Zwischen Koordinationsverbindungen und Clustern
Werbung

Zwischen Koordinationsverbindungen und Clustern – Prinzipien
und Konzepte zur Synthese hochkoordinierter, metallreicher
Moleküle
Dissertation
Spyros Thomas Cadenbach
Zwischen Koordinationsverbindungen und Clustern – Prinzipien
und Konzepte zur Synthese hochkoordinierter, metallreicher
Moleküle
Dissertation
Zur Erlangung der Doktorwürde
der Fakultät für Chemie und Biochemie
an der Ruhr-Universität Bochum
Vorgelegt von Diplom-Chemiker
Spyros Thomas Cadenbach
2009
Diese Arbeit wurde im Zeitraum von Oktober 2005 bis April 2009 am Lehrstuhl für
Anorganische Chemie II, Organometallics & Materials, der Ruhr-Universität Bochum
angefertigt.
Gutachter:
Prof. Roland A. Fischer
Prof. William S. Sheldrick
Mündliche Prüfung:
Sommer 2009
Hiermit erkläre ich, dass ich die vorliegende Dissertation selbst verfasst und mich dabei
keiner anderen als den von mir ausdrücklich bezeichneten Quellen und Hilfsmitteln bedient
habe. Weiterhin erkläre ich, dass ich an keiner anderen Stelle ein Prüfungsverfahren beantragt
bzw. die Dissertation in dieser oder anderer Form bereits anderweitig als Prüfungsarbeit
verwendet oder einer anderen Fakultät als Dissertation vorgelegt habe.
Thomas Cadenbach
Mein spezieller Dank gilt
Herrn Prof. Dr. Roland A. Fischer
für seine stete Ansprechbarkeit, sein großes Vertrauen, die gewährte wissenschaftliche
Freiheit und seine große Hilfsbereitschaft. Sie ermöglichte mir auch die Teilnahme an einem
unvergesslichen „Abenteuer mit abschließender Konferenzteilnahme“.
Mein besonderer Dank gilt:
Dr. Christian Gemel
Christian danke ich für die großartige Unterstützung und
Hilfsbereitschaft, für seine kreativen Ideen und den damit
verbundenen Anregungen und Diskussionen, die zum Erfolg
dieser Arbeit führten.
Timo Bollermann
Timo danke ich nicht nur für seine stete Hilfsbereitschaft,
sondern auch für die zahlreichen Gespräche über die Chemie,
den Laboralltag und die wirklich wichtigen Dinge – wie
König Fußball.
Sabine Pankau
Bei Sabine bedanke ich mich für ihre immerwährende,
riesige Hilfsbereitschaft und Freundlichkeit.
Malte Hellwig und
Malte und Denise danke ich für ihre Freundlichkeit und
Denise Zacher
Ehrlichkeit sowie dafür, dass sie stets ein offenes Ohr für
Frage- und Problemstellungen jeglicher
Art
haben.
Sie
haben stets für eine entspannte und lustige Atmosphäre
im
Büro gesorgt.
Saeed Amirjalayer
Mit Saeed habe ich eine unvergleichliche „Forschungsreise“
durch Kalifornien und Mexico gemacht, in der wir stets
gegen Chaos und Katastrophe ankämpften – mal gewonnen,
mal verloren. Ich möchte mich für eine unvergessliche Zeit
bedanken. Das waren gut angelegte 1000 Euro.
Dr. Rochus Schmid
Rochus danke ich für die geduldige und umfangreiche
Beratung und Unterstützung im Hinblick auf theoretische
Fragestellungen.
Natürlich möchte ich mich auch bei allen anderen Mitgliedern der Arbeitsgruppe Anorganische
Chemie II, Organometallics & Materials, für die großartige Arbeitsatmosphäre und
Unterstützung herzlich bedanken.
Ferner gilt mein Dank:
Prof. Dr. Gernot Frenking
Prof Dr. Frenking danke ich für die phantastische
Kooperation und die Durchführung der quantenchemischen
Analysen.
Hans-Jochen Hauswald,
Für ihre freundliche Beratung in NMR-Fragen.
Martin Gartmann und
Gregor Barchan
Fonds der Chemischen Industrie Dem Fonds der chemischen Industrie danke ich für die
finanzielle Unterstützung in Form eines Chemiefonds
Stipendiums.
Research School
Der Research School der Ruhr-Universität Bochum danke
ich
für
die
finanzielle
Unterstützung
meiner
Promotionsarbeit sowie für die Möglichkeit zur Teilnahme
an Seminaren und Konferenzen.
Ganz besonders danke ich meiner Familie, insbesondere meiner Mutter, Karin Folsche,
meiner Großmutter, Gisela Folsche sowie meinen Brüdern, Marcus und Martin Cadenbach.
Ihre Unterstützung, Hilfsbereitschaft, Geduld und Liebe ebneten den Weg, den ich im Laufe
meiner Promotion beschritt.
Zu guter Letzt danke ich meiner besten Freundin und großen Liebe, María José Benítez
Romero, die ich zu Anfang meines zweiten Promotionsjahres in einem Seminar kennenlernte.
Sie ist nicht nur mein Vorbild eines hervorragenden Wissenschaftlers, durch sie erfahre ich
jeden Tag die Sonnenseiten des Lebens. Ihre Liebe, ihr Lächeln, ihre Herzlichkeit und
Fürsorglichkeit gaben mir die Kraft und Motivation meine Promotion zu meistern. Te amo!
