Fragen Seminar + Fälle 21.03.2014
Werbung

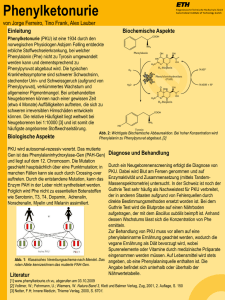
Lernfragen zum Thema Hormone I (21. März 2014) - Was ist der Unterschied zwischen glandulären und Gewebshormonen? Was sind Zytokine? Aus welchen Stoffklassen bestehen sie? - Wie wird Glucose vom Darmlumen in die Blutbahn aufgenommen (2 Systeme)? Was versteht man unter dem Begriff „Glucosesensor“? - Was ist die Funktion der Prä-Sequenz im Präpro-Insulin? - Erklären Sie den Mechanismus der Insulinsekretion in den beta-Zellen des Pankreas! Wieso wirken Sulfonylharnstoffe als Antidiabetika? - Woraus besteht Glucagon, wo wird es synthetisiert und welches ist das wichtigste Zielorgan? Gibt es physiologische Bedingungen unter denen gleichzeitig Insulin und Glucagon in das Blut ausgeschüttet werden? - Catecholamin-Synthese: Welches sind die Cofaktoren der involvierten MonoOxygenasen? Welche Reaktion wird durch die aromatische L-AminosäureDecarboxylase katalysiert? Wofür steht die Bezeichnung MAO? - Was sind beta -Blocker, wie wirken sie? - Adrenalin aktiviert Enzyme wie die Glykogen-Phosphorylase oder die Triacylglycerinlipase. Zeichnen Sie die Reaktionen auf, die von diesen Enzymen katalysiert werden. Problemfälle: Kohlenhydratreich und fettarm, immer gesund? Der Stoffwechsel der Katecholamine Verwertung von Phenylalanin Kohlenhydratreich und fettarm, immer gesund? Lesen Sie den aktuellen NZZ-Artikel (26.03.2008) und diskutieren Sie die untenstehenden Fragen! - Wie wirkt Insulin auf Leber-, Fett- und Muskelzellen? - Molekular: wie wirkt das anabole Hormon Insulin auf GlycogenSynthese/Glycogenolyse, Glycolyse/Gluconeogenese, Auf- und Abbau von Tryglyceriden (welche Enzyme werden aktiviert/inaktiviert)? - Beschäftigen Sie sich mit den Ursachen und Symptomen der Jugend-Diabetes (Typ 1) und der Altersdiabetes (Typ 2). Der Stoffwechsel der Catecholamine Ein häufig vorkommender Polymorphismus im Gen, das für Catechol-O-Methyltransferase (COMT) codiert, führt zur Substitution Val158Met. Diese Variante des Enzyms ist weniger stabil und deshalb ist deren Enzymaktivität um den Faktor 3-4x reduziert. Betroffen sind vor allem Menschen, die diese Mutation in beiden Allelen des Gens tragen (Homozygoten). Im Tiermodell führt eine erhöhte Catecholamin-Ausschüttung von präsynaptischen Zellen zu einer signifikanten Abnahme der Enkephalin-Menge und zu einer kompensatorischen Überproduktion von µ-Opiod-Rezeptoren in postsynaptischen Zellen. Das µ-Opiod- Rezeptor-System wird als lindernde Antwort auf Schmerzen, auf Stress und auf andere negative Umweltveränderungen aktiviert. Eine Arbeit von Wissenschaftlern der Universität Michigan (Zubieta et al.; 2003) zeigt, dass Menschen, die homozygotische Träger der Met-Variante der COMT sind, wesentlich empfindlicher und nachhaltiger auf Schmerzen und Stress reagieren als Heterozygoten oder homozygotische Träger der Val-Variante. Fragen - Was sind Enkephaline? Zu welcher Stoffgruppe gehören sie? - Welche Substanzen gehören zur Kategorie der Catecholamine? Welche enzymatische Reaktion katalysiert COMT? Welches Cosubstrat wird gebraucht, woraus wird dieses hergestellt? - Welche Wirkung haben Catecholamine? - In welcher Drüse werden Catecholamine synthetisiert? Wie wird ihre Synthese und Ausschüttung reguliert? M. Altmann Biochemie-VL März 2003 Mangel an Phenylalanin(PAH)-Hydroxylase / Phenylketonurie Lesen Sie den aktuellen NZZ-Artikel (16.02.2011; siehe Rückseite) und bearbeiten Sie die