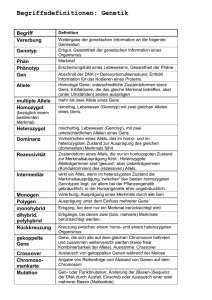
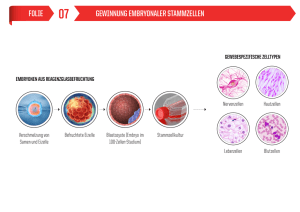
Mattern_Felix_Bos_taurus
Werbung

Alterungsbedingte Effekte auf DNA-Methylierungsprofile entwicklungsrelevanter Gene in Eizellen und Embryonen am Modellorganismus Bos taurus. Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Bayerischen Julius-Maximilians-Universität Würzburg vorgelegt von Felix Mattern geboren in Issyk, Kasachstan Würzburg, November 2016 Eingereicht am: Mitglieder der Promotionskommission: Vorsitzender: Gutachter: Univ.-Prof. Dr. med. Thomas Haaf Gutachter: Univ.-Prof. Dr. Christian Janzen Tag des Promotionskolloquiums: Doktorurkunde ausgehändigt am: Die vorliegende Arbeit wurde von Februar 2012 bis November 2016 am Institut für Humangenetik der Universität Würzburg unter Betreuung von Herrn Univ.-Prof. Dr. med. Thomas Haaf angefertigt. Eidesstattliche Versicherung Hiermit erkläre ich, dass ich diese Doktorarbeit selbständig und ohne Hilfe einer kommerziellen Promotionsberatung sowie ausschließlich unter Verwendung der angegebenen Quellen und Hilfsmittel verfasst habe. Die den herangezogenen Werken wörtlich oder sinngemäß entnommenen Stellen sind als solche gekennzeichnet. Weiterhin versichere an Eides statt, dass ich die Gelegenheit zum Promotionsvorhaben nicht kommerziell vermittelt bekommen habe und insbesondere nicht eine Person oder Organisation eingeschaltet habe, die gegen Entgelt Betreuer bzw. Betreuerinnen für die Anfertigung von Dissertationen sucht. Ich erkläre hiermit, dass die Regeln der Universität Würzburg über gute wissenschaftliche Praxis eingehalten wurden. Die Dissertation wurde bisher weder in gleicher noch in ähnlicher Form einer anderen Hochschule oder in einem anderen Prüfungsfach mit dem Ziel, einen akademischen Grad zu erwerben, vorgelegt. Am 21.10.2011 wurde mir von der Bayerischen Julius-Maximilians-Universität Würzburg der akademische Grad „Diplombiologe“ verliehen. Weitere akademische Grade habe ich weder erworben noch versucht zu erwerben. Würzburg, Oktober 2016 _______________________________ Felix Mattern INHALTSVERZEICHNIS Inhaltsverzeichnis ZUSAMMENFASSUNG ....................................................................................................... 1 ABSTRACT......................................................................................................................... 3 1 EINLEITUNG ....................................................................................................... 4 1.1 EPIGENETIK ...................................................................................................................... 4 1.1.1 DNA-Methylierung ..................................................................................................... 5 1.1.2 Genomische Prägung ................................................................................................. 7 1.1.2.1 Funktion und Regulation geprägter Gene .................................................... 8 1.1.3 Fehlregulation geprägter Gene................................................................................ 10 1.1.3.1 Prader-Willi-Syndrom und Angelman-Syndrom ......................................... 11 1.1.3.2 Silver-Russel-Syndrom und Beckwith-Wiedemann-Syndrom..................... 12 1.1.4 Epigenetische Genomreprogrammierung ............................................................... 13 1.2 OOGENESE UND OOZYTENMATURATION .............................................................................. 15 1.2.1 Allgemeine Oogenese .............................................................................................. 16 1.2.2 Follikulogenese beim Rind (Bos taurus) .................................................................. 17 1.2.3 Oozytenmaturation ................................................................................................. 21 1.2.4 Ovarielle Alterung .................................................................................................... 24 1.3 ASSISTIERTE REPRODUKTION ............................................................................................. 25 1.3.1 Assistierte Reproduktionstechniken in der humanen Reproduktionsmedizin ....... 26 1.3.2 Assistierte Reproduktionstechniken bei Nutztieren ............................................... 28 1.3.3 Assistierte Reproduktionstechniken und gesundheitliche Risiken ......................... 29 1.4 IN-VITRO-MATURATION VON EIZELLEN................................................................................ 32 1.4.1 IVM in der humanen Reproduktionsmedizin .......................................................... 32 1.4.2 IVM beim Rind (Bos taurus) ..................................................................................... 34 1.5 DAS HAUSRIND (BOS TAURUS) ALS MODELLORGANISMUS DER HUMANEN REPRODUKTION............ 35 1.6 ZIELSETZUNG DER ARBEIT ................................................................................................. 36 1.7 UNTERSUCHTE GENE ....................................................................................................... 38 1.7.1 Genset 1: Oozyten- und Maturations-spezifische Gene.......................................... 38 1.7.2 Genset 2: Oozyten- sowie Alters-spezifische Gene ................................................. 42 I INHALTSVERZEICHNIS 1.8 2 KONTAMINATIONSKONTROLLE ........................................................................................... 44 MATERIAL UND METHODEN ............................................................................ 45 2.1 MATERIAL ..................................................................................................................... 45 2.1.1 Biologisches Material ............................................................................................... 45 2.1.1.1 Unreife Eizellen aus Schlachttieren ............................................................ 45 2.1.1.2 Unreife Eizellen aus Kühen unterschiedlichen Alters ................................. 46 2.1.1.3 In vitro gereifte Eizellen .............................................................................. 47 2.1.1.4 Rinderembryonen ....................................................................................... 47 2.1.2 Puffer ....................................................................................................................... 48 2.1.3 Kommerzielle Kits und Reagenzien.......................................................................... 49 2.1.4 Geräte ...................................................................................................................... 49 2.2 METHODEN ................................................................................................................... 50 2.2.1 Natriumbisulfitbehandlung der DNA von Eizellen und Embryonen ........................ 50 2.2.2 Limiting Dilution ....................................................................................................... 52 2.2.3 Primersequenzen und PCR-Bedingungen ................................................................ 55 2.2.3.1 Genset 1: Oozyten- und Maturations-spezifische Gene ............................. 56 2.2.3.2 Genset 2: Oozyten- sowie Alters-spezifische Gene .................................... 58 2.2.4 Methoden zur Bisulfitsequenzierung ...................................................................... 61 2.2.4.1 Pyrosequenzierung ..................................................................................... 62 2.2.4.2 Direkte Bisulfitsequenzierung ..................................................................... 66 2.2.5 Computerprogramme und Datenbanken ................................................................ 68 3 ERGEBNISSE .................................................................................................... 70 3.1 DNA-METHYLIERUNGSPROFILE ENTWICKLUNGSRELEVANTER GENE IN UNREIFEN RINDEREIZELLEN .. 73 3.2 DNA-METHYLIERUNGSPROFILE ENTWICKLUNGSRELEVANTER GENE IN IN VITRO-GEREIFTEN RINDEREIZELLEN UND DARAUS GENERIERTEN RINDEREMBRYONEN........................................... 76 3.2.1 DNA-Methylierungsprofile entwicklungsrelevanter Gene in in vitro-gereiften Rindereizellen ....................................................................................................... 76 3.2.2 DNA-Methylierungsprofile entwicklungsrelevanter Gene in Rinderembryonen aus in vitro-gereiften Rindereizellen........................................................................... 79 3.3 DNA-METHYLIERUNGSPROFILE DER PROMOTORREGION BDNMT3LS IN UNREIFEN UND IN VITROGEREIFTEN RINDEREIZELLEN SOWIE DARAUS GENERIERTEN RINDEREMBRYONEN ......................... 84 II INHALTSVERZEICHNIS 3.4 DNA-METHYLIERUNGSPROFILE ENTWICKLUNGSRELEVANTER GENE IN UNREIFEN RINDEREIZELLEN AUS KÜHEN UNTERSCHIEDLICHEN ALTERS ................................................................................. 88 4 DISKUSSION .................................................................................................... 93 4.1 DNA-METHYLIERUNGSPROFILE ENTWICKLUNGSRELEVANTER GENE IN UNREIFEN UND IN VITROGEREIFTEN RINDEREIZELLEN SOWIE DARAUS GENERIERTEN RINDEREMBRYONEN ......................... 93 4.1.1 DNA-Methylierungsprofile der Gene bH19, bSNRPN, bZAR1, bDNMT3A, bOCT4 und DNMT3Lo in unreifen und in vitro-gereiften Rindereizellen sowie daraus generierten Rinderembryonen............................................................................. 93 4.1.2 DNA-Methylierungsprofile der Promotorregion bDNMT3Ls in unreifen und in vitrogereiften Rindereizellen sowie daraus generierten Rinderembryonen............... 97 4.2 DNA-METHYLIERUNGSPROFILE ENTWICKLUNGSRELEVANTER GENE IN UNREIFEN RINDEREIZELLEN AUS KÜHEN UNTERSCHIEDLICHEN ALTERS ............................................................................... 102 4.3 BEWERTUNG DER LIMITING DILUTION-BISULFITSEQUENZIERUNG ZUR UNTERSUCHUNG VON BOVINEN EIZELLEN UND EMBRYONEN........................................................................................... 106 4.4 5 SCHLUSSFOLGERUNG UND AUSBLICK ................................................................................. 109 REFERENZEN...................................................................................................112 5.1 6 LITERATURVERZEICHNIS .................................................................................................. 112 VERZEICHNISSE ...............................................................................................131 6.1 ABKÜRZUNGSVERZEICHNIS .............................................................................................. 131 6.2 ABBILDUNGSVERZEICHNIS ............................................................................................... 135 6.3 TABELLENVERZEICHNIS ................................................................................................... 136 7 PUBLIKATIONEN UND KONGRESSBEITRÄGE ....................................................139 7.1 PUBLIKATIONEN ............................................................................................................ 139 7.2 KONGRESSBEITRÄGE ...................................................................................................... 140 III ZUSAMMENFASSUNG Zusammenfassung Die postovulatorische Alterung sowie die ovarielle Alterung konnten bei der Anwendung assistierter Reproduktionstechniken (ARTs) als entscheidende Faktoren identifiziert werden, die den Reproduktionserfolg nachhaltig beeinträchtigen. Die postovulatorische Alterung tritt ein, sobald die reife Eizelle nicht mehr innerhalb ihres physiologischen Zeitfensters befruchtet wird. Die ovarielle Alterung beschreibt hingegen die Abnahme des Follikel-Vorrats mit zunehmendem Alter des weiblichen Individuums bzw. des Ovars. Sowohl die postovulatorische Alterung als auch die ovarielle Alterung führen u.a. zu einer reduzierten Oozytenqualität und einer geringeren Blastozystenrate. Die Zielsetzung dieser Arbeit bestand darin, den Einfluss der postovulatorischen Alterung und der ovariellen Alterung im Holstein-Rind (Bos taurus) auf die DNA-Methylierung entwicklungsrelevanter Gene in Eizellen und Embryonen zu untersuchen. Aus Schlachthof-Ovarien wurden Antralfollikeln unterschiedlicher Größe (<2 mm, 3-5 mm und >6 mm) isoliert. Eizellen aus Follikeln der Größe 3-5 mm wurden für 24h (physiologisch) und 48h (gealtert) in vitro gereift (IVM). Die gereiften Oozyten wurden anschließend in vitro fertilisiert und Embryonen im 4-6 Zellstadium generiert. Sowohl in den unreifen Eizellen aus Antralfollikeln unterschiedlicher Größe als auch in den gereiften Oozyten und den Embryonen wurde die Promotormethylierung der Gene bH19, bSNRPN, bZAR1, bDNMT3A, bOCT4, bDNMT3Lo und bDNMT3Ls analysiert. Zur Untersuchung der ovariellen Alterung wurden mittelgroßen Antralfollikel aus Ovarien lebender Rinder (in vivo) unterschiedlichen Alters (9-12 Monate, 37 Jahre und 8-11 Jahre) gewonnen. In den daraus isolierten unreifen Eizellen wurde die DNAMethylierung der Promotorregionen der Gene bTERF2, bREC8, bBCL-XL, bPISD, bBUB1, bDNMT3Lo, bH19 und bSNRPN bestimmt. Als Methode zur Analyse der Promotormethylierung wurde die Limiting Dilution Bisulfit-Sequenzierung angewendet. In unreifen Eizellen aus Antralfollikeln unterschiedlicher Größe (<2 mm, 3-5 mm und >6 mm) konnte ein erhöhtes Auftreten abnormal methylierter Allele in den geprägten Genen bH19 und bSNRPN von Eizellen kleiner Follikel (<2 mm) identifiziert werden. Dieses Ergebnis könnte eine mögliche Ursache einer bereits bekannten und mehrfach beschriebenen geringeren Entwicklungskompetenz von Eizellen kleiner Follikel (<2 mm) auf epigenetischer Ebene darstellen. Die verlängerte Reifungsdauer der IVM-Eizellen hatte eine signifikante Hypermethylierung in der Promotorregion des Gens DNMT3Lo von 48h-gereiften Eizellen zur Folge. Beim Übergang von 48h-gereiften Eizellen zum Embryo konnte eine signifikante Hypomethylierung von CpG7 des stammzellspezifischen Transkripts DNMT3Ls beobachtet werden. Diese CpG-Stelle wies ebenfalls einen signifikanten Anstieg von CpGs mit nicht-eindeutigem Methylierungszustand in unreifen Eizellen mit steigender Follikelgröße auf. Da sich die CpG-Position innerhalb eines Sequenz-Motivs einer Bindungsstelle des Transkriptionsfaktors CREB befindet, könnten die Methylierungsdaten auf eine Interaktion zwischen dem Transkriptionsfaktor CREB und der DNA-Methylierung während der Entwicklung und Reifung der Eizelle sowie der Transition von der Eizelle zum Embryo hindeuten. Die DNA-Methylierungsprofile der untersuchten Gene in unreifen Eizellen aus Kühen unterschiedlichen Alters (9-12 Monate, 3-7 Jahre und 8-11 Jahre) wiesen keine signifikanten Unterschiede zwischen den Altersgruppen auf. Die ovarielle Alterung bei Rindern zwischen 9 Monaten und 11 Jahren zeigte damit keinen Effekt auf die DNA-Methylierung der untersuchten Promotorregionen der Gene bTERF2, bREC8, bBCL-XL, bPISD, bBUB1, bDNMT3Lo, bH19 und bSNRPN. 1 Nach einer simulierten postovulatorischen Alterung durch eine in vitro Reifung für 48h konnte eine Veränderung der DNA-Methylierung der Oozyten-spezifischen (DNMT3Lo) und Stammzell-spezifischen (DNMT3Ls) Promotoren des katalytisch inaktiven Cofaktors von DNMT3A, DNMT3L, beobachtet werden. Die veränderte DNA-Methylierung von DNMT3Ls tritt dabei erst im frühen Embryo in Erscheinung und interagiert vermutlich mit dem Transkriptionsfaktor CREB. Die Veränderungen von DNMT3Lo in Eizellen und DNMT3Ls in den daraus generierten Embryonen lässt vermuten, dass es sich hierbei um eine dynamische Anpassung des Embryos auf äußere Umweltbedingungen der Eizelle über die Methylierung der DNA handelt. 2 ABSTRACT Abstract Postovulatory aging and ovarian aging have been identified as key factors in assisted reproductive techniques (ARTs) and have a lasting effect on reproductive success. Postovulatory aging occurs if the mature egg is not fertilized within its physiological time window. On the other hand, ovarian aging describes the decrease in the follicular reserve with increasing age of the female or the ovary, respectively. Both post-ovulatory aging and ovarian aging result in reduced oocyte quality and lower blastocyst rate. The aim of this thesis was to explore the effects of postovulatory aging and ovarian aging in Holstein cattle (Bos taurus) on the DNA methylation of developmentally important genes in oocytes and embryos. Antral follicles of different sizes (<2 mm, 3-5 mm and> 6 mm) were isolated from slaughterhouse ovaries. Female germ cells from middle-sized follicles (3-5 mm) were matured for 24h (physiological conditions) and 48h (aged conditions) in vitro (IVM). The IVMoocytes were subsequently fertilized in vitro and embryos at the 4-6 cell stage were generated. Promoter methylation of the genes bH19, bSNRPN, bZAR1, bDNMT3A, bOCT4, bDNMT3Lo and bDNMT3Ls was analysed in immature oocytes from antral follicles of different sizes as well as in matured oocytes and the respective embryos. For studying ovarian aging, middle-sized antral follicles were obtained in vivo from animals of different age groups (9-12 months, 3-7 years and 8-11 years). In the extracted immature gametes, the DNA methylation of the promoter regions of bTERF2, bREC8, bBCL-XL, bPISD, bBUB1, bDNMT3Lo, bH19 and bSNRPN was examined. The limiting dilution bisulfite (pyro)sequencing method was applied to determine the promoter methylation of the candidate genes at the single allele level. In immature oocytes from antral follicles of different diameters (<2 mm, 3-5 mm and> 6 mm) an increased occurrence of abnormally methylated alleles of the imprinted genes bH19 and bSNRPN was identified in small follicles (<2 mm). This failure to establish imprinting could be a possible cause of a well-known reduced developmental potential of small follicles (<2 mm) at the epigenetic level. The extended maturation time of the IVM-oocytes resulted in a significant hypermethylation in the promoter region of DNMT3Lo in 48h matured oocytes. After transition from 48h matured oocytes to embryos, a significant hypomethylation of CpG7 of the stem cell specific transcript DNMT3Ls was detected. The same CpG site showed a significant increase of CpGs with unclear methylation state in immature female germ cells with increasing follicular size. This CpG position is located within a potential binding site of the transcription factor CREB. Thus, the methylation data indicates an interaction between the transcription factor CREB and the DNA methylation during development and maturation of oocytes as well as during transition from the oocyte to the embryo. The DNA methylation profiles of the analysed genes in immature oocytes from cows of different age (9-12 months, 3-7 years and 8-11 years) showed no significant differences between age groups. Hence, the ovarian aging in cattle between 9 months and 11 years caused no effect on the DNA methylation of bTERF2, bREC8, bBCL-XL, bPISD, bBUB1, bDNMT3Lo, bH19 and bSNRPN. After a simulated postovulatory aging by in vitro maturation for 48h, a change in the DNA methylation of the oocyte-specific (DNMT3Lo) and stem cell-specific (DNMT3Ls) promoters of the catalytically inactive DNA-methyltransferase DNMT3L was observed. The altered DNA methylation of DNMT3Ls occurs in the early embryo and probably interacts with the transcription factor CREB. The changes of DNMT3Lo in oocytes and DNMT3Ls in the resulting embryos might represent a dynamic adaptation to external environmental conditions. 3 EINLEITUNG 1 Einleitung Lebende Organismen kennzeichnen sich neben einer Vielzahl von Eigenschaften insbesondere durch die Fähigkeit zur Evolution [1]. Evolution im biologischen Sinne kann verstanden werden, als „der Wandel von Merkmalen in Populationen von Organismen, der die Lebenszeit eines einzelnen Individuums überschreitet“[2]. Als molekulares Speichermedium zur Übertragung der zellulären Information hat sich bereits vor 3,5 Milliarden Jahren die Desoxyribonukleinsäure (DNA) etabliert [3]. Die DNA als Träger der Erbinformation des Organismus bzw. der Zelle erlaubt Aussagen über die Vergangenheit (bereits durchlaufene Evolution), Gegenwart (funktionelle Konstitution) sowie eine mögliche Zukunft (Prognose basierend auf der funktionellen Konstitution) der Zelle(n). Die Forschungsdisziplin der Genetik untersucht und erklärt, „wie Erbinformation weitergegeben wird, wie sie molekular aufgebaut ist und wie sie in den Zellen eines Organismus funktioniert“ [4]. Die Teildisziplin der „Molekularen Genetik“ „beschäftigt sich mit der stofflichen Grundlage der Gene, der DNA, ihrer Struktur sowie der Umsetzung ihrer Information und der Regulation der Genaktivität“ [4]. Nach der Identifizierung zahlreicher Gene und der Aufklärung ihrer Funktion tritt zunehmend die Analyse der Regulation und Organisation der Gene in den wissenschaftlichen Fokus. Mechanismen zur zeitlichen und räumlichen Koordination der Genaktivität sind essentiell, um den Organismus bzw. die Zelle flexibel und erfolgreich an sich verändernde äußere Umweltbedingungen anzupassen und letztlich die Chancen zur Weitergabe bzw. Erhaltung der gesamten oder eines Teils der eigenen Erbinformation durch Reproduktion zu erhöhen. Neben der DNA-Sequenz in den Promotobereichen der Gene selbst, werden einige essentielle Regulationsmechanismen der Genaktivität durch epigenetische Veränderungen gesteuert. 1.1 Epigenetik Der Begriff „Epigenetik“ wurde erstmals 1942 von Conrad Waddington verwendet und fasste die kausalen Interaktionen zwischen Genen und ihren Produkten zusammen, die eine phänotypische Expression ermöglichten [5]. Im Laufe der Zeit durchlief diese frühe Definition der Epigenetik einen Wandel und beschreibt heute allgemein alle vererbbaren Veränderungen des Phänotyps, welche nicht durch die primäre DNA-Sequenz verursacht werden [6]. Diese Ursachen umfassen im Wesentlichen die DNA-Methylierung, Histon- 4 EINLEITUNG Modifikationen sowie nicht-kodierende RNA [7]. Innerhalb dieser Mechanismen stellt die DNA-Methylierung die am intensivsten untersuchte epigenetische Modifikation dar [7]. 1.1.1 DNA-Methylierung Als DNA-Methylierung wird im Allgemeinen die kovalente Verknüpfung einer Methylgruppe (-CH3) an eine Nukleotidbase einer DNA bezeichnet. Bisher konnten 3 unterschiedliche Typen der DNA-Methylierung beschrieben werden: N6-Methyladenin (6mA), N4-Methylcytosin (4mC) und 5-Methylcytosin (5mC) [8-10]. Während 6mA und 4mC in Prokaryoten und einzelnen Eukaryoten vorkommen, stellt 5mC die vorherrschende Form der DNA-Methylierung in eukaryotischen Genomen dar [11]. In Säugern tritt 5mC überwiegend innerhalb von Cytosin-Phosphat-Guanin (CpG) Dinukleotiden auf [12]. Evolutionär hat sich die Methylierung der DNA vermutlich in Prokaryoten als Schutzmechanismus bakterieller Zellen vor der Invasion von Fremd-DNA entwickelt und etabliert [13, 14]. Innerhalb der prokaryotischen Restriktion-Modifikation-Systeme, die aus einem Restriktionsenzym (Restriktionsendonuklease) und einem modifizierenden Enzym (Methyltransferase) bestehen, wird die zelleigene DNA an spezifischen Stellen methyliert und damit markiert, wodurch die nicht-modifizierte Fremd-DNA erkannt und abgebaut werden kann [15-17]. Diese primitive Form eines Immunsystems richtet sich gegen für die Zelle potentiell gefährliche, virale Fremd-DNA. Mittels Restriktionsendonukleasen kann die Zelle diese parasitäre DNA unschädlich machen, bevor sie sich in das zelleigene Genom integrieren und die Expression von Virus-Proteinen induzieren kann [14]. Eine mögliche Adaption an dieses Abwehrsystem stellen Plasmide, Prophagen und Transposons dar, die in ihrem Genom eine Methyltransferase ohne dazugehörige Restriktionsendonuklease kodieren [18]. Diese viralen Elemente sind in der Lage ihre DNA zu methylieren und den Schutzmechanismus ihrer prokaryotischen Wirte zu umgehen. Die Existenz und die Vermehrung dieser eigennützigen Fremd-DNAs im Wirt bergen jedoch neben einer potentiellen Schädigung ebenfalls potentielle evolutive Vorteile für die Zelle. Neben direkten Selektionsvorteilen wie beispielsweise kodierten Antibiotikaresistenzen auf einem Plasmid [19], beschleunigen diese mobilen genetischen Elemente die Veränderung des Genoms insgesamt [20, 21]. Dieser „Infektion“ und Expansion mobiler DNA sind bzw. waren sowohl Prokaryoten als auch Eukaryoten ausgesetzt. Während der Anteil dieser parasitären DNA in Bakterien bei 0-21% liegt [22], enthalten eukaryotische Genome 0-65% dieser Sequenzen [21, 23]. Für das menschliche Genom liegen die Schätzungen zwischen 40% [21] bis 65% [24]. Der Großteil 5 EINLEITUNG dieser mobilen Sequenzen im humanen Genom besteht aus Retrotransposons (85%) [25]. Sowohl Retrotransposons als auch andere mobile genetische Elemente enthalten eigene, intrinsische regulatorische Sequenzen wie Promotoren, Spleißstellen, Terminatoren, Enhancer oder Insulatoren [26]. Diese DNA-Abschnitte ermöglichen es, die Expression spezifischer Gene besser zu koordinieren und zu regulieren und sich dynamisch an veränderte äußere Umweltbedingungen anzupassen. Aufgrund des damit einhergehenden Selektionsvorteils wird vermutet, dass die Wirtsgenome im Laufe der Evolution regulatorische Sequenzen von den mobilen Fremd-DNAs adaptierten. Für 20% der evolutionär konservierten regulatorischen Regionen konnten mobile genetische Elemente als Ursprung nachgewiesen werden [27-29]. Gleichzeitig stellen diese mobilen Elemente Abschnitte für Chromatininstabilitäten und genomische Veränderungen dar und bedürfen einer strickten Kontrolle [30, 31]. Als essentieller Mechanismus zur Repression von Transposons insbesondere in der Keimbahn und in undifferenzierten Zellen hat sich die DNAMethylierung etabliert [32, 33]. Die primäre Funktion der Cytosin-Methylierung im Genom von Säugetieren und anderen Wirbeltieren stellt die Suppression mobiler Sequenzabschnitte dar. Neben diesem Abwehrmechanismus übernimmt die DNA-Methylierung jedoch ebenfalls essentielle Funktionen in der Allel-spezifischen Genexpression wie bei der X-Inaktivierung und der genomischen Prägung [34] sowie der Regulation der Genexpression insbesondere während der fetalen Entwicklung (siehe Kapitel 1.1.4). Die chemische Modifikation der Cytosin-Nukleotide wird von der Enzym-Klasse der DNA-Methyltransferasen (DNMTs) übernommen. Bei den DNMTs in Säugern unterscheidet man 3 unterschiedliche Familien: DNMT1, DNMT2 und DNMT3 [35]. Während die Funktion von DNMT2 insbesondere in der Methylierung von Cytosinen in tRNA besteht [36], sind die Enzyme der Familien DNMT1 und DNMT3 maßgeblich an der Methylierung der genomischen DNA beteiligt [35]. DNMT1 übernimmt dabei die Funktion der Aufrechterhaltung der Methylierungsmuster während der Replikation [37] und der Reparatur [38]. Die initiale de novo-Etablierung der Methylierungsmuster wird durch DNMT3a, DNMT3b und DNMT3L realisiert [35, 39, 40]. DNMT3L (DNMT3-Like Protein) stellt hierbei einen katalytisch inaktiven, regulatorischen Faktor dar, der an DNMT3a bzw. DNMT3b bindet und sie unter Interaktion mit Histon H3 an die zu methylierende Stelle leitet [41]. 6 EINLEITUNG 1.1.2 Genomische Prägung Sexualität ist die dominante Form der Reproduktion bei vielzelligen Organismen. Der Unterschied zur nicht-sexuellen Fortpflanzung besteht in der zufälligen Verteilung der väterlichen und mütterlichen Erbanlagen auf die Nachkommen. Diese Durchmischung ermöglicht eine höhere Flexibilität mit der sich die Individuen an neue Umweltbedingungen, Krankheitserreger und Parasiten anpassen können und stellt damit vermutlich den entscheidenden evolutionären Vorteil der Sexualität dar. Ein typisches Merkmal der sexuellen Reproduktion höherer Pflanzen und Tiere ist die Ausbildung zweier unterschiedlicher Geschlechtszellen (Gameten) [42]. Bei der Verschmelzung einer mütterlichen und einer väterlichen Keimzelle zur Zygote werden die haploiden GametenGenome zu einem diploiden Genom der Zygote kombiniert. Durch TransplantationsExperimente mit Vorkernen von Mäusen konnte aufgedeckt werden, dass das väterliche und das mütterliche Genom in unterschiedlicher Weise zum Genom der Zygote beitragen [4346]. Nach Generierung von Embryonen mit zwei ausschließlich weiblichen Vorkernen (maternal uniparental) und zwei ausschließlich männlichen Vorkernen (paternal uniparental) zeigten sich Unterschiede in der Entwicklung der beiden Arten uniparentaler Embryonen. Während die maternal uniparentalen Zygoten überwiegend Gewebe embryonischen Ursprungs ausbildeten, bestanden die paternal uniparentalen Embryonen überwiegend aus extraembryonischem Gewebe [47]. Mit Beginn der 1990er Jahre konnte diese genomische Prägung (engl. „Imprinting“) anhand der ersten geprägten Gene, welche differenziell von maternalen und paternalen Chromosomen exprimiert wurden, bestätigt werden [48-50]. Bis heute konnte die genomische Prägung in Pflanzen (Bedecktsamer), Beuteltieren und höhere Säugetieren (Plazentatieren) nachgewiesen werden [51]. Ähnlich wie in Säugetieren beeinflusst die Expression der geprägten Gene in Pflanzen die Entwicklung des Endosperms, einem extraembryonischem Gewebe, welches die Entwicklung des Embryos unterstützt [47]. Der evolutionäre Grund zur Ausbildung der genomischen Prägung ist nicht bekannt. Weil die geprägten Gene sowohl bei Säugetieren als auch bei Pflanzen an der Versorgung des Embryos mit Nährstoffen beteiligt sind, wird vermutet, dass die genomische Prägung einen Regulationsmechanismus zur Verteilung mütterlicher Ressourcen darstellt [47, 52]. Da paternal exprimierte Gene das fetale Wachstum verstärken wohingegen maternal exprimierte Gene das Wachstum des Embryos abschwächen, wird angenommen, dass dieses System in einer antagonistischen Weise funktioniert. Während die paternal exprimierten 7 EINLEITUNG Gene zum Ziel haben der Mutter mehr Ressourcen zum Wohle der Entwicklung des Nachkommen zu entziehen, erhalten die maternal exprimierten Gene die Ressourcen der Mutter, um sie zwischen mehreren Nachkommen zu verteilen und den weibliche Reproduktionserfolg zu maximieren [51]. 1.1.2.1 Funktion und Regulation geprägter Gene In höheren Säugetieren konnten bisher etwa 100 geprägte Gene identifiziert werden [53]. Die Transkripte der meisten dieser Gene wurden in der Plazenta und dem Gehirn nachgewiesen [54, 55]. Darüber hinaus werden geprägte Gene auch in anderen Organen wie Muskeln, der Lunge, den Nieren und dem Auge exprimiert [47]. Zwischen den unterschiedlichen Arten der Säugetiere werden nicht zwingend gleiche Gene geprägt. Beispielsweise liegen in der Maus geprägte Gene vor, die im menschlichen Genom keine Prägung vorweisen [47]. Die im Gehirn exprimierten Gene mit Prägung stehen im Zusammenhang mit der Modulation von Stoffwechselprozessen, dem Verhalten, Lernen sowie der mütterlichen Brutpflege [47]. Charakteristisch für eine gestörte Prägung sind jedoch vorallem eine beeinträchtigte Entwicklung und Physiologie der Plazenta. Eine fehlende Expression des geprägten Gens Igf2 führt beispielsweise zu einem gestörten Nährstofftransport zum wachsenden Fötus [56]. Igf2 wird sowohl in der Maus als auch im Menschen paternal exprimiert und bildet einen Wachstumsfaktor der Plazenta und des Embryos [57]. Knockout-Mäuse ohne das Plazenta-spezifische Transkript von Igf2 sind zum Zeitpunkt der Geburt 40% kleiner als ihre normalen Artgenossen [58]. Die Regulation von Igf2 erfolgt über eine differenziell methylierte Region (DMR) auf Chromosom 7 bei der Maus und auf Chromosom 11 beim Menschen [51]. 8 CTCF 1 2 CTCF Igf2 CTCF CTCF EINLEITUNG 3 4 H19 Enhancers CH3 CH3 CH3 CH3 CH3 CH3 Igf2 1 2 3 4 H19 Enhancers Abbildung 1: Regulation des geprägten H19/Igf2-Lokus in der Maus. Die schwarzen Pfeile mit den Bezeichnungen der Gene zeigen die Position der Gene und die Richtung ihrer Transkription im aktiven Zustand. Pfeile von rechts nach links deuten die Interaktion zwischen den Enhancer-Sequenzen und den Genen an, die ihre Expression induziert. Der gelbe Balken markiert die ICR mit 4 Bindungsstellen für das Zinkfinger-Isolator-Protein (CTCF). Auf dem maternalen Chromosom liegt die ICR unmethyliert vor, wodurch CTCFs gebunden werden und die Interaktion zwischen Enhancern und Igf2 blockieren, was zur Expression von H19 führt. Auf dem paternalen Chromosom liegt die ICR methyliert vor. Dadurch können keine CTCFs binden, wodurch die Enhancer die Expression von Igf2 induzieren können, während H19 nicht exprimiert wird. Abbildung übernommen aus [59] und modifiziert. DMRs weisen eine hohe Dichte an CpG-Dinukleotiden (CpGs) auf, deren Cytosine einen methylierten oder unmethylierten Zustand einnehmen können. CpGs sind nicht gleichmäßig über das Genom verteilt, sondern treten gehäuft in CG-reichen Sequenzabschnitten auf, die als CpG-Inseln bezeichnet werden [60]. CpG-Inseln, die im Gegensatz zu DMRs für gewöhnlich unmethyliert vorliegen, sind vorwiegend mit Promotoren von sog. Haushaltsgenen (engl. „Housekeeping genes“) assoziiert [61]. Dabei handelt es sich um Gene, die ubiquitär exprimiert werden und selten gehemmt werden. Übernimmt eine DMR die Regulation mehrerer geprägter Gene wird diese Region auch als ICR (engl. „imprinting control region“) bezeichnet [62-67]. Dementsprechend treten etwa 80% der bekannten geprägten Gene in Clustern mit anderen geprägten Genen auf, die eine Region von 1 Mb oder mehr umfassen können [51, 59]. Während maternal methylierte ICRs am Promotor lokalisiert sind, befinden sich paternal methylierte ICRs in intergenischen Regionen [47]. Das paternal exprimierte Igf2 ist 90 kb entfernt von der maternal exprimierten Gensequenz von H19 [48, 49]. Beide Gene teilen sich eine Enhancer-Sequenz stromabwärts von H19 (Abbildung 1). Die ICR, die beide Gene reguliert, umspannt eine Region von 2-4 kb stromaufwärts der Transkriptionsstartstelle von H19 [68]. Sie ist paternal methyliert und liegt intergenisch zwischen Igf2 und H19. Darüber hinaus enthält sie mehrere Bindungsstellen für das Zinkfinger-Isolator-Protein (CTCF), welches ausschließlich an das 9 EINLEITUNG unmethylierte maternale Chromosom bindet [69, 70]. Die Bindung von CTCF auf dem maternalen Chromosom blockiert die Interaktion des Enhancers mit dem Promotor von Igf2, wodurch das Gen deaktiviert bleibt (Abbildung 1). Stattdessen interagiert der Enhancer auf dem maternalen Chromosom mit H19 und führt zur Expression des Gens. Auf dem paternalen Chromosom hingegen liegt die ICR methyliert vor, wodurch die Bindung von CTCF verhindert wird, die Interaktion des Enhancers mit dem Igf2 nicht blockiert wird und somit Igf2 exprimiert wird [47, 57] (Abbildung 1). Neben diesem Isolator-Model lassen sich die ICRs der Mehrheit der geprägten Gene in eine zweite Klasse einteilen, bei welcher das ICR als Promotor einer nicht-kodierenden RNA (ncRNA) fungieren [59]. Das am besten untersuchte Beispiel dieser Klasse von ICRs stellt das Igf2r-Cluster dar [71]. Das ICR befindet sich hier in einer CpG-Insel innerhalb eines Introns des Igf2r-Gens [72]. Exprimiert wird das Gen vom maternalen Chromosom, welches eine intronische Methylierung aufweist [50]. Auf dem paternalen Chromosom liegt das ICR dagegen unmethyliert vor und dient als Promotor einer langen nicht-kodierenden RNA (Airn), die in Antisense-Richtung zum Igf2r-Gen transkribiert wird [63, 73]. Diese AntisenseTranskription passiert den Igf2r-Promotor und hemmt die Expression des Gens vom paternalen Chromosom [74]. Neben Igf2r werden zwei weitere Gene stromabwärts ebenfalls vom maternalen Chromosom exprimiert und vom paternalen Chromosom gehemmt. Bei diesen Genen wird vermutet, dass Airn hemmende Histonmodifikationen induziert, welche den Promotor auf dem paternalen Chromosom inaktivieren [75]. Die Regulation geprägter Gene durch die Transkription einer nicht-kodierenden RNA in Abhängigkeit von DNAMethylierung ist ebenfalls für die geprägten Cluster Kcnq1 [67], Snrpn [76] und Gnas [65, 77] beschrieben. Das Wissen über die Funktion und Regulation geprägter Gene basiert zu einem erheblichen Teil auf Genotyp-Phänotyp-Studien von Patienten mit Störungen der genetischen Prägung sowie Experimenten in Mäusen. Prinzipiell stimmen dabei die Merkmale bzw. Symptome der Patienten überein mit denen der entsprechenden MausMutanten [47]. Welche Krankheiten und Krankheitssymptome durch eine Störung der genetischen Prägung verursacht werden, wird im Folgenden kurz erläutert. 1.1.3 Fehlregulation geprägter Gene Störungen der korrekten genomischen Prägung führen zu einer Veränderung der Expression der betreffenden Gene und zu einer pathologischen Veränderung des Phänotyps. 10 EINLEITUNG Erkrankungen dieser Art sind mit einer Inzidenz von 1-10 in 100.000 Kindern selten und kennzeichnen sich durch ein auffällig verändertes Größenwachstum, verschiedene Arten von Fehlbildungen, mentaler Retardierung und/oder Krebs im Kindesalter [78]. Dabei führen unterschiedliche Prägungen am gleichen Genlokus zu unterschiedlichen Imprintingerkrankungen mit deutlich verschiedenartigen Phänotypen. Die bekanntesten Erkrankungen, die mit einer gestörten genomischen Prägung assoziiert sind, sind das PraderWilli-Syndrom (PWS), Angelman-Syndrom (AS), Silver-Russell-Syndrom (SRS), BeckwithWiedemann-Syndrom (BWS), der transiente neonatale Diabetes mellitus, der Pseudohypoparathyroidismus Typ 1b sowie das paternale bzw. maternale UPD 14-Syndrom [47]. 1.1.3.1 Prader-Willi-Syndrom und Angelman-Syndrom Das Prader-Willi-Syndrom (PWS) und das Angelman-Syndrom (AS) sind die ersten bekannten Erkrankungen, die in Verbindung mit geprägten Genen gebracht wurden. Beide Krankheiten äußern sich durch neurogenetische Fehlfunktionen, die mit einer gestörten Prägung der Gene des langen Arms von Chromosom 15 assoziiert sind. In der chromosomalen Region 15q11.2 befindet sich ein Cluster geprägter Gene, die ausschließlich vom paternalen oder vom maternalen Chromosom exprimiert werden. Die Inzidenz beider Syndrome liegt bei 4-7 in 100000 Kindern.[79, 80] Das klinische Bild von PWS äußert sich durch ein geringes Geburtsgewicht, Trinkschwäche im ersten Lebensjahr, einer Hyperphagie und Adipositas im frühen Kindesalter, geistige Behinderung, muskuläre Hypotonie, Hypogonadismus und Kleinwuchs. Bei 25-30% der Patienten liegt eine maternale uniparentale Disomie des Chromosoms 15 [upd(15)mat] vor. Die Ursache dafür liegt in einer Fehlsegregation während der maternalen Meiose gefolgt von einem mitotischen Verlust des paternalen Chromosom 15 nach der Fertilisation. Dieser Verlust hat zur Folge, dass geprägte Gene, die vom paternalen Chromosom exprimiert werden, fehlen. Einige der dort lokalisierten Gene, die vermutlich maßgeblich zum Phänotyp von PWS beitragen, sind die C/D box small nucleolar (sno-)RNA SNORD116 Gene sowie SNURF-SNRPN, welches ebenfalls für snoRNA codiert. In welchem Ausmaß diese Gene zum Phänotyp von PWS beitragen ist nicht bekannt. 1% der PWS-Fälle ist auf Imprinting-Defekte zurückzuführen. [79, 80] 11 EINLEITUNG Die Symptome des AS weisen eine Mikrozephalie, Ataxie, Epilepsie, faziale Dysmorphien, ausbleibende Sprachentwicklung und geistige Behinderung auf. 2-5% der betroffenen ASPatienten zeigen eine paternale uniparentale Disomie des Chromosoms 15 [upd(15)pat]. Ursächlich dafür ist vermutlich in den meisten Fällen eine maternale Fehlsegregation mit einer anschließenden postzygotischen Duplikation des paternalen Chromosoms 15 [81]. Als Resultat fehlt eine aktive maternale Kopie des Gens UBE3A, was nachgewiesen zum ASPhänotyp führt. UBE3A kodiert eine E3 Ubiquitin-Ligase, die ausschließlich vom maternalen Chromosom im Gehirn exprimiert wird [82, 83]. Der Mechanismus, wie die gewebsspezifische Prägung von UBE3A reguliert wird, ist bisher nicht bekannt. Da das 3‘Ende des SNURF-SNRPN-Transkripts die Antisense-RNA von UBE3A darstellt, wird jedoch vermutet, dass das paternal exprimierte SNURF-SNRPN-sense/ UBE3A-anti-sense Transkript an der Hemmung des paternalen UBE3A-Allels beteiligt ist [84]. Zur diagnostischen Identifizierung von PWS als auch von AS stellt die Untersuchung der DNAMethylierung des SNURF-SNRPN-Lokus derzeit die sensitivste Methode dar. Während gesunde Individuen hier ein methyliertes und ein unmethyliertes Allel zeigen, lässt sich bei PWS nur das maternale methylierte Allel und bei AS nur das paternale unmethylierte Allel nachweisen [84]. 1.1.3.2 Silver-Russel-Syndrom und Beckwith-Wiedemann-Syndrom Die beiden Imprintingerkrankungen Silver-Russel-Syndrom (SRS) und Beckwith-WiedemannSyndrom (BWS) zeigen, ähnlich wie PWS und AS, ein gegensätzliches genetisches wie klinisches Bild. Ursächlich für beide Krankheiten ist in den meisten Patienten eine Veränderung der geprägten Genregion auf Chromosom 11p15.5 [85, 86]. Der Phänotyp des SRS kennzeichnet sich durch eine prä- und postnatale Wachstumsverzögerung mit altersentsprechendem Kopfumfang (relative Makrozephalie), ein kleines dreieckiges Gesicht, Asymmetrie und einer Entwicklungsverzögerung. Den ersten Nachweis einer Verbindung dieser Symptome mit der Expression geprägter Gene lieferte eine maternale uniparentale Disomie des Chromosoms 7 [upd(7)mat], die bei 5-10% der Patienten auftritt [79, 87]. Inwiefern die fehlende Expression der paternalen Gene bzw. die Überexpression der maternalen Gene zum Phänotyp des SRS führt ist jedoch derzeit noch Gegenstand der Forschung [86]. Molekulare Erkenntnisse im Zusammenhang zwischen Genotyp und Phänotyp von SRS konnte man jedoch bei der häufigsten Ursache der Krankheit 12 EINLEITUNG (35-60%), einer Veränderung des Chromosoms 11p15.5, gewinnen [79]. Chromosom 11p15 enthält zwei Imprintingkontrollregionen (ICR1 und ICR2), die die Expression von Genen steuern, welche entscheidende Funktionen im prä- und postnatalen Wachstum übernehmen. ICR1 reguliert die Expression des paternal exprimierten Igf2 und des maternal exprimierten H19. Die Funktion von Igf2 als fetaler Wachstumsfaktor ist bekannt, wohingegen die physiologische Rolle der nicht-translatierten RNA H19 unbekannt ist. Es wird jedoch vermutet, dass die Hypomethylierung von H19 zur Abschwächung der Expression von Igf2 führt und eine Wachstumsverzögerung verursacht. Das ausschließlich auf dem maternalen Allel methylierte ICR2 reguliert die reziproke Expression von CDKN1C, KCNQ1 und weiterer Gene [85, 86]. Während eine Hypomethylierung von ICR1 in etwa 40% der Fälle in SRS-Patienten nachgewiesen wird, zeigt ICR2 eine Hypomethylierung in 35-50% von BWSErkrankten [79, 86]. Die phänotypischen Merkmale von BWS sind ein prä- und postnataler Großwuchs (Makrosomie), Makroglossie, Ohrfurchen, Bauchwanddefekte und eine Viszeromegalie. Die Ausprägung des Phänotyps hängt hier vom molekularen Subtyp ab. Die genauen molekularen Interaktionen der betroffenen Genprodukte, die zum Phänotyp von BWS führen, konnten bisher nicht aufgeklärt werden [79, 85, 86]. Beide ICRs sind Gegenstand dieser Arbeit. Eine aberrante Methylierung der homologen Regionen beim Rind wurde nach SCNT mit dem sog. „Large offspring syndrome“ (LOS) (vgl. 1.3.3) beschrieben. 