„Grau im Leben ist alle Theorie,
entscheidend ist auf´m Platz.“
Alfred „Adi“ Preißler
(09.04.1921 – 17.07.2003)
Für meine Mutter
I. Inhaltsverzeichnis
1. Motivation ......................................................................................................................... 1
2. Einleitung .......................................................................................................................... 5
2.1 Synthese der niedervalenten Gruppe 13 Organyle EIR .................................................... 8
2.2 Ligandeneigenschaften von EIR im freien und gebundenen Zustand .............................. 9
2.3 Synthesen, Strukturen und Reaktivitäten von M-EIR Verbindungen ............................. 11
2.3.1 Substitution labiler Liganden ................................................................................... 11
2.3.2 Reaktivitäten der homoleptischen Komplexe [Ma(ECp*)b] (M = Pd, Pt)................ 15
2.3.3 Bindungsaktivierungen durch M(ECp*)n ................................................................ 17
2.3.4 Insertionsreaktionen von ECp* in Metall-Halogenid Bindungen ............................ 20
2.3.5 [Ga2Cp*][BArF] als Transferreagenz für „nacktes“ Ga+ ......................................... 22
3. Ergebnisse ....................................................................................................................... 26
3.1 Reaktivität von ECp* (E = Al, Ga) gegenüber basischen d8-Metall-OlefinKomplexen ...................................................................................................................... 26
3.2 Synthesen und Strukturen elektronenreicher ECp*-Ruthenium-PolyhydridKomplexe und Cluster (E = Al, Ga) ............................................................................... 28
3.3 Kontrolliertes Clusterwachstum durch Hydrogenolyse von koordinierten GaCp*:
Synthese und Struktur des „nackten“ Gallium Komplexes [Ru2(Ga)(GaCp*)7(H)3] ..... 31
3.4 Methylgallium als terminaler Ligand in dem all-Gallium koordinierten Rhodium
Kation [(Cp*Ga)4Rh(GaCH3)]+ ...................................................................................... 41
3.5 Über die Organometallchemie des Ga+ Kations: Ein- und Ausschalten des
Elektronenpaares des monovalenten Gallium in der Koordinationsumgebung von
Ruthenium....................................................................................................................... 50
3.6 Synthese und Struktur von [Mo(GaCp*)6] ..................................................................... 64
3.7 Zwölf Einelektronenliganden koordinieren ein Metallzentrum: Struktur und
Bindung von [Mo(ZnCH3)9(ZnCp*)3] ............................................................................ 66
i
3.8 Molekulare Ausschnitte von Mo/Zn Hume Rothery Phasen: Synthese und
Struktur von [{Mo(CO)4}4(Zn)6(μ-ZnCp*)4] ................................................................. 79
3.9 Die neue Stoffklasse der hochkoordinierten Zink-reichen Übergangsmetallkomplexe ......................................................................................................................... 84
3.9.1 Synthesen und Strukturen der M-ZnR Komplexe der d6-Metalle ........................... 85
3.9.2 Synthesen und Strukturen der M-ZnR Komplexe der d8-Metalle ........................... 89
3.9.3 Synthesen und Strukturen der M-ZnR Komplexe der d9-Metalle ........................... 93
3.9.4 Synthesen und Strukturen der M-ZnR Komplexe der d10-Metalle .......................... 96
3.9.5 Zusammenfassende Betrachtung zu den Synthesen und Strukturen der
Stoffklasse der hochkoordinierten Zink-reichen Übergangsmetallkomplexe ......... 98
3.9.6 Über die Bindungssituationen in den pseudohomoleptischen d8-d10 MetallZink-komplexen ..................................................................................................... 101
3.9.7 Synthesen und Strukturen von heteroleptischen Komplexen mit ZnR Liganden .. 115
4. Zusammenfassung ..................................................................................................... 121
4.1 Reaktivität von ECp* (E = Al, Ga) gegenüber Metall-Olefin-Komplexen .................. 121
4.2 Synthesen und Strukturen elektronenreicher ECp*-Ruthenium-PolyhydridKomplexe und Cluster (E = Al, Ga) ............................................................................. 122
4.3 Cp* als entfernbare Schutzgruppe ................................................................................ 123
4.3.1 Kontrolliertes Clusterwachstum durch Hydrogenolyse von koordinierten
GaCp* .................................................................................................................... 123
4.3.2 Methylgallium als terminaler Ligand in dem all-Gallium koordinierten
Rhodium Kation [(Cp*Ga)4Rh(GaCH3)]+ durch protolytische Spaltung von
Cp*H ...................................................................................................................... 125
4.4 Über die Organometallchemie des Ga+ Kations ........................................................... 126
4.5 Die neue Stoffklasse der hochkoordinierten Zink-reichen Übergangsmetallkomplexe ....................................................................................................................... 128
ii
I. Inhaltsverzeichnis
5. Ausblick......................................................................................................................... 134
6. Experimentelles .......................................................................................................... 137
6.1 Allgemeine Arbeitstechniken ....................................................................................... 137
6.2 Routineanalysemethoden .............................................................................................. 137
6.3 Ausgangsverbindungen ................................................................................................ 139
6.4 Arbeitsvorschriften und analytische Daten neuer Verbindungen ................................. 140
7. Ergänzende kristallographische Daten .............................................................. 163
8. Literaturverzeichnis ................................................................................................. 171
9. Anhang .......................................................................................................................... 181
9.1 Posterbeiträge und Vorträge ......................................................................................... 181
9.2 Publikationsliste............................................................................................................ 182
9.3 Lebenslauf .................................................................................................................... 186
iii
II. Abbildungsverzeichnis
II. Abbildungsverzeichnis
Abbildung 6.1.
Robert Bunsen (31.3.1811 – 16.8.1899).
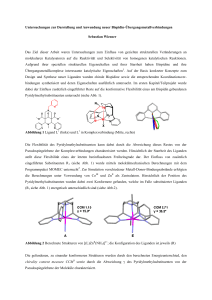
Abbildung 9.1.
Isolobalanalogie von EIR und CO.
Abbildung 10.1.
Wechselwirkungen in M-EIR Komplexen.
Abbildung 34.1.
Festkörperstruktur von 12.
Abbildung 36.1.
Berechnete Minima und Übergangszustände eines fluktionalen
Prozesses.
Abbildung 40.1.
Berechnete Reaktionspfade für die CpH Eliminierung, die zu IM3
führt.
Abbildung 43.1.
Molekülstruktur von [(Cp*Ga)4Rh(η1-Cp*GaCH3)] (14).
Abbildung 44.1.
