenachse dem Wert V = 0 und auf der Temperaturachse dem Wert T = -273,15 °C. Aus dieser Beobachtung wurde geschlossen, dass der Temperatur am gemeinsamen Schnittpunkt aller Ausdehnungskurven eine besondere physikalische Bedeutung zukommt und sie sich daher als Fixpunkt einer neuen Temperaturskala eignet. Weiterhin wurde festgestellt, dass zwar alle Gase in ihrem Ausdehnungsverhalten von dem idealen linearen Verlauf abweichen, dass aber unter bestimmten Umständen (z.B. niedriger Druck) ein gemeinsamer Verlauf angestrebt wird. Am besten funktioniert das bei Helium unter schrittweise absinkenden Drucken, dessen Verhalten sich für P → 0 zum idealen Verhalten extrapolieren lässt. Diese Erkenntnis diente zur Definition einer absoluten Temperaturskala in Kelvin: 1) Unterer Fixpunkt: Schnittpunkt der Volumenexpansionskurven „idealer“ Gase (z.B. Helium für den Grenzfall P→0): 0 Kelvin 2) Oberer Fixpunkt: Koexistenztemperatur von Eis, Wasser und Wasserdampf 273,16 Kelvin 3) Das Volumen eines „idealen“ Gases (z.B. Helium für den Grenzfall P→0) ist bei konstantem Druck proportional zur Temperatur und definiert die lineare Teilung der Temperaturskala Gemäß dieser Definition ist jede beliebige Temperatur unter Nutzung eines „idealen“ Gasthermometers auf der absoluten Kelvin-Skala eindeutig festgelegt. Die Verwendung der Kelvin-Skala ist gegenüber der Nutzung klassischer Temperatursysteme bei der Beschreibung physikalischer Vorgänge eindeutig von Vorteil. Vorgänge, bei denen die Temperatur konstant ist, nennt man isotherm. Mit der Definition der wichtigsten Zustandsparameter Teilchenzahl n, Volumen V, Druck P und Temperatur T besteht nun die Möglichkeit, das Verhalten makroskopischer Materie zu beschreiben. Am einfachsten gelingt das im Fall von Gasen. 8 2.3 Zustandsfunktionen Die somit eingeführten Größen Teilchenzahl, Volumen, Druck und Temperatur beschreiben den Zustand eines Systems. Man nennt sie deshalb Zustandsgrößen oder, falls sie als Funktionen anderer Parameter gegeben sind, Zustandsfunktionen. Sie besitzen eine Reihe von gemeinsamen Eigenschaften: 1) Der Wert einer Zustandsfunktion Z(x, y) hängt nur von der Größe der Variablen x und y ab, nicht von dem Weg, über den der entsprechende Zustand erreicht wurde. Beispiel: Eine Zustandsfunktion Z(V, T) unterscheidet sich von der Nicht-Zustandsfunktion Z’(V, T) dadurch, dass eine Veränderung der Parameter T und V um ∆T bzw. ∆V in jedem Fall reproduzierbar zu einer identischen Veränderung in Z führt (s. folgende Abb. links). Bei Z’ ist diese Veränderung abhängig vom Weg in T und V (s. folgende Abb. rechts). Zustandsfunktion: Nicht-Zustandsfunktion: Z’ Z Weg 1 Weg 1 Weg 2 Weg 2 Z Z’1 Z’2 Z+∆Z T T V V T T+∆T V T+∆T V+∆V V+∆V 2) T V Für die mathematische Formulierung der Zustandsfunktion gilt der ∂( Schwarzsche Satz: ∂Z( x, y ) ) ∂x ∂y ∂( = ∂Z( x, y ) ) ∂y ∂x das heißt, bei der Bildung der gemischten Ableitung ist es egal, ob zuerst nach x oder zuerst nach y abgeleitet wird. 3) Zustandsfunktionen besitzen ein Totales Differential. Innerhalb der Zustandsgrößen wird weiterhin zwischen sogenannten intensiven Größen und extensiven Größen unterschieden: 9 Intensive Zustandsgrößen sind von der Masse des Systems unabhängig. Greift man aus dem betrachteten System einen kleinen Ausschnitt heraus, so bleiben intensive Zustandsgrößen auch dort unverändert. Man kann diese Größen also messen, ohne das Gesamtsystem zu erfassen. Beispiele: Druck, Temperatur, Dichte. Extensive Zustandsgrößen sind abhängig vom Umfang des Systems. Im Allgemeinen sind sie proportional zur Größe eines betrachteten Teilsystems. Um sie zu messen, ist es nötig, das Gesamtsystem zu erfassen. Beispiele: Masse, Volumen, Teilchenzahl. Extensive Parameter können in intensive überführt werden, indem man sie auf die Stoffmenge bezieht. Beispiele: das Molvolumen Vm = V/n oder die Konzentration c = n/V oder die Molmasse M = m/n. 3 Beschreibung gasförmiger Systeme 3.1 Die Gasgesetze und das ideale Gas Mit den Parametern n, V, P und T sind die wichtigsten Zustandsgrößen zur Beschreibung eines gasförmigen Systems eingeführt. Es gilt nun, die grundlegenden Zusammenhänge zwischen diesen Messgrößen experimentell zu erfassen. Zunächst führte die Untersuchung des Zusammenhangs zwischen Volumen und Druck zur Formulierung des Boyle’schen Gesetzes (1660): 3.1.1 Das Boyle’sche Gesetz Auf eine Verringerung des Volumens reagieren Gase grundsätzlich mit einem Anstieg des Drucks. Der Physiker Robert Boyle stellte darüber hinaus fest, dass bei konstanter Temperatur das Produkt aus Druck und Volumen annähernd konstant ist. Bei der Auftragung von P gegen V in einem Diagramm ergibt sich somit für jede eingestellte Temperatur eine Hyperbel. Boyle interpretierte das Ergebnis fälschlich als Folge von intermolekularen Wechselwirkungen („spring of the air“), eine Vorstellung, die durch Mariotte unter Postulierung des kinetischen Gasmodells (s. 3.2) berichtigt wurde. 10 ∆V T = const. T = const. ∆P P T5 T4 T3 T2 T1 V Das Boyle’sche Gesetz lässt sich auf folgende einfache Aussage reduzieren: mit konstantem n und T: P.V = const. (Boyle) Untersuchungen bei variabler Temperatur führten 1787 schließlich zum Charles’schen Gesetz: 3.1.2 Das Charles’sche Gesetz Charles beobachtete, dass das Volumen eines Gases bei konstantem Druck annähernd linear mit der Temperatur ansteigt (Gasthermometer). P = const. ∆T P = const. ∆V 11 P3 V P2 P1 T Zusammengefasst lässt sich nach Charles festhalten: mit konstantem n und P: V = const..T (Charles) Beobachtungen hinsichtlich des Volumens von Teilchenzahl führten zum Avogadroschen Gesetz. 3.1.3 Gasen bei variabler Das Avogadro’sche Gesetz Bei konstanter Temperatur und konstantem Druck nimmt ein Mol eines beliebigen Gases stets ein Volumen von etwa 22 Liter ein (ideal: 22,4 L bei 273 K und 1,013 bar). Mit anderen Worten: das Volumen eines Gases (bei T, P = const.) ist proportional zur Teilchenzahl: mit konstantem T und P: V = const..n (Avogadro) Eine Kombination der drei Gasgesetze liefert schließlich das ideale Gasgesetz. 3.1.4 Die Verknüpfung der Gasgesetze - das ideale Gasgesetz Grafisch lassen sich die Gasgesetze nach Boyle und Charles kombinieren, indem man ein dreidimensionales Koordinatensystem einführt: P T V Mathematisch führt die Kombination aller Gasgesetze zu dem Ausdruck: (PV) / (nT) = const. 12 oder: PV = const. . nT Wie alle genannten Gasgesetze ist auch diese Kombination ein Grenzgesetz, d.h. es wird von allen Gasen zunächst nur annähernd erfüllt. Generell wird beobachtet, dass die genannte Gleichung um so genauer gilt, je niedriger der Druck P je höher die Temperatur T je geringer Wechselwirkungen zwischen den Gasteilchen Alle Gase streben unter diesen Bedingungen ein gemeinsames Grenzverhalten an. Die Konstante der Gleichung, auch allgemeine oder ideale Gaskonstante R genannt, beträgt dann: R = 8,314 J K-1 Mol-1 Diese Größe ist eine auch in anderer Hinsicht sehr wichtige Naturkonstante. Der hypothetische Grenzfall eines Gases, das exakt die oben genannte Beziehung erfüllt, wird Ideales Gas genannt. Die Gleichung PV = nRT heißt auch Ideale Gasgleichung. Festzuhalten ist die Tatsache, dass solch ein Gas nicht real existiert, sondern nur einen idealisierten Grenzfall beschreibt. Unter den real existierenden Gasen kommt wohl Helium bei niederen Drücken und hohen Temperaturen diesem Idealfall am nächsten. Mathematisch handelt es sich bei der Idealen Gasgleichung um eine Zustandsgleichung, die (gleichgültig, welche Größen als Variablen geführt werden) ein totales Differential besitzt. 3.2 Das Kinetische Gasmodell 3.2.1 Grundlagen Bei den im vorhergehenden Kapitel aufgeführten Gasgesetzen handelt es sich um mathematische Beschreibungen von makroskopisch beobachtbaren Vorgängen. Zur Interpretation der Gasgesetze auf molekularer Ebene wurden verschiedene Modelle vorgeschlagen. Das erfolgreichste unter ihnen war das sogenannte Kinetische Gasmodell. Es beruht auf der Vorstellung, dass ein Gas aus einer Vielzahl von Teilchen besteht, die folgende Bedingungen erfüllen: 1) Sie besitzen eine endliche Masse m, einen endlichen Durchmesser d und befinden sich in ständiger und ungeregelter Bewegung. 2) Die Größe der Teilchen ist im Verhältnis zum freien Volumen vernachlässigbar. 13 3) 3.2.2 Zwischen den Teilchen finden elastische Stöße statt. Ansonsten existieren keine weiteren Wechselwirkungen. Definition des Drucks im Kinetischen Gasmodell Nach der kinetischen Gastheorie besteht der Druck eines Gases aus der Summe aller Kräfte (pro Flächeneinheit), die durch auf eine Fläche aufprallende Gasteilchen ausgeübt werden. Die Impulsänderung ∆p, die jedes Teilchen dabei erfährt, beträgt ∆p = 2 mvx Dabei steht p für den Gesamtimpuls, vx für die Geschwindigkeitskomponente in x-Richtung und m für die Masse eines Teilchens. Die Kraft, die pro Teilchenstoß auf die Fläche wirkt, beträgt dann: Vx -Vx F = dp/dt ≅ 2 m vx / ∆t Das Volumen Vstoß, in dem sich Teilchen befinden, die potentiell innerhalb eines Zeitintervalls ∆t auf die Wand stoßen können, beläuft sich auf: Vx Vx Vx Vx Vx ∆t Vstoß = A vx ∆t wobei A der betrachteten Fläche entspricht. Die Zahl der Teilchen nStoß (in Mol), die pro Zeiteinheit auf die Fläche stoßen, beträgt dann: nStoß = ½ V stoß (n/V) = ½ A vx ∆t (n/V) Dabei wird zunächst davon ausgegangen, dass alle Teilchen die gleiche Geschwindigkeitskomponente vx aufweisen. Der Faktor ½ berücksichtigt, dass sich im Durchschnitt nur die Hälfte aller Teilchen aus dem betrachteten 14 Volumen in Richtung der Fläche A bewegt. Die Gesamtmasse mStoß der Teilchen, die pro Zeiteinheit auf die Wand stoßen, beläuft sich somit auf: = mStoß M nstoß = M ½ A vx ∆t (n/V) wobei M die Molmasse der Gasteilchen benennt. Die insgesamt ausgeübte Kraft beträgt also: ≅ F mStoß 2vx / ∆t = M ½ A vx (n/V) 2 vx und damit beläuft sich der Druck auf: P = F/A = M vx² (n/V) Bisher wurde der Einfachheit halber davon ausgegangen, dass alle Teilchen betraglich gleiche Geschwindigkeitskomponenten vx in x-Richtung aufweisen. Dieser Ansatz entspricht natürlich nicht den Gegebenheiten eines reellen Gases. Um der Realität gerecht zu werden, muss in der letztgenannten Gleichung die Größe vx² durch das entsprechende mittlere Geschwindigkeitsquadrat ⟨vx²⟩ ersetzt werden (wobei ⟨vx²⟩ ≠ ⟨vx⟩² !). Das mittlere Quadrat ⟨vx²⟩ der Geschwindigkeitskomponente in x-Richtung sollte, da alle Raumrichtungen gleichberechtigt sind, folgende Beziehung erfüllen: ⟨vx²⟩ = ⟨vy²⟩ = ⟨vz²⟩ Außerdem erfordert die Tatsache, dass sich die Gesamtgeschwindigkeit vektoriell aus den Komponenten zusammensetzt, die Gültigkeit folgenden Zusammenhangs: Vx V VY Vz ⟨v²⟩ = ⟨vx²⟩ + ⟨vy²⟩ + ⟨vz²⟩ = 3 ⟨vx²⟩ das mittlere Geschwindigkeitsquadrat ⟨v²⟩ der Teilchen wird im Allgemeinen als c² bezeichnet, es gilt: ⟨v²⟩ = c² Daraus ergibt sich abschließend folgender Ausdruck für den Druck nach dem kinetischen Gasmodell: 15 P = 1/3 M c² (n/V) oder, in der Schreibweise der idealen Gasgleichung: PV 3.2.3 = 1/3 n M c² Die Bewegungsgeschwindigkeit von Gasteilchen Zieht man nun das ideale Gasmodell hinzu und formuliert die ideale Gasgleichung entsprechend: PV = nRT so erhält man durch Koeffizientenvergleich: nRT = 1/3 n M c² oder: RT = 1/3 M c². Man kann unter Nutzung beider Gasmodelle so zu einem völlig neuen Verständnis des Phänomens Temperatur kommen: Die Temperatur eines Gases ist demnach direkt proportional zum mittleren Geschwindigkeitsquadrat der Gasteilchen: weiter gilt: T ∝ c ∝ c² T oder T ∝ ⟨Ekin ⟩ und c ∝ M−1 Die Wurzel aus dem mittleren Geschwindigkeitsquadrat, also die Größe c, liegt üblicherweise in der Größenordnung der Schallgeschwindigkeit (zum Beispiel für Stickstoff bei Raumtemperatur: c = 516 m/s). Nachdem das mittlere Geschwindigkeitsquadrat der Teilchen bekannt ist, stellt sich die Frage nach der Geschwindigkeitsverteilungsfunktion der Teilchen. Teilt man den Bereich der auftretenden Geschwindigkeiten in Intervalle auf, und zählt man die Teilchen, die gemäß ihrer Geschwindigkeit zu den einzelnen Intervallen zugeordnet werden müssen, so ergibt sich für die Geschwindigkeitsverteilung in vx und v folgendes Bild: n(vx) n(vx) Temperaturerhöhung - 0 + vx-Intervall - 0 + vx-Intervall 16 n(v) n(v) Temperaturerhöhung 0 + v-Intervall 0 + Der Mittelwert von vx (oder jeder anderen eindimensionalen Geschwindigkeitskomponente) ist grundsätzlich Null. Dagegen besitzt der Mittelwert von v stets eine endliche, von Null verschiedene Größe. Bei einer Erhöhung der Temperatur werden alle Verteilungsfunktionen breiter, der Mittelwert von v vergrößert sich. Die Verteilungsfunktionen in vx und v lauten (ohne Herleitung): f(vx) = [M/(2πRT)]1/2 exp [-Mvx²/(2RT)] f(v) = 4π [M/(2πRT)]3/2 v² exp [-Mv²/(2RT)] Die Temperatur eines Gases äußert sich also nicht nur im mittleren Geschwindigkeitsquadrat, sondern auch in der Form der Geschwindigkeitsverteilungsfunktion. Bei der Mischung von Gasen unterschiedlicher Temperatur muss, um die o.g. Forderung zu erfüllen, aus der einfachen Summe von zwei Verteilungsfunktionen eine neue, der Mischtemperatur entsprechende Verteilungsfunktion entstehen. Dies ist nur unter der Annahme möglich, dass ein Austausch kinetischer Energie unter den Teilchen erfolgen kann. Diese Tatsache bedingt die eingangs gestellte Forderung nach Teilchenstößen, also Wechselwirkungen zwischen den Teilchen. Darin besteht der wesentliche Unterschied zwischen einem Gas nach dem kinetischen Gasmodell und dem idealen Gas. 3.2.4 Stöße zwischen Gasteilchen Verschiedene makroskopisch beobachtbare Vorgänge belegen, dass Teilchenstöße ablaufen. Hierzu gehören neben der Geschwindigkeitsverteilung auch der Vorgang der Schallausbreitung und chemische Reaktionen zweier Gase. Die Beschreibung solcher Vorgänge setzt die quantitative Berechnung von Teilchenstößen voraus. Als wichtigste Kennzahl ist die Stoßzahl oder Stoßhäufigkeit z einzuführen, die angibt, wie viele Stöße ein einzelnes Teilchen pro Zeiteinheit „erleidet“. Dazu folgende Betrachtung: Ein Teilchen mit dem Durchmesser d stößt pro Zeiteinheit mit allen Teilchen zusammen, deren Schwerpunkte sich in einem gedachten Zylinder mit dem Durchmesser 2d und der Länge ⟨vrel⟩ ∆t befinden (s. Skizze). Die Größe ⟨vrel⟩ steht dabei für die mittlere Relativgeschwindigkeit der Teilchen. v-Intervall 17 d d Vrel ∆t Das Volumen dieses Zylinders beträgt Vz = πd² ⟨vrel⟩ ∆t in diesem Zylinder befinden sich damit (mit N als gesamte Teilchenzahl und V als Gesamtvolumen) insgesamt Nz = πd² ⟨vrel⟩ ∆t N/V Teilchen. Diese Teilchenzahl ist identisch mit der Zahl der Stöße eines Teilchens im Zeitintervall ∆t. Die mittlere Relativgeschwindigkeit ergibt sich aus der mittleren Geschwindigkeit ⟨v⟩ als: ⟨vrel⟩ = 2 ⟨v⟩ (Grund: mittlerer „Stoßwinkel“: 90°) Somit gilt für die Stoßzahl (Stöße eines einzelnen Teilchens pro Zeiteinheit, Einheit: 1/s): z = πd² 2 ⟨v⟩ N/V Die Gesamtzahl Z aller Stöße pro Volumen- und Zeiteinheit beträgt damit: Z = ½ z N/V = ½ πd² 2 ⟨v⟩ N²/V² mit der Einheit 1/(s⋅m³). Der Faktor ½ ist notwendig, damit nicht jeder Stoß doppelt gezählt wird. Stöße zwischen drei Teilchen sind sehr selten und werden vernachlässigt. Die Gesamtzahl der Stöße ist damit proportional zum Quadrat der Teilchendichte, was wichtige Konsequenzen für viele chemische Reaktionen hat. Ein weiterer bedeutender Parameter ist die Wegstrecke, die ein Teilchen durchschnittlich zurücklegt, ohne auf andere Teilchen zu stoßen. Für diese mittlere freie Weglänge λ gilt: λ = ⟨v⟩/z = ⟨v⟩ / (πd² 2 ⟨v⟩ N/V) = 1 / (πd² 2 N/V) mit der Einheit m. Die mittlere freie Weglänge ist somit umgekehrt proportional zur Teilchenzahl pro Volumen und zum Quadrat des Durchmessers, dagegen (bei konstantem Volumen) unabhängig von der Temperatur. 18 3.2.5 Mischungen von Gasen: Das Daltonsche Gesetz Das Daltonsche Gesetz bezieht sich auf eine Mischung aus zwei oder mehr Gasen. Die Aussage des Daltonschen Gesetzes lässt sich am ehesten über ein Gedankenexperiment darstellen: Man betrachte zwei Behälter, die zwei verschiedene Gase beinhalten. Alle physikalischen Zustandsparameter (also n, P, T, V) und die Masse der Gasteilchen seien bei beiden Proben identisch. Nun vereine man die Inhalte beider Behälter in einem dritten Behälter gleichen Volumens. Wenn nun das ideale Gasgesetz im Grenzfall für beide Gase gilt, so kann angesetzt werden: P1 P2 = = n1 RT / V n2 RT / V In der gemischten Probe gilt für die Teilchenzahl n n = n1 + n2 Welchen Gesamtdruck P erhält man nun in der gemischten Probe? Dalton legt bei der Beantwortung dieser Frage das kinetische Gasmodell zugrunde, demgemäß nach der Vereinigung der Gasvolumina genau die doppelte Anzahl an Stößen stattfindet und damit der doppelte Druck herrscht: P = P1 + P2 Dalton verallgemeinert diese Betrachtung als ein Grenzgesetz für Mischungen aus beliebig vielen verschiedenen Gasen. Für die Druckkomponente jedes einzelnen Gases führt er den Begriff Partialdruck ein. Es gilt: Pi = (ni/n). P woraus gemäß dem idealen Gasgesetz folgt: Pi = ni RT / V Außerdem gilt demnach für eine Mischung aus n verschiedenen Gasen: P = P1 + P2 + P3 + ... + Pn = Σ Pi Die Summe aller Partialdrücke entspricht also immer dem messbaren Gesamtdruck. Der Partialdruck eines Gases entspricht demjenigen messbaren Druck, den das Gas ausüben würde, wenn alle anderen Gase der Mischung nicht vorhanden wären. Dieser Zusammenhang wurde durch Dalton experimentell bestätigt und im Daltonschen Gesetz zusammengefasst: Der Druck, den eine Mischung von Gasen ausübt, entspricht der Summe aller Partialdrücke, also der Drücke, die jedes Gas für sich alleine ausüben würde. (Dalton) Auch diese Gesetzmäßigkeit ist ein Grenzgesetz, das im Realfall nur annähernd erfüllt wird. Das Daltonsche Gesetz gilt strenggenommen nur für 19 ideale Mischungen idealer Gase. In der Realität weichen die erhaltenen Drucke mehr oder weniger stark von dem so errechneten Pges ab. Besondere Bedeutung besitzt der Quotient ni/n. Er benennt den „partiellen Teilchenzahlanteil“ und wird als der Molenbruch xi bezeichnet: xi mit = (ni/n) x1 + x2 + x3 + ... = Σ xi = 1 Für den Fall einer idealen Mischung zweier idealer Gase können die Beziehungen zwischen Partialdrücken und Molenbrüchen folgendermaßen veranschaulicht werden: P Pges P2 0 Abhängigkeit der Partialdrücke und des Gesamtdrucks einer idealen Mischung zweier idealer Gase von dem Molenbruch x2 = P2/P. P1 1 x2 3.3 Reale Gase 3.3.1 Eigenschaften eines realen Gases Alle experimentell untersuchten Gase zeigen mehr oder weniger starke Abweichungen von den Eigenschaften eines idealen Gases oder eines Gases nach dem kinetischen Gasmodell. Der Grund liegt in der Gegenwart von Wechselwirkungen, die bei den bisher betrachteten Modellen entweder völlig vernachlässigt werden (ideales Gasmodell) oder nur unzureichend eingehen (kinetisches Gasmodell). Vergleicht man den im jeweiligen Modell vorgesehenen Verlauf der potentiellen Energie bei der Begegnung von Gasteilchen, so werden die wesentlichen Unterschiede deutlich: 8 Epot ideales Gas r Epot “kinetisches” Gas r Epot reales Gas r 20 Während für die Teilchen eines idealen Gases die potentielle Energie überall gleich ist (links), besitzt sie für Teilchen des „kinetischen“ Gases am Ort eines anderen Teilchens den Wert Unendlich (Mitte), wodurch mechanische Stöße beschrieben werden können. Der „wirkliche“ Potentialverlauf (reales Gas, rechts) beschreibt bei der Annäherung zweier Teilchen sowohl deren Abstoßung bei sehr kurzen Distanzen als auch deren Anziehung bei größeren Abständen in einer kontinuierlichen Funktion (Lennard-Jones-Potential). Aus dem realen Potentialverlauf lässt sich das Verhalten von realen Gasen im Unterschied zum idealen Gas ableiten. Trägt man beispielsweise den sogenannten Kompressibilitätsfaktor z = (PV) / (nRT) gegen P auf, so findet man für reale Gase Kurvenverläufe, die mehr oder weniger stark vom Idealwert z = 1,0 abweichen: C2 H4 H2 NH3 PV nRT 1,0 ideales Gas P Offensichtlich dominieren für viele reale Gase bei hohen Drücken die abstoßenden Wechselwirkungen (PV ist größer als ideal), bei kleinen Drücken dagegen die anziehenden Wechselwirkungen (PV ist kleiner als ideal). Nur bei Gasen mit sehr schwachen intermolekularen Wechselwirkungen (wie Wasserstoff oder Edelgasen) tritt der letztgenannte Effekt nicht auf (bzw. ist kaum messbar). Viele reale Gase besitzen neben dem Grenzfall P→0 noch einen weiteren Punkt im Diagramm, an dem sie sich „ideal“ verhalten. Dieser Punkt (z = 1 für P ≠ 0) entspricht dem Zustand, an dem sich anziehende und abstoßende Kräfte im Mittel aufheben. 