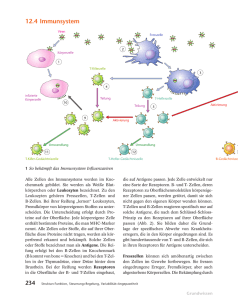
Antikörper-abhängige Immunantwort im SIV
Werbung

Tierärztliche Hochschule Hannover Antikörper-abhängige Immunantwort im SIV-Makakenmodell INAUGURAL-DISSERTATION zur Erlangung des Grades einer Doktorin der Veterinärmedizin - Doktor medicinae veterinariae (Dr. med. vet.) Vorgelegt von Katharina Raue aus Hannover Hannover 2012 Wissenschaftliche Betreuung: Univ.-Prof. Dr. med. vet. F.-J. Kaup Abt. Infektionspathologie, Deutsches Primatenzentrum, Göttingen 1. Gutachter: Univ.-Prof. Dr. med. vet. F.-J. Kaup Tierärztliche Hochschule Hannover Deutsches Primatenzentrum, Göttingen 2. Gutachter: Univ.-Prof. Dr. rer. nat. Georg Herrler Tierärztliche Hochschule Hannover Tag der mündlichen Prüfung: 21.11.2012 Meiner Familie Inhaltsverzeichnis 1. EINLEITUNG .................................................................................................................... 15 2. LITERATURÜBERSICHT............................................................................................... 17 2.1 Das Immunsystem.................................................................................................... 17 2.1.1 T-Zell-vermittelte Immunantwort ...................................................................... 17 2.1.2 Humorale Immunantwort ................................................................................... 18 2.1.2.1 B-Zellen .......................................................................................................... 18 2.1.2.2 Antikörper ....................................................................................................... 26 2.2 Humane und simiane Immundefizienzviren ......................................................... 32 2.2.1 HIV und AIDS ................................................................................................... 32 2.2.2 SIV ..................................................................................................................... 33 2.2.3 Klassifikation ..................................................................................................... 34 2.2.4 Vergleich von SIVsm, HIV-1 und HIV-2 .......................................................... 34 2.2.5 Struktur von HIV/SIV ........................................................................................ 35 2.2.6 Die Replikation von HIV/ SIV ........................................................................... 36 2.2.7 SIV-Infektion von Makaken als in vivo-Modell für die HIV-Infektion ............. 37 2.3 Pathophysiologie der Immundefizienzviren .......................................................... 38 2.3.1 Krankheitsverlauf ............................................................................................... 38 2.3.2 Immunantwort gegen HIV/ SIV ......................................................................... 40 2.3.2.1 Die zelluläre Immunantwort ........................................................................... 41 2.3.2.2 Die humorale Immunantwort.......................................................................... 41 2.3.3 Pathogenese der Immundefizienzviren .............................................................. 41 2.3.3.1 Auswirkungen auf das zelluläre Immunsystem .............................................. 42 2.3.3.2 Auswirkung auf das humorale Immunsystem ................................................ 42 3. MATERIAL UND METHODEN ..................................................................................... 44 3.1 Tiere .......................................................................................................................... 44 3.1.1 Narkotisierung der Versuchstiere ....................................................................... 44 3.1.2 Belastungsinfektion der Tiere mit SIV............................................................... 45 3.1.3 Blutentnahme bei den Versuchstieren ................................................................ 45 3.1.4 Bronchioalveoläre Lavage (BAL) ...................................................................... 45 3.1.5 Euthanasie .......................................................................................................... 46 3.2 Isolierung von mononukleären Zellen aus peripherem Blut und bronchoalveolärer Lavage ................................................................................................. 47 3.2.1 Isolierung von mononukleären Zellen aus peripherem Blut (peripheral blood mononuclear cells, PBMC) ................................................................................ 47 3.2.2 Isolierung von mononukleären Zellen aus BAL ................................................ 47 3.3 Vitalitäts- und Zellzahlbestimmung....................................................................... 48 3.4 Kryokonservierung von Probenmaterial............................................................... 48 3.4.1 Einfrieren von PBMC......................................................................................... 48 3.4.2 Einfrieren von zellfreiem Material ..................................................................... 48 3.4.3 Auftauen von PBMC .......................................................................................... 48 3.4.4 Auftauen von zellfreiem Material ...................................................................... 49 3.5 Immunologische Methoden zum Nachweis zellulärer und humoraler Immunantworten ........................................................................................................ 49 3.5.1 Nachweis bindender Antikörper im ELISA ....................................................... 49 3.5.2 Nachweis neutralisierender Antikörper gegen SIV mittels eines Luziferase Reportergensystems auf TZM bl-Zellen ............................................................ 50 3.5.2.1 Titration des Virusstocks SIVmac32H ........................................................... 50 3.5.2.2 Nachweis von neutralisierenden Antikörpern im Serum ................................ 52 3.5.3 Quantifizierung Antikörper-produzierender Zellen mit Hilfe der ELISpotTechnik ............................................................................................................... 53 3.5.3.1 Antikörper-produzierende Zellen im peripheren Blut .................................... 54 3.5.3.2 Antikörper-produzierende Zellen in BAL ...................................................... 55 3.5.3.3 Polyklonale Stimulation von PBMC und mononuklären Zellen aus BAL..... 56 3.5.4 Quantifizierung von IFNγ-sezernierenden Zellen mit Hilfe des ELISpotVerfahrens .......................................................................................................... 56 3.6 Charakterisierung von B-Zellen mittels polychromatischer Durchflusszytometrie…. ............................................................................................ 57 3.6.1 Markierung von Oberflächenantigenen von isolierten mononukleären Zellen aus peripherem Blut .................................................................................................. 58 3.6.2 Antikörper-Kombination zur Definition verschiedener B-Zellpopulationen ..... 59 3.6.3 Definition verschiedener Lymphozytenpopulationen ........................................ 59 3.7 Bestimmung der SIV-Last im peripheren Blut..................................................... 63 3.8 Erstellung eines Differentialblutbildes .................................................................. 64 3.9 Statistische Auswertung .......................................................................................... 64 3.10 Schutzmaßnahmen .................................................................................................. 64 4. ERGEBNISSE .................................................................................................................... 65 4.1 Retrospektiver Vergleich der humoralen und zellulären Immunantwort im SIVMakakenmodell mit Fokus auf die Langzeitüberlebenden .................................... 65 4.1.1 Virusbeladung .................................................................................................... 65 4.1.2 Bindende Antikörper gegen SIV ........................................................................ 67 4.1.3 Neutralisierende Antikörper gegen SIV ............................................................. 71 4.1.4 SIV-spezifische IFN-γ-Sekretion von T-Zellen ................................................. 74 4.2 Funktionale und phänotypische Charakterisierung der B-Zellpopulationen der Langzeitüberlebenden ................................................................................................ 76 4.2.1 Quantifizierung SIV-spezifischer Gedächtnis-B-Zellen .................................... 76 4.2.2 Durchflusszytometrische Analyse der B-Zellpopulationen ............................... 78 4.3 Longitudinale Untersuchung der humoralen Immunantwort von der akuten bis zur chronischen Phase der SIV-Infektion bei nicht-immunisierten Kontrolltieren ………………………………………………………………………………………...90 4.3.1 Virusbeladung bis 48 Wochen nach SIV- Infektion .......................................... 90 4.3.2 Bindende Antikörper in der frühen Phase der SIV-Infektion ............................ 91 4.3.3 Neutralisierende Antikörper gegen SIV ............................................................. 94 4.3.4 Quantifizierung SIV-spezifischer Antikörper-sezernierender Zellen ................ 95 4.3.5 Longitudinale durchflusszytometrische Analyse der B-Zellpopulationen ......... 97 5. DISKUSSION ................................................................................................................... 102 5.1 Die Viruslast in der akuten Plateauphase eignet sich als prognostischer Marker für den weiteren Krankheitsverlauf ....................................................................... 103 5.2 Vergleichende funktionale Untersuchungen der B-Zellimmunantwort von Langzeitüberlebenden und Progressoren............................................................... 104 5.2.1 Langzeitüberlebende waren durch höhere bindende Antikörpertiter gegen SIVgag im ELISA gekennzeichnet .................................................................. 104 5.2.2 Die SIV-spezifische IFN-γ-Sekretion von T-Zellen der Langzeitüberlebenden gegen SIVgag lag signifikant über der von Progressoren ................................ 106 5.2.3 Deutliche positive Korrelation zwischen viralen RNA-Kopien im Plasma und Höhe der Neutralisationstiter ........................................................................... 107 5.2.4 Beim Eintritt in die Plateauphase der SIV-Infektion bestand eine negative Korrelation zwischen Viruslast und SIV-spezifischen IgG-sezernierenden Zellen .......................................................................................................................... 109 5.2.5 Keine Korrelation zwischen Titern der bindenden Antikörper und SIVspezifischen ASC ............................................................................................. 110 5.2.6 Ohne vorherige Stimulation konnten keine SIV-spezifischen IgGsezernierenden Zellen im Blut der Langzeitüberlebenden nachgewiesen werden. .......................................................................................................................... 111 5.2.7 Der Anteil SIVenv-spezifischer Gedächtnis-B-Zellen im Blut der Langzeitüberlebende entsprach dem 6,5fachen des Anteils der SIVgagspezifischen Gedächtnis-B-Zellen .................................................................... 113 5.2.8 Der Nachweis von SIV-spezifischen ASC in der BAL sowie der Nachweis von IgA-sezernierenden Zellen im Blut der Langzeitüberlebenden fielen negativ aus .......................................................................................................................... 115 5.3 Phänotypische Charakterisierung der B-Zellsubpopulationen im Blut ........... 117 5.3.1 Eingeschränkte Vergleichbarkeit zwischen verschiedenen Studien ................ 117 5.3.2 SIV-negative Rhesusaffen besaßen einen höheren Anteil von CD27+B-Zellen als Menschen, nur ein kleiner Teil davon entsprach allerdings der Definition von ruhenden Gedächtnis-B-Zellen ........................................................................ 119 5.3.3 Der Anteil der B-Zellen an den Lymphozyten war bei chronisch infizierten Tieren mit mittlerer und hoher SIV-Beladung signifikant erhöht .................... 121 5.3.4 Eine hohe SIV-Last führte zu einer übersteigerten Aktivierung von B-Zellen 122 5.3.5 Elite controllers unterschieden sich nur hinsichtlich der ruhenden Gedächtnis-BZellen und der T-Helferzellen signifikant von den SIV-negativen Tieren ...... 124 5.3.6 Die SIV-Infektion bei Rhesusaffen hatte einen anderen Einfluss auf die untersuchten atypischen B-Zellsubpopulationen als die HIV-Infektion bei Menschen ......................................................................................................... 127 5.3.7 Die Anteile der CD20+Zellen an den Lymphozyten und der CD10-IgD-Zellen an den B-Zellen normalisierten sich innerhalb von 4 Wochen nach SIVInfektion ........................................................................................................... 128 5.4 Abschließende Bewertung und Ausblick ............................................................. 130 6. ZUSAMMENFASSUNG ................................................................................................. 133 7. SUMMARY....................................................................................................................... 136 8. LITERATURVERZEICHNIS ........................................................................................ 139 9. ABBILDUNGSVERZEICHNIS ..................................................................................... 164 10. TABELLENVERZEICHNIS ........................................................................................ 166 11. ANHANG ........................................................................................................................ 167 11.1 Tiertabelle............................................................................................................... 167 11.2 ELISA-Protokoll .................................................................................................... 173 11.3 Protokoll für Neutralisationstest auf TZM-bl mit Luziferase Read-out .......... 174 11.4 Protokoll für B-Zell-ELISpot ............................................................................... 176 11.5 Protokoll für IFN-γ-ELISpot ................................................................................ 179 Abkürzungsverzeichnis Abb. Abbildung Abt. Abteilung AIDS acquired immunodeficiency syndrome (erworbenes Immundefizienz-Syndrom ) Ak Antikörper ASC antibody-secreting cells (Antikörpersezernierende Zellen) Aqua dest. Aqua destillata BAL Bronchioalveoläre Lavage BSA Bovines Serumalbumin bzw. beziehungsweise °C Grad Celsius ca. circa CCR CC-Motiv-Chemokin-Rezeptor CD cluster of differentiation (Differenzierungsgruppen) CD 40L CD40 Ligand CMV Cytomegalievirus CpG Cytosin-Phosphat-Guanin CXCR CXC-Motiv-Chemokinrezeptor DMSO Dimethylsulfoxid DNA deoxyribonucleic acid (Desoxyribonukleinsäure) DPZ Deutsches Primatenzentrum dt. deutsch EBV Eppstein-Barr-Virus EDTA Ethylendiamintetraacetat ELISA enzyme linked immunosorbent assay (Enzymgekoppelter Immunabsorbationstest) ELISpot enzyme linked immunospot assay (Enzymgekoppelter Immunpunkttest) engl. englisch env envelope (Hüll-) et al. et alii Fa. Firma Fab fragment antigen binding (Antigen-bindendes Fragment) Fc fragment crystallizable (kristallinisierbares Fragment) FCS fetal calf serum (fetales Kälberserum) FSC forward-scatter (Forwärtsstreubereich) g Gramm G Gauge gag group specific antigen (Gruppen-spezifisches Antigen) GM II Göttinger Mischung II gp glycoprotein (Glykoprotein) h hour (Stunde) HAART highly active antiretroviral therapy (hochwirksame antiretrovirale Therapie) HIV Humanes Immundefizienz-Virus Ig Immunglobulin IL Interleukin IFN Interferon i.v. intravenös kg Kilogramm KGW Körpergewicht l Liter log Logarithmus LPS Lipopolysaccharide LTNP long-term nonprogressor M Molare Lösung mg Miligramm MHC Major Histocompatibility Complex (Haupthistokompatibilitätskomplex) min Minute ml Milliliter n Probengröße nAk neutralisierende Antikörper nef negative effective factor (negativer Effekt-Faktor) NHP nicht-humane Primaten NIBSC National Institute for Biological Standards and Control NK-Zellen Natürliche Killer-Zellen nm Nanometer ns nicht signifikant OD Optische Dichte p Protein p Signifikanzniveau PBMC peripheral blood mononuclear cells (periphere mononukleäre Zellen des Blutes) PBS phosphat buffered saline solution (Phosphatgepufferte isotonische Kochsalzlösung) PPR pattern recognition receptors (Mustererkennungsrezeptoren) pol Polymerase PWM pokeweed mitogen (Mitogen der Kermesbeere) r Korrelationskoeffizient nach Pearson rev regulator of expression of viral proteins (Expressionsregulator viraler Proteine) RLU relative luminescence units (relative Lumineszenzeinheiten) RNA ribonucleic acid (Ribonukleinsäure) rpm rounds per minute (Umdrehungen pro Minute) RT-qcPCR quantitative real-time polymerase chain reaction (quantitative Echtzeit-PolymeraseKettenreaktion) s. siehe SD Standardabweichung SHIV simian-human immunodeficiency virus (AffenMenschen Immundefizienz-Virus) SIV simian immunodeficiency virus (AffenImmundefizienz-Virus) SIVcpz simian immunodeficiency virus chimpanzee (Affen-Immundefizienz-Virus der Schimpansen) SIVmac simian immunodeficiency virus macaque (AffenImmundefizienz-Virus der Makaken) SIVsm simian immunodeficiency virus sooty mangabe (Affen-Immundefizienz-Virus der Rauchmangaben) SSC side-scatter (seitlicher Streubereich) STLV simian T cell leukemia virus (Affen-T-ZellLeukämie-Virus) tat transactivator of transcription (Auslöser der Transkription) TCID50 tissue culture infectious dose 50 (Gewebekulturinfektöse Dosis 50) TGF transforming growth factor (transformierender Wachstumsfaktor) TLR toll-like receptor (Toll-ähnlicher Rezeptor) TMB Tetramethylbenzidin TNF Tumor-Nekrose-Faktor u.a. und andere usw. und so weiter vif virion ineffectivity factor (Virion-IneffektivitätsFaktor) vpr/ vpu/ vpx viral protein r/u/x (virales Protein r/u/x) wpi weeks post infection z.B. zum Beispiel z.T. zum Teil µg Mikrogramm µl Mikroliter µm Mikrometer Einleitung S e i t e | 15 1. EINLEITUNG Mehr als 20 Jahre nach seiner Entdeckung war das Humane Immundefizienzvirus (HIV) 2009 immer noch für ca. 1,8 Millionen Tote weltweit (Global Report 2010, UNAIDS) verantwortlich. Trotz intensivster Bemühungen konnte bisher kein effektiver und sicherer Impfstoff entwickelt werden. Eine vollständige Heilung kann auch mit den heute verfügbaren wirksamen antiretroviralen Therapeutika nicht erzielt werden. Darüber hinaus sind letztere mit zahlreichen Nachteilen behaftet, wie starken Nebenwirkungen, Resistenzbildung und nicht zuletzt hohen Kosten, die ihren flächendeckenden Einsatz in wirtschaftlich schwachen Ländern unmöglich machen. Weitere Forschung, nicht nur klinisch-angewandter Natur, sondern auch im Bezug auf HIV/AIDS-Pathogenese, ist insofern unentbehrlich. Klinische Studien am Menschen weisen allerdings zahlreiche Hürden auf. Allen voran ethische Bedenken, aber auch praktische Gründe erschweren eine systematische Arbeit. Der genaue Infektionszeitpunkt eines Patienten ist selten bekannt, die latente Phase der HIV-Infektion erstreckt sich unter Umständen über mehrere Jahre und invasive Eingriffe zur Gewinnung von Probenmaterial, die über eine Blutentnahme hinausgehen, sind nur äußerst schwer realisierbar. Die Infektion von asiatischen Makaken mit dem Affenimmundefizienzvirus (engl.: simian immunodeficiency virus, SIV) stellt aufgrund der ähnlichen Immunreaktion und nahezu identischer, aber wesentlich kürzerer Krankheitsverläufe ein überaus wertvolles Modell dar, zumal sich kleine Labortiere wie Nager oder Kaninchen nicht mit HIV infizieren lassen (MORROW et al., 1987; FILICE et al., 1988). Durch die Arbeit mit diesem Tiermodell konnten bisher viele wertvolle Erkenntnisse über die AIDS-Pathogenese und Ansätze zur Entwicklung einer erfolgreichen Vakzine gewonnen werden. So ließen sich durch mehrere TZell-basierte Immunisierungsstrategien, die während des letzten Jahrzehnts im Fokus der wissenschaftlichen Anstrengungen standen, die Höhe der Viruslast in der akuten und chronischen Phase der Infektion senken und die Überlebenszeit verlängern, wenngleich die Infektion an sich nicht abgewendet werden konnte (BAROUCH et al., 2000; SHIVER et al., 2002; CASIMIRO et al., 2005; WILSON et al., 2006). Der passive Transfer von 16 | S e i t e Einleitung neutralisierenden Antikörpern kurz vor oder zum Zeitpunkt einer experimentellen Infektion blockierte diese aber und bewies damit auch die Relevanz der humoralen Immunantwort für die Bekämpfung der Immundefizienzviren (BABA et al., 2000; MASCOLA et al., 2000; PARREN et al., 2001; BURTON et al., 2011). Bisher getestete Impfstoffe erzielten jedoch nur ineffiziente oder schwach wirksame Antikörper, deren Titer rasch sanken. Im Rahmen dieser Dissertation sollte deswegen die Bildung und Ausprägung der humoralen Immunantwort nach SIV-Infektion während mehrerer Vakzineexperimente an Rhesusaffen analysiert werden, wobei das Hauptaugenmerk auf solche Tiere gerichtet wurde, die ohne jede Therapie über mehrere Jahre lang klinisch gesund blieben. Üblicherweise wird der Umfang der humoralen Immunantwort lediglich über eine Titration bindender oder neutralisierender SIV-spezifischer Antikörper mittels ELISA oder Neutralisationstest eingeschätzt. In dieser Arbeit soll darüber hinaus eine Methode etabliert werden, mit der das SIV-spezifische immunologische Gedächtnis beurteilt werden kann, da das Erzielen eines robusten immunologischen Gedächtnisses naturgemäß Ziel eines jeden Impfstoffes sein sollte. Die dafür nutzbare Technik des B-Zell-ELISpot, mit dem virus-spezifische Antikörpersezernierende Zellen und Gedächtnis-B-Zellen quantifiziert werden können, war zu Beginn der Dissertation noch nicht im SIV-Makakenmodell angewandt worden. Ergänzt wurden diese Untersuchungen durch die phänotypische Charakterisierung von Gedächtnis-B-Zellen und anderen für die Funktion der Antikörper-vermittelten Immunantwort wichtigen B-Zellsubpopulationen mittels Durchflusszytometrie. Daten in diesem Umfang existieren dazu weder von SIV-negativen Rhesusaffen noch von Tieren in unterschiedlichen Infektionsstadien. Dabei haben B-Zelldysfunktionen einen wesentlichen Anteil an AIDS-assoziierten Symptomen, wie z.B. Hypergammaglobulinämie (SHIRAI et al., 1992) und Anfälligkeit für bakterielle Infektionen (MIEDEMA et al., 1988; CHONG et al., 2004a). Darüber hinaus ist zu erwarten, dass sich eine Beeinträchtigungen des BZellkompartiments durch die HIV-/SIV-Infektion negativ auf die Wirksamkeit von Impfstoffen auswirkt, die auf dem Schutz durch Antikörper beruhen. Zusätzliche Kenntnisse über die B-Zellphysiologie und –pathologie sind somit dringend notwendig, sowohl für die Vakzineentwicklung als auch die AIDS-Therapie. Literaturübersicht S e i t e | 17 2. LITERATURÜBERSICHT 2.1 Das Immunsystem Die Fähigkeit eines Organismus, zwischen “körpereigen” und “körperfremd” zu unterscheiden und sich vor letzterem zu schützen, wird im Allgemeinen als Immunsystem definiert. Bei Vertebraten unterscheidet man zwischen dem angeborenen und dem erworbenen Immunsystem. Das angeborene oder auch unspezifischen Immunsystem wird häufig als erste Verteidigungslinie des Körpers bezeichnet, da es innerhalb von Minuten bis Stunden auf eine eindringendes Pathogen reagiert, ohne dass diese Reaktion speziell auf das betreffende Antigen abgestimmt ist. Das erworbene oder auch spezifische Immunsystem hingegen antwortet deutlich gezielter und damit effizienter auf ein Antigen. Dafür sind beim ersten Kontakt mit einem neuen Antigen allerdings mehrere Tage bis Wochen erforderlich. Beim zweiten Kontakt erfolgt eine Reaktion dagegen viel schneller und stärker. Dies ist einer weiteren Eigenart des adaptiven Immunsystems zu verdanken, dem immunologischen Gedächtnis. Darüber hinaus erfolgt eine funktionelle Unterteilung in zelluläre und humorale Immunantwort. Es darf aber nicht vergessen werden, dass eine effiziente Immunreaktion immer aus dem komplexen Zusammenspiel aus unspezifischer und spezifischer Immunantwort besteht, die sowohl von den Zellen des Immunsystems als auch durch Komplementfaktoren, Antikörper und Zytokine geformt wird. Die vorliegende Arbeit konzentriert sich jedoch vornehmlich auf das erworbene Immunsystem mit besonderem Fokus auf die humorale Immunantwort. 2.1.1 T-Zell-vermittelte Immunantwort Die Bekämpfung von Pathogenen, die im Zellinneren replizieren, wie alle Viren, aber auch einige Bakterien und Parasiten, ist die Hauptaufgabe der T-Lymphozyten. Trifft eine naive TZelle auf eine Antigen-präsentierende Zelle, die ihr spezielles Antigen als Peptid:MHCKomplex auf ihrer Oberfläche exprimiert, beginnt die T-Zelle zu proliferieren und zu Effektor-T-Zellen zu differenzieren. Dabei können zwei Hauptklassen von Effektorzellen 18 | S e i t e Literaturübersicht unterschieden werden, die beide auf Wirtszellen, die das entsprechende Antigen per MHCKomplex präsentieren, reagieren, nicht auf das Pathogen selbst: Die zytotoxischen T-Zellen, welche das Oberflächenprotein CD8 (engl.: Cluster of Differentiation) tragen und befallene Wirtszellen abtöten, und die T-Helferzellen mit dem Oberflächenprotein CD4, welche infizierte Makrophagen dazu stimulieren, das intrazelluläre Pathogen zu zerstören (Th1-Zellen) und die Antikörperproduktion spezifischer B-Zellen (Th2Zellen) aktivieren (JANEWAY, 2008). In der Anfangsphase einer Infektion findet eine explosionsartige Vermehrung antigenspezifischer T-Effektorzellen statt. Innerhalb der nächsten 7-30 Tage begehen die meisten der aktivierten T-Zellen in Reaktion auf den schwindenden Antigenstimulus Apoptose, nur ein kleiner Teil überlebt als inaktive Gedächtnis-T-Zellen, die ohne erneuten Antigenkontakt keine Effektorfunktion ausführen (AHMED u. GRAY, 1996). 2.1.2 Humorale Immunantwort Ein wesentlicher Bestandteil der adaptiven Immunantwort ist die Bildung von Antikörpern, die im Blut und extrazellulären Flüssigkeiten vorkommen und deren Entstehung und Wirkungsweise als humorales Immunsystem bezeichnet wird (lat. humor: Flüssigkeit). Dass die Wirksamkeit aller klinisch eingesetzten Vakzinen zumindest zum Teil auf der Erzeugung protektiver Antikörper basiert, macht die Relevanz der humoralen Immunantwort für die Infektionsforschung deutlich. Bevor auf die Antikörper als solche näher eingegangen werden kann, muss die Differenzierung von B-Zellen zu Plasmazellen erläutert werden, die für die Produktion von Antikörpern verantwortlich sind. Dieser Vorgang macht deutlich, wie eng zelluläre und humorale Immunantwort miteinander verzahnt sind. 2.1.2.1 B-Zellen Man unterscheidet eine antigenunabhängige Entwicklung zur naiven, reifen B-Zelle im Knochenmark und einer terminalen Differenzierung zu Gedächtnis-B-Zellen oder Antikörperproduzierenden Plasmazellen nach Antigenkontakt in sekundären lymphatischen Organen. Abbildung 1 veranschaulicht den Reifungsprozess schematisch. Literaturübersicht S e i t e | 19 B-Zellentwicklung Aus hematopoetischen Stammzellen entstehen im Knochenmark pro- und anschließend präB-Zellen, deren Entwicklung durch die Umgruppierung der Gensegmente, die für die schweren und leichten Ketten eines Immunglobulins (Ig) kodieren, gekennzeichnet ist. In jenem Vorgang liegt der Grundstein für die weitreichende Diversität der B-Zellrezeptoren. Die Vorläufer der humanen B-Zellen tragen nicht nur den Oberflächenrezeptor CD10, der auch von T-Zell-Vorstufen exprimiert wird, sondern außerdem schon die B-Zell-typischen Oberflächenmarker CD19 und CD20, die erst bei der terminalen Differenzierung zur Plasmazelle verloren gehen. Schließlich entsteht eine unreife B-Zelle, die ein intaktes Immunglobulin der Klasse M (IgM) mit einzigartiger Spezifität auf der Oberfläche trägt, den sogenannten B-Zellrezeptor. Der letzte Entwicklungsschritt im Knochenmark besteht aus der Testung auf Autoreaktivität, nur wenn der B-Zellrezeptor nicht an körpereigene Strukturen bindet, kann die unreife B-Zelle das Knochenmark verlassen (HARDY et al., 2000; LEBIEN, 2000). Unreife B-Zellen in der Peripherie werden als Übergangs-B-Zellen (engl.: transitional B cells) bezeichnet, da sie noch CD10 tragen, aber auch schon den Komplementrezeptor CD21 exprimieren, der auf allen späteren Entwicklungsstufen der B-Zellen vorkommt (SURYANI et al., 2010). Es ist erstaunlich, dass letztlich nur 3% der im Knochenmark gebildeten unreifen B-Zellen die peripheren sekundären lymphatischen Organe erreichen und sich zur reifen B-Zelle mit längerer Lebensdauer differenzieren. Dabei verlieren sie CD10 und bilden zusätzlich zu IgM noch membrangebundenes Immunglobulin D (IgD) aus (CAMPANA et al., 1985). Der Rest fällt der negativen Selektion auf Autoreaktivität zum Opfer oder der Tatsache, dass sie nicht innerhalb ihrer relativ kurzen Lebensspanne von 3 Tagen in der Lage sind, in Lymphfollikel einzutreten (RAJEWSKY, 1996; JANEWAY, 2008). Antigen-abhängige Aktivierung von B-Zellen Der B-Zell-Rezeptor-Komplex ist ein Transmembranprotein auf der Oberfläche von B-Zellen und setzt sich aus zwei funktionalen Teilen zusammen: Der Antigen-bindende Teil entspricht 20 | S e i t e Literaturübersicht Abbildung 1: B-Zellreihe (von B. Neumann; mod. nach MOIR u. FAUCI 2009). Literaturübersicht S e i t e | 21 dem Antigen-spezifischen Immunglobulin (im Falle einer naiven B-Zelle immer IgM), das abgesehen von seiner in die Zellmembran intergierten Bereich identisch mit seiner löslichen Form ist. Er ist mit dem Signal-vermittelnden Teil assoziiert, der aus zwei Antigenunspezifischen Molekülen, Igα und Igβ, besteht (SCHAMEL u. RETH, 2000). Jede B-Zelle trägt zahlreiche, identische B-Zell-Rezeptoren mit einer einzigartigen Spezifität auf ihrer Oberfläche. Trifft nun eine naive, reife B-Zelle im Lymphfollikel auf das Antigen, für das sie spezifisch ist, bindet dieses an den B-Zell-Rezeptor-Komplex und die B-Zelle erhält das erste für die Aktivierung notwendige Signal. Innerhalb von 24 Stunden beginnt sie zu proliferieren und teilt sich alle 6-8 Stunden (RUSH u. HODGKIN, 2001; DONAHUE u. FRUMAN, 2003). Man unterscheidet zwei Varianten der B-Zellaktivierung: Für die Aktivierung durch Proteinantigene ist die zusätzliche Hilfe von CD4+-T-Zellen notwendig, man spricht deshalb von Thymus-abhängigen Antigenen. Die andere Variante besteht in der Aktivierung durch Thymus-unabhängige Antigene. Ein Beispiel für letzteres sind bakterielle Polysaccharide und Lipopolysaccharide (LPS), die durch ihre Struktur gleichzeitig an mehrere B-Zell-Rezeptoren einer Zellen binden oder in hohen Konzentrationen zu einer polyklonalen B-Zellaktivierung führen und so das zweite notwendige Signal liefern. Die entstehenden Plasmazellen sind allerdings meist kurzlebig und produzieren Antikörper niedriger Affinität (FELDMANN u. EASTEN, 1971; SLOWE u. WALDMANN, 1975; DINTZIS et al., 1983). Um T-Zell-Hilfe zu erhalten, muss das an den B-Zell-Rezeptor gebundene Antigen vom Signal-vermittelnden Teil des Rezeptors internalisiert und als Peptid:MHC-Komplex wieder auf der Zelloberfläche präsentiert werden. Erkennt eine CD 4+-Zelle die dargebotenen Peptidfragmente, wird sie dadurch veranlasst, mehrere Effektormoleküle zu synthetisieren: Allen voran den Membran-gebundenen CD40 Liganden (CD 40L), der an den CD40 auf den B-Zellen bindet und einen wesentlichen Faktor für die B-Zell-Proliferation darstellt (RUSH u. HODGKIN, 2001; DONAHUE u. FRUMAN, 2003). Außerdem schützt die simultane Aktivierung von CD40 durch CD40L und des B-Zell-Rezeptors durch ein passendes Antigen die B-Zelle vor Apoptose (ROTHSTEIN et al., 1995). Weiterhin werden von der CD4+-TZelle die Zytokine Interleukin 4 (IL-4), IL-5 und IL-6 sezerniert. Auf diese Weise wird die B- 22 | S e i t e Literaturübersicht Zelle zu Proliferation, somatischer Hypermutation und zum Immunglobulin-Klassenwechsel angeregt (PARKER, 1993; MCHEYZER-WILLIAMS et al., 2009). Die Effizienz der B-Zell-Antwort auf ein Antigen kann durch die gleichzeitige Aktivierung des B-Zell-Korezeptor-Komplexes um das 10- bis 100-fache gesteigert werden (CARTER u. FEARON, 1992). Letzterer besteht aus drei Proteinen: Dem Komplementrezeptor CD21 und den signalvermittelnden Glykoproteinen CD19 und CD81. Ist das Antigen, welches vom BZell-Rezeptor erkannt wird, bereits komplementmarkiert, bindet auch CD21 und löst durch CD19 und CD81 eine intrazelluläre Signalkaskade aus. So wird die Differenzierung und Antikörperproduktion beschleunigt und die Produktion von ko-stimulierenden Molekülen eingeleitet, die wiederum T-Helfer-Zellen anregen (ENGEL et al., 1995; DEMPSEY et al., 1996; BARRINGTON et al., 2009). Proliferation, Genkonversion, Klassenwechsel und somatische Hypermutation der B-Zelle Die erste Phase der für die Diversität der B-Zellrezeptoren verantwortlichen Genumgruppierung erfolgt während der B-Zellentwicklung im Knochenmark und ist Antigenunabhängig. In der Proliferationsphase nach Antigen-abhängiger Aktivierung von reifen BZellen kann die Spezifität von Immunglobulinen und deren Effektorkapazität weiter gesteigert werden. Die dafür maßgeblichen Vorgänge nennt man somatische Hypermutation, Genkonversion und Klassen- oder Isotypenwechsel. Alle beinhalten Rekombination der DNA und sind deshalb irreversibel. Somatische Hypermutation geschieht durch Punktmutationen in den Genen, die die variable Region der schweren und leichten Ketten des Immunglobulins kodieren, und verändert die Affinität des Antikörpers für ein Antigen. Durch Genkonversion wird zusätzliche AntigenSpezifität erreicht, indem neue Gensequenzen in die variable Region eingefügt werden. Die Bedeutung der Genkonversion ist von Spezies zu Spezies unterschiedlich und dort besonders ausgeprägt, wo während der B-Zellentwicklung kaum Genumgruppierung stattfindet, wie zum Beispiel bei Vögeln, Kaninchen oder Rindern und Schafen. Führen die beschriebenen Mechanismen dazu, dass die mutierten B-Zellklone das Antigen besser binden als der ursprüngliche B-Zellrezeptor, werden diese bevorzugt dafür selektiert, zu Antikörper- Literaturübersicht S e i t e | 23 sezernierenden Zellen auszureifen. Ergebnis ist die Affinitätsreifung von Antikörpern im Laufe einer Immunantwort (RAJEWSKY, 1996). Während naive B-Zellen immer IgD und IgM exprimieren und die ersten sezernierten Antikörper einer Immunreaktion immer vom Typ IgM sind, kann nach der B-Zellaktivierung die Immunglobulinklasse gewechselt werden. Die ursprüngliche konstante Region der schweren Kette des Moleküls wird gegen eine andere ausgetauscht, die AntigenBindungsstelle bleibt aber erhalten. So können die produzierten Antikörper andere Effektorfunktionen ausführen und/ oder wie z.B. im Falle von IgA andere Kompartimente des Körpers erreichen. Im Gegensatz zur somatischen Hypermutation und Genkonversion geschieht der Klassenwechsel nicht willkürlich, sondern wird durch von T-Zellen sezernierte Zytokine oder mitogene Signale bestimmter Pathogene gesteuert. IL-4 induziert vor allem einen Wechsel zu IgG1 und IgE, während die Bildung von IgG2 und IgA vor allem durch TGF-β (engl.: transforming growth factor) gesteigert wird. Interferon-γ (IFN-γ) wiederum verursacht einen Isotypenwechsel zu IgG2 und IgG3 (STAVNEZER, 1996; PONE et al., 2010). Die Anwesenheit von T-Helferzellen ist aber nicht nur für die Affinitätssteigerung der B-Zelle notwendig. Proliferierende B-Zellen bedürfen ständiger Stimulation durch CD40 und den BZell-Rezeptor, um nicht der Apoptose zu erliegen (ARPIN et al., 1995). Die Plasmazelle Schon in der führen Phase der Immunantwort differenzieren einige der proliferienden BZellen in den Primärfoci zu Plasmablasten, welche schon begonnen haben, Antikörper zu sezernieren, sich aber immer noch teilen, Antigen aufnehmen und präsentieren können und empfänglich für Stimulation durch T-Zellen sind. Plasmablasten sind für die erste Welle gebildeter Antikörper als Reaktion auf ein neues Pathogen verantwortlich und stellen eine Art Übergangsform zu den ausdifferenzierten Plasmazellen dar. Letztere sind wegen niedriger Spiegel von membrangebunden Immunglobulinen und dem Fehlen von MHC-Klasse IIMolekülen nicht mehr in der Lage dazu, mit T-Helfer-Zellen zu interagieren. Dafür haben somatische Hypermutation und Klassenwechsel bereits stattgefunden und führen in der Regel dazu, dass die sezernierten Antikörper eine höhere Affinität zum Antigen aufweisen. Die 24 | S e i t e Literaturübersicht funktionale Gemeinsamkeit beider Zelltypen besteht in der Fähigkeit, Antikörper zu sezernieren, weswegen sie häufig als Antikörper-sezernierende Zellen (engl. antibody secreting cells, ASC) zusammengefasst werden. Phänotypisch stellen sich Plasmazellen durch eine im Vergleich zu Gedächtnis-B-Zellen höhere Expression von CD27 (CD27hi) dar, während die Ausbildung des B-Zell-typischen Oberflächenmoleküls CD20 gesenkt wird (CD20-/low). Weiterhin ist der Großteil IgD-negativ und 60-80% der humanen Plasmazellen expremieren das Oberflächenmolekül CD38 (AVERY et al., 2005; CARAUX et al., 2010). Da der Chemokinrezeptor CXCR5 in Plasmazellen herabgesetzt, dafür aber die Expression von CXCR4 gesteigert wird, verlassen die ausdifferenzierten Plasmazellen die Keimzentren in Lymphknoten oder Milz und wandern ins periphere Gewebe oder ins Knochenmark (MEDINA et al., 2002). Weiterhin finden sich besonders IgA-sezernierende Plasmazellen in der Lamina propria von Epithelien des Verdauungs- und Respirationstraktes (MEDINA et al., 2003; PEREZ-ANDRES et al., 2010). Während der überwiegende Teil der Plasmazellen eine Lebensspanne von 2-4 Tagen hat, persistieren einige über Monate oder Jahre und werden als Ursache für das Vorhandensein von Antikörpertitern lange nach erfolgreicher Elimination des Erregers angesehen. Auf diese Weise leisten auch Plasmazellen einen wesentlichen Beitrag zum immunologischen Gedächtnis. Ob dieser Pool langlebiger Plasmazellen durch gelegentliche, aber kontinuierliche Differenzierung von Gedächtnis-B-Zellen aufrecht erhalten wird und ob letztere von polyklonaler Stimulation durch unspezifische Antigene angeregt wird oder auf ungeklärte Weise persistierendes spezifisches Antigen von Nöten ist, ist eine aktuell diskutierte Hypothese (BERNASCONI et al., 2002; RADBRUCH et al., 2006). Literaturübersicht S e i t e | 25 Die Gedächtnis-B-Zelle Selbst wenn einige Zeit nach überstandener Infektion keine Antikörper mehr gegen das verantwortliche Pathogen nachgewiesen können, kann bei einer erneuten Infektion ein umgehender Anstieg spezifischer Immunglobuline beobachtet werden. Dieser Effekt wird durch sogenannte Gedächtnis- oder memory-B-Zellen verursacht, die ca. 40% der BZellpopulation im Blut eines erwachsenen Menschen ausmachen (KLEIN et al., 1998). Es handelt sich um langlebige Abkömmlinge von B-Zellen, die einst durch ein Antigen stimuliert wurden (KUROSAKI et al., 2010). Sie teilen sich, wenn überhaupt, sehr langsam und obwohl sie Oberflächenimmunglobulin exprimieren, sezernieren sie dieses nicht oder kaum. Da genetische Veränderungen wie die somatische Hypermutation und der Klassenwechsel stattgefunden haben, zeichnen sie sich, im Unterschied zu naiven B-Zellen, durch hochspezifische B-Zellrezeptoren aus und tragen in den meisten Fällen kein IgD, sondern vornehmlich IgG oder IgA (AHMED u. GRAY, 1996; ANDERSON et al., 2006). Sobald nachgewiesen worden war, dass das Transmembranprotein CD27, welches der TNFRezeptorfamilie angehört, und zuerst auf T-Zellen beschrieben wurde (VAN LIER et al., 1987), auch auf den ausdifferenzierten B-Zellabkömmlingen vorkommt, wurde die Beschreibung von Gedächtnis-B-Zellpopulationen vereinfacht (KLEIN et al., 1998; TANGYE et al., 1998). Von reifen B-Zellen unterscheiden sie sich durch eben dieses Protein, während sie im Gegensatz zu Plasmazellen noch positiv für den B-Zellmarker CD20 sind. Darüber hinaus bilden die Gedächtnis-B-Zellen größere Mengen von MHC-Klasse IIMolekülen auf ihrer Oberfläche aus und sind somit auch bei geringen Antigenkonzentrationen in der Lage, erfolgreich T-Zellhilfe zu erwirken. Eine kräftigere und schnellere Generation von Plasmazellen als nach der Primärantwort ist die Folge (TANGYE et al., 2003). Verantwortliche Stimuli sind, neben T-Zell-Hilfe, die Zytokine IL-2, IL-10 und IL-21 (AGEMATSU et al., 1999; AVERY et al., 2005; KUCHEN et al., 2007), wobei in vitro auch eine polyklonale Aktivierung durch mikrobielle Produkte, wie LPS oder CpG-Dinukleotide erreicht werden konnte (BERNASCONI et al., 2002). 