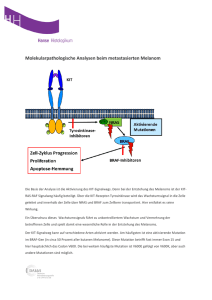
Molekulargenetische Klassifikation und klinische Charakterisierung
Werbung

Aus der Klinik für Innere Medizin / Abteilung IV (Schwerpunkt: Nephrologie und Allgemeinmedizin) der Albert-Ludwigs-Universität Freiburg i. Br. Molekulargenetische Klassifikation und klinische Charakterisierung der Paragangliom-Syndrome INAUGURAL – DISSERTATION zur Erlangung des Medizinischen Doktorgrades der Medizinischen Fakultät der Albert-Ludwigs-Universität Freiburg i. Br. Vorgelegt 2005 von Birke Bausch geboren in Karlsruhe Gewidmet meiner Familie, Hedera und Bernd Bausch und Traudl Koch. Dekan: Prof. Dr. med. J. Zentner 1. Gutachter Prof. Dr. med. H.P.H. Neumann 2. Gutachter PD. Dr. med. J. Kohlhase Promotionsjahr: 2005 Inhalt 1. Einleitung 1 1.1. Phäochromozytome, Paragangliome und Glomustumoren 3 1.1.1.Terminologie, klinische Diagnostik und Therapie 3 1.1.2.Phäochromozytom-assoziierte Syndrome 9 1.2. Die Succinatdehydrogenase 12 1.2.1.Das SDHB-Gen 13 1.2.2.Das SDHC-Gen 15 1.2.3.Das SDHD-Gen 17 1.3. Mutationen und Polymorphismen 17 1.4. Fragestellungen 19 2. Material und Methoden 20 2.1. Patientengut 20 2.2. Methoden 22 2.2.1.DNA-Extraktion 22 2.2.2.Polymerasekettenreaktion (PCR) 23 2.2.3.Agarosegel-Elektrophorese 27 2.2.4.Single Strand Conformation Polymorphism (SSCP) 27 2.2.5.Sequenzierung 30 2.2.6.Programm des klinischen Screenings 31 2.2.7.Statistik 32 3. Ergebnisse 33 3.1. Patientenkollektiv 33 3.2. Konstitutionelle Mutationen des SDHB-Gens 34 3.2.1.Molekulargenetisches Screening auf konstitutionelle Mutationen des SDHB-Gens 34 3.2.2.Molekulargenetische Testung von Verwandten 40 3.2.3.Klinische Charakterisierung der Träger von Mutationen des SDHB-Gens 44 3.2.4.Penetranz 49 3.3. Konstitutionelle Mutationen des SDHC-Gens 51 3.3.1.Molekulargenetisches Screening auf konstitutionelle Mutationen des SDHC-Gens 51 3.3.2.Klinische Charakterisierung der Träger von Mutationen des SDHC-Gens 53 3.4. Konstitutionelle Mutationen des SDHD-Gens 54 4. Diskussion 58 5. Zusammenfassung 73 6. Literaturverzeichnis 74 7. Danksagung 87 8. Lebenslauf 88 Teile dieser Arbeit wurden in 2 Publikationen veröffentlicht: Neumann HP, Bausch B, Mc Whinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K, Januszewicz A, Smith WM, Munk R, Manz T, Glaesker S, Apel TW, Treier M, Reincke M, Walz MK, Hoang-Vu C, Brauckhoff M, Klein-Franke A, Klose P, Schmidt H, Maier-Woelfle M, Peczkowska M, Szmigielski C, Eng C; The FreiburgWarsaw-Columbus Pheochromocytoma Study Group. (2002) Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 346:1459-66 Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, Buchta M, Franke G, Klisch J, Bley T, Hoegerle S, Boedeker C, Opocher G, Schipper J, Januszewicz A, Eng C. (2004) Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 292: 943-951 Abkürzungsverzeichnis A Adenin bp Basenpaare C Cytosin DNA Desoxyribonucleinsäure EDTA Ethylendiamintetraacetat FAD Flavin-Adenin-Dinucleotid G Guanin GT Glomustumor LOH Loss of Heterozygosity nt Nucleotid NTP Nucleosidtriphosphat PCR Polymerasekettenreaktion PGL 1- PGL 4 Paragangliom-Syndrom Typ 1-4 PGL-Syndrom Paragangliom-Syndrom Pheo Phäochromozytom SDH Succinatdehydrogenase SDHA, SDHB, SDHC, SDHD Succinatdehydrogenase Untereinheiten A-D SSCP Single strand Conformation Polymorphism T Thymin TBE Tris-Borsäure-EDTA 1 1. Einleitung Die Succinatdehydrogenase (SDH) ist ein Schlüsselenzym des Zitratzyklus (Krebszyklus) und der oxidativen Phosphorylierung (Atmungskette). Sie besteht aus 4 Proteinen, den Untereinheiten A-D. 4 Gene, das SDHA-, das SDHB-, das SDHC- und das SDHD-Gen, kodieren diese Untereinheiten. Sie wurden in der Dekade 1990-2000 identifiziert (Morris et al. 1994, Au et al. 1995, Elbehti-Green et al. 1998, Hirawake et al. 1997). 2000 berichteten B. Baysal und Mitarbeiter über eine Kopplung der Vererbung von Paragangliomen des Halses mit Markern auf Chromosom 11 Bandenregion 23 des kurzen Arms (11q23), dem chromosomalen Bereich des SDHD-Gens. Sie identifizierten in mehreren Paragangliomfamilien konstitutionelle Mutationen des SDHD-Gens, d.h. Keimbahnmutationen, die sich in Lymphozyten des peripheren Blutes nachweisen lassen (Baysal et al. 2000). Das Paragangliom-Syndrom Typ 1, PGL 1, war definiert. Ebenfalls 2000 entdeckte die Gruppe um U. Müller in Gießen eine konstitutionelle Mutation des SDHC-Gens bei einer Familie mit Paragangliomen des Halses (Niemann und Müller 2000). Damit war das Paragangliom-Syndrom Typ 3, PGL 3, definiert. 2001 beschrieb die Gruppe des englischen Genetikers und Molekularbiologen Eammon Maher erstmals konstitutionelle Mutationen des SDHB-Gens bei Patienten mit familiären Phäochromozytomen und Paragangliomen des Halses (Astuti et al. 2001). Das ParagangliomSyndrom Typ 4, PGL 4, war definiert. Die Freiburger Arbeitsgruppe von Herrn Prof. Neumann stellte sich die Frage, wie viele Patienten eines Registers als Träger einer Anlage, die zu dieser neuen Gruppe hereditärer Phäochromozytome und Glomustumoren prädisponiert, klassifiziert werden können. Die Bearbeitung dieser Frage erschien auf Grund des großen Patientengutes und den molekulargenetischen Vorraussetzungen des Labors erfolgversprechend. Kooperationen bestanden mit dem Team von Frau Prof. Eng, Ohio State University, Columbus, Ohio, USA -Schwerpunkt Cancer Genetics- und dem Team von Herrn Prof. Januszewicz, Institute of Cardiology, Warschau, Polen -Schwerpunkt klinische Diagnostik des Phäochromozytoms. Innerhalb des Freiburger Labors erfolgte eine personelle Aufteilung der Analysen des SDHB-, des SDHC-, und des SDHD-Gens. Die molekulargenetische Untersuchung des SDHB-Gens und des SDHC-Gens wurde schwerpunktmäßig von der Verfasserin im Rahmen dieser Arbeit bis 2004 durchgeführt. Die molekulargenetische Untersuchung des SDHD-Gens wurde in einer Dissertation, für die bis zum 1.12.2001 in 2 Freiburg registrierten Patienten, von Herrn Dr. Robin Munk bearbeitet. Die weitere Analyse des SDHD-Gens erfolgte im Rahmen dieser Arbeit. 3 1. 1. Phäochromozytome, Paragangliome und Glomustumoren 1.1.1. Terminologie, klinische Diagnostik und Therapie Der Sprachgebrauch der Begriffe Phäochromozytom, Paragangliom und Glomustumor ist uneinheitlich, teils vom klinischen Alltag und teils vom Bemühen um wissenschaftliche Korrektheit bestimmt. Der Terminus Paragangliom dient als Oberbegriff, der Phäochromozytome und Tumoren des extra-adrenalen paraganglionären Systems zusammenfasst (Abbildung 1) (Glenner und Grimley, AFIP, 1974). Es handelt sich um eine Gruppe unterschiedlicher und doch miteinander verwandter Neoplasien (Glenner und Grimley, AFIP, 1974 ). Ihre gemeinsame Basis bildet ihre Zugehörigkeit zum autonomen Nervensystem, ihre Abstammung von Neuralleistenzellen und ihre Fähigkeit Katecholamine zu speichern (Böck und Lassmann 1972, Chiocchio et al. 1971, Manger und Gifford 1995 und 2002). Sie unterscheiden sich jedoch durch ihre Funktion und Innervation (sympathisch oder parasympathisch). Phäochromozytome im engeren Sinne sind Tumoren des Nebennierenmarkes. Da sich ihre Zellen mit Dichromatsalzen anfärben lassen, werden sie auch als chromaffine Tumoren bezeichnet. Sie gehören zum autonomen sympathischen Nervensystem, speichern und sezernieren Katecholamine. Phäochromozytome zeigen Symptome einer dauerhaften oder intermittierenden Katecholaminintoxikation. Dies hat im klinischen Alltag zur Verwendung des Terminus Phäochromozytom als Überbegriff für alle paraganglionären Tumoren geführt, die Phäochromozytom-ähnliche Symptome zeigen. Um die Lokalisation des Tumors einzugrenzen, werden Zusätze wie extra-adrenal für die in der Regel abdominellen und thorakal für die seltenen Paragangliome des Brustkorbbereiches gebraucht. Neben extra-adrenalen paraganglionären Tumoren mit Phäochromozytom-ähnlicher Symptomatik und Histologie umfasst der Oberbegriff Paragangliom auch die selten symptomatisch werdenden Paragangliome des Halses und der Schädelbasis. Es handelt sich hierbei meist um Tumoren des autonomen parasympathischen Nervensystems. Die Benennung nach ihrem Sitz, zum Beispiel Glomus caroticum Tumor, hat zur Verwendung des Begriffs Glomustumor geführt. Im wissenschaftlich korrektem Sinne sind Glomustumoren Hauttumoren, die von glatten Muskelzellen abstammen (Hollingshead 1942). Mit Tumoren des paraganglionären Systems haben sie nichts gemein. Die Paraganglien von Hals und Schädelbasis haben die Funktion von Chemorezeptoren, sprechen auf pH-Wert- 4 Verschiebungen an und regulieren den Blutdruck. Dies führte zur Verwendung des Begriffs Chemodektom für Neoplasien dieser Paraganglien (Mulligan 1950). Im Gegensatz zu Phäochromozytomen und extra-adrenal abdominell oder thorakal gelegenen Paragangliomen lassen sich Glomustumoren nur sehr schwach oder gar nicht mit Dichromatsalzen anfärben (Watzka 1943). Somit unterscheidet man chromaffine von non-chromaffinen Paragangliomen. Abbildung 1: Sympathisches und Parasympathisches Nervensystem und Tumorprädilektionsstellen C Sympathische Nervenbahnen Extra-adrenale Phäochromozytome Parasympathische Nervenbahnen Abbildung A: Darstellung des Truncus sympathicus und seiner Ganglien; sie bilden die Prädilektionsstellen extraadrenaler Paragangliome, bzw. Phäochromozytome. Abbildung B: Darstellung der Lokalisationsmöglichkeiten extra-adrenaler, abdomineller und thorakaler Phäochromozytome bzw. Paragangliome im Verlauf des Truncus sympathicus. Abbildung C: Darstellung des zervikalen parasympathischen Nervensystems und seiner Ganglien anhand des Verlaufs entsprechender Hirnnerven. Diese Ganglien sind Prädilektionsort der meisten Glomustumoren. Aus: Glenner und Grimley, Tumors of the extra-adrenal paraganglion system (including chemoreceptors). Atlas of Tumor Pathology, 2nd Series, Fascicle 9. AFIP, 1974 Phäochromozytome treten meist im frühen Erwachsenenalter in Erscheinung und verursachen zahlreiche Symptome. Klinisch im Vordergrund stehen Kopfschmerzen, Herzklopfen (Palpitationen) und Schweißattacken. Durch phasenweise oder dauerhafte Sekretion erhöhter Mengen von Katecholaminen, entsteht eine intermittierende oder dauerhafte Hypertonie. Im Verlauf kann es zu hypertensiven Krisen mit akuter Linksherzinsuffizienz (Katecholamin- 5 Myokardiopathie), schweren Herzrhythmusstörungen und Insulten begleitet von permanenten neurologische Ausfällen kommen (Manger und Gifford 1995 und 2002). Die klinische Diagnostik beruht auf zwei Säulen: 1. dem Nachweis einer Raumforderung und 2. dem Nachweis einer erhöhten Katecholaminproduktion, -zirkulation und -ausscheidung. Abbildung 2 und 3 zeigen typische Befunde eines adrenalen Phäochromozytoms und eines Glomustumors des Glomus caroticum. Abbildung 2: Darstellung eines Phäochromozytoms mittels verschiedener radiologischer Verfahren A A: MRT eines Phäochromozytoms des rechten Nebennierenmarkes. Der Pfeil markiert den Tumor. B B: MIBG- Szintigraphie eines Phäochromozytoms des rechten Nebennierenmarkes. Der Pfeil markiert den Tumor C C: 18Fluor-DOPA-PET eines Phäochromozytoms des linken Nebennierenmarkes. Der Pfeil markiert den Tumor. Zusätzliche Akkumulation des Radiopharmakons im Nierenbecken, der Harnblase und dem Pankreas. 6 Abbildung 3: Darstellung eines Glomustumors mittels MRT und MR-Angiographie A B A: MRT eines Tumors des linken Glomus caroticum. B: MR-Angiographie eines Tumors des linken Glomus caroticum. Zu den bildgebenden Verfahren, die zur Darstellung eines Phäochromozytoms angewendet werden, gehören die Sonographie Kernspintomographie (MRT), die Jod (MIBG) und seit kurzem auch die 18 123 (US), die Computertomographie (CT), die bzw. Jod131- Metajodobenzylguanidinszintigraphie Fluor-DOPA-Positronenemissionstomographie (18Fluor- DOPA-PET). Die bislang einzige in diesem Zusammenhang durchgeführte prospektive Studie ermittelte die in Tabelle 1 wiedergegebenen Sensitivitäten der verschiedenen Verfahren (Neumann et al. 1993). Sensitivität in % Sonographie (US) 40% CT 76% MRT 95% MIBG 95% 18 Fluor-DOPA-PET* 100% Tabelle 1: Sensitivitäten für die sonographische, computertomographische, kernspinntomographische und szintigraphische Darstellung von Phäochromozytomen (Neumann et al. 1993, *Högerle et al. 2002) 7 Eine weitere im Jahr 2002 durchgeführte Studie, die die emissinstomographie 18 ( Fluor-DOPA Metajodobenzylguanidinszinitigraphie PET) (MIBG) mit 18 Jod123 der vergleicht, Fluor-DOPA Positronen- ermittelte bzw. ein Jod131- verbesserte Abbildungsqualität, ein verbessertes Auflösungsvermögen und eine Sensitivität von 100% für die 18 Fluor-DOPA-PET Untersuchung (Högerle et al. 2002). Für den Nachweis von Glomustumoren hat die DOPA-PET ebenfalls eine hohe Sensitivität und ist der Sonographie des Halses überlegen (Högerle et al. 2003). Die Bestimmung der Katecholamine Noradrenalin und Adrenalin sowie die Bestimmung des Katecholaminmetaboliten Vanillinmandelsäure im angesäuerten 24h Sammelurin gehören zur Standarddiagnostik. Noradrenalin und Adrenalin können auch im Plasma bestimmt werden. Die Sensitivität dieser Parameter ist in Tabelle 2 wiedergegeben (Neumann et al. 1993). Sensitivität in % Noradrenalin im Urin 86% Adrenalin im Urin 53% Vanillinmandelsäure im Urin 64% Noradrenalin im Plasma 58% Adrenalin im Plasma 33% Tabelle 2: Sensitivitäten für die Bestimmung von Noradrenalin, Adrenalin und Vanillinmandelsäure im Urin und im Plasma (Neumann et al. 1993). Metanephrine werden in Deutschland selten bestimmt. Studien aus USA zeigen eine Sensitivität von 99 % für die Bestimmung von Plasma-Metanephrinen und eine Sensitivität von 97 % für deren Messung im Urin (Eisenhofer et al. 1999, Lenders et al. 2002). Die Therapie des Phäochromozytoms ist die Resektion des Tumors nach entsprechender Vorbehandlung mit Alpha- und Betablockern zur Blockade der Katecholaminwirkung. Vier verschiedene Verfahren finden Anwendung: • Die klassische offene Operation mit Entfernung des Tumors mit kompletter Adrenalektomie • Die offene, organerhaltende Tumorresektion (Neumann et al. 1999) • Die lapraskopische Tumorresektion mit Adrenalektomie (Gagner et al. 1992) • Die lapraskopische, organerhaltende Tumorresektion (Janetschek et al. 1998, Walz et al. 2002) 8 Als Methode der Wahl wird heute die endoskopische, organerhaltende Resektion des Tumors angesehen, vor allem bei hereditären und bilateralen Phäochromozytomen. Besonders die Auswertung der Daten des Freiburger Projekts mit vielen hereditären Phäochromozytomen haben zu diesem Konzept geführt, das erstmals in Kooperation mit Prof. Janetschek, Innsbruck, umgesetzt wurde (Janetschek et al. 1998). Deutlich werden die Vorzüge dieser Operationsmethode nach Durchführung eines ACTH-Stimulationstests, der den Erhalt von funktionierendem Nebenierenrindengewebe nachweist (Neumann et al. 1999). Bedeutung hat dies vor allem für Patienten mit bilateralen Phäochromozytomen oder mit einer bereits entfernten Nebenniere. Maligne Phäochromozytome sind selten; es wird ein Anteil von 10 % (Manger und Gifford 1995) von den ohnehin schon seltenen Phäochromozytomen angenommen. Die Definition dieses Tumors als maligne oder benigne ist uneinheitlich. Einen eindeutigen Beweis stellen bisher nur Fernmetastasen dar, die bevorzugt in Lunge, Leber und im Skelettsystem lokalisiert sind. Infiltrationen in das umgebende Fettgewebe, obwohl von Manger und Gifford als Malignitätskriterium beschrieben (Manger und Gifford 1995), sind schwierig zu bewerten. Der Verlauf maligner Phäochromozytome ist meist langsam progredient. Die Patienten versterben nicht selten an interkurrenten Erkrankungen. Die Therapie maligner Phäochromozytome ist schwierig. Methode der Wahl ist die Tumorresektion. Alphablockern Symptome behandelt. der Auch Katecholaminüberproduktion eine Behandlung mit werden 123 Jod - palliativ bzw. mit Jod131- Metajodobenzylguanidin (MIBG) wird empfohlen, ist jedoch von sehr unterschiedlichem Erfolg. Eine weitere Therapiemöglichkeit besteht in einer Chemotherapie nach dem Averbuch-Schema: Cyclophosphamid, Vincristin, Dacarbacin (Averbuch et al. 1988). Auch hier ist der Erfolg schwer vorhersehbar und bleibt nicht selten aus. 9 1.1.2. Phäochromozytom-assoziierte Syndrome Hereditäre Phäochromozytome, Paragangliome und Glomustumoren sind Tumoren, die auf dem Boden konstitutioneller Mutationen (Keimbahnmutation) eines Gens entstehen. Meist treten diese hereditären Tumoren familiär gehäuft auf, da die jeweiligen Mutation an Nachkommen weitergegeben wird. Die Relevanz der Mutation für die Tumorgenese lässt sich anhand zweier Beobachtungen nachweisen: Zum einen durch die Cosegregation (Mitvererbung) von mutiertem Gen und Erkrankung in Familienstudien, zum anderen durch Veränderungen des korrespondierenden Wildtypallels (des gesunden Allels) im Tumorgewebe. Deletionen des Wildtypallels im Tumorgewebe wurden für das VHL-Gen, einem Tumorsuppressor-Gen, beschrieben (Bender et al. 2000). Duplikationen des mutierten Allels konnten für das RET-Gen, einem Protoonko-Gen, gezeigt werden (Huang et al. 2000). Hereditäre Phäochromozytome treten meist in Zusammenhang mit Tumorsyndromen auf, wie der von Hippel-Lindau Erkrankung (VHL), der Multiplen Endokrinen Neoplasie Typ 2 (MEN 2), der Neurofibromatose von Recklinghausen (periphere Neurofibromatose, Neurofibromatose Typ 1) sowie dem Paragangliom-Syndrom und der Carney-Triade. Die bekannteste Form ist die MEN 2 (Abbildung 4 und 5). Dominierende Läsion ist das medulläre Schilddrüsenkarzinom. Der Typ 2A ist zu 20% mit einem Hyperparathyreodismus vergesellschaftet, während der Typ 2B durch einen marfanoiden Habitus und Ganglioneuromatosen des Mundes, der Lippen, der Konjunktiven sowie des Darmes geprägt ist (Schimke 1990). Die MEN 2 entsteht auf dem Boden konstitutioneller Mutationen des 21 Exon umfassenden RET-Gens. Mutationen wurden in den Exons 10, 11 und 13-16 gefunden (Eng et al. 1996). Abbildung 4: 44 jährige Patientin mit MEN Typ 2. Darstellung eines Resektionspräparates eines medullären Schilddrüsenkarzinoms; Tumoren werden mittels weißer Pfeile markiert, Gewebebrücken mit Pfeilspitzen. 10 Abbildung 5: 44 jährige Patientin mit MEN Typ 2. CT eines bilateralen adrenalen Phäochromozytoms, markiert durch schwarze Pfeile. Es handelt sich um die Patientin deren Resektionspräparat eines medullären Schilddrüsenkarzinoms in Abbildung 4 dargestellt ist. Häufiger als bei der MEN 2 - Erkrankung kommen hereditäre Phäochromozytome beim von Hippel-Lindau Syndrom (VHL) vor. Es ist gekennzeichnet durch das Auftreten von Hämangioblastomen oder Angiomen der Retina, des Kleinhirns, der Medulla oblongata und dem Rückenmark sowie durch das Auftreten von Nierenkarzinomen und Pankreaszysten (Neumann 1987). Das Mutationsspektrum des VHL-Gens zeigt alle denkbaren Varianten bis hin zu größeren Deletionen (Zbar et al. 1996). VHL-assoziierte Phäochromozytome treten jedoch nahezu ausschließlich in Zusammenhang mit Missense oder Stopcodon Mutationen auf (Neumann et al. 2002). Eines der häufigsten Tumorsyndrome ist die Neurofibromatose von Recklinghausen. Das klinische Erscheinungsbild wird von Café-au-lait Flecken und Neurofibromen peripherer Nerven bestimmt (Riccardi 1981). Phäochromozytome entwickeln sich nur selten (Inzidenz 1%). Man nimmt an, das Veränderungen des NF1-Gens dieser Erkrankung zugrunde liegen. Ein weiteres sehr seltenes Syndrom, das durch das Auftreten von Glomustumoren des Halses und der Schädelbasis sowie Magensarkomen und Lungenchondromen gekennzeichnet ist, wird als Carney-Triade bezeichnet (Carney 1999). Die molekulargenetische Grundlage dieser Erkrankung ist noch unbekannt. Neu und Gegenstand dieser Arbeit sind die bisher nur wenig bekannten ParagangliomSyndrome (PGL). Typische Läsionen sind Glomustumoren des Halses und der Schädelbasis (Abbildung 6) sowie adrenale Phäochromozytome und abdominelle oder thorakale Paragangliome. Es werden 4 Entitäten unterschieden. Das PGL 1-Syndrom ist gekennzeichnet durch familiäre Glomustumoren, Phäochromozytome und abdominelle und thorakale 11 Paragangliome. Ursächlich liegen Mutationen des Gens der Succinatdehydrogenase Untereinheit D (SDHD-Gen) zugrunde (Astuti et al. 2001, Baysal et al. 2000, Gimm et al. 2000). Das PGL 2-Syndrom ist gekennzeichnet durch das Auftreten familiärer Glomustumoren. Ein bisher noch nicht identifiziertes, auf Chromosom 11q13 lokalisiertes Gen, steht in Zusammenhang mit der Entwicklung dieser Neoplasien (Mariman et al. 1995). Kürzlich wurde das PGL 3-Syndrom, gekennzeichnet durch familiäre Glomustumoren, beschrieben (Niemann et al. 1999). Mutationen des Gens der Succinatdehydrogenase Untereinheit C (SDHC-Gen) liegen dieser Erkrankung zugrunde (Niemann und Müller 2000, Niemann et al. 2003). Jüngste Entität ist das PGL 4-Syndrom. Es beruht auf Veränderungen des Gens der Succinatdehydrogenase Untereinheit B (SDHB-Gen) (Astuti et al. 2001). A B Abbildung 6: Darstellung von bilateralen Glomus caroticum Tumoren mittels verschiedener Techniken. A: MRT der Schädelbasis, B: 18Fluor-DOPA-PET. Die Pfeile kennzeichnen die Tumoren. Alle anderen Phäochromozytome werden als sporadisch bezeichnet. Die oft zitierte 10er Regel (10% extraadrenal, 10% bilateral, 10% maligne, 10% hereditär) wird auch für familiäre Phäochromozytome tradiert (Manger und Gifford 1995). Die Ergebnisse unter anderem dieser Arbeit zeigen, dass diese "Regel" nicht mehr gültig ist. 12 1.2. Die Succinatdehydrogenase Die Succinatdehydrogenase (SDH) ist ein Enzym, dem sowohl im Krebszyklus als auch in der Atmungskette wesentliche Bedeutung zu kommt (Saraste 1999). Im Zitratzyklus katalysiert es die Oxidation von Succinat zu Fumarat unter Übertragung der freiwerdenden Elektronen auf Flavin-Adenin Dinukleotide (FAD → FADH2). Als Komplex II der Atmungskette transferriert die Succinatdehydrogenase die an Flavin-Adenin-Dinukleotide gebundenen Elektronen auf Ubiquinon. Abbildung 7 stellt die Doppelfunktion der Succinatdehydrogenase im Zitratzyklus und in der Atmungskette dar. Abbildung 7: Darstellung der 4 Komplexe der Atmungskette mit ihrer Lokalisation in der inneren Mitochondrienmembran. Hauptschwerpunkt in diesem Zusammenhang liegt auf Komplex II der Succinatdehydrogenase, der als integraler Membrankomplex sowohl wichtige Aufgaben im Krebszyklus als auch in der Atmungskette zu kommen. Übernommen aus der Homepage des Natural Toxine Centers. Die Succinatdehydrogenase setzt sich aus vier verschiedenen Proteinuntereinheiten (A-D) zusammen (Scheffler 1998). Ein Flavoprotein (SDHA, 70 kD) und ein Eisensulfurprotein (SDHB, 27 kD) bilden zusammen die katalytische Einheit. Zwei Cytochrom-b-ähnliche lipophile Proteine, das größere cbL (SDHC, 15 kD) und das kleiner cybS (SDHD, 12 kD) verankern die katalytische Einheit in der inneren Mitochondrienmembran. Die vier Gene, SDHA, SDHB, SDHC, SDHD, die die vier Proteinuntereinheiten kodieren, sind in der KernDNA lokalisiert. Das SDHA-Gen befindet sich auf Chromosom 5p15, das SDHB-Gen auf Chromosom 1p36, das SDHC-Gen auf Chromosom 1q21-23 und das SDHD-Gen auf Chromosom 11q23 (Morris et al. 1994, Au et al. 1995, , Elbehti-Green et al. 1998, Leckschat et al. 1993, Hirawake et al. 1997). 