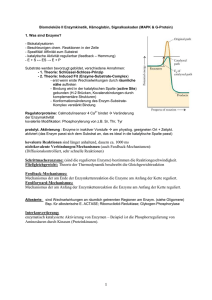
Inaktivierung der Ras-Proteine
Werbung

Wissenschaft intern 286 Habilitierte stellen sich vor Inaktivierung der Ras-Proteine: ein neuer Ansatz in der Tumortherapie Rainer Girgert, Universitätsfrauenklinik, Universität Ulm Bei der konventionellen Chemotherapie werden in erster Linie Zytostatika eingesetzt, die in den Stoffwechsel der DNAReplikation eingreifen und deshalb neben den Tumorzellen auch gesunde Gewebe schädigen, wodurch gravierende Nebenwirkungen ausgelöst werden, die dosislimitierend sind. In den letzten Jahren werden deshalb verstärkt Versuche unternommen, genspezifische Medikamente zu entwickeln, die veränderte Gene in Tumorzellen spezifisch inaktivieren (gene-targeted therapy). Mutierte ras-Gene sind an der Entstehung einer Vielzahl von Tumorerkrankungen beteiligt. Transformierende Punktmutationen treten bei den ras-Genen ausschließlich in den Codons 12, 13, 59 und 61 auf. 90 % aller Pankreaskarzinome und 50 % aller Colonkarzinome tragen ein mutiertes K-ras-Gen, ein mutiertes H-ras-Gen wird in 20 % der Blasenkarzinome gefunden und N-ras-Mutationen sind bei der akuten myeloischen Leukämie am häufigsten[1]. Dieses häufige Auftreten von transformierenden Mutationen in den ras-Genen von Tumoren ist ein eindeutiger Hinweis auf die wichtige Funktion der Ras-Proteine bei der Steuerung der Zellteilung. Die Wachstumssignale vieler Wachstumsfaktoren werden über die Adaptermoleküle SHC, Grb2 und SOS an die Ras-Proteine weitergeleitet. Die Ras-Proteine haben eine wichtige Relais-Funktion bei der Integration der Informationen, die von außen auf die Zelle einwirken. Die Ras-Proteine gehören zu einer großen Gruppe GTP-bindender Proteine, die sich in einem aktiven Zustand befinden, wenn sie GTP gebunden haben, und die inaktiv sind, wenn sie GDP gebunden haben. Durch Interaktion mit dem SOS-Protein wird das GDP-Molekül am Ras-Protein gegen ein GTP-Molekül ausgetauscht, und das Wachstumssignal in Richtung Zellkern weitergeben. Das Haupteffektormolekül der RasProteine ist die Serin/Threonin-Kinase Raf1. Weitere Effektormoleküle sind die Phosphatidylinositol-3-Kinase und der GuaninNukleotid-Austauschfaktor der GTPase Ral (RalGEF)[1]. Die Ras-Proteine sind ein gemeinsamer, integraler Bestandteil des Signaltransduktionswegs der Tyrosinkinase-Rezeptoren (Abb.1). Die Inaktivierung der Ras-Proteine stellt daher eine spezifische Möglichkeit dar, die Malignität der Tumore zu reduzieren. In den letzten Jahren wurden viele grundlegende Erkenntnisse über die spezifischen Eigenschaften der Ras-Proteine gewonnen. Diese bilden die Grundlage für die Entwicklung therapeutischer Strategien, die sich die Inaktivierung der Ras-Proteine zu Nutzen machen. Eine Möglichkeit, die Ras-Proteine zu inaktivieren, ergibt sich aus der Tatsache, dass die Ras-Proteine mit einem Farnesylrest gekoppelt werden müssen, mit dem sie an der Innenseite der Zellmembran verankert werden. Die Farnesylierung der Ras-Proteine geschieht enzymatisch durch eine FarnesylProtein-Transferase, die die C-terminale CAAX-Box erkennt[2]. Bei unseren ersten Versuche, die Farnesylierung der Ras-Proteine zu hemmen, wurde die