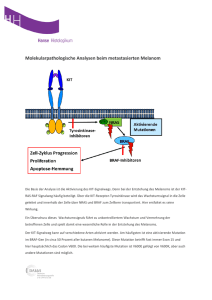
Mutationsanalyse ausgewählter neuromuskulärer und
Werbung

I Aus dem Labor der Neurologischen Klinik der Berufsgenossenschaftlichen Kliniken Bergmannsheil – Universitätsklinik – der Ruhr-Universität Bochum Direktor: Prof. Dr. med. J.-P. Malin Mutationsanalyse ausgewählter neuromuskulärer und metabolischer Erkrankungen Inaugural-Dissertation zur Erlangung des Doktorgrades der Medizin einer Hohen medizinischen Fakultät der Ruhr-Universität zu Bochum vorgelegt von Focke Ziemssen aus Dortmund 2002 II Dekan: Prof. Dr. Gerd Muhr Referent: PD Dr. M. Vorgerd Korreferent: Prof. Dr. J. Epplen Tag der mündlichen Prüfung: 11.02.2003 III Für meine Eltern und meinen Bruder Tjalf IV INHALTSVERZEICHNIS 1 EINLEITUNG 1.1 Mutationen und Pathogenese ............................................................................................ 1 1.2 Untersuchte metabolische Erkrankungen........................................................................ 2 1.2.1 Maturity Onset Diabetes of the Young (MODY) .................................................. 2 Überblick ........................................................................................................................... 2 Glucokinase und HNF1α................................................................................................... 3 Unterschiede im klinischen Phänotyp – MODY2 versus MODY3 .................................... 3 Unterschiede in der Prävalenz – MODY2 versus MODY3 ............................................... 4 Fragestellung .................................................................................................................... 5 1.2.2 Adulte Polyglucosan-Körper-Krankheit (Adult Polyglucosan Body Disease, APBD)............................................................................................................................... 5 Krankheitsbild.................................................................................................................... 5 Glykogenstoffwechsel und Polyglukosankörper ............................................................... 5 Glykogen Verzweigungsenzym (Glykogen Branching Enzyme, GBE) und vorbeschriebene Mutationen............................................................................................. 7 Fragestellung .................................................................................................................... 7 1.2.3 Phosphorylase-Kinase-Defizienz ......................................................................... 8 Phosphorylase-Kinase (PhK) und Phosphorylase-Kinase (PhK)-Defizienz ..................... 8 Lokalisation der Gene der γ1– und β-Untereinheit ........................................................... 8 Fragestellung .................................................................................................................... 8 1.2.4 Rippling Muskelerkrankung (Rippling muscle disease, RMD).......................... 9 Krankheitsbild.................................................................................................................... 9 Caveolin-3 ......................................................................................................................... 9 Fragestellung ..................................................................................................................10 1.3 Methoden der Mutationsanalyse ..................................................................................... 10 2 MATERIAL, METHODEN UND PATIENTEN..........................................................10 2.1 Material...............................................................................................................................10 2.1.1 Chemikalien.......................................................................................................... 10 2.1.2 Puffer und Medien/Lösungen ............................................................................. 12 2.1.3 Enzyme.................................................................................................................. 13 2.1.4 Kits ........................................................................................................................ 13 2.1.5 Oligonukleotide.................................................................................................... 13 2.1.6 Geräte.................................................................................................................... 17 2.2 Methoden ........................................................................................................................... 18 2.2.1 Grundlegende Methoden..................................................................................... 18 2.2.1.1 Extraktion von Nukleinsäuren ......................................................................... 18 Extraktion von DNA aus frischem Vollblut – Genome CleanTM-Procedure..................... 18 Extraktion von DNA aus frischem Vollblut – QIAamp® Blood Midi Kit ............................ 18 Extraktion von DNA aus älterem Vollblut (>48h) mittels Proteinase K ........................... 18 Extraktion von RNA nach Chomczynski und Sacchi ...................................................... 19 Extraktion von RNA mit Trizol® ....................................................................................... 19 DNA-Elution aus Agarose-Gelen Verfahren A................................................................ 20 DNA-Elution aus Agarose-Gelen Verfahren B................................................................ 20 V 2.2.1.2 Reverse Transkription ...................................................................................... 20 2.2.1.3 Polymerase-Ketten-Reaktion (PCR)................................................................ 21 2.2.1.4 Restriktion ......................................................................................................... 21 2.2.1.5 Anfärbung der Nukleinsäuren ......................................................................... 21 2.2.2 Direkt-Sequenzierung.......................................................................................... 22 2.2.3 Single Strand Conformation Polymorphism (SSCP) – Gele............................ 22 2.2.3.1 Horizontale Gelelktrophorese.......................................................................... 22 2.2.3.2 Silberfärbung von SSCP-Gelen ....................................................................... 22 2.2.4 Elektrophoretische Auftrennung auf denaturierenden Polyacrylamid-Gelen .................................................................................................... 23 2.2.4.1 Vertikale Gelelktrophorese .............................................................................. 23 2.2.4.2 Silberfärbung von dentaturierenden Polyacrylamid-Gelen.......................... 23 2.3 Patienten ............................................................................................................................ 24 2.3.1 Patienten mit MODY............................................................................................. 24 2.3.2 Patientin mit APBD .............................................................................................. 24 Anamnese .......................................................................................................................24 Histologie ........................................................................................................................ 25 Autopsie .......................................................................................................................... 26 Enzymaktivität .................................................................................................................26 2.3.3 Patienten mit PhK-Defizienz ............................................................................... 27 Anamnese .......................................................................................................................27 Muskelbiopsie und Glykogenstoffwechsel ...................................................................... 28 2.3.4 Patient mit sporadischer RMD............................................................................ 28 Anamnese .......................................................................................................................28 Körperliche Untersuchung .............................................................................................. 28 Muskelbiopsie ................................................................................................................. 29 3 ERGEBNISSE..........................................................................................................30 3.1 MODY.................................................................................................................................. 30 3.1.1 Etablierung SSCP-Methode ................................................................................ 30 3.1.2 Blindtestung der SSCP-Methode beim HNF1α Gen ......................................... 30 3.1.3 Optimierung unter Erhöhung des experimentellen Aufwands ....................... 32 3.1.4 SSCP-Analyse bei MODY-Patienten................................................................... 32 3.1.5 Direktsequenzierung bei MODY-Patienten........................................................ 33 3.1.6 Ausschluss einer mitochondrial vererbten Diabetesform (Mitochondrial Inherited Diabetes and Deafness, MIDD).......................................... 37 3.1.7 Klinischer Phänotyp ............................................................................................ 37 3.1.8 Überweisungsverhalten ...................................................................................... 38 3.2 APBD .................................................................................................................................. 39 3.2.1 Existenz von Spleißvarianten ............................................................................. 39 3.2.2 Direktsequenzierung des GBE-Gens (mRNA) .................................................. 39 VI 3.2.3 Single nucleotid polymorphisms (SNPs) innerhalb des BE-Gens ................. 40 3.3 PhK-Defizienz .................................................................................................................... 41 3.3.1 Kopplungs-Analyse einer Familie mit PhK-Defizienz ...................................... 41 Auswahl der Mikrosatelliten ............................................................................................ 41 Horizontale Gelelektrophorese und Auswertung des Bandenmusters........................... 41 Zuordnung der Haplotypen und Lod-Score-Berechnung................................................ 43 3.3.2 Ausschluss........................................................................................................... 43 3.4 RMD .................................................................................................................................... 44 3.4.1 Direktsequenzierung des Caveolin-3-Gens....................................................... 44 Caveolin-3 Mutation Arg26Gln bei einem Patienten mit sporadischer RMD................. 44 Nachweis de-novo-Mutation R26Q und Immunhistochemie .......................................... 44 4 DISKUSSION...........................................................................................................46 4.1 Erkrankungen .................................................................................................................... 46 4.1.1 Maturity Onset of the Young............................................................................... 46 4.1.1.1 Identifizierte Mutationen und ihre pathophysiologische Bedeutung .......... 46 Glucokinase ....................................................................................................................46 HNF1α ........................................................................................................................................ 48 Mutationsbiologische Besonderheiten ............................................................................ 50 4.1.1.2 Phänotyp............................................................................................................ 50 Gestationsdiabetes und MODY2 .................................................................................... 50 Adipositas........................................................................................................................ 51 Komplikationen und Krankheitsverlauf ........................................................................... 51 Rekrutierung, Überweisungsverhalten und Epidemiologie............................................. 52 4.1.1.3 Zukunftsperspektiven, Konsequenzen für die klinische Praxis .................. 53 4.1.2 Adult Polyglucosan Body Disease..................................................................... 54 Allelische Heterogenität .................................................................................................. 54 Modell-Mechanismen der allelischen Heterogenität ....................................................... 55 Klinischer Phänotyp der Patienten mit APBD ................................................................. 57 Mutationsanalyse ............................................................................................................ 59 Diagnostik der APBD ...................................................................................................... 61 4.1.3 PhK-Defizienz ....................................................................................................... 62 Krankheitsbild und Differentialdiagnose ......................................................................... 62 4.1.4 RMD ....................................................................................................................... 62 Nachweis der de-novo Mutation Arg26Gln im Caveolin-3-Gen bei RMD....................... 62 Heterogenität von Mutationen im Caveolin-3-Gen.......................................................... 63 Pathophysiologisches Modell.......................................................................................... 64 Perspektiven und Bedeutung der Caveolinopathien ...................................................... 66 4.2 Detektionsmethode........................................................................................................... 66 4.2.1 SSCP-Analyse ...................................................................................................... 66 4.2.1.1 Single Sequence Conformation Polymorphism – Methode ........................ 66 4.2.1.2 SSCP-Etablierung und Blind-Testung ............................................................ 67 4.2.1.2 Einflussfaktoren der SSCP-Analyse ............................................................... 68 Temperatur...................................................................................................................... 68 Gelmatrix......................................................................................................................... 68 VII Laufpufferkonzentration .................................................................................................. 69 Detergentienkonzentration.............................................................................................. 69 Fragmentlänge ................................................................................................................ 69 Variationen ...................................................................................................................... 69 4.2.1.3 Nachteile der SSCP-Methode .......................................................................... 70 Eingeschränkte Vorhersagbarkeit und unklarer theoretischer Hintergrund.................... 70 Notwendigkeit der visuellen Erfassung........................................................................... 71 Notwendigkeit einer Bestätigung durch eine spezifische Kontrollmethode .................... 71 4.2.1.3 Vorteile der SSCP-Methode ............................................................................. 71 Geschwindigkeit und hoher Probenumsatz .................................................................... 71 Geringe Toxizität............................................................................................................. 71 Kosten ............................................................................................................................. 72 Reproduzierbarkeit.......................................................................................................... 72 Erfahrungen und Ausblick............................................................................................... 72 4.2.2 Kopplungsanalyse ............................................................................................... 72 4.2.2.1 Repetitive DNA und Mikrosatelliten ................................................................ 72 Kopplung und Linkage .................................................................................................... 72 4.2.2.2 Praktische Durchführung und Probleme bei der Interpretation des Bandenmusters ............................................................................................................. 73 Berechnung des Lod Score ............................................................................................ 74 4.2.2.2 Theoretischer Hintergrund .............................................................................. 74 Anforderungen an die Familie......................................................................................... 74 Anforderungen an den Mikrosatelliten ............................................................................ 75 4.2.2.3 Beurteilung der Methode.................................................................................. 76 4.2.3 Direkt-Sequenzierung.......................................................................................... 76 4.2.3.1 Entwicklung der Methode ................................................................................ 76 4.2.3.2 Vorteile und Nachteile der Methode................................................................ 77 Nachteile ......................................................................................................................... 77 Vorteile ............................................................................................................................ 78 4.2.3.3 Vergleich der Sequenzierer ............................................................................. 79 4.2.4 SNP(single nucleotide polymorphismus) und RFLP (restriction fragment length polymorphism).................................................................................. 79 4.2.4.1 Entwicklung und Hintergründe der Methode ................................................. 79 4.2.4.2 Vorteile und Nachteile der Methode................................................................ 79 Vorteile ............................................................................................................................ 79 Nachteile ......................................................................................................................... 80 4.2.4.3 Beurteilung und Zukunftsperspektiven.......................................................... 81 5 ZUSAMMENFASSUNG...........................................................................................81 6 LITERATURVERZEICHNIS.....................................................................................82 7 ANHANG .................................................................................................................. A Danksagung...............................................................................................................................F Lebenslauf ................................................................................................................................ G VIII VERZEICHNIS DER ABKÜRZUNGEN A Adenin Abb Abbildung Aqua bidest. Bidestilliertes Wasser (Aqua bidestillata) ABI Applied Biosystems (Perkin-Elmer) APBD Adulte Polyglukosan-Körper-Krankheit APS Ammoniumperoxosulfat As Aminosäure(n) ATP Adenosintriphosphat Bp Basenpaar(e) BMI Body Mass Index BZ Blutzucker C Cytosin ca. circa cDNA Komplementäre Desoxyribonukleinsäure Ck Creatininkinase d Tag(e) Da, kDa Dalton, kiloDalton DGGE Denaturierende Gradienten-Gelelektrophorese (Denaturant Gradient Gel Electrophoresis) d.h. das heißt DNA Desoxyribonukleinsäure dNTP DeoxyNukleotidtriphosphat EDTA Ethylendiamintetraacetat et al. und andere e.t.c. und weitere G Guanin g, mg, µg Gramm, milligramm, mikrogramm g Vielfaches der Gravitationskonstante GBE Glykogen-Verzweigungsenzym (Glycogen Branching Enzyme) GSD Glykogen-Speicher-Krankheit (Glycogen Storage Disease) H Stunde(n) HbA1/HbA1c Glykiertes Hämoglobin A1/A1C Inkl. Inklusive l, ml, µl liter, milliliter, mikroliter M molar MIDD mitochondrial vererbte Diabetesform (Mitochondrial Inherited Diabetes and Deafness) Min Minute(n) Mio. Million(en) MODY Maturity Onset Diabetes of the Young IX mRNA mitochondriale Ribonukleinsäure N Anzahl N. Nervus nNOS neurale Stickoxydsynthase (Neuronal Nitrite Oxide Synthase) NO Nitritoxid Nt Nukleotid o.g. OMIM Oben genannt TM online mendelian inheritance in men OGGT Oraler Glucose-Toleranztest PCR Polymerase-Kettenreaktion PhK Phorsphorylase-Kinase PIC Polymorphismus Information Content PIRC Percussion-induced Rapid Muscle Contraction RFLP Restriktionsfragment-Längen-Polymophismus RMD Rippling Muskelerkrankung (Rippling muscle disease) RNA Ribonukleinsäure RT Raumtemperatur s.o., s.u. siehe oben, siehe unten SSCP Single-strand Conformation Polymorphism T Thymin T1DM / T2DM Diabetes mellitus Typ 1 / Typ 2 Tab Tabelle TAE Tris-Acetat EDTA gepufferte Lösung TBE Tris-Borat EDTA gepufferte Lösung TE Tris-EDTA Puffer TEMED Tetramethyl-Ethylendiamin Tris Trishydroxymethylaminomethan U Unit u.a. und andere UpM Umdrehungen pro Minute UV Ultraviolett V, kV Volt, kiloVolt Vgl. vergleiche Vol. Volumen z.B. zum Beispiel ZNS Zentrales Nervensystem z.T. zum Teil Bei den Abkürzungen der Aminosäuren werden die international üblichen Abkürzungen des Einbzw. Drei-Buchstaben Codes verwendet. X VERZEICHNIS DER ABBILDUNGEN UND TABELLEN Abb 1 Diskrepanz bei der Identifikation von MODY-Familien zwischen unterschiedlichen westeuropäischen Ländern Seite 4 Abb 2 Schematische Darstellung der Glykogen Biosynthese Seite 6 Abb 3 Genstruktur HNF1α und Lage der Primer Seite 14 Abb 4 Genstruktur des Glucokinase-Gens und Lage der Primer Seite 15 Abb 5 Genstruktur Caveolin-3 und Lage der Primer Seite 16 Abb 6 Optimierung der PCR am Beispiel der Fragmente mit Caveolin Exon 1 und 2 Seite 21 Abb 7 Nervus suralis Biopsie AD1 I.2, EM-Mikroskopie Seite 26 Abb 8 Stammbaum einer Familie mit PhK-Defizienz (PHK1) Seite 26 Abb 9 Stammbaum der Familie AD1 Seite 27 Abb 10 SSCP-Analyse verschiedener PCR-Fragmente, HNF1α Seite 31 Abb 11 Optimierung der SSCP-Analyse durch Restriktion für P291insC Seite 32 Abb 12 Mutation 3243 Leucin tRNA und ihr Nachweis durch Restriktion Seite 37 Abb 13 Zuordnung der MODY-Patienten zu geschlechts- und altersadaptierten BMIKategorien Seite 38 Abb 14 Überweisungsverhalten unterteilt nach MODY-Untertypen Seite 38 Abb 15 Expression des GBE-Gens in unterschiedlichen Geweben Seite 39 Abb 16 Direktsequenzierung Fragment B9-B10 der Patientin AD1 I.2 Seite 40 Abb 17 Nachweis der Mutationen R515H und R524Q im GBE-Gen Seite 41 Abb 18 Haplotypisierung der Familie PHK1 Seite 42 Abb 19 Mutation Arg26Gln im Caveolin-3-Gen Seite 44 Abb 20 Immunhistochemische Färbung der sarkolemmalen Proteine bei RMD Seite 45 Abb 21 Entscheidungsbaum zur Diagnostik bei juvenilen Diabetes-Formen Seite 54 Abb 22 Stufendiagnostik bei APBD Seite 61 Abb 23 Veränderungen der Proteinsequenz der Caveolin-3 Domänen bei drei verschiedenen Krankheitsbildern Seite 63 Abb 24 Pathophysiologisches Schema zur Interaktion zwischen nNOS und Caveolin-3 Seite 65 Abb 25 Theoretisches Modell zur Erklärung des Elektrophoresebilds bei der SSCP-Analyse Seite 67 Abb 26 Beispiel für die Zuordnung des Haplotyps eines Mikrosatelliten Seite 73 Abb 27 Direktsequenzierung Mutation S426delGC des Glucokinase-Gen Seite 78 Abb 28 Datenerhebungsbogen zur Erfassung der Diabetes-Komplikationen Seite A Tab 1 Bisher beschriebene MODY bezogene Gene und ihre Lokalisation Seite 2 Tab 2 Bisher identifizierte Mutationen im GBE-Gen Seite 7 Tab 3 Gemessene Enzymaktivitäten der Familie AD1 Seite 27 Tab 4 Gemessene CK-Werte der Familie mit Phosphorylase-Kinase-Defizienz Seite 27 Tab 5 Bedingungen der SSCP-Elektrophorese für die zehn PCR-Fragmente von HNF1α Seitw 30 Tab 6 Blindtestung von 15 Proben auf Mutationen im HNF1α-Gen Seite 31 Tab 7 Polymorphismen im HNF1α-Gen Seite 33 Tab 8 Patienten mit Mutationen im Glucokinase-Gen Seite 34 Tab 9 Patienten mit Mutationen im HNF1α-Gen Seite 36 Tab 10 Verwendete Mikrosatelliten-Marker und ihre Lokalisation Seite 41 Tab 11 Zwei-Punkte Lod-Score zur Bestimmung des Kopplungsungleichgewicht der untersuchten Mikrosatelliten Seite 43 XI Tab 12 Identifizierte Mutationen im Glucokinase-Gen Seite 46 Tab 13 Identifizierte Mutationen im HNF1α-Gen Seite 48 Tab 14 Übersicht über die bisher veröffentlichen APBD Fälle Seite 58 Tab 15 Lokalisation der untersuchten Mikrosatelliten Seite 75 Tab 16 Bisher beschriebene Mutationen im HNF1α-Gen Seite B Tab 17 Bisher beschriebene Mutationen im Glucokinase-Gen Seite C Tab 18 Übersicht untersuchter Proteine/Gene Seite E 1 1 Einleitung 1.1 Mutationen und Pathogenese An die Stelle des nosologischen Krankheitsmodels, nach dem die Krankheitsmanifestation das Ergebnis eines Prozesses darstellt, der eine Ursache hat und durch klinische Symptome charakterisiert ist, prägen nach heutiger Vorstellung vor allem drei Hauptfaktoren das Bild der Erkrankung; neben dem chronobiologischen und Umwelt-Faktor ist das vor allem auch der genetische Hintergrund. Inzwischen sind ca. 10.100 Gene bzw. Phänotyp-Loci in der OMIMTM Datenbank erfasst. Dabei können Gene pathogenetisch auf unterschiedliche Art und Weise zur Krankheitsmanifestation führen. So können schnell wachsende „triple repeats“ wie beim Morbus Huntington oder der myotonen Dystrophie, größere Deletionen wie bei der Duchenne´schen Dystrophie als auch Punktmutationen bzw. kleine Deletionen wie z.B. bei der zystischen Fibrose eine Erkrankung verursachen. Will man genetische Störungen identifizieren, ist dies mit geringerem Aufwand verbunden, wenn nur wenige distinkte Mutationen bekannt sind wie der Sichelzellanämie oder der α1-Antitrypsin Defizienz, als bei einer sehr heterogenen Verteilung von Mutationen wie bei den über 600 unterschiedlichen bekannten Mutationen des CFTR-Gens. Dabei kann sowohl eine Genmutation für die Entstehung verschiedener Syndrome verantwortlich sein (z.B. RET-Gen mit familiärem medullären Schildrüsen-Carcinom, Multipler Endokriner Neoplasie kurz MEN 2A und 2B, und Morbus Hirschsprung (van Heyningen 1994)), als auch erst das Zusammenspiel verschiedener genetischer Veränderungen für die Entstehung einer Krankheit verantwortlich sein (z.B. Hypercholesterinämie ). Dabei ist die experimentelle Untersuchung auf einen genetischen Defekt hin häufig von Bedeutung für die funktionelle Evidenz. Der Vorteil einer DNA-Analyse gegenüber einer Genprodukt- bzw. Metaboliten-Analyse ist z.B. dann gegeben, wenn ein gewebespezifischer Nachweis invasivere Maßnahmen erfordern würde. Die Methode ist auch überlegen bei Populationen mit FounderEffekten. Darüber hinaus erlaubt die Untersuchung auf DNA-Niveau eine pränatale Diagnostik und die frühzeitige Feststellung einer eventuellen Krankheitsprädisposition bei noch asymptomatischen Patienten. Die Diagnostik von somatischen Mutationen und Polymorphismen spielen u.a. im Bereich der Onkologie eine wichtige Rolle für eine sensitive Früherkennung und die Prognose bzw. die Therapieantwort. Letzten Endes muss bei den mittlerweise schon über 1000 bekannten monogen verursachten Erkrankungen im Einzelfall abgewogen werden, ob und welche Analyse in der Diagnostik zu bevorzugen ist. Eine genaue Charakterisierung etablierter und weit verbreiteter Methoden mit ihren Besonderheiten erfolgte im Rahmen dieser Doktorarbeit. Dabei sollten vor allem Aspekte der Effizienz und Reliabilität berücksichtigt werden, um bei der Auswahl der diagnostische Methode den Entscheidungsprozeß zu erleichtern. Die Notwendigkeit einer Diagnostik und die Relevanz der jeweils gewonnen Daten sollten für die untersuchten Krankheitsbilder einzeln kritisch beurteilt werden. 2 1.2 Untersuchte metabolische Erkrankungen 1.2.1 Maturity Onset Diabetes of the Young (MODY) Überblick Seit den frühen 70er Jahren wird in der Literatur eine familiäre Form der Volkskrankheit Diabetes beschrieben, die sich durch einen autosomal dominanten Erbgang und ein frühes Manifestationsalter abgrenzen läßt. Umfangreiche epidemiologische Untersuchungen zeigten, dass der Anteil der Patienten mit dieser Form des Diabetes mellitus Typ 2 (T2DM), dem sogenannten Maturity Onset Diabetes of the Young (MODY), ungefähr 2% aller Diabetiker ausmacht (68 000 Patienten in Deutschland) (Ledermann 1995). 1992 wurde die erste pathogene Mutation in einer MODY-Familie identifiziert (Stoffel, Froguel et al. 1992): Veränderungen in der Aminosäurensequenz der Glucokinase (ATP:D-hexose 6-phosphotransferase, EC 2.7.1.1), einem wichtigen Glucosesensor der pankreatischen β-Zelle, führen zu einem Diabetes mit relativ milden Komplikationen, der sich in einigen Fällen auch nur infektassoziiert oder als Gestationsdiabetes manifestiert. Mitte der 90er Jahre wurden dann weitere Gene mit pathogenen Mutationen identifiziert. Mutationen in den Genen der sogenannten „hepatischen nukleären Transkriptionsfaktoren“, die auch in β-Zellen expremiert werden und dort wesentliche regulatorische Funktionen ausüben (Hepatic Nuclear Factor 4α (HNF4α) als MODY1, HNF1α als MODY 3, HNF1β als MODY 5), verursachen im Unterschied zu den Glucokinase-Mutationen einen progredienten Verlust der Insulinsekretion und weisen eine hohe Komplikationsrate auf, die angesichts des frühen Manifestationsalters für die Prognose der jungen Patienten von großer Bedeutung ist. Tab 1: Bisher beschriebene MODY bezogene Gene und ihre Lokalisation. IPF: Insulin Promotor Faktor, NeuroD1 Neurogener Differenzierungsfaktor 1 Gen chromosomale klinische Charakteristika Lokalisation mikrovaskuläre Komplikationen erniedrigte Serumspiegel von Triglyzeriden, Apolipoprotein AII und CIII und Lipoprotein Lp(a) erhöhte Nüchtern-Glucose normale Proinsulin-Insulin-Ratio MODY1 HNF4α 20q MODY2 Glucokinase 7p MODY3 HNF1α 12q mikrovaskuläre Komplikationen renale Glucosurie, Sulfonylharnstoff-Sensitivität erhöhte Proinsulin-Insulin-Ratio MODY4 IPF1 13q homozygot Pancreas-Agenesie, selten MODY5 HNF1β 17q Nierenzysten, chronisches nichtdiabetisches Nierenversagen, innere genitale Missbildungen MODY6 NeuroD1 2q selten MODY7 Islet-1 5q selten Als MODY4 wird der Diabetes-Typ bezeichnet, der von Mutationen im Insulin-Promotor-Faktor 1 (IPF1) verursacht wird, einem Faktor für die Entwicklung und Differenzierung des Pancreas (Stoffers, Ferrer et al. 1997). Die seltenen Unterformen MODY 1, 4, 5, 6 und 7 wurden bisher nur in einzelnen Familien identifiziert. Deshalb beschränkten sich die Untersuchungen dieser Arbeit auf die beiden Gene der Glucokinase und des HNF1α, die sich bis heute als wichtigste Lokalisationen und häufigste Ursachen für die Manifestation des MODY herausgestellt haben. 3 Glucokinase und Hepatic Nuclear Factor 1α Die herausragende Stellung der Glucokinase innerhalb des Glucose-Stoffwechsels ist schon lange bekannt. Sie liegt in ihren zellulären und biochemischen Eigenschaften begründet: In Leber- und βZelle wird sie gewebespezifisch zusätzlich zu den übrigen Hexokinasen expremiert. Die hohe Michaelis-Menten Konstante ( kM 5-15 mM) bringt es mit sich, dass das Enzym je nach anfallender Glucose als erster entscheidender Schritt die Geschwindigkeit der nachfolgenden Glykolyse bestimmt, zumal gegenüber den übrigen Hexokinasen die allosterische Inhibition durch Glucose-6Phosphat entfällt und die Transportkapazität der Glut-2-Transporter in Hepatozyten und β-Zellen die Glucokinaseaktivität bei weitem übertrifft (kM 42 mmol/l). Gewebespezifische Promotoren und differentielles Splicen ermöglichen eine unterschiedliche Expression des Gens in Leber (wesentlich reguliert durch Plasmainsulinspiegel) und Pankreas (transkriptionell und posttranslational durch Glucose reguliert); das Enzym von Leber (Major-Form: Exon 1b und Exon 2 bis 10; Minor-Form: Exon 1c und Exon 2 bis 10) und Bauchspeicheldrüse (Exon 1a und Exon 2 bis 10) ist mit 466/464 bzw. 465 Aminosäuren und einem Molekulargewicht um 50 kDa auch kleiner als die anderen drei bekannten Hexokinasen. Der hepatische nukleäre Faktor 1α gehört zu den Homeo-Domänen enthaltenden Transkriptionsfaktoren und spielt eine wesentliche Rolle bei der Regulation der Expression verschiedener Proteine: In der Leber reguliert er zum Beispiel die Biosynthese von Gerinnungsfaktoren und Serumproteinen wie Albumin, Transthyretin, α-1 Antitrypsin, α und β Fibrinogen, α-Fetoprotein und Apolipoproteinen. In der β-Zelle finden sich Bindedomänen im Bereich der Promotoren des Glut-2 Gens (Takeda, Kayano et al. 1993) und der Pyruvatkinase (Yamada, Tomura et al. 1999), aber auch das humane Insulin-Gen ist Ziel einer Transaktivierung (Pontoglio, Sreenan et al. 1998; Okita, Yang et al. 1999). Dabei bindet HNF1α als Homo- oder zusammen mit HNF1β als Heterodimer an die Palindrom-Sequenz GTTAATNATTAAC. Kompletter Verlust des Faktors in der Maus führt zu Leberdysfunktion, Phenylketonurie und renalem FanconiSyndrom (Pontoglio, Barra et al. 1996). Die Homeo-Domäne ist eine spezielles DNA-bindendes Helix-turn-Helix Motiv (Codon 199-281) und enthält bei HNF1α eine Insertion von 21 Aminosäuren (Codon 238-258). Das gleichzeitige Vorkommen eines POU-Motivs weist eindeutig auf die Zugehörigkeit zu einer distinkten Gruppe von Proteinen hin, die eine wesentliche Bedeutung bei der embryologischen Differenzierung und Organogenese haben (Mendel, Khavari et al. 1991). Unterschiede im klinischen Phänotyp – MODY2 versus MODY3 Mutationen in beiden Genen, dem der Glucokinase und HNF1α, führen zur Manifestation des MODY. Funktionell ist MODY2 aber stärker durch eine leichte Rechts-Verschiebung der Glucose/Insulin Dosis-Antwort-Kurve geprägt (Bell, Pilkis et al. 1996), während bei den MODY3Patienten eher ein komplexeres und absolutes Insulin-Sekretionsdefizit auftritt (Hattersley 1998). So ist es auch zu erklären, dass mikrovaskuläre Komplikationen bei den MODY-Formen mit mutierten Transkriptionsfaktoren wesentlich häufiger sind als bei MODY2-Familien, die klinisch durch eine normales Proinsulin-Insulin-Verhältnis charakterisiert sind und deren GlucoseStoffwechsel meist durch Diät und Bewegung suffizient zu optimieren ist (Fajans, Bell et al. 2001). Charakteristisch für MODY3-Patienten ist darüber hinaus eine verstärkte renale Glucosurie 4 45 40 35 Familien 30 Frankreich (n=67) Großbritannien (n=55) Dresden (n=11) 25 20 15 10 5 0 MODY2 Abb 1: MODY3 Diskrepanz bei der Identifikation von MODY-Familien zwischen unterschiedlichen westeuropäischen Ländern (Menzel, Kaisaki et al. 1998) und ein verstärktes Ansprechen auf eine Pharmako-Therapie aus der Substansklasse der Sulfonylharnstoffe (Sovik, Njolstad et al. 1998). Unterschiede in der Prävalenz – MODY2 versus MODY3 Bezüglich der Verteilung der einzelnen MODY-Subtypen konnte eine Studie in Großbritannien bei 65% der betroffenen Familien einen MODY3-Typ identifizieren (siehe Abb 1), jedoch nur bei 11% einen MODY2-Typ. (Beards, Frayling et al. 1998). Dagegen machten in einer französischen Untersuchung die MODY2-Diabetiker einen Anteil von 63% aus, während der Anteil der MODY3Patienten nur 21% betrug (Chevre, Hani et al. 1998). Bei den einzigen Daten, die bisher für ein deutsches Patientengut veröffentlicht worden sind, wurden bei elf Patienten bzw. Familien vier Patienten mit MODY3, aber nur eine Familie mit MODY2 gefunden (Lindner, Cockburn et al. 1999). Unser Ziel war es nicht nur, durch die molekulargenetische Aufarbeitung eines größeren Kollektivs eine aussagekräftigere Zahl der Prävalenz der Subtypen in Deutschland zu erhalten, sondern auch eine Erklärung für die Diskrepanz der Verteilung in den benachbarten mitteleuropäischen Ländern zu finden. Die jugendlichen Typ 2 Diabetesformen insgesamt haben sich bisher als sehr heterogen in Krankheitsverlauf und Prognose erwiesen. Dabei ist unter den Kindern und Jugendlichen die Inzidenz des Diabetes mellitus Typ 2 insgesamt aktuell sprunghaft angestiegen (Rosenbloom, Joe et al. 1999). Die Diagnostik in Form der molekularbiologischen Mutationsanalyse ist von entscheidender Bedeutung, indem sie eine separate Betrachtung der eindeutig hereditären Formen (MODY2 und MODY3) erlaubt. Die Untersuchung ermöglicht so durch die Differenzierung bei Prognose und Therapie der Patienten auch Aussagen über alimentäre Einflüsse aller Untergruppen. 