Molekülstruktur
des
kationischen
Teils
des
Salzes
[(GaCp*)4Rh(GaCH3)][BArF] (15).
Abbildung 46.1.
Berechnete Ladungen am Galliumatom in GaMe, GaCp, GaMe2+ sowie
dem Modellkomplex [(CpGa)4Rh(GaCH3)]+ 15Cp (mit NBO = Natural
Bond Orbital).
Abbildung 48.1.
Molekülstruktur
des
kationischen
Teils
des
Salzes
[(GaCp*)4Rh(Ga(CH3)(py))][BArF] (16).
Abbildung 52.1.
Molekülstruktur von [Ru(GaCp*)3(TMM)] (13).
Abbildung 53.1.
Molekülstruktur von [Ru(PCy3)2(GaCp*)2(H)2] (17).
Abbildung 56.1.
Molekülstruktur des kationischen Teils von 18.
Abbildung 57.1.
Molekülstruktur des kationischen Teils von 19.
Abbildung 58.1.
Molekülstruktur des kationischen Teils von 20.
Abbildung 60.1.
Konturliniendiagramme der Laplace-Verteilungen 2ρ(r) von 19M.
Abbildung 62.1.
Molekülstruktur des kationischen Teils von 21.
Abbildung 65.1.
Molekülstruktur von [Mo(GaCp*)6] (22).
Abbildung 69.1.
Molekülstruktur von [Mo(ZnMe)9(ZnCp*)3] (23).
Abbildung 72.1.
Vergleiche
der
experimentellen
und
berechneten
[Mo(ZnR)12]
Strukturen.
Abbildung 73.1.
MO-Korrelationsdiagramm für [Mo(ZnH)12] (23H).
Abbildung 74.1.
HOMO-1 (hg) und HOMO-2 (ag) für [Mo(ZnH)12] (23H).
Abbildung 76.1.
(a) Schematische Darstellung der sd5 Hybridisierung.
(b) Chemische Bindungen in 23H aus der AIM Analyse.
iv
II. Abbildungsverzeichnis
Abbildung 77.1.
Konturliniendiagramme
der
Laplace-Verteilungen
2ρ(r)
von
[Mo(ZnH)12] (23H).
Abbildung 81.1.
Molekulare Struktur von [{Mo(CO)4}4(Zn)6(μ-ZnCp*)4] (27) wie sie
durch Röntgenstrukturanalyse bestimmt wurde.
Abbildung 82.1.
Der Mo4 Tetraeder (links) und der Zn6 Oktaeder (rechts) in der
molekularen Struktur von [{Mo(CO)4}4(Zn)6(μ-ZnCp*)4] (27).
Abbildung 83.1.
Ausschnitt aus einer (imaginären) Mo/Zn Legierugsstruktur, die aus
flächenverknüpften Mo4Zn6 super-Tetraedern (links) aufgebaut ist.
Abbildung 86.1.
Molekulare Struktur von [Mo(ZnCp*)4(GaMe)4] (24) wie sie durch
Röntgenstrukturanalyse bestimmt wurde.
Abbildung 87.1.
Molekulare Struktur von [Mo(ZnCp*)4(ZnMe)4(GaMe)2] (25) wie sie
durch Röntgenstrukturanalyse bestimmt wurde.
Abbildung 89.1.
Molekulare Struktur von [Mo(ZnCp*)2(ZnEt)10] (26) wie sie durch
Röntgenstrukturanalyse bestimmt wurde.
Abbildung 90.1.
Molekulare Struktur von [Ru(ZnCp*)4(ZnMe)6] (28) wie sie durch
Röntgenstrukturanalyse bestimmt wurde.
Abbildung 92.1.
Molekulare Struktur von [Ru(ZnCp*)4(ZnMe)4(H)2] (29) wie sie durch
Röntgenstrukturanalyse bestimmt wurde.
Abbildung 94.1.
Molekulare Struktur von [Rh(ZnCp*)3(ZnMe)6] (30) wie sie durch
Röntgenstrukturanalyse bestimmt wurde.
Abbildung 95.1.
Molekulare Struktur von [Rh(GaMe)(ZnCp*)4(ZnMe)3] (31) wie sie
durch Röntgenstrukturanalyse bestimmt wurde.
Abbildung 97.1.
Molekulare Struktur von [Pd(ZnCp*)4(ZnMe)4] (33) wie sie durch
Röntgenstrukturanalyse bestimmt wurde.
Abbildung 98.1.
Molekulare Struktur von [Pt(ZnCp*)4(ZnEt)4] (35) wie sie durch
Röntgenstrukturanalyse bestimmt wurde.
Abbildung 100.1.
Platonische Körper der pseudo-homoleptischen ausschließlich Zink
koordinierten
4d
Übergangsmetallkomplexe
wie
sie
durch
Röntgenstrukturanalyse bestimmt wurden.
Abbildung 104.1.
Geometrieoptimierte Minima der Modellverbindungen [M(ZnH)n]:
[Mo(ZnH)12] (Ih) (23H), [Ru(ZnH)10] (D4d) (28H), [Rh(ZnH)9] (D3h)
(30H) und [Pd(ZnH)8] (D4d) (33H).
Abbildung 105.1.
Ausgewählte besetzte Valenzorbitale von [Ru(ZnH)10] (28H).
v
II. Abbildungsverzeichnis
Abbildung 109.1.
Konturliniendiagramme der Laplace-Verteilungen 2ρ(r) der Komplexe
(a) [Mo(ZnH)12] (23H), (b) [Ru(ZnH)10] (28H), (c) [Pd(ZnH)8] (33H),
(d) [Rh(ZnH)9] (30H).
Abbildung 111.1.
Ausgewählte besetzte Valenzorbitale des Moleküls [Pd(ZnH)8] (33H).
Abbildung 113.1.
Ausgewählte besetzte Valenzorbitale des Moleküls [Rh(ZnH)9] (30H).
Abbildung 117.1.
Molekulare Struktur von [Mo(CO)3(ZnCp*)3(ZnMe)3] (36) wie sie
durch Röntgenstrukturanalyse bestimmt wurde.