3.3.2 Zustandsgleichungen für reale Gase: Die van-der-Waals-Gleichung Gesucht ist nun eine plausible Korrektur der idealen Gasgleichung, die sämtliche vorkommenden Abweichungen berücksichtigt. Diese neue Zustandsgleichung müsste dann das Verhalten aller realen Gase beschreiben können. Zunächst soll die Abstoßung zwischen den Teilchen berücksichtigt werden. In erster Näherung geschieht dies (analog zum kinetischen Gasmodell) unter Annahme unflexibler, aneinanderstoßender Teilchen mit einem sogenannten molaren Covolumen b. Das Covolumen beschreibt das Volumen des Gases bei ständigem mechanischem Kontakt zwischen jeweils zwei Teilchen (wegen der geringen Wahrscheinlichkeit von Dreierstößen kann die Bildung von Dreier- 21 gruppen ausgeschlossen werden) und entspricht damit dem vierfachen Eigenvolumen der Gasteilchen. Damit wird aus der idealen Gasgleichung: PV = nRT die erste korrigierte Version P (V - n b) = nRT Im zweiten Schritt soll berücksichtigt werden, dass die Anziehung zwischen den Teilchen für einen zusätzlichen, nach außen nicht messbaren „Binnendruck“ sorgt. Dieser Binnendruck ist proportional zum Quadrat der Teilchendichte (n/V)². Der zwischen den Teilchen tatsächlich wirkende, nach außen ebenfalls unmessbare Gesamtdruck ist dann gegeben als: Pgesamt (unmessbar) = P (messbar) + a (n/V)² mit einer für die anziehende Wechselwirkung charakteristischen Konstante a. Die danach korrigierte Version der Gasgleichung, die Zustandsgleichung für reale Gase nach van-der-Waals, lautet (P + a (n/V)² ) (V - nb) = nRT Die Konstanten a und b besitzen für jedes reale Gas charakteristische Werte, die dessen Eigenvolumen und intermolekularen Wechselwirkungen reflektieren. Beispiele: Gas Argon Kohlendioxid Helium Stickstoff a 0,1345 0,3592 0,0034 0,1390 b Pa m6/Mol² Pa m6/Mol² Pa m6/Mol² Pa m6/Mol² 3,22 4,267 2,37 3,913 10-5 m³/Mol 10-5 m³/Mol 10-5 m³/Mol 10-5 m³/Mol Der Parameter a, der die anziehenden Wechselwirkungen charakterisiert, unterscheidet sich bei den einzelnen Gasen dramatisch (man vergleiche beispielsweise die Werte für Helium und Kohlendioxid, die sich um mehr als zwei Größenordnungen unterscheiden). Dagegen sind die Unterschiede bei dem Parameter b, der die Abstoßung berücksichtigt, vergleichsweise klein (etwa Faktor zwei bei Helium und Kohlendioxid). 22 4 Der Erste Hauptsatz der Thermodynamik 4.1 Weiterführende Begriffe: Arbeit, Wärme und Energie Die ursprüngliche Formulierung des ersten Hauptsatzes basiert auf der Erfahrung, dass ein Perpetuum Mobile erster Art nicht existiert. Er lautet in seiner empirischen Form: Es ist unmöglich, eine Maschine zu konstruieren, die kontinuierlich Arbeit leistet, ohne dabei Energie zu verbrauchen. In dieser Formulierung tauchen die Begriffe Arbeit und Energie auf, die zunächst in physikalischer Hinsicht zu definieren sind. In engem Zusammenhang mit den Größen Arbeit und Energie steht die Wärme, die im Folgenden in die Betrachtung mit einbezogen wird. 4.1.1 Arbeit (w) Im Verlauf eines Vorgangs, bei dem die Position eines Gegenstands entgegen einer Kraft verschoben wird, wird Arbeit geleistet. Die dabei aufgebrachte Arbeit lässt sich (mit w als Arbeit, F als Kraft und x als Ortskoordinate) in folgender Weise formulieren: dw = -F dx Das negative Vorzeichen deutet an, dass bei positiver („am System geleisteter“) Arbeit der Kraftvektor und der Ortsvektor entgegengesetzte Ausrichtung aufweisen. Beispiele: Anheben einer Masse im Schwerefeld: dw = mg dh Spannung einer Feder mit der Federkonstante k: dw = kx dx Beschleunigung eines Körpers mit der Masse m: dw = m Arbeit, um n geladene Teilchen in einem elektrischen Feld mit dem Potential E zu verschieben: (Faraday-Konstante: F = 96 485 Coulomb / Mol) d w = F E dn ∂²x dx ∂t ² . Die Einheit der Arbeit beträgt in jedem Fall 1 [N m] oder 1 [Joule]. Sie ist definiert als diejenige Arbeit, die benötigt wird, um ein Objekt gegen die Kraft von einem Newton einen Meter weit zu verschieben. Eine im Umfeld der Thermodynamik besonders wichtige Form der Arbeit ist die Volumenarbeit. Die zu überwindende Kraft besteht hierbei in einer Druckkraft P⋅A, die Ortsveränderung in einer Bewegung des Kolbens entlang der Ortskoor- 23 dinate x. Damit gilt für die bei der Kompression geleistete (oder bei der Expansion maximal erhältliche) Arbeit: maximal erhältliche Volumenarbeit: x dwmax = - P dV p dw = -PA dx = -P dV Die gesamte, im Verlauf eines Kompressionsvorgangs entwickelte Arbeit ergibt sich aus dem Integral über dw bzw. -PdV zwischen den Grenzen V1 und V2. Im PV-Diagramm entspricht dies einer von Diagrammverlauf eingeschlossenen Fläche: dw = - P dV P 2 w= Arbeit w V1 V2 dw = 1 V - P dV V1 V2 V2 V1 Für die Kompression eines Gases wird immer die Arbeit dw = -PdV benötigt. Bei der Expansion eines Gases hängt das Maß an freigesetzter Arbeit dagegen von den äußeren Bedingungen ab. Sie entspricht nur dann der Kompressionsarbeit, wenn zu jedem Zeitpunkt der Expansion ein Kräftegleichgewicht zwischen Druckkraft und Gegenkraft am Kolben herrscht. In diesem Fall ist der Vorgang an jedem Punkt des Ausdehnungsprozesses umkehrbar. Man nennt eine so geführte Expansion deshalb reversibel. Bei allen anderen, irreversiblen Expansionen ist die erhaltene Arbeit kleiner, im Extremfall der freien Expansion gleich Null (s. folgende Abb.). Das Maß an zurückgewonnener Volumenarbeit ist also davon abhängig, wie viel Arbeit „abgeschöpft“ wird. Die im reversiblen Fall freigesetzte Arbeit entspricht dem erreichbaren Maximum, es gilt also: wrev = wmax und wirrev < wrev, wmax Für das Vorzeichen der Arbeit gilt stetes folgende Konvention: wird Arbeit w an einem betrachteten System geleistet, so wird sie mit positivem Vorzeichen angegeben (w > 0). Leistet dagegen ein System Arbeit an seiner Umgebung, so versieht man sie mit einem negativen Vorzeichen (w < 0). 24 Pex = Pint p p Pex = 0 Pex = const. p int pint int p reversibel Arbeit V p irreversibel gegen Pex =0 Arbeit =0 pex Arbeit V dw = 0 dwrev = - P dV irreversibel gegen konstanten Druck dw = -Pex dV Wichtig: die Größe Arbeit bezieht sich immer auf einen Vorgang, hier auf eine Volumenänderung. Sie ins wegabhängig und keine Zustandsfunktion. 4.1.2 Wärme (q) Ursprünglich stellte der Begriff der Wärme für die Naturwissenschaft ein ebenso großes Rätsel dar wie der Begriff der Temperatur. Die Naturforscher B. Thompson und R. Mayer stellten unabhängig voneinander fest, dass es eine gewisse Äquivalenz zwischen Arbeit und Wärme geben müsse. Ihre Beobachtungen wurden nachträglich durch J.P. Joule bestätigt und wesentlich verfeinert. Demnach lässt sich durch einfache mechanische Anordnungen (z.B. aufeinander reibende Flächen, mechanische Deformation oder ein Rührwerk in einer Flüssigkeit) ein bestimmtes Quantum Arbeit in ein genau entsprechendes Quantum Wärme umwandeln. Robert Mayer berichtete, dass man mittels einer bestimmten Portion Arbeit „ein Liter Wasser entweder um 365 Meter anheben oder um ein Grad erwärmen könne“ (heute weiß man, dass der genaue Wert für den genannten Höhenunterschied 426,4 m beträgt). Es liegt also nahe, Wärme (üblicherweise bezeichnet durch die Variable q) und Arbeit mit denselben Einheiten zu benennen. Die ursprüngliche Einheit der Wärme, die Kalorie, war definiert als diejenige Wärmemenge, die (unter Normalbedingungen) die Temperatur von einem Gramm Wasser um genau ein Grad Celsius (bzw. ein Kelvin) erhöht. Unter Nutzung der Äquivalenz von Arbeit und Wärme ergibt sich: 1 Kalorie ≅ 4,184 Joule 25 Damit ist die Definition einer eigenen Einheit für die Wärme überflüssig, dementsprechend sollten Wärmemengen nur noch in der SI-Einheit Joule und nicht mehr in Kalorien ausgedrückt werden. Beide Phänomene, sowohl Arbeit als auch Wärme, sind eng mit der Bewegung von Teilchen verknüpft. Der eigentliche Unterschied besteht darin, dass im Fall der Arbeit eine gerichtete, gleichsinnige Bewegung entsteht, während im Fall der Wärme chaotische, nur statistisch fassbare Bewegungsvorgänge erzeugt werden. Betrachtet man ein einzelnes Teilchen eines Systems, so existiert dieser Unterschied nicht mehr. Ein einzelnes Wassermolekül erhält durch einen freien Fall über 426,4 m (in 9.3 s auf etwa 91 m/s) genau so viel zusätzliche kinetische Energie wie durch eine Temperaturerhöhung um 1 Grad Celsius. Genau betrachtet ist eine Unterscheidung zwischen Arbeit und Wärme, zwischen kinetischer und thermischer Energie nur dann sinnvoll, wenn das betrachtete System aus einer Vielzahl nur statistisch zu erfassender Teilchen besteht. Das Phänomen der Wärme verknüpft in randomisierter Form alle in einem System möglichen Arten von Arbeit. Daher gibt es stets nur eine Erscheinungsform von Wärme, obwohl die Arbeit in sehr vielen verschiedenen Erscheinungsformen auftreten kann. Wie die Arbeit ist die Wärme ebenfalls eine wegabhängige Größe, die einen Vorgang beschreibt. Sie ist damit keine Zustandsgröße. 4.1.3 Energie Der dritte Begriff, der im Zusammenhang mit dem ersten Hauptsatz zu definieren ist, ist die Energie. Allgemein ließe sich aufgrund der Äquivalenz von Wärme und Arbeit folgendes festlegen: Energie ist die Fähigkeit, Arbeit zu leisten oder Wärme freizusetzen. Energie kann in einem System in verschiedenster Weise vorliegen, beispielsweise als kinetische Energie, thermische Energie, Gravitationsenergie, chemische Energie, oder elektromagnetische Energie. Um alle im Inneren eines Systems vorhandenen Formen der Energie zusammenzufassen, spricht man von ihrer Summe als der Inneren Energie U und definiert: U = Ekin + Etherm + Egrav + Echem + Eelmag + .... Zur Inneren Energie gehören dabei nicht solche Energieformen, die Lage und Bewegung des Systems als Ganzes betreffen, also nicht die Potentielle Energie des Behälters im Schwerefeld der Erde oder die mögliche Bewegungsenergie aufgrund der Bewegung des Systems als Ganzes. Die Größe U beschränkt sich daher ausdrücklich auf den inneren Zustand des Systems, nicht auf Zustände oder Vorgänge, die sich auf die Außenwelt beziehen. Bezugsmaßstab für alle Bewegungsvorgänge ist immer ein Koordinatensystem, das sich am Schwerpunkt des Systems orientiert. 4.2 Die Innere Energie und der Erste Hauptsatz Die Gesamtenergie eines Systems aus der Sicht einen systemfesten Koordinatensystems ist somit identisch mit dessen Innerer Energie U. Aus den 26 Beobachtungen bezüglich der Äquivalenz von Wärme und Arbeit sowie aus der Tatsache, dass beide Prozesse die Innere Energie eines Systems zu verändern vermögen, werden folgende Schlüsse gezogen: Die Innere Energie eines Systems erhöht sich, wenn man an dem System Arbeit leistet oder Wärme zufügt, sonst nicht. Die Innere Energie eines Systems vermindert sich, wenn das System an seiner Umgebung Arbeit leistet oder Wärme abgibt, sonst nicht. Aus diesem Postulat (das, wie alle Hauptsätze, nicht bewiesen werden kann) folgt schließlich die thermodynamische Formulierung des Ersten Hauptsatzes: Die Änderung der Inneren Energie ergibt sich aus der Summe der ausgetauschten Beträge von Wärme und Arbeit: ∆U dU = = w + q dw + dq Mit der gewählten Definition für die Systemeigenschaften „offen“, „geschlossen“ und „abgeschlossen“ (s. Abschnitt 2.1) folgt als weitere Formulierung des ersten Hauptsatzes: Die Innere Energie eines abgeschlossenen Systems ist konstant. Der erste Hauptsatz enthält in dieser Form zwei Aussagen: die Äquivalenz von Wärme und Arbeit und die Forderung nach der Erhaltung der Energie. Die Innere Energie ist eine Zustandsgröße, da sie einen energetischen Zustand eines Systems beschreibt. Arbeit und Wärme sind keine Zustandsgrößen, da sie nur Änderungen bedeuten, die am System vorgenommen werden. Die Summe aus Arbeit und Wärme beschreibt die Änderung der Zustandsgröße Innere Energie. Diese Beziehung zwischen den Größen Innere Energie, Arbeit und Wärme lässt sich beispielsweise mit der analogen Beziehung zwischen Kontostand, Überweisung und Barscheckeinzahlung verdeutlichen. Weder die Barscheckeinzahlungen noch die Überweisungen sagen allein etwas über die Zustandsgröße „Kontostand“ aus. Über die Summe beider Parameter kann jedoch auf die Veränderung des Zustands geschlossen werden. Arbeit und Wärme sind zwar selber keine Zustandsfunktionen, beschreiben aber in ihrer Summe die Veränderung einer Zustandsfunktion, nämlich die der Inneren Energie. Im allgemeinen Fall ist die Innere Energie eine Funktion aller grundlegenden Zustandsgrößen T, V, P und n. Im Sonderfall eines Gases nach dem kinetischen Gasmodell vereinfacht sich die Situation allerdings erheblich. Die einzige Energieform, die ideale Gasteilchen nach dem kinetischen Gasmodell beinhalten können, ist die kinetische Energie. Mit RT = 1/3 M c² (s. Kapitel 3.2.3) folgt mit Ekin = ½ mv² die Beziehung 27 RT = 2/3 ⟨Ekin⟩ oder ⟨Ekin⟩ = U = 3/2 R T Das bedeutet, dass die Innere Energie U(T) eines idealen Gases grundsätzlich allein von der Temperatur abhängt, nicht dagegen vom Druck oder vom Volumen. Bei einem realen Gas sind die Gegebenheiten komplizierter. Betrachtet man denjenigen Kompressionszustand eines realen Gases, bei dem die Teilchen sich jeweils im Minimum des Lennard-Jones-Potentials aufhalten (s. Kapitel 3.3.1), so führt sowohl eine Kompression als auch eine Expansion des Gases zu einem Anstieg der potentiellen Energie, auch wenn dabei die kinetische Energie der Gasteilchen (sprich die Temperatur) konstant bleibt. Das Phänomen ist vergleichbar mit der potentiellen Energie einer Feder, die sich sowohl auf Zug als auch auf Druck spannen lässt. Die molare Innere Energie U(T,P) bzw. U(T,V) ist damit bei realen Gasen auch eine Funktion des Drucks bzw. des Volumens. Sie ergibt sich aus der Summe der kinetischen Energie ⟨Ekin⟩ = 3/2RT und der potentiellen Energie nach dem Lennard-Jones-Potential. Anmerkung: Die Freisetzung von Energie durch Verminderung der Masse (oder umgekehrt) gemäß der relativistischen Äquivalenz von Masse und Energie nach E = mc² ist hierbei nicht berücksichtigt. Die hier dargestellte Form des Ersten Hauptsatzes lässt sich jedoch problemlos in diesem Sinne erweitern. Die Gleichung ∆U = q + w enthält dann einen dritten Term ∆mc², welcher dem Masseverlust bzw. Massezuwachs entspricht. Dieser Masseverlust tritt theoretisch auch bei chemischen Reaktionen auf, konnte allerdings bisher experimentell noch nicht nachgewiesen werden. Der Grund dafür liegt vor allem in den geringen Masseänderungen, die selbst bei energiereichen Reaktionen und leichten Reaktionspartnern kaum detektierbar sind. Eine Rekombination von 400 t atomaren Wasserstoffs ergibt beispielsweise nur einen Masseverlust von etwa einem Gramm. 4.3 Das Perpetuum mobile: ein Gedankenexperiment Der Zustand eines idealen oder realen Gases lässt sich (bei konstanter Teilchenzahl n) mit den Systemzustandsgrößen P, V und T eindeutig beschreiben. Da die drei Parameter gemäß einer realen Gasgleichung miteinander verknüpft sind, reichen schon zwei der Größen, beispielsweise T und V, zur Beschreibung des Systems vollkommen aus. Die Innere Energie ist eine Zustandsfunktion. Jeder denkbare Zustand des betrachteten Gases ist also eindeutig mit einem Wert für die Innere Energie verbunden, so dass man die Innere Energie als Funktion von T und V ausdrücken könnte (s. Abb.). Variiert man nun die Parameter T und V ausgehend von einem Startpunkt Tstart|Vstart derart, dass man nach einiger Zeit wieder an den Startpunkt zurückkehrt (Tend =Tstart, Vend = Vstart), so erhält man in der T-V-Ebene eine geschlossene Linie, die am Anfangs- und Endpunkt genau den gleichen Zustand des Systems markiert. Da Anfangs- und Endzustand identisch sind, müssen hier auch die zugeordneten Inneren Energien identisch sein; somit müssen, über den Zyklus hinweg, auch die Funktionswerte für die Innere Energie U(T,V) eine geschlossene Linie beschreiben: 28 U(V,T) T V Mathematisch formuliert lässt sich zusammenfassen: ∆T = 0 und ∆V = 0 ⇒ ∆U = 0 Die Summe aller Veränderungen im Verlauf der zyklischen Zustandsänderung ist also gleich Null. Mathematisch lässt sich diese Tatsache in folgender Schreibweise darstellen: ∫ dU = 0 Das bedeutet, dass im Verlauf eines vollständigen Zyklus des Systems weder Energie verbraucht noch Energie freigesetzt werden kann. Dem ersten Hauptsatz gemäß gilt: ∆U woraus folgt: = 0 = w + q w = -q Jeder Betrag an geleisteter Arbeit wird damit durch ein entsprechendes Quantum an zugeführter Wärme kompensiert. Es ist demnach also unmöglich, unter Nutzung eines idealen oder realen Gases eine Anordnung zu konstruieren, die in einem zyklischen Prozess Arbeit leistet, ohne Wärme aufzunehmen. Sinngemäß gilt diese Aussage ebenso für Flüssigkeiten, Festkörper und beliebige andere Phasenzustände. Sie entspricht der klassischen Formulierung des ersten Hauptsatzes der Thermodynamik: Es gibt kein Perpetuum mobile erster Art. Arbeitsleistung ohne Wärmeaufnahme ist nur in einem nichtzyklischen Prozess unter Verringerung der Inneren Energie möglich. 