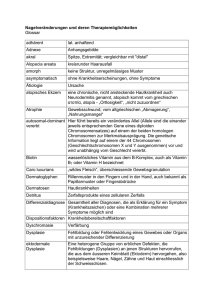
Klinisch-Pathologische Konferenz
Werbung

Skript zu den Veranstaltungen „Klinisch-Pathologische Konferenz“ Institut für Pathologie 2011 Inhalt KPK Mamma ……………………………………………………………… 3 KPK Entzündliche Lebererkrankung …………………………………. 9 KPK Knochen- und Weichteiltumoren ……………………………….. 14 KPK Entzündliche und vaskuläre Nierenerkrankung ……………… 21 KPK Neuropathologie ……………………………………………………. 27 KPK Lungenerkrankung …………………………………………………. 32 KPK Neoplasien der Cervix ……………………………………………... 41 KPK Lymphknoten- und Hämatopathologie …………………………. 49 KPK Prostata- und Harnblasentumoren …………………………….... 56 2 KPK Mamma Die Mamma gehört zu den Hautanhangsgebilden und entwickelt sich geschlechtsabhängig unter Östrogen- und Progesteroneinfluss. Ihre medizinische Bedeutung besteht vor allem in der häufigen Karzinomentwicklung und den tumorartigen Veränderungen, die hiervon differentialdiagnostisch abgegrenzt werden müssen. Die Histopathologie stellt hierbei die entscheidende Untersuchung dar. Epidemiologie des Mammakarzinoms Das Mammakarzinom ist in Nordamerika und Europa der häufigste maligne Tumor der Frau und deren häufigste Krebstodesursache. Die Inzidenz steigt bei sinkender Mortalität an. Inzidenz BRD Anteil an Krebserkrankungen bei Frauen Anteil an Krebstodesursachen b. Frauen Mittleres Erkrankungsalter Geographische Verteilung Inzidenz weiblich : männlich ca. 55.000/Jahr ca. 24% ca. 18% 63 Jahre, selten vor 25. Lebensjahr Häufig in U.S.A.und Westeuropa, seltener in Japan und Entwicklungsländern > 100 : 1 Ätiologie des Mammakarzinoms Die Ursachen des humanen Mammakarzinoms sind unbekannt. Es können lediglich Risikofaktoren benannt werden, wozu in erster Linie eine frühere und längere Östrogenexposition sowie eine genetische Belastung gehören. Gesicherte Risikofaktoren frühe Menarche (vor 12. Lebensjahr) Mögliche Risikofaktoren Fettreiche Nahrung (gesättigte Fettsäuren) späte Menopause Fettleibigkeit, besonders im höheren Alter. Hormonelle Antikonzeption späte erste Gravidität (nach dem 30. Lebensjahr) Postmenopausale Hormonsubstitution Positive Familienanamnese: mehr als doppeltes Risiko bei einer an Mammakarzinom erkrankten erstgradigen Verwandten, mehr als 5faches Risiko bei zwei Betroffenen. Noch höher bei bestimmten Keimbahndefekten (s. Pathogenese) Bestrahlung 3 Pathogenese des Mammakarzinoms Das Mammakarzinom entsteht hormonabhängig aus den Epithelien des ductulolobulären Übergangs unter dem Einfluss von Keimbahn- (ca. 10%) und somatischen Mutationen. Für die Entstehung der meisten Mammakarzinome sind Östrogen und Progesteron unverzichtbare Promotoren, 75-80% der Karzinome exprimieren die zugehörigen nukleären Rezeptoren und ihr Wachstum lässt sich durch Rezeptorantagonisten (Tamoxifen) oder Hemmer der Hormonsynthese (Aromatasehemmer) inhibieren. Bei etwa der Hälfte der Fälle von familiären Mammakarzinomen, d.h. ca. 5% aller Mammakarzinome, liegen bekannte und identifizierbare Keimbahnmutationen vor. Die häufigsten betreffen DNA-Reparaturgene (BRCAI und II) und erhöhen die Wahrscheinlichkeit von Mutationsträgerinnen, an einem Karzinom zu erkranken, auf bis zu 80%. BRCAI assoziierte Mammakarzinome treten gehäuft vor dem 40 Lebensjahr auf, eine Schwangerschaft erhöht anders als beim sporadischen Karzinom das Risiko und es handelt sich zumeist um wenig differenzierte, triple negative Karzinome (negativ für Östrogen- und Progesteronrezeptor und Her2). Familiäre Mammakarzinome insgesamt machen etwa 10% aus. Somatische Mutationen beim Mammakarzinom treten in großer Anzahl und unterschiedlicher Kombination auf. Die häufigsten sind: Inaktivierte Tumorsuppressorgene Aktivierte Protoonkogene CDH-1 Gen auf Chromosom 16q p53 Gen auf Chromosom 17p HER2-Gen auf Chr. 17q (amplifiziert) c-myc Gen auf Chr. 8p (amplifiziert) Invasive Karzinome entwickeln sich zu einem großen Teil der Fälle aus präinvasiven in-situ Carcinomen, die zumeist dieselben genetischen Veränderungen aufweisen. Reine In-situ Karzinome kommen in 10-20% vor (s. histologische Typen). Pathomorphologie des Mammakarzinoms Mammakarzinome, die in situ wachsen und nicht invasiv sind, werden nach dem Ort ihrer bevorzugten Ausbreitung als lobuläre Neoplasie oder als intraductales Karzinom bezeichnet. Im Gegensatz zu invasiven Karzinomen, die zumeist einen harten unregelmäßig begrenzten Tastbefund hervorrufen, sind sie nicht tastbar und nur durch Bildgebung nachzuweisen. Invasive Karzinome können aus In-situ Karzinomen entstehen oder kombiniert mit ihnen vorkommen, die häufigsten Formen sind das invasiv ductale (ca.75%) und das lobulär invasive (10-15%) Mammakarzinom. Zu den häufigsten differentialdiagnostisch abzugrenzenden benignen Raumforderungen gehören das Fibroadenom und die Mastopathie. Mammakarzinome weisen sehr unterschiedliche Malignitätspotentiale auf, die man versucht mit Prognosemarkern zu erfassen. 4 Makroskopie Häufigste Lokalisation ist der obere äußere Quadrant. Typisch, aber nicht beweisend, ist ein harter, strahlig begrenzter Knoten. Lobuläre invasive Karzinome sind gelegentlich schlechter tast- und abgrenzbar. In-situ Karzinome erzeugen i.d.R. keinen Tastbefund, eventuell sind intraduktale Komedonekrosen als gelbe Stippchen auf der Schnittfläche sichtbar. Histopathologie Mammakarzinome können in situ, invasiv oder in einer Kombination aus beidem wachsen. Das lobuläre Karzinom weist aufgrund einer somatischen Mutation ein defektes Adhäsionsmolekül (E-Cadherin) auf, so dass es verstreutzellig ohne Ausbildung von Epithelverbänden infiltriert. Präinvasive In-situ Karzinome Diese sind als Vorstadium eines invasiven Karzinoms aufzufassen, auch wenn nicht alle In-situ Karzinome in invasive Formen übergehen und bei vielen invasiven Karzinomen keine In-situ Komponente nachweisbar ist. Es werden die lobuläre Neoplasie (Carcinoma lobulare in situ) und das intraductale Karzinom voneinander unterschieden. Lobuläre Neoplasie Lobuli bis terminale Gänge Ausgefüllte Lumina in den normal weiten bis stark erweiterten Lobuli Graduierung in LIN1-3 Indikator eines ipsi- und kontralateral 7 bis 12-fach gesteigerten Risikos für invasive lobuläre und ductale Karzinome Häufig multizentrisch u. bilateral Überwachung, Exzision nur in Ausnahmen I.d.R. kein Befund in Bildgebung Intraductales Karzinom Terminale Gänge, Extension möglich in Lobuli und in große Gänge, evtl. sogar Epidermis (M.Paget). Mikropapilläre, cribriforme oder solide Epithelproliferate + Nekrosen Grad 1-3 n. Kerngröße und Nekrosen Übergang in invasive ductale Carcinome abhängig von Größe und Kerngrad Ausbreitung in einem Drüsenlappen Chirurgische Exzision Mikrokalk, feinkörnig oder amorph und gruppiert, amorph und verzweigt segmental 5 Durch die Einführung des flächendeckenden Mammographie-Screenings wird sich wahrscheinlich der Anteil reiner In-situ Karzinome von derzeit 5-7% auf bis zu 20% erhöhen. Invasive Karzinome Häufigster histologischer Subtyp ist das invasive nicht weiter spezifizierte Karzinom, das wegen des vermuteten Ausgangs von terminalen Gängen auch als ductales Carcinom bezeichnet wird. Alle Mammakarzinome werden in G1 bis G3 graduiert, wobei Tubulusbildung, nukleäre Pleomorphie und Mitosenfrequenz in einem Summationsscore berücksichtigt werden, der eine höhere prognostische Relevanz als der histologische Subtyp besitzt. Wenig Tubulusbildung, hohe Kernpleomorphie und hohe mitotische Aktivität führen zu einem hohen Grad 3, der eine gesteigerte Progressionsneigung anzeigt. Histologischer Typ Ductal Häufigkeit Wachstumsmuster Besonderheiten 75% Drüsig-trabekulär Lobulär 10% Verstreutzellig, Gänsemarsch Häufiger multizentrisch, bilateral Tubulär 5% Medullär <5% Sehr gute Prognose Bessere Prognose Muzinös 5% Papillär <5% >90% Tubuli, geringe Kernpolymorphie Rein trabekulär, syncytial ohne Tubuli, hohe Kernpolymorphie >75% extrazelluläre Schleimablagerung Papillär, intrazystisch Bessere Prognose Sehr gute Prognose Metastasierung Axilläre Lymphknoten, Lunge, Leber, Knochen, ZNS Axilläre Lymphknoten, Pleura, Lunge, Ovarien, Leber, Knochen selten Wie ductaler Typ Wie ductaler Typ selten Alle Karzinome metastasieren zunächst in axilläre Lymphknoten, weswegen neben der metrischen Tumorgrößenbestimmung deren Untersuchung durch komplette Dissektion oder als Wächterlymphknoten zu jeder pathologischen Bestimmung des Ausbreitungsstadiums invasiver Mammakarzinome nach dem TNM-System gehört. 6 Klinische Korrelation Präinvasive in-situ Karzinome erzeugen i.d.R. keinen Tastbefund, sondern fallen in der Mammographie durch feinkörnig-gruppierten, amorph-gruppierten oder amorphsegmental verzweigten Mikrokalk auf. Invasive Karzinome sind als unscharf begrenzte, derbe Verhärtung tastbar (Ausnahme medulläres Mammacarcinom) und erzeugen eine sternförmige Läsion im Röntgen mit sonographischer Schallauslöschung. Lobuläre Karzinome fallen röntgenologisch häufig nur als Architekturstörung auf. Differentialdiagnosen Es gibt eine Reihe von benignen Mammaveränderungen, die zumeist keine Beschwerden verursachen und deren einziger Krankheitswert darin besteht, dass sie von einem Mammacarcinom abgegrenzt werden müssen. - Mastopathie - intraductale Hyperplasie - sklerosierende Adenose - Fibroadenom - Papillom - Plasmazellmastitis Die Mastopathie wird durch eine hormonelle Dysregulation hervorgerufen und geht mit Zystenbildung, Faservermehrung, intraductaler Hyperplasie und nur gelegentlich mit Spannungsschmerz einher. Das Fibroadenom ist die häufigste benigne Neoplasie in der Adoleszenz und bei jungen Frauen und stellt neben der Mastopathie und dem Karzinom die dritthäufigste Erkrankung der Brustdrüse überhaupt dar. Fibroadenome sind hormonell induzierte benigne Tumoren, die vom Epithel und Stroma der terminalen ductulo-lobulären Einheit ausgehen mit einer epithelialen und mesenchymalen Proliferation. Fibroadenome werden selten größer als 2 cm. Vom Fibroadenom abzugrenzen ist der Phylloidestumor, der ebenfalls eine kombiniert mesenchymal-epitheliale Proliferation darstellt, aber zumeist zellreicher und größer als 3cm ist und ein malignes Potential besitzt, das im Einzelfall zu bestimmen ist. Papillome entstehen zumeist mamillennah und gehen häufiger als alle anderen Tumoren mit einer pathologischen Mamillensekretion einher. Sie sind i.d.R. gutartig, sollten aber komplett entfernt werden, da in ca. 10% der Fälle atypische Papillome oder In-situ Karzinome vorkommen. Prognosefaktoren Die meisten Mammakarzinome werden zusätzlich zu mit einer Operation (die bei ungefähr 70% der Patientinnen allein ausreichend wäre) zusätzlich noch („adjuvant“) medikamentös behandelt. Dabei stehen eine endokrine (Hormonhemmung), zytostatische und kombiniert endokrine/zytostatische Therapie zur Verfügung. Maßgeblich für die Entscheidung zur Chemotherapie ist die potentielle Aggressivität des Tumors. Folgende Tumoreigenschaften beeinflussen wesentlich die Aggressivität und Progressionsneigung eines Mammacarcinoms: 7 • • • • • Tumorgröße axilläre Lymphknotenmetastasen (pN0/pN1/N2/N3=0/bis 3/bis9/ >10 Lymphknoten) Histologischer Grad (hoher Grad/Proliferationsaktivität=ungünstig) Steroidhormonrezeptorexpression (positiv=günstig) HER2-Amplifikation (amplifiziert=ungünstig) Die Identifikation weiterer prognostischer Tumormarker, die eine risikoadaptiert Therapie ermöglichen ist Gegenstand der aktuellen Forschung. Insbesondere geht es um die Identifikation der Patientinnen, bei denen auf eine adjuvante Chemotherapie verzichtet werden kann und die mit einer antihormonellen Therapie ausreichend behandelt sind. Mehrere Genexpressionsprofile zur Erkennung von hoch- und niedrigrisikopatientinnen sind zurzeit in der Erprobung. Bei jedem invasiven Mammakarzinom werden durch die Pathologie folgende potentielle Zielstrukturen einer medikamentösen Therapie bestimmt: Molekül Östrogenrezeptor Untersuchungsverfahren Immunhistochemie Progesteronrezeptor Immunhistochemie Human Epidermal Growth Factor Receptor 2 (Her2) Immunhistochemie, In-Situ Hybridisierung Positiv Negativ Therapie bei Positivität ≥ 1% gefärbte Zellkerne invasiver Tumorzellen <1% Tamoxifen oder Aromatasehemmer über mindestens 5 Jahre ≥ 1% gefärbte Zellkerne invasiver Tumorzellen ≥ 30% stark und komplett gefärbte Membranen invasiver Tumorzellen (3+); ≥ 6 Her2 Gensignale pro Tumorzellkern <1% 0 – 1+, 2+ ohne Amplifikation Trastuzumab oder Lapatinib in Kombination mit Chemotherapie Seltene Tumore Phyllodestumor Tumore vom Speicheldrüsentyp Maligne Lymphome Sarkome (Angiosarkome) 8 KPK Entzündliche Lebererkrankungen Entzündliche Lebererkrankungen betreffen entweder das Parenchym (= Hepatitis), die intrahepatischen bzw. extrahepatischen Gallenwege (= Cholangitis) bzw. die Gefäße der Leber. Klinische Bedeutung haben vor allen Dingen die ersten beiden genannten Ursachen. Die durch diese Mechanismen ausgelösten Schäden können entweder folgenlos ausheilen (restitutio ad integrum) oder bei schweren chronischen bzw. akuten Schäden zu einem bindegewebigen Leberumbau (Leberzirrhose) mit Leberinsuffizienz und Ausbildung von venösen Umgehungskreisläufen (Portale Hypertension) führen. Als auslösende Ursachen sind vor allen Dingen Erreger (Viren, Bakterien, Parasiten, Pilze) bzw. toxische Faktoren (Alkohol) sowie gestörte Immunreaktionen zu nennen. 