Drosophila melanogaster: Ein Modellsystem zur
Werbung

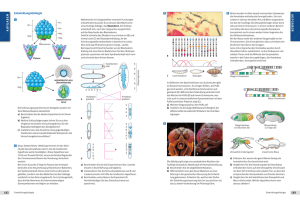
Überblick 626 Drosophila melanogaster: Ein Modellsystem zur Analyse neurodegenerativer Erkrankungen Jose A. Botella und Stephan Schneuwly Institut für Zoologie, Universität Regensburg Neurodegenerative Erkrankungen stellen in steigendem Umfang ein wichtiges gesundheits-ökonomisches Problem unserer Gesellschaft dar. Obwohl enorme Anstrengungen zur Klärung der Krankheitsursachen und zur Entwicklung neuer Therapien unternommen werden, liegt vieles noch im Dunkeln, insbesondere deshalb, weil adäquate Modellsysteme zur Untersuchung der pathophysiologischen Mechanismen fehlen. Die vollständige Sequenzierung mehrerer eukaryotischer Genome, vom Nematoden Caenorhabditis elegans über Drosophila melanogaster bis zum Menschen, ermöglichen es nun, vergleichende Untersuchungen an einfacheren Organismen zu unternehmen und neue Ansätze für die Aufklärung der Krankheitsursachen und möglicher Therapien zu entwickeln. Diese Arbeit soll einen Überblick über die verschiedenen Ansätze geben, mit denen an Drosophila melanogaster neue Erkenntnisse gewonnen werden. Abb. 1: Altersabhängige Neurodegeneration im Gehirn von Drosophila Mutanten. A) Horizontaler Schnitt durch ein Fliegengehirn, angefärbt mit einem Antikörper gegen Synapsen zeigt die typische Anordnung der Neuropil-Regionen des Drosophila-Gehirns. B) Ähnliche Schnittebene durch ein Gehirn einer gleichaltrigen Mutante vap (vacuolar pedunculi). Deutlich zu sehen ist die Vakuolisierung in großen Bereichen der Neuropil-Region. Der Phänotyp nimmt mit zunehmendem Alter der Fliegen zu. Neurodegenerative Erkrankungen sind mit zunehmender Überalterung unserer Gesellschaft ein ständig wachsendes Problem. Obwohl einige dieser Erkrankungen bereits in jungen Jahren ausbrechen können (z.B. ALS; Amyotrophe Lateralsklerose) ist das Alter der hauptsächliche Risikofaktor dieser Erkrankungen wie bei der Alzheimer Krankheit oder bei Parkinson. Altersabhängige neurodegenerative Erkrankungen sind im Zunehmen und die Krankheitsfälle werden in Zukunft noch dramatischer ansteigen, da man davon ausgehen kann, dass sich die Population der über 60-jährigen in den nächsten 50 Jahren annähernd verdoppeln wird. Trotz umfangreicher Forschungsansätze gibt es gegenwärtig weder eine Heilung noch eine ursächliche Therapie für diese Erkrankungen. Eine der Möglichkeiten, neue Entwicklungen in der Forschung anzustoßen, ist der Einsatz von einfachen Modellsystemen. Die Motivation, Invertebraten-Modelle wie Drosophila melanogaster zu nutzen, kommt aus dem hohen Verwandtschaftsgrad der bisher sequenzierten Genome. Im Jahre 2000 wurde die erste Fassung des Drosophila-Genoms veröffentlicht[1] und man rechnet mit ca. 14.000 Genen, dabei ist besonders interessant, dass von den etwa 1.000 Genen, die bereits mit humanen Erbkrankheiten assozi- iert sind, 77 % homologe Gene in Drosophila aufweisen[2]. Es wäre sicher irreführend, Invertebraten als die idealen Systeme zur Untersuchung humaner neurodegenerativer Krankheitsbilder zu bezeichnen, trotzdem gibt es einige wichtige Vorteile, die es zu berücksichtigen gilt. So besitzt die Fliege ein Zentrales Nervensystem welches nach den gleichen Prinzipien funktioniert wie bei höheren Organismen, nur mit einer wesentlich geringeren Anzahl an Neuronen und Glia-Zellen. Es teilt mit den Vertebraten die gleichen Neurotransmitter-Systeme wie z.B. GABA, Glutamat, Dopamin und Acetylcholin und es können komplexe Verhaltensmuster wie Lernen und Gedächtnis für die Untersuchungen herangezogen werden und dies bei wesentlich einfacheren Zuchtbedingungen als etwa bei Mäusen. Durch fast 