4.4 Messung von Änderungen der Inneren Energie Prinzipiell ist dem ersten Hauptsatz gemäß die Änderung der Inneren Energie einfach über die Summe der ausgetauschten Wärme und der ausgetauschten Arbeit zu bestimmen. Es sind jedoch vereinfachende experimentelle Ansätze denkbar: 29 a) Vollständige thermische Isolierung des Systems. Es gilt damit: q = 0, die Versuchsdurchführung ist adiabatisch. Daraus resultiert nach dem ersten Hauptsatz: ∆U = w + q = w. Die Änderung der Inneren Energie kann somit in Form der ausgetauschten Arbeit gemessen werden. b) Beibehaltung eines konstanten Volumens. Die Versuchsdurchführung ist isochor, es gilt: dV = 0. Somit verschwindet die Volumenarbeit gemäß dw = - P dV = 0. Daraus resultiert nach dem ersten Hauptsatz: ∆U = w + q = q. Die Änderung der Inneren Energie kann dann in Form der ausgetauschten Wärme bestimmt werden. Gewöhnlich wird die zweite, die isochore Variante genutzt, da Wärme in der Praxis einfacher messbar ist als Volumenarbeit. Hierzu dient das sogenannte Bombenkalorimeter, in dem eine abgewogene Probenmenge einer Reaktion unterzogen wird. Die frei werdende Wärmemenge führt zu einer entsprechenden Erwärmung des Druckbehälters und des umgebenden Wasserbads, die gemessen und ausgewertet werden kann (Abb.). + - Thermometer O2 Probe Bombe Zünddraht Wasserbad 4.5 Die Enthalpie Bei konstantem Volumen ist die Änderung der Inneren Energie gleich der ausgetauschten Wärme, es gilt ∆U = q ⇒ dV = 0 Dies ist eine für experimentelle Auswertungen sehr bequeme Beziehung, da sich nun jede Änderung der Inneren Energie in Wärme bzw. Temperaturänderung bemerkbar macht. Allerdings sind in der Praxis Prozesse mit konstantem Volumen eher selten. Chemische Umsetzungen werden zumeist in offenen Gefäßen oder in Gefäßen mit Druckausgleich durchgeführt. Deswegen ist eine Zustandsfunktion gesucht, die einen Bezug zur Inneren Energie hat, deren Änderung aber dem Wärmeaustausch bei konstantem Druck entspricht. Diese Funktion wurde in der Enthalpie H gefunden: H = U + PV 30 Diese auf den ersten Blick nicht unbedingt einleuchtende Definition führt zu der Beziehung ∆HP=const. = q Begründung: dH und, mit dP = 0: dH = = = = = = dU + dU + dw + - P dV dq + dq d (P V) P dV + V dP dq + P dV + V dP + dq + P dV + V dP V dP Die Enthalpie ist wie die Innere Energie eine Zustandsfunktion, da P und V (und somit auch PV) ebenfalls Zustandsfunktionen darstellen, und eine Summe von Zustandsfunktionen stets ebenfalls eine Zustandsfunktion ergibt. Der eigentliche Nutzen der Enthalpie besteht darin, dass Angaben über die Enthalpieänderung eines Systems direkte Aussagen über die bei konstantem Druck ausgetauschte Wärme zulassen: q = ∆U geschlossenes System Geschlossenes System dV=0 bei konstantem Volumen 4.6 q = ∆H offenes System Geschlossenes dP=0 System bei konstantem Druck Messung von Änderungen der Enthalpie Die Messung von ∆H erfolgt analog zu der von ∆U unter Bestimmung des Wärmeflusses, hier bei konstantem Druck, also unter isobaren Bedingungen. Dies könnte beispielsweise in einer Bombe mit Druckausgleich erfolgen. Bei Gasreaktionen (z.B. zwischen Luft und Erdgas) wird die Verwendung des so genannten Flammenkalorimeters bevorzugt: Zur Messung der Enthalpieänderung bei der Reaktion zwischen zwei Gasen werden die Gase in einer Kammer mit zwei Zuführungen zur Reaktion gebracht. Über einen Wärmetauscher gelangt die bei der Reaktion freigesetzte Wärme in das umgebende Kühlbad, dessen Temperaturänderung bestimmt wird. Diese Temperaturänderung ist eine Funktion der bei konstantem Druck freigewordenen Wärme, die wegen ∆HP=const = q genau der Enthalpieänderung entspricht. Ein in der physikalisch-chemischen Analytik sehr häufig praktiziertes Verfahren, bei dem unter anderem die Enthalpieänderung gemessen werden kann, ist die sogenannte Dynamische Differenzkalorimetrie (auch Differential Scanning 31 Calorimetry, DSC). Bei dieser Art der thermischen Analyse wird eine Probe zusammen mit einer Referenzzelle einem genau festgelegten Temperaturprogramm (z.B. ein Temperaturanstieg von 10 K/min) unterworfen und der zur Einhaltung der Temperaturänderung benötigte (oder freigesetzte) Wärmefluss gemessen. 4.7 Relation zwischen Innerer Energie und Enthalpie Aus der Definition der Enthalpie H = U + PV folgt zunächst, dass H immer größer ist als U (PV ist grundsätzlich positiv). Allerdings ist der Betrag, um den die Enthalpie die Innere Energie überschreitet, sehr stark von den Gegebenheiten abhängig. Bei der Betrachtung von Änderungen der Inneren Energie und der Enthalpie kann man im Wesentlichen zwei Fälle unterscheiden: a) Abläufe, bei denen keine Gase beteiligt sind. Hier ist, bei nicht allzu hohem Druck, ∆PV sehr klein. Daraus folgt, dass die Änderung der Enthalpie und die der Inneren Energie etwa den gleichen Betrag besitzen: ∆H ≅ ∆U. b) Abläufe, bei denen Gase beteiligt sind. Hier ist ∆PV im Allgemeinen nicht vernachlässigbar. Unter Ansatz des idealen Gasgesetzes als Näherung erhält man: ∆(PV) = ∆(nRT), woraus folgt: ∆H = ∆U + ∆(nRT). Unter der Voraussetzung, dass die Temperatur in etwa konstant ist, wird also die Veränderung der Teilchenzahl zum entscheidenden Kriterium. Die Enthalpie ist eine künstlich geschaffene Größe, die dem Chemiker, der im Allgemeinen unter konstantem Druck arbeitet, in der Praxis entgegenkommt. Sie erlaubt ihm, unter Verwendung tabellierter Enthalpiewerte direkt Aussagen über die zu erwartenden Wärmeflüsse zu machen. Bei veränderlichem Druck sind Enthalpiewerte lediglich abstrakte Größen ohne praktische Bedeutung. 4.8 Die totalen Differentiale der Inneren Energie und der Enthalpie Da es sich bei der Inneren Energie U und der Enthalpie H um Zustandsfunktionen handelt, kann man auch entsprechende totale Differentiale formulieren. Dabei muss zunächst festgelegt werden, welche Zustandsgrößen als variable Parameter angesetzt werden. Im Falle der Inneren Energie betrachtet man bevorzugt Prozesse bei konstantem Volumen. Es ist deswegen von Vorteil, als einen der beiden Parameter das Volumen zu wählen, da dann der entsprechende Term dV häufig entfällt. Als zweiten Systemparameter kann man beispielsweise die Temperatur verwenden (es wird sich später zeigen, dass ein anderer, noch einzuführender Zustandsparameter besser geeignet ist). Man erhält damit für das totale Differential zunächst folgenden Ausdruck: 32 dU(T,V) = ∂U dT ∂T V + ∂U dV ∂V T Im Falle der Enthalpie wird als zweiter Parameter sinnvollerweise der Druck verwendet, da hier häufig Prozesse bei konstantem Druck betrachtet werden, wobei der Term dP verschwindet. Man erhält entsprechend: dH(T,P) = ∂H dT ∂T P + ∂H dP ∂P T Alle genannten partiellen Differentiale sind für das Verständnis von prozessbedingten Veränderungen der Inneren Energie und der Enthalpie von großer Bedeutung. Auf sie wird deshalb in den folgenden Kapiteln eingegangen. 4.9 Die Temperaturabhängigkeit der Inneren Energie und der Enthalpie Bei konstantem Volumen, einer bei Verwendung der Inneren Energie häufig erfüllten Forderung, verschwindet in dem totalen Differential der Inneren Energie der zweite Summand und man erhält: dU(T,V) = ∂U dT ∂T V V = const. Das partielle Differential der Inneren Energie nach der Temperatur bei konstantem Volumen ist von großer Bedeutung und wird Wärmekapazität bei konstantem Volumen CV genannt. Es gilt also: CV = ∂U ∂T V Diese Wärmekapazität ist zunächst eine extensive, d.h. von der Stoffmenge abhängige Größe. Um intensive Größen zu erhalten, definiert man die molare Wärmekapazität CV,m = CV/n oder die spezifische Wärmekapazität CV,s= CV/m. Insgesamt sind also folgende Größen gebräuchlich: Wärmekapazität CV : ∂U CV = ∂T V molare Wärmekapazität CV,m : ∂U CV,m = ⋅1/n ∂T V Einheit: J/(K⋅mol) spezifische Wärmekapazität CV,s : ∂U CV,s = ⋅1/m ∂T V Einheit: J/(K⋅g) Einheit: J/K 33 In der grafischen Darstellung von U(T,V) entspricht CV der Steigung der Funktionswerte von U gegen eine Veränderung von T bei konstantem V (s. Abb. links): U U ( dU dT )V T V T T1 Im Allgemeinen ist CV temperaturabhängig. In den meisten Fällen steigt der Wert von CV mit der Temperatur an, die Temperaturabhängigkeit von U erhöht sich also (Abb. rechts). Bei konstantem Volumen (dV = 0) gilt gemäß dem ersten Hauptsatz: dU = dq + dw = dq - P dV = dq worauf sich die Definition der Wärmekapazität auch schreiben lässt: Cv = ∂U ∂T V ∂q ∂T V = In dieser Formulierung macht auch der Begriff Wärmekapazität einen Sinn. Anschaulich lässt sich also die Wärmekapazität folgendermaßen verstehen: Die Wärmekapazität bei konstantem Volumen CV ist diejenige Wärmemenge in J/K, die benötigt wird, um die Temperatur eines Systems bei konstantem Volumen um ein Grad zu erhöhen. Die Messung der Wärmekapazität geschieht üblicherweise unter Einbringung einer definierten Wärmemenge in Form von elektrischer Arbeit bei gleichzeitiger Temperaturmessung: dT V = const. + - el. Arbeit w = q e Um die Wärmekapazität bei konstantem Volumen zu messen, wird ein isochores System mit einer elektrischen Heizung versehen. Die elektrische Leistung der Heizung multipliziert mit der Heizdauer ergibt eine elektrische Gesamtarbeit in Joule (Watt.s). Diese Arbeit wird bei der gewählten Versuchsanordnung direkt in Wärme umgewandelt, die sich in Form einer Temperaturerhöhung des Systems äußert. Die Wärmekapazität wird dann als Steigung in einer Auftragung der Wärmemenge (= elektrische Arbeit) gegen die Temperatur bestimmt: wel = q Steigung = CV T 34 Völlig analog verläuft die Behandlung des entsprechenden partiellen Differentials der Enthalpie nach der Temperatur bei konstantem Druck: dH(T,P) = ∂H dT ∂T P = CP dT P = const. Die Größe CP heißt entsprechend Wärmekapazität bei konstantem Druck. Die Definition der entsprechenden intensiven Größen spezifische Wärmekapazität bei konstantem Druck CP,s und molare Wärmekapazität bei konstantem Druck CP,m erfolgt völlig analog zu CV,s und CV,m. Die Einheiten der Wärmekapazitäten bei konstantem Druck und bei konstantem Volumen sind identisch. Der Wert von CP entspricht der Steigung der Funktionsfläche von H gegen T bei konstantem P (Abb. links). Die Wärmekapazität bei konstantem Druck ist wie CV temperaturabhängig (Abb. rechts): H H ( dH dT )P T P T1 T Somit gilt folgende anschauliche Definition: Die Wärmekapazität bei konstantem Druck CP ist diejenige Wärmemenge in J/K, die benötigt wird, um die Temperatur eines Systems bei konstantem Druck um ein Grad zu erhöhen. Die Messung verläuft analog zur Messung von CV unter Einbringung von elektrisch erzeugter Wärme in ein Medium unter Bestimmung der Temperaturänderung, hier allerdings unter isobaren Bedingungen. Unter der letztgenannten Voraussetzung gilt (mit dHP=const. = dq): CP = ∂H ∂T P = ∂q ∂T P Die Differenz zwischen den Werten der Wärmekapazitäten bei konstantem Druck beziehungsweise bei konstantem Volumen resultiert aus den Definitionen von Innerer Energie und Enthalpie: CP − C V ∂H ∂U = − ∂T P ∂T V 35 ∂(U + PV ) ∂U = − P ∂T V ∂T ∂U ∂(PV ) ∂U = + − ∂T P ∂T P ∂T V Für ein ideales Gas gilt PV = nRT, somit lässt sich hier formulieren: CP − C V ∂U ∂(nRT) ∂U = + − ∂T P ∂T P ∂T V ∂U ∂U = + nR − ∂T P ∂T V Für ein ideales Gas ist die Innere Energie nur von der Temperatur abhängig, daher ist jedes partielle Differential der Inneren Energie nach der Temperatur identisch. Im Fall eines idealen Gases gilt damit: CP − C V = nR Diese Gesetzmäßigkeit wird von realen Gasen nicht exakt, aber doch in guter Näherung erfüllt. Allgemein gilt: Die Wärmekapazität bei konstantem Druck ist (vor allem bei Gasen) stets größer als die bei konstantem Volumen. Anschaulich erklärt sich dies aus der Tatsache, dass bei konstantem Druck während einer Erhöhung der Temperatur neben der reinen Erwärmung der Substanz auch ein gewisser Betrag an Volumenarbeit anfällt, für den ein zusätzliches Quantum an Wärme aufgewendet werden muss. 4.10 Die Volumenabhängigkeit der Inneren Energie Das Joule’sche Experiment Der Physiker James Prescott Joule beabsichtigte die Messung der Volumenabhängigkeit der Inneren Energie mittels folgender Versuchsanordnung: 1 2 Ein Gasbehälter (1, links) ist mit Gas unter Druck gefüllt, das bei Öffnen des Ventils in den rechten Behälter strömt (2). Der Vorgang ist exakt gleichbedeutend mit dem der irreversiblen Expansion gegen den Druck Null (s. Kapitel 4.1), das Gas leistet dabei keine Arbeit. Joule beobachtete keine Temperaturänderung eines umgebenden Wärmebades, ein Ergebnis, das für die Verhältnisse 36 bei idealen Gasen exakt zutrifft. Da keine Arbeit geleistet wird und offensichtlich auch kein Wärmeaustausch stattfindet, gilt dU = 0 und damit: ∂U =0 ∂V T (für ideale Gase) Bei realen Gasen und genaueren Messungen stellt man allerdings für gewöhnlich (bei nicht allzu hohen Drücken) fest, dass die Temperatur des Wasserbades nach dem Experiment geringer ist als vorher (T1 > T2). Das Gas hat also während des Expansionsvorgangs Wärme aufgenommen (somit ist die Expansion während des Joule’schen Experiments nicht adiabatisch, aber natürlich auch nicht exakt isotherm). Da nach außen keine Arbeit geleistet wird, gilt dU = dq. Die Wärmeaufnahme durch das System entspricht einem positiven Wert für q, damit gilt: ∂U >0 ∂V T (für reale Gase unter Normalbedingungen, außer Helium und Wasserstoff) Der Grund für die Erhöhung der Inneren Energie ist darin zu sehen, dass unter den gegebenen Voraussetzungen die anziehenden Wechselwirkungen zwischen den Teilchen überwiegen. Expandiert man nun das Gas, so wird eine zusätzliche potentielle Energie aufgebaut, die sich anschaulich mit dem Bild einer Vielzahl von auf Zug gespannten Federn interpretieren lässt. Bei einem realen Gas kann also Innere Energie nicht nur in Form von kinetischer Energie (als thermische Bewegung) sondern auch in Form von potentieller Energie bei gegenüber dem Idealwert vergrößertem Teilchenabstand vorliegen (s. LennardJones-Potential). Bei sehr hohen Drücken kann sich z.B. Stickstoff bei der Ausdehnung auch erwärmen, die Verhältnisse sind dann genau umgekehrt. In diesem Fall wird dann sozusagen eine auf Druck gespannte Feder entspannt, potentielle Energie wird freigesetzt, die Innere Energie vermindert sich. 4.11 Die Druckabhängigkeit der Enthalpie Der Joule-Thomson-Versuch Das Experiment wurde später von James Joule in Zusammenarbeit mit William Thomson (später Lord Kelvin) verfeinert. Sie verwendeten eine Versuchsanordnung, bei der ein Gas adiabatisch und irreversibel von einem Zustand P1, V1 und T1 in einen zweiten Zustand P2, V2 und T2 überführt wird (P1>P2): 37 m2 m1 P1 V1 T1 P2 V2 T2 m2 1 2 Der Vorgang ist exakt gleichbedeutend mit einer Expansion gegen einen konstanten Gegendruck (s. Kapitel 4.1). Da der Vorgang adiabatisch verläuft, wird keine Wärme ausgetauscht, es gilt zunächst q = q1 = q2 = 0. Weiterhin lässt sich die Arbeit w1, die bei dem Absenken des linken Kolbens am System geleistet wird, sowie die Arbeit w2, die beim Heben des rechten Kolbens vom System abgegeben wird, formulieren als: V2 0 w 2 = −P2 ∫ dV = −P2 V2 w 1 = −P1 ∫ dV = P1V1 0 V1 Mit ∆U = w+q muss dann gelten: ∆U = U 2 − U1 = q1 + q 2 + w 1 + w 2 = P1V1 − P2 V2 Durch Umstellen der Summanden erhält man: U1 + P1V1 = U 2 + P2 V2 H1 = H 2 oder Das überraschende Ergebnis dieser Betrachtung ist, dass der Prozess ohne Veränderung der Enthalpie abläuft, also ganz so, als wäre er unter adiabatischen Bedingungen isobar geführt worden (was nicht der Fall ist). Richtig ist dagegen, dass der Prozess entlang einer Linie gleicher Enthalpie (einer Isenthalpe, sozusagen) verläuft (Abb. links): “Isenthalpe” H T H = const. 1 1 2 2 H = const. T P P Die Schnittlinie mit der entsprechenden Ebene in T und P (Abb. rechts) gibt die Druckabhängigkeit der Temperatur bei konstanter Enthalpie wieder. Aus ihr lässt sich also entnehmen, wie stark die Temperatur im Verlauf eines JouleThomson-Versuchs mit fallendem Druck sinkt. Ihre Steigung, also die Größe (∂T/∂P)H, wird als Joule-Thomson-Koeffizient µ bezeichnet. Der Wert dieses Koeffizienten lässt sich aus dem Totalen Differential der Enthalpie berechnen. Da die Enthalpie bei dem betrachteten Vorgang konstant ist, muss gelten: 38 ∂H ∂H dH = dT + dP = 0 ∂T P ∂P T (∂H ∂P) T 1 ∂T =− =µ=− ∂P H CP (∂H ∂T) P oder ∂H ∂P T Der Wert für den gesuchten Koeffizienten (∂H/∂P)T ergibt sich daraus als ∂H ∂T = − C P = −C P µ ∂P T ∂P H Für ideale Gase ist die Innere Energie nur von der Temperatur abhängig, daher gilt in diesem Fall (∂H/∂P)T = (∂(PV)/∂P)T = (∂(nRT)/∂P)T = 0 und µ=0. Für reale Gase kann der Joule-Thomson-Koeffizient positive und negative Werte annehmen. Der Temperatureffekt bei dem Joule-Thomson-Versuch lässt sich nutzen, um ein Gas abzukühlen. Die Abkühlung kann so weit betrieben werden, dass das Gas sich verflüssigt (Linde-Verfahren). Voraussetzung für die Abkühlung während des Expansionsvorgangs ist ein positiver Wert für den JouleThomson-Koeffizienten µ. Der Wert für µ (der Steigung im T/P-Diagramm) hängt sowohl vom Druck als auch von der Temperatur ab (Abb.): µ<0 : Erwärmung bei Expansion T µ=0 : Keine Temperaturänderung 1 2 µ>0 : Abkühlung bei Expansion P Die meisten Gase besitzen bei Raumtemperatur einen positiven Wert für µ. Ausnahmen bilden Wasserstoff und Helium, die bei etwa 300 K auf Expansion mit Erwärmung reagieren. Die Temperatur, bei der sich das Vorzeichen des Joule-Thomson-Koeffizienten bei einem bestimmten Druck umkehrt, nennt man Inversionstemperatur. Bei µ>0 bedeutet eine Expansion eine Erhöhung der potentiellen Energie, da anziehende Wechselwirkungen bei der Expansion des Gases überwunden werden müssen und sich dadurch ein Spannungszustand aufbaut. Da die Innere Energie dabei konstant bleiben muss, wird der zusätzliche Energiebeitrag im adiabatischen Fall durch eine Abkühlung kompensiert. Umgekehrt gilt für µ<0, dass eine Expansion die potentielle Energie vermindert, da der durch abstoßende Wechselwirkungen bedingte Energiebeitrag abgebaut wird. Dabei erwärmt sich das Gas. Die mit einer adiabatischen Expansion ohne Austausch von Arbeit verbundenen Veränderungen der thermischen und potentiellen Energie sind in dem folgenden Diagramm zusammengefasst: 39 ideales Gas mit µ = 0: reales Gas mit µ > 0: Etherm Etherm E 0 reales Gas mit µ < 0: Etherm Epot Epot Epot V V V In allen Fällen ist die Summe beider Energien, die Innere Energie, konstant (dw = 0, dq = 0, dU = 0). 