1.1.4 Epigenetische Genomreprogrammierung Die genomische Prägung führt zu spezifischen epigenetischen Veränderungen der maternalen und paternalen Allele. Diese Prägungsmuster sind essentiell für die postzygotische Phase des sich entwickelnden Embryos nach der Fertilisation. Der neu entstehende Organismus bildet seinerseits ebenfalls Keimzellen, die seine geschlechtsspezifischen Prägungsmuster tragen. Um dies zu realisieren bedingt der Vorgang der Prägung einen Mechanismus, der das Epigenom der sich entwickelnden Keimzellen in einen neutralen Ausgangszustand zurückversetzt. Die Entfernung der parentalen Prägungsmuster ermöglicht anschließend die geschlechtsspezifische Prägung des neu entstandenen Organismus. Die Umsetzung dieses Prozesses erfolgt durch die epigenetische Genomreprogrammierung. Die Reprogrammierung betrifft neben den geprägten Regionen auch Keimzell-spezifische Gene, die es dem Genom des Spermiums und der Eizelle 13 EINLEITUNG ermöglichen, eine totipotente Zygote zu bilden. Da die epigenetische Genomreprogrammierung während der embryonalen Entwicklung erfolgt, liegen die meisten Daten zu diesem Mechanismus aus Studien im Maus-Modell vor. Maus-Embryonen zeigen zwei Phasen epigenetischer Reprogrammierung. Die erste frühe Phase beginnt unmittelbar nach der Befruchtung und findet in der Präimplantationsphase statt. Die zweite spätere Phase setzt nach dem embryonalen Stadium E7-E8 ein und betrifft ausschließlich die Primordialen Keimzellen (PGCs) (Abbildung 2). In beiden Entwicklungsphasen besteht der Reprogrammierungs-Prozess aus einer anfänglichen Demethylierung gefolgt von einer anschließenden Remethylierung. hoch Embryo DNA-Methylierung Befruchtung Methylierte Allele von DMRs Spermatozoen Gereifte Oocyten niedrig Primordiale Keimzellen Prospermatogonien Primäre Oocyten Unmethylierte Allele von DMRs E1 E4 - 5 E7 - 8 E15 - 16 GEBURT PUBERTÄT Entwicklungsphase Abbildung 2: Epigenetische Reprogrammierung in der Maus. Die epigenetische Reprogrammierung erfolgt in zwei Phasen. Sie beginnt unmittelbar nach der Befruchtung in der Präimplantationsphase. Zunächst werden das maternale (rot) und das paternale (blau) Genom demethyliert. Dabei entgehen die geprägten gDMRs der Demethylierung. Die Remethylierung erfolgt ab E4-E5. Etwa um den Zeitpunkt E7-E8 setzt die Demethylierung in den Primordialen Keimzellen (PGCs) ein und betrifft genomweit alle Regionen. Die Remethylierung in den PGCs erfolgt geschlechtsspezifisch. Bei männlichen Individuen (blau) wird die Etablierung der neuen DNA-Methylierungsmuster bis zur Geburt abgeschlossen. In der weiblichen Keimbahn erfolgt die Remethylierung ab der Geburt kontinuierlich mit der Entwicklung der Eizelle. Abbildung übernommen aus [8890] und modifiziert. Nach der Befruchtung setzt die Demethylierung der beiden Vorkerne ein und entfernt die DNA-Methylierungsmuster des väterlichen Genoms schneller als die des mütterlichen Genoms. Dabei werden die paternalen Regionen durch einen aktiven DemethylierungsMechanismus vermutlich in Verbindung mit der Oxidation durch Tet-Proteine demethyliert [91]. Beim maternalen Genom geht man davon aus, dass die DNA-Methylierungsmuster „passiv“ durch Replikation und ausbleibende Erhaltung der methylierten CpGs fortschreitend mit jeder neuen Zellteilung entfallen [89]. Diesem Prozess der Demethylierung nach der Befruchtung entgehen die geprägten Regionen des paternalen und maternalen Genoms und bleiben erhalten. Die daraus resultierende erhaltene parental-Allel-spezifische Methylierung 14 EINLEITUNG führt zu einer parental-Allel-spezifischen Expression der geprägten Gene im frühen Embryonalstadium. Die Demethylierung aller anderen DNA-Regionen mit Ausnahme der geprägten Sequenzen ist zum Zeitpunkt E4 bis E5 abgeschlossen. Die Remethylierung erfolgt im Zuge der Differenzierung der somatischen Zellen bzw. Zelllinien zwischen E4/E5 und der Geburt [89]. Die Demethylierung der PGCs beginnt parallel mit ihrer Proliferation und Migration in die Genitalleiste im Stadium E7.5. Dieser Vorgang betrifft global das gesamte Genom einschließlich der geprägten Regionen zeitgleich und unabhängig vom elterlichen Ursprung und dauert bis zum Zeitpunkt E15/E16 an. Die anschließende Etablierung der DNAMethylierungsmuster in den PGCs erfolgt geschlechts-spezifisch und betrifft sowohl die geprägten Gene als auch entwicklungsrelevante nicht-geprägte Gene. In männlichen Feten findet die Remethylierung und Prägung bereits pränatal vor der Meiose im mitotisch arretierten Stadium (G1-phase; Prospermatogonien) statt und ist bis zur Geburt abgeschlossen [89]. In weiblichen Feten treten die primären Oozyten nach der Demethylierung in die Meiose ein und verbleiben in der Prophase-I im Diplotän-Stadium. Die Etablierung der Eizell-spezifischen Methylierungsmuster setzt in den primären Oozyten erst nach der Geburt mit der zyklischen Rekrutierung der Eizellen (vgl. 1.2.2) ein und wird zeitlebens bis zur Menopause mit dem Prozess der Follikelreifung abgeschlossen [92]. Mit der Verschmelzung von Eizelle und Spermium bzw. der Befruchtung schließt sich der Reprogrammierungszyklus. Damit stellt die epigenetische Genomreprogrammierung in einer zyklischen Weise sicher, dass die essentielle, parental-spezifische Prägung in den somatischen Zellen erhalten bleibt, während sie in den PGCs geschlechts-spezifisch angepasst wird. 1.2 Oogenese und Oozytenmaturation Die Eizelle und das Spermium sind die zentralen Zellen der Reproduktion und Entwicklung in Säugern. Eine der wichtigsten Aufgaben der Eizelle besteht darin, sowohl das Genom der Eizelle selbst als auch das des Spermiums zu programmieren, um letztlich das neue totipotente embryonale Genom zu formen. Zusätzlich zu dieser Eigenschaft stellt die weibliche Keimzelle die einzige Zell-Art im erwachsenen Organismus dar, die in der Lage ist das Genom einer somatischen Zelle aus einem differenzierten Stadium in einen totipotenten Zustand zu versetzen [93]. Um diese herausragende Fähigkeit, die sog. 15 EINLEITUNG Entwicklungskompetenz, zu erlangen, durchlaufen die Vorläufer der befruchtungsfähigen Oozyte einen einzigartigen Entwicklungsprozess, der durch die Oogenese und Follikulogenese sowie die finale Eizellreifung repräsentiert wird. 1.2.1 Allgemeine Oogenese Die Oogenese beschreibt im Allgemeinen die Entwicklung einer Oogonie zu einer reifen, befruchtungsfähigen Oozyte [94]. Dabei erlangt die Eizelle die Fähigkeit, sich nach der Befruchtung durch ein Spermium zu einem Embryo und damit zu einem neuen Organismus zu entwickeln. Um dieses Entwicklungspotential zu gewinnen, muss die Oozyte eine Reihe von Veränderungen durchlaufen. Dies schließt insbesondere die Reduktionsteilung ein, durch welche der diploide Chromosomensatz zu einem haploiden reduziert wird. Dieser Vorgang ist nötig um die diploide Gendosis des adulten Organismus nach der Verschmelzung der Oozyte und des Spermiums aufrechtzuerhalten. Die Oogenese beginnt bereits pränatal im Mutterleib und wird erst mit Erlangung der Geschlechtsreife bzw. in der Pubertät abgeschlossen [94]. Die Oogonien teilen sich zunächst in der frühen Fetalphase mitotisch mit einer speziesspezifischen Anzahl von Teilungen. In der humanen Oogenese wird die maximale Anzahl an Oogonien mit geschätzt 5 Millionen Zellen etwa in der 20. Schwangerschaftswoche erreicht [95, 96]. Von diesem Zeitpunkt an reduziert sich die Anzahl der Zellen jedoch rapide und umfasst bei der Geburt etwa noch 295 000 [97]. Nach der mitotischen Teilungsphase gehen die Oogonien in die Prophase der Meiose I über und werden fortan als primäre Oozyten bezeichnet. Die Prophase von MI wird in 5 sequentielle Stufen unterteilt: Leptotän, Zygotän, Pachytän, Diplotän und Diakenese [94]. Nach Abschluss der homologen Rekombination der elterlichen Chromosomen, wird die Meiose im letzten Stadium der Prophase, dem Diplotän arretiert. Aufgrund der netzartigen Struktur des Zellkerns (griech. diktyon = Netz) wird dieses Stadium auch als Diktyotän bezeichnet. Der Zellkern der diplotänen Oozyte wird auch Keimbläschen oder Germinalvesikel (engl.: germinal vesicle, GV) genannt. Im Diktyotän verharren die primären Oozyten bis zum Eintritt der Pubertät. Mit Beginn der Geschlechtsreife wird die Prophase durch einen starken Anstieg des luteinisierenden Hormons (LH) etwa zwölf Stunden vor der Ovulation fortgesetzt. Dabei löst sich die Hülle des Kerns auf (engl.: germinal vesicle breakdown; GVBD) und die Chromosomen bilden eine Metaphaseplatte. Die Chromatiden werden dann paarweise voneinander getrennt und es 16 EINLEITUNG kommt zu einer asymmetrischen Zellteilung. Dabei wird ein Chromosomensatz von einer Kernmembran umschlossen und verbleibt mit nahezu dem gesamten Zytoplasma in der nun als sekundär bezeichneten Oozyte, während der andere Chromosomensatz in einer kleinen, funktionslosen Zelle, dem sog. ersten Polkörper, abgebaut wird. Die sekundäre Oozyte durchläuft eine zweite meiotische Teilung. Diese beginnt kurz vor der Ovulation und wird zunächst nur bis zum Stadium der Metaphase II (MII) passiert. Die Fortsetzung der zweiten Reifeteilung erfolgt erst, wenn die sekundäre Oozyte von einem Spermium befruchtet wird. Wie bereits bei der ersten Reifeteilung wird dann ein Chromosomensatz im sog. zweiten Polkörper abgeschnürt und abgebaut. Der verbliebende haploide Chromosomensatz wird in der befruchteten Oozyte von einer Kernmembran umgeben und bildet den weiblichen Vorkern, womit die Reifeteilung der Eizelle abgeschlossen ist [94]. 1.2.2 Follikulogenese beim Rind (Bos taurus) Während der bovinen Oogenese wird die Oozyte bereits zu Beginn der ersten Meiose etwa um den 90sten Tag nach der Befruchtung von einer einschichtigen Lage somatischer Zellen (Granulosazellen) umschlossen und bildet damit einen Ovarialfollikel bzw. Primordialfollikel [98]. Der Begriff Follikel bezeichnet eine Einheit aus einer Eizelle und mehreren sie umgebenden somatischen Zellen und stellt das funktionelle Grundelement des Ovars dar [99]. Die Oozyte verbleibt bis zur Ovulation im Follikel und steht dabei in einem intensiven Stoffaustausch mit den Follikelzellen, der essentiell für die Entwicklung und Reifung der Eizelle ist. Mit Bildung des Primordialfollikels beginnt die Follikulogenese, die die Entwicklung des Primordialfollikels zum ovulationsfähigen Graafschen Follikel beschreibt [100]. In den Ovarien von Rindern liegen grundsätzlich zwei unterschiedliche Follikelpopulationen vor [101]. Eine Kategorie bilden dabei die Primordialfollikel, die sich in einem ruhenden Stadium befinden und Follikel eines nicht-wachsenden Pools repräsentieren. Die zweite Klasse umfasst alle Follikelstadien, die sich über das Stadium des Primordialfollikels hinaus entwickeln und damit den wachsenden Pool bilden [101]. Der Übergang vom nicht-wachsenden in den wachsenden Pool erfolgt durch eine initiale Rekrutierung von Primordialfollikeln (Abbildung 3) [102]. 17 EINLEITUNG INITIALE REKRUTIERUNG ZYKLISCHE REKRUTIERUNG Ovulation Selektion & Dominanz Atresie Graafscher Follikel Maturation atretisch Antral Antral Sekundär atretisch Primär Primordial 20. SSW Erschöpfung der Follikelreserve (kontinuierliche initiale Rekrutierung) Geburt (kontinuierliche initiale Rekrutierung) (Depletion) Pubertät Abbildung 3: Follikulogenese. Ab der 20. Schwangerschaftswoche werden mehrere Primordialfollikel zeitgleich durch die zyklische Rekrutierung zur Weiterentwicklung aktiviert. Bis zur Pubertät entwickeln sich die Follikel nur bis zum Antralstadium und gehen in die Atresie über. Mit Eintritt der Pubertät setzt die zyklische Rekrutierung ein. Dadurch geht eine Kohorte von Antralfollikeln in die Phasen der Selektion und Dominanz über. Dabei entwickelt sich der dominante Follikel zum Graafschen Follikel, während alle anderen Foolikel atretisch werden. Abbildung übernommen aus [102] und modifiziert. In Rinder-Ovarien beginnt diese initiale Rekrutierung bereits in der 20. Schwangerschaftswoche [103]. Dabei veranlassen bisher unbekannte Faktoren einige Primordialfollikel zum Wachstum und zur Weiterentwicklung zum primären, sekundären und tertiären Follikelstadium (Abbildung 4). Ein Tertiär-Follikel wird aufgrund seines flüssigkeitsgefüllten Hohlraums, dem sog. Antrum, auch als Antralfollikel bezeichnet [99]. Antralfollikel gehen vor dem Einsetzen der Geschlechtsreife immer in die Atresie über und degenerieren [104]. Auch im geschlechtsreifen Rind stellt die Atresie bzw. die Rückbildung das „übliche“ Schicksal eines Antralfollikels dar [105]. Mit Beginn der Pubertät setzt jedoch zusätzlich zur initialen Rekrutierung die sog. zyklische Rekrutierung ein [102, 104]. Dabei wird eine Gruppe von 3 bis 10 Antralfollikeln ausgelöst durch einen Anstieg des follikelstimulierenden Hormons (FSH) zunächst vor der Atresie bewahrt und zur Weiterentwicklung geführt [106, 107]. Der zyklischen Rekrutierung schließt sich unter abnehmender FSH-Konzentration die Phase der Selektion an [108, 109]. Während dieser Selektionsphase beginnt ein Antralfollikel der Kohorte stärker und schneller zu wachsen als alle anderen Follikel, die zeitgleich von der zyklischen Rekrutierung aktiviert wurden. Die Auswahl dieses dominanten Follikels hängt insbesondere von der FSH- und LH-Sensitivität ab [106]. Die wachsenden Antralfollikel der Gruppe verursachen die Abnahme des FSH-Spiegels bei gleichzeitiger FSH-Abhängigkeit. Die größten Follikel produzieren Estradiol und Inhibin, die maßgeblich zur Reduktion von FSH beitragen [110]. Einzig der Follikel mit einer ausreichenden Anzahl an FSH-Rezeptoren ist in der Lage sich unter diesen reduzierten FSHBedingungen weiterzuentwickeln [106]. Etwa 8h vor der Selektion des dominanten Follikels 18 EINLEITUNG beginnen die Granulosazellen LH-Rezeptoren zu exprimieren. LH stimuliert zusätzlich die Poduktion von Estradiol und damit die Abnahme der FSH-Konzentration [111]. Dadurch nimmt die FSH-Abhängigkeit des dominanten Follikels stetig ab während gleichzeitig die LHSensitivität zunimmt [106, 112, 113]. Der sich unter diesen Bedingungen weiterentwickelnde dominante Follikel nimmt in der letzten Phase der Follikulogenese, der Dominanz, deutlich an Größe zu, während alle anderen Follikel der Gruppe atretisch werden [114-117]. Zu Beginn der zyklischen Rekrutierung treten Antralfollikel mit einem Durchmesser von 4-5 mm in die Selektionsphase ein, in der sie eine Größe von durchschnittlich 7,7-8,5 mm annehmen [104]. Der dominante Follikel wächst anschließend bis zu einem Durchmesser von 12 - 20 mm, wobei er die Fähigkeit zur Ovulation bei einer Größe von 10 mm erlangt und als Graafscher Follikel bezeichnet wird [104]. Die Abfolge aus zyklischer Rekrutierung, Selektion und Dominanz tritt in regelmäßigen Zeitabständen mehrfach innerhalb eines Menstruationszyklus im Rind auf. Da sich dabei stetig immer weniger Follikel der Kohorte weiterentwickeln und die Anzahl der sich entwickelnden Follikel sozusagen „abebbt“, wird die Abfolge dieser drei Phasen auch als Follikelwelle bezeichnet. Im Verlauf eines interovulatorischen Intervals konnten im Rind 2 bis maximal 4 auftretende Follikelwellen beobachtet werden, wobei 2 und 3 Wellen am häufigsten auftreten [104, 118-120]. Ausgelöst wird eine Follikelwelle mit Beginn der zyklischen Rekrutierung durch einen Anstieg des follikelstimulierenden Hormons (FSH). Nach Durchlaufen der Selektion und Dominanz erfolgt die Ovulation ausschließlich durch einen Anstieg des luteinisierenden Hormons (LH) [104]. Bleibt diese Ausschüttung von LH aus, beginnt der dominante Follikel sich zurückzubilden und erlaubt die Rekrutierung einer neuen Kohorte bzw. Welle [120, 121]. Kommt es zu einer LH-Spitze wird die Meiose I in der Eizelle des präovulatorischen Follikels wiederaufgenommen und die Ovulation ausgelöst [122]. Beim Eisprung expandieren die Kumuluszellen, der Kontakt zwischen der Corona radiata und der Oozyte wird gelöst und es kommt zur Bildung des Perivitellinen Spalts bevor letztlich die Eizelle in den Eileiter entlassen wird [123, 124]. Die im Ovar verbleibenden Follikelzellen (Granulosazellen und Thekazellen) bilden den Gelbkörper (Corpus luteum), der das für das Einsetzen und die Aufrechterhaltung der Schwangerschaft essentielle Hormon Progesteron produziert. Durch Progesteron wird u.a. ein Anstieg der LH-Konzentration unterdrückt, wodurch eine erneute Ovulation verhindert wird. Bleibt die Befruchtung der Eizelle aus, bildet sich der Gelbkörper 16 bis 18 Tage nach dem Eisprung unter dem Einfluss des luteolytischen Faktors Prostaglandin F 2α 19 EINLEITUNG (PGF2α) zurück und wird abgebaut. Die Luteolyse und die damit verbundene Absenkung des Progesteron-Spiegels erlaubt anschließend eine erneute Ovulation eines Graafschen Follikels. Innerhalb des Menstruationszyklus beginnt dann erneut die Follikelphase, die mit der Ovulation abgeschlossen wird und in die Lutealphase übergeht [125, 126]. Meiose 1. Reifeteilung (Meiose I) 2. Reifeteilung (Meiose II) OozytenWachstum Prophase I Oozytenmaturation Meiotischer Arrest Befruchtung Mitose 1. Ovulation Differenzierung Befruchtung (LH-Spitze) Demethylierung PGCs FSH Leptotän Zygotän Pachytän Diplotän Diktyotän Oogonien GV Metaphase I Metaphase II Follikulogenese Zeit ab Befruchtung 0 5 10 15 20 30 40 Wochen Geburt 10 Monate Pubertät 22 Tage (1. Zyklus) 2. Zyklus Präovulatorischer Graaf-Follikel Sekundärfollikel Antralfollikel 80-130 µm 0,25-10 mm Primärfollikel 12-20 mm 40-80 µm Primordialfollikel Diktyotän-Oozyte Thekazellen Granulosazellen Antrum Oozyte Zona pellucida <40 µm Remethylierung Follikelpool mit wachsenden Follikeln Follikelpool im Ruhestadium Zyklische Rekrutierung Initiale Rekrutierung Abbildung 4: Schematische Übersicht der bovinen Oogenese, Follikulogenese und Oozytenmaturation. Nach der Befruchtung und anschließender Differenzierung beginnen die primordialen Keimzellen (PGCs) etwa ab der 5. Schwangerschaftswoche (SSW) in die Genitalleiste zu wandern. Dabei teilen sich die PGCs ab der 9. SSW mitotisch. Die mitotischen Teilungen setzen sich im Stadium der Oogonien etwa bis zur 22. SSW fort [127]. Zirka 75 bis 82 Tage nach der Befruchtung (12. SSW) gehen die ersten Oogonien in die Prophase der Meiose I über und werden als primäre Oozyten bezeichnet [98, 128]. Bis zum Zeitpunkt der Geburt durchlaufen alle Eizellen im bovinen Ovar die Phasen des Leptotäns, Zygotäns und Pachytäns bis hin zum Diplotän der Prophase I, in dem sie arretiert werden (Meiotischer Arrest). Im Diplotän verharren die Oozyten bis zum Eintritt in die Pubertät mit 9 bis 10 Monaten. Mit Beginn der Pubertät nimmt jeweils eine Eizelle mit dem Menstruationszyklus ihre Meiose I unmittelbar vor dem ersten Eisprung erneut auf und geht zur Oozytenmaturation über [127]. Ab der 13. SSW lagert sich eine einschichtige Lage von Prä-Granulosazellen um jeweils eine Diplotäne-Oozyte an und bildet einen Primordialfollikel [98]. Die Primordialfollikel bilden einen Pool von Follikeln, die sich im Ruhestadium befinden und nicht wachsen. Follikel, die sich über das primordiale Stadium weiterentwickeln (Primär-, Sekundär und Antralfollikel sowie präovulatorischer Graaf-Follikel [127]) bilden den Pool wachsender Follikel [104]. Der Übergang vom ruhenden in den wachsenden Pool erfolgt durch die initiale Rekrutierung einer Kohorte von Primordialfollikeln [102]. Die erste initiale Rekrutierung erfolgt etwa in der 20. SSW und tritt dann regelmäßig 2- bis 3-mal innerhalb einer Zyklusphase bis zur Menopause auf [103]. Die durch die initiale Rekrutierung aktivierten Follikel können das Entwicklungsstadium des Antralfollikels erlangen, bevor sich in die Atresie übergehen. Erst mit Beginn der Pubertät und dem ersten Auftreten der Zyklischen Rekrutierung durch FSH wird eine Kohorte von Antralfollikeln zur Weiterentwicklung stimuliert, die nach dem Durchlaufen der Selektions- und Dominanzphasen zur Ovulation einer Eizelle ausgelöst durch eine LH-Spitze führt [102]. Bereits kurz vor der Ovulation beginnt die Oozytenmaturation mit Auflösung der Kernmembran (GVBD). Die Datenlage zu Demethylierungs- und Remethylierungsprozessen in bovinen Eizellen ist gegenwärtig limitiert. Insbesondere die frühen Phasen der Demethylierung unmittelbar nach der Befruchtung sind im Rind weitgehend unerforscht. Vermutlich erfolgt die Demethylierung jedoch während der Differenzierung der PGCs analog zur Maus (vgl. Kapitel 1.1.4). Die Remethylierung des Oozyten-Genoms findet kontinuierlich mit der Entwicklung der Eizelle bzw. während der Follikulogenese statt. Der Beginn sowie der Abschluss der Remethylierung sind bisher für bovine Eizellen ebenfalls nicht beschrieben. 20 EINLEITUNG Aus ethischen und technischen Gründen sind die Untersuchungsergebnisse sowie die daraus gewonnen Erkenntnisse der humanen Follikulogenese deutlich limitierter als die der bovinen Follikelentwicklung [129]. Dennoch konnte insbesondere die Entwicklung deutlich verbesserter Ultraschallgeräte und -methoden innerhalb der letzten Dekade zu einem Zugewinn wichtiger Daten auf dem Gebiet der humanen Follikulogenese führen [130]. Die Auswertung dieser Daten liefert zunehmend Hinweise darauf, dass die Mechanismen der Entwicklung und Reifung humaner Follikel in ähnlicher Weise ablaufen, wie die der beschriebenen bovinen Follikulogenese [129]. Wie beim Rind werden die Primordialfollikel durch eine initiale Rekrutierung aktiviert und entwickeln sich erst mit Beginn der Pubertät durch die zyklische Rekrutierung über das antrale Stadium hinaus [102]. Das Auftreten von Follikelwellen bzw. die Rekrutierung multipler Follikel-Kohorten wurde bereits seit den 1950er Jahren bei Frauen beobachtet [131-133]. Entscheidende Parallelen zum bovinen Modell konnten jedoch erst durch jüngere Studien gezeigt werden [134-137]. Dabei weisen etwa 68% der Frauen zwei und 32% drei Follikelwellen innerhalb eines interovulatorischen Intervals auf [135]. Die Dynamiken, die während der Rekrutierung, Selektion und Dominanz im humanen Ovar beobachtet werden, ähneln mit spezies-spezifischen Besonderheiten, jedoch ohne fundamentale Unterschiede, denen der Follikulogenese im Rind [134, 138]. Die zyklische Rekrutierung der Antralfollikel wird ebenfalls durch einen Anstieg von FSH ausgelöst, wobei humane Follikel bereits bei geringeren FSH-Dosen aufgrund einer erhöhten FSH-Sensitivität aktiviert werden [139]. Nach der Rekrutierung erfolgt die Selektion des dominanten Follikels wie im bovinen Ovar unter einem abnehmenden FSH-Spiegel [139141]. Dabei produzieren die rekrutierten, humanen Follikel gleichermaßen die FSH- Inhibitoren Estradiol und Inhibin wie die Follikel-Kohorte der Kuh [142, 143]. Auch der für die Ovulation benötigte Übergang von einer FSH-Abhängigkeit hin zu einer LH-Sensitivität während der Entwicklung des dominanten Follikels zum ovulationsfähigen Graafschen Follikel erfolgt wie beim Rind gleichermaßen auch bei der Frau [144, 145]. 1.2.3 Oozytenmaturation Als Oozytenmaturation bzw. Eizellreifung wird allgemein die Entwicklung von der arretierten diplotänen Oozyte mit Keimbläschen bzw. Germinalvesikel (GV) hin zur befruchtungsfähigen Eizelle bezeichnet [146, 147]. Diese Reifung beginnt mit der Fortsetzung der ersten Reifeteilung bzw. Meiose I und der Auflösung der Kernhülle (engl.: germinal vesicle breakdown; GVBD) (Abbildung 4). Im Rind (wie auch bei der Frau) tritt dies erstmalig mit 21 EINLEITUNG Eintritt der Geschlechtsreife bzw. Pubertät auf, ausgelöst durch einen starken Anstieg des luteinisierenden Hormons (LH) etwa 24 h bis 27 h vor der Ovulation [125]. Die Oozytenmaturation umfasst sowohl die nukleäre Reifung mit der Reduktion des diploiden Chromosomensatzes als auch die damit einhergehende zytoplasmatische Reifung mit molekularen und strukturellen Veränderungen der Eizelle [148]. Beide Prozesse sind miteinander verknüpft und werden während der Follikulogenese auch durch einen Stoffund Informationsaustausch mit den umgebenden Kumuluszellen reguliert. Die nukleäre Reifung beschreibt die Entwicklung des Eizellkerns vom Stadium des Germinalvesikels bis zum Erreichen der Metaphase II (MII). Dabei kommt es zur Auflösung der Kernmembran (GVBD), zur Kondensation der Chromosomen, der Ausbildung der Spindel, der Trennung der homologen Chromosomen, Ausschleusung des ersten Polkörpers und letztlich die Arretierung in MII [99]. Die nukleäre Reifung ist ein gradueller Prozess und benötigt in der bovinen Eizelle eine Dauer von 24 h [149]. Für die Auflösung der Kernhülle (GVBD) ist im Rind, im Gegensatz zu Nagern wie der Maus, eine aktive Proteinsynthese notwendig [150-156]. Mit der Abschnürung des ersten Polkörpers ist die Meiose I abgeschlossen und die Eizelle wird fortan als sekundäre Oozyte bezeichnet. Im Anschluss an die Meiose I geht die sekundäre Oozyte in die Meiose II über und wird im Stadium der Metaphase II arretiert. Damit ist die nukleäre Reifung abgeschlossen. Die Fortsetzung der Metaphase II und der Abschluss der Meiose II erfolgen einzig bei einer Befruchtung der reifen Eizelle durch ein Spermium [99]. Die Fähigkeit zur nukleären Maturation korreliert spezies-spezifisch mit der Größe der Antralfollikel bzw. der Größe der Oozyte [157]. In Rindern wird diese bei einem Durchmesser von 2-3 mm des Antralfollikels [158-160] und 110 µm der Eizelle [158, 161] erlangt. Die zytoplasmatische Reifung umfasst die Reorganisation des Zytoskeletts und der Organelle sowie die Produktion und Speicherung von mRNAs, Proteinen und Transkriptionsfaktoren, die am Reifungsprozess, der Befruchtung und der frühen Embryogenese beteiligt sind [162]. Diese Vorgänge sind zeitlich und räumlich miteinander koordiniert und schaffen die strukturellen Voraussetzungen für die Bildung einer funktionsfähigen Zygote aus zwei Gameten. Durch Mikrotubuli und Mikrofilamente des Zytoskeletts werden beim Übergang vom Germinalvesikel-Stadium zu MII neben der Separation der Chromosomen die gesamten Organellen einer Positionsänderungen innerhalb der Eizelle unterzogen [99, 163-165]. 22 EINLEITUNG Mitochondrien werden aus der Peripherie in der gesamten Zelle verteilt. Als Schlüsselkomponenten des Energiestoffwechsels [163, 166] erhöht sich ihre Zahl dabei von 6.000 in der primären Oozyte auf mehr als 50.000 bis zum Abschluss der Reifung [167-170]. Die Fragmente des Golgi-Apparats werden während des GVBD von Vesikeln umschlossen und bis zum MII-Stadium phosphoryliert an Stellen mit aktivem ER-Export transportiert [171, 172]. Die Membranen des ER enthalten Kalzium-Ionen, die gespeichert und freigesetzt werden. Diese Kalziumoszillationen stellen die Voraussetzung für die Aktivierung der Eizelle dar [173]. In enger Wechselwirkung mit dem Golgi-Komplex und dem ER stehen auch die Corticalgranula. Diese befinden sich im GV-Stadium über die gesamte Eizelle verteilt. Bis zur MII ordnen sie sich jedoch über die ganze innere Oberfläche nahe der Plasmamembran an. Diese Nähe der Corticalgranula zur Plasmamembran ist ebenfalls entscheidend für die Aktivierung der Eizelle und essentiell für die Verhärtung der Zona pellucida zur Verhinderung einer Polyspermie während des Befruchtungsvorgangs [174-176]. Die strukturellen Veränderungen der Eizelle während der Reifung schließen auch die Transkription, Speicherung und Prozessierung von mRNA-Molekülen ein. Das Anlegen dieser Reserve von Makromolekülen ist notwendig, da bis zur Aktivierung des embryonalen Genoms keine Transkription und Proteinsynthese stattfindet [162]. Der Zeitpunkt dieser Aktivierung ist spezies-spezifisch und erfolgt in der Maus im 2-Zellstadium, beim Menschen im 8-Zellstadium und beim Rind und Schaaf im 8-16-Zellstadium [177-179]. Die bei dieser molekularen Maturation generierten Transkripte werden in einer stabilen und zunächst inaktiven Form gespeichert. Durch die Prozessierung der mRNA-Moleküle zu Ribonukleoprotein-Partikeln wird die mRNA vor einer nukleolytischen Degradation geschützt bis sie im Verlauf der Reifung oder frühen Embryogenese translatiert wird [180-182]. Zu den Genen, die während der zytoplasmatischen Reifung sowohl transkribiert als auch translatiert werden gehören insbesondere regulatorische Faktoren des Zellzyklus. Zu erwähnen ist hier vorallem die Proteinkinase MPF (maturation promoting factor), die entscheidend an der Regulation des Reifungsprozesses beteiligt ist [99, 162]. MPF besteht aus der regulatorischen Komponente Cyclin B und der katalytischen Untereinheit p34 cdc2 [183]. Der MPF-Komplex scheint einen universellen Regulator der M-Phase sowohl für den mitotischen als auch für den meiotischen Zellzyklus darzustellen [184, 185]. Die Phosphorylierung bzw. Aktivierung des Proteins ist für den Übergang zu Meiose I und 23 EINLEITUNG Meiose II notwendig. Erst nach Aktivierung von MPF geht die Oozyte vom GV-Stadium zum GVBD-Stadium über und erreicht sein Aktivitätsmaximum in Meiose I [185, 186]. Während der Transition von Meiose I zu Meiose II fällt die Aktivität von MPF bevor sie in der Meiose II erneut ein Maximum erreicht [187]. 1.2.4 Ovarielle Alterung Zu Beginn der Oogenese teilen sich die Oogonien in der frühen Fetalphase mitotisch und erreichen ein spezies-spezifisches Maximum von Zellen. Die maximale Anzahl an Oogonien wird in der humanen Oogenese mit geschätzt 5 Millionen Zellen etwa in der 20. Schwangerschaftswoche erreicht [95, 96]. Nach Abschluss dieser mitotischen Teilungsphase nimmt die Anzahl dieser Vorläufer von Eizellen stetig ab. Die durchschnittlich etwa 600.000 Primordialfollikel zum Zeitpunkt der Geburt stellen den Vorrat bzw. das Reservoir dar, aus dem sich von der Pubertät bis zur Menopause der Frau befruchtungsfähige Oozyten entwickeln [97]. Mit Einsetzen der Follikulogenese beginnt dieser Vorrat bereits vor der Geburt ausgelöst durch regelmäßige Rekrutierungen von Follikel-Kohorten abzunehmen. Diese Abnahme des Follikel-Vorrats mit zunehmendem Alter des weiblichen Individuums bzw. des Ovars wird als ovarielle Alterung bezeichnet und kennzeichnet sich durch mehrere Eigenschaften [188]. Neben der Reduktion des Reservoirs von Primordialfollikeln ist die ovarielle Alterung charakterisiert durch eine abnehmende Oozytenqualität [189-191]. In Verbindung mit in-vitro-Fertilisationen (IVFs) [191-196] oder reproduktionsmedizinischen Behandlungen im Allgemeinen führt die reduzierte Eizell-Qualität zu geringeren Schwangerschaftsraten [194, 196, 197]. Gleichzeit wird ein erhöhtes Auftreten von Aneuploidien im Embryo [198-200] sowie eine erhöhte Fehlgeburtenrate [201, 202] beschrieben, die letztlich eine reduzierte Entbindungsrate nach spontanen Schwangerschaften und Schwangerschaften nach ART zur Folge haben [203]. Als Ursachen der ovariellen Alterung gelten vorallem genetische Faktoren und die Verkürzung der Telomere [204, 205]. Umweltfaktoren, wie die Ernährung oder oxidativer Stress, beeinflussen ebenfalls die Abnahme des Follikel-Vorrats mit zunehmendem Alter des Ovars [206]. Darüber hinaus konnten auch epigenetische Veränderungen in Eizellen von jüngeren im Vergleich zu älteren weiblichen Individuen identifiziert werden [207, 208]. Die genauen epigenetischen Mechanismen, die an der ovariellen Alterung beteiligt sind, sind jedoch bisher nicht bekannt [209]. 24 EINLEITUNG 1.3 Assistierte Reproduktion Die Reproduktion bei höheren Säugetieren hängt von dem Erfolg bzw. Misserfolg komplexer, teils wenig aufgeklärter, aufeinanderfolgender Prozesse ab, die zu einer Schwangerschaft und letztlich zur Geburt des gesunden Nachwuchses führen. Diese Prozesse beinhalten die Spermatogenese, die Oogenese, die Follikuliogenese, die Ovulation, den sexuellen Akt und den Transport der Keimzellen, die Migration des Embryos in den Uterus sowie seine darauffolgende Implantation und schließlich die interuterine Entwicklung des Fötus. Störungen können in jedem Schritt dieser Abfolge auftreten. Am häufigsten sind jedoch Fehler in den frühen Phasen zu beobachten. Die Gelegenheit eine Eizelle zu befruchten und eine Schwangerschaft einzuleiten ergibt sich regelmäßig mit jedem Menstruationszyklus. Beim Menschen zeigt sich für ein einzelnes Paar eine relativ konstante monatliche Wahrscheinlichkeit eine Schwangerschaft zu verursachen. Zwischen unterschiedlichen Paaren variiert diese Wahrscheinlichkeit jedoch stark zwischen 0% und 60%. Bei einer monatlichen Probabilität von 0% eine Schwangerschaft zu induzieren, spricht man auch von Sterilität, welche in 3%-5% aller Paare auftritt [210]. Während Sterilität das Unvermögen beschreibt, schwanger zu werden, bezeichnet man mit Infertilität das Unvermögen, ein lebendes Kind zu gebären. Beide Begriffe drücken einen absoluten Zustand aus, der für die meisten „sterilen“ bzw. „infertilen“ Paare nicht zutrifft. Der größte Teil dieser Paare ist nicht zu 100% unfruchtbar, sondern weist eine nicht genau quantifizierbare Einschränkung der Fertilität auf [211]. Obwohl für die letzten Jahrzehnte ein Anstieg der Rate an Paaren mit unerfülltem Kinderwunsch beschrieben wird, werden keine belastbaren Daten für diese Vermutung genannt. In Statistiken aus Australien um das Jahr 1900 wird bereits über eine ungewünschte Kinderlosigkeit von 11% berichtet [212]. Eine ähnliche Häufigkeit wird auch für das gegenwärtige Auftreten vermutet [213]. Lässt sich der Kinderwunsch auf natürlichem Wege nicht erfüllen, kann nach entsprechender Indikation die Verwendung assistierter Reproduktionstechniken (ART) zum Erfolg führen bzw. Hilfe leisten. Als ART bezeichnet man alle Methoden und Verfahren, die die in vitro-Behandlung von Eizellen und Spermien sowie Embryonen umfassen, die darauf abzielen eine Schwangerschaft bzw. einen Reproduktionserfolg herbeizuführen [214]. Darunter zählen u.a. die In-vitro-Maturation (IVM), die In-vitro-Fertilisation (IVF) einschließlich intrazytoplasmatische Spermieninjektion (ICSI), der Embryonentransfer (ET), der intratubare Gametentransfer (GIFT), der intratubare Zygotentransfer (ZIFT), die Kryokonservierung von Eizellen und Embryonen, die Eizell- und 25 EINLEITUNG Embryonenspende sowie die Leihmutterschaft [214]. ARTs werden neben der humanmedizinischen Anwendung auch zur Erzeugung von Nachkommen bei Zuchttieren genutzt. 1.3.1 Assistierte Reproduktionstechniken in der humanen Reproduktionsmedizin Eines der bedeutsamsten Ereignisse in der Geschichte der assistierten Reproduktion war die Geburt von Louise Brown, dem ersten nach in-vitro-Fertilisation (IVF) und Reimplantation geborenen Kind [215]. Die dadurch ausgelöste intensive Forschung auf diesem Gebiet führte 1992 zur Implementation der intrazytoplasmatische Spermieninjektion (ICSI; [216]). Seit Einführung der ICSI gab es bis heute kein Verfahren, das in vergleichbarer Weise die Konzeptionschancen in der Reproduktionsmedizin vervielfachen konnte [211]. Die Motivation zur Entwicklung der IVF war ursprünglich die Therapie von Patientinnen mit tubarer Sterilität. Durch die kontinuierliche Weiterentwicklung dieser Technik ist es heute möglich, eine Vielzahl von Ursachen für Infertilität mittels IVF erfolgreich zu behandeln [217]. Als IVF wird prinzipiell die Methode der assistierten Reproduktion beschrieben, bei der das männliche Sperma und die weibliche Eizelle außerhalb des Körpers bzw. extrakorporal miteinander kombiniert werden. Grundsätzlich besteht ein vollständiger IVFBehandlungszyklus aus einer ovariellen Stimulation, der Eizellgewinnung, der Fertilisation, der Kultivierung des Embryos sowie dem Embryonentransfer [217]. Die ovarielle Stimulation dient dazu, einen Eisprung oder eine Polyovulation zur Gewinnung mehrerer Eizellen für anschließende extrakorporale Behandlungen herbeizuführen. Prinzipiell wird dies durch eine indirekte oder direkte Erhöhung des Serum-FSH-Spiegels verursacht. Im Verlauf des natürlichen Zyklus kommt es durch die FSH- und LH-Wirkung zu einer zyklischen Rekrutierung von Follikeln im Ovar, die zur Selektion eines dominanten Follikels führt, der letztlich in einer Monoovulation in den Eileiter entlassen wird (siehe Kapitel 1.2.2). Der dominante Follikel stellt dabei den Follikel dar, der über die höchste FSH-Rezeptordichte und damit über die höchste FSH-Sensitivität verfügt. Wird bei der Ovulationsinduktion das Ziel einer Monoovulation forciert, wird der Serum-FSH-Spiegel minimal notwendig erhöht, um die Reifung eines einzelnen Follikels zu induzieren [218]. Für eine Polyovulation bzw. Superovulation wird der Serum-FSH-Spiegel gezielt so erhöht, dass sich neben dem dominanten Follikel auch mehrere, nicht-dominante Follikel weiterentwickeln, die unter natürlichen Bedingungen in die Atresie übergehen würden. Eine allgemein gültige Anzahl an Eizellen, die ein optimales Ergebnis einer ovariellen Stimulation kennzeichnet, existiert nicht. 26 EINLEITUNG Eine Studie, in der Nutzen und Risiken einer ovariellen Stimulation berücksichtigt wurden, proklamiert jedoch, dass dabei zwischen 5 und 15 Eizellen für die IVF gewonnen werden sollten [219]. Die Entnahme der reifen Follikel aus dem Ovar erfolgt in den meisten Fällen durch eine transvaginale ultraschallgesteuerte Follikelpunktion. Diese Technik wurde 1985 entwickelt [220] und hat sich in den vergangenen 30 Jahren als Methode der Wahl in der Reproduktionsmedizin etabliert. In der Regel werden alle Follikel punktiert, die punktiert werden können und einen Durchmesser von mehr als 10 mm aufweisen. Die so gewonnenen Follikel bzw. Kumulus-Eizell-Komplexe (COCs) werden anschließend einer visuellen Beurteilung unterzogen, um die qualitativ hochwertigsten Follikel bzw. Eizellen für die IVF zu identifizieren. Dabei kennzeichnet eine expandierte und luteinisierte Kumuluszellmatrix eine reife Eizelle (Metaphase II) während dichter gepackte Kumuluszellen auf eine unreife Oozyte hinweisen [221]. Ob die Befruchtung der Eizelle(n) durch eine IVF oder ICSI erfolgt entscheidet die Qualität der Spermien nach der Aufbereitung des Ejakulats. Bei der IVF werden ein oder mehrere COC in einem Kulturschälchen 2-3h nach der Punktion mit einer bestimmten Anzahl von Spermien versetzt. Dabei liegt die Konzentration der Samenzellen zwischen 20.000 und 25.000 Spermien pro Eizelle. Nach Inkubation über Nacht und der Entfernung der umgebenden somatischen Zellen (Denudation) wird die Gegenwart von 2 Vorkernen bzw. 2 Polkörpern als erfolgreiche Befruchtung gewertet. Für eine Befruchtung durch ICSI wird die Oozyte zunächst denudiert um zu ermöglichen, die Reife der sich entwickelnden Gameten zu kontrollieren. Nach der Selektion und einer in der Regel mechanischen Immobilisierung des Spermiums erfolgt die Injektion der männlichen Gamete in die Eizelle. Dabei ist zu beachten die Einstichstelle möglichst weit vom ersten Polkörper zu positionieren. Da sich die Teilungsspindel in unmittelbarer Nähe zum ersten Polkörper befindet soll dadurch verhindert werden, dass die sensitiven Mikrotubuli der Spindel mechanisch beschädigt werden [222]. Neben den beschriebenen standardisierten Abläufen einer IVF bzw. ICSI gibt es eine Vielzahl modifizierter Protokolle, die sich in ihren technischen Details unterscheiden und in den verschiedenen Kliniken und Laboratorien zur Anwendung kommen. Im Allgemeinen sollte in 60% (IVF) bis 80% (ICSI) der Eizellen eine erfolgreiche Befruchtung zu erwarten sein. Der Befruchtung schließt sich eine Inkubation in Kulturmedium an, wobei die Reifung des sich entwickelnden Embryos überwacht und bewertet wird. In Deutschland findet diese Bewertung der Embryonen nur nach lichtmikroskopischen Kriterien statt. Nur in Ausnahmefällen bei entsprechender Indikation 27 EINLEITUNG kann seit 2010 auch eine Präimplantationsdiagnostik (Trophektodermbiopsie) durchgeführt werden. Nach Auswahl der optimalen Embryonen werden diese in die Gebärmutter der Patientin transferiert. Während bis Ende der 1990er-Jahre die Embryonen üblicherweise etwa 2 bis 3 Tage nach der Insemination (4- bis 8-Zell-Stadium) in den Uterus transferiert wurden, hat sich seitdem eine längere, extrakorporale Kultur und der Blastozystentransfer als erfolgreichere Technik nach IVF und ICSI etabliert [223, 224]. Um das Risiko von Mehrlingsschwangerschaften zu reduzieren, ist die Anzahl der einzusetzenden Embryonen nach dem Deutschen Embryonenschutzgesetz auf maximal 3 beschränkt. Das Einsetzen der Embryonen in den Uterus erfolgt meist durch einen ultraschallgezielten oder freien Embryonentransfer (ET) mit anschließender Bettruhe für 24h. 1.3.2 Assistierte Reproduktionstechniken bei Nutztieren Die unter 1.3.1 genannten und beschriebenen Techniken der assistierten Reproduktion wurden vor ihrer Anwendung in humanen Patienten in domestizierten Tier(modell)en erprobt und entwickelt. Neben der Reproduktionsmedizinischen Nutzung im Menschen, wurden und werden assistierte Reproduktionstechniken zur Selektion genetisch favorisierter Zuchttiere insbesondere in Rindern, Schafen, Ziegen, Schweinen und Pferden eingesetzt. Die erste erfolgreiche artifizielle Insemination wurde 1784 durch Spallanzani an einer Hündin durchgeführt und beschrieben [225]. 1922 intensivierte Ivanoff die Untersuchungen dieser Technik an Nutztieren, Hunden, Füchsen, Karnickel und Federvieh [226]. Heutzutage gehört die artifizielle Insemination zu einer der am häufigsten angewandten Techniken in der Nutztierzucht und wird pro Jahr an 100 Millionen Rindern, 40 Millionen Schweinen, 3,3 Millionen Schafen und 0,5 Millionen Ziegen weltweit durchgeführt [227]. Der erste erfolgreiche Transfer eines Säuger-Embryos gelang 1890 Walter Heap [228]. Heap transferierte zwei 4-Zell-Embryonen von Angorakaninchen in ein weibliches Hasenkaninchen, welches zwei Angorakaninchen und vier Hasenkaninchen gebar. In den 1930er und 1940er Jahren wurden die ersten erfolgreichen Embryonentransfers in Schafen und Ziegen beschrieben [228, 229], bevor 1951 das erste, aus einem übertragenen Embryo entwickelte Kalb geboren wurde [229, 230]. Die erste Lebendgeburt eines Säugetiers nach IVF gelang 1959 M.C. Chang, welcher Oozyten von Kaninchen in vitro außerhalb des Organismus befruchtete [231]. 