5 Fragestellung Ziel der Untersuchungen sollte die Etablierung eines MODY-Screening sein. So sollten zum einen erstmals aussagekräftige Daten über die Bedeutung der Glucokinase und von HNF1α in einem deutschen Kollektiv gewonnen werden. Dabei stand insbesondere die relative Häufigkeit der beiden Subtypen, die Altersverteilung und die Zuweisung im Mittelpunkt des Interesses. Geklärt werden sollte auch, ob es weitere phänotypische Auffälligkeiten gibt oder die bisherigen Charakteristika der Subtypisierung bestätigt werden können. Die patientenspezifische Betrachtung der Mutation sollte darüber hinaus Zusammenhänge zwischen Genotyp und auftretendem Phänotyp ermöglichen und so zusätzliche Erkenntnisse über die Bedeutung einzelner funktioneller Enzymdomänen bzw. über die Interaktion der beiden Protein innerhalb des Glucose-Stoffwechsels liefern. Zum anderen sollte durch die Gründung einer deutschen MODY-Bank eine Längsschnitt-Erfassung der klinischen Daten ermöglicht werden, um prognostische Faktoren und die Bedeutung einer differenzierten Therapie in Zukunft besser abklären zu können. So sollte ein Datenerfassungsbogen entwickelt werden, der die schnelle Dokumentation der notwendigen Daten ermöglicht. Die Diagnosestellung eines MODY2 bzw. MODY3 sollte darüber hinaus die Grundlage dazu legen, bei den mutationsnegativen jungen Typ 2 Diabetikern weitere Kandidatengene an anderen Loci zu identifizieren, die auch für die große Gruppe der Patienten mit späterer Manifestation und ohne autosomal dominanten Erbgang von Bedeutung sein könnten. 1.2.2 Adulte Polyglucosan-Körper-Krankheit (Adult Polyglucosan Body Disease, APBD) Krankheitsbild Die adulte Polyglukosan-Körper-Krankheit (Adult Polyglukosan Body Disease, APBD) ist eine sehr seltene neurologische Erkrankung, die klinisch durch eine Tetraspastik, Ataxie, distale Sensibilitätsstörungen und Demenz mit einer Manifestation im späten Erwachsenenalter charakterisiert ist. Häufige und typische Erstsymptome sind Gangunsicherheit und/oder Inkontinenz. Die Erstbeschreibung geht auf Suzuki zurück (Suzuki, David et al. 1971), Robitaille führte erstmals den Ausdruck der Polyglukosankörper für die charakteristischen axonalen Ablagerungen ein (Robitaille, Carpenter et al. 1980), die mit der progressiven Degeneration des ersten und zweiten Motorneurons einhergehen. Histochemische Untersuchungen haben ergeben, dass der wesentliche Anteil von Polyglukosankörpern aus einer anormalen glykogenartigen Struktur besteht. Glykogenstoffwechsel und Polyglukosankörper Physiologisches Glykogen ist durch seine starken Verzweigungen gut wasserlöslich und bietet durch die große Anzahl von Seitenverzweigungen viele Angriffspunkte für eine schnelle Phosphorylyse bei Belastung (Newsholme, Zammit et al. 1978), um seine Funktion als rasch verfügbarer Energiespeicher in der Muskulatur zu erfüllen. Die Verzweigungen werden innerhalb des Glykogen-Stoffwechsels durch das Glykogen-Verzweigungsenzym (Glykogen Branching Enzyme, GBE) erzeugt, indem kurze Glykosylketten (bestehend aus ungefähr sieben 6 Glucose-6-P Phosphoglucomutase Glucose-1-P H C-OH H2C-OH 2 O UTP HO OPO3 OH Glucose-1-phosphatUTP-transferase OH HO - + PPi O - P H2C-OH O H2C-OH H2C-OH OH H2C-OH HO OH OH O OH OH OH [Glucose]n oder Glykogenin O O H2C-OH O OH HO OH OH H2C-OH O OH O [Glucose]n oder Glykogenin H2C-OH O O O OH H2C-OH H2C-OH OH OH O Amylo-1,6glucosidase O O OH O OH OH O UDP O O O OH O O OH OH H2C-OH H2C-OH H2C-OH O H2C-OH O Glykogensynthase [Glucose]n oder Glykogenin OH OH O HO HO O O UDP OH OH O GlykogenPhosphorylase - O H2C-OH OH O OH O OH UDP-Glucose HO H2C-OH O -- OH O H2C-OH O O OH OH O O [Glucose]n oder Glykogenin H2C-OH O OH OH OH OH OH OH OH HO OH H2C-OH Amylo-1,4-1,6transglucosylase (GBE) H2C-OH OH O HO OH O OH O OH OH HO 1,4-1,6-Glucantransferase OH O OH H2C-OH HC-OH O O OH O OH OH Abb 2: OH O OH O OH O [Glucose]n oder Glykogenin OH OH O O OH OH H2C-OH H2C-OH O O O O OH H2C-OH H2C-OH HC-OH O OH HO O O H2C-OH O O H2C-OH O OH OH O [Glucose]n oder Glykogenin Abkürzungen: P Phosphat PPi Pyrophosphat OH Schematische Darstellung der Glykogen Biosynthese (modifiziert nach Löffler und Petrides 1997) Glykosyleinheiten) in α-1,6-glycosidischer Bindung an die lineare periphere Glykogenkette mit α1,4-glycosidischer Bindungen angefügt werden (siehe Abb 2) Bei den Polyglukosankörper fehlt diese typische Verzweigungsstruktur. Die niedrigere Verzweigungsdichte führt zu der Bildung filamentöser und stabähnlicher Aggregate innerhalb des Zytoplasmas. Morphologisch imponieren diese Einschlüsse ultrastrukturell als granuläres Material (Elektronenmikroskopie siehe Abb 7), histochemisch sind die Perjodsäure(PAS)-positiven Ablagerungen durch eine Resistenz gegenüber Phosphorylase und Diastase charakterisiert. Insgesamt wurden ähnliche Strukturen bei der Glykogenose Typ II (Morbus Pompe) und Typ IV (Morbus Andersen), der APBD, einer Form der Myoklonus-Epilepsie (Lafora-Erkrankung), in einigen Fällen von Phosphofruktokinase-Defizienz (Morbus Tarui, GSD VII) und der BielschwoskyErkrankung beschrieben, die alle auch unter dem Begriff der Amylopectinosen zusammengefasst werden (Cavanagh 1999). Gegenüber den physiologisch, altersabhängig auftretenden Corpora amalycea grenzen sie sich durch einen geringeren Phosphatgehalt (Sakai, Austin et al. 1969), fehlende Metachromasie mit Toluidinblau und längere Außenketten des Glykogens ab. Die Laforabzw. Bielschwosky-Einschlußkörper zeigen gegenüber den Polyglukosankörpern zwar dieselbe Immunreaktivität zu bestimmten monoklonalen Antikörpern (Anti-KM-279), sind aber auch in den Perikarien lokalisiert (Reusche, Aksu et al. 1992). Anders als Corpora amalycea werden 7 Polyglukosankörper in Axonen gefunden und zeigen kaum subpiale und perivaskuläre Anreicherung (Bigio, Weiner et al. 1997). Histopathologisch sind die runden, basophilen und stark PAS-positiven, zytoplasmatischen Einschlüsse diagnostisch wegweisend, jedoch nicht pathognomonisch, da sie auch bei Gesunden mit zunehmendem Alter auftreten können, bei Diabetikern (Mancardi, Schenone et al. 1985), Hypoxie (Long, Mossakowski et al. 1972) oder Barbituratabusus (Phelps 1972). Glykogen Verzweigungsenzym (Glykogen Branching Enzyme, GBE) und vorbeschriebene Mutationen Für zehn Patienten mit APBD wurde die GBE-Aktivität in mehreren Geweben bestimmt, davon zeigten nur fünf Personen erniedrigte Aktivitäten in Leukozyten bzw. in Homogenaten des peripheren Nervs. Bei der APBD liegt also keine generelle Aktivitätsverminderung von GBE in den einzelnen Geweben vor (Cafferty, Lovelace et al. 1991). Auch bei der alleinigen Aktivitätsbestimmung in Muskelbiopsaten zeigte sich kein einheitliches Bild, so dass eine heterogene Entstehung postuliert wurde (Lossos, Barash et al. 1991, McDonald, Faust et al. 1993, Bruno, Servidei et al. 1993). Mutationen im GBE-Gen auf Chromosom 3 (Thon, Khalil et al. 1993) wurden bei fünf Patienten mit generalisierter Glykogenose Typ IV (GSD IV) angetroffen (siehe Tab 2). Bei fünf Ashkenazi-Patienten mit APBD wurde eine homozygote Missense Mutation innerhalb Tab 2: Bisher identifizierte Mutationen im GBE-Gen. Die Basenpositionen beziehen sich auf die Nummerierung der cDNA nach Thon, Khalil et al. (1993) Patient GSD IV Patient 1 GSD IV Patient 2 GSD IV Patient 3 GSD IV Patient 4 GSD IV xPatient 5 APBD 5 Familien Mutation Allel 1 Allel 2 262-331 Deletion 210 Bp Deletion 210 Bp Deletion von nt 873-1082 L224P Y329S CTA zu CCA TAC zu TCC 761 nt 1076 R515C nicht entdeckt CGT zu TGT nt 1633 F257L R524X TTT zu TTA CGA zu TGA nt 861 nt 1660 R524Q keine Expression CGA zu CAA des anderen Allels nt 1661 Y329S Y329S Phänotyp GSD IV kongenital, progressiv, neuromuskuläre Schwäche GSD IV, hepatische Verlaufsform Referenz (Bao, Kishnani et al. 1996) (Bao, Kishnani et al. 1996) GSD IV progressive, hepatische Verlaufsform (Bao, Kishnani et al. 1996) GSD IV progressive, hepatische Verlaufsform (Bao, Kishnani et al. 1996) GSD IV kongenital, neuromuskuläre und hepatische Symptome (Bruno, DiRocco et al. 1999) APBD (Lossos, Meiner et al. 1998) dieses Gens gefunden (Lossos, Meiner et al. 1998). Auch die wenigen Fälle mit einer Verminderung der GBE-Aktivität ohne nachgewiesene Mutation im GBE-Gen stammten ausschließlich aus diesem ethnischen Hintergrund. Fragestellung Bei einer deutschen Patientin mit klinisch manifester und bioptisch nachgewiesener APBD sollten nach Mutationen im GBE-Gen gesucht werden. Nach dem Versterben der Patienten sollten die bioptisch gewonnen Gewebe dazu genutzt werden, die Existenz von Isoformen und 8 gewebespezifische Expression des GBE zu untersuchen. Dadurch sollte die Pathogenese der APBD anhand der Befunde einer Familie weiter aufgeklärt werden. 1.2.3 Phosphorylase-Kinase-Defizienz Phosphorylase-Kinase (PhK) und Phosphorylase-Kinase(PhK)-Defizienz Die Phosphorylase-Kinase (ATP:phosphorylase-b phosphotransferase, EC 2.7.1.38, PhK) spielt eine wesentliche Rolle in der Regulation der Glykogenolyse durch Aktivierung der GlykogenPhosphorylasen in Leber, Skelettmuskulatur und anderen Geweben. Es handelt sich um ein großes Protein aus mehreren Untereinheiten mit einer hexadekamerischen (αβγδ)4-Struktur. Die kleine γ-Untereinheit (43 kDa) ist verantwortlich für die katalytische Aktivität, die δ-Untereinheit (16 kDa) steuert als Mitglied der Calmodulin-Familie die Ca2+-abhängige Regulation. Die großen αund β-Untereinheiten (138 bzw. 125 kDa) sind Substrate der cAMP-abhängigen Phosphorylierung durch Proteinkinase A. Unterschiedliche Isoformen von unterschiedlichen Genen (αM, αL, γM, γTL) führen ebenso wie differentielles Splicen (αM, αL und β) zur komplexen Struktur der PhK (Kilimann 1997). Die bisher beschriebenen Fälle von PhK-Defizienz unterscheiden sich im Vererbungsmodus und der Gewebeverteilung. Diese Heterogenität lässt sich durch die Mutationen erklären, die unterschiedliche Untereinheiten und Isoformen betreffen. Die häufige Phoyphorylase-KinaseDefizienz, der X-chromosomale Leber-PhK-Mangel, wird durch Mutationen der Leber-Isoform der α-Untereinheit hervorgerufen (Hendrickx, Dams et al. 1996; Burwinkel, Amat et al. 1998). Die Mutationen der ubiquitär exprimierten β-Untereinheit liegen der autosomal vererbten Form der PhK-Defizienz in Leber und Muskel zugrunde (Burwinkel, Maichele et al. 1997), wobei die Leber klinisch vordergründig betroffen ist (Burwinkel, Moses et al. 1997). Muskelspezifische PhKDefizienzen wurden bislang erst am Tiermodell (Schneider et al 1993) und bei 2 Patienten (Wehner et al. 1994; Bruns et al. 1998) durch den Nachweis von Mutationen im X-chromosomalen Gen der muskelspezifischen Isoformen von PhK-α-Untereinheiten genetisch aufgeklärt. Lokalisation der Gene der γ1- und β-Untereinheit Die exakte chromosomale Lokalisation der γ1-Untereinheit ist wegen der Existenz von Isoformen nicht eindeutig. In situ Hybridisierung mit Kaninchen cDNA ergab ein starkes Signal bei 7p11-p12 und schwächere Anfärbung bei 7q21 und 11p11-p14 (Jones, da Cruz e Silva et al. 1990). Jedoch legt die Kopplung des Mäuse PHKG1 Gens zum muralen Chromosom 5 eine Lokalisation beim humanen Chromosom 7q21-q22 analog der Synthänie bei den Genen Gus und Epo nahe (Maichele und Chamberlain 1994). Für die β-Untereinheit sind zusätzlich zwei homologe Pseudogene bekannt. Die chromosomale Zuordnung des eigentlichen Gens zu 16q12-q13 (Francke, Darras et al. 1989) konnte bei der Sequenzierung und Strukturanalyse durch chromosom-spezifische Gen-Banken bestätigt werden; die beiden Pseudogene mit 67% bzw. 72% Identität fanden sich nur in einem genomischen KlonPool (Wullrich-Schmoll und Kilimann 1996). 9 Fragestellung Die Untersuchung der Enzyme des Glykogenstoffwechsels zum Ausschluss einer MuskelGlykogenose erbrachte in einer deutschen Familie die Assoziation eines familiären Auftretens der Hyper-CKämie mit dem Befund einer erniedrigten Aktivität der PhK, die hier weiter untersucht werden sollte. Um die Ursache der enzymologisch gemessenen Aktivitätsverminderung festzustellen, lag daher eine molekularbiologische Untersuchung der muskelspezifischen γ1Untereinheit und der gewebespezifischen Exons der β-Untereinheit nahe, für die noch keine Mutation beschrieben war. Aufgrund der Komplexizität der Genstruktur der PhK (Wehner und Kilimann 1995), vor allem der β-Untereinheit mit 33 Exons und einem überspannten Bereich größer 140 kBp (Wullrich-Schmoll und Kilimann 1996) bestand die Absicht, über die Assoziation benachbarter Mikrosatelliten einen eventuellen Ausschluss vor der arbeitsintensiven Direktsequenzierung zu erbringen und so eine Aussage über die Bedeutung der γ1- und βUntereinheiten in einer autosomal-dominanten Hyper-Ckämie-Familie für das Krankheitsbild einer milden Muskelerkrankung treffen zu können. 1.2.4 Rippling-Muskelerkrankung (Rippling muscle disease, RMD) Krankheitsbild Die Rippling-Muskelerkrankung (RMD) ist eine seltene autosomal dominante, myotonieähnliche Erkrankung. Die Hauptsymptome sind Muskelsteifigkeit, belastungs-induzierbare Muskelschmerzen und krampfähnliche Beschwerden. Das Kennzeichen der RMD sind perkussions-induzierbare schnelle Muskelkontraktionen (PIRC), lokalisierte Muskelvorwölbungen sowie rollende Muskelbewegungen (Muskelwogen = “Rippling“). RMD wurde zuerst 1975 von Torbergsen in einer norwegischen Familie beschrieben (Torbergsen 1975). 1989 berichtete Ricker et al. von zwei deutschen RMD Familien und führte den Terminus “rippling muscle disease ein (Ricker, Moxley et al. 1989). Stephan charakterisierte 1994 eine große amerikanische Familie (Stephan, Buist et al. 1994), bevor bei zwei weiteren deutschen Familien diagnostische Kriterien von RMD und PIRC als zuverlässiges klinisches Charakteristikum des variablen Phänotyps herausgestellt wurden (Vorgerd, Bolz et al. 1999). Weitere Familien und sporadische Fälle sind beschrieben worden (Alberca, Rafel et al. 1980; Rao, Hafford et al. 1987; Dew, Verma et al. 1995; Ansevin und Agamanolis 1996; Muller-Felber, Ansevin et al. 1999). So, Zu et al. (2001) bestätigten kürzlich die Konstanz der PIRC bei einer weiteren amerikanischen RMDFamilie. Caveolin-3 Vor kurzem wurde die Kopplung innerhalb der norwegischen RMD- Familie und vier deutschen Familien vom klinischen Phänotyp zum Chromosom 3p25 nachgewiesen. Über eine positionelle Kandidatengensuche wurden vier unterschiedliche Missense Mutationen im Caveolin-3-Gen bei diesem Locus festgestellt (Betz, Schoser et al. 2001). Heterozygote Mutationen im Caveolin-3-Gen wurden auch schon bei Patienten mit Gliedergürteldystrophie (LGMD) der autosomal dominanten Form 1C beschrieben (McNally, de Sa Moreira et al. 1998; Minetti, Sotgia et al. 1998; Herrmann, 10 Straub et al. 2000) und bei Kindern mit sporadischem Auftreten einer Hyper-CKämie gefunden (Carbone, Bruno et al. 2000; Merlini, Carbone et al. 2002). Caveolin-3 ist die muskelspezifische 21-24 kDa Isoform der Caveolin-Familie (Tang, Scherer et al. 1996). In Skelett- und Herzmuskulatur ist Caveolin-3 eine der Hauptkomponenten der Membran der Caveolae, kleinen vesikulären Invaginationen der Plasmamembran. Caveolae bilden sich durch Selbstassoziation und Oligomerisierung des Caveolins. Nach der Synthese bildet Caveolin-3 im endoplasmatischen Retikulum Homo-Oligomere von 300-350 kDa, die jeweils ca. 14-16 einzelne Caveolin-3-Monomere enthalten. Nach dem Erreichen des Sarkolemms formieren sich aus den Oligomeren Cluster von 25-50 nm Durchmesser (Galbiati, Volonte et al. 1999). Die Mutationen führen zu einer Verminderung der Caveolin-3-Expression an der Oberfläche von Muskelzellen und Retention von Caveolin-3 im Golgi-Organ, wahrscheinlich über einen dominant negativen Mechanismus. Darüber hinaus wurden Sekundärveränderungen wie eine leicht erhöhte Aktivität von nNOS, einem wichtigen Interaktionspartner von Caveolin-3, für RMD nachgewiesen. Fragestellung Ziel dieser Untersuchung war es, bei einem sporadischen RMD-Fall das Caveolin-3-Gen auf Mutationen hin zu untersuchen. Ergänzende immunhistologische Untersuchungen der Muskelbiopsie dieses Patienten sollten weitere Erkenntnisse über die Pathogenese dieser Erkrankung zeigen. 1.3. Methoden Ziel der Untersuchungen war es ebenfalls, die Praktikabilität und Effizienz unterschiedlicher Methoden der molekularbiologischen Diagnostik kritisch zu überprüfen. Dabei sollte die Single-Strand-Conformation-Polymorphismen-Methode auf ihre Detektionsgrenzen und Einflussfaktoren getestet werden. Bei der Anwendung der Direkt-Sequenzierung wurden zwei verschiedene Sequenzierer (ABI Prism 310 und ABI PRISM 377 ) eingesetzt, um so ihre jeweiligen Vorteile feststellen zu können. Die Anwendung einer vereinfachten Kopplungsanalyse mittels polymorpher Mikrosatelliten sollte Möglichkeiten und Grenzen dieser Methode aufzeigen. Wegen der Zeit- und Hersteller-abhängigen Veränderungen und eines unterschiedlichen Einsatzbereiches der verschiedenen Methoden erschien ein direkter Kostenvergleich wenig sinnvoll. Eine vergleichende Einschätzung des finanziellen und zeitlichen Aufwands sollte jedoch durch die spätere Anwendung ermöglicht werden. 2 Material, Methoden und Patienten 2.1 Material 2.1.1 Chemikalien Chemikalie Hersteller (Bestellnummer) Sitz der Niederlassung Acrylamid für die Elektrophorese Merck (110784) 64271 Darmstadt Agarose Seakem LE FMC Bioproducts(50005) Rockland, Maine USA 11 Ammoniumperoxodisulfat Merck (101201) 64271 Darmstadt Bisacrylamid Serva (29196) 69115 Heidelberg Borsäure Merck (100165) 64271 Darmstadt Bromphenol Blue-Na-salt Serva (15375) 69115 Heidelberg Chloroform Merck (102445) 64271 Darmstadt Desoxynukleosidtriphosphate 100 mM dATP, dCTP, dGTP, dTTP Promega 68199 Mannheim Dichlordimethylsilan Merck (103014) 64271 Darmstadt MBI Fermentas (#SM0243) MBI Fermentas (#SM0223) 68789 St.Leon-Rot Essigsäure Merck (100063) 64271 Darmstadt Ethanol Merck (100983) 64271 Darmstadt Ethidiumbromid Sigma (E-8751) 89552 Steinheim Formamid Merck (109684) 64271 Darmstadt Glycerin Merck (818709) 64271 Darmstadt Guanidinium-Isothiocyanat Merck (104167) 64271 Darmstadt Hydroquinoline Sigma (H6878) 89552 Steinheim Isoamylalkohol Merck (100979) 64271 Darmstadt Kaliumchlorid Merck (109924) 64271 Darmstadt Kerosin Serva (26940) 69115 Heidelberg N-Lauryl-Sarcosine Sigma (L5125) 89552 Steinheim DNA Größenstandards 100 Bp Leiter pUC19 DNA/MspI Marker (N1240) TM 68789 St.Leon-Rot MDE Gel Solution High-Resolution Gel for FMC BioProducts SSCP Analysis (50620) Rockland Maine, USA gamma-Methacryloxypropyl-Trimethoxsilane Sigma (M-6514) 89552 Steinheim Magnesiumchlorid Merck (109949) 64271 Darmstadt Mercaptoethanol Merck (115433) 64271 Darmstadt Mineralöl Sigma (M-5904) 89552 Steinheim Natriumazetat Merck (6268) 64271 Darmstadt Natriumcarbonat Merck (106392) 64271 Darmstadt Natriumzitrat Merck (106448) 64271 Darmstadt Natriumchlorid Merck (101540) 64271 Darmstadt Natriumhydroxid-Plätzchen Merck (106495) 64271 Darmstadt Natriumthiosulfat Sigma (S-1648) 89552 Steinheim Orange G Merck (115925) 64271 Darmstadt Phenol Fluka (77613) 89555 Steinheim Ponceau S Merck (14275) 64271 Darmstadt 2-Propanol Merck (109634) 64271 Darmstadt Proteinase K Böhringer Mannheim (1000144) 79630 Grenzach-Wyhlen Recombinant RNAsin® Ribonuclease Inhibitor Promega (N2511) 68199 Mannheim Silbernitrat Merck )101512) 64271 Darmstadt 12 N, N, N’, N’-Tetramethylethylendiamine Sigma (T-8133) 68298 Mannheim Tris(hydroxymethyl)-aminomethan Merck (108380) 64271 Darmstadt Xylene Cyanole FF Sigma (X-4126) 89552 Steinheim 2.1.2 Puffer und Medien/Lösungen Ansatz (Menge) Bestandteile Agarose Ladepuffer jeweils Spatelspitze Bromphenolblau, Xylencyanol 558 ml H20, 200 ml Glycerin (86% (v/v)in H2O) AL-Puffer 3 M Guanidin-Thiocyanat, 10 mM Tris-HCl, 5% Ethanol (v/v), pH 6,6 AW-Puffer 20 mM NaCl, 10 mM Tris-HCl, 80% Ethanol (v/v) pH 7,5 Denaturierender Ladepuffer jeweils Spatelspitze Bromphenolblau, Xylencyanol 95% Formamid (v/v), 20mmol EDTA pH 8 Entwickler-Lösung 100 mM NaOH, 0,06% (v/v) Formaldehyd in Aqua bidest. Fixierungslösung 10% (v/v) Ethanol, 0,5% (v/v) Essigsäure in Aqua bidest. Gelladungspuffer jeweils Spatelspitze Cyanol FF, Bromphenolblau, Orange G 10mM EDTA pH7 (Endkonzentration), 25%Ficoll (w/v) Lösung D 4M Guanidiniumthiocyanat 25mM NaCitrat pH 7,0 0,5% N-lauryl-sarcosine (w/v) 0,1M 2-Mercaptoethanol NKM-Puffer 28ml 5M NaCl 1,5ml 1M MgCl2 30 ml 1M KCl Phenol/Tris Sättigung mit Pufferlösung (Hersteller), Zusetzen von 0,1% (w/v) 8-Hydroxychinolin Phenol/H2O Sättigung mit gleichem Volumen Aqua bidest. Puffer 1-3 AGS Kit siehe Hersteller Resusp – Puffer 20ml 5M NaCl 10ml 1M TrisCl pH 7,5 1,5ml 1M MgCl2 50xTAE 242g Tris (hydoxymethyl)aminomethan, 57,1 ml Eisessig, 100 ml 0,5M EDTA pH8 ad 1 l Aqua bidest. 10xTBE 55g Borsäure 108g Tris(hydoxymethyl)-aminomethan 40ml EDTA 0,5M pH8 TE 10mM TrisCl pH 7,5 1mM EDTA TEN Solution 10x 9ml 1M TrisCl pH 7,5 24ml 0,5M EDTA pH 8 9ml 5M NaCl ad 1 l Aqua bidest. ad 1 l Aqua bidest. ad 1 l Aqua bidest. 13 2.1.3 Enzyme Enzym Hersteller (Bestellnummer) Sitz der Niederlassung Apa I MBI Fermentas (#ER1411) 68789 St.Leon-Rot Bcl I MBI Fermentas (#ER0721) 68789 St.Leon-Rot Reverse Transcriptase M-MuLV Boehringer Mannheim (1062603) 79630 Grenzach-Wyhlen Taq DNA Polymerase Promega (M6101) GibcoBRL (18038-018) Taq I MBI Fermentas (#ER0671) 68789 St.Leon-Rot Vsp I GibcoBRL (15485-014) Foster City California, USA RQ1 Rnase freie Dnase 68199 Mannheim Foster City California, USA 2.1.4. Kits Extraktions-Kits Hersteller (Bestellnummer) Sitz der Niederlassung Genome CleanTM HighPure PCR Product Purification Kit Trizol® Reagent und Trizol® LS Reagent AGS (GC300) Heidelberg Boehringer Mannheim (1732676) 79630 Grenzach-Wyhlen Gibco BRL (10296) Foster City California, USA Quiagen (51104) 40724 Hilden QIAamp® Blood Mini Kits 2.1.5. Oligonukleotide HNF1α Primer Primer Sequenz MODY3 Fragment M3.1 GGC AGG CAA ACG CAA CCC ACG forward M3.2 GAA GGG GGG CTC GTT AGG AGC reverse M3.3 CAT GCA CAG TCC CCA CCC TCA forward M3.4 CTT CCA GCC CCC ACC TAT GAG reverse M3.5 GGG CAA GGT CAG GGG AAT GGA forward M3.6 CAG CCC AGA CCA AAC CAG CAC reverse M3.7 CAG AAC CCT CCC CTT CAT GCC forward M3.8 GGT GAC TGC TGT CAA TGG GAC reverse M3.9 GGC AGA CAG GCA GAT GGC CTA forward M3.10 GCC TCC CTA GGG ACT GCT CCA reverse M3.11 TGG AGC AGT CCC TAG GGA GGC forward M3.12 GTT GCC CCA TGA GCC TCC CAC reverse 1 Produktlänge: 483 Bp Annealing-Temperatur: 2 Produktlänge: 390 Bp Annealing-Temperatur: 3 63°C Produktlänge: 346 Bp Annealing-Temperatur: 6 63°C Produktlänge: 397 Bp Annealing-Temperatur: 5 61°C Produktlänge: 299 Bp Annealing-Temperatur: 4 61°C 62°C Produktlänge: 322 Bp Annealing-Temperatur: 62°C 14 M3.13 GGT CTT GGG CAG GGG TGG GAT forward M3.14 CTG CAA TGC CTG CCA GGC ACC reverse M3.15 GAG GCC TGG GAC TAG GGC TGT forward M3.16 CTC TGT CAC AGG CCG AGG GAG reverse M3.17 AGA GCC CCT CAC CCC CAC ATC forward M3.18 AGG GGG CAG GGA CAG TAA G reverse Annealing-Temperatur: M3.19 GTA CCC CTA GGG ACA GGC AGG forward 10 M3.20 ACC CCC CAA GCA GGC AGT ACA reverse Annealing-Temperatur: 7 Produktlänge: 347 Bp Annealing-Temperatur: 8 Produktlänge: 229 Bp Annealing-Temperatur: 9 62°C 63°C Produktlänge: 245 Bp 57°C Produktlänge: 248 Bp 63°C Abb 3: Genstruktur HNF1α und Lage der Primer. Die kursiven Zahlen geben die Position der Aminosäuren an. Glucokinase Primer Primer Sequenz MODY2 Fragment M2.1 TCC ACT TCA GAA GCC TAC TG forward M2.2 TCA GAT TCT GAG GCT CAA AC reverse M2.3 TGC AGA TGC CTG GTG ACA GC forward M2.4 CAC AGC TGC TTC TGG ATG AG reverse M2.5 TAA TAT CCG GCT CAG TCA CC forward M2.6 CTG AGA TCC TGC ATG CCT TG reverse M2.7 TAG CTT GGC TTG AGG CCG TG forward M2.8 TGA AGG CAG AGT TCC TCT GG reverse M2.9 GCA GCC ACG AGG CCT ATC TC forward M2.10 GAG AAA GGC AGG CAG TGC TG reverse M2.11 CCA GCA CTG CAG CTT CTG TG forward M2.12 GAG CCT CGG CAG TCT GGA AG reverse M2.13 AGT GCA GCT CTC GCT GAC AG forward M2.14 CAT CTG CCG CTG CAC CAG AG reverse M2.15 TGC CTG CTG ATG TAA TGG TC forward M2.16 TGA GAC CAA GTC TGC AGT GC reverse 1 Produktlänge: 195 Bp Annealing-Temperatur: 2 Produktlänge: 290 Bp Annealing-Temperatur: 3 62°C Produktlänge: 195 Bp Annealing-Temperatur: 6 61°C Produktlänge: 272 Bp Annealing-Temperatur: 5 65°C Produktlänge: 295 Bp Annealing-Temperatur: 4 60°C 62°C Produktlänge: 176 Bp Annealing-Temperatur: 62,5°C 7 Produktlänge: 285 Bp Annealing-Temperatur: 8 62°C Produktlänge: 263 Bp Annealing-Temperatur: 60,5°C 15 Produktlänge: 367 Bp M2.17 ACT GTC GGA GCG ACA CTC AG forward M2.18 CTT GGA GCT TGG GAA CCG CA reverse Annealing-Temperatur: 63,5°C M2.19 GTC GAC TGC GTG CAG GGC GC forward 10 M2.20 TGT GGC ATC CTC CCT GCG CT reverse Annealing-Temperatur: Abb 4: 9 Produktlänge: 263 Bp 60°C Genstruktur des Glucokinase-Gens und Lage der Primer. Die kursiven Zahlen geben das Codon der jeweiligen Aminosäure an. Mitochondriale Mutation 3243 Primer Primer Sequenz Mit3243 Fragment MIDD1 AAG GTT CGT TTG TTC AAC GA forward 3029-3610 MIDD2 GGC CTA GGT TGA GGT TGA CC reverse Produktlänge: 581 Bp Annealing-Temperatur: 55°C GBE Primer Primer Sequenz GBE-Fragment B1 ATT CGG TCC CAG CTA GAG C forward B2 CGT CCA CAG ATG TGC TGA reverse B3 CAT TGG AGA AAA TGA AGG TGG forward B4 CTA CCA AGA ATC AAA GGC CTT reverse B5 GAG TTT AAG CAT TCC AGA CC forward B6 GGA TAG CAG ATT GTT TGC CT reverse B7 TCA TTC TGG ACC TAG AGG G forward B8 CGC TAC CTT GAA AAG TGC AT reverse B9 AGA TGA AGA CTG GAA CAT GG forward B10 CCA CAG GCC TAC GTG AGT reverse B11 GGA AGA AAG ATA TGG TTG GC forward B12 GTT ATC ACT GTC ACA CAG CT reverse B13 CAA ACA TGA GTG TCC TGA CTCC forward B14 CTC ATT ACG CAT GGG CTT GGT reverse 1 Produktlänge: 331 Bp Annealing-Temperatur: 2 Produktlänge: 516 Bp Annealing-Temperatur: 3 60°C Produktlänge: 409 Bp Annealing-Temperatur: gen. 1 57°C Produktlänge: 462 Bp Annealing-Temperatur: 6 63°C Produktlänge: 479 Bp Annealing-Temperatur: 5 62°C Produktlänge: 432 Bp Annealing-Temperatur: 4 63°C 60°C Produktlänge: 92 Bp Annealing-Temperatur: 60°C 16 B15 CTG TCC CAT CAA ACA TAT TC forward B16 GAG CAT GCT TAC TAT GCC reverse quan. 1 Produktlänge: 191 Bp Annealing-Temperatur: 56°C β-Aktin Primer Sequenz Mikrosatellit BAC1 TCACCCACACTGTGCCCATCTACCA BAC2 CAGCGGAACCGCTCATTGCCAATGG reverse forward βAktin Produktlänge: 344 Bp Annealing-Temperatur: 78°C Mikrosatelliten Primer Loci 7p12-q21 und 16q12-13 Primer Sequenz Mikrosatellit Mi7.1 TGT GTC ATT ACG CTT TTC ATC forward D7S478 Mi7.2 TCA AAT GGT TCA GGA GAA AGA reverse Mi7.3 ATC TTG GAT TTA GGG TTG GC forward D7S492 Mi7.4 GGC TCT GCT CCA TCT TCA TA reverse M7.5 ATG CAA CCT ACC CTC AAA TG forward D7S669 M7.6 TAC GGA TTA CCC ACA TTG CTA T reverse Mi7.7 CAG TGC TGG AGT TGT TCA AG forward D7S2429 Mi7.8 CTG GGA GTC AAG TGT TTT GG reverse Produktlänge: 122 Bp Annealing-Temperatur: Produktlänge: 149 Bp Annealing-Temperatur: Mi16.2 AGT CAG TCT GTC CAG GTG reverse Mi16.3 CCG AGG TCA CAC CAT TGT forward D16S3093 Mi16.4 CCA AAA GGT TGA TTC TCT G reverse 61°C Produktlänge: 177 Bp Annealing-Temperatur: forward D16S409 57°C Produktlänge: 133 Bp Annealing-Temperatur: Mi16.1 TGA ATC TTA CAT CCC ATC CC 55°C 57°C Produktlänge: 139 Bp Annealing-Temperatur: 56°C Produktlänge: 306 Bp Annealing-Temperatur: 55°C Caveolin-3 Primer Primer Sequenz Caveolin Fragment Ca1 AGC TCG GAT CTC CTC CTG TGG forward Exon 1 Ca2 CAC GTC TCG CAA ACC TGA CAC reverse Ca3 GTG AGT TGA GGC TTC CCC TTG C forward Exon 2 Ca4 GTA TGG AGC AGT CCC TAA AGA G reverse Cav1 CTG GCA GGG ACA TAA GTC TGG forward 1 Cav2 ACT GTG TCT GCA GCA GAT ACC reverse Cav3 GAT TCT GAC ACA TGC ACG CAC forward 2 Cav4 CAG CCC CTG TGA AGA AGT CC reverse Abb 5: Exon-Intron-Struktur des Caveolin-3-Gens und Lage der Primer Produktlänge: 207 Bp Annealing-Temperatur: 62°C Produktlänge: 491 Bp Annealing-Temperatur: 61°C Produktlänge: 320 Bp Annealing-Temperatur: 62°C Produktlänge: 508 Bp Annealing-Temperatur: 62°C 17 Zur Suche der genomischen Sequenzen und zur Überprüfung der spezifischen Bindung wurde das Programm Blast unter http://www.ncbi.nlm.nih.gov/blast/ (Altschul, Madden et al. 1997) benutzt. Die Oligonukleotide wurden bei MWG Biotech (Anzinger Strasse 7a 85560 Ebersberg Deutschland) und Life Technologies Inc. (GIBCO BRL, 9800 Medical Center Dr. Rockville , MD 20849-6482) synthetisiert. 2.1.6 Geräte Gerät Hersteller Mastercycler Gradient® Eppendorf Robocycler 40 Stratagene TM Minicycler und Hot Bonnet Biozym Eismaschine Ziegra GelFix for PAG Serva GeneQuant RNA/DNA Calculator Pharmacia Hybridisator 400 HY (891634) Bachofer GmbH Imager Appligene Milli-Q Plus ZFMQ05001 Millipore Multiphor II (18-1018-06) Amersham Pharmacia HighVoltagePowerPack P30 Biometra Powersupply E452 Consort Power Supply ST 606 T Gibco Brl Schüttler WT 12 Biometra 373 DNA Sequencer mit Macintosh Quadra 650 Applied Biosystems Thermoprinter VP-1500 II Seikosha Typ TP 8/10, 20.000 UpM Ultra Turrax® Varioklav Dampfsterilisator Typ 500 EV-Z H+P Labortechnik Wasserkühler F10 C (9111027) Biometra GS Centrifuge Beckmann Biofuge pico Heraeus Zentrifuge GR2022 Jouan Zentrifuge 4123 Heraeus-Christ GmbH Zentrifuge 5402 Eppendorf Für die SSCP-Gele wurde das Multiphor II System (Amersham Pharmacia) mit den Glasplatten 125 x 260 x 0,5 mm und den Gel-Fix PAG-Folien von Serva (265 x 125 mm) als Gelträger verwendet. Gele wurden entweder mit handelsüblichen Scannern oder dem Personal Densitometer® SI von Molecular Dynamics eingescannt. Zur Auswertung der Sequenzierungen wurde ABI-Prism Sequencing Software 2.1.1 und ABI Genescan 672 1.1, außerdem Align Plus 3.0 für Windows von Scientific & Educational Software. 18 2.2 Methoden 2.2.1 Grundlegende Methoden 2.2.1.1. Extraktion von Nukleinsäuren Extraktion von DNA aus frischem Vollblut – Genome CleanTM-Procedure Zur Extraktion aus frischem Vollblut wurde zum einen die Genome CleanTM-Procedure (Angewandte Gentechnologie Systeme GmbH, Heidelberg) angewendet. Dazu wurden die VollblutMonovetten bei 3000 g für 15 min bei 4 °C zentrifugiert und die weiße Leukozyten-Interphase „Buffy coat“ in ein 2 ml Gefäß überführt. Nach erneuter Zentrifugation wurden nach Abnahme der zellreichen Zwischenschicht 0,3 ml dieser Zellsuspension mit Puffer 1 des Kits versetzt. Nach einer Inkubation bei 68°C für 10 min wurden 0,9 ml vorsichtig nach kurzem Abkühlen hinzugefügt und bis zur Bildung einer Emulsion aus den beiden Phasen vorsichtig gemischt. Die Phasen wurden daraufhin durch Zentrifugation bei 10.000 UpM und RT für 2 min wieder getrennt und die obere, wässrige Phase in ein neues 2 ml Gefäß überführt. Hier wurde die DNA durch Hinzufügen von 0,9 ml Aqua bidest. und 0,1 ml Puffer 2 gefällt. Nach vorsichtigem Schütteln bei Raumtemperatur wurde die präzipitierte DNA bei 10.000 UpM abzentrifugiert und der Überstand verworfen. Das Pellet wurde in 0,3 ml Puffer 3 aufgenommen. Durch 0,75 ml 96% (v/v) Ethanol wurde erneut eine Präzipitierung bewirkt. Die Probe wurde durch Kippen vorsichtig vermischt und bei 10.000 UpM und RT für 10 min zentrifugiert, der Überstand verworfen. Mit 0,5 ml 70% (v/v) Ethanol wurde das Pellet gewaschen. Nach Entfernung des Ethanols wurde die DNA in Aqua bidest. aufgenommen. Extraktion von DNA aus frischem Vollblut – QIAamp® Blood Midi Kit Als weiteres Verfahren wurde die DNA-Extraktion mit Silica-Säulchen benutzt (QIAamp® Kit). Dazu wurden je 200 µl Vollblut mit 25 µl Proteinase K und 200 µl AL-Puffer vermischt und für 10 min bei 70°C inkubiert. Nach Zugabe von 210 µl Ethanol (100%, -20°C) wurden die Proben gemischt und auf die vorbereiteten Säulen (Silicasäule in Auffanggefäß einsetzen) überführt. Die Säulen wurden bei 8.000 UpM für 1 min bei RT zentrifugiert, der Durchlauf verworfen und die Säulen in frische Auffanggefäße gesetzt. Anschließend wurde 500 µl AW-Puffer auf die Säulen pipettiert, zentrifugiert (1 min, 8.000 UpM), der Durchlauf verworfen und die Säulen wieder in frische Auffanggefäße gesetzt. Nach Zugabe von 500 µl AW-Puffer wurden die Proben 3 min bei 14.000 UpM zentrifugiert, der Durchlauf verworfen und die Säulen in frische 1,5 ml Reaktionsgefäße gesetzt. Zum Eluieren wurde 100 µl Aqua bidest. auf die Proben gegeben und nach Inkubation bei RT für 1 min bei 8.000 UpM zentrifugiert. Extraktion von DNA aus älterem Vollblut (>48h) mittels Proteinase K Diese Methode wurde hauptsächlich bei älteren Proben oder tiefgefrorenen Vollblutproben verwandt. 10 ml des Vollblutes wurden dazu zuerst in 40 ml kalten NKM-Puffers aufgenommen und gut durchmischt. Dieses Gemisch wurde anschließend 30 min bei 3500 g und 4º C zentrifugiert. Bis auf 10 ml wurde der Überstand entfernt. Das Pellet und der Rest wurde erneut in 40 ml NKMPuffer resuspendiert, gevortext und erneut für 30 min bei 3500 g und 4º C zentrifugiert. Dann wurde Überstand bis auf 4 ml abgenommen und 0,5 ml 10x TEN Lösung, 0,25 ml 2 mg/l Proteinase K und 0,5 ml 10%SDS hinzugefügt. Die Reaktionsgefäße wurden hierauf behutsam 19 geschüttelt und zur Inkubation bei 37º C über Nacht in einen Rührschüttler bei niedrigster Schüttelstufe gestellt. Daraufhin wurde zu gleichen Teilen Phenol und Chloroform hinzugefügt, das dann auf einem Schüttler 30 min bei Raumtemperatur gemischt wurde. Nach einer Zentrifugation bei Raumtemperatur für 15 min bei 1500 g wurde die organische Phase entfernt und verworfen. Zur wässrigen Phase wurde 5 M NaCl hinzufügen, um zur Proteinfällung eine Endkonzentration von 0,4 M zu erreichen. Nach Hinzufügen des zweifachen Volumens 100% Ethanols und leichtem Mischen wurde die Probe für 30 min bei 3500 g und 4º C zentrifugiert. Der Überstand wurde entfernt, das Pellet mir 5 ml 70% Ethanol gewaschen und noch einmal durch eine Zentrifugation für 30 min bei 3500 g aufgereinigt. Die DNA wurde im Lyophilisator/Exsikator kurz getrocknet und dann in Aqua bidest. bei Raumtemperatur über Nacht resuspendiert. Extraktion von RNA nach Chomczynski und Sachi Die RNA-Präparation aus tiefgefrorenem Vollblut erfolgte mit Guanidinium-Thiocyanat nach der Methode von Chomczynski und Sachi (Chomczynski und Sacchi 1987). 1-2 ml tiefgefrorenes EDTA Vollblut wurde in 9 ml Lösung D (250 mg Guanidium Thiocyanat, 312 ml Aqua bidest., 17,6 ml 0,75 M Natriumcitrat pH 7,0, 7,54 ml N-Lauroylsarcosin) aufgetaut. Es wurden 1 ml Natriumacetat pH 4,1 sowie 10 ml Phenol/H2O gesättigt (pH≈4,5) zugegeben und die Probe vorsichtig durch Kippen gemischt. Es folgte die Zugabe von 2 ml Chloroform/Isoamylalkohol (49:1) mit anschließendem vorsichtigen Kippen und 15minütiger Inkubation auf Eis. Die Phasentrennung erfolgte für 20 min durch Zentrifugation bei 2000 g bei 4°C. Die Phenol/Chloroform-Extraktion wurde zweimal wiederholt, bis die wässrige Phase absolut klar war. Diese wurde mit 1 Volumen Isopropanol versetzt und die RNA 1 h bei –20°C mit anschließender Zentrifugation für 30 min bei 10.000 g bei 4°C gefällt. Die RNA wurde in 400 µl Lösung D aufgenommen und mit 60 µl (0,15x Volumen) 2 M NaAcetat pH 4,1 und 1 ml (2,5x Volumen) Ethanol versetzt. Nach der Fällung für mindestens acht Stunden bei –20°C und 30minütiger Zentrifugation bei 10.000 g und 4°C wurde das Pellet mit 70% (v/v) Ethanol gewaschen, die RNA fast vollständig getrocknet und in 30 bis 40 µl Aqua bidest aufgenommen. Extraktion von RNA mit Trizol® Bei der Verwendung mit Trizol® (Life Technolgy, Foster City USA) wurde zu 100 mg bzw. 0,25 ml Gewebe anfangs 0,75 ml Trizol® gegeben und dieses Gemisch mit dem Turrax® zerkleinert. Tiefgefrorene Blutproben wurden vollständig aufgetaut, bevor 3 ml Blut zügig in einem 15 ml Reaktionsgefäß mit 9 ml Trizol® LS versetzt wurden. Nach 10minütigem gründlichen Mischen und Vortexen wurden 2,4 ml Chloroform hinzugefügt und erneut gut gemischt. Danach erfolgte eine Zentrifugation für 25 min bei 4 ºC und 3000 g. Der klare, wässrige Überstand wurde abgenommen und jeweils 1 ml in ein kleineres 2 ml Schraubdeckel-Reaktionsgefäß überführt. 1 ml Isopropanol wurde hinzugefügt und gut vermischt. Daraufhin folgte die Fällung bei - 20 ºC über Nacht bzw. mind. acht Stunden. Die Reaktionsgefäße wurden für 25 min bei höchster Umdrehungszahl und 2ºC auf einer herkömmlichen Tischzentrifuge zentrifugiert. Danach wurde unter Sichtkontrolle des Pellets der Überstand vorsichtig abgekippt und vollständig abpipettiert. 0,5 ml 75% (v/v) Ethanol wurden auf das Pellet gegeben und erneut für 25 min bei voller Umdrehung und 2 ºC zentrifugiert. 20 Der Überstand wurde wieder abgenommen und das Pellet kurz aber nicht vollständig getrocknet. Das Pellet wurde in 15 µl Aqua bidest. aufgelöst und die RNA nach Quantifizierung mittels Photometer(GeneQuant RNA/DNA Calculator) bei -20 ºC eingefroren. DNA-Elutions aus Agarose-Gelen Verfahren A Zuerst wurden 100 mg eines Agarose-Blockes, der aus einem 1,5% (w/v) Gel ausgeschnitten worden war, für 5 min auf 50°C erhitzt. Nach kurzem Abkühlen wurde die Probe nach Hinzufügen von 100 µl Phenol und 100 µl Chloroform für 5-10 s gründlich gevortext. Nach einer Zentrifugation bei einer Umdrehung von 14.000 UpM für 5 min wurde der wässrige Überstand vorsichtig abgenommen und erneut mit 100 µl Chloroform durchmischt. Den Überstand, den man nach der anschließenden Zentrifugation bei 14.000 UpM für 5 min erhielt, wurde mit einem 0,6fachen Volumen Isopropanol und einem 0,1fachem Volumen Natriumacetat (pH 5,2) versetzt. Nach intensivem Mischen auf dem Rührschüttler wurde die Probe über Nacht bei –20°C inkubiert. Nach einer Zentrifugation bei 14.000 UpM und 4°C für 15 min wurde der Überstand komplett verworfen. Das Pellet wurde mit 200 µl 70% (v/v) Ethanol aufgespült und nochmals bei14.000 UpM und 4°C für 15 Minuten zentrifugiert. Das Ethanol wurde danach vollständig abgenommen und das Pellet für 10 Minuten getrocknet und abhängig von der gewünschten Konzentration in einem festgesetzten Volumen Aqua bidest aufgenommen. DNA-Elutions aus Agarose-Gelen Verfahren B Hierbei wurde das High Pure PCR Product Purification Kit (Boehringer Mannheim, 79630 Grenzach-Wyhlen) verwendet. Zu 100 mg eines Agaroseblocks wurden 500 µl des Bindungspuffers gegeben. Bei größeren bzw. kleineren Volumina wurde die Menge proportional erhöht bzw. erniedrigt. Das Reaktionsgefäß wurde für 8 min bei 65°C und minütlichem Vortexen auf einem heizbarem Rührschüttler erwärmt. Hatte sich die Agarose verflüssigt und waren beide Anteile gleichmäßig vermischt, so wurde die Flüssigkeit in ein Filtrationsgefäß pipettiert, das in ein Auffanggefäß eingesetzt worden war. Nach Zentrifugation für 30 s bei RT und 13.000 g wurde der Durchlauf verworfen und das Filtrationsgefäß erneut in das Auffanggefäß eingesetzt. 