4.12 Reaktionsenergie und Reaktionsenthalpie 4.12.1 Vergleich zwischen Reaktionsenthalpie und Reaktionsenergie In der Chemie interessieren in erster Linie Energie- und Enthalpieänderungen, die mit chemischen Reaktionen verbunden sind. Je nach den gewählten Bedingungen ist, wie bereits begründet, die Reaktionsenergie (bei V = const.) oder die Reaktionsenthalpie (bei P = const.) die bevorzugte Größe. Da in der Praxis chemische Reaktionen zumeist bei konstantem Druck ausgeführt werden, besitzt die Reaktionsenthalpie die weitaus größte Bedeutung. Allgemein gelten folgende Definitionen: = ∆rU = Differenz der Gesamtenergien des Systems vor und nach dem Ablauf einer chemischen Reaktion Reaktionsenthalpie = ∆rH = Differenz der Gesamtenthalpien des Systems vor und nach dem Ablauf einer chemischen Reaktion Reaktionsenergie Bei konstantem Volumen entspricht der Betrag der Reaktionsenergie, bei konstantem Druck der Betrag der Reaktionsenthalpie der aufgenommenen Wärmemenge q. Wird bei einer chemischen Reaktion unter gegebenen Bedingungen Wärme frei, so spricht man von einer exothermen Reaktion, im umgekehrten Fall von einer endothermen Reaktion. Damit ist beispielsweise eine Reaktion mit negativem ∆rH bei konstantem Druck oder mit negativem ∆rU bei konstantem Volumen exotherm. Die Tatsache, dass ein Prozess bei konstantem Volumen exotherm ist, besagt jedoch nicht, dass die Reaktionsenthalpie negativ ist (und umgekehrt). Allgemein gilt: 40 ∆rH = ∆rU + ∆r(PV) Beispiel: Chlorknallgasreaktion in einem offenen bzw. geschlossenen System = H2 = Cl2 q = ∆H offenes System (P = const.) System mit Druckausgleich q = ∆U geschlossenes (V = const.) System ohne System Druckausgleich Findet eine chemische Reaktion nur zwischen Festkörpern und/oder Flüssigkeiten statt, so sind Reaktionsenergie und Reaktionsenthalpie einander sehr ähnlich. Sobald Gase an der Reaktion beteiligt sind, können sich beide Werte drastisch voneinander unterscheiden. 4.12.2 Standard-Enthalpieänderungen Bei chemischen Umsetzungen und den meisten anderen in der physikalischen Chemie betrachteten Vorgängen stehen nicht absolute Enthalpien, sondern lediglich Enthalpieänderungen im Vordergrund. Es ist deswegen sinnvoll, zur Referenzierung sogenannte Standardbedingungen festzulegen: Eine chemische Substanz liegt in ihrem Standardzustand vor, wenn: 1) 2) P = 1,000 bar Die Substanz in dem zu diesen Bedingungen stabilsten Phasenzustand bzw. in der stabilsten Modifikation vorliegt. Weiterhin wird definiert, dass alle Elemente im Standardzustand die Enthalpie Null besitzen. Die Temperatur wird nicht zu den Standardbedingungen gerechnet und stattdessen separat angegeben. Enthalpien zu den Standardbedingungen werden üblicherweise mit dem Kürzel H bezeichnet, die entsprechende Enthalpieänderung unter Standardbedingungen mit ∆H (unter Angabe der Temperatur im Index). Abhängig von dem betrachteten Vorgang definiert man eine Reihe von Standardenthalpien: S.-Phasenübergangsenthalpie ∆trsH : S.-Schmelzenthalpie ∆fusH S.-Verdampfungsenthalpie ∆vapH 41 S.-Reaktionsenthalpie ∆rH : S.-Sublimationsenthalpie ∆subH S.-Bildungsenthalpie ∆fH S.-Aktivierungsenthalpie ∆╪H S.-Verbrennungsenthalpie ∆cH Alle diese Enthalpiegrößen sind tabellarisch als Standard-Enthalpien (z.B. Standard-Reaktionsenthalpie bei 298 K: ∆rH 298) erfasst, wobei der StandardPhasenzustand und der Standarddruck vorausgesetzt, die Temperatur dagegen angegeben wird (zumeist 298 K). Wegen der extrem großen Zahl an denkbaren chemischen Reaktionen ist eine komplette Tabellierung aller Reaktionsenthalpien nicht möglich. Man behilft sich mit der Erfassung von Standard-Bildungsenthalpien ∆fH , die angeben, welche Reaktionsenthalpie mit der Bildungsreaktion von einem Mol der entsprechenden Verbindung aus Elementen bei Standardbedingungen und der angegebenen Temperatur verbunden ist: Die Standard-Bildungsenthalpie ∆fH einer Verbindung ist gleich der Reaktionsenthalpie der Bildungsreaktion der Verbindung unter der Voraussetzung, dass 1) 2) P = 1,000 bar die Bildung aus den Elementen verläuft, die jeweils in den unter den gegebenen Umständen stabilsten Zuständen vorliegen. Die jeweils gültige Temperatur wird für gewöhnlich angegeben. Diese StandardBildungsenthalpien werden dann genutzt, um im Einzelfall die Reaktionsenthalpien zu berechnen. Dazu bedient man sich des Hess’schen Satzes und eines Born-Haber’schen Kreisprozesses (s. folgender Abschnitt). 4.12.3 Berechnung von Reaktionsenthalpien Kann eine Reaktion gedanklich in Teilreaktionen zerlegt werden, so gilt unter Berücksichtigung des ersten Hauptsatzes und der Tatsache, dass H eine Zustandsfunktion darstellt, folgender Satz: Hess’scher Satz: Die Reaktionsenthalpie einer chemischen Reaktion entspricht der Summe der Reaktionsenthalpien jener Teilreaktionen, in welche die ursprüngliche Reaktion zerlegt werden kann. Die gedachten Teilreaktionen müssen dabei nicht unbedingt einem realistischen Reaktionsverlauf entsprechen. Der Hess’sche Satz lässt sich nun nutzen, indem man eine beliebige Reaktion in Teilschritte zerlegt, die alle der Bildung eines der Reaktionsteilnehmer aus den Elementen entsprechen. Durch Aufaddition dieser Teilschritte und der dazugehörigen Standard-Bildungsenthalpien (jeweils mit entsprechenden 42 positiven oder negativen Vorfaktoren) erhält man die Gesamtreaktion und deren Standard-Reaktionsenthalpie. Als einfaches Beispiel sei ein solcher sogenannter Born-Haber’scher Kreisprozess an der Reaktion A → B gezeigt: H H 0 Standardenthalpien der Elemente 0 Standardenthalpien der Elemente A A Born-Haber’scher Kreisprozess ∆ rH298 (A B)=? B B Hess’scher Satz: ∆rH298 (A B) = - ∆fH298 (A) +∆fH298 (B) Betrachtet man eine allgemeine Reaktion mit beliebigen Reaktionspartnern Ni, |ν1|N1 + |ν2|N2 + ... → ... |νi-1|Ni-1 + |νi|Ni so lässt sich zur Berechnung der Reaktionsenthalpie aus den Bildungsenthalpien demnach folgende allgemeingültige Beziehung heranziehen: ∆ rH = ∑ υ i ∆ f Hi Dabei sind die so genannten stöchiometrischen Koeffizienten der Reaktionspartner νi für alle Edukte negativ und für alle Produkte positiv. Dieses Vorgehen ermöglicht die Berechnung einer Standard-Reaktionsenthalpie bei derjenigen Temperatur, bei der die Referenzwerte angegeben wurden (gewöhnlich bei 298 K). Hat man es mit anderen Temperaturen zu tun, so ist die Temperaturabhängigkeit der Reaktionsenthalpie zu berücksichtigen. Man könnte, um die Abweichung von der Reaktionsenthalpie bei Standardtemperatur zu ermitteln, gedanklich folgende Reaktionsführung verfolgen (1-23-4 statt 1-4): T 1 4 ∆ rH T = ? ∆ HT ∆ H298 298 298 K T (Produkte) (Edukte) 2 3 ∆ rH298 Standardtemperatur 43 Um den ersten Schritt (1-2) vorzunehmen, müssen alle Edukte zunächst auf Standardtemperatur gebracht werden. Die damit verbundene Enthalpieänderung ergibt sich für jedes Edukt aus: T 298K dHEdukt = C P,Edukt dT oder ∆HEdukt = ∫ ∫ C P,Edukt dT = − C P,Edukt dT 298K T Der Reaktionsablauf (2-3) findet dann bei Standardbedingungen statt und ist mit der bekannten Enthalpieänderung ∆rH verbunden. Anschließend werden die Produkte der Reaktion von der Standardtemperatur zurück zur Ausgangstemperatur gebracht, (3-4) wobei für jedes Produkt folgende Enthalpieänderung eintritt: T dHPr odukt = C P,Pr odukt dT ∫ ∆HPr odukt = oder C P,Pr odukt dT 298K Die gesamte, temperaturbedingte Änderung der Reaktionsenthalpie erhält man damit als: ∑C d∆ r H = P,Pr odukt Pr odukte oder ∆∆ r H = T2 ∫ dT − ∑C Edukte P,Edukt dT = ∑υ C i P,i dT = ∆ r C P dT i ∆ r C P dT T1 oder, für annähernd temperaturunabhängige Wärmekapazitäten: ∆∆ r H = ∆ r C P ∆T Dieser Zusammenhang wird als das Kirchhoffsche Gesetz bezeichnet. Die Temperaturabhängigkeit der Reaktionsenthalpie hängt also stark von der Änderung der Wärmekapazität des Reaktionsgemisches im Verlauf der Reaktion ab. Besitzen Edukte und Produkte jeweils in der Summe die gleiche Wärmekapazität, so ist die Reaktionsenthalpie temperaturunabhängig. Ist die Wärmekapazität der Produkte insgesamt größer als die der Edukte (∆rCP > 0), so steigt die Reaktionsenthalpie mit steigender Temperatur an. Ist die Wärmekapazität der Produkte insgesamt kleiner als die der Edukte (∆rCP < 0), so verringert sich die Reaktionsenthalpie bei steigender Temperatur. Zusammen mit dem Hess'schen Wärmesatz ermöglicht das Kirchhoffsche Gesetz die Berechnung von beliebigen Reaktionsenthalpien unter Kenntnis der Standard-Bildungsenthalpien. So sind für Rektionen, die unter konstantem Druck ausgeführt werden, Abschätzungen der Wärmetönung, d.h. der dabei freiwerdenden oder verbrauchten Wärmemengen möglich. Die allgemeine Feststellung, dass dabei exotherme Reaktionen häufiger beobachtet werden als endotherme, verleitete viele Chemiker zu der Annahme, dass in einer negativen Reaktionsenthalpie die Triebkraft einer Reaktion zu suchen sei. Diese Annahme ist, wie an vielen Beispielen gezeigt werden kann, falsch. Die 44 Tatsache, dass eine gegebene chemische Reaktion freiwillig abläuft, hängt dies in keiner Weise davon ab, ob dabei Energie freigesetzt wird oder nicht. 5 Der Zweite Hauptsatz der Thermodynamik 5.1 Die Entropie Die klassische Formulierung des Zweiten Hauptsatzes sagt aus, dass ein Perpetuum mobile zweiter Art nicht existiert. Er lautet: Es ist unmöglich, eine Maschine zu konstruieren, die kontinuierlich Arbeit leistet und dabei nur von der Wärme eines Wärmebades gespeist wird. Der zweite Hauptsatz verbietet somit die Umwandlung von Wärme in Arbeit, solange nur ein Wärmereservoir zur Verfügung steht, ein Vorgang, der nach dem Ersten Hauptsatz durchaus zulässig wäre. Die Umkehrung, also eine vollständige Umwandlung von Arbeit in Wärme, wird dagegen sehr häufig beobachtet. Der Zweite Hauptsatz ordnet gewissen Prozessen somit eine bestimmte Richtung zu, insofern besitzt er die unter den bekannten Naturgesetzen einzigartige Eigenschaft einer Richtungsweisung. Für solche, anscheinend nur in eine Richtung ablaufende Prozesse gibt es in der alltäglichen Praxis viele Beispiele: 1) Ein Wärmefluss strebt immer einem Temperaturausgleich zu. Nie kommt es spontan zu einer Auftrennung in zwei Teilbereiche unterschiedlicher Temperatur. 2) Ein Gas strömt stets aus einem Behälter in ein Vakuum hinein. Nie kommt es dagegen zur spontanen Bildung eines Vakuums durch Ausströmen eines Gases in einen zweiten angeschlossenen Behälter. 3) Eine Kaffeetasse zerbricht beim Aufschlag in viele unzusammenhängende Scherben. Die spontane Bildung einer Kaffeetasse aus den Scherben wird nicht beobachtet. 4) Eine Kugel fällt auf eine Tischplatte, springt dabei mehrere Male auf und bleibt schließlich ruhig liegen. In diesem Fall hat sich die kinetische Energie der Kugel in Wärmeenergie verwandelt. Nach dem Ersten Hauptsatz der Thermodynamik könnte sich die dabei freigewordene Wärme spontan wieder in kinetische Energie verwandeln, wobei die ruhende Kugel plötzlich aufspringen würde. Ein solcher Vorgang wird nicht beobachtet. 45 5) Eine Uhr mit mechanischem Uhrwerk wird aufgezogen und läuft ab. Dabei wird die potentielle Energie der gespannten Uhrfeder allmählich in Wärme umgesetzt. Der umgekehrte Vorgang, also ein spontanes Rückwärtslaufen des Uhrwerks unter Wärmeentzug aus der Umgebung und Anspannen der Feder findet nicht statt. potentielle Energie Wärme Alle genannten Beispiele besitzen zwei Gemeinsamkeiten: a) Stets findet in einem abgeschlossenen System ein spontaner, zeitlich gerichteter und nicht umkehrbarer Prozess statt. b) Stets verläuft dieser spontane Prozess unter Zunahme von Unordnung. Letzteres, die Zunahme der Unordnung ist beispielsweise im Fall 3 (die zerbrochene Tasse) offensichtlich, in Fall 4 dagegen nicht unbedingt. Wo findet bei der Umwandlung von kinetischer Energie in Wärme die Bildung von Unordnung statt? Bei näherer Betrachtung ist die Entstehung von Chaos offensichtlich: jeder Aufprall der Kugel auf die Oberfläche führt zur Impulsübertragung auf die im Gitter der Oberfläche befindlichen Teilchen. Diese können keine geregelte, gemeinsame Bewegung ausführen, da sie, im Gegensatz zur Kugel, örtlich fixiert sind. Der Impuls führt daher zu gewöhnlich ungeregelten Schwingungsvorgängen im Gitter, also zu nichts anderem als einer Temperaturerhöhung des Oberflächenmaterials (s. Abbildung). 46 kinetische Energie kinetische Energie + Wärme Offensichtlich ist die kinetische und die potentielle Energie als „geordnete“, die thermische Energie dagegen als „ungeordnete“ Energieform zu betrachten. Damit lässt sich ein aus der ursprünglichen Formulierung des Zweiten Hauptsatzes verallgemeinertes Naturgesetz formulieren: Mit fortschreitender Zeit tendieren alle abgeschlossenen Systeme zu einer Erhöhung der Unordnung: →→→ →→→ →→→ →→→ Zeit Unordnung Die Zeit ist dabei als physikalische Größe leicht messbar. Problematisch erscheint die Quantifizierung des Begriffs „Unordnung“. Hierfür ist die Definition eines neuen Parameters notwendig. Dieser Parameter wird Entropie genannt und mit dem Kürzel S bezeichnet. Es gilt: Die Entropie ist ein Maß für die Unordnung in einem System vollständig geordnetes System: ungeordnetes System: S S = > 0 0 [Einheit ?] [Einheit ?] Über die thermodynamische Definition und die Einheit der Entropie wird zunächst eine Plausibilitätsbetrachtung angestellt: Aus mehreren der genannten Beispiele geht hervor, dass Wärme die Unordnung eines Systems und damit dessen Entropie erhöht. Also könnte man postulieren: dS ∝ dq Liegt in dem betrachteten System bereits ein hohes Maß an Unordnung (d.h., eine hohe Temperatur T) vor, so ist die Zunahme der Entropie pro Wärmeeinheit relativ gering. Umgekehrt wirkt sich die gleiche Wärmeeinheit auf ein extrem geordnetes System auf dessen Entropie sehr viel dramatischer aus. Man könnte diesen Zusammenhang formulieren als: dS ∝ 1/T 47 Die aus den genannten Zusammenhängen hervorgehende Definition der Entropie lautet: dS = dq / T Einheit: J/K oder: Die Entropieeinheit 1 J/K entspricht genau dem Maß an Unordnung, das die einfließende Wärmemenge 1 Joule anrichtet, wenn das System die Temperatur 1 Kelvin besitzt. Wärme q T System System Voraussetzung für diese Definition ist natürlich, dass nicht andere Weise Unordnung entsteht. Dies lässt sich durch die Reversibilität des betrachteten Vorgangs festschreiben, was Index „rev“ der Wärme markiert. Die exakte Definition der daher: dS = gleichzeitig auf Forderung nach man durch den Entropie lautet dqrev / T Die Entropie beschreibt die in einem System bestehende Unordnung, also einen Zustand des Systems. Es liegt deshalb nahe, in der Entropie eine Zustandsfunktion zu sehen (der Beweis folgt in Kapitel 5.3). Diese Tatsache wird genutzt, wenn eine nicht oder nicht nur durch Wärmeeinwirkung hervorgerufene (also irreversibel entstandene) Entropieänderung berechnet werden soll. In diesem Fall ist die Gleichung dS = dq/T nicht direkt anwendbar, da dq nicht mit dqrev identisch ist. Da aber S eine Zustandsfunktion darstellt, ist es völlig gleichgültig, auf welchem Wege man vom Anfangszustand zum Endzustand des Prozesses gelangt. Die Entropie ist hier also zu berechnen, indem man irgendeinen reversiblen Prozess sucht, der den Anfangs- in den Endzustand überführt: Anfangszustand irreversibler Prozess, ∆S = ? Endzustand reversibler Prozess, ∆S = dqrev/T ∫ Unter Nutzung des Entropiebegriffs lässt sich die Aussage des Zweiten Hauptsatzes mit Berücksichtigung der aus den genannten Beispielen gewonnenen Erkenntnisse folgendermaßen zusammenfassen (Clausius 1854): 48 In einem abgeschlossenen System nimmt die Entropie im Verlauf eines nicht umkehrbaren (irreversiblen) Prozesses grundsätzlich zu. Ergänzend lässt sich feststellen: In einem abgeschlossenen System bleibt die (Gesamt-) Entropie im Verlauf eines umkehrbaren (reversiblen) Prozesses konstant. Prozesse, die zur Verminderung der (Gesamt-) Entropie eines abgeschlossenen Systems führen, sind nicht möglich! Diese Aussagen bilden zusammen die thermodynamische Formulierung des Zweiten Hauptsatzes: Irreversibler Prozess Reversibler Prozess ⇔ ⇔ ∆Sges > 0 ∆Sges = 0 (Clausius’sche Ungleichung) Wie die Doppelpfeile in der schematischen Schreibweise andeuten, sind auch die Umkehrungen der genannten Formulierungen richtig. Eine wichtige Konsequenz dieser Aussagen ist die Tatsache, dass die Triebkraft sämtlicher (chemischer, physikalischer, oder sonstwie gearteter) Prozesse, die freiwillig (=irreversibel) ablaufen, einzig und allein in der Zunahme der Entropie des Universums besteht. So ist beispielsweise die Frage, ob eine chemische Reaktion abläuft oder nicht, nur durch die Frage nach einer eventuellen Entropiezunahme zu beantworten. Diese Frage muss bezogen auf ein abgeschlossenes Gesamtsystem gestellt werden, nicht nur für ein eventuell die Reaktion umgebendes, geschlossenes Teilsystem: Gesamtsystem beobachtetes System (geschlossen) Umgebung (abgeschlossen) freiwillig ablaufende Reaktion wenn ∆Sges > 0 statisches Gleichgewicht wenn ∆Sges = 0 Reaktion läuft nicht ab wenn ∆Sges < 0 ∆Sges = ∆Sbeobachtetes System + ∆SUmgebung Die Entropie des beobachteten Teilsystems kann bei einem spontan ablaufenden Prozess durchaus abnehmen, solange diese Abnahme durch eine entsprechend große Zunahme der Entropie der Umgebung überkompensiert wird. In diesem Fall muss eine Art „Entropieopfer“ gebracht werden. 