3. Enzymkatalyse - Online Media Server
Werbung

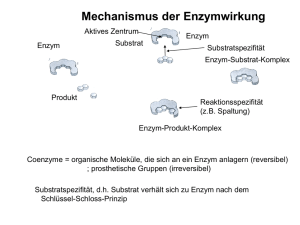
3. Enzymkatalyse 3.1 Allgemeines Enzyme katalysieren biochemische Reaktionen. Sie sind Unschlagbar in den drei entscheidenden Parametern der Katalyse: 1. Geschwindigkeit 2. Spezifität 3. Selektivität Enzyme katalysieren Reaktionen mit gewaltigen Ratenbeschleunigungen. So sind Werte von kcat/kuncat von 106-1014 keine Seltenheit. Das Beispiel der Spaltung einer glykosidischen Bindung (Abb. 1) soll das verdeutlichen. Bereits in Lösung befindliche Säure katalysiert die Spaltung der glykosidischen Bindung. Eine interne Säuregruppe beschleunigt die Reaktion noch weiter, da hier das Proton an „Ort und Stelle“ ist. Enzyme wie die β-Galaktosidase, hingegen sind in der Ratenbeschleunigung unschlagbar (kcat/kuncat = 2.5x107). Abb. 1 Raten der Hydrolyse einer glykosidischen Bindung mit unterschiedlichen Katalysevatrianten. 1 Die Selektivität der Enzyme hängt mit der selektiven molekularen Erkennung der Substrate durch die chirale aktive Tasche zusammen. Hierhinein passen nur bestimmte Substrate. Die Spezifität von Enzymen wird durch die präzise Anordnung der katalytisch aktiven Reste erreicht. Diese werden an eine bestimmte Stelle im Substrat gebracht, so dass nur dort die Reaktion ablaufen kann. Daraus folgt eine hohe Regio- und Stereoselektivität. Selektivität bezieht sich also auf die Auswahl des Substrates. Spezifität beschreibt wie Chemo-, Regio-, und Stereoselektiv die Reaktion abläuft. 3.1 Das thermodynamische Modell Ein Katalysator beschleunigt eine Reaktion, geht aber selber unverändert aus der Reaktion hervor. Ein Katalysator reduziert also Aktivierungsenergien. Hierbei kann entweder der Übergangszustand stabilisiert werden oder das Enzym ermöglicht einen anderen Reaktionsweg, der über energetisch niedrigere Übergangszustände verläuft. Abb. 2 Energieprofile enzymatischer Katalyse. 2 Im Beispiel a) (Abb. 2) wird verdeutlicht, dass bereits eine Protonierung zu einer leichteren Reaktion führen kann. Das Enzym protoniert sowohl das Substrat S als auch das Produkt und beschleunigt so die Reaktion. Allgemein gilt für Reaktionsgeschwindigkeiten die Arrhenius-Gleichung: k = A e Ratenbeschleunigung ergibt sich daraus: kcat/kuncat = e (-Eact/RT) . Für die (Euncat-Ecat/RT) . In diesen Gleichungen ist T die Reaktionstemperatur in K und R die universelle Gaskonstante mit 8.314 JK-1mol-1. Wenn man also den Übergangszustand um 10kJ/mol stabilisiert, so ergibt sich bei 25°C eine Ratenbeschleunigung von 55. Eine Stabilisierung von 20kJ/mol ergibt schon eine Ratenbeschleunigung von 3000 und 40kJ/mol erbringen die oft beobachten 107-fachen Beschleunigungen. Eine low-barrier H-bond (bis zu 100 kJ/mol), die den ÜZ stabilisiert, reicht also aus, um diese hohen Ratenbeschleunigungen zu realisieren. Wie in Abb. 2b gezeigt ist, verlaufen die enzymkatalysierten Reaktionen oft über viele einzelne Schritte. So wird oft ein kovalentes Enzym-Substrat-Intermediat gebildet, welches zum Enzym-Produkt-Intermediat umgesetzt wird. In jedem Fall wird aber der Übergangszustand stabilisiert. Einer der wichtigsten Faktoren hierbei ist der Proximitätseffekt. So wird aus einer intermolekularen Reaktion eine intramolekulare. 