500 µl Waschpuffer wurden in das obere Reservoir pipettiert und erneut zentrifugiert. Nach nochmaligem Verwerfen wurde dieser Schritt mit 200 µl Waschpuffer wiederholt. Einmal wurde das Filtrationsgefäß für 1 min „trocken“ zentrifugiert, bevor es in ein sauberes 1,5 ml Reaktionsgefäß gesetzt wurde. Zur Elution der Nukleinsäure wurden hierauf 100 µl Aqua bidest. mittig auf den Filter pipettiert. Nach Inkubation für 1 min bei RT wurde dieses bei 13.000 g und RT abzentrifugiert und das Filtrationsgefäß verworfen. 2.2.1.2 Reverse Transkription Zur reversen Transkription wurden jeweils 6 µg RNA, 2 µl RQ1 Puffer, 2 µl RQ1 Dnase und 1 µl RNAsin auf ein Gesamtvolumen von 20 µl mit Aqua bidest. angesetzt und für 30 min bei 37 °C zur Beseitigung restlicher DNA inkubiert. Nach Hinzufügen von 1,8 µl dN6-Primer, 24 µl RT-Puffer, 6 µl dNTP und 57,4 µl Aqua bidest. wurde die Probe zur Zerstörung möglicher Sekundärstrukturen für 5 min bei 94°C erwärmt und daraufhin sofort auf Eis gekühlt, um eine Renaturierung zu vermeiden. Nach kurzem Abzentrifugieren des Kondensats wurde die eigentliche Reaktion nach 21 Hinzupipettieren und Vermischen von 4,8 µl RNAsin und 6 µl MMulV Reverse Transkiptase gestartet. Mit einer zehnminütigen Erhitzung auf 65°C wurde die Reaktion nach 60 min beendet und die Produkte bei –20°C eingefroren. 2.2.1.3 Polymerase-Ketten-Reaktion (PCR) In einem Reaktionsansatz von 50 µl wurden für die PCR 1 µl 50 mmol/l Magnesium2+-Salz (Endkonzentration 1 mmol/l), 1 µl eines Gemisches von jeweils 10 mmol/l dATP, dCTP, dGTP und dTTP (Endkonzentration 200 µmol/l), 5 µl 10x PCR-Puffer, jeweils 1,25 µl 10 pmol/µl (Endkonzentration 250 nmol/l) für beide Primer und 0,25 µl Taq Polymerase mit 25 U/µl (Endaktivität 6,25 U/50 µl) eingesetzt. 200 ng der jeweiligen DNA wurden in einem Volumen von 2- Abb 6: Optimierung der PCR. Fragmente Caveolin Exon 1 und 2 (Primer Ca1-Ca2 bzw. Ca3-Ca4) bei den Temperaturen 61°C, 61.4°C, 62.2°C, 63,4°C, 64.8°C, 66.2°C, 67.6°C, 68.8°C, 69.8°C, 70.7°C von links nach rechts 5 µl hinzugefügt und das Volumen entsprechend mit Aqua bidest. ergänzt. Die optimalen Annealing-Temperaturen der verwendeten Primer wurden mittels eines Temperaturgradienten auf dem Mastercycler Gradient (Eppendorf) evaluiert und etabliert. Es wurde die höchstmögliche Temperatur mit einem spezifischen Produkt gewählt (siehe Abb 6). 2.2.1.5 Restriktion Die Restriktionsanalyse wurde in einem Ansatzvolumen von 10 µl durchgeführt. Die jeweils hinzugefügte Enzymaktivität wurde je nach der Größe des Fragments und der Anzahl der Schnittstellen innerhalb des λ-Vektors entsprechend vorliegender Standardprotokolle variiert. Die Pufferkonzentration entsprach den Herstellerangaben des verwendeten Enzyms. 2.2.1.6 Anfärbung der Nukleinsäuren Die in 1,2%igen (w/v) Agarose-Gelen oder 7,5%igen (w/v) Polyacrylamidgelen aufgetrennten Nukleinsäuren wurden – wenn nicht anders beschrieben - für 10 min in 100 ml einer Ethidiumbromid-Lösung (2,5 (Appligene) aufgenommen. µg/ml) inkubiert und mit einem UV-Dokumentationssystem 22 2.2.2 Direkt-Sequenzierung Zur Sequenzierung wurde das spezifische DNA-Fragment aus dem Agarose-Gel eluiert. Von diesem Eluat wurden 15 µl mit 1 µl (10 pmol) eines einzelnen Primers (unidirektional) und 4 µl der BigDyeTerminator Polymerase vermischt. Die PCR wurde ansonsten bei denselben Bedingungen, aber niedrigerer Zyklenzahl (32-33) durchgeführt. Nach der PCR wurde das Volumen mit 80 µl Aqua bidest. auf 100 µl aufgefüllt. Dazu wurden je 10 µl 3 M NaAcetat pH 4,8 und 250 µl 100% Ethanol hinzugefügt und dieses Gemisch nach kurzem intensiven Vortexen für 20 min bei 20.000 g und RT in einer Tischzentrifuge zentrifugiert. Der Überstand wurde verworfen und 100 µl 75% Ethanol zum Waschen hinzugefügt. Später wurde das Pellet für 5 min bei 20.000 g und RT ein weiteres Mal abzentrifugiert. Der Überstand wurde erneut entfernt und das Pellet entweder luftgetrocknet oder lyophilisiert. Anschließend wurde das Pellet je nach Sequenzierer entweder in 20 µl des Sequenzierpuffers (TSR, ABI PRISM 310) in ein spezielles Reaktionsgefäß überführt oder in 5 µl Laufpuffer (ABI PRISM 377) aufgenommen. 2.2.3 Single Strand Conformation Polymorphism (SSCP)-Gele 2.2.3.1 Horizontale Gelelektrophorese Für die SSCP-Gele wurde das Pharmacia Multiphor II System verwendet. Als Grundlage der Polymerisations-Lösung wurde dabei die MDE Gel Solution (FMC BioProducts) in variierenden Konzentrationen eingesetzt. Die Lösung wurde mit Aqua bidest. und 5x TBE auf 20 ml bei einer Endkonzentration des TBE von 0,6x ergänzt. Die Glasplatten wurden bei der Vorbereitung zu jedem dritten Lauf mit einer 2%igen (v/v) Dimethyldichlorsilan-Chloroform-Lösung benetzt, mit Aqua bidest. gespült und getrocknet. Die PAG-Folie (Serva) wurde mit einer kohäsiven Wasserschicht zu einer Glasplatte unter Vermeidung der Bildung von Luftblasen hin zwischen die zwei Platten geklemmt. Anschließend wurde Ammoniumperoxosulfat der Gellösung zugefügt und die gesamte Lösung für 5 Minuten mit Hilfe einer Wasserstrahlpumpe entgast, um den Gehalt an freien Radikalen zu verringern. Beim Beginn des Polymerisationsvorgangs mit TEMED wurde die Lösung mit einer 5 ml Spritze durch eine Kanüle zügig zwischen die Platten gegossen und mit 300 µl Isopropylalkohol überschichtet, um einen vollständigen Luftabschluss und eine gleichmäßige Polymerisation zu erreichen. Die Polymerisationszeit betrug 80 Minuten. Um eine Äquilibrierung des Gels zu ermöglichen, wurde ein einstündiger Vorlauf durchgeführt. Das Gel wurde auf der PAG-Folie mittig auf der wassergekühlten Platte positioniert. Dabei diente Kerosin als Kühlvermittler zwischen Trägerfolie des Gels und Kühlplatte und vermied durch seine Eigenschaft als hydrophobe Flüssigkeit im Falle des Auswaschens von Salzen auch Kurzschlüsse und Funkenschlag. Die Spannung wurde über TBE-Lösung getränktes Filterpapier von den Kammern gleichmäßig auf das Gel übertragen. Nach der Elektrophorese zu den weiter unten dargestellten Bedingungen wurde das Gel nach kurzem Abtropfen gefärbt. 2.2.3.2 Silberfärbung von SSCP-Gelen Das Gel auf der PAG-Folie wurde nach der Elektrophorese in einer Edelstahlschale (300 x 130 mm) für 30 min mit 100 ml einer 10% (v/v) Ethanol- und 0,5% (v/v) Essigsäure–Lösung fixiert. 23 Dieser Fixierungsschritt konnte bis zu mehreren Stunden verlängert werden, ohne dass dies Auswirkungen auf die Qualität des Geles hatte. Nach dreimaligem Auswaschen mit 100 ml Aqua bidest. für jeweils 180 s wurde das Gel für 20 min in einer 0,25% (w/v) AgNO3-Lösung inkubiert. Für 15 s wurde das Gel noch zweimal mit 100 ml Aqua bidest. gewaschen, bevor durch die Zugabe von 100 ml einer 0,07% (v/v) Formaldehyd / 1,5 M NaOH-Lösung der Entwicklungsprozess gestartet wurde. Kurz bevor nach visueller Einschätzung eine ausreichende Anfärbung bei möglichst optimalen Signal-Hintergrund-Verhältnis erreicht wurde, wurde die Entwicklung durch Inkubation mit Fixierungslösung gestoppt. Bei nicht ausreichender Anfärbung konnten die dargestellten Schritte der Entwicklung zur Nachfärbung wiederholt werden.. Zur Dokumentation und Lagerung wurde das Gel mit 100 ml einer 10% (v/v)Glycerinlösung inkubiert und mit einer handelsüblichen Haushalts-Zellophanfolie luftblasenfrei abgedeckt. 2.2.4 Elektrophoretische Auftrennung von DNA auf denaturierenden Polyacrylamid-Gelen 2.2.4.1 Vertikale Gelelektrophorese Vor der Polyacrylamid-Sequenzgel-Elektrophorese wurden zwei Glasplatten (Platte 1: 40 cm x 40 cm, Platte 2: 37 cm x .40 cm.) mit 100% (v/v) Ethanol mehrfach abgespült und gereinigt. Die kleinere Gelplatte wurde mit 20 ml einer 2%igen (v/v) Dimethyldichlorsilan-Lösung in Chloroform gleichmäßig benetzt, um später ein leichteres Ablösen des Gels zu erreichen. Nach vollständiger Verdunstung der Lösung wurde die Platte einmal mit Aqua bidest. abgespült und getrocknet. Die größere der beiden Platten wurde mit 10 ml einer Lösung (20 ml Ethanol, 120 µl γMethacryloxypropyltrimethoxysilan, 120 µl 10% (v/v) Essigsäure, 1,2 ml Aqua bidest.) behandelt, die eine feste Anheftung des Polyacrylamid-Gels sicherte. Die Platte wurde danach zweimal mit je 20 ml (100% (v/v) Ethanol abgespült und getrocknet. Daraufhin wurden die Platten an drei Seiten mit Dichtungen mit der gewünschten Dicke des späteren Gels zusammengeklemmt. Zu einer entgasten Lösung aus 7M Harnstoff, 30% (v/v) Formamid, 1x TBE und 6% (w/v) Acrylamid (Verhältnis Acrylamid/Bisacrylamid 38:2) wurden 500 µl einer 10% (w/v) AmmoniumperoxosulfatLösung und 50 µl TEMED hinzugefügt, um den Polymerisationsprozess zu starten. Die Lösung wurde zügig in den Platten-Zwischenraum eingefüllt. Die oberen Kante der Platten wurde mit einem Kamm luftdicht verschlossen. Zusammen mit der unteren Dichtung wurde dieser nach einer Stunde wieder entfernt. Zur Äquilibrierung des Gels wurde zuerst für 60 min in 1x TBE bei 70 W ein Vorlauf durchgeführt. Die Proben wurden mit einem einfachen Volumen Ladepuffer versetzt und für 90 s bei 90°C denaturiert. Der Gellauf fand dann unter ständiger Temperaturkontrolle statt und wurde bei entsprechender Position der Farbstoff-Banden beendet. 2.2.4.2 Silberfärbung der denaturierenden Polyacrylamid-Gele Die Glasplatten wurden vorsichtig nach kurzem Abkühlen voneinander getrennt und die größere Platte mit dem Gel in einer Wanne für 15 Minuten in 7,5% (v/v) Essigsäure fixiert. Danach wurde die Platte dreimal für jeweils 3 min mit entionisiertem Aqua bidest. (Milli-Q®) gespült, bis die Gelplatte nach Herausnehmen gleichmäßig von einer Wasserschicht bedeckt blieb. Danach wurde die Glasplatte genau horizontal ausgerichtet und mit einer Sprühflasche gleichmäßig mit einer 24 24 mM AgNO3 Lösung bedeckt. Diese wurde wiederholt alle 3 min während des 15minütigem Anfärbungsprozesses aufgebracht. Nach vorsichtigem Abgießen wurde das Gel einmal mit Aqua bidest. für 15 s gespült. In der Wanne wurden 100 ml Entwickler-Lösung vorsichtig auf das Gel gegossen, für 10 s unter schwenkenden Bewegungen inkubiert und schnell wieder abgegossen, um Ablagerungen von reduziertem Silber und so die Bildung eines dunklen Hintergrunds zu vermeiden. Anschließend wurden erneut 100 ml der Entwickler-Lösung vorsichtig und gleichmäßig auf die Oberfläche des Gels aufgebracht. Für einen guten Bildkontrast wurden möglichst geringe Mengen verwendet. Nach deutlicher Anfärbung der Banden wurde die Entwicklung durch Hinzufügen von 7,5% (v/v) Essigsäure gestoppt. Nach einer 3minütigen Inkubation mit Essigsäure wurde das Gel für 15 min mit Aqua bidest. gespült. In vertikaler Position und bei Raumtemperatur wurde das Gel getrocknet. 2.3 Patienten 2.3.1 Patienten mit MODY Die Patienten wurden elektiv von Ambulanzen, Tageskliniken und Diabetes-Zentren sowohl von internistischer als auch von pädiatrischer Seite aus in ganz Deutschland überwiesen. Die Auswahl richtete sich nach den klassischen MODY-Definitionen (Hattersley 1998). Für die Datenerhebung wurde ein Bogen (siehe Abb 28) entwickelt, in dem Felder zur aktuellen Diabetes-Therapie, zur Familienanamnese und schon vorhandenen Komplikationen vorgegeben waren. Die Proben wurden direkt nach dem Eintreffen anonymisiert und einer internen Nummerierung zugeordnet. Die Rücklaufquote der Datenerhebungsbögen betrug bei dem eingesandten Probematerial insgesamt 53,6%, wobei die Quote (32,9%) und Vollständigkeit insbesondere bei mutationsnegativen Patienten so niedrig war, dass eine statistische Zusammenstellung insgesamt nicht möglich oder sinnvoll war. Insgesamt wurde Material von 140 Patienten von 72 unterschiedlichen Familien in die Analyse einbezogen. Das Durchschnittsalter lag bei 31 ± 16 Jahren bei einer Spanne von 1 bis 84 Jahren. 2.3.2 Patienten mit APBD 2.3.2.1 Familie AD1 Anamnese Eine 46jährige deutsche Patientin entwickelte seit dem 43.Lebensjahr eine progrediente Gangstörung. Bei der neurologischen Untersuchung zeigte sich eine spastische Tetraparese mit gesteigerten Muskeldehnungsreflexen und beidseitigen Pyramidenbahnzeichen. Der Gang war breitbeinig-ataktisch bei ausgeprägter Hohlfußbildung beidseits. Das Vibrationsempfinden war an den Knien vermindert und an den Malleoli aufgehoben. Die Untersuchung ergab eine strumpfförmige Hypästhesie und Hypalgesie. Es bestand eine Dranginkontinenz, außerdem wurde eine Restharnmenge von 110 ml gemessen. Bei den testpsychologischen Untersuchungen (SIDAM) ergaben sich keine Hinweise auf eine Demenz. Im Liquor wurde ein Normalbefund mit lymphomonozytärem Zellbild bei einer Zellzahl von 1/µl im Liquor gefunden. Der Albuminquotient und IgG-Index waren unauffällig, es zeigten sich keine oligoklonale Banden. Eine intrathekale 25 Produktion erreger- oder virusspezifischer Immunglobuline war nicht nachweisbar. Eine unregelmäßige α-Grundaktivität mit Übergang zu einer leichten Allgemeinveränderung war im EEG nachweisbar. Die akustisch- und somatosensibel (N.medianus und N.tibialis) evozierten Potentiale ergaben verlängerte Latenzzeiten. Motorische und sensible Nervenleitgeschwindigkeiten waren herabgesetzt und die distalen Latenzen verzögert. Es fand sich eine Erniedrigung der motorischen Muskelsummenaktionspotentiale und der sensiblen Nervenaktionspotentiale. Die elektromyographischen Befunde ergaben Hinweise auf eine ältere neurogene Schädigung. Elektrophysiologisch zeigte sich somit eine sensomotorische Polyneuropathie. Signalintensive konfluierende Läsionen in der periventrikulären Substanz und fokale Herde in der Pons, Medulla und der subkortikalen weißen Substanz zeigten sich auf den T2-gewichteten Aufnahmen der cerebralen Kernspintomographie. Dabei fand sich eine innere und äußere Hirnatrophie über das altersentsprechende Maß hinaus. Die echokardiographische Untersuchung ergab eine deutliche linksventrikuläre Hypertrophie, im Sinne einer hypertrophen Kardiomyopathie. Die Oberbauchsonographie war ebenso unauffällig wie die Laboruntersuchungen, die Leber- und Muskelenzyme, ANA, AMA, ASMA, HMA und cANCA, Phytansäure, Lipidelektrophorese, Arylsulfatase A, β-Galaktosidase, β-Hexosaminidasen. Ausgeschlossen wurde auch eine Genträgerschaft für die spinocerebelläre Ataxie vom Typ1 (SCA1), Typ2 (SCA2), Typ3 (SCA3/MJD1) und Typ 6 (SCA6). Weil eine Muskelbiopsie abgelehnt wurde, wurden die Werte von Phosphocreatin, ATP, ADP, anorganisches Phosphat und intrazellulärer pH unter aerober und anaerober Muskelkontraktion mittels 31 Phosphor-Magnetresonanz-Spektroskopie gemessen. Hierbei fanden sich keine Auffälligkeiten des Glykogenmetabolismus. In einer neurologischen Rehabilitationsklinik wurden deutliche kognitive Defizite mit ausgeprägten Gedächtnisstörungen durch neuropsychologische Tests festgestellt. Im weiteren Krankheitsverlauf ereigneten sich mehrere Stürze, von denen einer zu einer medialen Schenkelhalsfraktur rechts führte. Die Mobilisierung nach der chirurgischen Versorgung mit einer dynamischen Hüftschraube war aufgrund der Tetraspastik und Polyneuropathie erheblich erschwert. Die Patientin wurde im 50. Lebensjahr unerwartet morgens tot im Bett liegend aufgefunden, nachdem 19 Tage zuvor Zeichen eines inferior-diaphragmalen Herzinfarkts im EKG aufgetreten waren. Histologie Die histologische Untersuchung von zehn exemplarischen Nervenfaszikel zeigte eine Reduktion der markhaltigen Nervenfasern Markscheidenabbauprodukten in und Relation frisch zur Altersnorm degenerierenden um 40%. Nervenfasern Neben waren Zwiebelschalenformationen und einige für den Axonquerschnitt zu dünn myelinisierte Nervenfasern anzutreffen. In Axonen waren zwölf Polyglukosankörper zu finden, während diese in Perineuralzellen nicht auffielen. Die elektronenmikroskopische Untersuchung der Körper zeigte eine Zusammensetzung aus teils filamentösen, teils granulären, teils amorphen Komponenten (siehe Abb 7). Dabei fanden sich auch Axone vor, die durch Einlagerungen von Neurofilamenten nach Art von Riesenaxonen aufgetrieben Schwannzellen ohne Axone. waren, und vereinzelte Polyglukosankörper auch in 26 Autopsie Die Autopsie wurde im Institut für Pathologie der BG-Kliniken Berg- mannsheil, Direktor Prof. Dr. Müller durchgeführt. Es zeigte sich eine globale Herzhypertrophie (Herz- gewicht 420 g) mit makroskopisch diffusen myokardialen Narbenbil- dungen ohne höhergradige Koronarsklerose. Die pulmonale Blutstauung (Lungengewicht 470 g) deutete auf ein terminales Linksherzversagen hin. Bei einem Hirngewicht von 1290 g Abb 7: Nervus suralis Biopsie. Elektronenmikroskopische Ansicht der imponierte ein deutlicher Polyglukosankörper (PB) in einer dünnen myelinisierten Nervenfaser zephalus (Vergrößerung x 9200 und Ausschnitt links unten x27800). Kleinhirnlager fanden sich beidseits internus e Hydro- vacuo. Im symmetrische 0,3 x 0,2 cm große Defekte. Histologisch wurde ein Faseruntergang im Großhirnmarklager zur Ventrikelwand hin mit spongiöser Auflockerung und reaktiven Astrozyten gefunden. Im gesamten ZNS stellten sich Ablagerungen von unterschiedlich großen, PAS positiven, überwiegend rundlichen Körpern, z.T. perivaskulär betont, dar. Bei fortgeschrittener Polyneuropathie in den peripheren Nerven mit Zeichen einer Faserregeneration in Form von „axonal clusters“ zeigte sich eine neurogene Atrophie der Skelettmuskulatur mit gruppenförmigen Atrophien ganzer Faszikelteile als Zeichen einer floriden Denervation. Die ultrastrukturelle Untersuchung des Myokards erbrachte auch hier den Nachweis einer subsarkolemmal und kernnah gelegenen Ablagerung scholliger Körper von feinfilamentöser Strukturen. Abb 8: Stammbaum Familie AD1 Enzymaktivität In der Familie der Indexpatientin wurden die Aktivitäten von Enzymen des Glykogenstoffwechsels von Frau Prof. Dr. Shin (Labor für Stoffwechselgenetik, München) bestimmt. Die Messung der GBE-Aktivität basiert auf der Synthese von Glykogen bei konstanter Glykogensynthase-Aktivität. Mit der angewandten Meßmethode wurde die Menge der UDP-[14C]-Glucose bestimmt, die in [14C]Glykogen eingebaut wurde. (Shin, Steiguber et al. 1988). Die Enzymaktivität war in den Leukozyten der Patientin deutlich und bei ihren klinisch gesunden Töchtern intermediär erniedrigt (siehe Tabelle 3). Bei der Indexpatientin wurden später in autoptisch entnommenem Gewebe folgende Enzymaktivitäten bestimmt. In Herzmuskel, Gehirn und peripherem Nerv fand sich keine 27 messbare GBE-Aktivität, im Skelettmuskel 0,35, in der Leber 0,03 nmol/min/mg Protein Aktivität von GBE (Normalbereich 0,2-1,5 0,03 nmol/min/mg Protein). Tab 3: Gemessene Enzymaktivitäten der Familie AD1 Kontrolle Enzym Patientin AD1 I.2 in Erythrozyten PhK in Leukozyten Phosphorylase a Phosphorylase a+b Glykogengehalt GBE Tochter AD1 II.1 in Leukozyten GBE Tochter AD1 II.2 in Leukozyten GBE Ehemann AD1 I.1 in Leukozyten GBE 218 100-300 mmol/min/mg Hb 5,7 19,3 2,7 0,06 5-20 10-50 0-10 0,2-1,5 nmol/min/mg Protein nmol/min/mg Protein mg/dl Gewebe nmol/min/mg Protein 0,08 0,2-1,5 nmol/min/mg Protein 0,08 0,2-1,5 nmol/min/mg Protein 0,22 0,2-1,5 nmol/min/mg Protein 2.3.3 Patienten mit PhK-Defizienz Anamnese Bei einem 38jährigen Patienten der Familie PHK1 (II.3) wurde im Rahmen eines chirurgischen Eingriffs als Zufallsbefund eine erhöhte CK im Serum gemessen (siehe Tab 4). Anamnestisch war ein weiteres Familienmitglied (III.2) nach einer Tonsillektomie mit CK-Werten um 400 U/l auffällig geworden. Dieses Kind III.2 war seit dem zweiten Lebensjahr intermittierend mit Muskelschmerzen auffällig geworden. An subjektiven Beschwerden gab der Indexpatient II.3 selbst lediglich leichte Muskelkrämpfe vor allem in den proximalen unteren Extremitäten an. Alle übrigen Laborwerte außer der CK, die bei Familienmitgliedern bestimmt wurden, lagen im Normbereich (abgesehen von Hyperurikämie bei I.2). Tab 4: Gemessene Ck-Werte der Familie PHK1 FamilienMitglied Alter Creatinkinase in U/l I.2 62 247 II.1 38 371 II.2 37 <80 II.3 36 944 II.4 40 <80 II.5 31 627 III.1 4 III.2 10 273 (zeitweise ~400) 312 Abb 9: Stammbaum Familie PHK1 28 Muskelbiopsie und Glykogenstoffwechsel Die durchgeführte Muskelbiopsie dem Patienten II.3 zeigte histologisch unauffällige Faszikel ohne irgendwelche Strukturveränderungen. Morphometrisch lagen die Querschnitte im Referenzbereich bei normaler Verteilung der Typ-1- und Typ-2-Fasern. Der in vitro-Test mit Exposition zu Koffein und Halothan ergab keine erhöhte Kontrakturneigung, so dass sich keine Hinweise auf eine Disposition zu einer malignen Hyperthermie ergaben (Neuromuskuläres Labor Bochum, Prof. Dr. W. Mortier). Die Analyse der Enzyme des Glykogenstoffwechsel (Labor für Stoffwechselgenetik München; Prof. Dr. Shin) zeigte schließlich eine stark erniedrigte Phosphorylase a-Aktivität 10,3 (Referenzbereich 200-600) nmol/min/mg Protein. Bei einem mit 2,1 (0,5-2,0) g /100 g Gewebe leicht erhöhten Glykogen-Gehalt und einer unauffälligen Aktivität der Gesamt-Phosphorylase-Aktivität a+b 1341 (Referenzbereich 500-2000) nmol/min/mg war hier vor allem die verminderte PhK-Aktivität mit 1,8 (Referenzbereich 3-20) nmol/min/mg auffällig. 2.3.4 Patient mit sporadischer RMD Anamnese Der 24jährige Patient berichtete über schmerzhafte Muskelkrämpfe seit dem siebten Lebensjahr, die sich vor allem im Bereich der Abdominalmuskulatur beim Aufstehen aus der sitzenden Position manifestierten. Bei einem 100m-Lauf im Alter von 13 Jahren traten erstmals Krämpfe und Verhärtungen in den Beinen auf. Mit 16 Jahren war die Extremitäten-Muskulatur vollständig betroffen. Die Muskelverhärtung trat betont nach einer kleinen Ruhepause auf. Kiefer- und Nackenmuskulatur war ebenfalls betroffen. So war der Patient z.B. nicht in der Lage, nach zehnminütigem Sitzen auf einem Stuhl unverzüglich aufzustehen und zu gehen. Es dauerte eine Minute, bevor sich die initiale Muskelsteifigkeit löste, und selbst dann konnte er die ersten Schritte nur auf seinen Zehenspitzen gehen. Er war in der Lage, die Tastatur seines Computers zu benutzen, berichtete aber über wellenartige Kontraktionen der Armmuskulatur bei dem Versuch, das Telefon abzunehmen. Kauen führte zu Schmerzen und Verhärtung der Kaumuskeln. In Ruhe traten manchmal sichtbare Krämpfe der Arm- und Beinmuskulatur auf. Nach Vorstellung in drei neuromuskulären Zentren wurde bei ihm eine unspezifische Muskelerkrankung, Neuromyotonie und myotone Störung diagnostiziert. Beide Eltern waren beschwerdefrei und die Untersuchung zeigte Normbefunde. Körperliche Untersuchung Die Untersuchung bei dem Indexpatienten zeigte normale Kraftentwicklung aller Muskelgruppen ohne Waden-Hypertrophie. Der Patient bewegte sich langsam, um Schmerzen, Verhärtung und Rippling seiner Muskulatur zu vermeiden. Dieses war einfach durch plötzliches Strecken oder Eindrücken der Muskeln zu induzieren, vor allem der Schulter- und Quadrizeps-Muskulatur. Rippling habituierte schnell. Perkussion oder sogar leichtes und schnelles Eindrücken der Unterarmmuskeln bewirkten eine schnelle Muskelkontraktion; diese PIRC habituierten nicht. Eine Verstärkung des Perkussions-Stimulus rief ein schmerzhaftes Muskelwogen hervor. Bei den ersten Schritten nach dem Aufstehen war nur Zehengang möglich. Beugung des Kopfes wurde wegen schmerzhafter Nackensteife vermieden. Loslassen nach dem Handgriff war verzögert. Alle 29 Sehnenreflexe erschienen normal. Nach Fixation waren initial keine Augenbewegungen möglich, danach wurde Doppelsehen und Schmerzen bei den Folgebewegungen angegeben. Therapieversuche mit Carbamazepin, Tizanidin und Mexiletin veränderten die Symptome nicht. Die CK war bei zweimaliger Bestimmung mit 700 und 698 U/l (normal < 80 U/l) erhöht. Das abgeleitete EMG und die Nervenleitungs-Untersuchungen waren normal. Eine Biopsie aus dem M.quadrizeps im Alter von 18 Jahren zeigte unspezifische myopathische Veränderungen wie auch eine Rebiopsie aus dem M.bizeps brachii im Alter von 22 Jahren. Muskelbiopsie Die Probe der Muskelbiopsie wurde nach Entnahme durch Immersion in Isopentan und Abkühlung in flüssigem Stickstoff tiefgefroren. Serienschnitte (6-8 µm dick) wurden bei -20°C mit einem Kryostaten angefertigt. Die Schnitte wurden für 5 Minuten mit 4% Paraformaldehyd fixiert und für 30 Minuten mit 2% BSA, 1% Ziege-Serum-IgG und 0.1% Triton X-100 in PBS inkubiert, bevor die Primär-Antikörper für zwei Stunden zugegeben wurden. Monoklonale oder polyklonale Primärantikörper waren gegen Caveolin-3, nNOS (Transduction, Lexington, UK), Dystrophin-2, αSarkoglykan, α-Dystroglykan und β-Dystroglykan (Novocastra, Newcastle-upon-Tyne, UK) gerichtet. Die Bindungen der Primärantikörper wurden durch Inkubation mit Fluoreszeinkonjugiertem IgG (Cy2) über zwei Stunden detektiert. Danach wurden die Biopsien mit einem Fluoreszenzmikroskop (Axiophot, Zeiss) untersucht. Diese immunhistologische Aufarbeitung wurde in Zusammenarbeit mit Dr.B.Schlatke, Neurologische Universitätsklinik Hamburg durchgeführt. 30 3 Ergebnisse 3.1 MODY 3.1.1 Etablierung der SSCP-Methode Zuerst wurden die Bedingungen der SSCP-Analyse etabliert. Die zehn Exone des HNF1α-Gens wurden in Polymerase-Ketten-Reaktionen mit den Primer-Paaren M3.1 bis M3.20 amplifiziert (siehe Abb 3). Die Primer wurden so gewählt, dass sie die vollständige codierende Region sowie die Splice-Akzeptor und -Donator-Stelle umfassten. In insgesamt 48 Gelläufen wurden die folgenden Elektrophorese-Bedingungen für die einzelnen Fragmente als aussagekräftig für schon beschriebene Mutationen und Polymorphismen bestätigt: Tab 5: Bedingungen der SSCP-Elektrophorese für die zehn PCR-Fragmente von HNF1α Fragment Laufzeit Spannun MDE-Konzentration /h g /V /x-fach Glycerinkonzentration /% Temperatur /°C 1 (M3.1-M3.2) 18 350 0,5 25 4 2 (M3.3-M3.4) 10,5 350 0,5 25 4 3 (M3.5-M3.6) 10 350 0,7 0 1 4 (M3.7-M3.8) 27 350 0,8 50 10 5 (M3.9-M3.10) 12 350 0,5 25 4 6 (M3.11-M3.12) 10 350 0,5 0 4 7 (M3.13-M3.14) 8 500 0,6 0 4 8 (M3.15-M3.16) 10 350 0,7 0 1 9 (M3.17-M3.18) 8 350 0,5 0 4 10 (M3.19-M3.20) 10 350 0,7 0 14 3.1.2 Blindtestung der SSCP-Methode bei HNF1α Nach der Untersuchung der Optimalbedingungen wurden zur Abschätzung der Sensitivität der SSCP-Methode Proben mit bereits nachgewiesenen Mutationen im HNF1α-Gen analysiert. 15 DNA-Proben, die uns freundlicherweise von Andrew Hattersley aus Exeter zur Verfügung gestellt worden waren, wurden unter den gewählten Bedingungen blind getestet, d.h. ohne dass bekannt war, ob und wenn ja welche Mutationen sich unter den Proben befanden. Dazu wurden die zehn Fragmente des HNF1α – Gens mittels Polymerase-Ketten-Reaktion (PCR) amplifiziert und auf einem SSCP-Gel entsprechend der etablierten Bedingungen aufgetrennt. Die standardisierte Auswertung des eingescannten Bandenmusters wurde daraufhin durch eine unbeteiligte Person mit den englischen Sequenzierungsergebnissen verglichen. Von 13 Proben mit bereits detektierter Mutation konnten vier nicht detektiert werden. Es handelt sich um eine Mutation im Exon 2 (P129T) und zwei verschiedene im Exon 4 (P291insC, W267R). Die Mutationen N127del, R131W, G132K, R159W, R200W, W267X, P379delCT, I414insCCdelTC, T620I wurden erkannt. 31 Abb 10: SSCP-Analyse verschiedener Mutationen (Wt Wildtyp-Kontrolle) 1: G31D 2: Wt 3: nt-12delG 4: I27L 5: A98V/nt12delG 6: Wt 7: H143Y 8: R159Q 9: S142P 10: A161T 11: DelN127 12: G132K 13: P129T 14: R159W 15: R131W 16: Wt 17: R200W 18: R229Q 19: Wt 20: I414insCCdelTC 21: P379delCT 22: Wt 23: L459L 24: S487N 25: T620I 26: Wt. Insgesamt wurden 19 Abweichungen im Laufverhalten festgestellt und als abklärungsbedürftig gedeutet, die nicht auf pathogene Mutationen zurückzuführen waren. Sie waren durch Polymorphismen begründet. Neben „stummen“ Mutationen ohne Auswirkung auf die Proteinsequenz innerhalb der kodierenden Sequenz handelte es sich vor allem um zahlreiche single nucleotide polymorphisms (SNPs) im intronischen Bereich der PCR-Fragmente. Vor allem in der 5´-Region von Fragment M3.1-2 (vgl. Tab 7 und Abb 10) war so die Spezifität bei festgestellten Auffälligkeiten im SSCP-Gel niedrig. Tab 6: Blindtestung von 15 Proben auf Mutationen im HNF1α-Gen Probe Mutation Exon Basen-austausch funktionelle Auswirkung Ergebnis: detektiert 1 P291insC 4 Insertion von C Frameshift nach Optimierung 2 R131W 2 CGG zu TGG Arg zu Trp Sofort 3 R159W 2 CGG zu TGG Arg zu Trp Sofort 4 P129T 2 CCA zu ACA Pro zu Thr nach Optimierung 5 P379delCT 6 Deletion von CT Frameshift Sofort 6 T620I 10 ACC zu ATC Thr zu Ile Sofort 7 W267X 4 TGG zu TAG Trp zu Stop Sofort 8 W267R 4 TGG zu TGT Trp zu Arg nach Optimierung 9 R200W 3 CGG zu TGG Arg zu Trp Sofort 10 I414insCCdelTC 6 Deletion von TC Frameshift Sofort 11 E132K 2 GAG zu AAG Glu zu Lys Sofort 12 DelN127 2 Deletion Deletion Sofort 13 P291insC 4 Insertion von C Frameshift nach Optimierung 14 nicht betroffen - - - - 15 nicht betroffen - - - - 32 3.1.3 Optimierung unter Erhöhung des experimentellen Aufwands der SSCP-Analyse Die Laufbedingungen der Proben bei nicht detektierten Mutationen konnten im Nachhinein - zum Teil auch erst durch nachträgliche Restriktion – so verändert werden, dass nach einer Abb 11: Optimierung der SSCP-Analyse durch Restriktion. Bei P291insC (1) werden gegenüber der Kontrolle (2) zusätzliche Banden sichtbar, die im ungeschnittenen Zustand (ohne Pst1-Verdau) nicht zu sehen waren. Optimierungsphase auch bei ihnen ein deviantes Laufverhalten festgestellt werden konnte. Die Mutation P129T im Exon 2 wurde erkannt, nachdem die MDE-Konzentration auf 75% (v/v) bei einer Laufzeit von 15 Stunden gesteigert wurde. Beim vierten PCR-Fragment (M3.7-8) waren nach der Restriktion mit Pst1 in zwei Fragmente der Wildtyp und P291insC zu differenzieren. Andere Verbesserungsmöglichkeiten wie ein zusätzlicher Aufreinigungsschritt der PCR-Proben, der einen als nachteilig beschriebenen Primer-Überschuss (Cai und Touitou 1993) verhindern soll, brachte in dieser Versuchsanordnung keine Verbesserung im Laufverhalten. Proben, die uns von P. Boutin aus Lille zur Verfügung gestellt wurden und die Mutationen G31D, Q7X, A161T, S142P, R159Q enthielten, wurden alle durch ein abweichendes Laufverhalten im SSCP-Gel detektiert. 3.1.4 SSCP-Analyse bei MODY-Patienten Für insgesamt 81 Personen aus 39 unterschiedlichen Familien wurden die zehn PCR-Produkte (M3.1-20) mit der SSCP-Analyse untersucht. Dadurch wurden zwar in drei Familien Mutationen im HNF1α-Gen identifiziert (G31D, R131W, R229Q, siehe Ergebnisse Direktsequenzierung). Die zahlreichen Polymorphismen (siehe Tab 7) reduzierten die Spezifität jedoch erheblich, so dass auf die Direktsequenzierung als Analyseverfahren gewechselt wurde. 33 Tab 7: Polymorphismen im HNF1α-Gen Exon 1 1 1 2 4 4 4 4 4 7 7 8 9 9 10 Intron 1 1 1 1 2 2 2 5 5 5 7 8 9 9 9 9 L17 I27L A98V H126H L254M G288G P290P P300P G319S L459L S487N T515T S550S R583Q M626K Basenaustausch CTC zu CTG ATC zu CTC GCC zu GTC CAC zu CAT CTC zu ATG GGG zu GGC CCC zu CCT CCC zu CCT GGT zu AGT CTG zu TTG/CTA/CTC AGC zu AAC ACG zu ACA GAG zu CAG CGG zu CGA ATG zu AAG (Yamagata, Oda et al. 1996) (Yamagata, Oda et al. 1996) (Yamagata, Oda et al. 1996) (Frayling, Bulamn et al. 1997) (Yamagata, Oda et al. 1996) (Yamagata, Oda et al. 1996) (Frayling, Bulamn et al. 1997) (Urhammer, Fridberg et al. 1997) (Hegele, Cao et al. 1999) (Yamagata, Oda et al. 1996) (Yamagata, Oda et al. 1996) (Yamagata, Oda et al. 1996) (Frayling, Bulamn et al. 1997) (Urhammer, Fridberg et al. 1997) (Lehto, Wipemo et al. 1999) nt –91 nt –52 nt –42 nt -12 nt +66 nt –51 nt –23 nt +9 nt -47 nt -42 nt +7 nt -15 nt +44 nt +65 nt -44 nt -24 A zu G T zu G G zu A Deletion von G G zu C T zu A C zu T C zu G C zu T G zu T G zu A G zu A C zu T Deletion von C C zu T T zu C (Yamagata, Oda et al. 1996) (Frayling, Bulamn et al. 1997) (Yamagata, Oda et al. 1996) (Lehto, Wipemo et al. 1999) (Ellard, Bulman et al. 1999) (Yamagata, Oda et al. 1996) (Yamagata, Oda et al. 1996) (Iwasaki, Oda et al. 1997) (Yamagata, Oda et al. 1996) (Iwasaki, Oda et al. 1997) (Yamagata, Oda et al. 1996) (Yamada, Nishigori et al. 1997) (Yamagata, Oda et al. 1996) (Lehto, Wipemo et al. 1999) (Yamada, Nishigori et al. 1997) (Yamagata, Oda et al. 1996) 3.1.5 Direktsequenzierung bei MODY-Patienten Bei 140 Patienten aus 72 unterschiedlichen Familien wurden das Gen der Glucokinase (MODY2) und von HNF1α (MODY3) untersucht. Dazu wurden die PCR Produkte M2 1-10 (Primer M2.1M2.20) und M3 1-10 (Primer M3.1-M3.20) zuerst auf einem Agarose-Gel auf die Spezifität der PCR und die richtige Größe der Fragmente hin geprüft. Die Sequenzierungsreaktion wurde direkt mit der aus dem Agarose-Gel eluierten DNA gestartet und eine Aufarbeitung des Sequenzierungsprodukts für die Auftrennung auf dem ABI PRISM 310 Kapillar-Sequenzierer durchgeführt. Mutationen, die mit der Manifestation und dem Auftreten von Diabetes assoziiert waren und cosegregierten, wurden in 19 Fällen identifiziert. Die Mutationen – sowohl innerhalb des HNF1αals auch innerhalb des Glucokinase-Gens – waren über die gesamte codierende Sequenz verteilt, es gab keinen Hinweis auf irgendeinen Hotspot. Es wurden 12 Familien mit Diabetes-assoziierten Mutationen der Glucokinase gefunden (siehe Tab 8). Die Mutationen R43H, W99X, G162delG, E216X, C220X, I225F, L304P, S383L, S426fsdelGC und R447del29 waren vorher noch nicht beschrieben worden. T228M und V203A sind schon in einem französischen Kollektiv nachgewiesen worden (Stoffel, Froguel et al. 1992). Unter den MODY2-Familien fand sich bei 25% ein Gestationsdiabetes. Bei sieben deutschen Familien wurden Mutationen im HNF1α-Gen detektiert (G31D, R131W, H143Y, R229Q, R272H, R238insG). Die Mutation (P291fsdelC) an der Position des vermeintlichen Hotspots (Kaisaki, Menzel et al. 1997) konnte nur einmal identifiziert werden (siehe Tab 9). Es fanden sich keine Mutationen bei Patienten, die nicht auch ein autosomal dominantes 34 Vererbungsmuster in ihren Familien-Stammbäumen zeigten. Sporadische Mutationen gab es nicht und die Penetranz der nachgewiesenen Mutationen war nach dem 25.Lebensjahr hoch. Tab 8: Patienten mit Mutationen im Glucokinase-Gen Mutation R43H Kurzanamnese Exon 2 Codon 43 CGC (Arg) zu CAC (His) Die Mutation R43H führte bei dem 8jährigen Indexpatienten zu einem Austausch der Aminosäure Arginin zu Histidin im Exon 2 der Glucokinase. Schon zwei Jahre zuvor war ein erhöhter Nüchtern-BZ-Wert aufgefallen. Nach mehrmaligen Kontrollen wurde eine gestörte Glucostoleranz bei bis jetzt unauffälligen HbA1cWerten gesichert. Auch die Stoffwechselstörung des Vaters zeigte eine ausgeprägt milde Verlaufsform; bei intermittierend erhöhten BZ-Werten ließ sich Stammbaum Familie MD19 W99X diätetisch eine stabile Einstellung erreichen. Exon 3 Codon 99 TGG (Trp) zu TGA (Stop) Bei der 49jährigen Patientin war seit 35 Jahren ein Diabetes bekannt, der diätetisch stabil eingestellt war. Der Basenaustausch führte im Codon 99 zum Kettenabbruch bei der Proteinbiosynthese (W99X). Beim Vater der Patientin war das Manifestationsalter von 63 Jahren fraglich, zumal er ein Jahr nach der Stammbaum Familie MD9 Diagnosestellung an diabetischen Komplikationen verstarb. G162delG Exon 5 Deletion von G Frameshift Die Deletion einer einzelnen Base bewirkte bei dem 11jährigen Patienten einen Verlust der Splice Akzeptor Site des Exon 5 der Glucokinase. Klinisch fiel er durch einen pathologischen Glucose-Toleranztest auf; bei einem Nüchtern-BZ von 124 mg/dl lag der 2 h-Wert bei 183 mg/dl. Der erhöhte HbA1c Wert von 5,7% (normal 4,1-4,6%) wies auf ein vorbestehendes Problem hin. Vater und Großvater Stammbaum Familie MD4 V203A entwickelten ebenfalls bereits in der Jugend einen Diabetes mellitus. Exon 6 Codon 203 GTG (Val) zu GCG (Ala) Ein Basenaustausch von Thymin zu Cytosin im Exon 6 der Glucokinase führte zu der Mutation . Bei dem zehnjährigen Indexpatienten lag bei einem Nüchtern-BZ von 102 mg/dl und einem HbA1c von 5,7% (normal 3,9-5,7 %) eine gestörte Glucose-Toleranz vor. Bei Vater und Großvater wurden bei rein diätetischer Stammbaum Familie MD4 E216X Einstellung keine Komplikationen angetroffen. Exon 6 Codon 216 GAA (Cys) zu TAA (Stop) Zwei Schwestern der Familie zeigten bei klassischer Familienanamnese initial im Alter von 10 bzw. 9 Jahren eine gestörte Glucose-Toleranz und entwickelten im Laufe von wenigen Jahren dann einen behandlungspflichtigen manifesten Diabetes. Die Mutation wurde auch bei der Mutter gesichert, bei der ursprünglich Stammbaum Familie MD8 V220X die Diagnose eines Gestationsdiabetes gestellt worden war. Exon 6 Codon 220, TGC (Cys) zu TGA (Stop) In der dritten Schwangerschaft manifestierte sich bei der Mutter dreier Kinder ein milder Diabetes. Alle drei Kinder zeigten bei jetzt normalen BZ-Werten niedrig pathologische HbA1c-Werte (6,1%, 6,6% und 6,7% normal kleiner 5,7%). Die gefundene Mutation V220X führt zur Bildung eines Stop-Codons. Stammbaum Familie MD10 35 I225F Exon 6 Codon 225 ATC(Ile) zu TTC (Phe) Die Transversion von Adenin zu Thymin bewirkte bei der Mutation I225F den Austausch der Aminosäure von Isoleucin zu Phenylalanin. Die sieben- und sechsjährigen Cousinen der jüngsten Generation fielen beide durch eine pathologische Glucose-Toleranz auf, während bei beiden Mütter erst später im 38.Lebensjahr bzw. in der Schwangerschaft im 28.Lebensjahr ein Diabetes Stammbaum Familie MD17 diagnostiziert wurde. T228M Exon 7 Codon 228 ACG (Thr) zu ATG (Met) Die zwölfjährige Patientin befand sich in pädiatrischer Behandlung wegen einer latenten Hypothyreose. Bei einem erhöhten Nüchtern-BZ von 154 mg/dl (normal <120 mg/dl) wurde aufgrund der positiven Familienanamnese ein oraler GlucoseToleranztest durchgeführt, der mit einem 2h-Wert von 77 mg/dl normal ausfiel. Wiederholte Kontrollen zeigten im Verlauf zunehmende HbA1c-Werte bei einem Fructosamin von 268 nmol/l (normal < 285 nmol/l). Bei dem 42jährigen Vater war Stammbaum Familie MD18 seit 12 Jahren ein Diabetes bekannt. Es wurde die Mutation T228M identifiziert, bei der im Exon 7 ein Aminosäure-Austausch von Threonin zu Methionin bestand. L304P Exon 8 Codon 304 CTT(Leu) zu CCT (Pro) Bei der Mutation L304P führte ein Basenaustausch zur Synthese von Prolin statt Leucin. Der erhöhte Nüchtern-BZ Wert des 30jährigen Patienten von 146 mg/dl wurde nur bei einer Routine-Labor-Kontrolle entdeckt. Das Manifestationsalter der Mutter des Patienten lag um das 40.Lebensjahr. Beide Patienten wurden rein Stammbaum Familie MD12 diätetisch behandelt. S383L Exon 9 Codon 383 TCG(Ser) zu TTG (Leu) Bis auf die 12jährige Tochter und ihre 34jährige Mutter war in der Familie mit der Mutation S383L niemand betroffen. Die nüchtern BZ-Werte waren mit 125 bzw. 127 mg/dl leicht erhöht. Eine medikamentöse Behandlung war nicht erforderlich. Stammbaum Familie MD11 S426delGC Exon 10 Codon 426 Deletion von GC Eine Deletion der zwei Basen GC führte in der Familie zur Änderung des Leserahmens bei der Translation und zur Bildung eines Stop-Codons (S426delGC). Die neunjährige Patientin hatte einen HbA1c Wert von 6,4 (normal kleiner 5,7%) mit intermittierend erhöhten BZ-Werten, ohne die klassische Stammbaum Familie MD13 R447del29 Symptomatik eines Typ1 Diabetes zu zeigen. Exon 10 Codon 447 Deletion von 29 Bp Eine Deletion von 29 Bp im Exon 10 der Glucokinase führte zum Verlust der Nterminalen Domäne der Glucokinase (R447del29). Insgesamt konnten Proben von neun Familienmitgliedern untersucht werden. In der jüngsten Generation waren alle Kinder (zwischen sechs und zwölf Jahre alt) klinisch betroffen, wobei zwei Stammbaum Familie MD2 Mädchen nur eine verminderte orale Glucosetoleranz Komplikationen waren bei den jungen Eltern (noch) nicht aufgetreten. zeigten. 