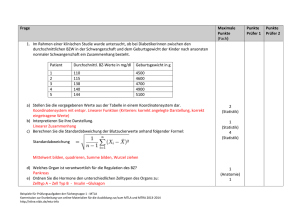
Molekulargenetische und biochemische
Werbung

Molekulargenetische und biochemische Untersuchungen zur Funktion und Struktur des Enzym IIMtl aus Escherichia coli K-12 Dissertation Zur Erlangung des Grades eines Doktors der Naturwissenschaften des Fachbereichs Biologie/Chemie der Universität Osnabrück Vorgelegt von Şevket Turgut Osnabrück 2003 Inhaltsverzeichnis I Inhaltsverzeichnis Inhaltsverzeichnis .......................................................................................... I Abbildungsverzeichnis .................................................................................. II Tabellenverzeichnis ..................................................................................... III Abkürzungsverzeichnis ................................................................................ IV I. Einleitung .............................................................................................1 II. Material und Methoden ....................................................................11 II.1. II.2. II.3. II.4. II.5. II.6. II.7. II.8. II.9. Bakterienstämme, Phagen, Plasmide und Oligonukleotide ....................... 11 Chemikalien, Isotope und Enzyme ......................................................... 19 Nährmedien ........................................................................................ 20 Anzuchtverfahren ................................................................................. 22 Herstellung von Zellextrakten ................................................................ 24 Genetische Techniken .......................................................................... 25 DNA-Techniken ................................................................................... 27 Chemische und biochemisch-analytische Techniken ................................. 34 Computerprogramme ........................................................................... 49 III. Ergebnisse .......................................................................................50 III.1. Selektion und Untersuchung entkoppelter Mutanten ....................... 50 III.1.1. Konstruktion von Stämmen zur Selektion entkoppelter Mutanten ......... 50 III.1.2. Entkoppelte Mutanten aus den Selektionen ....................................... 65 III.1.3. Entkoppelte Mutanten aus lokalisierter Mutagenese ........................... 69 III.1.4. Selektion von Suppressormutanten ................................................... 72 III.1.5. Charakterisierung der Mutanten ...................................................... 74 III.2. Topologieuntersuchungen am EIIMtl ................................................. 95 III.2.1. Konstruktion der erforderlichen Plasmide und Mutanten ..................... 96 III.2.2. Markierung der Einzel-Cystein MtlA-Mutanten ................................... 99 III.2.3. Reinigung und immunologischer Nachweis des MtlA-6His ............... 100 III.2.4. Ergebnisse der Biotinmaleimid-Markierungen ................................. 102 IV. Diskussion ......................................................................................107 IV.1. Für Transport und Phosphorylierung relevante AS im EIICMtl .......... 108 IV.1.1. Vergleich der Mutationen E218A, E218V und H256P ...................... 108 IV.1.2. Die AS 218 und 256 im Translokationsmodell ................................ 113 IV.2. Untersuchung der Sekundärstruktur des EIICMtl .............................. 121 IV.2.1. Mögliche Strukturen des EIICMtl ...................................................... 121 V. Zusammenfassung ..........................................................................133 VI. Literaturverzeichnis .......................................................................135 VII. Anhang .............................................................................................1 II Abbildungsverzeichnis Abbildungsverzeichnis I. Einleitung .............................................................................................1 Abb.I.1. Transport und Phosphorylierung eines Substrats über ein PTS ............... 4 Abb.I.2. Modell der Sekundärstruktur des IICMtl-Transporters aus E.coli .............. 7 Abb.I.3. Modell der essentiellen Strukturelemente des PTS Transport-Systems am Beispiel des EIIMtl ........................................................................ 9 II. Material und Methoden ....................................................................11 Abb.II.8.1. Substrataufnahme von D-Mtl ....................................................... 35 Abb.II.8.2. Schematische Darstellung eines Lineweaver-Burk-Diagramms ........ 37 Abb.II.8.3. Exemplarische Bindekurve ........................................................... 42 III. Ergebnisse .......................................................................................50 Abb.III.1.1. Wildtyp- und semisynthetischer Stoffwechselweg für den D-MtlAbbau in E.coli ......................................................................... 51 Abb.III.1.2. Schematische Darstellung der Konstruktion von F’lac::dalD ............. 52 Abb.III.1.3. Alternativer semisynthetischer Stoffwechselweg für den D-MtlAbbau in E.coli ......................................................................... 58 Abb.III.1.4. Sequenz des Gens mtlA aus E.coli K-12 ...................................... 67 Abb.III.1.5. mtlA-Plasmid pGJ9 ................................................................... 70 Abb.III.1.6. SDS-Gel der Vesikel nach der Überexpression ............................. 83 Abb.III.1.7. Phosphorylierungsaktivitäten der Vesikel mit 1mM [14C]-D-Mtl ....... 84 Abb.III.1.8. Phos.-Aktivitäten von H256P-Mutanten mit 500µM [14C]-D-Mtl ..... 85 Abb.III.1.9. KMapp-Wert Messungen des WT-Stamms LGS322/pGJ9 für D-Mtl ... 88 Abb.III.1.10. KMapp-Wert Messungen von LGS322/pGJT9-5 für D-Mtl ............... 88 Abb.III.1.11. KMapp-Wert Messungen von LGS322/pDSM1 für D-Mtl ................. 89 Abb.III.1.12. KMapp-Wert Messungen von LGS322/pDSM2 für D-Mtl ................. 89 Abb.III.1.13. Nichtlineare Kinetik der KMapp-Messung von LGS322/pDSM2 ........ 91 Abb.III.2.1. Abb.III.2.2. Abb.III.2.3. Abb.III.2.4. SDS-Gel einer MtlA(His)6-Reinigung ........................................... 100 SDS-Gel nach dem Proteintransfer auf Membran ........................ 101 Immunologischer Nachweis des MtlA(His)6 .................................. 101 Biotinmaleimidmarkierungen und Nachweise des MtlA(His)6 ........ 103 IV. Diskussion ......................................................................................107 Abb.IV.1. Abb.IV.2. Abb.IV.3. Abb.IV.4. Abb.IV.5. Abb.IV.6. Abb.IV.7. Abb.IV.8. Abb.IV.9. Interpretation der nichtlinearen Kinetik von LGS322/pDSM2 .......... 111 Vergleich der EIICMtl AS-Sequenzen aus E. coli und K. pneumoniae . 112 Vergleich der EIICMtl AS-Sequenzen verschiedener Stämme ............. 114 Bindung eines Phosphats bzw. Maleimids an ein Cystein ................ 122 Ergebnisse aus Sequenz- und Strukturuntersuchungen im Sekundärstrukturmodell des EIICMtl-Transporters aus E.coli ......... 123 Vergleich der postulierten transmembranen Helices von EIICMtl und EIICGlc ....................................................................... 126 TMHMM-Vorhersage der Topologien von EIICMtl und EIICGlc .......... 128 Mögliche 2D-Strukturen des IICMtl-Transporters aus E.coli .............. 130 Mögliche 3D-Strukturen des IICMtl-Transporters aus E.coli .............. 131 Tabellenverzeichnis III Tabellenverzeichnis II. Material und Methoden ....................................................................11 Tab.II.1.1. Tab.II.1.2. Tab.II.1.3. Tab.II.1.4. Tab.II.1.5. Tab.II.2. Tab.II.8.1. Tab.II.8.2. Bakterienstämme ......................................................................... 11 Phagen ....................................................................................... 12 Plasmide ..................................................................................... 13 Neukonstruierte und -isolierte Plasmide .......................................... 14 Oligonukleotide .......................................................................... 16 Häufig verwendete Puffer und Lösungen ......................................... 20 Zelltiter und Proteinmenge ............................................................ 34 Eingesetzte Zuckerkonzentrationen und dazugehörige cpm .............. 36 III. Ergebnisse .......................................................................................50 Tab.III.1.1. Tab.III.1.2. Tab.III.1.3. Tab.III.1.4. Tab.III.1.5. Tab.III.1.6. Tab.III.1.7. Tab.III.1.8. Tab.III.1.9. Tab.III.1.10. Tab.III.1.11. Tab.III.1.12. Tab.III.1.13. Tab.III.1.14. Tab.III.1.15. Tab.III.1.16. Tab.III.1.17. Tab.III.1.18. Tab.III.1.19. Tab.III.1.20. Tab.III.1.21. Tab.III.1.22. Tab.III.1.23. Plattentest zum Nachweis einer funktionellen DalD aus pHEXD ..... 54 Plattentest zum Nachweis einer funktionellen DalD aus pLSD101 . 55 Plattentest der Exkonjuganten aus der Kreuzung CSH36 /F’lac::dalD /pLSD101 /pPSO110 X S136∆ ................... 56 Plattentest des Selektionsstammes LTK31 .................................... 57 Plattentest LTK31-1 und LTK31-2 .............................................. 59 Plattenmarkertest LTK32 und LTK32-1 ....................................... 60 Ursprüngliche AtlR- und AtlS-Phänotypen ..................................... 61 Neu beobachtete AtlR- und AtlS-Phänotypen ................................ 62 Plattentest P1.∆(crr)::kan X LGS31 ........................................... 63 Entkoppelnde Mutationen aus LGS322-1 /pGJ9∆137 und LGS324-1 /pGJ9∆137 ...................................................... 68 Phänotypen der entkoppelten Mutanten ..................................... 69 Austausche bei pGAL3-1, pGAL3-2 und pGAL3-3 ...................... 71 Austausche bei pGAL3-4 und pGAL3-5 ..................................... 72 Mutationen aus LGS322 /pGJT9-3 und LGS322 /pGJT9-4 ......... 74 Markertest entkoppelnder Mutanten in LGS322 und LGS322-1 ... 76 Plattentest der Stämmen LGS322, LGS322-1 und LTK31-2 mit den „Suppressormutationen“ ............................................... 78 Zellerträge und Generationszeiten der Stämme LGS322 und LGS322-1 mit verschiedenen mtlA-Plasmiden ............................. 79 Mutanten aus dem Plasmid pMaHisMtlAPr ................................. 82 Gesamtproteinkonzentrationen der Vesikel ................................. 83 Spezifische Phosphorylierungsaktivitäten der Vesikel .................... 86 KMapp-Werte und V der mtlA-Mutanten für D-Mtl in LGS322 .......... 90 Stammabhängige Phänotypen von mtlA-Mutanten ...................... 92 Zusammengefasste Ergebnisse der EIIMtl-Mutanten im Stamm LGS322 .................................................................. 94 Tab.III.2.1. Plasmide der Reihen pMMX5-3x und pMMX5-4x .......................... 98 Tab.III.2.2. Aktivitäten der „Cys-Scanning“-MtlA-Derivate ............................... 98 Tab.III.2.3. Ergebnisse der Biotinmaleimid-Markierungen ............................. 104 IV Abkürzungsverzeichnis Abkürzungsverzeichnis α-Me-Glc AB Amp Atl AMHB1 B Biotinmaleimid bp CAA Cam Che cpm C-Quelle DMSO DOC DTT EI EII XY EDTA EK Fru α-Methyl-Glukose Antibiotikum Ampicillin D-Arabinitol Arginin/Methionin/Histidin/Thiamin-Lösung Bakterien „Nα-(3-maleimidylpropionyl) biocytin“ Basenpaare Casa Amino Acids Chloramphenicol Chemotaxisreaktion (auf Chemotaxisschwärmplatten) Counts (radioaktive Impulse) pro Minute Kohlenstoffquelle Dimethylsulfoxid Deoxycholic acid sodium salt monohydrate Dithiothreitol Enzym I der PTS Domäne X des Enzym II, spezifisch für Kohlenhydrat Y Ethylendiamintetraacetat Einzelkolonie D-Fruktose Gal Glc GLB Gly Gt D-Galaktose D(+)-Glucose Gel-Lade-Puffer („gel loading buffer“) Glycerin Generationszeit HPr Histidinprotein des PTS IPTG Isopropyl-β-D-Thiogalaktopyranosid Kan k.W. Kanamycin kein Wachstum Lac LKS D-Laktose Laborkultursammlung Abkürzungsverzeichnis Man McC MKS MM m.o.i. Mtl V D-Mannose MacConkey Multiple Klonierungsstelle Minimalmedium multiplicity of infection Mannitol n.b. n.g. n.m. n.z. nicht bestimmbar nicht getestet nicht messbar nicht zugänglich ODX Optische Dichte bei einer Wellenlänge von X nm PCR PEP PMSF PTS RT SDS Spc Stilben Str TEMED Tet Tp Polymerase-Kettenreaktion Phosphoenolpyruvat Phenylmethylsulfonylfluorid PEP-abhängiges Kohlenhydrat : Phosphotransferasesystem Raumtemperatur Natriumdodecylsulfat („Sodium dodecyl sulfat“) Spectinomycin “4-acetamido-4’-maleimidylstilbene-2,2’-disulfonic acid, disodium salt” Streptomycin N,N,N',N'-Tetramethylethylenediamine Tetracyklin Transport U ÜK ÜN UZ Units Übernachtkultur Über Nacht Ultra-Zentrifuge VM Vollmedium WB Wasserbad X-Gal Xul Xyl 5-bromo-4-chloro-indoyl-β-D-galaktosid D-Xylulose D-Xylose I. Einleitung 1 I. Einleitung I.1. Die Zelle Die Kompartimentierung von Reaktionsräumen war einer der ersten Schritte bei der Entstehung des Lebens. Dies wird sowohl bei Eukaryonten als auch bei Prokaryonten durch die Abgrenzung von Zellen von der Umwelt mit Hilfe der Zytoplasmamembran verwirklicht. In dieser Membran sorgen eine Lipid-Doppelschicht und Proteinmoleküle, die hauptsächlich durch nicht-kovalente, kooperative Wechselwirkungen zusammengehalten werden, nicht nur für die räumliche Trennung des Zellinneren von der Umgebung, sondern auch für eine selektive Permeabilität, welche den Zellen die Aufnahme und das Ausschleusen verschiedenster Substanzen ermöglicht. Die Nährstoffaufnahme und das Ausschleusen giftiger Substanzen sind essentielle Funktionen aber auch der bidirektionale Transfer von Proteinen, von DNA und von Signalmolekülen ist erforderlich. Hinzu kommen die Fähigkeiten, Signale aus der Umgebung zu erkennen und weiterzuleiten, Membran-assoziierte Reaktionen zu katalysieren und Zell-Zell Kontakte herzustellen. Zusätzlich zur Zytoplasmamembran findet man bei Bakterienzellen in der Regel noch die Zellwand, die allerdings für hydrophile Verbindungen wie Ionen, Vitamine, Kohlenhydrate, Aminosäuren und Peptide mit einem Molekulargewicht bis ca. 600 Dalton (Payne & Gilvarg, 1968; Decad & Nikaido, 1976) permeabel ist. Neben der zusätzlichen Stabilität erfüllt sie daher den Zweck eines Molekularsiebs, wobei die Substrattranslokation typischerweise durch erleichterte Diffusion über spezifische und unspezifische Porine erfolgt. Bei der Aufnahme über die Zytoplasmamembran gibt es nur in wenigen Fällen wie bei Glyzerin (Richey & Lin, 1972; Heller et al., 1980) oder langkettigen Fettsäuren (Overath et al., 1969; Frerman & Bennet, 1973; Maloy et al., 1980) Hinweise für passive erleichterte Diffusion über substratspezifische „Facilitatoren“. Der Grossteil an Substraten passiert die Zytoplasmamembran durch aktiven Transport. 2 I. Einleitung I.2. Aktive Transportsysteme Aktive Transportsysteme sind durch die Fähigkeit charakterisiert, unter Energieverbrauch entgegen eines Konzentrationsgefälles spezifische Substrate zu transportieren, wodurch z.B. Wachstum bei niedrigen Substratkonzentrationen ermöglicht wird. Bei Bakterien kann man drei Gruppen von aktiven Transportsystemen unterscheiden: Primäre Transportsysteme, zu denen die ABC-Transporter gehören, katalysieren die Aufnahme einer großen Zahl unterschiedlicher Substrate wie Oligopeptide, Aminosäuren, Kohlenhydrate und Vitamine (Übersicht bei Furlong, 1987), wobei Licht- oder chemische Energie in Konzentrationsgradienten aus Ionen oder gelösten Stoffen umgewandelt wird. Bei Bakterien ist die Abhängigkeit dieser Transportsysteme von einem periplasmatischen Bindeprotein, einer relativ hohen Substrataffinität und ATP als Energiequelle charakteristisch (Ames, 1986; Davidson, 2002). Systeme, die den Transport von Ionen ausschließlich mit der Energetisierung durch ATP betreiben, werden ebenfalls zu dieser Gruppe gezählt (Übersicht bei Pederson & Carafoli, 1987). Bei sekundären Transportsystemen ist die Translokation des Substrates mit dem Fluss von Ionen (H+, Na+, K+) entlang ihres elektrochemischen Gradienten gekoppelt (Mitchell, 1963; West, 1970). Dazu gehören Antiportsysteme, welche Substrat und Ionen in entgegengesetzter Richtung transportieren (z.B. Tetrazykline/H+Antiporter, TetA(C), Sutcliffe, 1978; TetA(A), Waters et al., 1983; Griffith et al., 1994). Die Laktose-Permease aus E. coli (West, 1970; West & Mitchell, 1973; Kaback, 1990; King et al., 1991) transportiert Substrat und Ionen in die gleiche Richtung und gehört damit zu den Symportsystemen. Charakteristisch für diese Gruppe ist die meist aus 12 transmembranen α-Helices bestehende Topologie. Beiden genannten Transportsystemen ist gemeinsam, dass das Substrat unmodifiziert in die Zelle gelangt. Bei dem Transport über ein PTS (PEP-abhängiges Kohlenhydrat : Phosphotransferasesystem) gelangt das Substrat phosphoryliert in die Zelle. Allerdings ist für den Transport des Substrats eine Phosphorylierung nicht zwingend notwendig. Vorangegangene Arbeiten sowie die vorliegende konnten zeigen, dass die chemische Modifikation erst nach der Translokation erfolgt und der Transport von der Phosphorylierung entkoppelt werden kann (Lolkema et al., 1990; Lolkema et al., 1991a; Postma et al., 1993; Lengeler & Jahreis, 1996; Otte, 2000). Der eigentliche I. Einleitung 3 Transportschritt zeigt Gemeinsamkeiten zwischen den PTSs, den ABC-Transportern sowie den sekundären Uniportern („Facilitatoren“; Lengeler et al., 1994). I.3. PTS vermittelter Transport in Bakterien Kundig, Gosh und Roseman (Kundig et al. 1964) konnten 1964 erstmals PTSs biochemisch beschreiben. Weiterführende genetische (Murphy & Rosenblum, 1964a, b; Egan & Morse, 1966; Tanaka & Lin, 1967) und biochemische Untersuchungen (Kundig et al., 1966) zeigten bei einer Vielzahl von Kohlenhydraten die Beteiligung dieser Systeme. Die der Phosphorylierung von Kohlenhydraten zu Grunde liegende allgemeine Gesamtreaktion lautet: Als weitere Besonderheit der PTSs wird deutlich, dass nicht ATP sondern Phosphonenolpyruvat (PEP) als Phophorylgruppendonor dient. PEP steht dabei am Anfang einer Phosphorylierungskaskade, an der folgend eine lösliche Histidinproteinkinase EnzymI (EI; Gen ptsI) und das Histidin-Protein (HPr; Gen ptsH) beteiligt sind. Die Gene ptsH und ptsI (und crr; Genprodukt EIIACrr) bilden ein Operon (De Reuse & Danchin, 1988). Sie werden mit Ausnahme des Fruktose-PTS von allen PTSs benötigt und daher zu den allgemeinen, substratunspezifischen Komponenten gezählt. Die Übertragung der Phosphatgruppe vom PEP auf EI erfolgt durch Autophosphorylierung des EI-Dimers an einem Histidin-Rest (Misset et al., 1980; Weigel et al., 1982), welches seinerseits die Phosphorylgruppe auf einen Histidin-Rest des HPr überträgt. Diesen allgemeinen PTS Komponenten stehen die Kohlenhydrat-spezifischen EnzymeII (EIIs) gegenüber, mit denen das P~HPr in der Folge der Phosphorylierungskaskade reagiert (Abb. I.1.). Die bakteriellen PTSs sind aber nicht nur Kohlenhydrat-Transportsysteme, vielmehr sind sie Teil eines zentralen Signaltransduktionssystems mit wichtigen regulatorischen Funktionen, die sich über Katabolitenrepression, Induktorausschluss, Chemotaxis und globale Regulation erstrecken (Postma et al., 1993; Lengeler & Jahreis, 1996). Bisher sind bei E. coli K-12 über 20 verschiedene PTSs beschrieben, die eine Vielzahl unterschiedlicher Substrate in die Zelle transportieren (Saier, 1993; Lengeler et al., 1994; Postma et al., 1996; Tchieu et al., 2001). 4 I. Einleitung Abb. I.1. Transport und Phosphorylierung eines Substrates über ein PTS In der Abbildung sind die biochemischen Reaktionen dargestellt, die an der Gruppentranslokation während des Transports und der Phosphorylierung eines Substrates über ein PTS beteiligt sind. Dargestellt sind ein membrangebundenes, substratspezifisches EnzymII, mit den speziellen Domänen IIC, IIB, IIA, sowie die allgemeinen PTSKomponenten, EnzymI und HPr, mit den entsprechenden Phosphorylierungsstellen (nach Lengeler & Jahreis, 1996). I.4. Strukturelle und funktionelle Domänen der EII-Permeasen Auf der Grundlage von Sequenzvergleichen und Komplementationsstudien lassen sich die Permeasen in fünf genetische Familien einteilen (Lengeler et al., 1994; Lengeler & Jahreis, 1996). Ihnen liegt ein modularer Aufbau zu Grunde, der die drei funktionell autonomen Domänen IIA, IIB und IIC (bzw. IIC und IID bei der Mannose Familie) umfasst. Die Expression der EIIBMtl-Domäne von E. coli zeigte, dass EIIBMtl als enzymatisch aktives Protein existieren kann und die in vitro Phosphorylierungsaktivität zusammen mit EIIAMtl und EIICMtl wiederherstellen kann (Robillard et al., 1993). Grundlegende Arbeiten hatte dabei vorher den einzelnen Domänen eine biochemische Funktion zugewiesen (Grisafi et al., 1989; Van Weeghel et al., 1991a; Van Weeghel et al., 1991b). In den jeweiligen Familien sind die Domänen in unterschiedlicher Reihenfolge angeordnet und können frei oder fusioniert bzw. durch „Linker“ verbunden vorliegen. Einander entsprechende Domänen aus einer PTS-Familie können sich funktionell ersetzen. Die funktionelle Abhängigkeit des EIIScr von Crr (Lengeler et al., 1982) sowie die Wiederherstellung des Glc+-Phänotyps in einer Crr--Mutante durch Synthese eines intakten EIINag (Vogler et al., 1988) gehören zu den ersten Beispielen funktioneller Übereinstimmung der strukturellen Domänen der PTS- I. Einleitung 5 Transportkomponenten. Ein Grund dafür ist, dass innerhalb einer Familie die strukturell und katalytisch relevanten Reste konserviert sind. Die Ähnlichkeit (% identische Aminosäuren) innerhalb einer Familie in den relevanten Regionen reicht von nahezu identisch (>98%) bis zu wenigen, lokal begrenzten Motiven. In funktionellen Bereichen der einzelnen Domänen können die hochkonservierten Bereiche teilweise durch ein bis zwei Aminosäuren definiert werden (Lengeler, 1990; Lengeler et al., 1994). Die aus etwa 100 AS bestehende hydrophile IIA-Domäne enthält einen konservierten HistidinRest, der durch P~HPr phosphoryliert werden kann. Ein konserviertes Cystein wird in der etwa gleichlangen hydrophilen IIB-Domäne von IIA phosphoryliert. Mit etwa 350 AS bildet die membrangebundene IIC-Domäne den größten Teil des EnzymII-Proteins. Der hydrophobe Bereich dieser Domäne kann 6-8 transmembrane Helices ausbilden, die eine große hydrophobe, zytoplasmatische Schleife flankieren. In dieser Schleife findet man oft ein GXXE-Motiv, wobei der Glutaminsäure-Rest (E) in allen bisher bekannten EII konserviert ist und in vergleichbarem Abstand immer durch einen hochkonservierten hydrophilen Rest (His, Asn oder Gln) begleitet wird. Im Gegensatz zu der variablen modularen Anordnung der Domänen untereinander, weist eine auffällig hohe Konservierung bestimmter Strukturen und Motive auf relevante Bereiche in dem Enzym hin (Lengeler & Jahreis, 1996). Es ist offensichtlich, dass man sich bei der Beantwortung der vielfältigen Fragen nach Substratspezifität, Mechanismus und Kopplung des PTSabhängigen Transports auf diese konservierten Bereiche konzentrieren sollte. I.5. Der EIIMtl-Transporter Das E. coli EIIMtl besteht aus einer einzigen Polypeptidkette von 637 Aminosäuren Länge, die durch das mtlA-Gen (Solomon & Lin, 1972; Jacobson et al., 1979; Lee & Saier, 1983) kodiert wird. Mit den Genen mtlD (Mtl 1-P-Dehydrogenase; Davis et al., 1988; Jiang et al., 1990) und mtlR (Repressor; Figge et al., 1994) bildet das mtlA-Gen ein Operon (Genreihenfolge mtlA, D, R), welches bei 80,7min auf dem E. coliChromosom kartiert (Solomon & Lin, 1972; Lengeler, 1975a). Nach Transport und Phosphorylierung des D-Mannitols (Mtl) durch das EIIMtl wird Mtl-1-P durch die NADabhängige Mtl-1-P-Dehydrogenase zu D-Fruktose-6-P umgewandelt (Wolff & Kaplan, 1956; Klungsöyr, 1966; Solomon & Lin, 1972). Die Regulation des Systems unterliegt dem Mtl-Repressor (MtlR), der molekulare Induktor ist ausschließlich Mtl (Tanaka et al., 1967; Lengeler & Steinberger, 1978). 6 I. Einleitung Das EIIMtl-Protein lässt sich in drei funktionelle Domänen unterteilen (Abb. I.1.). Die meist hydrophoben aminoterminalen AS der Position 1 bis 334 bilden die membranüberspannende EIIC-Domäne (Abb. I.2.) mit der Substratbindestelle und der Transporterfunktion (Grisafi et al., 1989; Lolkema et al., 1990; Briggs et al., 1992). Das für die Übertragung der Phosphorylgruppe essentielle Cys-384 (Pas & Robillard, 1988a; Pas & Robillard, 1988b; Grisafi et al., 1989; Pas et al., 1991) befindet sich in der folgenden hydrophilen EIIB-Domäne (AS 335 bis 457). Die AS 458 bis 637 gehören zur EIIA-Domäne, die ihrerseits das in der Phosphorylierungskette essentielle konservierte His-554 enthält (Pas & Robillard, 1988b; Pas et al., 1991; van Weeghel et al., 1991b, 1991c). Die exakte Topologie der EIIC-Domäne wurde bisher noch nicht bestimmt. Zahlreiche Untersuchungen deuten darauf hin, dass die funktionell aktive Form des Proteins eine dimere Konformation aufweist (Übersicht bei Jacobson, 1992; Boer et al., 1994; Boer et al., 1996; Koning et al., 1999; Heuberger et al., 2002). Hinsichtlich der Sekundärstruktur wurde anhand von Hydropathiestudien ein Model mit sieben transmembranen Segmente und einem periplasmatischen N-Terminus vorgeschlagen (Lee & Saier, 1983). Dagegen führten die Ergebnisse von MtlA-PhoA-Fusionsstudien zu einem Sekundärstrukturmodell mit sechs transmembranen α-Helices und einem zytoplasmatischen N-Terminus (Sugiyama et al., 1991). Ergebnisse aus TryptophanFluoreszenzstudien unterstützen dieses Modell (Dijkstra et al., 1996). Weitere Untersuchungen, deren Kern Hydrophobizitätsstudien und die Ermittlung einer Konsensussequenz aus den EIICMtl verschiedener Stämme bildete, führten unter der Berücksichtigung verschiedener Methoden zur Vorhersage von Membranproteinstrukturen (Kyte & Doolittle, 1982; Engelman et al., 1986; von Heijne & Gavel, 1988; von Heijne, 1992) zu einem EIIC-Modell aus 6 transmembranen Helices, drei kurzen periplasmatischen und zwei großen zytoplasmatischen Schleifen sowie einem zytoplasmatischen N-Terminus (Lengeler et al., 1994; Lengeler & Jahreis, 1996). Dieses Modell ist in Abbildung I.2., basierend auf einem Vergleich der aktiven und näher charakterisierten EIICMtl-Aminosäueresequenzen von E. coli (Lee & Saier, 1983), S. carnosus (Fischer & Hengstenberg, 1992; Reiche et al., 1996), K. pneumoniae (Otte & Lengeler, 2001), S. mutans (Honeyman & Curtiss, 2000) und B. stearothermophilus (Henstra et al. 1996), dargestellt. I. Einleitung 7 Abb. I.2. Modell der Sekundärstruktur des IICMtl-Transporters aus E. coli (nach Sugiyama et al., 1991 und Lengeler et al., 1994; verändert) Dem Modell liegen die Ergebnisse aus Sequenzvergleichen und mtlA-phoA Fusionsstudien zu Grunde. Periplasmatische und zytoplasmatische Schleifen sind durch arabische Zahlen gekennzeichnet. Die postulierten transmembranen α-Helices sind durch römische Zahlen gekennzeichnet. In den verglichenen Sequenzen identische Proteine sind schwarz unterlegt. Blaue Kreise zeigen Lücken bzw. Insertionen in den verglichenen AS-Sequenzen an. Wichtige am Transportmechanismus beteiligte AS sind rot hervorgehoben. Leere Kreise stellen Aminosäuresequenzen unbestimmter Länge dar. Weitere Erläuterungen sind im Text zu finden. Bereits die Betrachtung der Sequenzvergleiche lässt eine besondere Funktion der Schleife 5 aufgrund der sehr hohen Konservierung der AS vermuten. Die Ähnlichkeit liegt in dem 87 AS langen Bereich zwischen den vermuteten Helices IV und V bei 55,2% identischen AS, während die Konservierung über das gesamte EIICMtl (AS 1-344) bei 8 I. Einleitung 38,4% liegt. Zudem befinden sich in der Schleife 5 mehrere konservierte Aminosäuren die nachweislich eine wichtige Funktion beim Transportprozess haben. Dazu gehört im EIIMtl das His195, welches essentiell für die Substratbindung zu sein scheint (Weng & Jacobson, 1993). Darüber hinaus findet man in der Schleife 5 bei AS 254-257 mit der Aminosäure-Folge -G-I-H-E- das in EII-Transportern hochkonservierte GXXE-Motiv (Lengeler et al., 1990). Mutationen in diesem Bereich führten zum Verlust von Bindungs- und Phosphorylierungsaktivität (Saraceni-Richards & Jacobson, 1997), zum Verlust der Transportaktivität bei verbleibender Phosphorylierungsaktivität (Manayan et al., 1988) oder zu einer Entkopplung von Transport und Phosphorylierung (Otte, 2000; Turgut, diese Arbeit). Diese Ergebnisse deuten auf eine maßgebliche Beteiligung der Aminosäuren des GIHE-Motivs an den komplexen Transport- und Phosphorylierungsprozessen hin. Allerdings führten auch Austausche an dem konservierten E218 (E218A, E218V) des EIICMtl zu einer funktionellen Entkopplung von Transport und Phosphorylierung (Klawitter, 1992; Scholle, 1993; Heuel, 1997; Otte, 2000; Turgut, diese Arbeit), womit die gesamte Schleife 5 eine zentrale Struktur des gekoppelten Transportprozesses zu sein scheint. Da dieser Prozess neben Transport und Phosphorylierung auch die Bindung des Substrates beinhaltet, würde die Lokalisation der oben genannten wichtigen Aminosäuren in einer zytoplasmatischen Schleife widersinnig erscheinen. Überträgt man ein 1993 von Buhr & Erni für die Struktur vom EIIGlc vorgeschlagenes Modell auf das EIIMtl, so würde ein Teil der Schleife 5 zu einem transmembranen Bereich werden und das E257 des GIHE-Motivs im Periplasma liegen (Lengeler et al., 1994). Allerdings gibt es auch für das von Lengeler vorgeschlagene Sekundärstrukturmodell vom EIICMtl (Lengeler et al., 1994; Lengeler & Jahreis, 1996) ein dreidimensionales Modell, das den Ergebnissen aus Sequenzanalysen, Fusionsstudien und Hydrophobizitätstests entspricht. Nach diesem Modell von Lengeler (Lengeler, 1990; Lengeler et al., 1994) faltet sich die Schleife 5 in das Innere des EIICMtl-Transporters und bildet ein hydrophiles, katalytisches Zentrum (Abb. I.3.). Da eine genaue Kenntnis der Struktur des EIICMtl einen wichtigen Fortschritt im Verständnis der Funktion des Transportmechanismus bedeuten würde, wurde in dieser Arbeit versucht, mit weiteren Experimenten zusätzliche Informationen zur Topologie zu erhalten. Mit der gezielten Markierung von Cysteinen („Cystein-Scanning“) sollte vor allem die relative Lage der offensichtlich funktionell sehr bedeutsamen Schleife 5 ermittelt werden. I. Einleitung 9 Abb. I.3. Modell der essentiellen Strukturelemente des PTS Transport-Systems am Beispiel des EIIMtl (nach Lengeler, 1990) Es sind die allgemeinen Komponenten der PTS (EnzymI, HPr) sowie das substratspezifische IIMtl aus E. coli schematisch dargestellt. Für EIIC werden sechs transmembrane α-Helices mit einer Schleife 5 postuliert, die sich in den von den α-Helices gebildeten Kanal hineinfaltet. In dem hydrophilen Kern befinden sich die für den Transport essentiellen Aminosäuren (siehe Text). Aber auch die Selektion von Mutationen, die den Transport entkoppeln, kann für den Transport wichtige Aminosäuren identifizieren. Zusätzlich isolierte Suppressormutanten könnten zudem noch Informationen über die relative Lage einzelner Aminosäuren zueinander liefern. Wie bereits erwähnt, wurden schon einige Mutationen charakterisiert, die zur Entkopplung des Transports von der Phosphorylierung führten (Ruijter et al., 1992; Klawitter, 1992; Scholle, 1993; Heuel, 1997; Otte, 2000; Turgut, diese Arbeit). Ergänzend konnte gezeigt werden, dass das Vorhandensein der EIIC-Domäne ausreicht, um einen funktionellen Transporter mit intakter Substratbindestelle zu bilden (Grisafi et al., 1989; Briggs et al., 1992; Lolkema et al., 1990). Beide Beobachtungen entsprechen der gängigen Theorie, dass es sich beim Transport und der Phosphorylierung des Substrats über ein EII um zwei getrennte, nacheinander ablaufende Reaktionen handelt, die nicht strikt gekoppelt sind (Lolkema et al., 1990; Lolkema et al., 1991a; Postma et al., 1993; Lengeler & Jahreis, 1996). Niedrigaffine Substrate wie D-Arabinitol können auch ohne gleichzeitige Phosphorylierung durch das EIIMtl aufgenommen werden (Lengeler et al., 1994). So 10 I. Einleitung konnte nach dem Transport des niedrigaffinen Substrates D-Glucitol über EIIMtl sowie des hochaffinen Substrates D-Mannitol über ein mutiertes EIIMtl, intrazellulär freies Substrat nachgewiesen werden (Otte, 2000). Somit scheint der Grad der Kopplung von der Affinität der Bindestelle zum Substrat abhängig zu sein. Hochaffine Substrate werden im Wildtyp-EII so stark gekoppelt transportiert, dass ein Wachstum auf unphosphoryliertem Substrat nicht möglich ist (Postma & Stock, 1980). Die erleichterte Diffusion von niedrigaffinen Substraten wurde dagegen für die Aufnahme von Galaktose über EIIGlc (Kornberg & Riordan, 1976), für Glukose über EIIBgl (Schnetz et al., 1987) oder EIIScr (Schmid, 1988) sowie für D-Atl und Xyl über EIIGat (Reiner, 1977) beschrieben. Demnach vermitteln die entkoppelnden Mutationen eine Reduktion der Affinität der zum Substrat. Durch diesen strukturellen oder funktionellen Effekt arbeitet der Transporter als „Facilitator“ und katalysiert in einem energie-unabhängigen Prozess die erleichterte Diffusion des Substrats über die Membran. Das Hauptsubstrat verhält sich bei den veränderten Proteinen somit wie ein Nebensubstrat. Ein umgekehrt proportionales Verhältnis zwischen Affinitätskonstante und Flussrate ist denkbar (Lengeler et al., 1994; Lengeler & Jahreis, 1996), zumal entkoppelte EII einen teilweise drastisch erhöhten KM-Wert haben (Ruijter et al., 1991; Ruijter et al., 1992; Scholle, 1993; Lengeler et al., 1994; Otte, 2000). Neben den direkten strukturellen Untersuchungen mittels „Cystein-Scanning“ sollten in dieser Arbeit weitere EIIMtl-Varianten isoliert und charakterisiert werden, die den Transport von Mannitol entkoppeln. Durch die Konstruktion neuer Selektionssysteme wurde eine Selektion mit einem vollständigen EIIMtl (EIIMtlABC) ermöglicht. Darüber hinaus wurde die Selektion von Suppressormutationen durchgeführt. Durch die Kombination unterschiedlicher Mutationen mit Hilfe gezielter Mutagenese wurde versucht, die erhaltenen Mutanten weiter zu charakterisieren. Bei der Charakterisierung wurden verschiedene genetische und biochemische Methoden angewendet. II. Material und Methoden 11 II. Material und Methoden II.1. Bakterienstämme, Phagen, Plasmide und Oligonukleotide In den folgenden Tabellen (II.1.1. - II.1.5.) sind die in dieser Arbeit verwendeten Bakterienstämme, Phagen, Plasmide und Oligonukleotide beschrieben. Die Nomenklatur richtet sich nach Berlyn et al. (1996). Abweichend hiervon wurden die Gene der PTS-abhängigen Stoffwechselwege nach Postma et al. (1996) benannt. Tab. II.1.1. Bakterienstämme STAMM RELEVANTER GENO-/PHÄNOTYP REFERENZ Escherichia coli K-12 Derivate BMH71-18 thi-1 supE44 ∆(lac-proAB) [mutS::Tn10] /F‘ [proA+B+ lacIq Z∆M15] CSH36 /F’ thi-1 ∆(lac-pro) supE44 /F’lac CSH36-1 CSH36 /F’lac::TnLSD101 (dalD dalK‘) diese Arbeit HB101 F- ara-14 galK2 hsdS20 lacY1 mtlA1 proA2 recA13 rpsL20 supE44 thi-1 xyl-5 Leu- Thr- Boyer et al. (1969) JM109 thi-1 gutD::Tn10 ∆(lac-proA,B) recA1 endA1 gyrA1,96 hsdR17 supE44 relA1 /F’ [traD36 proA+B+ lacIq Z∆M15] Yanisch-Perron et al. (1985) JM109 (λDE3) JM109 int ::lacUV5- Yanisch-Perron et al. (1985) JWL191 F- thi-1 metB1 argG6 hisG1 galT6 galP63 lacY1 Mak0 malT1 ∆(mtlAP,O50) gatR49 gutAP,O49 ptsI191 rpsL104 supE44 tonA2 Lengeler (1980) JWL300 JWL191 Mak+ LGS31 F- thi-1 metB1 argG6 hisG1 galT6 lacY1 malT1 ∆(mtlAPA‘) MtlD+ gatR49 gatA50 gutAP,O49 gutA50 rpsL104 supE44 tonA2 [λDE3 (BamHI) Wallace et al. (1981); Kramer et al. (1984) Miller (1972) T7gene1.0] Aulkemeyer et al. (1991) Scholle (1993) 12 II. Material und Methoden Tab. II.1.1. Bakterienstämme (Fortsetzung) STAMM RELEVANTER GENO-/PHÄNOTYP REFERENZ LGS322 LGS31 ∆(gutR’MDBAP,O-recA) Scholle (1993) LGS324 LGS322 Atlr diese Arbeit LGS31-1 LGS31 /F‘lac::dalD diese Arbeit LGS322-1 LGS322 /F‘lac::dalD diese Arbeit LGS324-1 LGS323 /F‘lac::dalD diese Arbeit LLR103 F- thi-1 metB1 argG6 hisG1 galT6 lacY1 ∆(mtlAP,O50) gatR49 gutAP,O49 ∆(crr)::kan rpsL104 supE44 LGT31 LGS31 ∆(crr)::kann diese Arbeit LTK31 LGS31 ∆(ptsH ptsI crr)::kan diese Arbeit LTK31-1 LTK31 /F‘lac::dalD diese Arbeit LTK31-2 LTK31 /F‘lac::dalD /pUR404 diese Arbeit LTK32 LTK31 Mak+ diese Arbeit LTK32-1 LTK32 /F‘lac::dalD diese Arbeit S136 F- lac-85 malA1 mel-2 rpsL Mtl- S136-3 S136 ∆(ptsH ptsI crr)::kan STL136 S136-3 /F‘lac::dalD TP2811 F- xyl-5 argH1 ∆lacX74 aroB ilvA ∆(ptsH ptsI crr)::kann Lévy et al. (1990) XL1 Blue thi-1 recA1 endA1 gyrA96 hsdR17 supE44 relA1 /F‘lac [proAB lacIqZ∆M15 Tn10 (Tetr)] Bullock et al. (1987) Lux (1995) Schmitt (1968) Schmid (LKS) diese Arbeit Tab. II.1.2. Phagen Phagen Herkunft P1.kc Lennox (1955) R408 Dotto et al. (1981) II. Material und Methoden 13 Tab. II.1.3. Plasmide Aminosäureaustausche in WT-Proteinen sind in den Ein-Buchstaben- Abkürzungen der Aminosäuren sowie der numerischen Position der Aminosäure im Protein dargestellt (Bsp.: E218A = Glutaminsäure an Position 218 mit Alanin substituiert). Plasmide Relevanter Geno-/Phänotyp Referenz pALTER-1 Tcr Mutagenese-Vektor pGHL3 Apr dalD+ K+ T+ in pACYC177 pGJ9 Cmr pACYC184 mtlA+ (2,1kb SalI/EcoRV) Grisafi et al. (1989) pGJ9∆137 pGJ9 mtlA’ ∆137AS Grisafi et al. (1989) pHEX3/5 Cmr lacZP α-lacZ, Klonierungsvektoren (MCS aus pBlueskript KS+; SK+) Heuel (1997) pJOE637 Apr Tcr scrK+Y+A+B+R+ Schmid et al. (1988) pPSO110 Cmr Tcr tnpA Schmid (LKS) pSOL300 pHEX3 mtlAKAY2026 Otte (2000) pSOL313 pSOL300 mtlA E218A Otte (2000) pTIM101 Apr Scr, In vivo Klonierungsvektor pUR404 Tcr scrK+Y+A+B+R∆ pMaHisMtlAPr Überexpressionsvektor 6His-mtlA Smith (1985) Heilenmann, unveröffentlicht Zeppenfeld et al. (2000) Schmid et al. (1982) Montfort et al. (2001) 14 II. Material und Methoden Tab. II.1.4. Neukonstruierte und -isolierte Plasmide Bei den Eigenschaften der Plasmidserie pMMX5-x wurde zur Veranschaulichung der Mutation die Veränderung der vier WT-Cysteine von MtlA in eckigen Klammern angegeben: [CCCC] = [CAS110 CAS320 CAS384 CAS571]. Plasmid Eigenschaften pGAL3 pALTER-1 mtlA‘ pGAL3-1 pALTER-1 mtlA‘ E218A pGAL3-2 pALTER-1 mtlA‘ E218V pGAL3-3 pALTER-1 mtlA‘ H256P pGAL3-4 pALTER-1 mtlA‘ E218A, L229Q, H256P pGAL3-5 pALTER-1 mtlA‘ E218V, H256P pGJ9dEA pGJ9∆137 E218A (Selektion) pGJ9dEV pGJ9∆137 E218V (Selektion) pGJ9dHP pGJ9∆137 H256P (Selektion) pGJ9dHindIII pGJ9 ohne HindIII-Fragment in mtlA pGJT9-1 pGJ9 E218A (ortsspez. Mut.) pGJT9-1d pGJT9-1 mtlA‘ (∆ SnaBI-ClaI -Fragment) pGJT9-2 pGJ9 E218V (ortsspez. Mut.) pGJT9-2d pGJT9-2 