3.2.1 Der Proximitätseffekt Allgemein gilt das intramolekulare Reaktionen im Vergleich zu den intermolekularen sehr viel schneller und milder ablaufen. Das Konzept dahinter ist das der effektiven Konzentration. Diese erhält man durch den direkten Vergleich einer Reaktion einerseits als inter- und andererseits als intramolekulare Variante. Die effektive Konzentration ist die Konzentration, in der man das Reagenz in der intermolekularen Variante zugeben muss, damit man die gleiche Rate erhält wie in der intramolekularen Reaktion. Untersucht man die Hydrolyse von Phenylestern (Abb. 3), die eine interne Base in unterschiedlichen Positionen zur reaktiven Stelle aufweisen, so wird die Stärke des Proximitätseffektes deutlich. Referenz ist die Hydrolysereaktion in Gegenwart von Acetat als Base. Die im Molekül vorhandenen „Acetate“ bilden hierbei zunächst ein Anhydrid durch Angriff des Phenylesters durch das Carboxylat. In der intramolekularen Variante erfolgt dieser Schritt sehr viel schneller. Am schnellsten geht die Reaktion, wenn sich ein fünfgliederiges Intermediat bilden kann, gefolgt vom 3 sechsgliederigen Intermediat. Durch Vergleich mit der intermolekularen Reaktion lassen sich die effektiven Konzentrationen berechnen (k = v [Acetat] [S]). Abb. 3 Veranschaulichung des Konzepts der effektiven Konzentration am Beispiel der Hydrolyse von Phenylestern. Diese effektiven Konzentrationen sind so hoch, dass selbst eine intermolekulare Reaktion bei der das Substrat von Acetaten in Lösung umringt ist, langsamer verlaufen würde. Je besser das System in der intramolekularen Variante präorganisiert ist, umso schneller verläuft die Reaktion, weil die Wahrscheinlichkeit der Reaktion Präorganisation. wesentlich gesteigert Thermodynamisch wird. Enzyme bedeutet dies, sind dass also Meister der Aktivierungsentropie wesentlich erniedrigt wird. Ein Übergangszustand ist hoch geordnet. ΔS ist daher positiv, was ΔG positiver und damit ungünstiger macht. Erfolgt eine Präorganisation, muss dieser Entropieverlust nicht mehr aufgebracht werden. In der aktiven Tasche der Enzyme sind Substrate, Cofaktoren und reaktive Gruppe bereits so vororganisiert, dass kein Entropieverlust auftritt, wenn sich die Reaktion vom EnzymSubstrat-Komplex zum Übergangszustand bewegt. 4 3.2.2 Die thermodynamische Stabilisierung des Übergangszustandes Enzyme binden zunächst das Substrat, um dann einen Enzym-Substrat Komplex zu bilden. Hierbei ist entscheidend, dass das Substrat nicht zu fest gebunden wird, da andernfalls die Aktivierungsenergie angehoben und nicht abgesenkt wird. Dieser Zusammenhang wird in Abb. 4 verdeutlicht. Abb. 4 Einfluss der thermodynamischen Stabilität des Enzym-Substrat-Komplexes (ES) auf die Aktivierungsenergie (roter Pfeil). Der Ausgangszustand E + S ist immer der gleiche. Wird das Substrat zu fest gebunden, so erhöht sich die Energiebarriere bis zum Übergangszustand (c). Die gleichzeitige feste Bindung des Substrates S und des Übergangszustandes (ÜZ) senkt die Energie des ÜZ nicht ab (b). Das Enzym muss versuchen nur den ÜZ stark zu binden (d). Gleichzeitig ist es von Vorteil, wenn das Substrat nur schwach gebunden wird. Natürlich muss dabei die effiziente molekulare Erkennung gewährleistet bleiben. Tatsächlich beobachtet