untenstehenden Fragen! - Molekular: Beschreiben Sie den Funktionsmechanismus von PAH, zu welcher Gruppe von Enzymen gehört es, welches sind die Cosubstrate/Cofaktoren für die katalysierte Reaktion? Wieso kommt es zur Produktion von Phenylketonen? Wie wirkt die Tetrahydrobiopterin (BH4)-Therapie? - Mangelnde Hirnentwicklung / Fehlen von Tyrosin: Wie erklären Sie sich diesen Zusammenhang, um welche Substanzklasse handelt es sich, deren Synthese beeinträchtigt ist (Syntheseweg beschreiben)? - Phenylketonurie: Häufigste angeborene genetische Krankheit. Wie erklären Sie sich die Häufigkeit von 1/10000, wenn jeder 50. Träger ist? Neuö Zürcör Zäitung Mittwoch, 16. Februar 2011 ! Nr. 39 FORSCHUNG UND TECHNIK 57 Ausweg bei schwerer Stoffwechselstörung Die Phenylketonurie lässt sich neuerdings nicht mehr nur mit strenger Diät behandeln Die häufigste angeborene Stoffwechselstörung, die Phenylketonurie, konnte bisher nur mit einer rigorosen Diät behandelt werden. Seit kurzem gibt es ein Medikament, das zumindest bei einigen Patienten hilft. Ob ein Neugeborenes auf den seit etwas mehr als einem Jahr auf dem Markt befindlichen Wirkstoff BH4 (Medikamentenname: Kuvan) anspricht, zeigt ein einfacher Test. Dem Kind wird dazu BH4 in einer Konzentration von 20 mg pro Kilogramm Körpergewicht pro Tag verabreicht. Danach wird in seinem Blut die Phenylalanin-Konzentration nach 8, 16 und 24 Stunden bestimmt. Ronald D. Gerste Geografische Unterschiede Kleine Ursache, fatale Wirkung: Dieser Ausspruch trifft auf Neugeborene zu, denen ein bestimmtes Enzym im Körper fehlt: die Phenylalaninhydroxylase, kurz PAH genannt. Bei einem teilweisen oder vollständigen Mangel dieses Enzyms ist das Verdauungssystem nicht in der Lage, die mit der Nahrung aufgenommene, essenzielle Aminosäure Phenylalanin abzubauen, die Bestandteil von allen tierischen und pflanzlichen Eiweissen ist. Auf diese Weise entstehen unerwünschte Abbauprodukte, die Phenylketone. Zudem staut sich Phenylalanin an. Dadurch kann eine andere Aminosäure, Tyrosin – sie ist Vorstufe wichtiger Neurotransmitter im Gehirn –, nicht genügend gebildet werden. Gift für das Gehirn Statt den Körper über die Nieren zu verlassen, vergiften die überschüssigen Substanzen das Gehirn. Dies führt zu einer Beeinträchtigung der Hirnentwicklung mit geistiger Behinderung. Später kommen meist ausgeprägte Verhaltensstörungen wie Aggressivität hinzu. Das Krankheitsbild als Folge des PAH-Enzym-Defekts wird als Phenylketonurie bezeichnet. Im Vergleich zur erwähnten geistigen Retardierung scheint ein weiterer Effekt des PAH-Mangels nebensächlich: Da das nicht synthetisierte Produkt der Phenylalanin-Umwandlung, die Aminosäure Tyrosin, für die Bildung des Hautpigments Melanin nötig ist, leiden die Kinder unter einem Melaninmangel. Sie sind deshalb oft hellblond und von blasser Hautfarbe. Die Phenylketonurie gilt als häufigste angeborene Stoffwechselstörung, wobei «häufig» ein relativer Begriff ist. So tritt die Krankheit etwa einmal auf 10 000 Geburten auf. In Ländern mit vielen Ehen unter Blutsverwandten kann der Anteil auf 1 von 6500 Geburten ansteigen. Das hängt damit zusammen, dass die Erbanlage für eine Phenylketonurie nicht so selten ist: Etwa jede 50. Person trägt den Gendefekt in ihren Zellen – zumindest auf einem der beiden von der Mutter und dem Vater stammenden Chromosomen. Pflanzen sich nun ein Mann und eine Frau fort, die beide Träger des Gendefekts sind, dann besteht aufgrund des (autosomalrezessiven) Erbgangs ein statistisches Risiko von eins zu vier, dass ein Kind zwei defekte Gene erhält und damit an der Phenylketonurie erkrankt. Wegen der verheerenden Folgen gehört die Testung auf Phenylketonurie Ein Arzt entnimmt einem Baby Blut aus dem Fuss, um es auf die Stoffwechselstörung Phenylketonurie zu testen. heute in entwickelten Ländern zu den etablierten Screening-Untersuchungen. Der Test basiert dabei auf der Analyse eines Tropfens Blut, der dem Neugeborenen aus der Ferse entnommen wird. Das Blut wird im Labor auf Phenylketonurie und einige andere angeborene Stoffwechselerkrankungen untersucht. Ein Schwerpunktzentrum für die Auswertung solcher Proben ist das Kinderspital Zürich, das für seine Spezialanalysen zum Nachweis seltener Erkrankungen hohes Renommee geniesst. Hier geht biologisches Material aus ganz Europa ein – nicht nur zur Diagnose einer Phenylketonurie, sondern ................................................................................. PHENYLALANIN-BLUTSPIEGEL rdg. ! Die normale Phenylalanin-Konzentration im Blut beträgt zwischen 50 und 110 Mikromol pro Liter (!mol/l). Bei Werten von 120 bis 600 !mol/l spricht man noch nicht von einer Phenylketonurie, sondern von erhöhten Blutspiegeln (milde Hyperphenylalaninämie). Als milde Phenylketonurie werden Konzentrationen von 600 bis 1200 !mol/l bezeichnet; jenseits von 1200 !mol/l liegt eine klassische Phenylketonurie vor. Je höher die Blutkonzentration, umso ausgeprägter ist – bei fehlender Therapie – die geistige Retardierung des Kindes. Bei jenen Formen der Krankheit, bei denen das Enzym Phenylalaninhydroxylase vollständig fehlt, sind die Phenylalaninspiegel im Blut meist besonders hoch. auch zum Nachweis von wesentlich selteneren Stoffwechselstörungen. Dank dem flächendeckenden Screening sind inzwischen die Aussichten gut, einem Kind mit Phenylketonurie das Schicksal der geistigen Retardierung zu ersparen. So erlaubt die in den 1960er Jahren von Robert Guthrie eingeführte Analyse des Phenylalaninspiegels im Blut eine Einschätzung der Gefährdung (vgl. Kasten). Anspruchsvolle Diät Bisher gab es nur eine Möglichkeit, zu hohe Phenylalaninwerte und die damit verbundenen Schäden zu verhindern: eine Phenylalanin-freie Diät, wie sie der Kinderarzt Horst Bickel in den 1950er Jahren entwickelt hat. Diese Ernährung aufrechtzuerhalten, ist allerdings nicht einfach. Da die Aminosäure in allen Eiweissen zu finden ist, bedeutet eine Diät den Verzicht auf Fleisch, Fisch, Milchund Eierspeisen sowie auf Weizenprodukte. Für die Betroffenen gibt es speziell hergestellte Nahrungsmittel mit anderen Aminosäuren, die trotz den Fortschritten der Nahrungsmittelindustrie geschmacklich aber oft keine Gaumenfreude sind. Minimale Mengen von Phenylalanin, wie sie der Körper durchaus braucht, werden in Ergänzung zu dieser nutritiven Therapie in niedriger Konzentration zugeführt, etwa in Form eines Apfels. Auch jene Spurenelemente wie Kalzium, die bei einer Phenylalanin-freien Diät ungenügend aufgenommen werden, gilt es zu ersetzen. SPL / KEYSTONE Ähnlich wie beim Diabetes besteht auch bei der Phenylketonurie ein enger Zusammenhang zwischen der Behandlungsqualität und den klinischen Auswirkungen: In mehreren Studien zeigten an Phenylketonurie leidende Kinder eine gegenüber Gesunden signifikant schlechtere kognitive Leistungsfähigkeit, wenn ihre Phenylalaninspiegel im Blut über 400 !mol/l lagen. Für einen Teil der PhenylketonuriePatienten