mtlA‘ (∆ SnaBI-ClaI -Fragment) pGJT9-3 pGJ9 H256P (ortsspez. Mut.) pGJT9-3d pGJT9-3 mtlA‘ (∆ SnaBI-ClaI -Fragment) pGJT9-4 pGJ9 E218A, L229Q, H256P (ortsspez. Mut.) pGJT9-4d pGJT9-4 mtlA‘ (∆ SnaBI-ClaI -Fragment) pGJT9-5 pGJ9 E218V, H256P (ortsspez. Mut.) pGJT9-5d pGJT9-5 mtlA‘ (∆ SnaBI-ClaI -Fragment) pGJT9-6 pGJ9 H256A (ortsspez. Mut.) pDSM1 pGJT9-3 P256Q pDSM2 pGJT9-3 P256S pDSM3 pGJT9-4 P256A pDSM4 pGJT9-4 P256Q Beschreibung III.1.3.1. Seiten 69-71 III.1.2.1. S. 67-69 III.1.3.2. S. 72 III.1.5.3. S. 87 III.1.4.1. S. 73-74 II. Material und Methoden 15 Tab. II.1.4. Neukonstruierte und -isolierte Plasmide (Fortsetzung) Plasmid Eigenschaften Beschreibung pHEX5d pHEX5 ∆HindIII-Erkennungssequenz III.2.1.1. S. 96 pHEXD pHEX3 dalD dalK‘ III.1.1.1. S. 53 pLSD101 pTIM101 dalD dalK‘ III.1.1.1. S. 54 pMMX5 pHEX5d mtlA-6xHis [CCCC] pMMX5-1 pMMX5 mtlA-6xHis C320A [CACC] pMMX5-2 pMMX5-1 C110S [SACC] pMMX5-3 pMMX5-2 C571S [SACS] pMMX5-4 pMMX5-3 C384S [SASS] pMMX5-31 pMMX5-3 [SACS] S3C pMMX5-32 pMMX5-3 [SACS] S110C pMMX5-33 pMMX5-3 [SACS] S158C pMMX5-34 pMMX5-3 [SACS] S199C pMMX5-35 pMMX5-3 [SACS] S212C pMMX5-36 pMMX5-3 [SACS] S242C pMMX5-37 pMMX5-3 [SACS] S299C pMMX5-41 pMMX5-4 [SASS] S3C pMMX5-42 pMMX5-4 [SASS] S110C pMMX5-43 pMMX5-4 [SASS] S158C pMMX5-44 pMMX5-4 [SASS] S199C pMMX5-45 pMMX5-4 [SASS] S212C pMMX5-46 pMMX5-4 [SASS] S242C pMMX5-47 pMMX5-4 [SASS] S299C pMMX5-4HP pMMX5-4 H256P III.2.1.3. S.99 pMMX5-EA pMMX5 mtlA E218A III.1.5.3. S. 92 pRUG1 pMaHisMtlAPr mtlA E218A pRUG2 pMaHisMtlAPr mtlA E218V pRUG3 pMaHisMtlAPr mtlA H256P pRUG4 pMaHisMtlAPr mtlA E218A H256P III.2.1.1. S. 96-97 III.2.1.2. / III.2.1.3. S. 97-99 III.1.5.2. S. 81-82 16 II. Material und Methoden Tab. II.1.4. Neukonstruierte und -isolierte Plasmide (Fortsetzung) Plasmid Eigenschaften Beschreibung pRUG5 pMaHisMtlAPr mtlA E218A H256P pRUG6 pMaHisMtlAPr mtlA H256A pRUG7 pMaHisMtlAPr mtlA H256Q pRUG8 pMaHisMtlAPr mtlA H256S F‘lac::dalD F’lac::TnLSD101 dalD ScR III.1.5.2. S. 81-82 III.1.1.1. S. 53 Tab. II.1.5. Oligonukleotide In den Primern zur Mutagenese sind die Mutageneseziele farblich hervorgehoben. Rot bezeichnet das Triplet der mutagenisierten Aminosäure. Bei der HisTag-Konstruktion ist das Stop-Kodon dunkelrot, die Erkennungssequenzen für Restriktionsenzyme blau und die Kodone für die sechs His blaugrün gefärbt. Primer Sequenz Beschreibung mtlA-A-Cy5 5‘- ACC TCT GCC GCC ACA TTA AC -3‘ mtlA-2A-Cy5 5‘- GAT GAT CAC TTA TCT CCT GCC -3‘ mtlA-B-Cy5 5‘- TGC TGG TGA ATA ACT TCT CC -3‘ mtlA-2B-Cy5 5‘- GTT CTT TGG TCG TGG TAG CGC -3‘ „ mtlA-C-Cy5 5‘- GCT TAC TTC GCT AAC ATC GC -3‘ „ mtlA-2C-Cy5 5‘- GAG TCT AAA GGC GCA TCT CCG -3‘ „ mtlA-D-Cy5 5‘- TGA CCA ACT TCC TCG ACA GC -3‘ „ mtlA-2D-Cy5 5‘- AGC TGG TGA AAG GCG GTT ACG -3‘ „ mtlA-EH-Cy5 5‘- TCC TGT TCC TCA ACA ACG CC -3‘ „ mtlA-HE-Cy5 5‘- GAT AGT CAG CGT GAA CAC GC -3‘ „ mtlA-1-Cy5 5‘- ACC CCA CCT TCT CCA TGT GG -3‘ „ mtlA-2-Cy5 5‘- CTT TGC GAC CGA GGA AGA TG -3‘ „ mtlA-3-Cy5 5‘- TTC ATG TCC TGC ATA CGA CG -3‘ „ mtlA-4-Cy5 5‘- AAG AAC ATG TAC GCC AGC AG -3‘ „ mtlA-5-Cy5 5‘- CGA TAA CGC CCA TGG TGG TG -3‘ „ Cy5-markierte SequenzierPrimer II. Material und Methoden 17 Tab. II.1.5. Oligonukleotide (Fortsetzung) Primer Sequenz Beschreibung E218A (+) 5‘- CAA TCT TCT TCC TGA TTG CCG CTA ACC CAG GTC CAG G -3‘ E218A (-) 5‘- CCT GGA CCT GGG TTA GCG GCA ATC AGG AAG AAG ATT G -3‘ „ E218V (+) 5‘- CAA TCT TCT TCC TGA TTG TCG CTA ACC CAG GTC CAG G -3‘ „ E218A (-) 5‘- CCT GGA CCT GGG TTA GCG ACA ATC AGG AAG AAG ATT G -3‘ „ H256P (+) 5‘- CTT CCT GGG GGG TAT CCC AGA AAT CTA CTT CCC G -3‘ „ H256P (-) 5‘- CGG GAA GTA GAT TTC TGG GAT ACC CCC CAG GAA G -3‘ „ H256A (+) 5‘- CTT CCT GGG GGG TAT CGC CGA AAT CTA CTT CCC G -3‘ „ H256A (-) 5‘- CGG GAA GTA GAT TTC GGC GAT ACC CCC CAG GAA G -3‘ „ H256Q (+) 5‘- CTT CCT GGG GGG TAT CCA GGA AAT CTA CTT CCC G -3‘ „ H256Q (-) 5‘- CGG GAA GTA GAT TTC CTG GAT ACC CCC CAG GAA G -3‘ „ H256S (+) 5‘- CTT CCT GGG GGG TAT CAG CGA AAT CTA CTT CCC G -3‘ „ H256S (-) 5‘- CGG GAA GTA GAT TTC GCT GAT ACC CCC CAG GAA G -3‘ „ MtlA-HIS 1 5‘- GGA ATT CCC GTC GAC TGG ACA GTT AAC CGA TTC AGT G -3‘ MtlA-HIS 2 5‘- TTG GAT CCT TAG TGG TGG TGG TGG TGG TGC TTA CGA CCT GCC AGC AGT TCC AGC ACT TC -3‘ C110S (+) 5‘- CGC TGG GCG GCT GGT CTA TTA AGC ACT TCG AC -3‘ C110S (-) 5‘- GTC GAA GTG CTT AAT AGA CCA GCC GCC CAG CG -3‘ MutagenesePrimer Konstruktion eines mtlA6xHisTag „ MutagenesePrimer „ 18 II. Material und Methoden Tab. II.1.5. Oligonukleotide (Fortsetzung) Primer Sequenz Beschreibung C320A (+) 5‘- CAT CGC GGG TGT GGC TGC GGC GAT GGC TGT C -3‘ MutagenesePrimer C320A (-) 5‘- GAC AGC CAT CGC CGC AGC CAC ACC CGC GAT G -3‘ „ C384S (+) 5‘- CGT AAA ATC ATC GTT GCC TCT GAC GCC GGT ATG GG -3‘ „ C384S (-) 5‘- CCC ATA CCG GCG TCA GAG GCA ACG ATG ATT TTA CG -3‘ „ C571S (+) 5‘- GGG CGT CGT GTT CTC TCA GTA CCC GGA AGG CG -3‘ „ C571S (-) 5‘- CGC CTT CCG GGT ACT GAG AGA ACA CGA CGC CC -3‘ “ S3C (+) 5‘- GGT GTT TTT ATG TCA TGC GAT ATT AAG ATC AA -3‘ “ S3C (-) 5‘- TTG ATC TTA ATA TCG CAT GAC ATA AAA ACA CC -3‘ “ S110C (+) 5’- CTG GGC GGC TGG TGC ATT AAG CAC TTC G -3’ “ S110C (-) 5’- CGA AGT GCT TAA TGC ACC AGC CGC CCA G -3’ 5‘- GAT TGT TGA AGC CCT GTG CAA AAT GCT GGC TGC GG -3‘ “ S158C (-) 5‘- CCG CAG CCA GCA TTT TGC ACA GGG CTT CAA CAA TC -3‘ “ S199C (+) 5‘- CCA CGG TAT CTT CTG CCC GCT GGG TAT TCA G -3‘ “ S199C (-) 5‘- CTG AAT ACC CAG CGG GCA GAA GAT ACC GTG G -3‘ “ S212C (+) 5‘- CCA TGA ACT GGG TAA ATG CAT CTT CTT CCT GAT TG -3‘ “ S212C (-) 5‘- CAA TCA GGA AGA AGA TGC ATT TAC CCA GTT CAT GG -3‘ “ S242C (+) 5‘- GGT AGC GCT AAA CAG TGC GCG GGC GGT GCG GC -3‘ “ S242C (-) 5‘- GCC GCA CCG CCC GCG CAC TGT TTA GCG CTA CC -3‘ “ S158C (+) “ II. Material und Methoden 19 Tab. II.1.5. Oligonukleotide (Fortsetzung) Primer Sequenz Beschreibung S299C (+) 5‘- CGG CAT CTC CGG GTT GCA TCC TTG CTG TAC TGG -3‘ “ S299C (-) 5‘- CCA GTA CAG CAA GGA TGC AAC CCG GAG ATG CCG -3‘ “ II.2. Chemikalien, Isotope und Enzyme Handelsübliche Grundchemikalien, Detergenzien, Lösungsmittel, Puffersubstanzen, Kohlenhydrate, Vitamine, Antibiotika und Aminosäuren wurden von den Firmen Boehringer (Mannheim), Difco (Michigan, USA), Life Technologies und Roth (Karlsruhe), Merck (Darmstadt), Amersham Pharmacia (Freiburg, Braunschweig), Riedel de Haën (Seelze), Serva (Heidelberg) und Sigma (Deisenhofen) bzw. der Sigma-Aldrich Firmengruppe bezogen. Radioaktive Isotope stammten von Amersham LIFE SCIENCE (Buckinghamshire, England). DNA-modifizierende Enzyme und Feinchemikalien für DNA-Techniken stammten von den Firmen Genomed (Bad Oeynhausen), Boehringer (Mannheim) und Life Technologies (Karlsruhe). Bei der Sequenzierung wurden das “T7SequencingTM-Kit“, das ‘‘Cy5TM AutoRead Sequencing Kit‘‘ der Firma Pharmacia Biotech (Freiburg) und das „Thermo Sequenase fluorescent labelled primer cycle sequencing kit with 7-deaza-dGTP“ der Firma Amersham Pharmacia Biotech (Buckinghamshire, England) eingesetzt. Spezifische DNA-Primer für die Sequenzierung, PCR-Amplifikation und Mutagenese wurden von der Firma Interactiva (Ulm) und TIB MOLBIOL (Berlin) synthetisiert. Sulfhydrylreagenzien (Biotinmaleimid und Stilben) wurden von Molecular Probes Inc. (Eugene, OR 97402-0469) geliefert. Eingesetztes H2O stammte aus der hauseigenen VE-Anlage, zweifach entsalztes H2O wurde mit der „Seradest LFM 20“-Anlage von Seral (D-56235, Ransbach) hergestellt. 20 II. Material und Methoden Tab.II.2. Häufig verwendete Puffer und Lösungen ABKÜRZUNG ZUSAMMENSETZUNG GLB, 5fach Glyzerin TBE Bromphenolblau Xylencyanolblau TBE, 10fach Tris-Base Borat EDTA 0,5M pH 8,0 H2O TE 10.1 Tris-HCl pH 8,0 EDTA pH 8,0 KONZENTRATION 50% 0,5fach 0,25% 0,25% 108g 55g 40ml ad 1000ml 10mM 10mM KPi [X M, pH Y] Kaliumdihydrogenphosphat, X M di-Kaliumhydrogenphosphat, X M In der Tabelle sind häufig verwendeter Puffer und Lösungen und deren Zusammensetzung angegeben. II.3. Nährmedien II.3.1. Minimalmedium Als Minimalmedium wurde ein Standard-Phosphatmedium nach Tanaka et al. (1967) verwendet. Nach dem Autoklavieren wurde die entsprechende Kohlenstoffquelle, soweit nicht anders angegeben, in einer Endkonzentration von 0,2% (w/v) zugegeben. Bei Bedarf erfolgte eine Supplementierung mit Aminosäuren (20mg/l der L-Form; Methionin und Cystein 40mg/l) und Thiamin (B1) (50mg/l). Bei Derivaten des Stammes JWL181 wurde Thiamin mit 100mg/l eingesetzt, da eine Wachstumslimitierung bei den Stämmen LGS322 und LGS323 mit nur 50mg/l Thiamin beobachtet wurde. Vitamine (5mg/l) wurden je nach Verwendung dem Medium entsprechend zugegeben. MM-Platten enthielten zusätzlich 12g/l Gibco-Agar. II.3.2. Vollmedien (LB0, LB und 2xTY) LB0-Vollmedium setzte sich aus 10g Bacto-Trypton (Difco), 10g Hefeextrakt und 5g NaCl auf 1000ml H2O bidest zusammen. Zur Herstellung von LB-Medium (Lennox 1955) wurde LB0-Medium nach dem Autoklavieren zu einer Konzentration von II. Material und Methoden 21 0,2% (w/v) und CaCl2 (2,5 mM) steril zugesetzt. 2xTY-Medium bestand aus 13g BactoTrypton (Difco), 5g NaCl und 10g Hefeextrakt auf 1000ml H2O. Zur Herstellung von LB0-Platten wurden zu 1l LB0-Medium 12g Gibco-Agar zugegeben. II.3.3. MacConkey (McC) - Indikatorplatten Als Selektionsplatten wurden MacConkey-Indikatorplatten verwendet (MacConkey, 1905). Sie bestanden aus 40g MacConkey-Agar Base (Difco) in 1000ml H2O. Die zu testende Kohlenstoffquelle wurde nach dem Autoklavieren in einer Endkonzentration von 1% (w/v) steril zugegeben. Zuckeralkohole wurden in gleicher Konzentration vor dem Autoklavieren eingewogen. Die Konzentration bei D-Mannitol Platten lag dementsprechend bei 55mM. II.3.4. IPTG/X-Gal-Indikatorplatten In diesem Vollmedium war zusätzlich zu dem selektionierenden Antibiotikum 1mM IPTG und 0,02% (w/v) X-Gal enthalten. X-Gal-Platten können bei bestimmten Klonierungen das Vorhandensein eines Inserts anzeigen. Alle Vektoren, die ein α-lacZ mit einer integrierten multiplen Klonierungsstelle (mks) tragen, sind dafür geeignet. Auf X-Gal-Indikatorplatten erscheinen Kolonien, die einen Vektor mit Insert erhalten haben weiß. Diejenigen, die nur den Vektor tragen, erscheinen blau. II.3.5. P1-Weichagar P1-Weichagar diente zur Herstellung und Titration von P1-Phagenlysaten. Nach der Vorschrift von Arber (1960) setzte er sich aus 8g Nutrient Broth, 5g NaCl, 6,5g Difco-Agar auf 1000ml H2O zusammen. II.3.6. Chemotaxis-Schwärmplatten Die Herstellung von Chemotaxis-Schwärmplatten (ohne EDTA) erfolgte nach einer von Grübl (1989) modifizierten Vorschrift nach Adler (1973). Die Platten enthielten 3,9ml KH2PO4 (1M); 6,1ml K2HPO4 (1M); 1,0ml (NH4)2SO4 (1M); 1,0ml MgSO4 (1M; nach den Autoklavieren steril zugegeben) und 2,7g Difco-Agar auf 1l zweifach entsalztes H2O. Kohlenhydrate wurden in Endkonzentrationen zwischen 100µM und 1mM zugegeben. 22 II. Material und Methoden II.3.7. Medien mit Antibiotikazusatz Antibiotika wurden, wenn nicht anders angegeben, in folgenden Endkonzentrationen steril zugesetzt: Ampicillin (Amp/Ap) 100µg/ml; Chloramphenicol (Cam/Cp) 25µg/ml; Tetracyklin (Tet/Tc) 10µg/ml (alle in 50% Ethanol gelöst); Kanamycin (Kan/Kn) 25µg/ml; Spectinomycin (Spc/Sp) 100µg/ml; Streptomycin (Str/Sm) 50µg/ml (alle in H2O gelöst). II.3.8. Medium zum Anlegen von Dauerkulturen (nach Lengeler) Ein salzarmes Medium (Slant) enthielt 20g Bacto-Trypton, 10g Hefeextrakt und 0,2% (v/v) Glyzerin auf 1l Wasser. Eine getestete Einzelkolonie wurde ÜN in 8ml Medium angezogen. Die Kultur wurden abzentrifugiert und in 4ml Slant 50:50 (10g Bacto-Trypton, 5g Hefeextrakt in 500ml Wasser und 500ml Glyzerin 87%) resuspendiert. Je 2ml wurden in sterile Wheaton-Röhrchen überführt und getrennt bei 20°C gelagert. II.4. Anzuchtverfahren Die für die Anzucht von Stämmen der JWL-Stammreihe verwendeten Minimalmedien wurden mit Thiamin, L-Arginin, L-Methionin und L-Histidin (B1AMH) supplementiert. Sollten plasmidkodierte Funktionen erhalten bleiben, wurden entsprechende Antibiotika in den bereits genannten Konzentrationen zugegeben. II.4.1. Wachstumskurven Wachstumskurven wurden in Erlenmeyerkolben mit dem 10fachen Volumen des zu messenden Kulturvolumens durchgeführt. Aus einer gut gewachsenen ÜK wurden die Zellen zu einem Titer von 2,5-5x107 Bakterien/ml angeimpft und bei 37oC im Schüttelwasserbad inkubiert. Bei Wachstum in Minimalmedien erfolgte die Messung bei OD420 (1=5x108 B/ml), die Trübungszunahme in Vollmedien wurde bei OD650 (1=1x109 B/ml) gemessen. Die Generationszeiten wurden rechnerisch aus der Kurvengleichung der Wachstumskurve in der exponentiellen Phase ermittelt, wobei in der halb-logarithmischen Darstellung eine exponentielle Trendlinie mit der Gleichung II. Material und Methoden 23 y = m ∗ eb ∗x erstellt wurde. Der Wert b ergab durch G = ln2 / b den Wert für die Generationszeit G. II.4.2. Anzucht der Bakterien für Transportmessungen Die Anzucht der Stämme erfolgte aerob in LB0-Medium und falls erforderlich mit den entsprechenden Antibiotika. Nach dem Animpfen der Kultur aus einer ÜK zu einem Titer von 5x107 B/ml (OD420=0.1) wurden die Zellen bis zum Erreichen eines Titers von 5x108 B/ml im Schüttelwasserbad bei 37°C inkubiert. Eine Induktion des Transportsystems war nicht erforderlich, da nur mit konstitutiv exprimierten Systemen gearbeitet wurde. Anschließend wurden die Zellen abzentrifugiert (Hettich- Tischzentrifuge), in MM gewaschen und zu einem Titer von ca. 1x108 B/ml in MM aufgenommen. Nach Bestimmung der genauen Test-OD erfolgte der Transporttest. II.4.3. Anzucht der Bakterien für Proteinüberexpressionen Eine Überexpression wurde bei der Herstellung von EIIMtl-Membranvesikeln angewandt und in zwei unterschiedlichen Systemen durchgeführt: 1. An der Reichsuniversität in Groningen (RUG) wurde das Plasmid pMaHisMtlAPr benutzt, dass eine Induktion über Hitze ermöglichte. In diesem Plasmid ist das mtlA hinter den starken λ PR Promotor kloniert, der unter der Kontrolle des auf dem Plasmid vorhandenen thermolabilen (cI875) λ-Repressors steht (Davison et al., 1987). Hierzu wurde eine getestete EK in 2ml LB0-Medium+Cam unter Schütteln 7h bei 30°C im WB inkubiert um anschließend 100ml LB0-Medium+Cam anzuimpfen. Diese Kultur wurde ÜN bei 30°C im WB inkubiert. Mit je 25ml ÜK wurden 3x 1l LB0Medium+Cam angeimpft und bei 30°C und 250Upm im Inkubator (New Brunswick Scientific Co. Inc. - Model G25 Incubator Shaker) bis zu einer OD650=ca. 0,8 angezogen. Für die Hitzeinduktion wurde der Inkubator 10min auf 65°C, 20min auf 45°C und dann 2h auf 42°C eingestellt. Anschließend wurden die Zellen durch Zentrifugation geerntet. 24 II. Material und Methoden 2. In pMMX5-Derivaten wurde die Überexpression nach der veränderten Methode von Tabor und Richardson (1985) mit Hilfe des T7-RNA-Polymerasesystems von Studier und Moffat (1986) durchgeführt. Bei dieser Methode wird die Eigenschaft der T7-RNA-Polymerase genutzt, im Wesentlichen spezifische T7-Promotoren zu erkennen. Durch Insertion des mtlA-Genes hinter den T7-Promotor in pHEX5 (Heuel, 1997) und gezielter Induktion der T7-RNA-Polymerase-Synthese mit Isopropyl-ß-Dthiogalactopyranosid (IPTG) konnte MtlA in erhöhter Menge in dem Stamm JM109(λDE3) (Yanisch-Perron et al.,1985) produziert werden. Das Gen für die T7RNA-Polymerase liegt in Stamm JM109(λDE3) chromosomal unter Kontrolle des durch IPTG induzierbaren lacUV5-Promotors vor. Durch Induktion mit IPTG kommt es zur Expression der T7-RNA-Polymerase, welche dann die hinter den T7-Promotor klonierten Gene transkribiert. Mit dem entsprechenden Plasmid frisch transformierte JM109(λDE3)-Zellen wurden als ÜK bei 37°C angezogen und am nächsten Morgen zu einem Titer von ungefähr 5x107 B/ml in LB0-Medium+Cam angeimpft. Bei einer Zelldichte von ca. 7x108 B/ml erfolgte die Induktion der Überproduktion mit 2mM IPTG. Nach 1,5-2h wurden die Zellen geerntet. II.4.4. Anzucht der Bakterien zur Herstellung von Zellextrakten War eine Proteinüberexpression bei der Herstellung von Zellextrakten nicht möglich oder notwendig, wurde bei der Anzucht wie folgt verfahren. Aus einer ÜK in 2xTY-Medium wurde zu einem Titer von 3-5x107 B/ml angeimpft und bei 37°C im Schüttler bis zur spätexponentiellen Phase (ca. 1x109 B/ml) inkubiert. Anschließend wurde die Kultur durch Zentrifugation geerntet dann zweimal in 1%-igem NaCl gewaschen und direkt weiter verarbeitet oder bei -20°C aufbewahrt. II.5 Herstellung von Zellextrakten II.5.1. Präparation von Zellextrakten mit der „French Press“ Nach der Ernte wurden die Zellen mit Tris [20mM; pH 7,6], NaN3 [1mM] gewaschen und das Sediment mit 5ml Puffer (Tris [20mM; pH 7,6], NaN3 [1mM], DTT [5mM]) je Gramm Nassgewicht der Zellen überschichtet und auf Eis ÜN im Kühlraum gelagert. II. Material und Methoden 25 Anschließend wurde dem Puffer 1mM PMSF, 1mM MgCl2 und je eine Spatelspitze DNAse und RNAse zugegeben und die Zellen darin vorsichtig resuspendiert. Der Aufschluss der Zellen erfolgte bei >10.000psi in der „French Press“. Der Zellaufschluss wurde mit 2mM EDTA versetzt, anschließend 10min bei 15.000Upm zentrifugiert und der Überstand weitere 90min bei 48.000Upm zentrifugiert. Die Membranvesikel wurden in 2ml Puffer (Tris [25mM; pH 8,4], NaN3 [1mM], DTT [5mM]) je Nassgewicht der Zellen resuspendiert und in flüssigem N2 gelagert. II.5.2. Präparation von Zellextrakten durch Ultraschallbehandlung Die Anzucht der Kulturen erfolgte wie unter II.4. beschrieben. Nach Zentrifugation wurde das Sediment zweimal in 1/5 Volumen 1%iger (w/v) NaCl-Lösung gewaschen. Die Zellen wurden in gekühltem Tris [0,1M; pH 7,6] auf einen Titer von ca. 1x1011 B/ml eingestellt und während des Aufschlusses durch Ultraschall in EtOHEiswasser gekühlt. Beschallt wurde in Intervallen von 20s/20s (Ultraschall/Abkühlungsphase) bis das Extrakt klarer wurde. Nach der Kontrolle des Extraktes unter dem Mikroskop wurden Zelltrümmer und nicht aufgeschlossene Zellen durch 20-minütige Zentrifugation bei 14.000Upm/4°C entfernt. Zur weiteren Auftrennung von Membranen und Zellinhalt wurde der Überstand in der Mini-UZ 30min bei 80.000Upm/4°C zentrifugiert und das Membranpellet anschließend in 1/50 Volumen geeigneten Puffers resuspendiert. II.6. Genetische Techniken II.6.1. Strichkreuzungen Donor und Rezipient wurden in 5ml LB0-Medium angeimpft und in der exponentiellen Wachstumsphase geerntet. Nach zweimaligem Waschen in MM wurden die Zellen in 0,2ml MM aufgenommen. Mit einer Pipette wurde zuerst der Donor auf die entsprechende Selektionsplatte aufgetragen und nach dem Eintrocknen senkrecht dazu der Rezipient. 26 II. Material und Methoden II.6.2. Flüssigkreuzungen Donor und Rezipient wurden in Vollmedium angezogen und nach dem Erreichen der exponentiellen Phase in einem Verhältnis von 1:10 (Donor:Rezipient) in einem Erlenmeyerkolben unter sehr langsamem Schütteln gemeinsam inkubiert. Die Kreuzung wurde durch Resuspendieren in MM und eine kurze Vortexbehandlung unterbrochen. Entsprechende Verdünnungen wurden auf Selektionsplatten ausplattiert. II.6.3. Transformation von E. coli Plasmid-DNA wurde nach der CaCl2-Methode (Sambrook et al., 1989) in die Bakterienzellen transformiert. Alternativ dazu wurde eine vereinfachte Transformationsmethode eingesetzt, bei der 5ml 2xTy-Medium mit 0,2ml einer Übernachtkultur der zu transformierenden Bakterien angeimpft und 2 Stunden bei 37°C im Roller inkubiert wurde. Nach Zentrifugation der Zellen wurde das Sediment in 0,2ml kaltem CaCl2 (0,1M) resuspendiert und für 30min auf Eis gestellt. Mit den kompetenten Zellen wurde die Transformation wie bei Sambrook et al. (1989) beschrieben durchgeführt. II.6.4. Transformation von E. coli durch Elektroporation Bei Versuchen in denen eine höhere Transformationsrate erforderlich war, wurde die Elektroporation eingesetzt. Dazu wurden E.coli-Zellen wie in dem Handbuch zum ELEKTRO CELL MANIPULATOR 600 von BTX beschrieben in LB0-Medium bis zu einer OD650 von 0,5-1 bei 37°C unter Schütteln angezogen. Die Zellen wurden dann viermal mit kaltem, sterilem Wasser gewaschen und schließlich auf 1/2000 des Ausgangsvolumens in 10% (v/v) Glyzerin resuspendiert. Während des gesamten Vorgangs wurden die Zellen auf 4°C gehalten. Die Zellen konnten portioniert und bei 80°C aufbewahrt werden. 50µl der Zellsuspension wurden in eine Elektroporationsküvette mit 2mm Schlitz (Eurogenetech) gegeben. Nach der Zugabe der DNA wurde gemischt, das Gerät auf 2,5kV und R5 eingestellt und die Zellen dadurch für ungefähr 5ms der Spannung ausgesetzt. Anschließend wurden die Zellen sofort in eiskaltes Medium gegeben und dort für ca. 5min inkubiert. Dann wurden sie für 3090min unter Schütteln bei 37°C inkubiert und anschließend auf Selektionsplatten plattiert. II. Material und Methoden 27 II.6.5. P1-Transduktion Zur Durchführung von P1-Transduktionen wurde die von Lengeler (1966) modifizierte Methode nach Arber (1960) angewendet. II.6.5.1. Herstellung von Plattenlysaten P1-Weichagar wurde aufgekocht und im Wasserbad auf 55°C temperiert. Auf 3ml Weichagar wurden 0,25ml einer in LB-Medium angezogenen, gut gewachsenen ÜK des Donorstammes und etwa 106 P1.kc-Phagen gegeben. Der zur Infektion benutzte P1.kc sollte dabei möglichst auf einem WT-ähnlichen E. coli K-12-Stamm vermehrt worden sein. Dieser Ansatz wurde rasch gemischt, auf eine vorgewärmte LB-Platte gegossen und gleichmäßig verteilt. Die Platte wurde nach Erstarren des Weichagars bei 37°C bis zum Einsetzen der konfluenten Lyse inkubiert und dann zwecks Ernte der Phagen mit 3ml LB-Medium überschichtet. Abgeschwemmt wurden die Phagen indem der Ansatz ungefähr 30-60min vorsichtig bei RT geschwenkt oder ÜN bei 4°C aufbewahrt wurde. Die Phagensuspension wurde mit einer Pipette von der Platte abgezogen und zur Entfernung von Bakterien und Agarresten 15min bei 5.000Upm zentrifugiert. Das Phagenlysat wurde nach Zugabe von einigen Tropfen CHCl3 geschüttelt und anschließend bei 4°C aufbewahrt. Zur Bestimmung des Phagentiters wurden entsprechende Verdünnungen des Lysats (10-6, 10-7, 10-8, 10-9) wie oben beschrieben mit Bakterien und Weichagar gemischt und ÜN auf Platten inkubiert. II.6.5.2. Transduktion Für eine P1-Transduktion wird eine m.o.i. von 0,2 als geeignet angesehen. Dazu wurde eine in LB angezogene ÜK des Rezipientenstammes zunächst verdünnt (meistens 1:5) und mit 108 der auf dem Donorstamm vermehrten Phagen infiziert. Dieser Ansatz wurde 20min bei 37°C inkubiert. Eine Neuinfektion der Zellen wurde durch Zugabe von P1-Saline (7,5g Na-Citrat, 5g NaCl auf 1000ml H2O) und vortexen gestoppt. Die Bakterien wurden noch einmal mit P1-Saline gewaschen und - je nachdem welche Art von Markern übertragen werden sollte - direkt auf Selektionsplatten plattiert oder zur Etablierung von Antibiotikaresistenzen vorher 1h bei 37°C in 3-4ml LB0 inkubiert. 28 II. Material und Methoden II.7. DNA-Techniken II.7.1. Analytische Agarosegele Für die Auftrennung von DNA-Fragmenten wurden Agarosegel-Elektrophoresen nach Sambrook et al. (1989) durchgeführt. Je nach Größe der Fragmente wurden 0,7%ige (w/v) bzw. bei kleinen DNA-Fragmenten 1,5%ige (w/v) Agarosegele in 1xTBEPuffer benutzt. Die Elektrophorese erfolgte in einem mit 0,5µg/ml Ehtidiumbromid versetzten TBE-Puffer bei einer Spannung von 100V und einer Stromstärke von 100 bis 160mA. Das in die DNA interkalierende Ethidiumbromid konnte auf einem UVLeuchttisch bei Videodruckanlage 254nm (UVP, sichtbar Herolab, gemacht und Wiesloch) zur Auswertung dokumentiert mit einer werden. Zur Größenzuordnung der aufgetrennten Fragmente wurde ein Standard („1kb-ladder“) der Firma Life Technologies benutzt. II.7.2. Präparative Agarosegele und DNA-Fragmentisolierungen Die präparative Auftrennung von DNA-Fragmenten erfolgte wie oben beschrieben. Um Beschädigungen der DNA durch UV-Strahlung möglichst gering zu halten, wurden die gesuchten DNA-Fragmente mit einer UV-Handlampe (λ = 302nm) sichtbar gemacht und mit einem Skalpell aus dem Agarosegel ausgeschnitten. Die Isolation der aus den präparativen Gelen ausgeschnittenen DNA-Fragmente erfolgte mit Hilfe des Qiaex Gel Extraction Kits von Qiagen nach Angaben des Herstellers. II.7.3. Präparation von Plasmid-DNA Die Isolierung kleinerer Mengen von Plasmid-DNA (Miniplasmidisolierung) erfolgte durch alkalische Lyse (Birnboim &. Doly, 1979) nach dem Protokoll von Sambrook et al. (1989). Abweichend von dieser Vorschrift wurde der GET-Lösung (50mM Glukose; 10mM EDTA; 25mM Tris-HCl, pH 8,0) 4mg/ml Lysozym zugesetzt. Bei Plasmiden mit geringer Kopienzahl wurde bei gleichem Ansatzvolumen die doppelte Zellmenge eingesetzt. Zur Isolierung hochreiner Plasmid-DNA für Sequenzierungen wurden das „Jet Star-Kit“ (Genomed) oder das ‚‚Flexi-Prep-Kit‘‘ (Pharmacia Biotech) nach Angaben der Hersteller benutzt. II. Material und Methoden 29 II.7.4. Bestimmung der DNA-Konzentration Die Bestimmung der DNA-Konzentration erfolgte nach Sambrook et al. (1989) durch die photometrische Messung der Extinktion der DNA-Proben bei 260nm. Dabei entspricht OD260=1 etwa 50 µg/ml doppelsträngiger DNA. Um den Grad der Verunreinigung durch Proteine zu ermitteln, wurde die Extinktion der Proben bei einer Wellenlänge von 280nm gemessen. Reine Präparationen sollten einen Quotienten OD260 / OD280 von 1,8 bis 2,0 aufweisen. Werte unter 1,8 erlauben keine exakte Konzentrationsbestimmung. II.7.5. DNA-Restriktion Für die analytische oder präparative Spaltung von DNA wurden entsprechend den Herstellerangaben 1-3 Einheiten des Restriktionsenzyms pro µg DNA (DNAKonzentration einer Miniplasmidisolierung: 0,5 bis 1,5µg/µl) zugegeben und bei der für das Enzym optimalen Reaktionstemperatur im vom Hersteller empfohlenen Puffer inkubiert. Zur Analyse wurde etwa eine Stunde, für Präparationen 2-3 Stunden und bei Restriktionen mit chromosomaler DNA über Nacht verdaut. Bei Restriktionen mit mehr als einem Enzym wurde der für alle Enzyme günstigste Inkubationspuffer oder der ‘‘One-Phor-All‘‘-Puffer PLUS (Pharmacia Biotech) verwendet. Vor der gelelektrophoretischen Auftrennung wurde den Reaktionen 5xGLB zugegeben. II.7.6. “Klenowbehandlung“ von DNA-Fragmenten Zum Auffüllen 5’-überstehender Enden bzw. Abbau 3’-überstehender Enden wurde eine Behandlung der DNA mit der Klenow-Polymerase nach Ausubel et al. (1990) durchgeführt. Dieses Enzym besteht aus der großen Untereinheit der DNAPolymerase I (ohne 5’→3’ Exonuklease-Aktivität), die in Abwesenheit von dNTP überstehende 3’-Enden abverdaut und 5’-überstehende Enden bei Zugabe eines dNTPMixes auffüllen kann. Die entstehenden „glatten“ Enden wurden für Klonierungen von PCR-Produkten oder zur Ligation von DNA-Enden, die mit unterschiedlichen Enzymen geschnitten wurden, benötigt. Pro µg DNA wurden 0,5 Einheiten Klenow-Enzym und der vom Hersteller empfohlene Puffer zugegeben und bei 30°C für 5min inkubiert. Anschließend wurde der dNTP-Mix (50mM Tris-HCl, pH7,0 und je 125µM dATP, dCTP, dGTP sowie dTTP) in einer Endkonzentration von 6,25µM zugegeben und der Ansatz 30 II. Material und Methoden weitere 15min bei 30°C inkubiert. Nach Hitzeinaktivierung des Enzyms bei 70°C für 15min wurde die DNA direkt zur Ligation eingesetzt. II.7.7. Ligation von DNA-Fragmenten DNA-Fragmente wurden durch Zugabe von 1 Einheit T4-DNA-Ligase (Life Technologies oder Boehringer) je µg DNA und dem entsprechenden Ligase-Puffer ligiert. Das zu ligierende Fragment wurde bei 3’- oder 5’-überhängenden Enden in etwa 5-fachem, bei der Ligation „glatter“ Enden in ca. 10-fachem Überschuß gegenüber dem Vektor eingesetzt. Der Ligationsansatz, mit einem maximalen Volumen von 20µl, wurde über Nacht bei 14°C inkubiert und konnte danach direkt zur Transformation entsprechender Zellen eingesetzt werden. II.7.8. DNA-Sequenzierung Die Sequenzierung der DNA erfolgte entsprechend der Kettenabbruchmethode nach Sanger (Sanger et al., 1977; Sanger 1981). Dabei wurde nach den Angaben des Herstellers des „T7-SequencingTM-Kit“ der Firma Pharmacia vorgegangen. Zur radioaktiven Markierung der DNA wurde [35S]-ATP eingesetzt. Um Kompressionen zu beseitigen, wurde dem Ansatz falls erforderlich während der Primeranlagerung 1µl T4Gen32-Protein zugegeben (Kaspar et al. 1989). Je nach Stärke der radioaktiven Markierung erfolgte die Exposition des Röntgenfilms für 12-24h. Beim Einsatz der markierten Primer wurde das „Cy5TM AutoReadTM Sequencing Kit“ oder das „Thermo Sequenase fluorescent labelled primer cycle sequencing kit with 7-deaza-dGTP“ und beim Gebrauch spezifischer, unmarkierter Primer der „Cy5TM-dATP Labelling Mix“ verwendet. Die Sequenzreaktion für das „Thermo Sequenase fluorescent labelled primer cycle sequencing kit with 7-deaza-dGTP“ wurden in dem „1605 Air Thermo-Cycler“ der Firma Idaho Technology Inc. (Idaho Falls, USA) nach folgendem Ansatz durchgeführt: 1,6µl 2µl Cy5-Primer [5µM] BSA [2,5mg/ml] ca. 2ng DNA ad 24µl H2O II. Material und Methoden 31 Je 6µl wurden auf jeweils 2µl A-, C-, G-, T-Mix gegeben und in eine 10µl Glaskapillare gezogen. Die Glaskapillaren wurden in den Thermocycler getan und folgendes Programm gestartet. 4min 95°C Denaturierung 10sec 98°C Denaturierung 15sec X°C1 Anlagerung 30sec 72°C Elongation 4min 95°C Denaturierung 1 Zyklus 25 Zyklen 1 Zyklus 1 Die Anlagerungstemperatur X richtete sich nach der Tm (Schmelztemperatur) der Primer. Erfahrungsgemäß eignete sich als Anlagerungstemperatur die von Interaktiva angegebene Tm nach der „nearest neighbour“-Methode (Giesen et al. 1998). Anschließend wurde 6µl des Gel-Lade-Puffers zugegeben. Die Auftrennung der Cy5Proben erfolgte mit Hilfe des ALFExpressTM der Firma Pharmacia (Freiburg). II.7.9. Amplifikation von DNA mit Hilfe der Polymerase-Kettenreaktion Die Polymerase-Kettenreaktion (PCR) erfolgte nach einem Protokoll von Saiki et al. (1985). Hierfür wurde der „1605 Air Thermo-Cycler“ der Firma Idaho Technology Inc. (Idaho Falls, USA) verwendet. Dabei wurde nach Vorschrift der Firma gearbeitet und folgender Standardansatz (50µl Gesamtvolumen) gewählt: 5 µl 5 µl 5 µl 5 µl 5 µl 5 µl 5 µl 15 µl Template-DNA (1:50 verd. pGJ9 MPI) Reaktionspuffer (10-fach) MgCl2 (30mM) dNTP-Mix (je 2mM) Primer 1 (5µM) Primer 2 (5µM) Goldstar-Polymerase (0,4u) H2O Die Proben wurden in eine Glaskappilare gezogen, versiegelt und nach einmaliger Denaturierung für 2min bei 95°C wurde folgender Temperaturzyklus 30 Mal durchlaufen: 32 II. Material und Methoden 10sec 95°C 20sec 50/60°C Anlagerung der Primer 1,5min 72°C Denaturierung Elongation Nach Beendigung des Laufes wurden 2µl des Ansatzes auf ein Kontrollgel aufgetragen und Produkte bei dem 50°C- und 60°C-Ansatz festgestellt. Zur weiteren Verwendung des PCR-Produktes wurden eine Gelelektrophorese und anschließend eine Fragmentisolierung (II.7.2.) durchgeführt. Das isolierte Fragment wurde in einen entsprechend geschnittenen Vektor, wie unter II.7.7. beschrieben, kloniert. II.7.10. Entsalzen und Reinigen von DNA durch Ethanol-Fällung Um DNA zu entsalzen bzw. von Enzymen zu reinigen, wurde eine EthanolFällung nach Sambrook et al. (1989) durchgeführt. Dabei wurde zur DNA 0,1 Volumenteile (VT) 3M NaAc (pH6,0) und 2,5 VT 100% Ethanol gegeben, 30min zentrifugiert und das getrocknete Sediment in einer adäquaten Menge H2O aufgenommen. II.7.11. Inaktivierung von Enzymen Die Inaktivierung von Enzymen erfolgte einerseits durch 10min Hitzebehandlung bei 65°C oder, falls erforderlich, durch dreimaliges Schockgefrieren in Ethanol/Trockeneis mit anschließendem Auftauen in einem 70°C WB. Alternativ wurde eine Phenolextraktion mit anschließender Ethanolfällung durchgeführt. II.7.12. Primer-Phosphorylierung Wenn es erforderlich war wurden Primer nach folgender Methode mit der T4Polynukleotid-Kinase von Boehringer phosphoryliert: 10µl 2,5µl 0,5µl 2,5µl 9,5µl Primer [10pmol/µl] Kinase-Puffer [10fach] T4-Polynukleotid-Kinase [10U/µl] ATP [10mM] H2O II. Material und Methoden 33 Der 25µl-Ansatz wurde 30min bei 37°C im WB inkubiert. Die Reaktion wurde anschließend durch 10min bei 70°C im WB beendet und anschließend bei -20°C gelagert. II.7.13. Gerichtete „Altered Sites II“-Mutagenese Die Mutagenese mit dem „Altered Sites II in vitro Mutagenesis System“ von Promega erfolgte nach Angabe des Herstellers mit phosphorylierten MutagenesePrimern. Dabei wurde die Variante unter Einsatz der Einzelstrang-DNA mit dem Stamm BMH71-18 aus dem „Altered Sites I“-System verwendet. II.7.14. Gerichtete „QuikChange™“-Mutagenese Neuere Mutagenesen wurden mit dem „QuikChange™ Site-Directed Mutagenesis Kit“ der Firma Stratagene durchgeführt. Hierfür wurde der „T-Gradient Thermo-Cycler“ der Firma Biometra verwendet und nach Vorschrift der Firma folgender Standardansatz (50µl Gesamtvolumen) gewählt: 5µl 10xPCR-Puffer 1µl Plasmid [10ng/µl] 1µl Primer (+) [10µM] 1µl Primer (-) [10µM] 1µl dNTP [je 2,5mM] 1µl Pfu-Turbo-Polymerase [2,5Units/µl] 40µl H2O Für den Austausch von maximal zwei Basen wurde folgendes Programm in dem Thermocycler durchgeführt. 10min 94°C Denaturierung 1min 94°C Denaturierung 1min 50°C Anlagerung 16min 68°C Elongation 10min 68°C Elongation 4°C Kühlung ∞ 1 Zyklus 18 Zyklen 1 Zyklus 34 II. Material und Methoden Anschließend wurde durch Zugabe von 1µl des Restriktionsenzyms DpnI die methylierte Matrizen-DNA 1,5h bei 37°C im WB abgebaut und 50µl superkompetente XL1blue-Zellen mit 1µl der verdauten DANN transformiert. II.8 Chemische und biochemisch-analytische Techniken II.8.1. Bestimmung der Proteinkonzentration Der Proteingehalt einer Probe wurde mit Hilfe des „Bio-Rad Protein Assay Dye Reagent Concentrate“ nach Bradford (1976) den Angaben des Herstellers entsprechend durchgeführt. Die Eichkurve wurde mit im Probenpuffer gelöstem Rinderserumalbumin (BSA) aufgenommen. II.8.2. Bestimmung der Zellzahl Die Trübung des Mediums wurde photometrisch verfolgt. In MM wurde mit einer Wellenlänge von 420nm gemessen, in LB0 bei einer Wellenlänge von 650nm. Zellzahl und Proteinmenge ließen sich entsprechend einer Absorptionseinheit (OD=1) des Spektralphotometers bei der entsprechenden Wellenlänge wie folgt bestimmen: Tab.II.8.1. Zelltiter und Proteinmenge WELLENLÄNGE (nm) ZELLTITER PROTEINMENGE (mg/ml) 650 1 x 109 0,25 420 5 x 108 0,125 Angegeben ist das Verhältnis von Proteinmenge und Zelltiter zu einer Absorptionseinheit bei verschiedenen Wellenlängen für Kulturen von E. coli K-12. II.8.3. Bestimmung der spezifischen Transportaktivitäten Zur Bestimmung der Transportaktivitäten wurden Transporttests nach Schmid et al. (1982), durchgeführt. Die in vivo Bestimmungen der Transportaktivitäten wurden bei 25oC in Minimalmedium durchgeführt. Dazu wurden 1ml einer exponentiell gewachsenen Zellsuspension mit einem Titer von 5x108 B/ml und 100µl radioaktives Substrat in separaten Reagenzgläsern für 5min im Wasserbad bei 25oC äquilibriert. Die Reaktion wurde durch die Zugabe von 900ml Zellsuspension zum radioaktiven Substrat II. Material und Methoden 35 gestartet. Zu verschiedenen Zeitpunkten (in der Regel: 10s, 20s, 30s, 40s) wurden jeweils 200µl aus dem Teströhrchen entnommen, sofort über ein Membranfilter (Schleicher und Schuell, Porengröße 0,6µm) abgesaugt und zweimal mit 1ml raumtemperiertem MM+ gewaschen. Die Filter wurden anschließend getrocknet und in 5ml Szintillationsflüssigkeit Quicksafe N von Zinsser Analytic (Frankfurt/M) gegeben. Als Nullwert galten Zellen, die plasmidkodiert kein zu untersuchendes Transportsystem enthielten. Transportunabhängige, unspezifische Anlagerungen des radioaktiven Substrates an diese Zellen wurden auf Eis gemessen. Die Bestimmung der cpm-Rate erfolgte im Szintillationszähler „Liquid Scintillation Analyzer 1600TR“ von Canberra Packard (Illinois). Vier zeitabhängige Werte einer Probe wurden graphisch aufgetragen. Dabei zeigte sich, dass die Aufnahme des Substrates schon nach 10s nicht mehr linear erfolgte (s. Abb.II.8.1. Substrataufnahme von D-Mtl). Daher wurde der Wert nach zehn Sekunden für die Berechnung der spezifischen Transportaktivität auf eine Minute hochgerechnet. Die Auswertung erfolgte nach folgender Formel: spezifische Transportaktivität = ∆cpm = K= OD650 = t= 4 ⋅ VA ⋅ ∆cpm ⋅ K OD650 ⋅ t ⋅ VF ⋅ cpm ⋅ VB ml nmol min ⋅ mg Protein Gezählte Impulse pro Minute abzüglich Nullwert Substratkonzentration der radioaktiven Stammlösung [mol/l] gemessene Extinktion der eingesetzten Zellsuspension bei 650nm Zeitpunkt der Probenentnahme [min] VA = Volumen des Ansatzes [ml] (hier: 1) VB = Volumen der eingesetzten Bakteriensuspension [ml] (hier: 0,9) VF = auf den Filter gegebenes Probenvolumen [ml] (hier: 0,2) Z= radioaktive Zerfallsrate der Stammlösung [cpm/ml] 4= Umrechnungsfaktor für die Berechnung der Proteinmenge aus der Extinktion (OD650=1 entspricht 0,25mg Protein/ml) 36 II. Material und Methoden 18 16 -3 cpm [10 ] 14 12 10 8 6 4 2 0 0 10 20 t [sec] 30 40 Abb.II.8.1. Substrataufnahme von D-Mtl Exemplarisch ist die Aufnahme von Mannitol in LGS322/pGJ9 dargestellt ( = LGS322/pGJ9; = Nullwert). Es ist zu sehen, dass die lineare Aufnahme spätestens nach zehn Sekunden endet. Um aus den spezifischen Transportaktivitäten KM-Werte ermitteln zu können, wurden die Messungen bei unterschiedlichen Substratkonzentrationen durchgeführt. Die jeweiligen Konzentrationen und radioaktiven Zerfallsraten der in einem Verhältnis von 1:10 im Test eingesetzten Zuckerlösungen sind in der folgenden Tabelle dargestellt. Tab. II.8.2. Eingesetzte Zuckerkonzentrationen und dazugehörige cpm D-Mtl Konz. [µM] -3 cpm [x10 ] 3 5 7 10 15 20 30 50 100 31 53 78 107 168 210 197 210 236 II.8.4. Ermittlung des KM-Wertes und Vmax nach der „Lineweaver-Burk“Transformationsvorschrift Bei Enzymen variiert die Katalysegeschwindigkeit V mit der Substratkonzentration [S]. V ist definiert als die Anzahl der pro Sekunde entstehenden Mole eines Produkts. Die Maximalgeschwindigkeit Vmax wird erreicht, wenn alle Bindungsstellen am Enzym mit II. Material und Methoden 37 Substrat gesättigt sind. Die Bedeutung des KM-Wertes geht aus der Michaelis-MentenGleichung hervor: V = V max S S + KM Bei sehr kleinen Substratkonzentrationen – wenn [S] viel kleiner als KM – gilt: V = [S]Vmax/KM; das heißt, die Geschwindigkeit ist der Substratkonzentration direkt proportional. Bei hohen Substratkonzentrationen wird V = Vmax; das bedeutet, dass die Geschwindigkeit maximal ist, unabhängig von der Substratkonzentration. Bei [S] = KM ist V = Kmax/2. Der KM-Wert entspricht daher der Substratkonzentration, bei der die Reaktionsgeschwindigkeit die Hälfte ihres Maximalwertes erreicht. Um die MichaelisMenten-Beziehung in Form einer Geraden wiederzugeben, nimmt man von beiden Seiten der Gleichung den Kehrwert: 1 = 1 + KM ∗ 1 V V max V max [S ] Trägt man 1/V gegen 1/[S] auf, so ergibt sich ein Lineweaver-Burk-Diagramm mit dem Ordinatenabschnitt 1/Vmax und der Steigung KM/Vmax. 1/V = ng u ig Ste K /V m M a x y-Achsenabschnitt = 1/V m a x x-Achsenabschnitt = -1/K M 0 1/[S] Abb.II.8.2. Schematische Darstellung eines Lineweaver-Burk-Diagramms Der Schnittpunkt der x- und y-Achse entspricht dem Nullpunkt beider Achsen 38 II. Material und Methoden Durch Variation der Substratkonzentration bei den Transportmessungen konnten über die oben angegebenen Grundlagen die Größen KMapp und Vmax in den einzelnen Fällen ermittelt werden. Mittelwerte mehrerer Messungen wurden durch die Formel ∑x n ermittelt, wobei x die Werte der einzelnen Messungen und n die Anzahl der Messungen sind. Die Standardabweichung als Maß dafür, wie weit die jeweiligen Werte um den Mittelwert streuen, wurde nach folgender Formel berechnet: n∑ x 2 − (∑ x ) 2 n(n − 1) . II.8.5. Ermittlung des Kd-Wertes Die Ermittlung des Kd-Wertes der Bindekinetik von dem EIIMtl und dessen Varianten wurde mit der Fluss-Dialyse („flow dialysis“; Vos et al., nicht veröffentlicht) an der Reichsuniversität in Groningen (RUG) durchgeführt. Dabei wird das aus einer Dialysekammer austretende freie radioaktive D-Mtl in Abhängigkeit von vorhandenen EIIMtl-Vesikeln bei steigender D-Mtl-Konzentration gemessen. Das freie D-Mtl diffundiert bei 25°C durch eine Dialysemembran aus der Dialysekammer in die Flusskammer. Dort wird ein Fluss-Puffer (25mM Tris, pH 7,6) mit einer Rate von 230µl pro Minute durch die Flusskammer geleitet und in Fraktionen gesammelt. Die zu diesem Zweck eingesetzten Vesikel wurden mit der „French-Press“ hergestellt. Im Einzelnen wurde der Versuch wie folgt durchgeführt. Bindetest mit Vesikeln 430µl Reaktionsmix wurden mit 20µl Vesikeln zu einer Endkonzentration von 25mM Tris [pH 7,6], 5mM DTT, 5mM MgCl2 und 0,25% dPEG (v/v) gemischt und 10min bei 25°C equilibriert. Anschließend wurden 380µl des Reaktionsmix in die dreimal mit H2O gewaschene Dialysekammer pipettiert. Zu dem Zeitpunkt t=0min wurde 1µl [3H]-D-Mtl [10µM] zugegeben. Bei t=1min wurden in fünf Fraktionen jeweils 6 Tropfen aufgenommen. Im Folgenden wurde zu bestimmten Zeitpunkten weiteres [3H]-D-Mtl zugegeben und weitere Fraktionen aufgenommen: II. Material und Methoden 39 t = 3min + 2µl [3H]-D-Mtl [10µM] t = 4min je 6 Tropfen in 5 Fraktionen aufnehmen t = 6min + 2µl [3H]-D-Mtl [10µM] t = 7min je 6 Tropfen in 5 Fraktionen aufnehmen t = 9min + 2µl [3H]-D-Mtl [10µM] t = 10min je 6 Tropfen in 5 Fraktionen aufnehmen t = 12min + 2µl [3H]-D-Mtl [10µM] t = 13min je 6 Tropfen in 5 Fraktionen aufnehmen t = 15min + 4µl [3H]-D-Mtl [10µM] t = 16min je 6 Tropfen in 5 Fraktionen aufnehmen t = 18min + 8µl [3H]-D-Mtl [10µM] t = 19min je 6 Tropfen in 5 Fraktionen aufnehmen Die aufgenommenen Fraktionen wurden mit 2ml Scintillationsflüssigkeit versetzt und in dem Scintillationszähler gemessen. Über die kontinuierliche Zugabe erreicht man annähernd einen Konzentrationsgradienten von 26nM - 524nM [3H]-D-Mtl in der Dialysekammer. Allerdings muss die gleichzeitige unabhängige Diffusion des Mannitols aus der Kammer („leakage“) berücksichtigt werden. Zu diesem Zweck wurden zwei „Leerwertmessungen“ durchgeführt, die später in die Berechnung des Kd-Wertes eingebunden wurden. Ermittlung des Leerwertes Leerwert 1: 380µl Reaktionsmix ohne Vesikel (25mM Tris, pH 7,6; 5mM DTT; 5mM MgCl2; 0,25% (v/v) dPEG) wurden in die dreimal mit H2O gewaschene Dialysekammer pipettiert und 10min bei 25°C äquilibriert. Bei t.