1. Entzündliche Parenchymerkrankungen (akute und chronische Hepatitiden) 1.1 Akute (Virus-) Hepatitis Die akute Virushepatitis wird vorzugsweise durch klassische Hepatitisviren ausgelöst, daneben sind EBV-, CMV- bzw. das Gelbfiebervirus von untergeordneter Bedeutung. Die klassischen Hepatitisviren sind, mit Ausnahme der Hepatitis B, RNA Viren, die entweder fäkal - oral bzw. durch Blut/Körperflüssigkeiten auf den Menschen übertragen werden. Die Inkubationszeit bis zum Ausbruch der Erkrankung beträgt abhängig vom auslösenden Virus zwischen 15 bis etwa 180 Tagen. Die Viren bestehen, neben in der DNA (Hepatitis B) bzw. RNA hinterlegten Erbinformationen, aus Oberflächen- bzw. zentralen Anteilen, die als Antigene wirken und aus deren Persistenz bzw. Elimination neben dem Auftreten spezifischer Antikörper klinische Rückschlüsse auf die Ausheilung bzw. den Verlauf gezogen werden. Die morphologischen Veränderungen der einzelnen klassischen Hepatitis Viren ähneln einander und sind unter Tabelle 1 ausgeführt. Tabelle 1: Histologische Befunde bei akuter Hepatitis Parenchym Portalfeld Mesenchym Hydropische Schwellung der Hepatozyten Apoptosen -> Councilman Bodies Leberzellnekrosen Gallethromben in Canaliculi Infiltration Sternzellschwellung durch Lymphozyten und Histiozyten Lymphfollikel Sternzellknötchen Ceroidpigment Siderinpigment 9 Wesentlich unterscheidet man in der akuten Phase: 1. Parenchymveränderungen, insbesondere: - läppchenzentral betonte hydropische Schwellungen von Leberzellen (Ballonzellen), - apoptstisch Leberzelluntergänge mit Ausbildung von Councilman – Körpern, - wechselnd ausgedehnte konfluierte Leberzellnekrosen 2. bzw. Mesenchymreaktionen mit - läppchenzentralen lympozytären entzündlichen Infiltraten, - entzündlichen Infiltraten der Portalfelder bestehend aus Lymphozyten und Histiozyten mit teils follikelartiger Verdichtung dieser Infiltrate. Im Unterschied zur chronischen Hepatitis fehlen Nekrosen entlang der Grenzlamelle. Weiterhin findet sich eine Proliferation der von-Kupffer - Zellen, insbesondere entlang von Einzelzell- bzw. Nekrosen entlang der konfluierten Nekrosen nach Abräumungsreaktionen ( Residualknötchen etc.). Als Folgezustände nach akuter Virushepatitis können im Verlauf gefunden werden: - eine Ausheilung (restitutio ad integrum) - entzündliche Residuen, d. h. persistierende Aktivierung der Kupffer-Zellen, lymphohistiozytiozytäre entzündliche Portalfeld Infiltrate - eine posthepatitische Hyperbilirubinämie - eine Fibrose bzw. Zirrhose der Leber - eine chronische Hepatitis - ein hepatozelluläres Karzinom 1.2 Chronische Hepatitis Unter einer Chronischen Hepatitis versteht man eine Leberentzündung, die länger als sechs Monate anhält und klinische Symptome mehr oder weniger starker Ausprägung aufweist. Neben den klassischen akuten Hepatitis - Virus Infektionen (vorzugsweise Hepatitis B, C und D) können chronische Hepatitiden auch durch Störungen des Immunsystems (Autoimmunhepatitis) bzw. als Folge von Stoffwechselstörungen (Morbus Wilson, Alpha-1-Antitrypsin-Mangel) bzw. von Medikamentenüberempfindlichkeiten auftreten. Neben den auf virale Infektionen zurückzuführenden chronischen Hepatitiden macht die Autoimmunhepatitis einen Anteil von 5 bis 20 Prozent der chronischen Hepatitiden aus. Sie wird vorzugsweise bei jüngeren Frauen und nach der Menopause beobachtet. Klinisch ist sie mit dem Nachweis zirkulierender Auto-Antikörper, z.B. anti- Aktin- Antikörper, antinukleäre bzw. andere Antikörper assoziiert und es wird vermehrt bei den Patienten ein HLAB8- und -DR3- Status gefunden. Immunologisch lässt sich eine T-Zell-vermittelte Immunreaktion gegen die hepatozellulären Membranantigene auf Grund eines TSuppressor- Zell- Defektes nachweisen, weiterhin sollen auch Antikörper abhängige cytotoxische Reaktionen eine Rolle spielen. Histologisch ist die Autimmunhepatitis, neben einem entzündlichen Infiltrat der Portalfelder sowie der Lobuli durch Lymphozyten, insbesondere auch durch einen auffallend vermehrten Nachweis von Plasmazellen gekennzeichnet. 10 2. Alkohol-toxische Hepatitis Bei der Verstoffwechselung von Alkohol in der Leber kommt es über das Alkoholabbauprodukt Azetaldehyd zu einer toxischen Entschädigung der Leberzelle durch Bindung an Phospholipide, Aminosäuren, Hormone, Zellmembranen und Zellskelettkomponenten. Weiterhin steigert Azetaldehyd die Kollagensynthese, aktiviert das Komplement-System bzw. die Lipidperoxidation und stört den mitochondrialen Elektronentransport. Entscheidende Bedeutung bei der Entstehung entzündlicher Veränderungen haben hierbei reaktive Sauerstoffverbindungen und verschiedene Zytokine. Die alkoholische Hepatitis stellt hierbei ein Bindeglied zwischen der reversiblen Fettleber und der irreversiblen Leberzirrhose dar. Die morphologischen Veränderungen der alkoholische Hepatitis sind in Tabelle 2 ausgeführt. Tabelle 2: Histologie der alkoholtoxischen Leberschädigung Fettleber Alkohol. Hepatitis Zirrhose Grobtropfige Fettvakuolen Zentral -> diffus Leberzellnekrosen Kleinknotiger Umbau Apoptosen Maschendrahtfibrose Resorptionsknötchen, Mallory-Körperchen Grobtropfige bestehend aus Ballonierte Leberzellen Verfettung 1. v. Kupferzellen 2. Histiozyten 3. Lymphozyten 4. Neutroph. Granulozyten (Cholestase) Cholestase Zentrale Sklerose Maschendrahtfibrose Verfettung häufig > 50 % des Parenchyms 3. Entzündliche Gallenwegserkrankungen Entzündliche Gallenwegserkrankungen können ebenso wie die Parenchymerkrankungen sowohl akut als auch chronisch bzw. rezidivierend verlaufen und über eine Zerstörung der Gallengänge zu einem Leberumbau führen. Die wichtigsten Erkrankungen sind dabei 1. die akute eitrige Cholangitis 2. die primär biliäre Zirrhose (chronische nicht eitrige destruierende Cholangitis, Autoimmuncholangitis) 3. die sklerosierende Cholangitis. 3.1 Akute Cholangitis Die akute Cholangitis wird i. d. R. bakteriell ausgelöst, wobei sich die Entzündung entweder über die Gallengänge aufsteigend oder hämatogen über die Leberarterie bzw. die Pfortader bzw. lymphogen ausbreitet. Die in den Gallengänge aufsteigende Infektion wird häufig durch ein tiefsitzendes Galleabflusshindernis im Bereich der Papille oder des unteren Choledochus ausgelöst. 11 Die Histologie zeigt im Lumen, im Gallengangsepithel bzw. um die Gallengänge herum neutrophile Granulozyten, in den Portalfeldern findet sich ein Ödem, weiterhin zeigen sich auch Gallezylinder in den Leberepithelien bzw. bei schweren Entzündungen unter Beteiligung der großen Gänge Gallezylinder in den Cholangiolen. Aus langdauernden Verläufen können eine biliäre Fibrose bzw. eine sekundäre biliäre Leberzirrhose entstehen. 3.2 Primär biliäre Zirrhose (PBC) Bei der primär biliären Zirrhose handelt es sich um eine chronisch progrediente destruierende Cholangitis, die, auf Grund einer Zerstörung der intrahepatischen Gallenwege, schlussendlich zu einer Leberfibrose bzw. Leberzirrhose führten. Hierbei lassen sich histologisch vier Stadien der Progression unterscheiden (siehe Tabelle 3). Die überwiegende Zahl der Patienten (95 Prozent) sind Frauen im Alter zwischen 40 und 60 Jahren, die Erkrankung kommt vorwiegend in den entwickelten Staaten der Nordhalbkugel vor, in Afrika und Asien ist sie selten. Die Ätiologie der Erkrankung ist bisher noch nicht vollständig verstanden, es wird eine autoimmunologisch bedingte Gallengangsdestruktion durch zytotoxische TLymphozyten angenommen. Weiterhin soll auch eine Antikörper- abhängige zytotoxische Reaktion der Klasse III nach Coombs und Gell beteiligt sein. Bei diesen Patienten findet sich auch eine atypische Expression von Klasse-I- (HLA-A-, -B,-C) und Klasse-II (HLA-DR-) Histokompatibilitätsantigenen, die an normalen Gallengängen nicht nachzuweisen sind und die Ziele der T-Zell Attacken darstellen. Tabelle 3: Stadien der Entwicklung der Primär biliären Zirrhose (PBC) Stadium I II III IV Fokale Destruktion kleiner bzw. mittlerer Gallengänge (GG) mit Infiltration der GG durch Lymphozyten (Lymphoepitheliale Läsion), entzündliches Infiltrat aus Lymphozyten, Makrophagen und Plasmazellen, häufig epitheloid- und riesenzellhaltige Granulome zusätzlich Proliferation von Ductuli Entwicklung einer portalen Fibrose infolge der GGDestruktion und konsekutiven Leberparenchymschädigung Zirrhose 12 3.3 Sklerosierende Cholangitis Unter einer sklerosierenden Cholangitis versteht man eine chronische Entzündung mit Atrophie, Verschluss und Schwund der intrahepatischen und/oder der extrahepatischen Gallengänge. Man unterscheidet eine primär-sklerosierende Cholangitis unbekannter Ätiologie und eine sekundär sklerosierende Cholangitis mit bekannter Ursache. Bei der primär sklerosierende Cholangitis sind überwiegend Männer zwischen dem 25. und 50. Lebensjahr betroffen. Es findet sich eine häufige Vergesellschaftung der Erkrankung mit der Colitis ulcerosa, chronischer Thyreoiditis und Immundefizienzsyndromen. Histologisch findet sich in der Leber eine um die Gallengänge liegende Entzündung mit Lymphozyten, Plasmazellen und vereinzelten neutrophilen und eosinophilen Granulozyten, die mit einer zunehmenden periductalen Fibrose vergesellschaftet sind. Die Gallengänge verschwinden schließlich und werden durch Faserstränge ersetzt. Oberhalb der veränderten Gallengänge resultiert eine mechanische Cholestase mit Gallengangserweiterung sowie Vermehrung der Cholangiolen und Ablagerung von Gallethromben in denselben. Als Endstadium resultiert eine biliäre Zirrhose (s. Tabelle 4). Tabelle 4: Histologie der sklerosierenden Cholangitiden ¾ Zwiebelschalenartige progressive, destruierende Fibrose um die GG mit Einengung bzw. Verschluss und Ersatz der Gallengänge durch Faserstränge ¾ Entzündliches Infiltrat aus Lymphozyten, Plasmazellen und neutro- bzw. eosinophilen Granulozyten ¾ Proximal von Stenosen Cholestase, Gallethromben, GG-Proliferate in der Grenzzone ¾ Endstadium biliäre Zirrhose Die sekundär-sklerosierende Cholangitis zeigt histologisch weitgehend der primär sklerosierenden Cholangitis entsprechende Veränderungen. Die Erkrankung resultiert als Folge einer chronischen Galleabflussbehinderung (mechanische Cholestase) nach bakteriellen Infektionen des Gallengangssystems bzw. mangelnder Blutversorgung, außerdem wird sie nach Knochenmarkstransplantationen im Rahmen der Graft vs. Host- Reaktion bzw. nach Abstoßungsreaktionen nach Lebertransplantationen gefunden. Die Folgezustände entsprechen ebenfalls denen der primär sklerosierenden Cholangitis. 13 KPK Knochen- und Weichteiltumoren Einleitung Sarkome sind maligne Tumoren des Binde- und Stützgewebes, unterschieden wird dabei zwischen primären Sarkomen des Knochens u. des Weichgewebes. Hauptvertreter der Knochensarkome sind das Osteosarkom, Chondrosarkom und Ewingsarkom. Unter dem Begriff der "Weichgewebsarkome" wird eine inhomogene Gruppe von über 100 unterschiedlichen Tumoren zusammengefasst. Allen Tumoren ist eine niedrige Inzidenz gemein, weshalb Kenntnisse zu Tumorbiologie, Diagnostik & Therapie oft nicht ausreichend entwickelt sind. Die Behandlung sollte deshalb immer in spezialisierten Zentren durchgeführt werden. Weichteilsarkome Allgemeines & Epidemiologie Weichteilsarkome sind eine inhomogene Gruppe von seltenen Tumoren, die aus Vorläuferzellen des Bindegewebes entstehen. Sie werden nach dem Gewebe klassifiziert dessen Entwicklung sie rekapitulieren (Muskulatur, Fett, Gefäße, etc.). Für einige Weichteiltumoren gibt es jedoch keinen (erkennbaren) Bezug zu normalen ausdifferenzierten Gewebe. Da die meisten gutartigen Weichteiltumoren nicht reseziert werden, sind genaue Angaben zu ihrer Inzidenz schwierig, jedoch übertreffen sie maligne Vertreter wohl im Verhältnis 100:1. Maligne Weichteiltumoren, d.h. Sarkome treten mit einer Inzidenz von jährlich 2-3 Neuerkrankungen je 100000 Einwohner auf, so dass in Deutschland jährlich ca. 2000-3000 Weichteilsarkome neu diagnostiziert werden. Damit machen damit sie insgesamt nur ca. 1 % aller Tumoren im Erwachsenenalter aus. Die Häufigkeit ist bei Männern und Frauen ungefähr gleich. Weichgewebsarkome kommen in allen Altersstufen vor; im Erwachsenenalter bevorzugt vom 45. bis 55. Lebensjahr. Hinsichtlich der Primärlokalisationen überwiegt die untere Extremität mit 40 %, gefolgt vom Körperstamm, dem Retroperitoneum, der oberen Extremität und der Kopf-Hals-Region mit jeweils ca. 15 % der Fälle. Im Kindesalter treten 15% aller Sarkome auf, hier bestreiten sie die 4. häufigste Erkrankungsgruppe. Pathogenese Die Entstehungsursachen sind weitgehend unklar. Es gibt jedoch eine Reihe bekannter Assoziationen mit vorausgegangener Bestrahlung, chemischer o. thermischer Verbrennung. Die Mehrzahl der Sarkome tritt sporadisch, d.h. ohne Assoziation mit einer genetischen Vorbelastung, auf. Die seltenen Syndrome mit gehäuftem Auftreten von Sarkomen sind die Neurofibromatose (maligner Nervenscheidentumor), Gardner-Syndrom (Fibromatose), Li-Fraumeni Syndrom (alle Sarkome), Osler-Weber-Rendu (Teleangiektasie). 