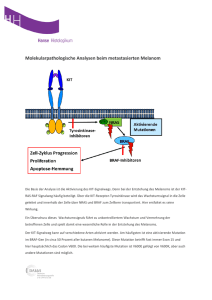
ZIP
Werbung

I Aus der Medizinischen Universitätsklinik - Abteilung Innere Medizin IV der Albert-Ludwigs-Universität Freiburg im Breisgau D Daass P maattoossee T Phhääoocchhrroom Tyypp 11 moozzyyttoom m bbeeii N Neeuurrooffiibbrroom K Klliinniikk uunndd M Moolleekkuullaarrggeenneettiikk INAUGURAL - DISSERTATION zur Erlangung des Medizinischen Doktorgrades der Medizinischen Fakultät der Albert-Ludwigs-Universität Freiburg im Breisgau Vorgelegt 2005 von Tomas Harenberg geboren in Heidelberg II Dekan Prof. Dr. R. Korinthenberg 1. Gutachter Prof. Dr. med. H.P.H. Neumann 2. Gutachter Prof. Dr. med. Ch. Bode Promotionsjahr 2006 III Meinen Eltern Anna und Job Meinem Bruder Daniel IV Inhalt Abkürzungen Vorbemerkung 1 2 3 4 Einleitung V VI 1 1.1 Geschichte des Phäochromozytoms und der Neurofibromatose Typ 1 1 1.2 Das Phäochromozytom 4 1.3 Die Neurofibromatose Typ 1 7 1.4 Genetische Grundlagen 12 1.5 Fragestellung 14 Patientengut und Methoden 15 2.1 Patientengut 15 2.2 Methoden 16 Ergebnisse 26 3.1 26 Patientenfallbeschreibungen 27 3.2 Zusammenfassung der klinischen Daten 76 3.3 Mutationen des NF1-Gens 80 3.4 Tabellarische Zusammenfassung der Mutationen 99 3.5 Tabellarische Zusammenfassung der Polymorphismen Diskussion 100 101 4.1 Klinische Ergebnisse 101 4.2 Molekulargenetische Ergebnisse 108 5 Zusammenfassung 115 6 Literaturverzeichnis 117 7 Danksagung 131 8 Curriculum vitae 133 V Abkürzungen A Adrenalin bp Basenpaare CT Computertomografie - Röntgenologisches Verfahren zur Erstellung von del Deletion DHPLC Denaturing High Performance Liquid Chromatography DNA Desoxyribonuclein Acid (Säure) DOPA Dihydroxyphenylalanin EDTA Ethylendiamin-Tetraessigsäure GAP GTPase-activating protein GDP Guanosindiphosphat GRD GAP-related Domain GTP Guanosintriphosphat ins Insertion M Metanephrine MAPK Mitogen-activated protein kinase MEN 2 Multiple Endokrine Neoplasie Typ 2 MIBG Metaiodobenzylguanidin MRT Magnetresonanztomografie; Bildgebungsverfahren auf dem Prinzip der magnetischen Kernresonanz, ohne ionisierende Strahlen NA Noradrenalin NF1 Neurofibromatose Typ 1 NM Normetanephrine PCR Polymerase chain reaction (Polymerasekettenreaktion) PET Positronenemissionstomographie PGL Paragangliom-Syndrom RET Rearranged during transfection RNA Ribonuclein Acid (Säure) SDH Succinatdehydrogenase Taq Thermus aquaticus, Enzym für die PCR VHL Von-Hippel-Lindau VI Vorbemerkung Das Phäochromozytom ist ein Tumor des Nebennierenmarkes, der sporadisch oder familiär auftritt. Die klinische und molekulargenetische Klassifikation der Phäochromozytome ist das langjährige Forschungsschwerpunkt der Freiburger Arbeitsgruppe von Prof. Neumann. In diesem Rahmen sind bereits Publikationen zum von Hippel-Lindau Syndrom, zur Multiplen Endokrinen Neoplasie Typ 2 und den Paragangliom-Syndromen 1-4 (Neumann et al. 2002A; Neumann et al. 2004) erschienen. Die Freiburger Gruppe hat daraufhin mit der klinischen und molekulargenetischen Charakterisierung der Phäochromozytome bei Neurofibromatose Typ 1 begonnen. Ursache der Krankheit ist eine Funktionsstörung des Proteins Neurofibromin. Das kodierende Gen ist das NF1-Gen auf Chromosom 17q11.2. Es ist ein Tumorsuppressorgen. Kooperationen bestanden mit dem Team von Herrn Prof. Opocher, Università degli Studi di Padova, Padua, Italien und dem Team von Herrn Prof. Januszewicz, Institute of Cardiology, Warschau, Polen. Innerhalb des Freiburger Labors erfolgte eine personelle Aufteilung der Analysen des NF1-Gens. Die Sequenzierung der Exons 1-8 und 28-49, d.h. von 30 der 57 Exons wurde vom Verfasser im Rahmen eines 10 monatigen Studienaufenthaltes bis 2004 an der Università degli Studi di Padova im Labor von Prof. Opocher begonnen und weitestgehend durchgeführt aber nicht beendet. Laborleiterin war Frau Dr. Schiavi. Die Untersuchung des kompletten NF1-Gens mittels der Denaturing High Performance Liquid Chromatography (DHPLC) und die Sequenzierung aller mittels dieser Methode als auffällig gefundener Exons wurde von Frau Dr. Bausch im Freiburger Labor etabliert und geleitet. In der Freiburger Arbeitsgruppe haben weiterhin Frau Buchta (MTA), Frau Bacher (MTA) und Herr Berisha (Hilfslaborant) am NF1-Projekt gearbeitet. Die in der DHPLC-Analyse auffälligen Proben wurden zum Sequenzieren in das Labor der Core Facility (Leiter: Dr. M. Hoffmann) der Medizinischen Universitätsklinik Freiburg gegeben und von Frau Dr. Bausch ausgewertet. Alle in dieser Arbeit abgebildeten molekulargenetischen Befunde, d.h. alle Mutationen wurden von Frau Dr. Bausch entdeckt. 6 Mutationen konnten durch die Sequenzierungen aus Padua bestätigt werden. Die klinischen Daten wurden im wesentlichen von Herrn Prof. Neumann und dem Verfasser der Arbeit mit den unmittelbar die Patienten betreuenden, ausserhalb Freiburgs tätigen Kollegen zusammengestellt. Die Auswertung der klinischen Daten bis Mai 2005 erfolgten von dem Verfasser der Arbeit. 1 1 Einleitung 1 Einleitung 1.1 Geschichte des Phäochromozytoms und der Neurofibromatose Typ 1 Der Begriff Phäochromozytom setzt sich aus dem griechischen Wort "phaios", das "grau" bedeutet, sowie den Wortteilen "chrom-" für "Farbe", "zyt" für "Zelle" und der Endung "-om" für "Geschwulst" zusammen. "Phäo-chromo-zyt-om" bedeutet also, wörtlich übersetzt, "graufarbene Zellgeschwulst". Der Begriff „Phäochromozytom“ geht auf den Berliner Pathologen Ludwig Pick (1868–1944) zurück, der Bezug auf die dunkelbraune Farbe der Zellen bei Kontakt mit Chromsalzen nimmt (Pick, 1912). Der erste Fall eines Phäochromozytoms wurde von Fränkel 1886 als „Ein Fall von doppelseitigem, völlig latent verlaufenen Nebennierentumor und gleichzeitiger Nephritis mit Veränderungen am Zirkulationsapparat und Retinitis“ beschrieben (Fraenkel, 1886). Die erste Verbindung von Phäochromozytom mit Neurofibromatose hatte 1910 Suzuki dargestellt 1910), (Suzuki, während die Koinzidenz mit anderen endokrinen Tumoren 1932 erstmalig von Eisenberg und Wallerstein erwähnt wurde (Eisenberg et al. 1932). Glushien et al. beschrieben 1953 eine familiäre Häufung der Erkrankung im Rahmen von Phakomatosen wie der Tuberösen Sklerose, dem SturgeWeber- und besonders dem von HippelLindau-Syndrom (VHL) (Glushien et al. 1953) und der Neurofibromatose Recklinghausen). erklärt sich Dieser durch den Typ 1 (M. Zusammenhang gemeinsamen neuroektodermalen Ursprung der chromaffinen Zellen und der oben genannten Syndrome und deren Tumoren. Abb. 1: Friedrich von Recklinghausen. Friedrich von Recklinghausen (1833-1910) (Abb. 1), Pathologe in Würzburg, gilt als der 2 1 Einleitung Erstbeschreiber der Neurofibromatose Typ 1 (NF1). 1882 erschien seine klassische Publikation "Über die multiplen Fibrome der Haut und ihre Beziehung zu den multiplen Neuromen", in welcher er den später benannten "Morbus Recklinghausen" detailliert an Hand der Autopsiebefunde zweier Patienten beschrieb ( von Recklinghausen, 1882). Zeichnungen und Beschreibungen der Krankheit lassen sich allerdings schon zu früheren Zeiten finden, so malte „Aldrovandi“ 1642 ein vermutliches NF-Abbild, das später als „monstrorum historica“ (Abb.2) bekannt wurde (Zanca et al. 1975). Abb. 2: Aldrovandis “monstrorum historica”, Opera Omnia. Bononiae, 1642 3 1 Einleitung 1773 beschrieb Tilesius einen Patienten mit multiplen fibrösen Hauttumoren, Café-au-lait Flecken, Makrozephalie und Skoliose (Abb.3) (Ruggieri und Pulizzi 2003). Grossen Verdienst an der Charakterisierung der Krankheit hatte auch der Lehrer Recklinghausens, Rudolf Virchow (1821-1902), der von 1847 - 1863 eine Reihe von klinischen und neuropathologischen Eigenschaften von Neuromen und Fibromen aufzeichnete und sogar Erblichkeit erkannte (Virchow, 1863): „Ein ausgezeichneter Fall dieser Art gab mir Gelegenheit, die Einzelheiten genau zu verfolgen. Eine 47jährige Frau trug auf ihrem gamzen Körper zerstreut eine grosse Masse kleinerer und grösserer Gewächse, welche sich seit Jahren langsam entwickelt hatten. Viele von ihnen waren ganz klein, erbsen- bis kirschkerngross, rund und von glatter Haut bedeckt; andere waren grösser, wallnussgross und darüber, übrigens von gleicher Beschaffenheit. Der grösste sass links in der unteren Rippengegend mit breiter Basis auf; es hatte 48 Zoll im Umfang und erstreckte sich von der Linea alba bis etwa 2 Zoll vom Rückgraht. Es hing von da tief nach unten über die Hüfte herab. An seiner Oberfläche und in seinem Umfange trug es mehrere kleine Secundärknoten; im Ganzen war die es bedeckende Haut aber glatt und verhältnissmässig dünn. Dabei fühlte es sich weich, fast fluktuierend an. Nachdem es [….] extirpiert war, wog es 32½ Pfund. Neun Jahre fürher war es Kindskopfgross gewesen. [….] Diese Vorstellung wird noch mehr dadurch begünstigt, dass Fälle von ausgemachter erblicher Übertragung fibromatöser Dispositionen vorkommen. Ich habe einen jungen Mann gesehen, dessen Körper ganz übersäet war mit Knoten von der Grösse eines Stecknadelknopfs bis zu der von Taubeneiern, und in dessen Familie diese Besonderheit schon in der Dritten Generation in erblicher Weise vorhanden war.” Abb. 3: “the wart man”, Tilesius von Tilenau : Historia Pathologica Singularis Cutis Turpitudinis Leipzig, Germany, 1793. Im Folgenden werden die klinischen und molekulargenetischen Grundlagen des Phäochromozytoms und der Neurofibromatose Typ 1 erläutert. 4 1 Einleitung 1.2 Das Phäochromozytom 1.2.1 Klinik Phäochromozytome sind neuroendokrine Tumoren aus chromaffinen Zellen des Nebennierenmarkes, die Katecholamine - vor allem Noradrenalin und Adrenalin - produzieren und sezernieren. Die 10% Regel (10% extraadrenal, 10% bilateral, 10% maligne, 10% hereditär, Manger und Gifford, 1993) wurde allerdings inzwischen weitgehend widerlegt: Die meisten Phäochromozytome kommen zwar sporadisch und unilateral vor, aber 24 % sind familiär (Neumann et al. 2002A). Maligne Entartung kommt selten vor (10%). Häufigste Lokalisation der Metastasen sind Lunge, Leber und Knochen. Bei adulten Patienten liegen etwa 10% der Phäochromozytome extraadrenal (Neumann et al. 1993) und werden in der Regel von Pathologen als Paragangliome bezeichnet. Paragangliome stammen aus den hauptsächlich retroperitoneal gelegenen Paraganglien des sympathischen Systems. Sie können aber auch in chromaffin negativen Zellen lokalisiert sein und dem parasympatischen System angehören (Neumann et al. 2002A). Paragangliome liegen am häufigsten paraaortal (75%), kommen aber auch in der Blase (10%), dem Thorax (10%), dem Kopf-, Nackenbereich (3%) und im Becken (2%) vor (Kaltsas et al. 2004). Sie können hormonaktiv und hormoninaktiv sein. Das Symptomenspektrum unterscheidet sich nicht von dem des Phäochromozytoms. Am häufigsten äußern sich Phäochromozytome mit einer klassischen Trias bestehend aus Kopfschmerzen, Palpitationen und Schweißausbrüchen. Andere Symptome sind Hypertonie, Blässe, psychische Störungen, Schmerzen und Übelkeit (Kaltsas et al. 2004). Differentialdiagnostisch kommen Angsterkrankungen, Schilddrüsenüberfunktion (Hyperthyreose), Kopfschmerzen anderer Genese, Alkoholentzugssymptomatik sowie andere sekundäre Hypertonieformen wie Hypercortisolismus, Nierenarterienstenose und ConnSyndrom (Hyperaldosteronismus) in Betracht. 1.2.2 Diagnostik und Therapie Bei klinischer Indikation sollte als Screeningverfahren eine Adrenalin-, Noradrenalin-, Metanephrin-, Normetanephrin- und Vanillinmandelsäurebestimmung im 24h-Urin, oder eine Adrenalin- und Noradrenalinbestimmung im Plasma erfolgen, um einen Hinweis auf evtl. erhöhte Katecholaminwerte zu erhalten (Eisenhofer et al. 2004). Im Plasma kann ein erhöhter Noradrenalin-Ruhewert auf ein Phäochromozytom hinweisen. Als radiologische bildgebende 5 Verfahren stehen Magnetresonanztomographie 1 Einleitung Ultraschallsonographie, (MRT), 123-Jod oder Computertomographie (CT), 131-Jod-Metaiodobenzylguanidin (MIBG)- Szintigraphie und die 18-Fluor-DOPA-Positronenemissionstomographie (DOPAPET) zur Verfügung. Die Sonographie ist trotz verbesserter Technik ungenau und erfordert einen versierten Untersucher. Die MRT ist der CT hinsichtlich der Sensitivität um ca. 15% überlegen. Sie erlaubt aufgrund der multiplanaren Schnittführung eine optimale Darstellung des Retroperitoneums, wo ca. 99% der Phäochromozytome lokalisiert sind. Die MIBGSzintigraphie (Van Gils et al. 1990; Campbell et al. 1996) nutzt den Katecholaminmetabolismus zum Uptake innerhalb 48 Stunden und hat eine Sensitivität von ca. 85 bis 97%. Bei der 18-FluorDOPA-Positronenemissionstomographie (PET) wird eine hochauflösende Bildgebung mit der Aufnahme des Radiopharmakons kombiniert. Nach einer Pilotstudie von Högerle et. al. (2002) ist die 18-Fluor-DOPA-PET mit einer Sensitivität und Spezifität von 100% der MRT gleichwertig und allen anderen Verfahren überlegen. Die derzeit zu empfehlenden Nachweisverfahren sind in Tabelle 1 zusammengefasst. Tab. 1: Sensitivität und Spezifität verschiedener Untersuchungen zur Diagnostik des Phäochromozytoms Untersuchung Adrenalin/Noradrenalin im Plasma Sensitivität Untersuchung 33%/58% Sensitivität Sonographie 40% Adrenalin im Urin 53% CT 76% Vanillinmandelsäure im Urin 64% MRT 95% Noradrenalin im 24h-Urin 86% MIBG-Szintigramm 95% Metanephrin/Normetanephrin im Urin 97% 18-Fluor-DOPA-PET 100% Angaben nach Neumann et al. (1993), Eisenhofer et al. (1999), Högerle et al. (2002), Lenders et al. (2002) Nach der bildgebenden und biochemischen Sicherung der Diagnose ist die Therapie der Wahl bei Phäochromozytomen die laparoskopische organerhaltende Operation. Als präoperative Medikation wird α- und β-Blockade mit Phenoxybenzamin und Metoprolol empfohlen (Neumann et al. 2002B). Die Behandlung maligner Phäochromozytome ist schwierig. Die Therapieoptionen sind Resektion, Chemotherapie (Averbuch et al. 1988), die Jod-131-MIBG in hoher Dosierung oder Oktreotid-Analoga sowie α-Blocker. Kriterien für ein Ansprechen der Therapie sind Reduktion von Tumorgewebe, Senkung des Blutdrucks, der Katecholamine, und der bestehenden Symptome. 1 Einleitung 6 1.2.3 Molekulargenetische Klassifikation Anders als bisher angenommen, sind mehr als 10% der Phäochromozytome hereditär. Zu den Tumorsyndromen, bei denen ein Phäochromozytom vorkommt, gehören: • Die multiple endokrine Neoplasie Typ 2 (MEN 2), ein autosomal dominantes Syndrom, das in drei Subtypen unterteilt werden kann, MEN 2A, MEN 2B und das familiäre medulläre Schilddrüsenkarzinom. Die Häufigkeit wird auf 1:35.000 geschätzt. Ca. 50% der MEN 2A und MEN 2B Patienten entwickeln ein Phäochromozytom, wobei sie auch multifokal und bilateral vorkommen. Ursache der Erkrankung sind Mutationen des RET-Protoonkogens auf Chromosom 10q11.2 (Neumann et al. 2002A). • Das von Hippel-Lindau Syndrom (VHL), ebenfalls autosomal dominant, kann in zwei Subtypen unterteilt werden: VHL Typ 1 (ohne Phäochromozytom) und VHL Typ 2 (mit Phäochromozytom). Die Prävalenz wird zwischen 1:36.000 und 1:85.000 angegeben. 20-30% der VHL Patienten entwickeln ein Phäochromozytom, bis zu 50% sind multifokal und/oder bilateral. Ursache sind Mutationen des VHL-Tumorsuppressorgens auf Chromosom 3p25-26 (Gimm et al.2004). • Hereditäre Phäochromozytome und Paragangliome, deren genetische Ursachen erst seit 2000 mit den SDHx Genen (Succinatdehydrogenase) entdeckt wurden. Die Paragangliom-Syndrome unterteilen sich in 4 Entitäten. Das PGL 1 Syndrom bedingt durch Mutationen des SDHD-Gens (Chromosom 11q23), das PGL 2 Syndrom, das im Zusammenhang mit Veränderungen im chromosomalen Bereich 11q13 steht, das PGL 3 Syndrom, bedingt durch Mutationen des SDHC-Gens (1q21) und das PGL 4 Syndrom, dessen genetische Ursache Mutationen des SDHB-Gens (1p36-1p35) sind. Im Zusammenhang mit dem PGL 3 Syndrom spricht man nicht von Phäochromozytomen sondern von HNPs („Head and Neck-Paragangliomas“). Mutationsanalysen ergaben bisher nur relevante Ergebnisse für SDHB und SDHD (Neumann et al. 2002A). • Die Neurofibromatose Typ 1 (Morbus von Recklinghausen) mit einer Prävalenz von 1:3500. Die Inzidenz von Phäochromozytomen wird von 0,1– 5,7 % in vivo und bis zu 13% bei Autopsie angegeben (Walther et al. 1999A) (Tab. 2). Für die Krankheit sind Mutationen des NF1-Tumorsuppresorgens verantwortlich. 7 1 Einleitung 1.3 Die Neurofibromatose Typ 1 1.3.1 Klinik, Diagnostik und Therapie Die Neurofibromatose Typ 1 ist eine autosomal dominante Krankheit, deren Inzidenz bei 1:3500 Lebendgeburten liegt (Huson et al. 1994). Die eine Hälfte der Fälle kommt familiär vor, die andere Hälfte ist sporadisch. Die Penetranz beträgt bei sehr variablen klinischen Erscheinungsbildern annähernd 100%. Für die Diagnose müssen die Patienten mindestens zwei der folgenden Merkmale aufweisen (Stumpf et al. 1988; Gutmann et al. 1997): • 6 oder mehr so genannte Café-au-lait Flecken, hellbraune Hautflecken, die präpubertal eine Größe von mindestens 5 mm, postpubertal über 15 mm aufweisen können. 95% der Patienten bilden Café-au-lait Flecken (Abb. 4a). • 2 oder mehr Neurofibrome, meist gutartige Geschwülste, die klinisch und histologisch in drei Typen unterteilt werden können: 1. Kutane Neurofibrome, benigne Tumoren in der Haut (Abb. 4b). 2. Noduläre Neurofibrome in peripheren Nerven, ubiquitär auftretend und nicht infiltrierend. 3. Plexiforme (netzartig wachsende) Neurofibrome (ca. 30 % der Patienten), die bei maligner Entartung die häufigste Todesursache der NF1 darstellen. • Sommersprossenartige Pigmentierung der Achselhöhlen und/oder der Leistengegend („Axillary Freckling“, „Inguinal Freckling“, Abb. 4c). • Ein Optikusgliom, ein Tumor am Sehnerv. • Mindestens 2 Irishamartome, so genannte „Lisch-Knoten“, die sich als Pigmentanreicherungen auf der Regenbogenhaut des Auges äußern (Abb. 4d). • Eine ausgeprägte Knochendysplasie des Sphenoids und/oder der langen Röhrenknochen. • Mindestens ein Verwandter ersten Grades mit der Diagnose Neurofibromatose Typ 1. 8 1 Einleitung a) b) c) d) Abb. 4 a-d: „Typischsten” Symptome der Neurofibromatose: a) Café-au-lait Fleck, Quelle: Internet b) Fibrome der Haut, Quelle: Internet. c) Freckling, axillär (wie in diesem Fall) oder inguinal, Quelle: Internet. d) Lisch-Knoten der Iris Quelle: Internet. Weitere klinische Symptome, die gehäuft bei Neurofibromatose Typ 1 auftreten, sind Skoliosen, mentale Retardierung mit Lern-, Leistungs- und Verhaltensstörungen, Minderwuchs, Makrozephalie, Hydrozephalus, Epilepsie, Hypertonie sowie Kopfschmerzen. Vermehrt treten auch Tumoren im Intestinum, maligne Gliome, juvenile chronische myeloische Leukämie und das Phäochromozytom auf (Hirsch et al. 2001). Eine ursächliche Therapie der Krankheit ist nicht möglich. Somit steht die symptomatische Behandlung im Vordergrund. Es können z.B. Neurofibrome der Haut laserchirurgisch auch in großer Zahl entfernt werden. Die Neurofibromatose Typ 1 erfordert eine Betreuung durch verschiedene Fachdisziplinen, wie durch Dermatologie, Genetik, Pädiatrie, Neurochirurgie, Orthopädie, etc.. Dies ist am besten in einem Neurofibromatose-Zentrum gewährleistet. 9 1 Einleitung 1.3.2 Molekulargenetik des NF1-Gens Das NF1-Gen (Abb. 5) wurde erst Anfang der 90er Jahre identifiziert (Cawthon et al. 1990, Viskochil et al. 1990). Es ist auf Chromosom 17q11.2 lokalisiert. Es ist mehr als 300 kb groß und besteht aus 60 Exons. Das mRNA-Transkript ist 8454 Basen lang. Sie werden zu 2818 Aminosäuren translatiert. Das Produkt ist das Protein Neurofibromin (Li et al. 1992). Abb. 5: 3D-Struktur des Neurofibromins (aus www.rcsb.org) Es gehört zur Gruppe der Tumorsuppressorgene (DeClue et al. 1992; Li et al. 1995). Eine im Protein zentral gelegene, 360 Aminosäuren lange Region zeigt dabei eine signifikante Ähnlichkeit mit der katalytischen Domäne des Ras GTPase-aktivierenden Proteins (p120GAP) der Säugetiere. Diese „GAP-related Domain“ (GRD) interagiert mit Ras und führt zur Hydrolyse des an Ras gebundenen aktiven GTP (Guanosintriphosphat) zum inaktiven GDP (Guanosindiphosphat) (Abb.6) (Xu et al. 1990; Feldkamp et al. 1999). Dadurch wird der klassische MAP-Kinase-Signaltransduktionsweg (Abb.7) und die daraus Abb. 6: Schematiche Darstellung des NF1-Gens, des mRNA Transkripts und des Proteinproduktes, dem Neurofibromin. Das NF1-Gen ist 350 kb, die mRNA 8-9 kb lang. Das große Protein Neurofibromin hat ein Molekulargewicht von 220 kDa. Blau markiert ist die “GAP-related-Domain” = GRD, nach Feldkamp et al. (1999). 1 Einleitung 10 entstehende Zellproliferation unterbrochen. Mutationen führen zur gestörten Expression des Neurofibromins und somit zu verminderten Inhibierung der Zellteilung, woraus Tumoren entstehen können. Das Ras-Protein hat 21 kDa und beeinflusst eine Reihe weiterer Mediatoren, meist Wachstumsfaktoren. Mutationen des Ras-Proteins sind bei 50% von Kolon-, 90% von Pankreas- und 25% von anderen bösartigen Tumoren beschrieben worden (Feldkamp et al. 1999). Abb. 7: Schematische Darstellung des Ras-Reaktionsweges. Ein Signalmolekül dockt an den Tyrosinkinaserezeptor an. Dessen Phosphorylisierung führt zur Bindung mehrerer Kinasen bis es zur Aktivierung des Ras-Proteins kommt. Über mehrere Phosphorylierungen kommt es dann zur Genexpression. Das Tumorsupressorgen Neurofibromin (NF) kann die Aktivierung des Ras-Proteins unterbinden. Verteilt im menschlichen Genom finden sich DNA-Sequenzen, die eine Homogenität von über 95% zu der NF1-Gensequenz aufweisen (Luijten et al. 2001). Diese so genannten Pseudogene spielen in der genetischen Untersuchung des NF1-Gens insofern eine Rolle, als dass ihre Amplifikation ausgeschlossen werden muss, um Fehldeutungen zu vermeiden. Diese Pseudogene können durch interchromosomale Konversion Ursprung mancher Mutationen im NF1-Gen sein (Marchuk et al. 1992). In der Tabelle 2 sind die bisher entdeckten Pseudogene aufgelistet. 11 1 Einleitung Tab. 2: Auflistung der bekannten Pseudogene. Accession Nr. bezieht sich auf die Datenbank des NCBI. Chromosom Exon(s) Accession Nr. Referenz 2 10b, 12a-19a, 27b AF232292-AF232302 2 12b-19a, 27b AC0009477 The sanger centre* 2 13-15* Y07858 Regnier et al. 1997 2 13 U35697 Purandare et al. 1995 12 16 U35686 Purandare et al. 1995 12 16, 17 AC024000 14 10b, 12a-19a, 27b AF232248-AF232258 Luijten et al. 2000 14 10b, 12a-19a, 27b AF232259-AF232269 Luijten et al. 2000 14 10b, 12a-19a, 27b AF232270-AF232280 Luijten et al. 2000 14 10b, 12a-19a, 27b AF232281-AF232291 Luijten et al. 2000 14 10b, 12a-19a, 27b AL512624 14 12b-19a, 27b AL512310 14 13-15 Y07854, Y07855 14 13, 15, 16, 18 U35696, U35684, U35687, U35690 15 13-15 Y07856, Y07857 15 14 AF011743 15 15 U35685 15 15 AF011746, AF011744 15 18 U35691 Purandare et al. 1995 15 19b U35693 Purandare et al. 1995 15 20-22 M84131 Legius et al. 1992 15 21 AF011748 15 23.1 U35694 Purandare et al. 1995 15 24 U35695 Purandare et al. 1995 15 24 AF011747, AF011745, AF011749 15 25-27b M84131 15 13-23-1, 24-27b AC021585, AC023191, AC037471 Birren et al. unpubliziert 15 13-23-1, 24-27b AC020679 Birren et al. unpubliziert 15 24-27b AC060814 Birren et al. unpubliziert 18 7-9, 11 AP001004 Hattori et al. unpubliziert 18 8 U35688 Purandare et al. 1995 Purandare et al. 1995 Luijten et al. 2000 Birren et al. unpubliziert Genoscope** Genoscope** Regnier et al. 1997 Purandare et al. 1995 Regnier et al. 1997 Kehrer-Sawatzki et al. 1997 Purandare et al. 1995 Kehrer-Sawatzki et al. 1997 Kehrer-Sawatzki et al. 1997 Kehrer-Sawatzki et al. 1997 Legius et al. 1992 18 9 U35689 21 7-9, 11 D26141, AC004527 Suzuki et al. 1994 22 10b, 12a-19a, 27b AC002471, AC003064, AC005374 Luijten et al. 2000 22 13-15 Y07859 Regnier et al. 1997 22 18 U35692 Purandare et al. 1995 * = siehe Literaturverzeichnis, ** Genoscope, Centre National de Sequencage (www.genoscope.cns.fr). Tabelle nach Luijten et al. (2001). 