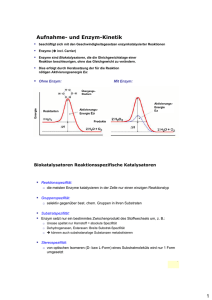
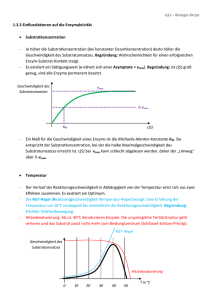
Enzyme in der organischen Synthese
Werbung

Enzyme in der organischen Synthese Wintersemester 2006-2007 Michael Müller Institut für Pharmazeutische Wissenschaften Tel. 203-6320 [email protected] Ziel der Vorlesung: Eine Grundlage für das Verständnis biokatalytischer Transformationen legen. 1. Grundlagen der Biokatalyse (3-4 Wochen) 2. Biosynthese (1 Woche) 3. Verwendung von Hydrolasen (2 Wochen) 4. Verwendung von Oxidoreduktasen (2-3 Wochen) 5. C-C-Bindungsknüpfung (2-3 Wochen) 6. Glycosyl-Transferasen (1 Woche) 7. Radikalreaktionen (1-2 Wochen) 8. State of the art und spezielle Aspekte der Biokatalyse (1-2 Wochen) 1 Enzyme als Katalysatoren • • • • • Sehr effiziente Katalysatoren Erhöhung Reaktionsgeschwindigkeit kcat/knon 108 – 1014 (teilweise bis 1017) Chem. Katalysatoren 0.1 – 1 mol % Enzyme 10-3 – 10-4 mol % Umweltverträglich Keine Schwermetallsalze (→ chem. Katalysatoren) Komplett biologisch abbaubar milde Bedingungen: pH 5 – 8, 20 – 40°C breites Substratspektrum + Lösungsmitteltoleranz (teilw.) + mögliche Immobilisierung => breite Anwendbarkeit breites Reaktionsspektrum teilweise Reaktionen, die mit chem. Methoden (noch) nicht möglich sind, z.B. selektive Oxidation nicht aktivierter C-H’s CH3 R CH3 CH3 R CH3 CH2OH CH3 R CH3 CHO R CH3 CH3 CO2H CH3 • sehr selektive Katalysatoren: Chemoselektivität Regio- und Diastereoselektivität Enantioselektivität • Bevorzugtes Lösungsmittel: H2O • Enzyme sind chiral (L-Aminosäuren [L-AA]) Wirkstoffe: Eutomer (höher aktives Enantiomer) Distomer (weniger aktiv oder unerwünschte Wirkung) „Eudismic ratio“ Aktivität E Aktivität D Nachteile Enzyme • Enzyme sind chiral (nur L-AA) chem. Katalysatoren lassen sich in aller Regel einfacher in beiden enantiomeren Formen darstellen • milde Bedingungen -78°C, 180°C, pH1, pH14 => nicht möglich mit Enzymen (Stabilitäts- oder Reaktivitätsverlust) 200 bar: möglich! • bevorzugtes Lösungsmittel: H2O (geringe Löslichkeit vieler organischer Substanzen in H2O) • können empfindlich auf Inhibition reagieren mögliche Allergene / biol. Wirkstoffe z.B. Prionen-Problem: Famulok et al, Angew. Chemie 1997, 109, 1748-1769. 2 Was ist ein Biokatalysator? Enzym = Protein (Protein: Peptid, Enzym: aktive Form) aber auch: - katalytische Antikörper (abzymes) - katalytische RNA (Ribozymes) - katalytische DNA (Deoxyribozymes) M. Famulok und P. Burgstaller, Synthetic Ribozymes and Deoxyribozymes, Organic Synthesis Highlights III. Seite 173 - 184. Sequenzinformation von Enzymen/Proteinen in DNA-Sequenz festgelegt DNA Transcription mRNA Translation Protein gilt uneingeschränkt nur teilweise DNA Introns Transcription mRNA Translation Protein Introns (z.B. in Eukaryonten) Beispiel für Posttranslationale Modifikation: Protein-Spleißen DNA Transcription mRNA Inteine Translation Protein posttranslationale Modifikation: Protein-Spleißen Exteine Enzym 3 Ein Enzym = ein Protein (absolut identische