14 Prognoseparameter der Sarkome Zunächst ist ein exaktes Typing wichtig, da die Tumorentitäten oft ein besonderes biologisches Verhalten aufweisen. Hierzu ist Erfahrung u. der Einsatz spezieller immunhistochemischer u. molekularbiologischer Verfahren nötig. Daher sollte die morphologische Diagnostik in der Hand erfahrener Pathologen liegen. Im Gegensatz zur Situation bei den meisten Karzinomen ist das Grading bei Sarkomen ein unverzichtbarer Bestandteil der Therapieplanung. Es wird durch eine Kombination aus Mitosehäufigkeit, Nachweis von Nekrosen u. dem histologischen Typing erstellt. Hochdifferenzierte Tumoren (G1) sind definitionsgemäß nicht metastasierungsfähig, können jedoch rezidivieren u. lokal aggressiv wachsen. Gering differenzierte Tumoren (G3) sind hochaggressiv u. zeigen in bis zu 40% Fernmetastasen (bes. Lunge). Mäßig differenzierte Sarkome liegen in ihrem biologischen Verhalten zwischen diesen beiden extremen Ausprägungen. Die Lage von Sarkomen ist ein wichtiger prognostischer Parameter, da Tumoren in epifaszialer (d.h. subkutaner) Lage eine wesentlich bessere Prognose als tief sitzende Tumoren haben. Wie bei allen malignen Tumoren ist die Beurteilung der Tumorausbreitung für die Therapieplanung u. Prognose von großer Bedeutung, dabei spielen die Tumorgröße, Lokalisation, Vorliegen von Metastasen die wichtigste Rolle. Wesentliche Risikofaktoren (ausgedrückt als relatives Risiko) für das Auftreten von Tumorkomplikation sind im Folgenden dargestellt. Ereignis Parameter Relatives Risiko Rezidiv bereits Rezidiv Inkomplette Resektion >50 Jahre 2.0 1.8 1.6 Metastase G3 subfasziale Lage Größe >5cm Leiomyosarkom 4.3 2.5 1.9 1.7 Überleben G3 subfasziale Lage Größe >10cm Inkomplette Resektion 4.0 2.5 2.1 1.7 Diagnostik der Sarkome Alle Verfahren der modernen bildgebenden Diagnostik werden hier eingesetzt, speziell die MRT-Diagnostik ist geeignet um die Tumorausdehnung gut zu beurteilen, die CT-Bildgebung spielt für den Nachweis von Metastasen eine Bedeutung. Radiologisch kann zur Dignität eine grobe Abschätzung abgegeben werden, eine genaue Typisierung ist naturgemäß nicht möglich. Hierfür ist die Histologie nötig. Nachdem früher meist offene Inzisionsbiopsien durchgeführt wurden, ist inzwischen der Einsatz gering invasiver Punktionsverfahren Standard. Allgemeine Therapieprinzipien der Sarkome Für hochdifferenzierte Sarkome ist die Therapie der Wahl die chirurgische Resektion mit ausreichendem Sicherheitsabstand. Eine Chemotherapie ist aufgrund der 15 fehlenden Metastasierungsfähigkeit kontraindiziert. Eine Bestrahlung kann ggf. zur Verbesserung der lokalen Tumorkontrolle (Vermeiden von Rezidiven) eingesetzt werden. Für mäßig & gering differenzierte Sarkome gilt auch das Primat der Chirurgie. Hier wird (fast) regelhaft die Bestrahlung prä- oder postoperativ zur lokalen Tumorkontrolle eingesetzt. Für die meisten Weichteilsarkome ist ein klarer Benefit der Patienten durch eine Chemotherapie bei (noch) nicht metastasierten Tumoren als adjuvante (postoperative) Therapie nicht belegt. Daher wird in der Regel eine Chemotherapie erst bei rasch progredienten, metastasierten & symptomatischen Patienten eingesetzt. Für die im Kindesalter auftretenden meist aggressiven Sarkome gilt dies nicht, hier wird zumeist eine Chemotherapie durchgeführt, mit hohen Heilungsraten. Klassifikation der Weichteilsarkome Grob können Weichteiltumoren in 8 Kategorien eingeteilt werden, die im Folgenden aufgeführt sind: - Fettgewebstumoren Liposarkom - Fibröse Tumoren Fibrosarkom - Fibrohistiozytäre Tumoren: Malignes fibröses Histiozytom - Tumoren der Skelettmuskulatur Rhabdomyosarkom - Tumoren der glatten Muskulatur Leiomyosarkom - Vaskuläre Tumoren Angiosarkom, Hämangioendotheliom, Kaposisarkom - Nerventumoren Maligner peripherer Nervenscheidentumor - Tumoren unklarer Histogenese Synovialzellarkom, Epitheloides Sarkom, Klarzellsarkom Für die Diagnostik sind neben dem histologischen Befund, die Immunphänotypisierung u. zunehmend der Nachweis spezifischer Translokationen relevant. Liposarkome Da eine genauere Darstellung der verschiedenen Weichteilsarkome den Rahmen dieses Skriptes sprengte, werden hier die lipomatösen Tumoren als häufigste Weichteiltumoren exemplarisch vorgestellt. Liposarkome sind mit 15% die häufigsten Sarkome. Ihre Lokalisationsverteilung ist wie folgt: untere Extremität 35% Stamm / Becken 22% obere Extremität 15% Retroperitoneum 15% Mediastinum 8% Samenstrang 5%. Lipomatöse Tumoren können nach ihrer Dignität u. biologischen Aggressivität wie folgt gruppiert werden. Benigne lipomatöse Tumoren Lipom Lipomatose Angiolipom 16 Intermedäre lipomatöse Tumoren (lokal aggressiv) Atypischer lipomatöser Tumor Maligne lipomatöse Tumoren gut differenziert & dedifferenziert myxoid/rundzellig pleomorph Hochdifferenzierte Liposarkome Hochdifferenzierte Liposarkome sind die häufigsten Sarkome der Extremitäten u. des Retroperitoneums, sie können in verschiedene histologische Typen unterteilt werden u. zeichnen sich alle durch eine Amplifikation des MDM2 Genlokus durch die Ausbildung sog. Ringchromosomen aus, was diagnostisch verwendet wird. Therapie ist die chirurgische Resektion, da insb. im Peritoneum o. Retroperitoneum eine vollständige Resektion schwierig ist, sind hier Rezidive häufiger als im Bereich der Extremitäten. Superfizielle (epifasziale) Tumoren zeigen Rezidive in 15%, Metastasen und tumorbedingte Todesfälle in <5% sowie eine 5-JÜLZ (%Jahresüberlebenszeit) von 95%. Tiefsitzende Tumoren rezidivieren in bis zu 45%, metastasieren nicht und führen in 25% zu tumorbedingten Todesfälle bei einer 5JÜLZ von 85%. Beim Auftreten von Rezidiven kann eine sog. Dedifferenzierung auftreten, d.h. sowohl morphologisch als auch biologisch entwickelt sich ein aggressiverer Klon, der auch zur Metastasierung fähig ist. Diese zeigen in 17% Metastasen und in bis zu 41% Rezidive. Myxoide/Rundzellige Liposarkome Beide Sarkome zeigen eine leicht differente Morphologie, sind jedoch durch eine Translokation t12;16 unter Beteiligung des CHOP-Gens gekennzeichnet. Die Lokalisationsverteilung ist wie oben beschrieben. Als klinische Besonderheit zeigen Sarkome der Extremitäten hier gehäuft den Nachweis intraperitonealer Metastasen. Das rundzellige Liposarkom ist aggressiver als die myxoide Variante u. zeigt in bis zu 50% der Fälle Metastasen bei einer 5-JÜLZ von unter 25%. Dedifferenzierte Liposarkome Diese Form der Liposarkome entsteht überwiegend sekundär aus gut differenzierten Liposarkomen. In bis zu 40% der Fälle liegen Metastasen vor bei einer 5-JÜLZ von 75%. Diese Entität ist nur zu diagnostizieren wenn nebeneinander die präexistente hochdifferenzierte Komponente u. der Dedifferenzierungsanteil vorliegen. Dieser imponiert meist wie ein undifferenziertes Sarkom. Pleomorphe Liposarkome Hierbei handelt es sich um de novo (also ohne Vorläuferläsion) entstandene gering differenzierte Sarkome mit komplexem Genotyp u. sehr pleomorpher Histologie. Metastasen liegen in bis zu 40% vor, bei einer 5-JÜLZ von 50%. Dies sind die seltensten Varianten des Liposarkoms. 17 Knochentumoren Allgemeines & Epidemiologie Knochentumoren sind eine sehr heterogene Erkrankungsgruppe, die biologisch von harmlosen Läsionen bis hochmalignen Tumoren mit hoher Mortalität reichen. Für die korrekte Diagnostik u. Therapie ist ein interdisziplinärer Ansatz unter Beteiligung der Radiologie, Orthopädie/Unfallchirurgie, Pathologie u. Strahlentherapie Primäre Knochentumoren leiten sich von mesenchymalen bzw. neuroektodermalen Vorläuferzellen ab. Es werden benigne und maligne Knochentumoren unterschieden. Sowohl hoch als auch niedrig maligne Knochentumoren zeichnen sich durch ein lokal aggressives, destruierendes Wachstum aus, wobei jedoch auch einige benigne oder tumorartige Läsionen des Knochens derartige Wuchsformen aufweisen können. Die Metastasierung dagegen ist ein charakteristisches Merkmal hochmaligner Knochentumoren. Die WHO listet folgende Läsionen auf. Maligne Tumoren sind sehr selten und werden pro Jahr ca. 700x in Deutschland diagnostiziert. Dennoch bestreiten Knochentumoren 4-5% aller malignen Tumoren im Kindesalter. 18 Diagnostik Der überwiegende Anteil der gutartigen Knochentumoren ist klinisch stumm. Sie werden häufig als Zufallsbefund im Rahmen von Diagnostiken anderer Intention gefunden. Gutartige Tumoren im aggressiven Stadium hingegen ähneln in ihrem klinischen Beschwerdebild den malignen Tumoren. Die beiden häufigsten primären Symptome sind der progrediente Schmerz und eine palpable Schwellung. Typischerweise besteht ein Ruhe- und Belastungsschmerz. In der Bildgebung sind CT, MRT u. die Szintigraphie als sich ergänzende Verfahren zu betrachten. Bei Verdacht auf einen malignen Tumor ist eine bioptische Klärung zwingend notwendig. Der Inzisionsbiopsie ist bei den Knochentumoren der Vorzug gegenüber der Stanzbiopsie zu geben. Die Biopsie sollte bereits in einem Zentrum mit Erfahrung in der Behandlung von Knochentumoren durchgeführt werden, da durch eine falsche Biopsietechnik die Resektion erschwert oder sogar unmöglich werden kann und das Risiko eines Lokalrezidivs erhöht ist. Therapie Bei Vorliegen eines primären malignen Knochentumors muss die Therapie interdisziplinär erfolgen. Das Hinzuziehen eines Onkologen nach Sicherung der Diagnose ist zwingend erforderlich. Abhängig von der Entität und dem Grading müssen eine neoadjuvante Chemotherapie und eine adjuvante Strahlentherapie erfolgen. Für einige Entitäten bestehen hier standardisierte Therapieoptimierungsstudien. So werden High-grade-Osteosarkome bis einschließlich des 40. Lebensjahrs im Rahmen des „European and American Osteosarcoma Study Group“-Protokolls (EURAMOS-1) behandelt. Ewing-Sarkome werden nach dem EURO-E.W.I.N.G.-99Protokoll behandelt. Zwischen dem 40. und 65. Lebensjahr sollten alle hochmalignen Knochensarkome nach dem Protokoll der „EUROpean-Bone Over 40 Sarcoma Study“ (EURO-B.O.S.S.) behandelt werden. Die enge Anbindung an einen Onkologen ist unabdingbar. Durch die interdisziplinäre Behandlung hat sich die Prognose der malignen Knochentumoren vor allem in Kindesalter in den letzten 30 Jahren signifikant verbessert. Osteosarkom Das Osteosarkom ist ein mesenchymaler Tumor mit verschiedenen Malignitätsgraden, der einen ersten Altersgipfel in der 1. u. 2. Lebensdekade u. eine zweiten Gipfel in der 6. u. 7. Dekade zeigt. Allen Osteosarkomen gemein ist die Bildung von Knochen und/oder Osteoid. Die Ätiologie der meisten Osteosarkome ist unbekannt. Es existiert eine kleine Gruppe sekundärer Osteosarkome auf dem Boden eines M. Paget, einer fibrösen Dysplasie oder aber einer lokalen Radiatio. Die wichtigste Unterscheidung für den Kliniker ist die Einteilung in ein Low-grade- oder High-grade-Osteosarkom. Das klassische Osteosarkom hat seinen Altergipfel um das 15. Lebensjahr. Das Osteosarkom ist der häufigste maligne Knochentumor im Kindesalter. Die häufigste Lokalisation ist die Metaphyse der langen Röhrenknochen (in absteigender Häufigkeit: distales Femur, proximale Tibia, proximaler Humerus). Das klassische High-grade-Osteosarkom wird durch Schmerzen und eine lokale Schwellung auffällig. Pathologische Frakturen sind möglich, jedoch eher selten. Das konventionelle Röntgenbild ist in der Diagnosestellung richtungsführend. Die typischen Zeichen sind Knochendestruktion, aggressive Periostreaktion (Sunburst, Lamellen, Codman-Dreieck), Weichteilinfiltration. 19 Das High-grade-Osteosarkom wird neoadjuvant chemotherapiert. Auch wenn im Staging keine Metastasen nachweisbar sind, liegt in vielen Fällen eine Mikrometastasierung vor. Nach den ersten Zyklen der Chemotherapie erfolgt die Operation. Am Operationspräparat kann durch Beurteilung des Ausmaßes der Regression der Erfolg der Chemotherapie beurteilt werden. Abhängig von diesem Befund wird die gleiche Chemotherapie fortgesetzt oder eine andere Substanzklasse gewählt. Nur die Kombination aus neoadjuvanter Chemotherapie und suffizienter Lokaltherapie hat zu einer signifikanten Verbesserung der Überlebenswahrscheinlichkeit geführt. Die Strahlentherapie spielt beim Osteosarkom eine untergeordnete Bedeutung. Ziel der Lokaltherapie ist die weite bzw. radikale Resektion. Verschiedene Rekonstruktionsmöglichkeiten des Defekts kommen in Betracht. Chondrosarkom Das klassische Chondrosarkom ist der zweithäufigste primäre Knochentumor. Der Altersgipfel liegt in der 5. bis 6. Lebensdekade. Das Chondrosarkom kann jedoch selten auch im juvenilen Alter auftreten. Nach der WHO werden 3 Malignitätsgrade unterschieden. Der klinische Verlauf der verschiedenen Grade differiert erheblich. G1- und G2-Läsionen zeigen typischerweise einen langen Krankheitsverlauf mit lang bestehender Schwellung und wenig Schmerzen. Aufgrund des langsamen asymptomatischen Verlaufs sind „Mega-Tumoren“ hier häufig. Aggressive Chondrosarkome hingegen zeigen ein rasantes Wachstum und metastasieren früh und häufig. Prädilektionsstellen des Chondrosarkoms sind das proximale Femur, der proximale Humerus, das Becken und die Skapula. Diagnostisch ist das konventionelle Röntgenbild wegweisend. Es finden sich als typische Zeichen der Malignität Knochendestruktion, endostale Einbuchtung und Kalzifizierungen („rings and arcs“). Die wichtigste Differenzialdiagnose des G1Chondrosarkoms ist das Enchondrom. Bei fehlender Weichteilkomponente ist die CT der MRT in der Diagnostik überlegen. Die Behandlung des Chondrosarkoms ist primär chirurgisch. Die (adjuvante) Strahlentherapie ist lediglich in Ausnahmefällen zu diskutieren. Bisher konnte keine Studie einen klaren Vorteil für eine neoadjuvante Chemotherapie zeigen. Die Indikation bei G3-Tumoren ist hier in Absprache mit dem erfahrenen Onkologen sehr kritisch zu diskutieren. Die klassische Therapie des Chondrosarkoms ist die weite En-bloc-Resektion. G1-Tumoren können in Abhängigkeit von der Lokalisation jedoch auch mit guter lokaler Kontrolle intraläsional reseziert werden. Eine Besonderheit stellt das dedifferenzierte Chondrosarkom dar, welches ca. 10% aller Chondrosarkome betrifft. Hier handelt es sich um einen Tumor, der zu weiten Anteilen einem gut bis mäßig differenzierten Chondrosarkom entspricht, jedoch Anteile eines nichtchondroiden High-grade-Sarkoms (Osteosarkom, Fibrosarkom, undifferenziertes Sarkom) enthält. Hierbei handelt es sich um einen hoch malignen Tumor mit sehr schlechter Prognose. Die Patienten werden häufig mit einer pathologischen Fraktur symptomatisch. Das dedifferenzierte Chondrosarkom entwickelt sich primär aus einem G1-Chondrosarkom oder als Lokalrezidiv eines resezierten G1-Chondrosarkoms. Die Prognose ist schlecht. Eine (neo)adjuvante Chemotherapie sollte in Erwägung gezogen werden. Der Nutzen der Chemotherapie wird jedoch weiterhin kontrovers diskutiert. 