man, dass die meisten Enzyme die Substrate nur mit milli- bis micromolarer Affinität binden. Die Michaelis-Konstanten, die in erster grober Nährung der Bindungskonstante entsprechen liegen typischerweise bei KM = 10-3-10-6M. Antikörper hingegen, die darauf optimiert sind Grundzustände zu binden 5 haben Assoziationskonstanten im Bereich Kass = 10-9-10-12M. Hieraus erklärt sich das Projekt der katalytischen Antikörper. Man synthetisiert Verbindungen, die die Struktur von Übergangszuständen möglichst genau nachstellen (Übergangszustands- analoga). Erzeugt man gegen diese Antikörper, so erhält man Proteine, die in der Tat die Reaktion katalysieren. Bis heute, sind die erhaltenen katalytischen Antikörper jedoch wesentlich schlechter als die natürlichen Enzyme. 3.2.3 Säure/Base-Katalyse Viele enzymatische Katalyseprozesse beruhen auf Säure/Base-Reaktionen. Die Substrate oder Intermediate werden protoniert oder deprotoniert. Hierbei arbeiten Enzyme unter physiologischen Bedingungen. Unter genereller Säurekatalyse versteht man, wenn das Substrat durch einen katalytisch aktiven Rest protoniert wird. Da die Enzymgruppe ein Proton verliert, muss der Rest zunächst protoniert vorliegen. Der pKa-Wert der Gruppe muss also knapp über 7 (7-10) liegen. Läge der pKa-Wert über 10, würde der Protonentransfer thermodynamisch ungünstig. Generelle Basekatalyse tritt auf, wenn entweder das Substrat oder Wasser deprotoniert wird, bevor das Substrat angegriffen wird. Hierzu muss der aktive Rest in der Enzymtasche deprotoniert sein. Der pKa-Wert der Gruppe muss daher knapp unter 7 (4-7) liegen. Der Imidazolring des Histidins kann mit einem pKa-Wert von ca. 7 sowohl als Säure als auch als Base fungieren. Abb. 5 Prinzipieller Mechanismus der enzymatischen Säure- (oben) und Base-Katalyse (unten). 6 Die unterschiedlichen Seitenketten der Aminosäuren liegen exakt im geforderten Bereich. In Abb. 6 sind die pKa-Werte einiger Aminosäureseitenketten aufgeführt. Die pKa-Werte können jedoch durch die Mikroumgebung im Protein stark verändert werden. Abb. 6 zeigt, über welchen Bereich die pKa-Werte verschoben sein können. So bewirkt eine protonierte Lysin-Seitenkette, dass der pKa-Wert eines benachbarten Lysins von 9 auf 5.9 verschoben wird. Abb. 6 pKa-Werte einiger Aminosäureseitenketten. Einen weiteren entscheidenden Punktgewinn verzeichnen Enzyme durch bifunctional catalysis. So findet an der einen Stelle eine Protonierung, an der anderen eine Deprotonierung statt. Ein Beispiel ist die Ketosteroid-Isomerase (Abb. 7). Asp38 fungiert hier als Base während Tyr14 gleichzeitig die Funktion einer Säure übernimmt. Diese bifunktionelle Katalyse ermöglicht auch die Deprotonierung in Fällen, wo das offensichtlich thermodynamisch eigentlich nicht möglich ist. So hat das H neben der Carbonylgruppe einen pKa-Wert von 18-20. Eine Deprotonierung findet statt mit einer Gruppe, die einen pKa-Wert von 3.7 hat. Nur durch die gleichzeitige O-Protonierung des Enolates wird diese Reaktion thermodynamisch möglich. In solchen Enzymen fungieren auch oft Metalle wie das Zn2+ als Lewissäuren (Elektronenpaarakzeptoren). 