Sequenz) ? z.B. Pig liver esterase PLE, Schweineleber Esterase: Besteht aus 5 Isoenzymen (sehr ähnliche aber nicht identische Enzyme) unterscheiden sich in Substratspektrum, Aktivität, Selektivität Untersuchung isolierter Enzyme (einzelne Proteinmoleküle) einzelne Enzyme unterscheiden sich bis zu Faktor 4 – 23 in ihrer Aktivität Sequenz alleine bestimmt nicht zwangsläufig Struktur und Aktivität Gründe: - unterschiedliche Faltungen - posttranslationale Modifikation Q. Xue, E. S. Yeung, Nature, 1995, 373, 681. Dorichi et al, JACS, 1996, 118, 5245 – 53. D. B. Craig, N. J. Dovichi, Can. J. Chem. 1998, 76, 623 – 26. vgl.hierzu: Famulok et al., Angew. Chem. 1997, 109, 1748 – 69. Zum tieferen Verständnis von Biokatalysatoren benötigte Grundlagen: • • • • • • • • Biosynthese Biochemie Katalyse-Mechanismus Natürliche Substrate / Produkte (nicht immer logisch) Stereochemie Unnatürliche Substrate / Org. Synthese Technische Chemie 3D-Struktur / Theoretische Chemie Interdisziplinarität Voraussetzung Regelmäßigkeiten erkennen => Ausnahmen generieren mit - genetic engineering (enzyme engineering) - substrate engineering - reaction engineering z.B. BAL - 2 Enantiomere - 1 Mutation im Vgl. zu BFD im active site ( Mutation umkehrbar) - C-C-Verknüpfung + C-C-Spaltung 4 Enzyme als Katalysatoren Allgemein: Enzymes function by lowering transition-state energies and energetic intermediates and by raising the ground state energy. Aber: Mindestens 21 verschiedene Hypothesen (Stand 2000) Mechanistische Aspekte: Katalysator (ganz allgemein): Stabilisierung des Übergangszustandes gegenüber Grundzustand → Abnahme der Aktivierungsenergie → dies führt zu rate accelaration Enzymmodelle: 1. Schlüssel-Schloß-Prinzip (lock and key) E. Fischer, Berichte dtsch. Chem. Gesellschaft 1894, 27, 2985. Voraussetzung: „feste“ (rigide) Enzymstruktur zu stark vereinfachend, bei Schlüssel-Schloß-Prinzip fehlt z.B. dass Substanzen die geringeren räumlichen Anspruch haben als die Substrate und den Substraten ähnlich sind, trotzdem nicht als solche akzeptiert werden (und umgekehrt!). 2. Induced-Fit Mechanismus Koshland und Neet, Ann. Rev. Biochem. 1968, 37, 359. Hand – Handschuh Substrat – Enzym Enzym ändert durch die Anbindung des (eines) Substrats seine Form →Aktivität. Flexibilität von Enzymen wurden schon früher von Linus Pauling als wichtiges Merkmal der Enzymkatalyse postuliert. Chem. Eng. News 1946, 24, 1375. Am. Sci. 1948, 36, 51. Vgl. 3. Desolvation- und Solvation-Substitution-Prinzinp M. J. S. Dewar, Enzyme 1986, 38, 8. Creighton et al., Proc. Natl. Acad. Sci. 1989, 86, 520. Wasser innerhalb active site wird durch Substrat ersetzt → formale Gas-Phasen-Reaktion. Wasserhülle um Substrat wird ersetzt (energetisch ungünstig) → Destabilisierung des Grundzustandes. 