5.2 Entropieänderungen bei ausgewählten Prozessen Im Folgenden soll zunächst einmal der Zusammenhang zwischen der Clausiusschen Formulierung des zweiten Hauptsatzes und dem Verbot des Perpetuum 49 mobiles zweiter Art herausgearbeitet werden. Anschließend werden Entropieänderungen berechnet, die mit verschiedenen reversiblen und irreversiblen Prozessen in abgeschlossenen Systemen verbunden sind. 5.2.1 Betrachtung eines Perpetuum mobiles zweiter Art In einem Perpetuum mobile zweiter Art soll Wärme aus einem einzelnen Wärmebad (T1) direkt in Arbeit umgewandelt werden: abgeschlossenes System Perpetuum Mobile zweiter Art: q T1 w Vorausgesetzt, dass es sich um einen kontinuierlich wiederholbaren Prozess handelt, also dass die Arbeit w nicht dazu verwendet wird, die Entropie eines anderen Systems zu erhöhen, gilt für die Änderung der Gesamtentropie: dS ges − = dq < T1 0 !! Der Prozess würde so zu einer Verminderung der Gesamtentropie führen und verstößt damit gegen die thermodynamische Formulierung des zweiten Hauptsatzes. Wie lässt sich dieser Mangel beheben? Die Entropieabnahme durch die Wärmeabfuhr aus dem betrachteten System müsste durch eine entsprechende Wärmezufuhr in ein anzuschließendes Teilsystem kompensiert, oder besser, überkompensiert werden: abgeschlossenes System w T1 q1 T2 q2 Die gesamte Entropieänderung errechnet sich nach: dS ges = dS1 + dS 2 = − dq1 T1 + dq 2 T2 Damit der Prozess die Forderung des Zweiten Hauptsatzes erfüllt, muss der zweite Summand betraglich größer sein als der erste. Nach dem ersten Hauptsatz der Thermodynamik (dU = 0) muss weiterhin gelten: 50 dw dq1 − dq 2 = Das bedeutet, dass der Temperaturunterschied zwischen den Teilsystemen letztlich darüber entscheidet, welches Quantum Arbeit abgeschöpft werden kann. 5.2.2 Temperaturausgleich zwischen zwei Teilsystemen Zwischen zwei geschlossenen Teilsystemen mit unterschiedlichen Temperaturen T1 und T2 (wobei T1 > T2), die zusammen ein abgeschlossenes Gesamtsystem bilden, finde ein Temperaturausgleich statt. Der Vorgang ist offensichtlich irreversibel und verläuft spontan: abgeschlossenes System q T1 > T2 T1 T2 Teilsystem 1 dS1 = - |dq| / T1 dS2 = |dq| / T2 Teilsystem 2 Wie groß ist die Entropieänderung des Gesamtsystems? Es gilt: dS ges = dS 1 + dS 2 = − = | dq| ( T1 − T2 ) dq T1 + dq T2 T1T2 Mit T1 > T2 ist der genannte Betrag immer positiv. Damit ist die Forderung des zweiten Hauptsatzes nach einer positiven Änderung der Gesamtentropie erfüllt. 5.2.3 Reversible, isotherme Expansion eines idealen Gases Bei der reversiblen isothermen Expansion eines idealen Gases ist zu beachten, dass das zunächst betrachtete System (das ideale Gas) kein abgeschlossenes System darstellt. Um die Einhaltung des zweiten Hauptsatzes zu überprüfen, muss ein übergeordnetes, abgeschlossenes System eingeführt werden. Aus der Tatsache, dass es sich insgesamt um einen reversiblen Vorgang handelt, folgt automatisch, dass sich die Entropie Sges dieses übergeordneten Gesamtsystems nicht ändert. Es gilt: dS ges = 0 Die Entropieänderung des Gases berechnet sich nach: 51 dS Gas dqrev = T 2 ∆S Gas = ∫ = 1 dq T dq T Da die Temperatur konstant ist, muss auch die Innere Energie des idealen Gases unveränderlich bleiben. Damit folgt aus dU = dw + dq = 0, dass |dq| = |dw|: 2 ∆S Gas ∫ = 1 dw T V2 V2 = PdV ∫V T 1 = nR ln = nR dV V V1 ∫ V2 V1 Das Gas erfährt somit bei einer Expansion (V2 > V1) eine positive Entropieänderung. Die Änderung der Entropie der Umgebung ist dementsprechend betraglich gleich, aber negativ. Insgesamt ergibt sich damit folgendes Bild: Gesamtsystem (abgeschlossen) w<0 - isotherm - reversibel ∆SGas = nR ln(V2/V1) ∆SUmg = -nR ln(V2/V1) ∆Sges = 0 ideales Gas (geschlossenes System) q>0 Umgebung 5.2.4 Irreversible, isotherme Expansion eines idealen Gases Betrachtet wird ein irreversibler Vorgang, bei dem das Gas im Verlauf der Ausdehnung keinerlei Arbeit leistet. Auch hier gilt die thermodynamische Definition der Entropieänderung, also der Ausdruck dS = dqrev/T. Allerdings ist in diesem Fall dqrev nicht bekannt. Da die Entropie eine Zustandsfunktion ist, genügt es, die Entropieänderung eines beliebigen reversiblen Prozesses zu berechnen, der mit dem irreversiblen Prozess den Anfangs- und den Endpunkt gemeinsam hat. Ein solcher Prozess ist die in 5.2.3 besprochene reversible Expansion. Für die Entropieänderung des Gases gilt also: dS Gas = dqrev T ∆SGas = nR ln V2 V1 52 Da in diesem Fall der irreversiblen Expansion vom Gas keine Arbeit geleistet wird, darf gemäß dem Ersten Hauptsatz auch keine Wärme in das Gas einfließen, wenn dessen Temperatur konstant sein soll. Folglich fließt von der Umgebung auch keinerlei Wärme ab. Sie bleibt, auch hinsichtlich ihrer Entropie, unverändert: dSUmg = 0. Die Entropie Sges des Gesamtsystems ändert sich damit genau wie die des Gases, d.h. sie steigt um den Betrag: dS ges = dqrev T V2 V1 ∆Sges = nR ln ∆SGas = nR ln(V2/V1) ∆SUmg = 0 ∆Sges = nR ln(V2/V1) Man erhält damit folgende Situation: Gesamtsystem (abgeschlossen) w=0 - isotherm - irreversibel ideales Gas (geschlossenes System) q=0 Umgebung 5.2.5 Reversible, adiabatische Expansion eines idealen Gases Im Verlauf einer adiabatischen Expansion fließt keine Wärme in das betrachtete Gas ein (q = 0). Da der Prozess weiterhin reversibel sein soll, ist damit q = qrev. Es gilt also: dS Gas = dqrev T = dq T = 0 Da die Umgebung keine Wärme abführt, ist außerdem: dS Umg = 0 und damit: Die Situation stellt sich also wie folgt dar: dS ges = 0. 53 Reversible adiabatische Expansion Gesamtsystem (abgeschlossen) w<0 - adiabatisch - reversibel ∆SGas = 0 ∆SUmg = 0 ∆Sges = 0 ideales Gas (geschlossenes System) q=0 Umgebung Obwohl das Gas seinen Zustand wesentlich verändert, tritt keinerlei Entropieänderung ein. Dieses zunächst überraschende Ergebnis erklärt sich dadurch, dass im adiabatisch reversiblen Fall die Entropieerhöhung durch die Volumenexpansion und die Entropieverminderung durch das Sinken der Temperatur einander genau kompensieren. 5.2.6 Irreversible, adiabatische Expansion eines idealen Gases Das Gas soll im Verlauf dieses Prozesses keine Arbeit leisten (dw = 0). Wie im vorangegangenen Beispiel fließt keine Wärme in das Gas ein, da der Prozess adiabatisch geführt wird (dq = 0). Der Erste Hauptsatz schreibt unter diesen Bedingungen vor, dass die Innere Energie konstant bleibt (dU = 0). Bei einem idealen Gas bedeutet dies, dass sich die Temperatur nicht ändert. Die irreversible, adiabatische Expansion ist damit bezüglich aller Zustandsparameter identisch mit der irreversiblen, isothermen Expansion (s. 5.2.4). Das Ergebnis lässt sich also völlig analog darstellen: Irreversible adiabatische Expansion Gesamtsystem (abgeschlossen) w=0 - adiabatisch - irreversibel ∆SGas = nR ln(V2/V1) ∆SUmg = 0 ∆Sges = nR ln(V2/V1) ideales Gas (geschlossenes System) q=0 Umgebung 5.3 Der Carnotsche Kreisprozess Der zweite Hauptsatz und damit der Begriff der Entropie sind direkt mit den technischen Möglichkeiten zur Umwandlung von Wärme in Arbeit verknüpft. Die Theorie der Wärmekraftmaschine, die sich letztlich auf eine Betrachtung der Entropieänderungen stützt, wurde von Sadi Carnot erarbeitet („Reflexions sur la Puissance Motrice du Feu“, 1824), lange bevor der Begriff der Entropie eingeführt war (Clausius, 1850). Carnot beschäftigte sich mit der grundsätzlichen 54 Darstellung der Funktion einer verallgemeinerten Wärmekraftmaschine in einem PV-Diagramm. Die von ihm erdachte Carnot-Maschine arbeitet mit einem idealen Gas, zyklisch und völlig reversibel. Der Vorgang beinhaltet vier Einzelschritte: isotherm 1) Isotherme, reversible Expansion im Wärmebad 1 adiabatisch 4 2) Adiabatische, reversible Expansion 4) Adiabatische, reversible Kompression 2 isotherm 3) Isotherme, reversible Kompression im Kältebad 3 In einem PV-Diagramm erhält man unter den gegebenen Voraussetzungen folgendes Bild: Adiabaten PVγ = const. P VA 1 4 Isothermen PV = const. VB 2 VD 3 Tw VC Tk V Carnot war bei der Betrachtung der Wärmekraftmaschine daran gelegen, einen reversiblen, zyklischen Prozess in einem PV-Diagramm möglichst einfach zu beschreiben. Seine Wahl fiel dabei auf eine einfache Kombination von je zwei isothermen und adiabatischen Prozessen, weil dabei sowohl die Änderungen der wichtigsten Zustandsparameter P, V, T, U und S als auch die ausgetauschten Beträge an Wärme und Arbeit rechnerisch am leichtesten zugänglich sind. Die Zustandsgrößen des in der Maschine eingeschlossenen idealen Gases sollen nun im Einzelnen aufgeführt werden: Schritt 1: Reversible, isotherme Expansion Druck: Volumen: Temperatur: Innere Energie: PA VA Tw Uw → PB → VB (konstant) (konstant, da ideales Gas bei T = const.) Arbeit: dw = − PdV 55 VB w1 = ∫ − PdV = − nRTw ln = nRTw ln VA Wärme: q1 = ∆U1 − w1 Entropie: ∆S1 = nR ln Schritt 2: VB VA VB VA VB VA <0 >0 >0 Reversible, adiabatische Expansion Druck: Volumen: Temperatur: Innere Energie: PB VB Tw Uw → → → → PC VC Tk Uk Arbeit: dw = − PdV w2 = (mit ∆U = w, da q = 0) = dU Tk ∆U 2 = ∫C V dT <0 Tw Wärme: Entropie: Schritt 3: q2 ∆S 2 = = 0 0 Reversible, isotherme Kompression Druck: Volumen: Temperatur: Innere Energie: PC VC Tk Uk → PD → VD (konstant) (konstant, da ideales Gas und T = const.) Arbeit: dw = − PdV VD w3 = ∫ − PdV q3 = ∆U3 − w 3 Entropie: ∆S 3 = nR ln Schritt 4: VD VC >0 VD VC <0 − nRTk ln = nRTk ln VC Wärme: VD VC = <0 Reversible, adiabatische Kompression Druck: Volumen: Temperatur: Innere Energie: PD VD Tk Uk → → → → PA VA Tw Uw Arbeit: dw = − PdV (mit ∆U = w, da q = 0) = dU 56 w4 = Tw ∆U 4 = ∫C V >0 dT Tk q4 ∆S 4 Wärme: Entropie: = = 0 0 Carnot war nun insbesondere daran interessiert, etwas über den Wirkungsgrad dieser hypothetischen Maschine aussagen zu können. Der Wirkungsgrad (häufig mit η oder ε bezeichnet) ist dabei definiert als Quotient aus der insgesamt geleisteten Arbeit (wges) und der vom System aufgenommenen Primärenergie (Wärme q1): η − w ges geleistete Arbeit = aufgenommene Wärme = q1 Dieser Term lässt sich unter Zusammenfassung der für die Schritte 1 bis 4 gefundenen Daten errechnen. Man erhält: ηCarnot = − w ges q1 − (w 1 + w 2 + w 3 + w 4 ) q1 = T nRTw ln = T k w VB V − ∫ C V dT + nRTk ln D − ∫ C V dT VA Tw VC Tk nRTw ln Tw ln = VB VA VB V + Tk ln D VA VC V Tw ln B VA Um diesen Ausdruck zu vereinfachen, wird die Adiabatengleichung zu Hilfe genommen: PV γ = const. nRT γ V = const. V nRTV γ −1 = const. γ −1 TV = const.’ Für die im Carnot-Prozess relevanten Adiabaten gilt also: Tw VB Tk VD γ −1 γ −1 = = Tk VC γ −1 Tw VA γ −1 ⇒ Tw Tk ⇒ Tw Tk = VC VB = VD VA γ −1 γ −1 57 Aus den beiden rechten Gleichungen folgt direkt, dass VC VB = VD VA VC VD bzw. = VB VA Mit dieser Beziehung lässt sich für den Wirkungsgrad schreiben: Tw ln ηCarnot = VB V − Tk ln B VA VA V Tw ln B VA oder: ηCarnot = Tw − Tk Tw Dieses Ergebnis, das zunächst nur für die Carnot-Maschine Gültigkeit besitzt, besagt Folgendes: Der Wirkungsgrad ist um so größer, je kleiner Tk bzw. je größer Tw ist. Für die Grenzfälle Tk → 0 und Tw → ∞ tendiert der Wirkungsgrad jeweils gegen den Wert eins. Darüber kann der Wirkungsgrad niemals steigen (ansonsten wäre auch der erste Hauptsatz der Thermodynamik verletzt). Neben der Berechnung des Wirkungsgrades ist auch die Betrachtung der Entropieänderungen interessant. Summiert man die Einzelbeiträge aller Einzelschritte auf, so erhält man: ∆S ges = ∆S 1 + ∆S 2 + ∆S 3 + ∆S 4 = nR ln VB V + 0 + nR ln D + 0 VA VC unter Berücksichtigung der Adiabatengleichung (s.o.) gilt: ∆S ges = nR ln VB V − nR ln B = VA VA 0 Das heißt, nach Ablauf eines vollständigen Zyklus der Carnot-Maschine haben sich sämtliche Änderungen der Entropie kompensiert. Diese treten im Einzelnen nur bei den isothermen Teilschritten auf. Die isotherme Expansion (Schritt 1) führt zu einer Entropieerhöhung, die isotherme Kompression (Schritt 3) zu einer betraglich exakt gleichen Entropieverminderung. Grafisch lässt sich der Ablauf durch ein neben das PV-Diagramm gestelltes TS-Diagramm verdeutlichen: 58 Adiabaten PVγ = const. P T A VA 4 1 1 B 4 2 VB 2 VD T1 D C 3 T2 3 VC V S Dabei entspricht die eingeschlossene Fläche im PV-Diagramm der insgesamt geleisteten Arbeit w1+w2+w3+w4. Die im TS-Diagramm eingeschlossene Fläche entspricht der dabei insgesamt ausgetauschten Wärme q1 + q3 und ist damit ebenso gleich der insgesamt geleisteten Arbeit w1+w2+w3+w4. Die Tatsache, dass die Entropieänderung bei einem kompletten Durchlauf des Prozesses verschwindet, ist eine notwendige Bedingung dafür, dass die Entropie eine Zustandsfunktion darstellt. Ließe sich zeigen, dass diese Bedingung für beliebige Kreisprozesse erfüllt ist, so wäre diese Tatsache, die bisher nur stillschweigend vorausgesetzt wurde, bewiesen (s. folgendes Kapitel). 5.4 Wirkungsgrade von allgemeinen Wärmekraftmaschinen Die große Leistung Carnots bestand darin, erkannt zu haben, dass seine ursprünglich auf die Carnot-Maschine bezogenen Ergebnisse auf alle Arten von Wärmekraftmaschinen zu verallgemeinern sind. Der grundsätzliche Denkansatz ist dabei folgender: Jede Zustandsänderung, die ein beliebiges Medium einer reversibel arbeitenden Wärmekraftmaschine durchläuft, muss irgendeinem zyklischen Weg in einem PV-Diagramm folgen (s. z.B. Abb. links). P P PV-Diagramm zu beliebiger Wärmekraftmaschine V V Andererseits lässt sich jeder beliebige zyklische Weg im PV-Diagramm durch einen Satz von gekoppelten Carnot-Prozessen so „ausfüllen“, dass sich diese im Inneren des geschlossenen Gebildes paarweise berühren und außen den Umriss des zyklischen Weges exakt nachzeichnen (Abb. rechts). Da an den Berührungsstellen zwischen einzelnen Carnot-Prozessen deren Ablauf entgegengesetzt, die betraglichen Änderungen aller Zustandsgrößen jedoch iden- 59 tisch sind, heben sich dort sämtliche Beiträge von w, q und S gegenseitig auf. Addiert man also alle Werte für ∆w, ∆q und ∆S der einzelnen Carnot-Schritte, so erhält man genau die Werte, die dem Ablauf des äußeren Umrisses entsprechen. Alle Befunde, die für den Carnot-Prozess bezüglich Wirkungsgrad und Entropieänderung gefunden wurden, gelten auch für eine Summe von Carnot-Prozessen und somit für jeden beliebigen Prozess im PV-Diagramm. Damit wird der maximale Wirkungsgrad ηmax für alle Wärmekraftmaschinen durch die Carnot'sche Formulierung festgelegt: ηmax = Tw − Tk Tw Ein größerer Wirkungsgrad als ηmax kann definitiv mit keiner Konstruktion erreicht werden. Um diesen Maximalwert zu erreichen, muss die betrachtete Wärmekraftmaschine, wie die Carnot-Maschine auch, reversibel betrieben werden. Dann, und nur dann, kann ηmax ausgeschöpft werden. Das bedeutet aber auch, dass in diesem Fall die Maschine nicht spontan arbeitet, da die Entropie konstant wäre. In der Praxis muss daher ein gewisser Wärmebetrag (qspontan) „geopfert“ werden, um Entropie zu erzeugen und damit einen spontanen Ablauf zu gewährleisten. Der Wirkungsgrad wird dadurch natürlich verkleinert. Die Situation stellt sich damit (am Beispiel der Carnot-Maschine) schematisch folgendermaßen dar: Warm q1 = ∆S.Tw CarnotMaschine reversibler Fall: T A 1 B 4 D 2 3 ∆S q3 = ∆S.Tk +qspontan w = q1 - q3 Kalt C S Die bereits bei der Carnot-Maschine angestellten Grenzwertbetrachtungen gelten für jede beliebige Wärmekraftmaschine. Ihr Wirkungsgrad tendiert gegen eins, wenn die obere Temperatur beliebig groß und/oder die untere Temperatur beliebig klein gewählt wird. Die Verallgemeinerung der Carnot-Maschine auf alle reversiblen Prozesse ermöglicht auch den abschließenden Nachweis, dass es sich bei der Entropie um eine Zustandsfunktion handelt. Wie bei der Carnot-Maschine selbst ist auch bei einem beliebigen zyklischen Durchlaufen eines Weges im PV-Diagramm die Entropie von Anfangs- und Endpunkt identisch. Damit ist jedem Punkt im PVDiagramm genau ein Entropiewert zuzuordnen, das Kennzeichen einer Zustandsfunktion. 60 Die Formulierung des maximalen Wirkungsgrads einer Wärmekraftmaschine bietet die Möglichkeit, sie zur Festlegung der absoluten Temperaturskala zu nutzen (s. Kapitel 2.2). Legt man einen Fixpunkt fest (z.B. Tw oder Tk als Tripelpunkt des Wassers), so kann man über die Beziehung ηmax = (Tw-Tk)/Tw eine zweite Temperatur (Tk oder Tw) über den maximalen Wirkungsgrad bestimmen. Diese Methode wurde von William Thomson (Lord Kelvin) genutzt um die thermodynamische Temperaturskala einzuführen. Der Zyklus einer Wärmekraftmaschine (beispielsweise der einer CarnotMaschine) kann auch in umgekehrter Richtung durchlaufen werden. Dabei wird, unter Aufwenden von Arbeit, Wärme von einem kalten Wärmebad in ein warmes transportiert: γ P Umkehrung des Carnot-Prozesses = Wärmepumpe Warm VA 2 umgekehrte CarnotMaschine 1 VB 4 VD 3 q3 T1 T2 VC q1 w = q1 - q3 Kalt V Eine nach diesem Prinzip arbeitende Maschine wird als Wärmepumpe bezeichnet. Sie kann genutzt werden, um tiefe Temperaturen Tk zu erreichen (Kühlmaschine), oder um die Temperatur Tw in einem warmen Raum auf Kosten der Umgebung weiter zu erhöhen (Wärmepumpe zur Heizung von Wohnräumen). Auch solche Aggregate besitzen Kenngrößen, die ihre Effektivitäten benennen. In diesen Fällen spricht man, da sie größere Werte als eins annehmen können, nicht von Wirkungsgraden (für die streng gilt: η≤1), sondern von Leistungskoeffizienten c (oder von dem "cop", dem „Coefficient of Performance“). Für Kühlmaschinen kann der Maximalwert von c analog zum Wirkungsgrad η abgeleitet werden: cmax = | q3 | = |w| Tk Tw − Tk Der maximal erreichbare Leistungskoeffizient einer Kühlmaschine ist also umso höher, je geringer der Temperaturunterschied Tw-Tk ist. Entsprechend erhält man für eine als Heizung verwendete Wärmepumpe den Ausdruck: | q1 | Tw cmax = = Tw − Tk |w| 61 Auch eine als Heizung verwendete Wärmepumpe arbeitet umso effektiver, je geringer der Temperaturunterschied zwischen den beiden Medien wird. Sie erreicht dabei sehr leicht maximale Leistungskoeffizienten, die ein Vielfaches von eins betragen. Bei Temperaturbedingungen von 20°C (innen) und 5°C (außen) erhält man beispielsweise einen theoretischen Maximalwert von cmax = 19,5. Die technisch tatsächlich erreichbaren Leistungskoeffizienten liegen weit darunter (etwa bei 2 bis 3), trotzdem wird dem Innenraum dabei eine Wärmemenge zugeführt, die immerhin dem Doppelten der verbrauchten (beispielsweise elektrischen) Energie entspricht. q1 w q3 Damit können solche Wärmepumpen als Raumheizung durchaus mit anderen Systemen konkurrieren. 