26 | S e i t e Literaturübersicht 2.1.2.2 Antikörper Die Bildung von Immunglobulinen ist die Hauptaufgabe der B-Zellen. In den vorangegangen Kapiteln wurden die Entwicklungsschritte der B-Zelle bis hin zur Antikörper-sezernierenden Plasmazelle dargestellt und die verschiedenen Erscheinungsformen von Immunglobulinen angedeutet: Sie können entweder als membrangebundener Bestandteil einer B-Zelle vorliegen und übernehmen damit die Aufgabe eines Antigen-Rezeptors, oder als lösliche Bestandteile im extrazellulären Raum, wo sie eine Fülle von Funktionen ausüben. Auf diese soll im Folgenden näher eingegangen werden. Des Weiteren werden Struktur und Vorkommen der verschiedenen Immunglobulinklassen erläutert. Aufbau eines Immunglobulins Ein Immunglobulinmolekül kann in zwei funktionelle Abschnitte unterteilt werden (s. Abb. 2): Der Antigen-bindende Teil ist Grundlage der Spezifität der humoralen Immunantwort und weist dementsprechend eine große Vielfalt auf. Er wird deshalb auch als variable bzw. VRegion bezeichnet. Der Effektor-vermittelnde Teil hat hingegen nur fünf Hauptformen entsprechend der Immunglobulinisotypen und wird folglich auch C-Region genannt (von engl.: constant region). Strukturell lässt sich das Molekül in je zwei Paar leichte und schwere Polypeptidketten (engl.: light-/ heavy-chains) gliedern. Die zwei schweren Ketten sind in der C-Region über eine Disulfid-Brücke miteinander verbunden, in der V-Region bindet jede schwere Kette eine leichte und formt damit zwei identische Antigen-Bindungsstellen. Jede Kette besteht aus einer Serie ähnlicher Aminosäuresequenzen, die zusammen eine bestimmte, festgepackte Proteinstruktur bilden, die Immunglobulin-Domäne. Die leichte Kette besteht aus zwei, die schwere aus vier Domänen. Die erste Domäne einer Kette ist sehr variabel und repräsentiert die V-Region eines Antikörpermoleküls (VH und VL, entsprechend der Domänen der schweren und leichten Ketten), der Rest entspricht der C-Region (CH und CL). Die drei konstanten Domänen der schweren Kette werden vom N-terminalen Ende her nummeriert: CH1-3. Literaturübersicht S e i t e | 27 Abbildung 2: Struktur eines Antikörpers (mod. nach JANEWAY, 2008). Zwei schwere Ketten (grün) und zwei leichte Ketten (gelb) bilden ein Immunglobulinmolekül. Die N-terminalen Domänen der Ketten bilden die konstante C-Region (blau), die ersten Domänen die variable V-Region (rot). Insgesamt entspricht das komplett gefaltete Molekül einer Y-ähnlichen Struktur mit drei ähnlich großen Teilen. Die zwei identischen Arme bestehen jeweils aus einer leichten Kette mit dem N-terminalen Ende einer schweren Kette und sind über die sogenannte hinge region (dt.: Scharnierregion) mit dem Stamm, bestehend aus dem C-terminalen Ende der beiden schweren Ketten, verbunden. Durch proteolytischen Verdau kann ein Antikörpermolekül in verschiedene Fragmente unterteilt werden: Die zwei Arme werden als Fab-Fragment (engl.: fragment antigen binding) bezeichnet und entsprechen dem Antigen-bindenden Teil des Immunglobulin, denn sie können zwar an das Antigen binden, vermitteln aber keine Effektorfunktion, während der Stamm Fc-Fragment genannt wird (engl.: fragment crystallizable) und über die Bindung an Fc-Rezeptoren unterschiedlicher Zellen die für die entsprechende Klasse typischen Immunreaktionen auslöst. Des Weiteren ist das Fc-Fragment verantwortlich für die Bindung von Komplementfaktoren und den aktiven Transport von Antikörpern in mukosale Sekrete, Milch oder über die Plazentaschranke (JANEWAY, 2008; SCHROEDER u. CAVACINI, 2010). 28 | S e i t e Literaturübersicht Immunoglobulinklassen und ihre Verteilung und Rollen im Körper Wie zuvor erläutert, wird die Klasse – und damit die Effektorfunktion – eines Antikörpers von der Struktur seiner schweren Kette definiert. Es gibt fünf Hauptklassen oder Isotypen, die zum Teil noch weiter in Unterformen eingeteilt werden: Immunglobulin M (IgM), Immunglobulin D (IgD), Immunglobulin G (IgG), beim Menschen mit den vier Sub-Klassen IgG 1-4, Immunglobulin A (IgA), mit den zwei Subklassen IgA 1-2, und Immunglobulin E (IgE). Ihre unterschiedlichen funktionalen Eigenschaften werden vom C-terminalen Ende der schweren Kette definiert, dort wo sie nicht an die leichte Kette bindet. Eine Übersicht über Vorkommen und Funktion der Immunglobulinklassen findet sich in Tabelle1. Die ersten Antikörper, die während einer Immunreaktion gebildet werden, sind immer vom Typ IgM, da für dessen Produktion kein Klassenwechsel erforderlich ist. Ihre Affinität ist normalerweise recht gering, da zu diesem Zeitpunkt noch keine somatische Hypermutation stattgefunden hat. Die Avidität insgesamt ist, bedingt durch die Struktur, aber relativ hoch: IgM formt normalerweise Pentamere, so dass 10 identische Antigen-Bindungsstellen vorliegen und die Bindungsstärke erhöht wird. Dies prädestiniert IgM für sich wiederholende Epitope, wie zum Beispiel die Polysaccharide bakterieller Zellwände. Durch die daraus resultierende Größe des Moleküls kommt es vornehmlich im Blut vor (RACINE u. WINSLOW, 2009). IgG ist das dominante Immunglobulin im menschlichen Körper und erreicht die höchste Konzentration in Serum und Lymphe. IgG-Antikörper besitzen gewöhnlich eine hohe Affinität. Sie sind in der Lage, Mikroorganismen effektiv zu neutralisieren, sie für die Phagozytose zu opsonisieren und die Komplementkaskade zu aktivieren (SCHROEDER u. CAVACINI, 2010). IgA liegt im Plasma als Monomer vor, während für den Transport durch Epithelien die dimerische Form notwendig ist. IgA ist vor allem an den Epithelien von Lunge und Verdauungstrakt zu finden. Zusammen mit der hohen Neutralisationskapazität lässt dies auf die Wichtigkeit von IgA als erste Verteidigungslinie gegen aus der Umwelt eindringende Pathogene schließen (SCHROEDER u. CAVACINI, 2010). Literaturübersicht S e i t e | 29 IgG1 IgG2 IgG3 IgG4 IgM IgA IgD IgE 9 3 1 0,5 1,5 2,1 0,03 5x10-5 21 20 7 21 10 6 3 2 Transport über Epithelien - - - - + +++ - - Diffusion in den extravaskulären +++ +++ +++ +++ -/+ ++ - + Komplementaktivierung ++ + +++ - ++++ - - - Aktivierung von Mastzellen und - - - - - - - +++ Neutralisation ++ ++ ++ ++ + ++ - - Opsonisierung +++ -/+ ++ + + + - - Sensibilisierung von NK-Zellen ++ - ++ - + - - - Serumlevel (in mg/ml-1) Halbwertszeit im Serum (in Tagen) Raum Basophilen Granulozyten Tab. 1: Immunglobulinklassen des Menschen (mod. nach JANEWAY, 2008; SCHROEDER u. CAVACINI, 2010) IgE schließlich kommt in sehr geringen Konzentrationen frei in Blut oder extrazellulären Flüssigkeiten vor. Es wird rasch von Mastzellen in der Haut oder Mukosa gebunden und hat in dieser Konfiguration eine weitaus höhere Halbwertszeit, als in Tabelle1 angegeben. Dort veranlasst es Mastzellen zur Chemokinausschüttung, die neben Inflammationsreaktionen auch Abwehrmaßnahmen des Körpers wie Husten, Niesen und Erbrechen auslösen können (STONE et al., 2010). Da IgD während der B-Zell-Ontogenese zusammen mit IgM der gleichen Spezifität exprimiert wird und reife B-Zellen nach Antigenkontakt die Ausbildung von IgD als Oberflächenmolekül herunterfahren, wurde der physiologischen Bedeutung dieses Isotypen lange Zeit wenig Beachtung geschenkt. Weil aber lösliches IgD in messbaren Konzentrationen im Serum vorhanden ist und in jüngerer Vergangenheit der Nachweis von reifen und Gedächtnis-B-Zellen gelang, die den Klassenwechsel von IgM zu IgD vollzogen 30 | S e i t e Literaturübersicht haben (KLEIN et al., 1998), liegt die Annahme nah, dass IgD vor allem in Geweben des oberen Respirationstraktes eine bedeutendere Rolle für das Immunsystem einnimmt, als ursprünglich angenommen. Diese Frage ist Thema gegenwärtiger Forschung (K. CHEN u. CERUTTI, 2010). Wirkungsweisen von Antikörpern Ebenso vielfältig wie die Pathomechanismen der Mikroorganismen, die den Körper bedrohen, sind auch die Strategien, mit denen das Immunsystem ihnen mit Hilfe von Antikörpern begegnet. Neben der Initiierung der Komplementkaskade und der Aktivierung von unterschiedlichsten Effektorzellen über deren Fc-Rezeptoren spielt die Neutralisation von Pathogenen eine große Rolle bei der Bekämpfung von Infektionskrankheiten. Klassische neutralisierende Antikörper (nAk) werden durch ihr Vermögen definiert, das schädliche Potential eines Agens ohne die Hilfe eines anderen Mitstreiters in vitro aufzuheben (BURTON, 2002). Zwar zeigen zahlreiche Studien in vivo, dass der passive Transfer von in vitro-neutralisierenden Antikörpern Schutz gegen intravenöse und mukosale Infektion mit SIV vermittelt (SHIBATA et al., 1999; MASCOLA et al., 2000; PARREN et al., 2001), nur darf nicht vergessen werden, dass im komplexen Immunsystem viele weitere Faktoren eine Rolle spielen können. Die antivirale Kapazität eines Antikörpers kann über viele verschiedene Mechanismen vermittelt werden, die sowohl virus- als auch antikörper- bzw. wirtsspezifisch sind. Abbildung 3 zeigt eine vereinfachte schematische Darstellung der im Folgenden erläuterten Methoden. Vor dem Eintritt in die Zielzelle (Abb. 3 a) können nAk, die an mehrere Viruspartikel zugleich binden, zu einer Agglutination dieser führen und so die Fusion mit der Wirtszelle verhindern. Durch das Imitieren der Struktur von Zellrezeptoren, die normalerweise die Fusion mit der Zellmembran bewirken, können die Bindungsstellen blockiert werden. Mittels Fc-Rezeptor-vermittelter Phagozytose oder Aktivierung des Komplementsystems werden diese Virus-Antikörper-Komplexe entfernt. Einige nAk sind in der Lage, die Fusion auch nach der Bindung an die Zellrezeptoren zu verhindern, indem sie deren Konformation ändern. Konnte die Infektion der Zelle nicht verhindert werden (Abb. 3 b), können nAk und nichtneutralisierende Antikörper an virale Proteine auf der Zelloberfläche binden und durch weitere Mechanismen die Ausbreitung des Virus verhindern: Auch in diesem Fall kann eine Literaturübersicht S e i t e | 31 Fc-vermittelte Lyse der infizierten Zelle stattfinden. Für einige Viren, zum Beispiel HIV-1, ist gezeigt worden, dass eine Bindung von nAk an den CD4-Rezeptor die reverse Transkriptase hemmen kann, obwohl der genaue Mechanismus noch nicht klar ist. Schließlich können nAk den Zusammenbau und die Freisetzung von neugebildeten Viruspartikeln an der Zelloberfläche unterbinden (BURTON, 2002; READING u. DIMMOCK, 2007). Abbildung 3: Wirkungsmechanismen von Antikörpern (modifiziert nach BURTON, 2002). 32 | S e i t e 2.2 Literaturübersicht Humane und simiane Immundefizienzviren 2.2.1 HIV und AIDS Nach dem ersten Auftreten des Acquired Immunodeficiency Syndrome (AIDS, engl. für Erworbenes Immunschwäche Syndrom) 1981 herrschten einige Jahre verschiedene Spekulationen für den Grund dieser Erkrankung, die sich durch das Auftreten von Lymphopenie, opportunistischen Infektionen wie z.B. Pneumocystis jirovecii -Pneumonien oder mukosalen Kandidosen und der Neigung zum Kaposi-Sarkom äußerte (GOTTLIEB et al., 1981; SIEGAL et al., 1981). 1983 wurde ein sich auf T-Zellen vermehrendes Retrovirus von Patienten isoliert, die an einem „Pre-AIDS-Syndrom“ litten (Barré-Sinoussi et al, 1983; Gallo et al, 1984). Bis 1986 wurde nicht nur das Humane Immundefizienz Virus Typ 1 (HIV1) isoliert, benannt und in Zellkultur vermehrt, sondern auch eine zweite Variante in westafrikanischen Patienten entdeckt, die als HIV-2 bezeichnet wurde (Alizon et al., 1984; Clavel et al., 1986). Gemäß dem Report über die gobale AIDS Epidemie UNAIDS von 2010 (ANONYMUS, 2010) ist die Rate der weltweiten Neuinfektionen mit HIV gegenüber 1997, welches als das Jahr angesehen wird, in dem die HIV-Epidemie ihren Höchststand erreichte, um 21% auf ca. 2,6 Millionen gesunken. In 33 Ländern ist die HIV-Inzidenz in den letzten Jahren um über 25% gefallen. Ein Großteil dieser Länder liegt in der Region südlich der Sahara, welche am stärksten von der HIV-Epidemie betroffen ist. Allerdings gibt es auch gegenläufige Trends. In sieben Ländern, davon fünf in Osteuropa und Zentralasien, stieg die Inzidenz um mehr als 25%. Dank antiretroviraler Therapie (ART) ist auch die Mortalität rückläufig, was aber im Gegenzug zu einer höheren Prävalenz führt: 2009 lebten weltweit 33,3 Millionen HIVinfizierte Menschen, 68% davon im südlichen Afrika. Beide Geschlechter sind nahezu gleich stark betroffen, der Anteil der weiblichen Infizierten ist leicht höher. Risikogruppen sind vor allem in den Industriestaaten intravenös Drogenabhängige, homosexuelle Männer und Prostituierte. Literaturübersicht S e i t e | 33 HIV wird hauptsächlich über sexuellen Kontakt übertragen und kann sowohl in infizierten mononukleären Zellen als auch zellfrei im Ejakulat, im Sekret der Vagina und in Zellen der Cervix gefunden werden. Dies bedeutet, dass eine Infektion durch heterosexuellen Geschlechtsverkehr sowohl von Mann zur Frau als auch umgekehrt möglich ist, wobei ersteres deutlich häufiger vorkommt (PADIAN et al., 1991). Das größte Risiko einer HIVÜbertragung besteht allerdings beim rezeptiven Analverkehr, wo die Wahrscheinlichkeit bei 1:20 – 1:300 pro Exposition liegt, verglichen mit 1:200 – 1:2000 bei der Übertragung von Mann zu Frau bei vaginalem Verkehr (HLADIK u. MCELRATH, 2008). Weiterhin ist eine Übertragung durch Blut und Blutprodukte möglich, wobei letzteres durch inzwischen standardmäßig ausgeführtes Screening seltener geworden ist (SCHREIBER et al., 1996; GLYNN et al., 2000). Epidemiologisch bedeutend ist hingegen immer noch die Übertragung durch gemeinsames Benutzen von Injektionsbestecken in der Drogenszene. Es konnte aber gezeigt werden, dass auch in diesem Bereich die Verbreitung von HIV durch Aufklärung und die Bereitstellung von kostenlosen Kanülen gemindert werden kann (DES JARLAIS, 1999). Obwohl das Risiko einer vertikalen Übertragung in den Industriestaaten durch entsprechende Vorsorge während der Schwangerschaft und Geburt stark verringert werden kann, stellte diese Infektionsroute in Drittweltländern fortwährend ein großes Problem dar, da antiretrovirale Therapie während der Schwangerschaft oder die Möglichkeit einer Sectio caesarea kaum in Anspruch genommen werden können. Zudem kann aus finanziellen und hygienischen Gründen kaum auf das Stillen verzichtet werden, um das Neugeborene vor einer Virusübertragung durch die Muttermilch zu schützen (CAVARELLI u. SCARLATTI, 2011). 2.2.2 SIV Es gibt viele deutliche Anhaltspunkte dafür, dass sich Affenimmundefizienzviren (engl.: simian immunodeficiency virus, SIV) nach einer Übertragung von ihren ursprünglichen Wirtsspezies auf den Menschen an diesen adaptiert haben und somit die Vorfahren von HIV-1 und HIV-2 darstellen. HIV-1 ist verwandt mit SIVcpz der Schimpansen (engl.: chimpanzee), während HIV-2 dem SIVsm entspringt, einem Virus der Rauchmangaben (engl.: sooty mangabey). Die drei Hauptgruppen von HIV-1 sind auf nur drei voneinander unabhängigen 34 | S e i t e Literaturübersicht Übertragungsereignissen von Schimpanse auf Mensch zurückzuführen. Diese geringe Zahl ist verwunderlich in Anbetracht dessen, dass mehr als 35 afrikanische Affenarten mit SIVVarianten infiziert sind und Menschen deshalb zahllosen Expositionen ausgesetzt gewesen sein müssen. Es scheint also so, als ob entscheidende Adaptionen stattfinden müssen, die zum einen die Infektion eines Menschen und darüber hinaus die Übertragbarkeit von diesem auf weitere Menschen ermöglichen. Die exakten Mechanismen sind noch nicht endgültig geklärt (HEENEY et al., 2006). Interessanterweise führt eine SIV-Infektion im natürlichen Wirt in den seltensten Fällen zu AIDS (MULLER-TRUTWIN et al., 1996; REY-CUILLE et al., 1998). In jüngster Vergangenheit wurden allerdings Hinweise auf einen möglichen pathogenen Verlauf der SIVcpz-Infektion wilder Schimpansen entdeckt (KEELE et al., 2009). 2.2.3 Klassifikation HIV und SIV gehören zu einer Untergruppe der Retroviridae, den Lentiviren. HIV-1 und HIV-2 sind die einzigen bekannten humanen Lentiviren. HIV-1 wird in 4 Gruppen klassifiziert: M (engl.: main, dt.: Haupt-), O (engl: outlier, dt.: Sonderfall), N (nicht M oder O) und die kürzlich identifizierte Gruppe P (PLANTIER et al., 2009). Die Hauptgruppe M wird weiter in die Stämme A bis K unterteilt. Die verschiedenen Stämme unterscheiden sich in ihrer geographischen Verbreitung. In Westeuropa, Nordamerika und Australien dominiert zum Beispiel Stamm B, während in Osteuropa sowohl A als auch B stark vertreten sind. Subtyp C herrscht in Südafrika, Indien und Brasilien vor (THOMSON u. NAJERA, 2005). HIV-2 wurde in zwei Gruppen unterteilt, Subtyp A, welcher fast ausschließlich in Westafrika anzutreffen ist, und Subtyp B, der in Frankreich, Portugal und der Elfenbeinküste vorkommt. Während die meisten HIV-1-Infektionen weltweit der Gruppe M zuzuordnen sind und vor allem bei Stamm C sowohl die Übertragungsrate als auch die Morbidität am höchsten ist, liegt die Transmission von HIV-2 deutlich niedriger. Diese Tatsache ist vermutlich auf niedrigere Virusreplikation und den geringeren Gehalt infektiöser Partikel in Körperflüssigkeiten zurückzuführen. Zusammen mit dem weniger pathogenen Verlauf der Erkrankung scheint die HIV-2-Infektion näher an der apathogenen Infektion der natürlichen Wirte zu liegen (LEVY, 2009). 2.2.4 Vergleich von SIVsm, HIV-1 und HIV-2 Virusstämme, die ursprünglich aus SIVsm entstanden sind, haben viele Ähnlichkeiten mit HIV-1 und -2. Insgesamt haben SIV und HIV ein sehr ähnliches Genom; tatsächlich Literaturübersicht S e i t e | 35 unterscheidet sich HIV-1 nicht mehr von SIVsm als von HIV-2. Alle drei Viren tendieren zu einer eher langen Inkubationszeit und sind durch die Immunantwort des Wirtes einem starken Selektionsdruck unterworfen, der sich in verschiedenen Antigenvariationen äußert. Der bedeutendste Unterschied ist aber, dass SIVsm-infizierte Mangaben kein AIDS entwickeln. Eine experimentelle Infektion von asiatischen Makaken führt hingegen zu einem Infektionsverlauf sehr ähnlich der humanen HIV-Infektion. Sowohl HIV als auch SIV nutzen hauptsächlich CD4+T-Zellen als Zielzellen und CCR5 als Ko-Rezeptoren für den Eintritt in diese (Z. CHEN et al., 1997). Beide Viren werden über den Reproduktionstrakt übertragen, allerdings findet in freier Wildbahn eine SIV-Verbreitung auch über Bisse statt. Ebenfalls ist der bei der HIV-Infektion typische initiale Verlust der CD4+ T-Zellen in der Darmmukosa, welcher den im Blut weit übertrifft, nach SIV-Infektion zu beobachten, genauso wie die Zunahme der aktivierten T-Zellen im Gastrointestinaltrakt, selbst wenn der natürliche Wirt keine AIDS-ähnlichen Symptome entwickelt (GORDON et al., 2007; GAUFIN et al., 2009). 2.2.5 Struktur von HIV/SIV Das komplette HIV-Virion misst ungefähr 100-120 nm im Durchmesser und enthält zwei identische Kopien von Ribonukleinsäure (engl. ribonucleic acid, RNA), die aus neun Genen besteht: Gag (engl.: group specific antigen), pol (engl.: polymerase), env (engl.: envelope), tat (engl.: transactivator of transcription), rev (engl.: regulator of expression of viral proteins), nef (engl.: negative effective factor), vif (engl.: virion ineffectivity factor), vpr und vpu (engl.: viral protein r/u) bei HIV-1 bzw. vpx (engl.: viral protein x) bei HIV-2 und SIV. Diese sind in einem kegelförmigen Kapsid aus p24 (respektive p27 bei SIV) eingeschlossen. Ebenfalls darin befindlich ist eine kleine Gruppe von Enzymen für die Virusreplikation, die virale Protease, die Reverse Transkriptase und die Integrase (GELDERBLOM, 1991). Die Lipid-Doppelmembran, die die gag-kodierte Matrix umgibt, entstammt der Membran der Wirtszelle und hat nach allen Seiten hervorstehende virale Glykoproteine, das transmembrane gp41 und das äußere gp120 (bzw. gp130 bei SIV). Diese werden von env kodiert. Da diese die einzigen vom Virus kodierten Proteine an der Oberfläche des Virions sind, sind sie entscheidend für die Fusion mit der Wirtszellmembran und wichtige Ziele für die Immunantwort des Wirtes (KWONG et al., 1998). 36 | S e i t e Literaturübersicht Abbildung 4: SIV-Partikel (modifiziert nach S. Norley). 2.2.6 Die Replikation von HIV/ SIV Sobald das Virus die physikalischen Barrieren an der Eintrittspforte überwunden hat, ist der erste Schritt zur Virusvermehrung der Eintritt in die Zielzelle. Dieser wird, sowohl im Falle von HIV-1/-2 als auch SIV, durch die Bindung des CD4-Rezeptors, welcher vor allem auf TLymphozyten, aber auch Makrophagen und Dendritischen Zellen zu finden ist, und dem viralen gp120 initiiert (SATTENTAU, 1988). Die Bindung verursacht konformative Änderungen innerhalb des gp120 und führt daraufhin zu einer Interaktion mit Korezeptoren, von denen die bedeutendsten Chemokinrezeptoren wie CCR5 und CXCR4 sind (BJORNDAL et al., 1997). Dies wiederum führt zu einer Konformationsänderung von gp41 und dadurch zu einer Fusion von Virion- und Wirtszellmembran (FREED et al., 1992). Innerhalb der Zelle wandelt die Reverse Transkriptase die virale RNA in provirale DNA um, die von der Integrase in die Wirts-DNA eingebaut wird. In diesem Stadium handelt es sich vorerst um eine latente Infektion, die, zumindest in T-Zellen, erst nach Aktivierung der Zelle produktiv Literaturübersicht S e i t e | 37 wird, aber auch, je nach Zelltyp, über Jahre verdeckt bestehen bleiben kann (WILLIAMS u. GREENE, 2007). Nach erfolgter Aktivierung wird die provirale DNA in messenger-RNA umgeschrieben und für die Translation ins Zytoplasma transportiert. Die Hüllproteine, die später auf der Virusoberfläche sitzen, gruppieren sich außen an der Zellmembran der Wirtszelle, und die Viruspartikel schnüren sich von der Zellmembran ab, während sich im Inneren viralen Proteine, Enzyme und RNA befinden. Das Virus reift, indem Gag- und GagPol-Vorläuferproteine von der viralen Protease in funktionale Proteine gespalten werden (FREED, 2001). Für die Transmission greift das Virus nicht nur auf die Freisetzung von freien Viruspartikeln zurück, ein bedeutender Mechanismus ist die Bildung von sog. „virologischen Synapsen“ zwischen infizierter und uninfizierter Zielzelle über gp120 und den CD4-Rezeptor und damit eine direkte Übertragung von Zelle zu Zelle (JOLLY et al., 2004). 2.2.7 SIV-Infektion von Makaken als in vivo-Modell für die HIV-Infektion Die Ähnlichkeit bezüglich Physiologie und Immunsystem von nicht-humanen Primaten (NHP) und Menschen macht erstere zu einem wertvollen Modell für Infektionskrankheiten. Auch wenn die Nutzung von NHP in der Forschung aus ethischen Gründen nach Möglichkeit vermieden werden sollte, gibt es in der HIV-Forschung und Impfstoffentwicklung bisher keine vollwertige Alternative. Versuche, kleinere Labortiere wie Kaninchen oder Nager mit HIV zu infizieren, scheiterten oder waren nicht reproduzierbar (MORROW et al., 1987; FILICE et al., 1988). Für die Pathogeneseforschung sind Modelle mit natürlichen Wirtsspezies interessant, die keine AIDS-Symptomatik entwickeln. So könnten zum Beispiel Unterschiede in der Immunantwort des natürlichen Wirtes bei einer apathogenen SIV-Infektion im Vergleich zu der HIV-Infektion im Menschen wertvolle Hinweise für neue Vakzine- oder Therapieansätze bieten (PANDREA et al., 2008). Eine andere Herangehensweise stellt das SIVMakakenmodell dar. Angesichts der Tatsache, dass Makaken nicht mit HIV-1 infiziert werden können (STREMLAU et al., 2004), wird das SIV-Model häufig genutzt, um die Immunantwort auf potentielle Impfstoffe einzuschätzen und ihren Effekt bei Virusexposition zu beurteilen oder die Wirksamkeit von antiretrovirale Therapien zu testen. Experimentelle 38 | S e i t e Literaturübersicht Infektionen mit SIVmac, einer Virusvariante, die durch Passage von SIVsm in asiatischen Makaken entstanden ist (APETREI et al., 2005), führen bei diesen Spezies zu einer AIDSähnlichen Erkrankung. SIV-infizierte Makaken erreichen wesentlich schneller als HIVinfizierte Menschen das AIDS-Stadium, üblicherweise innerhalb von sechs Monaten bis zwei Jahren (GERETTI, 1999). Um ähnliche Verhältnisse wie im Menschen zu schaffen, sind im Makakenmodell verschiedene Infektionsrouten etabliert, angefangen mit einer intravenösen Virusgabe bis hin zu rektaler, vaginaler oder oraler Applikation (CRANAGE et al., 1997; MILLER et al., 1997; STAHL-HENNIG et al., 1999). So können nicht nur lokale Immunreaktionen an der Eintrittspforte des Virus erforscht werden, wo bei menschlichen Patienten die Probengewinnung schwierig bis gar nicht durchführbar ist; es ist auch eine Verlaufskontrolle von Beginn an möglich, während die Charakteristik der frühen HIVInfektion im Menschen selten untersucht werden kann, da der eigentliche Infektionszeitpunkt meistens unbekannt ist. Unlängst wurde damit begonnen, neuartige Hybridviren aus SIV und HIV (engl.: simianhuman immunodeficiency virus, SHIV) zu entwickeln und einzusetzen, um auch die letzten Unterschiede in der Biologie und Pathogenese von HIV-1 und SIVmac zu überbrücken (NOMAGUCHI et al., 2008). 2.3 Pathophysiologie der Immundefizienzviren 2.3.1 Krankheitsverlauf Sowohl beim Menschen als auch bei der pathogenen SIV-Infektion von nicht-humanen Primaten verläuft die Erkrankung generell in drei Stadien. Während der kurzen, akuten Phase findet eine Immunaktivierung statt. Außerdem ist die akute Phase gekennzeichnet durch eine hohe Viruslast (engl.: peak viremia) und einen initialen, massiven Verlust von CD4+-T-Zellen vor allem in der Darmmukosa, aber auch in geringerem Maße im Blut. Es können Grippeähnliche Symptome auftreten (BARRE-SINOUSSI et al., 1983; COOPER et al., 1985). Daran schließt sich eine lange, klinisch asymptomatische, chronische Phase an, in der die CD4+Zahlen im Blut wieder ansteigen und sich auf einem leicht erniedrigten Spiegel stabilisieren. Die Menge der CD4+-Zellen im Darm bleibt hingegen dauerhaft niedrig. Trotz Abnahme der Viruslast stoppt die Replikation nicht völlig und auch die Immunaktivierung bleibt messbar. Obwohl häufig von der Latzenzphase gesprochen wird, darf das Fehlen von klinischen Literaturübersicht S e i t e | 39 Symptomen nicht für eine Stagnation der Erkrankung gehalten werden. Zuletzt folgt die finale Phase, in der sich die AIDS-Symptomatik manifestiert. Das Immunsystem ist durch die chronische Belastung erschöpft, die Viruslast steigt rasant an. Die CD4+-Zahlen fallen dadurch bedingt ab und opportunistische Erreger können nicht mehr beherrscht werden (GROSSMAN et al., 2006). Abbildung 5 zeigt den Krankheitsverlauf schematisch. Abbildung 5: Schematischer Verlauf der HIV-Infektion (mod. nach GROSSMAN et al., 2006). Die linke y-Achse stellt die relativen Veränderungen der Parameter des Immunsystems dar, die rechte y-Achse die logarithmische HIV-Last. Auf der x-Achse sind die Phasen der Infektion schematisch aufgetragen. PANTALEO u. FAUCI (1996) unterschieden vier mögliche Verlaufsformen der HIVErkrankung, die sowohl bei HIV-infizierten Menschen als auch bei SIV-infizierten Makaken beobachtet wurden: Typische Progressoren Zu dieser Gruppe gehören gut Dreiviertel der Erkrankten. Die Phase der klinischen Latenz dauert bei der HIV-1-Infektion 6-8 Jahre, im SIV-Makakenmodell progredieren die Tiere innerhalb von 6 Monate bis 2 Jahren zum klinisch manifesten Stadium (GERETTI, 1999). Die Zahl der CD4+-T-Zellen im Blut korreliert hierbei mit dem Auftreten von AIDS-Symptomen. Fällt sie unter 500 Zellen/µl, steigt das Risiko. 40 | S e i t e Literaturübersicht Schnelle Progressoren Bei ca. 10-15% der HIV-infizierten kann ein sehr schneller voranschreitender Krankheitsverlauf (engl.: rapid progression) beobachtet werden, bei der die Phase der Latenz und der damit verbundene Abfall der peripheren Viruslast kaum oder gar nicht eintritt. Auch die spezifische Immunantwort ist kaum ausgeprägt, bei einigen Individuen gar nicht messbar. Makaken mit einem derartigen Krankheitsverlauf erliegen innerhalb von 12 bis 26 Wochen der Infektion (SAUERMANN et al., 1997; DYKHUIZEN et al., 1998). Long-Term Nonprogressors (LTNP) Auch wenn die dafür verantwortlichen Mechanismen für dieses Phänomen noch nicht ermittelt werden konnten, gibt es Personen, deren Immunsystem trotz nachweislicher HIVInfektion kaum pathologische Veränderungen aufweist. Nach der akuten Infektion pendelt sich die Virusbeladung knapp oberhalb der Nachweisgrenze ein, gelegentlich fällt sie sogar darunter. Die T-Zellzahlen liegen stabil in einem annähernd physiologischen Bereich und die Funktion des Immunsystems bleibt erhalten, es treten folglich keine AIDS-assoziierten Erkrankungen auf. Mit einer Häufigkeit von höchstens 5% der beobachteten Kohorte, in Abhängigkeit vom Virusstamm, stellen diese Individuen eine Ausnahme dar. Langzeitüberlebende Im Unterschied zu den LTNP zeigt diese Gruppe sehr wohl eine deutlich verringerte CD4+Zahl und erhöhte Virusreplikation, ist aber in der Lage, diese über einen verlängerten Zeitraum hinweg zu kontrollieren (engl.: controllers). 2.3.2 Immunantwort gegen HIV/ SIV Obwohl das angeborenene Immunsystem deutlich auf die Infektion reagiert, indem es das Virus über bestimmte Strukturmerkmale, sog. PRR (engl.: pattern recognition receptors), erkennt, schützt dies den Körper nur unzureichend. Dadurch, dass die ausgeschütteten Chemokine neben NK-Zellen auch Virus-empfängliche Zellen wie Makrophagen, dendritische Zellen und T-Zellen an die Viruseintrittspforte locken, wird im Gegenteil die Ausbreitung des Virus noch erleichtert (BORROW et al., 2010). Wesentlich effizienter, wenn auch nicht ausreichend, wird die Infektion durch das adaptive Immunsystem bekämpft. Literaturübersicht S e i t e | 41 2.3.2.1 Die zelluläre Immunantwort Virus-spezifische zytotoxische T-Zellen lassen sich bereits etwa eine Woche nach Infektion nachweisen und ihr Anstieg korreliert mit dem Abfall der Viruslast (REIMANN et al., 1994). SCHMITZ et al. (1999) konnten die essenzielle Bedeutung von CD8+-T-Lymphozyten für die Kontrolle der SIV-Infektion mit einem Depletionsversuch beweisen: Nicht nur Rhesusaffen, deren zytotoxische T-Zellen vor der Infektion mit SIV depletiert wurden, zeigten einen rasanten Krankheitsverlauf ohne nennenswerten Abfall der Virusbeladung nach der primären Virämie, auch in Tieren, die sich schon in der chronischen Phase befanden, stieg die SIVLast nach der Depletion wieder an. Obwohl CD4+-T-Zellen die Zielzellen von HIV/ SIV sind, konnte auch hier ein Depletionsexperiment die Relevanz dieser Zellpopulation deutlich machen. Der typische Abfall nach der Peak-Virämie trat bei CD4+-depletierten Tieren nicht ein und sie zeigten das klinische Bild eines rapid progressors (ORTIZ et al., 2011). Der positive Effekt der T-HelferZellen auf die Bildung von zytotoxische T-Zellen und spezifischen B-Zellen überwiegt also ihre Kehrseite als Virusreplikator. 2.3.2.2 Die humorale Immunantwort Zwei bis drei Wochen nach erfolgter Infektion lassen sich die ersten Antikörper gegen HIV/ SIV nachweisen, die hauptsächlich gegen die Hüllproteine gerichtet sind. Ihre Titer steigen bei klassischem Krankheitsverlauf kontinuierlich an (REIMANN et al., 1994). Die Serokonversion korreliert also ebenfalls mit dem Abfall der initialen Spitzenvirämie. Da die in der frühen Phase der Infektion gebildeten Antikörper zwar von beträchtlicher Menge sind, ihre qualitativen Eigenschaften wie Spezifität, Avidität und Neutralisationskapazität sich aber erst in den folgenden Monaten steigern, wird die Bedeutung spezifischer Immunglobuline in der akuten Phase der Infektion kontrovers diskutiert (COLE et al., 1997; COLE et al., 1998; BAUM, 2010; MASCOLA u. MONTEFIORI, 2010). 2.3.3 Pathogenese der Immundefizienzviren Die Symptomatik von AIDS ähnelt sich bei Mensch und NHP stark. Häufig zu beobachten sind: Lymphadenopathie, chronische Diarrhoe und Auszehrung, opportunistische Infektionen (z.B. Pneumocystis jirovecii-Pneumonie, Candidiasis, Mycobakterium-Infektionen, Cytomegalievirus-[CMV]-Erkrankung), Enzephalitiden, Anämie und B-Zell-Lymphome. Bei 42 | S e i t e Literaturübersicht SIV-infizierten Makaken tritt außerdem die SIV-typische Riesenzellerkrankung auf (GERETTI, 1999). Allen klinischen Manifestationen liegen die Auswirkungen, die die Immundefizienzviren auf das Immunsystem des Wirtes haben, zu Grunde. 2.3.3.1 Auswirkungen auf das zelluläre Immunsystem Die deutlichste Konsequenz der HIV-/SIV-Infektion ist die massive Eliminierung von CD4+T-Zellen vor allem in den mukosalen Geweben. Als wichtigste Wirtszellen der Viren erfolgt einerseits eine Zerstörung durch Virusreplikation, zum anderen wird eine indirekte Induktion von Apoptose nicht-infizierter T-Zellen als Ursache für die CD4+-Zelldepletion verantwortlich gemacht (FINKEL et al., 1995). Obwohl sich zumindest die Population der Gedächtnis-T-Zellen im Blut nach der akuten Phase annähernd erholt, können die Verluste nur teilweise durch Proliferation und Differenzierung naiver T-Zellen ausgeglichen werden (SOPPER et al., 2003; GROSSMAN et al., 2006). Da auch CD4+-T-Zellen mit Spezifität gegen andere Antigene verloren gehen, wie z.B. gegen das CMV oder Tuberkulose (KOMANDURI et al., 2001; GELDMACHER et al., 2010), wirkt sich dieses Defizit der TZell-Hilfe für die Bekämpfung diverser Infektionskrankheiten als Immundefizienz aus. 2.3.3.2 Auswirkung auf das humorale Immunsystem Die abnorme B-Zellfunktion ist ein weiteres wichtiges Kennzeichen der HIV-/SIV-Infektion. Hierbei ist zu unterscheiden zwischen direkten Schäden, ausgelöst durch das Virus, und indirekten Schäden durch die T-Helferzellendysfunktion und –depletion. Obwohl es kaum Hinweise dafür gibt, dass die Immundefizienzviren in der Lage sind, effektiv in B-Zellen zu replizieren, konnte gezeigt werden, dass Komplement-markierte HIVVirionen an CD21 des B-Zell-Korezeptors binden. Auf diese Weise tragen B-Zellen zur Verbreitung des Virus bei, während der stimulatorische Effekt als eher gering einzuschätzen ist (MOIR et al., 2000; MALASPINA et al., 2002). Früh wurde ersichtlich, dass HIV in vitro eine direkte polyklonale Stimulation von B-Zellen verursacht (SCHNITTMAN et al., 1986). Entscheidend für die Hyperaktivierung der B-Zellen sind aber vor allem Zytokine wie INF-α, TNF, CD40-L und IL-6 und -10 (RIECKMANN et al., 1991; MULLER et al., 1998). Dies hat eine ausgeprägte Hypergammaglobulinämie, sowohl HIV-spezifischer als auch polyklonaler Antikörper zur Folge (SHIRAI et al., 1992). Literaturübersicht S e i t e | 43 Auch die Bildung von Autoantikörpern z.B. gegen CD8+-Zellen tritt in diesem Zusammenhang auf und könnte einen weiteren pathogenen Faktor darstellen (Z. WANG et al., 1999). Im Widerspruch zur Hyperaktivierung steht die verminderte Stimulierbarkeit der B-Zellen HIV-infizierter Patienten z.B. mit monoklonalen anti-CD40-Antikörpern, Staphylococcus aureus Cowan 1 Partikeln und Pokeweed Mitogen (PWM, Mitogene der Kermesbeere) (MIEDEMA et al., 1988; CONGE et al., 1998), die die erhöhte Suszeptibilität gegenüber bakteriellen Infektionen bedingen könnte. Folge der T-Zelldepletion ist eine Dysfunktion in der B-Zelldifferenzierung. So nimmt der Anteil an CD27+-Gedächtniszellen ab, während vermehrt naive B-Zellen auftreten (DE MILITO et al., 2001; CHONG et al., 2004b). Zudem kann ein gesteigertes Vorkommen von unreifen, CD10+B-Zellen im Blut festgestellt werden (MARTINEZ-MAZA et al., 1987). MOIR et al. (2008a) beschrieben bei HIV-Infizierten eine atypische Population von CD21CD27-B-Zellen, die zwar in der Lage sind, in gewissem Maße HIV-spezifische Antikörper zu sezernieren, aber aufgrund ihres verminderten Proliferationsvermögens und der erhöhten Expression inhibitorischer Marker als exhausted (dt.: erschöpft) memory B-Zellen definiert wurden. Die erhöhte Prävalenz von B-Zell-Lymphomen im Zusammenhang mit einer HIV-Infektion wird von einigen Autoren auf die pathologische Aktivierung der B-Zellen zurückgeführt (GRULICH et al., 2000). Andere Gruppen konnten aber sowohl bei der HIV- als auch der SIV-Infektion einen eindeutigen Zusammenhang zwischen dem Auftreten dieser Neoplasien und der Infektion mit Eppstein-Barr-Virus (EBV) herstellen (VAN BAARLE et al., 2001; KAHNT et al., 2002). 44 | S e i t e Material und Methoden 3. MATERIAL UND METHODEN 3.1 Tiere Sämtliche für diese Arbeit verwendeten Proben stammten von Rhesusaffen (Macaca mulatta) aus dem Bestand der Abteilung Infektionsmodelle. Sie wurden für Versuchszwecke in England, China oder dem Deutschen Primatenzentrum gezüchtet und waren zum Zeitpunkt ihrer Ankunft in der Abteilung Infektionsmodelle klinisch gesund und frei von SIV, Affen-TZell-Leukämievirus (engl. Simian T-cell Leukemia Virus, STLV) und Tuberkulose. Die Tiere wurden für verschiedene Vakzinestudien verwendet oder standen als nicht immunisierte und nicht infizierte Spendertiere für Kontroll- bzw. Vergleichszwecke zur Verfügung. Eine detaillierte Auflistung der Tierdaten hinsichtlich Alter, Geschlecht, Herkunft, stattgefundener Immunisierung und Infektionsstatus sind im Anhang 11.1 tabellarisch aufgeführt. Die Tiere wurden am DPZ gemäß den Richtlinien des Deutschen Tierschutzgesetzes gehalten und entsprechend den jeweiligen Versuchsanforderungen in Einzel- oder Gruppenkäfighaltung in vier voneinander unabhängigen Tierhaltungseinheiten untergebracht, die den biologischen und gentechnischen Sicherheitsstufen S-1 bis S-3 genügen. 3.1.1 Narkotisierung der Versuchstiere Übereinstimmend mit den Vorschriften zum Arbeits- und Tierschutz wurden die Rhesusaffen für alle Eingriffe sediert oder narkotisiert. Um die Kreislaufbelastung zu verringern, wurden die Tiere die vorhergehende Nacht nüchtern gehalten. Mit Hilfe der vorziehbaren Käfigrückwand erfolgte eine Fixierung und anschließend die intramuskuläre Applikation von 0,1 ml/kg KGW Ketavet® (Fa. Pfizer) für weitgehend schmerzfreie Eingriffe wie die Blutentnahme. Für schmerzhaftere Eingriffe oder solche, die eine stärkere Relaxation der Tiere erfordern, wie die bronchioalveoläre Lavage, wurden 0,2 ml/kg KGW GM II ( Göttinger Mischung II) appliziert. Ein Milliliter GM II enthält 5% Ketamin, 1% Xylazin (Fa. Bayer) und 0,1% Atropin sulfaricum solutum 1%ig (Fa. Eifelfango), Grundlage ist sterile isotonische Kochsalzlösung (Fa. Braun). Material und Methoden S e i t e | 45 3.1.2 Belastungsinfektion der Tiere mit SIV Die in dieser Arbeit untersuchten SIV-infizierten Rhesusaffen stammten aus verschiedenen Vakzinestudien und wurden zum Teil vor der Belastungsinfektion immunisiert. Für die Belastungsinfektion wurden entweder SIVmac239 (KESTLER et al., 1990; REGIER u. DESROSIERS, 1990), hergestellt im Labor der Abt. Infektionsmodelle, oder SIVmac251 (LE GRAND et al., 1992; NEILDEZ et al., 1998), zur Verfügung gestellt von Dr. A. Aubertin, Institut Pasteur, Straßburg, Frankreich, verwendet. Die Applikationsroute wurde entsprechend den Studienzielen gewählt. Entweder wurde eine einzelne hohe Dosis intravenös oder oral über die Tonsillen verabreicht oder, um die natürliche Übertragung von HIV im Menschen zu imitieren, eine steigende Dosis wiederholt rektal appliziert (engl.: repeated-low-dosechallenge). Letztere Methode hat zur Folge, dass die Tiere unter Umständen verschieden hohen Virusdosen ausgesetzt waren und die produktive Infektion mit SIV zu unterschiedlichen Zeitpunkten erfolgte. 3.1.3 Blutentnahme bei den Versuchstieren Blutproben wurden am sedierten Tier durch Punktion der rechten oder linken Vena femoralis mit einer 21 G Kanüle in der Leistengegend gewonnen. Für möglichst keimarmes Arbeiten wurde die Punktionsstelle vorher mit Sterillium Virugard® desinfiziert und das BD Vacutainer®-System eingesetzt. Je nach späterer Verwendung waren die Röhrchen mit Ethylendiamintetraacetat (EDTA) oder Zitrat zur Verhinderung der Koagulation des Blutes beschickt, für die Gewinnung von Serum wurden Röhrchen mit speziellem Trenngel verwendet, das sich während der Zentrifugation zwischen Überstand und Blutkuchen schiebt. 3.1.4 Bronchioalveoläre Lavage (BAL) Da für die eigentlich schmerzfreie Prozedur der bronchioalveoläre Lavage eine gute Relaxation der Tiere sowie eine Ausschaltung des Hustenreflexes nötig ist, wurden sie mit 0,2 ml GM II pro kg/ KGW narkotisiert. Die Lagerung der Tiere erfolgte in leicht sitzender Position. Eine abgeschnittene 1,0 mlSpritze wurde als Maulspreizer zwischen die oberen und unteren Canini geklemmt. Durch Herunterdrücken des Zungengrundes mit einem Laryngoskop wurde der Kehlkopf dargestellt und das Bronchoskop möglichst ohne Berührung des Kehldeckels während einer 46 | S e i t e Material und Methoden atmungsbedingten Öffnung der Stimmritze in die Trachea eingeführt. Eine lokale Anästhesie sowie die Verwendung von Gleitmittel war nicht erforderlich. Unter Sichtkontrolle wurde das Bronchoskop (R. Wolf, Flexible Fiberskope 7325.07) über die Bifurcatio trachea in einen der beiden Hauptbronchen bis in die wedge-Position (dt.: Verkeilungsposition) geschoben. Für die Isolierung von mononukleären Zellen wurden anschließend 55 ml körperwarme physiologische NaCl-Lösung in zwei Portionen langsam appliziert und mit einer 5 ml-Spritze sukzessive abgesaugt. Dabei wurde darauf geachtet, bei starkem Widerstand die Position des Bronchoskops minimal zu verändern, um eine Verletzung der Schleimhaut und die Kontamination der Probe mit Blut zu vermeiden. Die erlangte Probe wurde in ein 50 ml-Zentrifugationsröhrchen überführt und auf Eis gelagert. Die Rückgewinnungsrate lag bei 65 – 90% der applizierten Spülflüssigkeit. Wurden viele Zellen für die weitere Aufarbeitung benötigt, wurde das Bronchoskop unter Sichtkontrolle bis zu Bifurcatio trachea zurückgezogen, in den anderen Hauptbronchus bis zur wedge-Position vorgeschoben und auch dieser Lungenflügel wie oben beschrieben mit 55 ml NaCl-Lösung gespült. Diese Vorgehensweise war erforderlich, um die Zellausbeute zu erhöhen, das Waschen desselben Lungenflügels mit einer größeren Menge NaCl ergab keine Steigerung der Zellzahl. 3.1.5 Euthanasie Nach tiefer Narkose mit 0,2 – 0,3 ml/kg KGW GMII wurde die Bauchhöhle eröffnet und 100 – 150 ml Blut durch Punktion der Aorta abdominalis entnommen. Anschließend erfolgte die Euthanasie mit einer intravenösen Applikation von 1 -1,5 ml/kg KGW Narcoren® (Fa. Merial) in die Aorta abdominals. Die Euthanasie und Sektion wurde von den Pathologen des Deutschen Primatenzentrums durchgeführt. Material und Methoden S e i t e | 47 3.2 Isolierung von mononukleären Zellen aus peripherem Blut und bronchoalveolärer Lavage 3.2.1 Isolierung von mononukleären Zellen aus peripherem Blut (peripheral blood mononuclear cells, PBMC) Langjährige Erfahrungen in der Abteilung Infektionsmodelle belegten, dass sich Zitrat als Antikoagulanz für die Isolierung und anschließende Kultivierung von PBMC am besten eignet. Das gewonnene Blut wurde aus den Zitrat-Vacutainerröhrchen direkt auf 15ml-Leucosep®Röhrchen (Fa. Greiner) mit Trennscheibe gegeben, in dessen untere Kammer 3 ml Ficoll (Fa. PAA, Dichte 1,077 g/ml) vorgelegt wurden. Diese Art Röhrchen eignet sich für die Aufarbeitung von maximal 6 ml Blut, für größere Mengen wurden entsprechend mehr Röhrchen verwendet. Nach 25 min Zentrifugation bei 800 x g lag der Lymphozytensaum in der Interphase zwischen Ficoll und zellfreiem Plasma. Letzteres wurde vorsichtig abgenommen und verworfen oder portioniert und bei -20°C eingefroren. Anschließend wurde die Lymphozytenphase in ein 15 ml-Zentrifugationsröhrchen mit 9 ml Phosphat-gepufferter isotonischer Kochsalzlösung (PBS) überführt, hier konnten zwei Zellportionen desselben Tieres zusammengefasst werden. Nach 10 min Zentrifugation bei 1200 rpm und Dekantieren des Überstandes wurde das Pellet in 10 ml PBS resuspendiert. Nach Entnahme einer kleinen Menge zur Bestimmung der Zellzahl (s. Kapitel 3.3) wurden die PBMC erneut 10 min bei 1200 rpm zentrifugiert und in der für die folgenden Tests nötigen Konzentration in RPMI kompl. (Zellkulturmedium mit 10% Fetalem Kälberserum [FCS] und 1% Penicillin [10000 U/ml]/Streptomycin [10mg/ml] substituiert) resuspendiert. 