20 KPK Entzündliche und vaskuläre Nierenerkrankung Die folgende Zusammenstellung kann kein Lehrbuch ersetzen sondern kann nur als Orientierungshilfe und Leitfaden für die vertiefende Lektüre dienen. Einteilung nicht-neoplastischer Nierenerkrankungen: • • • Tubulo-interstitielle Erkrankungen Vaskuläre Erkrankungen Glomeruläre Erkrankungen • • Primäre nicht-neoplastische Nierenerkrankungen Beteiligung der Niere bei Systemerkrankungen Entzündliche interstitielle Nierenerkrankungen: Eitrige interstitielle Nephritis • Aszendierte Infektion (Pyelonephritis) • Septische Herd-Nephritis (sehr selten) • Malakoplakie Chronische Entzündungsreaktion mit schaumzelligen Makrophagen, inkomplett abgebaute Zellwandbestandteile gram-negativer Bakterien (Michaelis-GutmannKörper) bei immuninkompetenten Patienten, imponiert oft als Tumor Nicht-eitrige interstitielle Nephritis • Allergische Reaktion auf Medikamente (Antibiotika, NSAID, Paracetamol, Metamizol, Diuretika, Zytostatika, Lithium,…) • Viral (Hanta-Virus) • Chinese-herbs-Nephritis • Bestrahlungsfolge Sonderform: Granulomatöse interstitielle Nephritis Meistens Reaktion auf Medikamente DD: Infektiös (Tbc!), Sarkoidose Akute Erkrankungen des Tubulusapparates (akuter Tubulusepithelschaden): Meistens ischämisch • Dehydratation • Hypotension • Allgemeininfektion/ Sepsis • Post-/Perioperativ Toxisch • Kontrastmittel, Amhotericin B, Aminoglykoside… • Rhabdomyolyse • Bestrahlung 21 Vaskuläre Nierenerkrankungen: Benigne Nephrosklerose (Arteriolosklerose) • Im Rahmen physiologischer Alterungsprozesse • Arterieller Hypertonus • Diabetes mellitus (Diabetische Vaskulopathie) Thrombotische Mikroangiopathie Gruppe von Erkrankungen, die mit glomerulären und arteriolären Thromben einhergehen und auf morphologischer Basis kaum voneinander zu trennen sind • Typisches Hämolytisch-urämisches Syndrom (HUS), früher Diarrhoepositives HUS ausgelöst durch EHEC • Atypisches HUS, früher Diarrhoe-negatives HUS: Idiopathisch, genetischer Defekt im Komplementsystem • Bestrahlungsfolge, unter Chemotherapie, Schwangerschaft, Fruchtwasseremboliesyndrom, Calcineurininhibitor-Nebenwirkung, Sepsis/ Schock (DIC) • Verminderte ADAMTS13-Aktivität (TTP) • Sogenannte Maligne Nephrosklerose aufgepropft auf einen bestehenden Hypertonus oder bei Sklerodermie (Sklerodermale renale Krise) Cholesterinembolien Nach Gefäß-chirurgischen Eingriffen, Stent-Anlage, (Coronar)angiographie bei vorbestehender Atherosklerose Vaskulitis Die Vaskulitis der Niere ist eine Glomerulitis (s.u.). Selten Vaskulitis an ArcuataArterien-Ästen (Mikroskopische Polyangiitis, M. Wegener), Beteiligung der Nierenarterie bei Panarteriitis nodosa möglich (Glomerulitis schließt eine Panarteriitis nodosa aus) 22 Abb.1: Einteilung der Vaskulitiden Glomeruläre Erkrankungen: Einteilung: • Nach klinischer Symptomatik (nephrotisch, nephritisch, rapid-progressive) • Primär oder sekundär bei Systemerkrankungen (Hepatitis!, SLE…) • Ätiologisch (Stoffwechselerkrankungen, Vaskulitis, Immunkomplexerkrankung…) • Nach histomorphologischen „patterns“ (s.u.) Typische nephrotische/ proteinurische glomeruläre Erkrankungen: • Minimal Change Glomerulopathie (häufigste Ursache des nephrotischen Syndroms im Kindesalter) • Fokal segmentale Glomerulosklerose • Diabetische Glomerulopathie (kann nephrotisch sein) • Membranöse GN (häufigste Ursache des nephrotischen Syndroms im Erwachsenenalter) • Membranoproliferative GN (kann nephrotisch sein) • Amyloidose Formen der Proteinurie • Überlaufproteinurie (z.B. Bence-Jones-Proteinurie (CAVE: negativ im UrinStix), glomerulärer Filter selbst intakt) • Glomeruläre Proteinurie Selektiv (Mikroalbumin: Frühsymptom der diabetischen NP) Unselektiv • Tubuläre Proteinurie 23 Abb. 2 Relative Häufigkeit von nephrotischen/ proteinurischen glomerulären Erkrankungen Typische nephritische glomeruläre Erkrankungen: • • • • IgA-Glomerulonephritis (häufigste Glomerulonephritis überhaupt) Postinfektiöse Glomerulonephritis Halbmondbildende Glomerulonephritiden (im Rahmen einer systemischen Vaskulitis: M. Wegener, mikroskopische Polyangiitis, Churg-Strauss-Syndrom oder Anti-Basalmembran-AK-Glomerulonephritis/ Goodpasture-Syndrom) Membranoproliferative GN (kann nephritisch sein) „Glomerulopathien“ im Rahmen von Systemerkrankungen • • • • • Vaskulitiden o Immunkomplexvermittelt (SLE, Purpura Schönlein-Henoch) o ANCA-assoziiert (Mikroskopische Polyangiitis, M. Wegener, selten Churg-Strauss-Syndrom) Paraproteinämien, MGUS und Plasmozytom Amyloidose, Cast-Nephropathie Plasmozytomniere, Leichtkettenglomerulopathie Autoimmunerkrankungen/ Kollagenosen Goodpasture-Syndrom, SLE Infektionen Postinfektiöse Glomerulonephritis Amyloidose bei chronischen Infekten Stoffwechselerkrankungen 24 • Diabetes mellitus, Oxalose, Gicht, Morbus Fabry Genetische vererbbare Erkrankungen Alport-Syndrom, Syndrom der dünnen Basalmembranen (familiäre benigne Hämaturie) [Amyloidose: Selten familiäre Amyloidose mit Nierenbeteiligung, häufiger im Rahmen chronischer Infekte (AA-Amyloid) oder MGUS/ Plasmozytom (AL-Amyloid)] Histomorphologische Einteilung von Glomerulonephritiden: „Triple“-Diagnostik setzt sich zusammen aus • Lichtmikroskopie • Immunhistochemie für Immunglobuline und Komplementspaltprodukte • Elektronenmikroskopie Pathogenese glomerulärer Erkrankungen • Antikörper-Bindung in situ an Körpereigene glomeruläre Proteine Zuvor abgelagerte Proteine • Ablagerung von zuvor gebildeten Antigen-Antikörper-Komplexen in verschiedenen Kompartimenten des Glomerulus (mesangial, subepithelial, subendothelial) • ANCA-assoziiert (nicht Immunkomplex-vermittelt „pauci-immun“) Histomorphologische glomeruläre Reaktionsmuster • Mesangioproliferativ - Mesangiale Zellvermehrung - Mesangiale Ablagerung von Immunkomplexen Typischer Vertreter: IgA-Nephritis bzw. Purpura-Schönlein-Henoch als Systemerkrankung • Endokapillär proliferativ „Endokapilläre Zellvermehrung“ durch mesangiale Expansion oder intrakapilläre Entzündungszellen Mesangiale und subepitheliale Ablagerung von Immunkomplexen Typischer Vertreter: Postinfektiöse GN • Membranoproliferativ (Synonym: Mesangiokapillär) Mesangiale Zellvermehrung und Basalmembranverbreiterung Mesangiale und subendotheliale membranöse Ablagerung von Immunkomplexen - Primär idiopathisch oder - Sekundär: Hepatitis B und C, Lymphome, Endokarditis, Kryoglobuline • Extrakapillär proliferativ Extrakapilläre Zellvermehrung (Halbmondbildend, nekrotisierend) Korrelat der Rapid-progressiven GN 25 Typische Vertreter: Nierenbeteiligung bei M. Wegener, Mikroskopische Polyangiitis, Goodpasture-Syndrom - Keine Immunkomplexe (ANCA-assoziierte GN) - Bandförmige IgG-Ablagerungen (Goodpasture) • Membranöse GN Keine Zellvermehrung Subepitheliale Immunkomplexablagerungen Primär idiopathisch Sekundär: Hepatitis B und C, andere Malignome (paraneoplastisch) Ein SLE kann unter dem Bild jeder der genannten histomorphologischen Muster imponieren. Nierenbeteiligung bei Plasmozytom • • • Plasmozytom-Niere (Syn.: Cast-Nephropathie) Leichtkettenamyloidose (AL-Amyloidose) Leichtketten-/ Schwerkettenglomerulopathie (pathologische Ablagerung von Leichtketten (nicht Amyloid!) in Glomerula, andere Organe können betroffen sein) 26 KPK Neuropathologie Epilepsie als Symptom in der Neuropathologie Epilepsie = zerebraler Krampfanfall z Herdanfall - Herdförmige EEG-Veränderungen - Einfach partielle Anfälle, Komplex partielle Anfälle, Partielle Anfälle mit sekundärer Generalisierung z Primär generalisierter Anfall – Herdanfall / Fokale Epilepsie z Einfach partieller Anfall – z z z z z z – Grand mal / großer Krampfanfall mit Verlust der Statik Beginnt mit Initialschrei und Sturz Tonisches Stadium mit Apnoe Kurz danach klonisches Stadium – – – – Muskel-Zuckung des ganzen Körpers Speichelschaum, Zungenbiss Enuresis >Terminalschlaf Ohne Verlust der Statik z z z Epigastrische Aura mit aufsteigendem Wärmegefühl, Übelkeit, Engegefühl Sensationen: Geruch, Geschmack, akustisch Deja-vu Häufig Bewegungsautomatismen/Stereotypien, z.B. Kauen, Gehen Langsame Reorientierung Primär generalisierter Anfall z Einfache Absence Mit Bewustseinsstörungen z z z keine Bewustseinsstörungen, sensorisch, motorisch, kurze Dauer z z Primär generalisierter Anfall Komplex partieller (psychomotorischer) Anfall – „diffuse“ EEG-Veränderungen Sekunden-lange Abwesenheit Blickstarre tonische Blickwendung nach oben, Blinzeln Ursachen z Metabolische Störungen Alkohol Medikamente, Drogen – Alkohol-/Medikamenten-/Drogen-Entzug – Hypocalcämie oder Hypercalcämie – Hypoglykämie > Diabetes mellitus – Urämie > Nierenversagen -> psychogen > Hyperventilation > Alkalose > psychogen („simuliert“) – – 27 Ursachen z z z z z z Alle primären Hirntumoren + Hirn-Metastasen Ischämie / Blutung Perinatale (hypoxische) Hirnschädigung + Hirnfehlbildungen Degenerative Hirnerkrankungen > Hippocampus Post-traumatisch Meningoencephalitis = entzündlich Therapie z Medikamentöse Anfallsprophylaxe Serie von generalisierten Anfällen oder Status epilepticus: akute medikamentöse Therapie Nebenwirkungen / Unverträglichkeit z Ursachen-Behandlung z z Pathologisch-anatomisch darstellbare Veränderungen > Biopsie, Autopsie z z z z z z z Alle Hirntumoren + Hirn-Metastasen Ischämie / Blutung Perinatale (hypoxische) Hirnschädigung + Hirnfehlbildungen Degenerative Hirnerkrankungen > Hippocampus Post-traumatisch Meningo-encephalitis = entzündlich Metabolische Störungen nicht darstellbar Modellvorstellung > Ursache z z Eine sich ausbreitende unkontrollierte Nervenzellaktivierung Genaue molekular-physiologische Ursache nicht ganz geklärt Diagnostik z z z EEG > Hirnströme MRT, CT, konventionelles Röntgen Biopsie normalerweise nur, wenn die Therapie davon abhängt Graduierung der Gehirntumoren nach WHO Grad I: (benigne) Gut differenziert kaum proliferative / mitotische Aktivität verdrängend / nicht-infiltrativ wachsend Grad II: (benigne) Gut - mäßig differenziert kaum - geringe proliferative / mitotische Aktivität infiltrativ wachsend - häufig Rezidive Grad III: (maligne) mäßig - gering differenziert hohe proliferative / mitotische Aktivität Grad IV: gering differenziert (sehr maligne) sehr hohe proliferative / mitotische Aktivität sehr schneller, fataler Verlauf 28 Neuroepitheliale Tumoren: Astrogliale Tumoren (häufigste) Oligodendogliale Tumoren Ependymale Tumoren Neuronale Tumoren Embryonale Tumoren Gliome Mischgliome: astroglialer + oligodendroglialer Anteil > 60 % aller neuroepithelialen Tumoren sind maligne ! Astrogliale Hirntumore: Diffuses Astrozytom (WHO Grad II) Anaplastisches Astrozytom (WHO Grad III) Glioblastoma multiforme (WHO Grad IV) - Können jeweils de novo entstehen oder durch maligne Progression ineinander übergehen. - Unterscheiden sich “nur“ bezüglich der biologischen Wertigkeit / Malignität. - Bevorzugte Lokalisation: Großhirn (und hier: Marklager) Anaplastisches Astrozytom (WHO Grad III) - Durchschnittsalter bei Erstdiagnose: 45 Jahre - fast immer Rezidive, häufig maligne Progression - 5 - Jahres - Überlebensrate etwas höher als beim Glioblastoma multiforme (WHO Grad IV) Diffuses Astrozytom (WHO Grad II) - Durchschnittsalter bei Erstdiagnose: 35 Jahre - Aufgrund des diffus infiltrierenden Wachstumsmusters fast immer Rezidive, häufig maligne Progression Glioblastoma multiforme (WHO Grad IV) - Häufigster hirneigener Tumor ( 12-15% aller intrakranieller Tumoren, 60-75% aller astroglialer Tumoren) - Durchschnittsalter bei Erstdiagnose: 61 Jahre - Durchschnittliche Überlebenszeit nach Erstdiagnose: 6 Monate - 1 Jahr; 5 - Jahres-Überlebenrate: < 2% 29 Ursache > Mutationen z Wie bei jeder anderen Neoplasie auch Medulloblastom (WHO Grad IV) - Häufigster Tumor des Kindesalters - Durchschnittsalter der Patienten bei Erstdiagnose: 7 Jahre, nach dem 20. Lebensjahr selten - Per definitionem: kommt ausschließlich im Kleinhirn vor Überexpression des mutierten bzw. funktionslosen p53-Proteins - Hoch maligne, aber wegen guten Ansprechens auf Chemotherapie/Bestrahlung 5-Jahres-Überlebensrate bei 60-70 % - Histomorphologie: zelldicht, “klein-, rund- und blauzellig“, stark verschobene Kern-Plasma-Relation, sehr hohe Mitoseaktivität. Ependymom (WHO Grad II) Gangliogliom (WHO Grad I) - Auftreten in jedem Alter - Lokalisation: am häufigsten im Bereich des 4. Ventrikels und im Rückenmark - Die meisten Patienten zwischen 10 und 50 Jahre alt bei Erstdiagnose - Häufigster neuroepithelialer Tumor im Rückenmark - Mehr als 70% der Gangliogliome sind temporal gelegen - Prognose vor allem abhängig von Lokalisation. 10 - Jahres Überlebensrate (Erwachsene): ca. 45%. - Mit Epilepsie assoziiert -Histomorphologie: mäßig zelldicht bis zelldicht, ependymale Differenzierung, charakteristisch: kernfreie perivasale Manschetten (Pseudorosetten), manchmal auch echte Rosetten, geringe Mitoseaktivität - Histomorphologisch: Neuronale Tumorkomponente mit dysplastischen Ganglienzellen + astrogliale Tumorkomponente, kaum Mitoseaktivität, verdrängendes Wachstum - 94 % der Patienten nach 7,5 Jahren rezidivfrei. 