13 1.2.1. Das SDHB-Gen Das Gen der Succinatdehydrogenase Untereinheit B (SDHB-Gen) befindet sich auf Chromosom 1p35-36.1 (Leckschat et al. 1993). Die Sequenz des SDHB-Gens ist hochkonserviert und besteht aus 8 Exons und 7 Introns (Au et al. 1995). Tabelle 3 gibt die Größe der 8 Exons sowie deren Nukleotidbereich in der cDNA wieder. Exon Größe in bp Nukleotid 1 205 2-206 2 128 207-334 3 86 335-420 4 137 421-557 5 117 558-674 6 102 675-776 7 123 777-899 8 78 900-977 Tabelle 3: Zusammenstellung der 8 Exons des SDHB-Gens, der Exongröße in Basenpaaren (bp) und des Nukleotidbereiches in der cDNA. Die cDNA wird aus einer für die Aminosäuren des Proteins kodierenden Sequenz von 840 bp gebildet. Flankiert wird sie durch eine 133 bp große untranslatierte Region (UTR) am 5' Ende, die zu Exon 1 gehört und einer 123 bp großen untranslatierten Region (UTR) am 3' Ende, die einen Teil von Exon 8 ausmacht (Au et al. 1995). Die untranslatierte Region von 133 bp des Exon 1 ist Cytosin- und Guanin-reich und zeichnet sich durch die Bildung einer ausgeprägten Sekundärstruktur aus. Die Funktion ist jedoch nicht bekannt. Die Promotorregion des SDHBGens enthält zahlreiche Bindungsstellen für die Transkriptionsfaktoren NRF 1 und 2 (nuclear respiratory factor) sowie Sp 1 (Au et al. 1995). Sie regulieren die Expression des Gens und sind typisch für Gene, die für Enzyme der Atmungskette kodieren (Scarpulla 2002). Das SDHB-Gen kodiert ein 27 kD großes Eisensulfurprotein (Ip), das zusammen mit dem Flavoprotein (Fp), kodiert durch das SDHA-Gen (siehe Kapitel 1.2.), die katalytische Einheit der Succinatdehydrogenase (SDH) bildet. Die Struktur des Ip-Proteins zeigt als Besonderheiten drei gebundene Eisensulfurcluster S1 (2Fe-2S), S2 (4Fe-4S) und S3 (3Fe-4S), drei hochkonservierte Cystein-reiche Regionen und die Bindung instabiler Sulfide (Au et al. 1995). Es wird angenommen, dass das Eisensulfurprotein Ip als Brücke zwischen dem 14 Flavoprotein Fp und den beiden Cytochrom-b ähnlichen integralen Membranproteinen cybS (SDHD) und cybL (SDHC) liegt (Au et al. 1995). Abbildung 8 zeigt die cDNA des SDHB-Gens sowie die Aminosäuresequenz des Proteins. 1 ggcctcccac ttggttgctc gtacgcggct agtgggtcct cagtggatgt aggctgggcg 61 ccgcgatgtt cgacgggaca ccggcggaga gcgacctcgg ggttaagggg tggggctgga 121 cgtcaggagc caagatggcg M A 181 caacccttgg cggagcctgc T L G G A C 241 ctccccgtat caagaaattt P R I K K F 301 ctcatatgca gacttatgaa H M Q T Y K 361 taatcaagat taagaatgaa I K I K N E 421 gcatctgtgg ctcttgtgca I C G S C A 481 ggattgacac caacctcaat I D T N L N 541 taaaggatct tgttcccgat K D L V P D 601 atttgaagaa gaaggatgaa L K K K D E 661 agcgtgagaa actggacggg R E K L D G 721 gccccagcta ctggtggaac P S Y W W N 781 atcgctggat gattgactcc R W M I D S 841 acccattctc tctataccgc P F S L Y R 901 gtctgaatcc agggaaagct L N P G K A 961 agaaagcttc agtttaactg K A S V *** 1021 ataatttata tctaatttga gcggtggtcg A V V A ctgcaggcct L Q A S gccatctatc A I Y R gttgacctta V D L N gttgactcta V D S T atgaacatca M N I N aaggtctcaa K V S K ttgagcaact L S N F tctcaggaag S Q E G ctctacgagt L Y E C ggagacaaat G D K Y agagatgact R D D F tgccacacca C H T I attgcagaga I A E I tccatgctaa cactctcctt L S L cccgaggagc R G A gatgggaccc W D P ataaatgtgg K C G ctttgacctt L T F atggaggcaa G G N aaatctaccc I Y P tctatgcaca Y A Q gcaagcagca K Q Q gcattctctg I L C atctggggcc L G P tcacagagga T E E tcatgaactg M N C tcaagaaaat K K M acatgattta gaggcgccgg R R R ccagacagct Q T A agacaaggct D K A ccccatggta P M V ccgaagatca R R S cactctagct T L A tcttccacac L P H gtacaaatcc Y K S gtatctgcag Y L Q tgcctgctgt A C C tgcagttctt A V L gcgcctggcc R L A cacaaggacc T R T gatggcaacc M A T taaccagctc ttgccggcca L P A T gcagccacag A A T A ggagacaaac G D K P ttggatgctt L D A L tgcagagaag C R E G tgcacccgaa C T R R atgtatgtga M Y V I attgagcctt I E P Y tccatagaag S I E E agcaccagct S T S C atgcaggcct M Q A Y aagctgcagg K L Q D tgtcctaagg C P K G tataaggaga Y K E K agagctgaac gttcctttaa agatcttggt tttccatgaa tacagcatgt 1081 ataataaaaa ttttaagaaa taaatgttat tctactttat taacaaaaaa Abbildung 8: Darstellung der cDNA des SDHB-Gens. Die 8 verschiedenen Exons sind durch unterschiedliche Farben markiert. Rot: Exon 1, 205 bp groß. Orange: Exon 2, 128 bp groß. Gelb: Exon 3, 86 bp groß. Grün: Exon 4, 137 bp groß. Mintgrün: Exon 5, 117 bp groß. Blau: Exon 6, 102 bp groß. Hellblau: Exon 7, 123 bp groß. Rosa: Exon 8, 78 bp groß. Die nicht unterlegten fett gedruckten Großbuchstaben stellten die Aminosäuresequenz des Ip-Proteins dar (jeweils eine Aminosäure wird durch ein Basentriplet kodiert). Das Basentriplet 133-135 (atg) bildet das Startcodon. Es codiert die Aminosäure Methionin (M). Hier beginnt die Zählung der Aminosäuresequenz. Alle Basen vor dem Startcodon gehören zu Exon 1. Es handelt sich um einen 133bp großen untranslatierten Bereich (UTR), dessen Funktion noch ungeklärt ist. *** kennzeichnen das Stopcodon taa. Die cDNA ist von Au et al. 1995 beschrieben worden und unter folgenden Nummern in der GenBank sind die Einzelsequenzen der 8 Exons zu finden: U17248, U17296, U17880-U17886. 15 1.2.2. Das SDHC-Gen Das Gen der Succinatdehydrogenase Untereinheit C (SDHC-Gen) ist auf Chromosom 1q21 lokalisiert (Elbehti-Green et al. 1998). Zwei Pseudogene, die der Sequenz des aktiven SDHCGens sehr ähnlich sind und somit die Untersuchung auf konstitutionelle Mutationen stark erschweren, sind beschrieben worden (Elbehti-Green et al. 1998). Es handelt sich um ein in der Evolution nur geringfügig konserviertes Gen, das aus 6 Exons und 5 Introns besteht (Elbehti-Green et al. 1998). Tabelle 4 gibt die Größe der 6 Exons sowie deren Nukleotidbereich in der cDNA wieder. Exon Größe in bp Nukleotid 1 45 1-32 2 57 33-89 3 103 90-192 4 62 193-254 5 164 255-418 6 105 419-523 Tabelle 4: Zusammenstellung der 6 Exons des SDHC-Gens, der Exongröße in bp und des Nukleotidbereuches in der cDNA. Die cDNA, die für ein Protein von 169 Aminosäuren kodiert, wird von einer ca. 30 bp großen untranslatierten Region (UTR) am 5' Ende und einer 750 bp großen untranslatierten Region (UTR) am 3' Ende flankiert. Die untranslatierte Region des 5' Endes, die einen Großteil des Exon 1 bildet, enthält eine Bindungsstelle für den Transkriptionsfaktor NRF 2 (nuclear respiratory factor 2) (Elbehti-Green et al. 1998). Weitere Bindungsstellen für die Transkriptionsfaktoren NRF 1 und 2 sowie Sp 1 befinden sich im Bereich der Promotorregion. Ebenso wie beim SDHB-Gen wird auch beim SDHC-Gen über diese Faktoren die Expression des Gens reguliert (Scarpulla 2002). Das SDHC-Gen kodiert für ein 15 kD großes integrales Membranprotein, cybL oder CIII-3 genannt. Es bildet das größere zweier integraler Membranproteine, die die katalytische Einheit der Succinatdehydrogenase in der inneren Mitochondrienmembran verankern. Das cybL ist ebenso wie das kleinere cybS (SDHD) Protein, kodiert durch das SDHD-Gen (siehe Kapitel 1.2.) ein Cytochrom bähnliches Protein. In der Aminosäuresequenz des SDHC-Gens konnten an Position 55, 71, 127 und 134 hochkonservierte Histidine entdeckt werden. Es wird angenommen, dass diese die Bindungsstellen für die Häm-Gruppe bilden (Elbehti-Green et al. 1998). Diese Häm- 16 Gruppe bindet jedoch nicht nur am cybL- sondern auch am cybS-Protein und verbindet beide miteinander (Au et al. 1995). Abbildung 9 zeigt die cDNA des SDHC-Gens sowie die Aminosäuresequenz des Proteins. 1 ccggaaccca agatggctgc M A A 61 cactttagcc ctcagctctg H F S P Q L C 121 agagatggag cggttctgga E M E R F W N 181 tactatctac agttggtctc T I Y S W S L 241 tgctttgagt gcaggggtct A L S A G V S 301 tgagtcttat ttggaacttg E S Y L E L V 361 taagtttgca cttgtcttcc K F A L V F P 421 gtgggaccta ggaaaaggcc W D L G K G L 481 ggttcttact gtgttgtcct V L T V L S S 541 gcatcatctt cctacacatt gctgttgctg L L L tatcagaaat I R N ataagaatat K N I ttcccatggc P M A ctctttttgg L F G tgaagtccct K S L ctctcatgta L M Y tgaagattcc K I P ctatggggct M G L attacattca agacacgttg R H V G gctgttcctt A V P L aggttcaaac G S N gatgtccatc M S I catgtcggcc M S A gtgtctgggg C L G tcatacctgg H T W ccagctatac Q L Y ggcagccagt A A M cccatctttc gtcgtcattg R H C tgggaacca G T T cgtcctctgt R P L S tgccaccgtg C H R G ctgttactcc L L L P ccagcactga P A L I aatgggatcc N G I R cagtctggag Q S G V tgaagaaagg *** tgtttgtcat cctccgagcc L R A cggccaaaga A K E ctccccacat P H I gcactggtat T G I ctgggaactt G N F tccacacagc H T A gacacttgat H L M tggttgtcct V V L gaggctccca tcttatctcc 601 agcctgggaa aagttctcct tatttgttta gatccttttg tattttcaga tctcc Abbildung 9: Darstellung der cDNA des SDHC-Gens. Die sechs Exons des SDHC-Gens sind farbig gekennzeichnet. Rot: Exon 1, 45 bp groß. Orange: Exon 2, 57 bp groß. Gelb: Exon 3, 103 bp groß. Grün: Exon 4, 62 bp groß. Blau: Exon 5, 164 bp groß. Lila: Exon 6, 105 bp groß. Exon 1 ist nicht mit der vollständigen Anzahl von 45 bp dargestellt. Die nicht unterlegten, fett gedruckten Großbuchstaben beschreiben die Aminosäuresequenz des Proteins cybL (jeweils eine Aminosäure wird durch ein Basentriplet kodiert). Das Basentriplet 13-15 (atg) bildet das Startcodon. Es codiert die Aminosäure Methionin (M). Hier beginnt die Zählung der Aminosäuresequenz. Die 12 Basen vor dem Stopcodon gehören zu Exon 1. Sie beinhalten die Sequenz accggaaccc, an die NRF 2 bindet. *** kennzeichnen das Stopcodon tga. Die Sequenz ist der Arbeit von Elbehti-Green et al. 1998 entnommen. Die Einzelsequenzen der 6 Exons sind unter folgenden GenBank-Nummern registriert: AF039589, AF039590, AF039591, AF039592, AF039593, AF039594. 17 1.2.3. Das SDHD-Gen Das SDHD-Gen wurde ausführlich in der Dissertation von Herrn Dr. Robin Munk, Freiburg 2003, und einer Übersicht der Verfasserin dieser Arbeit beschrieben (Bausch et al. 2003). Seine Identifizierung erfolgte durch Hirawake und seine Mitarbeiter im Jahr 1997 (Hirawake et al. 1997). Die Relevanz von Mutationen des SDHD-Gens erkannten Baysal und Mitarbeiter 2000 (Baysal et al. 2000). Mutationen, die für Phäochromozytome prädisponieren, beschrieb erstmals eine Kooperationsarbeit aus Columbus, Ohio, und Freiburg (Gimm et al. 2000). Das SDHD-Gen ist auf Chromosom 11q23 lokalisiert (Hirawake et al. 1997). 4 Exons, Exon 1 = 52bp, Exon 2 = 117bp, Exon 3 = 145bp und Exon 4 = 163bp kodieren ein aus 159 Aminosäuren bestehendes, Cytochrom b-ähnliches Protein (Hirawake et al. 1997). 1.3. Mutationen und Polymorphismen Mutationen des SDHB-Gens, des SDHC-Gens und des SDHD-Gens sind krankheitsverursachende Veränderungen der DNA-Sequenz, die 1.) in einer veränderten Aminosäuresequenz bzw. in einem veränderten/verkürzten Protein resultieren oder einen vorzeitigen Abbau der mRNA bedingen, 2.) mit der Erkrankung cosegregieren, 3.) in der Regel nur bei Patienten nicht aber bei Kontrollpersonen vorkommen. Als Polymorphismen werden nicht pathogene Veränderungen der DNA-Sequenz bezeichnet, die 1.) häufig bei Kontrollpersonen vorkommen und eine Prävalenz von 1% aufweisen, 2.) meist, aber nicht immer, zu keiner Änderung der Aminosäuresequenz führen, 3.) nicht mit der Erkrankung cosegregieren. Die "Codesonne" (Abbildung 10) wird zur Übersetzung der kodierenden Basensequenz des SDHB-, des SDHC- und des SDHD-Gens in die Aminosäuresequenz des Proteins verwendet. Die Folgen von Mutationen und Polymorphismen dieser Gene lassen sich hieraus ableiten. 18 Abbildung 10: Darstellung der sogenannten "Codesonne". Sie ist von innen nach außen zu lesen und stellt die 64 möglichen Basentriplets der mRNA dar. 61 dieser Triplets kodieren 20 verschiedene Aminosäuren der Proteinsynthese. 3 Basentriplets, TAA = ochre, TAG = ambre und TGA = opal, dienen als sogenannte Stopcodons die zum Kettenabbruch der Proteinsynthese führen. Der genetische Code ist degeneriert und eindeutig. Dies bedeutet das mehrere Basentriplets eine einzige Aminosäure kodieren können aber ein Basentriplet immer nur für eine Aminosäure kodiert. Mutationen des SDHB-, des SDHC- und des SDHD-Gens haben unterschiedliche Auswirkungen auf die Aminosäuretranslation und -sequenz des Proteins. Man unterscheidet Missense-Mutationen, Stopcodon- und Frameshift-Mutationen sowie Splice-Site-Defekte. Missense-Mutationen führen durch einen Basenaustausch in der Nukleotidsequenz des Basentriplets zur Translation einer anderen Aminosäure. Die Folge ist die Synthese eines veränderten Proteins. Stopcodon-Mutationen bedingen den Abbruch des Translationsvorganges. Ein Basenaustausch in der Nukleotidsequenz des Basentriplets resultiert in der Bildung eines der 3 bekannten Stopcodons (TAA = ochre, TGA = opal, TAG = ambre). Die Folge ist die Synthese eines unvollständigen Proteins. Frameshift-Mutationen führen zu einer kompletten Verschiebung des Leserasters der Aminosäuretranslation. Ursache ist die Insertion oder Deletion einer unterschiedlichen Anzahl von Basen in der Nukleotidsequenz des Gens. Die Folge ist die Synthese eines veränderten oder unvollständigen Proteins. Ein Basenaustausch in der Nukleotidsequenz der Splice-Region eines Introns bedingt einen fehlerhaften Splice-Vorgang der mRNA und eine Verlust des folgenden Exons. Die Folge ist die Synthese eines unvollständigen, fehlerhaften Proteins. 19 1.4. Fragestellungen Die Fragestellungen dieser Arbeit waren: a) Sind Mutationen des SDHB-Gens und des SDHC-Gens genetische Grundlagen für die Tumorgenese von Phäochromozytomen, Paragangliomen und Glomustumoren? Wie häufig treten Mutationen dieser Gene auf? b) Welche Mutationstypen, d.h. Missense-Mutationen, Stopcodon- und FrameshiftMutationen, Insertionen und Deletionen treten auf? c) Welche Krankheitsausprägung ist typisch für Mutationen des SDHB-Gens, des SDHCGens und des SDHD-Gens? d) Gibt es Korrelationen zwischen Mutationstypus und phänotypischer Ausprägung der Erkrankung? e) Welcher Vererbungsmodus liegt den gefunden Mutationen zu Grunde? f) Welche Penetranz zeigen Mutationen des SDHB-, des SDHD-Gens? g) Bestehen Unterschiede für das Risikospektrum der Erkrankungen zwischen Trägern von Mutationen des SDHB-Gens, des SDHC-Gens und des SDHD-Gens? 20 2. Material und Methoden 2.1. Patientengut Die Patienten, die im Rahmen dieser Arbeit auf konstitutionelle Mutationen des SDHB-Gens, des SDHC-Gens und des SDHD-Gens untersucht wurden, sind Teil zweier verschiedener Register. Das in Freiburg und Warschau erstellte Phäochromozytom-Register enthielt 489 Patienten (Stand: Mai 2004), die an einem adrenalen oder extra-adrenalen Phäochromozytom erkrankt waren. Das Glomustumor-Register enthielt 84 Patienten (Stand: Mai 2004), die an einem Paragangliom (Glomustumor) des Kopfes oder Halses erkrankt waren. Ausschließlich Patienten, die nicht miteinander verwandt sind und deren Tumor histologisch gesichert war, sind Bestandteil dieser Register. Ausgeschlossen von den molekulargenetischen Untersuchungen dieser Arbeit wurden 153 Patienten mit klinischen Zeichen einer von Hippel-Lindau Krankheit, einer Multiplen Endokrinen Neoplasie Typ 2 und einer Neurofibromatose Typ 1 (von Recklinghausen) sowie Träger konstitutioneller Mutationen des VHL-Gens und des RET-Gens. 420 Patienten wurden im Rahmen dieser Arbeit auf konstitutionelle Mutationen des SDHBGens, des SDHC-Gens und des SDHD-Gens untersucht. 336 Patienten sind Teil des FreiburgWarschauer-Phäochromozytom-Registers, 84 Patienten sind Teil des Glomustumor-Registers. Folgende klinische Daten wurden mittels eines Fragebogens erhoben: Alter und Geschlecht, Diagnosejahr der Erkrankung, symptomatischer oder asymptomatischer Krankheitsverlauf, Lokalisation und Dignität des Tumors bzw. der Tumoren sowie die Familienanamnese. Das Ausmaß der Erkrankung wurde mittels folgender Verfahren erfasst: Sonographie, Computertomographie und Kernspintomographie des Halses, von Thorax und Abdomen sowie die Erfassung des endokrinologischen Status mit Bestimmung der Noradrenalin-, der Adrenalin- und der Vanillinmandelsäurewerte im 24h Sammelurin. Bei Patienten, deren DNA eine Mutation aufwies, wurde Verwandten ersten Grades ein molekulargenetisches Screening auf entsprechende Mutationen angeboten. Für die Patienteninformationen und Einverständniserklärungen lag ein positives Votum der Ethikkommission des Universitätsklinikums Freiburg vor. Alle getesteten Patienten hatten ihre Einwilligung gegeben. Als Kontrollproben diente DNA von 300 gesunden Probanden. Es waren, entsprechend der Herkunft der Patienten, Blutspender aus Deutschland, aus Polen und aus der Schweiz. 21 Die genannten 420 Patienten konnten durch Kooperation mit einer großen Zahl von Kliniken registriert werden, wobei von jedem Patienten Blut zur molekulargenetischen Untersuchung zugesandt wurde. Die Kooperationspartner waren: Prof. Dr. med. Adler und Prof. Dr. med. Luft, Klinikum Berlin-Buch; Prof. Dr. med. Allolio und Prof. Dr. med. Arlt, Universitätsklinikum Würzburg; Prof. Dr. med. Anlauf, Zentralkrankenhaus Reinkenheide, Bremerhaven; Prof. Dr. med. Brabant und Prof. Dr. med. Welkoborsky, Hochschule Hannover; Dr. med. Behrbohm, Klinikum Berlin-Weisensee; Prof. Dr. med. Berghaus, Prof. Dr. med. Dralle, Dr. med. Brauckhoff, Dr. med. Huong-Vu und Prof. Dr. med. Zumkeller, Universitätsklinikum Halle; Prof. Dr. med. Beyer, Kinderkrankenhaus, Evangelisches Krankenhaus, Oberhausen; Prof. Dr. med. Reincke, Prof. Dr. med. Peter, PD Dr. med. Schipper, Dr. med. Boedeker, Prof. Dr. med. Schultze-Seemann, Universitätsklinikum Freiburg; Dr. med. Caspari, Universitätsklinikum Bonn; Prof. Dr. med. Deitmer und Prof. Dr. med. Hausmann, Städtische Kliniken Dortmund; Dr. med. Diekmann, Städtisches Klinikum Ludwigshafen; Dr. med. Dörstelmann, Dr. med. Hartwein und Dr. med. Hudelmayer, Städtisches Klinikum Pforzheim; Prof. Dr. med. Draf und Prof. Dr. med. Fassbinder, Städtische Kliniken, Fulda; Prof. Dr. med. Esser, Universitätsklinikum Erfurt; Prof. Dr. med. Weber und Dr. med. Fottner, Universitätsklinikum Mainz; Prof. Dr. med. Frilling, Universitätsklinikum Hamburg; Dr. med. C. Fuentes Gomez, Hospital de Navarra, Pamplona, Spanien; Dr. med. Graubner, Dr. med. Jocham und Dr. med. Schmidt, Von Hauner'sches Kinderspital, München; Prof. Dr. med. Heidemann, Kinderkrankenhaus, Augsburg; Prof. Dr. med. Hetzer, Humboldt Kliniken, Charite, Berlin; Dr. med. Hofmockel, Medizinisches Zentrum, Wuerselen; Prof. Dr. med. Januszwewicz, Universitätsklinikum Warschau, Polen; Prof. Dr. med. Jung, Zentralkrankenhaus Bremen; Dr. med. Neumann, Charite, Berlin; Prof. Dr. med. Ganzer, Dr. med. Cupisti, Dr. med. Wagenmann, Dr. med. Willenberg, Universitätsklinikum Düsseldorf; Dr. med. Kaftan, Universitätsklinikum Greifswald; Prof. Dr. med. Klein und Dr. med. Muresan, Hospital de Barbois, Nancy, Frankreich; Prof. Dr. med. Klein-Franke und Dr. med. Steiner, Universitätsklinikum Göttingen; PD Dr. med. Klingenbiel, Universitätsklinikum Frankfurt; Dr. med. Klose, München; Prof. Dr. med. Kornley, Städtisches Klinikum Duisburg; Dr. med. Krämer, Warstein; Prof. Dr. med. Mann, Prof. Dr. med. Wenzel, Dr. med. Lederbogen, Universitätsklinikum Essen; Prof. Dr. med. Walz, Kliniken Essen-Mitte; Prof. Dr. med. Limacher, Universitätsklinikum Strasbourg; Universitätsklinikum Magdeburg; Prof. Dr. med. MacGregor, London; Prof. Dr. med. Mittler, Prof. Dr. med. Müller, München; Prof. Dr. med. Opocher; Universitätsklinikum Padua, Italien; Prof. Dr. med. Schinzel, Prof. Dr. med. Weber, Dr. med. Pichert, Universitätsklinikum Zürich; Dr. med. Pielken, Evangelisches Krankenhaus, Oberhausen; Dr. med. Riepe, Aahaus; Prof. Dr. med. Ritz, Heidelberg; Prof. Dr. med. Schilling, Klinikum Berlin-Neukölln; Dr. med. Schöniger, Städtisches Klinikum Schwabing, München; Dr. med. Schröder, Ruhr Universität Bochum; Dr. med. Uckun-Kitapci, Chapel Hill; Dr. med. Winquist, Regional Cancer Center, London Ontario, Kanada; Prof. Dr. med. Zerres, Universitätsklinikum Aachen; Dr. med. Zimmermann, Kinderkrankenhaus Chemnitz; Prof. Dr. med. Zimmerhackl, Universitätsklinikum Innsbruck. 