Abbildung 119.1.
Molekulare Struktur von [Ru(TMM)3(ZnCp*)3(ZnMe)3] (37) wie sie
durch Röntgenstrukturanalyse bestimmt wurde.
Abbildung 129.1.
Festkörperstrukturen der pseudo-homoleptischen Übergangsmetall-ZnR
Komplexe wie sie durch Röntgenstrukturanalyse bestimmt wurde.
Abbildung 130.1.
Festkörperstrukturen der gemischtmetallischen Ga/Zn Intermediate wie
sie durch Röntgenstrukturanalyse bestimmt wurde.
Abbildung 131.1.
Oben: Der Mo4 Tetraeder (links) und der Zn6 Oktaeder (rechts) in der
molekularen Struktur von [{Mo(CO)4}4(Zn)6(μ-ZnCp*)4] (27) wie sie
durch Einkristallröntgenstrukturanalyse bestimmt wurde.
Abbildung 134.1.
Hochkoordinierte Übergangsmetallkomplexe mit CdR Liganden.
Abbildung 135.1.
Molekülstrukur von [Mo{Au(PPh3)}8(AuCl)2(EMe)2] (E = Ga oder
Zn).
Abbildung 145.1.
Pulverdiffraktogramm einer Mischung aus RuGa2 und RuGa Partikeln.
Abbildung 145.2.
Pulverdiffraktogramm einer Mischung aus RuGa2 und Ru Partikeln.
Abbildung 146.1.
EDX Analysen der uncalcinierten (oben) und der calcinierten Probe
(unten), welche durch Co-Hydrogenolyse von [Ru(η4-COD)(η3-C4H7)2]
und GaCp* in einem molaren Verhältnis von 1:2 erhalten wurden (3 bar
H2, Mesitylen, 150 °C, 7 Tage).
Abbildung 147.1.
Berechnete parallele GaCp Assoziations-/Dissoziations-Gleichgewichte
in Lösung, die schließlich zu 1Cp führen.
Abbildung 148.1.
Berechnete parallele GaCp Assoziations-/Dissoziations-Gleichgewichte
in Lösung, die schließlich zu 1Cp führen.
Abbildung 148.2.
Berechnete Minima für einen fluktionalen Prozesses.
Abbildung 149.1.
Berechnete Minima für einen fluktionalen Prozesses.
vi
III. Schemenverzeichnis
III. Schemenverzeichnis
Schema 5.1.
Synthese der ersten organometallischen Verbindungen – Kakodyl und
Kakodyloxid.
Schema 12.1.
Synthese von [Mo2(CO)6{μ2-(GaCp*)}3] durch Verwendung des
molekularen Bausteins [fac-(GaCp*)3M(CO)3].
Schema 12.2.
Sterische Überladung in [Cp(CO)2M(μ-GaCp*)]2 (M=Mo, M=W) führt
zu einer Verschiebung der Haptizität der Cp* Gruppen.
Schema 13.1.
Synthese der homoleptischen Komplexe [M(EIR)4] durch Substitution
des labilen Olefinliganden COD.
Schema 14.1.
[M(GaCp*)4] als molekularer Bausteine in der Synthese von
zweikernigen Komplexen des Typs [MPt(GaCp*)2(μ2-GaCp*)3].
Schema 14.2.
Synthesen von [Pd2(GaCp*)2(μ2-GaCp*)3] und [Pd3(GaCp*)4(μ²GaCp*)4].
Schema 15.1.
Synthesen der homoleptischen Kationen [Rh(GaCp*)5]+, [M(GaCp*)4]+
(M = Cu, Ag) und [Zn(GaCp*)4]2+.
Schema 16.1.
Reaktivität von [M2(GaCp*)2(μ2-GaCp*)3].
Schema 17.1.
Synthesen von [Pd3(InCp*)3(PPh3)3] und [Pd3(InCp*)3(dppe)2]
Schema 18.1.
C-H und Si-H-Aktivierungen an [Ni(AlCp*)3].
Schema 20.1.
Reaktionsabfolge
für
die
C-C-Bindungsaktivierung
in
[Cp*Rh(GaCp*)(CH3)2].
Schema 21.1.
Insertionsreaktionen bei Umsetzung von [{RhCp*Cl2}2] mit ECp*]
Schema 23.1.
Synthese von [Ga2Cp*][BArF].
Schema 24.1.
Umsetzung
von
[Pt(GaCp*)4]
mit
[Ga2Cp*][BArF]
und
[H(OEt2)2][BArF].
Schema 25.1.
Oxidative Abspaltung von Cp* führt zu [Pt3(Ga)(GaCp*)6][BArF].
Schema 27.1.
Synthese der Ruthenium Komplexe 1-3.
Schema 28.1.
Synthese der Rhodium Komplexe 4- 6.
Schema 30.1.
Synthese von [Ru(COD)(H)(GaCp*)3][BArF] (7).
Schema 30.2.
Synthesen der Verbindungen 8 und 9.
Schema 31.1.
Synthesen der Verbindungen 10 und 11.
Schema 33.1.
Synthesewege der Verbindung 12 mit [Ru(η4-COD)(η3-C4H7)2] als RuQuelle.
vii
III. Schemenverzeichnis
Schema 33.2.
Synthesen der Verbindung 12 mit [Ru(η2,η2-COD)(η6-COT)] als RuQuelle.
Schema 38.1.
Arbeitshypothesen zur Bildung des Schlüsselintermediates in der
Synthese von 1Cp.
Schema 42.1.
Synthese von [(Cp*Ga)4Rh(η1-Cp*GaCH3)] (14).
Schema 44.1.
Synthese von 15 durch Protolyse von 14 in Fluorbenzol.
Schema 55.1.
Reaktionen von 13 mit [H(OEt2)2][BArF] und [Ga2Cp*][BArF] in
Fluorbenzol.
Schema 61.1.
Reaktion von 17 mit [Ga2Cp*][BArF] in Fluorobenzol.
Schema 64.1.