12 1 Einleitung 1.4 Genetische Grundlagen 1.4.1 Transkription und Translation Die genomische DNA (gen. DNA) wird im Zellkern in die messenger RNA (mRNA), auch codierende DNA (cDNA) genannt, übersetzt. Sie ist die Grundlage der Aminosäuren und somit der Proteinsynthese. Dabei werden die Nukleotide Adenin (A), Cytosin (C), Guanin (G) übernommen, Thymin (T) jedoch in Uracil (U) umgewandelt. Der genetische Code beinhaltet, dass von drei Abb. 8: Der genetische Code nach Bresch und Hausmann veranschaulicht die Verschlüsselung der 20 verschiedenen Aminosäuren durch die Basen der DNA. Das Triplett wird von innen nach außen gelesen. Bestimmte Dreierkombinationen von Basen signalisieren, wann die Proteinsynthese in den Ribosomen beginnen (Start-Codon) und wann sie enden soll (Stopp-Codon). Nukleotiden (Codon) eine Aminosäure codiert wird (Abb. 8). Der Code ist universell, das heißt, er ist bei Menschen, Tieren und Pflanzen identisch. Der Ausdruck „degeneriert“ bedeutet, dass eine Aminosäure durch verschiedene Tripletts codiert werden kann. Die mRNA wandert aus dem Zellkern zu den Ribosomen im Zellplasma und wird dort zu den Proteinen translatiert. Jedem Basen-Triplett wird eine Aminosäure zugeordnet. ATG codiert für das Start-Codon, somit beginnen alle Proteine mit der Aminosäure Methionin. Stopp-Codons (TGA, TAA, TAG) beenden die Proteinbiosynthese. Die 20 Aminosäuren sind in Tabelle 3 aufgelistet. Tab. 3: Abkürzungen der einzelnen Aminosäuren. Aminosäure 1. Alanin 2. Arginin 3. Asparagin 4. Asparaginsäure 5. Cytosin 6. Glutamin 7. Glutaminsäure 8. Glycin 9. Isoleucin 10. Histidin 3-Buchstaben Code Ala Arg Asp Asn Cys Glu Gln Gly Ile His 1-Buchstaben Aminosäure Code A R D N C Q E G I H 11. Leucin 12. Lysin 13. Methionin 14. Phenylalanin 15. Prolin 16. Serin 17. Threonin 18. Tryptophan 19. Tyrosin 20. Valin 3-Buchstaben 1-Buchstaben Code Code Leu Lys Met Phe Pro Ser Thr Trp Tyr Val L K M F P S T W Y V 13 1 Einleitung Bei Angabe von Mutationen können verschiedene Angaben gemacht werden: 1. Die Zahl nach Nukleotiden steht für die Position des Nukleotids, die Buchstaben für die Basen. Das Beispiel 1101 C/T bedeutet, dass im Nukleotid 1101 der codierenden DNA die Base Cytosin (C) durch die Base Thymin (T) ersetzt ist. 2. Die Zahl zwischen zwei Aminosäuren steht für die Codonnnummer. Der Buchstabe vor der Zahl steht für die Aminosäure in der Wildtypsequenz, der Buchstabe danach für die Aminosäure, die durch die Mutation resultiert. Im Beispiel der Mutation D112Y wird im Codon 112 die Aminosäure Asparagin (D) durch Tyrosin (Y) ersetzt. Alternativ kann auch die Aminosäure durch den dreibuchstabigen Code benannt werden (Asp 112 Tyr). 3. Handelt es sich bei der Mutation um eine Insertion oder Deletion von Basen, so führt dies zum so genannten Frameshift. Der Frameshift sagt aus, dass durch die Insertion oder die Deletion es zu einer Verschiebung des Leserasters kommt und die ursprüngliche Aminosäuresequenz nicht eingehalten wird. So werden „falsche“ Aminosäuren übersetzt, bis es zu einem Stopp-Codon kommt. Bei Insertionen und Deletionen wird die Nukleotidnummer genannt, nach der es zu einer Baseninsertion oder –deletion kommt. (z.B. 2706 ins C oder 1507 del A). 4. Mutationen, die sich 2 Basen vor oder 2 Basen nach dem Exon befinden, betreffen die AG/GT-Regel. Die AG/GT-Regel besagt, dass Introns - die nicht codierenden Sequenzen zwischen zwei Exons – am 5`Ende des Introns mit den Basen GT beginnen und mit der Basenfolge AG am 3`Ende des Introns enden (Breathnach et al. 1978; Mount SM, 1982). Bei der Darstellung dieser so genannten Splice site Mutationen wird entweder die Lokalisation des ersten Nukleotids des Exons genannt mit dem Zusatz -1 oder -2. Diese Angabe verdeutlicht die Entfernung zu dem Beginn des Exons. Bei einer Mutation im GT-Bereich nach dem Exon wird die Nukleotidnummer der letzten Base des Exons genannt. Es folgt der Zusatz +1 oder +2, um die genaue Position nach dem Exon anzugeben. 1 Einleitung 14 1.5 Fragestellung Die Neurofibromatose Typ 1 ist eine häufige Erbkrankheit, die Assoziation mit Phäochromozytomen aber selten. Von zentralem Interesse war die Charakterisierung der zugrunde liegenden Mutationen bei Patienten, die beide Merkmale aufweisen. Im Verlaufe stellten sich zwei Herausforderungen: Zum einen erschien es wichtig, die Zielgruppe möglichst groß zu gestalten, um repräsentative Ergebnisse zu erzielen. Zum anderen waren erhebliche Probleme bei den DNA-Analysen zu lösen. Vor diesem Hintergrund lässt sich die Fragestellung wie folgt fassen: 1. Klinische Charakterisierung von NF1-assoziierten Phäochromozytomen: Wie ist das Spektrum hinsichtlich Alter, Geschlecht, Produktion/Elimination der Katecholamine, Lokalisation und Zahl der Tumoren sowie Vorkommen maligner Tumoren? 2. Unter welchen methodischen Voraussetzungen lassen sich Mutationen nachweisen und welche Sensitivität wird dabei erreicht? 3. Wie häufig kommen intraexonische Mutationen des NF1-Gens als Ursache vor? 4. Lassen sich genetische Besonderheiten hinsichtlich der Lokalisation im Gen oder dem Mutationstypus (Missense Mutationen versus Protein-verkürzende Mutationen) feststellen? 5. Lässt sich die Untersuchung des NF1-Gens kostengünstig und schnell als Routinediagnostik durchführen, und welche Konsequenz und welcher Nutzen ergibt sich daraus für den Patienten und deren Angehörige? 15 2 Patientengut und Methoden 2 Patientengut und Methoden 2.1 Patientengut Grundlage für das Patientengut ist das Freiburger Phäochromozytomregister. Zu diesem Register haben im Wesentlichen folgende Erhebungen beigetragen: 1. Epidemiologische Erfassung aller Phäochromozytome in Südwestdeutschland, schwerpunktmäßig prospektiv seit 1983 bis 2005 und retrospektiv bis 1975. 2. Erfassung aller Phäochromozytome des Kindes- und Jugendalters in Deutschland, durchgeführt von 1998 bis 1999. 3. Erfassung aller Patienten mit Phäochromozytomen in Zentral-Polen, durchgeführt von Prof. W. Januszewicz und von Prof. A. Januszewicz, Warschau. 4. Weiterhin wurden zahlreiche Phäochromozytom-Patienten als Einzelfälle und kleinere Serien, unter anderem von den deutschen Universitäten in Lübeck und Essen sowie von italienischen Universitäten in Padua und Rom gemeldet. Einzelne Fälle lieferten Nancy, London und Basel. Von allen Patienten wurden folgende Daten erhoben: In • Geburtsdatum • Geschlecht • OP-Datum • Lokalisation und Größe des Tumors • Multiple oder solitäre Tumoren • Dignität der Tumoren • Tumoren in anderen Organen • Familienanamnese dieser Arbeit wurden alle Patienten aus dem Freiburger Internationalen Phäochromozytomregister berücksichtigt, die eine Neurofibromatose Typ 1 und ein Phäochromozytom aufwiesen. Die Neurofibromatose Typ 1 wurde klinisch, das Phäochromozytom histologisch diagnostiziert. Die Registrierung war auf den 15.05.2005 limitiert. Von jedem Patienten musste Blut oder Blut-DNA verfügbar sein. 16 2 Patientengut und Methoden 2.2 Methoden Die Exons 1-8 und 28-49 wurden vom Verfasser in einem Labor in Kooperation mit Herrn Prof. Opocher der endokrinologischen Abteilung der Università degli Studi di Padova (Padua) in Italien während eines 10 monatigen Forschungsaufenthaltes sequenziert. Die Untersuchungen konnten allerdings nicht komplett abgeschlossen werden. Die DHPLC und die Sequenzierung der Exons mit auffälligen DHPLC-Diagrammen wurde im Labor von Herrn Prof. H.P.H. Neumann unter Leitung von Frau Dr. Bausch durchgeführt. 2.2.1 Klinische Kasuistikdarstellung Die vorhandenen Akten und Informationen wurden gesammelt und im Detail ausgewertet. Für alle Patienten wurde ein Fragebogen erstellt, mit dem folgende Informationen eingeholt wurden: 1. Allgemeine Patientendaten 2. Detaillierte Anamnese 3. Familienanamnese 4. Klinischer Untersuchungsbefund 5. Diagnostik a) 24h-Blutdruckmessung präoperativ b) Katecholaminuntersuchungen im 24h-Urin und im Plasma c) Bildgebende Befunde: • Computertomogramm des Abdomens • Kernspintomogramm des Abdomens • MIBG-Szintigraphie • DOPA-PET 6. Therapie 7. Postoperativer Verlauf Diese Erhebungen gestalteten sich mehrheitlich mühevoll, da die Akten in den primär betreuenden Krankenhäusern erneut eingesehen werden mussten. Für eine gute Darstellung der Fälle in der vorgelegten Arbeit wurden alle verfügbaren Abbildungen zusammengetragen, ausgewertet und auszugsweise wiedergegeben. 17 2 Patientengut und Methoden 2.2.2 DNA-Extraktion aus EDTA-Blut Die DNA aus peripheren Blutleukozyten wurde mittels Extraktion durch Säulen von Qiagen (QIAamp DNA Blood Kit von Qiagen, Scherczinger et al. 1997) gewonnen. Das QIAamp DNA isolation blood kit beinhaltet verschiedene Puffer-Lösungen und eine Extraktionssäule. In ein 50 ml Falcon-Tube werden 5 ml EDTA antikoaguliertes Vollblut zu 5 ml eiskaltem Puffer C1 und 15 ml eiskaltem ddH2O gegeben und vorsichtig gemischt. Anschließend wird das Gemisch für 10 min auf Eis inkubiert. Die Lyse der Zellen erfolgt durch den Puffer C1, der Nukleus bleibt jedoch intakt. Das Lysat wird für 15 min bei 4°C und 2500 U/min zentrifugiert. Danach wird der Überstand verworfen. Das durch residuelles Hämoglobin rötlich gefärbte Pellet wird mit 1 ml eiskaltem Puffer C1 und 3 ml eiskaltem ddH2O resuspendiert und nochmals für 15 min bei 4°C und 2500 U/min zentrifugiert. Dadurch werden restliches Hämoglobin und weitere Zelltrümmer entfernt. Die Resuspension des weißen Pellets erfolgt durch Vortexen mit 5 ml Puffer G2. Darauf werden 95 μl Proteinase KLösung zugefügt und bei 50°C für 30 bis 60 min inkubiert. Der Puffer G2 bewirkt eine Lyse des Nukleus und eine Denaturierung von Proteinen, welche durch die Proteinase in kleinere Fragmente verdaut werden. Die Lösung sollte dabei klar werden, damit die mit Puffer QBT äquilibrierte Säule, auf die sie gegeben wird, nicht verstopft. Nachdem die Lösung die Säule passiert hat, werden 2 mal 7,5 ml Wasch-Puffer QC hinzugegeben. Durch 5 ml EluierungsPuffer wird die DNA von der Säule gelöst und mittels 3,5 ml Isopropanol präzipiert. Die DNA-Fäden können mit einer Pipettenspitze gefangen und in Tris-Borat-EDTA (TBE) gelöst werden. 2.2.3 Polymerasekettenreaktion (PCR) Einzelne spezifische DNA-Abschnitte können mittels PCR vervielfältigt werden. In mehreren Zyklen werden DNA-Einzelstrangabschnitte durch eine thermostabile DNA-Polymerase repliziert (Saiki et al. 1985). Jeder Zyklus besteht aus drei Teilschritten. Vor Beginn des ersten Zyklus erfolgt eine verlängerte Denaturierungsphase. Der erste der drei Teilschritte trennt nach jedem Zyklus die doppelsträngig vorliegende DNA in zwei Einzelstränge. Im zweiten Schritt folgt die Hybridisierung der Primer mit den DNA-Einzelsträngen. Jedes Primerpaar hat eine spezifische Annealing-Temperatur. Die beiden am 3` Ende zum Strang und Gegenstrang komplementären Primer-Oligonukleotide flankieren die zu replizierende DNA. Die kurzen Doppelstrangabschnitte, die von den Einzelsträngen und den daran gebundenen 18 2 Patientengut und Methoden Primern beim Annealing gebildet werden, dienen als Ausgangspunkt für die Replikation durch die thermostabile Taq-Polymerase. Dies geschieht im dritten Teilschritt. Die Replikation des zwischen den beiden Primern vorliegenden DNA-Abschnittes erfolgt bei jedem Zyklus genau so oft, wie Strang und Gegenstrang vorliegen. Die sechzig Exons des NF1-Gens wurden mit den in der Tabelle 5 aufgelisteten Primerpaaren und Untersuchungsbedingungen untersucht. Um die Amplifikation der vielen Pseudogene zu verhindern, wurden die Primer mit dem Programm Primer Express von Applied Biosystems erstellt. Das Programm stellt den Primer so her, dass er Abweichung am 3`-Terminus, eine zu den Sequenzen der Pseudogene aufweist (so genannte Allelspezifische PCR, Abb. 9) (Newton et al. 1989). Dadurch kommt am Pseudogen eine Basenfehlpaarung am 3`-Terminus zustande, wodurch die Taq-Polymerase unter optimierten Reaktionsbedingungen am 3`-Terminus des Primers, der sich an das Pseudogen heftet, nicht anbinden kann und so zu einer allel-spezifischen Amplifikation führt (Ugozzoli et al,. 1991). Alle verwendeten Primer sind in Tabelle 4 Abb. 9: Schematiche Darstellung des Funktionsprinzips einer Allel-spezifischen (AS) PCR. Der 3`-Terminus des Primers weist eine Base auf, die nur an die Wildtyp-Sequenz andockt. Somit kann die Polymerase am Pseudogen keine Amplifikation starten. Sind beide Pimer allelspezifisch, steigt die Spezifität des Produktes. aufgelistet. 2.2.4 Agarosegelelektrophorese Die Agarosegelelektrophorese dient der Kontrolle von erfolgter Amplifikation. Zusätzlich kann mittels der Agarosegelelektrophorese die Größe der Amplifikationsprodukte der Polymerasekettenreaktion bestimmt werden. Bei der Herstellung wird Agarose in TBE-Puffer (Tris-Borat-EDTA) gelöst und aufgekocht. Nach Abkühlung wird die noch flüssige Gelmasse in ein Geltablett gegossen. Das Geltablett wird mit Kämmen bestückt, um die Ladetaschen im Gel zu bilden. Die DNA-Proben werden vor dem Auftrag auf das Gel mit AgarosegelLadepuffer versetzt. Das Füllen der Taschen erfolgt nach dem Einlegen des Gels in die mit TBE gefüllte horizontale Elektrophoresekammer. Zum Abschätzen der Länge und Konzentration der Nukleinsäuren, wird ebenso ein DNA-Längenstandard aufgetragen. Der 19 2 Patientengut und Methoden Längenstandard definierte beinhaltet Menge Größen von und eine definierte DNA. Die Auftrennungszeit beträgt im Schnitt 30 min bei 100 Volt. Dabei werden die Fragmente im entsprechend elektrischen Größe und Feld Ladung aufgetrennt. Durch Färben des Gels mittels einer fluoreszierendes Abb. 10: Agarosegel mit aufgetragenem Marker V in Spalte 1, sowie Exon 2 ,3 ,4b, 34 und 35+36 in Spalten 2-16. Leere Spalten enthalten als Kontrolle Premix ohne DNA Zusatz. Lösung, die Ethidiumbromid enthält, können die Fragmente unter UV-Licht der Wellenlänge 366 nm sichtbar gemacht, mit einer PolaroidKamera fotografiert und dokumentiert werden. Auf dem Agarosegel in Abbildung 10 erkennt man in der ersten Spalte den Längenmarker und in den folgenden Spalten die Produkte der Exons. Der aufgetragene Marker vom Typ V zeigt 3 Bandenpakete mit definierten Größen, die das Abschätzen der Produktgrößen erleichtert. Deren Längenunterschiede sind deutlich zu erkennen. 2.2.5 Sequenzierung in Italien Das PCR-Amplifikat wurde für die Sequenzierung (Sanger et al. 1977) anhand eines Agarosegels durch Vergleich an einer mit Marker gefüllten Tasche quantifiziert und anschließend diluiert, um die richtige Menge DNA für die Sequenzreaktion zu erhalten. Die Sequenzierreaktion fand in einem 10 μl Ansatz statt. Darin enthalten waren 1 μl Primer, 5 μl verdünntes Amplifikat und 4 μl Big Dye (Applied Biosystems). Sie lief über 25 Zyklen bei 3 Teilschritten. Nach der Denaturierung des Amplifikats bei 96°C folgt das Annealing des Primers bei 50°C. Die Sequenzreaktion findet bei 60°C statt. Nach Aufreinigung des Produktes durch Zentrifugieren auf CentriSep-Säulen, wurden die Ergebnisse der Sequenzier-PCR in einem benachbarten Labor mittels Elektrophorese untersucht und als ABI-Datei bereitgestellt. Diese wurden danach mit dem Programm MTNavigator von Applied Biosystems bearbeitet. 2 Patientengut und Methoden 20 Tab. 4: Sequenz der verwendeten Primer. F steht für den Forward-Primer, R für den Reverse-Primer, MgCl2 für die Magnesiumchlorid-Konzentration in mM. TA bezeichnet die Annealing Temperatur des jeweiligen Primers in °C. Die drei Splice-Alternativen, Exons 9br, 23a und 48a, sind nicht aufgelistet, da sie nicht untersucht wurden. Primer 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 Exon Primersequenz ( 5´ - 3´) 1 2 3 4a 4b 4c 5 6 7 8 9 10a 10b 10c 11 12a 12b 13 14 15 16 17 18 19a 19b 20 21 22 23-1 23-2 24 F: CAG ACC CTC TCC TTG CCT CTT C R: CCA CTT ATCCCC CCT TCC C F: CGT CAT GAT TTT CAA TGG CAA G R: GCT CAC TGA ATC TAA AAC CCA GC F: GCC ATT TCT GTT TGC CTT AGA CT R: TCA AAT TCA CAA AGC CTG CC F: TTG TTC TGT GTG TGT GTT TG R: TTC AGT AGT CCC ATG TGG A F: GAG ATA CCA CAC CTG TCC CCT AA R: TTG ACC CAG TGA TTT TTT TCA GA F: GCT CTG AGT TGT ATT TGT GT R: ACA ACA GCA AAT TTT ACA TC F: GAA GGA AGT TAG AAG TTT GTG R: TCT TTT AAA TGC CTA CTT GTG F: CAT GTT TAT CTT TTA AAA ATG TTG CC R: GGA GGG CAA AAT TAT TTC CAT TAT F: CTG TTA ATT TGC TAT AAT ATT AGC R: CAT AAT ACT TAT GCT AGA AAA TTC F: GTA ATG TGT TGA TGT TAT TAC ATG R: GTC TTT TTG TTT ATA AAG GAT AAC A F: CTT TCT ATT TGC TGT TCT TTT TGG R: CCT TTT TGA AAA CCA AGA GTG CA F: GAT AAA ACT TAA AAC TAC AGT GAT AAA CAG R: ATG CAA TAG AAA GGA GGT GAG ATT C F: TCC TGA GTC TTA TGT CTG ATA CCA TG R: TTG GCG TTT CAG CTA AAC CC F: ATT GAA GTT TCC TTT TTT TCC TTG R: GTA TAG ACA TAA ACA TAC CAT TTC F: CTA TAT ATT GAA ACT ACA AAT GAA AG R: AGT TTT ACA ATG TAC TAA GTA CTA C F: TGC TAT TGA CAC TTT GAT AAC TGT TTC TC R: TCT CAC CAT TAC CAT TCC AAA TAT TCT T F: TTA TTC CTC TTG GTT GTC AGT GCT R: TTT ACC AAA TTT CAT TCA GAA AAC AAA C F: CCC AAG TTG CAA ATA TAT GTC R: GTG CTT TGA GGC AGA CTG AG F: TGA AGC ATT TGC TCT GCT CT R: GTT TCA AAC TTG ATG TAT ATT AAA F: CAA GTT TGA AAC TTG GCT GTA R: ATA TAT TTA GCA GAT CAG TTA AC F: GGA AGA AAT GTT GGA TAA AGC A R: AAA CAA GTC ACT CTA TTC ATA GA F: TAT CTG TAT GCT TAT TTG GCT CTA R: GTG CAG TAA AGA ATG GCC AG F: AAA CTT ACA TTT AAT TCG TTT TAC R: TCC TTT CTA CCA ATA ACC GC F: TTG TTC CCT TCT GGC TTT TAT R: ATC TCA AAA GTT TAA ATA CAC C F: ATT TGA GGG GAA GTG AAA GAA C R: GCT TTA TTT GCT TTT TGC TTT A F: CCA CCC TGG CTG ATT ATC G R: CTC TTA CAT GCC AGT TCT C F: TGG TCT CAT GCA CTC CAT A R: AAC ATC TTT CTT CTG GCT CTG F: TGC TAC TCT TTA GCT TCC TAC R: CAT ACC TCA GCA CAC ACA TA F: ATA TGG AGC AGG TAT AAT AAA C R: AAA ACA GCG GTT CTA TGT G F: CGT TGC ACT TGG CTT AAT GTC TG R: CCA TCA GCA GCT AGA TCC TTC TTT F: CTT ATA CTC AAT TCT CAA CTC C R: GAA TTT AAG ATA GCT AGA TTA TC [MgCl2] TA 1,5 mM 65°C 59°C 59°C 58°C 58°C 57°C 49°C 51°C 58°C 57°C 51°C 47°C 53°C 58°C 61°C 59°C 55°C 55°C 57°C 57°C 59°C 59°C 71°C 65°C 57°C 59°C 56°C 56°C 59°C 59°C 59°C 60°C 58°C 59°C 54°C 59°C 53°C 53°C 53°C 53°C 55°C 55°C 55°C 55°C 53°C 53°C 53°C 53°C 56°C 51°C 55°C 51°C 51°C 55°C 55°C 53°C 53°C 51°C 61°C 59°C 55°C 53°C 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 2,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 21 Primer Exon 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 25 26 27a 27b 28 29 30 31 32 33 34 35+36 37 38 39 40 41 42 43 44+45 46 47 48 49 Primersequenz ( 5´ - 3´) F: GAC TTC ATA CAA TAA ATA ATC TG R: TAT TTG ATT CAA ACA GAG CAA C F: CTC CAT ATT TGT AAT CTT AGT TA R: GGA GAG TGT TCA CTA TCC C F: AAT TTT TTT TCT AAG TAG TTT GCT G R: CAA CCA AAA AGA AAG CAA TCA ACT F: TAG ACA ACA TAA AGC CTC ATA A R: CCA TCT CTC TAT ATT TGC TAT A F: TGA ATC CAG ACT TTG AAG AAT TGT T R: CTA GGG AGG CCA GGA TAT AGT CTA GT F: GGT TGG TTT CTG GAG CCT TTT AGA R: CAA CAA ACC CCA AAT CAA ACT GA F: TTG GAA CTA TAA GGA AAA ATA CGT TT R: AGG GTT TTC TTT GAA TTC TCT TAG A F: CCA TTT TTT CCC CGA ATT CTT R: CCC AAT GTG GCA CCA GAT AAA F: TTG ATT AGG CTG TTC CAA TGA A R: CAA AAC AAA AAA CCT CCT GAT GAT F: GTG CTA AAA CTT TGA GTC CCA TGT R: ATA ATC TAT ATT GAT CAG GTG AAG TA F: GCA AGG AGC ATT AAT ACA ATG TAT C R: CCA TGC AAG TGT TTT TAT TTA AGC F: GGT AAC AGG TCA CTT AAT GAC ATC A R: GAC CTC AAA TTT AAA CGT CTT TTA GA F: TCA TTC CGA GAT TCA GTT TAG GAG R: GAT ACC CAA AAT GAA TGC ACT CA F: TCC CCA AAA GAG AAA ACA TGG R: AGC AAC AAG AAA AGA TGG AAG AGT F: TGT GTG AAC CTC ATC AAC CAT C R: GCC TGG CCT AGT TTG CAT TT F: GGA AGA AGA CCT CAG CAG ATG C R: CAA CCG CCA AAA AAC CTA TAG G F: TTT TTT AAC CTG CCA CCG TTT F: CCT CCA TTA GTT GGA AAA TTG AAA F: AAA CGA TGG TTG TAT TTG TCA CC R: TGC TAC ACT GAC ATG GAA AAT TTT F: GCT CCA GGG ATG TAT TAG AGC TTT R: TGA CTT TCA TGT ACT CTC CCA CCT F: TGA AGT GAT TAT CCA GGT GTT TGA R: AAA GAC AGG CAC GAA GGT GA F: AAT TTT GGC ACA TTA TTC TGG G R: AGC AAG TTC ATC AAC CAT CCT T F: AGG TTT TAG TTG CTT TGA CAC TCA R: GCA GGC ATA CTA ATT TGA ACA GAA F: ATA TTT TTG GCT TCA GAT GGG R: TTG GTG TCT TAT ATT GTT GCT CAA F: AAG CGA CAC ATG ACT GCA ATG R: TGG CTT TCA TCA CTG GCC A 2 Patientengut und Methoden [MgCl2] TA 1,5 mM 53°C 53°C 53°C 53°C 53°C 55°C 53°C 53°C 57°C 58°C 61°C 60°C 55°C 55°C 58°C 59°C 56°C 57°C 57°C 61°C 55°C 56°C 56°C 56°C 58°C 58°C 58°C 57°C 57°C 58°C 59°C 59°C 57°C 57°C 57°C 57°C 58°C 58°C 57°C 57°C 57°C 56°C 56°C 56°C 55°C 55°C 58°C 60°C 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 2,3 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 1,5 mM 2 Patientengut und Methoden 22 2.2.6 Denaturing High-Performance Liquid Chromatography Analyse (DHPLC) Für die Detektion von NF1-Keimbahnmutationen wurde die DHPLC Methode angewendet, auf der das WAVE® Nucleic Acid Fragment Analysis System (WAVE-System) von Transgenomic basiert. Die DHPLC-Analyse nach der Methode von Oefner (Oefner et al. 1998) basiert auf der unterschiedlichen Migrationsgeschwindigkeit von DNA-Homo- und Heteroduplices bei der Passage durch die Matrix einer Trennsäule. Diese werden von einer mobilen Phase (Laufpuffer) auf die DHPLC-Trennsäule aufgetragen, die aus einer hydrophoben Polymermatrix besteht. Dabei werden die DNA-Moleküle an die Säule gebunden, indem sich hydrophobe Ammonium-Kationen des Triethylammoniumacetats (TEAA) im Laufpuffer an die negativ geladenen Phosphatgruppen der DNA anlagern und die Alkylgruppen des TEAA an die Oberfläche der Polymermatrix binden. Danach werden die DNA-Fragmente über Ionenpaar-Umkehrphasen-Chromatographie freigegeben, wobei die DNA-Fragmente mittels einer kontinuierlich steigenden Acetonitril-Konzentration von der Polymermatrix eluiert werden. Längere Fragmente haften dabei stärker und damit länger an der Säule als kürzere Fragmente. Die Trennung verläuft bei einer nicht denaturierenden Temperatur daher streng nach Fragmentlänge und ist somit unabhängig von der Basenzusammensetzung. Anders verhält es sich bei der Mutationsanalyse. Hier werden PCR-Fragmente identischer Länge voneinander getrennt, die sich aufgrund einer Mutation in ihrer Basenzusammensetzung unterscheiden. Werden die PCR-Produkte kontrolliert denaturiert, können sich bei der Renaturierung bei einer vorhandenen heterozygoten Mutation vier mögliche DNA-Duplices generieren. (Wildtyp / Wildtyp, Wildtyp / Mutante(sense), Mutante(antisense) / Wildtyp, Mutante / Mutante). Abbildung 11 veranschaulicht die vier möglichen DNA-Duplices. Diese unterscheiden sich während der DHPLC-Analyse im Bereich ihrer Schmelztemperatur aufgrund minimaler struktureller Unterschiede in ihren Migrationsgeschwindigkeiten. So entsteht ein Elutionsprofil mit bis zu vier Elutionspeaks. Mit der Größe der Migrationsunterschiede zwischen den vier möglichen DNA-Duplices wächst auch der Abstand der einzelnen Elutionspeaks. Liegen die Migrationsgeschwindigkeiten jedoch aufgrund struktureller Besonderheiten der Hetero- bzw. Homoduplices nahe beieinander, so können die einzelnen Peakflächen miteinander verschmelzen, so dass weniger als vier Peaks im Elutionsprofil auftreten (Abb. 12). Im Minimalfall zeigt eine Mutation nur einen Peak mit einer „Schulter“. In Einzelfällen ist sie überhaupt nicht von der Wildtypsequenz zu unterscheiden. Hebt man die Temperatur über die Schmelztemperatur der DNA-Duplices an, 23 2 Patientengut und Methoden denaturieren diese vollständig, so dass ihre Einzelstränge nur mit einem Elutionspeak angezeigt werden. Auch wenn nur Homoduplices vorliegen, entsteht ein Einzelpeakprofil, z.B. beim Vorliegen einer homozygoten Wildtyp-Sequenz oder beim Vorliegen einer homozygoten Mutation. Durch Zumischen von Wildtyp-Amplifikat kann die homozygote Mutation detektiert werden. Für die DHPLC-Analyse werden die PCR-Produkte bei 95°C 20 min. denaturiert und zur Bildung der DNA-Duplices schrittweise (1°C/min) auf 65°C abgekühlt. Die spezifischen Schmelztemperaturen wurden mittels der Navigator™-Software ermittelt. Zur Ermittlung der optimalen Analysetemperatur wurden die Amplifikate um das von der Navigator™-Software berechnete Temperaturoptimum mit mindestens 3 verschiedenen Temperaturen analysiert, meist 2C° unter und über der vorgeschlagenen Temperatur. Die optimalen Temperaturbedingungen und die Konzentration des Puffers B sind in Tabelle 5 aufgelistet. Es wurden 5 µl PCR-Produkt auf die DNA Sep®Column Trennsäule injiziert und mit einem linearen Acetonitril-Gradienten aus Puffer A (0,1 M TEAA, ph 7,0) und Puffer B (0,1 M TEAA, 25% Acetonitril, 0,1 