22 2.2. Methoden 10 ml EDTA-Vollblut stand von allen 420 Patienten und 300 Kontrollprobanden zur molekulargenetischen Analyse zur Verfügung. Zunächst wurde aus diesen 10 ml Blut DNA extrahiert (Kapitel 2.2.1). Anschließend wurden alle 8 Exons des SDHB-Gens, alle 6 Exons des SDHC-Gens und alle 4 Exons des SDHD-Gens mittels Polymerasekettenreaktion amplifiziert (Kapitel 2.2.2). Die Kontrolle der Produktgröße der Amplifikate erfolgte unter Durchführung der Agarosegelelektrophorese (Kapitel 2.2.3). Dem Mutations- und Polymorphismusscreening diente die Methodik der SSCP (single strand conformation polymorphism) (Kapitel 2.2.4). Der Nachweis sowie die genaue Lokalisationsbestimmung der Mutationen und Polymorphismen erfolgte durch die Sequenzierung. Hierzu wurde DNAMaterial abnormer Banden entweder reamplifiziert oder die Amplifikate entsprechender Proben verwendet (Kapitel 2.2.5). Zur Bestätigung der Diagnose einer Mutation wurde eine zweite Blutprobe des entsprechenden Patienten angefordert. Diese wurde mittels SSCP unter Mitanalyse der ersten Probe erneut untersucht. Ohne eine erneute Sequenzierung durchzuführen, wurden identische Ergebnisse der Erst- und Zweitprobe als definitive Mutation gewertet. Auf die gleiche Weise wie mit der Zweitprobe wurde auch mit DNAProben von Verwandten verfahren. Das Gesamtprojekt wurde der Ethikkomission der Universität Freiburg vorgelegt, die ein positives Votum abgab. 2.2.1. DNA – Extraktion Aus 10 ml EDTA-Vollblut wurden 100 bis maximal 1000 µg chromosomale DNA gewonnen. Die angewendete Methode basiert auf einem patentierten Kitsystem der Firma Qiagen, das die Isolation von DNA in einer Größenordnung von 20 bis150 kb ermöglicht. Im ersten Schritt des Verfahrens wurden, unter Zugabe von 5-20 ml eines speziellen auf 2-8°C gekühlten Qiagen C1-Puffers und 15-30 ml eisgekühltem destillierten Wasser, durch zehnminütige Inkubation auf Eis die Zellmembranen der Blutzellen lysiert, Hämoglobin freigesetzt und die Nuklei stabilisiert. Durch Zentrifugation (15 Minuten) des lysierten Blutes bei 1300 x g und einer Temperatur von 4°C entstand am Boden des verwendeten 50 ml Falkons ein noch leicht durch Hämoglobin verunreinigtes, rötlich schimmerndes Pellet, das sich aus den Nuklei zusammensetzte, welche die genomischen DNA enthalten. Um letzte Zellbestandteile und zurückgebliebenes Hämoglobin auszuwaschen, wurde das Pellet erneut in 2 ml Qiagen C1-Puffer und 6 ml destilliertem Wasser gelöst und die Lösung bei 4°C und 1300 x g 15 Minuten zentrifugiert. Nach Dekantieren des Überstandes wurde das nun gereinigte Pellet in 10 ml Qiagen G2-Puffer aufgenommen und unter Zugabe 23 von 200 µl gelöster Qiagen Protease für 30-60 Minuten bei 50°C im Wasserbad inkubiert. Durch die Verwendung des G2-Puffers in Kombination mit der Protease wurden die Nuklei lysiert, die Proteine denaturiert und verdaut und die genomische DNA freigesetzt. Der G2-Puffers ist eine Lösung niedrigen pH-Wertes und niedriger Salzkonzentration und bietet somit eine gute Vorraussetzung für die Bindung der DNA an das Qiagen Genomic-tip-System. Dieses System ist so konzipiert, dass durch Anwendung von Puffern mit niedriger Salzkonzentration und niedrigem pH-Wert die DNA an die Säule bindet, Verunreinigungen wie RNA und Proteine mittels Puffer mittleren Salzgehaltes ausgewaschen werden und die DNA durch Verwendung von Puffern mit hohem Salzgehalt von der Säule gelöst werden kann. Die Grundlage dieses Qiagen Genomic-tipSystems ist ein Anionenaustauscher, dessen Prinzip auf Wechselwirkungen zwischen negativ geladenen Phosphatgruppen der DNA und positiv geladenen DEAE-Gruppen an der Oberfläche der Säule basiert. Im nächsten Schritt des angewendeten Verfahrens wurde das durch 10 ml QBT-Puffer äquillibrierte Genomic-tipSystem 500/G mit der DNA-Lösung beladen, durch Anwendung von 30 ml Qiagen QC-Puffers wurden RNA, Proteine und weitere Verunreinigungen ausgewaschen und verworfen, die gebundene DNA mittels 30 ml QFPuffers eluiert und in einem Isopropanol-haltigen Falkon, zur Ausfällung der DNA, aufgefangen. Zur weiteren Verarbeitung wurde die DNA in TE (Kapitel 2.1.4.) gelöst und im Kühlschrank gelagert. 2.2.2. Polymerasekettenreaktion (PCR) Die Polymerasekettenreaktion ist eine in vitro-Technik mit der DNA-Fragmente vervielfältigt werden können (Saiki et al. 1988) (Abbildung 11). Zwei bekannte DNA-Sequenzen, die Oligonukleotidprimer, bilden den Startpunkt der PCR. Die Verlängerung erfolgt über die TaqDNA-Polymerase. Die in dieser Arbeit gewählte Technik ist im folgenden näher beschrieben. Die Polymerasekettenreaktion durchläuft zyklisch drei verschieden Schritte: • Denaturierung der doppelsträngigen DNA • Anlagerung bzw. Annealing der Primer • Verlängerung der Primer durch die hitzestabile Taq-Polymerase Zur Denaturierung der komplexen doppelsträngigen DNA wird das Reaktionsgemisch, bestehend aus DNA, dNTP's, Oligonukleotidprimern, Taq-DNA-Polymerase und entsprechendem PCR-Puffer, auf 90°C erhitzt. Der zweite Schritt der Polymerasekettenreaktion, das Annealing der Primer, ist entscheidend für die Spezifität der Reaktion. Nach Abkühlen des Reaktionsgemisches auf die spezielle Annealing-Temperatur, meist in einem Bereich zwischen 40-65°C liegend, lagern sich die Primer, deren Länge nicht größer als 20-30 bp sein sollte und deren Sequenz jeweils komplementär zu den flankierenden Regionen am 3`-Ende von Strang und Gegenstrang ist, an die entstandenen Einzelstränge der Matritzen-DNA an. Die Annealing-Temperatur sollte für forward- und reverse-Primer ähnliche Werte besitzen und lässt sich mittels folgender Formel berechnen: (Anzahl von A+T) x 2°C + (Anzahl von G+C) x 4°C. Der erhaltene Wert muss experimentell optimiert werden. Im dritten Reaktionsschritt der PCR werden die Primer, gewöhnlich bei einer Temperatur von 72°C, der optimalen Temperatur der Taq-DNA-Polymerase, durch Anlagerung von Desoxynukleosidtriphosphaten verlängert. Die Taq-DNA-Polymerase ist ein hitzestabiles 24 Enzym, das ursprünglich aus dem Bakterium Thermus aquaticus stammt, heute jedoch synthetisch hergestellt wird. Sie benötigt die Primer als Starthilfe, um von dort aus einen komplementären Strang zum entsprechenden Matritzen-Strang in 5`→3`Richtung zu synthetisieren, d.h. ausgehend vom 5´-α Phosphat des Desoxynukleosidtriphosphats in Richtung der endständigen 3`-Hydroxylgruppe des wachsenden DNA-Stranges. Diese drei Reaktionsschritte werden 15-30 mal wiederholt um eine genügend hohe Ausbeute zu erreichen. Dabei stehen im jeweils nächsten Zyklus neben den ursprünglichen Strängen auch die neu synthetisierten Stränge zur Amplifikation zur Verfügung, d.h. es handelt sich um eine exponentielle Vervielfältigung (2x). In die Polymerasekettenreaktion wurde ein Reaktionsgemisch eingesetzt, bestehend aus 2 µl 1:10 verdünnter DNA (d.h. ca. 0,1 ng DNA) und 18 µl eines sogenannten Premix. Dieser setzt sich aus 10 µl ddH2O, 2 µl unverdünntem Taq spezifischem MgCl2-Puffer, 2 µl dNTP (d.h. 8 µM je dNTP), 2 µl jeweils von forward und reverse-Primer (je Primer eine Konzentration von 20 pM) und 0,2 µl Taq-DNA-Polymerase (5U/µl) zusammen. Zu Beginn jedes PCR-Zyklus wurde einmalig für 5-10 Minuten auf 95°C aufgeheizt um eine ausreichende Denaturierung zu gewährleisten. Danach wurden folgende Reaktionsschritte durchlaufen: • 30 sec, 95°C zur Denaturierung • 30 sec, spezifische Annealing-Temperatur • 30 sec, 72°C DNA-Synthese. Nach dem letzten Schritt wurde zusätzlich für 10 Minuten bei 72°C elongiert, um unvollständige Amplifikationsprodukte zu vermeiden. Insgesamt wurden die Zyklen 15-30 mal wiederholt, um eine genügend hohe Ausbeute für weiterführende Untersuchungen zu erhalten. Abbildung 11: Schematische Darstellung des zyklischen Reaktionsablaufs der Polymerasekettenreaktion. Schritt 1: Denaturierung der doppelstränigen DNA in zwei zueinander komplementäre Einzelstränge, die als Matritze dienen. Schritt 2: Anlagerung bzw. Annealing der Oligonukleotidprimer. Schritt 3: Verlängerung der Primer durch die hitzestabile Taq-DNA-Polymerase. Der eingeklammerte Bereich kennzeichnet die zyklische Wiederholung dieser Hauptschritte der Polymerasekettenreaktion, um eine genügend hohe Ausbeute des Amplifikationsproduktes sicherzustellen. 25 Alle 8 Exons des SDHB-Gens wurden mittels PCR amplifiziert. Angaben der Primersequenzen, der ermittelten PCR-Bedingungen und der Produktgröße der Amplifikate können Tabelle 5 entnommen werden. SDHB-Gen Exon Primersequenz (5´→3´) PCR-Bedingung Amplifikationsprodukt in bp 1 F: GGCGGAGAGCGACCT R: AGTCTCTGTGGCTTTCCT 60°C/ 15sec/ 30z 141 2 F : TCTTGTATTTCTAATTTTTTTTCCTT R : GTCCCTAAATCAAATCAAGAACTC 50°C/ 30sec/ 30z 186 3 F : TAAAGTGTAGGGAGGTTGAA R : GGCCAGCCCAAGC 60°C/ 15sec/ 30z 174 4 F : AAAGTATTTGGGGCAGGAC R : CCCCCATGCAAATAAAAAC 60°C/ 15sec/ 30z 208 5 F : ATCTGATCCTTTTCTTCTTCTTCTTC R : CACTCCTGGCAATCATCTTT 55°C/ 30sec/ 30z 183 6 F : GCAGAGTCTCTCCCGTCACA R : GGCTGGCTTACAGCAATCTAT 60°C/ 15sec/ 30z 197 7 F : CTCTGGCGCTGTTGATTG R : CATGCTACTTCTGGCGTGTC 58°C/ 30sec/ 30z 194 8 F : GTTTTCCCTTTCAGTTTCAGTTA R : CTCAAATTAGATATAAATTATGTTCAGC 60°C/ 15sec/ 30z 140 Tabelle 5: Verwendete Primersequenzen zur Amplifikation der 8 Exons des SDHB-Gens. Die angegebenen Temperaturen entsprechen den Annealing-Temperaturen der Primer; F steht für forward, R für reverse, sec für die Annealingdauer und z für die Anzahl der Zyklen. Generelle Magnesiumchloridkonzentration: 1,5 mM. Alle Primer wurden mit Hilfe des Programmes Primer-Select und entsprechenden Gensequenzen zusammengestellt. Um zu gewährleisten, dass die SSCP-Analyse (Kapitel 2.2.4.) als auch die anschließende Sequenzierung (Kapitel 2.2.5.) den vollständigen für die Aminosäuren kodierenden Bereich erfasst, werden ca.15-20 bp zwischen Primer und Exongrenze benötigt. So schließen die in Tabelle 5 genannten Primerpaare Intronabschnitte folgender Größe mit ein: Exon 1, 37 bp im 5' Bereich und 32 bp im 3' Bereich; Exon 2, 24 bp im 5' Bereich und 34 bp im 3' Bereich; Exon 3, 37 bp im 5' Bereich und 51 bp im 3' Bereich; Exon 4, 45 bp im 5' Bereich und 26 bp im 3' Bereich; Exon 5, 22 bp im 5' Bereich und 45 bp im 3' Bereich; Exon 6, 30 bp im 5' Bereich und 65 bp im 3' Bereich; Exon 7, 31 bp im 5' Bereich und 40 bp im 3' Bereich; Exon 8, 34 bp im 5' Bereich und 37 bp im 3' Bereich. 26 Alle 6 Exons des SDHC-Gens wurden mittels PCR amplifiziert. Angaben der Primersequenzen, der ermittelten PCR-Bedingungen und der Produktgröße der Amplifikate können Tabelle 6 entnommen werden. SDHC-Gen Exon Primersequenz (5´→3´) PCR-Bedingung Amplifikationsprodukt in bp 1 F: CACATGACACCCCCAACCCC R: CTGCCCAGGCACAGGATAAACA 65°C/ 15sec/ 30z 216 2 F : TACTTTTAATCTATCCCTTCAC R: TCTCCAGACTTAGAAACTTA 55°C/ 30sec/ 30z 176 3 F : ACGTTATGCAAAATATTAAACCAAGT R: AGCCTCTTCTCTGGCTCCA 55°C/ 15sec/ 30z 202 4 F : GTTTATATTTTTGCCAAGATAGACTC R : CCAAGTTTTTCAAAGAAGCACA 60°C/ 30sec/ 30z 195 5 F : TCATATTAGTTGTAACTTATGAGCAGC R : CTCCCCACTCCCTTCACAG 58°C/ 15sec/ 30z 269 6 F : TGTTAATGTCCTATTTACTGAA R : TAAACAAATAAGGAGAACTTTT 55°C/ 30sec/ 30z 230 Tabelle 6: Verwendete Primersequenzen zur Amplifikation der 6 Exons des SDHC-Gens. Die angegebenen Temperaturen entsprechen den Annealing-Temperaturen der Primer; F steht für forward, R für reverse, sec für die Annealingdauer und z für die Anzahl der Zyklen. Generelle Magnesiumchloridkonzentration: 1,5 mM. Die Primerpaaren von Exon 1-6 wurden mit Hilfe des Programmes Primer-Select und entsprechenden Gensequenzen zusammengestellt. Um zu gewährleisten, dass die SSCP-Analyse (Kapitel 2.2.4.) als auch die anschließende Sequenzierung (Kapitel 2.2.5.) den vollständigen für die Aminosäuren kodierenden Bereich erfasst, werden ca. 15-20 bp zwischen Primer und Exongrenze benötigt. So schließen die in Tabelle 6 genannten Primerpaare Intronabschnitte folgender Größe mit ein: Exon 1, 50 bp im 5' Bereich und 79 bp im 3' Bereich; Exon 2, 76 bp im 5' Bereich und 1 bp im 3' Bereich; Exon 3, 41 bp im 5' Bereich und 14 bp im 3' Bereich; Exon 4, 56 bp im 5' Bereich und 29 bp im 3' Bereich; Exon 5, 29 bp im 5' Bereich und 30 bp im 3' Bereich; Exon 6, 32 bp im 5' Bereich und 85 bp im 3' Bereich. Alle 4 Exons des SDHD-Gens wurden mittels PCR amplifiziert. Die Analysen des SDHDGens für 228 Patienten mit Phäochromozytomen wurden in der Dissertation von Herrn Dr. med. R. Munk, Freiburg 2003, beschrieben. Dort finden sich die entsprechenden methodischen Angaben. Die hier vorgelegte Arbeit bezieht die Analyse der Differenz von 192 Patienten ein. Es handelt sich um 108 Patienten mit einem Phäochromozytom und 84 Patienten mit einem Glomustumor. 27 2.2.3. Agarosegel-Elektrophorese Die Agarosegel-Elektrophorese diente der Kontrolle der Produktgröße der bei der PCR entstandenen Amplifikate. Ein 0,6%iges Agarosegel, hergestellt aus 1,6 g Agarose und 80 ml 1 x TBE wurde mit einem Gemisch, bestehend aus 5 µl 6 x Ladepuffer und 5 µl PCR-Produkt beladen und für 20 Minuten in einer dafür vorgesehenen, mit 1 x TBE gefüllten Elektrophoresekammer einer angelegten Spannung von 120V unterworfen. Unter dem Einfluss elektrischer Spannung zwischen Kathode und Anode beginnen die geladenen DNA- Fragmente im Agarosegel in Richtung Anode zu wandern. Je nach Größe des Fragments legen sie unterschiedlich lange Strecken zurück. Je größer das Fragment umso näher kommen sie bei der Kathode zum liegen. Zur Kontrolle der Produktgröße der Amplifikate wurde auf dem Gel parallel ein DNA-Längenstandart in Form eines sogenannten Marker V in einer separaten Bahn aufgetragen. Dieser Marker V stellt Produkte der Größenordnung 8-587 bp dar. Um die entstandenen Banden bzw. die DNA-Fragmente mittels UV-Licht einer Wellenlänge von 366 nm sichtbar zu machen, wurde das Agarosegel nach der Elektrophorese für 20 Minuten in ein Färbebad, hergestellt aus 200 ml destilliertem Wasser und 100 µl 1% Ethidiumbromidlösung, gelegt. Das Ergebnis wurde durch einen Alpha-Imager dokumentiert. 2.2.4. Single Strand Conformation Polymorphism (SSCP) Die SSCP-Analyse (single strand conformation polymorphism) ist eine Gelelektrophorese zur Detektion von Punktmutationen großer Gene (Orita et al. 1989) (Abbildung 12). DNA Doppelstränge werden durch starke Hitzeeinwirkung in ihre Einzelstränge denaturiert. Diese Einzelstränge werden durch Einwirkung einer elektrischen Spannung in einem Polyacrylamidgel aufgetrennt. Die aufgetrennten Banden werden mit einer Silbernitratfärbung schnell und hochempfindlich angefärbt und dargestellt. Ein Basenaustausch, kleine Deletionen oder Insertionen wirken sich auf das Laufverhalten der denaturierten Einzelstränge aus und resultieren in einem veränderten Bandenmustern. Die Analyse der Veränderung bleibt der Sequenzierung vorbehalten. Resultiert eine Sequenzveränderung in einer fehlerhaften Proteinsynthese wird eine pathogenetisch relevante Mutation angenommen. Diese darf bei der Untersuchung der Kontrollproben nicht nachgewiesen werden. Bleibt eine Sequenzveränderung ohne Folgen oder tritt diese gehäuft im Kontrollprobenkollektiv auf, wird von einem Polymorphismus gesprochen. Patienten- oder 28 DNA-Abschnitte einer Größe bis zu 400 bp können mittels der SSCP-Analyse untersucht werden. Jenseits dieser Größe nimmt die Sensitivität ab. Die SSCP-Analyse basiert auf dem Prinzip, dass die elektrophoretische Beweglichkeit der DNA-Fragmente von ihrer Größe, Ladung und ihrer Form abhängig ist. Nach der Denaturierung der komplexen doppelsträngigen DNA bei einer Temperatur von 95°C, unterstützt durch den Zusatz eines aus Formamid, Natriumhydroxid, Xylencyanol und Bromphenolblau zusammengesetzten SSCPPuffers und dem schnellen Abkühlen durch sofortige Lagerung in einem Eisbad nimmt jeder DNA-Einzelstrang eine seiner Sequenz entsprechende Konformation an, die sich in der entsprechenden Laufeigenschaft im Gel wiederspiegelt. Die Empfindlichkeit der Methode ist hoch, so dass ein einziger Unterschied in der Basensequenz, der zu einer Konformationsänderung des denaturierten Einzelstranges führt, ausreicht, um sich in einer veränderten Laufeigenschaft im Polyacrylamidgel und somit einem veränderten Bandenmuster darzustellen. In der vorliegenden Arbeit wurden 20 µl PCR-Produkt mit 30 µl SSCP-Puffer versetzt. Durch Erhitzen des Gemisches bei einer Temperatur von 95°C für 3 Minuten 40 sec wurde die komplexe doppelsträngige DNA denaturiert. Um eine Rehybridisierung zu verhindern, wurde das Gemisch anschließend sofort auf Eis gebettet. Auf die Polyacrylamidgelkammer wurde ein dünner Film Mineralöl aufgetragen, anschließend das Gel aufgelegt, mit dem eisgekühlten PCR-Produkt/SSCP-Puffer-Gemisch beladen und die Elektrophorese unter experimentell ermittelten Temperaturbedingungen für ca. 90 Minuten durchgeführt. Um die DNA-Fragmente, die sich durch die SSCP im Polyacrylamidgel als Banden zeigen, darstellen zu können, wurde das Gel nach abgeschlossener Elektrophorese mit Silbernitrat gefärbt. Dazu wurde es für 5 Minuten in 1% Salpetersäure (HNO3) gelegt und anschließend für 30 Minuten mit Silbernitrat (AgNO3) behandelt. Die an die DNA gelagerten Silberionen wurden mit Hilfe eines fünfminütigem Natriumcarbonat-Formaldehydbades (Na2CO3/HCHO) reduziert, so dass sich schwarze Banden im Acrylamidgel zeigten. Um die Redoxreaktion zu stoppen, wurde das Gel für weitere 5 Minuten in 10% Essigsäure (CH3COOH) gelegt. Abgeschlossen wurde die Silbernitratfärbung durch eine fünfminütige Konservierung des Gels mit 5% Glycerinlösung. Abgedeckt mit einer speziellen Folie stand es zur Auswertung zur Verfügung. Abbildung 12: Schematische Darstellung des Grundprinzips der SSCP-Analyse. Die Grundlage dieses Verfahrens bilden die elektrophoretische Beweglichkeit, die Größe, die Ladung und die Form des entsprechenden DNAFragmentes. Die Abbildung zeigt, dass bereits der Austausche einer Base zu einer Konformationsänderung des DNAFragmentes führt und in einem veränderten Laufverhalten im Polyacrylamid-Gel resultiert. 29 Tabelle 7 zeigt eine Übersicht der Bedingungen der SSCP-Analyse für alle 8 Exons des SDHB-Gens. SDHB-GEN Exon Bedingungen für GENEGEL EXCEL 12.5/24 kit 1 5 W / 20°C / 15min 5 W / 25°C / 1h 15min 2 6 W / 12°C / 1h 30min 3 6 W / 12°C / 1h 30min 4 6 W / 12°C / 1h 30min 5 6 W / 8°C / 1h 30min 6 6 W / 18°C / 1h 30min 7 5 W / 20°C / 15min 5 W / 25°C / 1h 15min 8 6 W / 12°C / 1h 30min Tabelle 7: Bedingungen der SSCP-Analyse; Die Angabe 5 W oder 6 W entspricht der angelegten Stromstärke Tabelle 8 zeigt eine Übersicht der Bedingungen der SSCP-Analyse für alle 6 Exons des SDHC-Gens. SDHC-GEN Exon Bedingungen für GENEGEL EXCEL 12.5/24 kit 5 W / 20°C / 15min 5 W / 25°C / 1h 15min 1 2 6 W / 12°C / 1h 30min 3 6 W / 12°C / 1h 30min 4 6 W / 12°C / 1h 30min 5 6 W / 8°C / 1h 30min 6 6 W / 18°C / 1h 30min Tabelle 8: Bedingungen der SSCP-Analyse; Die Angabe 5 W oder 6 W entspricht der angelegten Stromstärke SDHD-Gens Die Bedingungen der SSCP-Analyse für alle 4 Exons des SDHD-Gens finden sich in der Dissertation von Herrn Dr. med. R. Munk. 30 2.2.5. Sequenzierung Die in der SSCP-Analyse dargestellten Mutationen und Polymorphismen wurden mit Hilfe fluoreszenz-markierter Primer nach der Dideoxymethode in einem auswärtigen für Sequenzierungen spezialisiertem Labor analysiert (Sanger et al. 1977). Dieses Labor erhielt entweder direkt amplifizierte DNA, von Proben die in der SSCP Auffälligkeiten zeigten, oder Reamplifikate aberrierender Banden. Für die Reamplifikation wurden aberrante Banden aus dem SSCP-Gel ausgeschnitten und in 30 µl 10 mM Tris pH 8 aufgenommen. Über Nacht konnten ausreichende Mengen DNA in die Pufferflüssigkeit diffundieren. Für die zur Sequenzierung vorgesehene PCR (Kapitel 2.2.2) wurde folgender Ansatz gewählt. • 5 µl der DNA auffälliger Proben (ca. 2,5 ng DNA) bzw. 5 µl der oben beschriebenen Lösung aberrierender Banden • 25 µl ddH2O • 5 µl MgCl2-Puffer (1,5 mM) • 5 µl dNTPs (20 µM je dNTP) • je 5 µl forward- und reverse-Primer • 0,5 µl Taq-DNA-Polymerase Die PCR-Bedingungen für die jeweiligen Exons des SDHB- und des SDHC-Gens sind in Tabelle 5 und 6 aufgeführt. Abbildung 13 zeigt ein Beispiel des typischen Ergebnisses einer Sequenzierung. Abbildung 13: Fluoreszenz-markierte Primer werden mit Hilfe einer Taq-Polymerase und vier verschiedener dNTP-Moleküle verlängert. Durch Zugabe und Einbau von Didesoxynucleosidtriphosphaten (ddNTP's), einem dNTP-Analogon, kommt es zu eienm Abbruch der Sequenzierungsreaktion. Es Mutat: T A T G G A T G nt : 267 268 269 270 271 272 273 274 entsehen fluoreszenz-markierte ReaktionsWildtyp: T A T C G A T G produkte unterschiedlicher Kettenlänge. Durch die Größenauftrennung in einer Polyacrylamid-Gelelektrophorese und anschließendem Sichtbarmachen der einzelnen Banden mittels Fluorographie können die Positionen der Kettenabbrüche identifiziert werden. Im Anschluß an die Fluorographie und mit Hilfe einer speziellen DNA-Sequenzierungsmaschiene sowie eines entsprechenden Computerprogrammes entsteht die oben dargestellte farbige Kurve. Die unterschiedlichen farbigen Kurven kennzeichnen die am DNA-Aufbau beteiligten vier verschiedenen dNTP's, die nur mit den jeweiligen Basenkürzeln beschrieben sind. Rot steht für Thymin, grün steht für Adenin, blau steht für Cytosin und schwarz steht für Guanin. Peaks der jeweiligen Kurve kennzeichnen das Auftreten der entsprechende Base an der jeweiligen DNA-Position. 