6 Der Dritte Hauptsatz der Thermodynamik 6.1 Formulierung des Dritten Hauptsatzes Versucht man, mit physikalischen Methoden immer tiefere Temperaturen zu erreichen, so stellt man fest, dass der dazu nötige Aufwand umso größer wird, je näher man dem absoluten Nullpunkt der Temperatur kommt (T = -273,15°C bzw. 0 K). Da es trotz vieler Versuche nie gelang, diesen absoluten Nullpunkt zu erreichen oder gar zu unterschreiten, wurde postuliert, dass er einen natürlichen, unerreichbaren Grenzwert darstellt. Diese Erfahrung wurde zur Grundlage der empirischen Formulierung des Dritten Hauptsatzes der Thermodynamik (W. Nernst, 1912): Es ist unmöglich, den absoluten Nullpunkt der Temperatur zu erreichen. Bei der thermodynamischen Betrachtung des Problems wurde schnell klar, dass wieder die Entropie die entscheidende Rolle spielt, wenn es um eine präzise Begründung für dieses Phänomen geht. Dazu muss zunächst einmal festgestellt werden, welche Entropieänderungen im Verlauf einer systematischen Abkühlung stattfinden. Entzieht man einer Substanz reversibel Wärme, so lässt sich die damit verbundene Entropieänderung formulieren als: dS = dq T Wird dieser Vorgang bei konstantem Druck ausgeführt, so gilt mit dq = dH: 62 dS dH T = Finden dabei keine Phasenumwandlungen statt, so ist die Änderung der Enthalpie allein temperaturbedingt. Dann gilt dH = CP(T) dT und somit: dS CP (T ) dT = T Damit erhält man für die gesamte Entropieänderung von T1 nach T2, wenn in diesem Temperaturbereich keine Phasenumwandlungen stattfinden: T2 ∆S CP ( T ) dT T T1 ∫ = Am Ort von Phasenumwandlungen sieht die Situation anders aus. Die hier fließende Wärme wird durch die Phasenumwandlungsenthalpie ∆trH bestimmt, für die entsprechende Entropieänderung ergibt sich der Betrag: ∆ tr S ∆ trH = Ttr wobei Ttr für die Phasenumwandlungstemperatur steht. Der Entropiebetrag ∆trS wird sinnigerweise als Phasenumwandlungsentropie bezeichnet. Die abkühlungsbedingte Entropieänderung einer Substanz lässt sich also allgemein formulieren als: ∆S = ∆ tr1H C (T) ∫T PT dT + Ttr1 1 Ttr 1 Ttr 2 + C (T) ∫T PT dT tr 1 + ∆ tr 2H Ttr 2 Ttr 3 + CP ( T ) dT + ... T Ttr 2 ∫ Auf diese Weise lässt sich die Entropieänderung einer Substanz in einem Temperaturfenster zwischen der Umgebungstemperatur und dem absoluten Nullpunkt berechnen. Die mit dem Aufheizen und Abkühlen verbundenen Entropieänderungen sind betraglich gleich, besitzen aber unterschiedliche Vorzeichen. Bei zwei Phasenumwandlungen (Schmelzen und Verdampfen) erhält man beispielsweise für das Aufheizen zwischen 0 K und T: ∆S TSchmelz ∫ = 0 ∆ SchmelzH CP (T ) dT + + T TSchmelz ∆ Verdampf H CP (T ) dT + TVerdampf T TSchmelz TVerdampf ∫ T + CP ( T ) dT T TVerdampf ∫ was sich in einer Auftragung von S gegen T folgendermaßen darstellen lässt: 63 S ∆Verdampf.S ∆SchmelzS S0 TSchmelz TVerdampf. T Wäre die Größe S0 bekannt, so könnte man neben der relativen Änderung der Entropie auch deren Absolutbetrag bestimmen. Da bisher der Absolutwert einer Entropie nicht einmal definiert ist, müssen hierzu noch grundsätzliche Überlegungen angestellt werden. Diese beruhen insbesondere auf experimentellen Beobachtungen von T.W. Richards (um 1900). Richards beobachtete Systeme, bei denen jeweils zwei Zustände bei konstanter Temperatur miteinander im Gleichgewicht stehen, also isotherme B wie z.B.: Zustandsänderungen A Sn (grau) Sn (weiß) S (rhombisch) S (monoklin) 2 Ag + PbSO4 Ag2SO4 + Pb Er bestimmte dabei die Entropieänderungen im Verlauf solcher Umwandlungen und stellte fest, dass diese umso kleiner wurden, je niedriger er die Temperatur wählte. Eine Auftragung einer solchen Reaktionsentropie ∆rS gegen die Temperatur ergab etwa folgenden Verlauf: S A A B ∆S B S0 T Nernst zog aus dieser Beobachtung den Schluss, dass die Reaktionsentropien isothermer Prozesse bei Annäherung an den absoluten Nullpunkt gegen Null 64 tendieren. Er formulierte damit das Nernst’sche Wärmetheorem, das als eine thermodynamische Formulierung des Dritten Hauptsatzes gilt: Die mit einem isothermen Prozess verbundene Entropieänderung strebt gegen Null, wenn man sich dem absoluten Nullpunkt nähert. limT→ →0 ∆rS = 0 Später fand man heraus, dass dieses Theorem nur für Prozesse gilt, die sich in einem stabilen Gleichgewichtszustand befinden. Max Planck ging einige Jahre später (1912) noch einen Schritt weiter. Er postulierte, dass Entropieunterschiede nicht zwischen Reaktionspartnern eines chemischen Gleichgewichts, sondern zwischen allen Stoffen verschwinden. Das bedeutet, dass am absoluten Nullpunkt jedes System die gleiche absolute Entropie besitzt. Er definierte darüber hinaus diesen Punkt als den Nullpunkt der Entropieskala. So lautet die Planck’sche Formulierung des Dritten Hauptsatzes: Die Entropie einer chemisch homogenen Substanz strebt gegen Null, wenn man sich dem absoluten Nullpunkt nähert. limT→ →0 S = 0 Diese Formulierung kann als abschließende thermodynamische Formulierung des Dritten Hauptsatzes gelten, wenn man zwei Bedingungen hinzufügt: a) Die Substanz muss bei Annäherung an den absoluten Nullpunkt ideal kristallisieren, d.h. ihre Atome oder Moleküle müssen ideal geordnet vorliegen. b) Der Nullpunktsentropie der ideal kristallisierten Elemente muss der Wert Null zugeordnet werden. Das bedeutet, dass die trotz idealer Kristallstruktur noch vorhandene Unordnung der Elementarteilchen (z.B. Elektronenspins) vernachlässigt wird. Aus der Planck’schen Formulierung des Dritten Hauptsatzes (limT→0 S = 0) folgt automatisch die Nernst’sche Formulierung (limT→0 ∆rS = 0), nicht aber umgekehrt. Aus der Planck’schen Aussage resultiert weiterhin, dass absolute Entropien berechnet werden können. Besitzt eine Substanz bei niedrigen Temperaturen einen eindeutigen, maximal geordneten kristallinen Zustand, so kann man ihr am absoluten Nullpunkt die Entropie Null zuordnen, wie im folgenden Beispiel einer Substanz AB: A B S0 = 0 J/K 65 Verbleibt dagegen auch bei niedrigsten Temperaturen ein gewisses Maß an Unordnung, z.B. eine energetisch gleichwertige Umkehrung der Orientierung eines Moleküls im Kristallgitter, so erhält man eine positive Nullpunktsentropie: A B S0 > 0 J/K Ein Beispiel für den letztgenannten Fall bildet Kohlenmonoxid, in dessen Kristallgitter der Einbau des Moleküls CO offensichtlich gleichermaßen in beiden Orientierungen möglich ist. Die daraus resultierende Nullpunktsentropie des Kohlenmonoxids beträgt 4,2 J/(K Mol). Ähnliche Erscheinungen sind auch bei NO, N2O und bei Wasser zu beobachten, in diesen Fällen liegen die Nullpunktsentropien zwischen 3 und 6 J/(K Mol). Häufig wird, insbesondere beim raschen Abkühlen einer Substanz, die Unordnung des flüssigen Phasenzustandes „eingefroren“. Eine ungeregelte Struktur bleibt aus kinetischen Gründen erhalten, obwohl die geordnete Struktur energetisch günstiger wäre. Typische Vertreter solcher Zustände sind Gläser, beispielsweise glasartig erstarrtes Glycerin (Abb.). S TSchmelz TVerdampf. eit sigk Flüs -1 23,4 JK mol -1 Glas ∆ SchmelzS Kristall T Angesichts der nun sehr unterschiedlichen Formulierungen des Dritten Hauptsatzes („Der absolute Nullpunkt ist unerreichbar bzw. limT→0 S = 0) stellt sich die Frage, welcher Zusammenhang zwischen dem Erreichen des absoluten Nullpunkts und dem Wert der Nullpunktsentropie besteht. Im Folgenden wird deshalb näher auf die Methodik zum Erreichen tiefer Temperaturen eingegangen. 66 6.2 Erzeugung tiefer Temperaturen Die niedrigste Temperatur in der Natur beträgt 2,7 Kelvin, jede tiefere Temperatur muss künstlich erzeugt werden. Die technische Erzeugung tiefer Temperaturen bedeutet aber stets den Entzug von Wärme und damit die Vernichtung von Entropie. Diese Entropie muss zur Erfüllung des zweiten Hauptsatzes an anderer Stelle erzeugt werden. Dies geschieht beispielsweise bei der Verdunstung einer Flüssigkeit durch die Erzeugung von ungeordneten Zuständen auf Teilchenebene. Unter Nutzung der Verdunstungskälte kann mit Helium bei vermindertem Druck eine Temperatur von 0,7 K erreicht werden, darunter ist kein geeigneter Phasenübergang mehr zugänglich. Zur Erreichung noch tieferer Temperaturen wird auf Prozesse zurückgegriffen, die sich auf der Ebene der Elementarteilchen abspielen. Am bekanntesten ist dabei die adiabatische Entmagnetisierung. Voraussetzung für die Anwendung dieser Methode ist eine Substanz, die Elementarteilchen mit eigenen magnetischen Momenten besitzt, welche sich in einem äußeren Magnetfeld ausrichten (z.B. ungepaarte Elektronen bei paramagnetischen Substanzen wie Gadoliniumsulfat oder Manganchlorid). Die Versuchsanordnung besteht aus einer in einen Heliumstrom getauchten Probe, die sich zwischen den Polen eines Elektromagneten befindet: N + Probe (paramagnetischer Kristall) Helium S Der eigentliche Kühlvorgang besteht aus folgenden Einzelschritten: 1) Ausgangszustand: Die Probe befindet sich in einem Strom aus flüssigem Helium bei etwa 1 K, der Magnet ist abgeschaltet, die magnetischen Momente der Elementarteilchen sind regellos verteilt. 2) Isotherme Magnetisierung: Das Magnetfeld wird eingeschaltet und die magnetischen Momente ordnen sich parallel zum Magnetfeld. Da das System weiterhin in Kontakt mit dem flüssigen Helium steht, hat sich die Temperatur im Vergleich zu 1) nicht geändert, wohl aber die Entropie der Probe (sie sinkt bei gleichzeitiger Wärmeabfuhr an das Helium). 3) Adiabatische Entmagnetisierung: Das Helium wird entfernt, so dass der folgende Schritt adiabatisch verläuft. Das Magnetfeld wird abgeschaltet, wobei die magnetischen Momente spontan (unter Entropiegewinn) ihre unregelmäßige Anordnung zurückgewinnen. Dabei wird ihnen von der umgebenden Substanz (unter Entropieverlust) Wärme zugeführt. Insgesamt ist die Entropieänderung der Probe nahe Null, die Temperatur der Probe hat sich verringert (Abb. links). 67 S Probe He Probe S1 eti sie rt 1 gne er t t is i T2 gn un 3 ma q ma S0 eti s un Magnetfeld He q S2 ier ma t gn S Probe 2 T T1 S0 gn ma e t is ie r t T Der Vorgang kann mehrfach wiederholt werden, so dass die Temperatur schrittweise weiter absinkt (Abb. rechts). Dabei wird die Probenmenge stufenweise vermindert und flüssiges Helium eingesetzt, das bei dem jeweils vorhergehenden Schritt abgekühlt wurde. Der Temperaturrekord, der über ein derartiges Verfahren erreicht wurde, liegt bei 2.10-8 Kelvin. Trotz allem ist auf diese oder ähnliche Weise der absolute Nullpunkt nicht erreichbar. Gäbe es einen der Abbildung entsprechenden Zickzackkurs im STDiagramm, der wirklich zum absoluten Nullpunkt führen würde, so müsste sich dieser umgekehrt von T=0 aus geometrisch konstruieren lassen. Da gemäß der Formulierungen des Dritten Hauptsatzes nach Nernst und Planck alle Entropieunterschiede am absoluten Nullpunkt verschwinden, sähe die Startbedingung etwa folgendermaßen aus: un ma gn eti s ier t isotherm S Probe Startpunkt der Konstruktion eines umgekehrten Abkühlweges im ST-Diagramm gne ma tisi ert T adiabatisch T=0 Da wegen limT→0 ∆rS = 0 keine Möglichkeit für einen isothermen Prozess und wegen der mit steigender Temperatur ansteigenden Entropie keine Möglichkeit für einen adiabatischen Prozess besteht, kann dieser erste Schritt (bzw. der letzte Schritt bei der Abkühlung zum absoluten Nullpunkt) nicht formuliert werden. 68 7 Freie Energie und Freie Enthalpie 7.1 Definition und Bedeutung Die drei Hauptsätze der Thermodynamik stellen strenggenommen rein physikalische Gesetzmäßigkeiten dar. Es stellt sich die Frage, welche Relevanz für den Bereich der Chemie besteht. Im Fall des Dritten Hauptsatzes ist der Bezug zur Chemie, abgesehen von echter Tieftemperaturchemie, am geringsten. Der Erste Hauptsatz ist als Erhaltungssatz der Energie von tiefgreifender Bedeutung, in seiner Aussage für chemische Abläufe allerdings recht trivial und in seiner Anwendung denkbar unkompliziert. Anders dagegen der Zweite Hauptsatz: Er besitzt entscheidende Bedeutung für die Beschreibung von chemischen Reaktionen und Gleichgewichtszuständen. Auf seiner grundlegenden Aussage, der Tendenz zur Erhöhung der Gesamtentropie in abgeschlossenen Systemen, beruht letztlich die Triebkraft für alle chemischen Prozesse. Im allgemeinsten Fall kann man als übergeordnetes abgeschlossenes System das gesamte Universum betrachten und formulieren: dS Universum ≥ 0 Auf dieser Basis lassen sich alle spontan ablaufenden chemischen Reaktionen (dS > 0) und Gleichgewichtszustände (dS = 0) interpretieren. Der Ablauf einer chemischen Reaktion stellt sich thermodynamisch betrachtet folgendermaßen dar: q dSReakt. dSUmg. dSUniv. > 0 Die Änderung der Entropie des Universums, die mit dem Verlauf der Reaktion verbunden ist, besteht aus der Entropieänderung der Reaktionsmischung (im Folgenden einfach als dS bezeichnet) und der durch Wärmeaustausch bedingten Entropieänderung der Umgebung dSUmg.. Sie lässt sich damit schreiben als: dS Universum = dSReakt. ≥ + dS Umg. 0 oder, mit dS = dq/T für den reversiblen, wärmeflussbedingten Anteil der Entropieänderung und unter Berücksichtigung der Vorzeichenumkehr von q: dS Universum = dS - dq / T ≥ 0 Wieder bezieht sich dS > 0 auf eine spontan ablaufende Reaktion und dS=0 auf den Gleichgewichtszustand. Die Triebkraft einer chemischen Reaktion misst sich daran, welche Menge an „nutzbarer Energie“ der Reaktion zu ent- 69 nehmen wäre, ohne sie zu stoppen. Die nutzbare Energie einer chemischen Reaktion entspricht aber genau der Wärme qmax, die man theoretisch der Reaktion entziehen und in Arbeit umwandeln könnte, ohne durch die damit verbundene Entropieverminderung den zweiten Hauptsatz zu verletzen (s. Abbildung). q dS qmax wmax + dSUmg - dqmax/T =0 Die maximale theoretisch erzeugbare Arbeit, die unter den gegebenen Voraussetzungen zu erhalten wäre, steht in direkter Beziehung zu dem bereits formulierten Betrag von dSUniversum: dwmax/T oder = - dqmax / T = dwmax = - (dS + dSUmg) = - dS Universum - dqmax = - T dS Universum Die Größe (- TdSUniversum) stellt damit ein Maß für die Triebkraft einer Reaktion dar und ist in der Chemie von zentraler Bedeutung. Aus der Definition von dSUniversum folgt: - T dS Universum = - T dS + dq ≤ 0 Weiterhin gilt bei konstanter Temperatur und... ...bei konstantem Volumen: dq = -T dSUniversum = (und, mit SdT = 0) ...bei konstantem Druck: dU dq = dU - T dS -T dSUniversum = = dU - TdS - SdT (mit SdT = 0) = d (U-TS) = dF Freie Energie ≤ 0 F = U - TS dH dH - T dS = dH - TdS - SdT = d (H-TS) ≤ 0 = dG Freie Enthalpie G = H - TS (häufig auch mit dem Kürzel "A" statt "F" bezeichnet) Die Ausdrücke Freie Energie F und Freie Enthalpie G haben jeweils bei konstantem Volumen bzw. bei konstantem Druck die gleiche Bedeutung wie die Schlüsselgröße -TdSUniversum, sie charakterisieren damit unter den gewählten Bedingungen die Triebkraft einer chemischen Reaktion. Damit folgen aus der 70 thermodynamischen Formulierung des zweiten Hauptsatzes folgende Aussagen: 1) Eine Reaktion läuft dann und nur dann freiwillig ab, wenn die damit verbundene Änderung der Freien Energie (bei konstantem Volumen) bzw. der Freien Enthalpie (bei konstantem Druck) einen negativen Wert besitzt: dF < 0 bzw. ⇒ dG < 0 spontane Reaktion 2) Eine Reaktion läuft so lange ab, bis die Freie Energie (bei konstantem Volumen) oder die Freie Enthalpie (bei konstantem Druck) einen Minimalwert erreicht haben: dF = 0 bzw. ⇒ dG = 0 Gleichgewicht 3) Die maximale Arbeit, die aus einer chemischen Reaktion bezogen werden kann, entspricht bei konstantem Volumen der Änderung der Freien Energie. Die maximale Nicht-Volumenarbeit, die aus einer chemischen Reaktion gezogen werden kann, entspricht bei konstantem Druck der Änderung der Freien Enthalpie: wmax = ∆F bzw. wmax - wVol = ∆G Zusammenfassung: F spontane Reaktion spontane Reaktion G spontane Reaktion spontane Reaktion maximal erhältliche Nicht-Volumenarbeit maximal erhältliche Gesamtarbeit Gleichgewicht Gleichgewicht Wie Energien und Enthalpien sind auch Freie Energien und Freie Enthalpien umfassend tabelliert. Freie Enthalpien sind wegen der größeren Bedeutung isobarer Reaktionsbedingungen auch von größerem Interesse. Um Freie Reaktionsenthalpien ∆rG zu berechnen, wird (analog zu ∆rH) von Freien StandardBildungsenthalpien ausgegangen. Unter Nutzung der Beziehung G = H - TS können aus Reaktionsenthalpien und Freien Reaktionsenthalpien die Reaktionsentropien berechnet werden. Aufgrund der Definitionen von F und G ist ersichtlich, dass es sich jeweils um Zustandsfunktionen handelt, denn alle beteiligten Größen U, H, S und T sind ebenfalls Zustandsfunktionen. 71 Neben den Zustandsgrößen Innere Energie und Enthalpie sind somit zwei weitere Zustandsgrößen bekannt, die als „energetische“ Größe aufgefasst werden können. Alle vier Parameter stehen miteinander in Beziehung, zusammengefasst lauten ihre Definitionen: dU dH dF dG = = = = dU + d(PV) = dU - d(TS) = dH - d(TS) = dq dq dq dq + + + + dw dw + PdV + VdP dw - SdT - TdS dw + PdV + VdP - SdT - TdS Aus der Formulierung G = H - TS könnte man ableiten, dass die freie Enthalpie aus einem „Energieterm“ („H“) und einem „Entropieterm“ („-TS“) besteht. In Wirklichkeit repräsentiert auch H einen "Entropieterm", nämlich das Produkt aus Temperatur und der Entropiezunahme der Umgebung. Trotzdem wird das genannte Bild häufig herangezogen, um den Verlauf chemischer Reaktionen zu interpretieren. Man nennt Reaktionen, deren Triebkraft hauptsächlich auf dem Term „-TS“ beruht, „entropiegetrieben“. Der Einfluss des „Entropieterms“ wird durch die Temperatur bestimmt. Beispiele: a) Die Verdunstung einer Flüssigkeit: Der Übergang von der flüssigen Phase zur Gasphase vergrößert in starkem Maße das den Teilchen zur Verfügung stehende Volumen. Die dadurch bedingte Entropiezunahme treibt die Reaktion. b) Polykondensation von Polyestern oder Polyamiden: Die bei der Kondensationsreaktion entstehenden Wassermoleküle sorgen für einen großen positiven Entropiebeitrag. c) Bildung von Chelatkomplexen in wässriger Phase: Die bei dem Vorgang der Chelatbildung freigesetzten Wassermoleküle der Hydrathülle verursachen eine Entropiezunahme. Tatsächlich ist die Entropiezunahme die Triebkraft aller spontan ablaufenden Reaktionen, nur wird diese in vielen Fällen durch die Erwärmung der Umgebung verursacht. 