3.2.2 Isolierung von mononukleären Zellen aus BAL Das 50 ml-Zentrifugationsröhrchen mit der bronchioalveolären Lavage-Flüssigkeit wurde mit PBS/5% FCS auf 50 ml aufgefüllt und 6 min bei 450 x g zentrifugiert. Nach vorsichtigem Abgießen des Überstandes wurde das Pellet in 10 ml PBS/5% FCS aufgenommen und über ein 70 µm Zellsieb gegeben, um gröbere Partikel zu entfernen. Die filtrierte Probe wurde erneut auf 50 ml aufgefüllt und wie beschrieben zentrifugiert und das Zellpellet in 5 ml PBS/5% FCS wieder aufgenommen. Bevor die Probe weiterverarbeitet wurde, erfolgte die Berechnung der Anzahl lebender Zellen wie nachfolgend erläutert (s. Kapitel 3.3). 48 | S e i t e Material und Methoden 3.3 Vitalitäts- und Zellzahlbestimmung Zur Kalkulation der in der Probe enthaltenen lebenden, mononukleären Zellen wurde eine Zählprobe von 20µl Zellsuspension im Verhältnis 1:2 mit Trypanblaulösung verdünnt. Auf Grund veränderter Membrandurchlässigkeit abgestorbener Zellen kann der Farbstoff in die Zelle diffundieren, so dass sie bei mikroskopischer Betrachtung blau erscheinen, während unbeschädigte, lebende Zellen hell bleiben. Die anschließende Zählung der Zellen erfolgte mit Hilfe der Neubauer-Zählkammer 3.4 Kryokonservierung von Probenmaterial 3.4.1 Einfrieren von PBMC Nach Bestimmung von Vitalität und Zellzahl wurde die gewünschte Menge Zellen bei 1200 rpm 10 min zentrifugiert, das Pellet in 0,9 ml kaltem RPMI kompl. aufgenommen und in ein Einfrierröhrchen überführt, in das 0,9 ml kaltes Einfriermedium (70% FCS, 20% Dimethylsulfoxid [DMSO, Fa. Merk] und 10% RPMI) vorgelegt wurden. Wegen der Zelltoxizität des enthaltenen DMSO wurden die Proben während ihres Transports zum Gefrierschrank auf Eis gelagert. Mittels einer Isopropanol enthaltenden Einfrierbox (Fa. Nalgene) fand dort eine verlangsamte Abkühlung von 1°C/ min auf -80°C statt, bis die Proben zeitnah in den Stickstofftank überführt und dort bei -196°C gelagert wurden. 3.4.2 Einfrieren von zellfreiem Material Serum oder Plasma wurden in der gewünschten Menge in 0,5- bzw. 1,5 ml-Reaktionsgefäßen bei -20°C eingefroren. 3.4.3 Auftauen von PBMC Die Probe im Einfrierröhrchen wurde zügig im Wasserbad bei 37°C soweit aufgetaut, bis noch eine kleine Eisspindel zu erkennen war. Nach Überführung in ein 15 mlZentrifugationsröhrchen, in das 9 ml kaltes RPMI kompl. vorgelegt worden waren, wurde die Zellsuspension 10 min bei 1200 rpm zentrifugiert, der Überstand abgegossen und das Zellpellet erneut in 10ml RPMI kompl. resuspendiert und zentrifugiert. Nach diesem zweiten Waschschritt wurde das Zellpellet entsprechend seiner weiteren Verwendung zur Bestimmung der Zellzahl (s. Kapitel 3.3) in 5 ml PBS resuspendiert oder für die Durchflusszytometrie ohne vorherige Zellzahlbestimmung direkt in 50µl PBS aufgenommen. Material und Methoden S e i t e | 49 3.4.4 Auftauen von zellfreiem Material Plasma und Serum wurden bei Raumtemperatur aufgetaut. 3.5 Immunologische Methoden zum Nachweis zellulärer und humoraler Immunantworten 3.5.1 Nachweis bindender Antikörper im ELISA Für den Nachweis von Antikörpern mittels Enzyme Linked Immunoabsorbent Assay (ELISA), einem Test, bei dem der Nachweis einer stattgefundenen Antigen- Antikörperbindung durch Farbumschlag bzw. Fluoreszenzaktivierung erfolgt, wird ein lösliches Antigen auf einen festen Träger, zum Beispiel eine Mikrotiterplatte, gebunden. Die in der Probe vorhandenen spezifischen Antikörper binden an das Antigen, während unspezifische Antikörper im nachfolgenden Waschschritt entfernt werden. Anschließend werden Enzym-markierte Antikörper hinzugefügt, die an die noch vorhandenen AntigenAntikörperkomplexe binden. Nach erneutem Waschen wird im letzten Schritt ein Substrat beigegeben, welches von dem an die Sekundärantikörper gebundenem Enzym umgesetzt wird. Die Menge des entstehenden Produktes ist proportional zu der Anzahl von AntigenAntikörperkomplexen und somit zur Antikörperkonzentration in der Probe. Mittels photometrischer Messung erfolgt eine quantitative Bestimmung. Um einen Überblick über die Konzentration von gegen p27 und gp130 gerichtete bindende Antikörper der einzelnen Tiere im Infektionsverlauf zu erhalten, wurde Plasma in einer Verdünnung von 1: 200 eingesetzt und die optische Dichte (OD) bei 450nm (630nm Referenzwellenlänge) gemessen, wobei der Mittelwert aus einer Zweifachbestimmung als Ergebnis definiert wurde. Dieses Vorgehen ermöglicht ein relativ einfaches Screening einer größeren Menge von Proben, hat aber den Nachteil, dass, sobald der Sättigungsbereich erreicht wurde, die Verlaufskurve der ermittelten Werte ein Plateau bildete. Mittels einer ebenfalls als Zweifachbestimmung angesetzter Endpunkt-Titration wurde deshalb das Plasma von für die Fragestellung der Doktorarbeit interessanten Tiere zu ausgewählten Zeitpunkten im Infektionsverlauf soweit verdünnt, bis die gemessene OD knapp über dem Cut-off-Wert lag. Dieser wurde festgelegt, indem die durchschnittliche OD zweier autologer Negativplasmen verdoppelt wurde. Zur Sicherstellung der Funktionstüchtigkeit aller 50 | S e i t e Material und Methoden eingesetzten Reagenzien wurden auf jeder Mikrotiterplatte stark positiv reagierende, archivierte Plasmaproben als Positivkontrolle eingesetzt. Das seit langer Zeit im Labor der Abteilung Infektionsmodelle etablierte Protokoll sowie die verwendeten Antigene, Antikörper und Substrate finden sich in Anhang 11.2. 3.5.2 Nachweis neutralisierender Antikörper gegen SIV mittels eines Luziferase Reportergensystems auf TZM bl-Zellen Angelehnt an ein Protokoll der Duke Universität, USA, (LI et al., 2005; MONTEFIORI, 2009), wurde ein einfacher und doch sensitiver Neutralisationstest etabliert. Der Vorteil gegenüber dem bisher im Labor der Abteilung Infektionsmodelle verwendeten Neutralisationstest auf C8166-Zellen mit anschließendem p27-capture ELISA liegt in einer Dauer von nur zwei gegenüber sieben Tagen. Grundlage dieses Tests sind die TZM bl-Zellen. Diese adhärente Zellline wurde gentechnisch konstruiert, um mittels eines sensitiven und vielseitig einsetzbaren Reportergensystems eine gut reproduzierbare Alternative zur Virusquantifizierung mittels primärer Zellen zu schaffen. Ausgangspunkt war JC.53, ein Klon der HeLa-Zellinie, der stabil CXCR4, CD4 und CCR5 exprimiert und in dessen Genom Kopien der Photinus pyralis Luziferase- und E. coli βGalactosidasegene integriert wurden, die unter der Kontrolle des HIV-/SIV-Promoters stehen. Demzufolge reagieren TZM bl-Zellen bei Tat-Expression während einer HIV-/SIV-Infektion mit Luziferaseproduktion, deren Aktivität nach Zugabe entsprechender Substrate mit Hilfe eines Luminometers nachgewiesen werden kann. Die Luziferaseaktivität wird als relative Lichteinheiten (engl. relative luminescence units, RLU) quantifiziert und ist direkt proportional zur Anzahl infektiöser Viruspartikel in der Zellkultur. Das Vorhandensein von nAk kann durch eine Reduktion der RLU im Vergleich zu einer Antikörper-freien Kontrolle gezeigt werden. 3.5.2.1 Titration des Virusstocks SIVmac32H Für die Untersuchung im Rahmen der vorliegenden Dissertation wurde der Virusstock SIVmac32H verwendet. Hierbei handelt es sich um ein ex vivo-Isolat aus einem Tier, welches ursprünglich mit SIVmac251 infiziert worden war (CRANAGE et al., 1990). Material und Methoden S e i t e | 51 Da eine zu hohe TCID50 (tissue culture infectious dose 50, dt.: zellkultur-infektiöse Dosis 50) starke zytopathogene Effekte verursacht, die durch Zelltod ebenfalls eine Verringerung der RLU verursachen und somit zu einem falsch-positives Ergebnis führen, bei einer zu niedrigen TCID50 aber das Neutralisationsvermögen der Probe auf Grund zu geringer RLU nicht ausreichend genau bestimmt werden kann, war es vor Beginn der Versuchsreihe notwendig, die passende Virusverdünnung herauszufinden. Hierfür wurden die Zellen entsprechend den Vorgaben des eigentlichen Versuchsprotokolls (s. Anhang 11.3) zusammen mit Verdünnungen des Virusstocks inkubiert, anstelle von Serum wurde aber nur Medium hinzugefügt, um die gleichen Volumenverhältnisse wie im späteren Neutralisationstest zu schaffen. Jede Verdünnung wurde als Vierfachbestimmung angesetzt und der Mittelwert der gemessenen RLU gebildet. Dabei zeigte sich, dass die Luziferaseaktivität bis zu einer Virusverdünnung von 1:8 stetig ansteigt, danach aber wieder abnimmt. Anhand der Titrationskurve wurde für die Durchführung des Neutralisationstest eine Virusverdünnung von 1:25 gewählt, da hier der RLU-Wert noch mehr als 2 log-Stufen über dem der Negativkontrolle (uninfizierte Zellen) lag. Abbildung 6: Titration des Virusstocks SIVmac32H für den Nachweis neutralisierender Antikörper. Die y-Achse gibt die RLU wieder, auf der x-Achse sind die Verdünnungsstufen dargestellt. Schwarze Vertikalbalken verdeutlichen die Standardabweichung innerhalb dreier Wiederholungen des Vorversuches. 52 | S e i t e Material und Methoden 3.5.2.2 Nachweis von neutralisierenden Antikörpern im Serum Um das Vorhandensein von nAk gegen SIV im Blut zu untersuchen, ist Serum gegenüber Plasma vorzuziehen. Letzteres enthält Antikoagulatien, die bei niedrigen Plasmaverdünnungen zelltoxisch wirken und somit eine falsch-positives Ergebnis verursachen können. Sowohl für Plasma- als auch Serumproben ist eine Hitzeinaktivierung notwendig, um Komplementfaktoren zu inaktivieren. Dazu wurden die Serumproben bei Raumtemperatur aufgetaut, mittels eines Vortex gut durchmischt und anschließend wenige Sekunden zentrifugiert, um eventuelle Tropfen aus der Kappe des Probengefäßes zurückzuführen. Nach einer Inkubation von einer Stunde in einem auf 56°C erhitzen Wasserbades wurden die Aliquots erneut gemischt, zentrifugiert und anschließend direkt verwendet. Die nach SIV-Infektion gewonnenen Seren wurden in einer Endverdünnung von 1:20 bis 1:524.880 in Duplikaten aufgetragen. Der Mittelwert der gemessenen RLU wurde in folgender Formel für die Berechnung der Neutralisationskapazität der Probe eingesetzt: 1−� (RLU Probe – RLU Negativkontrolle) � = 𝑁𝑒𝑢𝑡𝑟𝑎𝑙𝑖𝑠𝑎𝑡𝑖𝑜𝑛 𝑖𝑛 % (RLU Positivkontrolle – RLU Negativkontrolle) Die in der vorangegangenen Formel genannte Negativkontrolle bestand aus uninfizierten TZM-bl-Zellen ohne Zugabe von Serum, die Positivkontrolle aus infizierten Zellen ohne Zugabe von Serum. Beide Kontrollen wurden auf jeder Mikrotiterplatte neu bestimmt. Des Weiteren wurde bei jedem Versuchsansatz eine Verdünnungsreihe des monoklonalen Antikörpers KK9 (bezogen vom National Institute for Biological Standards and Control [NIBSC], Großbritannien) durchgeführt, der ein hohes Neutralisationsvermögen besitzt. Die genaue Durchführung des Neutralisationstests ist in einem Protokoll in Anhang 11.3 dargestellt. Material und Methoden S e i t e | 53 3.5.3 Quantifizierung Antikörper-produzierender Zellen mit Hilfe der ELISpotTechnik Der Enzym-Linked-ImmunoSpot (ELISpot)-Test zum Nachweis von ASC wurde erstmals 1983 nahezu zeitgleich von zwei Arbeitsgruppen (CZERKINSKY et al., 1983; SEDGWICK u. HOLT, 1983) vorgestellt und basiert auf einem ähnlichen Verfahren wie der ELISA. Im Gegensatz zu letzterer Methode, die benutzt werden kann, um die Menge in einer Probe vorhandener Antikörper zu ermitteln, werden hier die Zellen quantifiziert, die diese produzieren. Je nach Fragestellung können dabei alle Zellen erfasst werden, die eine bestimmte Immunglobulinklasse abgeben, oder nur solche, die spezifische Antikörper gegen ein Antigen von Interesse bilden. Wie auch beim ELISA-Verfahren wird ein Antigen oder ein gegen die gesuchte Immunglobulinklasse gerichteter monoklonaler Antikörper an die Festphase einer Mikrotiterplatte gebunden (s. Abb. 7, Schritt 1). Nach Auswaschen der überschüssigen Antigene/ Antikörper und Blockierung der unspezifischen Bindungen werden PBMC oder auch aufgereinigte B- bzw. Plasmazellen für einen festgelegten Zeitraum in der Mikrotiterplatte inkubiert (Abb. 7, Schritt 2 und 3). Die von den Zellen sezernierten Antikörper bilden, Spezifität vorausgesetzt, einen Komplex mit den Festphaseassoziierten Antigenen bzw. –körpern (Abb. 7, Schritt 4) und können, nachdem die Zellen entfernt wurden, mit einem substratgekoppelten Antikörper und dem entsprechenden Enzym markiert werden (Abb.7, Schritt 5 – 6). Im Unterschied zum ELISAVerfahren entsteht aber kein lösliches Produkt, stattdessen bleibt es an die Immunkomplexe auf der Platte gebunden und wird in Form von dunklen Punkten sichtbar. Jeder Punkt repräsentiert eine Antikörper-sezernierende Zelle (Abb. 7, Schritt 8). Abbildung 7: Schematische Darstellung der ELISpot-Technik zum Nachweis Antikörper-sezernierender Zellen (von Fa. Mabtech) 54 | S e i t e Material und Methoden 3.5.3.1 Antikörper-produzierende Zellen im peripheren Blut Eine Anwendungsmöglichkeit des ELISpot-Verfahrens ist der Nachweis von vorhandenen ASC ohne vorherige in vitro-Stimulation. Vor allem nach einer in vivo-Aktivierung durch Vakzinierung oder der akuten Infektion mit einem Pathogen ist diese Herangehensweise sinnvoll, um den Impferfolg bzw. das Einsetzen der humoralen Immunantwort zu veranschaulichen und die Antikörper-sezernierenden Plasmazellen zu quantifizieren. Für die vorliegende Doktorarbeit wurde ein kommerziell erhältliches Testsystem der Fa. Mabtech zum Nachweis von IgG- und IgA-sezernierenden Zellen genutzt und dessen Anwendung auf die SIV-Antigene p27 und gp130 etabliert. Polyvinylidenfluorid-Mikrotiterplatten wurden eine Minute mit 35%igem Ethanol vorbehandelt, um die Bindungskapazität der Membran zu steigern. Vorversuche ergaben, dass diese Behandlung die Qualität der Punkte im Hinblick auf Färbeintensität und Abgegrenztheit deutlich steigerte. Anschließend wurden in die Kavitäten der Mikrotiterplatte Antikörper gegen humanes IgG oder IgA pipettiert, die alle sezernierten Immunglobuline der jeweiligen Klasse binden und benötigt wurden, um das Verhältnis der Antigen-spezifischen ASC zu berechnen. Gleichzeitig dienten sie als Positivkontrolle. Als Negativkontrolle wurde bovines Serumalbumin (BSA) benutzt. Um SIV-spezifische Antikörper zu binden, wurden weitere Vertiefungen mit p27 und gp130 beschickt. Bei der Etablierung der Testmethode wurde eine Titrationsreihe durchgeführt, um die notwendige Antigenkonzentration zu ermitteln. Zwar stieg die Qualität der Punkte mit der Menge des eingesetzten Antigens, da diese aber nur in begrenztem Rahmen zur Verfügung standen, wurde schließlich ein Konzentration von 0,2µg/Kavität als zufriedenstellend befunden. Nachdem die Antigene über Nacht an die Membran der Mikrotiterplatte binden konnten, wurden am Folgetag die frisch isolierten PBMC eingesetzt. Vorher stellte intensives Waschen und eine Behandlung der Membran mit RPMI kompl. sicher, dass alle überschüssigen Antigene entfernt und unspezifische Bindungen blockiert wurden. Für die Bestimmung von Gesamt-IgG-sezernierenden Zellen erbrachte der Einsatz von 50.000 - 100.000 Zellen pro Vertiefung die besten Ergebnisse, wurden mehr Zellen verwendet, bestand die Gefahr, dass die entstehenden Punkte konfluierten und somit nicht mehr akkurat erfasst werden konnten. Zur Quantifizierung von gegen SIV-Antigene gerichtetem IgG wurden, genauso wie in der Material und Methoden S e i t e | 55 Negativkontrolle, 200.000 Zellen pro Kavität eingesetzt, da der Anteil spezifischer ASC recht niedrig war. Für den Nachweis IgA-sezernierender Zellen musste die eingesetzte Zellzahl für eine valide Auswertung verdoppelt werden. Mit 200.000 PBMC pro Vertiefung für die Quantifizierung Gesamt-IgA-sezernierender Zellen bzw. 400.000 PBMC für SIV-spezifische ASC waren die Grenzen der Testmethode erreicht. Bei einer weitere Erhöhung der Zellzahl konnte nicht mehr davon ausgegangen werden, dass die Zellen in nur einer Schicht auf der Membran lagen. Nach einer Inkubation von 20 Stunden bei 37°C, 5% CO2, wurden die Platten gewaschen und in zwei Schritten mit Konjugat und Substrat gefärbt. Die entstandenen Punkte auf der Membran konnten mit Hilfe des ELISpot-Readers gezählt werden. Alle Bestimmungen wurden vorzugweise in Triplikaten, mindestens aber in Duplikaten ausgeführt. Nach der Berechnung des Mittelwertes der gezählten Punkte wurde dieser für die Bestimmung der Anzahl Gesamt-IgG/-IgA-sezernierender Zellen auf 1x106 PBMC/ml normiert, während der Anteil der SIV-spezifischen ASC mit folgender Formel berechnet wurde: � ( 𝐴𝑔 − 𝑠𝑝𝑒𝑧. 𝑃𝑢𝑛𝑘𝑡𝑒 − 𝑁𝑒𝑔𝑎𝑡𝑖𝑣𝑘𝑜𝑛𝑡𝑟𝑜𝑙𝑙𝑒) � 𝑥 100 𝐺𝑒𝑠𝑎𝑚𝑡 𝐼𝑔𝐺/𝐼𝑔𝐴 𝑃𝑢𝑛𝑘𝑡𝑒 𝑥2 = 𝐴𝑛𝑡𝑒𝑖𝑙 𝑑𝑒𝑟 𝐴𝑔 − 𝑠𝑝𝑒𝑧𝑖𝑓𝑖𝑠𝑐ℎ𝑒𝑛 𝐴𝑆𝐶 𝑖𝑛 % 𝑑𝑒𝑟 𝑔𝑒𝑠𝑎𝑚𝑡𝑒𝑛 𝐴𝑆𝐶 Das ausführliche Protokoll zur Durchführung des Immunglobulin-ELISpots ist in Anhang 11.4 beschrieben. 3.5.3.2 Antikörper-produzierende Zellen in BAL Der ELISpot kann darüber hinaus mit isolierten mononukleären Zellen aus anderen Kompartimenten des Körpers durchgeführt werden. Wegen der minimal-invasiven Gewinnungsart und der recht unkomplizierten Aufarbeitung wurden in der vorliegenden Arbeit mononukleäre Zellen aus der bronchioalveolären Lavage auf ihren Anteil an Immunglobulin-sezernierenden Zellen untersucht. Die Plattenvorbereitung, Durchführung und Auswertung geschah analog zum Protokoll für PBMC, allerdings erwies sich hier eine Verdoppelung der eingesetzten Zellzahl sowohl für die Bestimmung von IgA als auch IgG notwendig. 56 | S e i t e Material und Methoden 3.5.3.3 Polyklonale Stimulation von PBMC und mononuklären Zellen aus BAL Eine weitere Einsatzmöglichkeit des Immunglobulin-ELISpot besteht im Nachweis von ASC nach polyklonaler Stimulation in vitro. Mittels verschiedener Stimulationsprotokolle kann eine Differenzierung von vorhandenen Gedächtnis-B-Zellen zu Antikörper-sezernierenden Zellen erreicht und so das Vorhandensein eines immunologischen Gedächtnisses gezeigt werden. Die Quantifizierung der Antigen-spezifischen ASC lässt weiterhin eine gewisse Schlussfolgerung über dessen Ausmaß zu. In dieser Arbeit wurde das für den B-Zell-ELISpot empfohlene Stimulationsprotokoll der Firma Mabtech verwendet. Resiquimod (R848) ist ein Pharmakon mit antiviraler Wirkung und wird z.B. zur Behandlung bei lokaler Herpes simplex-Infektionen oder als Adjuvans in verschiedenen Vakzinen eingesetzt. Als potenter Toll-like-Rezeptor (TLR) 7/8-Agonist regt es, genau wie die einzelsträngige HIV-/SIV-RNA, die Aktivierung, Proliferation und Differenzierung von B-Zellen hin zu ACS an (TOMAI et al., 2000). IL-2 ist nicht nur verantwortlich für die Bildung von Gedächtnis-T-Zellen, sondern fungiert auch als kostimulatorischer Faktor bei der Bildung von Immunglobulin-sezernierenden Zellen (BANCHEREAU et al., 1994). Im Anschluss an die in Kapitel 3.2 und 3.3 beschriebenen Aufarbeitungsschritte wurden die frisch aus Blut oder BAL isolierten Zellen in einer Konzentration von 1x106/ml in RRMI kompl. mit einem Zusatz von 1µg R848 und 10ng rhIL-2 pro Milliliter aufgenommen und bei 37°C, 5% CO2, für 72 (PBMC) respektive 48 Stunden (Zellen aus BAL) kultiviert. Diese Stimulationszeiträume wurden nach einigen Vorversuchen als optimal erachtet, bei längerer Inkubation ließ die Vitalität der Zellen stark nach. Im weiteren Verlauf wurde mit den stimulierten Zellen wie in Anhang 11.4 erläutert verfahren. 3.5.4 Quantifizierung von IFNγ-sezernierenden Zellen mit Hilfe des ELISpotVerfahrens Die Technik des ELISpot kann nicht nur zum Nachweis und zur Quantifizierung Antikörpersezernierender Zellen eingesetzt werden, sondern auch für Zytokin-sezernierende Zellen nach Antigenstimulation (KUMAR et al., 2001). In dieser Studie wurden über die Produktion des Zytokins IFNγ antigenspezifische T-Zellen im Blut berechnet (SUH et al., 2006). Zur Stimulation dienten hierbei inaktiviertes Virus oder ein SIVgag-Peptid-Pool. Material und Methoden S e i t e | 57 Die Daten in dieser Arbeit wurden routinemäßig von Mitarbeitern der Abteilung Infektionsmodelle erhoben, das ausführliche Protokoll findet sich in Anhang 11.5. 3.6 Charakterisierung von B-Zellen mittels polychromatischer Durchflusszytometrie Mittels der Durchflusszytometrie ist es möglich, korpuskuläre Anteile einer heterogenen Suspension zu definieren. Am meisten verbreitet ist ihr Einsatz in der Analyse von Zellen oder Zellpopulationen. Indem die Zellen einzeln in einem laminaren Flüssigkeitsstrom durch die Messzelle des Durchflusszytometers geleitet und mit Laserlicht einer definierten Wellenlänge bestrahlt werden, können durch die unterschiedliche Intensität des von der Zelle gestreuten Lichtes Rückschlüsse auf die Gestalt der Zellen gezogen werden. Der ForwardScatter(FSC, Vorwärtsstreulicht)-Detektor misst dabei die Größe, der Side-Scatter(SSC, Seitwärtsstreulicht)-Detektor gibt Auskunft über die Granularität der Zelle. Diese Methode nennt man „morphologisches Gating“, wobei das englische Wort gate das Auswahlfenster bezeichnet, in dem die gesuchte Zellpopulation liegt. Eine weitergehende Differenzierung der Zellpopulationen kann durch den Einsatz von spezifischen, Fluorochrom-markierten Antikörpern gegen verschiedene Proteine der Zelle erreicht werden. Die Emission der Fluorochrome, welche durch das Laserlicht angeregt werden, wird durch verschiedene Filter aufgeteilt, detektiert und in elektrische Impulse umgewandelt. Durch die Analyse des Vorhandenseins oder Fehlens zeitgleich aufgenommener Fluoreszenzsignale können verschiedene Zellpopulationen voneinander abgegrenzt werden. Dieses Vorgehen wird als „immunologisches Gating“ bezeichnet. In dieser Arbeit wurde das Durchflusszytometer LSR II der Fa. BD verwendet, welches in der vorhandenen Konfiguration die gleichzeitige Analyse von zehn Fluorochromen mittels drei Lasern ermöglicht. Die Erzeugung von monochromatischem Licht der Wellenlänge 405 nm erfolgt mittels Coherent Vioflame Diodenlaser, die der Wellenlänge 488 nm mit einem Argonionenlaser und die von 635 nm mit einem JDS Uniphase Helium-Neon-Laser. Die optimale Antikörperkonzentration zum Färben der Zellen wurde durch Verdünnungsreihen ermittelt. Die Konzentration an Antikörper, die ein deutliches Signal der 58 | S e i t e Material und Methoden Positivpopulation und eine klare Abtrennung zur Negativpopulation ergab, wurde für die weiteren Versuche verwendet. Da es trotz entsprechender optischer Filter für die jeweiligen Wellenlängenbereiche zu einer Überlappung der Emissionsspektren kommt, wurde vor der eigentlichen Probenmessung immer eine Kompensationsmessung durchgeführt. Dazu wurden Kügelchen (Spherotec beads, Fa. BD) mit jeweils einem einzelnen Fluorochrom markiert und gemessen. Mit Hilfe der Kompensationselektronik der Software FACS Diva 5.X (Fa. BD) wurde das auftretende Signal in allen Detektoren ermittelt und der Anteil der Überlappung elektronisch subtrahiert. Alle Färbungen und Messungen wurden von den technischen Mitarbeitern der Abteilung Infektionsmodelle, DPZ, durchgeführt. Die Antikörperkombination und die Definition der Lymphozytensubpopulation erfolgten mit großer Unterstützung durch Dr. rer. nat. Sieghart Sopper, ehemaliger Mitarbeiter der Abt. Virologie und Immunologie, DPZ, und Dr. med. vet. Katharina Töpfer, ehemalige wissenschaftliche Mitarbeiterin der Abt. Infektionsmodelle, DPZ. 3.6.1 Markierung von Oberflächenantigenen von isolierten mononukleären Zellen aus peripherem Blut Für die Markierung von Oberflächenantigenen auf Lymphozyten wurden entweder frisches EDTA-Blut (Daten aus Kapitel 4.2.2) oder aus Zitratblut isolierte PBMC direkt nach dem Auftauen (Daten aus Kapitel 4.3.5) verwendet. Je nach Verfügbarkeit enthielten die eingefrorenen Aliquots 0,5 – 2 x 106 Zellen. Nach zweimaligem Waschen mit PBS wurden die aufgetauten Zellen in 50 µl PBS aufgenommen und in einem Rundbodenröhrchen (Fa. BD) mit 20 µl des unter 3.6.2 angegebenen Antikörpergemisches 30 min bei 4°C im Dunkeln inkubiert, anschließend 6 min bei 450 x g zentrifugiert und das Pellet in 1 ml PBS/0,5% BSAPuffer wieder aufgenommen. Bei der Verwendung von Vollblut entfiel der Waschschritt vor der Zugabe des Antikörpergemisches, 20 µl davon wurden direkt zu 50 µl EDTA-Blut gegeben. Allerdings war in diesem Fall nach der Färbung eine Erythrozytenlyse nötig. Dazu wurde jeder Probe vor der Weiterverarbeitung 1 ml Lysepuffer (Fa. BD) zugesetzt, sie wurde weitere 10 min inkubiert und zweimal wie oben angegeben gewaschen. Abschließend wurde der Überstand in beiden Fällen dekantiert, das Pellet in 50 µl 0,35%iger Formaldehydlösung in PBS resuspendiert und im Durchflusszytometer gemessen. Material und Methoden 3.6.2 S e i t e | 59 Antikörper-Kombination zur Definition verschiedener B-Zellpopulationen Fluorochrom Antikörper Klon Hersteller FITC CD 21 B-Ly4 IQ Products PerCP CD 27 O323 BioLegend Alexa F 700 CD 3 SP34-2 BD Bioscience APC-Cy7 CD 10 Hl 10a BioLegend V500/PO IgD biot Pooled sera SouthernBiotech E625 CD 20 2H7 eBioscience V450 CD 4 L200 BD Bioscience Tab. 2: Tabellarische Zusammenstellung der für die Durchflusszytometrie verwendeten Antikörper. 3.6.3 Definition verschiedener Lymphozytenpopulationen Die Darstellung der gewünschten Zellpopulation im Durchflusszytometer lässt sich anhand diverser Kriterien vollziehen. In der vorliegenden Arbeit wurde zuerst die Lymphozytenpopulation anhand der Morphologie definiert. So können Leukozyten in Lymphozyten, Monozyten und andere Zellen myeloiden Ursprungs (Granulozyten, Monozyten u.a.) unterschieden werden, wie Abbildung 8 a deutlich macht. Die y-Achse kennzeichnet hierbei die Stärke der Granularität, die x-Achse die Teilchengröße. Jeder Punkt (engl. event) in dieser Darstellungsweise (engl.: dot-plot) entspricht einem detektiertem Teilchen, je mehr Events in einem Bereich liegen und die gleiche Merkmalsausprägung besitzen, desto stärker verschiebt sich die Farbskala von blau über gelb bis in den roten Bereich. Mittels eines Analysefensters (engl.: gate), das der Betrachter bei der Auswertung einer repräsentativen Probe setzt, kann der Anteil von Events mit den definierten Eigenschaften an der Gesamtzahl der gemessenen Events bestimmt werden. Bei nachfolgenden Messungen übernimmt die Software automatisch diese Gates und gibt den Anteil der enthaltenen Events an. Dass ein Event nicht automatisch einer Zelle entspricht, zeigt Abb. 8 a. Solche von geringer Größe und Granularität sind erfahrungsgemäß Zelltrümmer. 60 | S e i t e Material und Methoden Um weiterführende Analysen durchführen zu können, ermöglicht die Software es, alle Events innerhalb eines Gates auf weitere Merkmale zu überprüfen. Im Hinblick auf die Zielsetzung dieser Arbeit wurden ausschließlich die Zellen innerhalb des Lymphozyten-Gates genauer untersucht. Anschließend an die Bestimmung der Lymphozytenpopulation durch morphologisches Gating wurden einzelne Zellen anhand ihrer Größe von Zellkonglomeraten abgegrenzt (Abb. 8 b), bevor diese mittels Fluorochrom-markierten Antikörpern genauer unterteilt wurden. Die Events in Abbildung 9 a entsprechen somit einzelnen Lymphozyten, welche durch ihre Expression von CD3 und CD20 aufgeteilt sind. Zellen, die keins der beiden Antigene auf ihrer Oberfläche tragen, liegen im linken unteren Quadranten, solche, die doppelt positiv sind, im rechten oberen. T-Zellen, die nur CD3, nicht aber CD20 exprimieren, liegen im rechten unteren Quadranten, die CD3-CD20+B-Zellen im linken oberen. Anhand der Expression von CD10 wurde zwischen reifen und unreifen CD20+Zellen unterschieden (Abb. 9 b), die jeweils auf ihre Expression von IgD untersucht wurden. Schließlich wurden mit Hilfe der Marker CD27 und CD21 die gewünschten BZellsubpopulationen definiert (Abb. 9 d). Dies geschah jeweils für die IgD-positiven als auch für die IgD-negativen CD20+Zellen (Abb. 9 c). Ein solches Vorgehen bezeichnet man als Gating-Kaskade. Material und Methoden S e i t e | 61 Abbildung 8: Definition der Lymphozytenpopulationen eines SIV-negativen Rhesusaffen im Durchflusszytometer. Der Dot-Plot a zeigt die Aufteilung der Leukozyten anhand ihrer Größe (yAchse) und Granularität (x-Achse). Die rote Linien markieren ein Gate, die Menge der darin enthaltenden Events ist als prozentualer Anteil an allen detektierten Teilchen angegeben. Aus der Lymphozytenpopulation wurden daraufhin Zellkonglomerate ausgegrenzt (b). 62 | S e i t e Material und Methoden Abbildung 9: Definition der T- und B-Zellpopulationen eines SIV-negativen Rhesusaffen im Durchflusszytometer. Der Dot-Plot a zeigt die Aufteilung der Lymphozyten anhand der Oberflächenantigene CD20 (y-Achse) und CD3 (x-Achse). Aus der CD20+Population wurden daraufhin CD10+B-Zellen ausgegrenzt (b). Mittels der Trennung von IgD+(y-Achse)CD20+(x-Achse) wurden B-Zellen mit und ohne Klassenwechsel definiert (c). Dot-Plot d veranschaulicht die Expression der Oberflächenmarker CD27 (y-Achse) und CD21 (x-Achse). Die roten Linien markieren ein Gate, die Menge der darin enthaltenden Events ist als prozentualer Anteil an allen detektierten Teilchen angegeben. Material und Methoden S e i t e | 63 3.7 Bestimmung der SIV-Last im peripheren Blut Es gibt mehrere Methoden zur Quantifizierung von Viruslasten, welche wiederum verschiedene Schlussfolgerungen zulassen. Diese Arbeit beschränkt sich auf die Darstellung der Menge viraler RNA-Kopien im Plasma als Maßstab für den Grad der Infektion. Dabei wurden Daten ausgewertet, die routinemäßig von Frau Dr. Sauermann aus der Abt. Infektionsmodelle, DPZ, erhoben wurden. Die Viruslast im peripheren Blut wurde über eine quantitative Bestimmung der viralen RNA-Kopien im Plasma mittels Echtzeit-PolymeraseKettenreaktion (engl.: quantitative real-time polymerase chain reaction, qRT-PCR,) ermittelt (NEGRI et al., 2004). Hierzu wurde das 7500 Real-Time PCR System von Applied Biosystems mit dem in Tabelle 3 aufgeführten Programm verwendet. a) Reagenz b) Plasma-RNA Gesamt RNA Reagenz Gesamt RNA Mastermix-Qiagen 12,5 µl/Ansatz 12,5 µl/Ansatz Mastermix-Qiagen 12,5 µl/Ansatz Gag sense 2,4 µM 2,4 µM GAPDH-for 2,4 µM Gag antisense 2,4 µM 2,4 µM GAPDH-rev 2,4 µM Taqman-Sonde 1,2 µM 1,2 µM SybrGreen 1,2 µM RT-Mix 0,5 U/Ansatz 0,5 U/Ansatz RT-Mix 0,5 U/Ansatz RNA 5 μl/Ansatz 500ng RNA 500ng H2O ad 25 μl ad 25 μl H2O ad 25 μl Tab. 3: Reaktionsansätze für die qRT-PCR. Zur Quantifizierung von viralen RNA-Genomkopien (a) und zur Überprüfung der RNA-Qualität (b). Die virale RNA aus 200 µl Serum wurde mit Hilfe des MagAttract® Virus Mini M48-Kit und des BioRobot M48 der Firma Qiagen isoliert. Mit dem QuantiTect™ Probe RT-PCR-Kit der Firma Qiagen wurde mit den in Tabelle 3 angegebenen Primern und Sonden eine qRT-PCR durchgeführt. Der Reaktionsansatz ist in Tab. 3 a zu finden und das entsprechende Programm ist in Tab 4 aufgeführt. Es wurde ein RNA-Standard mit 50, 102, 103, 104 und 106 viralen Kopien verwendet. Alle Bestimmungen erfolgten in Duplikaten und die Ergebnisse der qRTPCR wurden mit der 7500 System SDS-Software von Applied Biosystems ausgewertet. 64 | S e i t e Zyklusschritt Material und Methoden Temperatur (°C) Dauer (min) Zyklenzahl 50 30 1 Initiale Aktivierung 95 10 1 Denaturierung 95 0,15 45 Annealing und Elongation 60 1 Reverse Transkription PCR Tab. 4: PCR-Bedingungen für die qRT-PCR zur Quantifizierung viraler RNA-Kopien und zur Überprüfung der RNA-Qualität 3.8 Erstellung eines Differentialblutbildes Eine Analyse des Differentialblutbildes aus EDTA-Blut erfolgte bei den Versuchstieren in regelmäßigen Abständen in der Abteilung Hämatologie des Universitätsklinikums Göttingen gemäß aktueller Standards. 3.9 Statistische Auswertung Die statistische Auswertung erfolgte mit dem Programm Graph Pad Prism. Für Korrelationsanalysen wurden die nichtparametrische Spearman-Korrelation mit einer zweiseitigen Analyse und einem Konfidenzintervall von 95% verwendet. Für die vergleichenden Auswertungen wurde der nichtparametrische Mann-Whitney-Test genutzt. Auch bei diesem Test wurde eine zweiseitige Analyse mit einem Konfidenzintervall von 95% gewählt. Als statistisch signifikant wurden in beiden Tests Ergebnisse mit einem p-Wert kleiner als 0,05 und als hochsignifikant Ergebnisse mit einem p-Wert kleiner als 0,01 definiert. 3.10 Schutzmaßnahmen Alle Arbeiten mit potentiell infektiösem Material (Blut, BAL, Serum, Gewebe) sowie solche, die ein steriles Umfeld erforderten, fanden innerhalb eines Labors der gentechnischen Sicherheitsstufe II unter einer mikrobiologischen Werkbank der Sicherheitsstufe II statt. Ergebnisse S e i t e | 65 4. ERGEBNISSE 4.1 Retrospektiver Vergleich der humoralen und zellulären Immunantwort im SIV-Makakenmodell mit Fokus auf die Langzeitüberlebenden 4.1.1 Virusbeladung Im Rahmen der am Deutschen Primatenzentrum durchgeführten Vakzineexperimente waren immer wieder SIV-infizierte Tiere zu beobachten, die, unabhängig vom Immunisierungsstaus, in der Lage waren, nach der Spitzenvirämie die systemische Viruslast für einen sehr langen Zeitraum auf einem niedrigen Spiegel zu halten. In Abb. 10 sind die Unterschiede im Infektionsverlauf zwischen Langzeitüberlebenden und Progressoren dargestellt. Die Gruppe der Langzeitüberlebenden bestand zum Beginn der Untersuchungen aus 13 Tieren, deren Viruslast über einen Zeitraum von mindestens 3 Jahren unter 1x104 virale RNAKopien pro ml Plasma lag. Die höchste Anzahl viraler Kopien konnte während der zweiten Woche nach Infektion gemessen werden. Sie lag im Durchschnitt bei 1,6x106. Zur achten Woche nach Infektion, dem Eintritt in die sogenannte akute Plateauphase, hatte schon eine Virusreduktion um zwei Log-Stufen auf 1,8x104 virale RNA-Kopien/ml Plasma stattgefunden (s. Abb. 11) Im Vergleich dazu wurden 52 Tiere, die einen typisch progredienten Krankheitsverlauf erkennen ließen und an AIDS erkrankten, zu der Gruppe der Progressoren zusammengefasst. Deren mittlere SIV-RNA-Last im Plasma lag nach der Spitzenvirämie (3,8x106 virale RNA-Kopien/ml Plasma während der zweiten Woche nach Infektion) nahezu ständig über 1x105 Kopien pro ml Plasma. Bereits beim Eintritt in die akute Plateauphase unterschied sich die Viruslast deutlich von der der Langzeitüberlebenden, in der achten Woche nach SIV-Infektion lag sie bei 8,2x105 SIV-RNA-Kopien/ml Plasma. Zu beachten ist ferner die Tatsache, dass bei den Tieren 12536, 12543 und 2219 während der Datenerhebung zur vorliegenden Arbeit die periphere Virusbeladung graduell über 1x104 RNA-Kopien/ml Plasma anstieg und das erst genannte Tier sogar auf Grund von AIDSähnlicher Symptomatik euthanasiert werden musste. Die Auswertung der Vakzinestudien, aus der die in der vorliegenden Arbeit untersuchten Tiere stammten, zeigte, dass nur in der akuten Phase Unterschiede in der Viruslast zwischen 66 | S e i t e Ergebnisse immunisierten und nicht immunisierten Tieren bestanden, dies im weiteren Infektionsverlauf aber keinen Einfluss hatte (SCHULTE et al., 2009; SCHULTHEISS et al., 2011). Somit wurde die Einteilung in die Gruppen unabhängig vom Immunisierungsstatus durchgeführt. Abbildung 10: Viruslast von Langzeitüberlebenden und Progressoren im Zeitverlauf nach Belastungsinfektion. Die x-Achse gibt die Wochen nach SIV-Infektion wieder, die y-Achse die Viruslast als virale RNA-Kopien/ml Plasma. Zur besseren Übersicht während der frühen Phase der Infektion ist die x-Achse bis zur 25. Woche gedehnt. In Graph a ist die Virusbeladung der einzelnen Langzeitüberlebenden dargestellt. Die Legende führt die Tiernummern auf. Für die Tiere 8644 und 9794 sind keine Daten aus der akuten Phase der Infektion verfügbar. Die Virusbeladung der Progressoren ist in b dargestellt, die hellblaue Linie veranschaulicht den Mittelwert. Ergebnisse S e i t e | 67 Abbildung 11: Vergleich der mittleren Viruslast von Langzeitüberlebenden und Progressoren zum Zeitpunkt der Spitzenvirämie und zu Beginn der akuten Plateauphase im Vergleich. Graph a stellt die SIV-RNA-Kopien pro ml Plasma der Langzeitüberlebenden (rot) und Progressoren (blau) während der zweiten Woche nach Infektion einander gegenüber. Die Horizontalbalken bilden die Standardabweichung innerhalb der jeweiligen Gruppe ab, mit Querbalken und Sternen sind signifikante Unterschiede nach dem Mann-Whitney-Test (*** p < 0,001) gekennzeichnet. In Graph b wurden die viralen RNA-Kopien im Plasma von beiden Gruppen während der achten Woche nach Infektion miteinander verglichen. wpi = Wochen nach SIV-Infektion, n = Anzahl der Tiere 4.1.2 Bindende Antikörper gegen SIV Um eine erste Abschätzung der humoralen Immunantwort gegen SIV für die Langzeitüberlebenden und Progressoren durchführen zu können, wurden Plasmaproben aller Tiere zunächst bei einer konstanten Verdünnung von 1:200 in einem ELISA eingesetzt, der spezifische IgG-Antikörper gegen das Gag-kodierte Kapsidprotein p27 und gegen das Envkodierte Hüllprotein gp130 nachweist (Abb. 12). Während sich die Werte in der Gruppe der Langzeitüberlebenden als relativ homogen herausstellten, gab es bei den Progressoren deutliche Unterschiede in der Höhe der Antikörper vor allem gegen p27. Diese fielen im Krankheitsverlauf ab, wohin gegen die anti-gp130-Antikörper konstant blieben. Bereits 25 Wochen nach Infektion waren die gegen SIVgag gerichteten Antikörper bei Tieren mit progredientem Krankheitsverlauf signifikant niedriger, während kein signifikanter Unterschied zwischen beiden Kohorten bezüglich der Höhe der anti-gp-130-Antikörper bestand (Abb. 13). 68 | S e i t e Ergebnisse Abbildung 12: Bindende Antikörper von Langzeitüberlebenden und Progressoren im Infektionsverlauf gegen die SIV-Antigene p27 und gp130. Die x-Achse gibt die Wochen nach SIVInfektion wieder, die y-Achse die optische Dichte (OD) bei 450 nm als Maß der Antikörperkonzentration bei einer Plasmaverdünnung von 1:200. Zur besseren Übersicht während der frühen Phase der Infektion ist die x-Achse bis zur 25. Woche gedehnt. In rot sind die Langzeitüberlebenden, in blau die Progressoren dargestellt. Die Graphen a und b zeigen die Konzentration der bindenden Ak gegen p27, die Graphen c und b gegen gp130. Von den Tieren 8644 und 9794 waren keine Plasmaproben aus der frühen Phase der Infektion verfügbar. Ergebnisse S e i t e | 69 Da bei einem Großteil der Proben von Langzeitüberlebenden, aber auch bei einigen Progressoren, die OD bei einer Verdünnung von 1:200 den Sättigungsbereich des Testes erreichte, wurden Proben aller Langzeitüberlebenden und einer repräsentativen Auswahl der Progressoren titriert. Innerhalb der Gruppen der Langzeitüberlebenden ließ sich zu keinem Zeitpunkt eine signifikante Korrelation zwischen bindende Antikörpertiter gegen p27 (Daten nicht gezeigt) oder gp130 und der Viruslast herstellen, auch wenn sich ein Trend dahingehend andeutete, dass Tiere mit höherer Viruslast höhere Titer gegen gp130 aufwiesen (Abb. 14 a). Dieser Trend konnte bei den Progressoren nicht beobachtet werden (Daten nicht gezeigt). Hier zeigte sich hingegen zu Woche 25 und 50 eine signifikante negative Korrelation zwischen Titern gegen das Kapsidprotein und Virusbeladung (Abb. 14 b). Abbildung 13: Bindende Antikörper von Langzeitüberlenden und Progressoren zu den Wochen 25, 50 und 100 nach SIV-Infektion. Graph a stellt die Antikörperkonzentration gegen p27 als OD bei einer Plasmaverdünnung von 1:200 von Langzeitüberlebenden (rot) und Progressoren (blau) einander gegenüber, Graph b die gegen gp130. Die Querstriche bilden den Mittelwert der jeweiligen Gruppe ab, mit Querbalken und Sternen sind signifikante Unterschiede nach dem Mann-Whitney-Test (* p < 0,05) gekennzeichnet. Von den Tieren 8644 und 9794 waren keine Serumproben aus der frühen Phase der Infektion verfügbar. 70 | S e i t e Ergebnisse Abbildung 14: Beziehung von Viruslast und Antikörpertiter gegen gp130 und p27. Die x-Achse gibt den reziproken ELISA-Titer wieder, die y-Achse viralen RNA-Kopien/ml Plasma. Die Graphen der linken Spalte (a) zeigen das Verhältnis von Viruslast und bindenden Ak gegen gp130 in der Gruppe der Langzeitüberlebenden, die Graphen der rechten Spalte (b) das von viralen RNA-Kopien und Ak-Titern gegen p27 in der Gruppe der Progressoren. Liegt eine signifikante Korrelation vor, sind der Korrelationskoeffizient nach Pearson (r) und das Signifikanzniveau (p) angegeben. wpi = Woche nach SIV-Infektion Ergebnisse S e i t e | 71 4.1.3 Neutralisierende Antikörper gegen SIV Mittels eines Neutralisationstests wurde das Vermögen von Serumproben, SIVmac32H zu neutralisieren, überprüft. Diese funktionale Untersuchung wurde für alle Langzeitüberlebenden und eine Auswahl von Progressoren im Infektionsverlauf durchgeführt. Die Neutralisationstiter der Langzeitüberlebenden hielten über Jahre hinweg ein konstantes Niveau (Abb. 15 b), während die Titer der Progressoren anstiegen (Abb. 15 a). Zu den Zeitpunkten Woche 50 und Woche 100 nach SIV-Infektion waren sie signifikant höher als die der Langzeitüberlebenden. Abbildung 15: Neutralisierende Antikörper von Langzeitüberlebenden und Progressoren im Infektionsverlauf. Die x-Achse zeigt die Wochen nach SIV-Infektion, die y-Achse den reziproken Neutralisationstiter, bei dem eine Reduktion der Luziferaseaktivität der Zellen um 90% erreicht wurde. Rote Symbole repräsentieren die individuellen Werte der Langzeitüberlebende im Vergleich zu den der Progressoren (blau) zu den Wochen 25, 50 und 100 nach SIV-Infektion (a) und im weiteren Infektionsverlauf bis zu 300 Wochen nach Infektion (b). Die Querstriche bilden den Mittelwert der jeweiligen Gruppe ab, mit Querbalken und Sternen sind signifikante Unterschiede nach dem MannWhitney-Test (* p < 0,05; ** p < 0,01) gekennzeichnet, ns = nicht signifikant. 