31 2.2.6. Programm des klinischen Screenings Folgende Patientendaten wurden im Rahmen des klinischen Screenings erhoben: - Geburtsjahr - Geschlecht - Alter zum Zeitpunkt der Krankheitsmanifestation - Symptomatischer / asymptomatischer Krankheitsverlauf - Art des Tumors (Phäochromozytom, Paragangliom, Glomustumor) - Lokalisation des Tumors - Anzahl der Tumoren - Dignität - Detaillierte Familienanamnese Um eine genaue Beschreibung der Erkrankung zu erhalten, wurden entsprechende diagnostische Verfahren angewendet: - Magnetresonanztomographie des Halses und der Schädelbasis - Magnetresonanztomographie des Thorax - Magnetresonanztomographie des Abdomen - Bestimmung des Katecholaminspiegels im 24h Urin - Fakultativ: Ganzkörper DOPA-Positronenemissionstomographie (PET) Patienten die eine konstitutionelle Mutation des SDHB-Gens, des SDHC-Gens oder des SDHD-Gens zeigten, wurden erneut klinisch untersucht. Zunächst wurde der momentane Krankheitszustand erfragt, dann das oben bereits beschrieben diagnostische Screening wie die Bestimmung der Katecholamine sowie die Durchführung einer Magnetresonanztomographie durchgeführt. In die Auswertung wurden auch früher durchgeführte Untersuchungen mitttels Computertomographie und MIBG-Szintigraphie einbezogen. Positiv getesteten Patienten wurde des weiteren eine molekulargenetische Untersuchung von Verwandten angeboten und bei Einverständnis durchgeführt. Abschließend wurde positiv getesteten Verwandten die Durchführung einer klinischen Diagnostik empfohlen. 32 2.2.7. Statistik Der Fisher-Test für unverbundene, dichotome Stichproben wurde zur Überprüfung signifikanter Unterschiede klinischer Charakteristika des SDHB-Gens, des SDHC-Gens und des SDHD-Gens angewendet. Die Methode der Kaplan-Meier Schätzung diente der Ermittlung der Penetranzen SDHB-Gen, SDHC-Gen und SDHD-Gen assoziierter Tumoren. Der Parameter der Überlebenszeit wurde hierbei durch das Patientenalter ersetzt. Zum Vergleich der Altersunterschiede bei Patienten mit Mutationen des SDHB-Gens, des SDHCGens und des SDHD-Gens sowie der Ermittlung der altersabhängigen Penetranzen wurden der Wilcoxon-Test und der Log-Rank-Test angewendet. P-Werte < 0,05 wurden als signifikant angesehen. Die statistische Auswertung der Daten erfolgte unter freundlicher Mithilfe von Herrn Dr. med. Ch. Pawlu, Institut für Physiologie, Albert-Ludwigs-Universität, Freiburg. 33 3. Ergebnisse 3.1. Patientenkollektiv 420 nicht miteinander verwandte Patienten, die an einem adrenalen oder extra-adrenalen Phäochromozytom oder einem Paragangliom des Kopfes oder des Halses (Glomustumor) erkrankt waren, wurden auf konstitutionelle Mutationen des SDHB-Gens, des SDHC-Gens und des SDHD-Gens untersucht. 336 dieser Patienten hatten ein Phäochromozytom. 295 dieser Patienten hatten adrenal lokalisierte Phäochromozytome, 41 wiesen eine extra-adrenale Lokalisation auf. Von den Patienten mit extra-adrenaler Tumorlokalisation hatten 31 abdominelle und 10 thorakale Phäochromozytome. 84 der Patienten hatten einen Glomustumor. Alle Tumoren waren histologisch gesichert. 244 (58 %) weibliche und 176 (42 %) männliche Patienten bildeten das zu untersuchende Kollektiv. 84 % der 420 Patienten waren zwischen 19 und 65 Jahre alt. 8,4 % der Patienten waren jünger als 18 Jahre, 9,4 % waren älter als 65 Jahre. Alle 336 Phäochromozytom-Patienten stellten sich als nicht syndromale Fälle mit negativer Familienanamnese vor, während 3 der 84 GlomustumorPatienten eine positive Familienanamnese aufwiesen. Keiner der 420 Patienten zeigte Tumoren außerhalb der Nebenniere und des paraganglionären Systems. Tabelle 9 gibt eine detaillierte Übersicht des Patientenkollektives wieder. Patientendaten der Phäochromozytom-Patienten n = 336 Patientendaten der Glomustumor-Patienten n = 84 männlich 43 % männlich 38 % weiblich 57 % weiblich 62 % Durchschnittsalter 43 Jahre Durchschnittsalter 48 Jahre Altersspanne 4-83 Jahre Altersspanne 15-83 Jahre maligne 7% maligne 5% benigne 93 % benigne 95 % solitär 93 % solitär 86 % multiple 7% multiple 14 % adrenal 88 % Gl. caroticum 36 % extra-adrenal 12 % Gl. jugulare 30 % thorakal 3% Gl. tympanicum 21 % extra-adrenal abdominell 9% sonstige 3% Tabelle 9: Klinische Daten der 336 Patienten mit Phäochromozytomen und der 84 Patienten mit Glomustumoren. 34 3.2. Konstitutionelle Mutationen des SDHB-Gens 3.2.1. Molekulargenetisches Screening auf konstitutionelle Mutationen des SDHB-Gens Als Träger konstitutioneller Mutationen des SDHB-Gens wurden 25 nicht miteinander verwandte Patienten entdeckt. 16 dieser Mutationsträger sind Teil des FreiburgerPhäochromozytom-Registers sowie des deutschen Glomustumor-Registers. 5 Patienten sind Teil des Warschauer-Phäochromozytom-Registers. Spanischer, französischer und kanadischer Staatsangehörigkeit waren 4 Mutationsträger (Tabelle 10). Es wurden 18 verschiedene Mutationen des SDHB-Gens nachgewiesen. Diese finden sich in Exon 1, 2, 3, 4, 6 und 7 sowie in den für den Splice-Vorgang wichtigen Regionen des Introns 3, 4 und 7 (Abbildung 14). 271 G/A 270 C/G 221 ins CCAG 436 G/A 558-3 C/G 421-2 A/G 155 del C 213 C/T Exon1 Exon2 291 G/A 721 G/A 394 T/C Exon3 Exon4 708 T/C Exon5 859 G/A 847/849 del TCTC Exon6 899+1 G/A Exon7 Exon8 328 T/C 300 del CCTCA 881 C/A Abbildung 14: Verteilung der im Rahmen des molekulargenetischen Screenings des SDHB-Gens entdeckten konstitutionellen Mutationen. Das Mutationsspektrum der 18 verschiedenen Mutationen setzt sich aus 9 (50 %) MissenseMutationen, 2 (11 %) Stopcodon-Mutationen, 4 (22 %) Frameshift-Mutationen und 3 (17 %) Splice-Site-Defekten zusammen (Erklärungen siehe Kapitel 1.2.3. Mutationen und Polymorphismen, S.20). 35 Tabelle 10: Im Rahmen des molekulargenetischen Screenings entdeckte konstitutionelle Mutationen des SDHB-Gens. Nationalität Tumor Indexfall Mutation (Nukleotid) Resultat (Aminosäure) Mutationstypus Exon Deutsch Pheo B1 155 del C L7Frameshift Frameshift 1 Deutsch Pheo B2 213 C/T R27X Stopcodon 2 Polnisch Pheo B3 221 ins CCAG S30Frameshift Frameshift 2 Deutsch Pheo B4 270 C/G R46G Missense 2 Deutsch Pheo B5 270 C/G R46G Missense 2 Kanadisch GT B6 271 G/A R46Q Missense 2 Deutsch Pheo B7 271 G/A R46Q Missense 2 Deutsch Pheo B8 291 G/A G53R Missense 2 Spanisch Pheo B9 300 del CCTCA P56Frameshift Frameshift 2 Deutsch Pheo B10 328 T/C L65P Missense 2 Deutsch Pheo B11 394 T/C L87S Missense 3 Deutsch Pheo B12 394 T/C L87S Missense 3 Französisch Pheo B13 394 T/C L87S Missense 3 Deutsch GT B14 421–2 A/G - Splice-Defekt (3) Deutsch GT B15 421–2 A/G - Splice-Defekt (3) Deutsch Pheo B16 436 G/A C101Y Missense 4 Spanisch Pheo B17 558-3 C/G - Splice-Defekt (4) Polnisch Pheo B18 708 T/C C192R Missense 6 Polnisch Pheo B19 721 G/A C196Y Missense 6 Polnisch Pheo B20 847/849 del TCTC F238Frameshift Frameshift 7 Polnisch Pheo B21 847/849 del TCTC F238Frameshift Frameshift 7 Deutsch Pheo B22 859 G/A R242H Missense 7 Deutsch GT B23 859 G/A R242H Missense 7 Deutsch Pheo B24 881 C/A C249X Stopcodon 7 Deutsch GT B25 899+1 G/A - Splice-Defekt (7) Tabelle 10: Darstellung der im Rahmen des molekulargenetischen Screenings gefundenen Mutationen des SDHB-Gens. Pheo ist eine Abkürzung für Phäochromozytom, GT ist eine Abkürzung für Glomustumor des Kopfes oder des Halses. Aus Datenschutzgründen werden die Namen der Indexfälle mit den Ziffern B1-B25 verschlüsselt. Die Reihenfolge der Nummerierung orientiert sich an der Position der Mutation in der Nukleotidsequenz. Die Mutationen SDHB c. 270 C/G, SDHB c. 271 G/A, SDHB c. 421-2 A/G, SDHB c. 847/849 del TCTC und SDHB c. 859 G/A wurden bei jeweils 2 Patienten gefunden. Die Mutation SDHB c. 394 T/C wurde bei drei Patienten gefunden. Zur Frage einer verwandtschaftlichen Beziehung von Patienten mit gleichen Mutationen wurden Haplotyp-Analysen mit polymorphen Mikrosatellitenmarkern durchgeführt. Diese 36 Untersuchung erfolgte im Labor von Frau Prof. Eng, Ohio State University, Columbus, Ohio, USA. Es zeigten sich keine identischen Haplotypen. Eine Verwandtschaft der jeweiligen Indexfälle kann somit mit großer Wahrscheinlichkeit ausgeschlossen werden. Für Mutationen des SDHB-Gens wurde eine Prävalenz von 6 % (25/420) ermittelt. Dies gilt sowohl für die Gruppe der Phäochromozytom-Patienten (20/336) als auch für die Gruppe der Glomustumor-Patienten (5/84). Ein Beispiel der Darstellung der Mutationen in der SSCPAnalyse zeigt Abbildung 15. Die Sequenzierungsergebnisse sind in den Abbildungen 16-33 dargestellt. Beispiel einer SSCP-Analyse Exon 1: K Bahn1 Abbildung 15: K: steht für Kontrollprobe, dies bedeutet, dass die cDNA in Exon 1 dieser Probe keine Sequenzveränderung aufweist. Bahn 1: zeigt das SSCP-Bandenmuster der Sequenzabweichung 155 del C. Durch die Deletion der Base Cytosin (C) an Position 155 ändert sich das Laufverhalten im SSCP-Gel. Die mit Pfeilen markierten Extrabanden entstehen. Abbildungen 16-33: Darstellung aller im Rahmen dieser Arbeit identifizierten Mutationen als Sequenzdiagramme. Angegeben sind in Buchstaben die mutierte Sequenz (Mutat.) und die Wildtypsequenz (Wildtyp) sowie die entsprechenden Nukleotide (nt). SDHB c. 155 del C, entspricht L7frameshift SDHB c. 213 C/T, entspricht R27X Mutat: T nt: 210 Wildtyp: T C 211 C Codon 26 Abbildung 16: Das gelb markierte Kästchen kennzeichnet die Deletion der Base Cytosin (C) an Position 155 der Wildtypsequenz. Die Folge ist die Verschiebung des Leserasters der Aminosäuretranslation. Das ebenfalls markierte Codon 8 verdeutlicht die Veränderung. Die Wildtypsequenz des Codons 8 TCC wird nun zu CCT. C 212 C T 213 C G 214 G Codon 27 A G 215 216 A G G 217 G A 218 A Codon 28 Abbildung 17: Das gelb markierte Kästchen kennzeichnet das mutierte Codon 27. Die Wildtypsequenz (untere Zeile) der Nukleotide 213, 214 und 215 lautet CGA. Durch den Basenaustausch an Position 213 entseht die mutierte Sequenz (obere Zeile) TGA. 37 SDHB c. 221 ins CCAG, entspricht S30frameshift SDHB c. 270 C/G, entspricht R46G Mutat: T nt: 267 Wildtyp: T A 268 A Codon 45 T 269 T G 270 C G 271 G A 272 A Codon 46 T G 273 274 T G Codon 47 Abbildung 18: Das gelb markierte Kästchen kennzeichnet die Insertion der 4 Basen CCAG an Position 221 der Wildtypsequenz. Die Folge ist die Verschiebung des Leserasters der Aminosäuretranslation. Das ebenfalls markierte Codon 31 verdeutlicht die Veränderung. Die Wildtypsequenz des Codons 31 ACA wird nun zu CCA. Abbildung 19: Das gelb markierte Kästchen kennzeichnet das mutierte Codon 46. Die Wildtypsequenz (untere Zeile) der Nukleotide 270, 271 und 272 lautet CGA. Durch den Basenaustausch an Position 270 entsteht die mutierte Sequenz (obere Zeile) GGA. SDHB c. 271 G/A, entspricht R46Q SDHB c. 291 G/A, entspricht G53R Mutat: T nt: 267 Wildtyp: T A 268 A Codon 45 T 269 T C 270 C A 271 G Codon 46 A 272 A T 273 T G 274 G G 275 G Codon 47 Abbildung 20: Das gelb markierte Kästchen kennzeichnet das mutierte Codon 46. Die Wildtypsequenz (untere Zeile) der Nukleotide 270, 271 und 272 lautet CGA. Durch den Basenaustausch an Position 271 entseht die mutierte Sequenz (obere Zeile) CAA. Abbildung 21: Das gelb markierte Kästchen kennzeichnet das mutierte Codon 53. Die Wildtypsequenz (untere Zeile) der Nukleotide 291, 292 und 293 lautet GGA. Durch den Basenaustausch an Position 291 entseht die mutierte Sequenz (obere Zeile) AGA. SDHB c. 300 del CCTCA, entspricht P56frameshift SDHB c. 328 T/C, entspricht L65P Abbildung 22: Das gelb markierte Kästchen kennzeichnet die Deletion der 5 Basen CCTCA an Position 300 der Wildtypsequenz. Die Folge ist die Verschiebung des Leserasters der Aminosäuretranslation. Das ebenfalls markierte Codon 57 verdeutlicht die Veränderung. Die Wildtypsequenz des Codons 57 CAT wird nun zu GCA. Abbildung 23: Das gelb markierte Kästchen kennzeichnet das mutierte Codon 65. Die Wildtypsequenz (untere Zeile) der Nukleotide 327, 328 und 329 lautet CTT. Durch den Basenaustausch an Position 328 entseht die mutierte Sequenz (obere Zeile) CCT. 38 SDHB c. 421-2 A/G, entspricht Splice- Defekt SDHB c. 394 T/C, entspricht L87S Mutat: nt: Wildtyp: A 390 A C 391 C T 392 T T 393 T Codon 86 C 394 T G 395 G A 396 A Codon 87 C 397 C C 398 C Codon 88 T 434 T Codon 100 T 435 T A 436 G T 437 T G 438 G Codon 101 T T C G T T C A Splicesequ. G 420 G G 421 G C 422 C Codon 96 Abbildung 25: Das gelb markierte Kästchen kennzeichnet die mutierte Splicesequenz. Die Wildtypsequenz (untere Zeile) dieser Nukleotide lautet TCA. Durch den Basenaustausch an Position 421-2 entseht die mutierte Sequenz (obere Zeile) TCG. SDHB c. 558-3 C/G, entspricht Splice- Defekt SDHB c. 436 G/A, entspricht C101Y C 433 C T T Intron Abbildung 24: Das gelb markierte Kästchen kennzeichnet das mutierte Codon 87. Die Wildtypsequenz (untere Zeile) der Nukleotide 393, 394 und 395 lautet TTG. Durch den Basenaustausch an Position 394 entseht die mutierte Sequenz (obere Zeile) TCG. Mutat: T nt: 432 Wildtyp: T Mutat: C nt: Wildtyp: C C 439 C A 440 A Codon 102 Abbildung 26: Das gelb markierte Kästchen kennzeichnet das mutierte Codon 101. Die Wildtypsequenz (untere Zeile) der Nukleotide 435, 436, 437 lautet TGT. Durch den Basenaustausch an Position 436 entseht die mutierte Sequenz (obere Zeile) TAT. Abbildung 27: Das gelb markierte Kästchen kennzeichnet die mutierte Splicesequenz. Die Wildtypsequenz (untere Zeile) dieser Nukleotide lautet CAG. Durch den Basenaustausch an Position 558-3 entseht die mutierte Sequenz (obere Zeile) GAG. SDHB c. 708 T/C, entspricht C192R SDHB c. 721 G/A, entspricht C196Y Mutat: T nt: 705 Wildtyp: T G 706 G Codon 191 C 707 C C 708 T G 709 G Codon 192 T 710 T A 711 A G 712 G C 713 C Codon 193 Abbildung 28: Das gelb markierte Kästchen kennzeichnet das mutierte Codon 192. Die Wildtypsequenz (untere Zeile) der Nukleotide 708, 709 und 710 lautet TGT. Durch den Basenaustausch an Position 708 entseht die mutierte Sequenz (obere Zeile) CGT. Mutat: A nt: 717 Wildtyp: A G 718 G Codon 195 C T 719 720 C T A 721 G C C 722 723 C C Codon 196 C 724 C C 725 C Codon 197 Abbildung 29: Das gelb markierte Kästchen kennzeichnet das mutierte Codon 196. Die Wildtypsequenz (untere Zeile) der Nukleotide 720, 721 und 722 lautet TGC. Durch den Basenaustausch an Position 721 entseht die mutierte Sequenz (obere Zeile) TAC. 39 SDHB c. 847/849 del TCTC, entspricht F238frameshift Mutat: nt: Wildtyp: T 849 T C 850 C T 851 T A 852 C Codon 239 T 853 T A 854 A C 855 T Codon 240 C 856 A G 857 C Codon 241 Abbildung 30:.Das gelb markierte Kästchen kennzeichnet die Folge der Deletion der 4 Aminosäuren TCTC an Position 847/849 der Wildtypsequenz. Die Folge ist die Verschiebung des Leserasters für die Aminosäuretranslation. Das markierte Codon 240 und 241 kennzeichnet die Änderung. SDHB c. 881 C/A, entspricht C249X Mutat: A nt: 876 Wildtyp: A A 877 A Codon 248 C 878 C T 879 T G 880 G A 881 C Codon 249 SDHB c. 859 G/A, entspricht R242H Mutat: T nt: 855 Wildtyp: T A 856 A Codon 241 C 857 C C A 858 859 C G C 860 C Codon 242 T 861 T G 862 G C 863 C Codon 243 Abbildung 31: Das gelb markierte Kästchen kennzeichnet das mutierte Codon 242. Die Wildtypsequenz (untere Zeile) der Nukleotide 858, 859 und 860 lautet CGC. Durch den Basenaustausch an Position 859 entseht die mutierte Sequenz (obere Zeile) CAC. SDHB c. 899+1 G/A, entspricht Splice- Defekt A 882 A C 883 C A 884 A Codon 250 Abbildung 32: Das gelb markierte Kästchen kennzeichnet das mutierte Codon 249. Die Wildtypsequenz (untere Zeile) der Nukleotide 879, 880 und 881 lautet TGC. Durch den Basenaustausch an Position 881 entseht die mutierte Sequenz (obere Zeile) TGA. Abbildung 33: Das gelb markierte Kästchen kennzeichnet die mutierte Splicesequenz. Die Wildtypsequenz (untere Zeile) dieser Nukleotide lautet GTA. Durch den Basenaustausch an Position 899+1 entseht die mutierte Sequenz (obere Zeile) ATA. 40 Intraexonisch gelegenen Polymorphismen des SDHB-Gens wiesen 43 Patienten auf. 7 verschieden Polymorphismen wurden entdeckt (Tabelle 11). Sie finden sich in Exon 1, 4, 5 und 7. Für intraexonisch lokalisierte Polymorphismen des SDHB-Gens wurde eine Prävalenz von 10 % (43/420) ermittelt. Polymorphismus (Nukleotid) Resultat (Aminosäure) Anzahl der Patienten Anzahl positiver Kontrollproben Exon 104 T/A UTR* 0 1 1 152 A/C A6A 21 4 1 158 C/T S8S 3 0 1 434 T/C S100S 4 1 4 529 A/C** T101P 2 0 4 621 T/C S163P 12 2 5 809 C/T D225D 1 0 7 Tabelle 11: Darstellung der Polymorphismen des SDHB-Gens, sortiert nach Nukleotidnummern der cDNA, die in Kapitel 1.2.1. dargestellt ist. Es wurden nur Polymorphismen tabellarisch erfasst, die in den 8 verschiedenen Exons des SDHB-Gens gefunden wurden. *UTR steht für die 133bp große untranslatierte Region am 5'Ende des Exon 1. **Die Sequenzveränderung SDHB c. 529 A/C wurde als Polymorphismus interpretiert, da sie nicht mit der Erkrankung cosegregierte (zusammen vererbt wurden). 3.2.2. Molekulargenetische Testung von Verwandten Die Einwilligung zur molekulargenetischen Untersuchung auf konstitutionelle Mutationen des SDHB-Gens lag von 43 Verwandten von 15 Indexfällen vor. Nach entsprechender Aufklärung wurden jeweils 10 ml EDTA Blut zur Untersuchung auf die Mutation des entsprechenden Indexfalles zugesandt. Untersucht wurden Verwandte folgender Indexfälle: B2, B3, B4, B5, B9, B10, B13, B16, B17, B18, B19, B20, B21, B22 und B23. Von Verwandten der Indexfälle B1, B6, B7, B8, B11, B12, B14, B15, B24 und B25 stand kein Blut zur molekulargenetischen Untersuchung zur Verfügung. Als Träger konstitutioneller Mutationen des SDHB-Gens wurden 26 Personen identifiziert. Bei 17 Personen wurden die entsprechenden Mutationen ausgeschlossen. 41 Tabelle 12 fasst die Ergebnisse des molekulargenetischen Screenings der Verwandten zusammen. Familien Familie B2: Indexfall Mutter Familie B3: Indexfall Tochter Familie B4: Indexfall Mutter Vater Familie B5: Indexfall Mutter Vater Familie B9: Indexfall Mutter Familie B10: Indexfall Mutter Vater Bruder Familie B13: Indexfall Mutter Schwester Schwester Schwester Nichte Nichte Nichte Nichte Neffe Neffe Familie B16: Indexfall Mutter Vater Familie B17: Indexfall Tochter Tochter Familie B18: Indexfall Bruder Schwester Neffe Familie B19: Indexfall Mutter Vater Schwester Familie B20: Indexfall Sohn Nationalität Alter Geschlecht Testergebnis 13 - w w 213 C/T Normalbefund 48 - w w 221 ins CCAG Normalbefund 14 61 67 w w m 270 C/G 270 C/G Normalbefund 15 62 m w m 270 C/G 270 C/G Normalbefund 19 52 m w 300 del CCTCA 300 del CCTCA 15 48 48 14 m w m m 328 T/C Normalbefund 328 T/C 328 T/C 42 79 46 53 39 11 16 12 22 19 18 w w w w w w w w w m m 394 T/C Normalbefund Normalbefund 394 T/C 394 T/C Normalbefund Normalbefund 394 T/C 394 T/C 394 T/C Normalbefund 10 47 - w w m 436 G/A 436 G/A Normalbefund 65 33 35 m w w 558-3 C/G 558-3 C/G 558-3 C/G 26 54 52 25 w m w m 708 T/C 708 T/C 708 T/C Normalbefund 19 42 22 m w m w 721 G/A 721 G/A Normalbefund 721 G/A 34 12 w m 847-849 del TCTC Normalbefund Deutsch Polnisch Deutsch Deutsch Spanisch Deutsch Französisch Deutsch Spanisch Polnisch Polnisch Polnisch 42 Polnisch Familie B21: Indexfall Mutter Bruder Tochter Nichte Familie B22: Indexfall Schwester Sohn Sohn Sohn Tochter Neffe Familie B23: Indexfall Tochter Bruder 16 60 37 8 3 w w m w w 847-849 del TCTC Normalbefund 847-849 del TCTC 847-849 del TCTC 847-849 del TCTC 35 38 11 18 14 14 15 m w m m m w m 859 G/A 859 G/A 859 G/A 859 G/A Normalbefund 859 G/A Normalbefund 32 34 - m w m 859 G/A, 859 G/A 859 G/A Deutsch Deutsch Tabelle 12: Übersicht aller 43 getesteten Verwandten; Die Altersangabe bezieht sich entweder auf die Manifestation des Primärtumors oder den Zeitpunkt der molekulargenetischen Testung der Verwandten. Die letzte Spalte fasst alle gewonnenen Ergebnisse des molekulargenetischen Screenings zusammen. Konstitutionelle Mutationen des SDHB-Gens wurden sowohl über das väterliche als auch über das mütterliche Genom vererbt. Eine Manifestation der Erkrankung wurde bei beiden Vererbungswegen beobachtet. Das Vererbungsmuster entspricht einem autosomal- dominanten Erbgang. Abbildung 34 verdeutlicht anhand der Stammbäume der untersuchten Familien das Vererbungsmuster. Abbildung 34,Teil 1: Stammbaumanalysen der 15 Indexfälle Familie B4: SDHB c. 270 C/G, R46G - I:1 Familie B5: SDHB c. 270 C/G, R46 + - + I:2 I :1 I :2 I I :1 II:1 II:2 I:1 II:3 II:3 ist der Indexfall. Es handelt sich um eine Patientin, die im Alter von 14 Jahren an einem benignen, extaadrenal abdominellen Phäochromozytom erkrankte. Eine molekulargenetische Testung erfolgte für die Mutter und den Vater der Indexpatientin. Familie B9: SDHB c. 300 del CCTCA, P56frameshift + I:1 I:2 II:1 II:1 ist der Indexfall. Es handelt sich um einen Patienten, der im Alter von 19 Jahren an einem benignen, adrenalen Phäochromozytom erkrankte. Eine molekulargenetische Testung erfolgte für die Mutter des Indexpatienten. II:1 ist der Indexfall. Es handelt sich um einen Patienten, der im Alter von 15 Jahren an einem malignen, extra-adrenal thorakalen Phäochromozytom erkrankte. Eine molekulargenetische Testung erfolgte für die Mutter und den Vater des Indexpatienten.. Familie B10: SDHB c. 328 T/C, L65P + I:1 - I:2 + II:1 II:2 II:2 ist der Indexfall. Es handelt sich um einen Patienten, der im Alter von 15 Jahren an einem benignen, extraadrenal abdominellen Phäochromozytom erkrankte. Eine molekulargenetische Testung erfolgte für die Mutter, den Vater und den Bruder des Indexpatienten. 