1968 wurde diese Methode erfolgreich an Mäusen durchgeführt bevor 1978 das erste Kind nach einer in-vitro28 EINLEITUNG Fertilisation geboren wurde [232]. Ein durch IVF entstandenes Kalb wurde erstmals 1981 beschrieben [233]. Im Zuge der erfolgreichen IVF-Experimente wurden auch Methoden zur intrazytoplasmatische Spermieninjektion (ICSI) erprobt, welche 1976 erstmalig erfolgreich in einem Hamster durchgeführt werden konnten [234]. Die Anwendung assistierter Reproduktionstechniken bei Nutztieren konnte zu einer beschleunigten Vermehrung der Populationen genetisch favorisierter Zuchttiere von Rindern, Büffeln, Schafen, Ziegen, Pferden und Schweinen führen. Nach aktuellen Daten des „International Embryo Transfer Society Data Retrieval Committee“ umfasst die Anzahl der transferierten Embryonen pro Jahr weltweit etwa 800.000 in Rindern, 25.000 in Schafen, 7000 in Ziegen, 30.000 in Schweinen und 12.000 in Pferden, wobei etwa zwei Drittel in vivo gewonnen wurden und ein Drittel in vitro fertilisiert wurde [235]. Die Techniken zur in vitro-Produktion von Embryonen dienen nicht nur der Gewinnung von Nutztier-Populationen mit spezifisch gewünschten Eigenschaften sondern stellen auch eine verhältnismäßig einfache und geeignete Quelle von Embryonen für die biologische und biotechnologische Forschung dar. Biotechnologische Verfahren wie Klonierungstechniken und Zellkerntransfer-Experimente, die aus ethischen Gründen nicht an humanen Embryonen angewendet werden dürfen, können an tierischen Embryonen durchgeführt werden und dazu genutzt werden grundlegende entwicklungsbiologische Prozesse zu untersuchen. Unter den aufgeführten Arten von Zuchttieren zeigten insbesondere frühe embryonale Stadien von Rindern viele Ähnlichkeiten zu humanen Embryonen [236-239]. Diese sowie weitere Übereinstimmungen charakterisieren das Rind als geeigneten Modellorganismus der humanen Reproduktion, worauf im Kapitel 1.5 ausführlich eingegangen wird. 1.3.3 Assistierte Reproduktionstechniken und gesundheitliche Risiken Der gesamte Vorgang von der Entwicklung der Urkeimzellen bis zur Entstehung der Zygote stellt nicht nur ein sensibles Regelwerk von Entwicklungsschritten dar sondern beinhaltet vor allem zahlreiche notwendige Selektions- und Filtermechanismen, um einen Embryo mit höchstem Entwicklungspotential zu generieren. Die Eizelle steht bereits während der Entwicklung vom primordialen Stadium bis zur reifen, befruchtungsfähigen Oozyte unter enormem Selektionsdruck. Dies zeigt sich beispielsweise daran, dass von etwa 40.000 primären Oozyten zu Beginn der Pubertät nur etwa 400 Eizellen innerhalb der reproduktiven Lebensspanne ovulieren [240]. Unter Anwendung von ARTs werden in Abhängigkeit der verwendeten Technik einige dieser natürlichen Selektions- und Filtermechanismen verändert 29 EINLEITUNG bzw. umgangen. Diese Eingriffe bergen daher prinzipiell das Risiko, dass Keimzellen mit vermindertem Entwicklungspotential zur Generierung eines Embryos verwendet werden, die unter natürlichen Umständen bereits vor der Fertilisation aussortiert worden wären. Seit der Geburt des ersten Kindes nach durchgeführter in-vitro-Fertilisation (IVF) 1978 [215] wurden bis heute mehr als 5 Million IVF-Kinder weltweit geboren [241]. Innerhalb dieser 40 Jahre moderner humaner Reproduktionsmedizin wurden zahlreiche Studien und Metaanalysen durchgeführt, in denen die Auswirkung von ARTs auf die mütterliche und insbesondere auf die kindliche Gesundheit untersucht wurde. Den größten Risikofaktor sowohl für das Kind als auch für die Mutter in Verbindung mit ARTs stellen Mehrlingsschwangerschaften dar [242]. Um die Chancen einer Schwangerschaft zu erhöhen, werden mehrere, meist 3 Embryonen transferiert. Damit erhöhen sich allerdings gleichzeitig die Wahrscheinlichkeit einer Mehrlingsschwangerschaft und das damit einhergehende Risiko für Schwangerschaftskomplikationen. Der Transfer von 2 Embryonen kann diesen Risikofaktor jedoch bei unveränderter Schwangerschaftrate deutlich reduzieren [243, 244]. In den skandinavischen Ländern konnte sich die Übertragung von einzelnen Embryonen als Standardmethode durchsetzen, wodurch die Häufigkeit von Mehrlingsschwangerschaften erheblich gesenkt wurde [242, 245]. Eine weitere Beobachtung, die auch mit Mehrlingsschwangerschaften in Verbindung steht, ist das gehäufte Auftreten von Kindern mit niedrigem Geburtsgewicht nach ART [246, 247]. Als mögliche Ursache wird hier die Superovulation vermutet [248, 249]. Die hormonelle Behandlung während der Superovulation wird auch in Zusammenhang mit einem erhöhten Risiko von Implantationsversagen, Bluthochdruck, Blutungen, Placenta praevia und Imprinting-Fehlern gebracht [250-254]. Die Etablierung und Aufrechterhaltung der genomischen Prägung, die essentiell für eine normale Entwicklung des Embryos ist, könnten im besonderen Maße durch reproduktionsmedizinische Eingriffe beeinflusst werden. Da die Prägung verschiedener Gene in unterschiedlichen Entwicklungsphasen erfolgt, wird vermutet, dass durch unterschiedliche ARTs auch verschiedene Imprinting-Regionen betroffen sein könnten. Die Prägung von SNRPN, die vermutlich spät in der Follikulogenese etabliert wird [255, 256], könnte daher ebenfalls durch die Superovulation beeinträchtigt werden [254]. Das Auftreten von Imprinting-Fehlern wurde in etwa 1% bei Patienten mit Prader-Willi-Syndrom (PWS), 3% bei Patienten mit Angelman-Syndrom (AS), 50% bei Betroffenen mit Beckwith-WiedemannSyndrom (BWS) und 50% bei Patienten mit transientem neonatalem Diabetes mellitus 30 EINLEITUNG nachgewiesen [254]. AS und BWS zeigen dabei eine Hypermethylierung der ImprintingKontrollregion auf dem maternalen Chromosom [254]. Die publizierten Studien, in denen Imprinting-Fehler in ART-Kindern untersucht wurden, sind widersprüchlich. Bei den unterschiedlichen Vergleichen von Kindern, die nach einer ART-Behandlung geboren wurden, und denen, die auf natürlichem Weg entstanden, wird sowohl von erhöhten Fehlerraten [257-263] als auch von unveränderten Häufigkeiten von Imprinting-Fehlern bei ART-Kindern berichtet [254, 264, 265]. Aufgrund der geringen Inzidenz dieser Syndrome (110 in 100.000 Kindern, vgl. Kapitel 1.1.3) wird für eine solide Prüfung dieser Fragestellung eine deutlich höhere Fallzahl benötigt, als die bisher durchgeführten Studien umfassen. Die Anfälligkeit dieser Studien für Verzerrungen können bisher noch keine verlässlichen Aussagen gewährleisten. Hinweise auf einen Verbindung zwischen ARTs und Störungen der genomischen Prägung zeigen sich jedoch ebenfalls in Tiermodellen [266]. Das sog. „Large offspring syndrome“ (LOS) konnte bei Rindern, Schafen [267, 268] und Mäusen [269-271] nach Somatischem Zellkerntransfer (SCNT) und ARTs beobachtet werden und weist vergleichbare phänotypische wie epigenetische Auffälligkeiten zum humanen BeckwithWiedemann-Syndrom (BWS) auf [272, 273]. Trotz schwieriger Gesamtbewertung begründen die beschriebenen Einzelfälle den Verdacht einer Beeinträchtigung der genomischen Prägung durch ARTs und sollte daher weiterhin unter intensiver Beobachtung bleiben [254, 264]. Eine eingeschränkte Bewertung der Daten wurde ebenfalls für Studien beschrieben, die den Zusammenhang zwischen assistierten Reproduktionstechniken und großen Fehlbildungen untersuchten. Von drei Metaanalysen [274-276], denen diese Fragestellung zugrunde lag, konnten zwei eine signifikant erhöhte Fehlbildungsrate bei IVF- und ICSI-Kinder gegenüber natürlich gezeugten Kindern nachweisen [275, 276]. Die Ergebnisse aller drei Metaanalysen wiesen jedoch eine geringe Validität auf. Durch die Heterogenität der einbezogenen Einzelstudien in Bezug auf die Fehlbildungsklassifikationen, den Untersuchungsmodus, den Studienaufbau sowie die Ergebnisse kann eine Verzerrung der gesamten Analyse nicht ausgeschlossen werden [264]. Neben den geringen Fallzahlen und der fehlenden Homogenität der gegenwärtig veröffentlichten Studien, wird die Bewertung möglicher gesundheitlicher Risiken für das Kind durch ARTs allgemein durch den biologischen Hintergrund der infertilen Eltern erschwert [254, 277]. So wird beispielsweise vermutet, dass Eltern bereits eine genetische Prädisposition für eine genetische oder epigenetische Instabilität vorweisen und mögliche gesundheitliche Folgen für das Kind nicht 31 EINLEITUNG durch die verwendeten assistierten Reproduktionstechniken verursacht wurden [254]. Inwiefern ARTs die Entwicklung der Keimzellen und des Embryo bei vorliegender elterlicher Prädisposition sowohl negativ als auch positiv beeinflussen bleibt eine der wichtigen Fragen zukünftiger Forschung. Die Beobachtung und Dokumentation pathologischer Auswirkungen nach Verwendung von ARTs bleibt jedoch von elementarer Bedeutung, um mögliche Risiken für die Gesundheit des Kindes zu erkennen. 1.4 In-vitro-Maturation von Eizellen Als In-vitro-Maturation (IVM) bezeichnet man den Prozess, bei dem unreife Eizellen aus Antralfollikeln mittlerer Größe die Veränderungen der nukleären und zytoplasmatischen Reifung extrakorporal in einem Zellkultur-System durchlaufen [278, 279]. Der Vorteil dieser vergleichsweise jungen Methode der assistierten Reproduktionstechniken liegt insbesondere in der Vermeidung der ovariellen Stimulation und den damit verbundenen Gesundheitsrisiken für die Eizellspendering [211]. Die Grundlagen dafür lieferten Pincus und Kollegen in den 1930er Jahren. Sie beobachteten erstmals wie Eizellen von Kaninchen [280] und Menschen [281] spontan die Meiose fortsetzten, nachdem sie aus dem Antralfollikel isoliert wurden und in einem Medium kultiviert wurden. Die Untersuchungen an KaninchenEizellen wurden in den 50er Jahren von Chang intensiviert [282], bevor Bob Edwards Anfang der 60er Jahre die nukleäre Reifung an Oozyten außerhalb des Follikels bei mehreren Säuger-Spezies einschließlich dem Rind beobachten konnte [283]. 1969 gelang es ebenfalls Edwards erstmals eine humane Eizelle erfolgreich zu befruchten, die durch eine IVM gereift wurde [284]. Die erste humane Schwangerschaft nach IVM wurde 1991 von Cha et al. beschrieben, welche an Eizellen aus exstirpierten Ovarien durchgeführt wurde [285]. Trotz geringerer Schwangerschaftsraten im Vergleich zu in vivo gereiften Oozyten, konnte sich die IVM als nützliche Anwendung in der Produktion von Zuchttieren wie dem Rind [286] und als erfolgsversprechende Alternative für bestimmte Indikationsgruppen in der Reproduktionsmedizin [211] etablieren. 1.4.1 IVM in der humanen Reproduktionsmedizin Die ovarielle Gonadotropinstimulation bei der klassischen IVF/ICSI birgt bei bestimmten Patientengruppen wie Frauen mit polyzystischen Ovarsyndrom (PCOS) das Risiko eines ovariellen Überstimulationssyndroms (OHSS) (5% bei PCOS-Patientinnen [287]) [288]. Die Intention zur Entwicklung einer extrakorporalen Eizellreifung in der humanen 32 EINLEITUNG Reproduktionsmedizin bestand zunächst darin, die ovarielle Stimulation und das damit verbundene Risiko eines OHSS zu vermeiden. Neben den Risikogruppen des OHSS können auch Krebs-Patientinnen mit Kinderwunsch vor einer bevorstehenden zytotoxischen Therapie wie beispielsweise einer Chemotherapie von einer IVM profitieren [289]. Die technische Durchführung der humanen IVM beginnt mit einer dreitägigen Kurzzeitstimulation mit Gonadotropinen in niedriger Dosierung. Die Oozytenentnahme erfolgt durch eine Follikelaspiration unter Verwendung eines hochauflösenden Ultraschallgeräts. Die anschließende In-vitro-Maturation erfolgt durch Inkubation der Kumulus-Eizell-Komplexe (COCs) in einem Kulturmedium für 24 h. Die Zusammensetzung des Mediums variiert dabei zwischen unterschiedlichen Laboren in Abhängigkeit empirischer Erfahrungen. Häufig wird als Basismedium Hams F-10 oder TCM 199 verwendet. Dieses wird dann zumeist mit Glukose [290] sowie essenziellen und nicht essenziellen Aminosäuren und Vitaminen angereichert [211]. Weitere Zusätze stellen auch FSH [291], LH und humanes Choriongonadotropin (HCG) [292] sowie fetales Nabelschnurserum, humanes Serumalbumin, synthetische Präparate als auch Serum von der Patientin selbst dar. Der Inkubation schließt sich eine Beurteilung des Reifestadiums der Eizelle an, welche dann ggf. durch IVF oder ICSI befruchtet wird. Nach erfolgreicher Befruchtung und dem Embryonentransfer wird die Lutealphase mit Progesteron (und Östradiol ab Eizellgewinnung) bis zur 12. Schwangerschaftswoche unterstützt [211]. Die Schwangerschaftsraten pro Zyklus liegen zwischen 15% [293] und 24% [291, 294]. Dabei steigt die Schwangerschaftsrate mit der Größe des für die IVM verwendeten dominanten Follikels (<10 mm: 21,1%; 14 mm: 40,3%) [295]. Neben den genannten Vorteilen der IVM birgt diese Methode jedoch auch eine Reihe von Nachteilen. Dazu zählen die geringen Implantationsraten sowie der höhere technologische Aufwand. Zu erwähnen sind jedoch insbesondere mögliche ungeklärte epigenetische und anderweitige fetale Risiken verursacht durch die Reifung in einer unphysiologische In-vitroKultur. Aufgrund der geringen Anzahl der bisher nach IVM geborenen Kinder ist noch ungeklärt, ob IVM chromosomale Aberrationen, Genomreprogrammierungsfehler, Fehlbildungen oder gesundheitliche Risiken für die Mutter verursacht, obgleich die wenigen Studien bislang keine Hinweise darauf geben [254, 288, 296-301]. Gegenwärtig stellt die IVM in der humanen Reproduktionsmedizin eine erfolgsversprechende und nützliche Alternative zur konventionellen IVF/ICSI für eine spezifische Patientengruppe dar. Inwiefern die IVM 33 EINLEITUNG jedoch die IVF/ICSI mit ovarieller Stimulation ersetzen wird, werden die zukünftigen Erfahrungen aus den Kliniken sowie der Fortschritt der Forschung in den nächsten Jahren zeigen. 1.4.2 IVM beim Rind (Bos taurus) Ende der 60er und Anfang der 70er Jahre konnten Untersuchungen an Eizellen von Rindern und Schafen die Chronologie der Oozytenreifung aufdecken. Bei in vitro-Experimenten zeigte sich eine optimale Reifungsdauer von 24h, die dem natürlichen Intervall zwischen dem präovulatorischen LH-Anstieg und der Ovulation der reifen Oozyte in beiden Tieren entsprach. Der erste Bericht eines nach IVM und IVF geborenen Kalbs wurde 1986 veröffentlicht [302]. Das Ende der 80er Jahre etablierte Protokoll zur IVM von unreifen Rindereizellen unterscheidet sich nicht fundamental von dem Protokoll, das in der humanen Reproduktionsmedizin verwendet wird. Nach der Dissektion der Follikel aus dem Ovar wurden daraus die Kumulus-Oozyten-Komplexe (COCs) isoliert. Die Inkubation der COCs erfolgte dann für 24h in TCM 199, welchem zusätzlich Kumuluszellen und Granulosazellen sowie Antibiotika und oestrisches Kuhserum (OCS) zugesetzt wurden. Die Zugabe von Hormonen konnte keinen förderlichen Effekt für die Entwicklung des Embryos zeigen [303]. Ursächlich dafür könnten die Hormone des OCS sein, die die Eizellreifung bereits in ausreichendem Maße unterstützen [304]. Ein negativer Effekt auf die Blastozystenrate konnte hingegen bei einer Verlängerung der Reifungsdauer bzw. der Dauer der Inkubation über 24h hinaus beobachtet werden [305-308]. Dieser als postovulatorische Alterung bezeichneter Prozess wurde sowohl in vitro als auch in vivo neben dem Rind auch in der Maus [309], dem Schwein [310] und dem Menschen [311] beschrieben. In vivo tritt dieser Prozess ein, wenn das Spermium die Eizelle nach dem Eisprung verspätet und außerhalb des optimalen Zeitfensters erreicht. In vitro setzt die postovulatorische Alterung ein, wenn die Oozyte über die optimale Reifungsdauer hinaus inkubiert wird bevor bevor sie durch ein Spermium befruchtet wird [312]. Trotz einer deutlich geringeren Blastozytenrate der IVM im Vergleich zur Reifung in vivo (in vitro: 3%; in vivo: 40% [313]) bietet die IVM die einzigartige Möglichkeit Embryonen mit Eizellen von Schlachthof-Ovarien zu produzieren [314]. Von insgesamt 121.199 in vitro fertilisierten Oozyten in Europa 2014 wurden 37.414 Eizellen aus Schlachthof-Ovarien gewonnen [315]. Neben der industriellen Nutzung bietet die Produktion von Embryonen aus Schlachthof-Ovarien durch IVM eine günstige und einfache Quelle von Embryonen für Forschungszwecke. 34 EINLEITUNG 1.5 Das Hausrind (Bos taurus) als Modellorganismus der humanen Reproduktion Die Errungenschaften der Reproduktionsforschung ermöglichten es Paaren mit unerfülltem Kinderwunsch eine Behandlung anzubieten und diesen Wunsch gegenwärtig für etwa jedes 5. Paar (20%) mit einem Kind zu erfüllen [316]. Um diese Erfolgsrate zu erhöhen und die Grundlagen der humanen Reproduktionsmechanismen besser zu verstehen, ist das Fortschreiten der Forschung auf dem Gebiet der Reproduktion elementar. Im Gegensatz zu vielen anderen medizinischen Forschungsbereichen birgt die humane Reproduktionsmedizin jedoch eine besondere ethische Problematik, die insbesondere durch die Verwendung von Modellorganismen gelöst bzw. umgangen werden kann. Die Auswahl eines geeigneten Modellorganismus orientiert sich maßgeblich an der Fragestellung der Untersuchung. Auf dem Forschungsgebiet der Reproduktion und Embryologie stellt die Maus das bisher am häufigsten genutzte Modell dar [317]. Erkenntnisse über die bovine Reproduktion, die in den letzten Jahrzehnten gewonnen werden konnten, charakterisieren das Rind jedoch zunehmend als geeigneteres Modell für die humane Reproduktion als die Maus. Im Gegensatz zu Nagern sind Kühe und Frauen unipar und haben eine vergleichbare Dauer des Menstruationszyklus (durchschnittlich 21 bis 22 Tage bei Kühen [125] und 28 Tage bei Frauen [318]) sowie eine vergleichbare Tragzeit (etwa 280 Tage bei Kühen [125] und 266 Tage bei Frauen [319]). Neben einer ähnlichen Morphologie und Größe der Ovarien [118], zeigen beide Spezies auch eine gleichartige Follikelentwicklung und -dynamik (siehe Kapitel 1.2.2) und weisen wichtige Gemeinsamkeiten in der finalen Phase der Eizellreifung auf. Vor allem die Zeitspannen der Entwicklungsabläufe der Keimzellen und der Embyonen, die einen entscheidenden Faktor darstellen, sind zwischen Rind und Mensch vergleichbar, wohingegen sie bei Nagern deutlich beschleunigt ablaufen [317]. Die wellenförmige Entwicklung der Follikel im Ovar beispielsweise, die bereits während der letzten zwei Dekaden beim Rind beobachtet und beschrieben wurde, diente als Grundlage für die später entdeckten, ähnlichen Wellen im humanen Ovar [118, 134, 320]. Auch beim Übergang zur Zygote und der frühen embryonalen Entwicklung weisen bovine und humane Zellen Ähnlichkeiten auf. Diese Analogien betreffen die Ausbildung der Mikrotubuli während der Befruchtung, den Zeitpunkt der Genomaktivierung, die erforderlichen metabolischen Bedingungen, die Interaktionen mit dem Kulturmedium sowie den zeitlichen Ablauf der Entwicklung vor der Implantation [236-239]. Ein ebenfalls wesentlicher Unterschied zum Mausmodell stellt die Interaktion zwischen dem Embryo und dem Gelbkörper (Corpus luteum) dar, die 35 EINLEITUNG entscheidend für den Verlauf der Schwangerschaft ist. Bei Mäusen muss Prolactin zum Zeitpunkt des Koitus ausgeschüttet werden, um die Transformation vom zyklischen Gelbkörper (Corpus albicans) zum Gelbkörper, der bei einer Schwangerschaft vorliegt (Corpus luteum graviditatis), zu induzieren. Diese Umwandlung des Gelbkörpers erfolgt in Kühen durch Tau-Interferon und in Frauen durch Choriongonadotropin, deren Ausschüttung in beiden Organismen durch ein Signal des Embryos ausgelöst wird [317]. Neben physiologischen und biochemischen Parallelen zwischen Rind und Mensch im gesunden Zustand zeigen beide Organismen auch Ähnlichkeiten bei Entwicklungsstörungen, die durch epigenetische Veränderungen verursacht werden. Wie bereits in Kapitel 1.3.3 beschrieben weist das sog. „Large offspring syndrome“ (LOS) beim Rind vergleichbare phänotypische wie epigenetische Auffälligkeiten zum humanen Beckwith-Wiedemann-Syndrom (BWS) auf [272, 273]. Sowohl das LOS beim Rind als auch das BWS beim Menschen werden mit einer gestörten Keimzellentwicklung bzw. -reifung und der Anwendung assistierter Reproduktionstechniken assoziiert [257-263, 266-268]. Diese Analogie unterstreicht das Potential des Hausrinds als geeigneten Modellorganismus für die humane Reproduktion, insbesondere für epigenetische Fragestellungen. 1.6 Zielsetzung der Arbeit Die Prozesse der DNA-Methylierung, die während der Entwicklung der unreifen Eizelle im Antralstadium zur befruchtungsfähigen Oozyte ablaufen, stellen essentielle Vorgänge für die Entwicklung des Embryos dar. In dieser Phase erlangt die Eizelle ihre Entwicklungskompetenz, die eine umfassende epigenetische Restrukturierung des Genoms mittels DNA-Methylierung einschließt. Wie die gesamte Eizellentwicklung bzw. Follikulogenese folgt auch der epigenetische Umbau einem Zeitplan, der bestimmte Veränderungen innerhalb spezifischer Zeitfenster vorsieht. Das Ziel der vorliegenden Arbeit bestand darin, den Einfluss der Verweildauer in spezifischen Entwicklungsphasen auf die DNA-Methylierung entwicklungsrelevanter Kandidatengene in unreifen und reifen Eizellen zu untersuchen. Durch die Analyse von drei unterschiedlichen Eizell-Kohorten wurde der Effekt von drei verschiedenen Zeitfenstern dokumentiert. Oozyten aus Antralfollikeln verschiedener Größe (<2 mm, 3-5 mm und >6 mm) repräsentierten Eizellen, die sich für unterschiedliche Zeitspannen im Antralstadium befinden (Abbildung 5). Follikel der mittelgroßen Kategorie (3-5 mm) wurden für 24 h und 48 h in vitro gereift, um den Einfluss der Reifungsdauer bzw. den Einfluss der postovulatorischen Alterung zu untersuchen. Aus 36 EINLEITUNG gereiften Eizellen beider Gruppen wurden Embryonen generiert, um auch mögliche Auswirkungen der Reifungsdauer für den Embryo zu identifizieren. Sowohl in den unreifen Eizellen aus Antralfollikeln unterschiedlicher Größe als auch in den gereiften Oozyten und den Embryonen wurden die Promotormethylierung der Gene bH19, bSNRPN, bZAR1, bDNMT3A, bOCT4, bDNMT3Lo und bDNMT3Ls erfasst (Abbildung 5). Im dritten Teil der Arbeit wurden ebenfalls Oozyten aus mittelgroßen Antralfollikeln analysiert, die aus Rindern unterschiedlichen Alters gewonnen wurden (9-12 Monate, 3-7 Jahre und 8-11 Jahre). Aufgrund der verschiedenen Lebensalter der Tiere stellen ihre Oozyten Eizellen dar, die für unterschiedliche Zeitspannen im Diplotän der Meiose I arretiert wurden und sich damit in unterschiedlichen Stadien der ovariellen Alterung befinden. Die DNA-Methylierungsanalysen umfassten hier die Promotorregionen der Gene bTERF2, bREC8, bBCL-XL, bPISD, bBUB1, bDNMT3Lo, bH19 und bSNRPN (Abbildung 5). Die daraus gewonnenen Daten und Erkenntnisse sollen dazu dienen, den Einfluss zeitlicher Faktoren bei der Etablierung der DNA-Methylierungsmuster in unreifen und reifen Eizellen besser zu verstehen. Neben der Erweiterung des Basiswissens der DNA-Methylierungsprozesse während der Eizellentwicklung und -reifung, sollen die Schlussfolgerungen dieser Arbeit auch als Grundlage zur Optimierung assistierter Reproduktionstechniken in der humanen Reproduktionsmedizin genutzt werden. Antralfollikel aus in vivo-Ovarien Unreife Eizellen aus Kühen unterschiedlichen Alters: 9-12 Monate 5-7 Jahre Antralfollikel aus Schlachthof-Ovarien Unreife Eizellen aus Antralfollikeln unterschiedlicher Größe: 8-11 Jahre <2 mm 3-5 mm In vitro gereifte Eizellen unterschiedlicher Reifungsdauer: 24h 3-5 mm >6 mm 24h DNA-Methylierung In vitro fertilisierte Embryonen aus Eizellen unterschiedlicher Reifungsdauer: 48h 48h DNA-Methylierung H19 SNRPN DNMT3Lo TERF2 REC8 BCL-XL PISD BUB1 H19 SNRPN DNMT3Lo ZAR1 OCT4 DNMT3A DNMT3Ls Abbildung 5: Übersicht der untersuchten Proben und Gene. Es wurden Antralfollikel in vivo aus Kühen unterschiedlichen Alters (9-12 Monate, 3-7 Jahre und 8-11 Jahre) gewonnen. In den daraus isolierten unreifen Eizellen wurde die DNAMethylierung der Promotorregionen der Gene bH19, bSNRPN, bDNMT3Lo, bTERF2, bREC8, bBCL-XL, bPISD und bBUB1 37 EINLEITUNG untersucht. Aus Schlachthof-Ovarien wurden Antralfollikel unterschiedlicher Größe (<2 mm, 3-5 mm und >6 mm) isoliert. Eizellen von Antralfollikeln mittlerer Größe (3-5 mm) wurden für 24h und 48h in vitro gereift. Aus beiden Kategorien in vitro gereifter Eizellen wurden durch in vitro Fertilisation Embryonen generiert und bis zum 4-6 Zellstadium kultiviert. Sowohl in den unreifen und gereiften Eizellen als auch in den generierten Embryonen wurden die Promotormethylierung der Gene bH19, bSNRPN, bDNMT3Lo, bZAR1, bOCT4, bDNMT3A und bDNMT3Ls analysiert. Die rot unterlegten Gene (bH19, bSNRPN, bDNMT3Lo, und bOCT4) dienten u.a. als Kontrollgene zu Erkennung somatischer Kontaminationen. Schematische Abbildung eines Ovars entnommen aus [321]. 1.7 Untersuchte Gene Die DNA-Methylierungsanalysen der vorliegenden Arbeit konzentrierten sich auf Promotorregionen von ausgewählten Kandidatengenen. Die Anzahl der analysierbaren Gene wird dabei von der Menge des vorhandenen Probenmaterials bzw. der verwendeten Methode limitiert. Bei der in dieser Arbeit verwendeten Methode der Limiting Dilution (LD) wird die maximale Anzahl der untersuchten Regionen maßgeblich durch die in der MultiplexPCR eingesetzten Primer-Sequenzen bestimmt (siehe Kapitel 2.2.2). In der Multiplex-PCR werden mehrere DNA-Regionen gleichzeitig amplifiziert. Dabei besteht das Risiko, dass Primer-Moleküle unspezifisch binden und unspezifische Amplifikate produzieren, welche die Analyse der Zielregionen beeinträchtigen. Um störende Interaktionen der Primer-Moleküle auszuschließen, muss die Kompatibilität der Primer-Kombinationen empirisch getestet werden. Das Genset 1 beinhaltete sieben DNA-Regionen (bH19, bSNRPN, bZAR1, bDNMT3A, bOCT4, bDNMT3Lo und bDNMT3Ls) und das Genset 2 acht Promotorregionen (bTERF2, bREC8, bBCL-XL, bPISD, bBUB1, bDNMT3Lo, bH19 und bSNRPN). Die Gene H19, SNRPN und DNMT3Lo waren in beiden Gensets enthalten, da sie aufgrund ihrer Prägung zusätzlich als Kontrollgene zum Nachweis somatischer Kontamination dienten (siehe 1.8). Die Funktion der untersuchten Gene beider Gensets wird im Folgenden ausführlich beschrieben. 1.7.1 Genset 1: Oozyten- und Maturations-spezifische Gene Die Untersuchung mit dem Genset 1 zielte darauf ab, Veränderungen der DNAMethylierungsmuster zu erkennen, die während der Entwicklung und Reifung der Eizelle auftreten. Dazu wurden die DNA-Regionen von bH19, bSNRPN, bZAR1, bDNMT3A, bOCT4, bDNMT3Lo und bDNMT3Ls (Abbildung 6) bei unreifen Eizellen und in vitro gereiften Eizellen unterschiedlicher Reifungsdauer sowie den daraus generierten Embryonen untersucht. Bei den Genen bH19 und bSNRPN handelt es sich um geprägte Gene, welche nur von einem elterlichen Allel exprimiert werden. Die genaue Funktion von H19 konnte bisher nicht vollständig aufgeklärt werden. Es ist bekannt, dass H19 die Transkription des Gens IGF2 (insulin-like growth factor 2) hemmt. Darüber hinaus wird vermutet, dass H19 eine RNA 38 EINLEITUNG kodiert, welche als primärer Vorläufer einer microRNA fungiert, die spezifische mRNAMoleküle während der Entwicklung von Wirbeltieren herunterreguliert. In Eizellen liegt der Promotor von H19 unmethyliert vor, während er in Spermien methyliert ist [59, 322, 323]. SNRPN (small nuclear ribonucleoprotein N) kodiert einen Spleiß-Faktor und wird durch eine differentiell methylierte Region (DMR) reguliert. Diese DMR ist in Eizellen methyliert und wird im Falle einer abnormalen Methylierung mit dem Prader-Willi- und dem AngelmanSyndrom in Verbindung gebracht [324]. Die molekulare Funktion des Gens ZAR1 (zygote arrest 1) ist im Detail ebenfalls nicht bekannt. Studien mit Knockout-Mäusen beobachteten jedoch eine entscheidende Rolle beim Übergang von der Eizelle zum Embryo. Im Rind wird ZAR1 während der gesamten Phase der Präimplantation des Embryos exprimiert. Das Transkript von ZAR1 wurde auch in den Ovarien, den Hoden, dem Herzen und den Muskeln nachgewiesen. Das Protein dieses für die Eizelle essentiellen Gens enthält eine evolutionär konservierte “plant-homeo Domäne“ (PHD), welche eine generelle Rolle in der Regulation der Transkription vermuten lässt [325327]. Oct4 (Octamer-binding transcription factor 4) ist eines der validesten Marker-Gene für epigenetische Reprogrammierung und Pluripotenz. Das von OCT4 kodierte Protein verfügt über eine Pit-Oct-Unc (POU) Domäne, welche an ein Oktamer-Motiv als Erkennungssequenz innerhalb von Promotor- oder Enhancer-Regionen bindet und die Transkription der Zielgene reguliert. In pluripotenten Zellen wie embryonalen Stammzellen, embryonalen Krebszellen und Eizellen liegt Oct4 unmethyliert vor und wird exprimiert. Die Struktur, Sequenz und Lokalisation des Gens sowie die regulatorischen Regionen und Expressionsmuster sind zwischen den orthologen Genen der Maus, des Menschen sowie des Rinds hoch konserviert [328-330]. DNA-Methyltransferasen (DNMTs), wie DNMT1B, DNMT3A und DNMT3L, sind essentielle Enzyme zur Etablierung und Aufrechterhaltung der korrekten DNA-Methylierungsmuster an geprägten und nicht-geprägten DNA-Regionen während der Entwicklung. Dabei erfolgt durch DNMT3A in Kombination mit DNMT3L die Etablierung der initialen CpG- Methylierungsmuster (de novo-Methylierung) während der Gametogenese. DNMT1B hingegen ist vorrangig an der Aufrechterhaltung der Muster während der ChromosomenReplikation und –Reparatur beteiligt. DNMT3L hat im Komplex mit DNMT3A keine 39 EINLEITUNG katalytische Aktivität und dient vielmehr als ein navigierendes Coenzym, indem es an spezifische Histonmodifikationen bindet und DNMT3A rekrutiert. Studien in Mäusen zeigten, dass DNMT3L durch drei unterschiedliche Keimzelllinien-spezifische Promotoren reguliert wird und verschiedene Transkripte in Stammzellen (DNMT3Ls), Eizellen (DNMT3Lo) und Hoden (DNMT3Lat) exprimiert. Dabei wird das Transkript von DNMT3Lat nicht translatiert, wohingegen DNMT3Ls und DNMT3Lo Proteine produzieren, die durch DNA-Methylierung reguliert werden [331-333]. 40 Genset 1 EINLEITUNG 230 bp 18 CpGs bZAR1 211 bp 16 CpGs bDNMT3A 270 bp 8 CpGs bDNMT3Ls 52 bp 7 CpGs Genset 1 und 2 bOCT4 276 bp 30 CpGs bSNRPN bDNMT3Lo bTERF2 Genset 2 230 bp 20 CpGs bH19 bREC8 bBCL-XL 56 bp 5 CpGs 35 bp 7 CpGs 46 bp 5 CpGs 48 bp 8 CpGs 51 bp 6 CpGs bPISD bBUB1 38 bp 6 CpGs Abbildung 6: Schematische Darstellung der Promotorregionen des Gensets 1 und Gensets 2. Die untersuchten Promotorregionen von bZAR1, bDNMT3A, bDNMT3Ls, bOCT4, bH19, bSNRPN, bDNMT3Lo, bTERF2, bREC8, bBCL-XL, bPISD und bBUB1 sind als Lollipop-Diagramme dargestellt. Dabei respräsentiert eine schwarze Linie die untersuchte DNA-Region mit den darin enthaltenen CpG-Dinukleotiden als sog. Lollipops. Die Länge der schwarzen Linien und die Distanzen zwischen den Lollipops entsprechen den relativen Längen bzw. Abständen im Genom. bH19, bSNRPN und bDNMT3Lo sind in beiden Gensets (Genset 1 und Genset 2) enthalten. bZAR1, bDNMT3A, bDNMT3Ls, bH19, bSNRPN wurden mittels Sanger-Sequenzierung sequenziert. bOCT4, bDNMT3Lo, bTERF2, bREC8, bBCL-XL, bPISD und bBUB1 wurden mittels Pyrosequenzierung untersucht. Die Größe der untersuchten DNA-Region ist in Basenpaaren (bp) angegeben. 41 EINLEITUNG 1.7.2 Genset 2: Oozyten- sowie Alters-spezifische Gene Die in Genset 2 zusammengestellten DNA-Regionen wurden in unreifen in vivo gewonnen Eizellen von Rindern unterschiedlichen Alters untersucht. Ziel dieser Analyse war es, Veränderungen der DNA-Methylierung der Eizelle zu identifizieren, die durch Alterungsbedingte Effekte der Spendertiere verursacht wurden. Zur Untersuchung wurden daher insbesondere Gene ausgewählt, die im Zusammenhang mit Alterungs-bedingten Veränderungen stehen. Die mit Genset 2 analysierten Promotorregionen umfassen die Gene bTERF2, bREC8, bBCL-XL, bPISD, bBUB1, sowie die bereits in Abschnitt 1.7.1 beschriebenen Gene bDNMT3Lo, bH19 und bSNRPN (Abbildung 6). Terf2 (telomeric repeat binding factor 2) gehört zur Gruppe der Telomer-assoziierten Proteine des Shelterin-Komplexes, welche die Stabilität und Länge der Telomere regulieren [334]. Die Funktion des Proteins besteht darin, die Bildung des sogenannten T-Loops zu vermitteln und die Chromosomen vor einer Kopf-Kopf-Fusion zu schützen [335, 336]. Darüber hinaus ist Terf2 auch an DNA-Reparatur-Mechanismen beteiligt [337]. Die Expression eines dominant-negativen Allels von Terf2 führt zum Verlust des G-Überhangs, genomischer Instabilität, Kopf-Kopf-Fusion der Chromosomen und zur Aktivierung von Faktoren der DNA-Schadensantwort [338]. Studien im Rind konnten eine dynamische mRNAExpression für verschiedene Entwicklungsstadien von der Eizelle im Germinalvesikel-Stadium bis hin zum Blastozysten-Stadium nachweisen [339]. Im Ovar von Mäusen führt Methylisocyanat, ein Reagenz, das Schäden durch reaktive Sauerstoffspezies verursacht, zu einem Verlust der Expression von Terf2 und einer vorzeitigen Seneszenz der Epithelzellen [340]. Ein Hinweis auf eine mögliche epigenetische Regulation dieses Gens ist die Hypomethylierung einer CpG-Insel im Promotorbereich bei gleichzeitiger Hochregulierung der Transkription in humanen Magenkarzinomen. In diesem Gewebe korreliert die Abnahme der Transkription ebenfalls stark mit einer Hypermethylierung des Gen-Promotors [341]. Die Ursachen maternaler, altersbedingter Aneuploidie beruhen insbesondere auf Störungen der Rekombination, Defekte des Spindelapparat-Kontrollpunkts (spindle assembly checkpoint (SAC)) und vorzeitiger Verlust der Schwesterchromatid-Kohäsion [342, 343]. Während der Anordnung der Chromosomen in der Äquatorialebene (Metaphase) erkennt der SAC einzelne nicht verbundene Kinetochore und hemmt den Anaphase-fördernden Komplex (anaphase promoting complex, APC) bzw. das Zyklosom (C) und den Übergang zur Anaphase. 42 EINLEITUNG Dadurch wird eine Fehlsegregation der Chromosomen verhindert bevor die Schwesterchromatiden korrekt mit dem Spindelapparat verbunden sind. Das Kandidatengen BUB1 (budding uninhibited by benzimidazole 1) nimmt innerhalb dieses KontrollpunktSystems die molekulare Rolle einer Kinase ein und phosphoryliert den APC/C-Aktivator Cdc20 [344]. Maus-Studien zeigten, dass ein Abbau von BUB1 durch Mikroinjektion eines spezifischen Antikörpers (anti-BUB1) in Eizellen von Mäusen zu einer vorzeitigen Anaphase und einer erhöhten abnormalen Anordnung und Segregation von Chromosomen führt [345]. Es konnte ebenfalls in Mäusen nachgewiesen werden, dass Heterozygotie für ein verkürztes BUB1-Protein bei weiblichen Tieren zu einer altersabhängigen Verstärkung der Aneuploidie in Keimzellen führt, welche über die Zygote vererbt wird und das Überleben der Embryonen beeinträchtigt [346]. Die vollständige Deletion von BUB1 in Maus-Keimzellen führt zur Sterilität [347]. Das Kandidatengen REC8 ist ebenfalls in der Segregation von Chromosomen involviert und stellt die meiosespezifische Untereinheit des Cohesin-Komplexes dar. Es ist bekannt, dass Eizellen von Frauen im Alter von 40 Jahren und älter geringere Konzentrationen von Rec8 aufweisen als Oozyten von Frauen im Alter von 20 Jahren [348]. Ein ähnliches Ergebnis lieferte ein Vergleich von Eizellen aus jungen und alten Tieren bei Mäusen [349]. Die molekulare Verbindung zwischen Rec8 und Bub1 ist das Protein Shugoshin, welches durch BUB1 reguliert wird und Rec8 vor einer Spaltung durch die Separase schützt [350-352]. Bcl-XL gehört zur Gruppe der Apoptose-regulierenden Proteine der Bcl-2 Familie, benannt nach dem B-Zell Lymphom 2. Durch die Bindung pro-apoptotischer Proteine wie Bad, Bid und Bim erfüllt Bcl-XL eine anti-apoptotische Funktion in der äußeren Mitochondrienmembran [353, 354]. Bcl-XL wird in humanen, unreifen Eizellen exprimiert und dient dort als eines der wichtigsten anti-apoptotischen Proteine [355]. Die Phosphatidylserin Membranphospholipids Decarboxylase (PISD) Phosphatidylethanolamin katalysiert durch die die Bildung Decarboxylierung des von Phosphatidylserin. PISD ist auf der äußeren Oberfläche der inneren Mitochondrienmembran aktiv und nimmt eine zentrale Rolle im Phospholipid-Stoffwechsel ein. Die Inaktivierung von PISD in Mäusen ist letal und führt zwischen Tag 8 und Tag 10 der embryonalen Entwicklung zum Absterben der Frühstadien mit abnormal geformten Mitochondrien in den betroffenen 43 EINLEITUNG Embryonen [356]. Verringerte mRNA-Konzentrationen des Gens wurden in Eizellen von älteren Frauen nachgewiesen [357]. 1.8 Kontaminationskontrolle Molekularbiologische Untersuchungen von Keimzellen bergen stets das Risiko einer Kontamination durch somatische Zellen. Um zu gewährleisten, dass die generierten Daten ausschließlich von Keimzellen stammen, stellt der Einsatz einer geeigneten und verlässlichen Kontrolle zum Ausschluss somatischer Kontamination eine der größten Herausforderungen bei der molekularbiologischen Arbeit mit Keimzellen dar. Sowohl Genset 1 als auch Genset 2 enthielten die beiden geprägten Gene H19 und SNRPN sowie das entwicklungsrelevante, nicht-geprägte Gen DNMT3Lo. Zusätzlich dazu umfasste Genset 1 mit Oct4 ein weiteres nicht-geprägtes und für die Entwicklung entscheidendes Gen. H19 ist paternal geprägt und in der Eizelle unmethyliert, wohingegen SNRPN maternal geprägt ist und damit methyliert in der Oozyte vorliegt. Im Unterschied zur Eizelle besteht das Methylierungsmuster somatischer Zellen aus dem maternalen und dem paternalen Allel und zeigt eine Mischung aus methylierten und unmethylierten Molekülen für beide Gene. DNMT3Lo stellt den Eizellspezifischen Promotor des Gens DNMT3L dar. Sowohl DNMT3Lo als auch der PluripotenzMarker Oct4 liegen in der Eizelle unmethyliert vor und sind vollständig methyliert in somatischen Zellen. Die unterschiedlichen Methylierungszustände in der Eizelle und in somatischen Zellen machen alle 4 Genregionen zu geeigneten Kontrollgenen, um somatische Kontaminationen zu erkennen. Gleichzeitig können somatische Methylierungsmuster jedoch ebenfalls durch abnormal methylierte Allele (Epimutationen) der Eizelle verursacht werden. Zur Abgrenzung einer somatischen Kontamination von einer Epimutation wurde eine somatische Kontamination definiert als das Auftreten von mindestens einem abnormal methylierten Allel (>50% abnormal methylierte CpGs) in mindestens 2 dieser 3 (Genset 2) bzw. 4 (Genset 1) Promotorregionen innerhalb des gleichen Eizell-Pools. 44 MATERIAL UND METHODEN 2 Material und Methoden 2.1 Material 2.1.1 Biologisches Material Die untersuchten Eizellen und Embryonen stammen von Holstein-Rindern und wurden von Mitarbeitern des Instituts für Nutztiergenetik des Friedrich-Löffler-Instituts (FLI) in Mariensee/Hannover präpariert und bereitgestellt. 2.1.1.1 Unreife Eizellen aus Schlachttieren Die unreifen Eizellen, die nach unterschiedlichen Follikelgrößen gruppiert und für die in vitro Reifung (IVM), in vitro Fertilisation (IVF) sowie in vitro Kultivierung verwendet wurden, stammten aus Schlachttieren. Die Isolation und Präparation wurde nach etablierten Protokollen im FLI durchgeführt [358, 359]. Dazu wurden die Rinderovarien für den Transport vom Schlachthof zum Labor in 0.9%iger NaCl-Lösung mit 6 µg/ml Penicillin und 50 µg/ml Streptomycin aufbewahrt und anschließend mit frischer Lösung gleicher Zusammensetzung dreimal gewaschen. Die Gewinnung der unreifen Oozyten mitsamt den sie umgebenden Kumuluszellen (COCs) erfolgte durch behutsames Schneiden („Slicen“) der Ovarien in Dulbecco´s 1 x PBS - Medium (Sigma-Aldrich, München) versetzt mit 0,33 mM Natriumpyruvat (Sigma-Aldrich), 5,56 mM D-Glukose (Roth, Karlsruhe), 0,9 mM Kalziumchloriddihydrat (Fluka, Buchs, Schweiz), 50 µg/ml Streptomycinsulfat (AppliChem, Darmstadt), 6 µg/ml Penicillin G (AppliChem), 4 IU/l Heparin und 1 mg/ml BSA (Fraktion V) (Sigma-Aldrich). Die Auswahl der COCs für die nachfolgenden Analysen erfolgte unter einem Stereomikroskop bei 50 x Vergrößerung in TCM-air pH 7,2 bestehend aus TCM199 (SigmaAldrich), 50 µg/ml Gentamycinsulfat (Sigma-Aldrich), 0,2 mM Natriumpyruvat, 4,2 mM Natriumhydrogencarbonat (Roth) und 1 mg/ml BSA-FAF (Sigma-Aldrich). Um ein homogenes und gutes Entwicklungspotential der Oozyten zu gewährleisten, wurden nur COCs mit einem homogenen, gleichmäßig granulierten Zytoplasma und mindestens 3 Schichten kompakt umgebender Kumuluszellen ausgewählt (Klasse I-II nach [360, 361]). Zum Entfernen der Kumuluszellen wurden die COCs für 5 min bei 39 °C in 1 x PBS mit 1 mg/ml BSA (Fraktion V) und 0,1% Hyaluronidase (aus Rinderhoden; Sigma-Aldrich) inkubiert und anschließend für 5 min bei 1.400 rpm gevortext. Verbliebene, anhaftende Kumuluszellen wurden behutsam mit einer Pipette entfernt. Die isolierten, unreifen Oozyten wurden abschließend dreimal in 45 MATERIAL UND METHODEN PBS mit 0.1 % Polyvinylalkohol (PVA) gewaschen und in Gruppen von 5-10 Eizellen für die Analyse der DNA-Methylierung bei -80 °C aufbewahrt. 2.1.1.2 Unreife Eizellen aus Kühen unterschiedlichen Alters Die unreifen Eizellen aus Rindern unterschiedlichen Alters wurden durch eine ultraschallgesteuerte transvaginale Follikelpunktion („ovum pick up“, OPU) gewonnen. Die Kohorten der Spendertiere bestanden dabei aus 13 präpubertären Kühen (9-12 Monate alt), 8 erwachsenen weiblichen Rindern (3-7 Jahre alt) und 5 alten Kühen (>8-11 Jahre alt). Die OPU wurde zweimal pro Woche in einem Zeitintervall von 3 bis 4 Tagen durchgeführt. Vor der experimentellen Durchführung wurden die Tiere einer sorgfältigen Voruntersuchung unterzogen, um den allgemeinen Gesundheitszustand sowie den Entwicklungsstand und die allgemeine Konstitution der Reproduktionsorgane zu beurteilen. Die ultraschallgesteuerte transvaginale Follikelpunktion wurde mit wenigen Modifikationen nach etablierten Protokollen durchgeführt [323, 362-364]. Die Tiere wurden zunächst in einen Zwangsstand geführt und fixiert. Anschließend wurden die äußeren Geschlechtsorgane gereinigt und desinfiziert. Zur Visualisierung der Ovarien wurde ein Aloka Echtzeit B-Mode Ultraschallsystem (Aloka SSD-4000, Hitachi Aloka Medical Ltd., Tokyo, Japan) und eine 7,5 MHz Konvexsonde (Hitachi Aloka Medical Ltd., Tokyo, Japan) verwendet. Die Sonde wurde in einen aus POM (Polyoxymethylen) bestehenden und in Mariensee entwickelten Sondenträger eingebettet. Sondenträger und Sonde wurden mit einer EinmalSchutzfolie (32 mm x 190 mm, Servoprax® GmbH, Wesel) und Gleitgel (Bovivet Gel, Kruuse, Langeskov, Dänemark) versehen und in die Vagina eingeführt. Sichtbare Follikel mit einem Durchmesser von ≥ 3 mm wurden mit einer Einwegnadel 20G x 2¾” (0,9 x 70 mm, Terumo, Eschborn) punktiert, welche mit einem flexiblen Schlauchsystem (1,5 mm x 0,5 mm x 1,5 m, PTFE, BOLA, Grünsfeld) und einem sterilen 50 ml Zentrifugenröhrchen (Corning®, New York, USA) verbunden war. An dem konischen 50 ml Zentrifugenröhrchen wurde eine Vakuumpumpe (Aspirator 3, Labotect GmbH, Göttingen) angebracht und ein negativer Druck von 60 mmHg (20 ml/min) angelegt. Nach der Punktion von 4-5 Follikeln wurden die Punktionsnadel und das Schlauchsystem mit Dulbecco´s 1 x PBS - Medium (AppliChem, Darmstadt) versetzt mit 2,2 IU/ml Heparin (AppliChem) und 1% hitzeinaktiviertem, fetalem Kälberserum (PPA Laboratories, Coelbe) gespült. Nach der OPU wurden die COCs jedes Spendertieres einzeln filtriert, gewaschen und während der Identifikation in Dulbecco´s 1 x 46 MATERIAL UND METHODEN PBS - Medium (AppliChem, Darmstadt) klassifiziert. Ausschließlich COCs mit einem homogenen, gleichmäßig granulierten Zytoplasma und kompakt umgebenden Kumuluszellen wurden für die weitere Analyse verwendet. Die Kumuluszellen wurden entfernt, indem die COCs für 5 min bei 39 °C in 1 x PBS mit 1 mg/ml BSA (Fraktion V) und 0,1% Hyaluronidase (aus Rinderhoden; Sigma-Aldrich) inkubiert wurden und anschließend für 5 min bei 1.400 rpm gevortext wurden. Verbliebene, anhaftende Kumuluszellen wurden behutsam mit einer Pipette entfernt. Die isolierten, unreifen Oozyten wurden abschließend in Gruppen von 5-10 Eizellen für die Analyse der DNA-Methylierung bei -80 °C aufbewahrt. 