30 (Meningotheliomatöses) Meningeom (WHO Grad I) - Häufigster intrakranieller Tumor - Durchschnittsalter bei Erstdiagnose: ca. 60 Jahre - Von den Meningen ausgehend, verdrängend wachsend - Durch Bestrahlung induzierbar - 80 % der Patienten nach Resektion rezidivfrei Histomorphologie: mäßig zelldicht, meningeal differenziert, fokal “Lochkerne“, faszikuläre + wirbelförmige Histoarchtikturen, manchmal “zwiebelschalenförmige“ Histoarchitekturen, manchmal Psammonkörperchen, geringe Mitoseaktivität Neurinom (Schwannom) (WHO Grad I) - Patienten aller Altersklassen (nur bei Kindern selten) - Lokalisation: intrakraniell: meist vom Vestibularisanteil des VIII. Hirnnerven ausgehend (trotzdem “Akustikusneurinom“); intraspinal: meist von sensiblen Nervenwurzeln ausgehend - Multipel auftretend bei Neurofibromatose II -Sehr selten maligne Entartung, nach Resektion selten Rezidive -Histomorphologie: mäßig zelldicht, spindelförmige Tumorzellen, faszikuläre, fokal “fischzugartige“ Histoarchitekturen, geringe Mitoseaktivität Klinische Symptome von Hirntumoren z Fokal-neurologische Ausfälle: - motorische und sensible Ausfälle - psychische Veränderungen Sehstörungen Hörstörungen Sprachstörungen z Krampfanfälle z Hirndruckzeichen – – Was sind die lebensverkürzenden Faktoren? – Verdrängung > Hirndruckerhöhung> Einklemmung + Störung der Blutversorgung – Schädigung funktionell wichtiger Areale – (Blutungen) Kopfschmerzen Erbrechen / Übelkeit 31 KPK Lungenerkrankung Bronchialkarzinom Einleitung Bronchialkarzinome (BC; syn: Lungenkarzinom, bronchiogenes Karzinom, Lungenkrebs) sind alle vom Lungenparenchym abgeleiteten malignen epithelialen Tumoren. Das BC ist die häufigste bösartige Erkrankung des Menschen weltweit und die häufigste onkologische Todesursache (1.4 Mio. Patienten weltweit 2004). Hauptursache ist das inhalative Tabakrauchen. Jedoch sind auch durch Passivrauch entstandene BC inzwischen die 6. häufigsten Tumoren weltweit. Daneben gibt es einige Stoffe, die den Tumor auslösen können (beispielsweise Asbest oder Chrom), denen man beruflich bedingt ausgesetzt sein kann. Alle anderen Ursachen (wie zum Beispiel die Belastung durch die Umwelt) treten weit in den Hintergrund. Selbst das Lungenkrebsrisiko durch das natürlich vorkommende Edelgas Radon ist im Vergleich zum Tabakrauchen gering. Symptome der Erkrankung sind spät u. unspezifisch (z.B. chronische Heiserkeit oder Bluthusten), wenn aber Lungenkrebs frühzeitig entdeckt wird, ergibt sich eine verbesserte Heilungschance. Derzeit werden für das BC zahlreiche neue & vielversprechende Therapieverfahren basierend auf molekularen Veränderungen in Studien überprüft. Epidemiologie Die Zahl der Neuerkrankungen/Jahr liegt in Deutschland bei 46.000 (Bundesamt für Statistik 2005). Die Zahl der Neuerkrankungen weist dabei eine steigende Tendenz auf, was wesentlich der zunehmenden Inzidenz bei Frauen geschuldet ist (15-20% aller Krebsneuerkrankungen). Das Verhältnis von erkrankten Männern zu Frauen liegt noch bei etwa 3:1, wobei es auf Grund der Änderung im weiblichen Tabakkonsumverhalten immer mehr zu einer Angleichung kommt. Die Inzidenz in Mitteleuropa beträgt etwa 60 pro 100.000 Einwohner. Nach dem Mammakarzinom, Prostatakarzinom u. dem kolorektalen Karzinom ist es insgesamt die vierthäufigste Tumorerkrankung. Als Ursache von Krebssterbefällen mit rund 40.000 Todesfällen/Jahr liegt es aufgrund der hohen Mortalität jedoch auf Platz eins (30% aller Krebstodesfälle). Der Altersgipfel der Erkrankungen liegt um das 60. Lebensjahr. Die durchschnittliche 5Jahres-Überlebensrate aller Stadien kombiniert liegt derzeit bei nur 15%. Die Lebenserwartung des einzelnen Patienten hängt dabei wesentlich vom Stadium, dem histologischen Typ, Lebensalter und Allgemeinzustand des Patienten ab. Ätiologie & Pathogenese BC entstehen, wie die meisten Karzinome, durch die Kumulation genetischer Abnormalitäten. Für das Lungenkarzinom ist der wesentlich exogene Auslöser, das inhalative Zigarettenrauchen gut dokumentiert. Der Rauch enthält etwa 2000 Stoffe, von denen mindestens 100 krebserregend sind. 87% aller BC treten bei aktiven oder ehemaligen Rauchern auf, dabei sind die Dauer, die Dosis (Zahl der Zigaretten/Tag) u. die Inhalationstiefe entscheidend. Diese Risikofaktoren werden oft im Begriff der sog. „pack years“ (Zahl der Zigarettenpackungen/Tag/Jahr) zusammengefasst. 11% aller „schweren“ Raucher entwickeln ein Bronchialkarzinom (jedoch praktisch alle eine ausgeprägte Gefäßerkrankung u. ein Lungenemphysem). Frauen sind gegenüber den inhalativen Karzinogenen empfindlicher. Die zweithäufigste Ursache für ein Bronchialkarzinom ist das radioaktive Gas Radon. In Deutschland gibt es jährlich ca. 1900 Lungenkrebstodesfälle, die auf die Belastung durch Radon zurückzuführen sind. In einigen Gegenden Deutschlands, u.a. im 32 östlichen Bayern, in Sachsen und Thüringen sowie im Breisgau, wird empfohlen, den Keller gasdicht zu versiegeln, da durch das Gestein Radon aufsteigt. In der Schweiz gilt Radon als Verursacher von zehn Prozent aller Bronchialkarzinomfälle. Bei UranBergarbeitern ist Lungenkrebs eine anerkannte Berufskrankheit. Andere Giftstoffe wie Asbest, Uran, Chrom-Verbindungen, Senfgas, polyzyklische aromatische Kohlenwasserstoffe und Nickel gelten ebenfalls als karzinogen. Hierbei erhöht sich das Risiko einer Erkrankung um ein Vielfaches, wenn der Patient gleichzeitig Raucher ist. Bei Asbestexposition zum Beispiel erhöht sich das Erkrankungsrisiko von Nichtrauchern um das Fünffache, bei Rauchern um das Neunzigfache. Die Zahl der in einem Karzinom nachweisbaren Mutationen liegt bei 10-20 u. ist in Nichtraucherkarzinomen höher. Neben der Inaktivierung von Suppressorgenen (p53, RB1, p16) spielt die Aktivierung sog. Onkogene (c-myc, KRAS, EGFR, c-MET, EML4-ALK) eine entscheidende Rolle in der Karzinogenese. Nach einer in der frühen Entwicklung gemeinsamen molekularen Onkogenese sind für neuroendokrine Tumoren und die klassischen NSCLC unterschiedliche Onkogene relevant. .Für das kleinzellige Bronchialkarzinom sind Mutationen in C-KIT (40-70%), MYCN & MYCL (20-30%), p53 (90%) u. bcl2 (75-90%) nachgewiesen. Dagegen spielen EGFR (bis 25%), KRAS (25%), p53 50%) u. p16 (70%) beim NSCLC die größte Rolle. Klinische Symptome Es gibt keine für Lungentumoren spezifischen Symptome, zusätzlich sind mögliche Merkmale zumeist Spätsymptome. Zuerst zeigen sich Symptome wie Husten, Hämoptoe, Schmerzen des Brustkorbes und Atemschwierigkeiten (Dyspnoe), bei starken Rauchern sind dies selten auffällige oder neue Befunde. Lähmungen der Atemmuskulatur durch eine Phrenicusparese, Heiserkeit durch eine Recurrensparese, das Horner-Syndrom (Miosis, Ptosis, Enophtalmus) entstehen durch direkte Schädigung der Nerven. Teilweise dramatisch kann das Bild einer oberen Einflussstauung bei zentralem Tumor verlaufen. Bei neuroendokrinen Tumoren können paraneoplastische Syndrome (Cushing, Lambert-Eaton etc.) auftreten. Differentialdiagnosen der klinischen Symptome Für die 2 wesentlichen klinischen Symptome, Hämoptoe u. Dyspnoe, gibt es eine Vielzahl von Differentialdiagnosen: Hämoptoe - Blutung aus Bronchien und Trachea Entzündung: z.B. Bronchitis Neoplasie: z.B. Bronchialkarzinom , Metastase Bronchiektasen Fremdkörperaspiration - Blutung aus dem Lungenparenchym Entzündung: Tuberkulose, Pneumonie traumatische Lungenverletzung Vaskuläre Ursachen Arteriovenöse Malformationen Goodpasture-Syndrom Lungenembolie Pulmonale Hypertonie (z.B. bei Herzklappenfehlern, insbesondere Mitralstenose) 33 - Andere Ursachen Hämorrhagische Diathese Behandlung mit Antikoagulanzien Komplikation des Pulmonaliskatheters Osler-Syndrom Endometriose Morbus Wegener Systemischer Lupus erythematodes mit Lungenbeteiligung Myzetom Dyspnoe - Pulmonale Ursachen COPD Lungenemphysem Lungenödem Pneumonie Atelektasen Bronchialkarzinome Pleuramesotheliom Lymphangiosis carcinomatosa Lungenembolie Lungenfibrose Mukoviszidose Pneumothorax Pleuraerguss Asthma bronchiale Trachealstenose - Kardiale Ursachen Herzinsuffizienz Herzklappenerkrankungen (beispielsweise Mitralstenose) Myokarditis Myokardinfarkt Perikarderguss - Skelettale Ursachen Spondylitis ankylosans Kyphoskoliose Thoraxtrauma (z.B. Rippenfraktur oder Rippenprellung) - Neuromuskuläre Ursachen Amyotrophische Lateralsklerose Myasthenia gravis Poliomyelitis Rekurrensparese Tumorkachexie - Psychogene Ursachen Depression Stresssyndrom Angstsyndrome Hyperventilation 34 Diagnostik Am Anfang der Diagnostik steht eine lokale Ausbreitungsdiagnostik durch das CT. Standard ist heute zum Nachweis/Ausschluss von lymphogenen/hämatogenen Metastasen ein PET/CET, das durch den Nachweis der Stoffwechselaktivität sensitiver für den Nachweis von Tumormanifestationen als das reguläre CT ist. Nach Ausschluss hämatogener Fernmetastasen hängt das weitere Behandlungskonzept vom Lokalbefund u. insb. dem Vorliegen von mediastinalen Lymphknotenmetastasen ab. Bei Befall bestimmter mediastinaler Stationen (s.u.) profitieren Patienten nicht von einer Operation, weshalb präoperativ z.B. durch Mediastinoskopie die LK untersucht werden müssen. Klassifikation der Lungenkarzinome Ein genaues Typing (Festlegen der Tumorklassifikation) ist heute für die Behandlungsplanung wichtig, da verschiedene Typen des BC einer unterschiedlichen Behandlung zugeführt werden. Die wesentlichen histologischen Typen sind: Adenokarzinom (Männer 37%, Frauen 47%), Plattenepithelkarzinom (Männer 32%, Frauen 25%), Kleinzelliges Karzinom (Männer 14%, Frauen 18%), Großzelliges Karzinom (Männer 18%, Frauen 10%). Die Inzidenz des Adenokarzinoms nimmt dabei durch veränderte Rauchgewohnheiten bei allen Geschlechtern zu (Filterzigaretten, niedrige Teer- u. Kondensatwerte mit tieferer Inhalation). Maligne Lungentumoren sind in ca. 10% heterogen zusammengesetzt, d.h. bei ausreichendem Sampling sind häufig Tumoranteile verschiedener Differenzierung darzustellen. Aufgrund der schlechten Prognose und Operabilität kleinzelliger neuroendokriner Tumoren hat sich in der Klinik eine Aufteilung in klein- und nicht kleinzellige Tumoren etabliert. Da es eine Reihe kleinzelliger nicht neuroendokriner Tumoren und großzelliger neuroendokriner Tumoren gibt, erscheint diese Aufteilung unkorrekt und missverständlich und sollte deshalb vermieden werden. Dennoch ist der Begriff des nicht-kleinzelligen Bronchialkarzinoms (NSCLC) fest im klinischen Alltag etabliert. Sinnvollerweise sollte zwischen neuroendokrinen Tumoren, Tumoren mit neuroendokriner Differenzierung und nicht neuroendokrinen Tumoren unterschieden werden (Definitionen im Abschnitt neuroendokrine Tumoren). Problematik des Typing in der Biopsie Das Typing von Bronchialkarzinomen ist eine wichtige Grundlage der Therapieplanung und ein etablierter Prognosefaktor. Im Gegensatz zu den meisten anderen soliden Organtumoren kann nur bei 30% der Patienten eine Tumorresektion durchgeführt werden (s.u.), weshalb die Biopsie bei der Mehrzahl der Patienten wesentliche u. einzige Grundlage der Diagnose ist. Das Typing an der Biopsie weist einige Besonderheiten auf: die etablierten Definitionen der WHO-Klassifikation (1981, 1999 u. zuletzt 2004) basieren (fast) ausschließlich auf der Grundlage konventionell morphologischer Färbungen, d.h. die Immunhistochemie wird dabei nicht einbezogen, einzelne Entitäten (Großzelliges Karzinom) sind durch Ausschlusskriterien definiert, in bis zu 80% der Patienten mit einem Bronchialkarzinom steht ausschließlich bioptisch oder zytologisch gewonnenes Material für die Tumordiagnostik zur Verfügung, da ein resezierendes Verfahren klinisch nicht in Betracht kommt, oft nur kleine Biopsiegröße, 35 morphologische Heterogenität der Lungentumoren, die in bis zu 10% aus mehreren histologischen Komponenten bestehen, eine adenosquamöse Mischdifferenzierung ist dabei die häufigste Form, Gestaltwandel des Tumors unter Chemotherapie, Interobservervariabilität aufgrund unterschiedlich angewandter Kriterien in der Diagnostik. Zusammenfassend ist speziell in der bioptischen Diagnostik eine morphologische Grauzone vorhanden, aufgrund der teils nur herdförmig entwickelten definierenden Merkmale der Entitäten. Neuroendokrine Tumoren der Lunge Neuroendokrine Tumoren der Lunge stellen ein Spektrum dar, das von den gut differenzierten, niedrig malignen Karzinoiden bis zu den gering differenzierten, hochmalignen kleinzelligen neuroendokrinen Karzinomen reicht. Dabei gibt es jedoch keine Übergänge zwischen den gut u. den schlecht differenzierten Vertretern, die formale Pathogenese dieser Erkrankungen ist völlig different. Die Klassifikation innerhalb dieser Gruppe erfolgt nach Architektur, Zellbild, Nachweis von Nekrosen und mitotischer Aktivität. Karzinoide sind nicht mit dem Rauchen-assoziierte Tumoren i.d.R. guter Prognose, die zentral, häufig submukös im Bronchussystem auftreten. Der Altersgipfel liegt im 4.-5- Lebensjahrzehnt. Therapie der Wahl ist die Resektion. Dagegen sind kleinzellige u. großzellige neuroendokrine Tumoren hochaggressive Karzinome, die praktisch immer mit dem Rauchen assoziiert sind u. nur ein mittleres Überleben von 6 Monaten nach Diagnose zeigen. Bereits zum Diagnosezeitpunkt sind diese häufig metastasiert mit bevorzugter Lokalisation der Metastasen in Knochen, Gehirn u. Leber. Therapie der Wahl ist die kombinierte Radiochemotherapie, eine Resektion ist nur im Intervall bei guter Tumorkontrolle ggf. möglich. Die folgende Tabelle gibt die wesentlichen morphologischen Kriterien zur Abgrenzung der Tumoren wieder. Gut differenzierter neuroendokriner Tumor (typisches Karzinoid) Karzinoidmorphologie und >0,5cm und <2 Mitosen/10HPF und keine Nekrose Gut differenziertes neuroendokrines Karzinom (atypisches Karzinoid) Karzinoidmorphologie und Herdförmige Nekrosen oder 2-10/Mitosen/10HPF Gering differenziertes neuroendokrines Karzinom • Kleinzelliger Typ - kleinzelliger Tumor (<3 Lymphozytenkerne) - wenig Zytoplasma - feingranuläres Chromatin - fehlende oder wenig prominente Nukleolen - ≥11 Mitosen/10HPF - Nekrosen • Großzelliger Typ - neuroendokrine Morphologie (Rosetten, Trabekel) - ≥11 Mitosen/10HPF 36 - großzelliger Tumor (>3 Lymphozytenkerne) mit vesikulären Kernen mit meist prominenten Nukleolen - Nekrosen • neuroendokriner Immunphänotyp (mindestens 1 Marker zusätzlich zu NSE und/oder elektronenmikroskopischer Nachweis sekretorischer Granula) Plattenepithelkarzinom Das Plattenepithelkarzinom ist typischerweise ein Raucherkarzinom und entsteht zentral im Bronchialsystem über eine Vorläuferläsion, die Plattenepithelmetaplasie mit Dysplasie. Es kann polypös in das Bronchiallumen oder breit invasiv in das Parenchym wachsen. Zentral kann es zu Nekrosen mit Kavitation u. Einblutungen kommen. Bevorzugte Orte der Metastasierung sind die Lunge, Nebennieren Gehirn u. Knochen. Die wesentlichen histologischen, diagnostischen Kriterien sind im Folgenden aufgeführt. Histologische Kriterien: Interzelluläre Brückenbildung Keratinisierung Immunhistologie: Positiv: CK5/6 +++, Vimentin +, Desmin, +, CEA, +, HMFG-2 +, S-100 +, CD15 +, Synaptophysin + Allgemeine DD: floride Plattenepithelmetaplasie (Infarkt, Alveolarwandschaden) postinflammatorische Metaplasie Papillome, Condylome Adenokarzinom Das Adenokarzinom war bis vor 15 Jahren das klassische Karzinom des Nichtrauchers. Durch geänderte Rauchgewohnheiten (s.o.) nimmt es auch unter Rauchern inzwischen deutlich zu. Die Tumoren sind häufiger peripher in der Lunge, kleiner u. metastasieren früher u. ausgedehnter als Plattenepithelkarzinome. Für über 50% der Adenokarzinome ist die wesentliche aktivierende Mutation inzwischen bekannt. 