Verfügbarkeit des Farnesylpyrophosphats in der Zelle durch Hemmung der Cholesterinbiosynthese reduziert. Dadurch werden jedoch auch andere Metabolite, wie Ubichinon, Isopentenyl-Adenin und Do- lichol den Zellen entzogen, was unkontrollierbare Nebenwirkungen zur Folge hat[3]. Es wurden in den letzten Jahren große Anstrengungen unternommen, spezifische Hemmstoffe für die Inaktivierung von RasProteinen zu entwickeln. Mit dem Farnesylpyrophosphatanalogen, α-Hydroxyfarnesylphosphonat, konnte die Übertragung des Farnesylrestes auf die Ras-Proteine verhindert werden[4]. Tetrapeptide mit der CAAXSequenz erwiesen sich in vitro als sehr wirkungsvolle Hemmstoffe der Farnesylierung von rekombinantem H-ras-Protein. Eine Vielzahl von Wissenschaftlern und pharmazeutischen Unternehmen haben auf der Basis der CAAX-Peptide sogenannte Peptidomimetika entwickelt. Diese weisen in ihrer räumlichen Struktur große Ähnlichkeiten zu den Tetrapeptiden auf, besitzen jedoch keine spaltbaren Peptidbindungen und sind weniger polar, so dass ihre Permeation durch die Zellmembran verbessert ist[5]. In einer umfangreichen Untersuchung konnte das Wachstum von 70 % aller untersuchten Tumorzelllinien mit einem Farnesyltransferaseinhibitor gehemmt werden. Auch viele Tumorzelllinien, die keine rasMutation tragen, reagierten empfindlich auf die Hemmung der Farnesyl-Protein-Transferase. Diese Tumorzelllinien besaßen Rezeptoren für Wachstumsfaktoren, die sie selbst produzieren. Durch diese autokrine Stimulation lagen die Ras-Proteine permanent in einem aktivierten Zustand vor[6]. Farnesyltransferase-Inhibitoren eignen sich deshalb auch zur Behandlung von Tumoren, die ihr Wachstum autokrin stimulieren. Abb. 1: Unterbrechung der Signaltransduktion der Tyrosinkinase-Rezeptoren durch Hemmung der RasFarnesylierung durch Farnesyltransferase-Inhibitoren (FTI). FPTase: Farnesyl-Protein-Transferase; GF: Wachstumsfaktor; PI3K: Phosphatidylinositol-3-Kinase, TF: Transkriptionsfaktor BIOspektrum · 3/03 · 9. Jahrgang Wissenschaft intern 287 Wir haben beim Neuroblastom, einem soliden Tumor des Kindesalters, der trotz intensivster zytostatischer Behandlung sehr schlechte Heilungschancen aufweist, die Wachstumsfaktoren identifiziert, die autokrin wirken. Durch Genexpressionsanalysen wurden IGF-II, bFGF und BDNF als autokrine Wachstumsfaktoren in Neuroblastomzelllinien identifiziert[7,8]. Die Signaltransduktion der Rezeptoren für IGF-II und BDNF wurde durch Inaktivierung der Ras-Proteine unterbrochen. Drei verschiedene Wege zur Hemmung der Farnesylierung wurden untersucht. Die Hemmung der Cholesterinbiosynthese durch Lovastatin bewirkte in verschiedenen Zellen eine Reduktion des N-ras-Membrangehalts zwischen 30% und 36 %[3]. Mit α-Hydroxyfarnesylphosphonat wurde eine Verminderung der Ras-Membranverankerung um 60 % erreicht[4]. Die Anwendung des Peptidomimetikums FTI-277 führte zu einer Abnahme der Ras-Farnesylierung um 40 %. Mit FTI-277 wurden die geringsten unspezifischen Nebenwirkungen in der Zellkultur beobachtet. Die Auswirkungen der Hemmung der Farnesyltransferase mit FTI-277 auf den Ras-Signaltransduktionsweg wurden untersucht. Die Aktivität der MAP-Kinase