36 Tab 9: Patienten mit Mutationen im HNF1α-Gen Mutation Kurzanamnese G31D Exon 1 Codon 31 GGT (Gly) zu GAT (Asp) Bei der Familie mit der Mutation G31D war neben dem Diabetes noch eine familiäre Fettstoffwechselstörung bekannt. Diese war allerdings nicht mit der Mutation assoziiert. Bei der Tochter der Indexpatientin wurde die Mutation nicht gefunden, eine Fettstoffwechselstörung lag bei ihr aber vor. Stammbaum Familie MD1 R131W Exon 2 Codon 131 CGG (Arg) zu TGG (Trp) Eine Transversion von Cytosin zu Guanin im Exon 2 bewirkte einen Aminosäurenaustausch von Arginin zu Tryptophan (R131W). Das zehnjährige Mädchen wies trotz diätetischer Reduzierung und Therapie mit oralem Antidiabetikum einen HbA1-Wert von 8,9 % (normal < 7,8 %) auf. Stammbaum Familie MD6 H143Y Exon 2 Codon 143 CAC (His) zu TAC (Tyr) Die Mutation H143Y wurde bei einem schlanken 24jährigem Patienten gefunden, bei dem seit drei Jahren ein Diabetes bekannt war. Als Nebendiagnosen bestanden bei ihm eine Colitis ulcerosa und eine Nierenschädigung nach Medikamentenabusus. Bei einem HbA1c von 7,4% (Norm 4,9-6,0 %) war die Sekretionsleistung bei einer Insulinkonzentration im Blut von 14,8 mU/ml und einer Stammbaum Familie MD3 C-Peptid-Konzentration von 3,9 ng/ml gut. Bisher hatten sich sonst keine diabetischen Komplikationen entwickelt. R229Q Exon 3 Codon 229 CGA (Arg) zu CAA (Gln) Die 14jährige Indexpatienten mit der Mutation R229Q (Arginin zu Glutamin) entwickelte wie ihre Mutter und ihr Onkel im frühen Teenager-Alter einen Diabetes. Die initial erhöhten BZ-Werte (Nüchtern-BZ: 197 mg/dl) hatten sich unter einer Insulin-Therapie normalisiert. Stammbaum Familie MD7 R272H Exon 3 Codon 272 CGC (Arg) zu CAC (His) Bei dem 12jährigen Mädchen mit der Mutation R272H war bereits seit fünf Jahren ein Diabetes bekannt. Obwohl das Manifestationsalter der Mutter mit 35 Jahren höher als das der klassischen MODY-Definition lag, deuteten die familiäre Beteilung und das Auftreten bei anderen Verwandten schon in der frühen Jugend auf eine heriditäre Komponente hin. Komplikationen waren bei Mutter und Tochter Stammbaum Familie MD15 noch nicht bekannt, die Therapie erfolgte mit oralen Antidiabetika. R238insG Exon 4 Codon 238 Insertion von G Frameshift Die Insertion eines Guanin kurz vor der Splice Site am Ende des Exon 3 führte zum Verlust dieser Site bzw. zur Verschiebung des Leserahmens und Kettenabbruch(R238insG). Die 10jährige Patientin zeigte unter Diät eine Normalisierung ihrer diabetischen Stoffwechsellage. Die Mutter der Patientin war seit ihrem 18. Lebensjahr auch rein diätetisch therapiert worden, wurde dann aber Stammbaum Familie MD16 P291insC seit der Schwangerschaft im 22.Lebensjahr insulinpflichtig. Exon 4 Codon 291 Insertion von C Frameshift Lediglich bei einem Patienten wurde die bisher häufig beschriebene Mutation P291insC gefunden. Von dem Patienten stehen keine klinischen Daten zur Verfügung. 37 3.1.6 Ausschluss einer mitochondrial vererbten Diabetesform (MIDD) Alle Proben wurden auf die mitochondriale tRNA Leucin Mutation an der Position 3243 (Lehto, Wipemo et al. 1999) mittels einer RFLP nach van den Ouweland, Lemkes et al. (1994) untersucht. Der Restriktionsverdau mit dem Enzym Apa1 (Schnittstelle GGGCCC) in einem Ansatz des PCRAmplifikats MIDD1-MIDD2 hätte bei Vorliegen einer Mutation zu einer Spaltung des 581 BpProdukts in ein 214 Bp- und ein 367 Bp-Fragment geführt. Bei insgesant 140 analysierten Proben von den obigen Patienten aus 72 Familien konnte keine entsprechende Veränderung festgestellt werden (siehe Abb 12). Abb 12: Die Transition A zu G liegt bei nt 3243 innerhalb des Dihydrouridine-Rings der tRNA für Leucin, die mitochondrial lokalisiert ist. 1: Negativ-Ergebnis einer Probe ohne Mutation 2: Positiv-Kontrolle mit Mutation 3: Molekulargewichtsmarker pUC19 DNA/Msp1 3.1.7 Klinischer Phänotyp Bei der statistischen Auswertung wurde für eine entdeckte Mutation der klinische Phänotyp jeweils nur einmal berücksichtigt, um mutationsspezifische Besonderheiten nicht über- oder unter zu bewerten. Lagen die Daten von mehreren Familienmitgliedern mit der gleichen Mutation vor, so wurde ein Mittelwert aller Mutationsträger berechnet. In die Auswertung des Manifestationsalters wurden nur Patienten einbezogen, bei denen die Diagnose innerhalb der letzten fünf Jahre gestellt worden war, um Einflussfaktoren wie z.B. veränderte WHO-Richtlinien bei der Diagnosestellung und verändertes Früherkennungsverhalten aufgrund unterschiedlicher Ärztedichte und unterschiedlicher Screeningverfahren/Leitlininien zu minimieren. Dabei ergab sich bei den MODY2-Familien (n=12) ein durchschnittliches Manifestationsalter von 10,4 ± 4,4 und bei MODY3-Familien (n=6) von 16,1 ± 3,1 Jahren. Bei allen untersuchten Patienten wurden keine Antikörper gegen die Glutamatdecarboxylase oder andere Insellzellepitope gefunden, die auf eine akuten oder abgelaufene Insulitis im Sinne eines Typ 1 Diabetes hingewiesen hätten. Aufgrund der schlecht standardisierbaren dezentralen Erhebung wurde auf die Erfassung des Fettverteilungsmusters z.B. als Taillen-Hüft-Verhältnis (waist/hip ratio ) verzichtet. Der errechnete BMI betrug für die Gruppe der MODY2-Patienten (n=12) im Mittel 19,0 ± 4,4 kg/m2 und 23,1 ±.2,7 38 8 7 6 n 5 MODY2 4 MODY3 3 2 1 0 0 Abb 13: 1 2 3 Zuordnung der Patienten zu geschlechts- und altersadaptierten BMI-Kategorien 0 (Untergewicht) 1 (Normgewicht) 2 (leichtes Übergewicht) 3 (starkes Übergewicht) kg/m2.für das MODY3-Kollektiv (n=6). Weil sich aber viele Jugendliche und Kindern unter den Indexpatienten befanden, wurde dieser BMI mittels einer analogen Skala geschlechts- und altersbezogen gemäß den Leitlinien der Arbeitsgemeinschaft „Adipositas im Kindes- und Jugendalter“ gewichtet (Dietz und Robinson 1998; Ellis, Abrams et al. 1999). Danach zeigten sich keine signifikanten Unterschiede im Fettverteilungsmuster zwischen MODY2- und MODY3Patienten (siehe Abb 13). 3.1.8 Überweisungsverhalten Bei den MODY3-Patienten entsprach die Anzahl der von Internisten rekrutierten Patienten (n=3) in etwa der Anzahl der von Pädiatern zugewiesenen Mutationsträgern (n=4). Von den MODY2Patienten dagegen wurde nur der kleinere Anteil von 16,7% (n=2) von Internisten und Diabetologen zur Analyse gemeldet. Von den Patienten mit nachgewiesener Mutation (MODY2 und MODY3) insgesamt wurden so immerhin 73,7% (n=14) von Pädiatern zugewiesen. Von übrigen Familien, in denen keine Mutation innerhalb der untersuchten Sequenz gefunden wurde (n=53), wurden dagegen 71,7% (n=38) von Internisten/Diabetologen und 28,3% (n=15) von Kinderärzten überwiesen (siehe Abb 14). MODY3 MODY2 Internisten Pädiater insgesamt mit Mutation insgesamt ohne Mutation 0 10 20 30 40 50 Abb 14: Überweisungsverhalten unterteilt nach MODY-Untertypen 60 39 3.2 APBD 3.2.1 Existenz von Spleißvarianten Mit den RT-PCR-Reaktionen B1-B2 (nt 8 – nt 348), B3-B4 (nt 264 – nt 780), B5-B6 (nt 643 – nt 1075), B7-B8 (nt 1023 – nt 1511), B9-B10 (nt 1443 – nt 1905) und B11-B12 (nt 1860 – nt 2269) wurde der gesamte codierende Bereich des GBE Gens zuerst aus Muskelgewebe bzw. Vollblut später für die Patientin AD1 I.2 auch aus Leber, Herzmuskulatur, peripherem Nerv und Gehirn amplifiziert (Numerierung der Nukleotide der cDNA nach (Thon, Khalil et al. 1993)). Für die Suche nach Spleißvarianten, die zu Produkten unterschiedlicher Länge führen können, wurden die Amplifikate durch eine Elektrophorese auf einem Agarosegel aufgetrennt. Hier fanden sich sowohl bei der Indexpatientin als auch bei Normalpersonen keine zusätzlichen Banden. Auch bei Fragmenten, die durch unterschiedliche Kombinationen der Primerpaare größere Abschnitte – insbesondere auch die katalytische Domäne – überspannten, gab es keine Hinweise für gewebspezifische Isoformen oder Isoenzyme. Wegen der starken individuellen Schwankungen der postmortalen Gewebsproben musste auf eine Quantifizierung der Expression verzichtet werden. Abb 15: Amplifikation eines GBE-Fragments (B15-B16) aus jeweils vier unterschiedlichen Geweben (H: Herz, M: Muskel, G: Gehirn, N: Nerv), β-Aktin-Fragment zur semiquantitativen Standardisierung. 3.2.2 Direktsequenzierung des GBE-Gens (mRNA) Die cDNA der betroffenen Personen und ihrer Angehörigen wurde über den vollständigen codierenden Bereich des GBE-Gens hinweg durch Direktsequenzierung auf Mutationen hin analysiert. 40 Abb 16: Direktsequenzierung Fragment B9-B10 der Patientin AD1 I.2 Bei der Indexpatientin AD1 I.2 fanden sich die zwei noch nicht beschriebenen Missense Mutationen R515H und R524Q (siehe Abb 16). In der übrigen Sequenz wurden keine weiteren Veränderungen entdeckt. Zur Absicherung des Befundes wurde mit den Primern B13 und B14 ein 92 Bp-Fragment aus genomischer DNA amplifiziert und eine RFLP durchgeführt (siehe Abb 17). Die beiden Töchter AD1 II.1 und II.2 mit jeweils intermediär verminderter GBE-Aktivität in Leukozyten wurden als heterozygote Anlageträger identifiziert, eine bezüglich der R515H- und die andere bezüglich der R524Q-Mutation. Bei 50 Kontrollpersonen konnten diese Basenaustausche nicht gefunden werden. 3.2.3 Single nucleotid polymorphisms innerhalb des GBE-Gens In Übereinstimmung mit Bao, Kishnani et al. (1996) wurde in allen Sequenzierungen an der Position nt 353 ein Guanin anstatt eines Cytosins (Thon, Khalil et al. 1993) gefunden; damit wurde die Aminosäure Cystein an der Position des Codons 88 bestätigt. Der Polymorphismus R190G (A zu G bei nt 658) wurde bei 40% aller Allele detektiert, was ungefähr der beschriebenen Frequenz von 33% entspricht (Bao, Kishnani et al. 1996). Bei einem Sequenzabgleich mit dem genomischen Klon RP11-142L1 (Osoegawa, Woon et al. 1998) wurde eine vollkommene Homologität für die Exons bezüglich des kodierenden Bereichs festgestellt. Die Sequenzabschnitte, die zusätzlich im Klon RP11-142L1 enthalten sind und den Introns entsprechen dürften, deuten auf eine Genstruktur des BE-Gens mit mindestens 16 Exons hin. 41 A B Abb 17: A Nachweis der Transition G1634A (R515H) durch Restriktion mit BclI (Schnittstelle durch Basenaustausch bei Mutter I.2 und Tochter II.2 B Identifikation der G1661A Transition (R524Q) nach Verdau mit TaqI (Basenaustausch zerstört Schnittstelle bei Mutter I.2 und Tochter II.1). 3.3 PhK-Defizienz 3.3.1 Kopplungs-Analyse in einer Familie mit PhK-Defizienz Mikrosatelliten-Auswahl Es wurden Mikrosatelliten ausgewählt, die sich in vorherigen Untersuchungen (siehe Tab 10) als polymorph und hochinformativ herausgestellt hatten. Dabei wurde vor allem auf ihre chromosomale Nachbarschaft zu den Loci der beiden Kandidatengene PHKβ und PHKγ1 Wert gelegt, die mit zytogenetischen Karten und Locus Link (Pruitt und Maglott 2001) festgestellt wurde. Sämtliche verwendeten Marker waren nach dem Hardy-Weinberg-Äqulibrium verteilt (Allel-Prävalenzen siehe auch Genethone II, KIDD, CEPH). Tab 10: Verwendete Mikrosatelliten-Marker und ihre Lokalisation Primer WiederholungsEinheit Mi16.1-Mi16.2 (AC)n Mi16.3-Mi16.4 D16S3093 PhK γ1 7p12-q21 (AC)n D7S478 Mi7.1-Mi7.2 (AC)n D7S492 Mi7.3-Mi7.4 (AC)n D7S669 Mi7.5-Mi7.6 (AC)n D7S2429 Mi7.7-Mi7.8 (AC)n PhK β D16S409 u.a. schon verwendet bei folgenden Referenzen 16q12-13 (Cavanaugh, Callen et al. 1998), (Curran, Lau et al. 1998), (Mirza, Lee et al. 1998) (Sheu, Lin et al. 1999; Furuta, Ohira et al. 2000) (Tomita, Nagamitsu et al. 1999) (Yan, Guan et al. 2000),(Kozobolis, Detorakis et al. 1999), (Radhakrishna, Blouin et al. 1997) (Ishwad, Ferrell et al. 1995), (Palmer, Scherer et al. 1994) (Radhakrishna, Blouin et al. 1997), (Perez Jurado, Peoples et al. 1996), (Satsangi, Parkes et al. 1996), (Gunel, Awad et al. 1995) Horizontale Gelelektrophorese und Auswertung des Bandenmusters Mit Hilfe einer PCR wurden für acht Familienmitglieder mit PhK-Defizienz aus extrahierter genomischer DNA jeweils Fragmente amplifiziert, die die polymorphen Wiederholungssequenzen enthielten. Die Reaktionen wurden jeweils optimiert und im Vergleich mit MolekulargewichtStandards auf ihre Spezifität hin mit einem hochprozentigen Agarose-Gel überprüft. Die Proben wurden daraufhin auf oben beschriebene Weise in denaturierenden Polyacrylamid-Gelen und Vertikalelektrophorese aufgetrennt und durch eine optimierte hochsensitive Silberfärbung angefärbt. 42 Abb 18: Haplotypisierung der Familie PHK1 Die Gele wurden nach dem Trocknen mittels eines LASER-Densitometers eingescannt. Bei der Auswertung behinderten vor allem Stotterbanden eine einfache Interpretation. Außerdem führte die vollständige kodierenden Denaturierung und wegen komplementären der unterschiedlichen Strangs zu einer Basenzusammensetzung Auftrennung dieser des beiden 43 Nukleinsäurefragmente. Die mathematische Herleitung des relativen Unterschieds der Laufweite der beiden Banden erbrachte für jeden der untersuchten Mikrosatelliten aber eine eindeutige Zuordnung (siehe Abb 18). Insgesamt waren die Ergebnisse bei Wiederholung der PCR und Elektrophorese vollständig reproduzierbar. Zuordnung der Haplotypen und Lod-Score-Berechung Bei der Auswertung wurde zur Überprüfung einer eventuellen Kopplung zwischen Markerallelen und den Loci der Kandidatengenen eine vereinfachte Berechnung durchgeführt. Im Modell eines autosomal dominanten Erbgangs und unter der Annahme einer Frequenz von 1:1.000.000 wurde mit dem LINKAGE-Software Paket (FTP-Server: http://linkage.rockefeller.edu (Lathrop und Lalouel 1984)) der Zwei-Punkte Lod Score für das relative Risiko errechnet. Tab 11: Zwei-Punkte Lod-Score zur Bestimmung des Kopplungsungleichgewicht der untersuchten Mikrosatelliten Rekombinations- 0.0 0.001 0.05 0.1 0.15 0.20 0.30 0.40 0.5 D7S478 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 D7S492 -5.21 -1.87 -0.25 -0.04 0.05 0.08 0.07 0.02 0.00 D7S669 -5.62 -2.39 -0.72 -0.44 -0.29 -0.19 -0.08 -0.02 0.00 D7S2429 0.94 0.94 0.83 0.72 0.61 0.49 0.28 0.10 0.00 D16S409 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 D16S3093 -5.33 -1.93 -0.31 -0.09 0.00 0.04 0.04 0.01 0.00 fraktion θ Wie am Lod Score deutlich wird, erwiesen sich die Mikrosatelliten D7S478 und D16S409 in der Analyse als wenig informativ; innerhalb der Famlie bestand für diese beiden Marker eine zu geringe Heterozygotie, so dass die chromosomale Phase nicht bestimmt werden konnte. Insgesamt fand sich für keinen Marker eine Assoziation zwischen untersuchten Allelen und dem klinischen Phänotyp/Affektionsstatus (Lod Score >3). Dagegen sprachen die negativen Werte bei D7S492, D7S669 und D16S3093 eher gegen eine Kopplung. Ohne die Annahme einer stattgefundenen Rekombination (θ>0) konnte diese ausgeschlossen werden (Lod Score < -2). Wenn man von einer niedrigen intrafamilliären Wahrscheinlichkeit für ein Rekombinationsereignis zwischen den Loci der Mikrosatelliten und denen der Kandidatengene ausgeht, sprach der Befund insgesamt gegen eine Assoziation. 3.3.2 Ausschluss Obwohl die hier durchgeführte Analyse der Mikrosatelliten-Allele einen Ausschluss der Gene der γ1-Untereinheit und β-Untereinheit der PhK als Kandidatnegene für die hier beschriebene familiäre PhK-Defizienz nahelegte, wurde dennoch von Barbara Burwinkel (Institut für physiologische Chemie Ruhr-Universität Bochum, Prof.Dr.Dr. Kilimann) eine Direktsequenzierung der codierenden Sequenzen vorgenommen. In den untersuchten Abschnitten fand sich kein Hinweis für eine Mutation oder einen pathogenen Basenaustausch der beiden Untereinheiten der PhK. 44 3.4 RMD 3.4.1 Direktsequenzierung Caveolin-3-Gen Mutationsbefund für einen Patienten mit sporadischer RMD Die Direktsequenzierung der beiden codierenden Exone ergaben bei dem Patient eine heterozygote Mutation im Exon 1 vom Caveolin-3-Gen. Die Missense-Mutation ergab sich durch eine Transition von Guanin zu Adenin in einem Allel bei der Position nt 77 (Nummerierung ab Startcodon nach (Biederer, Ries et al. 1998)), die zu einem Austausch der positiv geladenen Aminosäure Arginin zu Glutamin führte (R26Q). Diese Mutation liegt in der N-terminal Domäne des Proteins. Diese Aminosäure ist bei verschiedenen humanen Caveolinen und in verschiedenen Spezies evolutionär konserviert (Minetti, Sotgia et al. 1998). Bei 200 Kontrollpersonen konnte diese Mutation nicht gefunden, was gegen einen seltenen Polymorphismus spricht. Die Arg26Gln Mutation fand sich weder bei Vater noch Mutter des Patienten, was auf eine de-novo-Mutation hindeutete. Abb 19: Mutation R26Q im Elektropherogramm. Direkt-Sequenzierung von Exon1 im Caveolin-3-Gen beim Patienten RMD1 Nachweis de-novo-Mutation R26Q und Immunhistochemie Eine falsche Paternität wurde durch die Genotypisierung polymorpher Mikrosatellitenmarker (Dr.Kress, Institut für Humangenetik der Universität Würzburg) ausgeschlossen. Während sich in Kontrollproben die Immunoreaktivität für Caveolin-3, nNOS, Dystrophin-2, αSarkoglycan, nNOS, Plasmamembran Dystrophin-2, zeigte, ergab sich α-Dystroglykan bei der und β-Dystroglykan immunhistochemischen entlang Untersuchung der der Muskelbiopsie des Patienten eine reduzierte Anfärbung des Sarkolemms für Caveolin-3. Darüber hinaus zeigte sich eine leicht Normalkontrolle (siehe Abb 20). reduzierte α-Dystroglykan-Anfärbung im Vergleich zur 45 Abb 20: Immunhistochemie beim Patienten mit sporadischer RMD (links) und einer Normalperson (rechts). A/B: Caveolin-3 C/D: α-Dystroglykan E/F: nNOS G/H: α2-Laminin I/J: β-Dystroglykan. Maßstabsbalken in A und B: jeweils 30 µm 46 4 Diskussion 4.1 Erkrankungen 4.1.1 MODY 4.1.1.1 Identifizierte Mutationen und ihre pathophysiologische Bedeutung Im folgenden sollen die identifizierten Mutationen des HNF1α- und Glucokinase-Gens vor dem Hintergrund der schon bekannten Mutationen (siehe Tab 16 und 17) eingeordnet werden. Dabei soll insbesondere auf die Bedeutung für die pathophysiologischen Zusammenhänge des MODYDiabetes eingegangen werden. Glucokinase Viele der bisher im Gen der Glucokinase identifizierten Mutationen (siehe Tab 17) sind bereits intensiv funktionell untersucht worden. Die daraus resultierende pathogene Wirkung mit einer veränderten Enzymaktivität ist vielfach in vitro eindeutig nachgewiesen worden. Die Rolle des Schrittmacherenzyms innerhalb des Glucose-Stoffwechsels hat zur Modellvorstellung der Glucokinase als Glucosesensor geführt, der die Einschleusung von Glucose in die Glycolyse reguliert. Die Änderung des ATP/ADP-Verhältnis bewirkt demnach dann eine Hemmung des ATP-empfindlichen K+-Kanals und so eine Depolarisierung der β-Zelle. Schließlich folgt die Insulinsekretion (Matschinsky 1996). Mutationen können über eine veränderte Enzymaffinität zu Glucose und somit eine veränderte Glucose-induzierte Insulinfreisetzung zu einer Verstellung des Sollwerts dieses Sensors führen. Das wird besonders deutlich an der einzigen bisher beschriebenen Mutation der Glucokinase, die zu einer erhöhten Substrataffinität der Glucokinase und somit bei den betroffenen Patienten zu chronischen Hypoglykämien führt (Glaser, Kesavan et al. 1998). Neben den Affinitätsveränderungen muss man bei den Hyperglykämie auslösenden Mutationen aber auch vor allem von einer Reduktion der Phosphorylierungsaktivität (vmax) ausgehen, die bis unter 25% der Totalphosphorilierungskapazität des Wildtyp-Enzyms liegt (Davis, Cuesta-Munoz et al. 1999). Bei den zu betrachtenden kinetischen Aktivitätsparameter muss man für die Glucokinase nicht nur die beiden Substrate Glucose und ATP-Mg gesondert betrachten, sondern auch einen kooperativen Effekt für Glucose berücksichtigen. Glucosebindung führt zu einer starken Konformationsänderung des Enzyms, was in der sigmoidalen Bindungskurve - ähnlich wie bei Tab 12: Exon 2 3 5 6 6 6 6 7 8 9 10 10 Identifizierte Mutationen im Glucokinase-Gen Mutation R43H W99X G162delG V203A E216X C220X I225F T228M L304P S383L S426delGC R447del29 Austausch funktionelle Auswirkung CGC zu CAC TGG zu TGA Deletion von G GTG zu GCG GAA zu TAA TGC zu TGA ATC zu TTC ACG zu ATG CTT zu CCT TCG zu TTG Deletion von GC Deletion von 29 Arg zu His Trp zu Stop Frameshift Val zu Ala Glu zu Stop Cys zu Stop Ile zu Phe Thr zu Met Leu zu Pro Ser zu Leu Frameshift Frameshift Familie MD19 MD9 MD14 MD4 MD8 MD10 MD17 MD18 MD12 MD11 MD13 MD2 47 Hämoglobin und O2- zum Ausdruck kommt. Der HILL-Koeffizient beträgt als Maß der Sigmoidizität der Glucose-Abhängigkeit beim Wildtyp-Enzym 1,7 (Matschinsky, Glaser et al. 1998). Die Mutationen lassen sich in unterschiedliche Gruppen unterteilen, je nachdem welche funktionelle Domäne durch den Aminosäureaustausch beeinflusst wird und so zu einer Änderung der maximalen spezifischen Aktivität kcat, der Glucose-Konzentration bei halbmaximalen Substratumsatz S0,5, der Michaelis-Konstanten Km für ATP oder des Hill-Koeffizienten führt. Von den hier identifizierten Mutationen ist die Mutation V203A am besten charakterisiert. In einem Transduktionsmodell mit rekombinanten Adenoviren wurde für diese Mutation eine stark verminderte katalytische Aktivität des Enzyms aufgrund verminderter Glucoseaffinität (Glucose S0,5 8fach erhöht) nachgewiesen (Burke, Buettger et al. 1999). Dieser ausgeprägte Effekt kann mit der unmittelbaren Nachbarschaft von V203 zum aktiven Zentrum erklärt werden. So sind Asn204 und Asp205 jeweils mit einer Wasserstoffbrückenbindung an der Substratbindung beteiligt (Xu, Weber et al. 1995). Zusätzlich tritt bei dieser Mutation eine veränderte Affinität für das Glucokinase Regulator-Protein auf (Veiga-da-Cunha, Xu et al. 1996), das in der Leber die Glucokinase kompetitiv hemmt (Vandercammen und Van Schaftingen 1991). Aufgrund der Kristallstruktur der Glucokinase müssen auch I225F und T228M in der unmittelbaren Nähe des katalytischen Zentrums angesiedelt werden. Sie sind Teil von Mg- (Codon 225-228) bzw. Glucose-bindenden (225-231) Domänen (Mahalingam, Cuesta-Munoz et al. 1999). Bei den Mutationen R43H, L304P und S383L kann man aufgrund der Struktur keine funktionelle Wirkungsbeziehung ableiten. Leu304 ist eine hochkonservierte Aminosäure, die auch in der entsprechenden Domäne des Hefe-Enzyms Hexokinase B vorhanden ist (Gidh-Jain, Takeda et al. 1993), Arg43 und Ser383 finden sich auch im Ratten- und Xenopus laevis-Enzym (Veiga-daCunha, Courtois et al. 1996). Für Mutationen außerhalb des aktiven Zentrums gibt es aber bereits verschiedene Erklärungen für eine verminderte Enzymaktivität der Glucokinase. Die Mutationen H173R, E300K und V367M führen bei unauffälliger Substrataffinität und Enzymaktivität zu einer drastisch erhöhten Thermolabilität der jeweiligen Proteine (Davis, Cuesta-Munoz et al. 1999). Die aufgereinigten Enzym-Proteine mit A53S und V367M zeigen keine veränderten kinetischen Parameter. Dagegen weisen sie eine erhöhte Sensitivität gegenüber den Inhibitoren Glucokinase Regulator-Protein und Palmitoyl-Coenzym A auf (Miller, Anand et al. 1999). Bei den Mutationen T168P und S336L konnte eine Aufhebung des kooperativen Effekts für Glucose (Hill-Koeffizient nahezu 1) nachgewiesen werden (Davis, Cuesta-Munoz et al. 1999). Die Hälfte der hier identifizierten Mutationen (W99X, G162delG, E216X, C220X, S426delGC und R447del29) führt zu einem vorzeitigen Kettenabbruch bei der Proteinsynthese des Enzyms, entweder durch Verschiebung des Leserahmens oder Erzeugung eines Stop-Codons. R447del29 verdeutlicht die Bedeutung, die auch periphere Aminosäuren-Reste des Enzyms für die Konformation und katalytische Aktivität haben. Mit 18 Aminosäuren fehlt bei dieser Mutation lediglich eine kleine α-Helix des carboxy-terminalen Endes (Gidh-Jain, Takeda et al. 1993). Im Tiermodell wurde nachgewiesen, dass bereits eine alleinige Nullmutation der pankreatischen Glucokinase für die Manifestation eines Diabetes ausreicht (Terauchi, Sakura et al. 1995). Eine Knock-Out-Maus brachte darüber hinaus aber die Erkenntnis, dass auch ein Aktivitätsverlust der 48 hepatischen Glucokinase zu einer signifikanten Steigerung des Blutglucosespiegels führt (Postic, Shiota et al. 1999). Von daher ist davon auszugehen, dass zur Genese dieses MODY-Typs beide Faktoren beitragen können. Die Glucokinase hat also nicht nur Bedeutung als Sensor innerhalb der β-Zelle. Über die hepatische Verwertung der portalen Glucose (Matschinsky, Glaser et al. 1998) ist sie auch an der Regulation der Blutglucosekonzentration beteiligt. Zusammen mit den hier erstmals beschriebenen Mutationen sind nun über 100 verschiedene Aminosäureveränderungen bekannt, die in ihrer Vielfalt für den MODY-Phänotyp verantwortlich sind. In vivo stellt sich der Pathomechanismus komplexer dar als die anfangs vermutete Rechtsverschiebung der Glucose-induzierten Insulinsekretion (Bell, Pilkis et al. 1996). Dabei spielen vor allem auch Kompensationsmechanismen bei stärkerer Verminderung der Enzymaktivität eine Rolle, für die Überexpression, unterschiedliche Beeinflussung der Inhibitoren Glucokinase Regulator-Protein und Palmitoyl-Coenzym A, aber auch die Isoenzyme (Epstein, Boschero et al. 1992) verantwortlich gemacht werden könnten. Die starke Heterogenität der Mutationen impliziert, dass weitere Analysen nicht auf Hotspots beschränkt bleiben können. Zusätzliche klinische Phänotypen durch Mutationen an verschiedenen Orten des Glucokinase-Gens sind nicht auszuschliessen. HNF1α Sechs der sieben hier identifizierten Mutationen sind inzwischen auch für MODY3-Patienten in anderen Ländern gefunden worden. Alle Veränderungen der Proteinsequenz betreffen evolutionär hochkonservierte Aminosäuren (Ratte, Maus, Hamster, Huhn und Xenopus) und liegen in funktionell bedeutenden Domänen: G31D in der Dimerisierungsdomäne (Codon 1-32) (Vaxillaire, Rouard et al. 1997), R131W, H143Y, R229Q und R272H im DNA-bindenden POU A - Motiv (Codon 82-172) (Ryffel 2001). Die beiden Frameshift Mutationen R238insG und P291insC führen schließlich dazu, dass das C-terminale Ende des Proteins (Codon 281-631) nicht synthetisiert wird, dem die transaktivierende Funktion des Transkriptionsfaktors zugeschrieben wird. HNF1α ist vor allem als Dimer aktiv, davon in Pankreas und Leber überwiegend als Homodimer (Rey-Campos, Chouard et al. 1991). So ist auch der dominant negative Effekt einiger Mutationen zu erklären, der durch Veränderungen innerhalb der funktionellen Domänen hervorgerufen wird. Zuerst konnte dieser Zusammenhang für die bei MD5 identifizierte Insertion gezeigt werden (Yamagata, Yang et al. 1998). Die Bindung eines funktionell inaktiven Monomer an seinen „intakten“ Partner kann zu dessen Inaktivierung führen. Eine Veränderung der Aminosäure Arg im Codon 272 der Familie MD15 führt zu einem Verlust der DNA-Bindung an der Zielsequenz innerhalb der jeweiligen Promotor-Regionen (Yoshiuchi, Yamagata et al. 1999). Letzten Endes Tab 13: Identifizierte Mutationen im HNF1α-Gens Exon 1 2 2 3 3 4 4 Mutation G31D R131W H143Y R229Q R238insG R272H P291insC Austausch GGT zu GAT CGG zu TGG CAC zu TAC CGA zu CAA Insertion von G CGC zu CAC Insertion von C funktionelle Auswirkung Familie Gly zu Asp Arg zu Trp His zu Tyr Arg zu Gln Frameshift Arg zu His Frameshift MD1 MD6 MD3 MD7 MD16 MD15 MD5 49 resultieren eine gestörte mitochondriale Funktion durch verminderte Transkription metabolischer Enzyme (s.o., z.B. GLUT 2 und Pyruvatkinase) wie auch eine Verringerung der Insulinexpression (Wang, Maechler et al. 1998). Die charakteristischen Eigenschaften der Mutation G31D wie auch die anderer Mutationen innerhalb der Dimerisierungsdomäne (z.B. Q7X) machen jedoch die negative Dominanz als alleiniges pathogenes Prinzip unwahrscheinlich. Der Austausch von Glycin zu Aspartat stellt nämlich hauptsächlich eine Veränderung der Polarität dar, die das Bindungsverhalten für die Dimerisierung im Sinne einer Erniedrigung und nicht einer Erhöhung der Bindungsaffinität verändert. Dementsprechend müsste man in diesem Fall eher von einem Gen-Dosis-Effekt ähnlich wie bei den Mutationen der Glucokinase ausgehen. Eine Haploinsuffizienz kann auch bei den Frameshift-Mutationen die entscheidende Rolle spielen, wenn man von einer schnellen Degradation der trunkierten Proteine ausgeht. Als weiterer Faktor für R238insG und P291insC muss ein fehlerhaftes intrazelluläres Targeting diskutiert werden, das für Deletionen im Cterminalen Ende beschrieben wurde (Sourdive, Chouard et al. 1993). Eine möglicherweise veränderte Interaktion mit einem aktivierenden Cofaktor (Mendel, Khavari et al. 1991) wurde bisher für keine Mutation untersucht. Die HNF1α-defiziente Maus zeigt in der homozygoten Form weniger β-Zellen und ein primäres Insulinsekretionsdefizit des Pankreas; der unauffällige Phänotyp der heterozygoten Variante macht jedoch auf die Grenzen dieses Modells für die humane Situation deutlich (Pontoglio, Sreenan et al. 1998). Die Zielstrukturen und Bindesequenzen in den Promotoren zahlreicher Gene bilden insgesamt - in der pankreatischen α- und β-Zelle wie auch in Leber und Niere ein unüberschaubares Funktionsgeflecht (Mendel und Crabtree 1991), dessen relevante, molekulare Beziehungen für MODY noch untersucht werden müssen. Die Kaskade der hepatischen Transkriptionsfaktoren ist durch Interaktionen (HNF3β reguliert HNF1α und HNF4α positiv, HNF3α negativ (Duncan, Navas et al. 1998)) und Rückkopplungen (HNF1α hemmt die Expression seines Regulators HNF4α (Ktistaki und Talianidis 1997)) charakterisiert. Wie in der Familie MD1 gibt es in einigen Fällen Hinweise auf einen Zusammenhang zwischen HNF1α und dem Cholesterin- und Fettsäure-Stoffwechsel. Expressionsanalysen weisen diesbezüglich auf eine wichtige Regulatorfunktion von HNF1α für die Gallenwegs-Transporter Slc10a1, Slc21a3 und Slc21a5 und Enzyme der Cholesterinsynthese ( z.B. Cholesterol-AcylTransferase, CYP7a1, Lecithin und Lipase) hin (Shih, Bussen et al. 2001). Ein möglicher Zusammenhang von MODY3 und Hypercholesterinämie sollte wegen der großen klinischen Bedeutung für die Komplikationsrate der betroffenen Patienten geklärt werden. Bisher gibt es jedoch weder bei unseren noch den bisher beschriebenen MODY-Patienten Anzeichen auf ein regelmäßiges Zusammentreffen. Größere Längsschnittuntersuchungen müssen diesbezüglich weitere Klärung bringen. HNF1α-Mutationen bewirken, dass die renale Glucose-Reabsorption wegen einer geringeren Anzahl von Na+-abhängigen Glucose Cotransportern reduziert ist (Pontoglio, Prie et al. 2000). Ein umfangreiches Screening für Diabetes mellitus Typ 2 durch Testung auf Glucosurie kann dadurch besonders auch bei jüngeren Menschen MODY3-Patienten erfassen. 50 Mutationsbiologische Besonderheiten Ungefähr 2/3 der veröffentlichten Mutationen in den Genen der Glucokinase und HNF1α sind Missense Mutationen. Davon abweichend wurden hier im Glucokinase-Gen zur Hälfte Nonsense und Frameshift Mutationen identifiziert. Beim HNF1α-Gen konzentrieren sich die Missense-Mutationen auf die DNA-bindenden Domänen, während Verschiebungen des Leserahmens vorwiegend am C-Terminus auftreten. Insertionen werden hier vor allem durch Sequenzbesonderheiten in Form von repetitiven Cytosin-Basen verursacht, so dass P291 nicht allein aus epidemiologischen Gründen, sondern auch mutationsbiologisch als Hotspot diskutiert werden muss (Glucksmann, Lehto et al. 1997; Kaisaki, Menzel et al. 1997). Hier lag bei drei Mutationen ein CG zu TG bzw. CG zu CA Basenaustausch vor, vereinbar mit einer Deaminierung von Methylcytosin in CpG Dinukleotiden. Die hier gefundene Rate der Deaminierungen auf die identifizierten Mutationen insgesamt (42%) entspricht damit dem Anteil solcher Punktmutationen an allen nachgewiesenen HNF1α-Mutationen insgesamt (43,5%) und liegt höher als in einer Analyse von 1262 Mutationen verschiedener Gene (30,4% (Cooper, Krawczak et al. 1995)). Aufgrund des hier nachgewiesenen Austauschs von Argininresten in Codon 131, 229 und 272, aber auch von Codon 159 und 200 scheint die Deaminierung bei CpG Dinukleotiden ein wichtiger Entstehungsmechanismus im HNF1α-Gen zu sein. 4.1.1.2 Phänotyp Gestationsdiabetes und MODY2 In immerhin drei untersuchten MODY2-Familien (E216X, V220X, I225F) fanden sich hier Frauen mit einem Gestationsdiabetes, die formal die diagnostischen Kriterien für MODY nicht erfüllen. Zum heterogenen Gestationsdiabetes rechnet man alle Fälle, die eine gestörte Glucosetoleranz unterschiedlicher Stärke mit Manifestation oder erster Diagnosestellung während der Schwangerschaft zeigen. Dabei werden allerdings vorher nicht erkannte Typ 1-Diabetikerinnen ebenso erfasst wie Patientinnen mit einer nur vorübergehenden Schwangerschafts-induzierten Stoffwechselstörung. Bisherige Untersuchungen gehen von einer Inzidenz des Gestationsdiabetes insgesamt um 2% aus, wobei die Diagnosestellung in der Regel im späten zweiten oder frühen dritten Trimenon erfolgt. Da die Prävalenz eines Gestationsdiabetes bei Frauen mit einer positiven Familienanamnese – insbesondere bei bekanntem Diabetes mellitus Typ 2 eines Verwandten ersten Grades – erhöht ist, spricht viel für eine genetische Beteiligung. Bisherige Untersuchungen haben dabei vor allem auf die Bedeutung der Glucokinase hingewiesen. In einer Untersuchung hatten 42% der Schwangeren mit Gestationsdiabetes einen Verwandten ersten Grades mit Diabetes; von diesen Frauen wiesen insgesamt 5% eine Mutation der Glucokinase auf (Stoffel, Bell et al. 1993). In einer aktuelleren Untersuchung konnte durch eine vorherige Selektion nach vier Kriterien (persistierende Hyperglykämie nach der Schwangerschaft, nur geringe BZ-Erhöhung während des oralen Glucose-Toleranz-Tests (OGTT), vorübergehende Insulintherapie mit späterer diätetischer Kontrolle und positive Familienanamnese) der Anteil der Trägerinnen einer Mutation im Glucokinase-Gen auf 80% erhöht werden (Ellard, Beards et al. 2000), obwohl diese Mütter die klinischen Diagnose-Kriterien des MODY nicht erfüllten. Eine Überarbeitung dieser Kriterien und die Aufnahme des Gestationsdiabetes zu den klinischen 51 Charakteristika des MODY2 scheint daher ratsam. Eine regelmäßige Mutationsanalyse des Glucokinase-Gens bei Gestationsdiabetikerinnen ist zur Zeit aus Kostengründen nicht zu realisieren, wobei auch eine routinemäßige Früherkennung per OGTT bei Schwangeren zur Zeit nicht durchgeführt wird. Bei einer hochgerechneten Prävalenz von 1 auf 2400 (Frequenz des Gestationsdiabetes 2%, davon 2,1% mit Mutation des Glucokinase-Gens), ist jedoch eine ähnliche Frequenz wie die der zystischen Fibrose erreicht (Boat und Cheng 1989). Betroffene Frauen sollten wegen des 50%igen Risikos einer Vererbung der Mutation auf eine konsequente und regelmäßige Früherkennung bei den Kindern hingewiesen werden. Eine enge Einbeziehung der Gynäkologen in diese Problematik erscheint dringend anzuraten. Erwähnt werden soll noch, dass entsprechend zu den übrigen Ergebnissen auch hier der Anteil der reinen Missense-Mutationen an den identifizierten Mutationen bei Gestationsdiabetes klein war. Mehr als die Hälfte der gefundenen Mutationen sind Nonsense-Mutationen bzw. Insertionen/Deletionen, die alle zu einem Kettenabbruch bzw. Ausfall eines Allels führen. Alle der hier gefundenen Mutationen liegen innerhalb des Exon 6. Für Interpretationen im Sinne eines Hotspots reichen die Zahlen noch nicht aus; es finden sich auch keine Hinweise hierfür in der Literatur. Adipositas Alle bisherigen Studien vernachlässigen in ihren Zusammenstellungen der MODY-Patienten, dass der BMI gerade in altersmäßig gemischten Kollektiven als interindividueller Vergleichsparameter für den Grad der Adipositas problematisch ist (O'Dea und Abraham 1995). Der BMI ist als intraindividueller Prognoseindikator schwach, im Hinblick auf Spezifität und Sensitivität in kaukasischen Kollektiven jedoch als aussagekräftigstes Maß bestätigt worden (Curtin, Morabia et al. 1997)– allerdings nur wenn er alters- und geschlechtskorrigiert wie in Abb 13 verwendet wird. Bei den von uns untersuchten Patienten ergeben sich keine signifikanten Unterschiede zwischen den beiden Gruppen; sie sind insgesamt eher der Gruppe der Normgewichtigen zuzuordnen. Ein direkter Vergleich mit den mutationsnegativen Patienten war leider aufgrund der schlechten Rücklaufquote der Datenerhebungsbögen nicht möglich. In den vorhandenen Daten zeichnet sich in dieser großen und sicher auch genetisch heterogenen Gruppe eine Tendenz zu stärkerer Adipositas ab, die den veröffentlichten Daten über Familien ohne MODY-Mutation entspricht (Doria, Yang et al. 1999). Komplikationen und Krankheitsverlauf Das Ausmaß der diabetischen Komplikationen war unter den Patienten mit bestätigten Mutationen sehr gering. Eine weitergehende Interpretation ist aber sicher nicht möglich, da es sich zum großen Teil um Patienten handelt, bei denen die Diagnosestellung nur kurze Zeit zurücklag. Bei familiärer Häufung und früher Manifestation eines Diabetes wird in den meisten Fällen eine weiterführende Diagnostik durchgeführt, die zu einer früheren Diagnosestellung beiträgt. Bei der in letzter Zeit beobachteten Tendenz zur früheren Manifestation der Erkrankung innerhalb einzelner Familien (Ng, Li et al. 2000) bleibt es schwer abzuschätzen, welchen Beitrag dazu veränderte Diagnosestellung oder veränderten Umweltfaktoren vor allem bezüglich der nutritiven Situation leisten. 