5 Zur Zeit (2000) 21 verschiedene Modelle für Emzymkatalyse; alle Enzymreaktionen werden initiiert durch allen gemeinsam: die Bildung eines Enzym-Substrat-Komplexes (ES). Bindungskräfte involviert in ES-Komplex-Bildung Kovalente-Bindung (-40 - -110 kcal/mol) Ionen-Paar-Bindung (elektrostatisch) (bis –5 kcal/mol) Ion-Dipol oder Dipol-Dipol-Bindung Wasserstoffbrücken (-1 - -3 kcal/mol) Charge-Transfer Komplex (z.B. π-π-stacking, edge-to face) Hydrophobe Wechselwirkungen (-0.5 kcal/mol) van-der-Waals-Kräfte (-0.5 kcal/mol) (Werte bezogen auf ∆G°) wichtig: viele kleine Kräfte können zusammen einen starken Effekt bewirken R. B. Silverman, The organic Chemistry of Enzyme-Catalyzed Reactions, Academic Press, 2000, S. 7. 6 Enzym-Mechanismen Enthalpie: Entropie: Änderung in Bindungsenergie (dominiert) Orientierung, Rotation, Translation, Konzentrations- und Lösungsmittel-Effekte 1. Annäherung in die räumliche Nähe bringen von (zwei) Reaktanten → Verlust an konformationeller Freiheitsgrade, fest vorgegebene Orientierung → Reaktion wird erster Ordnung statt zweiter Ordnung in Lösung ≅ Erhöhung der effektiven Konzentration der Reaktanten 2. Kovalente Katalyse vor allem bei proteolytischen Enzymen z.B. nukleophile Katalyse (entspricht dem anchimeren Effekt in der chemischen Katalyse) Beispiel: Serin-Protease (Serin als Nukleophil) 3. Allgemeine Säure/Base-Katalyse z.B. Serin-Protease R-OH schlechtes Nukleophil R-O- wesentlich besseres Nukleophil Katalytische Triade: Aspartat (Asp)-Histidin (His)-Serin (Ser) pka-Werte in Lösung unterscheiden sich deutlich von pka in active site (zum Teil wegen der niedrigeren Polarität innerhalb der Enzymtasche) → pka steigt (Säure) → pka sinkt (Base) Niedrigere Polarität innerhalb der Enzymtasche entspricht eher Benzol als Wasser (Dieleketrizitäts-Konstante innerhalb Proteinen ca. 2-3; im Vergleich dazu Benzol: 2.28, Wasser: 78.5) Eigentlich ist es nicht möglich, mit einem Carboxylat (Asp-CO2-) (pka 3.9) Histidin (Imidazol) zu deprotonieren (pka 6.1); ebenso kann Histidin Serin (pka 14) nicht deprotonieren. Dies gilt aber nur in Lösung. Aufgrund der pka-Werte wäre Enzymkatalyse nicht möglich. Aber: Protonen-Transfer-Schritte nicht einzeln betrachten. Gleichzeitiger Angriff von von SerO- an Carboxylgruppe → dadurch wird Gleichgewicht verschoben. 7 Beispiel für selektive Säure-Base-Katalyse: Enzyme können gleichzeitig mit Säure und Base aktivieren. Kann man diese Effekte addieren? Chemische Katalysatoren: resultieren immer bei pH 7 = Neutralisation Ausnahme: neue Katalysatorsysteme : Lewis-Säure + Base gleichzeitig. Schibasaki et al. J. Am. Chem. Soc. 1999, 121, 2641. 4. Low-barrier-hydrogen-bond Schwache H-Brücken-Bindung wird im Übergangszustand zu einer starken H-Brücken-Bindung. Übergangszustand wird durch 4-20 kcal/mol stabilisiert (vgl. normale H-Brücken bis zu –3 (-5) kcal/mol). Cleland et al. J. Biol. Chem. 1998, 273, 25529, Science 1995, 269, 104. 5. Elektrostatische Wechselwirkung z.B. Serin-Protease rate Acceleration Asp His Ser 109 Ala Ala Ala 103 (im Vgl. zur unkatalysierten Reaktion) Mutante: D32A, H64A, S221A → neben nukleophiler Katalyse, general base catalysis, elektrostatische Katalyse Dies ist ein wichtiges Beispiel dafür, dass das gesamte Enzym benötigt wird um die Katalse durchzuführen. Räumliche Annäherung des Substrats an Serin ist genauso wichtig, wie die katalytische Triade! 