Erk2 war nach IGF-II-Stimulation in den hemmstoffbehandelten Zellen um 30–70% gegenüber den Kontrollzellen reduziert. Die Aktivität von Erk2 nach Stimulation mit BDNF war in den FTI-behandelten Zellen um 50–60% verringert[7,8]. Die Induktion des für die Neuroblastomzellen wichtigen early-response-Gens Nmyc durch BDNF war nach Inaktivierung der Ras-Proteine fast vollständig verhindert, nach IGF-II-Stimulation war sie nur um 25 % niedriger als in den Kontrollzellen[9]. Rainer Girgert Jahrgang 1954; 1976 – 1982 Biochemiestudium an der Eberhardt-KarlsUniversität in Tübingen, 1982 – 1984 Diplomarbeit am Max-Planck-Institut für Entwicklungsbiologie in Tübingen bei Prof. Dr. Alfred Gierer, 1985 – 1990 Promotion bei Prof. Dr. Dietrich Niethammer in der Pädiatrischen Hämatologie der Eberhardt-KarlsUniversität, Tübingen, 1991 – 2000 Leiter des onkologischen Labor der Kinderchirurgie von Prof. Dr. Paul Schweizer an der Eberhardt-KarlsUniversität, Tübingen; seit 2001 wissenschaftlicher Angestellter bei Prof. Rolf Kreienberg in der Universitätsfrauenklinik Ulm, November 2001 Habilitation für das Fach: Experimentelle Onkologie IGF-II verwendet möglicherweise noch einen weiteren Signaltransduktionsweg für die Induktion der N-myc-Expression[10]. In den FTI-behandelten Neuroblastomzellen wurden sehr starke morphologische Veränderung beobachtet, die bereits auftraten, bevor die Ras-Proteine um 50 % inaktiviert waren. Diese Beobachtung lässt darauf schließen, dass von der Hemmung der Farnesyltransferase noch weitere zelluläre Strukturen betroffen sind, die eine kürzere Halbwertzeit als die Ras-Proteine besitzen und für das maligne Wachstum ebenfalls notwendig sind. Durch eine Genbank-Recherche, sind über 300 Proteine identifiziert worden, die eine Erkennungssequenz für die Farnesyltransferase tragen. Die von uns beobachteten morphologischen Veränderungen der FTI-behandelten Zellen weisen auf die Rho-Proteine als eine weitere potentielle Zielstruktur für die Farnesyltransferaseinhibitoren hin. Einige Rho-Proteine werden farnesyliert und besitzen eine wichtige Funktion bei der Aufrechterhaltung der morphologischen Integrität der Zellen[11]. Diese Beobachtungen zeigen, dass der Wirkungsmechanismus der Farnesyltransferase-Inhibitoren über eine Inaktivierung der Ras-Proteine hinausgeht. Bei der Anwendung verschiedener Farnesyltransferase-Inhibitoren in Mausversuchen sind jedoch keine systemisch toxischen Wirkungen beschrieben worden. Bevor eine klinische Anwendung dieser neuen Therapeutika gegen Neuroblastome und andere Tumore vertretbar wird, bedarf es noch der Aufklärung des gesamten Wirkungsmechanismus der Farnesyltransferaseinhibitoren. Die Beobachtung, dass auch Tumorzellen, die keine ras-Mutation tragen, von Farnesyltransferaseinhibitoren gehemmt werden, wirft die Frage auf, wieso dann gesundes Gewebe unempfindlich gegen die FTI-Wirkung ist. Die K-ras-Proteine sind besonders resistent gegen Farnesyltransferaseinhibitoren, dennoch lässt sich das Wachstum von Colon- und Lungencarcinomzellen, die durch ein mutiertes K-ras-Gen transformiert sind, mit FTI’s hemmen. Die nächsten Jahre werden sicherlich den Durchbruch bei