2.1.1.3 In vitro gereifte Eizellen Für die in vitro Reifung wurden unreife Eizellen mit anhaftenden Kumuluszellen verwendet, den sogenannten Kumulus-Eizell-Komplexen (COCs). Diese wurden wie in 2.1.1.1 beschrieben aus Ovarien von Schlachttieren gewonnen. Die Reifung der Eizellen erfolge somit vor dem Entfernen der Kumuluszellen. Dazu wurden die COCs in Gruppen von 15 in einem Tropfen von 100 μl TCM199 pH 7,4, versetzt mit 0,2 mM Natriumpyruvat , 25 mM Natriumhydrogencarbonat, 1 mg/ml BSA-FAF, 10 IU/ml equinem Choriongonadotropin (PMSG) und 5 IU/ml humanem Choriongonadotropin (Suigonan®, Intervet, Unterschleißheim) in einem Inkubator bei 39 °C, 5% CO2-Gehalt und gesättigter Luftfeuchtigkeit für 24h (Standard Protokoll) oder 48h (simulierte post-ovulatorische Alterung) gereift. Nach Abschluss der Maturation wurden die Kumuluszellen wie in 2.1.1.1 beschrieben mittels Hyaluronidasebehandlung entfernt und vereinzelt verbliebene Kumuluszellen mit einer Pipette beseitigt. Eine erfolgreiche Reifung wurde durch den Ausschluss des ersten Polkörpers bestimmt. Die gereiften Oozyten wurden ebenfalls abschließend dreimal in PBS mit 0.1 % Polyvinylalkohol (PVA) gewaschen und in Gruppen von 5-10 Eizellen für die Analyse der DNA-Methylierung bei -80 °C aufbewahrt. 2.1.1.4 Rinderembryonen Rinderembryonen wurden aus Eizellen bzw. COCs generiert, die wie in 2.1.1.3 beschrieben einer in vitro Reifung unterzogen wurden und anschließend in vitro fertilisiert und in vitro kultiviert wurden. Sowohl die in vitro Fertilisation (IVF) als auch die in vitro Kultivierung wurden mit geringfügigen Modifikationen nach bereits etablierten Protokollen durchgeführt [358, 359]. Für die Befruchtung wurde aufgetautes Tiefgefriersperma mit einer Konzentration von 1x106 Spermien/ml eines Bullen mit nachgewiesenem IVF-Erfolg 47 MATERIAL UND METHODEN verwendet. Das Sperma wurde zunächst bei 30 °C für 1 min aufgetaut und mittels BoviPureTM (Labotect) dem Standard-Protokoll entsprechend aufgereinigt. Die SpermienPellets wurden zwischen den Zentrifugationsschritten mit Fert.-TALP gewaschen [365, 366]. Nach dem letzten Zentrifugationsschritt wurden die Spermien-Pellets in 50 μl Fert.-TALP versetzt mit HHE (10 µM Hypotaurin (Sigma-Aldrich), 0,1 IU/ml Heparin (Serva), 1µM Epinephrin (Sigma-Aldrich) und 6 mg/ml BSA (Fraktion V)) resuspendiert. Zur Vorbereitung der IVF wurden die gereiften COCs zweimal mit 100 μl Fert.-TALP gewaschen und zur Befruchtung in Gruppen von 15 in 100 μl Fert.-TALP/HHE mit den resuspendierten Spermien bei 5 % CO2 und 39 °C für 19 h koinkubiert. Nach der Inkubation wurden die Kumuluszellen mechanisch durch vortexen bei 1.400 rpm für 5 min und anschließendes behutsames pipettieren entfernt. Die anhand eines exkludierten Polkörpers als befruchtet kategorisierten Eizellen wurden zweimal mit SOFaa-Kulturmedium versetzt mit 4 mg/ml BSA-FAF [359] gewaschen und in Gruppen von 5 Zellen in 30 μl Tropfen unter Silikonöl bei 5 % O2, 90 % N2 und 5 % CO2 in feuchter Atmosphäre bei 39 °C kultiviert. Eine Prüfung auf Furchungsstadien erfolgte nach 3 Tagen Kultivierung. 2.1.2 Puffer Die hergestellten Puffer umfassen den Denaturierungspuffer, den Waschpuffer und TAEPuffer. Der Denaturierungspuffer sowie der Waschpuffer wurden zur Immobilisierung und Aufreinigung der PCR-Produkte für die Pyrosequenzierung mittels PyroMark Q96 Vacuum Workstation verwendet. Der TAE-Puffer wurde zur Identifikation der PCR-Produkte mittels Gelelektrophorese benötigt. Die genaue Zusammensetzung der Puffer ist in Tabelle 1 aufgeführt. Tabelle 1: Zusammensetzung der hergestellten Puffer. Puffer Zusammensetzung Denaturierungspuffer 0,2 M NaOH TAE-Puffer (50 x) 2 M TRIS 1 M Essigsäure 0,05 M EDTA pH 8,0 48 MATERIAL UND METHODEN Waschpuffer 10 mM TRIS 4 M Essigsäure bis pH 7,6 2.1.3 Kommerzielle Kits und Reagenzien Alle kommerziell vertriebenen Kits sowie Reagenzien, die für diese Arbeit verwendet wurden, sind in Tabelle 2 aufgelistet. Tabelle 2: Verwendete Kits und Reagenzien. Produkt Hersteller Sitz BigDye® Terminator v1.1 Cycle Sequencing Kit Applied Biosystems/ Technologies CleanDTR Magnetic Beads GCbiotech Alphen aan den Rijn, Niederlande ExoSAP- IT® Affymetrix Santa Clara , CA/USA EZ DNA Methylation-Direct™ Kit Zymo Research Freiburg FastStart Taq DNA PolymeraseSystem Roche Diagnostics Mannheim Hi - Di™ Formamide Applied Biosystems/ Life Darmstadt Life Darmstadt Technologies PyroGold® SQA Reagent Kit Qiagen Hilden 2.1.4 Geräte Die für diese Arbeit benötigten Geräte mit der genauen Typenbezeichnung sowie dem Hersteller sind in Tabelle 3 aufgeführt. Tabelle 3: Verwendete Geräte. Gerät Typenbezeichnung Hersteller Elektrophoresekammer STARLAB Horizontal Gel System Starlab 49 MATERIAL UND METHODEN Gel-Dokumentation Intas Gel iX Imager Intas-Science-Imaging Instruments GmbH Heizblock Biometra Thermoblock TB2 Analytik Jena AG Kapillarsequenzierer Applied Biosystems 3130/3130xl Genetic Analyzer Applied Biosystems/ Technologies PCR Werkbank AirClean 600 PCR Workstation AirClean / Starlab Pipettier-Roboter JANUS® Automated Workstation PerkinElmer Inc. Pyrosequenzierer Pyromark Q96MD Qiagen Spannugsquellen Bio-Rad PowerPac 1000 Bio-Rad Thermocycler DNA Engine Cycler Thermal Bio-Rad Vakuum-Station PyroMark Q96 Vacuum Workstation Qiagen Tetrad®2 Life 2.2 Methoden Das Methodenspektrum, das zur Generierung der Daten der vorliegenden Arbeit genutzt wurde, umfasst im Wesentlichen die Natriumbisulfitbehandlung von DNA, die Limiting Dilution, die Polymerasekettenreaktion sowie die Sequenzierung der amplifizierten DNARegionen. Eine zentrale Rolle nimmt hierbei die Limiting Dilution ein, die wie die anderen Methoden im Folgenden detailliert beschrieben und erläutert wird. 2.2.1 Natriumbisulfitbehandlung der DNA von Eizellen und Embryonen Die chemische Behandlung von DNA mit Natriumbisulfit ermöglicht es, die sogenannte 5. Base, 5-Methylcytosin, nachzuweisen. Das Prinzip dieser Methode basiert auf der Eigenschaft des Natriumbisulfits, selektiv Cytosin-Reste zu deaminieren. Die spezifische Deaminierung von Cytosin durch Natriumbisulfit wurde erstmals 1970 von zwei Gruppen unabhängig voneinander beobachtet [367, 368] und 1992 erstmals zur Detektion von 5-Methylcytosin in DNA genutzt [369]. Die Bisulfit-vermittelte Deaminierung des Cytosins erfolgt in 3 Schritten (Abbildung 7) [370]. Im ersten Schritt erfolgt eine reversible Addition von Bisulfit (HSO3-) an Cytosin (Sulfonierung). Im zweiten Schritt kommt es dann zur 50 MATERIAL UND METHODEN Deaminierung durch eine hydrolytische Abspaltung von NH3. Der dritte Schritt führt zur Freisetzung von Bisulfit (HSO3-), wodurch sich erneut eine 5,6-Doppelbindung ausbildet und Uracil entsteht (Desulfonierung). Der geschwindigkeitsbestimmende Schritt der Gesamtreaktion ist die Deaminierung in Schritt 2. 1. Sulfonierung R Cytosin 2. Deaminierung R Cytosin-6-Sulphonat 3. Desulfonierung R Uracil-6-Sulphonat R Uracil R 5-Methylcytosin Abbildung 7: Bisulfit-vermittelte Deaminierung von Cytosin. Die Bisulfit-vermittelte Deaminierung des Cytosins erfolgt in 3 Schritten bestehend aus einer Sulfonierung (1), Deaminierung (2) und einer Desulfonierung (3). Die Deaminierung von 5-Methylcytosin vermittelt durch Natriumbisulfit erfolgt in einer zweihundertfach geringeren Geschwindigkeit als bei unmethyliertem Cytosin. Abbildung übernommen aus [370] und modifiziert. Das resultierende Produkt Uracil-6-Sulphonat ist unter neutralen Bedingungen eine stabile Verbindung. Im alkalischen Milieu wird die Desulfonierung von Uracil-6-Sulphonat unter Bildung von Uracil begünstigt, wohingegen die Konvertierung von Cytosin zu Uracil-6Sulphonat optimal im sauren pH-Bereich (pH 5-6) erfolgt. Die Deaminierung von 5-Methylcytosin unter dem Einfluss von Natriumbisulfit findet ebenfalls statt, jedoch erfolgt diese Reaktion in einer zweihundertfach geringeren Geschwindigkeit als bei unmethyliertem Cytosin [371]. Diese Eigenschaft von Natriumbisulfit, selektiv unmethylierte Cytosine zu deaminieren, ermöglicht es 5-Methylcytosine zu detektieren. Die Identifikation erfolgt dabei durch eine auf die Natriumbisulfitbehandlung folgende PCR und eine anschließende Sequenzierung des Produkts. Unmethylierte Cytosine werden durch die Bisulfitbehandlung zu Uracil konvertiert, welches in der PCR als Thymin amplifiziert wird. 5-Methylcytosine 51 MATERIAL UND METHODEN hingegen bleiben unverändert und treten im sequenzierten PCR-Produkt als Cytosine in Erscheinung (Abbildung 8). T C T C G A C G A G A G C T G C G A U G Denaturierung Natriumbisulfitbehandlung T U T C PCR T T T C G A T G A A A G C T A C Abbildung 8: Konvertierung von Cytosin zu Thymin durch eine Natriumbisulfitbehandlung mit anschließender PCR. Die unmethylierten Cytosine (C) werden durch Natriumbisulfit zu Uracil deaminiert und durch die PCR als Thymine amplifiziert. 5-Methylcytosin (C mit CH3-Gruppe) bleibt nach der Natriumbisulfitbehandlung und PCR unverändert. Der genaue Mechanismus, durch den die Hydrolyse bzw. Deaminierung von Cytosin durch Bisulfit katalysiert wird, ist nach wie vor nicht vollständig aufgeklärt [370, 372, 373]. Es wird vermutet, dass die Addition von Bisulfit an Cytosin durch das Radikal erfolgt, das aus einer Interaktion von Bisulfit und Sauerstoff generiert wird (•SO3-) [370]. Diese reaktive Spezies würde ebenfalls den hohen Grad an DNA-Degradation erklären (84-96%), der unter optimalen Reaktionsbedingungen mit Natriumbisulfit beobachtet wird [374, 375]. Trotz dieser bestehenden technischen Herausforderungen, stellt die Natriumbisulfitbehandlung bis heute die präziseste, effizienteste und am häufigsten genutzte Methode zur Identifizierung von 5-Methylcytosin in genomischer DNA dar [376]. 2.2.2 Limiting Dilution Zur Untersuchung der DNA-Methylierungsmuster boviner Eizellen und Embryonen wurde die Limiting-Dilution-Methode verwendet. Bei dieser Methode wird die DNA so stark verdünnt, dass im Reaktionsansatz zur Polymerasekettenreaktion (PCR) nur ein einzelnes DNA-Molekül bzw. Allel als Matrize dient [377]. Im Vergleich zu genomischer DNA birgt Natriumbisulfitbehandelte DNA ein höheres Risiko, die unterschiedlich modifizierten DNA-Moleküle der Zielregion asymmetrisch zu amplifizieren [376]. Die Natriumbisulfitbehandlung hat zur Folge, 52 MATERIAL UND METHODEN dass methylierte Moleküle Cytosin-reicher sind, als unmethylierte Allele, welche hingegen mehr Thymin-Basen enthalten. Dieses Gemisch von DNA-Molekülen mit unterschiedlichem chemischem Charakter führt in der PCR dazu, dass eine oder mehrere Populationen von Molekülen stärker amplifiziert werden als andere [378]. Da die Amplifikation exponentiell verläuft, können geringe Ungleichgewichte in der Amplifikation unterschiedlicher DNAMatrizen zu Beginn der PCR zu einer erheblichen Verschiebung des Gemisches von DNASpezies im endgültigen PCR-Produkt führen. Die Wahrscheinlichkeit solch einer Verzerrung nach Amplifikation Bisulfit-konvertierter DNA steigt, je weniger DNA in die PCR eingesetzt wird. Bei der Limiting-Dilution-Methode wird dieses Risiko einer asymmetrischen Amplifikation reduziert, indem nur ein einzelnes DNA-Molekül als Matrize in eine PCR eingebracht wird [379]. In Verbindung mit zwei aufeinander folgenden PCRs wird es ermöglicht, auch mehrere Regionen auf dem gleichen DNA-Molekül zu analysieren (Abbildung 9). Dazu werden alle zu untersuchenden Regionen in einer ersten Multiplex-PCR zunächst gleichzeitig amplifiziert. In einer zweiten, sogenannten Singleplex-PCR werden dann die Regionen der einzelnen Gene separat vervielfältigt. Das anschließende Auftragen der PCR-Produkte auf ein Gel liefert eine binäre Information über das Vorhandensein der spezifischen Ziel-DNA in den jeweiligen Wells. Dabei kann entweder kein PCR-Produkt (0) oder ein PCR-Produkt (1) vorliegen, weshalb diese Methode auch als digitale PCR bezeichnet wird [380]. Die Kombination aus Natriumbisulfitbehandlung, Limiting Dilution, Multiplex-PCR, Singleplex-PCR und einer finalen Sequenzierung der generierten Amplifikate wird als Limiting Dilution Bisulfite Sequencing (LD) bezeichnet [377]. Die LD eignet sich insbesondere zur Untersuchung von Einzelzellen bzw. Proben mit geringer Zellzahl. Der geringe DNA-Gehalt der Proben ermöglicht es die Verdünnung technisch umzusetzen und gleichzeitig ein repräsentatives Bild der DNA-Methylierungsmuster in der Auflösung einzelner Moleküle und damit einzelner Allele zu liefern. 53 MATERIAL UND METHODEN Eizellen / Embryonen Bisulfitbehandlung Verdünnung Multiplex-PCR Singleplex-PCR H19 SNRPN DNMT3A TERF2 Sequenzierung Abbildung 9: Schematischer Ablauf der Limiting-Dilution-Methode (LD). Pools von 5-10 Oozyten und vereinzelte Embryonen im 4- bis 8-Zell-Stadium wurden im ersten Schritt einer Bisulfitbehandlung unterzogen und anschließend bei der Präparierung des PCR-Ansatzes limitierend verdünnt. Der PCR-Ansatz eines Eizellpools bzw. eines Embryos wurde dann auf 20 Wells einer 96-Well-Mikrotiterplatte mit 4 Negativkontrollen verteilt. In einer ersten Multiplex-PCR wurden zunächst alle zu untersuchenden Regionen gleichzeitig amplifiziert. Eine darauffolgende zweite Singleplex-PCR vervielfältigte dann die Regionen der einzelnen Gene separat. Nach der Identifizierung der generierten PCR-Produkte mittels Gelelektrophorese erfolgte im letzten Schritt die Sequenzierung mittels Pyrosequenzierung oder direkter Sangersequenzierung. Abbildung übernommen aus [377] und modifiziert. Die LD wurde an Pools von 5-10 Oozyten und vereinzelten Embryonen im 4- bis 8-ZellStadium durchgeführt. Dazu wurde das biologische Material zunächst einer Natriumbisulfitbehandlung mittels EZ DNA Methylation Direct Kit (Zymo Research, USA) entsprechend dem Herstellerprotokoll unterzogen. Die konvertierte DNA wurde mit 14 μl Elutionspuffer in ein 1,5 ml LoBind tube (Eppendorf, Hamburg) eluiert. Das LoBind tube verfügt über eine spezielle Beschichtung der Innenfläche, die die Adhäsion zwischen Probe 54 MATERIAL UND METHODEN und Reaktionsgefäß reduziert. Da bei der LD mit sehr geringen DNA-Mengen gearbeitet wird soll die Verwendung des LoBind tubes den Verlust einzelner DNA-Moleküle durch das Anhaften an der Innenfläche des Reaktionsgefäßes minimieren bzw. vermeiden. Zu der eluierten konvertierten DNA wurden 486 μl des zuvor vorbereiteten Multiplex-PCRMastermix gegeben (Zusammensetzung und PCR-Bedingungen unter 2.2.3). Um eine gleichmäßige Verteilung der DNA-Moleküle in der Lösung zu gewährleisten, wurde das Gemisch durch intensives Auf- und Abpipettieren durchmischt. Dieser DNA-Multiplex-PCRAnsatz wurde anschließend in Volumina von 25 μl in 20 Wells einer 96-Well-Mikrotiterplatte verteilt. Sowohl zur Durchmischung als auch zur Verteilung der Lösung wurde die gleiche Pipettenspitze verwendet um den Verlust von DNA durch Adhäsion an der Innenfläche der Pipettenspitze zu reduzieren. Die Anzahl der Verdünnungsschritte wurde empirisch ermittelt. Die untersuchten Eizellen enthielten sowohl die Zona Pellucida als auch den Polarkörper. Damit ergibt sich eine maximal mögliche Anzahl von 4 amplifizierbaren Allelen für jedes Gen, die in einer Oozyte vorliegen. Bei einem Pool von 10 Eizellen würde man damit eine maximale Anzahl von 40 Allelen erwarten. Der hohe Grad an DNA-Degradation während der Natriumbisulfitbehandlung führt jedoch dazu, dass die Anzahl der analysierbaren Allele erfahrungsgemäß geringer als die Hälfte der eingesetzten Menge beträgt. Mit 1:20 verdünnter DNA und 4 Negativkontrollen je Oozyten-Pool bzw. Embryo konnten genau maximal 4 verschiedene Eizellpools auf eine 96-Well-Mikrotiterplatte aufgebracht werden (Abbildung 9). 1 μl des PCR-Produkts der Multiplex-PCR wurde als Matrize der nachfolgenden Gen-spezifischen Singleplex-PCR verwendet, in der alle zu untersuchenden DNA-Regionen separat vervielfältigt werden (Zusammensetzung und PCRBedingungen unter 2.2.3). Da bei der LD limitierend geringe DNA-Mengen eingesetzt werden, enthalten nicht alle Verdünnungen bzw. Wells DNA. Zur Identifikation der Wells, die ein Amplifikat vorweisen, wurden alle Verdünnungen und Negativkontrollen in einem 1,5 % Agarosegel mittels Gelelektrophorese analysiert. Im letzten Schritt werden die generierten Amplifikate aus den entsprechenden Wells isoliert und sequenziert. Die in dieser Arbeit angewandten Sequenzierungsmethoden umfassen die Pyrosequenzierung sowie die direkte Sangersequenzierung, die unter 2.2.4 detailliert beschrieben sind. 2.2.3 Primersequenzen und PCR-Bedingungen Alle Polymerasekettenreaktionen wurden mit dem FastStartTaq Polymerase-System (Roche, Mannheim) in Kombination mit Primern für Bisulfit-konvertierte DNA durchgeführt. Die 55 MATERIAL UND METHODEN Primer wurden mit Hilfe der Pyrosequencing Assay Design Software (Qiagen, Hilden) erstellt und von der metabion GmbH (Planegg) synthetisiert. Das Standard-Protokoll des FastStartTaq Polymerase-Systems besteht aus der Anfangsdenaturierung, Denaturierung, Primer-Anlagerung (Annealing), Elongation und Abschlusselongation sowie einer Anzahl von Zyklen, in denen sich Denaturierung, Annealing und Elongation wiederholen. Die PCRBedingungen der verschiedenen Gene bzw. Regionen unterscheiden sich dabei lediglich in der Annealing-Temperatur und der Zyklenzahl, welche maßgeblich vom eingesetzten PrimerPaar bestimmt werden. Mit beiden Gensets wurden Promotorregionen entwicklungsrelevanter Gene untersucht. Dabei waren die Gene H19, SNRPN und DNMT3Lo in beiden Gensets enthalten, da sie u.a. als Kontrollgene zum Nachweis somatischer Kontamination dienten (siehe 1.8). 2.2.3.1 Genset 1: Oozyten- und Maturations-spezifische Gene Mit dem Genset 1 wurden unreife Eizellen und unterschiedlich lange in vitro gereifte Eizellen sowie die daraus generierten Embryonen untersucht. Das Genset 1 beinhaltet die Promotorregionen der Gene bH19, bSNRPN, bZAR1, bDNMT3A, bOCT4, bDNMT3Lo und bDNMT3Ls. Die dafür verwendeten Primersequenzen sind in Tabelle 4 aufgelistet. Die detaillierte Zusammensetzung der Mastermixe der Multiplex-PCR sowie der Singleplex-PCRs ist in Tabelle 5 aufgeführt. Die entsprechenden PCR-Bedingungen sind in Tabelle 6 beschrieben. Tabelle 4: Primer des Gensets 1, die in der Multiplex-PCR, Singleplex-PCR und Pyrosequenzierung für die Promotorregionen der Gene, bH19, bSNRPN, bZAR1, bDNMT3A, bDNMT3Ls, bDNMT3Lo und bOCT4 in Rindereizellen und Rinderembryonen verwendet wurden. Die Sequenzierung der Regionen von bH19, bSNRPN, bZAR1, bDNMT3A und bDNMT3Ls erfolgte durch Sangersequencierung. Die PCR-Produkte von bDNMT3Lo und bOCT4 wurden durch Pyrosequenzierung analysiert. Der “inner forward” Primer von bOCT4 sowie der “inner reverse” Primer von bDNMT3Lo wurden am 5‘-Ende biotinyliert (Bio-). *Ensembl release 82 - September 2015. Gen Primer Sequenz (5’-3’) bH19 Outer forward GAGGGGTATTGAGAGGTTGT Outer reverse CAAACATAAAAATCCCTCAATATCCC Inner forward AGAGGTTGTGGGTGTGGAGATA Inner reverse TCCTCTCCCACCTTCAACAA Outer forward GGGGTGGGGTAGATATTATTTT Outer reverse AAAAAAAAAAAATATTACCCACCACAC Inner forward GGTTTTTTTGTTTGAGAGAG Amplikonlänge (bp) 279 Chromosomale Lokalisation* Anzahl der CpGStellen 29:5014781650148095 20 bSNRPN 230 29:5014782750148057 299 21:25818-26117 30 276 21:25818-26094 56 MATERIAL UND METHODEN bZAR1 Inner reverse AAAAAAAAAAAATATTACCCAC Outer forward ATTTGGGGTAAGTTTATTTTTAGATTAGT Outer reverse CCCCAAAACAACCATCAATA Inner forward GGGTGTGGAATATTTTTATATTAAGGT Inner reverse AAAACAACCATCAATATACCCCTAC Outer forward GGGTATTTAATTTTATTTGGATATTT Outer reverse CAAAACCCACTACACTCT Inner forward TTTTAAGGGAGGTTTAATTAATAGAGGT Inner reverse AACCCACTACACTCTACCT Outer forward TAGGGGGGTTTTTGAGAA Outer reverse CAACAAAAACACCCCACA Inner forward AGGGGGGTTTTTGAGAAGAAGA Inner reverse TCCACCCAAAACCCAATAACACTAA Outer forward GTTTAAGTTTAGGGGATGG Outer reverse CCTCCTTCTCCTTCAAAC Inner forward TAAGTTTAGGGGATGGATATAG Inner reverse Bio-TCCAAACCCCACTTCCTA Pyro GGGATGGATATAGTTGTTT Outer forward GGGAGGTTTTTGGAAGTTTAT Outer reverse TTCCCCCCACCCATCCAA Inner forward Bio-GGGAGGTTTTTGGAAGTTTAT Inner reverse AAATACCTTCCTTCCCCATA Pyro CCTTCCTTCCCCATAA 323 6:6882047868820801 18 bDNMT3A 230 6:6882056768820797 262 11:7396316073963422 16 bDNMT3Ls 211 11:7396320873963419 304 1:145717979145718283 8 bDNMT3Lo bOCT4 270 1:145717980145718250 158 1:145750153145750311 88 1:145750156- 5 145750244 262 23:2776975627770018 143 7:51481766- 7 51481909 Tabelle 5: Zusammensetzung der Multiplex-PCR und Singleplex-PCR des Genset 1. Konzentration der Stammlösung MultiplexPCR SingleplexPCR (52x) (51x) Puffer 10x 130 μl 127,5 μl 1x dNTPs 10 mM 26 μl 25,5 μl 200 μM Primer Fwd 10 μM 52 μl (7x) 51 μl 0,4 μM Komponente Endkonzentration 57 MATERIAL UND METHODEN Primer Rev 10 μM 52 μl (7x) 51 μl 0,4 μM Taq DNA Polymerase 5 U/μl 10,4 μl 10,2 μl 0,04 U/μl Wasser - 369,2 μl 958,8 μl - Tabelle 6: Ablauf und Bedingungen der Multiplex-PCR und der Singleplex-PCRs der untersuchten Regionen des Genset 1. Singleplex-PCR PCR-Schritt Dauer Multiplex-PCR bH19 1. Anfangsdenaturierung bSNRPN bDNMT3A bOCT4 bZAR1 bDNMT3Ls bDNMT3Lo 64 °C 54 °C 32 32 4 min 95 °C 2. Denaturierung 30 s 95 °C 3. Annealing 30 s 4. Elongation 45 s 72 °C 7 min 72 °C 5. Abschlusselongation Zyklenzahl (Wdh. der Schritte 2-4) 54 °C 35 65 °C 28 57 °C 31 2.2.3.2 Genset 2: Oozyten- sowie Alters-spezifische Gene Genset 2 kam bei unreifen, in vivo gewonnen Eizellen von Rindern unterschiedlichen Alters zur Anwendung. Die mit Genset 2 analysierten Promotorregionen beinhalten die Gene bTERF2, bREC8, bBCL-XL, bPISD, bBUB1, bDNMT3Lo, bH19 und bSNRPN. Die dafür eingesetzten Primersequenzen sind in Tabelle 7 aufgelistet. Die genaue Zusammensetzung der Mastermixe der Multiplex-PCR sowie der Singleplex-PCR sind in Tabelle 8 aufgeführt. Die entsprechenden PCR-Bedingungen sind Tabelle 9 zu entnehmen. 58 MATERIAL UND METHODEN Tabelle 7: Primer des Gensets 2, die in der Multiplex-PCR, Singleplex-PCR und Pyrosequenzierung für die Promotorregionen der Gene bTERF2, bREC8, bBCL-XL, bPISD, bBUB1, bDNMT3Lo, bH19 und bSNRPN in Rindereizellen verwendet wurden. Die Sequenzierung der Regionen von bH19 und bSNRPN erfolgte durch Sangersequencierung. Die PCRProdukte von bTERF2, bREC8, bBCL-XL, bPISD, bBUB1 und bDNMT3Lo wurden durch Pyrosequenzierung analysiert. Der “inner forward” Primer von bDNMT3Lo und bBCL-XL sowie der “inner reverse” Primer von bTERF2, bREC8, bPISD und bBUB1 wurden am 5‘-Ende biotinyliert (Bio-). *Ensembl release 82 - September 2015. Gen bTERF2 bREC8 bBCL-XL bPISD bBUB1 bDNMT3Lo Primer Sequenz (5’-3’) Outer forward AGTAGGTATTGYGGTTAGTATATTGG Outer reverse AAAACCRACCCTCACCCTACAT Inner forward GAGAGGGAGGTTGGGAGTTTT Inner reverse Bio-AAAACCRACCCTCACCCTACAT Pyro GGTTTTTTTTTTTTTAGAAGT Outer forward GTAGTTAATGGGGAATAGGGTTAAT Outer reverse AATCTCCTCCCCACTCATT Inner forward GTAGTTAATGGGGAATAGGGTTAAT Inner reverse Bio-AATCTCCTCCCCACTCATT Pyro GGGGAATAGGGTTAATT Outer forward AGAATGAATGGAAATGTGAATGTT Outer reverse AAAAAACCACATACCCCCTTC Inner forward Bio-AGAATGAATGGAAATGTGAATGTT Inner reverse ACCTCAATTTCCCTAAAAAAAC Pyro ACTTACAAACTCAATCAC Outer forward AGGTAGGTTTTTTAAATAGTTGTATTTG Outer reverse AAAACCTTAAACACATAACCCC Inner forward AGGTAGGTTTTTTAAATAGTTGTATTTG Inner reverse Bio-AAAACCTTAAACACATAACCCC Pyro GGGTAGTTATTTTAGATAT Outer forward GATTTATATTTATTGAAAAATATTTTT Outer reverse ACCCAAATAATTAACCAACTC Inner forward AGAGGAATTAGGGTAGTAGTA Inner reverse Bio-ACCCAAATAATTAACCAACTC Pyro ATTAGGGTAGTAGTAGTTT Outer forward GTTTAAGTTTAGGGGATGG Outer reverse CCTCCTTCTCCTTCAAAC Inner forward Bio-TAAGTTTAGGGGATGGATATAG Ampliconlänge (bp) 507 Chromosomale Lokalisation* Anzahl der CpGStellen 18:3664005736640564 467 18:36640057- 7 36640524 242 10:2081633420816576 242 10:20816334- 5 20816576 330 13:6181719861817528 198 13:61817330- 8 61817528 230 17:7245320372453433 230 17:72453203- 6 72453433 197 11:15591041559301 154 11:1559104- 6 1559258 158 1:145750153145750311 5 88 1:145750156145750244 59 MATERIAL UND METHODEN bH19 bSNRPN Inner reverse TCCAAACCCCACTTCCTA Pyro GGGATGGATATAGTTGTTT Outer forward GAGGGGTATTGAGAGGTTGT Outer reverse CAAACATAAAAATCCCTCAATATCCC Inner forward AGAGGTTGTGGGTGTGGAGATA Inner reverse TCCTCTCCCACCTTCAACAA Outer forward GGGGTGGGGTAGATATTATTTT Outer reverse AAAAAAAAAAAATATTACCCACCACAC Inner forward GGTTTTTTTGTTTGAGAGAG Inner reverse AAAAAAAAAAAATATTACCCAC 279 29:5014781650148095 230 20 29:5014782750148057 299 21:25818-26117 276 21:25818-26094 30 Tabelle 8: Zusammensetzung der Multiplex-PCR und Singleplex-PCR des Genset 2. Konzentration der Stammlösung MultiplexPCR SingleplexPCR (52x) (51x) Puffer 10x 130 μl 127,5 μl 1x dNTPs 10 mM 26 μl 25,5 μl 200 μM Primer Fwd 10 μM 52 μl (8x) 51 μl 0,4 μM Primer Rev 10 μM 52 μl (8x) 51 μl 0,4 μM Taq DNA Polymerase 5 U/μl 10,4 μl 10,2 μl 0,04 U/μl Wasser - 249,6 μl 958,8 μl - Komponente Endkonzentration 60 MATERIAL UND METHODEN Tabelle 9: Ablauf und Bedingungen der Multiplex-PCR und der Singleplex-PCRs der untersuchten Regionen des Genset 2. Singleplex-PCR PCR-Schritt 1. Anfangs- Dauer MultiplexPCR bTERF2 bREC8 bBUB1 bBCLXL bPISD 4 min 95 °C 2. Denaturierung 30 s 95 °C 3. Annealing 30 s 4. Elongation 45 s 72 °C 7 min 72 °C denaturierung 5. Abschlusselongation 54 °C Zyklenzahl (Wdh. der Schritte 2-4) 35 66 °C 56 °C 35 35 53 °C 35 bDNMT3Lo bH19 bSNRPN 52 °C 54 °C 65 °C 57 °C 35 32 28 31 2.2.4 Methoden zur Bisulfitsequenzierung Die Zielregionen Pyrosequenzierung der aus oder Bisulfit-DNA direkter generierten Amplifikate Sanger-Sequenzierung wurden analysiert. mittels Die Sequenzierungsmethode wurde dabei in Abhängigkeit der Dichte und Verteilung der CpGDinukleotide innerhalb der untersuchten DNA-Region gewählt. Neben den grundlegend unterschiedlichen technischen Prinzipien unterscheiden sich beide Methoden insbesondere in der Limitierung der Länge der zu analysierenden DNA-Fragmente. Während mit der Pyrosequenzierung Regionen bis maximal 70 Basenpaare untersucht werden können, ermöglicht die Sanger-Sequenzierung eine Analyse von bis zu 1000 Basenpaaren. Dementsprechend wurden geprägte Genpromotoren mit typischerweise hoher CpG-Dichte direkt Sanger-sequenziert, während nicht-geprägte Genpromotoren mit vereinzelten CpGs überwiegend pyrosequenziert wurden. Mittels Pyrosequenzierung wurden die PromotorRegionen der Gene bTERF2, bREC8, bBCL-XL, bPISD, bBUB1, bDNMT3Lo und bOCT4 untersucht. Die Sangersequenzierung kam bei der Untersuchung der Gene bH19, bSNRPN, bZAR1, bDNMT3A und bDNMT3Ls zur Anwendung. Im Folgenden wird die technische Funktionsweise und Durchführung beider Methoden erläutert. 61 MATERIAL UND METHODEN 2.2.4.1 Pyrosequenzierung Die Pyrosequenzierung ist eine Methode zur Sequenzierung von DNA-Fragmenten, die nach dem Prinzip der Sequenzierung durch Synthese erfolgt. Durch eine Kaskade enzymatischer Reaktionen wird dabei der Einbau eines Nukleotids in das synthetisierte DNA-Molekül in ein Lichtsignal umgesetzt, welches von einem Detektor erfasst wird [381]. Da sich die Pyrosequenzierung insbesondere zur Quantifizierung von Einzel-Nukleotid-Unterschieden (Single Nucleotide Polymorphism = SNPs) eignet, lassen sich damit ebenfalls MethylierungsUnterschiede bzw. Sequenz-Unterschiede (C oder T) an CpG-Dinukleotiden Natriumbisulfitbehandelter DNA untersuchen [382]. 1987 beschrieb P. Nyrén, erstmals wie die Aktivität der DNA-Polymerase in Form von Biolumineszenz beobachtet werden kann und legte damit den Grundstein für das Prinzip der Pyrosequenzierung [383, 384]. Nach 11 Jahren weiterer Entwicklungsarbeit wurde 1998 die Pyrosequenzierung in ihrer heutigen Funktionsweise ebenfalls von der Gruppe um P. Nyrén vorgestellt [381]. Die Enzym-Kaskade, die dabei vom eingebauten Nukleotid bis zum detektierbaren Lichtsignal durchlaufen wird, beinhaltet das Klenow-Fragment der DNAPolymerase I, die ATP-Sulfurylase, die Luciferase und die Apyrase [385]. Neben diesen 4 Enzymen enthält der Sequenzierungs-Ansatz darüber hinaus Adenosinphosphosulfat (APS), D-Luciferin sowie das zu sequenzierende PCR-Produkt mit dem spezifischen Sequenzierungs-Primer. Die 4 Nukleotide werden einzeln nacheinander zum Ansatz gegeben, während zeitgleich ein Sensor die emittierten Lichtsignale misst. Die Reaktionsfolge nach Zugabe eines Nukleotids ist in Abbildung 10 dargestellt. [DNA]n + dNTP Polymerase [DNA]n+1 + PPi PPi + APS dNTP Apyrase Luciferin Sulfurylase ATP Oxyluciferin Luciferase Licht Licht dNDP + dNMP + Phosphat dNTP = dATP-S, dTTP, dCTP, dGTP Zeit Abbildung 10: Enzymatische Reaktionsabfolge der Pyrosequenzierung. Der Reaktionsansatz der Pyrosequenzierung enthält das Klenow-Fragment der DNA-Polymerase I, die ATP-Sulfurylase, die Luciferase und die Apyrase sowie den nichtenzymatischen Bastandteile Adenosinphosphosulfat (APS), D-Luciferin und das zu sequenzierende PCR-Produkt mit angelagertem Sequenzierungs-Primer. Wird nach Zugabe einer Nukeotid-Sorte (dNTP) eine passende Base durch die Polymerase in den wachsenden DNA-Strang ([DNA]n) eingebaut, kommt es zur Freisetzung von Pyrophosphat (PPi), das wiederum als Substrat für die ATP-Sulfurylase dient, wodurch ATP gebildet wird. Luciferase setzt dieses ATP in Verbindung mit D-Luciferin zu Oxyluciferin und Licht um, welches von einem Lichtsensor detektiert wird. Die Apyrase baut anschließend 62 MATERIAL UND METHODEN Nukleotide, die nicht an der Reaktion beteiligt waren, sowie ATP ab bevor das nächste Nukleotid zum Ansatz gegeben wird und die Reaktionskaskade erneut ablaufen kann. Abbildung übernommen aus [385] und modifiziert. Das Klenow-Fragment der DNA-Polymerase I enthält die Domänen der Polymerase sowie der 3‘→5‘ Exonuklease-Aktivität. Zur Pyrosequenzierung wird ein Klenow-Fragment mit mutierter und inaktiver 3‘→5‘ Exonuklease verwendet und eine reine Polymerase-Aktivität aufweist. Dies ist entscheidend, um eine asynchrone DNA-Polymerisierung zu verhindern und einzig die Synthese der zu sequenzierenden Basenfolge sicherzustellen [385]. Bei Zugabe einer Sorte von Nukleotiden, die ein Basenpaar mit der DNA-Matrize bilden, erfolgt der Einbau des Nukleotids in den wachsenden DNA-Strang durch das Klenow-Fragment der DNAPolymerase. Das dabei abgespaltene Pyrophosphat (PPi) dient daraufhin als Substrat für die ATP-Sulfurylase, welche in Verbindung mit APS ATP generiert. Die finale Umsetzung des ATP in Kombination mit Luciferin zu Oxyluciferin durch die Luciferase erzeugt Licht, das durch einen Detektor erfasst wird. Nur der korrekte Einbau eines oder mehrerer passender Nukleotide führt zu einem Lichtsignal, dessen Intensität proportional zur Anzahl der eingefügten Nukleotide ist. Die übrigen, nicht eingebauten Nukleotid-Reste sowie ATP werden durch die Apyrase verdaut, womit sich der Ansatz erneut im Ausgangszustand für einen weiteren Reaktionsablauf mit neuen Nukleotiden befindet. Durch die wiederholte Zugabe unterschiedlicher Nukleotide wird die Synthese des wachsenden DNA-Stranges in Echtzeit dokumentiert bis die Synthese bzw. Sequenzierung der Zielregion vollständig abgeschlossen ist (Abbildung 11) [385]. 63 MATERIAL UND METHODEN dATP-S Polymerase Template 3‘ Primer 5‘ A dCTP TGAA A PPi A A 5‘ A A Polymerase Template 3‘ Primer 5‘ C TGAA AC A C ATP 5‘ PPi C Sulfurylase Apyrase dGTP C C Polymerase Template 3‘ Primer 5‘ G TGAA AC A ATP Luciferase Luciferase Licht Licht 5‘ G G Sulfurylase Apyrase dTTP G G Polymerase Template 3‘ Primer 5‘ T TGAA ACTT AC Sulfurylase Apyrase 2 PPi T T 5‘ T T Sulfurylase 2 ATP Apyrase Luciferase Luciferase Licht Lichtintensität ACTT TGAA Sequenz der synthetisierten DNA Sequenz der Matrizen-DNA Dispersions-Folge der Nukleotide Pyrogram Abbildung 11: Schematische Darstellung des Prinzips der Pyrosequenzierung. Paart eine Base nach Zugabe eines Nukleotids mit der DNA-Matrize, wird das Nukleotid durch das Klenow-Fragment der DNA-Polymerase I eingebaut, Pyrophosphat wird abgespalten, was die ATP-Sulfurylase zur Bildung von ATP nutzt, wodurch die Luciferase Licht generiert. Die Intensität des detektierten Lichtsignals ist dabei proportional zur Anzahl der eingebauten Nukleotide. Die Apyrase baut vor der Zugabe einer Nukleotid-Sorte noch im Ansatz verbliebene Nukleotide und ATP ab und versetzt das enzymatische System somit in den Ausgangszustand zurück. Abbildung übernommen aus [385] und modifiziert. Voraussetzung für eine erfolgreiche Pyrosequenzierung ist das Vorliegen des zu sequenzierenden PCR-Produkts als Einzelstrang mit angelagertem Sequenzierungs-Primer vor der Zielregion. Durch Verwendung eines am 5‘-Ende biotinylierten Primers in der PCR ist es möglich mittels Streptavidin-Beads einen DNA-Strang selektiv zu isolieren und mit einem spezifischen Sequenzierung-Primer zu hybridisieren [386]. Das durchgeführte Protokoll zur Präparation des PCR-Produkts für die Pyrosequenzierung wird im Folgenden kurz beschrieben: 1. Zunächst wurde ein Mix aus Streptavidin Sepharose High Performance Beads (GE Healthcare, München) (2 μl / Probe), Binding-Puffer (Qiagen, Hilden) (40 μl / Probe) und Reinstwasser (28 μl / Probe) vorbereitet und 70 μl des Mixes in eine 96-WellMikrotiterplatte ohne Rand für jede Probe vorgelegt. 2. Nach Zugabe von 10 μl des PCR-Produkts zu dem vorgelegten Mix in der 96-WellMikrotiterplatte ohne Rand wurde die Wells der Platte mit einer selbstklebenden 64 MATERIAL UND METHODEN Folie verschlossen und für 5 min bei 14.000 rpm geschüttelt (Stufe 5 auf VortexGenie). 3. In die PyroMark® Q96 HS Pyrosequenzierplatte (Qiagen, Hilden) wurden für jede Probe 11,5 μl Annealing-Puffer (Qiagen, Hilden) mit 0,5 μl des jeweiligen Sequenzierungs-Primer entsprechend der Anordnung der Proben auf der Mikrotiterplatte vorgelegt. 4. Nach der Inkubation des PCR-Produkts mit den Streptavidin Sepharose High Performance Beads wurde die Mikrotiterplatte auf die Position 1 der PyroMark® Q96 Vacuum Workstation (Qiagen, Hilden) überführt und der Kopf der Station unter angelegtem Vakuum für 10 Sekunden in destilliertem Wasser gewaschen. 5. Der Vakuumkopf wurde anschließend auf die Mikrotiterplatte gesetzt, sodass die Metallstifte des Kopfes in die Wells der Platte eingeführt wurden. Durch das Vakuum wurde die Flüssigkeit in den Wells abgesaugt, während die Beads an der Membran der Stifte haften blieben. 6. Der Kopf mit immobilisierten Streptavidin Sepharose Beads und gebundenem PCRProdukt wurde anschließend vorsichtig in ein Becken mit 70 % Ethanol überführt (10 s). 7. Diesem Waschvorgang schloss sich eine chemische Denaturierung in einer 0,2 M NaOH-Lösung (Denaturierungspuffer, siehe Kapitel 2.1.2) (10 s) an. Dies diente dazu, den zu sequenzierenden Strang vom komplementären Strang zu lösen und durch das Vakuum zu entfernen. 8. Nach einem finalen Waschschritt im Waschpuffer (siehe Kapitel 2.1.2) (20 s) wurde der Kopf vorsichtig über der Pyrosequenzierplatte mit vorgelegtem Annealing-Puffer und Sequenzierungs-Primer positioniert. Unmittelbar vor dem Einführen der Metallstifte in die Wells wurde das Vakuum abgeschaltet und die Beads durch sanftes Schütteln des Kopfes im Volumen der Wells gelöst. 9. Die gereinigte Proben-DNA wurde für 2 min bei 80 °C auf einem Heizblock denaturiert und anschließend für 5 min bei Raumtemperatur abgekühlt. In der Phase 65 MATERIAL UND METHODEN der Abkühlung erfolgte die spezifische Anlagerung der Sequenzierungs-Primer, womit die Präparation des PCR-Produkts für die Pyrosequenzierung abgeschlossen wurde. Die Pyrosequenzierung wurde mit dem PyroMark® Q96 MD System (Qiagen, Hilden) und dem PyroGold® SQA Reagent Kit (Qiagen, Hilden) durchgeführt [387]. Die Pyroassays bzw. alle verwendeten Primer wurden mittels PyroMark® Assay Design 2.0 Software (Qiagen, Hilden) entworfen. Die Datenauswertung erfolgte mit der Pyro Q-CpG Software (Qiagen, Hilden). Vor Beginn der Sequenzierung wurden die einzelnen Dispensierküvetten mit dem Enzymmix (DNA-Polymerase, ATP-Sulfurylase, Luciferase und Apyrase), dem Substrat (APS und Luciferin) und den einzelnen Nukleotiden (PyroGold® SQA Reagent Kit) befüllt. Die benötigten Volumina der Reagenzien wurden nach Eingabe der Proben bzw. verwendeten Assays durch die Pyro Q-CpG Software berechnet. Um Dispensierungsfehler von Beginn an auszuschließen, wurde vor jedem Start der Pyrosequenzierung ein Dispensierungs-Test durchgeführt. Nach der erfolgreichen Prüfung der korrekten Dispensierung aller Reagenzien wurde die Pyrosequenzierplatte mit den zu untersuchenden Proben in das PyroMark® Q96 MD Sequenziergerät überführt und die Sequenzierung gestartet. 2.2.4.2 Direkte Bisulfitsequenzierung Bei der direkten Bisulfitsequenzierung wird das aus Natriumbisulfit-behandelter DNA generierte PCR-Produkt mittels Sanger-Sequenzierung analysiert. Die Sanger-Sequenzierung wurde von Frederick Sanger 1977 entwickelt und basiert auf dem Prinzip der KettenabbruchSynthese [388]. Dabei werden einem normalen Standard-PCR-Ansatz aus DNA-Polymerase, Sequenzierungs-Primer und normalen Desoxyribonucleosidtriphosphaten (dNTPs), zusätzlich fluoreszenzmarkierte Didesoxyribonucleosidtriphosphate (ddNTPs) beigefügt. ddNTPs besitzen keine 3‘-Hydroxylgruppe, die für die Verknüpfung der Nukleotide benötigt wird. Nach Einbau eines ddNTPs ist eine weitere Verlängerung des DNA-Stranges nicht mehr möglich, was einen Kettenabbruch zur Folge hat. Die zufällige statistische Verteilung dieser Einbau- bzw. Abbruch-Stellen führt zu einem Gemisch aus unterschiedlich langen, synthetisierten DNA-Fragmenten. Die fluoreszenzmarkierten Nukleotide am Ende dieser DNA-Fragmente decken in ihrer Gesamtheit den vollständigen, zu sequenzierenden Bereich der DNA-Matrize ab. Die Analyse dieser PCR-Produkte erfolgt in einem Sequenziergerät. Zur Auflösung der Zielsequenz werden die Sequenzprodukte unterschiedlicher Länge kapillarelektrophoretisch nach Größe aufgetrennt und photometrisch analysiert. Dabei regt 66 MATERIAL UND METHODEN ein Laser die Fluoreszenzfarbstoffe an, deren emittiertes Licht von einer Kamera detektiert wird. Aus dem resultierenden Elektropherogramm lässt sich dann die Basenfolge ablesen. Die praktische Durchführung der direkten Bisulfitsequenzierung wurde zunächst mit einer Aufreinigung des PCR-Produkts mittels ExoSAP-IT® (Affymetrix, USA) begonnen. Dabei wurden verbliebene Reste von Primern und Nukleotiden im PCR-Produkt enzymatisch abgebaut. Dazu wurde in einer PCR-Platte 1 μl PCR-Produkt mit 0,5 μl ExoSAP- IT® und 5,5 μl Reinstwasser versetzt. Der Verdau erfolgte in einem Thermocycler bei 37 °C für 30 min, gefolgt von 85 °C für 15 min und 95 °C für 2 min. Die darauf folgende Sequenzierreaktion wurde in dieser Arbeit mit dem BigDye® Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Damstadt) durchgeführt. Dafür wurde zunächst ein Mastermix bestehend aus 1 μl BigDye® Terminator v1.1 Cycle Sequencing Ready Reaction Mix, 2 μl BigDye® Terminator v1.1/3.1 Sequencing Buffer (5x) und 1 μl Sequenzierprimer (M13/Stuffer-Reverse) pro Probe vorbereitet und jeweils 4 μl des Mastermixes zum aufgereinigten PCR-Produkt dispensiert. Die Sequenzierreaktion erfolgte in einem Thermocycler. Die dabei durchgeführten CyclerSchritte sind in Tabelle 10 aufgeführt. Tabelle 10: Ablauf und Bedingungen der Sequenzierreaktion. Cycler-Schritt Dauer Temperatur 1. Anfangsdenaturierung 2 min 96 °C 2. Denaturierung 20 s 96 °C 3. Annealing 20 s 58 °C 4. Elongation 3 min 60 °C 5. Abschlusselongation 3 min 60 °C Zyklenzahl (Wdh. der Schritte 2-4) 25 Der Sequenzierreaktion schloss sich eine Ethanolfällung (85%) mit den CleanDTR Magnetic Beads (GCbiotech, Niederlande) mit Hilfe eines Pipettier-Roboters (JANUS® Automated Workstation, Perkin Elmer, Rodgau) an. Die in 40 μl Hi-Di™ Formamide (Applied Biosystems, 67 MATERIAL UND METHODEN Damstadt) gelöste DNA wurde auf einem ABI 3730 oder ABI 3130 xl Genetic Analyzer (Applied Biosystems, Damstadt) sequenziert. 