8 6. Desolvation und Solvation-Substitutionsprinzip Entfernen von Wassermolekülen von geladenen Gruppen im active site durch Substrat. → Destabilisierung des Grundzustandes. z.B. Rucker und Byers J. Am. Chem. Soc. 2000, 122, 8365. Beitrag von Desolvation eines Dianions resultiert in 105-fache Reaktionsbeschleunigung. Desolvation nonoanionischer Nukleophile resultiert nur in 100-facher Beschleunigung. Bei neutralen Nukleophilen kann Desolvation (z.B. Ersatz von Wasser-Hülle durch DMSO) inhibitorische Wirkung haben. 7. (Ring)-Spannung und „distortion“ am Enzym → hoch energetischer Zustand am Substrat → Erhöhung der Energie des Grundzustandes durch Destabilisierung. Dies führt zu einer höheren Reaktivität vgl. Ether – Epoxide, Epoxide sind deutlich reaktiver als andere Ether. Ringspannung und „distortion“ bezieht sich auf Substrat und Enzym. Vgl. induced-fit-Mechanismus bei Koshland → Enzym kann in einen hoch energetischen Zustand überführt werden. 8. Negative Katalyse (Reaktionsselektivität) Review: J. Rétey Angew. Chem. 1990, 102, 373. Zitat: Die Selektivität solcher Reaktionen wird daher eher durch das Verhindern unerwünschter Reaktionen als durch die Förderung der eigentlichen Zielreaktion bedingt. 9 Zusammenfassung Für „rate-enhancement“ sind verschiedene Faktoren zuständig. Der jeweilige Beitrag der verschiedenen Mechanismen hängt stark von der Natur der zu katalysierenden Reaktion (Substrate, Intermediate, Produkte, basisch, nukleophil ...) ab. → allgemeine Erklärung nicht einfach möglich. Bindungsenergie führt zu einer Stabilisierung des Übergangszustandes (ÜZ), aber Grundzustand (ES bzw. EP) wird auch abgesenkt → Destabilisierung ist notwendig. z.B. durch - strain - distortion - Entropie-Verlust Stabilisierung des Übergangszustandes (ÜZ) wichtiger als Destabilisierung des Grundzustandes. Enzyme No Enzyme S P + Binding Energy + Destabilization + Binding Energy E+S E+P E+S ES EP E+P ∆GD -T∆S ES A EP B C 10 Gründe für Selektivität Drei-Punkte-Regel (stark vereinfachend, gilt zum Teil nicht) - Chiralitätszentrum D A' D A' A A C B B C C' B' C' B' nicht bevorzugt optimaler fit Enantiotopic discrimination: (Enantiotope Diskriminierung) Gründe für Selektivität - Prochiralität pro S pro R A A' A A' A A B C C' B' optimaler fit C B C' B' nicht bevorzugt A ersetzen durch A*, A* > A > B > C R: A ist pro R S: A Ist pro S 11 Enantioface discrimination (Enantiofaciale Diskriminierung) Re-face Si-face A' A A' B x C x C' B C C' A B' B' nicht bevorzugt optimaler fit Enantioface Discrimination (Enantiofaciale Diskriminierung) z.B. prochivales Molekül H3C OH ∧ = HO Beispiel für meso-Verbindung CH3 H3C CH3 * * H3CO2C H3CO2C CO2CH3 H3CO2C PLE H3C OH CO2CH3 H3C CH3 * * H3CO2C CO2CH3 CO2CH3 PLE PLE HO ∧ = CH3 H3C CH3 * * H3CO2C CO2H H3CO2C CO2H HO2C CO2CH3 