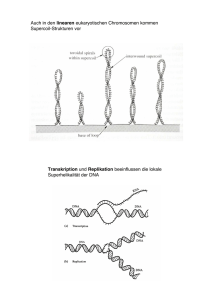
Die Rolle von DNA-Gyrase und Efflux für die
Werbung

Die Rolle von DNA-Gyrase und Efflux für die Antibiotikaempfindlichkeit, die Fitness und den DNA-Superspiralisierungsgrad eines Salmonella-Lebendimpfstammes Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades im Fachbereich Chemie der Universität Hamburg vorgelegt von Antje Preisler aus Gera - Hamburg 2006 - Die vorliegende Arbeit wurde in der Zeit von März 2002 bis Februar 2006 unter der Leitung von Herrn Prof. Dr. Peter Heisig in der Abteilung für Pharmazeutische Biologie und Mikrobiologie am Institut für Pharmazie der Universität Hamburg angefertigt. 1. Gutachter: Prof. Dr. P. Heisig (Universität Hamburg, Abteilung für Pharmazeutische Biologie und Mikrobiologie) 2. Gutachter: Prof. Dr. U. Hahn (Universität Hamburg, Abteilung für Biochemie und Molekularbiologie) Disputationstermin: 25.09.2006 Meinem Mann Sascha, meiner Schwester Andrea und meinen Eltern gewidmet Inhalt 1 Einleitung...................................................................................................................1 1.1 Salmonella als Infektionserreger .......................................................................1 1.1.1 Die Gattung Salmonella.....................................................................................1 1.1.2 Das Auftreten von Salmonellosen......................................................................2 1.1.3 Die Pathogenität von Salmonella.......................................................................4 1.1.3.1 Die Salmonella-Pathogenitätsinseln...........................................................4 1.1.3.2 Adhäsion und Invasion von Salmonella und die Funktion ………………….. des Typ III-Sekretionssystems....................................................................5 1.1.3.3 Überlebensmechanismen von Salmonella in den Wirtszellen ……………… und die Auswirkungen der Besiedlung .......................................................6 1.2 Das Auftreten und die Verbreitung resistenter Salmonellen...........................8 1.3 Fluorchinolone – Wirkung und Resistenzmechanismen...............................10 1.3.1 Die Topoisomerasen als Zielstrukturen für Chinolone ..................................... 11 1.3.1.1 Die DNA-Topologie in der Zelle ................................................................ 11 1.3.1.2 Die Wirkung von Chinolonen ....................................................................16 1.3.1.3 Die Auswirkung von Mutationen in den Genen der Topoisomerasen .......17 1.3.2 Verringerung der Akkumulation von Antibiotika – Die Bedeutung ………………. von Porinen und Efflux-Systemen ...................................................................19 1.3.3 Neue Resistenzmechanismen und enzymatische Inaktivierung ………………… von Fluorchinolonen ........................................................................................22 2 1.4 Die Entwicklung von Salmonella-Impfstämmen.............................................24 1.5 Zielstellung der Arbeit.......................................................................................28 Material und Methoden .........................................................................................29 2.1 Material...............................................................................................................29 2.1.1 Bakterienstämme.............................................................................................29 2.1.2 Vektoren...........................................................................................................30 2.1.3 Oligonukleotide ................................................................................................31 2.1.4 Nährmedien .....................................................................................................34 2.1.5 Verwendete Kits...............................................................................................35 2.1.6 Puffer und Lösungen .......................................................................................35 2.1.7 Enzyme............................................................................................................37 2.1.8 Antibiotika ........................................................................................................38 2.1.9 Chemikalien .....................................................................................................38 2.1.10 Geräte und Materialien.................................................................................40 Inhalt 2.2 Methoden ........................................................................................................... 41 2.2.1 Mikrobiologische Methoden............................................................................. 41 2.2.1.1 Herstellung von Übernachtkulturen (ÜNK)............................................... 41 2.2.1.2 Herstellung und Verwendung von Dauerkulturen..................................... 41 2.2.1.3 Bestimmung der Lebendzellzahl (KBE) ................................................... 41 2.2.1.4 Bestimmung der Generationszeit............................................................. 42 2.2.1.5 In-vitro-Selektion resistenter Keime ......................................................... 43 2.2.1.6 Bestimmung der minimalen Hemmkonzentration (MHK) ......................... 44 2.2.1.6.1 MHK-Wert-Bestimmung mittels selbst angelegter Platten................... 45 2.2.1.6.2 MHK-Wert-Bestimmung mittels Fertigplatten ...................................... 45 2.2.1.6.3 MHK-Wert-Bestimmung in Anwesenheit eines …………………………… Effluxpumpeninhibitors ........................................................................ 46 2.2.1.6.4 MHK-Wert-Bestimmung über Agardilutionsverfahren.......................... 46 2.2.1.7 Methoden des DNA-Transfers.................................................................. 47 2.2.1.7.1 Calciumchlorid (CaCl2)-Methode ......................................................... 47 2.2.1.7.2 Elektroporation .................................................................................... 48 2.2.1.7.3 Konjugation.......................................................................................... 49 2.2.1.8 Komplementationstest.............................................................................. 50 2.2.1.9 Bestimmung des DNA-Superspiralisierungsgrades ................................. 51 2.2.1.9.1 Verwendung der Luciferase-Reportergensysteme .............................. 52 2.2.1.9.2 Messung der Luciferase-Aktivität ........................................................ 52 2.2.1.9.3 Berechnung des Qsc-Wertes .............................................................. 53 2.2.1.10 In-vitro-Mutagenese mittels homologer Rekombination ....................... 54 2.2.1.10.1 Homologe Rekombination unter Verwendung des Vektors pKOV ..... 54 2.2.1.10.2 Sequenzspezifische Rekombination mit dem Lambda Red-System . 57 2.2.1.10.3 System zur Selektion hergestellter Rekombinanten .......................... 58 2.2.2 Molekularbiologische Methoden...................................................................... 61 2.2.2.1 Isolierung chromosomaler DNA ............................................................... 61 2.2.2.2 Isolierung von Plasmid-DNA .................................................................... 61 2.2.2.2.1 Plasmid-Minipräparation nach Birnboim und Doly (modifiziert) ........... 61 2.2.2.2.2 Plasmid-Minipräparation-Kits............................................................... 62 2.2.2.2.3 Plasmid-Midipräparation ...................................................................... 62 2.2.2.3 Konzentrationsbestimmung und Reinheitsprüfung von DNA ................... 63 2.2.2.4 Konzentrationsbestimmung von Oligonukleotiden ................................... 63 2.2.2.5 Amplifikation von DNA ............................................................................. 64 2.2.2.5.1 Polymerase-Kettenreaktion (PCR) ...................................................... 64 2.2.2.5.2 PCR-Fusions-Technik (SOEing) .......................................................... 65 2.2.2.5.3 Sequenzspezifische PCR (ARMS) ...................................................... 66 2.2.2.6 Gelelektrophoretische Auftrennung von DNA-Fragmenten ...................... 67 2.2.2.7 Extraktion von DNA-Fragmenten aus Agarosegelen ............................... 67 Inhalt 2.2.2.8 Aufreinigung und Konzentrierung von DNA..............................................68 2.2.2.8.1 Nutzung von Silika-Säulen...................................................................68 2.2.2.8.2 Ethanol-Präzipitation und Phenol-Chloroform-Extraktion.....................68 2.2.2.9 Enzymatische Modifikation von DNA........................................................69 2.2.2.9.1 Restriktionsspaltung.............................................................................69 2.2.2.9.2 Restriktionsfragmentlängenpolymorhismus (RFLP) ............................69 2.2.2.9.3 Dephosphorylierung.............................................................................70 2.2.2.9.4 Erzeugen von glatten Enden für die Klonierung ..................................70 2.2.2.9.5 Ligation mittels T4-DNA-Ligase ...........................................................71 2.2.2.9.6 Ligation mittels Topoisomerase I inklusive Blau-Weiß-Selektion .........71 2.2.2.10 Sequenzierung von DNA ......................................................................73 2.2.3 Proteinbiochemische Methoden ......................................................................74 2.2.3.1 Probenvorbereitung ..................................................................................74 2.2.3.2 Bestimmung der Proteinkonzentration .....................................................75 2.2.3.3 2D-Gelelektrophorese ..............................................................................76 2.2.3.3.1 Erste Dimension: Isolektrische Fokussierung (IEF) .............................76 2.2.3.3.2 Zweite Dimension: SDS-PAGE ............................................................77 2.2.3.3.3 Färbemethoden: Coomassiefärbung und Silberfärbung ......................78 2.2.3.3.4 Imaging, Detektion und Bildauswertung mittels PDQuest® ..................80 3 Ergebnisse ............................................................................................................. 81 3.1 Charakterisierung der Ausgangsstämme .......................................................81 3.1.1 Bestimmung der Generationszeiten ................................................................82 3.1.2 Bestimmung der Empfindlichkeiten gegenüber verschiedenen Antibiotika .....83 3.1.3 Bestimmung der DNA-Superspiralisierungsgrade ...........................................86 3.1.4 DNA-Sequenzanalyse der Gene der Fluorchinolon-Zielstrukturen und cpxA..88 3.1.5 In-Vitro-Selektionsversuche.............................................................................90 3.1.5.1 Selektionsversuch mit S. Typhimurium M415...........................................90 3.1.5.2 Selektionsversuch mit TAD Salmonella vacT ...........................................93 3.1.6 Komplementationstest mittels eines plasmidcodierten gyrA+-Gens.................96 3.1.6.1 Bestimmung der Fluorchinolonempfindlichkeiten in Anwesenheit …………. von gyrA+ ..................................................................................................96 3.1.6.2 Untersuchung von Wachstum und Qsc-Werten in Anwesenheit …………… von gyrA+ ..................................................................................................98 3.1.7 Optimierung der Methode der 2D-Gelelektrophorese....................................100 3.1.7.1 Optimierung der Probenaufbereitung und der Bestimmung ………………... der Proteinkonzentration ........................................................................100 3.1.7.2 Optimierung der Bedingungen für die isoelektrische Fokussierung ……….. und die SDS-PAGE ................................................................................101 3.1.7.3 Optimierung der Färbemethode .............................................................103 Inhalt 3.2 Untersuchung der Rolle der im Impfstamm auftretenden ……………………… gyrA-Mutationen.............................................................................................. 105 3.2.1 Überblick über die Systeme zur Rekombination ........................................... 105 3.2.1.1 Insertion eines gyrA-Gens mit Mutationen ins Chromosom von M415 .. 106 3.2.1.2 Knock out des chromosomalen gyrA-Gens und Verwendung ……………… eines Rescueplasmides ......................................................................... 107 3.2.1.3 System zur Selektion nach positiver homologer Rekombination ........... 109 3.2.2 Auswahl eines geeigneten Rekombinationssystems .................................... 110 3.2.3 Klonierungsstrategien zur In-vitro-Mutagenese mittels …………………………… homologer Rekombination ............................................................................ 113 3.2.3.1 Mutagenese mittels SOEing................................................................... 113 3.2.3.2 Klonierung der Mutagenese-Fragmente in pCR2.1TOPO®.................... 115 3.2.3.3 Klonierung von gyrA in pT7-6................................................................. 116 3.2.3.4 Klonierung der Rekombinationsvektoren ausgehend von pKOV ........... 117 3.2.4 Homologe Rekombination, Selektion und Screening der Mutanten .............. 121 3.2.4.1 Entwicklung eines Screeningsystems zum Nachweis …… ………………… der gyrA-Mutationen (ARMS) ................................................................. 121 3.2.4.2 Herstellung und Selektion der Rekombinanten...................................... 123 3.2.5 Charakterisierung der Rekombinanten.......................................................... 126 3.2.5.1 Bestimmung der Fluorchinolon-MHK-Werte........................................... 127 3.2.5.2 Komplementationstest mittels eines plasmidcodierten gyrA+ ................. 128 3.2.5.3 Bestimmung der DNA-Superspiralisierungsgrade ................................. 129 3.2.5.4 Untersuchungen zum Wachstumsverhalten........................................... 131 3.3 Untersuchung des Einflusses von Effluxpumpen ....................................... 131 3.3.1 MHK-Wert-Bestimmung des Effluxpumpeninhibitors .................................... 132 3.3.2 MHK-Wert-Bestimmung unter Verwendung eines Effluxpumpeninhibitors ... 132 4 Diskussion.................................................................................................. 135 4.1 Charakterisierung der Untersuchungsstämme............................................ 136 4.1.1 Mutationen in den Topoisomerasegenen....................................................... 136 4.1.2 Untersuchung des cpxA-Gens ...................................................................... 138 4.1.3 Bestimmung der Empfindlichkeiten gegenüber verschiedenen …………………. Antibiotikaklassen.......................................................................................... 140 4.1.4 Veränderungen im DNA-Superspiralisierungsgrad ....................................... 143 4.1.5 Wachstum als Fitnessparameter ................................................................... 145 4.1.6 Proteomics als globaler Ansatz ..................................................................... 150 Inhalt 4.2 Untersuchungen der im Impfstamm auftretenden gyrA-Mutationen ..........153 4.2.1 Komplementationstest mittels eines plasmidcodierten gyrA+ ........................153 4.2.2 In-vitro-Selektion von gyrA-Mutanten ............................................................154 4.2.3 In-vitro-Mutagenese von gyrA-Mutanten mittels homologer Rekombination .156 4.2.3.1 Einfluss der gyrA-Mutationen auf die MHK-Werte ..................................156 4.2.3.2 Beeinflussung des DNA-Superspiralisierungsgrades.............................158 4.2.3.3 Einfluss der untersuchten gyrA-Mutationen auf die Fitness ...................159 4.3 Der Einfluss von Effluxpumpen .....................................................................161 4.3.1 Der Einfluss eines Effluxpumpeninhibitors ....................................................162 4.3.2 Expressionsstudien der Pumpen-Systeme AcrAB- und AcrEF-TolC .............164 4.3.3 Der Einfluss defekter Effluxpumpen auf die Virulenz.....................................165 4.4 Ausblick ...........................................................................................................167 5 Zusammenfassung ............................................................................................. 169 6 Literatur................................................................................................................. 173 7 Abkürzungsverzeichnis .................................................................................... 189 8 Anhang / Gefahrstoffliste ……………………………………………………........ 191 Danksagung Lebenslauf / Veröffentlichungen Einleitung 1 Einleitung 1.1 Salmonella als Infektionserreger 1.1.1 Die Gattung Salmonella Die Salmonellose, hervorgerufen durch zur Gattung Salmonella gehörende Bakterien, stellt weltweit eine der häufigsten bakteriellen Infektionskrankheiten dar [120,131,283]. Bei Salmonellen handelt es sich um gramnegative, stäbchenförmige, peritrich begeißelte und fakultativ anaerobe Bakterien von 0,7-1,5 x 2,0-5,0 µm Größe, welche in der Lage sind, sich in einem weiten pH- und Temperaturbereich zu vermehren. Der Gattungsname geht auf den amerikanischen Veterinär Daniel E. Salmon (1850-1914) zurück, in dessen Labor 1885 die Schweinecholerabakterien S. Choleraesuis entdeckt wurden. Erstmals wurden Bakterien der Gattung Salmonella 1880 als „Typhusbakterien“ von Eberth in mikroskopischen Präparaten von Gewebeproben an Typhus Verstorbener gefunden. Als Verursacher einer Fleischvergiftung wurden Salmonellen 1888 erstmals von Gärtner nachgewiesen [131]. Die Gattung Salmonella zählt zur Familie der Enterobacteriaceae und wird in die beiden Species enterica und bongori unterteilt, welche sich erneut in verschiedene Subspecies gliedern. 99,5% der isolierten Salmonella-Stämme werden Salmonella enterica ssp. enterica zugeordnet. Einzelne Subspecies werden aufgrund unterschiedlicher Kapsel-, Zellwand-Antigene (O-Antigene) und Flagella-Antigene (HAntigene) nach dem Kauffmann-White-Schema [174] wiederum in ca. 2500 Serovare (Serotypen) differenziert. Häufig wird als verkürzte Form nur die Serovarbezeichnung angegeben, z.B. Salmonella Typhimurium statt Salmonella enterica ssp. enterica Serovar Typhimurium [35]. Einige humanmedizinisch bedeutsame Serovare wie Typhimurium, Enteritidis und Paratyphi werden außerdem noch weiter in Lysotypen unterteilt. Für diese weitere epidemiologische Charakterisierung verschiedender Stämme innerhalb eines Serovars nutzt man das phage-typing oder Lysotypie. Darunter versteht man die Möglichkeit, Phagen zur Typendifferenzierung (Lysotyp) von Bakterien einzusetzen. Aufgrund spezifischer Rezeptoren auf der Bakterienoberfläche (die auch bei Stämmen des gleichen Serovars unterschiedlich sein können) und der Wirtsspezifität der Phagen, können bestimmte Phagen nur bestimmte Bakterienstämme lysieren. Die zu untersuchenden Isolate werden mit Phagen infiziert und produzieren ein definiertes Lysemuster, anhand dessen die Isolate einem bestimmten Phagentyp zugeordnet werden. Epidemiologisch bedeutsame Lysotypen von S. Typhimurium sind z.B. DT 204c und DT 104 (DT = defined phage type). Trotz der starken Differenzierung der Salmonellen werden sie als genetisch sehr einheitlich eingestuft. 1 Einleitung 1.1.2 Das Auftreten von Salmonellosen Nur ca. 20 bis 30 Salmonella-Serovare sind human- oder tierpathogen. Dazu zählen unter anderem die Erreger von Typhus und Paratyphus, aber auch die Erreger akuter Durchfallerkrankungen (Gastroenteritis). Die durch Salmonella spec. ausgelösten Krankheitsbilder lassen sich also in „typhöse“ und „enteritische“ Salmonellosen unterscheiden. Die Erreger „typhöser“ Salmonellosen sind S. Typhi und S. Paratyphi, welche nach einer geringen Infektionsdosis (100-1000 Bakterien) und nach einer Inkubationsdauer von 3 bis 30 Tagen die als Typhus bekannte Kranhkeit auslösen. Nach einem Stadium mit stufenweise ansteigendem Fieber und unspezifischen Symptomen wie Übelkeit, Kopfund Gliederschmerzen folgt unbehandelt ein bis zu 3 Wochen dauerndes Stadium mit kontinuierlich hohem Fieber, breiartigen Stuhlentleerungen und beeinträchtigtem Bewusstsein (griechisch typhos: Nebel, Rauch) des Patienten [258]. Das Reservoir der Typhus-Erreger ist der Mensch und eine Infektion erfolgt über die Aufnahme von Wasser und Lebensmitteln, die durch Ausscheidungen kontaminiert sind. Präventiv kann dieser Krankheit durch Impfung vorgebeugt werden: zum einen durch einen oralen Lebendimpfstoff und zum anderen durch einen parental zu verabreichenden Impfstoff, der aus hochgereinigten Antikörpern besteht [258]. Eine „enteritische“ Salmonellose nimmt einen wesentlich leichteren Verlauf. Vor allem die Serovare S. Typhimurium und S. Enteritidis haben als Verursacher lebensmittelbedingter Infektionen große Bedeutung erlangt [120]. Die infektiöse Dosis liegt je nach Beschaffenheit des Immunsystems des betroffenen Menschen zwischen 103 und 106 Bakterien [82,269]. Nach einer Inkubationszeit von 5 bis 72 h, abhängig von der Infektionsdosis, setzen Leibschmerzen und wässriger Stuhl ein, oft verbunden mit Fieber, Übelkeit und Kopfschmerzen. Die Symptome dauern in der Regel nur einige Tage oder Stunden, allerdings kann bei Älteren oder Kleinkindern schnell eine Dehydration eintreten. In seltenen Fällen kommt es zu Schüttelfrost, hohem Fieber bis zum Kollaps oder es treten andere Komplikationen auf, die bis zum Tod führen können. In schweren Fällen erfolgt eine Behandlung der „enteritischen“ Salmonellose mit Antibiotika. Im folgenden Text ist mit dem allgemeinen Begriff „Salmonellose“ die Infektionskrankheit hervorgerufen durch „enteritische“ Salmonella gemeint. Andere Erreger bakterieller Gastroenteritiden sind z.B. Campylobacter spec., Yersinia enterocolitica oder Shigella spec. [131]. Insbesondere Geflügel, Schweine und Rinder gelten als primäre Salmonella- Infektionsquellen. Dabei werden die Bakterien hauptsächlich über kontaminierte Lebensmittel wie Fleisch und Speisen, welche Rohei enthalten, übertragen. Salmonellosen sind Zoonosen, d.h. Erkrankungen, die vom Tier auf den Menschen übertragbar sind. Präventive Maßnahmen zur Salmonellosenbekämpfung haben deshalb die Verhinderung einer Übertragung vom Nutztier zum Menschen zum Ziel. Die Überwachung und Bekämpfung wird über verschiedene nationale und europäische 2 Einleitung Richtlinien und Verordnungen, z.B. Zoonose-Verordnung oder Hühnereiverordnung, geregelt [269]. Außer in Nutztieren kommt Salmonella spec. auch in Wildvögeln, Nagetieren, Reptilien und Insekten vor. Als weitere Reservoire gelten Gewässer oder das Erdreich. Diese werden häufig über Abwässer oder Düngung durch Fäkalien aus Mastbetrieben kontaminiert [7,170,269]. Eine weitere Kontaminationsquelle stellen Dauerausscheider dar. Das sind Menschen, die nach einer Salmonella-Infektion weiterhin in Darm, Leber oder Gallenblase Salmonellen beherbergen und diese dauernd oder schubweise ausscheiden, ohne selbst Symptome zu zeigen. Auch der Konsum getrockneter oder in ihrem Wasserwert reduzierter Lebensmittel wie Trockenmilch, Gewürze, Tee oder Nahrungsergänzungsmittel führte wiederholt zum Ausbruch von Salmonellosen. Salmonellen weisen aufgrund ihrer Kapselbildung eine hohe Trockenresistenz auf. Nach Schätzungen der WHO treten jährlich weltweit ca. 1,3 Milliarden Fälle von durch enteritische Salmonellosen verursachte Diarrhö auf. Das führt im Jahr zu ca. 3 Millionen Todesfällen. Insbesondere in Entwicklungsländern Asiens und Afrikas sind Salmonellosen sehr stark verbreitet. Jedoch stellen Salmonella-Infektionen nach wie vor auch in industrialisierten Ländern ein erhebliches Problem dar. So haben Salmonellen auch in Deutschland eine große epidemiologische und damit wirtschaftliche Bedeutung [120,133]. Deshalb sind die Überwachung und Bekämpfung seuchenrechtlich vorgeschrieben und Erkrankungen unterliegen der Meldepflicht. Insgesamt wurde in den 80er Jahren ein erheblicher Anstieg der Salmonellosen beim Menschen, sowohl in Europa als auch in Amerika, registriert. In dieser Zeit nahm die Massentierhaltung zu. Das verstärkte Vorkommen von Salmonellen aufgrund der guten Verbreitungsbedingungen und aufgrund von kontaminiertem Tierfutter wird als Ursache für den Anstieg angesehen [105,283]. Die Meldezahlen für die Salmonella-Infektionen sind seit 1992 zwar rückläufig (Abbildung 11), aber noch immer ist sie eine der häufigsten gemeldeten lebensmittelbedingten Infektionskrankheiten [120]. In Deutschland wurden im Jahr 2004 ca. 57.000 Salmonellose-Krankheitsfälle im Vergleich zu ca. 77.000 Meldungen aus dem Jahr 2001 an das Robert-Koch-Institut im Rahmen des Infektionsschutzgesetzes übermittelt [259]. Die Dunkelzahl liegt jedoch schätzungsweise zehnmal höher. Die meisten Salmonellosefälle sind nach wie vor durch Stämme der beiden Salmonella-Serovare Typhimurium und Enteritidis bedingt (Abbildung 1-1). Bis in die 80er Jahre wurde S. Typhimurium als das häufigste Serovar als Krankheitserreger nachgewiesen. Dieses wird allerdings seitdem durch S. Enteritidis Phagentyp 4 (Europa) und Phagentyp 8 (Nordamerika) verdrängt. 3 Einleitung Abbildung 1-1: Salmonellosen bei Menschen (Quelle: Robert Koch-Institut [120]) 1.1.3 Die Pathogenität von Salmonella Pathogenität (griech. pathos: Krankheit) ist die Fähigkeit eines Organismus, seinen Wirt nachhaltig zu schädigen oder zu töten. Als Virulenz (lat. virulentus: voller Gift) wird der Grad, d.h. die quantitative Ausprägung, der Pathogenität bezeichnet. Dementsprechend werden Proteine, welche die Virulenz infektiöser Bakterien fördern als Pathogenitäts- oder Virulenzfaktoren bezeichnet. Dazu zählen Toxine, Adhäsine sowie Proteine, die das Bakterium vor den Immunreaktionen der Wirtszelle schützen [50]. All diese Faktoren ermöglichen es den Bakterien, sich an die Zellen des Zielorganismus anzuheften, in diese einzudringen und sie zu besiedeln. Pathogene Mikroorganismen wie Salmonella spec. verursachen exogene Infektionen. 1.1.3.1 Die Salmonella-Pathogenitätsinseln Das Pathogenitätsprinzip von Salmonella ist noch nicht bis ins Detail aufgeklärt und verstanden. Allerdings wurde eine große Anzahl an Virulenzgenen, die an der Pathogenese von Salmonella-Infektionen beteiligt sind, mittlerweile identifiziert. Zwei wichtige Kennzeichen der Salmonella-Pathogenese sind (1) die Fähigkeit zur Invasion in nicht-phagozytische Zellen sowie (2) das intrazellulare Überleben und Replizieren [42]. Diese Eigenschaften werden zum größten Teil von Virulenzgenen vermittelt, die auf bestimmten Genclustern lokalisiert sind. Diese werden als Salmonella- Pathogenitätsinseln (SPI) bezeichnet. Die SPI sind große DNA-Fragmente (10-40 Kilobasenpaare), die nach der Aufnahme von anderen Bakterien in das eigene bakterielle Chromosom von Salmonella integriert wurden [65,278]. Diese Insertionen zeichnen sich in ihrer Sequenz im Vergleich zum übrigen Chromosom durch einen höheren G+C Gehalt aus. Der Erwerb der SPI erfolgte mit hoher Wahrscheinlichkeit durch horizontalen 4 Einleitung Gentransfer. In Salmonella spec. sind fünf Pathogenitätsinseln beschrieben, wobei SPI1 und SPI5 vor allem bei der Invasion und die SPI2 bis 4 bei dem Überleben, der Replikation und der Ausbreitung von Salmonella im Wirtsorganismus eine Rolle spielen. SPI1 konnte sowohl in S. enterica, als auch in der phylogenetisch älteren Species S. bongori nachgewiesen werden. SPI2 hingegen ist nur in S. enterica-Isolaten gefunden worden. Dies ist ein Hinweis, dass SPI1 weitaus früher erworben wurde, als SPI2 [42]. 1.1.3.2 Adhäsion und Invasion von Salmonella und die Funktion des Typ III-Sekretionssystems Salmonellen werden hauptsächlich oral mit der Nahrung aufgenommen und gelangen so als enteroinvasive Erreger rasch in den Magen-Darm-Trakt. Im Bereich des Dünndarms werden die Bakterien primär über die Peyer’schen Plaques, lymphatisches Gewebe auf der Dünndarm-Oberfläche, in das Gewebe eingeschleust. Der initiale Schritt im Infektionszyklus ist die Anlagerung von Salmonella spec. an das Darmepithel. Durch den Zellkontakt wird die Expression verschiedener spezifischer Gene induziert. So kommt es zur Ausbildung von Fimbrien (Adhäsionsfaktoren). Die spezifische Bindung von bakteriellen Adhäsinen an Wirtzellrezeptoren (z.B. EGF-Rezeptor = Epidermal-GrowthFactor-Receptor) wird Adhäsion genannt. Die Adhäsion ist essentiell für den Eintritt der Bakterien in die Wirtszellen und wird daher als wichtiger Virulenzmechanismus angesehen. Der eigentliche Invasionsprozess beinhaltet die Freisetzung von Invasionsproteinen (inv-Gene) und anderen Pathogenitätsfaktoren vor allem von der Salmonella-Pathogenitätsinsel 1 (SPI1) in das Cytoplasma der Wirtszelle. Die SPI1 beinhaltet u.a. Gene für ein Typ III-Sekretionssystem. Durch das Typ III- Sekretionssystem werden Substratproteine aus dem Cytoplasma der Bakterienzelle in das umgebende Medium ausgeschleust (Sekretion) oder aus dem Bakterium in die Zielzelle transportiert (Translokation). Typ III-Sekretionssysteme sind komplexe Multiproteinapparate, welche in zahlreichen gramnegativen pathogenen Bakterien vorkommen. Diese Systeme sind augrund hoher Sequenzhomologie und struktureller Ähnlichkeit eng verwandt mit dem Flagellen-Apparat. Sie sind aus über 20 Strukturproteinen aufgebaut, welche die Cytoplasmamembran, das Periplasma und die äussere Membran durchdringen. Die Injektion von Effektorproteinen in die Wirtszelle wird nach Zellkontakt aktiviert, aber auch z.B. durch eine pH-Wert-Änderung in den alkalischen Bereich. Die Regulation der Invasion scheint also von Umweltfaktoren beeinflusst zu werden. Die Expression des invasiven Phänotyps wird z.B. durch SirA, einem Transkriptionsaktivator, positiv und durch das Zweikomponentensystem PhoP/Q negativ beeinflusst. Der Export von Invasionsproteinen (z.B. SipA, SipB) und anderen Virulenzfaktoren von der Bakterien- in die Epithelzelle ruft eine Änderung von Signaltransduktionswegen in der Wirtszelle hervor, was eine Veränderung der Plasmamembranstruktur der Wirtszelle bewirkt. Durch dadurch entstehende Membranausstülpungen, so genannten „membrane 5 Einleitung ruffles“ (morphologische, filamentöse Veränderung des Wirts-Cytoskeletts), wird die Aufnahme des Bakteriums in die Darmepithelzelle ermöglicht (Abbildung 1-2). Vor allem die M-Zellen, aber auch normale Epithelzellen stellen die Eingangspforte für die Erreger dar [42,65,278]. Abbildung 1-2: S. Typhimurium (rot) beim Eindringen in kultivierte Humanzellen Gefärbte Elektronenmikroskopaufnahme (Quelle: [291]) 1.1.3.3 Überlebensmechanismen von Salmonella in den Wirtszellen und die Auswirkungen der Besiedlung Die Bakterien werden innerhalb großer Vakuolen in die Zelle aufgenommen, in denen sie persistieren und sich replizieren können. Innerhalb der Zelle werden die Bakterien von der apikalen zur basolateralen Seite der Zelle transloziert, wo sie von Gewebe-Makrophagen, die eng mit der Lamina propria der M-Zellen assoziiert sind, aufgenommen werden. Salmonella repliziert sich innerhalb der Zelle und kann der Immunantwort der Wirtszelle widerstehen. Eine Invasion und das Überleben in den Makrophagen wird vor allem durch die Expression von Pathogenitätsfaktoren des Genclusters SPI2 ermöglicht [69]. Eine Voraussetzung der Expression der SPI2-Gene ist die Ansäuerung des Phagosoms. SPI2 ist für die systematische Ausbreitung des Bakteriums und die Kolonisation von Wirtsorganen wie Leber und Milz von Bedeutung. Auch auf SPI2 befinden sich Gene für ein weiteres Typ III-Sekretionssystem. Eine Hauptfunktion der Genprodukte von SPI2 ist der Schutz der Bakterienzelle vor den intrazellulären antimikrobiellen Abwehrmechanismen (u.a. Defensine) der Wirtszelle und den Stressfaktoren (saurer pH, Sauerstoffradikale). Die molekularen Wirkungsmechansimen der in der SPI2 codierten Proteine sind erst in wenigen Details aufgeklärt. Nach der Invasion der Bakterien antworten die Makrophagen mit der Entwicklung einer oxidativen metabolischen Reaktion in deren Verlauf reaktive Oxidantien freigesetzt werden. Salmonella reagiert auf den oxidativen Stress mit einer gesteigerten Produktion z.B. der Katalsae KatG und der Alkyl-Hydroperoxid-Reduktase Ahp sowie RecA und RecBC zur Reparatur von den durch die oxidative Reagentien entstandenen DNA- 6 Einleitung Schäden (siehe auch Abschnitt 1.3.1.2). Weitere Proteine, die an der Regulation auf den oxidativen Stress beteiligt sind, sind SlyA (Transkriptionsregulator) und RpoS (Sigmafaktor). Letzteres spielt auch eine wichtige Rolle bei der Resistenz auf niedrige pHWerte. Salmonella ist im Laufe der Infektion verschiedentlich auch niedrigen pH-Werten ausgesetzt und ist in der Lage, sich durch ein Säure-Toleranz-System zu schützen. Dieses beeinhaltet z.B. das CadA/CadB-System, durch welches H+-Ionen an Lysin gebunden und entfernt werden. Bei Säureschock wird die Expression von rpoS induziert. RpoS (37,9 kDa) ist eine σ-Untereinheit (Sigmafaktor) der bakteriellen RNA-Polymerase [42]. Sigmafaktoren vermitteln die Promotorerkennung und wirken als Regulatoren der Transkription Veränderte für spezifische Anpassungsreaktionen Umweltbedingungen führen dazu, auf dass bestimmte die Umweltreize. RNA-Polymerase einen alternativen Sigmafaktor bindet und dadurch an andere Promotoren bindet. Die Bildung von RpoS wird durch Aufnahme von Salmonella in Makrophagen oder Epithelzellen bzw. in vitro nach Erreichen der stationären Vermehrungsphase der Kultur, durch Mangel an Nährstoffen, durch Erhöhung der Temperatur auf 42°C oder Senkung des pH-Werts induziert. induziert Salmonella in den Makrophagen und Epithelzellen Apoptose, einen programmierten Zelltod. Der apoptotische Zelltod ist ein hoch regulierter, aktiver Prozess, der durch komplexe Signalkaskaden reguliert wird und es dem Organismus ermöglicht, defekte Zellen zu entfernen. Durch die Induktion der Apoptose (unter Beteiligung des Typ III-Sekretionssystems) entgehen die Salmonella-Keime zum einen der effektiven Abwehr der Wirtszelle und werden zum anderen nach erfolgter intrazellulärer Replikation freigesetzt. Sie können so extrazelluläre Räume erreichen und benachbarte Zellen befallen. So gelangen die Salmonellen auch in mesenteriale Lymphknoten [162]. Wichtige weitere Virulenzfaktoren von Salmonellen, welche bei der Infektion eine große Rolle spielen, sind das Lipoid A (Endotoxin) und das O-Antigen, als Teile des Lipopolysaccharids (LPS) der äußeren Membran, sowie weitere verschiedene Toxine, wie ein Enterotoxin mit ADP-Ribosyltransferaseaktivität und ein Cytolysin mit proteinsynthesehemmender Wirkung. Aufgrund der u.a. durch das bakterielle Endo- und Enterotoxin hervorgerufenen Entzündungsreaktion wird der Flüssigkeit- und Elektrolyttransport im Körper gestört und es kommt zu Durchfällen, Erbrechen und Fieber [269,278]. In verschiedenen Salmonella-Stämmen wurden Plasmide isoliert, welche dem Stamm Virulenzeigenschaften verliehen [269]. Proteine von Virulenzplasmiden sind vor allem für das Wachstum der Bakterien innerhalb der Makrophagen notwendig. Im Rahmen der Entwicklung von Lebendimpfstoffen wird eine Attenuation, d.h. eine Reduktion der Virulenz eines Stammes, z.B. durch das Ausschalten einzelner Faktoren des Pathogenitätsprinzipes des Bakterienstammes, erreicht. Die Entwicklung von Salmonella-Impfstoffen gewinnt zunehmend an Bedeutung. Zur Prävention von TyphusErkrankungen werden bereits Impfstämme eingesetzt, aber zur Behandlung der Krankheit ist eine Antibiotika-Therapie unumgänglich. Bei der harmloseren enteritischen 7 Einleitung Salmonellose erfolgt eine Prävention durch eine Behandlung der Nutztiere (z.B. Hühner) zur Vermeidung der Übertragung einer Salmonellen-Infektion auf den Menschen. Auch hier spielt der Einsatz von Impfstämmen eine immer größere Rolle. Auf die Entwicklung von Salmonella-Impfstämmen wird ausführlich im Abschnitt 1.4 eingegangen. Die meisten Erkrankungen des Gastrointestinaltraktes beim Menschen, die enteritischen Salmonellosen, verlaufen harmlos und selbstlimitierend. In schweren Fällen kann jedoch eine Gabe von Antibiotika indiziert sein. Vor allem bei immunsupprimierten Patienten ist eine Antibiotika-Therapie oft lebensrettend [278]. 1.2 Das Auftreten und die Verbreitung resistenter Salmonellen Trotz der rückläufigen Meldezahlen der enteritischen Salmonellosen stellen vor allem resistente Erreger ein großes Problem dar [18]. Bereits in den 60er Jahren traten erste Salmonella-Stämme auf, die auf bestimmte Antibiotika nicht mehr ansprachen. In den 70er und 80er Jahren kamen multiresistente Stämme dazu. Besonderes Interesse erweckte der in den 90er Jahren erstmals in Dänemark aufgetretene multiresistente Stamm S. Typhimurium DT104. Dieser war klinisch resistent gegenüber sechs verschiedenen Antibiotikagruppen [52,206]. Fast die Hälfte aller seitdem identifizierten Salmonellen ist gegenüber mehreren Antibiotika unempfindlich, z.T. sogar gegenüber Fluorchinolonen, die bisher als Reserveantibiotika eingesetzt wurden [18]. Dänische Salmonella-Isolate im Jahr 2004 in aus Deutschland importierten Puten, die auf 16 von 17 heute verfügbaren Antibiotika-Klassen resistent sind, alarmieren. Das einzig wirksame Antibiotikum Florfenicol ist in der Humanmedizin nicht zugelassen. Das Auftreten antibiotikaresistenter Salmonellen erhöht das Risiko für eine aggressive Infektion, die leicht zum Tod führen kann [130-132,196,206,292]. Als Ursache für das Auftreten und die Verbreitung resistenter Keime wird der jahrzehntelange großzügige Einsatz von Antibiotika verantwortlich gemacht [18]. Im Veterinärbereich wurden Antibiotika lange Zeit nicht nur zur Therapie bereits bestehender, sondern auch zur Prophylaxe von Erkrankungen und zusätzlich als Leistungsförderer angewendet, wobei sie in subtherapeutischen Konzentrationen als Zusatzstoffe dem Futter beigemischt wurden. Aufgrund der veränderten Darmflora und einer Erhöhung der Aktivität der Verdauungsenzyme kommt es zu einer verbesserten Futterverwertung [269]. Der Einsatz von Antibiotika als Leistungsförderer ist ab 2006 in der EU komplett verboten. Insgesamt muss der Einsatz von Antibiotika in der Veterinärmedizin als Risikofaktor bei der Resistenzentwicklung von Bakterien angesehen werden [170]. Dies ist von besonderer Bedeutung, da es sich bei Salmonellen um pathogene Zoonoserreger handelt und eine Entwicklung resistenter Keime im Tier auch zur potentiellen Gefahr für den Menschen wird [7,170]. 8 Einleitung Enstandene Resistenzmechanismen können auf unterschiedliche Weise weitergegeben werden: (1) Speicherung der Resistenzinformation auf Chromosomen und Weitergabe durch Teilung von Mutter- an Tochterzelle, (2) Speicherung auf Plasmiden und Weitergabe über Zell-Zell-Kontakt (Konjugation) oder (3) über Bakteriophagen (Transduktion). Darüber hinaus können Resistenzgene innerhalb einer Zelle vom Chromosom auf ein Plasmid und umgekehrt „springen“, wenn sie Bestandteil von mobilen genetischen Elementen, wie Transposons oder Integrons sind. Eine Untersuchung des Nationalen Referenzzentrums für Salmonellen am Bundesinstitut für Risikobewertung zur „Erfassung phänotypischer und genotypischer Resistenzeigenschaften bei Salmonella- und E. coli-Isolaten vom Tier, Lebensmitteln, Futtermitteln und aus der Umwelt“ verdeutlicht die aktuelle Resistenzlage [133]. In den Jahren 2000 bis 2003 waren durchschnittlich 69 % der aus Lebensmitteln (Rind, Schwein, Geflügel) stammenden Salmonella-Isolate resistent gegen antimikrobiell wirksame Substanzen. 52 % der vom Nutztier und 42 % der von Lebensmitteln stammenden Keime waren sogar multiresistent. Als Ursachen für die Multiresistenz wurden, wie auch schon in anderen Studien, vor allem auf Class 1-Integrons lokalisierte Resistenzgene identifiziert [248]. Die am weitesten verbreiteten Resistenzgene in den Salmonella-Isolaten waren unter anderem das floR-Gen (Efflux-Protein) verantwortlich für die Florfenicol- und Chloramphenicolresistenz, das Kanamycin/Neomycinresistenz, aphA1-Gen das aadA-Gen (Phosphotransferase) für die (Adenyltransferase) für die Streptomycinresistenz, das tet(G)-Gen (Efflux-Protein) für die Tetracyclinresistenz, das pse-1-Gen (ß-Lactamase) für die Ampicillinresistenz sowie das sul1-Gen (DihydropteroatSynthase) für die Sulfonamidresistenz [133]. Speziell bei Geflügel ist eine zunehmende Resistenz gegenüber Chinolonen zu beobachten. Im Untersuchungszeitraum 2000 bis 2003 nahm die Chinolonresistenz signifikant von 5,3 % auf 26 % zu [133]. Bei Kleinkindern, Älteren oder immunsuprimierten Menschen ist eine antibakterielle Therapie von Salmonellosen oft lebensnotwendig [257,269]. Nach dem Auftreten multiresistenter Stämme mussten die bis dahin verwendeten Antibiotika wie Ampicillin oder Chloramphenicol durch andere Chemotherapeutika ersetzt werden. Zur Bekämpfung von Salmonellosen werden derzeit hauptsächlich Fluorchinolone eingesetzt, welche sich durch eine hohe antibakterielle Aktivität gegenüber Salmonella spec. auszeichnen [257,269,278]. Demzufolge lässt sich feststellen, dass chinolonresistente Salmonellen ein großes gesundheitliches Risiko für den Menschen darstellen [55]. 9 Einleitung 1.3 Fluorchinolone – Wirkung und Resistenzmechanismen Fluorchinolone sind synthetische, niedermolekulare, antibakterielle Substanzen. 1962 stand die Basissubstanz Nalidixinsäure, welche als Nebenprodukt des Malariamittels Chloroquin entstand, erstmals zur Verfügung [175]. Prinzipiell besitzen alle Chinolone einen Chinolingrundkörper (Ausnahme: Enoxacin und Gemifloxacin mit einem Naphthyridingrundkörper), an dem sich an C7 ein Piperazinrest, an C3 eine Carbonsäuregruppe und an C4 eine Oxogruppe befinden. Nach Einführung eines Fluorsubstituenten an C6 entstand 1978 Norfloxacin und somit der erste Vertreter der Fluorchinolone (Abbildung 1-3), welche sich durch eine erhöhte Wirksamkeit auszeichnen [74]. Durch weitere Substitutionen oder Modifikationen von Seitenketten folgten 1985 Ofloxacin, 1987 Ciprofloxacin (Cyclopropylgruppe), 1996 Sparfloxacin (Addition einer NH2Gruppe), 1997 Grepafloxacin (Addition einer CH3-Gruppe), 1998 Trovafloxacin und Levofloxacin sowie 1999 Moxifloxacin (Azabizyclosubstituent) [49,73,87]. (Fluor)chinolone wirken bakterizid. Bereits 1965 postulierten Cook und Mitarbeiter einen Einfluss von Chinolonen auf die DNA-Synthese [106,107]. Gellert und Mitarbeiter konnten zeigen, dass die Gyrase Zielstruktur der Chinolone ist [98]. Auf die Wirkungen zwischen DNA, Topoisomerasen und Chinolonen wird an anderer Stelle ausführlich eingegangen (Abschnitt 1.3.1.2). Der Wirkungsmechanismus der einzelnen Chinolone ist zwar sehr ähnlich, allerdings unterscheiden sich die Substanzen bezüglich der antibakteriellen Aktivität, ihrer Pharmakokinetik und klinischen Wirksamkeit. Abbildung 1-3: Ausgewählte Vertreter der (Fluor)chinolone (modifiziert nach [233]) 10 Einleitung Bakterien sind jedoch in der Lage, Resistenzen gegenüber Chemotherapeutika zu entwickeln. Die Empfindlichkeiten von Bakterien gegenüber antibakteriellen Substanzen werden durch Bestimmung der minimalen Hemmkonzentrationen (MHK) des Antibiotikums in µg/ml ermittelt [72]. Ein Überschreiten eines für die entsprechende Species und das entsprechende Antibiotikum angegebenen Wertes, der eine Therapie verhindert, wird als klinische Resistenz bezeichnet. Prinzipiell lässt sich zwischen natürlicher und erworbener Resistenz unterscheiden. Bei der natürlichen Resistenz handelt es sich um eine in der Species bereits vorhandene Unempfindlichkeit gegenüber einer Substanz, wie z.B. die natürliche Resistenz von E. coli und Salmonella spec. gegenüber Makroliden [314]. Durch den Erwerb von Mutationen oder den Transfer von Resistenzgenen können Mikroorganismen Resistenzen erwerben. Es existieren drei Möglichkeiten zur Ausbildung von Resistenzmechanismen [301]: (1) Veränderung der Zielstruktur des Antibiotikums. So bewirken z.B. Mutationen in den Genen für die Gyrase eine verringerte Affinität der Chinolone zu ihrer Zielstruktur (Target). (2) Verringerung der Konzentration des Wirkstoffes am Zielort. Beispielsweise beeinflusst die verränderte Expression von Porinen oder Effluxpumpen die Akkumulation von Substanzen in der Zelle. (3) Enzymatische Inaktivierung des Antibiotikums (Abbau oder Modifizierung). Das findet man z.B. bei den ß-Lactamen, welche durch ß-Lactamasen inaktiviert werden. Für die Entwicklung einer Resistenz auf die Antibiotikagruppe der Flourchinolone wurden bis vor kurzem nur die Mechanismen (1) und (2) verantwortlich gemacht. Neuere Erkenntnisse zeigen aber auch die Möglichkeit der enzymatischen Inaktivierung (3) als Möglichkeit der Entwicklung reduzierter Empfindlichkeiten [260]. Im Folgenden sollen die bekannten Resistenzmechanismen erklärt und am Beispiel der Fluorchinolone näher beleuchtet werden. 1.3.1 Die Topoisomerasen als Zielstrukturen für Chinolone Im folgenden Abschnitt soll zunächst auf die Veränderung der Zielstruktur, als sehr gut untersuchter Resistenzmechanismus gegen Fluorchinolone, eingegangen werden. 1.3.1.1 Die DNA-Topologie in der Zelle Gellert et al. identifizierten 1977 die DNA-Gyrase als Zielstruktur der Chinolone [98]. Die Gyrase ist eine Topoisomerase, welche für die räumliche Anordnung der DNA in der Zelle (Topologie) verantwortlich ist. Sie ermöglicht die Komprimierung der DNA. So ist in E. coli die Unterbringung eines Chromosoms einer Länge von ca. 1,4 mm in eine Zelle von nur ca. 2 µm Länge und einem Durchmesser von ca. 1 µm möglich. In der relaxierten energiearmen Form, der physiologischen B-Konfiguration, sind die beiden Einzelstränge der DNA-Doppelhelix alle 10,4 bp einmal umeinander gewunden und einmal pro Umwindung miteinander verknüpft. In dieser Form befindet sich die Helixachse in einer 11 Einleitung Ebene. Verringert sich die Verdrillung der DNA, so kommt es aufgrund von Torsionsspannung Verdrillung bewirkt zu einer hingegen negativen eine Superspiralisierung. positive Eine Superspiralisierung. zunehmende Durch den Torsionsstress wird die Helixachse aus der Ebene herausgedrückt und es bilden sich so genannte Superhelices [251]. Beide Formen der Superspiralisierung (positiv und negativ) würden zu einer Komprimierung der DNA führen, allerdings weisen fast alle bislang untersuchten Organismen eine negativ superspiralisierte DNA auf [44]. Neben der räumlichen Komprimierung der DNA führt die negative Superspiralisierung auch zu einer Speicherung von Energie, die für Vorgänge wie Replikation, Rekombination oder Transkription benötigt wird [97,295]. Diese zellulären Prozesse ziehen eine Änderung der lokalen Topologie der DNA nach sich. Es kommt bei der fortschreitenden Trennung der DNA-Stränge sowohl durch die DNA-Helikase während der Replikation als auch durch die RNA-Polymerase während der Transkription zu einer lokalen Akkumulation positiven Superhelices vor und negativen Superhelices hinter dem jeweiligen Enzym [182,295]. Diese positiven Superhelices müssen aufgelöst werden, um ein Fortschreiten der EnzymKomplexe zu gewährleisten und den globalen negativ-superspiralisierten Status der DNA aufrechtzuerhalten. Ein weiteres topologisches Problem in der Bakterienzelle, das durch die Topoisomerasen gelöst wird, ist die Trennung der beiden miteinander verbundenen Chromosomen nach der Replikation. Dieser Vorgang ist für die Zellteilung essentiell. Die Bedeutung der einzelnen Topoisomerasen ist in Abbildung 1-4 gezeigt. Abbildung 1-4: Die verschiedenen Aktivitäten der Topoisomerasen (Topo) I, II (Gyrase) und IV (Abbildung nach [68]) (A) Relaxierung der DNA durch die Topo I (partiell) und die Topo IV (vollständig) und Einführung negativer Superspiralisierung durch Gyrase (B) Trennung der bei Replikation oder Rekombination entstandenen DNA-Catenane durch die Topo IV (C) Entknotung der DNA durch die Topo IV Topoisomerasen kommen in Pro- und Eukaryonten vor. Es werden verschiedene Typen von Topoisomerasen unterschieden [43]. Zu den Typ I-Topoisomerasen, welche energieunabhängig Einzelstrangbrüche verursachen, gehören in E. coli die Topoisomerasen I und III. Weiterhin existieren Typ II-Topoisomerasen, die ATP-abhängig vorübergehend einen Doppelstrangbruch einführen. Typ II-Topoisomerasen in E. coli sind Topoisomerase II (Gyrase) und Topoisomerase IV [295]. Je nach der Bindung der geschnittenen DNA (5`- oder 3`-Ende) während der Reaktionszyklen an die Tyrosinreste im aktiven Zentrum werden noch Subtypen unterteilt [43]. Die Anzahl der vorkommenden Topoisomerasen ist für den jeweiligen Organismus spezifisch. So besitzt z.B. die Bäckerhefe Saccharomyces cerevisiae zwei verschiedene Topoisomerasen (Typ I und 12 Einleitung Typ II) und der Mensch fünf verschiedene Topoisomerasen (drei von Typ I und zwei von Typ II). In E. coli wurden vier verschiedene Topoisomerasen (jeweils zwei von Typ I und Typ II) identifiziert, die im Folgenden näher beschrieben werden. Die Topoisomerase I ist ein Monomer und wird durch das topA-Gen codiert. Sie trennt einen Einzelstrang des DNA-Doppelstranges und kann die DNA dadurch relaxieren [200,277]. Durch die Relaxierungsaktivität der Topoisomerase I wird der globale Superspiralisierungsgrad in der Zelle reguliert und die Anhäufung von negativen Überspiralisierungen, welche im Verlaufe der Transkription entstehen, verhindert. Die Topoisomerase II, auch Gyrase genannt, ist ein Heterotetramer, das aus jeweils zwei Untereinheiten A und B besteht (A2B2), die durch die Gene gyrA und gyrB codiert werden. Die Gyrase ist in der Lage, einen DNA-Doppelstrang zu spalten und kann ringförmig geschlossene DNA-Moleküle, wie das bakterielle Chromosom oder Plasmide, aus einem energiearmen relaxierten in einen energiereichen negativ superspiralisierten Zustand überführen. Bei der Einführung negativer Superspiralisierung wird die Verwindungszahl des DNA-Moleküls um zwei reduziert und Energie in Form von ATP zur Konformationsänderung und für die Strangpassage verbraucht. Der genaue Prozess der Einführung der negativen Superspiralisierung ist der Abbildung 1-5 zu entnehmen. Die Topoisomerase III ist wie die Topoisomerase I ein Monomer und wird durch das Gen topB codiert [71]. Es wird vermutet, dass die Topoisomerase III eine Rolle bei der Entwindung einzelsträngiger DNA-Knoten spielt, die während der Replikation oder Rekombination auftreten können. Dabei verwendet sie hypernegativ superspiralisierte DNA als Substrat [71]. Die Topoisomerase III ist nicht essentiell für die Zelle [266]. Die Topoisomerase IV ist ein Heterotetramer, das aus zwei C- und zwei E-Untereinheiten (C2E2) besteht, welche durch die Gene parC und parE codiert werden (in Staphylococcus aureus grlA und grlB). Die Topoisomerase IV weist auf Ebene der Aminosäuresequenz sehr hohe Homologie mit der Topoisomerase II auf [157,231]. Allerdings besteht die Hauptaufgabe der Topoisomerase IV in der Trennung der durch die Replikation entstandenen Catenane. Neben dieser Decatenierungsaktivität, welche für die Segregation der Tochterchromosomen notwendig ist [157,231], ist sie außerdem in der Lage, superspiralisierte DNA zu relaxieren. Allerdings spielt bei der Relaxierung der Superspiralen die Topoisomerase I die wichtigere Rolle. Vergleiche der Relaxierungsaktivität von Topoisomerase I und IV ergaben, dass die Topoisomerase I die negativ superspiralisierte DNA sechsmal schneller relaxiert als Topoisomerase IV, wobei letztere, im Gegensatz zu Topoisomerase I, die DNA hingegen fast vollständig relaxieren kann [312]. Vermutlich ähnelt der Reaktionsmechanismus dem der Gyrase, allerdings wird das gebundene DNA-Segment nicht um das Enzym gewunden, sondern es wird eine intermolekulare Strangpassage favorisiert [311]. Die Topoisomerase IV ist, wie auch die Gyrase, für die Zelle essentiell. Bakterien mit Defektmutationen in Gyrase und Topoisomerase IV sind nicht lebensfähig [187,312]. 13 Einleitung Abbildung 1-5: Einführung der negativen Superspiralisierung durch die Gyrase (modifiziert nach [28,289]) 1 Ausgangssituation ist eine relaxierte oder positiv superspiralisierte DNA und das Heterotetramer Gyrase mit zwei A-Untereinheiten [blau] und zwei B-Untereinheiten [rot] mit freien ATPaseDomänen [gelb]. Zunächst wird die doppelsträngige DNA um die Gyrase gewunden. Ein 140 bp langes DNA Fragment, das G-Segment („gate“) [grau], bindet dabei an die freie Gyrase. 2 Aufgrund dieser Bindung kommt es zu einer Konformationsänderung des Enzyms, vermutlich unterstützt durch die B-Untereinheit [97]. An den freien ATPase-Domänen der B-Untereinheit kommt es zu einer Bindung von ATP. Dadurch ausgelöste Konformationsänderungen des Enzyms führen zur Spaltung des G-Segmentes. 3 Es handelt sich dabei um einen um 4 bp versetzten DNA-Doppelstrangbruch. Dabei werden 5’Phosphatgruppen frei, welche je an einen Tyrosinrest (Tyr-122) der A-Untereinheit gebunden werden [149]. Die Fixierung der freien 3’-OH-Gruppen erfolgt vermutlich über nicht kovalente Bindung an das Enzym. 4 Weiterhin kommt es zur Bindung eines weiteren DNA-Fragmentes, des T-Segmentes („transfer“) [grün], das durch die Lücke im G-Segment geführt wird. 5 Durch Wiederherstellung der Phosphodiesterbindung wird der DNA-Doppelstrangbruch wieder religiert [251]. Die DNA löst sich von dem Enzym und durch ATP-Hydrolyse kehrt die Gyrase in ihre Ausgangskonformation zurück. 14 Einleitung Für die Rekombination, Initiation der Replikation und Transkription vieler Gene ist die Aufrechterhaltung eines globalen Superspiralisierungsgrades innerhalb enger Grenzen essentiell [194]. Die Regulation des DNA-Superspiralisierungsgrades wird vor allem durch die Topoisomerasen I und II (Gyrase) auf den Ebenen der antagonistischen Enzymaktivität, der Substratspezifität und der Genexpression sichergestellt. Während die Gyrase negative Superspiralisierung einführt, relaxiert die Topoisomerase I hingegen die DNA. Die negativ superspiralisierte DNA ist das bevorzugte Substrat der Topoisomerase I. Im Gegensatz dazu zeigt die Gyrase eine hohe Affinität zu positiv superspiralisierter und relaxierter DNA, während die Affinität zu negativ superspiralisierter DNA gering ist [99,200,251,312]. Weiterhin wird die Genregulation der Topoisomerasegene über den Superspiralisierungsgrad selbst reguliert. So führte eine stark superspiralisierte DNA zur Expression der Topoisomerase I und das Vorliegen relaxierter DNA zur Erhöhung der Topoisomerase II-Expression [204,214]. Die Transkription der Topoisomerase I (topA) wird dabei durch mehrere Promotoren reguliert, die auf Veränderungen in der DNASuperspiralisierung reagieren [287,288]. Diese Regulation der negativen Superspiralisierung auf unterschiedlichen Ebenen wurde von Menzel und Gellert als homöostatische Kontrolle bezeichnet [204]. Sie garantiert eine schnelle Anpassung des DNA-Superspiralisierungsgrades an interne und externe Einflüsse und hält ihn in engen Grenzen [273]. An der Komprimierung der DNA und der Aufrechterhaltung der dreidimensionalen Struktur sind weiterhin „histonähnliche Proteine“ beteiligt. Es handelt sich dabei um kleine basische oder neutrale Proteine, die in ihrer Funktion als Strukturproteine mit den Histonen in den Nukleosomen vergleichbar sind. Diese Proteine werden u.a. eingeteilt in H-NS (histon-like nukleotid structuring protein), welches DNA-Biegungen induziert und den Zustand der negativen Superspiralisierung erhält, HU (heat-unstable nucleotid protein), das als Dimer unspezifisch an die DNA bindet und eine dynamische Stabilität vermittelt sowie multifunktionelle DNA-Biegeproteine wie IHF (integration host factor) und FIS (factor for inversion stimulation) [80,234]. IHF und FIS können eine Biegung der Helixachse bewirken und damit ebenfalls die Topologie der DNA beeinflussen [232]. Neben den intrazellulären Prozessen können aber auch externe Faktoren den Superspiralisierungsgrad der DNA verändern. So zeigten verschiedene Studien einen deutlichen Einfluss von Umweltbedingungen wie Temperatur, Nährstoffgehalt, Osmolarität und Sauerstoffgehalt. Außerdem ändert sich der Superspiralisierungsgrad in unterschiedlichen Phasen während des Wachstums von Bakterienzellen [19,76,77,104]. 15 Einleitung 1.3.1.2 Die Wirkung von Chinolonen Da die Topoisomerasen eine essentielle Funktion in der Bakterienzelle erfüllen, eignen sie sich als Zielstrukturen zur Inhibition und somit als Ansatz zur Bekämpfung pathogener Mikroorganismen. So können natürliche oder synthetische Substanzen zu Wachstumsdefiziten oder letaler Zellschädigung des Bakteriums führen. Als synthetische und therapeutisch eingesetzte Inhibitoren haben die Fluorchinolone große Bedeutung erlangt. Seit der Identifizierung der Gyrase als Zielstruktur der Chinolone wurden zahlreiche Verbindungen eingesetzt, um Bakterien durch eine Hemmung der essentiellen Typ II-Topoisomerasen zu bekämpfen. Möglich ist dies aufgrund der sehr geringen Wirkung der meisten Chinolone auf eukaryontische Zellen [21,86]. Für eine Hemmung eukaryontischer Typ II-Topoisomerasen ist eine bis zu 1000fach höhere Konzentration notwendig. Somit sind hauptsächlich die Topoisomerasen der Bakterien Zielstruktur für die Fluorchinolone, nicht die des Wirtsorganismus. Jedoch ist auch die Affinität von Chinolonen zu den bakteriellen Enzymen unterschiedlich aufgrund von strukturellen Unterschieden der Typ II-Topoisomerasen. Chinolone haben eine bakterizide Wirkung gegenüber sich teilenden Bakterien mit aktiver Proteinbiosynthese. Dabei binden die Chinolone an den Topoisomerase-DNA-Komplex und stabilisieren dadurch den Zustand im Reaktionszyklus des Enzyms, in dem die DNA geschnitten und über den Tyrosinrest kovalent an das Enzym gebunden ist [81]. Vermutlich wird die DNA durch die Inhibitoren in der Nähe der Schnittstelle so deformiert, dass die Interaktion zwischen DNA und Enzym gestört ist [194]. Dadurch wird die DNA zum Cleavage-Substrat für die Spaltungsreaktion des Enzyms („positional poison model“) [139,160,195,221]. Durch Bindung der Chinolone an den DNA-Gyrase-Komplex entsteht ein stabiler ternärer spaltbarer Komplex (Cleavage-Komplex) [39]. So können essentielle Prozesse wie die DNA-Replikation und Transkription nicht mehr stattfinden, da der Cleavage-Komplex eine Barriere für die replikativen Helikasen und RNA-Polymerasen darstellt [79,270,302]. Normalerweise besteht eine Gleichgewichtsreaktion zwischen der Einführung (Cleavage) und dem Schließen (Religation) eines Doppelstrangbruchs. Eine Verschiebung dieses Gleichgewichtes in Richtung der Cleavage-Reaktion verhindert die Religation der DNA. Die so entstehenden Doppelstrangbrüche als Ergebnis der Chinolonwirkung induzieren die SOS-Antwort in der Zelle [199,235]. Durch die Induktion von über 30 Genen (SOSRegulon), die u.a. die DNA-Reparatur sowie die DNA-Rekombination betreffen, wird die Zellteilung verhindert und die Produktion von DNA-Reparaturenzymen verstärkt [140]. RecA als SOS-Reparaturprotein bindet an einzelsträngige DNA-Fragmente. Der RecBCDEnzymkomplex hingegen spielt eine wichtige Rolle bei der Erkennung von Doppelstrangbrüchen in der DNA [45]. RecBCD baut, beginnend am Doppelstrangbruch, ein Stück eines DNA-Stranges von 3’- in 5’-Richtung ab. Das entstehende einzelsträngige DNA-Segment stellt das Induktionssignal für das zentrale SOS-Regulatorprotein RecA dar. Somit fungiert RecA als eine Art Sensor für DNA-Schäden. Das durch die DNA16 Einleitung Bindung aktivierte RecA spaltet LexA, den Repressor der SOS-Gene. Dies führt zur Derepression der SOS-Gene [181]. Sind die DNA-Schäden repariert, sinkt die Konzentration an aktiviertem RecA. Dadurch akkumuliert LexA wieder und reprimiert die SOS-Gene [265]. Durch die Wirkung der Chinolone und die permanent vorhandenen DNA-Schäden wird das SOS-System allerdings konstitutiv induziert. Durch mutagene SOS-Reparaturmechanismen kommt es zu einer Akkumulation von Mutationen im Genom. Weiterhin wird die Zellteilung verhindert und die Bildung von Autolysinen führt letztendlich zum Tod der Bakterienzelle [151]. Neben dem SOS-System ist die homologe Rekombination als Mechanismus zur Beseitigung der durch die Chinolone induzierten Schäden beschrieben [151]. Prinzipiell wirken Chinolone aufgrund der Blockierung der Replikation nur auf sich teilende Zellen. Bakterienkulturen mit verlängerter Generationszeit oder teilungsinaktive Zellen zeigen daher unter Chinoloneinwirkung eine Lebensfähigkeit. 1.3.1.3 Die Auswirkung von Mutationen in den Genen der Topoisomerasen Die Anzahl der gebildeten ternären Komplexe aus Gyrase, DNA und Chinolon bestimmt die Empfindlichkeit einer Bakterienzelle gegenüber Chinolonen. Dabei ist zur Ausbildung und Stabilität dieser Komplexe, neben der intrazellulären Konzentration, die Affinität des Chinolons zu Gyrase oder der Topoisomerase IV entscheidend. Diese Affinität kann durch Aminosäureaustausche in den Untereinheiten beeinflusst sein [81]. Der Nachweis, dass Chinolonresistenz über gyrA-Mutationen vermittelt wird, wurde in verschiedenen In-vitroSelektionsversuchen u.a. mit E. coli und verschiedenen Salmonella-Stämmen erbracht [24,48,51,118,128,253,263]. Bei den In-vitro-Selektionsversuchen unter Einwirkung von Fluorchinolonen wurden im ersten und oft auch im zweiten Selektionsschritt ausschließlich gyrA-Mutationen gefunden. Das zeigt, dass die Gyrase das primäre Target der Chinolone ist. Weiterhin zeigten auch In-vitro-Versuche mit aufgereinigter DNA und Gyrase eine Hemmung des DNA-Superspiralisierungsgrades unter Chinolonzusatz (Cleavage-Assays). Diese Hemmung wurde reduziert, wenn eine mutierte Gyrase vorlag [2,100,127,263]. Die Topoisomerase IV wird als sekundäres Target bezeichnet, da bei Invitro-Selektion frühestens im zweiten oder dritten Selektionsschritt Mutationen im Gen für die A-Untereinheit dieses Enzyms auftraten [17,118,124]. Die Topoisomerase IV erlangt also erst Bedeutung, wenn das primäre Target Gyrase durch Mutationen unempfindlich gegenüber Chinolonen ist. Bei grampositiven Keimen, z.B. Staphylococcus aureus, ist allerdings die Topoisomerase IV (Gene grlA und grlB) das primäre und die Gyrase das sekundäre Target [89-91,145,146]. Bei Streptococcus pneumonia wurde beobachtet, dass das Target von der eingesetzten Substanz abhängt. So ist bei Verwendung von Ciprofloxacin parC und von Sparfloxacin gyrA das primäre Target. Bei chinolonresistenten Keimen wurden in Einschrittmutanten Mutationen vor allem in der QRDR (quinolone resistance determining region) der Gene für die Untereinheiten der Gyrase (gyrA und gyrB) und der Topoisomerase IV (parC und parE) identifiziert. Die 17 Einleitung QRDR erstreckt sich bei E. coli in gyrA von der Aminosäureposition 67 bis 106 [305] und in gyrB von 426 bis 447 [306]. Reese und Maxwell postulierten eine Beteiligung der QRDR an der Ausbildung der Bindetasche für Chinolone [251]. In chinolonresistenten Keimen wurden vermehrt Mutationen in den Genen gyrA und parC und in geringerem Maße in gyrB und parE gefunden. [48,110,118,124,126,128,253]. Bestimmte Positionen innerhalb der QRDR zeichnen sich wiederum dadurch aus, dass sie häufig von einem Aminosäureaustausch betroffen sind, wenn die Stämme reduzierte Empfindlichkeiten gegenüber Chinolonen zeigen, so vor allem die Aminosäuren an Position 83 und 87 in gyrA und 80 und 84 in parC (Nummerierung von E. coli) [24,48,89,108,124,126,135,159]. Insgesamt können aber auch andere Mutationen in den Genen gyrA, gyrB, parC und parE die Empfindlichkeiten gegenüber Chinolonen reduzieren. Bei der Chinolonbindung sind daher vermutlich sowohl die A- als auch die B-Untereinheiten der Topoisomerasen beteiligt [305,306]. Während für die Ausbildung einer klinischen Resistenz gegenüber älteren Chinolonen wie Nalidixinsäure meist eine Mutation ausreicht, ist für eine klinische Resistenz gegenüber Fluorchinolonen das Auftreten von mehreren Mutationen notwendig. Vor allem die gyrA-Doppelmutation an den Positionen 83 und 87 ist in E. coli und Salmonella spec. als Fluorchinolonresistenz vermittelnd bekannt [17,48,118,128,293]. Auch in anderen Species wie Klebsiella pneumoniae, Enterobacter aerogenes, Streptococcus pneumoniae, Staphylococcus aureus sind gyrA-Doppelmutationen mit klinischen Resistenz assoziiert [67,156,264,298]. Durch die verringerte Empfindlichkeit gegenüber Antibiotika ergibt sich für das Bakterium ein Selektionsvorteil unter Einwirkung von Fluorchinolonen. Allerdings führen einige Mutationen in den Topoisomerasegenen auch zu einer eingeschränkten Enzymaktivität. Das bedeutet, dass gyrA-Mutationen nicht nur eine verringerte Affinität zu Chinolonen, sondern auch in eine reduzierte Bindung der Gyrase mit der DNA bewirken können. Das kann wiederum zu einer Änderung des negativen Superspiralisierungsgrades führen [17]. Prinzipiell kann der DNA Superspiralisierungsgrad als ein globaler Regulator für die Expression vieler Gene betrachtet werden [75]. Durch eine räumliche Veränderung der DNA kann sich z.B. die Zugänglichkeit der –10- und –35-Promotorregionen so verändern, dass eine topologisch günstigere oder ungünstigere Anordnung erreicht wird. Diese ermöglicht dann eine effizientere oder ineffizientere Bindung der RNA-Polymerase [137,222]. Solche Veränderung der Genexpression könnte auch Virulenzgene von pathogenen Keimen betreffen [75,76,94]. So beschrieb Bagel [16] veränderte Adhärenzeigenschaften von E. coli, bei denen eine globale Verminderung des DNASuperspiralisierungsgrades Fimbrienexpression abhängigen von die E. coli Phasenwechsel Typ 1-Fimbrienexpression wird reguliert. durch Der einen vom reduziert. Die Superspiralisierungsgrad Phasenwechsel ist ein effizienter Adaptionsmechanismus an unterschiedliche Wirtsbereiche, der das Überleben zumindest eines Teils der Population ermöglicht. Ein verringerter negativer Superspiralisierungsgrad kann also möglicherweise Virulenz- oder Fitnessreduktion zur Folge haben. 