100 Jahre Genetik mit Drosophila stehen nicht nur Tausende von Mutanten zur Verfügung, sondern auch eine Vielzahl genetischer Methoden zur Analyse komplexer Prozesse. Zudem ist, wie bereits erwähnt, eine große Zahl von Genen und Signaltransduktionskaskaden von Fliegen bis zum Menschen konserviert. Aus diesen Gründen hat sich Drosophila in den letzten Jahren als wichtiger Modellorganismus etabliert, um neue Hypothesen zu überprüfen oder neue KrankBIOspektrum · 5/04 · 10. Jahrgang Überblick 627 Der reverse Genetik-Ansatz Tab. 1: Einige Beispiele der genetischen Ansätze in Drosophila Krankheit Gene Methode Literatur Adenoleukodystrophie(ALD) VLCFA acyl CoA synthase Forward-Genetik Min and Benzer, 1999 Alzheimer und Tauopathien Drosophila APPL Humanes APP TAU GAL4/UAS Gunawardena and Goldstein, 2001 Wittmann et al., 2001 Parkinson Human α-synuclein GAL4/UAS Feany and Bender, 2000 Polyglutamin Erkrankungen Huntington Spinozerebelläre Ataxie Typ 1 Spinozerebelläre Ataxie Typ 3 Huntingtin SCA-1 SCA-3 GAL4/UAS Jackson et al., 1998 Fernández-Fúnez et al., 2000 Warrick et al., 1998 heits-relevante Gene mit Methoden zu identifizieren, die in anderen Organismen wie z.B. der Maus nicht in der Einfachheit zugänglich sind. Es gibt im Wesentlichen drei verschiedene Ansätze zur Untersuchung neurodegenerativer Phänotypen bei Drosophila: „forward“-Genetik, reverse Genetik und die Übertragung spezifischer Konstrukte (Tab. 1). von Nervenzellen und damit zu Neurodegeneration führt. Auch bei ALD-Patienten (wie bei bubblegum Mutanten) kommt es zu einer Erhöhung des Anteils dieser langkettigen Fettsäuren und zeigt damit die Parallelität der Mechanismen der Pathogenität auf. Der „forward“-Genetik-Ansatz Die klassische Art mit Drosophila zu arbeiten läuft über die Isolierung neuer Mutanten in einem „forward“-Genetik Ansatz. Diese neuen Mutanten werden aufgrund eines neurodegenerativen Phänotyps im ZNS selektioniert, der meist altersabhängig auftritt. Abbildung 1 zeigt ein Beispiel aus unserem Labor, wo es uns gelungen ist mit der Isolierung und Charakterisierung der Mutante vap (vacuolar pedunculi), ein wichtiges Gen zu entdecken, welches für die Stabilität der Neurone eine entscheidende Bedeutung hat. Deutlich sichtbar ist in Abbildung 1 die altersabhängige Degeneration des Zentralen Nervensystems. Mit dieser Mutante (Abb. 2) konnte auch aufgedeckt werden, dass Signaltransduktionswege, die in der Entwicklungsbiologie von Drosophila eine wichtige Rolle spielen, auch im ausgewachsenen Nervensystem eine entscheidende Bedeutung für die Stabilität der Nervenzellen haben[3]. Solche neu identifizierten Gene können als wichtige Kandidaten-Gene angesehen werden und die Identifizierung entsprechend homologer Gene beim Menschen ermöglicht damit neue Forschungsansätze bis hin zur Therapie. Eines der bekanntesten Beispiele in Drosophila ist die Mutation bubblegum[4]. bubblegum Fliegen zeigen eine altersabhängige Degeneration des ZNS, die mit einer Mutation in der überlangkettigen Fettsäure Coenzym A-Synthetase gekoppelt ist und zu einer Akkumulation dieser überlangkettigen Fettsäuren führt. Interessant ist die Analogie zu einer humanen Erbkrankheit, der Adrenoleukodystrophie (ALD), die u. a. zu einer Demyelinisierung BIOspektrum · 5/04 · 10. Jahrgang Abb. 2: Die Rolle der vap-Mutante in der RASMAPK-Signaltransduktionskaskade. Vereinfachtes Schema der EGF-Rezeptorsignalkaskade und der Bedeutung von vap. Ein extrazellulärer Ligand aktiviert den EGF-Rezeptor, der wiederum zur Aktivierung von Ras führt, welches über die MAP-Kinasekaskade ein Signal in den Kern sendet. Vap kodiert für ein RasGAP-Protein, welches mit einer GTPase-Aktivität