52 Die bisher beschriebenen Unterschiede der klinischen Progredienz zwischen MODY2 und MODY3 (Chevre, Hani et al. 1998; Isomaa, Henricsson et al. 1998) sind bezüglich ihrer Aussage nur vorsichtig zu interpretieren, weil es sich ausschließlich um retrospektive Daten ohne MODYUntergruppen-spezifische Therapie handelt. Diese unterschiedliche Therapie ist heute aber gerade wegen der bekannten pathophysiologischen Unterschiede zu fordern. Die funktionelle und klinische Charakterisierung der Mutationen (siehe oben) deutet eindeutig auf ein größeres Risikopotential der MODY3-Mutationen durch ihr absolutes Insulin-Sekretionsdefizit und die daraus resultierende, höhere Rate diabetischer Komplikationen hin. Für diese Untergruppe ist seit der UKPDS34-Studie (Nathan 1998) daher ein straffes und konsequentes Therapie-Regime zu fordern, während für die MODY2-Patienten meist eine rein diätetische Behandlung ausreichen dürfte. Rekrutierung, Überweisungsverhalten und Epidemiologie Bezogen auf die epidemiologischen Unterschiede in verschiedenen europäischen Ländern entsprechen die hier gefundenen Ergebnisse von der Anzahl der identifizierten Mutationen und dem Verhältnis der beiden MODY-Untergruppen zueinander eher den französischen Ergebnissen. Bei Vergleich dieser epidemiologischen Unterschiede müssen aber die Besonderheiten der Rekrutierung beachtet werden. So sind in den englischen Studien und der kleinen deutschen Analyse nur Internisten, dagegen keine Pädiater beteiligt gewesen. Die Ergebnisse legen nahe, dass die interdisziplinäre Rekrutierung für die Verteilung der Mutationen von großer Bedeutung ist. In Deutschland ist so erstmalig durch zwei unabhängige Screenings eine unterschiedliche Verteilung aufgrund populationsabhängigen der Rekrutierung Faktoren und gefunden unabhängig worden. von geographischen Interpretationen im Sinne oder von länderspezifischen Besonderheiten, wie sie auch von Pädiatern in einer aktuellen italienischen Untersuchung mit hoher MODY2-Frequenz angestellt werden (Massa, Meschi et al. 2001) erscheinen daher fragwürdig. Für eine wichtige Rolle der Rekrutierung sprechen auch die gefundenen, tendenziellen Unterschiede im Manifestationsalter (MODY2 < MODY3) und die Sonderstellung des Gestationsdiabetes. Letzten Endes muss man festhalten, dass eine genaue epidemiologische Aussage nach wie vor nicht möglich ist. Einer statistischen Hochrechnung auf die Gesamtbevölkerung stehen nicht nur die niedrigen Fallzahlen entgegen. Aus Kostengründen wird eine Mutationsanalyse nach wie vor in der Regel nur bei vorselektionierten Kollektiven durchgeführt, die eindeutige klinische und anamnestische Hinweise auf einen Mutationsträger-Status haben. Rückschlüsse auf die Frequenz der Mutationen unter allen Diabetikern oder der Gesamtbevölkerung sind somit nicht möglich. Es gibt nur wenige Untersuchungen, in denen Typ 1 (Chiu, Tanizawa et al. 1993; Yamada, Nishigori et al. 1997; Yoshiuchi, Yamagata et al. 1999) oder Typ 2 Diabetiker (Stoffel, Patel et al. 1992; Glucksmann, Lehto et al. 1997; Iwasaki, Oda et al. 1997; Urhammer, Rasmussen et al. 1997; Cox, Southam et al. 1999) ohne Vorauswahl molekulargenetisch untersucht wurden. Diese weisen nur punktuelle, vereinzelte Mutationsnachweise auf, was in Übereinstimmung zu frühen MikrosatellitenAnalysen steht, die von keiner generellen Bedeutung der MODY-Loci für den Typ 2 Diabetes ausgehen (Tanizawa, Matsutani et al. 1992; Hattersley, Saker et al. 1993). 53 Studien, in denen nach einigen oder den vollständigen klinisch diagnostischen MODY-Kriterien (Stoffel, Patel et al. 1992; Hager, Blanche et al. 1994; Yamagata, Furuta et al. 1996; Dussoix, Vaxillaire et al. 1997; Frayling, Bulamn et al. 1997; Glucksmann, Lehto et al. 1997; Iwasaki, Oda et al. 1997; Kaisaki, Menzel et al. 1997; Behn, Wasson et al. 1998; Chevre, Hani et al. 1998; Lehto, Wipemo et al. 1999; Yoshiuchi, Yamagata et al. 1999) eine Vorauswahl getroffen worden ist, sind eindeutig in der Überzahl. Ein typisches Beispiel für die Gefahren dieser Vorgehensweise bietet die bisher einzige deutsche Untersuchung (Lindner, Cockburn et al. 1999); bei einer ohnehin schon kleinen Fallzahl (11 Familien) wurde das MODY2 Screening auf milde Verlaufsformen (3 Familien) beschränkt. Dass diese Einschränkung für ein MODY2-Scrrening nicht zu verallgemeinern ist, zeigen die Beispiele der Familien MD8 und MD14. Innerhalb dieser Familien befanden sich Patienten mit Glucokinase-Mutationen, die insulinpflichtig waren. Nachteil unseres Screenings ist, dass bisher keine Untersuchung weiterer Gene oder Kandidatengene stattgefunden hat, für die in vereinzelten Fällen schon MODY-assoziierte Mutationen nachgewiesen wurden. Dazu zählen die Promotoren der beiden untersuchten Gene, aber auch die „seltenen“ MODY-Gene HNF4α, IPF1, HNF1β, NeuroD und Islet 1. Durch die Art der Mutationsanalyse konnten auch keine Veränderungen der Basensequenz erfasst werden, die innerhalb eines Introns gelegen sind und über zusätzliche Splice-Sites zu pathogenen Änderungen führen könnten. Der Ausschluss der mitochondrialen Mutation an Position 3243 wurde für alle Patienten durchgeführt. Diese Mutation wurde in 5-10% einer skandinavischen und japanischen Untersuchung angetroffen (Katagiri, Asano et al. 1994; Lehto, Wipemo et al. 1999). Dass sie nicht innerhalb unseres Patientenkollektivs nachgewiesen werden konnte, unterstützt Untersuchungen, die auf ein sporadisches Auftreten hindeuten (Dussoix, Vaxillaire et al. 1997; Saker, Hattersley et al. 1997). Selbstkritisch muss allerdings auf die eingeschränkte Sensitivität bei fraglicher Heteroplasmie hingewiesen werden, die bei der Gewinnung der DNA aus peripheren Leukozyten nicht zu vernachlässigen ist. Theoretisch ist bei der Pathogenese des MODY auch ein „Two-hit Modell“ wie bei der Kanzerogenese denkbar und in Erwägung zu ziehen. Eine somatische Mutation innerhalb der Glucokinase und des HNF1α könnte dabei zusammen mit einer familiär vererbten Prädisposition zur Manifestation führen. Ein Nachweis hierfür könnte aber nur durch Detektion von spezifischen βzellulären Mutationen selbst erbracht werden, die bisher noch nicht erfolgt ist. 4.1.1.3 Phänotyp Zukunftsperspektiven, Konsequenzen für die klinische Praxis Insgesamt hat die Prävalenz der Kinder und Jugendlichen mit Typ2 Diabetes stark zugenommen (Guazzarotti, Bartolotta et al. 1999; Libman und Arslanian 1999); in wieweit für diese „Epidemie“ (Rosenbloom, Joe et al. 1999) alimentäre und sozioökonomische Faktoren verantwortlich sind, bleibt offen. Die molekularbiologische Diagnostik vorselektionierter Kollektive erlaubt es jedenfalls, eine distinkte Gruppe hiervon abzutrennen, bei der spezifische Therapiemöglichkeiten aufgrund der pathophysiologischen Zusammenhänge angewendet werden können. Als klinische Selektionskriterien für eine molekularbiologische Untersuchung bei Kindern und Jugendlichen mit T2DM können dabei das fehlende Übergewicht, das Manifestationsalter und der Verlauf der Erstmanifestation wichtig sein (siehe Abb 21). Das Verteilungsmuster der bisher identifizierten 54 Mutationen zeigt, dass die molekularbiologische Analyse über den gesamten codierenden Bereich zu erfolgen hat. Sie muss sich eng an der Familienanamnese orientieren und in enger interdisziplinärer Zusammenarbeit von Pädiatern, Gynäkologen und Internisten erfolgen. Abb 21: Entscheidungsbaum zur Diagnostik bei juvenilen Diabetes-Formen 4.1.2 APBD Allelische Heterogenität Durch das charakteristische Auftreten der Polyglukosankörper ist die APBD eng mit der Glykogenose Typ IV verknüpft. Bei näherer Betrachtung besteht sogar ein fliessender Übergang zwischen diesen beiden Krankheitsbildern, so dass nicht erst seit der Detektion pathogener Mutationen in demselben Gen die Forderung nach einer gemeinsamen und differenzierenden Betrachtung ihre Berechtigung hat. Die Glykogenose Typ IV ist mit ihrer primären GBE-Defizienz ausgesprochen variabel: 1) Klassische Verlaufsform mit progressivem Leberversagen Die typische hepatische Manifestationsform der Glykogenose Typ IV manifestiert sich in der Regel in den ersten Lebensmonaten durch Hepatosplenomegalie und Entwicklungsrückstand. Leider entwickeln sich meist schnell Zirrhose und Leberversagen mit portaler Ösophagusvarizen und Aszites, was zum Tod vor dem fünften Lebensjahr führt. Hypertension, 55 2) Hepatische Verlaufsform mit geringer Progredienz Bei Diagnosestellung zum ähnlichen Zeitpunkt gibt es eine kleine Untergruppe, die trotz bestehender Leberschäden einen stabilen Krankheitsverlauf hat. Die Prognose ist hier wesentlich besser (Bao, Kishnani et al. 1996). 3) Varianten mit zusätzlicher multisystemischer Beteiligung Es sind auch weniger progressive Varianten bekannt, bei denen das neuromuskuläre System oder eine Kardiomyopathie im Vordergrund stehen. Innerhalb dieses Beteiligungsmusters ist die Ausprägung von kongenitaler, generalisierter Hypotonie und frühem Tod bis zur milden und späten Manifestation sehr variabel. 4) Myopathische Form Scharf abgrenzen lässt sich die Untergruppe der juvenilen GSD IV-Ausprägungen, bei denen die Muskelbeteiligung im Vordergrund steht. Die Patienten mit der auch adulte PolyglukosankörperMyopathie genannten Störung zeigen durchgehend bei allen Messungen eine normale Enzymaktivität in den Zellen des peripheren Bluts, während die Muskulatur (Skelettmuskulatur, glatte Muskulatur und teilweise auch Herzmuskulatur) zahlreiche Ablagerungen von Polyglukosankörpern und einen völligen Verlust der Enzymaktivität aufweist (Greene, Weldon et al. 1987, Weis und Schroder 1988, . Die typische Manifestationszeit liegt hierbei gegen Ende der zweiten Dekade. 5) APBD Die APBD ist vor allem durch ihre späte Manifestation frühestens ab der fünften Lebensdekade und die neurologische Symptomatik charakterisiert. Widersprüchliche Angaben über das Auftreten erster Symptome schon im frühen Kindesalter (Felice, Grunnet et al. 1997) sind am ehesten durch Probleme bei der Diagnosestellung (Diagnose post mortem, keine Biopsie oder Enzymbestimmung zu Lebzeiten) und Fehldiagnosen begründet. Weitere weniger charakteristische Krankheitsbilder mit mildem Verlauf und Enzymdefizienz in verschiedenen Organen sind beschrieben, die sich schwer einer der aufgeführten Unterformen zuordnen lassen (z.B. Scotto, de Barsy et al. 1981). Die drei wesentlichen Faktoren, die im Grenzbereich der beiden Entitäten GSD IV und APBD variieren, sind Manifestationsalter, betroffene Organe und die GBE-Aktivität in den einzelnen Organen. Dabei zeigen Übersichten der bestimmten Enzymaktivitäten (McConkie-Rosell, Wilson et al. 1996, van Noort, Straks et al. 1993), dass sich keine eindeutige Korrelation zwischen Aktivitätsmuster und Krankheitsverlauf herstellen lässt. Lediglich bei den kongenitalen, schnell progressiven Verlaufsformen (Formen 1 und 2) findet sich fast durchgehend ein vollständiger Verlust der GBE-Aktivität in allen untersuchten Geweben. Vor allem aber bei den milden GSD IVVarianten zeigt sich bei der Analyse der GBE-Aktivität von Fibroblasten kein homogenes Bild (Guerra, van Diggelen et al. 1986, Greene, Brown et al. 1988). Modell-Mechanismen der allelischen Heterogenität Die molekularen Mechanismen der allelischen und klinischen Heterogenität sind ungeklärt. Verschiedene Erklärungsansätze bzw. Hypothesen sind zu diskutieren: 1) gewebsspezifische Isoformen bzw. gewebsspezifisch regulierte Isoenzyme 56 Die selektive Defizienz eines organspezifischen oder altersabhängigen Isoenzyms ist eher unwahrscheinlich (Zimmerman und Gold 1983), weil es bisher keinen Hinweis auf die Existenz von Isoenzymen gibt. Auch in unserer Analyse wurden keine gewebespezifischen Auffälligkeiten gefunden. Unterschiedliche Isoformen des GBE-Gens könnten auch unterschiedliche gewebespezifische Expression erklären. Dabei zeigen die Ergebnisse der Analyse auf mRNAEbene keinen Hinweis dafür, stellen jedoch auch keinen hinreichenden Ausschluss dafür dar. 2) gewebsspezifische posttranslationale Modifikation Bei der isoelektrischen Auftrennung des Proteins ließen sich unterschiedliche Mikroformen nachweisen, die durch Deamidierung im Sinne einer nichtenzymatische Veränderungen erklärt werden könnten (Zimmerman und Gold 1982). Eine posttranslationale Modifizierung wie Glucosidierung oder Phosphorylierung sind eher unwahrscheinlich (Zimmerman und Gold 1983). Dagegen wurde eine gesteigerte Aktivität einer RNA-assoziierten Form beschrieben (Satoh und Sato 1982). Die Empfindlichkeit gegenüber Proteasen oder die thermische Stabilität könnten aber auch durch zusätzliche extrinsische und genetische Faktoren beeinflusst werden. 3) Zusammenspiel mit anderen Enzymen des Glykogen-Stoffwechsels Das oben angesprochene Modell (Raben, Danon et al. 2001) des Gleichgewichts zwischen GBE und Glykogensynthase stellt einen weiteren Erklärungsansatz dar, wieso eine alleinige Betrachtung der Aktivität des GBE nicht ausreicht. Dass es sich in vivo um ein fein reguliertes System zwischen Verzweigung und Kettenverlängerung des Glykogens handelt, wird auch durch weitere Beobachtungen deutlich. So führt die Akkumulation von Glucose-6-Phosphat in einigen Fällen von Phosphofructokinase-Defizienz zu einer bevorzugten Hemmung des GBE im Vergleich zur normalen Glykogensynthase-Aktivität; diese Imbalance bewirkt die Synthese längerer Glykogenketten und das Auftreten Polyglukosan-ähnlicher Ablagerungen, die Ursache einer Fehldiagnose sein könnten (Agamanolis, Askari et al. 1980). 4) Mutationstypus bzw -position Ein Modell für eine spezifischere Genotyp-Phänotyp Relation der APBD und GSD IV soll kurz erwähnt werden. Die Ashkenazi-Patienten mit APBD waren homozygot für die Y329S MissenseMutation, die zu einer 50%igen Reduktion der BE-Aktivität führte (Lossos, Meiner et al. 1998). Die hier beschriebene Patientin hat ebenfalls eine Missense-Mutation des GBE Gens. Expressionsversuche zur Bestimmung der Restaktivität des mutierten Proteins wurden jedoch bisher noch nicht unternommen. Interessanterweise hatte ein Patient mit relativ milder, nicht progressiver hepatischer Ausprägung von GSD IV auch zwei Missense-Mutationen (L224P und Y329S). Im Gegensatz dazu zeigten zwei schwere Fälle eine Translation-terminierende bzw. eine Mutation mit Verlust der Splice-Stelle (Bao, Kishnani et al. 1996). So erscheint es, dass Veränderungen der Aminosäuresequenz an bestimmten Positionen APBD und die hepatischen und neuromuskulären Ausprägungsformen von GSD IV hervorrufen; hingegen könnten die Translation terminierende oder disruptive Mutationen für die multisystemischen schweren Ausprägungen verantwortlich sein. Weitere Untersuchungen des GBE-Gens und Expressionsstudien können dabei helfen, die Genotyp-Phänotyp-Korrelation bei GSD IV und APBD besser zu verstehen. 57 Klinischer Phänotyp der Patienten mit APBD Beim vorliegenden Krankheitsfall ist das klinische Vollbild der APBD erfüllt. Tetraspastik, Ataxie und distale Sensibilitätsstörungen mit einer Manifestation im späten Erwachsenenalter lagen vor. Auch wenn bei der Testung initial keine Demenz bestand, wurden bei der Patientin später die Gedächtnisstörungen und kognitiven Defizite nachgewiesen. Das Krankheitsbild der Patientin AD I.2 stellt sich vor allem durch die in der Herzmuskulatur nachgewiesenen Polyglukosankörper als Sonderfall innerhalb der APBD dar. Trotz des Befalls dieses Organs ist wegen des typischen Manifestationsalters in der fünften Dekade und der geringen Beteiligung der Skelettmuskulatur eine Zugehörigkeit zur Gruppe der myopathischen Glykogenose Typ IV auszuschließen. Die zusätzliche Beteiligung der Herzmuskulatur, die als Todesursache klinisch im Vordergrund stand, ist bisher nur für GSD IV beschrieben worden (Servidei, Riepe et al. 1987); erstmalig bestand somit bei der APBD eine signifikante Kardiomyopathie. Eine falsche Zuordnung ist bei unserer Patientin unwahrscheinlich, auch wenn von der morphologischen Struktur der Polyglukosankörper selbst her keine Differenzierung zwischen Lafora disease, GSD IV und APBD möglich ist (Ishihara, Yokota et al. 1987). Schwierigkeiten der Differenzierung sind hier auch in der Veterinärmedizin beschrieben (wie z.B. Hunden (Yamanami, Ishihara et al. 1992), Katzen (Kamiya und Suzuki 1989; Fyfe 1995), Kühen(Yanai, Masegi et al. 1994) und Schweinen (O'Toole, Wells et al. 1994)). Doch auch wenn man die APBD für sich betrachtet, bringt es der variable Phänotyp mit sich, dass Fehldiagnosen gestellt werden. Die Diagnosen einer Multiplen Sklerose (Bruno, Servidei et al. 1993) und einer Amyotrophen Lateralsklerose (Bruno, Servidei et al. 1993; McDonald, Faust et al. 1993) mussten retrospektiv bei einer positiven Familienanamnese zur APBD korrigiert werden. In der klinischen Variabilität liegt es auch begründet, dass sich bei bisherigen Versuchen einer Übersichtsdarstellung große Unterschiede bezüglich der Gesamtzahl der bisher beschriebenen Fälle finden (21 (Bigio, Weiner et al. 1997), 27 (Boulan-Predseil, Vital et al. 1995), 12 (Gray, Gherardi et al. 1988), 25 (Robertson, Wharton et al. 1998), 20 (Busard, Gabreels-Festen et al. 1991), 26 (Sindern, Patzold et al. 1999)). Nach strengen Kriterien der Diagnosestellung stellt der hier beschriebene Fall den 48. Fall dar (siehe Tab 14). Bei unserer Patientin fanden sich keine Hinweise auf eine gesteigerte Empfindlichkeit für Drucklähmungen (Gil-Neciga, Pareja et al. 1995). Es konnte auch kein Zusammenhang zwischen der Anzahl und Lokalisation der Ablagerungen und mechanischer Beanspruchung der einzelnen Muskelgruppen (Matsumuro, Izumo et al. 1993) hergestellt werden. 58 Tab 14: Übersicht über die bisher veröffentlichen APBD Fälle, Gewebe BE-aktivität in Herzmuskulatur(C), Leber (L), Skelettmuskel (M), peripherem Nerv (N), Leukozyten (L), ZNS (Z) Referenz (Suzuki, David et al. 1971) (Carpenter und Karpati 1976) (Carpenter und Karpati 1976) (Peress, DiMauro et al. 1979) (Stam, Wigboldus et al. 1980) (Robitaille, Carpenter et al. 1980) (Robitaille, Carpenter et al. 1980) (Robitaille, Carpenter et al. 1980) (Robitaille, Carpenter et al. 1980) (Okamoto, Llena et al. 1982) (Vos, Joosten et al. 1983) (Vos, Joosten et al. 1983) (Karpati und Carpenter 1983) (Baudrimont, Gherardi et al. 1986) (Baudrimont, Gherardi et al. 1986) (Cafferty, Hays et al. 1986) (Cafferty, Hays et al. 1986) (Taboada, Suzuki et al. 1986) (Gray, Gherardi et al. 1988) (Gray, Gherardi et al. 1988) (Gray, Gherardi et al. 1988) (Sugiyama, Budka et al. 1989) (Sugiyama, Budka et al. 1989) (Wisniewski, Goldman et al. 1990) (Cafferty, Lovelace et al. 1991) (Cafferty, Lovelace et al. 1991) (Lossos, Barash et al. 1991) (Lossos, Barash et al. 1991) (Negishi und Sze 1992) (McDonald, Faust et al. 1993) (McDonald, Faust et al. 1993) (Chretien, Louarn et al. 1993) (Matsumuro, Izumo et al. 1993) (Bruno, Servidei et al. 1993) (Bruno, Servidei et al. 1993) Alter Krankheitsdauer /Jahre 62 3 51 0 51 0 55 8 67 8 64 21 67 20 59 8 65 14 64 20 Autopsie Todesursache < GeDeschlecht menz Motori- Sensibilische tätsverStörung lust Pyramidenbahn Syndrom Inkontinenz FamiliEnzymdiag enanamostik (BE) nese + + - M + + + - (N.suralis Biopsie) F - + + - + - (N.suralis Biopsie) F - + + - + < M + + + + + < M + + + + + < M + + + + + M + + + + + + - (N.suralis Biopsie) F - - + + - (N.suralis Biopsie) F - + + + < F + + + + + - F - + + + + + F - + + + + + F - + + + - M + + + + + - F + + + + + - ? ? - + + + + + + + + + - + + + Sepsis, Pyelonephritis Sepsis, Pyelonephritis Pneumonie < Sepsis Sepsis - (N.suralis und Muskel-Biopsie) - (N.suralis und Muskel-Biopsie) M nor N red 46 10 57 6 63 9 73 17 78 15 44 54 0 0 - (N.peroneus Biopsie) - (N.peroneus Biopsie) - (N.suralis Biopsie) - (N.suralis Biopsie) 74 14 < M 8 < F - + + + + - M + - + + + - M + + + + - F + + - + - F - - - - - < M + - - - - 63 63 74 8 1 Bronchopneumonie < Pyelonephritis <(N.suralis Biopsie) Pneumonie < 71 47 - (N.suralis Biopsie) 0 - M nor N red 53 9 - (N.suralis Biopsie) F - + + + + - 66 0 - (N.suralis Biopsie) F - + + + + + 58 3 - (N.suralis Biopsie) F + L red 64 13 - (N.suralis Biopsie) M + L red 67 0,7 < M + + + + + - 52 2,3 kardiopulmonale Insuffizienz F + + + + - - 68 2 - (N.suralis Biopsie) F + + + + + - < M + + - + + - M - - + - - - F + + + + + - M - + + + - - 78 0,5 75 1 46 8 50 0 Pneumonie < Apoplex, Herzversagen - (N.suralis und Muskel-Biopsie) - (N.suralis und Muskel-Biopsie) - (N.suralis Biopsie) L nor M nor L red M red M erh N nor M nor N nor 59 Alter Referenz (Bruno, Servidei et al. 1993) Krank -heitsdauer /Jahre Autopsie Todesursache Geschlec ht Deme nz Motori -sche Störun g Sensibi litätsverlust Pyramidenbahn Syndrom Inkontinenz Familienanam -nese - 56 14 - (N.suralis Biopsie) M + + + + + (Bruno, Servidei et al. 1993) (Wierzba-Bobrowicz und Stroinska-Kus 1994) (Rifai, Klitzke et al. 1994) (Boulan-Predseil, Vital et al. 1995) (Vital, Lamarche et al. 1995) (Inoue, Ishii et al. 1996) (Bigio, Weiner et al. 1997) 87 3 M + + + + + 45 1,5 - (N.suralis Biopsie) < F + + + + + - 56 14 + + + + + - 1 - (N.suralis Biopsie) - (N.suralis und cerebrale Biopsie) M 65 F + - - - + - 79 0 - (N.suralis Biopsie) M 51 15 - + + + + - 9 - (N.suralis Biopsie) < M 72 M + + - + + + (Bigio, Weiner et al. 1997) 69 20 F + + + + + + (Lossos, Meiner et al. 1998) (Lossos, Meiner et al. 1998) (Lossos, Meiner et al. 1998) 59 60 58 5 10 3 - (N.suralis Biopsie) - (N.suralis Biopsie) - (N.suralis Biopsie) M M M + + + + + + + + + + + + + + + + + - Eigener 50 4 < F + + + + + - Pneumonie Knochenkrebs < Sepsis,Bronchopneumonie akuter Herztod Enzymdia gostik (BE) M red N nor L red L red L red C red L red P red Z red H red M? Therapeutisch stehen bei APBD kaum spezifische Ansätze zur Verfügung. Theoretisch kann man zwar wie bei der GSD IV ableiten, dass zur Vermeidung von Hypoglykämien und eines hohen Glykogen-Umsatzes eine kohlenhydratreiche Diät und ein Verzicht auf körperliche Anstrengungen sinnvoll sind (Greene, Ghishan et al. 1988). Allerdings wurde bei GSD IV jedoch in der Praxis ein positiver Einfluss einer proteinreichen, kohlenhydratarmen Diät beobachtet (Goldberg und Slonim 1993). Der Erfolg einer Lebertransplantation, die bei GSD IV anfangs zu einzelnen Besserungen des Krankheitsverlaufs führte (Selby, Starzl et al. 1993), musste mittelfristig in der Bewertung revidiert werden (Rosenthal, Podesta et al. 1995). Positive Therapieeffekte einer Lebertransplantation sind vor allem für zentralnervöse oder peripherneurale Ablagerungen von Polyglukosankörpern bei APBD unwahrscheinlich. Es sind bisher keine äußeren Faktoren bekannt, die zu einer Progredienz und Verschlechterung der neurologischen Symptomatik führen. Eine Intubationsanästhesie bei APBD wurde problemlos vertragen und hatte keine Auswirkungen auf den Krankheitsverlauf (Inoue, Ishii et al. 1996). Die Kernspintomographie hat sich bisher als probate Methode herausgestellt, das Ausmaß des zentralnervösen Krankheitsbefalls abzuschätzen (Negishi und Sze 1992; Rifai, Klitzke et al. 1994). Auch bei der hier beschriebenen Patientin war die Bildgebung ante mortem mit dem autoptischen Befund kongruent. Mutationsanalyse Durch Direkt-Sequenzierung konnten die beiden Mutationen R515H und R524Q bei der APBDPatientin identifiziert und durch RFLP bestätigt werden. Die Zusammenhänge deuten darauf hin, dass die detektierten Basenaustausche pathogene Mutationen und für die Manifestation der APBD verantwortlich zu sein scheinen. Dafür spricht der negative Befund bei den Kontrollpersonen genauso wie der Nachweis jeweils einer der beiden Missense-Mutationen bei den heterozygoten 60 Töchtern, die eine intermediäre Enzymaktivität vereinbar mit einem autosomal rezessiven Vererbungsmuster aufweisen. Beide Positionen der Aminosäure-Sequenz sind hochkonserviert zwischen der humanen Sequenz und Saccharomyces cerevisiae (Thon, Khalil et al. 1993). Sie liegen in einer Region, die nicht das katalytisch aktive Zentrum des GBE darstellt (Baba, Kimura et al. 1991). Deshalb könnte man vermuten, dass beide Missense-Mutationen über eine Veränderung der Tertiär-Struktur des Proteins dessen Funktion beeinträchtigen. Die Untersuchung fügt zwei neue Mutationen zu den schon vorher bekannten sechs Mutationen des GBE-Gens hinzu (L224P, F257L, Y329S, R515C, R524X, 210-bp Deletion). Damit konnte bestätigt werden, dass APBD assoziiert mit einer GBE-Defizienz ist und APBD und GSD IV allelische Erkrankungen, hervorgerufen durch Mutationen innerhalb desselben Gens sind. Mittlerweile ist die Mutation R524Q (siehe Tab 2) auch bei einem Patienten mit neuromuskulärer Form der GSD IV nachgewiesen worden (Bruno, DiRocco et al. 1999). Auch für das Codon 515 ist bereits eine Missense-Mutation bei einem Glykogenose-Patienten beschrieben worden (Bao, Kishnani et al. 1996). Es könnte sich also bei dieser Region um einen Hotspot handeln, der mit dem beschriebenen Primerpaar bei weiteren Patienten schnell untersucht werden kann. Bisher sind Veränderungen im GBE-Gen mit Verminderung der GBE-Aktivität lediglich für APBDPatienten beschrieben worden, die der Ashkenazi-Population angehörten (Motulsky 1995). Die einzige Mutation Y325S wurde jedoch lediglich in Homozygotie detektiert und hätte auch einen seltenen Polymorphismus innerhalb der Population darstellen können (Lossos, Barash et al. 1991). Diese Arbeit beschreibt den ersten Fall außerhalb der Ashkenazi-jüdischen Population und bestätigt die Bedeutung der Mutationen im GBE-Gen für ABPD. Ganz eindeutig ist das Krankheisbild nicht nur zwingend mit Y325S assoziiert. Eine sekundäre Enzymhemmung durch Akkumulation einer inhibitorischen Komponente, die von (Lossos, Barash et al. 1991) als Möglichkeit in Betracht gezogen wurde, erscheint durch die vorliegenden Ergebnisse unwahrscheinlich, weil die intermediäre Aktivität bei den heterozygoten Töchtern der Patientin in peripheren Leukozyten gemessen wurde. Dass die Überaktivierung der Glykogensynthase im transgenen Tiermodell zur Formation und Ablagerung der oben beschriebenen Polyglukosankörper führt (Raben, Danon et al. 2001), weist darauf hin, dass der Verzweigungsgrad letzten Endes zu der Ablagerung der Polyglukosankörper führen kann und nicht allein nur durch das GBE beeinflusst wird. Vieles spricht daher dagegen, dass die Polyglukosankörper nur einen Nebeneffekt einer parallelen metabolischen Störung der Neurone darstellen. Vielmehr deutet die in distalen Abschnitten gefundene Waller Degeneration darauf hin (Gray, Gherardi et al. 1988), dass erst diese Ablagerungen von Polyglukosankörpern die Axone schädigen. Die gefundenen Rosenthal-Fasern und zentralnervösen Befunde sprechen zusätzlich für einen gestörten Stoffaustausch zwischen zentralen Neuronen und Gefäßen. Diese Zusammenhänge sprechen insgesamt für eine kausalen Zusammenhang zwischen Mutationen im GBE-Gen, gefundenen Ablagerungen und beobachteten Krankheitssymptomen. 61 MuskelBiopsie N.suralis Biopsie Mutationsanalyse Bestimmung der GBE-Aktivität in Leukozyten Bildgebung, cerebrales MRT EKG/UKG, urologische Diagnostik Basislabor (CK, TSH, BSG), NLG, EMG Anamnese, klinische Untersuchung Abb 22: Stufendiagnostik bei APBD (NLG Nervenleitgeschwindigkeit, UKG Herzechokardiographie, EMG Elektromyographie, TSH Thyreoidea stimulierendes Hormon, BSG Blutsenkungsgeschwindigkeit) Diagnostik der APBD Aus den gewonnen Erkenntnissen sind für die Diagnostik verschiedene Konsequenzen zu fordern. Wegen der zusätzlichen Möglichkeit der nicht invasiven Mutationsanalyse ist diese gemäß einer sinnvollen Stufendiagnostik vor den invasiven Verfahren zu fordern (siehe Abb 22). Nach initialer Basisdiagnostik sollte bei Krankheitsverdacht neben einer urologischen Zusatzdiagnostik unbedingt eine echo- und elektrokardiographische Abklärung erfolgen. Der hier beschriebene Fall hat die Möglichkeit einer Beteiligung der Herzmuskulatur ähnlich wie bei GSD IV (Servidei, Riepe et al. 1987) eindrucksvoll unterstrichen. Parallel dazu scheint die Kernspintomographie nicht nur zum Ausschluss anderer Systemerkrankungen sinnvoll, sondern hat sich auch in der Diagnostik der APBD bewährt. Schließlich steht die wenig invasive GBEAktivitäsbestimmung aus peripheren Leukozyten zur Verfügung. Mit der hier angewandten Bestimmungsmethode der Synthese 14 C-markierten Glykogens ist (Shin, Steiguber et al. 1988) kann auch eine Pränataldiagnose aus Amniozyten oder Chorionvilli durchgeführt werden (Brown und Brown 1989). Eine vorsichtige Interpretation insbesondere falsch negativer Ergebnisse ist aufgrund des indirekten Charakters der Bestimmungsmethode (Brown et al. 1983) dringend notwendig. Obwohl mit der Verwendung peripheren Bluts eine elegante und wenig invasive Methode zur Verfügung steht, wurde in der Vergangenheit häufig darauf verzichtet (WierzbaBobrowicz und Stroinska-Kus 1994; Vital, Lamarche et al. 1995; Bigio, Weiner et al. 1997; Felice, Grunnet et al. 1997; Robertson, Wharton et al. 1998). Als neue Möglichkeit schließt sich hier unmittelbar die Mutationsanalyse an. Erst dann erscheint die Durchführung einer Biopsie primär im N.suralis (Gray, Gherardi et al. 1988) sinnvoll. In einigen Fällen war ebenfalls eine axilläre Hautbiopsie diagnoseweisend (Busard, Gabreels-Festen et al. 1991). Jedoch haben vor allem die Ergebnissse der Muskelbiopsien und Autopsien (Weis et al. 62 1988) darauf hingewiesen, dass trotz vorliegender generalisierter Erkrankung Unterschiede in der Ausprägung der Krankheit oder dem Beteiligungsmuster zwischen einzelnen Muskelgruppen bzw. Körperabschnitten bestehen können. Diese mosaikartige Beteiligung erfordert die vorsichtige Interpretation negativer Befunde. Die Befunde zeigen, dass die GBE-assoziierte Form der APBD keineswegs auf Ashkenazi-Juden beschränkt ist. Von daher ist die Bestimmung der Enzymaktivität bei Krankheitsverdacht auch bei kaukasischen Patienten sinnvoll. Der beobachtete Krankheitsverlauf unserer Patientin zeigt aber auch, dass nach eindeutiger Diagnosestellung (z.B. positivem Nachweis von Polyglukosankörpern) in Erwägung gezogen werden muss, ob weitere Gewebeuntersuchungen und Aktivitätsbestimmungen durchgeführt werden sollten. Denn die Zuordnung des Verteilungsmusters des Organbefalls scheint für die Prognose des Krankheitsverlaufs eine wichtige Rolle zu spielen. 4.1.3 PhK-Defizienz Krankheitsbild und Differentialdiagnose Die molekulargenetische Ursache für die gefundene Defizienz der PhK bleibt auch nach der hier durchgeführten Analyse letzten Endes unklar. Das hier beschriebene Krankheitsbild war vor allem durch einen sehr benignen Phänotyp geprägt. Die CK-Erhöhung ging nur mit Muskelbeschwerden einher, so dass sie auch als familiäre „idiopathischen Hyper-CKämie“ (erstmals Rowland (1980)) eingeordnet werden könnte. Die HyperCKämie als solches stellt hier eine Ausschlussdiagnose dar, bei der Muskelerkrankungen wie maligne Hyperthermie, Myopathien im Rahmen von Endokrinopathien (z.B. Hypothyreose), toxische Myopathien durch Medikamente ( wie z.B. durch β-Blocker, Antipsychotika, Cholesterin-Senker), metabolische Myopathien (z.B. bei Hypokaliämie) und Muskeltrauma/-belastung ausgeschlossen werden müssen. Einheitlich erscheint lediglich der symptomarme Verlauf (Reijneveld, Notermans et al. 2001), der auch in der hier typisierten Familie gefunden wurde. Das besondere Charakteristikum jedoch der vorliegenden HyperCKämie ist die familiäre Assoziation mit der festgestellten PhK-Defizienz. Die PhK-Defizienz macht eine Erklärung der HyperCKämie als Caveolinopathie (siehe unten) bei der beschriebenen Familie unwahrscheinlich. Weil das Vererbungsmuster einem autosomal dominanten Erbgang entspricht, standen die β- und γ1-Untereinheit der PhK als mögliche Kandidaten für die PhK-Defizienz im Vordergrund. Die durchgeführte Kopplungsanalyse, v.a. aber die anschließende Sequenzierung schlossen eine Bedeutung dieser Untereinheiten für die familiäre HyperCKämie hier aus. Bei der PhK-Defizienz ist eine artefizielle, fehlerhafte Messung oder transportbedingte Proteindegradation bei den sensiblen Größenordnungen der Essays gleichfalls zu bedenken. 4.1.4 RMD Nachweis der de-novo Mutation R26Q im Caveolin-3-Gen bei RMD Der untersuchte Patient erfüllte die klinischen Kriterien der RMD ( PIRC in jeweils mindestens zwei Muskeln der oberen und unteren Extremität ohne gesteigerte Sehnenreflexe und mindestens eine der folgenden Eigenschaften: Muskelvorwölbungen, Muskelwogen („Rippling“) oder CK-Erhöhung). 63 Es gab keine positive Familienanamnese bezüglich neuromuskulärer Symptome. Der typische Phänotyp führte dazu, nach einer Caveolin-3-Mutation zu suchen, wobei die Arg26Gln-Mutation im Exon 1 nachgewiesen werden konnte. Für diese Mutation wurde bereits beschrieben, mit einem entsprechenden Phänotyp zu segregieren. (Vorgerd, Bolz et al. 1999). Darüber hinaus wurde derselbe Basenaustausch als eine de novo Mutation bei zwei nicht verwandten, asymptomatischen Kindern detektiert, die eine CK-Erhöhung und einen Verlust des Caveolin-3 Proteins in der Western-blot-Analyse ihrer Skelettmuskulatur zeigten (Carbone, Bruno et al. 2000). Die familiäre Analyse und der Paternitätsnachweis zeigten hier im Zusammenhang, dass es sich um die erste de novo Mutation im Caveolin-3-Gen handelt, die mit RMD einhergeht. Somit sollte auch bei Fehlen einer positiven Familienanamnese die Diagnose RMD nicht von vorneherein ausgeschlossen werden. Dagegen zeigt die Existenz von sporadischen Mutationen und Krankheitsfällen, dass RMD eventuell häufiger ist als bisher angenommen. Mutationsanalysen des relativ kleinen Caveolin-3-Gens (151 Aminosäuren) stellen bei entsprechendem Verdacht eine einfache Möglichkeit dar, RMD bei Patienten molekularbiologisch zu bestätigen. Heterogenität von Mutationen im Caveolin-3-Gen Allelischen Mutationen des Caveolin-3-Gens liegen drei distinkten Erkrankungen der Skelettmuskulatur zugrunde, einer idiopathischen Erhöhung der CK (HyperCKämie), der autosomal dominanten Form der Gliedergürtel-Muskeldytrophie (LGMD1C) und der milden nicht-dystrophen RMD. Klinische Untersuchungen dieser drei unterschiedlichen Caveolinopathien zeigen variable Symptome wie Muskelkrämpfe, Steifheit, Muskelschmerzen, wiederkehrende Rhabdomyolysen, Wadenhypertrophie, Schwäche und Zeichen der muskulären Hyperexzitabilität (PIRC, „Rippling“). CK-Erhöhungen scheinen die überlappende Gemeinsamkeit dieser verschiedenen Erkrankungen zu sein. Abb 23: Veränderungen der Proteinsequenz der Caveolin-3 Domänen bei drei verschiedenen Krankheitsbildern Sechs unterschiedliche Mutationen des Caveolin-3-Gens sind bis jetzt beschrieben (siehe Abb 23). Zwei Veränderungen, G55S und C71W, die bei LGMD1C- Patienten gefunden wurden (McNally, de Sa Moreira et al. 1998), stellten sich als Polymorphismen und nicht pathogen heraus (de Paula, Vainzof et al. 2001). Insgesamt konnten Missense-Mutationen für vier Codons (Codon 26, 28, 45 und 104) identifiziert werden, was auf ihre Rolle als eventuelle genetische „Hotspots“ der hereditären Caveolinopathien hinweist. Interessanterweise können dieselben Mutationen sowohl zur HyperCKämie als auch zur RMD führen – wie R26Q – oder zu RMD und LGMD1C – wie P104L 64 und A45T. Dieser Sachverhalt führt zu der Annahme, dass zusätzliche epigenetische Faktoren oder zusätzliche Gene für die Erkrankungen von Bedeutung sind. Die Frage einer möglichen Herzbeteiligung bei RMD wurde durch die nachgewiesene Expression von Caveolin-3 auch in der Herzmuskulatur (Tang, Scherer et al. 1996) neu aufgeworfen. Gegen eine generelle kardiale Beteiligung spricht, dass bei einer Familie die diagnostizierte Kardiomyopathie nicht mit einer zugehörigen Mutation kosegregierte (Ricker, Moxley et al. 1989). Andererseits ist beim Krankheitsbild RMD eine Herzbeteiligung möglich, so stehen z.B. bei einem autosomal rezessiven Phänotyp mit multisystemischen Verlauf kardiale Ereignisse klinisch im Vordergrund (Koul, Chand et al. 2001 ). Pathophysiologisches Modell Caveolin-3 stellt das konstituierende Protein der muskulären Caveolae dar, deren Rolle als endozytotische Vesikel und bei Prozessen der Signaltransduktion diskutiert wird (Kurzchalia und Parton 1999). Die hier beschriebene Mutation führt über eine Änderung der N-terminalen Domäne zu einer Veränderung der Proteinfunktion. Die heterozygote R26Q Mutation könnte einen dominant-negativen Effekt ausüben, indem sie die Bildung instabiler Aggregate von Wildtyp und mutiertem Caveolin-3 bewirkt, die innerhalb des Golgi-Komplex verbleiben und die Plasmamembran nicht erreichen können. Ein ähnlicher Mechanismus wurde für die P104L und die T63-65 Deletion des Caveolin-3-Gens (Galbiati, Volonte et al. 1999) nachgewiesen. Solche intrazellulären Aggregate würden auch in Übereinstimmung zu den immunhistologischen Untersuchungen beim untersuchten Patienten stehen und sowohl das reduzierte MembranExpressionsmuster als auch die punktförmige Anfärbung des Sarkoplasmas erklären. Neben vielen anderen Signalmolekülen der Skelettmuskulatur, die in den Caveolae lokalisiert sind, interagiert Caveolin-3 mit nNOS. nNOS ist die muskelspezifische Isoform der Nitritoxid-Synthasen und wird durch Caveoline gehemmt (Michel, Feron et al. 1997). Stickoxid (kurz NO) kann die Kontraktion der Skelettmuskulatur auf komplexe Art und Weise beeinflussen (Stamler und Meissner 2001). Die nNOS Expression war in der Muskel-Biopsie des Patienten nicht verändert. Das bestätigt bisherige Studien, die eine normale nNOS Expression innerhalb der muskulären Plasmamembran in stabil transfizierten C2C12-Mäusezellen (Galbiati, Volonte et al. 1999) und der Muskelbiopsie eines anderen Patienten mit autosomal dominanter RMD gefunden haben. Die reduzierte Caveolin-3 Expression an der Oberfläche war mit einer gesteigerten Induzierbarkeit der nNOS in mutierten C2C12-Zellen assoziiert, was in Zusammenhang mit der Muskelübererregbarkeit bei RMD stehen könnte(Betz, Schoser et al. 2001). Sunada et al. (Sunada, Ohi et al. 2001) berichten über einen Anstieg der nNOS Aktivität in der Skelettmuskulatur einer transgenen Maus, deren Caveolin-3 die P104L Mutante trägt. Im Gegensatz dazu wurde bei einem Patienten mit LGMD1C (Herrmann, Straub et al. 2000) und einem Patienten mit X-chromosomaler Duchenne’scher Muskeldystrophie (Brenman, Chao et al. 1995) eine starke Verminderung der nNOS-Aktivität nachgewiesen, auch im Tiermodell fanden sich entsprechende Ergebnisse (Galbiati, Engelman et al. 2001). Es ist also denkbar, dass die unterschiedliche nNOS-Expression bei primären Caveolinopathien von funktioneller Relevanz ist und den Phänotyp der Erkrankung beeinflussen kann. Primäre Caveolin-3 Defizienz mit normaler nNOS Expression und gesteigerter 65 Induzierbarkeit der nNOS könnte zum milden RMD-Phänotyp ohne dystrophe Veränderungen führen, während eine starke Reduktion von Caveolin-3 mit Verlust von nNOS den dystrophen LGMD Phänotyp hervorrufen könnte. Extrazelluläre Matrix Laminin-2 γ α Dystroglykan-Komplex β δ Sarkoglykan-Komplex Sarkospan Sarkolemm Syntrophine nNOS Dystrophin Caveola Zytosol Caveolin-3 F-aktin Abb 24: Pathophysiologisches Schema zur Interaktion zwischen nNOS und Caveolin-3 In Skelettmuskelfasern sind Caveolin-3 und nNOS mit dem Dystrophin-Glycoprotein Komplex assoziiert, der eine Verbindung zwischen Laminin-2 der extrazellulären Matrix und dem F-Aktin des Zytoskeletts darstellt (Abb 24). Der Membran-Anker Dystroglykan besteht aus einer extrazellulären α- und einer transmembranären β-Untereinheit. α-Dystroglykan verbindet das BasalmembranProtein Laminin-2 mit ß-Dystroglykan. Caveolin-3 interagiert direkt mit dem C-terminalen Ende von ß-Dystroglykan (Sotgia, Lee et al. 2000). In der Muskelbiopsie des untersuchten RMD-Patienten fand sich eine moderate Reduktion des α-Dystroglykan am Sarkolemm. Dieser Befund unterscheidet sich von einem Patienten mit LGMD1C und einer Ala45Thr-Mutation im Caveolin-3Gen (Herrmann, Straub et al. 2000) und von verschiedenen anderen Muskeldystrophien (Lim und Campbell 1998, Ohlendieck, Matsumura et al. 1993), bei denen eine drastische Reduktion des αDystroglykan nachgewiesen wurde. Obwohl die β-Dystroglykan Expression bei unserem Patienten erhalten war, könnte dieses integrale Membranprotein in der Relation zum extrazellulären Ankerprotein α-Dystroglykan nicht richtig positioniert sein. Eine Reduktion von von α-Dystroglykan ist dabei nicht ausschließlich auf Muskeldystrophien beschränkt, sondern kann auch als sekundäre Veränderung bei nicht dystrophen Muskelerkrankungen vorkommen. Es könnte in solchen Fällen ein unspezifisches Phänomen aufgrund einer sekundären Protein-Degradation darstellen. 