mM EDTA) eluiert. Die Durchflussrate des Elutionspuffers durch die Säule lag konstant bei 1,5 ml/min. Die Abb. 11: Schematische Darstellung des Funktionsprinzips einer DHPLCAnalyse. Gesamtanalysezeit betrug 2,5 min. Abb. 12: Beispiel eines DHPLC Diagramms. Nach mV 0 1.6 1.7 1.8 m Denaturierung der PCR-Produkte können sich bei Mutationen während der Renaturierung bis zu vier verschiedene DNA-Duplices bilden. Diese unterscheiden sich aufgrund kleinster struktureller Unterschiede in ihrer Schmelztemperatur und führen zu minimalen Migrationsunterschieden durch die Polymermatrix. Die gestrichelte Kurve gilt als Referenz und kennzeichnet das Wildtyp-DNADuplex. Jeder weitere Peak oder jede weitere „Schulter“ zeigt das Vorhandensein eines NichtWildtyp-Duplices an. Diese müssen zur Verifizierung der Mutation oder des Polymorphismus sequenziert werden. Die X-Achse kennzeichnet die Dauer in Minuten, die Y-Achse gibt die elektrische Spannung in Millivolt an. 24 2 Patientengut und Methoden Tab. 5: Temperaturen der Trennsäule und Konzentration des Puffers B bei der DHPLC-Analyse. Größe (bp) Temp. Größe (bp) Temp. Start Puffer B(%) Exon 1 323 65°C 57,4 Exon 23-1 253 56,6 55,1 Exon 2 438 55°C 59,7 Exon 23-2 327 54°C 57,5 Exon 3 324 55°C 57,4 Exon 24 226 56.2°C 54 Exon 4a 314 56°C 57,1 Exon 25 195 55°C 52,5 Exon 4b 215 54.5°C 53,5 Exon 26 298 55.6°C 56,7 Exon 4c 190 54.9°C 52,2 Exon 27a 223 54°C 53,9 Exon 5 210 53°C 53,3 Exon 27b 223 52.3°C 53,9 Exon 6 301 56°C 56,8 Exon 28 644 54.9°C 62 Exon 7 328 55.2°C 57,5 Exon 29 467 55.3°C 60,1 Exon 8 273 54.5°C 56 Exon 30 321 53.3°C 57,3 Exon 9 264 54°C 55,6 Exon 31 350 56.3°C 58 Exon 10a 267 58.4°C 55,7 Exon 32 298 56.0°C 56,7 Exon 10b 248 54.7°C 55 Exon 33 385 54°C 58,8 Exon 10c 275 58.7°C 53,3 Exon 34 507 53.0°C 60,7 Exon 11 308 51.9°C 57 Exon 35+36 512 52.5°C 60,7 Exon 12a 341 53.9°C 57,8 Exon 37 219 54.0°C 53,7 Exon 12b 283 53.6°C 56,2 Exon 38 334 56.0°C 57,6 Exon 13 336 56.8°C 57,7 Exon 39 303 54.0°C 56,8 Exon 14 347 58.6°C 58 Exon 40 303 54.7°C 56,8 Exon 15 215 55.4°C 53,5 Exon 41 233 55.0°C 54,3 Exon 16 576 54.8°C 64,4 Exon 42 335 51.9°C 57,7 Exon 17 385 55.5°C 58,8 Exon 43 325 55.3°C 57,4 Exon 18 321 57°C 57,3 Exon 44+45 506 56.1°C 60,6 Exon 19a 365 55°C 58,4 Exon 46 290 54.4°C 56,4 Exon 19b 237 50.4°C 54,5 Exon 47 263 54.2°C 55,5 Exon 20 390 58.1°C 58,9 Exon 48 339 54.8°C 57,8 Exon 21 476 56.5°C 60,3 Exon 49 571 56.4°C 61,4 Exon 22 279 57.5°C 56,1 Produkt Start Produkt Puffer B(%) 25 2 Patientengut und Methoden 2.2.7 Sequenzierung Bei den in der DHPLC-Analyse auffälligen Proben wurden zum Sequenzieren in das Labor der Core Facility (Leiter: Dr. Hoffmann) der Medizinischen Universitätsklinik Freiburg gegeben. Die Sequenzierung erfolgte mit fluoreszenz-markierten Primern nach der Dideoxymethode in einem halbautomatischen Sequenzierer. Die Abbildung 13 zeigt ein Diagramm einer Sequenzierung. Wildtyp-Sequenz: GAAGC TAT AAGTA Mutierte Sequenz: GAAGC TGT AAGTA Nukleotid: 1466 Substitution: A/G Codon 489 Abb. 13: Beispiel eines Sequenz-Diagramms: Der komplementäre Strang zu einer einzelsträngigen Matrize wird von der Taq-Polymerase synthetisiert. Es werden Desoxynukleosidtriphosphate (dNTP`s) als Bausteine benutzt. Der Kettenabbruch erfolgt durch hinzugefügte fluoreszenzmarkierte Didesoxynukleosidtriphosphate (ddNTP`s), die im Vergleich zu den dNTP`s eine weitere OH-Gruppe an ihrer Ribose besitzen und nicht verlängert werden können. Dadurch entstehen Produkte unterschiedlicher Kettenlänge, die durch Polyacrylamid-Gelelektrophorese aufgetrennt und druch Fluorographie sichtbar gemacht werden. Mittels einer speziellen DNA-Sequenzierungsmaschine und eines entsprechenden Programms entsteht die oben dargestellte farbige Kurve. Anhand der unterschiedlichen Farben der Kurven läßt sich die Sequenz ablesen. Rot steht für Thymin (T), grün steht für Adenin (A), blau steht für Cytosin (C) und schwarz steht für Guanin (G). Peaks der jeweiligen Kurve kennzeichnen das Auftreten der entsprechenden Base an der jeweiligen DNA-Position. Das Textfeld beschreibt die Wildtyp- und die mutierte Sequenz, sowie die Nukleotidnummer und die Substitution. Das rot eingerahmte Kästchen markiert das mutierte Codon 489. 2.2.8 Mutationen und Polymorphismen Die intraexonischen Abweichungen der DNA von der Normalsequenz (=Wildtyp) wurden wie folgt bewertet: Stopp Codon, Insertionen, Deletionen und Splice site Mutationen führen zu verkürztem (Neurofibromin) Protein und sind damit Mutationen, d.h. Auslöser der Erkrankung. Missense Varianten wurden anhand mindestens 100 gesunder anonymisierter Blutspender überprüft. Wenn sie hierbei nicht auftraten, wurden sie als Mutationen, andernfalls als Polymorphismen bezeichnet. Als Polymorphismen wurden auch Varianten bezeichnet, die trotz Änderung des Codons für dieselbe Aminosäure codieren. 3 Ergebnisse 26 3 Ergebnisse Zusammengetragen wurden insgesamt 26 Fälle. Die Herkunft ist in Abbildung 14 dargestellt. Das Durchschnittsalter liegt bei 41 Jahren. Die Geschlechtsverteilung beträgt 15 weibliche zu 11 männlichen Patienten. Im Folgenden werden alle Fälle einzeln dargestellt. Danach werden die wesentlichen Daten in Tabellen 36-39 zusammengefasst. Schließlich folgt der molekulargenetische Teil mit den Ergebnissen der Untersuchung des NF1-Gens aller Patienten. Diese Ergebnisse sind in den Tabellen 40-42 zusammengefasst. Abb. 14: Herkunft der Fälle. Hierbei sind die primär betreuenden Zentren genannt. 27 3 Ergebnisse 3.1 26 Patientenfallbeschreibungen Kasuistik 1 Quelle: Phäochromozytom-Register Universitätsklinik Freiburg, Prof. Dr. Neumann Initialen: C.S. Jahr der Operation: 1994 Geburtsjahr: 1951 Alter bei Operation: 43 Jahre Geschlecht: weiblich Anamnese: Bei der Patientin waren schon immer disseminierte Veränderungen der Haut im Sinne einer Neurofibromatose von Recklinghausen bekannt. 1995 präsentierte sich die 43jährige Patientin beim Hausarzt mit Oberbauchbeschwerden und Druckgefühl im Kopf. Daraufhin wurde die Patientin stationär aufgenommen. Familienanamnese: Bekannt ist eine Neurofibromatose Typ 1 beim Vater und bei der Tochter der Patientin. Klinischer Untersuchungsbefund: Guter Allgemeinzustand, Puls 112/min, RR 155/90 mmHg. Multiple kutane und subkutane Fibrome am ganzen Körper (Abb. 15). Am Integument 5 diskrete Café-au-lait Flecken. Abb. 15: Klinisches Bild zu Fall 1 mit multiplen Fibromen. 28 3 Ergebnisse Diagnostik und Befunde: Adrenalin und Noradrenalin zeigten erhöhte Werte. In allen radiologischen Verfahren waren die Raumforderungen sichtbar, zudem fanden sich ein Neurofibrom der Galea (2cm) und des Hinterhauptes (1cm) (Tab.6). Tab. 6: Diagnostik und Befunde zu Fall 1. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 18 Sonographie: Raumforderung in beiden Nebennieren CT: Raumforderung rechte (4 cm) und linke Nebenniere(2 cm) MRT-Abdomen: • A (μg/die) Norm: <20 42 NA (μg/die) Norm: <80 82 M (μg/die) Norm: <80 k. A. NM (μg/die) Norm: <120 k. A. Phäochromozytom typische adrenale Raumforderung rechts und links von 4/2 cm Größe (Abb. 16 a und b) • MIBG-Szintigraphie: Multiple kutane Neurofibrome thorakal und abdominal Aufnahme des Radiopharmakons in beiden Nebennierentumoren Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe. a) b) a) b) Abb. 16 a und b: Fall 1: MRT-Abdomen mit sichtbaren Raumforderungen beidseits (Pfeile). a) Horizontale- b) transversale Projektion. 29 3 Ergebnisse Therapie: In der Chirurgischen Universitätsklinik Freiburg wurde eine Laparatomie mit bilateraler subtotaler Adrenalektomie durchgeführt. Gleichzeitig wurden Fibrome aus der linken Thoraxwand und dem M. psoas links entfernt. Der intraoperative Verlauf war komplikationslos. Die Phäochromozytome wurden histologisch bestätigt. Verlauf: Katecholamine im 24h-Urin waren zwei Monate nach OP normal (Tab.7). Es ist postoperativ keine Raumforderung erkennbar. Tab. 7: Verlauf zu Fall 1. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 7 CT: Kein Tumorrezidiv MRT-Abdomen: Kein Tumorrezidiv A (μg/die) Norm: <20 4 NA (μg/die) Norm: <80 52 M (μg/die) Norm: <80 k. A. NM (μg/die) Norm: <120 k. A. Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe. Der Hormonstatus zur Nebennierenrinde der Patientin erwies sich zwei Jahre nach OP weiter im Normbereich: Cortisol 112 ng/ml, Aldosteron 79 pg/ml, Renin-Aktivität 2,0 ng/ml/h. Ein ACTH-Test zur Kontrolle der Nebennierenrindenfunktionsreserve drei Jahre nach OP ergab ein gutes Ansprechen der Nebennierenrinde: bei 0 min.: 80ng/ml Cortisol. bei 30 min.: 205ng/ml Cortisol. bei 60 min.: 252ng/ml Cortisol. 3 Ergebnisse 30 Kasuistik 2 Quelle: Praxis Dr. Ising, Sindelfingen Initialen: R. B. Alter bei Operation: 46 Jahre Geburtsjahr: 1957 Jahr der Operation: 2003 Geschlecht: männlich Anamnese: Bei dem Patienten waren schon immer disseminierte Veränderungen der Haut im Sinne einer Neurofibromatose von Recklinghausen bekannt. 2002 präsentierte sich der 44jährige Patient beim Arzt wegen Nackenschmerzen. Dieser stellte einen erhöhten Blutdruck fest. Der Patient war asymptomatisch. 2004 wurde der Patient zum Ausschluss einer neuroendokrinen Ursache der schwer einstellbaren arteriellen Hypertonie stationär in Stuttgart aufgenommen. Familienanamnese: Bei der Tochter ist eine Neurofibromatose Typ 1 bekannt. Klinischer Untersuchungsbefund: Patient in gutem Allgemeinzustand, Puls 78/min, RR 150/100 mmHg, Multiple Neurofibrome, Café-au-lait Flecken. Diagnostik und Befunde: Die Metanephrine und Normetanephrine waren im 24h-Urin massiv erhöht. Sowohl in der MRT, als auch in der Szintigraphie waren die Raumforderungen sichtbar (Tab. 8). 31 3 Ergebnisse Tab. 8: Diagnostik und Befunde zu Fall 2. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 k. A. 13 50 1255 1717 Sonographie: Kein sicherer Anhalt für einen Nebennierentumor MRT-Abdomen: Phäochromozytomtypische Raumforderung beidseits von 2 und 3 cm mittlerem Durchmesser ( Abb. 17 a und b) Beidseits adrenale phäochromozytomtypische MIBG-Speicherung ( Abb. 18) MIBG-Szintigraphie: Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine. k. A. =keine Angabe; Stark erhöhte Werte für Metanephrine und Normetanephrine im 24h-Urin. b) a) Abb. 17 a und b: MRT zu Fall 2: Darstellung der Tumoren in der a) linken und b) rechten Nebenniere in transversaler Projektion. Mit freundlicher Genehmigung von Dr. Ising, Sindelfingen. Therapie: Die Einstellung des Blutdrucks auf zufriedenstellende Bereiche erfolgte mit Carvediol 25-025 mg und Phenoxybenzamin retroperitoneoskopische 20-10-20 Nebennierenresektion mg. Anschließend beidseits wurde durchgeführt. Phäochromozytome wurden histologisch bestätigt. Verlauf: Der postoperative Verlauf war unauffällig. Die Blutdruckwerte liegen um 120/80 mmHg. eine Die 3 Ergebnisse 32 1. 2. 3. Abb. 18: Fall 2: MIBG-Szintigraphie mit Phäochromozytom-typischer Speicherung links. Reihe 1 transaxiale Projektion, Reihe 2 coronale, Reihe 3 sagittale Projektion. Mit freundlicher Genehmigung von Dr. Ising, Sindelfingen. 33 3 Ergebnisse Kasuistik 3 Quelle: Universitäts-Kinderklinik Essen, Herr PD Dr. Kremens Initialen: N.T. Jahr der Operation: 1994 Geburtsjahr: 1980 Alter bei Operation: 13 Jahre Geschlecht: männlich Anamnese: Der Patient berichtete seit über einem Jahr ständig über Kopfschmerzen und Schwindel. Dabei bestand auch verstärkter Husten und Leistungsverminderung. Der Blutdruck lag bei 170/110 mmHg. Eine erste Röntgen-Thorax Aufnahme zeigte multiple noduläre Verschattungen in beiden Lungenflügeln. Eine durchgeführte Sonographie- und Echokardiographie Untersuchung blieb ohne pathologischen Befund. Eine Kontrolle der multiplen Verschattungen ließ eine Progredienz erkennen. Im Rahmen eines Krankenaufenthaltes wurde schließlich eine Blutdruckspitze von 250/180 mmHg gemessen, worauf der Patient in die Universitätsklinik Essen verlegt wurde. Klinischer Untersuchungsbefund: Patient in gutem Allgemeinzustand, Blutdruck: 140/95 mmHg. Multiple subkutane Fibrome, Café-au-lait Flecken und Hypopigmentierungen. Inguinales und axilläres „Freckling“. LischKnoten an der linken Iris. Der Patient hat überstreckbare Gelenke und eine schwache Rückenmuskulatur mit Rundrücken und beginnender Skoliose. Diagnostik und Befunde: Im 24h-Urin zeigen sich die Vanillinmandelsäure und die Metanephrine, sowie die Normetanephrine stark erhöht. In der bildgebenden Diagnostik waren die Tumoren überall erkennbar und zeigten vermehrte Speicherung in der Szintigraphie (Tab. 9). Die Diagnose der Neurofibromatose Typ 1 wurde erst im Zusammenhang mit dem Auftreten des metastasierenden Phäochromozytoms gestellt. Die Metastasen waren zu diesem Zeitpunkt in Lunge und Knochen nachweisbar. 34 3 Ergebnisse Tab. 9: Diagnostik und Befunde zu Fall 3. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 458 CT: Raumforderung im Bereich der linken Nebenniere (7x8x6cm) (Abb. 19 a und b) MRT-LWS, BWS: Teils diffuse, teils umschriebene Metastasierung in zahlreichen Brust- und A (μg/die) Norm: <20 k. A. NA (μg/die) Norm: <80 k. A. M (μg/die) Norm: <80 ↑↑ NM (μg/die) Norm: <120 ↑↑ Lendenwirbeln MIBG-Szintigraphie: Disseminierte, speichernde Metastasierung als Hinweis auf ein Phäochromozytom Multiple pulmonale und ossäre Metastasierung (Abb. 20 a-d) PET Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe; ↑↑ = stark erhöht, ohne genaue Angabe. a) b) Abb. 19 a und b: Fall 3: a und b): CT-Bilder der Lunge des Patienten. Beide Flügel sind mit massiven Raumforderungen durchsetzt. Mit freundlicher Genehmigung von Herrn PD Dr. Kremens, Essen. Therapie: 1994 wurde der Patient laparatomiert und der Tumor Nebennieren-organerhaltend entfernt. 1995 wurde eine Therapie mit 4,5 Gbq 131-Jod-MIBG durchgeführt. 1999 ist der Patient mit 7,0 GBq 131-Jod-MIBG therapiert worden. Der Hypertonus wurde zufrieden stellend mit Dibenzyran 20-10-20 mg (Phenoxybenzamin) und Beloc-Zok 190 mg/die (Metoprololsukzinat) eingestellt (RR: 140/85 mmHg). 35 3 Ergebnisse Verlauf: Nach der 2. MIBG-Therapie wurden folgende Untersuchungen durchgeführt (Tab.10): Tab. 10: Diagnostik und Befunde des Verlaufs zu Fall 1. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 80 Plasma: A (μg/die) Norm: <20 k. A. NA (μg/die) Norm: <80 k. A. M (μg/die) Norm: <80 k. A. NM (μg/die) Norm: <120 k. A. A (ng/l); Norm: <150 NA (ng/l); Norm: <450 1302 4170 Röntgen-Thorax: Multiple Lungenmetastasen in beiden Lungenflügeln Ganzkörper-MRT: Ossäre, pulmonale und hepatische Herde Ganzkörper-PET: Speichernde Herde in Schädel, Schultern, Humerus, Lungen, Rippen, Brust-, Lenden-, Sakralwirbel und Femura ( Abb. 19) Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe. Die pulmonalen und ossären Metastasen zeigen jedoch keinen Anhalt auf Progredienz. Der Patient fühlte sich unbeeinträchtigt, so dass sich von einer onkologische Tumortherapie keine Verbesserung versprochen wurde. Der Patient verstarb schließlich im Alter von 24 Jahren an seinem Leiden. a) c) b) d) Abb. 20 a-d: PET-Sequenzen zu Fall 3. Die Pfeile markieren die multiplen Metastasen. a) multiple Metastasen im Thorax b) große Metastase in der Lunge. c) Metastasen in einer Rippe. d) große Metastase im Becken. Mit freundlicher Genehmigung von Herrn PD Dr. Kremens, Essen. 36 3 Ergebnisse Kasuistik 4 Quelle: Kliniken Essen-Mitte, Prof. Dr. Walz Initialen: K.-A.D. Jahr der Operation: 2004 Geburtsjahr: 1972 Alter bei Operation: 32 Geschlecht: weiblich Anamnese: Der Patient hat eine Neurofibromatose von Recklinghausen. Es besteht eine Hypertension seit 5 Jahren und ein irritables Kolon. An Symptomen sind gelegentliche Dyspnoe, Hitzewallungen, Kopfschmerzen und Palpitationen sowie Tremor bekannt. Klinischer Untersuchungsbefund: Disseminierte Zeichen der Neurofibromatose Typ 1 am Rumpf. RR: 150/100 mmHg. Diagnostik und Befunde: Adrenalin und Noradrenalin im Urin waren erhöht. Der Tumor war in Sonographie, MRTAbdomen und MIBG-Szintigraphie darstellbar (Tab. 11). Tab. 11: Diagnostik und Befunde zu Fall 4. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 k. A. Sonographie: Raumforderung im Bereich der linken Nebenniere MRT-Abdomen: • 6x5 cm großer Tumor der linken Nebenniere • Mehrere große Steine in der Gallenblase MIBG-Szintigraphie: A (μg/die) Norm: <20 186 NA (μg/die) Norm: <80 340 M (μg/die) Norm: <80 k. A. NM (μg/die) Norm: <120 k. A. Linksseitig adrenale phäochromozytomtypische MIBG-Speicherung. Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe. 37 3 Ergebnisse Therapie: Der Bluthochdruck wurde präoperativ mit 3x40 mg Phenoxybenzamin behandelt. Die Entfernung des Tumors der linken Nebenniere erfolgte endoskopisch in Form einer Adrenalektomie. Ein Teil des Tumors wurde unmittelbar nach der Entnahme asserviert (Abb. 21 a und b). Die Gallenblase wurde laparoskopisch entfernt. Das Phäochromozytom wurde histologisch bestätigt. a) b Abb. 21 a und b: Fall 4: Operatives Resektat. a): Phäochromozytom nach Entnahme. b): Resektat aufgeschnitten. Gut erkennbar die zentrale zystisch-nekrotische Form. Am oberen Bildrand ist ein Zentimetermaß angefügt. Mit freundlicher Genehmigung von Prof. Walz, Essen. Verlauf: Der Blutdruck lag postoperativ im Normbereich (120/80 mmHg). Die Patientin stammte aus Kuwait. Deshalb sind keine Daten zum weiteren Verlauf bekannt. 3 Ergebnisse 38 Kasuistik 5 Quelle: Universitätskinderklinik Leipzig, Prof. Dr. Pfäffle Initialen: S.H. Jahr der Operation: 2002 Geburtsjahr: 1985 Alter bei Operation: 16 Jahre Geschlecht: weiblich Anamnese: Bei der Patientin ist seit ihrer Kindheit eine Neurofibromatose von Recklinghausen bekannt. Zudem leidet sie an einer Epilepsie. 1995 war die Patientin wegen eines Optikus-Glioms in onkologischer Behandlung. Bei einer ambulanten Routinekontrolle fiel ein erhöhter Blutdruck auf. Es bestanden gelegentlicher Kopfschmerz und eine Flush-Symptomatik. Daraufhin erfolgte zur Abklärung eine stationäre Aufnahme. Familienanamnese: Die Mutter und die Großmutter leiden an einer Neurofibromatose Typ 1. Klinischer Untersuchungsbefund: Blutdruck: 130/80 mmHg, Hydrozephalus (Kopfumfang 59 cm; >97. Perzentile). Mehrere Café-au-lait Flecken. Diagnostik und Befunde: Die Werte für Noradrenalin, Metanephrine und Normetanephrine im 24h-Urin waren stark erhöht. Im Plasma zeigte sich eine massiv vermehrte Sekretion von Noradrenalin. In allen radiologischen Bildgebungen ließ sich der Tumor darstellen (Tab. 12). 39 3 Ergebnisse Tab. 12: Diagnostik und Befunde zu Fall 5. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 k. A. k. A. 1465 286 8879 Plasma: A (ng/l); Norm: <150 NA (ng/l); Norm: <450 92 17914 Sonographie: Tumor im Bereich der rechten Nebenniere CT-Thorax: Pulmonale Rundherde MRT-Abdomen: • Bestätigung des Tumors der Nebenniere • Multiple Läsionen im Bereich der Lendenwirbelsäule MIBG-Szintigraphie: Aufnahme des Radiopharmakons in der rechten Nebenniere (Abb. 22 a und b) Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe. Therapie: Die Patientin wurde laparatomiert und der Tumor entfernt. Die Nebenniere konnte erhalten werden. Das Phäochromozytom wurde histologisch bestätigt. Die Behandlung der Metastasen erfolgte durch 131-Jod MIBG Therapie. Danach zeigte sich in der Ganzkörperszintigraphie jedoch ein unverändertes Bild mit multiplen Herden überwiegend in der Brust- und Lendenwirbelsäule. Danach wurde eine chemotherapeutischen Behandlung begonnen, die aber zu keinem Rückgang der Metastasen führte. Daraufhin wurde eine Bestrahlung der Metastasen durchgeführt. Der Hypertonus wurde mit Captohexal und Dibenzyran eingestellt. Verlauf: Vereinzelte kurze Blutdruckspitzen bis 230/120 mmHg, die bei der Patientin eine Aura erzeugten. Das MRT erbrachte einen unauffälligen Befund im Nebennierenbereich und unveränderte Darstellung der bekannten metastatischen Herde. 2004 entwickelte die Patientin eine Akute Myeloische Leukämie, die chemotherapeutisch nach dem Pädiatrischen Rezidiv-AML 2001 Protokoll bis zur kompletten Remission therapiert wurde. 3 Ergebnisse a) 40 b) Abb. 22 a und b: MIBG-Szintigraphie zu Fall 5. a) Ansicht von anterior. b) Ansicht von posterior. Man sieht multiple Metastasen in der Schädelkalotte, im Brust und Lendenwirbelbereich, pulmonal, sowie im Becken und im Femur. Mit freundlicher Genehmigung von Prof. Dr. Pfäffle, Universitätsklinik Leipzig. 41 3 Ergebnisse Kasuistik 6 Quelle: Phäochromozytom-Sammlung Lübeck, Dr. Klingenberg Initialen: M.-L.M. Jahr der Operation: 1999 Geburtsjahr: 1949 Alter bei Operation: 50 Jahre Geschlecht: weiblich Anamnese: Bei der Patientin ist die Diagnose der Neurofibromatose Typ 1 im zweiten Lebensjahrzent gestellt worden. Klinische Zeichen waren Café-au-lait Flecken. Sie wurde wegen einem seit vier Jahren bestehenden Hypertonus mit Blutdruckkrisen stationär aufgenommen. Klinischer Untersuchungsbefund: Multiple kutane Café-au-lait Flecken. Diagnostik und Befunde: Es fällt der leicht erhöhter Wert von Adrenalin in Plasma und 24h-Urin im Gegensatz zur massiven Vermehrung des Noradrenalin im 24h-Urin und Plasma auf. CT, MRT und die Szintigraphie ließen den Tumor erkennen (Tab. 13). Tab. 13: Diagnostik und Befunde zu Fall 6. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 k. A. 45 1392 426 2585 Plasma: CT: A (ng/l); Norm: <150 NA (ng/l); Norm: <450 180 10392 • Zystisch zerfallener Tumor der rechten Nebenniere • Vergrößerte mediastinal gelegene LK, szintigraphisch kein Uptake MRT-Abdomen: Zystisch zerfallener Tumor der rechten Nebenniere MIBG-Szintigraphie: Eindeutiges Uptake des Radiopharmakons in der rechten Nebenniere (Abb. 23) Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe. 42 3 Ergebnisse a) b) Abb. 23 a und b: Fall 6: MIBG-Szintigraphie. Deutliche Mehranreicherung (Pfeile) im Bereich der rechten Nebenniere. a) Ansicht von anterior. b) Ansicht von posterior. Mit freundlicher Genehmigung von Herrn Dr. Klingenberg, Universitätsklinikum, Schleswig-Holstein, Lübeck. Therapie: An der Universitätsklinik Lübeck wurde der Tumor transabdominell reseziert (Abb. 24). Intraoperativ war eine Zwerchfellinfiltration zu erkennen. Diese wurde entfernt. Das Phäochromozytom wurde histologisch bestätigt (Abb. 25 a-c). Die regionalen Lymphknoten zeigten keinen Tumorbefall. Verlauf: Postoperativ lagen im Urin und im Plasma die Katecholamine im Normbereich (Tab. 14). Ein MRT, das wegen der Zwerchfellinfiltration und der damit verbundenen Frage nach Malignität durchgeführt wurde, zeigte keine Tumorformationen. Sechs Monate nach Operation erfolgte ein Kontrolluntersuchung: MRT, Thorax-CT und MIBG-Szintigraphie ergaben keinen Hinweis auf ein Tumorrezidiv. 43 3 Ergebnisse Tab. 14: : Verlauf zu Fall 6: Biochemische Werte der Katecholamine und Metaboliten in Urin und Plasma sechs Monate postoperativ. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 k. A. 9 51 29 34 Plasma: A (ng/l); Norm: <150 NA (ng/l); Norm: <450 31 262 Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe. Abb. 24 Fall 6: Operatives Resektat. Partiell zystischnekrotisch verändertes Phäochromozytom. Mit freundlicher Genehmigung von Dr. Klingenberg, Universitätsklinik Schleswig-Holstein, Lübeck. a) b) c) Abb. 25 a-c: Fall 6: Histologie a) HE-Färbung, gut sichtbare Mitose (Pfeil). b) HE-Färbung,Tumor mit einem Gefäßeinbruch am oberen Bildrand. c) Färbung Chromogranin A. Mit freundlicher Genehmigung von Dr. Klingenberg, Universitätsklinikum Schleswig-Holstein, Lübeck. 44 3 Ergebnisse Kasuistik 7 Quelle: Phäochromozytom-Sammlung Lübeck, Dr. Klingenberg Initialen: R. P. Jahr der Operation: 1999 Geburtsjahr: 1939 Alter bei Operation: 60 Jahre Geschlecht: weiblich Anamnese: Bei der Patientin sind seit dem zweiten Lebensjahrzehnt disseminierte Veränderungen der Haut im Sinne einer Neurofibromatose von Recklinghausen bekannt. Weiterhin Zustand nach Strumektomie bei Struma multinodosa. Es besteht eine beidseitige Gonarthrose. Stationär wurde sie zur Abklärung intermittierender hypertensiver Krisen und Blutdruckwerten im Intervall aufgenommen. Klinischer Untersuchungsbefund: Multiple kutane Fibrome, Café-au-lait-Flecken (Abb. 26 a und b). a) b) Abb. 26 a und b: klinisches Bild zu Fall 7: Klinisches Bild der multiplen Fibrome und Café-au-lait Fleck am Rumpf (Pfeil). Mit freundlicher Genehmigung von Dr. Klingenberg, Universitätsklinikum Schleswig-Holstein, Lübeck. hypotonen 45 3 Ergebnisse Diagnostik und Befunde: Im 24-Urin waren die Metanephrine erhöht. Der Tumor war in MRT und in der Szintigraphie sichtbar (Tab. 15). Tab. 15: Diagnostik und Befunde zu Fall 7. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 k. A. 28 36 504 102 Plasma: A (ng/l); Norm: <150 NA (ng/l); Norm: <450 16 432 MRT-Abdomen: Raumforderung der linken Nebenniere MIBG-Szintigraphie: Verstärkte Anreicherung des Radiopharmakons in der linken Nebenniere Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe. Therapie: Die Patientin wurde an der Universitätsklinik Lübeck laparatomiert, der Tumor extirpiert und in flüssigem Stickstoff schockgefroren. Mittels HPLC (High Performance Liquid Chromatography) konnte ein Exzess der Katecholamine und der Metabolite nachgewiesen werden. Damit ist von einem Metanephrin produzierendem Phäochromozytom auszugehen. Das Phäochromozytom wurde histologisch bestätigt. Verlauf: Postoperativ lagen die 24h-Urin und die Plasmawerte bis auf Noradrenalin im Normbereich. 6 Monate postoperativ zeigten sich im Urin und im Plasma die Katecholamine und deren Metaboliten im Normbereich (Tab. 16). Tab. 16: Diagnostik und Befunde des Verlaufes zu Fall 7. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 k. A. 23 57 36 75 Plasma: A (ng/l); Norm: <150 NA (ng/l); Norm: <450 18 669 Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe. 46 3 Ergebnisse Kasuistik 8 Quelle: Phäochromozytom-Sammlung Lübeck, Dr. Klingenberg Initialen: A. S. Jahr der Operation: 2000 Geburtsjahr: unbekannt Alter bei Operation: 41 Jahre Geschlecht: weiblich Anamnese: Bei der Patientin wurde die Diagnose der Neurofibromatose von Recklinghausen im 16. Lebensjahr gestellt. Seitdem sind mehrere Fibrome entfernt worden. Die 40-jährige Patientin präsentierte sich beim Hausarzt wegen Schwindel, Kopfschmerzen und starkem Schwitzen. Die Blutdruckwerte waren erniedrigt. Sonographisch fand sich eine Raumforderung der linken Nebenniere. Die Patientin wurde darauf zur weiteren Abklärung stationär aufgenommen. Klinischer Untersuchungsbefund: Multiple kutane und subkutane Fibrome am ganzen Körper. Hypotone Blutdruckwerte (RR <100/60 mmHg). Diagnostik und Befunde: Katecholamine waren im 24h-Urin und im Plasma erhöht. Radiologisch ließ sich der Tumor darstellen. Im MRT fanden sich des Weiteren paravertebrale Neurofibrome (Tab. 17). Tab. 17: Diagnostik und Befunde zu Fall 8. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 k. A. 88 195 255 199 Plasma: A (ng/l); Norm: <150 NA (ng/l); Norm: <450 227 1581 MRT-Abdomen: Bestätigung der Raumforderung der linken Nebenniere MIBG-Szintigraphie: Verstärktes Uptake des Radiopharmakons in der linken Nebenniere Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe. 47 3 Ergebnisse Therapie: An der Universitätsklinik Lübeck wurde der Patient mittels Flankenschnitt-Zugang adrenalektomiert. Das Phäochromozytom wurde histologisch bestätigt. Verlauf: Postoperativ zeigten sich im Urin und im Plasma eine Normalisierung der Katecholamine (Tab. 18). Die Patientin wurde 6 Monate später mit der gleichen Symptomatik wieder aufgenommen. Tumorrezidive ließen sich aber mit Laboruntersuchungen und 131-J-MIBG nicht nachweisen. Die Beschwerden persistierten bis zur nächsten stationären Aufnahme 10 Monate nach der Operation. Wiederum lagen die Laborwerte des Urins und des Plasmas im Normbereich. Eine durchgeführte FDG-Positronenemissionstomographie (PET) ergab schließlich ein verstärktes Enhancement im Beckenbereich. Der Bereich speicherte jedoch kein Jod-MIBG, so dass nicht von einem katecholaminproduzierenden Tumor auszugehen ist. Eine weitere Operation mit Resektion des Befundes wurde von der Patientin abgelehnt. Tab. 18: Verlauf zu Fall 8. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 k. A. 18 32 35 k. A. Plasma: A (ng/l); Norm: <150 NA (ng/l); Norm: <450 10 397 Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe. 48 3 Ergebnisse Kasuistik 9 Quelle: Department für Endokrinologie, Universitätsspital Basel, Dr. Walter Initialen: J.F. Jahr der Operation: 1990 Geburtsjahr: 1949 Alter bei Operation: 40 Jahre Geschlecht: weiblich Anamnese: Bei dem Patienten ist eine Neurofibromatose von Recklinghausen bekannt. Eine Hypertonie besteht seit 12 Jahren. Episoden mit Hitzegefühl, Kaltschweißigkeit und Tachykardien sind seit 2 Jahren bekannt. Klinischer Untersuchungsbefund: Blutdruck: 180/120 mmHg, Vereinzelte Café-au-lait Flecken und multiple Neurofibrome. Diagnostik und Befunde: Katecholamine im 24h-Urin stark erhöht, der Tumor war in Sonographie, CT und in der Szintigraphie sichtbar (Tab. 19). Tab. 19: Diagnostik und Befunde zu Fall 9. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 18 548 939 k. A. k. A. Sonographie: Raumforderung in der rechten Nebenniere CT: Bestätigung des Tumors von 5x5 cm Größe MIBG-Szintigraphie: Verstärkte Anreicherung im Bereich der rechten Nebenniere Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe. Therapie und Verlauf: Die Laparotomie mit Entfernung des Tumors an der rechten Nebenniere verlief komplikationslos. Das Phäochromozytom wurde histologisch bestätigt. Der Verlauf war unauffällig. 49 3 Ergebnisse Kasuistik 10 Quelle: Department für Endokrinologie, Universitätsklinik Padua, Prof. Opocher Initialen: M.-C.A. Jahr der Operation: 1988 Geburtsjahr: 1952 Alter bei Operation: 36 Jahre Geschlecht: weiblich Anamnese: Bei dem Patienten ist eine Neurofibromatose von Recklinghausen bekannt. Familienanamnese: Bei dem Vater, der Schwester und dem Neffen ist eine Neurofibromatose Typ 1 bekannt. Klinischer Untersuchungsbefund: Vereinzelte Café-au-lait Flecken am Gesicht, wenige Neurofibrome. Die Patientin wurde wegen erhöhten Blutdrucks stationär aufgenommen. Diagnostik und Befunde: Es liegen keine Informationen zur Diagnostik, Bildgebung und deren Befunde vor. Therapie: Der Blutdruck wurde mit ß-Blockern kontrolliert. Die Entfernung des Tumors der rechten Nebennieren erfolgte laparotomisch und verlief komplikationslos. Das Phäochromozytom wurde histologisch bestätigt. 50 3 Ergebnisse Kasuistik 11 Quelle: Department für Endokrinologie, Universitätsklinik Padua, Prof. Opocher Initialen: M.-M.C. Jahr der Operation: 2002 Geburtsjahr: 1954 Alter bei Operation: 48 Jahre Geschlecht: weiblich Anamnese: Bei Geburt bestanden bei der Patientin Deformationen des Gesichtes. Die Diagnose der Neurofibromatose Typ 1 wurde bei der Patientin erst mit 23 Jahren durch Auftreten von Neurofibromen gestellt. Aufgrund einer akuten Pankreatitis wurde die Patientin 2002 stationär aufgenommen. Familienanamnese: Bei der Mutter ist eine Neurofibromatose Typ 1 bekannt. Klinischer Untersuchungsbefund: Die Patientin war asymptomatisch. Vereinzelte Neurofibrome und Café-au-lait Flecken. Diagnostik und Befunde: Das Adrenalin im 24h-Urin war leicht erhöht, der Tumor ließ sich in MRT und in der Szintigraphie darstellen (Tab. 20). Tab. 20: Diagnostik und Befunde zu Fall 11. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 k. A. 32 79 k. A. k. A. MRT-Abdomen: Bestätigung des Tumors im Bereich beider Nebennieren von 2 cm (links) und 6 cm Größe (rechts) MIBG-Szintigraphie: Verstärkte Anreicherung im Bereich beider Nebennieren Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe. 51 3 Ergebnisse Therapie: Laparoskopische Entfernung des Tumors an beiden Nebennieren verlief komplikationslos. Die Phäochromozytome wurden histologisch bestätigt. Verlauf: Alle Kontrolluntersuchungen waren unauffällig. Bei der Patientin besteht weiterhin eine Tachykardie. 3 Ergebnisse 52 Kasuistik 12 Quelle: Department für Endokrinologie, Universitätsklinik Padua, Prof. Opocher Initialen: I.-R.C. Jahr der Operation: 1996 Geburtsjahr: 1953 Alter bei Operation: 43 Jahre Geschlecht: weiblich Anamnese: Die Neurofibromatose von Recklinghausen wurde im Alter von 13 Jahren bei einer zervikalen Fibromektomie diagnostiziert. Im Laufe der Jahre erfolgten mehrere Entfernungen von Neurofibromen. Weitere Krankheitsanamnese: 1. 1969 Spontanpneumothorax 2. 1973 Gastroduodenale Ulzera 3. 1982 Diskushernie L4-L5 An die Universitätsklinik Padua kam die Patientin 1996 schließlich mit akut aufgetretenem Herzklopfen, Vertigo und Kopfschmerzen. Familienanamnese: Der Vater und der Bruder wiesen eine Neurofibromatose Typ 1 und Hypertonie auf. Der Vater ist 59-jährig an einem Blasenkarzinom verstorben, der Bruder 30-jährig an einem zervikalen Fibrosarkom. Klinischer Untersuchungsbefund: Am Intugement typische Zeichen einer Neurofibromatose Typ 1, Blutdruck: 165/110 mmHg. Diagnostik und Befunde: Auffällig im 24h-Urin war die starke Erhöhung der Metanephrine. Im CT und in der Szintigraphie war der Tumor zu erkennen (Tab. 21). 53 3 Ergebnisse Tab. 21: Diagnostik und Befunde zu Fall 12. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 k. A. 150 82 1800 k. A. CT: 4 cm großer Tumor der linken Nebenniere MIBG-Szintigraphie: Vermehrte Speicherung im Bereich der linken Nebenniere Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe. Therapie: Die Einstellung der RR-Werte auf zufrieden stellende Bereiche erfolgte mit Cardula (aBlocker). Der Patient wurde laparatomiert und der Tumor mittels Adrenalektomie entfernt. Das Phäochromozytom wurde histologisch bestätigt. Verlauf: Die Katecholamine des 24h-Urins befanden sich postoperativ im Normbereich. 54 3 Ergebnisse Kasuistik 13 Quelle: Department für Endokrinologie, Universitätsklinik Padua, Prof. Opocher Initialen: A.C. Jahr der Operation: 2002 Geburtsjahr: 1967 Alter bei Operation: 35 Jahre Geschlecht: weiblich Anamnese: Die Patientin leidet an einer Neurofibromatose von Recklinghausen. Weiterhin sind bei der Patientin Granulosazelltumoren der Haut und der Blase bekannt. Klinischer Untersuchungsbefund: Die Patientin war asymptomatisch. Café-au-lait Flecken und multiple Neurofibrome am Intugement. Diagnostik und Befunde: Die Untersuchungen ergaben erhöhte Werte für Metanephrine und Normetanephrine im 24-hUrin. CT und MRT zeigten den Tumor (Tab. 22). Tab. 22: Diagnostik und Befunde zu Fall 13. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 k. A. k. A. k. A. ↑ ↑ CT: Raumforderung im Bereich der rechten Nebenniere MRT-Abdomen: Bestätigung des Tumors im Bereich der rechten Nebenniere (2,5cm) Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe. Therapie und Verlauf: Die laparoskopische Entfernung des Tumors der rechten Nebenniere verlief komplikationslos. Das Phäochromozytom wurde histologisch bestätigt. Der Patientin geht es postoperativ gut. Alle Katecholaminkontrolluntersuchungen waren unauffällig. 55 3 Ergebnisse Kasuistik 14 Quelle: Department für Endokrinologie, Universitätsklinik Rom, Prof. Letizia Initialen: L.d.A. Jahr der Operation: 2001 Geburtsjahr: 1958 Alter bei Operation: 43 Jahre Geschlecht: männlich Anamnese: Bei dem Patienten besteht eine Neurofibromatose von Recklinghausen. Zudem besteht eine chronische Autoimmunthyreoiditis. Er wurde wegen einer hypertensiven Krise (RR: 200/110 mmHg) eingeliefert. An Symptomen sind Kopfschmerzen, Palpitationen und starkes Schwitzen bekannt. Familienanamnese: Beim Vater besteht eine Neurofibromatose Typ 1. Klinischer Untersuchungsbefund: Adipöser Patient (BMI 33,4) mit leichter Hypertonie (150/90mmHg) mit paroxysmalen Krisen. Café-au-lait Flecken und multiple Neurofibrome am ganzen Körper. „Freckling“ im Bereich der Achselhöhlen. An den Augen sind Lisch-Knoten vorhanden. Diagnostik und Befunde: In allen radiologischen Bildgebungen ließ sich den Tumor darstellen (Tab.23). Tab. 23: Diagnostik und Befunde zu Fall 14. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 17 k. A. k. A. 460 k. A. Sonographie: Raumforderung in der rechten Nebenniere CT: Bestätigung des Tumors von 2 cm Größe in der rechten Nebenniere MRT-Abdomen: Bestätigung des Tumors von 2 cm Größe in der rechten Nebenniere MIBG-Szintigraphie: Anreicherung im Bereich der rechten Nebenniere (Abb. 27) Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe; 3 Ergebnisse 56 Abb. 27 MIBG-Szintigraphie zu Fall 14. Ansicht von hinten. Der weiße Pfeil markiert das Phäochromozytom, der rote Pfeil die Leber und der gelbe Pfeil die Milz.Mit freundlicher Genehmigung von Prof. Letizia, Universitätsklinik Rom, Italien. Therapie: Präoperativ wurden 2 mg/die Doxazosin zur Behandlung des Hypertonus verabreicht. Darauf laparoskopische Entfernung des Tumors der rechten Nebenniere. Das Phäochromozytom wurde histologisch bestätigt. Verlauf: Alle Kontrolluntersuchungen ergaben keine Auffälligkeiten. 57 3 Ergebnisse Kasuistik 15 Quelle: Department für Endokrinologie, Universitätsklinik Rom, Prof. Letizia Initialen: I.C. Jahr der Operation: 2004 Geburtsjahr: 1971 Alter bei Operation: 33 Jahre Geschlecht: männlich Anamnese: Bei dem Patienten ist eine Neurofibromatose von Recklinghausen bekannt. Er wurde von seinem Dermatologen in die Ambulanz wegen seit einem Monat bestehenden Phasen mit massiver Blutdruckerhöhung überwiesen. Es bestanden Palpitationen. Klinischer Untersuchungsbefund: Blutdruck: 175/105 mmHg, Puls 80/min. Café-au-lait Flecken und multiple Neurofibrome am ganzen Körper. „Freckling“ im Bereich der Achselhöhlen. Diagnostik und Befunde: Sonographie und CT ließen den Tumor erkennen (Tab. 24). Tab. 24: Diagnostik und Befunde zu Fall 15. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 15 k. A. k. A. 388 k. A. Sonographie: Raumforderung in der rechten Nebenniere CT: Bestätigung des Tumors von 3,5x3 cm Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe. Therapie und Verlauf: Präoperativ wurden 4 mg/die Doxazosin zur Behandlung des Hypertonus verabreicht. Die laparoskopische Entfernung des Tumors an der rechten Nebenniere verlief komplikationslos. Das Phäochromozytom wurde histologisch bestätigt. Alle Kontrolluntersuchungen ergaben keine Auffälligkeiten. 58 3 Ergebnisse Kasuistik 16 Quelle: Department für Endokrinologie, Universitätsklinik Rom, Prof. Letizia Initialen: M.M. Jahr der Operation: 2004 Geburtsjahr: 1962 Alter bei Operation: 42Jahre Geschlecht: männlich Anamnese: Bei dem Patienten ist eine Neurofibromatose von Recklinghausen bekannt. Es besteht eine Osteoporose. Er trägt seit einer Amputation des rechten Unterschenkels wegen einer Pseudarthrose des Kniegelenkes eine Prothese. Er wurde von seinem Dermatologen in die Ambulanz mit seit sechs Monaten bestehender asymptomatischer Hypertonie (RR: 150/90 mmHg) überwiesen. Klinischer Untersuchungsbefund: Blutdruck: 145/90 mmHg. Café-au-lait Flecken und multiple Neurofibrome am Rumpf, sowie „Freckling“ im Bereich der Achselhöhlen. An den Augen sind Lisch-Knoten und eine hypertensive Retinopathie Grad 2 vorhanden. Diagnostik und Befunde: Die VMA und die Metanephrine waren im 24h-Urin leicht erhöht. MRT und Szintigraphie zeigten eine Raumforderung (Tab. 25). Tab. 25: Diagnostik und Befunde zu Fall 16. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 12 k. A. k. A. 398 k. A. MRT-Abdomen: Tumor der linken Nebenniere von 2x3 cm Größe MIBG-Szintigraphie: Deutliches Uptake im Bereich der linken Nebenniere (Abb. 28) Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe. 59 3 Ergebnisse Therapie: Präoperativ wurden 4 mg Doxazosin und 80 mg/die Propanolol zur Behandlung des Hypertonus verabreicht. Der Patient wurde laparatomiert und der Tumor der linken Nebenniere entfernt. Intraoperativ wurde ein intestinales Neurofibrom entdeckt (Abb. 29). Perioperativ gab es keine Komplikationen. Das Phäochromozytom wurde histologisch bestätigt. Verlauf: Alle Kontrolluntersuchungen ergaben keine Auffälligkeiten. Abb. 28 MIBG-Szintigraphie zu Fall 16. Ansicht posterior-anterior. Sichtbare Mehranreicherung im Bereich der linken Nebenniere (weißer Pfeil). Der schwarze Pfeil markiert die Leber. Mit freundlicher Genehmigung von Prof. Letizia, Universitätsklinik Rom, Italien. Abb. 29: Neurofibrom des Dünndarms von Fall 16, das während Operation entdeckt wurde. Mit freundlicher Genehmigung von Prof. Letizia, Universitätsklinik Rom, Italien. 3 Ergebnisse 60 Kasuistik 17 Quelle: Department für Endokrinologie, Universitätsklinik Rom, Prof. Letizia Initialen: F.P. Jahr der Operation: 2001 Geburtsjahr: 1964 Alter bei Operation: 37 Jahre Geschlecht: männlich Anamnese: Bei dem Patienten ist eine Neurofibromatose von Recklinghausen bekannt. Zudem leidet der Patient an einer multiplen endokrinen Neoplasie Typ 2A (MEN 2A; Sipple-Syndrom) mit primärem Hyperparathyreoidismus (bedingt durch ein Adenom der Nebenschilddrüse) und einem C-Zellkarzinom. Er wurde von seinem Dermatologen in die Ambulanz wegen seit einem Monat bestehender Hypertension überwiesen. Paroxysmale Krisen, Palpitationen, Kopfschmerzen, sowie Angstund Schweißzustände sind bekannt. Der Dermatologe hatte den Bluthochdruck mit ßBlockern und ACE-Hemmern nicht senken können. Klinischer Untersuchungsbefund: Patient mit konstanter Hypertension (160/120 mmHg), Puls 120/min. Café-au-lait Flecken und multiple Neurofibrome am ganzen Körper, sowie „Freckling“ im Bereich der Achselhöhlen (Abb. 30). Die Augenuntersuchung zeigte Lisch-Knoten und eine hypertensive Retinopathie Grad 2. Abb. 30: Links zu sehen ist das klinisches Bild zu Patient 17. Man sieht multiple Neurofibrome, einen Café-au-Lait Fleck. Mit freundlicher Genehmigung von Prof. Letizia, Universitätsklinik Rom, Italien. 61 3 Ergebnisse Diagnostik und Befunde: Die Werte für Vanillinmandelsäure und Metanephrine waren im 24h-Urin erhöht. Alle bildgebenden Untersuchungen ließen die Raumforderung erkennen (Tab. 26). Tab. 26: Diagnostik und Befunde zu Fall 17. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 15 k. A. k. A. 415 k. A. Sonographie: Raumforderung in der linken Nebenniere CT: Bestätigung des Tumors von 4,5x3 cm Größe MRT-Abdomen: Bestätigung des Tumors von 4,5x3 cm Größe MIBG-Szintigraphie: • Verstärkte Anreicherung der linken Nebenniere (Abb. 31 a) • Adenom des rechten Schilddrüsenlappens (Abb. 31 b) Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe. a) b) Abb. 31 a und b: a) MIBG-Szintigraphie zu Fall 17. Aufnahme posterior-anterior. Verstärkte Anreicherung in der linken Nebenniere (weißer Pfeil). Der rote Pfeil zeigt die Leber. b) MIBISzintigraphie der Schilddrüse (grün umrahmt). Der rote Fleck im rechten Drüsenlappen entspricht dem Adenom (Pfeil). Der helle Bereich am unteren Bildrand stellt die Lunge dar. Mit freundlicher Genehmigung von Prof. Letizia, Universitätsklinik Rom, Italien. Therapie und Verlauf: Präoperativ wurden 4 mg/die Doxazosin und 80 mg/die Propanolol zur Behandlung des Hypertonus verabreicht. Die laparoskopische Entfernung des Tumors an der linken Nebenniere verlief komplikationslos. Das Phäochromozytom wurde histologisch bestätigt. Der weitere Verlauf war unauffällig. 3 Ergebnisse 62 Kasuistik 18 Quelle: Department Endokrinologie, Hopital Barbois, Universität Nancy, Dr. Muresan Initialen: L.F. Jahr der Operation: 2004 Geburtsjahr: 1956 Alter bei Operation: 47 Jahre Geschlecht: weiblich Anamnese: Bei der Patientin waren schon immer disseminierte Veränderungen der Haut im Sinne einer Neurofibromatose von Recklinghausen bekannt (Abb. 32). Im Laufe der Jahre erfolgten mehrere Entfernungen von Neurofibromen. Weitere Krankheitsanamnese: • 1969 Operation einer großen Meningocele • 1977 Entfernung eines gutartigen Tumors aus der Lunge (Histologie unbekannt) • 1979 Sectio caesarea • Seit 2001 insulinpflichtiger Typ 2 Diabetes • Hyperlipidämie • Hypertonie Familienanamnese: Es sind Hautveränderungen im Sinne einer Neurofibromatose Typ 1 bei der Tochter bekannt. Klinischer Untersuchungsbefund: Multiple Neurofibrome am ganzen Körper, vereinzelte Café-au-lait Flecken, inguinales „Freckling“ (Abb. 32 a und b). Die Patientin präsentierte sich mit der Trias Kopfschmerzen, Schweißzuständen und Palpitationen. Zusätzlich bestand starke Hypertonie. 63 a) 3 Ergebnisse b) Abb. 32 a und b: Klinisches Bild zu Fall 18 mit multiple Neurofibromen und vereinzelten Caféau-lait Flecken. Mit freundlicher Genehmigung von Dr. Muresan, Universität Nancy, Frankreich. Diagnostik und Befunde: Katecholamine und deren Metabolite waren im 24h-Urin stark erhöht. Die CT und die Szintigraphie zeigten Tumoren (Tab.27). Tab. 27: Diagnostik und Befunde zu Fall 18. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 k. A. ↑↑ ↑↑ ↑↑ ↑↑ CT: 4 cm große Raumforderung in rechten Nebenniere (Abb. 33a) MRT-Abdomen: 3,5 cm große Raumforderung in linken Nebenniere (Abb.33b) MIBG-Szintigraphie: Aufnahme des Radiopharmakons in beiden Nebennieren Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe. 3 Ergebnisse a) 64 b) Abb. 33 a und b: Fall 18: a) CT-Abdomen mit Raumforderung im Bereich der rechten Nebenniere (Pfeil). b) MRT-Abdomen zu deutlichen Raumforderung im Bereich der linken Nebenniere (Pfeil). Mit freundlicher Genehmigung von Dr. Muresan, Universität Nancy, Frankreich. Therapie: Im Juli 2004 Operation des linken Nebennierentumors, wonach die erhöhten Katecholaminund Calcitoninwerte persistierten. Im November 2004 erfolgte die Exzision des rechten Nebennierentumors. Die Phäochromozytome wurden jeweils histologisch bestätigt. Verlauf: Nach Entfernung beider Phäochromozytome normalisierten sich der Blutdruck, die Katecholamine, sowie das Calcitonin. Zusätzlich kam es zu einer starken Reduktion des Bedarfs an Insulin. 65 3 Ergebnisse Kasuistik 19 Quelle: Department Endokrinologie St. Gorge Hospital London, Prof. MacGregor Initialen: D.K. Jahr der Operation: 1987 Geburtsjahr: 1944 Alter bei Operation: 43 Jahre Geschlecht: weiblich Anamnese: Bei dem Patienten waren schon immer disseminierte Veränderungen der Haut im Sinne einer Neurofibromatose von Recklinghausen bekannt. 9 Jahre nach dem Phäochromozytom wurde ein linksseitiges Mammakarzinom diagnostiziert. Familienanamnese: Screening der Familie ergab erhöhte Vanillinmandelsäure bei der Mutter 37 mg/die (Norm: <8 mg/die). Klinischer Untersuchungsbefund: Multiple Neurofibrome, Café-au-lait Flecken. Diagnostik und Befunde: Erhöhte Werte für VMA, Adrenalin und Noradrenalin im 24h-Urin. Die Sonographie und die CT ließen den Tumor erkennen (Tab.28). Tab. 28: Diagnostik und Befunde zu Fall 19. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 14 185 321 k. A. k. A. Sonographie: Raumforderung im Bereich der rechten Nebenniere CT: Bestätigung des Tumors der rechten Nebenniere von 3 cm Größe Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe. 66 3 Ergebnisse Therapie: Der Tumor wurde mittels Laparatomie und Adrenalektomie entfernt. Verlauf: Es wurde post-operativ jährlich eine Kontrolluntersuchung anhand 24h-Urins durchgeführt (Tab. 29): Tab. 29: Verlauf zu Fall 19. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 7 7 86 k. A. k. A. Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe 67 3 Ergebnisse Kasuistik 20 Quelle: Phäochromozytom-Register Warschau, Prof. Januszewicz Initialen: J.K. Jahr der Operation: 1997 Geburtsjahr: 1955 Alter bei Operation: 42 Jahre Geschlecht: männlich Anamnese: Bei dem Patienten waren disseminierte Veränderungen der Haut im Sinne einer Neurofibromatose von Recklinghausen bekannt. 1997 wurde der 42-jährige Patient wegen paroxysmaler Hypertonie (bis 240/110 mmHg) Herzpalpitationen und Schweißausbrüchen stationär aufgenommen. Der Patient ist seit seiner frühen Kindheit am rechten Auge blind. Klinischer Untersuchungsbefund: Patient mit multiplen subkutanen Fibromen am Kopf, Rumpf und der unteren Extremität. Am Integument mehrere diskrete Café-au-lait Flecken und Vitiligo. Lisch-Knoten der Iris im linken Auge. Großer Tumor (ca. 30 cm) über dem rechten Becken (Abb. 34). Diagnostik und Befunde: Adrenalin und Noradrenalin waren im 24h-Urin erhöht. In Sonographie, CT und in der Szintigraphie war der Tumor sichtbar (Tab. 30). Tab. 30: Diagnostik und Befunde zu Fall 20. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 k. A. 377 331 k. A. k. A. Sonographie: Raumforderung (37x31x32 mm) oberhalb der linken Niere CT: Bestätigung des Tumors der linken Nebenniere von 3cm Größe MIBG-Szintigraphie: Erhöhte Akkumulation des Radionukleotids in der linken NN Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe. 68 3 Ergebnisse Abb. 34 a-c: Klinisches Bild zu Fall 20. Am Intugement Fibrome, multiple Café-au-lait Flecken und Vitiligo. Deutlich sichtbarer Tumor über der rechten Hüfte. Mit freundlicher Genehmigung von Prof. Januszewicz, Warschau, Polen. Therapie: Im Juni 1997 wurde eine Laparotomie mit linksseitiger Adrenalektomie durchgeführt. Das Phäochromozytom wurde histologisch bestätigt. Verlauf: Es liegen keine Angaben zum Verlauf vor. 69 3 Ergebnisse Kasuistik 21 Quelle: Phäochromozytom-Register Warschau, Prof. Januszewicz Initialen: W.K. Jahr der Operation: 1990 Geburtsjahr:1944 Alter bei Operation: 46 Jahre Geschlecht: weiblich Anamnese: Bei dem Patienten ist eine Neurofibromatose von Recklinghausen bekannt. Es sind keine weiteren Informationen vorhanden. Klinischer Untersuchungsbefund: Der Patient weist Zeichen der Neurofibromatose Typ 1 auf. RR:124/78 mmHg. Diagnostik und Befunde: Es liegen keine Informationen zur Katecholaminbestimmung, zu radiologischen oder nuklearmedizinischen Diagnostik vor. Therapie und Verlauf: 1990 wurde das Phäochromozytom mittels Laparatomie und Adrenalektomie entfernt. Der Tumor war in der linken Nebenniere lokalisiert. Das Phäochromozytom wurde histologisch gesichert. Der Patient stellte sich zur Kontrolluntersuchung 1996 in Warschau vor. 24h-Blutdruck: 130/80 mmHg (Tab. 31). Tab. 31: Postoperativer Verlauf zu Fall 21. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 k. A. 2 12 k. A. k. A. CT: ohne pathologischen Befund Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; k. A. = keine Angabe. 70 3 Ergebnisse Kasuistik 22 Quelle: Phäochromozytom-Register Warschau, Prof. Januszewicz Initialen: K.P. Jahr der Operation: 1992 Geburtsjahr:1940 Alter bei Operation: 52 Jahre Geschlecht: weiblich Anamnese: Bei dem Patienten ist eine Neurofibromatose von Recklinghausen bekannt. Es sind keine weiteren Informationen vorhanden. Klinischer Untersuchungsbefund: Der Patient hat Zeichen der Neurofibromatose Typ 1. RR: 110/70 mmHg. Diagnostik und Befunde: Es liegen keine Informationen zur Katecholaminbestimmung, zu radiologischen oder nuklearmedizinischen Diagnostik vor. Therapie: 1992 wurde das Phäochromozytom mittels Laparatomie und Adrenalektomie entfernt. Der Tumor war in der linken Nebenniere lokalisiert. Verlauf: 24h-Blutdruck: 113/75 mmHg. Die Kontrolluntersuchungen waren unauffällig (Tab. 32). Tab. 32: Postoperativer Verlauf zu Fall 22. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 k. A. 4 31 k. A. k. A. CT: ohne pathologischen Befund Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; k. A. = keine Angabe. 71 3 Ergebnisse Kasuistik 23 Quelle: Phäochromozytom-Register Warschau, Prof. Januszewicz Initialen: W.S. Jahr der Operation: 2003 Geburtsjahr: 1942 Alter bei Operation: 61 Jahre Geschlecht: männlich Anamnese: Bei dem Patienten sind disseminierte Veränderungen der Haut im Sinne einer Neurofibromatose von Recklinghausen bekannt. 2003 wurde der 61jährige Patient im Department of Hypertension, Institute of Cardiology, Warschau, wegen Hypertonie aufgenommen. Sonographisch war zuvor ein Tumor der linken Nebenniere festgestellt worden. Die Hypertonie war seit 10 Jahren bekannt. Der Patient gab zudem intermittierende Kopfschmerzen, Schwitzen und Palpitationen an. Familienanamnese: Die Tochter hat Hautveränderungen im Sinne einer Neurofibromatose Typ 1. Klinischer Untersuchungsbefund: RR: 230/170 mmHg. Multiple kleine, subkutane Fibrome am ganzen Körper. Am Integument mehrere, mehr als 15 mm große, diskrete Café-au-lait Flecken. Axillär und inguinal deutlich sichtbares „Freckling“. Diagnostik und Befunde: Auffällig ist die enorme Erhöhung der Metanephrine im 24h-Urin. Sonographie, CT und Szintigraphie ließen den Tumor erkennen (Tab. 33). 72 3 Ergebnisse Tab. 33: Diagnostik und Befunde zu Fall 23. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 k. A. 102 459 34201 k. A. Sonographie: Bestätigung der Raumforderung in der Nebenniere CT: • 13 cm große, kalzifizierende Raumforderung der linken Nebenniere • vergrößerte Prostata mit Verdacht eines Prostata-Karzinoms MIBG-Szintigraphie: starke Aufnahme des Radiopharmakons im Bereich der linken Nebenniere Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; Plasma: A = Adrenalin, NA = Noradrenalin; k. A. = keine Angabe. Therapie: Präoperativ wurde für zwei Wochen die Kombination eines α-Adrenoblockers (Phenoxybenzamin 40 mg/die) mit einem β-Blocker (Metoprolol 75 mg/die) eingesetzt. In Warschau wurde dann eine Laparatomie mit linksseitiger Adrenalektomie durchgeführt. Dabei wurde ein zweiter Tumor exzidiert, der sich histologisch als Lymphknotenmetastase des Prostata-Karzinoms herausstellte. Das Phäochromozytom wurde histologisch bestätigt. Verlauf: Postoperativ ergaben sich keine Komplikationen. Die Katecholamine des 24h-Urins lagen nach der Operation bis auf die Metanephrine im Normbereich (Tab. 34). Tab. 34: Diagnostik und Befunde zu Fall 23. Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 k. A. 3 10 245 k. A. Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; k. A. = keine Angabe. 73 3 Ergebnisse Kasuistik 24 Quelle: Phäochromozytom-Register Warschau, Prof. Januszewicz Initialen: J.S. Jahr der Operation: 1993 Geburtsjahr: 1955 Alter bei Operation: 38 Jahre Geschlecht: männlich Anamnese: Es liegen keine Informationen zur Anamnese vor. Familienanamnese: Es liegen keine Informationen zur Familienanamnese vor. Klinischer Untersuchungsbefund: Es liegen keine Informationen zum Untersuchungsbefund vor. Diagnostik und Befunde: Es liegen keine Informationen zur Diagnostik, Bildgebung und deren Befunde vor. Therapie: 1993 erfolgte die Laparatomie mit Entfernung des Tumors und der rechten Nebenniere. Verlauf: Es liegen keine Informationen zum Verlauf vor. 74 3 Ergebnisse Kasuistik 25 Quelle Phäochromozytom-Register Warschau, Prof. Januszewicz Initialen: J.W. Jahr der Operation: 1986 Geburtsjahr:1936 Alter bei Operation: 50 Jahre Geschlecht: männlich Anamnese: Bei dem Patienten ist eine Neurofibromatose von Recklinghausen bekannt. Es sind keine weiteren Informationen vorhanden. Klinischer Untersuchungsbefund: Der Patient hat Zeichen der Neurofibromatose Typ 1. RR 140/80 mmHg. Diagnostik und Befunde: Es liegen keine Informationen zur Katecholaminbestimmung, zur radiologischen oder nuklearmedizinischen Diagnostik vor. Therapie: 1986 wurde das Phäochromozytom mittels Laparatomie und Adrenalektomie entfernt. Der Tumor war in der linken Nebenniere lokalisiert. Verlauf: Der Patient stellte sich zur Kontrolluntersuchung 1996 in Warschau vor. Die Katecholamine befanden sich im Normbereich (Tab. 35). 24h-Blutduck: 150/85 mmHg. Tab. 35: Verlauf zu Fall 25: Diagnostik Befund 24h-Urin: VMA (mg/die) Norm: <8 A (μg/die) Norm: <20 NA (μg/die) Norm: <80 M (μg/die) Norm: <80 NM (μg/die) Norm: <120 k. A. 2 12,3 k. A. k. A. Urin: VMA = Vanillinmandelsäure, A = Adrenalin, NA = Noradrenalin, M = Metanephrine, NM = Normetanephrine; k. A. = keine Angabe. 75 3 Ergebnisse Kasuistik 26 Quelle: Phäochromozytom-Register Warschau, Prof. Januszewicz Initialen: W.W. Jahr der Operation: 1990 Geburtsjahr: 1957 Alter bei Operation: 33 Jahre Geschlecht: männlich Anamnese: Bei dem Patienten ist eine Neurofibromatose von Recklinghausen bekannt. Es sind keine weiteren Informationen vorhanden. Klinischer Untersuchungsbefund: Es liegen keine Informationen zum Untersuchungsbefund vor. Diagnostik und Befunde: Es liegen keine Informationen zur Katecholaminbestimmung, zur radiologischen oder nuklearmedizinischen Diagnostik vor. Therapie: 1990 wurden dem damals 33jährigen Patienten zwei Phäochromozytome mittels Laparatomie und beidseitiger Adrenalektomie entfernt. Verlauf: Es liegen keine Informationen zum Verlauf vor. 76 3 Ergebnisse 3.2 Zusammenfassung der klinischen Daten Tab. 36: Allgemeine Patientendaten. Fall Geschl. Geb.Jahr OP Jahr OP Alter s/f Herkunftsland Ort Klinik 1 w 1951 1995 44 f Deutschland Freiburg 2 m 1957 2003 46 f Deutschland Freiburg 3 m 1980 1994 14 s Deutschland Essen 4 w 1972 2004 32 s Deutschland Essen 5 w 1985 1985 16 f Deutschland Leipzig 6 w 1949 1999 52 s Deutschland Lübeck 7 w 1939 1999 60 s Deutschland Lübeck 8 w 1959 2000 41 s Deutschland Lübeck 9 w 1949 1990 40 s Schweiz Basel 10 w 1952 1988 36 f Italien Padua 11 w 1954 2002 48 f Italien Padua 12 w 1953 1996 43 s Italien Padua 13 w 1967 2002 35 s Italien Padua 14 m 1958 2001 43 f Italien Rom 15 m 1971 2004 33 s Italien Rom 16 m 1962 2004 42 s Italien Rom 17 m 1964 2001 37 s Italien Rom 18 w 1956 2004 48 f Frankreich Nancy 19 w 1944 1987 43 s Großbritannien London 20 m 1955 1997 42 s Polen Warschau 21 w 1944 1990 46 s Polen Warschau 22 w 1940 1992 52 k. A. Polen Warschau 23 m 1942 2003 61 f Polen Warschau 24 m 1955 1993 38 k. A. Polen Warschau 25 m 1936 1986 50 k. A. Polen Warschau 26 m 1957 1990 33 k. A. Polen Warschau s = sporadisch, f = familiär; k. A. = keine Angabe. 77 3 Ergebnisse Tab. 37: Typische NF1 Zeichen und Phäochromozytom-Symptomatik. Fall Café-auKutane Freckling Augen lait Fibrome Flecken RR Palpit. Schwitzen Kopf schmerz. 1 pos pos neg neg pos neg neg neg 2 pos pos neg neg pos neg neg neg 3 pos pos pos Lisch pos neg neg pos 4 pos pos pos k. A. pos pos pos pos 5 pos neg neg Gliom pos neg neg pos 6 pos neg neg neg pos pos pos pos 7 pos pos neg neg pos pos pos pos 8 pos pos neg neg neg pos pos pos 9 pos pos neg neg pos pos pos neg 10 pos pos k. A. neg pos k. A. k. A. k. A. 11 pos pos k. A. neg neg neg neg neg 12 pos pos neg neg pos pos neg pos 13 pos pos pos neg neg neg neg neg 14 pos pos pos Lisch pos pos pos pos 15 pos pos pos neg pos pos neg neg 16 pos pos pos Lisch pos neg neg neg 17 pos pos pos Lisch pos pos pos pos 18 pos pos pos neg pos pos pos pos 19 pos pos pos neg neg neg pos pos 20 pos pos pos Lisch pos pos pos pos 21 k. A. k. A. k. A. k. A. k. A. k. A. k. A. k. A. 22 k. A. k. A. k. A. k. A. k. A. k. A. k. A. k. A. 23 pos pos k. A. neg pos pos pos pos 24 k. A. k. A. k. A. k. A. k. A. k. A. k. A. k. A. 25 k. A. k. A. k. A. k. A. k. A. k. A. k. A. k. A. 26 k. A. k. A. k. A. k. A. k. A. k. A. k. A. k. A. k. A. = keine Angabe. 78 3 Ergebnisse Tab. 38: Phäochromozytom-spezifische Befunde I. Fall 1 CT MRT MIBG pos 2 U-A <20 μg/d UNA <80 VMA 1-8 mg/d U-M <80 μg/d U-NM <120 μg/d Pl-A <150 ng/l Pl-NA <450 ng/l pos pos 42 81 18 - - - - pos pos 13 50 - 1255 1717 - - 3 pos pos pos ↑ ↑ 458 ↑ ↑ 5000 2700 4 pos pos pos 186 340 - - - - - 5 pos pos pos - 1465 - 286 8879 92 17914 6 pos pos pos 45 1392 - 426 2585 180 10392 7 pos pos pos 28 36 - 504 102 16 432 8 pos pos pos 88 195 - 255 199 227 1581 9 pos - pos 548 939 18 - - 4 8 10 k. A. k. A. k. A. k. A. k. A. k. A. k. A. k. A. k. A. k. A. 11 pos pos pos k. A. k. A. k. A. k. A. k. A. 32 79 12 pos - pos 150 82 5 1800 - - - 13 pos pos k. A. k. A. k. A. k. A. ↑ ↑ k. A. k. A. 14 pos pos pos - - 17 460 - - - 15 pos pos - - - 16 388 - - - 16 - pos pos - - 12 398 - - - 17 pos pos pos - - 15 415 - - - 18 pos pos pos ↑ ↑ 2692 926 ↑ ↑ 19 pos - - 185 321 14 - - - - 20 pos k. A. pos 377 331 k. A. k. A. k. A. k. A. k. A. 21 k. A. k. A. k. A. k. A. k. A. k. A. k. A. k. A. k. A. k. A. 22 k. A. k. A. k. A. k. A. k. A. k. A. k. A. k. A. k. A. k. A. 23 pos - pos 102 459 - 34201 - - - 24 k. A. k. A. k. A. k. A. k. A. k. A. k. A. k. A. k. A. k. A. 25 k. A. k. A. k. A. k. A. k. A. k. A. k. A. k. A. k. A. k. A. 26 k. A. k. A. k. A. k. A. k. A. k. A. k. A. k. A. k. A. k. A. 24-Urin: Adrenalin (U-A), Noradrenalin (U-NA), Vanillinmandelsäure (VMA), Metanephrine (U-M) und Normetanephrine (U-NM); Plasma: Adrenalin (Pl-A) und Noradrenalin (Pl-NA). ↑ = erhöht ohne genaue Angabe, k. A. = keine Angabe, - = nicht durchgeführt. 79 3 Ergebnisse Tab. 39: Phäochromozytom-spezifische Befunde II. Fall Zahl der Lokalisation Art der Größe Metastasen Histol. Dignität Tumoren adrenal Operation cm Lokalisation 1 2 beidseits Laparatomie 4/2 pos keine benigne 2 2 beidseits Endoskopie 4/3 pos keine benigne 3 1 links Laparatomie 7 pos Lunge, Knochen maligne 4 1 links Endoskopie 6 pos keine benigne 5 1 rechts Laparatomie 5 pos Lunge, Knochen maligne 6 1 rechts Laparatomie 9 pos keine benigne 7 1 links Laparatomie 4 pos keine benigne 8 1 links Laparatomie 3 pos keine 9 1 rechts Laparatomie 5 pos keine Frgl. infiltrativ benigne 10 1 rechts Laparatomie 2 pos keine benigne 11 2 beidseits Endoskopie 6/2 pos keine benigne 12 1 links . Laparatomie 4 pos keine benigne 13 1 rechts Endoskopie 3 pos keine benigne 14 1 rechts . Endoskopie 2 pos keine benigne 15 1 rechts Endoskopie 3 pos keine benigne 16 1 links Laparatomie 3 pos keine benigne 17 1 links Endoskopie 4 pos keine benigne 18 2 beidseits Laparatomie 4/3 pos keine benigne 19 1 rechts Laparatomie 3 pos keine benigne 20 1 links Laparatomie 3 pos keine benigne 21 1 links Laparatomie k. A. pos keine benigne 22 1 links Laparatomie k. A. pos keine benigne 23 1 links Laparatomie 13 pos keine benigne 24 1 links Laparatomie k. A. pos keine benigne 25 1 rechts Laparatomie k. A. pos keine benigne 26 2 beidseits Laparatomie k. A. pos keine benigne k. A. = keine Angabe. 80 3 Ergebnisse 3.3 Mutationen des NF1-Gens Im Folgenden werden die Kurven und Sequenzen, die eine Mutation identifizieren ließen, abgebildet und erläutert. Sie sind nach den Fällen sortiert. Dabei sind Missense Mutationen rot eingekreist, Splice site Mutationen blau, Deletionen und Insertionen grün gekennzeichnet. Die Polymorphismen werden lediglich in einer zusammenfassenden Tabelle aufgelistet. Fall 1: Die DHPLC-Analyse zeigte für das Exon 10b ein von der Norm abweichendes Diagramm. Die Sequenzierung des Exons ergab folgendes Ergebnis: Mutation: NF1 c. 1466 A/G entspricht Y489C Das DHPLC-Diagramm zu Exon 10b ist in Abbildung 35a wiedergegeben. Die dazugehörige Sequenzierung ist in Abbildung 35b dargestellt. Die Basensequenz zeigt eine Änderung an Nukleotid-Position 1466. Die Base Adenin (A) ist durch Guanin (G) ersetzt. Das Codon 489, zu dem diese Base gehört, lautet in der Wildtyp-Sequenz TAT und codiert für die Aminosäure Tyrosin (Y). Die mutierte Basensequenz lautet somit TGT und codiert eigentlich für Cystein (C). Diese Mutatione ist nur formal eine Missense Mutation, weil sich durch die neue Nukleotidsequenz eine neue Splice site AG-GT bildet, die zu einem Verlust von 62 bp führt. Dies wurde von Messiaen et al. (1999) durch RNA-Analyse gezeigt. a) b) mV 1 Wildtyp-Sequenz: GAAGC TAT AAGTA Mutierte Sequenz: GAAGC TGT AAGTA Nukleotid: 1466 0 1.9 2 2.1 m Substitution: A/G Codon 489 Abb. 35 a und b: a) DHPLC Diagramm zu Fall 1 mit gestrichelter Kurve als Referenz. Die X-Achse kennzeichnet die Dauer in Minuten, die Y-Achse gibt die elektrische Spannung in Millivolt an. Der zusätzliche Peak beginnt bei 1,9 min. b) Dazugehörige Sequenzierung mit rot eingekreister 1466 A/G Substitution. Das Textfeld zeigt Wildtyp-Sequenz, mutierte Sequenz und gibt Codon-, sowie Nukleotidnummer und die Substitution an. 81 3 Ergebnisse Diese Mutation ist in der Literatur bereits beschrieben (Messiaen et al. 1999). Es ist eine Mutation, deren Sequenz der eines Pseudogenes entspricht (Luijten et al. 2001). In Abbildung 36 ist die Wildtyp-Sequenz mit der Pseudogensequenz als „Blast“ vergleichend dargestellt. „Blast“ ist ein Computerprogramm des National Center for Biotechnology Information (NCBI), mit dem sich Sequenzen vergleichen lassen. Abb. 36: Blast-Sequenz von Wildtyp-Exon 10b (obere Sequenz) und Pseudogen ähnlich Exon 10b auf Chromosom 22 (AC002471). Das so genannte Blast ist ein Computerprogramm des National Center for Biotechnology Information (NCBI), mit dem sich Sequenzen vergleichen lassen. Rot markiert ist die in Fall 1 und 17 gefundene Mutation, die in der Sequenz des Pseudogens vorkommt. Grün markiert sind alle weiteren Unterschiede, die jedoch in der Sequenzierung der beiden Fälle unauffällig waren, womit eine versehentliche Amplifikation ausgeschlossen ist. Fall 2: Die DHPLC-Analyse zeigte für das Exon 2 ein von der Norm abweichendes Diagramm. Die Sequenzierung des Exons ergab folgendes Ergebnis: Mutation: NF1 c. 61-1 G/A (IVS2-1 G/A) entspricht einem Basenaustausch der Splice-Region am Ende von Intron 1/Anfang Exon 2 Das DHPLC-Diagramm zu Exon 10b ist in Abbildung 37a wiedergegeben. Die dazugehörige Sequenzierung ist in Abbildung 37b dargestellt. Das Exon 2 beginnt am Nukleotid 61. Die Basensequenz zeigt eine Änderung an Nukleotid-Position 61-1. Dies betrifft die Splice-Region des Exons 2. Die Base Guanin (G) ist durch Adenin (A) ersetzt. Dadurch kommt es zu einem inkorrekten Splice-Vorgang der mRNA, wodurch eine veränderte Synthese des Proteins resultiert. Dies stellt daher eine Mutation dar. 82 3 Ergebnisse mV 1 a) b) Wildtyp-Sequenz: TTTTCAG CTTCCA Mutierte Sequenz: TTTTCAA CTTCCA Nukleotid: 61-1 0 Substitution: G/A 1.6 1.7 1.8 1.9 AG/GT-Regel Codon 21 m Abb. 37 a und b: a) DHPLC Diagramm zu Fall 2 mit gestrichelter Kurve als Referenz. Die X-Achse kennzeichnet die Dauer in Minuten, die Y-Achse gibt die elektrische Spannung in Millivolt an. Ein Peak fehlt bei 1,83 min. b) Dazugehörige Sequenzierung mit blau eingekreister 61-1 G/A Splice site Mutation. Das Textfeld zeigt Wildtyp-Sequenz, mutierte Sequenz und gibt Codon-, sowie Nukleotidnummer und die Substitution an. Fall 3: Die DHPLC-Analyse zeigte für das Exon 3 ein von der Norm abweichendes Diagramm. Die Sequenzierung des Exons ergab folgendes Ergebnis: Mutation: NF1 c. 277 T/C entspricht C93R Das DHPLC-Diagramm zu Exon 3 ist in Abbildung 38a wiedergegeben. Die dazugehörige Sequenzierung ist in Abbildung 38b dargestellt. Die Basensequenz zeigt eine Änderung an Nukleotid-Position 277. Die Base Thymin (T) ist durch Cytosin (C) ersetzt. Das Codon 277, zu dem diese Base gehört, lautet in der Wildtyp-Sequenz TGT und codiert für die Aminosäure Cystein (C). Die mutierte Basensequenz lautet somit CGT und codiert für Arginin (R). Diese Basenänderung fand sich bei keinem der Kontrollproben. Sie stellt daher eine Mutation dar. mV 1 a) b) Wildtyp-Sequenz: GAAAAA TGT CTTG Mutierte Sequenz: GAAAAA CGT CTTG Nukleotid: 277 0 Substitution: T/C 1.4 1.5 1.6 m Codon 93 Abb. 38 a und b: a) DHPLC Diagramm zu Fall 3 mit gestrichelter Kurve als Referenz. Die X-Achse kennzeichnet die Dauer in Minuten, die Y-Achse gibt die elektrische Spannung in Millivolt an. Der zusätzliche Peak beginnt bei 1,4 min. b) Dazugehörige Sequenzierung mit rot eingekreister 277 T/C Substitution. Das Textfeld zeigt Wildtyp-Sequenz, mutierte Sequenz und gibt Codon-, sowie Nukleotidnummer und die Substitution an. 83 3 Ergebnisse Fall 4: Die DHPLC-Analyse zeigte für das Exon 37 ein von der Norm abweichendes Diagramm. Die Sequenzierung des Exons ergab folgendes Ergebnis: Mutation: NF1 c. 6795 ins C entspricht E2255Frameshift Das DHPLC-Diagramm zu Exon 37 ist in Abbildung 39a wiedergegeben. Die dazugehörige Sequenzierung ist in Abbildung 39b dargestellt. Die Basensequenz zeigt eine Änderung nach Nukleotid-Position 6795. Es ist die Base Cytosin (C) eingefügt. Durch die Insertion kommt es ab dem Codon 2266, in dem die Base eingefügt ist, zu einer Verschiebung des Leserasters der Aminosäurentranslation, dem so genannten Frameshift. Dies stellt somit eine Mutation dar. mV 1 a) b) Wildtyp-Sequenz: TACAAC AGT CAAG Mutierte Sequenz: TACAAC CAG TCAA Nukleotid: 6795 0 Insertion: C 1.8 1.9 Codon 2266 m Abb. 39 a und b: a) DHPLC Diagramm zu Fall 4 mit gestrichelter Kurve als Referenz. Die X-Achse kennzeichnet die Dauer in Minuten, die Y-Achse gibt die elektrische Spannung in Millivolt an. Der zusätzliche Peak beginnt bei 1,83 min. b) Dazugehörige Sequenzierung mit grün eingekreister Cytosin-Insertion am Nukeotid 6795. Das Textfeld zeigt WildtypSequenz, mutierte Sequenz und gibt Codon-, sowie Nukleotidnummer und die Insertion an. Dadurch werden 19 falsche Aminosäuren übersetzt, bis es im 20. Triplett zu einem StoppCodon kommt (Abb. 40). Dies führt zum Abbruch der Proteinbiosynthese. Abb. 40: Rot eingerahmt ist das Codon, in dem die homozygote TT-Insertion zum Frameshift führt. Zum Vergleich sind Wildtyp-DNA- und Aminosäurensequenz angegeben. Der Frameshift findet in Exon 37 statt und beginnt in Codon 2266. 84 3 Ergebnisse Fall 5: Die DHPLC-Analyse zeigte für das Exon 16 ein von der Norm abweichendes Diagramm. Die Sequenzierung des Exons ergab folgendes Ergebnis: Mutation: NF1 c. 2849 ins TT homo entspricht Q950Frameshift Das DHPLC-Diagramm zu Exon 16 ist in Abbildung 41a wiedergegeben. Die dazugehörige Sequenzierung ist in Abbildung 42b dargestellt. Die Basensequenz zeigt eine Änderung nach Nukleotid-Position 2849. Es sind zwei zusätzliche Basen Thymin (T) eingefügt. Durch die Insertion kommt es ab dem Codon 2849, in dem die erste Base eingefügt ist, zu einer Verschiebung des Leserasters der Aminosäurentranslation, dem so genannten Frameshift. Dies stellt somit eine Mutation dar. mV 1 a) b) Wildtyp-Sequenz: GGA CAG GTAAAGT Mutierte Sequenz: GGA CAT TGGTAAA Nukleotid: 2849 0 Insertion: TT homo 1.8 1.9 Codon 950 m Abb. 41 a und b: a) DHPLC Diagramm zu Fall 5 mit gestrichelter Kurve als Referenz. Die X-Achse kennzeichnet die Dauer in Minuten, die Y-Achse gibt die elektrische Spannung in Millivolt an. Der zusätzliche Peak beginnt bei 1,78 min. b) Dazugehörige Sequenzierung mit grün eingekreister Thymin-Insertion am Nukleotid 2849. Das Textfeld zeigt WildtypSequenz, mutierte Sequenz und gibt Codon-, sowie Nukleotidnummer und die Insertion an. Dadurch werden 4 falsche Aminosäuren übersetzt, bis es im 5. Triplett zu einem Stopp-Codon kommt. Dies ist in Abbildung 42 gezeigt. Abb. 42: Homozygote TT Insertion am Ende von Exon 16 (rot eingerahmt). In der mutierten Sequenz (5. Reihe) verschieben sich die weiteren Basen zur Wildtyp-Sequenz (Reihe 3) um 2 Basen nach rechts. In der untersten Reihe erkennt man die zur normalen Aminosäurensequenz (Reihe 4) veränderte Sequenz. Nach vier Aminosäuren kommt es im Triplett zu einem Stopp-Codon (X). 85 3 Ergebnisse Fall 6: Die DHPLC-Analyse zeigte für das Exon 22 ein von der Norm abweichendes Diagramm. Die Sequenzierung des Exons ergab folgendes Ergebnis: Mutation: NF1 c. 3826 C/T entspricht R1276X Das DHPLC-Diagramm zu Exon 22 ist in Abbildung 43a wiedergegeben. Die dazugehörige Sequenzierung ist in Abbildung 43b dargestellt. Die Basensequenz zeigt eine Änderung an Nukleotid-Position 3826. Die Base Cytosin (C) ist durch Thymin (T) ersetzt. Das Codon 1276, zu dem diese Base gehört, lautet in der Wildtyp-Sequenz CGA und codiert für die Aminosäure Arginin (R). Die mutierte Basensequenz lautet somit TGA und codiert für ein Stopp-Codon (X). Dadurch kommt es zum Abbruch des Translationsvorganges. Die Basenänderung stellt daher eine Mutation dar. mV 1 a) b) Wildtyp-Sequenz: CTCTTC CGA GGCA Mutierte Sequenz: CTCTTC TGA GGCA Nukleotid: 3826 0 Substitution: C/T 1.8 1.9 Codon 1276 m Abb. 43 a und b: a) DHPLC Diagramm zu Fall 6 mit gestrichelter Kurve als Referenz. Die X-Achse kennzeichnet die Dauer in Minuten, die Y-Achse gibt die elektrische Spannung in Millivolt an. Der zusätzliche Peak beginnt bei 1,8 min. b) Dazugehörige Sequenzierung mit rot eingekreister 3826 C/T Substitution. Das Textfeld zeigt Wildtyp-Sequenz, mutierte Sequenz und gibt Codon-, sowie Nukleotidnummer und die Substitution an. Diese Mutation ist von Heim et al. (1995) schon identifiziert worden. Sie entspricht der Sequenz eines Pseudogens auf Chromosom 15. Um eine fehlerhafte Amplifikation auszuschließen, ist in Abbildung 44 die Wildtyp-Sequenz von Exon 22 mit der PseudogenSequenz als Blast vergleichend dargestellt. 