=.0min wurden 10µl [3H]-D-Mtl [10µM] zu einer Endkonzentration von 256nM zugegeben. Nach 20sec wurden in fünf Fraktionen jeweils sechs Tropfen aufgenommen. Die aufgenommenen Fraktionen wurden mit 2ml Scintillationsflüssigkeit versetzt und in dem Scintillationszähler gemessen. Leerwert 2: 380µl Reaktionsmix ohne Vesikel (25mM Tris, pH 7,6; 5mM DTT; 5mM MgCl2; 0,25% dPEG) wurden in die dreimal mit H2O gewaschene Dialysekammer pipettiert und 10min bei 25°C äquilibriert. Bei t.=.0min wurden 20µl [3H]-D-Mtl [10µM] zu einer Endkonzentration von 500nM zugegeben. Nach 20sec wurden in fünf 40 II. Material und Methoden Fraktionen jeweils sechs Tropfen aufgenommen. Die Aufnahme der Fraktionen wurde jeweils nach 5min:20sec und 10min:20sec wiederholt. Die aufgenommenen Fraktionen wurden mit 2ml Scintillationsflüssigkeit versetzt und in dem Scintillationszähler gemessen. Berechnung des Kd-Wertes (pers. Mitteilung E. Voss / RUG) Für die mathematische Umsetzung der erhaltenen Werte wurden drei Gleichungen zu Grunde gelegt, die in ihrer Kombination mit Hilfe eines Näherungsprogramms zu der Berechnung des Kd-Wertes führen: c n = n0 ∗ (s0 - x) s0 n = Konzentration der Bindestellen in Dialysekammer [nM] n0 = Anfängliche Konzentration der Bindestellen in Dialysekammer [nM] s0 = Konzentration der [3H]-D-Mtl Stocklösung [nM] x = Menge des zugegebenen [3H]-D-Mtl [nM] Diese Gleichung korrigiert die Konzentration der vorhandenen Bindestellen, die durch Zugabe von weiterem Mannitol verdünnt wird. Je mehr Mannitol (x) zugegeben wird, desto kleiner wird die Konzentration der freien Bindestellen. d (k + x + n) 2 - 4 ∗ n ∗ x (k + x + n) b= 2 2 (1) b = Menge des an das Enzym gebundenen [3H]-D-Mtl [nM] k = Kd-Wert des Enzyms x = Menge des zugegebenen [3H]-D-Mtl [nM] n = Konzentration der Bindestellen in Dialysekammer [nM] Dieser Gleichung liegt das Verhältnis zwischen Enzym (E), Substrat (S) und Enzym-Substrat-Komplex (ES) zugrunde. Im chemischen Gleichgewicht gilt: E + S ⇔ ES II. Material und Methoden 41 Für den Kd-Wert ergibt sich folgende Gleichung: Kd = [E] [S] [ES] (2) In der Gleichung entspricht die Menge des an das Enzym gebundenen [3H]-DMtl dem Enzym-Substrat-Komplex: [ES]x=xb. Die Anzahl der Bindestellen in der Dialysekammer ergibt sich aus der Summe von freiem Enzym und mit Substrat gebundenem Enzym: [E]x+x[ES]x=xn. Ähnlich verhält es sich bei der Substartkonzentration. Sie ist die Summe aus freiem Substrat und Enzym-SubstratKomplex: [S]x+x[ES]x=xx. Ersetzt man in den letzten beiden Gleichungen [ES] durch b, so ergibt sich für Ex=xn-b und Sx=xx-b. Wenn man die neuen Parameter in die Gleichung für den Kd-Wert (1) einsetzt; erhält man: k= (n - b) (x - b) b (3) Über die Umstellung der Gleichung (3) nach null ergibt sich: b 2 - (x + n + k) ∗ b + n ∗ x = 0 (4) Die Auflösung der Gleichung (4) nach b ergibt wiederum die Gleichung (1) mit der es möglich ist, die Menge des an das Enzym gebundenen [3H]-D-Mtl zu berechnen. e y = r1 + r2 ∗ (x - b) + r3 ∗ (x - b) 2 y = freies [3H]-D-Mtl, dass durch die Membran dialysiert ist [cpm] x = Menge des zugegebenen [3H]-D-Mtl [nM] b = Menge des an das Enzym gebundenen [3H]-D-Mtl [nM] r1 = 0 r2 = wird über die Leerwerte ermittelt r3 = 0 Diese Gleichung berechnet die Menge an freiem [3H]-D-Mtl [nM], dass durch die Membran dialysiert ist. Dieser Wert steht natürlich in Zusammenhang mit dem in der Dialysekammer vorhandenen Enzym, dass eine Bindung eingehen kann (b) und dem zugegebenen [3H]-D-Mtl (x). Weiterhin wird die Hintergrundstrahlung durch r1 42 II. Material und Methoden einbezogen, wurde aber hier gleich null gesetzt, da dies schon bei den Messwerten berücksichtigt wurde. Gleiches gilt für den Wert r3, der die Bindung an die Dialysekammer darstellt. Diese Bindung konnte nicht festgestellt werden und somit wurde dieser Wert ebenfalls gleich null gesetzt. Der Wert r2 ergibt sich aus der Auswertung der Leerwertmessungen. Aus den Werten der zweiten Leerwertmessung wurde eine lineare Regression erstellt aus deren Geradengleichung (y=mx+b) die „leakage“-Rate (-m/b ∗ 100%) berechnet wurde. Um r2 zu ermitteln, wurde in einer Kombination aller drei Gleichungen unter Eingabe der ersten Leerwertmessung und des ersten Wertes der zweiten Leerwertmessung eine Näherungsrechnung durchgeführt. Alle enzymabhängigen Werte (n, n0, k, b) wurden in dieser Rechnung gleich null gesetzt, da cpm kein Enzym vorhanden ist. 10000 9000 8000 7000 6000 5000 4000 3000 2000 1000 0 0 100 200 300 400 3 H-Mtl [nM] 500 600 Abb. II.8.3. Exemplarische Bindekurve Dargestellt ist eine Bindekurve für die Bindung von [3H]-D-Mtl an das 6His-Tag EIIMtl (H6wt). (z = Leerwertmesspunkte; gepunktete Linie = lineare Regression der Leerwertmesspunkte; = Bindekurve H6wt). Mit Hilfe der „leakage rate“ und des r2-Wertes konnte die Referenzgerade für die enzymunabhängigen [3H]-D-Mtl-Dialyse erstellt werden. Diese grafische Darstellung der [3H]-D-Mtl-Dialyse verläuft nicht mehr linear, wenn bindendes Enzym vorhanden ist (siehe Abb. II.8.3.). Die Bindekurve wird unter Berücksichtigung der gemessenen cpm und den Werten r2 und „leakage-rate“ abgeleitet. Aus der Abweichung dieser Kurve II. Material und Methoden 43 von der Leerwertgeraden lassen sich über ein Computerprogramm die Konzentration und der Kd-Wert des untersuchten Enzyms berechnen. II.8.6. Test der PTS-abhängigen in vitro Phosphorylierung Die in den Kd-Wert-Bestimmungen eingesetzten Vesikel wurden vorher auf ihre Phosphorylierungsaktivität getestet. Dazu wurde an der RUG der PEP-abhängige Phosphorylierungs-Test nach Robillard und Blaauw (1987) durchgeführt. Die spezifischen Phosphorylierungsaktivitäten der Vesikel wurden in 25mM Tris-HCl (pH 7,6), 5mM MgCl2, 5mM DTT, 5mM PEP, 0,25% (v/v) decylPEG, 10µM HPr und 0,1µM EnzymI bei 30°C gemessen. Die Menge an [14C]-D-Mtl und Enzym variierten in den einzelnen Versuchsansätzen. Die Versuchsansätze wurden 10min bei 30°C inkubiert bevor die Reaktion mit radioaktivem D-Mannitol gestartet wurde. Das Volumen der Ansätze betrug 100µl wovon in verschiedenen Zeitabständen vier mal jeweils 20µl Probe auf eine Dowex AG1-X2-Säule geladen wurde. 10µl des Ansatzes wurden zur Bestimmung der Radioaktivität in dem Ansatz verwendet. Anschließend wurden die Säulen dreimal mit H2O gespült und das gebundene D-Mannitol-P mit zwei mal 1,8ml HCl [0,2M] eluiert. Nach Zugabe der „Scint-Flüssigkeit“ wurde die Radioaktivität in einem Scintillationszähler gemessen. Die spezifische Aktivität wurde nach folgender Formel berechnet: spez. Akt. = ∆ cpm ∗ F t ∗ VA ∗ P nmol min ⋅ mg Protein ∆cpm = gezählte Impulse pro min abzüglich Nullwert F = spez. Radioaktivität des Substrates [nmol/cpm] t = Reaktionszeit [min] P = eingesetzte Proteinmenge [mg/ml] VA = Gesamtvolumen des Ansatzes [ml] II.8.7. SDS-Polyacrylamid-Gel-Elektrophorese Zur Auftrennung von Proteinen wurde eine SDS-Gelelektrophorese nach Laemmli (1970) durchgeführt, bei der die durch SDS denaturierten Proteine 44 II. Material und Methoden entsprechend ihrem Molekulargewicht aufgetrennt werden. Die Proteingele hatten folgende Zusammensetzung: Trenngel: 4ml 4,9ml 3ml Acrylamid-Bisacrylamid (30%/0,8% (w/v))-Lösung H2O 1,5M Tris pH8,8 120µl 10% (w/v) SDS 120µl 10% (w/v) Ammoniumpersulfat Sammelgel: 1ml 6,3ml 3ml 100µl 65µl Acrylamid-Bisacrylamid (30%/0,8% (w/v))-Lösung H2O 1,5M Tris pH8,8 10% (w/v)SDS 10% (w/v) Ammoniumpersulfat Die Lösungen wurden vermischt und 5min entgast. Nach Zugabe von 40 bzw. 5µl TEMED-Lösung zum Trenn- bzw. Sammelgel wurden 10x8cm große und 1,5mm dicke Gele gegossen. Die Proben wurden in einem Puffer nach Laemmli (1970) (60mM Tris-HCl, pH6,8; 1% (w/v) SDS; 1% (v/v) 2-Mercaptoethanol; 10% (v/v) Glyzerin; 0,01% (w/v) Bromphenolblau) resuspendiert und vor dem Auftragen 3min bei 95oC hitzedenaturiert. Als Größenstandart wurde ein nichtradioaktiver Proteinstandard (Biorad low range, Biorad Laboratories) verwendet. Der verwendete Laufpuffer bestand aus 25mM Tris-HCl; 0,2% (v/v) Glyzerin und 0,1% (w/v) SDS mit einem pH-Wert von 6,8. Bei der Elektrophorese erfolgte im Sammelgel bei 15 bis 20mA die Fokussierung der Proteinproben auf eine scharfe Lauffront und im Trenngel bei 25 bis 30mA die Auftrennung der Proben. Hatte die Lauffront das untere Gelende erreicht wurde der Lauf beendet. Das Trenngel wurde mit einer Lösung (0,1% (w/v) Serva BlauG; 10% (v/v) Essigsäure; 24% (v/v) Methanol) gefärbt, entfärbt (10% (v/v) Essigsäure, 25% (v/v) Methanol) bis der Standard zu erkennen war und unter Vakuum bei 65°C getrocknet (LKB Bromma, 2003 Slab Gel Dryer). Um radioaktiv markierten Substanzen sichtbar zu machen, wurde nach dem Trocknen eine Autoradiographie durchgeführt. II. Material und Methoden 45 II.8.8. Gezielte Markierung von Cysteinen („Cystein-Scanning“) Die Methode des Cystein-Scannings, wie sie hier angewendet wurde, dient zur Lokalisierung der relativen Lage von definierten Aminosäuren zur Zytoplasmamembran in Membranproteinen. Dazu wird ein Membranprotein konstruiert, das an einer definierten Position einen einzelnen Cysteinrest enthält. Durch den Einsatz von membranpermeablen (hier: Biotinmaleimid) und membranimpermeablen Sulfhydrylreagenzien (hier: Stilben) kann im Idealfall ermittelt werden, ob dieser definierte Cysteinrest im Peri- oder Zytoplasma lokalisiert ist. Dabei wird mit einem membranimpermeablen Sulfhydrylreagenz vorinkubiert, um im Periplasma befindliche Cysteine zu identifizieren. Im Zytoplasma und unter Umständen in der Membran liegende Cysteine werden bei diesem Vorgang nicht belegt. Eine anschließende Markierung mit dem membranpermeablen Biotinmaleimid erreicht theoretisch alle Cysteine unabhängig von ihrer Lage. Es kann aber nicht an Cysteinen binden, die bereits mit Stilben abgedeckt oder membranständig sind. Allerdings ist die Membranpermeabilität von Biotinmaleimid stark von dem pH-Wert, der Temperatur, Konzentration und Inkubationszeit abhängig (Loo und Clarke, 1994). In der hier beschriebenen Versuchsdurchführung erwies sich das Biotinmaleimid als weitgehend membranimpermeabel. Folglich können im Zytoplasma oder in der Membran liegende Cysteine in keinem Fall markiert werden. Im Periplasma befindliche Cysteine werden nach einer Vorinkubation mit Stilben nicht durch Biotinmaleimid markiert, wohl aber bei alleiniger Inkubation mit Biotinmaleimid. Da in diesem Test gebundenes Biotinmaleimid nachgewiesen wird, kann durch unterschiedliche Kombination der eingesetzten Sulfhydrylreagenzien in ganzen Zellen und in Membranpräparationen die allgemeine Zugänglichkeit von Cysteinen und deren Lage im Membranprotein ermittelt werden. II.8.8.1. Markierung ganzer Zellen mit Biotinmaleimid 300ml Kultur des Stammes JM109(λDE3) mit den entsprechenden pMMX5Derivaten wurden wie in II.4.3. beschrieben angezogen und zentrifugiert. Das Sediment wurde aufgeteilt wobei ein Teil eingefroren und der andere in 3ml KPi-Puffer [100mM; pH 7,5] resuspendiert wurde. Die 3ml Kultur wurden wiederum in je 1,5 ml aufgeteilt. Ein Teil wurde mit 1mM Stilben aus einer 50mM Stocklösung 15min bei Raumtemperatur vorinkubiert. Nach der Inkubation wurde der Ansatz zweimal mit KPiPuffer [100mM; pH 7,5] gewaschen. Anschließend wurden beide Teile 30min mit 46 II. Material und Methoden 200µM Biotinmaleimid aus einer 30mM Stocklösung bei RT markiert. Die Reaktion wurde mit 50mM β-Mercapto-EtOH abgestoppt, die Kulturen zweimal mit KPi-Puffer [100mM; pH 7,5] gewaschen und die Sedimente eingefroren. II.8.8.2. Markierung von Membranvesikeln mit Biotinmaleimid In diesem Schritt wurde die allgemeine Zugänglichkeit der Cysteine im Membranprotein überprüft. Zur Herstellung von Membranvesikeln wurde das unmarkierte Sediment aus II.8.7.1. in je 3ml KPi-Puffer [100mM; pH 7,5] resuspendiert. In der praktischen Durchführung wurden aus den beiden markierten Sedimenten parallel Membranvesikel hergestellt. Die somit vier Ansätze wurden jeweils in COREXRöhrchen überführt und mit je 10µl Protease-Inhibitor-Cocktail (SIGMA, P-8849) versetzt. Die folgende Ultraschallbehandlung der Zellen wurde wie in II.5.2. beschrieben durchgeführt. Die Sedimente wurden in 150µl KPi-Puffer [100mM; pH 7,5] resuspendiert. Die bereits als ganze Zellen markierten Membranvesikel wurden in der Zwischenzeit auf Eis gelagert. 150µl der unmarkierten Membranvesikel wurde wieder mit 1mM Stilben 15min bei RT vorinkubiert. Um eine Permeabilität der Vesikel zu gewährleisten, wurde der Ansatz zweimal in EtOH-Trockeneis gefroren und in dem Ultraschallbad (RT) aufgetaut. Im nächsten Schritt wurden beide Ansätze 30min mit 200µM Biotinmaleimid bei RT markiert. Im Folgenden wurden alle vier Ansätze in Lösung gebracht („solubilisiert“). II.8.8.3. Solubilisierung von Membranvesikeln Die vier Ansätze wurden in den Zentrifugenröhrchen mit DOC- Solubilisierungspuffer [20mM Tris, pH8; 300mM NaCl; 10mM β-Mercapto-EtOH; 0,5% (w/v) DOC (Deoxycholic acid sodium salt monohydrate (SIGMA-ALDRICH; D89552) 100µM D-Mannitol] auf 1500µl aufgefüllt und 45min auf Eiswasser gerührt. Der Anteil an DOC wurde in vier Portionen alle fünf Minuten hinzugegeben. Nach einer Zentrifugation von 45min bei 45.000Umin/4°C wurde das solubilisierte Membranprotein über eine Ni-NTA-Säule gereinigt. Dies war erforderlich, damit die übrigen ebenfalls am Cystein markierten Proteine der Zelle möglichst vollständig aus dem Hintergrund entfernt wurden. II. Material und Methoden 47 II.8.9. Reinigung von MtlA über eine Ni-NTA-Säule Da das MtlA für die Reinigung mit einem His-Tag versehen wurde, konnte es über Ni-NTA-Säulen gereinigt werden. Alle Zentrifugationsschritte mit den Säulen wurden bei 2.000Upm 2min mit geschlossenem Deckel durchgeführt. Vor dem Beladen der Säulen mit dem Überstand wurden diese mit 600µl DOC-Solubilisierungspuffer equilibriert. Der Überstand aus II.8.7.3. wurde mit 10mM Imidazol versehen und mit zweimal 600µl wurden die Säulen beladen. Der Waschschritt erfolgte mit 600µl DOCWaschpuffer [20mM Tris, pH8; 300mM NaCl; 30mM Imidazol; 0,1% (w/v) DOC; 100µM D-Mannitol]. Das Protein wurde mit 200µl DOC-Elutionspuffer [20mM Tris, pH7,6; 200mM Imidazol; 100µM D-Mannitol] eluiert und das Eluat bei -20°C gelagert. II.8.10. Immuno-Nachweis von Proteinen mit spezifischen Antikörpern Nach der Reinigung wurden die Proteine und deren Markierung mit Biotinmaleimid in einer Western-Hybridisierung nachgewiesen. Die Durchführung und die verwendeten Antikörper werden folgend beschrieben. II.8.10.1. Nachweis des His-Tag Um festzustellen, ob mit den Ni-NTA-Säulen das richtige Protein gereinigt wurde, wurde eine „Semidry“-Western-Hybridisierung in der „TRANS-BLOT SD SEMI-DRY TRANSFER CELL“ von BIO-RAD (USA, CA 54547) mit Anti-His Antikörpern durchgeführt. Der Aufbau der Hybridisierung erfolgte mit folgenden auf Gelgröße zugeschnittenen Komponenten: [Deckel (Kathode)] 2x Filterpapier (GB 004 GEL-BLOTTING Paper. Schleicher & Schuell, D-37582) SDS-Gel Nitrocellulose Transfer-Membran (PROTRAN. Schleicher & Schuell, D-37582) 2x Filterpapier (GB 004 GEL-BLOTTING Paper. Schleicher & Schuell, D-37582) [Boden (Anode)] Die Nitrocellulose wurde 5sec in Wasser inkubiert, bevor sie wie alle anderen Hybridisierungs-Komponenten 30min bei RT in dem Hybridisierungspuffer (25mM Tris, 192mM Glycin, 20% (v/v) Methanol [pH 8,3]) equilibriert wurde. Die Hybridisierung 48 II. Material und Methoden wurde wie oben beschrieben aufgebaut und die Transferparameter abhängig von der Gelgröße eingestellt. Bei zwei kleinen Gelen (jeweils ca. 8,5cm x 5cm x 1,5mm) wurden 60min Transfer bei 20V gewählt. Die Membran wurde anschließend mit Ponceau S (0,2% (w/v) in 3% (v/v) TCA) gefärbt um die Übertragung zu bestätigen. Nachdem die Membran mit H2O entfärbt wurde, erfolgte ein 20minütiger Waschschritt in TNT (20mM Tris/HCl [pH 7,5], 0,5M NaCl, 0,05% (v/v) Tween 20) bei RT. Anschließend wurde die Membran ÜN bei 4°C in TNT/3% (w/v) BSA (Albumine Bovine Fraction V, BIOMOL, D22769) geblockt. In dem folgenden Schritt wurde die Reaktion mit dem Penta-His Antikörper von QIAGEN (mouse anti-(H)5, Cat. No. 34660) durchgeführt. Dazu wurde die Membran 1h bei RT mit 6ml Antikörper-Lösung (TNT, 1% (w/v) BSA, 0,067µg/ml Penta-His AK) in einer verschweißten Plastiktasche inkubiert. Die Membran wurde dann drei mal 10min bei Raumtemperatur in TNT gewaschen. Anschließend wurde die Membran mit der Meerrettich-Peroxidase („Horseradishperoxidase“, N31430. Pierce, Rockford, IL 61105), die 1:10000 in TNT/1% (w/v) BSA verdünnt wurde, 1,5h bei RT in einer verschweißten Plastiktasche inkubiert. Die Membran wurde dann wieder drei mal 10min bei Raumtemperatur in TNT gewaschen. Die darauf folgende Chemilumineszens-Reaktion wurde nach Angaben des Herstellers mit der „Western Blot Chemiluminescence Reagent“ (NEN Life Science Products, Boston, MA 02118-2512) durchgeführt. Der Nachweis der Chemilumineszens wurde bei unterschiedlichen Expositionszeiten zwischen 10 und 60sec auf dem „Hyperfilm ECL“ (Amersham Pharmacia Biotech UK Limited) durchgeführt. II.8.10.2. Nachweis der Biotinmaleimid-Markierung Der Nachweis der Biotinmaleimid-Markierung erfolgte ähnlich wie unter II.8.10.1. beschrieben. Es wurden lediglich andere spezifische Antikörper eingesetzt. Da bei der Methode des Cystein-Scanning das Biotinmaleimid den ersten Antikörper darstellt und die Bindung schon während der Markierung erfolgte, fiel dieser Inkubationsschritt weg. In der zweiten Antikörperreaktion wurde das StreptavidinMeerrettich-Peroxidase-Konjugat (RPN 1231)“ (SHRPC) von Amersham Pharmacia Biotech UK Limited statt HRP eingesetzt. Die Membran wurde in 10ml Antikörperlösung II. Material und Methoden 49 (TNT, 1% (w/v) BSA, 2µl SHRPC) bei 4°C in einer verschweißten Plastiktasche inkubiert. Alle folgenden Schritte wurden wie beschrieben durchgeführt. II.9. Computerprogramme Die Auswertung der DNA- und Protein-Sequenzen erfolgte mit Hilfe folgender Programme: BLAST, The NCBI BLAST family of programs (Altschul et al., 1997) DNASIS for Windows, Version 2 (Hitachi Software; San Bruno) AlLIGN, Version 1.01 (Scientific u. Educational Software, 1989) Vector NTI Suite, Version 6.0 (InforMax, Inc.; No. Bethesda, MD 20852) TMHMM (Transmembrane Hidden Markov Model, Sonnhammer et al., 1998) 50 III. Ergebnisse III. Ergebnisse III.1. Selektion und Untersuchung entkoppelter Mutanten Während des Transportvorganges eines hochaffinen Substrates über das Phosphoenolpyruvat-abhängige Kohlenhydrat:Phosphotransferasesystem wird das Substrat normalerweise durch eine Phosphorylierung chemisch modifiziert. Dabei wird davon ausgegangen, dass bei hochaffinen Substraten der Transport über die Zytoplasmamembran und die anschließende Phosphorylierung gekoppelt ablaufen. Es sind sowohl bei Escherichia coli als auch bei Klebsiella pneumoniae entkoppelte EIIMtl Mutanten beschrieben worden (Klawitter, 1992; Scholle 1993; Heuel, 1997; Otte, 2000), bei denen Transport und Freisetzung des Substrates in das Zytoplasma unabhängig von der Phosphorylierung ablaufen. Diese Mutanten geben Hinweise auf wichtige am Transportprozess beteiligte Aminosäuren und deren Einfluss auf die Kopplung von Translokation des Substrates und Energetisierung des Transporters. Daher war es von großem Interesse, die Selektionssysteme zu optimieren, um weitere Mutanten zu selektionieren und diese anschließend zu charakterisieren. III.1.1. Konstruktion von Stämmen zur Selektion entkoppelter Mutanten Die früheren und in dieser Arbeit verwendeten Selektionssysteme benutzen einen semisynthetischen Stoffwechsel, der es E. coli ermöglicht, unphosphoryliertes („freies“) D-Mannitol zu verstoffwechseln. Da die Mtl.1P-Dehydrogenase nicht in der Lage ist, unphosphoryliertes Mtl umzusetzen, wurde in früheren Ansätzen der Stamm LGS323 (∆(mtlAPA‘) MtlD+) mit den Plasmiden pGJ9∆137 (mtlAPA’) und pASL14 (dalD+K+) transformiert (Scholle, 1993). Die carboxyterminale Deletion des mtlA in dem Plasmid pGJ9∆137 beinhaltet die Nukleotidsequenz, welche für die AS His554 der IIAMtlDomäne kodiert, wodurch ein gekoppelter Transport von Mtl verhindert wird. Eine direkte Phosphatgruppenübertragung von HPr auf das Cys384 der IIBMtl-Domäne ist nicht möglich (Scholle, 1993). Das Gen dalD codiert für die D-ArabinitolDehydrogenase (DalD) aus Klebsiella oxytoca M5a1, welche sowohl unphosphoryliertes D-Arabinitol (Atl) als auch unphosphoryliertes D-Mannitol (Mtl) als Substrat erkennt und III. Ergebnisse 51 eine Umsetzung zu D-Xylulose (Xul) bzw. D-Fructose (Fru) katalysiert (Wood et al., 1961; Tanaka et al., 1967; Charnetzky & Mortlock, 1974; Lengeler 1975b; Heuel et al., 1998). Bei der Selektion auf Wachstum mit D-Mtl erhält man Mutanten, die den entkoppelten Transport von D-Mtl ermöglichen (siehe Abb. III.1.1.). Die Reproduzierbarkeit dieses Systems scheiterte jedoch an der starken Kurierung des Plasmides pASL14. Um eine Kurierung und somit den Verlust der Fähigkeit, freies Mannitol umzusetzen, zu verhindern, musste dalD auf stabilere Plasmide kloniert werden. Abb.III.1.1. Wildtyp- und semisynthetischer Stoffwechselweg für den D-Mtl-Abbau in E. coli Unter I. WT ist der D-Mtl-Abbau im WT und unter II. mtlA∆137 der semisynthetische DMtl-Abbau in LGS322-1 /pGJ9∆137 dargestellt. Rote Pfeile: Stoffwechselweg für den normalen, d.h. gekoppelten Transport von D-Mtl über ein Wild-Typ II.CBAMtl. Unterbrochene rote Pfeile (): kein Transport. Grüne Pfeile: Stoffwechselweg bei entkoppeltem Transport von D-Mtl. Die IIC-Domäne mit entkoppeltem Transport ist mit einem Stern versehen (IICMtl*). Orangefarbene Pfeile: Phosphatgruppenübertragung über allgemeine PTS-Komponenten. Unterbrochene orangefarbene Pfeile (): keine Phosphatgruppenübertragung. Grau schattiert: Deletierte Domäne des II.CBAMtl. Ⓟ~AS: An der Phosphatgruppenübertragung beteiligte Aminosäuren. MtlD: Mtl.1P-Dehydrogenase. DalD: D-Arabinitol-Dehydrogenase. Weitere Erläuterungen siehe Text und Abkürzungsverzeichnis. 52 III. Ergebnisse Abb. III.1.2. Schematische Darstellung der Konstruktion von F’lac::dalD Die einzelnen Schritte der Konstruktion werden im nachfolgendenText näher beschrieben. Die grafisch hervorgehobenen genetischen Elemente sind nicht maßstabsgetreu dargestellt. bla = Gen für eine β-Lactamase (Ampicillin-Resistenz, ApR). IR = zwei sich invertiert wiederholende Sequenzen (ermöglichen die Transposition von TnLSD101). mks = multiple Klonierungsstelle. ori = Replikationsursprung des Plasmides pBR322. spc = Gen für eine Spectinomycin-Resistenz (SpR). III. Ergebnisse 53 III.1.1.1. In vivo Klonierung von dalD auf ein F’-Plasmid In einem ersten Ansatz wurde das Gen dalD in den Niedrigkopien-Vektor pHEX3 (Heuel, 1997) kloniert. Das resultierende Plasmid pHEXD zeigte jedoch eine für das Selektionssystem zu hohe Kurierungsrate. Da F’-Plasmide eine hohe Stabilität in Bakterien zeigen, wurde anschließend das F’lac als Träger des Gens dalD gewählt. Bei der Konstruktion des F’lac::dalD -Plasmides wurde nach Schmid et. al. (1991) und Zeppenfeld et. al. (2000) das Gen zwischen zwei sich invertiert wiederholende Sequenzen („inverted repeats“ = IR) kloniert, um es so als Transposon springen zu lassen. Das verwendete Plasmid pTIM101 erlaubt zusätzlich eine „Blau/Weiß-Selektion“ durch α-Komplementation, trägt eine multiple Klonierungsstelle aus dem pHEX3, ein ΩFragment zur Verhinderung unphysiologischer Expression durch einen fremden, stromaufwärts liegenden Promotor und eine Spectinomycin-Resistenz. Das Gen für die Transposase (tnpA), welche das Springen des Transposons ermöglicht, wird von einem weiterem Plasmid pPSO110 exprimiert, wodurch eine Stabilisierung des Transposons in einem pPSO110-freien Stamm gewährleistet ist. Im folgenden werden die Einzelschritte beschrieben, welche zur Konstruktion des Plasmides F’lac::dalD durchgeführt wurden (siehe Abb. III.1.2.). Konstruktion des Plasmids pHEXD (dalDK’) Das erste für den semisynthetischen Stoffwechsel benötigte Gen dalD stammt aus Klebsiella oxytoca M5a1. Es kodiert für eine D-Arabinitol-Dehydrogenase (DalD) und wurde aus dem Plasmid pGHL3 (Heilenmann, unveröffentlicht) in die mks des Niedrigkopie-Vektors pHEX3 subkloniert. Ein Doppelverdau von pGHL3 mit den Restriktionsenzymen HindIII und EcoRI ergab u.a. ein 2,7kb großes dalD/dalK‘Fragment, welches anschließend mit dem entsprechend verdauten Vektor pHEX3 zum neuen Plasmid pHEXD ligiert wurde. Die Funktionalität der D-Arabinitol-Dehydrogenase (DalD) aus pHEXD wurde in dem Stamm LGS322 getestet. Der Stamm LGS322 kann über das Glukose-PTS (II.CBAGlc) freies D-Arabinitol transportieren (Budde, 1991), welches ohne DalD nicht verstoffwechselt wird. 54 III. Ergebnisse Tab. III.1.1. Plattentest zum Nachweis einer funktionellen DalD aus pHEXD Platten1 McC Atl MM Atl LB0 LGS322 /pHEX3 s - + LGS322 /pHEXD 2+ + + Stamm 1 Supplementation mit den entsprechenden Antibiotika, Kohlenhydraten und Aminosäuren (siehe Abkürzungsverzeichnis) wie im Teil II der Arbeit angegeben. Phänotyp der Kolonien auf McC-Platten: 3+ = dunkelrot mit trübem Hof; 2+ = dunkelrot; + = rot; (+) = rosa ; (w) = rosa mit weißem Rand; w = weiß; s = sensitiv; - = kein Wachstum. Phänotyp der Kolonien auf MM-Platten: + = Wachstum; (+) = schwaches Wachstum, auf den Platten erst nach 2-3 Tagen sichtbar; - = kein Wachstum. Phänotyp auf LB0-Platten: + = Wachstum; (+) = schwaches Wachstum; - = kein Wachstum. Der Markertest zeigt, dass dalD vom Plasmid pHEXD in ausreichender Menge exprimiert wird, und somit ein schnelles Wachstum auf freiem D-Arabinitol ermöglicht (Phänotyp Atl2+). LGS322/pHEX3 ist ohne dalD auf D-Arabinitol sensitiv Phänotyp, was vermutlich auf eine intrazelluläre Anhäufung von D-Atl 5-P zurückzuführen ist. Konstruktion des Plasmids pLSD101 mit dem Transposon TnLSD101 Um das dalD+/dalK‘-Fragment des Plasmids pHEXD auf ein F’lac-Plasmid springen zu lassen, musste es vorher in ein künstliches Transposon kloniert werden. Dieses Transposon wurde durch die beiden IRs auf dem Plasmid pTIM101 gebildet. Das dalD+/dalK‘-Fragment wurde folglich mit den Restriktionsenzymen ClaI und SacI (SstI) aus dem Plasmid pHEXD geschnitten und in die entsprechenden Stellen der mks des Plasmides pTIM101 kloniert. Das resultierende Plasmid wurde pLSD101 genannt. Die Funktionalität von pLSD101 wurde in dem Stamm LGS322/pGJ9∆137 E218A überprüft. Der Austausch E218A, der in mtlA∆137 kodiert wird, ermöglicht den Transport von freiem D-Mannitol in die Zelle (Klawitter, 1992; Scholle 1993; Otte, 2000) und eine funktionelle DalD den zum Wachstum notwendigen Abbau von D-Mannitol. Folglich wurde das Plasmid in den benannten Stamm transformiert und das Wachstum auf D-Mtl und D-Atl getestet (Tab. III.1.2.). III. Ergebnisse 55 Tab. III.1.2. Plattentest zum Nachweis funktioneller DalD aus pLSD101 Platten1 McC Mtl Cam/Spc McC Mtl Cam McC Mtl Spc /pGJ9∆137 E218A - w - - - /pLSD101 - - w - - 2+ 2+ 2+ + + Stamm LGS322 /pGJ9∆137 E218A /pLSD101 1 MM Mtl MM Atl Cam/Spc Cam/Spc Für Symbole und sonstige Erklärungen siehe Tab. III.1.1. Der neue Stamm LGS322 /pGJ9∆137 E218A /pLSD101 zeigt ausreichende Expression von DalD, um eine entkoppelte IIMtl-Mutante auf freiem D-Mannitol bzw. D-Arabinitol wachsen zu lassen. Konstruktion des Kreuzungs-Stammes STL136 mit dem Plasmid F’lac::dalD Der Stamm CSH36 mit den Plasmiden F’lac, pLSD101 (TnLSD101) und pPSO110 (tnpA+) wurde benötigt, um das Transposon TnLSD101 mit dalD+/dalK‘ auf das F’lac -Plasmid springen zu lassen. Kompetente Zellen des Stammes CSH36 /F’lac wurden parallel mit den beiden Plasmiden pLSD101 und pPSO110 transformiert und die Selektion auf LB0Cam/Spc-Platten durchgeführt. Gereinigte und getestete Kolonien der gesuchten Exkonjuganten (CSH36 /F’lac /pLSD101 /pPSO110) wurden in LB0Cam/Spc angeimpft, über Nacht bei 37°C und anschließend für 3 Tage jeweils bei RT oder im Kühlschrank inkubiert. In dieser Zeit sollte die Transposition F’lac::TnLSD101 ermöglicht werden. Darauffolgend wurden die Kulturen in LB0 gewaschen und eine Flüssigkreuzung mit dem Stamm S136-3 durchgeführt. Das Ausstreichen auf LB0Spc/Kan - Platten selektionierte die S136-3-Stämme, welche das F’lac::TnLSD101 erhalten hatten. 56 Exkonjuganten wurden auf LB0-Platten gereinigt und anschließend getestet, wovon sechs Kolonien den folgenden Phänotyp zeigten (Tab. III.1.3.). 56 III. Ergebnisse Tab. III.1.3. Plattentest der Exkonjuganten aus der Kreuzung CSH36 /F’lac::dalD /pLSD101 /pPSO110 X S136∆ Platten1 McC Glc McC Lac McC Mtl LB0 Amp LB0 Cam LB0 Spc LB0 Kann 2+ + 2+ + + + - S136-3 w w w - - - + S136-3 F’lac::TnLSD101 w + w - - + + Stamm CSH36 /F’lac::TnLSD101 /pPSO110 /pLSD101 1 Für Symbole und sonstige Erklärungen siehe Tab. III.1.1. Angegeben sind die Ergebnisse vom Donor, Rezipienten und Exkonjuganten. Der neue Stamm S136-3/F’lac::TnLSD101 zeigt die gewünschten Phänotypen und wurde STL136 genannt. Mit Hilfe dieses Stammes konnte das F’lac::TnLSD101 (im folgenden kurz F’lac::dalD genannt) in die verschiedenen Stämme zur Mutantenselektion und -Charakterisierung gekreuzt werden. III.1.1.2. Konstruktion der Selektions-Stämme LGS31-1, LGS322-1 und LGS323-1 Für die Verstoffwechselung von D-Mannitol wurde das F’lac::dalD in die gewünschten Selektionsstämme LGS31, LGS322 und LGS323 gekreuzt. Zur Gegenselektion wurden die Rezipientenstämme mit dem Plasmid pGJ9∆137E218A transformiert. Dies ermöglichte in Verbindung mit F’lac::dalD eine Gegenselektion auf MM Mtl-Platten. Zudem kodiert das Plasmid eine Chloramphenicol-Resistenz, die ebenfalls zur Selektion genutzt werden konnte. Die Stämme LGS31/pGJ9∆137 E218A, LGS322/pGJ9∆137 E218A und LGS322/pGJ9∆137E218A wurden jeweils mit dem Stamm STL136 gekreuzt und auf MM-Mtl.Cam- und MM-Mtl.Cam/Spc-Platten ausgestrichen. Gereinigte und getestete Exkonjuganten wurden in MM.Gly-Medium angeimpft und die ÜK dreimal in neues Medium überimpft. Anschließend wurde eine geeignete Verdünnung der Kulturen auf LB0-Platten ausgestrichen. Dabei zeigte sich bei etwa 2% der getesteten EK ein Verlust des Plasmides. Die kurierten Stämme LGS31./F’lac::dalD, LGS322-/F’lac::dalD und LGS323./F’lac::dalD wurden entsprechend LGS31-1, LGS322-1 und LGS323-1 genannt. III. Ergebnisse 57 III.1.1.3. Konstruktion des Selektions-Stammes LTK31 (∆mtlA MtlD+ recA+ ∆(ptsHIcrr)::kan) In einem zweiten Selektionssystem wurde die Phosphorylierungskette nicht in dem EIIMtl-Transporter sondern an den allgemeinen PTS-Komponenten des Stammes LGS31 unterbrochen (Abb.III.1.3.). Mit Hilfe einer P1-Transduktion wurde eine ∆(ptsH ptsI crr)::kan-Kasette aus dem Stamm TP2811 in den Stamm LGS31 gekreuzt. Die Integration der ∆(ptsH ptsI crr)::kan-Kasette in das Chromosom von LGS31 bewirkt eine Deletion der Gene ptsH, ptsI und crr. Folglich werden das HPr (ptsH) und EI (ptsI) der allgemeinen PTS-Komponenten sowie das EIIAGlc (crr) nicht mehr exprimiert. Da die Methode eine Rekombination in vivo erfordert, wurde der Stamm LGS31 (recA+) den ∆recA-Stämmen LGS322 und LGS323 vorgezogen. Die Selektion erfolgte auf LB0Kan/Str-Platten und ergab eine für diese Methode hohe Transduktionsrate. Nach der Reinigung auf LB0-Platten wurden 56 EK in einem Plattentest überprüft, wovon 51 das gewünschte Ergebnis zeigten (Tab. III.1.4.). Tab. III.1.4. Plattentest des Selektionsstammes LTK31 Stamm Platten1 McC Mtl McC Glc McC Fru McC Gal LB0 Str LB0 Kan LB0 Tet LB0 Spc LGS31 w 3+ 3+ s + - - - LTK31 w w w s + + - - 1 Für Symbole und sonstige Erklärungen siehe Tab. III.1.1. Die gewünschten Transduktanten zeigten einen KnR/Pts--Phänotyp (Glc-, Fru-). Der resultierende Stamm wurde LTK31 genannt. Konstruktion der Selektions-Stämme LTK31-1 und LTK31-2 mit den Plasmiden F’lac::dalD (DalD+ SpcR) u. pUR404 (scr+ ScrR- TetR) Für die Selektion von entkoppelten Mutanten war es erforderlich, dass im Stamm LTK31 die Gene dalD und scrK vorhanden waren (Abb.III.1.3.). Nach der Reduktion von D-Mannitol durch DalD muss die entstandene D-Fruktose für eine weitere Verstoffwechselung phosphoryliert werden. Da das EIIFru als Folge der Deletion der Gene für die allgemeinen PTS-Komponenten inaktiviert wurde, erhielt LTK31 alternativ das konjugierbare Plasmid pUR404. Dieses exprimiert aufgrund einer Deletion in scrR 58 III. Ergebnisse konstitutiv scrK, welches für die ATP-abhängige D-Fruktose-Kinase ScrK kodiert (Schmid et al. 1988, Aulkemeyer et al. 1991). Abb.III.1.3. Alternativer semisynthetischer Stoffwechselweg für den D-Mtl-Abbau in E. coli III. WT ∆ptsHIcrr: Semisynthetischer D-Mtl-Abbau in LTK31-1/pGJ9. Unterbrochene rote Pfeile (): kein normaler Transport. Grüne Pfeile(¼): Stoffwechselweg bei entkoppeltem Transport von D-Mtl. IICMtl*: IICMtl mit entkoppeltem Transport. Grau schattiert: Allgemeine PTS-Komponenten, deren Gene deletiert wurden. DalD: DArabinitol-Dehydrogenase. ScrK: D-Fruktose-Kinase. Weitere Erläuterungen siehe Text und Abkürzungsverzeichnis. In den Stamm LTK31 wurde deshalb zunächst das F‘lac::dalD aus LGS31-1 gekreuzt und auf LB0Kan/Spc-Platten selektioniert. Gereinigte und getestete Exkonjuganten (LTK31./F‘lac::dalD) wurden LTK31-1 genannt. Der Rezipient LTK31-1 wurde anschließend mit dem Donorstamm PS5 /pUR404 gekreuzt und auf LB0Kan/Spc/Tet-Platten ausgestrichen. Nach Reinigung auf LB0-Platten wurde der Exkonjugations-Stamm LTK31-1./pUR404 in einem Plattentest identifiziert und LTK31-2 genannt. III. Ergebnisse 59 Tab. III.1.5. Plattentest LTK31-1 und LTK31-2 Platten1 Stamm MM Fru MM Scr LB0 Kan LB0 Spc LB0 Tet PS5 /pUR404 ( scr+ ScrR- TetR ) + + - - + LTK31 ( ∆(ptsHI crr)::kan ) - - + - - LTK31-1 ( DalD+ SpcR ∆(ptsHI crr)::kan ) - - + + - LTK31-2 ( DalD+ SpcR ∆(ptsHI crr)::kan scr+ ScrR- TetR ) - - + + + 1 Für Symbole und sonstige Erklärungen siehe Tab. III.1.1. Der Stamm LTK31-1 zeigte die durch das F‘lac::dalD vermittelte SpectinomycinResistenz und LTK31-2 zusätzlich die von pUR404 kodierte Tetracyklin-Resistenz. Das Plasmid pUR404 vermittelte keinen Scr+-Phänotyp in LTK31-2, da das EIIScr die EIIAGlcDomäne (crr) benötigt (Tab. III.1.5.). Konstruktion des Stammes LTK32-1 ( ∆(ptsHI crr)::kan Mak+ ) Der Stamm JWL300 zeigt eine erhöhte Expression (Mak+) des normalerweise kryptisch vorliegenden mak-Lokus. Diese Mutation ermöglicht dem PtsI--Stamm JWL300 das Wachstum auf D-Fruktose (Aulkemeyer et al. 1991). Ein P1-Phagenlysat aus dem Donor JWL300 wurde zur Transduktion in den Rezipienten LTK31 eingesetzt und die Transduktanten auf MM Fru-Platten selektioniert. Dabei waren nach sechs Tagen ca. 50 Kolonien auf der Selektionsplatte zu sehen. Auf der Platte mit der negativen Kontrolle wuchsen ebenfalls nach sechs Tagen 3 Kolonien, die als Spontanmutationen klassifiziert aber nicht genauer charakterisiert wurden. Nach Reinigung und Identifizierung wurde der LTK31 Mak+-Stamm LTK32 genannt. Über eine Konjugation mit LGS322-1 wurde das F‘lac::dalD in den Rezipienten LTK32 gekreuzt. LTK32-/F‘lac::dalD wurde LTK32-1 genannt. Die nachfolgende Tabelle zeigt die Phänotypen, die bei der Konstruktion und Kontrolle der Stämme betrachtet wurden. 