12 Kinetische Gründe für Selektivität E: Enzym A: Substrat B: Substrat (Stereoisomer von A) A EA E + P1 B EB E + P2 E EB ∆G ∆∆G++ EA E + A,B E + P2 E + P1 G++ sein für ee 99%? Wie groß muß G++ = G++ - T · G++ = - RT ln S++ K1 K2 G++ (kcal/mol) ee K1/K2 10 1.22 0.118 50 3 0.651 90 19 1.74 95 39 2.17 99 199 3.14 99.9 1999 4.50 Stereoselektivität E = k cat Km L k cat Km D ee = P1 − P2 ⋅ 100 P2 + P2 13 Kinetik Michaelis-Menten [E] E ++ [S] S K1 K -1 [ES] ES [EP] EP K2 [E]++P[P] E [S] v = vmax Km + [S] vmax = K2 [E] Annahme: K1 >> K2, v = K2 [E] [S] Km + [S] K2 = kcat (turnover number) (first order) Km: Michaelis-Menten Konstante (bei ½ vmax) (Dissoziationskonstante des ES-Komlexes) v vmax 1v max 2 [S] Km (M) Km: ist unabhängig von der Enzymkonzentration üblich: 10-6 – 10-2 M vmax: max. Reaktionsgeschwindigkeit wenn jedes Enzym mit Substrat gesättigt ist (Substrat-Sättigung) üblich: 103 – 104 sec-1 katalytische Aktivität: 1U ≅ katalysiert die Umsetzung von 1 mol/min bei spez. Bedingungen (pH, Temp.) unkatalysiert Energie kcat + [ES]+ Km kcat E+S E+P ES EP k cat Km = spezifische Konstante bezieht sich auf Reactions rate von freiem Enzym und Substrat für obiges Beispiel: kcat K ⋅K = 1 2 Km K−1 + K−2 (second order) dient dazu, verschiedene Substrate auf Effizienz hin zu vergleichen 14 Bestimmung der kinetischen Konstante dient dazu, optimale Reaktionsbedingungen zu generieren (Produktivität, Selektivität) Durchführung: Anfangsreaktionsgeschwindigkeit in Abh. von [S] bei konstantem pH, [E], … Verschiedene Möglichkeiten der Auftragung: - Michaelis-Menten - Lineweaver-Burk - plot - procedure (häufig benutzt) 1 Km 1 = + v vmax ⋅ [S] vmax Nachteil: Werte für hohe [S] fallen in engen Bereich 1 1 Plot : gegen v [S] 1 v Steigung = Km vmax vmax 1 - Km 0 1 [S] 15 Enzym-Inhibierung Definition: Abnahme der enzymatischen Aktivität aufgrund geänderter Reaktionsparameter A: Reversible Inhibierungen 1) competitive 2) noncompetitive 3) uncompetitive 4) mix 1-3 v ohne Inhibitor vmax nonspecific inactivation competitive noncompetitive [S] Ganzzellbiotransformation Enzymatische Katalyse Chemische Katalyse homogen / heterogen Availability gut ++ oft limitierend +/– Bedingungen mild ++ mild ++ oft drastisch (+) / – +/– Reproduzierbarkeit variabel – konstant ++ konstant ++ Nebenaktivitäten hoch – keine ++ keine ++ Permeabilitätsprobleme limitierend – keine ++ keine ++ Cofaktorregenerierung intrazellulär + Katalysatorkonzentration unbekannt limitierend (+) / – keine ++ niedrig (10-3-10-4) + hoch (0,1-1 %) – TTN unbekannt hoch ++ niedrig – TOF (turn over frequency) unbekannt hoch ++ niedrig – Preis Katalysator niedrig + mittel +/– mittel - hoch (+ ) / – Substratkonzentration niedrig – mittel +/– hoch ++ Produktrecovery schwierig – einfach + einfach + ee mittel - hoch +/– hoch ++ mittel - hoch +/– Produktivität (Atomökonomie) niedrig – hoch + mittel (+) / – Raum-Zeit-Ausbeute (space time yield) niedrig - mittel (+) / – niedrig - mittel (+) / – hoch ++ 16