72 | S e i t e Ergebnisse Wurden beide Tierkohorten zusammengefasst, offenbarte sich, wie in Abbildung 16 ersichtlich, in der 25. Woche nach Infektion eine hoch signifikante positive Korrelation zwischen Neutralisationstitern und bindenden Antikörpern sowohl gegen p27 wie auch gp130. Zu Woche 50 hingegen bestand nur noch eine leichte positive Korrelation zwischen neutralisierenden Antikörpern und bindenden Antikörpern gegen das Hüllprotein, nicht jedoch gegen das Kapsidprotein. Zu späteren Zeitpunkten wurde keine Beziehung zwischen diesen Parametern ersichtlich (Daten nicht dargestellt). Abbildung 16: Beziehung von Neutralisationstitern und bindenden Antikörpern gegen p27 und gp130. Die x-Achse gibt den reziproken ELISA-Titer wieder, die y-Achse den reziproken Neutralisationstiter, bei dem eine Reduktion der Luziferaseaktivität der Zellen um 90% erreicht wurde. Grüne Kreise stellen das Verhältnis von Neutralisationstitern und denen von bindenden Antikörpern gegen p27 dar, violette Vierecke das von Neutralisationstitern und bindenden Antikörpern gegen gp130 zu den Wochen 25 (a) und 50 (b) nach SIV-Infektion. r ist der Korrelationskoeffizient nach Spearman, p das Signifikanzniveau. wpi = Woche nach SIV-Infektion. Ergebnisse S e i t e | 73 Während innerhalb der Gruppen der Langzeitüberlebenden bzw. Progressoren keine signifikante Korrelation zwischen Neutralisationstitern und SIV-RNA-Last hergestellt werden konnte, zeigte sich eine deutliche, gruppenübergreifende positive Korrelation von Viruslast und Neutralisationstitern zu allen untersuchten Zeitpunkten (Abb. 17) Abbildung 17: Beziehung von Neutralisationstitern und Virusbeladung im Plasma. Auf der xAchse sind die viralen RNA-Kopien/ml Plasma aufgetragen, die y-Achse gibt den reziproken Neutralisationstiter wieder, bei dem eine Reduktion der Luziferaseaktivität der Zellen um 90% erreicht wurde. Rote Kreise stellen das Verhältnis von Neutralisationstitern und Viruslast der Langzeitüberlebenden dar, blaue Vierecke das der Progressoren zu den Wochen 25 (a), 50 (b) und 100 (c) nach SIV-Infektion. r ist der Korrelationskoeffizient nach Spearman, p das Signifikanzniveau. wpi = Woche nach SIV-Infektion. 74 | S e i t e Ergebnisse 4.1.4 SIV-spezifische IFN-γ-Sekretion von T-Zellen Für den Vergleich von humoraler Immunantwort mit SIV-spezifischer T-Zellantwort wurden während der Immunisierungsstudien durchgeführte IFN-γ-ELISpot-Tests retrospektiv mit besonderem Fokus auf die Langzeitüberlebenden analysiert. Analog zur humoralen Immunantwort gegen das Kapsidprotein p27 (s. Kapitel 4.1.2) war auch die zelluläre Immunantwort gegen einen SIVgag-Peptidpool zu Woche 50 und 100 nach SIV-Infektion bei den Langzeitüberlebenden signifikant höher. Die Anzahl von IFN-γausschüttenden T-Zellen als Reaktion auf einen Pool aus SIVenv-Peptiden hingegen fiel in beiden Gruppen deutlich geringer aus (Abb. 18). Innerhalb der Gruppen gab es keine signifikante Korrelation zwischen der Virusbeladung und der Menge SIV-spezifischer IFN-γsezernierender Zellen (Daten nicht gezeigt). Ergebnisse S e i t e | 75 Abbildung 18: Quantifizierung von IFN-γ-sezernierenden T-Zellen nach Stimulation mit SIVgag oder SIVenv. Die x-Achse zeigt die Anzahl IFN-γ-sezernierender Zellen/ 1x106 PBMC nach Stimulation mit einer Mischung aus SIVgag-Peptiden (a) oder SIVenv-Peptiden (b), die y-Achse die Wochen nach SIV-Infektion bei Langzeitüberlebenden (rot) und Progressoren (blau). Die Querstriche bilden den Mittelwert der jeweiligen Gruppe ab, Querbalken und Sternen kennzeichnen signifikante Unterschiede nach dem Mann-Whitney-Test (* p < 0,05; ** p < 0,01), ns = nicht signifikant. 76 | S e i t e Ergebnisse 4.2 Funktionale und phänotypische Charakterisierung der B-Zellpopulationen der Langzeitüberlebenden 4.2.1 Quantifizierung SIV-spezifischer Gedächtnis-B-Zellen Mittels der ELISpot-Technik wurde das Vorhandensein SIV-spezifischer Antikörpersezernierender Zellen nach polyklonaler Stimulation in Blut und BAL untersucht. Da für diesen Test kein geeignetes Material aus dem Infektionsverlauf zur Verfügung stand, konnte diese Untersuchung nur als Momentaufnahme bei den Langzeitüberlebenden durchgeführt werden. In Abb. 19 sind die Mittelwerte dreier unabhängiger Untersuchungen von PBMC aufgetragen, die im Abstand von 4-6 Wochen durchgeführt wurden. Die Tiere waren zu diesen Zeitpunkten zwischen 174 und 598 Wochen mit SIV infiziert. Der Anteil an Zellen, die gegen p27 gerichtetes IgG sezernierten, lag im Durchschnitt bei 0,80% der IgG-sezernierenden Zellen, wobei die Spannbreite bei den einzelnen Tieren zwischen 0,025 – 1,64% lag. Mit einem Mittelwert von 5,243% war der Anteil gp130-spezifisches IgG-sezernierender Zellen deutlich höher, hier bewegten sich die Individualwerte zwischen 0,65 und 9,46%. Weder zur Anzahl der viralen SIV-RNA-Kopien noch zu den ELISA-Titern gegen p27 und gp130 konnte eine signifikante Korrelation hergestellt werden. Ohne vorherige in vitro-Stimulation fiel der B-Zell-ELISpot im Bezug auf SIV-spezifisches IgG negativ aus, auch SIV-spezifische IgA-sezernierende Zellen konnten im Blut weder mit noch ohne Stimulation mit R848 und IL-2 detektiert werden. Obwohl nach der in vitro-Stimulation von isolierten mononukleären Zellen aus der BAL IgGund IgA-sezernierende Zellen nachweisbar waren, war deren Menge sehr gering. SIVspezifische ASC konnten mit dieser Technik nicht sichtbar gemacht werden (Daten nicht gezeigt). Ergebnisse S e i t e | 77 Abbildung 19: Quantifizierung IgG-sezernierender Zellen im Blut der Langzeitüberlebenden nach dreitägiger in vitro-Stimulation mit R848 und IL-2. Die y-Achse gibt den Anteil SIVspezifischer ASC als Prozentzahl der gesamten IgG-sezernierenden Zellen wieder, auf der x-Achse sind die individuellen Werte der einzelnen Langzeitüberlebenden dargestellt. Schwarze Vertikalbalken verdeutlichen die Standardabweichung innerhalb der drei Untersuchungen. 78 | S e i t e Ergebnisse 4.2.2 Durchflusszytometrische Analyse der B-Zellpopulationen Als Ergänzung zu der im vorherigen Abschnitt beschriebenen funktionalen Untersuchung der Gedächtnis-B-Zellen der Langzeitüberlebenden wurden die B-Zellpopulationen im Blut mittels Durchflusszytometrie in einer Querschnittsanalyse genauer charakterisiert. Die Tiere waren zu diesem Zeitpunkt zwischen 273 und 648 Wochen mit SIV infiziert. Zum Vergleich wurden Proben von SIV-negativen Tieren und Progressoren aus aktuellen Studien (29. bis 81. Woche nach SIV-Infektion) herangezogen. Alle Messungen erfolgten mit frischem EDTABlut. Abbildung 20 veranschaulicht die gating-Strategie, anhand der die nachfolgenden Lymphozytenpopulationen bestimmt wurden. Bedingt durch individuelle Eigenschaften, wie vermindertes Bindungsvermögen oder erhöhte Autofluoreszenz der Zellen, ließen sich bei den Proben einzelner Tiere im Bezug auf bestimmte Antikörperkombinationen keine distinkten Zellpopulationen abgrenzen. Aufgrund der geringen Gruppengrößen wurden diese Tiere jedoch nicht völlig, sondern nur aus den betreffenden Auswertungen ausgeschlossen. Bei der anschließenden Analyse der Daten fiel auf, dass sich der Anteil von gesamten B- und T-Zellen an den Lymphozyten bei den Progressoren stark signifikant von den beiden anderen Gruppen unterschied, die erhobenen Daten innerhalb beider infizierten Gruppen im Vergleich zu SIV-negativen Tieren aber deutlich stärker schwankten (s. Abb. 21). So lag der Mittelwert der CD20+Lymphozyten in der Gruppe der Langzeitüberlebenden bei 17,0 ± 6,4% der Lymphozyten und in der Gruppe der SIV-negativen Tiere bei 22,5± 6,5%, in der Gruppe der Progressoren hingegen war der Anteil der B-Zellen mit 35,3 ± 12,0% deutlich höher. Im Vergleich dazu zeigten die CD3+Lymphozyten ein umgekehrtes Bild, diese Population entsprach bei den Progressoren mit 38,6 ± 13,0% nur etwa Zweidrittel der der Langzeitüberlegenden und SIV-negativen mit 65,7 ± 14,4% bzw. 61,9 ± 5,2%. S e i t e | 79 Ergebnisse Abbildung 20: Gatingschema der durchflusszytometrischen Analyse der B-Zellsubpopulationen. Zuerst wurde die Lymphozytenpopulation mittels Forward(Größe)- und Side-Scatter (Granularität) definiert (a). Im Anschluss daran wurden doppelte Partikel ausgeschlossen (b). Anhand der Oberflächenmarker CD3 (x-Achse) und CD20 (y-Achse) wurden die T- bzw. B-Zellen ermittelt (c). Die CD20+-B-Zellen konnten daraufhin mit dem Oberflächenantigen CD10 (x-Achse) in reife und unreife B-Zellen differenziert werden (d) oder durch die Expression von IgD (y-Achse) in B-Zellen mit oder ohne Ig-Klassenwechsel unterteilt werden (e). Abschließend wurden sowohl die IgD- positive als auch die IgD-negativeB-Zellpopulationen durch CD27 (y-Achse) und CD21(x-Achse) weiter charakterisiert (g). 80 | S e i t e Ergebnisse Abbildung 21: Anteil der gesamten B- und T-Zellpopulation an den Lymphozyten. Die y-Achse gibt den Anteil CD20+- (a) bzw. CD3+-Zellen (b) als Prozentzahl der Lymphozyten wieder. Die Querstriche bilden den Mittelwert der jeweiligen Gruppe ab, mit Querbalken und Sternen sind signifikante Unterschiede nach dem Mann-Whitney-Test (***p < 0,001) gekennzeichnet, ns = nicht signifikant. Da auch die Virusbeladung innerhalb der Progressoren-Kohorte deutlich variierte und überdies zwei Tiere aus der Gruppe der Langzeitüberlebenden zu diesem Zeitpunkt schon über 1x104 SIV-RNA-Kopien/ml Plasma lagen, wurde entschieden, für die folgenden Auswertungen eine Gruppeneinteilung nach Viruslast zu Grunde zu legen. Zu der Gruppe mit niedriger Virusbeladung wurden alle Tiere mit einer Viruslast bis 9x102 virale RNA-Kopien gezählt (7 Langzeitüberlebende und 3 Tiere aus aktuellen Experimenten mit einer Infektionszeit von 72-81 Wochen). Im englisch-sprachigen Raum werden HIVinfizierte Individuen mit diesen Eigenschaften häufig als elite controllers (dt.: Elitekontrollierende) bezeichnet. Die Gruppe mit mittlerer Virusbeladung bestand aus 7 Tieren mit einer Viruslast zwischen 1x103- 9x104 Kopien (4 Langzeitüberlebende, 3 Tiere 40. - 69. Woche nach Infektion), controllers (dt.: Kontrollierende) genannt. Die Gruppe mit hoher Virusbeladung enthielt 12 Tiere mit einer Viruslast zwischen 1x105 - 3x107 (ein Langzeitüberlebender und 11 Tiere mit einer Infektionszeit von 29 bis 74 Wochen) und Ergebnisse S e i t e | 81 entspricht somit klassischen Progressoren (engl.: progressors). In den weiteren Ausführungen werden analog zur Fachliteratur die englischen Begriffe gewählt. Während sich der Anteil der Gesamt-B-Zellen (CD20+Lymphozyten) der elite controllers und der SIV-negativen Tiere nicht signifikant voneinander unterschied, lag der prozentuale Anteil der Gesamt-B-Zellen der progressors signifikant höher (Abb. 22). Auch die controllers wiesen eine Tendenz zu erhöhten B-Zellwerten auf, selbst wenn in diesem Fall keine statistische Signifikanz erreicht wurde. Im Vergleich zu Abbildung 21 beweist diese Tatsache, dass für die weitere Diskussion eine Aufteilung nach Virusbeladung der nach Infektionszeit überlegen ist. Abbildung 22: Anteil der gesamten B-Zellpopulation an den Lymphozyten in Abhängigkeit vom Infektionsstatus. Die y-Achse gibt den Anteil CD20+-Zellen als Prozentzahl der Lymphozyten wieder. Die Querstriche bilden den Mittelwert der jeweiligen Gruppe ab, mit Querbalken und Sternen sind signifikante Unterschiede nach dem Mann-Whitney-Test (*p < 0,05, **p< 0,01) gekennzeichnet, ns = nicht signifikant. 82 | S e i t e Ergebnisse Die gebräuchlichste Darstellung von Gedächtnis-B-Zellen in der Literatur erfolgt durch den Oberflächenmarker CD27. Häufig wird diese noch ergänzt durch die Charakterisierung der Immungloblinklasse mittels IgD. Auf diese Weise lassen sich naive B-Zellen, ohne Antigenkontakt und Klassenwechsel (CD20+CD27-IgD+), von aktivierten B-Zellen mit Klassenwechsel (CD20+CD27-IgD-) und memory-B-Zellen (CD20+CD27+IgD+/-) unterscheiden. Die in Abbildung 23 dargestellten Daten demonstrieren, dass sich die B-Zellsubpopulationen in Abhängigkeit von der Viruslast verschoben. Auf der einen Seite standen die elite controllers mit sehr niedriger SIV-RNA-Beladung, die sich nicht signifikant von den SIVnegativen unterschieden. Einzig der Prozentsatz IgD+-Gedächtnis-B-Zellen war gegenüber dem der SIV-negativen leicht erniedrigt (Abb. 23 d). Bei Tieren mit einer höheren Viruslast auf der anderen Seite änderten sich die Verhältnisse eindeutig zu Lasten der naiven B-Zellen (Abb. 23 a). Sie entsprachen nur 21,16 ± 6,8% (controllers) bzw. 18,55 ± 10,01% (progressors) der gesamten B-Zellen im Gegensatz zu 33,91 ± 11,84% (elite controllers) und 30,18 ± 9,89% (SIV-negative). Durch die hohen Standardabweichungen erreichte der Unterschied zwischen controllers einerseits und elite controllers und SIV-negativen andererseits keine statistische Signifikanz. Diese wurde hingegen besonders deutlich bei CD27+-B-Zellen, bei denen ein Klassenwechsel stattgefunden hatte (Abb. 23 c): controllers und progressors wiesen 64,28 ± 11,64% bzw. 69,69 ± 13,79% IgD-negative Gedächtniszellen auf. Dem gegenüber standen 39,16 ± 13,37% und 38,4 ± 10,48% bei den elite controllers respektive SIV-negativen Tieren. Ergebnisse S e i t e | 83 Abbildung 23: Relative CD27+B-Zellpopulationen in Abhängigkeit von der Viruslast. Dargestellt sind die Individualwerte der elite controllers (rot), controllers (hellblau), progressors (dunkelblau) und SIV-negativen Tiere (grün) .Die y-Achse gibt den Anteil der Zellen als Prozentzahl der CD20+Lymphozyten wieder. Die Querstriche bilden den Mittelwert der jeweiligen Gruppe ab, mit Querbalken und Sternen sind signifikante Unterschiede nach dem Mann-Whitney-Test (*p < 0,05; **p< 0,01; ***p < 0,001) gekennzeichnet, ns = nicht signifikant. 84 | S e i t e Ergebnisse Neuere Erkenntnisse (DAS et al., 2011) deuten darauf hin, dass eine differenziertere Analyse der B-Zellen notwendig ist, um eine Einteilung zu erreichen, die ihren funktionellen Eigenschaften entspricht. Im Kapitel 5.3.1 wird diese Theorie ausführlich diskutiert. Mithilfe der Oberflächenmarker CD10 und CD21 wurden die Definitionen der B- Zellsubpopulationen erweitert, die entsprechende schematische Darstellung findet sich in Kapitel 2.1.2.1 der Literaturübersicht. Die Mittelwerte der verschiedenen Lymphozytensubpopulationen sind für die drei SIV-infizierten Kohorten und die SIVnegativen Tiere in Tabelle 5 zusammengefasst, die entsprechenden Graphen finden sich in Abbildung 23, 24 und 25. Den größten Anteil an B-Lymphozyten bilden bei allen vier Tiergruppen die aktivierten, reifen B-Zellen (CD10-CD21-CD27+IgD-). Im Gegensatz zu den elite controllers und den SIV-negativen Tieren, bei denen diese Population etwa 35% der B-Zellen entsprach, unterschieden sich die Tiere mit mittlerer und hoher Virusbeladung davon deutlich. Mit 60,97% respektive 66,96% lag der Anteil nahezu doppelt so hoch (Abb. 24 b). Kaum Unterschiede zeigten sich im Bezug auf die naiven B-Zellen (CD10-CD21+CD27IgD+). Zwar war diese Population bei den elite controllers und den SIV-negativen Tieren etwas höher, signifikant war diese Differenz aber nur zwischen progressors und SIVnegativen Tieren (p = 0,019) (Abb. 24 a). Gedächtnis-B-Zellen (CD10-CD21+CD27+) machten etwa 6% der B-Zellen der uninfizierten Tiere aus. Anhand der Oberflächenimmunglobuline ließ sich dieser Zelltyp weiter differenzieren. Der Anteil IgD+Gedächtnis-B-Zellen war bei SIV-negativen Tieren mehr als dreimal so hoch wie bei den infizierten Tierkohorten. Innerhalb dieser Gruppe wiesen die elite controllers den höchsten Anteil an IgD+Gedächtnis-B-Zellen auf, allerdings war der Unterschied nur im Vergleich zu den controllers leicht signifikant (Abb. 24 c). Die Prozentsätze der IgD-Gedächtnis-B-Zellen grenzten sich bei den infizierten Tiergruppen nicht signifikant von einander ab und waren nur leicht signifikant niedriger als die der SIVnegativen Tiere (Abb. 24 d). Ergebnisse S e i t e | 85 Zusätzlich wurden die Verhältnisse zweier atypischer B-Zellpopulationen untersucht: Übergangs-B-Zellen (CD10+CD21+CD27-) stellen eine unreife Zellform dar, da sie weiterhin den Oberflächenmarker CD10 tragen, und sind im peripheren Blut nur minimal vertreten. Sie lagen bei elite controllers und SIV-negativen Tieren in einem ähnlichen Bereich (0,41% bzw. 0,51%), bei progressors mit 0,14% deutlich darunter. Die Standardabweichung von ca. 0,2 war bei allen Gruppen angesichts der geringen Prozentsätze sehr hoch (Abb. 24 e). Die zweite atypische B-Zellform entsprach, unabhängig vom Infektionsstatus, einem Anteil von 3-4,7% der CD20+Lymphozyten. In der englischsprachigen Literatur wird sie als exhausted tissue-like (dt.: erschöpfte Gewebe-ähnliche) Gedächtnis-B-Zelle bezeichnet, da nahezu alle für B-Zellen charakteristischen Oberflächenmarker verloren gegangen sind (CD21-CD27-), obwohl ein Klassenwechsel stattgefunden haben musste (IgD-) (Abb. 24 f). Im Gegensatz zu den B-Zellen liegt der Anteil der T-Zellen an den Lymphozyten bei den progressors deutlich unterhalb dem der anderen Gruppen, die sich nicht signifikant voneinander unterschieden (s. Abb. 25 a). Die Analyse der CD4+T-Zellsuppopulation zeigte ein anderes Bild (s. Abb. 25 b). Die Tiere mit mittlerer und hoher Virusbeladung wiesen die geringsten relativen Anteile auf und unterschieden sich nicht signifikant voneinander. Der relative Anteil der T-Helferzellen lag bei elite controllers deutlich höher (p = 0,0028), aber immer noch klar unterhalb dem der SIV-negativen Tiere (p = 0,0098). T-Helferzellen sind notwendig für die Aktivierung von naiven und Gedächtnis-B-Zellen. Aufgrund dieser funktionalen Zusammenhänge wurde untersucht, ob sich diese auch anhand der Populationsgrößen von T-Helferzellen einerseits und aktivierten bzw. Gedächtnis-BZellen andererseits widerspiegelten. Wurden alle vier Tierkohorten zusammengefasst, offenbarte sich im Bezug auf IgD-negative ruhende Gedächtniszellen eine leichte Tendenz zu einer positiven Korrelation mit CD4+Lymphozyten (Abb. 26 a), die IgD-positiven ruhenden Gedächtniszellen zeigten sogar eine starke positive Korrelation (Abb. 26 b). Der relative Anteil aktivierter reifer B-Zellen hingegen korrelierte hochsignifikant invers mit dem der THelferzellen (Abb. 26 c). Bei der Einzelauswertung der Gruppen blieben diese Tendenzen erhalten, die statistische Signifikanz ging allerdings verloren. 86 | S e i t e Gesamt-BZellen Ergebnisse elite controllers controllers progressors SIV-negative MW MW MW SD MW SD SD SD 18,47% 8,67 23,42% 9,21 35,53% 10,66 21,94% 6,59 18,23% 8,97 11,95% 5,86 11,04% 6,05 20,80% 8,82 35,87% 13,23 60,97% 10,97 66,96% 13,93 34,74% 10,22 0,99% 0,43 0,47% 0,35 0,61% 0,39 3,14% 1,23 1,72% 0,91 2,28% 1,38 1,34% 0,74 2,72% 0,90 0,41% 0,29 0,32% 0,20 0,14% 0,12 0,51% 0,22 4,73% 2,00 4,13% 4,74 3,00% 1,68 3,19% 0,92 CD20+ naive B-Zellen CD10-CD21+CD27IgD+ aktivierte reife B-Zellen CD10-CD21CD27+IgD- ruhende Gedächtnis-BZellen (IgD+) CD10CD21+CD27+IgD+ ruhende Gedächtnis-BZellen (IgD-) CD10CD21+CD27+IgD- Übergangs-BZellen CD10+CD21+CD27IgD+/- exhausted tissue-like Gedächtnis-BZellen CD10-CD21-CD27IgD- Gesamt-TZellen 74,24% 9,61 67,03% 11,38 39,07% 13,38 70,08% 4,43 25,22% 5,26 26,25% 8,10 60,91% 3,82 CD3+ Helfer-T-Zellen 44,09% 13,21 CD3+CD4+ Tab. 5: Mittelwerte und Standardabweichungen verschiedener B- und T-Zellpopulationen. Aufgeführt sind die relativen Anteile der B- und T-Zellen an den Lymphozyten bzw. deren Subpopulationen als Anteil an den Gesamt-B- bzw. -T-Zellen. MW = Mittelwert der Gruppe, SD = Standardabweichung Ergebnisse S e i t e | 87 Abbildung 24: Relative B-Zellsubpopulationen in Abhängigkeit von der Viruslast. Dargestellt sind die Individualwerte der elite controllers (rot), controllers (hellblau), progressors (dunkelblau) und SIV-negativer Tiere (grün). Die y-Achse gibt den Anteil der Zellen als Prozentzahl der CD20+Lymphozyten wieder. Die Querstriche bilden den Mittelwert der jeweiligen Gruppe ab. Zur besseren Übersicht sind lediglich statistisch signifikante Unterschiede nach dem Mann-Whitney-Test (*p < 0,05; **p< 0,01; ***p < 0,001) mit Querbalken und Sternen gekennzeichnet. 88 | S e i t e Ergebnisse Abbildung 25: Relative T-Zellsubpopulationen in Abhängigkeit von der Viruslast. Dargestellt sind die Individualwerte der elite controllers (rot), controllers (hellblau), progressors (dunkelblau) und SIV-negativer Tiere (grün). Die y-Achse gibt den Anteil der CD3+Zellen als Prozentzahl der Lymphozyten (a) bzw. den Anteil CD3+CD4+Zellen an den CD3+Lymphozyten (b) wieder. Die Querstriche bilden den Mittelwert der jeweiligen Gruppe ab, mit Querbalken und Sternen sind signifikante Unterschiede nach dem Mann-Whitney-Test (**p< 0,01; ***p < 0,001) gekennzeichnet, ns = nicht signifikant. Ergebnisse S e i t e | 89 Abbildung 26: Beziehung von T-Helferzellen und verschiedenen B-Zellpopulationen. Auf der xAchse ist der Anteil CD4+-Zellen an CD3+Lymphozyten aufgetragen, die y-Achse gibt den Anteil von IgD- ruhenden Gedächtniszellen (a), von IgD+ ruhenden Gedächtniszellen (b) und aktivierten reifen B-Zellen (c) an CD20+Lymphozyten wieder. r ist der Korrelationskoeffizient nach Spearman, p das Signifikanzniveau. 90 | S e i t e Ergebnisse 4.3 Longitudinale Untersuchung der humoralen Immunantwort von der akuten bis zur chronischen Phase der SIV-Infektion bei nicht-immunisierten Kontrolltieren 4.3.1 Virusbeladung bis 48 Wochen nach SIV- Infektion Die im Kapitel 4.2. dargestellten Ergebnisse stammen aus der chronischen Phase der SIVInfektion, da während der Anfangsphase kein entsprechendes Material asserviert worden war. Um Besonderheiten zu ermitteln, die einen Grundstein für die klinische Ausprägung des späteren Infektionsverlaufs legen, wurden dieselben Techniken auf Proben angewandt, die von Tieren aus zwei kürzlich gestarteten Vakzinestudien stammten. Da es sich bei diesen 12 Tieren um Kontrolltiere handelte, konnten Einflüsse einer Immunisierung ausgeschlossen werden. In Abb. 27 ist die Virusbeladung dieser 12 Tiere in Bezug auf die Anzahl viraler RNAKopien im Plasma dargestellt. In der untersuchten Gruppe gab es sowohl ein Tier (2272), das über den Beobachtungszeitraum hinweg durchgehend eine hohe Virusbeladung aufwies und als erstes Tier dieser Kohorte in der 52. Woche nach SIV-Infektion wegen AIDS-ähnlicher Symptomatik euthanasiert werden musste, als auch drei Tiere, die innerhalb weniger Wochen nach der initialen Spitzenvirämie in der Lage waren, die Viruslast in einem Bereich knapp oberhalb der Nachweisgrenze zu kontrollieren (13906, 13907 und 13913). Im Durchschnitt lag die Anzahl viraler RNA-Kopien/ml Plasma während der Spitzenvirämie bei 2,23x106. Ergebnisse S e i t e | 91 Abbildung 27: Virusbeladung bis 48 Wochen nach SIV-Infektion in unbehandelten Kontrolltieren. Die x-Achse kennzeichnet die Wochen nach SIV-Infektion, die y-Achse gibt die Menge der SIV-RNA-Kopien/ml Plasma wieder. Zur besseren Übersichtlichkeit während der akuten Phase der Infektion ist die x-Achse bis zur 8. Woche gedehnt. Die Legende führt die Nummern der einzelnen Tiere auf, deren individuellen Werte in hell-violett dargestellt sind, die gestrichelte Linie entspricht dem Mittelwert. 4.3.2 Bindende Antikörper in der frühen Phase der SIV-Infektion Die Analyse von Plasmaproben auf bindende Antikörper mittels ELISA ergab, dass eine schwache Antikörperantwort gegen das Hüllprotein gp130 bereits zwei Wochen nach SIVInfektion vorhanden war, diese aber erst 4 Wochen nach Infektion deutlich anstieg. Zu diesem Zeitpunkt waren auch Antikörper gegen das Kapsidprotein p27 erstmalig nachweisbar. In der akuten Phase der Infektion stiegen die Antikörpertiter gegen p27 kontinuierlich an, bis die Mittelwerte ein Plateau zwischen 1x105 und 1x106 erreichten. Die Antikörpertiter gegen gp130 verhielten sich ähnlich, allerdings lagen die Mittelwerte eine Log-Stufe niedriger und auch die Standardabweichung war deutlich geringer. Auffällig ist, dass die Titer des Tieres 2272, welches nach der initialen Spitzenvirämie nicht in der Lage war, die Viruslast nennenswert zu reduzieren, deutlich unterhalb der Mittelwerte lagen (Abb. 28). 92 | S e i t e Ergebnisse Es ließ sich zu keinem Zeitpunkt eine Korrelation zwischen Viruslast und Antikörpern gegen gp130 herstellen (Daten nicht gezeigt). Es bestand jedoch eine signifikant positive Korrelation zwischen Antikörpern gegen p27 und viralen RNA-Kopien zu Woche 4 nach SIV-Infektion. Diese ging später nicht nur verloren, sondern entwickelte sogar einen Trend hin zur negativen Korrelation. Dieser Trend war allerdings nicht, wie die in Kapitel 4.1.2 dargestellten Daten, signifikant (Abb. 29). Abbildung 28: Bindende Antikörper gegen p27 und gp130 bis 42 Wochen nach SIV-Infektion in unbehandelten Kontrolltieren. Die x-Achse kennzeichnet die Wochen nach SIV-Infektion, die yAchse gibt den reziproken ELISA-Titer bindender Antikörper gegen p27 (a) oder gp130 (b) wieder. Zur besseren Übersichtlichkeit während der akuten Phase der Infektion ist die x-Achse bis zur 8. Woche gedehnt. Die Legende führt die Nummern der einzelnen Tiere auf, deren individuellen Werte in hell-violett dargestellt sind, die dunkel-violette Linie entspricht dem Mittelwert. Ergebnisse S e i t e | 93 Abbildung 29: Beziehung von SIV-Last und Antikörpertiter gegen p27 nach SIV-Infektion in unbehandelten Kontrolltieren. Die x-Achse gibt die Anzahl SIV-RNA-Kopien/ ml Plasma wieder, die y-Achse den reziproken ELISA-Titer. Die Graphen stellen die Beziehungen dieser Parameter zu Woche 4 (a), 8 (b), 16 (c) und 32 (d) nach SIV-Infektion dar. Liegt eine signifikante Korrelation vor, sind der Korrelationskoeffizient nach Spearman (r) und das Signifikanzniveau (p) angegeben. 94 | S e i t e Ergebnisse 4.3.3 Neutralisierende Antikörper gegen SIV Analog zu den bindenden Antikörpern war eine deutliche Immunantwort in Form von neutralisierenden Antikörpern im Serum der nicht-immunisierten Tiere erst in der vierten Woche nach SIV-Infektion nachweisbar. Auch in diesem Fall erreichte der mittlere Titer ein ein Plateau (Abb. 30). Im vorherigen Kapitel wurde ersichtlich, dass die bindenden Antikörper des Tieres 2272 deutlich unterhalb des Mittelwertes lagen und jene gegen gp130 erst zu Woche 16 messbar waren. Im Gegensatz dazu unterschieden sich dessen Neutralisationstiter nicht von denen der restlichen Kohorte. Es bestand keine signifikante Korrelation zwischen RNA-Last und Neutralisationstiter (Daten nicht gezeigt). Abbildung 30: Verlauf der Neutralisationstiter bis 42 Wochen nach SIV-Infektion in unbehandelten Kontrolltieren. Die x-Achse kennzeichnet die Wochen nach SIV-Infektion, die yAchse gibt den reziproken Neutralisationstiter wieder. Zur besseren Übersichtlichkeit während der akuten Phase der Infektion ist die x-Achse bis zur 8. Woche gedehnt. Die Legende führt die Nummern der einzelnen Tiere auf, deren individuellen Werte in hell-violett dargestellt sind, die dunkel-violette Linie entspricht dem Mittelwert. Ergebnisse S e i t e | 95 4.3.4 Quantifizierung SIV-spezifischer Antikörper-sezernierender Zellen Zwei, acht und vierzig Wochen nach erfolgter SIV-Infektion wurden frisch isolierte PBMC ohne vorherige in vitro-Stimulation im B-Zell-ELISpot eingesetzt und das Vorhandensein von Zellen überprüft, die p27- bzw. gp130-spezifische IgG-Antikörper sezernierten. Sowohl der Prozentsatz der p27-spezifischen als auch der gp130-spezifischen ASC an den Gesamt-IgG-sezernierenden Zellen stieg im Infektionsverlauf an, allerdings war nur die Differenz p27-spezifischer ASC zwischen 2. und 40.wpi statistisch signifikant (s. Abb. 31). Die mittleren Anteile von p27-spezifischen ASC unterschieden sich zu keinem Zeitpunkt signifikant von denen der gp130-spezifischen (Daten nicht gezeigt). Abbildung 31: SIV-spezifische ASC im Infektionsverlauf. Die x-Achse kennzeichnet die Wochen nach SIV-Infektion, die y-Achse gibt die mittleren relativen Anteile der p27-spezifischer (a) und gp130-spezifischer (b) IgG-sezernierender Zellen an den Gesamt-IgG-sezernierenden Zellen wieder. Vertikalbalken verdeutlichen die Standardabweichung, Horizontalbalken kennzeichnen die signifikanten Unterschiede nach dem Mann-Whitney-Test zwischen Woche 2 und Woche 8 bzw. Woche 40 nach SIV-Infektion. * p< 0,05; ns = nicht signifikant. 96 | S e i t e Ergebnisse Während im Blut der Langzeitüberlebenden ohne vorherige Stimulation keine SIVspezifischen ASC nachgewiesen werden konnten (s. Kapitel 4.2.1), fiel diese Untersuchung innerhalb der ersten Monate der SIV-Infektion positiv aus. Bemerkenswert war überdies, dass 40 Wochen nach Infektion der mittlere Anteil von Zellen, die gegen p27-gerichtete Antikörper sezernierten, bei 3,53% lag und damit mehr als viermal so hoch wie der mittlere Wert der Gedächtnis-B-Zellen bei den Langzeitüberlebenden war. Im Bezug auf gp130spezifische ASC zeigte sich ein umgekehrtes Bild. Mit einem Mittelwert von 2,56% war der Anteil an den IgG-sezernierenden Zellen nur etwa halb so hoch wie bei den Langzeitüberlebenden (5,24%). Analog zu den Analysen, die bei der Charakterisierung der Langzeitüberlebenden unternommen wurden, sind auch für diese Tierkohorte mögliche Korrelationen zwischen den SIV-spezifischen ASC und Viruslast überprüft worden. Im Gegensatz zu den Zeitpunkten 2. und 40. Woche nach Infektion, zu denen der Anteil SIV-spezifischer ASC nicht signifikant mit der Anzahl viraler RNA-Kopien pro ml Plasma korrelierte, bestand in der 8. Woche eine hochsignifikante negative Korrelation zwischen Virusbeladung einerseits und dem Anteil der p27- bzw. gp130-spezifischen ASC andererseits (s. Abb. 32 a). Darüber hinaus wiesen Tiere mit einem hohen Anteil von spezifischen ASC acht Wochen nach Infektion auch niedrigere Viruslasten in der 40. Woche auf (s. Abb. 32 b), während die Viruslast zu Woche 8 keinen statistisch messbare Beziehung zur Menge der ASC 40 Wochen nach SIV-Infektion hatte (Daten nicht gezeigt). Die Titer der bindenden Antikörper, die mittels ELISA-Technik bestimmt wurden, und der Anteil der ASC korrelierten nicht miteinander (Daten nicht gezeigt). Ergebnisse S e i t e | 97 Abbildung 32: Beziehung von Viruslast und SIV-spezifischen ASC. Die x-Achse gibt die Anzahl SIV-RNA-Kopien/ml Plasma wieder, die Ordinate den Anteil p27- (türkis) und gp130- (violett)spezifischer ASC an den Gesamt-IgG-sezernierenden Zellen. Graph a stellt die beiden Parameter zur Woche 8 dar, Graph b die des Anteils der SIV-spezifischen ASC zur Woche 8 und der Viruslast zur Woche 40 nach SIV-Infektion. Korrelationskoeffizient nach Spearman (r) und das Signifikanzniveau (p) sind angegeben. wpi = Wochen nach SIV-Infektion 4.3.5 Longitudinale durchflusszytometrische Analyse der B-Zellpopulationen Die phänotypische Untersuchung der Lymphozytenpopulation mittels Durchflusszytometrie erfolgte, im Gegensatz zu dem in Abschnitt 4.2.2 dargestellten Probenmaterial, anhand von tiefgefrorenen, isolierten mononukleären Zellen. Ein direkter Vergleich bestimmter Populationen zwischen den zwei Schwerpunkten der vorliegenden Arbeit war deswegen nur unter Vorbehalt möglich, da eine Beeinflussung der Oberflächenantigene durch die FrierTauprozesse oder die Isolationsschritte nicht ausgeschlossen werden konnte. Der mittlere Anteil von CD20+B-Zellen und CD3+T-Zellen an den Lymphozyten lag vor SIVInfektion der 12 Tiere bei 15,6 ± 5,2% bzw. 53,4 ± 9,6% und somit in beiden Fällen niedriger als die mittleren Anteile von B- und T-Zellen der SIV-negativen Tiere aus Kapitel 2.2.2.2, die in frischem EDTA-Blut bestimmt wurden (21,9% respektive 70,1%, s. Tab. 5). Zwei Wochen nach erfolgreicher SIV-Infektion waren der relative Anteil der B-Lymphozyten auf 9,2 ± 3,7% gefallen und unterschied sich somit signifikant vom Präwert. Schon in der dritten Woche nach Infektion stieg der Mittelwert wieder an und erreichte im Infektionsverlauf ein Plateau oberhalb des Präwertes, wobei der Unterschied nicht signifikant war (s. Abb. 33 a). Die individuellen Werte der einzelnen Tiere lagen z.T. deutlich darüber oder darunter, eine signifikante Korrelation zur SIV-RNA-Last konnte aber zu keinem Zeitpunkt nachgewiesen 98 | S e i t e Ergebnisse werden. Auch die Höhe der Virusbeladung zum Zeitpunkt der Spitzenvirämie konnte nicht in einen statistischen Zusammenhang zur Höhe der B-Zellanteile 20 Wochen nach Infektion gebracht werden. Die Schwankungen der mittleren relativen T-Zellzahlen während des Infektionsverlaufes unterschieden sich zu keinem Zeitpunkt signifikant von dem mittleren Präwert (s. Abb. 33 b). Des Weiteren bestand keine signifikante Korrelation zwischen der Menge der viralen RNAKopien und dem relativen Anteil der T-Zellen an den Lymphozyten. Da für die Verlaufsuntersuchung CD27 als Gedächtnis-B-Zell-Marker nicht zur Verfügung stand, musste in diesem Fall die Population der memory-B-Zellen anders definiert werden. Die Analyse der Daten aus Kapitel 4.2.2 zeigte, dass im Mittel 86,4 ± 8,9% der CD10-IgD-BZellen auch CD27 trugen. Auch in der Literatur wird von einer deutliche inversen Korrelation von IgD- und CD27-Expression berichtet (DE MILITO et al., 2001; CHONG et al., 2004a). Mit der Darstellung dieser Zellpopulation kann demnach die Veränderung der CD27+BZellanteile näherungsweise beschrieben werden, allerdings war eine Unterscheidung von aktivierten und ruhenden Gedächtniszellen auf Grund des Fehlens von CD21 nicht möglich. Obwohl, wie in der Abbildung 34 a ersichtlich, der mittlere prozentuale Anteil der CD10-IgDZellen an den B-Zellen in der zweite und dritten Woche nach Infektion abfiel (40,4% bzw. 32,9%), erreichte dieser Unterschied keine statistische Signifikanz im Vergleich zum mittleren Präwert 44,3 ± 6,9%. Auch der durchschnittliche Wert 20 Wochen nach SIVInfektion (46,4 ± 14,5%) unterschied sich nicht signifikant vom Präwert. Da sich die individuellen Werte der Tiere z.T. stark voneinander unterschieden, wurde auch in diesem Fall die Korrelation mit der Viruslast überprüft, konnte aber nicht festgestellt werden. In Abbildung 34 b sind die T-Helferzellen als Anteil der T-Zellen dargestellt. Im Unterschied zu der B-Zellsubpopulation sank der Mittelwert dieser Zellgruppe im Vergleich zum mittleren Präwert (65,0 ± 6,9%) signifikant und lag auch nach 20 Wochen deutlich darunter (48,6 ± 14,5%). Es bestand eine starke negative Korrelation zwischen der Anzahl viraler RNAKopien während der Spitzenvirämie zum relativen Anteil der T-Helferzellen, nicht nur zum selben Zeitpunkt, sondern auch zum Wert 20 Wochen nach SIV-Infektion (s. Abb. 35). Ergebnisse S e i t e | 99 Abbildung 33: Änderungen der B- und T-Zellpopulationen bis zum Beginn der Plateauphase der SIV-Infektion in unbehandelten Kontrolltieren. Auf der y-Achse dargestellt sind die relativen Anteile der CD10-20+- (a) bzw. CD3+-Zellen (b) an den Lymphozyten, die x-Achse kennzeichnet die Wochen nach SIV-Infektion. Die individuellen Werte der einzelnen Tiere sind in hell-violett dargestellt, die dunkel-violette Linie entspricht dem Mittelwert. Schwarze Querbalken und Sterne kennzeichnen signifikante Unterschiede nach dem Mann-Whitney-Test zwischen Präwert und Woche 2 bzw. 20 nach Infektion. ** p< 0,01; ns = nicht signifikant. 100 | S e i t e Ergebnisse Abbildung 34: Änderungen der B- und T-Zellsubpopulationen bis zum Beginn der Plateauphase der SIV-Infektion in unbehandelten Kontrolltieren. Auf der y-Achse dargestellt sind die relativen Anteile der CD10-IgD-Zellen an CD20+B-Zellen (a) bzw. CD4+-Zellen an den CD3+Lymphozyten (b) , die x-Achse kennzeichnet die Wochen nach SIV-Infektion. Die individuellen Werte der einzelnen Tiere sind in hell-violett dargestellt, die dunkel-violette Linie entspricht dem Mittelwert. Schwarze Querbalken und Sterne kennzeichnen signifikante Unterschiede nach dem Mann-Whitney-Test zwischen Präwert und Woche 2 bzw. 4, 14 oder 20 nach Infektion. * p<0,05; **p< 0,01; ns = nicht signifikant Ergebnisse S e i t e | 101 Abbildung 35: Beziehung der Viruslast zum Zeitpunkt der Spitzenvirämie und CD4+TZellspiegel während der Spitzenvirämie und in der frühen Plateauphase der SIV-Infektion unbehandelter Kontrolltiere. Die x-Achse gibt die Anzahl SIV-RNA-Kopien/ml Plasma wieder, die y-Achse den Anteil CD4+T-Zellen an CD3+-Lymphozyten. Graph a stellen die Beziehungen dieser Parameter zu Woche 2 dar, Graph b die Beziehung zwischen Viruslast zwei Wochen und CD4+TZellen 20 Wochen nach SIV-Infektion. Angegeben sind der Korrelationskoeffizient nach Spearman (r) und das Signifikanzniveau (p). wpi = Wochen nach SIV-Infektion 102 | S e i t e Diskussion 5. DISKUSSION Die zentrale Bedeutung der humoralen Immunantwort bei der Bekämpfung von diversen Infektionskrankheiten ist unumstritten. Auch auf eine HIV- und SIV-Infektion reagiert der Körper mit der Bildung von spezifischen Antikörpern. Obwohl mehrere Studien belegen, dass der passive Transfer von Antikörpern Makaken vor einer SIV-Infektion schützen kann (BABA et al., 2000; MASCOLA et al., 2000; PARREN et al., 2001; BURTON et al., 2011), scheint die Primärantwort des Immunsystems allerdings zu spät einzusetzen, um eine Ausbreitung von Retroviren und eine Integration der viralen RNA ins Wirtsgenom zu verhindern. Bei den meisten Individuen ist eine chronische Infektion mit voranschreitender Zerstörung des Immunsystems die Folge. Der Versuch ist also naheliegend, mittels vorangehender Immunisierung das immunologische Gedächtnis zu aktivieren, so dass die viel schneller einsetzende Sekundärantwort die Infektion verhindern kann. Leider ist dies bisher nicht gelungen. Grund dafür könnten neben den speziellen viralen Eigenschaften, wie dem Vorkommen diverser Stämme, ungewöhnlich hohen Mutationsraten und geringer Immunogenität, die bisher unzureichenden Kenntnisse über die Wirkungsweise der Antikörper und die für ihre Bildung zuständigen Zellen der B-Zellreihe sein. Weitere Pathogeneseforschung in diesem Bereich ist also unerlässlich. Hinzu kommt, dass die Auswirkungen der HIV-/SIV-Infektion auf die B-Zellphysiologie gravierend sind. Folgen sind zum Beispiel Hypergammaglobulinämie, verminderte Stimulierbarkeit und Abnahme der Gedächtnis-B-Zellpopulationen, unabhängig von der Spezifität. So tragen auch die B-Zelldysfunktionen zur Ausbildung des Immunschwächesyndroms bei, obwohl das Studium dieser Veränderungen bisher zugunsten des T-Zellsystems als Wirtszellen des Virus etwas vernachlässigt wurde. Differenzierteres Wissen könnte einen wichtigen Beitrag für bessere Therapiemöglichkeiten von AIDS bedeuten. In der vorliegenden Dissertation wurde die Reaktion des B-Zell-abhängigen Arms des Immunsystems von Rhesusmakaken auf eine Infektion mit SIV als Modell für die HIVInfektion des Menschen untersucht. Dies geschah einerseits anhand funktioneller, auf den Nachweis von Antikörper basierender Tests, die z.T., wie der B-Zell-ELISpot, zu Beginn der Diskussion S e i t e | 103 Arbeit noch nicht auf SIV-Infektion von Makaken angewendet worden waren. Der zweite Schwerpunkt bestand in der phänotypischen Charakterisierung von B-Zellsubpopulationen in verschiedenen Infektionsstadien mittels Durchflusszytometrie, die in diesem Umfang noch nicht im SIV-Makakenmodell beschrieben worden war. Durch den glücklichen Umstand, dass sich die Abteilung Infektionsmodelle des DPZ im Besitz einer außergewöhnlich großen Kohorte von mit SIV-infizierten, langzeitüberlebenden Makaken befindet, konnte dieses Phänomen untersucht und mit Individuen mit typisch progredientem Krankheitsverlauf verglichen werden. Insgesamt flossen Daten von 100 Tieren in diese Arbeit, 13 davon zu Beginn der Studien mit dem Status von Langzeitüberlebenden. Derart groß angelegte Studien sind in der biomedizinischen Forschung mit Primaten selten anzutreffen. 5.1 Die Viruslast in der akuten Plateauphase eignet sich als prognostischer Marker für den weiteren Krankheitsverlauf Die retrospektive Auswertung der Menge an SIV-RNA-Kopien pro ml Plasma als Maß der Virusbeladung von insgesamt 65 Tieren aus verschiedenen Vakzinestudien ergab, dass 13 Individuen über einen Zeitraum von mindestens 3 Jahren in der Lage waren, die Viruslast unter 1x104 virale Kopien/ml Plasma zu kontrollieren (s. Abb. 10, Kapitel 4.1.1). Vorherige Analysen in der Arbeitsgruppe belegten, dass dieser Effekt unabhängig von einer vorhergehenden Immunisierung war (SCHULTE et al., 2009; SCHULTHEISS et al., 2011). Hinsichtlich der Höhe der Spitzenvirämie zwei Wochen nach SIV-Infektion unterschieden sich diese Tiere nicht von denen, die später an AIDS erkrankten. Die Höhe der Spitzenvirämie eignete sich in dieser Studie also nicht als prognostischer Marker für den Infektionsverlauf. Bei dem Eintritt in die akute Plateauphase (engl. set-point) in der achten Woche nach Infektion lag hingegen die Viruslast bei den Langzeitüberlebenden mit 1,8x104 RNAKopien/ml Plasma um zwei Log-Stufen niedriger als zur Spitzenvirämie, während sich die mittlere Viruslast der Progressoren zu diesem Zeitpunkt zwar auch deutlich verringert hatte, aber immer noch hoch signifikant über der der Langzeitüberlebenden lag. Diese Ergebnisse decken sich mit denen von BEER et al. (1996) und WATSON et al. (1997) und lassen die Reaktion des erworbenen Immunsystems als Ursache für die Kontrolle der Infektion wahrscheinlich erscheinen. Dem gegenüber stehen die Untersuchungen einer anderen Arbeitsgruppe, die allerdings eine andere Inokulationsroute und einen anderen Virusstamm 104 | S e i t e Diskussion verwendet hatte (LIFSON et al., 1997). Hier konnte bereits eine Woche nach Infektion eine Korrelation zwischen der Anzahl viraler RNA-Kopien und der Höhe der set-point-Virämie hergestellt werden. Dies würde das erworbene Immunsystem als treibende Kraft ausschließen und spräche für genetische Faktoren oder Mechanismen des angeborenen Immunsystems. Bei der in Kapitel 4.1 untersuchten Kohorte von 12 Tieren lag die durchschnittliche Spitzenvirämie mit 2,23x106 virale RNA-Kopien/ml Plasma in dem gleichen Bereich wie die der in Kapitel 4.3 dargestellten Tiere. Da die erstgenannte Kohorte aus den nichtimmunisierten Kontrollgruppen zweier Vakzineexperimente bestand, konnte der Einfluss einer vorhergehenden Immunisierung naturgemäß ausgeschlossen werden. 