66 Hinweise auf altersabhängige Expressionsveränderungen (Kawabe et al. 2001) von Caveolin-3 deuten auf eine mobile Homeostase der Interaktion mit nNOS und verschiedenen Signalmolekülen [Proteinkinase C (Oka, Yamamoto et al. 1997), G-Proteine (Li, Okamoto et al. 1995), Adenylatcyclasen (Schwencke, Okumura et al. 1999)] hin. Geringe Krankheitsprogredienz und frühe Manifestation sprechen jedoch eher gegen starke altersabhängige Effekte in vivo beim Menschen. Perspektiven und Bedeutung der Caveolinopathien Die neuen genetischen Befunde werfen Fragen nach einer speziellen Funktion von Caveolin-3 innerhalb des Muskelkontraktionsablaufes auf. Verminderung von Caveolin-3 assoziiert mit einer gesteigerten nNOS-Induzierbarkeit könnten zur Übererregbarkeit der Muskulatur bei RMD führen. Eine gesteigerte Synthese von NO und/oder eine sekundär gesteigerte Ca2+-Freisetzung nach mechanischer Muskelstimulation könnten funktionell bedeutend sein. Der genaue Mechanismus bleibt jedoch weiterhin unklar. Zukünftige Untersuchungen der Caveoline und der Proteine, die mit ihnen interagieren, könnten diese pathophysiologischen Zusammenhänge des eigentümlichen Phänotyps von RMD weiter erhellen. 4.2 Methoden 4.2.1 SSCP-Analyse 4.2.1.1 Single Sequence Conformation Polymorphism – Methode Die Erkennung einer Punktmutation innerhalb eines DNA-Fragments durch die veränderte Konformation des zugehörigen DNA-Einzelstrangs erfolgte erstmals durch Kanazawa bei der H+ATPase in Escherichia coli (Kanazawa, Noumi et al. 1986). Orita führte diese Methode den Begriff der Single Sequence Conformation Polymorphism – (kurz SSCP) Detektion ein (Orita, Iwahana et al. 1989). Die Methode beruht auf dem theoretischen Prinzip, dass unter bestimmten Bedingungen Nukleinsäuren, genauer die Einzelstränge von Nukleinsäuren, bestimmte Sekundärstrukturen in Lösungen annehmen. Die Sekundärstruktur hängt von der Basenzusammensetzung ab und kann durch Austausch einzelner Basen verändert werden, die elektrophoretische Mobilitäts- Unterschiede bei nichtdenaturierenden Bedingungen bewirken (siehe Abb 25). In der Original-Arbeit untersuchte Orita restringierte DNA-Fragmente, die er durch eine alkalischen Lösung zu Einzelsträngen denaturierte. Nach der Elektrophorese auf einem neutralen, nichtdenaturierenden Polyacrylamidgel wurden die Einzelstränge durch Hybridisierung dargestellt. In der zweiten Arbeit fand die radioaktive Labelung der Primer bei einer Polymerase-Kettenreaktion Verwendung, die die Methode insgesamt schnell und handlich machte und zu ihrer weiten Verbreitung und Anwendung führte. Die SSCPs wurden zur Charakterisierung hochrepetitiver Alu repeats, Detektion von Punktmutationen in verschiedenen Onkogenen (Mashiyama, Murakami et al. 1991), Gerinnungsfaktoren (Sarkar, Yoon et al. 1992), der Tay-Sachs-Krankheit (Ainsworth, Surh et al. 1991), zystischen Fibrose (Ravnik-Glavac, Glavac et al. 1994), dem LDL-Rezeptor (Humphries, Gudnason et al. 1997), u.a. eingesetzt. 67 Dabei wurde bei der Herstellung der Einzelstränge der DNA die Verwendung von alkalischen Detergentien durch die einfachere Hitzedenaturierung abgelöst. Bei der Auswertung der Polyacrylamidgele wurde auf Ethidiumbromid- bzw. Silberfärbung sowie auch auf Fluoreszenzmarkierung anstelle der toxischeren Autoradiographie zurückgegriffen. Am Grundprinzip der Renaturierung denaturierter Einzelstränge innerhalb einer PolyacrylamidGelmatrix änderte sich jedoch nichts. Deshalb ergibt sich dasselbe elektrophoretische Bild: Ein Abb 25: Modell zur Erklärung des Elektrophoresebilds bei der SSCP-Analyse kleiner Anteil der Einzelstränge bildet wieder Doppelstränge und läuft den nachfolgenden Einzelsträngen voran (Ainsworth, Surh et al. 1991). Die Einzelstränge haben ihr morphologisches Substrat in mindestens zwei distinkten Banden des Elektropherogramms, die für die Konformation je eines Stranges – Sense und Antisense – stehen. Befindet sich jetzt ein monoallelischer Basenaustausch innerhalb des untersuchten Sequenzabschnitts, so führt dies in der Regel zu zwei zusätzlichen, also zu insgesamt vier Banden. Sowohl im Sense- als auch im Anti-Sense-Strang befindet sich eine Veränderung der Primärstruktur, die auch eine geänderte Konformation der Sekundärstruktur gegenüber dem Wildtyp bewirkt. 4.2.1.2 SSCP-Etablierung und Blind-Testung In unserem Blindtest der SSCP-Analyse konnten wir drei verschiedene Mutationen von 13 Mutationen zuerst nicht detektieren (P129T, W267R, P291FsinsC). Weil alle diese Mutationen auf zwei PCR-Fragmenten (Exon 2 und 4) verteilt sind, sind zwei Erklärungen möglich. Zum einen handelt es sich beim vierten PCR-Fragment aufgrund dessen überdurchschnittlich hohen GC-Gehalt (lokaler GC-Gehalt ±40 Bp: 76% vgl. (Lander, Linton et al. 2001)) um einen DNA-Abschnitt mit besonders hoher Schmelztemperatur. Die besondere Stabilität von Sekundärstrukturen führt seltener zu Konformationsänderungen und somit zu sequenz-abhängigen Sensitivitätseinbußen. Die relativ niedrige Sensitivität ist in solchen Bereichen daher nur durch eine 68 hohe Variation der Laufparameter und einem damit verbundenen erhöhten experimentellen und finanziellen Aufwand zu verbessern. Außerdem ist diese Optimierung erst möglich, wenn die Mutationen bekannt sind. Zum anderen ist eine Abnahme der Sensitivität für PCR-Fragmente beobachtet worden, die eine Länge von über 150 bis 200 Basenpaaren haben (Sheffield, Beck et al. 1993). Bei unserem Ansatz zeigten Veränderungen von Exon 2 und 4 die niedrigste Sensitivität. Diese beiden Fragmente stellten mit 390 und 397 Bp auch zwei der längsten PCR-Produkte dar. Die Fragmentlänge hätte also bei der Auswahl der Primer stärker in Betracht gezogen werden müssen oder hätte den zusätzlichen Verdau durch ein Restriktionsenzym erfordert. Die niedrige Spezifität war ein Hauptproblem unserer Methode. Sie wird durch die hohe Anzahl der „falsch positiven“ Mutationen verursacht und ist aufgrund der eigentlich überflüssigen Sequenzierungen, die zur Validierung der Diagnose doch noch letzten Endes nötig sind, für einen deutlich erhöhten finanziellen Aufwand verantwortlich. Problematisch stellte sich hierbei das Primer-Design in Bezug auf die Position der Exon-Intron-Grenzen dar, weil eine große Anzahl der Polymorphismen des HNF1α Gens (siehe Tab 7) nahe dieser Übergänge lokalisiert ist, die codierende Sequenz aber in der Sequenzierung vollständig lesbar sein sollte. 4.2.1.3 Beobachtete Einflussfaktoren der SSCP-Analyse Temperatur Abgesehen von dem Effekt, den die Temperatur auf die Stabilisierung bestimmter Konformationen der DNA-Fragmente hat (Orosz und Wetmur 1977), ist in der Literatur von einer zunehmenden Schärfe der Banden der SSCP-Auftrennung bei niedriger Temperatur berichtet worden (Glavac und Dean 1993). Zumindest aber aus Gründen der Reproduzierbarkeit ist eine standardisierte Kontrolle der Temperatur zu fordern (Hayashi 1992). Dabei wurden trotz intensiver Konvektion klimatisierter Raumluft Schwankungen gemessen. Hier führten diese zur Benutzung eines wassergekühlten Elektrophoresesystems mit Kerosin zur Isolation in der Interphase. Temperaturkontrollen während der Elektrophorese zeigten eine Temperaturkonstanz ±0,5 °C. Gelmatrix Untersuchungen mit komplementären Einzelsträngen haben ergeben, dass eine bessere Auftrennung in einer Polyacrylamid-Matrix mit einem geringen Anteil von Quervernetzungen, d.h. wenig N,N’-methylenbisacrylamid- gegenüber Acrylamid-Monomeren gegeben ist (Hayashi und Yandell 1993). Die Polyacrylamid Fasern in einem Gel mit 5% Quervernetzung haben die festeste Zwischenraumanordnung, so dass die Fasern mehr Bewegungsfreiheit in einer Matrix niedriger oder höherer Quervernetzung haben (Chrambach und Rodbard 1971). Daher wurde wegen der größeren Flexibilität hier eine niedrigere Konzentration von Quervernetzern verwendet. Die fertige MDE-Lösung bot neben der leichteren Handhabung noch den Vorteil der im Ferguson-Plot nachgewiesenen Volumenradiuskonstanz bei hoher Ladungsdichte (Pulyaeva, Zakharov et al. 1994). Die Entgasung des Ansatzes vor der Polymerisation und eine geübte Technik des Gießens stellte sich nach Durchschreiten einer Lernkurve als wichtige Voraussetzung für ein gleichmäßiges Laufmuster heraus. 69 Laufpufferkonzentration Die Ionenkonzentration des TBE-Laufpuffers hatte in den Vorversuchen nachweislichen Einfluss auf das Bandenmuster, die Trennschärfe und Laufweite. Weil Veränderungen der Salzkonzentration mit Veränderungen der Konduktivität und so der Hitzeproduktion einhergehen, handelt es sich um eine sperrige und unhandliche Variable bei der Optimierung, die wiederum indirekte Auswirkungen auf die übrigen Einflussfaktoren hat. Daher wurde der TBE-Puffer in der Kammer nach drei Gelläufen regelmäßig erneuert, um Veränderungen durch Verdunstung des Wassers oder Auswaschen von Salzen auszuschließen. Detergentienkonzentration Der Zusatz von neutralen Bestandteilen wurde in den bisherigen Anwendungen der SSCPMethode häufig variiert (Yip, Hopkinson et al. 1999). Dabei wurde für Glycerin eine bessere Auftrennung beschrieben. Für die Veränderungen des Trennverhaltens wird hauptsächlich die schwach denaturierende Wirkung der Hydroxyl-Gruppen verantwortlich gemacht. Im Vergleich dazu erscheinen Glucose, Butanol und Harnstoff weniger geeignet (Glavac und Dean 1993), während Sucrose einen leichten Vorteil zu haben scheint (Danenberg, Horikoshi et al. 1992). In den Vorversuchen erbrachte Glycerin keinen generellen Vorteil, so dass es nur vereinzelt bei bestimmten Fragmenten verwendet wurde. Fragmentlänge Kein Einflussfaktor der SSCP-Analyse ist so klar erwiesen wie die Länge des zu untersuchenden Fragments. Statistisch gesehen trifft eine Sequenz von vier Basenpaaren alle 256 Nukleotide auf ihr komplementäres Korrelat, wenn man ungleichmäßige Verteilungen der Basen vernachlässigt. Darüber erklärt sich von der Theorie her der stabilisierende Einfluss auf die Konformation und die sinkende Sensitivität mit steigender Fragmentlänge. Unterschiede im Größen/Ladungsverhältnis spielen bei größeren Fragmenten eine immer kleinere Rolle. Experimentell gibt es Untersuchungen mit unterschiedlich gelegenen Primern (Sheffield, Beck et al. 1993), wobei sich auf diesem Weg der Längeneffekt nie vollständig von der Sequenzabhängigkeit trennen lässt. Auch die Trennzeit hängt von der Fragmentgröße ab, wobei durch die problemlos automatisierte Elektrophorese über Nacht in unserem Versuchsaufbau kein größerer Nachteil hieraus resultierte. Die Art und Position der Mutation spielt im individuellen Fall wahrscheinlich eine gewisse Rolle. Ein genereller Trend – unabhängig von der untersuchten Sequenz – ist jedoch diesbezüglich genauso wenig beobachtet worden wie für die Art des Basenaustauschs (Miterski, Kruger et al. 2000). Variationen Der in einer Arbeit beschriebene, negative Einfluss eines Primer-Überschusses (Cai und Touitou 1993) konnte nicht bestätigt werden. Ein direkter Vergleich der Probe vor und nach einer Aufreinigung erbrachte keine Unterschiede im Laufverhalten; bei der Etablierung und Optimierung der PCR wurde jedoch ohnehin auf eine effiziente und spezifische Synthese geachtet. Die Detektion mittels noch sensitiverer Färbemethoden kann in Einzelfällen Vorteile bieten. Die Verwendung von Radioaktivität (S35 oder P32) oder Fluoreszenz kann Nebenkonformationen mit geringer Abundanz zeigen. Nach Möglichkeit sollte dann aber die endständige radioaktive Markierung der internen Inkorporation bevorzugt werden, um eventuelle Einflüsse durch 70 ungleichmäßigen Einbau zu vermeiden. Die Silberfärbung bietet dagegen eine gleichmäßige Anfärbung und hat keinen Einfluss auf die Elektrophorese (Merril 1990). Die Detektion auf RNA-Ebene verspricht wegen des größeren Repertoirs an Sekundärstrukturen (zusätzliche 2’-Hydroxyl-Gruppe) Vorteile (Sarkar, Yoon et al. 1992). Sie ist jedoch wie die Kombination mit anderen Methoden mit einer Erhöhung des experimentellen Aufwands verbunden, weshalb hier darauf verzichtet wurde. 4.2.1.3 Nachteile der SSCP-Methode Eingeschränkte Vorhersagbarkeit und unklarer theoretischer Hintergrund Ein Nachteil der hier verwendeten SSCP-Methode ist, dass es im Moment noch nicht möglich ist, vorherzusagen, ob und unter welchen Bedingungen ein PCR-Fragment mit einer Mutation eine andere Konformation einnimmt. Es existiert kein adäquates theoretisches Model zur Vorhersage der dreidimensionalen Struktur der Einzelstrang-DNA; die gefaltete dreidimensionale Struktur ist wesentlich komplexer als die bekannte Organisation einer einfachen Stammspirale oder die durch Kristallographie und NMR-Untersuchungen bestimmte Struktur der tRNA oder einzelner Nukleinsäure-Schleifen (Baxter, Greizerstein et al. 1993). Ähnliche Sequenzen können trotz relativ homologer Primärstruktur zu bemerkenswerten Unterschieden im Laufverhalten führen, wobei die Unterschiede nachweislich auf unterschiedliche Sekundärstrukturen zurückzuführen sind (Nishigaki, Husimi et al. 1986). Am ehesten ist von einer strukturellen Dynamik der Einzelstränge auszugehen. Die Struktur der Einzelstrang-DNA in Lösung kann annähernd als Hybrid von Helizes beschrieben werden, bei denen die Fehlpaarungen der Watson-Crick-Basen-Paarung Unterbrüche, Wölbungen und Loops hervorrufen können. Danach entfalten und konvertieren sich diese semiund unstabilen Strukturen innerhalb relativ kurzer Zeit in andere mögliche Strukturen. Das Spektrum der unterschiedlichen Konformationen ist letzten Endes durch die DNA-Sequenz bestimmt (Nakabayashi und Nishigaki 1996). Dafür spricht auch die Beobachtung, dass das Ausmaß der Verschiebung (Shift) stärker von der Struktur der benachbarten Sequenzen als von der Art des Basenaustausches beeinflusst wird (Glavac und Dean 1993). Es kann immer nur versuchsweise gelingen, über Sequenzmuster-Analysen Annäherungen für stabile Sekundärstrukturen (Kanehisa und Goad 1982) oder mit Hilfe im Rahmen der DGGE entwickelter Algorithmen Indikatoren für die jeweilige intrinsische Stabilität zu finden (Lerman und Silverstein 1987). In Übereinstimmung zu frühen Beobachtungen, dass intramolekulare Interaktionen neben der Watson-Crick-Paarung wichtige Faltungsdeterminanten darstellen, ist die Wirkung der Mutationen auf die Mobilität im Gel oft zwischen den beiden komplementären Strängen sehr unterschiedlich. Darüber hinaus spielen intermolekulare Wechselwirkungen eine wichtige Rolle, die schwer zu berechnen sind. Selbst wenn die dreidimensionale Struktur verlässlich vorhergesagt werden kann, so ist eine Abschätzung der daraus resultierenden elektrophoretische Mobilität nicht möglich. Dabei werden bei aktuellen Modellen der Gelelektrophorese noch Faktoren wie die Steifheit der geladenen Partikelketten und die hydrodynamische, inelastische Interaktion zwischen Gel und Polymersegmenten vernachlässigt (Burlatsky, Oshanin et al. 1990). Aus der beschränkten Vorhersagbarkeit folgt zwangsläufig die Beschränkung auf Empirie bei der Etablierung einer 71 SSCP-Analyse, so dass die experimentelle Erfahrung mit der Methode eine große Rolle spielt. So ergeben sich Bedenken für die Anwendung bei der Suche nach noch nicht bekannten Mutationen und noch wenig untersuchten Sequenzen bei nicht zu vernachlässigender Sequenzabhängigkeit. Notwendigkeit der visuellen Erfassung Den densitometrischen Gel-Auswertungen war und ist die direkte visuelle Auswertung bezüglich der Sensitivität überlegen. Sie stellte eine Einschränkung der Automatisierbarkeit dar und stieß bei der Auswertung komplexer Bandenmuster an ihre Grenzen. Die bisherigen Erfahrungen zeigen, dass häufig auch mehr als eine thermodynamisch stabile Konformation für ein DNA-Fragment existieren, wobei anscheinend keine Mengen/Dosis-Relation über die Bande hergestellt werden kann (Hayashi und Yandell 1993) Die von uns verwandte Silberfärbung ist ein hochsensitives Verfahren, die beschriebene Auflösung kleiner 0,03 ng DNA pro mm2 (Goldman, Dickey et al. 1982) konnte experimentell bestätigt werden. Dennoch befand sich oft die zusätzliche Bande direkt neben der des Wildtyps und war daher nur sehr schwer optisch aufzulösen. Eine Detektion über Fluoreszenz (Boutin, Hani et al. 1997) mit anschließender Auswertung des Elektropherogramms über ein Computerprogramm könnte hier für die Zukunft noch Verbesserungsmöglichkeiten beinhalten. Notwendigkeit einer Bestätigung durch eine spezifische Kontrollmethode Die bisherigen theoretischen Modelle (s.o.) bringen es mit sich, dass eine eindeutige und spezifische Zuordnung einer Mutation nicht möglich ist, weil dasselbe Bandenmuster auch durch eine andere Mutation oder einen Polymorphismus fraglicher Relevanz hätte hervorgerufen werden können. Bei den forensischen Konsequenzen, die für den Probanden und den Analytiker mit einer positiven Mutationsdetektion verbunden sind, ist vor der Diagnosestellung einer hereditären Erkrankung eine Kontrolle mit einer Methode höherer Spezifität zu fordern. Ein ausschließlicher Einsatz dieser Methode im klinischen Bereich muss durch die schwankenden Zahlen der Sensitivität nach unten bis zu lediglich 30% ((Sarkar, Yoon et al. 1992)) in Frage gestellt werden. 4.2.1.4 Vorteile der SSCP-Methode Geschwindigkeit und hoher Probenumsatz Die Arbeitszeit pro SSCP-Gel betrug ca. 2,5 Stunden, die Laufzeit 12, 9 ± 6,7 Stunden. Dabei konnten pro Lauf mindestens 30 Proben aufgetragen werden. In der Beurteilung konnte trotz dieses hohen Probenumsatzes eine schnelle visuelle Auswertung durchgeführt werden. Theoretisch ist es auch denkbar, größere Probenmengen in einer Multiplex-Analyse zu poolen. Bei einer beschriebenen Sensitivitätsgrenze von 3% der Gen-Kopien (Hongyo, Buzard et al. 1993) erscheint dieses Vorgehen nicht nur bei der Suche nach somatischen Mutationen in Onkogenen sinnvoll. Geringe Toxizität Aufgrund der Methode der DNA-Detektion war die Methode nur mit einer geringen toxischen Belastung verbunden. Doch auch bei Verzicht auf Radioaktivität und kanzerogenes Ethidiumbromid ist die Belastung mit dem neurotoxischen Acrylamid unvermeidbar. Durch die Verwendung einer vorgefertigten Lösung ließ sich die Kontaminationsgefahr aber auf ein Minimum reduzieren. 72 Kosten Die laufenden Kosten unserer Methode waren gegenüber anderen Detektionsmethoden gering. Bei der Etablierung der Methode fallen je nach der vorhandenen Ausstattung mit wassergekühlten Elektrophorese-Systemen höhere Investitionen an. Reproduzierbarkeit Bei genauer Kontrolle und Berücksichtigung aller oben genannten Einflussfaktoren war eine 100% Reproduzierbarkeit der Ergebnisse im Intra- und Inter-Gel-Vergleich möglich. Dies bedeutet für etablierte und optimierte DNA-Fragmente eine hohe Sensitivität. Viele Schritte der SSCP-Analyse sind dabei problemlos automatisierbar und standardisierbar bis hin zur densitometrischen Auswertung der Elektropherogramme. Erfahrungen und Ausblick Vor allem gegen Mitte/Ende der 90ziger Jahre erlebte die SSCP-Methode einen Boom, so dass in der Literatur zahlreiche Erfahrungen und Anwendungsbeispiele zu finden sind. Es handelt sich um eine Methode, die für einen großen Probenumsatz geeignet ist. Aufgrund zahlreicher Alternativen, die vor allem in Hinblick auf Kosten und Geschwindigkeit stark aufgeholt haben, ist ihr Einsatz jedoch gegenüber anderen Methoden im Einzelfall abzuwägen. Bei der Frage der Eignung für ein Anwendungsgebiet ist zum einen das Verteilungsmuster der Mutationen innerhalb der codierenden Sequenz oder des zu untersuchenden PCR-Fragments zu berücksichtigen. Eine diffuse Verteilung der pathogenen Mutationen über das gesamte Gen wie z.B. beim HNF1α-Gen verlangt für nahezu jede Basenposition eine separate Optimierung. Wegen des damit verbundenen experimentellen Aufwands scheint vorerst die Direkt-Sequenzierung für das HNF1α-Gen Methode der Wahl zu sein. Bei einzelnen Fragmenten und Sequenzveränderungen dürfte die SSCP-Analyse dagegen hohe Reliabilität und Detektionsraten unabhängig von der Art des Basenaustausch bieten. 4.2.2 Kopplungsanalyse 4.2.2.1 Repetitive DNA und Mikrosatelliten Nach Dichtegradientenzentrifugation lässt sich die DNA in eine Hauptbande und drei zusätzliche Satelliten-Banden auftrennen. Neben den klassischen Satelliten (100-6500 Bp Repeats) und Minisatelliten (20-100 Bp Repeats) stellen die Mikrosatelliten mit 1 bis 10 Bp Repeats die häufigste Form repetitiver DNA dar. Davon sind die meisten tandemartig aufgebaut und enthalten Di-, Trioder Tetranukleotide. Eine genaue Funktion dieser einfachen Tandemwiederholungen (simple tandem repeats kurz STS) ist noch nicht bekannt. Kopplung und Linkage Weil Mikrosatelliten eine mittlere Heterozygotie über 70% und ein durchschnittliches Auftreten alle 0,7 cM aufweisen, stellen sie hochinformative Marker dar. Damit bieten sie nicht nur die Möglichkeit der Identifikation eines Individuums (Forensik, Paternitäts-Nachweis) und der Erforschung genetischer Distanz unterschiedlicher Organismen, sondern ermöglichen auch die Erkennung krankheitsrelevanter Gene. Bei der Linkage-Analyse prüft man innerhalb einer Population den Zusammenhang (allelische Assoziation) zwischen bestimmten Allelen des Markers und dieser Krankheit. Dagegen ist bei der Kopplungsanalyse die gemeinsame Transmission von 73 Allelen eines genetischen Markers und der interessierenden Krankheit innerhalb eines Familienstammbaums Ziel der Untersuchung. Dazu muss die Position des Gens (Locus) auf dem Chromosom bekannt sein. Zur Wahl des geeigneten Mikrosatelliten stehen umfassende genetische Karten des humanen Genoms zur Verfügung (Dib, Faure et al. 1996) (Broman, Murray et al. 1998). Liegt in einer Familie eine nichtzufällige Assoziation von Genen und Allelen in Abweichung ihrer nach dem Hardy-Weinberg-Äquilibrium erwarteten Frequenz vor, so spricht man vom sogenannten Kopplungsungleichgewicht oder Linkage Disäqulibrium. Über dieses Ungleichgewicht für einen Locus können Rückschlüsse auf seine Bedeutung für die Krankheitsentwicklung gezogen werden. 4.2.2.2 Praktische Durchführung und Probleme bei der Interpretation des Bandenmusters Bei der Auswertung der Gele musste zum einen der Effekt von sogenannten Stotterbanden berücksichtigt werden. Der Effekt, der bei der Replikation überhaupt zur Polymorphie und damit zum hohen Informationsgehalt der Tandem-Repeats führt (Stephan 1989), bewirkt bei der Polymerase-Kettenreaktion in vitro eine Amplifikation verschiedener Nebenprodukte, hauptsächlich mit niedrigerer Wiederholungszahl der Repeats. Man erklärt sich diesen ungewollten Effekt über ein Verrutschen der Polymerase entlang des Matrizen-Strangs. Die vollständige Denaturierung durch Harnstoff führt dazu, dass der kodierende und komplementäre Strang wegen ihrer unterschiedlichen Basenzusammensetzung und des daraus resultierenden Molekulargewichts bzw. Ladung/Masse-Verhältnisses voneinander aufgetrennt wurden. Setzt man für jede Base des jeweiligen Strangs die genaue Masse (Adenin: 312,2 Da, Cytosin: 288,2 Da, Guanin: 328,2 Da und Tyrosin: 303,2 Da) ein, kann man den absoluten Unterschied des Molekulargewichts der Einzelstränge berechnen. Über das durchschnittliche Gewicht einer Base von 307,95 Da wurde der relativen Laufunterschied der beiden Fragmente bestimmt; vier Hauptbanden konnten so zwei Allelen zugeordnet werden (siehe Abb 26). Nur bei relativen Unterschieden, die ein Vielfaches der Wiederholungseinheit eines Mikrosatelliten betrugen, ergaben sich Probleme bei der Interpretation des Bandenmusters, weil hier die Kombination von Stotterbanden, Allelen und unterschiedlichen Strängen eine genaue Zuordnung erschwerte und die Abschätzung allein aufgrund der Intensität der Banden wegen der mangelnden Linearität nicht immer möglich ist. In diesem Fall war aber oft eine asymmetrische PCR hilfreich, um die Zuordnung eines Einzelstrangs herbeizuführen. Prinzipiell ist für die Zuordnung der Allele auch die Interpretation eines Strangs ausreichend. Abb 26: Beispiel für die Zuordnung des Haplotyps. Die beiden Stränge haben hier einen physikalischen Gewichtsunterschied von 1217 Dalton, was einem mittleren Basengewicht von 3,95 Basen entspricht. 74 Berechnung des Lod Score Zur Überprüfung eines familiären Kopplungsungleichgewichts (Linkage Disäquilibrium) stehen verschiedene statistische Auswertungstests zur Verfügung. Hier wurde auf die aufwendigere Berechnung des TDT (transmission disequilibrium test nach (Spielman, McGinnis et al. 1993)) oder eines Multipoint-Lod Score verzichtet, weil angesichts der wenigen Familienmitglieder und ungesicherter Marker-Gen Linkage die Aussagekraft quantitativer Berechnungen von vorneherein vermindert war. So wurde stattdessen mit einem einfachen Modell eines Mendel’schen Erbgangs und geringerer Rechner-Leistung das familienbasierte Haplotyp-Risiko bestimmt (Zwei-Punkte Lod Score nach Lathrop und Lalouel (1984)). Um mit einer Fehleinschätzung der Allelfrequenzen eine der Hauptfehlerquellen zu vermeiden, wurden die Daten mit großen kaukasischen Populationen abgeglichen (s.o.). Auf die weitere Charakterisierung des Krankheitsstatus – wie z.B. bei der Duchenne´schen Muskeldystrophie erfolgreich mittels der CK-Werte angewandt - wurde verzichtet, weil die CK-Erhöhungen der betroffenen Familienmitglieder vergleichbar waren. Aufgrund des hohen Anteils von Betroffenen an den Familienmitgliedern und des Vererbungsmusters innerhalb der Familie wurde ein autosomal dominantes Modell mit hoher Penetranz (0.99) zugrunde gelegt. 4.2.2.3 Theoretischer Hintergrund Anforderungen an die Familie Die Familie stellt bedingt die Voraussetzungen zur Durchführbarkeit einer Kopplungsanalyse zur Verfügung. Da es für die Fragestellung auf die meiotische Kopplung ankommt, ist die Anzahl der Meiosen ein ganz wichtiges Kriterium; bei der einzelnen Meiose ist letzten Endes die Bestimmung der chromosomalen Phase entscheidend. Es müssen also möglichst viele betroffene und nicht betroffene Familienmitglieder typisiert werden. Für D16S3093 war durch die Charakterisierung von I.2 auch ohne die Daten von I.1 bei II.5 das väterliche und mütterliche (= das mit dem Phänotyp kosegregierende) Allel zuzuordnen. Vorteil der familiären Erfassung ist die niedrige Generationszeit. So ist eine wesentlich niedrigere Rekombinationswahrscheinlichkeit anzunehmen als in intrafamiliären Untersuchungen. Außerdem ist von einer niedrigeren Heterogenität der Erkrankung auszugehen; die Wahrscheinlichkeit für dieselbe Form einer Erkrankung ist sehr hoch. Wegen der Größe der Familie darf insbesondere bei einer seltenen Erkrankung Unilinealität angenommen werden. Aus demselben Grund kam eine konditionale Simulation (Feststellung beobachteter Linkage-Ereignisse unter der Annahme einer Linkage) nicht in Frage. Bei nur acht charakterisierten Familienmitgliedern muss von einer großen Anfälligkeit gegenüber Fehldiagnosen und Genotypisierungs-Fehlern ausgegangen werden. Ein negatives Ergebnis wie hier im Sinne eines Ausschlusses hat eine größere Aussagekraft als die Möglichkeit einer Kopplung, weil innerhalb eines 3-Generationen-Stammbaums die Wahrscheinlichkeit für eine zufällig gefundene Kosegregation höher ist als für ein rekombinatorisches Ereignis ohne Trennung von Risikoallel und Phänotyp. Auch wenn bei so einem seltenen Befund (HyperCKämie bei nachgewiesener PhK-Defizienz) von keiner Familien-Auswahl im eigentlichen Sinne mehr gesprochen werden kann, müssen BiasEffekte berücksichtigt werden. Aussagen über ähnliche Krankheitsbilder sind nicht möglich. 75 Anforderungen an den Mikrosatelliten Die möglichst große physikalische Nähe der Loci von Marker und Gen kann heute als eines der Hauptkriterien nicht mehr nur aus indirekten zytogenetischen Rekombinationskarten (Distanz in cM angegeben) entnommen werden (Lander, Linton et al. 2001). Mit Fortschreiten des Humanen Genomprojekts stehen immer detailliertere Informationen über die exakte Lokalisation der Gene und short tandem repeats zur Verfügung. Bei der Planung wurden zwar nicht nur Karten nach der Radiation-Hybrid-Methode [GeneMap 99-G3 (Deloukas, Schuler et al. 1998), NCBI RH (Agarwala, Tab 15: Lokalisation der untersuchten Mikrosatelliten Genethon (Dib, Faure et al. 1996) (Abstand von Spitze) PHK β D16S409 D16S3093 PHK γ1 D7S478 D7S492 D7S669 D7S2429 Contig (LocusLink) 56,2 cM 50,8 cM 44.225-44.469 kBp 115.000 kBp 25.705 kBp 69,4 cM 100,5 cM 90,9 cM 77,7 cM 57.273-57.285 kBp 46.102 kBp nicht bekannt 75.852 kBp 60.420 kBp Applegate et al. 2000)] verwendet, sondern mit Locus Link (Pruitt und Maglott 2001) über die zytogenetischen Banden Mikrosatelliten ausgewählt [ Genethon (Dib, Faure et al. 1996), (Broman, Murray et al. 1998)]. Die aktuellsten Informationen nach Erstellen einer physikalischen Karte (Lander, Linton et al. 2001) und Zuordnung einzelner benachbarter Klone (Contigs) nach Sequenzierung weisen für diese Anwendung auf beträchtliche Distanzen der Marker zueinander hin. Dadurch wird die Aussage einer Kopplungsanalyse wesentlich eingeschränkt. Zu bemängeln ist, dass für die benutzten Mikrosatelliten jeweils keine Linkage zu den Genen der PhKUntereinheiten nachgewiesen wurde. Für sämtliche hier angewendeten Polymorphismen war ein hoher Informationsgehalt (polymorphism information content kurz PIC als Maß für die Heterozygotie) bekannt. Fast alle Mikrosatelliten wiesen eine starke Multiallelität auf, die als wichtiges Marker-Kriterium zu einer hohen Aussagekraft beiträgt (Sham, Zhao et al. 2000). Die niedrige Heterozygotie innerhalb der charakterisierten Familie führte bei nur drei gefundenen Allelen von D7S478 und D16S409 dazu, dass keine Aussage bezüglich einer Assoziation dieser beiden Mikrosatelliten getroffen werden konnte. Die Mutationsrate der hier amplifizierten Mikrosatelliten war nicht bekannt; bei der Berechnung wurde jedoch von einem durchschnittlichen Wert ausgegangen. 4.2.2.4 Beurteilung Die Anwendung einer intrafamilliären Kopplungsanalyse ist in der praktischen Durchführung mit geringem, experimentellen Aufwand verbunden, wenn die Bedingungen für den Mikrosatelliten etabliert sind und Hintergrundinformationen über Allelfrequenzen, PIC und Vorliegen eines HardyWeinberg Äquilibriums zur Verfügung stehen. Die Methode ist dann vor allem der direkten Analyse überlegen, wenn es sich bei dem Kandidatengen um eine komplexe Genstruktur handelt oder eine Charakterisierung des Gens selbst überhaupt noch nicht erfolgt ist. Dabei würden auch intronische Veränderungen ein Disäquilibrium ergeben, die bei einer alleinigen Betrachtung des kodierenden Bereichs, auf den in der Regel Analyse beschränkt bleibt, übersehen würden. 76 Nachteilig zu beurteilen sind dagegen die hohen Anforderungen an die Familie und verwendbaren Marker. Der sensible Umgang mit zahlreichen Fehlerquellen ist angesichts der unhandlichen, mathematischen Modelle essentiell. Ohne in großen Kollektiven nachgewiesene Linkage zwischen Marker und Gen ist die Aussagekraft einer Kopplungsanalyse stark eingeschränkt. Bei dem Kenntniszugewinn und den wachsenden Ressourcen der Sequenzierung verliert die Methode zunehmend an Bedeutung. Bei bestimmten Anwendungen stellt die indirekte Diagnostik (Epplen, Buitkamp et al. 1995) über ein Kopplungsungleichgewicht dennoch weiterhin eine attraktive Alternative dar – vor allem, wenn ohnehin eine größere Menge von Mitgliedern einer Familie charakterisiert werden soll und die Methode für einen polymorphen Mikrosatelliten mit nachgewiesener Linkage zu einem unbekannten Gen oder Gen mit komplexer Genstruktur etabliert ist. Diesbezüglich bieten die neu gewonnen Informationen der physikalischen Kartierung (Lander, Linton et al. 2001) eine stark verbesserte Ausgangsposition bei der Auswahl der Marker. 4.2.3 Direktsequenzierung 4.2.3.1 Direktsequenzierung – Entwicklung der Methode Die Bestimmung der Sequenz der DNA hat sich zum wesentlichen Grundstein der modernen Molekularbiologie und –medizin entwickelt. Von den ursprünglich zwei Verfahren hat sich die enzymatische Methode von (Sanger, Nicklen et al. 1977) gegenüber der chemischen Degradation der DNA nach (Maxam und Gilbert 1977) durchgesetzt, spätestens seit der Einführung der Polymerasekettenreaktion (PCR). Die Methode beruht auf dem Prinzip, dass bei der Synthese Didesoxy-Nukleotide in den wachsenden Strang inkorporiert werden, die keine Bindungsstelle für Hinzufügen weiterer an die Kette durch die Polymerase besitzen und so zum Abbruch der Verlängerung führen. Technische Innovationen wie die auf Fluoreszenz-Farbstoffen basierte Detektion führten zu stärkerer Automatisierung gegenüber dem radioaktiven Nachweis (Smith, Sanders et al. 1986). Die Entwicklung thermisch stabiler Polymerasen ermöglichten eine zyklische Erhitzung in einer PCR-Maschine (Griffin und Griffin 1993). Nach Steigerung der Effizienz des Einbaus fluoreszenzmarkierter Analoga (Tabor und Richardson 1995) mit Verwendung vier verschiedener Farbstoffe in einer Reaktion, die durch einen Laser automatisiert erfasst werden, ließen sich Veränderungen sensitiver und standardisiert erfassen. Die Sensitivität ließ sich durch die Verwendung von Rhodamin-Farbstoffen noch weiter steigern (Parker, Zakeri et al. 1996), so dass die direkte Sequenzierung eines PCR-Amplifikats aus genomischer DNA ohne zusätzliche Klonierungs- oder Aufreinigungsschritte möglich wurde (Kilger und Paabo 1997). Bei dieser Mutationsanalyse sollten zwei unterschiedliche Sequenzierer eingesetzt und verglichen werden. Der ABI PRISM 377 ist der am häufigsten benutzte Sequenzierer und beruht auf einer vertikalen PAGE-Elektrophorese von bis zu 40 verschiedener Proben gleichzeitig, während der ABI PRISM 310 auf dem Prinzip der Kapillarelektrophorese durch ein Biopolymer (POP6) beruht. Bei der Sequenzierung wurden cDNA- als auch genomische DNA-Amplifikate als Template eingesetzt unter Verwendung desselben Sequenzierungs-Kits (Big Dye Terminator Kit, Applied Biosystems). 77 4.2.3.2 Vorteile und Nachteil der Methode Nachteile Die Nachteile der Direktsequenzierung sind zum einen methodisch bedingt. Fehlerraten bei der PCR bedingen Einschränkungen - hauptsächlich der Spezifität; abhängig von der Menge des Ausgangsmaterials (10-100 ng), der Fragmentlänge (hier 175-508 Bp), der Zyklenzahl (hier 30-35) und der Fehlerrate der Polymerase (10-7) können artifizielle Substitutionen erfolgen (Reiss, Krawczak et al. 1990). Statistische Fehler dieser Art konnten durch Wiederholung der Reaktion oder Überprüfung mittels einer zweiten Methode z.B. RFLP minimiert werden. Zur Kontrolle wurde eine Mutation immer durch Sequenzierung in beide Richtungen überprüft. Polymorphismen innerhalb der Primer-Bindungs-Sequenz, die selbst eine pathogene Mutation darstellen oder mit einer pathogenen Veränderung kosegregieren, können eine Amplifikation des mutierten Allels verhindern, so dass eine Verringerung der Sensitivität resultiert. Dieser Sachverhalt ist für HNF1α beschrieben („allelic drop-out“ (Ellard, Bulman et al. 1999)), hier aber nicht nachgewiesen worden. Bei der Analyse von cDNA ist die Abhängigkeit einer effizienten Amplifikation beider Allele von besonders großer Bedeutung. Zeichen der Heterozygotie konnten hier dazu genutzt werden, eine Erfassung beider Allele in der Analyse zu überprüfen. Ganz wesentliche Voraussetzungen der Direktsequenzierung war die Kenntnis der Sequenz und mögliche Etablierung einer spezifischen PCR. In dieser Grundvoraussetzung können großer experimenteller Aufwand bei einer komplexen Genstruktur oder fehlende Durchführbarkeit bei hochpolymorphen Sequenzen begründet sein. Theoretisch muss auch die Existenz von Pseudogenen berücksichtigt werden. Probleme der Lesbarkeit des Elektropherogramms ergeben sich insbesondere bei Insertionen oder Deletionen. Diese Veränderungen führten zu einem Überlappen der Signale beider Allele (siehe Abb 27), was bei einer Verschiebung um einige Bp die Auswertung sehr erschwerte. Hier stellte sich die Primer-Wahl als wichtiges Kriterium heraus. So erfasste das Primerpaar Cav1-Cav2 eine intronische Sequenz polymorpher Repetitionen, was durch die diffusen Signale den Ausschluss eines möglichen Basenaustausches unmöglich machte. Wesentlich für den zeitlichen Bedarf, den die Mutationsdetektion mittels Direktsequenzierung in Anspruch nahm, war die Auswertung der Elektropherogramme. Auch wenn Auswertungs-Software (Align Plus 3.0, ABI Genescan 672 1.1) zur Verfügung stand, musste für eine ausreichende Sensitivität die Sequenzierungs-Qualität manuell und die detektierten Signale einzeln visuell beurteilt werden. In bisherigen Vergleichen führt dieser Punkt zu einem Nachteil bei der Kosteneffizienz gegenüber anderen Methoden in der Routineanwendung (Boutin, Vasseur et al. 2001). Ansonsten sind die Kosten wegen der unterschiedlichen Grundausstattung und dem wichtigen Faktor der Personalkosten nur schwer zu vergleichen. In unseren Untersuchungen konnten die Kosten der Direktsequenzierung durch Einsetzen der Hälfte des Sequenzierungs-Kits reduziert werden. 