7 Abb. 7 Bifunktionelle Katalyse im Fall der Ketosteroid-Isomerase. Prinzipiell lassen sich bei der Säure/Base-Katalyse zwei Fälle unterscheiden: spezifische und generelle Säure/Base-Katalyse. Abb. 8 Spezifische (a) und generelle (b) Säure/Base-Katalyse. Man spricht von spezifischer Säure/Base-Katalyse (Abb. 8a), wenn die Katalyse von der Konzentration an H+ und OH- direkt abhängt, d.h. abhängig ist vom pH-Wert, wie z. B. die Hydrolyse von Essigsäurethylester. Die Hydrolyse ist langsam im neutralen 8 nimmt aber im sauren (mehr H+ zur Aktivierung der C=O Gruppe) und im basischen (mehr OH-) stark zu. Man spricht von genereller Säure/Base-Katalyse (Abb. 8b), wenn die Rate bei konstantem pH mit steigender Pufferkonzentration, d.h. mit zunehmender Ionenstärke zunimmt. Die Konzentrationen an H+ und OH- bleiben hier konstant. Der Katalysator ist der Puffer(!), wie z. B. Imidazol-Puffer. 3.2.4 Nukleophile Katalyse Hierbei handelt es sich um eine kovalente Katalyse. Eine nukleophile Gruppe greift am Substrat an und bildet einen kovalenten Komplex mit dem Substrat. Vor allem in unpolaren Enzymtaschen kann ein „nacktes“ Nukleophil sehr aktiv sein. Potentielle Nukleophile sind: -CH2-OH Serin Serin-Proteasen, Lipasen, Esterasen -CH(CH3)-OH Threonin Phosphotransferase -CH2-SH Cystein Cycstein-Protease, Acyl-Transferase -(CH2)n-COOH Glutamin-/Asparaginsäure Epoxid-Hydrolase, Dehalogenase -(CH2)4-NH2 Lysin Aldolase class 1 Imidazol-NH Imidazol Phosphotransferase Ar-OH Tyrosoin DNA-Topoisomerase Schema 1 Nukleophile Katalyse am Beispiel der Acetoacetat-Decarboxylase. 9 Ein Beispiel ist die Acetoacetat-Decarboxylase (Schema 1). In diesem Enzym ist der pKa-Wert des Lysins 5.9. Daher ist das Lysin unter physiologischen Bedingungen deprotoniert und kann als Nukleophil fungieren. 3.2.5 Die Verwendung von Spannungsenergie Es handelt sich um ein Phänomen, das sich nicht so einfach experimentell fassen lässt. Manche Enzyme scheinen die Substrate in einer „verzerrten“ Konformation zu binden. Das Substrat wird in Richtung Geometrie des Übergangszustandes verbogen. Dadurch nimmt die Energie zwischen dem gebundenen Grundzustand und dem Übergangszustand entscheidend ab. Es wird also Bindungsenergie verwendet, um die Aktivierungsenergie zu reduzieren. In Abb. 10 ist die Verzerrung des Substrates im Fall des Enzyms Carboxypeptidase A gezeigt. Hier wird die Amidbindung out-of-plane verzerrt und damit für den Angriff des Nukleophils vorbereitet. Ein Zn2+ fungiert als Lewissäure. Abb. 10 Verzerrung der Amidbindung im Fall der Carboxypeptidase A. 3.2.6 Maximale Ratenbeschleunigung Wie stark kann ein Enzym maximal eine Rate Beschleunigen. Gibt es eine Obergrenze? Die Antwort ist ja. Die Grenze ist erreicht, wenn die Rate diffusionskontrolliert wird. Dieses Diffusionslimit wird von der Stosszahl bestimmt, die eine Maß für die Wahrscheinlichkeit ist, mit der zwei Teilchen zusammen stoßen können. Sie liegt bei ca. 108M-1s-1. Das ist gleichzeitig die maximale biomolekulare Rate, die erreichbar ist. Für die Reaktion eines freien Enzyms mit einem frei in Lösung befindlichen Substrat ist die bimolekulare Rate kcat/KM. Diese bimolekulare Rate nennt man auch die katalytische Effizienz. Die meisten Enzyme habe Effizienzen zwischen 106-107 M-1s-1. 