Synthese von [Mo(GaCp*)6] (22).
Schema 68.1.
Herstellung der Verbindungen 23 - 26.
Schema 80.1.
Synthese von Verbindung 27.
Schema 90.1.
Synthesen der hochkoordinierten Komplexe [Ru(ZnCp*)4(ZnMe)6] (28)
und [Ru(ZnCp*)4(ZnMe)4(H)2] (29).
Schema 93.1.
Darstellung der hochkoordinierten Komplexe [Rh(ZnCp*)3(ZnMe)6]
(30) und [Rh(GaMe)(ZnCp*)4(ZnMe)3] (31).
Schema 96.1.
Synthese der pseudo-homoleptischen Komplexe [M(ZnCp*)4(ZnMe)4]
(M = Ni (32), Pd (33), Pt (34).
Schema 116.1.
Synthese des heteroleptischen Komplexes [Mo(CO)3(ZnCp*)3(ZnMe)3]
(36).
Schema 120.1.
Synthese
der
heteroleptischen
Komplexe
[Ru(TMM)3(ZnCp*)3(ZnMe)3] (37) und [Ru(TMM)3(ZnCp*)2(ZnMe)4]
(38).
Schema 124.1.
Clusterwachstum durch Hydrogenolyse von koordinierten Liganden
Schema 126.1.
Terminales Methylgallium durch Protolyse eines koordinierten (η1Cp*GaCH3) Liganden.
Schema 127.1.
Oben: Das Ga+ Transferreagenz [Ga2Cp*][BArF] als Synthon für
ungewöhnliche Komplexe des Typs [LnM-GaR’].
viii
IV. Tabellenverzeichnis
IV. Tabellenverzeichnis
Tabelle 47.1.
Resultate der EDA auf BP86/TZ2P Niveau.
Tabelle 70.1.
Überblick
der
Bindungslängen
in
Å
ermittelt
aus
Röntgenstrukturanalysen.
Tabelle 75.1.
EDA Ergebnisse von [Mo(ZnH)12] (23H) auf BP86/TZ2P Niveau.
Tabelle 100.1.
Elektronenanzahl in den synthetisierten metallreichen Molekülen.
Tabelle 102.1.
Vergleich der experimentellen Bindungslängen mit den berechneten
Daten, wobei in den Rechnungen Cp* gegen Cp substituiert wurde.
Tabelle 103.1.
Berechnete Bindungslängen in den Modellverbindungen.
Tabelle 107.1.
Ergebnisse der EDA Analysen von [Ru(ZnH)10] (28H) auf
BP86/TZ2P+ Niveau mit unterschiedlichen elektronischen Zuständen
der interagierenden Fragmente.
Tabelle 108.1.
Die berechneten atomaren Partialladungen der Modellverbindungen
Tabelle 112.1.
EDA Ergebnisse auf BP86/TZ2P Niveau für die Komplexe [M(ZnH)8]
(M = Ni, Pd, Pt) mit den Fragmenten M (s0d10) und (ZnH)10 im
elektronischen Singulett-Zustand.
Tabelle 115.1.
EDA Ergebnisse auf BP86/TZ2P+ Niveau für den Komplex
[Rh(ZnH)9]
(30H)
Berücksichtigung
mit
D3h
Symmetrie
unterschiedlicher
BP86/TZ2P+
elektronischer
Zustände
unter
der
Fragmente.
Tabelle 133.1.
Überblick
über
die
neu
synthetisierten
und
charakterisierten
Verbindungen.
Tabelle 163.1.
Ergänzende kristallographische Daten zu Verbindungen 1, 3-6.
Tabelle 164.1.
Ergänzende kristallographische Daten zu Verbindungen 7-11.
Tabelle 165.1.
Ergänzende kristallographische Daten zu Verbindungen 12-16.
Tabelle 166.1.
Ergänzende kristallographische Daten zu Verbindungen 17-20.
Tabelle 167.1.
Ergänzende kristallographische Daten zu Verbindungen 21-25.
Tabelle 168.1.
Ergänzende kristallographische Daten zu Verbindungen 26-29.
Tabelle 169.1.
Ergänzende kristallographische Daten zu Verbindungen 30-33.
Tabelle 170.1.
Ergänzende kristallographische Daten zu Verbindungen 34-37.