18 Einleitung 1.3.2 Verringerung der Akkumulation von Antibiotika – Die Bedeutung von Porinen und Efflux-Systemen Zur Entwicklung einer klinischen Resistenz gegenüber Fluorchinolonen ist eine Akkumulation von mehreren Mutationen notwendig [17,124,159,169]. Neben Punktmutationen in den Genen der Topoisomerasen ist auch oft eine veränderte Antibiotika-Akkumulation an der Ausprägung der Resistenz beteiligt. Eine verringerte Akkumulation von Antibiotika, welche zu einer reduzierten Empfindlichkeit des Bakteriums führt, beruht auf einem verringerten Einstrom (Influx) der Antibiotika in die Zelle oder einem erhöhten Ausstrom (Efflux) aus der Zelle. Bei gramnegativen Bakterien ist neben der Cytoplasmamembran und der Zellwand noch zusätzlich die äußere Membran zu überwinden. Viele Substanzen gelangen durch die LPS-Schicht oder/und durch Porine in die Zelle. Dabei penetrieren in erster Linie hydrophilere Moleküle wie Norfloxacin oder Ciprofloxacin durch Porine. Hydrophobe Moleküle wie Nalidixinsäure gelangen sowohl durch die LPS-Schicht als auch durch Porine in die Zelle [141]. Eine Influx-Reduktion wird vor allem durch eine reduzierte Anzahl an Porinen hervorgerufen. Porine sind Transmembrankanäle, die durch Omps (outer membrane proteins) gebildet werden. Besonders einer Reduktion von OmpFProteinen wird eine Bedeutung bei der Resistenzentwicklung bei E. coli zugeschrieben [58,141,142,148,205]. OmpF liegt in der äußeren Membran von E. coli (homologe Proteine mit anderen Namen finden sich bei anderen Gramnegativen) und bildet ein relativ großes Porin, wodurch viele Nährstoffe in die Zelle gelangen [217]. Unter nährstoffarmen Bedingungen wird ompF vermehrt exprimiert. Eine Regulation der OmpFExpression wird u.a. über den übergeordneten Regulator OmpR und auf posttranskriptionaler Ebene durch die antisense-RNA micF gesteuert [9]. Durch Bindung von micF-RNA an die OmpF-RNA bildet sich ein RNA-Doppelstrang, der die Translation der OmpF-RNA verhindert. Ein ähnliches System zur Regulation der Proteinsynthese findet man in eukaryontischen Zellen mit der Antisense-RNA. Auch hier kommt es zur Zusammlagerung zweier RNA-Stränge. Allerdings lagern sich dabei zwei zueinander komplementäre RNA-Stränge zusammen, denn es wird von dem gleichen Gen neben der mRNA auch eine Antisense-RNA vom komplementären DNA-Strang hergestellt. MicF wird dann permanent exprimiert, wenn der globale Aktivator MarA durch den Funktionsverlust des Repressors MarR konstitutiv exprimiert wird. Die permanente Anwesenheit von micF führt dann zur vollständigen Blockade der OmpF-Translation und somit zu einem Verlust der OmpF-Porine [70,147]. Dies zieht einen verminderten Influx von Antibiotika nach sich und infolgedessen eine reduzierte Empfindlichkeit gegenüber verschiedenen Substanzen [59,141]. Der Aktivator MarA ist neben der indirekt negativen Regulation der Porinexpression auch an der direkt positiven Regulation von Effluxpumpen beteiligt [20,113]. Effluxpumpen besitzen elementare Transportfunktionen für die Bakterienzelle. Neben Pumpen mit hoher 19 Einleitung Substratspezifität, wie z.B. Tetracyclin-Pumpen [203] transportieren so genannte multidrug efflux (MDE) Transporter unterschiedlicher Substanzen. ein breites Aufgrund der Spektrum auch verschiedenen für strukturell den sehr Transport verwendeten Energiequellen werden zwei Hauptgruppen unterschieden. Pumpen, die ihre Energie über die ATP-Hydrolyse beziehen, werden der ABC-Superfamilie (ATP-binding cassette) zugeordnet [144,172]. ABC-Transporter spielen vor allem in eukaryontischen Zellen eine große Rolle. In Bakterien ist vor allem die zweite Gruppe von Effluxpumpen von Bedeutung, die ihre Energie über Kopplung des Transportes an den Protonengradienten beziehen [229,230]. In letztere Gruppe fallen Pumpen der Familien MFS (major facilitator superfamily), SMR (small multidrug resistance), MATE (multidrug and toxic compound extrusion) und RND (resistance nodulation cell division) (siehe Abbildung 1-6). Abbildung 1-6: Schema der fünf Familien der Effluxpumpen (aus [238]) Als eine Hauptpumpe bei der Entwicklung klinischer Resistenz von E. coli ist die AcrABTolC-Pumpe beschrieben [188,219,225]. Sie wird der Familie der RND-Pumpen zugeordnet, welche in gramnegativen Bakterien Antibiotika transportieren können [243]. Die RND-Pumpen sind aus drei Teilen aufgebaut [184]. Das eigentliche Pumpenprotein AcrB durchspannt die Cytoplasmamembran [310] und das Membranfusionsprotein AcrA stellt die Verbindung zu TolC, dem Porin in der äußeren Membran her [92,163]. Neben dem AcrAB-System von E. coli gehören auch z.B. die MexAB-Pumpe von Pseudomonas aeruginosa, die CmeABC-Pumpe von Campylobacter jejuni und die MtrCDE-Pumpe von Neisseria gonorrhoeae zur RND-Familie und weisen in ihren Pumpenproteinen hohe Homologie auf [138,238]. Eine Regulation der Pumpenaktivität ist für die Zelle lebensnotwendig, da die Pumpe ein sehr breites Substratspektrum besitzt und sonst auch essentielle Stoffwechselprodukte aus der Zelle hinaus transportiert [243]. Die AcrAB-TolC-Pumpe transportiert verschiedene Antibiotika 20 wie Chloramphenicol, Tetracycline, ß-Lactame, Novobiocin, Linezolid, Einleitung Nalidixinsäure und Fluorchinolone, aber auch andere Substanzen wie SDS, Triton X-100, Gallensalze und kationische Farbstoffe [216,243]. Die Regulation der Effluxpumpe AcrAB erfolgt über lokale und globale Regulatoren. Als globaler Regulator, der neben der Porinexpression auch Effluxpumpen reguliert, ist MarA zusammen mit dem Repressor MarR als ein Bestandteil des marRAB-Operons in E. coli identifiziert worden [4,57,238,242]. MarR reprimiert die Expression des Operons. Mutationen in marO oder marR führen über den Verlust der reprimierenden Wirkung zur konstitutiven Überexpression des Operons und somit von MarA. Dies wiederum bewirkt durch Bindung des Transkriptionsaktivators MarA an konservierte DNA-Bereiche (mar-Boxen) im Promotorbereich verschiedener Gene die Überexpression u.a. der AcrAB-Effluxpumpe und der micF-RNA. Letztere wiederum bewirkt eine Reduktion von OmpF (siehe oben) [15,176]. Für eine Vielzahl von Antibiotika wird also gleichzeitig der Influx reduziert und der Efflux erhöht. Somit führen z.B. Mutationen in marR zum MAR-Phänotyp (multiple Antibiotikaresistenz) der betroffenen Zelle. Eine Induktion der Effluxpumpe AcrAB-TolC kann in vitro durch die Bindung von Salicylat an MarR erfolgen [4,5,249,280]. Neben MarA sind auch SoxRS und Rob als weitere globale Regulatoren von AcrAB-TolC und möglicherweise auch anderer Effluxpumpen in E. coli beschrieben. Das soxRS-System reguliert mit dem Transkriptionsaktivator SoxS und dem Sensor SoxR vor allem die Antwort der Zelle auf oxidativen Stress. Sekundär wird aber auch Antibiotikatransport über Effluxpumpen beeinflusst [8,165,166,224]. Das soxRS-Regulon ist in vitro z.B. durch Stickstoffoxid oder Paraquat stark induzierbar [6,8,53]. Rob (right oriC binding) kann, wie auch SoxS, an die Mar-Promotor-Bindungsstellen (mar-Boxen) binden und somit ebenfalls die Pumpenexpression positiv regulieren [27,300]. Zusätzlich wird die Expression der AcrAB-Effluxpumpe aber auch über den lokalen Repressor AcrR reguliert [48,225]. Defektmutationen in acrR führen ebenfalls zur Überexpression der AcrABPumpe und reduzieren die Empfindlichkeit gegenüber Fluorchinolone [48,226,297]. Die Identifizierung von Effluxpumpen und die Aufklärung ihrer Regulation bei Bakterien werden von vielen Arbeitsgruppen vorangetrieben. Das Wissen über die Effluxpumpen in Salmonella spec. ist bisher nur begrenzt [24,26,48,51,103]. Aufgrund der nahen Verwandtschaft mit E. coli wird aber davon ausgegangen, dass es sich um sehr ähnliche Pumpensysteme und Regulationen derselben handelt [238]. Als weitere Pumpen neben AcrAB [24,26] ist mit hoher Homologie zu E. coli in Salmonella spec. z.B. auch das AcrEFTolC-System beschrieben. Diesem kommt aber wahrscheinlich, ähnlich wie in E. coli eine untergeordnete Bedeutung zu [84]. Auch das marRAB- und das soxRS-Operon sind im Genom von S. enterica mit hoher Homologie zu E. coli lokalisiert und die gleiche Wirkungsweise beschrieben worden [84,165,249,280]. Bei klinischen Isolaten von S. Typhimurium konnte eine erhöhte Expression von acrB durch eine Mutation in acrR als Ursache für den MAR-Phänotyp identifiziert werden [26,103,226,240]. Konkrete Hinweise auf eine breite Beteiligung von marR Mutationen an der Fluorchinolonresistenz in Salmonella spec. gibt es jedoch bislang nicht. 21 Einleitung Neben Fluorchinolonen sind bzw. waren auch ß-Lactame und früher Chloramphenicol bei der Behandlung von Salmonellosen von Bedeutung. Diese Substanzen sind Substrate von MDR-Effluxpumpen. Somit kommt der Erforschung der Effluxpumpen als Resistenzmechanismus bei Salmonella spec. eine besondere Bedeutung zu. Für die Entwicklung klinischer Resistenz haben Effluxpumpen eine starke Bedeutung. Die Überexpression von Effluxpumpen lässt zwar, auch in Kooperation mit Porinverlust, die minimalen Hemmkonzentrationen der Antibiotika meist nur um zwei bis vier Verdünnungsstufen steigen. Das bedeutet bei sehr empfindlichen Bakterien wie E. coli oder Salmonella spec. nicht notwendigerweise klinische Resistenz. Dazu sind meistens noch zusätzliche Resistenzmechanismen, wie z.B. Targetgenmutationen, notwendig. Allerdings wird vermutet, dass die durch den verstärkten Efflux leicht erhöhten MHKWerte der Zelle ein verlängertes Überleben auch unter erhöhten Substanzkonzentrationen sichern. Das ermöglicht es dem Bakterium, weitere Resistenzmutationen zu akkumulieren. 1.3.3 Neue Resistenzmechanismen und enzymatische Inaktivierung von Fluorchinolonen Die Veränderungen der Zielstrukturen der Fluorchinolone und die reduzierte Akkumulation der Substanzen in der Zelle wurden lange Zeit als einzige Resistenzmechanismen gegenüber Fluorchinolonen angesehen. Eine Verbreitung plasmid-codierter Chinolonresistenz war zunächst nicht beobachtet worden. Allerdings stellten 1998 Martinez-Martinez et al. [198] fest, dass die Transformation des Multiresistenz-Plasmides pMG252 aus ciprofloxacinresistenten klinischen Isolaten von Klebsiella pneumoniae in sensible E. coli-Stämme eine MHK-Wert-Erhöhung von Ciprofloxacin und Nalidixinsäure bewirkte. 2002 amplifizierten und klonierten Tran & Jacoby [285] das plasmidcodierte qnrA-Gen und fanden in einem DNA-Gyrase-Assay heraus, dass das Genprodukt QnrA die DNA-Gyrase von E. coli vor der Inhibition durch Ciprofloxacin schützt. Nach Tran et al. [284] interagiert QnrA vermutlich noch vor der DNA mit der Gyrase, denn es war eine reduzierte Affinität der DNA zur Gyrase zu beobachten, wenn QnrA gebunden war. Vermutlich ist der Mechanismus ähnlich dem des aus Mycobacterium tuberculosis stammenden Proteins MfpA, das die DNA-Helixstruktur imitiert. Dadurch bindet die Gyrase an das Protein MfpA statt an die DNA und ist somit vor Chinolonhemmung geschützt [122]. Da Chinolone an den Gyrase-DNA-Komplex binden, bewirkt die aufgrund der QnrA-Wirkung reduzierte Anzahl Gyrase-DNA-Verbindungen auch eine geringere Anzahl Targets für die Chinolone [284]. Eine Interaktion zwischen QnrA und Topoisomerase IV ist beschrieben, allerdings sind die genauen Mechanismen aber noch nicht endgültig verstanden [286]. Bei dem beschriebenen Resistenzmechanismus wird demzufolge das Target der Chinolone so durch eine Protein-Interaktion geschützt, dass die Chinolone nur eingeschränkte Wirkung ausüben können. Neben dem QnrResistenzmechanismus, 22 war auch die Erkenntnis neu, dass reduzierte Einleitung Fluorchinolonempfindlichkeit mobil über Plasmide übertragbar ist. Das Integron des Plasmides pMG252 enthält neben qnrA auch Gene für die Resistenz gegenüber ßLactamen, Chloramphenicol, Streptomycin, Sulfonamiden und Tremethoprim. Die Anwesenheit von qnrA löst zwar nur reduzierte Empfindlichkeiten gegenüber Fluorchinolonen aus, die Gefahr liegt jedoch in der Entwicklung von multiresistenten Keimen aufgrund der Kombination mehrerer Resistenzmechanismen. Das konnten Martinez-Martinez und Mitarbeiter bereits in vitro zeigen [197]. In den letzten zwei Jahren wurde bereits in mehreren Publikationen das Auftreten klinischer Isolate in den USA, China und Europa beschrieben, die eine reduzierte Chinolonempfindlichkeit besaßen und in denen das qnr-Gen nachgewiesen werden konnte [155,191,260,296]. Ein völlig anderer neuer Resistenzmechanismus gegen Fluorchinolonen wurde von Robicsek et al. [260] beschrieben. Sie fanden ein Enzym, welches Fluorchinolone modifizierenden kann. Die enzymatische Inaktivierung eines Antibiotikums als Mechanismus bakterieller Resistenz kann generell sowohl hydrolytische Spaltungen, als auch enzymatische Modifikationen durch die Übertragung einer funktionellen Gruppe umfassen. Bekannt ist dieser Resistenzmechanismus von den ß-Lactam-Antibiotika, Aminoglykosidantibiotika sowie Chloramphenicol. Alle drei Substanzklassen haben die Isolierung aus Mikroorganismen als gemeinsames Merkmal. Das Auftreten von inaktivierenden Enzymen wurde bisher ausschließlich bei natürlich vorkommenden Antibiotika gefunden und damit begründet, dass sich im Laufe der Evolution neben der Entwicklung antibakterieller Substanzen auch die Entwicklung von entsprechenden Gegenstrategien in Form von Resistenzmechanismen vollziehen konnte. Aufgrund der synthetischen Herstellung der Fluorchinolone wurde eine Co-Evolution von inaktivierenden Enzymen innerhalb eines relativ kurzen Zeitraums von wenigen Jahrzehnten seit Einführung des ersten Chinolons als unwahrscheinlich angesehen. Neuere Studien bestätigen aber das Auftreten von Enzymen, welche durch Modifikation der Chinolone eine reduzierte Empfindlichkeit der Zelle bewirken. Robicsek et al. [260] beschrieben das Auftreten der Aminoglykosid-Acetyltransferase AAC(6')-Ib-cr in klinischen Isolaten, die eine reduzierte Empfindlichkeit gegenüber Ciprofloxacin besaßen. Es konnten in den Stämmen Plasmide isoliert werden, die neben qnrA auch eine Variante des aac(6`)-Ib Gens mit zwei Mutationen enthielten, die zu Änderungen in den Codons Trp102 Æ Arg, Asp179 Æ Tyr führen. In Tests konnte AAC(6')-Ib-cr für die reduzierte Empfindlichkeit gegenüber Ciprofloxacin verantwortlich gemacht werden. Als inaktivierendes Enzym für antimikrobielle Substanzen waren Acetyltransferasen bisher lediglich für Chloramphenicol und Aminoglykoside beschrieben. Diese mutierte Variante einer Aminoglykosid-Acetyltransferase reduziert zwar die vermittelte Aminoglykosidresistenz, bewirkt aber zusätzlich eine Chinolonmodifikation. Allerdings konnte festgestellt werden, dass AAC(6`)-Ib-cr nicht alle Fluorchinolone modifiziert. Es werden nur Ciprofloxacin und Norfloxacin angegriffen, welche an C7 eine unsubstituierte Piperazinylgruppe besitzen, an der eine N-Acetylierung stattfinden kann [260]. Mit dieser 23 Einleitung Acetyltransferase-Variante wurde zum einen ein bisher unbekannter Resistenzmechanismus gegenüber Fluorchinolonen beschrieben und zum anderen ein Enzym identifiziert, welches zwei Antibiotikaklassen gleichzeitig modifiziert und somit eine Co-Selektion von Resistenzen ermöglicht. Die aktuellen Entwicklungen und Berichte über plasmidvermittelte Fluorchinolonresistenz und die Möglichkeit der Modifizierung der Chinolone durch eine Acetyltransferase lassen eine Abnahme der Wirksamkeit der Fluorchinolone befürchten, die bisher immer als Reserveantibiotika der neueren Generation galten. 1.4 Die Entwicklung von Salmonella-Impfstämmen Wie bereits einleitend beschrieben, stellen Infektionen mit Salmonella spec. nach wie vor weltweit ein großes Gesundheitsproblem dar (Abschnitt 1.1.2). Aufgrund der Zunahme des Auftretens von resistenten Salmonella spec. (Abschnitt 1.2) und die Entwicklung und Verbreitung neuer Resistenzmechanismen gegenüber Fluorchinolonen (Abschnitt 1.3.3) ist die Wirksamkeit einer Antibiotikatherapie als wirksame Behandlungsmethode bei schweren Fällen von Salmonellosen (Abschnitt 1.1.2) zunehmend reduziert. Die effizienteste und kosten-effektivste Möglichkeit gegen Infektionskrankheiten vorzugehen ist die prophylaktische Immunisierung gegen Pathogene. Deshalb ist die Entwicklung neuer und sicherer Impfstämme eine der größten Herausforderungen der modernen Medizin. Im Gegensatz zu Totimpfstoffen, die im Körper eine humorale Immunität auslösen, sind Lebendimpfstoffe wesentlich effizienter. Sie enthalten vermehrungsfähige Viren oder Bakterien mit abgeschwächter Virulenz (Attenuation), welche im Körper zusätzlich eine zelluläre Immunantwort auslösen. Die Attenuation kann durch zufällige oder gezielte Mutationen erreicht werden. Eine Mutation wird entweder chemisch induziert oder über Transposons bzw. gezielt durch Rekombinationstechniken eingeführt. Zur Attenuierung eignen sich vor allem Gene, die für essentielle Stoffwechselleistungen codieren oder die für die Virulenz des Stammes entscheidend sind. Bei der Impfstammentwicklung werden weiterhin Mutationen als epidemiologische Marker verwendet. Generell muss ein bakterieller Impfstamm verschiedene Sicherheitsanforderungen erfüllen [269]: (1) das Vorhandensein von einem oder mehreren unabhängigen Attenuationsmarkern, welche einerseits eine Virulenzabschwächung bewirken und andererseits eine sichere Unterscheidung des Impfstammes von Feldstämmen und eine molekularbiologischen Charakterisierung ermöglichen, (2) Stabilität der Attenuation mit Vermeidung der Reversion zum virulenten Wildtyp, (3) Vermehrungsfähigkeit und „Restvirulenz“ zur Auslösung der Immunität im Zielorganismus, (4) Unschädlichkeit für Menschen und immunsupprimierte Tiere. 24 Einleitung Durch die ursprüngliche Methode der chemisch induzierten Mutagenese mittels MethylNitro-Nitrosoguanidin und die Einführung von Auxotrophiemarkern wurden einige Salmonella-Impfstoffe entwickelt. Auxotrophie bedeutet ein Stoffwechseldefizit für den Bakterienstamm. Die Stämme wurden auf die gewünschten phänotypischen Marker vorselektiert und dann erfolgte eine Einführung zusätzlicher Marker durch ungezielte Mutagenese sowie eine tierexperimentelle Selektion auf verminderte Virulenz [177-179]. Die Einstellung des Attenuierungsgrades erfolgte dabei über die Kopplung von Mutationen, welche als Stoffwechseldrift-Ereignisse in essentiellen Enzymen bzw. Stoffwechselsystemen sowohl Antibiotikaresistenz als auch verringerte Virulenz vermitteln. Ein Stoffwechseldrift-Ereignis bedeutet eine durch Mutation hervorgerufene graduell abgestufte Veränderung von bestimmten Leistungsparametern, wie z.B. der Generationszeit. Stoffwechselsysteme mit essentiellen Funktionen können zum Beispiel Replikations-, Transkriptions-, Translations- oder Permeationssysteme sein. Mutationen können an verschiedenen Genloci auftreten und einen neuen Phänotyp bedingen, der sich durch eine verlängerte Generationszeit und entsprechende Virulenzreduktion auszeichnet. Im Gegenteil dazu versteht man unter Stoffwechselshift eine qualitative Änderung im Leistungsspektrum, wie z.B. die Aufnahme neuer genetischer Information in Form von Plasmiden. Die Stoffwechseldrift-Theorie besagt, dass mutative Veränderungen in essentiellen Enzymen bzw. Zellkompartimenten in vivo eine evolutionäre Anpassung an die sich ständig verändernde Umwelt ermöglichen [177,179]. So sind zum Beispiel pathogene Bakterien darauf angewiesen, auf Veränderungen in der Wirtspopulation zu reagieren und sich anzupassen. Diese genetische Variabilität, die eine Veränderung bestimmter Leistungsparameter und vor allem der Virulenzeigenschaften eines Stammes nach sich zieht, ermöglicht die gezielte Ausnutzung bei der Impfstammentwicklung. Da essentielle Stoffwechselkompartimente auch oft Angriffspunkte für Antibiotika sind, wird bei der Mutagenese der Zusammenhang zwischen verlängerter Generationszeit, reduzierter Virulenz und Bindungsverlust für Antibiotika und somit Resistenzentwicklung genutzt. Eine Kopplung verschiedener Marker resultiert in einem durch seine verlängerte Generationszeit optimal an den gewünschten Wirt attenuierten Lebendimpfstamm. Folgende Proteine eignen sich als Angriffspunkte bei der Impfstammentwicklung [179]: (1) Die RNA-Polymerase spielt eine entscheidende Rolle bei der Transkription und Mutationen z.B. in rpoB können eine Resistenz gegen Rifampicin vermitteln. (2) Die Gyrase steuert als Topoisomerase II den DNA-Superspiralisierungsgrad und beeinflusst v.a. die DNA-Replikation und Transkription. Mutationen in den Genen der Gyrase bedingen oft eine Nalidixinsäureresistenz. (3) Mutationen in den Genen für Ribosomenproteine, welche für eine exakte Polypeptidsynthese mitverantwortlich sind, bewirken meistens Resistenz gegen Streptomycin. 25 Einleitung Um attenuierte Lebendimpfstämme zu entwickeln, wurden stabile Mutanten aus S. enterica erzeugt [41,63,88]. Die eingeführten Mutationen beeinflussten vor allem die Auxotrophien für bestimmte Aminosäuren und Purine oder bewirken Veränderungen des Stoffwechsels. Dadurch ist die Fähigkeit dieser Stämme reduziert, in den eukaryontischen Wirtzellen zu überleben und sich auszubreiten [177-179]. Im Folgenden sind einige beispielhafte Mutationen aufgeführt, welche sich aufgrund der daraus resultierenden Virulenz- und Wachstumsreduktion als vorteilhaft bei der Impfstammentwicklung erwiesen haben: Als Impfstamm einsetzbar wären z.B. Salmonella-Stämme mit Mutationen in aroA. Das Gen aroA codiert für die 3-Enolpyruvylshikimat-5-phosphat-Synthetase, welche die Umsetzung von Shikimat-3-phosphat und Phosphoenolpyruvat in 5-Enolpyruvylshikimat3-phosphat katalysiert. S. Typhimurium Mutanten mit defektem aroA-Gen sind z.B. nicht mehr in der Lage bestimmte Synthesewege für einige aromatische Verbindungen, z.B. für die aromatische Aminosäuren Tryptophan, Tyrosin und Phenylalanin, oder bestimmte Metabolite als Vorstufen des Vitamins Folsäure zu durchlaufen [60,185]. Aufgrund des Fehlens dieser Metabolite im Säugetiergewebe können sich die betroffenen Mutanten dort nicht vermehren und sind somit attenuiert. Stämmen mit Mutationen in galE hingegen fehlt das Enzym Galactose-Epimerase, welches für die Synthese der bakteriellen Zellwand essentiell ist. Die Störung der Synthese des LPS führt zum Fehlen des vollständigen O-Antigens, wodurch die Bakterien in ihrer Virulenz abgeschwächt sind. Attenuation in Folge einer stark verlängerten Generationszeit weisen Mutanten mit Defekten in den Genen cya und crp auf. Durch Inaktivierung der Adenylatcyclase (cya) bzw. des cAMP-Rezeptorproteins (crp) kann entweder keine Umwandlung von ATP zu cAMP in den Zellen stattfinden bzw. keine Bindung des cAMPs an das cAMPRezeptorprotein erfolgen. Dieser Komplex ist in der Zelle als Transkriptionsaktivator für zahlreiche Gene notwendig, die für den Transport und den Abbau von Kohlenhydraten und Aminosäuren benötigt werden. Ein weiteres Beispiel für die Attenuation in SalmonellaStämmen stellen Mutationen in phoP dar. Da PhoP zusammen mit PhoQ ein Zweikomponentensystem bildet, was u.a. für die Expression des invasiven Phänotyps verantwortlich ist (Abschnitt 1.1.3.3), sind entsprechende Mutanten nicht in der Lage, den Intestinaltrakt zu kolonisieren [88]. Die Entwicklung von Salmonella-Impfstämmen erlangt ebenfalls wichtige Bedeutung, da Salmonellen als Vektoren für fremde Antigene in Frage kommen [41]. Der Einsatz von attenuierten Mikroorganismen als Vektoren für die Impfung gegen verschiedene Pathogene ist ein weiterer wichtiger Ansatz bei der Entwicklung von neuen Impfstoffen [164]. Dabei ermöglicht die Präsentation fremder Antigene durch replikationsfähige und in ihrer Virulenz abgeschwächte bakterielle Erreger eine Impfung auch gegen andere Organismen als den Impfstamm. 26 Einleitung Zur Bekämpfung von Salmonellosen wird neben den Anstrengungen die hygienischen Bedingungen bei der Lebensmittelgewinnung zu verbessern, auch der Verwendung von Lebendimpfstoffen eine große Bedeutung beigemessen. Das Ziel ist es, die Infektionskette zwischen Huhn und Mensch zu unterbrechen und schon präventiv im Tier Salmonellosen zu unterbinden [22,313]. Während eine Antibiotikatherapie nur auf den Zeitraum der Behandlung beschränkt ist, hält eine Immunisierung für einen (lebens)langen Zeitraum an. In Deutschland gibt es derzeit mehrere Salmonella-Lebendimpfstoffe zur Anwendung beim Huhn, Rind, Schwein oder bei der Taube. In allen Fällen hat eine Kombination von Selektion auf bestimmte Phänotypen (Antibiotikaresistenz oder Auxotrophien) und Behandlung mit N-Methyl-N´-Nitrosoguanidin stattgefunden und zur Einführung weiterer Mutationen geführt. Bei den Ausgangsstämmen dieser Impfstoffe handelt es sich z.B. um die Salmonella-Serovare Enteritidis, Dublin (Impfstamm Bovisaloral zum Einsatz bei Rindern) und Choleraesuis (Impfstoff Suisaloral zum Einsatz bei Schweinen). Bei Hühnern werden die Impfstoffe Zoosaloral und TAD Salmonella vacT (S. enterica-Serovar Typhimurium) eingesetzt. Beide Stämme mit reduzierter Virulenz wurden aus dem S. Typhimurium Wildstamm M415 durch Behandlung mit N-Methyl-N´-Nitrosoguanidin erzeugt [177,179].Im Impfstamm Zoosaloral sind Veränderungen der Auxotrophien zu finden. Dieser Stamm ist eine Doppelmutante (his-155/ade-4), welche auxotroph für Histidin und Adenin ist [93]. Der attenuierte Lebendimpfstoff „TAD Salmonella vacT“ (Lohmann Animal Health GmbH & Co. KG) ist zur Immunisierung von Legehennen zugelassen. Bei diesem Stamm handelt es sich um eine antibiotikaresistente Doppelmutante (Nal2/Rif9) des S. Typhimurium Wildstammes M415. Der Stamm wurde auf Nährmedien mit 400 µg/ml Nalidixinsäure und 100 µg/ml Rifampicin isoliert und weist eine verlängerte Generationszeit [38]. Darüber hinaus besitzt der Stamm einen Marker, der eine Sensibilität gegenüber MakrolidAntibiotika vermittelt und als antiepidemischer Marker dient. Dieser Marker soll die Überlebensfähigkeit der Bakterien in der Umwelt reduzieren. Somit besitzt vacT neben der gewünschten Virulenzreduktion, einerseits therapeutisch unbedenkliche Antibiotikaresistenzen, welche als Erkennungs- und Selektionsmarker nutzbar sind, und andererseits zusätzliche Sicherheitsmarker, die die Impfstammausscheidung und sein Überleben in der Außenwelt verkürzen und eine Sensibilität gegen therapeutisch relevante Antibiotika vermitteln. Die Marker werden wie folgt bezeichnet: Erkennungsmarker Nal und Rif (Resistenz gegenüber Nalidixinsäure und Rifampicin) und die Sicherheitsmarker rtt (reversion to tensid tolerance) und ssq (super susceptibility to quinolones). 27 Einleitung 1.5 Zielstellung der Arbeit Der Impfstamm vacT ist durch ungezielte Mutagenese entstanden. Er besitzt neben der Nalidixinsäure- und Rifampicinresistenz, die als Selektionsmarker dienen, auch Sicherheitsmarker in Form von Sensibilitäten gegenüber Fluorchinolonen und Makroliden. Da die Ursache dieser phänotypischen Merkmale und der Attenuation unbekannt sind, ist es das Ziel dieser Arbeit, die molekularen Ursachen der Marker des Impfstammes vacT zu erforschen. Der Ausgangspunkt der Arbeit war die Beobachtung der Diskrepanz zwischen der Nalidixinsäureresistenz und der Supersensibilität gegenüber Fluorchinolonen. Das Vorhandensein der gyrA-Mutation D87G in vacT, welche die Nalidixinsäureresistenz erklären könnte, war bekannt. Diese gyrA-Mutation vermittelt normalerweise aber ebenfalls reduzierte Empfindlichkeiten gegenüber Fluorchinolonen. Im Rahmen dieser Arbeit sollen sowohl der Ausgangsstamm S. Typhimurium M415 als auch der in einem Selektionsschritt bei der Impfstammentwicklung entstandene Stamm Nal2ori und dessen Derivat Nal2passage sowie der Impfstamm vacT geno- und phänotypisch charakterisiert werden. Es sollen die Empfindlichkeiten gegenüber zahlreichen Antibiotika Superspiralisierungsgrad, aus sowie verschiedenen die Fitness Substanzklassen, der Stämme der (in Form DNAder Wachstumsgeschwindigkeit) erfasst werden. Durch diese Charakterisierung sollen die für den Impfstamm beschriebenen Marker überprüft werden. Ein weiteres Ziel der Arbeit ist der Vergleich der Expressionsmuster der Proteine in den Untersuchungsstämmen, um so auf mögliche Stoffwechseldefekte im Impfstamm zurückschließen zu können. Außerdem soll überprüft werden, wie sich der Ausgangsstamm M415 und der Impfstamm vacT in vitro unter Chinolon-Selektionsdruck verhalten. Für die Kompensation der Fluorchinolonresistenz im Impfstamm vacT kommen intraoder extragenische Mutationen in Frage. Im Impfstamm vacT neu identifizierte Mutationen in gyrA sollen auf ihre mögliche intragenische Kompensationsfähigkeit untersucht werden. Durch eine Charakterisierung von isogenen Stämmen, die über homologe Rekombination hergestellt wurden, können die Auswirkungen der einzelnen Mutationen auf die entsprechenden phänotypischen Eigenschaften, wie dem DNA-Superspiralsierungsgrad, der Wachstumsgeschwindigkeit und der Fluorchinolonempfindlichkeit, untersucht werden. Aufgrund der ungezielten Mutagenese des Impfstammes vacT ist das Auftreten von Mutationen in unbekannten Genen möglich und sogar wahrscheinlich. Aufgrund des rttMarkers wird dem Transport durch die Membran, v.a. dem aktiven Efflux, Bedeutung zugemessen. Ein Defizit im Efflux kommt als extragenische Kompensation der normalerweise durch gyrA-Mutation hervorgerufenen reduzierten Empfindlichkeit gegenüber Fluorchinolonen in Frage. Die Funktionstüchtigkeit der Effluxpumpen im Impfstamm vacT im Vergleich zum Ausgangsstamm soll also ebenfalls überprüft werden. 28 Material und Methoden 2 Material und Methoden 2.1 Material 2.1.1 Bakterienstämme Die Tabellen 2-1 und 2-2 enthalten die im Rahmen der Arbeit verwendeten Bakterienstämme einschließlich der über Selektions- bzw. Rekombinationsversuche hergestellten Stämme. Tabelle 2-1: Bakterienstämme der Species Escherichia coli Stammbezeichnung Genetische Marker Herkunft / Referenz - DH5α F , Φ80dlacZ∆M15, ∆(lacZYA-argF), U169, recA1, endA1, + hsdR17(rk , mk ), phoA, supE44, λ , thi-1, gyrA96, relA1 Invitrogen GmbH TOP10F’ q R F’, lacI , TN10 (Tet ), mcrA ∆ (mrr-hsdRMS-mcrBC), Φ80lacZ ∆M15, ∆lacX74, recA1, araD139 ∆(araleu)7697, galU, galK, rpsL (StrR), endA1, nupG Invitrogen GmbH K-12 KNK402 F+, gyrA43(ts), polA1, endA2, thyA755, rpsL, tsx-79, phx [167] K-12 C600SN mit Plasmid RP1H thr1, leuB6, thi1, lacY, tonA, supE44, hsdR, rpsL, recA, gyrAR [129] Tabelle 2-2: Bakterienstämme der Species Salmonella spec. Stammbezeichnung bekannte Mutationen S. Typhimurium ATCC14028 (Referenzstamm) keine Dr. W. Rabsch, Robert-Koch-Institut Wernigerode S. Typhimurium M415 keine Dr. W. Rabsch, Robert-Koch-Institut Wernigerode S. Typhimurium Nal2ori gyrA: G75A, D87G, A866S Dr. J. Beer, Universität Leipzig S. Typhimurium Nal2passage gyrA: G75A, D87G, A866S Dr. J. Beer, Universität Leipzig S. Typhimurium TAD Salmonella vacT gyrA: W59R, G75A, D87G, A866S Lohmann Animal Health, Cuxhaven S. Typhimurium vacTE1 gyrA: W59R, G75A, D87G, A866S H. Hansen / aus Selektionsversuch S. Typhimurium vacTE2 gyrA: W59R, G75A, D87G, A866S H. Hansen / aus Selektionsversuch S. Typhimurium vacTE3 gyrA: W59R, G75A, D87G, A866S H. Hansen / aus Selektionsversuch S. Typhimurium SM415-81 gyrA: G81D Diese Arbeit / aus Selektionsversuch S. Typhimurium SM415-83 gyrA: S83F Diese Arbeit / aus Selektionsversuch S. Typhimurium SM415-87 gyrA: D87Y Diese Arbeit / aus Selektionsversuch S. Typhimurium RM415 keine Diese Arbeit / aus Rekombinationsversuch S. Typhimurium RM415-87 gyrA: D87G Diese Arbeit / aus Rekombinationsversuch S. Typhimurium RM415-59-87 gyrA: W59R, D87G Diese Arbeit / aus Rekombinationsversuch S. Typhimurium RM415-75-87 gyrA: G75A, D87G Diese Arbeit / aus Rekombinationsversuch Serovar Herkunft / Referenz 29 Material und Methoden 2.1.2 Vektoren In Tabelle 2-3 sind alle in dieser Arbeit verwendeten bzw. konstruierten Vektoren inklusive ihrer Größe, relevanten genetischen Marker und Herkunft aufgeführt. Tabelle 2-3: Vektoren Vektor* Größe in bp Antibiotikaresistenzeigenschaften** relevante Charakteristika Herkunft / Referenz pBAD102/D 4417 Amp, Kan araC-Parab Invitrogen GmbH pBP507 9198 Gen, Ami, Kan - [123] pBP517 12355 Gen, Ami, Kan gyrA+ aus E. coli [123] pCR2.1TOPO® 3931 Amp lacZα Invitrogen GmbH Amp, Clm repA101-ts, FLP (flp), Expressionsplasmid [46] pCP20 pT7-6 2208 Amp pT7 [282] pKD4 3267 Amp, Kan Kan-FRT-Templateplasmid [66] pKD46 6329 Amp repA101-ts, araC-Parab, λ RedRecombinaseExpressionsplasmid(exo, β, γ) [66] pKOV 8673 Clm repA101-ts, sacB [180] pPHB90 6531 Tet ptopA-luc aus E. coli [1] pPHB91 6269 Tet ptopA-luc aus E. coli [1] pPHB94 11368 Gen, Ami ptopA-luc aus E. coli [1] pPHB95 11106 Gen, Ami ptopA-luc aus E. coli [1] pPHB451 4694 Amp, Kan lacZα, gyrA-Fragment + W59R aus S. Typhimurium M415 Diese Arbeit* pPHB452 4694 Amp, Kan lacZα, gyrA-Fragment + G75A aus S. Typhimurium M415 Diese Arbeit* pPHB453G 4694 Amp, Kan lacZα, gyrA-Fragment + D87G aus S. Typhimurium M415 Diese Arbeit pPHB454 4694 Amp, Kan lacZα, gyrA-Fragment + W59R, G75A aus S. Typhimurium M415 Diese Arbeit* pPHB455 4694 Amp, Kan lacZα, gyrA-Fragment + W59R, D87G aus S. Typhimurium M415 Diese Arbeit* 10563 Clm, Kan repA101-ts, sacB, gyrA aus S. Typhimurium M415 Diese Arbeit * pPHB650 10563 Clm, Kan repA101-ts, sacB, gyrA + D87G aus S. Typhimurium M415 Diese Arbeit * pPHB653G 10563 Clm, Kan repA101-ts, sacB, gyrA + W59R, D87G aus S. Typhimurium M415 Diese Arbeit * pPHB655 10563 Clm, Kan repA101-ts, sacB, gyrA + G75A, D87G, A866S aus S. Typhimurium Diese Arbeit * pPHB658 30 * Material und Methoden * 10563 Clm, Kan repA101-ts, sacB, gyrA + W59R, G75A, D87G, A866S aus S. Typhimurium vacT Diese Arbeit pPHB659 pPHB750 5059 Amp pT7, gyrA aus S. Typhimurium M415 Diese Arbeit* 5059 Amp pT7, gyrA + D87Y aus S. Typhimurium M415 Diese Arbeit * pPHB753Y 5059 Amp pT7, gyrA + G75A, D87G, A866S aus S. Typhimurium Nal2ori Diese Arbeit * pPHB758 5059 Amp pT7, gyrA + W59R, G75A, D87G, A866S aus S. Typhimurium vacT Diese Arbeit * pPHB759 * Nomenklatur der im Rahmen dieser Arbeit konstruierten Plasmide: p = Plasmid; PHB = Institut für Pharm. Biologie u. Mikrobiologie; 1. Zahl = Bezeichnung des Ausgangsvektors: 3 Æ pBR322, 4 Æ pCR2.1TOPO, 6 Æ pKOV, 7 Æ pT7-6; 2. Zahl = 5 = Projekte von Antje Preisler; 3. Zahl = Schlüssel der im Plasmid vorhandenen gyrA-Mutationen: 0 Æ keine, 1 Æ W59R, 2 Æ G75A, 3 Æ D87 gefolgt von Angabe des Aminosäureaustausches = G Æ Glycin oder Y Æ Tyrosin, 4 Æ W59R+G75A, 5 Æ W59R+D87G, 8 Æ aus Nal2ori: G75A+D87G+A866S, 9 Æ aus vacT: W59R+G75A+D87G+A866S; ** Antibiotikaresistenteigenschaften: Ami = Aminkacin, Amp = Ampicillin, Clm = Chloramphenicol, Gen = Gentamicin, Kan = Kanamycin 2.1.3 Oligonukleotide Die in dieser Arbeit eingesetzten Oligonukleotide wurden mit dem DNA-Synthesizer Modell 391 PCR-Mate im Labor selbst hergestellt oder von den Firmen Sigma Aldrich Chemie GmbH bzw. Invitrogen GmbH bezogen. In den Tabellen 2-4 bis 2-8 sind alle verwendeten Oligonukleotide mit ihrer Sequenz und der Lage (Nukleotidposition) relativ zum Genanfang aufgeführt. Die Sequenz der Oligonukleotide ist standardmäßig in 5`Æ 3`Richtung angegeben. Die Primer sind mit Hilfe der Referenzsequenz von S. Typhimurium LT2 (GenBank accession number NC_003197) erstellt [202]. Tabelle 2-4 zeigt alle Primer, welche hauptsächlich für die Sequenzierung der kompletten Targetgene gyrA, gyrB, parC und parE und für das Gen cpxA verwendet wurden. Tabelle 2-4: Oligonukleotide zur Sequenzierung Bezeichnung Sequenz (5’ Æ 3’) (Nukleotidposition) Länge [bp] gyrA-5-1 AS GTTTACCAGTACGTGGGCGAATG (-76- -54) 23 gyrA-3-1 AS GATACCACGACGACCGTTGATGA (687-665) 23 gyrA-5-2 AS GACAACGAAGACATCAGCATTGA (595-617) 23 gyrA-3-2 AS TTCAGTCAGGTAGTACTGACCGT (1356-1334) 23 gyrA-5-3 AS ATCTGGGCAACGTTGCTGC (1247-1265) 19 gyrA-3-3 AS GAATTCGGTCAGGCGCGTT (1987-1969) 19 gyrA-5-4 AS AGGCGTCAACGTCTTTATGG (1926-1945) 20 gyrA-3-4 AS TTACTCGTCAGCGTCATCCG (2637-2618) 20 gyrA-5-1S GAATCCGGGATACAGTAGAGGGATA (-33- -25) 25 gyrA-3-1S GGAATTTTGGTCGGCATGACGTCCG (491-467) 25 gyrB-5-1S CCGCTTAAAAGCCACGCAAT (-149- -129) 20 31 Material und Methoden gyrB-3-1S TTATCGGTATCGCCAGTGACGG (485-464) 22 gyrB-5-2S GTCTCGGTAGTCAACGCTCTGT (358-379) 22 gyrB-3-2S CCATGTAGGCGTTCAGCGTA (892-873) 20 gyrB-5-3S GCTGCAGTGGAACGATGGTT (765-784) 20 gyrB-3-3S CACCAGGTACAGTTCGGACA (1269-1250) 20 GYR5B2S(I) GTCTGCCGGGCAAACTGGC (1205-1223) 19 (I) GYR3B2S GCCGTCGACGTCCGCATCG (1509-1491) 19 gyrB-5-4S CGCGCTTCGACAAGATGCTT (1367-1386) 20 AACTTCCACTGACTGCCGTGCT (1946-1925) 22 gyrB-5-5S TCTGATGAGCAGACTGTAACGC (1861-1882) 22 gyrB-3-5S(I) CCAATTAGCGACCTGTTCG (2590-2572) 19 parC-5-1A ATGAGCGATATGGCAGAGCGC (1-21) 21 parC-3-1 AGCTCGGCGTATTTGGACAGG (392-372) 21 parC-5-1S ATGGCGCAGCCGTTCTCTTA (265-284) 20 parC-3-1S ATCGTCCACCATCGGCAGTT (852-833) 20 parC-5-2S TATGGACCAAAGAAGACGGCG (731-751) 21 parC-3-2S TCATTTTGCGTTCGGACG (1372-1355) 18 parC-5-3S CATCTCGCCAAACTGGAAGAGA (1270-1291) 22 parC-3-3S TCTGGTCATCGCCTTCCAT (1777-1759) 19 parC-5-4S ATATGCTGATGGAAGGCGAT (1751-1770) 20 parC-3-4S TCCGATCTGAAACCGTAGGC (2370-2349) 21 parE-5-1S TGTCGATTTTCTTGGACTGC (-58- - 38) 20 parE-3-1S GCTCGCTGTTATTCACTTCATC (631-610) 22 parE-5-2S GATGAAGTGAATAACAGCGAGC (610-631) 22 gyrB-3-4S (I) parE-3-2S CTTCATCGGAAGAGACCTCC (1363-1344) 20 (I) ACCGAGCTGTTCCTTGTGGAAG (1286-1307) 22 PARE3-2-S(I) ACAGATCTTGCCGTAGCGCAGC (1489-1510) 22 parE-5-3S AAGTGCTGGCCTCGCAAGAAGT (1361-1382) 22 parE-3-3S CGTCATGTCGGATTCGCTTAT (1956-1936) 21 cpxA5-1-SalTyph TCACCCTGCTCTATTTGCTG (-211- -192) 20 cpxA-3-1-SalTyph TGCGAGGTCATCATCCGTT (716-697) 19 cpxA-5-2-SalTyph GGTCAGTTCGCCGCTCTTGCTA (519-540) 22 cpxA3-2-SalTyph GATGGCAAAATTACCCGGAT (1499-1480) 20 PARE5-5-S (I) [117] Alle Oligonucleotide, die als Primer für die SOEing-PCR-Technik (siehe Abschnitt 2.2.2.5.2) verwendet wurden (Einführung der Nukleotidaustausche zur zielgerichteten Mutagenese), sind in Tabelle 2-5 aufgelistet. 32 Material und Methoden Tabelle 2-5: Oligonukleotide für die zielgerichtete Mutagenese (SOEing) Bezeichnung Sequenz (5’ Æ 3’) (Nukleotidposition) Länge [bp] gyrA-5-59-AS GCAATGACAGGAACAAAGCCT (167-187) 21 gyrA-3-59-AS AGGCTTTGTTCCTGTCATTGC (187-167) 21 gyrA-5-75-AS GACGTAATCGCTAAATACCATC (214-235) 22 gyrA-3-75-AS GATGGTATTTAGCGATTACGTC (235-214) 22 gyrA-5-87AS GCAGTGTATGGCACCATCGT (250-269) 20 gyrA-3-87-AS ACGATGGTGCCATACACTGC (269-250) 20 fettes Nukleotid = eingeführter Basenaustausch zur Erzeugung der Mutationen In Tabelle 2-6 sind die Reverse-Primer für die sequenzspezifische PCR (ARMS) aufgeführt (siehe Abschnitt 2.2.2.5.3). Tabelle 2-6: Oligonukleotide für die sequenzspezifische PCR Bezeichnung Sequenz (5’ Æ 3’) (Nukleotidposition) Länge [bp] gyrA-59-orig-AS ACGGGCAGATTTTTTATAGGCTTTGTTACA (204-175) 30 gyrA-59-mut-AS ACGGGCAGATTTTTTATAGGCTTTGTTACT (204-175) 30 gyrA-75-orig-AS TGCGGAATCGCCGTGGGGATGGTATTGAC (252-224) 29 gyrA-75-mut-AS TGCGGAATCGCCGTGGGGATGGTATTGAG (252-224) 29 gyrA-87-orig-AS GAATGGCTGCGCCATACGAACGATGGGGT (288-260) 29 gyrA-87-mut-AS GAATGGCTGCGCCATACGAACGATGGGGC (288-260) 29 fettes Nukleotid = zusätzlich eingeführter Basenaustausch; unterstrichenes Nucleotid = Basenaustausch Tabelle 2-7 beinhaltet die Oligonukleotide für die Amplifikation der Gene (inklusive Einführung von Restriktionsschnittstellen und homologen Flanken) für die verschiedenen Klonierungen. Tabelle 2-7: Oligonukleotide für die verschiedenen Klonierungen Bezeichnung Sequenz (5’ Æ 3’) (Nukleotidposition) Länge [bp] Einsatz* gyrA-5-SalI-AS TCTCCCCTGTGTCGACTCTGAAG (-181- -160) 23 A gyrA-3-BamHI-AS GTAAATGACGGATCCCGCAACG (2704-2682) 22 A gyrA-Bam-fw TGAGTGAGTCACGGATCCGAGGTATTGATTTTCTAAC (-241-204) 37 B gyrA-Not-rv TATGACTGCGGCCGCTAAGGTAGCGGTAAATGACG (2729-2697) 35 B HF-5-Sal TCATGCCTAGGTCGACTGAAGAACCGATTCATCCAT (-888- -852) 36 C HF-3-pkD4 CTAAGGAGGATATTCATATGCGGGAGGCATAAACCATAAT (-355- -394) 40 C pKD4-5-BamHI AGTCCAGATCGGATCCGTGTAGGCTGGAGCTGCTTC (-444- -408) 36 D pKD4-3-HF ATTATGGTTTATGCCTCCCGCATATGAATATCCTCCTTAG (1069-1030) 40 D * A = Klonierung von gyrA in pT7-6; B = Klonierung von gyrA in pKOV; C = Amplifikation der homologen Flanke vor gyrA liegend; D = Amplifikation der Kanamycinresistenz-Kassette aus pKD4; unterstrichen = Primer-Bindungsstelle; fett = Enzym-Erkennungssequenz 33 Material und Methoden Die als Primer zur Kontrolle des Erfolges der homologen Rekombination (Abschnitt 3.2.4.2) eingesetzten Oligonukleotide sind in Tabelle 2-8 zusammengefaßt. Tabelle 2-8: Oligonukleotide für Screening-PCRs Bezeichnung Sequenz (5’ Æ 3’) (Nukleotidposition) Länge [bp] Einsatz* pKOV-L(II) AGGGCAGGGTCGTTAAATAGC 22 E pKOV-R(II) TTAATGCGCCGCTACAGGGCG 21 E k2(III) CGGTGCCCTGAATGAACTGC (162-181) 20 F kt(III) CGGCCACAGTCGATGAATCC (632-613) 20 F TGCGGCTCTTGACGATGGAT (-1142-1122) 20 G gyrA-1142fw (II) (III) * [180] [66]; E = Nachweis des Plasmides pKOV; F = Nachweis der Kanamycinresistenz-Kassette; G = Primer vor der homologen Flanke liegend 2.1.4 Nährmedien Tabelle 2-9 enthält die Zusammensetzung der verschiedenen flüssigen und festen Nährmedien. Soweit nicht anders angegeben, beziehen sich die Einwaage-Angaben auf 1 Liter Gesamtvolumen. Die Medien wurden unmittelbar nach der Einwaage in dH2O gelöst und autoklaviert. Tabelle 2-9: Nährmedien Bezeichnung Herstellung Bq 10 x M9-Salze 5 g NaCl, 112,27 g Na2HPO4 x 7H2O, 10 g NH4Cl, 30 g KH2PO4; pH 7,4 - Chinablau-Lactose-Agar nach Herstellerangaben 1 LB-Bouillon 10 g Trypton, 5 g Hefeextrakt, 10 g NaCl; pH 7,4 - M9-Minimalmedium + Glucose 750 ml dH2O autoklavieren, auf 50°C abkühlen; plus 2 ml 1M MgSO4 + 0,1 ml 1M CaCl2 + 100 ml 10 x M9-Salze + 10 ml 20% Glucoselösung - M9-Minimalagar + Glucose 18 g Agar in 800 ml dH2O autoklavieren, auf 50°C abkühlen lassen; Zugabe von 1 ml 1M MgSO4 + 0,1 ml 1M CaCl2 + 100 ml 10 x M9-Salze + 10 ml 20% Glukoselösung - Mueller-Hinton-Bouillon nach Herstellerangaben 2 SOB 0,5 % Hefeextrakt, 2,0 % Trypton, 10 mM NaCl, 2,5 mM KCl, 10 mM MgCl2 , 10 mM MgSO4 - SOC 0,5 % Hefeextrakt, 2 % Trypton, 10 mM NaCl, 2,5 mM KCl, 10 mM MgCl2 , 20 mM MgSO4, 20 mM Glucose - Standard NI-Nährbouillon nach Herstellerangaben 3 Standard NI-Nähragar nach Herstellerangaben 3 Terrific Broth nach Herstellerangaben 2 TYE-Agar 3 g Trypton, 5 g Glucose, 5 g Hefeextrakt, 20 g Agar pH 7 - Bq = Bezugsquelle: 1 = Oxoid GmbH, England; 2 = Becton Dickinson; 3 = Merck KGaA 34 Material und Methoden 2.1.5 In Verwendete Kits dieser Arbeit wurden Kits unterschiedlicher Firmen verwendet. Die Pufferzusammensetzungen (soweit angegeben), die Protokolle und Hinweise zur Durchführung sind in den jeweiligen Handbüchern zu finden. Tabelle 2-10 zeigt die verwendeten Kits, deren Anwendung und Hersteller. Tabelle 2-10: Verwendete Kits Anwendung Kit-Bezeichung Hersteller QIAquick PCR-Purification Kit QIAGEN GmbH NucleoSpin® Extract II MACHEREY-NAGEL QIAquick Gel Extraction Kit QIAGEN GmbH NucleoSpin® Extract II MACHEREY-NAGEL QIAGEN Plasmid Midi Kit QIAGEN GmbH NucleoSpin® Plasmid MACHEREY-NAGEL Perfectprep® Plasmid Kit Eppendorf AG pCR2.1TOPO-Klonierung TOPO TA Cloning® Kit Invitrogen GmbH® pBAD-Klonierung pBAD Directional TOPO Expression Kit Invitrogen GmbH® Sequencer Kit Applied Biosystems Proteinaufbereitung ReadyPrep protein extraction kit Bio-Rad Laboratories Proteinkonzentrationsbestimmung RC DC Protein Assay Bio-Rad Laboratories Silver Stain Plus Bio-Rad Laboratories SilverQuest Silver Staining Kit Invitrogen GmbH® Aufreinigung von Nukleinsäuren DNA-Aufreinigung Gelextraktion Plasmidpräparation Plasmid-DNA-Aufreinigung Klonierung Sequenzierung Sequenzierkit 2D-Gelelektrophorese Silberfärbung 2.1.6 Puffer und Lösungen Tabelle 2-11 enthält alle verwendeten Puffer und Lösungen und ihre Zusammensetzung. Soweit nicht anders angegeben, beziehen sich die Einwaagen auf 1 Liter Gesamtvolumen. 35 Material und Methoden Tabelle 2-11: Verwendete Puffer und Lösungen Puffer / Lösung Zusammensetzung Beschwererlösung 0,25 % Bromphenolblau, 0,25 % Xylencyanol, 20 % Ficoll-400 Puffer EB (Elutionspuffer) 10 mM Tris-HCL (pH 8,5) Isotonische Kochsalzlösung, 0,9 % 9 g NaCl ad dH2O, autoklavieren 10 x Ligationspuffer 660 mM Tris-HCl, 50 mM MgCl2, 10 mM DTT, 10 mM ATP ad 10 ml; pH 7,5; aliquotieren und bei -20°C lagern TAE-Puffer (50 x) 2 M Tris-Base, 50 mM EDTA, mit Essigsäure auf pH 8,0 einstellen Herstellung kompetenter Zellen zur Lagerung bei -80°C TFB1 0,5 g K-Acetat, 1 g MgCl2, 0,75 g KCl, 1,6 ml 1M CaCl2, 17 ml Glycerin (87 %) ad 100 ml H2O TFB2 0,1 g MOPS, 3,8 ml CaCl2, 0,0375 g KCl, 8,5 ml Glycerin (87 %) ad 50 ml H2O Luciferase-Assay-System K2HPO4 / EDTA-Puffer 1 M K2HPO4 (pH 7,8), 20 mM EDTA Cell Culture Lysis Reagent (1 x) 25 mM Trisphosphat (pH 7,8), 5 vmM DDT, 2 mM CDTA, 10 % Glycerol, 1 % Triton X-100 Lysis Mix 1 x CCLR, 5 mg/ml BSA, 5 mg/ml Lysozym Plasmid Minipräparation (alkalisch Lyse modifiziert nach [31]) GTE-Lösung 50 mM Glucose, 25 mM Tris-HCl (pH 8,0), 10m M EDTA in dH2O, autoklavieren, bei 4 °C lagern Lysepuffer 0,2 N NaOH, 1 % SDS in dH2O, jeweils frisch ansetzen 5 M Kaliumacetatlösung 29,5 ml Eisessig + KOH-Pellets bis pH 8,0, mit H2O auf 100 ml auffüllen, nicht autoklavieren, Lagerung bei RT Elutionspuffer 10 mM Tris-HCl (pH 8,0), 1 mM EDTA in dH2O, autoklavieren Proteinbestimmung nach Lowry Alkalisches Kupferreagenz 2 % CuSO4, 4 % K-Na-Tartrat, 3 % Na2CO3 in 0,1 M NaOH Lösungen für 2D-Gelelektrophorese 10 x PBS-Puffer 0,58 M Na2HPO4, 0,17 M NaH2PO4, 0,68 M NaCl, pH 7,4 Lysepuffer 9 M Harnstoff (Urea), 1 % DTT, 2 % CHAPS, 0,8 % Bio-Lyte 3/10 Rehydrierungspuffer 8 M Harnstoff, 10 mM DTT, 1 % CHAPS, 0,25 % Bio-Lyte pH 3-10 Äquilibrierungslösung 8 M Harnstoff, 30 % Glycerin, 2 % SDS (in 0,05 M Tris/HCL pH 8,8), 1 % DTT APS 40 % 400 mg Ammoniumpersulfat ad 1 ml dH2O Trenngelpuffer 1,5 M Tris/HCL (pH 8,8) 10 x Elektrodenpuffer (Laufpuffer) 0,025 M Tris , 0,192 M Glycin, 0,1 % SDS (pH 8,3 nicht mit HCl einstellen) Coomassie-Färbelösung 0,08 % Coomassie in 1,6 %Phosphorsäure, 8 % Ammoniumsulfat, 20 % Methanol 36 Material und Methoden Fixierlösung 1 für Silberfärbung 30 % Ethanol, 10 % Eisessig Fixierlösung 2 für Silberfärbung 30 % Ethanol, 0,5 M Na-Acetat, 0,5 % Glutardialdehyd, 0,2 % NaThiosulfat x 5 H20 ReadyStrip IPG Strips pH 4–7; 12 cm Silbernitratlösung 0,1 % Silbernitrat, 0,02 % Formaldehyd Entwicklerlösung für Silberfärbung 2,5 % Natriumcarbonat, 0,01 % Formaldehyd Stopplösung für Silberfärbung 0.05 M EDTA (Titriplex) 2.1.7 Enzyme In Tabelle 2-12 sind alle verwendeten Enzyme und deren Bezugsquelle aufgelistet. Tabelle 2-12: Verwendete Enzyme Enzym Bezugsquelle Alkalische Phosphatase (Shrimp) Roche Diagnostics GmbH DNA Polymerase I, Large (Klenow) Fragment New England Biolabs TM Expand High Fidelity Polymerase Roche Diagnostics GmbH Lysozym Sigma Aldrich Chemie GmbH Pefabloc Roche Diagnostics GmbH Phusion™ High-Fidelity DNA Polymerase New England Biolabs Restriktionsendonukleasen New England Biolabs bzw. Fermentas GmbH RNAse A QIAGEN GmbH Taq DNA Polymerase New England Biolabs T4-DNA-Ligase Roche bzw. New England Biolabs T4-DNA-Polymerase Roche Diagnostics GmbH 37 Material und Methoden 2.1.8 Antibiotika Tabelle 2-13 enthält die für die In-vitro-Selektionsversuche (Abschnitt 2.2.1.5), Empfindlichkeitsbestimmungen mittels Mikrodilutionsverfahren (Abschnitt 2.2.1.6) und zur Selektion rekombinanter Bakterien verwendeten Chemotherapeutika. Tabelle 2-13: Verwendete Antibiotika Antibiotika Bezugsquelle Amikacin Bristol-Myers Squibb Ampicillin Bayer AG, Ciprofloxacin Bayer AG Gentamicin Merck KGaA Kanamycin Sigma Aldrich Chemie GmbH Nalidixinsäure Sterling Winthrop Inc. Sparfloxacin Bayer AG Tetracyclin Sigma Aldrich Chemie GmbH Weiterhin wurden für Empfindlichkeitsbestimmungen verschiedene Mikrotitrationsplatten der Firma MERLIN Diagnostika GmbH verwendet. Diese Platten enthalten die Antibiotika in dehydratisierter Form. Die in diesen Platten enthaltenen Substanzen sind in Tabelle 216 (Abschnitt 2.2.1.6.2) aufgeführt. 2.1.9 Chemikalien Alle verwendeten Chemikalien sind in Tabelle 2-14 alphabetisch aufgeführt. Tabelle 2-14: Verwendete Chemikalien Bezeichnung Bezugsquelle Acrylamidlösung 30 %, 37.5:1 Bio-Rad Laboratories Agar-Agar, granuliert Merck KGaA Agarose, Seakem® LE, für die Elektrophorese Biozym Diagnostik GmbH Ammoniumpersulfat Bio-Rad Laboratories Bacto Trypton Becton Dickinson Bio-Lyte 3/10 Bio-Rad Laboratories Bromphenolblau, für die Elektrophorese Bio-Rad Laboratories sec-Butanol Sigma Aldrich Chemie GmbH Calciumchlorid-dihydrat Merck KGaA CHAPS Bio-Rad Laboratories Coomassie G-250 Bio-Rad Laboratories dNTPs, 100 mM Lithium-Salz-Lösungen, für die PCR Roche Diagnostics GmbH 38 Material und Methoden MOPS Sigma Aldrich Chemie GmbH Natriumchlorid Merck KGaA di-Natriumhydrogenphosphat Merck KGaA Dimethylsulfoxid (DMSO) Merck KGaA Dithiothreitol (DTT) Bio-Rad Laboratories Essigsäure, 100 % Acros Organics Ethanol, absolut Merck KGaA Ethidiumbromid, 1 %ige Lösung in H2O Merck KGaA Ethylendiamin-tetra-acetat EDTA (Titriplex) Merck KGaA Ficoll-400, 20 % Sigma Aldrich Chemie GmbH Formaldehyd (37 %) Merck KGaA Glucose-Monohydrat Merck KGaA Glutardialdehyd Merck KGaA Glycerin, 87 % Merck KGaA Glycerin, 99 % SERVA Electrophoresis GmbH Hefeextrakt, granuliert Merck KGaA Isopropanol Merck KGaA Kaliumacetat Merck KGaA Kaliumchlorid Merck KGaA Kaliumhydroxid Merck KGaA Magnesiumchlorid Merck KGaA Methanol Merck KGaA Natriumacetat Merck KGaA Natriumcarbonat Merck KGaA Natriumchlorid Merck KGaA Natriumdodecylsulfat (SDS), 10 % und 20 % Bio-Rad Laboratories Natriumthiosulfat Merck KGaA Phenylalanyl-Arginyl-β-Naphthylamid Sigma Aldrich Chemie GmbH Phosphorsäure Merck KGaA Rinderserumalbumin (BSA) SERVA Electrophoresis GmbH Salzsäure, 37 % Merck KGaA Silbernitrat Merck KGaA Smart Ladder (DNA-Längenstandard) Eurogentec TEMED (Tetramethylethylenediamin) Bio-Rad Laboratories trans-1,2-Diaminocyclohexan-N,N,N’,N’,Tetraacetat Sigma Aldrich Chemie GmbH Tris(hdroxymethyl)-aminomethan (Tris-Base) Merck KGaA Urea (Harnstoff) Bio-Rad Laboratories Xylencyanol Merck KGaA 39 Material und Methoden 2.1.10 Geräte und Materialien Tabelle 2-15 enthält alle Geräte und Materialien, die in dieser Arbeit verwendet wurden. Tabelle 2-15: Verwendete Geräte und Materialen Gerät / Material Bezugsquelle Brutschränke Heraeus DNA-Sequencer ABI-Prism 310 Applied Biosystems DNA-Syntheziser 391 PCR-Mate Applied Biosystems Einmalspritzen (5 ml, 10 ml) Merck KGaA Elektrophorese Power Supply Biozym Diagnostik GmbH Elektrophorese Power Pac1000 Bio-Rad Laboratories Elektroporationsküvetten Bio-Rad Laboratories bzw. Eurogentic Eppendorf-Caps (0,2 ml, 1,5 ml, 2,0 ml) Eppendorf AG Gelelektrophorese-Apparatur Protean II Bio-Rad Laboratories Gelkammern Comphor Mini, Midi, Maxi Biozym Diagnostik GmbH Gilson Kolbenhubpipetten, variabel ABIMED Analysen-Technik GmbH Elektroporationsgerät Genepulser II und Pulse Controller II Bio-Rad Laboratories IEF Cell Bio-Rad Laboratories Küvetten (Halbmikro) CARL ROTH GmbH & Co KG Luminometer (Lucy 2) anthos Mikrosysteme GmbH Mikrotiterplatten: Luminometer-Platten DYNATECH MicroFOUR Mikrotiterplatten: MHK-Platten Merck KGaA Multistep-Pipette (8-Kanal) Hirschmann Laborgeräte GmbH & Co.KG pH-Meter WTW GmbH Petrischalen SARSTEDT AG & Co Reinstwasseranlage (Milli-Q synthesis) Millipore GmbH Schüttelinkubatoren: Certomat R+H Schüttelinkubator (Serie 25) und NEW BRUNSWICK SCIENTIFIC & CO, Inc und Braun Biotech Sicherheitswerkbank, Hera safe Heraeus Savant Speed Vac SDC 100 Global Medical Instrumentation, Inc Spektralphotometer (Cary 50) Varian GmbH Sterilfilter, 0,2 µm, 7 bar Sartorius AG Sterilwerkbank, Hera gard Heraeus Thermocycler T3, TGradient, TPersonal Biometra GmbH Thermopapier, Sony UPP-110HP Biometra GmbH Thermo-Printer, Sony UP-D890 Biometra GmbH Ultraschallbad Sonorex TK 52 BANDELIN electronic GmbH & Co. KG Video-Dokumentationsanlage BioDoc Biometra GmbH Vortex Genie 2 Scientific Industries, Inc. Wasserbad JULABO Labortechnik GmbH Zentrifugen: Biofuge pico, Biofuge fresco, Biofuge stratos (Rotoren #3331, #3332, #3335, #3046) Heraeus 40 Material und Methoden 2.2 Methoden 2.2.1 Mikrobiologische Methoden 2.2.1.1 Herstellung von Übernachtkulturen (ÜNK) Um eine Übernachtkultur (ÜNK) herzustellen, wurden ein bis drei Bakterienkolonien von einer Agarplatte mit Hilfe eines Glasstabes abgenommen und in 3 ml entsprechende Bouillon eingerieben. Für die Selektion plasmidtragender Stämme wurde der Bouillon das entsprechende Antibiotikum zugesetzt. Anschließend wurde die Übernachtkultur für 1418 h bei der entsprechenden Temperatur bei 130 upm schüttelnd inkubiert. 2.2.1.2 Herstellung und Verwendung von Dauerkulturen Glycerindauerkulturen: Um eine langfristige Lagerung und damit auch eine Sicherung der gewünschten Bakterienstämme zu gewährleisten, wurden die Stämme als Glycerindauerkulturen angelegt und bei -80 °C gelagert. Dazu wurden Probenröhrchen mit Schraubverschluss (Kryoröhrchen) mit 0,5 ml Standard-No.I-Nährbouillon-Glycerin- Lösung (Verhältnis 1:1) und 10 Glasperlen gefüllt und autoklaviert. Dann wurden von einer über Nacht inkubierten Agarplatte zwei bis drei Bakterienkolonien mit einem Glasstab abgenommen und in dem Medium suspendiert. Nach 30 min Inkubation bei Raumtemperatur (RT) wurde die Flüssigkeit oberhalb der Glasperlen abgenommen und die Glycerinkultur bei –80 °C gelagert. Zum Ausimpfen der Glycerindauerkultur wurde dem Röhrchen eine Perle entnommen und auf eine Agarplatte gebracht und ausgestrichen. Stichkulturen: Als Alternative zur langfristigen Lagerung wurden Stichkulturen angelegt. Diese werden bei RT gelagert und bieten den Vorteil des einfachen schnellen Ausimpfens ohne Auftauen. Allerdings sind die Kulturen nicht so lange haltbar wie Glycerindauerkulturen bei -80 °C. Zur Vorbereitung wurde ein Probenröhrchen mit Weichagar (bestehend aus 10 mg NII-Nähragar, 3 mg NI-Nährbouillon und 1 ml dH2O) gefüllt und autolaviert. Zum Anlegen einer Stichkultur wurden eine oder mehrere Bakterienkolonien von einer über Nacht bebrüteten Agarplatte abgenommen und der Agar im Röhrchen mittels einer Impfnadel angeimpft. Die Dauerkultur wurde über Nacht bei der entsprechenden Wachstumstemperatur inkubiert, dann fest verschlossen und bei RT gelagert. Zum Ausimpfen wurde mit einer Impfnadel in den mit Bakterien bewachsenen Agar gestochen und etwas Material auf eine Agarplatte ausgestrichen. 2.2.1.3 Bestimmung der Lebendzellzahl (KBE) Für viele Versuche (z.B. Generationszeitbestimmung, In-vitro-Selektion) ist es wichtig, die genaue Lebendzellzahl der verwendeten Bakterien zu kennen. Bestimmt wird diese Lebendzellzahl durch Auszählen der Kolonie-bildenden Einheiten (KBE). 41 Material und Methoden Für eine KBE-Bestimmung wurde die zu untersuchende Bakteriensuspension in physiologischer Kochsalzlösung 1:10 verdünnt. Entsprechend nach der erwarteten Zellzahl wurde eine adäquate Verdünnungsreihe über mehrere Zehnerpotenzen hergestellt. Von den unterschiedlichen Verdünnungsstufen wurden jeweils 50 µl auf eine Agarplatte pipettiert und mit einem Glasstab gleichmäßig verteilt. Bei Stämmen, welche ein Plasmid enthielten, wurde den Platten das entsprechende Selektionsantibiotikum zugesetzt. Zur Materialersparnis wurde die Agarplatte geviertelt, so dass auf einer Agarplatte vier verschiedene Verdünnungsstufen ausplattiert werden konnten. Die Inkubation der Agarplatten erfolgte über 24 h bei 37 °C im Brutschrank. Am nächsten Tag wurden alle Kolonien ausgezählt und die KBE pro ml errechnet. In die Auswertung wurden die Verdünnungsstufen einbezogen, deren Koloniezahlen zwischen 10 und 100 lagen. Die Berechnung erfolgte: KBE / ml = 2.2.1.4 Zur Koloniezahl Verdünnungsstufe × aufgetragenes Volumen [ml ] Bestimmung der Generationszeit Berrechnung der Generationszeit wurde eine Wachstumskurve des zu untersuchenden Stammes erstellt: (1) durch Auftragen der optischen Dichte (OD) gegen die Zeit oder (2) durch Auftragen der logKBE/ml gegen die Zeit. Bei (1) geht man davon aus, dass die optische Dichte einer Bakteriensuspension mit der Bakteriendichte über einen bestimmten Zeitbereich korreliert. Eine etwas aufwendigere Methode ist die Möglichkeit der Bestimmung über die Lebendzellzahl (2). Diese Methode gibt aber ein genaueres Bild der Zellzahlen, da nur die lebenden Zellen in die Berechnung eingehen. Zur Berechnung der Generationszeiten wurde die OD546 bzw. logKBE/ml gegen die Zeit aufgetragen. Aus den Daten der exponentiellen Wachstumsphase wurde dann die Regressionsgerade für diesen Graphen berechnet. Aus dem Anstieg dieser Regressionsgerade (m) ergibt sich die Generationszeit (g) nach: υ= lg N − lg N 0 lg 2(t − t0 ) ⇒ υ × lg 2 = m lg N − lg N 0 =m ⇒ υ= (t − t0 ) lg 2 lg N − lg N 0 (t − t0 ) g= 1 υ υ = Teilungsrate; t = Zeit; N = Zellzahl zum Zeitpunkt t; N0 = Zellzahl zum Zeitpunkt 0; m = Steigung der Regressionsgeraden im Bereich der exponentiellen Wachstumsphase bei halblogarithmischer Skalierung der Achse; g = Generationszeit 42 Material und Methoden Generationszeitbestimmung über KBE-Bestimmung: Um die Generationszeit über KBEBestimmung zu ermitteln, wurde von dem zu untersuchenden Stamm eine Übernachtkultur in 3 ml LB-Bouillon angesetzt. Am folgenden Tag wurde diese ÜNK 1:1000 in ein Gesamtvolumen von 400 ml LB-Bouillon verdünnt. Um eine ausreichende Sauerstoffversorgung der Bakterienkultur zu gewährleisten, wurden 1000 ml Kolben genutzt. Die Bakterienkultur wurde bei 37 °C mit 225 upm schüttelnd inkubiert. In regelmäßigen Zeitabständen wurden Proben entnommen. Ein Teil wurde bei einer Wellenlänge von 546 nm photometrisch vermessen, um eine ungefähre Einordnung der Zellzahl vorzunehmen und die Verdünnungsstufen zum Ausplattieren festzulegen. Ein weiterer Teil (0,5 ml) wurde genutzt, um die Verdünnungsreihe in 4,5 ml physiologischer Kochsalzlösung herzustellen. Die KBE-Bestimmung wurde wie im Abschnitt 2.2.1.3 beschrieben durchgeführt. Nach Berechnung der KBE/ml für die einzelnen Zeitpunkte wurde logKBE/ml gegen die Zeit aufgetragen. Aus dem Anstieg der berechneten Regressionsgeraden wurde wie oben beschrieben die Teilungsrate υ und daraus die Generationszeit g bestimmt. Generationszeitbestimmung über die optische Dichte: Dazu wurde eine ÜNK des zu untersuchenden Stammes am folgenden Tag 1:1000 in ein Gesamtvolumen von 50 ml LBBouillon verdünnt. Die Bakterienkultur wurde bei 150 upm bei 37 °C schüttelnd inkubiert. In regelmäßigen Zeitabständen wurde eine Probe entnommen und bei einer Wellenlänge von 546 nm photometrisch vermessen. Zur Auswertung wurden die OD-Werte gegen die Zeit aufgetragen. Für die Werte aus der exponentiellen Phase wurde eine Regressionsgerade berechnet und aus deren Anstieg die Teilungsrate υ sowie die Generationszeit g bestimmt. 2.2.1.5 Um In-vitro-Selektion resistenter Keime Aussagen über Selektionsbedingungen das Auftreten bestimmter (Antibiotikazugabe) treffen Mutationen zu unter können, spezifischen wurden In-vitro- Selektionen durchgeführt. Zunächst erfolgte eine Mutantenselektion. Zur Absicherung, ob die aufgetretenen Mutationen in dem Stamm stabil sind, wurden die selektierten Mutanten einem Stabilitätstest unterzogen. Mutantenselektion: Dazu (Agarplatten unterschiedlichen mit wurde eine Bakteriensuspension auf Antibiotikakonzentrationen) Selektionsplatten ausplattiert. Zur Bestimmung der Selektionskonzentration wurden die durch Mikrodilutionsverfahren (siehe Abschnitt 2.2.1.6) ermittelten MHK-Werte zugrundegelegt. Die spontane Mutationsrate für einen Nukleotidaustausch liegt etwa zwischen 10-6 und 10-10 [158]. Um eine ausreichend hohe Zellzahl (108-1010 Bakterien) auf die Selektionsplatten aufbringen zu können, wurde eine konzentrierte Bakteriensuspension hergestellt. Dazu wurde eine Übernachtkultur in 100 ml NI-Bouillon angesetzt und am nächsten Tag 15 min bei 5.300 upm und 4 °C abzentrifugiert. Das Pellet wurde dann in 1/20 des Ausgangsvolumens in NI-Bouillon 43 Material und Methoden resuspendiert. Von dieser konzentrierten Bakteriensuspension wurden 100 µl auf jeweils 10 Agarplatten einer Selektionskonzentration ausgebracht. Die Agarplatten wurden bis zu 96 h bei 37 °C inkubiert. Nach 24, 48, 72 und 96 h wurde die Gesamtzahl der Kolonien auf den 10 Selektionsplatten ermittelt. Parallel zur Selektion wurde auch eine KBEBestimmung durchgeführt (siehe Abschnitt 2.2.1.3), um die eingesetzte Bakterienzahl genau zu ermitteln. Die Kolonie-bildenden Einheiten (KBE) wurden nach 24 h Inkubationszeit ausgezählt, die Lebendkeimzahl in KBE/ml berechnet und die Mutationsfrequenz mit Hilfe der folgenden Formel bestimmt: Mutationsfrequenz = Anzahl selektierter Mutanten pro ml KBE pro ml Ein Teil der aufgetretenen Mutanten wurden durch Ausstreichen auf frischen Agarplatten mit der gleichen Selektionskonzentration gereinigt. Die gereinigten Mutanten wurden durch MHK-Wert-Bestimmung über Agardilutionsverfahren (siehe Abschnitt 2.2.1.6.4) einer ersten Charakterisierung unterzogen. Stabilitätstest: Dazu wurden von ausgewählten Mutanten 5 ml Übernachtkulturen hergestellt (NI-Bouillon ohne Antibiotikum) und nach 24 stündiger Inkubation (37 °C, 130 upm) 1:100 verdünnt. Nach 3 bis 4 derartiger Passagen in Flüssigmedium wurden Zellen aus Zwischenpassagen auf Agarplatten ausplattiert. So konnten evtl. Kontaminationen erkannt werden und Zwischenstadien der Stämme aufbewahrt werden. Nach mindestens 10 Passagen wurden die Mutantenkulturen wiederum auf Agarplatten mit dem Selektionsantibiotikum ausgebracht. Durch das Wachstum wurde sichergestellt, dass die erworbene Mutation, welche zur reduzierten Empfindlichkeit gegen das Antibiotikum führte, im Stamm stabil war. Im Anschluss wurden die Mutanten weiteren Charakterisierungen wie RFLP, Sequenzierungen und MHK-Wert-Bestimmungen unterzogen. 2.2.1.6 Bestimmung der minimalen Hemmkonzentration (MHK) Um die Empfindlichkeit der Bakterienstämme gegenüber verschiedenen Antibiotika zu testen, wurde eine Bestimmung der minimalen Hemmkonzentration (MHK) durchgeführt. Die minimale Hemmkonzentration [µg/ml] ist definiert als die niedrigste Konzentration eines Antibiotikums, bei der kein sichtbares Wachstum erfolgt. Die meisten Bestimmungen der MHK-Werte im Rahmen dieser Arbeit wurden mittels Mikrodilutionsverfahren durchgeführt [213]. Die Kavitäten einer Mikrotiterplatte, welche serielle zweifache Verdünnungsstufen ausgehend von einer Antibiotikakonzentration enthielten, wurden mit einer Bakteriensuspension befüllt. Das Gesamtvolumen der Kavitäten betrug dabei jeweils 100µl. Jede Platte enthielt als Wachstumskontrolle jeweils eine Kontroll-Kavität mit Medium ohne Antibiotikum. Die Platten wurden für 18-20 h bei der entsprechenden Wachstumstemperatur der Stämme bebrütet und dann abgelesen. Das Ablesen erfolgte visuell im Vergleich mit der mitgeführten, antibiotikumfreien Wachstumskontrolle. 44 Material und Methoden 2.2.1.6.1 MHK-Wert-Bestimmung mittels selbst angelegter Platten Zunächst wurde eine Stammlösung des zu testenden Antibiotikums entsprechend der Vorschriften und daraus dann eine Verdünnungsreihe (1:1) in Mueller-Hinton-Bouillon hergestellt. Jeweils 50 µl einer Verdünnungsstufe wurden in die Kavitäten einer Mikrotiterplatte pipettiert. Pro Reihe in der Mikrotiterplatte wurde jeweils eine Kavität als Wachstumskontrolle nur mit Bouillon befüllt. Im Anschluss wurden jeweils 50µl einer eingestellten Bakteriensuspension in die Kavitäten pipettiert. Die Einstellung der Konzentration einer Bakteriensuspension erfolgte nach folgendem Standard: Von einer frisch bebrüteten Agarplatte wurden mit einem Glasstab Kolonien abgenommen und in 4,5ml sterile physiologische Kochsalzlösung (0,9 % NaCl-Lösung) eingerieben. Diese Bakteriensuspension wurde photometrisch auf eine Trübung (OD550nm = 0,125 +/- 0,02) eingestellt, die einem McFarland Standard 0,5 entsprach (ca. 1 x 108 KBE/ml). Die Suspension wurde daraufhin 1:100 in Mueller-Hinton-Bouillon verdünnt, so dass sich eine Keimzahl von ungefähr 1 x 106 KBE/ml ergab. Da 50 µl Bakteriensuspension pro Kavität verwendet wurden, ergab das eine resultierende Keimzahl pro Kavität von ca. 5 x 104 KBE/ml. Nach der Bebrütung der Mikrotiterplatten erfolgte eine Auswertung nur bei positiver Wachstumskontrolle. 