3 Ergebnisse 86 Abb. 44: Blast Sequenz zwischen Wildtyp-Exon 22 (obere Reihe) und dem Pseudogen auf Chromosom 15 (untere Reihe) (Accession Nr. AC021585, AC020679). Das so genannte Blast ist ein Computerprogramm des National Center for Biotechnology Information (NCBI), mit dem sich Sequenzen vergleichen lassen. Die in Fall 6 gefundene Mutation ist rot markiert. Sie entspricht der Sequenz des Pseudogens. Alle weiteren Unterschiede, die jedoch in der Sequenzierung durchgehend unauffällig waren, sind grün eingerahmt, womit eine versehentliche Amplifikation trotz der allel-spezifischen PCR ausgeschlossen werden kann. Fall 7: Die DHPLC-Analyse zeigte für das Exon 15 ein von der Norm abweichendes Diagramm. Die Sequenzierung des Exons ergab folgendes Ergebnis: Mutation: NF1 c. 2409+1 G/C (IVS15+1 G/C) entspricht einem Basenaustausch der Splice-Region am Ende von Exon 15/Anfang von Intron 15 Das DHPLC-Diagramm zu Exon 15 ist in Abbildung 45a wiedergegeben. Die dazugehörige Sequenzierung ist in Abbildung 45b dargestellt. Das Exon 15 endet am Nukleotid 2409. Die Basensequenz zeigt eine Änderung an Nukleotid-Position 2409+1. Dies betrifft die Splice-Region des Exons 15. Die Base Guanin (G) ist durch Cytosin (C) ersetzt. Dadurch kommt es zu einem inkorrekten SpliceVorgang der mRNA, wodurch eine veränderte Synthese des Proteins resultiert. Dies stellt somit eine Mutation dar. 87 mV 1 a) 3 Ergebnisse b) Wildtyp-Sequenz: GGCCAG GT AAGTC Mutierte Sequenz: GGCCAG CT AAGTC Nukleotid 2409+1 0 2.1 2.2 2 m Substitution G/C Codon 803 AG/GT-Regel Abb. 45 a und b: a) DHPLC Diagramm zu Fall 7 mit gestrichelter Kurve als Referenz. Die X-Achse kennzeichnet die Dauer in Minuten, die Y-Achse gibt die elektrische Spannung in Millivolt an. Der zusätzliche Peak beginnt bei 1,9 min. b) Dazugehörige Sequenzierung mit blau eingekreister 2409+1 G/C Splice site Mutation. Das Textfeld zeigt WildtypSequenz, mutierte Sequenz und gibt Codon-, sowie Nukleotidnummer und die Substitution an. Fall 9: Die DHPLC-Analyse zeigte für das Exon 35 ein von der Norm abweichendes Diagramm. Die Sequenzierung des Exons ergab folgendes Ergebnis: Mutation: NF1 c. 6641+1 G/A (IVS35+1 G/A) entspricht einem Basenaustausch der Splice-Region am Ende von Exon 35/Anfang von Intron 35 Das DHPLC-Diagramm zu Exon 35 ist in Abbildung 46a wiedergegeben. Die dazugehörige Sequenzierung ist in Abbildung 46b dargestellt. Das Exon 35 endet am Nukleotid 6641. Die Basensequenz zeigt eine Änderung an Position 6641+1. Dies betrifft die Splice-Region des Exons 35. Die Base Guanin (G) ist durch Adenin (A) ersetzt. Dadurch kommt es zu einem inkorrekten Splice-Vorgang der mRNA, wodurch eine veränderte Synthese des Proteins resultiert. Dies stellt somit eine Mutation dar. mV 1 a) b) Wildtyp-Sequenz: TCAAAG GT ATGAC Mutierte Sequenz: TCAAAG AT ATGAC Nukleotid: 6641+1 0 Substitution: G/A 1.6 1.7 1.8 1.9 Teilcodon 2214 AG/GT-Regel 2 m Abb. 46 a und b: a) DHPLC Diagramm zu Fall 9 mit gestrichelter Kurve als Referenz. Die X-Achse kennzeichnet die Dauer in Minuten, die Y-Achse gibt die elektrische Spannung in Millivolt an. Der zusätzliche Peak beginnt bei 1,77 min. b) Dazugehörige Sequenzierung mit blau eingekreister 6641+1 G/A Splice site Mutation. Das Textfeld zeigt WildtypSequenz, mutierte Sequenz und gibt Codon-, sowie Nukleotidnummer und die Substitution an. 88 3 Ergebnisse Fall 10: Die DHPLC-Analyse zeigte für die Exons 7 und 45 von der Norm abweichende Diagramme. Die Sequenzierung der Exons ergab folgende Ergebnisse: 1. Mutation: NF1 c. 1062+2 T/C (IVS7+2 T/C) entspricht einem Basenaustausch der Splice-Region am Ende von Exon 7/Anfang von Intron 7 Das DHPLC-Diagramm zu Exon 7 ist in Abbildung 47a wiedergegeben. Die dazugehörige Sequenzierung ist in Abbildung 47b dargestellt. Das Exon 7 endet am Nukleotid 1062. Die Basensequenz zeigt eine Änderung an Nukleotid-Position 1062+2. Dies betrifft die Splice-Region des Exons 7. Die Base Thymin (T) ist durch Cytosin (C) ersetzt. Dadurch kommt es zu einem inkorrekten SpliceVorgang der mRNA, wodurch eine veränderte Synthese des Proteins resultiert. Sie stellt daher eine Mutation dar. mV 1 a) b) Wildtyp-Sequenz: TTAAG GT AACATG Mutierte Sequenz: TTAAG GC AACATG Nukleotid: 1062+2 0 1.4 m Substitution: T/C Codon 354 AG/GT-Regel Abb. 47 a und b: a) DHPLC Diagramm zu Fall 10 mit gestrichelter Kurve als Referenz. Die X-Achse kennzeichnet die Dauer in Minuten, die Y-Achse gibt die elektrische Spannung in Millivolt an. Der zusätzliche Peak beginnt bei 1,37 min. b) Dazugehörige Sequenzierung mit blau eingekreister 1062+2 T/C Splice site Mutation. Das Textfeld zeigt WildtypSequenz, mutierte Sequenz und gibt Codon-, sowie Nukleotidnummer und die Substitution an. 2. Mutation: NF1 c. 9833 T/A entspricht D2611E Das DHPLC-Diagramm zu Exon 45 ist in Abbildung 48a wiedergegeben. Die dazugehörige Sequenzierung ist in Abbildung 48b dargestellt. Die Basensequenz zeigt eine Änderung an Nukeosid-Position 7833. Die Base Thymin (T) ist durch Adenin (A) ersetzt. Das Codon 2611, zu dem diese Base gehört, lautet in der Wildtyp-Sequenz GAT und codiert für die Aminosäure Asparagin (D). Die mutierte Basensequenz lautet somit GAA und codiert für Glutamin (E). Diese Basenänderung fand sich bei keinem der Kontrollproben. Sie stellt daher eine Mutation dar. 89 a) b) mV 1 3 Ergebnisse Wildtyp-Sequenz: CACA GAT GAGTTT Mutierte Sequenz: CACA GAA GAGTTT Nukleotid: 7833 0 1.7 1.8 1.9 m Substitution: T/A Codon 2611 Abb. 48 a und b: a) DHPLC Diagramm zu Fall 10 mit gestrichelter Kurve als Referenz. Die X-Achse kennzeichnet die Dauer in Minuten, die Y-Achse gibt die elektrische Spannung in Millivolt an. Der zusätzliche Peak beginnt bei 1,73 min. b) Dazugehörige Sequenzierung mit rot eingekreister 7833 T/A Substitution. Das Textfeld zeigt Wildtyp-Sequenz, mutierte Sequenz und gibt Codon-, sowie Nukleotidnummer und die Substitution an. Fall 14: Die DHPLC-Analyse zeigte für das Exon 23-2 ein von der Norm abweichendes Diagramm. Die Sequenzierung des Exons ergab folgendes Ergebnis: Mutation: NF1 c. 4077 del T entspricht P1359Frameshift Das DHPLC-Diagramm zu Exon 23-2 ist in Abbildung 49a wiedergegeben. Die dazugehörige Sequenzierung ist in Abbildung 49b dargestellt. Die Basensequenz zeigt eine Änderung an Nukleosid-Position 4077. Das Nukleotid Cytosin (C) ist deletiert. Dadurch kommt es ab dem Codon 1359, in dem die Base fehlt, zu einer Verschiebung des Leserasters der Aminosäurentranslation, dem so genannten Frameshift. Dies stellt somit eine Mutation dar. mV 1 a) b) Wildtyp-Sequenz: CCCC CTC ACTTC Mutierte Sequenz: CCCC CCA CTTCG Nukleotid: 4077 0 Deletion: T 1.8 1.9 Codon 1359 m Abb. 49 a und b: a) DHPLC Diagramm zu Fall 14mit gestrichelter Kurve als Referenz. Die X-Achse kennzeichnet die Dauer in Minuten, die Y-Achse gibt die elektrische Spannung in Millivolt an. Der Peak ist verbreitert und beginnt bei 1,78 min. b) Dazugehörige Sequenzierung mit grün eingekreister Thymin-Deletion am Nukeotid 4077. Das Textfeld zeigt Wildtyp-Sequenz, mutierte Sequenz und gibt Codon-, sowie Nukleotidnummer und die Deletion an. 3 Ergebnisse 90 Dadurch werden 25 falsche Aminosäuren übersetzt, bis es im 26. Triplett zu einem StoppCodon kommt. Dies ist in Abbildung 50 gezeigt. Abb. 50: Deletion in Exon 10c. In der Ersten Zeile ist jeweils das Exon genannt. Die zweite Zeile beschreibt das Codon. Unter der Wildtyp-Nukleotid-Sequenz ist die Aminosäuresequenz dargestellt. Es folgt die mutierte Sequenz von Fall 14. Rot eingerahmt ist das Codon der Deletion. Durch die Leserasterverschiebung werden “falsche” Aminosäuren übersetzt, bis es zu einem Stopp-Codon kommt (X). Fall 15: Die DHPLC-Analyse zeigte für das Exon 10c ein von der Norm abweichendes Diagramm. Die Sequenzierung des Exons ergab folgendes Ergebnis: Mutation: NF1 c. 1580 del C entspricht Q950Frameshift Das DHPLC-Diagramm zu Exon 10c ist in Abbildung 51a wiedergegeben. Die dazugehörige Sequenzierung ist in Abbildung 51b dargestellt. Die Basensequenz zeigt eine Änderung an Nukleosid-Position 1580. Das Nukleotid Cytosin (C) ist deletiert. Dadurch kommt es ab dem Codon 950, in dem die Base fehlt, zu einer Verschiebung des Leserasters der Aminosäurentranslation, dem so genannten Frameshift. Dies stellt somit eine Mutation dar. 91 mV 1 a) 3 Ergebnisse b) Wildtyp-Sequenz: TAATT ACA GGGCT Mutierte Sequenz: TAATT AAG GGCTC Nukleotid: 1580 0 1.7 1.8 1.9 m Deletion: C Codon 527 Abb. 51 a und b: a) DHPLC Diagramm zu Fall 15 mit gestrichelter Kurve als Referenz. Die X-Achse kennzeichnet die Dauer in Minuten, die Y-Achse gibt die elektrische Spannung in Millivolt an. Der zusätzliche Peak beginnt bei 1,7 min. b) Dazugehörige Sequenzierung mit grün eingekreister Cytosin-Deletion am Nukeotid 1580. Das Textfeld zeigt WildtypSequenz, mutierte Sequenz und gibt Codon-, sowie Nukleotidnummer und die Deletion an. Dadurch codieren die folgenden 28 Tripletts für falsche Aminosäuren, bis es im 29. Triplett zu einem Stopp-Codon kommt. Dies ist in Abbildung 52 gezeigt. Abb. 52: Frameshift-Sequenz der Deletion in Exon 10c. In der ersten Zeile ist jeweils das Exon genannt. Die zweite Zeile zeigt das Codon der Wildtyp-Sequenz. Darunter ist die Wildtyp-Aminosäuresequenz dargestellt. Darunter steht die in Fall 15 beobachtete Basensequenz. Rot eingerahmt ist das Codon der Deletion. Durch die Leserasterverschiebung werden “falsche” Aminosäuren übersetzt, bis es zum Stopp-Codon kommt (X). 92 3 Ergebnisse Fall 17: Die DHPLC-Analyse zeigte für das Exon 10c ein von der Norm abweichendes Diagramm. Die Sequenzierung des Exons ergab folgendes Ergebnis: Mutation: NF1 c. 1466 A/G entspricht Y489C Das DHPLC-Diagramm zu Exon 10b ist in Abbildung 53a wiedergegeben. Die dazugehörige Sequenzierung ist in Abbildung 53b dargestellt. Die Basensequenz zeigt eine Änderung an Nukleotid-Position 1466. Die Base Adenin (A) ist durch Guanin (G) ersetzt. Das Codon 489, zu dem diese Base gehört lautet in der Wildtyp-Sequenz TAT und codiert für die Aminosäure Tyrosin (Y). Die mutierte Basensequenz lautet somit TGT und codiert eigentlich für Cystein (C). Diese Mutation ist nur formal eine Missense Mutation, weil sich durch die neue Nukleotidsequenz eine neue Splice site AG-GT bildet, die zu einem Verlust von 62 bp führt. Dies wurde von Messiaen et al. (1999) durch RNA-Analyse gezeigt. mV 1 a) b) Wildtyp-Sequenz: GAAGC TAT AAGTA Mutierte Sequenz: GAAGC TGT AAGTA Nukleotid: 1466 0 Substitution: A/G 2 2.1 2.2 m Codon 489 Abb. 53 a und b: a) DHPLC Diagramm zu Fall 17 mit gestrichelter Kurve als Referenz. Die X-Achse kennzeichnet die Dauer in Minuten, die Y-Achse gibt die elektrische Spannung in Millivolt an. Der zusätzliche Peak beginnt bei 1,9 min. b) Dazugehörige Sequenzierung mit rot eingekreister 489 A/G Substitution. Das Textfeld zeigt Wildtyp-Sequenz, mutierte Sequenz und gibt Codon-, sowie Nukleotidnummer und die Substitution an. Diese Mutation ist in der Literatur bereits beschrieben (Messiaen et al. 1999). Es ist eine Mutation, deren Sequenz der eines Pseudogenes entspricht (Luijten et al. 2001). In Abbildung 36 ist die Wildtyp-Sequenz mit der Pseudogensequenz als Blast vergleichend dargestellt. 93 3 Ergebnisse Fall 19: Die DHPLC-Analyse zeigte für das Exon 41 ein von der Norm abweichendes Diagramm. Die Sequenzierung des Exons ergab folgendes Ergebnis: Mutation: NF1 c. 7337 C/G entspricht S2446X Das DHPLC-Diagramm zu Exon 41 ist in Abbildung 54a wiedergegeben. Die dazugehörige Sequenzierung ist in Abbildung 54b dargestellt. Die Basensequenz zeigt eine Änderung an Nukleosid-Position 7337. Die Base Cytosin (C) ist durch Guanin (G) ersetzt. Das Codon 2446, zu dem diese Base gehört, lautet in der Wildtyp-Sequenz TCA und codiert für die Aminosäure Serin (S). Die mutierte Basensequenz lautet somit TGA und codiert für ein Stopp-Codon (X). Dadurch kommt es zum Abbruch des Translationsvorganges. Dies stellt somit eine Mutation dar. mV 1 a) b) Wildtyp-Sequenz: ATATT TCA ATGGA Mutierte Sequenz: ATATT TGA ATGGA Nukleotid: 7337 0 Substitution: C/G 1.7 1.8 m Codon 2446 Abb. 54 a und b: a) DHPLC Diagramm zu Fall 19 mit gestrichelter Kurve als Referenz. Die X-Achse kennzeichnet die Dauer in Minuten, die Y-Achse gibt die elektrische Spannung in Millivolt an. Der zusätzliche Peak beginnt bei 1,9 min. b) Dazugehörige Sequenzierung mit rot eingekreister 7337 C/G Substitution. Das Textfeld zeigt Wildtyp-Sequenz, mutierte Sequenz und gibt Codon-, sowie Nukleotidnummer und die Substitution an. Fall 20: Die DHPLC-Analyse zeigte für das Exon 29 von der Norm abweichendes Diagramm. Die Sequenzierung des Exons ergab folgendes Ergebnis: Mutation: NF1 c. 5537+1 G/T (IVS29+2 G/T) entspricht einem Basenaustausch der Splice-Region am Ende von Exon 29/Anfang von Intron 29 Das DHPLC-Diagramm zu Exon 29 ist in Abbildung 55a wiedergegeben. Die dazugehörige Sequenzierung ist in Abbildung 55b dargestellt. Das Exon 29 endet am Nukleotid 5537. Die Basensequenz zeigt eine Änderung an Nukleotid-Position 5537+1. Dies betrifft die Splice-Region des Exons 29. Die Base 94 3 Ergebnisse Guanin (G) ist durch Thymin (T) ersetzt. Dadurch kommt es zu einem inkorrekten SpliceVorgang der mRNA, wodurch eine veränderte Synthese des Proteins resultiert. Dies stellt somit eine Mutation dar. mV a) b) Wildtyp-Sequenz: TTTACG GT AGGTT Mutierte Sequenz: TTTACG TT AGGTT Nukleotid: 5537+1 0 1.6 1.7 1.8 m Substitution: G/T Teilcodon 1849 AG/GT-Regel Abb. 55 a und b: a) DHPLC Diagramm zu Fall 20 mit gestrichelter Kurve als Referenz. Die X-Achse kennzeichnet die Dauer in Minuten, die Y-Achse gibt die elektrische Spannung in Millivolt an. Zwei zusätzliche Peaks beginnen bei 1,58 min. und 1,64 min. b) Dazugehörige Sequenzierung mit blau eingekreister 5537 G/T Splice site Mutation. Das Textfeld zeigt Wildtyp-Sequenz, mutierte Sequenz und gibt Codon-, sowie Nukleotidnummer und die Substitution an. Fall 21: Die DHPLC-Analyse zeigte für das Exon 2 ein von der Norm abweichendes Diagramm. Die Sequenzierung des Exons ergab folgendes Ergebnis: Mutation: NF1 c. 269 T/C entspricht L90P Das DHPLC-Diagramm zu Exon 2 ist in Abbildung 56a wiedergegeben. Die dazugehörige Sequenzierung ist in Abbildung 56b dargestellt. Die Basensequenz zeigt eine Änderung an Nukleotid-Position 269. Die Base Thymin (T) mV 1 a) b) Wildtyp-Sequenz: ATACA CTG GAAAA Mutierte Sequenz: ATACA CCG GAAAA Nukleotid: 269 0 Substitution: T/C 1.4 1.5 1.6 Codon 90 m Abb. 56 a und b: a) DHPLC Diagramm zu Fall 21 mit gestrichelter Kurve als Referenz. Die X-Achse kennzeichnet die Dauer in Minuten, die Y-Achse gibt die elektrische Spannung in Millivolt an. Der zusätzliche Peak beginnt bei 1,48 min. b) Dazugehörige Sequenzierung mit rot eingekreister 269 T/C Substitution. Das Textfeld zeigt Wildtyp-Sequenz, mutierte Sequenz und gibt Codon-, sowie Nukleotidnummer und die Substitution an. 95 3 Ergebnisse ist durch Cytosin (C) ersetzt. Das Codon 90, zu dem diese Base gehört, lautet in der Wildtyp-Sequenz CTG und codiert für die Aminosäure Leucin (L). Die mutierte Basensequenz lautet somit CCG und codiert für Prolin (P). Diese Basenänderung fand sich bei keinem der Kontrollproben. Sie stellt daher eine Mutation dar. Fall 24: Die DHPLC-Analyse zeigte für das Exon 37 ein von der Norm abweichendes Diagramm. Die Sequenzierung des Exons ergab folgendes Ergebnis: Mutation: NF1 c. 6858+2 T/C (IVS37+2 T/C) entspricht einem Basenaustausch der Splice-Region am Ende von Exon 37/Anfang von Intron 37 Das DHPLC-Diagramm zu Exon 37 ist in Abbildung 57a wiedergegeben. Die dazugehörige Sequenzierung ist in Abbildung 57b dargestellt. Das Exon 37 endet am Nukleotid 6858. Die Basensequenz zeigt eine Änderung an Nukleotid-Position 6858+2. Dies betrifft die Splice-Region des Exons 37. Die Base Thymin (T) ist durch Cytosin (C) ersetzt. Dadurch kommt es zu einem inkorrekten SpliceVorgang der mRNA, wodurch eine veränderte Synthese des Proteins resultiert. Die Basenänderung stellt daher eine Mutation dar. mV 1 a) b) Wildtyp-Sequenz: ATAAG GT AATTAC Mutierte Sequenz: ATAAG GC AATTAC Nukleotid: 6858+2 0 1.8 1.9 m Substitution: T/C Codon 2286 AG/GT-Regel Abb. 57 a und b: a) DHPLC Diagramm zu Fall 24 mit gestrichelter Kurve als Referenz. Die X-Achse kennzeichnet die Dauer in Minuten, die Y-Achse gibt die elektrische Spannung in Millivolt an. Der zusätzliche Peak beginnt bei 1,76 min. b) Dazugehörige Sequenzierung mit blau eingekreister 6858+2 T/C Splice site Mutation. Das Textfeld zeigt WildtypSequenz, mutierte Sequenz und gibt Codon-, sowie Nukleotidnummer und die Substitution an. 96 3 Ergebnisse Fall 25: Die DHPLC-Analyse zeigte für das Exon 13 ein von der Norm abweichendes Diagramm. Die Sequenzierung des Exons ergab folgendes Ergebnis: Mutation: NF1 c. 2023 ins C entspricht G675Frameshift Das DHPLC-Diagramm zu Exon 13 ist in Abbildung 58a wiedergegeben. Die dazugehörige Sequenzierung ist in Abbildung 58b dargestellt. Die Basensequenz zeigt eine Änderung nach Nukleotid-Position 2023. Es ist die Basen Cytosin (C) eingefügt. Dadurch kommt es ab dem Codon 675, in dem die Base eingefügt ist, zu einer Verschiebung des Leserasters der Aminosäurentranslation, dem so genannten Frameshift. Dies stellt somit eine Mutation dar. mV 1 a) b) Wildtyp-Sequenz: CCCC CCG ATTTGC Mutierte Sequenz: CCCC CCC GATTTG Nukleotid 2033 0 1.6 1.7 1.8 1.9 m Insertion C Codon 678 Abb. 58 a und b: a) DHPLC Diagramm zu Fall 25 mit gestrichelter Kurve als Referenz. Die X-Achse kennzeichnet die Dauer in Minuten, die Y-Achse gibt die elektrische Spannung in Millivolt an. Der zusätzliche Peak beginnt bei 1,83 min. b) Dazugehörige Sequenzierung mit grün eingekreister Cytosin-Insertion nach Nukeotid 2033. Das Textfeld zeigt WildtypSequenz, mutierte Sequenz und gibt Codon-, sowie Nukleotidnummer und die Insertion an. Hierdurch kommt es zur Verschiebung des Leserasters und der Translation von 24 falschen Aminosäuren, bis das Triplett TGA, das für ein Stopp-Codon codiert, zum Abbruch der Proteinsynsthese führt. Dies ist in Abbildung 59 gezeigt. 97 3 Ergebnisse Abb. 59: Sequenzen zur Insertion von Fall 25. In Exon 13 (Reihe 1) findet sich im Codon 675 (Reihe 2) in der mutierten Sequenz (Reihe 5) eine Insertion von Guanin (rot eingerahmt). Dadurch kommt es in der Aminosäuresequenz zu einem Frameshift (Reihe 6) bis ein Stopp-Codon (X) auftritt. Zum Vergleich sind Wildtyp-Seqzenz (Reihe 3) und die Aminosäurenfolge des Wildtyp-Proteins (Reihe 4) dargestellt. Fall 26: Die DHPLC-Analyse zeigte für das Exon 44 ein von der Norm abweichendes Diagramm. Die Sequenzierung des Exons ergab folgendes Ergebnis: Mutation: NF1 c. 7739 C/G entspricht L234S Das DHPLC-Diagramm zu Exon 44 ist in Abbildung 60a wiedergegeben. Die dazugehörige Sequenzierung ist in Abbildung 60b dargestellt. Die Basensequenz zeigt eine Änderung an Position 7739. Die Base Cytosin (C) ist durch Guanin (G) ersetzt. Das Codon 234, zu dem diese Base gehört, lautet in der WildtypSequenz TCA und codiert für die Aminosäure Leucin (L). Die mutierte Basensequenz lautet somit TGA und codiert für Serin (S). Diese Basenänderung fand sich bei keinem der Kontrollproben. Sie stellt daher eine Mutation dar. 98 3 Ergebnisse mV 1 a) b) Wildtyp-Sequenz: CTGAA TCA AATGT Mutierte Sequenz: CTGAA TGA AATGT Nukleotid: 7739 0 Substitution: C/G 1.8 1.9 Codon 2580 m Abb. 60 a und b: a) DHPLC Diagramm zu Fall 26 mit gestrichelter Kurve als Referenz. Die X-Achse kennzeichnet die Dauer in Minuten, die Y-Achse gibt die elektrische Spannung in Millivolt an. Die Kurve stellt sich über den ganzen Verlauf dünner dar. b) Dazugehörige Sequenzierung mit rot eingekreister 7739 C/G Substitution. Das Textfeld zeigt Wildtyp-Sequenz, mutierte Sequenz und gibt Codon-, sowie Nukleotidnummer und die Substitution an. Nach Abschluß der Auswertungen in Freiburg wurden die Sequenzierungen, die von 17 Fällen für die Exons 1-8 und 28-49 in Padua durchgeführt wurden, noch vorgenommen. Es konnten dabei folgende Mutationen bestätigt werden: Fall 2: Mutation: NF1 c. 61-1 G/A (IVS2-1 G/A) entspricht einem Basenaustausch der SpliceRegion am Ende von Intron 1/Anfang Exon 2. Fall 4: Mutation: NF1 c. 6795 ins C entspricht E2255Frameshift in Exon 37. Fall 10: Mutation: NF1 c. 1062+2 T/C (IVS7+2 T/C) entspricht einem Basenaustausch der Splice-Region am Ende von Exon 7/Anfang von Intron 7. Fall 21: Mutation: NF1 c. 269 T/C entspricht L90P in Exon 3. Fall 24: Mutation: NF1 c. 6858+2 T/C (IVS37+2 T/C) entspricht einem Basenaustausch der Splice-Region am Ende von Exon 37/Anfang von Intron 37. Fall 26: Mutation: NF1 c. 7739 C/G entspricht L234S. 99 3 Ergebnisse 3.4 Tabellarische Zusammenfassung der Mutationen Tab. 40: Tabellarische Zusammenfassung der Mutationen aufgelistet in aufsteigender Zahl der Exons. Fall Codon Genomische Mutation Konsequenz MutationsTypus Exon 2 2 - 61-1 G/A - Splice-Defekt Exon 3 21 90 269 T/C L90P Missense Exon 3 3 93 277 T/C C93R Missense Exon 7 10 - 1062+2 T/C - Splice-Defekt Exon 10b 1 489 1466 A/G Y489C Missense Exon 10b 17 489 1466 A/G Y489C Missense Exon 10c 15 527 1580 del C T527 Frameshift Deletion Exon 13 25 675 2023 ins G G675 Frameshift Insertion Exon 15 7 - 2409+1 G/C - Splice-Defekt Exon 16 5 950 2849 ins TT homo Q950 Frameshift Insertion Exon 22 6 1276 3826 C/T R1276X Stopp Exon 23-2 14 1359 4077 del T P1359 Frameshift Deletion Exon 29 20 - 5537+1 G/T - Splice-Defekt Exon 35 9 - 6641+1 G/A - Splice-Defekt Exon 37 4 2266 6795 ins C E2255 Frameshift Insertion Exon 37 24 - 6858+2 T/C - Splice-Defekt Exon 41 19 2446 7337 C/G S2446X Stopp Exon 44 26 2580 7739 C/G S2580A Missense Exon 45 10 2611 7833 T/A D2611E Missense Lokalisation s = sporadisch, f = familiär. ins = Insertion, del = Deletion. 100 3 Ergebnisse 3.5 Tabellarische Zusammenfassung der Polymorphismen Tab. 41: Tabellarische Zusammenfassung I der exonischen Polymorphismen aufsteigend nach der Lokalisation. Vorkommen in dieser Studie Heterozygot Exon 2 4% 1/26 (4%) Exon 5 58% 5/26 (19%) Exon 13 15% 4/26 (15%) Lokalisation Homozygot 10/26 (38%) NukleotidKonsequenz Substitution 168 C/T S56S 702 G/A L234S 2034 G/A P678P IVS = intervening sequenze (Introns). Tab. 42: Tabellarische Zusammenfassung II der intronischen Polymorphismen aufsteigend nach der Lokalisation. Lokalisation Vorkommen in dieser Studie Heterozygot Homozygot NukleotidKonsequenz Substitution IVS 2 8% 2/26 (8%) 204+51 G/C Intron IVS 10c 15% 4/26 (15%) 1641+39 T/C Intron IVS 11 4% 1/26 (4%) 1642-88 A/G Intron IVS 11 4% 1/26 (4%) 1721+73 C/T Intron IVS 17 4% 1/26 (4%) 2851-16 T/C Intron IVS 21 19% 4/26 (15%) 3497-88 C/A Intron IVS 26 19% 5/26 (19%) 4368-46 G/C Intron IVS 28 15% 4/26 (15%) 5205+23 T/C Intron IVS 29 23% 6/26 (23%) 5537+19 T/A Intron IVS 39 96% 3/26 (12%) 2/26 (8%) 7126+37 C/G Intron IVS 39 96% 6/26 (23%) 19/26 (73% 7126+74 T/C Intron IVS 42 92% 6/26 (23%) 18/26 (69%) 7395-29 G/A Intron IVS = intervening sequenze (Introns). 1/26 (4%) 101 4 Diskussion 4 Diskussion Die klinische und molekulargenetische Klassifikation von Phäochromozytomen ist das langjährige Forschungsleitthema der Freiburger Arbeitsgruppe von Prof. Neumann. In diesem Rahmen ist die vorgelegte Arbeit entstanden. Ziel war es, ein repräsentatives Patientengut mit Neurofibromatose Typ 1 und Phäochromozytomen zusammenzustellen und mit den verfügbaren molekulargenetischen Methoden zu charakterisieren. 