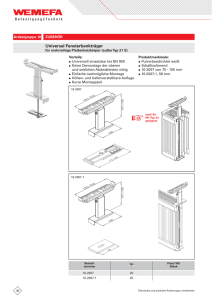
Phenylkentonurie (PKU)
Werbung
")
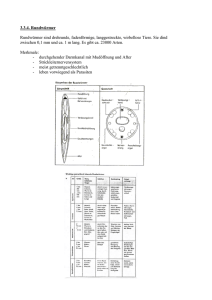
Phenylkentonurie(PKU) Häufigkeit:1:10.000,Heterozygotenfrequenz1:50 Genetik:PAH-Gen(Chromosom12q,13Exone) >400Mutationenbeschrieben 25%homozygotfüreineMutation,75%compound-heterozygot Diagnostik: Neugeborenen-Screening(2.–3.Lebenstag) Früher:Guthrie-Test(bakteriellerInhibitionstest),heute:TandemMassenspektrometrie Klinik(unbehandelt): StörungderPhenylalanin-HydroxylasemassiverAnstiegvonPhenylalaninim KörperprogredienteHirnschädigung Neugeborene:keineAuffälligkeiten Entwicklung:progredienteVerzögerungderpsychmotorischenEntwicklungab 3.LM Krankheitsbild:blaueAugen,blondeHaare,blassHaut(Tyrosinmangel), „muffiger“GeruchwegenAusscheidungvonPhenylessigsäureinSchweißund Urin Therapie:PhenylalninarmeDiät(Phenylalanin<90µmol/l),wenignatürlichesEiweiß, andereAminosäureundVitaminesowieSpurenelementealsSupplemente, RegelmäßigeKontrolle Prognose:SehrgutePrognosebeifrühemBehandlungsbeginnundguterEinstellung