2.2.1.6.2 MHK-Wert-Bestimmung mittels Fertigplatten Für zahlreiche MHK-Wert-Bestimmungen im Rahmen dieser Arbeit wurden Fertigplatten (MICRONAUT-S-Mikrotitrationsplatten) der Firma MERLIN Diagnostika GmbH verwendet (Tabelle 2-16). Diese Platten enthalten Antibiotika in dehydratisierter Form. Die Sensitivitätsprüfung in den Mikrotitrationsplatten beruht auf der Rehydratisierung der Antibiotika durch die Zugabe von Bakteriensuspension. In jede Kavität der Mikrotiterplatte wurden 100 µl der Bakteriensuspension pipettiert. Ablesung und Auswertung erfolgten wie oben beschrieben. Tabelle 2-16: Plattenbezeichnung und enthaltene Antibiotika (inklusive verwendete Abkürzung und Konzentrationen) in den MICRONAUT-S-Mikrotitrationsplatten der Firma MERLIN Diagnostika Antibiotika verwendete Abkürzung Konzentration in µg/ml Quinolone I Antibiotika verwendete Abkürzung Konzentration in µg/ml Quinolone II Nalidixinsäure NDS 1 – 1024 Ciprofloxacin CIP 0,015 – 32 Norfloxacin NOR 0,06 – 128 Gatifloxacin GAT 0,015 – 32 Ofloxacin OFL 0,015 – 32 Sitafloxacin SIT 0,015 – 32 Levofloxacin LEV 0,015 – 32 Moxifloxacin MOX 0,015 – 32 Ciprofloxacin CIP 0,015 – 32 Trovafloxacin TFX 0,015 – 32 Sparfloxacin SPA 0,015 – 32 Grepafloxacin GFX 0,015 – 32 Flexoxacin FLEX 0,015 – 32 Gemifloxacin GMF 0,015 – 32 Pefloxacin PEF 0,06 - 128 Clinafloxacin CLIF 0,015 – 32 45 Material und Methoden Natürliche Resistenz Makrolide u.a. Natürliche Resistenz Tetracycline u.a. Erythromycin ERY 0,03 - 64 Tetracyclin TET 0,03 - 64 Roxithromycin ROXI 0,03 - 64 Doxycyclin DOX 0,03 - 64 Azithromycin AZIT 0,03 - 64 Minocyclin MIN 0,03 - 64 Clarithromycin CLAR 0,03 - 64 Sulfamethoxazol SULF 0,25 - 512 Clindamycin CLIN 0,015 – 32 Trimethoprim TRI 0,03 - 64 Lincomycin LIN 0,015 – 32 Cotrimoxazol COT 0,125 – 256 Livithromycin LIV 0,06 – 128 Nitrofurantoin NFT 0,125 – 256 Synercid RPC 0,03 - 64 Chloramphenicol CHA 0,125 - 128 Natürliche Resistenz Aminoglycoside u.a. Natürliche Resistenz NEU Amikacin AMIK 0,125 - 256 Linezolid LIZO 0,03 - 64 Gentamicin GEN 0,06 – 128 Teicoplanin TEI 0,06 – 128 Kanamycin KANA 0,125 – 256 Vancomycin VAN 0,03 - 64 Netlimicin NET 0,06 – 128 Fusidinsäure FDS 0,015 – 32 Neomycin NEO 0,125 – 256 Fosfomycin FOS 0,125 – 256 Tobramycin TOB 0,06 – 128 Telithromycin TEL 0,03 - 64 Streptomycin STRE 0,125 – 256 ABT-773 Ketolid ABT 0,03 - 64 Apramycin APRA 0,06 – 128 Rifampicin RIF 0,03 - 32 2.2.1.6.3 MHK-Wert-Bestimmung in Anwesenheit eines Effluxpumpeninhibitors Ein Effluxpumpeninhibitor (EPI) wird bei der MHK-Wert-Bestimmung eingesetzt, um den Anteil des Einflusses von Effluxpumpen bei der Ausprägung der Antibiotikaempfindlichkeit eines Stammes zu testen. Wird als Ursache für erhöhte MHK-Werte eine Überexpression von Effluxpumpen vermutet, so ist zu erwarten, dass die MHK-Werte in EPI-Anwesenheit stärker sinken als bei einem Stamm ohne erhöhten aktiven Efflux. Bei der Durchführung der MHK-Wert-Bestimmungen unter Verwendung eines Effluxpumpeninhibitors wurde genauso vorgegangen wie unter 2.2.1.6.1 beschrieben. Zusätzlich wurde hier allerdings zu der Bakteriensuspension der EPI in der doppelten Konzentration zugesetzt, um den späteren Verdünnungseffekt im Testansatz durch den Zusatz der Antibiotkumlösung auszugleichen. Als Effluxpumpeninhibitor wurde in dieser Arbeit die Substanz Phenylalanyl-Arginyl-β-Naphthylamid in einer Konzentration zwischen 4 und 64 µg/ml verwendet. 2.2.1.6.4 MHK-Wert-Bestimmung über Agardilutionsverfahren Um eine Empfindlichkeitsbestimmung für eine größere Anzahl an Bakterienstämmen, z.B. Mutanten aus der In-vitro-Selektion, material- und zeitsparend durchzuführen, wurde das Agardilutionsverfahren verwendet. Dafür wurden Agarplatten mit dem jeweiligen Antibiotikum in steigenden Konzentrationen gegossen. Für die Bestimmung wurden die einzelnen Kavitäten einer Mikrotiterplatte mit jeweils 100 µl NI-Medium beschickt und 46 Material und Methoden mittels eines Glasstabes mit einzelnen Bakterienklonen inokuliert. Eine Kavität wurde als Sterilitätskontrolle nicht beimpft. Nach Übernachtinkubation bei 37°C wurde von einer Häfte einer Mikrotiterplatte (6 x 8 Kavitäten) jeweils ein Aliquot Übernachtkultur mittels Replikastempel („Igel“) auf die Agarplatten mit unterschiedlichen Antibiotikakonzentrationen übertragen. Dafür wurde der Replikastempel in einem ersten Schritt in die beimpfte Mikrotiterplatte getaucht und ein Volumen von etwa 1-2 µl Bakteriensuspension in Kavitäten mit 100 µl physiologischer Kochsalzlösung einer zweiten Mikrotiterplatte überführt. Anschließend wurde dieser Verdünnung wiederum durch einen sterilisierten Replikastempel auf die Agarplatten ausgestempelt. Dabei wurde davon ausgegangen, dass die ÜNK ca. 5 x 108 KBE/ml enthielt und bei der Verdünnung (ca. 1:100) eine KBE/ml von ca. 5 x 106 entstand. Dieser Verdünnungsschritt war notwendig, um eine In-Vitro-Selektion resistenter Bakterien durch zu hohe Baktrenkonzentrationen zu vermeiden. Mit dem Stempel wurde dann ein ungefähres Inokulum von 5 x 104 auf die Agarplatten übertragen. Nach 24 h Inkubation bei 37 °C wurden die Platten durch visuelle Beurteilung des Wachstums unter Beachtung der Sterilitätskontrolle ausgewertet. 2.2.1.7 Methoden des DNA-Transfers Um DNA-Moleküle in Bakterienzellen einzuschleusen, wurden im Rahmen dieser Arbeit die Methoden der Transformation und der Konjugation angewendet. Für eine Transformation müssen die Bakterienzellen aufnahmebereit (kompetent) gemacht werden [153]. Für die Transformationen wurden entweder chemisch (durch CaCl2-Methode) oder elektrisch (für Elektroporation) kompetente Zellen eingesetzt. 2.2.1.7.1 Calciumchlorid (CaCl2)-Methode Die CaCl2-Methode [56] beruht darauf, die DNA-Kompetenz durch eine Vorbehandlung mit bivalenten Kationen (z.B. mit Ca2+) und Aufbewahrung in Kälte zu erhöhen. Dies führt zu einer Veränderung der Zelloberfläche bzw. zu einer porösen Zellwand, wodurch die DNA-Aufnahme ermöglicht wird [29]. Zur Herstellung kompetenter Zellen nach der CaCl2Methode wurde eine Übernachtkultur des Bakterienstammes 1:100 in NI-Bouillon in ein Gesamtvolumen von 50 ml verdünnt und bei bei der entsprechenden Wachstumstemperatur (30 o. 37°C) bei 130 upm bis zu einer OD546nm von 0,4 bis 0,6 schüttelnd inkubiert. Nach Erreichen der OD wurde die Zellsuspension kurze Zeit auf Eis gelagert, bevor die Suspension bei 4 °C und 5.300 upm für 5 min abzentrifugiert, der Überstand dekantiert und das Zellpellet auf Eis gelagert wurde. Kompetente Zellen zur sofortigen Verwendung: Das Zellpellet wurde in 10 ml eiskalter 100 mM CaCl2-Lösung resuspendiert (5:1-Konzentrierung) und für 30 min auf Eis inkubiert. Nach erneuter Zentrifugation (4 °C, 5.300 upm, 5 min) wurde das Pellet in 1 ml kalter 100 mM CaCl2-Lösung aufgenommen (insgesamt 50:1 Konzentrierung) und bei 47 Material und Methoden 4 °C auf Eis aufbewahrt. Eine Lagerung über Nacht kann abhängig vom Stamm die Kompetenz und somit die Effizienz der anschließenden Transformation erhöhen. Im Durchschnitt lassen sich nach chemischer Kompetenz Transformationsraten von 106 bis 107 Kolonien pro µg transformierter DNA erreichen. Bei einer Lagerungszeit über 24 h nimmt die Effizienz jedoch deutlich ab [64]. Kompetente Zellen zur Lagerung bei -80 °C: Nach Abzentrifugieren der Bakterienkultur wurde das Zellpellet in 10 ml eiskaltem TFB1-Puffer resuspendiert, 10 min auf Eis inkubiert, dann wiederum für 5 min bei 5.300 upm und 4 °C abzentrifugiert. Das Pellet wurde dann in 2 ml eiskaltem TFB2-Puffer resuspendiert, 10 min auf Eis inkubiert und anschließend in Aliquots von je 200 µl Zellen bei -80 °C eingefroren. Zur Verwendung wurden die Zellen langsam auf Eis aufgetaut. Transformation durch Hitzeschock nach Hanahan [115]: Zu 100 µl kompetenten Zellen wurde der Ligationsansatz bzw. die Plasmid-DNA (ca. 50 ng) gegeben und für ca. 30 min auf Eis gelagert. In dieser Zeit kann sich die freie DNA an die Cytoplasmamembran anlagern. Im nächsten Schritt wurde der Transformationsansatz für 45 bis 90 Sekunden bei 42 °C inkubiert. Der Hitzeschritt erhöht die Membrandurchlässigkeit und ermöglicht den DNA-Transfer. Danach wurden die Zellen wieder abgekühlt. Dann wurde 1 ml SOCMedium zugegeben und bei der entsprechenden Wachstumstemperatur (30 °C oder 37 °C) bei 130 upm ca. 1 h schüttelnd inkubiert. Im Anschluss wurden je nach erwarteter Transformationsrate 50 µl, 100 µl oder 200 µl Zellsuspension auf antibiotikahaltige Selektionsplatten ausplattiert und über Nacht bei der entsprechenden Temperatur inkubiert. Die CaCl2-Methode wurde ausschließlich für die Transformation von DNA in E. coli-Zellen angewendet. Für die Transformation des pCR2.1-TOPO Vektors wurden in dieser Arbeit kompetente E. coli TOP10F’ Zellen von Invitrogen GmbH verwendet. 2.2.1.7.2 Elektroporation Eine alternative Methode zur Transformation von DNA in chemisch kompetente Bakterien stellt die Elektroporation dar. Das Prinzip besteht darin, dass das durch eine angelegte Spannung entstehende elektrische Feld eine erhöhte Permeabilität der Membran erzeugt [78]. Die Elektroporation liefert prinzipiell höhere Transformationsraten als die CaCl2Methode und wurde im Rahmen der Arbeit angewendet, um DNA in die SalmonellaStämme zu transformieren. Um Kurzschlüsse während der Elektroporation zu vermeiden, wurde immer darauf geachtet, dass die zu transformierende DNA-Lösung salzfrei vorlag. Gegebenenfalls wurde die DNA-Lösung durch Ethanol-Präzipitation (Abschnitt 2.2.2.8.2) und anschließende Aufnahme in Wasser von den Salzen befreit. 48 Material und Methoden Herstellung kompetenter Zellen: Von dem zu transformierenden Stamm wurde eine Übernachtkultur in SOB-Medium angesetzt. Nach einer 1:100 Verdünnung am nächsten Tag in ein Gesamtvolumen von 50 ml SOB-Medium wurde die Bakterienkultur bei 30 bzw. 37 °C und ca. 130-200 upm bis zu einer OD600 von ca. 0,5 bis 0,6 inkubiert. Nach Erreichen der entsprechenden optischen Dichte wurde die Zellsuspension in FalconTubes überführt, 30 min auf Eis gelagert und anschließend bei 7.000 upm für 15 min bei 4 °C abzentrifugiert. Das Pellet wurde dann zweimal in eiskaltem dH2O gewaschen (Aufnahme erst in 5 ml und dann in 2 ml dH2O und jeweils Zentrifugation 15 min bei 4 °C und 7.000 upm). Zum Schluss wurde das Zellpellet in 500 µl eiskaltem dH2O aufgenommen. Für eine Lagerung bei -80 °C kann das Zellpellet auch mit einer 10 % Glycerinlösung gewaschen werden. Eine höhere Kompetenz wurde aber durch Waschen in dH2O und sofortige Verwendung erreicht. Elektroporation: 50 µl kompetente Zellen wurden mit der zu transormierenden DNA gemischt und nach 10 min Inkubation auf Eis in eine Elektroporationsküvette (2 mm Elektrodenabstand) gefüllt. Der elektrische Impuls erfolgte im GenePulser der Firma BioRad Laboratories unter folgenden Bedingungen: 2,5 kV Spannung, Kapazität von 25 µF und Widerstand von 200 Ω. Danach wurde mit einer Pasteurpipette zügig 1 ml SOCMedium in die Küvette gefüllt, um den Transformationsansatz herauszuspülen. Das gesamte Volumen wurde anschließend entnommen und in ein steriles Röhrchen gefüllt. Der Transformationsansatz wurde dann für ca. 1 h schüttelnd bei der entsprechenden Wachstumstemperatur inkubiert. Im Anschluss wurden je nach erwarteter Transformationsrate 50 µl, 100 µl oder 200 µl Zellsuspension auf antibiotikahaltige TYEAgarplatten ausplattiert und über Nacht bei der entsprechenden Temperatur inkubiert. Die Elektroporationsküvette wurde zur Wiederverwendung mit 0,1 % HCL-Lösung gespült, über Nacht mit ca. 70 % Ethanol bedeckt gelagert und dann getrocknet. Die Küvetten wurden maximal zweimal wieder verwendet. 2.2.1.7.3 Konjugation Die Konjugation ermöglicht den DNA-Transfer von einer Bakterienzelle zu einer anderen über eine cytoplasmatische Verbindung (F- oder Sexpilus). Im Rahmen dieser Arbeit wurde diese Methode verwendet, um den Transfer von pBP507-Konstukten aus dem E. coli Stamm C600SN (enthält das Helferplasmid RP1H) in Salmonella-Stämme zu ermöglichen (siehe Abschnitt 2.2.1.8 Komplementationstest) [123,116]. Der Stamm E. coli C600SN diente somit als Donor des Plasmides, die Salmonella-Stämme als Rezipienten. Voraussetzung zur Anwendung dieser Methode ist die Möglichkeit, Donor und Rezipient mit und ohne Plasmid zu unterscheiden. Eine geeignete Selektionsmöglichkeit zur Unterscheidung zwischen E. coli C600SN und den Salmonella-Stämmen, ist die Auxotrophie der E. coli K12-Donorzellen, die auf M9 + Glu-Agar nicht zu wachsen vermögen. Für die Durchführung des Komplementationstestes wurden zunächst je 100 µl 49 Material und Methoden NI-Bouillon in jede Kavität einer Mikrotiterplatte pipettiert. In diese wurden mit dem Glasstab Kolonien der einzusetzenden Stämme resuspendiert. Das Auftragsschema wurde so gewählt, dass zwischen der Auftragsspalte mit Zellen des Rezipienten und denen des Donors, der das zu konjugierende Plasmid trägt, eine Spalte frei blieb, die nur mit Medium gefüllt war. In die Vertiefungen für die Donor-Stämme, die die pBP507- Konstrukte enthielten, wurde Kanamycin (50 µg/ml) zur Selektion des Plasmides zugesetzt. Nachdem nach Übernachtinkubation bei 37°C Donor und Rezipient ausreichend gewachsen waren, wurden jeweils 5 µl Rezipient und 10 µl Donor in die mittlere Spalte pipettiert, gemischt und über Nacht bei 37 °C zum konjugativen Plasmidtransfer inkubiert. Zur Selektion von Transkonjuganden sowie der Trennung von Donor, Rezipient und Transkonjugant wurde ein Aliquot aus den Kavitäten der Mikrotiterplatte per Replikastempel („Igel“) auf geeignete Selektionsplatten übertragen (siehe Abschnitt 2.2.1.6.4). Eine Übersicht gibt Abbildung 2-2 im folgenden Abschnitt. 2.2.1.8 Komplementationstest Bei dem Komplementationstest wird das Dominanzprinzip eines sensiblen Wildtyp gyrA+Gens über ein unsensibleres (mutiertes) gyrA-Gen genutzt. In einem Stamm mit zwei Kopien des gyrA-Gens wird die Chinolonempfindlichkeit durch die sensiblere Gyrase (gyrA+) bestimmt. Für diese Untersuchungen wird das Plasmid pBP517 verwendet, welches das gyrA-Wildtyp-Gen ohne Mutationen aus E. coli enthält [123]. Als Plasmidkontrolle dient der Vektor pBP507 ohne gyrA-Gen. Die Plasmide sind ursprünglich abgeleitete vom natürlichen Plasmid RSF1010 (Inkompatibilitätsklasse Q) [111]. Die verwendeten Vektoren sind in Abbildung 2-1 dargestellt. Abbildung 2-1: Für den Komplementationstest verwendete Plasmide [123] links: Kontrollplasmid pBP507 ohne gyrA-Gen; rechts: pBP517 mit gyrA+ aus E. coli; aacA = Amikacinresistenzgen; aadB = Gentamicinresistenzgen; aphA = Kanamycinresistenzgen; oriV = vegetativer Replikationsursprung; oriT = Transferreplikationsursprung; mob = Mobilisierungsstelle 50 Material und Methoden Die Plasmide wurden über Elektroporation oder Konjugation (Abschnitt 2.2.1.7) in die zu testenden Stämme transferiert. Als Selektionsmarker für die Plasmide konnten, aufgrund der in den pBP507-Konstrukten enthaltenen Resistenzgene, Amikacin, Gentamicin und Kanamycin verwendet werden. Ein Schema der Durchführung und der Selektion beim Komplementationstest über Konjugationstransfer zeigt Abbildung 2-2. Im Anschluss an den Transfer der Plasmide pBP507 bzw. pBP517 in die Stämme konnten verschiedene Versuche wie MHK-Wert-Bestimmung, Wachstumsuntersuchungen oder Messung des Superspiralisierungsgrades durchgeführt werden, um die Rolle der Mutationen im gyrA-Gen aufzuklären. Abbildung 2-2: Schema der Durchführung des Komplemetionstestes mittels Konjugation und geeignete Selektionsmarker zur Unterscheidung von Donor, Rezipient und Transkonjugant 2.2.1.9 Bestimmung des DNA-Superspiralisierungsgrades Um eine Aussage über Veränderungen im DNA-Superspiralisierungsgrad in einer Zelle zu treffen, wurden mit Hilfe von Reportergensystemen die Änderungen der Promotoraktivität der Topoisomerasegene bestimmt. 51 Material und Methoden 2.2.1.9.1 Die Verwendung der Luciferase-Reportergensysteme von Mobarak Abu Mraheil [1] konstruierten Plasmide enthalten die Reportergensysteme. Tabelle 2-17 enthält eine Übersicht über die verwendeten Plasmidderivate in dieser Arbeit. Der natürliche Promotor des Luciferasegens wurde durch die Promotoren der Gene der Topoisomomerase I (ptopA) bzw. Gyrase (pgyrA) ersetzt. Tabelle 2-17: Übersicht über die verwendeten Reportergenplasmide Plasmidbezeichnung BasisKlonierungsvektor mit luc-Gen fusionierter Topoisomerasepromotor Antibiotikaresistenzeigenschaften pPHB90 pBR322 ptopA Tet pPHB91 pBR322 pgyrA Tet pPHB94 pBP507 ptopA Gen, Ami pPHB95 pBP507 pgyrA Gen, Ami Als Reportergen wurde das Luciferasegen (luc) aus dem nordamerikanischen Feuerschwanz Photinus pyralis („fire fly“) verwendet. Das Substrat Luciferin wird von der exprimierten Luciferase in der Anwesenheit von ATP, Mg2+ und O2 abgebaut, wodurch u.a. Oxiluciferin und Licht (Biolumineszenz) entsteht. Luciferase Luciferin + ATP + O2 + Mg 2+ Oxyluciferin + AMP + PPi + CO2 + Licht Das Luciferase-System bietet gegenüber anderen Reportergenen wie z.B. ß-Laktamase oder Chloramphenicol-Acetyltransferase den Vorteil (1) hoher Sensitivität (Nachweis von weniger als 1 Femtogramm ≈10-20 Mol) [303], (2) eines großen linearen Messbereiches und (3) des fehlenden endogenen Vorkommens in Bakterienzellen, wodurch das Entstehen von Hintergrundaktivität verhindert wird. Eine Änderung der Promotoraktiviät der Topoisomerasegene kann also direkt über die Änderung der Lichtintensitäten (RLUs = relative light units) gemessen werden. 2.2.1.9.2 Messung der Luciferase-Aktivität Im Luminometer Lucy2 der Firma Anthos erfolgte die Messung der Luciferase-Aktivität mit dem „Luciferase Assay System“ der Firma Promega. Zunächst wurden die zu untersuchenden Stämme mit den entsprechenden Reportergenplasmiden transformiert. Die Anzucht der Stämme mit Plasmid erfolgte unter Zusatz des entsprechenden Selektionsantibiotikums. Aus der ÜNK wurde eine 1:100 Verdünnung in 3 ml NI-Bouillon in einem Reagenzglas hergestellt und bei 37 °Cund 130 upm bis zu einer OD546nm zwischen 0,3 und 0,8 (mittlere exponentielle Phase) inkubiert. Aus der Bakterienkultur wurden dann je 3 Proben á 90 µl entnommen und mit 10 µl Puffer (1M K2HPO4, 2 mM EDTA) gemischt. Dieser Puffer stabilisiert die Luciferase in den Zellen. Die Proben wurden dann bei -80 °C eingefroren, um die Membran zu permeabilisieren. Zu den wieder aufgetauten Proben 52 Material und Methoden wurden dann jeweils 300 µl frisch präparierter Lyse-Mix gegeben, zusammengesetzt aus einerseits 200 µl CCLR und andererseits 100 µl, bestehend aus Lysozym (5 mg/ml) und BSA (5 mg/ml),. Für die Lyse wurden die Zellen 10 bis 30 min bei RT inkubiert. Für die Messung wurden dann pro lysierter Probe dreimal je 2,5 µl entnommen und zu 25 µl LAR (Luciferase Assay Reagent) in Kavitäten spezieller, für luminometrische Messungen geeignete Mikrotiterplatten pipettiert. Anschließend wurde die Mikrotiterplatte in dem Luminometer positioniert und die Lichtintensität bei 560nm mit Hilfe des Softwareprogramms Lucysoft (Version 1.6) gemessen. Aus den drei Biolumineszenz-Einzelmesswerten einer Probe wurde ein Mittelwert gebildet. Die spezifische Aktivität wurde durch Division des Mittelwertes durch den OD546nm-Wert der Kultur zum Zeitpunkt der Probenentnahme errechnet: spez. Luciferaseaktivität [RLU/OD546nm ] = OD546nm x RLU`s einer Probe zum Zeitpunkt der Probenentnahme Die errechnete spezifische Luciferaseaktivität entspricht der spezifischen Aktivität des jeweiligen Topoisomerase-Promotors unter den entsprechenden Versuchsbedingungen. 2.2.1.9.3 Berechnung des Qsc-Wertes Der negative DNA Superspiralisierungsgrad in einer Zelle wird in engen Grenzen konstant gehalten. Diese homöostatische Kontrolle [204] wird u.a. durch die antagonistischen Aktivitäten der beiden Enzyme Gyrase und Topoisomerase I gewährleistet, deren Expression in reziproker Weise durch den DNA-Superspiralisierungsgrad beeinflusst wird. Deshalb kann die Änderung des Superspiralisierungsgrades über einen Quotienten der spezifischen Aktivitäten der jeweiligen Promotoren (ptopA und pgyrA) ausgedrückt werden. Bei der Verwendung eines Paares von Reportergenplasmiden gibt der Superspiralisierungsquotient Qsc (Quotient of supercoiling) ein relatives Maß für den DNA-Superspiralisierungsgrad. Qsc = spezifische Aktivität von ptopA spezifische Aktivität von pgyrA Um abzusichern, dass es sich bei Abweichungen der Qsc-Werte um keine zufälligen Streuungen handelt, wurde die absolute Standardabweichung (s) berechnet: n ∑ x 2 − (∑ x ) 2 s= Bei prozentualer Angabe der n(n − 1) Qsc-Werte erfolgt eine Angabe der relativen Standardabweichung (s %): s% = s × 100 x 53 Material und Methoden 2.2.1.10 In-vitro-Mutagenese mittels homologer Rekombination Die Methode der homologen Rekombination wird genutzt, um das zu untersuchende chromosomale Gen in der gewünschten Form zu verändern und über die entstehenden phänotypischen Änderungen eine Aussage zur Rolle des Gens zu treffen. Das allgemeine Prinzip der homologen Rekombination beruht darauf, dass sich zwei DNA-Moleküle aneinanderlagern und durch den Austausch von Nucleotiden interagieren. Die Anlagerung der zwei DNA-Moleküle geschieht aufgrund hoher Sequenz-Homologie zueinander. Der Austausch der Nucleotidfragmente beruht auf Einzelstrangbrüchen, freien DNA-Enden oder Einzelstrangregionen. Von biologischer Bedeutung ist die homologe Rekombination für die Variation des genetischen Materials während der Evolution und bei der SOSAntwort in der DNA-Reparatur. Die Rekombination wird in E. coli und Salmonella spec. durch den RecA-RecBCD Komplex vermittelt. Dabei bindet RecBCD an ein lineares DNA-Doppelstrangende. Die DNA wird entwunden und der RecBCD-Proteinkomplex wirkt als Endonuklease und schneidet einen Einzelstrang. RecA lagert sich an das entstandene Einzelstranende an und begünstigt so eine Invasion in einen anderen, homologen Doppelstrang. Somit bilden Einzelstrang, homologer Doppelstrang und RecA-Protein einen Komplex, in dem es zu Strangunterbrechnungen, Strangverdrängung und Ligation kommt [208]. 2.2.1.10.1 Homologe Rekombination unter Verwendung des Vektors pKOV Eine grundlegende Methode der homologen Rekombination beruht auf dem nach Hamilton et al. [114] entwickeltem Verfahren. Dabei wird ein Vektor (pMAK705) genutzt, der sich nur bei Temperaturen bis 30 °C selbständig repliziert (orits). In diesen Vektor wird das einzukreuzende Fragment einkloniert, das von Sequenzen von mindestens 500 bp Länge flankiert wird. Diese Flankensequenzen weisen eine Homologie zu dem chromosomalen Bereich auf, an dem die Rekombination erfolgen soll. Von pMAK705 sind verschiedene Vektoren abgeleitet worden. In dieser Arbeit wurde zur Herstellung von gyrA-Mutanten der Vektor pkOV verwendet, der von Link et al. [180] konstruiert wurde. Der Vektor pKOV repliziert sich temperatursensitiv, besitzt eine Chloramphenicolresistenz und das für die Levansucrase codierende Gen sacB. Letzteres wird dazu verwendet, nach erfolgter Rekombination Zellen mit Plasmid aus der Population zu eliminieren. Zunächst wurde das einzukreuzende Fragment mit den homologen Flanken und einem Selektionsmarker in den Vektor pKOV kloniert und das so hergestellte pKOV-Derivat mittels Elektroporation in den Stamm M415 transformiert. Aufgrund der als Selektionsmarker für die Rekombination einklonierten Kanamycinresistenz-Kassette (siehe Abschnitt 2.2.1.10.3) besitzen alle für die Rekombination verwendeten Vektoren auch eine Kanamycinresistenz. Die Selektion der rekombinanten Stämme erfolgte bei 30 °C auf TYE-Agarplatten mit Kanamycin (50 µg/ml). Die Transformanden wurden zur Aufreinigung auf frische Selektionsplatten überimpft. Von diesen frisch bewachsenen 54 Material und Methoden Agarplatten wurde von einem Einzelklon eine ÜNK in 3 ml LB-Medium (mit Chloramphenicolzusatz von 15 µg/ml zur Plasmidselektion) hergestellt und bei 30 °C inkubiert. Am nächsten Morgen erfolgte eine 1:1000 Verdünnung in 50 ml LB-Medium mit 15 µg/ml Chloramphenicol. Diese Kultur wurde bei 30 °C mit 130 upm schüttelnd für ca. 2 h inkubiert, bis eine leichte Trübung erkennbar war. Danach wurden die Kolben in ein auf 42 °C vorgeheiztes Wasserbad umgesetzt und dort bei 130 upm für ca. 4 h weiter inkubiert. Nach der Anlagerung der homologen Flanken an die entsprechenden chromosomalen Bereiche findet eine homologe Rekombination zwischen dem klonierten Fragment und dem Bakterienchromosom über den RecA-RecBCD-Komplex statt. Das zirkuläre Plasmid wird dabei durch die Endonukleaseaktivität von RecBCD geöffnet und in das Chromosom integriert (Cointegratbildung). Nur Zellen, in welchen diese Integration des Plasmides in das Chromosom stattgefunden hat, können unter Chloramphenicolzugabe, aber unabhängig von der Plasmidreplikation, d.h. bei 42 °C, weiter wachsen (Abbildung 2-3). Aus diesen Bakterienkulturen wurde eine Verdünnungsreihe bis 10-4 in 0,9 % NaCl-Lösung erstellt. Von den Verdünnungsstufen wurden jeweils 100µl auf LB-Agarplatten mit Chloramphenicol (15 µg/ml) ausplattiert. Aus diesem Doppelansatz wurde je eine der Agarplatten bei 30 °C und eine bei 42 °C für 24 h. Auf der 42 °C-Agarplatte wuchsen vereinzelt die Cointegrate. Nach dem ersten Rekombinationsschritt konnten die Stämme als Cointegrate gelagert werden. Von den 42 °C-Agarplatten wurden große Einzelkolonien (Cointegrate) mit einem Glasstab in 4,5 ml 0,9 % NaCl-Lösung inokuliert. Von dieser Bakteriensuspension wurden je 100 µl auf LB-Agarplatten mit Kanamycin (50 µg/ml) und Saccharose (5 %) ausplattiert und bei 30 °C inkubiert, um die Rekombinanten ohne Plasmid zu selektieren. Durch Senken der Temperatur auf 30 °C, wodurch die Replikation des Plasmides wieder möglich ist, erfolgte die Excision (das Auskreuzen) des Plasmides durch eine zweite Rekombination. Je nachdem wo die zweite Rekombination (Crossing over) stattfindet, enthält das Chromosom nach dem Auskreuzen des Plasmides entweder die Wildtypsequenz oder die veränderte Sequenz vom Plasmid (Abbildung 2-4). Die Excision des Plasmides aus dem Chromosom und die Eliminierung des Plasmides aus der Zellen geschahen also während eines Selektionsschrittes. Durch die in den Selektionsplatten enthaltene Saccharose werden die Zellen, welche ein pKOV-Derivat mit sacB-Gen enthalten, in ihrem Wachstum stark gehemmt. Das plasmidcodierte und ursprünglich aus Bacillus subtilis stammende Gen sacB codiert für die Levansucrase. Dieses Enzym katalysiert die Vernetzung der Saccharose zu Levan, einem Fructose-Polymer mit hohem Molekulargewicht. Dieses in Gegenwart von Saccharose entstehende Polymer akkumuliert im Periplasma der Zellen, die das Plasmid tragen und wirkt sich toxisch auf die Bakterien aus. Der genaue Mechanismus der Toxizität ist noch nicht genau verstanden, allerdings wurde die Wirkung in vielen gramnegtiven Bakterien wie E. coli, Pseudomonas aeruginosa oder Klebsiella pneumoniae nachgewiesen [95,173,180]. Das Gen sacB wird daher häufig als Selektionsmarker genutzt [255]. 55 Material und Methoden Abbildung 2-3: Erster Rekombinationsschritt: Cointegratbildung Durch Paarung homologer Bereiche des Gens von Plasmid (P_Gen) u. von Chromosom (C_Gen) wird das Plasmid geöffnet und in das Chromosom integriert. Durch Cointegratbildung befindet sich die Chloramphenicolresistenz-Kassette (cat) im Chromosom u. ist unabhängig von der Plasmidreplikation (ori ts). Abbildung 2-4: Zweiter Rekombinationsschritt: Auskreuzen des Plasmides Durch wiederholte Paarung homologer Bereiche findet eine zweite Rekombination statt, welche das Auskreuzen des Plasmides aus dem Chromosom bewirkt. Je nachdem wo der 2. Rekombinationsschritt stattfindet, entsteht im Chromosom ein Gen mit (2) oder ohne (1) Mutation. 56 Material und Methoden Nach spätestens 3 Tagen wurden die entstandenen kanamycinresistenten Klone (Rekombinanten) durch Überimpfen auf frische Kanamycin-Selektionsplatten gereinigt und durch Replikaplattieren auf den Verlust der Chloramphenicolresistenz und somit auf Verlust des Rekombinationsvektors überprüft. Die Inkubation erfolgte in diesem Fall bei 37 °C. Es schloss sich eine Untersuchung auf die Integration des Selektionsmarkers (Abschnitt 2.2.1.10.3) und das Vorhandensein der eingekreuzten Mutationen an (Abschnitt 2.2.2.5.3). 2.2.1.10.2 Sequenzspezifische Rekombination mit dem Lambda Red-System Eine weitere Möglichkeit, mittels Rekombination eine Veränderung des Chromosoms zu bewirken, ist die Zuhilfenahme von phagenbasierten Enzymen [61,66,209- 211,244,245,308,309]. Murphy und Poteete [209,211,244,245] entwickelten die Nutzung des phageneigenen Rekombinationssystems. Prinzipiell beruht die entwickelte Methode auf dem Lambda-Red-System welches es erlaubt, dass kleine DNA-Fragmente, die nach Elektroporation in die Zelle gelangen, durch Rekombination in das Bakterienchromosom integriert werden. Normalerweise werden kurze DNA-Fragmente in Bakterien von zelleigenen Exonukleasen abgebaut. Einige Phagen können diese Exonukleasen inhibieren. Zusätzlich produzieren Phagen Rekombinationsproteine, welche – im Gegensatz zu den zelleigenen Rekombinationsproteinen (mind. 500 bp) – schon sehr kurze homologe Sequenzen (ca. 20 bis 50 bp) erkennen und eine spezifische Rekombination durchführen. Bei dieser Methode findet die Rekombination also unabhängig vom zelleigenen RecABC-System, sondern über die Phagenenzyme Exo, Beta und Gam (γ, β, ε) statt. Dabei codiert γ für Gam, welches die Wirts-RecBCDExonuklease inhibiert und β und ε codieren für Beta (ssDNA Annealing Protein) und Exo (5`-3`-Exonuklease), welche mit der transformierten DNA interagieren und die sequenzspezifische Rekombination durchführen. Sogar die Verwendung von einzelsträngigen Oligonukleotiden soll zur spezifischen Rekombination ausreichend sein [66]. Die beiden Arbeitsgruppen Court et al. [85,308] und Datsenko & Wanner [66] erhöhten die Effizienz des Red-Systems dadurch, dass sie die Phagengene in Plasmiden unter die Kontrolle regulierbarer Promotoren setzten. Court et al. nutzten den temperatursensitiven Lambda cI857 Repressor, welcher bei 42 °C inaktiv ist, wodurch eine Induktion der Expression von Genen bewirkt wird, die unter der Kontrolle von Lambda PL- und PRPromotoren stehen. Der Ansatz von Datsenko und Wanner beinhaltet die Verwendung des para-Promotors inklusive des AraC Aktivators, wodurch eine Induktion der Gene bei Zugabe von L-Arabinose auslöst wird. Datsenko & Wanner [66] konstruierten das Helferplasmid pKD46 (siehe Abbildung 2-5), welches neben den Phagengenen mit paraPromotor, einen temperatursensitiven Replikationsursprung und eine AmpicillinresistenzKassette zur Selektion enthält. 57 Material und Methoden Abbildung 2-5: pKD46: Red Rekombinase Expressionsvektor [66] Helferplasmid pKD46 für die Red vermittelte Rekombination: bla = Ampicillinresistenz-Kassette; araC = Aktivator für Arabinosepromotor; ParaB/C = Arabinosepromotor; gam, beta, exo = Phagengene; oriR101 / repA101ts = temperatursensitiver Replikationsursprung Zunächst wurde der Salmonella-Stamm M415 mit diesem Helferplasmid pKD46 per Elektroporation transformiert. Die Transformanden wurden bei 30 °C auf TYE-Agarplatten mit Ampicillin (50 µg/ml) selektiert. Von M415-Zellen mit pKD46 wurden erneut kompetente Bakterien hergestellt (Abschnitt 2.2.1.7.2). Um die Phagengene zu exprimieren, wurde während der Anzucht der kompetenten Zellen 10 mM L-Arabinose zum Medium gegeben. Im Anschluss erfolgte die Elektroporation eines zuvor hergestellten linearen DNA-Fragmentes. Dieses Fragment enthielt den in das Zielgen einzukreuzenden DNA-Abschnitt, die Kanamycinresistenz-Kassette als Selektionsmarker und flankierende Sequenzabschnitte für das Lambda Red-System. Nach der Elektroporation wurden die Zellen in SOC-Medium bei 37 °C für ca. 1 h regeneriert. Im Anschluss wurde der Transformationsansatz in 200 µl-Aliquots auf TYE-Agarplatten mit Kanamycin (50 µg/ml) ausplattiert und bei 37 °C inkubiert. Nach spätestens 2 Tagen Inkubation wurden die gewachsenen Klone auf frische Kanamycin-Selektionsplatten überimpft und auf Integration der Kanamycinresistenz-Kassette und die gewünschten eingekreuzten Mutationen untersucht. 2.2.1.10.3 System zur Selektion hergestellter Rekombinanten Die Integration einer Kanamycinresistenz-Kassette ist ein geeigneter Selektionsmarker, um eine erfolgreiche Rekombination zu erkennen. Um eine Insertion der ResistenzKassette zu erreichen, wurden homologe Flanken genutzt (siehe Abbildung 2-6). Die erste homologe Flanke umfasste die Sequenz vor dem gyrA-Gen liegend und die zweite homologe Flanke war das gyrA-Gen selbst, welches z.T. die einzukreuzenden Mutationen enthielt. Es wurde davon ausgegangen, dass eine Insertion der KananmycinResisitenzkassette zusammen mit dem Einkreuzen der gewünschten gyrA-Mutationen erfolgt. Das macht eine positive Selektion der Rekombinanten auf Kanamycin-haltigen Agarplatten 58 möglich. Neben der phänotypischen Resistenzeigenschaft der Material und Methoden Rekombinanten konnte die korrekte Integration der Kanamycinkassette mittels PCRs festgestellt werden (siehe Abbildung 2-6). Die im Rahmen dieser Arbeit verwendeten Primer (P1, P2) sind Abschnitt 3.2.4.1 gezeigt. Abbildung 2-6: Verwendung der Kanamycinresistenz-Kassette als Selektionsmarker für die erfolgreiche Rekombination Die Insertion der Kanamycinresistenz-Kassette inklusice FRT-Sites (KanaR) findet mit hoher Wahrscheinlichkeit gemeinsam mit der gewünschten gyrA-Mutation (schwarzes Rechteck) statt. Die erfolgreiche Insertion kann mittels PCR nachgewiesen werden: Verwendung der Primer kt und k2 zum Nachweis der Resistenz-Kassette; Primer P1 liegt vor der vorderen homologen Flanke (HF) und ergibt in Kombination mit Primer k2 ein PCR-Produkt, ebenso wie die Kombination kt und P2. Dies liefert den Nachweis der richtigen Lokalisation der Kassette. Um eventuelle Beeinflussungen der phänotypischen Eigenschaften durch die ResistenzKassette auszuschließen, ist es möglich, diese nach erfolgreicher Selektion wieder zu eliminieren. Die Resistenz-Kassette wird von zwei Elementen flankiert, die später von einer in trans exprimierten Rekombinase erkannt werden, die den Selektionsmarker wieder ausschneiden kann. In dieser Arbeit wurde die Flippase (Flp), ein Mitglied der Superfamilien der Lambda-Integrasen, als Rekombinase zum nachträglichen Ausschneiden der Resistenz-Kassette verwendet. Es handelt sich dabei um eine sequenzspezifische Integrase [14], welche die DNA an bestimmten Erkennungssequenzen, so genannten FRT-Sites (Flp recombination target), schneidet und dann mit einem DNA-Fragment ligiert, welches von einer anderen sequenzidentischen Stelle stammt. Die die FRT-Sites erkennende Rekombinase wird über ein weiteres Plasmid (pCP20) in die Zelle eingeführt [46]. Die Zielsequenz für die Flippase ist mit einer Größe von 34 bp groß genug, um auszuschließen, dass dieses Element zufällig im Genom auftritt. Die Kanamycinresistenz-Kassette (inklusive FRT-Sites) stand durch das Templateplasmid pKD4 (siehe Abbildung 2-7) zur Verfügung, von wo sie über PCR amplifiziert werden konnte [66]. Die für diese PCR verwendeten Primer wurden so gestaltet, dass sie entweder Schnittstellen für Restriktionsenzyme zur Klonierung, homologe Bereiche für weiterführende SOEing-PCR oder direkt die homologen Flanken zur Rekombination enthielten. 59 Material und Methoden Abbildung 2-7: Templateplasmid pKD4 zur Amplifikation der Kanamycinresistenz-Kassette inklusive flankierender FRT-Sites [66] bla = Ampicillinresistenz-Kassette; ori = Replikationsursprung; aphA = KanamycinresistenzKassette; FRT site = FLP recognition target Abbildung 2-8 zeigt das Prinzip der Resistenz-Kassette als Selektionsmarker und das nachträgliche Eliminieren durch Ausschneiden mittels Flippase. Sowohl das Vorhandensein der Resistenz-Kassette, als auch ihre Eliminierung können mittels PCR und der Verwendung der entsprechenden Primer nachgewiesen werden. Der Wegfall der Bindungsstellen für die Primer k2 und kt lassen nach der Eliminierung keine PCRProdukte mehr entstehen. Diese PCRs als Nachweismethode wurden bei der Selektion der Rekombinanten genutzt (siehe Abschnitt 3.2.4). Abbildung 2-8: Eliminierung der Resistenz-Kassette mittels der FLP-Rekombinase Die Kanamycinresistenz-Kassette wird flankiert von FRT-Sites (KanaR). Diese Sequenzen werden von der FLP-Rekombinase erkannt und der Bereich zwischen diesen ausgeschnitten und wieder ligiert; HF = homologe Flanke; P1/2 = Primer, kt und k2 = KanaR-interne Primer 60 Material und Methoden 2.2.2 Molekularbiologische Methoden 2.2.2.1 Isolierung chromosomaler DNA Die Isolierung chromosomaler DNA aus Salmonella spec.- oder E. coli-Zellen erfolgte durch die „Kochmethode“. Ein bis drei Kolonien des Bakterienstammes wurden mittels eines Glasstabes in 100 µl dH2O suspendiert und bei 95 °C für 15 min inkubiert. Im Anschluss wurde die chromosomale DNA von den Zellresten durch Zentrifugation für 5 min bei 13.000 upm getrennt. Der die chromosomale DNA enthaltende Überstand wurde in ein Eppendorfgefäß überführt. Zur Überprüfung der DNA-Isolierung wurden 5-10 µl des Überstandes auf ein 1 %iges Agarosegel aufgetragen. Die DNA-Lösung wurde bei –20 °C bis zur weiteren Verwendung gelagert. 2.2.2.2 Isolierung von Plasmid-DNA 2.2.2.2.1 Plasmid-Minipräparation nach Birnboim und Doly (modifiziert) Für eine schnelle und einfache Plasmid-Minipräparation wurde die Methode nach Birnboim und Doly [31] modifiziert eingesetzt. Die Methode dieser Präparation beruht auf der alkalischen Lyse und dem Prinzip der Trennung der DNA aufgrund der Konformation. Zur Plasmidisolierung wurden rekombinante Klone in geeignetem Medium (low-copy Plasmide z.B. in Terific Broth® für erhöhte Ausbeute) mit dem entsprechenden Selektionsantibiotikum inokuliert. Jeweils 1,5 bis 3 ml ÜNK wurden bei 13.000 upm für 1 min abzentrifugiert und das Zellpellet in 100 µl GTE-Puffer oder Resuspensionspuffer (QIAGEN) resuspendiert. Durch die Zugabe von 200 µl frisch angesetztem alkalischem Lysepuffer erfolgte die Lyse der Zellen. Durch die Behandlung mit SDS (im Lysepuffer) werden Proteine und RNA zum großen Teil unlöslich und können später abzentrifugiert werden. Die entstehende trübe Lösung wurde mehrmals invertiert und so lange bei RT inkubiert bis sie klarer wurde. Unter den stark alkalischen Bedingungen denaturiert die DNA (chromosomale und Plasmid-DNA) vollständig. Bei der Plasmid-DNA bleiben die DNA-Einzelstränge aber durch den Ring kovalent geschlossen und daher topologisch miteinander verknüpft. Durch die Zugabe von RNase (maximal 100 µg/ml) wurde die störende RNA aus der Probe eliminiert und damit an der DNA anheftende Komplexe aus mRNA und Ribosomen abgetrennt. Der Ansatz wurde anschließend durch die Zugabe von 150 µl Kaliumacetat-Lösung und Inkubation für ca. 5 min auf Eis neutralisiert und damit die DNA renaturiert. So können die Plasmid-DNA-Einzelstränge wieder korrekt zueinander finden (s.o.), während die denaturierten chromosomalen DNA-Einzelstrangfragmente sich zu einer unlöslichen Masse zusammenlagern. Die Verwendung von Kaliumacetat sorgt dafür, dass das SDS ein unlösliches Präzipitat bildet und gut entfernt werden kann. Dieses wurde durch Zentrifugation sedimentiert. Die Plasmid-DNA befindet sich nun im Überstand. Die Zentrifugation erfolgte bei 13.000 upm für 5 min. Der Überstand wurde in ein neues Gefäß überführt, mit 800 µl absolutem Ethanol versetzt und 2 min bei RT inkubiert. Nach mindestens 1 minütiger Zentrifugation bei 13.000 upm wurde der 61 Material und Methoden Überstand verworfen und das DNA-Pellet mit 1 ml 70 %igem Ethanol gewaschen. Wiederum wurde mindestens 1 min bei 13.000 upm zentrifugiert, der Überstand wiederum verworfen und das DNA-Pellet im Brutschrank bei 37 °C maximal 30 min getrocknet. Zum Schluss wurde das DNA-Pellet in 30 µl dH2O (bei sofortiger Verwendung) oder EB-Puffer (für längere Aufbewahrung) aufgenommen. Die Plasmid-DNA-Lösung wurde bei -20 °C gelagert. 2.2.2.2.2 Plasmid-Minipräparation-Kits Für größere Ausbeuten bei der Plasmidpräparation sowie saubererer Abtrennung von Proteinen und vor allem von RNA wurden Plasmid-Minipräparationen über Silika-Säulen durchgeführt. Das Prinzip dieser Methoden beruht ebenfalls auf der alkalischen Lyse. Das Bakterienpellet wurde in entsprechendem Puffer resuspendiert, unter alkalischen Bedingungen lysiert und anschließend neutralisiert. Der Neutralisierungspuffer schafft gleichzeitig die Bedingungen unter denen die Plasmid-DNA an der Silika-Membran der „Minipräp-Säulen“ bindet. Nach einem Waschschritt wurde die DNA durch einen Elutionspuffer durch pH-Wert-Änderung wieder von der Membran gelöst und eluiert. Durch diese Bindung wird die Plasmid-DNA von störenden Komponenten entfernt. Im Rahmen dieser Arbeit wurden Kits der Firmen Eppendorf AG und MACHEREY-NAGEL eingesetzt. Weiterhin wurde von der Firma NexttecTM das nexttecTM „Plasmid DNA Isolation Kit for Bacteria“ verwendet, welches entgegengesetzt zu den Silika-basierten Systemen funktioniert. Es wurden Proteine und andere störende Komponenten von einem in einer Säule vorhandenen Sorbent gebunden, wobei die ungebundene DNA die Säule passierte und dadurch abgetrennt wurde. Die DNA-Konzentration war aufgrund des hohen Elutionsvolumens gering und es war direkt im Anschluss durch die Zusammensetzung des Eluats (welches im Grunde dem Lysepuffer entsprach) keine Restriktionsspaltung möglich. Deshalb wurde die eluierte DNA-Lösung im Anschluss über Ethanolfällung konzentriert. Vorteil dieses Systems ist die sehr kurze Präparationszeit. 2.2.2.2.3 Plasmid-Midipräparation Um größere Mengen an Plasmid-DNA (z.B. für Klonierungen) zu erhalten, sind größere Ausbeuten nötig als die der Plasmid-Minipräparation. Bei der Plasmid-Midipräparation wird von höheren Kulturvolumina ausgegangen. Im Rahmen dieser Arbeit wurde vor allem das Plasmid-Midikit der Firma QIAGEN GMBH verwendet. Das Prinzip dieses Kits beruht ebenfalls auf der alkalischen Lyse und es werden Silika-Säulen zur DNA-Bindung durch Anionen-Austausch unter den entsprechenden Salz- und pH-Bedingungen verwendet. RNA, Proteine und niedermolekulare Verunreinigungen werden durch Waschschritte mit mittlerem Salzgehalt entfernt und die DNA wird durch einen salzreichen Puffer eluiert. Im Anschluss erfolgt eine Isopropanol-Fällung zur Konzentrierung und Entsalzung der Probe. 62 Material und Methoden Das QIAGEN-Protokoll ist optimiert für LB-Medium und eine Ausgangsbakterienkultur mit einer Zelldichte von 3 bis 4 x 109 Zellen/ml (ÜNK). Die erwarteten Ausbeuten sind bis zu 100 µg Plasmid-DNA. Die Durchführung erfolgte wie im Protokoll von QIAGEN angegeben: Abzentrifugation von 100 ml ÜNK für 15 min bei 4 °C und 5300 upm, Resuspension des Zellpellets in 4 ml Puffer P1, Zugabe von 4 ml Lysepuffer P2, Inkubation für 5 min bei RT, Neutralisation mit 4 ml eiskaltem Puffer P3, 15 min Inkubation auf Eis, Zentrifugation für 30 min bei 4 °C und 15.000 upm, Equilibrierung der Säule (QIAGEN-tip 100) mit QBT-Puffer, Zugabe des Überstandes auf die Säule, zweimaliges Waschen der Säule mit 10 ml QC-Puffer und Eluieren der DNA von der Säule mit 5 ml QFElutionspuffer. Anschließend erfolgte eine Isopropanol-Fällung durch Zugabe von 3,5 ml Isopropanol, 30 min Zentrifugation bei 15.000 upm bei RT, Waschen des Pellet mit 70 %igem Ethanol und Zentrifugation bei 15.000 upm für 10 min. Das für 10-20 min bei 37 °C getrocknete Pellet wurde in 50 µl EB-Puffer gelöst und bei -20 °C bis zur Verwendung gelagert. Weiterhin wurde das Perfectprep Kit der Firma Eppendorf zur Plasmidaufreinigung nach Vorgabe durch den Hersteller verwendet, welches auf dem gleichen Prinzip und sehr ähnlicher Durchführung beruht. 2.2.2.3 Konzentrationsbestimmung und Reinheitsprüfung von DNA Um eine rasche Konzentrationsbestimmung von DNA vorzunehmen, wurde vorzugsweise eine Abschätzung über ein Agarosegel nach Gelelektrophorese vorgenommen. Als Referenz diente der Größen- und Mengenstandard Smart Ladder (Eurogentec). Über einen visuellen Vergleich der auftretenden Bandenintensität der Probe und der entsprechenden Bande des Smart Ladders wurde die Menge abgeschätzt. Alternativ wurde die DNA-Konzentration einer Probe durch photometrische Messung bestimmt. Dabei wurde ein Aliquot der gelösten DNA gegen den entsprechenden Leerwert spektralphotometrisch vermessen. Es gilt für einen DNA-Doppelstrang gilt: OD260nm = 1 entspricht ungefähr 50 µg/ml DNA. Für reine Doppelstrang-DNA gilt weiterhin: OD260nm = 1,8 . OD280nm Nimmt der Quotient einen Wert größer als 1,8 an, so deutet dies auf Verunreinigungen durch RNA hin. Liegt der Wert unterhalb von 1,8, so sind Proteine in der Lösung enthalten. 2.2.2.4 Konzentrationsbestimmung von Oligonukleotiden Der größte Teil der in dieser Arbeit verwendeten Oligonukleotide wurde über die Firmen Invitrogen GmbH und Sigma-Genosys bezogen. Einige der in dieser Arbeit verwendeten Primer wurden aber auch mit Hilfe des im Labor vorhandenen DNA-Synthesizers PCRMate 391 (Applied Biosystems) hergestellt. Das automatisierte Verfahren beruht auf der 63 Material und Methoden Festphasensynthese nach Merrifield. Die getrockneten Primer wurden bei -20 °C gelagert und bei Bedarf in 50 µl dH2O aufgenommen. Zur Bestimmung der hergestellten Primerkonzentration wurde eine 1:200 Verdünnung mit dH2O erstellt und die Lösung bei einer Wellenlänge von 260 nm in Glasküvetten photometrisch vermessen. Die Konzentration C errechnet sich nach folgender Formel: CPrimer [pmol/µl] = OD260nm × Verdünnung bp × 0,01 Für den PCR-Einsatz wurde eine Primer-Stammlösung mit einer Konzentration von 10 pmol/µl eingestellt. 2.2.2.5 Amplifikation von DNA 2.2.2.5.1 Polymerase-Kettenreaktion (PCR) Prinzipiell ist die PCR (polymerase chain reaction – Polymerase-Kettenreaktion) eine Methode zur enzymatischen Vervielfältigung von DNA. Die PCR dient zum Nachweis, zur Amplifikation und Modifikation von DNA. Die DNA-Amplifikation findet zwischen zwei an die denaturierten DNA-Ausgangsstränge (Template) komplemetär angelagerten Oligonukleotiden (Primer) statt. Hierbei dienen die freien 3´-OH-Enden der Primer als Startpunkt für eine thermostabile DNA-Polymerase. Die Reaktion besteht aus der Wiederholung von Denaturierung des DNA-Doppelstranges, Hybridisierung der Oligonukleotid-Primer und Synthese durch die Polymerase (Tabelle 2-18). Durch eine mehrfache Wiederholung der Zyklen entstehen bei der Kettenreaktion vielfache Kopien des DNA-Abschnittes zwischen den Primern, wobei amplifizierte DNA-Abschnitte in nachfolgenden Reaktionszyklen als Template dienen, so dass sich eine exponentielle Vermehrung ergibt. Tabelle 2-18: Standardtemperaturprofil für eine PCR Schritt Temperatur Zeit Wiederholung 1. Initialdenaturierung 95 °C 3 min 1x 2. Denaturierung 95 °C 30 sec 3. Primeranlagerung TA°C 30 sec 4. Synthese TP°C tV 5. verlängerte Synthesephase TP°C 5-10 min 25 – 30x 1x TA = Anlagerungstemperatur der Primer abhängig von der Basensequenz, TP = Synthesetemperatur abhängig von eingesetzter Polymerase, tV = Synthesezeit abhängig von der Länge des zu synthetisierenden Fragmentes (bei Taq-Polymerase ca. 1kb pro min) Die Länge der Primer wird zwischen 15 und 25 Basen gewählt. Die Basensequenz des Primers ist zugleich verantwortlich für die Annlagerungstemperatur TA, bei der der Primer an das DNA-Template hybridisiert. Die ungefähre Anlagerungsstemperatur kann man aus der Schmelztemperatur (Tm) des Hybrids aus Primer und DNA-Template ableiten. Die 64 Material und Methoden Annlagerungsstemperatur TA liegt etwas unter der Schmelztemperatur Tm, die sich wie folgt aus der Sequenz eines Primers berechnet: Tm [°C] = 2 × (ΣdATP + ΣdTTP) + 4 × (ΣdGTP + ΣdCTP) Die Schmelztemperaturen für die beiden in einer PCR eingesetzten Primer sollten in einem ähnlichen Bereich liegen. Für eine erfolgreiche PCR sind neben dem DNA-Template, dem Primerpaar (je 10 pmol/ml) und der Polymerase (ca. 5 U/µl) mit entsprechendem Puffer auch die richtigen Mengen an dNTP`s (2 mM) und MgCl2 (25 mM) wichtig. Optional, z.B. bei der Amplifikation von Plasmiden, wurde dem PCR-Ansatz auch DMSO zugesetzt. Die Durchführung der PCR erfolgte in automatischen Thermocyclern. Die Überprüfung des PCR-Amplifikationsproduktes erfolgte mit Hilfe der Agarose-Gelelektrophorese. 2.2.2.5.2 PCR-Fusions-Technik (SOEing) Mit der PCR-Fusions-Technik SOEing (splicing by overlap extension) ist es möglich, DNAFragmente unterschiedlicher Herkunft zu fusionieren oder bzw. und über die Primer Veränderungen in das zu amplifizierende Fragment einzuführen [150]. Im Rahmen dieser Arbeit wurde die SOEing-Technik eingesetzt, um die gewünschten Mutationen als Basenaustausche in das gyrA-Fragment einzuführen (Abschnitt 3.2.3.1). Außerdem wurde die SOEing-Technik genutzt, um die homologe Flanke und die KanamycinresistenzKassette für die Klonierung zum Einsatz bei der homologen Rekombination zu fusionieren (Abschnitt 3.2.3.4). Abbildung 2-9: Prinzip der SOEing-Technik (Beschreibung erfolgt im Text) Prinzipiell beruht SOEing darauf, dass die Primer im ersten PCR-Schritt für zwei zu fusionierende DNA-Fragmente jeweils am 5’- bzw. 3’-Ende des Überlappungsbereiches komplementäre Bereiche zum jeweils anderen DNA-Fragment einführen. Bei Mischung beider Amplifikate miteinander hybridisierten diese dann an den zueinander komplementären Bereichen. Der zweite PCR-Schritt beinhaltete also die Fusion der 65 Material und Methoden beiden Fragmente, wobei in der 2. Reaktion zunächst keine Primer eingesetzt wurden, sondern jeweils die Fragmente selbst als Startermoleküle dienten. Es wurde eine PCR mit 10 Zyklen durchgeführt. Die durch die Hybridisierung resultierenden 3’OH-Enden dienten nun als Startermoleküle und wurden durch die Expand High Fidelity Polymerase® mit Desoxynukleotiden verlängert, wobei das erwünschte rekombinante DNA-Molekül erzeugt wurde. Erst in der 3. Reaktion wurden 10 µl (= 1/10) aus der 2. Reaktion als Template eingesetzt und die Außenprimer zur Amplifikation zugesetzt. In diesem PCR-Schritt erfolgte dann eine Amplifikation des gesamten Fusionsproduktes mit den Außenprimern (siehe Abbildung 2-9). Die Verwendung der Expand High Fidelity Polymerase® mit Proofreading-Funktion statt Taq-Polymerase garantierte eine höhere Genauigkeit beim Einbau der Nukleotid-Bausteine, so dass die Zahl der während der PCR auftretenden Austausche reduziert wurde. 2.2.2.5.3 Sequenzspezifische PCR (ARMS) Die sequenzspezifische PCR wird auch als ARMS (amplification refractory mutation system) bezeichnet und ist zum Nachweis von Punktmutationen geeignet [215]. Das Prinzip dieser PCR ist, die nach einem Basenaustausch veränderte DNA-Sequenz für unterschiedliche Primerbindung zu nutzen und durch eine bewusste Paarung bzw. Fehlpaarung am 3`-Ende eines Primers die Bindung zu beeinflussen. Zur Untersuchung einer möglichen Mutante wurden zwei verschiedene PCR-Ansätze hergestellt. Der Forward-Primer (5`) war in beiden Fällen der gleiche und als Reverse-Primer (3`) wurde zum einen einer mit der Originalsequenz und zum anderen einer mit der veränderten Sequenz eingesetzt. Die Rerverse-Primer variieren entsprechend der erwarteten Sequenz. Dabei wurde die variierende Base an das 3`-Ende des Primers gelegt und durch einen zusätzlichen Mismatch verstärkt. Passt die Sequenz des Primers also nicht zur Templatesequenz kommt es zu keiner Anlagerung im 3`-Bereich des Primers. Dann kann die DNA-Polymerase keine Synthese durchführen und es entsteht kein PCRProdukt. Das Vorhandensein eines PCR-Produktes gibt also über die Sequenz des eingesetzten Primers, die Nukleotidsequenz des Templates bekannt. Prinzipiell wurde bei allen PCR-Ansätzen ein unabhängiges Primerpaar als Positivkontrolle dazu gegeben. Dadurch konnten für das Ausbleiben eines ARMS-PCRProduktes methodische Ursachen ausgeschlossen werden. In dieser Arbeit wurden die Primer PARE5-5-S und PARE3-2-S als Kontrolle verwendet. Entscheidungskriterien für die Wahl der Kontroll-PCR-Primer waren: (1) weit entfernte Bindung im Chromosom ohne Störung der ARMS-PCR, (2) stabile PCR auch bei unterschiedlichen Temperaturen sowie (3) dass die Größe des entstehenden PCR-Produkts von ARMS-Produkten abweicht, um eine gute Auftrennung der Produkte voneinander bei der Agarose-Gelelektrophorese zu erreichen. Die für die Kontrolle verwendeten parE-Primer erfüllten diese Kriterien. Sie lieferten ein PCR-Produkt der Länge von 241 bp. 66 Material und Methoden 2.2.2.6 Gelelektrophoretische Auftrennung von DNA-Fragmenten Mit Hilfe der Agarose-Gelelektrophorese können DNA-Fragmente unterschiedlicher Größe getrennt werden [192]. Das Prinzip der Elektrophorese beruht auf der Wanderung geladener Teilchen in einem elektrischen Feld. Das Agarosegel wirkt als poröse Matrix und hält die DNA-Moleküle in Abhängigkeit von ihrer Größe bei der Wanderung im elektrischen Feld unterschiedlich stark zurück. Dabei werden größere Moleküle im Gel stärker zurückgehalten als kleinere Moleküle. Das Agarosegel befindet sich bei der Elektrophorese in 0,5 x TAE-Puffer, durch dessen pH-Werte die aufzutrennende DNA aufgrund ihrer Phosphatgruppen negativ geladenen ist und somit zur Anode wandert. Der Agaroseanteil der Gele betrug 0,8 % bis 2 %. Die Prozentualität wurde abhängig von der erwarteten Größe der aufzutrennenden DNA gewählt. Die Agarose wurde in 0,5 x TAEPuffer durch Aufkochen gelöst. Nach dem Abkühlen auf ca. 55 °C wurde Ethidiumbromid zu einer ungefähren Endkonzentration von 0,1 µg/ml zugegeben. Dies geschah, um die DNA nach der Trennung sichtbar zu machen, da Ethidiumbromid aufgrund seiner planaren Struktur in die DNA-Doppelstränge interkaliert und so unter UV-Licht sichtbar ist. Das Agarosegel wurde in einer Horizontalapparatur gegossen und nach der Polymerisation mit 0,5 x TAE-Puffer überschichtet. Die aufzutrennenden Proben wurden mit ca. 1/6 Vol. Ladepuffer (Bromphenolblaulösung) versetzt und in die Geltaschen einpipettiert. Zum Einlaufen der Proben aus den Taschen in das Gel wurde eine Feldstärke von ca. 3 V/cm angelegt und anschließend auf ca. 5 V/cm erhöht. Nach der Elektrophorese wurden die DNA-Banden durch UV-Licht sichtbar gemacht und mit Hilfe einer Videoanlage dokumentiert und ausgewertet. Die Größe und Konzentration der aufgetrennten DNA-Banden ließ sich mit Hilfe eines während der Elektrophorese mitgelaufenden Standards (Smart Ladder) bestimmen. 2.2.2.7 Extraktion von DNA-Fragmenten aus Agarosegelen Bei zahlreichen Versuchen war es notwendig, nach der Auftrennung ein Teil der DNAFragmente zur weiteren Verarbeitung aus dem Gel zu reisolieren. Dabei wurden Kits der Firmen QIAGEN (QIAquick Gel Extraction Kit) bzw. MACHEREY-NAGEL (NucleoSpin® Extract II) verwendet. Die Durchführung geschah entsprechend dem Protokoll des Herstellers. Zunächst wurden die gewünschten DNA-Fragmente aus dem Agarosegel unter UV-Licht mit einem sauberen Skalpell ausgeschnitten und das Gewicht bestimmt. Um das Gelstück zum Schmelzen zu bringen, wurde ein entsprechender Volumenanteil an Puffer dazugegeben und bei 50 °C im Wasserbad bis zur Auflösung des Agarosegels inkubiert. Im Anschluss wurde die Lösung über eine Silika-Säule gereinigt. Das Prinzip dieser Aufreinigung beruht auf der selektiven Bindung von Nukleinsäuren an Silika-GelMembranen, wenn hohe Konzentrationen eines chaotropen Salzes vorliegen. Störende Komponenten wie Salze und Agarosereste werden nicht gebunden und dadurch ausgewaschen. Nach Bindung der DNA an die Silika-Membran wurde die Säule durch 67 Material und Methoden Spülen und anschließender Zentrifugation gewaschen. Zum Schluss wurde die DNA durch den entsprechenden pH-Wert des Elutionspuffers wieder von der Säule gelöst. 2.2.2.8 Aufreinigung und Konzentrierung von DNA 2.2.2.8.1 Nutzung von Silika-Säulen Wie bereits beschrieben, erfolgte die Aufreinigung von DNA hauptsächlich über Silika-GelMembranen. Auf diesem Weg wurden nicht nur Plasmid-DNA nach der Präparation (Abschnitt 2.2.2.2) bzw. DNA nach der Isolation aus dem Agarosegel gereinigt (2.2.2.7), sondern auch DNA aus PCR-Ansätzen oder nach Restriktionsspaltung von den Salzen, Primern, dNTPs, Polymerase oder Restriktionsenzymen getrennt, aufgereinigt und konzentriert. Es wurden die Kits der Firmen QIAGEN (QIAquick PCR-Purification Kit) und MACHEREY-NAGEL (NucleoSpin® Extract II) verwendet und die Protokolle der Hersteller befolgt. Die Methode besteht prinzipiell aus den Schritten: (1) Bindung der aufzureinigenden DNA an die Silika-Membran der Säule, (2) Waschen der Säule und dadurch Ausspülen der störenden Komponenten, (3) Elution der DNA von der Säule. Die Elution erfolgte mit EB-Puffer oder dH2O, um nachfolgende Reaktionen nicht zu stören. Alle Schritte wurden durch 1 min Zentrifugation bei 13.000 upm durchgeführt. 2.2.2.8.2 Ethanol-Präzipitation und Phenol-Chloroform-Extraktion Eine weitere Möglichkeit der Aufreinigung und Konzentrierung von DNA ist die Anwendung einer Ethanol-Präzipitation und Phenol-Chloroform-Extraktion. Das Prinzip der Ethanolfällung beruht darauf, dass in Gegenwart von einwertigen Kationen wie Na+ absolutes Ethanol Nukleinsäuren sehr wirksam ausfällt. Durch Zentrifugieren kann die DNA pelletiert und im Anschluss mit 70 % Ethanol gewaschen werden. Zu 100µl DNALösung wurden 10 µl NaAcetat-Lösung (3M, pH 4,6) und 250 µl 96 % Ethanol gegeben, maximal 25 min bei 15.000 upm zentrifugiert, das Pellet mit 400 µl 70 % Ethanol gewaschen und 5 min zentrifugiert. Das getrocknete Pellet wurde in dH2O oder EB-Puffer aufgenommen. Die besondere Reinigung der DNA durch Phenol-Chloroform-Extraktion war nach der Plasmidpräparation aus Salmonella spec.-Stämmen nötig, um störende Nukleasen zu entfernen. Dazu wurde die DNA-Lösung mit einem Volumen Phenol/Chloroform/Isoamylalkohol (25:24:1, v/v) gründlich ausgeschüttelt und 5 min bei 13.000 upm zentrifugiert. Die obere wässrige Phase (unter Vermeidung der Berührung der Interphase) wurde in ein neues Gefäß pipettiert und wiederum ausgeschüttelt und zentrifugiert. Alle Verschmutzungen wurden so denaturiert und befinden sich in der unteren organischen Phase und die Nukleinsäuren in der oberen wässrigen Phase. Zur Entfernung aller Phenolreste wurde eine Ethanolfällung angeschlossen. 68 Material und Methoden 2.2.2.9 Enzymatische Modifikation von DNA 2.2.2.9.1 Restriktionsspaltung Durch den Einsatz von Typ II-Restriktionsendonukleasen lässt sich DNA an spezifischen Stellen spalten. Ursprünglich schützen sich die Bakterien durch die Endonukleasen vor dem Eindringen fremder DNA. Die Enzyme erkennen spezifische palindrome Basensequenzen und spalten innerhalb dieser den DNA-Strang durch Hydrolyse. Es entstehen je nach Enzym glatte (blunt) oder kohäsive (sticky) Enden. So wird artfremde DNA zerschnitten, während die bakterieneigene DNA durch Modifikationen vor dem Endonuklease-Abbau geschützt ist. Durch das Erkennen spezifischer Sequenzen und das Entstehen bestimmter Enden können die Restriktionsenzyme des Typs II in der Gentechnik zum Klonieren rekombinanter DNA benutzt werden. Setzt man zueinander kompatible Enden ein, lassen sich zwei vorher gespaltene DNA-Fragmente wieder verknüpfen. Die Menge an Restriktionsenzym für eine vollständige DNA-Spaltung ist abhängig von der Größe und Menge der DNA, der Anzahl der vorhandenen Schnittstellen sowie der Inkubationsdauer und kann nach folgender Formel berechnet werden: Unit Enzym = µg × (5 × 10 4 ) × Sv bp DNA × S λ × h SV = Schnittstellen in zu spaltender DNA, Sλ = Schnittstellen in λ-DNA, h = Inkubationsdauer Um eine möglichst komplette Restriktion zu erhalten, wurde für die Spaltung das Zwei- bis Dreifache der errechneten Enzymmenge eingesetzt. In der Regel wurde ein Gesamtansatz von 20 µl gewählt. Neben der DNA wurde die entsprechende Unitmenge an Enzym(en), der entsprechende Puffer und z.T. auch BSA zugegeben. Um eine Reaktionshemmung durch einen zu hohen Glycerinanteil (in Enzymlösung) zu vermeiden, darf der Enzymanteil 1/10 Vol % des Gesamtansatzes nicht überschreiten. Um ein Fragment mit zwei Restriktionsenzymen zu spalten, wurde ein Puffer gewählt, in dem beide Enzyme eine ausreichende Aktivität (Angaben des Herstellers) aufweisen. Waren die Reaktionsbedingungen Spaltungsreaktionen beider nacheinander mit Enzyme einem zu verschieden, zusätzlichen wurden die zwischengeschaltetem Reinigungsschritt durchgeführt oder die Pufferbedingungen für das zweite Enzyms z.B. durch Salzzugabe angepasst. In der Regel wurden die Restriktionsansätze 1 bis 3 h bei 37 °C inkubiert und die Enzyme im Anschluss durch Erhitzen auf 65 °C inaktiviert. 2.2.2.9.2 Restriktionsfragmentlängenpolymorhismus (RFLP) Diese Methode ermöglicht ein leichtes und schnelles Screening auf das Vorhandensein einer Nukleotidsequenzänderung im Fall einer Mutation. Durch den daraus resultierenden Verlust oder das Entstehen einer Enzym-Erkennungssequenz ändert sich die Größe und Anzahl der entstehenden Fragmente nach der Restriktionsspaltung. Dies kann als 69 Material und Methoden Nachweis einer Sequenzänderung, z.B. durch einen Basenaustausch dienen. Ein Basenaustausch in den Codons Asp82/Ser83 kann mittels einer Restriktionsspaltung mit dem Enzym HinfI detekiert werden, da dieser Bereich eine Erkkennungssequenz für dieses Restriktionsenzym beeinhaltet. Zum Screening einer Mutation in dieser Region erfolgte zunächst eine PCR mit den Primern gyrA-5-1S und gyrA-3-1S (siehe Tabelle 2-4) zur Amplifikation eines ca. 530bp großen DNA-Fragmentes aus dem gyrA-Gen. Im Anschluss wurde das PCR-Produkt mit Hilfe des Enzyms HinfI einer Restriktionsspaltung unterzogen. Bei einem Basenaustausch in den Codons Asp82/Ser83 geht eine der zwei in diesem PCR-Produkt vorhandenen Erkennungssequenzen für das Restriktionsenzym HinfI verloren. Als Wildtypsituation (keine Mutation) fanden sich also nach der Restriktionsspaltung drei Fragmente der ungefähren Größe 290bp, 140bp und 100bp. Im Fall einer Mutation an Position Ser83 entstanden durch den Verlust einer Schnittstelle nur zwei Fragmente der ungefähren Größe 390 bp und 140 bp (vergleiche auch Abschnitt 3.1.5.1, Abbildung 3-3). 2.2.2.9.3 Dephosphorylierung Klonierung besteht darin, ein DNA-Fragment (Insert) und einen vorher durch Restriktionsenzyme gespaltenen Vektor zu ligieren. Durch die Phosphatreste an den 5`Enden, welche bei der Restriktionsspaltung entstehen, kann es zu einer Verknüpfung mit dem 3'-Hydroxylende des gleichen Vektors kommen. Um diese Religation des Vektors zu verhindern und das Inserieren eines DNA-Fragmentes in einen Klonierungsvektor zu ermöglichen, wurden die Phosphatreste am Vektor abgespalten. Dazu wurde der linearisierte Klonierungsvektor mit einer alkalischen Phosphatase behandelt. Die Dephosphorylierung erfolgte auch bei einer gerichteten Klonierung über zwei verschiedene Restriktionsschnittstellen, wenn die Enzym-Erkennungssequenzen sehr eng beieinander lagen und das entstehende sehr kleine Fragment bei der AgaroseGelelektrophorese nicht nachweisbar war. Die möglicherweise nur mit einem Enzym gespaltene linearisierte Plasmide sollten so aus der Klonierung ausgeschlossen werden. Es erfolgte eine Zugabe von 1 µl (= 1 U) Phosphatase und 10 x Dephosphorylierungspuffer (Endkonzentration 1 x) zur Vektor-DNA und eine Inkubation von 30 min bei 37 °C sowie anschließende Inaktivierung des Enzyms bei 65 °C für 15 min. 2.2.2.9.4 Erzeugen von glatten Enden für die Klonierung Waren keine zwei kompatiblen Restriktionsschnittstellen für eine Klonierung vorhanden, wurde mit den möglichen unterschiedlichen Restriktionsenzymen gespalten. Die inkompatiblen überhängenden Fragmentenden (sticky ends) wurden anschließend geglättet, d.h. in glatte Enden (blunt ends) umgewandelt. Dieses ist mit Hilfe von T4 DNA Polymerase oder dem Klenow-Fragment der Polymerase I möglich. Zum Auffüllen von 5`Überhängen wurde zum Reaktionsansatz 1 µl Klenow-Fragment und 100 µM dNTP`s gegeben, 15 min bei 25 °C inkubiert und die Reaktion mit Zugabe von 25 mM EDTA und 70 Material und Methoden 20 min Erhitzen auf 75 °C gestoppt. Das Glätten (Abbau) der 3`-Überhänge wurde durch Zugaben von 1 µl T4 DNA-Polymerase, 100 µM dNTP`s, 5 x Puffer (Endkonzentration 1 x), 15 min Inkubation bei 37 °C und Abstoppen der Reaktion durch Zugabe von 25 mM EDTA und Erhitzen erreicht. 2.2.2.9.5 Zur Ligation mittels T4-DNA-Ligase Verknüpfung der 5’-Phosphatgruppe eines DNA-Fragmentes und der 3’- Hydroxylgruppe eines anderen DNA-Fragmentes für eine erfolgreiche Klonierung wurde vor allem die T4-DNA-Ligase verwendet. Durch DNA-Ligase wird in einer ATP-abhängigen Reaktion die Bildung von Phosphodiesterbindungen katalysiert. Für eine Schnell-Ligation wurden neben dem Vektor und dem zu klonierenden Insert, Ligationspuffer sowie 1-2 Units Ligase zugesetzt. Die Inkubation erfolgte für 10 min (sticky ends) bis 60 min (blunt ends) bei RT. Als Ligationspuffer wurde entweder ein Fertigpuffer der Firmen Roche oder New Englang Biolabs oder ein selbst angesetzter Puffer (siehe Tabelle 2-11) verwendet. Bei der Verwendung des selbst angesetzten Ligationspuffers wurde eine Beschleunigung der Reaktion durch Zugabe von 10 mM Hexamincobalttrichlorid und ein Abstoppen durch 80 mM Kaliumchlorid erreicht. Zur Klonierung wurde ein molares Verhältnis von Vektor zu Insert je nach Fragmentgröße von 1:3 bis 1:5 gewählt. Der Gesamtansatz sollte ein Gesamtvolumen von 25 µl nicht überschreiten, um eine optimale Reaktion zu gewährleisten. Im Anschluss an die Reaktion konnte der Ligationsansatz direkt transformiert werden. 