ix
V. Liste der verwendeten Abkürzungen
V. Liste der verwendten Abkürzungen
AAS
Atom-Absorptions-Spektroskopie
AIM
Atoms in Molecules
AO
Atomorbital
BArF
[B{C6H3(CF3)2}4]-Anion
COE
Norbornadien
COD
cis,cis-1,5-Cyclooctadien
COT
cis,cis,cis-1,3,5-Cyclooctatrien
Cp
Cyclopentadienyl-anion
CpH
1,3-Cyclopentadien
Cp*
1,2,3,4,5-Pentamethylcyclopentadienyl-anion
Cp*H
1,2,3,4,5-Pentamethylcyclopentadien
Cy
Cyclohexyl
t
Bu
tert.-butyl
d
Doublet
DDP
2-Diisopropylphenylamino-4-diisopropylphenylimino-2-penten
DFT
Dichtefunktionaltheorie
dmso
Dimethylsulfoxid
dppe
Bis(diphenylphospino)ethan
dvds
1,3-Divinyl-1,1,3,3-tetramethyldisiloxan
E
Gruppe 13 Metall, Erdmetall
EA
Elementaranalyse
EDA
Energiedekompositionsanalyse
Et
Ethyl
FT-IR
Fourier Transform Infrarotspektroskopie
HF
Hartree-Fock
IR
Infrarotspektroskopie
L
Ligand
m
Multiplet
M
Übergangsmetall
Me
Methyl
Mesitylene
1,3,5-Trimethylbenzene,
x
V. Liste der verwendeten Abkürzungen
Mes*
2,6-Bis(2,4,6-triisopropylphenyl)phenyl
MO
Molekülorbital
NMR
Nuclear Magnetic Resonance
NBD
Norbornadien
NBO
Natürliches Bindungsorbital
NHC
N-heterocyclische Carbene
Ph
Phenyl
Pr
Propyl
i
Pr
iso-Propyl
py
Pyridin
s
Singlet
RT
Raumtemperatur
THF
Tetrahydrofuran
tmeda
Tetramethyledthylendiamin
TMM
Trismethylenmethan
VE
Valenzelektronen
XRD
X-Ray Diffraction
xi
1. Motivation
1. Motivation
„Ein grundlegendes Gebiet der Chemie ist die chemische Bindung, der Grundstein auf dem
ein
ganzes
chemisches
Universum
errichtet
ist.“1
Seit
der
Definition
einer
Elektronenpaarbindung2 durch Lewis im Jahre 1916 entwickelte sich das Prinzip einer
chemischen Bindung durch zahlreiche experimentelle und theoretische Arbeiten stets weiter
und bleibt bis heute ein zentrales Forschungsgebiet vieler Arbeitsgruppen. Bindungen
zwischen fast allen nur erdenklichen Elementkombinationen wurden bis ins Detail
beschrieben und „nur ein kleiner Teil von Element-Element Bindungen warten auf ihre
Entdeckung.“3 Der Hauptbestandteil der chemischen Elemente im Periodensystem sind
Metalle. Daher ist das Studium von Verbindungen mit Metall-Metall-Bindungen aus
bindungstheoretischer
Sichtweise
heraus
eine
der
Forschungsgebiete der modernen anorganischen Chemie.4,
wichtigsten
5
und
reizvollsten
Hierbei liegt der Fokus unter
anderem auf der Generierung von Bindungen neuer Elementkombinationen, der Erzeugung
von Metall-Metall-Mehrfachbindungen sowie auf der Synthese von definierten Molekülen mit
einer größtmöglichen Anzahl von Metall-Metall- oder, verallgemeinert im Kontext der
Übergangsmetall-Komplex-Chemie, von Metall-Ligand-Kontakten. Bei den Entdeckungen
von CdI-CdI,6 ZnI-ZnI,7 HgI-HgI,8 MgI-MgI,9 oder Pd-Zn3 Bindungen handelt es sich nur um
ausgewählte Glanzlichter von Molekülen mit neuartigen Elementkombinationen. Ein neues
Gebiet der anorganischen Chemie eröffnete der 1964 von Cotton formulierte Vorschlag, dass
eine Vierfachbindung zwischen den Rheniumatomen des Anions [Re2Cl8]2- vorliegt.10-12 Da
die
Verfügbarkeit
von
d-Orbitalen
bei
Übergangsmetallen
zu
einer
maximalen
Bindungsordnung von sechs führt, wurde die bisher höchste Mehrfachbindung zwischen zwei
Elementen kürzlich in der von Power und Mitarbeitern synthetisierten stabilen
Übergangsmetallverbindung Cr2Ar’2 (Ar’=C6H3-2,6-(C6H3-2,6-iPr2)2) beobachtet, in der eine
Fünfachbindung zwischen den beiden Chrom-Zentren vorliegt.13-17 Zu Verbindungen, in
denen eine Vielzahl von Metall-Metall-Bindungen vorliegen, zählen die sogenannten ClusterMoleküle. Der Begriff Cluster wurde 1966 durch Cotton definiert als „Moleküle, in denen
eine endliche Gruppe von Metallatomen ausschließlich oder zumindest in beträchtlichem
Maße von Metall-Metall-Bindungen zusammengehalten wird, wobei zusätzlich auch
Nichtmetallatome assoziiert sein können.“18 Typischerweise bilden die Metallatome hier
cyclische oder käfigartige Strukturen, wobei die Bindungsverhältnisse zwischen den Metallen
häufig nicht mit konventionellen 2e2c-Bindungen beschreibbar sind. Da der von Cotton
verwandte Begriff so weit gefasst ist, dass er auf viele Verbindungen zutrifft, führte
1
1. Motivation
Schnöckel 1999 den Begriff des metalloiden Clusters ein. Metalloide (metallähnliche) Cluster
sollten „sich dadurch auszeichnen, dass in ihnen die Zahl der Metall-Metall-Kontakte die der
Metall-Ligand-Kontakte übersteigt und dass es Metallatome gibt, die ausschließlich MetallMetall-Wechselwirkungen eingehen.“19,
20
Dazu zählen beispielsweise die ligandfreien,
„nackten“ Metallatomcluster, darunter das von Pyykkö im Jahr 2002 vorhergesagte21 und
kurze Zeit später von Wang hergestellte22 Molekül [WAu12]. Meilensteine im Bereich der
ligandenstabilisierten, metalloiden Cluster sind u.a. die von Schmid hergestellten
Verbindungen M55L12Clx (M = Rh, Ru, Pt, Au, L = PR3, AsR3, x = 6, 20)23, das von Dahl
synthetisierte Molekül [Pd145(CO)x(PEt3)30]24 (x ≈ 60), und die von Schnöckel isolierten
Hauptgruppenmetallcluster
[Al77{N(SiMe3)2}20{Li(OEt2)3(μ-I)Li(OEt2)2}2]25
[Ga84{N(SiMe3)2}20Li6Br20(thf)20]26,
wobei
letzterer
hinsichtlich
seiner
64
sowie
nackten
Galliumatome einen Rekord darstellt. Der Komplexbegriff steht dem des Clusters gegenüber.