78 Abb 27: Durch Direktsequenzierung in beide Richtungen lässt sich die Deletion S426delGC im Glucokinase Gen gut lokalisieren. Vorteile Der große Vorteil der Direktsequenzierung ist, dass es sich um eine direkte Methode zur Mutationsdetektion handelt. Dabei ist durch die Information über die Art der Mutation schon eine Abschätzung der pathophysiologischen Auswirkung möglich. Bei sogenannten „neutralen“ oder „stummen“ Veränderungen ist die unveränderte Proteinexpression aus der Primärstruktur der Nukleinsäure festzustellen. Veränderungen der Aminosäuresequenz deuten über ihren Konservierungsgrad und die Position innerhalb einer bestimmten Funktionsdomäne auf eine funktionelle Auswirkung hin, auch wenn im Zweifelsfall der eindeutige Funktionsausfall noch nachgewiesen werden muss. Die Sequenzierung der cDNA hatte bei komplexeren Genstrukturen wie der des GBE-Gens den Vorteil der größeren Geschwindigkeit und Erfassung möglicher SpliceMutationen. Voraussetzung ist dann aber eine ausreichende Expression unter Berücksichtigung gewebespezifischer Effekte. Gegenüber neueren Methoden, die über Hybridisierung von Oligonukleotiden auf DNA-Chips (Lipshutz, Fodor et al. 1999) oder fluoreszenzmarkierten Nukleotiden spezifisch und punktgenau Mutationen im großen Maßstab erkennen können, bestehen bei der Direktsequenzierung eine größere Erfahrung und zahlreichere Ressourcen. Für die etablierte Methode sind weniger bioinformatische Grundlagen und mathematische Weiterverarbeitung der Daten nötig. Die neueren Fluoreszenz-Farbstoffe und moderne Argon-Ionen-Laser-Generation bieten eine hohe Detektionsgrenze, so dass eine Template-Menge von 10-20 ng bei der einfachen und schnellen Ethanol-Elution (Hogdall, Boye et al. 1999) ausreichende Signale lieferte. Ein Vorteil der direkten Sequenzierung bei der praktischen Durchführung lag in der Beschränkung auf zwei Reaktionsgefäße. Dieser Umstand ist deshalb von Bedeutung, als die Kontamination von DNA, das Vertauschen von Proben und die fehlerhafte Zuordnung zu Patienten als die wesentlichen Fehlerquellen in der Analyse gelten und somit minimiert werden sollte. 79 4.2.3.3 Vergleich der Sequenzierer Der Sequenzierer ABI PRISM 310, der auf dem Prinzip der Kapillarelektrophorese durch ein Biopolymer beruht (Hogdall, Boye et al. 1999), bot vor allem den Vorteil der höheren Geschwindigkeit. Eine einzelne Probe konnte ohne Vorbereitung eines Gels innerhalb einer guten Stunde anhand des Elektropherogramms analysiert werden. Dagegen war der mögliche Probenumsatz pro Zeit mit dem ABI PRISM 377 wesentlich höher. Hier zeichnete sich die Qualität und das Signal/Hintergrund-Verhältnis durch eine höhere Zuverlässigkeit aus. Nach einer bestimmten Anzahl von Elektrophorese-Läufen ließ die eindeutige Interpretierbarkeit des BasenMusters bei den Kapillaren nach, so dass diese regelmäßig erneuert werden mussten. Der Nutzen einer geringeren, benötigten Probenmenge, die zur Detektion benötigt wurde, machte keinen bedeutenden Vorteil aus. So überstieg nämlich auch beim ABI PRISM 377 Probenmenge diese Größenordnung kaum. Das verbleibende Eluat wurde nur in Einzelfällen verwandt, und seine Menge stellte nur in Einzelfällen einen limitierenden Faktor dar. 4.2.4 SNP(single nucleotide polymorphismus) und RFLP (restriction fragment length polymorphism) 4.2.4.1 Entwicklung und Hintergründe der Methode 90% der DNA Polymorphismen stellen Single nucleotide polymorphisms (SNPs) dar (Collins, Brooks et al. 1998), die statistisch ca. alle 500 Bp im humanen Genom auftauchen. Pro Individuum entstehen durchschnittlich 100 Basenaustausche neu (Crow 1995), von denen einige durch genetischen Drift eliminiert werden und andere als seltene Varianten (<1% Allelfrequenz) vererbt werden. Die Seltenheit (10-8 auf das gesamte Genom pro Generation) und die Zufälligkeit dieses Ereignisses führen zu einer Stabilität dieser Veränderungen einzelner Basen. So sind 85% in unterschiedlichen Frequenzen in allen Populationen anzutreffen, während 15% populationsspezifisch sind. (Barbujani, Magagni et al. 1997). Trotz wechselnder Verteilungsdichte entspricht die Frequenz innerhalb des codierenden Bereichs insgesamt der in den untersuchten nicht-codierenden Regionen (Cargill, Altshuler et al. 1999). SNPs, die zu einer nicht synonymen Codierung innerhalb der codierenden Sequenz führen, haben durch die Selektion hierbei einen wesentlich kleineren Anteil und niedrigere Allelfrequenzen. 4.2.4.2 Vorteile und Nachteile der Methode Vorteile Der SNP, der in der hier angewendeten Form mit dem sogenannten RFLP (restriction length polymorphism) übereinstimmt, erfüllt in der Mutationsdetektion eine doppelte Funktion. Zum einen können SNPs als Marker der Genotypisierung und Lokalisierung von Genen verwendet werden. Für diesen Zweck besteht Zugriff auf über 7500 SNPs in ständig wachsenden Datenbanken (NIH, HUGO, Whitehead Institut, SNP Konsortium, u.a. (Schork, Fallin et al. 2000) mit einem Intermarker-Abstand, der mit 500 kB eine wesentlich größere Dichte als Mikrosatelliten bietet (Broman, Murray et al. 1998). Eine niedrigere Mutationsrate als bei anderen Formen von Polymorphismen (s.o.) bewirkt eine Erfassung von Locus-Assoziationen, genetischer Distanzen und Kosegregationsphänomene mit höherer Reliabilität (Brookes 1999). 80 Zum anderen können SNPs im kodierenden Bereich direkte biologische Bedeutung besitzen – ganz unabhängig davon, ob mit einer Frequenz unter 1% ihre eigentliche Definition als Polymorphismen nicht mehr erfüllt ist (wie z.B. bei G zu A [R26Q] im Caveolin-3-Gen). Viele Mutationen stellen in diesem Sinne SNPs dar. Durch die Fortschritte des humanen Genomprojekts sind SNPs im kodierenden Bereich identifizierter Gene direkt auf ihre Krankheitsassoziation zu testen. Die hier untersuchten Veränderungen der Nukleotidsequenz waren mit geringem experimentellem Aufwand und schnell auf ihre Frequenz hin in größeren Kollektiven zu testen. Dabei scheinen für die Polymorphismen C zu T [L8] und C zu T [N32] im Caveolin-3-Gen aufgrund einer mit dem Kontrollkollektiv übereinstimmenden Frequenz nicht pathologisch relevant zu sein. Die Transitionen G zu A [R515H] und G zu A [R524Q] im GBE-Gen konnten dagegen bei Normalpersonen nicht identifiziert werden und deuten somit eindeutig auf einen selektionierenden Effekt und auch ohne einen proteinanalytischen Nachweis eines Funktionsverlusts auf einen pathogenen Charakter dieser Mutationen hin. Sämtliche Basenaustausche befanden sich in der Erkennungssequenz eines Restriktionsenzyms, so dass die RFLP-Analyse auch ohne die artifizielle Bildung einer Schnittstelle mittels PCR etabliert werden konnte. Großer Vorteil der Restriktionsanalyse ist die hohe Spezifität. Deshalb kommt die Methode bei häufigen und distinkten Veränderungen der Nukleotidsequenz bevorzugt zur Anwendung. Bei der mitochondrialen Mutation 3243 war die Sensitivität dabei von der Heteroplasmie abhängig, bei bis zu 10% Heteroplasmie konnte mit der verwendeten RFLP-Analyse eine Mutante nachgewiesen werden. Die Berechnung eines Risikos oder die Bestimmung einer indirekten SNP-Assoziation hat in der pränatalen Diagnostik den Vorteil, dass sie die Fragestellung nach der Gefährdung des Kindes beantworten kann, ohne damit automatisch eine ethisch fragwürdige Zuweisung des AnlageträgerStatus an ein Elternteil zu verbinden. Nachteile Die Charakterisierung der SNPs bleibt in der Praxis meist indirekt. So kann bei Verlust einer Restriktions-Schnittstelle keine Aussage über die Art des Basenaustauschs getroffen werden. Ohne die vorherige Kenntnis der Mutation und ihrer Position ist ein Mutationsscreening mittels RFLP nicht möglich, nach Hochrechnungen entdeckt die RFLP-Analyse weniger als 50% der SNPs (Landegren, Kaiser et al. 1988). So können wie hier in der Regel nur vorbeschriebene SNPs auf ihre Abundanz überprüft werden. Die SNPs stellen zum überwiegenden Anteil (<80%) lediglich einen biallelischen Marker dar, obwohl sie theoretisch bis zu vier verschiedene Allele an einer Position aufweisen könnten. Die geringe Anzahl von Allelen erfordert größere Kollektive oder die Verwendung zahlreicher SNPs, um denselben Informationsgehalt wie bei einem hochpolymorphen Mikrosatellit zu erhalten (Sham, Zhao et al. 2000) oder einen Individualitätsnachweis (Chakraborty, Stivers et al. 1999) zu führen. Mit hier benutzten PCR-abhängigen Methoden ist der experimentelle Geschwindigkeitsvorteil marginal; neuere Techniken hätten hier eine schnellere und einfachere Charakterisierung ermöglicht (Lyamichev, Mast et al. 1999). Geht man bei einem SNP– anders als bei den beiden SNPs des Caveolin-3-Gens – von einer Veränderung mit Auswirkung auf Proteinebene und damit von einer für die Krankheit relevanten 81 und kausalen Veränderung aus, ist die Risikoberechnung und Durchtestung jedes einzelnen Polymorphismus ohne Kenntnis dieser Relevanz mit bisherigen Methoden (ANOVA-Test, SNPbasiertes Risiko) nicht ohne weiteres möglich. Das gilt für kodierende SNPs genauso wie für nicht kodierende in potentiell regulativen Elementen. Auch lässt sich die Aussagekraft durch eine höhere Anzahl von SNPs nur begrenzt steigern, so dass bei heterogenen und häufigeren Erkrankungen ohne starke Founder-Effekte die Haplotypisierung großer Kollektive erforderlich ist (Bader 2001). 4.2.4.3 Beurteilung und Zukunftsperspektiven Effektivere Verfahren der SNP-Detektion wie Massenspektrometrie und High performance liquid chromatography (Gut 2001) eröffnen zur Zeit durch eine hohe Effizienz und großen Probenumsatz der schnellen Genotypisierung großer DNA-Banken neue Möglichkeiten. Inzwischen gibt es einige kommerzielle Anbieter, die die Typisierung in großem Maßstab durchführen. Wenn eine Charakterisierung der SNPs z.B. bezüglich der Frequenzen in Subpopulationen in den wachsenden Datenbanken erfolgt ist, werden sie schon allein aufgrund ihrer hohen Dichte (Chakravarti 2001) für repetitive Marker-Elemente eine attraktive Alternative darstellen. 5 Zusammenfassung: Fragestellung: In dieser Arbeit wurden bei vier unabhängigen Krankheitsbildern, Maturity Onset Diabetes of the Young (MODY), Adult Polyglucosan Body Disease (APBD), idiopathischer HyperCKämie und Rippling Muscle Disease (RMD), verschiedene Techniken der Mutationsdetektion evaluiert. Dabei standen Fragen der Verteilung von einzelnen Mutationen, der Genotyp-Phänotyp-Korrelation sowie mutationsbiologische und krankheitsspezifische Besonderheiten im Vordergrund. Methodik: Die auf unterschiedliche Weise extrahierten Nukleinsäuren wurden mittels Polymerasekettenreaktion amplifiziert und mittels Single Strand Conformation Polymorphism (SSCP), Restriction Fragment Length Polymorphism (RFLP) oder Direktsequenzierung auf Mutationen hin untersucht. Bei einer Familie mit Phosphorylase-Kinase-Defizienz wurde eine Kopplungsanalyse mit zwei Phosphorylase-Kinase(PhK)-Untereinheiten durchgeführt. Ergebnisse: In der SSCP-Analyse wurden 17 bekannte MODY-Mutationen detektiert, davon drei erst nach Optimierung der Bedingungen. In einer Blindtestung der Methode war die Spezifität aufgrund von 19 beobachteten, nicht relevanten Polymorphismen reduziert. Bei 12 MODY-Familien wurden Mutationen im Glucokinase-Gen gefunden, davon zehn vorher noch nicht beschriebene (R43H, W99X, G162delG, E216X, C220X, I225F, L304P, S383L, S426fsdelGC und R447del29 zu den bekannten T228M und V203A) und bei 25% in Assoziation mit einem Gestationsdiabetes. Bei sieben deutschen Familien wurden Mutationen im HNF1α-Gen detektiert (G31D, R131W, H143Y, R229Q, R272H, R238insG, P291fsdelC). Beide Untergruppen zeigten keine deutlichen Abweichungen bezüglich des Körpergewichts. Das durchschnittliche Manifestationsalter lag bei den MODY2-Patienten bei 10,4 ± 4,4 im Gegensatz zu 16,1 ± 3,1 Jahren bei MODY3. Die Zuweisung von MODY2-Patienten erfolgte mit 73,7% überdurchschnittlich häufig durch Pädiater. Das Vorliegen der mitochondrialen Mutation mt 3243 wurde mittels RFLP für alle übrigen MODYPatienten ausgeschlossen. In dieser Arbeit wurden erstmals zwei verschiedene Mutationen 82 (R515H und R524Q) innerhalb des Glykogen-Branching-Enzyme(GBE)-Gens bei einer NichtAshkenazi-jüdischen Patientin mit APBD identifiziert. Die beiden Töchter der Patientin mit intermediärer Enzymaktivität erwiesen sich als heterozygote Anlageträger der Erkrankung. Bei einem Patienten mit sporadischer RMD wurde erstmals eine dominante de-novo-Mutation des Caveolin-3-Gens (R26Q) nachgewiesen. Die indirekte Immunfluoreszenz in der Muskelbiopsie zeigte neben der Caveolin-3-Verminderung eine Reduktion der α-Dystroglykan-Anfärbung bei unveränderter Expression von der neuralen Stickoxydsynthase (nNOS) an der Muskelmembran. Die Analyse der polymorphen Mikrosatelliten bei der Familie mit Phosphorylase-Kinase-Defizienz sprach gegen eine Assoziation dieses Phänotyps mit den Genen der β- und γ1-Untereinheit der Phosphorylase-Kinase. Schlussfolgerungen: Die bisher beobachteten, länderabhängigen Prävalenzunterschiede von MODY2- und MODY3Patienten sind durch Selektionierung bei der Rekrutierung begründet. Eine MODY2-Diagnostik erscheint bei Patientinnen mit Gestationsdiabetes sinnvoll. Die nachgewiesenen Mutationen im GBE-Gen wiesen auf dessen pathogene Bedeutung für die Entstehung der Polyglukosankörper hin. Wegen der grundlegenden Bedeutung scheint bei RMD eine Analyse des Caveolin-3-Gens dringend erforderlich; eine Bedeutung der Interaktion von Caveolin-3 und nNOS für die Pathogenese der RMD erscheint wahrscheinlich. Die angewandten Methoden stellten - mit Einschränkungen - bei differenzierter Anwendung ein ausreichendes Armentarium der Mutationsanalyse zur Verfügung. 6 Literaturliste 1) Agamanolis, D. P., A. D. Askari, et al. (1980). Muscle phosphofructokinase deficiency: two cases with unusual polysaccharide accumulation and immunologically active enzyme protein. Muscle Nerve 3(6): 456-67. 2) Agarwala, R., D. L. Applegate, et al. (2000). A fast and scalable radiation hybrid map construction and integration strategy. Genome Res 10(3): 350-64. 3) Ainsworth, P. J., L. C. Surh, et al. (1991). Diagnostic single strand conformational polymorphism, (SSCP): a simplified non-radioisotopic method as applied to a Tay-Sachs B1 variant. Nucleic Acids Res 19(2): 405-6. 4) Alberca, R., E. Rafel, et al. (1980). Increased mechanical muscle irritability syndrome. Acta Neurol Scand 62(4): 250-4. 5) Altschul, S. F., T. L. Madden, et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17): 3389-402. 6) Ansevin, C. F. and D. P. Agamanolis (1996). Rippling muscles and myasthenia gravis with rippling muscles. Arch Neurol 53(2): 197-9. 7) Baba, T., K. Kimura, et al. (1991). Sequence conservation of the catalytic regions of amylolytic enzymes in maize branching enzyme-I. Biochem Biophys Res Commun 181(1): 87-94. 8) Bader, J. S. (2001). The relative power of SNPs and haplotype as genetic markers for association tests. Pharmacogenomics 2(1): 11-24. 83 9) Bao, Y., P. Kishnani, et al. (1996). Hepatic and neuromuscular forms of glycogen storage disease type IV caused by mutations in the same glycogen-branching enzyme gene. J Clin Invest 97(4): 941-8. 10) Barbujani, G., A. Magagni, et al. (1997). An apportionment of human DNA diversity. Proc Natl Acad Sci U S A 94(9): 4516-9. 11) Baudrimont, M., R. Gherardi, et al. (1986). Adult Polyglucosan body disease (APBD). Report of 3 cases [Abstract]. J Neuropathol Exp Neurol 45: 366. 12) Baxter, S. M., M. B. Greizerstein, et al. (1993). Conformational properties of DNA hairpins with TTT and TTTT loops. Biochemistry 32(33): 8702-11. 13) Beards, F., T. Frayling, et al. (1998). Mutations in hepatocyte nuclear factor 1beta are not a common cause of maturity-onset diabetes of the young in the U.K. Diabetes 47(7): 1152-4. 14) Behn, P. S., J. Wasson, et al. (1998). Hepatocyte nuclear factor 1alpha coding mutations are an uncommon contributor to early-onset type 2 diabetes in Ashkenazi Jews. Diabetes 47(6): 967-9. 15) Bell, G. I., S. J. Pilkis, et al. (1996). Glucokinase mutations, insulin secretion, and diabetes mellitus. Annu Rev Physiol 58: 171-86. 16) Betz, R. C., B. G. Schoser, et al. (2001). Mutations in CAV3 cause mechanical hyperirritability of skeletal muscle in rippling muscle disease. Nat Genet 28(3): 218-9. 17) Biederer, C., S. Ries, et al. (1998). Molecular cloning of human caveolin 3. Biochim Biophys Acta 1406(1): 5-9. 18) Bigio, E. H., M. F. Weiner, et al. (1997). Familial dementia due to adult polyglucosan body disease. Clin Neuropathol 16(4): 227-34. 19) Boat, T. F. and P. W. Cheng (1989). Epithelial cell dysfunction in cystic fibrosis: implications for airways disease. Acta Paediatr Scand Suppl 363: 25-9. 20) Bornemann, A., R. Besser, et al. (1996). A mild adult myopathic variant of type IV glycogenosis. Neuromuscul Disord 6(2): 95-9. 21) Boulan-Predseil, P., A. Vital, et al. (1995). Dementia of frontal lobe type due to adult polyglucosan body disease. J Neurol 242(8): 512-6. 22) Boutin, P., E. H. Hani, et al. (1997). Automated fluorescence-based screening for mutation by SSCP: use of universal M13 dye primers for labeling and detection. Biotechniques 23(3): 358-62. 23) Boutin, P., F. Vasseur, et al. (2001). Routine mutation screening of HNF-1alpha and GCK genes in MODY diagnosis: how effective are the techniques of DHPLC and direct sequencing used in combination? Diabetologia 44(6): 775-8. 24) Brenman, J. E., D. S. Chao, et al. (1995). Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 82(5): 743-52. 25) Broman, K. W., J. C. Murray, et al. (1998). Comprehensive human genetic maps: individual and sex-specific variation in recombination. Am J Hum Genet 63(3): 861-9. 26) Brookes, A. J. (1999). The essence of SNPs. Gene 234(2): 177-86. 27) Brown, B. I. and D. H. Brown (1989). Branching enzyme activity of cultured amniocytes and chorionic villi: prenatal testing for type IV glycogen storage disease. Am J Hum Genet 44(3): 37881. 28) Brown, D. H. and B. I. Brown (1983). Studies of the residual glycogen branching enzyme activity present in human skin fibroblasts from patients with type IV glycogen storage disease. Biochem Biophys Res Commun 111(2): 636-43. 84 29) Bruno, C., M. DiRocco, et al. (1999). A novel missense mutation in the glycogen branching enzyme gene in a child with myopathy and hepatopathy. Neuromuscul Disord 9(6-7): 403-7. 30) Bruno, C., S. Servidei, et al. (1993). Glycogen branching enzyme deficiency in adult polyglucosan body disease. Ann Neurol 33(1): 88-93. 31) Burke, C. V., C. W. Buettger, et al. (1999). Cell-biological assessment of human glucokinase mutants causing maturity-onset diabetes of the young type 2 (MODY-2) or glucokinase-linked hyperinsulinaemia (GK-HI). Biochem J 342(Pt 2): 345-52. 32) Burlatsky, S. F., G. S. Oshanin, et al. (1990). Direct energy transfer in polymer systems. Physical Review Letters 65(25): 3205-3208. 33) Burns, R. J., A. H. Bretag, et al. (1994). Benign familial disease with muscle mounding and rippling. J Neurol Neurosurg Psychiatry 57(3): 344-7. 34) Burwinkel, B., L. Amat, et al. (1998). Variability of biochemical and clinical phenotype in Xlinked liver glycogenosis with mutations in the phosphorylase kinase PHKA2 gene. Hum Genet 102(4): 423-9. 35) Burwinkel, B., A. J. Maichele, et al. (1997). Autosomal glycogenosis of liver and muscle due to phosphorylase kinase deficiency is caused by mutations in the phosphorylase kinase beta subunit (PHKB). Hum Mol Genet 6(7): 1109-15. 36) Burwinkel, B., S. W. Moses, et al. (1997). Phosphorylase-kinase-deficient liver glycogenosis with an unusual biochemical phenotype in blood cells associated with a missense mutation in the beta subunit gene (PHKB). Hum Genet 101(2): 170-4. 37) Busard, H. L., A. A. Gabreels-Festen, et al. (1991). Adult polyglucosan body disease: the diagnostic value of axilla skin biopsy. Ann Neurol 29(4): 448-51. 38) Cafferty, M. S., R. E. Lovelace, et al. (1991). Polyglucosan body disease. Muscle Nerve 14(2): 102-7. 39) Cai, Q. Q. and I. Touitou (1993). Excess PCR primers may dramatically affect SSCP efficiency. Nucleic Acids Res 21(16): 3909-10. 40) Carbone, I., C. Bruno, et al. (2000). Mutation in the CAV3 gene causes partial caveolin-3 deficiency and hyperCKemia. Neurology 54(6): 1373-6. 41) Cargill, M., D. Altshuler, et al. (1999). Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet 22(3): 231-8. 42) Cavanagh, J. B. (1999). Corpora-amylacea and the family of polyglucosan diseases. Brain Res Brain Res Rev 29(2-3): 265-95. 43) Cavanaugh, J. A., D. F. Callen, et al. (1998). Analysis of Australian Crohn's disease pedigrees refines the localization for susceptibility to inflammatory bowel disease on chromosome 16. Ann Hum Genet 62(Pt 4): 291-8. 44) Chakraborty, R., D. N. Stivers, et al. (1999). The utility of short tandem repeat loci beyond human identification: implications for development of new DNA typing systems. Electrophoresis 20(8): 1682-96. 45) Chakravarti, A. (2001). To a future of genetic medicine. Nature 409(6822): 822-3. 46) Chevre, J. C., E. H. Hani, et al. (1998). Mutation screening in 18 Caucasian families suggest the existence of other MODY genes. Diabetologia 41(9): 1017-23. 47) Chiu, K. C., Y. Tanizawa, et al. (1993). Glucokinase gene variants in the common form of NIDDM. Diabetes 42(4): 579-82. 85 48) Chomczynski, P. and N. Sacchi (1987). Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162(1): 156-9. 49) Chrambach, A. and D. Rodbard (1971). Polyacrylamide gel electrophoresis. Science 172(982): 440-51. 50) Collins, F. S., L. D. Brooks, et al. (1998). A DNA polymorphism discovery resource for research on human genetic variation. Genome Res 8(12): 1229-31. 51) Cooper, D. N., M. Krawczak, et al. (1995). The nature and mechanisms of human gene mutation. Scriver C.R., Beaudet A.L.,Sly W.S., Valle D. (eds): The metabolic and molecular basis of inherited disease. New York: McGraw-Hill, Inc.: 259-291. 52) Cox, R. D., L. Southam, et al. (1999). UKPDS 31: Hepatocyte nuclear factor-1alpha (the MODY3 gene) mutations in late onset Type II diabetic patients in the United Kingdom. United Kingdom prospective diabetes study. Diabetologia 42(1): 120-1. 53) Crow, J. F. (1995). Spontaneous mutation as a risk factor. Exp Clin Immunogenet 12(3): 121-8. 54) Curran, M. E., K. F. Lau, et al. (1998). Genetic analysis of inflammatory bowel disease in a large European cohort supports linkage to chromosomes 12 and 16. Gastroenterology 115(5): 1066-71. 55) Curtin, F., A. Morabia, et al. (1997). Body mass index compared to dual-energy x-ray absorptiometry: evidence for a spectrum bias. J Clin Epidemiol 50(7): 837-43. 56) Danenberg, P. V., T. Horikoshi, et al. (1992). Detection of point mutations in human DNA by analysis of RNA conformation polymorphism(s). Nucleic Acids Res 20(3): 573-9. 57) Davis, E. A., A. Cuesta-Munoz, et al. (1999). Mutants of glucokinase cause hypoglycaemiaand hyperglycaemia syndromes and their analysis illuminates fundamental quantitative concepts of glucose homeostasis. Diabetologia 42(10): 1175-86. 58) de Paula, F., M. Vainzof, et al. (2001). Mutations in the caveolin-3 gene: When are they pathogenic? Am J Med Genet 99(4): 303-7. 59) Deloukas, P., G. D. Schuler, et al. (1998). A physical map of 30,000 human genes. Science 282(5389): 744-6. 60) Dew, M. S., A. Verma, et al. (1995). Electromyographic variability and pathology of rippling muscle disease. [abstract]. Neurology 45(suppl 4): A447. 61) Dib, C., S. Faure, et al. (1996). A comprehensive genetic map of the human genome based on 5,2c64 microsatellites. Nature 380(6570): 152-4. 62) Dietz, W. H. and T. N. Robinson (1998). Use of the body mass index (BMI) as a measure of overweight in children and adolescents. J Pediatr 132(2): 191-3. 63) Doria, A., Y. Yang, et al. (1999). Phenotypic characteristics of early-onset autosomal-dominant type 2 diabetes unlinked to known maturity-onset diabetes of the young (MODY) genes. Diabetes Care 22(2): 253-61. 64) Duncan, S. A., M. A. Navas, et al. (1998). Regulation of a transcription factor network required for differentiation and metabolism. Science 281(5377): 692-5. 65) Dussoix, P., M. Vaxillaire, et al. (1997). Diagnostic heterogeneity of diabetes in lean young adults: classification based on immunological and genetic parameters. Diabetes 46(4): 622-31. 66) Ellard, S., F. Beards, et al. (2000). A high prevalence of glucokinase mutations in gestational diabetic subjects selected by clinical criteria. Diabetologia 43(2): 250-3. 86 67) Ellard, S., M. P. Bulman, et al. (1999). Allelic drop-out in exon 2 of the hepatocyte nuclear factor-1alpha gene hinders the identification of mutations in three families with maturity-onset diabetes of the young. Diabetes 48(4): 921-3. 68) Ellis, K. J., S. A. Abrams, et al. (1999). Monitoring childhood obesity: assessment of the weight/height index. Am J Epidemiol 150(9): 939-46. 69) Epplen, J. T., J. Buitkamp, et al. (1995). Indirect DNA/gene diagnoses via electrophoresis--an obsolete principle? Electrophoresis 16(5): 683-90. 70) Epstein, P. N., A. C. Boschero, et al. (1992). Expression of yeast hexokinase in pancreatic beta cells of transgenic mice reduces blood glucose, enhances insulin secretion, and decreases diabetes. Proc Natl Acad Sci U S A 89(24): 12038-42. 71) Fajans, S. S., G. I. Bell, et al. (2001). Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med 345(13): 971-80. 72) Felice, K. J., M. L. Grunnet, et al. (1997). Childhood-onset spinocerebellar syndrome associated with massive polyglucosan body deposition. Acta Neurol Scand 95(1): 60-4. 73) Francke, U., B. T. Darras, et al. (1989). Assignment of human genes for phosphorylase kinase subunits alpha (PHKA) to Xq12-q13 and beta (PHKB) to 16q12-q13. Am J Hum Genet 45(2): 27682. 74) Frayling, T. M., M. P. Bulamn, et al. (1997). Mutations in the hepatocyte nuclear factor-1alpha gene are a common cause of maturity-onset diabetes of the young in the U.K. Diabetes 46(4): 7205. 75) Furuta, S., M. Ohira, et al. (2000). Analysis of loss of heterozygosity at 16p12-p13 (familial neuroblastoma locus) in 470 neuroblastomas including both sporadic and mass screening tumors. Med Pediatr Oncol 35(6): 531-3. 76) Fyfe, J. C. (1995). Glycogen storage disease in cats. J Am Vet Med Assoc 206(3): 286. 77) Galbiati, F., J. A. Engelman, et al. (2001). Caveolin-3 null mice show a loss of caveolae, changes in the microdomain distribution of the dystrophin-glycoprotein complex, and t-tubule abnormalities. J Biol Chem 276(24): 21425-33. 78) Galbiati, F., D. Volonte, et al. (1999). Targeted down-regulation of caveolin-3 is sufficient to inhibit myotube formation in differentiating C2C12 myoblasts. Transient activation of p38 mitogenactivated protein kinase is required for induction of caveolin-3 expression and subsequent myotube formation. J Biol Chem 274(42): 30315-21. 79) Galbiati, F., D. Volonte, et al. (1999). Phenotypic behavior of caveolin-3 mutations that cause autosomal dominant limb girdle muscular dystrophy (LGMD-1C). Retention of LGMD-1C caveolin-3 mutants within the golgi complex. J Biol Chem 274(36): 25632-41. 80) Gidh-Jain, M., J. Takeda, et al. (1993). Glucokinase mutations associated with non-insulindependent (type 2) diabetes mellitus have decreased enzymatic activity: implications for structure/function relationships. Proc Natl Acad Sci U S A 90(5): 1932-6. 81) Gil-Neciga, E., J. A. Pareja, et al. (1995). [Multiple entrapments neuropathy in adult polyglucosan body disease]. Neurologia 10(4): 167-70. 82) Glaser, B., P. Kesavan, et al. (1998). Familial hyperinsulinism caused by an activating glucokinase mutation. N Engl J Med 338(4): 226-30. 83) Glavac, D. and M. Dean (1993). Optimization of the single-strand conformation polymorphism (SSCP) technique for detection of point mutations. Hum Mutat 2(5): 404-14. 87 84) Glucksmann, M. A., M. Lehto, et al. (1997). Novel mutations and a mutational hotspot in the MODY3 gene. Diabetes 46(6): 1081-6. 85) Goebel, H. H., Y. S. Shin, et al. (1992). Adult polyglucosan body myopathy. J Neuropathol Exp Neurol 51(1): 24-35. 86) Goldberg, T. and A. E. Slonim (1993). Nutrition therapy for hepatic glycogen storage diseases. J Am Diet Assoc 93(12): 1423-30. 87) Goldman, A. S., W. D. Dickey, et al. (1982). Lymphocyte uropod inhibitory protein: an overview. Surv Immunol Res 1(1): 24-9. 88) Gray, F., R. Gherardi, et al. (1988). Adult polyglucosan body disease (APBD). J Neuropathol Exp Neurol 47(4): 459-74. 89) Greene, G. M., D. C. Weldon, et al. (1987). Juvenile polysaccharidosis with cardioskeletal myopathy. Arch Pathol Lab Med 111(10): 977-82. 90) Greene, H. L., B. I. Brown, et al. (1988). A new variant of type IV glycogenosis: deficiency of branching enzyme activity without apparent progressive liver disease. Hepatology 8(2): 302-6. 91) Greene, H. L., F. K. Ghishan, et al. (1988). Hypoglycemia in type IV glycogenosis: hepatic improvement in two patients with nutritional management. J Pediatr 112(1): 55-8. 92) Griffin, H. G. and A. M. Griffin (1993). DNA sequencing. Recent innovations and future trends. Appl Biochem Biotechnol 38(1-2): 147-59. 93) Guazzarotti, L., E. Bartolotta, et al. (1999). Maturity-onset diabetes of the young (MODY): a new challenge for pediatric diabetologists. J Pediatr Endocrinol Metab 12(4): 487-97. 94) Guerra, A. S., O. P. van Diggelen, et al. (1986). A juvenile variant of glycogenosis IV (Andersen disease). Eur J Pediatr 145(3): 179-81. 95) Gut, I. G. (2001). Automation in genotyping of single nucleotide polymorphisms. Hum Mutat 17(6): 475-92. 96) Hager, J., H. Blanche, et al. (1994). Six mutations in the glucokinase gene identified in MODY by using a nonradioactive sensitive screening technique. Diabetes 43(5): 730-3. 97) Hattersley, A. T. (1998). Maturity-onset diabetes of the young: clinical heterogeneity explained by genetic heterogeneity [published erratum appears in Diabet Med 1998 May;15(5):437]. Diabet Med 15(1): 15-24. 98) Hattersley, A. T., P. J. Saker, et al. (1993). Linkage of maturity-onset diabetes of the young to the glucokinase gene--evidence of genetic heterogeneity. Biochem Soc Trans 21(1): 24S. 99) Hayashi, K. (1992). PCR-SSCP: a method for detection of mutations. Genet Anal Tech Appl 9(3): 73-9. 100) Hayashi, K. and D. W. Yandell (1993). How sensitive is PCR-SSCP? Hum Mutat 2(5): 338-46. 101) Hendrickx, J., E. Dams, et al. (1996). X-linked liver glycogenosis type II (XLG II) is caused by mutations in PHKA2, the gene encoding the liver alpha subunit of phosphorylase kinase. Hum Mol Genet 5(5): 649-52. 102) Herrmann, R., V. Straub, et al. (2000). Dissociation of the dystroglycan complex in caveolin-3deficient limb girdle muscular dystrophy. Hum Mol Genet 9(15): 2335-40. 103) Hogdall, E., K. Boye, et al. (1999). Simple preparation method of PCR fragments for automated DNA sequencing. J Cell Biochem 73(4): 433-6. 104) Hongyo, T., G. S. Buzard, et al. (1993). 'Cold SSCP': a simple, rapid and non-radioactive method for optimized single-strand conformation polymorphism analyses. Nucleic Acids Res 21(16): 3637-42. 88 105) Humphries, S. E., V. Gudnason, et al. (1997). Single-strand conformation polymorphism analysis with high throughput modifications, and its use in mutation detection in familial hypercholesterolemia. International Federation of Clinical Chemistry Scientific Division: Committee on Molecular Biology Techniques. Clin Chem 43(3): 427-35. 106) Inoue, S., R. Ishii, et al. (1996). Sevoflurane anaesthesia for a patient with adult polyglucosan body disease. Can J Anaesth 43(12): 1257-9. 107) Ishihara, T., T. Yokota, et al. (1987). Comparative study of the intracytoplasmic inclusions in Lafora disease and type IV glycogenosis by electron microscopy. Acta Pathol Jpn 37(10): 1591601. 108) Ishwad, C. S., R. E. Ferrell, et al. (1995). Molecular and cytogenetic analysis of chromosome 7 in uterine leiomyomas. Genes Chromosomes Cancer 14(1): 51-5. 109) Isomaa, B., M. Henricsson, et al. (1998). Chronic diabetic complications in patients with MODY3 diabetes. Diabetologia 41(4): 467-73. 110) Iwasaki, N., N. Oda, et al. (1997). Mutations in the hepatocyte nuclear factor-1alpha/MODY3 gene in Japanese subjects with early- and late-onset NIDDM. Diabetes 46(9): 1504-8. 111) Jones, T. A., E. F. da Cruz e Silva, et al. (1990). Localisation of the gene encoding the catalytic gamma subunit of phosphorylase kinase to human chromosome bands 7p12-q21. Biochim Biophys Acta 1048(1): 24-9. 112) Kaisaki, P. J., S. Menzel, et al. (1997). Mutations in the hepatocyte nuclear factor-1alpha gene in MODY and early-onset NIDDM: evidence for a mutational hotspot in exon 4 [published erratum appears in Diabetes 1997 Jul;46(7):1239]. Diabetes 46(3): 528-35. 113) Kamiya, S. and Y. Suzuki (1989). Polyglucosan bodies in the brain of the cat. J Comp Pathol 101(3): 263-7. 114) Kanazawa, H., T. Noumi, et al. (1986). Analysis of Escherichia coli mutants of the H(+)transporting ATPase: determination of altered site of the structural genes. Methods Enzymol 126: 595-603. 115) Kanehisa, M. I. and W. B. Goad (1982). Pattern recognition in nucleic acid sequences. II. An efficient method for finding locally stable secondary structures. Nucleic Acids Res 10(1): 265-78. 116) Katagiri, H., T. Asano, et al. (1994). Mitochondrial diabetes mellitus: prevalence and clinical characterization of diabetes due to mitochondrial tRNA(Leu(UUR)) gene mutation in Japanese patients. Diabetologia 37(5): 504-10. 117) Kawabe, J. I., B. S. Grant, et al. (2001). Changes in caveolin subtype protein expression in aging rat organs. Mol Cell Endocrinol 176(1-2): 91-5. 118) Kilger, C. and S. Paabo (1997). Direct exponential amplification and sequencing (DEXAS) of genomic DNA. Biol Chem 378(2): 99-105. 119) Kilimann, M. W. (1997). Glycogen storage disease due to phosphorylase kinase deficiency. Swallow D.M., Edwards Y.T. (eds): Protein dyfunction in human genetic disease, BIOS Scientific Publishers Oxford: 57-75. 120) Koul, R. L., R. P. Chand, et al. (2001). Severe autosomal recessive rippling muscle disease. Muscle Nerve 24(11): 1542-7. 121) Kozobolis, V. P., E. T. Detorakis, et al. (1999). Loss of heterozygosity in pseudoexfoliation syndrome. Invest Ophthalmol Vis Sci 40(6): 1255-60. 122) Ktistaki, E. and I. Talianidis (1997). Modulation of hepatic gene expression by hepatocyte nuclear factor 1. Science 277(5322): 109-12. 89 123) Kurzchalia, T. V. and R. G. Parton (1999). Membrane microdomains and caveolae. Curr Opin Cell Biol 11(4): 424-31. 124) Landegren, U., R. Kaiser, et al. (1988). A ligase-mediated gene detection technique. Science 241(4869): 1077-80. 125) Lander, E. S., L. M. Linton, et al. (2001). Initial sequencing and analysis of the human genome. Nature 409(6822): 860-921. 126) Lathrop, G. M. and J. M. Lalouel (1984). Easy calculations of lod scores and genetic risks on small computers. Am J Hum Genet 36(2): 460-5. 127) Ledermann, H. M. (1995). Is maturity onset diabetes at young age (MODY) more common in Europe than previously assumed? [letter] [see comments]. Lancet 345(8950): 648. 128) Lehto, M., C. Wipemo, et al. (1999). High frequency of mutations in MODY and mitochondrial genes in Scandinavian patients with familial early-onset diabetes. Diabetologia 42(9): 1131-7. 129) Lerman, L. S. and K. Silverstein (1987). Computational simulation of DNA melting and its application to denaturing gradient gel electrophoresis. Methods Enzymol 155: 482-501. 130) Li, S., T. Okamoto, et al. (1995). Evidence for a regulated interaction between heterotrimeric G proteins and caveolin. J Biol Chem 270(26): 15693-701. 131) Libman, I. and S. A. Arslanian (1999). Type II diabetes mellitus: no longer just adults. Pediatr Ann 28(9): 589-93. 132) Lim, L. E. and K. P. Campbell (1998). The sarcoglycan complex in limb-girdle muscular dystrophy. Curr Opin Neurol 11(5): 443-52. 133) Lindner, T. H., B. N. Cockburn, et al. (1999). Molecular genetics of MODY in Germany [letter]. Diabetologia 42(1): 121-3. 134) Lipshutz, R. J., S. P. Fodor, et al. (1999). High density synthetic oligonucleotide arrays. Nat Genet 21(1 Suppl): 20-4. 135) Löffler, G., P. E. Petrides (1997) Biochemie und Pathobiochemie. Fünfte Auflage, ISBN 3540-59006-4., Springer-Verlag, New York, 1997. 136) Long, D. M., M. J. Mossakowski, et al. (1972). Glycogen accumulation in spinal cord motor neurons due to partial ischemia. Acta Neuropathol 20(4): 335-47. 137) Lossos, A., V. Barash, et al. (1991). Hereditary branching enzyme dysfunction in adult polyglucosan body disease: a possible metabolic cause in two patients. Ann Neurol 30(5): 655-62. 138) Lossos, A., Z. Meiner, et al. (1998). Adult polyglucosan body disease in Ashkenazi Jewish patients carrying the Tyr329Ser mutation in the glycogen-branching enzyme gene. Ann Neurol 44(6): 867-72. 