60 III. Ergebnisse Tab. III.1.6. Plattenmarkertest LTK32 und LTK32-1 Platten1 Stamm JWL300 Mak+ MM Fru MM Scr LB0 Kann LB0 Spc (+) - - - - - + - LTK32 ∆(ptsHI crr) Mak+ (+) - + - LGS322-1 F‘lac::dalD + - - + LTK32-1 F‘lac::dalD ∆(ptsHI crr) Mak+ (+) - + + LTK31 ∆(ptsHI crr) 1 Für Symbole und sonstige Erklärungen siehe Tab. III.1.1. LTK32 zeigt die Fähigkeit auf Fructose (MM Fru = (+)) zu wachsen, welche aus dem Stamm JWL300 in LTK31 tranzduziert werden konnte. Es bestätigte sich das erwartete sehr langsame Wachstum auf dieser Kohlenstoffquelle. Das für die Selektion erforderliche DalD konnte mit dem Plasmid F‘lac::dalD aus LGS322-1 in LTK32 gekreuzt werden (LTK32-1 auf LB0Spc = +), (Tab. III.1.6.). III.1.1.4. Vergleich der eingesetzten Selektionssysteme In erster Linie sollte bei der Konstruktion der neuen Systeme eine hohe Reproduzierbarkeit der Ergebnisse erreicht werde, die bei pASL14 als Folge starker Plasmidkurierung nicht gegeben war. Das stabile Einzelkopieplasmid F‘lac::dalD erlaubte sowohl die Selektionen neuer Mutationsarten bzw. -orte als auch die Bestätigung der erhalten Mutationen. Vergleich der Stämme LGS322 und LGS323 (nach Scholle, 1993) Der Transport von Atl über das EIIMtl weist auf einen funktionellen Einbau der Translokationsdomäne des EIIMtl hin. Dieser Hinweis ist besonders bei Mtl--Mutanten hilfreich, die den Transport betreffen. Mit dem Stamm LGS323 sollte der Transport von D-Arabinitol über ein plasmidkodiertes, verkürztes EIIMtl getestet werden. Dazu musste der WT-Transport von D-Atl in LGS322 ausgeschaltet werden. Der Stamm LGS323 wurde als AtlR Variante von LGS322 selektioniert (Scholle, 1993). LGS322 ist auf McC Atl sensitiv, kann aber mit einem DalD+-Plasmid zu Atl+ transformiert werden. Die Sensitivität von LGS322 wird auf eine cytoplasmatische Anhäufung von D-Atl-P III. Ergebnisse 61 zurückgeführt, die mit der Verstoffwechselung von D-Atl oder D-Atl-P durch DalD verhindert werden kann (Scholle, 1993). Der Stamm LGS323 erschien dagegen sowohl mit als auch ohne DalD auf McC Atl weiß. 1991 zeigte Budde, dass auch das GlukosePTS (IICBAGlc) Atl transportieren kann, wohingegen alle anderen bekannten AtlTransportsysteme (Gat-PTS, Gut-PTS, Fru-PTS, Mtl-PTS) in LGS322 inaktiviert sind. Der Stamm LGS323 zeigte bei Transporttests mit α-Methyl-Glukose, welches bei einer Testkonzentration von 25µl über ein aktives Glc-PTS aufgenommen wird, keine Transportaktivität, wohingegen der Elternstamm LGS322 eine Transportaktivität von 14 [nmol pro mg Protein pro Minute] aufwies. Weiterhin war LGS322/pJOE637 Scr+ während LGS323/pJOE637 einen Scr—Phänotyp zeigte. Das Plasmid pJOE637 (scr+) vermittelt nur bei Vorhandensein eines intakten EIIAGlc (crr+) einen Scr+-Phänotyp. Die AtlR von LGS323 wurde daraufhin auf eine Mutation in crr (crr-) zurückgeführt. Tab. III.1.7. Ursprüngliche AtlR- und AtlS-Phänotypen (nach Scholle, 1993) Platten1 Stamm Relevanter Genotyp LGS322 ∆mtlA ptsG+crr+ s w 3+ 14 LGS323 ∆mtlA ptsG+ w w 2-3+ 0 LGS322 /pASL14 ∆mtlA ptsG+crr+ /dalD 3+ n.g. 3+ n.g. LGS323 /pASL14 ∆mtlA ptsG+ /dalD w n.g. 2-3+ n.g. LGS322 /pJOE637 ∆mtlA ptsG+crr+ /scr+ s 3+ n.g. n.g. LGS323 /pJOE637 ∆mtlA ptsG+ /scr+ w w n.g. n.g. McC Atl McC Scr McC Glc α-Me-Glc Tp2 1 Für Symbole und sonstige Erklärungen siehe Tab. III.1.1. Die Anzucht der Zellen für den α-Me-Glc-Transporttest erfolgte zur Messung des uninduzierten Basalniveaus in MM CAA (0,1%) ohne Zugabe von Glukose. 14C-α-MeGlc wurde in einer Endkonzentration von 25µM zugegeben. Die Messwerte sind in [nmol × min-1 × mg Protein-1] angegeben. 2 Der in dieser Arbeit eingesetzte LGS323 aus der Glyzerinkultur von Scholle zeigte jedoch auf Atl und Scr einen abweichenden Phänotyp. Diese Variante von LGS323 wurde LGS324 genannt. 62 III. Ergebnisse LGS324 zeigte mit DalD einen 2+ Phänotyp auf McC Atl, war aber ohne DalD noch AtlR. Zudem war LGS324./pJOE637 Scr+. Eine Mutation in crr konnte damit weitestgehend ausgeschlossen werden (WT-Expression von NagE vorausgesetzt; Vogler et al., 1988). Um die Auswirkungen einer authentischen crr--Mutation auf den AtlPhänotyp festzustellen, wurden mit Hilfe einer Genkassette eine crr-Deletionen in den Stamm LGS31 eingeführt. In den RecA--Stämmen LGS322 und LGS324 konnte keine eindeutige Insertion der Genkassette erreicht werden (Tab. III.1.8.). Tab. III.1.8. Neu beobachtete AtlR- und AtlS-Phänotypen Stamm Genotyp LGS322 LGS324 Platten1 McC Atl McC Scr ptsG+crr+ s w ptsG+crr+ w w LGS322 /pHEXD ptsG+crr+ /dalD 2+ w LGS324 /pHEXD ptsG+crr+ /dalD 2+ w LGS322 /pJOE637 ptsG+crr+/scr+ s 3+ LGS324 /pJOE637 ptsG+crr+/scr+ w 3+ 1 Für Symbole und sonstige Erklärungen siehe Tab. III.1.1. Mit Hilfe einer P1-Transduktion wurde die ∆(crr)::kan-Kasette aus LLR103 in LGS31 gekreuzt. Die Selektion erfolgte auf LB0Kan/Str-Platten. Nach Ausplattierung des halben Transduktionsansatzes und Inkubation ÜN wurden 400 EK gezählt. Nach der Reinigung auf LB0-Platten wurden 14 EK in einem Plattentest auf Kan- und Str-Resistenz überprüft. Der transduzierte Stamm LGS31 wurde LGT31 genannt. Außerdem wurden die Transduktanten mit den Plasmiden pJOE637 (scr+) bzw. pHEXD (dalD) transformiert, um eine Kontrolle auf den Scr- und Atl-Phänotyp zu ermöglichen. Der Stamm LTK31 (∆(ptsH ptsI crr)::kan) wurde ebenfalls eingesetzt. Aus dem Markertest (Tab. III.1.9.) ergaben sich folgende Beobachtungen: Eine crr-Deletion konnte keine vollständige Atl-Resistenz bewirken. LGT31/pJOE637 (scr+) war Scr-, was auf eine korrekte Insertion der Genkassette hinweist. LGT31 war weniger sensitiv als der Elternstamm LGS31, wies aber mit dalD einen Atl+-Phänotyp auf. Die crr-Deletion reduzierte die Sensitivität, wobei allerdings immer noch Atl in die Zelle III. Ergebnisse 63 transportiert werden konnte. Folglich war die Atl-Resistenz von LLR103 nicht alleine auf ∆crr zurückzuführen. Tab. III.1.9. Plattentest P1.∆(crr)::kan X LGS31 Platten Stamm LGS322 ∆mtlA ∆recA 1 McC Atl MM Xyl LB0Kan + dalD 3 + scr 2 McC Scr Tet McC Atl Cam s 2+ - 3+ 2+ w 2+ - w W LGS324 ∆mtlA ∆recA Atlr w 2+ - 3+ 2+ LLR103 ∆crr ∆mtlAP,O w 2+ + (w) W ss 5 2+ - 3+ 2+ LGT31 ∆mtlA ∆crr s 2+ + (w) 2+ LTK31 ∆mtlA ∆(ptsHI crr) w 2+ + w 2+ LGS323 ∆mtlA ∆recA Atlr LGS31 ∆mtlA 4 1 Für Symbole und sonstige Erklärungen siehe Tab. III.1.1. 2 Die Stämme der jeweiligen Zeile wurden mit dem Plasmid pJOE637 (scr+) transformiert. 3 Die Stämme der jeweiligen Zeile wurden mit dem Plasmid pHEXD (dalD) transformiert (Ausnahme: 4 Die Daten für LGS323 wurden aus der Tabelle III.1.8. übernommen). 5 ss = supersensitiv: kein Wachstum auf der Platte, kein Papillenwachstum. Die Phänotypen aller Stämme auf McC Atl mit und ohne dalD zeigten, dass der Transport von Atl nicht zwangsläufig zu einer Sensitivität führte. Die Stämme LGS324 und LTK31 waren ohne dalD AtlR aber mit dalD Atl+. Aus den Beobachtungen geht hervor, dass mehrere Faktoren zu einer Resistenz bzw. Sensitivität führen. In Bakterien ist die Anhäufung von phosphorylierten Zwischenprodukten häufig die Ursache toxischer Effekte (Ferenci und Kornberg, 1973; Lengeler, 1975; Solomon und Lin, 1972). D-Atl-P kann von E. coli K-12 nicht weiter verstoffwechselt werden und wirkt so auf die Zellen toxisch. Der Transport von D-Atl kann über das EIIGlc erfolgen. Ob bei dem Transport eine teilweise oder vollständige Phosphorylierung des Substrates stattfindet, ist nicht bekannt. Die Phosphorylierung von D-Atl könnte daher durch das EIIGlc, der Xylulose-Kinase XylB (Scangos und Reiner, 1979) oder eine weitere unbekannten Kinase erfolgen. Eine Deletion von crr verhindert in jedem Fall den gekoppelten Transport von D-Atl über das EIIGlc durch die Repression der Expression des EIIGlc. Ein entkoppelter Transport würde aber immer noch freies D-Atl in die Zelle bringen. Bei einer Anhäufung von Arabinitol kann XylB D-Atl phosphorylieren und zu einer Sensitivität führen (Scangos und Reiner, 1979). Dagegen 64 III. Ergebnisse wird in Gegenwart von dalD D-Atl zu D-Xylulose (Charnetzky und Mortlock, 1974a) und D-Xylulose durch XylB zu D-Xylulose-5iP verstoffwechselt (Lawlis et al., 1984), welches in den Pentosephosphat-Zyklus eingespeist werden kann. Eine Resistenz kann vorliegen, wenn der Transport von D-Atl oder dessen Phosphorylierung verhindert wird. Eine XylB-Mutante mit dalD würde dementsprechend zu einer nicht toxischen Anhäufung von DXylulose und einem Atl--Phänotyp führen. Alle AtlR- und AtlS-Stämme zeigten jedoch den gleichen Xyl+-Phänotyp. Eine durch die beschriebenen Ergebnisse nicht ersichtliche Kombination dieser Faktoren ist in dem Stamm LGS323 zu erwarten. Für die Selektionssysteme konnte kein Stamm konstruiert werden, der die relevanten Eigenschaften von LGS323 besaß. Die weiteren Versuche wurden hauptsächlich mit dem Stamm LGS322 durchgeführt. LGS322-1 (∆mtlA ∆gut DalD+) /pGJ9∆137 (mtlAPA’) Von den oben genannten Selektionssystemen wurde im Lauf der Arbeit hauptsächlich der Stamm LGS322-1./pGJ9∆137 benutzt. Parallel durchgeführte Selektionen mit den Stämmen LGS31-1.(∆mtlA dalD+)./pGJ9∆137 und LGS324-1 (∆mtlA ∆gut dalD+ AtlR) /pGJ9∆137 ergaben keine neuen Mutationsarten. LTK32-1 (∆mtlA ∆(ptsH ptsI crr)::kan DalD+ Mak+) /pGJ9 (mtlA) Im Stamm LTK32-1./pGJ9 erfolgt die Phosphorylierung der D-Fruktose über die Mannofruktokinase (Mak). Der Stamm LTK32-1-/pGJT9-1 zeigte wie erwartet nur sehr schlechtes Wachstum auf MM-Mtl-Cam-Platten und keinen deutlichen + Phänotyp auf McC-Mtl-Cam-Platten. Die Aktivität der Mannofruktokinase war in diesem System nur sehr gering zumal sie schon dem Donorstamm JWL300 nur eine Generationszeit von 160min auf MM mit 1% Fru ermöglichte (Aulkemeyer et al., 1991). Um die Selektion zu beschleunigen, wurde in einem weiteren System die Fruktokinase aus dem SucroseOperon eingesetzt. LTK31-2 (∆mtlA ∆(ptsH ptsI crr)::kan DalD+ ScrK+) /pGJ9 (mtlA) Um festzustellen, ob das System LTK31-2-/pGJ9 grundsätzlich zu einer Selektion von entkoppelten mtlA-Mutanten geeignet war, wurde der Stamm LTK31-2 mit dem Plasmid pGJT9-1 transformiert. Dieses Plasmid entstand aus pGJ9, in das die III. Ergebnisse 65 entkoppelnde Mutation E218A mit Hilfe der lokalisierten Mutagenese eingeführt wurde (siehe III.1.3.2.). Der Stamm LTK31-2-/pGJT9-1 zeigte gutes Wachstum auf MM-MtlCam-Platten. In einer 1:107 Mischung mit LGS31 (Mtl-) wuchs der Stamm mit deutlichem 2+ Phänotyp auf McC-Mtl-Cam-Platten durch den Bakterienrasen. Damit konnte gezeigt werden, dass dieses System das Wachstum von entkoppelten Mutanten der E218A-Art ermöglicht. III.1.2. Entkoppelte Mutanten aus den Selektionen Die Selektion der entkoppelten Mutanten erfolgte in den beschriebenen Selektions-Systemen nach gleichem Vorgehen. Eine EK des eingesetzten Stammes wurde in einem Reagenzglas in 5ml LB0Cam angeimpft und über Nacht bei 37°C in einem Roller (bzw. Schüttelwasserbad bei 30°C und 42°C) inkubiert. Von der ÜK wurden 0,3ml (≈ 6 x 108 Zellen bei einer durchschnittlichen OD650.=.2 der ÜN-Kulturen) direkt auf einer Selektionsplatte ausgestrichen. Die Selektion konnte sowohl auf MM-MtlCam- als auch auf McC-Mtl-Cam-Platten durchgeführt werden. Aufgrund der deutlicheren Identifikation von Mutanten wurden hauptsächlich MM-Platten verwendet. Die Zellen wurden vor dem Ausplattieren nicht gewaschen, um durch das LB0-Medium noch etwas Wachstum auf dem MM-Medium und damit weitere Mutationen zu ermöglichen. Um ein Austrocknen des Agars zu verhindern, wurden die Platten in Plastiktüten luftdicht verschlossen und anschließend mehrere Tage inkubiert bis Einzelbzw. rote Kolonien zu sehen waren. Die Inkubationstemperatur wurde je Ansatz zusätzlich variiert (30°C, 37°C bzw. 42°C). Die so erhaltenen Kolonien wurden gereinigt und anschließend noch einmal auf ihren Mtl-Phänotyp überprüft. Bei einem positiven Phänotyp wurde eine Plasmidisolierung durchgeführt und der entsprechende Selektionsstamm mit dem erhaltenen Plasmid transformiert. Damit konnte sichergestellt werden, dass der beobachtete Phänotyp nicht chromosomal kodiert war. Aus den mutierten Plasmiden wurde das HindIII-SnaB1-Fragment des mtlA-Gens ausgeschnitten (siehe Abb. III.1.4.) und an Stelle des entsprechenden Fragments in das Plasmid pGJ9∆137 kloniert. Nach Retransformation des entsprechenden Selektionsstamms konnte bei einem Mtl+Phänotyp davon ausgegangen werden, dass die Mutation in dem HindIII-SnaB1Fragment des mtlA-Gens lag. Dieses Fragment wurde anschließend doppelsträngig sequenziert, um die Mutation zu ermitteln. 66 III. Ergebnisse Die erhaltenen Mutanten und Ihre Phänotypen werden in Teil III.1.5. dieser Arbeit näher beschrieben. Zum besseren Verständnis folgender Punkte des Ergebnisteils wird nachstehend die mtlA-Sequenz aus E. coli mit für diese Arbeit relevanten Informationen angegeben. Abb. III.1.4. Sequenz des Genes mtlA aus E. coli K-12 -35 -10 SalI TTGACA TAT AAT +1 CCCGTCG ACTGGACAGT TAACCGATTC AGTGCCAGAT TTCGCAGTAT CTACAAGGTC CGGCTACCTC RBS AGGAG G TGCCGCCACA TTAACAAAAA ACCTCGGGCT TCCAGCCTGC GCGACAGCAA ACATAAGAAG GGGTGTTTTT mtlA-Start 1 S3 20 HindIII 40 50 60 70 ATGTCATCCG ATATTAAGAT CAAAGTGCAA AGCTTTGGTC GTTTCCTCAG CAACATGGTG ATGCCAAATA 80 90 100 110 120 130 140 TCGGCGCGTT TATCGCGTGG GGTATCATCA CCGCGTTATT TATTCCAACA GGGTGGTTAC CGAACGAGAC 150 160 170 180 190 200 210 GCTGGCGAAG CTGGTCGGGC CGATGATCAC TTATCTCCTG CCGCTGCTGA TCGGTTATAC CGGTGGTAAG 220 230 240 250 260 270 280 CTGGTAGGCG GCGAACGTGG CGGCGTAGTC GGTGCCATCA CCACCATGGG CGTTATCGTC GGCGCAGACA 290 300 310 320 C110 340 350 TGCCGATGTT CCTCGGTTCT ATGATTGCAG GTCCGCTGGG CGGCTGGTGC ATTAAGCACT TCGACCGCTG 360 370 380 390 400 410 420 GGTAGACGGT AAGATCAAAT CCGGTTTTGA GATGCTGGTG AATAACTTCT CCGCAGGCAT CATCGGGATG 430 440 450 460 470 S158 480 490 ATCCTCGCTA TTCTGGCATT CCTCGGCATT GGCCCGATTG TTGAAGCCCT GTCCAAAATG CTGGCTGCGG 500 510 520 530 540 550 560 GCGTTAACTT CATGGTTGTC CATGACATGC TGCCGCTGGC GTCTATCTTT GTTGAACCGG CGAAAATCCT 570 580 590 S199 610 620 630 GTTCCTCAAC AACGCCATTA ACCACGGTAT CTTCTCGCCG CTGGGTATTC AGCAGTCCCA TGAACTGGGT S212 650 E218 660 670 680 690 700 AAATCAATCT TCTTCCTGAT TGAAGCTAAC CCAGGTCCAG GTATGGGCGT GCTGCTGGCG TACATGTTCT 710 720 S242 740 750 760 H256 TTGGTCGTGG TAGCGCTAAA CAGTCTGCGG GCGGTGCGGC AATCATCCAC TTCCTGGGGG GTATCCACGA 780 790 800 810 820 830 840 AATCTACTTC CCGTATGTGC TGATGAATCC GCGTCTGATC CTCGCAGTCA TCCTCGGCGG TATGACTGGC 850 860 870 880 890 S299 910 GTGTTCACGC TGACTATCCT GGGCGGTGGT CTGGTTTCTC CGGCATCTCC GGGTTCTATC CTTGCTGTAC 920 930 940 950 C320 970 980 TGGCGATGAC ACCAAAAGGT GCTTACTTCG CTAACATCGC GGGTGTGTGT GCGGCGATGG CTGTCTCCTT 990 1000 1010 1020 1030 1040 1050 CGTTGTCTCT GCTATTTTGC TGAAAACCAG CAAAGTGAAA GAAGAAGATG ATATTGAAGC AGCAACTCGT 1060 1070 1080 1090 1100 1110 1120 CGTATGCAGG ACATGAAAGC TGAGTCTAAA GGCGCATCTC CGCTGTCTGC TGGCGATGTG ACTAACGACC III. Ergebnisse 67 SnaBI 1140 C384 1160 1170 1180 1190 TGAGCCACGT ACGTAAAATC ATCGTTGCCT GTGACGCCGG TATGGGTTCC AGTGCGATGG GCGCAGGCGT 1200 1210 1220 1230 1240 1250 1260 TCTGCGTAAG AAAATTCAGG ATGCAGGTCT GTCGCAGATT TCTGTTACTA ACAGCGCGAT CAACAACCTG 1270 1280 1290 1300 1310 1320 1330 CCGCCAGATG TGGACCTCGT CATCACTCAC CGTGACCTGA CCGAACGCGC TATGCGCCAG GTTCCGCAGG 1340 1350 1360 1370 1380 1390 1400 CACAGCATAT TTCGCTGACC AACTTCCTCG ACAGCGGCCT GTACACCAGC CTGACCGAAC GTCTGGTTGC 1410 1420 1430 1440 1450 1460 1470 TGCCCAACGC CACACGGCAA ACGAAGAGAA AGTAAAAGAC AGCCTGAAAG ACAGCTTTGA CGATTCCAGT 1480 1490 1500 1510 1520 1530 ∆137 GCTAACCTGT TCAAGCTAGG CGCGGAGAAC ATCTTCCTCG GTCGCAAAGC GGCAACCAAA GAAGAAGCGA 1550 1560 1570 1580 1590 1600 1610 TTCGTTTTGC TGGCGAGCAG CTGGTGAAAG GCGGTTACGT TGAGCCGGAA TACGTTCAGG CGATGCTGGA 1620 1630 1640 1650 H554 1670 1680 TCGTGAAAAA CTGACCCCGA CTTATCTGGG TGAGTCTATC GCGGTGCCAC ACGGTACGGT TGAAGCGAAA 1690 1700 1710 C571 1720 1730 1740 1750 GATCGCGTAC TGAAAACGGG CGTCGTGTTC TGCCAGTACC CGGAAGGCGT GCGCTTCGGT GAAGAAGAAG 1760 1770 1780 1790 1800 1810 1820 ATGACATTGC CCGTCTGGTG ATTGGTATTG CTGCCCGTAA CAACGAGCAC ATTCAGGTTA TCACCAGCCT 1830 1840 1850 1860 1870 1880 1890 GACCAATGCA CTGGATGATG AGTCCGTCAT CGAGCGTCTG GCACACACCA CCAGCGTGGA TGAAGTGCTG 1900 1910 mtlA-Stopp GAACTGCTGG CAGGTCGTAA GTAATCCAAT CCCACCCTCT CCACATGGAG AAGGTGGGGT TAATTGCCTG BamHI ATGCGCTACG CTTATCAGGA TCCCAGGATG CATCACAATT TGTTGAATTT GCACGTTCTT GTAGG Abb. III.1.4. Sequenz des Genes mtlA aus E. coli K-12 (nach Lee & Saier, 1983 - korrigiert 1993) Dargestellt ist die Nukleotidsequenz in dem Bereich deslA in E. coli K-12. Die vermuteten -35 und -10-Regionen (Davis et al., 1988; Jiang et al., 1990) und die mögliche Ribosomenbindestelle (RBS) sind in Boxen mit darüber angegebenen Konsensussequenzen dargestellt. Der mögliche Transkriptionsstartpunkt der mRNA-Synthese ist mit +1 gekennzeichnet (Davis et al., 1988). Die Zählung beginnt mit dem Adenin des ATGStartcodons. Farblich hervorgehoben sind folgende Sequenzteile: blau = Start- und Stoppkodon; grün = Erkennungssequenzen relevanter Restriktionsenzyme; rot = relevante Tripletts für wichtige AS; orange = Ende des mtlA∆137-Fragmentes. III.1.2.1. Entkoppelte Mutanten aus den Selektionen LGS322-1 (∆mtlA ∆gut DalD+) /pGJ9∆137 (mtlAPA’) und LGS324-1 (∆mtlA ∆gut DalD+ AtlR) /pGJ9∆137 (mtlAPA’) Bei den Selektionen in den beiden oben genannten Stämmen waren nach 4-6 Tagen wenige (0-5) mögliche Mutanten auf den Platten zu erkennen. Erwartungsgemäß dauerte es bei 30°C und 42°C 2-3 Tage länger bis Kolonien zu erkennen waren. Mit 68 III. Ergebnisse Ausnahme der Mutation E218V wurden die erhaltenen Mutationsarten bei allen Temperaturen isoliert. Die Variante E218V konnte nur einmal bei 37°C isoliert werden. Temperaturabhängige Wachstumsunterschiede stellten sich in allen untersuchten Fällen als chromosomal codiert heraus. Bei 22 unabhängig voneinander isolierten Mutanten wurden folgende entkoppelte Mutationen identifiziert: Tab. III.1.10. Entkoppelnde Mutationen aus LGS322-1 /pGJ9∆137 und LGS324-1 /pGJ9∆137 Phänotyp E218A Selektionstemp. E218V H256P Mutation in bp 653 bp 653 bp 767 GAA Ö GCA GAA Ö GTA CAC Ö CCC 30 [°C] 2 - 3 37 [°C] 5 (2) 1 6 (3) 42 [°C] 2 - 3 Gesamt 9 1 12 Die Tabelle gibt die Art und Anzahl der erhaltenen Mutationen bei unterschiedlichen Temperaturen wieder. Die betroffenen Nukleotide und deren Nummern sind blau hervorgehoben (Zählung wie in Abb. III.1.4.). In Klammern ist der Anteil der Mutanten aus LGS324-1 angegeben. In neun Fällen führte die Transversion des zweiten Adenins in dem Triplett 218 (GAA) zu Cytosin zu dem Aminosäureaustausch E218A (Glutaminsäure zu Alanin). Eine weitere Transversion in demselben Nukleotid zu Thymin führte zu dem AS-Austausch E218V (Glutaminsäure zu Valin). Zwölf Mutationen betrafen das Adenin in dem Triplett 256 (CAC). Diese bewirkten eine Transversion zu Cytosin und damit den AS-Austausch H256P (Histidin zu Prolin). Bei der Selektion mit den beiden Stämmen zeigte sich, dass die Inkubationstemperatur vermutlich keinen Einfluss auf die Art der Mutation hatte. Die Plasmide mit den jeweiligen Mutationen wurden folgendermaßen benannt: Plasmid resultierender AS-Austausch pGJ9∆EA = pGJ9∆EV = pGJ9∆HP = E218A E218V H256P Die Eigenschaften der Plasmide sind nachfolgend in der Tab. III.1.11. dargestellt. III. Ergebnisse 69 III.1.2.2. Entkoppelte Mutanten aus der Selektion LTK31-2 (∆mtlA ∆(ptsH ptsI crr)::kan DalD+ ScrK+) /pGJ9 (mtlA) Die Selektionen mit diesem Stamm wurden bei 37°C auf MM-Mtl-Cam-Platten durchgeführt. Dabei wurden nach 3 Tagen 3-6 Kolonien je Platte beobachtet. Von acht unabhängig isolierten und untersuchten Mutanten zeigten sechs den Austausch E218A (Transversion GAA Ö GCA) und zwei den Austausch H256P (Transversion CAC Ö CCC). Somit wurde mit diesem System keine neue Mutation selektioniert, allerdings konnten die Ergebnisse aus dem vorangegangenen System bestätigt werden. Die Phänotypen aller entkoppelten Mutanten sind in der folgenden Tabelle III.1.11. dargestellt. Die verwendeten Plasmide sind wie oben beschrieben aus den originalen Mutanten konstruiert worden und entsprechen den Phänotypen der ursprünglich isolierten Mutanten. Tab. III.1.11. Phänotypen der entkoppelten Mutanten Markerplatten1 Stamm McC Mtl MM Mtl LB0Cam LB0Spc LGS322-1 w - - + LGS322-1 /pGJ9∆137 w - + + LGS322-1 /pGJ9∆EA 2+ + + + LGS322-1 /pGJ9∆EV 2+ + + + LGS322-1 /pGJ9∆HP 2+ + + + 1 Für Symbole und sonstige Erklärungen siehe Tab. III.1.1. III.1.3. Entkoppelte Mutanten aus lokalisierter Mutagenese Um auf einem weiteren Weg zeigen zu können, dass die beobachteten Phänotypen von der beschriebenen Mutation abhängen, wurden die entsprechenden Mutationen durch lokalisierte Mutagenese in das Plasmid pGJ9 eingeführt. Dadurch konnten auch die Auswirkungen der Mutationen in einem nicht deletierten mtlA (EIIMtlABC) untersucht werden. Um ein mit dem pGJ9∆137 vergleichbares Deletionsplasmid zu erhalten, wurde anschließend über vorhandene RestriktionsenzymSchnittstellen eine Deletion eingeführt (siehe III.1.3.2.). Weiterhin bot sich die Möglichkeit einen Doppelaustausch in den betroffenen Tripletts einzuführen, um bei Wachstumsuntersuchungen und der Selektion von Suppressormutanten Reversionen zu erschweren. 70 III. Ergebnisse III.1.3.1. Konstruktion des Plasmides pGAL3 (mtlA’) und seiner mutierten Derivate Die lokalisierte Mutagenese wurde nach dem „Altered Sites“ in vitro Mutagenese System von Promega ausgeführt. Dazu musste ein Fragment aus mtlA, welches das zu mutierende Triplett enthält, in den Mutagenese-Vektor pALTER-1 kloniert werden. Das Plasmid pGJ9 (Abb. III.1.5.) wurde mit HindIII verdaut und das resultierende 2,1kb-mtlA’-Fragment in die entsprechende HindIII-Restriktionsschnittstelle von pALTER-1 kloniert. Die korrekte Ausrichtung des Fragments wurde mit einer BamHIRestriktionsanalyse überprüft und die beiden Enden des Fragments über Sequenzanalyse identifiziert. Das Plasmid wurde pGAL3 genannt. Das System mit dem MutageneseVektor pALTER-1 verwendet zur Erhöhung der Mutagenese-Ausbeute ein AmpicillinReperatur-Oligonukleotid. Nach durchgeführter Mutagenese besitzt der Vektor die Antibiotikaresistenzen gegen Tetracyklin und Ampicillin. Während der Mutagenese wurde festgestellt, dass es eine Tandem-Duplikation einer 11bp langen Sequenz (5’GGCGTGCTGCT-3’) in mtlA (Basen 676-686) und in der Tet-Resistenz von pALTER-1 (876-886) gibt, die häufiger zu Deletionen führte. Daher wurde im Folgenden immer eine Selektion mit Tetracyklin durchgeführt, um das Vorkommen deletierter Plasmide zu mtlA tet’ p15A ori (5746) BamHI (1978) HindIII (2135) ClaI (2141) ∆137 (1532) SnaBI (1132) HindIII (30) pGJ9 SalI (5750) unterdrücken. cat ‘tet ATG (1/5882) Abb. III.1.5. mtlA-Plasmid pGJ9 (nach Grisafi et al., 1989) Schematische Darstellung des mtlA-Plasmids pGJ9. Die Position der Erkennungssequenzen der relevanten Restriktionsenzyme und die Deletion ∆137 sind in Klammern angegeben. Die Zählung beginnt mit dem Adenin des ATG-Startcodons (1/5882). III. Ergebnisse 71 Konstruktion der Plasmide pGAL3-1, pGAL3-2 und pGAL3-3 mit zweifachem Basenaustausch Die Konstruktion der Plasmide erfolgte durch den Einsatz der Mutagenese-Primer E218A (+), E218V (+) und H256P (+). Bei der Auswahl der neuen Codone wurde nach zwei Kriterien vorgegangen: 1. Es mussten mindestens zwei Basenaustausche im Codon für eine Reversion zu der WT-Aminosäure an der jeweiligen Stelle erforderlich sein. 2. Von den verbleibenden Codonen wurde das mit der häufigsten Verwendung („codon usage“) in mtlA in E. coli gewählt. In der folgenden Tabelle sind die aus der Mutagenese resultierenden Doppelaustausche dargestellt, wie sie durch einzelsträngige Sequenzierung nachgewiesen wurden. Tab. III.1.12. Austausche bei pGAL3-1, pGAL3-2 und pGAL3-3 Plasmidname MutagenesePrimer 1 Basenaustausch Basennummern2 pGAL3-1 E218A (+) GAA Ö GCC 652-654 pGAL3-2 E218V (+) GAA Ö GTC 652-654 pGAL3-3 H256P (+) CAC Ö CCA 766-768 1 Phänotypen in Teil III.1.5.1. 2 Zählung wie in Abb. III.1.4. Konstruktion der Plasmide pGAL3-4 und pGAL3-5 mit zweifachen AS-Austauschen Die Konstruktion dieser Plasmide erfolgte durch den gleichzeitigen Einsatz zweier Mutagenese-Primer in einem Mutageneseansatz. Damit konnten die unterschiedlichen Mutationen, die einen entkoppelten Transport von D-Mannitol ermöglichen, kombiniert und ihr Phänotyp untersucht werden. Dabei wurden die Kombinationen, die für die ASAustausche E218A (+)/H256P(+) und E218V(+)/H256P(+) kodieren, gewählt. In der folgenden Tabelle sind die resultierenden Nukleotid-Austausche dargestellt, wie sie durch einzelsträngige Sequenzierung nachgewiesen wurden. 72 III. Ergebnisse Tab. III.1.13. Austausche bei pGAL3-4 und pGAL3-51 Plasmidname MutagenesePrimer H256P (+) GAA Ö GCC (CTG Ö CAG)3 CAC Ö CCA 652-654 685-687 766-768 E218V (+) H256P (+) GAA Ö GTC CAC Ö CCA 652-654 766-768 E218A (+) pGAL3-4 pGAL3-5 1 3 Basenaustausch Basennummern2 .Phänotypen in Teil III.1.5.1. 2.Zählung wie in Abb. III.1.4. -unbeabsichtigter Austausch L229Q (Leucin zu Glutamin). III.1.3.2. Klonierung der Plasmidreihe pGJT9 mit einem vollständigen mtlA-Gen Die mutierten mtlA-Fragmente mussten aus den vorangehend beschriebenen pGAL3-Plasmiden über einen HindIII-Verdau zurück in den mtlA-Vektor pGJ9 kloniert werden. Dafür wurde zur Vereinfachung das Plasmid pGJ9dHindIII konstruiert, dem das entsprechende HindIII-Fragment fehlte. Die mtlA HindIII-Fragmente aus den mutierten pGAL3-Plasmiden wurden nach der Klonierung in pGJ9dHindIII einzelsträngig sequenziert und die neu konstruierten wie in Tab.II.1.4. Plasmide benannt. Um ein dem pGJ9∆137 entsprechendes Deletionsplasmid zu erhalten, wurden die neuen Plasmide mit SnaBI und ClaI verdaut. Anschließend wurde der um ca. 1kb verkürzte Vektor nach einer Klenow-Behandlung religiert. Die dadurch entstandenen Plasmide trugen mtlA’-Gene, die eine Deletion des für das Cys384 aus Domäne IIBMtl und das His554 aus IIAMtl kodierenden Bereiches aufwiesen (Vgl. Abb. III.1.4.). Die Klonierungen ergaben folgende Plasmide. pGJT9-1 = pGJ9∆HindIII + mtlA HindIII-Fragmente aus pGAL3-1 = pGJT9-1d = pGJT9-1 mtlA‘ (SnaBI/ClaI-Deletion) pGJT9-2 = pGJ9∆HindIII + mtlA HindIII-Fragmente aus pGAL3-2 = pGJT9-2d = pGJT9-2 mtlA‘ (SnaBI/ClaI-Deletion) pGJT9-3 = pGJ9∆HindIII + mtlA HindIII-Fragmente aus pGAL3-3 = pGJ9 H256P pGJT9-3d = pGJT9-3 mtlA‘ (SnaBI/ClaI-Deletion) pGJT9-4 = pGJ9∆HindIII + mtlA HindIII-Fragmente aus pGAL3-4 = pGJ9 E218A, L229Q, H256P pGJ9 E218A pGJ9 E218V III. Ergebnisse 73 pGJT9-4d = pGJT9-4 mtlA‘ (SnaBI/ClaI-Deletion) pGJT9-5 = pGJ9∆HindIII + mtlA HindIII-Fragmente aus pGAL3-5 = pGJ9 E218V, H256P pGJT9-5d = pGJT9-5 mtlA‘ (SnaBI/ClaI-Deletion) III.1.4. Selektion von Suppressormutanten Aus der Selektion von Suppressormutanten in Stämmen mit entkoppeltem aber vollständigem EIIMtl sollten Mutationen an anderer Stelle im mtlA gesucht werden, die entkoppelte Mutanten wieder in den Mtl+-Phänotyp umwandeln. Die entkoppelnden Mutationen E218A und E218V (pGJT9-1 und pGJT9-2) zeigten in dem vollständigen MtlA keine Beeinträchtigung des Wachstums auf D-Mannitol (siehe Teil III.1.5.1.), können also nicht zur Suppressor-Selektion eingesetzt werden. Dagegen verursachten die Mutationen H256P, E218A/L229Q/H256P und E218V/H256P im Stamm LGS322 einen Mtl--Phänotyp. Mit den Stämmen LGS322 /pGJT9-3 /pGJT9-4 und /pGJT9-5 konnte so eine Selektion auf Supressormutanten durchgeführt werden. Eine ÜK des jeweiligen Stammes wurde auf MM Mtl-Cam-Platten ausgestrichen und bei 30°C bzw. 37°C inkubiert. Erhaltene Kolonien wurden gereinigt, getestet, Zellen der entsprechenden Kolonien mit dem Plasmid transformiert, das HindIII-SnaB1-Fragment neu kloniert und anschließend sequenziert. III.1.4.1. Suppressormutanten aus den Stämmen LGS322 /pGJT9-3 (H256P), /pGJT9-4 (E218A/H256P) und /pGJT9-5 (E218V/H256P) Nach vier Tagen Inkubation waren bei den Selektionen mit LGS322 /pGJT9-3 unabhängig von der Inkubationstemperatur 8-16 Kolonien pro Platte zu sehen. Es wurden jeweils sechs bzw. fünf (30°C/37°C) Kolonien untersucht und die Mutationen identifiziert (siehe Tab. III.1.14.). Die Selektionen von Suppressormutationen aus LGS322 /pGJT9-4 ergaben 3 Kolonien pro Platte, wobei die Kolonien erst nach sieben Tagen Inkubation unabhängig von der Inkubationstemperatur zu sehen waren. Zwei Kolonien aus der 30°C- und eine Kolonie aus der 37°C-Selektion wurden untersucht. Die folgenden Tabellen III.1.14a. und III.1.14b. fassen die erhaltenen Mutationen zusammen. 74 III. Ergebnisse Tab. III.1.14. Mutationen aus LGS322 /pGJT9-3 und LGS322 /pGJT9-4 LGS322 /pGJT9-3 Genotyp der Mutation Phänotyp der Mutation Anzahl der Mutationen bei 30°C 37°C C766CA Ö T766CA Pro256 Ö Ser256 3 0 CC767A Ö CA767A Pro256 Ö Gln256 3 5 LGS322 /pGJT9-4 C766CA Ö G766CA Pro256 Ö Ala256 1 0 CC767A Ö CA767A Pro256 Ö Gln256 1 1 Die Tabelle gibt Ort, Art und Anzahl der Mutationen bei 30°C und 37°C wieder. Die mutierten Basen und resultierenden AS der betroffenen Tripletts sind nach Abb. III.1.4. nummeriert. Mit dem Ansatz LGS322-/pGJT9-5 wurden keine Mutanten gefunden. Die neuen Plasmide wurden folgendermaßen benannt: pDSM1 = pGJT9-3 P256Q pDSM2 = pGJT9-3 P256S pDSM3 = pGJT9-4 E218A/L229Q/P256A pDSM4 = pGJT9-4 E218A/L229Q/P256Q III.1.5. Charakterisierung der Mutanten Anhand der Selektion in den jeweiligen Stämmen wurde lediglich die Fähigkeit des Wachstums auf D-Mannitol ohne DalD festgestellt. Die entkoppelten Mutanten waren darauf angewiesen, freies, das heißt unphosphoryliertes D-Mannitol zu transportieren und zu verstoffwechseln. Bei den „Supressormutanten“ dagegen erforderte die Selektion in Abwesenheit des DalD eine Wiederherstellung des Transports und der Phosphorylierung. Dabei war nicht auszuschließen, dass Anteile des D-Mannitols unphosphoryliert in die Suppressor-Zelle gelangen. Um einen Vergleich des Wachstums aller Mutanten und des WT auf D-Mannitol zu erstellen, wurden weitere III. Ergebnisse 75 Untersuchungen mit den Mutanten in unterschiedlichen Stamm/Plasmid-Kombinationen durchgeführt. Dabei wurden der Transport und einzelne Komponenten des Transports wie Phosphorylierung und Substratbindung untersucht. Weil die relevanten Phänotypen der Mutanten aus der ortspezifischen Mutagenese und den Selektionen keine ersichtlichen Unterschiede zeigten, wurde, wenn nicht anders beschrieben, bei allen Tests mit Plasmiden aus der gerichteten Mutagenese gearbeitet. III.1.5.1. Phänotypen der verschiedenen Mutanten Der von den Mutationen vermittelte Phänotyp wurde zunächst mit Hilfe von Plattentests und Wuchskurven untersucht. Dabei war es für die Feststellung der Ursache und des Grades der Entkopplung erforderlich, in unterschiedlichen Stammhintergründen und mit unterschiedlichen Plasmidkonstruktionen zu arbeiten. Vor allem der Einfluss des F’dalD auf das Wachstum sollte Hinweise auf freies D-Mannitol in der Zelle geben. In der Darstellung der Markertests wurde versucht, die Ergebnisse so gut wie möglich zu gliedern bzw. zu bündeln, um den Vergleich der verschiedenen Mutanten untereinander zu erleichtern. Markertests und Wachstumskurven entkoppelter Mutanten in den Stämmen LGS322 und LGS322-1 Im folgenden wurden in den Stämmen LGS322 und LGS322-1 (F’lac::dalD+) nur Plasmide aus der gerichteten Mutagenese verwendet, womit weitere unerkannte Mutationen ausgeschlossen werden konnten. Unterschiede in der Fähigkeit, D-Mannitol in Abhängigkeit des Vorhandenseins von dem F’lac::dalD zu verstoffwechseln, fielen bereits bei diesem, in der Tab. III.1.15. dargestellten, qualitativen Test auf. Im Allgemeinen verbessert DalD das Wachstum der Mutanten auf D-Mannitol. Eine Ausnahme stellten die Mutationskombinationen E218A/L229Q/H256P und E218V/H256P sowie das deletierte WT-Protein dar, welche kein Wachstum auf D-Mtl ermöglichten. Zellen mit dem WT-Plasmid pGJ9 zeigten auf McC Mtl mit und ohne DalD einen 3+-Phänotyp. Eventuelle Wachstums-Unterschiede sind in diesem Fall auf McC-Platten nicht ersichtlich. Auf D-Arabinitol unterdrückte DalD bei allen Stämmen die Sensitivität und ermöglichte die Verstoffwechslung des Zuckeralkohols. 76 III. Ergebnisse Tab. III.1.15. Markertest entkoppelnder Mutanten in LGS322 und LGS322-1 Stamm LGS322 LGS322-1 LGS322 LGS322-1 LGS322 LGS322-1 LGS322 LGS322-1 LGS322 LGS322-1 LGS322 LGS322-1 LGS322 LGS322-1 LGS322 LGS322-1 LGS322 LGS322-1 LGS322 LGS322-1 LGS322 LGS322-1 LGS322 LGS322-1 Plasmid relevante Austausche Markerplatten1,2 McC Mtl MM Mtl McCAtl /pGJ9∆137 w - S ” w - 2+ /pGJ9 3+ + S ” 3+ + 2+ /pGJT9-1∆ w - S + + 2+ 2+ + S ” 3+ + 2+ /pGJT9-2∆ w - S + + 2+ + + S ” 3+ + 2+ /pGJT9-3∆ w - S + + 2+ w - S ” 2+ + 2+ /pGJT9-4∆ w - S w - 2+ w - S ” w - 2+ /pGJT9-5∆ w - S w - 2+ w - S w - 2+ ” /pGJT9-1 ” /pGJT9-2 ” /pGJT9-3 ” /pGJT9-4 ” /pGJT9-5 ” E218A E218V H256P E218A/ L229Q/ H256P E218V/ H256P 1 Für Symbole und sonstige Erklärungen siehe Tab. III.1.1. 2Phänotyp aller Stämme auf McC Glc = 3+, McC Gut = w, LB0Str = +, LB0Cam = +. Die AS-Austausche E218A, E218V und H256P ermöglichten im verkürzten EIIMtl (LGS322-1 /pGJT9-1∆, /pGJT9-2∆, pGJT9-3∆) nur mit DalD Wachstum auf D-Mtl. In diesen Mutanten fand entkoppelter Transport statt. In einem vollständigen EIIMtl ohne DalD konnten nur die Mutanten mit den Austauschen E218A und E218V auf D-Mtl wachsen (LGS322 /pGJT9-1, /pGJT9-2). Das Wachstum war in beiden Fällen III. Ergebnisse 77 schwächer als beim WT. Mit DalD zeigen die Mutanten wieder WT-Aktivität. In einem vollständigen EIIMtl wurde das Substrat durch die Mutationen E218A und E218V nur teilweise phosphoryliert und somit ein unterschiedlich großer Anteil an D-Mtl unphosphoryliert transportiert. Die Mutation H256P verhinderte in einem vollständigen EIIMtl ohne DalD das Wachstum der Zellen auf D-Mtl vollständig (LGS322 /pGJT9-3), während mit DalD ein Mtl+-Phänotyp erreicht wurde (LGS322-1 /pGJT9-3). Mit dem AS-Austausch H256P war der gekoppelte Transport für ein Wachstum auf MM Mtl nicht ausreichend, es fand hauptsächlich oder nur entkoppelter Transport statt. Die Mutations-Kombinationen E218A/L229Q/H256P und E218V/H256P verhinderten mit und ohne DalD Wachstum auf D-Mtl. Diese Kombinationen hatten entweder eine große Einwirkung auf Bindung und/oder Transport, oder eine funktionelle Faltung der Translokationsdomäne wurde beeinträchtigt. Markertest der „Suppressormutanten“ in LGS322 (∆mtlA), LGS322-1 (∆mtlA DalD+) und LTK31-2 (∆mtlA ∆(ptsH ptsI crr)::kan DalD+ ScrK+) In der folgenden Tabelle (Tab. III.1.16.) sind die relevanten Markertesteigenschaften der Suppressormutanten in dem Stamm LGS322 und LGS322-1 dargestellt. Um festzustellen, ob diese Aminosäureaustausche auch zu einer Entkopplung führen können, wurden die Mutationen zusätzlich in einem Stamm ohne allgemeine PTS-Komponenten getestet. Hierfür wurde der Stamm LTK31-2 eingesetzt. Als Referenz wurden die Stämmen LTK31-2, LGS322 bzw. LGS322-1 mit den Plasmiden pGJ9 und pGJT9-1 angeführt. In den Stämmen LGS322 und LGS322-1 vermittelten die Plasmide pDSM1 (P256Q) und pDSM2 (P256S) wieder eine WT-Aktivität auf McC Mtl-Platten. In LTK31-2 war mit diesen Plasmiden keine Entkoppelung mehr zu erkennen. Dagegen zeigten die Plasmide pDSM3 (E218A/L229Q/P256A) und pDSM4 (E218A/L229Q/P256Q) in LTK31-2 entkoppelten Transport sowie einen schwächeren Phänotyp in dem Stamm LGS322, was auf die noch vorhandene entkoppelnde Mutation E218A zurückgeführt werden kann. Die bessere Verstoffwechselung von DMannitol in dem Stamm LGS322-1 zeigte, dass auch bei diesen Mutanten entkoppelter und gekoppelter Transport stattfindet. Allerdings führten beide Plasmide in den Stämmen LGS322 und LGS322-1 zu schwächeren Phänotypen auf D-Mannitol als das 78 III. Ergebnisse Plasmid pGJT9-1 mit der einfachen E218A-Mutation. Dadurch wird ein weiterer Einfluss der Mutationen L229Q und/oder P256A bzw. P256Q beim Wachstum auf D-Mannitol deutlich. Besonders im Hinblick auf die scheinbaren WT-Aktivitäten der einfachen Mutationen P256Q und P256S sind weitere Untersuchungen erforderlich. Grundsätzlich fielen bei entkoppelten Mutanten die Phänotypen der D-Mannitol-Verstoffwechselung in dem Stamm LTK31-2 schwächer aus als in dem Stamm LGS322-1. Die Ursache hierfür ist aber eher im grundsätzlich abweichenden Stammhintergrund als in funktionellen Unterschieden in dem Transportmechanismus zu suchen. Tab. III.1.16. Plattentest der Stämme LGS322, LGS322-1 und LTK31-2 mit den „Suppressor-Mutationen“ Stamm Plasmid LTK31-2 /pGJ9 LGS322 /pGJT9-1 relevante Austausche E218A Markerplatten1,2 McC Mtl MM Mtl Mc Atl w - 2+ 2+ + S 3+ + 2+ LGS322-1 ” LTK31-2 ” + + 2+ LGS322 /pDSM1 3+ + S 3+ + 2+ P256Q LGS322-1 “ LTK31-2 ” w - 2+ LGS322 /pDSM2 3+ + S 3+ + 2+ w - 2+ (+) (+) S 2+ + 2+ + + 2+ (+) (+) S 2+ + 2+ + + 2+ LGS322-1 “ LTK31-2 ” LGS322 /pDSM3 LGS322-1 “ LTK31-2 ” LGS322 /pDSM4 LGS322-1 “ LTK31-2 ” 1,2 P256S E218A/ L229Q/ P256A E218A/ L229Q/ P256Q siehe Tabelle III.1.15. Zellerträge und Generationszeiten mit verschiedenen mtlA-Plasmiden Über Wuchskurven können die Wachstums-Vergleiche der unterschiedlichen Mutanten auf D-Mannitol in quantitativer Weise bestimmt werden. Da die Ergebnisse der Plattentests schon zeigten, dass es nach dem Transport über die veränderten III. Ergebnisse 79 IIC*BA-Domänen zu einer Freisetzung von teilweise und nicht phosphoryliertem DMannitol kommen kann, wurden das Wachstum von Zellen mit und ohne DalD in einem vollständigem EIIMtlC*BA verglichen. Tab. III.1.17. Zellerträge und Generationszeiten der Stämme LGS322 und LGS322-1 mit verschiedenen mtlA-Plasmiden Stamm LGS322-1 (F’lac::dalD+) LGS322 Gt1 [min] Zellertrag2 [∗108 B/ml] Gt1 [min] Zellertrag2 [∗108 B/ml] 83 - 88 8 - 8,5 81 - 87 7,5 - 8 k.W. k.W. k.W. k.W. /pGJT9-1 E218A 180 - 200 4 - 4,4 110 - 125 6–8 /pGJT9-2 E218V 230 - 380 2,5 - 3 105 - 115 6 - 7,5 /pGJT9-3 H256P k.W. k.W. 220 - 345 3,5 – 4,3 /pGJT9-4 E218A/L229Q/H256P k.W. k.W. k.W. k.W. /pGJT9-5 E218V/H256P k.W. k.W. k.W. k.W. /pDSM1 P256Q 82 - 87 7,5 - 8 86 - 88 7 /pDSM2 P256S 84 - 87 7-8 84 - 86 7 /pDSM3 E218A/L229Q/P256A 580 - 900 0,3 - 1 140 - 150 5,5 - 6 /pDSM4 E218A/L229Q/P256Q 330 - 360 2 - 2,5 145 - 155 5 - 5,5 /Plasmid relevante Austausche /pGJ9 /pGJ9∆HindIII Die Stämme wurden in MM Mtl/Glc Cam angeimpft und über Nacht bei 37°C in einem Roller inkubiert. Die ÜK wurden in MM+ gewaschen und in vorgewärmtes Medium (MM Mtl [0,2%] Cam) zu einer Zellzahl von 5x107 B/ml überimpft. Alle Wuchskurven wurden im Schüttelwasserbad bei 37°C mindestens dreimal unabhängig voneinander aufgenommen. 1Generationszeiten (Gt) wurden aus der Steigung der Wuchskurven in der exponentiellen Wachstumsphase berechnet. 