10 Einige wenige schaffen jedoch auch das Diffusionslimit: Triosephosphat-Isomerase 3.0 x 108 M-1s-1 Acetylcholinesterase 1.4 x 108 M-1s-1 Ketosteroid-Isomerase 1.3 x 108 M-1s-1 β-Lactamase 1.0 x 108 M-1s-1 3.3 Michaelis-Menten-Kinetik Das Michaelis-Menten-Modell für die Enzymkatalyse nimmt an, dass die Enzymreaktion über die folgenden Stufen verläuft: E + S k1 ES k2 E + P k-1 Dieses Modell beruht auf einer ganzen Reihe von (meistens nicht stimmenden) Annahmen. So soll nach dem Modell das Enzym nur ein Substrat binden. Der ESKomplex zerfällt in einem Schritt zum Produkt. Die Produktbildung ist irreversibel. Für die kinetische Analyse geht man nun von einem Gleichgewichtszustand aus (steadystate). Bei konstantem turn-over ist die Konzentration an ES konstant. Daraus ergibt sich, dass die Rate der ES Bildung und die Rate für den ES Verbrauch gleich groß sind. k1 [E] [S] = k2 [ES] + k-1[ES] mit [E0] = [E] + [ES] k1 [E0] [S] - k1 [ES] [S] = k2 [ES] + k-1[ES] [ES] = [E0] [S] Km + [S] Produktbildungsrate = k2 [ES] = k-1 + k2 mit [Km] = k1 k2 [E0] [S] KM + [S] 11 Misst man nun die Enzymreaktion in Abhängigkeit von der Substratkonzentration, so ergibt sich im Idealfall die folgende Kurve: Aus einer solchen Messung lassen sich nun die zwei kinetischen Konstanten kcat und KM ermitteln. kcat ist die turn-over Konstante. Es handelt sich um eine unimolekulare Ratenkonstante, die in min-1 oder s-1 angegeben wird. Die Konstante sagt aus, wie viel μmol Substrat pro μmol Enzym pro Sekunde umgesetzt wird. Oder in molekularen Worten: Die Anzahl der Moleküle, die durch ein Enzym pro Sekunde umgesetzt werden. Typische Werte liegen zwischen 0.1-100 s-1. KM ist die Michaelis-Konstante in (M). Der Wert gibt an bei welcher Substratkonzentration die Enzymgeschwindigkeit halbmaximal (vmax/2) ist. Der Wert ist ein grobes Maß dafür, wie stark das Enzym das Substrat bindet. Schwach gebundene Substrate haben einen großen KM-Wert. Stark bindende Enzyme haben ein sehr kleines KM. KM ist jedoch nicht die tatsächliche Dissoziationskonstante! Typische Werte liegen bei 1 μM – 1 mM. Bei sehr hohen Substratkonzentrationen [S]>>KM reduziert sich die Ratengleichung zu: v = kcat [E]. Man kann nun die Substratkonzentration weiter erhöhen. Der Umsatz bleibt immer der gleiche, da das gesamte vorhandene Enzym mit Substrat gesättigt ist. Bei sehr niedrigen Substratkonzentrationen gilt v = (kcat/KM)[E][S]. Nun ist die Rate proportional zur Substratkonzentration. Die beobachtete Rate ist nun wirklich bimolekular. Diese Rate (kcat/KM) ist die katalytische Effizienz. Sie hängt davon ab, wie stark das Enzym das Substrat bindet. Abb. 11 verdeutlicht die beiden Regime: Bei hoher Substratkonzentration ([S]>>KM) ist das Enzym immer gesättigt. Die Energiedifferenz zwischen ES und dem ÜZ bestimmt nun die Reaktionsgeschwindigkeit. Am Punkt [S] = KM ist das Enzym 12 halbgesättigt. Bei niedrigen Substratkonzentrationen ([S]<<KM) liegt vorwiegend freies Enzym vor. Nun ist die Energie zwischen dem Zustand [E] + [S] und dem [ÜZ] Raten bestimmend. Abb. 11 Veranschaulichung der einzelnen Grenzfälle in der Michaelis-Menten-Kinetik. 