Der Begriff „Komplex“ bezieht sich dabei auf ein Molekül [MLm] mit einem Zentralmetall M,
das über Ligatoratome E einen Liganden L durch Donor-Akzeptor-Wechselwirkungen bindet,
was zu einer Kernstruktur MEn führt.27 Seit der Begründung der Koordinationschemie durch
Alfred Werner im Jahre 189327,
28
ist der Stand der Forschung und damit auch des
Lehrbuchwissens, dass Koordinationszahlen n ≥ 10 in diskreten Molekülen bzw. Komplexen
[MLn] für einzähnige Liganden L unbekannt sind. Zu Liganden, in denen die Ligatoratome E
aus Metallen bestehen, zählen die niedervalenten Gruppe 13 Organyle des Typs EIR (E =
Gruppe 13 Metall). Die Koordinationschemie dieser exotischen Ligandensysteme „ist ein
hochaktuelles Gebiet der modernen Hauptgruppenchemie, das in den letzten Jahren
beträchtliche Fortschritte verzeichnete.“29-34 Jüngste Glanzlichter in diesem Bereich stellen
die Synthesen der ersten Nd-Ga,35 U-Ga36 oder U-Al37 Bindungen dar. Dieser Aufschwung
wird unter anderem durch die elektronisch und sterisch stabilisiernde Wirkung der Gruppen R
begründet, da sie für die Stabilität und Isolierbarkeit der Liganden EIR (E = Al, Ga, In; R =
Cp*, C(SiMe3)3, CH(SiMe3)2, Si(SiMe3)3, CMe4, Amide, β-Diketiminate, Amidinate,
Guanidinate) verantwortlich sind. Die σ-Donor- und π-Akzeptoreigenschaften der EIR
Liganden wurden ausführlich und kontrovers diskutiert.38-43 Dabei gilt es zu beachten, dass
die Bindungseigenschaften stark von den organischen Substituenten R abhängen. Während
beispielsweise GaCp* ein sehr starker σ-Donor-Ligand, aber aufgrund der konkurrierenden πDonoreigenschaften des Cp*-Fragments, ein schlechter π-Akzeptor-Ligand ist, wird bei
Komplexen mit Beteiligung von alkylsubstituierten EIR Verbindungen ein größerer Beitrag
zur M-E-Bindung gefunden. Dieses Verhalten wird auf eindrucksvolle Weise durch die ersten
Komplexe mit terminal gebundenen, „nackten“ Ga+ und In+ Atomen bestätigt. Dabei handelt
2
1. Motivation
es sich um die Verbindungen [(Ga)Pt(L)4][BArF] (L = GaCp*, PR3; BArF =
[B{C6H3(CF3)2}4]) und [(In)Pt(PPh3)3][BArF],29, 44 die aus den Umsetzungen von [Pt(L)4] mit
den „nackten“ E+-Transferreagenzien [Ga2Cp*][BArF]45 bzw. [InBArF]44 resultieren.
Quantenchemische Analysen belegen, dass es sich bei den substituentenfreien E+ Liganden
ausschließlich um σ- und π-Akzeptorliganden ohne jegliche Donoreigenschaften handelt. Bei
den Komplexen [(Ga)Pt(L)4][BArF] handelte es sich um die ersten und bis zu Beginn dieser
Arbeit auch um die einzigen Produkte, welche bei einer Umsetzung mit dem Ga+Transferreagenz [Ga2Cp*][BArF] resultieren. Im Rahmen der vorliegenden Arbeit wurde nun
eine charakteristische Organometallchemie des Ga+-Kations untersucht. Dabei wurden zwei
zentrale Fragestellungen wie folgt definiert: Erstens, wenn sich Ga+ als starkes Elektrophil
gegenüber Lewis-basischen Übergangsmetallzentren verhält, wie sieht dann dessen Verhalten
gegenüber nukleophilen π-Liganden aus? Zweitens, was kann man über dessen Reaktivität
gegenüber M-X Einheiten berichten? Dabei ist zu berücksichtigen, dass es bereits zahlreiche
Reaktionen von GaIR Verbindungen mit Komplexen des Typs [LnM-X] gibt.
Während über die Struktur- und Bindungseigenschaften der Komplexe [LaMb(ER)c] im Laufe
der letzten 15 Jahre viele Arbeiten entstanden, ist bisher sehr viel weniger über ihre
Organometallchemie und den damit verbundenen potentiellen Anwendungen berichtet
worden. Durch Verwendung von ECp* und ausschließlich Kohlenwasserstoff-Liganden
beinhaltenden Systemen [MLn] als Präkursoren konnte unter sanften hydrogenolytischen
Synthesebedingungen M/E Hume Rothery Phasen gezielt dargestellt werden.46-48 Da sich
GaCp* inert gegenüber H2 verhält, werden GaCp*-haltige Koordinationsverbindungen als
Intermediate in dieser M/E Materialsynthese vermutet. In diesem Zusammenhang ist das
Abspalten einer Cp* Gruppe von koordinierten ECp* Liganden als Initialisierung eines M/E
Clusterwachsums ein wichtiger Aspekt, wie auch die Synthese des galliumverbrückten
Dimers [Pt2H(Ga)(GaCp*)7]2+ über selektive Protolyse von koordiniertem GaCp* in
[Pt(GaCp*)4] durch [H(OEt2)2][BArF] belegt.44 Eine selektive, oxidative Abspaltung von Cp*
unter Bildung von Fulvalen tritt in der Reaktion von [Pt(GaCp*)4] mit [Cp2Fe][BArF] auf, bei
dem der trinuklearen Cluster [Pt3(Ga)(GaCp*)6]+ entsteht.49 Mit dem Ziel frühe Intermediate
in
Legierungsbildungsprozessen
zu
charakterisieren,
wurden
exemplarische
Hydrogenolyseversuche unter Verwendung verschiedener Rutheniumquellen durchgeführt.
Dabei konnte unter selektiver hydrogenolytischer Spaltung eines Cp* Liganden aus einem
kooridinierten
GaCp*
der
neutrale,
„nackte“
Gallium
verbrückte
Komplex
[(GaCp*)4(H)Ru(μ-Ga)Ru(H)2(GaCp*)] isoliert werden. Dabei handelt es sich um eine frühe
molekulare Stufe auf dem Weg zu Ru/Ga Nanopartikeln in einer nasschemischen Synthese.
3
1. Motivation
Die Besonderheit von Cp* als entfernbare Schutzgruppe führte bei der Protolyse von
[(Cp*Ga)4Rh(η1-Cp*GaCH3)] durch [H(OEt2)2][BArF] zu der Synthese des Komplexes
[(GaCp*)4Rh(GaMe)][BArF], welcher eine terminale GaMe Einheit enthält. Diese Ergebnisse
tragen zu einem tieferen Verständnis für die Entwicklung einer sehr kontrollierten
molekularen Bausteinchemie der Hume Rothery artigen intermetallischen M/E Cluster und
Nanomaterialien bei.