2Zellerträge wurden aus der Differenz der anfänglichen (t0) und der maximal gemessenen OD420 in einem Zeitraum von 24h ermittelt. k.W. = kein Wachstum. Als Kontrolle für das Wachstum unter WT-Bedingungen wurden die Messungen mit den Stämmen LGS322/pGJ9, LGS322-1/pGJ9 durchgeführt. Die 80 III. Ergebnisse Generationszeiten für Zellen mit dem WT-MtlA (pGJ9) in einem Stamm mit und ohne DalD (LGS322-1 bzw. LGS322) lagen bei den Messungen in beiden Fällen reproduzierbar in einem Bereich zwischen 80 und 90 Minuten. Ein etwas geringerer Zellertrag wurde lediglich bei den Stämmen mit dem F’-Plasmid beobachtet (ca. 0,5∗108 B/ml weniger Zellen). Um einen pleiotropen Effekt der Mutationen auf das Wachstum ausschließen zu können, wurden Wachstumsmessungen in MMGlc [0,2%] mit den entsprechenden Antibiotika und Aminosäuren durchgeführt. Dadurch sollte der Einfluss des F’-Plasmids, eines zusätzlichen Plasmids und des Chloramphenicols berücksichtigt werden. Die Messungen wurden jeweils dreimal durchgeführt. Der Stamm LGS322 zeigte dabei eine Generationszeit von 66.±3 min., LGS3221 wuchs mit 70.±3 min. etwas langsamer. Auch der Zellertrag war beim LGS322-1 etwas schwächer (9.±0,5 ∗108 B/ml) als beim LGS322 (9,5.±0,5 ∗108 B/ml). Beide Effekte könnten auf das zusätzliche F’-Plasmid in dem Stamm LGS322-1 zurückgeführt werden. Wuchsen die beiden Stämme mit dem Plasmid pGJ9∆HindIII, welches um den Großteil von mtlA deletiert ist, auf Chloramphenicol, verlangsamte sich das Wachstum in beiden Fällen um etwa 7min. (LGS322: 73.±3 min.) bzw. 5 min. (LGS322-1: 75.±3 min.) und auch der Zellertrag reduzierte sich (LGS322: 9.±0,5 ∗108 B/ml; LGS322-1: 7,5.±0,5 ∗108 B/ml). Im Vergleich zu diesen Ergebnissen wurde bei allen anderen in der Tab. III.1.17. aufgeführten Plasmiden in dem Stamm LGS322 eine Generationszeit von 75.±3 min (Zellertrag 7,5 ±0,5 ∗108 B/ml) und in dem Stamm LGS322-1 eine Generationszeit von 80.±3 min (Zellertrag 7 ±0,5 ∗108 B/ml) gemessen. Lediglich bei Stämmen mit dem Plasmid pGJT9-2 (E218V) wurde eine verlängerte Anlaufphase im Wachstum festgestellt, die sich allerdings nicht in der Generationszeit bemerkbar machte. Aufgrund der lediglich geringfügigen Abweichungen im Wachstum der Stämme mit den verschiedenen mtlA-Plasmiden wurde ein pleiotroper Effekt der Mutationen in mtlA ausgeschlossen. Bei der Betrachtung des Wachstums von Zellen mit den unterschiedlichen MtlAVarianten auf D-Mannitol lassen sich die Phänotypen in einer ersten Betrachtung in vier Gruppen unterteilen. Dabei wird die erste Gruppe aus Mutanten mit den Mutationen P256Q, und P256S gebildet, die mit und ohne DalD eine Generationszeit und einen Zellertrag im Bereich des Wildtyps zeigten. Die Messungen deuten darauf hin, dass die Mutationen III. Ergebnisse 81 bei der untersuchten D-Mtl-Konzentration keinen großen Einfluss auf das Wachstum haben. Ferner scheint der Anteil an freiem D-Mannitol in der Zelle in allen Fällen so gering zu sein, dass ein Vorteil eines Wachstums mit DalD nicht zu erkennen ist. Bei der zweiten Gruppe von Mutationen (E218A, E218V, E218A/L229Q/P256Q) war das Wachstum auf D-Mannitol in dem Stamm LGS322 deutlich langsamer als das Wachstum des Wildtyps. Der Zellertrag reduzierte sich in diesen Fällen entsprechend der sich verlängernden Generationszeit. Das Vorhandensein von DalD führte dagegen zu einer deutlichen Verbesserung des Zellertrags und der Generationszeiten. Der Zellertrag steigerte sich in allen Fällen um etwa 100%. Eine Sonderstellung nimmt in dieser Gruppe die Mutation in dem Plasmid pDSM3 (E218A/L229Q/P256A) ein. Diese Mutation ermöglichte auch Wachstum in LGS322, dieses war jedoch drastisch verlangsamt und auch der Zellertrag war reduziert. In dem Stamm LGS322-1 kam es wiederum zu einer Verbesserung des Wachstums, wie es schon bei den anderen Mutationen dieser Gruppe beobachtet wurde. Der Ertrag steigerte sich von 0,3-1 auf 5,5-6 [∗108 B/ml]. Dies deutet auf einen sehr hohen Anteil unphosphorylierten, dass bedeutet entkoppelt transportierten D-Mannitols hin. Die dritte Gruppe wird durch das Plasmid pGJT9-3 (H256P) gebildet. In dem Stamm LGS322 ermöglichte es kein Wachstum auf D-Mannitol, wogegen mit DalD Wachstum zu sehen war. Die Mutation H256P ermöglichte lediglich Wachstum auf freiem D-Mannitol durch entkoppelten Transport. Die Mutationen der vierten Gruppe (E218A/H256P, E218V/P256A) schalteten jeden Transport aus. Es war weder Wachstum im Stamm LGS322 noch im Stamm LGS322-1 möglich. III.1.5.2. Bindekinetik der Mutanten Alle Daten zu den Bindekinetiken wurden Dank der freundlichen Unterstützung von G.T. Robillard und seinen Mitarbeitern (E. Vos, J. Broos, R.H. Duurkens, G.K. Schuurman-Wolters) in deren Laboratorien an der Reichsuniversität Groningen selbstständig ermittelt. Ortsspezifische Mutagenese in dem Plasmid pMaHisMtlAPr Um eine Überexpression von mtlA mit Hilfe der Hitzeinduktion durchzuführen, mussten die bekannten Mutationen in das über Hitze induzierbare Plasmid 82 III. Ergebnisse pMaHisMtlAPr eingeführt werden. Die Mutagenesen wurden mit dem „QuikChange™ Site-Directed Mutagenesis Kit“ der Firma Stratagene und den im Ergebnisteil erwähnten Mutagenese-Primern (.E218A(+/-), E218V(+/-), H256P(+/-), H256A(+/-), H256Q(+/-), H256S(+/-.)) durchgeführt. Die daraus resultierenden Derivate des Plasmides pMaHisMtlAPr (WT) sind in der folgenden Tabelle dargestellt. Tab. III.1.18. Mutanten aus dem Plasmid pMaHisMtlAPr Plasmid Phänotyp (AS-Austausch in MtlA) pRUG1 pMaHisMtlAPr mtlA E218A pRUG2 pMaHisMtlAPr mtlA E218V pRUG3 pMaHisMtlAPr mtlA H256P pRUG4 pMaHisMtlAPr mtlA E218A/H256P pRUG5 pMaHisMtlAPr mtlA E218V/H256P pRUG6 pMaHisMtlAPr mtlA H256A pRUG7 pMaHisMtlAPr mtlA H256Q pRUG8 pMaHisMtlAPr mtlA H256S Bei der weiterführenden Untersuchung der „Supressormutanten“ wurden lediglich die Auswirkungen der AS-Austausche in dem His256 untersucht. Das bedeutet, dass der zusätzliche Austausch E218A (sowie der unspezifische Austausch L229Q), wie er in pDSM3 und pDSM4 kodiert vorkommt, nicht eingefügt wurde. Die Phänotypen der Mutationen mit den neuen Konstrukten entsprachen den Phänotypen der zuvor beschrieben pGJ9-Reihe. Dabei zeigte der Stamm LGS322./pRUG4 (E218A/H256P) den gleichen Phänotyp auf D-Mtl wie LGS322./pGJT9-4 (E218A/L229Q/H256P). Bei der Untersuchung der einfachen AS-Austausche H256A, H256Q und H256S mit dem Stamm LTK31-2 wurde keine Entkopplung festgestellt. Herstellung der Vesikel Die Phosphorylierungstests und Bindestudien wurden mit Vesikeln durchgeführt. Als Teststamm wurde LGS322 verwendet, der mit den oben beschriebenen Plasmiden der Reihe pRUGx transformiert wurde. Die eingesetzten Vesikel wurden mit der „French Press“ wie in dem Teil II.5.1. der Arbeit beschrieben hergestellt. Die Abb.III.1.6. zeigt III. Ergebnisse 83 ein Coomassie-gefärbtes SDS-Gel, dass mit den Vesikeln nach der Überexpression beladen wurde. 1 2 3 4 5 6 7 8 9 116 kDa 80 kDa Í MtlA(His)6 52,5 kDa 34,9 kDa Abb. III.1.6. SDS-Gel der Vesikel nach der Überexpression 10µl der Vesikel wurden in einer 1:10 Verdünnung auf ein 12,5%iges Acryl-Amid-Gel aufgetragen. Auf das Gel wurden die Vesikel der jeweiligen Mutanten aufgetragen (LGS322/pRUGx). Bahn 1= E218A (13,6); 2= E218V; 3= H256P; 4= E218A/H256P; 5= E218V/H256P; 6= LGS322; 7= H256Q; 8= H256A; 9= H256S; letzte Bahn= „BIORAD SDS-PAGE Standard, Low Range“. Die Größen der Standard-Banden sind neben dem Bild angegeben. Mit einem Pfeil ist die Laufhöhe des MtlA(His)6 angegeben (~60 kDa. Montfort et al., 2001). In Bahn 6 ist das Gesamtmembranprotein aus dem Stamm LGS322 ohne Plasmid aufgetragen und zeigt daher keine Expression von MtlA. Die Gesamtproteinkonzentrationen der Vesikel wurden wie in Teil II.8.1. beschrieben gemessen. Bei der Messung wurden jeweils 10µl und 30µl einer 1:50 Verdünnung der Vesikel eingesetzt. Die folgende Tabelle III.1.19. gibt die durchschnittlich gemessenen Werte wieder. Tab. III.1.19. Gesamtproteinkonzentrationen der Vesikel Gesamtproteinkonzentration der Vesikel [mg/ml] E218A E218V H256P E218A H256P E218V H256P 13,6 20,5 13,7 12,5 14,1 H256S H256Q H256A LGS322 18,4 16,7 15,7 5,1 WT 21,9 84 III. Ergebnisse Phosphorylierungstests mit den Vesikeln Mit den Vesikeln wurden Tests der PTS-abhängigen in vitro Phosphorylierung durchgeführt um festzustellen, ob und wie viel aktives Protein isoliert werden konnte. Eine Testreihe untersuchte die Phosphorylierungsaktivität bei einer D-Mtl Konzentration von 1mM nach 220sec, 440sec, 660sec und 880sec. Jeder Ansatz von 100µl enthielt ca. 33000cpm. Die Vesikel wurden abhängig von den eingesetzten Mutanten in unterschiedlichen Verdünnungen eingesetzt. Als Kontrollen wurde ein Ansatz mit Vesikeln aus dem ∆mtlA-Stamm LGS322 und ein Ansatz mit H2O getestet. Die Kontrollen überschritten unabhängig von der Vesikelverdünnung (bei LGS322) nie den Wert von 200cpm. Die folgende Abb. III.1.7. zeigt typische Phosphorylierungsaktivitäten der verschiedenen MtlA-Derivate in Vesikeln nach rechnerischem Abgleich der unterschiedlichen Verdünnungen. 140.000 120.000 100.000 cpm E218A E218V 80.000 H256P H256Q 60.000 H256A H256S WT 40.000 20.000 0 0 200 400 600 800 1000 t [sec] Abb. III.1.7. Phosphorylierungsaktivitäten der Vesikel mit 1mM [14C]-D-Mtl Die ursprünglichen Verdünnungen (1:4000 bei dem WT; 1:1000 bei E218A, E218V, H256Q, H256S und H256A; 1:250 bei H256P, E218A/H256P und E218V/H256P) sind rechnerisch auf 1:250 angeglichen worden. Die Kontrollen durch den ∆mtlA-Stamm LGS322 und dem Ansatz mit Wasser sind nicht dargestellt. III. Ergebnisse 85 Um die scheinbar nicht vorhandene Phosphorylierungsaktivität der Mutante H256P näher zu untersuchen, wurden Phosphorylierungstests bei geringerer Verdünnung der Vesikel (1:10) und einer Konzentration von 500µM [14C]-D-Mtl durchgeführt. Jeder Ansatz von 100µl enthielt ca. 120000cpm. In diesem Test wurden auch die Doppelmutanten E218A/H256P und E218V/H256P untersucht. Die folgende Abb. III.1.8. zeigt einen beispielhaften Verlauf der Phosphorylierung. 8000 7000 6000 cpm 5000 H256P 4000 EA/HP EV/HP 3000 H2O 2000 1000 0 0 5 10 15 20 25 30 t [min] Abb. III.1.8. Phos.-Aktivitäten von H256P-Mutanten mit 500µM [14C]-D-Mtl Die Vesikel sind in dem Test in einem Verhältnis von 1:10 verdünnt worden. Zusätzlich ist der Leerwert (H2O) dargestellt, bei dem statt der Vesikel Wasser eingesetzt wurde. Anhand der gemessenen Phosphorylierungsaktivitäten wurden die spezifischen Aktivitäten der Mutanten bei einer Konzentration von 1mM bzw. 500µM D-Mtl berechnet. Die spezifischen Aktivitäten beziehen sich hierbei auf die gemessenen Gesamtproteinkonzentrationen. Die nur minimal über dem Leerwert liegenden Aktivitäten der Doppelmutanten E218A/H256P und E218V/H256P wurden nicht als Phosphorylierungsaktivität gewertet. 86 III. Ergebnisse Tab. III.1.20. Spezifische Phosphorylierungsaktivitäten der Vesikel nmol Spezifische Phosphorylierungsaktivitäten der Vesikel min ⋅ mg Gesamtprotein WT LGS322 E218A E218V H256P E218A/ H256P E218V/ H256P H256S H256Q H256A 278 0 307 32 2 0 0 339 303 253 Die Ergebnisse lassen die Mutanten hinsichtlich der Phosphorylierungsaktivität in drei Klassen einteilen. Die Mutationen E218A, H256S, H256Q und H256A lagen in einem Aktivitätsbereich, die dem WT-Protein entsprechen. Keine Aktivität zeigten die Doppelmutanten E218A/H256P und E218V/H256P. Eine nur geringe Aktivität zeigten die Mutanten E218V und H256P. Während der AS-Austausch E218V eine ca. 10%ige WT-Aktivität zeigte, lag der AS-Austausch H256P mit <1% Aktivität nahe der Inaktivität. Allerdings konnte in einem direkten Vergleich mit den Doppelmutanten und den Leerwerten diese Minimalaktivität bestätigt werden. Für die weiteren Bindestudien konnte allgemein gezeigt werden, dass mit der Methode aktives Protein in den Vesikeln isoliert wurde. Die nicht vorhandene oder schwache Aktivität bei einzelnen Mutanten (E218V, H256P, E218A/H256P, E218V/H256P) deckte sich mit den vorherigen Beobachtungen (Tab. III.1.15. - III.1.17.) und ist nicht auf die Art der VesikelPräparation zurückzuführen. Bindestudien der MtlA-Derivate in Vesikeln Bei diesen Bindestudien wurde die Bindung von D-Mtl an das jeweilige EIIMtl in einem Konzentrationsbereich von 26nM - 524nM untersucht. Für die Vesikel mit WTMtlA wurde ein Kd-Wert von ~40nM ermittelt. In mehrfachen Versuchen konnte für alle getesteten Mutanten (E218A, E218V, H256P, E218A/H256P, E218V/H256P, H256S, H256Q, H256A) keine Bindeaktivität gemessen werden. Dieses Ergebnis war für die in den Phosphorylierungsuntersuchungen nicht (E218A/H256P, E218V/H256P) oder nur schwach aktiven (E218V, H256P) Mutanten mit Einschränkungen zu Erwarten gewesen. Jedoch war eine fehlende Bindeaktivität bei den Mutanten, die bei der Phosphorylierung im Bereich der WT-Aktivität lagen (E218A, H256S, H256Q, H256A), überraschend. Folglich musste eine mit dieser Methode messbare Bindeaktivität in einem Konzentrationsbereich von 26nM - 524nM für alle Mutanten ausgeschlossen werden. III. Ergebnisse 87 III.1.5.3. Transport-Kinetiken der Mutanten Um weitere Aussagen über die Auswirkungen der AS-Austausche auf den Transportprozess von D-Mtl machen zu können, wurden anhand von Transportmessungen bei unterschiedlichen Konzentrationen (0,3µM; 0,5µM; 0,7µM; 1µM; 1,5µM; 2µM; 3µM; 5µM; 10µM) die KMapp-Werte [µM] und Vmax [nmol∗min-1∗mg Protein-1] (in den Abbildungen = [nmol]*) ermittelt. Die Testkulturen wurden wie unter II.4.2. beschrieben in LB0Cam angezogen. Die spezifischen Transportaktivitäten der einzelnen Mutanten wurden für die jeweiligen Konzentrationen aus einer Kultur in demselben Versuchsansatz gemessen. Die Messungen sind mit mindestens fünf (0,5µM; 1µM; 2µM; 5µM; 10µM) bzw. allen neun unterschiedlichen Konzentrationen durchgeführt worden. In den folgenden Abbildungen (Abb.III.1.9-12) sind nur die Messpunkte dargestellt, welche bei der Berechnung auch berücksichtigt wurden. Alle Mutanten wurden mindestens dreimal unabhängig getestet. Der Plasmidhintergrund der Mutationen war in allen Fällen pGJ9 und der Wirtsstamm LGS322. Die Mutation für den Austausch ortsspezifische beschriebenen H256A wurde Mutagenese Fällen mit den (pGJT9-6) wurden die Primern konstruiert. KMapp-Werte H256A(+)/H256A(-) In einzelnen, zusätzlich mit durch gesondert anderen Plasmidhintergründen und Stämmen gemessen. In dem Stamm LGS322 mit den Plasmiden pGJT9-1 (E218A), pGJT9-2 (E218V), pGJT9-3 (H256P), pGJT9-4 (E218A/H256P) oder pGJT9-5 (E218V/H256P) konnte keine Transportaktivität gemessen werden. Die anderen aus den folgenden „Lineweaver-Burk“-Diagrammen ermittelten Werte sind im Anschluss in der Tabelle III.1.21. zusammengefasst. 88 III. Ergebnisse LGS322/pGJ9 1/V [nmol]* 0.2 WT I WT II 0.2 WT III 0.1 0.1 0.0 -0.1 -0.5 0 0.5 1 1.5 2 2.5 1/S [µM] Abb. III.1.9. KMapp-Wert Messungen des WT-Stamms LGS322/pGJ9 für D-Mtl Dargestellt sind drei „Lineweaver-Burk“-Diagramme unabhängiger Messungen. Zu den Messpunkten der D-Mannitol-Konzentrationen wurde eine lineare Regressiongerade ermittelt und dargestellt. Die KMapp-Werte und V wurden anschließend wie in Teil II.8.4. beschrieben berechnet. 1/V [nM]* LGS322/pGJT9-5 (H256A) 0.4 0.4 0.3 0.3 H256A I H256A II H256A III 0.2 0.2 0.1 0.1 0.0 -0.1 -0.1 -0.5 0 0.5 1 1.5 2 2.5 3 3.5 1/S [µM] Abb. III.1.10. KMapp-Wert Messungen von LGS322/pGJT9-5 für D-Mtl siehe Abb. III.1.7. III. Ergebnisse 89 LGS322/pDSM1 (P256Q) 1/V [nM]* 0.2 P256Q I 0.2 P256Q II P256Q III 0.1 0.1 0.0 -0.1 -0.5 0 0.5 1 1.5 2 2.5 3 3.5 1/S [µM] Abb. III.1.11. KMapp-Wert Messungen von LGS322/pDSM1 für D-Mtl siehe Abb. III.1.7. 1/V [nM]* LGS322/pDSM2 (P256S) 1.2 1.0 P256S I P256S II 0.8 P256S III 0.6 0.4 0.2 0.0 -0.2 -0.5 0 0.5 1 1.5 2 2.5 3 3.5 1/S [µM] Abb. III.1.12. KMapp-Wert Messungen von LGS322/pDSM2 für D-Mtl siehe Abb. III.1.7. 90 III. Ergebnisse Tab. III.1.21. KMapp-Werte und V der MtlA-Mutanten für D-Mtl in LGS322 V [nmol∗min-1∗mg Protein-1] KMapp [µM]1 1 ASAustausch I II III Ø2 I II III Ø2 - 3,9 4 4 4 ±0,1 102 92 53 82 ±26 pGJT9-6 H256A 7,4 10,9 8,2 8,2 ±1,8 78 107 100 95 ±15 pDSM1 P256Q 3,8 6,4 5,5 5,2 ±1,3 76 135 103 105 ±30 pDSM2 P256S 5,1 7 7,2 6,4 ±1,2 25 22 28 25 ±3 Plasmid pGJ9 1 Angegeben sind die jeweiligen Werte der einzelnen Messungen (I-III). 2 Ø = Mittelwerte der drei Messungen mit Angabe der Standardabweichungen (±) wie in II.8.4. beschrieben. Bei der Betrachtung der Daten fällt auf, dass die Werte für V innerhalb einer Mutation voneinander abwichen. Dies ist auf verschiedene MtlA-Konzentrationen innerhalb der Testkulturen zurückzuführen. Da die Expression von mtlA in dem verwendeten Testsystem konstitutiv erfolgte, liegt eine variierende Plasmidkopienzahl nahe. Bei Überprüfungen von EK getesteter Kulturen auf McMtl und LB0cam (Daten nicht angegeben) wurde eine 30-70%ige Kurierung des Plasmides (bzw. des Mtl+Phänotyps) festgestellt. Die KMapp-Werte zeigten in der Tendenz, dass alle Mutationen einen gegenüber dem WT erhöhten KMapp-Wert bewirken. Diese Werte sprechen für eine geringfügig verringerte Affinität von D-Mtl zu der Bindestelle in MtlA, die allerdings nicht für die in III.1.5.2. beschriebene fehlende Bindung ausschlaggebend sein kann. Trotz der eingeschränkten Bewertungsmöglichkeiten der V-Werte, fällt der konstant bis zu einem Faktor 6 niedrigere V der Variante P256S auf. Mit diesem Protein wurde bei sieben Messungen immer ein V zwischen 19 und 28 [nM] und eine KMappWert zwischen 4,9 und 7,2 [µM] gemessen. Bei weiteren sieben Messungen wurden mit dem Stamm LGS22/pDSM2 nichtlineare Kinetiken erhalten (siehe Abb. III.1.13.). In verschiedenen Versuchsansätzen konnte nicht festgestellt werden welche Parameter lineare und nichtlineare Kinetiken verursachen. Variierende MtlA-Konzentrationen bei den Transportmessungen sind als Ursache auszuschließen, da diese keine Auswirkung auf den KM-Wert haben. III. Ergebnisse 91 1/V [nM]* LGS322/pDSM2 (P256S) 0.6 0.5 0.4 0.3 0.2 0.1 0.0 -0.1 -0.5 0 0.5 1 1.5 2 2.5 1/S [µM] Abb. III.1.13. Nichtlineare Kinetik der KMapp-Messung von LGS322/pDSM2 Die Abbildung zeigt die Messpunkte einer nichtlinear verlaufenden KMappMessung. Die fehlende Transportaktivität in dem Stamm LGS322 mit den Plasmiden pGJT9-1 (E218A) (McCMtl = 2+;) und LGS322/ pRUG1 (E218A) (Phosphorylierungsaktivität [1mM D-Mtl] = 307 [nmol], WT= 278 [nmol]) ist aufgrund der beschriebenen Ergebnisse unerwartet. Zudem konnte für die gleiche Mutation in aus mtlA K. pneumoniae Transport (KM = 2,9µM, V = 59nmol) gemessen werden (Otte, 2000). Diese Messungen wurde allerdings in dem Stamm LGS324 und mit dem NiedrigkopienVektor pHEX3 als mtlA-Plasmid durchgeführt. Daraufhin wurde in dieser Arbeit untersucht ob der Stamm- oder Plasmidhintergrund Auswirkung auf die Messbarkeit der Transportaktivitäten des MtlA haben könnte. In einem Markertest wurde festgestellt wie sich der Stammhintergrund auf den Phänotyp auswirkt. Dazu wurden die äquivalenten Plasmide pSOL300 (mtlAKAY2026) und pSOL313 (mtlAKAY2026 E218A), bzw. pGJ9 und pGJT9-1 (E218A) in den Stämmen LGS322 und LGS324 auf McCMtl getestet und die spezifische Transportaktivität der entsprechenden Proteine erneut gemessen. 92 III. Ergebnisse Tab. III.1.22. Stammabhängige Phänotypen von mtlA-Mutanten Stamm LGS322/ LGS324/ McC Mtl1 spez. Transportaktivität2 pGJ9 3+ 5,2 pGJT9-1 2+ 0 pSOL300 3+ 2,5 pSOL313 1+ 0 pGJ9 3+ 5,7 pGJT9-1 2+ 0 pSOL300 3+ 3,8 pSOL313 3+ 3,4 Plasmid 1. Für Symbole und sonstige Erklärungen siehe Tab. III.1.1. 2.Angaben in [nmol∗min-1∗mg Protein-1], gemessen mit 10µM [14C]-D-Mtl. Die Testkulturen wurden in LB0Cam angezogen. Aus den Tests geht hervor, dass der Stammhintergrund in dem Fall des pSOL313 (mtlAKAY2026 E218A) sowohl einen Einfluss auf den Phänotyp auf Platte (LGS322 1+, LGS324 3+) als auch auf die messbare Transportaktivität hat. Keine stammabhängigen Unterschiede wurden bei den beiden Wildtypen (pGJ9, pSOL300) und bei der E218AMutante aus E. coli (pGJT9-1) festgestellt. Daraufhin wurde mit den Primern E218A (+/-) durch ortspezifische Mutagenese erneut die E218A-mtlA-Mutation in das Plasmid pMMX5 eingefügt (Um eventuelle Reversionen oder Suppressionen in pSOL313 auszuschließen, wurde zur Kontrolle die E218A auch in pSOL300 eingefügt. Die Ergebnisse aus der Tab. III.1.22. konnten mit diesem Plasmid bestätigt werden). Das Plasmid pMMX5 ist ein Niedrigkopieplasmid mit einem mtlA(His)6. Dadurch sollte festgestellt werden, welchen Einfluss das Plasmid auf die Transportaktivität hat. Das resultierende Plasmid pMMX5-EA zeigte in dem Stamm LGS324 auf McMtl einen 3+Phänotyp und eine spezifische Transportaktivität von 11,3 und 11,8 [nmol]. In dem Stamm LGS322 war der Phänotyp auf McMtl unverändert 3+, allerdings konnte wiederum keine Transportaktivität gemessen werden. Weitere Transportmessungen mit dem Stamm LGS324 /pMMX5-EA erwiesen sich dennoch als schwierig, da die Transportaktivität nicht regelmäßig zu messen war. Die erfolgreiche Messung einer Transport-Kinetik ergab einen V von 25 [nmol∗min-1∗mg Protein-1] und einen KMapp von 4,9 [µM] für LGS324./pMMX5-EA. Die Mindestvoraussetzungen für eine erfolgreiche III. Ergebnisse 93 Transportmessung scheinen in dem beschriebenen Fall der Stamm LGS323 und ein Niedrigkopienvektor wie der pHEX3/5 zu sein. Was die genauen Gründe für diese Beobachtungen sein können, war im Rahmen dieser Arbeit nicht mehr zu bestimmen. Erschwerend ist dabei der nicht genau bekannte Genotyp des Stammes LGS324 im Vergleich zu dem Stamm LGS322 (siehe III.1.1.4.). An den Mutationen E218A und P256S wird ersichtlich, dass sich E. coli MtlA in diesen Fällen in Grenzbereichen der Messbarkeit bewegt. Inwiefern dies an der Konformation, Dimerisierung, oder der Expressionsstärke liegt, lässt sich anhand der vorliegenden Daten nicht genau bestimmen. In der Kinetik scheint die E218A-Mutation gegenüber dem WT (LGS322/pMMX5: V 83-84 [nmol∗min-1∗mg Protein-1], KMapp 4,8-6,3.[µM]) einen niedrigeren V (25 [nmol∗min-1∗mg Protein-1]) und einen ähnlichen KMapp (4,9 [µM]) zu haben. In der Tabelle III.1.23. sind die wesentlichen Ergebnisse der molekularbiologischen und biochemischen Untersuchungen an den mtlA-Mutanten zusammengefasst. Die Messungen zeigten, dass mit einem Selektionsprinzip (Wachstum auf DMannitol mit unterbrochener Phosphorylierungskette) drei entkoppelte Mutationen (E218A, E218V, H256P) mit unterschiedlichen biochemischen Eigenschaften isoliert werden konnten. Alle drei Mutanten zeigten in MtlA’ (EIIC) den gleichen Phänotyp auf McC Mtl. Die Menge des entkoppelt transportierten D-Mtl scheint gleich zu sein. Die Mutationen E218A und E218V führten in einem vollständigem MtlA (EIICBA) zu gleichzeitigem ge- und entkoppelten Transport. Dabei hatte E218V im Vergleich mit E218A nur 10% Phosphorylierungsaktivität, erreichte aber mit DalD gleiche Generationszeiten auf D-Mtl. Das weist auf einen höheren Anteil an entkoppeltem Transport mit der Mutation E218V hin. Mit der Mutation H256P wurde die Phosphorylierungsaktivität fast vollständig ausgeschaltet, Wachstum mit gekoppeltem Transport fand nicht mehr statt. Es wart nur noch Wachstum mit DalD möglich, welches aber schwächer als bei den Mutationen E218A und E218V ausfiel. Die Mutation H256P entkoppelte den Transport von D-Mtl vollständig. Die Kombination der Austausche (E218A/H256P und E218V/H256P) führte zu einem vollständigen Ausfall der Phosphorylierungs- und Transportaktivitäten. 94 III. Ergebnisse Tab. III.1.23. Zusammengefasste Ergebnisse der EIIMtl-Mutanten im Stamm LGS322 IIMtl I IICBA (WT) IICBA +DalD IIC IIC +DalD IICBA IICBA +DalD IIC IIC +DalD IICBA IICBA +DalD IIC IIC +DalD IICBA IICBA +DalD IIC IIC +DalD IICBA IICBA +DalD IICBA IICBA +DalD IIC IIC +DalD IICBA IICBA +DalD IIC IIC +DalD IICBA IICBA +DalD IICBA IICBA +DalD IICBA IICBA +DalD IICBA IICBA +DalD IICBA IICBA +DalD ASAustausch II WT E218A E218V H256P E218A/ H256P E218A/ L229Q/ H256P E218V/ H256P H256A P256Q P256S E218A/ L229Q/ P256A E218A/ L229Q/ P256Q Gt [min] IV 83-88 81-87 k.W. k.W. 180-200 110-125 Tp KMapp [µM] V 4 ±0,1 Tp V [nM]* VI 82 ±26 Pho [nM]* VII 278 Kd [nM]* VIII 40 [4,9]1 [25]1 307 n.m. 230-380 105-115 n.m. n.m. 32 n.m. k.W. 220-345 n.m. n.m. 2 n.m. k.W. k.W. k.W. k.W. n.m. n.m. 0 n.m. n.m. n.m. k.W. k.W. n.m. n.m. 0 n.m. 8,2 ±1,8 95 ±15 253 n.m. 82-87 5,2 ±1,3 105 ±30 86-88 84-87 6,4 ±1,2 25 ±3 84-86 580-900 n.m. n.m. 140-150 303 n.m. 339 n.m. McC Mtl III 3+ 3+ w w 2+ 3+ w + + 3+ w + w 2+ w + w w w w w w w w w w 3+ 3+ 3+ 3+ 3+ 3+ (+) 2+ (+) 2+ 330-360 145-155 n.m. n.m. Kopplung IX + + k.T. k.T. +/+/+/+/k.T. k.T. k.T. k.T. k.T. k.T. k.T. k.T. k.T. k.T. + + + + + + +/+/+/+/- Die Tabelle fasst wesentliche Ergebnisse der Mutanten-Untersuchungen zusammen. Die Mutationen wurden in verschiedenen Plasmid-Hintergründen gemessen. Für schattierte Felder liegen keine Messungen vor. [nM]*=[nmol∗min-1∗mg Protein-1]. Spalte I: IICBA=MtlA, IIC=MtlA’. II: AS-Austausche. III: Phänotyp auf McC Mtl (siehe Tab.III.I.15. u.16.). IV: Gt=Generationszeit auf D-Mtl (s. Tab.III.I.17.). V u VI: Tp=Transport (s. Tab.III.I.21.). VII: Pho=spez. Phosphorylierungsaktivität. (s. Tab.III.I.20.). IX: Art des DMtl-Transports: +=gekoppelter Tp, -=entkoppelter Tp, +/-=ge- und entkoppelter Tp, k.T.=kein Tp. [ ]1 = Messung im Stamm LGS324. III. Ergebnisse 95 Bei der Selektion von Supressormutanten wurden nur Mutationen der AS256 gefunden. Die daraus resultierenden Derivate H256A, P256Q und P256S stellten in den Zellen annähernd den WT-Phänotyp auf D-Mtl wieder her. Eine zusätzliche den Transport entkoppelnde Mutation E218A, wie sie in den Kombinationen E218A/L229Q/P256A und E218A/L229Q/P256Q untersucht wurde, bewirkt wie auch allein ge- und entkoppelten Transport. Das Wachstum auf D-Mtl fiel allerdings in beiden Fällen schwächer aus als mit der E218A-Mutation. Die Selektionen und Messungen zeigen, dass die Aminosäuren E218 und H256 maßgeblich am Transport- und Phosphorylierungsprozess beteiligt sein müssen. Mutationen wurden nur an diesen Stellen gefunden und beeinflussten den D-MtlTransport auf unterschiedliche Weise. III.2. Topologieuntersuchungen am EIIMtl Die oben gezeigten Ergebnisse haben eine deutliche Beteiligung der Aminosäuren E218 und H256 am eigentlichen Transportprozess von D-Mtl durch das EIIMtl gezeigt. In dem bisherigen 2D-Modell von MtlA (Abb. I.3.) sind diese in der vermuteten zytoplasmatischen Schleife 5 zu finden. Die in dem WT-Transporter beobachtete Kopplung von Transport und Phosphorylierung setzt eine lokale Nachbarschaft zwischen den an der Bindung und Phosphorylierung beteiligten Aminosäuren voraus. Da Mutationen/Austausche der genannten AS eine Entkopplung bewirken, ist es wahrscheinlich, dass diese in der Nähe oder in der Bindestelle liegen. Daher war die genauere Kenntnis der Topologie der Schleife 5 des EIIMtl wichtig. Das bisherige Modell stützt sich überwiegend auf Untersuchungen von Sugiyama et al. (1991), in denen ausschließlich über φ(mtlA’-phoA)-Fusionen Teile des MtlA dem Zytooder Periplasma zugeordnet wurden. Die Methode beruht darauf, dass PhoA im Periplasma Aktivität zeigt, nicht aber in dem Zytoplasma. Dabei fehlten jedoch die notwendigen φ(mtlA’-lacZ), deren Aktivitäten sich hinsichtlich der Lage genau gegensätzlich verhalten, sowie Versuche mit sogenannten „Sandwich“-Fusionen (φ(mtlA’-lacZ-mtl’A)) und vollständigen, korrekt gefalteten IICBAMtl-Komplexen. Solche langen AS-Insertionen können allerdings die Faltung beeinflussen und falsch positive sowie falsch negative Ergebnisse hervorrufen. Im Gegensatz dazu bietet das „Cystein- 96 III. Ergebnisse Scanning“ die Möglichkeit, mit einem aktiven Protein zu arbeiten, dass zudem nur in wenigen Aminosäuren verändert ist. III.2.1. Konstruktion der erforderlichen Plasmide und Mutanten Die Voraussetzung für das „Cystein-Scanning“ sind MtlA-Komplexe, welche jeweils an definierter Stelle nur ein Cystein besitzen. Die erforderliche T7Überexpression des Membran-Proteins wurde mit dem Niedrigkopieplasmid pHEX5 als Klonierungs- und Überexpressionsvektor in dem Stamm JM109(λDE3) durchgeführt. Für die Trennung der zu untersuchenden Proteine von den restlichen markierten Proteinen in der Zelle war zudem eine effektive MtlA-Reinigung notwendig. In dieser Arbeit wurden Proteine mit carboxyterminalem „His-Tag“ aus sechs Histidinen verwendet. Die Reinigung erfolgte mit Hilfe von Ni-NTA-Säulen. III.2.1.1. Konstruktion der Plasmide pMMX5 und pMMX5-1 bis pMMX5-4 Um eventuelle Klonierungen über die HindIII-Erkennungssequenz in mtlA zu ermöglichen, wurde zuerst die HindIII-Erkennungssequenz in pHEX5 über einen HindIIIVerdau mit anschließender Klenow-Auffüllung und Ligation entfernt. Das resultierende Plasmid wurde pHEX5d genannt. Mit einer PCR wurde das vollständige mtlA von dem Plasmid pGJ9 amplifiziert. Der erste verwendete Primer MtlA-HIS1 (5‘- GGA ATT CCC GTC GAC TGG ACA GTT AAC CGA TTC AGT G -3‘; in blau: EcoRI, in rot: SalI) enthielt an seinem 5’-Ende die Erkennungssequenz für das Restriktionsenzym EcoRI. Dadurch entstand hinter der natürlich vorkommenden SalI-Schnittstelle eine BamHISchnittstelle. Der Primer lagert sich ca. 135bp stromaufwärts des ATG-Startcodons von mtlA an und amplifiziert somit auch die Promotorregion von mtlA. Der zweite Primer MtlA-HIS2 (5‘- TTG GAT CCT TAG TGG TGG TGG TGG TGG TGC TTA CGA CCT GCC AGC AGT TCC AGC ACT TC -3‘; blau: BamHI, rot: Stopp, grün: His-Tag) hängt hinter dem letzten Codon von mtlA sechs Codone für Histidin, ein Stopp-Codon und eine Erkennungssequenz für das Restriktionsenzym BamHI an. Die zusätzlichen Nukleotide an den 5’-Enden der beiden Primer sind für eine effektivere Funktion der Restriktionsenzyme nötig. Die PCR wurde mit dem Plasmid pGJ9 als Matrize wie in II.7.9. beschrieben durchgeführt, das PCR-Produkt mit den entsprechenden Restriktionsenzymen geschnitten und in den ebenso geschnittenen Vektor pHEX5d III. Ergebnisse 97 kloniert. Die korrekte Sequenz wurde durch einzelsträngige Sequenzierung festgestellt und das neue Ausgangsplasmid pMMX5 genannt. Die Plasmide pMMX5-1 bis pMMX5-4 wurden durch nacheinander erfolgte Mutagenesen der vier Kodone für die im WT vorhandenen Cysteine (C110, C320, C384, C571) hergestellt. Das Ziel dieser Mutagenesen war ein Cystein-freies MtlA aus dem die weiteren „Einzel-Cystein“-Mutanten abgeleitet wurden. Die Cysteine wurden durch Serine ersetzt, lediglich das vermutlich in einer transmembranen Helix liegende Cys320 wurde gegen ein Alanin ausgetauscht. Über ortspezifische Mutagenese (Primer in runden Klammern) entstanden, ausgehend von pMMX5, nacheinander folgende Plasmide (In eckigen Klammern ist zur Vereinfachung der veränderte Phänotyp hinsichtlich der vier Cysteine angegeben): pMMX5 WT = [CCCC] xx¬pMMX5-1 (C320A (+/-)) = [CACC] xxxx¬pMMX5-2 (C110S (+/-)) = [SACC] xxxxxx¬pMMX5-3 (C571S (+/-)) = [SACS] xxxxxxxx¬pMMX5-4 (C384S (+/-)) = [SASS] III.2.1.2. Konstruktion der zu untersuchenden „Einzel-Cys“-Mutanten Die zu untersuchenden „Einzel-Cystein“-Mutanten wurden auch in Kombination mit dem Cys384 für die Aktivitätskontrolle konstruiert. Dabei wurden die Mutageneseansätze parallel mit dem Plasmid pMMX5-3 [SACS] und dem Plasmid pMMX5-4 [SASS] als Matrize durchgeführt. Für die gezielten Austausche zu Cystein wurden Serine an unterschiedlichen Stellen auch außerhalb der zytoplasmatischen Schleife 5 ausgewählt. Die Austauschstellen, benutzten Primer und neuen Konstrukte sind in der folgenden Tabelle III.2.1. aufgelistet und wurden durch einzelsträngige Sequenzierung des mtlA überprüft. Die Auswahl der Untersuchungsorte stützte sich auf das 2D-Modell von Sugiyama et al. (1991) und Lengeler et al. (1994), dargestellt in Abb.I.2. Als sichere Innenkontrolle wurde das Plasmid pMMX5-3 [SACS] eingesetzt, da das Cystein384 in der zytoplasmatischen Domäne EIIBMtl liegt (Stephan und Jacobson, 1986). Die negative Kontrolle stellte das cysteinfreie MtlA (pMMX5-4 [SASS]) dar. 98 III. Ergebnisse Tab. III.2.1. Plasmide der Reihen pMMX5-3x und pMMX5-4x in pMMX5-3 [SACS] in pMMX5-4 [SASS] ⇓ ⇓ zp Schleife 1 (IK) pMMX5-31 pMMX5-41 S110C (+/-) zp Schleife 3 (IK) pMMX5-32 pMMX5-42 S158C S158C (+/-) pp Schleife 4 (AK) pMMX5-33 pMMX5-43 S199C S199C (+/-) zp Schleife 5 pMMX5-34 pMMX5-44 S212C S212C (+/-) zp Schleife 5 pMMX5-35 pMMX5-45 S242C S242C (+/-) zp Schleife 5 pMMX5-36 pMMX5-46 S299C S299C (+/-) pp Schleife 6 (AK) pMMX5-37 pMMX5-47 verwendete Primer postulierte Topologie der Austauschstelle 1 S3C (+/-) S110C Mutation S3C 1 zp = zytoplasmatisch, pp = periplasmatisch, IK = Innenkontrolle, AK = Außenkontrolle III.2.1.3. Untersuchung der Funktionalität der MtlA-Derivate Da Transportaktivität eine korrekte Faltung der Proteine voraussetzt, wurden zur Überprüfung die resultierenden Phänotypen der Plasmide im Stamm LGS322 auf McMtl und die Transportaktivitäten mit 10µM D-Mtl untersucht (Tab. III.2.2.). MtlA-Varianten mit dem Austausch C384S (pMMX5-4 und seine Derivate) zeigten erwartungsgemäß keine Aktivität. Tab. III.2.2. Aktivitäten der „Cys-Scanning“-MtlA-Derivate Plasmid relevante Austausche McMtl1 spezifische Transportaktivität2 pMMX5d WT 3+ 26 pMMX5-4 Cys-frei [SASS] W 0 pMMX5-31 [SACS]+ S3C 3+ 23,6 pMMX5-32 [SACS]+ S110C 3+ 21,3 pMMX5-33 [SACS]+ S158C 3+ 30,5 pMMX5-34 [SACS]+ S199C 3+ 14,2 pMMX5-35 [SACS]+ S212C 3+ 20,6 pMMX5-36 [SACS]+ S242C 3+ 27,2 pMMX5-37 [SACS]+ S299C 3+ 15,2 1. Für Symbole und sonstige Erklärungen siehe Tab. III.1.1. 2.Werte in [nmol∗min-1∗mg Protein-1], gemessen mit 10µM [14C]-D-Mtl. Die LGS322-Testkulturen wurden in 2xTY Cam angezogen. III. Ergebnisse 99 Um eine stark abweichende Faltung des cysteinfreien MtlA (pMMX5-4) auszuschließen, wurde durch ortspezifische Mutagenese mit den Primern H256P (+/-) der entkoppelnde Austausch H256P eingefügt. Zellen des Stammes LTK31-2 mit dem daraus resultierenden Plasmid pMMX5-4HP zeigten einen 1+-Phänotyp auf McMtl und Wachstum auf MM Mtl Cam (Alle anderen Derivate wurden ebenfalls auf Entkopplung getestet, diese wurde aber in keinem Fall festgestellt). Dies ist ein guter Hinweis darauf, dass die Faltung im cysteinfreien MtlA Transport erlaubt und vermutlich der des WT entspricht. Die Transportaktivitäten der „Cys-Scanning“-MtlA-Derivate (Tab. III.2.2.) und der Nachweis, dass eine MtlA-Mutante mit C384S-Austausch immer noch mit hoher Affinität (Kd = 107nM, WT Kd = 45nM) D-Mannitol bindet (Boer et al. 1995), stützen diese Annahme. III.2.2. Markierung der Einzel-Cystein MtlA-Derivate Die Markierung der Cysteine erfolgte wie unter Material und Methoden beschrieben. Dazu wurde der mit den jeweiligen „Einzel-Cystein“-Plasmiden transformierte Stamm JM109(λDE3) in Flüssigkultur angezogen. Nach Induktion der T7-Polymerase wurde mtlA überexprimiert und das Protein in ganzen Zellen zu einem Teil mit Stilben und folgend mit Biotinmaleimid sowie nur mit Biotinmaleimid markiert. Aus den markierten Zellen wurden Membranpräparationen gewonnen, die anschließend gelöst wurden. Das MtlA(His)6 wurde aus den Lösungen gereinigt und bei den SDS-Gelen und folgenden immunologischen Nachweisen eingesetzt. Diese beiden Proben aus den Markierungen mit ganzen Zellen (Probe1: Stilben.+.Biotinmaleimid; Probe 2: Biotinmaleimid) ergaben die Aussage über die Lokalisation der Cysteine. Aus dem anderen Teil der ganzen Zellen wurde ebenfalls mittels Ultraschall Membranpräparationen erstellt, die danach zu einem Teil mit Stilben + Biotinmaleimid sowie nur mit Biotinmaleimid markiert wurden. Die Präparationen wurden gelöst und das MtlA(His)6 gereinigt. Diese Proben 3 (Stilben + Biotinmaleimid) und 4 (Biotinmaleimid) überprüften die allgemeine Zugänglichkeit der Cysteine für die Sulfhydrylreagenzien. 100 III. Ergebnisse III.2.3. Reinigung und immunologischer Nachweis des MtlA-6His Da bei der Markierung der Cysteine auch alle anderen cysteinhaltigen Proteine einer Zelle markiert werden, war eine Reinigung des Zielproteins notwendig. Dazu wurde das MtlA(His)6 aus der gelösten Gesamtproteinfraktion mit Hilfe von Ni-NTASäulen gereinigt. Die Abbildung III.2.1. zeigt das Coomassie-gefärbte SDS-Gel einer Reinigung. St 1.1 5.1 1.2 5.2 1.3 5.3 1.4 5.4 St 116 kDa 80 kDa Í MtlA(His)6 52,5 kDa 34,9 kDa Abb. III.2.1. SDS-Gel einer MtlA(His)6-Reinigung Auf dem Gel sind die Proben 1-4 zu sehen wobei jeweils das gelöste Gesamtmembranprotein vor der Beladung der Säulen (Bahn: 1.1-1.4) und nach der Elution (Bahn: 5.1-5.4) aufgetragen worden sind. Bahn St.: „BIO-RAD SDS-PAGE Standard, Low Range“. Die Größen der Standard-Banden sind neben dem Bild angegeben. Mit einem Pfeil ist die Laufhöhe des MtlA(His)6 angegeben (~60 kDa, Montfort et al., 2001). Die Laufhöhe von ca. 60 kDa des gereinigten Proteins entspricht der beschriebenen Höhe für MtlA(His)6 in Laemmli (1970)-Gelsystemen (Montfort et al., 2001). Im weiteren Verlauf des Versuchs wurden die Eluate der Proben 1-4 aus zwei unterschiedlichen Markierungsansätzen auf ein SDS-Gel aufgetragen. Um die Wirksamkeit der Bindung des MtlA(His)6 an die Ni-NTA-Säule zu testen, wurde die Gesamtproteinfraktion nach dem ersten Säulendurchlauf mit auf das Gel aufgetragen. Zur genauen Identifizierung und ungefähren Quantifizierung des MtlA(His)6 wurden bei dem ersten Gel in einem Western-Blot die aminoterminalen (His)6 immunologisch nachgewiesen (Abb.III.2.3.). Die Abb.III.2.2. zeigt das mit Coomassie-Lösung nachgefärbte Gel im Anschluss an den Proteintransfer. III. Ergebnisse 101 Ansatz 1 GP 1 2 3 4 Ansatz 2 GP 1 2 3 4 Í MtlA(His)6 Abb. III.2.2. SDS-Gel nach dem Proteintransfer auf Membran Abgebildet ist das nachgefärbte SDS-Gel nach dem Proteintransfer auf die Nitrocellulose-Membran. GP: Gesamtprotein nach dem ersten Säulendurchlauf. 1-4: die jeweiligen Proben 1-4 der Markierungsansätze 1 und 2. Mit einem Pfeil ist die Laufhöhe des MtlA(His)6 angegeben (~60 kDa, Montfort et al., 2001). Die noch deutlich zu erkennenden MtlA(His)6-Banden weisen darauf hin, dass ein großer Teil des gereinigten Proteins nicht auf die Nitrocellulose-Membran übertragen wurde. Allerdings ist in Abbildung III.2.3. zu sehen, dass für den Nachweis im „Western-Blot“ ausreichend Protein auf der Membran vorhanden war. Ansatz 1 GP 1 2 3 Ansatz 2 4 GP 1 2 3 4 Í MtlA(His)6 Abb. III.2.3. Immunologischer Nachweis des MtlA(His)6 Abgebildet ist ein Film, der die Chemilumineszens-Reaktion nach dem Western-Blot gegen das (His)6 nachweist. GP: Gesamtprotein nach dem ersten Säulendurchlauf. 1-4: die jeweiligen Proben 1-4 der Markierungsansätze 1 und 2. Mit einem Pfeil ist die Laufhöhe des MtlA(His)6 angegeben (~60 kDa, Montfort et al., 2001). 