7.2 Die Fundamentalgleichung Nach dem ersten Hauptsatz gilt folgender, bereits geläufiger Zusammenhang: dU = dq + dw Für reversible Prozesse gilt dw = -PdV und, gemäß dem zweiten Hauptsatz, dS = dq/T; also folgt: dUrev = TdS - PdV Diese Formulierung ist damit zunächst auf reversible Vorgänge beschränkt. Aber: gilt sie für einen reversiblen Weg von 1 nach 2 (s. Abb.), dann muss sie auch für jeden beliebigen irreversiblen Weg von 1 nach 2 gelten, denn U(S,V) ist als Zustandsfunktion unabhängig vom zurückgelegten Weg: 72 irreversibler Weg U S 1 re W ler sib r e v eg U 2 2 S 1 V V Es gilt also grundsätzlich für alle Systeme das Gesetz dU = TdS - PdV Diese Gleichung wird, ihrer großen Bedeutung wegen, Fundamentalgleichung genannt. Die Innere Energie U lässt sich statt in S und V auch als Funktion vieler anderer Zustandsgrößen darstellen. In keinem anderen Fall könnte man dann jedoch einen ähnlichen Zusammenhang formulieren wie dies in Form der Fundamentalgleichung mit S und V möglich ist. Man nennt deshalb S und V die natürlichen Variablen der Inneren Energie. Das dazugehörige totale Differential dU = ∂U ∂U dS + dV ∂S V ∂V S führt mit der Fundamentalgleichung dU = T dS - P dV über Koeffizientenvergleich zu: ∂U =T ∂S V und ∂U = −P ∂V S Auf diese Weise werden völlig neue thermodynamische Zusammenhänge gewonnen, die für sich gesehen wenig anschaulich sind, aber trotzdem eine wichtige Rolle spielen können. So stellt beispielsweise die linke Gleichung eine weitere thermodynamische Definition der Temperatur dar. Es liegt nun nahe, für die verwandten Zustandsgrößen H, F und G nach ähnlichen Zusammenhängen zu suchen. Die Ansätze dazu entsprechen der beschriebenen Herleitung für die Innere Energie. Fundamentalgleichung für die Enthalpie: Es gilt nach der Definition der Enthalpie und unter Nutzung der Fundamentalgleichung der Inneren Energie dH = dU + PdV + VdP dH = TdS - PdV + PdV + VdP dH = TdS + VdP 73 Fundamentalgleichung für die Freie Energie: Man erhält mit der Definition der Freien Energie und unter Nutzung der Fundamentalgleichung der Inneren Energie dF = dU - TdS - SdT dF = TdS - PdV - TdS - SdT dF = - SdT - PdV Und schließlich die Fundamentalgleichung für die Freie Enthalpie: Über die Definition der Freien Enthalpie und unter Nutzung der Fundamentalgleichung der Enthalpie erhält man dG = dH - TdS - SdT dG = TdS + VdP - TdS - SdT dG = - SdT + VdP An sich sind diese Herleitungen nicht unbedingt aufwändig; trotzdem kursiert ein Merkschema, das die zugeordneten natürlichen Variablen und die Fundamentalgleichungen relativ einprägsam zusammenfasst: S + H + U G V P A(F) T Die vier Funktionen U, H, F (bzw. A) und G stehen dabei auf den Seiten des Quadrats, jeweils in direkter Nachbarschaft dazu die entsprechenden natürlichen Variablen (z.B. S und V für U). Die Vorfaktoren der in den Fundamentalgleichungen als Differentialausdrücke verwendeten natürlichen Variablen stehen jeweils diagonal gegenüber (z.B. T für dS und P für dV). Die Differentialausdrücke, deren Variablen im Diagramm mit „⊕„ markiert sind (dS und dP) werden stets mit positivem Vorzeichen, die anderen (dV und dT) mit negativem Vorzeichen versehen (z.B. +TdS und -PdV). Im englischsprachigen Raum kennt man für dieses Schema die Eselsbrücke „SHiP UG VAT“, was zugegebenermaßen nicht besonders viel Sinn macht, sich aber vielleicht gerade deswegen gut einprägen lässt. 74 7.3 Die Maxwellschen Gleichungen Die Herleitung der Maxwellschen Gleichungen beruht auf dem Schwarzschen Satz, der allgemein für Zustandsfunktionen und somit auch für die Funktionen U, H, F und G gelten muss. Dieser lautete (zur Erinnerung): ∂f ( x, y) ∂ ∂x y ∂y x = ∂f ( x, y) ∂ ∂y x ∂x y oder fxy = fyx Damit kann man beispielsweise für die Innere Energie U formulieren: USV = UVS Mit der Fundamentalgleichung dU = TdS - PdV, nach der gilt: ∂U Us = =T ∂S V folgt demnach: ∂T ∂V S ∂U UV = = −P ∂V S und = ∂P − ∂S V Ein solcher Zusammenhang läuft unter der Bezeichnung Maxwellsche Gleichung. Entsprechend kann man nun für die Zustandsfunktionen H, F und G vorgehen, wobei jeweils der Satz der dazugehörigen natürlichen Variablen und die Fundamentalgleichung verwendet wird. Für die Enthalpie H gilt: HS,P = HP,S mit dH = TdS + VdP folgt: ∂H HS = =T ∂S P folgt demnach: ∂T ∂P S ∂H HP = =V ∂P S und = ∂V ∂S P = FV,T Für die Freie Energie F (A) gilt: FT,V 75 mit dF = -SdT - PdV folgt: ∂F FT = = −S ∂T V folgt demnach: ∂S ∂V T ∂F FV = = −P ∂V T und = ∂P ∂T V = GT,P Für die Freie Enthalpie G gilt: GP,T mit dG = -SdT + VdP folgt: ∂G GT = = −S ∂T P folgt demnach: ∂S ∂P T und = ∂G GP = =V ∂P T ∂V − ∂T P Zusammengefasst lauten damit die vier Maxwellschen Gleichungen: ∂T ∂V S ∂T ∂P S ∂S ∂V T ∂S ∂P T = = = = ∂P − ∂S V ∂V ∂S P ∂P ∂T V ∂V − ∂T P Daraus ergeben sich einige interessante Relationen. So kann beispielsweise mittels der dritten und vierten Maxwellschen Gleichung eine Aussage über die Abhängigkeit der Entropie von Volumen und Druck (jeweils bei konstanter Temperatur) abgeleitet werden. Auch die Maxwellschen Gleichungen sind aus dem bei den Fundamentalgleichungen erwähnten Merkschema ableitbar. 7.4 Die Abhängigkeit der Freien Enthalpie von Temperatur und Druck Die Frage nach der Temperatur- und Druckabhängigkeit von G wird direkt durch die Fundamentalgleichung für die Freie Enthalpie beantwortet: dG = -SdT + VdP 76 Für die Temperaturabhängigkeit bei konstantem Druck gilt daher zunächst die bereits bekannte Formulierung: ∂G dT P = -S Da die Entropie stets einen positiven Wert besitzt, muss die Temperaturabhängigkeit zwangsläufig negativ sein, d.h. die Freie Enthalpie nimmt bei konstantem Druck mit der Temperatur ab. Aus diesem Ergebnis folgt auch, dass die Temperaturabhängigkeit der Freien Reaktionsenthalpie durch die Reaktionsentropie beschrieben wird: ∂∆ R G dT P = -∆RS Das wiederum bedeutet, dass eine Reaktion mit steigender Temperatur dann an Triebkraft gewinnt, wenn die dazugehörige Reaktionsentropie positiv ist (und umgekehrt). Da die Reaktionsentropie aus der Differenz zwischen einfacher Reaktionsenthalpie ∆RH und freier Reaktionsenthalpie ∆RG hervorgeht, kann man diese Gleichung umformen und erhält (ohne Herleitung): ∂( ∆ R G / T) P ∂T = − ∆ RH T2 Diese Gibbs-Helmholtz-Gleichung ermöglicht eine direkte Abschätzung der Temperaturabhängigkeit der Triebkraft der Reaktion aus der Wärmetönung (die im Allgemeinen bekannt oder leicht messbar ist). Ist die Reaktion exotherm (∆RH < 0), so steigt mit steigender Temperatur auch der Quotient ∆RG/T an. Ein entsprechendes Gesetz ist auch bezüglich der Druckabhängigkeit bekannt. Um dies thermodynamisch zu untermauern, wird zunächst die Druckabhängigkeit der Freien Enthalpie zugrunde gelegt: ∂G ∂P T = V Das bedeutet, dass die Freie Enthalpie mit steigendem Druck grundsätzlich (V ist immer positiv) ansteigt. Daraus lässt sich für chemische Reaktionen leicht ableiten: ∂∆ R G ∂P T = ∆RV Im Gegensatz zu ∆RS ist diese Größe in der Praxis entweder zu vernachlässigen (z.B. bei ausschließlich festen bzw. flüssigen Reaktionspartnern) oder leicht messbar. Eine der Gibbs-Helmholtz-Gleichung entsprechende Berechnungsmethode für die Druckabhängigkeit ist also nicht notwendig. Die Konsequenzen dieses Zusammenhangs treten insbesondere dann zutage, wenn gasförmige Komponenten abreagieren oder entstehen: Steigt dabei das Volu- 77 men des Reaktionsgemisches (∆RV > 0), so nimmt die Triebkraft der Reaktion bei steigendem Druck ab. Sinkt das Volumen (∆RV < 0), so steigt die Triebkraft der Reaktion bei steigendem Druck an. Dies entspricht wieder exakt der Aussage des Gesetzes des kleinsten Zwangs. Gleichgültig, ob die Temperatur oder der Druck verändert wird: eine chemische Reaktion zeigt in der Regel die Tendenz, einem äußeren Zwang auszuweichen, indem sie stets den Reaktionsablauf bevorzugt, bei dem die ursprünglichen Gegebenheiten bezüglich T und P angestrebt werden. Ein solches Verhalten scheint in gewissem Sinne naturgegeben. Die anschauliche Betrachtung sollte jedoch nicht darüber hinwegtäuschen, dass der eigentliche Grund für dieses „Ausweichen“ des Systems wieder in einer Maximierung der Entropie des Universums zu sehen ist. 7.5 Das Chemische Potential Die bisher betrachteten Abhängigkeiten der Freien Enthalpie beziehen sich auf rein physikalische Größen, nämlich auf die Temperatur und den Druck. Zur Betrachtung einer chemischen Reaktion muss ein Parameter eingebracht werden, der die Zusammensetzung des Reaktionsgemisches beschreibt. Unter den Möglichkeiten, die sich hierfür anbieten, ist die Nutzung der Molzahl n sicherlich die praktikabelste. Dieser Parameter wurde bereits bei der Formulierung des idealen Gasgesetzes erwähnt, trat danach jedoch in den Hintergrund, da zwischenzeitlich nur reine Systeme betrachtet wurden. Um die Zusammensetzung eines Gemisches zu beschreiben, werden Molzahlen eingeführt, die sich auf die einzelnen Komponenten (1, 2, 3, ... k) in jeweils einem gegebenen Phasenzustand beziehen. Dann gilt: k nges = ∑n i i =1 Mit der Einführung dieser Parameter wird die Freie Enthalpie auch eine Funktion der Größen ni: G(T, P, n1, n2, n3, ... nk) Damit erweitert sich folgerichtig auch das Totale Differential von G zu: dG = ∂G ∂G ∂G dT + dP + ∂T P,n1,n2 ,n3 ,...nk ∂P T,n1,n2 ,n3 ...nk ∂n1 T,P,n ∂G dn1 + dn 2 +... ∂ n 2 T,P,n1,n3 ,...nk 2 ,n3 ,...nk ∂G ∂G = − S dT + V dP + dn1 + dn 2 +... ∂n1 T,P,n2 ,n3 ,...nk ∂n 2 T,P,n1,n3 ,...nk Somit hätte man die komplette Beschreibung aller Abhängigkeiten der Freien Enthalpie, die nun auch die Zusammensetzung eines Reaktionsgemisches berücksichtigt. Etwas unbefriedigend dabei ist, dass zwar die partiellen 78 Differentiale nach T und P, nicht jedoch die partiellen Differentiale nach den Molzahlen durch andere physikalische Größen zu ersetzen sind. Die partielle Ableitung der Freien Enthalpie nach der Molzahl einer Komponente k ist eine Größe, die stark stoffspezifisch ist. Sie beschreibt den Beitrag der jeweiligen Komponente i an der gesamten Freien Enthalpie, und ist insbesondere in der Chemie von großer Bedeutung. Sie wird deswegen Chemisches Potential genannt und üblicherweise mit dem Kürzel µi bezeichnet: µi = ∂ G ∂ n i T,P,n k ≠i Damit ist eine angenehme Vereinfachung des totalen Differentials von G möglich, es lautet nun: dG = -S dT + V dP + µ1 dn1 + µ2 dn2 + ... Die Größen µi erscheinen etwas unanschaulich, besitzen jedoch eine konkrete Bedeutung. Komponenten, die (im positiven oder negativen Sinne) stark zur gesamten freien Enthalpie beitragen, besitzen ein betraglich großes (positives oder negatives) chemisches Potential und umgekehrt. Bei Komponenten mit stark positiven Beiträgen zum chemischen Potential erfährt das Reaktionsgemisch die Tendenz, den Anteil der Komponente abzubauen. Dagegen werden Komponenten mit stark negativen Beiträgen zum chemischen Potential in einer gegebenen Reaktionsmischung bevorzugt angereichert. 7.6 Das chemische Gleichgewicht Ausschlaggebende Bedeutung besitzt das chemische Potential bei der Betrachtung von chemischen Gleichgewichten. Bei konstanter Temperatur und konstantem Druck gilt: k dG = ∑ µi dni i =1 Gleichzeitig repräsentiert ein eingestelltes, stabiles Gleichgewicht den Fall eines statischen Zustands mit dSUniversum = 0 , damit gilt auch: dG = 0 Daraus resultiert, dass bei Einstellung des Gleichgewichts ein Zustand vorliegt, bei dem sich die Beträge von µi dni gegenseitig genau kompensieren. Gleichzeitig stellt dieser Punkt das Minimum von G(ni) dar, da dieser Zustand von jedem anderen Punkt aus spontan angestrebt wird. Um dies anschaulicher darzustellen, betrachten wir ein einfaches Gleichgewicht des Typs A←→B, zum Beispiel das Gleichgewicht zwischen „Sessel-„ und „Wannenform“ des Cyclohexans in der Gasphase: 79 Cyclohexan “Sesselform” “Wannenform” G Gleichgewichtszustand 100% “Sessel” 100% “Wanne” Dazu sei ein Verlauf der chemischen Potentiale postuliert, der etwa folgende Gestalt besitzt: µWanne µSessel µi Gleichgewichtszustand 100% “Sessel” 100% “Wanne” Befindet man sich an einem Punkt außerhalb des Gleichgewichtszustands (beispielsweise bei 100% „Sessel“), so ist das chemische Potential der Sesselform offensichtlich höher als das der „Wanne“. Jedes Molekül, das aus der Sesselform in die Wannenform übergeht, sorgt dann für eine Verminderung der gesamten Freien Enthalpie. Es resultiert ein spontaner Ablauf der Reaktion. Dabei ändern sich die chemischen Potentiale von „Wanne“ und „Sessel“ bis zu einem Punkt, wo diese gleich sind: µSessel = µWanne Mit dGP,T=const. = µSessel dnSessel+ µWanne dnWanne und dnSessel = - dnWanne folgt: dG = µSessel(dnSessel + dnWanne) = 0 An diesem Punkt kann also ein Molekül „Sessel“ in ein Molekül „Wanne“ übergehen, ohne dass sich G verändert. Somit ist hier der statische Zustand eines Gleichgewichts erreicht, der dem Minimum der Enthalpiekurve entspricht. Der Gleichgewichtszustand, der durch dG = 0 gekennzeichnet ist, bedeutet also auch die Gleichheit der chemischen Potentiale. Verallgemeinert kann man sagen, dass sich ein chemisches Gleichgewicht durch die Gleichheit der aufaddierten chemischen Potentiale der Edukte i mit den aufaddierten chemi- 80 schen Potentialen der Produkte j auszeichnet, wenn man dabei die stöchiometrischen Koeffizienten νi berücksichtigt. Im Gleichgewicht gilt dann: k ∑ |νi| µi i =1 m = ∑ j =1 m |νj| µj ∑ νi µi oder = 0 i =1 In diesem Zustand erhält man unter Ansatz der Konzentrationsabhängigkeit des chemischen Potentials (ohne Herleitung) für eine beliebige chemische Gleichgewichtsreaktion vom Typ: n1 A1 + n2 A2 + ... → ← m1 B1 + m2B2 + ... das so genannte Massenwirkungsgesetz: K c B1m1 ⋅ c B2 m 2 ⋅... n1 n2 c A1 ⋅ c A 2 ⋅... ≈ Dabei steht jedes cX für die dimensionslose „relative“ Konzentration des Reaktionspartners X nach der Definition cX = cX / (1Mol/L). Das bedeutet, dass sich im Gleichgewicht eine feste Relation zwischen den Konzentrationen aller Reaktionspartner einstellt. 7.7 Die Elektromotorische Kraft Bei der Einführung der freien Enthalpie in Kapitel 7.1 wurde festgehalten, dass die negative Änderung der freien Enthalpie, die mit einem betrachteten Vorgang verbunden ist, mit der maximalen Nicht-Volumenarbeit gleichgesetzt werden kann. Eine sehr wichtige Art von Nicht-Volumenarbeit ist die elektrische Arbeit wel, die sich zum Beispiel bei der Verschiebung von Ionen in einem elektrischen Feld ergibt. Diese elektrische Arbeit errechnet sich aus dem Produkt aus der Ladung (n z F) und der Potentialdifferenz E. Es gilt: wel = nzFE Dabei steht z für die (dimensionslose) Ladungszahl, n für die Zahl der Ionen (in Mol), E für die angelegte Spannung (in Volt) und F für die Faraday-Konstante. Letztere bemisst die Gesamtladung von einem Mol einer einfach geladenen Ionensorte und beträgt F = 96 485 Coulomb / Mol Die Einheit „Coulomb“ für die elektrische Ladung wird generell mit „C“ abgekürzt. Es gilt: 1C = 1 A·s = 1J/V Die letztgenannte Einheit bedeutet eigentlich „Arbeit pro Spannung“. Man kann also sagen, dass 1 Coulomb diejenige Ladung bedeutet, die bei der Verschie- 81 bung gegen eine Potentialdifferenz von einem Volt eine Arbeit von einem Joule erfordert. Stoff 2 Stoff 1 Angenommen, eine chemische (Redox-) Reaktion zwischen zwei Elektroden, an der zwei verschiedene z-fach positiv geladene Kationen 1 und 2 beteiligt sind, führe zu einer Ladungstrennung, die sich in einer Spannung E äußert. Dabei sei der Stoff 1 derjenige, der bei der Reaktion oxidiert wird und damit den negativen Pol bildet. Eine typische experimentelle Anordnung dieser Art ist in der folgenden Abbildung wiedergegeben. Man spricht dabei von einer elektrochemischen Zelle, die elektrische Spannung E zwischen den Elektroden bezeichnet man in diesem Zusammenhang als Elektromotorische Kraft. Stromschlüssel Kation1n+ Kation2n+ Unter Standardbedingungen (die Konzentrationen c1 und c2 beider Ionensorten besitzen den Wert 1 Mol/L) erhält man dann unter Ansatz von wel = -∆rG0 die Gleichung: z F E0 = - ∆rG0 wobei der erhaltene Wert auf ein Mol Stoffumsatz bezogen ist und die Einheit J/Mol besitzt. Die Vorzeichenumkehr ergibt sich aus der Tatsache, dass die Reaktionsenthalpie negativ ist (Triebkraft!) und die Spannung im Normalfall positiv angegeben wird. Der Wert der Spannung E0 wird dann als Normalpotential der Zelle bezeichnet. Weichen die Konzentrationen der Ionensorten von der Standardkonzentration 1 Mol/L ab, so ergibt sich ein zusätzlicher Beitrag wkonz für diejenige Arbeit, die für die Aufkonzentration der Ionen benötigt wird (bei einer Verringerung der Konzentration kehrt sich entsprechend das Vorzeichen dieses Beitrags um). Dieser Beitrag errechnet sich in Analogie zur in Kapitel 5.3 eingeführten integrierten Volumenarbeit w = - RT ln (V2/V1) als wkonz = V - RT ln 2 V1 = c - RT ln 1 c2 Die neue Spannungsdifferenz ergibt sich dann wie folgt: zFE = z F E0 - c RT ln 1 c2 = c - ∆rG0 - RT ln 1 c2 Daraus ergibt sich schließlich die Nernstsche Gleichung für die Berechnung der Elektromotorischen Kraft als: 82 E = E 0 c RT ln 1 zF c2 - Die Spannung der Zelle kann also dadurch erhöht werden, dass man entweder die Ionenkonzentration desjenigen Partners, der oxidiert wird (Stoff 1), verkleinert, oder die Konzentration desjenigen Partners, der reduziert wird (Stoff 2), vergrößert. Durch Einsetzen der Konstanten R und F, Ansatz von T = 298 K als Raumtemperatur und unter Berücksichtigung des Faktors zwischen natürlichem Logarithmus und dekadischem Logarithmus erhält man daraus die bekannte und praxisgerechte Form der Nernstschen Gleichung: E = E0 c (1/z) (0,059 V) log10 1 c2 - Misst man die Normalpotentiale E0 verschiedener Kationensorten gegen eine so genannte Normalwasserstoffelektrode, X H2 Xn+ H+ (alle Werte gegen H2 gemessen und in Volt) Li K Ca Na Mg Al Mn Zn Cr Fe Lithium Kalium Calcium Natrium Magnesium Aluminum Mangan Zink Chrom Eisen - 2.96 - 2.92 - 2.76 - 2.71 - 2.34 - 1.33 - 1.10 - 0.76 - 0.51 - 0.44 Schwache Reduktionsmittel Starke Reduktionsmittel so erhält man die bekannte Spannungsreihe der Kationen, welche die Stärke der Reduktionswirkung durch die entsprechenden ungeladenen Stoffe (meistens Metalle) bzw. die Oxidationswirkung der dazugehörigen Kationen wiedergibt: Cd Co Ni Sn Pb H2 Cu Ag Hg Au Pt Cadmium Cobalt Nickel Zinn Blei Wasserstoff Kupfer Silber Quecksilber Gold Platin - 0.40 - 0.28 - 0.23 - 0.16 - 0.12 0 0.34 0.79 0.85 1.36 1.60 Die Differenz zwischen