2.2.2.9.6 Ligation mittels Topoisomerase I inklusive Blau-weiß-Selektion Zur Herstellung der pCR2.1-Derivate (Abschnitt 3.2.3.2) wurde auf das TOPO TA Cloning® Kit der Firma Invitrogen verwendet. Dieses beruht darauf, dass statt einer Ligase für die Ligation ein mit Vaccinia Topoisomerase I „aktivierter“ Vektor eingesetzt wird. Mittels TaqPolymerase wurde ein PCR-Produkt erzeugt, welches ein Nukleotid am 3’-Ende, meistens ein Adenosin, enthält [207]. Der linearisierte Vektor pCR2.1-TOPO besitzt komplementär dazu jeweils einen Desoxythymidin-Überhang. Beide DNA-Enden des Vektors sind durch Topoisomerase blockiert, was eine Selbstligation des Vektors ausschließt. Bei der Ligation von Fragment und Vektor wird die Phosphotyrosyl-Bindung zwischen der Vektor-DNA und der Topoisomerase I durch das 5’-OH Ende des PCR-Fragments nucleophil angegriffen, was zur Freisetzung der Topoisomerase [272] und zur Ligation von Fragment und Vektor führt. Der pCR2.1TOPOVektor besitzt neben der Ampicillinresistenz-Kassette einen lacZPromotor und ein lacZ-Gen (codiert für β-Galactosidase). Diese Variante macht eine Blauweiß-Selektion bei der Klonierung möglich. Klone mit intaktem lacZ-Gen erscheinen auf LB-Platten mit X-Gal und IPTG (Induktor der β-Galactosidase-Genexpression) durch Spaltung des chromogenen Substrats X-Gal zu 5-Brom-4-Chlor-Indigo blau. Durch 71 Material und Methoden Insertion eines DNA-Fragmentes wird der Leserahmen des lacZ-Gens zerstört und die Bildung von β-Galactosidase unterbunden. Klone mit rekombinantem Plasmid erscheinen demnach weiß. Nach der Transformation in kompetente TOP10F’ Zellen (exprimieren den lac-Repressor LacI, besitzen jedoch kein aktives lacZ-Gen) konnte die Selektion positiver Klone also durch einen Farbtest (Blau-weiß-Selektion) auf LB-Platten mit Ampicillin (50 µg/ml) X-Gal (40 µg/ml) und IPTG (100 µg/ml) erfolgen. Zur Klonierung des Rescueplasmides (siehe Abschnitt 3.2.1.2) wurde als Klonierungsvektor das Plasmid pBAD/D102 der Firma Invitrogen gewählt. Dieser Vektor beinhaltet einen induzierbaren Arabinosepromotor zur Steuerung der Expression des klonierten Genes. Auch hier kann das gewünschte Gen über TOPO®-Klonierung unter Verwendung der Topoisomerase I anstelle einer Ligase mit Hilfe eines einfachen PCRProduktes in den Vektor kloniert werden. Das Prinzip des pBAD Directional TOPO® Expression Kits der Firma Invitrogen besteht allerdings darin, ein blunt-end PCR-Produkt (hergestellt mittels Phusion™ High-Fidelity DNA Polymerase) über TOPO®-Klonierung in den Vektor einzuführen. Um eine gerichtete Klonierung zu gewährleisten, welche eine notwendige Voraussetzung für die Expression des Gens über den Arabinosepromotor ist, wurde über den Forward-Primer ein Anhang von vier Basen (CACC) eingeführt. Der Vektor enthält einen Überhang aus den Basen GTGG, so dass sich die vier Basen aufgrund der komplementären Sequenz anlagern und das PCR-Produkt in der richtigen Orientierung stabilisiert wird (siehe Abbildung 2-10). Abbildung 2-10: Prinzip des pBAD Directional TOPO® Expression Kits der Firma Invitrogen Prinzip der TOPO-Klonierung unter der Verwendung von Überhängen zur Bestimmung der Orientierung des einzuklonierenden blunt-end PCR-Produktes 72 Material und Methoden 2.2.2.10 Sequenzierung von DNA Die automatische Sequenzierung von DNA wurde im Labor mit Hilfe des DNA-Sequenzer ABI PRISMTM 310 und dem Sequencer Kit der Firma Applied Biosystems durchgeführt. Das Prinzip beruht auf dem von Sanger et al. entwickelten Kettenabbruchverfahren [262]. Voraussetzung für das automatisierte Verfahren ist das Cycle Sequencing. Hierbei wird bei einer PCR-Reaktion ein DNA-Template mit nur einem Primer amplifiziert. Dafür werden neben den vier dNTPs kleine Mengen von 2’-3’- Didesoxyribonukleosidtriphosphaten (ddNTPs) mit unterschiedlicher Fluoreszenzfarbstoffmarkierung zugesetzt. Nach dem Einbau eines ddNTPs kommt es auf Grund der fehlenden OH-Gruppe am C3-Atom der Desoxyribose, die für die Bildung einer Phosphodiesterbindung benötigt wird, zum Kettenabbruch. Da der Kettenabbruch einer statistischen Verteilung folgt, entsteht ein Gemisch von Fragmenten mit unterschiedlichen Kettenlängen und je nach Base entsprechender Farbmarkierung. Im DNA-Sequenzer ABI PRISMTM 310 werden diese Fragmente mittels einer Kapillarelektrophorese nach ihrer Größe aufgetrennt. Die unterschiedlichen Fluoreszenzfarbstoffmarkierungen der ddNTPs werden mit einem Laser detektiert. Die Basensequenz der untersuchten DNA-Probe ergibt sich aus der Abfolge der auftretenden basenspezifischen Farbsignale. Die Farbsignale werden anschließend mit Hilfe einer Software (ABI-CE, Version 3.0, Perkin Elmer Corporation, Foster City, USA) in die entsprechende Basenreihenfolge umgesetzt. Die Sequenzierung bestand insgesamt aus 3 Schritten. Im ersten Schritt wurde das zu sequenzierende Fragment über eine einfache PCR (Abschnitt 2.2.2.5.1) amplifiziert. Dieses Fragment wurde mittels des PCR-Purification Kit der Firma QIAGEN aufgereinigt (Abschnitt 2.2.2.8.1) und über ein Agarosegel wurde eine Konzentrationsbestimmung (Abschnitt 2.2.2.6) vorgenommen. Der zweite Schritt beinhaltete das Cycle Sequencing. Der Standardansatz von 20 µl bestand neben der DNA, dem Sequenzierungspuffer, dem Big Dye (Mix aus Polymerase und ddNTP`s) aus einem Sequenzierprimer (10 pmol/µl). Bei der Sequenzierung von Plasmiden wurde zusätzlich DMSO zugesetzt und ohne Big Dye für 5 min bei 96 °C vorinkubiert, um eine bessere Denaturierung zu erreichen. Nach Zugabe von Big Dye wurde das untere Temperaturprofil gestartet. Tabelle 2-19: Standardtemperaturprofil für Cycle Sequencing Schritt Temperatur Zeit Wiederholung Initialdenaturierung 96 °C 2 min 1x Denaturierung 96 °C 10 min Primeranlagerung TA°C 5 sec Synthese 60 °C 4 min 25x TA = Anlagerungstemperatur der Primer abhängig von der Basensequenz Æ bei Plasmiden wurde prinzipiell eine Anlagerungstemperatur von 60 °C gewählt. Im dritten Schritt erfolgte eine Aufreinigung der Cycle-Ansätze mittels EthanolPräzipitation. Dafür wurden zu jedem 20 µl Ansatz 80 µl dH2O, 10 µl Natriumacetat (3M, pH 4,6) und 250 µl 100 %iger Ethanol (p.A.) gegeben. Anschließend wurde die Probe 73 Material und Methoden 25 min bei 15.000 upm und RT zentrifugiert. Der Überstand wurde mittels Vakuumpumpe vorsichtig abgesaugt und das Pellet mit 400 µl frischem 70 %igem Ethanol gewaschen. Nach einer erneuten Zentrifugation für 5 min bei 15.000 upm und RT wurden die DNAPellets für 30 min lichtgeschützt bei 37 °C getrocknet. Vor Beladung der Kapillare wurden die DNA-Pellets in 20 µl HiDi (reines Formamid) aufgenommen. Die im DNA-Sequenzer ABI PRISMTM 310 erstellten Basensequenzen wurden über die Software Vector NTI (InforMax) ausgewertet. 2.2.3 Proteinbiochemische Methoden Ein Ansatz, um qualitative und quantitative Veränderungen im Proteinexpressionsmuster von Zellen zu vergleichen, bietet die Methode der 2D-Gelelektrophorese. Dabei werden die Proteine einer Zelle in zwei Dimensionen aufgetrennt: zum einen nach ihrem isoelektrischen Punkt und zum anderen nach ihrem Molekulargewicht (Masse). Die Methode beruht auf Arbeiten von O´Farrel und Klose [228]. 2.2.3.1 Probenvorbereitung Ziel der Probenvorbereitung ist es, alle Proteine möglichst komplett zu lösen, zu desaggregieren, zu denaturierten und zu reduzieren. Weiterhin sollte das Protokoll so einfach wie möglich sein, um die Reproduzierbarkeit zu erhöhen. Wichtig ist, die Proteinmodifikationen möglichst zu minimieren, um die Artefaktbildung im Gel zu verhindern. Für die Isoelektrische Fokussierung müssen die Proteine ihre Ladung behalten. Die Probenaufbereitung besteht aus drei Schritten: (1) Zerstörung der Zelle (2) Inaktivierung oder Entfernung störender Substanzen und (3) Lösen der Proteine. Probenvorbereitung nach Schulte (2001) [268]: Für die Bakterienanzucht wurde eine Übernachtkultur des zu testenden Stammes in 3 ml NI-Bouillon angesetzt. Diese wurde dann 1:100 in einem Gesamtvolumen von 50 ml NI-Bouillon verdünnt und bei 37 °C und 250 upm bis OD546= 0,7 schüttelnd inkubiert. Die Bakteriensuspension wurde in 50 ml Falcon überführt und bei 5.300 upm und 4 °C für 10 min zentrifugiert. Das Zellpellet wurde dreimal mit 10 ml PBS (1 x Puffer) gewaschen und nach jedem Waschschritt zentrifugiert (5.300 upm, 4 °C, 10 min). Im Anschluss wurde das Zellpellet ausgewogen und mit Lysispuffer (1 ml Lysispuffer auf 120 mg Zellen) versetzt. Erst kurz vor Gebrauch wurden dem 1 ml Lysispuffer die 10 µl Pefabloc (Proteinaseinhibitor) zugesetzt. Die Lösung wurde 30 min geschüttelt und dann für 5 min in ein Ultraschallbad gegeben. Nach neuer Zentrifugation (15.000 upm, 4 °C, 30 min) wurde der klare Überstand, in dem sich die Proteine befanden, abgenommen. Die Proteinlösung wurde aliquotiert und bei –80 °C gelagert. 74 Material und Methoden Probenvorbereitung mit ReadyPrep protein extraction kit „Total Protein“ von Bio-Rad Laboratories: Um eine höhere Proteinausbeute zu erlangen und das präparierte Proteinspektrum vergleichbar zu halten, wurde eine Probenaufarbeitung mit dem ReadyPrep protein extraction kit „Total Protein“ von Bio-Rad Laboratories genutzt. Dafür wurde die Bakterienanzucht wie oben beschrieben durchgeführt und die Zellen mit Phosphatpuffer gewaschen. Zu 0,05 ml abzentrifugiertem nassem Zellpellet wurde je 1 ml Sample-Puffer gegeben, der 10 µl Pefabloc als Proteaseinhibitor enthielt. Die Probe wurde dann auf Eis gestellt. Danach wurde sie 3- bis 4-mal für je 30 sec Ultraschall ausgesetzt und anschließend bei 16.000 g bei 18-20 °C für 20 bis 30 min zentrifugiert. Der Überstand wurde dann in ein neues Gefäß überführt und bei -80 °C eingefroren. Bei beiden Durchführungen wurden die Zellen durch Lyse und anschließende Ultraschallbehandlung zerstört, wobei Harnstoff und CHAPS im Lysepuffer als Detergenzien wirken. Das Bio-Rad Kit beinhaltet weiterhin ASB-14 als stakes Detergenz. DTT bzw. TBP wirken als reduzierende Agenzien. Pefabloc inhibiert die Proteasen. Das Bio-Rad Kit enthält weiterhin Benzonase als Endonuklease, um störende DNA abzubauen. 2.2.3.2 Bestimmung der Proteinkonzentration Für eine optimale Durchführung der 2D-Gelelektrophorese ist es unabdingbar, die genaue Proteinkonzentration der eingesetzten Probe zu kennen. Zunächst wurde die Proteinkonzentrationsbestimmung nach Lowry durchgeführt. Nachdem die Ergebnisse aber nicht befriedigend waren, da die Detergenzien im Lysepuffer die Farbreaktionen stören, wurde der RC DC Protein Assay von Bio-Rad Laboratories verwendet. Lowry-Test: Die Methode [186] beruht auf der Bindung der Aminosäuren mit dem Stickstoffatom ihrer Aminogruppe an Cu2+, wobei ein farbiger Chelat-Komplex entsteht. Zusätzlich reduzieren bestimmte Aminosäuren (Tyrosin, Tryptophan, Cystein, Histidin) das Folin-Ciocalteu-Reagenz, was zu einer Blaufärbung der Lösung führt. Es wurde zunächst eine Verdünnungsreihe von BSA-Proben mit bekanntem Proteingehalt (Endkonzentration 0-240 µg/ml) erstellt. Für die Reaktion wurden 100 µl der entsprechenden BSAVerdünnungsstufe und die zu vermessenden Proben mit 1 ml alkalischem Kupferreagenz vermischt und 10 min bei RT inkubiert. Anschließend wurden 100 µl Folin-CiocalteuReagenz (frisch angesetzt 1:3 in dH2O) zugegeben und der Ansatz gut gemischt. Nach 20 min Inkubation bei RT wurde die Extinktion bei 578 nm gegen den Leerwert (ohne Protein) photometrisch gemessen. Aus den bekannten Proteinkonzentrationen wurde eine Eichgerade erstellt, anhand derer eine Errechnung des Proteingehaltes der unbekannten Probe erfolgen konnte. RC DC Protein Assay von Bio-Rad Laboratories: Auch dieser Assay beruht auf der Farbreaktion nach Lowry. Allerdings wurde hier aufgrund der starken Detergenzien und reduzierenden Agenzien im Probenpuffer (ASB-14 und TBP) zusätzlich ein Schritt 75 Material und Methoden (Fällung) vorgeschaltet. Die Durchführung erfolgte wie im Protokoll des Kits angegeben. Bereits nach 15 min erreicht die Farbreaktion ihr Maximum, ist für mind. 1 h stabil und verliert selbst nach 2 h nur 10 % der Intensität. Die gefärbten Proben werden bei einer Wellenlänge von 750 nm vermessen. Als Protein-Standard wird BSA vermessen. 2.2.3.3 2D-Gelelektrophorese Die Zwei-Dimensionale-Gelelektrophorese (2D-GE) besteht, wie der Name schon sagt, aus zwei Dimensionen. In der ersten Dimension erfolgte eine elektrophoretische Auftrennung der Proteine nach ihrem isolektrischen Punkt (isolektrische Fokussierung). In der zweiten Dimension kommt es in einem SDS-Polyacrylamid-Gel zu einer Auftrennung der Proteine nach ihrer Molekülgröße [228]. Durch die beiden Dimensionen lassen sich die Proteine einer Probe separieren, wodurch ein Vergleich der Proteinexpressionsspektren unterschiedlicher Zellen in Qualität und Quantität möglich ist. 2.2.3.3.1 Erste Dimension: Isolektrische Fokussierung (IEF) Nur am isoelektrischen Punkt (pI) ist die Nettoladung eines Proteins aufgrund seines Charakters als Ampholyt (Zwitterion) gleich Null. In einem Gel mit pH-Gradienten wandern die Proteine bei angelegter Spannung aufgrund ihrer Ladung zu dem Punkt (pH-Wert), der ihrem isoelektrischen Punkt entspricht. Ziel des ersten Schrittes der 2D-GE ist eine Konzentrierung (Fokussierung) der Proteine an ihren isoelektrischen Punkten (Abbildung 2-11). Abbildung 2-11: Prinzip der isoelektrischen Fokussierung oben: verschiedene Nettoladungen eines Proteins; unten: Veränderung der Nettoladung eines Proteins in Abhängigkeit vom pH-Wert der Umgebung (Abbildung verändert aus [30]) 76 Material und Methoden Um das Problem der Wanderung der den pH-Gradienten bildenden Ampholyte bei längerer Fokussierungszeit zu verhindern, wurden immobilisierte pH-Gradienten (IPG) in Form von IPG-Strips, d.h. im Gel fixierte Ampholyte, verwendet. Der IPG entsteht durch Kopolymerisation der Immobiline (Acrylamidderivate mit puffernden Gruppen) mit Acrylamidmonomeren [30]. Die IPG-Streifen lagen getrocknet vor und mussten zuächst rehydriert werden. Die Menge des eingesetzten Proteins richtete sich nach dem gewählten IPG-Gradienten, dessen Länge und vor allem der Proteinfärbung. Es wurden Streifen von 17cm Länge und einem pH-Gradienten zwischen 4 und 7 gewählt. Für eine anschließende Silberfärbung der Gele wurden 50-80 µg Protein und für eine Coomassiefärbung wurden 200-400 µg Protein eingesetzt. Nach Auftauen der Probe wurde sie in Rehydrierungspuffer aufgenommen. Bei den 17 cm langen Streifen wurden 330 µl Rehydratisierungslösung eingesetzt. Die Lösung wurde für 1 h bei 30.000 g zentrifugiert und der Überstand verwendet. Dieser wurde in die Kammer der IEF Cell von Bio-Rad pipettiert und darauf der IPG-Streifen mit der Gelseite nach unten gelegt. An die Enden der Streifen wurden kleine Filterstücke (wicks) gelegt, um die Salze aus der Lösung zu sammeln. Um oxidative Prozesse und Verdunstung zu verhindern, wurden die Streifen mit Mineralöl überschichtet. Die Rehydrierung der Streifen wurde aktiv durch das Anlegen einer leichten Spannung (50 µA/Streifen für 12 h) erreicht. Die isoelektrische Fokussierung erfolgte sofort im Anschluss durch folgendes Fokussierungsprogramm: (1) 20 min bei 250 V (2) linearer Anstieg der Spannung (2,5 h auf 10.000 V) (3) Halten der Spannung 4 h bei 10.000 V (= 40.000 V/h - Voltstunden). Nach der Fokussierung konnten die Streifen bei -20 °C gelagert werden. 2.2.3.3.2 Zweite Dimension: SDS-PAGE In der zweiten Dimension erfolgte eine Auftrennung der Proteinkomplexe in einer porösen Polyacrylamidmatrix nach ihrem Stokes-Radius und damit nach ihrer Masse. Um eine Wanderung der Proteine während der Elektrophorese zu erreichen, wurden die IEFStreifen nach der Fokussierung äquilibriert, d.h. mit SDS gesättigt. Proteine binden SDS (Sodiumdodecylsulfat) und es entstehen negativ geladene SDS-Protein-Komplexe mit konstantem Ladungs-zu-Masse-Verhältnis. Außerdem denaturiert SDS die Proteine (nach vorheriger Reduktion mit Mercaptoethanol oder DTT) und unterbindet Protein-Protein Wechselwirkungen (Quartärstrukturen). Die IEF-Streifen wurden nach dem Auftauen und dem Abfluss des Öls in 10 ml Äquilibrierungslösung I (enthält Harnstoff, Glycerin, SDS, Tris-HCL und DTT) gelegt, etwas Bromphenolblau dazugegeben und 10 min bei RT schüttelnd inkubiert. Nach Abgießen der ersten Lösung wurden 10 ml Äquilibrierungslösung II (enthält statt DTT Iodoacetamid zur Blockierung der freien –SHGruppen) dazugegeben und noch einmal 10 min bei RT geschüttelt. Die äquilibrierten IEFStreifen wurden anschließend auf die obere Gelkante eines SDS-Gels gelegt. Das verwendete SDS-Gel wurde nach dem Puffersystem von Laemmli [171] ohne Sammelgel hergestellt. Die Funktion des Sammelgels wird bei der 2D-Gelelektrophorese vom IEF- 77 Material und Methoden Streifen übernommen. In der Gelelektrophorese-Apparatur Protean II von Bio-Rad konnten immer 2 Gele gleichzeitig gefahren werden. Es wurden 12 % Polyacrylamidgele verwendet. Die Rezeptur ist in Tabelle 2-20 beschrieben. Tabelle 2-20: Zusammensetzung eines 12 % SDS-Gels Bestandteil Volumen 30 % Acrylamid/Bis-Lösung (37,5:1) 40 ml Glycerin 10 ml Trenngelpuffer pH 8,8 25 ml dH20 24 ml 10 % SDS-Lösung 1 ml Endvolumen 100 ml Die Lösung wurde gemischt und für 10 min gerührt. Kurz vor dem Gießvorgang wurden als Katalysatoren 50 µl Temed (100 %) und 150 µl APS (40 %) dazugegeben. Nach dem Einfüllen des Gels in die Gieskassette wurde das Gel mit 1 ml Butanol überschichtet. Die gegossenen Gele polymerisierten mindestens 1 bis optimal ca. 3 h. Vor Gebrauch wurde das Butanol abgezogen und die Gelkante mit Wasser gewaschen. Der IEF-Streifen wurde luftblasenfrei auf die horizontale Gelkante des SDS-Gels gelegt und mit 0,5 % bromphenolblauhaltiger Agarose (in 1 x Laufpuffer) fixiert. Nach dem Auspolymerisieren der Agarose wurden die Gele in die Gelelektrophoreseapparatur eingesetzt und der Laufpuffer in die Kammern gefüllt. Die Gelelektrophorese erfolgte bei 15 mA für 20 h. 2.2.3.3.3 Färbemethoden: Coomassiefärbung und Silberfärbung Nach Beendigung der Gelelektrophorese wurden die Gele aus der Laufkammer entnommen und durch verschiedene Färbemethoden sichtbar gemacht. Für die Optimierung der Gelelektrophorese wurde eine Coomassiefärbung verwendet. Für die analytischen Gele wurde eine Silberfärbung verwendet. Die Zusammensetzung der Lösungen ist Tabelle 2-11 zu entnehmen oder entsprach den Herstellerangaben. Die Gele wurden in Glaswannen schüttelnd inkubiert und die Lösungen nacheinander dazugegeben und abgegossen. Coomassiefärbung: Der Farbstoff Coomassie-Blau hat eine Nachweisgrenze von ca. 8 ng Protein. Die Färbemethode beruht darauf, dass saure (anionische) Farbstoffe an die basischen Aminosäureseitenketten von Lysin oder Arginin binden. Die Gele wurden in Glaswannen überführt, welche 500 ml Coomassie-Färbelösung enthielten und schüttelnd über Nacht (max. 24 h) gefärbt. Im Anschluss wurden sie in dH2O entfärbt, bis der Hintergrund hell war [268]. 78 Material und Methoden Silberfärbung modifiziert nach Heukeshofen et al. [134]: Bei der konventionellen Silberfärbung (Tabelle 2-21) werden wesentlich niedrigere Nachweisgrenzen (< 0,3 ng) erreicht als bei der Coomassiefärbung. Die Methode beruht darauf, dass die funktionellen Gruppen im Proteinmolekül die Reduktion von Silberionen zu elementarem Silber katalysieren. Pro Gel wurde ein Volumen von 500 ml der Lösungen eingesetzt. Die Gesamtfärbezeit betrug ca. 3 h. Tabelle 2-21: Silberfärbung nach Heukeshofen et al. [134] Lösung Inkubationszeiten Fixierlösung 1 40 min Fixierlösung 2 40 min dH20 3 x 5 min Silbernitratlösung 40 min dH20 1 x 2 min Entwicklerlösung 3-7 min (bis Spots zu sehen sind) Stopplösung 15 min dH20 15 min Silberfärbung Silver Stain Plus®: Es handelt sich dabei um ein Färbekit der Firma Bio-Rad, welches eine schnelle und einfache Proteindetektion über Silberfärbung möglich macht (Tabelle 2-22). Es ist 30 bis 50fach sensitiver als die Coomassiefärbung. Tabelle 2-22: Silberfärbung Silver Stain Plus® (Bio-Rad Laboratories) Lösung Inkubationszeiten Fixierlösung (Fixative Enhancer Solution) 40 min dH20 2 x 10 min Färbe- und Entwicklerlösung (bestehend aus Silver Complex Solution, Reduction Moderator Solution, Development Reagent, Development Accelerator) 20 min Stoppen durch Zugabe von 5 % Eisessig Lösung 15 min dH2O 5 min Silberfärbung SilverQuest®: Dieses Kit der Firma Invitrogen zeichnet sich dadurch aus, dass es zum einen die Sensitivität der Silberfärbung und zum anderen die Kompatibilität der Massenspektrometrie vereint. Zunächst wurden die Proteine im Gel fixiert und Detergenzien und störenden Ionen entfernt. Anschließend erfolgte zur Erhöhung der Sensitivität und des Kontrastes, gefolgt vom Waschen zum Entfernen der Rückstände. Die Silberionen werden im Färbeschritt durch Bindung der Silberionen an die Proteine zu metallischem Silber reduziert. Danach wurde die Reaktion gestoppt und die freien Silberkomplexe wurden entfernt. Die Durchführungszeit kann von 2 h auf 1 h durch die Anwendung eines Mikrowellenprogrammes reduziert werden. 79 Material und Methoden Tabelle 2-23: Silberfärbung SilverQuest® (Invitrogen GmbH) Lösung Inkubationszeiten Wasser ganz kurz spülen Fixierlösung 20 min (ggf. über Nacht) Waschen (30 % Ethanol) 10 min Sensitizing solution 10 min Waschen (30 % Ehtanol) 10 min Wasser 10 min Staining Solution 15 min Wasser 20 – 60 sec Developing solution 4 – 8 min (bis Spots zu sehen sind) 10 ml Stopper direkt zum Gel in Entwicklerlösung geben 10 min (Farbwechsel von pink zu klar) Wasser 10 min 2.2.3.3.4 Imaging, Detektion und Bildauswertung mittels PDQuest® Unabhängig von der Färbemethode wurden alle Gele mit einem Büroscanner eingescannt und die Bilder im TIF-Format gespeichert. Anschließend konnten die Gelbilder mit der Software PDQuest® der Firma Bio-Rad verarbeitet werden. Die Auswertung besteht aus folgenden Schritten: (1) Datenimport, (2) Bildbearbeitung (Zuschneiden, Rotation usw.), (3) Spotdetektion, (4) Spoteditierung, (5) Matchset, d.h. Vergleich der Gele untereinander sowie (6) Datenanalyse. 80 Ergebnisse 3 Ergebnisse 3.1 Charakterisierung der Ausgangsstämme Die in dieser Arbeit verwendeten Untersuchungsstämme waren der Ausgangsstamm S. Typhimurium M415 (M415), die Stämme Nal2ori und Nal2passage und der Impfstamm TAD Salmonella vacT (vacT). Der Impfstamm ging durch ungezielte Mutagenese aus dem Ausgangsstamm S. Typhimurium DT009 M415 hervor und enthält im Vergleich zu M415 eine unbekannte Anzahl nicht charakterisierter Mutationen. Die Stämme Nal2ori und Nal2passage werden im Folgenden als Zwischenderivate bezeichnet, da sie in unterschiedlichen Stadien während der Entwicklung des Impfstammes entstanden sind. Dabei stammt die Mutante Nal2ori direkt aus der Selektion während der Entwicklung des Impfstammes. Der Stamm Nal2passage ist in einem gesonderten Versuch aus einem Seitenzweig aus Nal2ori entstanden. Der Impfstamm vacT ist nach mehreren Selektionsschritten aus dem Zwischenderivat Nal2ori entwickelt worden. Als Ausgangspunkt für die weiteren Untersuchungen erfolgte zunächst eine phänotypische Charakterisierung der Salmonella-Stämme, die Bestimmungen der Generationszeiten (Abschnitt 3.1.1), Bestimmungen der minimalen Hemmkonzentrationen (MHK) von Vertretern verschiedener Antibiotika-Klassen (Abschnitt 3.1.2) und Messungen der Superspiralisierungsgrade der DNA (Abschnitt 3.1.3) beinhaltete. Im Anschluss wurden für eine genotypische Charakterisierung der Untersuchungsstämme verschiedene Gene sequenziert (Abschnitt 3.1.4). Um eine genauere Aussage über das mögliche Auftreten von Mutationen unter Selektionsdruck im Stamm M415 und die Rolle der im Impfstamm vacT vorkommenden Mutationen treffen zu können, wurden mittels In-vitroSelektion Einschrittmutanten aus dem Ausgangsstamm M415 hergestellt. Mit dem Impfstamm vacT wurde ebenfalls versucht eine In-vitro-Selektion durchzuführen, mit dem Ziel, Aussagen über die Sicherheit und das Verhalten des Impfstammes zu treffen, wenn er in der Umwelt weiterem Selektionsdruck ausgesetzt ist (Abschnitt 3.1.5). Um zu zeigen, welche Rolle die bei der genotypischen Charakterisierung gefundenen gyrA-Mutationen spielen, wurde ein Komplementationstest mit einem plasmidcodierten sensiblen gyrAWildtyp-Gen durchgeführt Charakterisierung der (siehe Abschnitt Untersuchungsstämme 3.1.6). wurde Als globaler weiterhin Ansatz zur begonnen, die Proteinexpressionsmuster der Stämme mittels 2D-Gelelektrophorese zu vergleichen (Abschnitt 3.1.7). 81 Ergebnisse 3.1.1 Bestimmung der Generationszeiten Die Wachstumsgeschwindigkeit kann als ein Maß für die Fitness der Stämme angesehen werden. Bestimmte Mutationen, vor allem in essentiellen Genen, können sich auf die Wachstumseigenschaften der Bakterien auswirken und so deren Fitness und Konkurrenzfähigkeit in der Umwelt beeinflussen. Im Rahmen dieser Arbeit wurden die Generationszeiten der Stämme M415, Nal2ori, Nal2passage und des Impfstammes vacT, wie in Abschnitt 2.2.1.4 beschrieben, mittels Lebendzellzahl-Bestimmung ermittelt. Für jeden Stamm erfolgte eine Doppelbestimmung. Die Abbildung 3-1 zeigt die berechneten Regressionsgeraden (logKBE/ml gegen die Zeit). Abbildung 3-1: Vergleich der Regressionsgeraden für die untersuchten Stämme Für alle Stämme wurde eine Doppelbestimmung (1) und (2) durchgeführt. Aus der bestimmten logKBE/ml gegen die Zeit während der exponentiellen Phase wurden die Regressionsgeraden berechnet, deren Anstieg (m) der Berechnung der Generationszeit (g) zugrunde liegt. Die Generationszeit g (in Minuten) wurde über den Anstieg der Regressionsgeraden errechnet, die durch Auftragung der logKBE/ml über der Zeit bestimmt wurden (Tabelle 31). Die einbezogenen Daten stammen aus der exponentiellen Wachstumsphase. 82 Ergebnisse Tabelle 3-1: Generationszeit der untersuchten Stämme* in Minuten * M415 Nal2ori Nal2passage vacT 1. Versuch 14,69 44,17 20,12 30,34 2. Versuch 13,17 42,66 18,43 32,7 gerundeter Durchschnitt 14 43 19 32 Prozent [%] 100 307 136 229 Untersuchungsbedingungen: 400 ml Versuchsansatz, LB-Medium, 37 °C, 225 upm Die vier untersuchten Stämme Wachstumsgeschwindigkeit. Unter unterscheiden den sich durchgeführten z.T. sehr stark in ihrer Untersuchungsbedingungen (siehe Abschnitt 2.2.1.4) wies der Ausgangsstamm M415 eine Generationszeit von ca. 14 min auf. Mit 32 min besaß der Impfstamm eine ungefähr doppelt so lange Generationszeit wie der Ausgangsstamm. Die längste Generationszeit von 43 min (Verdreifachung im Vergleich zum M415) wurde für das Zwischenderivat Nal2ori ermittelt. Interessanterweise zeigt der Stamm Nal2passage mit einer Generationszeit von 19 min im Gegensatz zu Nal2ori (43 min) eine Fitnesskompensation. Die Anpassung der Generationszeit spielt für die Entwicklung eines Impfstammes eine zentrale Rolle. Die Generationszeit des Impfstammes vacT wurde während der Entwicklung an die Wirtsspecies Huhn angepasst. 3.1.2 Bestimmung der Empfindlichkeiten gegenüber verschiedenen Antibiotika Zur Ermittlung der Empfindlichkeit der Stämme M415, Nal2ori, Nal2passage und des Impfstammes vacT unterschiedlichen wurden die Vertretern minimalen Hemmkonzentrationen verschiedener (MHKs) Antibiotika-Klassen von mittels Mikrobouillondilutionsmethode (Abschnitt 2.2.1.6) bestimmt. Tabelle 3-2 gibt die MHK-Werte in µg/ml von ausgewählten Chinolonen und Fluorchinolonen wieder. Es handelt sich dabei um Antibiotika, die als Hemmer der DNAGyrase wirken und somit die DNA-Replikation stören. Die MHK-Werte für den Ausgangsstamme M415 wichen nicht von denen des Referenzstammes ATCC14028 ab. Nal2ori und Nal2passage zeigen gegenüber den meisten Fluorchinolonen wie z.B. Ciprofloxacin, Gatifloxacin, Levofloxacin oder Norfloxacin leicht reduzierte Empfindlichkeiten. 83 Ergebnisse Tabelle 3-2: Minimale Hemmkonzentrationen [µg/ml] ausgewählter Gyrasehemmer Antibiotikum ATCC14028 M415 Nal2ori Nal2passage vacT Nalidixinsäure 2 4 512 64 32 Norfloxacin 0,125 0,125 0,5 0,25 0,125 Ofloxacin 0,125 0,125 0,25 0,25 0,06 Levofloxacin 0,06 0,03 0,125 0,125 0,03 Ciprofloxacin 0,03 0,03 0,125 0,125 0,03 Sparfloxacin 0,03 0,03 0,125 0,06 0,006 Fleroxacin 0,06 0,06 0,5 0,5 0,125 Pefloxacin 0,125 0,125 0,5 0,5 0,125 Gatifloxacin 0,06 0,03 0,125 0,125 0,03 Sitafloxacin 0,03 0,015 0,06 0,125 ≤0,015 Moxifloxacin 0,125 0,125 0,25 0,25 0,06 Trovafloxacin 0,06 0,06 0,25 0,25 0,03 Grepafloxacin 0,06 0,03 0,25 0,5 ≤0,015 Gemifloxacin 0,06 0,03 0,125 0,125 ≤0,015 Clinafloxacin 0,03 0,03 0,06 0,06 0,03 In der Tabelle 3-3 sind die MHK-Werte für eine Auswahl von Antibiotika zusammengefasst, deren Wirkung auf der Hemmung der Proteinbiosynthese beruht. Bei diesen Antibiotika war erkennbar, dass der Impfstamm im Vergleich zu seinem Ausgangsstamm M415 und dem Vorgängerstamm Nal2ori empfindlich gegenüber Makroliden und Ketoliden reagierte. Salmonella spec., als gramnegative Erreger, besitzen durch die äußere Membran, die eine Permeationsbarriere für größere Moleküle darstellt, eine natürliche Resistenz gegenüber Makrolidantibiotika (vgl. ATCC14028 und M415). Die MHK-Werte für vacT waren gegenüber dem Ausgangsstamm M415 für Makrolide um mindestens vier Verdünnungsstufen erniedrigt. MHK-Wert-Schwankungen um +/- eine Verdünnungsstufe werden als Fehlerbereich gewertet. 84 Ergebnisse Tabelle 3-3: Minimale Proteinbiosynthese Hemmkonzentrationen [µg/ml] ausgewählter Hemmer der Antibiotikum Gruppe ATCC14028 M415 Nal2ori Nal2passage vacT Erythromycin Makrolid 64 ≥64 32 ≥64 4 Roxithromycin Makrolid ≥64 ≥64 ≥64 ≥64 4-8 Azithromycin Makrolid 2-4 4 1 2 0,25 Clarithromycin Makrolid 64 32 16 64 1 Tylosin Makrolid 128 256 32 32 8 Telithromycin Ketolid 8 8 4 4 0,125 ABT-773 Ketolid 4 8 2 4 0,25 Linezolid Oxazolidinon ≥64 ≥64 64 ≥64 8 Clindamycin Lincosamin ≥32 ≥32 ≥32 ≥32 1 Lincomycin Lincosamin ≥32 ≥32 ≥32 ≥32 32 Synercid Streptogramin 64 64 32 64 4 Tetracyclin Tetrayclin 1 1 1 1 0,25 Doxycyclin Tetrayclin 2 2 1 1 0,125 Minocyclin Tetrayclin 2 2 1 1 0,125 Chloramphenicol Chloramphenicol 4 4 2 2 1 Amikacin Aminoglykosid 1 1 2 2 2 Gentamicin Aminoglykosid 0,5 0,5 0,5 1 1 Kanamycin Aminoglykosid 2 2-4 2 2 2 Neomycin Aminoglykosid 1 1 1 1 1 Tobramycin Aminoglykosid 0,25 0,25 1 0,5 1 Streptomycin Aminoglykosid 8 16 16 8 32 Apramycin Aminoglykosid 2 4 4-8 2 2 Fusidinsäure Fusidinsäure ≥32 ≥32 ≥32 ≥32 16 In der letzten Gruppe der getesteten Antibiotika (Tabelle 3-4) finden sich z.B. Glykopeptide und Fosfomycin, welche die Mureinsynthese in der Zellwand der Bakterien stören. Einen anderen Wirkungsmechanismus, nämlich die Hemmung des Enzyms Dihydropteroatsynthetase, und damit die Hemmung der Synthese von Folsäure in Bakterien findet man bei Sulfonamiden. Rifampicin als Vertreter der Ansamycine hemmt die DNA-abhängige RNA-Polymerase und damit den Transkriptionsvorgang. In dieser Gruppe der getesteten Antibiotika gab es nur im Fall der Empfindlichkeit gegenüber Rifampicin eine Veränderung der MHK-Werte zwischen Impfstamm und Ausgangsstamm. Der Impfstamm vacT zeigte eine MHK-Wert-Erhöhung um mindestens zwei Verdünnungsstufen gegenüber dem Ausgangsstamm. 85 Ergebnisse Tabelle 3-4: Minimale Hemmkonzentration [µg/ml] ausgewählter Hemmer der Zellwand- und Folsäuresynthese und Sonstige Antibiotikum Gruppe ATCC 14028 M415 Nal2ori Nal2passage vacT Teicoplanin Glykopeptid ≥128 ≥128 ≥128 ≥128 ≥128 Vancomycin Glykopeptid ≥64 ≥64 64 ≥64 ≥64 Fosfomycin Fosfomycin 0,05 8 8 32 128 Sulfamethoxazol Sulfonamid ≥512 ≥512 ≥512 ≥512 ≥512 Trimethoprim Sulfonamid 0,25 0,125 0,25 0,125 0,25 Cotrimoxazol Sulfonamid 1 0,5 1-2 1 1 Rifampicin Ansamycin 8 16 8 8 ≥32 Zusammenfassend läßt sich sagen, dass der Impfstamm sich durch eine Makrolidsensibilität auszeichnete und eine Rifampicin- und Nalidixinsäureresistenz besaß. Bei den Fluorchinolonen zeigten die Zwischenderivate leicht erniedrigte Empfindlichkeiten. Der Impfstamm vacT hingegen war mindestens ebenso sensibel wie der Ausgangsstamm M415. In Einzelfällen (z.B. Sparfloxacin) zeigte der Impfstamm aber MHK-Werte, die unter denen des Ausgangsstammes lagen. Dies zeigt die Supersensibilität des Impfstammes vacT auf Fluorchinolone. 3.1.3 Bestimmung der DNA-Superspiralisierungsgrade Veränderungen in den Topoisomerase-Genen, d.h. Mutationen in den Genen gyrA, gyrB, parC und parE, senken nicht nur die Empfindlichkeit der Mutante gegenüber (Fluor)chinolonen, sondern können auch veränderte Enzymaktivität zur Folge haben. Veränderte Enzymaktivitäten der Gyrase ziehen Abweichungen des DNA- Superspiralisierungsgrades nach sich. Um diese zu messen, wurden plasmidbasierte Reportergensysteme verwendet. Im Rahmen dieser Arbeit wurden zwei Systeme genutzt: zum einen das System basierend auf dem Klonierungsvektor pBR322 mit den Reportergenplasmiden pPHB90 und pPHB91 und zum anderen das System ausgehend vom Vektor pBP507 mit den Plasmiden pPHB94 und pPHB95 [1]. Alle Plasmide enthalten das luc-Gen, welches mit den entsprechenden Promotoren der Gene der A-Untereinheiten der Topoisomerase I oder II aus E. coli fusioniert wurde. Eine Aktivierung des entsprechenden Promotors führt zur Expression des plasmid-codierten Luciferase-Gens (Abschnitt 2.2.1.9). Dessen Aktivität, als Maß der Promotoraktivität, wurde durch Reaktion mit zugesetztem Luciferin in Form des entstehenden Lichtes als RLUs (relativ light units) im Luminometer gemessen. Aus dem Verhältniss der spezifischen Aktivitäten (RLU / OD546nm) der 86 Ergebnisse jeweiligen Promotoren (RLU ptopA / OD546 zu RLU pgyrA / OD546) wurde der Qsc-Wert (Quotient of supercoiling) berechnet. Um den Qsc-Wert sicher zu bestimmen, wurden pro Stamm die Werte in drei unabhängigen Experimenten bestimmt. Für jedes einzelne Experiment wurden jeweils drei Proben pro Versuchskultur dreimal unabhängig voneinander vermessen. Von den ermittelten Qsc-Werten wird der Mittelwert mit Standardabweichung und zum besseren Vergleich die Werte in Prozent (inklusive relativer Standardabweichung) angegeben, wobei der Wert des Ausgangsstammes M415 100 % gesetzt wurde. Tabelle 3-5 enthält die mit den Reportergenplasmiden pPHB90 und pPHB91 gemessenen spezifischen Aktivitäten und die errechneten Qsc-Werte für die verwendeten Stämme. Tabelle 3-5: Daten aus den DNA-Superspiralisierungsmessungen Reportergenplasmiden pPHB90 und pPHB91 Stamm M415 Nal2ori Nal2pass vacT spezifische Aktivität ptopA* spezifische Aktivität pgyrA* Qsc** 1762,34 1149,55 1,53 2150,68 1194,40 1,80 8880,77 4252,14 2,08 2493,05 2309,66 1,08 2440,23 2632,86 0,93 3572,32 3808,83 0,94 2134,21 2408,38 0,89 1700,33 1799,63 0,94 3565,94 3071,50 1,16 3605,40 3631,58 0,99 5935,70 5316,16 1,12 3754,60 3179,16 1,18 mit den Mittelwert Qsc (± s) Prozent (± s %) 1,80 100 % (±0,28) (± 15,5) 0,98 54,44 % (± 0,08) (± 8,16) 0,99 55 % (± 0,14) (± 14,14) 1,09 60,55 % (± 0,09) (± 8,26) * spezifische Aktivität ptopA bzw. pgyrA = RLU / OD546nm; ** Qsc = spezifische Aktivität ptopA / spezifische Aktivität pgyrA; s = Standardabweichung, s % = relative Standardabweichung Zur Absicherung der Abweichungen der Superspiralisierungsgrade zwischen dem Ausgangsstamm und dem Impfstamm wurden beide Stämme mit weiteren Reportergenplasmiden (pPHB94 und pPHB95) vermessen (Tabelle 3-6). 87 Ergebnisse Tabelle 3-6: Daten aus den DNA-Superspiralisierungsmessungen Reportergenplasmiden pPHB94 und pPHB95 Stamm M415 vacT spezifische Aktivität ptopA* spezifische Aktivität pgyrA* Qsc** 8490,87 2508,21 3,39 8325,09 2166,59 3,84 4718,64 1376,42 3,43 4365,32 1687,38 2,59 5382,35 2008,07 2,68 3531,69 1217,35 2,90 mit den Mittelwert Qsc (± s) Prozent (± s %) 3,55 100 % (±0,25) (±7,04) 2,72 76,62 % (±0,16) (±5,88) * spezifische Aktivität ptopA bzw. pgyrA = RLU / OD546nm; ** Qsc = spezifische Aktivität ptopA / spezifische Aktivität pgyrA; s = Standardabweichung, s % = relative Standardabweichung 3.1.4 DNA-Sequenzanalyse der Gene der Fluorchinolon-Zielstrukturen und cpxA Die Ergebnisse aus den Empfindlichkeitsmessungen gegenüber verschiedenen Antibiotika und die veränderten Superspiralisierungswerte weisen auf mögliche Mutationen in den Genen für die Typ II-Topoisomerasen Gyrase (gyrA und gyrB) und Topoisomerase IV (parC und parE) hin, die aus diesem Grund vollständig sequenziert wurden. Weiterhin wurde das Gen cpxA sequenziert, da in einer Publikation ein Hinweis auf Mutation(en) in diesem Gen im Stamm vacT gegeben war [261]. Die verwendeten Primer sind der Tabelle 2-4 im Abschnitt 2.1.3 zu entnehmen. Tabelle 3-7 enthält die Daten aus den Sequenzierungen für die Stämme M415, Nal2ori, Nal2passage und vacT. Das Primerdesign und die Auswertung der Sequenzen über Alignments wurden im Vergleich mit der Referenzsequenz des Stammes S. Typhimurium LT2 (GenBank accession number NC_003197) unter Verwendung der Software Vector NTI (Informax) durchgeführt. Bei der Sequenzierung der Targetgene des Impfstammes vacT konnten im Vergleich zum Ausgangsstamm M415 nur im Gen für die A-Untereinheit der Gyrase gyrA vier Basenaustausche festgestellt werden, welche auch alle zu einem Aminosäureaustausch führen. Von den vier in gyrA gefundenen Aminosäureaustauschen liegen zwei (Gly-75 Æ Ala und Asp-87 Æ Gly) innerhalb der QRDR. Zwei weitere Mutationen (Trp-59 Æ Arg und Ala866 Æ Ser) liegen außerhalb der QRDR. In den Stämmen Nal2ori und Nal2passage konnten im gyrA-Gen nur drei der vier im Impfstamm auftretenden Mutationen gefunden werden. Die Mutation Trp-59Æ Arg tritt erst im Impfstamm vacT auf. In Tabelle 3-7 sind alle in den entsprechenden Untersuchungsstämmen gefundenen Sequenzabweichungen aufgeführt. 88 Ergebnisse betroffene Aminosäureposition vacT 2713 bp Basenaustausch Nal2pass gyrA Größe des sequenzierten Fragmentes Nal2ori Gen M415 Tabelle 3-7: Sequenzabweichungen von der Referenzsequenz von S. TyphiumuriumLT2 TGG Æ AGG Trp-59 Æ Arg - - - x GGT Æ GCT Gly-75 Æ Ala - x x x GAC Æ GGC Asp-87 Æ Gly - x x x GCG Æ TCG Ala-866 Æ Ser - x x x gyrB 2739 bp - - - - - - parC 2370 bp - - - - - - parE 2014 bp - - - - - - 1710 bp CTA Æ CTG Leu-250 Æ Leu x x x x cpxA CTA Æ ATA Leu-313 Æ Ile x x x x In Abbildung 3-2 sind die drei Mutationen in bzw. nahe der QRDR anhand der ermittelten Sequenzdaten von M415 und vacT dargestellt. In den Genen gyrB, parC und parE wurden keine Sequenzabweichungen bei vacT im Vergleich zu M415 festgestellt. Abbildung 3-2: Darstellung der abweichenden Sequenzdaten der Stämme M415 und vacT in bzw. nahe der QRDR von gyrA (Mutationen an Aminosäureposition 59, 75 bzw. 87) Im Gen cpxA wurden im Vergleich zur Referenzsequenz (S. Typhimurium LT2) zwei Basenaustausche in allen vier Untersuchungsstämmen, also auch dem Ausgangsstamm M415, gefunden. Nur einer dieser beiden Basenaustausche führte auch zu einem Aminosäureaustausch. 89 Ergebnisse 3.1.5 In-Vitro-Selektionsversuche 3.1.5.1 Selektionsversuch mit S. Typhimurium M415 M415 diente als Ausgangsstamm für die Entwicklung des Impfstammes vacT. Bei der Sequenzierung des gyrA-Gens von vacT wurden vier Mutationen gefunden, von der die Mutation an Aminosäureposition Aspartat-87 als Chinolonresistenz vermittelnd bekannt ist [24,48,108,118,128,236,237,253]. Der Impfstamm vacT zeigte aber eine Supersensibilität auf einige Fluorchinolone (siehe Abschnitt 3.1.2 Tabelle 3-2). Da der Impfstamm über ungezielte Mutagenese entstanden ist, sollte in diesem In-vitro-Versuch untersucht werden, welche Mutationen in M415 unter Selektionsdruck im Labor auftreten würden. Zentrale Frage war, ob die aus dem Impfstamm bekannten Mutationen durch In-vitroSelektion hergestellt werden könnten und wie sich selektierte Mutanten verhalten. Um chinolonresistente Keime zu isolieren, wurde die Selektion mit M415 als Ausgangsstamm auf Ciprofloxacin-haltigen Agarplatten durchgeführt (siehe Abschnitt 2.2.1.5). Neben der Ermittlung der Mutationsfrequenz wurden im Anschluss der Resistenzgenotyp (Sequenzierung) und der Resistenzphänotyp (MHK-Wert-Bestimmung) bestimmt. Zur Selektion wurden Konzentrationen verwendet, die der 2-, 4-, 6-, 8- bzw. 12-fachen MHK des Ausgangsstammes M415 entsprachen. Das bedeutet Ciprofloxacin- Konzentrationen von 0,06 µg/ml, 0,12 µg/ml, 0,18 µg/ml, 0,24 µg/ml bzw. 0,36 µg/ml. Für diesen Selektionsversuch wurde eine Ausgangskeimzahl von 1,71 x 1011 eingesetzt, mit Hilfe derer die Mutationsfrequenzen (Berechnung siehe Abschnitt 2.2.1.5) ermittelt werden konnten. Tabelle 3-8 zeigt die Mutationsfrequenzen dieses In-vitro-Selektionsversuches. Tabelle 3-8: Mutationsfrequenzen aus der In-vitro-Selektion von M415 auf Ciprofloxacin (CIP) * xfacher MHK-Wert* Selektionskonzentration CIP [µg/ml] Gesamtkeimzahl Summe selektierter Mutanten** Mutationsfrequenz 2x 0,06 1,71 x 1011 272 1,27 x 10-9 4x 0,12 1,71 x 1011 57 2,67 x 10-10 6x 0,18 1,71 x 1011 32 1,49 x 10-10 8x 0,24 1,71 x 1011 50 2,34 x 10-10 12x 0,36 1,71 x 1011 28 1,31 x 10-10 bezogen auf die MHK für den Ausgangsstamm M415; ** bezogen auf Wachstum nach 48 h Ausgewählte Mutanten wurden gereinigt und einem Stabilitätstest (siehe Abschnitt 2.2.1.5) unterzogen. Insgesamt wurden 14 Mutanten weiter untersucht. Es folgte zunächst eine Empfindlichkeitsbestimmung gegenüber Ciprofloxacin mittels Agardilutionsverfahren (siehe Abschnitt 2.2.1.6.4). Alle 14 untersuchten Mutanten zeigten MHK-Werte von Ciprofloxacin von ≥ 0,12 µ/ml. Alle genauen Werte sind der Tabelle 3-9 zu entnehmen. 90 Ergebnisse Tabelle 3-9: Minimale Hemmkonzentration [mg/ml] von Ciprofloxacin ausgewählter Selektionsmutanten von M415 SK * MHK * ** 1 2 3 4 5 6 7 8 9 10 11 12 13 14 0,06 0,06 0,06 0,06 0,12 0,12 0,12 0,12 0,18 0,18 0,18 0,24 0,24 0,24 0,18 0,18 0,18 0,18 0,24 0,18 0,72 0,12 0,12 0,24 0,24 0,18 0,18 0,24 SK = Selektionskonzentration CIP [µg/ml]; ** MHK bestimmt mittels Agardilutionsverfahren Mittels Restriktionsfragmentlängenpolymorhismus (RFLP) wurde über eine HinfIRestriktionsspaltung ein schnelles Screening in Hinsicht darauf vorgenommen, ob bei den selektierten Mutanten ein Basenaustausch an den Codons Asp82/Ser83 vorliegt (siehe Abschnitt 2.2.2.9.2). Im Fall einer Mutation an Position Ser83 entstanden durch den Verlust einer Schnittstelle nur zwei Fragmente der ungefähren Größe 390 bp und 140 bp. Abbildung 3-3 zeigt das Ergebnis des RFLP-Screening von 14 selektierten Mutanten. Die Wildtyp-Kontrolle (Verwendung des Ausgangsstammes M415) erfolgte in einem separaten Agarosegel (Daten nicht gezeigt) und zeigte nach der Restriktionsspaltung DNAFragmente der Größe 290 bp, 140 bp und 100 bp. Abbildung 3-3: Ergebnis des RFLP-Screenings der selektierten Mutanten aus der In-vitroSelektion von M415 auf Ciprofloxacin Pfeile kennzeichnen die Mutanten 7 und 14, welche nach der Restriktionsspaltung mit dem Enzym HinfI durch Verlust einer Erkennungssequenz aufgrund eines Basenaustausches im Codon Ser83 Fragmentgrößen von ca. 390 bp und 140 bp aufweisen; die übrigen Mutanten zeigen die Wildtypsituation mit Fragmentgrößen von ca. 290 bp, 140 bp und 100 bp; SL = : Smart Ladder Es konnten von den 14 getesteten Selektionsmutanten zwei Mutanten identifiziert werden, die mit hoher Wahrscheinlichkeit eine Mutation im Codon Ser83 besitzen, welches als „Hotspot“ für Mutationen zur Fluorchinolonresistenz bekannt ist. Diese Vermutung konnte durch Sequenzanalyse der QRDRs der gyrA-Gene bestätigt werden. Die untersuchten 91 Ergebnisse Mutanten enthielten Mutationen in der QRDR von gyrA. Betroffene Codons waren Gly81Asp, Ser83Phe und Asp87Tyr (Tabelle 3-10). Tabelle 3-10: Ergebnisse aus den Sequenzierungen der gyrA-QRDR ausgewählter Selektionsmutanten von M415 auf Ciprofloxacin Mutante 3 7 14 Sonstige Gly81Asp Ser83Phe Ser83Phe Asp87Tyr GGCÆ GAC TCCÆ TTC TCCÆ TTC GACÆ TAC Codon Basenaustausch Im Folgenden werden die entsprechenden Mutanten nach ihren Mutationen benannt und mit einem „S“ versehen, das ihre Herkunft aus dem Selektionsversuch kennzeichnet: Mutante 1 (Asp87Tyr) = SM415-87, Mutante 3 (Gly81Asp) = SM415-81 und Mutante 7 (Ser83Phe) = SM415-83. Nach der Genotypbeschreibung der Selektionsmutanten wurde zur näheren Bestimmung des Phänotyps eine Empfindlichkeitsbestimmung auf unterschiedliche (Fluor)chinolone durchgeführt (Tabelle 3-11). Tabelle 3-11: Minimale Hemmkonzentrationen [µg]ml] verschiedener (Fluor)chinolone der Selektionsmutanten Antibiotikum ATCC14028 M415 SM415-81 SM415-83 SM415-87 gyrA-Mutation keine keine Gly81Asp Ser83Phe Asp87Tyr Nalidixinsäure 2 4 64 512 64 Norfloxacin 0,125 0,125 1 2 1 Ofloxacin 0,125 0,125 0,5 1 0,25 Levofloxacin 0,06 0,03 0,25 1 0,125 Ciprofloxacin 0,03 0,03 0,25 0,5 0,25 Sparfloxacin 0,03 0,03 0,25 1 0,06 Fleroxacin 0,06 0,06 1 2 0,5 Pefloxacin 0,125 0,125 1 8 0,5 Gatifloxacin 0,06 0,03 n.b. n.b. 0,06 Sitafloxacin 0,03 0,015 n.b. n.b. 0,03 Moxifloxacin 0,125 0,125 n.b. n.b. 0,125 Trovafloxacin 0,06 0,06 n.b. n.b. 0,125 Grepafloxacin 0,06 0,03 n.b. n.b. 0,125 Gemifloxacin 0,06 0,03 n.b. n.b. 0,125 Clinafloxacin 0,03 0,03 n.b. n.b. 0,06 n.b. = nicht bestimmt, SM415 = Mutante aus M415 durch Selektion hervorgegangen 92 Ergebnisse Alle drei untersuchten gyrA-Mutanten zeigten erniedrigte Empfindlichkeiten gegenüber Fluorchinolonen. Zusammenfassend lässt sich festhalten, dass bei der In-vitro-Selektion mit S. Typhimurium M415 auf Ciprofloxacin Mutanten mit reduzierter Empfindlichkeit gegenüber (Fluor)chinolonen selektiert werden konnten. Alle untersuchten Mutanten wiesen eine Mutation in der QRDR von gyrA auf. Die betroffenen Codons waren Gly81Asp, Ser83Phe und Asp87Tyr. Diese Aminosäurepositionen sind aus früheren Selektionsversuchen bereits als Fluorchinolonresistenz vermittelnd bekannt. Keine der im Impfstamm vorkommenden gyrA-Mutationen konnte selektiert werden. Allerdings besitzen sowohl der Impfstamm als auch die Selektionsmutante SM415-87 eine Veränderung an Aminosäureposition Asp87, wobei aber das Aspartat beim Impfstamm durch Glycin und bei der Selektionsmutante durch Tyrosin ausgetauscht ist. Die Mutante SM415-87 wurde in den folgenden Versuchen als Referenz mitgeführt. 3.1.5.2 Selektionsversuch mit TAD Salmonella vacT Der Impfstamm TAD Salmonella vacT wird als Lebendimpfstoff für Hühner eingesetzt. Bei der genotypischen Charakterisierung wurden vier Mutationen im gyrA-Gen bestimmt (Abschnitt 3.1.4). Trotz dieses gehäuften Auftretens von Mutationen in gyrA, inklusive der Mutation an Aminosäureposition Ser87, welche als Fluorchinolonresistenz vermittelnd bekannt ist, ergab die phänotypische Charakterisierung des Impfstammes keine erhöhten MHK-Werte der Fluorchinolone im Vergleich zu dem Ausgangsstamm M415. Lediglich der MHK-Wert der Nalidixinsäure ist erhöht. Diese Ergebnisse werfen die Frage auf, wie sich der Impfstamm unter Selektionsdruck in der Umwelt verhalten würde. Um die Frage zu beantworten, ob sich aus dem Impfstamm ein Stamm mit Fluorchinolonresistenz entwickeln könnte (durch ein Auftreten entweder weiterer gyrA-Mutationen oder Mutationen in anderen Genen), wurde ein Versuch der In-vitro-Selektion von TAD Salmonella vacT auf verschiedenen (Fluor)chinolonen durchgeführt. Im Rahmen dieser Arbeit wurde mehrfach versucht, den Impfstamm auf Ciprofloxacin (2 bis 8 x MHK) zu selektieren. Bei den Experimenten wurde vorgegangen wie unter Abschnitt 2.2.1.5 beschrieben. Allerdings konnte auch nach mehreren Tagen Inkubation kein Mutantenwachstum festgestellt werden. Der Versuch der In-vitro-Selektion auf Ciprofloxacin mit vacT wurde zur Absicherung dreimal durchgeführt. In allen Fällen konnten keine Mutanten selektiert werden. Auch der Versuch der In-vitro-Selektion von vacT auf 64 bis 256 µg/ml Nalidixinsäure (2 bis 8 x MHK) war in früheren Experimenten (H. Hansen, pers. Mitteilung) erfolglos geblieben. Es gelang Hansen allerdings die Selektion von Mutanten bei dem Einsatz von Enrofloxacin (pers. Mitteilung). Ausgewählte zur Verfügung gestellte Mutanten (vacTE-1 bis -3, Tabelle 2-2) wurden im Rahmen dieser Arbeit näher charakterisiert. Die Mutanten wurden dazu wie folgt benannt: Mutante 1 (selektiert auf 0,125 µg/ml) = vacTE1, Mutante 2 (selektiert auf 0,125 µg/ml) = vacTE2, 93 Ergebnisse Mutante 3 (selektiert auf 0,06 µg/ml) = vacTE3 (siehe Tabelle 3-12). Zunächst wurden die MHK`s für eine Auswahl von (Fluor)chinolonen bestimmt (Tabelle 3-12). Tabelle 3-12: Minimale (Fluor)chinolon-Hemmkonzentrationen Selektionsmutanten von vacT auf Enrofloxacin vacT Nr. der Mutante - Name* SK [µg/ml] ausgewählter vacT Enro vacT Enro vacT Enro vacT Enro vacT Enro vacT Enro vacT Enro vacT Enro 0,125-1 0,125-2 0,06-1 0,06-2 0,06-3 0,06-4 0,06-5 0,06-6 1 2 3 vacTE1 vacTE2 vacTE3 0,125 0,125 4 5 6 7 8 0,06 0,06 0,06 0,06 0,06 0,06 NAL 32 128 128 64 64 64 64 64 64 NOR 0,125 1 0,5 0,5 0,5 0,5 0,25 0,5 0,5 LEV 0,03 0,25 0,25 0,125 0,125 0,125 0,125 0,125 0,25 CIP 0,03 0,125 0,125 0,125 0,06 0,125 0,125 0,125 0,125 SPA ≤0,015 0,125 0,125 0,06 0,06 0,06 0,06 0,06 0,06 * Name = Benennung der im Rahmen dieser Arbeit verwendeten Stämme; SK = Selektionskonzentration Enrofloxacin in µg/ml; NAL = Nalidixinsäure; NOR = Norfloxacin; LEV = Levofloxacin: CIP = Ciprofloxacin; SPA = Sparfloxacin; Wie aus der Tabelle 3-12 ersichtlich, konnten durch eine Selektion auf Enrofloxacin Stämme mit reduzierter Empfindlichkeit gegenüber Fluorchinolonen aus dem Impfstamm vacT erzeugt werden. Die Sequenzierung der QRDRs der Targetgene gyrA, gyrB und parC dieser Stämme ergaben keine abweichenden Mutationen im Vergleich zum Selektionsausgangsstamm vacT (H. Hansen, pers. Mitteilung). Im Rahmen dieser Doktorarbeit erfolgte eine Sequenzierung der QRDR von parE, die aber ebenfalls keine Abweichung von vacT ergab. Demzufolge konnten Veränderungen in den Targetgenen als Ursache für die erhöhten MHK-Werte gegenüber den Fluorchinolonen weitestgehend ausgeschlossen werden. Eine weitere Möglichkeit der Veränderungen der Empfindlichkeiten gegenüber bestimmten Antibiotika ist die veränderte Expression von Effluxpumpen. Aus diesem Grund erfolgte in dieser Arbeit eine Bestimmung der minimalen Hemmkonzentrationen mit und ohne Effluxpumpeninhibitor (EPI) Phenylalanyl-Arginyl-β-Naphthylamid (Tabelle 3-13). Die Bestimmung der MHK-Werte für die Selektionsmutanten umfasste auch Makrolidantibiotika. Auffallend war, dass die vom Impfstamm bekannte Makrolidsensibilität bei den Selektionsmutanten wieder revertiert war. Die drei getesteten Selektionsmutanten besaßen eine Makrolidresistenz (siehe z.B. Erythromycin und Roxithromycin). Aus der Tabelle 3-13 ist auch zu entnehmen, dass die MHK-Werte für die Selektionsmutanten mit Zugabe des Effluxpumpeninhibitors auf das Niveau des Impfstammes vacT zurückgingen, der als Ausgangsstamm für die Selektion diente. 94 Ergebnisse Tabelle 3-13: Minimale Hemmkonzentrationen [µg/ml] mit und ohne EPI* vacT vacTE1 vacTE2 vacTE3 ohne EPI ohne EPI mit EPI ohne EPI mit EPI ohne EPI mit EPI ERY 4 64 4-16 32 0,5 16 2 ROXI 4-8 64 4-16 64 0,125 32 4 AZIT 0,25 2 0,125 0,5 ≤0,03 0,5 0,06 CLAR 1 32 2 32 ≤0,03 8 1 CLIN 1 ≥32 2 ≥32 1 8-32 4 LIN 32 ≥32 ≥32 ≥32 ≥32 ≥32 ≥32 LIV 4 4 2 4 2 2-4 2 RPC 4 64 4 64 0,125 32 2 NDS 32 128 16 32 4 32-64 16 NOR 0,25 0,5 0,5 0,5 0,25 0,25 0,5 OFL 0,125 0,5 0,125 0,25 0,06 0,25 0,125 LEV 0,06 0,25 0,06 0,125 0,03 0,125 0,06 CIP 0,03 0,25 0,06 0,125 0,03 0,125 0,06 SPA ≤0,015 0,125 ≤0,015 0,03 ≤0,015 0,06 ≤0,015 FLEX 0,25 0,5 0,25 0,5 0,03 0,25 0,25 PEF 0,125 1 0,25 0,5 0,125 0,5 0,125 * EPI = Effluxpumpeninhibitor Phenylalanyl-Arginyl-β-Naphthylamid zugesetzt in einer Konzentration von 8µg/ml; Antibiotika-Abkürzungen siehe Tabelle 2-16 95 Ergebnisse 3.1.6 Komplementationstest mittels eines plasmidcodierten gyrA+-Gens Ziel dieses Versuches war es zu zeigen, welche möglichen phänotypischen Auswirkungen die identifizierten gyrA-Mutationen in den Stämmen Nal2ori, Nal2passage und vacT haben. Das Prinzip dieses Experimentes war es, dass in einem Stamm mit zwei Kopien des gyrA-Gens der Phänotyp, wie z.B. die Chinolonempfindlichkeit, durch die sensiblere Gyrase (gyrA+) bestimmt wird. Das heißt, es liegt Dominanz der sensiblen (gyrA+) über der weniger sensiblen (gyrA) Gyrase vor. Sind Mutationen im gyrA-Gen für erhöhte MHKWerte der Chinolone verantwortlich, werden durch die Expression eines nicht mutierten – sensiblen – gyrA-Gens die MHK-Werte wieder erniedrigt. Für diese Untersuchungen wurde das Plasmid pBP517 [123] verwendet, welches das gyrA-Wildtyp-Gen aus E. coli enthält. Es wurde also in den Salmonellen chromosomal ein mutiertes gyrA-Gen und plasmidcodiert ein unmutiertes gyrA+-Gen exprimiert. Als Kontrolle diente der Vektor pBP507 ohne gyrA-Gen. Die verwendeten Vektoren sind in Abbildung 2-1 Abschnitt 2.2.1.8 dargestellt. Die Plasmide pBP517 (gyrA+) und pBP507 (Vektorkontrolle) wurden durch Konjugation oder Transformation (Abschnitt 2.2.1.7) in die zu untersuchenden Salmonella-Stämme Antibiotikaempfindlichkeit, Superspiralisierungsgrade die für eingebracht. Anschließend Wachstumsgeschwindigkeiten die merodiploiden Zellen wurden sowie bestimmt. Um die die den Selektionsdruck für die Plasmide aufrecht zu erhalten, wurden alle Untersuchungen in Gegenwart von Kanamycin durchgeführt. 3.1.6.1 Bestimmung der Fluorchinolonempfindlichkeiten in Anwesenheit von gyrA+ Um sicher zu gehen, dass nicht nur durch die Verwendung eines gyrA+-Gens aus E. coli in den Salmonella-Stämmen eine MHK-Wert-Veränderung hervorgerufen wird und das Dominanzprinzip funktioniert [123], wurde der Selektionsstamm SM415-87 (siehe Abschnitt 3.1.5.1) als Kontrolle mitgeführt. Dieser Stamm besitzt leicht erhöhte MHKWerte der Fluorchinolone durch die selektierte gyrA-Mutation Asp87Tyr. Durch die dominante plasmidcodierte Gyrase müssen die MHK-Werte der Chinolone auf das Niveau des Ausgangsstammes M415 zurückgehen. In der folgenden Tabelle 3-14 sind alle MHK-Werte für die Stämme ohne Plasmid, mit der Plasmidkontrolle pBP507 und mit dem gyrA+-Plasmid pBP517 dargestellt. 96 - 4 0,125 0,125 0,03 0,03 0,03 0,06 0,125 0,03 0,015 0,125 0,06 0,03 0,03 0,03 Plasmide NDS NOR OFL LEV CIP SPA FLEX PEF GAT SIT MOX TFX GFX GMF CLIF Mutationen 0,03 0,03 0,06 0,125 0,06 0,03 0,03 0,125 0,06 0,06 0,03 0,06 0,125 0,125 4 pBP507 keine M415 0,03 0,06 0,06 0,06 0,125 0,015 0,03 0,125 0,06 0,03 0,03 0,06 0,125 0,125 4 pBP517 0,06 0,125 0,125 0,125 0,125 0,03 0,06 0,5 0,5 0,06 0,25 0,125 0,25 1 64 - n.b. n.b. n.b. n.b. n.b. n.b. n.b. 0,5 0,5 0,06 0,125 0,125 0,25 0,5 64 pBP507 Asp87Tyr n.b. n.b. n.b. n.b. n.b. n.b. n.b. 0,125 0,06 0,015 0,03 0,03 0,06 0,125 2 pBP517 SM415-87 0,06 0,125 0,25 0,25 0,25 0,06 0,125 0,5 0,5 0,125 0,125 0,125 0,25 0,5 512 - 0,06 0,125 0,25 0,25 0,25 0,06 0,125 0,5 0,5 0,125 0,125 0,125 0,25 0,5 512 pBP507 ≤0,015 0,015 0,015 0,03 0,03 ≤0,015 ≤0,015 0,06 0,03 ≤0,015 ≤0,015 0,015 0,03 0,06 4 pBP517 Gly75Ala, Asp87Gly Nal2ori 0,06 0,125 0,5 0,25 0,25 0,125 0,125 0,5 0,5 0,06 0,125 0,125 0,25 0,25 64 - 0,06 0,125 0,25 0,125 0,125 0,06 0,125 0,5 0,25 0,125 0,125 0,25 0,25 0,5 128 pBP507 ≤0,015 0,06 ≤0,015 0,06 0,06 0,03 0,06 0,06 0,06 ≤0,015 ≤0,015 0,06 0,125 0,125 16 pBP517 Gly75Ala, Asp87Gly Nal2passage 32 - vacT 0,03 ≤0,015 ≤0,015 0,03 0,06 ≤0,015 0,03 0,125 0,125 0,006 0,03 0,03 0,06 0,03 ≤0,015 ≤0,015 0,03 0,06 ≤0,015 ≤0,015 0,25 0,25 0,003 0,03 0,06 0,125 0,25 16 pBP507 <0,015 ≤0,015 ≤0,015 0,03 ≤0,015 ≤0,015 ≤0,015 ≤0,015 0,03 0,0007 0,0015 ≤0,015 0,03 0,015 0,5 pBP517 Trp59Arg, Gly75Ala, Asp87Gly 0,125 Tabelle 3-14: Ergebnisse der MHK-Wert-Bestimmungen der (Fluor)chinolone innerhalb des Komplementationstestes Ergebnisse Der Ausgangsstamm M415 zeigte in allen Fällen (mit und ohne Plasmide) die gleichen Empfindlichkeiten. Dies diente als Kontrolle dafür, dass die MHK-Werte durch die Anwesenheit der Plasmide nicht beeinflusst wurden. Die Werte des Stammes SM415-87 dienten zur Kontrolle der Funktionsweise des Komplementationstestes. Die Empfindlichkeiten dieser gyrA-Mutante lagen ohne und mit Kontrollplasmid je nach Fluorchinolon um zwei (z.B. Levofloxacin) bis drei (z.B. Flerofloxacin) Verdünnungsstufen über den Werten des Ausgangsstammes M415. Bei Nalidixinsäure war ein Anstieg um vier Verdünnungsstufen zu finden. Mit pBP517 (gyrA+) sanken die Werte auf das Niveau des Ausgangsstammes. GyrA+ aus E. coli zeigte also auch im Salmonella-Stamm den erwarteten Dominanzeffekt (vgl. [123,126]. Auch bei den Zwischenderivaten Nal2ori und Nal2passage, in denen ohne und mit Kontrollplasmid einen maximalen MHK-Anstieg von zwei bis drei Verdünnungsstufen zu finden war (nur bei Nalidixinsäure bei Nal2ori um sieben und Nal2passage um vier bis fünf Stufen), sanken die MHK-Werte mit pBP517 (gyrA+) auf das Ausgangsniveau zurück. Einige Werte lagen um eine Verdünnungsstufe unter dem Niveau des Ausgangsstammes, was hier aber als Schwankung in den MHK-Wert-Bestimmungen bewertet wurde. Wie bereits aus den MHK-Wert-Bestimmungen bekannt (Abschnitt 3.1.2), zeigte der Impfstamm vacT im Gegensatz zu den Zwischenderivaten keine Erhöhung der Fluorchinolon-MHK-Werte. Nach der Einführung von gyrA+ waren aber in fast allen Fällen die Empfindlichkeiten gegenüber den (Fluor)chinolonen im Vergleich zum Ausgangsstamm M415 nochmals stark erhöht. Zum Beispiel lagen die MHK-Werte der Nalidixinsäure, Norfloxacin, Levofloxacin, und Sparfloxacin um drei bis fünf Verdünnungsstufen unter dem Niveau des Ausgangsstammes M415. Mit Hilfe des Komplementationstestes konnte also gezeigt werden, dass die leicht reduzierten Empfindlichkeiten der Zwischenderivate Nal2ori und Nal2passage gegenüber Fluorchinolonen auf den gyrA-Mutationen beruhen, da mit Einführung von gyrA+ ein Absinken der Werte mindestens auf das Niveau des Ausgangsstammes erreicht werden konnte. Im Fall des Impfstammes vacT, der z.T. sogar leicht erhöhte Empfindlichkeiten gegenüber verschiedenen Fluorchinolonen zeigte, brachte die Einführung von gyrA+ aber noch ein zusätzliches Absinken der MHK-Werte sogar unter das Niveau des Ausgangsstammes M415. 3.1.6.2 Untersuchung von Wachstum und Qsc-Werten in Anwesenheit von gyrA+ Es wurde versucht, auch die Superspiralisierungsgrade und Generationszeiten im Rahmen des Komplementationstest zu bestimmen. Sollten die Reduktionen im Superspiralisierungsgrad (siehe Abschnitt 3.1.3) und der Wachstumsgeschwindigkeit (Abschnitt 3.1.1) auf den Mutationen im gyrA-Gen beruhen, würden in Anwesenheit einer sensiblen Gyrase die Werte wieder erhöht sein. 98 Ergebnisse Die Wachstumsgeschwindigkeiten wurden über die Messung der optischen Dichte in Abhängigkeit von der Zeit bestimmt (Abschnitt 2.2.1.4). Um sicher zu stellen, dass die Plasmide pBP507 (Kontrolle) und pBP517 (gyrA+) in den Stämmen präsent sind, wurde zum Medium Kanamycin (50µg/ml) zugesetzt. Der Stamm M415 wurde als interner Standard mitgeführt. Zur Auswertung wurden die ermittelten Generationszeiten prozentual ins Verhältnis gesetzt, wobei der Wert des Ausgangsstammes M415 jeweils auf 100 % gesetzt wurde. Die Generationszeiten unterschieden sich in den einzelnen Versuchen sehr stark. Die Wachstumskurven ließen sich schlecht auswerten. In Tabelle 3-15 ist der Trend dargestellt, der den Wachstumskurven entnommen werden konnte. Es ist zu erkennen, dass das Vorhandensein des sensiblen gyrA+-Gens (exprimiert von pBP517) zu keiner Wachstumsverbesserungen führte. Tabelle 3-15: Wachstumsverhalten unter Einfluss von gyrA+ M415 Plasmid g* [%] * Nal2ori Nal2passage vacT - - pBP507 pBP517 - pBP507 pBP517 - pBP507 pBP517 100 174 171 200 190 165 165 168 135 195 g = Generationszeit Auch die Ermittlung des Superspiralisierungsgrades unter den Bedingungen des Komplementationstestes erwies sich als problematisch. Offenbar stellte die Replikation zweier Plasmide in einer Zelle ein Problem dar. Bereits die Transformation mit den Reportergensystem-Plasmiden für die Qsc-Wert-Bestimmung war für die Stämme, welche die Plasmide für den Komplementationstest (pBP507 und pBP517) enthielten, schwierig. Um eine gemeinsame Replikation von zwei Plasmiden in der Zelle zu gewährleisten, müssen Plasmide mit unterschiedlichem Replikationsursprung verwendet werden. Deshalb wurden für die Bestimmung des Superspiralisierungsgrades pBR322-Derivate benutzt, da im Komplementationstest pBP507-Derivate verwendet wurden. Wegen der trotzdem aufgetretenen Schwierigkeiten bei der Transformation konnten nur wenige Stämme mit beiden Plasmiden erhalten werden. Von diesen wurden die spezifischen Aktivitäten gemessen und der Qsc-Wert berechnet (siehe Abschnitt 2.2.1.9.3). Die Ergebnisse sind in Tabelle 3-16 dargestellt. Zur Einschätzung der Werte sind auch die Qsc-Werte der Stämme aus der vorangegangenen Bestimmung (Abschnitt 3.1.3) aufgeführt. 99 Ergebnisse Tabelle 3-16: Daten aus den DNA-Superspiralisierungsmessungen Reportergenplasmiden pPHB90 und pPHB91 M415 Plasmid Nal2pass mit den vacT - - pBP507 pBP517 - pBP517 Qsc (± s) 1,80 (± 0,28) 0,99 (± 0,14) 0,84 (±0,3) 1,12 (±0,28) 1,09 (± 0,09) 1,17 Qsc % (± s %) 100 (± 15,5) 55 (± 14,14) 41,2 (±35,7) 54,9 (±25) 60,55 (± 8,26) 57,35 Qsc = spezifische Aktivität ptopA / spezifische Aktivität pgyrA, s = Standardabweichung, s% = relative Standardabweichung Der Tabelle ist zu entnehmen, dass die Qsc-Werte bei den Bestimmungen mit den Stämmen aus dem Komplementationstest großen Schwankungen unterworfen waren (große Standardabweichungen), was eine richtige Auswertung unmöglich machte. Eine deutliche Kompensation der Qsc-Werte in Anwesenheit von gyrA+ war aber nicht zu erkennen. Die Experimente des Komplementationstestes konnten erste Hinweise auf die Ursachen des vorhandenen Phänotyps der Zwischenderivate und des Impfstammes geben. Es konnte festgestellt werden, dass die gyrA-Mutationen einen Einfluss auf die MHK-Werte der Fluorchinolone haben und es gibt Hinweise darauf, dass Efflux beteiligt ist. Eine genaue Aussage über die Rolle der gyrA-Mutationen bei den Wachstumsgeschwindigkeiten und bei den Superspiralisierungsgraden konnte nicht getroffen werden. 3.1.7 Optimierung der Methode der 2D-Gelelektrophorese Als globaler Ansatz zur weiteren Charakterisierung der Stämme und um Unterschiede in der Proteinexpression festzustellen, bietet sich die 2D-Gelelektrophorese an. Die Methode ermöglicht es, Expressionsspektren der Proteine verschiedener Stämme untereinander zu vergleichen und Unterschiede in der Qualität und Quantität der Proteinexpression zu finden. Im Rahmen dieser Arbeit wurden die Bedingungen der Durchführungen der Probenaufbereitung, der isoelektrischen Fokussierung, der SDS-PAGE und der Färbemethode optimiert. 3.1.7.1 Optimierung der Probenaufbereitung und der Bestimmung der Proteinkonzentration Ziel der Optimierung der Probenaufbereitung war es, ein einfaches Protokoll mit erhöhter Reproduzierbarkeit zu entwickeln. Zunächst wurde die Probenaufbereitung nach Schulte [268] vorgenommen (Abschnitt 2.2.3.1). Mit dieser Methode konnten auch Proteine von S. Typhimurium Stämmen aufgearbeitet werden. Einer der Hauptnachteile bestand jedoch darin, dass in Anwesenheit des Lysepuffers keine Bestimmung der 100 Ergebnisse Proteinkonzentration nach Lowry erfolgen konnte. Durch die Detergenzien bzw. reduzierende Agenzien im Lysepuffer wurde bereits eine Farbreaktion hervorgerufen, die eine Konzentrationsbestimmung der Proben unmöglich machte. Der Einsatz einer definierten Proteinmenge wurde dadurch verhindert, und es konnte nur mit Schätzwerten gearbeitet werden. Die optimierte Probenvorbereitung wurde letztendlich mit dem ReadyPrep protein extraction kit „Total Protein“ der Firma Bio-Rad durchgeführt. Der Vorteil dieses Kits besteht u.a. darin, dass mit fertigen Puffern gearbeitet wurde, was die Reproduzierbarkeit beim Aufarbeiten erhöht. Außerdem beinhaltet das Kit neben Harnstoff und Thioharnstoff das starke Detergenz ASB-14, das eine bessere Solubilisierung der Proteine ermöglicht als Harnstoff. Das reduzierende Agens der Standardmethode zum Auflösen der Disulfidbrücken DTT ist im Kit durch das stärkere TBP (Tributylphosphin) ersetzt. Im Anschluss an die Probenaufbereitung wurde der RC DC Protein Assay von Bio-Rad Laboratories zur Konzentrationsbestimmung von Proteinen in der Probe durchgeführt. Dieser Assay beruht auf einem kolorimetrischen Verfahren zur Konzentrationsbestimmung (Prinzip der Methode nach Lowry [186]) und funktioniert auch in Anwesenheit von reduzierenden Agenzien (RC = reducing agent compatible) und Detergenzien (DC = detergent compatible). Der Assay ist kompatibel mit Reagenzien wie DTT (350 mM), Mercaptoethanol (10 %), TBP (2 mM), CHAPS (2 %), Triton X-100 (2 %), EDTA (100 mM), Tris pH 8,4 (500 mM) und anderen. Ein weiterer Vorteil besteht in der langen Stabilität des Farbkomplexes (mind. 1 h). Es wurden von den Stämmen S. Typhimurium M415, Nal2ori, Nal2passage und vacT Proteinaufbereitungen mit dem ReadyPrep protein extraction kit „Total Protein“ vorgenommen und im Anschluss die Konzentration mit dem RC DC Protein Assay bestimmt. Es wurde eine Verdünnungsreihe bekannter BSA-Konzentrationen vermessen, eine Regressionsgerade erstellt und daraus die Konzentration der Proben bestimmt. Die ermittelten Proteinkonzentrationen der aufgearbeiteten Stämme lagen zwischen 1,4 und 2,8 mg/ml Protein. 