der Identifizierung des essentiellen Zielproteins der Farnesyltransferaseinhibitoren bringen. Obwohl der Wirkungsmechanismus der Farnesyltransferaseinhibitoren noch nicht vollständig aufgeklärt ist, sind bereits erste klinische Phase-I- und Phase-II-Studien durchgeführt worden. Bei den behandelten Patienten wurden erst bei sehr hohen Dosierungen myelotoxische und neurotoxische Nebenwirkungen beobachtet, so dass die Farnesyltransferaseinhibitoren ein günstigeres Wirkprofil zeigen als konventionelle Zytostatika[12]. Literatur [1] Bos, J.L. (1995): p21ras: an oncoprotein functioning in growth factor-induced signal transduction. Eur. J. Cancer. 31A: 1051–1054 [2] Goldstein, J.L., Brown, M.S., Stradley, S.J., Reiss, Y. & Gierasch, L.M. (1991): Nonfarnesylated tetrapeptide inhibitors of protein farnesyltransferase. J. Biol. Chem. 266: 15575–15578 [3] Girgert, R., Marini, P., Janessa, A., Bruchelt, G., Treuner, J. & Schweizer, P. (1994): Inhibition of membrane localization of ras-p21-protein in N-ras transformed tumor cells by Lovastatin. Oncology 51: 320–322 [4] Girgert, R., Hohnecker, A., Wittrock, J. & Schweizer, P. (1999): Inhibition of farnesyl-protein- transferase in neuroblastoma cells by α-hydroxyfarnesylphosphonate. Anticancer Res. 19: 2959–2962 [5] Sebti, S.M. & Hamilton, A.D. (2000): Farnesyltransferase and geranylgeranyltransferase I inhibitors and cancer therapy: lessons from mechanism and benchto-bedside translational studies. Oncogene 19: 6584–6593 [6] Sepp-Lorenzino, L., Ma, Z., Rands, E., Kohl, N.E., Gibbs, J.B., Oliff, A. & Rosen, N. (1995): A peptidomimetic inhibitor of farnesyl:protein transferase blocks the anchorage-dependent and -independent growth of human tumor cell lines. Cancer Res. 55: 5302–5309 [7] Wittrock, J., Schweizer, P. & Girgert, R. (2002): Induction of N-myc expression in neuroblastoma by autocrine IGF-II depends on farnesylated Ras. Potential application of farnesyltransferase inhibitors. Anticancer Res. 22: 4205–4210 [8] Girgert, R., Wittrock, J., Pfister, S. & Schweizer, P. (2003): Farnesyltransferase inhibitor FTI277 prevents autocrine growth stimulation of neuroblastoma by BDNF. J. Canc. Res. Clin. Oncol. (im Druck) [9] Schwäble, J., Wittrock, J., Schweizer, P. & Girgert, R. (1998): Detection of rare target genes on Northern-Blots with cDNA probes labeled by RT-PCR and simultaneous Digoxigenin incorporation. Anal. Biochem. 262:77–79 [10] Girgert, R., Wittrock, J. & Schweizer, P. (2001): Basical science in pediatric surgery. Neuroblastoma: Part II: Inhibition of progression. Eur. J. Ped. Surg. 11: 363–367 [11] Girgert, R., Janessa, A. & Schweizer, P. (2002): Activity of farnesyltransferase inhibitors against neuroblastoma cells depends on loss of adhesion. Onkologie 25 Suppl.4: 185 [12] Karp, J.E., Kaufmann, S.H., Adjei, A.A., Lancet, J.E., Wright, J.J. & End, D.W. (2001): Current status of clinical trials of farnesyltransferase inhibitors. Curr. Opin. Oncol. 13: 470–476 Korrespondenzadresse: Priv.-Doz. Dr. Rainer Girgert Universitätsfrauenklinik Ulm Prittwitzstraße 43 D-89075 Ulm Tel.: 0731-50033088 Fax: 0731-5026674 [email protected] BIOspektrum · 3/03 · 9. Jahrgang