2.2.5 Computerprogramme und Datenbanken Zur Erstellung der Primersequenzen (Primerdesign), der Literaturrecherche, der Datenauswertung sowie der Datenverarbeitung wurden unterschiedliche Programme sowie öffentlich zugängliche Internetseiten verwendet. Eine detaillierte Auflistung der genutzten Software und Internetadressen mit den dazugehörigen Herstellern bzw. Betreibern sowie dem Verwendungszweck sind in Tabelle 11 aufgeführt. Tabelle 11: Verwendete Computerprogramme und Datenbanken. Programm / Internetseite Hersteller / Betreiber Max-Planck-Institut für BiQ Analyzer v2.0 Informatik, Saarbrücken [389] Verwendungszweck Datenauswertung der direkten Bilsufitsequenzierungen Institute of Enzymology, Bisearch http://bisearch.enzim.hu/ BRC, Hungarian Academy of Sciences, Budapest Primerdesign [390, 391] Technelysium Pty Ltd, Chromas Lite 2.1.1 South Brisbane, Australien DNA-Baser 3.5.4.2 Datenauswertung der direkten Bilsufitsequenzierungen Heracle BioSoft SRL, Konvertierung der AB1- Rumänien Dateien zu FASTA-Dateien The European Bioinformatics Institute (EMBL-EBI), Cambridge, Ensembl http://www.ensembl.org/index.html UK Primerdesign und Datenauswertung The Wellcome Trust Sanger Institute, Cambridge, UK LASAGNA-Search 2.0 Department of Computer Identifizierung von http://biogrid- Science and Engineering, Transkriptionsfaktor- 68 MATERIAL UND METHODEN lasagna.engr.uconn.edu/lasagna_search/ USA [392] bindestellen Textverarbeitung, Microsoft-Office 2010 Microsoft Corporation Datenauswertung und Erstellung von Abbildungen National Center for NCBI http://www.ncbi.nlm.nih.gov/ Biotechnology Primerdesign und Information, Bethesda, Datenauswertung USA Pyro Q-CpG 1.0.9 Pyro-Mark Assay Design 2.0 Biotage, Uppsala, Datenauswertung der Schweden Pyrosequenzierungen Qiagen, Hilden Primerdesign 69 ERGEBNISSE 3 Ergebnisse Zur Unterscheidung einer somatischen Kontamination von einer Epimutation wurde eine somatische Kontamination definiert als das Auftreten von mindestens einem abnormal methylierten Allel (>50% abnormal methylierte CpGs) in mindestens 2 der 3 Promotorregionen bH19, bSNRPN und bDNMT3Lo bei Genset 2 bzw. 2 der 4 Promotorregionen bH19, bSNRPN, bDNMT3Lo und bOCT4 bei Genset 1 innerhalb des gleichen Eizell-Pools. Unter Anwendung dieser Definition wurden 7 von insgesamt 118 untersuchten Eizell-Pools aufgrund potentieller Kontamination durch somatische Zellen aus der Analyse ausgeschlossen. Eine detaillierte Auflistung der betroffenen Eizell-Gruppen ist Tabelle 12 zu entnehmen. Tabelle 12: Auflistung der Anzahl der untersuchten Eizell-Pools sowie der somatisch kontaminierten Eizell-Pools. Unreifen Eizellen aus Antralfollikeln unterschiedlicher Größe Anzahl der untersuchten EizellPools Anzahl der somatisch kontaminierte EizellPools Eizellen von Kühen unterschiedlichen Alters IVM-Eizellen unterschiedlicher Reifungsdauer Σ <2 mm 3-5 mm >6 mm 9-12 Monate 3-7 Jahre 8-11 Jahre 24h 48h 19 11 8 15 17 13 18 17 118 2 (10%) 0 0 0 1 (6%) 0 4 (22%) 0 7 (6%) Allele wurden als vollständig methyliert bewertet, wenn sie mehr als 50% methylierte CpGs enthielten. DNA-Moleküle, die weniger als 50% methylierte CpGs aufwiesen wurden als vollständig unmethyliert kategorisiert. Auf der Ebene einzelner CpG-Stellen wurden aufgrund der Verwendung zweier unterschiedlicher Sequenzierungstechniken Methoden-spezifische Definitionen für die Methylierungszustände formuliert. Die Pyrosequenzierung liefert quantitative Rohdaten in Form eines Messwerts zwischen 1% und 100% für einzelne CpGStellen. Als vollständig methyliert wurden CpG-Stellen gewertet, die eine Methylierung von mehr als 80% aufwiesen. CpGs, die geringer als 20% methyliert waren, wurden als vollständig unmethyliert klassifiziert. Methylierungswerte einzelner CpGs zwischen 20% und 80% konnten weder als vollständig methyliert noch als vollständig unmethyliert gewertet werden und wurden als CpGs mit uneindeutigem Methylierungszustand kategorisiert. Unter 70 ERGEBNISSE der Annahme, dass die LD Sequenzdaten einzelner DNA-Moleküle bzw. Allele liefert, wurden Methylierungswerte von 0 bzw. 100% an einzelnen CpG-Positionen erwartet. Unter Berücksichtigung der Messungenauigkeit wurde der Toleranzbereich auf <20% bzw. >80% ausgedehnt. Bei Messwerten zwischen 20% und 80% Methylierung einzelner CpGs konnte jedoch nicht mehr davon ausgegangen werden, dass es sich um die Sequenzdaten eines einzelnen DNA-Moleküls handelt. Werte dieser Art können das Ergebnis eines überlagerten Signals von mindestens zwei Allelen mit CpGs unterschiedlicher Methylierung sein. Eine unvollständige Natriumbisulfit-Konvertierung kann jedoch ebenfalls als mögliche Ursache für diese Messwerte nicht ausgeschlossen werden. Die direkte Bisulfitsequenzierung bzw. Sanger-Sequenzierung lieferte qualitative Rohdaten in Form eines Chromatogramms. Deutliche heterozygote Signale an einer CpG-Stelle wurden als CpG-Stellen mit uneindeutigem Methylierungszustand definiert. Im Chromatogramm stellte sich dieser Zustand durch zwei überlagerte Peaks für Cytosin und Thymin dar. Eine Gegenüberstellung von Rohdaten der Pyrosequenzierung sowie der Sanger-Sequenzierung für die aufgeführten Methylierungszustände sowie der im späteren Verlauf verwendeten vereinfachten Darstellungsform sind in Abbildung 12 veranschaulicht. 71 ERGEBNISSE Sanger-Sequenzierung Darstellung Pyrosequenzierung 0% Methylierung 50% Methylierung und/oder 100% Methylierung Abbildung 12: Exemplarische Darstellung von Chromatogrammen (Sanger-Sequenzierung, bDNMT3Ls) und Pyrogrammen (Pyrosequenzierung, bDNMT3Lo) sowie die verwendete schematische Darstellung der CpG-Stelen. Die CpG-Stellen sind mit schwarzen Pfeilen in den Chromatogrammen der Sanger-Sequenzierungen und grau hinterlegt in den Pyrogrammen markiert. Rote Pfeile zeigen die CpG-Stellen mit uneindeutiger (50% Methylierung) Methylierung. Ein einzelnes Quadrat repräsentiert ein einzelnes CpG. Aneinander angrenzende Quadrate befinden sich auf dem gleichen DNA-Molekül. Ein weißes Quadrat zeigt ein unmethyliertes CpG (0% Methylierung), ein schwarzes Quadrat ein methyliertes CpG (100% Methylierung) und ein graues Quadrat ein CpG mit uneindeutigem Methylierungszustand (50% Methylierung). Als CpGs mit uneindeutigem Methylierungszustand wurden bei der Pyrosequenzierung Messwerte zwischen 20% und 80% einzelner CpGs bewertet sowie ein deutliches heterozygotes Signal an einer CpG-Stellen in der Sanger-Sequenzierung (mittlere Zeile; 50% Methylierung). Zur Beurteilung der Effizienz der Limiting Dilution wurde die Ausbeute der identifizierten Allele als Verhältnis der Anzahl sequenzierter DNA-Moleküle zu der Anzahl eingesetzter Allele bestimmt. Da die Eizellen noch im Besitz der Zona pellucida und des Polkörpers waren, lagen 4 amplifizierbare Allele für jedes Gen in einer einzelnen Eizelle vor. Damit waren für einen Pool von 10 Eizellen maximal 40 detektierbare Allele zu erwarten. Aufgrund des hohen Grads an DNA-Degradation verursacht durch die Natriumbisulfitbehandlung lag der empirische Wert jedoch unter der Hälfte der Anzahl eingesetzter Allele. Die ausführliche Auflistung der Ausbeuten für die einzelnen Gene in den untersuchten Eizellen bzw. 72 ERGEBNISSE Embryonen sind Tabelle 13, Tabelle 14, Tabelle 15 und Tabelle 19 zu entnehmen (AllelAusbeute). 3.1 DNA-Methylierungsprofile entwicklungsrelevanter Gene in unreifen Rindereizellen Die Untersuchung der DNA-Methylierung in unreifen Rindereizellen umfasste die Promotorregionen der Gene bH19, bSNRPN, bZAR1, bDNMT3A, bOCT4, bDNMT3Lo und bDNMT3Ls (Genset 1). Die untersuchten Eizellen wurden aus Antralfollikeln von Schlachttieren isoliert und nach Follikelgrößen von <2 mm, 3-5 mm und >6 mm gruppiert (siehe 2.1.1.1). Die Mehrzahl der detektierten Allele (>95%) zeigte für die Gene bH19, bZAR1, bOCT4, bDNMT3A und bDNMT3Lo ein vollständig unmethyliertes Muster und für bSNRPN ein hypermethyliertes Muster (Abbildung 13). Insgesamt konnten für diese 6 Gene 466 DNAMoleküle mit 6318 CpGs aus 343 Eizellen analysiert werden (Tabelle 13). Dabei zeigten 3 der 466 untersuchten Allele (<1%) ein abnormales Methylierungsmuster (>50% abnormal methylierte CpGs). Alle 3 abnormal methylierten DNA-Moleküle wurden in Eizellen aus Follikeln <2 mm nachgewiesen und enthielten eine aberrante Methylierung in bH19 und 2 abnormal methylierte Allele in bSNRPN. Die detektierten Allele der Promotorregion des Gens DNMT3Ls zeigten nach direkter SangerSequenzierung eine Mischung aus methylierten und unmethylierten CpGs sowie eine erhöhte Häufigkeit von CpGs mit uneindeutigem Methylierungszustand (Abbildung 17A). Da sich das Methylierungsmuster von DNMT3Ls deutlich von denen der anderen Gene des Gensets (bH19, bSNRPN, bZAR1, bDNMT3A, bOCT4 und DNMT3Lo) unterschied, werden die Ergebnisse für die Promotorregion von DNMT3Ls gesondert dargestellt (siehe Kapitel 3.3). 73 ERGEBNISSE bDNMT3Lo bH19 <2 mm 3-5 mm >6 mm <2 mm 3-5 mm >6 mm <2 mm bOCT4 bSNRPN <2 mm 3-5 mm bDNMT3A >6 mm <2 mm 3-5 mm 3-5 mm >6 mm bZAR1 >6 mm <2 mm 3-5 mm >6 mm Abbildung 13: Ergebnisse der Analyse der DNA-Methylierung für die Promotorregionen der Gene bH19, bSNRPN, bZAR1, bOCT4, bDNMT3A und bDNMT3Lo in unreifen Rindereizellen aus Antralfollikeln unterschiedlicher Größe (>2 mm, 3-5 mm und >6 mm). Ein einzelnes Quadrat repräsentiert ein einzelnes CpG. Eine Reihe verbundener Quadrate stellt ein einzelnes Allel dar. Ein weißes Quadrat zeigt ein unmethyliertes CpG, ein schwarzes Quadrat ein methyliertes CpG und ein graues Quadrat ein CpG mit uneindeutigem Methylierungszustand. Allele, die CpGs mit uneindeutigem Methylierungszustand (graue Quadrate) enthalten, wurden bei der statistischen Auswertung ausgeschlossen. Rote Pfeile markieren die als abnormal kategorisierten Allele. 74 ERGEBNISSE Tabelle 13: Gesamtübersicht der Ergebnisse zur Analyse der DNA-Methylierung für die Promotorregionen der Gene bH19, bSNRPN, bOCT4, bDNMT3A, bDNMT3Lo und bZAR1 in unreifen Rindereizellen aus Antralfollikeln unterschiedlicher Größe (>2 mm, 3-5 mm und >6 mm). Die ausgeschlossenen Allele beinhalten Allele, die CpG-Stellen mit einem uneindeutigen Methylierungszustand enthalten (vgl. graue Quadrate, Abbildung 12). Diese Allele tragen nicht zur Anzahl der Einzel-CpGFehler bei und wurden bei der statistischen Auswertung ausgeschlossen. Signifikante Unterschiede zwischen den Follikeln verschiedener Größen in der Methylierung von vollständigen Allelen und Einzel-CpG-Fehlern wurden für keines der untersuchten Gene detektiert. Gen bH19 bSNRPN bOCT4 bDNMT3A bDNMT3Lo bZAR1 Σ Anzahl der CpG-Stellen im Assay <2 mm 3-5 mm >6 mm Σ 18 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 33 19 (58%) 1 (7%) 3 (1%) 0,05 40 26 (65%) 0 1 (1%) 0,10 16 10 (63%) 0 1 (1%) 0,06 89 55 (62%) 1 (3%) 5 (1%) 0,06 30 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 15 2 (13%) 2 (15%) 2 (1%) 0,02 8 6 (75%) 0 1 (2%) 0,02 8 4 (50%) 0 2 (2%) 0,03 31 12 (39%) 2 (11%) 5 (1%) 0,02 7 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 23 1 (4%) 0 6 (4%) 0,03 25 3 (12%) 0 5 (3%) 0,06 28 3 (11%) 0 3 (2%) 0,10 76 7 (9%) 0 14 (3%) 0,06 16 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 16 6 (38%) 0 2 (1%) 0,02 37 18 (49%) 0 8 (3%) 0,09 21 11 (52%) 0 4 (3%) 0,08 74 35 (47%) 0 14 (2%) 0,05 5 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 104 12 (12%) 0 4 (1%) 0,15 28 0 0 0 0,07 26 6 (23%) 0 2 (2%) 0,09 158 18 (11%) 0 6 (1%) 0,12 18 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 2 2 (100%) 0 0 0,003 27 11 (41%) 0 1 (0%) 0,07 9 7 (78%) 0 0 0,03 38 20 (53%) 0 1 (0%) 0,03 94 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 193 42 (22%) 3 (2%) 17 (1%) 0,05 165 64 (39%) 0 16 (1%) 0,07 108 41 (38%) 0 12 (1%) 0,07 466 147 (32%) 3 (1%) 45 (1%) 0,06 75 ERGEBNISSE 3.2 DNA-Methylierungsprofile entwicklungsrelevanter Gene in in vitro-gereiften Rindereizellen und daraus generierten Rinderembryonen Um mögliche Veränderungen der DNA-Methylierung verursacht durch in vitro-Reifung zu identifizieren, wurden Eizellen aus 3-5 mm großen Antralfollikeln von Schlachttieren für 24h (Standard Protokoll) und 48h (simulierte post-ovulatorische Alterung) gereift (siehe 2.1.1.3). Zur Generierung von Rinderembryonen wurden die gereiften Eizellen einer in vitro Fertilisation (IVF) mit anschließender in vitro Kultivierung unterzogen (siehe 2.1.1.4). Wie bereits bei unreifen Rindereizellen, umfasste die Untersuchung der DNA-Methylierung sowohl bei den in vitro-maturierten (IVM) Eizellen als auch bei den daraus generierten Rinderembryonen die Promotorregionen der Gene bH19, bSNRPN, bZAR1, bDNMT3A, bOCT4, bDNMT3Lo und bDNMT3Ls (Genset 1). Mehr als 98% der detektierten Allele der Eizellen und Embryonen zeigten für die Gene bH19, bZAR1, bDNMT3A, bOCT4 und bDNMT3Lo ein vollständig unmethyliertes Profil und für bSNRPN ein vollständig methyliertes Profil (Abbildung 14, Abbildung 15 und Abbildung 16). Ähnlich wie bei den analysierten Allelen der unreifen Eizellen, wiesen die DNA-Moleküle der Promotorregion des Gens DNMT3Ls auch bei IVM-Oozyten und Embryonen ein Methylierungsmuster aus methylierten und unmethylierten CpGs auf (Abbildung 17B). Die Ergebnisse dieser Promotorregion von DNMT3Ls in IVM-Eizellen und Embryonen werden gemeinsam mit den Resultaten der unreifen Eizellen separat beschrieben (siehe Kapitel 3.3). 3.2.1 DNA-Methylierungsprofile entwicklungsrelevanter Gene in in vitro-gereiften Rindereizellen In den analysierten IVM-Eizellen wurden 7 abnormal methylierte Allele identifiziert (Abbildung 14 und Tabelle 14). Alle 7 DNA-Moleküle mit aberranter Methylierung wurden in Eizellen nachgewiesen, welche für 48h reiften. Dabei gehört 1 abnormal methyliertes Allel dem geprägten Gen bSNRPN an und 6 aberrante Allele der Promotorregion von DNMT3Lo. Das Auftreten der abnormal methylierten DNA-Moleküle von DNMT3Lo repräsentiert einen signifikanten Gruppenunterschied zwischen in vitro-gereiften Eizellen für 24h und 48h (p<0.03). Für die Gene bH19, bSNRPN, bZAR1, bDNMT3A und bOCT4 konnten keine signifikanten Gruppenunterschiede in der DNA-Methylierung vollständiger Allele sowie beim Auftreten von Einzel-CpG-Fehlern in IVM-Eizellen detektiert werden. 76 ERGEBNISSE bH19 24h bSNRPN 48h 24h 48h * 24h 48h 24h 48h bDNMT3A bDNMT3Lo 24h bOCT4 bZAR1 48h 24h 48h Abbildung 14: Ergebnisse der Analyse der DNA-Methylierung für die Promotorregionen der Gene bH19, bSNRPN, bZAR1, bDNMT3Lo, bDNMT3A und bOCT4 in gereiften Rindereizellen unterschiedlicher Reifungsdauer (24h und 48h).Ein einzelnes Quadrat repräsentiert ein einzelnes CpG. Eine Reihe verbundener Quadrate stellt ein einzelnes Allel dar. Ein weißes Quadrat zeigt ein unmethyliertes CpG, ein schwarzes Quadrat ein methyliertes CpG und ein graues Quadrat ein CpG mit uneindeutigem Methylierungszustand. Allele, die CpGs mit uneindeutigem Methylierungszustand (graue Quadrate) enthalten, wurden bei der statistischen Auswertung ausgeschlossen. Rote Pfeile markieren die als abnormal kategorisierten Allele. Eizellen, die für 48h gereift wurden zeigten für die Promotorregion von bDNMT3Lo eine signifikant erhöhte Anzahl abnormal methylierter Allele im Vergleich zu den 24h-gereiften Oozyten (*). 77 ERGEBNISSE Tabelle 14: Gesamtübersicht der Ergebnisse zur Analyse der DNA-Methylierung für die Promotorregionen der Gene bH19, bSNRPN, bDNMT3A, bZAR1, bOCT4 und bDNMT3Lo in gereiften Rindereizellen unterschiedlicher Reifungsdauer (24h und 48h). Die ausgeschlossenen Allele beinhalten Allele, die CpG-Stellen mit einem uneindeutigen Methylierungszustand enthalten (vgl. graue Quadrate, Abbildung 14). Diese Allele tragen nicht zur Anzahl der Einzel-CpG-Fehler bei und wurden bei der statistischen Auswertung ausgeschlossen. Eizellen, die für 48h gereift wurden zeigen für die Promotorregion von bDNMT3Lo eine signifikant erhöhte Anzahl abnormal methylierter Allele im Vergleich zu den 24h-gereiften Oozyten (*). Gen bH19 bSNRPN bDNMT3A bZAR1 bOCT4 bDNMT3Lo Σ Anzahl der CpG-Stellen im Assay 24h 48h Σ 20 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 20 2 (10%) 0 0 0,03 55 24 (44%) 0 3 (1%) 0,08 75 26 (35%) 0 3 (1%) 0,05 30 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 20 6 (30%) 0 6 (1%) 0,03 16 7 (44%) 1 (6%) 5 (2%) 0,02 36 13 (36%) 1 (3%) 11 (2%) 0,03 16 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 39 1 (3%) 0 19 (3%) 0,06 38 4 (11%) 0 7 (1%) 0,06 77 5 (6%) 0 26 (2%) 0,06 18 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 14 10 (71%) 0 2 (3%) 0,02 2 2 (100%) 0 0 0,003 16 12 (75%) 0 2 (3%) 0,01 7 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 31 11 (35%) 0 2 (1%) 0,04 45 3 (7%) 0 5 (2%) 0,07 76 14 (18%) 0 7 (2%) 0,06 5 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 93 14 (15%) 0* 10 (3%) 0,13 103 10 (10%) 6* (6%) 12 (3%) 0,15 196 24 (12%) 6 (3%) 22 (3%) 0,14 96 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 217 44 (20%) 0 39 (1%) 0,05 259 50 (19%) 7 (3%) 32 (1%) 0,06 476 94 (20%) 7 (2%) 71 (1%) 0,06 78 ERGEBNISSE 3.2.2 DNA-Methylierungsprofile entwicklungsrelevanter Gene in Rinderembryonen aus in vitro-gereiften Rindereizellen Die Embryonen, die aus in vitro-gereiften Eizellen unterschiedlicher Dauer (24h und 48h) generiert wurden, wiesen für die Gene bH19, bSNRPN, bZAR1, bDNMT3A, bOCT4 und bDNMT3Lo keine signifikanten Gruppenunterschiede in der Promotormethylierung auf (Abbildung 15, Abbildung 16 und Tabelle 15). Die beiden Embryonengruppen zeigten weder einen signifikanten Unterschied in der Methylierung vollständiger Allele noch im Auftreten einzelner aberrant methylierter CpGs. In der 24h-Gruppe der Embryonen wurde ein abnormal methyliertes Allel des Gens DNMT3Lo nachgewiesen. Unabhängig von der Reifedauer der Eizellen wurde in den Embryonen für die mittels Sanger-Sequenzierung analysierten Allele von DNMT3A ein Anstieg von CpG-Stellen mit undefinierbarem Methylierungszustand (7.9%) gegenüber den anderen Genen (bZAR1: 2.4%, bOCT4: 2.0%, bDNMT3Lo: 1.0%) beobachtet. 79 ERGEBNISSE bZAR1 24h bDNMT3A 48h 24h 48h bOCT4 24h bDNMT3Lo 48h 24h 48h Abbildung 15: Ergebnisse der Analyse der DNA-Methylierung für die Promotorregionen der Gene bZAR1, bDNMT3A, bOCT4 und bDNMT3Lo in Embryonen aus gereiften Rindereizellen unterschiedlicher Reifungsdauer (24h und 48h). Ein einzelnes Quadrat repräsentiert ein einzelnes CpG. Eine Reihe verbundener Quadrate stellt ein einzelnes Allel dar. Ein weißes Quadrat zeigt ein unmethyliertes CpG, ein schwarzes Quadrat ein methyliertes CpG und ein graues Quadrat ein CpG mit uneindeutigem Methylierungszustand. Allele, die CpGs mit uneindeutigem Methylierungszustand (graue Quadrate) enthalten, wurden bei der statistischen Auswertung ausgeschlossen. Rote Pfeile markieren die als abnormal kategorisierten Allele. 80 ERGEBNISSE Tabelle 15: Gesamtübersicht der Ergebnisse zur Analyse der DNA-Methylierung für die Promotorregionen der Gene bH19, bSNRPN, bDNMT3A, bZAR1, bOCT4 und bDNMT3Lo in Embryonen aus gereiften Rindereizellen unterschiedlicher Reifungsdauer (24h und 48h). Die ausgeschlossenen Allele beinhalten Allele, die CpG-Stellen mit einem uneindeutigen Methylierungszustand enthalten (vgl. graue Quadrate, Abbildung 15). Diese Allele tragen nicht zur Anzahl der Einzel-CpGFehler bei und wurden bei der statistischen Auswertung ausgeschlossen. Es wurden keine signifikanten Unterschiede in der Methylierung von vollständigen Allelen oder Einzel-CpG-Fehlern zwischen den beiden Gruppen von Embryonen für die untersuchten Gene detektiert. Gen bH19 bSNRPN bDNMT3A bZAR1 bOCT4 bDNMT3Lo Σ Anzahl der CpG-Stellen im Assay 24h 48h Σ 20 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 35 17 (48%) 0,14 21 16 (76%) 0,10 56 33 (59%) 0,12 30 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 18 5 (28%) 0,07 14 9 (64%) 0,06 32 14 (44%) 0,07 16 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 50 29 (58%) 0 10 (3%) 0,20 48 40 (83%) 0 14 (11%) 0,22 98 69 (70%) 0 24 (5%) 0,21 18 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 21 4 (19%) 0 4 (1%) 0,08 22 8 (36%) 0 1 (1%) 0,10 43 12 (28%) 0 5 (1%) 0,09 7 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 23 1 (4%) 0 4 (3%) 0,09 28 3 (11%) 0 9 (5%) 0,13 51 4 (8%) 0 13 (4%) 0,11 5 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 34 1 (3%) 1 (3%) 4 (3%) 0,13 43 2 (5%) 0 5 (2%) 0,20 77 3 (4%) 1 (1%) 9 (3%) 0,16 96 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 181 57 (32%) 1 (1%) 22 (1%) 0,12 176 78 (44%) 0 29 (1%) 0,13 357 135 (38%) 1 (1%) 51 (1%) 0,13 Das embryonale Genom enthält nach der Verschmelzung des paternalen Spermien- und maternalen Eizellgenoms für die geprägten Gene bH19 und bSNRPN eine Kombination aus methylierten und unmethylierten Allelen für beide Imprinting-Gene. Zur besseren Übersicht 81 ERGEBNISSE der Verteilung der gewonnen Allele in den einzelnen Embryonen wurden die analysierten DNA-Moleküle von bH19 und bSNRPN aufgetrennt nach den einzelnen Embryonen dargestellt (Abbildung 16). Das paternal geprägte Gene bH19 und das maternal geprägte Gen bSNRPN zeigten in 7 von 15 untersuchten Embryonen unabhängig von der Reifedauer der Eizellen ein gemischtes Methylierungsprofil mit einer Kombination aus vollständig methylierten und vollständig unmethylierten Allelen (Abbildung 16; 24h: E7, E8, E12, E13; 48h: E1, E3, E4). Für 3 Proben konnten nur DNA-Moleküle von bH19 amplifiziert werden (E9, E6, E23). In beiden Reifungsgruppen konnten jeweils zwei deutlich hypomethylierte Embryonen identifiziert werden, die ausschließlich unmethylierte Allele der beiden geprägten Gene aufwiesen (24h: E10, E15; 48h: E5, E24). Da zum Zeitpunkt der experimentellen Durchführung keine Sequenzinformationen zu Einzelnukleotid- Polymorphismen in der untersuchten bovinen DNA-Region von bH19 und bSNRPN vorlag, konnte nicht bestimmt werden, ob die analysierten Allele väterlichen oder mütterlichen Ursprungs sind. 82 ERGEBNISSE 24h Embryo bH19 48h bSNRPN Embryo E7 E1 E8 E2 E9 E3 E10 E4 E12 E5 E13 E6 E15 E23 bH19 bSNRPN E24 Abbildung 16: Ergebnisse der Analyse der DNA-Methylierung für die Promotorregionen der Gene bH19 und bSNRPN in Embryonen aus gereiften Rindereizellen unterschiedlicher Reifungsdauer (24h und 48h). Ein einzelnes Quadrat repräsentiert ein einzelnes CpG. Eine Reihe verbundener Quadrate stellt ein einzelnes Allel dar. Ein weißes Quadrat zeigt ein unmethyliertes CpG, ein schwarzes Quadrat ein methyliertes CpG und ein graues Quadrat ein CpG mit uneindeutigem Methylierungszustand. Die detektierten Allele für bH19 und bSNRPN sind separat für die einzelnen Embryonen dargestellt. 83 ERGEBNISSE 3.3 DNA-Methylierungsprofile der Promotorregion bDNMT3Ls in unreifen und in vitro-gereiften Rindereizellen sowie daraus generierten Rinderembryonen Die detektierten Allele der Promotorregion des Gens DNMT3Ls zeigten nach direkter SangerSequenzierung eine Mischung aus methylierten und unmethylierten CpGs in Eizellen aus Antralfollikeln unterschiedlicher Größe (<2 mm, 3-5 mm und >6 mm) (Abbildung 17A). Zusätzlich zu diesen beiden Zuständen konnte eine erhöhte Häufigkeit von CpGs mit uneindeutigem Methylierungszustand beobachtet werden, welche sich im Chromatogramm durch eine heterozygote Überlagerung von Cytosin und Thymin an den uneindeutigen CpGStellen zeigte (vgl. Beispiel in Abbildung 12). Insbesondere CpG7 und CpG8 wiesen in allen analysierten Allelen von Eizellen der größten Follikelkategorie (>6 mm) ausschließlich Signale uneindeutiger Methylierungszustände auf. Die statistische Auswertung bestätigte einen signifikanten Anstieg dieser Signale für CpG2, CpG7 und CpG8 in Eizellen aus großen Antralfollikeln (>6 mm) im Vergleich zu Oozyten aus kleinen (<2 mm: pCpG2 < 0.01, pCpG7 < 0.0001, pCpG8 < 0.0001) und mittelgroßen Follikeln (3-5 mm: pCpG2 < 0.004, pCpG7 < 0.0001, pCpG8 < 0.0001) (Tabelle 16). Die untersuchte Promotorregion von DNMT3Ls schließt den Transkriptionsstartpunkt mit einem Bereich von 134 bp stromaufwärts und 169 bp stromabwärts ein (Abbildung 17C). CpG1 bis CpG4 befinden sich im ersten Exon während CpG5 bis CpG8 110 bp stromaufwärts des Transkriptionsstartpunkts lokalisiert sind. Um zu prüfen, ob sich die Methylierung der CpG-Stellen des ersten Exons von der Methylierung des Bereichs stromaufwärts des Transkriptionsstartpunkts unterscheidet, wurden die Messwerte der benachbarten 4 CpGs miteinander kombiniert und separat für beide Regionen des geteilten Amplikons analysiert. Dieser Vergleich zeigte für beide Bereiche CpG 1-4 und CpG 5-8 eine signifikante Zunahme der uneindeutigen Methylierung in Eizellen großer Follikel (>6 mm) gegenüber mittelgroßen Follikeln (3-5 mm) (pCpG1-4 < 0.002, pCpG5-8 < 0.0001) (Abbildung 17A). Eine signifikante Zunahme dieser Methylierungszustände konnte ebenfalls zwischen Eizellen kleiner Follikel (<2 mm) und Oozyten großer Follikel (>6 mm) für CpG5 bis CpG8 (pCpG5-8 < 0.0001) nachgewiesen werden (Abbildung 17A). Eine Sequenzanalyse nach möglichen Transkriptionsfaktorbindestellen innerhalb der untersuchten Sequenz von DNMT3Ls führte zu einer Bindestelle des „cAMP response element (CRE)-binding protein“ (CREB) an CpG7 der untersuchten Promotorregion von bDNMT3Ls (Abbildung 17C). 84 ERGEBNISSE A 3-5mm 100 40 20 0 60 40 20 0 1 2 3 4 5 6 7 8 20 0 5-8 80 60 40 20 80 60 40 20 0 1-4 24h 5-8 1-4 100 100 100 80 80 80 60 60 60 60 40 40 40 40 20 20 20 0 0 1 2 3 4 5 6 7 8 20 0 1 2 3 4 5 6 7 8 0 1 2 3 4 5 6 7 8 1 2 3 4 5 6 7 8 * 100 100 100 100 80 80 80 80 60 60 60 60 40 40 40 40 20 20 20 20 0 0 1-4 5-8 5-8 0 0 1-4 5-8 1-4 5-8 1-4 5-8 * * * 48h 80 * 100 0 1-4 20 48h * Methylation [%] Methylation [%] 40 40 Embryonen 100 1 2 3 4 5 6 7 8 100 60 60 0 * 80 80 1 2 3 4 5 6 7 8 100 Methylierung [%] 60 100 80 24h Methylierung [%] 80 Gereifte Eizellen >6mm Methylation [%] 100 Methylation [%] Methylierung [%] <2mm Methylierung [%] B Unreife Eizellen C bDNMT3Ls 1 +169 2 12 bp 3 4 19 bp 1 bp 56 137 bp +1 7 8 0 bp 8 bp 12 bp -134 CGCGCAGGATGACGCAGGGATGAGAGCG CREB Bindungsstelle Abbildung 17: Zusammenfassung der Ergebnisse zur Analyse der DNA-Methylierung für die Promotorregion des Gens bDNMT3Ls in unreifen Rindereizellen aus Antralfollikeln unterschiedlicher Größe (>2 mm, 3-5 mm und >6 mm)(A) und gereiften Rindereizellen unterschiedlicher Reifungsdauer (24h und 48h) sowie den daraus generierten Embryonen (B). A/B: Ein einzelnes Quadrat repräsentiert ein einzelnes CpG. Jede Reihe stellt ein einzelnes Allel dar. Ein weißes Quadrat zeigt ein unmethyliertes CpG, ein schwarzes Quadrat ein methyliertes CpG und ein graues Quadrat ein CpG mit uneindeutigem Methylierungszustand (heterozygot T/C in direkter Sanger-Sequenzierung). Die Balkendiagramme stellen die relative Methylierung für die einzelnen CpG-Positionen sowie für die Kombination der exonischen CpGs (CpG 1-4) und der Promotorregion (CpG 5-8) von DNMT3Ls dar. A: Das Auftreten von CpGs mit einem uneindeutigen Methylierungszustand ist signifikant erhöht für die CpG-Positionen 2, 7 und 8 in Eizellen aus großen Follikeln (>6 mm) im Vergleich zu Oozyten aus kleineren Follikeln (<2 mm und 3-5 mm)(*). B: CpG5 und CpG7 sind in Embryonen von 48h 85 ERGEBNISSE gereiften Eizellen signifikant hypomethyliert im Vergleich zu Embryonen, die aus Oozyten generiert wurden, die nur 24h gereift wurden (*, durchgezogene Linie). CpG7 zeigt zusätzlich eine signifikante Hypermethylierung der 24h-Embryonen gegenüber 24h gereiften Eizellen (*, gestrichelte Linie) B: Untersuchte Region von DNMT3Ls mit der Anordnung der CpGs auf dem Amplikon. CpG1 bis CpG4 befinden sich im ersten Exon. CpG5 bis CpG8 liegen in der Promotorregion von DNMT3Ls. An CpG7 befindet sich eine CREB Bindungsstelle. Tabelle 16: Übersicht der Ergebnisse zur Analyse der DNA-Methylierung für die Promotorregion des Gens bDNMT3Ls in unreifen Rindereizellen aus Antralfollikeln unterschiedlicher Größe (>2 mm, 3-5 mm und >6 mm). Die CpG-Positionen 2, 7 und 8 zeigten einen signifikanten Anstieg von CpGs mit uneindeutigem Methylierungszustand in Eizellen aus großen Antralfollikeln (>6 mm, ab) im Vergleich zu Oozyten aus a: kleinen (<2 mm) (p(CpG2) < 0.01, p(CpG7) < 0.0001, p(CpG8) < 0.0001) und b: mittelgroßen Follikeln (3-5 mm) (p(CpG2) < 0.004, p(CpG7) < 0.0001, p(CpG8) < 0.0001). Follikelgröße CpG-Kategorie <2 mm CpG-Position 1 2 3 4 5 6 7 8 methylierte CpGs uneindeutige CpGs Unmethylierte CpGs 5 2 15 16 a 1 5 1 0 21 3 1 18 8 1 13 11 3 8 7 a 4 11 13 a 2 7 3-5 mm methylierte CpGs uneindeutige CpGs unmethylierte CpGs 11 0 20 22 b 1 8 0 0 31 2 1 28 10 0 21 14 0 17 7 b 11 13 13 b 0 18 >6 mm methylierte CpGs uneindeutige CpGs unmethylierte CpGs 1 1 10 5 ab 5 2 0 1 11 3 0 9 3 1 8 4 2 6 0 ab 12 0 0 ab 12 0 Tabelle 17: Übersicht der Ergebnisse zur Analyse der DNA-Methylierung für die Promotorregion des Gens bDNMT3Ls in gereiften Rindereizellen unterschiedlicher Reifungsdauer (24h und 48h). Es konnten keine signifikanten Unterschiede in der Häufigkeit von methylierten bzw. unmethylierten CpGs zwischen den 24h und 48h gereiften Eizellen identifiziert werden. Reifungsdauer CpG-Kategorie 24h 48h CpG-Position 1 2 3 4 5 6 7 8 methylierte CpGs unmethylierte CpGs 8 24 26 6 2 30 7 25 9 23 16 16 7 25 12 20 methylierte CpGs unmethylierte CpGs 7 16 15 8 4 19 7 16 12 11 14 9 7 16 10 13 Tabelle 18: Übersicht der Ergebnisse zur Analyse der DNA-Methylierung für die Promotorregion des Gens bDNMT3Ls in Embryonen aus gereiften Rindereizellen unterschiedlicher Reifungsdauer (24h und 48h). Die CpG-Positionen 5 und 7 zeigten eine signifikante Abnahme an methylierten CpGs in Embryonen, die aus Eizellen generiert wurden, die für 48h gereift wurden, im Vergleich zu Embryonen aus 24h gereiften Eizellen (*; p(CpG5) < 0.05, p(CpG7) < 0.05). Reifungsdauer CpG-Kategorie 24h 48h CpG-Position 1 2 3 4 5 6 7 8 methylierte CpGs unmethylierte CpGs 16 27 22 21 21 22 16 27 22* 21* 23 20 27* 16* 25 18 methylierte CpGs unmethylierte CpGs 13 25 12 26 20 18 18 20 10* 28* 16 22 15* 23* 18 20 86 ERGEBNISSE In IVM-Eizellen und den daraus entwickelten Embryonen bildete die Promotorregion von DNMT3Ls ein Methylierungsprofil ab, das fast ausschließlich methylierte und unmethylierte CpGs in einzelnen Allelen enthielt (Abbildung 17B). CpGs mit uneindeutigem Methylierungszustand traten im Vergleich zu den detektierten Allelen in unreifen Oozyten deutlich seltener auf. Um mögliche Muster der auftretenden Methylierungszustände zu erkennen, wurden die Daten in ähnlicher Weise ausgewertet wie die Ergebnisse der unreifen Eizellen. Bei der statistischen Auswertung wurden zunächst Gruppenvergleiche (24h vs. 48h) innerhalb der Eizellen sowie der Embryonen durchgeführt (Eizellen24h vs. Eizellen48h; Embryonen24h vs. Embryonen48h). Zusätzlich dazu wurden ebenfalls die Gruppen gleicher Kategorie zwischen den Eizellen und den Embryonen getestet (Eizellen 24h vs. Embryonen24h; Eizellen48h vs. Embryonen48h). Diese Vergleiche wurden sowohl für die einzelnen CpGPositionen durchgeführt als auch für die kombinierten Daten von CpG1 bis CpG4 und CpG5 bis CpG8. Da CpGs mit uneindeutigen Methylierungszuständen nur vereinzelt ohne statistische Relevanz (p>0,05) auftraten, wurden die betroffenen Allele aus der Analyse ausgeschlossen und nur Moleküle mit eindeutig methylierten und unmethylierten CpGs berücksichtigt. Die Testung der beiden Maturationsgruppen von Eizellen zeigte weder für einzelne CpG-Positionen noch für die kombinierten Daten des geteilten Amplikons signifikante Unterschiede in der DNA-Methylierung (Abbildung 17B und Tabelle 17). Der Vergleich der beiden Reifungsgruppen von Embryonen wies hingegen eine signifikante Hypomethylierung in der 48h-Gruppe auf, welche sowohl die einzelnen CpG-Positionen 5 (CpG5; p<0.03) und 7 (CpG7; p<0.05) als auch den kombinierten exonischen Bereich dieser CpGs von CpG5 bis CpG8 (p<0.002) betraf (Abbildung 17B und Tabelle 18). Die Gegenüberstellung der unterschiedlich lange gereiften Eizellen mit Embryonen, die sich aus Eizellen gleicher Reifedauer entwickelten, ergab keine signifikanten Methylierungsunterschiede zwischen den 48h-Gruppen. In Embryonen, die aus 24h gereiften Eizellen generiert wurden, zeigten jedoch sowohl das CpG7 als auch die kombinierten Methylierungszustände von CpG5 bis CpG8 eine signifikante Hypermethylierung von p<0.0005 bzw. p<0.0002 gegenüber den 24h-Eizellen (Abbildung 17B). 87 ERGEBNISSE 3.4 DNA-Methylierungsprofile entwicklungsrelevanter Gene in unreifen Rindereizellen aus Kühen unterschiedlichen Alters Um Veränderungen der DNA-Methylierung in der Eizelle zu identifizieren, die mit dem steigenden Lebensalter des weiblichen Individuums auftreten, wurden Oozyten aus Rindern unterschiedlichen Alters untersucht (9-12 Monate, 3-7 Jahre und 8-11 Jahre). Die Kandidatengene, die in den Eizellen unterschiedlich alter Spendertiere analysiert wurden, umfassen bTERF2, bREC8, bBCL-XL, bPISD, bBUB1, bDNMT3Lo, bH19 und bSNRPN (Genset 2). Die detektierten Allele (>95%) wiesen für die Gene bTERF2, bBCL-XL, bPISD, bBUB1, bDNMT3Lo und bH19 ein vollständig methyliertes Muster auf und für bREC8 und bSNRPN ein vollständig unmethyliertes Muster (Abbildung 18 und Abbildung 19). Die Ausbeute der Daten beinhaltet insgesamt für alle 8 Gene 1044 Allele mit 88740 CpGs von 372 Rindereizellen. 9 der 1044 DNA-Moleküle (<1%) zeigten eine abnormale Methylierung (>50% abnormal methylierte CpGs), die in 3 der 8 Kandidatengene (bREC8, bDNMT3Lo und bH19) auftraten. Für bREC8 wurden 2 aberrante Allele in Eizellen von 8-11 Jahre alten Kühen identifiziert. DNMT3Lo war abnormal methyliert in einem DNA-Molekül aus Kühen der jüngsten (9-12 Monate) und der ältesten Gruppe (8-11 Jahre) sowie 2 Allelen aus Kühen mittleren Alters (37 Jahre). Abnormal methylierte Allele von bH19 wurden mit einem Molekül in Eizellen aus jungen Kühen (9-12 Monate) und mit 2 Sequenzen in Oozyten von alten Tieren (8-11 Jahre) beobachtet (Tabelle 19). Einzelne, abnormal methylierte CpGs traten für die Gene bREC8, bBCL-XL, bDNMT3Lo, bH19 und bSNRPN auf (insgesamt 68, <1%). bDNMT3Lo, bH19 und bSNRPN enthielten in allen drei Gruppen von Eizellen CpG-Fehler, bREC8 nur in der Kohorte junger Kühe und bBCL-XL in Eizellen junger und alter Tiere. Die statistische Auswertung zwischen den drei Kategorien von Oozyten ergab für kein Gen weder für das Auftreten abnormal methylierter Allele noch für die Häufigkeit von CpG-Fehlern signifikante Gruppenunterschiede. 88 ERGEBNISSE bBCL-XL bPISD 9-12 Monate 3-7 Jahre 8-11 Jahre 9-12 Monate 3-7 Jahre bBUB1 8-11 Jahre 9-12 Monate 3-7 Jahre 8-11 Jahre bTERF2 9-12 Monate 3-7 Jahre 8-11 Jahre bREC8 9-12 Monate 3-7 Jahre 8-11 Jahre Abbildung 18: Ergebnisse der Analyse der DNA-Methylierung für die Promotorregionen der Gene bREC8, bPISD, bBUB1, bTERF2 und bBCL-XL in Rindereizellen von Kühen unterschiedlichen Alters (9-12 Monate, 3-7 Jahre und 8-11 Jahre). Ein einzelnes Quadrat repräsentiert ein einzelnes CpG. Eine Reihe verbundender Quadrate stellt ein einzelnes Allel dar. Ein weisses Quadrat zeigt ein unmethyliertes CpG, ein schwarzes Quadrat ein methyliertes CpG und ein graues Quadrat ein CpG mit uneindeutigem Methylierungszustand. Allele, die CpGs mit uneindeutigem Methylierungszustand (graue Quadrate) enthalten, wurden bei der statistischen Auswertung ausgeschlossen. Rote Pfeile markieren die als abnormal kategorisierten Allele. 89 ERGEBNISSE bDNMT3Lo 9-12 Monate 3-7 Jahre 8-11 Jahre 9-12 Monate bH19 3-7 Jahre bSNRPN 8-11 Jahre 9-12 Monate 3-7 Jahre 8-11 Jahre Abbildung 19: Ergebnisse der Analyse der DNA-Methylierung für die Promotorregionen der Gene bDNMT3Lo, bH19 und bSNRPN in Rindereizellen von Kühen unterschiedlichen Alters (9-12 Monate, 3-7 Jahre und 8-11 Jahre). Ein einzelnes Quadrat repräsentiert ein einzelnes CpG. Eine Reihe verbundender Quadrate stellt ein einzelnes Allel dar. Ein weisses Quadrat zeigt ein unmethyliertes CpG, ein schwarzes Quadrat ein methyliertes CpG und ein graues Quadrat ein CpG mit uneindeutigem Methylierungszustand. Allele, die CpGs mit uneindeutigem Methylierungszustand (graue Quadrate) enthalten, wurden bei der statistischen Auswertung ausgeschlossen. Rote Pfeile markieren die als abnormal kategorisierten Allele. 90 ERGEBNISSE Tabelle 19: Gesamtübersicht der Ergebnisse zur Analyse der DNA-Methylierung für die Promotorregionen der Gene bTERF2, bREC8, bBCL-XL, bPISD, bBUB1, bDNMT3Lo, bH19 und bSNRPN in Rindereizellen von Kühen unterschiedlichen Alters (9-12 Monate, 3-7 Jahre und 8-11 Jahre). Die ausgeschlossenen Allele beinhalten Allele, die CpG-Stellen mit einem uneindeutigen Methylierungszustand enthalten (vgl. graue Quadrate, Abbildung 18 und Abbildung 19). Diese Allele tragen nicht zur Anzahl der Einzel-CpG-Fehler bei und wurden bei der statistischen Auswertung ausgeschlossen. Signifikante Unterschiede zwischen den verschiedenen Altersgruppen in der Methylierung von vollständigen Allelen und Einzel-CpGFehlern wurden für keines der untersuchten Gene detektiert. Gen bTERF2 bREC8 bBCL-XL bPISD bBUB1 bDNMT3Lo bH19 bSNRPN Anzahl der CpG-Stellen im Assay 9-12 Monate 3-7 Jahre 8-11 Jahre Σ 7 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 23 0 0 0 0,04 16 1 (6%) 0 0 0,03 26 0 0 0 0,06 65 1 (2%) 0 0 0,05 5 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 18 1 (6%) 0 1 (1%) 0,03 4 0 0 0 0,01 21 0 2 (10%) 0 0,05 43 1 (2%) 2 (5%) 1 (1%) 0,03 8 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 66 0 0 3 (1%) 0,13 31 0 0 0 0,07 54 1 (2%) 0 1 (1%) 0,13 151 1 (1%) 0 4 (1%) 0,11 6 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 26 0 0 0 0,05 23 2 (9%) 0 0 0,05 19 1 (5%) 0 0 0,04 68 3 (4%) 0 0 0,05 6 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 111 0 0 0 0,21 63 0 0 0 0,13 81 1 (1%) 0 0 0,19 255 1 (1%) 0 0 0,18 5 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 103 5 (5%) 1 (1%) 8 (2%) 0,2 47 2 (4%) 2 (4%) 8 (4%) 0,10 67 5 (8%) 1 (2%) 10 (3%) 0,16 217 12 (6%) 4 (2%) 26 (3%) 0,15 18 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 77 14 (18%) 1 (1%) 7 (1%) 0,15 39 2 (5%) 0 2 (1%) 0,08 60 9 (15%) 2 (3%) 5 (1%) 0,14 176 25 (14%) 3 (2%) 14 (1%) 0,12 30 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 25 10 (40%) 0 8 (2%) 0,05 25 5 (20%) 0 9 (2%) 0,05 19 0 0 6 (1%) 0,04 69 15 (22%) 0 23 (1%) 0,05 91 ERGEBNISSE Σ 85 Analysierte Allele Ausgeschlossene Allele Abnormale Allele CpG-Fehler Allel-Ausbeute 449 30 (7%) 2 (1%) 27 (1%) 0,11 248 12 (5%) 2 (1%) 19 (1%) 0,07 347 17 (5%) 5 (1%) 22 (1%) 0,10 1044 59 (6%) 9 (1%) 68 (1%) 0,09 92 DISKUSSION 4 Diskussion 4.1 DNA-Methylierungsprofile entwicklungsrelevanter Gene in unreifen und in vitro-gereiften Rindereizellen sowie daraus generierten Rinderembryonen Die korrekte Entwicklung des Spermiums und der Eizelle stellen die Grundlage einer normalen, gesunden Embryogenese dar. Insbesondere epigenetische Prozesse repräsentieren entscheidende Schritte sowohl in der Entwicklung der Keimzellen als auch in der anschließenden embryonalen Entwicklung [393]. Fehler im epigenetischen Profil von Keimzellen führen zu Erkrankungen wie dem Beckwith-Wiedemann-Syndrom beim Menschen bzw. dem „Large Offspring Syndrome“ (LOS) in Wiederkäuern [272, 273, 394]. Die Untersuchung von unreifen und in vitro-gereiften Rindereizellen sowie den daraus generierten Rinderembryonen hat zum Ziel, die Entwicklung vom Frühstadium der Eizelle bis hin zum Frühstadium des Embryos abzudecken. Sowohl beim Rind als auch beim Menschen liegt die optimale Zeitspanne der Eizellreifung bei 24 h (siehe Kapitel 1.4). Damit stellt das Rind ein geeignetes Modell insbesondere für die post-ovulatorische Reifung bzw. postovulatorische Alterung der Oozyte im humanen Organismus dar. Die abnehmende Entwicklungskompetenz bei einer Reifung der Eizelle über 24 h hinaus könnten durch epigenetische Veränderungen verursacht werden. Durch die Auswahl unterschiedlicher Stadien von unreifen Eizellen bzw. durch gereifte Oozyten unterschiedlicher Reifedauer sollen natürliche Veränderungen sowie Veränderungen induziert durch eine verlängerte Maturation im DNA-Methylierungsprofil entwicklungsrelevanter Gene identifiziert werden. 4.1.1 DNA-Methylierungsprofile der Gene bH19, bSNRPN, bZAR1, bDNMT3A, bOCT4 und DNMT3Lo in unreifen und in vitro-gereiften Rindereizellen sowie daraus generierten Rinderembryonen Aus früheren Studien ist bekannt, dass bei der IVM humaner Eizellen die Erfolgsraten mit der Größe des Antralfollikels der Oozyte korrelieren [295]. Die in dieser Arbeit untersuchten unreifen Rindereizellen aus Antralfollikeln unterschiedlicher Größe (<2 mm, 3-5 mm und >6 mm) repräsentierten Eizellen, die für unterschiedliche Zeitspannen im Antralstadium der zyklischen Follikelreifung des geschlechtsreifen Rind verweilten. Die Mehrzahl der amplifizierten Allele der Gene bH19, bSNRPN, bZAR1, bDNMT3A, bOCT4 und bDNMT3Lo weisen ein normales DNA- Methylierungsmuster in allen drei Kategorien von Eizellen auf. 3 93 DISKUSSION von 466 (<1%) detektierten DNA-Molekülen zeigten ein abnormales Methylierungsprofil. Alle 3 abnormal methylierten Allele wurden in geprägten Genen von Eizellen aus kleinen Antralfollikeln (<2 mm) nachgewiesen (Abbildung 13). Diese Beobachtung stellt keinen signifikanten Anstieg abnormaler Methylierung in dieser Gruppe weiblicher Keimzellen dar. Dieses Ergebnis kann jedoch dennoch auf ein potentiell höheres Risiko für MethylierungsFehler in Eizellen von Antralfollikeln <2 mm hinweisen. Frühere Studien haben gezeigt, dass bovine Eizellen aus Antralfollikeln <2 mm signifikant geringere Erfolgsraten bei der in vitroReifung und Fertilisation aufweisen und nicht im Stande sind sich über das 8-Zellstaium hinaus zu teilen [395]. Eine höhere Entwicklungskompetenz wurde jedoch für Eizellen aus Follikeln >6 mm im Vergleich zu Oozyten aus Follikeln <4 mm nachgewiesen [396]. Ähnliche Resultate wurden bei der Bewertung der Entwicklungskompetenz von Eizellen unterschiedlicher Größen gewonnen. Bei Oozyten mit einem Durchmesser von <100 μm, 100-110 μm, 110-120 μm und >120 μm wurde untersucht, in wie fern sie über das Potential verfügen die Stadien der Meiose II, der Furchung und der Bildung einer Blastozytse zu erreichen. Der Anteil an Eizellen, die diese Entwicklungsstadien erreichten, stieg graduell mit dem Durchmesser der Oozyten. Der höchste Anstieg wurde dabei zwischen Eizellen mit einem Durchmesser von 100-110 μm und 110-120 μm beobachtet. Dieser Anstieg könnte die Oozyten-Größe von 100-120 μm als entscheidenden Punkt zur Erlangung der Entwicklungskompetenz der Eizelle kennzeichnen [397]. Bovine weibliche Keimzellen erlangen die volle meiotische Kompetenz bei einem Durchmesser von etwa 110 μm und zeigen bei einer Größe >110 μm keine transkriptionelle Aktivität [158]. Die Gruppe um Fair konnte zeigen, dass es eine schwache Korrelation zwischen der Oozyten-Größe und der Größe des Follikel gibt (r=0.32, p<0.0001) [158]. Dabei enthalten 10 bis 15% der großen Follikel kleine Eizellen und umgekehrt. Die Eizelle stellt ihr Wachstum bei einem Durchmesser von 120 μm ein wenn der Follikel eine Größe von 3 mm erreicht hat. Die kritische Größe von 110 μm erlangt die Eizelle bei einem Follikeldurchmesser von etwa 2 mm [158]. Somit bestätigen die Ergebnisse dieser Arbeit mit einer erhöhten abnormalen Methylierung in Keimzellen von Follikeln <2 mm die reduzierte Entwicklungskompetenz von Oozyten dieser Kategorie auf epigenetischer Ebene. O’Doherty et al. untersuchten die Etablierung der DNA-Methylierung geprägter Gene in bovinen Eizellen unterschiedlicher Größe [398]. Analysiert wurde die Methylierung von SNRPN, MEST, IGF2R, PEG10 und PLAGL1 in Oozyten mit einem Durchmesser von 101-110 μm, 110-120 μm und ≥120 μm. Die 94 DISKUSSION Ergebnisse zeigten einen graduellen genspezifischen Anstieg der Methylierung in Abhängigkeit der Größe der Eizelle. Die größte Veränderung der DNA-Methylierung konnte bei SNRPN, PEG10 und PLAGL1 zwischen Eizellen kleiner und größer als 110 μm beobachten. Diese Resultate bestätigen unsere Daten für SNRPN. Bovine Eizellen mit einem Durchmesser unter 110 μm haben eine reduzierte Entwicklungskompetenz und weisen ein vermutlich noch nicht vollständig etabliertes DNA-Methylierungsprofil bei geprägten Genen auf. Eine mögliche Erklärung dieser Beobachtung könnte sein, dass Eizellen aus Follikeln <2 mm bzw. Oozyten mit einem Durchmesser <110 μm kritische Zwischenstadien sich entwickelnder Eizellen repräsentieren. Es ist anzunehmen, dass die reduzierte Entwicklungskompetenz von Eizellen <110 μm in Verbindung mit DNA-Methylierungsprofilen geprägter Gene steht, die zu diesem Zeitpunkt der Entwicklung noch nicht vollständig etabliert sind. Die Präsenz abnormal methylierter Allele geprägter Gene in dieser Wachstumsphase stellt vermutlich ein natürliches Zwischenstadium der sich entwickelnden Eizelle und keinen pathologischen Zustand dar. Da dieser Prozess der epigenetischen Reprogrammierung noch nicht vollständig abgeschlossen ist, haben Eizellen dieser Größe weiterhin das Potential die korrekten Methylierungsprofile und die vollständige Entwicklungskompetenz zu erlangen. Mit Antralfollikeln der Größe 3-5 mm wurde eine in vitro-Reifung für 24h (Standard Protokoll) und 48h (simulierte post-ovulatorische Alterung) durchgeführt. Follikel dieser Größenordnung sollten 100-120 μm große Eizellen mit potentiell hoher Entwicklungskompetenz enthalten. Das Risiko möglicher Entwicklungsdefizite, die bereits in der unreifen Eizelle vorliegen, wird damit reduziert und die Untersuchung des Einflusses der Reifedauer auf die Entwicklung der Oozyte und des Embryos ermöglicht. Zur Simulation der post-ovulatorischen Alterung bei Ausbleiben der Befruchtung wurden die Eizellen experimentell über die üblichen 24 h hinaus im Reifungsmedium belassen. Erst nach 48 h wurden diese post-ovulatorisch gealterten Eizellen befruchtet und die gewonnenen Embryonen epigenetisch untersucht. Die Gen-spezifische Methylierungsanalyse der postovulatorisch gealterten Eizellen zeigte signifikant mehr abnormal methylierte Allele für die Oozyten-spezifische Form von DNMT3L (DNMT3Lo). DNMT3L stellt einen essentiellen Cofaktor für die korrekte Funktionsweise von DNMT3A bei der de novo Methylierung während der Embryonalentwicklung dar. Die signifikante Hypermethylierung von DNMT3Lo in der 48h-Gruppe könnte das Protein als Schlüsselmolekül identifizieren, das durch die post95 DISKUSSION ovulatorische Alterung beeinflusst wird. Die Hypermethylierung von DNMT3Lo könnte zu einer reduzierten Expression von DNMT3L in der Eizelle führen, was zu einer fehlerhaften oder verminderten de novo Methylierung geprägter und nicht-geprägter Gene in der Oozyte führt. So ist aus Studien in Mäusen bekannt, dass eine veränderte Expression von DNMT3L verursacht durch eine Hypermethylierung die Entwicklung der Plazenta und der Nachkommenschaft beeinträchtigt [399, 400]. Während eine Hypermethylierung von DNMT3Lo in post-ovulatorisch gealterten Eizellen vorlag, konnte in der vorliegenden Arbeit hingegen keine Hypermethylierung von DNMT3Lo in Embryonen detektiert werden, die aus dieser Kategorie von Eizellen generiert wurden. Die aberrante Methylierung von DNMT3Lo scheint damit während der Reprogrammierungsprozesse im Embryo eine Veränderung zum normalen Methylierungsmuster durchlaufen zu haben. Der Einfluss einer post-ovulatorischen Alterung auf geprägte Gene ist noch weitgehend unerforscht. In einer Studie im Mausmodell von Imamura et al. wurde das geprägte Gen Peg1/Mest in ovulierten Eizellen, in vitro-gealterten Oozyten sowie Präimplantationsembryonen untersucht [401]. Die Ergebnisse zeigten eine dynamische Veränderung der Methylierung. Eizellen in der Metaphase II wiesen ein heterogenes Methylierungsmuster auf, das nach 22h in vitro-Kultivierung hypermethyliert wurde und nach weiteren 20h in vitro-Kultur (insgesamt 42h) erneut hypomethyliert in den Eizellen vorlag. Diese für 42 h gereiften Oozyten besaßen damit ein ähnliches Methylierungsprofil wie Embryonen. In einer weiteren Studie mit Mäusen von Liang et al. wurde die DNAMethylierung von Peg1/Mest und Snrpn in post-ovulatorischen Eizellen analysiert, die für 13h, 21h und 29h in vivo gereift wurden sowie in Oozyten mit Kumuluszellen („cumulusenclosed oocytes“ (CEOs)) und ohne Kumuluszellen („denuded oocytes“ (DOs)), die für 21h und 29h in vitro-kultiviert wurden [402]. Peg1/Mest zeigte keine Demethylierung in allen Gruppen unterschiedlicher Reifung, während Snrpn sowohl in den in vivo- als auch in vitrogereiften DOs nach 29h demethyliert wurde. Die ungleichen Ergebnisse beider Studien hinsichtlich der Peg1-Demethylierung könnten durch die unterschiedlichen Methoden der Reifung (in vivo- und in vitro) aber auch durch die verschiedenen Reifungszeiträume erklärt werden. Die Methylierungsprofile von Peg1/Mest nach 21h (Liang) und 22h (Imamura) weisen unabhängig von der Art der Reifung einen vollständig methylierten Zustand auf. Die 29 h in vitro-gereiften CEOs in Liangs Studie lassen nur eine leichte Demethylierung erkennen und deuten damit einen Trend zur Hypomethylierung von Peg1 an, was in Einklang 96 DISKUSSION mit den 42h in vitro-kultivierten Eizellen in Imamuras Studie steht. Obwohl Peg1/Mest hier nicht als Kandidatengen untersucht wurde, konnte für das geprägte Gen SNRPN ein abnormal methyliertes Allel detektiert werden, was einen ähnlichen Trend wie bei Peg1/Mest in Imamuras Studie wiederspiegelt. Dieses Ergebnis ist nicht zwingend widersprüchlich zu den Daten von Liang für SNRPN, da für die in vitro-Reifung der vorliegenden Arbeit ausschließlich Eizellen mit Kumuluszellen (CEOs) verwendet wurden, die keine Demethylierung bei Liang aufwiesen. Zudem wurden die bovinen Eizellen mit 48 h länger gealtert als die Eizellen bei Liang mit 29 h, was das Auftreten abnormaler Methylierung für SNRPN erklären könnte. 