5.2 Vergleichende funktionale Untersuchungen der B-Zellimmunantwort von Langzeitüberlebenden und Progressoren 5.2.1 Langzeitüberlebende waren durch höhere bindende Antikörpertiter gegen SIVgag im ELISA gekennzeichnet Bedingt durch den Replikationszyklus von HIV/SIV sind die einzigen viralen Proteine, die auf der Oberfläche eines intakten Viruspartikels für das Immunsystem frei zugänglich sind, die Env-kodierten Glykoproteine. Ansonsten besteht die Oberfläche aus der Lipidmembran infizierter Wirtszellen. Dies macht die viralen Glykoproteine wie gp130 zu einem naheliegenden Ziel für Antikörper. Die in Kapitel 4.1.2 dargestellten Ergebnisse belegen, dass die Antikörperantwort gegen gp130 gleichzeitig mit der Spitzenvirämie exponentiell anstieg und beim Großteil der Tiere über den Infektionsverlauf auf einem konstant hohen Niveau lag. Umso erstaunlicher ist es, dass die Höhe der anti-Env-Antikörper im Gegensatz zu den antiGag-Antikörpern kaum eine prognostische Aussage hat. Letztere fielen bei den Progressoren im Krankheitsverlauf ab, während die Langzeitüberlebenden schon zur 25. Woche nach SIVInfektion signifikant höhere anti-p27-Antikörpertiter aufwiesen. Dieser Sachverhalt wurde sowohl für das SIV-Makakenmodell (DYKHUIZEN et al., 1998; SMITH et al., 1999) als auch für die HIV-Infektion des Menschen beschrieben (TEEUWSEN et al., 1991; FENOUILLET et al., 1993; ZWART et al., 1994; HOGERVORST et al., 1995; BINLEY et al., 1997). Die auch in der vorliegenden Arbeit beobachtete negative Korrelation von anti-p27Antikörpern und Viruslast legt die Vermutung nahe, dass anti-Gag-Antikörper durch Diskussion S e i t e | 105 zirkulierendes Gag-Protein gebunden werden und deshalb nicht mehr nachweisbar sind. Allerdings ist nicht nachvollziehbar, warum anti-Env-Antikörper davon nicht betroffen sind. Zudem wurde diese Theorie von FENOUILLET et al. (1993) widerlegt, die beweisen konnten, dass durch eine in vitro-Säuredissoziation der Immunkomplexe aus dem Plasma HIV-Infizierter die Menge von p24-Protein nicht stieg. Ungeklärt ist, ob die Höhe der anti-Gag-Antikörpertiter eine Ursache für die unterschiedlichen Krankheitsverläufe ist, oder ob sie lediglich eine Begleiterscheinung der stärker oder schwächer ausgeprägten Beeinträchtigung des Immunsystems darstellt. Letztere These wird z.B. durch die Beobachtungen von DYKHUIZEN et al. (1998) und STEGER et al. (1998) unterstützt, die bei rapid progressors im SIV-/SHIV-Modell eine positive Korrelation zwischen niedrigen Antikörpertitern und dem Verlust von B-Zellen feststellen konnten. Da für die in Kapitel 4.1 untersuchten Langzeitüberlebenden und Progressoren retrospektiv keine Analysen der B-Zellzahlen durchgeführt werden konnten, kann hier nur versucht werden, anhand der Daten der 12 Tiere aus Kapitel 4.3 eine Aussage zu treffen. Das Tier 2272 fiel durch deutlich niedrigere Antikörpertiter und eine persistierende hohe Viruslast auf. Obwohl der relative Anteil der CD20+-Lymphozyten dieses Tieres nach SIV-Infektion deutlich unter dem Mittelwert der Kohorte lag, konnte insgesamt keine Korrelation zwischen Viruslast und B-Zellen hergestellt werden (s. Kapitel 4.3.5). Die funktionelle Untersuchung der B-Zellen mittels der ELISpot-Technik zeigte allerdings (s. Kapitel 4.3.4), dass zum Zeitpunkt der setpoint-Virämie der Anteil p27- und gp130-spezifischer Antikörper-sezernierenden Zellen hochsignifikant negativ mit der Viruslast korreliert war. Da aber der Anteil der CD4+-TZellen ebenfalls deutlich negativ mit der der Viruslast korreliert, stellt sich die Frage, ob die niedrigen Antikörpertiter bzw. die beeinträchtigte Produktion von Antikörpern durch die BZellen der Progressoren auf mangelnde T-Zellhilfe zurückzuführen ist (BINLEY et al., 1997; BONSIGNORI et al., 2009). Ein Paradoxon scheint jedoch zu sein, dass nur die Antikörperantwort gegen Gag beeinträchtigt ist, denn gp130 erfüllt eigentlich nicht die Kriterien eines typischen, T-Zell-unabhängigen Antigens (s. Kapitel 2.1.2.1). Möglicherweise könnte aber die hohe Dichte von dem in trimerischer Form auf Virionen und infizierten Zellen präsentierten gp130 zu einer ähnlichen Kreuzvernetzung von BZellrezeptoren führen, die sonst nur von bakteriellen Polysacchariden bekannt ist (BINLEY et al., 1997). In jüngerer Vergangenheit konnten zwei Arbeitsgruppen Hinweise für die Wirkung 106 | S e i t e Diskussion des HIV-/SIV-Glykoproteins als T-Zell-unabhängiges Antigen liefern: HIV-1 gp120 konnte in vitro direkt an CD40 auf B-Zellen binden und so Proliferation und Ig-Klassenwechsel hervorrufen (HE et al., 2006) und in einem Transfermodell mit immundefizienten Mäusen wurde deutlich, dass zwar die Primärantwort gegen SIVgag T-Zell-abhängig war, GedächtnisB-Zellen hingegen durch gp130 zur Differenzierung in spezifische ASC angeregt werden konnten, ohne dass aktivierende Signale von T-Helferzellen nötig waren (NABI et al., 2012). Selbst wenn diese Argumente auf eine unterschiedliche Regulierung der humoralen Immunantwort gegen die Kapsid- und Hüllproteine hindeuten, sagen sie per se nichts über die Qualität der Antikörper aus. Die ELISA-Technik erlaubt lediglich eine Quantifizierung der bindenden Antikörper. Neben der später diskutierten Überprüfung der Neutralisationsfähigkeit (Kapitel 4.1.3) sind weitere funktionale Test wie die Untersuchung auf Antikörper-vermittelte Zelltoxizität oder Antikörper-vermittelte virale Inhibition wünschenswert, um die Dimension der humoralen Immunantwort der Langzeitüberlebenden genauer zu beschreiben. Ein weiteres Indiz für die Relevanz der Immunantwort gegen SIVgag bei der Kontrolle der Virusreplikation liefert die Beobachtung mit der sich der folgende Abschnitt auseinandersetzt. 5.2.2 Die SIV-spezifische IFN-γ-Sekretion von T-Zellen der Langzeitüberlebenden gegen SIVgag lag signifikant über der von Progressoren Während die IFN-γ-Ausschüttung als Reaktion auf die Stimulation mit SIVenv-Peptiden in beiden Tiergruppen als minimal bezeichnet werden konnte, lag die T-Zellantwort gegen SIVgag-Peptide deutlich höher und stieg im Infektionsverlauf bei den Langzeitüberlebenden über das Niveau der Progressoren. In einer umfangreichen Studie, die über 500 HIV-infizierte Individuen ohne HAARTTherapie einschloss, stellte sich ebenfalls heraus, dass die Anzahl IFN-γ-sezernierender Zellen nach einer Stimulation mit Gag-Peptiden mit einer effektiveren Kontrolle der Virusreplikation assoziiert war, Env-spezifische T-Zellen hingegen ineffizient zu sein schienen (KIEPIELA et al., 2007). Die Autoren stellten die Hypothese auf, dass eine escapeMutation in den hoch konservierten Gag-Epitopen eher zu einer Beeinträchtigung der viralen Replikationsfähigkeit führt, während Mutationen der sehr variablen Env-Epitope nicht zu Diskussion S e i t e | 107 Lasten der viralen Fitness fallen. Dies würde den Nutzen einer hohen Gag-spezifischen TZellantwort in der chronischen Phase erklären. In einer in vitro-Studie mit anschließender Übertragung auf das SIV-Makakenmodell konnte darüber hinaus bewiesen werden, dass Gag-Epitope des eindringenden Virions 2-6 Stunden nach Infektion der Zielzelle auf deren Oberfläche als MHC-Komplex präsentiert wurden und CD8+T-Zellen aktivierten. Indessen wurden Env-Epitope frühestens nach 18 Stunden erkannt. Im Hinblick auf den ca. 24 Stunden dauernden Replikationszyklus von HIV/SIV ist diese Entdeckung insofern bemerkenswert, als dass Gag-Epitope vor der Integration des viralen Genoms in die Wirts-DNA eine Reaktion der zytotoxischen T-Zellen auslösten, Env-Epitope aber erst neu synthetisiert werden mussten und CD8+T-Zellen eine Freisetzung von infektiösen Viruspartikeln nicht mehr verhindern konnten (SACHA et al., 2007). 5.2.3 Deutliche positive Korrelation zwischen viralen RNA-Kopien im Plasma und Höhe der Neutralisationstiter Obwohl es Berichte über den günstigen Effekt neutralisierender Antikörper während der HIV/SIV-Infektion gibt (BEER et al., 1996; SCHMITZ et al., 2003; MILLER et al., 2007), konnten andere Studien, genau wie die vorliegende Arbeit, keinen Zusammenhang zwischen der Höhe der Neutralisationstiter und Kontrolle der Viruslast bzw. milderem Krankheitsverlauf feststellen (REIMANN et al., 1994). Im Gegenteil, die Titer der in Kapitel 4.1.3 untersuchten Progressoren stiegen mit voranschreitender Infektionsdauer an und lagen letztlich über denen der Langzeitüberlebenden. Ähnliche Beobachtungen gab es bei HIVinfizierten Menschen (BAILEY et al., 2006; PEREYRA et al., 2008; PIANTADOSI et al., 2009). Somit stellt sich die Frage, ob die Bildung von nAk überhaupt einen Nutzen für das betroffene Individuum birgt, oder ob sie angesichts der signifikanten Korrelation zwischen Neutralisationstitern und Viruslast gar schädlich ist. PIANTADOSI et al. (2009) bewiesen aber anhand statistischer Tests, dass, zumindest in der von ihnen untersuchten Kohorte, Neutralisationsvermögen und Krankheitsverlauf zwar beide von der Viruslast beeinflusst wurden, voneinander jedoch unabhängig waren. Vielmehr scheint eine hohe Virusreplikation erforderlich zu sein, um zu einer entsprechenden Stimulation und Reifung von B-Zellen und daraus resultierenden Antikörpertitern zu führen. Durch die Bildung von escape-Mutationen entstehen bei Progressoren zudem neutralisierende Antikörper gegen ein breites Repertoire von Isolaten, das sich stetig verändert (SATHER et al., 2009). Dies deutet darauf hin, dass 108 | S e i t e Diskussion nAk sehr wohl eine Bürde für das Virus darstellen, letzteres aber aufgrund der hohen Mutationsrate des Env-Proteins der Immunantwort des Wirtes immer einen Schritt voraus ist (WILLEY u. AASA-CHAPMAN, 2008). So konnte hitzeinaktiviertes Plasma von Makaken, die sich in der Phase der set-point-Virämie befanden, die Infektiösität von Virionen, die vor der Spitzenvirämie aus den selben Tieren isoliert wurden, senken und eine Infektion von naiven Tieren mit dem Isolat verhindern. Die Autoren sehen neutralisierende Antikörper im Plasma als eine mögliche Ursache an (MA et al., 2009). In Experimenten zweier anderer Arbeitsgruppen wurden die B-Zellen von Rhesusaffen vor der Belastungsinfektion mit SIV temporär depletiert (SCHMITZ et al., 2003; MILLER et al., 2007). Obwohl die von SCHMITZ et al. (2003) beschriebenen Tiere deutlich niedrigere Antikörpertiter als die Kontrollgruppe aufwiesen, unterschied sich der typische Verlauf der Kurve der Plasma-SIVRNA-Spiegel nicht voneinander. Neutralisierende Antikörper schienen demnach keinen Einfluss auf die Kontrolle der Virusreplikation während der Spitzenvirämie zu haben. 28 Tage nach Infektion stiegen die Titer der nAk gleichzeitig mit dem Wiederauftreten von BZellen und korrelierten invers mit der Viruslast. Bei MILLER et al. (2007) offenbarte sich darüber hinaus auch während der akuten Phase der Infektion ein deutlicher Vorteil durch das Vorhandensein von SIV-spezifischen Antikörpern. In der vorliegenden Dissertation wurde lediglich das neutralisations-sensitive SIVmac32H im Neutralisationstest verwendet, ein ex vivo-Isolat aus einem Tier, welches ursprünglich mit SIVmac251 infiziert worden war (CRANAGE et al., 1990). Es konnten demnach keinerlei Aussagen über die Breite der Antikörperantwort der Langzeitüberlebenden gemacht werden. In der Literatur finden sich diesbezüglich widersprüchliche Aussagen. Viele Wissenschaftler gehen davon aus, dass die niedrige Virusreplikationsrate bei Langzeitüberlebenden nicht ausreicht, um die Bildung potenter, kreuzreagierender neutralisierender Antikörper zu stimulieren (DORIA-ROSE et al., 2008; PEREYRA et al., 2008; DORIA-ROSE et al., 2009; LAMBOTTE et al., 2009). Andere Kohorten von Langzeitüberlebenden wiesen ein breites Spektrum von nAk auf (PILGRIM et al., 1997; Q. WANG et al., 2008; MAHALANABIS et al., 2009; MEDINA-RAMIREZ et al., 2011). Angesichts der Tatsache, dass in den genannten Studien zum Teil Langzeitüberlebenden unterschiedliche gewählt wurden, Kriterien die für die Statusdefinition Neutralisationsfähigkeit in Bezug von auf unterschiedliche Virusvarianten getestet wurde und die Individuen darüber hinaus mit Diskussion S e i t e | 109 unterschiedlichen Stämmen infiziert waren, sind die Ergebnisse schwer vergleichbar. Eine interessante Theorie ist außerdem, dass möglicherweise bei Progressoren mit HIVassoziierten B-Zellabnormalitäten starke Antigenstimuli in Form von erheblicher Virusreplikation für hohe und umfassende Neutralisationstiter notwendig sind. Bei Langzeitüberlebenden oder Individuen unter HAART mit nahezu unbeeinträchtigtem Immunsystem könnten diese unter Umständen gar nicht erforderlich sein (MEDINARAMIREZ et al., 2011). Bei der Analyse der Zusammenhänge zwischen bindenden und neutralisierenden Antikörpern in Kapitel 4.1.3 ergab sich angesichts der Tatsache, dass nAk gegen das Hüllprotein gerichtet sind, eine scheinbare Diskrepanz: In der frühen chronischen Phase 25 Wochen nach SIVInfektion bestand eine starke positive Korrelation zwischen den bindenden Antikörpertitern gegen gp130 und den neutralisierenden Antikörpertitern, die später aber abnahm und schließlich völlig verschwand. In Kapitel 4.3.3 unterschied sich das Tier 2272, welches vorher durch deutlich niedrigere ELISA-Titer aufgefallen war, bezüglich der Neutralisationstiter nicht von den übrigen. An diesem Punkt werden erneut die Grenzen der Testsysteme erkennbar. Manche nAk binden nur an die trimerische Form des Hüllproteins oder an den transmembranen Teil, während im ELISA nur Monomere verwendet werden (MOORE u. SODROSKI, 1996; WALKER et al., 2009). So kann Plasma, wie in Kapitel 4.3.3 dargestellt, trotz niedriger bindender Antikörper gegen Env dennoch Neutralisationskapazität besitzen. Umgekehrt sind viele Antikörper gegen nicht-funktionelle Teile des Glykoproteins gerichtet und haben somit keinen protektiven Effekt (WILLEY u. AASA-CHAPMAN, 2008). 5.2.4 Beim Eintritt in die Plateauphase der SIV-Infektion bestand eine negative Korrelation zwischen Viruslast und SIV-spezifischen IgG-sezernierenden Zellen Anhand der ELISpot-Technik konnten, wie in Kapitel 4.3.4 beschrieben, schon zwei Wochen nach SIV-Infektion erste Zellen, die spontan p27- und gp130-spezifisches IgG sezernierten, detektiert werden. Während der Spitzenvirämie bestand allerdings, genauso wie in der späteren chronischen Phase 40 Wochen nach Infektion, kein messbarer Zusammenhang mit der entsprechenden Viruslast. Zum Zeitpunkt der set-point-Virämie acht Wochen nach Infektion wiesen Tiere mit niedrigerer Viruslast jedoch deutlich höhere Anteile an SIVspezifischen ASC auf. Auch in diesem Fall muss, genau wie in den vorhergehenden Kapiteln, Ursache und Wirkung diskutiert werden. Denkbar ist auch in diesem Fall eine 110 | S e i t e Diskussion Beeinträchtigung der Funktion des B-Zellsystems durch mangelnde T-Zellhilfe bei Tieren mit hoher Virusreplikation. Allerdings ergaben sich keine Hinweise auf die in Abschnitt 4.2.1 angesprochene unterschiedliche Steuerung von Antikörperantworten gegen SIVgag und SIVenv, denn die mittleren Anteile der p27- und gp130-spezifischen ASC unterschieden sich nicht signifikant voneinander. Da außerdem die ASC in der 8. Woche negativ mit der Viruslast 40 Wochen nach Infektion korrelierte, ist eine zumindest unterstützende Wirkung der Antikörper auf die Kontrolle der Virusreplikation denkbar. In einer kürzlich veröffentlichten Studie wurde ebenfalls geschildert, dass höhere Anteile von Env-spezifischen ASC an den IgG-sezernierenden Zellen nach der Belastungsinfektion mit einer niedrigeren SIV-RNA-Last in den Wochen 8-36 einhergingen (BROCCA-COFANO et al., 2011). Im Gegensatz zur vorliegenden Arbeit wurden Rhesusaffen untersucht, die zuvor immunisiert worden waren, und die eingesetzten PBMC in vitro stimuliert. Es handelt sich in diesem Fall also um einen Nachweis von SIV-spezifischen Gedächtnis-B-Zellen. Nichtsdestotrotz stärkt diese Beobachtung die These eines positiven Effekts durch spezifische B-Zellen. Es wäre interessant gewesen, wenn jene Arbeitsgruppe zusätzlich die spontan Antikörper-sezernierenden Zellen untersucht hätte. 5.2.5 Keine Korrelation zwischen Titern der bindenden Antikörper und SIVspezifischen ASC Es gibt zwei Untersuchungen an gp120-immunisierten Freiwilligen und HIV-Infizierten (TEEUWSEN et al., 1991; BONSIGNORI et al., 2009), in welchen der prozentuale Anteil an HIV-spezifischen ASC mit den im ELISA gemessenen Antikörpertitern korrelierte, wobei die Korrelation in letztgenannter nur knapp statistische Signifikanz erreichte. Im Gegensatz dazu konnte in der vorliegenden Arbeit kein messbarer Zusammenhang bezüglich der bindenden Antiköper gegen p27 und gp130 hergestellt werden, weder mit (bei den Langzeitüberlebenden) als auch ohne vorherige polyklonale Stimulation (bei den in Abschnitt 4.3 untersuchten Tieren). Andere Studien bestätigen diese Ergebnisse sowohl für HIV (MORRIS et al., 1998; GUAN et al., 2009) als auch SIV (BROCCA-COFANO et al., 2011). Neben dem sehr heterogenen Probenmaterial und den unterschiedlichen Nachweismethoden für Antigen-spezifische B-Zellen in den genannten Publikationen, die einen direkten Vergleich der Ergebnisse kaum zulassen, gibt es mehrere logische Erklärungsansätze, die Diskussion S e i t e | 111 diesen scheinbaren Widerspruch auflösen: Einer davon ist die Grundlage der ELISpotTechnik. Mittels dieses Nachweissystems werden Antikörper-sezernierende Zellen quantifiziert, jeder Punkt auf der Membran entspricht einer Zelle. Es ist aber nicht möglich, Rückschlüsse auf die Menge der abgegebenen Immunglobuline zu ziehen. Wenige Zellen können also eine große Menge Antikörper abgeben et vice versa. Werden die eingesetzten Zellen darüber hinaus in vitro stimuliert, differenzieren memory-B-Zellen erst dann zu Plasmazellen, während sie im Körper gar keine Antikörper abgaben und somit nicht zu den im Plasma gemessenen Titern beitragen konnten. Zudem wurden nur die Anteile der ASC im Blut untersucht. Eine Vielzahl von Plasmazellen befindet sich aber in anderen Kompartimenten wie Lymphknoten, Milz oder Knochenmark, während sich die gebildeten Immunglobuline über das Blut im Körper verteilen (SLIFKA et al., 1995; AHMED u. GRAY, 1996; RADBRUCH et al., 2006; MAMANI-MATSUDA et al., 2008). Im folgenden Kapitel soll ausführlich auf diesen Sachverhalt eingegangen werden. 5.2.6 Ohne vorherige Stimulation konnten keine SIV-spezifischen IgG-sezernierenden Zellen im Blut der Langzeitüberlebenden nachgewiesen werden. Trotz deutlich messbarer Antikörpertiter in Serum oder Plasma traten bei keinem der untersuchten Langzeitüberlebenden ohne Stimulation Reaktionen in den mit p27 oder gp130 beschichteten Kavitäten der Mikrotiterplatte auf, die die Menge der als Hintergrund betrachteten unspezifischen Reaktionen in der Negativkontrolle überstiegen. Es kann also davon ausgegangen werden, dass zu den Zeitpunkten der Blutentnahmen im Blut der Langzeitüberlebenden keine SIV-spezifischen Plasmazellen vorkamen. Auch BROCCACOFANO et al. (2011) beschrieben dies für drei Langzeitüberlebende im SIVMakakenmodell und MORRIS et al. (1998) für humane LTNP. Darüber hinaus belegten deren Untersuchungen, dass die Frequenz Env-spezifischer ASC bei kontinuierlicher HAART sank. Diese Beobachtungen liefern Beweise für zwei Thesen. Erstens scheint für das Vorhandensein von Plasmazellen im Blut ein gewisser Antigenstimulus notwendig zu sein. Erst dadurch können naive B-Zellen oder Gedächtnis-B-Zellen zur Differenzierung angeregt werden. Eine chronische Generierung von Plasmazellen durch ungerichtete Zytokine, die eine Aktivierung von Gedächtnis-B-Zellen unabhängig vom Antigen bewirkt (BERNASCONI, 2002), ist zumindest bei den nahezu physiologischen Verhältnissen in Langzeitüberlebenden offensichtlich nicht der Fall. Zweitens werden die Antikörpertiter augenscheinlich über Jahre 112 | S e i t e Diskussion hinweg durch Plasmazellen konstant gehalten, die sich in anderen Kompartimenten des Körpers als dem Blut befinden müssen. Dass diese extrem langlebig sein können, wird anhand des Beispiels von Pocken deutlich. Obwohl Pocken seit 1980 als ausgerottet gelten und deswegen ein späterer Antigenkontakt bei geimpften Personen ausgeschlossen werden kann, wiesen diese Individuen z.T. über 40 Jahre nach der letzten Impfung noch messbare Antikörpertiter auf (CROTTY et al., 2003). Für die Theorie einer Antigen-unabhängigen Persistenz der langlebigen Plasmazellen sprechen weitere Untersuchungen: In einem Mausmodell konnten von zwei Arbeitsgruppen durch den Transfer von Plasmazellen aus dem Knochenmark immunisierter Tiere spezifische Antikörpertiter in Antigen-naiven Tieren erzielt werden. Bei einem Transfer von Gedächtnis-B-Zellen hingegen war eine erneute Stimulation durch Antigen nötig, um spezifische Antikörper zu erzeugen (MANZ et al., 1998; SLIFKA et al., 1998). Darauf, dass Gedächtnis-B-Zellen nicht für die Erhaltung der langlebigen Plasmazellen erforderlich sind, deuten Studien an B-Zell-depletierten Mäusen und Menschen hin (AHUJA et al., 2008; MAMANI-MATSUDA et al., 2008). Obwohl durch die Behandlung mit anti-CD20-Antikörpern alle B-Zellstufen mit Ausnahme der Plasmazellen, bei denen das Oberflächenantigen CD20 mit ihrer terminalen Differenzierung verschwindet, verloren gingen, blieben Antikörpertiter erhalten. Leider war es in der vorliegenden Arbeit nur möglich, das Vorhandensein von Antikörpersezernierenden Zellen im Blut (und als Korrelat der mukosalen Immunantwort in der BAL) zu untersuchen. Zu den Verhältnissen in Organen, die als Hauptsitz von Plasmazellen und Gedächtnis-B-Zellen angesehen werden, wie Knochenmark und Milz (RADBRUCH et al., 2006; MAMANI-MATSUDA et al., 2008), kann für Langzeitüberlebende im SIVMakakenmodell noch keine Aussage getroffen werden. Zusätzlich wäre eine Analyse von anderen immunologisch wichtigen Organen wie Lymphknoten oder Darmmukosa aufschlussreich. Zusammengefasst legen die genannten Ergebnisse die Schlussfolgerung nahe, dass im Blut zirkulierende ASC keinen oder nur einen geringen Beitrag zu der Produktion von spezifischen Antikörpern während der HIV-/SIV-Infektion von Langzeitüberlebenden leisten. Das Erscheinen von größeren Mengen ASC im Blut könnte sogar nur eine pathologische Konsequenz der Virusinfektion sein (MORRIS et al., 1998). Diskussion S e i t e | 113 5.2.7 Der Anteil SIVenv-spezifischer Gedächtnis-B-Zellen im Blut der Langzeitüberlebende entsprach dem 6,5fachen des Anteils der SIVgagspezifischen Gedächtnis-B-Zellen Zur Analyse der Gedächtnis-B-Zellen im Blut der Langzeitüberlebenden wurde das Stimulationsprotokoll der Firma Mabtech, die das verwendete ELISpot-Testsystem vertreibt, übernommen (s. Kapitel 3.5.3 und Anhang 11.4). Da die Differenzierung von memory-BZellen hin zu Plasmazellen ein komplexer Vorgang ist, der durch eine Vielzahl von Faktoren gesteuert wird, gibt es in der Literatur diverse Stimulationsprotokolle. Im Bezug auf die HIV/SIV-Forschung häufig verwendet wurden einzeln oder in wechselnder Kombination und unterschiedlichen Dosierungen: als polyklonale Stimulatoren PWM, CpG- Oligodesoxynukleotide und/oder Staphylokokkus aureus Protein A Cowan; die Zytokine IL-4, IL-10, IL-15 und/oder IL-21, sowie CD40-L um T-Zellhilfe zu simulieren (FONDERE et al., 2003; BONSIGNORI et al., 2009; BUSSMANN et al., 2009; DOUAGI et al., 2010; SUNDLING et al., 2010; BROCCA-COFANO et al., 2011). Es ist nicht auszuschließen, dass so Gedächtnis-B-Zellen von unterschiedlicher Spezifität und Menge zur Differenzierung angeregt wurden. Eine vergleichende Prüfung der verschieden Protokolle überstieg den Rahmen dieser Doktorarbeit, so dass die im Folgenden diskutierten Ergebnisse immer unter diesem Vorbehalt zu bewerten sind. Eine zusätzliche Varianz stellt die Dauer der in vitro-Stimulation dar. Vorversuche für die Etablierung des Tests zeigten, dass nach 72 Stunden Stimulation von PBMC mit R848 und IL-2 die Zahl der ASC ein Maximum erreichte, während sie bei einer längeren Stimulation von 4 oder 5 Tagen wieder sank und die Vitalität der PBMC insgesamt nachließ. Diese Vorgehensweise war konform mit der zweier anderer Arbeitsgruppe, die den B-Zell-ELISpot im SIV-Makakenmodel anwendeten (DOUAGI et al., 2010; BROCCA-COFANO et al., 2011), es wurden aber auch 4 Tage (SUNDLING et al., 2010) stimuliert, bzw. 5 (FONDERE et al., 2003) oder 6 Tage bei humanen PBMC (BONSIGNORI et al., 2009; BUSSMANN et al., 2009). Der Anteil von p27-spezifischen ASC an den Gesamt-IgG-sezernierenden Zellen lag bei den Langzeitüberlebenden nach in vitro-Stimulation im Durchschnitt bei 0,8%. Da es keine veröffentlichten Studien gibt, die p27-spezifische memory-B-Zellen im SIV-Makakenmodel untersuchten, kann nur ein Vergleich aus der humanmedizinischen Forschung herangezogen 114 | S e i t e Diskussion werden. Eine Gruppe von 10 HIV-Infizierten mit einer Viruslast unter 1x104 HIV-RNAKopien/ml Plasma und CD4+-Zellzahlen über 350 pro µl, insofern also ähnlich den langzeitüberlebenden Affen, wies im Durchschnitt einen Anteil von 0,5% p24-spezifischen Gedächtnis-B-Zellen auf (BUSSMANN et al., 2009) und lag somit in einem nahezu identischen Bereich. Mit 5,2% lag der Anteil gp130-spezifischer ASC bei den Langzeitüberlebenden 6,5mal so hoch wie der der p27-spezifischen. Bezüglich Env-spezifischer Gedächtnis-B-Zellen ergaben Literaturrecherchen mehr Vergleichswerte. Bei drei Rhesusaffen, die in der Lage waren, die SIV-Replikation effizient zu kontrollieren, entsprachen sie ebenfalls einem Anteil von 5,4% der gesamten IgG-memory-B-Zellen (BROCCA-COFANO, 2011). Zwei Vakzineexperimente erzielten hingegen sehr verschiedenartige Daten: SUNDELING et al. (2010) berichteten, dass der Anteil Env-spezifischer Gedächtniszellen nach Abschluss der Immunisierungsphase zwischen 10-20% lag, während DOUAGI et al. (2010) mit dem von ihnen verwendeten Impfstoff nur sehr niedrige Spiegel erzeugen konnten (0,1%). Leider wurden die GedächtnisB-Zellen in beiden Fällen nicht nach einer Belastungsinfektion mit SIV untersucht. Bei HIVinfizierten Menschen sind ebenfalls Anteile von unter einem Prozent beschrieben (BONSIGNORI et al., 2009; BUSSMANN et al., 2009). Dass verschiedene Immunisierungsstrategien die Bildung von Gedächtnis-B-Zellen unterschiedlich stark fördern, ist nicht weiter verwunderlich. Warum aber der prozentuale Anteil Env-spezifischer Gedächtnis-B-Zellen bei Rhesusaffen um den Faktor 10 höher ist als bei Menschen, die die Virusreplikation ebenfalls effizient kontrollieren, kann nur vermutet werden. BUSSMANN et al. (2009) bewiesen, dass Progressoren mit hoher HIV-Last und niedrigen CD4+-Zellzahlen extrem wenig gegen das Hüllprotein gerichtete memory-B-Zellen hatten (0,002% im Vergleich zu 0,111% bei den Kontrollierenden). Sie führten dies auf den Verlust der HIV-spezifischen T-Helferzellen zurück, denn unter HAART normalisierte sich bei ihreen Untersuchungen zwar der Gedächtnis-B-Zellspiegel insgesamt, HIV-spezifische waren aber irreversible verloren. Denkbar wäre also, dass die Rhesusaffen in der akuten Phase der Infektion einen schwächeren Abfall der CD4+-Zellzahl und/oder der Gedächtnis-B-Zellen erlitten als die HIV-Infizierten in der untersuchten Kohorte. Ebenfalls möglich wäre, dass die T-Zellhilfe der langzeitüberlebenden Rhesusaffen die Bildung des immunologischen Diskussion S e i t e | 115 Gedächtnisses auf welche Art auch immer effizienter unterstützt. Eine gänzlich andere Herangehensweise eröffnet eine zweite Studie mit HIV-Infizierten unter anti-retroviraler Therapie (FONDERE et al., 2003). Diese ergab, dass die Zahl HIV-spezifischer GedächtnisB-Zellen sogar sank, nachdem die Viruslast unter die Nachweisgrenze gefallen war. Der Autor schlussfolgert, dass HIV-spezifische Gedächtnis-B-Zellen eher kurzlebiger Natur sind und ein gelegentlicher Antigenstimulus notwendig ist, um einen gewissen Spiegel zu halten. Wahrscheinlich entspricht eine Kombination aus beidem der Wirklichkeit. Führt man sich die in Kapitel 5.2.1 erläuterte Theorie erneut vor Augen, nach der die Gagspezifische humorale Immunantwort stärker auf T-Zellhilfe angewiesen ist, während EnvProteine eventuell in einer eher polyklonalen Weise die Antikörperbildung stimulieren, könnte dies die Tatsache erklären, weswegen die Langzeitüberlebenden deutlich höhere Anteile gp130- als p27-spezifischer Gedächtnis-B-Zellen besitzen. Durch T-Zellverluste während der akuten Phase der Infektion wird die Bildung von Gag-spezifischen memory-BZellen gravierender gestört, stabilisieren sich die CD4+-Zahlen durch Kontrolle der Viruslast wieder, fehlt der Anreiz durch das Antigen. 5.2.8 Der Nachweis von SIV-spezifischen ASC in der BAL sowie der Nachweis von IgA-sezernierenden Zellen im Blut der Langzeitüberlebenden fielen negativ aus Nicht nur im Blut und im Knochenmark, sondern auch in der Mukosa kommen Plasma- und Gedächtnis-B-Zellen vor, die die Schleimhautbarriere durch die Sekretion von Immunglobulinen (hauptsächlich IgA) vor dem Eindringen von Mikroorganismen schützen (MEDINA et al., 2003; PEREZ-ANDRES et al., 2010). Um die SIV-spezifischen ASC in mukosalen Kompartimenten zu evaluieren, wurde die BAL als minimal invasives Verfahren ausgewählt. Obwohl der Nachweis von Gesamt-IgG- und -IgA-sezernierenden Zellen unter den mononukleären Zellen der BAL gelang, konnten keine Env- oder Gag-spezifischen ASC detektiert werden, selbst wenn die im ELISpot eingesetzte Zellzahl im Vergleich zum Blut verdoppelt wurde. Zudem war die Vitalität der gewonnenen Zellen unter den angewendeten Kultur- und Stimulationsbedingungen schon nach 24 Stunden äußerst schlecht. Dies deutet daraufhin, dass technische Mängel für die negativen Ergebnisse verantwortlich sein könnten. Zwar konnten in der Literatur keine Hinweise auf das Vorkommen von HIV-/SIVspezifischen ASC in der BAL gefunden werden, auf der anderen Seite ist der Nachweis von SIV-spezifischen Antikörpern (HIDAJAT et al., 2009) und auch von B-Zellen 116 | S e i t e Diskussion (SCHULTHEISS et al., 2010) in der BAL prinzipiell möglich. Unter Umständen stellen mononukleäre Zellen aus der Lunge andere Ansprüche an die Kultivierung oder Stimulation. Leider konnte eine diesbezügliche Optimierung im Rahmen der Doktorarbeit nicht mehr durchgeführt werden. Im Blut der Langzeitüberlebenden traten sowohl vor als auch nach in vitro-Stimulation IgAsezernierende Zellen auf. SIV-spezifische ASC lagen allerdings, wenn vorhanden, unter dem Sensitivitätsbereich des Tests. Auch die Steigerung der eingesetzten Zellmenge erzielte keine Erfolge. Um auszuschließen, dass mangelnde Kreuzreaktivität der verwendeten Konjugate die Ursache war, wurde darüber hinaus alternativ ein anti-Rhesus-IgA-Konjugat getestet, dass allerdings keine Zunahme der gemessenen ASC verursachte. Infolgedessen wurde auf eine Analyse SIV-spezifischer IgA-sezernierender Zellen bei der Tiergruppe aus Abschnitt 4.3.4 verzichtet. In einer kürzlich erschienenen Veröffentlichung (BROCCA-COFANO et al., 2011) gelang der Nachweis von SIVenv-spezifischen IgA-sezernierenden Zellen im Blut jedoch, sowohl bei drei untersuchten Langzeitüberlebenden als auch bei Tieren aus zwei Immunisierungsstudien. Gründe für diese divergenten Ergebnisse könnten entweder in der unterschiedlichen Ausführung des B-Zell-ELISpot oder tatsächlich in einer gesteigerten Frequenz der IgA-sezernierenden Zellen liegen. Für die Beschichtung der Platten wurde in der zitierten Studie anderes rekombinantes EnvAntigen verwendet, an das unter Umständen mehr Antikörper banden. Das Anti-IgAKonjugat war allerdings das gleiche, das auch in der vorliegenden Arbeit als Alternative verwendet wurde und zu keiner Verbesserung der Sensitiviät führte. Es ist nicht auszuschließen, dass durch die Stimulation mit CpG Oligonukleotiden, CD40-L und IL-21 mehr IgA-Gedächtnis-B-Zellen zur Differenzierung angeregt worden sein könnten, als durch die in dieser Arbeit verwendete Stimulation mit R848 und IL-2. Ein Mechanismus, der dafür verantwortlich wäre, ist jedoch nicht offensichtlich. Die Langzeitüberlebenden aus dieser Dissertation stammten aus Vakzineexperimenten, die eine mukosale Immunisierung in Form einer oropharyngealen Sprayapplikation mit der intramuskulären Injektion verglichen (SCHULTE et al., 2009), während die von BROCCA- Diskussion S e i t e | 117 COFANO et al. (2011) untersuchten Tiere alle oral oder nasal immunisiert wurden. Unabhängig von der Route und der applizierten Vakzine fiel der Nachweis SIV-spezifischer IgA-Gedächtnis-B-Zellen aber bei keinem der 13 Tiere dieser Arbeit positiv aus. Es kann also auch nicht davon ausgegangen werden, dass eine mukosale Immunisierung gegenüber einer systemischen Applikation generell zu einer Steigerung der IgA-memory-B-Zellen führt. Bei einer der beiden von BROCCA-COFANO et al. (2011) analysierten Immunisierungsstudien wurde die anschließende Belastungsinfektion in Form einer wiederholten, rektalen Applikation von niedrigen Virusdosen durchgeführt, die andere Tiergruppe wurde intravenös infiziert. Nach der rektalen repeated-low-dose-Infektion sanken die Titer IgA-sezernierender Zellen im Blut im Vergleich zu den Präinfektionswerten, was von den Autoren auf einer Abwanderung der Zellen in den Darm, der Viruseintrittspforte, zurückgeführt wurde. Bei den i.v.-infizierten Tieren hingegen stiegen die IgA-sezernierenden Zellen im Blut an. Auch 11 der 13 in dieser Doktorarbeit untersuchten Langzeitüberlebenden wurden über eine mukosale Route infiziert. Da kein Material aus der Immunisierungsphase für den B-Zell-ELISpot vorlag, konnten keine Aussagen darüber getroffen werden, ob evtl. vorher SIV-spezifische IgA-sezernierende Zellen in messbaren Mengen vorhanden waren und diese erst nach der Infektion unter die Nachweisgrenze des Testsystems fielen. Für künftige Untersuchungen sollten deshalb zum einen Optimierungen der Kultivierungsbedingungen für mononukläre Zellen aus der BAL angestrebt werden, um die B-Zellantwort in der Lunge charakterisieren zu können. Zum anderen sollten unterschiedliche Stimulationsprotokolle vor allem im Hinblick auf IgA-sezernierende Zellen getestet werden. Wenn dazu Proben von Tieren zur Verfügung stünden, die sowohl i.v. als auch mukosal infiziert wurden, würde das die Aussagekraft der Analysen noch verbessern. 5.3 Phänotypische Charakterisierung der B-Zellsubpopulationen im Blut 5.3.1 Eingeschränkte Vergleichbarkeit zwischen verschiedenen Studien Für die durchflusszytometrische Analyse der B-Zellen unter Einfluss der SIV-Infektion wurden Populationsdefinitionen aus der HIV-Forschung übernommen (MOIR u. FAUCI, 2009, s. auch Kapitel 2.1.2.1). Wegen der engen genetischen Verwandtschaft von Makaken und Menschen kann davon ausgegangen werden, dass die funktionellen und phänotypischen Eigenschaften weitestgehend übereinstimmen, einen Garant dafür gibt es jedoch nicht. 118 | S e i t e Diskussion So ist das signalvermittelnde Glykoprotein CD19 als Teil des B-Zell-Korezeptorkomplexes in der humanmedizinischen Forschung Mittel der Wahl für den durchflusszytometrischen Nachweis von B-Zellen. Die kommerziell erhältlichen Antikörper gegen humanes CD19 kreuzreagieren allerdings nicht verlässlich mit dem von Rhesusaffen (SOPPER et al., 1997). Deswegen wurde in der vorliegenden Arbeit, wie im SIV-Makakenmodell üblich, auf CD20 für die Definition von B-Zellen ausgewichen, welches ebenfalls auf allen Zellen der BZellreihe mit Ausnahme der Pro-B-Zellen und Plasmazellen vorkommt. In einer jüngeren Publikation wurde durch umfangreiche Untersuchungen bestätigt, dass CD27 in Kombination mit CD20 auch bei Rhesusaffen Gedächtniszellen beschreibt (KUHRT et al., 2011). CD20+CD27+Zellen sind, wie beim Menschen, größer als naive B-Zellen, weisen typische Aktivierungsmarker auf, zeigen durch somatische Hypermutation, dass Antigenkontakt stattgefunden hat und kommen im Blut von Neugeborenen in verschwindend geringen Anteilen vor. Allerdings konnte eine andere Veröffentlichung im gleichen Jahr deutlich machen, dass die Verwendung von CD21 für eine differenzierte Beschreibung von memory-B-Zellen notwendig ist (DAS et al., 2011). Nach Stimulation durch Antigen steigerten nur CD21+CD27+B-Zellen die Expression des Plasmazellmarkers CD138 signifikant im Vergleich zur unstimulierten Kontrolle. Zusätzlich entsprach die Menge an sezerniertem IgG der dreifachen der CD21-CD27+-Zellen, was dafür spricht, dass erstere eine stärkere Neigung zur Differenzierung in Antikörper-sezernierende Zellen hatten. Darüber hinaus wurden nur naive B-Zellen mit dem Phänotyp CD21+CD27- und die vermeintlichen Gedächtnis-B-Zellen mit dem Phänotyp CD21+CD27+ zur Proliferation angeregt. Obwohl also eine Definition von naiven und Gedächtnis-B-Zellen, die üblicherweise einzig über die Marker CD20 und CD27 geschieht, ungenau ist, wurde sie hier dennoch einer detaillierteren Beschreibung vorangestellt, um Vergleiche mit anderen Untersuchungen zu ermöglichen. Der Einsatz unterschiedlicher Oberflächenmarker und Gatingstrategien erschwert es, Referenzwerte zu finden. So ändern sich die prozentualen Anteile der CD20+-Zellen an den Lymphozyten in Abhängigkeit davon, wie letztere definiert werden. Eine Möglichkeit, dieses Diskussion S e i t e | 119 Problem zu umgehen, besteht in der Darstellung der absoluten Zellzahlen. Dafür werden die mittels Differentialblutbild erhobenen Lymphozytenzahlen als Berechnungsgrundlage für die entsprechenden Subpopulationen genutzt. Allerdings ist die inter- und intraindividuelle Variabilität recht hoch (REIMANN et al., 1994; SOPPER et al., 1997). Deswegen wurde in dieser Dissertation davon abgesehen. Auch das Probenmaterial beeinflusst die Daten. Vergleicht man die in Abschnitt 4.2.2 aus frischem EDTA-Vollblut gewonnenen Daten der SIV-negativen Tiere mit den retrospektiv analysierten PBMC, die den Tieren aus Kapitel 4.3.5 vor Infektion entnommen, isoliert und eingefroren wurden, fällt auf, dass die Anteile von CD20- und CD3-positiven Zellen an den Lymphozyten im zweiten Fall signifikant niedriger waren. Da hinsichtlich Haltung, Alter und vorhergehenden etwaigen Behandlungen keine Unterschiede zwischen den beiden Tierkohorten bestanden und die Verwendung von frischem Vollblut und frisch isolierten PBMC vergleichbare Ergebnisse erzielt (MORBACH et al., 2010), liegt die Ursache dafür wahrscheinlich in einem partiellen Verlust der Oberflächenmarker durch den Einfrier- und Tauprozess. Diese Gegebenheit muss bei der Gegenüberstellung von Daten aus der Literatur immer beachtet werden. SIV-negative Rhesusaffen besaßen einen höheren Anteil von CD27+B-Zellen als Menschen, nur ein kleiner Teil davon entsprach allerdings der Definition von ruhenden Gedächtnis-B-Zellen In frischem EDTA-Blut der SIV-negativen Tiere entsprachen im Mittel 30,2 ± 9,9% der B- 5.3.2 Zellen dem Phänotyp naiver B-Zellen (CD20+CD27-IgD+) und 58,1 ± 11,2% dem von Gedächtnis-B-Zellen (CD20+CD27+IgD+/-). Damit lagen die prozentualen Anteile der Gedächtnis-B-Zellen dieser Tierkohorte etwas höher als bei der von KUHRT et al. (2011) untersuchten (44,9%). Bei den nah verwandten Javaneraffen (Macaca fascicularis) hingegen sind Werte von 58-68% CD27+- und 11-68% CD27-B-Zellen beschrieben (PERUCHON et al., 2009; KLING et al., 2011). Abgesehen von potentiellen Unterschieden zwischen den beiden Spezies sind außerdem Faktoren wie Krankheitsvorgeschichte und Alter möglicherweise für diese Abweichungen verantwortlich (AGEMATSU et al., 1997; KLEIN et al., 1998; MORBACH et al., 2010). Während PERUCHON et al. (2009) und KLING et al. (2011) für ihre Studien adulte Tiere verwendeten, wie es auch in der vorliegenden Arbeit geschah, machten KUHRT et al. (2011) keine Angaben dazu. Bei erwachsenen Menschen 120 | S e i t e Diskussion werden niedrigere Mittelwerte von 28%-40% CD27+B-Zellen beschrieben. Ähnlich wie bei Rhesusmakaken ist der Anteil IgD-Gedächtnis-B-Zellen größer als der der IgDexprimierenden (AGEMATSU et al., 1997; KLEIN et al., 1998; MORBACH et al., 2010; PEREZ-ANDRES et al., 2010). Durch die differenziertere Aufteilung der B-Zellsubpopulationen unter Zuhilfenahme von CD21 und CD10 sank der Anteil naiver B-Zellen (CD10-CD21+CD27-IgD+) auf 20,8±8,8% der gesamten B-Zellen. Dieser Wert ist konform mit den beiden Studien, die ebenfalls die BZellen von Rhesusaffen auf eine ähnliche Weise charakterisierten, allerdings ohne die Immunglobulinklassen zu bestimmen (TITANJI et al., 2010; DAS et al., 2011). Bezüglich der ruhenden Gedächtnis-B-Zellen (CD10-CD21+CD27+IgD-/+) und der aktivierten B-Zellen (CD10-CD21-CD27+IgD-) bestanden ebenfalls Übereinstimmungen mit einer der beiden Veröffentlichungen (TITANJI et al., 2010). Die vorliegenden Untersuchungen ergaben Anteile von 5,9% ruhender Gedächtnis-B-Zellen und 34,7% aktivierter B-Zellen an den CD20+Lymphozyten, während die erwähnte Arbeitsgruppe 8% und 38% beschreibt. DAS et al. (2011), berichteten allerdings von 20% ruhender Gedächtnis-B-Zellen. Die veröffentlichten Ergebnisse bei Javaneraffen liegen mit 31,1% ruhende Gedächtnis-B-Zellen noch höher (VUGMEYSTER et al., 2004). Bei dem Vergleich mit Referenzwerten aus der Humanmedizin (TITANJI et al., 2010; PALLIKKUTH et al., 2011) fällt auf, dass zwar ein höherer Anteil der B-Zellen von Rhesusaffen den Oberflächenmarker CD27 trägt, allerdings nur ein relativ kleiner Teil dieser den ruhenden Gedächtniszellen zuzuordnen ist. Beim Menschen zeigt sich eine umgekehrte Tendenz: 18-25% der B-Zellen sind positiv für CD21 und CD27, während ein sehr kleiner Prozentsatz dem Phänotyp der aktivierten B-Zellen entspricht (3-8%). Eine Erklärung dafür könnte sein, dass bei Menschen aufgrund der höheren Lebensdauer mehr Gedächtnis-BZellen akkumulieren (KLEIN et al., 1998). Eine andere Hypothese ist, dass die B-Zellen von Rhesusaffen physiologischer Weise auf einem höheren Aktivierungsniveau als die der Menschen liegen. Die experimentelle Haltung von Primaten jedoch entspricht selbstverständlich nicht den natürlichen Verhältnissen. Haltungseinflüsse oder veränderter Keimdruck könnten also ursächlich sein. Um diese Theorie zu belegen, wären durchflusszytometrische Analysen von Rhesusaffen in freier Wildbahn oder zumindest in Diskussion S e i t e | 121 Außenhaltung mit annähernd gleichen Umweltreizen angebracht. Bei Rauchmangaben allerdings, die unter den gleichen Haltungsbedingungen wie Rhesusaffen gehalten wurden, machten aktivierte B-Zellen nur 20% der B-Zellen aus, während naive B-Zellen und exhausted tissue-like-B-Zellen (CD21-CD27-) mit jeweils knapp 40% die vorherrschenden Zelltypen sind (TITANJI et al., 2010). Speziesspezifische Unterschiede innerhalb der BZellsubpopulationen naiver Individuen sind demzufolge vorhanden und müssen bei der Interpretation von Daten und der Übertragung von Ergebnissen aus dem SIV-Modell auf den Menschen beachtet werden. 