30-35% der Tumoren zeigen eine KRAS-Mutation, 10-15% eine EGFR Mutation u. jeweils 2-5% eine Mutation der Onkogene BRAF, EML4-ALK. Diese Mutationen ereignen sich früh in der Karzinogenese und sind gegenseitig exklusiv, d.h. in der Regel kann nur eine dieser Mutationen auftreten. Inzwischen sind für Patienten mit einer aktivierenden Mutation von EML4-ALK u. EGFR spezifische Kinaseinhibitoren verfügbar, die (zumindest für 1-2 Jahre) auch bei fortgeschrittener Erkrankung klinisch ein gutes Ansprechen zeigen. Deshalb ist eine Testung auf die genannten Mutationen inzwischen im klinischen Alltag fest verankert. Eine Mutationsfrequenz von 2-3% mag sich zunächst unbedeutend anhören, aufgrund der hohen Zahl an Neuerkrankungen bedeutet dies jedoch, dass z.B. jährlich in Deutschland 1500 Adenokarzinome mit EML4-ALK Translokation neu auftreten. Dies entspricht z.B. der Zahl an Patienten mit neu aufgetretenem M, Hodgkin. Aufgrund des guten klinischen Ansprechens der neuen Therapeutika ist eine Testung auch für niederfrequent auftretende Mutationen lohnend. Die wesentlichen histologischen Kriterien sind im Folgenden aufgeführt. Histologische Kriterien: - Glanduläres Muster: papillär oder azinär - Solides Muster mit Schleimnachweis - Mucinablagerung (PAS n. Diastase, Mucicarmin, Alcianblau): - intra- oder extrazelluläre Lumina - diffus zytoplasmatisch Immunhistologie: Positiv: CK7 +++, Vimentin +, Desmin, +, TTF-1 +, CEA, ++, HMFG-2 +, S-100 +, CD15 +, Synaptophysin -/+ Negativ: CK20, bcl-2, ER7PR, S-100, CA19-9 37 Histologische Subtypen: - Azinär azinäre und tubuläre Proliferate aus schleimbildenden atypischen Epithelien oft an Bronchialdrüsen erinnernd - Papillär - Solides Adenokarzinom mit Schleimbildung - Bronchioloalveoläres Karzinom Wachstum kolumnärer Epithelien auf präexistenten Alveolarwänden, d.h. Lungenstruktur ist erhalten ohne Infiltration - keine anderweitige Differenzierung erlaubt - teils papilläres Wachstum Allgemeine DD: - florider Alveolarwandschaden - Radio-Chemotherapie-Effekt - Reaktive Atypie bei inflammatorischen Prozessen - Benigne Tumoren mit glandulären Merkmalen: - Sklerosierendes Hämangiom - Alveoläres Adenom - Bronchioalveoläres Adenom - Metastasen (siehe Metastasen) - Diffuses Mesotheliom Großzelliges Karzinom Das großzellige Karzinom ist im Wesentlichen keine molekular selbstständige Tumorgruppe, sondern ein Sammelbecken schlecht differenzierter Karzinome, die keine eindeutigen plattenepithelialen oder adenoiden Merkmale mehr tragen. Daher ist es eine Ausschlussdiagnose, die an der kleinen Biopsie nicht zuverlässig gestellt werden kann. Die wesentlichen Kriterien sind: - Ultrastrukturell: ¾ aller großzelligen Karzinome zeigen ultrastrukturell plattenepitheliale (Desmosomen, Tonofilamente), adenoide (Lumina, Mikrovilli, tight junctions) oder neuroendokrine (Sekretgranula) Differenzierungen. - Histologische Kriterien: - großzellig (>33µm = 3 Lymphozytendurchmesser) - vesikuläre Kerne mit prominenten Nukleolen - zytoplasmareich - distinkte Zellgrenzen - Immunhistologie: Positiv: PanCK +++, EMA ++, CEA ++, Vimentin ++, NSE +, CD15 -/+, Synaptophysin -/+, Chromogranin -/+, Bombesin -/+. Je diskohäsiver das Wachstum, desto eher keine Expression epithelialer Marker 30% aller Fälle sind ohne immunhistochemische Differenzierungsmarker Stadien des NSCLC Grundlage für die Einteilung von nicht-kleinzelligem Lungenkrebs in Tumorstadien ist die sogenannte TNM-Klassifikation. Hierbei werden berücksichtigt: - die Größe des Tumors bzw. seine Ausdehnung in der Lunge (T) - ein möglicher Befall von Lymphknoten (Nodi lymphatici) (N) - das eventuelle Vorhandensein von Metastasen in anderen Organen (M). Diese Parameter werden im sog. klinischen Stadium zusammengefasst. So sind für das T-Stadium die Tumorgröße u. der Bezug zur Pleura u. dem Hauptbronchus die Hauptdeterminanten. Für das N-Stadium ist der Befall hoch- u. tief mediastinaler LKGruppen von besonderer Bedeutung. Der Befall dieser LK-Gruppen entspricht einem N2-Stadium u. wird in der Regel mit einer Inoperabilität gleichgesetzt. Details sind z.B. auf www.lungenkrebs.de nachzulesen. 38 Übersicht der Stadien & Therapien des NSCLC Die an die Stadien der Erkrankung angepasste Behandlung wird im Folgenden tabellarisch wiedergegeben (modifiziert n. Wikipedia): Bezeichnung Stadieneinteilung Therapieoptionen/Standardbehandlungen NSCLC Stadium I NSCLC Stadium II NSCLC Stadium IIIA NSCLC Stadium IIIB T1, N0, M0 = IA T2, N0, M0 = IB 1. Anatomisch korrekte Lobektomie. 2. Strahlentherapie in kurativer Intention (für potentiell resektable Patienten mit medizinischen Kontraindikationen für eine Resektion). 3. Klinische Studien zur adjuvanten Chemotherapie nach Resektion. 4. Studien zur adjuvanten Chemoprävention. 5. Endoskopische photodynamische Therapie (derzeit in klinischer Prüfung; besonders geeignet für ausgewählte Patienten im Stadium T1, N0, M0, keine Standardtherapie). T1, N1, M0 T2, N1, M0 T3, N0, M0 1. Lobektomie; Pneumonektomie oder segmentale, Keil-, oder Manschettenresektion je nach Einschätzung der Chirurgen. 2. Strahlentherapie in kurativer Absicht (für potentiell operable Patienten, die medizinische Kontraindikationen gegen eine Operation aufweisen). 3. Klinische Studien zur adjuvanten Chemotherapie mit oder ohne andere Behandlungsarten (Chemo-, Strahlentherapie) nach kurativer Operation. 4. Klinische Studien zur Strahlentherapie nach vollständiger Entfernung des sichtbaren Tumors. T1, N2, M0 T2, N2, M0 T3, N1, M0 T3, N2, M0 1. alleinige Operation bei operablen Patienten ohne große Tumormasse („bulky disease“) 2. alleinige Strahlentherapie bei Patienten, die keine neoadjuvante Chemotherapie plus Bestrahlung erhalten können 3. Chemotherapie in Kombination mit anderen Modalitäten jedes T, N3, M0 T4, jedes N, M0 1. Alleinige Strahlentherapie 2. Chemotherapie in Kombination mit Strahlentherapie 3. Chemotherapie und gleichzeitige Strahlentherapie 4. Alleinige Chemotherapie. 39 NSCLC Stadium IV Jedes T, jedes N, M1 1. Strahlentherapie, primär in palliativer Intention bei lokalem Tumorwachstum 2. Chemotherapie. Die folgenden Therapieprotokolle sind mit ähnlichen Überlebensraten assoziiert: Cisplatin oder Carboplatin jeweils in Kombination 3. Pemetrexed (Alimta®) oder Erlotinib (Tarceva®) 40 KPK Neoplasien der Cervix KPK Cervixcarcinom und seine Vorstufen Zusammenfassung Humane Papillomvirus-Typen (HPV) sind u.a. für die Entstehung von Genitalwarzen (Condylomata acuminata) und für Gebärmutterhalskrebs (CxCa) verantwortlich. Das CxCa ist weltweit gesehen das zweithäufigste Carcinom überhaupt. Eine chronische HPV Infektion ist nahezu immer die Voraussetzung zur Entstehung von CxCa. (>99%) und seiner Vorstufen. Man unterscheidet High-risk-(CxCa)und Low-risk- (Kondylome) HPV-Subtypen. HPV ist der häufigste sexuell übertragene Erreger. Nach Infektion (oder Impfung) besteht typenspezifische Immunität, d.h. syn-/metachrone Infektionen anderer Subtypen sind möglich. Infektionen verlaufen meist asymptomatisch und heilen aus. Die Unfähigkeit des Immunsystems infizierte Zellen zu eliminieren führt zur Persistenz und Progression der Infektion, dies ist ein entscheidender Risikofaktor für CxCa und seiner Vorstufen. Die Häufigkeit von CxCa (Plattenepithel-Ca < Adeno-Ca) in entwickelten Ländern ist durch zytologische Vorsorgeprogramme dramatisch reduziert worden. Frauen, die in den letzten 10 Jahren am CxCa starben, lebten zu > 80% in Entwicklungsländern. Zytologisches Screening, Differential-Kolposkopie mit Biopsie sowie ggf. Konisation mit pathologischer Untersuchung stellen den klassischen Weg zur Erkennung und Sanierung risikoreicher Vorstufen des CxCa (Cervicale intraepitheliale Neoplasie=CIN 1/2/3 & AdenoCa in situ=AIS) dar. Seit Einführung von zwei Impfstoffen („Cervarix®“: Subtyp 16/18 bzw. „Gardasil®“: Subtyp 6/11/16/18) ist erstmals eine primäre Prävention einer malignen Erkrankung möglich! Ätiologie Humane Papillomviren (HPV: doppelstrang DNA Virus) HPV-Familie umfasst > 180 Genotypen, > 30 davon führen zu Infektionen im Genitalbereich. Primärer Infektionsort sind nur basale, proliferationsfähige Keratinozyten und Drüsenzellen, Prädilektionsstelle der Infektion ist deshalb der Übergang Ekto-Endocervix. In Kondylomen finden sich Infektion mit Typen 6 oder 11 (daher „Low-risk“), in Vorstufen und CxCa finden sich Typ 16, 18, 31, 33 und 35 und weitere (daher „High-risk“, Nennung nach Häufigkeit in Deutschland). 41 Tab. 1: Papillomvirustypen, die an verschiedenen humanen Tumoren beteiligt sind Tumorlokalisation HPV-Typ % HPV + Zervix 16, 18, 31, 33, 35, 39, 45, 51, 52, >95 56, 58, 59, 66 (26, 68, 73, 82) >50 16, 18 Basal >50 16, 18 Warzig Vulva <10 Keratinisierend 16 Vagina 16, 18 >50 Anus 16, 18 >70 Mundhöhle / Tonsille 16, 18, 33 <20 Nagelbett 16 ~75 Risikofaktoren HPV ist eine sehr häufige, sexuell übertragbare Infektion. Das Risiko steigt mit der Anzahl der Geschlechtspartner. Kondome stellen keinen sicheren Schutz dar. Außer durch Geschlechtsverkehr ist eine Übertragung durch Schmierinfektionen, durch Körperkontakt bei gemeinsamem Baden und möglicherweise auch durch kontaminierte Gegenstände möglich. Kofaktoren (zusätzlich zu HPV Infektion) o lang dauernde Einnahme von oralen Kontrazeptiva (>5 Jahre), o Rauchen, o hohe Parität, o Immunsuppression, o HIV-Infektion, o andere genitale Infektionen (Chlamydien, Herpes). Die Zirkumzision des Mannes wurde als protektiver Faktor identifiziert. Infektionsablauf Lokale Infektion verläuft ohne Virämie und meist auch ohne klinische Symptomatik. Typabhängig kommt es zur natürlichen Elimination der infizierten Zellen (50%), zur Persistenz (28%) oder zur Progression (22%) und ggf. zu Zell-/Gewebeveränderungen (Prozentzahlen für HPV 16). Lebensrisiko für eine HPV-Infektion und deren Folgen: o HPV Exposition 75% o HPV-HR Exposition 50% o Persistierende CIN 2+ 10% o CxCa < 1,3%* o Versterben an CxCa < 0,4* * Regionsabhängig 42 Primärprävention Information und Erziehung Zielgruppe der Primärprävention sind besonders Jugendliche (vor Infektion!). Ärzte haben die Aufgabe, als Multiplikatoren zu wirken o für Eltern: Beratung über Prävention von HPV bei ihren Kindern: Impfempfehlung! o für die Schule: Sexualaufklärung und Schulung von Pädagogen. o für die Medien: Beratung z.B. für Jugendzeitschriften, Internetadressen u. ä. Verhütung Die konsequente Verwendung von Kondomen vermindert das Übertragungsrisiko. Sicheren Schutz bietet nur Abstinenz bzw. Monogamie beider Partner ab dem ersten Sexualkontakt. Impfung Pro Jahr werden 33.000 neue Fälle von CxCa in Europa diagnostiziert, 2002 starben 14.638 Frauen in Europa am CxCa. 70% der CxCa werden von 2 Typen - HPV 16 und 18 - verursacht. HPV-Impfstoffe bestehen aus VLPs („virus like particles“), d.h. synthetische, leere Viruskapside ohne HPV-DNA, daher auch ohne onkogene bzw. infektiöse Gefahr. Ein tetravalenter Impfstoff zur Prophylaxe von Infektionen mit HPV 6, 11, 16 und 18 (Gardasil®) und ein zweiter, bivalenter Impfstoff gegen die HPV-Typen 16 und 18 (Cervarix®) sind in Deutschland zugelassen. Wirksamkeit in Studien >90% (je nach Endpunkt: inzidente Infektion, 16/18 Persistenz, CIN2/3 Läsionen, AIS; aber: noch keine Daten für CxCa!) Sekundärprävention, Früherkennung und Diagnostik Vorsorgeuntersuchung nach den Krebsfrüherkennungsrichtlinien Frauen >20 J. haben Anspruch auf Früherkennungsuntersuchung 1x pro Jahr (GKV). Die Vorsorgeuntersuchung (Inspektion, Zytologie von der Portiooberfläche und Zervixkanal) sollte drei Jahre nach Aufnahme vaginalen Geschlechtsverkehrs beginnen, spätestens jedoch mit dem 20. Lebensjahr. In anderen Ländern sind unterschiedliche zytologische Untersuchungsintervalle möglich. Da in Deutschland in der Vorsorge nach den Krebsfrüherkennungrichtlinien zur Zeit keine HPV-Testung vorgesehen ist, sollte eine jährliche zytologische Testung beibehalten werden. Die Kolposkopie bei der Vorsorgeuntersuchung zur Steuerung der Abstrichentnahme ist wegen Verbesserung der Qualität sinnvoll. Die Differentialkolposkopie mit Biopsie (nicht die Konisation) ist das Goldstandardverfahren zur minimal invasiven histologischen Abklärung von Auffälligkeiten und zur Therapieplanung bei histologisch gesicherten Neoplasien. 