102 III. Ergebnisse Das gereinigte Protein konnte eindeutig als das MtlA(His)6 identifiziert werden. Unterhalb der ~60 kDa MtlA-Bande sind drei schwächere scharfe Banden zu erkennen, die möglicherweise aus dem Abbau der zytoplasmatischen Domänen IIB und IIA stammen. Die untere Bande entspricht dem 34 kDa-Fragment, die von Stephan und Jacobson (1986) als carboxyterminaler, zytoplasmatischer Teil des EIIMtl identifiziert wurde. Da das (His)6 carboxyterminal inseriert wurde, geben diese Abbauprodukte ebenfalls ein Signal. In der Gesamtproteinfraktion wurde kein MtlA(His)6 nachgewiesen, was für eine effiziente Bindung an die Säulenmatrix spricht. III.2.4. Ergebnisse der Biotinmaleimid-Markierungen Ein zweites Gel, dass die identischen Proben 1-4 des immunologischen Nachweises von MtlA(His)6 enthielt, wurde parallel ebenfalls einem „Western-Blot“ unterzogen, und eine Biotinmaleimid-Markierung mit „Streptavidin-Meerrettich Peroxidase Konjugat“ und anschließendem Chemilumineszens-Nachweis detektiert. Das an die Sulfhydrylgruppe des Cysteins gebundene Biotinmaleimid zeigte so ein Signal, wobei die Auswertung der Signale qualitativ erfolgte. In Abbildung III.2.4. sind die Ergebnisse der Markierungen zusammengefasst. Die mehrfach durchgeführten Versuche ergaben für die jeweiligen MtlA-Derivate vergleichbare Ergebnisse. Die Auswertungen setzten Membraninpermeabilität des Biotinmaleimids unter den gewählten Bedingungen voraus. Unabhängig davon wurde in den Membranvesikeln der Kontrollen zur allgemeinen Zugänglichkeit der Sulfhydrylreagenzien immer ein negatives Signal bei der Probe 3 und ein positives bei der Probe 4 angenommen. In jedem Fall sollte das Stilben bei der Probe 3 nach der Vorinkubation ein weiteres Binden des Biotinmaleimids verhindern. Ebenso sollte das Biotinmaleimid in der Probe 4 ohne Blockierung der Cysteine durch Stilben eine Bindung und somit ein positives Signal bewirken. Dementsprechend bedeuten die fehlenden Signale der Proben 1 und 2 bei den Markierungen in den ganzen Zellen, dass das untersuchte Cystein im Zytoplasma liegen muss, da das Biotinmaleimid unabhängig von einer Vorinkubation mit Stilben das Cystein nicht erreichen und somit auch nicht daran binden kann. Kein Signal bei der Probe 1 und eines bei der Probe 2 deuten darauf hin, dass das Cystein in dem Periplasma liegt. In dem Fall ist es für beide Sulfhydrylreagenzien zugänglich, so dass das gleiche Muster wie bei der allgemeinen Zugänglichkeit zu erwarten ist. III. Ergebnisse 103 Cys-frei 1 2 3 S3C 4 1 2 S110C 3 4 1 2 A) A) A) B) B) B) S158C 1 2 3 S199C 4 1 2 3 4 1 2 A) A) B) B) B) 1 2 3 S299C 4 1 2 3 4 S212C A) S242C 3 3 4 C384 4 1 A) A) A) B) B) B) 2 3 4 Abb. III.2.4. Biotinmaleimid-Markierungen und Nachweise des MtlA(His)6 Die Markierungen sind einmal für die „Einzel-Cysteine“ S110C und S299C, zweimal für die Mutanten S3C, S158C, S199C, S212C und S242C und dreimal für die cysteinfreie Mutante ([SASS]) sowie die Einzel-Cys384-Mutante ([SACS]) durchgeführt worden. Abgebildet sind die Filme zu dem Nachweis der Chemilumineszens-Reaktionen. Die jeweiligen Tests sind nach den relevanten Mutationen betitelt. 1-4: Proben 1-4. Film A) Nachweis der BiotinmaleimidBindung. Film B) Nachweis des MtlA(His)6. Weitere Erläuterungen im Text. 104 III. Ergebnisse Die Ergebnisse aus den Biotinmaleimid-Markierungen sind in der Tabelle III.2.3. und den folgenden Erläuterungen zusammengefasst. Tab. III.2.3. Ergebnisse der Biotinmaleimid-Markierungen Markierung der ganzen Zellen MembranPräparationen relevante AS SB B SB B (1) (2) (3) (4) pMMX5-4 Cys-frei - - - - pMMX5-41 S3C - - - + Zytoplasma pMMX5-42 S110C - - - - n. z. pMMX5-43 S158C - + - + Periplasma pMMX5-44 S199C - + - + Periplasma pMMX5-45 S212C - + - + Periplasma pMMX5-46 S242C - - - + Zytoplasma pMMX5-47 S299C - - - - n. z. pMMX5-3 C384 + + - + n. b. Plasmid Lokalisation In der Tabelle sind die eingesetzten Plasmide und die relevanten AS-Austausche dargestellt. Die Ergebnisse der Markierungen von ganzen Zellen und Membranpräparationen mit Stilben und Biotinmaleimid (SB) sowie nur mit Biotinmaleimid (B) sind wie die Proben 1-4 in den Versuchen geordnet (Zahlen in Klammern). + = Markierung durch Biotinmaleimid. - = keine Markierung durch Biotinmaleimid. Aus den Markierungen wurden die Lokalisationen der AS oder andere Schlussfolgerungen abgeleitet. n.z. = nicht zugänglich. n.b. = nicht bestimmbar. 1. Cys-frei (pMMX5-4): A) Erwartungsgemäß ist bei keiner Probe ein Signal zu erkennen. Bei den Proben 3 und 4 sind lediglich sehr schwache Banden zu sehen, die auf unspezifische Anlagerungen des Biotinmaleimid zurückzuführen sind. B) Der Nachweis des Proteins über das (His)6 zeigt, dass ausreichend Protein vorhanden war. Mit den gewählten Versuchsbedingungen waren keine unspezifischen Signale zu erwarten. 2. S3C (pMMX5-41): A) Bei den Proben 1 und 2 ist kein Signal zu erkennen. Die schwache Bande bei der Probe 1 ist als unspezifisch anzusehen, zumal bei einer Vorinkubation mit Stilben eher schwächere Banden zu erwarten sind. Die Proben 3 und 4 zeigen eine allgemeine Zugänglichkeit für beide Reagenzien. B) Bei allen Proben war III. Ergebnisse 105 eine vergleichbare Menge Protein vorhanden. Das Cys3 wurde dem Modell entsprechend im Zytoplasma nachgewiesen. 3. S110C (pMMX5-42): A) Keine der Proben gab ein Signal. B) Der Nachweis des Proteins zeigt, dass ausreichend Protein vorhanden war. Daher ist davon auszugehen, dass das Cys110 zumindest für Biotinmaleimid nicht zugänglich ist. Die Ursache kann eine unzugängliche Faltung des Proteins oder eine Lokalisation des Cys110 in einer transmembranen Helix sein. 4. S158C (pMMX5-43): A) In der ersten Probe ist kein und in der zweiten ist ein Signal zu sehen. Die allgemeine Zugänglichkeit wird mit den Proben 3 und 4 bestätigt. B) Bei allen Proben war eine vergleichbare Menge Protein vorhanden. Anhand dieser Ergebnisse liegt das Cys158 im Periplasma. 5. S199C (pMMX5-44): A) In der ersten Probe ist kein und in der zweiten ist ein Signal zu sehen. Die allgemeine Zugänglichkeit wird mit den Proben 3 und 4 bestätigt. B) Bei allen Proben war eine vergleichbare Menge Protein vorhanden. Die Ergebnisse sprechen dafür, dass das Cys199 in einer periplasmatischen Schleife liegen muss. 6. S212C (pMMX5-45): A) Eine schwache Bande ist bei Probe 1 zu sehen und eine deutliches Signal bei der Probe 2. Ähnlich verhält es sich bei den Proben 3 und 4. B) Die Proben 1 und 3 enthielten scheinbar mehr Protein als die Proben 2 und 4. Dadurch lassen sich die schwachen Banden bei den Proben 1 und 3 erklären. Diese können eindeutig als negativ bewertet werden, wodurch das Cys212 einer periplasmatischen Schleife zugeordnet werden muss. 7. S242C (pMMX5-46): A) Die Proben 1 und 2 sind negativ, bei einer in den Proben 3 und 4 gezeigten allgemeinen Zugänglichkeit für die Sulfhydrylreagenzien. B) Die Probe 1 hatte deutlich weniger Protein als die anderen Proben, beeinträchtigt in diesem Fall aber nicht das Ergebnis. Das Cys242 liegt dementsprechend im Zytoplasma. 8. S299C (pMMX5-47): A) Keine der Proben zeigt ein Signal. B) Der Nachweis des Proteins zeigt, dass bei allen Proben ausreichend Protein vorhanden war. Folglich ist davon auszugehen, dass das Cys299 zumindest für Biotinmaleimid nicht zugänglich ist. Die Ursache kann eine unzugängliche Faltung des Proteins oder eine Lokalisation des Cys299 in einer transmembranen Helix sein. 106 III. Ergebnisse 9. C384 (pMMX5-3): A) Die Proben 1 und 2 zeigen ein Signal, während die Proben 3 und 4 die allgemeine Zugänglichkeit für Stilben und Biotinmaleimid bestätigen. B) Bei allen Proben war eine vergleichbare Menge an Protein vorhanden. Dieses Ergebnis wäre bei einer Membranpermeabilität von Biotinmaleimid zu erwarten. Dies konnte jedoch in mehreren Versuchen mit den gewählten Bedingungen nicht gezeigt werden. Eine Erklärung für diese Abweichung könnte eher in der Sonderstellung des Cys384 liegen. Es ist Teil der Phosphorylierungskette, die dafür verantwortlich ist, dass das Phosphat von dem PEP auf das D-Mtl übertragen wird (Pas & Robillard, 1988a und 1988b). IV. Diskussion 107 IV. Diskussion Beim substratspezifischen Transport über ein EnzymII werden der Transportschritt und die darauf folgende Phosphorylierung als voneinander unabhängig ablaufende Reaktionen betrachtet (Lolkema et al., 1990; Lolkema et al., 1991a; Postma et al., 1993; Lengeler & Jahreis, 1996). Die erfolgreiche Teilung des EII in eine aktive membrangebundene EIICMtl-Domäne und zwei aktive Phosphatüberträger EIIAMtl und EIIBMtl zeigten, dass Transport und Phosphorylierung als zwei aufeinander folgende Prozesse zu sehen sind, die voneinander getrennt werden können (Grisafi et al., 1989; Lolkema et al., 1990). Unphosphorylierte EII transportieren ihr Substrat nicht in Mengen, die Wachstum ermöglichen (Postma & Stock, 1980). Folglich müssen Transport und Phosphorylierung gekoppelt sein. Das EIICMtl kann als isolierter Transporter seine Substrate mit normaler Affinität binden (Grisafi et al., 1989; Briggs et al., 1992) und einen aktiven transmembranen Transporter bilden (Lolkema et al., 1990), aber das hochaffine D-Mannitol (KMapp 1-2µM) nicht transportieren. Dagegen wird das niedrigaffine C5-Isomer D-Arabinitol (KMapp ≥ 500µM) in einer für das Wachstum ausreichenden Menge durch EIICMtl transportiert (Klawitter, 1992; Otte et al., 2003). Die Stärke der Kopplung von Transport und Phosphorylierung scheint durch die Affinität der Bindestelle zum Substrat beeinflusst zu werden (Otte, 2000, Otte et al. 2003). In ∆ptsHI-Stämmen von Salmonella enterica serovar typhimurium und Escherichia coli wurden Mutanten isoliert, die durch erleichterte Diffusion freie Glucose über das EIIGlc aufnehmen konnten (Postma, 1981; Ruijter et al., 1992). Das Wachstum konnte in diesen Mutanten mit einer ATP-abhängigen Glukokinase ermöglicht werden. Transport und Phosphorylierung des Substrats waren demnach entkoppelt. Alle mutierten EIIGlc zeigten eine stark verringerte Affinität für Glukose (≥100fach). Die erfolgreiche Isolierung von diversen entkoppelten EIIGlc- und EIIMtl-Mutanten (EIIGlc: Postma, 1981; Ruijter et al., 1990; Ruijter et al., 1992. EIIMtl: Klawitter, 1992; Scholle, 1993; Heuel, 1997; Otte, 2000; Turgut, diese Arbeit) ist ein weiterer Beweis dafür, dass diese Kopplung nicht als strikt anzusehen ist. Ein Transport ohne gleichzeitige Phosphorylierung ist scheinbar immer dann möglich, wenn ein geeignetes niedrigaffines Substrat durch das EIIC transportiert werden soll, oder Mutationen die Affinität des 108 IV. Diskussion Transporters für das hochaffine Substrat verringern. Um den Mechanismus der Entkopplung weiterführend untersuchen zu können, wurden in dieser Arbeit entkoppelte Mutanten mit verschiedenen Systemen isoliert. IV.1. Für Transport und Phosphorylierung relevante AS im EIICMtl Von 30 untersuchten entkoppelnden Mutationen zeigten 15 den Austausch E218A, eine den Austausch E218V und 14 den Austausch H256P. Untersuchungen von Otte (Otte, 2000; Otte et al., 2003) ergaben weitere 40 aus K. pneumoniae, die die Austausche E218A (16 Mutanten), H256P (21 Mutanten) und H256Y (3 Mutanten) zeigten. Dadurch ergeben sich vier Substitutionen in zwei Aminosäuren, die eine Entkopplung hervorrufen und sowohl in dem verkürzten (EIIC) als auch dem vollständigen (EIICBA) MtlA zusammen mit DalD ein Wachstum auf D-Mannitol ermöglichen (Tab. III.1.23.). Im Rahmen der Untersuchungen konnten keine Unterschiede der Mutationsarten in Abhängigkeit des Selektionssystems beobachtet werden. Weder die Selektionstemperatur, noch die Selektion mit vollständigem oder verkürztem mtlA ergaben abweichende Mutationen. Lediglich das seltene Auftreten des Austausches E218V konnte festgestellt werden. Die Tatsache, dass das Vorhandensein der Domänen EIIA und EIIB keine Auswirkungen auf die Art der Mutationen hatte, unterstützt die Feststellung, dass EIIC die für Bindung und Transport entscheidende Domäne ist. IV.1.1. Vergleich der Mutationen E218A, E218V und H256P Anhand der in der Tabelle III.1.23. zusammengefassten Daten lassen sich folgende Beobachtungen machen. Alle drei AS-Austausche (E218A, E218V und H256P) führen zu einer Entkopplung des Transports von D-Mtl. In einem phosphorylierten EIIMtl (IICBA) ist jedoch der Anteil des gekoppelten Transports sehr verschieden. Das verbesserte Wachstum in Anwesenheit von DalD ist ein Hinweis darauf, dass unphosphoryliertes D-Mtl in die Zelle transportiert wird und erst von einer Dehydrogenase genutzt werden kann. Dieser Anteil ist beim Derivat mit dem Austausch E218A am geringsten. Die Generationszeit auf D-Mtl wird jedoch um ca. 40% reduziert und die spezifische Phosphorylierungsaktivität liegt im Bereich des WT. Der KMapp IV. Diskussion 109 entspricht ebenfalls WT-Werten, lediglich V ist auf etwa 25% reduziert. Die TransportWerte waren jedoch nicht reproduzierbar und wurden in anderen Systemen gemessen. Die starke Abhängigkeit der Transportmessungen von Vektor und Stamm (Teil III.1.5.3. / Tab. III.1.22.) zeigt, dass ein exakter Vergleich der einzelnen Messungen nur in identischen Systemen möglich ist. Für diese Arbeit wird deshalb bei EIIMtlE218A von einem erhöhten KM-Wert außerhalb der Messbarkeit und einer Phosphorylierungsaktivität im Bereich des WT ausgegangen. Deutlich größer ist der Anteil an freiem D-Mtl bei E218V. Die Reduktion der Generationszeit liegt bei 52 bis 70% und die spezifische Phosphorylierungsaktivität erreicht nur noch ca. 12% des WT. Der gekoppelte Transport ist nicht mehr messbar. Bei den Austauschen E218A und E218V sieht man vergleichbare Effekte, die lediglich in Ihrer Intensität bei E218V stärker sind. Bei dem Austausch H256P fällt sofort die nicht mehr zum Wachstum auf D-Mtl ausreichende Phosphorylierung (<1% im Vergleich zum WT) auf. Ein Wachstum auf Grund eines gekoppelten Transports ist nicht mehr möglich und auch mit DalD wird lediglich eine Gt von 220-345min auf D-Mtl gemessen. In einem Vergleich mit den vorher genannten MtlA-Derivaten kann man den Austausch H256P als neue Klasse für die Entkopplung des Transports sehen oder als weitere Verstärkung der vorher beobachteten Effekte beschreiben. Die in K.-pneumoniae charakterisierten Mutationen der AS H256 (H256P und H256Y. Otte, 2000; Otte et al., 2003) zeigten im Gegensatz zu der hier beschriebenen Mutante immer noch Phosphorylierungsaktivität in einem vollständigen MtlA. Das K. pneumoniae-Derivat mit der Substitution H256P liegt im Vergleich zum WT bei 14%, das mit dem Austausch H256Y jedoch noch bei nahezu 100%. Möglicherweise kann dies auf unterschiedliche Grade der Entkopplung zurückgeführt werden, die durch die Messungen der Bindeaktivität nicht differenziert werden konnten. Bei dem Vergleich von AS-Austauschen an zwei unterschiedlichen Stellen des Proteins sollte man aber auch völlig abweichende Mechanismen der Entkopplung in Betracht ziehen. Die Konzentration der isolierten AS-Substitutionen auf zwei Stellen im Protein weist jedoch auf eine maßgebliche Beteiligung der AS 218 und 256 an dem Transport oder an der Substraterkennung hin. Kombiniert man die entkoppelnden AS-Austausche der AS218 und AS256, so schaltet man jeglichen Transport und Phosphorylierung aus. Hier besteht allerdings noch die Möglichkeit, dass durch die Doppelmutationen das Protein nicht mehr mit der richtigen Faltung in der 110 IV. Diskussion Membran vorliegt. Dass es in der Membran zu finden ist, zeigten die Überexpressionsgele der Vesikel (Abb.III.1.6.). Auf der Suche nach Suppressormutationen mit den Mutanten P256 und A218/P256 ergab die natürliche Selektion nur Mutationen kodierend für die AS256. Dies kann ein Hinweis darauf sein, dass unter den gewählten Bedingungen keine Suppressormutationen selektioniert werden können. Ebenfalls möglich ist, dass eine essentielle Funktion der AS256 keine Suppressormutationen erlaubt. Wachstum mit gekoppeltem Transport wurde bei A218/P256 durch die Austausche P256A und P256Q wiederhergestellt. Aufgrund der noch vorhandenen Substitution E218A zeigten Zellen mit diesen Kombinationen noch Wachstum mit entkoppeltem Transport. Jedoch war das Wachstum sowohl mit gekoppeltem als auch entkoppeltem Transport schwächer als bei einem alleinigem E218A-Austausch, was an dem ungewollten zusätzlichen Austausch L229Q liegen kann oder ein Hinweis darauf ist, dass das Alanin bzw. Glutamin an Position 256 die WT-Aktivität nicht wiederherstellt. Die einzelnen Austausche Alanin, Glutamin sowie Serin an der AS-Position 256 führten annähernd zur Wiederherstellung der WT-Eigenschaften. Bei der Substitution H256A wurden auch von Weng et al. (1992) nach einem gezielten AS-Austausch keine signifikanten Unterschiede der Transport- und Phosphorylierungsaktivitäten festgestellt. Daraufhin wurde diesem Histidinrest keine besondere katalytische Funktion im EIIMtl zugesprochen. Ein Glutamin an dieser Position führte ebenfalls zu keinen nennenswerten Veränderungen, bei einem Serin fällt lediglich der niedrige V-Wert auf. Wie schon im Ergebnisteil beschrieben (III.1.5.3.) wurden bei KM-Messungen mit MtlA*H256S auch nichtlineare Kinetiken beobachtet. Tests mit unterschiedlichen Versuchsansätzen konnten jedoch nicht darlegen, auf Grund welcher Parameter lineare und nichtlineare Kinetiken auftreten. Variierende MtlA-Konzentrationen innerhalb der Testkulturen waren auszuschließen, jedoch könnte der Grund in einer Beeinflussung der Dimerisierung von MtlA liegen (Lolkema, pers. Mitteilung). Möglicherweise liegt so bei hohen Konzentrationen eine erleichterte Diffusion vor, welche der grünen Gerade in der Abb.IV.1. entsprechen würde. Eine Bindestelle für geringe Konzentrationen könnte durch eine erhöhte Affinität die rote Gerade hervorrufen, was einem reduzierten V-Wert und KM-Wert entspräche. In jedem Fall spiegelt sich der reduzierte V-Wert bei den linearen Messergebnissen wieder. Sollte in Fällen linearer Messergebnisse die Dimerisierung nicht gestört sein, würde auch eine Reduktion des KMapp-Werts durch Diffusion entfallen. IV. Diskussion 111 LGS322/pDSM2 [P256S] 0.6 1/V [nM] P256S 0.5 0.4 0.3 0.2 0.1 0.0 -0.1 -0.5 1/S [µM] 0 0.5 1 1.5 2 2.5 Abb. IV.1. Interpretation der nichtlinearen Kinetik von LGS322/pDSM2 Die Abbildung zeigt die Messpunkte einer nichtlinear verlaufenden KMappMessung. Die rote und die grüne Linie deuten zwei unterschiedliche Kinetiken an, die sich zu den abgebildeten Punkten überlagern. Folglich könnte diese Mutation sowohl die Affinität der Bindestelle, als auch die Verbindung der Monomere beeinflussen. Diese Beobachtungen sind ein weiteres Indiz dafür, dass es sich bei H256, wie bei E218, durchaus um katalytisch und/oder strukturell relevante AS handelt. Die Ergebnisse der Suche nach Suppressormutanten bestärken durch das ausschließliche Vorkommen von AS-Austauschen in der der AS256 diese Aussage. Allerdings besitzt das MtlA*H256P aus K.-pneumoniae noch genügend Phosphorylierungsaktivität, um ein Wachstum ohne DalD zu ermöglichen (Otte, 2000). Die Ursache für diese Abweichung kann in den unterschiedlichen Stammhintergründen und Vektoren liegen, so wie es im Ergebnisteil für die Variante E218A beschrieben worden ist (Teil III.1.5.3. / Tab. III.1.22.). Denkbar ist auch eine AS- Sequenzabweichung in MtlAK.ipn. außerhalb der AS256, welche zu einem anderen Phänotypen führt, da der Austausch H256P in E.-coli die Phosphorylierungsaktivität fast vollständig verhindert. Im Vergleich zu dem MtlA aus E.vcoli (637AS) weist MtlA aus K.pneumoniae (635AS) 94% identische Aminosäuren auf. Dieser Wert steigt für die Transportdomäne EIIC (AS 1-360) auf 96% (siehe Abb. IV.2.). 112 Ec Kp IV. Diskussion 1 TM1 TM2 70 MSSDIKIKVQSFGRFLSNMVMPNI GAFIAWGIITALFIPTGWLP NETLAK LVGPMITYLLPLLIGYTGG K MSSDIKIKVQSFGRFLSNMVMPNI GAFIAWGIITALFIPTGWLP NETLAK LVGPMITYLLPLLIGYTGG K Kon XSSXIKVKVQXFGRFLSNMVMPNI GAFIAWGIITALFIPTGWLP NETLAK LVGPMITYLLPLLIGYTGG K Ec Kp 140 LVGGERGGVVGAITTMGVIVGADMPMFLGSMIAGPLGGWCIKHFDRWVDGKIKSGFEMLVNNFS AGIIGM LVGGERGGVVGAITTMGVIVGADMPMFLGSMIAGPLGGYCIKKFDNWVDGKIKSGFEMLVNNFS AGIIGM Kon LIGGERGGVVGAIATMGVIVGADIPMFLGAMIXGPLGGWLIKKFDKXVDGKIKSGFEMLVNNFS AGIIGM Ec Kp TM3 TM4 210 ILAILAFLGIGPIV EALSKMLAAGV NFMVVHDMLPLASIFVEPA KILFLNNAINHGIFSPLGIQQSHELG ILAILAFLGIGPAV EVLSKILAAGV NFMVAHDMLPLASIFVEPA KILFLNNAINHGIFSPLGIQQSHELG Kon ILAILAFXGIGPVV XXLSKXLAAGV EXIVXXXLLPLASIFVEPA KILFLNNAINHGIFSPLGIQQXXEXG Ec Kp 280 KSIFFLIEANPGPGMGVLLAYMFFGRGSAKQSAGGAAIIHFLGGIHEIYFPYVLMNPRLIL AVILGGMTG KSIFFLIEANPGPGMGVLLAYMFFGRGSAKQSAGGAAIIHFLGGIHEIYFPYVLMNPRLIL AVILGGMTG Kon KSIFFLIEANPGPGLGVLLAYMLFGKGSAKQSAGGAAIIHFLGGIHEIYFPYVLMNPRLIL AVIAGGMTG Ec Kp TM5 TM6 350 VFTLTILGGGL VSPASPGSILAVLAMTPKGAYF ANIAGVCAAMAVSFVVSAIL LKTSKVKEEDDIEAATR VFTLTILNGGL VSPASPGSILAVLAMTPKGAYF ANIAAIIAAMAVSFVVSAVL FKTSKVKERSDIEAATR Kon. VFTLTILNAGL VAPASPGSIIAVLAMTPKGAYL AVLAGVXVAAAVSFVVSAII LKTSKXXEEDDIEAATR Ec Kp 360... RMQDMKAESK... RMHDMKAESK... Kon RMXXMKAXSK Abb. IV.2. Vergleich der EIICMtl AS-Sequenzen aus E. coli und K. pneumoniae Dargestellt sind die abgeleiteten EIIC-Aminosäuresequenzen der Gene mtlA aus Escherichia coli K12 (Ec) (Lee & Saier, 1983) und Klebsiella pneumoniae (Kp) (Otte, 2000) und die Konsensussequenz (Kon) aus einem Vergleich mit weiteren MtlA-Sequenzen (siehe Abb. IV.3.). Die identischen AS aus der Konsensussequenz sind gelb unterlegt. Vermutete transmembrane Helices (TM) sind eingerahmt und nummeriert. Abweichungen in den beiden Sequenzen sind grau oder orangefarben hervorgehoben, dabei markiert Orange die Austausche, die relevant sein könnten (weiteres siehe Text und Abb. IV.3.). Im Bereich von EIIC gibt es 16 voneinander abweichende AS. Konservative ASAustausche (AS 318, 319, 332) und Austausche in Aminosäuren, die in der Konsensussequenz verschiedener MtlA (Abb. IV.3.) keine Berücksichtigung finden (AS 156, 160, 170, 320, 353) haben mit geringerer Wahrscheinlichkeit Auswirkungen auf IV. Diskussion 113 das Transportverhalten. Danach bleiben acht Aminosäuren (109, 113, 116, 153, 288, 334, 342, 343), die am ehesten für veränderte katalytische Merkmale verantwortlich sein könnten, ohne die übrigen Aminosäuren völlig auszuschließen. Es fällt aber auf, dass in dem hoch konservierten Bereich zwischen TM4 und TM5 keine Austausche zu finden sind. Für eine genaue Beurteilung der unterschiedlichen Phänotypen sind weitere Untersuchungen der Stammhintergründe, Vektoren und der hier benannten ASAustausche erforderlich. IV.1.2. Die AS 218 und 256 im Translokationsmodell Für die in dieser Arbeit näher untersuchten AS218 und AS256 lässt sich feststellen, dass unterschiedliche Aminosäuren an diesen Stellen unterschiedliche Einflüsse auf den gekoppelten Transport, die Phosphorylierung und Bindung haben. Beide Aminosäuren liegen nach dem aktuellen Strukturmodell in der hochkonservierten Schleife 5 (Abb.IV.3. und IV.5.) und sind in den MtlA-Sequenzen konserviert. Das H256 liegt zudem in dem GIHE-Motiv, welches aufgrund der sehr starken Konservierung schon vorher als essentiell angesehen wurde (Lengeler, 1990). In einem Modell für den katalytischen Mechanismus von EIIMtl (Lolkema et al., 1991a; 1992) wird eine Kopplung zwischen Transport und Phosphorylierung mit einer Rate <50% festgelegt. Untersuchungen mit Vesikelpräparationen des EIIMtl zeigten, dass freies und phosphoryliertes D-Mannitol mit gleicher Wahrscheinlichkeit von der Bindestelle freigesetzt werden (Lolkema et al., 1991b). Diese in vitro gemachten Beobachtungen widersprechen den Ergebnissen aus in vivo Untersuchungen. Anhand dünnschichtchromatographischer Versuche konnte kein freies D-Mannitol nach einem Transport durch EIIMtl nachgewiesen werden (Otte, 2000). Das geringe Vorkommen von freiem D-Mtl wurde auf einen schwachen Transport durch das Gut-PTS sowie der Hydrolyse von Mtl1P zurückgeführt. Untersuchungen mit EIIMtl und EIIMtl+DalD zeigten dementsprechend keine relevanten Unterschiede im Wachstum von Zellen auf D-Mtl (Tab. III.1.23.). Allerdings könnte das freie D-Mtl sofort wieder an der Membraninnenseite gebunden und phosphoryliert werden, was mit den genannten Methoden nicht nachweisbar wäre. Ist dagegen die interne Phosphorylierung beeinträchtigt, steigt der Anteil des freien Substrates in der Zelle. Der resultierende Anteil unphosphorylierten Substrats ist dadurch von der Phosphorylierungsaktivität bzw. der Affinität der intrazellulären Bindestellen des EIIMtl-Komplexes abhängig. So zeigt sich 114 IV. Diskussion bei den Mutationen dieser Arbeit, dass die am stärksten entkoppelten MtlA-Varianten die geringste Phosphorylierungsaktivität zeigten (Tab. III.1.23.). Ähnliche Effekte wurden auch bei den entkoppelten Derivaten aus EIIGlc beobachtet. Die entkoppelten EIIGlcMutanten zeigten eine Phosphorylierungsaktivität, die 0 bis 100% des Wildtyps entsprach (Ruijter et al., 1992). Eine direkte Beziehung zu den KM-Werten der verschiedenen Proteine zeigte sich hier jedoch nicht. Abb. IV.3. Vergleich der EIICMtl AS-Sequenzen verschiedener Stämme K12 O157:H7 CFT073 S.fle S.ent K.pne Y.pes V.cho P.mul C.ace S.mut L.lac S.car B.hal B.ste B.sub (1) (1) (1) (1) (1) (1) (1) (1) (1) (1) (1) (1) (1) (1) (1) (1) 1 70 ----------MSSDIKIKVQSFGRFLSNMVMPNIGAFIAWGIITALFIPTGWLPNETLAK-LVGPMITYL ----------MSSDIKIKVQSFGRFLSNMVMPNIGAFIAWGIITALFIPTGWLPNETLAK-LVGPMITYL ----------MSSDIKIKVQSFGRFLSNMVMPNIGAFIAWGIITALFIPTGWLPNETLAK-LVGPMITYL ----------MSSDIKIKVQSFGRFLSNMVMPNIGAFIAWGIITALFIPTGWLPNETLAK-LVGPMITYL ----------MSSDIKIKVQSFGRFLSNMVMPNIGAFIAWGIITALFIPTGWLPNETLAK-LVGPMITYL ----------MSSDIKIKVQSFGRFLSNMVMPNIGAFIAWGIITALFIPTGWLPNETLAK-LVGPMITYL ---------MFSPDAKVRVQNFGRFLSNMVMPNIGAFIAWGIITALFIPTGWLPNETLAK-LVGPMITYL ---------MISSDAKVKIQNFGRFLSNMVMPNIGAFIAWGFITALFIPTGWVPNETLAS-LVGPMITYL MVILSTRLTMLSANAKVKVQNFGRFLSNMVMPNIGAFIAWGFITALFIPTGWLPNETLAK-LVGPMITYL METYKDSTQVSKSSLKKKIQGFGGFLSGMVMPNIGAFIAWGLITALFIKTGWLPNDNLSK-LVDPMIHYM --------MERKSSLKVRVQKLGTSLSNMVMPNIGAFIAWGVAASLFIATGYLPNKALDTNVVGPMNKYV --------MTDTVSLKVRVQKLGTALSNMVMPNIPVLIAWGVLTMFFIPDGFTPNKTFAA-MVSPMLPFL -----MSNSQQNKGIGRKVQAFGSFLSSMIMPNIGAFIAWGFIAAIFIDNGWFPNKDLAQ-LAGPMITYL --------MNNQPSFRARVQKFGSFLSGMIMPNIGAFIAWGLITALFIPTGWWPNEQLAE-LVGPMITYL ----MTHTSENQAGFRVKIQRFGSYLSGMIMPNIGAFIAWGIITALFIPTGWLPNETFAK-LVGPMITYL ----MQQQEQQQGGMKVKVQRFGSYLSGMIMPNIGAFIAWGIITALFIPAGWFPNEQLNT-LVSPMITYL xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx xxxxxxxxxTM1xxxxxxxx xxxxxxx xxxxxTM2x Kons (1) MVTYMXXXXXXSSXIKVKVQXFGRFLSNMVMPNIGAFIAWGIITALFIPTGWLPNETLAKNLVGPMITYL K12 O157:H7 CFT073 S.fle S.ent K.pne Y.pes V.cho P.mul C.ace S.mut L.lac S.car B.hal B.ste B.sub (60) (60) (60) (60) (60) (60) (61) (61) (70) (70) (63) (62) (65) (62) (66) (66) 71 140 LPLLIGYTGGKLVGGERGGVVGAITTMGVIVGA------------DMPMFLGSMIAGPLGGWCIKHFDRW LPLLIGYTGGKLVGGERGGVVGAITTMGVIVGA------------DMPMFLGSMIAGPLGGWCIKHFDRW LPLLIGYTGGKLVGGERGGVVGAITTMGVIVGA------------DMPMFLGSMIAGPLGGWCIKHFDRW LPLLIGYTGGKLVGGKRGGVVGAITTMGVIVGA------------DMPMFLGSMIAGPLGGWCIKHFDRW LPLLIGYTGGRLVGGERGGVVGAITTMGVIVGA------------DMPMFLGSMIAGPLGGYCIKKFDSW LPLLIGYTGGKLVGGERGGVVGAITTMGVIVGA------------DMPMFLGSMIAGPLGGYCIKKFDNW LPLLIGFTGGRLVGGDRGGVVGAITTMGVIVGA------------DMPMFLGAMIVGPLGGWAIKHFDRW LPLLIGYTGGKLAGGERGAVVGAITTMGVIVGT------------DIPMFMGAMIVGPMGGWAIKAFDKK LPLLIGYSGGKLIAGERGAVVGAIATAGVIVGT------------DIPMFLGAMIAGPTGGWAIKRFDKW LPMLIGYQGGKLVYDTRGGVVGAIATMGMIVGA------------SIPMFLGGMIIGPLGGYVIKKFDKA LPLLIGYTGGYNIHKQRGGVIGAIASFGAIAGS------------TVTMFIGAMIMGPLSAWILKKFDEK IPLMIGYTGGKNIYEHRGGVVGAIATFGSIIATASISVGGLNAKGNVPMILGAMILGPFGAWLIKKFDEY IPLLIAFSGGRLIHDLRGGIIAATATMGVIVALP-----------DTPMLLGAMIMGPLVGWLMKKTDEF LPLLIGYTGGKMIYDVRGGVVGAAATMGVVVGA------------DIPMFIGAMIMGPLGGFLIKKVDQV LPLLIGYTGGKMIYDVRGGVVGATATMGVIVGS------------DIPMFLGAMIMGPLGGYLIKKFDQQ LPLLIAYTGGKMIYDHRGGVVGATAAIGVIVGS------------DIPMFLGAMIMGPLGGYLIKQTDKL xxxxxxxxxxxxxx xTM2xxxxxx xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx Kons (71) LPLLIGYTGGKLIGGERGGVVGAIATMGVIVGAPSISVGGLNAKGDIPMFLGAMIXGPLGGWLIKKFDKX IV. Diskussion K12 O157:H7 CFT073 S.fle S.ent K.pne Y.pes V.cho P.mul C.ace S.mut L.lac S.car B.hal B.ste B.sub (118) (118) (118) (118) (118) (118) (119) (119) (128) (128) (121) (132) (124) (120) (124) (124) 115 141 210 VDGKIKSGFEMLVNNFSAGIIGMILAILAFLGIGPIVEALSKMLAAGVNFMVVHDMLPLASIFVEPAKIL VDGKIKSGFEMLVNNFSAGIIGMILAILAFLGIGPIVETLSKMLAAGVNFMVVHDMLPLASIFVEPAKIL VDGKIKSGFEMLVNNFSAGIIGMILAILAFLGIGPVVEALSKMLAAGVNFMVVHDMLPLASIFVEPAKIL VDGKIKSGFEMLVNNFSAGIIGMILAILAFLGIGPIVEALSKMLAAGVNFMVVHDMLPLASIFVEPAKIL VDGKIKSGFEMLVNNFSAGIIGMILAILAFLGIGPAVEVLSKILAAGVNFMVAHDMLPLASIFVEPAKIL VDGKIKSGFEMLVNNFSAGIIGMILAILAFLGIGPAVEVLSKILAAGVNFMVAHDMLPLASIFVEPAKIL VEGKIKSGFEMLVNNFSSGIIGMLLALLAFMAIGPLVEVLSKGLAYGVDVMVQNNLLPLASIFVEPAKIL IDGKVRSGFEMLVNNFSAGIIGMLCAIIAFFLIGPFVKVLSGALAAGVNFLVTAHLLPLTSIFVEPAKIL ADGKIKSGFEMLVNNFSSGIIGMILAILFFWLIGPAVKALSTMLAAGVDILVKAHLLPLTSIFVEPAKIL IENKIPTGFEMLVNNFSAGILGAALAIISYVAVGPVVAGASTGLGSIALAITNQGLLPLIAVVVEPAKIL VQPKIRTGFEMLVNNFSLGLIGFALMVLAFFVIGPVVAQLTEWVGIGVEAIVKVHLLPLANLIIEPAKIL IQPHIKAGLEMLINNFSAGLVGFGLSLFAVKVVGPLVAWLTDVMGHGVDFLISNHLIPLANLFIEPAKIL VQPRTPQGFEMLFNNFSAGILGFIMTIFGFEVLAPIMKFIMHILSVGVEALVHAHLLPLVSILVEPAKIV LQPKVRSGFEMLVNNFSAGILAAILAIVAFLGIGPVVVSFSNVLASGVEVIIGAGLLPLASIFIEPAKVL IQGKVKQGFEMLVNNFSAGIIGGLLTLAAFKGVGPVVSAISKTLAAGVEKIVDLHLLPLANIFIEPGKVL FKDKVKQGFGIRINNFTAGIVGAALTILAFYAIGPVVLTLNKLLAAGVEVIVHANLLPVASVFLEPAKVL xxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx xxxxxxxxTM3xxxxxxxxx xxxxxxxxxxx xxxxxxxxTM4xxxxxxxx xxx Kons (141) VDGKIKSGFEMLVNNFSAGIIGMILAILAFXGIGPVVXXLSKXLAAGVEXIVXXXLLPLASIFVEPAKIL 211 280 FLNNAINHGIFSPLGIQQSHELGKSIFFLIEANPGPGMGVLLAYMFFGRGSAKQSAGGAAIIHFLGGIHE FLNNAINHGIFSPLGIQQSHELGKSIFFLIEANPGPGMGVLLAYMFFGRGSAKQSAGGAAIIHFLGGIHE FLNNAINHGIFSPLGIQQSHELGKSIFFLIEANPGPGMGVLLAYMFFGRGSAKQSAGGAAIIHFLGGIHE FLNNAINHGIFSPLGIQQSHELGKSIFFLIEANPGPGMGVLLAYMFFGRGSAKQSAGGAAIIHFLGGIHE FLNNAINHGIFSPLGIQQSHEMGKSIFFLIEANPGPGMGVLLAYMFFGRGSAKQSAGGAAIIHFLGGIHE FLNNAINHGIFSPLGIQQSHELGKSIFFLIEANPGPGMGVLLAYMFFGRGSAKQSAGGAAIIHFLGGIHE FLNNAINHGIFSPLGVQQAAETGKSIFFLIEANPGPGLGVLMAYMFFGKGNAKQSAGGAAIIHFFGGIHE FLNNAINHGIFSPLGIQQASETGQSIFFLIEANPGPGLGILLAYMVFGKGTARQTAGGATIIHFFGGIHE FLNNAINHGIFSPLGIQQSQEFGQSIFFLIEANPGPGLGVLLAYIIFGKGTAKQTAGGATIIHFFGGIHE FLNNAINHGVFSPLGIEQVQHLGKSVFFLLEADPGPGLGILLAYSLYGKGSAKNSAPGAVIIHFLGGIHE FLNNALNHGIFTPLGTEQVAKVGKSVLFLLEANPGPGLGVLIAYAMFGKGSAKSSSWGAMIIHFFGGIHE FLNNAINQGILSPLGIQQVSENGKSLLFLLEANPGPGLGILIAFMLFGKGSAKATAPGAILIQFVGGIHE FLNNAINHGVFTPLGADQAAHAGQSILYTIESNPGPGIGVLIAYMIFGKGTAKATSYGAGIIQFFGGIHE FLNNAINHGILSPIGIDQAASAGKSILFLLETNPGPGLGVLLAFMVFGKGMAKQSAPGAAVIHFAGGIHE FLNNAINHGILSPLGIEQAAKTGKSILFLLEPNPGPGLGILLAYWLFGKGMAKQSAPGAIIIHFLGGIHE FLNNAINHGILSPIGIEQASQTGKSILFLVEANPGPGLGIFLAYMFFGKGSSKSTAPGAAIIHFFGGIHE K12 O157:H7 CFT073 S.fle S.ent K.pne Y.pes V.cho P.mul C.ace S.mut L.lac S.car B.hal B.ste B.sub (188) (188) (188) (188) (188) (188) (189) (189) (198) (198) (191) (202) (194) (190) (194) (194) Kons H195 E218 GIHE (211) FLNNAINHGIFSPLGIQQXXEXGKSIFFLIEANPGPGLGVLLAYMLFGKGSAKQSAGGAAIIHFLGGIHE K12 (258) O157:H7 (258) CFT073 (258) S.fle...(258) S.ent (258) K.pne (258) Y.pes (259) V.cho (259) P.mul (268) C.ace (268) S.mut (261) L.lac (272) S.car (264) B.hal (260) B.ste (264) B.sub (264) 281 350 IYFPYVLMNPRLILAVILGGMTGVFTLTILGGGLVSPASPGSILAVLAMTPK------GAYFANIAGVCA IYFPYVLMNPRLILAVILGGMTGVFTLTILGGGLVSPASPGSILAVLAMTPK------GAYFANIAGVCA IYFPYVLMNPRLILAVILGGMTGVFTLTILGGGLVSPASPGSILAVLAMTPK------GAYFANIAGVCA IYFPYVLMNPRLILAVILGGMTGVFTLTILGGGLVSPASPGSILAVLAMTPK------GAYFANIAGVCA IYFPYVLMNPRLILAVILGGMTGVFTLTILNGGLVSPASPGSILAVLAMTPK------GAYFANIAAIVA IYFPYVLMNPRLILAVILGGMTGVFTLTILNGGLVSPASPGSILAVLAMTPK------GAYFANIAAIIA IYFPYVLMNPRLLLAVILGGMTGVFTLTLLNGGLVSAASPGSILAILAMTPK------GAYFANIAAVAT IYFPYILMNPRLILAAIAGGMTGVFTLTVFNAGLVSPASPGSIFAVLLMTNK------GSILGVVCSIFA IYFPYVLMNPRLLLAVIAGGVSGVFTLVLFNAGLVAPASPGSIIAVLLMTPQ------NAIVGVLASVAI IYFPYVLMKPFLLLAVIAGGICADLTFVLLKAGLVAAASPGSIIAILAMSPK------GGQLPVLAGVAV IYFPYVMMKPAMFLAVIAGGLTGTFTFQTLGAGLTAPASPGSIIAIMGMSPK----GWGPHLVVLAGVFA IYFPYVMMKPALFFAVMAGGVSGTLTNNLLGSGLAAPASPGSIIAITGMASGKSAGGFANVLCVWAGIAV IYFPYVLMRPLLFVSVILGGMTGVATYSLLDFGFKTPASPGSIIVYAINAPK------GEFLHMLTGVVL IYFPYILMKPTLILAVIAGGMSGVFTFVLFNAGLVAVPSPGSIFALLAMTPR------GEYAGVLAGVII IYFPYVLMRPILILAAIAGGVSGVLTFTIFDAGLVAVPSPGSIFALLAMTPK------GNYLGVLAGVLV IYFPYILMKPGPDSRSHCRRSKRTLNITIFNAGLVAAASPGSIIALMAMTPR------GGYFGVLAGVLV xxxxxxxxxxxxxxxxxxxxxxxxxxxx xxxxxxxxTM5xxxxxxxxx xxxxxxxxxxxxxxxxxxxxxxxxxxxx xxxxTM6x Kons (281) IYFPYVLMNPRLILAVIAGGMTGVFTLTILNAGLVAPASPGSIIAVLAMTPKKSAGGWGAYLAVLAGVXV 116 IV. Diskussion K12 O157:H7 CFT073 S.fle S.ent K.pne Y.pes V.cho P.mul C.ace S.mut L.lac S.car B.hal B.ste B.sub (322) (322) (322) (322) (322) (322) (323) (323) (332) (332) (327) (342) (328) (324) (328) (328) 351 390... AMAVSFVVSAILLKTSKVKE-EDDIEAATRRMQDMKAESK...(360) AMAVSFVVSAILLKTSKVKE-EDDIEAATRRMQDMKAESK...(360) AMAVSFVVSAILLKTSKVKE-EDDIEAAARRVQEMKAESK...(360) AMAVSFVVSAILLKTSKVKE-EDDIEAATRRMQDMKAESK...(360) AMAVSFVVSAILLKTSKVKE-EDDIEAATRRMHDMKAESK...(360) AMAVSFVVSAVLFKTSKVKE-RSDIEAATRRMHDMKAESK...(360) AFAVSFVVSAILLKSSKAKDDEEGLEGATRRMQDMKAQSK...(362) AAAVSFTVAALLMKAQTSTEQDGDKDALVKATSIMQEMKA...(362) AATVSFVIASFFLKIQ----KEENGHSLEKMQAASKAMKS...(367) GAIVSFVVASIILKGSKE-KSKDNFEEAQNKMKEMKKESK...(370) AAVASFLVASIILKSDN--SDDDSLETAQAVTQAAKAESK...(364) AAIVSFLVAAFILKRDKSMIDDSAFENAKVGVATDKAVSK...(381) AALVSFVVSALILKFTKDP--KQDLAEATAQMEATKGKKS...(365) ATVVSFVIASIILKTSKAT--AEDLTEATSKMEGLKGKES...(361) ATAVSFFVASIFLKSAKNN--EEDITKATEKMQQLKGKKS...(365) AAAVSFIVSAVILKSSKAS--EEDLAAATEKMQSMKGKKS...