5.3.3 Der Anteil der B-Zellen an den Lymphozyten war bei chronisch infizierten Tieren mit mittlerer und hoher SIV-Beladung signifikant erhöht Obwohl verschiedene Publikationen einen Verlust von CD20+Lymphozyten im Rahmen einer HIV- (MOIR et al., 2008b; BUSSMANN et al., 2009; DORIA-ROSE et al., 2009) oder SIVInfektion (DYKHUIZEN et al., 1998; PERUCHON et al., 2009; KUHRT et al., 2010; TITANJI et al., 2010) beschreiben, andere diesbezüglich wiederum keine signifikanten Unterschiede feststellen konnten (DE MILITO et al., 2001; CHONG et al., 2004b; D'ORSOGNA et al., 2007), wiesen die hier untersuchten Tiere mit mittlerer und hoher Virusreplikation gesteigerte B-Zellprozentsätze auf. Auch in diesem Fall muss dem Umstand Rechnung getragen werden, dass in der vorliegenden Arbeit eine Analyse der relativen Anteile an den Lymphozyten durchgeführt wurde, während einige der obengenannten Veröffentlichungen die absoluten Zellzahlen vergleichen (DYKHUIZEN et al., 1998; DE MILITO et al., 2001; CHONG et al., 2004b; MOIR et al., 2008b; BUSSMANN et al., 2009; KUHRT et al., 2010). Durch den virusbedingten starken Abfall der T-Zellen steigt naturgemäß der prozentuale Anteil der übrigen Zellpopulationen, selbst wenn tatsächlich eine Reduktion der absoluten B-Zellzahlen eintritt, diese aber unterhalb der der T-Zellen liegt. Eine Studie mit SHIV-infizierten Javaneraffen verglich die prozentualen und absoluten Veränderungen der B-Zellen und belegte, dass die Prozentsätze der CD20+-Zellen ein Jahr nach Infektion ihre Ausgangswerte erreichten, während die reellen Zellzahlen weiterhin erniedrigt waren (KLING et al., 2011). Dass in jener Studie keine Erhöhung der prozentualen B-Zellanteile beobachtet wurde, könnte darauf zurückzuführen sein, dass die mittlere Virusbeladung nach der akuten Virämie nahezu dauerhaft zwischen 10² und 10³ viralen RNAKopien/ml Plasma lag und die Tierkohorte nach den Definitionen dieser Arbeit eher den elite 122 | S e i t e Diskussion controllers zuzuordnen wäre. Der Beobachtungszeitraum ist ein weiterer Faktor, der die unterschiedlichen Ergebnisse verursachen könnte. PERUCHON et al. (2009), TITANJI et al. (2010) und KUHRT et al. (2010) verfolgten die Veränderungen der B-Zellen nur 4 -15 Wochen nach SIV-Infektion, während die Tiere der Vergleichsstudie aus Kapitel 4.2.2 mindestens 29 Wochen infiziert waren. In allen drei genannten Publikationen war nach einem deutlichen Abfall ein Wiederanstieg der CD20+-Zellen zu erkennen. Es ist also möglich, dass die Absenkung nur transient war. Für diese Dissertation wurde ebenfalls eine longitudinale Untersuchung der B-Zellen in der akuten Phase der SIV-Infektion durchgeführt, deren Ergebnisse im Anschluss an die der Vergleichsstudie diskutiert werden sollen (s. Kapitel 5.3.7). 5.3.4 Eine hohe SIV-Last führte zu einer übersteigerten Aktivierung von B-Zellen Bezüglich der Anteile von CD27+B-Zellen finden sich in der Literatur Angaben, die den Resultaten dieser Doktorarbeit scheinbar widersprechen. Eine Reihe von Studien, in denen die Veränderungen der Gedächtnis-B-Zellen während der HIV-Infektion anhand der Oberflächenantigene CD20 und CD27 untersucht wurden, beschreiben einen mit der Höhe der Viruslast assoziierten Abfall der Gedächtnis-B-Zellen (DE MILITO et al., 2001; NAGASE et al., 2001; CHONG et al., 2004a; DE MILITO et al., 2004; DORIA-ROSE et al., 2009; RICHARD et al., 2010). In der vorliegenden Arbeit hingegen war der Anteil von CD27+Zellen an den B-Zellen bei Tieren mit mittlerer oder hoher Plasmavirämie im Vergleich zu SIV-negativen deutlich erhöht. Nutzt man einzig diesen Marker zur Definition von Gedächtnis-B-Zellen, schien dieser Zelltyp im Gegensatz zu den Ergebnissen aus der Humanmedizin prozentual anzusteigen. Bei der erweiterten Analyse der B- Zellsubpopulationen wurde aber deutlich, dass in erster Linie eine Verdoppelung der aktivierten B-Zellen ( CD20+CD10-CD21-CD27+IgD-) für die Verschiebung verantwortlich war, während die prozentualen Anteile der ruhenden Gedächtnis-B-Zellen und naiven BZellen bei SIV-infizierten Tieren verringert waren. Auch bei SIV-infizierten Javaneraffen (TITANJI et al., 2010) und der HIV-Infektion des Menschen (MOIR et al., 2001; HO et al., 2006; MOIR et al., 2008b; FONTAINE et al., 2011) ist eine Zunahme der aktivierten BZellen dieses Phänotyps beschrieben. Selbst wenn deren Anteile an den Gesamt-B-Zellen bei Menschen in einem viel niedrigeren Bereich lagen, wie in Kapitel 5.3.2 erläutert, war bei Diskussion S e i t e | 123 humanen Progessoren ebenfalls eine Steigerung der aktivierten, reifen B-Zellen um nahezu 100% zu beobachten (FONTAINE et al., 2011). Eine Bestimmung aktivierter Zelltypen mittels Durchflusszytometrie lässt sich zudem über eine Vielzahl anderer Oberflächenmarker erreichen. Schon früh entdeckten MARTINEZMAZA et al. (1987), dass die B-Zellen HIV-infizierter eine erhöhte Expression des Transferrinrezeptors zeigten, welcher als Aktivierungsmarker angesehen wird. Im Gegensatz dazu ist der Inhibitionsrezeptor LAIR-1 herunterreguliert (DE MILITO et al., 2004). Auch eine Steigerung der Ki-67- und CD95-positiven B-Zellen ist mehrfach sowohl für die HIV(MOIR et al., 2004; TITANJI et al., 2005; HO et al., 2006) als auch SIV-Infektion (KUHRT et al., 2010; KLING et al., 2011) beschrieben worden. Dies weist auf eine verstärkte Proliferation und Aktivierung sowie eine vergrößerte Neigung zur Apoptose hin. Dementsprechend ist auch der Zellumsatz von CD20+-Zellen bei SIV-infizierten Makaken erhöht (MOHRI et al., 1998), bei natürlichen Wirten wie den Rauchmangaben hingegen nicht, ein weiteres Indiz für die pathologische Aktivierung der B-Zellen (KAUR et al., 2008). Letztere scheinen sich also, trotz der massiven Verluste an T-Helferzellen im Rahmen der chronischen Virusinfektion, vermehrt zu kurzlebigen Plasmablasten zu differenzieren und so die HIV-/SIV-assoziierte, unspezifische Hypergammaglobulinämie zu verursachen (DE MILITO et al., 2001; MOIR et al., 2001). Interessanterweise stiegen in der Studie mit Javaneraffen die aktivierten B-Zellen nur bei normalen Progressoren, bei rapid progressors hingegen sanken die Anteile (TITANJI et al., 2010). Diese Beobachtung liegt auf einer Linie mit dem Unvermögen der rapid progressors, SIV-spezifische Antikörper in nennenswertem Umfang zu bilden. Lange war unklar, ob die übersteigerte Aktivierung unmittelbar auf eine Stimulation durch die Virusreplikation zurückzuführen ist, oder ob es sich vielmehr um einen Nebeneffekt anderer pathologischer Veränderungen, wie z.B. denen des T-Zellkompartiments, handelt. Auch in der vorliegenden Arbeit wurden diese Zusammenhänge überprüft (s. Kapitel 4.2.2). Der Anteil aktivierter B-Zellen an den CD20+Lymphozyten korrelierte hochsignifikant invers mit dem der CD4+Zellen an den T-Zellen und Tiere mit mittlerer und hoher Virusbeladung besaßen nahezu doppelt so viele aktivierte B-Zellen wie die elite controllers und SIVnegativen Tiere. Widersprüchliche Ergebnisse darüber, ob ART zu einer Normalisierung 124 | S e i t e Diskussion dieses hyperaktivierten Zustandes führt (DE MILITO et al., 2001; TITANJI et al., 2005; MOIR et al., 2008b), sprechen allerdings gegen einen direkten Einfluss der Virusvermehrung. FONTAINE et al. (2011) legten unlängst Beweise für die interessante Theorie vor, dass dendritische Zellen maßgeblich an den B-Zellfehlsteuerungen während der Retrovirusinfektion beteiligt sind. Physiologischerweise beeinflussen dendritische Zellen durch die Produktion von B-Zellwachstumsfaktoren wie B-Lymphozyten-Stimulator (B-LyS) und einem proliferations-verursachendem Liganden (engl.: a proliferation-inducing ligand, APRIL) die Regulation der B-Zellreihe. Schon in der frühen Phase der HIV-Infektion wird deren Produktion massiv gesteigert, obwohl es zu einem Verlust der dendritischen Zellen im Blut kommt, und auch in der chronischen Phase trotz ART und sinkender Viruslast bleibt diese aufrecht erhalten. Selbst wenn der ursächliche Mechanismus dafür nicht aufgedeckt werden konnte, macht dieser Sachverhalt doch erneut deutlich, wie komplex die Pathomechanismen der Immundefizienzviren sind. 5.3.5 Elite controllers unterschieden sich nur hinsichtlich der ruhenden Gedächtnis-BZellen und der T-Helferzellen signifikant von den SIV-negativen Tieren Einer der wesentlichen Befunde dieser Doktorarbeit ist, dass Tiere mit einer Viruslast von unter 1x103 SIV-RNA-Kopien/ml Plasma annähernd physiologische Werte innerhalb der untersuchten Lymphozytenpopulationen aufwiesen. Einzig die T-Helferzellen und die ruhenden Gedächtnis-B-Zellen waren gegenüber den Normalwerten signifikant erniedrigt, wobei der Unterschied hinsichtlich der ruhenden Gedächtniszellen ohne Ig-Klassenwechsel (CD10-CD20+CD21+CD27+IgD+) am deutlichsten war. Bisher gibt es keine veröffentlichten Studien, in denen die Gedächtnis-B-Zellen von SIV-infizierten Makaken untersucht wurden, die in der Lage sind, die Viruslast effektiv zu kontrollieren. Eine vergleichbare Gruppe aus dem Bereich der HIV-Forschung könnten Patienten darstellen, die mit ART behandelt wurden und bei denen es zu einer nahezu kompletten Reduktion der Virusreplikation gekommen war. Mehrere Gruppen beschrieben, dass die memory-B-Zellanteile trotz erfolgreicher + antiretroviraler Therapie nicht völlig wiederhergestellt wurden und die IgD CD27+B-Zellen ebenfalls stärker betroffen waren als die IgG/IgA-exprimierenden (DE MILITO et al., 2001; CHONG et al., 2004a; TITANJI et al., 2005; D'ORSOGNA et al., 2007; HART et al., 2007; MOIR et al., 2008b). Auch SIV- bzw. SHIV-infizierte Makaken litten während ART Diskussion S e i t e | 125 weiterhin unter erniedrigten IgD+Gedächtnis-B-Zellen (KUHRT et al., 2010; KLING et al., 2011). Als mögliche Ursache für die Verluste der Gedächtnis-B-Zellen wird eine verstärkte Differenzierung zu Plasmazellen diskutiert, entweder wegen einer direkten polyklonalen Stimulation durch das Virus oder wegen einer indirekten durch aktivierte T-Helferzellen oder Zytokine (DE MILITO et al., 2001; NAGASE et al., 2001). Nach einer anderen Theorie, die durch die verstärkte Expression von Apoptosemarkern unterstützt wird, könnten memory-BZellen vermehrt dem Zelltod unterliegen (TITANJI et al., 2010). Angesichts der Korrelation zwischen Gedächtnis-B-Zellen und T-Helferzellen wäre zudem die Möglichkeit denkbar, dass eine Neubildung von memory-B-Zellen erschwert ist, wenn die CD4+-Zellzahlen aufgrund hoher Virusreplikation fallen. Bedingt durch die Auswirkungen auf die Architektur der Keimzentren im Lymphknoten, die die HIV-/SIV-Infektion hat, könnte die Kontaktzone zwischen B- und T-Zellen, welche für eine einwandfreie Reifung von Gedächtnis-B-Zellen notwendig ist, beschädigt sein (CAGIGI et al., 2008). Da in den meisten Fällen ausschließlich eine Analyse der B-Zellen im peripheren Blut stattfand, ist natürlich auch eine Abwanderung der Gedächtnis-B-Zellen in andere Kompartimente des Körpers wie Milz, Knochenmark oder Lymphknoten nicht auszuschließen. Denn auch die Ausbildung von Zytokinrezeptoren, welche für die Wanderung der Zellen in verschiedene Zielorgane (engl.: homing) verantwortlich sind, werden durch die HIV-Infektion verändert (MOIR u. FAUCI, 2008). Die Konsequenzen des Gedächtnis-B-Zellverlustes in der frühen Phase der Infektion sind gravierend und unter Umständen irreversibel, wie die Beispiele von Langzeitüberlebenden und der zumindest nach virologischen Kriterien erfolgreich Therapierten zeigen. Folgende Beispiele verdeutlichen, dass das humorale Gedächtnis sowohl gegenüber Antigenen, denen das Individuum vor der Infektion ausgesetzt war, als auch solchen, denen es später begegnet, beeinträchtigt ist und dies das Auftreten von opportunistischen Erkrankungen wesentlich begünstigt: Erstens belegten die Bestimmung von Antikörpertitern und Untersuchungen mittels B-ZellELISpot, dass das immunologische Langzeitgedächtnis gegen Pathogene wie Masern und Tetanus nicht nur bei Progressoren, sondern auch bei Langzeitüberlebenden und Patienten unter ART beeinträchtigt ist (DE MILITO et al., 2004; HART et al., 2007). Zweitens zeigte 126 | S e i t e Diskussion die Arbeitsgruppe von MALSPINA et al. (2005), dass die Neubildung von Gedächtnis-BZellen eingeschränkt ist. Sie verglich die Reaktion auf eine Influenzaimpfung von HIVinfizierten unter HAART mit der von gesunden Kontrollen. Sieben Tage nach Immunisierung war die Bildung von Antikörper-produzierenden Zellen im B-Zell-ELISpot gleich stark. Wurde der Test aber 54 Tage später mit einer fünftägigen in vitro-Stimulation durchgeführt, wiesen HIV-negative Personen deutlich mehr Influenza-spezifische Gedächtnis-B-Zellen auf. Drittens ist auch die Bildung von HIV-spezifischen Gedächtniszellen in Abhängigkeit von den CD27+-Anteilen gestört (BUSSMANN et al., 2011). Bei der Bekämpfung genannter Erreger spielen vornehmlich Antikörper der Immunglobulinklassen A und G eine Rolle, Gedächtnis-B-Zellen ohne Klassenwechsel sind allerdings stärker von den HIV-/SIV-assoziierten Verlusten betroffen. IgD+CD27+Zellen stellen eine Art Brücke zwischen angeborenem und erworbenem Immunsystem dar, indem sie hochaffines IgM produzieren und somit äußerst wichtig für die Bekämpfung von T-Zellunabhängigen Antigenen, wie z.B. der Polysaccharidhülle von Bakterien, sind (WELLER et al., 2005). Dies könnte erklären, warum bei humanen HIV-Patienten unter ART trotz sehr niedriger Viruslast und physiologischen CD4+-Zellzahlen eine höhere Prävalenz für invasive Pneumokokkeninfektionen besteht (BARRY et al., 2006). Laut Hart, 2007, reagieren HIVinfizierte Individuen mit geringen IgM+Gedächtniszellspiegeln schlechter auf eine Impfung gegen diesen Erreger. Fazit dieser Beobachtungen könnte sein, dass eine antiretrovirale Therapie möglichst früh nach erfolgter Infektion einsetzen sollte, um die Beeinträchtigungen des humoralen Gedächtnisses zu minimieren. Zumindest bei vertikal infizierten Kindern konnte diese Annahme bestätigt werden: Pädiatrische Patienten, die innerhalb von einem Jahr nach Geburt mit HAART behandelt wurden, unterschieden sich bezüglich ihrer Reaktionen auf Masernschutzimpfungen nicht von gesunden Kontrollen gleichen Alters, während spät- oder nichtbehandelte Kindern merkliche Defizite in ihrer B-Zellentwicklung und der Antwort auf Vakzinierungen aufwiesen (PENSIEROSO et al., 2009). Diskussion S e i t e | 127 5.3.6 Die SIV-Infektion bei Rhesusaffen hatte einen anderen Einfluss auf die untersuchten atypischen B-Zellsubpopulationen als die HIV-Infektion bei Menschen Zusätzlich zu den bisher diskutierten Veränderungen der B-Zellen treten bei der HIVInfektion des Menschen zwei Zellarten verstärkt auf, die physiologischerweise nur sehr geringe Anteile am Gesamt-B-Zellkontingent stellen. Sie werden deshalb als atypische BZellpopulationen bezeichnet. CD21-CD27-IgD-B-Zellen weisen keine typischen Gedächtnismarker mehr auf, ihre Immunglobulinklasse beweist aber, dass ein Antigenkontakt stattgefunden haben muss. Erstmals wurden diese Zellen in humanen Tonsillen beschrieben. Da sie zudem ein Muster aus hemmenden und homing-Rezeptoren exprimieren, welches denen von antigenspezifischen T-Zellen sehr ähnelt, die durch persistierende Virusreplikation entkräftet sind, werden sie in der englischsprachigen Literatur als exhausted tissue-like (dt.: erschöpfte Gewebe-ähnliche) Gedächtnis-B-Zelle bezeichnet. Zudem proliferieren sie deutlich schwächer als Reaktion auf übliche B-Zellstimuli. Bei HIV-negativen und –avirämischen Personen entsprechen sie weniger als 4% der Gesamt-B-Zellen, während ihr Anteil bei Progressoren auf 19% steigt (MOIR et al., 2008a). Die Autoren dieser Studie konnten zusätzlich beweisen, dass exhausted tissue-like-B-Zellen von HIV-positiven Patienten nach polyklonaler Stimulation zu einem signifikant höheren Anteil zu gp120-spezifischen ASC differenzierten, als naive oder klassische Gedächtnis-B-Zellen, welche eher Antikörper anderer Spezifität bildeten. Die durchflusszytometrische Bestimmung der CD21-CD27-IgD-B-Zellen in dieser Arbeit ließ, im Gegensatz zu den Ergebnissen aus der humanmedizinischen Forschung, keine signifikanten Unterschiede zwischen den vier Tierkohorten erkennen. Die Anteile der exhausted tissue-like-B-Zellen lagen unabhängig vom SIV-Status in einem ähnlichen Bereich wie die von gesunden Menschen. Gleichartige Resultat erzielte eine Arbeitsgruppe bei Rhesusaffen, die bis zur 12. Wochen nach SIV-Infektion beobachtet worden waren. Die Anteile der exhausted tissue-like-B-Zellen änderten sich bei typischen Progressoren nicht, bei rapid progressors sanken sie sogar (TITANJI et al., 2010). Eine hinreichend begründete Erklärung für diese konträren Ergebnisse zwischen Menschen und NHP scheint es leider nicht zu geben. Angesichts der sonstigen massiven Störungen der B-Zelldifferenzierung darf aber 128 | S e i t e Diskussion keinesfalls geschlussfolgert werden, dass die SIV-Infektion diese weniger stark beeinträchtigt als die HIV-Infektion. Vielmehr ist anzunehmen, dass der Phänotyp der exhausted tissue-likeB-Zelle bei Rhesusaffen eine andere Funktion und/oder Regulierung hat als beim Menschen. Die zweite atypische B-Zellpopulation entspricht unreifen B-Zellen, die zwar noch CD10positiv sind, das Knochenmark aber schon verlassen haben. Daraus resultiert die Bezeichnung Übergangs-B-Zellen. Mehrere Studien berichteten davon, dass jener Zelltyp im Rahmen der HIV-Infektion vermehrt auftritt (MARTINEZ-MAZA et al., 1987; HO et al., 2006; MALASPINA et al., 2006; HART et al., 2007). So wurde in dieser Arbeit das Vorhandensein von Übergangs-B-Zellen in SIV-infizierten Rhesusaffen überprüft. Dabei wurde deutlich, dass im SIV-Makakenmodell gegensätzliche Verhältnisse bestehen. SIV-negative Tiere besaßen im Mittel einen Anteil von 0,5% Übergangs-B-Zellen im peripheren Blut, controllers und progressors hingegen signifikant niedrigere. Auch in diesem Fall ist eine unterschiedliche funktionelle Bedeutung dieses B-Zellsubtyps wahrscheinlich Hauptursache der Diskrepanz. Schon die physiologischen Verhältnisse bei Mensch und Rhesusaffen unterscheiden sich außerordentlich voneinander. Der B-Zellpool im Blut gesunder, adulter Menschen enthält ca. 10% Übergangs-B-Zellen, was dem 20-fachen der Situation im Rhesusaffen entspricht. Der Anstieg während der HIV-Infektion wird auf eine homöostatische Kompensation der Lymphopenie zurückgeführt, da er direkt mit CD4+Zellverlusten und steigenden IL-7Spiegeln korreliert (HO et al., 2006; MALASPINA et al., 2006). Auch bei der idiopathischen CD4+-Lymphopenie sind IL-7-Spiegel und Übergangs-B-Zellen erhöht (MALASPINA et al., 2007). Es wäre denkbar, dass bei Rhesusaffen andere Wechselbeziehungen zu Grunde liegen und der Einfluss der SIV-Infektion deshalb ein anderer ist. 5.3.7 Die Anteile der CD20+Zellen an den Lymphozyten und der CD10-IgD-Zellen an den B-Zellen normalisierten sich innerhalb von 4 Wochen nach SIV-Infektion Angesichts der in den vorangegangenen Abschnitten diskutierten massiven Auswirkungen, die die SIV-Infektion bei chronischen Progressoren auf nahezu alle B-Zellpopulationen hat, sind die Ergebnisse aus Kapitel 4.3.5, in dem 12 nicht-immunisierte Tiere bis zur 20. Woche nach SIV-Infektion untersucht wurden, erstaunlich. Nach einem initialen Abfall während der zweiten Woche nach SIV-Infektion stiegen sowohl die mittleren Anteile der B-Zellen an den Lymphozyten als auch die der CD10-IgD-Zellen an den B-Zellen wieder auf das Niveau der Präwerte an. Bei letzteren, die zum größten Teil CD27-positiven B-Zellen entsprechen Diskussion S e i t e | 129 dürften, wie in Kapitel 4.3.5 erläutert, erreichte der Tiefpunkt nicht einmal statistische Signifikanz im Vergleich zum mittleren Präwert. Obwohl keine signifikante Korrelation zur Viruslast bestand, muss darauf hingewiesen werden, dass sich die individuellen Werte der Tiere zum Teil stark unterschieden. Darüber hinaus zeigten 5 von 10 Tieren eine ungewöhnlich niedrige peak-Virämie zu Woche 2 nach Infektion und 10 von 12 Tieren hatten zu Woche 20 nach Infektion eine virale RNA-Last von unter 1x104 Kopien/ml Plasma. Rückblickend kann festgestellt werden, dass nur zwei dieser Tiere bis zur Fertigstellung der Dissertation wegen klinischer Symptomatik euthanasiert werden mussten. Vermutlich ist dieser ungewöhnlich hohe Anteil von Tieren, die die SIVInfektion effizient kontrollierten, auf den repeated-low-dose-challenge zurückzuführen, obwohl die Gründe für diese Beobachtung trotz intensiver Diskussionen unklar sind. Bei der Vergleichsuntersuchung von Langzeitüberlebenden und Progressoren wurde deutlich, dass die aktuelle SIV-Beladung einen größeren Einfluss auf die B-Zellprozentsätze hatte als die Krankheitsdauer (s. Kapitel 4.2.2). Therapiestudien, sowohl bei HIV-Patienten als auch im SIV-Makakenmodell, belegen dies (TITANJI et al., 2005; MOIR et al., 2008b; KUHRT et al., 2010; KLING et al., 2011). Deshalb ist durchaus anzunehmen, dass sich die annähernd physiologischen Verhältnisse der B-Zellpopulationen auf die verhältnismäßig niedrige Viruslast zurückführen lassen, sie sich aber bei einer längeren Beobachtungsperiode in Abhängigkeit von dieser ändern könnten. Verschiedene Arbeitsgruppen untersuchten die Veränderungen der B-Zellanteile im Makakenmodel bis 12 – 53 Wochen nach SIV-Infektion (PERUCHON et al., 2009; KUHRT et al., 2010; TITANJI et al., 2010; KLING et al., 2011). Alle dokumentierten, wie die vorliegende Arbeit, einen signifikanten Abfall der Gesamt-B-Zellen, der seinen Tiefpunkt synchron zur Spitzenvirämie hatte. Auch ein Wiederanstieg wurde beschrieben, allerdings variierten die Zeitpunkte, zu denen Präinfektionswerte erreicht wurden, von 4 (PERUCHON et al., 2009) bis zu 20 Wochen (KUHRT et al., 2010) nach SIV-Infektion. Mit Ausnahme der Arbeitsgruppe von KLING et al. (2011), die ebenfalls nur tendenzielle Veränderungen bei den Gedächtnis-B-Zellen beobachten konnte, folgten die Kurven der Gedächtnis-B-Zellanteile in den genannten Veröffentlichungen einem übereinstimmenden Verlauf. 130 | S e i t e Diskussion Zusammen mit den Ergebnissen der vergleichenden durchflusszytometrischen Untersuchung zwischen Langzeitüberlebenden, Progressoren und SIV-naiven Tieren in der vorliegenden Untersuchung unterstreichen die Beobachtungen der zitierten Arbeitsgruppen die Schwierigkeiten bei dem Versuch, die HIV-/SIV-Pathogenese zu begreifen. Um die Veränderungen in der Zusammensetzung der B-Zellsubpopulationen in ihrer Gänze zu durchschauen, muss deshalb eine begleitende Untersuchung vom Beginn der Infektion bis zum Einsetzen von AIDS durchgeführt werden. Dies ist bei HIV-infizierten Menschen nicht möglich, da der genaue Infektionszeitpunkt selten bekannt ist und sich eine Verzögerung des Therapiebeginns bis zum symptomatischen Ausbruch der Erkrankung aus ethischen Gründen verbietet. Leider kann auch im Tiermodell ein derart langer Beobachtungszeitraum nur vereinzelt gewährleistet werden. 5.4 Abschließende Bewertung und Ausblick Trotz umfangreicher Analysen beantworten die Ergebnisse dieser Arbeit die Frage, welches Gewicht Antikörper und Gedächtnis-B-Zellen bei der Kontrolle der SIV-Infektion haben, nicht eindeutig. Es konnte gezeigt werden, dass Langzeitüberlebende im Vergleich zu Progressoren signifikant höhere Immunantworten gegen SIV-gag im ELISA und IFN-γELISpot ausgebildet hatten und dass sich die B-Zellsubpopulationen von Tieren, die die Viruslast auf einem sehr niedrigen Spiegel kontrollieren konnten, kaum von denen SIVnegativer Tiere unterschieden. Tiere mit mittlerer oder hoher Viruslast hingegen wiesen diesbezüglich deutliche Unterschiede auf. Ob diese Resultate allerdings die Ursache für einen milden bis scheinbar apathogenen Verlauf der SIV-Infektion darstellen, bleibt weiterhin unklar. Die Hypothese einer unterschiedlichen Steuerung der Immunantwort gegen SIVgag (T-Zellabhängig) und SIVenv (T-Zellunabhängig) deutet darauf hin, dass die höheren Antikörpertiter der Langzeitüberlebenden gegen p27 lediglich eine Begleiterscheinung der konservierten T-Helferzellzahlen sein könnten. Gleiches gilt für die Veränderungen der BZellzusammensetzungen. Durch die nur im Tiermodell mögliche experimentelle Depletion von B-Zellen versuchten mehrere Arbeitsgruppen, die Bedeutung des humoralen Arms der Immunabwehr für den Verlauf der SIV-Infektion aufzuklären. Trotz der Gradlinigkeit dieser Herangehensweise war die Antwort auch in diesem Fall nicht klar. Eine Depletion der CD20+Zellen hatte entweder keinen (GAUFIN et al., 2009), mäßigen (SCHMITZ et al., 2003) Diskussion S e i t e | 131 oder einen deutlichen (MILLER et al., 2007) Einfluss auf die Virusreplikation und den Krankheitsverlauf. Bei der Bewertung der Relevanz von Antikörpern und dem humoralem Gedächtnis für die HIV-/SIV-Erkrankung muss allerdings zwischen ihren Effekten nach erfolgter Infektion mit Integration in das Wirtsgenom und ihrer prophylaktischen Nutzung durch Impfstoffe oder Mikrobizide unterschieden werden. Selbst wenn die Bildung von neutralisierenden Antikörpern im Rahmen der primären Immunantwort zu spät erfolgt, um eine Infektion zu verhindern und bei der Kontrolle der Virusreplikation die zelluläre Immunantwort eine größere Rolle spielen sollte, konnte mehrfach bewiesen werden, dass das Vorhandensein ausreichender Mengen neutralisierender Antikörper vor Eintritt des Virus in den Körper eine Manifestation der Infektion verhindern kann (BABA et al., 2000; MASCOLA et al., 2000; PARREN et al., 2001; BURTON et al., 2011). Bis heute besteht aber eine der größten Schwierigkeiten bei der Entwicklung einer wirksamen Vakzine in der Erzeugung einer robusten Antikörperantwort und langanhaltendem immunologischen Gedächtnis. Die in dieser Arbeit etablierten Methoden des SIV-spezifischen B-Zell-ELISpot und der durchflusszytometrische Analyse der B-Zellsubpopulationen stellen eine hervorragende Technik dar, diese Faktoren zu bewerten und sollten deshalb bei künftigen Impfstoffstudien eine Anwendung bereits während der Immunisierung finden. Abgesehen von der Bedeutung der humoralen Immunantwort für die Bekämpfung der HIV/SIV-Infektion ist unstrittig, dass die Hyperaktivierung der B-Zellen und der Verlust der Gedächtnis-B-Zellen eine wesentliche Rolle bei der Ausbildung des Immundefizienzsyndroms spielen. Zur weiteren Erforschung dieser Pathomechanismen vor allem in der frühen Phase der Infektion bleibt die SIV-Infektion von Makaken momentan das wertvollste Modell. Beim Menschen ist der genaue Infektionszeitpunkt selten bekannt und die Rahmenbedingungen wie Virusstamm, Infektionsroute, Alter und Lebensumfeld der Betroffenen variieren stark. Außerdem ist das Blut bei humanen Patienten meist das einzige Kompartiment des Immunsystems, das für Analysen zugänglich ist. In der vorliegenden Arbeit wurde aber deutlich, dass z.B. die Bildung von SIV-spezifischen Antikörpern in der chronischen Phase der Infektion weniger im Blut, sondern vermutlich im Knochenmark stattfindet. Eine phänotypische Charakterisierung der B-Zellpopulationen in diesem, sowie 132 | S e i t e Diskussion anderen immunologisch wichtigen Organen wie Lymphknoten, Milz und Darmmukosa, anhand der Durchflusszytometrie sowie eine funktionelle Untersuchung mittels B-ZellELISpot werden in einem kürzlich begonnenen Dissertationsvorhaben durchgeführt und gestatten es hoffentlich, weitere Rückschlüsse auf die Situation der humanen HIVErkrankung zu ziehen. Allerdings offenbarte die vorliegende Arbeit auch einige Unterschiede bezüglich der phänotypischen Eigenschaften von B-Zellen bei Rhesusaffen einerseits und Menschen andererseits, ein Umstand, der bei der Interpretation von Daten aus Tiermodellen immer in Betracht gezogen werden muss. Eine Separierung der einzelnen Subpopulationen und anschließenden funktionellen Tests könnten weitere Aufschlüsse darüber liefern, welche Zelltypen einander entsprechen und sollten im Sinne einer wissenschaftlichen Erkenntnisgewinns dringend durchgeführt werden. Verbesserung des Zusammenfassung S e i t e | 133 6. ZUSAMMENFASSUNG Katharina Raue (2012) Antikörper-abhängige Immunantwort im SIV-Makakenmodell Trotz jahrzehntelanger Bemühungen steht noch kein zuverlässiger Impfstoff zum Schutz vor der HIV-Infektion des Menschen zur Verfügung. Für das Erreichen dieses Zieles sind weitere Untersuchungen über die Bedeutung und Funktionsweise der humoralen Immunantwort einschließlich der B-Zellentwicklung und -pathogenese unerlässlich. Hierfür stellt die Infektion von asiatischen Makaken mit dem Simian Immunodeficiency Virus (SIV) aufgrund von deutlichen virologischen, immunologischen und klinischen Analogien ein hervorragendes Tiermodell dar. Ziel dieser Arbeit war es, die Bedeutung der antikörperabhängigen Immunantwort im SIVMakakenmodell evaluieren. Dazu wurden in einer retrospektiven Studie Tiere mit klassisch progredientem Infektionsverlauf (Progressoren) mit solchen verglichen, die ohne Therapie dazu in der Lage waren, die Virusreplikation über einen ungewöhnlich langen Zeitraum hinweg zu kontrollieren und klinisch gesund blieben (Langzeitüberlebende). Für die Bewertung der humoralen Immunantwort im Laufe der Infektion wurden als Parameter die Titer bindender Antikörper gegen SIVgag und SIVenv und die Titer neutralisierender Antikörper genutzt, welche mittels konventionellem ELISA bzw. anhand des TZM-blNeutralisationstest mit Luziferase-read-out bestimmt wurden. Zum Vergleich mit der zellulären Immunantwort wurden Daten eines IFN-γ-ELISpot ausgewertet. Schließlich erfolgte eine Analyse der SIV-spezifischen Gedächtnis-B-Zellen im Blut und der BAL von Langzeitüberlebenden anhand des B-Zell-ELISpots. In einer Vergleichsstudie wurden die gegenwärtigen phänotypischen B-Zellsubpopulationen der Langzeitüberlebenden durchflusszytometrisch charakterisiert und denen von SIVnegativen Tieren und solchen mit hoher Virusbeladung gegenübergestellt. Um die Einflüsse der B-Zellpathogenese in der Phase der frühen SIV-Infektion auf die Entstehung der antikörperabhängigen Immunantwort zu bewerten, wurden schließlich 12 134 | S e i t e Zusammenfassung nicht-immunisierte Tiere, die als Kontrolltiere in zwei Impfstoffstudien dienten, von der Belastungsinfektion bis hin zur chronischen Phase der SIV-Infektion mit den genannten Testmethoden untersucht. Die Ergebnisse lassen sich wie folgt zusammenfassen: 1) Langzeitüberlebende besaßen signifikant höhere Antikörpertiter gegen SIVgag und auch mehr T-Zellen, die IFN-γ als Reaktion auf SIVgag ausschütteten, als Progressoren. Die humorale und zelluläre Immunantwort gegen SIVenv unterschied sich nicht zwischen Langzeitüberlebenden und Progressoren. 2) Progressoren hatten in Abhängigkeit von der Viruslast höhere Titer von neutralisierenden Antikörpern als Langzeitüberlebende. 3) Ohne vorherige Stimulation konnten keine SIV-spezifischen IgG- oder IgAsezernierenden Zellen im Blut und der BAL von Langzeitüberlebenden nachgewiesen werden. 4) Im Blut der Langzeitüberlebenden konnten nach in vitro-Stimulation mehr SIVenvspezifische IgG-Gedächtnis-B-Zellen als SIVgag-spezifische IgG-Gedächtnis-BZellen detektiert werden. Der Nachweis von SIV-spezifischen IgA-Gedächtnis-BZellen im Blut sowie der von SIV-Spezifischen IgG- und IgA-Gedächtnis-B-Zellen in der BAL blieb erfolglos. 5) Die phänotypischen B-Zellpopulationen von SIV-negativen Rhesusaffen unterschieden sich von denen gesunder Menschen. So bestand der größte Anteil der BZellen von Rhesusaffen aus aktivierten B-Zellen, welche bei Menschen weniger als 10% ausmachen. Umgekehrt besitzen Menschen deutlich höhere Prozentsätze von ruhenden Gedächtnis-B-Zellen als Rhesusaffen. 6) Während die Anteile von aktivierten B-Zellen bei Tieren mit mittlerer und hoher Virusbeladung signifikant anstiegen und die Anteile naiver B-Zellen und ruhender Gedächtnis-B-Zellen abfielen, unterschieden sich Tiere mit sehr niedriger Virusbeladung nur hinsichtlich der ruhenden Gedächtnis-B-Zellen von SIV-negativen Tieren. Zusammenfassung S e i t e | 135 7) Im Unterschied zu der Situation während der HIV-Infektion des Menschen fiel der Anteil von Übergangs-B-Zellen durch die SIV-Infektion ab, während sich die Anteile der exhausted tissue-like Gedächtnis-B-Zellen nicht änderten. 8) Nach einem initialen Abfall zum Zeitpunkt der Spitzenvirämie zwei Wochen nach SIV-Infektion erholten sich sowohl die Anteile der Gesamt-B-Zellen an den Lymphozyten, als auch die der Gedächtnis-B-Zellen an den Gesamt-B-Zellen wieder. 9) Weder in der akuten noch in der chronischen Phase der SIV-Infektion bestand eine Korrelation zwischen den Titern SIV-spezifischer bindender Antikörper und den SIVspezifischen antikörper-sezernierenden Zellen. 10) Die SIV-spezifischen antikörper-sezernierenden Zellen korrelierten beim Eintritt in die Plateauphase negativ sowohl mit der derzeitigen Viruslast als auch mit jener 40 Wochen nach SIV-Infektion. Die Resultate der vorliegenden Arbeit konnten deutliche Unterschiede zwischen Langzeitüberlebenden und Progressoren sowohl bezüglich der antikörperabhängigen Immunantwort als auch bezüglich der B-Zellsubpopulationen aufzeigen. Eine weiterführende Untersuchung der Ursachen und Konsequenzen dieser Unterschiede sollte im Hinblick auf zukünftige Immunisierungsstrategien und für die verbesserte Therapie von B-Zell-assoziierten Symptomen der HIV-Erkrankung durchgeführt werden. 136 | S e i t e Summary 7. SUMMARY Katharina Raue (2012) Antibody-dependent immune response in the SIV-macaque-model Despite comprehensive efforts, there is still no safe vaccine against the human HIV infection available. To achieve this goal further analysis of the impact of humoral immune responses on HIV as well as of B cell development and –pathogenesis are needed. The infection of Asian macaques with the Simian Immunodeficiency Virus (SIV) provides an excellent animal model for this due to its analogies regarding viral replication, immune responses and clinical aspects. The aim of this study was to evaluate the impact of antibody-dependent immune defense in the SIV-macaque-model. Therefore, in a retrospective study, animals with a classical progressive course of disease (progressors) where compared to those, which were able to control viral replication for an unusually long period of time and remained clinically healthy (long-term survivors). To assess the humoral immune response during the course of infection, binding and neutralizing antibody titers were determined by ELISA and TZM-bl neutralization assay with luciferase read-out. For comparison with the cellular immune response, data of an INF-γ-ELISpot were analyzed. Additionally, the existence of SIVspecific memory B cells in blood and BAL was tested by B cell-ELISpot. In a cross-sectional study the current phenotypic B cell subpopulations were characterized by polychromatic flow cytometry and compared to those of SIV-negative animals and animals with high viral load. Finally, 12 animals, which served as naïve controls in two immunization experiments, were monitored from the day of SIV infection to its chronic phase by the assays mentioned afore. The purpose was to evaluate the impact of B cell pathogenesis during the early phase of SIVinfection on the development of the antibody-dependent immunity. Summary S e i t e | 137 The following results were obtained: 1) Long-term survivors had significantly higher binding antibody titers against SIVgag as well as more SIVgag-specific IFN-γ-secreting T cells than progressors. There was no difference in the humoral and cellular immune responses against SIVenv. 2) Neutralizing antibodies were higher in progressors depending on viral load levels. 3) Without stimulation, neither in blood nor in BAL of the long-term survivors were SIV-specific IgG- or IgA-secreting cells detectable. 4) After in vitro-stimulation, there were more SIVenv-specific than SIVgag-specific IgGmemory B cells in the blood of the long-term survivors. There was no evidence for SIV-specific IgA-memory B cells in blood and for SIV-specific IgG- and IgAmemory B cells in BAL. 5) The phenotypic B cell populations of SIV-negative Rhesus monkeys differed from those of healthy humans. The main percentage of B cells in Rhesus macaques consisted of activated B cells, which represented less than 10% of the human B cell pool. By contrast, humans had a considerably higher fraction of resting memory B cell subset. 6) While activated B cells increased significantly in animals with medium and high viral load and the percentages of naïve and resting memory B cells significantly decreased, animals with low viral loads differed from SIV-naïve animals only with regard to the resting memory B cells. 7) In contrast to the situation in human HIV infection, transitional B cells decreased after SIV infection but the percentages of exhausted tissue-like B cells remained normal. 8) After an initial decrease during peak viremia, total B cells as well as the fraction of memory B cells recovered. 9) There was no correlation between titers of SIV-specific antibodies and SIV-specific antibody-secreting cells, neither during acute nor during chronic phase of SIV infection. 10) SIV-specific antibody-secreting cells during the beginning of the plateau phase inversely correlated with the current viral load as well as with the viral load 40 weeks after SIV infection. 