43 HPV Diagnostik Ein HPV-Test bei unauffälliger Zytologie ist als Ergänzung ab dem 30. Lebensjahr sinnvoll. Der HPV-Nachweis (In-Situ Hybridisierung oder PCR) detektiert hochgradige Präkanzerosen und Karzinome mit signifikant höherer Sensitivität, aber schlechterer Spezifität als die Zytologie. Tabelle 2: Empfehlungen zur weiteren Diagnostik je nach Befund (Zytologie/HPV) Zytologischer Befund Pap I / II Pap II w Pap III** / III D erstmalig HPVZytologische Befund Kontrolle HR-negativ Routineintervall HR-positiv 12 Monate HR-negativ 12 Monate HR-positiv 6 Monate HR-negativ 6 Monate HR-positiv 3 - 6 Monate HR-negativ 6 Monate Pap III** / III D wiederholt HR-positiv Pap IV a und höher unabhängig Weitere Diagnostik Gleichzeitig HPVKontrolle. Falls wieder HR-positiv oder zytologisch auffällig: Dysplasiesprechstunde* + erneute HPV-Testung Gleichzeitig HPVKontrolle. Falls wieder HR-positiv oder zytologisch auffällig: Dysplasiesprechstunde* + erneute HPV-Testung Falls erneut HPV-HRpositiv: Dysplasiesprechstunde*. + erneute HPV-Testung. In jedem Fall Dysplasiesprechstunde* nach 12 Monaten Dysplasiesprechstunde* Dysplasiesprechstunde* HR = high-risk-Viren (S. 41) * Dysplasiesprechstunde = Differentialkolposkopie mit Biopsie eventueller Herdbefunde. ** Pap III mit dringendem V. a. höhergradige Atypie immer rasche diagnostische Abklärung. Intraepitheliale Neoplasien Onkogene oder High-risk-HPV-Typen (z.B. HPV 16, 18 u.a.) können Läsionen verursachen, die die Histologie einer intraepithelialen Neoplasie (IN; s.u.) aufweisen. Diese werden in drei Grade (Grad I - III) unterteilt. Im Genitalbereich unterscheidet man je nach Lokalisation: o Vulväre intraepitheliale Neoplasie VIN (nach WHO) o Vaginale intraepitheliale Neoplasie VAIN o Zervikale intraepitheliale Neoplasie CIN o Perianale intraepitheliale Neoplasie PAIN o Anale intraepitheliale Neoplasie AIN 44 Verlauf 10-40% der HR-Infektionen werden zu persistierenden Infektionen. Daraus wiederum wird in 10-50% nach fünf bis 10 Jahren eine CIN3. Die Progressionsrate von CIN3 zum CxCa >12%. Am CxCa erkranken häufiger jüngere Frauen; mittleres Erkrankungsalter 50,4 Jahre, damit liegt es ca. 19 Jahre unter dem das mittlere Erkrankungsalter aller Krebserkrankungen. Pathomorphologische Untersuchung Allgemeine Grundsätze Mit dem Gewebe sollte dem Pathologen mitgeteilt werden: Patientendaten (Name, Geburtsdatum, Geschlecht), wesentliche anamnestische Angaben/Vorbefunde, klinische Informationen (u.a. makroskopischer, ggf. kolposkopischer Aspekt), Entnahmelokalisation (ggf. räumliche Orientierung), Entnahmeart (z.B. Punch-Biopsie, Knipsbiopsie, Exzisat, Konisat, Messer-/Schlingenabtragung), klin. Verdachtsdiagnose. Nomenklatur: Intraepitheliale Neoplasie Der Begriff der intraepithelialen Neoplasie (IN; der ehemalige Begriff Dysplasie ist obsolet) wird für die Beschreibung von nicht-invasiven, präkanzerösen Epithelveränderungen verschiedener, auch extragenitaler Organe, verwendet, die mit einem erhöhten Krebsrisiko einhergehen. Gemeint sind histo- und zytomorphologisch fassbare Veränderungen, die im Rahmen einer atypisch gesteigerten Zellproliferation zu einer Störung der normalen Epitheldifferenzierung (d.h. fehlende Ausreifung des Plattenepithels) und zu zellulären Atypien (in Form von Hyperchromasie, Pleomorphie, Verschiebung der Kern-Plasma-Relation, atypische Mitosen) führen. Die IN des Plattenepithels der Cervix wird in drei Stufen graduiert: CIN 1 (leichte oder geringgradige intraepitheliale Neoplasie) CIN 2 (mittelschwer oder mäßiggradig) CIN 3 (schwer oder hochgradig, inklusive dem früher bezeichneten „Carcinoma in situ“) 45 Tabelle 3: Vergleich zytologischer (Münchener, Bethesda) und histologischer (WHO) Nomenklaturen plattenepithelialer Läsionen Münchner Nomenklatur (Pap) I II III IIID WHONomenklatur; CIN normales Zellbild leichte entzündliche, degenerative oder metaplastische Veränderungen unklarer Befund: schwere entzündliche oder degenerative Veränderungen, auffällige Drüsenzellen; eine Dysplasie, ein Carcinoma in situ oder (in seltenen Fällen) ein Malignom können nicht ausgeschlossen werden Leichte bzw. mäßige Dysplasie CIN 1 CIN 2 IVa IVb V schwere Dysplasie /Carcinoma in CIN 3 situ schwere Dysplasie oder Carcinoma in situ, invasives Karzinom nicht auszuschließen invasives Karzinom Bethesda-System; Squamous intraepithelial lesion ASC-US: atypische plattenepitheliale Zellen unbestimmter Signifikanz ASC-H: atypische plattenepitheliale Zellen, HSIL nicht auszuschliessen LSIL low-grade squamous intraepithelial lesion HSIL high-grade squamous intraepithelial lesion Mögliche Differentialdiagnosen zur CIN (zytologisch, kolposkopisch, histologisch) o Atrophie o Regeneratepithel o Unreife Plattenepithelmetaplasie Intraepitheliale Neoplasien des endozervikalen Zylinderepithels Eine Graduierung entsprechend der IN des Plattenepithels wurde versucht, ist aber schlecht reproduzierbar und bislang umstritten, verschiedene Nomenklaturen sind in Gebrauch (ICGN: intraepitheliase cervicale glanduläre Neoplasie; syn.: GCIN; GIN). Laut WHO werden zwar eine glanduläre Dysplasie und das Adenocarcinoma in situ (AIS) unterschieden, dies ist aber nicht generell akzeptiert, da die Abgrenzung Schwierigkeiten bereitet und meist ohne klinische Konsequenz ist. ICGN sind meist HPV-assoziiert und treten auch in Kombination mit CIN auf. Abgegrenzt werden muss die (entzündlich) reaktive glanduläre Atypie, ggf. über Zusatzuntersuchungen (z.B. p16, Ki-67, s.u.) 46 Pathologisch-anatomische Aufarbeitung Stanz- und Knipsbiopsien Formalin fixiert, Paraffineinbettung Schnittstufen Portiokonisate Orientiert an die Pathologie einsenden (z.B. Faden bei 12 Uhr), vollständige Einbettung mit radiärer bzw. segmentaler Aufarbeitungstechnik. Beurteilung des endozervikalen Absetzungsrandes (sinnvoll: Tuschemarkierung), dieser ist ggf. separat zu untersuchen. Ergänzende histomorphologische Untersuchungen o Ki-67 (Mib 1) o p16ink4a (Cyclin-Kinase Inhibitor: CKI) o L1-Capsid-Proteins o Eine (erneute) In-Situ Hybridisierung auf HPV Subtypen ist am histopathologischen Gewebe wenig sinnvoll. Operative Therapie CIN Die Differentialkolposkopie mit Biopsie (und nicht die Konisation) ist das Goldstandardverfahren zur minimal invasiven histologischen Abklärung von Auffälligkeiten bei der Vorsorgeuntersuchung und zur Therapieplanung. Das therapeutische Vorgehen ist abhängig vom Befund, Alter und Wünschen(!) der Patientin (Tabelle 4). Ziel ist die vollständige Entfernung der Transformationszone mit allen neoplastischen Läsionen. Cave: in Abhängigkeit von der Größe des Konus/Exzidates steigt das Risiko für Frühgeburtlichkeit und zervikale Stenosen an. Schlingenkonisation (LEEP / LLETZ) Die Resektion der Transformationszone (TZ) mittels Hochfrequenzschlingen wird als Schlingenkonisation (LEEP, Loop electrosurgical excisional procedure, oder LLETZ, Large loop excision of the transformation zone) bezeichnet. Die TZ wird mit einer entsprechend geformten Drahtschlinge möglichst in einem Stück reseziert. Die Schlingenkonisation ist die chirurgische Methode der Wahl. Frühund Spätkomplikationen treten seltener auf als nach Messerkonisationen, weshalb letztere Methode nicht mehr empfehlenswert ist. Tabelle 4: Management von zervikalen intraepithelialen Neoplasien. Management OP-Verfahren Konservatives Management Bis zu 24 Schlingenkonisation, CIN 1 KolposkopischLaserkonisation/Vaporisation Monaten (nur bei zytologische HPV-HR(bei Befundpersistenz, HPVKontrolle alle 6 HR-Positivität und Wunsch der Positivität Monate (nur bei relevant) HPV-HR-Positivität) Patientin) CIN 2 KolposkopischSchlingenkonisation, Bis zu 12 zytologische Laserkonisation/Vaporisation Monaten 47 (bei Befundpersistenz, HPVKontrolle alle 6 HR-Positivität und Wunsch der (nur bei HPVMonate (nur bei HR-Positivität HPV-HR-Positivität) Patientin) relevant) Konisation (Schlinge, Laser, CIN 3 Therapie In graviditate Nadel, Messer) Konisation (Schlinge, Laser Bei CIN 1 Ausdehnung Kolposkopischoder Messer) möglich (nur bei zytologische in die tiefe HPV-HREndozervix Kontrolle Positivität relevant) Alternative Therapieverfahren bei CIN - Therapie mit rekombinantem humanen Interferon Ggf. bei Rezidiven oder ausgedehnter Erkrankung, Datenlage zum Nutzen widersprüchlich - Vitamin A Daten für die Wirksamkeit widersprüchlich, Einsatz nicht empfohlen. - Cyclooxigenase-2-Inhibitoren Regression von CIN2/3-Läsionen unter der Therapie mit Cyclooxigenase-2Inhibitoren. Nach kardiovaskulären Zwischenfällen wurde Rofecoxib vom Markt genommen. - Leukozytenultrafiltrat 5 E. i.m. Derzeit fehlen prospektiv randomisierte Studien, die eine Wirksamkeit aufzeigen könnten. In Kasuistiken und Einzelfallberichten wurde die Regression von Läsionen beschrieben. - Folsäure, Riboflavin, Thiamin und Vitamin B12 Die ausreichende Zufuhr von Folsäure, Riboflavin, Thiamin und Vitamin B12 kann das Risiko für zervikale Dysplasien (CIN 1-3) um 50 bis 90% reduzieren. Dieses zeigte eine Fall-Kontroll-Studie mit 485 Frauen. Kein Therapieansatz bei bestehender CIN. Referenzliteratur • WHO Bluebook: Tumours of the Breast and Female Genital Organs (www.iarc.fr/who-bluebooks) • Blaustein’s pathology of the female genital tract. 5th Edition. ISBN 0-38795203-9 • Leitlinien der Deutschen Gesellschaft für Gynäkologie und Geburtshilfe (DGGG), AWMF Leitlinienregister 015/027. 48 KPK Lymphknoten- und Hämatopathologie Neoplastische Erkrankungen des hämatopoietischen Systems Die Zellen des peripheren Blutes sowie der lymphatischen Organe entstammen dem Knochenmark. Sie differenzieren sich aus gemeinsamen pluripotenten Stammzellen für die lymphatischen bzw. myeloischen Zellreihen unter Stimulation durch Zytokine. Hierbei entstehen aus den myeloischen pluripotenten Stammzellen in mehreren Differenzierungsschritten im Knochenmark reife Granulozyten (so genannte Segmentkernige), Monozyten, Thrombozyten und Erythrozyten. Aus den lymphatischen Stammzellen differenzieren sich im Knochenmark B-Lymphozyten, die die Antikörper vermittelte Immunität tragen, und im Thymus die T-Lymphozyten, die die zytotoxische Immunität vermitteln. Durch genetische Veränderungen (z. B. Genmutationen, Chromosomenbrüche mit Translokationen, etc.) der Stammzellen kommt es zu einer gestörten Differenzierung der hämatopoietischen bzw. lymphatischen Zellreihen, die neben den Erkrankungen des Myelodysplastischem Syndromes bzw. der aplastischen Anämie auch zu neoplastischen Erkrankungen des hämatopoietischen sowie lymphatischen Systems führen. Diese Veränderungen betreffen immer alle aus der veränderten Stammzelle hervorgegangenen Zellen, weshalb Leukämien und Lymphome als monoklonale Tumore bezeichnet werden. 1. Bösartige Erkrankungen des hämatopoietischen Systems Die bösartige Erkrankungen des hämatopoietischen Systems werden auch als Leukämie bezeichnet, wobei man zwischen akuten und chronischen Leukämien sowie myeloischen und lymphatischen Leukämien unterscheidet. Akute Leukämien sind durch eine Anhäufung unreifer Vorstufen der Hämatopoiese bzw. Lymphopoiese, sogenannten Blasten, infolge von Differenzierungsblockaden gekennzeichnet, wodurch es zu einer Verdrängung der normalen Hämatopoiese kommt. Auf Grund des Fehlens ausdifferenzierter Granulozyten resultiert eine Störung der unspezifischen Abwehr mit erhöhter Infektanfälligkeit und daraus resultierenden septischen Allgemeininfektionen bzw. Septikopyämien. Die Verdrängung der Megakaryopoiese führt zu einem Mangel an Thrombozyten und hierdurch zu einer erhöhten Blutungsneigung mit der Möglichkeit von Hirnmassenblutungen bzw. einem Verbluten aus Erosionen bzw. Ulcera des Gastro49 Duodenal-Traktes. Die durch eine Verdrängung der Erythropoiese bedingte Anämie kann gerade bei älteren Patienten zu einer Verschlechterung von vorbestehenden chronischen Erkrankungen, z. B. des Herz-Kreislaufsystems und hierdurch zu einem vorzeitigen Tod des Patienten führen. Man unterscheidet insgesamt sieben verschiedene Formen akuter myeloischer Leukämien sowie drei verschiedene Untertypen von akuten lymphatischen Leukämien (siehe Tabelle 1). Tabelle 1: WHO-Klassifikation der akuten myeloischen und lymphatischen Leukämien Subtyp Häufigkeit (%) AML M0 AML, undifferenziert 5 - 10 AML M1 Akute Myeloblastische Leukämie 10 - 20 ohne Differenzierung AML M2 Akute Myeloblastische Leukämie mit 30 - 45 Differenzierung AML M3 Akute Promyelozytenleukämie 5 - 10 AML M4 Akute myelomonozytäre Leukämie 5 AML M5a,b Akute Monoblasten (5a) bzw. 5 Monozyten (5b) Leukämie AML M6 Akute Erythroleukämie 5 AML M7 Akute Megakaryblastenleukämie 5 Man unterscheidet bei der chron. Leukämie, bei denen man die chronisch lymphatische Leukämie von den myeloproliferativen Neoplasien (MPN) unterscheidet, zu denen die chronisch myeloische Leukämie, die Polyzythämia vera, die essenzielle Thrombozythämie sowie die so genannte primäre Myelofibrose neben den seltenen Formen der chronischen Neutro- bzw. Eosinophilenleukämie gerechnet werden. Die chronischen Leukämien zeigen im Unterschied zu den akuten Leukämien eine übermäßige Produktion reifer Zellenlinien, z. B. der Granulopoiese (chronisch myeloische Leukämie), der Thrombozyten (essenzielle Thrombozythämie) bzw. aller zellulären Blutbestandteile (Polyzythämia Vera). Die Patienten sind hier bei fehlender Behandlung insbesondere durch eine Störung der Durchblutung der kleinen Gefäße (Kapillaren, Venolen) und der dadurch bedingten Störung von Organfunktionen wie z.B. des Gehirns (Koma) gefährdet. Im Verlaufe der Erkrankung kann es durch weitere Störungen im Genom zu einem Übergang in eine akute 50 myeloische bzw. lymphatische Leukämie kommen, was als sogenannte Blastenkrise bezeichnet wird. Dies wird insbesondere bei der chronisch myeloischen Leukämie beobachtet. Diese Blastenkrisen stellen häufig die tödliche Komplikation der chronisch myeloischen Leukämie dar. Auf Grund der entscheidenden Rolle genetischer Veränderungen bei der Auslösung hämatopoietischer Neoplasien kommt dem Nachweis chromosomaler Veränderungen bei der Diagnostik der akuten und chronischen myeloischen Leukämien eine besondere Bedeutung zu. Bei der chronisch myeloischen Leukämie konnte bereits in den sechziger Jahren ein konstanter Chromosomendefekt, das so genannte Philadelphia-Chromosom nachgewiesen werden, welches bei allen Patienten mit chronisch myeloischer Leukämie zu finden ist und deswegen als Markerchromosom bezeichnet wird. Es handelt sich hierbei um eine reziproke Translokation zwischen den Chromosomen 9 und 22, wobei es zu einer Ausbildung eines neuen bcr-abl-Fusionsgens kommt, welches für ein 210 kd großes Protein codiert. Dieses besitzt die Eigenschaften einer Thyrosinkinase und fungiert in der Membran als Rezeptor, welcher die Proliferation des neoplastischen Klons bewirkt. Durch die Blockade dieses Rezeptorproteins durch das Medikament Imatinib sind eine Unterdrückung des neoplastischen Klons und hierdurch eine Heilung der Patienten möglich geworden. Auch bei der Diagnostik akuter myeloischer Leukämien kommt dem Nachweis chromosomaler Aberrationen eine besondere Bedeutung bei. Hier finden sich für die einzelnen Subtypen zwar nur bedingt charakteristische Markerchromosomen wie bei der chronischen myeloischen Leukämie, jedoch hat hier der Nachweis einfacher bzw. sogenannter komplexer chromosomaler Aberrationen bzw. Karyotypen eine prognostische Bedeutung, d. h. Patienten mit mehrfachen Chromosomenbrüchen (komplexe Karyotypen) zeigen ein schlechteres Ansprechen bzw. ein kürzeres Überleben auf eine Chemotherapie als Patienten ohne chromosomale Aberrationen. 