43 Abbildung 34, Teil 2:Stammbaumanalyse der 15 Indexfälle Familie B16: SDHB c. 436 G/A, C101Y Familie B13: SDHB c. 394 T/C, L87S - I:1 II:1 - I:2 - II:2 + + II:3 II:4 - + III:1 III:2 - + I:1 I:2 II:1 + + III:3 III:4 - III:5 III:6 II:1 ist der Indexfall. Es handelt sich um eine Patientin, die im Alter von 42 Jahren an einem benignen, extraadrenal abdominellen Phäochromozytom erkrankte. Eine molekulargenetische Testung erfolgte für die Mutter, 3 Schwestern, 4 Nichten und 2 Neffen der Indexpatientin. Ein Neffe (III:1) erkrankte im Alter von 19 Jahren an einem benignen, extra-adrenal abdominellen Phäochromozytom II:1 ist der Indexfall. Es handelt sich um eine Patientin, die im Alter von 10 Jahren an einem benignen, extraadrenal abdominellen Phäochromozytom erkrankte. Eine molekulargenetische Testung erfolgte für die Mutter und den Vater der Indexpatientin. Familie B17: SDHB c. 558-3 C/G, Splice- Defekt Familie B18: SDHB c. 708 T/C, C192R I:1 I:1 I:2 + + II:1 II:2 II:4 I:2 + - + II:1 II:2 II:3 III:1 I:1 ist der Indexfall. Es handelt sich um einen Patienten, der im Alter von 65 Jahren an einem malignen, extraadrenal abdominellen Phäochromozytom erkrankte. Eine molekulargenetische Testung erfolgte für 2 Töchter des Indexpatienten. Eine Tochter (II:1) erkrankte im Alter von 33 Jahren an einem benignen, extra-adrenal abdominellen Phäochromozytom II:3 ist der Indexfall. Es handelt sich um eine Patientin, die im Alter von 26 Jahren an einem benignen, extraadrenal abdominellen Phäochromozytom erkrankte. Eine molekulargenetische Testung erfolgte für die Schwester, den Bruder und 1 Neffen der Indexpatientin. Familie B19 SDHB c. 721 T/C, C196Y Familie B21: SDHB c. 847-849 del TCTC, F238Frameshift - + I:1 I:2 II:1 II:2 I:1 + + + II:1 III:2 II:1 ist der Indexfall. Es handelt sich um einen Patienten, der im Alter von 19 und 21 Jahren an einem benignen, extra-adrenal abdominellen Phäochromozytom der Harnblase erkrankte. Eine molekulargenetische Testung erfolgte für die Mutter, den Vater und die Schwester des Indexpatienten. - I:2 II:2 + II:3 III:1 II:2 ist der Indexfall. Es handelt sich um eine Patientin, die im Alter von 16 Jahren an einem benignen, extraadrenal abdominellen Phäochromozytom erkrankte. Rezidive des Primärtumors traten im Alter von 19, 21 und 26 Jahren auf. Im Alter von 24 Jahren wurde zusätzlich ein Nierenzellkarzinom diagnostiziert. Eine molekulargenetische Testung erfolgte für die Mutter, den Bruder, die Tochter und die Nichte der Indexpatientin. Der Bruder erkrankte im Alter von 26 Jahren an einem Nierenzellkarzinom und im Alter von 37 Jahren an einem extra-adrenal thorakalem Phäochromozytom. 44 Abbildung 34, Teil 3:Stammbaumanalyse der 15 Indexfälle Familie B22: SDHB c. 859 G/A, R242H I:1 Familie B23: SDHB c. 859 G/A, R242H I:1 I:2 + + II:1 II:2 II:3 II:4 I:2 II:1 II:5 II:2 + II:6 III:1 - III:7 + + III:1 III:2 - + III:3 III:4 III:5 III:6 II:5 ist der Indexfall. Es handelt sich um einen Patienten, der im Alter von 35 Jahren an einem benignen, adrenalen Phäochromozytom erkrankte. Rezidive des Primärtumors traten im Alter von 39 und 40 Jahren auf. Eine molekulargenetische Testung erfolgte für die Schwester, die Tochter, 3 Söhne und den Neffen des Indexpatienten. II:2 ist der Indexfall. Es handelt sich um einen Patienten, der im Alter von 32 Jahren an einem malignen Tumor des Glomus caroticum erkrankte. Der Patient erlag im Alter von 64 Jahren den Folgen seiner Krankheit. Eine molekulargenetische Testung erfolgte für den Bruder und die Tochter des Indexpatienten. Anamnestisch konnte evaluiert werden, dass der bereits verstorbene Bruder ebenfalls an einem malignen Tumor des Glomus caroticum erkrankt war. Abbildung 34: Darstellung der Stammbäume der molekulargenetisch untersuchten Indexfamilien. Der Stammbaum der Familie B2, B3 und B20 ist nicht dargestellt. Bei diesen Indexpatienten stand DNA-Material nur jeweils eines Angehörigen zur Verfügung und dieser war kein Träger der entsprechenden Mutation. Männer werden durch Vierecke, Frauen durch Kreise symbolisiert. Durchgestrichene Symbole kennzeichnen bereits verstorbene Verwandte. Ausgefüllte Symbole stellen die jeweiligen Indexfälle dar, die an einem Phäochromozytom oder Glomustumor erkrankt waren. Symbole die ein Plus besitzen, kennzeichnen im Rahmen des Screenings der Verwandten entdeckte Mutationsträger. Symbole die ein Minus besitzen, kennzeichnen molekulargenetisch getestete Verwandte, die keine Mutationsträger sind. 3.2.3. Klinische Charakterisierung der Träger von Mutationen des SDHB-Gens Die molekulargenetische Untersuchung auf konstitutionelle Mutationen des SDHB-Gens der 420 Patienten und der 43 Verwandten ergab eine Gesamtzahl von 53 Mutationsträgern. Diese sind Träger 18 verschiedener Mutationen des SDHB-Gens. Sie setzten sich aus 25 Indexfällen und 26 positiv getesteten Angehörigen zusammen. Zusätzlich wurden 2 weitere Personen als Mutationsträger angenommen. Es handelt sich um 2 Angehörige des Indexfalles B6, die im Alter von 45 bzw. 55 Jahren an einem Tumor des Glomus caroticum erkrankten. Der Indexfall B6, Träger der Mutation SDHB c. 271 G/A erkrankte im Alter von 50 Jahren an einem malignen, adrenalen Phäochromozytom. DNA der Angehörigen stand bisher zur molekulargenetischen Untersuchung nicht zur Verfügung. Von 42 Mutationsträger liegen Daten klinischer Untersuchungen vor. Dazu gehörten Bestimmungen von Adrenalin und Noradrenalin im 24h Sammelurin sowie Ergebnisse der 45 Computertomographie / Magnetresonanztomographie von Thorax und Abdomen, Hals und Schädelbasis. 10 Mutationsträger waren asymptomatisch. Das klinische Screening ergab keine Hinweise auf das Vorliegen eines Phäochromozytoms, eines Paraganglioms oder eines Glomustumors. Von 11 Mutationsträgern stehen die Ergebnisse des klinischen Screenings noch aus. Sie wurden von der Auswertung der klinischen Daten ausgeschlossen. 32 Mutationsträger (25 Indexfälle + 5 positiv getestete Verwandte + 2 Angehörige mit angenommenen Mutationen des SDHB-Gens) waren symptomatisch. Bei der Auswertung der klinischen Daten wurden Patienten mit mehreren, verschiedenen Tumoren, z.B. einem Phäochromozytom und einem Glomustumor, doppelt berücksichtigt. Ebenso wurden Patienten mit unterschiedlichen Tumorlokalisationen, z.B. einem adrenalen und einem extraadrenalen Tumor, doppelt berücksichtigt. Das durchschnittliche Manifestationsalter der 32 symptomatischen Mutationsträger lag bei 31,3 Jahren. Bei Patienten mit einem Glomustumor manifestierte sich die Erkrankung durchschnittlich mit 43,7 Jahren, bei Patienten mit der Diagnose eines Phäochromozytom bei durchschnittlich 27,8 Jahren. 88 % (28/32) der Patienten zeigten ein Phäochromozytom. 31 % (10/32) der Patienten zeigten einen Glomustumor. 2 Patienten (6,3 %) entwickelten im Verlauf ihrer Erkrankung sowohl ein Phäochromozytom als auch einen Glomustumor. Ein adrenal lokalisiertes Phäochromozytom wiesen 28 % (9/32) der Mutationsträger auf. Ein extra-adrenal adominelles Phäochromozytom zeigten 50 % (16/32) der Patienten (Abbildung 35) und 9 % (3/32) zeigten ein extra-adrenal thorakales Phäochromozytom. A B Abbildung 35. CT des Abdomens und Jod131 MIBG- Szintigraphie des Indexpatienten B19. Der Indexpatient B19, Träger der Mutation SDHB c. 721 T/C, erkrankte im Alter von 19 und 21 Jahren an einem benignen, extra-adrenal abdominell lokalisierten Phäochromozytom. Die Besonderheit dieses Falls liegt in der außergewöhnlichen Lokalisation des Phäochromozytoms. Es manifestierte sich an der Wand der Harnblase. A: CT des Abdomens. Der Pfeil markiert das benigne, extraadrenal abdominelle Phäochromozytom der Harnblase. B: Jod131 MIBG- Szintigraphie. Der Pfeil markiert das Phäochromozytom der Harnblase. 46 Ein multifokales Wachstum fand sich bei 9 der 32 (28 %) Patienten. 11 von 32 (34 %) Patienten waren an einem malignen Phäochromozytom oder einem Glomustumor erkrankt; ein malignes Phäochromozytom lag bei 8 Patienten vor, einen malignen Glomustumor zeigten 3 Patienten. Sie wiesen bei Diagnosestellung bzw. im Krankheitsverlauf vor allem ossäre und pulmonale Metastasen auf (Abbildung 36). 5 dieser Personen erlagen in unterschiedlichem Alter ihrer Grunderkrankung. Tabelle 13 fasst die klinischen und molekulargenetischen Daten dieser Patienten zusammen. Fall Geschlecht Lokalisation Status 2004 Krankheitsdauer Mutation B1 B5 B7 B6 B7 B11 B17 B20 B22 B23 B25 w m m m w w m w m m m thorakal extra-adrenal, abdominal Glomustumor adrenal extra-adrenal, abdominal adrenal extra-adrenal, abdominal adrenal Glomustumor Glomustumor extra-adrenal, abdominal L 34 L 35 L 45 L 56 T 45 T 28 L 68 T 36 T 64 T 64 L 66 3 20 11 6 32 11 3 2 32 2 6 155 del C 270 C/G 271 G/A 271 G/A 300 del CCTCA 394 T/C 558-3 C/G 847/849 del TCTC 859 G/A 859 G/A 899+1 G/A Tabelle 13: Übersicht aller Patienten, die an einem malignen Phäochromozytom, Paragangliom oder einem malignen Glomustumor erkrankt sind oder waren. Die Spalte "Fall" gibt die verschlüsselten Ziffern der betroffenen Indexfamilien wieder. Die Spalte "Status 2004" gibt an, ob die Patienten noch leben (L) oder bereits verstorben (T) sind. Die zusätzliche Zahl gibt das Alter der Patienten im Jahr 2004 an oder das Alter bei Tod des Patienten. A Abbildung 36: Darstellung einer Jod131-MIBG-Szintigraphie des Indexfalles B23. Der Indexpatient B23, Träger der Mutation SDHB c. 859 G/A erkrankte im Alter von 31 Jahren an einem malignen Tumor des Glomus caroticum. Beweisend für die Malignität des Tumors galten vor allem Wirbelkörpermetastasen. Im Alter von 64 Jahren erlag der Patient seiner malignen Erkrankung. In beiden Abbildungen sind die Befund der Jod131 MIBG-Szintigraphie dargestellt. Deutlich zu sehen ist eine ausgeprägte Metastasierung in die Wirbelkörper. A: Ganzkörperansicht von vorne (links) und hinten (rechts). B: Seitansicht von links, der unteren Thorax und Abdomen-Region. B 47 Maligne Zweiterkrankungen in Form eines papillären Schilddrüsenkarzinoms, einer akuten myeloischen Leukämie und zweier Nierenzellkarzinome wurden bei 4 Patienten diagnostiziert. Der Indexpatient B10, Träger der Mutation SDHB c. 328 T/C, erkrankte im Alter von 14 Jahren an einem papillären Schilddrüsenkarzinom. Mit 15 Jahren zeigte der Patient ein benignes, extra-adrenal abdominell lokalisiertes Phäochromozytom. Die Mutter des Indexpatienten hatte ebenfalls ein papilläres Schilddrüsenkarzinom, war aber keine Trägerin der Mutation. Die Indexpatientin B11, Trägerin der Mutation SDHB c. 394 T/C erkrankte im Alter von 17 Jahren an einem malignen, adrenalen Phäochromozytom. Sie verstarb mit 28 Jahren an einer akuten myeloischen Leukämie. Die Indexpatientin B21, Trägerin der Mutation SDHB c. 847/849 del TCTC, erkrankte im Alter von 16 Jahren an einem benignen, extra-adrenal abdominell lokalisierten Phäochromozytom. Rezidive des Primärtumors traten im Alter von 19, 21 und 26 Jahren auf. Mit 24 Jahren zeigte sie zusätzlich ein Nierenzellkarzinom (Abbildung 37). Der Bruder der Indexpatientin B21, ebenfalls Träger der Mutation SDHB c. 847/849 del TCTC, zeigte im Alter von 26 Jahren ein Nierenzellkarzinom. 11 Jahre später, mit 37 Jahren, erkrankte er an einem extra-adrenal thorakal lokalisierten Phäochromozytom. A A B B Abbildung 37: CT-Darstellung des Nierenzellkarzinoms der linken Niere der Indexpatientin B21. Sie ist Trägerin der Mutation 847/849 del TCTC, entspricht F238Frameshift. Im Alter von 16 Jahren erkrankte sie erstmals an einem extra-adrenal abdominellen Phäochromozytom. Rezidive des Primärtumors traten mit 19, 21 und 26 Jahren auf. Im Alter von 24 wurde das oben dargestellte Nierenzellkarzinom diagnostiziert. Abbildungen A und B: Darstellung des Tumors in unterschiedlicher Schnittebene. Die Pfeile markieren den Tumor. 48 Tabelle 14 zeigt klinische und molekulargenetische Daten der 32 erkrankten, symptomatischen Träger konstitutioneller Mutationen des SDHB-Gens. Nationalität Indexfall Alter Pheo / GT Anzahl der Tumore Lokalisation Pheo Dignität Deutsch B1 31 Pheo 1 thorakal maligne Maligne Zweiterkrankung Mutation 155 del c Deutsch B2 13 Pheo 1 abdominell 213 C/T Polnisch B3 48 Pheo 1 adrenal 221 ins CCAG Deutsch B4 14 Pheo 1 abdominell 270 C/G Deutsch B5 15 Pheo 1 1 thorakal maligne adrenal maligne GT Kanadier B6 50 Pheo 1 Kanadier Angehörige 45 GT 1 Kanadier Angehörige 55 GT 1 Deutsch B7 34 GT 1 270 C/G 271 G/A 271 G/A 271 G/A maligne 271 G/A Deutsch B8 36 Pheo 1 adrenal 291 G/A Spanisch B9 19 Pheo 1 adrenal 300 del CCTCA Spanisch Mutter 13 Pheo 1 abdominell Deutsch B10 15 Pheo 1 abdominell Deutsch B11 17 Pheo 1 adrenal maligne maligne 300 del CCTCA papilläres SD-CA 328 T/C AML 394 T/C Deutsch B12 54 Pheo 1 abdominell 394 T/C Französisch B13 42 Pheo 1 abdominell 394 T/C Französisch Neffe 13 Pheo 1 abdominell Deutsch B14 43 GT 1 Deutsch B15 45 GT 2 Deutsch B16 10 Pheo 1 abdominell Spanisch B17 65 Pheo 1 abdominell Spanisch Tochter 33 Pheo 1 abdominell 558-3 C/G Polnisch B18 26 Pheo 1 abdominell 708 T/C Polnisch B19 19 Pheo 2 abdominell Polnisch B20 34 Pheo 1 adrenal 394 T/C 421-2 A/G 421-2 A/G 436 G/A maligne 558-3 C/G 721 G/A maligne 847/849 del TCTC Polnisch B21 16 Pheo 3 abdominell RCC 847/849 del TCTC Polnisch Bruder 37 Pheo 1 thorakal RCC 847/849 del TCTC Deutsch B22 35 Pheo 1 adrenal Deutsch B23 31 GT 1 Deutsch Bruder 53 GT 1 Deutsch B24 12 Pheo 1 adrenal Deutsch B25 21 Pheo GT 1 1 adrenal 859 G/A maligne 859 G/A maligne 859 G/A 881 C/A maligne 899+1 G/A Tabelle 14: Klinische und molekulargenetische Daten der 32 symptomatischen Träger konstitutioneller Mutationen des SDHB-Gens. Das Alter der genannten 32 symptomatischen Mutationsträger bezieht sich auf den Zeitpunkt der Manifestation des Primärtumors. Dieser ist fett gedruckt markiert. Pheo steht als Abkürzung für Phäochromozytom, GT steht als Abkürzung für Glomustumor. Alle nicht mit "maligne" gekennzeichneten Patienten, wiesen Tumoren benigner Dignität auf. 49 Tabelle 15 fasst die wesentlichen Daten der 32 erkrankten, symptomatischen Träger konstitutioneller Mutationen des SDHB-Gens zusammen. Klinische Charakteristika Symptomatische SDHB- Mutationsträger n = 32 Phäochromozytome: adrenal extra-adrenal abdominell extra-adrenal thorakal 28 % 50 % 9% (9/32) (16/32) (3/32) Glomustumoren 31 % (10/32) multifokale Tumoren 28 % (9/32) maligne Tumoren 34 % (11/32) maligne Zweiterkrankungen 13 % (4/32) Tabelle 15: Übersicht der erhobenen klinischen Daten der Träger konstitutioneller Mutationen des SDHB-Gens. Von den ursprünglichen 53 Mutationsträgern sind 21 Mutationsträger ausgeschlossen. Von diesen 21 Personen gelten 10 als asymptomatisch und von 11 Personen standen die Befunde und Ergebnisse des klinischen Screenings noch aus. Patienten mit mehreren unterschiedlichen Tumoren und Tumorlokalisationen wurden mehrfach gewertet. 3.2.4. Penetranz Die altersabhängige Penetranz von Mutationen des SDHB-Gens wurde mittels einer KaplanMeier-Berechnung dargestellt (Abbildung 38, 39 und 40). Grundlage der Darstellung ist das Manifestationsalter. Die Berechnung basiert auf den Daten von 42 Mutationsträgern. Diese setzten sich aus 32 symptomatischen Patienten mit konstitutionellen Mutationen des SDHBGens und 10 asymptomatischen Mutationsträgern zusammen. Von der Analyse ausgenommen sind 11 positiv getestete Verwandte der Indexfälle, bei denen Ergebnisse und Befunde des klinischen Screenings noch ausstehen. Abbildung 38 zeigt die Penetranzdarstellung für Patienten mit Phäochromozytomen, Paragangliomen und Glomustumoren. Abbildung 39 zeigt die Penetranzdarstellung für Patienten mit adrenalen Phäochromozytomen. Abbildung 40 zeigt die Penetranzdarstellung für Patienten mit Glomustumoren des Kopfes und des Halses. 50 Abbildung 38: Altersabhängige Penetranzdarstellung für Patienten mit Phäochromozytomen, Paragangliomen und Glomustumoren. Abbildung 39: Altersabhängige Penetranzdarstellung für Patienten mit adrenalen Phäochromozytomen. Abbildung 40: Altersabhängige Penetranzdarstellung für Patienten mit Glomustumoren des Kopfes und des Halses. 51 Für Mutationen des SDHB-Gens zeigte sich eine Penetranz von 50 % mit Erreichen des 35. Lebensjahres. Dies bedeutet dass 50 % aller SDHB- Mutationsträger bis zum Alter von 35 Jahren ein Phäochromozytom, ein Paragangliom oder einen Glomustumor entwickeln. Die altersabhängige Penetranz steigt auf 77 % mit Erreichen des 50. Lebensjahres an (Abbildung 38). Die altersabhängige Penetranz für die Manifestation adrenaler Phäochromozytome bei Patienten mit Mutationen des SDHB-Gens liegt mit Erreichen des 48. Lebensjahres bei 25 % (Abbildung 39). Für die Manifestation von Glomustumoren liegt die Penetranz bei 25 % mit Erreichen des 45. Lebensjahres und mit Erreichen des 55. Lebensjahres bei 50 % (Abbildung 40). 3.3. Konstitutionelle Mutationen des SDHC-Gens 3.3.1. Molekulargenetisches Screening auf konstitutionelle Mutationen des SDHC-Gens Als Träger konstitutioneller Mutationen des SDHC-Gens wurden 3 nicht miteinander verwandte Patienten entdeckt. Eine Patientin ist Teil des deutschen Glomustumor-Registers. Sie ist Trägerin der Mutation SDHC c. 39 C/A, entspricht C13X. Eine Patientin wurde aus Spanien eingebracht. Sie ist Trägerin der Mutation SDHC c. 1 A/G, entspricht M1V. Eine Patientin wurde aus Polen eingebracht. Sie ist Trägerin der Mutation SDHC c. 214 C/T, entspricht R72C (Tabelle 16). Nationalität Tumor Indexfall Alter Mutation (Nukleotid) Resultat (Aminosäure) Mutationstypus Exon Spanisch GT C1 52 1 A/G M1V Verlust des Startcodons 1 Deutsch GT C2 37 39 C/A C13X Stopcodon 2 Polnisch GT C3 62 214 C/T R72C Missense 4 Tabelle 16: Darstellung der im Rahmen des molekulargenetischen Screenings gefundenen Mutationen des SDHC- Gens. GT ist eine Abkürzung für Glomustumor. Aus Datenschutzgründen werden die Namen der Indexfälle mit den Ziffern C1, C2 und C3 verschlüsselt. Die Reihenfolge der Nummerierung orientiert sich an der Position der Mutation in der Nukleotidsequenz. Die Mutationen finden sich in Exon 1, 2 und 4. Das Mutationsspektrum setzt sich aus einer Missense-Mutation, die einen Verlust des Startcodons ATG der Aminosäuretranslation bedingt, einer Missense-Mutationen, die in einem Aminosäureaustausch resultiert und einer Stopcodon-Mutation zusammen (Erklärungen siehe Kapitel 1.2.3. Mutationen und Polymorphismen, S.20). 52 Für die Gesamtzahl der Indexfälle, d.h. für Patienten mit Phäochromozytomen und / oder Glomustumoren, wurde eine Prävalenz von 0,7 % (3/420) ermittelt. Da alle 3 Patienten einen Glomustumor zeigten, wurde bei dieser Patientengruppe eine Prävalenz von 3,5 % (3/84) für Mutationen des SDHC-Gens ermittelt. Die wesentlichen Ergebnisse des molekulargenetischen Screenings sind in den Abbildungen 41 bis 46, als Befunde der SSCP-Analyse und als Befunde der Sequenzierung dargestellt. K Bahn 1 Abbildung 41: K steht für Kontrollprobe, dies bedeutet, dass die cDNA in Exon 1 dieser Probe keine Sequenzveränderung aufweist. Bahn 1: zeigt das SSCPBandenmuster der Sequenzabweichung SDHC c. 1 A/G. Durch den Basenaustausch an Position 1 ändert sich das Laufverhalten im SSCP-Gel. Die mit einem Pfeil markierte Extrabande entseht. K Bahn 1 Abbildung 42: K steht für Kontrollprobe, dies bedeutet, dass die cDNA in Exon 2 dieser Probe keine Sequenzveränderung aufweist. Bahn 1: zeigt das SSCPBandenmuster der Sequenzabweichung SDHC c. 39 C/A. Durch den Basenaustausch an Position 39 ändert sich das Laufverhalten im SSCP-Gel. Die mit einem Pfeil markierte Extrabande entseht. SDHC c. 1 A/G, entspricht M1V K Bahn 1 Abbildung 43: K steht für Kontrollprobe, dies bedeutet, dass die cDNA in Exon 4 dieser Probe keine Sequenzveränderung aufweist. Bahn 1: zeigt das SSCPBandenmuster der Sequenzabweichung SDHC c. 214 C/T. Durch den Basenaustausch an Position 214 ändert sich das Laufverhalten im SSCP-Gel. Die mit einem Pfeil markierte Extrabande entseht. SDHC c. 39 C/A, entspricht C13X Codon 12 Abbildung 44: Das gelb markierte Kästchen kennzeichnet das mutierte Codon 1. Die Wildtypsequenz (untere Zeile) der Nukleotide 1, 2 und 3 lautet ATG. Durch den Basenaustausch an Position 1 entseht die mutierte Sequenz (obere Zeile) GTG. Codon 13 Codon 14 Abbildung 45: Das gelb markierte Kästchen kennzeichnet das mutierte Codon 13. Die Wildtypsequenz (untere Zeile) der Nukleotide 37, 38 und 39 lautet TGC. Durch den Basenaustausch an Position 39 entseht die mutierte Sequenz (obere Zeile) TGA. SDHC c. 214 C/T, entspricht R72C Abbildung 46: Das gelb markierte Kästchen kennzeichnet das mutierte Codon 72. Die Wildtypsequenz (untere Zeile) der Nukleotide 214, 215 und 216 lautet CGT. Durch den Basenaustausch an Position 214 entseht die mutierte Sequenz (obere Zeile) TGT. Codon 71 Codon 72 Codon 73 53 3.3.2. Klinische Charakterisierung der Träger von Mutationen des SDHC-Gens Die Auswertung der klinischen Daten von Trägern konstitutioneller Mutationen des SDHCGens ergab folgende Ergebnisse. Die Indexpatientin C1, Trägerin der Mutation SDHC c. 1 A/G, erkrankte im Alter von 52 Jahren an einem benignen Tumor des Glomus caroticum. Anamnestisch gab sie an, dass ihr Neffe ebenfalls an einem Glomustumor der Kopf-Hals-Region erkrankt war. Blut des Neffen stand nicht zur molekulargenetischen Untersuchung auf konstitutionelle Mutationen des SDHC-Gens zur Verfügung. Eine Nachuntersuchung beider Personen steht noch aus. Die Indexpatientin C2, Trägerin der Mutation SDHC c. 39 C/A, erkrankte im Alter von 38 Jahren an einem benignen Tumor des Glomus jugulare rechts. Diese Patientin wurde 2004 nachuntersucht. Die Katecholamine im 24h Sammelurin waren im Normbereich. Das MRT von Thorax und Abdomen zeigte keine pathologischen Befunde. Das MRT der SchädelbasisHals-Region zeigte den einen, bereits bekannten Tumor des Glomus jugulare rechts (Abbildung 47). Die Größe von 4 x 2,5 cm war identisch mit dem Vorbefund von 2002. Blut von Verwandten der Indexpatientin stand bislang nicht zur molekulargenetischen Untersuchung auf konstitutionelle Mutationen des SDHC-Gens zur Verfügung. A B Abbildung 47: Darstellung des Glomus jugulare-Tumors rechts der Indexpatientin C2 mittels MR-Angiographie und MRT der Schädelbasis-Hals-Region. A: MR-Angiographie. Die Pfeile markieren den Tumor. B: MRT der HalsSchädelbasis-Region. Die Pfeile markieren den Tumor. 