138 | S e i t e Summary These results show a clear difference between long-term survivors and progressors regarding antibody-dependent immune defense and B cell subpopulations. Additional research on the cause and effect should be carried out in terms of future immunization strategies and to improve the therapy of B cell-associated symptoms of HIV-disease. Literaturverzeichnis S e i t e | 139 8. LITERATURVERZEICHNIS K. AGEMATSU, S. HOKIBARA, H. NAGUMO, K. SHINOZAKI, S. YAMADA u. A. KOMIYAMA (1999): Plasma cell generation from B-lymphocytes via CD27/CD70 interaction. Leukemia & lymphoma 35, 219-225 K. AGEMATSU, H. NAGUMO, F. C. YANG, T. NAKAZAWA, K. FUKUSHIMA, S. ITO, K. SUGITA, T. MORI, T. KOBATA, C. MORIMOTO u. A. KOMIYAMA (1997): B cell subpopulations separated by CD27 and crucial collaboration of CD27+ B cells and helper T cells in immunoglobulin production. European journal of immunology 27, 2073-2079 R. AHMED u. D. GRAY (1996): Immunological memory and protective immunity: understanding their relation. Science (New York, N.Y.) 272, 54-60 A. AHUJA, S. M. ANDERSON, A. KHALIL u. M. J. SHLOMCHIK (2008): Maintenance of the plasma cell pool is independent of memory B cells. Proceedings of the National Academy of Sciences of the United States of America 105, 4802-4807 S. M. ANDERSON, M. M. TOMAYKO u. M. J. SHLOMCHIK (2006): Intrinsic properties of human and murine memory B cells. Immunological reviews 211, 280-294 C. APETREI, M. J. METZGER, D. RICHARDSON, B. LING, P. T. TELFER, P. REED, D. L. ROBERTSON u. P. A. MARX (2005): Detection and partial characterization of simian immunodeficiency virus SIVsm strains from bush meat samples from rural Sierra Leone. Journal of virology 79, 2631-2636 C. ARPIN, J. DECHANET, C. VAN KOOTEN, P. MERVILLE, G. GROUARD, F. BRIERE, J. BANCHEREAU u. Y. J. LIU (1995): Generation of memory B cells and plasma cells in vitro. Science (New York, N.Y.) 268, 720-722 D. T. AVERY, J. I. ELLYARD, F. MACKAY, L. M. CORCORAN, P. D. HODGKIN u. S. G. TANGYE (2005): Increased expression of CD27 on activated human memory B cells correlates with their commitment to the plasma cell lineage. Journal of Immunology 174, 4034-4042 T. W. BABA, V. LISKA, R. HOFMANN-LEHMANN, J. VLASAK, W. XU, S. AYEHUNIE, L. A. CAVACINI, M. R. POSNER, H. KATINGER, G. STIEGLER, B. J. BERNACKY, T. A. RIZVI, R. SCHMIDT, L. R. HILL, M. E. KEELING, Y. LU, J. E. WRIGHT, T. C. CHOU u. R. M. RUPRECHT (2000): Human neutralizing monoclonal antibodies of the IgG1 subtype protect against mucosal simian-human immunodeficiency virus infection. Nature medicine 6, 200-206 140 | S e i t e Literaturverzeichnis J. R. BAILEY, K. G. LASSEN, H. C. YANG, T. C. QUINN, S. C. RAY, J. N. BLANKSON u. R. F. SILICIANO (2006): Neutralizing antibodies do not mediate suppression of human immunodeficiency virus type 1 in elite suppressors or selection of plasma virus variants in patients on highly active antiretroviral therapy. Journal of virology 80, 4758-4770 J. BANCHEREAU, F. BRIERE, Y. J. LIU u. F. ROUSSET (1994): Molecular control of B lymphocyte growth and differentiation. Stem cells (Dayton, Ohio) 12, 278-288 D. H. BAROUCH, S. SANTRA, J. E. SCHMITZ, M. J. KURODA, T. M. FU, W. WAGNER, M. BILSKA, A. CRAIU, X. X. ZHENG, G. R. KRIVULKA, K. BEAUDRY, M. A. LIFTON, C. E. NICKERSON, W. L. TRIGONA, K. PUNT, D. C. FREED, L. GUAN, S. DUBEY, D. CASIMIRO, A. SIMON, M. E. DAVIES, M. CHASTAIN, T. B. STROM, R. S. GELMAN, D. C. MONTEFIORI, M. G. LEWIS, E. A. EMINI, J. W. SHIVER u. N. L. LETVIN (2000): Control of viremia and prevention of clinical AIDS in rhesus monkeys by cytokine-augmented DNA vaccination. Science (New York, N.Y.) 290, 486-492 F. BARRE-SINOUSSI, J. C. CHERMANN, F. REY, M. T. NUGEYRE, S. CHAMARET, J. GRUEST, C. DAUGUET, C. AXLER-BLIN, F. VEZINET-BRUN, C. ROUZIOUX, W. ROZENBAUM u. L. MONTAGNIER (1983): Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science (New York, N.Y.) 220, 868-871 R. A. BARRINGTON, T. J. SCHNEIDER, L. A. PITCHER, T. R. MEMPEL, M. MA, N. S. BARTENEVA u. M. C. CARROLL (2009): Uncoupling CD21 and CD19 of the B-cell coreceptor. Proceedings of the National Academy of Sciences of the United States of America 106, 14490-14495 P. M. BARRY, N. ZETOLA, J. C. KERULY, R. D. MOORE, K. A. GEBO u. G. M. LUCAS (2006): Invasive pneumococcal disease in a cohort of HIV-infected adults: incidence and risk factors, 19902003. AIDS (London, England) 20, 437-444 L. L. BAUM (2010): Role of humoral immunity in host defense against HIV. Current HIV/AIDS reports 7, 11-18 B. BEER, S. NORLEY, K. CICHUTEK u. R. KURTH (1996): Control of initial viraemia coincides with neutralizing antibody response after SIVmac infection of rhesus macaques. AIDS (London, England) 10, 681-682 N. L. BERNASCONI, E. TRAGGIAI u. A. LANZAVECCHIA (2002): Maintenance of serological memory by polyclonal activation of human memory B cells. Science (New York, N.Y.) 298, 2199-2202 Literaturverzeichnis S e i t e | 141 J. M. BINLEY, P. J. KLASSE, Y. CAO, I. JONES, M. MARKOWITZ, D. D. HO u. J. P. MOORE (1997): Differential regulation of the antibody responses to Gag and Env proteins of human immunodeficiency virus type 1. Journal of virology 71, 2799-2809 A. BJORNDAL, H. DENG, M. JANSSON, J. R. FIORE, C. COLOGNESI, A. KARLSSON, J. ALBERT, G. SCARLATTI, D. R. LITTMAN u. E. M. FENYO (1997): Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. Journal of virology 71, 7478-7487 M. BONSIGNORI, M. A. MOODY, R. J. PARKS, T. M. HOLL, G. KELSOE, C. B. HICKS, N. VANDERGRIFT, G. D. TOMARAS u. B. F. HAYNES (2009): HIV-1 envelope induces memory B cell responses that correlate with plasma antibody levels after envelope gp120 protein vaccination or HIV-1 infection. Journal of Immunology 183, 2708-2717 P. BORROW, R. J. SHATTOCK u. A. VYAKARNAM (2010): Innate immunity against HIV: a priority target for HIV prevention research. Retrovirology 7, 84 E. BROCCA-COFANO, K. MCKINNON, T. DEMBERG, D. VENZON, R. HIDAJAT, P. XIAO, M. DALTABUIT-TEST, L. J. PATTERSON u. M. ROBERT-GUROFF (2011): Vaccine-elicited SIV and HIV envelope-specific IgA and IgG memory B cells in rhesus macaque peripheral blood correlate with functional antibody responses and reduced viremia. Vaccine 29, 3310-3319 D. R. BURTON (2002): Antibodies, viruses and vaccines. Nature reviews 2, 706-713 D. R. BURTON, A. J. HESSELL, B. F. KEELE, P. J. KLASSE, T. A. KETAS, B. MOLDT, D. C. DUNLOP, P. POIGNARD, L. A. DOYLE, L. CAVACINI, R. S. VEAZEY u. J. P. MOORE (2011): Limited or no protection by weakly or nonneutralizing antibodies against vaginal SHIV challenge of macaques compared with a strongly neutralizing antibody. Proceedings of the National Academy of Sciences of the United States of America 108, 11181-11186 B. M. BUSSMANN, S. REICHE, B. BIENIEK, I. KRZNARIC, F. ACKERMANN u. C. JASSOY (2009): Loss of HIV-specific memory B-cells as a potential mechanism for the dysfunction of the humoral immune response against HIV. Virology 397, 7-13 A. CAGIGI, F. MOWAFI, L. V. PHUONG DANG, K. TENNER-RACZ, A. ATLAS, S. GRUTZMEIER, P. RACZ, F. CHIODI u. A. NILSSON (2008): Altered expression of the receptor-ligand pair CXCR5/CXCL13 in B cells during chronic HIV-1 infection. Blood 112, 4401-4410 142 | S e i t e Literaturverzeichnis D. CAMPANA, G. JANOSSY, M. BOFILL, L. K. TREJDOSIEWICZ, D. MA, A. V. HOFFBRAND, D. Y. MASON, A. M. LEBACQ u. H. K. FORSTER (1985): Human B cell development. I. Phenotypic differences of B lymphocytes in the bone marrow and peripheral lymphoid tissue. Journal of Immunology 134, 1524-1530 A. CARAUX, B. KLEIN, B. PAIVA, C. BRET, A. SCHMITZ, G. M. FUHLER, N. A. BOS, H. E. JOHNSEN, A. ORFAO u. M. PEREZ-ANDRES (2010): Circulating human B and plasma cells. Age-associated changes in counts and detailed characterization of circulating normal CD138- and CD138+ plasma cells. Haematologica 95, 1016-1020 R. H. CARTER u. D. T. FEARON (1992): CD19: lowering the threshold for antigen receptor stimulation of B lymphocytes. Science (New York, N.Y.) 256, 105-107 D. R. CASIMIRO, F. WANG, W. A. SCHLEIF, X. LIANG, Z. Q. ZHANG, T. W. TOBERY, M. E. DAVIES, A. B. MCDERMOTT, D. H. O'CONNOR, A. FRIDMAN, A. BAGCHI, L. G. TUSSEY, A. J. BETT, A. C. FINNEFROCK, T. M. FU, A. TANG, K. A. WILSON, M. CHEN, H. C. PERRY, G. J. HEIDECKER, D. C. FREED, A. CARELLA, K. S. PUNT, K. J. SYKES, L. HUANG, V. I. AUSENSI, M. BACHINSKY, U. SADASIVAN-NAIR, D. I. WATKINS, E. A. EMINI u. J. W. SHIVER (2005): Attenuation of simian immunodeficiency virus SIVmac239 infection by prophylactic immunization with dna and recombinant adenoviral vaccine vectors expressing Gag. Journal of virology 79, 15547-15555 M. CAVARELLI u. G. SCARLATTI (2011): Human immunodeficiency virus type 1 mother-to-child transmission and prevention: successes and controversies. Journal of internal medicine 270, 561-579 K. CHEN u. A. CERUTTI (2010): New insights into the enigma of immunoglobulin D. Immunological reviews 237, 160-179 Z. CHEN, P. ZHOU, D. D. HO, N. R. LANDAU u. P. A. MARX (1997): Genetically divergent strains of simian immunodeficiency virus use CCR5 as a coreceptor for entry. Journal of virology 71, 2705-2714 Y. CHONG, H. IKEMATSU, K. KIKUCHI, M. YAMAMOTO, M. MURATA, M. NISHIMURA, S. NABESHIMA, S. KASHIWAGI u. J. HAYASHI (2004a): Selective CD27+ (memory) B cell reduction and characteristic B cell alteration in drug-naive and HAART-treated HIV type 1-infected patients. AIDS research and human retroviruses 20, 219-226 Y. CHONG, H. IKEMATSU, M. YAMAMOTO, M. MURATA, K. YAMAJI, M. NISHIMURA, S. NABESHIMA, S. KASHIWAGI u. J. HAYASHI (2004b): Increased frequency of CD27- (naive) B cells and their phenotypic alteration in HIV type 1-infected patients. AIDS research and human retroviruses 20, 621-629 Literaturverzeichnis S e i t e | 143 K. S. COLE, M. MURPHEY-CORB, O. NARAYAN, S. V. JOAG, G. M. SHAW u. R. C. MONTELARO (1998): Common themes of antibody maturation to simian immunodeficiency virus, simian-human immunodeficiency virus, and human immunodeficiency virus type 1 infections. Journal of virology 72, 7852-7859 K. S. COLE, J. L. ROWLES, B. A. JAGERSKI, M. MURPHEY-CORB, T. UNANGST, J. E. CLEMENTS, J. ROBINSON, M. S. WYAND, R. C. DESROSIERS u. R. C. MONTELARO (1997): Evolution of envelope-specific antibody responses in monkeys experimentally infected or immunized with simian immunodeficiency virus and its association with the development of protective immunity. Journal of virology 71, 5069-5079 A. M. CONGE, K. TARTE, J. REYNES, M. SEGONDY, J. GERFAUX, M. ZEMBALA u. J. P. VENDRELL (1998): Impairment of B-lymphocyte differentiation induced by dual triggering of the B-cell antigen receptor and CD40 in advanced HIV-1-disease. AIDS (London, England) 12, 1437-1449 D. A. COOPER, J. GOLD, P. MACLEAN, B. DONOVAN, R. FINLAYSON, T. G. BARNES, H. M. MICHELMORE, P. BROOKE u. R. PENNY (1985): Acute AIDS retrovirus infection. Definition of a clinical illness associated with seroconversion. Lancet 1, 537-540 M. P. CRANAGE, COOK, N., JOHNSTONE, P., GREENAWAY, P.J., KITCHIN, P.A., STOTT, E.J., ALMOND, N., BASKERVILLE, A. (1990): SIV Infection of rhesus macaques; in vivo titration of infectivity and development of an experimental vaccine. In: Animal Models in AIDS Elsevier, Amsterdamm, S. pp. 103-113 M. P. CRANAGE, A. M. WHATMORE, S. A. SHARPE, N. COOK, N. POLYANSKAYA, S. LEECH, J. D. SMITH, E. W. RUD, M. J. DENNIS u. G. A. HALL (1997): Macaques infected with live attenuated SIVmac are protected against superinfection via the rectal mucosa. Virology 229, 143-154 S. CROTTY, P. FELGNER, H. DAVIES, J. GLIDEWELL, L. VILLARREAL u. R. AHMED (2003): Cutting edge: long-term B cell memory in humans after smallpox vaccination. Journal of Immunology 171, 4969-4973 C. C. CZERKINSKY, L. A. NILSSON, H. NYGREN, O. OUCHTERLONY u. A. TARKOWSKI (1983): A solid-phase enzyme-linked immunospot (ELISPOT) assay for enumeration of specific antibodysecreting cells. Journal of immunological methods 65, 109-121 L. J. D'ORSOGNA, R. G. KRUEGER, E. J. MCKINNON u. M. A. FRENCH (2007): Circulating memory B-cell subpopulations are affected differently by HIV infection and antiretroviral therapy. AIDS (London, England) 21, 1747-1752 144 | S e i t e Literaturverzeichnis A. DAS, H. XU, X. WANG, C. L. YAU, R. S. VEAZEY u. B. PAHAR (2011): Double-positive CD21+CD27+ B cells are highly proliferating memory cells and their distribution differs in mucosal and peripheral tissues. PloS one 6, e16524 A. DE MILITO, C. MORCH, A. SONNERBORG u. F. CHIODI (2001): Loss of memory (CD27) B lymphocytes in HIV-1 infection. AIDS (London, England) 15, 957-964 A. DE MILITO, A. NILSSON, K. TITANJI, R. THORSTENSSON, E. REIZENSTEIN, M. NARITA, S. GRUTZMEIER, A. SONNERBORG u. F. CHIODI (2004): Mechanisms of hypergammaglobulinemia and impaired antigen-specific humoral immunity in HIV-1 infection. Blood 103, 2180-2186 P. W. DEMPSEY, M. E. ALLISON, S. AKKARAJU, C. C. GOODNOW u. D. T. FEARON (1996): C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science (New York, N.Y.) 271, 348-350 D. C. DES JARLAIS (1999): Needle exchange. AIDS policy & law 14, 12 R. Z. DINTZIS, M. H. MIDDLETON u. H. M. DINTZIS (1983): Studies on the immunogenicity and tolerogenicity of T-independent antigens. Journal of Immunology 131, 2196-2203 A. C. DONAHUE u. D. A. FRUMAN (2003): Proliferation and survival of activated B cells requires sustained antigen receptor engagement and phosphoinositide 3-kinase activation. Journal of Immunology 170, 5851-5860 N. A. DORIA-ROSE, R. M. KLEIN, M. G. DANIELS, S. O'DELL, M. NASON, A. LAPEDES, T. BHATTACHARYA, S. A. MIGUELES, R. T. WYATT, B. T. KORBER, J. R. MASCOLA u. M. CONNORS (2008): Breadth of human immunodeficiency virus-specific neutralizing activity in sera: clustering analysis and association with clinical variables. Journal of virology 84, 1631-1636 N. A. DORIA-ROSE, R. M. KLEIN, M. M. MANION, S. O'DELL, A. PHOGAT, B. CHAKRABARTI, C. W. HALLAHAN, S. A. MIGUELES, J. WRAMMERT, R. AHMED, M. NASON, R. T. WYATT, J. R. MASCOLA u. M. CONNORS (2009): Frequency and phenotype of human immunodeficiency virus envelope-specific B cells from patients with broadly cross-neutralizing antibodies. Journal of virology 83, 188-199 I. DOUAGI, M. N. FORSELL, C. SUNDLING, S. O'DELL, Y. FENG, P. DOSENOVIC, Y. LI, R. SEDER, K. LORE, J. R. MASCOLA, R. T. WYATT u. G. B. KARLSSON HEDESTAM (2010): Influence of novel CD4 binding-defective HIV-1 envelope glycoprotein immunogens on neutralizing antibody and T-cell responses in nonhuman primates. Journal of virology 84, 1683-1695 Literaturverzeichnis S e i t e | 145 M. DYKHUIZEN, J. L. MITCHEN, D. C. MONTEFIORI, J. THOMSON, L. ACKER, H. LARDY u. C. D. PAUZA (1998): Determinants of disease in the simian immunodeficiency virus-infected rhesus macaque: characterizing animals with low antibody responses and rapid progression. The Journal of general virology 79 ( Pt 10), 2461-2467 P. ENGEL, L. J. ZHOU, D. C. ORD, S. SATO, B. KOLLER u. T. F. TEDDER (1995): Abnormal B lymphocyte development, activation, and differentiation in mice that lack or overexpress the CD19 signal transduction molecule. Immunity 3, 39-50 M. FELDMANN u. A. EASTEN (1971): The relationship between antigenic structure and the requirement for thymus-derived cells in the immune response. The Journal of experimental medicine 134, 103-119 E. FENOUILLET, N. BLANES, A. COUTELLIER u. J. C. GLUCKMAN (1993): Relationship between anti-p24 antibody levels and p24 antigenemia in HIV-infected patients. AIDS research and human retroviruses 9, 1251-1255 G. FILICE, P. M. CEREDA u. O. E. VARNIER (1988): Infection of rabbits with human immunodeficiency virus. Nature 335, 366-369 T. H. FINKEL, G. TUDOR-WILLIAMS, N. K. BANDA, M. F. COTTON, T. CURIEL, C. MONKS, T. W. BABA, R. M. RUPRECHT u. A. KUPFER (1995): Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nature medicine 1, 129-134 J. M. FONDERE, M. F. HUGUET, H. YSSEL, V. BAILLAT, J. REYNES, P. VAN DE PERRE u. J. P. VENDRELL (2003): Detection of peripheral HIV-1-specific memory B cells in patients untreated or receiving highly active antiretroviral therapy. AIDS (London, England) 17, 2323-2330 J. FONTAINE, J. CHAGNON-CHOQUET, H. S. VALCKE, J. POUDRIER u. M. ROGER (2011): High expression levels of B lymphocyte stimulator (BLyS) by dendritic cells correlate with HIVrelated B-cell disease progression in humans. Blood 117, 145-155 E. O. FREED (2001): HIV-1 replication. Somatic cell and molecular genetics 26, 13-33 E. O. FREED, E. L. DELWART, G. L. BUCHSCHACHER, JR. u. A. T. PANGANIBAN (1992): A mutation in the human immunodeficiency virus type 1 transmembrane glycoprotein gp41 dominantly interferes with fusion and infectivity. Proceedings of the National Academy of Sciences of the United States of America 89, 70-74 146 | S e i t e Literaturverzeichnis T. GAUFIN, M. PATTISON, R. GAUTAM, C. STOULIG, J. DUFOUR, J. MACFARLAND, D. MANDELL, C. TATUM, M. H. MARX, R. M. RIBEIRO, D. MONTEFIORI, C. APETREI u. I. PANDREA (2009): Effect of B-cell depletion on viral replication and clinical outcome of simian immunodeficiency virus infection in a natural host. Journal of virology 83, 10347-10357 H. R. GELDERBLOM (1991): Assembly and morphology of HIV: potential effect of structure on viral function. AIDS (London, England) 5, 617-637 C. GELDMACHER, N. NGWENYAMA, A. SCHUETZ, C. PETROVAS, K. REITHER, E. J. HEEREGRAVE, J. P. CASAZZA, D. R. AMBROZAK, M. LOUDER, W. AMPOFO, G. POLLAKIS, B. HILL, E. SANGA, E. SAATHOFF, L. MABOKO, M. ROEDERER, W. A. PAXTON, M. HOELSCHER u. R. A. KOUP (2010): Preferential infection and depletion of Mycobacterium tuberculosis-specific CD4 T cells after HIV-1 infection. The Journal of experimental medicine 207, 2869-2881 A. M. GERETTI (1999): Simian immunodeficiency virus as a model of human HIV disease. Reviews in medical virology 9, 57-67 S. A. GLYNN, S. H. KLEINMAN, G. B. SCHREIBER, M. P. BUSCH, D. J. WRIGHT, J. W. SMITH, C. C. NASS u. A. E. WILLIAMS (2000): Trends in incidence and prevalence of major transfusion-transmissible viral infections in US blood donors, 1991 to 1996. Retrovirus Epidemiology Donor Study (REDS). Jama 284, 229-235 S. N. GORDON, N. R. KLATT, S. E. BOSINGER, J. M. BRENCHLEY, J. M. MILUSH, J. C. ENGRAM, R. M. DUNHAM, M. PAIARDINI, S. KLUCKING, A. DANESH, E. A. STROBERT, C. APETREI, I. V. PANDREA, D. KELVIN, D. C. DOUEK, S. I. STAPRANS, D. L. SODORA u. G. SILVESTRI (2007): Severe depletion of mucosal CD4+ T cells in AIDS-free simian immunodeficiency virus-infected sooty mangabeys. Journal of Immunology 179, 3026-3034 M. S. GOTTLIEB, R. SCHROFF, H. M. SCHANKER, J. D. WEISMAN, P. T. FAN, R. A. WOLF u. A. SAXON (1981): Pneumocystis carinii pneumonia and mucosal candidiasis in previously healthy homosexual men: evidence of a new acquired cellular immunodeficiency. The New England journal of medicine 305, 1425-1431 Z. GROSSMAN, M. MEIER-SCHELLERSHEIM, W. E. PAUL u. L. J. PICKER (2006): Pathogenesis of HIV infection: what the virus spares is as important as what it destroys. Nature medicine 12, 289-295 Literaturverzeichnis S e i t e | 147 A. E. GRULICH, X. WAN, M. G. LAW, S. T. MILLIKEN, C. R. LEWIS, R. J. GARSIA, J. GOLD, R. J. FINLAYSON, D. A. COOPER u. J. M. KALDOR (2000): B-cell stimulation and prolonged immune deficiency are risk factors for non-Hodgkin's lymphoma in people with AIDS. AIDS (London, England) 14, 133-140 Y. GUAN, M. M. SAJADI, R. KAMIN-LEWIS, T. R. FOUTS, A. DIMITROV, Z. ZHANG, R. R. REDFIELD, A. L. DEVICO, R. C. GALLO u. G. K. LEWIS (2009): Discordant memory B cell and circulating anti-Env antibody responses in HIV-1 infection. Proceedings of the National Academy of Sciences of the United States of America 106, 3952-3957 R. R. HARDY, Y. S. LI, D. ALLMAN, M. ASANO, M. GUI u. K. HAYAKAWA (2000): B-cell commitment, development and selection. Immunological reviews 175, 23-32 M. HART, A. STEEL, S. A. CLARK, G. MOYLE, M. NELSON, D. C. HENDERSON, R. WILSON, F. GOTCH, B. GAZZARD u. P. KELLEHER (2007): Loss of discrete memory B cell subsets is associated with impaired immunization responses in HIV-1 infection and may be a risk factor for invasive pneumococcal disease. Journal of Immunology 178, 8212-8220 B. HE, X. QIAO, P. J. KLASSE, A. CHIU, A. CHADBURN, D. M. KNOWLES, J. P. MOORE u. A. CERUTTI (2006): HIV-1 envelope triggers polyclonal Ig class switch recombination through a CD40-independent mechanism involving BAFF and C-type lectin receptors. Journal of Immunology 176, 3931-3941 J. L. HEENEY, A. G. DALGLEISH u. R. A. WEISS (2006): Origins of HIV and the evolution of resistance to AIDS. Science (New York, N.Y.) 313, 462-466 R. HIDAJAT, P. XIAO, Q. ZHOU, D. VENZON, L. E. SUMMERS, V. S. KALYANARAMAN, D. C. MONTEFIORI u. M. ROBERT-GUROFF (2009): Correlation of vaccine-elicited systemic and mucosal nonneutralizing antibody activities with reduced acute viremia following intrarectal simian immunodeficiency virus SIVmac251 challenge of rhesus macaques. Journal of virology 83, 791-801 F. HLADIK u. M. J. MCELRATH (2008): Setting the stage: host invasion by HIV. Nature reviews 8, 447-457 J. HO, S. MOIR, A. MALASPINA, M. L. HOWELL, W. WANG, A. C. DIPOTO, M. A. O'SHEA, G. A. ROBY, R. KWAN, J. M. MICAN, T. W. CHUN u. A. S. FAUCI (2006): Two overrepresented B cell populations in HIV-infected individuals undergo apoptosis by different mechanisms. Proceedings of the National Academy of Sciences of the United States of America 103, 19436-19441 148 | S e i t e Literaturverzeichnis E. HOGERVORST, S. JURRIAANS, F. DE WOLF, A. VAN WIJK, A. WIERSMA, M. VALK, M. ROOS, B. VAN GEMEN, R. COUTINHO, F. MIEDEMA u. ET AL. (1995): Predictors for non- and slow progression in human immunodeficiency virus (HIV) type 1 infection: low viral RNA copy numbers in serum and maintenance of high HIV-1 p24-specific but not V3specific antibody levels. The Journal of infectious diseases 171, 811-821 C. M. JANEWAY, K.; TRAVERS, P.; WALPORT, M. (2008): Janeway's Immunobiology. Garland Science, Taylor & Francis Group, LLC, C. JOLLY, K. KASHEFI, M. HOLLINSHEAD u. Q. J. SATTENTAU (2004): HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. The Journal of experimental medicine 199, 283-293 K. KAHNT, K. MATZ-RENSING, P. HOFMANN, C. STAHL-HENNIG u. F. J. KAUP (2002): SIV-associated lymphomas in rhesus monkeys (Macaca mulatta) in comparison with HIV-associated lymphomas. Veterinary pathology 39, 42-55 A. KAUR, M. DI MASCIO, A. BARABASZ, M. ROSENZWEIG, H. M. MCCLURE, A. S. PERELSON, R. M. RIBEIRO u. R. P. JOHNSON (2008): Dynamics of T- and B-lymphocyte turnover in a natural host of simian immunodeficiency virus. Journal of virology 82, 1084-1093 B. F. KEELE, J. H. JONES, K. A. TERIO, J. D. ESTES, R. S. RUDICELL, M. L. WILSON, Y. LI, G. H. LEARN, T. M. BEASLEY, J. SCHUMACHER-STANKEY, E. WROBLEWSKI, A. MOSSER, J. RAPHAEL, S. KAMENYA, E. V. LONSDORF, D. A. TRAVIS, T. MLENGEYA, M. J. KINSEL, J. G. ELSE, G. SILVESTRI, J. GOODALL, P. M. SHARP, G. M. SHAW, A. E. PUSEY u. B. H. HAHN (2009): Increased mortality and AIDS-like immunopathology in wild chimpanzees infected with SIVcpz. Nature 460, 515-519 H. KESTLER, T. KODAMA, D. RINGLER, M. MARTHAS, N. PEDERSEN, A. LACKNER, D. REGIER, P. SEHGAL, M. DANIEL, N. KING u. ET AL. (1990): Induction of AIDS in rhesus monkeys by molecularly cloned simian immunodeficiency virus. Science (New York, N.Y.) 248, 1109-1112 P. KIEPIELA, K. NGUMBELA, C. THOBAKGALE, D. RAMDUTH, I. HONEYBORNE, E. MOODLEY, S. REDDY, C. DE PIERRES, Z. MNCUBE, N. MKHWANAZI, K. BISHOP, M. VAN DER STOK, K. NAIR, N. KHAN, H. CRAWFORD, R. PAYNE, A. LESLIE, J. PRADO, A. PRENDERGAST, J. FRATER, N. MCCARTHY, C. BRANDER, G. H. LEARN, D. NICKLE, C. ROUSSEAU, H. COOVADIA, J. I. MULLINS, D. HECKERMAN, B. D. WALKER u. P. GOULDER (2007): CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nature medicine 13, 46-53 U. KLEIN, K. RAJEWSKY u. R. KUPPERS (1998): Human immunoglobulin (Ig)M+IgD+ peripheral blood B cells expressing the CD27 cell surface antigen carry somatically mutated variable region genes: CD27 as a general marker for somatically mutated (memory) B cells. The Journal of experimental medicine 188, 1679-1689 Literaturverzeichnis S e i t e | 149 H. M. KLING, T. W. SHIPLEY u. K. A. NORRIS (2011): Alterations in peripheral blood B-cell populations in SHIV89.6P-infected macaques (Macacca fascicularis). Comparative medicine 61, 269-277 K. V. KOMANDURI, J. FEINBERG, R. K. HUTCHINS, R. D. FRAME, D. K. SCHMIDT, M. N. VISWANATHAN, J. P. LALEZARI u. J. M. MCCUNE (2001): Loss of cytomegalovirus-specific CD4+ T cell responses in human immunodeficiency virus type 1infected patients with high CD4+ T cell counts and recurrent retinitis. The Journal of infectious diseases 183, 1285-1289 S. KUCHEN, R. ROBBINS, G. P. SIMS, C. SHENG, T. M. PHILLIPS, P. E. LIPSKY u. R. ETTINGER (2007): Essential role of IL-21 in B cell activation, expansion, and plasma cell generation during CD4+ T cellB cell collaboration. Journal of Immunology 179, 5886-5896 D. KUHRT, S. FAITH, A. HATTEMER, A. LEONE, D. SODORA, L. PICKER, L. BORGHESI u. K. S. COLE (2011): Naive and memory B cells in the rhesus macaque can be differentiated by surface expression of CD27 and have differential responses to CD40 ligation. Journal of immunological methods 363, 166-176 D. KUHRT, S. A. FAITH, A. LEONE, M. ROHANKEDKAR, D. L. SODORA, L. J. PICKER u. K. S. COLE (2010): Evidence of early B-cell dysregulation in simian immunodeficiency virus infection: rapid depletion of naive and memory B-cell subsets with delayed reconstitution of the naive B-cell population. Journal of virology 84, 2466-2476 A. KUMAR, W. WEISS, J. A. TINE, S. L. HOFFMAN u. W. O. ROGERS (2001): ELISPOT assay for detection of peptide specific interferon-gamma secreting cells in rhesus macaques. Journal of immunological methods 247, 49-60 T. KUROSAKI, Y. AIBA, K. KOMETANI, S. MORIYAMA u. Y. TAKAHASHI (2010): Unique properties of memory B cells of different isotypes. Immunological reviews 237, 104-116 P. D. KWONG, R. WYATT, J. ROBINSON, R. W. SWEET, J. SODROSKI u. W. A. HENDRICKSON (1998): Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393, 648-659 O. LAMBOTTE, G. FERRARI, C. MOOG, N. L. YATES, H. X. LIAO, R. J. PARKS, C. B. HICKS, K. OWZAR, G. D. TOMARAS, D. C. MONTEFIORI, B. F. HAYNES u. J. F. DELFRAISSY (2009): Heterogeneous neutralizing antibody and antibody-dependent cell cytotoxicity responses in HIV-1 elite controllers. AIDS (London, England) 23, 897-906 150 | S e i t e Literaturverzeichnis R. LE GRAND, G. VOGT, B. VASLIN, P. ROQUES, F. THEODORO, A. M. AUBERTIN u. D. DORMONT (1992): Specific and non-specific immunity and protection of macaques against SIV infection. Vaccine 10, 873-879 T. W. LEBIEN (2000): Fates of human B-cell precursors. Blood 96, 9-23 J. A. LEVY (2009): HIV pathogenesis: 25 years of progress and persistent challenges. AIDS (London, England) 23, 147-160 M. LI, F. GAO, J. R. MASCOLA, L. STAMATATOS, V. R. POLONIS, M. KOUTSOUKOS, G. VOSS, P. GOEPFERT, P. GILBERT, K. M. GREENE, M. BILSKA, D. L. KOTHE, J. F. SALAZARGONZALEZ, X. WEI, J. M. DECKER, B. H. HAHN u. D. C. MONTEFIORI (2005): Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. Journal of virology 79, 10108-10125 J. D. LIFSON, M. A. NOWAK, S. GOLDSTEIN, J. L. ROSSIO, A. KINTER, G. VASQUEZ, T. A. WILTROUT, C. BROWN, D. SCHNEIDER, L. WAHL, A. L. LLOYD, J. WILLIAMS, W. R. ELKINS, A. S. FAUCI u. V. M. HIRSCH (1997): The extent of early viral replication is a critical determinant of the natural history of simian immunodeficiency virus infection. Journal of virology 71, 9508-9514 Z. M. MA, M. STONE, M. PIATAK, JR., B. SCHWEIGHARDT, N. L. HAIGWOOD, D. MONTEFIORI, J. D. LIFSON, M. P. BUSCH u. C. J. MILLER (2009): High specific infectivity of plasma virus from the pre-ramp-up and ramp-up stages of acute simian immunodeficiency virus infection. Journal of virology 83, 3288-3297 M. MAHALANABIS, P. JAYARAMAN, T. MIURA, F. PEREYRA, E. M. CHESTER, B. RICHARDSON, B. WALKER u. N. L. HAIGWOOD (2009): Continuous viral escape and selection by autologous neutralizing antibodies in drug-naive human immunodeficiency virus controllers. Journal of virology 83, 662-672 A. MALASPINA, S. MOIR, D. G. CHAITT, C. A. REHM, S. KOTTILIL, J. FALLOON u. A. S. FAUCI (2007): Idiopathic CD4+ T lymphocytopenia is associated with increases in immature/transitional B cells and serum levels of IL-7. Blood 109, 2086-2088 A. MALASPINA, S. MOIR, J. HO, W. WANG, M. L. HOWELL, M. A. O'SHEA, G. A. ROBY, C. A. REHM, J. M. MICAN, T. W. CHUN u. A. S. FAUCI (2006): Appearance of immature/transitional B cells in HIV-infected individuals with advanced disease: correlation with increased IL-7. Proceedings of the National Academy of Sciences of the United States of America 103, 2262-2267 Literaturverzeichnis S e i t e | 151 A. MALASPINA, S. MOIR, D. C. NICKLE, E. T. DONOGHUE, K. M. OGWARO, L. A. EHLER, S. LIU, J. A. MICAN, M. DYBUL, T. W. CHUN, J. I. MULLINS u. A. S. FAUCI (2002): Human immunodeficiency virus type 1 bound to B cells: relationship to virus replicating in CD4+ T cells and circulating in plasma. Journal of virology 76, 8855-8863 M. MAMANI-MATSUDA, A. COSMA, S. WELLER, A. FAILI, C. STAIB, L. GARCON, O. HERMINE, O. BEYNE-RAUZY, C. FIESCHI, J. O. PERS, N. ARAKELYAN, B. VARET, A. SAUVANET, A. BERGER, F. PAYE, J. M. ANDRIEU, M. MICHEL, B. GODEAU, P. BUFFET, C. A. REYNAUD u. J. C. WEILL (2008): The human spleen is a major reservoir for long-lived vaccinia virus-specific memory B cells. Blood 111, 4653-4659 R. A. MANZ, M. LOHNING, G. CASSESE, A. THIEL u. A. RADBRUCH (1998): Survival of long-lived plasma cells is independent of antigen. International immunology 10, 1703-1711 O. MARTINEZ-MAZA, E. CRABB, R. T. MITSUYASU, J. L. FAHEY u. J. V. GIORGI (1987): Infection with the human immunodeficiency virus (HIV) is associated with an in vivo increase in B lymphocyte activation and immaturity. Journal of Immunology 138, 3720-3724 J. R. MASCOLA u. D. C. MONTEFIORI (2010): The role of antibodies in HIV vaccines. Annual review of immunology 28, 413-444 J. R. MASCOLA, G. STIEGLER, T. C. VANCOTT, H. KATINGER, C. B. CARPENTER, C. E. HANSON, H. BEARY, D. HAYES, S. S. FRANKEL, D. L. BIRX u. M. G. LEWIS (2000): Protection of macaques against vaginal transmission of a pathogenic HIV-1/SIV chimeric virus by passive infusion of neutralizing antibodies. Nature medicine 6, 207-210 L. J. MCHEYZER-WILLIAMS, N. PELLETIER, L. MARK, N. FAZILLEAU u. M. G. MCHEYZER-WILLIAMS (2009): Follicular helper T cells as cognate regulators of B cell immunity. Current opinion in immunology 21, 266-273 M. MEDINA-RAMIREZ, V. SANCHEZ-MERINO, S. SANCHEZ-PALOMINO, A. MERINOMANSILLA, C. B. FERREIRA, I. PEREZ, N. GONZALEZ, A. ALVAREZ, J. M. ALCOCERGONZALEZ, F. GARCIA, J. M. GATELL, J. ALCAMI u. E. YUSTE (2011): Broadly cross-neutralizing antibodies in HIV-1 patients with undetectable viremia. Journal of virology 85, 5804-5813 F. MEDINA, C. SEGUNDO, A. CAMPOS-CARO, I. GONZALEZ-GARCIA u. J. A. BRIEVA (2002): The heterogeneity shown by human plasma cells from tonsil, blood, and bone marrow reveals graded stages of increasing maturity, but local profiles of adhesion molecule expression. Blood 99, 2154-2161 152 | S e i t e Literaturverzeichnis F. MEDINA, C. SEGUNDO, A. CAMPOS-CARO, I. SALCEDO, A. GARCIA-POLEY u. J. A. BRIEVA (2003): Isolation, maturational level, and functional capacity of human colon lamina propria plasma cells. Gut 52, 383-389 F. MIEDEMA, A. J. PETIT, F. G. TERPSTRA, J. K. SCHATTENKERK, F. DE WOLF, B. J. AL, M. ROOS, J. M. LANGE, S. A. DANNER, J. GOUDSMIT u. ET AL. (1988): Immunological abnormalities in human immunodeficiency virus (HIV)-infected asymptomatic homosexual men. HIV affects the immune system before CD4+ T helper cell depletion occurs. The Journal of clinical investigation 82, 1908-1914 C. J. MILLER, M. GENESCA, K. ABEL, D. MONTEFIORI, D. FORTHAL, K. BOST, J. LI, D. FAVRE u. J. M. MCCUNE (2007): Antiviral antibodies are necessary for control of simian immunodeficiency virus replication. Journal of virology 81, 5024-5035 C. J. MILLER, M. B. MCCHESNEY, X. LU, P. J. DAILEY, C. CHUTKOWSKI, D. LU, P. BROSIO, B. ROBERTS u. Y. LU (1997): Rhesus macaques previously infected with simian/human immunodeficiency virus are protected from vaginal challenge with pathogenic SIVmac239. Journal of virology 71, 1911-1921 H. MOHRI, S. BONHOEFFER, S. MONARD, A. S. PERELSON u. D. D. HO (1998): Rapid turnover of T lymphocytes in SIV-infected rhesus macaques. Science (New York, N.Y.) 279, 1223-1227 S. MOIR u. A. S. FAUCI (2008): Pathogenic mechanisms of B-lymphocyte dysfunction in HIV disease. The Journal of allergy and clinical immunology 122, 12-19; quiz 20-11 S. MOIR, J. HO, A. MALASPINA, W. WANG, A. C. DIPOTO, M. A. O'SHEA, G. ROBY, S. KOTTILIL, J. ARTHOS, M. A. PROSCHAN, T. W. CHUN u. A. S. FAUCI (2008a): Evidence for HIV-associated B cell exhaustion in a dysfunctional memory B cell compartment in HIV-infected viremic individuals. The Journal of experimental medicine 205, 1797-1805 S. MOIR, A. MALASPINA, J. HO, W. WANG, A. C. DIPOTO, M. A. O'SHEA, G. ROBY, J. M. MICAN, S. KOTTILIL, T. W. CHUN, M. A. PROSCHAN u. A. S. FAUCI (2008b): Normalization of B cell counts and subpopulations after antiretroviral therapy in chronic HIV disease. The Journal of infectious diseases 197, 572-579 S. MOIR, A. MALASPINA, Y. LI, T. W. CHUN, T. LOWE, J. ADELSBERGER, M. BASELER, L. A. EHLER, S. LIU, R. T. DAVEY, JR., J. A. MICAN u. A. S. FAUCI (2000): B cells of HIV-1-infected patients bind virions through CD21-complement interactions and transmit infectious virus to activated T cells. The Journal of experimental medicine 192, 637-646 Literaturverzeichnis S e i t e | 153 S. MOIR, A. MALASPINA, K. M. OGWARO, E. T. DONOGHUE, C. W. HALLAHAN, L. A. EHLER, S. LIU, J. ADELSBERGER, R. LAPOINTE, P. HWU, M. BASELER, J. M. ORENSTEIN, T. W. CHUN, J. A. MICAN u. A. S. FAUCI (2001): HIV-1 induces phenotypic and functional perturbations of B cells in chronically infected individuals. Proceedings of the National Academy of Sciences of the United States of America 98, 10362-10367 S. MOIR, A. MALASPINA, O. K. PICKERAL, E. T. DONOGHUE, J. VASQUEZ, N. J. MILLER, S. R. KRISHNAN, M. A. PLANTA, J. F. TURNEY, J. S. JUSTEMENT, S. KOTTILIL, M. DYBUL, J. M. MICAN, C. KOVACS, T. W. CHUN, C. E. BIRSE u. A. S. FAUCI (2004): Decreased survival of B cells of HIV-viremic patients mediated by altered expression of receptors of the TNF superfamily. The Journal of experimental medicine 200, 587-599 D. C. MONTEFIORI (2009): Measuring HIV neutralization in a luciferase reporter gene assay. Methods in molecular biology (Clifton, N.J 485, 395-405 J. P. MOORE u. J. SODROSKI (1996): Antibody cross-competition analysis of the human immunodeficiency virus type 1 gp120 exterior envelope glycoprotein. Journal of virology 70, 1863-1872 H. MORBACH, E. M. EICHHORN, J. G. LIESE u. H. J. GIRSCHICK (2010): Reference values for B cell subpopulations from infancy to adulthood. Clinical and experimental immunology 162, 271-279 L. MORRIS, J. M. BINLEY, B. A. CLAS, S. BONHOEFFER, T. P. ASTILL, R. KOST, A. HURLEY, Y. CAO, M. MARKOWITZ, D. D. HO u. J. P. MOORE (1998): HIV-1 antigen-specific and -nonspecific B cell responses are sensitive to combination antiretroviral therapy. The Journal of experimental medicine 188, 233-245 W. J. MORROW, M. WHARTON, D. LAU u. J. A. LEVY (1987): Small animals are not susceptible to human immunodeficiency virus infection. The Journal of general virology 68 ( Pt 8), 2253-2257 M. C. MULLER-TRUTWIN, S. CORBET, M. D. TAVARES, V. M. HERVE, E. NERRIENET, M. C. GEORGES-COURBOT, W. SAURIN, P. SONIGO u. F. BARRE-SINOUSSI (1996): The evolutionary rate of nonpathogenic simian immunodeficiency virus (SIVagm) is in agreement with a rapid and continuous replication in vivo. Virology 223, 89-102 F. MULLER, P. AUKRUST, I. NORDOY u. S. S. FROLAND (1998): Possible role of interleukin-10 (IL-10) and CD40 ligand expression in the pathogenesis of hypergammaglobulinemia in human immunodeficiency virus infection: modulation of IL-10 and Ig production after intravenous Ig infusion. Blood 92, 3721-3729 154 | S e i t e Literaturverzeichnis G. NABI, V. TEMCHURA, C. GROSSMANN, S. KUATE, M. TENBUSCH u. K. UBERLA (2012): T cell independent secondary antibody responses to the envelope protein of simian immunodeficiency virus. Retrovirology 9, 42 H. NAGASE, K. AGEMATSU, K. KITANO, M. TAKAMOTO, Y. OKUBO, A. KOMIYAMA u. K. SUGANE (2001): Mechanism of hypergammaglobulinemia by HIV infection: circulating memory B-cell reduction with plasmacytosis. Clinical immunology (Orlando, FL) 100, 250-259 D. R. NEGRI, S. BARONCELLI, S. CATONE, A. COMINI, Z. MICHELINI, M. T. MAGGIORELLA, L. SERNICOLA, F. CROSTAROSA, R. BELLI, M. G. MANCINI, S. FARCOMENI, Z. FAGROUCH, M. CICCOZZI, S. BOROS, P. LILJESTROM, S. NORLEY, J. HEENEY u. F. TITTI (2004): Protective efficacy of a multicomponent vector vaccine in cynomolgus monkeys after intrarectal simian immunodeficiency virus challenge. The Journal of general virology 85, 1191-1201 O. NEILDEZ, R. LE GRAND, A. CHERET, P. CAUFOUR, B. VASLIN, F. MATHEUX, F. THEODORO, P. ROQUES u. D. DORMONT (1998): Variation in virological parameters and antibody responses in macaques after atraumatic vaginal exposure to a pathogenic primary isolate of SIVmac251. Research in virology 149, 53-68 M. NOMAGUCHI, N. DOI, K. KAMADA u. A. ADACHI (2008): Species barrier of HIV-1 and its jumping by virus engineering. Reviews in medical virology 18, 261-275 A. M. ORTIZ, N. R. KLATT, B. LI, Y. YI, B. TABB, X. P. HAO, L. STERNBERG, B. LAWSON, P. M. CARNATHAN, E. M. CRAMER, J. C. ENGRAM, D. M. LITTLE, E. RYZHOVA, F. GONZALEZ-SCARANO, M. PAIARDINI, A. A. ANSARI, S. RATCLIFFE, J. G. ELSE, J. M. BRENCHLEY, R. G. COLLMAN, J. D. ESTES, C. A. DERDEYN u. G. SILVESTRI (2011): Depletion of CD4(+) T cells abrogates post-peak decline of viremia in SIV-infected rhesus macaques. The Journal of clinical investigation 121, 4433-4445 N. S. PADIAN, S. C. SHIBOSKI u. N. P. JEWELL (1991): Female-to-male transmission of human immunodeficiency virus. Jama 266, 1664-1667 S. PALLIKKUTH, S. PILAKKA KANTHIKEEL, S. Y. SILVA, M. FISCHL, R. PAHWA u. S. PAHWA (2011): Upregulation of IL-21 receptor on B cells and IL-21 secretion distinguishes novel 2009 H1N1 vaccine responders from nonresponders among HIV-infected persons on combination antiretroviral therapy. Journal of Immunology 186, 6173-6181 I. PANDREA, D. L. SODORA, G. SILVESTRI u. C. APETREI (2008): Into the wild: simian immunodeficiency virus (SIV) infection in natural hosts. Trends in immunology 29, 419-428 Literaturverzeichnis S e i t e | 155 G. PANTALEO u. A. S. FAUCI (1996): Immunopathogenesis of HIV infection. Annual review of microbiology 50, 825-854 D. C. PARKER (1993): The functions of antigen recognition in T cell-dependent B cell activation. Seminars in immunology 5, 413-420 P. W. PARREN, P. A. MARX, A. J. HESSELL, A. LUCKAY, J. HAROUSE, C. CHENG-MAYER, J. P. MOORE u. D. R. BURTON (2001): Antibody protects macaques against vaginal challenge with a pathogenic R5 simian/human immunodeficiency virus at serum levels giving complete neutralization in vitro. Journal of virology 75, 8340-8347 S. PENSIEROSO, A. CAGIGI, P. PALMA, A. NILSSON, C. CAPPONI, E. FREDA, S. BERNARDI, R. THORSTENSSON, F. CHIODI u. P. ROSSI (2009): Timing of HAART defines the integrity of memory B cells and the longevity of humoral responses in HIV-1 vertically-infected children. Proceedings of the National Academy of Sciences of the United States of America 106, 7939-7944 F. PEREYRA, M. M. ADDO, D. E. KAUFMANN, Y. LIU, T. MIURA, A. RATHOD, B. BAKER, A. TROCHA, R. ROSENBERG, E. MACKEY, P. UEDA, Z. LU, D. COHEN, T. WRIN, C. J. PETROPOULOS, E. S. ROSENBERG u. B. D. WALKER (2008): Genetic and immunologic heterogeneity among persons who control HIV infection in the absence of therapy. The Journal of infectious diseases 197, 563-571 M. PEREZ-ANDRES, B. PAIVA, W. G. NIETO, A. CARAUX, A. SCHMITZ, J. ALMEIDA, R. F. VOGT, JR., G. E. MARTI, A. C. RAWSTRON, M. C. VAN ZELM, J. J. VAN DONGEN, H. E. JOHNSEN, B. KLEIN u. A. ORFAO (2010): Human peripheral blood B-cell compartments: a crossroad in B-cell traffic. Cytometry 78 Suppl 1, S47-60 S. PERUCHON, N. CHAOUL, C. BURELOUT, B. DELACHE, P. BROCHARD, P. LAURENT, F. COGNASSE, S. PREVOT, O. GARRAUD, R. LE GRAND u. Y. RICHARD (2009): Tissue-specific B-cell dysfunction and generalized memory B-cell loss during acute SIV infection. PloS one 4, e5966 A. PIANTADOSI, D. PANTELEEFF, C. A. BLISH, J. M. BAETEN, W. JAOKO, R. S. MCCLELLAND u. J. OVERBAUGH (2009): Breadth of neutralizing antibody response to human immunodeficiency virus type 1 is affected by factors early in infection but does not influence disease progression. Journal of virology 83, 10269-10274 A. K. PILGRIM, G. PANTALEO, O. J. COHEN, L. M. FINK, J. Y. ZHOU, J. T. ZHOU, D. P. BOLOGNESI, A. S. FAUCI u. D. C. MONTEFIORI (1997): Neutralizing antibody responses to human immunodeficiency virus type 1 in primary infection and long-term-nonprogressive infection. The Journal of infectious diseases 176, 924-932 156 | S e i t e Literaturverzeichnis J. C. PLANTIER, M. LEOZ, J. E. DICKERSON, F. DE OLIVEIRA, F. CORDONNIER, V. LEMEE, F. DAMOND, D. L. ROBERTSON u. F. SIMON (2009): A new human immunodeficiency virus derived from gorillas. Nature medicine 15, 871-872 E. J. PONE, H. ZAN, J. ZHANG, A. AL-QAHTANI, Z. XU u. P. CASALI (2010): Toll-like receptors and B-cell receptors synergize to induce immunoglobulin class-switch DNA recombination: relevance to microbial antibody responses. Critical reviews in immunology 30, 1-29 R. RACINE u. G. M. WINSLOW (2009): IgM in microbial infections: taken for granted? Immunology letters 125, 79-85 A. RADBRUCH, G. MUEHLINGHAUS, E. O. LUGER, A. INAMINE, K. G. SMITH, T. DORNER u. F. HIEPE (2006): Competence and competition: the challenge of becoming a long-lived plasma cell. Nature reviews 6, 741-750 K. RAJEWSKY (1996): Clonal selection and learning in the antibody system. Nature 381, 751-758 S. A. READING u. N. J. DIMMOCK (2007): Neutralization of animal virus infectivity by antibody. Archives of virology 152, 1047-1059 D. A. REGIER u. R. C. DESROSIERS (1990): The complete nucleotide sequence of a pathogenic molecular clone of simian immunodeficiency virus. AIDS research and human retroviruses 6, 1221-1231 K. A. REIMANN, K. TENNER-RACZ, P. RACZ, D. C. MONTEFIORI, Y. YASUTOMI, W. LIN, B. J. RANSIL u. N. L. LETVIN (1994): Immunopathogenic events in acute infection of rhesus monkeys with simian immunodeficiency virus of macaques. Journal of virology 68, 2362-2370 M. A. REY-CUILLE, J. L. BERTHIER, M. C. BOMSEL-DEMONTOY, Y. CHADUC, L. MONTAGNIER, A. G. HOVANESSIAN u. L. A. CHAKRABARTI (1998): Simian immunodeficiency virus replicates to high levels in sooty mangabeys without inducing disease. Journal of virology 72, 3872-3886 Y. RICHARD, C. AMIEL, V. JEANTILS, D. MESTIVIER, A. PORTIER, G. DHELLO, J. FEUILLARD, R. CREIDY, J. C. NICOLAS u. M. RAPHAEL (2010): Changes in blood B cell phenotypes and Epstein-Barr virus load in chronically human immunodeficiency virus-infected patients before and after antiretroviral therapy. The Journal of infectious diseases 202, 1424-1434 Literaturverzeichnis S e i t e | 157 P. RIECKMANN, G. POLI, C. H. FOX, J. H. KEHRL u. A. S. FAUCI (1991): Recombinant gp120 specifically enhances tumor necrosis factor-alpha production and Ig secretion in B lymphocytes from HIV-infected individuals but not from seronegative donors. Journal of Immunology 147, 2922-2927 T. L. ROTHSTEIN, J. K. WANG, D. J. PANKA, L. C. FOOTE, Z. WANG, B. STANGER, H. CUI, S. T. JU u. A. MARSHAK-ROTHSTEIN (1995): Protection against Fas-dependent Th1-mediated apoptosis by antigen receptor engagement in B cells. Nature 374, 163-165 J. S. RUSH u. P. D. HODGKIN (2001): B cells activated via CD40 and IL-4 undergo a division burst but require continued stimulation to maintain division, survival and differentiation. European journal of immunology 31, 1150-1159 J. B. SACHA, C. CHUNG, E. G. RAKASZ, S. P. SPENCER, A. K. JONAS, A. T. BEAN, W. LEE, B. J. BURWITZ, J. J. STEPHANY, J. T. LOFFREDO, D. B. ALLISON, S. ADNAN, A. HOJI, N. A. WILSON, T. C. FRIEDRICH, J. D. LIFSON, O. O. YANG u. D. I. WATKINS (2007): Gag-specific CD8+ T lymphocytes recognize infected cells before AIDS-virus integration and viral protein expression. Journal of Immunology 178, 2746-2754 D. N. SATHER, J. ARMANN, L. K. CHING, A. MAVRANTONI, G. SELLHORN, Z. CALDWELL, X. YU, B. WOOD, S. SELF, S. KALAMS u. L. STAMATATOS (2009): Factors associated with the development of cross-reactive neutralizing antibodies during human immunodeficiency virus type 1 infection. Journal of virology 83, 757-769 Q. J. SATTENTAU (1988): The role of the CD4 antigen in HIV infection and immune pathogenesis. AIDS (London, England) 2 Suppl 1, S11-16 U. SAUERMANN, M. KRAWCZAK, G. HUNSMANN u. C. STAHL-HENNIG (1997): Identification of Mhc-Mamu-DQB1 allele combinations associated with rapid disease progression in rhesus macaques infected with simian immunodeficiency virus. AIDS (London, England) 11, 1196-1198 W. W. SCHAMEL u. M. RETH (2000): Monomeric and oligomeric complexes of the B cell antigen receptor. Immunity 13, 5-14 J. E. SCHMITZ, M. J. KURODA, S. SANTRA, V. G. SASSEVILLE, M. A. SIMON, M. A. LIFTON, P. RACZ, K. TENNER-RACZ, M. DALESANDRO, B. J. SCALLON, J. GHRAYEB, M. A. FORMAN, D. C. MONTEFIORI, E. P. RIEBER, N. L. LETVIN