3.1.7.2 Optimierung der Bedingungen für die isoelektrische Fokussierung und die SDS-PAGE Zunächst erfolgte die Festlegung der einzusetzenden Proteinkonzentrationen. Für eine anschließende Silberfärbung der Gele sollten 50 bis 80 µg Protein für die 2D-GE eingesetzt werden. Die Wahl der richtigen Konzentration ist für eine Auswertung des Gels von großer Bedeutung. Eine zu geringe Konzentration führt zum Fehlen von Proteinspots, da diese nicht mehr im Gel nachweisbar sind. Eine zu hohe Konzentration stört durch Überladung sowohl die isoelektrische Fokussierung als auch die SDS-PAGE. Die 101 Ergebnisse Abbildung 3-4 zeigt zwei Beispiele nach 2D-Gelelektrophorese mit unterschiedlichen Proteinkonzentrationen. Abbildung 3-4: Optimierung der Proteinkonzentration links: A mit 80 µg überladenes Gel mit unvollständiger Fokussierung und starker Streakbildung; rechts: B mit 50 µg beladenes 2D-Gel mit gut ausgebildeten Spots Ein weiteres Ergebnis der Optimierung der Durchführung der IEF war die Feststellung, dass bessere Ergebnisse erzielt wurden, wenn die Probe vor der Durchführung der isoelektrischen Fokussierung 1 h bei 30.000 g abzentrifugiert wurde. Ziel dieses Schrittes war eine Entsalzung der Lösung, um die Störungen in der isoelektrischen Fokussierung zu minimieren. Schlechte Fokussierungen aufgrund hohen Salzgehaltes waren u.a. als „streak“ (horizontale Linien) im SDS-Gel erkennbar. Um die restlichen Salze bei der Fokussierung zu entfernen, wurden kleine Filterstücke („wicks“) an die Elektroden gelegt. Weiterhin unterstützte die Zugabe einer Ampholyt-Lösung (Bio-Lyte) zum Lysepuffer die Fokussierung. Die Auswahl eines optimalen pH-Gradienten verbesserte die Auftrennung der Proteine. Bei der Wahl eines zu engen pH-Bereiches gehen u.U. sehr viele Proteine für die Analyse verloren. Ein zu breiter pH-Bereich löst die große Menge von Proteinen, welche einen sehr ähnlichen isoelektrischen Punkt haben, nur unzureichend auf. Abbildung 3-5 zeigt die 2D-Gele mit verschiedenen pH-Gradienten in der ersten Dimension. Ein weiterer Punkt bei der Optimierung war die Anpassung der Äquilibrierungszeiten der IEF-Streifen. Eine nicht ausreichende Beladung der Proteine mit SDS führte im SDS-Gel zu keiner ausreichenden Trennung, da die Moleküle ohne Ladung nicht richtig wandern konnten. Um dies zu vermeiden, wurde die Äquilibrierungszeit von 10 min auf 15 min 102 Ergebnisse erhöht. Für die zweite Dimension brachte die Verwendung von 12 % Polyacrylamidgelen für die SDS-PAGE sehr gute Ergebnisse. Abbildung 3-5: Optimierung des pH-Bereiches für die IEF oben: zu breiter pH Bereich zwischen 3 und 10 löst den größten Anteil der Proteine zu wenig auf; unten: pH-Bereich zwischen 4 und 7 deckt den Hauptbereich der Proteine ab und schafft eine gute Auflösung 3.1.7.3 Optimierung der Färbemethode Bei der Färbung der 2D-Gele wurde die Silberfärbung gewählt, da sie wesentlich sensitiver ist als die Coomassiefärbung. Die Reaktion beruht auf der Reduktion der Silberionen zu metallischem Silber. Die in der Färbelösung enthaltenen Ag+-Ionen bilden Komplexe mit Glu-, Asp- und Cys-Resten der Proteine im Gel. Anschließend werden diese Komplexe durch alkalisches Formaldehyd zu Ag reduziert. Für die Silberfärbung wurde das Kit SilverQuest® von der Firma Invitrogen verwendet. Dies bietet im Gegensatz zur Standard Silberfärbung und der Silver Stain Plus® Färbung (Bio-Rad Laboratories) den Vorteil der Kompatibilität mit der Massenspektroskopie. Mit einem Destainer können relevante Spots entfärbt und massenspektrometrisch untersucht werden. Außerdem ist die SilverQuest® Färbung schnell und einfach durchführbar. Die Abbildung 3-6 zeigt die unterschiedlichen Sensitivitäten der Silberfärbungen. Es wurde dafür die gleiche Proteinprobe (vacT aufgearbeitet mit dem ReadyPrep protein extraction kit® „Total Protein“ der Firma Bio-Rad) verwendet und die erste Dimension (IEF), inklusive Äquilibrierung, unter den gleichen Bedingungen durchgeführt. 103 Ergebnisse Abbildung 3-6: Vergleich von 2D-Gelen mit unterschiedlichen Silberfärbungen links: A 2D-Gel mit Standard-Silberfärbung; rechts: B 2D-Gel gefärbt mit SilverQuest® Die Färbung mit dem SilverQuest® Kit der Firma Invitrogen zeigte ein sehr gutes Färbeergebnis. Auch wenn der Hintergrund leicht gelb verfärbt war, war die Sensitivität und somit die detektierbare Spotzahl höher als bei der konventionellen Färbemethode. Im Rahmen der Arbeit konnte die Methode der 2D-Gelelektrophorese von der Probenaufbereitung, Proteinkonzentrationsbestimmung über die isoelektrische Fokussierung bis zur Färbung der Gele in den einzelnen Schritten optimiert werden. Es wurden exemplarisch 2D-Gele der Proteinspektren der Stämme M415 und vacT hergestellt. Eine Auswertung mit der Software PDQuest® ergab eine gute Auflösung der Proteinspots in den Gelen. Es konnte nach Digitalisierung der Bilder und der Bildbearbeitung eine Spotdetektion vorgenommen werden. Je nach eingestellten Parametern konnten mindestens 800 bis zu ca. 2.000 Spots pro Gel detektiert werden. 104 Ergebnisse 3.2 Untersuchung der Rolle der im Impfstamm auftretenden gyrA-Mutationen Aufgrund der Entstehung des Impfstammes durch ungezielte Mutagenese ist das Vorhandensein weiterer unbekannter Mutationen in anderen Genen wahrscheinlich. Diese nicht charakterisierten Mutationen können unbekannte Effekte haben und die Wirkungen der gyrA-Mutationen verändern, so dass die phänotypischen Eigenschaften des Stammes durch das Zusammenwirken verschiedener Komponenten entstehen können. Zur Charakterisierung der Rolle der auftretenden gyrA-Mutationen müssen die Auswirkungen anderer Mutationen ausgeschaltet werden. Um eine Aussage über die genaue Rolle der gyrA-Mutationen im Impfstamm vacT treffen zu können, war es erforderlich, isogene Stämme herzustellen, die lediglich die zu untersuchenden Mutationen tragen. Das Ziel der Arbeit war es, ausgewählte Mutationen in das gyrA-Gen des Ausgangsstammes M415 einzuführen. Die zu untersuchenden Mutationen sollten einzeln bzw. als Kombination in das gyrA-Gen von M415 eingebracht werden. Dabei sollten die Aminosäureaustausche den Mutationen des Impfstammes vacT entsprechen. Nur mittels solcher hergestellter isogener Stämme, welche ausschließlich die eingeführte(n) gyrAMutatione(n) im Vergleich zum Ausgangsstamm enthalten, ist eine Analyse der Rolle der gyrA-Mutationen des Impfstammes möglich. Als Methode zum Einbringen der gewünschten Aminosäureaustausche in den Ausgangsstamm wurde die homologe Rekombination gewählt. Diese Methode ermöglicht es, das Chromosom des Ausgangsstammes in gewünschter Form zu verändern. Es existieren verschiedene Möglichkeiten homologe Rekombinationen durchzuführen (siehe Abschnitt 2.2.1.10). Mit zwei verschiedenen Methoden konnten im Rahmen dieser Arbeit Rekombinanten hergestellt werden. Dieses Kapitel beinhaltet die Beschreibung der Optimierung und der Ergebnisse aus den einzelnen Versuchen zur homologen Rekombination. Weiterhin wird beschrieben, wie die Rekombinanten selektiert und anschließend auf den Erfolg des Einkreuzens der Mutationen gescreent werden konnten. Außerdem sind die Ergebnisse aus der Charakterisierung der hergestellten Rekombinanten dargestellt. 3.2.1 Überblick über die Systeme zur Rekombination Im Rahmen dieser Arbeit wurden verschiedene Strategien verfolgt, um die Rolle der im Impfstamm auftretenden gyrA-Mutationen zu untersuchen. Zum einen wurde über homologe bzw. sequenzspezifische Rekombination ein vorher mit den Mutationen versehenendes gyrA-Gen in das Chromosom von M415 eingekreuzt (Abschnitt 3.2.1.1). Zum anderen wurde eine Strategie entwickelt, nach dem knock out des chromosomalen Wildtyp-Gens in M415, das gyrA-Gen mit entsprechenden Mutationen über ein „Rescue“plasmid zu exprimieren (Abschnitt 3.2.1.2). 105 Ergebnisse 3.2.1.1 Insertion eines gyrA-Gens mit Mutationen ins Chromosom von M415 Beide zur homologen Rekombination verwendeten Systeme (über das zelleigene bzw. über das phagengenbasierte Rekombinationssystem) erzeugten genotypisch das gleiche Ergebnis: In das chromosomale gyrA-Gen des Stammes M415 wurde ein neues konstruiertes gyrA-Gen mit den gewünschten Mutationen eingekreuzt. Viele Protokolle zur Durchführung der Methode der homologen Rekombination in E. coli [114,180] nutzen das zelleigene Rekombinationssystem „RecABCD“ der Zelle (siehe Abschnitt 2.2.1.10.1). Mit Hilfe der Methode nach Hamilton unter Verwendung von pMAK705-Derivaten konnten bereits früher einige gyrA-Mutanten hergestellt werden [109]. Ein Nachteil bei der Verwendung dieses Plasmides ist, dass nach dem Auskreuzen des Plasmides keine Selektionsmöglichkeit für den Verlust des Plasmides existiert, d.h. es mussten viele Passagen durchgeführt werden, um das Plasmid aus der Zelle zu eliminieren. Das Problem der schwierigen Eliminierung des Plasmides lösten Link et al. [180] mit der Einführung des Gens sacB aus Bacillus subtilis in den für die Rekombination verwendeten Vektor pKO3. Zur leichteren Klonierung wurde aus dem Vektor pKO3 durch Insertion eines 3 kb DNA-Fragmentes (Puffer) zwischen die Schnittstellen NotI und BamHI der Vektor pKOV konstruiert. Weiterhin besitzt dieser Vektor eine Chloramphenicolresistenz-Kassette und einen temperatursensitiven Replikationsursprung. Der Vektor pKOV wurde im Rahmen dieser Arbeit zur Durchführung der homologen Rekombination als „Rekombinationsvektor“ eingesetzt, um spezielle Aminosäuren in der GyrA-Untereinheit des Stammes M415 auszutauschen. Das gyrA-Gen mit den einzukreuzenden Mutationen wurde dafür komplett (inklusive natürlichen Promotor) in den Vektor pKOV kloniert. Mit diesem Vektor pKOV wurden verschiedene gyrA-Mutanten hergestellt. Die Klonierungsstrategie ist im Abschnitt 3.2.3 und die genaue Durchführung der Methode der homologen Rekombination mit Hilfe des Systems nach Link et al. [180] mit dem Vektor pKOV in Abschnitt 2.2.1.10.1 beschrieben. Eine weitere und sehr häufig für eine gezielte Mutagenese des Chromosoms verwendete Methode der sequenzspezifischen Rekombination ist das phagengenbasierte System mit Hilfe des Helferplasmides pKD46 (siehe Abschnitt 2.2.1.10.2). Im Rahmen dieser Arbeit konnte die Methode nach Datsenko und Wanner [66] mit Einsatz des Helferplasmides pKD46 ebenfalls erfolgreich für das Einkreuzen von gyrA-Mutationen in den Stamm S. Typhimurium M415 angewandt werden (siehe Abschnitt 3.2.4.2). Die Durchführung der Methode ist in Abschnitt 2.2.1.10.2 beschrieben. 106 Ergebnisse 3.2.1.2 Knock out des chromosomalen gyrA-Gens und Verwendung eines Rescueplasmides Die Prinzipien der beiden bisher beschriebenen Methoden bestanden darin, das gyrA-Gen im Chromosom als funktionstüchtig zu erhalten und nur in einzelnen Aminosäuren zu verändern. Eine alternative Idee für die Untersuchung der Rolle der gyrA-Mutationen beinhaltete die Strategie, das chromosomale gyrA-Gen auszuschalten (knock out) und ein plasmidcodiertes gyrA in die Zelle einzubringen. Die für die Zelle essentielle Enzymaktivität der Gyrase bliebe also durch dieses Rescueplasmid erhalten. Versuche mit solchen Rescueplasmiden wurden auch bereits in anderen Arbeitsgruppen zur Untersuchung verschiedener Gene genutzt [36,112]. Das Rescueplasmid muss das komplette gyrA-Gen unter der Kontrolle eines funktionierenden natürlichen oder künstlich eingeführten Promotors enthalten. Da es sich bei der Gyrase um ein essentielles Gen handelt, muss dieses Rescueplasmid bereits in der Zelle sein, bevor der knock out des chromosomalen gyrA-Gens durchgeführt werden kann. Um den knock out von gyrA durchzuführen, bietet sich das Red-RekombinaseSystem an [66]. Durch eine Insertion der Kanamycinresistenz-Kassette (Selektionsmarker) ist die Expression des Genes nicht mehr möglich. Man nutzt ein PCRProdukt, welches den Selektionsmarker mit FRT-Sites und an beiden Seiten ca. 50 bp lange homologe Flanken, welche den Ort der Rekombination bestimmen, beinhaltet. Zu beachten ist dabei, dass die Lokalisation der vorderen (vor gyrA liegend) homologen Flanke für die Rekombination so gewählt wird, dass keine Rekombination mit der Plasmidsequenz stattfinden kann. Zunächst wurde über PCR die KanamycinresistenzKassette inklusive FRT-Sites vom Templateplasmid pKD4 amplifiziert. Durch die Primer wurden die homologen Flanken für die Rekombination eingeführt, wobei eine Flanke homolog ist zu einer gyrA-Sequenz stromaufwärts der einzukreuzenden Mutationen und die andere Flanke chromosomal weit vor gyrA lokalisiert ist. Durch diese zweite Flanke, welche keine Homologie zu Bereichen auf dem Rescueplasmid aufweist, ist die Rekombination mit der Sequenz auf dem Plasmid unmöglich. Zur Herstellung des Rescueplasmides mit dem gyrA-Gen wurde der Vektor pBAD/D102 gewählt, der eine Ampicillinresistenz-Kassette zur Selektion enthält. Zunächst wurde das komplette gyrA-Gen aus M415 amplifiziert. Dabei wurden mit dem Forward-Primer die CACC-Basen eingeführt, die im Anschluss die gerichtete Klonierung erlaubten (siehe Abschnitt 2.2.2.9.6). Abbildung 3-7 zeigt den prinzipiellen Aufbau des Rescueplasmides. 107 Ergebnisse Abbildung 3-7: Aufbau des Rescueplasmides pBAD102_gyrA (mit gyrA aus S. Typhimurium) gyrA = gyrA-Gen aus M415; bla = Ampicillinresistenz-Kassette; pBR322 origin = Replikationsursprung aus pBR322; araBAD Promotor (inkl. AraC) = Arabinosepromotor; RBS = Ribosomenbindungsstelle Voraussetzung für den Einsatz des Rescueplasmides war die Kontrolle der biologischen Aktivität des einklonierten gyrA-Gens. Die einwandfreie Funktionsweise der Arabinoseinduzierten gyrA-Expression und der biologischen Aktivität des entstehenden Proteins wurde durch die Verwendung des E. coli Stammes KNK402 überprüft. Dieser Stamm trägt eine Mutation im Gen für die A-Untereinheit der Gyrase, wodurch das Protein bei höheren Temperaturen instabil wird. Bei 30 °C zeigt der Stamm normales Wachstum, aber bei 42 °C ist das Wachstum sehr stark beeinträchtigt. Zur Kontrolle der Funktion der von pBAD102_gyrA exprimierten Gyrase wurde das Rescueplasmid in den Stamm E. coli KNK402 transformiert. Dieser rekombinante Stamm exprimiert nun zwei verschiedene Gyrasen: die chromosomalcodierte Form und die plasmidcodierte Form. Im Fall einer Temperaturerhöhung kann die plasmidcodierte Gyrase den Ausfall der chromosomalen mutierten Gyrase kompensieren und das Wachstum der Zellen sichern. Funktioniert die Expression vom Rescueplasmid nicht, dann würde auch bei 42 °C kein normales Wachstum zu beobachten sein. Die Selektion erfolgte zunächst bei 30 °C unter Zugabe von Ampicillin, um das Vorhandensein des Rescueplasmides sicherzustellen. Um die Expression des Genes vom Plasmid zu induzieren, wurde 1mM L-Arabinose zugesetzt. Abbildung 3-8 zeigt die Ergebnisse aus dem Wachstumsversuch des Stammes E. coli KNK402 mit dem Rescueplasmid pBAD102_gyrA. Als Kontrollplasmid wurde ein Konstrukt genutzt, was nur ein kleines Fragment vom Startbereich des gyrA-Gens (kein funktionstüchtiges Gen) enthielt. 108 Ergebnisse Abbildung 3-8: Wachstumsversuch von E. coli KNK402 zur Überprüfung der Funktionsweise des Rescueplasmides pBAD102_gyrA Es wurden jeweils vier Klone aus der Transformation pro Agarplatte (Ampicillin 50 µg/ml) ausgestrichen und 24 h bei den entsprechenden Temperaturen inkubiert; oben: Der Stamm E. coli KNK402 zeigt mit pBAD-Kontrollplasmid nur bei 30°C ein gutes Wachstum, bei 42 °C ist nur ein schlechtes Wachstum zu erkennen. unten: Mit Rescueplasmid pBAD102_gyrA kann der Stamm auch bei 42 °C sehr gut wachsen. Der Wachstumstest zeigte, dass die Expression des plasmidcodierten gyrA-Gens von pBAD102_gyrA funktionierte und das exprimierte Protein die Funktion der defekten chromosomalcodierten A-Untereinheit der Gyrase übernehmen kann. Das Gen auf dem Plasmid kann nun so verändert werden, dass es die entsprechenden Mutationen, deren Rolle untersucht werden sollte, enthält. Nach einem knock out des chromosomalen gyrAGens würde dann ausschließlich das plasmidcodierte gyrA exprimiert werden und mögliche Veränderungen im Phänotyp des Stammes würden auf die Mutationen zurückzuführen sein. 3.2.1.3 System zur Selektion nach positiver homologer Rekombination Homologe Rekombination wird sehr häufig genutzt, um eine Insertion in ein Gen einzuführen und dieses dadurch auszuschalten. Ziel ist es, durch den knock out eines Gens, dessen Funktion zu untersuchen. Weiterhin kann getestet werden, ob Veränderungen des Genes einen bestimmten Phänotyp bedingen. Zum Beispiel führt der knock out eines Effluxpumpen-Repressors zur Überexpression der Effluxpumpe und das wiederum zur Erhöhung der MHK-Werte bestimmter Antibiotika. Ein so veränderter Phänotyp eröffnet die Möglichkeit, den Erfolg der Rekombination durch diesen Phänotyp zu testen, z.B. durch Selektion auf Agarplatten, die ein entsprechendes Antibiotikum in 109 Ergebnisse erhöhter Konzentration beinhalten. Auch Veränderungen des gyrA-Gens können unter Umständen zu einer solchen, für die Selektion der erfolgreichen Rekombination nutzbare, Veränderung des Phänotyps führen. So ist bekannt, dass Aminosäureaustausche an den Positionen 83 oder 87 zu einer reduzierten Empfindlichkeit gegenüber (Fluor)chinolonen führen. Diese Tatsache konnte z.B. Grüger [109] bei der Detektion von Rekombinanten nutzen. Das Ziel dieser Arbeit war es, die Rolle bestimmter z.T. bisher unbekannter gyrAMutationen zu untersuchen. Von diesen Mutationen war nicht bekannt, ob und - wenn ja welche Veränderung des Phänotyps sie bedingen. Da ein Rekombinationsereignis sehr selten ist (abhängig vom Grad der Homologie, der Länge des homologen Fragmentes sowie der chromosomalen Lage), war es nötig ein Selektionssystem zu entwickeln, welches die Erkennung von positiven Rekombinanten ermöglicht. Aus diesem Grund wurde eine Resistenz-Kassette als Selektionsmarker zusätzlich mit dem gyrA-Gen in das Chromosom eingekreuzt. Unabhängig von der zur homologen Rekombination genutzten Methode wurde eine Kanamycinresistenz-Kassette als Selektionsmarker genutzt, um positive Rekombinanten zu erkennen (siehe Abschnitt 2.2.1.10.3). Die Resistenz-Kassette befand sich immer zwischen den beiden für die Rekombination notwendigen homologen Flanken. Dabei war die erste Flanke eine Sequenz, die homolog zu einem Bereich des Chromosoms ist, der vor dem gyrA-Gen liegt und die zweite homologe Flanke war das gyrA-Gen selbst. Da sich die einzukreuzenden Aminosäureaustausche (im Bereich der Positionen 59 bis 87) relativ weit im 5’-Bereich des Gens befinden, wurde davon ausgegangen, dass statistisch gesehen die meisten Rekombinationsereignisse im 3’Bereich, d.h. hinter den Mutationen, stattfinden und somit das Einkreuzen der Mutationen gemeinsam mit der Kanamycinresistenz-Kassette wahrscheinlich ist. Bei dem Knock-outVersuch wurden links und rechts der Kanamycinresistenz-Kassette einfach die homologen Flanken über die Primer bei der Amplifikation angehängt. 3.2.2 Auswahl eines geeigneten Rekombinationssystems Es standen nun verschiedene Systeme zur Verfügung, um die Rolle der im Impfstamm auftretenden gyrA-Mutationen mit Hilfe der homologen Rekombination zu untersuchen. Zunächst wurde mit dem Lambda Red System begonnen, da es am wenigsten aufwendig erschien und in S. Typhimurium LT2 bereits beschrieben war [121]. Benötigt wurde für diese Methode ein PCR-Produkt, welches neben dem gyrA-Fragment mit den Mutationen auch den Selektionsmarker mit den durch die Primer eingeführten homologen Flanken enthielt. Vorteil dieser Rekombinationsmethode war, dass PCR-Produkte leicht, schnell und in beliebiger Menge hergestellt werden konnten. Es erfolgte eine Transformation mit dem Helferplasmid pKD46 in den Ausgangsstamm M415. Diese Kombination von M415 und pKD46 konnte dann für alle Rekombinationsversuche genutzt werden. Vom Helferplasmid wurden durch Arabinosezugabe die Expression der 110 Ergebnisse Phagengene induziert. Nach Transfer des PCR-Produktes mittels Elektroporation sollten dann die kanamycinresistenten Rekombinanten selektierbar sein (Abschnitt 2.2.1.10.2). Zunächst war es jedoch auf diesem Weg unter den beschriebenen Bedingungen auch nach mehrfachen Wiederholungen nicht möglich, Rekombinanten herzustellen. Vermutlich führte einer der folgenden Punkte zum Misserfolg dieser Variante: (1) Da es sich bei dem Stamm S. Typhimurium M415 um keinen Laborstamm, sondern ein klinisches Isolat handelte, enthielt der Stamm möglicherweise Nukleasen in der Menge oder der Stärke, dass sie nicht von den Phagenenzymen inhibiert werden konnten. Ein Abbau des PCR-Produktes macht die Rekombination unmöglich. (2) Die Kanamycinresistenz-Kassette sollte als Selektionsmarker zusammen mit den Mutationen in das Chromosom gekreuzt werden. Um eine schädigende Auswirkung eines knock outs unbekannter Bereiche des Chromosoms zu verhindern, wurden die homologen Bereiche sehr eng beieinander gewählt (Abstand ca. 150 nt). Möglicherweise verhinderte eine Loop-Bildung der zu inserierenden Kassette (ca. 1.500 nt) aus sterischen Gründen eine Rekombination. (3) Der Erfolg der Rekombination ist stark von der Lokalisation der homologen Flanken abhängig. Eventuell war die Lage der homologen Bereiche ungünstig gewählt. Veränderungen der Rekombinationsbedingungen führten nachfolgend aber zu einer erfolgreichen Anwendung dieser Methode: Bei diesem späteren Versuch wurde ein Fragment, bestehend aus der Kanamycinresistenz-Kassette mit einer 500 bp homologen Flanke und dem gyrA-Gen als zweite homologe Flanke, mittels Elektroporation in den Stamm M415 transferiert. Dieses Fragment stammte aus pPHB650 (siehe Abschnitt 3.2.3.4) und wurde mittels Restriktionsspaltung unter Verwendung der Enzyme NotI und SalI nach Gelextraktion gewonnen. Die homologe Flanke war in einem größeren Abstand von ca. 800 nt vor gyrA lokalisiert. Dies bewirkte den knock out eines Teils des Chromosoms von M415, was sich aber in den anschließenden Charakterisierungen als unproblematisch erwies. Die sequenzspezifische Rekombination mit diesem Fragment mit Hilfe der Phagenproteinen war erfolgreich und führte zu einer nachweisbaren Insertion der Kanamycinresistenz-Kassette. In diesem Experiment wurden die oben genannten Probleme umgangen, indem die neuen homologen Bereiche weiter voneinander entfernt waren, was eine sterische Behinderung minimierte. Die homologen Flanken waren mit mindestens 500 bp Länge wesentlich länger, als für die phagenbasierte Vermittlung der Rekombination nötig. Somit schadete auch ein eventueller Angriff von zelleigenen Exonukleasen nicht, da die verbleibende Flanke immer noch eine ausreichende Länge hätte. Somit konnte der Beweis erbracht werden, dass diese Methode auch in klinischen Isolaten von S. Typhimurium zur Anwendung kommen kann (Durchführung siehe 2.2.1.10.2). 111 Ergebnisse Wie bereits beschrieben (3.2.1.2), gab es weiterhin den Ansatz, über einen knock out des chromosomalen gyrA-Gens und das Einbringen eines Rescueplasmides, welches das mit gyrA-Gen den zu Aminosäureaustausches untersuchenden zu untersuchen. Mutationen Die enthält, Vorversuche die Rolle zeigten, eines dass das Rescueplasmid seine Funktion erfüllte. Nachteil dieses Systems war, dass für jede Untersuchung der entsprechenden Mutation ein eigenes Rescueplasmid kloniert werden muss. Dieses Rescueplasmid muss zusammen mit dem Helferplasmid in der Zelle präsent sein. Das wäre bei pBAD102_gyrA und pKD46 möglich, da sie verschiedene Replikationsstartregionen (ori) haben. Trotzdem kann die Anwesenheit von zwei Plasmiden in der Zelle zu Problemen führen, wie die Versuche im Komplementationstest (Abschnitt 3.1.6) zeigten. Eine weitere Schwierigkeit bestand in der Selektionsmöglichkeit: Das Helferplasmid, das Rescueplasmid und der Rekombinations-Selektionsmarker müssen unterschiedliche Resistenz-Kassetten enthalten. Als Rekombinations- Selektionsmarker wurde die Kanamycinresistenz-Kassette gewählt (Abschnitt 3.2.1.3). Helfer- und Rescueplasmid enthielten beide eine Ampicillin-Kassette. Da verschiedene Rescueplasmide kloniert werden müssten, welche die unterschiedlichen Mutationen enthalten, bot es sich an, die Resistenz-Kassette des Helferplasmides pKD46 zu verändern. Als Klonierungsstrategie wurde geplant, in die PvuI-Schnittstelle innerhalb des Ampicillinresistenzgens eine Tetracyclinresistenz-Kassette zu klonieren. Damit würde zum einen die Ampicillinresistenz-Kassette zerstört und das Helferplasmid wäre mit Tetracyclin selektierbar. Dieser Ansatz wurde nicht weiter verfolgt, da andere Rekombinationsmethoden im Laufe der Arbeit zum Erfolg geführt hatten. Die später weiter charakterisierten Rekombinanten aus M415, welche die im Impfstamm vorkommenden gyrA-Mutationen beinhalten, wurden mit dem System unter der Verwendung des Vektors pKOV (Abschnitt 2.2.1.10.1) nach Court und Link hergestellt. Der Vorteil dieses Systems war, dass es in der Zelle zu keinem Abbau des Plasmides durch Nukleasen – wie bei den PCR-Produkten – kommt. Weiterhin war es möglich, trotz geringer Rekombinationsraten, durch eine Anreicherung der gewünschten Zellen (z.B. die Cointegrate) das Auffinden von Rekombinanten zu erleichtern. Allerdings hatte das System den Nachteil, in der Vorarbeit etwas aufwendiger zu sein. Zunächst mussten Vektoren für jede einzukreuzende gyrA-Mutation kloniert werden (siehe Abschnitt 3.2.3.4). Die Klonierung wurde vor allem dadurch erschwert, dass es sich bei pKOV um einen Lowcopy-Vektor mit temperatursensitiven Replikationsursprung (30 °C) handelt. Dies zieht langsames Wachstum und geringe Ausbeuten bei der Plasmidpräparation nach sich. Die klonierten Vektoren wurden jeweils in den Ausgangsstamm M415 transformiert. Für die Rekombination waren mehrere Passagen unter verschiedenen Temperaturbedingungen notwendig (siehe Abschnitt Klonierungsstrategien Rekombinanten und der der 2.2.1.10.1). verwendeten In den Vektoren, gyrA-Mutationen sowie Charakterisierungen der Rekombinanten beschrieben. 112 folgenden das die Kapiteln Screening der Ergebnisse sind die positiven aus den Ergebnisse 3.2.3 Klonierungsstrategien zur In-vitro-Mutagenese mittels homologer Rekombination Ziel der Arbeit war die Herstellung von isogenen Stämmen aus dem Ausgangsstamm M415, welche die im Impfstamm enthaltenen gyrA-Mutationen einzeln oder in Kombination enthalten. Als besonders interessant wurde dabei die Mutation an Aminosäureposition 87 eingeschätzt. Diese war aus vorherigen Arbeiten [117,128] als Fluorchinolonresistenz vermittelnd bekannt. Allerdings zeigte der Impfstamm in den MHKWert-Bestimmungen (Abschnitt 3.1.2) keine reduzierten Empfindlichkeiten gegenüber Fluorchinolonen. Der im Impfstamm erstmalig aufgetretenen Mutation an Aminosäureposition 59 wurde große Bedeutung für eine intragenische Kompensation zugemessen. Das Ziel war es, isogene Stämme mit der Mutation an Aminosäureposition 87 und die auftretenden Kombinationen mit den Mutationen an den Positionen 59 bzw. 75 herzustellen und zu untersuchen. Um die Mutanten über homologe Rekombination herzustellen, mussten zunächst die Mutationen im gyrA-Gen über SOEing-PCR hergestellt werden, da sich keine Schnittstellen für Restriktionsenzyme für eine Umklonierung der Mutationen anboten und da die zu untersuchenden Mutationen (an Positionen 59, 75 und 87) sehr eng beieinander liegen. Die durch PCR erzeugten SOEing-Produkte konnten ohne das Vorhandensein von Restriktionsschnittstellen mittels TOPO® TA Klonierung in den Vektor pCR2.1TOPO® der Firma Invitrogen kloniert werden (siehe Abschnitt 2.2.2.9.6). Somit konnten die PCRProdukte eingefroren und später weiter genutzt Umklonierungen aus den Vektoren pCR2.1TOPO ® werden. Durch verschiedene und pT7-6 konnten die für die Rekombination benötigten Vektoren auf der Basis des Vektors pKOV hergestellt und die Rekombinationen durchgeführt werden. 3.2.3.1 Mutagenese mittels SOEing Die Herstellung der einzelnen Mutationen erfolgte durch SOEing-PCR. Das Prinzip und die Methode dieser PCR sind in Abschnitt 2.2.2.5.2 beschrieben. Für eine Einführung einer Mutation in gyrA wurden ein Vorwärts- und ein Rückwärtsprimer gewählt, welche komplementär zueinander sind und den gewünschten Basenaustausch einführen (siehe Tabelle 2-5). Abbildung 3-9 zeigt die Auswahl der SOEing-Primer für die Mutagenese anhand der Nukleotidsequenz von gyrA. 113 Ergebnisse Abbildung 3-9: Auswahl der Primer für die SOEing-PCR schwarz-fett = Bindungsbereiche der Primer; rot-fett = Basen, welche durch die Primer ausgetauscht wurden Æ Positionen: Trp-59 Æ Arg = TGG Æ AGG; Gly-75 Æ Ala = GGT Æ GCT; Asp-87 Æ Gly = GAC Æ GGC Abbildung 3-10 zeigt beispielhaft die Herstellung eines gyrA-Fragmentes mit einem Aminosäureaustausch an Position 59. Als erstes wurden zwei PCR-Produkte hergestellt, welche am 5` bzw. 3`-Ende einen durch die Primer (gyrA-5-59 AS und gyrA-3-59 AS) definierten Bereich mit Mutation enthielten. Diese PCR-Produkte waren 263 bzw. 521 bp lang. Die Längen der PCR-Produkte im ersten PCR-Schritt waren abhängig von der Position der einzuführenden Mutation. Der zweite Schritt war eine Fusion dieser beiden Fragmente mit Hilfe der komplementären Bereiche sowie eine Amplifikation des gesamten Fusionsproduktes mit den Außenprimern gyrA-5-1-AS und gyrA-3-1-AS zu einer Gesamtlänge von 763 bp. Abbildung 3-10: Darstellung der SOEing-PCR zum Einführen der Basenaustausche in das gyrA-Fragment Pfeile stellen die verwendeten Primer für die PCRs dar; in der Zeichnung finden sich die erwarteten PCR-Fragmentgrößen; der Erfolg der PCR und die richtigen Fragmentgrößen sind den Fotos für die Agarose-Gelelektrophoresen zu entnehmen. 114 Ergebnisse Auf dem beschriebenen Weg wurden über SOEing-Technik verschiedene PCR-Produkte hergestellt, welche die zu untersuchenden Mutationen einzeln bzw. in verschiedenen Kombinationen enthielten. Diese Mutagenese-Fragmente stellten die Grundlage für die weitere Klonierungsarbeit dar. 3.2.3.2 Klonierung der Mutagenese-Fragmente in pCR2.1TOPO® Um diese PCR-Produkte für weitere Versuche zu erhalten, wurden sie in den Klonierungsvektor pCR2.1TOPO® der Firma Invitrogen mittels TOPO® TA Klonierung eingebracht (siehe Abschnitt 2.2.2.9.6). Dadurch standen die Mutagenese-Fragmente für das weitere Vorgehen als PCR-Template oder für Restriktionsspaltung in beliebiger Menge auch zu späteren Zeitpunkten zur Verfügung. Die Klonierung erfolgte ungerichtet und die Klone wurden über Ampicillinzugabe (50 µg/ml) selektiert. Die Überprüfung des Erfolges bei der Klonierung eines Fragmentes in den pCR2.1TOPO®-Vektor konnte über Blau-Weiß-Selektion erfolgen (Abschnitt 2.2.2.9.6). Weiße Klone wurden durch Überimpfen gereinigt, per Plasmid-Minipräparation (Abschnitt 2.2.2.2.1) isoliert und mittels Restriktionsspaltung auf die korrekte Größe geprüft. Abbildung 3-11 zeigt die erwarteten Fragmentgrößen nach einer Spaltung mit den Restriktionsenzymen XbaI und HindIII bei erfolgreicher Klonierung. Abbildung 3-11: Überprüfung der Klonierung der Mutagenese-Fragmente in den Vektor pCR2.1TOPO® durch Spaltung mit XbaI und HindIII oben: Größenangaben der entstehenden Fragmente bei erfolgreicher Klonierung; unten: Agarosegelfoto zur Auftrennung der entstandenen Fragmente nach der Spaltung vier zu testender Plasmide Æ alle vier Plasmide zeigen die erwarteten Fragmentgrößen 115 Ergebnisse Klone mit positiven Inserts wurden als Dauerkultur gesichert. Da die Klonierung der Mutagenese-Fragmente in den pCR2.1TOPO®-Vektor ungerichtet erfolgt, sind zwei Orientierungen des gyrA-Fragmentes im Plasmid möglich. Da die Orientierung aber für weitere Versuche irrelevant ist, wurde sie nicht überprüft. Um zu gewährleisten, dass die Mutationen korrekt in den Fragmenten vorliegen, wurde ein Teil der QRDR von gyrA mit dem Primer gyrA-5-1-AS direkt aus dem Plasmid sequenziert. In Tabelle 3-17 sind die klonierten pCR2.1TOPO®-Derivate und die enthaltenen gyrA-Mutationen aufgeführt. Unter Beachtung der in der Arbeitsgruppe eingeführten Nomenklatur erhalten die im Rahmen dieser Arbeit klonierten Derivate des pCR2.1TOPO-Vektors die Namen pPHB451 bis 455. Tabelle 3-17: Bezeichnung der pCR2.1TOPO®-Derivate Bezeichnung im gyrA-Fragment enthaltene Mutation(en) pPHB451 Trp-59 Æ Arg pPHB452 pPHB453G Gly-75 Æ Ala * Asp-87 Æ Gly pPHB454 Trp-59 Æ Arg; Gly-75 Æ Ala pPHB455 Trp-59 Æ Arg; Asp-87 Æ Gly * die 3 als dritte Ziffer der Bezeichnung weist auf eine Mutation an Position 87 hin; das G kennzeichnet den Aminosäureaustausch von Aspartat zu Glycin 3.2.3.3 Klonierung von gyrA in pT7-6 Über SOEing-PCR wurden alle Einzelmutationen bzw. die Mutationskombinationen hergestellt, die nicht so im Impfstamm oder den Zwischenderivaten vorhanden waren. Um aber auch die in den Zwischenderivaten vorkommende Doppelmutation und die im Impfstamm auftretende Dreifachmutation untersuchen zu können, wurden die kompletten gyrA-Gene aus diesen Stämmen kloniert. Weiterhin wurde das gyrA-Gen aus dem Ausgangsstamm M415 und aus der Selektionsmutante SM415-87 kloniert, um diese als Kontrollen für weitere Versuche nutzen zu können. Als Klonierungsvektor bot sich pT7-6 an, da er eine geringe Größe von 2208 bp hat und das gyrA-Gen bei Bedarf über den T7Promotor exprimiert werden kann. Die gyrA-Gene aus den Stämmen wurden per PCR amplifiziert. Über die Primer (siehe Tabelle 2-7) wurden am 5`-Ende eine Erkennungssequenz für das Restriktionsenzym SalI und am 3`-Ende für BamHI eingeführt. Sowohl das entstehende PCR-Produkt (2885 bp), als auch der Vektor wurden dann mit beiden Restriktionsenzymen gespalten. Bei dem Vektor pT7-6 gingen durch die Doppelrestriktion 12 bp verloren. Da jeweils nur kleine Reste abgespalten wurden, konnte eine Aufreinigung über Silikasäulchen des QIAquick® Purification Kit (Abschnitt 2.2.2.8.1) durchgeführt werden, weil Fragmente unter ca. 100 bp nicht an die Matrix binden. Es erfolgte dann die Ligation von pT7-6 und den PCR-Fragmenten über die Schnittstellen der Enzyme SalI und BamHI sowie die anschließende Transformation (unter Ampicillinselektion 50µg/ml) in DH5α. Entstandene Klone wurden überimpft und die Plasmide einer Minipräparation unterzogen. Das entstandene Plasmid-Konstrukt musste eine Größe von 5059 bp haben (siehe Abbildung 3-17). Der Erfolg der Klonierung wurde 116 Ergebnisse überprüft, indem die entstandenen Plasmide wiederum einer Doppelrestriktion mit SalI und BamHI unterzogen wurden. Bei einer positiven Klonierung mussten zwei Fragmente der ungefähren Größe von 2200 bp und 2850 bp entstanden sein (siehe Abbildung 3-12). Abbildung 3-12: pPHB750: Klonierung der gyrA-Gene in den pT7-6-Vektor links: Vektorkarte des entstehenden Konstruktes pPHB750; bla = Ampicillinresistenz-Kassette; rechts: Überprüfung des Klonierungserfolges nach Doppelrestriktion (SalI und BamHI): 1 = positiver Klon mit erwarteten Fragmentgrößen von 2850 bp und 2200 bp; 2 = negativer Klon, 3 = Kontrolle: linearisierter pT7-6-Vektor (ca. 2200 bp) Klone, die positive Plasmide enthielten, wurden als Glycerindauerkultur gesichert. Die entstandenen Konstrukte mit den kompletten gyrA-Genen inklusive der unterschiedlichen Mutationen und ihre Bezeichnungen sind der Tabelle 3-18 zu entnehmen. Tabelle 3-18: Bezeichnung der pT7-6-Derivate Bezeichnung enthaltene gyrA- Mutation(en) Herkunft pPHB750 keine M415 pPHB753Y * Asp-87 Æ Tyr SM415-87 pPHB758 Gly-75 Æ Ala; Asp-87 Æ Gly; Ala-866 Æ Ser Nal2ori pPHB759 Trp-59 Æ Arg; Gly-75 Æ Ala; Asp-87 Æ Gly; Ala-866 Æ Ser vacT * die 3 als dritte Ziffer der Bezeichnung weist auf eine Mutation an Position 87 hin; das Y kennzeichnet den Aminosäureaustausch von Aspartat zu Tyrosin 3.2.3.4 Klonierung der Rekombinationsvektoren ausgehend von pKOV Um die homologe Rekombination durchführen zu können, mussten verschiedene Vektoren kloniert werden. Diese sollten eine Kanamycinresistenz-Kassette (flankiert von FRT-Sites), eine homologe Flanke von ca. 500 bp sowie das S. Typhimurium gyrA-Gen mit den einzukreuzenden Mutationen enthalten. Als Ausgangsvektor wurde pKOV genutzt. Zunächst wurde das mutationslose Gen aus M415 kloniert. Da bei der späteren Rekombination die Kanamycinresistenz-Kassette zusammen mit dem Anfangsbereich des gyrA-Gens in das Chromosom integriert wird, wird durch diese Insertion das 117 Ergebnisse chromosomale gyrA ausgeschaltet. Somit muss vor dem einzukreuzenden gyrA der natürliche Promotor erhalten werden. Bei der PCR von gyrA aus M415 wurde darauf geachtet, einen Bereich von ca. 240 bp vor dem Startcodon mit zu amplifizieren. Die Klonierungsstrategie ist in Abbildung 3-13 dargestellt. Die Restriktionsschnittstellen wurden so genutzt bzw. bei der PCR eingeführt, dass die Kanamycinresistenz-Kassette und das gyrA-Gen in entgegengesetzter Richtung exprimiert wurden. Zunächst wurde das gesamte gyrA-Gen unter Einführung der Schnittstellen NotI (3`-Ende) und BamHI (5`Ende) durch die Primer gyrA-Bam-fw und gyrA-Not-rv aus M415 mittels Phusion®Polymerase amplifiziert und in pKOV kloniert. Im entstandenen Vektor wurde das komplette gyrA-Gen sequenziert, um sicherzugehen, dass durch Fehler während der PCR keine unbeabsichtigten Basenaustausche eingeführt wurden. Anschließend wurde über SOEing-PCR die homologe Flanke aus M415 (Primer: HF-5-Sal und HF-3-pkD4) und die Kanamycinresistenz-Kassette inklusive der FRT-Sites (Primer: pKD4-5-BamHI und pKD4-3-HF) amplifiziert und fusioniert. Bei der Fusions-PCR wurden durch die Außenprimer Erkennungssequenzen für die Restriktionsenzyme SalI und BamHI eingeführt, welche eine anschließende Einklonierung des Fusionsprodukts aus homologer Flanke und Kanamycinresistenz-Kassette in das pKOV-gyrA-Konstrukt erlaubten. Der so konstruierte Rekombinationsvektor enthält eine Kanamycin- und Chloramphenicolresistenz-Kassette zur Selektion, einen temperatursensitiven Replikationsursprung, eine homologe Flanke für einen Rekombinationsschritt und das gesamte gyrA-Gen als zweite homologe Flanke. Nach der Ligation und Transformation in DH5α unter Kanamycinselektion (50 µg/ml) wurden Klone durch Überimpfen auf neue Selektionsplatten aufgereinigt, die Plasmide mittels Minipräparation isoliert und auf die richtige Größe überprüft. Der Erfolg der Klonierung wurde anhand der entstehenden Fragmentgrößen nach Doppelrestriktion mit den Restriktionsenzymen BamHI und SalI (erwartete Fragmentgrößen: 1980 bp und 8583 bp) sowie BamHI und NotI (erwartete Fragmentsgrößen: 2944 bp und 7619 bp) überprüft. Der Stamm mit dem korrekten Plasmid wurde als Dauerkultur angelegt und für weitere Klonierungen verwendet. Der nächste Klonierungsschritt beinhaltete das Einbringen der gewünschten Aminosäureaustausche in das gyrA-Gen. Die Mutationen in den Codons 59, 75 und 87 liegen im gyrA-Gen zwischen den Erkennungssequenzen für die Enzyme SacI und AatII. SacI spaltet die DNA 39 Codons vor der Mutation an Position 59 und AatII spaltet die DNA 71 Codons hinter der Mutation an Position 87. Eine Doppelrestriktion mit diesen beiden Enzymen ergab ein 415 bp großes Fragment, in dem die Mutationen enthalten waren. Die Klonierungsstrategie zur Einführung der Mutationen beinhaltete folgende Schritte: 118 Ergebnisse (1) Doppelrestriktion des Ausgangsvektors pPHB650 mit SacI und AatII zur Isolierung eines 415 bp-Fragmentes aus gyrA, (2) Gelextraktion des 10158 bp-Fragmentes (Plasmid-Anteil), (3) Dephosphorylierung des Plasmid-Anteils zur Verhinderung der Religation von unvollständig (d.h. nur mit einem Restriktionsenzym) gespaltenen Anteilen, (4) Doppelrestriktion der Vektoren pPHB451 bis 455 (siehe Abschnitt 3.2.3.2) mit SacI und AatII, wodurch ein 415 bp-Fragment aus gyrA ausgeschnitten wurde, (5) Gelextraktion des 415 bp-Fragementes (gyrA-Anteil) und (6) Ligation des Plasmid-Anteiles pPHB650 mit den gyrA-Anteilen, die die Mutationen enthalten. Abbildung 3-13: Klonierungsstrategie zur Herstellung von pPHB650 A Æ Amplifikation von gyrA mit Einführung der Schnittstellen für BamHI und NotI und Klonierung in pKOV; B Æ Amplifikation der Kanamycinresistenz-Kassette (aphA) inklusive der FRT-Sites aus pKD4 mit Einführung einer Erkennungssequenz für BamHI und eines Überhanges - homologe Flanke (HF); Amplifikation der homologen Flanke für die Rekombination aus dem Chromosom von M415 unter Einführung einer Erkennungssequenz für SalI und eines Überhanges, der homolog zur Kanamycinresistenz-Kassette ist; SOEing-PCR zur Fusion der homologen Flanke und der Kanamycinresistenz-Kassette; C Æ Einklonierung des Fusionsproduktes aus B in das pKOV-gyrAKonstrukt über die Schnittstellen BamHI und SalI; bla = Ampicillinresistenz-Kassette; cat = Chloramphenicolresistenz-Kassette; sacB = Gen für Levansucrase; ori / RepA = PlasmidReplikationsursprung 119 Ergebnisse Anschließend wurden DH5α-Zellen mit den Ligationsansätzen transformiert und auf kanamycinhaltigen (50 µg/ml) Agarplatten selektiert. Ein Anteil der gewachsenen Klone wurde durch Überimpfen auf Selektionsplatten gereinigt und die Plasmide durch Minipräparation isoliert. Als nachteilig bei dieser Klonierung erwies sich das Fehlen einer einfachen Kontrolle für den Klonierungserfolg. Durch den intragenischen Austausch der 415 bp-Fragmente wurde die Größe des Plasmides nicht verändert. Somit kam zur Kontrolle des Klonierungserfolges nur die Sequenzierung des betroffenen gyrA-Bereiches in Frage. Mit Hilfe des Primers gyrA-5-1-AS konnte direkt aus dem Plasmid sequenziert werden. Mittels der beschriebenen Klonierungsstrategie konnten zwei Plasmide hergestellt werden: pPHB653G, welches das gyrA-Gen mit dem Aminosäureaustausch Asp87 Æ Gly enthielt und pPHB655, mit den Mutationen Trp59 Æ Arg und Asp87 Æ Gly. Damit waren zwei wichtige Vektoren geschaffen, mit denen mittels Rekombination Mutanten erzeugt werden konnten. Diese Mutanten ermöglichen eine Bewertung der Rolle der Mutation an Position 87 und eine Untersuchung der denkbaren intragenischen Kompensation der Wirkungen der 87er Mutation durch die Mutation an Position 59. Um auch die Vektoren herzustellen, welche die Doppelmutation aus dem Zwischenderivat Nal2ori und die Dreifachmutation aus dem Impfstamm vacT enthalten, wurde auf die pT7-6-Derivate pPHB758 und pPHB759 (siehe Abschnitt 3.2.3.3) zurückgegriffen. Die Strategie beinhaltete die Verwendung der Restriktionsenzyme SacI (Erkennungssequenz im Gen vor Mutationen) und BamHI (ursprünglich zum Einklonieren des gyrA-Gens in pT7-6 über Primer an das 3`-Ende des Gens eingeführt). Dieser ca. 2650 bp große gyrA-Anteil mit den Mutationen wurde ausgetauscht gegen den gleichen gyrA-Anteil aus pPHB650. Das gyrA-Gen in pPHB650 besitzt am 3`-Ende eine NotI-Erkennungssequenz (eingeführt bei der Einklonierung von gyrA in pKOV, siehe Abbildung 3-13). Um eine Klonierung über diese unterschiedlichen Schnittstellen der Restriktionsenzyme NotI und BamHI zu ermöglichen, wurden beide Überhänge, die bei der Restriktionsspaltung entstehen, mittels DNA Polymerase I, Large (Klenow) Fragment aufgefüllt. So konnte eine blunt-end Ligation erfolgen. Die Klonierung wurde wie folgt durchgeführt: (1) erste Restriktion der Plasmide pPHB650 (pKOV-Derivate) mit NotI sowie pPHB758 und pPHB759 (pT7-6-Derivate) mit BamHI, (2) Auffüllen der durch die Spaltung der Restriktionsenzyme entstandenen 5`-Überhänge mittels DNA Polymerase I, Large (Klenow) Fragment, (3) Aufreinigung der Fragmente mittels QIAquick® PCR Purification Kit, (4) zweite Restriktion beider linearisierten Plasmide mit SacI, (5) Gelextraktion des Plasmid-Anteiles von pPHB650 (Größe 7897 bp) und der gyrAAnteile von pPHB758 bzw. pPHB759 (Größe: 2640 bp) und (6) Ligation der Plasmid-Anteile von pPHB650 und der gyrA-Anteile von pPHB758 bzw. pPHB759 über die SacI-Schnittstelle und blunt-ends. 120 Ergebnisse Nach der Transformation des Ligationsansatzes in DH5α und Selektion der Klone unter Zugabe von Kanamycin (50 µg/ml), wurde von ausgewählten Klonen eine Minipräparation durchgeführt. Anschließend wurde mit Hilfe des Primers gyrA-5-1-AS ein Teil des gyrAGens von den Plasmiden sequenziert. Zwei der sequenzierten Plasmide enthielten die gewünschten Mutationskombinationen. Alle hergestellten Rekombinationsvektoren sowie ihre Bezeichnung und die enthaltenen Mutationen sind in Tabelle 3-19 zusammengestellt. Tabelle 3-19: Rekombinationsvektoren basierend auf pKOV Bezeichnung enthaltene gyrA- Mutation(en) Herkunft pPHB650 keine M415 pPHB653G * Asp-87 Æ Gly pPHB453G pPHB655 Trp-59 Æ Arg; Asp-87 Æ Gly pPHB455 pPHB658 Gly-75 Æ Ala; Asp-87 Æ Gly; Ala-866 Æ Ser Nal2ori pPHB659 Trp-59 Æ Arg; Gly-75 Æ Ala; Asp-87 Æ Gly; Ala-866 Æ Ser vacT * die 3 als dritte Ziffer der Bezeichnung weist auf eine Mutation an Position 87 hin; das G kennzeichnet den Aminosäureaustausch von Aspartat zu Glycin Alle Vektoren basieren auf dem Plasmid pKOV und enthalten eine Chloramphenicolresistenz-Kassette, eine homologe Flanke (weit vor gyrA liegend), eine Kanamycinresistenz-Kassette als Selektionsmarker für die Rekombination und das komplette gyrA-Gen inklusive des natürlichen Promotors. Das Herstellen weiterer Vektoren auf Grundlage der vorhandenen Plasmide und Klonierungsstrategien ist jederzeit möglich. 3.2.4 Homologe Rekombination, Selektion und Screening der Mutanten Nachdem nun ausgewählte Rekombinationsvektoren zur Verfügung standen, wurde die homologe Rekombination unter Verwendung des zelleigenen Rekombinationssystems durchgeführt (Abschnitt 2.2.1.10.1). Augrund der Verwendung einer KanamycinresistenzKassette, welche zusammen mit den Mutationen eingekreuzt wurde (Abschnitt 3.2.1.1), war es möglich, die positiven Rekombinanten zu selektieren. Ohne aufwendige Sequenzierung würde es aber nicht möglich sein, zu kontrollieren, ob auch die gewünschten Mutationen eingekreuzt worden sind. Aus diesem Grund wurde ein Screeningsystem entwickelt, welches es ermöglichte, das Vorhandensein der Basenaustausche mittels einer einfachen PCR zu überprüfen. 3.2.4.1 Entwicklung eines Screeningsystems zum Nachweis der gyrAMutationen (ARMS) Die homologe Rekombination in der Zelle ist ein zufälliger und seltener Prozess. Das zusätzliche Einkreuzen der Kanamycinresistenz-Kassette eröffnet die Möglichkeit sofort diejenigen Zellen zu selektieren, bei denen eine Rekombination zwischen Vektor und Chromosom stattgefunden hat. Das eigentliche Ziel der homologen Rekombination war 121 Ergebnisse das Einkreuzen von Mutationen aus dem gyrA-Gen des pkOV-Vektors in das chromosomale Gen. Die homologe Rekombination wird über zwei homologe Flanken vermittelt. Für das zelleigene Rekombinationssystem sind homologe Flanken von ca. 500 bp Länge erforderlich, damit ein Austausch vermittelt wird. Im Rekombinationsvektor liegen mindestens 414 bp (für die Aminosäureposition 59) bzw. bis zu 500 bp (für die Mutation an Position 87) zwischen den einzukreuzenden Mutationen und der Kanamycinresistenz-Kassette. Ein Strangbruch und eine Rekombination vor den Mutationen (also kein Einkreuzen der gewünschten Mutationen) sind daher prinzipiell denkbar. Nach der Rekombination gefundene kanamycinresistente Klone müssen also auf das Vorhandensein der gewünschten Basenaustausche getestet werden. Im Rahmen dieser Arbeit wurde ein Screeningsystem für die erwünschten Basenaustausche basierend auf einer sequenzspezifischen PCR entwickelt (siehe Abschnitt 2.2.2.5.3). Es bestehen prinzipiell zwei Möglichkeiten der Nukleotidsequenzabfolge: Entweder die Rekombination war nicht erfolgreich, dann entspricht die Sequenz der des Ausgangsstammes M415 oder der gewünschte Basenaustausch ist vorhanden. Für jede PCR wurde der gleiche Forward-Primer gyrA-51-AS eingesetzt. Es wurden allerdings verschiedene Reverse-Primer entwickelt, die sich in ihrer Sequenz unterschieden. Die in der sequenzspezifischen PCR detektierbaren Basenaustausche betreffen die Aminosäureposition 59 (TGGÆAGG), 75 (GGTÆGCT) und 87 (GACÆGGC). Die für die ARMS-PCR verwendeten Primer sind in Tabelle 2-6 aufgelistet. Das Prinzip der sequenzspezifischen PCR wird exemplarisch in Abbildung 314 gezeigt. Die sequenzspezifische PCR wurde mit den Stämmen M415, Nal2ori, Nal2passage und vacT optimiert. Die optimierten Annealingtemperaturen für die PCRs lagen zwischen 56 und 65 °C. Prinzipiell sollte eine hohe Annealingtemperatur gewählt werden, um eine möglichst spezifische Anlagerung der Primer abhängig von den Sequenzänderungen zu erhalten. Die ARMS-PCR konnte somit sehr gut verwendet werden, um das Vorhandensein der Basenaustausche zu kontrollieren. Da sie als Alternative zur DNASequenzierung wesentlich billiger und schneller ein Ergebnis liefert, ließ sie sich sehr gut als Screeningmethode einsetzen. Nachteil der sequenzspezifischen PCR war, dass für jede zu testende Nukleotidposition optimalerweise zwei PCRs (für Original- und Mutantensequenz) durchzuführen waren, da es unter Umständen auch durch Bindung eines Primers trotz Mismatch zu falsch positiven PCR-Produkten kam. Eine negative PCR zeigte aber immer an, dass die Templatesequenz von der Primersequenz abwich. 122 Ergebnisse Abbildung 3-14: Prinzip der sequenzspezifischen PCR (ARMS) und beispielhafte Darstellung am Stamm Nal2ori oben: Prinzip der sequenzspezifischen PCR Æ Entstehung eines PCR-Produktes in Abhängigkeit der Basensequenz und entsprechender Bindung der Primer (P_ori, P_mut) unten: Gelfoto nach Auftrennung der ARMS-PCR-Produkte für den Stamm Nal2ori (G75A, D87G) nur bei Verwendung der Primer 59_orig, 75_mut und 87_mut entsteht ein PCR-Produkt; die Kontroll-PCR eines Fragmentes aus parE ist in allen Fällen positiv 3.2.4.2 Herstellung und Selektion der Rekombinanten Die Herstellung der Rekombinanten erfolgte durch die Verwendung des zelleigenen Rekombinationssystems mit Nutzung von Rekombinationsvektoren (2.2.1.10.1). Zunächst wurde der Vektor mit der einzukreuzenden gyrA-Mutation in den Stamm M415 transformiert und bei 30 °C angezogen. Durch den Wechsel der Wachstumstemperatur auf 42 °C und Chloramphenicolzugabe zum Medium erfolgte eine Selektion von Cointegraten, welche im ersten Rekombinationsschritt durch das Einkreuzen der Plasmide entstanden waren. Nach Absenken der Temperatur auf 30 °C, erfolgte das Auskreuzen der Plasmide und somit der zweite Rekombinationsschritt. Anschließend wurde durch ein Ausplattieren auf Agarplatten, welche Kanamycin (50 µg/ml) und Saccharose (5 %) enthielten, Kanamycinresistenz-Kassette, auf aber Rekombinanten kein Plasmid selektiert, mehr welche enthielten. die Durch Replikaplattierung auf Agarplatten mit Chloramphenicol (15 µg/ml) und Kanamycin (50 µg/ml) wurde der Verlust des Plasmides kontrolliert. Als nächstes erfolgte ein 123 Ergebnisse Screening mittels PCR zum Nachweis der Integration der Kanamycinresistenz-Kassette und der eingekreuzten Mutation(en). Abbildung 3-15 zeigt die verwendeten Primer, die erwarteten Fragmentgrößen der PCR und beispielhafte Ergebnisse aus verschiedenen Agarose-Gelelektrophoresen als Nachweis der Durchführung und Erfolge der PCRs. Abbildung 3-15: Screening-PCRs zum Nachweis der Integration der KanamycinresistenzKassette als Marker für eine erfolgreiche Rekombination oben: schematische Darstellung der erwarteten Fragmente nach PCR mit den entsprechenden Primern (Pfeile) im Vergleich die Rekombinante mit der eingekreuzten KanamycinresistenzKassette inklusive FRT-Sites (KanaR) vor dem gyrA-Gen zum Ausgangsstamm; HF = Homolge Flanke; unten: Auswahl von Gelfotos nach Auftrennung der PCR-Fragmente zur Unterscheidung der positiven (Rekombinanten) und negativen Klone (Situation wie im Ausgangsstamm) Weiterhin konnte durch die Verwendung von Primern, deren Bindung spezifisch für die Sequenz des Plasmides pKOV ist, nachgewiesen werden, ob das Plasmid wirklich eliminiert wurde. Die Kombinationen der für den Eliminierungstest verwendeten Primer sind: pKOV-R zusammen mit k2 (erwartete PCR-Fragmentgröße: 1434 bp) und pKOV-L in Kombination mit gyrA-5-4-AS (erwartete PCR-Fragmentgröße: 858 bp). In allen Fällen, in denen die Insertion der Kanamycinresistenz-Kassette nachgewiesen werden konnte, war der pKOV-Nachweis, wie erwartet, negativ. Nachdem die Rekombinanten auf eine positive Insertion der Kanamycinresistenz-Kassette getestet wurden, erbrachte die sequenzspezifische PCR den Nachweis, ob die gewünschten Basenaustausche vorhanden waren. Nicht bei allen Rekombinanten, welche die KanamycinresistenzKassette inseriert hatten, konnten die gewünschten Basenaustausche nachgewiesen 124 Ergebnisse werden. Zum Abschluss wurden mit dem Primer gyrA-5-1-AS die entsprechenden gyrAAbschnitte sequenziert, um die Anwesenheit der Mutationen zu bestätigen. Von den die Mutationen tragenden Rekombinanten wurden Dauerkulturen angelegt. Im Rahmen dieser Arbeit konnten verschiedene gyrA-Mutanten aus dem Ausgangsstamm M415 hergestellt werden. Tabelle 3-20 zeigt alle hergestellten Stämme mit den entsprechenden Basenaustauschen. Die Stämme erhalten den Namen „RM415“ zur Verdeutlichung, dass sie aus der homologen Rekombination hervorgegangen sind und im Anschluss die Nummer der Aminosäureposition, die verändert wurde. Tabelle 3-20: Über homologe Rekombination hergestellte gyrA-Mutanten Bezeichnung enthaltene gyrA-Mutation RM415 keine RM415-87 Asp-87 Æ Gly RM415-59-87 Trp-59 Æ Arg; Asp-87 Æ Gly RM415-75-87 Gly-75 Æ Ala; Asp-87 Æ Gly In allen Rekombinanten war die Kanamycinresistenz-Kassette im Chromosom enthalten. Um zu zeigen, dass ein Ausschneiden dieser Kassette über die FRT-Sites möglich ist (Abschnitt 3.2.1.1), wurde die Eliminierung exemplarisch für die Rekombinante RM41559-87 durchgeführt. Dafür wurde der Stamm mit dem Plasmid pCP20 transformiert und bei 30 °C unter Ampicillinzugabe (50 µg/ml) selektiert. Von diesem Plasmid wurde die FLP-Rekombinase exprimiert, welche die FRT-Sites erkannte und das Ausschneiden der Kassette bewirkte. Dabei blieb eine „Narben“sequenz. Durch anschließende Kultivierung des Stammes bei 37 °C ließ sich pCP20 leicht eliminieren. Über eine PCR mit den in Abbildung 3-20 gezeigten Primern, konnte die Eliminierung der KanamycinresistenzKassette über Fragmentgröße (bei Einsatz der Primer gyrA-1142fw und gyrA-3-1AS) bzw. Fehlen eines PCR-Produktes (bei Einsatz der Primer gyrA-1142fw und k2, k2 und kt oder kt und gyrA-3-1AS) bewiesen werden. Dies konnte am Beispiel des Stammes RM415-5987 durchgeführt und bewiesen werden. Zusätzlich zu dem System basierend auf den Vektor pKOV, konnten auch mit dem Helferplasmid pKD46 Rekombinanten hergestellt werden (Abschnitt 2.2.1.10.2). Mittels Restriktionsspaltung mit den Enzymen NotI und SalI wurde ein 5638 bp großes Fragment aus pPHB650 (Abbildung 3-13) ausgeschnitten. Dieses Fragment enthielt die Kanamycinresistenz-Kassette mit der 500 bp langen homologen Flanke und dem gyrAGen. Nach Gelextraktion dieses Fragmentes wurde der Stamm M415, welcher das Helferplasmid pKD46 enthielt, mittels Elektroporation mit dem DNA-Fragment transformiert. Von pKD46 wurden bei der vorherigen Herstellung der kompetenten Zellen durch Zugabe von 10 mM L-Arabinose die Phagengene exprimiert. Die sequenzspezifische Rekombination mit dem Restriktionsfragment war erfolgreich und führte zu einer nachweisbaren Insertion der Kanamycinresistenz-Kassette. Somit konnte die Rekombinante RM415 auf zwei verschiedenen Wegen hergestellt werden. 125 Ergebnisse Mehrere Versuche mit dem Vektor pPHB659 eine gyrA-Dreifachmutante (Mutationskombination aus dem Impfstamm) herzustellen schlugen fehl. Die nach der homologen Rekombination entstandenen Stämme enthielten zwar alle die Kanamycinresistenz-Kassette, aber keine der einzukreuzenden gyrA-Mutationen. Die Integration der Kanamycinresistenz-Kassette konnte über die Screening-PCRs nachgewiesen werden (Abbildung 3-15). Es konnte ebenfalls über PCR gezeigt werden, dass der Vektor eliminiert wurde. Es lagen also weder Cointegrate noch plasmidtragende Stämme vor. Eine Überprüfung des Plasmides pPHB659 auf das Vorhandensein der Dreifachmutation über DNA-Sequenzierung brachte wiederholt ein positives Ergebnis. Der für die homologe Rekombination eingesetzte Vektor besaß alle Mutationen. Trotz dreimaliger Wiederholung des Rekombinationsversuches konnten die Mutationen nicht in das Chromosom eingekreuzt werden bzw. ein Stamm mit den drei gyrA-Mutationen nicht selektiert werden. Als alternativer Versuch wurde die homologe Rekombination mit dem phagengenbasierten System wie oben beschrieben mit einen Fragment aus pPHB659 (nach NotI und SalI Restriktionsspaltung) durchgeführt. Dieser Versuch ergab ebenfalls positive Rekombinanten, d.h. es erfolgte ein Einkreuzen der KanamycinresistenzKassette. Eine Selektion eines Stammes, welcher alle drei Mutationen in das Chromosom eingekreuzt hat, war aber auch mit dieser Strategie nicht möglich. Diese Ergebnisse zeigen, dass die Rekombination vor den gyrA-Mutationen stattfindet. Eine Rekombination irgendwo im gyrA-Gen hinter den Mutationen ist aufgrund der Länge des homologen Bereiches zwar viel wahrscheinlicher, wenn aber eine Gyrase mit einer Dreifachmutation weniger aktiv und die Zelle damit eingeschränkt oder gar nicht lebensfähig ist, können nur die Zellen selektiert werden, wo die Rekombination vor den Mutationen stattgefunden hat. Somit ließen sich nur Zellen selektieren, wo zwar eine Integration der Kanamycinresistenz-Kassette aber kein Einkreuzen der Mutationen erfolgt war. 3.2.5 Charakterisierung der Rekombinanten Die über homologe Rekombination hergestellten Mutanten wurden charakterisiert, um die Rolle der gyrA-Mutationen bei der Ausprägung bestimmter Eigenschaften aufzuklären. Zunächst wurden die minimalen Hemmkonzentrationen für ausgewählte (Fluor)chinolone bestimmt. Um zu beweisen, dass eine Veränderung der Antibiotikaempfindlichkeiten der rekombinanten Stämme auf die eingekreuzten gyrA-Mutationen zurückzuführen ist, wurde ein Komplementationstest mit der Einführung von gyrA+ durchgeführt (Abschnitt 2.2.1.8). Im Anschluss wurden zusätzlich das Wachstumsverhalten Superspiralisierungsgrad für jede Rekombinante untersucht. 126 und der Ergebnisse 3.2.5.1 Bestimmung der Fluorchinolon-MHK-Werte Ein Hauptschwerpunkt der Arbeit war es herauszufinden, ob bzw. welche der gyrAMutationen bei den Mutanten Nal2ori und Nal2passage für die reduzierten Empfindlichkeiten gegenüber Fluorchinolonen verantwortlich sind. Weiterhin sollte eine mögliche Rolle des Aminosäureaustausches W59R im Impfstamm vacT für die Kompensation der erhöhten Fluorchinolon-MHK-Werte untersucht werden. Tabelle 3-21 zeigt die ermittelten MHK-Werte. Als Vergleich wurden die Werte der Ausgangsstämme M415, Nal2ori und vacT mit aufgeführt. Tabelle 3-21: Minimale Hemmkonzentrationen [µg/ml] ausgewählter Fluorchinolone für die Ausgangsstämme und die Rekombinanten M415 Nal2ori vacT RM415 RM41587 RM41559-87 RM41575-87 4 512 32 4 256 64 256 Norfloxacin 0,125 0,5 0,125 0,125 0,5 0,25 0,5 Ofloxacin 0,125 0,25 0,06 0,125 0,5 0,25 0,5 Levofloxacin 0,03 0,125 0,03 0,06 0,25 0,125 0,5 Ciprofloxacin 0,03 0,125 0,03 0,03 0,25 0,06 0,25 Sparfloxacin 0,03 0,125 0,006 0,03 0,125 0,06 0,125 Fleroxacin 0,06 0,5 0,125 0,06 0,5 0,25 1 Pefloxacin 0,125 0,5 0,125 0,125 1 0,5 1 Nalidixinsäure Den Daten der MHK-Wert-Bestimmung ist zu entnehmen, dass alle Rekombinanten mit gyrA-Mutationen erniedrigte Empfindlichkeiten gegenüber Fluorchinolonen zeigten. Der Stamm RM415 diente als Kontrolle. In diesem Stamm war auch eine Integration der Kanamycinresistenz-Kassette erfolgt, aber es wurden keine gyrA-Mutationen eingekreuzt, sondern das Gen des Ausgangsstammes M415 genutzt. Der Kontrollstamm RM415 zeigte die gleichen MHK-Werte wie der Ausgangsstamm M415. So konnte gezeigt werden, dass eine Integration des Selektionsmarkers keine Auswirkungen auf die Empfindlichkeiten gegenüber Fluorchinolonen hat. Erst durch Einführung der gyrA-Mutationen kommt es zu einer Erhöhung der Fluorchinolon-MHKs. Die Mutation an Position 87 erniedrigte die Empfindlichkeit der entsprechenden Mutante gegenüber den meisten untersuchten Fluorchinolonen um zwei bis drei Verdünnungsstufen, gegenüber Nalidixinsäure sogar um sechs Verdünnungsstufen. Die Rekombinante RM415-75-87 mit gyrA-Mutationen an den Positionen 75 und 87 verhielt sich genauso wie RM415-87. Dagegen zeigte Mutante RM415-59-87 eine geringere Erhöhung der MHK-Werte als die anderen beiden gyrARekombinanten. Die Empfindlichkeiten gegenüber den meisten getesteten Fluorchinolone waren nur um ein bis maximal zwei Verdünnungsstufen, gegenüber Nalidixinsäure um vier Verdünnungsstufen erniedrigt. Das heißt, die MHK-Werte der Chinolone für RM415-59-87 lagen im Durchschnitt mindestens eine Stufe unter den Werten für die Rekombinanten 127 Ergebnisse RM415-87 und RM415-75-87. Alle Rekombinanten zeigten aber prinzipiell erhöhte (Fluor)chinolon-MHK-Werte. 3.2.5.2 Komplementationstest mittels eines plasmidcodierten gyrA+ Um den Beweis zu erbringen, dass die eingekreuzten gyrA-Mutationen für die Erniedrigung der Empfindlichkeit der Rekombinanten gegenüber Fluorchinolonen verantwortlich sind, wurde ein Komplementationstest mit einem Wildtyp-Gen von gyrA (gyrA+) durchgeführt (Abschnitt 2.2.1.8). Durch Elektroporation wurden die verschiedenen Stämme mit dem Plasmid pBP517 transformiert. Aufgrund des Dominanzeffekts des plasmidcodierten gyrA+-Gens über ein mutiertes gyrA-Gen ist ein Absinken der MHKWerte der Fluorchinolone auf das Niveau des Ausgangsstammes M415 zu erwarten. In Tabelle 3-22 sind die Ergebnisse der MHK-Bestimmungen für die Stämme ohne Plasmid, mit Kontrollplasmid pBP507 (ohne gyrA-Gen) und mit Plasmid pBP517 (gyrA+) aufgelistet. Nach der Einführung eines plasmidcodierten gyrA+-Gens in die Zellen erreichten alle Stämme die MHK-Werte für den Ausgangsstamm M415. Somit konnte gezeigt werden, dass die Erniedrigung der Fluorchinolon-Empfindlichkeiten auf dem Einkreuzen der gyrAMutationen durch homologe Rekombination beruhte. Die Kontrollen (Stämme mit Plasmid pBP507) zeigten keine Veränderungen der MHK-Werte im Vergleich zum gleichen Stamm ohne Plasmid. M415 RM415 Tabelle 3-22: Ergebnisse aus dem Komplementationstest für die Rekombinanten im Vergleich zum Ausgangsstamm M415 Plasmid - - - pBP 507 pBP 517 - pBP 507 pBP 517 - pBP 507 pBP 517 NDS 4 4 256 256 4 64 128 4 256 256 2 NOR 0,125 0,125 0,5 0,5 0,06 0,25 0,5 0,125 0,5 1 0,125 OFL 0,125 0,125 0,5 0,5 0,25 0,25 0,5 0,125 0,5 0,5 0,125 LEV 0,03 0,06 0,25 0,25 0,06 0,125 0,125 0,06 0,5 0,5 0,06 CIP 0,03 0,03 0,25 0,25 0,015 0,06 0,06 0,03 0,25 0,25 0,03 SPA 0,03 0,03 0,125 0,125 0,06 0,06 0,06 0,06 0,125 0,125 0,06 FLEX 0,06 0,06 0,5 0,5 0,06 0,25 0,25 0,125 1 1 0,125 PEF 0,125 0,125 1 1 0,06 0,5 0,5 0,125 1 1 0,25 128 RM415-87 RM415-59-87 RM415-75-87 Ergebnisse 3.2.5.3 Bestimmung der DNA-Superspiralisierungsgrade Eine Untersuchung der DNA-Superspiralisierungsgrade der hergestellten Rekombinanten sollte Aufschluss darüber geben, ob die eingeführten gyrA-Mutationen eine Veränderung des DNA-Superspiralisierungsgrades bewirken. In die rekombinanten Stämme wurden mittels Elektroporation jeweils die Reportergensystem-Plasmide pPHB90 bzw. pPHB91 transformiert, die Luciferaseaktivitäten gemessen und daraus die jeweiligen Qsc-Werte, als Parameter für den DNA-Superspiralisierungsgrad, berechnet (Tabelle 3-23). Zum Vergleich wurde der Stamm SM415-87 aus dem Selektionsversuch (Abschnitt 3.1.5.1) mitgeführt. Abbildung 3-16 zeigt die Qsc-Werte in Prozent von allen untersuchten Stämmen. Dabei wurde Superspiralisierungsgrad der der Ausgangsstamm Selektionsmutante M415 100 % SM415-87 gesetzt. wich Der nicht vom Ausgangsstamm M415 ab. In der GyrA-Untereinheit der Selektionsmutante ist die Aminosäure Asparagin Aminosäureaustausch an Position bewirkte Superspiralisierungsgrades 87 aber gegenüber gegen keine M415. Im Tyrosin ausgetauscht relevante Gegensatz Dieser Änderung dazu des zeigte die Rekombinante RM415-87, welche eine Mutation an Codon Aspartat 87 zu Glycin aufweist, einen deutlich reduzierten Qsc-Wert. Die gyrA-Mutante RM415-87 (D87G) besaß einen reduzierten DNA-Superspiralisierungsgrad. Tabelle 3-23: Daten aus den DNA-Superspiralisierungsmessungen für die Rekombinanten Stamm M415 SM415-87 RM415 RM415-87 RM41559-87 RM41575-87 spezifische Aktivität ptopA* spezifische Aktivität pgyrA* Qsc** - - - 3051,54 1494,56 2,04 2045,82 989,30 2,07 2237,00 1316,17 1,70 2129,03 1125,91 1,89 2805,81 1826,54 1,54 2537,26 1537,38 1,65 1353,47 1616,60 0,84 2332,41 2144,87 1,09 4981,81 4529,66 1,10 2593,87 2224,04 1,17 970,48 1030,08 0,94 2179,40 2198,94 0,99 1727,48 1427,16 1,21 2244,71 2240,81 1,00 2933,63 2780,54 1,06 Mittelwert Qsc (± s) Prozent (± s %) 1,80 100 % (±0,28) (± 15,5) 1,93 107,22 % (±0,21) (±10,88) 1,69 93,98 % (± 0,18) (± 10,65) 1,01 56,11 % (±0,15) (± 14,85) 1,03 57,22 % (±0,12) (±11,65) 1,09 60,55 % (±0,11) (±10,09) * spezifische Aktivität ptopA bzw. pgyrA = RLU / OD546nm; ** Qsc = spezifische Aktivität ptopA / spezifische Aktivität pgyrA; s = Standardabweichung, s % = relative Standardabweichung 129 Ergebnisse Der Kontrollstamm RM415 zeigte einen sehr ähnlichen Qsc-Wert (ca. 1,7) wie der Ausgangsstamm M415 (ca. 1,8). Unter Berücksichtigung der Standardabweichung konnte gezeigt werden, dass der DNA-Superspiralisierungsgrad des Kontrollstammes RM415 dem des Ausgangsstammes entspricht. Somit spielt die integrierte KanamycinresistenzKassette auch für den Qsc-Wert keine Rolle. Die übrigen Rekombinanten RM415-87, RM415-59-87 und RM415-75-87 zeigten alle die gleiche Reduktion des Qsc-Wertes auf ca. 56-61 %. Im Vergleich zum Ausgangsstamm M415 waren alle Superspiralisierungsgrade der Rekombinanten signifikant um fast die Hälfte erniedrigt. Berücksichtigt man die relativen Standardabweichungen, gab es aber zwischen den untersuchten Rekombinanten sowie Nal2ori und vacT keine signifikanten Unterschiede (siehe Abbildung 3-16). Abbildung 3-16: Vergleich der Superspiralisierung aller Stämme (Ausgangsstamm M415 = 100 %) inklusive relativer Standardabweichung in Prozent blau: Ausgangsstämme M415, Nal2ori, Nal2passage und Impfstamm vacT; türkis: SM415-87 aus Selektionsversuch; rot: hergestellte Rekombinanten (gyrA-Mutanten) 130 Ergebnisse 3.2.5.4 Untersuchungen zum Wachstumsverhalten Es wurden die Generationszeiten für die Rekombinanten bestimmt, indem die Entwicklung der optischen Dichte über die Zeit gemessen wurde (Abschnitt 2.2.1.4). Ziel war es, die Unterschiede im Wachstumsverhalten der Rekombinanten zueinander und im Vergleich zum Ausgangsstamm M415 zu beurteilen. Es sollte untersucht werden, ob bei den Stämmen das Auftreten der gyrA-Mutationen zu einer Fitnessreduktion bzw. – kompensation, gemessen als Veränderung der Generationszeit, führen würde. Tabelle 3-24: Wachstumsuntersuchung der Rekombinanten M415 RM415 RM415-87 RM415-59-87 RM415-75-87 g in min 36 36 38 34 37 g in % 100 100 106 94 103 g = Generationszeit Die Generationszeiten der Rekombinanten im Vergleich untereinander und mit dem Ausgangsstamm M415 zeigten keine großen Abweichungen. Als Vergleich können die Werte aus Tabelle 3-1 (Abschnitt 3.1.1) und Tabelle 3-15 (Abschnitt 3.1.6) herangezogen werden, in denen die ermittelten Unterschiede zwischen den Stämmen M415, Nal2ori, Nal2passage und vacT dargestellt sind. Dabei zeigte der Impfstamm bei der Bestimmung über die OD-Messung im Vergleich zum Ausgangsstamm eine Verlängerung der Generationszeit um ca. 35% sowie Nal2ori und Nal2passage um ca. 74 bis 90%. Legt man die Ergebnisse der KBE-Bestimmung zugrunde, waren die Werte sogar noch höher. Die prozentualen Abweichungen von bis zu 6% bei den Rekombinanten liegen im Bereich der methodischen Schwankung der Messung. 