zwei der hier aufgelisteten Spannungswerten ergibt die Spannung, die aus einer Kombination von den betreffenden zwei Halbzellen zu erzielen ist. 83 84 7.4 Die Abhängigkeit der Freien Enthalpie von Temperatur und Druck Die Frage nach der Temperatur- und Druckabhängigkeit von G wird direkt durch die Fundamentalgleichung für die Freie Enthalpie beantwortet: dG = -SdT + VdP Für die Temperaturabhängigkeit bei konstantem Druck gilt daher zunächst die bereits bekannte Formulierung: ∂G dT P = -S Da die Entropie stets einen positiven Wert besitzt, muss die Temperaturabhängigkeit zwangsläufig negativ sein, d.h. die Freie Enthalpie nimmt bei konstantem Druck mit der Temperatur ab. Aus diesem Ergebnis folgt auch, dass die Temperaturabhängigkeit der Freien Reaktionsenthalpie durch die Reaktionsentropie beschrieben wird: ∂∆ R G dT P = -∆RS Das wiederum bedeutet, dass eine Reaktion mit steigender Temperatur dann an Triebkraft gewinnt, wenn die dazugehörige Reaktionsentropie positiv ist (und umgekehrt). Häufig ist die mit einer chemischen Reaktion verbundene Entropieänderung nicht bekannt. Hierzu stellten W.J. Gibbs und H. von Helmholtz ausgehend von einer Größe G/T folgende Überlegung an: ∂ (G / T ) ∂T P = = = und schließlich: ∂(G / T) ∂T P = oder, angewandt auf chemische Reaktionen: ∂G T−G ∂T P T2 − ST − G T2 − ST − (H − TS) T2 −H T2 85 ∂( ∆ R G / T) P ∂T = − ∆ RH T2 Diese Gibbs-Helmholtz-Gleichung ermöglicht somit eine direkte Abschätzung der Temperaturabhängigkeit der Triebkraft der Reaktion aus der Wärmetönung (die im Allgemeinen bekannt oder leicht messbar ist). Ist die Reaktion exotherm (∆RH < 0), so steigt mit steigender Temperatur auch der Quotient ∆RG/T an. Folglich muss ∆RG zunehmen, wodurch die Reaktion an Triebkraft verliert. Für eine endotherme Reaktion gilt mit gewissen Einschränkungen (bei sehr kleinen Änderungen von ∆RG könnte der Einfluss der Temperatur im Nenner überwiegen und d∆RG/dT könnte trotz positivem ∆RH ebenfalls positiv sein) die umgekehrte Schlussfolgerung. Dieser Zusammenhang ist die Grundlage des aus der Chemie bekannten Gesetzes des kleinsten Zwangs bei Variation der Temperatur. Ein entsprechendes Gesetz ist auch bezüglich der Druckabhängigkeit bekannt. Um dies thermodynamisch zu untermauern, wird zunächst die Druckabhängigkeit der Freien Enthalpie zugrunde gelegt: ∂G ∂P T = V Das bedeutet, dass die Freie Enthalpie mit steigendem Druck grundsätzlich (V ist immer positiv) ansteigt. Daraus lässt sich für chemische Reaktionen leicht ableiten: ∂∆ R G ∂P T = ∆RV Im Gegensatz zu ∆RS ist diese Größe in der Praxis entweder zu vernachlässigen (z.B. bei ausschließlich festen bzw. flüssigen Reaktionspartnern) oder leicht messbar. Eine der Gibbs-Helmholtz-Gleichung entsprechende Berechnungsmethode für die Druckabhängigkeit ist also nicht notwendig. Die Konsequenzen dieses Zusammenhangs treten insbesondere dann zutage, wenn gasförmige Komponenten abreagieren oder entstehen: Steigt dabei das Volumen des Reaktionsgemisches (∆RV > 0), so nimmt die Triebkraft der Reaktion bei steigendem Druck ab. Sinkt das Volumen (∆RV < 0), so steigt die Triebkraft der Reaktion bei steigendem Druck an. Dies entspricht wieder exakt der Aussage des Gesetzes des kleinsten Zwangs. Gleichgültig, ob die Temperatur oder der Druck verändert wird: eine chemische Reaktion zeigt in der Regel die Tendenz, einem äußeren Zwang auszuweichen, indem sie stets den Reaktionsablauf bevorzugt, bei dem die ursprünglichen Gegebenheiten bezüglich T und P angestrebt werden. Ein solches Verhalten scheint in gewissem Sinne naturgegeben. Die anschauliche Betrachtung sollte jedoch nicht darüber hinwegtäuschen, dass der eigentliche Grund für dieses „Ausweichen“ des Systems wieder in einer Maximierung der Entropie zu sehen ist. 86 7.5 Das Chemische Potential Die bisher betrachteten Abhängigkeiten der Freien Enthalpie beziehen sich auf rein physikalische Größen, nämlich auf die Temperatur und den Druck. Zur Betrachtung einer chemischen Reaktion muss ein Parameter eingebracht werden, der die Zusammensetzung des Reaktionsgemisches beschreibt. Unter den Möglichkeiten, die sich hierfür anbieten, ist die Nutzung der Molzahl n sicherlich die praktikabelste. Dieser Parameter wurde bereits bei der Formulierung des idealen Gasgesetzes erwähnt, trat danach jedoch in den Hintergrund, da zwischenzeitlich nur reine Systeme betrachtet wurden. Um die Zusammensetzung eines Gemisches zu beschreiben, werden Molzahlen eingeführt, die sich auf die einzelnen Komponenten (1, 2, 3, ... k) in jeweils einem Phasenzustand beziehen. Dann gilt: k nges = ∑n i i =1 Mit der Einführung dieser Parameter wird die Freie Enthalpie auch eine Funktion der Größen ni: G(T, P, n1, n2, n3, ... nk) Damit erweitert sich folgerichtig auch das Totale Differential von G zu: dG = ∂G ∂G ∂G dT + dP + ∂T P,n1,n2 ,n3 ,...nk ∂P T,n1,n2 ,n3 ...nk ∂n1 T,P,n ∂G dn1 + dn 2 +... ∂n 2 T,P,n1,n3 ,...nk 2 ,n3 ,...nk ∂G ∂G = − S dT + V dP + dn1 + dn 2 +... ∂n1 T,P,n2 ,n3 ,...nk ∂n 2 T,P,n1,n3 ,...nk Somit hätte man die komplette Beschreibung aller Abhängigkeiten der Freien Enthalpie, die nun auch die Zusammensetzung eines Reaktionsgemisches berücksichtigt. Etwas unbefriedigend dabei ist, dass zwar die partiellen Differentiale nach T und P, nicht jedoch die nach den Molzahlen durch andere physikalische Größen zu ersetzen sind. Die partielle Ableitung der Freien Enthalpie nach der Molzahl einer Komponente k ist eine Größe, die stark stoffspezifisch ist. Sie beschreibt den Beitrag der jeweiligen Komponente i an der gesamten Freien Enthalpie, und ist insbesondere in der Chemie von großer Bedeutung. Sie wird deswegen Chemisches Potential genannt und üblicherweise mit dem Kürzel µi bezeichnet: µi = ∂ G ∂ ni T,P,nk ≠ i 87 Damit ist eine angenehme Vereinfachung des totalen Differentials von G möglich, es lautet nun: dG = -S dT + V dP + µ1 dn1 + µ2 dn2 + ... Die Größen µi erscheinen etwas unanschaulich, besitzen jedoch eine konkrete Bedeutung. Komponenten, die (im positiven oder negativen Sinne) stark zur gesamten freien Enthalpie beitragen, besitzen ein betraglich großes (positives oder negatives) chemisches Potential und umgekehrt. Bei Komponenten mit positiven Beiträgen zum chemischen Potential besitzt das Reaktionsgemisch die Tendenz, den Anteil der Komponente abzubauen. Dagegen werden Komponenten mit negativen Beiträgen zum chemischen Potential in einer gegebenen Reaktionsmischung bevorzugt angereichert. 7.6 Das chemische Gleichgewicht Ausschlaggebende Bedeutung besitzt das chemische Potential bei der Betrachtung von chemischen Gleichgewichten. Bei konstanter Temperatur und konstantem Druck gilt: k dG = ∑ µi dni i =1 Gleichzeitig repräsentiert ein eingestelltes, stabiles Gleichgewicht den Fall eines statischen Zustands mit dSUniversum = 0 , damit gilt auch: dG = 0 Daraus resultiert, dass bei Einstellung des Gleichgewichts ein Zustand vorliegt, bei dem sich die Beträge von µi dni gegenseitig genau kompensieren. Gleichzeitig stellt dieser Punkt das Minimum von G(ni) dar, da dieser Zustand von jedem anderen Punkt aus spontan angestrebt wird. Um dies anschaulicher darzustellen, betrachten wir ein einfaches Gleichgewicht des Typs A←→B, zum Beispiel das Gleichgewicht zwischen „Sessel-„ und „Wannenform“ des Cyclohexans in der Gasphase: Cyclohexan “Sesselform” “Wannenform” G 100% “Sessel” Gleichgewichtszustand 100% “Wanne” 88 Dazu sei ein Verlauf der chemischen Potentiale postuliert, der etwa folgende Gestalt besitzt: µWanne µSessel µi Gleichgewichtszustand 100% “Sessel” 100% “Wanne” Befindet man sich an einem Punkt außerhalb des Gleichgewichtszustands (beispielsweise bei 100% „Sessel“), so ist das chemische Potential der Sesselform offensichtlich höher als das der „Wanne“. Jedes Molekül, das aus der Sesselform in die Wannenform übergeht, sorgt dann für eine Verminderung der gesamten Freien Enthalpie. Es resultiert ein spontaner Ablauf der Reaktion. Dabei ändern sich die chemischen Potentiale von „Wanne“ und „Sessel“ bis zu einem Punkt, wo diese gleich sind: µSessel = µWanne Mit dGP,T=const. = µSessel dnSessel+ µWanne dnWanne und dnSessel = - dnWanne folgt: dG = µSessel(dnSessel + dnWanne) = 0 An diesem Punkt kann also ein Molekül „Sessel“ in ein Molekül „Wanne“ übergehen, ohne dass sich G verändert. Somit ist hier der statische Zustand eines Gleichgewichts erreicht, der dem Minimum der Enthalpiekurve entspricht. Die Frage ist nun, warum und in welcher Weise sich das chemische Potential der beteiligten Komponenten mit Ablauf der Reaktion ändert. Betrachtet man den Punkt des Prozesses, an dem nur die Sesselform vorliegt, so gilt: dG = -S dT + V dP + µSessel dnSessel beziehungsweise, bei konstanter Temperatur und konstantem Druck: dG = µSessel dnSessel Die Freie Enthalpie einer Menge von n Mol an reiner Sesselform errechnet sich aus deren molarer Freier Enthalpie GmSessel nach: G = GmSessel nSessel Dann gilt bei konstantem Druck und konstanter Temperatur aber auch: 89 dG = G m dnSessel Sessel wonach sich durch Koeffizientenvergleich ergibt: µSessel = G m Sessel Das heißt, dass für eine reine Substanz das chemische Potential gleich der molaren freien Enthalpie ist. Dann folgt für die reine Komponente, die man der Einfachheit halber zunächst in der Gasphase betrachtet: ∂µ Sessel ∂PSessel T ∂Gm Sessel ∂PSessel T = = V m Sessel mit PSessel als Partialdruck des Cyclohexans in der Sesselform. Dieser Ausdruck gibt nun an, wie sich das chemische Potential verändert, wenn der Partialdruck des sesselförmigen Cyclohexans bzw. sein Mengenanteil sinkt. Das chemische Potential der Sesselform lässt sich dann an einem beliebigen Punkt des Reaktionsverlaufs berechnen: dµSessel VmSessel dPSessel = PSessel µSessel - µ0Sessel ∫V = m Sessel dPSessel P0 mit µ Sessel als dem chemischen Potential des Ausgangszustands mit P0. Unter Ansatz des idealen Gasgesetzes (PVm = RT) ergibt die Integration: 0 µSessel - µ 0 Sessel = P RT ln Sessel P0 Definiert man den relativen Partialdruck PSessel über die Relation PSessel/P0 als dimensionslose Größe, so erhält man schließlich: µSessel = µ0Sessel = µ0i + RT ln PSessel oder allgemein: µi + RT ln Pi Diese Gleichung gibt die Abhängigkeit des chemischen Potentials einer Komponente von deren relativem Partialdruck an; sie gilt jedoch strenggenommen nur für die gasförmige Phase. Die experimentelle Erfahrung zeigt allerdings, dass für verdünnte Lösungen in guter Näherung eine entsprechende Formulierung unter Verwendung der relativen Konzentration anwendbar ist: µi ≈ µ0i + RT ln ci 90 wobei sich µ dann auf die Konzentration 1 Mol/l bezieht. Man kann diese Beziehung exakt formulieren, wenn man statt der Konzentration die sogenannte (relative) Aktivität a einführt: 0 µi µ0i = + RT ln ai Der Wert der Aktivität wird dem der Konzentration umso ähnlicher, je verdünnter die Lösung ist: lim a = c c→0 Mit Hilfe der soeben hergeleiteten Ausdrücke für die chemischen Potentiale in Abhängigkeit von Partialdruck und Aktivität bzw. Konzentration lässt sich nun die abschließende Gleichgewichtsbedingung formulieren. Aus dem Zusammenhang µSessel µWanne = folgt damit: µ + RT ln PSessel = µ0Wanne + RT ln PWanne µ - µ = RT ln Pwanne - RT ln PSessel µ - µ = 0 Sessel 0 Sessel 0 Sessel 0 Wanne 0 Wanne PWanne RT ln PSessel Die Forderung nach Verwendung von dimensionslosen Relativgrößen für die Partialdrucke kann hier entfallen, da ein Quotient aus zwei dieser Größen gebildet wird. Auf der linken Seite der letzten Gleichung steht die Differenz der chemischen Potentiale, wenn jeweils die reinen Komponenten Sessel bzw. Wanne betrachtet werden. Diese ist identisch mit der Differenz der molaren Freien Enthalpien beider Zustände unter den gewählten Standardbedingungen: µ 0 Sessel - µ0Wanne = G0Sessel - G0Wanne = -∆rG0Sessel→Wanne Folglich gilt: ∆rG0Sessel→Wanne = PWanne - RT ln PSessel Diese Gleichung, an dem Beispiel der Konformationsänderung des Cyclohexans exemplarisch abgeleitet, ist der zentrale Zusammenhang zur Beschreibung chemischer Gleichgewichte. Der Quotient der Partialdrucke besitzt bei festgelegter Temperatur einen konstanten Wert, der die Lage des Gleichgewichts beschreibt. Dieses Ergebnis gilt entsprechend für alle Gleichgewichtsreaktionen des Typs A←→B. Im Fall einer allgemeinen Reaktion 91 n1 A1 + n2 A2 + ... → ← m1 B1 + m2B2 + ... ist zu berücksichtigen, dass die chemischen Potentiale der einzelnen Komponenten A und B bei der Bilanzierung jeweils mit ihren stöchiometrischen Faktoren n und m multipliziert werden müssen. Am Punkt des Gleichgewichts gilt dort: n1 µA1 + n2 µA2 + ... = m1 µB1 + m2 µB2 + ... oder: 0 A1 n1 (µ 0 A2 + RT ln PA1) + n2 (µ + RT ln PA2) + ... = m1 (µ0B1 + RT ln PB1) + m2 (µ0B2 + RT ln PB2) + ... Alle chemischen Potentiale mit dem Index „0“ lassen sich samt ihrer Vorfaktoren wieder unter Bildung der Freien Reaktionsenthalpie zusammenfassen und es gilt: -∆rG0 = RT (-n1 ln PA1 - n2 ln PA2 - ... + m1 ln PB1 + m2 ln PB2 + ... ) und damit die allgemeine Beziehung zwischen der Freien Reaktionsenthalpie und den Partialdrucken der Reaktionspartner im Gleichgewicht: ∆rG 0 = P B1m1 ⋅ P B2 m2 ⋅... - RT ln n1 n2 P A1 ⋅ P A 2 ⋅... Der Quotient der Partialdrucke ist damit unter gewählten Temperatur- und Druckbedingungen konstant und wird als Gleichgewichtskonstante K bezeichnet: K = P B1m1 ⋅ P B2 m2 ⋅... n1 n2 P A1 ⋅ P A 2 ⋅... Wie erwähnt, können die Partialdrücke durch die Aktivitäten, oder, in mehr oder weniger guter Näherung, durch Konzentrationen ersetzt werden: K K = aB1m1 ⋅ aB2 m2 ⋅... n1 n2 a A1 ⋅ a A 2 ⋅... ≈ c B1m1 ⋅ c B2 m 2 ⋅... n1 n2 c A1 ⋅ c A 2 ⋅... jeweils mit ∆rG0 = - RT ln K. Die Gleichgewichtskonstante enthält stets die Edukte im Nenner, die Produkte im Zähler. Die Tatsache, dass bei eingestelltem chemischem Gleichgewicht 92 eine feste Relation zwischen den Anteilen der Reaktionspartnern vorliegt, ist in der Chemie von weitreichender Bedeutung. Die Gleichgewichtskonstante K ist allerdings, je nach Reaktion, mehr oder weniger stark temperatur- und druckabhängig, was sich notwendigerweise aus der Abhängigkeit der Freien Reaktionsenthalpie von T und P ergibt. Mit der Gibbs-Helmholtz-Gleichung ∂( ∆ r G 0 / T) ∂T P = − ∆ rH0 T2 0 ergibt sich (wegen ∆rG = - RT ln K) für die Temperaturabhängigkeit der Gleichgewichtskonstante die van’t Hoffsche Beziehung: ∂ lnK ∂T P = ∆ rH0 RT 2 Bei Erwärmung des Reaktionsgemisches verschiebt sich somit für exotherme Reaktionen (∆rH0 < 0) das Gleichgewicht zu den Edukten (K wird kleiner), für endotherme Reaktionen zu den Produkten (K wird größer). Dieser Zusammenhang stellt die Grundlage für das Gesetz des kleinsten Zwangs unter Variation der Temperatur dar. Ähnliches gilt für die Druckabhängigkeit der Lage eines chemischen Gleichgewichts. Allerdings führt ein entsprechender Ansatz zu dem Ergebnis, dass bei konstanter Temperatur die Abhängigkeit der Gleichgewichtskonstante vom Druck verschwindet. Dies ist eigentlich zu erwarten, da im Unterschied zur Temperaturabhängigkeit die Druckabhängigkeit bereits in der Definition der Gleichgewichtskonstante berücksichtigt ist. Betrachtet man beispielsweise eine Reaktion des Typs A+B →← C, wobei alle Reaktionsteilnehmer gasförmig vorliegen (z.B. die Reaktion von 2 NO2 zu N2O4), so gilt die Gleichgewichtskontante K = PC/(PA⋅PB). Verdoppelt man den Gesamtdruck durch Kompression des Reaktionsgemisches, so verändert sich der Quotient der Partialdrucke zunächst zu 2PC/(2PA⋅2PB). Er wird damit halb so groß wie nach der Gleichgewichtsbedingung gefordert. Dies wird unter erneutem Anstreben des Gleichgewichts dadurch kompensiert, dass sich die Partialdrucke der Edukte (im Nenner) verringern, wobei der Partialdruck des Produkts (im Zähler) ansteigt. Aus jeweils zwei Molen Gas der Edukte (A und B) wird dabei je ein Mol C gebildet. Die Reaktion verläuft damit in diejenige Richtung, die zum teilweisen Abbau des von außen erzwungenen Druckanstiegs führt. Somit gilt auch im Fall der Druckvariation das Gesetz des kleinsten Zwangs, wonach das Reaktionsgleichgewicht stets dazu tendiert, die von außen auferlegte Veränderung zu kompensieren. Anhang: reversible Expansionsprozesse 93 Sowohl der Joule’sche als auch der Joule-Thomson’sche Versuch sind mit irreversiblen Vorgängen verbunden. Der erstgenannte Prozess entspricht dabei einer Expansion gegen den Druck Null, der zweite einer Expansion gegen einen konstanten Außendruck. Im Folgenden wird die Änderung der Inneren Energie an einem idealen Gas unter völlig reversiblen Bedingungen betrachtet. Dabei sind, im Vergleich zwischen dem isothermen und dem adiabatischen Fall, folgende Unterschiede erkennbar: isotherme, reversible Expansion: P adiabatische, reversible Expansion: P Isotherme PV = const. Isotherme PV = const. Abkühlung, dadurch Druckminderung Adiabate V V Im isothermen Fall gilt für für die Volumenarbeit: Im adiabatischen Fall gilt die Volumenarbeit: V2 w = − ∫ PdV U=q+w und q=0 V1 V2 1 dV V V1 = −nRT ∫ wisoth = −nRT ln ⇒ V2 V1 V2 2 V1 1 w = − ∫ PdV = ∫ dU wadiab = T2 ∫C V dT T1 Somit resultieren, abhängig von der Prozessführung (isotherm oder adiabatisch) zwei völlig verschiedene Ausdrücke für die Volumenarbeit. Für ideale Gase kann man die mathematische Form der Isotherme im PV-Diagramm aus T = const. und dem idealen Gasgesetz ableiten. Man erhält P = const./V oder PV = const.. Für die Adiabaten idealer Gase ist die Herleitung einer Funktion P(V) wesentlich komplizierter. Sie setzt an der Definition der Volumenarbeit und an dem gefundenen Ausdruck für wadiab an, man erhält: dU = dw C V dT = − PdV mit PV = nRT: C V dT = − nRT Variablentrennung: CV Integration: 1 dT T T C V ln 2 T1 = = 1 dV V 1 − nR dV V V − nR ln 2 V1 94 T2 T1 T ln 2 T1 T ln 2 T1 ln mit CV -CP = -nR: mit γ = CP/CV: T2 T1 mit PV = const. ⋅T: = = = = nR V2 ln C V V1 C V − C P V2 ln CV V1 V (1 − γ )ln 2 V1 − V2(1− γ ) V1(1− γ ) V2(1− γ ) P2 V2 P1V1 = V1(1− γ ) P2 V2γ = P1V1γ Daraus folgt, in Analogie zur Isothermengleichung PV = const., die Adiabatengleichung: PVγ = const.. Die Konstante γ heißt Adiabatenkonstante und besitzt den Wert γ = CP/CV. Da CP immer einen größeren Wert besitzt als CV, ist die Adiabatenkonstante stets größer als Eins. Aufgrund dieser Tatsache ist, bei gleichem P und V, die Steigung einer Adiabaten betraglich größer als die einer Isothermen.