139) Lyamichev, V., A. L. Mast, et al. (1999). Polymorphism identification and quantitative detection of genomic DNA by invasive cleavage of oligonucleotide probes. Nat Biotechnol 17(3): 292-6. 140) Mahalingam, B., A. Cuesta-Munoz, et al. (1999). Structural model of human glucokinase in complex with glucose and ATP: implications for the mutants that cause hypo- and hyperglycemia. Diabetes 48(9): 1698-705. 141) Maichele, A. J. and J. S. Chamberlain (1994). The gamma phosphorylase kinase gene, Phkg, maps to mouse chromosome 5 near Gus. Mamm Genome 5(1): 15-8. 142) Mancardi, G. L., A. Schenone, et al. (1985). Polyglucosan bodies in the sural nerve of a diabetic patient with polyneuropathy. Acta Neuropathol 66(1): 83-6. 90 143) Mashiyama, S., Y. Murakami, et al. (1991). Detection of p53 gene mutations in human brain tumors by single-strand conformation polymorphism analysis of polymerase chain reaction products. Oncogene 6(8): 1313-8. 144) Massa, O., F. Meschi, et al. (2001). High prevalence of glucokinase mutations in Italian children with MODY. Influence on glucose tolerance, first-phase insulin response, insulin sensitivity and BMI. Diabetes Study Group of the Italian Society of Paediatric Endocrinology and Diabetes (SIEDP). Diabetologia 44(7): 898-905. 145) Matschinsky, F. M. (1996). Banting Lecture 1995. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm. Diabetes 45(2): 223-41. 146) Matschinsky, F. M., B. Glaser, et al. (1998). Pancreatic beta-cell glucokinase: closing the gap between theoretical concepts and experimental realities. Diabetes 47(3): 307-15. 147) Matsumuro, K., S. Izumo, et al. (1993). Chronic demyelinating neuropathy and intra-axonal polyglucosan bodies. Acta Neuropathol 86(1): 95-9. 148) Maxam, A. M. and W. Gilbert (1977). A new method for sequencing DNA. Proc Natl Acad Sci U S A 74(2): 560-4. 149) McConkie-Rosell, A., C. Wilson, et al. (1996). Clinical and laboratory findings in four patients with the non-progressive hepatic form of type IV glycogen storage disease. J Inherit Metab Dis 19(1): 51-8. 150) McDonald, T. D., P. L. Faust, et al. (1993). Polyglucosan body disease simulating amyotrophic lateral sclerosis. Neurology 43(4): 785-90. 151) McNally, E. M., E. de Sa Moreira, et al. (1998). Caveolin-3 in muscular dystrophy. Hum Mol Genet 7(5): 871-7. 152) Mendel, D. B. and G. R. Crabtree (1991). HNF-1, a member of a novel class of dimerizing homeodomain proteins. J Biol Chem 266(2): 677-80. 153) Mendel, D. B., P. A. Khavari, et al. (1991). Characterization of a cofactor that regulates dimerization of a mammalian homeodomain protein. Science 254(5039): 1762-7. 154) Menzel, R., P. J. Kaisaki, et al. (1998). A low renal threshold for glucose in diabetic patients with a mutation in the hepatocyte nuclear factor-1alpha (HNF-1alpha) gene. Diabet Med 15(10): 816-20. 155) Merlini, L., C. Carbone, et al. (2002). Familial isolated hyperCKaemia with a new mutation in the caveolin (CAV-3) gene. J Neurol Neurosurg Psychiatry 73: 65-67. 156) Merril, C. R. (1990). Silver staining of proteins and DNA. Nature 343(6260): 779-80. 157) Michel, J. B., O. Feron, et al. (1997). Caveolin versus calmodulin. Counterbalancing allosteric modulators of endothelial nitric oxide synthase. J Biol Chem 272(41): 25907-12. 158) Miller, S. P., G. R. Anand, et al. (1999). Characterization of glucokinase mutations associated with maturity-onset diabetes of the young type 2 (MODY-2): different glucokinase defects lead to a common phenotype. Diabetes 48(8): 1645-51. 159) Minetti, C., F. Sotgia, et al. (1998). Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat Genet 18(4): 365-8. 160) Mirza, M. M., J. Lee, et al. (1998). Evidence of linkage of the inflammatory bowel disease susceptibility locus on chromosome 16 (IBD1) to ulcerative colitis. J Med Genet 35(3): 218-21. 161) Miterski, B., R. Kruger, et al. (2000). PCR/SSCP detects reliably and efficiently DNA sequence variations in large scale screening projects. Comb Chem High Throughput Screen 3(3): 211-8. 91 162) Motulsky, A. G. (1995). Jewish diseases and origins. Nat Genet 9(2): 99-101. 163) Muller-Felber, W., C. F. Ansevin, et al. (1999). Immunosuppressive treatment of rippling muscles in patients with myasthenia gravis. Neuromuscul Disord 9(8): 604-7. 164) Nakabayashi, Y. and K. Nishigaki (1996). Single-strand conformation polymorphism (SSCP) can be explained by semistable conformation dynamics of single-stranded DNA. J Biochem (Tokyo) 120(2): 320-5. 165) Nase, S., K. P. Kunze, et al. (1995). A new variant of type IV glycogenosis with primary cardiac manifestation and complete branching enzyme deficiency. In vivo detection by heart muscle biopsy. Eur Heart J 16(11): 1698-704. 166) Nathan, D. M. (1998). Some answers, more controversy, from UKPDS. United Kingdom Prospective Diabetes Study. Lancet 352(9131): 832-3. 167) Negishi, C. and G. Sze (1992). Spinal cord MRI in adult polyglucosan body disease. J Comput Assist Tomogr 16(5): 824-6. 168) Newsholme, E. A., V. A. Zammit, et al. (1978). The role of glucose and glycogen as fuels for muscle. Biochem Soc Trans 6(3): 512-20. 169) Ng, M. C., J. K. Li, et al. (2000). Nature or nurture: an insightful illustration from a Chinese family with hepatocyte nuclear factor-1 alpha diabetes (MODY3). Diabetologia 43(6): 816-8. 170) Nishigaki, K., Y. Husimi, et al. (1986). Detection of differences in higher order structure between highly homologous single-stranded DNAs by low-temperature denaturant gradient gel electrophoresis. J Biochem (Tokyo) 99(3): 663-71. 171) O'Dea, J. and S. Abraham (1995). Should body-mass index be used in young adolescents? Lancet 345(8950): 657. 172) Ohlendieck, K., K. Matsumura, et al. (1993). Duchenne muscular dystrophy: deficiency of dystrophin-associated proteins in the sarcolemma. Neurology 43(4): 795-800. 173) Oka, N., M. Yamamoto, et al. (1997). Caveolin interaction with protein kinase C. Isoenzymedependent regulation of kinase activity by the caveolin scaffolding domain peptide. J Biol Chem 272(52): 33416-21. 174) Okita, K., Q. Yang, et al. (1999). Human insulin gene is a target gene of hepatocyte nuclear factor-1alpha (HNF-1alpha) and HNF-1beta. Biochem Biophys Res Commun 263(2): 566-9. 175) Orita, M., H. Iwahana, et al. (1989). Detection of polymorphisms of human DNA by gel electrophoresis as single-strand conformation polymorphisms. Proc Natl Acad Sci U S A 86(8): 2766-70. 176) Orosz, J. M. and J. G. Wetmur (1977). DNA melting temperatures and renaturation rates in concentrated alkylammonium salt solutions. Biopolymers 16(6): 1183-99. 177) Osoegawa, K., P. Y. Woon, et al. (1998). An improved approach for construction of bacterial artificial chromosome libraries. Genomics 52(1): 1-8. 178) O'Toole, D., G. Wells, et al. (1994). Ultrastructural pathology of an inherited lower motor neuron disease of pigs. J Vet Diagn Invest 6(2): 230-7. 179) Parker, L. T., H. Zakeri, et al. (1996). AmpliTaq DNA polymerase, FS dye-terminator sequencing: analysis of peak height patterns. Biotechniques 21(4): 694-9. 180) Perez Jurado, L. A., R. Peoples, et al. (1996). Molecular definition of the chromosome 7 deletion in Williams syndrome and parent-of-origin effects on growth. Am J Hum Genet 59(4): 78192. 92 181) Phelps, C. H. (1972). Barbiturate-induced glycogen accumulation in brain. An electron microscopic study. Brain Res 39(1): 225-34. 182) Pontoglio, M., J. Barra, et al. (1996). Hepatocyte nuclear factor 1 inactivation results in hepatic dysfunction, phenylketonuria, and renal Fanconi syndrome. Cell 84(4): 575-85. 183) Pontoglio, M., D. Prie, et al. (2000). HNF1alpha controls renal glucose reabsorption in mouse and man. EMBO Rep 1(4): 359-65. 184) Pontoglio, M., S. Sreenan, et al. (1998). Defective insulin secretion in hepatocyte nuclear factor 1alpha- deficient mice. J Clin Invest 101(10): 2215-22. 185) Postic, C., M. Shiota, et al. (1999). Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem 274(1): 305-15. 186) Pruitt, K. D. and D. R. Maglott (2001). RefSeq and LocusLink: NCBI gene-centered resources. Nucleic Acids Res 29(1): 137-40. 187) Pulyaeva, H., S. F. Zakharov, et al. (1994). Detection of a single base mismatch in doublestranded DNA by electrophoresis on uncrosslinked polyacrylamide gel. Electrophoresis 15(8-9): 1095-100. 188) Raben, N., M. Danon, et al. (2001). Surprises of genetic engineering: a possible model of polyglucosan body disease. Neurology 56(12): 1739-45. 189) Radhakrishna, U., J. L. Blouin, et al. (1997). Mapping one form of autosomal dominant postaxial polydactyly type A to chromosome 7p15-q11.23 by linkage analysis. Am J Hum Genet 60(3): 597-604. 190) Rao, K. R., J. M. Hafford, et al. (1987). Electrically silent muscle contractions, myoedema and myopathy [abstract]. Electroenceph Clin Neurophysiol 66: S85. 191) Ravnik-Glavac, M., D. Glavac, et al. (1994). Screening for CF mutations in adult cystic fibrosis patients with a directed and optimized SSCP strategy. Hum Mutat 3(3): 231-8. 192) Reijneveld, J. C., N. C. Notermans, et al. (2001). Hyper-CK-aemia revisited. Neuromuscul Disord 11(2): 163-4. 193) Reiss, J., M. Krawczak, et al. (1990). The effect of replication errors on the mismatch analysis of PCR-amplified DNA. Nucleic Acids Res 18(4): 973-8. 194) Reusche, E., F. Aksu, et al. (1992). A mild juvenile variant of type IV glycogenosis. Brain Dev 14(1): 36-43. 195) Rey-Campos, J., T. Chouard, et al. (1991). vHNF1 is a homeoprotein that activates transcription and forms heterodimers with HNF1. Embo J 10(6): 1445-57. 196) Ricker, K., R. T. Moxley, et al. (1989). Rippling muscle disease. Arch Neurol 46(4): 405-8. 197) Rifai, Z., M. Klitzke, et al. (1994). Dementia of adult polyglucosan body disease. Evidence of cortical and subcortical dysfunction. Arch Neurol 51(1): 90-4. 198) Robertson, N. P., S. Wharton, et al. (1998). Adult polyglucosan body disease associated with an extrapyramidal syndrome. J Neurol Neurosurg Psychiatry 65(5): 788-90. 199) Robitaille, Y., S. Carpenter, et al. (1980). A distinct form of adult polyglucosan body disease with massive involvement of central and peripheral neuronal processes and astrocytes: a report of four cases and a review of the occurrence of polyglucosan bodies in other conditions such as Lafora's disease and normal ageing. Brain 103(2): 315-36. 200) Rosenbloom, A. L., J. R. Joe, et al. (1999). Emerging epidemic of type 2 diabetes in youth. Diabetes Care 22(2): 345-54. 93 201) Rosenthal, P., L. Podesta, et al. (1995). Failure of liver transplantation to diminish cardiac deposits of amylopectin and leukocyte inclusions in type IV glycogen storage disease. Liver Transpl Surg 1(6): 373-6. 202) Rowland, L. P. (1980). Approaches in the membrane theory of Duchenne muscular dystrophy. Angelini C., Danielli G.A. (eds) Amsterdam, Excerpta Medica [Muscular dytrophy research.]: 3-20. 203) Ryffel, G. U. (2001). Mutations in the human genes encoding the transcription factors of the hepatocyte nuclear factor (HNF)1 and HNF4 families: functional and pathological consequences. J Mol Endocrinol 27(1): 11-29. 204) Sakai, M., J. Austin, et al. (1969). Studies of corpora amylacea. I. Isolation and preliminary characterization by chemical and histochemical techniques. Arch Neurol 21(5): 526-44. 205) Saker, P. J., A. T. Hattersley, et al. (1997). UKPDS 21: low prevalence of the mitochondrial transfer RNA gene (tRNA(Leu(UUR))) mutation at position 3243bp in UK Caucasian type 2 diabetic patients. Diabet Med 14(1): 42-5. 206) Sanger, F., S. Nicklen, et al. (1977). DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 74(12): 5463-7. 207) Sarkar, G., H. S. Yoon, et al. (1992). Dideoxy fingerprinting (ddE): a rapid and efficient screen for the presence of mutations. Genomics 13(2): 441-3. 208) Sarkar, G., H. S. Yoon, et al. (1992). Screening for mutations by RNA single-strand conformation polymorphism (rSSCP): comparison with DNA-SSCP. Nucleic Acids Res 20(4): 8718. 209) Satoh, K. and K. Sato (1982). Purification of RNA associated and unassociated forms of 1,4alpha-glucan branching enzyme from rat liver. J Biochem (Tokyo) 91(4): 1129-37. 210) Satsangi, J., M. Parkes, et al. (1996). Two stage genome-wide search in inflammatory bowel disease provides evidence for susceptibility loci on chromosomes 3, 7 and 12. Nat Genet 14(2): 199-202. 211) Schork, N. J., D. Fallin, et al. (2000). Single nucleotide polymorphisms and the future of genetic epidemiology. Clin Genet 58(4): 250-64. 212) Schwencke, C., S. Okumura, et al. (1999). Colocalization of beta-adrenergic receptors and caveolin within the plasma membrane. J Cell Biochem 75(1): 64-72. 213) Scotto, J. M., T. de Barsy, et al. (1981). [A study of the abnormal polysaccharide in a child with type IV glycogen storage disease (author's transl)]. Arch Fr Pediatr 38 Suppl 1: 837-41. 214) Selby, R., T. E. Starzl, et al. (1993). Liver transplantation for type I and type IV glycogen storage disease. Eur J Pediatr 152(Suppl 1): S71-6. 215) Servidei, S., R. E. Riepe, et al. (1987). Severe cardiopathy in branching enzyme deficiency. J Pediatr 111(1): 51-6. 216) Sham, P. C., J. H. Zhao, et al. (2000). The effect of marker characteristics on the power to detect linkage disequilibrium due to single or multiple ancestral mutations. Ann Hum Genet 64(Pt 2): 161-9. 217) Sheffield, V. C., J. S. Beck, et al. (1993). The sensitivity of single-strand conformation polymorphism analysis for the detection of single base substitutions. Genomics 16(2): 325-32. 218) Sheu, J. C., Y. W. Lin, et al. (1999). Loss of heterozygosity and microsatellite instability in hepatocellular carcinoma in Taiwan. Br J Cancer 80(3-4): 468-76. 219) Shih, D. Q., M. Bussen, et al. (2001). Hepatocyte nuclear factor-1alpha is an essential regulator of bile acid and plasma cholesterol metabolism. Nat Genet 27(4): 375-82. 94 220) Shin, Y. S., H. Steiguber, et al. (1988). Branching enzyme in erythrocytes. Detection of type IV glycogenosis homozygotes and heterozygotes. J Inherit Metab Dis 11(Suppl 2): 252-4. 221) Sindern, E., T. Patzold, et al. (1999). [Adult polyglucosan antibody disease. Case report with predominant involvement of the central and peripheral nervous system and branching enzyme defect in leukocytes]. Nervenarzt 70(8): 745-9. 222) Smith, L. M., J. Z. Sanders, et al. (1986). Fluorescence detection in automated DNA sequence analysis. Nature 321(6071): 674-9. 223) So, Y. T., L. Zu, et al. (2001). Rippling muscle disease: evidence for phenotypic and genetic heterogeneity. Muscle Nerve 24(3): 340-4. 224) Sotgia, F., J. K. Lee, et al. (2000). Caveolin-3 directly interacts with the C-terminal tail of beta dystroglycan. Identification of a central WW-like domain within caveolin family members. J Biol Chem 275(48): 38048-58. 225) Sourdive, D. J., T. Chouard, et al. (1993). The HNF1 C-terminal domain contributes to transcriptional activity and modulates nuclear localisation. C R Acad Sci III 316(4): 385-94. 226) Sovik, O., P. Njolstad, et al. (1998). Hyperexcitability to sulphonylurea in MODY3 [letter]. Diabetologia 41(5): 607-8. 227) Spielman, R. S., R. E. McGinnis, et al. (1993). Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet 52(3): 506-16. 228) Stamler, J. S. and G. Meissner (2001). Physiology of nitric oxide in skeletal muscle. Physiol Rev 81(1): 209-237. 229) Stephan, D. A., N. R. Buist, et al. (1994). A rippling muscle disease gene is localized to 1q41: evidence for multiple genes. Neurology 44(10): 1915-20. 230) Stephan, W. (1989). Tandem-repetitive noncoding DNA: forms and forces. Mol Biol Evol 6(2): 198-212. 231) Stoffel, M., K. L. Bell, et al. (1993). Identification of glucokinase mutations in subjects with gestational diabetes mellitus. Diabetes 42(6): 937-40. 232) Stoffel, M., P. Froguel, et al. (1992). Human glucokinase gene: isolation, characterization, and identification of two missense mutations linked to early-onset non-insulin-dependent (type 2) diabetes mellitus [published erratum appears in Proc Natl Acad Sci U S A 1992 Nov 1;89(21):10562]. Proc Natl Acad Sci U S A 89(16): 7698-702. 233) Stoffel, M., P. Patel, et al. (1992). Missense glucokinase mutation in maturity-onset diabetes of the young and mutation screening in late-onset diabetes. Nat Genet 2(2): 153-6. 234) Stoffers, D. A., J. Ferrer, et al. (1997). Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat Genet 17(2): 138-9. 235) Sunada, Y., H. Ohi, et al. (2001). Transgenic mice expressing mutant caveolin-3 show severe myopathy associated with increased nNOS activity. Hum Mol Genet 10(3): 173-8. 236) Suzuki, K., E. David, et al. (1971). Presenile dementia with &quot;Lafora-like&quot; intraneuronal inclusions. Arch Neurol 25(1): 69-80. 237) Tabor, S. and C. C. Richardson (1995). A single residue in DNA polymerases of the Escherichia coli DNA polymerase I family is critical for distinguishing between deoxy- and dideoxyribonucleotides. Proc Natl Acad Sci U S A 92(14): 6339-43. 238) Takeda, J., T. Kayano, et al. (1993). Organization of the human GLUT2 (pancreatic beta-cell and hepatocyte) glucose transporter gene. Diabetes 42(5): 773-7. 95 239) Tang, Z., P. E. Scherer, et al. (1996). Molecular cloning of caveolin-3, a novel member of the caveolin gene family expressed predominantly in muscle. J Biol Chem 271(4): 2255-61. 240) Tanizawa, Y., A. Matsutani, et al. (1992). Human glucokinase gene: isolation, structural characterization, and identification of a microsatellite repeat polymorphism. Mol Endocrinol 6(7): 1070-81. 241) Terauchi, Y., H. Sakura, et al. (1995). Pancreatic beta-cell-specific targeted disruption of glucokinase gene. Diabetes mellitus due to defective insulin secretion to glucose. J Biol Chem 270(51): 30253-6. 242) Thon, V. J., M. Khalil, et al. (1993). Isolation of human glycogen branching enzyme cDNAs by screening complementation in yeast. J Biol Chem 268(10): 7509-13. 243) Torbergsen, T. (1975). A family with dominant hereditary myotonia, muscular hypertrophy, and increased muscular irritability, distinct from myotonia congenita thomsen. Acta Neurol Scand 51(3): 225-32. 244) Urhammer, S. A., S. K. Rasmussen, et al. (1997). Genetic variation in the hepatocyte nuclear factor-1 alpha gene in Danish Caucasians with late-onset NIDDM. Diabetologia 40(4): 473-5. 245) van den Ouweland, J. M., H. H. Lemkes, et al. (1994). Maternally inherited diabetes and deafness is a distinct subtype of diabetes and associates with a single point mutation in the mitochondrial tRNA(Leu(UUR)) gene. Diabetes 43(6): 746-51. 246) van Heyningen, V. (1994). Genetics. One gene--four syndromes. Nature 367(6461): 319-20. 247) van Noort, G., W. Straks, et al. (1993). A congenital variant of glycogenosis type IV. Pediatr Pathol 13(5): 685-98. 248) Vandercammen, A. and E. Van Schaftingen (1991). Competitive inhibition of liver glucokinase by its regulatory protein. Eur J Biochem 200(2): 545-51. 249) Vaxillaire, M., M. Rouard, et al. (1997). Identification of nine novel mutations in the hepatocyte nuclear factor 1 alpha gene associated with maturity-onset diabetes of the young (MODY3). Hum Mol Genet 6(4): 583-6. 250) Veiga-da-Cunha, M., S. Courtois, et al. (1996). Amino acid conservation in animal glucokinases. Identification of residues implicated in the interaction with the regulatory protein. J Biol Chem 271(11): 6292-7. 251) Veiga-da-Cunha, M., L. Z. Xu, et al. (1996). Effect of mutations on the sensitivity of human beta-cell glucokinase to liver regulatory protein. Diabetologia 39(10): 1173-9. 252) Vital, A., J. Lamarche, et al. (1995). [Significance of polyglucosan bodies in neuropathology. Clinico-pathologic study of seven cases]. Ann Pathol 15(4): 269-75. 253) Vorgerd, M., H. Bolz, et al. (1999). Phenotypic variability in rippling muscle disease. Neurology 52(7): 1453-9. 254) Wang, H., P. Maechler, et al. (1998). Dominant-negative suppression of HNF-1alpha function results in defective insulin gene transcription and impaired metabolism-secretion coupling in a pancreatic beta-cell line. Embo J 17(22): 6701-13. 255) Wehner, M. and M. W. Kilimann (1995). Human cDNA encoding the muscle isoform of the phosphorylase kinase gamma subunit (PHKG1). Hum Genet 96(5): 616-8. 256) Weis, J. and J. M. Schroder (1988). Adult polyglucosan body myopathy with subclinical peripheral neuropathy: case report and review of diseases associated with polyglucosan body accumulation. Clin Neuropathol 7(6): 271-9. 96 257) Wierzba-Bobrowicz, T. and B. Stroinska-Kus (1994). Adult polyglucosan body disease. Folia Neuropathol 32(1): 37-41. 258) Wullrich-Schmoll, A. and M. W. Kilimann (1996). Structure of the human gene encoding the phosphorylase kinase beta subunit (PHKB). Eur J Biochem 238(2): 374-80. 259) Xu, L. Z., I. T. Weber, et al. (1995). Sugar specificity of human beta-cell glucokinase: correlation of molecular models with kinetic measurements. Biochemistry 34(18): 6083-92. 260) Yamada, S., H. Nishigori, et al. (1997). Identification of mutations in the hepatocyte nuclear factor (HNF)-1 alpha gene in Japanese subjects with IDDM. Diabetes 46(10): 1643-7. 261) Yamada, S., H. Tomura, et al. (1999). Identification of mutations in the hepatocyte nuclear factor-1alpha gene in Japanese subjects with early-onset NIDDM and functional analysis of the mutant proteins. Diabetes 48(3): 645-8. 262) Yamagata, K., H. Furuta, et al. (1996). Mutations in the hepatocyte nuclear factor-4alpha gene in maturity- onset diabetes of the young (MODY1) [see comments]. Nature 384(6608): 458-60. 263) Yamagata, K., Q. Yang, et al. (1998). Mutation P291fsinsC in the transcription factor hepatocyte nuclear factor-1alpha is dominant negative. Diabetes 47(8): 1231-5. 264) Yamanami, S., T. Ishihara, et al. (1992). Comparative study of intraneuronal polyglucosan bodies in brains from patients with Lafora disease and aged dogs. Acta Pathol Jpn 42(11): 787-92. 265) Yan, W. L., X. Y. Guan, et al. (2000). Childhood-onset schizophrenia/autistic disorder and t(1;7) reciprocal translocation: identification of a BAC contig spanning the translocation breakpoint at 7q21. Am J Med Genet 96(6): 749-53. 266) Yanai, T., T. Masegi, et al. (1994). Polyglucosan bodies in the brain of a cow. Acta Neuropathol 88(1): 75-7. 267) Yip, S. P., D. A. Hopkinson, et al. (1999). Improvement of SSCP analysis by use of denaturants. Biotechniques 27(1): 20-2, 24. 268) Yoshiuchi, I., K. Yamagata, et al. (1999). Three new mutations in the hepatocyte nuclear factor-1alpha gene in Japanese subjects with diabetes mellitus: clinical features and functional characterization. Diabetologia 42(5): 621-6. 269) Zimmerman, C. P. and A. M. Gold (1982). Glycogen branching enzyme in Lafora myoclonus epilepsy. Biochem Med 28(1): 83-93. 270) Zimmerman, C. P. and A. M. Gold (1983). Isolation and characterization of glycogen branching enzyme from rabbit liver. Biochemistry 22(14): 3387-92. A Anhang Abb 28: Datenerhebungsbogen zur Erfassung der Diabetes-Komplikationen B Tab 16: Exon Bisher beschriebene Mutationen im HNF1α-Gen Mutation 1 1 1 1 1 Q7X L12H G31D E48K R55G56delGAGGG 1 L107R L107I K117E Y122C N127del I128V I128N P129T R131Q R131W E132K S142F H143Y K158N R159Q R159W A161T R171X G191D R200W R200Q K205Q R229Q R229P R229X E324X G237A C241G C241R T260M R263C R263G W267X W267R R271W R272C R272H P289R P291insC P291delC G292delG C241G T354M G375delT P379delCT 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 3 3 3 3 3 3 3 3 3 4 4 4 4 4 4 4 4 4 4 4 4 4 4 5 6 6 6 6 6 6 6 6 7 7 8 8 9 P379insC P379delT T392delA Q401delC I414G415insCCdelT C G415R T433I A443delCA P447L Q486X P519L T537R T547E548delTG Austausch CAG zu TAG CTC zu CAC GGT zu GAT GAG zu AAG Deletion von GAGGG CTT zu CGT CTT zu ATT AAG zu GAG TAC zu TGC Deletion ATC zu GTC ATC zu AAC CCA zu ACA CGG zu CAG CGG zu TGG GAG zu AAG TCC zu TTC CAC zu TAC AAG zu AAT CGG zu CAG CGG zu TGG GCC zu CCC CGA zu TGA GGT zu GAT CGG zu TGG CGG zu CAG AAG zu CAG CGA zu CAA CGA zu CCA CGA zu TGA GAG zu TAG GGT zu GCT TGC zu GGC TGC zu CGC ACA zu ATA CGT zu TGT CGT zu TGT TGG zu TAG TGG zu TGT CGG zu TGG CGC zu TGC CGC zu CAC CCC zu CGC Insertion von C Deletion von C Deletion von G TGC zu GGC ACG zu ATG Deletion von G Deletion von CT Insertion von C Deletion von T Deletion von A Deletion von C Deletion von TC GGG zu CGG ACC zu ATC Deletion von CA CCG zu CTG CAG zu TAG CCG zu CTG ACG zu AGG Deletion von TG funktionelle Auswirkung Gln zu Stop Leu zu His Gly zu Asp Glu zu Lys Frameshift Leu zu Arg Leu zu Iso Lys zu Glu Tyr zu Cys veröffentlicht (Vaxillaire, Rouard et al. 1997) (Iwasaki, Oda et al. 1997) (Chevre, Hani et al. 1998) (Moller, Dalgaard et al. 1998) (Vaxillaire, Rouard et al. 1997) Ala zu Val Ile zu Asn Pro zu Thr Arg zu Gln Arg zu Trp Glu zu Lys Ser zu Phe His zu Tyr Lys zu Asn Arg zu Gln Arg zu Trp Ala zu Thr Arg zu Stop Gly zu Asp Arg zu Trp Arg zu Gln Lys zu Gln Arg zu Gln Arg zu Pro Arg zu Stop Glu zu Stop Gly zu Ala Cys zu Gly Cys zu Arg Thr zu Met Arg zu Cys Arg zu Gly Trp zu Stop Trp zu Arg Arg zu Trp Arg zu Cys Arg zu His Pro zu Arg Frameshift Frameshift Frameshift Cys zu Gly Thr zu Met Frameshift Frameshift (Glucksmann, Lehto et al. 1997) (Lehto, Wipemo et al. 1999) (Ellard, Bulman et al. 1999) (Vaxillaire, Rouard et al. 1997) (Hattersley 1998) (Urhammer, Fridberg et al. 1997) (Hansen, Eiberg et al. 1997) (Frayling, Bulamn et al. 1997) (Yamagata, Oda et al. 1996) (Frayling, Bulamn et al. 1997) (Hattersley 1998) (Vaxillaire, Rouard et al. 1997) (Hansen, Eiberg et al. 1997) (Yamada, Tomura et al. 1999) (Vaxillaire, Rouard et al. 1997) (Frayling, Bulamn et al. 1997) (Chevre, Hani et al. 1998) (Vaxillaire, Rouard et al. 1997) (Iwasaki, Oda et al. 1997) (Chevre, Hani et al. 1998) (Hattersley 1998) (Iwasaki, Oda et al. 1997) (Lindner, Cockburn et al. 1999) (Dalgaard et al. 1998) (Kaisaki, Menzel et al. 1997) (Dalgaard et al. 1998) (Cox, Southam et al. 1999) (Kaisaki, Menzel et al. 1997) (Hattersley 1998) (Glucksmann, Lehto et al. 1997) (Iwasaki, Oda et al. 1997) (Gragnoli, Lindner et al. 1997) (Hattersley 1998) (Hattersley 1998) (Chevre, Hani et al. 1998) (Yoshiuchi, Yamagata et al. 1999) (Glucksmann, Lehto et al. 1997) (Dalgaard et al. 1998) (Yamagata, Oda et al. 1996) (Vaxillaire, Rouard et al. 1997) (Vaxillaire, Rouard et al. 1997) (Kaisaki, Menzel et al. 1997) (Bellanet-Chantelot et al. 1998) (Lehto, Wipemo et al. 1999) (Yamagata, Oda et al. 1996) Frameshift Frameshift Frameshift Frameshift Frameshift (Vaxillaire, Rouard et al. 1997) (Hansen, Eiberg et al. 1997) (Iwasaki, Oda et al. 1997) (Kaisaki, Menzel et al. 1997) (Hattersley 1998) Gly zu Arg Thr zu Iso Frameshift (Yoshiuchi, Yamagata et al. 1999) (Yagi et al. 1999) (Frayling, Bulamn et al. 1997) Pro zu Leu Gln zu Stop Pro zu Leu Thr zu Arg Frameshift (Yamagata, Oda et al. 1996) (Ordonez 1999) (Frayling, Bulamn et al. 1997) (Elbein, Teng et al. 1998) (Yamagata, Oda et al. 1996) C 9 9 9 Frameshift Arg zu Gly Frameshift (Hansen, Eiberg et al. 1997) (Yamada, Nishigori et al. 1997) (Iwasaki, Oda et al. 1997) E619K T620I Insertion von A CGG zu CAG Insertion von TC GAG zu AAG ACC zu ATC Glu zu Lys Thr zu Ile (Elbein, Teng et al. 1998) (Frayling, Bulamn et al. 1997) IVS2ntdelGinsA AG zu AA (Ng, Li et al. 2000) I5 IVS5ntdelAinsG AG zu GG I9 IVS9ntdelGinsAA GT zu AT SpliceAkzeptor Verlust SpliceAkzeptor Verlust Splice Donor Verlust 10 10 Intron I2 A559insA R583G L584S585insTC (Yamagata, Oda et al. 1996) (Yamagata, Oda et al. 1996) Tab 17: Bisher beschriebene Mutationen im Glucokinase-Gen Exon Mutation Austausch 1 1 1 1 D4N E9delG A11T K15insGdelT GAC zu AAC Deletion GCC zu CCC GT zu GG 2 2 2 2 2 2 2 2 Q26X R36W Q38P E40ins21 H50Y H50R A53S V62A CAG zu TAG CGG zu TGG CAG zu CCG Insertion von 21 CAT zu TAT CAT zu CGC GGC zu TGC GTG zu GCT 2 3 3 3 3 3 3 3 3 3 3 E70K G72R D73G G80A G80S G81S L88del10 E93del10 Q98X T103N M121insGdelT GAA zu AAA GGG zu CGG GAC zu GGC GGT zu GCT GGT zu AGT GGC zu AGC Deletion von 10 Deletion von 10 CAG zu TAG ACC zu AAC TG zu GG 4 L122insTdelG AG zu AT 4 4 4 4 4 4 4 4 4 4 4 4 4 L122insGdelT L122V Y125del C129Y S131P L134P Q138P H137R F150del F150S S151delCC E157K D158A GCTC zu GGCC CTC zu GTC Deletion TGC zu TAC TCC zu CCC CTG zu CCG CAG zu CCG CAT zu CGT Deletion von TT TTC zu TCC Deletion von CC GAA zu AAA GAC zu GCC 4 4 4 K161ins2del10 K161ins2del15 K161delT Insertion von 2 Insertion von2 Deletion von T 5 5 5 5 5 5 5 T168P G175R G175V A176S N180K V182M R186X ACC zu CCC GGA zu CGA GGA zu GTA GCA zu TCA AAT zu AAG GTG zu ATG CGA zu TGA funktionelle Auswirkung Asp zu Asn Frameshift Ala zu Thr SpliceDonor Verlust Gln zu Stop Arg zu Trp Gln zu Pro Frameshift His zu Tyr His zu Arg Ala zu Ser Val zu Ala Glu zu Lys Gly zu Arg Asp zu Gly Gly zu Ala Gly zu Ser Gly zu Ser Frameshift Frameshift Gln zu Stop Thr zu Asn SpliceDonor Verlust SpliceAkzeptor Verlust Frameshift Leu zu Val Frameshift Cys zu Tyr Ser zu Pro Leu zu Pro Gln zu Pro His zu Arg Frameshift Phe zu Ser Frameshift Glu zu Lys Asp zu Ala Frameshift Frameshift SpliceDonor Verlust Thr zu Pro Gly zu Arg Gly zu Val Ala zu Ser Asp zu Lys Val zu Met Arg zu Stop Veröffentlicht (Bell, Pilkis et al. 1996) (Massa, Meschi et al. 2001) (Bell, Pilkis et al. 1996) (Massa, Meschi et al. 2001) (Massa, Meschi et al. 2001) (Hager, Blanche et al. 1994) (Massa, Meschi et al. 2001) (Froguel 1998) (Massa, Meschi et al. 2001) (Massa, Meschi et al. 2001) (Velho, Blanche et al. 1997) (Njolstad, Cockburn et al. 1998) (Burke, Buettger et al. 1999) (Froguel 1998) (Massa, Meschi et al. 2001) (Velho, Blanche et al. 1997) (Guazzini, Gaffi et al. 1998) (Massa, Meschi et al. 2001) (Hager, Blanche et al. 1994) (Massa, Meschi et al. 2001) (Bell, Pilkis et al. 1996) (Massa, Meschi et al. 2001) (Massa, Meschi et al. 2001) (Velho, Blanche et al. 1997) (Hattersley 1998) (Massa, Meschi et al. 2001) (Massa, Meschi et al. 2001) (Massa, Meschi et al. 2001) (Stoffel, Bell et al. 1993) (Massa, Meschi et al. 2001) (Massa, Meschi et al. 2001) (Velho, Blanche et al. 1997) (Massa, Meschi et al. 2001) (Hattersley 1998) (Massa, Meschi et al. 2001) (Massa, Meschi et al. 2001) (Davis, Cuesta-Munoz et al. 1999) (Hattersley 1998) (Bell, Pilkis et al. 1996) (Velho, Blanche et al. 1997) (Velho, Blanche et al. 1997) (Bell, Pilkis et al. 1996) (Moises, Reis et al. 2001) (Froguel 1998) (Ellard, Beards et al. 2000) (Bell, Pilkis et al. 1996) (Velho, Blanche et al. 1997) D 5 5 5 A188T R191W G193del33 GCT zu ACT CGG zu TGG Deletion von 33 6 6 6 6 6 6 6 6 6 6 6 6 7 V203A T206M T209M M210T C213R Y215X E221K G223S M224T I225M V226M V226delTinsAA G227delAAinsT GTG zu GCG ACG zu ATG ACG zu ATG ATG zu ACG TGC zu CGC TAC zu TAA GAG zu AAG GGC zu AGC ATG zu ACG ATC zu ATG GTG zu ATG Deletion von T Deletion von AA 7 7 7 7 7 7 7 7 7 7 7 7 7 7 7 7 7 8 G227C T228M N231delA M238delT E248X M251I E256K E257R A259T G261R G261E G264S E265X L271del22 E279X E279Q S281F L288delGinsA GGT zu TGT ACG zu ATG Deletion von A Deletion von T GAG zu TAG ATG zu ATC GAG zu AAG TGG zu TGA GCC zu CCC GGG zu AGG GGG zu GAG GGC zu AGC GAG zu TAG Deletion von 22 GAG zu TAG GAG zu CAG TCT zu TTT AG zu AA 8 8 8 8 8 9 9 9 9 9 9 9 9 9 9 9 10 G299R E300Q E300K L309P S336L S360X V367M C382Y S383delC A384T G385X E395-R403del V401fsdelT R403dsdelC K414E L415V S418delGinsC GGC zu CGC GAG zu CAG GAG zu AAG CTC zu CCC TCG zu TTG TCG zu TAG GTG zu ATG TGC zu TAC Deletion von C GCG zu CCG GGG zu TGG Deletion GTA zu TAA Deletion von C AAG zu GAG CTG zu GTG Deletion von G 10 10 10 S441W S453X V455M TCG zu TGG TCG zu TAG GTG zu ATG Ala zu Thr Arg zu Trp SpliceDonor Verlust Val zu Ala Thr zu Met Thr zu Met Met zu Thr Cys zu Arg Tyr zu Stop Glu zu Lys Gly zu Ser Met zu Thr Ile zu Met Val zu Met Frameshift SpliceAkzeptor Verlust Gly zu Cys Thr zu Met Frameshift Frameshift Glu zu Stop Met zu Ile Glu zu Lys Trp zu Arg Ala zu Thr Gly zu Arg Gly zu Glu Gly zu Ser Gly zu Stop Frameshift Glu zu Stop Glu zu Gln Ser zu Phe SpliceDonor Verlust Gly zu Arg Glu zu Gln Glu zu Lys Leu zu Pro Ser zu Leu Ser zu Stop Val zu Met Cys zu Tyr Frameshift Ala zu Thr Gly zu Stop Deletion Stop Frameshift Lys zu Glu Leu zu Val SpliceAkzeptor Verlust Ser zu Trp Ser zu Stop Val zu Met (Shimada, Makino et al. 1993) (Ellard, Beards et al. 2000) (Hager, Blanche et al. 1994) (Burke, Buettger et al. 1999) (Massa, Meschi et al. 2001) (Hager, Blanche et al. 1994) (Velho, Blanche et al. 1997) (Velho, Blanche et al. 1997) (Ellard, Beards et al. 2000) (Massa, Meschi et al. 2001) (Massa, Meschi et al. 2001) (Massa, Meschi et al. 2001) (Massa, Meschi et al. 2001) (Velho, Blanche et al. 1997) (Costa, Bescos et al. 2000) (Hattersley 1998) (Guazzini, Gaffi et al. 1998) (Stoffel, Froguel et al. 1992) (Massa, Meschi et al. 2001) (Costa, Bescos et al. 2000) (Velho, Blanche et al. 1997) (Massa, Meschi et al. 2001) (Bell, Pilkis et al. 1996) (Bell, Pilkis et al. 1996) (Hattersley 1998) (Stoffel, Froguel et al. 1992) (Hager, Blanche et al. 1994) (Massa, Meschi et al. 2001) (Stoffel, Bell et al. 1993) (Massa, Meschi et al. 2001) (Bell, Pilkis et al. 1996) (Bell, Pilkis et al. 1996) (Massa, Meschi et al. 2001) (Ellard, Beards et al. 2000) [(Stoffel, Patel et al. 1992) (Bell, Pilkis et al. 1996) (Burke, Buettger et al. 1999) (Bell, Pilkis et al. 1996) (Velho, Blanche et al. 1997) (Velho, Blanche et al. 1997) (Velho, Blanche et al. 1997) (Froguel) (Massa, Meschi et al. 2001) (Hattersley 1998) (Costa, Bescos et al. 2000) (Massa, Meschi et al. 2001) (Velho, Blanche et al. 1997) (Froguel) (Bell, Pilkis et al. 1996) (Massa, Meschi et al. 2001) (Bell, Pilkis et al. 1996) (Massa, Meschi et al. 2001) (Massa, Meschi et al. 2001) (Glaser, Kesavan et al. 1998) E Tab 18: Übersichten der Identifikationsnummern untersuchter Gene GCK Glucokinase Synonyme: Hexokinase IV OMIM: 138097 McCusick: Größe: 465 As Molekulargewicht: 52 kDa HNF1α Synonyme: 125851 McCusick: Größe: 631 As Molekulargewicht: 600496 McCusick: Größe: 702 As Molekulargewicht: GDB ID: 67 kDa 263570 125297 EC: 138442 EC: GDB ID: Größe: 1092 As Molekulargewicht: 120286 OMIM: 172470 CAV3 EC: GDB ID: Synonyme: 2.4.1.18 LocusLink ID: 22632 2.7.1.38 LocusLink ID: 5257 124 kDa Genomische Lokalisation: Phosphorylase Kinase Unterinheit γ1 Molekulargewicht: LocusLink ID: 6927 Genomische Lokalisation: 3p21 Phosphorylase Kinase Unterinheit β Größe: 386 As LocusLink ID: 2645 Genomische Lokalisation: 12q24.2 GDB ID: 80 kDa OMIM: 172490 PhKG1 127550 Genomische Lokalisation: 7p15.1-3 Glycogen Branching Enzyme OMIM: 232500 PhKB 2.7.1.1 TCF1, LF-B1, APF OMIM: 142410 GBE GDB ID: EC: 44 kDa 16q12-13 2.7.1.38 LocusLink ID: 5260 Genomische Lokalisation: 7q21-q22 m-Caveolin OMIM: 601253 McCusick : 606072 Größe: 151 As Molekulargewicht: GDB ID: 17 kDa 9837402 LocusLink ID: 859 Genomische Lokalisation: 3p25 F 7 Danksagung Für die Überlassung des Themas, die intensive Betreuung während meiner Experimente, die gute und erfolgreiche Zusammenarbeit bei bisherigen Veröffentlichungen und die ständige Diskussionsbereitschaft möchte ich Herrn PD Dr. Vorgerd ganz herzlich danken. Seine klaren Vorstellungen und die gute Planung der Projekte ermöglichten überhaupt erst die Bearbeitung mehrerer Krankheitsbilder. Bedanken möchte ich mich bei Professor Dr. Pfeiffer (inzwischen Deutsches Institut für Ernährungsforschung Potsdam, Universitätsklinikum Benjamin Franklin, Berlin) für die Betreuung des Screenings der MODY-Patienten und die vielen gemeinsamen Gespräche und Kongressaufenthalte. Ohne ihn wäre die intensive Probenbeschaffung und experimentelle Durchführung nicht möglich gewesen. Herrn Professor Dr. Malin und Herrn Professor Dr. Schatz danke ich für die großzügige Bereitstellung der Räumlichkeiten und Geräte. Ich danke Frau Professor Dr. Shin (Labor für Stoffwechselgenetik, München) für die gute Zusammenarbeit bei der Bestimmung der Enzymaktivitäten, Herrn Professor Dr. Müller (Pathologisches Institut, Berufsgenossenschaftliche Kliniken Bergmannsheil Bochum) und Herrn Professor Dr. Schröder histopathologischen (Pathologisches Untersuchungen. Frau Institut, Dr. Universitätsklinik Bellanét-Chantelot Aachen) (Centre für d’ Ètude die du Polymorphisme humain, Paris) bin ich für die gute Zusammenarbeit zum Nutzen der Patienten dankbar. Andrew Hattersley und Philip Boutin danke ich für die prompte und uneigennützige Bereitstellung der Proben und Prof. Dr. Gerbitz für die Bereitstellung einer Positiv-Kontrolle zur MIDD-Analyse. Gerne denke ich zurück an die gute Atmosphäre des Endokrinologischen Labors. Hier möchte ich besonders Frau Salami-Shojaie und Herrn Dr. Bruno Voss für die gute Zusammenarbeit sowie Herrn Dr. Osterhoff und Dr. Assert für die wertvollen Hilfestellungen und Kommentare danken. Dankbar bin ich für die gemeinsamen Gespräche bei der Etablierung der Silberfärbung mit Herrn Dr. Laszdins. Ich bedanke mich bei Herrn Prof. Dr. Kunau und Herrn Prof. Dr. Duntze; durch die Mitarbeit in ihren Laboren war es mir möglich, grundlegende Techniken der Molekularbiologie zu erlernen. Hier hat mich insbesondere Dr. Garnett Will durch ihre geduldige und gründliche Vorgehensweise vorbildlich eingeführt. Vielen Dank auch an meinen Bruder für die Beseitigung letzter orthographischer Stolpersteine. G 8 Lebenslauf Name: Focke Ziemssen Geburtstag: 9. Mai 1975 Geburtsort: Dortmund Eltern: Dr.Malte Ziemssen (Augenarzt) Ulrike Ziemssen geb.Kuithan (Sonderschullehrerin) Konfession: evangelisch Staatsangehörigkeit: deutsch Schulausbildung: 1981-85 Libori-Grundschule Dortmund 1986-94 Stadtgymnasium Dortmund mit Erwerb der allgemeinen Hochschulreife 1987 Sieger des altsprachlichen Wettbewerbs des Altphilologenvereins 1989 Bundessieger im Europäischen Zeichen-Wettbewerb 1994 Hans-Uhde-Preis Wehrdienst: 1994-95 Wehrdienst (Stabsdienstsoldat InstRegiment 7, Hauptgefreiter) Universitätsausbildung: 01.01.1995 Immatrikulation an der Ruhr-Universität Bochum als Student der Humanmedizin Aug 1997 Ärztliche Vorprüfung Aug 1998 1. Abschnitt der Ärztlichen Prüfung Aug 2000 2. Abschnitt der Ärztlichen Prüfung Okt 2001 3. Abschnitt der Ärztlichen Prüfung Seit Dez 2001 Arzt im Praktikum an der Universitätsaugenklinik Tübingen