4.1.2 DNA-Methylierungsprofile der Promotorregion bDNMT3Ls in unreifen und in vitrogereiften Rindereizellen sowie daraus generierten Rinderembryonen Die Promotorregion des Stammzell-spezifische Transkript des Gens bDNMT3L (bDNMT3Ls) wies im Vergleich zu allen anderen untersuchten DNA-Bereichen dieser Arbeit ein auffällig heterogenes Methylierungsmuster bestehend aus methylierten und unmethylierten CpGs sowie CpGs mit uneindeutigem Methylierungszustand auf. Die statistische Auswertung des Auftretens der drei Methylierungszustände an einzelnen CpG-Positionen konnte signifikante Gruppenunterschiede zwischen den einzelnen Kohorten aufdecken (Abbildung 17). Die gereiften Eizellen verschiedener Reifungsdauer wiesen keine signifikanten Unterschiede in bDNMT3Ls auf. In den daraus generierten Embryonen konnte jedoch eine signifikante Hypomethylierung der Positionen 5 und 7 in der 48h-Gruppe nachgewiesen werden (Abbildung 17B). Der Vergleich der Gruppen gleicher Kategorie zwischen den Eizellen und den Embryonen (Eizellen24h vs. Embryonen24h; Eizellen48h vs. Embryonen48h) identifizierte eine signifikante Hypermethylierung von Position 7, die ausschließlich zwischen den 24hgereiften Eizellen und den entsprechenden Embryonen auftrat. Da die in vitro-Reifung der Eizellen für 24h die physiologischen Bedingungen simulieren bzw. repräsentieren, lassen diese Ergebnisse darauf schließen, dass die Hypermethylierung der CpG-Stelle von DNMT3Ls an Position 7 nach erfolgter Befruchtung einen normalen, physiologischen Zustand darstellt. Diese Hypermethylierung beim Übergang von der Eizelle zum Embryo trat nicht auf, wenn die Eizellen eine verlängerte Reifungsdauer von 48h durchliefen. Die verlängerte in vitroMaturation der Oozyte hat somit einen Einfluss auf die physiologischen MethylierungsVeränderungen der CpG-Stelle an Position 7 der Promotorregion von bDNMT3Ls beim 97 DISKUSSION Übergang von der Eizelle zum Embryo. Eine solche spezifische Veränderung einzelner CpGStellen könnte ein Hinweis auf eine funktionelle Rolle der betreffenden Stelle(n) sein. Eine Suche nach möglichen Transkriptionsfaktorbindestellen innerhalb der analysierten Sequenz von DNMT3Ls führte zu einer Bindestelle des „cAMP response element (CRE)-binding protein“ (CREB) an CpG7 der untersuchten Promotorregion von bDNMT3Ls (Abbildung 17C). CREB stellt einen entscheidenden Transkriptionsfaktor dar, welcher eine Vielzahl biologischer Prozesse reguliert mit dem Ziel einer korrekten Zelldifferenzierung und eines gesunden Zellwachstums [403]. Ursprünglich wurde CREB als Zielprotein des zyklischen AMP (cAMP) Signalwegs identifiziert. Es wird darüber hinaus von mehreren, verschiedenen Signalwegen angesteuert, die ihrerseits durch zahlreiche unterschiedliche Stimuli aktiviert werden [404]. Die Regulation der Expression wird dabei durch die Bindung an ein „cAMP response element“ (CRE) im Promotorbereich des Gens vermittelt. Dabei bindet der Transkriptionsfaktor an zwei unterschiedliche Klassen von CREs. Beide Klassen unterscheiden sich lediglich in der Symmetrie bzw. Asymmetrie des enthaltenen DNA-Motivs CGTCA. Bei der asymmetrischen Form ist diese Sequenz nur jeweils einmal enthalten, während die symmetrische Form zwei dieser Motive in palindromischer Anordnung aufweist (TGACGTCA) [405]. Eine methylierungssensitive Regulation der Expression durch CREB konnte für die Matrix-Metalloproteinase (MMP) 13 nachgewiesen werden. Indem CREB methylierungssensitiv an den Promotor von MMP13 bindet, sobald die CpG-Stelle unmethyliert vorliegt, wird die Expression des Gens induziert [406]. CREB stellt u.a. ein essentielles Protein für die Eizelle und die frühe embryonale Entwicklung dar, was in Studien an Eizellen von Fröschen (Xenopus laevis) und Fischen (Oreochromis niloticus and Clarias gariepinus) gezeigt wurde [407, 408]. Untersuchungen an Eizellen und Blastozysten von Xenopus laevis konnten eine essentielle Rolle von maternalem CREB der Eizelle für die Gastrulation nachweisen [407]. In Oreochromis niloticus konnten drei unterschiedliche Formen von CREB identifiziert werden. CREB1 und CREB2 konnten dabei neben dem Ovar auch in anderen Geweben nachgewiesen werden, wohingegen CREB3 ausschließlich im Ovar und den Follikeln exprimiert wurde. Eine Gonadotropin-induzierte Maturation der Eizelle führte zu einer Steigerung der Expression sowohl von CREB2 (Faktor 1,5) als auch CREB3 (Faktor 3). Diese Ergebnisse lassen vermuten, dass CREB2 und CREB3 entscheidende Funktionen während der Reifung von Eizellen ausführen, während CREB1 eine wichtige Rolle im Wachstum der Oozyte zugeschrieben wird [408]. Clarias gariepinus wies nur eine Form 98 DISKUSSION von CREB auf. Die Steigerung der Expression durch Gonadotropin lässt jedoch ebenfalls vermuten, dass der Transkriptionsfaktor eine essentielle Aufgabe während der Maturation der Eizelle übernimmt [408]. Diese entscheidende Rolle von CREB in der Eizelle sowie die Fähigkeit zur methylierungssensitiven Regulation der Expression könnten aufgrund der vorliegenden Methylierungsdaten für CpG7 der analysierten Promotorregion von bDNMT3Ls auf einen ähnlichen Regulationsmechanismus von bDNMT3Ls in bovinen Eizellen und Embryonen hinweisen. Neben den gereiften Eizellen und Embryonen weist CpG7 auch signifikante Unterschiede in den Methylierungsprofilen der ungereiften Eizellen unterschiedlicher Größe auf. Bei den unreifen Eizellen unterschiedlicher Größe konnte eine erhöhte Frequenz von CpGs mit uneindeutigem Methylierungszustand beobachtet werden. Im Gegensatz zu methylierten und unmethylierten CpGs, zeigte das Auftreten der unklaren CpGs signifikante Unterschiede an den Positionen 2, 7 und 8 zwischen großen Eizellen (>6mm) und Eizellen der beiden kleineren Kategorien (<2mm und 3-5mm) (Abbildung 17A). Dies entspricht einem graduellen Anstieg an CpGs mit uneindeutigem Methylierungszustand an Position 7 mit zunehmender Oozyten-Größe (Abbildung Sangersequenzierung 17A). Dieser Methylierungszustand ist in der durch ein heterozygotes Signal eines Cytosins und Thymins gekennzeichnet. Empirisch ist bekannt, dass diese Muster selten auftreten. Für gewöhnlich wurden diese Muster als technische Artefakte interpretiert, da vermutet wurde, dass sie durch eine unvollständige Bisulfit-Konvertierung oder eine fehlgeschlagene Separation mehrerer Allele verursacht wurden. Vor dem Hintergrund der Resultate für bDNMT3Ls in in vitro-gereiften Eizellen und Embryonen könnten diese uneindeutigen Methylierungszustände an CpG7 in unreifen Eizellen auf eine biologische Ursache hinweisen. Als eine mögliche physiologische Erklärung für das signifikant erhöhte Auftreten dieser Signale an spezifischen CpG-Stellen könnte die molekulare Maschinerie darstellen, die für die epigenetische Regulation verantwortlich ist. Das gemeinhin verwendete Standard-Protokoll für die Bisulfitsequenzierung enthält zwischen dem Schritt der DNA-Isolierung und der Zugabe des Bisulfitreagenz einen Schritt zur Entfernung von Proteinen. In den meisten Protokollen, einschließlich des verwendeten Protokolls, wird dieser Arbeitsschritt durch eine Behandlung mit Proteinase K durchgeführt. In einer Studie von Warnecke und Kollegen wurde untersucht, welchen Einfluss Proteine auf die Bisulfitsequenzierung ausüben wenn sie vor der Bisulfitbehandlung nicht entfernt wurden [376]. Dazu wurde eine Region des GSTP199 DISKUSSION Promotors in DU145 Zellen mit und ohne Proteinase K Verdau vor der Bisulfitbehandlung kloniert und sequenziert. Ein Vergleich der Sequenzen beider Behandlungen zeigte sowohl einen hohen Grad von nicht-CpG-Methylierung als auch eine partielle Konvertierung beliebiger Cytosin-Nukleotide in Sequenzen, die aus DNA generiert wurde, die nicht mit Proteinase K behandelt wurde. Dieses extreme Beispiel zeigt, dass Proteine, die an der DNA gebunden sind, tatsächlich mit dem Bisulfitreagenz interagieren und die Qualität der Konvertierung beeinflussen. Obwohl bei der Generierung der vorliegenden Daten ein Proteinase K Vedau durchgeführt wurde und keine nicht-CpG Methylierung beobachtet werden konnte, liegt die Vermutung nahe, dass die uneindeutigen Signale an spezifischen CpGs durch Faktoren verursacht werden, die mit der DNA-Methylierung in Verbindung stehen und nicht ausreichend durch Proteinase K entfernt werden konnten. Der Methylierungsstatus der CpGs an den Positionen 2, 7 und 8 innerhalb der untersuchten Region von DNMT3Ls scheinen eine entscheidende Rolle für die Regulation des Gens zu spielen. Diese Stellen molekularer Aktivität durchlaufen Methylierungs-Veränderungen während der Entwicklung der unreifen Eizelle. Diese molekulare Aktivität verstärkt sich mit zunehmender Follikelgröße bzw. Oozytengröße und erreicht sein Maximum in Eizellen aus Follikeln mit einem Durchmesser >6mm. Als Folge der molekularen Interaktion der DNAMethylierungs-Maschinerie mit den regulatorischen CpGs kommt es zu einer Beeinträchtigung der Bisulfitreaktion, wodurch die unklaren Signale an den betreffenden CpG-Positionen verursacht werden. DNMT3L als katalytisch inaktives und navigierendes Coenzym von DNMT3A nimmt eine zentrale Rolle in DNA-Methylierungsprozessen von Eizellen und frühen embryonalen Zellen ein. Während für DNMT3A keine signifikanten Unterschiede in der DNA-Methylierung unreifer und in vitro-gereifter Rindereizellen sowie den daraus generierten Embryonen identifiziert werden konnten, zeigten sowohl DNMT3Lo als auch DNMT3Ls Veränderungen in den Methylierungsprofilen der untersuchten Stadien. Diese Ergebnisse könnten ein Hinweis darauf sein, dass DNMT3L neben der räumlichen Koordination auch einen entscheidenden Faktor bei der zeitlichen Abfolge der Methylierungsprozesse von der Entwicklung der Eizelle bis hin zum frühen Embryonalstadium darstellt. Die zeitliche Steuerung dieser Prozesse erfolgt dabei vermutlich durch eine Promotorbereiche sowie unterschiedlicher Steuerungsmechanismen Kombination Transkriptionsfaktoren von wie könnte DNA-Methylierung CREB. es dem Diese der Kombination komplexen DNA100 DISKUSSION Methylierungssystem ermöglichen dynamisch und flexibel auf äußere Störfaktoren vor der Fertilisation zu reagieren und die Methylierungsprofile des entstehenden Organismus stabil aufrecht zu erhalten. Die vorliegenden Ergebnisse in vitro-gereifter Eizellen sowie den daraus erzeugten Embryonen unterstützen diese Theorie. Neben bDNMT3Lo weisen auch die Gene bZAR1, bOCT4, bDNMT3A, bH19 und bSNRPN keine signifikanten Unterschiede in der DNAMethylierung der beiden untersuchten Gruppen von Embryonen auf. Damit scheint die abnormale Methylierung von DNMT3Lo in den post-ovulatorisch gealterten Eizellen keinen Einfluss auf die Methylierungsprofile von DNMT3Lo, ZAR1, OCT4, DNMT3A, H19 und SNRPN in den resultierenden 4-8-Zell-Embryonen zu haben. Einzig DNMT3Ls zeigt eine abnormale Veränderung der Methylierung einer potentiellen Transkriptionsfaktorbindetelle in Embryonen aus post-ovulatorisch gealterten Eizellen. Diese Veränderung könnte als balancierende Regulation interpretiert werden, um das System an den äußeren Einfluss der verlängerten Reifung anzupassen und eine stabile Etablierung der DNA-Methylierungsprofile des Embryos zu gewährleisten. Ähnliche Ergebnisse lieferte eine Studie im Mausmodell, in der die DNA- Mtehylierungsmuster verschiedener Gene (SCP3, DAZL, CDKN1C, IGF2RDMR1, IGF2 DMR1, OCT4, DNMT3Lo, DNMT3Ls und 4 unterschiedlichen repetitiven Sequenzen) in Embryonen DNMT3Ls-difizienter Mäuse untersucht wurden [409]. Mit Ausnahme von OCT4 und IGF2R DMR1 konnten keine signifikanten Unterschiede bzw. nur Unterschiede im E6.5 Stadium nicht jedoch zum späteren E9.5 Stadium zwischen dem Wildtyp und den DNMT3Lsdifizienten Embryonen gemessen werden. Gleichzeitig beobachtete man eine signifikante Steigerung der Expression von DNMT3A in E6.5 DNMT3Ls-defizienter Embryonen. Die Schlussfolgerung aus diesen Ergebnissen war, dass die Expression von DNMT3L nicht essentiell für die de novo Methylierung im frühen Embryo zu sein scheint und das Fehlen von DNMT3L durch eine Verstärkte Bildung von DNMT3A ausgeglichen werden könnte [409]. Neben den DNA-Methylierungsdaten der vorliegenden Arbeit wurde zusätzlich als Teil der Studie die RNA-Expression einiger entwicklungsrelevanter Gene von Mitarbeitern des Instituts für Nutztiergenetik des Friedrich-Löffler-Instituts in Mariensee/Hannover bestimmt. Dabei wurde u.a. eine signifikante Abnahme der Expression von DNMT3A in Embryonen aus post-ovulatorisch gealterten Eizellen identifiziert, die nicht auf Veränderungen der DNAMethylierung zurückzuführen ist. Obgleich in der vorliegenden Arbeit frühere embryonale Stadien im Rind untersucht wurden und keine DNMT3Ls-Defizienz vorlag, gleichen sich 101 DISKUSSION sowohl der Studienentwurf als auch die Ergebnisse in Teilen mit der Studie im Mausmodell. Als Schlussfolgerung aus beiden Studien lässt sich festhalten, dass die beiden für die Etablierung der DNA-Methylierung essentiellen Proteine DNMT3A und DNMT3L sowohl direkt über molekulare Interaktion als auch indirekt über DNA-Methylierung und RNAExpression in einer komplexen Verbindung stehen. Diese Verbindung ermöglicht es dem biologischen System bei der sensiblen Transition von der Eizelle zum Embryo flexibel auf äußere Störfaktoren zu reagieren um letztendlich die für das Überleben des Embryos essentiellen korrekten DNA-Methylierungsmuster stabil zu etablieren. 4.2 DNA-Methylierungsprofile entwicklungsrelevanter Gene in unreifen Rindereizellen aus Kühen unterschiedlichen Alters Der Prozess der Alterung biologischer Systeme ist charakterisiert durch mindestens neun zelluläre und molekulare Kennzeichen, einschließlich genomischer Instabilität, Telomerverkürzung, Proteostase und epigenetischen Veränderungen [410]. Während entscheidende Faktoren der Alterung somatischer Zellen identifiziert wurden, ist wenig über die reproduktive Alterung bzw. die ovarielle Alterung bekannt. Vergleiche der Erfolgsquoten nach IVF und Embryonentransfer bei jüngeren und älteren Frauen zeigten ein höheres Auftreten von Implantationsversagen und Spontanaborten bei älteren Patientinnen [200, 411, 412]. Als Ursache für die Abnahme der weiblichen Fertilität mit steigendem Lebensalter konnte die Eizellqualität identifiziert werden [195]. Die erhöhte Häufigkeit von Aneuploidien in Eizellen älterer Frauen (50%) [413, 414] im Vergleich zu Oozyten jüngerer Frauen (20%) [342] deutet auf eine fehlerhafte Separation der Chromosomen in alternden Ovarien hin [415, 416]. Ein Nachlassen der Kohäsion könnte für die durch Alterung verursachte, fehlerhafte Trennung der Chromosomen während der Meiose verantwortlich sein [417-419]. Cohesin beispielsweise als Schlüsselprotein, welches die Separation der Chromosomen reguliert und für die Stabilisierung der Chiasmata benötigt wird, steht im Verdacht mit dem Alter auftretende Aneuploidien zu verursachen [420, 421]. Neben einer fehlerhaften Verteilung der Chromosomen beeinflussen auch apoptotische Prozesse [422-424], die Veränderung der Telomere [425-427] sowie die Funktion der Mitochondrien [428-434] die Qualität der Eizelle in alternden Ovarien. In der vorliegenden Arbeit wurde das epigenetische Profil von 8 entwicklungsrelevanten Genen in bovinen Eizellen untersucht, welche in vivo aus Spendertieren von drei unterschiedlichen Alterskategorien (9-12 Monate, 3-7 Jahre und >8- 102 DISKUSSION 11 Jahre) gewonnen wurden. Zur Untersuchung altersbedingter Effekte wurde neben den Genen bDNMT3Lo, bH19 und bSNRPN, die u.a. als Kontrollgene dienten, Promotorbereiche von Genen analysiert, die Funktionen in der meiotischen Separation der Chromosomen (bREC8 [349, 350, 352, 419], Bub1 [344-347, 350, 352]), in der Apoptose (Bcl-XL [353-355]), in der Protektion der Telomere (bTERF2 [334-341]) und der Mitochondrienmembran (bPISD [356, 357]) übernehmen. Die Ergebnisse zeigten eine geringe Häufigkeit abnormal methylierter Allele (<1%) ohne signifikante Unterschiede zwischen den Eizellen unterschiedlicher Altersgruppen. Dies deutet daraufhin, dass eine abnormale Methylierung eines vollständigen Allels (>50% abnormal methylierte CpGs) in Rindereizellen der untersuchten Altersspanne für die 8 selektierten Gene ein seltenes Ereignis darstellt. In einer früheren Studie wurden die DNAMethylierung und mRNA-Expression in Eizellen untersucht, welche aus präpubertären Kälbern und erwachsenen Kühen isoliert wurden. Für die entwicklungsrelevanten, nicht geprägten Gene SLC2A1, PRDX1 und ZAR1 konnten keine signifikanten Unterschiede identifiziert werden, während die Methylierung der Satelliten-DNA „bovine testis satellite I“ (BTS) eine signifikante Abnahme mit steigendem Alter der Spendertiere zeigte [362]. Ähnliche Ergebnisse für die globale Methylierung konnten in unterschiedlich alten Eizellen von Mäusen nachgewiesen werden [207]. In einer Studie von Malhi et al. wurde die Entwicklungskompetenz boviner Eizellen während der reproduktiven Alterung untersucht [435]. Dazu wurden die gewonnenen Embryonen sowie die Schwangerschaftsraten von 13 bis 16 Jahre alten Kühen mit denen ihrer 3 bis 6 Jahre jungen Töchter verglichen. Alle Tiere beider Altersgruppen wurden zu diesem Zweck zunächst einer Superovulation unterzogen. Die generierten Embryonen wurden anschließend in 2 bis 5 Jahre alte Empfänger-Kühe transferiert. Die Auswertung der Ergebnisse zeigte eine geringere Ausbeute an Embryonen bzw. einen größeren Anteil an unbefruchteten Eizellen bei den Oozyten der älteren Kühe (13 bis 16 Jahre). Keinen Unterschied zwischen alten und jungen Rindern gab es jedoch in den Schwangerschaftsraten nach dem Morula- bzw. Blastozystenstadium. Erickson et al. konnte beobachten, dass etwa die Hälfte einer Herde von 14 bis 15 Jahre alten Kühen infertil war [436] (bei einer durchschnittlichen Lebenserwartung von 20 Jahren [437]). Damit stellen 13 bis 16 Jahre alte Kühe eine Altersgruppe dar, in der die reproduktive Alterung bereits vorangeschritten ist wodurch die reduzierte Entwicklungskompetenz der Eizellen verursacht wird. Die ältesten Spender-Kühe der in dieser Arbeit verwendeten Eizellen stehen mit 8-11 103 DISKUSSION Jahren möglicherweise noch nicht am Ende ihrer reproduktiven Lebenspanne. Die Untersuchung von Eizellen älterer Spendertiere (>13 Jahre) könnte aufgrund der fortgeschrittenen reproduktiven Alterung eine erhöhte abnormale DNA-Methylierung aufzeigen. Betrachtet man jedoch die vorliegenden Methylierungs-Ergebnisse aller Gene einer Altersgruppe, weisen die Eizellen der ältesten Spendertiere (8-11 Jahre) die höchste Anzahl abnormal methylierter Allele (n=5) auf, verglichen mit denen der beiden jüngeren Alterskategorien (jeweils n=2). 2 der 5 abnormal methylierten Allele der Eizellen von 8-11 Jahre alten Kühen traten für das Gen bREC8 auf. Inwiefern bREC8 durch einen epigenetischen Mechanismus reguliert wird ist bisher nicht bekannt. Die gezeigten Ergebnisse stellen die ersten DNA-Methylierungsdaten für bREC8 dar und weisen für diese Promotorregion in bovinen Eizellen einen vollständig methylierten Zustand nach. Neben bREC8 wiesen ausschließlich bDNMT3Lo und bH19 abnormal methylierte Allele auf. Für bDNMT3Lo konnte jeweils 1 abnormal methyliertes Allel in der jüngsten und ältesten Altersgruppe identifiziert werden (9-12 Monate: 1%; 8-11 Jahre: 2%) und 2 aberrant methylierte DNA-Moleküle in 3-7 Jahre alten Spendertieren (4%). bH19 zeigte 1 abnormales Allel in Eizellen von jungen Kühen (9-12 Monate: 1%) und 2 aberrante Moleküle in Oozyten der ältesten Rinder (8-11 Jahre: 3%). Zusätzlich zu abnormal methylierten Allelen traten auch Einzel-CpG-Fehler in den Genen bREC8, bDNMT3Lo und bH19 sowie bBCL-XL und bSNRPN. Auffällig dabei ist, dass die drei entwicklungsrelevanten Gene bDNMT3Lo, bH19 und bSNRPN von abnormalen Veränderungen der DNA-Methylierung betroffen sind. In den anderen 5 untersuchten Genen (bTERF2, bREC8, bBCL-XL, bPISD und bBUB1) traten hingegen in 3 dieser 5 Regionen weder ein aberrant methyliertes Allel noch ein Einzel-CpG-Fehler auf. Eine genauere Betrachtung der Funktionen der Kandidatengene lässt erkennen, dass sich die ausgewählten Gene nach ihrer biologischen Aufgabe grundsätzlich in zwei Gruppen einteilen lassen. bDNMT3Lo, bH19 und bSNRPN sind primär für die Entwicklung des Embryo entscheidend, wohingegen bTERF2 (telomeric repeat binding factor 2), bREC8 (meiosespezifische Untereinheit des Cohesin-Komplexes), Bcl-XL (Apoptose-Regulator), bPISD (Phospholipid Metabolismus) und bBUB1 (Teil des Spindelapparat-Kontrollpunkts) vorwiegend essentiell für die korrekte Entwicklung der Eizelle sind. Ausgehend von dieser Gruppierung zeigte statistische Testung eine Tendenz für ein erhöhtes Auftreten abnormal methylierter Allele (p=0,09) und eine signifikant höhere Häufigkeit von Einzel-CpG-Fehlern 104 DISKUSSION (p=0,0001) in bDNMT3Lo, bH19 und bSNRPN im Vergleich zu bTERF2, bREC8, bBCL-XL, bPISD und bBUB1. Neben dem signifikanten Unterschied in der Häufigkeit von Einzel-CpG-Fehlern stellt auch das Auftreten von ausgeschlossenen Allelen bedingt durch einen nichteindeutigen Metylierungszustand einzelner CpGs einen signifikanten Unterschied zwischen den beiden Gruppen von Genen dar (p=0,0001). Die Tatsache, dass die Untersuchungen von bDNMT3Lo, bH19 und bSNRPN über 10fach mehr Allele generierten, welche ausgeschlossen werden mussten (12,6%) als die Messungen der anderen 5 Gene (1,2%) könnte theoretisch auf einen technisch bedingtes Artefakt hindeuten. Ein Vergleich der Häufigkeiten ausgeschlossener Allele bzw. Allele mit nicht-eindeutigen Metylierungszuständen zwischen den verschiedenen untersuchten Eizell-Kategorien zeigt jedoch eine deutliche Variation innerhalb des gleichen Gens für bDNMT3Lo, bH19 und bSNRPN (Follikeln unterschiedlicher Größe: bDNMT3Lo: 11,4%; bH19: 61,8%; bSNRPN:38,7%; IVM: bDNMT3Lo: 12,3%; bH19 (ohne die letzten beiden CpGs): 4,0%; bSNRPN: 36,1%; Follikeln aus Kühen unterschiedlichen Alters: bDNMT3Lo: 5,5%; bH19: 14,0%; bSNRPN: 21,75%). Diese dynamischen Veränderungen der Methylierungszustände dieser drei Assays (experimentell definierter Untersuchungsmethoden) zwischen verschiedenen Entwicklungsstadien von Eizellen spiegeln eher eine biologische Ursache als ein technisches Artefakt wieder. Betrachtet man die erhöhte Methylierungsdynamik der Gene (bDNMT3Lo, bH19 und bSNRPN im Vergleich zu bTERF2, bREC8, bBCL-XL, bPISD und bBUB1), wären drei unterschiedliche Erklärungen dafür möglich: (i) bTERF2, bREC8, bBCL-XL, bPISD und bBUB1 werden nicht primär durch DNAMethylierung reguliert und damit nicht beeinflusst. (ii) Die Methylierungsprofile dieser zwei Gruppen von Genen werden zu unterschiedlichen Zeitpunkten während der Entwicklung etabliert. (iii) bDNMT3Lo, bH19 und bSNRPN, als essentielle Gene für die Entwicklung des Embryo, sind weniger widerstandsfähig gegen Methylierungs-Veränderungen als die anderen 5 Gene, welche entscheidende Funktionen für die Entwicklung der Eizelle ausüben. Die Ergebnisse der vorliegenden Arbeit beinhalten erstmals DNA-Methylierungsdaten für die Promotorregionen der Gene bTERF2, bREC8, bBCL-XL, bPISD und bBUB1 in bovinen Eizellen. Obwohl eine Genregulation durch DNA-Methylierung nur für bTERF2 vermutet wird [341], lässt sich nicht ausschließen, dass bREC8, bBCL-XL, bPISD und bBUB1 epigenetisch reguliert werden. Im Gegensatz zu diesen Regionen wird die epigenetische Regulation der zwei mitunter am umfassendsten untersuchten geprägten Gene bH19 und bSNRPN sowie dem Eizell-spezifischen Promotor von DNMT3L (DNMT3Lo) in mehreren Studien beschrieben [51, 105 DISKUSSION 323, 324, 332, 409]. Aus Studien in Mäusen ist ebenfalls bekannt, dass die Methylierung geprägter Gene in unreifen Eizellen in einer Gen-spezifischen zeitlichen Abfolge stattfindet [438-440]. O’Doherty und Kollegen untersuchten in einer früheren Studie die Etablierung von DNA-Methylierungsmustern von 5 maternal geprägten Genen (SNRPN, MEST, IGF2R, PEG10 und PLAGL1) in unreifen bovinen Eizellen unterschiedlicher Größe [398]. Ein wesentliches Ergebnis dieser Untersuchung war, dass diese Prägungen in einer von der Eizellgröße abhängigen Form etabliert werden, wobei der Grad der Methylierung mit zunehmender Oozytengröße ansteigt. Diese genspezifische zeitliche Abfolge zeigt sich durch unterschiedliche Methylierungsgrade der 5 geprägten Gene innerhalb des gleichen Entwicklungsstands repräsentiert durch die Oozytengröße. Diese Daten bestätigen die vorliegenden Ergebnisse, dass bSNRPN und bH19 als geprägte Gene und vermutlich auch das nicht-geprägte Gen bDNMT3Lo Regionen darstellen, welche dynamischen Methylierungsveränderungen während der Eizellentwicklung ausgesetzt sind. Diese Unterschiede in der DNA-Methylierung deuten nicht notwendigerweise auf eine abnormale Entwicklung hin sondern könnten Variationen der normalen Entwicklung repräsentieren. Inwiefern bTERF2, bREC8, bBCL-XL, bPISD und bBUB1 Regionen geringerer MethylierungsAktivität in der Eizelle darstellen, muss durch weitere Studien an unterschiedlichen Entwicklungsstadien untersucht werden. Diese 5 Gene zeigten jedoch eine signifikant geringere Anzahl an abnormalen Methylierungsveränderungen als bDNMT3Lo, bH19 und bSNRPN. Diese Unterschiede lassen die Annahme zu, dass DNA-Regionen, welche entscheidend für die Entwicklung nach der Fertilisation sind, wie geprägte Gene oder Gene wie DNMT3L stärker Methylierungsveränderungen unterworfen sind als Gene die primär essentiell für die Entwicklung der Eizelle sind. Damit könnten Gene, welche entscheidend für die Entwicklung des Embryos sind, anfälliger bzw. sensitiver für MethylierungsVeränderungen sein als Gene, die Aufgaben bei der Entwicklung der Eizelle selbst übernehmen. 4.3 Bewertung der Limiting Dilution-Bisulfitsequenzierung zur Untersuchung von bovinen Eizellen und Embryonen Die Limiting Dilution-Bisulfitsequenzierung (LD-BS) ist eine Methode zur Sequenzierung geringer DNA-Mengen auf Einzel-Allel-Ebene bzw. in der Auflösung einzelner DNA-Moleküle. Dabei verbindet diese Technik die Anwendung der digitalen PCR mit einer Folge aus 106 DISKUSSION Multiplex- und Singelplex-PCR um mehrere Regionen geringer Mengen von Bisulfit-konvertierter DNA zu untersuchen. Die digitale PCR wurde ursprünglich entwickelt, um seltene Mutationen innerhalb eines Gemisches von Wildtyp-Sequenzen zu identifizieren und allelische Imbalancen in Tumorgewebe zu bewerten [380, 441, 442]. Dazu werden einzelne DNA-Moleküle durch Verdünnung separiert und isoliert, bevor sie individuell durch PCR amplifiziert werden. Dadurch wird das exponentielle, analoge Signal der konventionellen PCR in ein lineares, digitales Signal umgewandelt. Dies ermöglicht statistische Analysen des PCR-Produkts und verbessert gleichzeitig das Signal-RauschVerhältnis der PCR. Da sich im Reaktionsansatz nur ein einzelnes DNA-Molekül als Matrize in der PCR befindet wird das Risiko einer unspezifischen Bindung der Primer an HintergrundDNA reduziert und die Generierung von Hintergrund-Amplifikaten verhindert bzw. minimiert [443]. Die unspezifische Bindung der Primer erhöht sich mit abnehmender Komplexität der DNA-Sequenz. Bisulfit-konvertierte DNA enthält außerhalb von Regionen mit methylierten Cytosinen nur drei Basen. Aufgrund dieser erhöhten Redundanz stellt die unspezifische Bindung der Primer eine entscheidende Schwierigkeit bei PCR-basierten Untersuchungen von Bisulfit-konvertierter DNA dar [443]. Die Analyse der DNA-Methylierung nach erfolgter Natriumbisulfitbehandlung stellt daher ein ideales Anwendungsgebiet der digitalen PCR dar. In Kombination mit einer Multiplex- und Singelplex-PCR ermöglicht diese Technik die präzise Identifizierung unterschiedlicher Epiallele mehrerer DNA-Regionen. Alternative Methode zur Sequenzierung einzelner Epiallele stellen die klassische Bisulfitsequenzierung nach vorangegangener Klonierung [444] sowie moderne NGS-basierte Verfahren [445] dar. Verglichen mit diesen beiden Techniken wird bei der LD-BS die aufwendige Klonierung vermieden und die klassische Sanger-Sequenzierung bzw. Pyrosequenzierung genutzt. Trotz Vermeidung der Klonierung stellt die LD-BS verglichen mit NGS-basierten Ansätzen eine aufwendige Methode dar. So wurden beispielsweise im Rahmen dieser Arbeit etwa 17.520 PCRs durchgeführt um 2343 Allele zu identifizieren (Tabelle 20). Dabei lag die Allel-Ausbeute (Anzahl identifizierter Allele pro Gen / Anzahl Allele) zwischen 0,05 (<2 mm) und 0,11 (9-12 Monate) bei Eizellen sowie bei 0,12 (24h) und 0,13 (48h) in Embryonen. Die vergleichsweise geringen Allel-Ausbeuten sind auf die starke DNA-Degradation während der Natriumbisulfitbehandlung in Kombination mit dem hohen Verdünnungsgrad der geringen DNA-Mengen zurückzuführen [377]. In den Sequenzen Bisulfit-konvertierter DNA treten CpGs, die nicht methyliert sind, als Thymine auf, wohingegen methylierte CpGs als Cytosine 107 DISKUSSION bestehen bleiben. Nach der Amplifikation eines einzelnen DNA-Moleküls sollte damit in der Sequenz nur ein einzelnes Signal für Thymin oder Cytsoin für jedes einzelne CpG zu detektieren sein. In den identifizierten Allelen wurden jedoch ebenfalls Überlagerungen beider Signale an CpG-Stellen gemessen. Da man beim Auftreten solcher Messergebnisse nicht mehr davon ausgehen kann, dass es sich dabei um das PCR-Produkt eines einzelnen DNA-Moleküls handelt, wurden die betroffenen Allele aus der statistischen Auswertung ausgeschlossen. Eine mögliche Ursache einer solchen Amplifizierung von mehreren Allelen könnte eine unzureichende Trennung von DNA-Molekülen sein. Durch ein Verklumpen der DNA (engl. „DNA-Clumping“) können DNA-Moleküle trotz intensiven Mischens nicht voneinander getrennt werden. Nach der PCR treten bei der Sequenzierung die überlagerten Signale der amplifizierten DNA-Moleküle auf. Der Anteil solcher Sequenzen liegt in Eizellen zwischen 5% (3-7 Jahre, 8-11 Jahre) und 39% (3-5 mm) sowie bei 32% (24h) und 44% (48h) in Embryonen (Tabelle 20). Insgesamt mussten etwa 19% der detektierten Allele aufgrund von nicht eindeutigen Messwerten ausgeschlossen werden. Dieser Wert erscheint zunächst hoch. NGS-basierte Methoden zur Sequenzierung von Bisulfit-behandelter DNA generieren ebenfalls Sequenzdaten einzelner DNA-Moleküle. Vor der Datenanalyse werden dabei die Rohdaten des Sequenzierungslaufs zunächst gefiltert. Bei dieser Filterung werden etwa 30% der Rohdaten von der weiteren Datenanalyse ausgeschlossen (von 14,15 Millionen generierten Sequenzen wurden 10 Millionen analysiert [445]). Wie hoch dabei der Anteil von Sequenzen mit heterozygoten Signalen für einzelne CpGs ist, ist nicht bekannt. Dennoch ist anzunehmen, dass der relative Wert dieser unerwarteten Messwerte bei NGS-basierten Methoden in einer ähnlichen Größenordnung wie bei der hier angewendeten LD-BS liegt. Obgleich man mittels NGS eine deutlich höhere Menge an Sequenzdaten in kürzerer Zeit generieren kann als mit LD-BS, bietet LD-BS die einzigartige Möglichkeit mit vergleichsweise simpler Methodik einzelne DNA-Moleküle zu amplifizieren und zu sequenzieren. In den im Rahmen dieser Arbeit durchgeführten Untersuchungen erwies sich die LD-BS als geeignete Methode zur Sequenzierung Bisulfit-konvertierter DNA aus bovinen Eizellen und Embryonen. Insbesondere die hohe Präzision und Reproduzierbarkeit kennzeichnen diese Technik als zuverlässige Methode zur Untersuchung der DNA-Methylierung geringer DNA-Mengen. 108 DISKUSSION Tabelle 20: Auflistung unterschiedlicher Daten der untersuchten Proben sowie der durchgeführten Analysen zur Berwertung der Limiting Dilution-Bisulfitsequenzierung. Eizellen Embryonen Σ <2 mm 3-5 mm >6 mm 9-12 Monate 3-7 Jahre 8-11 Jahre 24h 48h Σ 24h 48h Σ Anzahl der untersuchten Pools/Embryonen 19 11 8 18 17 15 17 13 118 14 14 28 146 Anzahl der enthaltenen Zellen 173 101 69 176 169 132 118 107 1045 64 55 119 1164 Anzahl der enthaltenen Allele 692 404 276 704 676 528 472 428 4180 256 220 476 4656 Anzahl der identifizierten Allele pro Gen 32.2 27.5 18.0 36.2 43.2 56.1 31.0 43.4 287.5 30.2 29.3 59.5 347.0 Allel-Ausbeute 0.05 0.07 0.07 0.05 0.06 0.11 0.07 0.10 0.07 0.12 0.13 0.13 0.10 Anzahl der durchgeführten PCRs 2280 1320 960 2160 2040 1800 2040 1560 14160 1680 1680 3360 17520 Anzahl der identifizierten Allele aller Gene 193 165 108 217 259 449 248 347 1986 181 176 357 2343 Ausgeschlossene Allele 42 (22%) 64 (39%) 41 (38%) 44 (20%) 50 (19%) 30 (7%) 12 (5%) 17 (5%) 300 (15%) 35 (32%) 53 (44%) 88 (38%) 435 (19%) 4.4 Schlussfolgerung und Ausblick Die Zielsetzung dieser Arbeit war, die bereits bestehenden Erkenntnisse zu DNAMethylierungsprofilen entwicklungsrelevanter Gene während der Entwicklung und Reifung von Eizellen sowie der Transition zum Embryo zu erweitern. Der Schwerpunkt lag dabei auf dem Einfluss der postovulatorischen Alterung und der ovariellen Alterung auf die DNAMethylierung der Eizelle. Die gewonnenen Fakten und Analysen dieser Arbeit sollen darüber hinaus auch als Grundlage zur Optimierung assistierter Reproduktionstechniken in der humanen Reproduktionsmedizin dienen. Aufgrund der Ähnlichkeit der bovinen Follikulogenese und Embryogenese zur humanen Follikel- und Embryonalentwicklung wurde als Modell für diese Studie das Holstein-Rind (Bos taurus) gewählt. 109 DISKUSSION Die Daten der vorliegenden Arbeit liefern wichtige Erkenntnisse zum Verständnis von DNAMethylierungsprozessen, die während der Entwicklung der unreifen Eizelle hin zur befruchtungsfähigen Oozyte ablaufen und die DNA-Methylierung des Embryos beeinflussen. Die DNA-Methylierungsprofile der untersuchten Gene in unreifen Eizellen aus Kühen unterschiedlichen Alters (9-12 Monate, 3-7 Jahre und 8-11 Jahre) wiesen keine signifikanten Unterschiede zwischen den Altersgruppen auf. Die Methylierungsmuster der untersuchten Promotorregionen der Gene bTERF2, bREC8, bBCL-XL, bPISD, bBUB1, bDNMT3Lo, bH19 und bSNRPN wurden damit nicht von der ovariellen Alterung der weiblichen Rinder zwischen 9 Monaten und 11 Jahren beeinträchtigt. In unreifen Eizellen aus Antralfollikeln unterschiedlicher Größe (<2 mm, 3-5 mm und >6 mm) zeigten lediglich Oozyten kleiner Follikel (<2 mm) ein erhöhtes Auftreten abnormal methylierter Allele in den geprägten Genen bH19 und bSNRPN. Insgesamt stellt sich jedoch in den Eizellen aus unterschiedlich großen und unterschiedlich alten Antralfollikeln eine stabile Etablierung der Methylierungsprofile der untersuchten Gene dar. Nach einer simulierten postovulatorischen Alterung durch eine in vitro Reifung für 48h konnte eine signifikante Hypermethylierung in der Promotorregion des Gens DNMT3Lo in 48h-gereiften Eizellen detektiert werden. Beim Übergang von 48h-gereiften Eizellen zum Embryo konnte eine signifikante Hypomethylierung von CpG7 des stammzellspezifischen Transkripts DNMT3Ls beobachtet werden. Diese CpG-Stelle wies ebenfalls einen signifikanten Anstieg von CpGs mit nicht-eindeutigem Methylierungszustand in unreifen Eizellen mit steigender Follikelgröße auf und befindet sich innerhalb eines Bindungs-Motivs des Transkriptionsfaktors CREB. Durch Studien im Maus-Model wurde bereits gezeigt, dass DNMT3L durch unterschiedliche Promotoren bzw. Transkripte Oozyten-spezifisch (DNMT3Lo) und Stammzell-spezifisch (DNMT3Ls) reguliert bzw. exprimiert wird [332]. Die vorliegende Arbeit liefert erstmals Daten, die einen Zusammenhang zwischen der DNA-Methylierung von DNMT3Lo und DNMT3Ls in Verbindung mit einer verlängerten Reifungsdauer von Eizellen und den daraus generierten Embryonen zeigt. Diese dynamisch methylierten DNA-Regionen erlauben dem entstehenden Organismus möglicherweise flexibel auf äußere Störfaktoren zu reagieren und eine stabile Etablierung der essentiellen DNA-Methylierungmuster zu gewährleisten. Das System der DNA-Methylierung stellt sich somit nicht als starrer, strickt regulierter und 110 DISKUSSION dadurch störungsanfälliger Mechanismus dar, sondern weist eine Fähigkeit zur flexiblen Anpassung auf, die maßgeblich zur Stabilität des Systems beitragen könnte. Eine entscheidende Rolle bei dieser Anpassungsfähigkeit in der Eizelle und dem Embryo scheinen die Gene und Promotoren des katalytisch inaktiven Cofaktors von DNMT3A, DNMT3L einzunehmen. Das System der DNA-Methylierung scheint sich dabei durch eine Feinjustierung dieser DNMT3L-Promotoren in einer Art Selbstregulation in Kombination mit Transkriptionsfaktoren aktiv an die veränderten Umstände anzupassen, um die Methylierungsmuster entwicklungsrelevanter Gene stabil zu etablieren und aufrechtzuerhalten. Inwiefern diese Vermutung bzw. Hypothese tatsächlich den realen Mechanismen in der Zelle bzw. dem Organismus entspricht, müssen weitere Untersuchungen aufklären. Zukünftige Studien sollten neben der DNA-Methylierung auch Transkriptionsfaktoren und weitere epigenetische Modifikationen wie Histon- Modifikationen und nicht-kodierende RNA in ihren Fokus aufnehmen. Eine der entscheidenden Herausforderungen bei der Arbeit mit Eizellen und frühen Embryonen stellt nach wie vor die Analyse des begrenzten Probenmaterials dar. Eine mögliche Lösung dieses Problems stellen die Einzel-Zell-Untersuchungsmethoden dar, die auf der Sequenzierung der nächsten Generation (engl. next generation sequencing, NGS) basieren. Mit dieser Technik ist es bereits möglich eine genomweite Bisulfitsequenzierung von Einzelzellen durchzuführen [446]. Zukünftig wird diese Methode es jedoch ermöglichen, mehrere integrierte, auf DNASequenzierung basierende Analysen von Einzelzellen zu realisieren [447]. In Anbetracht der gegenwärtigen rasanten Entwicklung dieser Einzel-Zell-Untersuchungsmethoden werden vermutlich bald die technischen Voraussetzungen geschaffen sein, um tiefere Einblicke in die Epigenetik des biologischen Phänomens der Reproduktion zu gewinnen. 111 REFERENZEN 5 Referenzen 5.1 Literaturverzeichnis 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. 24. 25. Mayr, E., Das ist Biologie: Die Wissenschaft des Lebens. 1998: Spektrum Akademischer Verlag. Evolutionsbiologie. 2013: Birkhäuser Basel. Lemmon, R.M., Chemical Evolution. Chemical Reviews, 1970. 70(1): p. 95-&. Janning, W. and E. Knust, Genetik: Allgemeine Genetik - Molekulare Genetik Entwicklungsgenetik. 2008: Thieme. Waddington, C.H., The epigenotype. 1942. Int J Epidemiol, 2012. 41(1): p. 10-3. Waterland, R.A. and R.L. Jirtle, Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol, 2003. 23(15): p. 5293-300. Tollefsbol, T., Handbook of Epigenetics: The New Molecular and Medical Genetics. 2010: Elsevier Science. Fu, Y., et al., N6-methyldeoxyadenosine marks active transcription start sites in Chlamydomonas. Cell, 2015. 