gehört die Abhängigkeit von der Diät der Vergangenheit an. Denn seit kurzem gibt es eine medikamentöse Therapie der Störung mit dem Kofaktor BH4, auch Tetrahydrobiopterin genannt, wie Nenad Blau vom Kinderspital Zürich erklärt. BH4, eine körpereigene Substanz, die in der Nahrung nur in Spuren vorkommt, erhöht die Aktivität des bei vielen Patienten noch in geringem Masse vorhandenen Enzyms PAH. Das führt dazu, dass mehr Phenylalanin in der Leber umgewandelt werden kann, als es dem Stoffwechsel ohne diese Unterstützung möglich wäre. Je mehr BH4 zu dieser Umwandlung beitragen kann, desto geringer werden die Phenylalaninspiegel im Blut und damit die Gefahr einer mentalen Schädigung. Bei Patienten mit PAH-Restaktivität wird der Kofaktor BH4 zusammen mit der Diät oder alleine zugeführt. Letzteres stellt eine grosse Verbesserung der Lebensqualität der Patienten und ihrer Familien dar. Bei Patienten ohne jegliche PAH-Aktivität – das sind meist jene mit sehr hohen Blutspiegeln – wirkt der Kofaktor indes nicht. Eine Normalisierung der Messgrösse nach nur 8 Stunden deutet dabei auf einen BH4-Mangel hin, wie Blau sagt. Keine oder eine nur geringe Reduktion der Phenylalanin-Konzentration spreche hingegen dafür, dass die Phenylketonurie nicht auf BH4 anspreche und nicht genügend Rest-PAH-Aktivität vorliege. Rund 30 Prozent der Patienten scheinen auf die neue Therapieform anzusprechen, wobei es geografische Unterschiede gibt. So finden sich bei Personen in Nord- und Osteuropa eher die schwerere Form der Krankheit ohne PAH-Restaktivität und eine entsprechend tiefe Rate des Ansprechens auf Kuvan. In Südeuropa sprechen je nach untersuchtem Kollektiv zwischen 70 und 80 Prozent der Erkrankten auf das neue Medikament an. Inzwischen mehren sich auch in der medizinischen Literatur die Berichte über die Erfahrungen mit Kuvan. Angesichts der Seltenheit der Krankheit basieren sie aber meist auf kleinen Patientengruppen. So erhielten etwa in einer multinationalen Studie 89 Kinder ergänzend zur herkömmlichen Diät entweder das Medikament oder ein Placebo. Wie sich zeigte, reagierten 44 Prozent der Patienten, die BH4 erhalten hatten, mit einem Sinken des Phenylalaninspiegels um 30 Prozent und mehr. Die Einführung der medikamentösen Therapie mit Kuvan könnte für Patienten mit Phenylketonurie der Beginn einer neuen Epoche sein, in der Möglichkeiten jenseits der oft als trist empfundenen Diät zur Verfügung stehen. Andere Optionen befinden sich noch in der Frühphase der Erforschung, etwa ein aus Bakterienkulturen gewonnenes Enzym, die Phenylalanin-AmmoniumLyase, die bei Mäusen mit Phenylketonurie die Konzentrationen der Aminosäure und ihrer Abbauprodukte reduziert. Die Substanz befindet sich derzeit in klinischer Erprobung, wo erste Ergebnisse nach Blaus Informationen nicht auf einen Durchbruch hindeuten. Eine Heilung der Phenylketonurie mag vielleicht in ferner Zukunft möglich sein, wenn eine Gentherapie mit der Transplantation von Zellen mit funktionierendem PAH/BH4-Mechanismus und deren Nutzbarmachung im Körper des Patienten Realität wird. Dieser Ansatz wird derzeit an einigen Zentren an Mäusen erforscht. Solange es noch keine überzeugenden Resultate gibt, bleibt BH4 nebst der Diät die einzige Therapie bei Phenylketonurie. Träge Proteinforschung Die Wissenschaft beschäftigt sich mehrheitlich immer noch mit den Eiweissen, die schon vor dem «Human Genome Project» bekannt waren Die Sequenzierung des menschlichen Erbguts