54 Die Indexpatientin C3, Trägerin der Mutation SDHC c. 214 C/T, erkrankte im Alter von 62 Jahren an einem benignen Tumor des Glomus tympanicum. Eine Nachuntersuchung der Indexpatientin steht noch aus. Das durchschnittliche Erkrankungsalter der Mutationsträger lag bei 50,7 Jahren. Kein Patient mit konstitutioneller Mutation des SDHC-Gens war jünger als 21 Jahre alt. Nur Personen mit der Diagnose eines Glomustumors des Kopfes oder des Halses wiesen konstitutionelle Mutationen des SDHC-Gens auf. Aussagen über den Vererbungsmodus lassen sich wegen der geringen Zahl identifizierter Mutationsträger nicht machen. Der Indexfall C1 spricht am ehesten für einen autosomaldominanten Erbgang. 3.4. Konstitutionelle Mutationen des SDHD-Gens Die Ergebnisse von 228 Patienten mit Phäochromozytomen (Stand 1.12.2001) wurden in der Dissertation von Herrn Dr. med. Robin Munk zusammengefasst. 11 Indexfälle zeigten konstitutionelle Mutationen. Bis Mai 2004 wurden weitere 108 Patienten mit Phäochromozytomen und 84 Patienten mit Glomustumoren untersucht. Konstitutionelle Mutationen fanden sich bei weiteren 13 Indexfällen. Tabelle 17 zeigt Nationalität, Tumorform und Mutationen aller 24 Indexfälle. Nationalität Tumor Indexfall Mutation (Nukleotid) Resultat (Aminosäure) Mutationstypus Exon Deutsch Pheo D1 14 G/A* W5X Stopcodon 1 Deutsch Pheo D2 14 G/A W5X Stopcodon 1 Deutsch GT D3 14 G/A W5X Stopcodon 1 Polnisch Pheo D4 33 C/A* C11X Stopcodon 1 Polnisch Pheo D5 33 C/A* C11X Stopcodon 1 Polnisch Pheo D6 33 C/A* C11X Stopcodon 1 Deutsch Pheo D7 33 C/A* C11X Stopcodon 1 Polnisch GT D8 33 C/A C11X Stopcodon 1 Polnisch GT D9 33 C/A C11X Stopcodon 1 Polnisch GT D10 33 C/A C11X Stopcodon 1 Deutsch Pheo D11 36/37 del TG* G12Frameshift Frameshift 1 55 Deutsch Pheo D12 52+2 T/G* Splice-Defekt Splice-Defekt (1) Deutsch Pheo D13 112 C/T* R38X Stopcodon 2 Französisch GT D14 112 C/T R38X Stopcodon 2 Polnisch Pheo D15 112 C/T* R38X Stopcodon 2 Marokkanisch Pheo D16 204-216 del 13bp S68Frameshift Frameshift 3 Deutsch GT D17 242 C/T P81L Missense 3 Französisch GT D18 252 T/G Y84X Stopcodon 3 Polnisch Pheo D19 274 G/T* D92Y Missense 3 Deutsch GT D20 337-340 del GACT D113Frameshift Frameshift 4 Deutsch GT D21 341 A/G Y114C Missense 4 Deutsch Pheo D22 361 C/T* Q121X Stopcodon 4 Deutsch Pheo D23 441 del G V147Frameshift Frameshift 4 Deutsch GT D24 443 G/T G148V Missense 4 Tabelle 17: Darstellung der im Rahmen des molekulargenetischen Screenings gefundenen Mutationen des SDHD-Gens. Pheo ist eine Abkürzung für Phäochromozytom, GT ist eine Abkürzung für Glomustumor des Kopfes oder des Halses. Aus Datenschutzgründen werden die Namen der Indexfälle mit den Ziffern D1-D24 verschlüsselt. Die Reihenfolge der Nummerierung orientiert sich an der Position der Mutation in der Nukleotidsequenz. Die mit * gekennzeichneten Mutationen wurden im Rahmen der Dissertation von Herrn Dr. Med. Robin Munk entdeckt. Es sind insgesamt 11 Träger 7 verschiedener konstitutioneller Mutationen des SDHD-Gens. Die Mutationen fanden sich in allen 4 Exons des SDHD-Gens. Das Mutationsspektrum setzte sich aus 4 (27 %) Missense-Mutationen, 5 (33 %) Stopcodon-Mutationen, 4 (27 %) Frameshift-Mutationen und einem Splice-Site-Defekt zusammen. Haplotyp-Analysen von Indexfällen mit identischen Mutationen des SDHD-Gens, durchgeführt von Frau Prof. Eng Ohio State University, Columbus, Ohio, ergaben keine Hinweise für interfamiliäre Verwandtschaften. 60 Angehörige der 24 Indexfälle gaben ihre Einwilligung zu einer molekulargenetischen Testung. Es wurden 21 weitere Mutationsträger identifiziert. Zusätzlich wurden 2 weitere Personen als Mutationsträger angenommen. Es handelte sich um je einen Angehörigen des Indexfalles D13 und D15, die im Alter von 52 und 68 Jahren an einem benignen Tumor des Glomus caroticum erkrankt waren. Der Indexfall D13, Träger der Mutation SDHD c. 112 C/T erkrankte im Alter von 31 Jahren an einem benignen Glomustumor. Der Indexfall D15, Träger der Mutation SDHD c. 253 C/T erkrankte im Alter von 49 Jahren an einem benignen Glomustumor. DNA der Angehörigen stand in beiden Fällen bisher nicht zur molekulargenetischen Untersuchung zur Verfügung. Zur Auswertung der klinischen Daten betrug somit die Gesamtzahl der Mutationsträgern 47. Von diesen 47 Personen lagen klinische Befunde von 34 Personen vor. Bei 13 Mutationsträgern stehen die Ergebnisse des klinischen Screenings noch aus. Sie wurden von 56 der Auswertung der klinischen Daten ausgeschlossen. Alle 34 untersuchten Träger konstitutioneller Mutationen des SDHD-Gens waren symptomatisch. Keiner wies ein malignes Phäochromozytom oder einen malignen Glomustumor auf. Die Zusammenfassung der wesentlichen klinischen Daten zeigt Tabelle 18. Klinische Charakteristika Symptomatische SDHD- Mutationsträger n = 34 Phäochromozytome: adrenal extra-adrenal abdominell extra-adrenal thorakal 53 % 21 % 18 % (18/34) ( 7/34) (6/34) Glomustumoren 79 % (27/34) multifokale Tumoren 74 % (25/34) maligne Tumoren 0% (0/34) maligne Zweiterkrankungen 3% (1/34) Tabelle 18: Übersicht der erhobenen klinischen Daten der Träger konstitutioneller Mutationen des SDHD-Gens. Von den ursprünglichen 47 Mutationsträgern sind 13 Mutationsträger ausgeschlossen. Von diesen 13 Personen standen die Befunde und Ergebnisse des klinischen Screenings noch aus. Patienten mit mehreren unterschiedlichen Tumoren und Tumorlokalisationen wurden mehrfach gewertet. Die Berechnung der altersabhängigen Penetranz erfolgte wie beim SDHB-Gen mittels einer Kaplan-Meier-Berechnung. Grundlage der Darstellung ist das Manifestationsalter. Die Berechnung basiert auf den Daten von 34 Mutationsträgern. Alle Mutationsträger waren symptomatisch. Abbildung 48 zeigt die Penetranzdarstellung für Patienten mit Phäochromozytomen, Paragangliomen und Glomustumoren. Abbildung 49 zeigt die Penetranzdarstellung für Patienten mit adrenalen Phäochromozytomen. Abbildung 50 zeigt die Penetranzdarstellung für Patienten mit Glomustumoren des Kopfes und des Halses. Für Mutationen des SDHD-Gens zeigte sich eine Penetranz von 50 % mit Erreichen des 31. Lebensjahres. Die altersabhängige Penetranz steigt auf 86 % mit Erreichen des 50. Lebensjahres an. Die altersabhängige Penetranz für die Manifestation adrenaler Phäochromozytme bei Patienten mit Mutationen des SDHD-Gens liegt mit Erreichen des 26. Lebensjahres bei 25 % und mit Erreichen des 44. Lebensjahres bei 50 % (Abbildung 49). Für die Manifestation von Glomustumoren liegt die Penetranz bei 25 % mit Erreichen des 29. Lebensjahres und mit Erreichen des 39. Lebensjahres bei 50 % (Abbildung 50). 57 Abbildung 48: Altersabhängige Penetranzdarstellung für Patienten mit Phäochromozytomen, Paragangliomen und Glomustumoren. Abbildung 49: Altersabhängige Penetranzdarstellung für Patienten mit adrenalen Phäochromozytomen. Abbildung 50: Altersabhängige Penetranzdarstellung für Patienten mit Glomustumoren des Kopfes und des Halses. 58 4. Diskussion In den Jahren 2000 und 2001 wurden von verschiedenen Arbeitsgruppen in Pittsburgh/Ohio, in Gießen und in Birmingham/England Berichte publiziert, die zeigten, dass bei Patienten mit Paragangliomen (Glomustumoren) der Schädelbasis-Hals-Region und bei Patienten mit Phäochromozytomen sog. konstitutionelle Mutationen (d.h. Keimbahnmutationen, die sich in Lymphozyten des peripheren Blutes nachweisen lassen) in einer Gruppe von neuen Genen, dem SDHB-Gen, dem SDHC-Gen und dem SDHD-Gen vorkommen können. Es handelt sich dabei um Gene, die für Untereinheiten der Succinatdehydrogenase kodieren: Succinatdehydrogenase Untereinheit B (SDHB), Succinatdehydrogenase Untereinheit C (SDHC) und Succinatdehydrogenase Untereinheit D (SDHD) (SDHB: Au et al. 1995, Astuti et al. 2001, SDHC: Elbehti-Green et al. 1998, Niemann und Müller 2000, SDHD: Hirawake et al. 1997, Baysal et al. 2000). Die familiären Paragangliom- Syndrome Es werden 4 familiäre Paragangliom-Syndrome unterschieden: Das PGL 1-Syndrom, dessen molekulargenetische Grundlage Mutationen des SDHD-Gens sind (Astuti et al. 2001, Baysal et al. 2000). Das PGL 2-Syndrom ist ursächlich ungeklärt. Nach der Studie einer großen holländischen Familie besteht eine Kopplung mit Markern des langen Arms von Chromosom 11 (11q13) (Mariman et al. 1995). Das PGL 3-Syndrom, das durch Mutationen des SDHC-Gens bedingt ist (Niemann et al. 1999, Niemann und Müller 2000, Niemann et al. 2003). Das PGL 4-Syndrom, dessen molekulargenetische Grundlage Mutationen des SDHB-Gens sind (Astuti et al. 2001, Baysal et al. 2002). Syndrome assoziiert mit Genen von Succinatdehydrogenase-Untereinheiten Mutationen von Genen der Succinatdehydrogenase sind mit folgenden Syndrome assoziiert: SDHA-Gen: Homozygote Mutationen des SDHA-Gens führen zur Manifestation typischer Erkrankungen mitochondrialer Dysfunktionen. Das Leigh-Syndrom, eine subakut nekrotisierende Enzephalomyopathie des Kindesalters sowie Wachstumsretardierungen und Kardiomyopathien werden durch Defekte dieses Gens und dem damit verbundenen kompletten Verlust der Enzymaktivität hervorgerufen (Bourgeron et al. 1995, Kleist-Retzow et al. 1998, Parfait et al. 2000 und Rustin et al. 1997). 59 SDHB- Gen: Paragangliom Syndrom Typ 4 (PGL 4): siehe oben SDHC- Gen: Paragangliom Syndrom Typ 3 (PGL 3): siehe oben SDHD- Gen: Paragangliom Syndrom Typ 1 (PGL 1): siehe oben Molekulargenetische und klinische Charakterisierung der Paragangliom-Syndrome Mit dieser Arbeit wird eine systematische Untersuchung eines großen Patientengutes mit bis dato als sporadisch zu klassifizierenden Phäochromozytomen und Glomustumoren vorgelegt. Ausgeschlossen wurden Patienten mit anderen hereditären Phäochromozytom-assoziierten Syndromen, d.h. mit klinischen Hinweisen für eine von Hippel-Lindau Krankheit (VHL), eine Multiple Endokrine Neoplasie Typ 2 (MEN 2) und eine Neurofibromatose Typ 1 (von Recklinghausen, NF 1) sowie Träger konstitutioneller Mutationen des VHL-Gens und des RET-Gens. Von allen eingeschlossenen Patienten lagen klinische Daten und DNA aus peripherem Blut vor. Alle DNA Proben wurden hinsichtlich Mutationen in der Gesamtzahl von 18 Exons der 3 Gene analysiert. Nach Identifikation von Mutationen wurde den Indexfällen das Ergebnis mitgeteilt und angeboten, dass Verwandte getestet werden. Ziel war es, Risikopersonen molekulargenetisch zu charakterisieren und mit einem klinischen Untersuchungsprogramm Tumoren im Frühstadium zu erkennen und eventuell zu entfernen. Die klinischen Daten beinhalteten Alter bei Erkrankung bzw. Untersuchung sowie Zahl, Lokalisation und Dignität der Tumoren, wobei früher durchgeführte Untersuchungen und aktuell erfolgte Screeninguntersuchungen ausgewertet wurden. Zur Beurteilung wurden herangezogen: CT oder MRT der Schädelbasis-Hals-Region, CT oder MRT des Thorax und des Abdomens, Ganzkörperdarstellungen mittels Jod123- oder Jod131 - MIBG - Szintigraphie und 18 Fluor-DOPA-PositronenEmissionsTomographie, 18 Fluor-DOPA-PET, sowie Messungen der Katecholamine Noradrenalin, Adrenalin und Vanillinmandelsäure im 24h Urin. Die Gesamtszahl der Patienten betrug 420. Dieses Register-gestütze Kollektiv bestand aus 2 Subregistern: dem Freiburg-Warschau-Phäochromozytom-Register mit 336 Patienten und dem Glomustumor-Register mit 84 Patienten. 52 Patienten (12 %) der insgesamt 420 Indexfälle wiesen Mutationen auf. 25 Patienten hatten Mutationen des SDHB-Gens, 3 Patienten hatten Mutationen des SDHC-Gens und 24 Patienten hatten Mutationen des SDHD-Gens. Das Mutationsspektrum zeigte für das SDHB-Gen 9 Missense-Mutationen, 2 Stopcodon- und 4 Frameshift-Mutationen sowie 3 60 Splice-Site-Defekt. Mutationen des SDHC-Gens waren 2 Missense- und eine StopcodonMutation. Das Mutationsspektrum des SDHD-Gens setzte sich aus 4 Missense-, 5 StopcodonMutationen, 4 Fameshift-Mutationen und einem Splice-Site-Defekt zusammen. Alle in Freiburg gefundenen Varianten und die bis 2004 beschriebenen Varianten sind in den Tabellen 19, 20 und 21 zusammengefasst. In Freiburg wurden 18 der bekannten 33 SDHBMutationen, 3 der 7 SDHC-Mutationen und 8 der 34 SDHD-Mutationen festgestellt. 16 SDHB-Mutationen, 3 SDHC-Mutationen und 8 SDHD-Mutationen wurden erstmals anhand der Freiburger Analysen beschrieben. Das Spektrum der Mutationen unterscheidet sich nicht wesentlich zwischen den 3 Genen: das SDHB-Gen zeigte 15 Missense- und 18 andere Mutationen (Stopcodon, Frameshift-Mutationen und Splice-Site-Defekte), das SDHC-Gen zeigte 5 Missense- und 2 andere Mutationen und das SDHD-Gen zeigte 12 Missense- und 22 andere Mutationen. Konstitutionelle Mutationen des SDHB- Gens: Exon Resultat ( Aminosäure ) L7Frameshift Mutationstypus 1 Mutation ( cDNA Nukleotid ) 155 del C Referenz Frameshift diese Arbeit 2 213 C/T R27X Stopcodon diese Arbeit 2 221 ins CCAG S30Frameshift Frameshift diese Arbeit 2 222 del C Q30Frameshift Frameshift Benn et al. 03 2 261 G/C A43P Missense Gimenez- Roqueplo et al. 03 2 270 C/G R46G Missense diese Arbeit, GimenezRoqueplo et al. 03 Benn et al. 03 2 270 C/T R46X Stopcodon 2 271 G/A R46Q Missense 2 291 G/A G53R Missense 2 300 del CCTCA P56Frameshift Frameshift diese Arbeit 2 309 C/T Q59X Stopcodon Baysal et al. 02 2 328 T/C L65P Missense diese Arbeit 2 328 T/A L65H Missense Benn et al. 03 diese Arbeit, Benn et al. 03, Gimenez- Roqueplo et al. 03 diese Arbeit Splice-Site IVS2+3 G/C Splice-Site Splice-Defekt Benn et al. 03 3 345/347 ins C M71Frameshift Frameshift Baysal et al. 02 3 394 T/C L87S Missense diese Arbeit, Bauters et al. 03 3 402 C/T R90X Stopcodon Astuti et al. 01, Benn et al. 03 Splice-Site 421-2 A/G Splice-Site Splice-Defekt diese Arbeit Splice-Site IVS4-1 G/A Splice-Site Splice-Defekt Benn et al. 03 4 436 G/A C101Y Missense diese Arbeit 4 526 C/G P131R Missense Baysal et al. 02 4 529 A/C H132P Missense Maier-Woelfle et al. 04 Splice-Site 558-3 C/G Splice-Site Splice-Site diese Arbeit 6 708 T/C C192R Missense diese Arbeit 6 721 G/A C196Y Missense diese Arbeit 6 724 C/G P197R Missense Astuti et al. 01 6 725 del C P197Frameshift Frameshift Gimenez- Roqueplo et al. 03 6 754/755 del TG L205Frameshift Frameshift Gimenez- Roqueplo et al. 03 61 7 822 C/T R230C Missense Gimenez- Roqueplo et al. 03 7 847/849 del TCTC F238Frameshift Frameshift diese Arbeit 7 859 G/A R242H Missense diese Arbeit, Young et al. 02 7 881 C/A C249X Stopcodon diese Arbeit Splice-Site 899+1 G/A Splice-Site Splice-Site diese Arbeit Tabelle 19: Übersicht aller bisher beschriebenen konstitutionellen Mutationen des SDHB-Gens bei Patienten mit familiären und sporadischen Phäochromozytomen, Paragangliomen und Glomustumoren. Stand: Juni 04. Dick gedruckt hervorgehoben sind die in dieser Dissertationsarbeit gefundenen konstitutionellen Mutationen des SDHB- Gens. Konstitutionelle Mutationen des SDHC- Gens: Exon Mutationstypus Referenz Missense diese Arbeit Verlust des Startcodons Missense Niemann et al. 2000 L4L / Promotor Region Missense Bauters et al. 2003 Mutation ( cDNA Nukleotid ) 1 A/G Resultat ( Aminosäure ) M1V / Verlust des Startcodons 1 3 G/A 1 12 G/A 1 2 39 C/A C13X Stopcodon diese Arbeit 4 214 C/T R72C Missense diese Arbeit Splice-Site IVS5+1 G/T Splice-Site Splice-Site Niemann et al. 2003 6 473 T/C L158D Missense Bauters et al. 2003 Tabelle 20: Übersicht aller bisher beschriebenen konstitutionellen Mutationen des SDHC-Gens bei Patienten mit familiären und sporadischen Glomustumoren. Stand: Juni 04. Dick gedruckt hervorgehoben sind die in dieser Dissertationsarbeit gefundenen konstitutionellen Mutationen des SDHC- Gens. Konstitutionelle Mutationen des SDHD- Gens: Exon Mutation ( cDNA Nukleotid ) Resultat ( Aminosäure ) Mutationstypus Referenz 1 3 G/C M1I Missense Badenhop et al. 2001 1 14 G/A W5X Stopcodon Neumann et al. 2002 1 33 C/A C11X Stopcodon Neumann et al. 2002 1 34 G/A G12S Missense 1 36/37 del TG G12Frameshift Frameshift Gimm et al. 2000, Gimenez-Roqueplo et al. 2003 Neumann et al. 2002 Splice-Site IVS1+2 T/G Splice-Site Splice-Site Gimm et al. 2000 2 54 ins C A18Frameshift Frameshift Taschner et al. 2001 2 64 C/T R22X Stopcodon 2 94 del TC S32Frameshift Frameshift Taschner et al. 2001, Gimenez-Roqueplo et al. 2001 Astuti et al. 2001 2 95 C/G oder 95 C/A S32X Stopcodon Milunsky et al. 2001 2 106 C/T Q36X Stopcodon Baysal et al. 2000, Astuti et al. 2001 2 112 C/T R38X Stopcodon 2 120 ins C I40Frameshift Frameshift Gimm et al. 2000, Astuti et al. 2001, Baysal et al. 2000 Taschner et al. 2001 2 129 G/A W43X Stopcodon Cascon et al. 2002 2 160 A/G H50R Missense Gimenez-Roqueplo et al. 2003 3 192/193 del TC0 L64Frameshift Frameshift Badenhop et al. 2001 3 204-216 del 13 bp S68Frameshift Frameshift Neumann et al. 2004 3 208 A/G R70G Missense Taschner et al. 2001 3 242 C/T P81L Missense Baysal et al. 2002, Badenhop et al. 2001, Milunsky et al. 2001, Gimm et al. 2000 62 3 252 T/G Y84S Missense Neumann et al. 2004 3 274 G/T D92Y Missense Baysal et al. 2000, Dannenberg et al. 2002 3 277 del TAT Del Y93 Deletion Badenhop et al. 2001 3 284 T/C L95P Missense Taschner et al. 2001, Dannenberg et al. 2002 3 305 A/T H102L Missense Baysal et al. 2000 4 325 C/T Q109X Stopcodon Baysal et al. 2002 4 337 ins T Frameshift Frameshift Milunsky et al. 2001 4 337 del GACT Frameshift Frameshift Cascon et al. 2002 4 341 A/G Y114C Missense Milunsky et al. 2001 4 361 C/T Q121X Stopcodon Neumann et al. 2002 4 381/383 del G G127Frameshift Frameshift Baysal et al. 2002 4 416 T/C L139P Missense Taschner et al. 2001, Dannenberg et al. 2002 4 441 del G V147Frameshift Frameshift Neumann et al. 2004 4 443 G/T G148V Missense Neumann et al. 2004 4 443 del G G148Frameshift Frameshift Milunsky et al. 2001 Tabelle 21: Übersicht aller bisher beschriebenen konstitutionellen Mutationen des SDHD-Gens bei Patienten mit familiären und sporadischen Phäochromozytomen, Paragangliomen und Glomustumoren. Stand: Juni 04. Die genetische Testung von Familienangehörigen ergab für das SDHB-Gen bei 26 Verwandten und für das SDHD-Gen bei 21 Verwandten einen positiven Mutationsnachweis. Bei weiteren 2 Verwandten von SDHB-Indexfällen, 2 Verwandten von SDHD-Indexfällen und einem Verwandten eines SDHC-Indexfalls sind Tumorerkrankungen, bzw. Glomustumoren, bekannt. Somit konnte das Risikoprofil anhand von 104 Personen charakterisiert werden. Ausgeschlossen werden konnte eine Mutationsträgerschaft für das SDHB-Gen bei 17 und des SDHD-Gens bei 39 Verwandten der Indexfälle. Klinische Untersuchungen von Mutationsträgern des SDHB-, des SDHC- oder des SDHD-Gens mittels eines Screening-Programms sind aus der Literatur unbekannt. Dagegen wurde dem hier zugrunde liegenden Patientengut ein solches Untersuchungsprogamm angeboten und systematisch durchgeführt. Unter Mitberücksichtigung von Voruntersuchungen liegen Befunde von Serienschnittuntersuchungen von folgenden Patientenzahlen vor: SDHB-Mutationsträger: Schädelbasis-Hals n = 32, Thorax n = 29, Abdomen n = 32. Nicht untersucht wurden weitere 11 bislang tumorfreie, neu entdeckte Mutationsträger. Das Manifestationsspektrum umfasst Glomustumoren und Phäochromozytome. 31 % der Patienten entwickelten Glomustumoren, 9 % thorakale und 50 % abdominelle Phäochromozytome. 