u. K. A. REIMANN (1999): Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science (New York, N.Y.) 283, 857-860 158 | S e i t e Literaturverzeichnis J. E. SCHMITZ, M. J. KURODA, S. SANTRA, M. A. SIMON, M. A. LIFTON, W. LIN, R. KHUNKHUN, M. PIATAK, J. D. LIFSON, G. GROSSCHUPFF, R. S. GELMAN, P. RACZ, K. TENNER-RACZ, K. A. MANSFIELD, N. L. LETVIN, D. C. MONTEFIORI u. K. A. REIMANN (2003): Effect of humoral immune responses on controlling viremia during primary infection of rhesus monkeys with simian immunodeficiency virus. Journal of virology 77, 2165-2173 S. M. SCHNITTMAN, H. C. LANE, S. E. HIGGINS, T. FOLKS u. A. S. FAUCI (1986): Direct polyclonal activation of human B lymphocytes by the acquired immune deficiency syndrome virus. Science (New York), N.Y 233, 1084-1086 G. B. SCHREIBER, M. P. BUSCH, S. H. KLEINMAN u. J. J. KORELITZ (1996): The risk of transfusion-transmitted viral infections. The Retrovirus Epidemiology Donor Study. The New England journal of medicine 334, 1685-1690 H. W. SCHROEDER, JR. u. L. CAVACINI (2010): Structure and function of immunoglobulins. The Journal of allergy and clinical immunology 125, S41-52 R. SCHULTE, Y. S. SUH, U. SAUERMANN, W. OCHIENG, S. SOPPER, K. S. KIM, S. S. AHN, K. S. PARK, N. STOLTE-LEEB, G. HUNSMANN, Y. C. SUNG u. C. STAHL-HENNIG (2009): Mucosal prior to systemic application of recombinant adenovirus boosting is more immunogenic than systemic application twice but confers similar protection against SIV-challenge in DNA vaccineprimed macaques. Virology 383, 300-309 T. SCHULTHEISS, R. SCHULTE, U. SAUERMANN, W. IBING u. C. STAHL-HENNIG (2011): Strong mucosal immune responses in SIV infected macaques contribute to viral control and preserved CD4+ T-cell levels in blood and mucosal tissues. Retrovirology 8, 24 T. SCHULTHEISS, N. STOLTE-LEEB, S. SOPPER u. C. STAHL-HENNIG (2010): Flow cytometric characterization of the lymphocyte composition in a variety of mucosal tissues in healthy rhesus macaques. Journal of medical primatology 40, 41-51 J. D. SEDGWICK u. P. G. HOLT (1983): A solid-phase immunoenzymatic technique for the enumeration of specific antibody-secreting cells. Journal of immunological methods 57, 301-309 Literaturverzeichnis S e i t e | 159 R. SHIBATA, T. IGARASHI, N. HAIGWOOD, A. BUCKLER-WHITE, R. OGERT, W. ROSS, R. WILLEY, M. W. CHO u. M. A. MARTIN (1999): Neutralizing antibody directed against the HIV-1 envelope glycoprotein can completely block HIV1/SIV chimeric virus infections of macaque monkeys. Nature medicine 5, 204-210 A. SHIRAI, M. COSENTINO, S. F. LEITMAN-KLINMAN u. D. M. KLINMAN (1992): Human immunodeficiency virus infection induces both polyclonal and virus-specific B cell activation. The Journal of clinical investigation 89, 561-566 J. W. SHIVER, T. M. FU, L. CHEN, D. R. CASIMIRO, M. E. DAVIES, R. K. EVANS, Z. Q. ZHANG, A. J. SIMON, W. L. TRIGONA, S. A. DUBEY, L. HUANG, V. A. HARRIS, R. S. LONG, X. LIANG, L. HANDT, W. A. SCHLEIF, L. ZHU, D. C. FREED, N. V. PERSAUD, L. GUAN, K. S. PUNT, A. TANG, M. CHEN, K. A. WILSON, K. B. COLLINS, G. J. HEIDECKER, V. R. FERNANDEZ, H. C. PERRY, J. G. JOYCE, K. M. GRIMM, J. C. COOK, P. M. KELLER, D. S. KRESOCK, H. MACH, R. D. TROUTMAN, L. A. ISOPI, D. M. WILLIAMS, Z. XU, K. E. BOHANNON, D. B. VOLKIN, D. C. MONTEFIORI, A. MIURA, G. R. KRIVULKA, M. A. LIFTON, M. J. KURODA, J. E. SCHMITZ, N. L. LETVIN, M. J. CAULFIELD, A. J. BETT, R. YOUIL, D. C. KASLOW u. E. A. EMINI (2002): Replication-incompetent adenoviral vaccine vector elicits effective anti-immunodeficiency-virus immunity. Nature 415, 331-335 F. P. SIEGAL, C. LOPEZ, G. S. HAMMER, A. E. BROWN, S. J. KORNFELD, J. GOLD, J. HASSETT, S. Z. HIRSCHMAN, C. CUNNINGHAM-RUNDLES, B. R. ADELSBERG u. ET AL. (1981): Severe acquired immunodeficiency in male homosexuals, manifested by chronic perianal ulcerative herpes simplex lesions. The New England journal of medicine 305, 1439-1444 M. K. SLIFKA, R. ANTIA, J. K. WHITMIRE u. R. AHMED (1998): Humoral immunity due to long-lived plasma cells. Immunity 8, 363-372 M. K. SLIFKA, M. MATLOUBIAN u. R. AHMED (1995): Bone marrow is a major site of long-term antibody production after acute viral infection. Journal of virology 69, 1895-1902 A. SLOWE u. H. WALDMANN (1975): The 'intrinsic adjuvanticity' and immunogenicity of trinitrophenylated lipopolysaccharide. Immunology 29, 825-834 S. M. SMITH, B. HOLLAND, C. RUSSO, P. J. DAILEY, P. A. MARX u. R. I. CONNOR (1999): Retrospective analysis of viral load and SIV antibody responses in rhesus macaques infected with pathogenic SIV: predictive value for disease progression. AIDS research and human retroviruses 15, 1691-1701 160 | S e i t e Literaturverzeichnis S. SOPPER, D. NIERWETBERG, A. HALBACH, U. SAUER, C. SCHELLER, C. STAHL-HENNIG, K. MATZ-RENSING, F. SCHAFER, T. SCHNEIDER, V. TER MEULEN u. J. G. MULLER (2003): Impact of simian immunodeficiency virus (SIV) infection on lymphocyte numbers and T-cell turnover in different organs of rhesus monkeys. Blood 101, 1213-1219 S. SOPPER, C. STAHL-HENNIG, M. DEMUTH, I. C. JOHNSTON, R. DORRIES u. V. TER MEULEN (1997): Lymphocyte subsets and expression of differentiation markers in blood and lymphoid organs of rhesus monkeys. Cytometry 29, 351-362 C. STAHL-HENNIG, R. M. STEINMAN, K. TENNER-RACZ, M. POPE, N. STOLTE, K. MATZRENSING, G. GROBSCHUPFF, B. RASCHDORFF, G. HUNSMANN u. P. RACZ (1999): Rapid infection of oral mucosal-associated lymphoid tissue with simian immunodeficiency virus. Science (New York, N.Y.) 285, 1261-1265 J. STAVNEZER (1996): Immunoglobulin class switching. Current opinion in immunology 8, 199-205 K. K. STEGER, M. DYKHUIZEN, J. L. MITCHEN, P. W. HINDS, B. L. PREUNINGER, M. WALLACE, J. THOMSON, D. C. MONTEFIORI, Y. LU u. C. D. PAUZA (1998): CD4+-T-cell and CD20+-B-cell changes predict rapid disease progression after simian-human immunodeficiency virus infection in macaques. Journal of virology 72, 1600-1605 K. D. STONE, C. PRUSSIN u. D. D. METCALFE (2010): IgE, mast cells, basophils, and eosinophils. The Journal of allergy and clinical immunology 125, S73-80 M. STREMLAU, C. M. OWENS, M. J. PERRON, M. KIESSLING, P. AUTISSIER u. J. SODROSKI (2004): The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427, 848-853 Y. S. SUH, K. S. PARK, U. SAUERMANN, M. FRANZ, S. NORLEY, D. WILFINGSEDER, H. STOIBER, Z. FAGROUCH, J. HEENEY, G. HUNSMANN, C. STAHL-HENNIG u. Y. C. SUNG (2006): Reduction of viral loads by multigenic DNA priming and adenovirus boosting in the SIVmac-macaque model. Vaccine 24, 1811-1820 C. SUNDLING, M. N. FORSELL, S. O'DELL, Y. FENG, B. CHAKRABARTI, S. S. RAO, K. LORE, J. R. MASCOLA, R. T. WYATT, I. DOUAGI u. G. B. KARLSSON HEDESTAM (2010): Soluble HIV-1 Env trimers in adjuvant elicit potent and diverse functional B cell responses in primates. The Journal of experimental medicine 207, 2003-2017 Literaturverzeichnis S e i t e | 161 S. SURYANI, D. A. FULCHER, B. SANTNER-NANAN, R. NANAN, M. WONG, P. J. SHAW, J. GIBSON, A. WILLIAMS u. S. G. TANGYE (2010): Differential expression of CD21 identifies developmentally and functionally distinct subsets of human transitional B cells. Blood 115, 519-529 S. G. TANGYE, D. T. AVERY, E. K. DEENICK u. P. D. HODGKIN (2003): Intrinsic differences in the proliferation of naive and memory human B cells as a mechanism for enhanced secondary immune responses. Journal of Immunology 170, 686-694 S. G. TANGYE, Y. J. LIU, G. AVERSA, J. H. PHILLIPS u. J. E. DE VRIES (1998): Identification of functional human splenic memory B cells by expression of CD148 and CD27. The Journal of experimental medicine 188, 1691-1703 V. J. TEEUWSEN, J. M. LANGE, R. KEET, J. K. SCHATTENKERK, C. DEBOUCK, R. VAN DEN AKKER, J. GOUDSMIT u. A. D. OSTERHAUS (1991): Low number of functionally active B lymphocytes in the peripheral blood of HIV-1-seropositive individuals with low p24-specific serum antibody titers. AIDS (London, England) 5, 971-979 M. M. THOMSON u. R. NAJERA (2005): Molecular epidemiology of HIV-1 variants in the global AIDS pandemic: an update. AIDS reviews 7, 210-224 K. TITANJI, F. CHIODI, R. BELLOCCO, D. SCHEPIS, L. OSORIO, C. TASSANDIN, G. TAMBUSSI, S. GRUTZMEIER, L. LOPALCO u. A. DE MILITO (2005): Primary HIV-1 infection sets the stage for important B lymphocyte dysfunctions. AIDS (London, England) 19, 1947-1955 K. TITANJI, V. VELU, L. CHENNAREDDI, M. VIJAY-KUMAR, A. T. GEWIRTZ, G. J. FREEMAN u. R. R. AMARA (2010): Acute depletion of activated memory B cells involves the PD-1 pathway in rapidly progressing SIVinfected macaques. The Journal of clinical investigation 120, 3878-3890 M. A. TOMAI, L. M. IMBERTSON, T. L. STANCZAK, L. T. TYGRETT u. T. J. WALDSCHMIDT (2000): The immune response modifiers imiquimod and R-848 are potent activators of B lymphocytes. Cellular immunology 203, 55-65 D. VAN BAARLE, E. HOVENKAMP, M. F. CALLAN, K. C. WOLTHERS, S. KOSTENSE, L. C. TAN, H. G. NIESTERS, A. D. OSTERHAUS, A. J. MCMICHAEL, M. H. VAN OERS u. F. MIEDEMA (2001): Dysfunctional Epstein-Barr virus (EBV)-specific CD8(+) T lymphocytes and increased EBV load in HIV-1 infected individuals progressing to AIDS-related non-Hodgkin lymphoma. Blood 98, 146-155 162 | S e i t e Literaturverzeichnis R. A. VAN LIER, J. BORST, T. M. VROOM, H. KLEIN, P. VAN MOURIK, W. P. ZEIJLEMAKER u. C. J. MELIEF (1987): Tissue distribution and biochemical and functional properties of Tp55 (CD27), a novel T cell differentiation antigen. Journal of Immunology 139, 1589-1596 Y. VUGMEYSTER, K. HOWELL, A. BAKSHI, C. FLORES, O. HWANG u. K. MCKEEVER (2004): B-cell subsets in blood and lymphoid organs in Macaca fascicularis. Cytometry A 61, 69-75 L. M. WALKER, S. K. PHOGAT, P. Y. CHAN-HUI, D. WAGNER, P. PHUNG, J. L. GOSS, T. WRIN, M. D. SIMEK, S. FLING, J. L. MITCHAM, J. K. LEHRMAN, F. H. PRIDDY, O. A. OLSEN, S. M. FREY, P. W. HAMMOND, S. KAMINSKY, T. ZAMB, M. MOYLE, W. C. KOFF, P. POIGNARD u. D. R. BURTON (2009): Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science (New York, N.Y.) 326, 285-289 Q. WANG, H. SHANG, X. HAN, Z. ZHANG, Y. JIANG, Y. WANG, D. DAI u. Y. DIAO (2008): High level serum neutralizing antibody against HIV-1 in Chinese long-term non-progressors. Microbiology and immunology 52, 209-215 Z. WANG, H. W. HOROWITZ, T. ORLIKOWSKY, B. I. HAHN, V. TREJO, A. S. BAPAT, R. S. MITTLER, R. J. RAYANADE, S. Y. YANG u. M. K. HOFFMANN (1999): Polyspecific self-reactive antibodies in individuals infected with human immunodeficiency virus facilitate T cell deletion and inhibit costimulatory accessory cell function. The Journal of infectious diseases 180, 1072-1079 A. WATSON, J. RANCHALIS, B. TRAVIS, J. MCCLURE, W. SUTTON, P. R. JOHNSON, S. L. HU u. N. L. HAIGWOOD (1997): Plasma viremia in macaques infected with simian immunodeficiency virus: plasma viral load early in infection predicts survival. Journal of virology 71, 284-290 S. WELLER, C. A. REYNAUD u. J. C. WEILL (2005): Vaccination against encapsulated bacteria in humans: paradoxes. Trends in immunology 26, 85-89 S. WILLEY u. M. M. AASA-CHAPMAN (2008): Humoral immunity to HIV-1: neutralisation and antibody effector functions. Trends in microbiology 16, 596-604 S. A. WILLIAMS u. W. C. GREENE (2007): Regulation of HIV-1 latency by T-cell activation. Cytokine 39, 63-74 Literaturverzeichnis S e i t e | 163 N. A. WILSON, J. REED, G. S. NAPOE, S. PIASKOWSKI, A. SZYMANSKI, J. FURLOTT, E. J. GONZALEZ, L. J. YANT, N. J. MANESS, G. E. MAY, T. SOMA, M. R. REYNOLDS, E. RAKASZ, R. RUDERSDORF, A. B. MCDERMOTT, D. H. O'CONNOR, T. C. FRIEDRICH, D. B. ALLISON, A. PATKI, L. J. PICKER, D. R. BURTON, J. LIN, L. HUANG, D. PATEL, G. HEINDECKER, J. FAN, M. CITRON, M. HORTON, F. WANG, X. LIANG, J. W. SHIVER, D. R. CASIMIRO u. D. I. WATKINS (2006): Vaccine-induced cellular immune responses reduce plasma viral concentrations after repeated lowdose challenge with pathogenic simian immunodeficiency virus SIVmac239. Journal of virology 80, 5875-5885 G. ZWART, L. VAN DER HOEK, M. VALK, M. T. CORNELISSEN, E. BAAN, J. DEKKER, M. KOOT, C. L. KUIKEN u. J. GOUDSMIT (1994): Antibody responses to HIV-1 envelope and gag epitopes in HIV-1 seroconverters with rapid versus slow disease progression. Virology 201, 285-293 164 | S e i t e Abbildungsverezichnis 9. ABBILDUNGSVERZEICHNIS Abbildung 1: B-Zellreihe. ........................................................................................................ 20 Abbildung 2: Struktur eines Antikörpers. ............................................................................... 27 Abbildung 3: Wirkungsmechanismen von Antikörpern. ......................................................... 31 Abbildung 4: SIV-Partikel. ..................................................................................................... 36 Abbildung 5: Schematischer Verlauf der HIV-Infektion. ....................................................... 39 Abbildung 6: Titration des Virusstocks SIVmac32H für den Nachweis neutralisierender Antikörper. ............................................................................................................................... 51 Abbildung 7: Schematische Darstellung der ELISpot-Technik zum Nachweis Antikörpersezernierender Zellen. ............................................................................................................. 53 Abbildung 8: Definition der Lymphozytenpopulationen eines SIV-negativen Rhesusaffen im Durchflusszytometer. ............................................................................................................... 61 Abbildung 9: Definition der T- und B-Zellpopulationen eines SIV-negativen Rhesusaffen im Durchflusszytometer. ............................................................................................................... 62 Abbildung 10: Viruslast von Langzeitüberlebenden und Progressoren im Zeitverlauf nach Belastungsinfektion. ................................................................................................................. 66 Abbildung 11: Vergleich der mittleren Viruslast von Langzeitüberlebenden und Progressoren zum Zeitpunkt der Spitzenvirämie und zu Beginn der akuten Plateauphase im Vergleich. .... 67 Abbildung 12: Bindende Antikörper von Langzeitüberlebenden und Progressoren im Infektionsverlauf gegen die SIV-Antigene p27 und gp130…………………………………..68 Abbildung 13: Bindende Antikörper von Langzeitüberlenden und Progressoren zu den Wochen 25, 50 und 100 nach SIV-Infektion............................................................................ 69 Abbildung 14: Beziehung von Viruslast und Antikörper-Titer gegen gp130 und p27. ........... 70 Abbildung 15: Neutralisierende Antikörper von Langzeitüberlebenden und Progressoren im Infektionsverlauf. ..................................................................................................................... 71 Abbildung 16: Beziehung von Neutralisationstitern und bindenden Antikörpern gegen p27 und gp130. ................................................................................................................................ 72 Abbildung 17: Beziehung von Neutralisationstitern und Virusbeladung im Plasma............... 73 Abbildung 18: Quantifizierung von IFN-γ-sezernierenden T-Zellen nach Stimulation mit SIVgag oder SIVenv. ............................................................................................................... 75 Abbildungsverzeichnis S e i t e | 165 Abbildung 19: Quantifizierung IgG-sezernierender Zellen im Blut der Langzeitüberlebenden nach dreitägiger in vitro-Stimulation mit R848 und IL-2. ....................................................... 77 Abbildung 20: Gatingschema der durchflusszytometrischen Analyse der BZellsubpopulationen. ................................................................................................................ 79 Abbildung 21: Anteil der gesamten B- und T-Zellpopulation an den Lymphozyten. ............. 80 Abbildung 22: Anteil der gesamten B-Zellpopulation an den Lymphozyten in Abhängigkeit vom Infektionsstatus. ............................................................................................................... 81 Abbildung 23: Relative CD27+B-Zellpopulationen in Abhängigkeit von der Viruslast. ........ 83 Abbildung 24: Relative B-Zellsubpopulationen in Abhängigkeit von der Viruslast. ............. 87 Abbildung 25: Relative T-Zellsubpopulationen in Abhängigkeit von der Viruslast. .............. 88 Abbildung 26: Beziehung von T-Helferzellen und verschiedenen B-Zellpopulationen. ......... 89 Abbildung 27: Virusbeladung bis 48 Wochen nach SIV-Infektion in unbehandelten Kontrolltieren. .......................................................................................................................... 91 Abbildung 28: Bindende Antikörper gegen p27 und gp130 bis 42 Wochen nach SIV-Infektion in unbehandelten Kontrolltieren. .............................................................................................. 92 Abbildung 29: Beziehung von SIV-Last und Antikörpertiter gegen p27 nach SIV-Infektion in unbehandelten Kontrolltieren.. ................................................................................................. 93 Abbildung 30: Verlauf der Neutralisationstiter bis 42 Wochen nach SIV-Infektion in unbehandelten Kontrolltieren. .................................................................................................. 94 Abbildung 31: SIV-spezifische ASC im Infektionsverlauf...................................................... 95 Abbildung 32: Beziehung von Viruslast und SIV-spezifischen ASC. ..................................... 97 Abbildung 33: Änderungen der B- und T-Zellpopulationen bis zum Beginn der Plateauphase der SIV-Infektion in unbehandelten Kontrolltieren. ................................................................ 99 Abbildung 34: Änderungen der B- und T-Zellsubpopulationen bis zum Beginn der Plateauphase der SIV-Infektion in unbehandelten Kontrolltieren. ........................................ 100 Abbildung 35: Beziehung der Viruslast zum Zeitpunkt der Spitzenvirämie und CD4+TZellspiegel während der Spitzenvirämie und in der frühen Plateauphase der SIV-Infektion unbehandelter Kontrolltiere.................................................................................................... 101 166 | S e i t e Tabellenverzeichnis 10. TABELLENVERZEICHNIS Tabelle 1: Immunglobulinklassen des Menschen. ...................................................................29 Tabelle 2: Tabellarische Zusammenstellung der für die Durchflusszytometrie verwendeten Antikörper. ...............................................................................................................................59 Tabelle 3: Reaktionsansätze für die qRT-PCR. .......................................................................63 Tabelle 4: PCR-Bedingungen für die qRT-PCR zur Quantifizierung viraler RNA-Kopien und zur Überprüfung der RNA-Qualität. .................................................................................64 Tabelle 5: Mittelwerte und Standardabweichungen verschiedener B- und T-Zellpopulationen. ..................................................................................................................86 Anhang S e i t e | 167 11. ANHANG 11.1 Tiertabelle Tier Virus Inokulationsroute Immunisiert Zeitraum der Untersuchung (wpi) ELISA 2107 SIVmac239 tonsillär ja bis 151 x 2118 SIVmac239 tonsillär ja bis 215 x 2121 SIVmac239 tonsillär ja bis 48 x 2139 SIVmac239 tonsillär nein bis 331 x 2141 SIVmac239 tonsillär ja bis 130 x 2145 SIVmac239 tonsillär ja bis 77 x 2151 SIVmac239 tonsillär ja bis 331 2153 SIVmac239 tonsillär ja 2155 SIVmac239 tonsillär 2161 SIVmac239 2165 ELISA Titration B-ZellELISpot NT Durchflusszytometrie x x x x x x x x x bis 331 x x x x x nein bis 331 x x x x x tonsillär ja bis 101 x SIVmac239 tonsillär nein bis 102 x 2168 SIVmac239 tonsillär ja bis 114 x 2169 SIVmac239 tonsillär ja bis 27 x 2171 SIVmac239 tonsillär nein bis 25 x 2172 SIVmac239 tonsillär ja bis 334 x x 2175 SIVmac239 tonsillär nein bis 14 x x x 2178 SIVmac239 tonsillär ja bis 28 x 2180 SIVmac239 tonsillär ja bis 27 x 2187 SIVmac239 tonsillär ja bis 52 x 2188 SIVmac239 tonsillär ja bis 138 x x x 2191 SIVmac239 tonsillär nein bis 155 x x x x x x x Anhang S e i t e | 168 Tier Virus Inokulationsroute Immunisiert Zeitraum der Untersuchung (wpi) ELISA Titration B-ZellELISpot NT Durchflusszytometrie 2192 SIVmac251 i.v. nein bis 89 x 2194 SIVmac239 tonsillär nein bis 110 x 2201 SIVmac251 i.v. ja bis 273 x x x x x 2208 SIVmac251 i.v. ja bis 38 x 2219 SIVmac251 i.v. ja bis 273 x x x x x 2223 SIVmac239 tonsillär nein bis 54 x x 2231 SIVmac239 tonsillär nein bis 48 x 2233 SIVmac251 i.v. nein bis 44 x 2250 2247 SIVmac251 SIVbis LoxLTR i.v. nein bis 96 x i.v. nein bis 40 x 2249 SIVmac251 rektal (repeatedlow-dose) nein bis 48 x x x x 2272 SIVmac251 rektal (repeated-lowdose) nein bis 48 x x x x 2284 SIVmac251 rektal (repeatedlow-dose) nein bis 48 x x x x 2290 SIVmac251 rektal (repeatedlow-dose) nein bis 48 x x x x 2294 SIVmac251 rektal (repeatedlow-dose) nein bis 48 x x x x 2302 SIVmac251 rektal (repeated-lowdose) nein bis 48 x x x x SIVmac251 rektal (repeatedlow-dose) bis 48 x x x x 2305 nein ELISA x x Anhang Tier Virus 2311 SIVbis LoxLTR S e i t e | 169 Inokulationsroute Immunisiert Zeitraum der Untersuchung (wpi) ELISA Titration B-ZellELISpot NT Durchflusszytometrie nein bis 29 rektal (repeated-lowdose) nein bis 48 i.v. nein bis 29 x i.v. nein bis 29 x x i.v. nein bis 29 x i.v. nein bis 29 x i.v. nein bis 29 x i.v. nein bis 29 x i.v. nein bis 29 x i.v. ja bis 72 x 2385 2392 2411 2413 2415 2416 SIVmac251 SIVbis LoxLTR negativ negativ negativ negativ negativ i.v. nein bis 29 x x x x x x 8644 SIVmac251 i.v. nein bis 648 x x x x x 9794 SIVmac239 tonsillär ja bis 414 x x x x x 2325 2331 2334 2342 2345 2347 2353 2358 2359 2366 SIVmac251 SIVbis LoxLTR SIVbis LoxLTR negativ SIVbis LoxLTR SIVbis LoxLTR SIVbis LoxLTR SIVbis LoxLTR SIVbis LoxLTR i.v. ELISA x x x x x Anhang S e i t e | 170 Tier Virus Inokulationsroute Immunisiert Zeitraum der Untersuchung (wpi) ELISA ELISA Titration B-ZellELISpot 10425 SIVmac239 tonsillär ja bis 150 x 11139 SIVmac251 i.v. ja bis 148 x 11612 SIVmac251 i.v. ja bis 51 x 12056 SIVmac251 i.v. ja bis 80 x 12149 SIVmac251 rektal (repeated-lowdose) ja 12529 SIVmac239 tonsillär nein 12530 SIVmac239 tonsillär ja bis 30 x x 12531 SIVmac239 tonsillär ja bis 155 x x x 12532 SIVmac239 tonsillär ja bis 64 x x x 12533 SIVmac239 tonsillär nein bis 153 x 12534 SIVmac239 tonsillär ja bis 60 x 12535 SIVmac239 tonsillär ja bis 156 x 12536 SIVmac239 tonsillär ja bis 297 x 12537 SIVmac239 tonsillär ja bis 155 x x x 12538 SIVmac251 i.v. nein bis 146 x 12539 SIVmac239 tonsillär ja bis 138 x 12540 SIVmac239 tonsillär nein bis 19 x x x 12541 SIVmac239 tonsillär ja bis 115 x 12542 SIVmac239 tonsillär nein bis 31 x 12543 SIVmac239 tonsillär ja bis 334 x 12544 SIVmac239 tonsillär nein bis 101 x 12670 SIVmac239 tonsillär nein bis 25 x 12671 SIVmac239 tonsillär nein bis 334 x x NT Durchflusszytometrie x 69 x x x x x x x x x Anhang S e i t e | 171 Tier Virus Inokulationsroute Immunisiert Zeitraum der Untersuchung (wpi) ELISA ELISA Titration B-ZellELISpot NT Durchflusszytometrie 12672 SIVmac239 tonsillär nein bis 334 x x x x x 12673 13147 SIVmac239 negativ tonsillär nein bis 45 x 13248 SIVmac251 i.v. ja bis 22 x 13249 SIVmac251 i.v. ja bis 30 x 13250 SIVmac251 i.v. ja bis 150 x 13251 SIVmac251 i.v. ja bis 146 x 13252 SIVmac251 i.v. ja bis 45 x 13253 SIVmac251 i.v. ja bis 85 x 13255 SIVmac251 i.v. ja bis 36 x 13256 SIVmac251 i.v. ja bis 37 x 13257 SIVmac251 i.v. nein bis 141 x 13258 SIVmac251 i.v. ja bis 104 x 13260 SIVmac251 i.v. nein bis 102 x 13261 SIVmac251 i.v. ja bis 51 x 13262 13441 13676 13677 13721 SIVmac251 negativ negativ negativ negativ i.v. nein bis 37 x 13906 SIVmac251 rektal (repeated-lowdose) nein bis 73 x x x x 13907 SIVmac251 rektal (repeated-lowdose) nein bis 73 x x x x x x x x x x x x x x Anhang Tier Virus 13913 SIVmac251 rektal (repeatedlow-dose) nein 13918 13921 S e i t e | 172 Inokulationsroute Immunisiert Zeitraum der Untersuchung (wpi) ELISA ELISA Titration B-ZellELISpot NT Durchflusszytometrie bis 81 x x x x SIVmac251 rektal (repeated-lowdose) nein bis 48 x x x x SIVmac251 rektal (repeated-lowdose) nein bis 48 x x x x Anhang S e i t e | 173 11.2 ELISA-Protokoll Material: - Mikrotiterplatte: Sterile 96-well-Platten mit mittlerer Bindungskapazität (Fa. Greiner) - Antigene: p27 / gp130, Ausgangskonzentration 1mg/ml (NIBSC) - Coating-Puffer: Carbonatpuffer (0,32g/l Na2CO3; 5,9g/l NaHCO3; 0,4 g/l Natriumazid, pH 9,6) - Waschpuffer: PBS mit 0,05% Tween 20 (Fa. Sigma) - Blocking-Puffer: PBS mit 5% Milchpulver (Fa. Roth) - Konjugat: Anti-Human-IgG Peroxidase-Konjugat (Fa. Jackson Bio-Lab) in einer Verdünnung von 1:3000 in Blocking-Puffer - Substrat: Tetramethylbenzidin-Lösung (TMB, Fa. Sigma) - Stopplösung: 1,3M H2SO4 Plattenbeschichtung: (am Vortag) 1. 3µg p27 bzw. 2µg gp130 pro Platte zu 5ml Coating-Puffer geben 2. 50µl je Vertiefung pipettieren (entspricht 0,03 bzw. 0,02µg pro Kavität) 3. Inkubation bei 4°C über Nacht Durchführung: 1. Mikrotiterplatte 2 x mit Waschpuffer waschen 2. Je Vertiefung 200µl Blocking-Puffer zugeben 3. Inkubation für 1 ½ h bei Raumtemperatur 4. Währenddessen Probenverdünnungen in Blocking-Puffer ansetzen 5. Mikrotiterplatte 2x mit Waschpuffer waschen 6. 50µl Probenverdünnung pro Vertiefung pipettieren 7. Inkubation für 3 h bei Raumtemperatur 8. Mikrotiterplatte 4x mit Waschpuffer waschen 9. 100µl verdünntes Konjugat pro Kavität pipettieren 10. Inkubation für 1 ½ h bei Raumtemperatur 11. Mikrotiterplatte 6x mit Waschpuffer waschen 174 | S e i t e Anhang 12. 50µl TMB pro Vertiefung pipettieren 13. Inkubation für 2 min bei Raumtemperatur 14. 50µl Stopplösung pro Vertiefung pipettieren 15. Messung mit ELISA-Reader Sunrise® (Fa. Tecan) bei 450 nm 11.3 Protokoll für Neutralisationstest auf TZM-bl mit Luziferase Read-out Material: - Platten: o Sterile 96-well Zellkulturplatte mit flachem Boden (Fa. Corning) o Weiße unsterile 96-well Platten mit flachem Boden für die Bestimmung der Luziferaseaktivität (Fa. Costar) - Medium: DMEM (Fa. PAN) supplementiert mit 10% FCS und 1% Penicillin (10000 U/ml)/Streptomycin (10mg/ml) - Virusstock: SIVmac32H/C8166 vom 27.08.04 - Zellen: TZM-bl-Zelllinie (NIBSC) - Färbesubstrat: Bright-Glo® Luciferase-Testsystem (Fa. Promega) - Sonstige Reagenzien: o PBS o Trypsin (0,05%)-EDTA (0,02%) (Fa. PAN) o DEAE Dextran (Fa. Sigma) Stocklösung mit 3,7g/ml Durchführung: (Tag 1, sterile Bedingungen) 1. Serumproben bei Raumtemperatur auftauen lassen 2. Mittels Vortex gut mischen, kurz herunterzentrifugieren 3. Hitzeinaktivierung der Proben im Wasserbad bei 56°C für 1 h 4. Währenddessen 150µl Medium pro Vertiefung in Reihe 1 der Zellkulturplatte (Zellkontrolle) vorlegen 5. 100µl Medium pro Vertiefung in Reihe 2 (Viruskontrolle) vorlegen 6. Proben nach Hitzeinaktivierung mittels Vortex gut mischen 7. Bei einer gewünschten Startverdünnung der Proben von 1:20 Anhang S e i t e | 175 a. 140µl Medium pro Vertiefung in Zeile H in Reihe 3-12 vorlegen b. 11µl Serumprobe 1 in die beiden Vertiefungen Zeile H Reihe 3 und 4 pipettieren c. 11µl Serumprobe 2 in die beiden Vertiefungen Zeile H Reihe 5 und 6 pipettieren usw. Bei einer gewünschten Startverdünnung der Proben von 1:240: a. Für jede Probe 45µl Medium in ein 0,5ml-Reaktionsgefäß vorlegen b. Jeweils 5µl Serumprobe pipettieren, gut mischen (entspricht einer Vorverdünnung von 1:10) c. 150µl Medium pro Vertiefung in Zeile H in Reihe 3-12 vorlegen d. 10µl der vorverdünnten Serumprobe 1 in die beiden Vertiefungen Zeile H Reihe 3 und 4 pipettieren e. 10µl der vorverdünnten Serumprobe 1 in die beiden Vertiefungen Zeile H Reihe 5 und 6 pipettieren usw. 8. Mit einer Mehrkanalpipette Proben in Zeile H mischen (dabei Reihe 1 & 2 auslassen!) und 50µl in Zeile G überführen, erneut mischen und 50µl in Zeile F überführen, auf diese Art weiterverfahren bis zu Zeile A, dort nach dem Mischen 50µl entnehmen und verwerfen 9. Virusstock auftauen und 1:25 mit Medium verdünnen 10. 50µl Virusverdünnung in Vertiefungen der Reihen 2-12 pipettieren (Achtung, Verschleppung der Probe vermeiden!) 11. Inkubation für 1 h bei 37°C, 5% CO2 12. In der Zwischenzeit Zellen in einer Dichte von 1x105/ml Medium mit DEAE Dextran, vorbereiten. Dafür die DEAE Dextran-Stocklösung 1:1000 verdünnen (die Endverdünnung beträgt 15µg/ml). 13. 100µl Zellsuspension pro Vertiefung (entspricht 10 000 Zellen/Vertiefung) pipettieren, dabei eine Verschleppung von Probe oder Virus vermeiden 14. Platte für 48 h bei 37°C, 5% CO2 inkubieren 176 | S e i t e Anhang Färbung: (Tag 3, unsterile Bedingungen) 15. Kontrolle der Kultur unter dem Mikroskop (kein zytopathologischer Effekt in der Zellkontrolle, gleichmäßige Verteilung der Zellen, keine Kontamination) 16. Medium entfernen, indem die Platte vorsichtig auf Ethanol-getränkten Tüchern ausgeklopft wird 17. 2 x mit PBS waschen, vorsichtig ausklopfen, einen Moment ablaufen lassen 18. 25µl PBS pro Vertiefung pipettieren 19. 50µl Bright-glo pro Vertiefung pipettieren, 7 min inkubieren 20. Mit einer Multikanalpipette 50µl/well in die weiße Platte übertragen, dabei mehrmals auf- und abpipettieren 21. Sofortige Bestimmung der Luziferaseaktivität mit dem Detektionsgerät Plate Chameleon® (Fa. Hiddex) 11.4 Protokoll für B-Zell-ELISpot Material: - Platte: 96-well Filtrationsplatten MultiScreen HTS (Fa. Millipore) - Antigene: o p27 / gp130 (NIBSC), Ausgangskonzentration 1mg/ml o sterile BSA-Stocklösung (Fa. Sigma), Ausgangskonzentration 1mg/ml - Coating-Antikörper: o Monoklonaler Anti-IgG-Antikörper MT91/145, Ausgangskonzentration 0,5mg/ml (Fa. Mabtech) o Anti-IgA-Antikörper, Ausgangskonzentration 0,5mg/ml (Fa. Mabtech) - Medium: RPMI kompl. - Capture-Antikörper: o Biotinylierter monoklonaler Anti-IgG-Antikörper MT78/145, Ausgangskonzentration 0,5mg/ml (Fa. Mabtech) o Biotinylierter Anti-IgA-Antikörper, Ausgangskonzentration 0,5mg/ml (Fa. Mabtech) Anhang S e i t e | 177 - Konjugat: Streptavidin-Alkaline Phosphatase (Fa. Mabtech) - Substrat: BCIP/NBT (Fa. Mabtech) - Sonstige Reagenzien: o PBS o PBS/0,5% FCS o 35%iges Ethanol o Steriles Aqua dest. Plattenbeschichtung: (am Vortag, sterile Bedingungen) 1. Verdünnung der Antigene und des Coating-Antikörpers in sterilem PBS: a. p27/ gp130 → 2µg/ml b. BSA → 10µg/ml c. MT91/145 bzw. Anti-IgA-Ak → 15µl/ml 2. Vorbehandlung der Plattenmembran mit 15µl 35%igem Ethanol für eine Minute 3. 5 x Waschen mit 200µl sterilem Aqua dest. pro Vertiefung 4. 100µl der Antigen- bzw. Antikörperverdünnung pro Vertiefung pipettieren 5. Inkubation über Nacht bei 4-8°C Durchführung: (Tag 1, sterile Bedingungen) 1. 5 x Waschen mit 200µl PBS pro Vertiefung 2. Blockierung unspezifischer Bindungsstellen mit 200µl RMPI kompl. pro Vertiefung 3. Inkubation für mindestens 45 min bei 37°C 4. Medium entfernen 5. Zellen 2 x mit PBS waschen 6. Zählen der Zellen und Einstellung der Zellzahl auf a. 1x106/ml RMPI kompl. für die Bestimmung von IgG-sezernierenden Zellen in PBMC b. 2x106/ml für die Bestimmung von IgA-sezernierenden Zellen in PBMC c. 2x106/ml für die Bestimmung von IgG- und IgA-sezernierenden Zellen aus BAL 178 | S e i t e Anhang 7. Vorlage von a. 150µl RPMI kompl. in die Vertiefungen mit Coating-Antikörper gegen IgG b. 100µl RPMI kompl. in die Vertiefungen mit Coating-Antikörper gegen IgA c. 100µl RPMI kompl. sowohl in die Vertiefungen mit Coating-Antikörpern gegen IgG als auch in solche mit Coating-Antikörper gegen IgA beim Einsatz von mononukleären Zellen aus BAL 8. Zellen nach folgendem Schema pipettieren: a. Für die Bestimmung von Gesamt-IgG-sezernierenden Zellen in PBMC 50µl/Vertiefung → entspricht 50.000 Zellen pro Vertiefung b. Für die Bestimmung von Gesamt-IgA-sezernierenden Zellen in PBMC 100µl/Vertiefung → entspricht 200.000 Zellen pro Vertiefung c. Für die Bestimmung von Gesamt-IgG- oder –IgA-sezernierender Zellen in BAL 100µl/Vertiefung → entspricht 200.000 Zellen pro Vertiefung d. Für die Bestimmung antigen-spezifischer ACS 200µl/Vertiefung → entspricht bei der Bestimmung antigen-spezifischer IgG-sezernierender Zellen aus PBMC 200.000 Zellen pro Vertiefung, ansonsten 400.000 Zellen pro Vertiefung 9. Inkubation für 20 h bei 37°C, 5% CO2. Während dieser Zeit die Platte nicht bewegen und vor Austrocknung schützen (z.B. durch Verpackung in Alufolie) Färbung: (Tag 2, unsterile Bedingungen) 10. 5 x Waschen mit 200µl PBS pro Vertiefung 11. Verdünnung des Capture-Antikörpers auf 1µg/ml in PBS/0,5% FCS, davon 50µl pro Vertiefung pipettieren 12. Inkubation für 2 h bei Raumtemperatur 13. 5 x Waschen mit 200µl PBS pro Vertiefung 14. Verdünnung des Konjugats 1:1000 in PBS/0,5% FCS, davon 50µl pro Vertiefung pipettieren 15. Inkubation für 1 h bei Raumtemperatur 16. 5 x Waschen mit 200µl PBS pro Vertiefung 17. Substrat durch ein 0,45µm Filter filtrieren, 50µl pro Vertiefung pipettieren Anhang S e i t e | 179 18. Inkubation bei Raumtemperatur bis sich klar erkennbare Punkte entwickeln 19. Plattenboden entfernen, Entwicklung durch gründliches Waschen beider Seiten mit Leitungswasser unterbrechen, Platte trocknen lassen 20. Zählung der Punkte mittels Lupe oder ELISpot-Reader Bio-Reader 2000® (Fa. BioSys) 11.5 Protokoll für IFN-γ-ELISpot Material: - Platte: 96-well Filtrationsplatten MultiScreen HTS (Fa. Millipore) - Antigene: o SIVgag/env-Peptidpool, Ausgangskonzentration 1mg/ml o steriler BSA-Stock (Fa. Sigma), Ausgangskonzentration 1mg/ml - Coating-Antikörper: o Monoklonaler Anti-Mensch/Affe-IFN-γ-Antikörper mAb GZ-4, Ausgangskonzentration 1mg/ml (Fa. Mabtech) - Medium: RPMI kompl. - Capture-Antikörper: o Biotinylierter monoklonaler Anti-Mensch-IFN-γ-Antikörper mAb 7-B6-1Biotin, Ausgangskonzentration 1mg/ml (Fa. Mabtech) - Konjugat: Streptavidin-Alkaline Phosphatase (Fa. Mabtech) - Substrat: BCIP/NBT (Fa. Dunnlab) - Sonstige Reagenzien: o PBS o PBS/0,5% FCS Plattenbeschichtung: (am Vortag, sterile Bedingungen) 1. Coating-Antikörpers 1:100 in sterilem PBS verdünnen 2. 100µl pro Vertiefung pipettieren 3. Inkubation über Nacht bei 4-8°C 180 | S e i t e Anhang Durchführung: (Tag 1, sterile Bedingungen) 4. 5 x Waschen mit 200µl PBS pro Vertiefung 5. Blockierung unspezifischer Bindungsstellen mit 200µl RMPI kompl. pro Vertiefung 6. Inkubation für 2 h bei 37°C 7. Peptide verdünnen, Endkonzentration soll 2µg/ml pro Vertiefung betragen 8. Zählen der Zellen und Einstellung der Zellzahl auf 2x106 Zellen/ml RPMI kompl. 9. Medium aus der Platte entfernen 10. Vorlage von 50µl Peptidverdünnung pro Vertiefung 11. 50µl Zellen pro Vertiefung pipettieren 12. Inkubation für 16 h bei 37°C, 5% CO2. Während dieser Zeit die Platte nicht bewegen und vor Austrocknung schützen (z.B. durch Verpackung in Alufolie) Färbung: (Tag 2, unsterile Bedingungen) 13. 5 x Waschen mit 200µl PBS pro Vertiefung 14. Verdünnung des Capture-Antikörpers auf 1:1000 in PBS/0,5% FCS, davon 100µl pro Vertiefung pipettieren 15. Inkubation für 2 h bei Raumtemperatur 16. 5 x Waschen mit 200µl PBS pro Vertiefung 17. Verdünnung des Konjugats 1:1000 in PBS/0,5% FCS, davon 100µl pro Vertiefung pipettieren 18. Inkubation für 1 h bei Raumtemperatur 19. 5 x Waschen mit 200µl PBS pro Vertiefung 20. Substrat durch ein 0,45µm Filter filtrieren, 50µl pro Vertiefung pipettieren 21. Inkubation bei Raumtemperatur bis sich klar erkennbare Punkte entwickeln 22. Plattenboden entfernen, Entwicklung durch gründliches Waschen beider Seiten mit Leitungswasser unterbrechen, Platte trocknen lassen 23. Zählung der Punkte mittels Lupe oder ELISpot-Reader Bio-Reader 2000® (Fa. BioSys) Hannover, 25.09.2012 Eidesstattliche Erklärung Hiermit erkläre ich, dass ich die Dissertation mit dem Titel: „Antikörper-abhängige Immunantwort im SIV-Makakenmodell“ selbstständig verfasst habe. Bei der Anfertigung wurden folgende Hilfsmittel und Hilfen Dritter in Anspruch genommen: - Technisch-methodische Einweisung durch die technischen Angestellten und das Tierpflegepersonal der Abt. Infektionsmodelle, Deutsches Primatenzentrum. - Redaktionelle Überarbeitung durch wissenschaftliche Mitarbeiter und Leiter der Abt. Infektionsmodelle, Deutsches Primatenzentrum. - Bereitstellung der Rohdaten der SIV-RNA-Beladung der untersuchten Tiere durch Frau Dr. rer. nat. Ulrike Sauermann, Abt. Infektionsmodelle, Deutsches Primatenzentrum. - Bereitstellung der Rohdaten des IFN-γ-ELISpot der untersuchten Tiere durch die Abteilungsleiterin der Abt. Infektionsmodelle, Deutsches Primatenzentrum. - Beratung bei der Zusammenstellung des Antikörpergemisches und der Definition der Lymphozytenpopulationen im Durchflusszytometer durch Herrn Prof. Dr. rer. nat. Sieghart Sopper, ehemaliger Mitarbeiter der Abt. Virologie und Immunologie, Deutsches Primatenzentrum, und durch Frau Dr. med. vet. Katharina Töpfer, ehemalige Mitarbeiterin der Abt. Infektionsmodelle, Deutsches Primatenzentrum, sowie Unterstützung bei der Datenerhebung durch die technischen Mitarbeiter der Abt. Infektionsmodelle, Deutsches Primatenzentrum. Ich habe keine entgeltliche Hilfe von Vermittlungs- bzw. Beratungsdiensten (Promotionsberater oder andere Personen) in Anspruch genommen. Niemand hat von mir unmittelbar oder mittelbar entgeltliche Leistungen für Arbeiten erhalten, die im Zusammenhang mit dem Inhalt der vorliegenden Dissertation stehen. Die Dissertation wurde in der Abt. Infektionsmodelle des Deutschen Primatenzentrums in Göttingen unter der Leitung von Frau Dr. med. vet. Christiane Stahl-Hennig durchgeführt und bisher nicht für eine Prüfung oder Promotion oder für einen ähnlichen Zweck zur Beurteilung eingereicht. Ich versichere, dass ich die vorstehenden Angaben nach bestem Wissen vollständig und der Wahrheit entsprechend gemacht habe. Katharina Raue Danksagung Bis zur erfolgreichen Beendigung dieser Arbeit kreuzten viele Menschen meinen Weg, ohne die ich mit Sicherheit nicht an dem Punkt angekommen wäre, an dem ich jetzt bin. Allein dafür, dass ich sie alle kennenlernen durfte, hat sich die Promotion gelohnt und zumindest einigen davon möchte ich im Folgenden namentlich erwähnen, um ihnen zu danken: Herrn Prof. Dr. med. vet. Franz-Josef Kaup danke ich für die Übernahme der Betreuung sowie das Entgegenkommen und die Freundlichkeit, die er mir gegenüber stets gezeigt hat. Besonderen Dank schulde ich Frau Dr. med. vet. Christiane Stahl-Hennig für die Bereitstellung des Themas, die Unterstützung bei dessen Bearbeitung und die Freiheit, meine eigenen Ideen in die Dissertation einzubringen. Darüberhinaus eröffnete sie mir durch den Arbeitsplatz in der Abteilung Infektionsmodelle Einblicke in ein für mich neues und spannendes Berufsfeld und schenkte mir schlussendlich sogar die Möglichkeit und das Vertrauen, frühzeitig eine verantwortungsvolle Position in der Arbeitsgruppe einzunehmen. Die Erfahrungen, die ich sammeln durfte, werden mit Sicherheit hilfreich für mein weiteres (Berufs-)Leben sein. Herrn Prof. Dr. rer. nat. Sieghart Sopper danke ich für die unentbehrliche Hilfe bei der Durchflusszytometrie, eine faszinierende Methode, deren Tücken ich zur Genüge kennenlernen durfte. Für die Bereitstellung der Daten zur Viruslast danke ich Frau Dr. rer. nat. Ulrike Sauermann. Dr. rer. nat. Tina Schultheiß und Kelly da Costa haben einen großen Teil zum Gelingen dieser Arbeit beigetragen, zum einen durch fruchtbare fachliche Diskussionen und wertvolle Anregungen, zum anderen durch ihre seelische Unterstützung und ihre Freundschaft, die einen wohltuenden Ausgleich zum Arbeitsalltag bildete. Ich hoffe, ich konnte ihnen Gleiches bieten. Auch die anderen Doktoranden und wissenschaftlichen Mitarbeiter, mit denen ich in den letzten Jahren zusammenarbeiten durfte, allen voran Dr. rer. nat. Ann-Christin Schmädicke, Wiebke Ibing, Kathrin Donde, Dr. med. vet. Katharina Töpfer, Dr. med. vet. Nicole StolteLeeb, Antonina Klippert und Dr. rer. nat. Berit Neumann, haben meine Zeit in Göttingen (sowohl während der Arbeit als auch außerhalb) sehr bereichert. Dafür ein herzliches Dankeschön! Großer Dank gilt den technischen Mitarbeiterinnen der Abteilung Infektionsmodelle Sandra Heine, Judith Hampe und Kerstin Eckelmann, für die Einarbeitung in die Laborarbeit, die Bereitschaft, mir unter die Arme zu greifen, wenn ich zu verzweifeln drohte und so manchen geretteten Versuch, der ohne ihre wachsamen Blicke wahrscheinlich misslungen wäre. Und natürlich für die angenehme Gesellschaft in den Mittags- und Kaffeepausen. Kathleen Listermann danke ich für die Durchführung der durchflusszytometrischen Messungen. Durch die Tierpfleger Peter Müller, Thorsten Eggers, Gabi Marschhausen, Vanessa Böning und Henning Mascher sowie meine ehemalige Kollegin Dr. med. vet. Monika Franz wurde meine Tätigkeit im Tierhaus sehr erleichtert. Sie haben mir Arbeit abgenommen, wo sie konnten und es herrschte immer eine angenehme, lockere Stimmung. Bei ihnen möchte ich mich bedanken, genauso wie bei den Tierärzten der Primatenhaltung Dr. med. vet. Annette Schrod und Dr. med. vet. Tamara Becker, die mir bei Fragen und Notfällen immer zur Seite standen. Den Mitarbeitern der Abteilung Infektionspathologie Dr. med. vet. Kerstin Mätz-Rensing, Dr. med. vet. Eva Gruber-Durjardin, Dr. med. vet. Martina Zöller, Marius Kunze und Wolfgang Henkel danke ich für ihre Hilfe bei den Sektionen und die warme und freundliche Atmosphäre. Herzlicher Dank gebührt meiner Familie, ohne deren liebevolle Unterstützung ich nie soweit gekommen wäre. Mir standen stets alle Möglichkeiten offen und ich hoffe, ich habe diese Großzügigkeit in ihrem Sinne genutzt. Zu guter Letzt möchte ich meinem Mann Jonathan aus tiefstem Herzen danken, der die Höhen und Tiefen der letzten Jahre wohl am deutlichsten miterlebt hat. Er hat mir immer beigestanden und dafür große Belastungen auf sich genommen.