hätte nach der Prognose von Experten die Erforschung neuer Proteine und die Entwicklung neuer Medikamente beflügeln sollen. Nach zehn Jahren zieht ein Kritiker jedoch eine ernüchternde Bilanz. Stephanie Lahrtz Vor zehn Jahren wurde in den Fachzeitschriften «Nature» und «Science» die Buchstabenfolge des menschlichen Erbguts publiziert. Während Genomforscher die dadurch angestossenen technologischen Fortschritte in der Gentechnik feiern, weist der Proteinexperte Aled Edwards von der Universität Toronto in einem nun in «Nature» publizierten Kommentar auf ein in vielen Publikationen zwar immer wieder gemachtes, aber noch nicht eingelöstes Versprechen hin.1 Seine Analyse der Fachpublikationen der letzten Jahrzehnte habe ergeben, dass sich mehr als 75 Prozent der Proteinforschung mit denjenigen 10 Prozent der Proteine befassten, die bereits vor dem Genomprojekt bekannt gewesen seien, schreibt Edwards. So kenne man beispielsweise mittlerweile mehr als 500 sogenannte Proteinkinasen (Vertreter einer Klasse von Enzymen). Doch obwohl mehrere hundert davon bei Krankheiten eine Rolle spielten, hätten sich noch im Jahre 2009 zwei Drittel der Kinase-Veröffentlichungen mit jenen 50 Enzymen befasst, die bereits vor der Entzifferung des Genoms bekannt gewesen seien. Seiner Meinung nach liegt das zum einen an der eher konservativen Haltung der Geldgeber. Oft gebe es nur finanzielle Unterstützung, wenn man schon handfeste Hinweise für die Bedeutung eines Proteins habe. Und viele Forscher würden lieber ein bekanntes Problem vertiefen, als sich komplett neuen Gebieten mit ungewissen Erfolgsaussichten zuzuwenden. Er fordert von Forschern und Geldgebern eine grössere Risikobereitschaft. Zum anderen gebe es auch technische Gründe für das Verharren im Bekannten. Denn man könne nur diejenigen Proteine erforschen, für die bereits das biologische Handwerkszeug für die Analysen vorhanden sei, schreibt Edwards. Auch andere Experten stimmen seiner Einschätzung zu. Sie geben aber zu bedenken, dass viele Pharmafirmen durchaus umfangreiche Analysen von bisher unerforschten Proteinen durchführten, diese jedoch wegen des Geschäftsgeheimnisses nicht veröffentlichten. Zudem seien zehn Jahre in der Forschung eine kurze Zeit, sagt Markus Ralser vom Max-Planck-Institut für molekulare Genetik in Berlin zu der von Edwards monierten Diskrepanz zwischen Anspruch und Realität. Die Analyse eines Proteins, seiner Aufgaben in der Zelle und seiner möglichen Rolle bei Erkrankungen sei ein längerfristiges Projekt. Ausserdem sei es deutlich einfacher, eine Gensequenz zu entschlüsseln, als die Funktion eines Proteins zu verstehen, sagt Ralser. Denn die DNA sei chemisch gesehen einfacher aufgebaut als Proteine; Struktur und Verhalten seien leichter zu analysieren. Erst in den letzten drei bis vier Jahren habe man Methoden entwickelt, um in grossem Massstab nach denjenigen Proteinen zu suchen, die als neue Angriffspunkte für Medikamente in Frage kommen könnten. Stefan Wiemann, Genomforscher am Deutschen Krebsforschungszentrum in Heidelberg, fügt hinzu, man wisse heutzutage, dass ein Protein in der Zelle Teil eines äusserst komplexen Netzwerks aus Hunderten von miteinander agierenden Komponenten sei. Diese Erkenntnis sei durch die Entzifferung des Genoms entscheidend beeinflusst worden. Gerade für Medikamente gegen komplexe Erkrankungen könne man neue Angriffspunkte nur finden, wenn man diese Netzwerke verstehe. 1 Nature 470, 163–165 (2011).