die aktive Form von Ras wieder inaktiviert und damit das Signal abschaltet. Bei fehlendem RasGAP in vap Mutanten wird der Signalweg überaktiviert und führt zur Neurodegeneration. Im Gegensatz zum „forward“-Genetik Ansatz macht sich der reverse Genetik-Ansatz die rasanten Fortschritte auf dem Gebiet der vergleichenden Genomuntersuchungen zu Nutze. Dabei werden in Drosophila die zum humanen System verwandten Gene identifiziert und zur funktionellen Analyse in Drosophila entweder entsprechende Mutanten erzeugt oder Defekte mittels gezielter Überexpression induziert. Mit diesen Methoden können wichtige Erkenntnisse über die Funktion einzelner Gene in Drosophila erlangt werden, welchen zwar eine Beteiligung an humanen Erkrankungen nachgesagt wird, aber über deren biologische Bedeutung man im Wesentlichen noch im Unklaren ist (siehe unten, z.B. das APP-Gen). Der Einsatz dominanter Konstrukte in Drosophila Der dritte Ansatz basiert auf der Übertragung einer toxischen Wirkung von dominanten Konstrukten. Bei vielen neurodegenerativen Erkrankungen wie z.B. bei Chorea Huntington, Spinozerebellärer Ataxien (Poly-Glutamin-Erkrankungen), Synucleinopathien (Parkinson) oder Tauopathien (Fronto-temporale Demenz) liegen dominante Gendefekte vor. In diesen experimentellen Ansätzen wird die krankheitsrelevante Form des menschlichen Gens in Drosophila mittels dem Hefe UAS/GAL-4 System ektopisch in definierten Geweben exprimiert[5] (siehe Abb. 3). Gerade dieser Ansatz hat sich bei Drosophila äußerst erfolgreich etabliert und soll anhand einiger Beispiele erläutert werden. Bei der Alzheimer Erkrankung kommt es zum Verlust von Neuronen, der von extrazellulären abnormen Ablagerungen der amyloiden Plaques begleitet wird, die durch die Aggregation des Aβ-Peptids, einem Prozessierungsprodukt von APP (amyloid precursor-Protein) entstehen. Zusätzlich sind auch intrazelluläre neurofibrilläre Strukturen (Tangles) zu finden die aus hyperphosphoryliertem TAU-Protein bestehen. Obwohl bereits sehr viele Hinweise für die Beteiligung von APP an der Alzheimer Erkrankung bestehen, ist über die physiologische Bedeutung dieses Gens noch wenig bekannt. Drosophila besitzt ebenfalls ein zu APP ähnliches Gen (APPL; APP-like). Eine Analyse von appl-Mutanten und Überexpressionsstudien mit verschiedenen Mutanten der humanen- und Fliegen-APP-Gene hat gezeigt, dass APP eine wichtige Rolle beim axonalen Transport spielt[6]. Diese in vivo Phänotypen zeigen neben den axonalen Transportdefekten auch eine Erhöhung des larvalen Zelltods im ZNS. Obwohl keine amyloiden Überblick 628 Abb. 3: Das Drosophila GAL4-UAS-System. In der Enhancer-trap-Fliege wird der Hefe-Transkriptionsfaktor gewebespezifisch exprimiert. Durch Kreuzung mit einer Fliege, die das zur Expression vorgesehene Gen hinter der Kontrollsequenzen UAS (upstream activating sequence) trägt, wird dieses Gen durch die Gewebe-spezifische Expression des GAL4 angeschaltet. Plaques gefunden werden konnten, wurde beschrieben, dass Drosophila humanes APP prozessieren und das Aβ-Peptid nachgewiesen werden kann[7]. Außerdem ist dieser Prozessierungsweg identisch mit der Prozessierung von Notch, einem außerordentlich wichtigen Protein, welches an verschiedenen Entwicklungsprozessen bei Drosophila beteiligt ist. Damit können in Drosophila wichtige Erkenntnisse über die biologische und pathophysiologische Bedeutung von APP gewonnen werden. In ähnlicher Weise wird auch die Bedeutung des TAU-Proteins für die Destabilisierung von Neuronen in Drosophila untersucht[8]. Das Parkinson-Syndrom umfasst verschiedene familiäre und spontane Erkrankungen mit motorischen Störungen, wobei dopaminerge Neurone in einem