(365) xxxxxxxxxxxxxx xTM6xxxxxxxx xxxxxxxxxxxxxxxxxxxxxxxxxxxx Kons (351) AAAVSFVVSAIILKTSKXXEDEDDIEAATRRMXXMKAXSK Abb. IV.3. Vergleich der EIICMtl AS-Sequenzen verschiedener Stämme Dargestellt sind die abgeleiteten EIIC-Aminosäuresequenzen der mtlA-Gene folgender Organismen: K12, Escherichia coli K12 (IICBA; Lee & Saier, 1983); OH157:H7, E. coli OH157:H7 (IICBA; Hayashi et al., 2001); CFT073, E. coli CFT073 (EIICBA; Welch et al., 2002); S.fle, Shigella flexneri (IICBA; Jin et al., 2002); S.ent, Salmonella enterica serovar typhimurium (EIICBA; McClelland et al., 2001); K.pne, Klebsiella pneumoniae (EIICBA; Otte, 2000); Y.pes, Yersinia pestis (EIICBA; Parkhill et al., 2001); V.cho, Vibrio cholerae (EIICBA; Heidelberg et al., 2000); P.mul, Pasteurella multocida (EIICBA; May et al., 2001); C.ace, Clostridium acetobutylicum (EIICB, EIIA; Behrens et al. 2001); S.mut, Streptococcus mutans (EIICB, EIIA; Honeyman & Curtiss III., 2000); L.lac., Lactococcus lactis (Mansour et al., 2001); S.car, Staphylococcus carnosus (IICB, IIA; Reiche et al., 1988; Fischer et al., 1989; Fischer & Hengstenberg, 1992); B.hal, Bacillus halodurans (IICB, IIA; Takami et al., 2000); B.ste, Bacillus stearothermophilus (IICB, IIA; Henstra et al., 1996); B.sub, Bacillus subtilis (IICBA; Akagawa et al., 1995, geändert nach Henstra et al., 1996). Die Konsensussequenz (Kons) wurde mit Hilfe des Programms „VectorNTI Suite“ ermittelt. Mit „X“ wurden Bereiche der Konsensussequenz markiert, in denen keine AS zugeordnet werden konnte. Gruppen ähnlicher AS wurden farblich hervorgehoben. Konservierte Aminosäuren sind gelb unterlegt. AS, die der Konsensussequenz entsprechen, sind in Blau, dazu konservative Austausche in Rot dargestellt. Als konservative Austausche wurden zugelassen: A¥G, D¥E, F¥Y, K¥R, N¥Q, S¥T, und Austausche innerhalb der Gruppe I¥L¥V. Bei der Gesamtanalyse wurde nur die E.coliSequenz von K12 berücksichtigt, um die Aussage nicht durch drei fast identische E.coliSequenzen zu beeinträchtigen. Ein weiterer Punkt des Modells besagt, dass nur phosphoryliertes EIIMtl eine erleichterte Diffusion mit hoher Rate katalysiert. Dies bedeutet, dass der Grad der Phosphorylierung der IIBAMtl-Domänen die Aktivität der Translokator-Domäne EIICMtl beeinflusst, indem wahrscheinlich der Translokationsvorgang durch eine drastische Senkung der Aktivierungsenergie erreicht wird. Das entspricht der Tatsache, dass ein WT-EIIC nicht dazu in der Lage ist, D-Mtl ausreichend für ein Wachstum zu transportieren. Dagegen sind in den entkoppelten Mutanten weder Phosphorylierung IV. Diskussion 117 noch das Vorhandensein der EIIAB-Domänen für einen ausreichenden Transport notwendig. Der Grund hierfür könnte eine der Phosphorylierung analoge Senkung der Aktivierungsenergie durch die AS-Austausche sein. Es ist schon länger bekannt, dass das niedrigaffine C5-Isomer D-Arabinitol (KMapp ≥ 500µM) mit einer hohen Rate unphosphoryliert sowohl durch das WT- als auch das entkoppelte EIICMtl transportiert wird, unabhängig von dem Phosphorylierungszustand des EIICMtl und dem Vorhandensein der EIIBAMtl-Domänen (Klawitter, 1992; Scholle, 1993; Otte 2000). Dies ist vergleichbar mit anderen nicht-PTS-Substraten, welche niedrigaffine StereoIsomere natürlicher PTS-Substrate sind und unphosphoryliert durch EII-Domänen transportiert werden. Beispiele hierfür ist der Transport von D-Galaktose und Trehalose durch EIIMan in S. enterica (Postma, 1976; Postma et al., 1986), der Transport von DGalaktose, D-Arabinitol, Ribitol und D-Fruktose durch EIIGlc in E. coli (Kornberg & Riordan, 1976; Kornberg et al., 2000;; Zeppenfeld et al., 2000) und der Transport von D-Arabinitol und Xylitol durch EIIGat (Lengeler, pers. Mitteilung). Da auch entkoppelnde Mutationen oft die Affinität des Transporters zu seinen Hauptsubstraten verringern (Ruijter et al., 1992; Otte, 2000; diese Arbeit), zeigt sich ein großer Einfluss der Stärke der Substratbindung auf die Kopplung von Transport und Phosphorylierung (Lengeler et al., 1994). Man geht davon aus, dass im WT eine hohe Phosphorylierungsaktivität dazu führt, dass freies intrazelluläres D-Mtl sofort wieder gebunden und phosphoryliert wird. Effektiv ist in der Zelle nur D-Mtl-Phosphat vorhanden (Otte, 2000). Wie kann es aber zum Transport von freiem D-Mtl in die Zelle kommen? Für ein EII-Dimer wird das Vorhandensein zweier hochaffiner, zur periplasmatischen Seite orientierten Bindestellen postuliert (Pas et al., 1988; Briggs et al., 1992; Lolkema et al., 1992). Durch die Bindung eines Substrates an diese hochaffine Bindestelle könnte eine Konformationsänderung induziert werden, die die Bindestelle in einen geschlossenen Zustand überführt. Danach würde die Umwandlung in eine, dem Zytoplasma zugewandte, niedrigaffine Bindestelle erfolgen. Bindestudien mit Vesikeln (Lolkema et al., 1992) und Tryptophan Phosphoreszenz-Studien (Broos et al., 2000) lieferten Hinweise auf solche substratbindungs- und phosphorylierungsabhängige Strukturveränderungen im EIIMtl. Nach dem Modell von Lolkema moduliert der Phosphorylierungszustand der zytoplasmatischen EIIB-Domäne die Aktivität der Translokationsdomäne EIIC (Lolkema 118 IV. Diskussion et al., 1991b). In vitro Untersuchungen hatten gezeigt, dass sich die Transportrate in Anwesenheit von Phospho-IIB drastisch erhöht (Elferink, et al., 1990; Lolkema et al., 1991a). Demnach bewirke die Phosphorylierung von IIB eine Verringerung der Aktivierungsenergie für den eigentlichen Translokationsschritt, also die Umlagerung der geschlossenen in die niedrigaffine Bindestelle. Die Phosphorylierung von IIB würde damit zu einer signifikanten Steigerung der Isomerisierungsrate der Bindestelle und somit zum Anstieg der Transportraten führen. Nach diesem Modell wäre entkoppelter Transport in einem nicht phosphorylierten oder isolierten EIIC nicht denkbar. Nach dem Modell von Lengeler (Lengeler et al. 1994) gibt es im EIIMtl eine Bindestelle mit drei Zuständen (hochaffin / geschlossen / niedrigaffin) zwischen denen umgeschaltet werden kann. Das Substrat wird außen mit hoher Affinität gebunden und nach einer durch die Substratbindung induzierten Konformationsänderung, die zu einer Umlagerung der Bindestellen in einen „geschlossenen“ Zustand führt, erfolgt in einem zweiten Schritt die Phosphorylierung des gebundenen Substrates. Die Affinität der Bindestelle für das phosphorylierte Substrat ist so gering, dass es nun freigesetzt werden kann. Somit würde durch die Phosphorylierung nicht die Mobilisierung der Bindestelle, sondern das Ablösen des hochaffinen Substrates von der Bindestelle beschleunigt. Die Ergebnisse aus in vitro Untersuchungen in Vesikeln weisen hingegen darauf hin, dass freies und phosphoryliertes D-Mannitol mit gleicher Wahrscheinlichkeit von der Bindestelle freigesetzt wird (Lolkema et al., 1991b). Die Durchlässigkeit von Vesikeln für D-Mtl stellt jedoch ein Problem bei der Auswertung der Ergebnisse dar. In vivo durchgeführte DC- und Wachstumsuntersuchungen haben gezeigt, dass nach dem Transport von D-Mtl über EIIMtl das intrazelluläre Verhältnis von Substrat : SubstratPhosphat ganz auf der Seite des phosphorylierten Substrates liegt (Otte, 2000; diese Arbeit). Für eine im Transport entkoppelte Mutante wäre in dem Lengeler-Modell denkbar, dass die Affinität der Bindestelle durch den AS-Austausch grundsätzlich herabgesetzt und freies D-Mtl in die Zelle abgegeben wird. Unterschiedliche Klassen von Mutationen könnten durch zwei Faktoren entstehen: 1. Die Stärke bzw. Schwäche der Bindestellen-Affinität und 2. Die Stärke der internen Phosphorylierung des freien D-Mtl. Anhand der in dieser Arbeit ermittelten Generationszeiten (Tab III.1.23.) wird deutlich, dass das Wachstum von Zellen auf DMtl mit EIIMtlE218A und DalD verbessert wird, was wiederum auf intrazelluläres freies DMtl hinweist. Das Transporttests mit den E218A-Derivatern nicht reproduzierbar waren, IV. Diskussion 119 ist ein Hinweis darauf, dass der Austausch E218A im vollständigen EIIMtl den WTPhänotyp nicht wiederherstellt. Dennoch war eine hohe Phosphorylierungsrate messbar. Bei diesem Protein ist die Entkopplung durch ein Herabsetzen der Bindestellen-Affinität entstanden, wobei die Phosphorylierungsaktivität nicht eingeschränkt war. Ähnlich verhält es sich bei MtlA*E218V, welches Zellen mit DalD fast eine WT-Generationszeit auf D-Mtl vermittelt, aber nur noch 10% der Phosphorylierungsaktivität. Hier scheinen beide Faktoren für eine Entkopplung vorzuliegen. In der Mutante H256P scheint die Affinität der Bindestelle nicht wie bei E218A oder E218V herabgesetzt zu sein. Das Wachstum von Zellen mit DalD ist im Vergleich mit beiden Mutanten schwächer und eine Phosphorylierungsaktivität ist kaum mehr vorhanden. Dies weist auf eine stärkere Entkopplung auf der Ebene der internen Phosphorylierung des freien D-Mtl hin. In der Arbeit von Otte (2000) wurden entkoppelte Transporter aus K.vpneumoniae und E. coli, sowie plasmidkodierte entkoppelte EIIGlc-Mutanten, die sich bezüglich ihrer KM-Werte und der Phosphorylierungsaktivitäten unterscheiden (Ruijter et al., 1992; Weng et al., 1992; Saraceni-Richards & Jacobson, 1997; Kessels, 1999), beschrieben. EIIMtlE218AK.pn., EIIMtlH195N, EIIMtlE257D, EIIGlcV206A und EIIGlcR203S zeigen eine hohe Phosphorylierungsaktivität, der KM-Wert ist nicht deutlich erhöht und in Anwesenheit von EIIBC~P findet kein entkoppelter Transport statt. EIIMtlH256YKK.pn., Phosphorylierungsaktivität, EIIMtlH195A einen und erhöhten EIIGlcI296N KM-Wert und haben gekoppelten noch sowie entkoppelten Transport. Die in dieser Arbeit beschriebenen Mutationen EIIMtlE218AE.coli und EIIMtlE218VE.coli zeigen entsprechende Werte und scheinen ähnliche Auswirkungen auf den gekoppelten Transport zu haben. Die Mutationen EIIMtlH256PxK.pn., EIIMtlE257A, EIIMtlE257Q und EIIGlcK257N vermitteln wie die Mutation EIIMtlH256PxE.coli drastisch verringerte bzw. nicht vorhandene Phosphorylierungsaktivitäten und erhöhte KM-Werte. Auch bei der Selektion von Mutanten mit entkoppeltem EIIGlc aus Salmonella typhimurium (Postma, 1981; Ruijter et al., 1991) sowie E. coli (Ruijter, 1992) wurde in Pts--Stämmen einige gefunden, die sich durch einen erhöhten KM-Wert auszeichneten. Die Isolierung weiterer Mutanten und die Untersuchung der Phänotypen mit den Haupt- und Nebensubstraten sind für eine weiterführende Aufklärung des gekoppelten 120 IV. Diskussion und entkoppelten Transports notwendig. Dabei müssen weiterhin die einzelnen Komponenten aus Transport, Bindung, KM-Wert und Phosphorylierungsaktivität in Zusammenhang gebracht werden. Die dafür benötigten Methoden sind in dieser Arbeit beschrieben worden und sollten durch die von Otte (2000) durchgeführten dünnschichtchromatographischen Versuche erweitert werden. Damit ist eine weitere Methode gegeben, relativ einfach die Verhältnisse zwischen phosphoryliertem und unphosphoryliertem Substrat darzustellen. Auch die Klassifizierung der PTSs neben den ionenabhängigen Sym- und Antiportern sowie den ATP-abhängigen ABC-Transportern ist zu überdenken. Unter Berücksichtigung der beschriebenen Untersuchungen werden im Transportvorgang immer mehr Ähnlichkeiten mit den sehr substratspezifischen, erleichterte Diffusion vermittelnden Uniportern und sogar den ABC-Transportern deutlich (Lengeler et al., 1994). Bei den ABC-Transportern wird die freie Energie des ATPs dazu verwendet, die Energiebarriere des an die hochaffine Bindestelle gebundenen Substrats zu verringern und unter Bildung von ADP und Phosphat das Substrat in der Zelle anzuhäufen (Davidson, 2002). Die Energiebarriere eines im EII gebundenen Substrats muss ebenfalls durch die hohe freie Energie eines His~P oder Cys~P verringert werden, wobei hier die Phosphorylgruppe direkt auf das Substrat übertragen wird. Dies ermöglicht eine bessere Energieausbeute und eine nahezu irreversible Anhäufung von Substrat~P. Die Ergebnisse dieser Arbeit bestätigen bei der Suche nach Mutanten die Fokussierung auf konservierte Bereiche im EIIMtl, insbesondere in der im Vergleich zum gesamten EIIC (26%x=xkonserviert, 39%x=xkonserviert +xkonservativexAustausche) stark konservierten Schleife 5 (44%x=xkonserviert, 63%x=xkonserviert +xkonservative Austausche; Abb. IV.3. und IV.5.). Bisher wurden alle entkoppelnden Mutationen im EIIMtl und EIIGlc (Buhr et al., 1992; Ruijter et al., 1992) in dieser Schleife und dem GIHEbzw. GITE-Motiv gefunden. Fluoreszenz-Studien zeigten zudem in zwei Bereichen Reaktionen auf die Bindung von Mannitol. Dazu gehörten das Trp30 (Dijkstraa et al., 1996) und die Aminosäuren im Bereich der Position 196 in der Schleife 5 (Boer et al., 1996; Broos et al., 1999). Zudem führten zahlreiche ortgerichtete Mutagenesen auf der Suche nach transportrelevanten AS ausschließlich zu dem AS-Bereich, welcher der Schleife 5 und deren direkte Umgebung im EIIMtl-Modell entspricht (Buhr et al., 1992; Weng et al., 1992; Saraceni-Richards & Jacobson, 1997; Lanz & Erni, 1998). IV. Diskussion 121 IV.2. Untersuchung der Sekundärstruktur des EIICMtl Für ein umfassendes Verständnis des Translokationsmechanismus im EIIMtl ist ein genaues Wissen um die Sekundär- bzw. Tertiärstruktur unumgänglich. Aufgrund zahlreicher Untersuchungen geht man heute von einer dimeren aktiven Konformation des Proteins aus (Übersicht bei Jacobson, 1992, Boer et al., 1994; Boer et al., 1996; Koning et al., 1999; Heuberger et al., 2002), die durch nicht-kovalente, hydrophobe Wechselwirkung der C-Domänen vermittelt wird (Lolkema et al., 1993). Ein weiterer Einfluss der AS378-465 auf die Dimerisierung ist aber nicht auszuschließen (Briggs et al., 1992). Die Sekundärstruktur entwickelte sich aus den Erkenntnissen der Arbeiten von Lee & Saier (1983), Sugiyama et al. (1991), Dijkstra et al. (1996) und Lengeler (Lengeler et al., 1994; Lengeler & Jahreis, 1996) zu einem Modell aus mindestens sechs transmembranen Helices, drei kurzen periplasmatischen und zwei großen zytoplasmatischen Schleifen sowie einem zytoplasmatischen N-Terminus. Für das EIIGlc wurde mittels PhoA-Fusionsstudien ein Modell mit acht transmembranen Helices ermittelt (Buhr & Erni, 1993), obwohl die Hydrophobizitätsmuster von EIIGlc und EIIMtl sehr ähnlich sind. Die Strukturprojektion eines 2D-EIIMtl-Kristalls mit einer Auflösung von 5 Å gibt Hinweise auf das EIIMtl-Modell mit sechs transmembranen Helices und schließt dabei die Assoziation einer transmembranen Helix mit extramembranen Strukturen nicht aus (Koning et al., 1999). Bei der Betrachtung des Sekundärstrukturmodells von EIICMtl unter Berücksichtigung der Untersuchungen entkoppelter Transporter ist dies auch zu fordern. Wichtige an der Kopplung beteiligte Aminosäuren (H195, E218, H256) liegen in der zytoplasmatischen Schleife 5, während das Substrat ursprünglich im Periplasma zu finden ist. Eine Assoziation der Schleife 5 mit transmembranen Elementen, wie es im Modell von Lengeler (Lengeler 1990; Lengeler et al. 1994) vorgeschlagen wird, ist daher durchaus denkbar. Unter Berücksichtigung dessen, wurde in dieser Arbeit eine strukturelle Untersuchung mit dem Schwerpunkt auf der Schleife 5 durchgeführt. IV.2.1. Mögliche Strukturen des EIICMtl Mit der Methode der gezielten Biotinmaleimid-Markierung von Cysteinen in dem Protein wurde die Lokalisation der Aminosäuren S3, C110, S158, S199, S212, S242, S299 und C384 in EIICMtl untersucht (siehe III.2.4. und Abb. III.2.4.). Als positive Innenkontrolle wurde anfänglich das Cys384 gewählt, das als essentielle Aminosäure 122 IV. Diskussion bei der Phosphatgruppenübertragung in der EIIBMtl-Domäne fungiert (Pas & Robillard, 1988a, 1988b). Aus den Kontrollen geht hervor, dass Cys384 allgemein für Stilben und Biotinmaleimid zugänglich ist. Unter den gewählten Bedingungen erwies sich Biotinmaleimid jedoch in allen anderen Versuchen als membranimpermeabel und die Kontrollmarkierungen mit einer cysteinfreien Mutante lassen unspezifische Bindungen ausschließen. Dabei sollte in der Diskussion die Sonderstellung des Cys384 im EIIMtl berücksichtigt werden. Man könnte zum einen eine schwächere Bindung für Sulfhydrylreagenzien erwarten, da bei einem Wachstum in einem Medium ohne D-Mtl das EIIMtl an der Phosphorylierungsstelle Cys384 größtenteils phosphoryliert ist. Abb. IV.4. Bindung eines Phosphats bzw. Maleimids an ein Cystein A) Bindung einer Phosphorylgruppe (grün) an die Sulfhydrylgruppe des Cysteins (blau). B) Bildung des Thioethers durch Bindung des Maleimids (rot) an die Sulfhydrylgruppe des Cysteins (blau). Eine Blockierung der Bindestelle für Maleimid wäre denkbar (Abb. IV.4.), zeigte sich jedoch nicht bei den Kontrollen auf allgemeine Zugänglichkeit. Zum anderen sollte das Cys384 in der Nähe des D-Mtl-Transports durch die Membran sein, da es das Phosphat auf das Substrat überträgt. Dort wären Membranstrukturen denkbar, die es Biotinmaleimid, nicht aber Stilben ermöglichen, Cys384 zu erreichen, zumal die Membranpermeabilität bzw. -impermeabilität von Biotinmaleimid zeitabhängig ist (Loo und Clarke, 1994). Ein nahe oder in der Membran liegendes Cys384 könnte noch von Biotinmaleimid erreicht werden, während zytoplasmatische Cysteine innerhalb der Inkubationszeit unmarkiert bleiben. In jedem Fall ist es als sicher anzusehen, dass Cys384 vom Zytoplasma erreichbar sein muss. Für eine „positive Innenkontrolle“ wurde zudem der Austausch S3C gewählt, der nach dem derzeitigen Sekundärstrukturmodell im Zytoplasma liegen soll (Abb. IV.5.). IV. Diskussion 123 Abb.IV.5. Ergebnisse aus Sequenz- und Strukturuntersuchungen im Sekundärstrukturmodell des IICMtl-Transporters aus E. coli (nach Sugiyama et al., 1991 und Lengeler et al., 1994; verändert) Das Modell entspricht der Abb.I.2. erweitert durch den Sequenzvergleich aus Abb. IV.3. Die Aminosäuren sind wie folgt markiert: In dem Sequenzvergleich konservierte AS haben einen schwarzen Kasten, AS mit lediglich konservativen Austauschen einen schwarzen Kreis, AS mit Konsensussequenz einen leeren Kreis und AS ohne Konsensus keine Markierung. AS deren Position im Sekundärstrukturmodell in dieser Arbeit oder durch aktuelle Struktur-Untersuchungen bestätigt worden sind, sind in Grün dargestellt. Abweichende Positionen sind in Rot, unbestimmte Positionen in Blau dargestellt. Die Messungen der PhoA-Aktivitäten einzelner AS (Sugiyama et al., 1991) sind in den Farben Gelb (<20 Einheiten pro OD Einheit (= EpOD)), Orange (20-75 EpOD) und Braun (>75 EpOD) angegeben. Blaue Kreise zeigen Lücken bzw. Insertionen in den verglichenen AS-Sequenzen an. Leere Kreise stellen AS-Sequenzen unbestimmter Länge dar. Weitere Erläuterungen sind im Text zu finden. 124 IV. Diskussion Für die AS-Austausche S110C und S299C konnte keine allgemeine Zugänglichkeit für Sulfhydrylreagenzien festgestellt werden. Im Falle des S110C sind unzugängliche Strukturen denkbar, die durch eine Anlagerung der EIIBMtl-Domäne an die Schleife 3 entstehen. So konnte gezeigt werden, dass zwischen zwei EIIMtl-Domänen Disulfidbrücken durch Cys124-Cys124, Cys384-Cys384, Cys124-Cys384 sowie innerhalb einer Domäne durch Cys124-Cys384 gebildet werden können (van Montfort et al., 2001). Dadurch ergibt sich ein geringer Abstand der zwei Cys124 und Cys384 in der B/C-Domäne und der Dimer-Verbindung, der mit maximal 5 Å vorgeschlagen wird. Die Vielzahl der möglichen Disulfidkombinationen spiegelt hier die dynamischen Prozesse der einzelnen Domänen in der Substrattranslokation wieder. Die EIIB-Domäne muss dafür mit dem Cys384 in die Nähe der EIIA-Domäne gelangen, um die Phosphorylgruppe vom His554 zu übernehmen und dann zur Phosphorylierung des Substrats mit der EIIC-Domäne in Wechselwirkung treten. Des Weiteren kann die Phosphatgruppe im Dimer zwischen den jeweiligen Monomeren von der EIIA- zur EIIBDomäne bzw. der EIIB-Domäne zum Substrat übertragen werden (van Weghel et al., 1991a; Weng et al., 1992; Boer et al., 1996; Saraceni-Richards & Jacobson, 1997). Dadurch wird deutlich, dass das aktive Zentrum der EIIB-Domäne unterschiedliche Konformationen durchlaufen muss, die verschiedene Disulfidbrücken ermöglichen. Der Grund für die Unzugänglichkeit des S299C-Austausches könnte eine Lokalisation in der Membran sein. In diesem Bereich der Schleife 6 gibt es zwar drei phoA-Fusionen, die für die Lage von I300, G310 und A311 im Periplasma sprechen (Sugiyama et al., 1991). Hohe phoA-Aktivität kann aber auch erwartet werden, wenn die drei AS in die Helix 6 gelegt werden würden. Für eine genaue Einordnung fehlen in diesem Bereich weitere phoA-Fusionen. Grundsätzlich haben Deletionsfusionen den Nachteil, dass das Protein nicht aktiv ist und carboxyterminale, für eine genaue Insertion wichtige AS-Sequenzen deletiert sein können (Calamia & Manoil, 1990 und 1992). Die Markierungs-Ergebnisse für die Austausche S158C, S199C und S212C weisen deutlich auf periplasmatische Aminosäuren hin. Im Fall des Ser158 stützt dieses Ergebnis das aktuelle Sekundärstruktur-Modell und die Messungen aus phoA-Fusionen in diesem Bereich (Sugiyama et al., 1991), die Ser158 in die periplasmatische Schleife 4 legen. Widersprüchlich sind dagegen die Ergebnisse für S199C und S212C. Beide Aminosäuren (Ser199 und Ser212) liegen in dem EIIMtl-Modell in der zytoplasmatischen Schleife 5 und die Werte zahlreicher phoA-Fusionen bestätigen dies (Sugiyama et al., IV. Diskussion 125 1991). Es ist jedoch schon im ersten Teil der Arbeit auf die Besonderheit der Schleife 5 hingewiesen worden, die sich aus der relativ hohen Konservierung und den vielen Mutationen, welche den Transport beeinflussen, ergibt. Dynamische Prozesse, die keine Insertion von Teilen der Schleife 5 in die Membran erfordern, und Strukturen wie der von Lengeler beschriebene hydrophile Kern vom EIICMtl (Lengeler 1990; Lengeler et al. 1994) könnten unter Umständen mit Deletionsfusionen nicht nachgewiesen werden. Dies wäre der Fall, wenn die Schleife 5 für eine Faltung in die Membran die Stabilisierung von weiteren, in dem jeweiligen Protein aber deletierten Strukturen benötigte. Die nach den Markierungsversuchen periplasmatische Anordnung des Ser199 und Ser212 ist ein starker Hinweis auf eine Faltung von Teilen der Schleife 5 in die Membran, da die AS S242 nach Ergebnissen dieser Arbeit wiederum zytoplasmatisch lokalisiert ist. Das bisherige Sekundärstrukturmodell stützt sich lediglich auf die phoA-Fusionen (Sugiyama et al., 1991) und mathematische und empirische Methoden zur Vorhersage von Membranstrukturen in Proteinen (Kyte & Doolittle, 1982; Engelman et al., 1986; von Heijne & Gavel, 1988; von Heijne, 1992; Lengeler et al., 1994). Weitere gezielte Untersuchungen zur Lokalisation einzelner Aminosäuren im Protein gibt es nur wenige. Als gesichert ist die Bestimmung des Ser124 in unmittelbarer Nähe zum Cys384 anzusehen (van Montfort et al., 2000). Ergebnisse aus Tryptophan- Phosphoreszensstudien geben starke Hinweise auf das Vorkommen der AS Trp30 und Trp42 in einer Membran-Helix, was der Helix 1 im Modell entsprechen sollte (Broos et al., 2000). Die Studie gibt zudem noch gute Hinweise darauf, dass Trp109 und Trp117 im Zytoplasma liegen, so wie es im Modell vorgesehen ist. Auf Grund der Ähnlichkeiten im Hydropathie-Muster und der Domänenstruktur, wurden für das EIIMtl und EIIGlc ähnliche Topologien vermutet. Für EIIGlc wird allerdings in einer Studie mit phoA- und lacZ-Deletionsfusionen ein Modell mit acht transmembranen Helices vorgeschlagen (Buhr & Erni, 1993). Vergleicht man die Anordnung der einzelnen Helices zwischen EIIMtl und EIIGlc (Abb. IV.6.), so sieht man annähernde Übereinstimmungen bei den Schleifen Mtl1-Glc1, Mtl2-Glc2, Mtl3-Glc5, Mtl4-Glc6 und Mtl5-Glc8. Abweichungen gibt es durch die Schleifen Glc3, Glc4, Glc7 und Mtl6. Glc3 und Glc4 stellen zwei Schleifen dar, die im Sekundärstrukturmodell von EIIMtl nicht berücksichtigt werden. 126 IV. Diskussion 1 70 EIIGlcxxxxxxxxxxxxxxxxIIIIIIIIIIIIIIIIIIIIIII xxxxxxxGlc1xxxxxxxxxx AAAAAAAAAAAAAAAAAAAAAAAAAAA xxxxGlc2x TMHMMxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxAxxxxxxxxxxxxxxxxxxxxxxxxxxxxBx K12 (1) ----------MSSDIKIKVQSFGRFLSNMVMPNIGAFIAWGIITALFIPTGWLPNETLAK-LVGPMITYL TMHMMxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxAxxxxxxxxxxxxxxxxxxxxxxxxxxxBx EIIMtlxxxxxxxxxxxxxxxxIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII xxxxxxxxxMtl1xxxxxxx AAAAAAAAAAA xxxxMtl2x xxxxxxxxxxxxxxxxxxxxxxxS3x xxxx xxxxxxxxxxxxxxxxxxW30xxxxxxxxxW42 71 140 EIIGlcxxxxxx xGlc2xxxxx II xxxxxxxGlc3xxxxxxxx AAAAAAAAAAAAAAAAAAAAAAAAAAA xxxxxxxxxGlc4xxxxxxx IIII TMHMMxxxxxxxBxxxxxxxxxxxxxxxxxxxxxxCxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxDxxxxxxxxxxxxx K12 (60) LPLLIGYTGGKLVGGERGGVVGAITTMGVIVGA------------DMPMFLGSMIAGPLGGWCIKHFDRW TMHMMxxxxxxxBxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxC/Dxxxxxxxxxxxxxxxxxxxxxxxxx EIIMtlxxxxxx xMtl2xxxxx IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII W109/C110 W117 141 210 EIIGlcxxxxxxIIIIIIIIIIIIII xxxxxxxxxGlc5xxxxxxxxx AAAAAAAAAAAAAAAAAAA xxxxxxxGlc6xxxxxxxx IIIIIIIIIIIIIII TMHMMxxxxxxxxxxxxxxxxxxxxxxxxxxxxExxxxxxxxxxxxxxxxxxxxxxxxxxxxxFxxxxxxxxxxxxxxxxx K12 (118) VDGKIKSGFEMLVNNFSAGIIGMILAILAFLGIGPIVEALSKMLAAGVNFMVVHDMLPLASIFVEPAKIL TMHMMxxxxxxxxxxxxxxxxxxxxxxxxxxxxExxxxxxxxxxxxxxxxxxxxxxxxxxxxxxFxxxxxxxxxxxxxxxx EIIMtlxxxxxxIIIIIIIIIIIIIIIIIIIIIIIIIIIII xxxxxxxxMtl3xxxxxxxx AAAAAAAAAAAAAAAAAA xxxxxxxxMtl4xxxxxxx IIIII xxxxxxxxxxxxxxxxxS124 S158 211 280 EIIGlc xxxxxIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII xxxxxxxGlc7xxxxxx AA TMHMMxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxGxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxHxx K12 (188) FLNNAINHGIFSPLGIQQSHELGKSIFFLIEANPGPGMGVLLAYMFFGRGSAKQSAGGAAIIHFLGGIHE TMHMMxxxxxxxxxxxxxxxxxxxxxxxxxxxxxixxxxxxxxxxxGxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxHx EIIMtlxxxxxxIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII S199 S212 S242 281 350 EIIGlcxxxxxxAAAAAAA xxxxxxxxGlc8xxxxxxxxx IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII TMHMMxxxxxxxHxxxxxxxxxxxxxxxxxxxIxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxJxxxxxxxxx K12 (258) IYFPYVLMNPRLILAVILGGMTGVFTLTILGGGLVSPASPGSILAVLAMTPK------GAYFANIAGVCA TMHMMxxxxxxxHxxxxxxxxxxxxxxxxxxxxxIxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxJx EIIMtlxxxxxxIIIIIIIIIIIIIIIIIIIIIIII xxxxxxxxMtl5xxxxxxxx AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA xxxMtl6x S158 351 390... EIIGlcxxxxxxIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII TMHMMxxxxxxxJxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx K12 (322) AMAVSFVVSAILLKTSKVKE-EDDIEAATRRMQDMKAESK...(360) TMHMMxxxxxxxJxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxx EIIMtlxxxxxx xMtl6xxxxxxx IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII Abb. IV.6. Vergleich der postulierten transmembranen Helices von EIICMtl und EIICGlc Die Abbildung zeigt die relative Lage postulierter Helices aus EIICGlc (Buhr & Erni, 1993) in der AS-Sequenz von EIICMtl (K12; Lee & Saier, 1983). Die Helices aus EIICGlc sind mit einem weißen Kasten versehen (Glc1 - Glc8), die Helices aus EIICMtl mit einem grauen Kasten (Mtl1 - Mtl6). AS-Bereiche im Zytoplasma sind mit grünen Is, die im Periplasma mit roten As gekennzeichnet. Die dargestellte AS-Sequenz ist aus der Abb.IV.3. mit den Markierungen übernommen worden. Transmembrane Bereiche nach der Vorhersage des Programms TMHMM (Sonnhammer et al., 1998; Abb.V.7.) sind mit blauen (EIIMtl) bzw. grünen (EIIGlc) Balken und alphabetisch markiert. Relevante AS sind hervorgehoben und nummeriert. Der Abbildung liegt ein Sequenzvergleich zwischen EIICMtl und EIICGlc zu Grunde (weiteres siehe Text). IV. Diskussion 127 Die Tryptophan-Phosphoreszensstudien an W109 und W117 (Broos et al., 2000), sowie die Cystein-Kreuzverbindungsstudien (van Montfort et al., 2000) schränken das Vorkommen der Schleife Glc4 in EIICMtl stark ein. Damit entfällt aus topologiesymmetrischen Gründen auch eine mögliche Schleife 3, zumal S158 in dieser Arbeit im Periplasma lokalisiert wurde. Die Abweichungen der Schleifen Glc7 und Mtl6 erscheinen danach als eine Verschiebung der zwei aufeinander folgenden Helices in den jeweiligen Proteinen. Neben den experimentellen Möglichkeiten, die Topologie eines transmembranen Proteins aufzuklären, gibt es noch zahlreiche Computerprogramme, die entsprechende Vorhersagen berechnen. Allen Programmen ist gemein, dass die grundsätzlichen Regeln der Ladungsverteilung und des bevorzugten Vorkommens bestimmter Aminosäuren berücksichtigt wird (Kyte & Doolittle, 1982; Engelman et al., 1986; von Heijne & Gavel, 1988; von Heijne, 1992). Das Programm TMHMM („transmembrane hidden Markov model“) von Sonnhammer et al. (1998) hat in einem Vergleich mehrerer Vorhersageprogramme am besten abgeschnitten (Möller et al., 2001). Das Prinzip von TMHMM liegt in der dynamischen Berücksichtigung biologischer Besonderheiten bei der Entstehung transmembraner Strukturen. So können z.B. Bereiche geringer Hydrophobizität dennoch als transmembrane Helix beurteilt werden, wenn umliegende topogene Strukturen dies unterstützen. Solche Helices kommen oft vor, wenn transmembrane Helices untereinander hydrophile Wechselwirkungen ausüben. Dennoch ist bei der Arbeit mit Vorhersage-Programmen immer zu berücksichtigen, dass mögliche Strukturen nur vorgeschlagen werden und die angegebenen transmembranen Bereiche variabel anzusehen sind. So wird eine Vorhersage als korrekt beurteilt, wenn der angegebene Bereich für eine Helix mit dem der realen Helix überlappt. Daher können die transmembranen Bereiche in variablen Größen angegeben werden. Die Ergebnisse von TMHMM (Abb. IV.6. und IV.7.) bestätigen das Sekundärstrukturmodell von EIIGlc in den acht postulierten transmembranen Helices, insgesamt werden zehn mögliche Bereiche für Helices vorhergesagt. Diese zwei zusätzlichen Helices würden in der Schleife 5 und hinter der Schleife 8 (Glc8) am Ort der Schleife 6 (Mtl6) von EIICMtl liegen. Auch die Vorhersage für EIIMtl bestätigt das entsprechende Sekundärstrukturmodell, obwohl die Helix 4 (Mtl4; hellblauer Balken in Abb. IV.6.) nur bei der Berechnung für die Konsensussequenz von EIICMtl erkannt wird (Für die Berechnung wurde die Konsensussequenz ohne Insertionen verwendet). 128 IV. Diskussion Abb. IV.7. TMHMM-Vorhersage der Topologien von EIICMtl und EIICGlc Die Abbildung zeigt die graphische Darstellung der Wahrscheinlichkeitsberechnungen des Programms TMHMM (Sonnhammer et al., 1998; Abb.V.7.) für EIICGlc und EIICMtl. Den Berechnungen für transmembrane, zytoplasmatische (innen) und periplasmatische (außen) Bereiche liegen die Konsensussequenzen für EIICMtl (Abb. IV.3.), EIICBMtl (Lee & Saier, 1983) und EIICBGlc (Erni & Zanolari, 1986) zu Grunde. Über den jeweiligen transmembranen Bereichen sind die ersten und letzten AS angegeben. Die zugehörigen Helices der aktuellen Topologiemodelle (Sugiyama et al., 1991; Buhr & Erni, 1993) sind mit römischen Ziffern gekennzeichnet. Einander im Sequenzvergleich entsprechende transmembrane Bereiche (Abb. IV.6.) sind alphabetisch gekennzeichnet (Näheres siehe Text). IV. Diskussion 129 Die Ergebnisse für EIIMtl enthalten ebenfalls mehr Helices als das Modell beinhaltet. Eine davon liegt in der Schleife 3 und zwei in der Schleife 5 des Sekundärstrukturmodells von EIICMtl. Im Vergleich zwischen EIICGlc und EIICMtl fällt auf, dass bei der Vorhersage, beiden Modellen entsprechend, mehr transmembrane Bereiche für EIIGlc vorgeschlagen werden. Vieles weist darauf hin, dass EIIGlc und EIIMtl Transporter gleichen Ursprungs sind, deren Funktion und Hydropathie-Muster ähnlicher sind als die Topologie. Der Bereich für eine mögliche Helix in der Schleife 3 von EIIMtl liegt dabei in einem Bereich, der im Sequenzvergleich eine Deletion bzw. Insertion aufweist. Das kann auf eine Deletion im Bereich zweier transmembraner Helices in der Schleife 3 von EIIMtl hinweisen. Mögliche transmembrane Bereiche in Schleife 5 von EIICMtl weisen auf eine Membranassoziation der Schleife 5 hin. Sowohl in EIICMtl als auch in EIICGlc gibt es auf die Schleife 5 folgend drei Bereiche für transmembrane Helices in denen jeweils zwei Helices aus den Modellen eingeordnet werden. Die Ergebnisse aus den zugehörigen phoA- bzw. phoA- und lacZ-Untersuchungen sprechen für die Zuordnung wie sie in den Modellen erfolgt. Es sind allerdings bei dem Modell für EIIMtl auf Grund weniger Daten in diesem Bereich Verschiebungen der transmembranen Bereiche möglich. Anhand der in dieser Arbeit durchgeführten und vorgestellten Untersuchungen sind verschiede Strukturen in EIICMtl denkbar (Abb. IV.8.). Dabei wurden folgende Punkte gesondert berücksichtigt. Die Maleimid-Markierungen des Ser3 bestätigt den zytoplasmatischen Aminoterminus von EIIMtl. Die Helix 1 wird durch Tryptophan Phosphoreszensstudien (Broos et al., 2000) gestützt. Die AS W109 und W117 in der Schleife 3 werden durch dieselben Phosphoreszensstudien zytoplasmatisch lokalisiert. Kreuzverbindungsstudien am S124 weisen auf eine unmittelbare Nähe zum C384 hin (van Montfort et al., 2000). Die Maleimid-Markierungen des C110 zeigten eine Unzugänglichkeit der Aminosäure. Ein Vergleich der postulierten Helices in EIIMtl und EIIGlc sowie die phoA-Untersuchungen an den AS G91, A92 und D93 geben Hinweise auf deletierte Membran-Helices. Die Helices 2 und 3 resultieren aus den vorangegangenen Ergebnissen sowie der periplasmatische Zuordnung des Ser158 in der Schleife 4. Die Helix 4 fällt in der TMHMM-Vorhersage schwach aus und die Bestimmung von Ser199 und Ser212 im Periplasma spricht für eine Helix nach den AS 199 und 212. Allerdings sind die Werte der phoA-Studien in diesem Bereich sehr gut, womit die Helix 4 beibehalten wird. Die Daten aus den Mutantenselektionen und die Bestimmung von Ser199 und Ser212 130 IV. Diskussion sprechen für eine Faltung der Schleife 5 „in die Membran“, wie sie von Lengeler (Lengeler 1990; Lengeler et al. 1994) vorgeschlagen wurde. Dabei sind die dargestellten hellblauen Strukturen nicht zwingend als Helix anzusehen. Die in dieser Arbeit nachgewiesene Relevanz von Glu218 für die Kopplung bestärkt die Verlegung der AS in die Membran. Gleiches gilt für die AS His195 und Umgebung. Dabei wurde die von TMHMM in der Schleife 5 vorgeschlagene Helix (AS212-234) eingebaut. Das Ser242 liegt danach den Markierungen entsprechend im Zytoplasma. Das GIHE-Motiv wird „nahe“ der Membran angeordnet, da seine zentrale Bedeutung in der Kopplung von Transport und Phosphorylierung gezeigt werden konnte. Die Helices 5 und 6 werden dem ursprünglichen Modell entsprechend beibehalten. Abb.IV.8. Mögliche 2D-Strukturen des IICMtl-Transporters aus E. coli Die Darstellung entspricht der Abb. IV.5. Abweichend sind AS, die relevant am Transportprozess beteiligt sind rot markiert. Mögliche Membran-Strukturen sind entsprechend der Abb. IV.7. alphabetisch gekennzeichnet. Der hellblaue Bereich in der Membran stellt eine hydrophile Struktur dar, in der nicht zwingend Helices liegen. IV. Diskussion 131 Auf Grund der neuen Struktur, die eine Zurückfaltung der Schleife 5 nur im kompletten Protein ermöglicht, lassen sich auch die PhoA-Aktivitäten der Deletionsfusion L209 erklären, da in diesen Konstrukten die Faltung in die Membran nicht erfolgt. Letztlich wurde in der Abbildung die nachgewiesene Nachbarschaft der AS Ser124 aus der Schleife 3 und dem Cys384 aus EIIBMtl dargestellt. Das abgebildete Modell (Abb. IV.8.) stellt nur mögliche Strukturen im EIICMtl dar. Außerdem ist zu berücksichtigen, dass es sich dabei um eine von vielen möglichen Konformationen handeln kann. Es sei aber darauf hingewiesen, dass das Modell alle bisherigen Erkenntnisse über das EIIMtl vereinigt. Eine mögliche dreidimensionale Anordnung der einzelnen Elemente zeigt die Abb.xIV.9. Dabei soll die Formung eines hydrophilen Kerns wie in Abb.xI.3. dargestellt werden. Abb.IV.9. Mögliche 3D-Strukturen des IICMtl-Transporters aus E. coli Die Nummerierung der begrenzenden AS einer Struktur entspricht der Abb. IV.8. In rot sind wichtige am Transportprozess beteiligte AS sowie das dem C384 benachbarte S124 hervorgehoben. Das C384 wurde in der phosphorylierten Form dargestellt (C384~P). 132 IV. Diskussion Ein Dimer aus zwei solchen Anordnungen würde einen Ring aus stabilisierenden transmembranen Helices formen, deren hydrophiler Kern das reaktive Zentrum bilden würde. Dieses reaktive Zentrum entspricht dabei dem hochkonservierten Bereich zwischen den AS L174 und P267 und würde nicht in der Membran, sondern in einem Ring von transmembranen Helices liegen. In diesem Bereich befinden sich alle Aminosäuren, die anhand der Mutantenselektion einen Einfluss auf den gekoppelten Transport haben. Weiterführende Untersuchungen sind erforderlich, um die vorgeschlagenen Strukturen in dem Modell genauer bestimmen zu können. In dieser Arbeit konnte die Methode der Biotinmaleimid Markierung in geeigneter Weise etabliert werden. Bei folgenden Untersuchungen ist zu berücksichtigen, dass in dem aktiven Zentrum dynamische Strukturen zu erwarten sind. Markierungen an einem phosphoryliertem EIIMtl könnten diese aufzeigen. V. Zusammenfassung 133 V. Zusammenfassung Die Konstruktion des Plasmids F’lac::dalD ermöglichte die Entwicklung stabiler Systeme zur Selektion entkoppelter EIIMtl-Mutanten. Semisynthetische Stoffwechselwege für den D-Mtl-Abbau wurden durch die Stämme LGS322-1/pGJ9∆137 und LTK31-2/pGJ9 etabliert und erfolgreich zur Selektion von Mutanten eingesetzt. Von 30 untersuchten entkoppelnden Mutationen zeigten 15 den Austausch E218A, eine den Austausch E218V und 14 den Austausch H256P. Die Phänotypen der Mutanten wurden nach ortsspezifischer Mutagenese bestätigt und in unterschiedlichen Stammhintergründen untersucht. Alle Mutationen ermöglichten im EIICMtl nur noch entkoppelten Transport. Im vollständigen MtlA (EIICBAMtl) wurde bei dem Austausch E218A gekoppelter und entkoppelter Transport gemessen. Mit DalD lag die Generationszeit bei ~118min, ohne DalD bei ~190min (WT-Gt = ~85min mit und ohne DalD). Kinetische Transportmessungen waren bei der Mutation E218A nicht reproduzierbar und zeigten eine starke Abhängigkeit vom Plasmid- und Stammhintergrund. Die Phosphorylierungsaktivität lag mit 307 [nM]1 im Bereich des WT (278 [nM]1). Die Mutation E218V zeigte stärkere Auswirkungen auf den Phänotyp. Im EIICBAMtl wurde auch gekoppelter und entkoppelter Transport festgestellt. Mit DalD lag die Generationszeit bei ~110min, ohne bei ~305min. Transportmessungen waren nicht möglich, die Phosphorylierungsaktivität lag bei 32 [nM]1. Mit der Mutation H256P war im EIICBAMtl nur noch entkoppelter Transport möglich (Gt mit DalD = 283min / ohne DalD = k.W.). Transport war nicht mehr messbar, die Phosphorylierungsaktivität lag bei 2 [nM]1. Die Ergebnisse geben Hinweise darauf, dass die Kopplung von Transport und Phosphorylierung durch die Affinität des EIICMtl zum Substrat beeinflusst wird. Durch ortsspezifische Mutagenese konstruierte Doppelmutationen E218A/H256P und E218V/H256P konnten D-Mtl in keinem Stammhintergrund verstoffwechseln. Die Proteine mit den Doppelmutationen wurden in der Membran nachgewiesen, nicht aber deren korrekte Faltung. 1 [nM]= [nmol∗min-1∗mg Protein-1] 134 V. Zusammenfassung Die Selektion von Suppressormutanten ergab lediglich Aminosäureaustausche in der AS256. Die Mutationen H256A, P256Q und P256S zeigten in Generationszeit, Transport- und Phosphorylierungsmessungen annähernd WT-Werte. Besonderheiten bei den Messungen von P256S lassen jedoch eine Beeinflussung der Dimerisierung durch Mutationen in H256 vermuten. Aufgrund des ausschließlichen Auftretens von entkoppelten Mutanten in E218 und H256 und des starken Selektionsdrucks, der keine Suppressormutationen in anderen AS ermöglichte, ergibt sich eine essentielle Funktion dieser AS in der Kopplung von Transport und Phosphorylierung. Zur Analyse der Sekundärstruktur des EIIMtl mittels „Cystein-Scanning“ wurde ein MtlA(His)6 in einem Überexpressionsvektor konstruiert. Nach ortsspezifischer Mutagenese konnte die Aktivität des EIIMtl mit den Austauschen S3C, S110C, S158C, S199C, S212C, S242C oder S299C gezeigt werden. Aus den BiotinmaleimidMarkierungen ergab sich die Lokalisation von C3 und C242 im Zytoplasma sowie von C158, C199 und C212 im Periplasma. Keine oder unbestimmbare Zugänglichkeiten für Sulfhydrylreagenzien zeigten die AS C110, C299 und C384. Aus diesen Ergebnissen ergibt sich eine Anordnung von Teilen der ursprünglichen Schleife 5 in der Membran. Diese Schleife 5 bildet das aktive Zentrum, in der sich alle AS befinden, deren direkte Beteiligung an Transport und Phosphorylierung nachgewiesen wurden. Es ist nahe liegend, dass solche Strukturen in der Nähe der Substratbindung und des Transports liegen sollten. Ein Modell, das diesen Anforderungen und den bisherigen Ergebnissen entspricht, wurde vorgeschlagen. Es stellt aber lediglich eine Anregung für weitere Untersuchungen dar. VI. Literaturverzeichnis 135 VI. Literaturverzeichnis Akagawa, E., Kurita, K., Sugawara, T., Nakamura, K., Kasahara, Y., Ogasawara, N., and Yamane, K. 1995. Detremination of a 17,484 bp nucleotid sequence around the 39 degrees region of the Bacillus subtilis chromosom and similarity analysis of the products of putative ORF’s. Microbiology 141: 3241-3245. Altschul, S.F., Madden, T.L., Schaffer, A.A., Zahng, J., Zahng, Z., Miller, W., and Lipman, D.J. 1997. Grapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25: 3389-3402. Ames, G.F.L. 1986. Bacterial periplasmic transport systems: Structure, mechanism, and evolution. Ann. Rev. Biochem. 55: 397-425. Arber, W. 1960. Transduction of chromosomal genes and episomes in E.coli. Virologie 11: 273-288. Aulkemeyer, P., Ebner, R., Heilenmann, G., Jahreis, K., Schmid, K., Wrieden, S., and Lengeler, J.W. 1991. Molecular analysis of two fructokinases involved in sucrose metabolism of enteric bacteria. Molecular Microbiol. 5 (12): 2913-2922. Ausubel, F.A., Brent, R., Kingston, R.E., Moore, D.D., Seidmann, J.G., Smith, J.A., and Struhl, K. (eds.) 1990. Current protocols in molecular biology. Greene Publishing and Wiley-Interscience, New York. Bachmann, B.J. 1990. Linkage map of Escherichia coli K12. Edition 8. Microbiol. Rev. 54: 130-197. Behrens, S., Mitchell, W., and Bahl, H. 2001. Molecular analysis of the mannitol operon of Clostridium acetobutylicum encoding a phosphotransferase system and a putative PTS-modulated regulator. Microbiology 147(1): 75-86. Berlyn, M.K., Brooks Low, K., Rudd, K., and Singer, M. 1996. Linkage map of Escherichia coli K-12. In Frederick C. Neidhardt (editor in chief): Escherichia coli and Salmonella: Cellular and molecular biology - 2nd ed.. ASM Press, Washington D.C.: 1715-1902. Birnboim, H.C. and Doly, J. 1979. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucl. Acids. Res. 7: 1513. Boer, H., ten Hoeve-Duurkens, R.H., Schuurman-Wolters, G.K., Dijkstra, A., and Robillard, G.T. 1994. Expression, purification, and kinetic characterization of the mannitol transport domain of the phosphoenolpyruvate-dependent mannitol phosphotransferase system of Escherichia coli. Kinetic evidence that the E. coli mannitol transport protein is a functional dimer. J. Biol. Chem. 269 (27): 17863-17871. 136 VI. Literaturverzeichnis Boer, H., ten Hoeve-Duurkens, R.H., Lolkema, J.S., and Robillard, G.T. 1995. Phosphorylation site mutants of the mannitol transport protein enzyme IImtl of Escherichia coli: studies on the interaction between the mannitol translocating Cdomain and the phosphorylation site on the energy-coupling B-domain. Biochemistry 34 (10): 3239-3247. Boer, H., ten Hoeve-Duurkens, R.H., and Robillard, G.T. 1996. Relation between the oligomerization state and the transport and phosphorylation function of the Escherichia coli mannitol transport protein: interaction between mannitol-specific enzyme II monomers studied by complementation of inactive site-directed mutants. Biochemistry 35: 12901-12908. Boyer, H.W., and Roulland-Dussoix, D. 1969. A complementation analysis of the restriction and modification of DANN in Escherichia coli. J. Mol. Biol. 41: 459-472. Bradford, MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72: 248-54. Briggs, C.E., Khanderkar, S.S., and Jacobson, G.R. 1992. Structure/function relationships in the Escherichia coli mannitol permease: identification of regions important for membrane insertion, substrate binding oligomerization. Res. Microbiol. 