4.1 Klinische Ergebnisse Orientierende Eckdaten ergaben sich aus der Perspektive der Neurofibromatose Typ 1 aus der viel beachteten Übersichtsarbeit von Riccardi aus dem New England Journal of Medicine von 1981, der für das Zusammentreffen beider Erkrankungen eine Prävalenz von Phäochromozytomen bei Patienten mit Neurofibromatose Typ 1 von einem Prozent angibt (Riccardi et al. 1981). Hierdurch ist klar ein überzufälliges Vorkommen der Phäochromozytome belegt. Eine gemeinsame Ursache lässt sich weiterhin daraus ableiten, dass beide Erkrankungen neuroektodermalen Ursprungs sind. Die Neurofibromatose Typ 1 ist eine der häufigsten hereditären Erkrankungen. Die Prävalenz in der Gesamtbevölkerung wird mit 1:3500 angegeben (Huson et al. 1994). Die Durchsicht der medizinischen Literatur mittels der elektronischen Suchsysteme Medline und Pubmed ergab allerdings für die letzten 20 Jahre keine Original-Darstellung eines größeren Patientengutes von Neurofibromatose Typ 1 Patienten mit Phäochromozytomen, vielleicht weil für eine anstrebenswerte Zahl von ca. 20 Fällen eine Untersuchung von etwa 2000 Patienten erfordert. Somit stellte sich die Frage nach der Prävalenz von Phäochromozytomen bei Patienten mit Neurofibromatose Typ 1. Als Eckdaten stehen Schätzungen im Raum, die von einer Forschergruppe von den National Institutes of Health in Bethesda mit bis zu 5,7 Prozent angegeben werden (Walther et al. 1999A). Es lässt sich ableiten, dass für eine Fallzahl von 20 Patienten mit Phäochromozytom und Neurofibromatose Typ 1 eine Sammlung von mindestens 400 Neurofibromatose-Patienten notwendig ist, um 20 Fälle zu finden. Da solche Registerzahlen bislang nur von Fahsold et al. (2000) bekannt sind, dürfte dies die Erklärung sein, dass eine umfassende Darstellung dieser Erkrankungskombination bislang nicht existiert. Möglicherweise sind die angegebenen Eckdaten auch zu hoch angesetzt. Das Freiburger Internationale Phäochromozytom Register hat sich von einer regionalen Sammlung der in Freiburg operierten Patienten über ein überregionales Register (Neumann et al. 1993) zu einem internationalen Register entwickelt, das bis 2002 neben familiären Fällen 274 102 4 Diskussion nicht syndromale Phäochromozytome umfasste (Neumann et al. 2002A). Das Register wurde in der Folgezeit durch Meldung von zahlreichen Zentren ergänzt, wobei systematische epidemiologische Arbeiten in Zentral-Polen einen wesentlichen Anteil ausmachten. Damit waren gute Voraussetzungen für die Thematik dieser Dissertation gegeben. Beim Start dieser Arbeit waren 12 Fälle von Neurofibromatose Typ 1 und Phäochromozytom registriert. Durch gezielte Weitergabe des neuen Interessenschwerpunktes konnten in der Folge 8 Fälle aus Italien, 3 Fälle von einer kasuistischen Publikation aus Lübeck und 1 Fall aus der Schweiz aufgenommen werden. Die regelmäßig weiter erfolgten Meldungen erbrachten weitere 2 Patienten aus Behandlungszentren in Lothringen und Deutschland, wobei letztere Patientin aus Kuwait stammte. Damit gelang es, zum Aufnahmeschlussdatum 15.05.05 insgesamt 26 Fälle von Phäochromozytomen bei Neurofibromatose Typ 1 zu registrieren. Der Registerstand aller Phäochromozytome, d.h. aller Patienten mit funktionell aktiven Tumoren von Nebennierenmark und der Paraganglien in Abdomen und Thorax betrug zu diesem Zeitpunkt 624 Fälle. Zieht man hiervon die Fälle mit syndromalen Phäochromozytomen ab, so verbleiben 397 Fälle. Bezogen auf dieses Patientengut beträgt die Prävalenz von Neurofibromatose Typ 1 - Phäochromozytomen im Freiburger Internationalen Phäochromozytom-Register 26/397 und somit 6,5%. Dieser Wert ist aufgrund der gezielten Anfragen nach solchen Patienten als zu hoch einzuschätzen und lässt sich somit nicht verallgemeinern. Betrachtet man den Zeitpunkt der Diagnose der Neurofibromatose Typ 1Phäochromozytom Fälle unter der Prämisse, dass in einem gegebenen geographischen Raum die jährliche Erkrankungsrate konstant sein sollte (siehe Fallbeschreibungen und Tab. 43), so zeigen sich zwar keine überdurchschnittlichen Schwankungen pro Jahr, aber unklare geographische Grenzziehungen, womit die Aussagekraft der Prävalenzangabe von 6,5% weiter relativiert werden muss. Tab. 43: Diagnose und Herkunftsland der Patienten Fall 1 2 3 4 5 6 7 8 9 10 11 12 13 Diagnosejahr Herkunftsland 1995 2003 1994 2004 1985 1999 1999 2000 1990 1988 2002 1996 2002 Deutschland Deutschland Deutschland Kuwait Deutschland Deutschland Deutschland Deutschland Schweiz Italien Italien Italien Italien Fall 14 15 16 17 18 19 20 21 22 23 24 25 26 Diagnosejahr Herkunftsland 2001 2004 2004 2001 2004 1987 1997 1990 1992 2003 1993 1986 1990 Italien Italien Italien Italien Frankreich Großbritannien Polen Polen Polen Polen Polen Polen Polen 103 4 Diskussion Zusammenfassend konnten für diese Arbeit 26 Patienten mit Neurofibromatose Typ 1 und Phäochromozytom zusammengestellt werden. Es sind 15 Frauen und 11 Männer. Das Alter bei Diagnose rangiert zwischen 14 und 61 Jahren und lag im Mittel bei 41 Jahren. Alle Patienten zeigten adrenale Tumoren. Kein Patient hatte extraadrenale Tumoren. Multiple Tumoren wiesen 5 Patienten auf. Maligne Tumoren lagen bei 2 Patienten vor. Die Metastasen waren in Lungen und Knochen lokalisiert. Ein Patient erlag dem Tumorleiden. Im Folgenden werden die Ergebnisse als „Freiburg-Studie“ bezeichnet. Wie oben ausgeführt existieren ähnliche Fallsammlungen als Originalpublikation eines Zentrums oder eines Forschungskonsortiums nicht. Allerdings haben Walther et al (1999A) 148 Fallberichte der Literatur ausgewertet, nachdem sie eine große elektronische Suche für die Stichworte „Neurofibromatose“ und „von Recklinghausen“ der Jahre 1966 bis 1999 durchführten („Walther-Studie“ im Folgenden genannt). Die Ergebnisse werden im Vergleich mit der Freiburg-Studie in der Tabelle 44 gegenübergestellt. Das Durchschnittsalter der Patienten lag bei der Walther-Studie mit 42 Jahren im gleichen Bereich wie in der Freiburg-Studie (41 Jahre). In der Freiburg-Studie waren 9% der Patienten symptomfrei. Eine Hypertonie fand sich in der Walther-Studie nur bei 61%, während in der Freiburg-Studie 85% der Patienten erhöhte Blutdruckwerte aufwiesen. 61% der Patienten der Walther-Studie gaben Symptome an, die dem Phäochromozytom zugerechnet werden können; in der Freiburg-Studie waren es 74%. 22% der Patienten der Walther-Studie wiesen keine Phäochromozytom-assoziierten Symptome auf, in der Freiburg-Studie waren es 25%. In der Walther-Studie hatten 84 % unilaterale und 10% bilaterale adrenale Phäochromozytome. In der Freiburg-Studie wiesen 81% unilaterale und 19% bilaterale Phäochromozytome auf. Diese Unterschiede sind nicht erheblich. Sie könnten aber auch durch die Entwicklung der diagnostischen Möglichkeiten erklärt werden. Das Review von Walther et al. (1999A) geht bis in das Jahr 1966 zurück. Die MRT, die MIBG-Szintigraphie und die PET, die die höchste Sensitivität in der Phäochromozytom-Diagnostik aufweisen, wurden erst ab den 90er Jahren in größeren medizinischen Zentren regelmäßig bei den gegebenen Fragestellungen eingesetzt. Extraadrenale Phäochromozytome werden in der Walther-Studie mit 6%, in der Freiburg-Studie aber gar nicht beobachtet. Dies lässt vermuten, dass extraadrenale Phäochromozytome bei Neurofibromatose Typ 1 sehr selten vorkommen. 104 4 Diskussion Erwähnt werden soll in diesem Zusammenhang die klinische Studie zu Neurofibromatose Typ 1 und Phäochromozytom von Kalff et al. (1982). Sie untersuchten Neurofibromatose Typ 1 – Patienten, die einen Hypertonus aufwiesen. Bei 10 von 18 hypertensiven Patienten diagnostizierte man ein Phäochromozytom. In der Kalff-Studie sind Angaben zu Durchschnittsalter, Lokalisation, Dignität und Symptomen ähnlich wie in der Freiburg-Studie (Tab. 44). Eine maligne Entartung zeigten in der Freiburg-Studie 8%, in der Walther-Studie 12% und in der Kalff-Studie 10% der Patienten. Tab. 44: Tabellarischer Vergleich der Ergebnisse der Freiburg-Studie und zwei anderen Studien. Patienten (n) Alter bei Diagnose Durchschnittsalter Multi-fokal Unilateral adrenal Bilateral adrenal Extraadrenal Malignität Symptomfrei Hypertonus Phäochromozytomassoziierte Symptome NF1 (diese Arbeit) NF 1 (Walther et al. 1999A) NF 1 (Kalff et al. 1982) 26 14-61 Jahre 41 Jahre 19% 81% 19% 0% 8% 9% 85% 74% 148 2-74 Jahre 42 Jahre 17% 84% 10% 6% 12% 22% 61% 61% 10 15-62 Jahre 42 Jahre 20% 80% 20% 0% 10% 10% 90% 80% Zu den Phäochromozytom-assoziierten Symptomen zählen Kopfschmerz, Palpitationen und Schweißausbrüche. Angaben nach Walther et al. (1999A) und Kalff et al. (1982). Zusammenfassend lässt sich sagen, dass sich für mittleres Alter bei Diagnose, multifokale Phäochromozytome und Malignität nur sehr geringe Abweichungen zeigen. Dagegen sind in der Walther-Studie 6% extraadrenale Phäochromozytome aufgeführt, die weder in der eigenen Serie noch in der Kalff-Studie vorkamen. Bemerkenswert erscheint, dass Phäochromozytome lange keine Symptome verursachen können; der Anteil der Patienten lag in den Studien zwischen 9% und 22%. Die Einteilung der Neurofibromatose Typ 1 der WHO nach Schwergraden von Riccardi und Eichner (1992) in minimal (Grad 1), mild (Grad 2), moderat (Grad 3) und schwer (Grad 4) wurde im eigenen Patientengut wegen weitgehend fehlender entsprechender Angaben nicht durchgeführt. 105 4 Diskussion 4.1.1 Phäochromozytom-assoziierte Syndrome Phäochromozytome kommen sporadisch oder familiär vor. Die familiären Phäochromozytome haben somit eine genetische Ursache. Die Begriffe familiär bedingte und genetisch verursachte Phäochromozytome werden oft synonym verwendet. Man muss jedoch beachten, dass auch als Einzelfälle beobachtete Patienten mit Phäochromozytomen durchaus eine entsprechende Anlage aufweisen können. So zeigte die Freiburger Arbeitsgruppe, dass von 274 nicht syndromalen, d.h. sporadischen Phäochromozytomen bei 24% eine Mutation im Blut nachweisbar war (Neumann et al. 2002A). Dies sind sog. Keimbahnmutationen oder konstitutionelle Mutationen, die sich mit molekulargenetischen Techniken, ähnlich wie in dieser Arbeit beschrieben, in den Genen VHL, RET, SDHB oder SDHD (siehe unten) nachweisen lassen. Mutationen in den genannten Genen liegen in der Regel nicht nur den Phäochromozytomen sondern komplexen Krankheitsbildern zugrunde, die im Folgenden kurz beschrieben werden. Im von Hippel-Lindau Syndrom (VHL) kommen - neben Phäochromozytomen - retinale Angiome, Hämangioblastome des Zentralnervensystems (vorwiegend Kleinhirn, Hirnstamm und Rückenmark), Nierenkarzinome, Nierenzyste, Pankreaszyste, Inselzelltumore und Nebenhodenzystadenome vor. Andere zystische oder neoplastische Läsionen sind selten. Das Phäochromozytom tritt nicht bei allen VHL-Patienten auf; die Angaben schwanken; im Mittel werden 20% angegeben. Das zugrunde liegende Gen wird VHL genannt. Die Charakteristika der VHL-assoziierten Phäochromozytome werden in Tab. 45 gezeigt. Die Multiple Endokrine Neoplasie Typ 2 (MEN 2) ist charakterisiert durch das Auftreten von C-Zelltumoren der Schilddrüse (medulläres Schilddrüsenkarzinom) kombiniert mit Phäochromozytomen in ca. 50 % der Fälle. Die MEN 2 entsteht auf dem Boden konstitutioneller Mutationen des 21 Exons umfassenden RET-Gens. Bei Patienten mit Phäochromozytom wurden Mutationen nur in den Exons 10, 11 und 16 gefunden (Eng et al. 1996, Neumann et al. 2002A). Die häufigste Form MEN 2A zeigt in 15% einen Hyperparathyreoidismus. Bei der seltenen MEN 2B fehlt die Nebenschilddrüsenbeteiligung, dafür treten eine Schleimhautganglioneuromatose und ein marfanoider Habitus hinzu. Der dritte Subtypus, familiäres medulläres Schilddrüsenkarzinom (FMTC), zeigt keine weiteren Endokrinopathien. Die Charakteristika der MEN 2-assoziierten Phäochromozytome werden in Tab. 45 gezeigt. Das Paragangliom Syndrom Typ 1 (PGL 1) wurde anhand familiär auftretender Paragangliome der Hals-Schädelbasisregion beschrieben. Es entsteht auf dem Boden von Mutationen des so genannten SDHD Gens. Dabei steht SDH für das im Zitratzyklus und in 106 4 Diskussion der Atmungskette zentral bedeutsame Enzym Succinatdehydrogenase und D für die nach Buchstaben benannten Untereinheiten, hier also für die Untereinheit D (Baysal et al. 2000). Kurz nach dieser Beschreibung wurden von der Freiburg-Columbus/Ohio-Kooperationsgruppe Mutationen des SDHD Gens auch bei Patienten mit Phäochromozytomen beschrieben (Gimm et al. 2000). Andere Organe sind beim PGL 1 nicht involviert. Die Charakteristika der PGL 1- assoziierten Phäochromozytome werden in Tab. 45 gezeigt. Das Paragangliom Syndrom Typ 4 (PGL 4) beruht auf Mutationen der Succinatdehydrogenase Untereinheit B (Astuti et al. 2001). Die PGL 4 stellt die jüngste Entität dar und korreliert nach neuesten Erkenntnissen neben dem Phäochromozytom in seltenen Fällen auch mit dem Auftreten von Nierenkarzinomen (Vanharanta et al. 2004). Die Charakteristika der PGL 4-assoziierten Phäochromozytome werden in Tabelle 45 gezeigt. Tab. 45: Vergleich der Phäochromozytom-assoziierten Syndrome MEN 2 VHL SDHD (PGL1) SDHB (PGL4) 36 Jahre 18 Jahre 29 Jahre 26 Jahre 41 Jahre 44 67% 58% 74% 28% 19% 5% Adrenal fast immer adrenal 88% 53% 28% 100% 78% Extraadrenal Abdominal sehr selten 12% 21% 50% 0% 6% Thorakal sehr selten selten 18% 9% 0% 1% Paragangliome des Halses und der Schädelbasis sehr selten sehr selten 79% 31% 0% 0% 4% sehr selten sehr selten 35% 8% 10% Durchschnittsalter Multifokal Malignität Assoziierte Tumoren Gen-Name Gen-Lokalisation Nr. Exons Hämangiome medulläres HämangioSchilddrüsen blastome karzinom Nieren-, HyperparaPankreasthyreoidiszysten mus Nierenkarzino me NF1 Nicht(diese Studie) Syndromal keine Neurofibrome, Assoziation NierenzellIrishamartome, mit anderen karzinom Optikusgliome Tumoren - RET VHL SDHD SDHB NF1 ohne 10q11.2 3p25-26 11q23 1p36 17q11.2 - 21 3 4 8 57 - Angaben nach Neumann et al. (2002A), Pawlu et al. (2005) und Manger und Gifford (2002) 107 4 Diskussion Alle vier Syndrome werden autosomal dominant vererbt. Als Besonderheit gibt es beim Paragangliom Syndrom Typ 1 (PGL 1) den „parent of origin-effect“: Es entwickeln sich nur Tumoren bei Personen, die die Mutation vom Vater erben. Mutationsträger, deren Mütter die Mutation weitergaben, bleiben gesund. Allen diesen Syndromen ist das Auftreten der Phäochromozytome gemeinsam. Die Unterschiede lassen sich aus Tabelle 45 ableiten. Das Alter bei Diagnose des Phäochromozytoms liegt bei der MEN 2 unter 30 Jahren (Nguyen et al. 2001), bei VHL Patienten um 30 Jahre (Bauters et al. 2003) und bei PGL 1 und PGL 4 im Bereich von 31 Jahren (Neumann et al. 2002A). Bei der Neurofibromatose Typ 1 liegt nach der Freiburg-Studie der Durchschnitt bei 41 Jahren. Damit ist das mittlere Alter ähnlich hoch wie bei Patienten mit sporadischem Phäochromozytom (44 Jahre, Tab. 45). Die Lokalisation der Phäochromozytome ist bei der MEN 2 fast immer adrenal (Bryant et al. 2003). Multifokale Tumoren kommen bei der MEN 2 in 67% der Patienten vor (Modigliani et al. 1995). Im VHL-Syndrom überwiegen mit 88% adrenale Phäochromozytome, wovon 58% multifokal auftreten (Walther et al. 1999B). Dagegen sind beim PGL 4 nur etwa 28% der Phäochromozytome adrenal lokalisiert. Extraadrenale Tumoren fanden sich abdominal bei 50% und thorakal bei 9% der Patienten mit PGL 4 (Neumann et al. 2002A). Beim PGL1 treten extraadrenale abdominale Phäochromozytome bei 21% und thorakale bei 18% der Patienten auf. Multiple Tumoren finden sich beim PGL1 in 74% der Patienten (Neumann et al. 2002A). Differierend hierzu zeigen Patienten mit Neurofibromatose Typ 1 seltener multiple Phäochromozytome (in der Freiburg-Studie 19%). Die Malignität der Tumoren liegt bei MEN 2 in 4% (Modigliani et al. 1995). Auch beim VHLSyndrom entarten die Phäochromozytome selten. Bei PGL 4 hingegen kommt es in 35% zu maligner Entartung (Neumann et al. 2002A). Für die PGL 1 sind maligne Phäochromozytome nicht beschrieben. Bei der Neurofibromatose Typ 1 traten in der Freiburg-Studie bei 8% der Patienten maligne Phäochromozytome auf . Von besonderem Interesse ist Fall 17. Es besteht zweifelsfrei eine Neurofibromatose Typ 1 und ein adrenales Phäochromozytom. Darüber hinaus wurde ein medulläres Schilddrüsenkarzinom operiert und histologisch gesichert. Des Weiteren wurde ein Nebenschilddrüsenadenom bei Hyperparathyreoidismus entfernt und ebenfalls histologisch gesichert. Es liegt also das eigentümliche Zusammentreffen einer Neurofibromatose Typ 1 und einer Multiplen Endokrinen Neoplasie Typ 2 vor. Ähnliche Beobachtungen sind in der verfügbaren Literatur nicht zu finden. 4 Diskussion 108 4.2 Molekulargenetische Ergebnisse Die molekulargenetischen Untersuchungen, die den Ergebnissen dieser Arbeit zugrunde liegen, erfolgten mit der derzeit modernsten Technik unter Berücksichtigung einer Optimierung der notwendigen Arbeitszeit und der vertretbaren Kosten. Die wesentlichen Schritte waren DNA-Extraktion aus Blutproben und ein Suchverfahren (Mutationsscreening), um die teure und hinsichtlich Auswertung aufwendige Sequenzierung auf das Nötigste zu minimieren. Als Screeningverfahren wurde die Denaturing High Performance Liquid Chromatography (DHPLC) eingesetzt (Gerät WAVE der Firma Transgenomics). Mit diesem Verfahren werden zuvor durch PCR amplifizierte DNA-Abschnitte durch Schmelzkurven abgebildet und DNASequenzvarianten in Kurvenabweichungen erkennbar. Im Vergleich zur bislang im Labor der Arbeitsgruppe eingesetzten Single Strand Confirmation Polymorphism (SSCP) Methode arbeitet das WAVE-Gerät vollautomatisch und wesentlich schneller. Der programmierte Ablauf erlaubt ein Beladen des Gerätes mit Probenserien mehrfach am Tag, womit auch die Nachtstunden genutzt werden können. Als grober Anhalt kann die Zeitersparnis auf ein Fünftel des bisherigen Zeitaufwandes genannt werden. Die Trefferquoten für die Detektion bekannter Mutationen (Sensitivität) werden für die DHPLC-Methode mit etwa 99% angegeben (Oefner et al. 1998). Durch diese technischen Neuerungen wurde es möglich, große Gene in einer Serie von Probanden zu untersuchen, was zuvor von führenden Gruppen anhand der SSCP und vergleichbaren Methoden nicht für durchführbar gehalten wurde. Zu derart großen Genen zählt das NF1-Gen mit insgesamt 57 Exons. Ein weiteres großes Problem der Analyse des NF1-Gens ist das Vorkommen vieler so genannter Pseudogene. Hierunter versteht man Sequenzen der DNA, die weitestgehende Übereinstimmungen mit Abschnitten des NF1-Gens aufweisen. Es besteht somit die Möglichkeit, dass bei der DNA-Amplifikation – in der Regel eines Exons – mit entsprechenden Primern Abschnitte von Pseudogenen mitamplifiziert werden und die Ergebnisse somit verfälscht sind. Die Zahl der Pseudogene des NF1-Gens liegt bei 36 (Luijten et al. 2001). Im Labor in Padua hat der Verfasser in Zusammenarbeit mit Frau Dr. Schiavi die Analyse des NF1-Gens begonnen, aber nicht vollständig abschließen können. Im Freiburger Labor wurde schließlich eine Wissenschaftlerin als Hauptmitarbeiterin und Volltagskraft, Frau Dr. Bausch, gewonnen, die die molekulargenetischen Untersuchungen mittels der neuen Methode, der 109 4 Diskussion DHPLC, durchgeführt hat und dabei auffällige Exons sequenziert hat. Nach Fertigstellung der Analysen von Frau Dr. Bausch hat der Verfasser die Sequenzierungsdaten des Labors aus Padua noch auswerten können. Dies ermöglichte einen Vergleich bzw. eine Bestätigung der Freiburger Befunde. Die Ergebnisse der Analysen der 26 Patienten mit Neurofibromatose Typ 1 und Phäochromozytom zeigten in 18 Fällen 19 Mutationen. Eine Mutation kam zweimal vor. Diese 18 verschiedenen Mutationen sind in Abbildung 61 graphisch hinsichtlich ihrer Lokalisation im NF1-Gen dargestellt (vgl. auch Tab. 40). Es zeigt sich, dass die Mutationen auf das gesamte Gen verteilt sind. Dabei liegen in 13 Exons eine Mutation und in 3 Exons 2 Mutationen. Protein-verkürzende Mutationen kamen 13x vor. Dabei handelte es sich 2x um Stopp-Codons, 3x um Insertionen von einem oder zwei Nukleotiden, 2x um Deletionen von einem Nukleotid und 6x um Splice site Mutationen. 6 Mutationen waren Missense Mutationen (einschließlich der Mutation NF1 c. 1466 A/G, siehe unten) nach dem Abgleich mit den Befunden von 100 gesunden Probanden (siehe Methodik). Folgende Besonderheiten wurden beobachtet: 1. Ein Fall (Fall 10) wies eine Splice site Mutation (NF1 c. 1062+2 T/C) und eine Missense Mutation (NF1 c. 7833 T/A) auf. Die Interpretation ist schwierig. Es ist sehr ungewöhnlich, dass neben einer Splice site Mutation eine Missense Mutation beim selben Patienten vorkommt. Es kann ein sehr seltener Polymorphismus nicht ausgeschlossen werden. Die Entscheidung bleibt bis auf weiteres offen. 2. Die Mutation NF1 c. 1466 A/G (Fall 1 und 17) ist nur formal eine Missense Mutation, weil das neue Codon für eine Aminosäure codieren könnte. Jedoch bildet sich durch die neue Nukleotidsequenz eine Splice site AG-GT, die zu einem Verlust von 62 bp führt. Dies wurde von Messiaen et al. (1999) durch RNA-Analyse gezeigt. 3. Die Insertion NF1 c. 2849 ins TT ist homozygot. Homozygote Mutationen kommen in der Regel nur bei autosomal-rezessiven Erkrankungen vor. Die Analyse müsste auf die Eltern ausgedehnt werden, um weitere Erkenntnisse zu erhalten. Leider war dies nicht möglich. Das Verständnis dieser Veränderung bleibt somit offen. Phänotypisch unterscheidet sich der zugehörige Fall 5 von den anderen 25 Fällen nicht. Nur drei der festgestellten Mutationen sind in der Literatur beschrieben: 1. NF1 c. 61-1 G/A von Fahsold et al. (2000) 2. NF1 c. 1466 A/G von Fahsold et al. (2000) 3. NF1 c. 3826 C/T von Heim et al. (1995) Somit sind 15 Mutationen in dieser Dissertation neu beschrieben. 4 Diskussion 110 Abb. 61: Schematische Darstellung der Lokalisation der in dieser Arbeit detektierten Mutationen. Grau unterlegt ist die Serin-Cystein-reiche Domäne zwischen Exon 11 und 17. Grün unterlegt die “GAP-related Domain” (GRD). * = in der Literatur beschrieben. 111 4 Diskussion 3 DNA-Varianten innerhalb der Exons 2, 5 und 13 waren auch bei 100 Normalkontrollen vorhanden; dies sind somit Polymorphismen. Die nachträglich erfolgte Auswertung der Sequenzierungsdaten aus Padua ergab, dass 6 Mutationen bestätigt werden können. Die anderen in Freiburg gefundenen Mutationen lagen in Exons, die in Padua nicht analysiert wurden, oder sie fanden sich bei Fällen, die in Padua nicht sequenziert wurden. Die Detektionsrate der Freiburg-Studie für intraexonische Mutationen lässt sich mit 18/26 = 69% angeben (18 Mutationen bei 26 Patienten). Warum 31% der Mutationen nicht gefunden wurden, kann drei Gründe haben. Am wahrscheinlichsten ist, dass Deletionen größerer DNAAbschnitte vorliegen. Solche Deletionen entziehen sich der DNA-Amplifikation und somit der Analyse mit der eingesetzten Methodik. Zweitens kann die DHPLC nicht zur Aufdeckung geführt haben, weil die Sensitivität „nur“ 99%, nicht 100% beträgt. Drittens ist es möglich, dass Patienten mit Neurofibromatose Typ 1 ein Mosaik aufweisen (genomische Unterschiede verschiedener Körperzellen), so dass in den untersuchten Blut-Lymphozyten, aus denen die DNA stammt, die Mutation nicht vorgelegen hat und somit nicht erkennbar war. Zahlreiche Publikationen wurden inzwischen vorgelegt, in denen über Mutationen des NF1Gens berichtet wurde. Hieraus wurden acht Arbeiten herausgegriffen, die einen ähnlichen Ansatz wie die Freiburg-Studie hatten. Dies war die komplette Analyse des NF1-Gens auf intraexonische Mutationen in einem größeren Patientengut. In Tabelle 46 sind diese Arbeiten aufgelistet mit Zahl der untersuchten Probanden, Methodik und wesentlichen Ergebnissen. Ähnlich wie in der Freiburg-Studie wurde die DHPLC bislang nur in den Studien von Han et al. (2001), de Luca et al. (2003) und Upadhyaya et al. (2004) zum Mutationsscreening eingesetzt, während die anderen Studien auf weniger sensitive Methoden wie SSCP, Sequenzierung (DGS), Fluoreszenz In Situ Hybridisierung (FISH), Heteroduplex-Analyse (HDA) oder Temperature Gradient Gel Electrophoresis (TGGE), sowie Protein Truncating Test (PTT) beruhten. 