28 % wiesen multiple Tumoren auf. 34 % der Patienten hatten maligne Tumoren, wobei die Primärtumoren sowohl Glomustumoren als auch Phäochromozytome waren. Extraparaganglionäre Tumoren hatten 4 Patienten: 2 Patienten hatten ein 63 Nierenzellkarzinom und jeweils ein Patient eine Akute Myeloische Leukämie und ein papilläres Schilddrüsenkarzinom. SDHC-Mutationsträger: Schädelbasis-Hals n = 4, Thorax n = 1, Abdomen n = 1. Alle in Freiburg klassifizierten und in der Literatur berichteten, einschließlich ihrer betroffenen Angehörigen insgesamt 4 + 4 Fälle zeigten Glomustumoren (Niemann und Müller 2000, Niemann et al. 2003, Bauters et al. 2003). SDHD-Mutationsträger: Schädelbasis-Hals n = 35, Thorax n = 27, Abdomen n = 29. Nicht untersucht sind 13 weitere bislang tumorfreie, neu entdeckte Mutationsträger. Das Manifestationsspektrum umfasst Glomustumoren und Phäochromozytome. 79 % der Patienten entwickelten Glomustumoren, 18 % thorakale und 21 % abdominelle Phäochromozytome. 74 % wiesen multiple Tumoren auf. Kein Patient hatte einen malignen Tumor. Vererbung Der Erbgang der Paragangliom Syndrome ist für Personen mit SDHB-, SDHC- und SDHDMutationen unterschiedlich. Bei SDHB-Mutationsträgern ist der Erbgang autosomal-dominant. Dies ist anhand eigener Beobachtungen und von Literaturmitteilungen gut belegt (Astuti et al. 2001, Baysal et al. 2002, Benn et al. 2003) . Bei SDHC-Mutationsträgern ist der Erbgang autosomal-dominant. Dies ist belegt durch die Mitteilungen der Giessener Arbeitsgruppe sowie der Familie eines eigenen Indexfalles (Niemann et al. 1999, Niemann und Müller 2000, Niemann et al. 2003). Bei SDHD-Mutationsträgern ist der Erbgang autosomal-dominant mit der Besonderheit eines geschlechts-abhängigen Vererbungsmusters. Nur Kinder, deren Väter Mutationsträger sind, erkranken am Paragangliom-Syndrom Typ 1. Kinder mutations-tragender Mütter bleiben tumorfrei. Publikationen, basierend auf klinischen Daten, zeigten dies bereits 1989 bei Patienten mit hereditären Glomustumoren (van der Mey et al. 1989). Die ursprüngliche Annahme eines "Maternalen Imprintings" konnte wiederlegt werden. Mutationsträger mit und ohne PGL 1-Syndrom wiesen eine biallelische Expression des SDHD-Gens auf (Baysal et al. 2000). Nicht nur ein Verlust des Wildtypallels des SDHD-Gens in Tumorzell-DNA, sondern ein kompletter Verlust des mütterlichen Chromosom 11, scheint für die Tumorgenese der Paragangliome und Phäochromozytome verantwortlich zu sein (Hensen et al. 2004). Eine ektope Imprinting-Region auf Chromosom 11 (11p15.5) wird angenommen (Baysal et al. 2000, Hensen et al. 2004). 64 Tumorgenese Das SDHB-Gen und das SDHD-Gen sind Tumorsuppressorgene. Sie bilden den Anfang einer Kette von Inhibitionssignalen, die hemmend und regulierend in die Zellteilung eingreifen. Beide Allele eines solchen Gens müssen funktionsunfähig sein, bevor ein kanzerogenes Wachstum resultiert. Nach der sogenannten "two-hit"- Theorie führen zwei unabhängige Mutationsereignisse zu einem solchen Funktionsverlust (Knudson et al. 1971). Eine erste, erblich bedingte Keimbahnmutation und eine zweite somatische Mutation führen zu einer kanzerogenen Zellproliferation. Molekulargenetische Analysen auf konstitutionelle Mutationen des SDHB- und des SDHD-Gens, wie in dieser Arbeit durchgeführt, sowie LOHAnalysen entsprechender Tumorzell-DNA zeigen sowohl Keimbahn-Mutationen dieser Gene als auch einen Verlust des Wildtypallels in Tumorzell-DNA (Gimenez-Roqueplo et al. 2002, Gimenez-Roqueplo et al. 2001, Astuti et al. 2001). Heterozygote Mutationen des SDHB- und des SDHD-Gens resultieren in einer herabgesetzten Enzymaktivität bzw. einem kompletten Funktionsverlust der Succinatdehydrogenase (Gimenez-Roqueplo et al. 2002, Gimenez-Roqueplo et al. 2001). Diese Dysfunktion scheint zu mitochondrialen Energiedefiziten, einer damit einhergehenden Hypoxie und zum Auftreten freier Radikale zu führen. Eine Aktivierung sogenannter "hypoxia-inducible factors", z.B. HIF-1α und HIF-2α, ist die Folge (Gimenez-Roqueplo et al. 2002, Gimenez-Roqueplo et al. 2001). HIF-1α und HIF-2α bewirken eine vermehrte Expression bestimmter Gene mit antiapoptotischer, proliferativer Wirkung. Produkt eines dieser Gene ist der vascular endothelial growth factor (VEGF), der einen wesentlichen Indikator der Tumorgenese darstellt (Chandel et al. 1998). Ein Anstieg von HIF-1α und HIF-2α sowie VEGF konnte auch in Zusammenhang mit Phäochromozytomen und Paragangliomen im Rahmen des PGl 1- und des PGL 4-Syndroms nachgewiesen werden (Gimenez-Roqueplo et al. 2002, GimenezRoqueplo et al. 2001). 65 Vergleich der Risikospektren von SDHB- und SDHD-Mutationsträgern (PGL 4 versus PGL 1) Gemeinsamkeiten zeigen das PGL 1- und das PGL 4-Syndrom bezüglich ihres Mutationsspektrums, ihres durchschnittlichen Manifestationsalters und ihrer Prävalenz. Wesentliche Unterschiede weisen ihr Vererbungsmodus, ihre Klinik und ihr Manifestationsrisiko auf. Die unterschiedlichen Vererbungsmodi sind oben erläutert. Mutationen des SDHB-Gens werden sowohl über das mütterliche als auch das väterliche Genom vererbt. (Astuti et al. 2001, Baysal et al. 2002, Benn et al. 2003). Mutationen des SDHD-Gens hingegen zeigen einen autosomal-dominanten Erbgang mit dem Effekt des "Maternalen Imprintings" (van der Mey et al. 1989). SDHB- und SDHD-Gen Mutationen sind mit gleicher Häufigkeit für das Auftreten von Phäochromozytomen, Paragangliomen und Glomustumoren verantwortlich. Eine Prävalenz von 6 % (25/420) wurde für das SDHB-Gen ermittelt. Dieses Ergebnis der bislang größten Studie reiht sich in die Werte bisher publizierter Daten von 9,5 % (6/84) und 3 % (1/33) ein (Gimenez-Roqueplo et al. 2003, Baysal et al. 2002, Astuti et al. 2001). Die Prävalenz für Mutationen des SDHD-Gens liegt ebenfalls bei 6 % (24/420). Eine weitere Gemeinsamkeit zeigt das durchschnittliche Alter betroffener Indexpatienten, erhoben bei Diagnosestellung der Erkrankung. Patienten mit Mutationen des SDHB-Gens erkranken durchschnittlich mit 29,8 Jahren. Betroffene, die Mutationen des SDHD-Gens aufweisen, erkranken durchschnittlich im Alter von 30,6 Jahren (p = 0,77). Bezüglich der Penetranz bei Trägern von Mutationen des SDHB- und des SDHD-Gens zeigt sich statistisch kein signifikanter Unterschied. Abbildung 51 stellt die altersabhängige Penetranz beider Gene graphisch dar. Für Mutationen des SDHB-Gens zeigt sich eine Penetranz von 50 % mit Erreichen des 35. Lebensjahres, während dies für Mutationen des SDHD-Gens mit Erreichen des 31. Lebensjahres (p = 0,67) vorliegt. Trotz dieser Daten scheinen Mutationen des SDHB-Gens durch eine unvollständige Penetranz gekennzeichnet zu sein. 10 der oben genannten 42 Mutationsträger (24 %) entwickelten kein Phäochromozytom, Paragangliom oder einen Glomustumor, während alle 34 SDHD-Mutationsträger an einem Tumor erkrankten (p = 0,0015). Mit einem hohen Risiko einer Tumormanifestation ist somit vor allem bei Patienten mit Mutationen des SDHD-Gens zu rechnen. Des weiteren zeigte die Auswertung der altersabhängigen Penetranz, dass Träger konstitutioneller Mutationen des 66 SDHD-Gens signifikant häufiger und in jüngeren Jahren adrenale Phäochromozytome (p = 0,03) und Glomustumoren (p = 0,007) entwickeln. Abbildung 51: A: Darstellung der altersabhängigen Penetranz des SDHB-Gens, bezogen auf 42 Mutationsträger (32 symptomatische Mutationsträger + 10 asymptomatische Mutationsträger). Nur Patienten von denen alle Ergebnisse des klinischen Screenings vorlagen wurden in die Auswertung eingeschlossen. 11 SDHB-Mutationsträger sind von der Auswertung ausgeschlossen. Darstellung der altersabhängigen Penetranz des SDHD-Gens, bezogen auf 34 Mutationsträger (24 symptomatische Indexfälle + 10 symptomatische, positiv getestete Angehörige). 13 SDHD-Mutationsträger sind von der Auswertung ausgeschlossen. B + C: Auswertung der altersabhängigen Penetranzen des SDHB- und des SDHD- Gens nach Unterteilung der 42 bzw. 34 Mutationsträger in folgende Subgruppen: 1.) Patienten mit adrenal lokalisierten Phäochromozytomen (SDHB- Gen Mutationen n = 9; SDHD- Mutationen n = 18) und 2.) Patienten mit Glomustumoren des Kopfes und des Halses (SDHB- Gen Mutationen n = 10; SDHD- Mutationen n = 27) Das Manifestationsspektrum ist beim PGL 1- und beim PGL 4-Syndrom unterschiedlich. Phäochromozytome, die sich im Rahmen des PGL 1-Syndroms entwickeln, sind signifikant häufiger adrenal lokalisiert (SDHD-Gen 53 % (18/34), SDHB-Gen 28 % (9/32), p = 0,0487). Ebenso wurde ein multifokales Tumorwachstum bei 74 % (25/34) der SDHDMutationsträger aber nur bei 28 % (9/32) der SDHB-Mutationsträger beobachtet. Dies bedeutet, dass multifokale Tumoren signifikant häufiger durch Mutationen des SDHD-Gens als durch Mutationen des SDHB-Gens bedingt sind (p = 0,00046). Vor allem Mutationen des 67 SDHD-Gens prädisponieren zur Entwicklung von Glomustumoren. Im Gegensatz zu 79 % der SDHD-Mutationsträger hatten nur 31 % der SDHB-Mutationsträger einen Glomustumor (p = 0,00015). Phäochromozytome, die sich im Rahmen des PGL 4 -Syndroms entwickeln, sind signifikant häufiger extra-adrenal abdominell lokalisiert (p = 0,019). Maligne Tumoren wurden bei insgesamt 34 % (11/32) der Patienten mit Mutationen des SDHB-Gens diagnostiziert. Keiner der 34 untersuchten Patienten mit Mutationen des SDHD-Gens wies Metastasen auf. Somit haben Tumoren, die auf der molekulargenetischen Grundlage von Mutationen des SDHBGens entstehen, ein signifikant höheres Risiko einer malignen Entartung (p = 0,00012). In einer im Jahr 2003 publizierten Studie zeichnete sich bereits eine solche Tendenz ab (Gimenez-Roqueplo et al. 2003). 8 von insgesamt 84 Patienten mit der Diagnose eines Phäochromozytoms oder eines Paraganglioms wiesen konstitutionelle Mutationen des SDHBGens auf. 5 dieser Patienten hatten maligne Tumoren und 5 Patienten waren an einem extraadrenal abdominellen Phäochromozytom erkrankt. Die aggressivere Natur von Dysfunktionen des SDHB-Gens verdeutlicht sich auch durch die Beobachtung der Manifestation maligner Zweiterkrankungen im Sinne zweier Nierenzellkarzinoms und einer Akuten Myeloischen Leukämie im Verlaufe der Krankheit. Eine Indexpatientin, Trägerin der Mutation SDHB c. 394 T/C, entwickelte im Alter von 17 Jahren ein malignes adrenales Phäochromozytom. 11 Jahre später wurde eine Akute Myeloische Leukämie diagnostiziert, an der die Patientin verstarb. Unklar bleibt, inwieweit die Therapie mit Jod131-Metajodobenzylguanidin und diversen Chemotherapeutika die Akute Myeloische Leukämie induziert haben kann. Eine weitere Indexpatientin, Trägerin der Mutation SDHB c. 847/849 del TCTC, erkrankte im Alter von 16 Jahren an einem benignen extra-adrenalen Phäochromozytom. Im Alter von 24 Jahren wurde bei dieser Patientin ein Nierenzellkarzinom festgestellt. Der Bruder der Patientin, ebenfalls Träger der genannten Mutation, erkrankte im Alter von 26 Jahren an einem Nierenzellkarzinom und im Alter von 37 Jahren an einem thorakal lokalisierten Phäochromozytom. Eine im Jahr 2004 publizierte Untersuchung griff diesen Fall auf und ging der Assoziation von Mutationen des SDHB-Gens mit der Entwicklung von Nierenzellkarzinomen weiter nach (Vanharanta et al. 2004). Tumorzell- DNA des Nierenzellkarzinoms sowie des thorakalen Phäochromozytoms des Bruders der Indexpatientin zeigten einen Verlust des entsprechenden Wildtypallels, einen LOH-Effekt. Ausschließlich das mutierte Allel (SDHB c. 847/849 del TCTC) wurde im Tumor exprimiert. Ein weiterer 68 Patient, der Teil des finnischen Krebs-Registers ist, wies die Mutation SDHB c. 213 C/T auf. Er erkrankte im Alter von 28 Jahren an einem Nierenzellkarzinom. Seine Mutter, ebenfalls Trägerin der oben genannten Mutation, hatte ein extra-adrenales Phäochromozytom. Die DNA Proben auch dieser beiden Tumoren zeigten einen Verlust des entsprechenden Wildtypallels. Somit konnte ein Zusammenhang von Mutationen des SDHB- Gens und der Entwicklung von Nierenzellkarzinomen gezeigt werden. Ein anderer Indexpatient, Träger der Mutation SDHB c. 328 T/C, erkrankte im Alter von 14 Jahren an einem papillären Schilddrüsenkarzinom und im Alter von 15 Jahren an einem extra-adrenal abdominellen Phäochromozytom. Als Besonderheit entwickelte auch die Mutter ein papilläres Schilddrüsenkarzinom. Sie ist jedoch keine Trägerin der Mutation SDHB c. 328 T/C. Daher ist ein Zusammenhang zwischen der Mutation und diesem Tumor unwahrscheinlich. Tabelle 22 stellt in einer Übersicht die unterschiedliche Klinik des PGL 1- und des PGL 4Syndroms dar. Prävalenz Verebungsmodus Manifestationsalter (Jahre) altersabhänige Prävalenz adrenal Phäo extra-adrenal abdominell extra-adrenal thorakal multiple Tumoren Glomustumoren Tumoren maligner Dignität Maligne Zweit erkrankungen PGL 4-Syndrom (Mutationen des SDHB-Gens) n = 32 6% autosomal-dominant 29,8 PGL 1-Syndrom p-Werte (Mutationen des SDHD-Gens) n = 34 6% autosomal-dominant mit dem Effekt des maternalen Imprintings P = 0,77 30,6 50 % im Alter von 35 Jahren 28 % (9/32) 50 % (16/32) 9 % (3/32) 28 % (9/32) 31 % (10/32) 50 % im Alter von 31 Jahren 53 % (18/34) 21 % (7/34) 18 % (6/34) 74 % (25/34) 79 % (27/34) 34 % (11/32) 0 % (0/34) 4 1 P = 0,67 P = 0,0487 P = 0,0194 P = 0,48 P = 0,00046 P = 0,00015 P = 0,0008 - Tabelle 22: Vergleich des PGL 4-Syndroms, bedingt durch Mutationen des SDHB-Gens und des PGL 1-Syndroms, bedingt durch Mutationen des SDHD-Gens. Gemeinsamkeiten bestehen bezüglich der Prävalenz, des durchschnittlichen Manifestationsalters und rein statistisch gesehen bezüglich der altersabhängigen Penetranz. Wesentliche Unterschiede finden sich hinsichtlich der klinischen Ausprägung und des Vererbungsmodus. p-Werte < 0,05 gelten als signifikant 69 Genetische Testung bei Glomustumor- und Phäochromozytom-assoziierten TumorSyndromen Die definierten hereditären Glomustumor- und Phäochromozytom-assoziierten TumorSyndrome zeigt in einer Übersicht Tabelle 23. Syndrom Gen Chromosom Adrenales Phäo PGL 1 SDHD 11q23 PGL 2 unbekannt 11q13 PGL 3 SDHC 1q21 - PGL 4 SDHB 1p36 + VHL VHL 3p25-26 + MEN2 A+B RET 10q11.2 NF 1 NF 1 unbekannt Carney Triade Extra-adrenales, thorakales Phäo + GL + Extra-adrenales, abdominales Phäo + ? ? ? + - - + + + + + ↓ ↓↓ + ↓↓ - ↓↓ 17q11 + ↓ - ↓↓ unbekannt - - - + + Tabelle 23: Übersicht aller bis dato bekannten Tumor-assoziierten Syndrome, die mit der Manifestation eines Phäochromozytoms, eines Paraganglioms oder eines Glomustumors einhergehen. Phäo ist eine Abkürzung für Phäochromozytom, GL ist eine Abkürzung für Glomustumor. + bedeutet, das sich der entsprechende Tumor manifestiert hat, ? bedeutet das keine Kenntnisse über eine entsprechende Manifestation vorliegen, ↓ bedeutet eine seltene Manifestation des entsprechenden Tumors, ↓↓ bedeutet eine sehr seltene Manifestation des Tumors, - bedeutet, das sich der entsprechende Tumor nicht manifestiert. Wesentliche Hinweise sind eine Manifestation in jungen Jahren, eine extra-adrenale Lokalisation und ein multifokales oder bilaterales Tumorwachstum (Neumann et al. 1993, Eng et al. 1996, Eng et al. 1995, Woodward et al. 1997). Bevor eine genetische Testung veranlasst wird, müssen die klinischen Informationen überprüft werden, um die Testung effizient zu gestalten, d.h. das Gen bzw. die Gene, die mit Wahrscheinlichkeit mutiert sind, zu selektieren. Das extraparaganglionäre Manifestationsspektrum ist von vorrangiger Bedeutung. Bei den Paragangliom-Syndromen ist bislang nur beim Typ 4 (mit SDHB-Mutationen) über Nierenzellkarzinome berichtet worden (Vanharanta et al. 2004). Die Multiple Endokrine Neoplasie Typ 2 ist in über 90% mit einem medullären Schilddrüsenkarzinom vergesellschaftet. Beim Typ 2A tritt in 20% ein Hyperparathyroidismus, beim Typ 2B treten in aller Regel ein marfanoider Habitus und Schleimhaut-Ganglioneurome auf. Bei der von Hippel-Lindau Erkrankung finden sich als häufigste Manifestationen und in variabler Ausprägung retinale, cerebelläre oder spinale Hämangioblastome, Klarzellkarzinome der Nieren, multiple Zysten oder Inselzelltumoren des Pankreas. Bei der Neurofibromatose von Recklinghausen finden sich kutane Neurofibrome und Irisveränderungen. 70 Das Alter bei Erstmanifestation ist gegenüber Patienten mit sporadischen Phäochromozytomen in der Regel 15 Jahre niedriger aber sehr variabel. Es ist am niedrigsten bei der von Hippel -Lindau Erkrankung, gefolgt von den Paragangliom-Syndromen und der Multiplen Endokrinen Neoplasie Typ 2 (Neumann et al. 2002) und liegt bei der Neurofibromatose Typ 1 am höchsten. Die paraganglionären Tumoren sind wegweisend. Glomustumoren kommen nahezu ausschließlich bei den Paragangliom-Syndromen vor. Extraadrenale Phäochromozytome finden sich häufig bei der von Hippel-Lindau Erkrankung und den Paragangliom-Syndromen. Maligne Glomustumoren oder Phäochromozytome werden häufig beim ParagangliomSyndrom Typ 4 (PGL 4 (mit SDHB-Mutationen)) beobachtet, selten auch bei der von HippelLindau Erkrankung und der Neurofibromatose Typ 1. Bei der MEN 2 sind Phäochromozytome extrem selten maligne ( Koch et al. 2001, Neumann et al. 1993 und 2002, Woodward et al. 1997, Gimenez-Roqueplo et al. 2003, Maier-Woelfle et al. 2004). Prädisponierende Regionen in den Kandidaten-Genen. Phäochromozytom-prädisponierende Regionen für Mutationen des 21 Exon umfassenden RET-Gens sind hauptsächlich Exon 10 (Codon 609, 611, 618) und Exon 11 (Codon 620 und 634) sowie das Exon 13 und das Exon 16 (Codon 918). Es handelt sich jeweils um MissenseMutationen (Eng et al. 1996, Neumann et al. 2002). Die Mutationen im VHL-Gen erstrecken sich über alle 3 Exons des VHL-Gens und zeigen alle denkbaren Varianten. In Zusammenhang mit dem Auftreten hereditärer Phäochromozytome wurden nahezu immer Punktmutationen vom Missense- und StopcodonTyp festgestellt (Neumann et al. 1996, Zbar et al. 1996). Häufigkeit hereditärer Phäochromozytome Die Entdeckung und Analyse der molekulargenetischen Grundlage zweier neuer mit dem Auftreten von Phäochromozytomen assoziierten Tumor-Syndrome, des PGL 1-Syndroms und des PGL 4-Syndroms, zeigt, dass Phäochromozytome häufiger als angenommen hereditär bedingt sind. Bislang galten vor allem Mutationen des VHL-Gens und des RET-Gens als prädisponierende Faktoren. Der tradierte Grundsatz, die sogenannte "10er-Regel", nach der 10 % aller Phäochromozytome hereditär bedingt, 10 % maligne und 10 % extra-adrenal lokalisiert sind, ist nunmehr ungültig (Manger und Gifford 1995, Dluhy 2002: Pheochromocytoma - The death of an axioma). Die Analyse von 271 nicht verwandten Patienten ergab 24 % hereditäre Phäochromozytome (Neumann et al. 2002). Berücksichtigt 71 man ein metachrones Auftreten von Symptomen bei Verwandten, steigt der Anteil von Trägern von VHL-, RET-, SDHB- und SDHD-Mutationen auf 50 % (Bauters et al. 2003). Nicht eingerechnet sind dabei Patienten mit Neurofibromatose Typ 1. In Register-gestützten großen Studien ist somit das Erreichen von etwa 40 % realistisch. Relevanz der Ergebnisse für Diagnostik und Behandlung betroffener Patienten Die Kenntnis typischer klinischer Merkmale der einzelnen Tumor-assoziierten Syndrome, sowie die Kenntnis ihrer molekulargenetischen Grundlage eröffnet bessere Möglichkeiten in der Vorsorge und Behandlung betroffener Patienten und eine effizientere Durchführung molekulargenetischer Untersuchungen. Mutationen des VHL-Gens werden vor allem bei Patienten, die vor dem 18. Lebensjahr erkranken, die ein multifokales, bilaterales Tumorwachstum zeigen, wahrscheinlich. SDHB- und SDHD-Gen Mutationen finden sich dagegen häufiger bei Patienten um das 30. Lebensjahr. Mutationen des SDHD-Gens prädisponieren vor allem für Glomustumoren und zeigen ein erhöhtes Risiko, an einem der typischen Tumoren zu erkranken. Mutationen des SDHB-Gens sind durch eine niedrigere Penetranz als Mutationen des SDHD-Gens gekennzeichnet und prädisponieren vor allem für extra-adrenal lokalisierte Phäochromozytome. Sie scheinen aggressiverer Natur zu sein. Hoch ist das Risiko für eine maligne Entartung. Zweiterkrankungen mit malignen Nierentumoren kommen offenbar vor. Neben der molekulargenetischen Abklärung betroffener Patienten und deren Familien, wird für asymptomatische Mutationsträger die Durchführung folgender Diagnostik empfohlen: MRT von Schädelbasis und Hals, MRT des Thorax und MRT des Abdomen oder die 18 Fluor-Dopa-PET sowie die Messung der Katecholamine im Plasma oder im 24h Sammelurin. Schlusswort Nicht mehr 10 % sondern ein geschätzter Anteil von bis zu 24 % aller Phäochromozytome ist hereditär bedingt. Prädisponierende konstitutionelle Mutationen finden sich im VHL-Gen, im RET-Gen, im SDHB- und im SDHD-Gen. Allein ein Anteil von ca. 12 % tritt im Rahmen der familiären Paragangliom-Syndrome, respektiv im Rahmen des PGL 1- und des PGL 4Syndroms auf. Das PGL 1-Syndrom ist vor allem durch adrenal lokalisierte Phäochromozytome oder Glomustumoren des Kopfes und des Halses gekennzeichnet. Ein multifokales Tumorwachstum und ein hohes Erkrankungsrisiko sind typisch. Das PGL 4Syndrom ist mit einer erhöhten Gefahr einer malignen Entartung der Tumoren und dem Auftreten maligner Zweiterkrankungen assoziiert. Mutationen des SDHB-Gens 72 prädisponieren für die Entwicklung maligner Nierentumore. Die Erkenntnisse dieser Arbeit ermöglichen eine effizientere molekulargenetische Untersuchung von Patienten mit Phäochromozytomen, Paragangliomen und Glomustumoren und eine Optimierung der klinischen Betreuung und Diagnostik Betroffener. 