3.3 Untersuchung des Einflusses von Effluxpumpen An der Ausprägung der Empfindlichkeit eines Stammes gegenüber Antibiotika können verschiedene Faktoren beteiligt sein. Im Falle der Fluorchinolone kommt neben Variationen in den Genen für die Topoisomerasen (Zielstrukturveränderung) vor allem der Antibiotikakonzentration am Wirkort eine große Bedeutung zu. Neben passivem Influx über Porine, spielen vor allem die Aktivitäten und der Expressionsstatus von Membranpumpen für den aktiven Efflux eine entscheidende Rolle (siehe Abschnitt 1.3.2). Eine reduzierte Empfindlichkeit gegenüber bestimmten Antibiotika kann somit durch Targetgenmutationen aber auch durch eine erhöhte Expression von Effluxpumpen hervorgerufen sein. Die veränderte Expression der Effluxpumpen kann auf Defekten der Strukturgene oder Veränderungen in den Regulatoren der Effluxpumpen beruhen. Durch eine MHK-Wert-Bestimmung unter Zugabe eines Effluxpumpeninhibitors (EPI) lässt sich der Einfluss von Effluxpumpen auf die Antibiotikaempfindlichkeit bestimmen. Dieser würde sich dann in einer Reduktion der MHK-Werte ausdrücken. 131 Ergebnisse 3.3.1 MHK-Wert-Bestimmung des Effluxpumpeninhibitors Um den Einfluss des aktiven Efflux zu untersuchen, wurden die minimalen Hemmkonzentrationen unter Zugabe eines Effluxpumpeninhibitors bestimmt. Die Konzentration des zugesetzten EPIs ist entscheidend für die Veränderung der MHK-Werte des untersuchten Stammes. Eine zu hohe Konzentration des eingesetzten EPIs würde ein Ausschalten zu vieler Effluxpumpen bewirken, was sich für die Zelle letal auswirkt. Bereits bei Vorversuchen wurde festgestellt, dass die Stämme M415, Nal2ori, Nal2passage und vacT sehr unterschiedlich auf Zugabe hoher EPI-Konzentrationen reagierten. Um die Konzentration des eingesetzten EPIs optimal anzupassen, wurde die minimale Hemmkonzentration des Effluxpumpeninhibitors für die einzelnen Stämme zunächst in Abwesenheit eines Antibiotikums bestimmt (Tabelle 3-25). Der Stamm ATCC14028 wurde als Referenzstamm und die selektierte Mutante SM415-87 als Kontrollstamm, welcher nur eine gyrA-Mutation besitzt, mitgeführt. Tabelle 3-25: Bestimmung der EPI-MHK [µg/ml] EPI-MHK ATCC14028 M415 SM415-87 Nal2ori Nal2passage vacT 256 256 256 128 64 16 Die Werte aus Tabelle 3-25 zeigen, dass der Impfstamm vacT die höchste Empfindlichkeit gegenüber dem Effluxpumpeninhibitor aufwies (16µg/ml). Die Empfindlichkeit nahm in der Reihe M415 Æ Nal2ori Æ Nal2passage Æ vacT ab. Aus den ermittelten EPI-MHK-Werten wurden die Konzentrationen abgeleitet, welche in den folgenden Antibiotika-MHK-WertBestimmungen eingesetzt wurden. 3.3.2 MHK-Wert-Bestimmung unter Verwendung eines Effluxpumpeninhibitors Im Folgenden wurden für ausgewählte Antibiotika MHK-Werte mit und ohne die Zugabe des Effluxpumpeninhibitors Phenylalanyl-Arginyl-β-Naphthylamid bestimmt (Tabelle 3-26). Die für die einzelnen Stämme eingesetzten EPI-Konzentrationen leiteten sich aus den vorher bestimmten MHK-Werten des EPIs ab und sind ebenfalls der Tabelle 3-26 zu entnehmen. 132 Ergebnisse Tabelle 3-26: Bestimmung der minimalen Hemmkonzentrationen [µg/ml] verschiedener Antibiotika mit und ohne Effluxpumpeninhibitor M415 SM415-87 +EPI Nal2ori +EPI Nal2passage +EPI vacT +EPI +EPI EPI-Konz.* 0 64 0 64 0 32 0 16 0 8 ERY 64 1 64 0,5 32 0,5 64 0,5 4 0,5 ROXI ≥64 0,5 32 0,25 ≥64 2 ≥64 0,5 4-8 0,5 CLAR 32 0,125 32 0,25 16 0,5 64 0,125 1 0,125 TYL 256 4 n.b. n.b. 32 1 32 8 8 2 TET 1 0,5 1 1 1 0,25 1 0,25 0,5 0,25 NDS 4 0,25 64 2 512 32 64 4 32 4 NOR 0,125 0,03 0,5 0,125 0,5 0,125 0,25 0,125 0,125 0,125 CIP 0,03 0,007 0,125 0,015 0,125 0,015 0,125 0,015 0,03 0,015 SPA 0,03 0,007 0,06 0,0015 0,125 0,003 0,06 0,003 0,007 0,003 GAT 0,06 n.b. n.b. n.b. 0,125 0,003 0,125 0,003 0,03 0,003 MOX 0,125 n.b. n.b. n.b. 0,25 0,015 0,25 0,015 0,06 0,015 * für die Bestimmungen eingesetzte EPI-Konzentration in µg/ml; EPI = Effluxpumpeninhibitor = PhenylalanylArginyl-β-Naphthylamid; n.b. = nicht bestimmt Beim Ausgangsstamm M415 war zu erkennen, dass die MHK-Werte unter Einfluss des Effluxpumpeninhibitors stark absinken. Bei den Makroliden waren Unterschiede in den MHK-Werten von mindestens sechs Verdünnungsstufen festzustellen, für Nalidixinsäure vier und für die getesteten Fluorchinolone zwei Verdünnungsstufen. Für den Stamm SM415-87 zeigten sich ohne EPI die durch die Gyrasemutation erhöhten MHK-Werte der (Fluor)chinolone, welche jedoch in Anwesenheit des EPIs um zwei bis drei Verdünnungsstufen, bei Nalidixinsäure um fünf Stufen absanken. Auch bei den Stämmen Nal2ori und Nal2passage sanken die erhöhten MHK-Werte der Fluorchinolone in Anwesenheit des Effluxpumpeninhibitors auf sehr niedrige Werte (von z.T. 0,003 µg/ml) stark ab. Bei den Makroliden zeigten die Stämme SM415-87, Nal2ori und Nal2passage sowie der Ausgangsstamm M415 eine natürliche Resistenz, welche unter dem Einfluss des EPIs auf ein sensibles Niveau um bis zu sieben Verdünnungsstufen absank. Der Stamm vacT, welcher bereits ohne Effluxpumpeninhibitor eine Makrolidsensibilität aufwies, zeigte mit EPI noch einmal eine Erniedrigung um drei Verdünnungsstufen. Auch bei den Fluorchinolonen zeigte sich ein ähnliches Bild: Das Niveau auf welches die MHK-Werte unter maximalem EPI-Einfluss sanken, war für jedes getestete Antibiotikum ähnlich, auch wenn die Ausgangswerte vorher unterschiedlich hoch waren. Eine Ausnahme stellte Tetracyclin dar, dessen MHK-Werte für alle Stämme mit und ohne Effluxpumpeninhibitor kaum verändert waren. 133 Ergebnisse Tabelle 3-27 zeigt die Ergebnisse eines Versuches, der die Beobachtungen bestätigt. Es ist deutlich zu erkennen, dass die MHK`s der getesteten Antibiotika auf das gleiche Niveau zurückgehen, wenn die maximale für den Stamm zu tolerierende Menge an Effluxpumpeninhibitor zugesetzt wird. Bei dem Makrolid Erythromycin betrugen die Differenzen bei den Stämmen M415, Nal2ori und Nal2passage sechs bis sieben Verdünnungsstufen, da die Stämme vorher mit MHK-Werten von 32-64 µg/ml eine Resistenz gegenüber Erythromycin zeigten. Der Impfstamm vacT reagierte bereits ohne EPI sensibel auf Erythromycin und es war nur noch eine geringe Menge an EPI nötig, den Wert maximal abzusenken. Bei dem Fluorchinolon Sparfloxacin erkennt man, dass die Stämme Nal2ori und Nal2passage, da sie vorher leicht reduzierte Empfindlichkeiten besaßen, auch die höchste MHK-Stufen-Absenkung bei dem Zusatz von Effluxpumpeninhibitor zeigten. Die MHK-Werte von Tetracyclin waren durch den Effluxpumpeninhibitor nur schwach beeinflusst. Bei allen Stämmen zeigten sich mit und ohne EPI ähnliche Werte. Tabelle 3-27: Absinken der MHK-Werte in Abhängigkeit der zugesetzten Konzentration des Effluxpumpeninhibitors (EPI-Konz.) in µg/ml ohne 4 8 16 32 64 MHK-StufenDifferenz M415 64 n.b. 32 n.b. n.b. 1 6 Nal2ori 32 n.b. 4 n.b. 0,5 k.W. 6 Nal2passage 64 n.b. 4 0,5 k.W. k.W. 7 vacT 4 0,5 0,5 k.W. k.W. k.W. 3 M415 0,03 n.b. 0,015 n.b. n.b 0,007 2 Nal2ori 0,125 n.b. 0,015 n.b. 0,003 k.W. 5 Nal2passage 0,06 n.b. 0,007 0,003 k.W. k.W. 4 vacT 0,007 0,015 0,003 k.W. k.W. k.W. 2 M415 1 n.b. 1 n.b. n.b 0,5 1 Nal2ori 1 n.b. 0,5 n.b. 0,25 k.W. 2 Nal2passage 1 n.b. 0,5 0,25 k.W. k.W. 2 0,5 0,25 0,25 k.W. k.W. k.W. 1 EPI-Konz. Erythromycin Sparfloxacin Tetracyclin vacT n.b. = nicht bestimmt; k.W. = kein Wachstum Zusammenfassend lässt sich sagen, dass in der Reihe M415 Æ Nal2ori Æ Nal2passage Æ vacT immer geringere Mengen an Effluxpumpeninhibitor benötigt wurden, um die MHKWerte auf das gleiche Niveau zu senken. 134 Diskussion 4 Diskussion Salmonellen stellen als Erreger zoonotischer Infektionen nach wie vor ein großes Gesundheitsrisiko dar. Infektionsquellen sind vor allem mit Salmonella spec. kontaminierte Lebensmittel, wie Fleisch, aber auch Eier und Milchprodukte. Eine Therapie mit Antibiotika kommt für Menschen nur bei schweren Verläufen oder für Risikopatienten (Ältere Personen, Kleinkinder) in Frage. Eine sinnvolle Maßnahme zur Bekämpfung von Salmonellosen stellt eine Immunprophylaxe der Nutztiere mit Hilfe von Lebendimpfstoffen dar. Der Vorteil der Immunisierung von Tieren ist der präventive und oft lebenslange Schutzeffekt. Besonders vor dem Hintergrund der in den letzten Jahren stark zunehmenden Resistenzentwicklung bei Salmonella spec. [18,120] kommt der Lebendimpfstämme von Impfstammentwicklung eine immer größere Bedeutung zu [22,313]. Zwischen 1994 und 2002 sind in Deutschland 10 S. Typhimurium, Enteritidis und Choleraesuis zur Anwendung bei Hühnern, Schweinen und Rindern zugelassen worden [290]. Der Lebendimpfstamm „TAD Salmonella vacT“ (vacT) wird seit 1994 zur Prophylaxe von Salmonellosen bei Hühnern eingesetzt [38,290]. Er wurde aus dem Wildstamm S. Typhimurium M415 durch Selektion auf Nalidixinsäure und Rifampicin und anschließend durch eine zusätzliche Behandlung mit N-Methyl-N´Nitrosoguanidin erzeugt [177,179]. Die Entwicklung des Impfstammes beruhte auf der Theorie des Stoffwechseldriftes. Diese besagt, dass Mutationen in essentiellen Genen den gesamten Stoffwechsel der Zelle so verändern, dass sich das phänotypisch auf die Empfindlichkeiten gegenüber Antibiotika, die Generationszeit sowie die Reduktion der Virulenz auswirkt [178] (Abschnitt 1.4). Der attenuierte Lebendimpfstamm vacT besitzt vier verschiedene genetische Marker, welche durch Resistenz bzw. Sensibilität gegenüber bestimmten Antibiotika charakterisiert sind: (1) „Rif“ (2) „Nal (3) „rtt“ und (4) „ssq“. Die ersten beiden Marker kennzeichnen Resistenz gegen Rifampicin (1) und Nalidixinsäure (2) und dienen der Selektion von Isolaten des Impfstammes sowie ihrer Unterscheidung von Wildstämmen. Der rtt Marker (reversion to tensid tolerance) ist assoziiert mit Makrolidsensibilität (3) und der ssq Marker (super susceptible to quinolones) mit Fluorchinolonsensibilität (4). Beide fungieren als Sicherheitsmarker. Die molekularen Grundlagen der Marker waren zu Beginn der Arbeit weitgehend unbekannt. Für die Makrolidsensibilität wurde eine veränderte Durchlässigkeit der Zellwand verantwortlich gemacht. Eine Mutation im rpoB-Gen, dem Gen für die βUntereinheit der RNA-Polymerase, könnte Rifampicinresistenz bedingen. Die Nalidixinsäureresistenz wurde mit einer Mutation (D87G) im gyrA-Gen, dem Gen für die AUntereinheit der Gyrase, begründet [38,179]. Nicht mit bisherigen Erfahrungen im Einklang steht in dieser Beziehung das Auftreten des ssq Markers (Fluorchinolonsensibilität): Mutationen in der QRDR (quinolone resistance determining region) von gyrA sind als Fluorchinolonresistenz vermittelnd bekannt [48,51,118,128,253] (Abschnitt 1.3.1.3). Die Nalidixinsäureresistenz bei gleichzeitiger Fluorchinolonsensibilität 135 Diskussion in einem Stamm mit der gyrA-Mutation D87G bildete somit einen Widerspruch, dessen Aufklärung das Hauptanliegen dieser Arbeit war. Eine Kompensation der Wirkung der resistenzvermittelnden gyrA-Mutation könnte einerseits intragenisch durch eine weitere gyrA-Mutation oder andererseits extragenisch durch Veränderungen anderer Genloci erfolgen. Für die Untersuchung der Bedeutung der im Impfstamm auftretenden Mutationen wurden durch homologe Rekombination isogene Stämme mit entsprechenden Veränderungen im gyrA-Gen hergestellt. Neben der Charakterisierung der Rekombinanten mit den aus dem Impfstamm identifizierten gyrA-Mutationen wurden auch Untersuchungen zum Einfluss des aktiven Efflux im Impfstamm vacT durchgeführt. 4.1 Charakterisierung der Untersuchungsstämme 4.1.1 Mutationen in den Topoisomerasegenen Vordringliches Ziel dieser Arbeit war es, die molekulare Grundlage für die Nalidixinsäureresistenz bei gleichzeitiger Fluorchinolonsensibilität des Impfstammes TAD Salmonella vacT zu untersuchen. Bekannte Ursachen für eine Chinolonresistenz sind neben Veränderungen der Expression von Porinen [141,142,148] oder Effluxpumpen [24,26,238] vor allem Mutationen in den Genen für Typ II Topoisomerasen (Gyrase, Topoisomerase IV). So wurden z.B. in chinolonresistenten Salmonella spec. und E. coli vor allem Austausche von Aminosäuren an den Codons Serin-83 bzw. Aspartat-87 in der Chinolonresistenz-bestimmenden Region (QRDR) des Gens gyrA, aber auch Mutationen in gyrB beschrieben [17,48,108,118,125,128,135,253]. Weiterhin stellt die Topoisomerase IV ein sekundäres Target für die Fluorchinolone (FQ) dar und in den Genen der Untereinheiten (parC, parE) auftretende Mutationen wurden als bedeutend für die Resistenzentwicklung beschrieben [17,48,124,125]. Um alle Mutationen in den Genen für beide Zielstrukturen zu identifizieren, erfolgte eine Sequenzierung der kompletten Gene gyrA, gyrB, parC und parE der Untersuchungsstämme M415, Nal2ori, Nal2passage und vacT (Abschnitt 2.2.2.10). Durch den Vergleich der Nukleotidsequenzen aller vier Gene von vacT mit M415 konnten ausschließlich im Gen für die A-Untereinheit der Gyrase vier Mutationen identifiziert werden (Abschnitt 3.1.4). Alle diese Mutationen führen auch zum Austausch einer Aminosäure. Von diesen vier gyrA-Mutationen liegen zwei (Gly-75ÆAla und Asp-87ÆGly) innerhalb und zwei (Trp-59ÆArg und Ala866ÆSer) außerhalb der QRDR. In den Stämmen Nal2ori und Nal2passage wurden nur drei Mutationen im gyrA-Gen gefunden, der Aminosäureaustausch Trp-59ÆArg fehlte. Die Mutation Asp-87ÆGly (D87G) scheint für die Nalidixinsäureresistenz von besonderer Bedeutung zu sein, denn Änderungen dieser Aminosäure sind in den gyrA-Genen chinolonresistenter Stämme verschiedener Species sowohl bei klinischen Isolaten [48,108,237,239] als auch bei Labormutanten von E. coli und Salmonella spec. 136 Diskussion [118,128,253] identifiziert worden. Neben Glycin findet an dieser Aminosäureposition sehr häufig ein Wechsel zu Tyrosin oder Asparagin statt [102,108,239,253,293]. Unklar war, ob neben der Mutation D87G die in allen drei Mutanten (Nal2ori, Nal2passage und vacT) vorkommenden Mutationen Gly-75ÆAla (G75A) und Ala-866ÆSer (A866S) eine Bedeutung für die Resistenz gegen Nalidixinsäure haben. In bisherigen Studien wurde bislang keine Mutation an Codon 75 beschrieben. Lediglich ein Basenaustausch in Codon 72 (D72G), dessen Bedeutung aber nicht geklärt ist, wurde bei einer in vitro selektierten Mutante von E. coli nach vier Selektionsschritten gefunden [162]. Reyna et al. [253] identifizierten eine gyrA-Doppelmutante an den Positionen 67 (AlaÆPro) und 81 (GlyÆSer) in S. Typhimurium. Diese Autoren wiesen aber nur dem Aminosäureaustausch an Position 81 eine Bedeutung für die reduzierte FQ-Empfindlichkeit zu. Auch der Aminosäureaustausch an Codon 866 (AlaÆSer) ist bisher unbekannt. Er befindet sich relativ weit am 3’-Ende des Gens. Cullen et al. [62] beschrieben eine Ser-828 Æ Ala Mutation, welche in ihren Studien aber ohne Konsequenz für den Stamm blieb. Über den möglichen Einfluss der Mutation Ala-866ÆSer lässt sich daher nur spekulieren. Denkbar wäre eventuell, dass sie entweder für die Interaktion der A- und B-Untereinheiten des Gyrase-Enzyms eine Rolle spielt oder die DNA-Bindung an das Enzym beeinflussen könnte [252]. Alle drei für Nal2ori und Nal2passage beschriebenen Mutationen (G75A, D87G, A866S) wurden auch bei der Sequenzierung von gyrA im Impfstamm vacT gefunden. Allerdings wurde erstmalig im Impfstamm eine vierte, ebenfalls bisher unbekannte Mutation, in Codon Trp-59ÆArg (W59R) identifiziert. Auch sie liegt außerhalb der QRDR, welche für E. coli zwischen den Aminosäuren 67 bis 106 beschrieben ist [305]. Piddock et al. erweiterten allerdings die gyrA QRDR von Salmonella spec. auf die Aminosäuren 67 bis 122. Da sich die in vacT identifizierte Mutation W59R relativ nah (in Bezug auf die Aminosäureposition) an der QRDR befindet, kann ein Einfluss auf die Empfindlichkeit gegenüber Fluorchinolonen nicht ausgeschlossen werden. Möglicherweise muss die definierte QRDR bei Identifizierung weiterer Chinolonresistenz vermittelnder Mutationen zukünftig noch weiter ausgedehnt werden. Eine Aussage über die Lage der Substitution W59R im gyrA-Molekül und somit einer Beteiligung an Bindungen der DNA oder der Chinolone kann aufgrund des Fehlens eines Salmonella gyrA-Strukturmodells nicht getroffen werden. Alternativ besteht allerdings auch die Möglichkeit, dass dieser nur im Impfstamm gefundene Aminosäureaustausch eine Rolle bei der intragenischen Kompensation der Auswirkung der Mutation D87G (Chinolonresistenz vermittelnd) spielt. Generell könnte das Auftreten der Mutation an Position W59R in vacT für spezielle, nur dem Impfstamm eigene Charakteristika verantwortlich gemacht werden. Diese Mutation ist die einzige bekannte genotypische Abweichung des Impfstammes vom Vorgänger Nal2ori. Die Anwendung von N-Methyl-N´-Nitrosoguanidin während der Impfstammentwicklung kann zahlreiche Mutationen ausgelöst haben. Trotz der Akkumulation von immerhin vier 137 Diskussion gyrA-Mutationen in einem für die Zelle essentiellen Gen, wurde in den weiteren Targetgenen gyrB, parC und parE sowohl in Nal2ori, Nal2passage als auch im Impfstamm vacT überraschenderweise keine Mutation gefunden. Die möglichen Ursachen für diese Anhäufung von Mutationen im gyrA-Gen werden im Abschnitt 4.2.3.3 diskutiert. Ein Auftreten von Mutationen in gyrB, parC und parE wurde vor der Sequenzierung nicht ausgeschlossen. Bei chinolonresistenten Mutanten sind mitunter auch Aminosäuren im Gen der B-Untereinheit der Gyrase ausgetauscht [17,128]. Bei hochresistenten klinischen Isolaten findet man, wie bei Mehr-Schritt-Mutanten aus In-vitro-Selektionsversuchen, auch Mutationen in den Genen parC bzw. parE für Topoisomerase IV [17,23,48,124]. Dadurch konnte diese Topoisomerase als sekundäre Zielstruktur für Fluorchinolone bei gramnegativen Bakterien identifiziert werden (Abschnitt 1.3.1.3). Möglicherweise erklärt das Fehlen weiterer Mutationen in den Topoisomerasegenen auch die Sequenzierung der Fluorchinolonempfindlichkeit von vacT. Zusammenfassend lässt sich sagen, dass durch die Topoisomerasegene vier verschiedene gyrA-Mutationen im Impfstamm vacT identifiziert werden konnten, darunter die aus der Literatur bekannte Mutation an Position D87G. Diese ist als Chinolonresistenz vermittelnd beschrieben und kann den Nal-Marker (Nalidixinsäureresistenz) von vacT erklären. Inwieweit die anderen gyrA-Mutationen eine Rolle bei der Kompensation der Wirkung von der Mutation D87G im Impfstamm spielen und den ssq-Marker in vacT darstellen, sollte mit Hilfe weiterer Untersuchungen geklärt werden: Einerseits wurde ein gyrA-Komplementationstest (Abschnitt 4.2.1) durchgeführt, andererseits sollten durch homologe Rekombinationstechnik isogene Stämme aus dem Impfstamm erzeugt werden, welche die identifizierten gyrA-Mutationen einzeln oder in Kombination im genetischen Hintergrund des Ausgangsstammes M415 enthielten (Abschnitt 4.2.3). Prinzipiell können Mutationen in den Genen der Gyrase auch eine reduzierte Enzymaktivität bewirken. So ist für einige gyrA-Mutationen eine Korrelation mit einem reduzierten Superspiralisierungsgrad in der betroffenen Zelle beschrieben [17]. Eine Vierfachmutation im Impfstamm könnte in einer stark eingeschränkten Gyraseaktivität resultieren und möglicherweise die Attenuation und die veränderte Fitness erklären. Aus diesem Grund wurde eine Bestimmung des Superspiralisierungsgrades der Stämme durchgeführt (Abschnitt 4.1.4). 4.1.2 Untersuchung des cpxA-Gens Das Gen cpxA wurde sequenziert, da Roesler et al. [261] den Impfstamm vacT als cpxAMutante beschrieben hatten. Das Genprodukt CpxR bildet zusammen mit CpxA ein Zweikomponenten-Regulationssystem, bestehend Sensorkinase einem (CpxA) und aus einer Transmembran- Response-Regulator (CpxR). Signaltransduktionssysteme, die extrazelluläre Reize wahrnehmen und durch die Bakterienmembran z.B. an die Transkriptionsfaktoren weiterleiten, spielen z.B. eine entscheidende Rolle für die Interaktion mit Oberflächen. Für Salmonellen als pathogene 138 Diskussion Bakterien hat dies eine große Bedeutung bei der Besiedlung verschiedener Habitate oder des Wirtes [65]. Salmonellen können bei der Besiedlung einer abiotischen Oberfläche Biofilme ausbilden, während der Kontakt zu einer Wirtszelle zur Invasion führt. Bei beiden Prozessen ist eine initiale, über Fimbrien vermittelte Adhäsion essentiell [227]. Bei der Ausbildung eines Biofilms erfolgt die Produktion einer extrazellulären Matrix, z.B. aus Cellulose [274]. Für die Invasion der Bakterien in die Wirtszelle werden Effektorproteine mittels des Typ III-Sekretionssystems in das Cytosol der Wirtszellen transloziert (Abschnitt 1.1.3.2). Die unterschiedlichen Reaktionen der Bakterien nach Kontakt mit einer Oberfläche machen deutlich, dass regulatorische Mechanismen existieren müssen, die verschiedene extrazelluläre Signale wahrnehmen und an den Transkriptionsapparat im Cytoplasma weiterleiten, um eine dem Reiz entsprechende Antwort zu generieren. Das Cpx-System reagiert aber auch auf extracytoplasmatischen Stress (z.B. hervorgerufen durch falsche Faltung der Proteine der Bakterienmembran) mit der Regulation von Genen, die an der Faltungskatalyse, der Ausbildung von Disulfidbrücken und dem Abbau falsch gefalteter Proteine beteiligt sind [241]. CpxA ist die Sensor- oder auch Histidinkinase (HKS), ein in der cytoplasmatischen Membran verankertes Protein, das den Reiz wahrnimmt und in ein intrazelluläres Signal umwandelt. CpxR ist ein cytoplasmatisches Protein und fungiert als Antwortregulator, welcher das Signal von der Histidinkinase empfängt und dadurch aktiviert wird [246,247]. Unter nicht induzierten Bedingungen wird die Aktivität des Sensors CpxA durch Bindung des periplasmatischen Proteins CpxP inhibiert. Unter induzierenden Bedingungen wird die CpxA-CpxP-Interaktion aufgehoben und dadurch CpxA aktiviert. Es wird auch eine Rolle des Cpx-Systems bei der Virulenz pathogener Bakterien diskutiert [152]. Allerdings gibt es dazu bisher nur In-vitro-Studien [152]. So könnte bei S. Typhimurium die Expression von Invasionsgenen bei Mutationen in cpxA gestört und die Fähigkeit zur Adhäsion und Invasion reduziert sein. Mutationen in Cpx-assoziierten Genen könnten also prinzipiell eine Attenuation des Impfstammes vacT erklären. Die Ergebnisse der Sequenzierung des cpxA-Gens im Rahmen dieser Arbeit weisen aber keine Abweichungen zwischen dem Ausgangsstamm M415 und dem Impfstamm auf. In der Tabelle 3-7 im Abschnitt 3.1.4 sind die Unterschiede der Nukleotidsequenz zwischen den Untersuchungsstämmen und dem Referenzstamm S. Typhimurium LT2 (LT2) aufgeführt. Da der Stamm LT2 der am nächsten verwandte Stamm ist, von dem das komplette Genom bekannt ist [202], wurde dieser Stamm als Referenz bei Sequenzvergleichen und als Vorlage für die Erstellung von Primersequenzen verwendet. Alle vier Untersuchungsstämme (M415, Nal2ori, Nal2passage, vacT) zeigen in cpxA die gleichen zwei Nukleotidaustausche in den Codons 250 und 313 im Vergleich mit LT2. Allerdings führt nur der zweite Basenaustausch auch zu einem Aminosäureaustausch (Leu-313 zu Ile). Diese Unterschiede beziehen sich aber lediglich auf den Vergleich der Untersuchungsstämme mit der Sequenz von LT2. Es wurden keine Abweichungen zwischen dem Ausgangsstamm M415 und Nal2ori, Nal2passage oder dem Impfstamm 139 Diskussion festgestellt. Der Impfstamm vacT ist demnach nach der Sequenzanalyse aus dieser Arbeit keine cpxA-Mutante. 4.1.3 Bestimmung der Empfindlichkeiten gegenüber verschiedenen Antibiotikaklassen Eine Änderung der Empfindlichkeit der Stämme M415, Nal2ori, Nal2passage und des Impfstammes vacT gegenüber verschiedenen Antibiotika könnte Hinweise auf das Vorhandensein möglicher Defekte oder Veränderungen in den Stämmen ergeben. Daher wurden alle vier Untersuchungsstämme umfangreichen Empfindlichkeitsbestimmungen unterzogen. Als weiterer Referenzstamm neben M415 wurde der S. Typhimurium Stamm ATCC14028 bei den MHK-Wert-Untersuchungen mitgeführt. Aus der Sequenzanalyse für die Stämme Nal2ori und Nal2passage sind die Mutationen G75A, D87G und A866S im gyrA-Gen bekannt (Abschnitt 3.1.4). Mutationen in gyrA bewirken je nach betroffener Aminosäureposition eine verringerte Empfindlichkeit gegenüber Chinolonen [17,51,125,128,253,305]. Die Bestimmungen der minimalen Hemmkonzentrationen für diese beiden Stämme zeigten wie erwartet erhöhte MHK-Werte sowohl der Nalidixinsäure als auch der verschiedenen Fluorchinolone im Vergleich zum Ausgangsstamm M415 (Tabelle 3-2 Abschnitt 3.1.2). Die MHK-Werte der Fluorchinolone waren sowohl für Nal2ori als auch für Nal2passage gleichermaßen um bis zu zwei Verdünnungsstufen bei Ciprofloxacin (0,125 µg/ml), Norfloxacin (0,5 µg/ml), Sparfloxacin (0,125 µg/ml) sowie Gatifloxacin (0,15 µg/ml) erhöht. Unterschiede in beiden Stämmen wurden jedoch bezüglich der Empfindlichkeit gegenüber Nalidixinsäure festgestellt: Hier zeigte Nal2ori mit einer MHK von 512 µg/ml eine deutliche Erhöhung gegenüber dem Ausgangsstamm M415 (4 µg/ml) um sieben Verdünnungsstufen. Bei der Variante Nal2passage war jedoch nur noch ein MHK-Wert-Anstieg von vier Verdünnungsstufen (64 µg/ml) im Vergleich mit M415 zu beobachten. Der Wirkmechanismus dieser Antibiotika beruht, wie in Abschnitt 1.3.1.2 beschrieben, auf der Bindung an den DNA-GyraseKomplex und der daraus resultierenden Hemmung des Enzyms. Diese beeinträchtigt einen einwandfreien Ablauf der Replikation, was sich schädlich bis letal für die Zelle auswirkt [79,270,302]. Typisch für Stämme mit gyrA-Einzelmutationen sind klinische Nalidixinsäureresistenz (Grenzwert 32 µg/ml) und verringerte Empfindlichkeit gegenüber Fluorchinolonen [118,128]. Das Auftreten weiterer Mutationen führt meistens zu klinischer Resistenz gegenüber Fluorchinolonen. Neben einer weiteren gyrA-Mutation handelt es sich dabei oft um eine Mutation, die zu einer reduzierten Fluorchinolonkonzentration in der Zelle führt. Alternativ kann die zweite Zielstruktur Topoisomerase IV verändert sein. Die Stämme Nal2ori und Nal2passage verhalten sich aufgrund ihrer MHK-Werte somit wie Einzelmutanten. Daraus resultiert die Hypothese, dass die Mutation D87G die größte Bedeutung für die Nalidixinsäureresistenz und reduzierte Fluorchinolonempfindlichkeit hat. 140 Diskussion Der Impfstamm vacT besitzt vier gyrA-Mutationen: W59R, G75A, D87G und A866S. Die Empfindlichkeitsbestimmung ergab jedoch im Vergleich zu M415 nur im Fall von Nalidixinsäure eine MHK-Wert-Erhöhung von drei Verdünnungsstufen, die geringer ausfällt als bei Nal2ori (sechs Stufen). Der Impfstamm vacT zeigte mit einer MHK von 32 µg/ml klinische Nalidixinsäureresistenz. Diese lässt sich durch das Auftreten der Mutation an Codon 87 im gyrA-Gen erklären. Im Impfstamm gibt es aber anscheinend einen Faktor, der die Empfindlichkeit gegenüber Nalidixinsäure im Vergleich mit Nal2ori etwas reduziert. Außerdem zeigte vacT im Gegensatz zu Nal2ori keine reduzierte Empfindlichkeit gegenüber Fluorchinolonen, wie z.B. Ciprofloxacin (vacT: MHKCip = 0,03 µg/ml). Bei einigen Fluorchinolonen wie Sparfloxacin oder Sitafloxacin wies der Impfstamm mit MHK-Werten von 0,006 µg/ml sogar eine „Supersensibilität“ auf. Die MHK-Werte der Fluorchinolone lagen also in einigen Fällen unter dem Niveau des Ausgangsstammes M415. Im Impfstamm wird offenbar die Wirkung der Resistenz vermittelnden gyrA-Mutation D87G kompensiert. Als Ursache dafür kommt die im Impfstamm neu identifizierte gyrA-Mutation an Position 59 (W59R) in Frage (intragenische Kompensation), was mittels eines über Rekombination hergestellten isogenen Stammes mit den Mutationen W59R und D87G überprüft werden sollte (Abschnitt 4.2.3.1). Eine weitere mögliche Erklärung für die reduzierten Fluorchinolon-MHK-Werte könnte aber auch eine verstärkte Akkumulation von Antibiotika in der Zelle sein. Dies kann durch einen reduzierten Efflux begründet sein, z.B. hervorgerufen durch das Ausschalten von Effluxpumpen (Abschnitt 4.3). Es kommt aber auch ein erhöhter Influx in Frage, ausgelöst durch eine erhöhte Anzahl an Porinen (z.B. OmpF; siehe Abschnitt 1.3.2) oder Störungen in der LPS-Schicht. Eine Akkumulation der Fluorchinolone würde trotz verringerter Empfindlichkeit der Gyrase (aufgrund der Mutation D87G) einen erniedrigten MHK-Wert aufgrund der hohen Wirkkonzentration in der Zelle bewirken. Bei weiteren MHK-Wert-Bestimmungen wurden verschiedene Vertreter aus der Klasse der Makrolide getestet (Tabelle 3-3 Abschnitt 3.1.2). Der Wirkmechanismus der Makrolide beruht auf der reversiblen Bindung an die ribosomale 50S-Untereinheit (bevorzugt an Adenin 2058 in der Domäne V der 23S-rRNA) und der Inhibition der Translokation der mRNA [96,299]. Der Stamm M415 zeigte eine natürliche Makrolidresistenz aufgrund der Barrierefunktion der äußeren Membran sowie der Aktivität von mdr-Effluxpumpen [314]. Nal2ori und Nal2passage zeigten im Vergleich zu M415 keine oder nur geringe Veränderungen der Empfindlichkeiten gegenüber Makroliden. Abweichungen in den MHKWerten der Makrolide Azithromycin oder Clarithromycin für Nal2ori und Nal2passage lagen im Fehlerbereich der Messungen. Nur im Fall von Tylosin, einem in der Veterinärmedizin eingesetztem Makrolid, waren die MHK-Werte für Nal2ori und Nal2passage im Vergleich zum Ausgangsstamm um drei Verdünnungsstufen erniedrigt. Auffällig war die deutliche Erhöhung der Empfindlichkeit gegenüber Makroliden beim Impfstamm vacT um mindestens vier (Erythromycin, Roxithromycin, Azithromycin) bis fünf Verdünnungsstufen (Clarithromycin, Tylosin). Der Impfstamm vacT zeigte demzufolge im 141 Diskussion Gegensatz zum Ausgangsstamm M415 eine Makrolidsensibilität. Nah verwandt mit den Makroliden sind die Ketolide, welche sich durch einen dualen Wirkmechanismus auszeichnen, indem sie zusätzlich auch an das Adenin 752 in der Domäne II der 23S rRNA binden [119]. Wie auch bei den Makroliden zeigten Nal2ori und Nal2passage gegenüber Ketoliden nur geringfügige Änderung der Empfindlichkeit. Der Impfstamm hingegen wies eine MHK-Wert-Erniedrigung von vier bis fünf Verdünnungsstufen im Vergleich zum M415 auf. Die natürliche Makrolid- und Ketolidresistenz des Ausgangsstammes beruht wie bei den meisten gramnegativen Bakterien auf der geringen Konzentration der Makrolide am Wirkungsort. Für die im Impfstamm gefundene Sensibilität kann möglicherweise eine verstärkte Akkumulation von Makroliden aufgrund der veränderten Durchlässigkeit der Membran verantwortlich sein. Eine mögliche Erklärung dafür ist der bereits bei den Chinolonen diskutierte reduzierte Efflux. Dieses Argument kann ebenso für die gegenüber dem Ausgangsstamm reduzierten MHK-Werte des Linezolids (Oxazolidinon), Clindamycins (Lincosamin) und Synercids (Streptogramin) für den Impfstamm dienen (Tabelle 3-3 Abschnitt 3.1.2). Wie die Makrolide und Ketolide inhibieren auch die Oxazolidinone die Proteinbiosynthese, und zwar die Ausbildung des ternären Komplexes aus Ribosom (30S und 50S), m-RNA und Formyl-methionin-t-RNA [281]. Durch Interaktion mit der 50S-Untereinheit wirken die Lincosamine und Streptogramine [54]. Veränderungen in verschiedenen Zielstrukturen aufgrund der ungezielten Mutagenese während der Herstellung des Impfstammes sind als Ursache der veränderten Antibiotikaempfindlichkeiten neben der verstärkten Akkumulation jedoch auch nicht auszuschließen. Keine oder nur sehr geringfügige Änderungen gegenüber dem Ausgangsstamm fanden sich für Nal2ori, Nal2passage oder vacT in den MHK-Werten der getesteten Sulfonamide, Aminogykoside, Glykopeptide oder der Fusidinsäure. Gegenüber Chloramphenicol und den Tetracyclinen war nur der Impfstamm vacT etwas empfindlicher als der Ausgangsstamm M415. Im Falle von Doxycyclin zeigte sich bei vacT eine MHK-Wert-Erniedrigung um vier Verdünnungsstufen. Auch bei diesen beiden Antibiotika kann der Einfluss eines defekten Efflux-Systems vermutet werden, da sie Substrate für Effluxpumpen sind [240]. Die aufgeführten Antibiotika sind in der Humanmedizin therapeutisch relevante Antibiotika. Eine erhöhte Empfindlichkeit stellt somit einen zusätzlichen Sicherheitsmarker für den Impfstamm vacT dar. Im Falle von Rifampicin und Fosfomycin wurden für den Impfstamm erhöhte MHKWerte festgestellt (Tabelle 3-4 Abschnitt 3.1.2). Rifampicinresistenz wird durch Mutationen in dem Gen für die ß-Untereinheit der RNA-Polymerase vermittelt. Der Impfstamm vacT war als rpoB-Mutante beschrieben [261]. Diese Mutation wurde aufgrund der Selektion auf Rifampicin bewusst bei der Entwicklung des Impfstammes selektiert. Fosfomycin ist ein Inhibitor der Zellwandsynthese. Neben plasmidcodierten Resistenzgenprodukten wie FosA, welches Fosfomycin inaktiviert [13], konnten bei In-vitro-Selektion fosfomycinresistenter Keime (E. coli) verschiedenste Mutationen identifiziert werden in 142 Diskussion Genen, die hauptsächlich den Transportweg von Fosfomycin betreffen wie ptsI, cyaA, glpT oder uhpA/T [220]. Die beiden ersten Mutationen wurden aber bislang nicht bei klinischen Isolaten gefunden. Alle Mutationen bedingen eine starke Wachstumsbeeinträchtigung der selektierten Stämme. Dieser Fitnessverlust erklärt wahrscheinlich, weshalb fosfomycinresistente klinische Isolate nur selten gefunden werden. Diese können sich vermutlich durch die Verlängerung der Generationszeit z.B. in der Blase nicht gegenüber schnell wachsenden Keimen durchsetzen und sich dort etablieren. Außerdem wurde beschrieben, dass bedingt durch die Entwicklung der Fosfomycinresistenz die Adhäsion an die Epithelzellen der Bakterien herabgesetzt ist. Das wäre eine weitere Begründung für eine erschwerte Etablierung der Stämme [220]. Die Ursache für die Fosfomycinresistenz in vacT ist unbekannt, aber ein Zusammenhang zur Veränderung der Fitness oder Attenuation kann nicht ausgeschlossen werden. 4.1.4 Veränderungen im DNA-Superspiralisierungsgrad Mutationen in den Topoisomerasegenen, vor allem an bestimmten Hotspots in gyrA, reduzieren die Empfindlichkeit der Mutante gegenüber (Fluor)chinolonen (siehe Abschnitt 1.3.1.3). Aleixandre et al. [3] isolierten gyrA-Mutanten, welche zwar Veränderungen des Superspiralisierungsgrades aber keine Fluorchinolonresistenz bewirkten und Bagel et al. [17] wiederum beschrieben chinolonresistente gyrA-Mutanten, welche in Abhängigkeit von den betroffenen gyrA-Mutationen z.T Veränderungen im Superspiralisierungsgrad oder der Fitness zeigten. Mutationen in gyrA können also eine veränderte Enzymaktivität der Gyrase bewirken [17]. Derartige Änderungen des Superspiralisierungsgrades wurden im Rahmen dieser Arbeit mittels eines von Abu Mraheil [1] konstruierten plasmidbasierten Reportergensystems detektiert (Abschnitt 2.2.1.9). Das verwendete System besteht aus einem Fusionsprodukt aus den Promotoren der Gene für die Topoisomerase I sowie der A-Untereinheit der Gyrase (ptopA und pgyrA) mit dem Luciferasegen (luc). Mit jeweils einem Paar von Plasmiden, das aus ptopA-luc bzw. pgyrA-luc Reportergenen besteht, können Änderungen in der Aktivität der Promotoren gemessen werden. Der daraus errechnete Quotient (Abschnitt 2.2.1.9.3) gibt Hinweise auf Veränderungen im Superspiralisierungsgrad. Mit dem verwendeten Reportergensystem auf Basis des Plasmides pBR322 (Plasmidpaar pPHB90 und pPHB91) wurde für den Ausgangsstamm M415 ein Qsc-Wert von 1,8 bestimmt (Tabelle 3-5, Abschnitt 3.1.3). Im Vergleich dazu zeigten die Derivate Nal2ori und Nal2passage mit 0,98 und 0,99 nur einen Qsc-Wert von ca. 55 % des Ausgangsstammes. VacT wies ebenfalls nur ca. 61 % (1,09) des QscWertes von M415 auf (Abschnitt 3.1.3). Auch bei der Messung mit den alternativen Reportergenplasmiden pPHB94 und pPHB95 (pBP507-System) zeigte sich eine signifikante Reduktion des Superspiralisierungsgrades bei vacT gegenüber M415. Der Qsc-Wert von vacT betrug ca. 77 % von M415 (Tabelle 3-6). Bereits in früheren Arbeiten konnten in gyrA-Mutanten Veränderungen im DNASuperspiralisierungsgrad nachgewiesen werden. Dies geschah größtenteils mittels 143 Diskussion Auftrennung isolierter Plasmid-DNA in chloroquinhaltigen Agarosegelen [17]. Abu Mraheil konnte diese Ergebnisse mit der Reportergensystem-basierten Methode bestätigen [1]. Er ermittelte z.B. für einen E. coli Stamm mit einer gyrA-Doppelmutation (S83L, D87G) ein Absinken des Ausgangsstamm Qsc-Wertes [1]. auf Aufgrund 79 % der im Vergleich zu homöostatischen dem entsprechenden Kontrolle des DNA- Superspiralisierungsgrades [204] (siehe Einleitung Abschnitt 1.3.1.1) bewirkt die Abnahme des negativen Superspiralisierungsgrades eine Zunahme der Aktivität von pgyrA. Das spiegelt sich bei der Berechnung des Quotienten in einer Erniedrigung des Qsc-Wertes wider. Grund für die reduzierte Superspiralisierungsaktivität in den Mutanten könnte eine möglicherweise durch die Aminosäureaustausche hervorgerufene Veränderung der Molekülstruktur in der Gyrase sein. Dadurch könnte z.B. die Anlagerung der DNA erschwert sein. Ob starke Veränderungen des Moleküls verursacht werden, ist allerdings fraglich, da Gyrase ein hoch konserviertes und für die Zelle essentielles Enzym ist. Ein in silico 3D-Strukturmodell des gyrA-Gens aus E. coli (Zusammenarbeit mit Jörn Lenz, Daten nicht gezeigt) lieferte Hinweise darauf, dass durch die veränderten Aminosäuren die Molekülstruktur des GyrA-Proteins so verändert sein könnte, dass das Tyrosin an Position 122 verschoben ist. Diese Aminosäure ist das aktive Zentrum des Enzyms, an das die DNA vorübergehend kovalent bindet [149] (siehe Einleitung Abschnitt 1.3.1.1). Eine Verschiebung des Tyrosins könnte möglicherweise die Bindungsaffinität so verändern, dass die Gyrase ihrer Funktion als Topoisomerase nicht mehr vollständig gerecht werden kann und der negative Superspiralisierungsgrad der Zelle reduziert ist. Da aufgrund der veränderten Zugänglichkeit der Promotoren bestimmte Gene nicht in der ursprünglichen Stärke exprimiert werden können, beeinflusst eine Veränderung im DNASuperspiralisierungsgrad die Expression einer Reihe von Genen [75,76,94]. Solche Störungen in der Genexpression können wichtige Proteine für den Stoffwechsel oder auch Virulenzgene betreffen [94,276]. Derartige Effekte wurden bereits z.B. für die Expression von Typ1-Fimbrien in E. coli beschrieben [16]. Typ1-Fimbrien spielen eine wichtige Rolle bei der initialen Phase einer Harnwegsinfektion. Die Ausprägung der Fimbrien bei Wirtskontakt wird über einen Phasenwechsel gesteuert, der über ein invertierbares DNAFragment reguliert wird. Auf diesem DNA-Fragment befindet sich der Promotor des Gens für FimA, einem Hauptstrukturprotein der Typ1-Fimbrien. Je nach Orientierung des Fragmentes werden Fimbrien exprimiert („ON“) oder nicht („OFF“). Die Inversion erfolgt aufgrund von Rekombination an zwei flankierenden homologen Flanken - „inverted repeats“ - und wird durch die Integrasen FimB und FimE gesteuert. Die Transkription von fimB ist über einen vom Superspiralisierungsgrad abhängigen Promotor gesteuert. Ein reduzierter Superspiralisierungsgrad führt zur Überexpression von FimB, das die „inverted repeats“ besetzt, wodurch eine Reduktion der fimA-Expression und somit eine verminderte Typ1-Fimbrien-Expression bewirkt wird. Generell kommen demzufolge gyrAMutationen, aufgrund des möglichen negativen Einflusses auf die Expression von 144 Diskussion Virulenzgenen durch einen veränderten Superspiralisierungsgrad, als mögliche Attenuationsmarker in Frage. Es bedarf aber noch weiterer zukünftiger Untersuchungen, um konkrete Zusammenhänge herzustellen. In allen drei untersuchten Stämmen (Nal2ori, Nal2passage und vacT) weist der ähnlich stark reduzierte Qsc-Wert (55-60 %) auf eine beeinträchtigte Gyraseaktivität hin. In allen Stämmen ist deshalb vermutlich die gleiche Ursache für den reduzierten Qsc-Wert verantwortlich. Als Grund für die verringerte DNA-Superspiralisierung kommt daher die Mutation D87G in Frage. Für Stämme mit dieser Mutation ist eine Veränderung der Enzymaktivität der Gyrase bereits beschrieben [17]. Wie die Ergebnisse der Messungen des Superspiralisierungsgrades für vacT zeigen (Abschnitt 3.1.3, Tabelle 3-5 und 3-6), verursacht die zusätzliche Mutation W59R keine intragenische Kompensation der Wirkung der anderen gyrA-Mutation(en). Der konkrete Nachweis, ob die bzw. welche der bereits in Nal2ori identifizierten Mutationen D87G, G75A oder A866S für den veränderten Superspiralisierungsgrad verantwortlich sind oder ob einzelne Mutationen z.B. noch einen verstärkenden Effekt haben, kann erst durch eine Untersuchung von isogenen Stämmen erfolgen, welche ausschließlich diese Mutationen enthalten (Abschnitt 4.2.3). Den genauen Zusammenhang zwischen gyrA-Mutationen, reduziertem Superspiralisierungsgrad, Veränderungen in der Fitness sowie in den MHK-Werten für die untersuchten Stämme sollten weitere Experimente im Rahmen dieser Arbeit zeigen (Abschnitt 4.2). 4.1.5 Wachstum als Fitnessparameter Die Wachstumsgeschwindigkeit kann als ein experimentell leicht bestimmbarer Parameter für die Fitness eines Bakterienstammes angesehen werden. Mutationen in essentiellen Genen haben oft, aufgrund eines veränderten Stoffwechsels, Auswirkungen auf die Fitness der Bakterien [177-179]. Unter Selektionsdruck entstehen Mutationen in essentiellen Stoffwechselgenen, die oft auch Zielstrukturen für Antibiotika darstellen. Die durch die Mutationen verursachte Antibiotikaresistenz sichert das Überleben der Bakterien in Anwesenheit des Antibiotikums. Allerdings ziehen Mutationen in essentiellen Genen, aufgrund der dadurch oft entstehenden Beeinflussung des Stoffwechsels, häufig auch einen Fitnessverlust für den betroffenen Stamm nach sich (Einleitung Abschnitt 1.4). Durch die Beeinträchtigung der Fitness kommt es in der Folge auch zu einer herabgesetzten Konkurrenzfähigkeit in der Umwelt. Deshalb ging man früher davon aus, dass resistente Keime einen Konkurrenznachteil gegenüber sensiblen und schnell wachsenden Keimen haben. So entstand die Theorie, dass durch einen reduzierten Einsatz von Antibiotika die Frequenz von resistenten Bakterien in der Population sinken würde, da sie von den sensiblen Keimen mit besserer Fitness verdrängt würden. Allerdings wurde eine Persistenz von resistenten Keimen auch in antibiotikafreier Umgebung festgestellt [12] und viele resistente klinische Isolate zeigten trotz auftretender Stoffwechselmutation keine Fitnessreduktion. Das beruht auf der Fähigkeit, der Bakterien, 145 Diskussion durch kompensatorische Mutationen ihre Fitness partiell oder sogar vollständig wieder herzustellen und konkurrenzfähig gegenüber sensiblen Keimen zu sein [10,11]. Björkmann & Andersson [32] stellten prinzipiell fest, dass die Frequenz von Mutationen zur Resistenzentwicklung in einer Bakterienkultur sowohl von (1) der Menge der eingesetzten Antibiotika, (2) den Fitnesskosten für die Resistenz als auch (3) der Möglichkeit eines Bakteriums diese Fitnesskosten zu kompensieren, abhängt. Das Auftreten kompensatorischer Mutationen und die Durchsetzungsfähigkeit der entsprechenden Mutanten sind dabei abhängig von der Populationsgröße, der Mutationsfrequenz und dem Fitnessgewinn für die Mutante [10,12]. Die kompensatorischen Mutationen können sowohl intra- als auch extragenisch lokalisiert sein. Intragenische Kompensation kann als Reversion, d.h. genaue Wiederherstellung des Ausgangsgenotyps erfolgen oder durch eine weitere Mutation an anderer Stelle im Gen, welche die Wirkung der ersten Mutation kompensiert. Wie bereits in der Einleitung (Abschnitt 1.4) beschrieben, führen vor allem Mutationen in Genen für Proteine, die für die Replikation, Transkription oder Translation wichtig sind und welche eine Antibiotikaresistenz vermitteln, auch zu Fitnesseinbußen für den betroffenen Stamm. Beispielsweise bewirken Mutationen im rpsL-Gen ein Wachstumsdefizit in streptomycinresistenten Mycobacterium tuberculosis [34] oder E. coli [267]. Die Streptomycinresistenz beruht auf Mutationen im Gen rpsL, welches für das ribosomale Protein S12 codiert. Eine Veränderung dieses Proteins resultiert in einer verminderten Bindung von Aminoglykosiden und aufgrund der Veränderungen des Ribosoms auch in einer Fitnessreduktion. Weiterhin führen in chinolonresistenten Streptococcus pneumoniae [101], Pseudomonas aeruginosa [168] und S. Typhimurium [33] nicht näher charakterisierte Mutationen in den Genen der Topoisomerasen zu einem Fitness- und Virulenzverlust. Für Stämme mit der Akkumulation von Mutationen, die verantwortlich für eine Norfloxacinresistenz sind (Topoisomerasegene und Effluxpumpen betreffend), wurde ebenfalls ein Wachstumsdefizit beschrieben [162]. Vor allem Mutationen in rpoB (Gen für die ß-Untereinheit der RNA-Polymerase), die im Bakterienstamm eine Rifampicinresistenz vermitteln, sind aufgrund von Transkriptions- und Elongationsproblemen mit Fitnesskosten für die betroffenen Stämme verbunden [193]. Auch fusidinsäureresistente Salmonella mit Mutationen im Gen fusA, das für den Elongationsfaktor G codiert, zeigen aufgrund von Einbußen bei der Funktionalität des Elongationsfaktors G Wachstumsreduktion [32,189]. Generell wurden sowohl in In-vitro-Versuchen als auch in Labortieren und in klinischen Isolaten Mutationen zur Kompensation des Fitnessverlustes gefunden [33,34,190,212,254,267,271]. Allerdings unterschieden sich z.T. das Auftreten und die Art der kompensatorischen Mutationen (z.B. Reversion, intra- oder extragenische Mutation). Dies war auf der einen Seite von dem betroffenen Gen abhängig und auf der anderen Seite davon, ob die Mutanten in vitro in LB-Medium oder in vivo in der Maus passagiert wurden. Streptomycin- und fusidinsäureresistente S. Typhimurium wurden auf Unterschiede in dem Auftreten von fitnesskompensatorischen Mutationen untersucht, 146 Diskussion abhängig davon, ob sie in der Maus oder in LB-Medium passagiert wurden [34,189]. Die streptomycinresistenten Mutanten mit Ausgangsmutationen im Gen rpsL zeigten unabhängig von der Passage in LB-Medium oder in der Maus sowohl intra- (im Gen rpsL) als auch extragenische (in den Genen rpsD und rpsE) Mutationen. Die fusidinsäureresistente fusA-Mutanten zeigten im Gegensatz dazu grundsätzlich nur intragenische Kompensation, in seltenen Fällen sogar direkte Reversionen [34]. Weiterhin wurde festgestellt, dass die kompensatorischen Mutationen, die in Salmonellen auftraten, welche in vitro in LB-Medium passagiert wurden, z.T. keine Fitnesskompensation in der Maus bewirkten. Erklärt wurde dies dadurch, dass die Fitness von Bakterien in vivo sowohl durch die Wachstumsgeschwindigkeit, als auch durch die Virulenz beeinflusst wird [189]. So zeigen fusA-Mutanten aufgrund der Mutationen im Elongationsfaktor G Störungen der Proteinsynthese als auch Reduktion der Virulenz aufgrund der Reduktion der ppGpp-Produktion. Das Molekül ppGpp (Guanosintetraphophat) ist ein globaler Regulator. Es bindet an die RNA-Polymerase und beeinflusst normalerweise z.B. die Regulation von rpoS positiv. RpoS als Sigmafaktor wiederum beeinflusst u.a. die Expression von Virulenzgenen und ist bei der Infektion wichtig (Abschnitt 1.1.3.3). Der Unterschied im Auftreten der kompensatorischen Mutationen in fusA-Mutanten in vitro bzw. in vivo werden folgendermaßen erklärt: Die Translationseffizienz kann in vitro offenbar einfach durch verschiedene intragenische Mutationen kompensiert werden. Das reicht den Bakterien in LB-Medium zur Kompensation der Wachstumsgeschwindigkeit. Eine Wachstumskompensation mit Erhalt der Resistenz kann also prinzipiell auch unabhängig von der Wiederherstellung der Virulenz stattfinden. In vivo in der Maus sind aber andere, ganz spezielle intragenische Mutationen nötig, die auch die verminderte Expression von RpoS und die damit verbundene Virulenzreduktion kompensieren. Diese wurden dann auch bei aus der Maus isolierten S. Typhimurium gefunden [34,189]. Bestätigt wurden diese Beobachtungen der intragenischen Mutationen von fusA auch in klinischen Isolaten von Staphylococcus aureus [212]. Fitnesskompensation durch extragenische Mutationen wurde beispielsweise in Untersuchungen von klinisch isolierten isoniazidresistenten Mycobacterium tuberculosis beschrieben [136]. Diese Mykobakterien besaßen Mutationen im katG-Gen, codierend für eine Katalase-Peroxidase. Diese gilt als Virulenzfaktor, weil KatG die Bakterien gegen die im Makrophagen befindlichen oxidativen Intermediate schützt und dadurch ihr Überleben im Wirt ermöglicht (siehe Abschnitt 1.1.3.3). Die Zellen müssen über ein alternatives Peroxidase-System das Defizit ausgleichen. So bewirken kompensatorische Mutationen in der regulatorischen Region von ahpC eine Überexpression von AhpC, einer HyperoxidPeroxidase. Die vollständige Virulenz wird aber nicht in allen Fällen wiederhergestellt [136,271]. Zusammenfassend lässt sich sagen, dass bei pathogenen Bakterien häufig eine Beeinträchtigung des Wachstums, welche durch Antibiotikaresistenz vermittelnde 147 Diskussion Mutationen ausgelöst wurde, auch mit Einbußen in der Virulenz verbunden ist. Deshalb kann eine verlängerte Generationszeit auch als ein Aspekt der Attenuation angesehen werden [177-179]. Die Wachstumsbeeinträchtigung kann durch intra- oder extragenische Mutationen oft leicht wieder kompensiert werden, was aber nicht in allen Fällen auch eine Wiederherstellung der Virulenz des Stammes bedeutet. Um in den Untersuchungsstämmen die Zusammenhänge zwischen gyrA-Mutationen, Antibiotikaresistenz und Fitness näher beleuchten zu können, wurden die Generationszeiten der Stämme M415, Nal2ori, Nal2passage und des Impfstammes vacT mittels KBE-Bestimmung ermittelt (Abschnitt 2.2.1.3). Der Ausgangsstamm M415 weist unter den gewählten Untersuchungsbedingungen eine Generationszeit von ungefähr 14 min auf (Tabelle 3-1, Abschnitt 3.1.1). Bei der Entwicklung des Impfstammes vacT wurde bewusst eine Selektion auf Antibiotika gewählt (Nalidixinsäure / Rifampicin), bei denen eine Resistenzentwicklung aufgrund von Veränderungen in essentiellen Enzymen (Gyrase / RNA-Polymerase) erfolgt. Das während der Impfstammentwicklung entstandene Zwischenderivat Nal2ori weist unter den durchgeführten Bedingungen eine im Vergleich zum Ausgangsstamm sehr stark verlängerte Generationszeit von ca. 43 min auf. Es ist bekannt, dass Stämme mit mehreren gyrA-Mutationen eine verringerte Wachstumsgeschwindigkeit besitzen können [17]. Es ist nicht auszuschließen, dass die Akkumulation von drei gyrA-Mutationen (G75A, D87G, A866S) in Nal2ori eine Veränderung der Generationszeit des Stammes bewirkt. Die Messungen des DNASuperspiralisierungsgrades ergaben eine starke Reduktion des Qsc-Wertes in Nal2ori (Abschnitt 3.1.3). Der reduzierte DNA-Superspiralisierungsgrad könnte möglicherweise aufgrund der veränderten Promotoraktivität (Abschnitt 1.3.1.3) eine Änderung der Expression von Proteinen bewirken, die für eine verlängerte Generationszeit und eine Virulenzreduktion in Frage kommen. Der Stamm Nal2passage ist durch In-Vitro-Selektion, unabhängig von der Impfstammentwicklung, aus dem Stamm Nal2ori hervorgegangen. Interessanterweise besitzt Nal2passage nur noch eine Generationszeit von 19 min. Es hat also eine Kompensation der Generationszeit bis fast auf das Niveau des Ausgangsstammes stattgefunden. In den untersuchten Genen (gyrA, gyrB, parC, parE, cpxA) wurden keine Unterschiede zwischen den beiden Stämmen Nal2ori und Nal2passage festgestellt. Der Grund für die Fitnesskompensation ist nicht bekannt. Auch der Impfstamm vacT besitzt eine, wenn auch nicht so starke, Reversion der Wachstumsgeschwindigkeit im Vergleich zum Vorgänger Nal2ori. VacT besitzt eine Generationszeit von 32 min. Das bedeutet, er wächst schneller als Nal2ori aber langsamer als M415. Dies zeigt auch die gewünschte und während der Selektion beabsichtigte Anpassung der Generationszeit des Impfstammes an den Wirt. Der Anspruch an die Wachstumsgeschwindigkeit eines Lebendimpfstammes richtet sich nach dem gewünschten Wirt, in dem der Stamm eingesetzt werden soll. Der Impfstamm vacT wird in Hühnern eingesetzt und die Generationszeit ist durch eine Kopplung von Markern 148 Diskussion optimal an den Wirt angepasst. Eine genau angepasste Wachstumsgeschwindigkeit ist wichtig für das optimale Wirken des Salmonella-Stammes im Tier. Vermutlich ist ein in vacT im Vergleich zu Nal2ori neu eingeführter Marker die Ursache für die Veränderung der Generationszeit. Auch in vacT hat also im Vergleich zum Vorgänger Nal2ori eine Fitnesskompensation stattgefunden, obwohl die Generationszeit im Vergleich zum Ausgangsstamm M145 verlängert bleibt. Die Ursachen für die Fitnesskompensation in den Stämmen Nal2ori und vacT sind aufgrund der unterschiedlichen Wachstumsgeschwindigkeiten wahrscheinlich verschieden. Eine Wiederherstellung der Virulenz hat aber in diesem Zusammenhang nicht statt gefunden. Das Konzept der Impfstammentwicklung ist eine Kopplung verschiedener Marker und dadurch eine schrittweise Attenuierung des Stammes. Dazu wurde der Stamm M415 auf die gewünschten Marker selektiert (NalR, RifR), die Virulenzreduktion in Tierversuchen überprüft und durch Mutagenese mit N-Methyl-N´-Nitrosoguanidin weitere Marker (rtt, ssq) eingeführt [179]. Generell kommt als Ursache der Fitnesskompensation, wenn sie z.B. durch eine verringerte Gyraseaktivität ausgelöst ist, eine Wiederherstellung des Superspiralisierungsgrades in Frage, wie z.B. von Kugelberg et al. [168] für fluorchinolonresistente Pseudomonas aeruginosa oder von Schulte für fluorchinolonresistente E. coli [268] beschrieben. In beiden Fällen ist die Ursache der Kompensation des Superspiralisierungsgrades unbekannt. Mutationen in den Zielstrukturen der Chinolone, d.h. in den Genen für die Topoisomerasen, konnten in diesen Arbeiten ebenso ausgeschlossen werden, wie Veränderungen in den Genen für wichtige DNA-Bindeproteine. Im Impfstamm vacT konnte im gyrA-Gen neben den drei auch in Nal2ori gefundenen Mutationen eine neue Mutation an Position 59 (W59R) identifiziert werden. Möglicherweise bewirken die drei Mutationen an den Positionen 75, 87 und 866 eine eingeschränkte Enzymaktivität, was sich in der stark verlängerten Generationszeit von Nal2ori widerspiegelt. Als intragenische kompensatorische Mutation bezüglich der Wiederherstellung der Enzymaktivität kam der nur im Impfstamm vacT gefundene Aminosäureaustausch an Position 59 in Frage. Diese ursprüngliche Hypothese konnte jedoch durch die Ergebnisse der Bestimmung des Superspiralisierungsgrades widerlegt werden. In den Stämmen Nal2passage und vacT wurden keine Kompensationen im Superspiralisierungsgrad gefunden. Unterschiede in den Stämmen Nal2ori, Nal2passage und vacT können nicht mit Veränderungen im Superspiralisierungsgrad erklärt werden, da dieser in allen drei Stämmen sehr ähnlich ist (Tabelle 3-5, Abschnitt 3.1.3). Da eine Fitnesskompensation aufgrund einer Änderung des DNA- Superspiralisierungsgrades ausgeschlossen werden kann, muss überprüft werden, ob die Fitnesseinbußen überhaupt durch den reduzierten Superspiralisierungsgrad bedingt sind. Neben Veränderungen der Gyrase kommen auch andere Faktoren als Ursache einer reduzierten Fitness von Nal2ori in Frage. Eine Veränderung der RNA-Polymerase (rpoB149 Diskussion Mutationen) könnte prinzipiell eine verlängerte Generationszeit im Gegensatz zu M415 erklären. Da aber der langsam wachsende Stamm Nal2ori keine Rifampicinresistenz besitzt, ein Hinweis auf fehlende rpoB-Mutation, wird eine veränderte RNA-Polymerase als Grund für die verlängerte Generationszeit in diesem Stamm ausgeschlossen. Ein weiterer möglicher Defekt für die Fitnesseinbuße könnte eine Veränderung der Zellmembran sein. Zum Beispiel kann reduzierter Efflux zu einer Anreicherung von toxischen Stoffen in der Zelle führen. Andererseits kann sich auch eine schlechte Versorgung mit Nährstoffen durch gestörten Influx negativ auf die Fitness der Zelle auswirken. Es konnten außer der Nalidixinsäureresistenz aber keine großen Abweichungen in den MHK-Werten zwischen Nal2ori und M415 gefunden werden. Das ist ein starker Hinweis auf die gyrA-Mutation(en) als Ursache für die Fitnessreduktion. Die genauen Gründe der Fitnessreduktion in Nal2ori und der Fitnesskompensation in Nal2passage und vacT sind unbekannt. Um die Ursachen der Veränderungen bezüglich der MHK- und Qsc-Werte sowie des Wachstums in den untersuchten Stämmen näher untersuchen zu können, wurden drei Ansätze verfolgt: (1) Als globaler Ansatz wurde die 2D-Gelelektrophorese gewählt, um die Expression der Proteine in den Untersuchungsstämmen zu untersuchen (Abschnitt 4.1.6), um bisher noch unbekannte Einflüsse aufzudecken. (2) Weiterhin wurde der Einfluss der gyrA-Mutationen näher betrachtet, indem entsprechende Mutanten mittels homologer Rekombination hergestellt wurden (Abschnitt 4.2.3). (3) Außerdem wurde die Rolle von Efflux im Impfstamm untersucht (Abschnitt 4.3). 4.1.6 Proteomics als globaler Ansatz Jede phänotypische Veränderung in der Zelle beruht auf einer Veränderung der Expressionsstärke oder der Struktur von Proteinen. Einen globalen Ansatz zur Untersuchung der Proteinexpression verschiedener Stämme bietet die 2D- Gelelektrophorese (2D-GE). Durch die Anwendung der 2D-GE konnte beispielsweise in E. coli und Salmonella spec. die Regulation der Proteinexpression durch SlyA, einem an der Virulenzausprägung beteiligtem Transkriptionsregulator (siehe Abschnitt 1.1.3.3), untersucht werden [275]. Auch der Einfluss von Fis (factor for inversion stimulation), einem am Pathogenitätsprinzip von S. Typhimurium beteiligtem DNA-Bindeprotein (siehe Abschnitt 1.3.1.1), auf die Proteinexpression wurde mittels 2D-GE erforscht [304]. Ziel war es, die Methode der 2D-GE zum Nachweis von Unterschieden in den Proteinmustern im Ausgangsstamm, den Zwischenderivaten und dem Impfstamm einzusetzen. Im Rahmen dieser Arbeit wurden mittels 2D-GE verschiedene Proteingele hergestellt, um verschiedene Parameter wie Probenaufbereitung, eingesetzte Proteinkonzentration, Färbemethode usw. zu optimieren (Abschnitt 3.1.7). Im Laufe der Optimierung der 2D-GE-Methode zeigte sich, dass aufgrund der höheren Reproduzierbarkeit sowie einer schnelleren und einfacheren Durchführung das Kit (z.B. 150 Diskussion „Total Protein“ der Firma Bio-Rad) für die Probenaufbereitung genutzt werden sollte. Der erste Schritt sollte zunächst ein allgemeiner Vergleich eines möglichst großen Proteinanteils der Zelle sein. Mit dem oben genannten Kit konnte ein großer Anteil des Gesamtproteoms der Zelle aufgearbeitet werden, was sich in der hohen Anzahl der Spots im 2D-Gel zeigte Membranproteinen, (weit würden über eine 1000 Spots). Anpassung Genauere und Analysen, Spezialisierung z.B. von in der Probenaufbereitung erfordern. Die einzusetzende Proteinmenge richtet sich nach der gewünschten Methode zur Färbung der Proteingele. Bei der Auswahl der Färbemethode ist die Kompatibilität zur Massenspektroskopie für eine anschließende Proteinanalyse zu berücksichtigen. Aus diesem Grund wurde das „SilverQuest Kit“ der Firma Invitrogen gewählt, da es die Sensitivität der Silberfärbung (Abbildung 3-6, Abschnitt 3.1.7.3) und die Kompatibilität zur Massenspektroskopie vereint. Für diese Färbemethode hat sich der Einsatz einer Proteinmenge von ca. 50 µg bei der Optimierung bewährt (Abbildung 3-4, Abschnitt 3.1.7.2). Im Rahmen dieser Arbeit sollten möglichst viele Proteine in den 2DGelen dargestellt werden, um die Expressionsmuster der Untersuchungsstämme untereinander zu vergleichen. Um dieses Ziel zu erreichen, wurde der zu verwendende pH-Bereich für die isoelektrische Fokussierung optimiert. Der zunächst sehr breit gewählte pH-Gradient von 3 bis 10 erwies sich aber als ungünstig. Da die größte Menge der aufgereinigten Proteine im pH-Bereich zwischen 4 und 7 lagen, wurde für die weitere Arbeit der kleinere pH-Gradient (pH 4 bis 7) gewählt (Abbildung 3-5, Abschnitt 3.1.7.2). Der Verlust von wenigen Proteinen im sehr sauren oder sehr alkalischen Bereich wurde in Kauf genommen, um den Hauptanteil der isolierten Proteine in der 2. Dimension im SDSGel gut aufzulösen. Im Rahmen dieser Arbeit wurden zahlreiche, aber bezüglich der Proteinmenge und der Färbemethode sehr unterschiedliche Proteingele hergestellt. Die Optimierungsarbeit erwies sich als sehr arbeits- und zeitintensiv. Prinzipiell sollten bei der Anwendung der 2DGelelektrophorese die Möglichkeiten, aber auch deren Grenzen und Nachteile, bedacht werden. Aufgrund fehlender Automatisierung ist die Reproduzierbarkeit beschränkt. Abweichungen von einigen Prozent in der Position und bis zu 20 % in den relativen Mengen einzelner Proteine müssen einkalkuliert werden. Trotz starker Verbesserungen, wie z.B. die immobilisierten pH-Gradienten-Gele [30] für die isoelektrische Fokussierung, ist die technische Durchführung schwierig und zeitaufwendig. Weiterhin gilt zu beachten, dass der Transfer von der ersten (IEF) auf die zweite Dimension (SDS-PAGE) oft nicht vollständig ist und die Gefahr von Proteinverlusten birgt. Eine weitere Schwierigkeit besteht in der nicht ausreichenden Dynamik. Sehr selten vorkommende Proteine können nicht gleichzeitig neben sehr häufigen Proteinen dargestellt werden. Mittels Vergleich der Bilder der Proteingele mit der Software PDQuest® kann ein Vergleich der Proteinexpression in den Stämmen erfolgen. Für eine befriedigende Auswertung im Rahmen dieser Arbeit stand jedoch keine ausreichende Menge an vergleichbaren Gelen pro Stamm zur Verfügung. Ein Vergleich (Matching) von Gelen mit Proteinproben aus 151 Diskussion unterschiedlichen Probenaufbereitungen oder Färbemethoden ergab Unterschiede zwischen M415 und vacT von mehr als 200 Spots. Ein auswertbarer Vergleich der Proteinspektren der Stämme M415 und vacT erfordert eine hohe Anzahl von Gelen (zur Absicherung auch mehrfache biologische Aufarbeitungen) mit identischer Durchführung. Für die eigentliche Analytik müssen die Proteine nach der 2D-GE als Spots aus der Gelmatrix entfernt und u.U. entfärbt werden. Deshalb ist eine gute Optimierung der einzelnen Schritte essentiell für die Auswertung. Da vacT mittels ungezielter Mutagenese entstanden ist, ist das Vorhandensein zahlreicher Mutationen in verschiedenen Genen wahrscheinlich. Der Ansatz der 2D-GE könnte prinzipiell Aufschluss darüber geben, ob Proteine durch Mutationen in ihrer Struktur oder ihrer Expression verändert sind (Lage und Intensität der Spots). Eine anschließende Analyse der Proteinspots mittels Massenspektroskopie ermöglicht eventuell die Identifizierung von Faktoren, die an der Veränderung der Generationszeit, der Attenuation oder den rtt-Marker im Impfstamm beteiligt sind. Weiterhin könnten solche Untersuchungen Rückschlüsse auf eine DNA Superspiralisierungsgrad abhängige Genexpression aufgrund der im Impfstamm akkumulierten vier gyrA-Mutationen zulassen. Dem Ansatz der 2D-GE wird zum Vergleich der Proteinspektren verschiedener Stämme große Bedeutung zugemessen. Durch diese Methode kann der Einfluss bisher unbekannter und nicht beachteter Faktoren identifiziert werden. Insgesamt konnten die einzelnen Schritt zur Durchführung der 2D-GE bis zur Spotdetektion optimiert werden und somit steht diese Methode für die weitere Forschungsarbeit zur Verfügung. Ziel weiterführender Arbeit wäre die Erstellung von Proteingelen mittels 2D-GE mit Proteinen aus den Untersuchungsstämmen M415, Nal2ori, Nal2passage und vacT unter gleichen Versuchsbedingungen Färbemethode). 