3.3.1 Reversible Enzyminhibition: Die Enzyminhibition kann kompetitiv und nicht-kompetitiv erfolgen. Bei der kompetitiven Inhibition konkurriert der Inhibitor mit dem Substrat um die Bindungsstelle. Nicht-kompetitive Inhibition bedeutet, dass der Inhibitor an einer andere Stelle des Enzyms bindet und ein unproduktiver ternärer Komplex EIS gebildet wird. 3.3.2 Irreversible Enzyminhibition: Eine zweite Möglichkeit der Enzyminhibition stellt die irreversible Inhibition dar. Hier bindet der Inhibitor in der aktiven Tasche und bildet dann eine kovalente Bindung mit dem Enzym aus (E-I). Hierdurch wird die aktive Tasche vollständig und irreversibel blockiert. 13 Die meisten Enzymkinetiken werden unter steady-state Bedingungen durchgeführt. Eine Enzymkinetik wird in diesem Fall über 1 – 10 min gemessen. Kann man Raten vor der Einnahme des steady-state messen, so erhält man Informationen über einzelne Enzymschritte. Man spricht von pre-steady-state Kinetik. Ist kcat = 10 s-1 so wird unter Sättigungsbedingungen ein Molekül Substrat pro 0.1 s umgesetzt. Um nun einen Umsatz verfolgen zu können muss man im Bereich 0-100 ms messen. Das wird in der Regel mit stopped-flow Apparaturen durchgeführt (Abb. 12). Die Zeitauflösung einer solchen Apparatur liegt im Bereich Mikrosekunden/Millisekunden. Abb.12 Schematische Darstellung einer stopped-flow Anlage. 3.4 Stereochemische Aspekte Enzymreaktionen sind in der Regel stereoselektiv und stereospezifisch. Stereoselektiv heißt, Enzyme setzen aus einem Isomerengemisch nur ein Isomer um. Enantioselektiv heißt, dass aus einem Razemat nur ein Enantiomer als Substrat akzeptiert wird. Stereospezifisch bedeutet, dass ein spezifischer Reaktionsweg beschritten wird, um nur ein Produktisomer zu generieren. Die Grundlage hierfür ist, dass die Reaktion in einer chiralen Enzymkavität ausgeführt wird. Als Beispiel ist in Schema 3 die Fructose-1,6-diphosphat-Aldolase gezeigt. Die Aldolase katalysiert eine Aldolreaktion bei der nur ein Stereoisomer entsteht. Schema 3 Katalysierte Reaktion der Fructose-1,6.diphosphat-Aldolase. 14 Enzyme sind auch in der Lage pro-chirale Atome zu unterscheiden. So sind die zwei H-Atome der CH2-Gruppe im Ethanol pro-chiral, d. h. ersetzt man ein H durch ein D so entsteht eine chirale Substanz. Das H, bei dessen Ersatz die R-Verbindung entsteht, nennt man pro-R, dasjenige nach dessen Ersatz das s-Isomer entstehen würde, nennt man pro-S. Diese Differenzierung ist wieder nur möglich, weil das Substratmolekül in der Enzymtasche fest eingebunden vorliegt. Der Angriff erfolgt dann entweder von oben oder von unten. Entscheidend ist die Fixierung in einer chiralen Umgebung. Um die pro-Chiralitätselektivität zu ermitteln, müssen Substrate verwendet werden, in denen einer der pro-chiralen Atome durch ein Isotop ersetzt ist. Wichtige Isotope sind: H: 1H, 2H, 3H C: 12C, 13C, 14C N: 14N, 15N O: 16O, 17O, 18O Entfernt das Enzym ein H-Atom von einer Methylgruppe, so kann erneut nur entschieden werden, ob das stereoselektiv erfolgt, wenn entsprechend gelabelt wird. Hierzu muss eine Methylgruppe hergestellt werden, die ein H, ein D und ein T enthält. Man muss das Produkt anschließend auf die chirale Essigsäure zurückführen. Für diese Verbindung hat D. Arigoni und J. W. Cornforth eine Methode zu Bestimmung, ob die Essigsäure R oder S konfiguriert ist, entwickelt (Schema 4). Schema 4 Bestimmung der Stereokonfiguration von chiraler Essigsäure nach D. Arigoni und J. W. Cornforth. 15