51 2. Bösartige Erkrankungen des lymphatischen Systems (maligne Lymphome) Maligne Lymphome werden ähnlich wie die Leukämien durch genetisch bedingte Störungen der Vorläuferzellen ausgelöst, wobei virale Infektionen, insbesondere mit Epstein- Barr-Virus (EBV), eine besondere Rolle zu spielen scheinen. Die malignen Lymphome resultieren in den ersten Stadien in lokalen Tumorbildungen des lymphoretikulären Parenchyms, wobei man ein nodales, (d. h. Wachstum in einem Lymphknoten), bzw. ein extranodales Wachstum, (d. h. den Befall des lymphoretikulären Parenchyms in Schleimhäuten bzw. Organen Mukosa assoziierten lymphatischen Gewebe,[MALT] oder in Bronchus assoziierten lymphatischen Gewebe [BALT]) unterscheidet. Später generalisieren die malignen Lymphome durch Ausbreitung in andere Lymphknotenstationen oberhalb bzw. unterhalb des Zwerchfelles bzw. in andere Organe wie Lunge, Knochenmark etc. Als Staging- System wird die Ann- Arbor Klassifikation für alle maligne Lymphome angewendet (s. Abb 1). Weiterhin unterscheidet man bei der Einteilung der malignen Lymphome den Morbus Hodgkin von den später beschriebenen nicht Hodgkin-Lymphomen (NonHodgkin-Lymphom; NHL), wobei letztere nochmals nach dem Ort der Prägung der Vorläuferzellen in B- bzw. T-NHL aufgetrennt werden. Die einzelnen Subtypen der malignen Non-Hodgkin-Lymphome korrelieren, insbesondere beim B-NHL, mit den jeweiligen zytologischen Formen der peripheren Differenzierungsstadien von naiven B-Lymphozyten zum (nach Antigenkontakt determinierten) B-GedächtnisLymphozyten bzw. der entsprechenden Plasmazelle, determiniert nach Antigenkontakt. 52 Abbildung 1: Staging maligner Lymphome nach dem Ann-Arbor-Staging-System Das Staging maligner NHL erfolgt in Analogie zum Ann Arbor- Staging System(1971) für das Hodgkin Lymphom in vier verschiedene Stadien: • Stadium I: Befall einer Lymphknotenstation bzw. eines extralymphatischen Organes oder Gewebes (Stadium IE) • Stadium II: Befall zweier oder mehrerer Lymphknotenstationen auf derselben Seite des Zwerchfelles oder eines extralymphatischen Organes oder Gewebes (Stadium IIE). • Stadium III: Befall von Lymphknotenstationen auf verschiedenen Seiten des Durch die Entwicklung immunhistologischer Techniken seit den 70er Jahren konnten den Subtypen des Morbus Hodgkin bzw. den Non-Hodgkin-Lymphomen charakteristische Gruppen positiver Antikörper nach der CD-Klassifikation zugeordnet werden, die diese Subtypen neben der reinen Morphologie weiter als eigenständige Kategorien lymphatischer Erkrankungen kennzeichnen. So lässt sich bei den B-NHL als gemeinsames Kennzeichen das Antigen CD 20 nachweisen, während bei den T-NHL das Pan-T-Zell-Antigen CD 3 zu finden ist. Das klassische Hodgkin Lymphom wird durch den Nachweis von CD 15 und CD 30 auf den für diese Erkrankung charakteristischen Hodgkin- bzw. Reed-Sternberg-Zellen gekennzeichnet. Die insbesondere für die Differential-Diagnostik der klinisch wichtigen bzw. häufigeren niedrig malignen B-NHL geltenden Profile zeigt die nachstehende Tabelle 2. 53 Immunhistologisches Profil von B-NHL Subtyp CD 5 CD23 CD10 CD43 Bcl-6 IGM CycD1 B-CLL/SLL + + - + - -/+ - Mantelzelllymphom + - - + - (+) + Marginalzonen Lymphom - - - - - - - Follikuläres Lymphom - - + - + - - Lymphoplasma.Lymphom - - - +/- - + - DLBCL - -/+ +/- +/- (+)/- - -/+ Auch bei den malignen Non Hodgkin Lymphomen haben sich in den cytogenetischen Untersuchungen bei bestimmten Subtypen bei etwa 30 bis 40 Prozent der Patienten konstante Chromosomenaberrationen gefunden, die für diese Subtypen zusätzliche diagnostische Bedeutung erlangt haben (siehe Tabelle 3). Tabelle 3: Chromosomale Aberrationen maligner Lymphome Translokation NHL - Subtyp t(11;14) Mantelzelllymphom t(14;18) Follikuläres Lymphom t(11;18) MALT-Lymphom t(8;14) Burkitt-Lymphom 54 Die neue WHO Klassifikation der malignen Lymphome beruht deswegen neben morphologischen, insbesondere auch auf immunhistologischen und cytogenetischen Kriterien für die einzelnen Subtypen. Im Unterschied zu älteren Klassifikationssystemen wie z. B. der Kiel-Klassifikation, werden jetzt der Morbus Hodgkin sowie die Non-Hodgkin-Lymphome und auch das Plasmozytom unter einem Dach als maligne Lymphome zusammengefasst und die klassische Unterscheidung zwischen Hodgkin- bzw. Non-Hodgkin-Lymphome aufgegeben. Die von der KielKlassifikation geläufige Graduierung in hoch-und niedrig maligne Lymphome war zunächst aufgegeben worden, findet sich jetzt aber unter den Bezeichnungen indolent (= niedrig maligne) bzw. aggressiv bzw. hoch aggressiv (= hoch maligne) wieder. Tabelle 3: WHO Klassifikation der NHL nach klinischem Aggressivitätsgrad WHO Klassifikation der NHL nach klinischem Aggressivitätsgrad Indolente Lymphome B Kleinzelliges lymphozytisches BZell-Lymphom / B-CLL T Aggressive Lymphome Hoch aggressive Lymphome B T B T Adult T-Zell Lymphom / Leukämie T-Zell LGL Follikuläres Lymphom (Grad III) Peripheres TZell Lymphom Lymphoplasmacytoides Mycosis Lymphom /I fungoides mmunocytom Diffuses großzelliges B-Zell Lymphom Anaplastisches T-ALL / TB-ALL / Bgroßzelliges lymphoblastisches lymphoblastisches Lymphom, T -/ Lymphom Lymphom null-Zell-Typ Plasmozytom Haarzellenleukämie Follikuläres Lymphom (Grad I und II) Marginalzonenlymphom Mantelzelllymphom* T-Zell Prolymphozyten Mantelzelllymphom* Leukämie NK-Zell LGL Burkitt Lymphom * kann klinisch indolent oder aggressiv verlaufen 55 KPK Prostata- und Harnblasentumoren Harnblasenneoplasien: Epidemiologie: Die Inzidenz von Harnblasentumoren liegt bei ca. 35/100.000 in der Altersgruppe über 40 Jahre. Das Urothelcarcinom ist das zweithäufigste urologische Malignom. Vornehmlich sind Männer im Alter von 50-70 Jahren betroffen. Risikofaktoren sind Rauchen, Exposition gegenüber bestimmten Chemikalien (Farbstoffe, Lacke) und eine positive Familienanamnese. Klinik: Makrohämaturie, Mikrohämaturie, Urgesymptomatik, in fortgeschrittenen Fällen Schmerzen, Harnstauungsniere, Blasenentleerungsstörungen, Gewichtsverlust. Diagnostik: Zytologie oder Spülzytologie als Screening. Ausscheidungsurographie. Zystoskopie oder Ureterorenoskopie mit Biopsie zur definitiven histologischen Diagnosesicherung. Unter Umständen mit Fluoreszenzfarbstoff Hexvix (Hexaminolaevulinat). Pathologie: Harnblasencarcinome imponieren makroskopisch als papilläre Herde oder solide, ulcerierte, weiße Herde bei invasiven Carcinomen. Dysplasien und Carcinomata in situ sind makroskopisch schlecht sichtbar. Bei Harnblasencarcinomen handelt es sich zumeist um Urothelcarcinome (Synonym: Übergansepithelcarcinom, Transitionalzellcarcinom). Adenocarcinome sind Raritäten, Plattenepithelcarcinome (Verlangt wird eine reine plattenepitheliale Differenzierung, Partielle Plattenepitheldifferenzierung ist beim Urothelcarcinom häufig!) sind hierzulande selten, in Ägypten beispielsweise aber häufig wegen der Schistosomiasis, die offenbar eine Metaplasie induziert. Verwirrend für den Anfänger ist die Gruppe der nichtinvasiven papillären Urothelcarcinome. Eigentlich sind Carcinome ja per definitionem invasiv! Entität Aufbau Kerne Dysplasie flach Carcinoma in situ flach Papillom papillär Nucleolen, Pleomorph Schwere Pleomorphie unauffällig Nichtinv. pap. papillär geringe Atypie Progressrisiko (5 Jahre) ca. 20% ca. 36% 0% ca. 10% 56 Carcinom G1 Nichtinv. pap. Carcinom G2 Nichtinv. pap. Carcinom G3 Invasives Urothelcarcinom papillär mittlere Atypie ca. 25% papillär hohe Atypie ca. 60% invasiv hohe Atypie - Weitere Verwirrung wurde durch eine neue WHO-Einteilung 2004 geschaffen, die im Schema unten dargestellt ist, sich aber in der Praxis nicht durchsetzen konnte. Wichtig ist, dass die Zuordnung der WHO 1973 Entitäten zu denen von 2004 keine Projektion ist, d. h. beispielsweise sind alle G3-Tumoren nach 1973 hochgradige papilläre nichtinvasive Urothelcarcinome nach WHO 2004, umgekehrt gilt das aber nicht. Beachte die Stufen in der Einteilung! WHO 1973 Papillom Papilläres nichtinvasives Urothelcarcinom G1 Papilläres nichtinvasives Urothelcarcinom G2 Papilläres nichtinvasives Urothelcarcinom G3 WHO 2004 Papillom Papilläre urotheliale Neoplasie mit niedrigem malignen Potential (PUNLMP) Niedriggradiges papilläres Nichtinvasives Urothelcarcinom Hochgradiges papilläres nichtinvasives Urothelcarcinom Urothelcarcinome weisen in unterschiedlichem Ausmaß eine Tendenz zur Rekurrenz und zur Progression auf (siehe Tabelle). Die Tendenz zur Rekurrenz hängt mit einem sog. Feldeffekt zusammen, wie er auch bei den Plattenepithelcarcinomen der oberen Atemwege beobachtet wird. Feldeffekt bedeutet, dass die gesamte Schleimhaut (bedeutet beim Urothel bis hoch in das Nierenbeckenkelchsystem) einem stark erhöhten Risiko neoplastischer Transformation ausgesetzt ist. Mit Progression bezeichnen wir die Entwicklung zu einer höhergradigen Neoplasie oder das Auftreten einer rekurrenten Neoplasie mit höherem Grad, beispielsweise das Auftreten eines urothelialen Carcinoma in situ nach einer initialen Dysplasie, oder das Auftreten eines papillären nichtinvasiven Urothelcarcinoms G3 nach einem vorhergehenden G2-Befund. Das Grading von invasiven Urothelcarcinomen hat keinerlei prognostische Bedeutung, daher wird jedes invasive Urothelcarcinom von den meisten Pathologen als G3 (schlecht differenziert) eingeordnet. 57 Therapie: Niedrigrisikotumoren (pTa G1, solitärer Tumor) - 1x TUR (Transurethrale Resektion) - keine weitere Therapie Intermediärrisikotumoren (Andere) - 1x TUR (Transurethrale Resektion) - Frühinstillation mit Mitomycin - Wiederholte TUR nach 4-6 Wochen - Intravesikale Chemotherapie für 1 Jahr Hochrisikotumoren (Carcinoma in situ, pTa G3, pT1 G3) - 1x TUR (Transurethrale Resektion) - Frühinstillation mit Mitomycin - Wiederholte TUR nach 4-6 Wochen - BCG Instillationstherapie für mindestens ein Jahr - Zystektomie bei Therapieversagen und/oder lokal nicht zu kontrollierendem Tumor Invasive Urothelcarcinome ab pT2 (Muskelinvasion) -Radikale Zystoprostatektomie. Prostatacarcinom Epidemiologie: Häufigste Tumorerkrankung über dem 65. Lebensjahr. 28.000 Neuerkrankunge pro Jahr in Deutschland. Inzidenz steigt altersabhängig. 50/100.000 bei 60 Jahren, 400/100.000 bei 75 Jahren. In Autopsieserien latentes Prostatacarcinom bei 10% der 50 Jahre alten Männer, bei den 80 Jahre alten Männern 70%. Risikofaktoren sind das Lebensalter, androgene Stimulation, hochkalorische Ernährung, und ethnische Zugehörigkeit (häufiger bei Afroamerikanern). Klinik: Häufig inapparent, später Harnabflussstörungen. Diagnostik: Prostataspezifisches Antigen im Serum, digital-rektale Untersuchung, transrektale Stanzbiopsie. Pathologie: Makroskopisch lassen sich Prostatacarcinome nur in etwa 2/3 aller Fälle als weiße, solide Herde abgrenzen. Sie finden sich zumeist in der peripheren Zone dorsolateral. Prostatacarcinome sind zumeist azinäre Adenocarcinome, duktale Prostatacarcinome sind selten. Für die Diagnose eines Prostatacarcinoms ist die Immunhistochemie sehr hilfreich. Mit den Markern hochmolekulares Zytokeratin oder besser noch p63 können wir 58 Basalzellen oder ein Fehlen derselben nachweisen. Hierbei gilt aber zu bedenken, dass einige Läsionen, die eine Prostatacarcinom vortäuschen können, zwar nicht komplett, dennoch aber über weite Bereiche eine fehlende Basalzellschicht aufweisen können. Dies kann bei kleinen Proben (die typische Prostatastanze) problematisch sein. Ein invasives Prostatacarcinom und auch die hochgradige intraepitheliale Neoplasie färben sich mit dem Marker Razemase (im englischen Sprachraum auch AMACR genannt) an. Eine Kombination aus beiden Markern hat sich im diagnostischen Alltag als sehr zuverlässig bewährt. Azinäre Prostatacarcinome werden nach Gleason graduiert (siehe diagnostischer Atlas). Beim Gleason-Grading ist im Unterschied zu anderen Graduierungssystemen ausschließlich das Wachstumsmuster, nicht aber die Kernmorphologie Kriterium der Einteilung. Das prädominante Wachstumsmuster bildet den ersten Summanden einer Addition, das sekundäre den zweiten. Beispiel: Gleason 5+3=8. Je höher der Gleason-Grad, desto schlechter differenziert ist das Carcinom. Es zählen nur sekundäre Wachstumsmuster, die mehr als 5% des Tumors ausmachen. Tertiäre Gleasonmuster werden im Diagnosekommentar erwähnt. Die seltenen duktalen Adenocarcinome sollten als 4+4=8 graduiert werden. Therapieoptionen: - Watchful waiting: PSA-Wert-Bestimmung, Abwarten bis palliativer Therapie nötig. Vorteile: Nicht invasiv. Nachteile: Kein kurativer Ansatz. - Active Surveillance: PSA-Wert-Bestimmung, abwarten bis kurative Therapie nötig (beispielsweise PSA-Anstieg oder Anstieg des Gleason-Grades (Rebiopsie). Vorteil: zunächst nebenwirkungsarm und wenig invasiv. Nachteil: Rebiopsien nötig. - Radikale Prostatektomie: operative Entnahme der Prostata, der Samenblasen und der Samenleiterenden, Lymphadenektomie kleines Becken. Vorteil: kurativer Ansatz. Abschließende Histologie mit Nodalstatus. Nachteil: Impotenzrisiko (bis 50%, gemildert durch Viagra etc.), Inkontinenzrisiko (5%). Invasiv. - Radiatio extern: transdermale Strahlenapplikation. Kurativ. Vorteil: auch Beherrschung ausgedehnter Tumoren möglich. Kein Narkoserisiko (alte, multimorbide Patienten). Nachteil: Strahlenschäden, kein Staging, kein Lymphknotenstatus. Häufige Wiederholung nötig. - Radiatio intern: Transperineale Applikation von Strahlenquellen. Kurativ. Vorteil: kurze Krankenhausverweildauer (1 Tag), minimal invasiv, anfangs kein Impotenzrisiko, kein Inkontinenzrisiko. - Chemotherapie: palliative Therapie nach Hormonrefraktärität. Vorteil: letzte Option. Nachteil: Nebenwirkungen der Chemotherapie, nicht kurativ. 59 Diagnostischer Atlas: Nichtinvasives papilläres Urothelcarcinom G1 Harnblasenneoplasien: Normalbefund Nichtinvasives papilläres Urothelcarcinom G2 Urotheliale Dysplasie Nichtinvasives papilläres Urothelcarcinom G3 Urotheliales Carcinoma in situ Invasives Urothelcarcinom Urotheliales Papillom Prostatacarcinom Prominente Nukleolen und Mitosefigur (Pfeil) in Carcinomdrüsen, rechts zum Vergleich normale Drüsen Normalbefund Beispiele für azinäre Prostatcarcinome Gleason Grading Irrguläre, dichtgelagerte Drüsenkomplexe im Zentrum Perineuralscheideninfiltration (rechte Bildhälfte) Hochgradige Prostatische intraephiteliale Neoplasie (PIN). Prominente Nukleolen