73 5. Zusammenfassung Konstitutionelle Mutationen der Succinatdehydrogenase Untereinheiten B (SDHB), C (SDHC) und D (SDHD) bilden die molekulargenetische Grundlage der familiären Paragangliom-Syndrome Typ 4 (PGL 4), Typ 3 (PGL 3) und Typ 1 (PGL 1). Typische Läsionen sind Phäochromozytome, Paragangliome und Glomustumoren von Hals und Schädelbasis. Die molekulargenetischen Grundlage der familiären Paragangliom-Syndrome ist weitgehend unbekannt. Schwerpunkt dieser Arbeit ist die molekulargenetische Klassifikation und die klinische Charakterisierung der Paragangliom-Syndrome. 420 nicht miteinander verwandte Patienten, mit der Diagnose eines Phäochromozytoms, Paraganglioms und /oder Glomustumors wurden auf konstitutionelle Mutationen des SDHB-, des SDHC- und des SDHD-Gens untersucht. Der molekulargenetischen Analyse der Indexpatienten folgte ein genetisches Screening von Familienangehörigen. Alle Patienten wurden vor und nach Entdeckung einer Mutation klinisch mittels CT oder MRT von Hals und Schädelbasis, Thorax und Abdomen, fakultativ einer 18 Fluor-DOPA-PET sowie der Bestimmung der Katecholaminausscheidung im 24h Sammelurin untersucht. 104 Personen wiesen Mutationen auf. 25 Indexpatienten und 28 Angehörige hatten 18 verschiedene Mutationen des SDHB-Gens. 3 Indexpatienten und ein Familienangehöriger hatten 3 verschiedene Mutationen des SDHC-Gens. 14 verschiedene Mutationen des SDHDGens wiesen 24 Indexpatienten und 23 Verwandte auf. Eine Prävalenz von 6 % (25/420 bzw. 24/420) wurde für Mutationen des SDHB-Gens und des SDHD-Gens ermittelt. Mutationen des SDHC-Gens sind selten und weisen eine Prävalenz von 0,7 % auf. Das PGL 1-Syndrom manifestiert sich ähnlich wie das PGL 4-Syndrom mit durchschnittlich 30,6 Jahren (PGL 4-Syndrom: 29,8 Jahren; p = 0,77). Wesentliche Charakteristika sind adrenal lokalisierte Phäochromozytome und Glomustumoren des Halses und der Schädelbasis. Ein multifokales Tumorwachstum und ein hohes Erkrankungsrisiko sind typisch. Das PGL 4-Syndrom ist durch eine hohe Rate maligner Tumoren und die Manifestation maligner Zweiterkrankungen gekennzeichnet. Das PGL 3-Syndrom ist selten und ist nur durch Glomustumore gekennzeichnet. Die Kenntnis der molekulargenetischen Grundlage und der klinischen Charakteristika der familiären Paragangliom-Syndrome eröffnet bessere Möglichkeiten in der Vorsorge und Behandlung betroffener Patienten und eine effizientere Durchführung molekulargenetischer Untersuchungen. 74 4. Literaturverzeichnis Astuti D, Douglas F, Lennard TW, Aligianis IA, Woodward ER, Evans GR, Eng C, Latif F, Maher ER. (2001) Germline SDHD mutation in familial pheochromocytoma. Lancet. 357: 1181-82 Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, George E, Sköldberg F, Husebye ES, Eng C, Maher ER. (2001) Gene mutations in the succinate dehydrogenase subunit B cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 69: 49-54 Au HC, Ream-Robinson D, Bellew LA, Broomfield E, Saghbini M, Scheffler IE. (1995) Structural organization of the gene encoding the human iron-sulfur subunit of succinate dehydrogenase. Gene. 159: 249-53 Averbuch SD, Steakley CS, Young RC, Gelmann EP, Goldstein DS, Stull R, Keiser HR. (1988) Malignant pheochromocytoma: effective treatment with a combination of cyclophosphamide, vincristine, and dacarbazine. Ann Intern Med. 109: 267-73 Badenhop RF, Cherian S, Lord RS, Baysal BE, Taschner PE, Schofield PR. (2001) Novel mutations in the SDHD gene in pedigrees with familial carotid body paraganglioma and sensorineural hearing loss. Gens Chromosomes Cancer. 31 : 255-63 75 Bausch B, Munk R, Schipper J, Hoegerle S, Berger DP, Böhm N, Neumann HP. (2003) SDHD mutations in carotid body tumors and pheochromocytomas: paraganglioma syndrome type 1. Current Opinion in Endocrinology & Diabetes. 10: 197-204 Bauters C, Vantyghem M, Leteurtre E, Odou M, Mouton C, Porchet N, Wemeau J, Proye C, Pigny P. (2003) Hereditary phaeochromocytomas and paragangliomas: a study of five susceptibility genes. J Med Genet. 40: e75 Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, Richard CW, Cornelisse CJ, Devilee P, Devlin B. (2000) Mutations in SDHD, a mitochondrial complexe II gene, in hereditary paraganglioma. Science. 287: 848-51 Baysal BE, Willett-Brozick JE, Lawrence EC, Drovdlic CM, Savul SA, McLeod DR, Yee HA, Brackmann DE, Slattery WH 3rd, Myers EN, Ferrell RE, Rubinstein WS. (2002) Prevalence of SDHB, SDHC, and SDHD germline mutations in clinic patients with head and neck paragangliomas. J Med Genet. 39: 178-83 Bender BU, Gutsche M, Gläsker S, Muller B, Kirste G, Eng C, Neumann HP. (2000) Differential genetic alterations in von Hippel-Lindau syndrome-associated and sporadic pheochromocytomas. J Clin Endocrinol Metab. 85: 4568-74 76 Benn DE, Croxson MS, Tucker K, Bambach CP, Richardson AL, Delbridge L, Pullan PT, Hammond J, Marsh DJ, Robinson BG. (2003) Novel succinate dehydrogense subunit B (SDHB) mutations in familial phaeochromocytomas and paragangliomas, but an absence of somatic SDHB mutations in sporadic phaeochromocytomas. Oncogene. 22. 1358-64 Böck P, Lassmann H. (1972) Histochemical studies on the action of reserpine on glomus caroticum. Verh Anat Ges. 67: 229-32 Bourgeron T, Rustin P, Chretien D, Birch-Machin M, Bourgeois M, Viegas-Pequignot E, Munnich A, Rötig A. (1995) Mutation of nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat Genet. 11: 144-49 Carney JA. (1999) Gastric stromal sarcoma, pulmonary chondrom and extra-adrenal paraganglioma (Carney Triad): natural history, adrenocortical component and possible familial occurence. Mayo Clin Proc. 74: 543 –52 Cascon A, Ruiz-Llorente S, Cebrian A, Telleria D, Rivero JC, Diez JJ, Lopez-Ibarra PJ, Jaunsolo MA, Benitez J, Robledo M. (2002) Identification of novel SDHD mutations in patients with phaeochromocytoma and / or Paraganglioma. European J Hum Genet. 10:457-61 Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. (1998) Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA. 95: 11715-11720 77 Chiocchio SR, King MP, Angelakos ET. (1971) Monoamines in the carotid body cells of the cat. J Histochem Cytochem. 19: 621-26 Dannenberg H, Dinjens WN, Abbou M, van Urk H, Pauw BK, Mouwen D, Mooi WJ, Krijger RR. (2002) Frequent germline succinate dehydrogenase subunit D gene mutations in patients with apparently sporadic parasympatic paraganglioma. Clinical Cancer Res. 8: 2061-66 Dluhy RG. (2002) Pheochromocytoma – Death of an axiom. N Engl J Med. 346: 1486-1488 Eisenhofer G, Lenders JW, Linehan WM, Walther MM, Goldstein DS, Keiser HR. (1999) Plasma metanephrine and metanephrine for detecting pheochromocytoma in von HippleLindau disease and multiple endocrine neoplasia type 2. N Engl J Med. 340: 1872-9 Elbehti-Green A, Au HC, Mascarello JT, Ream-Robinson D, Scheffler IE. (1998) Characterization of the human SDHC gene, encoding one of the integral membrane proteins of succinate-quinone oxidoreductase in mitochondria. Gene. 213: 133-40 Eng C, Clayton D, Schuffenecker I, Lenoir G, Cote G, Gagel RF, van Amstel HK, Lips CJ, Nishisho I, Takai SI, Marsh DJ, Robinson BG, Frank-Raue K, Raue F, Xue F, Noll WW, Romei C, Pacini F, Fink M, Niederle B, Zedenius J, Nordenskjöld M, Komminoth P, Hendy GN, Gharib H, Thibodeau N, Lacroix A, Frilling A, Mulligan LM. (1996) The relationship between specific RET-proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation cosortium analysis. JAMA. 276: 1575-79 78 Eng C, Crossey PA, Mulligan LM, Healey SC, Houghton C, Prowse A, Chew SL, Dahia PL, O'Riordan JL, Toledo SP, Smith DP, Maher ER, Ponder BA. (1995) Mutations in the RET proto-oncogene and the von Hippel-Lindau disease tumour suppressor gene in sporadic and syndromic phaeochromocytomas. J Med Genet. 32: 934-37 Gagner M, Lacrix A, Bolte E. (1992) Laparoscopic adrenalectomy in Cushing´s syndrome and pheochromocytoma. N Engl J Med. 327: 1033 Gimenez-Roqueplo AP, Favier J, Rustin P Mourad JJ, Plouin PF, Corvol P, Rötig A, Jeunemaitre X. (2001) The R22X mutation of the SDHD gene in hereditary paraganglioma abolishes the enzymatic activity of complex II in the mitochondrial respiratory chain and activates the hypoxia pathway. Am J Hum Genet. 69: 1186-97 Gimenez-Roqueplo AP, Favier J, Rustin P, Rieubland C, Crespin M, Nau V, van Kien PK, Corvol P, Plouin PF, Jeunemaitre X. (2003) Mutations in the SDHB gene are associated with extra-adrenal and/or malignant pheochromocytomas. Cancer Res. 63: 5615-21 Gimm O, Armanios M, Dziema H, Neumann HP, Eng C. (2000) Somatic and occult germline mutations in SDHD, a mitochondrial complex II gene, in non-familial pheochromocytoma. Cancer Res. 60: 6822-25 Glenner GG, Grimley PM. (1974) Tumors of the extra-adrenal paraganglion system (including chemoreceptors). Atlas of Tumor Pathology. 2nd Series, Fascicle 9. Armed Forces Institute of Pathology. 79 Hensen EF, Jordanova ES, Van Minderhout IJ, Hogendoorn CW, Taschner PE, Van der Mey AG, Devilee P, Cornelisse CJ. (2004) Somatic loss of maternal chromosom 11 causes parent-of-origin-dependent inheritance in SDHD-linked paraganglioma and pheochromocytoma families. Oncogene. 23: 4076-4083 Hirawake H, Taniwaki M, Tamura A, Kojima S, Kita K. (1997) Cytochrome b in human complex II (succinate-ubiquinone oxidoreductase): cDNA cloning of the components in liver mitochondria and chromosome assignment of the genes for the large (SDHC) and the small (SDHD) subunits to 1q21 nd 11q23. Cytogenet Cell Genet. 79: 132-38 Hoegerle S, Ghanem N, Altehoefer C, Schipper J, Brink I, Moser E, Neumann HP. (2003) 18 F-DOPA positron emission tomography for the detection of glomus tumors. Eur J Nucl Med Mol Imaging. 30: 689-94 Hoegerle S, Nitzsche E, Altehoefer C, Ghanem N, Manz T, Brink I, Reincke M, Moser E, Neumann HP. (2002) Pheochromocytomas: detection with 18F-DOPA whole body PET—initial results. Radiology. 222: 507-12 Hollingshead WS. (1942) A comparative study of the glomus coccygeum and the carotid body. Anat Rec. 84: 1-16 Huang SC, Koch CA, Vortmeyer AO, Pack SD, Lichtenauer UD, Mannan P, Lubensky IA, Chrousos GP, Gagel RF, Pacak K, Zhuang Z. (2000) Duplication of the mutant RET allele in trisomy 10 or loss of wild type allele in multiple endocrine neoplasia type 2-associated pheochromocytomas. Cancer Res. 60: 6223-26 80 Janetschek G, Finkenstedt G, Gasser R, Waibel UG, Peschel R, Bartsch G, Neumann HP. (1998) Laparoscopic surgery for pheochromocytoma: adrenalectomy, partial resection, excision of paragangliomas. J Urol. 160: 330-34 Kleist-Retzow JC, Cormier-Daire V, de Lonlay P, Parfait B, Chretien D, Rustin P, Feingold J, Rötig A, Munnich A. (1998) A high (20-30%) rate of parental consanguinity in cytochrome-oxidase deficiency. Am J Hum Genet. 63: 428-435 Knudson AG, Jr. (1971) Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA. 68: 820-3 Koch CA, Vortmeyer AO, Huang SC, Alesci S, Zhuang Z, Pacak K. (2001) Genetic aspects of pheochromocytoma. Endocrine Regulations. 35: 43-52 Leckschat S, Ream-Robinson D, Scheffler IE. (1993) The gene for the iron sulfur protein of the succinate dehydrogenase (SDH-IP) maps to human chromosome 1p35-36.1. Somat Cell Mol Genet. 19: 505-11 Lenders JW, Pacak K, Walther MM, Linehan WM, Mannelli M, Friberg P, Keiser HR, Goldstein DS, Eisenhofer G. (2002) Biochemical diagnosis of pheochromocytoma: which test is the best? JAMA. 287: 1427-34 81 Maier-Woelfle M, Brändle M, Komminoth P, Saremaslani P, Schmid S, Locher T, Heitz P, Krull I, Galeazzi RL, Schmid C, Perren A. (2004) A novel succinate dehydrogenase subunit B gene mutation, H132P, causes familial malignant sympathetic extraadrenal paragangliomas. J Endocrinol Metab. 89: 362-367 Manger WM, Gifford RW. (1995) Pheochromocytoma: a clinical overview. In: Laragh JH, Brenner BM, eds. Hypertension: pathophysiology, diagnosis, and management. 2nd ed. Vol.2. New York: Raven Press, 2225-44 Manger WM, Gifford RW. (2002) Pheochromocytoma. J Clin Hypertens. 4: 62-72 Mariman EC, van Beersum SE, Cremers CW, Struycken PM, Ropers HH. (1995) Fine mapping of a putatively imprinted gene for familial non-chromaffin paragangliomas to chromosome 11q13.1: evidence for genetic heterogeneity. Hum Genet. 95: 56-62 Milunsky JM, Maher TA, Michels VV, Milunsky A. (2001) Novel mutations and the emergence of a common mutation in the SDHD gene causing familial paraganglioma. Am J Genet. 100: 311-14 Morris AA, Farnsworth L, Ackrell BA, Turnbull DM, Birch-Machin MA. (1994) The cDNA sequence of the flavoprotein subunit of human heart succinate dehydrogenase. Biochem. Biophys. Acta. 1185: 125-128 Mulligan RM. (1950) Chemodectoma in the dog. Am J Pathol. 26: 680 82 Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K, Januszewicz A, Smith WM, Munk R, Manz T, Glaesker S, Apel TW, Treier M, Reincke M, Walz MK, Hoang-Vu C, Brauckhoff M, Klein-Franke A, Klose P, Schmidt H, Maier-Woelfle M, Peczkowska M, Szmigielski C, Eng C; The Freiburg-Warsaw-Columbus Pheochromocytoma Study Group. (2002) Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 346:1459-66 Neumann HP, Bender B, Zauner I, Berger DP, Eng C, Brauch H, Zbar B. (1996) Monogenetic hypertension and pheochromocytoma. Am J Kidney Dis. 28: 329-333 Neumann HP, Bender BU, Reincke M, Eggstein S, Laubenberger J, Kirste G. (1999) Adrenal-sparing surgery for pheochromocytoma. Br J Surg. 86: 94-97 Neumann HP, Berger DP, Sigmund G, Blum U, Schmidt D, Parmer PJ, Volk B, Kirste G. (1993) Pheochromocytomas, multiple endocrine neoplasia type 2, and von Hippel-Lindau disease. N Engl J Med. 329: 1531-38 Neumann HP, Reincke M, Bender BU, Elsner R, Janetschek G. (1999) Preserved adrenocortical function after laparoscopic bilateral adrenal sparing surgery for hereditary pheochromocytoma. J Clin Endocrinol Metab. 84: 2608-10 Neumann HP. (1987) Basic criteria for clinical diagnosis and genetic counselling in von Hipple-Lindau syndrome. Vasa. 16: 220-26 83 Niemann S, Müller U, Engelhardt D, Lohse P. (2003) Autosomal dominant malignant and catecholamine-producing paraganglioma caused by a splice donor site mutation in SDHC. Hum Genet. 113: 92-94 Niemann S, Müller U. (2000) Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. 26: 268-270 Niemann S, Steinberger D, Müller U. (1999) PGL 3, a third, not maternally imprinted locus in autosomal dominant paraganglioma. Neurogenetics. 2: 167-70 Orita M, Iwahana H, Kanazawa H, Hayashi K, Sekiya T. (1989) Detection of polymorphisms of human DNA by gel electrophoresis as single-strand conformation polymorphisms. Proc Natl Acad Sci USA. 86: 2766-70 Parfait B, Chretien D, Rötig A, Marsac C, Munnich A, Rustin P. (2000) Compound heterozygous mutations in the flavoprotein gene of the respiratory chain complex II in a patient with Leigh syndrome. Hum Mol Genet. 106: 236-243 Riccardi VM. (1981) Von Recklinghausen neurofibromatosis. N Engl J Med. 305: 1617-27 Rustin P, Bourgeron T, Parfait B, Chretien D, Munnich A, Rötig A. (1997) Inborn errors of the Krebs cycle: a group of unusual mitochondrial diseases in human. Biochim. Biophys. Acta. 1361: 185-197 84 Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, Mullis KB, Erlich HA. (1988) Primer-directed enzymatic amplification of DNA with t. thermostable DNA polymerase. Science 239: 487-91 Sanger F, Nicklen S, Coulson AR. (1977) DNA-sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 74: 5463-7 Saraste M. (1999) Oxidative phosphorylation at the fin du siecle. Science. 283: 1488-93 Scarpulla RC. (2002) Transcriptional activators and coactivators in the nuclear control of mitochondrial function in mammalian cells. Gene. 286: 81-89 Scheffler IE. (1998) Molecular genetics of succinate:quinone oxidoreductase in eukaryotes. Prog Nuclei Acid Res Mol Biol. 60: 267-315 Schimke RN. (1990) Multiple endocrine neoplasia: how many syndromes? Am J Med Genet. 37: 375-83 Taschner PE, Jansen JC, Baysal BE, Bosch A, Rosenberg EH, Bröcker-Vriends AH, van der Mey AG, van Ommen GJ, Cornelisse CJ, Devilee P. (2001) Nearly all hereditary paragangliomas in the Netherlands are caused by two founder mutations in the SDHD gene. Genes Chromosomes Cancer. 31: 274-281 85 Van der Mey AG, Maaswinkel-Mooy PD, Cornelisse CJ, Schmidt PH, Van de Kamp J. (1989) Genomic Imprinting in hereditary glomus tumours: evidence for new genetic theory. Lancet. 2: 1291-1294 Vanharanta S, Buchta M, McWhinney SR, Virta SK, Peczkowska M, Morrison CD, Lehton R, Januszewicz A, Järvinen H, Juhola M, Mecklin JP, Pukkala E, Herva R, Kiuru M, Nupponen N, Aaltonen LA, Neumann HP, Eng C. (2004) Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHBassociated heritable paraganglioma. Am J Hum Genet. 74: 153-159 Walz MK, Peitgen K, Neumann HP, Janssen OE, Philipp T, Mann K. (2002) Endoscopic treatment of solitary, bilateral, multiple, and recurrent pheochromocytomas and paragangliomas. World J Surg. 26: 1005-12 Watzka M. (1943) Die Paraganglien. Einführung; Wesen un Einteilung der Paraganglien, pp. 262-308. In: Handbuch der Mikroskopischen Anatomie des Menschen, Vol 6, Part 4 (Ed) Möllendorf, W. V. Berlin: Springer Verlag Woodward ER, Eng C, McMahon R, Voutilainen R, Affara NA, Ponder BA, Maher ER. (1997) Genetic predisposition to pheochromocytoma: analysis of candidate genes GDNF, RET and VHL. Hum Mol Genet. 6: 1051-56 Young AL, Baysal BE, Deb A, Young WF Jr. (2002) Familial malignant catecholamine-secreting paraganglioma with prolonged survival associated with mutation in the succinate dehydrogenase B gene. J Clin Endocrinol Metab. 87: 4101-4105 86 Zbar B, Kishida T, Chen F, Schmidt L, Maher ER, Richards FM, Crossey PA, Webster AR, Affara NA, Ferguson-Smith MA, Brauch H, Glavac D, Neumann HP, Tisherman S, Mulvihill JJ, Gross DJ, Shuin T, Whaley J, Seizinger B, Kley N, Olschwang S, Boisson C, Richards S, Lips CH, Lerman M. (1996) Germline mutations in the von Hippel-Lindau (VHL) gene in families from North America, Europe, and Japan. Hum Mutat. 8: 348-57 87 7. Danksagung Mein besonderer Dank gilt Herrn Prof. Neumann. Für die Überlassung des Themas und die Unterstützung bedanke ich mich ebenso wie für die Bereitstellung des Materials. Erst die jahrelange Sammlung von Patientenproben und die genaue Dokumentation von Patientendaten haben eine solch umfangreiche Untersuchung ermöglicht. Weiterhin danke ich ihm für das Einbeziehen in wissenschaftliche Tätigkeiten über den Rahmen der Dissertation hinaus und für die Unterstützung und Beteiligung an hervorragenden Publikationen. Herrn Dr. Pawlu danke ich für die freundliche Hilfe und Unterstützung bei der statistischen Auswertung der gewonnenen Daten. Für die Mithilfe bei der grafischen Gestaltung möchte ich meinem Bruder Dr. Dirk Bausch danken. Für die tatkräftige Unterstützung bei der Durchführung des experimentellen Teils meiner Dissertation danke ich dem Laborteam, Frau Franke und Frau Salzmann, Frau Bohnert-Iwan, Frau Buchta, Frau Bacher und Herr Berisha. 88 Lebenslauf Angaben zur Person Name: Wohnort: Birke Bausch Herrenstr. 49 79098 Freiburg 0761/2020802 12.09.1977 Karlsruhe ledig deutsch Telefon: Geburtsdatum: Geburtsort: Familienstand: Nationalität: Schulischer Werdegang 1984-1988 1988-1997 17.06.1997 Grundschule in Karlsruhe-Durlach Fichte-Gymnasium in Karlsruhe Abitur Universitärer Werdegang WS 1997–SS 2004 Studium der Humanmedizin an der Albert-Ludwigs- Universität Freiburg 07.09.1999 29.08.2000 28.03.2003 28.04.2003-14.03.2004 12.05.2004 Physikum Erster Abschnitt der Ärztlichen Prüfung Zweiter Abschnitt der Ärztlichen Prüfung Praktisches Jahr mit dem Wahlfach Neurochirurgie Akademisches Lehrkrankenhaus VS: Chirurgie Akademisches Lehrkrankenhaus VS: Innere Medizin Kantonspital St-Gallen, Schweiz: Neurochirurgie Dritter Abschnitt der Ärztlichen Prüfung Studienbegleitende Tätigkeiten Famulaturen: Frühjahr 2000 Sommer 2000 Sommer 2001 Herbst 2002 Anästhesie, Städtisches Klinikum, Karlsruhe Innere Medizin, Gynäkologie, Victoria Hospital, Mahé, Republic of the Seychelles Innere Medizin, Krankenhaus der Barmherzigen Brüder, München Chirurgie, St-Josefs Krankenhaus, Freiburg 89 Publikationen: Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K, Januszewicz A, Smith WM, Munk R, Manz T, Glaesker S, Apel TW, Treier M, Reincke M, Walz MK, Hoang-Vu C, Brauckhoff M, Klein-Franke A, Klose P, Schmidt H, Maier-Woelfle M, Peczkowska M, Szmigielski C, Eng C; The Freiburg-Warsaw-Columbus Pheochromocytoma Study Group. (2002) Germ-line mutations in nonsyndromic pheochromocytoma N Engl J Med. 346:1459-66 Bausch B, Munk R, Schipper J, Hoegerle S, Berger S, Böhm N, Neumann HP. (2003) SDHD mutations in carotid body tumors and pheochromocytomas: parganglioma syndrome type 1. Current Opinion in Endocrinology & Diabetes Hoegerle S, Bausch B, Moser E, Neumann HPH. (2003) Nuklearmedizinische Diagnostik des Nebennierenmarks. Der Nuklearmediziner 26: 25-32 Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, Buchta M, Franke G, Klisch J, Bley T, Hoegerle S, Boedeker C, Opocher G, Schipper J, Januszewicz A, Eng C. (2004) Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 292: 943-951