152 (Probenaufarbeitung, Proteinkonzentration, pH-Gradient und Diskussion 4.2 Untersuchungen der im Impfstamm auftretenden gyrA-Mutationen Das Hauptinteresse dieser Arbeit galt der Aufklärung der Bedeutung der im Impfstamm vacT identifizierten gyrA-Mutationen. Die Einführung eines gyrA-Wildtyp-Gens durch einen Komplementationstest und eine anschließende phänotypische Charakterisierung der Transformanden können Aufschluss über die Rolle der Mutationen geben, z.B. bei der Ausprägung der Empfindlichkeiten gegenüber speziellen Antibiotika,. Um aber den Einfluss weiterer, nicht charakterisierter Mutationen auszuschließen, bestand die Notwendigkeit, über In-vitro-Mutagenese isogene Stämme herzustellen, welche ausschließlich die gyrA-Mutationen des Impfstammes einzeln bzw. in Kombination besitzen. Weiterhin sollten über Ergebnisse aus In-vitro-Selektionsversuchen des Ausgangsstammes und des Impfstammes Aussagen über das prinzipielle Auftreten von gyrA-Mutationen bzw. die Impfstammsicherheit getroffen werden. 4.2.1 Komplementationstest mittels eines plasmidcodierten gyrA+ Der Komplementationstest dient dem Nachweis der Auswirkung von gyrA-Mutationen auf den Phänotyp eines Stammes. Durch den Dominanzeffekt eines plasmidcodierten chinolonempfindlichen gyrA+-Gens über das chromosomale chinolonresistente gyrA-Gen erlangen gyrA-Mutanten wieder Chinolonempfindlichkeit (Abschnitt 3.1.6) [116,129]. Für diesen Versuch wurden die Plasmide pBP507, als Kontrollplasmid, und pBP517, mit dem gyrA+-Gen aus E. coli, genutzt. Um den Nachweis zu erbringen, dass der Test mit dem Gen aus E. coli in Salmonella vergleichbar funktioniert, wurde im Komplementationstest der Stamm SM415-87 als Kontrolle mitgeführt. Dieser Stamm war aus M415 während eines Selektionsversuches (Abschnitt 3.1.5.1) entstanden und besitzt die gyrA-Mutation D87Y. Diese Mutation bewirkt im Vergleich zu M415 einen Anstieg der MHK der Nalidixinsäure um vier (von 4 µg/ml auf 64 µg/ml) und des Ciprofloxacins um drei Verdünnungsstufen (von 0,03 µg/ml auf 0,25 µg/ml) (Abschnitt 3.1.5.1, Tabelle 3-11). Eine Transformation des Kontrollplasmides pBP507 verursachte keine Änderung der MHKWerte. Nach Einführung des Plasmides pBP517 bewirkte die Expression des gyrA+-Gens ein Absinken der MHK auf den Wert für den Ausgangsstamm. An diesem Beispiel konnte bestätigt werden, wie bereits von Heisig [123] gezeigt, dass das Dominanzprinzip mit einem aus E. coli stammenden gyrA-Gen einwandfrei in S. Typhimurium funktioniert und die MHK-Wert-Erhöhung für den Stamm SM415-87 durch die gyrA-Mutation D87Y hervorgerufen wurde. Nach Transformation der Dominanzplasmide in die Untersuchungsstämme Nal2ori, Nal2passage und vacT wurde eine MHK-Wert-Bestimmung mit zahlreichen Fluorchinolonen durchgeführt (Abschnitt 3.1.6.1). Bei allen untersuchten Vertretern dieser Antibiotikaklasse wurde die MHK durch Einführung von gyrA+-Gens mindestens auf den Wert des Ausgangsstammes gesenkt. Mit Hilfe des Komplementationstests konnte 153 Diskussion demzufolge gezeigt werden, dass die leicht reduzierten Empfindlichkeiten der Zwischenderivate Nal2ori und Nal2passage gegenüber Nalidixinsäure und den Fluorchinolonen auf einer oder mehreren gyrA-Mutationen beruhen. Im Fall des Impfstammes vacT, der z.T. sogar leicht erhöhte Empfindlichkeiten gegenüber verschiedenen Fluorchinolonen zeigte, brachte die Einführung von gyrA+ noch ein zusätzliches Absinken der MHK-Werte. Das bedeutet, dass die gyrA-Mutationen im Impfstamm grundsätzlich auch eine reduzierte Empfindlichkeit gegenüber Fluorchinolonen bewirken. Dieses ist aber phänotypisch nicht ausgeprägt. Im Impfstamm vacT muss daher ein weiterer Effekt für die Kompensation der reduzierten Empfindlichkeit gegenüber den Fluorchinolonen verantwortlich sein. Ein Einfluss der gyrA-Mutationen auf die Generationszeit oder den DNA- Superspiralisierungsgrad konnte mittels des Komplementationstests nicht nachgewiesen werden (Abschnitt 3.1.6.2). Um deutlichere Ergebnisse zur Bedeutung der gyrAMutationen sowohl bei der Ausprägung der (Fluor)chinolonempfindlichkeit als auch bei der Wachstumsgeschwindigkeit und dem DNA-Superspiralisierungsgrad zu erhalten, wurden aus M415 Stämme mit den relevanten gyrA-Mutationen erzeugt und charakterisiert. 4.2.2 In-vitro-Selektion von gyrA-Mutanten Die Herstellung von gyrA-Mutanten aus dem Ausgangsstamm M415 erfolgte durch zwei verschiedene Methoden. Zum einen wurde eine In-vitro-Selektion des Stammes auf Ciprofloxacin vorgenommen (Abschnitt 4.2.2) und zum anderen wurden die gewünschten Mutationen mittels homologer Rekombination in das Chromosom von M415 (Abschnitt 4.2.3) eingekreuzt. Die In-vitro-Selektion des Stammes M415 diente der Kontrolle, welche Mutationen in M415 unter Selektionsdruck auftreten. Es wurde überprüft, ob die aus dem Impfstamm bekannten Mutationen nach Selektion in vitro auftreten bzw. welche anderen Aminosäureaustausche in M415 erfolgen und wie sich die Mutanten phänotypisch verhalten. Bei einer In-vitro-Selektion handelt es sich um eine Methode, die das Auffinden von Mutationen ermöglicht, die dem Stamm eine veränderte Empfindlichkeit gegenüber dem zur Selektion genutztem Antibiotikum verleihen. Mutationen treten spontan und zufällig auf. Da Gyrase das primäre Target für Chinolone darstellt, tragen chinolonresistente Mutanten aus dem ersten Selektionsschritt fast immer Mutationen im gyrA-Gen. Nach Selektion von M415 auf ciprofloxacinhaltigen Agarplatten (Konzentration von Ciprofloxacin: 2 bis 8 x MHK) Mutationsfrequenz von ca. 2 x 10 -10 konnten resistente Mutanten mit einer -9 bis 1 x 10 isoliert werden (Abschnitt 3.1.5.1). Die im Rahmen dieser Arbeit selektierten gyrA-Mutanten zeigten reduzierte Empfindlichkeiten gegenüber Fluorchinolonen und waren resistent gegen Nalidixinsäure (Tabelle 3-11, Abschnitt 3.1.5.1). Es wurden Mutationen identifiziert, die zu Aminosäureaustauschen Gly81Asp, Ser83Phe und Asp87Tyr führen (Abschnitt 3.1.5.1). Die Ergebnisse bestätigen also die aus der Literatur bekannten Aminosäureaustausche an Hotspots innerhalb der QRDR von gyrA in chinolonresistenten Stämmen [48,51,83,118,237,253,294]. In E. coli 154 Diskussion sind abstufende Empfindlichkeiten gegenüber Fluorchinolonen in Abhängigkeit von der von dem Austausch betroffenen Aminosäure beschrieben [305]. Danach bewirkt eine Veränderung an der Position 83 den größten Effekt auf die MHK-Werte, was auch im Selektionsversuch im Rahmen dieser Arbeit beobachtet werden konnte. Der Stamm SM415-83 (Ser83Phe) wies die höchsten Fluorchinolon-MHK-Werte mit Steigerungen gegenüber dem Ausgangsstamm von z.B. drei (Ofloxacin), vier (Ciprofloxacin) oder bis zu sechs Verdünnungsstufen (Pefloxacin) auf. Die MHK der Nalidixinsäure für SM415-83 betrug 512 µg/ml im Vergleich zu 4 µg/ml von M415. Die selektierten Stämme mit Aminosäureaustauschen an den Positionen Gly81Asp und Asp87Tyr reagierten untereinander sehr ähnlich, jedoch im Vergleich zu SM415-83 insgesamt etwas empfindlicher auf die Gyrasehemmer. Die Werte gegenüber dem Ausgangsstamm waren nur um z.B. ein bis zwei (Ofloxacin), drei (Ciprofloxacin) oder zwei bis drei Verdünnungsstufen (Pefloxacin) erhöht. Cambau et al. [40] beschrieben einen E. coliStamm mit der gyrA-Mutation Gly81Asp, welcher zwar verringerte Empfindlichkeiten gegenüber Fluorchinolonen, aber nicht gegenüber Nalidixinsäure zeigte. Diese Beobachtung kann für den Salmonellenstamm SM415-81 nicht bestätigt werden. Wie auch SM415-87 wies der Stamm SM415-81 mit einem MHK-Wert von 64µg/ml eine Nalidixinsäureresistenz auf. Der Selektionsversuch von S. Typhimurium M415 in Anwesenheit von Ciprofloxacin bestätigte das Auftreten von bereits in der Literatur beschriebenen Aminosäureaustauschen. Folglich stellt die Entwicklung reduzierter Fluorchinolonresistenz und das Auftreten spezifischer gyrA-Mutationen in dem Stamm M415 keine Besonderheit dar. Auch eine Mutante mit einem Aminosäureaustausch an Position 87 konnte selektiert werden. Allerdings ist bei dieser Mutante Aspartat-87 durch Tyrosin ausgetauscht. Im Impfstamm vacT befindet sich an der Aminosäureposition 87 allerdings Glycin. Eine Mutante mit diesem Aminosäureaustausch konnte bei der Selektion in vitro ebenso wie die anderen aus dem Impfstamm bekannten Mutationen an Aminosäurepositionen 59, 75 oder 866 nicht selektiert werden. Hierbei gilt es zu beachten, dass die Mutationen in vacT nicht durch Selektion sondern durch ungezielte Mutagenese entstanden sind. Somit konnten auch Basenaustausche auftreten, die nicht unmittelbar an der Ausprägung des Phänotyps beteiligt sind. Das fehlende Auftreten von gyrA-Mutanten mit den aus vacT bekannten Mutationen legt nahe, dass diese Mutationen entweder mit einer sehr geringen Mutationsfrequenz, durch ein anderes Fluorchinolon als Ciprofloxacin selektiert oder erst in späteren Selektionsschritten auftreten würden oder nicht an der Entwicklung der verringerten Fluorchinolonempfindlichkeit beteiligt sind. Um letzteren Punkt zu be- oder widerlegen wurden über gezielte Mutagenese mittels homologer Rekombination entsprechende Mutanten hergestellt und untersucht. 155 Diskussion 4.2.3 In-vitro-Mutagenese von gyrA-Mutanten mittels homologer Rekombination Die phänotypischen Eigenschaften des Impfstammes vacT beruhen offensichtlich auf dem Zusammenwirken verschiedener genotypischer Veränderungen. Aufgrund der Entstehung des Impfstammes durch ungezielte Mutagenese ist davon auszugehen, dass noch weitere Mutationen in anderen Genen als gyrA wahrscheinlich sind. Um die Bedeutung der identifizierten gyrA-Mutationen zu bestimmen, ist es notwendig, die Auswirkungen anderer Mutationen auszuschließen. Das Ziel der Experimente zur In-vitro-Mutagenese war daher ausgewählte Mutationen in das gyrA-Gen des Ausgangsstammes M415 einzeln bzw. in Kombination einzuführen. 4.2.3.1 Einfluss der gyrA-Mutationen auf die MHK-Werte Alle über homologe Rekombination aus M415 hergestellten Stämme enthielten die Mutation D87G. Neben der Einzelmutante wurden weiterhin Stämme erzeugt, die zusätzlich jeweils eine der Mutationen W59R bzw. G75A tragen. Die hergestellten isogenen Stämme wurden phänotypisch hinsichtlich ihrer Fluorchinolonempfindlichkeit untersucht. Sowohl die Einzel- (D87G) als auch die beiden Doppelmutanten (D87G + W59R und D87G + G75A) zeigten Nalidixinsäureresistenz und reduzierte Empfindlichkeiten gegenüber Fluorchinolonen (Tabelle 3-21, Abschnitt 3.2.5.1). Der im Anschluss durchgeführte Komplementationstest mit gyrA+ (Tabelle 3-22, Abschnitt 3.2.5.2) bestätigte, dass die eingekreuzte(n) Mutation(en) die Ursache der MHK-Wert-Erhöhungen sind. In Anwesenheit des empfindlichen gyrA+-Genproduktes, codiert von Plasmid pBP517, sanken die MHK-Werte aufgrund des Dominanzprinzipes auf das Niveau des Ausgangsstammes M415 (Abschnitt 2.2.1.8). Die Ergebnisse aus den MHK-Wert-Bestimmungen bestätigen die Hypothese, dass die Mutation D87G in einem Stamm zu einer verringerten (Fluor)chinolonempfindlichkeit führt. Alle Rekombinanten, welche diese Mutation allein oder in Kombination mit W59R oder G75A tragen, zeigten sehr ähnliche MHK-Wert-Erhöhungen. Die Rekombinanten RM41587 und RM415-75-87 verhielten sich bei den Empfindlichkeitsbestimmungen identisch. Die minimalen Hemmkonzentrationen der Fluorchinolone waren um zwei bis drei Verdünnungsstufen erhöht (Tabelle 3-21 Abschnitt 3.2.5.1). Die MHK-Werte des Sparfloxacin bzw. Ciprofloxacin stiegen von 0,03 µg/ml für den Ausgangsstamm auf 0,125 bzw. 0,25 µg/ml für die beiden Rekombinanten. Der MHK-Wert der Nalidixinsäure lag für beide Stämme bei 256 µg/ml. Das entspricht einer Erhöhung von sechs Verdünnungsstufen bezogen auf den Ausgangsstamm M415 (MHKNal 4 µg/ml). Daraus folgt, dass die Mutation D87G für die Erhöhung der minimalen Hemmkonzentrationen von Nalidixinsäure und Fluorchinolonen verantwortlich ist. Die Mutation G75A hat weder einen verstärkenden noch kompensierenden Einfluss auf die Empfindlichkeit gegenüber den Fluorchinolonen oder der Nalidixinsäure, obwohl sie 156 Diskussion innerhalb der QRDR liegt. Die Reduktion der Empfindlichkeit der Stämme Nal2ori und Nal2passage wird also allein durch die Mutation D87G hervorgerufen. Da keine Unterschiede in den MHK-Werten zwischen Nal2ori und RM415-75-87 festzustellen waren, wird auch der in Nal2ori zusätzlich vorkommenden Mutation an Position A866S keine weitere Bedeutung bei der verringerten Chinolonempfindlichkeit zugemessen. Die Rekombinante RM415-59-87 zeigte im Vergleich zum Stamm M415 ebenfalls erhöhte MHK-Werte der Fluorchinolonen und Nalidixinsäure. Allerdings stiegen in diesem Stamm die MHK-Werte z.B. des Sparfloxacin bzw. Ciprofloxacin nur um eine Verdünnungsstufe (auf 0,06 µg/ml) und der Nalidixinsäure um vier Verdünnungsstufen (auf 64 µg/ml). Die Unterschiede zwischen den Stämmen RM415-87 bzw. RM415-75-87 einerseits und RM415-59-75 andererseits betrugen nur eine bis maximal zwei Verdünnungsstufen, was gegebenenfalls als Fehlerbereich der MHK-Wert-Bestimmung gewertet werden kann. Diese Unterschiede wurden aber in allen Wiederholungen der Messung beobachtet. Die Mutation W59R bewirkt also nur eine geringe Kompensation des Effektes der Mutation D87G im Bezug auf die Chinolonempfindlichkeit. Eine Erklärung dafür könnte eine mögliche Änderung der Molekülstruktur sein: Der Austausch eines sauren Aspartats durch ungeladenes Glycin an der Position 87 könnte eine veränderte Affinität der Gyrase zu Fluorchinolonen verursachen. Aber auch die Mutante SM415-87 (D87Y) zeigte erhöhte MHK-Werte der Fluorchinolone. Bei diesem Austausch scheint die Position des Austausches entscheidend zu sein. Allerdings fehlen bislang durch Kristallstrukturen abgesicherte molekulare Modelle für die Chinolonbindung an den DNA-Komplex. Yoshida et al. [307] gehen aus ihren Beobachtungen mit selektierten Mutanten davon aus, dass die vordringlich gefundenen Aminosäureaustausche an Position 83 und 87 möglicherweise aufgrund von Veränderungen in einer postulierten Bindungstasche der Gyrase die Affinität der Chinolone reduzieren. Bei der Doppelmutante RM415-75-87 wurden nun zusätzlich an Position 75 mit Glycin zu Alanin zwei unpolare, aliphatische und ähnlich große Aminosäuren gegeneinander ausgetauscht. Dadurch ist vermutlich keine allzu starke Änderung der Molekülstruktur der Gyrase zu erwarten. Die Mutation G75A hat somit wahrscheinlich keinen Einfluss auf die Empfindlichkeit gegenüber Chinolonen, wie bei der Empfindlichkeitsbestimmung auch ermittelt. In der Doppelmutante RM415-59-87 hingegen wurde mit Tryptophan zu Arginin an Position 59 eine unpolare, aromatische Aminosäure gegen eine basische, aliphatische und damit ganz anders strukturierte Aminosäure ausgetauscht. Es ist denkbar, dass die Mutation W59R durch die unmittelbare Nähe zur für die Chinolonbindung relevanten QRDR zu einer Änderung der Struktur des Gyrasemoleküls führt. Dies ermöglicht vielleicht eine wieder minimal verbesserte (wenn auch immer noch sehr eingeschränkte) Bindung der Chinolone an die Gyrase. Die Ergebnisse aus den MHK-Wert-Bestimmungen der Rekombinanten zeigen aber generell die Relevanz der Mutation D87G für eine reduzierte Empfindlichkeit sowohl gegenüber Nalidixinsäure als auch gegenüber den Fluorchinolonen. Eine Wirkung der 157 Diskussion gyrA-Mutationen A866S und G75A konnte weitestgehend ausgeschlossen werden. Die Mutation W59R in Kombination mit der Mutation D87G bewirkt nur eine sehr leichte Reduktion der Empfindlichkeit gegenüber Fluorchinolonen im Vergleich zur Einzelmutante mit D87G oder der Doppelmutante mit D87G + G75A. Das Auftreten der Mutation W59R kann aber nicht die für den Impfstamm vacT beschriebene Supersensibilität gegenüber Fluorchinolon erklären. Die Kompensation der Wirkung der gyrA-Mutation im Impfstamm vacT ist somit nicht intragenisch, sondern eher extragenisch, d.h. in Veränderungen anderer Genloci begründet. 4.2.3.2 Beeinflussung des DNA-Superspiralisierungsgrades Die ermittelten DNA-Superspiralisierungsgrade der drei erzeugten Rekombinanten zeigten keinen Unterschied zwischen der gyrA-Einzelmutante mit D87G und den gyrADoppelmutanten mit D87G + G75A bzw. D87G + W59R (Abschnitt 3.2.5.3). Die Qsc-Werte aller drei Rekombinanten betrugen zwischen ca. 56 und 60 % des Wertes des Ausgangsstammes M415 (100 %). Als Kontrolle wurden die Stämme SM415-87 (Abschnitt 3.1.5.1) und die Rekombinante RM415 bei der Bestimmung des Superspiralisierungsgrades mitgeführt. Beide Stämme zeigten keine signifikanten Änderungen des Qsc-Wertes im Vergleich zum Ausgangsstamm M415 (Tabelle 3-23, Abschnitt 3.2.5.3). Die Rekombinante RM415 enthält keine gyrA-Mutationen, aber eine integrierte Kanamycinresistenz-Kassette (Abschnitt 3.2.4.2). Aufgrund des unveränderten Qsc-Wertes dieses Stammes im Vergleich zu M415 kann die Insertion der ResistenzKassette als Einflussfaktor auf den Superspiralisierungsgrad der Rekombinanten ausgeschlossen werden. Der im Vergleich zum Ausgangsstamm unveränderte Qsc-Wert der gyrA-Mutante SM415-87 (D87Y) zeigt, dass nicht jeder beliebige Aminosäureaustausch an Position 87 zu einer veränderten Gyraseaktivität und somit einem reduzierten DNA-Superspiralisierungsgrad führt. Die Rekombinanten RM415-87 und RM415-75-87 besaßen mit 1,09 bzw. 1,01 sehr ähnliche und gegenüber dem Ausgangsstamm M415 (~1,8) stark reduzierte Qsc-Werte. Hier zeigt sich, dass die Mutation G75A keinen Einfluss auf den Superspiralisierungsgrad hat. Ähnliche Qsc-Werte wurden für Nal2ori und Nal2passage gemessen (0,98 bzw. 0,99), in denen ebenfalls die gyrA-Mutationen D87G+G75A vorliegen. Zusätzlich besitzen die beiden letzteren Stämme eine weitere gyrA-Mutation an Position A866S, welche den DNA-Superspiralisierungsgrad demzufolge aber nicht beeinflusst. Auch das Auftreten der Mutation W59R bewirkt keine Änderung des Qsc-Wertes (Tabelle 3-23, Abschnitt 3.2.5.3). Die Bestimmung der Superspiralisierungsgrade zeigte keine signifikanten Unterschiede zwischen RM415-87 und RM415-59-87. Möglicherweise erlaubt die Methode des Reportergensystems aber auch nicht das Erkennen von sehr feinen Unterschieden, wie sie bei den MHK-Wert-Bestimmungen festgestellt wurden, da Schwankungen von ca. 1015 % als Fehlerbereich angesehen werden müssen (s. Tab. 3-23). Im direkten Vergleich der Qsc-Werte zwischen der Einzelmutante RM415-87 und der Vierfachmutante vacT 158 Diskussion zeigten sich keine Unterschiede im DNA-Superspiralisierungsgrad. In allen durch Rekombination erzeugten gyrA-Mutanten und den Untersuchungsstämmen war der DNASuperspiralisierungsgrad ähnlich stark reduziert, was dafür spricht, dass die verringerte Gyraseaktivität in allen Fällen als Ursache anzusehen ist. Ähnlich wie bei der Ausprägung der Chinolonempfindlichkeit scheint also auch im Fall der Reduktion des DNASuperspiralisierungsgrades die Mutation D87G der maßgebliche Faktor für die Veränderung zu sein. Ein anderer Aminosäureaustausch an dieser Position, wie z.B. D87Y, bewirkt keine signifikante Qsc-Wert-Änderung. Allein die Anwesenheit der Mutation D87G reduziert den Qsc-Wert des Stammes RM415-87 auf 56,11 %. Zusätzlich gefundene Mutationen wie W59R, G75A oder A866S beeinflussen die Gyraseaktivität nicht. 4.2.3.3 Ein Einfluss der untersuchten gyrA-Mutationen auf die Fitness Vergleich der Generationszeiten der Rekombinanten im Vergleich zum Ausgangsstamm M415 (Tabelle 3-24 Abschnitt 3.2.5.4) zeigte keine Abweichungen. Der Stamm Nal2ori (gyrA: G75A, D87G) wies allerdings eine erhebliche Fitnessreduktion mit einer ungefähr dreimal so langen Generationszeit wie der Ausgangsstamm auf (Abschnitt 4.1.5). Da aber weder die Rekombinante RM415-87 noch RM415-75-87 ein Wachstumsdefizit zeigten, muss die Ursache für die Generationszeitenverlängerung in Nal2ori in anderen Effekten begründet sein. Es wurden keine Unterschiede in den QscWerten zwischen den langsam wachsenden Stämmen Nal2ori und vacT und den normal wachsenden Rekombinanten festgestellt. Deshalb kann der DNA- Superspiralisierungsgrad und die daraus resultierende veränderte Genexpression als Grund für die Abweichung der Wachstumsgeschwindigkeit weitestgehend ausgeschlossen werden. Allerdings muss berücksichtigt werden, dass keine Rekombinante existiert, welche die Mutation A866S in Kombination mit anderen Mutationen aufweist. Möglicherweise spielt diese Mutation, wie bereits diskutiert (Abschnitt 4.1.1), eine Rolle bei der Interaktion der Untereinheiten oder der Bindung der DNA. Als auffällig problematisch erwies sich die Herstellung einer Rekombinanten mit der gyrA-Dreifach-Kombination W59R, G75A und D87G. Diese Kombination entspricht, bis auf die Abwesenheit der Mutation A866S, der Situation im Impfstamm. Rekombinationssystemen Trotz konnte mehrerer diese Versuche Dreifachmutante mit nicht unterschiedlichen erzeugt werden. Möglicherweise ist ein Stamm mit einer Gyrase mit dieser Dreifachmutation ohne die Mutation A866S nicht lebensfähig und letztere nötig, um das Enzym zu stabilisieren. Ein ähnlicher Zusammenhang wurde bisher allerdings nicht beschrieben. Cullen et al. [62] beschrieben eine Ser-828 Æ Ala Mutation. Diese Mutante enthielt weiterhin gyrAMutationen in den Codons 83 und 678. Der untersuchte E. coli Stamm mit diesen gyrAMutationen war lebensfähig. Aber auch in diesem Fall ist nicht klar, welche Rolle die Mutationen im C-Terminus von gyrA spielen. Auch der Stamm vacT ist mit allen vier gyrA159 Diskussion Mutationen lebensfähig, was aber nicht ausschließt, dass eine Gyrase mit nur drei Mutationen eine eingeschränkte Aktivität zeigt, was durch die Mutation A866S möglicherweise kompensiert wird. Um diese Hypothese zu testen, müsste man zunächst eine Rekombinante mit der Mutation A866S erzeugen und in diese die drei weiteren gyrAMutationen einkreuzen. Die These, dass eine Mutation nötig ist, um die Stabilität des Proteins mit den drei weiteren Aminosäureaustauschen zu gewährleisten, könnte erklären, warum eine Akkumulation von vier Mutationen in gyrA erfolgt ist, wohingegen in den anderen Targetgenen gyrB, parC und parE keine Mutationen zu finden waren. Weiterhin wäre dies auch eine mögliche Erklärung für die schlechte Mutationsfrequenz (2 x 10-10 bis 6 x 10-11) des Impfstammes vacT im Selektionsversuch mit Fluorchinolonen (Abschnitt 3.1.5.2). Eine Reduktion der Fluorchinolonempfindlichkeit oder gar Ausbildung von Resistenz würde beispielsweise eine weitere gyrA-Mutation erfordern, z.B. an Position 83 [17,129]. Da aber eine weitere Mutation möglicherweise auch eine weitere Schwächung der essentiellen Enzymaktivität in vacT bewirken würde und die Zelle möglicherweise nicht mehr lebensfähig wäre, konnten keine weiteren gyrA-Mutationen selektiert werden. Das Auftreten von Doppelmutationen in den Codons 83 und 87 konnte in In-Vitro-Selektionsversuchen mehrfach beobachtet werden [17,129]. Allerdings weist der Impfstamm mit den neu identifizierten Mutationen W59R, G75A und A866S einen genetischen Hintergrund auf, zu dem es noch keine Untersuchungen gibt. Somit kann keine Aussage über die mögliche Lebensfähigkeit des Stammes mit einer zusätzlichen Fluorchinolonresistenz vermittelnden gyrA-Mutation getroffen werden. Dies drückt möglicherweise auch eine hohe Sicherheit des Impfstammes vacT aus. Auch unter Selektionsdruck ist die Entwicklung zu einem fluorchinolonresistenten Stamm aufgrund des bestehenden Genotyps kaum möglich. Es konnten nur vereinzelt Mutanten mit erhöhten Fluorchinolon-MHK-Werten selektiert (vacT-E1-3) werden, welche aber keine Fluorchinolonresistenz zeigen. Diese Stämme weisen aber keine vom Ausgangsstamm vacT abweichenden Mutationen in den Targetgenen auf. In diesen Stämmen wird eine Veränderung des aktiven Effluxes als Grund für die reduzierte Empfindlichkeit gegenüber Fluorchinolonen vermutet (Abschnitt 4.3). 160 Diskussion 4.3 Der Einfluss von Effluxpumpen Bereits die Ergebnisse Komplementationstestes möglicherweise nicht der MHK-Wert-Bestimmungen (Abschnitt nur die 3.1.6.1) ergaben gyrA-Mutationen an im Hinweise der Rahmen des darauf, dass Ausprägung der Fluorchinolonempfindlichkeit beteiligt sind. Im Impfstamm vacT, der sogar eine erhöhte Fluorchinolonempfindlichkeit besitzt, konnten durch die Einführung eines + chinolonsensiblen gyrA -Gens die MHK-Werte der Fluorchinolone noch weiter gesenkt werden. Dieser Effekt spricht dafür, dass eine oder mehrere gyrA-Mutationen die Fluorchinolonempfindlichkeit reduzieren. Ein anderer Effekt ist aber dafür verantwortlich, dass dies phänotypisch nicht zur Ausprägung kommt. Auch die bei dem Impfstamm vacT festgestellte Makrolidsensibilität weist auf einen unabhängig von den gyrA-Mutationen existierenden Defekt in vacT hin. Beide Ergebnisse geben einen Hinweis auf eine verstärkte Akkumulation verschiedener Antibiotika in der Zelle. Dafür kommt sowohl ein reduzierter Efflux, als auch ein verstärkter Influx in Frage. Bisher sind in zahlreichen Studien die Transportwege durch die Membran v.a. in Zusammenhang mit einer verringerten Akkumulation der Substanzen in der Zelle untersucht. Im Rahmen der Resistenzentwicklung von Bakterien ist ein verstärkter Efflux, ausgelöst durch eine Überexpression von Effluxpumpen [26,92,103,201,243], und verringerter Influx durch die Reduktion von „Outer Membrane Proteins“ (Omps) beschrieben [141]. Überexpression von Effluxpumpen bietet der Zelle in einem Habitat mit Antibiotika einen ökologischen Vorteil. Das Pumpen von toxischen Produkten aus der Zelle sichert dem Bakterium ein Überleben unter entsprechenden biologischen Bedingungen bzw. das Erschließen neuer verschiedener natürlicher Habitate, ökologischer Nischen und das Überleben im Wirt. Im Gegensatz zu Veränderungen der Zielstrukturen, die zu einer Resistenz gegenüber spezifischen Antibiotika führen, bewirken Veränderungen der Transportwege, aufgrund des breiten Substratspektrums der meisten Pumpen, eine veränderte Empfindlichkeit gegenüber verschiedenen Antibiotikaklassen. Eine Überexpression von Effluxpumpen ist deshalb oft gekennzeichnet durch einen „multidrug efflux“ (mde), was zu einem MARPhänotyp (multiple antibiotic resistance) des betroffenen Stammes führt. Im Gegensatz dazu zeichnen sich allerdings bestimmte Pumpen auch durch ein spezifisches Spektrum an Substraten aus. Spezielle Pumpen, wie z.B. der Tetracyclin / H+ - Antiporter transportieren sogar nur Vertreter einer speziellen Antibiotikaklasse [203]. Da der aktive Efflux über Pumpen ein wichtiger Faktor bei der Resistenzentwicklung ist, berichten verschiedene Arbeitsgruppen über die Identifizierung von Pumpen und deren Regulation (Überblick siehe [238]). Obwohl schon einige Ergebnisse über Effluxpumpen von Salmonella spec. existieren [23,24,26,48,51,103], fehlt noch immer ein umfassendes Wissen über gattungsspezifische Pumpen und vor allem über deren Regulation. Da Salmonella nah verwandt mit E. coli ist, geht man davon aus, dass zahlreiche Regulationsmechanismen und Pumpen sehr ähnlich in beiden Gattungen funktionieren. So wird z.B. den AcrAB/EF-Pumpen-Systemen auch in Salmonella spec. eine große 161 Diskussion Bedeutung zugemessen [24]. Eine Überexpression dieses Pumpensystems zieht eine reduzierte Empfindlichkeit gegenüber z.B. Fluorchinolone, Chloramphenicol oder Tetracyclin nach sich. Hervorgerufen wird eine erhöhte Expression der AcrAB-Pumpe z.B. durch Veränderungen des lokalen Regulators AcrR oder des globalen Regulators MarA (siehe Einleitung Abschnitt 1.3.2). Im Gegensatz zur Resistenz durch Überexpression der Effluxpumpe, z.B. aufgrund des Ausfalls lokaler oder globaler Repressoren, zeigen Mutanten mit Deletionen in acrAB den gegenteiligen Effekt und es kommt zu einer Überempfindlichkeit gegenüber verschiedenen Chemotherapeutika [23,24,103,218,223,225]. Eaves et al. [84] und andere [23] beschrieben acrF- oder acrB-, aber auch tolC-Mutanten von S. Typhimurium mit erhöhten Sensibilitäten gegenüber Fluorchinolonen, Tetracyclinen oder Chloramphenicol. Der größte Effekt auf die MHK der Fluorchinolone war bei Ausfall von AcrB zu erkennen und wurde durch acrF-Deletion nicht wesentlich verstärkt. Dies ist ein Hinweis darauf, dass die AcrAB-TolC-Pumpe auch in Salmonella spec. eine Hauptfunktion übernimmt [23]. Aus In-Vitro-Selektionsversuchen mit acrB-Mutanten in Gegenwart von Fluorchinolonen ergaben sich Stämme mit wieder erhöhten MHK-Werten der Fluorchinolone. Ursache war der Anstieg der acrEF-Expression aufgrund der Integration von Insertionselementen mit eigenen starken Promotoren in den Regulationsbereich 5’ von acrEF [161,226]. Dies zeigt die Möglichkeit, wie der Ausfall einzelner Pumpenproteinen möglicherweise kompensiert werden kann. 4.3.1 Der Einfluss eines Effluxpumpeninhibitors Als globaler Ansatz zur Prüfung der Rolle von Effluxpumpen im Impfstamm vacT wurde im Rahmen dieser Arbeit eine Empfindlichkeitsbestimmung unter Zugabe eines Effluxpumpeninhibitors (EPI) durchgeführt. Im Rahmen von Untersuchungen zur Resistenzentwicklung, die durch eine Überexpression von Effluxpumpen hervorgerufen ist, gilt das Absinken der MHK-Werte der spezifischen Antibiotika unter Einfluss des EPIs als Hinweis auf die Beteiligung von Effluxpumpen. Effluxpumpen beziehen ihre Energie entweder aus der ATP-Hydrolyse oder dem Protonengradienten [229]. In Bakterien spielen vor allem Pumpensysteme eine Rolle, die durch Energie aus dem Protonengradienten betrieben werden. Für die Untersuchungen in dieser Arbeit wurde der Inhibitor Phenylalanyl-Arginyl-β-Naphthylamid verwendet. Diese Substanz ist dafür bekannt, Effluxpumpen zu hemmen, die den Protonengradienten als Energiequelle nutzen [24,48,183,201]. Der genaue Mechanismus des EPIs ist allerdings noch umstritten [238]. Möglicherweise zeigt der EPI eine hohe Affinität zu Substraten von MDR-Pumpen. Dieser Substrat-Inhibitor-Komplex kann nicht mehr von den Pumpen aus der Zelle transportiert werden [315]. Aber auch andere Vermutungen, dass der EPI ein Substrat der Pumpe ist und an MDR-Transporter bindet, konnte durch Kristallstrukturen von AcrB in Gegenwart verschiedener Substanzen und u.a. Phenylalanyl-Arginyl-β-Naphthylamid noch nicht bestätigt werden [310]. 162 Diskussion Zunächst wurden die minimalen Hemmkonzentrationen des Effluxpumpeninhibitors für die Untersuchungsstämme M415, Nal2ori, Nal2passage und vacT bestimmt (Tabelle 325 Abschnitt 3.3.1). Dabei war die hohe Sensibilität des Impfstammes auf den EPI auffällig. Während Nal2ori und Nal2passge mit 128 und 64 µg/ml noch relativ unempfindlich gegenüber dem Inhibitor waren, führten bei dem Impfstamm vacT bereits 16µg/ml zum Absterben des Stammes. Dieses Ergebnis lässt darauf schließen, dass dem Impfstamm eine geringere Menge an funktionierenden Effluxpumpen zur Verfügung steht. Die Funktionalität einer ausreichenden Menge an Pumpen ist für das Bakterium essentiell, denn nur so ist eine Entsorgung toxischer Substanzen und schädlicher Stoffwechselprodukte möglich. Eine zu starke Limitierung der Effluxpumpen durch eingeschränkte Funktionalität oder fehlende Energieversorgung kann zur Vergiftung der Zelle und zu einem eingeschränkten Wachstum der Bakterien bis hin zu deren Tod führen. Die Ergebnisse aus den Bestimmungen der minimalen Hemmkonzentrationen unter Zugabe des Effluxpumpeninhibitors (Tabelle 3-26 und Tabelle 3-27 Abschnitt 3.3.2) gaben weitere Hinweise auf eine Limitierung des aktiven Effluxes im Impfstamm vacT. Es zeigte sich ein Absinken der MHK-Werte unter Einfluss des EPIs. Allerdings waren Unterschiede bei den einzelnen Stämmen zu beobachten. Das Beispiel Erythromycin zeigte deutlich, dass im Impfstamm vacT eine wesentlich geringere Menge an EPI (8 µg/ml) eingesetzt werden muss, um den MHK-Wert auf den Wert von 0,5 µg/ml zu senken, als im Stamm Nal2ori (32 µg/ml), in Nal2passage (16 µg/ml) oder gar im Ausgangsstamm M415 (64 µg/ml). Eine höhere Konzentration von EPI führte in allen Fällen zu einer Wachstumshemmung der Stämme. Die angegebenen EPI- Konzentrationen spiegeln also die notwendige maximale Inhibition wider, um die MHKWerte des Makrolids Erythromycin auf das minimale Niveau zu senken, dem Stamm aber noch ein Überleben zu ermöglichen. Auch bei dem Fluorchinolon Sparfloxacin war dieser Effekt zu beobachten. In diesem Fall war der Impfstamm vacT aber bereits so sensibel (MHKSpar 0.007 µg/ml), dass kein weiteres Absinken des MHK-Wertes durch die Zugabe des EPIs erreicht werden konnte. Das Beispiel Tetracyclin zeigte bei allen eingesetzten EPI-Konzentrationen keine starken Änderungen der MHK-Werte. Diese lagen in allen Fällen zwischen 0,25 und 1 µg/ml. Dies spricht dafür, dass der Transportweg von Tetracyclin aus der Zelle nicht messbar beeinflusst ist. Neben den Fluorchinolonen war der Efflux der Makrolide ebenfalls stark beeinträchtigt. Im Ausgangsstamm M415 ist die Makrolidresistenz durch ein gut funktionierendes Efflux-System gewährleistet. Der Einfluss der AcrAB-TolC-Pumpe bei der Makrolidresistenz wurde bereits von Chollet et al. [47] beschrieben, die Clarithromycin- und Erythromycin-Empfindlichkeit bei E. coli- Mutanten mit Defekten im AcrAB-TolC-Pumpensystem beobachteten. Eine Inhibition der Effluxpumpen durch Phenylalanyl-Arginyl-β-Naphthylamid führte wieder zu einer Makrolidsensibilität der Stämme. Diese Makrolidsensibilität durch EPI-Behandlung konnte im Rahmen dieser Arbeit auch für den Ausgangsstamm M415 gezeigt werden. Somit könnte sowohl die Makrolid- als auch die Fluorchinolonsensibilität des Impfstammes vacT 163 Diskussion in einem Defizit in den Effluxpumpen begründet sein. Oethinger et al. [223] zeigten, dass in E. coli eine funktionsuntüchtige AcrAB-Pumpe den Stämmen Hypersensibilität gegenüber Fluorchinolon verlieh, trotz vorhandener Gyrase-Mutationen. Einen weiteren Hinweis auf eine mögliche Mutation in einem Efflux-System des Impfstammes brachten die Ergebnisse des Selektionsversuches von vacT auf Enrofloxacin. Die wenigen selektierten Mutanten (vacT-E1-3) zeigten im Gegensatz zu vacT eine leicht reduzierte Fluorchinolonempfindlichkeit und interessanterweise wieder die Makrolidresistenz des Ausgangsstammes M415 (Tabelle 3-13, Abschnitt 3.1.5.2). Nach einer Sequenzierung der QRDR der Targetgene konnte das Auftreten der Mutationen in den Genen gyrA, gyrB, parC Fluorchinolonempfindlichkeit oder parE ausgeschlossen als Ursache werden. für die verringerte Untersuchungen mit Effluxpumpeninhibitor zeigten ein Absinken der Makrolid-MHK-Werte in einen sensiblen Bereich. Bei diesem Selektionsversuch waren also offensichtlich Mutanten aufgetreten, welche eine erhöhte Expression von Effluxpumpen als Ursache für die reduzierten Antibiotikaempfindlichkeiten besitzen. Bisher ist die molekulare Ursache dafür nicht geklärt. So könnte bei vacTE1-3 z.B. eine Reversion der in vacT zum Pumpendefekt führenden Mutation (intragenische Mutation) oder eine extragenische Kompensation durch Mutation in einem anderen Genlocus stattgefunden haben. Auch die geringen Mutationsraten bei diesem In-vitro-Selektionsversuch könnten als weiterer Hinweis auf Effluxdefekte gewertet werden. Auch andere Arbeitsgruppen hatten Schwierigkeiten bei der Selektion ciprofloxacinresistenter Salmonellen ausgehend von acrB- und tolCMutanten [256]. 4.3.2 Expressionsstudien der Pumpen-Systeme AcrAB- und AcrEF-TolC Der anschließende Schritt im weiteren Verlauf der Arbeit zielte darauf ab, das betroffene Pumpensystem in vacT und vacTE1-3 zu identifizieren. Da das AcrAB-TolC Pumpensystem in E. coli als Haupteffluxsystem beschrieben wurde, dem auch in Salmonella spec. eine wichtige Rolle zugesprochen wird, wurden von Matthiessen (Dissertation in Vorbereitung) Untersuchungen zur Regulation der Pumpensysteme AcrAB- und AcrEF mit Hilfe eines plasmidbasierten Reportergensystems durchgeführt. Ähnlich wie bei der Bestimmung des Superspiralisierungsgrades (Abschnitt 2.2.1.9) wurden dabei die Promotoren für die Strukturgene der Effluxpumpen mit dem Luciferasegen fusioniert. Veränderungen der Ansprache der Promotoren dieser Gene beeinflussen die Expression des Luciferasegens. So führen z.B. Mutationen in den Regulatoren AcrR [297] und MarR oder der Induktion von MarA durch Salicylat [280] zu einer verstärkten Expression der Strukturgene acrAB, was sich in dem beschriebenen Reportergensystem in erhöhten RLU-Werten ausdrückt. Bei den bekannten Regulatoren der Effluxpumpen in E. coli oder Salmonella spec. handelt es sich aber vor allem um Repressoren. 164 Da Regulationsmutanten einen gegenüber Chinolonen weniger Diskussion empfindlichen oder sogar resistenten Phänotypen besitzen, der Impfstamm im Gegensatz dazu aber sehr empfindlich reagiert, wurden Veränderungen in den Regulatoren ausgeschlossen. Die Ergebnisse der Untersuchungen der acrAB-Expression im Impfstamm vacT zeigten aber im Vergleich zum Ausgangsstamm M415 eine sehr geringfügige, jedoch signifikante Aufregulation der Expression der AcrAB-TolC-Pumpe. Die Expression von acrEF lag in vacT hingegen unter dem Wert von M415 (Daten nicht gezeigt). Eaves et al. [84] beobachteten z.B. auch bei Versuchen mittels real-time PCR einen Anstieg der acrBExpression in acrF-Deletionsmutanten. Möglicherweise wird so die Pumpenaktivität kompensiert. Möglicherweise ist also die AcrEF-Pumpe im Impfstamm vacT in ihrer Aktivität reduziert. Das würde die beobachtete leichte Aufregulation der acrAB-Expression erklären. Die minimal erhöhte Promotoraktivität der Gene acrAB reicht aber nicht aus, um den Pumpendefekt wirklich zu kompensieren und die Akkumulation der Fluorchinolone in der Zelle zu senken, denn der Stamm zeigt phänotypisch Fluorchinolonsensibilität. Zu beachten gilt weiterhin, dass die gemessene Promotoraktivität letztendlich keine konkrete Aussage über die wirkliche Proteinexpression und somit die tatsächliche Anzahl funktionstüchtiger Pumpen in der Zellmembran zulässt. In der Selektionsmutante vacT-E1 hingegen lag sowohl die Expression von AcrAB als auch AcrEF unter dem Niveau von M415 (Daten nicht gezeigt). Eine mögliche Erklärung dafür ist die Wiederherstellung der Funktionstüchtigkeit von AcrEF in vacT-E1 z.B. durch eine kompensatorische Mutation. Dies korreliert mit den leicht erhöhten MHK-Werten in diesem Stamm (Abschnitt 3.1.5.2). Eine Sequenzanalyse der Strukturgene oder Expressionsstudien mittels real-time PCR in vacT und vacT-E1 könnten Veränderungen aufdecken, die möglicherweise zu einer Störung in der Ausbildung von betroffenen Pumpensystemen führen (Abschnitt 4.4). Wie Defekte in den Efflux-Systemen, so ändern auch Störungen von passiven Transportsystemen durch die Zellmembran über Porine oder einfache Diffusion durch die LPS-Schicht die Zugänglichkeit der Antibiotika in die Zelle [142,205]. Untersuchungen zur Reduktion von Influx z.B. aufgrund von Veränderungen in der LPS-Schicht oder der Porinexpression könnten die Ursache für eine erniedrigte Akkumulation von Antibiotika in der Zelle konkretisieren. 4.3.3 Der Einfluss defekter Effluxpumpen auf die Virulenz Auch die Beteiligung von Effluxpumpen-Systemen an der Ausprägung der Pathogenität von Salmonellen wird nicht ausgeschlossen [280]. So fanden Stone und Miller [279] reduzierte Virulenz von S. Enteritidis gegenüber Mäusen aufgrund einer Tn5 Insertionsmutation in tolC. Auch in anderen Organismen wie Pseudomonas aeruginosa oder Neisseria gonorrhoeae führten Mutationen in den Hauptpumpensystemen zu einer abgeschwächten Virulenz [143,154]. Dabei wird postuliert, dass die Bakterien nur mit 165 Diskussion funktionierenden Efflux-Systemen in Gegenwart von hydrophoben Substanzen in der Schleimhaut des Wirtes überleben und Infektionen auslösen können. Baucheron et al. [25] zeigten, dass tolC-Mutanten von S. Typhimurium unfähig sind, Milz oder Leber von Hühnern zu besiedeln. Neuere Erkenntnisse von Mitarbeitern um Piddock [37,238] zeigten, dass sowohl acrB- als auch tolC-Mutanten von S. Typhimurium nicht in der Lage waren, den Darm von Hühnern zu kolonisieren und auch keine Invasion in Makrophagen stattfand. In dieser Studie beeinträchtigten aber z.B. acrD- oder acrF-Mutationen die Kolonisation und Persistenz der Bakterien im Geflügel nicht. Es wird aber vermutet, dass möglicherweise schon eine eingeschränkte Expression von acrB durch das Ausschalten von MarA für eine reduzierte Kolonisationsfähigkeit ausreicht [250]. Infolgedessen kann man davon ausgehen, dass die Efflux-Systeme eine Rolle beim Überleben in der Umgebung, bei der Besiedlung des Wirtes sowie bei der Adhäsion und Invasion der Bakterien in die Wirtszellen spielen. Durch aktiven Efflux können auch Virulenzfaktoren (Toxine etc.) aus der Bakterienzelle transportiert werden. So wird z.B. bei E. coli ein Hämolysin durch eine Effluxpumpe aus der Zelle geschleust. Somit kommt der Untersuchung von möglichen Defekten in den Efflux-Systemen des Impfstammes vacT eine große Bedeutung zu. Möglicherweise kommen Mutationen in den AcrAB- oder AcrEF-Systemen sogar als Attenuationsmarker für Lebendimpfstämme in Frage. 166 Diskussion 4.4 Ausblick Ziel dieser Arbeit war die Suche nach molekularen Ursachen für die Marker des Impfstammes vacT und vor allem die Aufklärung der Diskrepanz zwischen der Nalidixinsäureresistenz und der Fluorchinolonsensibilität. Da der Impfstamm vacT aufgrund der ungezielten Mutagenese wahrscheinlich zahlreiche unbekannte Mutationen aufweist, ist ein genaues Abgrenzen einzelner Effekte sehr schwierig. Die Aufklärung der Zusammenhänge im Impfstamm könnte aber das Verständnis bisher weniger erforschter Bereiche, wie den Effluxpumpen, bei Salmonella spec. erleichtern und z.B. durch die Aufklärung der Ursache der Attenuation einen Beitrag zu neuen Ansätzen bei der Impfstammentwicklung leisten. Der Hauptschwerpunkt dieser Arbeit lag in der Aufklärung der Bedeutung der gyrAMutationen. Um die Rolle jeder einzelnen gyrA-Mutation im Impfstamm vacT abschließend zu untersuchen, wäre die Herstellung weiterer gyrA-Mutanten über homologe Rekombination notwendig. So könnte die möglicherweise notwendige stabilisierende Wirkung der Mutation A866S geklärt werden, da eine Gyrase mit den drei Mutationen W59R, D75A und D87G nicht aktiv zu sein scheint (Abschnitt 4.2.3.3). Auch eine Untersuchung von Rekombinanten mit den Einzelmutationen W59R oder D75A und der entsprechenden Doppelmutation könnte die Rolle dieser Mutationen in bzw. nahe der QRDR konkretisieren. Weiterhin könnte ein in silico Struktur-Modell der Gyrase unter Berücksichtigung der Nukleotidsequenz von S. Typhimurium hergestellt werden. Die genauen Veränderungen dieser Molekülstruktur durch die Einführung der im Impfstamm identifizierten Aminosäureaustausche, z.B. in Bezug auf die Verschiebungen des Tyrosin 122, könnten so beschrieben und untersucht werden. Zur Identifizierung der vom Defizit betroffenen Effluxpumpe im Impfstamm vacT könnten neben dem Vergleich der Membranproteine über SDS-PAGE oder der gesamten Proteinmuster mittels 2D-Gelelektrophorese auch Expressionsstudien über real-time PCR beitragen. Aus Salmonella spec. oder E. coli bekannte Pumpensysteme könnten vergleichend in M415 und vacT untersucht werden. Die Gene acrA/B/E/F und tolC sollten sequenziert werden, da Mutationen in diesen Genen für einen veränderten Efflux verantwortlich gemacht werden. Auch ein Zusammenhang zwischen dem Defizit im Efflux und der Generationszeit als Parameter für die Fitness des Stammes sollte zukünftig näher untersucht werden. Vergleichende Untersuchungen des gesamten Proteinmusters über 2D- Gelelektrophorese könnten Hinweise zu bisher nicht identifizierten Mechanismen geben. Zusammenhänge zwischen dem reduzierten Superspiralisierungsgrad und z.B. der Virulenz oder der veränderten Fitness können so aufgeklärt werden. Aus diesem Grund wird der 2D-Gelelektrophorese eine große Bedeutung zugemessen. Diese Methode 167 Diskussion ermöglicht einen globalen Ansatz bei der Untersuchung der Quantität und Qualität der tatsächlich exprimierten Proteine. Unterstützend könnte mit Hilfe von Phagentransduktion der Versuch unternommen werden, die Ursache (bzw. Lage im Chromosom) für den rtt-Marker, d.h. dem reduzierten Transport durch die Membran, zu finden. Der Zusammenhang zwischen dem Defizit im Efflux und der Attenuation im Impfstamm vacT, wie er bereits von anderen Arbeitsgruppen beschrieben wurde, muss genauer überprüft werden. Die rekombinanten Salmonella-Stämme mit den eingeführten gyrAMutationen und den unterschiedlichen DNA-Superspiralisierungsgraden müssen auf ihre Virulenz geprüft werden. Ein Zusammenhang zwischen gyrA-Mutationen, verändertem DNA-Superspiralisierungsgrad und daraus resultierender veränderter Virulenz würde so aufgeklärt. Eine Fortsetzung der Untersuchung des Zusammenhanges zwischen Targetgenmutationen, verändertem DNA-Superspiralisierungsgrad, Effluxdefizit, Fitness und Attenuation ist von großem Interesse für die weitere Impfstammentwicklung und das Verständnis der Resistenzentwicklung bzw. -vermeidung bei Salmonella spec.. 168 Zusammenfassung / Summary 5 Zusammenfassung TAD Salmonella vacT (vacT) ist ein durch Selektion auf Nalidixinsäure und Rifampicin sowie durch zusätzliche Behandlung mit N-Methyl-N´-Nitrosoguanidin aus dem Ausgangsstamm M415 entwickelter attenuierter Lebendimpfstamm zur Prävention von Salmonellosen bei Hühnern. Weitere Untersuchungsstämme dieser Arbeit waren Nal2ori (stammend aus einem Selektionsschritt bei der Impfstammentwicklung) und Nal2passage (eine fitnesskompensierte Variante von Nal2ori). Der Impfstamm vacT besitzt vier verschiedene genetische Marker, die phänotypisch durch Resistenz bzw. Sensibilität gegenüber bestimmten Antibiotika charakterisiert sind. Im Rahmen dieser Arbeit wurden die molekularen Grundlagen dieser Marker untersucht. Dazu wurden die Stämme M415, Nal2ori, Nal2passage sowie vacT hinsichtlich ihrer Antibiotikaempfindlichkeit, ihrer Superspiralisierungsgrades analysiert. Nal2passage mit dem Fitness Im Ausgangsstamm Empfindlichkeitsbestimmung deutlich sowie Vergleich M415 erhöhte bezüglich der ihres Mutanten zeigten die Nalidixinsäure- und DNA- Nal2ori und Ergebnisse der leicht erhöhte Fluorchinolon-MHK-Werte. Der Impfstamm vacT hingegen zeigte unter anderem Rifampicin- und Nalidixinsäureresistenz sowie Makrolid- und Fluorchinolonempfindlichkeit. Die DNA-Sequenzanalyse ergab keine Mutationen in den Genen für die beiden Untereinheiten der Topoisomerase IV (parC und parE), für die B-Untereinheit der Gyrase (gyrB) und für die Sensorkinase cpxA für Nal2ori, Nal2passage und vacT im Vergleich mit M415. Allerdings wurden in vacT vier Mutationen in gyrA gefunden, dem Gen für die AUntereinheit der Gyrase. Zwei dieser Mutationen liegen innerhalb (G75A, D87G) und zwei außerhalb (W59R, A866S) der QRDR, welche als wesentlicher Genabschnitt für die Resistenzentwicklung gegenüber Chinolonen beschrieben ist. In den Stämmen Nal2ori und Nal2passage konnten im gyrA-Gen nur drei Mutationen identifiziert werden. Der Aminosäureaustausch W59R tritt nur im Impfstamm vacT auf. Nal2ori wies eine deutliche Verlängerung der Generationszeit im Vergleich zum Ausgangsstamm auf. Der Impfstamm vacT zeigte gegenüber Nal2ori wieder eine kompensierte Generationszeit, wuchs aber langsamer als M415. Der DNA- Superspiralisierungsgrad betrug sowohl in Nal2ori, Nal2passage als auch in vacT 55-60 % des Ausgangsstammes M415. Der reduzierte Superspiralisierungsgrad und eine daraus resultierende veränderte Proteinexpression kommen als möglicher Grund für das Wachstumsdefizit in Nal2ori und für eine Virulenzreduktion in Frage. Die Ursachen für die Fitnesskompensation sind nicht bekannt. Die Wiederherstellung des Superspiralisierungsgrades durch eine intragenische Mutation konnte als Grund ausgeschlossen werden. Weiterhin Mutationen wurden aus über vacT homologe einzeln und Rekombinationstechniken in Kombination in verschiedene das Chromosom gyrAdes 169 Zusammenfassung / Summary Ausgangsstammes M415 eingeführt. Die resultierenden isogenen Stämme wurden ebenfalls phänotypisch charakterisiert. Zusammenfassend lässt sich sagen, dass durch die im Impfstamm auftretende gyrA-Mutation D87G allein oder auch in Kombination mit einer der anderen Mutationen die Gyrase so verändert ist, dass (1) die Empfindlichkeit der Gyrase gegenüber Chinolone erniedrigt ist. (2) die Aktivität des Enzyms beeinträchtigt und dadurch der Superspiralisierungsgrad reduziert ist (evt. Zusammenhang zu Attenuation). (3) das Auftreten der Mutation G75A keinen und das Auftreten der Mutation W59R nur einen minimalen kompensatorischen Einfluss hat. Letzterer erklärt jedoch nicht die Fluorchinolonsensibilität des Impfstammes. Im Impfstamm vacT kommt es zu einer Unterdrückung des durch die Mutation D87G bedingten Fluorchinolon-unempfindlichen Phänotyps. Eine Erklärung dafür ist wahrscheinlich eine intra- oder extragenische Kompensation dieser Mutation. Eine intragenische Kompensation aufgrund der allein im Impfstamm vacT identifizierten Mutation W59R erscheint durch die Ergebnisse der Untersuchung der Rekombinanten RM415-59-87 unwahrscheinlich. Als extragenische Kompensation kommt ein reduzierter Efflux oder verstärkter Influx und somit eine erhöhte Antibiotikakonzentration in der Zelle in Frage. Dies würde neben der Supersensibilität des Impfstammes auf Fluorchinolone auch die erhöhten Empfindlichkeiten gegenüber Makroliden, Ketoliden bzw. Linezolid erklären. Die Ergebnisse aus den MHK-Wert-Bestimmungen mit einem Effluxpumpeninhibitor und Messungen der Promotoraktivitäten der Effluxpumpen-Systeme AcrAB/EF-TolC geben Hinweise auf ein Defizit im aktiven Efflux. Die Kombination aus Targetgenmutationen und herabgesetzten Efflux macht die Ausprägung der MHK-Werte für den Impfstamm aus. Als Attenuationsmarker und Ursache für die Fitnessreduktion in vacT kommen sowohl gyrA-Mutationen, rpoB-Mutation aber auch Mutationen in den Genen für die Effluxpumpen in Frage. Aufgrund der ungezielten Mutagenese während der Impfstammentwicklung können auch Defekte in weiteren Genen, die z.B. mit der Fosfomycinresistenz in Zusammenhang stehen, als Ursache für die Attenuation nicht ausgeschlossen werden. Die Erforschung der Zusammenhänge zwischen Superspiralisierungsgrad-abhängiger Genexpression, Fitness und Attenuation bietet interessante Ansätze für die weitere Entwicklung von Salmonella-Lebendimpfstoffen. Aber auch die Möglichkeit der Kompensation der Chinolonresistenz, die durch Mutationen in den Genen für die Zielstruktur Gyrase hervorgerufen wird, z.B. durch das Ausschalten von Effluxpumpen, eröffnet neue Forschungsansätze bezüglich der Untersuchung von Resistenzentwicklung oder –unterdrückung. 170 Zusammenfassung / Summary Summary Live vaccine strain TAD Salmonella vacT was derived from Salmonella Typhimurium M415 by isolating mutants which have been selected on nalidixic acid and rifampicin and also have been treated with N-Methyl-N´-Nitrosoguanidine. Strain Nal2ori is taken from a selection step during vaccine development. Nal2passage is a variant of Nal2ori. Vaccine strain TAD Salmonella vacT is used for active immunization in chicken. VacT carries four different genetic markers which are characterized by resistance or sensitivity to different antibiotics. In this study the molecular basis of these markers were analysed. The four strains were analysed regarding their fitness, DNA supercoiling degree and susceptibility to different antibiotics. The results of the susceptibility testing of Nal2ori and Nal2passage showed increased MICs of nalidixic acid and fluoroquinolones compared to M415. Live vaccine strain vacT showed resistance to rifampicin and nalidixic acid and sensitivity to macrolides and fluoroquinolones. DNA sequence analyses showed no mutations in the genes encoding both subunits of topoisomerase IV (parC and parE), subunit B of gyrase (gyrB) and sensor kinase cpxA comparing strains Nal2ori, Nal2passage and vacT to M415. However, in the mutants four mutations in subunit A of gyrase were found. Two of them (G75A, D87G) are located within the QRDR and two of them (W59R, A866S) are outside. The QRDR is a highly conserved region and described as the area most important for the development of fluoroquinolone resistance. In strains Nal2ori and Nal2passage only three out of the four mentioned mutations were identified. Mutation W59R was unique to vacT. Nal2ori showed a prolonged generation time compared to M415. In Nal2passage and vacT the generation time was compensated, but the growth rate of the live vaccine strain was still reduced compared to M415. The DNA supercoiling degree of Nal2ori, Nal2passage and vacT averaged only 55-60 % to that of M415. This reduced supercoiling degree (putatively resulting in a change of protein expression) could be responsible for prolonged generation time and attenuation. The reasons for fitness compensation in Nal2passage and vacT are unknown, but restoration of the supercoiling degree can be excluded. Using homologous recombination, different gyrA mutations - known from vacT – alone and in combination were introduced into the chromosome of M415. The resulting strains were characterized phenotypically. Recapitulatory, mutation D87G alone and also in combination with the other gyrA mutations leads to the following consequences: 171 Zusammenfassung / Summary (1) The susceptibility of gyrase to quinolones is reduced. (2) The activity of the enzyme is impaired and, thus, the DNA supercoiling degree is reduced (putative reason for reduction of virulence). (3) There is no compensatory effect (MICs, supercoiling degree) of mutation G75A and none to minimal compensatory effect of mutation W59R. The latter does not explain the hypersusceptibility to fluoroquinolones in the vaccine strain. The normally decreased fluoroquinolone susceptibility caused by gyrA mutation D87G in the vaccine strain is obviously suppressed somehow. A plausible explanation might be an intra- or extragenic compensatory mutation. An intragenic compensation caused by mutation W59R can be excluded because of the investigations done with mutant RM41559-87. A possible extragenic compensation is decreased efflux or increased influx resulting in increased accumulation of antibiotics. Increased accumulation could not only explain the fluoroquinolone hypersusceptibility of vacT, but also the increased susceptibilities to macrolides, ketolides and linezolide. Determination of MICs in presence of an efflux pump inhibitor as well as measurement of acrAB/EF-promoter activities indicates a deficit in active efflux of vacT. It is assumed that the MICs of vacT caused by a combined effect of target gene mutations and decreased efflux. Mutations in gyrA, rpoB, or efflux pump genes are possible attenuation markers and responsible for fitness reduction. Due to chemical mutagenesis during vaccine strain development defects in other genes (for example in genes which play a role in fosfomycin resistance) could not be excluded as reason for attenuation. Examining the connections between supercoiling degree-dependent protein expression, fitness, attenuation as well as compensation of a fluoroquinolone resistant phenotype resulting from gyrA mutations (for example caused by reduced activity of efflux pumps) offers interesting possibilities for designing safe and stable Salmonella live vaccine strains. 172 Literatur 6 Literatur 1. Abu Mraheil, M. 2004. Entwicklung eines molekularbiologischen Testsystems zum Nachweis von Inhibitoren bakterieller Topoisomerasen, deren Wirkung und des DNA-Superspiralisierungsgrades in vivo. Dissertation. Universität Hamburg. 2. Aleixandre, V., G. Herrera, A. Urios, and M. Blanco. 1991. Effects of ciprofloxacin on plasmid DNA supercoiling of Escherichia coli topoisomerase I and gyrase mutants. The clinical use of fluoroquinolones for the treatment of mycobacterial diseases. Antimicrobial Agents and Chemotherapy 35[1]:20-23. 3. Aleixandre, V., A. Urios, G. Herrera, and M. Blanco. 1989. New Escherichia coli gyrA and gyrB mutations which have a graded effect on DNA supercoiling. The clinical use of fluoroquinolones for the treatment of mycobacterial diseases. Mol.Gen.Genet. 219[1-2]:306-312. 4. Alekshun, M. N. and S. B. Levy. 1997. Regulation of chromosomally mediated multiple antibiotic resistance: the mar regulonThe clinical use of fluoroquinolones for the treatment of mycobacterial diseases. Antimicrob Agents Chemother 41[10]:2067-2075. 5. Alekshun, M. N. and S. B. Levy. 1999. Alteration of the repressor activity of MarR, the negative regulator of the Escherichia coli marRAB locus, by multiple chemicals in vitro. J.Bacteriol. 181[15]:4669-4672. 6. Alekshun, M. N. and S. B. Levy. 1999. Characterization of MarR superrepressor mutants. J.Bacteriol. 181[10]:3303-3306. 7. Alexy, R. 2003. Antibiotika in der aquatischen Umwelt: Eintrag, Elimination und Wirkung auf Bakterien. Dissertation.Albert-Ludwigs-Universiät Freiburg 8. Amabile-Cuevas, C. F. and B. Demple. 1991. Molecular characterization of the soxRS genes of Escherichia coli: two genes control a superoxide stress regulon. Nucleic Acids Res 19[16]:44794484. 9. Andersen, J., S. A. Forst, K. Zhao, M. Inouye, and N. Delihas. 1989. The function of micF RNA. micF RNA is a major factor in the thermal regulation of OmpF protein in Escherichia coli. J Biol Chem 264[30]:17961-17970. 10. Andersson, D. I. 2003. Persistence of antibiotic resistant bacteria. Curr.Opin.Microbiol 6[5]:452-456. 11. Andersson, D. I., J. Bjorkman, and D. Hughes. 1998. Antibiotic resistance here to stay? Compensatory mutations restore virulence of resistant bacteria. Lakartidningen 95[37]:3940, 39433940, 3944. 12. Andersson, D. I. and B. R. Levin. 1999. The biological cost of antibiotic resistance. Curr.Opin.Microbiol 2[5]:489-493. 13. Arca, P., G. Reguera, and C. Hardisson. 1997. Plasmid-encoded fosfomycin resistance in bacteria isolated from the urinary tract in a multicentre survey. J Antimicrob.Chemother. 40[3]:393-399. 14. Argos, L., A. Landy, A. Abremski, J. B. Egan, E. Haggard-Ljungquist, R. H. Hoess, M. L. Kahn, B. Kalionis, S. V. Narayana, L. S. Pierson, and et al. 1986. The integrase family of site-specific recombinases: regional similarities and global diversity. EMBO J 5[2]:433-440. 15. Asako, H., H. Nakajima, K. Kobayashi, M. Kobayashi, and R. Aono. 1997. Organic solvent tolerance and antibiotic resistance increased by overexpression of marA in Escherichia coli. Appl.Environ.Microbiol. 63[4]:1428-1433. 16. Bagel, S. 1998. Adhärenzeigenschaften chinolonresistenter E.coli-Isolate. Dissertation.Rheinische Friedrich-Wilhelm-Universität Bonn 17. Bagel, S., V. Hullen, B. Wiedemann, and P. Heisig. 1999. Impact of gyrA and parC mutations on quinolone resistance, doubling time, and supercoiling degree of Escherichia coli. Antimicrob Agents Chemother 43[4]:868-875. 18. Bager, F. and R. Helmuth. 2001. Epidemiology of resistance to quinolones in Salmonella. Vet.Res. 32[3-4]:285-290. 173 Literatur 19. Balke, V. L. and J. D. Gralla. 1987. Changes in the linking number of supercoiled DNA accompany growth transitions in Escherichia coli. J.Bacteriol. 169[10]:4499-4506. 20. Barbosa, T. M. and S. B. Levy. 2000. The impact of antibiotic use on resistance development and persistence. Drug Resist.Updat. 3[5]:303-311. 21. Barrett, J. F., T. D. Gootz, P. R. McGuirk, C. A. Farrell, and S. A. Sokolowski. 1989. Use of in vitro topoisomerase II assays for studying quinolone antibacterial agents. Antimicrobial Agents and Chemotherapy 33[10]:1697-1703. 22. Barrow, P. A. 1997. Novel approaches to control of bacterial infections in animals. Acta Vet.Hung. 45[3]:317-329. 23. Baucheron, S., E. Chaslus-Dancla, and A. Cloeckaert. 2004. Role of TolC and parC mutation in high-level fluoroquinolone resistance in Salmonella enterica serotype Typhimurium DT204. J.Antimicrob.Chemother. 53[4]:657-659. 24. Baucheron, S., H. Imberechts, E. Chaslus-Dancla, and A. Cloeckaert. 2002. The AcrB multidrug transporter plays a major role in high-level fluoroquinolone resistance in Salmonella enterica serovar Typhimurium phage type DT204. Microb.Drug Resist. 8[4]:281-289. 25. Baucheron, S., C. Mouline, K. Praud, E. Chaslus-Dancla, and A. Cloeckaert. 2005. TolC but not AcrB is essential for multidrug-resistant Salmonella enterica serotype Typhimurium colonization of chicks. J Antimicrob.Chemother. 55[5]:707-712. 26. Baucheron, S., S. Tyler, D. Boyd, M. R. Mulvey, E. Chaslus-Dancla, and A. Cloeckaert. 2004. AcrAB-TolC directs efflux-mediated multidrug resistance in Salmonella enterica serovar Typhimurium DT104. Antimicrobial Agents and Chemotherapy 48[10]:3729-3735. 27. Bennik, M. H., P. J. Pomposiello, D. F. Thorne, and B. Demple. 2000. Defining a rob regulon in Escherichia coli by using transposon mutagenesis. J Bacteriol 182[13]:3794-3801. 28. Berger, J. M. and J. C. Wang. 1996. Recent developments in DNA topoisomerase II structure and mechanism. Curr.Opin.Struct.Biol. 6[1]:84-90. 29. Bergmans, H. E., I. M. van Die, and W. P. Hoekstra. 1981. Transformation in Escherichia coli: stages in the process. J Bacteriol 146[2]:564-570. 30. Berkelman, T. and T. Stenstedt. 1998. 2-D Electrophoresis using immobilized pH gradients. Principles and Methods. Amersham Biosciences 31. Birnboim, H. C. and J. Doly. 1979. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 7[6]:1513-1523. 32. Bjorkman, J. and D. I. Andersson. 2000. The cost of antibiotic resistance from a bacterial perspective. Drug Resist Updat. 3[4]:237-245. 33. Bjorkman, J., D. Hughes, and D. I. Andersson. 1998. Virulence of antibiotic-resistant Salmonella Typhimurium. Proc.Natl.Acad.Sci.U.S.A 95[7]:3949-3953. 34. Bjorkman, J., I. Nagaev, O. G. Berg, D. Hughes, and D. I. Andersson. 2000. Effects of environment on compensatory mutations to ameliorate costs of antibiotic resistance. Science 287[5457]:1479-1482. 35. Brenner, F. W., R. G. Villar, F. J. Angulo, R. Tauxe, and B. Swaminathan. 2000. Salmonella nomenclature. J Clin Microbiol 38[7]:2465-2467. 36. Brown, E. D., E. I. Vivas, C. T. Walsh, and R. Kolter. 1995. MurA (MurZ), the enzyme that catalyzes the first committed step in peptidoglycan biosynthesis, is essential in Escherichia coli. J Bacteriol 177[14]:4194-4197. 37. Buckley, A. M., M. A. Webber, S. Cooles, L. P. Randall, R. M. La Ragione, M. J. Woodward, and L. J. Piddock. 2006. The AcrAB-TolC efflux system of Salmonella enterica serovar Typhimurium plays a role in pathogenesis. Cell Microbiol 8[5]:847-856. 38. Bundesamt für Verbraucherschutz und Lebensmittelsicherheit (BVL). 1996. Stellungnahme der ZKBS zur Einstufung von zwei Salmonella "enterica"-Serovar Typhimurium-Impfstämmen für Hühner (Impfstoffe Zoosaloral H bzw. TAD Salmonella vac T), einem Salmonella „enterica"-Serovar Choleraesuis-Impfstamm für Schweine (Impfstoff Suisaloral) sowie einem Salmonella „enterica"Serovar Dublin-Impfstamm für Rinder (Impfstoff Bovisaloral)als Empfängerorganismen bei gentechnischen Arbeiten. Az.: 6790-10-33 http://www.bvl.bund.de 174 Literatur 39. Cabral, J. H., A. P. Jackson, C. V. Smith, N. Shikotra, A. Maxwell, and R. C. Liddington. 1997. Crystal structure of the breakage-reunion domain of DNA gyrase. Nature 388[6645]:903-906. 40. Cambau, E., F. Bordon, E. Collatz, and L. Gutmann. 1993. Novel gyrA point mutation in a strain of Escherichia coli resistant to fluoroquinolones but not to nalidixic acid. Antimicrobial Agents and Chemotherapy 37[6]:1247-1252. 41. Cardenas, L. and J. D. Clements. 1992. Oral immunization using live attenuated Salmonella spp. as carriers of foreign antigens. Clin Microbiol Rev. 5[3]:328-342. 42. Castringius, A. 2000. In-vitro-Untersuchungen zur Adhäsivität, Invasivität und intrazellulärer Persistenz von Salmonella-enterica-Impfstämmen. Dissertation.Justus-Liebig-Universität Gießen 43. Champoux, J. J. 2001. DNA topoisomerases: structure, function, and mechanism. Annu.Rev.Biochem. 70:369-413. 44. Charbonnier, F. and P. Forterre. 1994. Comparison of plasmid DNA topology among mesophilic and thermophilic eubacteria and archaebacteria. J.Bacteriol. 176[5]:1251-1259. 45. Chaudhury, A. M. and G. R. Smith. 1985. Role of Escherichia coli RecBC enzyme in SOS induction. Mol Gen Genet 201[3]:525-528. 46. Cherepanov, P. P. and W. Wackernagel. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9-14. 47. Chollet, R., J. Chevalier, A. Bryskier, and J. M. Pages. 2004. The AcrAB-TolC pump is involved in macrolide resistance but not in telithromycin efflux in Enterobacter aerogenes and Escherichia coli. Antimicrobial Agents and Chemotherapy 48[9]:3621-3624. 48. Chu, C., L. H. Su, C. H. Chu, S. Baucheron, A. Cloeckaert, and C. H. Chiu. 2005. Resistance to fluoroquinolones linked to gyrA and parC mutations and overexpression of acrAB efflux pump in Salmonella enterica serotype Choleraesuis. Microb Drug Resist 11[3]:248-253. 49. Chu, D. T. and P. B. Fernandes. 1989. Structure-activity relationships of the fluoroquinolones. Antimicrobial Agents and Chemotherapy 33[2]:131-135. 50. Clark, P. D. 2006. Molecular Biology. Understanding the Genetic Revolution. Elsevier GmbH.Spektrum Akademischer Verlag :783 S51. Cloeckaert, A. and E. Chaslus-Dancla. 2001. Mechanisms of quinolone resistance in Salmonella. Vet Res. 32[3-4]:291-300. 52. Cloeckaert, A. and S. Schwarz. 2001. Molecular characterization, spread and evolution of multidrug resistance in Salmonella enterica Typhimurium DT104. Vet.Res. 32[3-4]:301-310. 53. Coban, A. Y. and B. Durupinar. 2003. The effect of nitric oxide combined with fluoroquinolones against Salmonella enterica serovar Typhimurium in vitro. Mem.Inst.Oswaldo Cruz 98[3]:419-423. 