112 4 Diskussion Tab. 46: Zusammenfassung der im Text beschriebenen Studien zur Mutationssuche im NF1-Gen. NF1Patienten Methodik Kern-Ergebnisse Upadhyaya et al. (1997)* 320 HDA, SSCP 16 Mutationen in der GRD, 14 neue Mutationen Abernathy et al. (1997) 67 HDA, SSCP Detektionsrate 19%; 13 Mutationen in 45 Exons Messiaen et al. (1999)** 232 PTT 9 Mutationen in Exon 10b Messiaen et al. (2000)** 67 PTT, FISH Detektionsrate aller Methoden zusammen 95%; 32 neue Mutationen, Fahsold et al. (2000) 521 TGGE, DGS Detektionsrate 53%; 161 neue Mutationen u.a. der GRD Han et al. (2001)* 111 DHPLC Sensitivität DHPLC 97%; Detektionsrate 68%; 22 neue Mutationen De Luca et al. (2003) 40 DHPLC Detektionsrate 72%; 18 neue Mutationen Upadhyaya et al. (2004)* 43 DHPLC Detektionsrate 58%; 11 neue Mutationen Freiburg-Studie (2005) 26 DHPLC Nur Patienten mit NF1 und Phäochromozytom; Detektionsrate 69%; 15 neue Mutationen Autoren * = Ein Zentrum, daduch Überschneidungen der NF1-Patienten möglich; ** = Überschneidungen der NF1-Patienten möglich. HDA = Heteroduplex-Analyse; SSCP = Single Strand Confirmation Polymorphism; PTT = Protein Truncation Test; FISH = Fluoreszenz in situ Hybridisierung; TGGE = Temperature Gradient Gel Electrophoresis; DGS = Direct Genome Sequencing. All diese Serien wurden darauf geprüft, ob es Abschnitte im NF1-Gen gibt, in denen Mutationen gehäuft auftreten. Dies ist z.B. für das RET-Gen, das der MEN 2 zugrunde liegt (siehe oben), bekannt. Über 90% der Mutationen liegen im Exon 11, Codon 634. Solche „Hotspots“ sind allein schon aus praktischen Gesichtspunkten interessant, weil bei gegebener klinischer Befundkonstellation die Analyse auf Hotspots konzentriert werden kann. Hotspots im NF1-Gen wurden diskutiert für Exon 31 und 37 von Upadhyaya und Cooper (1998), Exon 10b von Messiaen et al. (1999), Exon 4b und Exon 37 von Fahsold et al. (2000) und Exons 10a-10c und 37 zusammen geben Messiaen et al 30 % an. In der Freiburg-Studie sind es 5/19 Mutationen, d.h. 26%. Ein anderer interessanter Aspekt ist die Frage, ob Mutationen gehäuft in Regionen des NF1-Gens vorkommen, denen nach derzeitigem Wissenstand besondere Funktionen zugeschrieben werden. Hier sind zu nennen die so genannte GAP-related Domain (GRD) in Exons 20-27a (vergleiche Einleitung) und eine Cystein/Serin-reiche Domäne in den Exons 11 bis 17, die ATP-Bindung anregen kann (Fahsold et al. 2000). Fahsold et al. (2000) identifizierten 28 verschiedene Mutationen (10%) in der GRD und in der Cystein/Serinreichen Domäne. De Luca et al. (2003) beschrieben 4 Mutationen (19%) in der GRD und 9 in der Cystein/Serin-reichen Region (31%). In der Freiburg-Studie sind es in der GRD 2/19 Mutationen (11%) und in der Cystein/Serin-reichen Region 3/19 Mutationen (16%). 113 4 Diskussion Alle diese Studien und weitere wie die von Carey et al. (1999) und von Rasmussen et al. (2000) suchten nach Geno-Phänotyp-Beziehungen. Es ließen sich aber keine Besonderheiten herauskristallisieren. Wie schon eingangs dieser Diskussion gesagt, findet sich zur Thematik Neurofibromatose Typ 1 und Phäochromozytom nur die Studie von Kalff et al. (1982) mit 10 Fällen. Eine entsprechende molekulargenetische Analyse wird mit der Freiburg-Studie erstmals präsentiert. Spezielle Regionen des NF1-Gens, die ursächlich mit der Entstehung des Phäochromozytoms in Zusammenhang gebracht werden können, lassen sich nach Abgleich mit den Mutationen der diskutierten Studien und der Zusammenfassung aller bekannten Mutationen unter http://www.hgmd.org, den über 600 Mutationen der Human Genome Mutation Database des Institutes für Medizinische Genetik der Universität Cardiff, nicht erkennen. Mit einer Detektionsrate von 69% liegt die Freiburg-Studie international methodisch an der Spitze. Diese Quote wird sicherlich noch ansteigen, wenn die im Aufbau begriffene Analyse des NF1-Gens mittels Real Time PCR auf größere Deletionen ausgewertet werden kann. Wie das überzufällige Zusammentreffen von Neurofibromatose Typ 1 und Phäochromozytom zu erklären ist, muss daher anderen Studien überlassen werden. 114 115 5 Zusammenfassung 5 Zusammenfassung Phäochromozytome sind Tumoren des Nebennierenmarkes, die Katecholamine produzieren und sezernieren. Die Tumorsyndrome, bei denen ein Phäochromozytom vorkommt, sind: Multiple endokrine Neoplasie Typ 2 (MEN 2), von Hippel-Lindau Syndrom (VHL), die Paragangliom-Syndrome Typ 1 und Typ 4 sowie die Neurofibromatose von Recklinghausen (NF1); die Inzidenz von Phäochromozytomen bei NF1 beträgt 0,1 – 5,7 %. Die NF1 ist eine autosomal dominante Erkrankung, deren Inzidenz bei 1:3500 Lebendgeburten liegt. Sie wird klinisch diagnostiziert. Ziel der Arbeit war es, eine hohe Zahl an Fällen klinisch zu charakterisieren und Mutationen des NF1-Gens zu bestimmen. Aufnahmekriterium war die klinisch gesicherte Neurofibromatose Typ 1 und das Vorhandensein eines Phäochromozytom. Die klinischen Daten der Patienten wurden gesammelt und dargestellt. Bis zum 15. Mai 2005 sind 26 Fälle in die Studie aufgenommen worden. 8 Fälle stammen aus Deutschland, ebenso viele aus Italien, 7 aus Polen, ein Fall jeweils aus Frankreich, der Schweiz und England. Als relevante klinische Daten wurden die allgemeinen Patientendaten, die Eigen-, die Familien-, sowie die aktuelle Anamnese, die klinischen Untersuchungsbefunde, die Diagnostik, sowie die Therapie und der Verlauf erhoben. 5/26 Patienten (19%) hatten bilaterale Phäochromozytome. Es fanden sich keine extraadrenalen Phäochromozytome. In 2 Fällen (8%) kam es zur malignen Entartung. Molekulargenetisch wurden die Analysen des NF1-Gens mittels „Denaturing High Performance Liquid Chromatography“ (DHPLC) und Sequenzierung durchgeführt. Die Amplifikation der vielen Pseudogene des NF1-Gens wurde durch Allel-spezifische PCR ausgeschlossen. In den 26 Fällen wurden bei 18 Patienten 19 Mutationen identifiziert, wovon 15 erstmals beschrieben wurden. Darunter waren 8 Basensubstitutionen; davon führten 2 zum Abbruch der Proteinbiosynthese, während 6 einen Aminosäureaustausch bewirkten. Weiterhin fanden sich 2 Deletionen und 3 Insertionen von 1 oder 2 Nukleotiden, die zum Verschieben des Leserasters führen. Splice Defekte wurden in 6 Fällen identifiziert. Die Mutationen fanden sich in den Exons 2, 3(2x), 7, 10b(2x), 10c, 13, 15, 16, 22, 23-2, 29, 37(2x), 41 und 44. Dies ist die bisher einzige Studie einer größeren Serie von Phäochromozytomen bei Neurofibromatose Typ 1. Die Detektionsrate der NF1-Mutationen mittels DHPLC ist 69% und liegt damit international an der Spitze. 116 117 6 Literaturverzeichnis 6 Literaturverzeichnis Abernathy CR, Rasmussen SA, Stalker HJ, Zori R, Driscoll DJ, Williams CA, Kousseff BG, Wallace MR (1997) NF1 mutation analysis using a combined heteroduplex/SSCP approach. Hum Mutat 9: 548-54 Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, George E, Skoldberg F, Husebye ES, Eng C, Maher ER (2001) Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet 69: 49-54 Averbuch SD, Steakley CS, Young RC, Gelmann EP, Goldstein DS, Stull R, Keiser HR (1988) Malignant pheochromocytoma: effective treatment with a combination of cyclophosphamide, vincristine, and dacarbazine. Ann Intern Med 109: 267-73 Bauters C, Vantyghem MC, Leteurtre E, Odou MF, Mouton C, Porchet N, Wemeau JL, Proye C, Pigny P (2003) Hereditary phaeochromocytomas and paragangliomas: a study of five susceptibility genes. J Med Genet 40: e75 Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, Richard CW 3rd, Cornelisse CJ, Devilee P, Devlin B (2000) Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 287: 848-51 6 Literaturverzeichnis 118 Breathnach R, Benoist C, O'Hare K, Gannon F, Chambon P (1978) Ovalbumin gene: evidence for a leader sequence in mRNA and DNA sequences at the exon-intron boundaries. Proc Natl Acad Sci U S A 75: 4853-7 Bryant J, Farmer J, Kessler LJ, Townsend RR, Nathanson KL (2003) Pheochromocytoma: the expanding genetic differential diagnosis. J Natl Cancer Inst 95: 1196-204 Campbell L, Mouratidis B, Sullivan P (1996) Improved detection of disseminated pheochromocytoma using post therapy I-131 MIBG scanning. Clin Nucl Med 21: 960-3 Carey JC, Viskochil DH (1999) Neurofibromatosis type 1: A model condition for the study of the molecular basis of variable expressivity in human disorders. Am J Med Genet 89: 7-13 Cawthon RM, Weiss R, Xu GF, Viskochil D, Culver M, Stevens J, Robertson M, Dunn D, Gesteland R, O'Connell P (1990) A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell 13: 193-201 De Luca A, Buccino A, Gianni D, Mangino M, Giustini S, Richetta A, Divona L, Calvieri S, Mingarelli R, Dallapiccola B (2003) NF1-Gene analysis based on DHPLC. Hum Mutat 21: 171-2 119 6 Literaturverzeichnis DeClue JE, Papageorge AG, Fletcher JA, Diehl SR, Ratner N, Vass WC, Lowy DR (1992) Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell 69: 265-73 Eisenberg AA, Wallerstein H (1932) Pheochromocytoma of the suprarenal medulla (paraganglioma). Arch Pathol 14: 818–836 Eisenhofer G, Lenders JW, Pacak K (2004) Biochemical diagnosis of pheochromocytoma. Front Horm Res 31: 76-106 Eisenhofer G, Lenders JWM, Linehan WM, Walther MM, Goldstein DS, Keiser HR (1999) Plasma normetanephrine and metanephrine for detecting pheochromocytoma in von Hippel-Lindau disease and multiple endocrine neoplasia type 2. New Engl J Med 340: 1872-9 Eng C, Clayton D, Schuffenecker I, Lenoir G, Cote G, Gagel RF, van Amstel HK, Lips CJ, Nishisho I, Takai SI, Marsh DJ, Robinson BG, Frank-Raue K, Raue F, Xue F, Noll WW, Romei C, Pacini F, Fink M, Niederle B, Zedenius J, Nordenskjold M, Komminoth P, Hendy GN, Mulligan LM (1996) The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA 276: 1575-9 Fahsold R, Hoffmeyer S, Mischung C, Gille C, Ehlers C, Kucukceylan N, Abdel-Nour M, Gewies A, Peters H, Kaufmann D, Buske A, Tinschert S, Nurnberg P (2000) Minor lesion mutational spectrum of the entire NF1-gene does not explain its high mutability but points to a functional domain upstream of the GAP-related domain. Am J Hum Genet 66: 790-818 6 Literaturverzeichnis 120 Feldkamp MM, Angelov L, Guha A (1999) Neurofibromatosis type 1 peripheral nerve tumors: aberrant activation of the Ras pathway. Surg Neurol 51: 211-8 Fraenkel F (1886) Ein Fall von doppelseitigem, völlig latent verlaufenen Nebennierentumor und gleichzeitiger Nephritis mit Veränderungen am Circulationsapparat und Retinitis. Arch Pathol Anat Physiol Klin Med 103: 244-263 Gimm O, Armanios M, Dziema H, Neumann HP, Eng C (2000) Somatic and occult germ-line mutations in SDHD, a mitochondrial complex II gene, in nonfamilial pheochromocytoma. Cancer Res 60: 6822-5 Gimm O, Koch CA, Januszewicz A, Opocher G, Neumann HP (2004) The genetic basis of pheochromocytoma. Front Horm Res 31: 45-60 Glushien AS, Mansuy MM, Littman DS (1953) Pheochromocytoma. Its relationship to the neurocutaneous syndromes. Am J Med 14: 318 Gutmann DH, Aylsworth A, Carey JC, Korf B, Marks J, Pyeritz RE, Rubenstein A, Viskochil D (1997) The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 278: 51-7 Han SS, Cooper DN, Upadhyaya MN (2001) Evaluation of denaturing high performance liquid chromatography (DHPLC) for the mutational analysis of the neurofibromatosis type 1 ( NF1) gene. Hum Genet 109: 487-97 121 6 Literaturverzeichnis Heim RA, Kam-Morgan LN, Binnie CG, Corns DD, Cayouette MC, Farber RA, Aylsworth AS, Silverman LM, Luce MC (1995) Distribution of 13 truncating mutations in the neurofibromatosis 1 gene. Hum Mol Genet 4: 975-81 Hirsch NP, Murphy A, Radcliffe JJ (2001) Neurofibromatosis: clinical presentations and anaesthetic implications. Br J Anaesth 86: 555-64 Hoegerle S, Nitzsche E, Altehoefer C, Ghanem N, Manz T, Brink I, Reincke M, Moser E, Neumann HP (2002) Pheochromocytomas: detection with 18F DOPA whole body PET-initial results. Radiology 222: 507-12 Huson SM, Hughes RAC (1994) The neurofibromatoses: a pathogenetic and clinical overview. London: Chapman&Hall Kalff V, Shapiro B, Lloyd R, Sisson JC, Holland K, Nakajo M, Beierwaltes WH (1982) The spectrum of pheochromocytoma in hypertensive patients with neurofibromatosis. Arch Intern Med 142: 2092-8 Kaltsas GA, Papadogias D, Grossman AB (2004) The clinical presentation (symptoms and signs) of sporadic and familial chromaffin cell tumours (phaeochromocytomas and paragangliomas). Front Horm Res 31: 61-75 Kehrer-Sawatzki H, Schwickardt T, Assum G, Rocchi M, Krone W (1997) A third neurofibromatosis type 1 (NF1) pseudogene at chromosome 15q112 Hum Genet 100: 595-600 6 Literaturverzeichnis 122 Legius E, Marchuk DA, Hall BK, Andersen LB, Wallace MR, Collins FS, Glover TW (1992) NF1-related locus on chromosome 15. Genomics 13: 1316-1318 Lenders JW, Pacak K, Walther MM, Linehan WM, Mannelli M, Friberg P, Keiser HR, Goldstein DS, Eisenhofer G (2002) Biochemical diagnosis of pheochromocytoma: which test is best? JAMA 287: 1427-34 Li Y, Bollag G, Clark R, Stevens J, Conroy L, Fults D, Ward K, Friedman E, Samowitz W, Robertson M (1992) Somatic mutations in the neurofibromatosis 1 gene in human tumors. Cell 69: 275-81 Li Y, O'Connell P, Breidenbach HH, Cawthon R, Stevens J, Xu G, Neil S, Robertson M, White R, Viskochil D (1995) Genomic organization of the neurofibromatosis 1 gene (NF1). Genomics 25: 9-18 Luijten M, Fahsold R, Mischung C, Westerveld A, Nurnberg P, Hulsebos TJ (2001) Limited contribution of interchromosomal gene conversion to NF1-Gene mutation. J Med Genet 38: 481-5 Luijten M, Wang Y, Smith BT, Westerveld A, Smink LJ, Dunham I, Roe BA, Hulsebos TJ (2000) Mechanism of spreading of the highly related neurofibromatosis type 1 (NF1) pseudogenes on chromosomes 2, 14 and 22. Eur J Hum Genet 8: 209-214 Manger WM, Gifford RW (1993) Pheochromocytoma: current diagnosis and management. Cleve Clin J Med 60: 365-78 123 6 Literaturverzeichnis Manger WM, Gifford RW (2002) Pheochromocytoma. J Clin Hypertens 4: 62-72 Marchuk DA, Saulino AM, Tavakkol R, Swaroop M, Wallace MR, Andersen LB, Mitchell AL, Gutmann DH, Boguski M, Collins FS (1991) cDNA cloning of the type 1 neurofibromatosis gene: complete sequence of the NF1-gene product. Genomics 11: 931-40 Messiaen LM, Callens T, Mortier G, Beysen D, Vandenbroucke I, Van Roy N, Speleman F, De Paepe A (2000) Exhaustive mutation analysis of the NF1-gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum Mutat 15: 541-555 Messiaen LM, Callens T, Roux KJ, Mortier GR, De Paepe A, Abramowicz M, Pericak-Vance MA, Vance JM, Wallace MR (1999) Exon 10b of the NF1-Gene represents a mutational hotspot and harbors a recurrent missense mutation Y489C associated with aberrant splicing. Genet Med 1: 248-253 Modigliani E, Vasen HM, Raue K, Dralle H, Frilling A, Gheri RG, Brandi ML, Limbert E, Niederle B, Forgas L (1995) Pheochromocytoma in multiple endocrine neoplasia type 2: European study. The Euromen Study Group. J Intern Med 238: 363-7 Mount SM (1982) A catalogue of splice junction sequences. Nucleic Acids Res 22: 459-72 6 Literaturverzeichnis 124 Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K, Januszewicz A, Eng C, Smith WM, Munk R, Manz T, Glaesker S, Apel TW, Treier M, Reineke M, Walz MK, Hoang-Vu C, Brauckhoff M, Klein-Franke A, Klose P, Schmidt H, Maier-Woelfle M, Peczkowska M, Szmigielski C, Eng C for The Freiburg-Warsaw-Columbus Pheochromocytoma Study Group (2002A) Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 346: 1459-66 Neumann HP, Hoegerle S, Manz T, Brenner K, Iliopoulos O (2002B) How many pathways to pheochromocytoma? Semin Nephrol 22: 89-99 Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, Buchta M, Franke G, Klisch J, Bley TA, Hoegerle S, Boedeker CC, Opocher G, Schipper J, Januszewicz A, Eng C for the European-American Paraganglioma Study Group (2004) Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 292: 943-51 Neumann HPH, Berger DP, Sigmund G, Blum U, Schmidt D, Parmer RJ, Volk B, Kirste G (1993) Pheochromocytoma, multiple endocrine neoplasia type 2, and Von-Hippel-Lindau disease. N Engl J Med 329: 1531-8 Newton CR, Graham A, Heptinstall LE, Powell SJ, Summers C, Kalsheker N, Smith JC, Markham, AF (1989) Analysis of any point mutation in DNA. The amplification refractory mutation system. Nucleic Acids Res 17: 2503-16 125 6 Literaturverzeichnis Nguyen L, Niccoli-Sire P, Caron P, Bastie D, Maes B, Chabrier G, Chabre O, Rohmer V, Lecomte P, Henry JF, Conte-Devolx B; French Calcitonin Tumors Study Group (2001) Pheochromocytoma in multiple endocrine neoplasia type 2: a prospective study. Eur J Endocrinol 144: 37-44 Oefner PJ, Underhill PA (1998) DNA mutation detection using denaturing highperformance liquid chromatography (DHPLC). Current Protocols in Human Genetics Wiley & Sons, New York, Supplement 19:7.10.17.10.12. Pawlu C, Bausch B, Reisch N, Neumann HP (2005) Genetic testing for pheochromocytoma-associated syndromes. Ann Endocrinol 66: 178-85 Pick L (1912) Das Ganglioma embryonale sympathicum - eine typisch bösartige Geschwulstform des sympathischen Nervensysthems. Berl Klin Wochenschr 49: 16-22 Purandare SM, Huntsman Breidenbach H, Li Y, Zhu XL, Sawada S, Neil SM, Brothman A, White R, Cawthon R, Viskochil D (1995) Identification of neurofibromatosis 1 (NF1) homologous loci by direct sequencing, fluorescence in situ hybridization, and PCR amplification of somatic cell hybrids. Genomics 30: 476-485 Rasmussen SA, Friedman JM (2000) NF1-gene and neurofibromatosis 1. Am J Epidemiol 151: 33-40 126 6 Literaturverzeichnis Régnier V, Meddeb M, Lecointre G, Richard F, Duverger A, Nguyen VC, Dutrillaux B, Bernheim A, Danglot G (1997) Emergence and scattering of multiple neurofibromatosis (NF1)- related sequences during hominoid evolution suggest a process of pericentromeric interchromosomal transposition. Hum Mol Genet 6: 9-16 Riccardi VM (1981) Von Recklinghausen neurofibromatosis. N Engl J Med 305: 1617-27 Riccardi VM, Eichner JE (1992) Neurofibromatosis: phenotype, natural history and pathogenesis. 2nd ed. Baltimore, MD. John Hopkins University Press Ruggirei M, Pulizzi A (2003) From Aldrovandi’s "Homuncio" (1592) to Buffon’s girl (1749) and the "Wart Man" of Tilesius (1793): antique illustrations of mosaicism in neurofibromatosis? J Med Genet 40: 227-232 Saiki RK, Scharf S, Faloona F, Mullis KB, Horn GT, Erlich HA, Arnheim N (1985) Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 230: 1350–4 Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 74: 5463-7 Scherczinger CA, Bourke MT, Ladd C, Lee HC (1997) DNA extraction from liquid blood using QIAamp. J Forensic Sci 42: 893-6 127 6 Literaturverzeichnis Stumpf DA, Alksne JF, Annegers JF, Brown SS, Conneally PM, Housman D, Leppert MF, MillerJP, Moss ML, Pileggi AJ, Rapin I, Strohman RC, Swanson LW, Zimmerman A (1988) Neurofibromatosis: conference statement. Arch Neurol 45: 575-578 Suzuki H, Ozawa N, Taga C, Kano T, Hattori M, Sakaki Y (1994) Genomic analysis of a NF1-related pseudogene on human chromosome 21. Gene 147: 277-280 Suzuki S (1910) Über zwei Tumoren aus Nebennierenmarkgewebe. Berlin Klin Wchnschr 47: 1623 The Sanger Centre, The Washington University Genome Sequencing Center Toward a complete human genome sequence. Genome Res 8: 1097-108 Ugozzoli L, Wallace RB (1991) Allele-specific polymerase chain reaction. Methods Enzymol 2: 42-48 Upadhyaya M, Cooper DN (1998) Neurofibromatosis type 1: from genotype to phenotype. Oxford BIOS Scientific Publ Upadhyaya M, Han S, Consoli C, Majounie E, Horan M, Thomas NS, Potts C, Griffiths S, Ruggieri M, von Deimling A, Cooper DN (2004) Characterization of the somatic mutational spectrum of the neurofibromatosis type 1 (NF1) gene in neurofibromatosis patients with benign and malignant tumors. Hum Mutat 23: 134-46 6 Literaturverzeichnis 128 Upadhyaya M, Osborn MJ, Maynard J, Kim MR, Tamanoi F, Cooper DN (1997) Mutational and functional analysis of the neurofibromatosis type 1 (NF1) gene. Hum Genet 99: 88-92 van Gils AP, van der Mey AG, Hoogma RP, Falke TH, Moolenaar AJ, Pauwels EK, van Kroonenburgh MJ (1990) Iodine-123-metaiodobenzylguanidine scintigraphy in patients with chemodectomas of the head and neck region. J Nucl Med 31: 1147-55 Vanharanta S, Buchta M, McWhinney SR, Virta SK, Peczkowska M, Morrison CD, Lehtonen R, Januszewicz A, Jarvinen H, Juhola M, Mecklin JP, Pukkala E, Herva R, Kiuru M, Nupponen NN, Aaltonen LA, Neumann HP, Eng C (2004) Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHBassociated heritable paraganglioma. Am J Hum Genet 74: 153-9 Virchow R (1863) Die krankhaften Geschwülste. Berlin:: Hirschwald: 325-327 Viskochil D, Buchberg AM, Xu G, Cawthon RM, Stevens J, Wolff RK, Culver M, Carey JC, Copeland NG, Jenkins NA, et al. (1990) Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell 62: 187-92 von Recklinghausen FD (1882) Über die multiplen Fibrome der Haut und ihre Beziehung zu den multiplen Neuromen. Berlin: Hirschwald: 3-18 129 6 Literaturverzeichnis Walther MM, Herring J, Enquist E, Keiser HR, Linehan WM (1999A) von Recklinghausen's disease and pheochromocytomas. J Urol 162: 1582-6 Walther MM, Reiter R, Keiser HR, Choyke PL, Venzon D, Hurley K, Gnarra JR, Reynolds JC, Glenn GM, Zbar B, Linehan WM (1999B) Clinical and genetic characterization of pheochromocytoma in von Hippel-Lindau families: comparison with sporadic pheochromocytoma gives insight into natural history of pheochromocytoma. J Urol 162: 659-64 Xu GF, O'Connell P, Viskochil D, Cawthon R, Robertson M, Culver M, Dunn D, Stevens J, Gesteland R, White R (1990) The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 62: 599-608 Zanca A (1975) Iconografia dermatologica del XVI secolo. Un caso di neurofibromatosi multipla illustrato da Ulisse Aldrovandi. Arch Ital Dermatol Sessuol 40: 119-123 130 131 7 Danksagung 7 Danksagung Mein besonderer Dank gilt Herrn Prof. Neumann. Für die Überlassung des Themas und die Unterstützung bedanke ich mich ebenso wie für die Bereitstellung des Materials. Erst die jahrelange Sammlung von Patientenproben und die genaue Dokumentation von Patientendaten haben diese Studie ermöglicht. Weiterhin danke ich ihm für das Vertrauen und die Unterstützung, mit der er es mir in Zusammenarbeit mit Herrn Prof. Opocher ermöglicht hat, einen Teil der Arbeit in Italien absolvieren zu können. Er hat mir damit neben der beruflichen Weiterbildung ein wundervolles Jahr geschenkt. An dieser Stelle seien all diejenigen, die daran Teil hatten, gegrüßt. Frau Dr. Birke Bausch, Mary Buchta und Gani Berisha, sowie dem ganzen Laborteam danke ich für die Überlassung ihrer Ergebnisse. Vor allem Frau Dr. Bausch trägt einen großen Anteil am Zustandekommen dieser Arbeit. Ein großes „Grazie“ für Herrn Professor Opocher und Dottoressa Francesca Schiavi. Francesca Schiavi war in Italien meine Ansprechpartnerin für jegliche Probleme und stand mir mit Rat und Tat beiseite. Sie hat mein molekulargenetisches Verständnis und das molekulargenetische Arbeiten in vielen Stunden geprägt. Meiner Freundin Rena Hönlein danke ich für ihre große mentale Unterstützung, für die vielen entspannenden Stunden und das Verständnis sowie die Geduld, die sie aufbrachte. Meinen Eltern Anna Trapanesi und Job Harenberg danke ich für die finanzielle Stütze, die sie während des gesamten Studiums leisteten. Des Weiteren sei ihnen und meinem Bruder Daniel Harenberg für die ständige Unterstützung und Liebe gedankt. Daniel Paul, Martin Koeppel, Johanna Feindt und dem Tenno danke ich für die immerwährende Freundschaft. 132 133 8 Lebenslauf 8 Curriculum vitae Persönliche Daten Name Tomas Harenberg Geburtstag 27. Juni 1979 Geburtsort Heidelberg Nationalität deutsch und italienisch Familienstand ledig Akademische Ausbildung Juni 1998 Abitur am Kurfürst Friedrich Gymnasium, Heidelberg Okt. 1999 - Aug. 2001 Studium der Humanmedizin an der Ruprecht-Karls-Universität, Heidelberg Aug. 2001 Vordiplom (“Physikum”), Ruprecht-Karls-Universität, Heidelberg Seit Okt. 2001 Studium der Humanmedizin an der Albert-Ludwigs-Universität, Freiburg Aug. 2002 1. Staatsexamen, Albert-Ludwigs-Universität, Freiburg März 2003 Okt. 2003 - Aug. 2004 Beginn der Doktorarbeit bei Prof. Dr. med. H.P.H. Neumann, Abteilung Innere Medizin IV Universitätsklinikum Freiburg Forschungs- und Studienaufenthalt, Università degli Studi di Padova, Italien Stipendium der Baden-Württemberg-Stiftung März 2005 2. Staatsexamen, Albert-Ludwigs-Universität, Freiburg Oktober 2006 3. Staatsexamen, Albert-Ludwigs-Universität, Freiburg Praktische Tätigkeiten Okt. 1998 - Aug. 1999 Wehrdienst, Marineversorgungsschule, Sylt, Sanitätsgrundausbildung, Sanitäter, Führungsakademie der Bundeswehr, Hamburg Feb. 2001 - Juni 2001 Hilfswissenschaftler, Max-Planck-Institut für med. Forschung, Heidelberg Sept. 2002 Famulus, Universitätsklinik, Freiburg, internistische Station Sept. 2003 Famulus, Universität von Stellenbosch, Südafrika, Thoraxchirurgie Sept. 2004 Famulus, Universitätsklinik, Freiburg, orthopädische Ambulanz Okt. 2004 Famulus, Frauenklinik, Gerolstein, Gynäkologie und plastische Chirurgie 134