161(4): p. 879-92. Zhang, G., et al., N6-methyladenine DNA modification in Drosophila. Cell, 2015. 161(4): p. 893-906. Greer, E.L., et al., DNA Methylation on N6-Adenine in C. elegans. Cell, 2015. 161(4): p. 868-78. Zemach, A., et al., Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science, 2010. 328(5980): p. 916-9. Jones, P.A. and D. Takai, The role of DNA methylation in mammalian epigenetics. Science, 2001. 293(5532): p. 1068-70. Mruk, I. and I. Kobayashi, To be or not to be: regulation of restriction-modification systems and other toxin-antitoxin systems. Nucleic Acids Res, 2014. 42(1): p. 70-86. Makarova, K.S., Y.I. Wolf, and E.V. Koonin, Comparative genomics of defense systems in archaea and bacteria. Nucleic Acids Res, 2013. 41(8): p. 4360-77. Kovall, R.A. and B.W. Matthews, Type II restriction endonucleases: structural, functional and evolutionary relationships. Curr Opin Chem Biol, 1999. 3(5): p. 578-83. Williams, R.J., Restriction endonucleases: classification, properties, and applications. Mol Biotechnol, 2003. 23(3): p. 225-43. Orlowski, J. and J.M. Bujnicki, Structural and evolutionary classification of Type II restriction enzymes based on theoretical and experimental analyses. Nucleic Acids Res, 2008. 36(11): p. 3552-69. Murphy, J., et al., Bacteriophage orphan DNA methyltransferases: insights from their bacterial origin, function, and occurrence. Appl Environ Microbiol, 2013. 79(24): p. 7547-55. Smillie, C., et al., Mobility of plasmids. Microbiol Mol Biol Rev, 2010. 74(3): p. 434-52. Asakura, Y., H. Kojima, and I. Kobayashi, Evolutionary genome engineering using a restrictionmodification system. Nucleic Acids Res, 2011. 39(20): p. 9034-46. Werren, J.H., Selfish genetic elements, genetic conflict, and evolutionary innovation. Proc Natl Acad Sci U S A, 2011. 108 Suppl 2: p. 10863-70. Newton, I.L.G. and S.R. Bordenstein, Correlations Between Bacterial Ecology and Mobile DNA. Current Microbiology, 2011. 62(1): p. 198-208. Blumenstiel, J.P., Evolutionary dynamics of transposable elements in a small RNA world. Trends Genet, 2011. 27(1): p. 23-31. de Koning, A.P.J., et al., Repetitive Elements May Comprise Over Two-Thirds of the Human Genome. Plos Genetics, 2011. 7(12). Padeken, J., P. Zeller, and S.M. Gasser, Repeat DNA in genome organization and stability. Current Opinion in Genetics & Development, 2015. 31: p. 12-19. 112 REFERENZEN 26. 27. 28. 29. 30. 31. 32. 33. 34. 35. 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. 46. 47. 48. 49. 50. Rebollo, R., M.T. Romanish, and D.L. Mager, Transposable Elements: An Abundant and Natural Source of Regulatory Sequences for Host Genes. Annual Review of Genetics, Vol 46, 2012. 46: p. 21-42. Faulkner, G.J., et al., The regulated retrotransposon transcriptome of mammalian cells. Nature Genetics, 2009. 41(5): p. 563-571. Sundaram, V., et al., Widespread contribution of transposable elements to the innovation of gene regulatory networks. Genome Research, 2014. 24(12): p. 1963-1976. Lindblad-Toh, K., et al., A high-resolution map of human evolutionary constraint using 29 mammals. Nature, 2011. 478(7370): p. 476-482. Prak, E.T.L. and H.H. Kazazian, Mobile elements and the human genome. Nature Reviews Genetics, 2000. 1(2): p. 134-144. Beck, C.R., et al., LINE-1 Elements in Structural Variation and Disease. Annual Review of Genomics and Human Genetics, Vol 12, 2011. 12: p. 187-215. Crichton, J., et al., Defending the genome from the enemy within: mechanisms of retrotransposon suppression in the mouse germline. Cellular and Molecular Life Sciences, 2014. 71(9): p. 1581-1605. Li, Z.G., et al., Distinct roles of DNMT1-dependent and DNMT1-independent methylation patterns in the genome of mouse embryonic stem cells. Genome Biology, 2015. 16. Yoder, J.A., C.P. Walsh, and T.H. Bestor, Cytosine methylation and the ecology of intragenomic parasites. Trends in Genetics, 1997. 13(8): p. 335-340. Bestor, T.H., The DNA methyltransferases of mammals. Human Molecular Genetics, 2000. 9(16): p. 2395-2402. Goll, M.G., et al., Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science, 2006. 311(5759): p. 395-8. Chen, T. and E. Li, Establishment and maintenance of DNA methylation patterns in mammals. Curr Top Microbiol Immunol, 2006. 301: p. 179-201. Mortusewicz, O., et al., Recruitment of DNA methyltransferase I to DNA repair sites. Proc Natl Acad Sci U S A, 2005. 102(25): p. 8905-9. Qiu, C., et al., The PWWP domain of mammalian DNA methyltransferase Dnmt3b defines a new family of DNA-binding folds. Nat Struct Biol, 2002. 9(3): p. 217-24. Lukasik, S.M., et al., High resolution structure of the HDGF PWWP domain: a potential DNA binding domain. Protein Sci, 2006. 15(2): p. 314-23. Ooi, S.K., et al., DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature, 2007. 448(7154): p. 714-7. Junker, T., Die Evolution des Menschen. 2016: C.H.Beck. Surani, M.A.H. and S.C. Barton, Development of Gynogenetic Eggs in the Mouse - Implications for Parthenogenetic Embryos. Science, 1983. 222(4627): p. 1034-1036. Surani, M.A., S.C. Barton, and M.L. Norris, Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature, 1984. 308(5959): p. 54850. McGrath, J. and D. Solter, Nuclear transplantation in mouse embryos. J Exp Zool, 1983. 228(2): p. 355-62. McGrath, J. and D. Solter, Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell, 1984. 37(1): p. 179-83. Bartolomei, M.S. and A.C. Ferguson-Smith, Mammalian Genomic Imprinting. Cold Spring Harbor Perspectives in Biology, 2011. 3(7). DeChiara, T.M., E.J. Robertson, and A. Efstratiadis, Parental imprinting of the mouse insulinlike growth factor II gene. Cell, 1991. 64(4): p. 849-59. Bartolomei, M.S., S. Zemel, and S.M. Tilghman, Parental Imprinting of the Mouse H19 Gene. Nature, 1991. 351(6322): p. 153-155. Barlow, D.P., et al., The Mouse Insulin-Like Growth-Factor Type-2 Receptor Is Imprinted and Closely Linked to the Tme Locus. Nature, 1991. 349(6304): p. 84-87. 113 REFERENZEN 51. 52. 53. 54. 55. 56. 57. 58. 59. 60. 61. 62. 63. 64. 65. 66. 67. 68. 69. 70. 71. 72. 73. 74. Reik, W. and J. Walter, Genomic imprinting: parental influence on the genome. Nat Rev Genet, 2001. 2(1): p. 21-32. Reik, W., et al., Regulation of supply and demand for maternal nutrients in mammals by imprinted genes. J Physiol, 2003. 547(Pt 1): p. 35-44. Al Adhami, H., et al., A systems-level approach to parental genomic imprinting: the imprinted gene network includes extracellular matrix genes and regulates cell cycle exit and differentiation. Genome Res, 2015. 25(3): p. 353-67. Wilkinson, L.S., W. Davies, and A.R. Isles, Genomic imprinting effects on brain development and function. Nat Rev Neurosci, 2007. 8(11): p. 832-43. Coan, P.M., G.J. Burton, and A.C. Ferguson-Smith, Imprinted genes in the placenta--a review. Placenta, 2005. 26 Suppl A: p. S10-20. Sibley, C.P., et al., Placental-specific insulin-like growth factor 2 (Igf2) regulates the diffusional exchange characteristics of the mouse placenta. Proc Natl Acad Sci U S A, 2004. 101(21): p. 8204-8. Miozzo, M. and G. Simoni, The role of imprinted genes in fetal growth. Biol Neonate, 2002. 81(4): p. 217-28. Reik, W., et al., Igf2 imprinting in development and disease. Int J Dev Biol, 2000. 44(1): p. 14550. Ideraabdullah, F.Y., S. Vigneau, and M.S. Bartolomei, Genomic imprinting mechanisms in mammals. Mutat Res, 2008. 647(1-2): p. 77-85. Antequera, F., Structure, function and evolution of CpG island promoters. Cell Mol Life Sci, 2003. 60(8): p. 1647-58. Bird, A., DNA methylation patterns and epigenetic memory. Genes Dev, 2002. 16(1): p. 6-21. Yang, T., et al., A mouse model for Prader-Willi syndrome imprinting-centre mutations. Nat Genet, 1998. 19(1): p. 25-31. Wutz, A., et al., Imprinted expression of the Igf2r gene depends on an intronic CpG island. Nature, 1997. 389(6652): p. 745-9. Thorvaldsen, J.L., K.L. Duran, and M.S. Bartolomei, Deletion of the H19 differentially methylated domain results in loss of imprinted expression of H19 and Igf2. Genes & Development, 1998. 12(23): p. 3693-3702. Williamson, C.M., et al., Identification of an imprinting control region affecting the expression of all transcripts in the Gnas cluster. Nature Genetics, 2006. 38(3): p. 350-355. Lin, S.P., et al., Asymmetric regulation of imprinting on the maternal and paternal chromosomes at the Dlk1-Gtl2 imprinted cluster on mouse chromosome 12. Nature Genetics, 2003. 35(1): p. 97-102. Fitzpatrick, G.V., P.D. Soloway, and M.J. Higgins, Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat Genet, 2002. 32(3): p. 426-31. Tremblay, K.D., K.L. Duran, and M.S. Bartolomei, A 5' 2-kilobase-pair region of the imprinted mouse H19 gene exhibits exclusive paternal methylation throughout development. Mol Cell Biol, 1997. 17(8): p. 4322-9. Bell, A.C. and G. Felsenfeld, Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature, 2000. 405(6785): p. 482-5. Hark, A.T., et al., CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature, 2000. 405(6785): p. 486-489. Koerner, M.V., et al., The function of non-coding RNAs in genomic imprinting. Development, 2009. 136(11): p. 1771-1783. Stoger, R., et al., Maternal-Specific Methylation of the Imprinted Mouse Igf2r Locus Identifies the Expressed Locus as Carrying the Imprinting Signal. Cell, 1993. 73(1): p. 61-71. Lyle, R., et al., The imprinted antisense RNA at the Igf2r locus overlaps but does not imprint Mas1. Nature Genetics, 2000. 25(1): p. 19-21. Sleutels, F., R. Zwart, and D.P. Barlow, The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature, 2002. 415(6873): p. 810-813. 114 REFERENZEN 75. 76. 77. 78. 79. 80. 81. 82. 83. 84. 85. 86. 87. 88. 89. 90. 91. 92. 93. 94. 95. 96. 97. 98. 99. 100. Nagano, T., et al., The Air Noncoding RNA Epigenetically Silences Transcription by Targeting G9a to Chromatin. Science, 2008. 322(5908): p. 1717-1720. Horsthemke, B. and J. Wagstaff, Mechanisms of imprinting of the Prader-Willi/Angelman region. American Journal of Medical Genetics Part A, 2008. 146a(16): p. 2041-2052. Chotalia, M., et al., Transcription is required for establishment of germline methylation marks at imprinted genes (vol 23, pg 105, 2008). Genes & Development, 2009. 23(19): p. 23582358. Lidegaard, O., A. Pinborg, and A.N. Andersen, Imprinting disorders after assisted reproductive technologies. Curr Opin Obstet Gynecol, 2006. 18(3): p. 293-6. Moog, U. and O. Rieß, Medizinische Genetik für die Praxis: Diagnostik, Beratung, Fallbeispiele. 2014: Thieme. Buiting, K., Prader-Willi syndrome and Angelman syndrome. Am J Med Genet C Semin Med Genet, 2010. 154C(3): p. 365-76. Robinson, W.P., et al., The mechanisms involved in formation of deletions and duplications of 15q11-q13. J Med Genet, 1998. 35(2): p. 130-6. Vu, T.H. and A.R. Hoffman, Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat Genet, 1997. 17(1): p. 12-3. Matsuura, T., et al., De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet, 1997. 15(1): p. 74-7. Buiting, K., C. Williams, and B. Horsthemke, Angelman syndrome - insights into a rare neurogenetic disorder. Nat Rev Neurol, 2016. 12(10): p. 584-93. Eggermann, T., Silver-Russell and Beckwith-Wiedemann syndromes: opposite (epi)mutations in 11p15 result in opposite clinical pictures. Horm Res, 2009. 71 Suppl 2: p. 30-5. Eggermann, T., Russell-Silver syndrome. Am J Med Genet C Semin Med Genet, 2010. 154C(3): p. 355-64. Kotzot, D., et al., Uniparental disomy 7 in Silver-Russell syndrome and primordial growth retardation. Hum Mol Genet, 1995. 4(4): p. 583-7. Reik, W., W. Dean, and J. Walter, Epigenetic reprogramming in mammalian development. Science, 2001. 293(5532): p. 1089-93. Smallwood, S.A. and G. Kelsey, De novo DNA methylation: a germ cell perspective. Trends Genet, 2012. 28(1): p. 33-42. Ungerer, M., J. Knezovich, and M. Ramsay, In Utero Alcohol Exposure, Epigenetic Changes, and Their Consequences. Alcohol Research-Current Reviews, 2013. 35(1): p. 37-46. Gu, T.P., et al., The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature, 2011. 477(7366): p. 606-10. Sasaki, H. and Y. Matsui, Epigenetic events in mammalian germ-cell development: reprogramming and beyond. Nature Reviews Genetics, 2008. 9(2): p. 129-140. Meissner, A. and J. Walter, Epigenetic mechanisms in cellular reprogramming. Epigenetics and human health. 2014, Berlin ; London: Springer. Moore, K.L., T.V.N. Persaud, and C. Viebahn, Embryologie: Entwicklungsstadien, Frühentwicklung, Organogenese, Klinik. 2007: Elsevier, Urban & Fischer. Baker, T.G., A Quantitative and Cytological Study of Germ Cells in Human Ovaries. Proc R Soc Lond B Biol Sci, 1963. 158: p. 417-33. Mamsen, L.S., et al., Germ cell numbers in human embryonic and fetal gonads during the first two trimesters of pregnancy: analysis of six published studies. Hum Reprod, 2011. 26(8): p. 2140-5. Wallace, W.H. and T.W. Kelsey, Human ovarian reserve from conception to the menopause. PLoS One, 2010. 5(1): p. e8772. Erickson, B.H., Development and Radio-Response of Prenatal Bovine Ovary. Journal of Reproduction and Fertility, 1966. 11(1): p. 97-&. Gordon, I., Laboratory Production of Cattle Embryos. 2003: CABI. Eppig, J. and C. Hegele-Hartung, The Future of the Oocyte: Basic and Clinical Aspects. 2013: Springer Berlin Heidelberg. 115 REFERENZEN 101. 102. 103. 104. 105. 106. 107. 108. 109. 110. 111. 112. 113. 114. 115. 116. 117. 118. 119. 120. 121. 122. 123. 124. Kanitz, W., et al., Comparative aspects of follicular development, follicular and oocyte maturation and ovulation in cattle and pigs. Archiv Fur Tierzucht-Archives of Animal Breeding, 2001. 44: p. 9-23. McGee, E.A. and A.J. Hsueh, Initial and cyclic recruitment of ovarian follicles. Endocr Rev, 2000. 21(2): p. 200-14. Yang, M.Y. and J.E. Fortune, The capacity of primordial follicles in fetal bovine ovaries to initiate growth in vitro develops during mid-gestation and is associated with meiotic arrest of oocytes. Biol Reprod, 2008. 78(6): p. 1153-61. Kanitz, W., Follicular dynamic and ovulation in cattle - a review. Archiv Fur Tierzucht-Archives of Animal Breeding, 2003. 46(2): p. 187-198. Hsueh, A.J., et al., Gonadal cell apoptosis. Recent Prog Horm Res, 1996. 51: p. 433-55; discussion 455-6. Ginther, O.J., et al., Follicle selection in monovular species. Biol Reprod, 2001. 65(3): p. 63847. Ginther, O.J., et al., Follicle Selection in Cattle: Relationships among Growth Rate, Diameter Ranking, and Capacity for Dominance. Biol Reprod, 2001. 65(2): p. 345-50. Austin, E.J., et al., Alterations in intrafollicular regulatory factors and apoptosis during selection of follicles in the first follicular wave of the bovine estrous cycle. Biol Reprod, 2001. 64(3): p. 839-48. Evans, A.C., et al., Changes in androgen secretion and luteinizing hormone pulse amplitude are associated with the recruitment and growth of ovarian follicles during the luteal phase of the bovine estrous cycle. Biol Reprod, 1997. 57(2): p. 394-401. Ginther, O.J., et al., Selection of the dominant follicle in cattle: role of estradiol. Biol Reprod, 2000. 63(2): p. 383-9. Ginther, O.J., et al., Follicle selection in cattle: role of luteinizing hormone. Biol Reprod, 2001. 64(1): p. 197-205. Ginther, O.J., Selection of the dominant follicle in cattle and horses. Anim Reprod Sci, 2000. 60-61: p. 61-79. Ginther, O.J., et al., Selection of the dominant follicle in cattle: establishment of follicle deviation in less than 8 hours through depression of FSH concentrations. Theriogenology, 1999. 52(6): p. 1079-93. Adams, G.P., et al., Selection of a Dominant Follicle and Suppression of Follicular-Growth in Heifers. Animal Reproduction Science, 1993. 30(4): p. 259-271. Ginther, O.J., J.P. Kastelic, and L. Knopf, Composition and Characteristics of Follicular Waves during the Bovine Estrous-Cycle. Animal Reproduction Science, 1989. 20(3): p. 187-200. Ginther, O.J., J.P. Kastelic, and L. Knopf, Intraovarian Relationships among Dominant and Subordinate Follicles and the Corpus-Luteum in Heifers. Theriogenology, 1989. 32(5): p. 787795. Spicer, L.J. and S.E. Echternkamp, Ovarian Follicular-Growth, Function and Turnover in Cattle a Review. Journal of Animal Science, 1986. 62(2): p. 428-451. Adams, G.P. and R.A. Pierson, Bovine Model for Study of Ovarian Follicular Dynamics in Humans. Theriogenology, 1995. 43(1): p. 113-120. Sirois, J. and J.E. Fortune, Ovarian follicular dynamics during the estrous cycle in heifers monitored by real-time ultrasonography. Biol Reprod, 1988. 39(2): p. 308-17. Knopf, L., et al., Ovarian follicular dynamics in heifers: test of two-wave hypothesis by ultrasonically monitoring individual follicles. Domest Anim Endocrinol, 1989. 6(2): p. 111-9. Pierson, R.A. and O.J. Ginther, Follicular Populations during the Estrous-Cycle in Heifers .1. Influence of Day. Animal Reproduction Science, 1987. 14(3): p. 165-176. Browder, L.W., Oogenesis. 2012: Springer US. Hyttel, P., H. Callesen, and T. Greve, Ultrastructural features of preovulatory oocyte maturation in superovulated cattle. J Reprod Fertil, 1986. 76(2): p. 645-56. Hyttel, P., T. Greve, and H. Callesen, Ultrastructural aspects of oocyte maturation and fertilization in cattle. J Reprod Fertil Suppl, 1989. 38: p. 35-47. 116 REFERENZEN 125. 126. 127. 128. 129. 130. 131. 132. 133. 134. 135. 136. 137. 138. 139. 140. 141. 142. 143. 144. 145. 146. 147. 148. 149. Pond, W.G., Encyclopedia of Animal Science (Print). 2004: Taylor & Francis. Dairyman, H., Dairy Cattle Fertility & Sterility. 1996: W.D. Hoard & Sons. Hopper, R.M., Bovine Reproduction. 2014: Wiley. Russe, I., Oogenesis in cattle and sheep. Bibl Anat, 1983. 24: p. 77-92. Baerwald, A.R., G.P. Adams, and R.A. Pierson, Ovarian antral folliculogenesis during the human menstrual cycle: a review. Hum Reprod Update, 2012. 18(1): p. 73-91. Jokubkiene, L., et al., Assessment of changes in volume and vascularity of the ovaries during the normal menstrual cycle using three-dimensional power Doppler ultrasound. Human Reproduction, 2006. 21(10): p. 2661-2668. Block, E., Quantitative morphological investigations of the follicular system in women; variations in the different phases of the sexual cycle. Acta Endocrinol (Copenh), 1951. 8(1): p. 33-54. Hackeloer, B.J., et al., Correlation of ultrasonic and endocrinologic assessment of human follicular development. Am J Obstet Gynecol, 1979. 135(1): p. 122-8. Queenan, J.T., et al., Ultrasound Scanning of Ovaries to Detect Ovulation in Women. Fertility and Sterility, 1980. 34(2): p. 99-105. Baerwald, A.R., G.P. Adams, and R.A. Pierson, Characterization of ovarian follicular wave dynamics in women. Biology of Reproduction, 2003. 69(3): p. 1023-1031. Baerwald, A.R., G.P. Adams, and R.A. Pierson, A new model for ovarian follicular development during the human menstrual cycle. Fertil Steril, 2003. 80(1): p. 116-22. Hale, G.E., et al., Endocrine features of menstrual cycles in middle and late reproductive age and the menopausal transition classified according to the Staging of Reproductive Aging Workshop (STRAW) staging system. J Clin Endocrinol Metab, 2007. 92(8): p. 3060-7. Bentov, Y., et al., An ongoing pregnancy from two waves of follicles developing during a long follicular phase of the same cycle. Fertil Steril, 2010. 94(1): p. 350 e8-11. Mihm, M. and A.C. Evans, Mechanisms for dominant follicle selection in monovulatory species: a comparison of morphological, endocrine and intraovarian events in cows, mares and women. Reprod Domest Anim, 2008. 43 Suppl 2: p. 48-56. van Santbrink, E.J., et al., Decremental follicle-stimulating hormone and dominant follicle development during the normal menstrual cycle. Fertil Steril, 1995. 64(1): p. 37-43. Schipper, I., W.C. Hop, and B.C. Fauser, The follicle-stimulating hormone (FSH) threshold/window concept examined by different interventions with exogenous FSH during the follicular phase of the normal menstrual cycle: duration, rather than magnitude, of FSH increase affects follicle development. J Clin Endocrinol Metab, 1998. 83(4): p. 1292-8. Hohmann, F.P., et al., Low-dose exogenous FSH initiated during the early, mid or late follicular phase can induce multiple dominant follicle development. Hum Reprod, 2001. 16(5): p. 846-54. Schneyer, A.L., et al., Dynamic changes in the intrafollicular inhibin/activin/follistatin axis during human follicular development: relationship to circulating hormone concentrations. J Clin Endocrinol Metab, 2000. 85(9): p. 3319-30. Laven, J.S. and B.C. Fauser, Inhibins and adult ovarian function. Mol Cell Endocrinol, 2004. 225(1-2): p. 37-44. Zeleznik, A.J., Follicle selection in primates: "many are called but few are chosen". Biol Reprod, 2001. 65(3): p. 655-9. Filicori, M., et al., Stimulation and growth of antral ovarian follicles by selective LH activity administration in women. J Clin Endocrinol Metab, 2002. 87(3): p. 1156-61. Guraya, S.S., Ovarian Follicles in Reptiles and Birds. 2013: Springer Berlin Heidelberg. Nieuwkoop, P.D., A.G. Johnen, and B. Albers, The Epigenetic Nature of Early Chordate Development: Inductive Interaction and Competence. 1985: Cambridge University Press. Gosden, R. and B. Lee, Portrait of an oocyte: our obscure origin. Journal of Clinical Investigation, 2010. 120(4): p. 973-983. Sirard, M.A., et al., Timing of Nuclear Progression and Protein-Synthesis Necessary for Meiotic Maturation of Bovine Oocytes. Biology of Reproduction, 1989. 40(6): p. 1257-1263. 117 REFERENZEN 150. 151. 152. 153. 154. 155. 156. 157. 158. 159. 160. 161. 162. 163. 164. 165. 166. 167. 168. 169. Hunter, A.G. and R.M. Moor, Stage-Dependent Effects of Inhibiting Ribonucleic-Acids and Protein-Synthesis on Meiotic Maturation of Bovine Oocytes Invitro. Journal of Dairy Science, 1987. 70(8): p. 1646-1651. Tatemoto, H. and T. Horiuchi, Requirement for Protein-Synthesis during the Onset of Meiosis in Bovine Oocytes and Its Involvement in the Autocatalytic Amplification of MaturationPromoting Factor. Molecular Reproduction and Development, 1995. 41(1): p. 47-53. Tatemoto, H. and T. Terada, Time-Dependent Effects of Cycloheximide and Alpha-Amanitin on Meiotic Resumption and Progression in Bovine Follicular Oocytes. Theriogenology, 1995. 43(6): p. 1107-1113. Tatemoto, H. and T. Terada, Involvement of cyclic AMP-dependent protein kinase in chromatin condensation before germinal vesicle breakdown in bovine oocytes. Animal Reproduction Science, 1996. 44(2): p. 99-109. Tatemoto, H. and T. Terada, Activation of p34(cdc2) kinase around the meiotic resumption in bovine oocytes cultured in vitro. Theriogenology, 1996. 45(2): p. 427-437. Kastrop, P.M.M., et al., Changes in Protein-Synthesis and Phosphorylation Patterns during Bovine Oocyte Maturation Invitro. Journal of Reproduction and Fertility, 1990. 90(1): p. 305310. Kastrop, P.M.M., et al., The Effects of Alpha-Amanitin and Cycloheximide on Nuclear Progression, Protein-Synthesis, and Phosphorylation during Bovine Oocyte Maturation Invitro. Molecular Reproduction and Development, 1991. 28(3): p. 249-254. Wickramasinghe, D. and D.F. Albertini, Cell cycle control during mammalian oogenesis. Curr Top Dev Biol, 1993. 28: p. 125-53. Fair, T., P. Hyttel, and T. Greve, Bovine oocyte diameter in relation to maturational competence and transcriptional activity. Mol Reprod Dev, 1995. 42(4): p. 437-42. Lonergan, P., et al., Effect of follicle size on bovine oocyte quality and developmental competence following maturation, fertilization, and culture in vitro. Mol Reprod Dev, 1994. 37(1): p. 48-53. Motlik, J. and J. Fulka, Factors Affecting Meiotic Competence in Pig Oocytes. Theriogenology, 1986. 25(1): p. 87-96. Otoi, T., et al., Bovine oocyte diameter in relation to developmental competence. Theriogenology, 1997. 48(5): p. 769-774. Ferreira, E.M., et al., Cytoplasmic maturation of bovine oocytes: structural and biochemical modifications and acquisition of developmental competence. Theriogenology, 2009. 71(5): p. 836-48. Stojkovic, M., et al., Mitochondrial distribution and adenosine triphosphate content of bovine oocytes before and after in vitro maturation: correlation with morphological criteria and developmental capacity after in vitro fertilization and culture. Biol Reprod, 2001. 64(3): p. 904-9. Albertini, D.F., Cytoplasmic microtubular dynamics and chromatin organization during mammalian oogenesis and oocyte maturation. Mutat Res, 1992. 296(1-2): p. 57-68. Fan, H.Y. and Q.Y. Sun, Involvement of mitogen-activated protein kinase cascade during oocyte maturation and fertilization in mammals. Biology of Reproduction, 2004. 70(3): p. 535-547. Krisher, R.L. and B.D. Bavister, Responses of oocytes and embryos to the culture environment. Theriogenology, 1998. 49(1): p. 103-14. Trimarchi, J.R., et al., Oxidative phosphorylation-dependent and -independent oxygen consumption by individual preimplantation mouse embryos. Biol Reprod, 2000. 62(6): p. 1866-74. Cummins, J.M., The role of mitochondria in the establishment of oocyte functional competence. Eur J Obstet Gynecol Reprod Biol, 2004. 115 Suppl 1: p. S23-9. Tarazona, A.M., et al., Mitochondrial activity, distribution and segregation in bovine oocytes and in embryos produced in vitro. Reprod Domest Anim, 2006. 41(1): p. 5-11. 118 REFERENZEN 170. 171. 172. 173. 174. 175. 176. 177. 178. 179. 180. 181. 182. 183. 184. 185. 186. 187. 188. 189. 190. 191. 192. Shoubridge, E.A. and T. Wai, Mitochondrial DNA and the mammalian oocyte. Curr Top Dev Biol, 2007. 77: p. 87-111. Hyttel, P., et al., Ultrastructure of in-vitro oocyte maturation in cattle. J Reprod Fertil, 1986. 78(2): p. 615-25. Payne, C. and G. Schatten, Golgi dynamics during meiosis are distinct from mitosis and are coupled to endoplasmic reticulum dynamics until fertilization. Dev Biol, 2003. 264(1): p. 5063. Machaca, K., Ca2+ signaling differentiation during oocyte maturation. J Cell Physiol, 2007. 213(2): p. 331-40. Hosoe, M. and Y. Shioya, Distribution of cortical granules in bovine oocytes classified by cumulus complex. Zygote, 1997. 5(4): p. 371-6. Wessel, G.M., et al., The biology of cortical granules. Int Rev Cytol, 2001. 209: p. 117-206. Thibault, C., D. Szollosi, and M. Gerard, Mammalian oocyte maturation. Reprod Nutr Dev, 1987. 27(5): p. 865-96. Dobson, A.T., et al., The unique transcriptome through day 3 of human preimplantation development. Hum Mol Genet, 2004. 13(14): p. 1461-70. Meirelles, F.V., et al., Genome activation and developmental block in bovine embryos. Anim Reprod Sci, 2004. 82-83: p. 13-20. Adjaye, J., et al., Conserved molecular portraits of bovine and human blastocysts as a consequence of the transition from maternal to embryonic control of gene expression. Physiol Genomics, 2007. 31(2): p. 315-27. Tomek, W., H. Torner, and W. Kanitz, Comparative analysis of protein synthesis, transcription and cytoplasmic polyadenylation of mRNA during maturation of bovine oocytes in vitro. Reprod Domest Anim, 2002. 37(2): p. 86-91. Sirard, M.A., Resumption of meiosis: mechanism involved in meiotic progression and its relation with developmental competence. Theriogenology, 2001. 55(6): p. 1241-54. Fulka, J., N.L. First, and R.M. Moor, Nuclear and cytoplasmic determinants involved in the regulation of mammalian oocyte maturation. Molecular Human Reproduction, 1998. 4(1): p. 41-49. Jones, K.T., Turning it on and off: M-phase promoting factor during meiotic maturation and fertilization. Mol Hum Reprod, 2004. 10(1): p. 1-5. Fissore, R.A., C.L. He, and G.F. Vande Woude, Potential role of mitogen-activated protein kinase during meiosis resumption in bovine oocytes. Biol Reprod, 1996. 55(6): p. 1261-70. Wu, B., et al., Expression of Mos proto-oncoprotein in bovine oocytes during maturation in vitro. Biol Reprod, 1997. 56(1): p. 260-5. Levesque, J.T. and M.A. Sirard, Resumption of meiosis is initiated by the accumulation of cyclin B in bovine oocytes. Biol Reprod, 1996. 55(6): p. 1427-36. Wehrend, A. and B. Meinecke, Kinetics of meiotic progression, M-phase promoting factor (MPF) and mitogen-activated protein kinase (MAP kinase) activities during in vitro maturation of porcine and bovine oocytes: species specific differences in the length of the meiotic stages. Anim Reprod Sci, 2001. 66(3-4): p. 175-84. Gleicher, N., A. Weghofer, and D.H. Barad, Defining ovarian reserve to better understand ovarian aging. Reprod Biol Endocrinol, 2011. 9: p. 23. Ireland, J.J., et al., Variation in the ovarian reserve is linked to alterations in intrafollicular estradiol production and ovarian biomarkers of follicular differentiation and oocyte quality in cattle. Biol Reprod, 2009. 80(5): p. 954-64. Broekmans, F.J., M.R. Soules, and B.C. Fauser, Ovarian aging: mechanisms and clinical consequences. Endocr Rev, 2009. 30(5): p. 465-93. La Marca, A., et al., Anti-Mullerian hormone (AMH) as a predictive marker in assisted reproductive technology (ART). Hum Reprod Update, 2010. 16(2): p. 113-30. Barad, D.H., A. Weghofer, and N. Gleicher, Age-specific levels for basal follicle-stimulating hormone assessment of ovarian function. Obstet Gynecol, 2007. 109(6): p. 1404-10. 119 REFERENZEN 193. 194. 195. 196. 197. 198. 199. 200. 201. 202. 203. 204. 205. 206. 207. 208. 209. 210. 211. 212. 213. 214. 215. 216. 217. 218. Barad, D.H., A. Weghofer, and N. Gleicher, Utility of age-specific serum anti-Mullerian hormone concentrations. Reprod Biomed Online, 2011. 22(3): p. 284-91. Lau, W.N., et al., The effect of ageing on female fertility in an assisted reproduction programme in Hong Kong: retrospective study. Hong Kong Med J, 2000. 6(2): p. 147-52. Navot, D., et al., Poor oocyte quality rather than implantation failure as a cause of agerelated decline in female fertility. Lancet, 1991. 337(8754): p. 1375-7. Tarlatzis, B.C. and L. Zepiridis, Perimenopausal conception. Ann N Y Acad Sci, 2003. 997: p. 93-104. Frank, O., P.G. Bianchi, and A. Campana, The end of fertility: age, fecundity and fecundability in women. J Biosoc Sci, 1994. 26(3): p. 349-68. Yamamoto, M., et al., Chromosome studies in 500 induced abortions. Humangenetik, 1975. 29(1): p. 9-14. Hodges, C.A., et al., Experimental evidence that changes in oocyte growth influence meiotic chromosome segregation. Hum Reprod, 2002. 17(5): p. 1171-80. Warburton, D., et al., Does the karyotype of a spontaneous abortion predict the karyotype of a subsequent abortion? Evidence from 273 women with two karyotyped spontaneous abortions. Am J Hum Genet, 1987. 41(3): p. 465-83. Haadsma, M.L., et al., The predictive value of ovarian reserve tests for miscarriage in a population of subfertile ovulatory women. Hum Reprod, 2009. 24(3): p. 546-52. Haadsma, M.L., et al., Miscarriage risk for IVF pregnancies in poor responders to ovarian hyperstimulation. Reprod Biomed Online, 2010. 20(2): p. 191-200. Hoesli, I.M., et al., Spontaneous fetal loss rates in a non-selected population. Am J Med Genet, 2001. 100(2): p. 106-9. Velarde, M.C. and R. Menon, Positive and negative effects of cellular senescence during female reproductive aging and pregnancy. J Endocrinol, 2016. 230(2): p. R59-76. Li, Q., et al., Current understanding of ovarian aging. Sci China Life Sci, 2012. 55(8): p. 659-69. Amanvermez, R. and M. Tosun, An Update on Ovarian Aging and Ovarian Reserve Tests. Int J Fertil Steril, 2016. 9(4): p. 411-5. Yue, M.X., et al., Abnormal DNA methylation in oocytes could be associated with a decrease in reproductive potential in old mice. J Assist Reprod Genet, 2012. 29(7): p. 643-50. Guglielmino, M.R., et al., TAp73 is downregulated in oocytes from women of advanced reproductive age. Cell Cycle, 2011. 10(19): p. 3253-6. Ge, Z.J., et al., Oocyte ageing and epigenetics. Reproduction, 2015. 149(3): p. R103-14. Habbema, J.D.F., et al., Towards less confusing terminology in reproductive medicine: a proposal. Human Reproduction, 2004. 19(7): p. 1497-1501. Diedrich, K., M. Ludwig, and G. Griesinger, Reproduktionsmedizin. 2013: Springer Berlin Heidelberg. Cummins, J., Evolutionary forces behind human infertility. Nature, 1999. 397(6720): p. 557-8. Snick, H.K., et al., The spontaneous pregnancy prognosis in untreated subfertile couples: the Walcheren primary care study. Hum Reprod, 1997. 12(7): p. 1582-8. Zegers-Hochschild, F., et al., The International Committee for Monitoring Assisted Reproductive Technology (ICMART) and the World Health Organization (WHO) Revised Glossary on ART Terminology, 2009. Human Reproduction, 2009. 24(11): p. 2683-2687. Steptoe, P.C. and R.G. Edwards, Birth after the reimplantation of a human embryo. Lancet, 1978. 2(8085): p. 366. Palermo, G., et al., Pregnancies after intracytoplasmic injection of single spermatozoon into an oocyte. Lancet, 1992. 340(8810): p. 17-8. A Guide for Patients. 15.09.2016]; Available from: http://www.asrm.org/BOOKLET_Assisted_Reproductive_Technologies/. van Weissenbruch, M.M., et al., Pharmaco-dynamics of human menopausal gonadotrophin (HMG) and follicle-stimulating hormone (FSH). The importance of the FSH concentration in initiating follicular growth in polycystic ovary-like disease. Hum Reprod, 1993. 8(6): p. 813-21. 120 REFERENZEN 219. 220. 221. 222. 223. 224. 225. 226. 227. 228. 229. 230. 231. 232. 233. 234. 235. 236. 237. 238. 239. 240. 241. 242. Popovic-Todorovic, B., et al., A prospective randomized clinical trial comparing an individual dose of recombinant FSH based on predictive factors versus a 'standard' dose of 150 IU/day in 'standard' patients undergoing IVF/ICSI treatment. Hum Reprod, 2003. 18(11): p. 2275-82. Wikland, M., L. Enk, and L. Hamberger, Transvesical and transvaginal approaches for the aspiration of follicles by use of ultrasound. Ann N Y Acad Sci, 1985. 442: p. 182-94. Veeck, L.L., An Atlas of Human Gametes and Conceptuses: An Illustrated Reference for Assisted Reproductive Technology. 1999: Taylor & Francis. Nagy, Z.P., et al., The influence of the site of sperm deposition and mode of oolemma breakage at intracytoplasmic sperm injection on fertilization and embryo development rates. Hum Reprod, 1995. 10(12): p. 3171-7. Keck, C., Kinderwunschbehandlung in der gynäkologischen Praxis: Sinnvolle Diagnostik- und Therapiestrategien für Frauenärzte. 2013: Thieme. Krüssel, J.S., J. Hirchenhain, and H.G. Bender, Blastocyst culture. Der Gynäkologe, 2004. 37(8): p. 696-700. Spallanzani, L. and C. Bonnet, Dissertations Relative to the Natural History of Animals and Vegetables. 1784: J. Murray. Ivanoff, E.I., On the use of artificial insemination for zootechnical purposes in Russia. The Journal of Agricultural Science, 1922. 12(03): p. 244-256. Boa-Amponsem, K.M., G., The State of Development of Biotechnologies as They Relate to the Management of Animal Genetic Resources and Their Potential Application in Developing Countries. Background Study Paper, 33., 2006. Betteridge, K.J., A history of farm animal embryo transfer and some associated techniques. Anim Reprod Sci, 2003. 79(3-4): p. 203-44. Betteridge, K.J., An historical look at embryo transfer. J Reprod Fertil, 1981. 62(1): p. 1-13. Willett, E.L., et al., Successful transplantation of a fertilized bovine ovum. Science, 1951. 113(2931): p. 247. Chang, M.C., Fertilization of rabbit ova in vitro. Nature, 1959. 184(Suppl 7): p. 466-7. Edwards, R.G., P.C. Steptoe, and J.M. Purdy, Establishing full-term human pregnancies using cleaving embryos grown in vitro. Br J Obstet Gynaecol, 1980. 87(9): p. 737-56. Brackett, B.G., et al., Normal development following in vitro fertilization in the cow. Biol Reprod, 1982. 27(1): p. 147-58. Uehara, T. and R. Yanagimachi, Microsurgical injection of spermatozoa into hamster eggs with subsequent transformation of sperm nuclei into male pronuclei. Biol Reprod, 1976. 15(4): p. 467-70. Verma, O.P., et al., Assisted Reproductive Techniques in Farm Animal - From Artificial Insemination to Nanobiotechnology. Vet World, 2012. 5(5): p. 301-310. Navara, C.S., C. Simerly, and G. Schatten, Imaging motility during fertilization. Theriogenology, 1995. 44(8): p. 1099-114. Anderiesz, C., et al., Effect of recombinant human gonadotrophins on human, bovine and murine oocyte meiosis, fertilization and embryonic development in vitro. Hum Reprod, 2000. 15(5): p. 1140-8. Menezo, Y.J., A. Veiga, and J.L. Pouly, Assisted reproductive technology (ART) in humans: facts and uncertainties. Theriogenology, 2000. 53(2): p. 599-610. Niemann, H. and C. Wrenzycki, Alterations of expression of developmentally important genes in preimplantation bovine embryos by in vitro culture conditions: implications for subsequent development. Theriogenology, 2000. 53(1): p. 21-34. Moore, K.L., T.V.N. Persaud, and M.G. Torchia, The Developing Human: Clinically Oriented Embryology. 2015: Elsevier Health Sciences. Hyrapetian, M., E.M. Loucaides, and A.G. Sutcliffe, Health and disease in children born after assistive reproductive therapies (ART). Journal of Reproductive Immunology, 2014. 106: p. 21-26. Fauser, B.C., P. Devroey, and N.S. Macklon, Multiple birth resulting from ovarian stimulation for subfertility treatment. Lancet, 2005. 365(9473): p. 1807-16. 121 REFERENZEN 243. 244. 245. 246. 247. 248. 249. 250. 251. 252. 253. 254. 255. 256. 257. 258. 259. 260. 261. 262. 263. 264. Bhattacharya, S. and A. Templeton, In treating infertility, are multiple pregnancies unavoidable? N Engl J Med, 2000. 343(1): p. 58-60. Templeton, A. and J.K. Morris, Reducing the risk of multiple births by transfer of two embryos after in vitro fertilization. N Engl J Med, 1998. 339(9): p. 573-7. Bergh, C., Single embryo transfer: a mini-review. Hum Reprod, 2005. 20(2): p. 323-7. Schieve, L.A., et al., Low and very low birth weight in infants conceived with use of assisted reproductive technology. N Engl J Med, 2002. 346(10): p. 731-7. Katalinic, A., et al., Pregnancy course and outcome after intracytoplasmic sperm injection: a controlled, prospective cohort study. Fertil Steril, 2004. 81(6): p. 1604-16. Keizer, M., et al., Birth weight of singletons born after modified natural cycle IVF compared with singletons born after IVF with ovarian hyperstimulation. Fertility and Sterility, 2004. 82: p. S48-S48. Mitwally, M.F.M., et al., Estradiol production during controlled ovarian hyperstimulation correlates with the risk of pregnancy loss in pregnancies achieved after IVF-ET. Fertility and Sterility, 2004. 82: p. S198-S198. Ertzeid, G. and R. Storeng, Adverse-Effects of Gonadotropin Treatment on Preimplantation and Postimplantation Development in Mice. Journal of Reproduction and Fertility, 1992. 96(2): p. 649-655. Tanbo, T., et al., Obstetric Outcome in Singleton Pregnancies after Assisted Reproduction. Obstetrics and Gynecology, 1995. 86(2): p. 188-192. Maman, E., et al., Obstetric outcome of singleton pregnancies conceived by in vitro fertilization and ovulation induction compared with those conceived spontaneously. Fertility and Sterility, 1998. 70(2): p. 240-245. Shi, W. and T. Haaf, Aberrant methylation patterns at the two-cell stage as an indicator of early developmental failure. Molecular Reproduction and Development, 2002. 63(3): p. 329334. Horsthemke, B. and M. Ludwig, Assisted reproduction: the epigenetic perspective. Human Reproduction Update, 2005. 11(5): p. 473-482. El-Maarri, O., et al., Maternal methylation imprints on human chromosome 15 are established during or after fertilization. Nature Genetics, 2001. 27(3): p. 341-344. Geuns, E., et al., Methylation imprints of the imprint control region of the SNRPN-gene in human gametes and preimplantation embryos. Human Molecular Genetics, 2003. 12(22): p. 2873-2879. Manipalviratn, S., A. DeCherney, and J. Segars, Imprinting disorders and assisted reproductive technology. Fertil Steril, 2009. 91(2): p. 305-15. DeBaun, M.R., E.L. Niemitz, and A.P. Feinberg, Association of in vitro fertilization with Beckwith-Wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am J Hum Genet, 2003. 72(1): p. 156-60. Maher, E.R., et al., Beckwith-Wiedemann syndrome and assisted reproduction technology (ART). J Med Genet, 2003. 40(1): p. 62-4. Gicquel, C., et al., In vitro fertilization may increase the risk of Beckwith-Wiedemann syndrome related to the abnormal imprinting of the KCN1OT gene. Am J Hum Genet, 2003. 72(5): p. 1338-41. Ludwig, M., et al., Increased prevalence of imprinting defects in patients with Angelman syndrome born to subfertile couples. J Med Genet, 2005. 42(4): p. 289-91. Cox, G.F., et al., Intracytoplasmic sperm injection may increase the risk of imprinting defects. Am J Hum Genet, 2002. 71(1): p. 162-4. Lucifero, D., J.R. Chaillet, and J.M. Trasler, Potential significance of genomic imprinting defects for reproduction and assisted reproductive technology. Hum Reprod Update, 2004. 10(1): p. 3-18. Bertelsmann, H., et al., Fehlbildungsrisiko bei extrakorporaler Befruchtung. Dtsch Arztebl International, 2008. 105(1-2): p. 11-7. 122 REFERENZEN 265. 266. 267. 268. 269. 270. 271. 272. 273. 274. 275. 276. 277. 278. 279. 280. 281. 282. 283. 284. 285. 286. 287. 288. Bowdin, S., et al., A survey of assisted reproductive technology births and imprinting disorders. Hum Reprod, 2007. 22(12): p. 3237-40. Urrego, R., N. Rodriguez-Osorio, and H. Niemann, Epigenetic disorders and altered gene expression after use of Assisted Reproductive Technologies in domestic cattle. Epigenetics, 2014. 9(6): p. 803-15. Wilmut, I., et al., Viable offspring derived from fetal and adult mammalian cells. Nature, 1997. 385(6619): p. 810-3. Wilmut, I., et al., Somatic cell nuclear transfer. Nature, 2002. 419(6907): p. 583-6. Wakayama, T., et al., Full-term development of mice from enucleated oocytes injected with cumulus cell nuclei. Nature, 1998. 394(6691): p. 369-74. Eggan, K., et al., Hybrid vigor, fetal overgrowth, and viability of mice derived by nuclear cloning and tetraploid embryo complementation. Proc Natl Acad Sci U S A, 2001. 98(11): p. 6209-14. Fernandez-Gonzalez, R., et al., Long-term effect of in vitro culture of mouse embryos with serum on mRNA expression of imprinting genes, development, and behavior. Proc Natl Acad Sci U S A, 2004. 101(16): p. 5880-5. Chen, Z., et al., Large offspring syndrome: a bovine model for the human loss-of-imprinting overgrowth syndrome Beckwith-Wiedemann. Epigenetics, 2013. 8(6): p. 591-601. Robbins, K.M., et al., Expression of KCNQ1OT1, CDKN1C, H19, and PLAGL1 and the methylation patterns at the KvDMR1 and H19/IGF2 imprinting control regions is conserved between human and bovine. J Biomed Sci, 2012. 19: p. 95. Lie, R.T., et al., Birth defects in children conceived by ICSI compared with children conceived by other IVF-methods; a meta-analysis. Int J Epidemiol, 2005. 