54. Cocito, C., M. Di Giambattista, E. Nyssen, and P. Vannuffel. 1997. Inhibition of protein synthesis by streptogramins and related antibiotics. J.Antimicrob.Chemother. 39 Suppl A:7-13.:7-13. 55. Cohen, M. L. and R. V. Tauxe. 1986. Drug-resistant Salmonella in the United States: an epidemiologic perspective. Science 234[4779]:964-969. 56. Cohen, S. N., A. C. Chang, and L. Hsu. 1972. Nonchromosomal antibiotic resistance in bacteria: genetic transformation of Escherichia coli by R-factor DNA. Proc.Natl.Acad.Sci.USA 69[8]:2110-2114. 57. Cohen, S. P., H. Hachler, and S. B. Levy. 1993. Genetic and functional analysis of the multiple antibiotic resistance (mar) locus in Escherichia coli. J Bacteriol 175[5]:1484-1492. 58. Cohen, S. P., D. C. Hooper, J. S. Wolfson, K. S. Souza, L. M. McMurry, and S. B. Levy. 1988. Endogenous active efflux of norfloxacin in susceptible Escherichia coli. Antimicrobial Agents and Chemotherapy 32[8]:1187-1191. 59. Cohen, S. P., L. M. McMurry, D. C. Hooper, J. S. Wolfson, and S. B. Levy. 1989. Cross-resistance to fluoroquinolones in multiple-antibiotic-resistant (Mar) Escherichia coli selected by tetracycline or chloramphenicol: decreased drug accumulation associated with membrane changes in addition to OmpF reduction. Antimicrobial Agents and Chemotherapy 33[8]:1318-1325. 175 Literatur 60. Cooper, G. L., L. M. Venables, M. J. Woodward, and C. E. Hormaeche. 1994. Vaccination of chickens with strain CVL30, a genetically defined Salmonella enteritidis aroA live oral vaccine candidate. Infect Immun. 62[11]:4747-4754. 61. Court, D. L., J. A. Sawitzke, and L. C. Thomason. 2002. Genetic engineering using homologous recombination. Annu.Rev.Genet. 36:361-388. 62. Cullen, M. E., A. W. Wyke, R. Kuroda, and L. M. Fisher. 1989. Cloning and characterization of a DNA gyrase A gene from Escherichia coli that confers clinical resistance to 4-quinolones. Antimicrobial Agents and Chemotherapy 33[6]:886-894. 63. Curtiss, R., III, S. M. Kelly, and J. O. Hassan. 1993. Live oral avirulent Salmonella vaccines. Vet Microbiol 37[3-4]:397-405. 64. Dagert, M. and S. D. Ehrlich. 1979. Prolonged incubation in calcium chloride improves the competence of Escherichia coli cells. Gene 6[1]:23-28. 65. Darwin, K. H. and V. L. Miller. 1999. Molecular basis of the interaction of Salmonella with the intestinal mucosa. Clin Microbiol Rev. 12[3]:405-428. 66. Datsenko, K. A. and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc.Natl.Acad.Sci.USA 97[12]:6640-6645. 67. Deguchi, T., A. Fukuoka, M. Yasuda, M. Nakano, S. Ozeki, E. Kanematsu, Y. Nishino, S. Ishihara, Y. Ban, and Y. Kawada. 1997. Alterations in the GyrA subunit of DNA gyrase and the ParC subunit of topoisomerase IV in quinolone-resistant clinical isolates of Klebsiella pneumoniae. Antimicrobial Agents and Chemotherapy 41[3]:699-701. 68. Deibler, R. W., S. Rahmati, and E. L. Zechiedrich. 2001. Topoisomerase IV, alone, unknots DNA in E. coli. Genes Dev. 15[6]:748-761. 69. Deiwick, J., T. Nikolaus, S. Erdogan, and M. Hensel. 1999. Environmental regulation of Salmonella pathogenicity island 2 gene expression. Mol Microbiol 31[6]:1759-1773. 70. Delihas, N. and S. Forst. 2001. MicF: an antisense RNA gene involved in response of Escherichia coli to global stress factors. J Mol Biol 313[1]:1-12. 71. DiGate, R. J. and K. J. Marians. 1989. Molecular cloning and DNA sequence analysis of Escherichia coli topB, the gene encoding topoisomerase III. J.Biol.Chem. 264[30]:17924-17930. 72. DIN (Deutsches Institut für Normung e.V.). 2002. Medizinische Mikrobiologie Empfindlichkeitsprüfung von mikrobiellen Krankheitserregern gegen Chemotherapeutika - Teil 8: Mikrodilution; Allgemeine methodenspezifische Anforderungen. DIN 58940-8 Ausgabe:2002-10 73. Domagala, J. M. 1994. Structure-activity and structure-side-effect relationships for the quinolone antibacterials. J.Antimicrob.Chemother. 33[4]:685-706. 74. Domagala, J. M., L. D. Hanna, C. L. Heifetz, M. P. Hutt, T. F. Mich, J. P. Sanchez, and M. Solomon. 1986. New structure-activity relationships of the quinolone antibacterials using the target enzyme. The development and application of a DNA gyrase assay. J Med Chem. 29[3]:394-404. 75. Dorman, C. J. 1991. DNA supercoiling and environmental regulation of gene expression in pathogenic bacteria. Infect Immun. 59[3]:745-749. 76. Dorman, C. J. 1995. DNA topology and the global control of bacterial gene expression: implications for the regulation of virulence gene expression. Microbiology 141:1271-1280. 77. Dorman, C. J., G. C. Barr, N. N. Bhriain, and C. F. Higgins. 1988. DNA Supercoiling and the Anaerobic and Growth Phase Regulation of tonB Gene Expression. J Bacteriol. 170:2816-2826. 78. Dower, W. J., J. F. Miller, and C. W. Ragsdale. 1988. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 16[13]:6127-6145. 79. Drlica, K. 1999. Mechanism of fluoroquinolone action. Curr.Opin.Microbiol 2[5]:504-508. 80. Drlica, K. and J. Rouviere-Yaniv. 1987. Histonelike proteins of bacteria. Microbiol Rev. 51[3]:301319. 81. Drlica, K. and X. Zhao. 1997. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol.Mol.Biol.Rev. 61[3]:377-392. 176 Literatur 82. D`Aoust, J. Y. 1985. Infective dose of Salmonella Typhimurium in cheddar cheese. Am J Epidemiol 122[4]:717-720. 83. Eaves, D. J., E. Liebana, M. J. Woodward, and L. J. Piddock. 2002. Detection of gyrA mutations in quinolone-resistant Salmonella enterica by denaturing high-performance liquid chromatography. J Clin.Microbiol 40[11]:4121-4125. 84. Eaves, D. J., V. Ricci, and L. J. Piddock. 2004. Expression of acrB, acrF, acrD, marA, and soxS in Salmonella enterica serovar Typhimurium: role in multiple antibiotic resistance. Antimicrobial Agents and Chemotherapy 48[4]:1145-1150. 85. Ellis, H. M., D. Yu, T. DiTizio, and Court DL. 2001. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc.Natl.Acad.Sci.U.S.A 98[12]:6742-6746. 86. Elsea, S. H., P. R. McGuirk, T. D. Gootz, M. Moynihan, and N. Osheroff. 1993. Drug features that contribute to the activity of quinolones against mammalian topoisomerase II and cultured cells: correlation between enhancement of enzyme-mediated DNA cleavage in vitro and cytotoxic potential. Antimicrobial Agents and Chemotherapy 37[10]:2179-2186. 87. Emmerson, A. M. and A. M. Jones. 2003. The quinolones: decades of development and use. J.Antimicrob.Chemother. 51 Suppl 1:13-20.:13-20. 88. Everest, P., P. Griffiths, and G. Dougan. 1995. Live Salmonella vaccines as a route towards oral immunisation. Biologicals 23[2]:119-124. 89. Ferrero, L., B. Cameron, and J. Crouzet. 1995. Analysis of gyrA and grlA mutations in stepwiseselected ciprofloxacin- resistant mutants of Staphylococcus aureus. Antimicrobial Agents and Chemotherapy 39[7]:1554-1558. 90. Ferrero, L., B. Cameron, B. Manse, D. Lagneaux, J. Crouzet, A. Famechon, and F. Blanche. 1994. Cloning and primary structure of Staphylococcus aureus DNA topoisomerase IV: a primary target of fluoroquinolones. Mol.Microbiol. 13[4]:641-653. 91. Fournier, B., X. Zhao, T. Lu, K. Drlica, and D. C. Hooper. 2000. Selective targeting of topoisomerase IV and DNA gyrase in Staphylococcus aureus: different patterns of quinolone-induced inhibition of DNA synthesis. Antimicrobial Agents and Chemotherapy 44[8]:2160-2165. 92. Fralick, J. A. 1996. Evidence that TolC is required for functioning of the Mar/AcrAB efflux pump of Escherichia coli. J.Bacteriol. 178[19]:5803-5805. 93. Frech, G., M. Weide-Botjes, E. Nussbeck, W. Rabsch, and S. Schwarz. 1998. Molecular characterization of Salmonella enterica subsp. enterica serovar Typhimurium DT009 isolates: differentiation of the live vaccine strain Zoosaloral from field isolates. FEMS Microbiol Lett. 167[2]:263-269. 94. Galan, J. E. and R. Curtiss. 1990. Expression of Salmonella Typhimurium genes required for invasion is regulated by changes in DNA supercoiling. Infect.Immun. 58[6]:1879-1885. 95. Gay, P., D. Le Coq, M. Steinmetz, T. Berkelman, and C. I. Kado. 1985. Positive selection procedure for entrapment of insertion sequence elements in gram-negative bacteria. J Bacteriol 164[2]:918-921. 96. Gaynor, M. and A. S. Mankin. 2003. Macrolide antibiotics: binding site, mechanism of action, resistance. Curr.Top.Med.Chem. 3[9]:949-961. 97. Gellert, M. 1981. DNA topoisomerases. Annu.Rev.Biochem. 50:879-910.:879-910. 98. Gellert, M., K. Mizuuchi, M. H. O'Dea, T. Itoh, and J. I. Tomizawa. 1977. Nalidixic acid resistance: a second genetic character involved in DNA gyrase activity. Proc.Natl.Acad.Sci.U.S.A 74[11]:47724776. 99. Gellert, M., K. Mizuuchi, M. H. O'Dea, and H. A. Nash. 1976. DNA gyrase: an enzyme that introduces superhelical turns into DNA. Proc.Natl.Acad.Sci.U.S.A 73[11]:3872-3876. 100. Gellert, M., M. H. O'Dea, T. Itoh, and J. Tomizawa. 1976. Novobiocin and coumermycin inhibit DNA supercoiling catalyzed by DNA gyrase. Proc.Natl.Acad.Sci.U.S.A 73[12]:4474-4478. 101. Gillespie, S. H., L. L. Voelker, and A. Dickens. 2002. Evolutionary barriers to quinolone resistance in Streptococcus pneumoniae. Microb Drug Resist 8[2]:79-84. 177 Literatur 102. Giraud, E., A. Brisabois, J. L. Martel, and E. Chaslus-Dancla. 1999. Comparative studies of mutations in animal isolates and experimental in vitro- and in vivo-selected mutants of Salmonella spp. suggest a counterselection of highly fluoroquinolone-resistant strains in the field. Antimicrob Agents Chemother 43[9]:2131-2137. 103. Giraud, E., A. Cloeckaert, D. Kerboeuf, and E. Chaslus-Dancla. 2000. Evidence for active efflux as the primary mechanism of resistance to ciprofloxacin in Salmonella enterica serovar Typhimurium. Antimicrob Agents Chemother 44[5]:1223-1228. 104. Goldstein, E. and K. Drlica. 1984. Regulation of bacterial DNA supercoiling: plasmid linking numbers vary with growth temperature. Proc.Natl.Acad.Sci.U.S.A 81[13]:4046-4050. 105. Gordon, R. F. and J. F. Tucker. 1965. The epizootiology of Salmonella menston infection of fowls and the effect of feeding poultry food artificially infected with salmonella. Br.Poult.Sci. 6[3]:251-264. 106. Goss, W. A., W. H. Deitz, and T. M. Cook. 1964. Mechanism of action of nalidixic acid on Escherichia coli (I). J Bacteriol. 88[4]:1112-1118. 107. Goss, W. A., W. H. Deitz, and T. M. Cook. 1965. Mechanism of Action of Nalidixic Acid on Escherichia coli. II. Inhibition of Deoxyribonucleic Acid Synthesis. J Bacteriol. 89[4]:1068-1074. 108. Griggs, D. J., K. Gensberg, and L. J. Piddock. 1996. Mutations in gyrA gene of quinolone-resistant Salmonella serotypes isolated from humans and animals. Antimicrob Agents Chemother 40[4]:10091013. 109. Grüger, T. 2004. Die Rolle von Topoisomerase IV als Zielstruktur für Inhibitoren in E.coli sowie die Transkriptionskontrolle ihrer Strukturgene parC und parE. Dissertation.Universität Hamburg 110. Guerra, B., B. Malorny, A. Schroeter, and R. Helmuth. 2003. Multiple resistance mechanisms in fluoroquinolone-resistant salmonella isolates from Germany. Antimicrobial Agents and Chemotherapy 47[6]:2059111. Guerry, P., J. Embden, and S. Falkow. 1974. Molecular nature of two nonconjugative plasmids carrying drug resistance genes. J Bacteriol. 117[2]:619-630. 112. Guzman, L. M., D. Belin, M. J. Carson, and J. Beckwith. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177[14]:41214130. 113. Hächler, H., S. P. Cohen, and S. B. Levy. 1991. marA, a regulated locus which controls expression of chromosomal multiple antibiotic resistance in Escherichia coli. J Bacteriol. 173[17]:5532-5538. 114. Hamilton, C. M., M. Aldea, B. K. Washburn, P. Babitzke, and S. R. Kushner. 1989. New method for generating deletions and gene replacements in Escherichia coli. J Bacteriol 171[9]:4617-4622. 115. Hanahan, D. 1983. Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166[4]:557-580. 116. Hane, M. W. and T. H. Wood. 1969. Escherichia coli K-12 mutants resistant to nalidixic acid: genetic mapping and dominance studies. J Bacteriol. 99[1]:238-241. 117. Hansen H. 2000. Bedeutung von Topoisomerase IV für die Chinolonresistenz bei Salmonella enterica subspecies enterica. Diplomarbeit. Rheinische Friedrich-Wilhelms-Universität Bonn. 118. Hansen, H. and P. Heisig. 2003. Topoisomerase IV mutations in quinolone-resistant salmonellae selected in vitro. Microb.Drug Resist. 9[1]:25-32. 119. Hansen, L. H., P. Mauvais, and S. Douthwaite. 1999. The macrolide-ketolide antibiotic binding site is formed by structures in domains II and V of 23S ribosomal RNA. Mol.Microbiol. 31[2]:623-631. 120. Hartung, M. H. 2004. Epidemiologische Situation der Zoonosen in Deutschland im Jahr 2003. Bundesinstitut für Risikobewertung (BfR) 121. Havemann, G. D., E. M. Sampson, and T. A. Bobik. 2002. PduA is a shell protein of polyhedral organelles involved in coenzyme B(12)-dependent degradation of 1,2-propanediol in Salmonella enterica serovar Typhimurium LT2. J Bacteriol 184[5]:1253-1261. 122. Hegde, S. S., M. W. Vetting, S. L. Roderick, L. A. Mitchenall, A. Maxwell, H. E. Takiff, and J. S. Blanchard. 2005. A fluoroquinolone resistance protein from Mycobacterium tuberculosis that mimics DNA. Science 308[5727]:1480-1483. 178 Literatur 123. Heisig, P. 1993. High-level fluoroquinolone resistance in a Salmonella Typhimurium isolate due to alterations in both gyrA and gyrB genes. J Antimicrob Chemother 32[3]:367-377. 124. Heisig, P. 1996. Genetic evidence for a role of parC mutations in development of high-level fluoroquinolone resistance in Escherichia coli. Antimicrob Agents Chemother 40[4]:879-885. 125. Heisig, P. 2001. Inhibitors of bacterial topoisomerases: mechanisms of action and resistance and clinical aspects. Planta Med 67[1]:3-12. 126. Heisig, P., B. Kratz, E. Halle, Y. Graser, M. Altwegg, W. Rabsch, and J. P. Faber. 1995. Identification of DNA gyrase A mutations in ciprofloxacin- resistant isolates of Salmonella Typhimurium from men and cattle in Germany. Microb Drug Resist. 1[3]:211-218. 127. Heisig, P., H. Schedletzky, and H. Falkenstein-Paul. 1993. Mutations in the gyrA gene of a highly fluoroquinolone-resistant clinical isolate of Escherichia coli. Antimicrob Agents Chemother 37[4]:696701. 128. Heisig, P. and R. Tschorny. 1994. Characterization of fluoroquinolone-resistant mutants of Escherichia coli selected in vitro. Antimicrob Agents Chemother 38[6]:1284-1291. 129. Heisig, P. and B. Wiedemann. 1991. Use of a broad-host-range gyrA plasmid for genetic characterization of fluoroquinolone-resistant gram-negative bacteria. Antimicrob Agents Chemother 35[10]:2031-2036. 130. Helms, M., J. Simonsen, and K. Molbak. 2004. Quinolone resistance is associated with increased risk of invasive illness or death during infection with Salmonella serotype Typhimurium. J Infect.Dis. 190[9]:1652-1654. 131. Helms, M., J. Simonsen, and K. Molbak. 2006. Foodborne bacterial infection and hospitalization: a registry-based study. CID 42:498-506. 132. Helms, M., P. Vastrup, P. Gerner-Smidt, and K. Molbak. 2002. Excess mortality associated with antimicrobial drug-resistant Salmonella Typhimurium. Emerg.Infect.Dis. 8[5]:490-495. 133. Helmuth, R., B. Guerra, B. Malorny, A. Miko, and A. Schroeter. 2004. Erfassung phänotypischer und genotypischer Resistenzeigenschaften bei Salmonella- und E.coli-Isolaten vom Tier, Lebensmittteln, Futtermitteln und der Umwelt. Abschlussbericht zum Forschungsvorhaben. Bundesinstitut für Risikobewertung (BfR) 134. Heukeshofen, J. and R. Dernick. 1988. Electrophoresis 9:335-362. 135. Heurtin-Le Corre, C., P. Y. Donnio, M. Perrin, M. F. Travert, and J. L. Avril. 1999. Increasing incidence and comparison of nalidixic acid-resistant Salmonella enterica subsp. enterica serotype Typhimurium isolates from humans and animals. J Clin Microbiol 37[1]:266-269. 136. Heym, B., E. Stavropoulos, N. Honore, P. Domenech, B. Saint-Joanis, T. M. Wilson, D. M. Collins, M. J. Colston, and S. T. Cole. 1997. Effects of overexpression of the alkyl hydroperoxide reductase AhpC on the virulence and isoniazid resistance of Mycobacterium tuberculosis. Infect Immun. 65[4]:1395-1401. 137. Higgins, C. F., C. J. Dorman, D. A. Stirling, L. Waddell, I. R. Booth, G. May, and E. Bremer. 1988. A physiological role for DNA supercoiling in the osmotic regulation of gene expression in S. Typhimurium and E. coli. Cell 52[4]:569-584. 138. Higgins, M. K., E. Bokma, E. Koronakis, C. Hughes, and V. Koronakis. 2004. Structure of the periplasmic component of a bacterial drug efflux pump. Proc.Natl.Acad.Sci.U.S.A 101[27]:9994-9999. 139. Hill, S. A., D. S. Samuels, J. H. Carlson, J. Wilson, D. Hogan, L. Lubke, and R. J. Belland. 1997. Integration host factor is a transcriptional cofactor of pilE in Neisseria gonorrhoeae. Mol.Microbiol. 23[4]:649-656. 140. Hill, T. M., B. Sharma, M. Valjavec-Gratian, and J. Smith. 1997. sfi-independent filamentation in Escherichia coli is lexA dependent and requires DNA damage for induction. J Bacteriol 179[6]:19311939. 141. Hirai, K., H. Aoyama, T. Irikura, S. Iyobe, and S. Mitsuhashi. 1986. Differences in susceptibility to quinolones of outer membrane mutants of Salmonella Typhimurium and Escherichia coli. Antimicrob Agents Chemother 29[3]:535-538. 179 Literatur 142. Hirai, K., H. Aoyama, S. Suzue, T. Irikura, S. Iyobe, and S. Mitsuhashi. 1986. Isolation and characterization of norfloxacin-resistant mutants of Escherichia coli K-12. Antimicrob Agents Chemother 30[2]:248-253. 143. Hirakata, Y., R. Srikumar, K. Poole, N. Gotoh, T. Suematsu, S. Kohno, S. Kamihira, R. E. Hancock, and D. P. Speert. 2002. Multidrug efflux systems play an important role in the invasiveness of Pseudomonas aeruginosa. J Exp.Med. 196[1]:109-118. 144. Holland, I. B. and M. A. Blight. 1999. ABC-ATPases, adaptable energy generators fuelling transmembrane movement of a variety of molecules in organisms from bacteria to humans. J Mol Biol 293[2]:381-399. 145. Hooper, D. C. 2000. Mechanisms of action and resistance of older and newer fluoroquinolones. Clin.Infect.Dis. 31 Suppl 2:S24-S28. 146. Hooper, D. C. 2001. Emerging mechanisms of fluoroquinolone resistance. Emerg.Infect.Dis. 7[2]:337-341. 147. Hooper, D. C., J. S. Wolfson, M. A. Bozza, and E. Y. Ng. 1992. Genetics and regulation of outer membrane protein expression by quinolone resistance loci nfxB, nfxC, and cfxB. Antimicrobial Agents and Chemotherapy 36[5]:1151-1154. 148. Hooper, D. C., J. S. Wolfson, K. S. Souza, E. Y. Ng, G. L. McHugh, and M. N. Swartz. 1989. Mechanisms of quinolone resistance in Escherichia coli: characterization of nfxB and cfxB, two mutant resistance loci decreasing norfloxacin accumulation. Antimicrobial Agents and Chemotherapy 33[3]:283-290. 149. Horowitz, D. S. and J. C. Wang. 1987. Mapping the active site tyrosine of Escherichia coli DNA gyrase. J Biol Chem. 262[11]:5339-5344. 150. Horton, RM. C. ZL. H. SN. P. LR. 1990. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques 8[5]:528-535. 151. Howard, B. M., R. J. Pinney, and J. T. Smith. 1993. Function of the SOS process in repair of DNA damage induced by modern 4- quinolones. J.Pharm.Pharmacol. 45[7]:658-662. 152. Humphreys, S., G. Rowley, A. Stevenson, M. F. Anjum, M. J. Woodward, S. Gilbert, J. Kormanec, and M. Roberts. 2004. Role of the two-component regulator CpxAR in the virulence of Salmonella enterica serotype Typhimurium. Infect.Immun. 72[8]:4654-4661. 153. Inoue, H., H. Nojima, and H. Okayama. 1990. High efficiency transformation of Escherichia coli with plasmids. Gene 96 (1):23-28. 154. Jerse, A. E., N. D. Sharma, A. N. Simms, E. T. Crow, L. A. Snyder, and W. M. Shafer. 2003. A gonococcal efflux pump system enhances bacterial survival in a female mouse model of genital tract infection. Infect.Immun. 71[10]:5576-5582. 155. Jonas, D., K. Biehler, D. Hartung, B. Spitzmuller, and F. D. Daschner. 2005. Plasmid-mediated quinolone resistance in isolates obtained in german intensive care units. Antimicrobial Agents and Chemotherapy 49[2]:773-775. 156. Jones, M. E., D. F. Sahm, N. Martin, S. Scheuring, P. Heisig, C. Thornsberry, K. Kohrer, and F. J. Schmitz. 2000. Prevalence of gyrA, gyrB, parC, and parE mutations in clinical isolates of Streptococcus pneumoniae with decreased susceptibilities to different fluoroquinolones and originating from Worldwide Surveillance Studies during the 1997-1998 respiratory season. Antimicrobial Agents and Chemotherapy 44[2]:462-466. 157. Kato, J., Y. Nishimura, R. Imamura, H. Niki, S. Hiraga, and H. Suzuki. 1990. New topoisomerase essential for chromosome segregation in E. coli. Cell 63[2]:393-404. 158. Kayser, F. H., K. A. Bienz, J. Eckert, and R. M. Zinkernagel. 1998. Probleme mit der Resistenz in: Medizinische Mikrobiologie. 193-196. 159. Khodursky, A. B., E. L. Zechiedrich, and N. R. Cozzarelli. 1995. Topoisomerase IV is a target of quinolones in Escherichia coli. Proc.Natl.Acad.Sci.U.S.A. 92[25]:11801-11805. 160. Kingma, P. S. and N. Osheroff. 1997. Spontaneous DNA damage stimulates topoisomerase IImediated DNA cleavage. J Biol Chem 272[11]:7488-7493. 180 Literatur 161. Kobayashi, K., N. Tsukagoshi, and R. Aono. 2001. Suppression of hypersensitivity of Escherichia coli acrB mutant to organic solvents by integrational activation of the acrEF operon with the IS1 or IS2 element. J Bacteriol 183[8]:2646-2653. 162. Komp, L. P., L. L. Marcusson, D. Sandvang, N. Frimodt-Moller, and D. Hughes. 2005. Biological cost of single and multiple norfloxacin resistance mutations in Escherichia coli implicated in urinary tract infections. Antimicrobial Agents and Chemotherapy 49[6]:2343-2351. 163. Koronakis, V. 2003. TolC--the bacterial exit duct for proteins and drugs. FEBS Lett. 555[1]:66-71. 164. Kotton, C. N. and E. L. Hohmann. 2004. Enteric pathogens as vaccine vectors for foreign antigen delivery. Infect.Immun. 72[10]:5535-5547. 165. Koutsolioutsou, A., E. A. Martins, D. G. White, S. B. Levy, and B. Demple. 2001. A soxRSconstitutive mutation contributing to antibiotic resistance in a clinical isolate of Salmonella enterica (Serovar Typhimurium). Antimicrob Agents Chemother 45[1]:38-43. 166. Koutsolioutsou, A., S. Pena-Llopis, and B. Demple. 2005. Constitutive soxR mutations contribute to multiple-antibiotic resistance in clinical Escherichia coli isolates. Antimicrobial Agents and Chemotherapy 49[7]:2746-2752. 167. Kreuzer, K. N., K. McEntee, A. P. Geballe, and N. R. Cozzarelli. 1978. Lambda transducing phages for the nalA gene of Escherichia coli and conditional lethal nalA mutations. Mol Gen Genet 167[2]:129-137. 168. Kugelberg, E., S. Lofmark, B. Wretlind, and D. I. Andersson. 2005. Reduction of the fitness burden of quinolone resistance in Pseudomonas aeruginosa. J.Antimicrob Chemother 55[1]:22-30. 169. Kumagai, Y., J. I. Kato, K. Hoshino, T. Akasaka, K. Sato, and H. Ikeda. 1996. Quinolone-resistant mutants of Escherichia coli DNA topoisomerase IV parC gene. Antimicrobial Agents and Chemotherapy 40[3]:710-714. 170. Kümmerer, K. P. 2002. Antibiotikaresistenz - Vorkommen und Übertragung in Abwasser, Oberflächenwasser und Trankwasser Teil 2: Resistenzausbildung und Verbreitung durch Antibiotikaeintrag in Abwasser und Kläranlagen: Untersuchungen mittels Chemotaxonomie und Kläranlagensimulation. Abschlussbericht.Universität Freiburg 171. Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227[259]:680-685. 172. Lage, H. 2003. ABC-transporters: implications on drug resistance from microorganisms to human cancers. Int.J Antimicrob.Agents 22[3]:188-199. 173. Lalioti, M. and J. Heath. 2001. A new method for generating point mutations in bacterial artificial chromosomes by homologous recombination in Escherichia coli. Nucleic Acids Res 29[3]:E14174. Le Minor, L. and M. Y. Popoff. 1988. Kauffmann-White-Schema. Antigen-Formeln der SalmonellaSerovare, 5.Revision.WHO Referenz- und Forschungszentrum für Salmonella.Behring Diagnostika 175. Lesher, G. Y., E. J. Froelich, M. D. Gruett, J. H. Bailey, and R. P. Brundage. 1962. 1,8Naphthyridine derivatives, a new class of chemotherapy agents. J.Med.Chem. 5:1063-1065. 176. Linde, H. J., F. Notka, M. Metz, B. Kochanowski, P. Heisig, and N. Lehn. 2000. In vivo increase in resistance to ciprofloxacin in Escherichia coli associated with deletion of the C-terminal part of marR. Antimicrob Agents Chemother 44[7]:1865-1868. 177. Linde, K., Beer.J., and B. Pless. 2002. Live vaccine constituting minor risk for humans. United States Patent: 6,479,056.Lohman Animal Health GmbH & Co.KG (Cuxhaven,DE) Appl.No.:686113 178. Linde, K., G. C. Fthenakis, and A. Fichtner. 1998. Bacterial live vaccines with graded level of attenuation achieved by antibiotic resistance mutations: transduction experiments on the functional unit of resistance, attenuation and further accompanying markers. Vet.Microbiol. 62[2]:121-134. 179. Linde, K., I. Hahn, and E. Vielitz. 1996. Entwicklung von optimal für das Huhn attenuierten Salmonella-Lebendimpfstoffen. Tierärztl.Umschau 51[1]:23-31. 180. Link, A. J., D. Phillips, and G. M. Church. 1997. Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: application to open reading frame characterization. J Bacteriol. 179[20]:6228-6237. 181 Literatur 181. Little, J. W. 1993. LexA cleavage and other self-processing reactions. J Bacteriol. 175[16]:49434950. 182. Liu, L. F. and J. C. Wang. 1987. Supercoiling of the DNA template during transcription. Proc.Natl.Acad.Sci.U.S.A. 84[20]:7024-7027. 183. Lomovskaya, O. and W. Watkins. 2001. Inhibition of efflux pumps as a novel approach to combat drug resistance in bacteria. J.Mol.Microbiol.Biotechnol. 3[2]:225-236. 184. Lomovskaya, O., H. I. Zgurskaya, and H. Nikaido. 2002. It takes three to tango. Nat.Biotechnol. 20[12]:1210-1212. 185. Lowe, D. C., T. C. Savidge, D. Pickard, L. Eckmann, M. F. Kagnoff, G. Dougan, and S. N. Chatfield. 1999. Characterization of candidate live oral Salmonella typhi vaccine strains harboring defined mutations in aroA, aroC, and htrA. Infect.Immun. 67[2]:700-707. 186. Lowry, O. H., N. J. Rosenbrough, A. Farr, and R. J. Randall. 1951. Protein measurement with folin phenol reagent. J Biol Chem. 193:265-275. 187. Luttinger, A. 1995. The twisted 'life' of DNA in the cell: bacterial topoisomerases. Mol.Microbiol. 15[4]:601-606. 188. Ma, D., D. N. Cook, M. Alberti, N. G. Pon, H. Nikaido, and J. E. Hearst. 1995. Genes acrA and acrB encode a stress-induced efflux system of Escherichia coli. Mol Microbiol 16[1]:45-55. 189. Macvanin, M., J. Bjorkman, S. Eriksson, M. Rhen, D. I. Andersson, and D. Hughes. 2003. Fusidic acid-resistant mutants of Salmonella enterica serovar Typhimurium with low fitness in vivo are defective in RpoS induction. Antimicrobial Agents and Chemotherapy 47[12]:3743-3749. 190. Maisnier-Patin, S., O. G. Berg, L. Liljas, and D. I. Andersson. 2002. Compensatory adaptation to the deleterious effect of antibiotic resistance in Salmonella Typhimurium. Mol Microbiol 46[2]:355366. 191. Mammeri, H., L. M. Van De, L. Poirel, L. Martinez-Martinez, and P. Nordmann. 2005. Emergence of plasmid-mediated quinolone resistance in Escherichia coli in Europe. Antimicrobial Agents and Chemotherapy 49[1]:71-76. 192. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1989. Molecular cloning, a laboratory manual. Cold Spring Harbor Laboratory Press, New York 193. Mariam, D. H., Y. Mengistu, S. E. Hoffner, and D. I. Andersson. 2004. Effect of rpoB mutations conferring rifampin resistance on fitness of Mycobacterium tuberculosis. Antimicrobial Agents and Chemotherapy 48[4]:1289-1294. 194. Marians, K. J. 1987. DNA gyrase-catalyzed decatenation of multiply linked DNA dimers. J Biol Chem. 262[21]:10362-10368. 195. Marians, K. J. and H. Hiasa. 1997. Mechanism of quinolone action. A drug-induced structural perturbation of the DNA precedes strand cleavage by topoisomerase IV. J.Biol.Chem. 272[14]:94019409. 196. Martin, L. J., M. Fyfe, K. Dore, J. A. Buxton, F. Pollari, B. Henry, D. Middleton, R. Ahmed, F. Jamieson, B. Ciebin, S. A. McEwen, and J. B. Wilson. 2004. Increased burden of illness associated with antimicrobial-resistant Salmonella enterica serotype Typhimurium infections. J Infect.Dis. 189[3]:377-384. 197. Martinez-Martinez, L., A. Pascual, I. Garcia, J. Tran, and G. A. Jacoby. 2003. Interaction of plasmid and host quinolone resistance. J.Antimicrob.Chemother. 51[4]:1037-1039. 198. Martinez-Martinez, L., A. Pascual, and G. A. Jacoby. 1998. Quinolone resistance from a transferable plasmid. Lancet 351[9105]:797-799. 199. Mascellino, M. T., S. Farinelli, F. Iegri, and E. Iona. 1998. The influence of the SOS response on the activity of 4-quinolones and zidovudine against some strains of Enterobacteria. New Microbiol. 21[1]:31-40. 200. Masse, E. and M. Drolet. 1999. Relaxation of transcription-induced negative supercoiling is an essential function of Escherichia coli DNA topoisomerase I. J.Biol.Chem. 274[23]:16654-16658. 182 Literatur 201. Mazzariol, A., Y. Tokue, T. M. Kanegawa, G. Cornaglia, and H. Nikaido. 2000. High-level fluoroquinolone-resistant clinical isolates of Escherichia coli overproduce multidrug efflux protein AcrA. Antimicrob Agents Chemother 44[12]:3441-3443. 202. McClelland, M., K. Sanderson, J. Spieth, S. Clifton, P. Latreille, L. Courtney, S. Porwollik, J. Ali, M. Dante, F. Du, S. Hou, D. Layman, S. Leonard, C. Nguyen, K. Scott, A. Holmes, N. Grewal, E. Mulvaney, E. Ryan, H. Sun, L. Florea, W. Miller, T. Stoneking, M. Nhan, R. Waterston, and R. Wilson. 2001. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 413[6858]:852-856. 203. McMurry, L., R. E. Petrucci, Jr., and S. B. Levy. 1980. Active efflux of tetracycline encoded by four genetically different tetracycline resistance determinants in Escherichia coli. Proc.Natl.Acad.Sci.U.S.A 77[7]:3974-3977. 204. Menzel, R. and M. Gellert. 1983. Regulation of the genes for E. coli DNA gyrase: homeostatic control of DNA supercoiling. Cell 34[1]:105-113. 205. Mitsuyama, J., Y. Itoh, M. Takahata, S. Okamoto, and T. Yasuda. 1992. In vitro antibacterial activities of tosufloxacin against and uptake of tosufloxacin by outer membrane mutants of Escherichia coli, Proteus mirabilis, and Salmonella Typhimurium. Antimicrobial Agents and Chemotherapy 36[9]:2030-2036. 206. Molbak, K., D. L. Baggesen, F. M. Aarestrup, J. M. Ebbesen, J. Engberg, K. Frydendahl, P. Gerner-Smidt, A. M. Petersen, and H. C. Wegener. 1999. An outbreak of multidrug-resistant, quinolone-resistant Salmonella enterica serotype Typhimurium DT104. N.Engl.J Med 341[19]:14201425. 207. Mülhardt, C. 2002. Klonierung von PCR-Produkten in: Der Experimentator, Molekularbiologie/Genomics. Spektrum Akademischer Verlag Heidelberg - Berlin :120-123. 208. Munk, K. H. 2001. Genetik und Evolution in: Mikrobiologie. Spektrum Akademischer Verlag Heidelberg - Berlin :6-12-6-13. 209. Murphy, K. C. 1998. Use of bacteriophage lambda recombination functions to promote gene replacement in Escherichia coli. J.Bacteriol. 180[8]:2063-2071. 210. Murphy, K. C. and K. G. Campellone. 2003. Lambda Red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Molecular Biology 4[11] 211. Murphy, K. C., K. G. Campellone, and A. R. Poteete. 2000. PCR-mediated gene replacement in Escherichia coli. Gene 246[1-2]:321-330. 212. Nagaev, I., J. Bjorkman, D. I. Andersson, and D. Hughes. 2001. Biological cost and compensatory evolution in fusidic acid-resistant Staphylococcus aureus. Mol Microbiol 40[2]:433-439. 213. National Committee for Clinical Laboratory Standards. 2001. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. Approved Standard M7-A5. NCCLS, USA, 2001 214. Neumann, S. and A. Quinones. 1997. Discoordinate gene expression of gyrA and gyrB in response to DNA gyrase inhibition in Escherichia coli. J.Basic.Microbiol. 37[1]:53-69. 215. Newton, C. R., A. Graham, L. E. Heptinstall, S. J. Powell, C. Summers, N. Kalsheker, J. C. Smith, and A. F. Markham. 1989. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res 17[7]:2503-2516. 216. Nikaido, H. 1996. Multidrug efflux pumps of gram-negative bacteria. J Bacteriol 178[20]:5853-5859. 217. Nikaido, H. 1996. Outer Membrane. in. Escherischia coli and Salmonella Typhimurium.Neidhardt, F.et al. ASM Press. American Society for Microbiology:29-47. 218. Nikaido, H., M. Basina, V. Nguyen, and E. Y. Rosenberg. 1998. Multidrug efflux pump AcrAB of Salmonella Typhimurium excretes only those beta-lactam antibiotics containing lipophilic side chains. J Bacteriol. 180[17]:4686-4692. 219. Nikaido, H. and H. I. Zgurskaya. 2001. AcrAB and related multidrug efflux pumps of Escherichia coli. J.Mol.Microbiol.Biotechnol. 3[2]:215-218. 220. Nilsson, A. I., O. G. Berg, O. Aspevall, G. Kahlmeter, and D. I. Andersson. 2003. Biological costs and mechanisms of fosfomycin resistance in Escherichia coli. Antimicrobial Agents and Chemotherapy 47[9]:2850-2858. 183 Literatur 221. Noble, C. G., F. M. Barnard, and A. Maxwell. 2003. Quinolone-DNA Interaction: SequenceDependent Binding to Single-Stranded DNA Reflects the Interaction within the Gyrase-DNA Complex. Antimicrobial Agents and Chemotherapy 47[3]:854-862. 222. O'Byrne, C. P., N. N. Bhzriain, and C. J. Dorman. 1992. The DNA supercoiling-sensitive expression of the Salmonella Typhimurium his operon requires the his attenuator and is modulated by anaerobiosis and by osmolarity. Molecular Microbiology 6[17]:2467-2476. 223. Oethinger, M., W. V. Kern, A. S. Jellen-Ritter, L. M. McMurry, and S. B. Levy. 2000. Ineffectiveness of topoisomerase mutations in mediating clinically significant fluoroquinolone resistance in Escherichia coli in the absence of the AcrAB efflux pump. Antimicrob Agents Chemother 44[1]:10-13. 224. Oethinger, M., I. Podglajen, W. V. Kern, and S. B. Levy. 1998. Overexpression of the marA or soxS regulatory gene in clinical topoisomerase mutants of Escherichia coli. Antimicrob Agents Chemother 42[8]:2089-2094. 225. Okusu, H., D. Ma, and H. Nikaido. 1996. AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-antibiotic-resistance (Mar) mutants. J Bacteriol 178[1]:306-308. 226. Olliver, A., M. Valle, E. Chaslus-Dancla, and A. Cloeckaert. 2005. Overexpression of the multidrug efflux operon acrEF by insertional activation with IS1 or IS10 elements in Salmonella enterica serovar Typhimurium DT204 acrB mutants selected with fluoroquinolones. Antimicrobial Agents and Chemotherapy 49[1]:289-301. 227. Otto, K. and T. J. Silhavy. 2002. Surface sensing and adhesion of Escherichia coli controlled by the Cpx-signaling pathway. Proc.Natl.Acad.Sci.U.S.A 99[4]:2287-2292. 228. O`Farrell, P. H. 1975. High resolution two-dimensional electrophoresis of proteins. J Biol Chem 250[10]:4007-4021. 229. Paulsen, I. T. 2003. Multidrug efflux pumps and resistance: regulation and evolution. Curr.Opin.Microbiol 6[5]:446-451. 230. Paulsen, I. T., M. H. Brown, and R. A. Skurray. 1996. Proton-dependent multidrug efflux systems. Microbiol.Rev. 60[4]:575-608. 231. Peng, H. and K. J. Marians. 1993. Escherichia coli topoisomerase IV. Purification, characterization, subunit structure, and subunit interactions. J Biol.Chem. 268[32]:24481-24490. 232. Perez-Martin, J. and V. de Lorenzo. 1997. Clues and consequences of DNA bending in transcription. Annu.Rev.Microbiol. 51:593-628:593-628. 233. Petersen, U. 2001. Die Evolution der Chinolone: Von der Nalidixinsäure zu den Chinolonen der dritten Generation. Pharmazie in unserer Zeit 5:376-381. 234. Pettijohn, D. E. 1988. Histone-like proteins and bacterial chromosome structure. J Biol Chem. 263[26]:12793-12796. 235. Phillips, I., E. Culebras, F. Moreno, and F. Baquero. 1987. Induction of the SOS response by new 4-quinolones. J Antimicrob.Chemother. 20[5]:631-638. 236. Piddock, L. J. 1999. Mechanisms of fluoroquinolone resistance: an update 1994-1998. Drugs 58 Suppl 2:11-8:11-18. 237. Piddock, L. J. 2002. Fluoroquinolone resistance in Salmonella serovars isolated from humans and food animals. FEMS Microbiol Rev. 26:3-16. 238. Piddock, L. J. 2006. Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clin.Microbiol Rev. 19[2]:382-402. 239. Piddock, L. J., V. Ricci, I. McLaren, and D. J. Griggs. 1998. Role of mutation in the gyrA and parC genes of nalidixic-acid-resistant Salmonella serotypes isolated from animals in the United Kingdom. J Antimicrob Chemother 41[6]:635-641. 240. Piddock, L. J., D. G. White, K. Gensberg, L. Pumbwe, and D. J. Griggs. 2000. Evidence for an efflux pump mediating multiple antibiotic resistance in Salmonella enterica serovar Typhimurium. Antimicrob Agents Chemother 44[11]:3118-3121. 184 Literatur 241. Pogliano, J., A. S. Lynch, D. Belin, E. C. Lin, and J. Beckwith. 1997. Regulation of Escherichia coli cell envelope proteins involved in protein folding and degradation by the Cpx two-component system. Genes Dev. 11[9]:1169-1182. 242. Poole, K. 2000. Efflux-mediated resistance to fluoroquinolones in gram-positive bacteria and the mycobacteria. Antimicrobial Agents and Chemotherapy 44[10]:2595-2599. 243. Poole, K. 2004. Efflux-mediated multiresistance in Gram-negative bacteria. Clin.Microbiol Infect. 10[1]:12-26. 244. Poteete, A. R. and A. C. Fenton. 1993. Efficient double-strand break-stimulated recombination promoted by the general recombination systems of phages lambda and P22. Genetics 134[4]:10131021. 245. Poteete, A. R. and A. C. Fenton. 2000. Genetic requirements of phage Lambda Red-mediated gene replacement in Escherichia coli K-12. J Bacteriol 182[2]:2336-2340. 246. Raivio, T. L., M. W. Laird, J. C. Joly, and T. J. Silhavy. 2000. Tethering of CpxP to the inner membrane prevents spheroplast induction of the cpx envelope stress response. Mol Microbiol 37[5]:1186-1197. 247. Raivio, T. L. and T. J. Silhavy. 1997. Transduction of envelope stress in Escherichia coli by the Cpx two-component system. J Bacteriol 179[24]:7724-7733. 248. Randall, L. P., S. W. Cooles, M. K. Osborn, L. J. Piddock, and M. J. Woodward. 2004. Antibiotic resistance genes, integrons and multiple antibiotic resistance in thirty-five serotypes of Salmonella enterica isolated from humans and animals in the UK. J Antimicrob.Chemother. 53[2]:208-216. 249. Randall, L. P. and M. J. Woodward. 2001. Multiple antibiotic resistance (mar) locus in Salmonella enterica serovar Typhimurium DT104. Appl.Environ.Microbiol 67[3]:1190-1197. 250. Randall, L. P. and M. J. Woodward. 2001. Role of the mar locus in virulence of Salmonella enterica serovar Typhimurium DT104 in chickens. J Med.Microbiol 50[9]:770-779. 251. Reece, R. J. and A. Maxwell. 1991. DNA gyrase: structure and function. Crit.Rev.Biochem.Mol.Biol. 26[3-4]:335-375. 252. Reece, R. J. and A. Maxwell. 1991. The C-terminal domain of the Escherichia coli DNA gyrase A subunit is a DNA-binding protein. Nucleic.Acids.Res. 19[7]:1399-1405. 253. Reyna, F., M. Huesca, V. Gonzalez, and L. Y. Fuchs. 1995. Salmonella Typhimurium gyrA mutations associated with fluoroquinolone resistance. Antimicrob Agents Chemother 39[7]:16211623. 254. Reynolds, M. G. 2000. Compensatory evolution in rifampin-resistant Escherichia coli. Genetics 156[4]:1471-1481. 255. Reyrat, J. M., V. Pelicic, B. Gicquel, and R. Rappuoli. 1998. Counterselectable markers: untapped tools for bacterial genetics and pathogenesis. Infect Immun. 66[9]:4011-4017. 256. Ricci, V., P. Tzakas, A. Buckley, and L. J. Piddock. 2006. Ciprofloxacin-resistant Salmonella enterica serovar Typhimurium strains are difficult to select in the absence of AcrB and TolC. Antimicrobial Agents and Chemotherapy 50[1]:38-42. 257. Robert Koch-Institut. 2002. Salmonellose - Merkblatt für Ärzte. Erstveröffentlichung im Bundesgesundheitsblatt 01/1997 - aktualisiert 2002 http://www.rki.de/cln_011/nn_225576/DE/Content/Infekt/EpidBull/Merkblaetter/Mbl__Salmonell ose.html 258. Robert Koch-Institut. 2005. Typhus abdominalis, Paratyphus - RKI-Ratgeber Infektionskrankheiten – Merkblätter für Ärzte. URL: http://www.rki.de/cln_011/nn_225576/DE/Content/Infekt/EpidBull/Merkblaetter/Ratgeber__Mbl__Typh us__Paratyphus.html 259. Robert Koch-Institut. 2005. Zur Situation bei wichtigen Infektionskrankheiten in Deutschland: Bakterielle Gastroenteritiden – Focus Salmonellosen und Schweinefleisch-assoziierte Ausbrüche (2001–1. Halbjahr 2005). Epidemiologisches Bulletin 33 260. Robicsek, A., J. Strahilevitz, G. A. Jacoby, M. Macielag, D. Abbanat, C. H. Park, K. Bush, and D. C. Hooper. 2006. Fluoroquinolone-modifying enzyme: a new adaptation of a common aminoglycoside acetyltransferase. Nat.Med. 12[1]:83-88. 185 Literatur 261. Roesler, U., H. Marg, I. Schröder, S. Mauer, H. Sigmarsson, T. Arnold, J. Lehmann, U. Truyen, and A. Hensel. 2004. Oral vaccination of pigs with an invasive gyrA-cpxA-rpoB Salmonella Typhimurium mutant. Vaccine 23[5]:595-603. 262. Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA sequencing with chain-terminating inhibitors. Proc.Natl.Acad.Sci.U.S.A 74[12]:5463-5467. 263. Schedletzky, H., B. Wiedemann, and P. Heisig. 1999. The effect of moxifloxacin on its target topoisomerases from Escherichia coli and Staphylococcus aureus. J Antimicrob Chemother 43 Suppl B:31-7:31-37. 264. Schmitz, F. J., M. E. Jones, B. Hofmann, B. Hansen, S. Scheuring, M. Luckefahr, A. Fluit, J. Verhoef, U. Hadding, H. P. Heinz, and K. Kohrer. 1998. Characterization of grlA, grlB, gyrA, and gyrB mutations in 116 unrelated isolates of Staphylococcus aureus and effects of mutations on ciprofloxacin MIC. Antimicrobial Agents and Chemotherapy 42[5]:1249-1252. 265. Schnarr, M., P. Oertel-Buchheit, M. Kazmaier, and M. Granger-Schnarr. 1991. DNA binding properties of the LexA repressor. Biochimie 73[4]:423-431. 266. Schofield, M. A., R. Agbunag, M. L. Michaels, and J. H. Miller. 1992. Cloning and sequencing of Escherichia coli mutR shows its identity to topB, encoding topoisomerase III. J.Bacteriol. 174[15]:5168-5170. 267. Schrag, S. J., V. Perrot, and B. R. Levin. 1997. Adaptation to the fitness costs of antibiotic resistance in Escherichia coli. Proc.R.Soc.Lond.B.Biol.Sci. 264[1386]:1287-1291. 268. Schulte, A. 2001. Stabilität hoher Fluorchinolonresistenz bei Enterobakterien ohne Selektionsdruck. Dissertation.Rheinische Friedrich-Wilhelm-Universität Bonn 269. Selbitz, H. J., H. J. Sinell, and A. Sziegoleit. 1995. Das Salmonellen-Problem - Salmonellen als Erreger von Tierseuchen und Zoonosen. Gustav Fischer Verlag Jena Stuttgart :196 S270. Shea, M. E. and H. Hiasa. 2000. Distinct effects of the UvrD helicase on topoisomerase-quinoloneDNA ternary complexes. J.Biol.Chem. 275[19]:14649-14658. 271. Sherman, D. R., K. Mdluli, M. J. Hickey, T. M. Arain, S. L. Morris, C. E. Barry, III, and C. K. Stover. 1996. Compensatory ahpC gene expression in isoniazid-resistant Mycobacterium tuberculosis. Science 272[5268]:1641-1643. 272. Shuman, S. 1992. Two classes of DNA end-joining reactions catalyzed by Vaccinia topoisomerase I. J.Biol.Chem. 267[24]:16755-16758. 273. Snoep, J. L., C. C. Van Der Weijden, H. W. Andersen, H. V. Westerhoff, and P. R. Jensen. 2002. DNA supercoiling in Escherichia coli is under tight and subtle homeostatic control, involving geneexpression and metabolic regulation of both topoisomerase I and DNA gyrase. Eur.J Biochem. 269[6]:1662-1669. 274. Solano, C., B. Garcia, J. Valle, C. Berasain, J. M. Ghigo, C. Gamazo, and I. Lasa. 2002. Genetic analysis of Salmonella enteritidis biofilm formation: critical role of cellulose. Mol Microbiol 43[3]:793808. 275. Spory, A., A. Bosserhoff, C. von Rhein, W. Goebel, and A. Ludwig. 2002. Differential regulation of multiple proteins of Escherichia coli and Salmonella enterica serovar Typhimurium by the transcriptional regulator SlyA. J Bacteriol 184[13]:3549-3559. 276. Steck, T. R., R. J. Franco, J. Y. Wang, and K. Drlica. 1993. Topoisomerase mutations affect the relative abundance of many Escherichia coli proteins. Mol.Microbiol. 10[3]:473-481. 277. Sternglanz, R., S. DiNardo, K. A. Voelkel, Y. Nishimura, Y. Hirota, K. Becherer, L. Zumstein, and J. C. Wang. 1981. Mutations in the gene coding for Escherichia coli DNA topoisomerase I affect transcription and transposition. Proc.Natl.Acad.Sci.U.S.A 78[5]:2747-2751. 278. Stock I. 2000. Infektionen durch Salmonella enterica. MMP 7:214-226. 279. Stone, B. J. and V. L. Miller. 1995. Salmonella enteritidis has a homologue of tolC that is required for virulence in BALB/c mice. Mol.Microbiol. 17[4]:701-712. 280. Sulavik, M. C., M. Dazer, and P. F. Miller. 1997. The Salmonella Typhimurium mar locus: molecular and genetic analyses and assessment of its role in virulence. J Bacteriol. 179[6]:1857-1866. 186 Literatur 281. Swaney, S. M., H. Aoki, M. C. Ganoza, and D. L. Shinabarger. 1998. The oxazolidinone linezolid inhibits initiation of protein synthesis in bacteria. Antimicrobial Agents and Chemotherapy 42[12]:3251-3255. 282. Tabor, S. and C. C. Richardson. 1985. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Biotechnology 24:280-284. 283. Thorns, C. J. 2000. Bacterial food-borne zoonoses. Rev.Sci.Tech. 19[1]:226-239. 284. Tran, J. H., G. Jacoby, and D. C. Hooper. 2002. Interaction of the Plasmid-Mediated QuinoloneResistance Protein Qnr with DNA Gyrase. ASM 102nd General Meeting, Salt Lake City :Abstract A137285. Tran, J. H. and G. A. Jacoby. 2002. Mechanism of plasmid-mediated quinolone resistance. Proc.Natl.Acad.Sci.U.S.A 99[8]:5638-5642. 286. Tran, J. H., G. A. Jacoby, and D. C. Hooper. 2005. Interaction of the plasmid-encoded quinolone resistance protein Qnr with Escherichia coli DNA gyrase. Antimicrob Agents Chemother 49[1]:118125. 287. Tse-Dinh, Y. C. 1985. Regulation of the Escherichia coli DNA topoisomerase I gene by DNA supercoiling. Nucleic Acids Res. 13[13]:4751-4763. 288. Tse-Dinh, Y. C. and R. K. Beran. 1988. Multiple promoters for transcription of the Escherichia coli DNA topoisomerase I gene and their regulation by DNA supercoiling. J.Mol.Biol. 20[4]:735-742. 289. URL. 2006. http://138.192.68.68/bio/Courses/biochem2/DNA/DNAStructure.html. website 290. URL. 2006. http://www.pei.de/cln_042/nn_433396/DE/arzneimittel/vet-mittel/vet-mittelnode.html__nnn=true. website 291. URL. 2006. www3.niaid.nih.gov/biodefense/public/images.htm. website 292. Varma, J. K., K. D. Greene, J. Ovitt, T. J. Barrett, F. Medalla, and F. J. Angulo. 2005. Hospitalization and antimicrobial resistance in Salmonella outbreaks, 1984-2002. Emerg.Infect.Dis. 11[6]:943-946. 293. Vila, J., J. Ruiz, F. Marco, A. Barcelo, P. Goni, E. Giralt, and T. Jimenez de Anta. 1994. Association between double mutation in gyrA gene of ciprofloxacin- resistant clinical isolates of Escherichia coli and MICs. Antimicrobial Agents and Chemotherapy 38[10]:2477-2479. 294. Walker, R. A., N. Saunders, A. J. Lawson, E. A. Lindsay, M. Dassama, L. R. Ward, M. J. Woodward, R. H. Davies, E. Liebana, and E. J. Threlfall. 2001. Use of a LightCycler gyrA mutation assay for rapid identification of mutations conferring decreased susceptibility to ciprofloxacin in multiresistant Salmonella enterica serotype Typhimurium DT104 isolates. J Clin Microbiol 39[4]:14431448. 295. Wang, J. C. 1996. DNA topoisomerases. Annu.Rev.Biochem. 65:635-92:635-692. 296. Wang, M., J. H. Tran, G. A. Jacoby, Y. Zhang, F. Wang, and D. C. Hooper. 2003. Plasmid-mediated quinolone resistance in clinical isolates of Escherichia coli from Shanghai, China. Antimicrob Agents Chemother 47[7]:2242-2248. 297. Webber, M. A., A. Talukder, and L. J. Piddock. 2005. Contribution of mutation at amino acid 45 of AcrR to acrB expression and ciprofloxacin resistance in clinical and veterinary Escherichia coli isolates. Antimicrobial Agents and Chemotherapy 49[10]:4390-4392. 298. Weigel, L. M., C. D. Steward, and F. C. Tenover. 1998. gyrA mutations associated with fluoroquinolone resistance in eight species of Enterobacteriaceae. Antimicrobial Agents and Chemotherapy 42[10]:2661-2667. 299. Weisblum, B. 1998. Macrolide resistance. Drug Resist.Updat. 1:29-41. 300. White, D. G., J. D. Goldman, B. Demple, and S. B. Levy. 1997. Role of the acrAB locus in organic solvent tolerance mediated by expression of marA, soxS, or robA in Escherichia coli. J.Bacteriol. 179[19]:6122-6126. 301. Wiedeman, B. and Heisig P. 1999. Bakterielle Resistenz gegenüber Chinolonen (Ciprofloxacin). Chemotherapie Jounal 8.Jahrgang[Heft 3]:99-107. 187 Literatur 302. Willmott, C. J., S. E. Critchlow, I. C. Eperon, and A. Maxwell. 1994. The complex of DNA gyrase and quinolone drugs with DNA forms a barrier to transcription by RNA polymerase. J.Mol.Biol. 242[4]:351-363. 303. Wood, K. V. 1998. The Chemistry of Bioluminescent Reporter Assays. Promega Notes 65:14-20. 304. Yoon, H., S. Lim, S. Heu, S. Choi, and S. Ryu. 2003. Proteome analysis of Salmonella enterica serovar Typhimurium fis mutant. FEMS Microbiol Lett. 226[2]:391-396. 305. Yoshida, H., M. Bogaki, M. Nakamura, and S. Nakamura. 1990. Quinolone resistance-determining region in the DNA gyrase gyrA gene of Escherichia coli. Antimicrobial Agents and Chemotherapy 34[6]:1271-1272. 306. Yoshida, H., M. Bogaki, M. Nakamura, L. M. Yamanaka, and S. Nakamura. 1991. Quinolone resistance-determining region in the DNA gyrase gyrB gene of Escherichia coli. Antimicrobial Agents and Chemotherapy 35[8]:1647-1650. 307. Yoshida, H., M. Nakamura, M. Bogaki, H. Ito, T. Kojima, H. Hattori, and S. Nakamura. 1993. Mechanism of action of quinolones against Escherichia coli DNA gyrase. Antimicrobial Agents and Chemotherapy 37[4]:839-845. 308. Yu, D., H. M. Ellis, E. C. Lee, N. A. Jenkins, N. G. Copeland, and D. L. Court. 2000. An efficient recombination system for chromosome engineering in Escherichia coli. PNAS 97[11]:5978-5983. 309. Yu, D., J. A. Sawitzke, H. Ellis, and Court DL. 2003. Recombineering with overlapping singlestranded DNA oligonucleotides: testing a recombination intermediate. Proc.Natl.Acad.Sci.U.S.A 100[12]:7207-7212. 310. Yu, E. W., J. R. Aires, G. McDermott, and H. Nikaido. 2005. A periplasmic drug-binding site of the AcrB multidrug efflux pump: a crystallographic and site-directed mutagenesis study. J Bacteriol 187[19]:6804-6815. 311. Zechiedrich, E. L. and N. R. Cozzarelli. 1995. Roles of topoisomerase IV and DNA gyrase in DNA unlinking during replication in Escherichia coli. Genes Dev. 9[22]:2859-2869. 312. Zechiedrich, E. L., A. B. Khodursky, S. Bachellier, R. Schneider, D. Chen, D. M. Lilley, and N. R. Cozzarelli. 2000. Roles of Topoisomerases in Maintaining Steady-state DNA Supercoiling in Escherichia coli. J Biol.Chem.2000.Mar.17.;275.(11.):8103.-8113. 275[11]:8103-8113. 313. Zhang-Barber, L., A. K. Turner, and P. A. Barrow. 1999. Vaccination for control of Salmonella in poultry. Vaccine 17[20-21]:2538-2545. 314. Zhong, P. and V. D. Shortridge. 2000. The role of efflux in macrolide resistance. Drug Resist.Updat. 3[6]:325-329. 315. Zloh, M., G. W. Kaatz, and S. Gibbons. 2004. Inhibitors of multidrug resistance (MDR) have affinity for MDR substrates. Bioorg.Med.Chem Lett. 14[4]:881-885. 188 Abkürzungsverzeichnis 7 Abkürzungsverzeichnis A Ac Ala APS Arg Asp ATP bla bp BSA bzw. °C CCLR cm D ddNTP DIN DMSO DNA dNTP DTT EDTA EPI et al. EtBr F FQ g G Gly h IEF IPTG KBE l kb LB LPS M MHK min mm mM MOPS µg µl ml nm mRNA Alanin Acetat Alanin Ammoniumpersulfat Arginin Asparaginsäure Adenosintriphosphat ß-Lactamase Basenpaare Bovines Serumalbumin, Rinderserumalbumin beziehungsweise Grad Celsius Cell Culture Lysis Reagent Zentimeter Asparaginsäure Didesoxynukleosidtriphosphat Deutsches Institut für Normung Dimethylsulfoxid Desoxyribonukleinsäure (englisch: desoxyribonucleic acid) Desoxynukleosidtriphosphat Dithiothreitol Ethylendiamintetraacetat Effluxpumpeninhibitor et alii (lateinisch: und andere) Ethidiumbromid Phenylalanin Fluorchinolon (englisch: fluorquinolone) Gramm Glycin Glycin Stunde Isoelektrische Fokussierung Isopropyl β-D-Thiogalactopyranoside koloniebildende Einheiten Liter Kilobasenpaar(e) Luria Broth (Medium) Lipoploysaccharidschicht Molar Minimale Hemmkonzentration Minute Millimeter Millimolar (Millimol/l) 3-(N-Morpholino)propansulfonsäure Mikrogramm Mikroliter Milliliter Nanometer Boten-Ribonukleinsäure (englisch: mesenger ribonucleic acid) 189 Abkürzungsverzeichnis n.b. NCCLS ng nt ODnm p.A. PAGE PBS PCR Phe pmol R RFLP QRDR Qsc RBS RNA RnaseA RLU RT S SDS sec Ser TAE TEMED Topo Tris Trp ts Tyr U u.a. ÜNK upm UV V V/h Vol W X-Gal Y z.B. ∆ nicht bestimmt National Commitee for Clinical Laboratory Standards Nanogramm Nukleotide optische Dichte (Index gibt die Wellenlänge an) zu Analyse Polyacrylamidgelelektrophorese phosphatgepufferte Kochsalzlösung Polymerase-Kettenreaktion (englisch: polymerase chain reaction) Phenylalanin Pikomol Arginin Restriktionsfragment-Längenpolymorphismus quninolone resistance determining region quotient of supercoiling Ribosomenbindungsstelle Ribonukleinsäure (englisch: ribonucleic acid) Ribonuklease A Relative Light Units Raumtemperatur Serin Natriumdodecylsulfat (englisch: sodium dodecyl sulfate) Sekunde Serin Tris-Acetat-EDTA-Puffer Tetramethylethylenediamin Topoisomerase Tris(hydroxymethyl)aminimethan Tryptophan temperatursensitiv Tyrosin Unit unter anderem Übernachtkultur Umdrehungen in der Minute Ultraviolett Volt Voltstunden Volumen Tryptophan 5-Brom-4-Chlor-3-indolyl-β-D-galactosid Tyrosin zum Beispiel Deletion Die Abkürzungen für die verwendeten Antibiotika sind der Tabelle 2-16 zu entnehmen. 190 Ethanol Essigsäure DTT CalciumchloridDihydrat Butanol Ammoniumpersulfat Acrylamid-Bis Fertiglösung 30% (37,5:1) Gefahrstoff F C Xn Xi Xn Xn O T Gefahrensymbol R 22-36/38 Gesundheitsschädlich beim Verschlucken. Reizt die Augen und die Haut. R 10-35 Entzündlich. Verursacht schwere Verätzungen. R 11 Leichtentzündlich. R 36 Reizt die Augen. R 10-22-37/38-41-67 Entzündlich. Gesundheitsschädlich beim Verschlucken. Reizt die Atmungsorgane und die Haut. Gefahr ernster Augenschäden. Dämpfe können Schläfrigkeit und Benommenheit verursachen. S 53-36/37-45 Exposition vermeiden - Vor Gebrauch besondere Anweisungen einholen. Bei der Arbeit geeignete Schutzhandschuhe und Schutzkleidung tragen. Bei Unfall oder Unwohlsein sofort Arzt hinzuziehen R 45-46-E22-43-E48/23/24/25-62 Kann Krebs erzeugen. Kann vererbbare Schäden verursachen. Sensibilisierung durch Hautkontakt möglich. Gefahr ernster Gesundheitsschäden bei längerer Exposition durch Einatmen, Berührung mit der Haut und durch Verschlucken. Kann möglicherweise die Fortpflanzungsfähigkeit beeinträchtigen. R 8-22-36/37/38-42/43 Feuergefahr bei Berührung mit brennbaren Stoffen. Gesundheitsschädlich beim Verschlucken. Reizt die Augen, Atmungsorgane und die Haut. Sensibilisierung durch Einatmen und Hautkontakt möglich. S 23.2-26-45 Dampf nicht einatmen. Bei Berührung mit den Augen sofort gründlich mit Wasser abspülen und Arzt konsultieren. S 7-16 Behälter dicht geschlossen halten. Von Zündquellen fernhalten - Nicht rauchen. S 7/9-13-26-37/39-46 Behälter dicht geschlossen an einem gut gelüfteten Ort aufbewahren. Von Nahrungsmitteln, Getränken und Futtermitteln fernhalten. Bei Berührung mit den Augen sofort gründlich mit Wasser abspülen und Arzt konsultieren. Bei der Arbeit geeignete Schutzkleidung tragen. Bei Verschlucken sofort ärztlichen Rat einholen. S 22-24 Staub nicht einatmen. Berührung mit der Haut vermeiden. S 22-24-26-37 Staub nicht einatmen. Berührung mit der Haut vermeiden. Bei Berührung mit den Augen sofort gründlich mit Wasser abspülen und Arzt konsultieren. Geeignete Schutzhandschuhe tragen. Sicherheitsratschläge - S-Sätze Gefahrenhinweise - R-Sätze Tabelle 8 – 1 Wichtige in der Arbeit verwendete Gefahrstoffe mit Gefahrensymbol und R- sowie S-Sätze 8 Anhang Natrium-carbonat Methanol Kaliumhydroxid Isopropanol Glutardialdehyd Formaldehyd EDTA Ethidiumbromid Xi T F C Xi F N T T Xi Xn R 11-23/24/25-39/23/24/25 Leichtentzündlich. Giftig beim Einatmen, Verschlucken und Berührung mit der Haut. Ernste Gefahr irreversiblen Schadens durch Einatmen, Berührung mit der Haut und durch Verschlucken. R 36 Reizt die Augen. R 22-35 Gesundheitsschädlich beim Verschlucken. Verursacht schwere Verätzungen. R 68 Irreversibler Schaden möglich. Erbgutverändernd R 36-52/53 Reizt die Augen. Schädlich für Wasserorganismen, kann in Gewässern längerfristig schädliche Wirkungen haben. R 20/21/22-36/37/38-39/23/24/25-40-43 Reizt die Augen, Atmungsorgane und die Haut.Ernste Gefahr irreversiblen Schadens durch Einatmen, Berührung mit der Haut und durch Verschlucken. Verdacht auf krebserzeugende Wirkung. R 22-23-34-42/43-50 Gesundheitsschädlich beim Verschlucken. Giftig beim Einatmen. Verursacht Verätzungen. Sensibilisierung durch Einatmen und Hautkontakt möglich. Sehr giftig für Wasserorganismen. R 11-36-67 Leichtentzündlich. Reizt die Augen. Dämpfe können Schläfrigkeit und Benommenheit verursachen. S 22-26 Staub nicht einatmen. Bei Berührung mit den Augen sofort gründlich mit Wasser abspülen und Arzt konsultieren. S 26-36/37/39-45 Bei Berührung mit den Augen sofort gründlich mit Wasser abspülen und Arzt konsultieren. Bei der Arbeit geeignete Schutzkleidung tragen. Bei Unfall oder Unwohlsein sofort Arzt hinzuziehen. S 7-16-36/37-45 Behälter dicht geschlossen halten. Von Zündquellen fernhalten Nicht rauchen. Bei der Arbeit geeignete Schutzhandschuhe und Schutzkleidung tragen. S 26-36/37/39-45-61 Bei Berührung mit den Augen sofort gründlich mit Wasser abspülen und Arzt konsultieren. Bei der Arbeit geeignete Schutzkleidung tragen. Bei Unfall oder Unwohlsein sofort Arzt hinzuziehen. Freisetzung in die Umwelt vermeiden. Besondere Anweisungen einholen/ Sicherheitsdatenblatt zu Rate ziehen. S 7-16-24/25-26 Behälter dicht geschlossen halten. Von Zündquellen fernhalten Nicht rauchen. Berührung mit den Augen und der Haut vermeiden. Bei Berührung mit den Augen sofort gründlich mit Wasser abspülen und Arzt konsultieren. S 26-36/37-45-51 Bei Berührung mit den Augen sofort gründlich mit Wasser abspülen und Arzt konsultieren. Bei der Arbeit geeignete Schutzhandschuhe und Schutzkleidung tragen.Bei Unfall oder Unwohlsein sofort Arzt hinzuziehen. Nur in gut gelüfteten Bereichen verwenden. S 36/37 Bei der Arbeit geeignete Schutzhandschuhe und Schutzkleidung tragen. S 61 Freisetzung in die Umwelt vermeiden. Besondere Anweisungen einholen/Sicherheitsdatenblatt zu Rate ziehen. Xi Xi C F N C C C Xn F R 36 Reizt die Augen. R 36/38 Reizt die Augen und die Haut. R 36/38 Reizt die Augen und die Haut. R 11-20/22-34 Leichtentzündlich. Gesundheitsschädlich beim Einatmen und Verschlucken. Verursacht Verätzungen. R 34-50/53 Verursacht Verätzungen. Sehr giftig für Wasserorganismen, kann in Gewässern längerfristig schädliche Wirkungen haben. R 34-37 Verursacht Verätzungen. Reizt die Atmungsorgane. R 34 Verursacht Verätzungen. R 11-21/22-36/37/38 Leichtentzündlich. Gesundheitsschädlich bei Berührung mit der Haut und beim Verschlucken. Reizt die Augen, Atmungsorgane und die Haut. Xi Quelle: http://www.merck.de/servlet/PB/menu/1409670/index.html Xylencyanol trans-1,2Diaminocyclo-hexanN,N,N',N'-tetraacetat Tris-Base TEMED Silbernitrat Salzsäure Phosphorsäure SDS S 24 Berührung mit der Haut vermeiden. S 16-26-36/37/39-45 Von Zündquellen fernhalten - Nicht rauchen. Bei Berührung mit den Augen sofort gründlich mit Wasser abspülen und Arzt konsultieren. Bei der Arbeit geeignete Schutzkleidung, Schutzhandschuhe und Schutzbrille/ Gesichtsschutz tragen. Bei Unfall oder Unwohlsein sofort Arzt hinzuziehen. S 26-45 Bei Berührung mit den Augen sofort gründlich mit Wasser abspülen und Arzt konsultieren. Bei Unfall oder Unwohlsein Arzt hinzuziehen. S 26-36/37/39-45 Bei Berührung mit den Augen sofort gründlich mit Wasser abspülen und Arzt konsultieren. Bei der Arbeit geeignete Schutzkleidung tragen. Bei Unfall oder Unwohlsein sofort Arzt hinzuziehen. S 26-45-60-61 Bei Berührung mit den Augen sofort gründlich mit Wasser abspülen und Arzt konsultieren. Bei Unfall oder Unwohlsein sofort Arzt. Dieser Stoff und sein Behälter sind als gefährlicher Abfall zu entsorgen. Freisetzung in die Umwelt vermeiden. S 26-36/37 Bei Berührung mit den Augen sofort gründlich mit Wasser abspülen und Arzt konsultieren. Bei der Arbeit geeignete Schutzhandschuhe und Schutzkleidung tragen. Danksagung Herrn Prof. Peter Heisig danke ich für die Bereitstellung des sehr spannenden und schönen Themas und die Möglichkeit, diese Arbeit in seiner Arbeitsgruppe anfertigen zu können. Vielen Dank für die Betreuung der Dissertation, die stete Diskussionsbereitschaft und Anregungen, sowie die Möglichkeit der Teilnahme an zahlreichen Kongressen und Seminaren. Herrn Prof. Hahn danke ich für die freundliche Übernahme des Korreferats. Bei Herrn PD Jörg Beer (Universität Leipzig) bedanke ich mich für das Überlassen der S. Typhimurium-Stämme Nal2ori und Nal2passage und Herrn Dr. W. Rabsch (Robert Koch-Institut, Wernigerode) danke ich für die Bereitstellung von S. Typhimurium M415. Mein Dank gilt weiterhin Frau Dr. Anke Heisig für die anregenden Gespräche und eine schöne Zeit während der Betreuung des Biochemie-Praktikums. Ich danke ihr und Antje Schnasse für die Unterstützung bei der DNA-Sequenzierung. Bei allen Mitarbeitern unserer Arbeitsgruppe und bei Sabine Badziong bedanke ich mich ganz herzlich für die Hilfsbereitschaft und die sympathische Atmosphäre in unserem Labor. Besonders danke ich meinen „Weggefährtinnen“ Dr. Barbara Körber-Irrgang und Silke Schmidt, die mir stets freundschaftlich mit Rat und Tat zur Seite standen und großes Interesse am Fortgang meiner Arbeit zeigten. Danke für die schöne Zeit, die wir zusammen hatten. Silke danke ich besonders für das Korrekturlesen des Manuskriptes. Tatjana Claußen und Antje Schnasse danke ich für ihre Freundschaft und die gemeinschaftliche Lösung all der großen und kleinen Probleme und für ihre uneingeschränkte Hilfsbereitschaft in allen fachlichen und nichtfachlichen Fragen. Nina Matthiessen danke ich für die Bereitstellung der Daten der Messungen der Promotoraktivitäten. Bei Dr. Mobarak Abu Mraheil bedanke ich mich für die Bereitstellung der Reportergenplasmide zur Bestimmung des DNA-Superspiralisierungsgrades. Dr. Antje Pieconka danke ich für die so freundliche Aufnahme in die Arbeitsgruppe und die Einführung in die Geheimnisse des Laboralltages, für ihre „Fürsorge“ und viel Humor sowie für die Vermittlung der Mehoden der 2D-Gelelektrophorese. Dr. Nadja Auer danke ich für die hilfreichen und humorvollen Gespräche in der ersten Phase meiner Doktorarbeit. Ein sehr großer Dank gilt meinen Eltern und meiner „großen“ Schwester, ohne deren Liebe und stete Unterstützung in jeder Hinsicht diese Arbeit nicht möglich gewesen wäre. Mein besonderer Dank gebührt meinem Mann Sascha. Seine Liebe, sein Verständnis und seine Geduld haben vieles einfacher gemacht. Sie waren ein Ausgleich für den Stress und die schweren Zeiten während der gesamten Arbeit und haben mich immer wieder aufgebaut und motiviert. Lebenslauf • Persönliche Daten: o Antje Preisler, geb. Schulz o geboren am 09. November 1975 in Gera • Schulbildung: o 1982 - 1984: Polytechnische Oberschule Gera o 1984 - 1990: Polytechnische Oberschule Potsdam o 1990 - 1995: Humboldt-Gymnasium Potsdam • Hochschulbildung: o 1995 - 2001: Studium der Biologie an der Freien Universität Berlin o 1997 - 1999: Zusatzstudium Umweltmanagement (FU Berlin) o 2001: Abschluss Diplom in Biologie mit „sehr gut“ o 2001 - 2002: Molekularbiologisches Praktikum bei mice & more GmbH & Co. KG Hamburg Transgenic Services & Animal Models o 2002 – 2006: Promotion in der Abteilung „Pharmazeutische Biologie und Mikrobiologie“ an der Universität Hamburg unter der Leitung von Prof. Heisig Poster und Veröffentlichungen o o o o o Preisler, A. and P. Heisig. 2006. Impact of novel gyrA mutations and multiple drug resistance efflux on susceptibility to fluoroquinolones of live vaccine strain TAD Salmonella vacT (vacT). Presentation number C1-0045. 46th ICAAC. American Society for Microbiology, San Francisco, USA Preisler, A., M.A. Mraheil, P. Heisig. 2006. Role of novel gyrA mutations in the suppression of the fluoroquinolone resistance genotype of vaccine strain Salmonella Typhimurium vacT (gyrA D87G). J Antimicrob Chemother. 57(3): 430-6 Preisler, A. and P. Heisig. 2005. Detection of a quinolone resistance-mediating gyrA mutation in a fluoroquinolone susceptible Salmonella live vaccine strain. P1425. Clinical Microbiology and Infection 11 (Suppl. 2): 459-460. 15th European Congress of Clinical Microbiology and Infectious Diseases, Kopenhagen, Dänemark Schulz, A., H. Hansen, P. Heisig. 2003. Identification of new target-associated mutations in quinolone-resistant salmonellae. PB8. DPhG-Jahrestagung Würzburg Schulz, A., H. Hansen, P.Heisig. 2003. Identifizierung neuer Topoisomerase IVMutationen in fluorchinolonresistenten Salmonella ssp. P130. Infection 31 (Suppl. 1):130. 7. Kongress für Infektionskrankheiten und Tropenmedizin, Berlin Hiermit versichere ich an Eides statt, dass ich die vorliegende Arbeit selbstständig verfasst und keine anderen als die von mir angegebenen Quellen und Hilfsmittel verwendet habe. Alle Stellen, die im Wortlaut oder dem Sinn nach anderen Arbeiten entnommen sind, habe ich unter genauer Angabe der Quelle als Entlehnung kenntlich gemacht. Hamburg, den 11.07.2006 .............................................. Antje Preisler