definierten Bereich des Gehirns in einer Alters-abhängigen Weise verloren gehen. Dieser Verlust neuronaler Zellen wird begleitet durch das Erscheinen von zellulären Einschlüssen, den Lewy-Körperchen. Eine wichtige Komponente dieser Einschlüsse ist das Protein αSynuclein. Mehrere Mutationen im α-Synuclein-Gen konnten in den seltenen familiären Formen von Parkinson bereits aufgedeckt werden, jedoch sind die Proteinaggregate in allen, insbesondere der spontanen (idiopath.) Form von Parkinson zu finden. α-Synucleine scheinen deshalb eine zentrale Rolle in der Pathogenese dieser Erkrankung zu spielen. Vor wenigen Jahren konnte das erste Drosophila Modell für Parkinson etabliert werden[9]. Dabei wurde das humane α-Synuclein im Fliegen-Gehirn überexprimiert. Überraschenderweise wurde im ZNS der Fliegen ebenfalls nur eine spezifische Neurodegeneration des dopaminergen Systems beobachtet, obwohl α-Synuclein in allen Neuronen exprimiert wurde. Die befallenen Neurone zeigen ebenfalls mit Lewy-Körperchen vergleichbare Einschlüsse wie sie im Gehirn von Patienten zu finden sind. Neben α-Synuclein befinden sich in den Lewy Körperchen auch andere Proteine wie z.B. Chaperone. So konnte kürzlich in Drosophila gezeigt werden, dass Chaperone eine schützende Wirkung vor Degeneration der dopaminergen Neurone haben[10]. Eine weitere Gruppe von Erkrankungen betrifft die Polyglutamin-Erkrankungen. Dabei werden durch genetische Defekte in den betroffenen Genen bestimmte Trinukleotid-Wiederholungen (CAG), die für die Aminosäure Glutamin kodieren, vermehrt. Eine damit verbundene Erhöhung der Anzahl Wiederholungen führt zu einer pathologischen Akkumulation des mutierten Proteins, welches dadurch auf die Zellen toxisch wirkt. Auch hier kommt es also zu intrazellulären Einschlüssen und zu einer spezifischen Degeneration eines Teils des Zentralen Nervensystems. Dabei konnte am Beispiel der Expression von unterschiedlich langen Polyglutaminwiederholungen im Drosophila Auge sowohl für Chorea Huntington, wie auch für die Spinozerebelläre Ataxie (SCA) ein klarer Zusammenhang zwischen der Länge des Segmentes und der Stärke des Defektes hergestellt werden[11,12]. Das Drosophila Auge ermöglicht nun in ei- nem weiteren Ansatz die Suche nach genetischen Modifikatoren, d.h. nach Mutationen, die den Defekt abschwächen und verstärken[13,14]. Damit können wiederum neue Faktoren identifiziert werden, die an der Pathogenese beteiligt sind oder die möglicherweise neue potenzielle therapeutische Ziele darstellen können. Diesbezüglich sind die Ergebnisse mit der Histon-Acetylierung von besonderem Interesse. So konnten mehrere Mutationen identifiziert werden, die die Histon-Acetylierung beeinflussen, und den Phänotyp im SCA-Drosophila-Modell wie auch im Huntington-Modell modifizieren[15,16]. Interessanterweise konnte in dieser Studie auch durch Füttern eines Inhibitors eines der Enzyme (SAHA; superoylanilide hydroxamic acid als Inhibitor der HistonAcetyltransferase) die durch diese Konstrukte induzierte Toxizität reduzieren. Ähnliche Effekte wurden auch in der Maus beschrieben[17]. SAHA und andere HistonDeacetylasen-Inhibitoren werden gegenwärtig in verschiedenen klinischen Tests untersucht. Damit stellt Drosophila auch hier ein sehr interessantes Modell dar, mit welchem einerseits grundlegende biologische Vorgänge in solchen Erkrankungen am Modell untersucht werden können, andererseits aber auch wie im Falle der Histon-Acetyl-Transferasen-Inhibitoren neue interessante therapeutische Ansätze identifiziert werden können. Literatur [1] Adams, M. D., Celniker, S. E., Holt, R. A., Evans, C. A., Gocayne, J. D., et al. (2000): The genome sequence of