143: 139-149. Broos, J., ter Veld, F., Robillard, G.T. 1999. Membrane protein-ligand interactions in Escherichia coli vesicles and living cells monitored via a biosynthetically incorporated tryptophan analogue. Biochemistry 38(31): 9798-803. Broos, J., Strambini, G.B., Gonnelli, M., Vos, E.P., Koolhof, M., and Robillard, G.T. 2000. Sensitive monitoring of the dynamics of a membrane-bound transport protein by tryptophan phosphorescence spectroscopy. Biochemistry 39(35): 10877-83. Budde, A. 1991. Genetische und biochemische Analyse der Pentitol- und Hexitoltransportsysteme und -stoffwechselwege in Stämmen von Escherichia coli. Diplomarbeit, Universität Osabrück. Buhr, A., Daniels, G.A., Erni, B. 1992. The glucose transporter of Escherichia coli. Mutants with impaired translocation activity that retain phosphorylation activity. J. Biol. Chem. 267(6): 3847-51. Buhr, A., and Erni, B. 1993. Membrane topology of the glucose transporter of Escherichia coli. J. Biol. Chem. 268: 11599-11603. Bullock, W.O., Fernandez, J.M., and Short, J.M. 1987. Xl1-blue: A High Efficiency Plasmid Transforming Reca Es. Biotechniques 5: 376-379. Calamia, J., and Manoil, C. 1990. lac permease of Escherichia coli: topology and sequence elements promoting membrane insertion. Proc. Natl. Acad. Sci. USA 87(13): 4937-41. VI. Literaturverzeichnis 137 Calamia, J., and Manoil, C. 1992. Membrane protein spanning segments as export signals. J Mol Biol 224(3): 539-43. Charnetzky, W.T., and Mortlock, R.P. 1974a. D-arabitol catabolic pathway in Klebsiella aerogenes. J. Bacteriol. 119: 170-175. Charnetzky, W.T., and Mortlock, R.P. 1974b. Close genetic linkage of the determinants of the ribitol and D-arabinitol catabolic pathways in Klebsiella aerogenes. J. Bacteriol. 119: 176-182. Davidson, A.L. 2002. Mechanism of coupling of transport to hydrolysis in bacterial ATPbinding cassette transporters. J. Bacteriol. 184(5): 1225-33. Davis, T., Yamada, M., Elgort, M., and Saier Jr., M.H. 1988. Nucleotide sequence of the mannitol (mtl) operon in Escherichia coli. Molecular Microbiol. 2: 405-412. Davison, J., Heusterpreute, M., Chevalier, N., and Brunel, F. 1987. A ‘phase-shift’ fusion system for the regulation of foreign gene expression by lambda repressor in Gram-negative bacteria. Gene 60: 227-235. Decad, G.M., and Nikaido, H. 1976. Outer membrane of gram-negative bacteria. XII. Molecular-sieving function of cell wall. J. Bacteriol. 128: 325-336. De Reuse, H., and Danchin, A. 1988. The ptsH, ptsI and crr genes of the Escherichia coli phosphoenolpyruvate-dependent phosphotransferase system: a complex operon with several modes of transcription. J. Bacteriol. 170: 3827-3837. Dijkstra, D.S., Broos, J., Lolkema, J.S., Enequist, H., Minke, W., and Robillard, G.T. 1996. A fluorescence study of a singel tryptophan-containing mutans of enzyme IImtl of the Escherichia coli phosphoenolpyruvate-dependent mannitol transport system. Biochem. 35: 6628-6634. Dotto, G.P. Enea, V., and Zinder, N.D. (1981). The Morphogenetic Signal of Bacteriophage f1. Virology 130: 252-256. Egan, J.B., and Morse, M.L. 1966. Carbohydrate transport in Staphylococcus aureus. III. Studies of the transport process. Biochim. Biophys. Acta 112: 63-73. Engelman, D.M., Steitz, T.A., Goldman, A. 1986. Identifying nonpolar transbilayer helices in amino acid sequences of membrane proteins. Annu. Rev. Biophys. Biophys. Chem. 15: 321-53. Erni, B., Zanolari, B. 1986. Glucose-permease of the bacterial phosphotransferase system. Gene cloning, overproduction, and amino acid sequence of enzyme IIGlc. J. Biol. Chem. 261(35): 16398-403. Ferenci, T., and Kornberg, H.L. 1973. The utilization of fructose by Escherichia coli. Properties of a mutant defective in fructose 1-phosphate kinase activity. Biochem J. 132(2):341-7. 138 VI. Literaturverzeichnis Fischer, R., Eisermann, R., Reiche, B., and Hengstenberg, W. 1989. Cloning, sequencing and overexpression of the mannitol-specific enzyme-III-encoding gene of Staphylococcus carnosus. Gene 82:249-257. Fischer, R., and Hengstenberg, W. 1992. Mannitol-specific enzyme II of the phosphoenolpyruvate-dependent phosphotransferase system of Staphylococcus carnosus. Sequence and expression in Escherichia coli and structural comparison with the enzyme II mannitol of Escherichia coli. Eur. J. Biochem. 204: 963-969. Frerman, F.E., and Bennet, W. 1973. Studies on the uptake of fatty acids by Escherichia coli. Arch. Biochem. Biophys. 159: 434-443. Furlong, C.E. 1987. Osmotic-shock sensitive transport systems. In Neidhardt, F.C., et al. (eds.): Escherichia coli and Salmonella typhimurium. ASM Press, Washington D.C.: 768-796. Giesen, U., Kleider, W., Berding, C., Geiger, A., Ørum, H., and Nielsen , P.E. 1998. A formula for thermal stability (Tm) prediction of PNA/DNA duplexes. Nucleic Acids Res. 26 (21): 5004-5006. Griffith, J.K., Cuellar, D.H., Fordyce, C.A., Hutchings, K.G., and Mondragon, A.A. 1994. Structure and function of the class C tetracycline/H+ antiporter: Three independent groups of phenotypes are conferred by TetA(C) in Escherichia coli. Mol. Membr. Biol. 11: 271-277. Grisafi, P.L., Scholle, A., Sugiyama, J., Briggs, C., Jacobson, G.R., and Lengeler, J.W. 1989. Deletion Mutants of the Escherichia coli K-12 Mannitol Permease: Dissection of Transport-Phosphorylation, Phospho-Exchange, and Mannitol-Binding Activities. J. Bacteriol. 171: 2719-2727. Grübl, G. 1989. PTS-Chemotaxis von Escherichia coli K-12: Untersuchungen zum Mechanismus der Signaltransduktion. Diplomarbeit, Universität Osnabrück. Hayashi, T., Makino, K., Ohnishi, M., Kurokawa, K., Ishii, K.,Yokoyama, K., Han, C.G., Ohtsubo, E., Nakayama, K., Murata, T.,Tanaka, M., Tobe, T., Iida, T., Takami, H., Honda, T., Sasakawa, C.,Ogasawara, N., Yasunaga, T., Kuhara, S., Shiba, T., Hattori, M. and Shinagawa, H. 2001. Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 8(1): 11-22. Heidelberg, J.F., Eisen, J.A., Nelson, W.C., Clayton, R.A., Gwinn, M.L., Dodson, R.J., Haft, D.H., Hickey, E.K., Peterson, J.D., Umayam, L.A., Gill, S.R., Nelson, K.E., Read, T.D., Tettelin, H., Richardson, D., Ermolaeva, M.D., Vamathevan, J., Bass, S., Qin, H., Dragoi, I., Sellers, P., McDonald, L., Utterback, T., Fleishmann, R.D., Nierman, W.C., White, O., Salzberg, S.L., Smith, H.O., Colwell, R.R., Mekalanos, J.J., Venter, J.C. and Fraser, C.M. 2000. DNA Sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406: 477-483. VI. Literaturverzeichnis 139 Heller, K.B., Lin, E. C.C., and Wilson, T.H. 1980. Substrate specificity and transport properties of the glycerol facilitator of Escherichia coli. J. Bacteriol. 144: 274-278. Henstra, S.A., Tolner, B., ten Hoeve-Duurkens, R.H., Konings, W.N., and Robillard, G.T. 1996. Cloning, expression and isolation of the mannitol transport protein from the thermophilic bacterium Bacillus stearothermophilus. J. Bacteriol. 178: 5586-5591. Heuberger, E.H., Veenhoff, L.M., Duurkens, R.H., Friesen, R.H., Poolman, B. 2002. Oligomeric state of membrane transport proteins analyzed with blue native electrophoresis and analytical ultracentrifugation. J Mol Biol 317(4): 591-600. Heuel, H.J. 1997. Struktur und Funktion bakterieller Polyalkohol-Transportsysteme: Molekularbiologische und genetische Untersuchungen zum Transport und Stoffwechsel von D-Arabinitol, Ribitol und D-Mannitol in Enterobakterien. Dissertation, Universität Osnabrück. Heuel, H., Shakeri-Garakani, A., Turgut, Ş., and Lengeler, J.W. 1998. Genes for Darabinitol and ribitol catabolism from Klebsiella pneumoniae. Microbiology 144: 16311639. Honeyman, A.L., and Curtiss III, R. 2000. The mannitol-specific enzyme II (mtlA) gene and the mtlR gene of the PTS of Streptococcus mutans. Microbiology 146( 7):15651572. Jiang, W., Wu, L.-F., Tomich, J., Saier Jr., M.H., and Niehaus, W.G. 1990. Corrected sequence of the mannitol (mtl) operon in Escherichia coli. Molecular Microbiol. 4: 2003-2006. Jin, Q., Yuan, Z., Xu, J., Wang, Y., Shen, Y., Lu, W., Wang, J., Liu, H., Yang, J., Yang, F., Zhang, X., Zhang, J., Yang, G., Wu, H., Qu, D., Dong, J., Sun, L., Xue, Y., Zhao, A., Gao, Y., Zhu, J., Kan, B., Ding, K., Chen, S., Cheng, H., Yao, Z., He, B., Chen, R., Ma, D., Qiang, B., Wen, Y., Hou, Y., Yu, J. 2002. Genome sequence of Shigella flexneri 2a: insights into pathogenicity through comparison with genomes of Escherichia coli K12 and O157. Nucleic Acids Res. 30(20):4432-41. Jacobson, G.R. 1992. Interrelationships between protein phosphorylation and oligomerization in transport and chemotaxis via the Escherichia coli mannitol phosphotransferase system. Res. Microbiol. 143: 113-116. Jacobson, G.R., Tanney, L.E., Kelly, D.M., Palman, K.B., and Corn, S.B. 1983. Substrate and phospholipid specifity of the purifield mannitol permease of Escherichia coli. J. Cell. Biochem. 23: 231-240. Jacobson, G.R., Lee, C.A. and Saier, M.H., Jr. 1979. Purification of the mannitolspecific enzyme II of the Escherichia coli phosphoenolpyruvate:sugar phosphotransferase system. J. Biol. Chem. 254: 249-252. Kaback, H.R. 1990. Lac permease of Escherichia coli: on the path of the proton. Phil. Trans. R. Soc. Lond. B. 326: 425-436. 140 VI. Literaturverzeichnis Kessels, K. 1999. Molekulare Analyse und Selektion von Mutanten des Enzyms IIMtl in Escherichia coli K-12. Diplomarbeit, Universität Osnabrück. King, S.C., and Wilson, T.H. 1990. Identification of valine 177 as a mutation altering specifity for transport of sugars by Escherichia coli lactose carrier. J. Biol. Chem. 17: 9638-9644. Klawitter, S. 1992. Molekulare Charakterisierung und Sequenzierung von mtlAMutanten des Stammes E. coli K-12. Diplomarbeit, Universität Osnabrück. Klungsöyr, L. 1966. Purification of the mannitol-1-phosphate dehydrogenase of Escherichia coli. Biochim. Biophys. Acta. 128: 55-62. Koning, R.I., Keegstra, W., Oostergetel, G.T., Schuurman-Wolters, G., Robillard, G.T., Brisson, A. 1999. The 5 Å projection structure of the transmembrane domain of the mannitol transporter enzyme II. Mol. Biol. 287(5): 845-51. Kornberg, H.L., Lambourne, T.M., and Sproul, A.A. 2000. Facilitated diffusion of fructose via the phosphoenolpyruvate/glucose phosphotransferase system of Escherichia coli. Proc. Natl. Acad. Sci. USA 97: 1808-1812. Kornberg, H.L. and Riordan, C. 1976. Uptake of galactose into Escherichia coli by facilitated diffusion. J. Gen. Microbiol. 94: 75-89. Krämer, R., 1994. Functional principles of solute transport systems: concepts and perspectives. Biochim. Biophys. Acta. 1185: 1-34. Kramer, B., Kramer, W., and Fritz, H.J. 1984. Different base/base mismatches are corrected with different efficiencies by the methyl-directed DANN mismatch-repair system of E. coli. Cell 38: 879. Kundig, W., Gosh, S., and Roseman, S. 1964. Phosphate bound to histidine in a protein as an intermediate in a novel phosphotransferase system. Proc. Natl. Acad. Sci. USA 52: 1067-1074. Kundig , W., Kundig, F.D., Anderson, B., and Roseman, S. 1966. Restoration of active transport of glycosides in Escherichia coli by a component of a phosphotransferase system. J. Biol. Chem. 241 (13): 3243-3246. Kyte, J., Doolittle, R.F. 1982. A simple method for displaying the hydropathic character of a protein. J Mol Biol 157(1): 105-32. Laemmli, U.K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680-685. Lanz, R., Erni, B. 1998. The glucose transporter of the Escherichia coli phosphotransferase system. Mutant analysis of the invariant arginines, histidines, and domain linker. J. Biol. Chem. 273(20): 12239-43. VI. Literaturverzeichnis 141 Lawlis, V.B., Dennis, M.S., Chen, E.Y., Smith, D.H., and Henner, D.J. 1984. Cloning and sequencing of the xylose isomerase and xylulose kinase genes of Escherichia coli. Appl. Environ. Microbiol. 47 (1): 15-21. Lee, C.A., Jacobson, G.R., and Saier, M.H. 1981. Plasmid-directed synthesis of enzymes required for D-mannitol transport and utilization in Escherichia coli. Proc. Natl. Acad. Sci. USA 78: 7336-7340. Lee, C.A., and Saier, M.H., Jr. 1983. Mannitol-specific enzyme II of the bacterial phosphotransferase system. III. The nucleotide sequence of the permease gene. J. Biol. Chem. 258: 10761-10767. Lengeler, J.W. 1966. Untersuchungen zum Glucoseeffekt bei der Synthese der Galaktose-Enzyme von Escherichia coli. Z. Vererbungsl. 98: 203-229. Lengeler, J.W. 1975a. Mutations affecting transport of the hexitols D-mannitol, Dglucitol and galactitol in Escherichia coli K-12: isolation and mapping. J. Bacteriol. 124: 26-38. Lengeler, J.W. 1975b. Nature and properties of hexitol transport systems in Escherichia coli. J. Bacteriol. 124: 39-47. Lengeler, J.W., and Steinberger, H. 1978. Analysis of the regulatory mechanisms controlling the synthesis of the hexitol transport systems in Escherichia coli K-12. Mol. Gen. Genet. 164: 163-169. Lengeler, J.W. 1980. Polyhydric alcohol transport by bacteria. Meth. in Enzym. 125: 473-485. Lengeler, J.W., Mayer, R.J., Schmid, K. 1982. Phosphoenolpyruvate-dependent phosphotransferase system enzyme III and plasmid-encoded sucrose transport in Escherichia coli K-12. J. Bacteriol. 151(1): 468-471. Lengeler, J.W., and Vogler, A.P. 1989. Molecular mechanisms of bacterial chemotaxis towards PTS-carbohydrates. FEMS Microbiology Reviews 63: 81-92. Lengeler, J.W. 1990. Molecular analysis of the Enzyme II-complexes of the bacterial phosphotransferase system (PTS) as carbohydrate transport systems. Biochim. Biophys. Acta 1018: 155-159. Lengeler, J.W., Titgemeyer, F., Vogler, A.P., and Wöhrl, B.M. 1990. Structure and homologies of carbohydrate:phosphotransferase system (PTS) proteins. Phil. Trans. R. Soc. Lond. ser. B 326: 489-504. Lengeler, J.W., Jahreis, K., and Wehmeier, U.F. 1994. Enzymes II of the phosphoenolpyruvate-dependent phosphotransferase systems: their structure and function in carbohydrate transport. Biochim. Biophys. Acta 1188: 1-28. 142 VI. Literaturverzeichnis Lengeler, J.W., and Jahreis, K. 1996. Phosphotransferase Systems or PTSs as Carbohydrate Transport and as Signal Transduction Systems. Handbook of Biol. Phys. 2: 573-598. Lennox, E.S. 1955. Transduction of linked genetic characters of the host by bacteriophage P1. Virology 1: 190-206. Lévy, S., Zeng, G.Q., and Danchin, A. 1990. Cyclic AMP synthesis in Escherichia coli strains bearing known deletions in the pts phosphotransferase operon. Gene 86: 2733. Lolkema, J.S., Dijkstra, D.S., ten Hoeve-Duurkens, R.H., and Robillard, G.T. 1990. The membran-bound domain of the phosphotransferase enzymeIIMtl of Escherichia coli constitutes a mannitol translocating unit. Biochemistry 29: 10659-10663. Lolkema, J.S., ten Hoeve-Duurkens, Ria H., Dijkstra, D.S., and Robillard, G.T. 1991a. Mechanistic Coupling of Transport and Phosphorylation Activity by Enzyme IImtl of the Escherichia coli Phosphoenolpyruvate-Dependent Phosphotransferase System. Biochem. 30: 6716-6721. Lolkema, J.S., Dijkstra, D.S., ten Hoeve-Duurkens, R.H., and Robillard, G.T. 1991b. Interaction between the cytoplasmic and membran-bound domain of enzymeIIMtl of the Escherichia coli phosphoenolpyruvat-dependet phosphotransferase system. Biochem. 30: 6721-6726. Lolkema, J.S., Dijkstra, D.S., and Robillard, G.T. 1992. Mechanics of Solute Translocation Catalyzed by Enzyme IImtl of the Phosphoenolpyruvate-Dependent Phosphotransferase System of Escherichia coli. Biochem. 31: 5514-5521. Loo, W., and Clarke, M., 1995. Membrane Topology of a Cysteine-less Mutant of Human P-glycoprotein. J. Biol. Chem. 270 (2): 843-848. Lux, R. 1995. PTS-vermittelte Chemotaxis: Molekulare Analyse der Signaltransduktion bei Escherichia coli K-12. Dissertation, Universität Osnabrück. MacConkey, A. 1905. Lactose-fermenting bacteria in faeces. J. Hyg. 8: 333-379. Maloy, S.R., Bohlander, M., and Nunn, W.D. 1980. Elevated levels of glyoxylate shunt enzymes in Escherichia coli strains constitutive for fatty acid degradation. J. Bacteriol. 143: 720-725. Manayan, R., Tenn, G., Yee, H.B., Desai, J.D., Yamada, M., and Saier, M.H., Jr. 1988. Genetic analyses of the mannitol permease of Escherichia coli: isolation and characterization of a transport-deficient mutant which retains phosphorylating activity. J. Bacteriol. 170: 1290-1296. Mansour, N.M., Shearman, C.A. and Gasson, M.J. 2001. The mtlA gene of Lactococcus lactis MG1363. Unpublished Direct Submission (13-JUL-2001): DBSOURCE AF399759. VI. Literaturverzeichnis 143 May, B.J., Zhang, Q., Li, L.L., Paustian, M.L., Whittam, T.S. and Kapur, V. 2001. Complete genomic sequence of Pasteurella multocida, Pm70. Proc. Natl. Acad. Sci. U.S.A. 98(6): 3460-3465. McClelland, M., Sanderson ,K.E., Spieth, J., Clifton, S.W., Latreille, P., Courtney, L., Porwollik, S., Ali, J., Dante, M., Du, F., Hou, S., Layman, D., Leonard, S., Nguyen, C., Scott, K., Holmes, A., Grewal, N., Mulvaney, E., Ryan, E., Sun, H., Florea, L., Miller, W., Stoneking, T., Nhan, M., Waterston, R. and Wilson, R.K 2001. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 413(6858): 852856. Miller, J.H. 1972. Experiments in Molecular Genetics. Cold Spring Harbour Laboratory, New York. Misset, O., Brouwer, M., and Robillard, G.T. 1980. Escherichia coli phosphoenolpyruvat-dependet phosphotransferase system. Evidence that the dimer is the active form of Enzyme II. Biochemistry 19: 883-890. Mitchell, P. 1963. Molecule, group and electron transfer through natural membranes. Biochem. Soc. Symp. 22: 142-169. Möller, S., Croning, M.D., Apweiler, R. 2001. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics (7): 646-53. Montfort, B.A., Schuurman-Wolters, G.K., Duurkens, R.H., Mensen, M., Poolman, B., and Robillard, G.T. 2001. Cysteine cross-linking defines part of the dimer and B/C domain interface of the Escherichia coli mannitol permease. J. Biol. Chem. 276(16): 12756-12763. Murphy, W.H. and Rosenblum, E.D. 1964 a. Mannitol catabolism by Staphylococcus aureus. Arch. Biochem. Biophys. 107: 292-297. Murphy, W.H. and Rosenblum, E.D. 1964 b. Genetic recombination by transduction between mannitol negativ mutans of Staphylococcus aureus. Proc. Soc. Exp. Biol. Med. 116: 544 -548. Neumann, B., Pospiech, A., and Schairer, H.U. 1992. Rapid isolation of genomic DNA from gram-negative bacteria. Trends in Genetics 8: 332-333. Nuoffer, C., Zanolari, B., and Erni, B. 1988. Glucose permease of Escherichia coli. The effect of cysteine to serine mutations on the function, stability, and regulation of transport and phosphorylation. J. Biol. Chem. 263:6647-6655. Otte, S. 2000. Untersuchungen zum molekularen Mechanismus der Substrattranslokation über das Enzym IIMtl und Analyse der mtl-Gene aus Klebsiella pneumoniae. Dissertation, Universität Osnabrück. 144 VI. Literaturverzeichnis Otte, S., Lengeler J.W. 2001. The mtl genes and the mannitol-1-phosphate dehydrogenase from Klebsiella pneumoniae KAY2026. FEMS Microbiol Lett 194 (2): 221-227. Otte, S., Scholle, A., Turgut, S., and Lengeler, J.W. 2003. Mutations Which Uncouple Transport and Phosphorylation in the D-Mannitol Phosphotransferase System of Escherichia coli K-12 and Klebsiella pneumoniae 1033-5P14. J. Bacteriol. 185(7): 2267-2276. Overath, P., Pauli, G., and Schairer, H.U. 1969. Fatty acid degradation in Escherichia coli: induction and localization. J. Bacteriol. 132: 532-540. Parkhill, J., Wren, B.W., Thomson, N.R., Titball, R.W., Holden, M.T.G., Prentice, M.B., Sebaihia, M., James, K.D., Churcher, C., Mungall, K.L., Baker, S., Basham, D., Bentley, S.D., Brooks, K., Cerdeno-Tarraga, A.M., Chillingworth, T., Cronin, A., Davies, R.M., Davis, P., Dougan, G., Feltwell, T., Hamlin, N., Holroyd, S., Jagels, K., Leather, S., Karlyshev, A.V., Moule, S., Oyston, P.C.F., Quail, M., Rutherford, K., Simmonds, M., Skelton, J., Stevens, K., Whitehead, S. and Barrell, B.G. 2001. Genome sequence of Yersinia pestis, the causative agent of plague. Nature 413(6855): 523-527. Pas, H.H., and Robillard, G.T. 1988 a. Enzyme IIMtl of the phosphoenolpyruvatdependet phosphotransferase system: identification of the activity-linked cysteine on the mannitol carrier. Biochemistry 27: 5515-5519. Pas, H.H., and Robillard, G.T. 1988 b. S-phosphocysteine and phosphohistidine are the intermediates in the phosphoenolpyruvate-dependent mannitol transport catalyzed by Escherichia coli EIImtl. Biochemistry 27: 5835-5839. Pas, H.H., Meyer, G.H., Kruizinga, W.H., Tamminga, K.S., van Weeghel, R.P., and Robillard, G.T. 1991. 31Phospho-NMR demonstration of phosphocysteine as a catalytic intermediate on the Escherichia coli phosphotransferase system EIIMtl. J. Biol. Chem. 266: 6690-6692. Payne, J.W., and Gilvarg, C. 1968. Size restriction on peptide utilization in Escherichia coli. J. Biol. Chem. 243: 6291-6299. Pederson, P.L., and Carafoli, E. 1987. Ion motive ATPases. I. Ubiquity, properties and significance to cell function. Trends Biochem. Sci. 12: 146-150. Postma, P.W. 1976. Involvement of the phosphotransferase system in galactose transport in Salmonella typhimurium. FEBS Lett. 61:49-53. Postma, P,W., and J.B., Stock 1980. Enzymes II of the phosphotransferase system do not catalyze sugar transport in the absence of phosphorylation. J. Bacteriol. 114: 476484. Postma, P.W. 1981. Devective enzyme II-BGlc of the phosphoenolpyruvat:sugar phosphotransferase system leading to uncoupling of transport and phosphorylation in Salmonella typhimurium. J. Bacteriol. 147: 382-389. VI. Literaturverzeichnis 145 Postma, P.W., Keizer, H.G., and Koolwijk, P. 1986. Transport of Trehalose in Salmonella typhimurium. J. Bacteriol. 168:1107-1111. Postma, P.W., and Lengeler, J.W. 1985. Phosphoenolpyruvat:carbohydrate phosphotransferase system of bacteria. Microbiol. Rev. 49 (3): 232-269. Postma, P.W., Lengeler, J.W., and Jacobson, G.R. 1993. Phosphoenolpyruvate: carbohydrate phosphotransferase systems of bacteria. Microbiol. Rev. 57: 543-594. Postma, P.W., Lengeler, J.W., and Jacobson, G.R. 1996. Phosphoenolpyruvate: carbohydrate phosphotransferase systems. In Frederick C. Neidhardt (editor in chief): Escherichia coli and Salmonella: Cellular and molecular biology - 2nd ed.. ASM Press, Washington D.C.: 1149-1174. Reiche, B., Frank, R., Deutscher, J., Meyer, N., and Hengstenberg, W. 1988. The staphylococcal phosphoenolpyruvate-dependent phosphotransferase system: mtl purification and characterization of the mannitol-specific enzyme II of Staphylococcus aureus and Staphylococcus carnosus and homology with the enzyme IImtl of Escherichia coli. Biochemistry 27 (17): 6512-6516. Reiner, A.M. 1977. Xylitol and D-Arabitol toxicities due to depressed fructose, galactitol, and sorbitol phosphotransferase of Escherichia coli. J. Bacteriol. 132: 166173. Richey, D.P., and Lin, E.C.C. 1972. Importance of facilitated diffusion for effective utilization of glycerol by Escherichia coli. J. Bacteriol. 112: 784-790. Robillard, G.T., and Blaauw, M. 1987. Enzyme II of the Escherichia coli phosphoenolpyruvate-dependent phosphotransferase system: protein-protein and protein-phospholipid interactions. Biochemistry 26 (18): 5796-5803. Robillard, G.T., Boer. H., van Weeghel, R.P., Wolters, G., and Dijkstra, A. 1993. Expression and characterization of a structural and functional domain of the mannitolspecific transport protein involved in the coupling of mannitol transport and phosphorylation in the phosphoenolpyruvate-dependent phosphotransferase system of Escherichia coli. Biochemistry 32: 9553-9562. Ruijter, G.J.G., Postma, P.W., and van Dam, K. 1991. Energetics of glucose uptake in Salmonella typhimurium mutants containing uncoupled enzyme IIGlc. Arch. Microbiol. 155: 234-237. Ruijter, G.J.G., van Meurs, G., Verwey, M.A., Postma, P.W., and van Dam, K. 1992. Analysis of Mutations That Uncouple Transport from Phosphorylation in Enzyme IIGlc of the Escherichia coli Phosphoenolpyruvate-Dependent Phosphotransferase System. J. Bacteriol. 174: 2843-2850. 146 VI. Literaturverzeichnis Ruijter, G.J.G., Verwey, M.A., Postma, P.W., and van Dam, K. 1992a. Characterization of uncoupled IIMan of the phosphotransferase system in Escherichia coli. Coupling of transport to phosphorylation and control of glucose metabolism. Academische proeschrift, Universiteit van Amsterdam: 73-79. Saier, M.H. Jr. 1993. Regulatory interactions involving the proteins of the phosphotransferase system in enteric bacteria. J. Cell. Biochem. 51: 62-68. Saiki, R., Scharf, S., Faloona, F., Mullis, K.B., Horn, G.T., Ehrlich, H.A., and Arnheim, M. 1985. Enzymatic amplification of β-globin genomic sequences and restriction sites of sickle cell anemia. Science 230: 1350-1354. Sambrook, J., Fritsch, E.F., and Maniatis, T. 1989. Molecular cloning. A laboratory manual. Cold Spring Harbor Laboratory. Sanger, F., Nicklen, S., and Coulson, A.R. 1977. DNA sequencing with chain terminating inhibitors. Proc. Natl. Acad. Sci. USA 74: 5463-5467. Sanger, F.S. 1981. Determination of nucleotid sequences in DNA. Science 214: 12051210. Saraceni-Richards, C.A., and Jacobson, G.R. 1997. A conserved Glutamate residue, Glu-257, is important for substrate binding and transport by the Escherichia coli mannitol permease. J. Bacteriol. 179: 1135-1142. Schnetz, K., Toloczyki, C., Rak, B. 1987. Beta-glucoside (bgl) operon of Escherichia coli K-12: nucleotide sequence, genetic organization, and possible evolutionary relationship to regulatory components of two Bacillus subtilis genes. J Bacteriol 169 (6): 25792590. Schmid, K., Schupfner, M., and Schmitt, R. 1982. Plasmid-mediated uptake and metabolism of sucrose by Escherichia coli K-12. J. Bacteriol. 151: 68-76. Schmid, K., Ebner, R., Altenbuchner, J., Schmitt, R., and Lengeler, J.W. 1988. Plasmidmediated sucrose metabolism in Escherichia coli K12: mapping of the scr genes of pUR400. Mol. Microbiol. 2: 1-8. Schmid, K., Ebner, R., Jahreis, K., Lengeler, J.W., and Titgemeyer, F. 1991. A sugarspecific porin, ScrY, is involved in sucrose uptake in enteric bacteria. Mol. Microbiol. 5 (4): 941-950. Schmitt, R. 1968. Analysis of melibiose mutants deficient in α-galactosidase and thiomethylgalaktosidase permease II in Escherichia coli K-12. J. Bacteriol. 69: 462471. Scholle, A. 1993. Molekulare Analyse der Transportschritte des Mannitol-TransportSystems von Escherichia coli K-12. Dissertation, Universität Osnabrück. Smith, M. 1985. In vitro mutagenesis. Ann. Rev. Genet. 19: 423-462. VI. Literaturverzeichnis 147 Solomon, E., and Lin, E.C.C. 1972. Mutations affecting the dissimilation of mannitol by Escherichia coli K-12. J. Bacteriol. 111: 566-574. Sonnhammer, E.L., von Heijne, G., Krogh, A. 1998. A hidden Markov model for predicting transmembrane helices in protein sequences. In J. Glasgow, T. Littlejohn, F. Major, R. Lathrop, D. Sankoff, and C. Sensen, (edts.), Proceedings of the Sixth International Conference on Intelligent Systems for Molecular Biology: 175-182. Menlo Park, CA, 1998. AAAI Press. Sprenger, G.A. 1993. Two open reading frames adjacent to the Escherichia coli K-12 Transketolase (tkt) gene show high similarity to the mannitol phosphotransferase system enzymes from Escherichia coli and various gram-positive Bacteria. Biochim. Biophys. Acta 1158: 103-106. Sprenger, G.A., and Lengeler, J.W. 1984. L-sorbose metabolism in Klebsiella pneumoniae and Sor+ derivatives of Escherichia coli K-12 and chemotaxis toward sorbose. J. Bacteriol. 157: 39-45. Stephan, M.M., and Jacobson, G.R. 1986. Subunit interaction of the Escherichia coli mannitol permease: correlation with enzymic activities. Biochemistry 25: 4046-4051. Stephan, M.M., and Jacobson, G.R. 1986. Membrane Disposition of the Escherichia coli Mannitol Permease: Identification of Membrane-Bound and Cytoplasmic Domains. Biochemistry 25: 8230-8234. Studier, F.W., and B.A. Moffat 1986. Use of the bacteriophage T7 RNA polymerase to selective high level expression of cloned genes. J. Mol. Biol. 189: 113. Sugiyama, J.E., Mahmoodian, S., and Jacobson, G.R. 1991. Membrane topology analysis of Escherichia coli mannitol permease by using a nested-deletion method to create mtlA-phoA fusions. Proc. Natl. Acad. Sci. USA 88: 9603-9607. Sutcliffe, J.G. 1978. Complete nucleotide sequence of the Escherichia coli plasmid pBR322. Cold Spring Harbor Symposium on Quantitative Biology. 43: 77-90. Tabor, S., and Richardson, C.C. 1985. A bacteriophage T7 RNA polymerase/ promoter system for controlled exclusive expression of specific genes. Proc. Natl. Acad. Sci. USA 82: 1074-1078. Takami, H., Nakasone, K., Takaki, Y., Maeno, G., Sasaki, R., Masui, N., Fuji, F., Hirama, C., Nakamura, Y., Ogasawara, N., Kuhara, S., Horikoshi, K. 2000. Complete genome sequence of the alkaliphilic bacterium Bacillus halodurans and genomic sequence comparison with Bacillus subtilis. Nucleic Acids Res 28(21):4317-31. Tanaka, S., and Lin, E.C.C. 1967. Two classes of pleiotropic mutants of Aerobacter aerogenes lacking components of a phosphoenolpyruvate-dependent phosphotransferase system. Proc. Natl. Acad. Sci. USA 57: 913-919. 148 VI. Literaturverzeichnis Tanaka, S., Lerner, S.A., and Lin, E.C.C. 1967. Replacement of a phosphoenolpyruvate-dependent phosphotransferase by a nicotinamide adenine dinucleotide-linked dehydrogenase for the utilization of mannitol. J. Bacteriol. 93: 642-648. Tchieu, J.H., Norris, V., Edwards, J.S., Saier, M.H. Jr. 2001. The complete phosphotranferase system in Escherichia coli. J. Mol. Microbiol. Biotechnol. 3 (3): 329346. van Montfort, B.A., Schuurman-Wolters, G.K., Duurkens, R.H., Mensen, R., Poolman, B., and Robillard, G.T. 2001. Cysteine Cross-linking Defines Part of the Dimer and B/C Domain Interface of the Escherichia coli Mannitol Permease. J. Biol. Chem. 276 (16): 12756-12763. van Weeghel, R.P., G. Meyer, W. Keck, and Robillard, G.T. 1991a. Phosphoenolpyruvate-dependent mannitol phosphotransferase system of Escherichia coli: overexpression, purification, and characterisation of the enzymatically active Cterminal domain of Enzyme IIMtl equivalent to Enzyme IIIMtl. Biochem. 30: 1768-1773. van Weeghel, R.P., Meyer, G., Pas, H.H., Keck, W., and Robillard, G.T. 1991b. Cytoplasmic phosphorylating domain of the mannitol-specific transport protein of the phosphoenolpyruvate-dependent phosphotransferase system in Escherichia coli: overexpression, purification, and functional complementation with the mannitol binding domain. Biochem. 30: 9478-9485. van Weeghel, R.P., van der Hoek, Y.Y., Pas, H.H., Elferink, M., Keck, W., and Robillard, G.T. 1991 c. Details of mannitol transport in Escherichia coli elucidated by site-specific mutagenesis and complementation of phosphorylation site mutants of the phosphoenolpyruvate-dependent mannitol-specific phosphotransferase system. Biochem. 30: 1768-1773. Vogler, A.P., and Lengeler, J.W. 1991. Comparison of the sequences of the nagE operons from Klebsiella pneumoniae and Escherichia coli K-12: enhanced variability of the enzyme IINag in regions connecting functional domains. Mol. Gen. Genet. 230: 270-276. Vogler, A.P., Broekhuizen, C.P., Schuitema, A., Lengeler, J.W., and Postma, P.W. 1988. Suppression of IIIGlc-defects by enzymes IINag and IIBgl of the PEP:carbohydrate phosphotransferase system. Mol. Microbiol. 2 (6): 719-726. Vogler, A. P., and Lengeler, J. W. 1988. Complementation of a truncated membranebound Enzyme IINag from Klebsiella pneumoniae with a soluble Enzyme III in Escherichia coli K12. Mol. Gen. Genet. 213: 175-178. von Heijne, G., Gavel, Y. 1988. Topogenic signals in integral membrane proteins. Eur. J. Biochem. 174(4): 671-8. von Heijne, G. 1992. Membrane protein structure prediction. Hydrophobicity analysis and the positive-inside rule. J.Mol.Biol. 225: 487-494. VI. Literaturverzeichnis 149 Vos, E., Broos, J., and Poolman, B. Unveröffentlicht - Referenz siehe Van Montfort et al. (2001). Wallace, R.B., Johnson M.J., Suggs, S.V., Miyoshi, K., Bhatt, R., and Itakura, K. 1981. A set of synthetic oligodeoxyribonucleotide primers for DNA sequencing in the plasmid vector pBR322. Gene 16(1-3): 21-26. Waters, S.H., Rogowsky, P., Grinstead, J., Altenbuchner, J., and Schmitt, R. 1983. The teracycline-resistance determinants of RP1 and Tn1721: nucleotide sequence analysis. Nucleic Acid Res. 11: 6089-6105. Weigel, N., Kukuruzinska, M.A., Nakazawa, T., Waygood, E.B., and S. Roseman 1982. Sugar transport by the bacterial phosphotransferase system. Phosphoryl transfer reactions catalyzed by Enzyme I of Salmonella typhimurium. J. Biol. Chem. 257: 14477-14491. Welch, R.A., Burland, V., Plunkett, G. III., Redford, P., Roesch, P., Rasko, D., Buckles, E.L., Liou, S.R., Boutin, A., Hackett, J., Stroud, D., Mayhew, G.F., Rose, D.J., Zhou, S., Schwartz, D.C., Perna, N.T., Mobley, H.L., Donnenberg, M.S., Blattner, F.R. 2002. Extensive mosaic structure revealed by the complete genome sequence of uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. U S A 99(26):17020-17024. Weng, Q.-P., Elder, J., and Jacobson, G.R. 1992. Site-specific Mutagenesis of Residues in the Escherichia coli Mannitol Permease That Have Been Suggested to Be Important for its Phosphorylation and Chemoreception Functions. J. Biol. Chem. 267 (27): 19529-19535. Weng, Q.-P., and Jacobson, G.R. 1993. Role of a Conserves Histidine Residue, His195, in the Activities of the Escherichia coli Mannitol Permease. Biochem. 32: 1121111216. West, I.C. 1970. Lactose transport coupled to proton movements in Escherichia coli. Biochem. Biophys. Res. Commun. 41: 655-661. West, I.C., and Mitchell, P. 1972. Proton-coupled ß-galactoside translocation in nonmetabolizing Escherichia coli. Bioenergetics 3: 445-462. White, D.W., and Jacobson, G.R. 1990. Molecular cloning of the C-terminal domain of Escherichia coli D-mannitol permease: expression, phosphorylation, and complementation with C-terminal permease deletion proteins. J. Bacteriol. 172: 15091515. Wolff, Br. J.B., and Kaplan, N.O. 1956. D-mannitol 1-phosphate dehydrogenase from Escherichia coli. J. Biol. Chem. 218: 849-869. Wood, W.A., McDohough, M.J., and Jacobs, L.B. 1961. Ribitol ans D-arabitol utilization by Aerobacter aerogenes. J. Biol. Chem. 236: 2190-2195. 150 VI. Literaturverzeichnis Yanisch-Perron, C., Vieira, J., and Messing, J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of M13mp 18 and pUC19 vectors. Gene 33: 103-119. Zeppenfeld, T., Larisch, C., Lengeler, J.W., and Jahreis, K. 2000. Glucose Transporter Mutants of Escherichia coli K-12 with Changes in Substrate Recognition of IICBGlc and Induction Behavior of the ptsG Gene. J. Bacteriol. 182 (16): 4443-4452. VII. Anhang 151 LEBENSLAUF UND AUSBILDUNGSGANG Persönliche Daten Şevket Turgut geb. 16.03.1968 in Zürich ledig seit 07.1999 deutsche Staatsangehörigkeit, davor türkische von - bis Schulbildung 1974 - 1978 Grundschule in Bad Pyrmont 1978 - 1988 Humboldt-Gymnasium in Bad Pyrmont Abschluss: allgemeine Hochschulreife am 27.05.1988 von - bis Hochschulstudien 10.1988 - 09.1989 Studium der Chemie an der Universität Paderborn, FB Chemie Studium der Biologie an der Universität Osnabrück, FB Biologie / Chemie Abschluss: Dipl. biol. Titel der Diplomarbeitarbeit in der AG Genetik bei Herrn Professor J.W. 10.1989 - 07.1995 Lengeler: „Molekulargenetische Untersuchungen des rbt-Operons für den Ribitol-Transport und -Stoffwechsel aus dem Bakterium Klebsiella pneumoniae KAY2026“ 10.1995 - 2003 Promotionsstudium in der AG Genetik an der Universität Osnabrück. Thema der Arbeit: „Molekulargenetische und biochemische Untersuchungen zur Funktion und Struktur des Enzym IIMtl aus Escherichia coli K-12“ Veröffentlichungen 1997 Heuel, H., Turgut, S., Schmid, K., and Lengeler, J.W. Substrate Recognition Domains As Revealed by Active Hybrids between the DArabinitol and Ribitol Transporters from Klebsiella pneumoniae. J. Bacteriol. 179: 6014-6019. 1998 Heuel, H., Shakeri-Garakani, A., Turgut, S., and Lengeler, J.W. Genes for D-arabinitol and ribitol catabolism from Klebsiella pneumoniae. Microbiology 144: 1631-1639. 2003 Otte, S., Scholle, A., Turgut, S., and Lengeler, J.W. Mutations Which Uncouple Transport and Phosphorylation in the D-Mannitol Phosphotransferase System of Escherichia coli K-12 and Klebsiella pneumoniae 1033-5P14. J. Bacteriol. 185(7): 2267-2276. 152 VII. Anhang Danksagung Mein besonderer Dank gilt Herrn Prof. Dr. Joseph W. Lengeler für die Bereitstellung der Promotionsmöglichkeit an seinem Lehrstuhl. Ohne seine überdurchschnittliche Unterstützung bei der Fertigstellung und die vielen anregenden und informativen Gespräche wäre diese Arbeit nicht möglich gewesen. Nicht zu vergessen ist die Schulung der persönlichen Kritikfähigkeit, die sich auch außerhalb der Universität als sehr hilfreich erweist. Herrn Dr. Kurt Schmid danke ich ebenfalls für die Unterstützung während dieser Arbeit und die vielen praktischen Tipps sowie unterhaltsamen und hilfreichen Gespräche. Bei Herrn Prof. Dr. Karlheinz Altendorf bedanke ich mich für die freundliche Übernahme des Koreferats. Herrn Prof. Dr. George T. Robillard danke ich für Ermöglichung der Versuche an seinem Lehrstuhl der Reichsuniversität Groningen unter der freundlichen Mithilfe von Erwin Vos. Herrn Dr. Heinrich Jung danke ich für die informativen und sehr hilfreichen Gespräche rund um das „Cystein-Scanning“ sowie seinen Einsatz in der Prüfungskomission. Allen Mitgliedern der Arbeitsgruppe danke ich für die hilfreichen Gespräche und das sehr angenehme Arbeitsklima, insbesondere der Besatzung des Nordseite Angi, Dana, Hedwig, Jens, Jörg und Knut. Bei Georg Zimmermann möchte ich mich auch im Namen der Arbeitsgruppe bedanken, da er mich jederzeit bei meiner Arbeit als Computerbeauftragter unterstützt hat und die Systeme trotz Mr. Gates am Laufen gehalten wurden. Nancy Tholema danke ich für die „Starthilfe“ beim Cystein-Scanning und das sehr gewisenhafte Korrekturlesn dre Arbeit. Ganz besonderer Dank gilt Dette, die nicht nur durch ihre Hilfe bei den Markierungsversuchen zum Gelingen dieser Arbeit beigetragen hat. VII. Anhang 153 Eidesstattliche Erklärung Ich versichere, dass die vorliegende Arbeit mein erster Promotionsversuch ist und ich die beschriebenen Arbeiten selbst durchgeführt und keine anderen als die beschriebenen Quellen und Hilfsmittel benutzt habe. Osnabrück, März 2003 ............................................. (Şevket Turgut)