34(3): p. 696-701. Rimm, A.A., et al., A meta-analysis of controlled studies comparing major malformation rates in IVF and ICSI infants with naturally conceived children. J Assist Reprod Genet, 2004. 21(12): p. 437-43. Hansen, M., et al., Assisted reproductive technologies and the risk of birth defects - a systematic review. Human Reproduction, 2005. 20(2): p. 328-338. Sutcliffe, A.G. and M. Ludwig, Outcome of assisted reproduction. Lancet, 2007. 370(9584): p. 351-9. Dahan, M.H., et al., Clinical definition paper on in vitro maturation of human oocytes. Hum Reprod, 2016. 31(7): p. 1383-6. Coticchio, G., IVM in need of clear definitions. Hum Reprod, 2016. 31(7): p. 1387-9. Pincus, G. and E.V. Enzmann, The Comparative Behavior of Mammalian Eggs in Vivo and in Vitro : I. The Activation of Ovarian Eggs. J Exp Med, 1935. 62(5): p. 665-75. Pincus, G. and B. Saunders, The comparative behavior of mammalian eggs in vivo and in vitro. VI. The maturation of human ovarian ova. The Anatomical Record, 1939. 75(4): p. 537545. Chang, M.C., Fertilization and normal development of follicular oocytes in the rabbit. Science, 1955. 121(3155): p. 867-9. Edwards, R.G., Maturation in vitro of mouse, sheep, cow, pig, rhesus monkey and human ovarian oocytes. Nature, 1965. 208(5008): p. 349-51. Edwards, R.G., B.D. Bavister, and P.C. Steptoe, Early stages of fertilization in vitro of human oocytes matured in vitro. Nature, 1969. 221(5181): p. 632-5. Cha, K.Y., et al., Pregnancy after in vitro fertilization of human follicular oocytes collected from nonstimulated cycles, their culture in vitro and their transfer in a donor oocyte program. Fertil Steril, 1991. 55(1): p. 109-13. Hasler, J.F., The current status and future of commercial embryo transfer in cattle. Anim Reprod Sci, 2003. 79(3-4): p. 245-64. Heijnen, E.M., et al., A meta-analysis of outcomes of conventional IVF in women with polycystic ovary syndrome. Hum Reprod Update, 2006. 12(1): p. 13-21. Rao, G.D. and S.L. Tan, In vitro maturation of oocytes. Semin Reprod Med, 2005. 23(3): p. 242-7. 123 REFERENZEN 289. 290. 291. 292. 293. 294. 295. 296. 297. 298. 299. 300. 301. 302. 303. 304. 305. 306. 307. 308. 309. 310. De Vos, M., Fertility preservation in women with cancer: in vitro maturation of oocytes. Expert Review of Quality of Life in Cancer Care, 2016. 1(2): p. 127-135. Chian, R.C., et al., Maturational and developmental competence of immature oocytes retrieved from bovine ovaries at different phases of folliculogenesis. Reprod Biomed Online, 2002. 4(2): p. 127-32. Chian, R.-C., W.M. Buckett, and S.-L. Tan, In-vitro maturation of human oocytes. Reproductive BioMedicine Online, 2004. 8(2): p. 148-166. Hreinsson, J., et al., Recombinant LH is equally effective as recombinant hCG in promoting oocyte maturation in a clinical in-vitro maturation programme: a randomized study. Hum Reprod, 2003. 18(10): p. 2131-6. Benkhalifa, M., et al., Natural cycle IVF and oocyte in-vitro maturation in polycystic ovary syndrome: a collaborative prospective study. Reprod Biomed Online, 2009. 18(1): p. 29-36. Chian, R.C., et al., Prospective randomized study of human chorionic gonadotrophin priming before immature oocyte retrieval from unstimulated women with polycystic ovarian syndrome. Hum Reprod, 2000. 15(1): p. 165-70. Son, W.Y., et al., Comparison of in-vitro maturation cycles with and without in-vivo matured oocyte retrieved. Reproductive Biomedicine Online, 2008. 17(1): p. 59-67. Zhang, X.Y., et al., Chromosome abnormality rates in human embryos obtained from in-vitro maturation and IVF treatment cycles. Reprod Biomed Online, 2010. 21(4): p. 552-9. Cha, K.Y., et al., Pregnancies and deliveries after in vitro maturation culture followed by in vitro fertilization and embryo transfer without stimulation in women with polycystic ovary syndrome. Fertil Steril, 2000. 73(5): p. 978-83. Mikkelsen, A.L., et al., Basal concentrations of oestradiol may predict the outcome of in-vitro maturation in regularly menstruating women. Hum Reprod, 2001. 16(5): p. 862-7. Buckett, W.M., et al., Obstetric outcomes and congenital abnormalities after in vitro maturation, in vitro fertilization, and intracytoplasmic sperm injection. Obstetrics and Gynecology, 2007. 110(4): p. 885-891. Shu-Chi, M., et al., Growth and development of children conceived by in-vitro maturation of human oocytes. Early Hum Dev, 2006. 82(10): p. 677-82. Soderstrom-Anttila, V., et al., Favourable pregnancy results with insemination of in vitro matured oocytes from unstimulated patients. Hum Reprod, 2005. 20(6): p. 1534-40. Hanada, A., In vitro fertilization in cattle with particular reference to sperm capacitation by ionophore. A23187, Jpn. J. Anim. Reprod. 31:56–61; 1985, 1985. Sirard, M.A., et al., The culture of bovine oocytes to obtain developmentally competent embryos. Biol Reprod, 1988. 39(3): p. 546-52. Greve, T. and V. Madison, In vitro fertilization in cattle: a review. Reproduction Nutrition Development, 1991. 31(2): p. 147-157. Ward, F., et al., Optimization of in vitro bovine embryo production: effect of duration of maturation, length of gamete co-incubation, sperm concentration and sire. Theriogenology, 2002. 57(8): p. 2105-17. Rizos, D., et al., Effect of duration of oocyte maturation on the kinetics of cleavage, embryo yield and sex ratio in cattle. Reprod Fertil Dev, 2008. 20(6): p. 734-40. Koyama, K., et al., Aging-related changes in in vitro-matured bovine oocytes: oxidative stress, mitochondrial activity and ATP content after nuclear maturation. J Reprod Dev, 2014. 60(2): p. 136-42. Koyama, K., et al., Estimation of the optimal timing of fertilization for embryo development of in vitro-matured bovine oocytes based on the times of nuclear maturation and sperm penetration. J Vet Med Sci, 2014. 76(5): p. 653-9. Huang, J.C., et al., Changes in histone acetylation during postovulatory aging of mouse oocyte. Biol Reprod, 2007. 77(4): p. 666-70. Cui, M.S., et al., Acetylation of H4K12 in porcine oocytes during in vitro aging: potential role of ooplasmic reactive oxygen species. Theriogenology, 2011. 75(4): p. 638-646. 124 REFERENZEN 311. 312. 313. 314. 315. 316. 317. 318. 319. 320. 321. 322. 323. 324. 325. 326. 327. 328. 329. 330. 331. 332. 333. 334. 335. 336. 337. Wilcox, A.J., C.R. Weinberg, and D.D. Baird, Post-ovulatory ageing of the human oocyte and embryo failure. Human Reproduction, 1998. 13(2): p. 394-397. Heinzmann, J., et al., Extended in vitro maturation affects gene expression and DNA methylation in bovine oocytes. Mol Hum Reprod, 2015. 21(10): p. 770-82. Leibfried-Rutledge, M.L., et al., Development potential of bovine oocytes matured in vitro or in vivo. Biol Reprod, 1987. 36(2): p. 376-83. Galli, C. and G. Lazzari, Practical aspects of IVM/IVF in cattle. Animal Reproduction Science, 1996. 42(1-4): p. 371-379. George and Perry, Data Retrieval Committee Report. 2015. Rauprich, O., Die Kosten des Kinderwunsches: interdisziplinäre Perspektiven zur Finanzierung reproduktionsmedizinischer Behandlungen. 2012: Lit. Menezo, Y.J. and F. Herubel, Mouse and bovine models for human IVF. Reprod Biomed Online, 2002. 4(2): p. 170-5. Jones, R.E. and K.H. Lopez, Human Reproductive Biology. 2013: Elsevier Science. Stinson, S., B. Bogin, and D. O'Rourke, Human Biology: An Evolutionary and Biocultural Perspective. 2012: Wiley. Ginther, O.J., J.P. Kastelic, and L. Knopf, Composition and characteristics of follicular waves during the bovine estrous cycle. Animal Reproduction Science, 1989. 20(3): p. 187-200. Senger, P.L., Pathways to pregnancy and parturition. 1997: Current Conceptions. Cai, X. and B.R. Cullen, The imprinted H19 noncoding RNA is a primary microRNA precursor. RNA, 2007. 13(3): p. 313-6. Hansmann, T., et al., Characterization of differentially methylated regions in 3 bovine imprinted genes: a model for studying human germ-cell and embryo development. Cytogenet Genome Res, 2011. 132(4): p. 239-47. Lucifero, D., et al., Bovine SNRPN methylation imprint in oocytes and day 17 in vitro-produced and somatic cell nuclear transfer embryos. Biol Reprod, 2006. 75(4): p. 531-8. Wu, X., et al., Zygote arrest 1 (Zar1) is a novel maternal-effect gene critical for the oocyte-toembryo transition. Nat Genet, 2003. 33(2): p. 187-91. Wu, X., et al., Zygote arrest 1 (Zar1) is an evolutionarily conserved gene expressed in vertebrate ovaries. Biol Reprod, 2003. 69(3): p. 861-7. Uzbekova, S., et al., Zygote arrest 1 gene in pig, cattle and human: evidence of different transcript variants in male and female germ cells. Reprod Biol Endocrinol, 2006. 4: p. 12. Nordhoff, V., et al., Comparative analysis of human, bovine, and murine Oct-4 upstream promoter sequences. Mamm Genome, 2001. 12(4): p. 309-17. Kurosaka, S., S. Eckardt, and K.J. McLaughlin, Pluripotent lineage definition in bovine embryos by Oct4 transcript localization. Biol Reprod, 2004. 71(5): p. 1578-82. Kuhtz, J., et al., Human in vitro oocyte maturation is not associated with increased imprinting error rates at LIT1, SNRPN, PEG3 and GTL2. Hum Reprod, 2014. 29(9): p. 1995-2005. Suetake, I., et al., DNMT3L stimulates the DNA methylation activity of Dnmt3a and Dnmt3b through a direct interaction. J Biol Chem, 2004. 279(26): p. 27816-23. O'Doherty, A.M., et al., DNA methylation plays an important role in promoter choice and protein production at the mouse Dnmt3L locus. Dev Biol, 2011. 356(2): p. 411-20. Saitou, M., S. Kagiwada, and K. Kurimoto, Epigenetic reprogramming in mouse preimplantation development and primordial germ cells. Development, 2012. 139(1): p. 15-31. de Lange, T., Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev, 2005. 19(18): p. 2100-10. Smogorzewska, A., et al., Control of human telomere length by TRF1 and TRF2. Molecular and Cellular Biology, 2000. 20(5): p. 1659-1668. Matsutani, N., et al., Expression of telomeric repeat binding factor 1 and 2 and TRF1interacting nuclear protein 2 in human gastric carcinomas. Int J Oncol, 2001. 19(3): p. 507-12. Tanaka, H., et al., DNA damage-induced phosphorylation of the human telomere-associated protein TRF2. Proc Natl Acad Sci U S A, 2005. 102(43): p. 15539-44. 125 REFERENZEN 338. 339. 340. 341. 342. 343. 344. 345. 346. 347. 348. 349. 350. 351. 352. 353. 354. 355. 356. 357. 358. 359. 360. De Boeck, G., et al., Telomere-associated proteins: cross-talk between telomere maintenance and telomere-lengthening mechanisms. J Pathol, 2009. 217(3): p. 327-44. Gilchrist, G.C., et al., Telomere length and telomerase activity in bovine pre-implantation embryos in vitro. Reprod Domest Anim, 2015. 50(1): p. 58-67. Raghuram, G.V. and P.K. Mishra, Stress induced premature senescence: a new culprit in ovarian tumorigenesis? Indian J Med Res, 2014. 140 Suppl: p. S120-9. Dong, W., et al., Upregulation and CpG island hypomethylation of the TRF2 gene in human gastric cancer. Dig Dis Sci, 2010. 55(4): p. 997-1003. Hassold, T., H. Hall, and P. Hunt, The origin of human aneuploidy: where we have been, where we are going. Human Molecular Genetics, 2007. 16: p. R203-R208. Jones, K.T., Meiosis in oocytes: predisposition to aneuploidy and its increased incidence with age. Human Reproduction Update, 2008. 14(2): p. 143-158. Kang, J.S., et al., Structure and Substrate Recruitment of the Human Spindle Checkpoint Kinase Bub1. Molecular Cell, 2008. 32(3): p. 394-405. Yin, S., et al., Bub1 prevents chromosome misalignment and precocious anaphase during mouse oocyte meiosis. Cell Cycle, 2006. 5(18): p. 2130-2137. Leland, S., et al., Heterozygosity for a Bub1 mutation causes female-specific germ cell aneuploidy in mice. Proceedings of the National Academy of Sciences of the United States of America, 2009. 106(31): p. 12776-12781. McGuinness, B.E., et al., Regulation of APC/C activity in oocytes by a Bub1-dependent spindle assembly checkpoint. Curr Biol, 2009. 19(5): p. 369-80. Tsutsumi, M., et al., Age-Related Decrease of Meiotic Cohesins in Human Oocytes. Plos One, 2014. 9(5). Chiang, T., et al., Evidence that Weakened Centromere Cohesion Is a Leading Cause of AgeRelated Aneuploidy in Oocytes. Current Biology, 2010. 20(17): p. 1522-1528. Wang, X., Y. Yang, and W. Dai, Differential subcellular localizations of two human Sgo1 isoforms: implications in regulation of sister chromatid cohesion and microtubule dynamics. Cell Cycle, 2006. 5(6): p. 635-40. Tang, Z., et al., Human Bub1 protects centromeric sister-chromatid cohesion through Shugoshin during mitosis. Proc Natl Acad Sci U S A, 2004. 101(52): p. 18012-7. Kitajima, T.S., et al., Human Bub1 defines the persistent cohesion site along the mitotic chromosome by affecting Shugoshin localization. Curr Biol, 2005. 15(4): p. 353-9. Murphy, E., K. Imahashi, and C. Steenbergen, Bcl-2 regulation of mitochondrial energetics. Trends Cardiovasc Med, 2005. 15(8): p. 283-90. Stauffer, S.R., Small molecule inhibition of the Bcl-X(L)-BH3 protein-protein interaction: proofof-concept of an in vivo chemopotentiator ABT-737. Curr Top Med Chem, 2007. 7(10): p. 9615. Boumela, I., et al., Involvement of BCL2 family members in the regulation of human oocyte and early embryo survival and death: gene expression and beyond. Reproduction, 2011. 141(5): p. 549-61. Steenbergen, R., et al., Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects. J Biol Chem, 2005. 280(48): p. 4003240. Trounson, A., R.G. Gosden, and U. Eichenlaub-Ritter, Biology and pathology of the oocyte : role in fertility, medicine and nuclear reprogramming. Second edition. ed. xiv, 454 pages. Heinzmann, J., et al., Epigenetic profile of developmentally important genes in bovine oocytes. Mol Reprod Dev, 2011. 78(3): p. 188-201. Wrenzycki, C., et al., Effects of culture system and protein supplementation on mRNA expression in pre-implantation bovine embryos. Hum Reprod, 2001. 16(5): p. 893-901. Looney, C.R., et al., Commercial Aspects of Oocyte Retrieval and in-Vitro Fertilization (Ivf) for Embryo Production in Problem Cows. Theriogenology, 1994. 41(1): p. 67-72. 126 REFERENZEN 361. 362. 363. 364. 365. 366. 367. 368. 369. 370. 371. 372. 373. 374. 375. 376. 377. 378. 379. 380. 381. 382. Goodhand, K.L., et al., In vivo oocyte recovery and in vitro embryo production from bovine donors aspirated at different frequencies or following FSH treatment. Theriogenology, 1999. 51(5): p. 951-961. Diederich, M., et al., DNA methylation and mRNA expression profiles in bovine oocytes derived from prepubertal and adult donors. Reproduction, 2012. 144(3): p. 319-30. Zaraza, J., et al., Developmental competence and mRNA expression of preimplantation in vitro-produced embryos from prepubertal and postpubertal cattle and their relationship with apoptosis after intraovarian administration of IGF-1. Theriogenology, 2010. 74(1): p. 75-89. Oropeza, A., et al., Improvement of the developmental capacity of oocytes from prepubertal cattle by intraovarian insulin-like growth factor-I application. Biology of Reproduction, 2004. 70(6): p. 1634-1643. Parrish, J.J., et al., Capacitation of bovine sperm by heparin. Biol Reprod, 1988. 38(5): p. 117180. Parrish, J.J., et al., Bovine in vitro fertilization with frozen-thawed semen. Theriogenology, 1986. 25(4): p. 591-600. Shapiro, R., R.E. Servis, and M. Welcher, Reactions of Uracil and Cytosine Derivatives with Sodium Bisulfite . A Specific Deamination Method. Journal of the American Chemical Society, 1970. 92(2): p. 422-&. Hayatsu, H., Y. Wataya, and K. Kai, Addition of Sodium Bisulfite to Uracil and to Cytosine. Journal of the American Chemical Society, 1970. 92(3): p. 724-&. Frommer, M., et al., A genomic sequencing protocol that yields a positive display of 5methylcytosine residues in individual DNA strands. Proc Natl Acad Sci U S A, 1992. 89(5): p. 1827-31. Hayatsu, H., Discovery of bisulfite-mediated cytosine conversion to uracil, the key reaction for DNA methylation analysis-A personal account. Proceedings of the Japan Academy Series BPhysical and Biological Sciences, 2008. 84(8): p. 321-329. Hayatsu, H. and M. Shiragami, Reaction of Bisulfite with the 5-Hydroxymethyl Group in Pyrimidines and in Phage Dnas. Biochemistry, 1979. 18(4): p. 632-637. Sono, M., Y. Wataya, and H. Hayatsu, Role of Bisulfite in Deamination and Hydrogen IsotopeExchange of Cytidylic Acid. Journal of the American Chemical Society, 1973. 95(14): p. 47454749. Shapiro, R., V. Difate, and M. Welcher, Deamination of Cytosine Derivatives by Bisulfite Mechanism of Reaction. Journal of the American Chemical Society, 1974. 96(3): p. 906-912. Inoue, M., H. Tanooka, and H. Hayatsu, Concentration Effect of Bisulfite on Inactivation of Transforming Activity of DNA. Chemico-Biological Interactions, 1972. 5(2): p. 85-&. Grunau, C., S.J. Clark, and A. Rosenthal, Bisulfite genomic sequencing: systematic investigation of critical experimental parameters. Nucleic Acids Res, 2001. 29(13): p. E65-5. Warnecke, P.M., et al., Identification and resolution of artifacts in bisulfite sequencing. Methods, 2002. 27(2): p. 101-7. El Hajj, N., et al., Limiting dilution bisulfite (pyro)sequencing reveals parent-specific methylation patterns in single early mouse embryos and bovine oocytes. Epigenetics, 2011. 6(10): p. 1176-88. Warnecke, P.M., et al., Detection and measurement of PCR bias in quantitative methylation analysis of bisulphite-treated DNA. Nucleic Acids Res, 1997. 25(21): p. 4422-6. Chhibber, A. and B.G. Schroeder, Single-molecule polymerase chain reaction reduces bias: application to DNA methylation analysis by bisulfite sequencing. Anal Biochem, 2008. 377(1): p. 46-54. Vogelstein, B. and K.W. Kinzler, Digital PCR. Proc Natl Acad Sci U S A, 1999. 96(16): p. 923641. Ronaghi, M., M. Uhlen, and P. Nyren, A sequencing method based on real-time pyrophosphate. Science, 1998. 281(5375): p. 363-+. Dupont, J.M., et al., De novo quantitative bisulfite sequencing using the pyrosequencing technology. Anal Biochem, 2004. 333(1): p. 119-27. 127 REFERENZEN 383. 384. 385. 386. 387. 388. 389. 390. 391. 392. 393. 394. 395. 396. 397. 398. 399. 400. 401. 402. 403. 404. 405. 406. Nyren, P., Enzymatic Method for Continuous Monitoring of DNA-Polymerase-Activity. Analytical Biochemistry, 1987. 167(2): p. 235-238. Hyman, E.D., A New Method of Sequencing DNA. Analytical Biochemistry, 1988. 174(2): p. 423-436. Ahmadian, A., M. Ehn, and S. Hober, Pyrosequencing: History, biochemistry and future. Clinica Chimica Acta, 2006. 363(1-2): p. 83-94. Marimuthu, C., et al., Single-stranded DNA (ssDNA) production in DNA aptamer generation. Analyst, 2012. 137(6): p. 1307-1315. Tost, J., J. Dunker, and I.G. Gut, Analysis and quantification of multiple methylation variable positions in CpG islands by Pyrosequencing (TM). Biotechniques, 2003. 35(1): p. 152-+. Sanger, F., S. Nicklen, and A.R. Coulson, DNA Sequencing with Chain-Terminating Inhibitors. Proceedings of the National Academy of Sciences of the United States of America, 1977. 74(12): p. 5463-5467. Bock, C., et al., BiQ Analyzer: visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics, 2005. 21(21): p. 4067-8. Aranyi, T., et al., The BiSearch web server. BMC Bioinformatics, 2006. 7: p. 431. Tusnady, G.E., et al., BiSearch: primer-design and search tool for PCR on bisulfite-treated genomes. Nucleic Acids Res, 2005. 33(1): p. e9. Lee, C. and C.H. Huang, LASAGNA-Search: an integrated web tool for transcription factor binding site search and visualization. Biotechniques, 2013. 54(3): p. 141-53. Monk, D., Germline-derived DNA methylation and early embryo epigenetic reprogramming: The selected survival of imprints. Int J Biochem Cell Biol, 2015. 67: p. 128-38. Amor, D.J. and J. Halliday, A review of known imprinting syndromes and their association with assisted reproduction technologies. Hum Reprod, 2008. 23(12): p. 2826-34. Pavlok, A., A. Lucas-Hahn, and H. Niemann, Fertilization and developmental competence of bovine oocytes derived from different categories of antral follicles. Mol Reprod Dev, 1992. 31(1): p. 63-7. Lequarre, A.S., et al., Poly(A) RNA is reduced by half during bovine oocyte maturation but increases when meiotic arrest is maintained with CDK inhibitors. Biol Reprod, 2004. 71(2): p. 425-31. Fair, T., Follicular oocyte growth and acquisition of developmental competence. Anim Reprod Sci, 2003. 78(3-4): p. 203-16. O'Doherty, A.M., L.C. O'Shea, and T. Fair, Bovine DNA methylation imprints are established in an oocyte size-specific manner, which are coordinated with the expression of the DNMT3 family proteins. Biol Reprod, 2012. 86(3): p. 67. Liang, X.W., et al., Effect of postovulatory oocyte aging on DNA methylation imprinting acquisition in offspring oocytes. Fertil Steril, 2011. 96(6): p. 1479-84. Liang, X.W., et al., The effects of postovulatory aging of mouse oocytes on methylation and expression of imprinted genes at mid-term gestation. Molecular Human Reproduction, 2011. 17(9): p. 562-567. Imamura, T., et al., Dynamic CpG and non-CpG methylation of the Peg1/Mest gene in the mouse oocyte and preimplantation embryo. J Biol Chem, 2005. 280(20): p. 20171-5. Liang, X.W., et al., Loss of methylation imprint of Snrpn in postovulatory aging mouse oocyte. Biochem Biophys Res Commun, 2008. 371(1): p. 16-21. Steven, A. and B. Seliger, Control of CREB expression in tumors: from molecular mechanisms and signal transduction pathways to therapeutic target. Oncotarget, 2016. Shaywitz, A.J. and M.E. Greenberg, CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem, 1999. 68: p. 821-61. Nichols, M., et al., Phosphorylation of CREB affects its binding to high and low affinity sites: implications for cAMP induced gene transcription. EMBO J, 1992. 11(9): p. 3337-46. Bui, C., et al., cAMP response element-binding (CREB) recruitment following a specific CpG demethylation leads to the elevated expression of the matrix metalloproteinase 13 in human articular chondrocytes and osteoarthritis. FASEB J, 2012. 26(7): p. 3000-11. 128 REFERENZEN 407. 408. 409. 410. 411. 412. 413. 414. 415. 416. 417. 418. 419. 420. 421. 422. 423. 424. 425. 426. 427. 428. 429. 430. 431. Sundaram, N., et al., The role of maternal CREB in early embryogenesis of Xenopus laevis. Dev Biol, 2003. 261(2): p. 337-52. Senthilkumaran, B., et al., Expression Patterns of CREBs in Oocyte Growth and Maturation of Fish. PLoS One, 2015. 10(12): p. e0145182. Guenatri, M., et al., Plasticity in Dnmt3L-dependent and -independent modes of de novo methylation in the developing mouse embryo. Development, 2013. 140(3): p. 562-72. Lepez-Otin, C., et al., The Hallmarks of Aging. Cell, 2013. 153(6): p. 1194-1217. Romeu, A., et al., Results of Invitro Fertilization Attempts in Women 40 Years of Age and Older - the Norfolk Experience. Fertility and Sterility, 1987. 47(1): p. 130-136. Lim, A.S. and M.F. Tsakok, Age-related decline in fertility: a link to degenerative oocytes? Fertil Steril, 1997. 68(2): p. 265-71. Fragouli, E., et al., The cytogenetics of polar bodies: insights into female meiosis and the diagnosis of aneuploidy. Mol Hum Reprod, 2011. 17(5): p. 286-95. Handyside, A.H., et al., Multiple meiotic errors caused by predivision of chromatids in women of advanced maternal age undergoing in vitro fertilisation. Eur J Hum Genet, 2012. 20(7): p. 742-7. Hassold, T. and P. Hunt, Maternal age and chromosomally abnormal pregnancies: what we know and what we wish we knew. Curr Opin Pediatr, 2009. 21(6): p. 703-8. Nagaoka, S.I., T.J. Hassold, and P.A. Hunt, Human aneuploidy: mechanisms and new insights into an age-old problem. Nat Rev Genet, 2012. 13(7): p. 493-504. Petronczki, M., M.F. Siomos, and K. Nasmyth, Un menage a quatre: the molecular biology of chromosome segregation in meiosis. Cell, 2003. 112(4): p. 423-40. Revenkova, E., et al., Cohesin SMC1 beta is required for meiotic chromosome dynamics, sister chromatid cohesion and DNA recombination. Nat Cell Biol, 2004. 6(6): p. 555-62. Tsutsumi, M., et al., Age-related decrease of meiotic cohesins in human oocytes. PLoS One, 2014. 9(5): p. e96710. Lister, L.M., et al., Age-related meiotic segregation errors in mammalian oocytes are preceded by depletion of cohesin and Sgo2. Curr Biol, 2010. 20(17): p. 1511-21. Jessberger, R., Age-related aneuploidy through cohesion exhaustion. EMBO Rep, 2012. 13(6): p. 539-46. Perez, G.I. and J.L. Tilly, Cumulus cells are required for the increased apoptotic potential in oocytes of aged mice. Hum Reprod, 1997. 12(12): p. 2781-3. Matsumine, M., et al., Pentosidine accumulation in human oocytes and their correlation to age-related apoptosis. Acta Histochem Cytochem, 2008. 41(4): p. 97-104. Santonocito, M., et al., The apoptotic transcriptome of the human MII oocyte: characterization and age-related changes. Apoptosis, 2013. 18(2): p. 201-211. Keefe, D.L., K. Marquard, and L. Liu, The telomere theory of reproductive senescence in women. Curr Opin Obstet Gynecol, 2006. 18(3): p. 280-5. Keefe, D.L. and L. Liu, Telomeres and reproductive aging. Reprod Fertil Dev, 2009. 21(1): p. 10-4. Ghosh, S., et al., Telomere length is associated with types of chromosome 21 nondisjunction: a new insight into the maternal age effect on Down syndrome birth. Hum Genet, 2010. 127(4): p. 403-9. Muller-Hocker, J., et al., Morphological-cytochemical and molecular genetic analyses of mitochondria in isolated human oocytes in the reproductive age. Mol Hum Reprod, 1996. 2(12): p. 951-8. Eichenlaub-Ritter, U., et al., Spindles, mitochondria and redox potential in ageing oocytes. Reprod Biomed Online, 2004. 8(1): p. 45-58. Iwata, H., et al., Effect of maternal age on mitochondrial DNA copy number, ATP content and IVF outcome of bovine oocytes. Reprod Fertil Dev, 2011. 23(3): p. 424-32. Bentov, Y., et al., The contribution of mitochondrial function to reproductive aging. J Assist Reprod Genet, 2011. 28(9): p. 773-83. 129 REFERENZEN 432. 433. 434. 435. 436. 437. 438. 439. 440. 441. 442. 443. 444. 445. 446. 447. Eichenlaub-Ritter, U., et al., Age related changes in mitochondrial function and new approaches to study redox regulation in mammalian oocytes in response to age or maturation conditions. Mitochondrion, 2011. 11(5): p. 783-96. Rambags, B.P., et al., Advancing maternal age predisposes to mitochondrial damage and loss during maturation of equine oocytes in vitro. Theriogenology, 2014. 81(7): p. 959-65. May-Panloup, P., et al., Ovarian ageing: the role of mitochondria in oocytes and follicles. Hum Reprod Update, 2016. Malhi, P.S., et al., Oocyte developmental competence in a bovine model of reproductive aging. Reproduction, 2007. 134(2): p. 233-239. Erickson, B.H., R.A. Reynolds, and R.L. Murphree, Ovarian Characteristics and ReproductivePerformance of Aged Cow. Biology of Reproduction, 1976. 15(4): p. 555-560. Nowak, R.M., Walker's Mammals of the World. 1999: Johns Hopkins University Press. Hara, S., et al., Forced expression of DNA methyltransferases during oocyte growth accelerates the establishment of methylation imprints but not functional genomic imprinting. Hum Mol Genet, 2014. 23(14): p. 3853-64. Lucifero, D., et al., Gene-specific timing and epigenetic memory in oocyte imprinting. Hum Mol Genet, 2004. 13(8): p. 839-49. Hiura, H., et al., Oocyte growth-dependent progression of maternal imprinting in mice. Genes Cells, 2006. 11(4): p. 353-61. Pohl, G. and M. Shih Ie, Principle and applications of digital PCR. Expert Rev Mol Diagn, 2004. 4(1): p. 41-7. Weisenberger, D.J., et al., DNA methylation analysis by digital bisulfite genomic sequencing and digital MethyLight. Nucleic Acids Res, 2008. 36(14): p. 4689-98. Weisenberger, D.J., et al., Analysis of repetitive element DNA methylation by MethyLight. Nucleic Acids Res, 2005. 33(21): p. 6823-36. Zhang, Y., et al., DNA methylation analysis by bisulfite conversion, cloning, and sequencing of individual clones. Methods Mol Biol, 2009. 507: p. 177-87. Bernstein, D.L., et al., The BisPCR(2) method for targeted bisulfite sequencing. Epigenetics Chromatin, 2015. 8: p. 27. Smallwood, S.A., et al., Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat Methods, 2014. 11(8): p. 817-20. Shapiro, E., T. Biezuner, and S. Linnarsson, Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat Rev Genet, 2013. 14(9): p. 618-30. 130 VERZEICHNISSE 6 Verzeichnisse 6.1 Abkürzungsverzeichnis 6mA N6-Methyladenin 4mC N4-Methylcytosin 5mC 5-Methylcytosin A Adenin (DNA-Base) APC engl. anaphase promoting complex APS Adenosinphosphosulfat AR Assistierte Reproduktion ART Assistierte Reproduktionstechniken AS Angelman-Syndrom ATP Adenosintriphophat b bovin (lat. Rind-/ Rinder-) Bio- biotinyliert bp Basenpaar(e) BSA Bovines Serum-Albumin BSA-FAF essentially fatty acid-free BSA BWS Beckwith-Wiedmann-Syndrom bzw. beziehungsweise C Cytosin (DNA-Base) ca. circa (etwa, ungefähr) CEO engl. cumulus-enclosed oocytes 131 VERZEICHNISSE CpG Cytosin-Phosphat-Guanin-Dinukleotid CTCF CCCTC-binding factor DMR differenziell methylierte Region DNA Desoxyribonukleinsäure (engl. desoxyribonucleic acid) DNMT DNA-Methyltransferase DO engl. denuded oocytes dNTP Desoxyribonukleosidtriphosphat ddNTP Didesoxyribonukleosidtriphosphat EDTA Ethylendiamintetraacetat engl. englisch ET Embryonentransfer et al. et alii (lateinisch: und andere, weitere) FLI Friedrich-Löffler-Institut für Nutztiergenetik (Mariensee) FSH Follikelstimulierendes Hormon G Guanin (DNA-Base) GIFT intratubare Gametentransfer griech. griechisch GV Germinales Vesikel GVBD Germinal vesicle breakdown h Stunde(n) HCG humanes Choriongonadotropin ICR Imprinting-Kontroll-Region (engl. Imprinting Control Region) 132 VERZEICHNISSE ICSI Intrazytoplasmatische Spermieninjektion IVF in vitro-Fertiliation IVM in vitro-Maturation COC Kumulus-Oozyten-Komplex LD Limiting Dilution LD-BS Limiting Dilution-Bisulfitsequenzierung LH Luteinisierendes Hormon LOS Large Offspring Syndrome MII Metaphase II MPF engl. maturation promoting factor MS-PCR methylierunssensitive PCR ncRNA nicht-kodierende RNA OCS oestrisches Kuhserum OHSS ovarielles Hyperstimulationssyndrom OPU ultrasound guided ovum pick up (transvaginale Follikelpunktion) PBS phosphatgepufferte Salzlösung (engl. phosphate buffered saline) PCOS Polyzystisches Ovarialsyndrom PCR Polymerasekettenreaktion (engl. polymerase chain reaction) PGC primordiale Keimzelle (engl. primordial germ cell) PGF2α Prostaglandin F2α PHD plant-homeo Domäne PMSG Pregnant mare serum gonadotropin 133 VERZEICHNISSE PPi Pyrophosphat PVA Polyvinylalkohol PWS Prader-Willi-Syndrom RNA Ribonukleinsäure (engl. ribonucleic acid) rpm Umdrehungen pro Minute (engl. revolutions per minute) SAC engl. spindle assembly checkpoint SCNT somatic cell nuclear transfer SNP Single Nucleotide Polymorphism snoRNA engl. small nucleolar RNA SRS Silver-Russell-Synd SSW Schwangerschaftswoche T Thymin (DNA-Base) Taq Thermus aquaticus TCM Tissue Culture Medium TRIS Trishydroxymethylaminomethan u.a. und andere/ unter anderem UPD Uniparentale Disomie vgl. vergleiche Wdh. Widerholung ZIFT intratubare Zygotentransfer rom 134 VERZEICHNISSE 6.2 Abbildungsverzeichnis Abbildung 1: Regulation des geprägten H19/Igf2-Lokus in der Maus. ...................................... 9 Abbildung 2: Epigenetische Reprogrammierung in der Maus. ................................................ 14 Abbildung 3: Follikulogenese. .................................................................................................. 18 Abbildung 4: Schematische Übersicht der bovinen Oogenese, Follikulogenese und Oozytenmaturation........................................................................................................... 20 Abbildung 5: Übersicht der untersuchten Proben und Gene. ................................................. 37 Abbildung 6: Schematische Darstellung der Promotorregionen des Gensets 1 und Gensets 2. ........................................................................................................................................... 41 Abbildung 7: Bisulfit-vermittelte Deaminierung von Cytosin. ................................................. 51 Abbildung 8: Konvertierung von Cytosin zu Thymin durch eine Natriumbisulfitbehandlung mit anschließender PCR. ................................................................................................... 52 Abbildung 9: Schematischer Ablauf der Limiting-Dilution-Methode (LD). .............................. 54 Abbildung 10: Enzymatische Reaktionsabfolge der Pyrosequenzierung. ................................ 62 Abbildung 11: Schematische Darstellung des Prinzips der Pyrosequenzierung. ..................... 64 Abbildung 12: Exemplarische Darstellung von Chromatogrammen (Sanger-Sequenzierung, bDNMT3Ls) und Pyrogrammen (Pyrosequenzierung, bDNMT3Lo) sowie die verwendete schematische Darstellung der CpG-Stelen........................................................................ 72 Abbildung 13: Ergebnisse der Analyse der DNA-Methylierung für die Promotorregionen der Gene bH19, bSNRPN, bZAR1, bOCT4, bDNMT3A und bDNMT3Lo in unreifen Rindereizellen aus Antralfollikeln unterschiedlicher Größe (>2 mm, 3-5 mm und >6 mm). ........................................................................................................................................... 74 Abbildung 14: Ergebnisse der Analyse der DNA-Methylierung für die Promotorregionen der Gene bH19, bSNRPN, bZAR1, bDNMT3Lo, bDNMT3A und bOCT4 in gereiften Rindereizellen unterschiedlicher Reifungsdauer (24h und 48h). ..................................... 77 135 VERZEICHNISSE Abbildung 15: Ergebnisse der Analyse der DNA-Methylierung für die Promotorregionen der Gene bZAR1, bDNMT3A, bOCT4 und bDNMT3Lo in Embryonen aus gereiften Rindereizellen unterschiedlicher Reifungsdauer (24h und 48h). ..................................... 80 Abbildung 16: Ergebnisse der Analyse der DNA-Methylierung für die Promotorregionen der Gene bH19 und bSNRPN in Embryonen aus gereiften Rindereizellen unterschiedlicher Reifungsdauer (24h und 48h). .......................................................................................... 83 Abbildung 17: Zusammenfassung der Ergebnisse zur Analyse der DNA-Methylierung für die Promotorregion des Gens bDNMT3Ls in unreifen Rindereizellen aus Antralfollikeln unterschiedlicher Größe (>2 mm, 3-5 mm und >6 mm)(A) und gereiften Rindereizellen unterschiedlicher Reifungsdauer (24h und 48h) sowie den daraus generierten Embryonen (B). A/B: ......................................................................................................... 85 Abbildung 18: Ergebnisse der Analyse der DNA-Methylierung für die Promotorregionen der Gene bREC8, bPISD, bBUB1, bTERF2 und bBCL-XL in Rindereizellen von Kühen unterschiedlichen Alters (9-12 Monate, 3-7 Jahre und 8-11 Jahre). ................................ 89 Abbildung 19: Ergebnisse der Analyse der DNA-Methylierung für die Promotorregionen der Gene bDNMT3Lo, bH19 und bSNRPN in Rindereizellen von Kühen unterschiedlichen Alters (9-12 Monate, 3-7 Jahre und 8-11 Jahre). .............................................................. 90 6.3 Tabellenverzeichnis Tabelle 1: Zusammensetzung der hergestellten Puffer. .......................................................... 48 Tabelle 2: Verwendete Kits und Reagenzien............................................................................ 49 Tabelle 3: Verwendete Geräte. ................................................................................................ 49 Tabelle 4: Primer des Gensets 1, die in der Multiplex-PCR, Singleplex-PCR und Pyrosequenzierung für die Promotorregionen der Gene, bH19, bSNRPN, bZAR1, bDNMT3A, bDNMT3Ls, bDNMT3Lo und bOCT4 in Rindereizellen und Rinderembryonen verwendet wurden............................................................................................................ 56 Tabelle 5: Zusammensetzung der Multiplex-PCR und Singleplex-PCR des Genset 1. ............. 57 136 VERZEICHNISSE Tabelle 6: Ablauf und Bedingungen der Multiplex-PCR und der Singleplex-PCRs der untersuchten Regionen des Genset 1. .............................................................................. 58 Tabelle 7: Primer des Gensets 2, die in der Multiplex-PCR, Singleplex-PCR und Pyrosequenzierung für die Promotorregionen der Gene bTERF2, bREC8, bBCL-XL, bPISD, bBUB1, bDNMT3Lo, bH19 und bSNRPN in Rindereizellen verwendet wurden. .............. 59 Tabelle 8: Zusammensetzung der Multiplex-PCR und Singleplex-PCR des Genset 2. ............. 60 Tabelle 9: Ablauf und Bedingungen der Multiplex-PCR und der Singleplex-PCRs der untersuchten Regionen des Genset 2. .............................................................................. 61 Tabelle 10: Ablauf und Bedingungen der Sequenzierreaktion. ............................................... 67 Tabelle 11: Verwendete Computerprogramme und Datenbanken. ........................................ 68 Tabelle 12: Auflistung der Anzahl der untersuchten Eizell-Pools sowie der somatisch kontaminierten Eizell-Pools. ............................................................................................. 70 Tabelle 13: Gesamtübersicht der Ergebnisse zur Analyse der DNA-Methylierung für die Promotorregionen der Gene bH19, bSNRPN, bOCT4, bDNMT3A, bDNMT3Lo und bZAR1 in unreifen Rindereizellen aus Antralfollikeln unterschiedlicher Größe (>2 mm, 3-5 mm und >6 mm). ...................................................................................................................... 75 Tabelle 14: Gesamtübersicht der Ergebnisse zur Analyse der DNA-Methylierung für die Promotorregionen der Gene bH19, bSNRPN, bDNMT3A, bZAR1, bOCT4 und bDNMT3Lo in gereiften Rindereizellen unterschiedlicher Reifungsdauer (24h und 48h). ................. 78 Tabelle 15: Gesamtübersicht der Ergebnisse zur Analyse der DNA-Methylierung für die Promotorregionen der Gene bH19, bSNRPN, bDNMT3A, bZAR1, bOCT4 und bDNMT3Lo in Embryonen aus gereiften Rindereizellen unterschiedlicher Reifungsdauer (24h und 48h). .................................................................................................................................. 81 Tabelle 16: Übersicht der Ergebnisse zur Analyse der DNA-Methylierung für die Promotorregion des Gens bDNMT3Ls in unreifen Rindereizellen aus Antralfollikeln unterschiedlicher Größe (>2 mm, 3-5 mm und >6 mm). .................................................. 86 137 VERZEICHNISSE Tabelle 17: Übersicht der Ergebnisse zur Analyse der DNA-Methylierung für die Promotorregion des Gens bDNMT3Ls in gereiften Rindereizellen unterschiedlicher Reifungsdauer (24h und 48h). .......................................................................................... 86 Tabelle 18: Übersicht der Ergebnisse zur Analyse der DNA-Methylierung für die Promotorregion des Gens bDNMT3Ls in Embryonen aus gereiften Rindereizellen unterschiedlicher Reifungsdauer (24h und 48h). ............................................................. 86 Tabelle 19: Gesamtübersicht der Ergebnisse zur Analyse der DNA-Methylierung für die Promotorregionen der Gene bTERF2, bREC8, bBCL-XL, bPISD, bBUB1, bDNMT3Lo, bH19 und bSNRPN in Rindereizellen von Kühen unterschiedlichen Alters (9-12 Monate, 3-7 Jahre und 8-11 Jahre). ....................................................................................................... 91 Tabelle 20: Auflistung unterschiedlicher Daten der untersuchten Proben sowie der durchgeführten Analysen zur Berwertung der Limiting Dilution-Bisulfitsequenzierung. ......................................................................................................................................... 109 138 PUBLIKATIONEN UND KONGRESSBEITRÄGE 7 Publikationen und Kongressbeiträge 7.1 Publikationen Extended in vitro maturation affects gene expression and DNA methylation in bovine oocytes. Heinzmann J, Mattern F, Aldag P, Bernal-Ulloa SM, Schneider T, Haaf T, & Niemann H. Mol Hum Reprod. 2015 Oct;21(10):770-82. Development of a New Antileishmanial Aziridine-2,3-Dicarboxylate-Based Inhibitor with High Selectivity for Parasite Cysteine Proteases. Schad C, Baum U, Frank B, Dietzel U, Mattern F, Gomes C, Ponte-Sucre A, Moll H, Schurigt U, Schirmeister T. Antimicrob Agents Chemother. 2015 Nov 23;60(2):797-805. DNA methylation and mRNA expression of developmentally important genes in bovine oocytes collected from donors of different age categories. Mattern F, Herrmann D, Heinzmann J, Hadeler KG, Bernal-Ulloa SM, Haaf T, Niemann H. Mol Reprod Dev. 2016 Sep;83(9):802-814. Gene specific profiling of DNA methylation and mRNA expression in bovine oocytes derived from follicles of different size categories Mattern F, Heinzmann J, Herrmann D, Lucas-Hahn A, Haaf T, Niemann H. Reproduction, Fertility and Development. Eingereicht am 19. August 2016. 139 PUBLIKATIONEN UND KONGRESSBEITRÄGE 7.2 Kongressbeiträge Epigenetic and developmental effects of extended in vitro maturation-time in bovine oocytes. Mattern F, Heinzmann J, Aldag P, Wrenzycki C, Niemann H, Haaf T. Conference of the German Society of Human Genetics, Essen, Germany (2014) Epigenetic determinants of resilience - Implications of positive environmental factors during pregnancy. Mattern F, Pliushch G, Post A, Reif A, Haaf T. Conference of the German Society of Human Genetics, Lübeck, Germany (2016) 140 PUBLIKATIONEN UND KONGRESSBEITRÄGE Danksagung An erster Stelle möchte ich mich bei Herrn Prof. Dr. Thomas Haaf bedanken, der mir die Möglichkeit gab, mich im Rahmen meiner Promotion wissenschaftlich zu verwirklichen. Seine geduldige und großzügige Betreuung erlaubte es mir Projekte eigenverantwortlich zu entwickeln und in die Realität umzusetzen. Bei Herrn Prof. Dr. Christian Janzen möchte ich mich recht herzlich für die Übernahme der Zweitkorrektur als Gutachter der Fakultät für Biologie bedanken. Besonderer Dank gebührt Dr. Tamara Schneider für das Korrekturlesen meiner Dissertation und ihre erfahrenen und wichtigen Ratschläge, auf die ich stets zählen konnte. Bedanken möchte ich mich auch bei Herrn Prof. Dr. Heiner Niemann, Dr. Julia Heinzmann und Doris Herrmann vom Institut für Nutztiergenetik des Friedrich-Löffler-Instituts (FLI) in Mariensee/Hannover für die Bereitstellung der bovinen Eizellen sowie Embryonen und die gute Zusammenarbeit während meiner Promotion. Des Weiteren danke ich Dr. Eberhard Schneider, Anna Maierhofer und Dr. Nady El Hajj sowie allen anderen Mitgliedern der Arbeitsgruppe Haaf, auf deren fachlichen Rat ich immer bauen konnte und die zur guten Stimmung im Labor beitrugen. Ganz besonders bedanken möchte ich mich aber an dieser Stelle bei meinen Eltern, Helene und Oleg Mattern, für die finanzielle, aber vor allem moralische Unterstützung während meiner gesamten Studienzeit. Sowohl biologisch, finanziell und moralisch wäre diese Arbeit ohne euch nicht möglich gewesen! 141