Drosophila melanogaster. Science 287: 2185–2195. [2] Reiter, L. T., Potocki, L., Chien, S., Gribskov, M., and Bier, E. (2001): A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res 11: 1114–1125. [3] Botella, J. A., Kretzschmar, D., Kiermayer, C., Feldmann, P., Hughes, D.A., Schneuwly, S. (2003) Deregulation of the Egfr/Ras Signaling pathway induces age-related brain degeneration in the Drosophila mutant vap. Mol. Biol. Cell 14:241–250 [4] Min, K.T., and Benzer, S. (1999): Preventing neurodegeneration in the Drosophila mutant bubblegum. Science 284: 1985–1988. [5] Brand, A.H., and Perrimon, N. (1993): Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118: 401–415. [6] Gunawardena, S., and Goldstein, L.S.B. (2001): Disruption of axonal transport and neuronal viability by amyloid precursor protein mutation in Drosophila. Neuron 32: 389–401. [7] Fossgreen, A., Bruckner, B., Czech, C., Masters, C.L., Beyreuther, K., et al. (1998): Transgenic BIOspektrum · 5/04 · 10. Jahrgang Überblick Drosophila expressing human amyloid precursor protein show gamma-secretase activity and a blistered-wing phenotype. Proc Natl Acad Sci U S A. 95:13703–13708. [14] Kazemi-Esfarjamni, P., and Benzer, S. (2000): Genetic suppression of polyglutamine toxicity in Drosophila. Science 287:137–1840. [8] Wittmann, C. W., Wszolek, M. F., Shulman, J. M., Salvaterra, P. M., Lewis, J., et al. (2001): [15] Steffan, J. S., Kazantsev, A., Spasic- Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293: 711–714. (2000): The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA. 97:6763–6768. [9] Feany, M. B., and Bender, W. W. (2000): A Drosophila model of Parkinson´s disease. Nature 404: 394–398. [10] Auluck, P. K., Chan, H. Y. E., Trojanowski, J. Q., Lee, V. M. Y., and Bonini, N. M. (2001): Chaperone suppression of a-synuclein toxicity in a Drosophila model for Parkinson´s disease. Science 295: 865–868. [11] Warrick, J. M., Paulson, H. L., Gray-Board, G. L., Bui, Q. T., Fischbeck, K. H., et al. (1998): Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell 93:939–49. [12] Jackson, G. R., Salecker, I., Dong, X., Yao, X., Arnheim, N., et al., (1998): Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron 21:633–42. [13] Fernandez-Funez, P., Nino-Rosales, M. L., de Gouyon, B., She, W. C., Luchak, J. M., et al. (2000): Identification of genes that modify ataxin-1–induced neurodegeneration. Nature 408: 101–106. José A. Botella Jahrgang 1966; Studium der Biologie und Pro- Stephan Schneuwly Jahrgang 1959, Studium der Biologie am Biozentrum der Universität Basel, Promotion 1986 bei motion an der University of Murcia in Spanien. 1996–97 Postdoc an der University of Kent, UK, 1997–2001 Postdoc und seit 2001 wissenschaftlicher Assistent am Institut für Zoologie Universität Regensburg. Prof. Dr. W. Gehring, Biozentrum der Universität Basel. 1986–1989 Postdoc am Dep. of Biological Sciences, Purdue University. 1990–94 Nachwuchsgruppenleiter einer BMBF-Nachwuchsgruppe bei Prof. Dr. M. Heisenberg am Lehrstuhl für Genetik, Universität Würzburg. Seit 1995 Professor für Zoologie an der Universität Regensburg. BIOspektrum · 5/04 · 10. Jahrgang Boskovic, O., Greenwald, M., Zhu, Y.Z., et al. [16] Steffan, J. S., Bodai, L., Pallos, J., Poelman, M., McCampbell, A. (2001): Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 413: 739–743. [17] Hockly. E., Richon, V. M., Woodman, B., Smith, D. L., Zhou, X. (2003): Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 100:2041–2046. Korrespondenzadresse: Prof. Dr. Stephan Schneuwly Lehrstuhl für Biologie VI Institut für Zoologie Universitätsstr. 31 Universität Regensburg D-93040 Regensburg [email protected]