Die oben diskutierten Ergebnisse führten zu der Vermutung, dass auch weitere
Transformationen von koordinierten Liganden in Übergangsmetallkomplexen möglich seien.
In dem Versuch, die Cp* Gruppen in [Mo(GaCp*)6] mit Hilfe von ZnMe2 in
Transmetallierungsreaktionen
durch
[Mo(ZnMe)9(ZnCp*)3]
Prototyp
der
Methylgruppen
einer
neuen
zu
substituieren,
Verbindungsklasse
wurde
mit
gefunden.
Quantenchemische Analysen zeigen, dass es sich bei dem zwölffach koordinierten
[Mo(ZnMe)9(ZnCp*)3] um ein hyperkoordiniertes aber gleichzeitig hypoelektronisches
Molekül
handelt,
das
weder
die
Bindungscharakteristika
einer
typischen
Koordinationsverbindung aufweist, noch korrekt als Cluster zu beschreiben ist. In der
Reaktion
von
[Mo(CO)4(GaCp*)2]
mit
ZnMe2
resultiert
der
verwandte
Cluster
[{Mo(CO)4}4(Zn)6(μ-ZnCp*)4], der sechs „nackte“ Zinkatome in oktaedrischer, unmittelbarer
Nachbarschaft zueinander als Kern enthält. Beide Moleküle, [Mo(ZnMe)9(ZnCp*)3] und
[{Mo(CO)4}4(Zn)6(μ-ZnCp*)4], stellen molekulare Ausschnitte aus einer intermetallischen
Hume-Rothery-Phase dar. Der leichte Zugang zu den Ausgangsmaterialen [LaMb(GaCp*)c]
sowie die allgemeine Anwendbarkeit einen GaCp* Liganden gegen zwei ZnR Liganden
auszutauschen, ermöglichte die Synthese einer neuen Molekülfamilie hochkoordinierter,
metallreicher Moleküle, die den Raum zwischen Koordinationsverbindungen und Clustern
füllen und zur weiteren Verknüpfung der Chemie und Physik von Molekülen und
Festkörpermaterialien beitragen werden.
4
2. Einleitung
2. Einleitung
„Because it is there!“
(George Herbert Leigh Mallory (18.6.1886 – Juni 1924), auf die Frage, warum er den Mount
Everest besteigen wolle)
Die Organometallchemie (Metallorganik) ist die Lehre von Verbindungen, die direkte MetallKohlenstoffbindungen aufweisen. Ihre Wiege befindet sich in einer Pariser Militärapotheke
als im Jahre 1757 der französische Apotheker Louis Claude Cadet de Gassicourt (1731 1799) Versuche durchführte, unsichtbare Tinten aus Co-Erzen herzustellen.4,
50
Allerdings
wusste er zunächst nicht, dass die von ihm verwendeten Co-Salze auch Arsenik, also As2O3,
enthielten und so entstand beim Aufheizen seiner Erze in Gegenwart von Kaliumacetat eine
stark rauchende Flüssigkeit von extrem durchdringenden, knoblauch- bis kotartigen Geruch.
„Weder Essig noch andere stark riechende Substanzen können den Gestank vernichten, der in
den Reaktionsgefäßen zurückbleibt.“51 Der Geruch war so intensiv, dass nur ein offenes
Lagern der Kolben und Reagenzgläser an frischer Luft über mehrere Monate ihn beseitigen
konnte. Bei der Synthese bildete sich eine Mischung der hochgiftigen Substanzen [(CH3)2AsAs(CH3)2] und [((CH3)2As)2O], welche später von Friherre Jöns Jacob Berzelius (20.8.1779 –
7.8.1848) die Trivialnamen Kakodyl und Kakodyloxid (griech.: κακωδης = stinkend)
erhielten.52
As2O3
+ 4 CH3COOK
Δ
H3C
CH3
As
H3C
H3C
+
As
CH3
CH3
O
As
As
CH3
CH3
Schema 5.1. Synthese der ersten organometallischen Verbindungen – Kakodyl und
Kakodyloxid
Da Cadet sich entschied seiner Entdeckung nicht weiter nachzugehen, dauerte es ca. 21 Jahre
bis drei französische Chemiker, Guyton de Morveau, Maret und Durande sich wagten,
„Cadet´s rauchende Flüssigkeit“ zu reproduzieren, um sie genauer zu untersuchen.53 Obwohl
auch sie den penetranten Geruch sehr schnell bemerkten und zudem eine „sehr unangenehme
Irritation des Halses“ auftrat, setzten sie ihre Analysen fort. Dabei beobachteten sie, dass nur
wenige Tropfen einen starken Rauch verursachten und dass das bei ihren Versuchen
5
2. Einleitung
„Because it is there“
verwendete Filterpapier sich spontan entzündete und „in einer wunderschönen rosafarbenen
Flamme“ verbrannte. Auch sie konnten keine genaue Zusammensetzung dieser Flüssigkeit
liefern. So dauerte es weitere 59 Jahre bis der deutsche Chemiker Robert Wilhelm Eberhard
Bunsen (31.3.1811 – 16.8.1899) sich der Analyse widmete.54-61 Bunsen war nicht nur ein
außergewöhnlicher Experimentator, er war auch ein begabter Glasbläser, der seine Glasgeräte
selbst herstellte. Dies war der Grundstein für eine erfolgreichen Analyse an Cadet´s
Flüssigkeit. Trotz seiner neu entwickelten Glasgeräte blieb
auch ihm der äußerst unangenehme Charakter der
untersuchten Substanz nicht verborgen. So warnte Bunsen,
dass die bei der Reaktion auftretenden Gase und Produkte
bereits in kleinen Mengen extrem tränenreizend sind und
das Atemwegssystem bis hin zur Bewusstlosigkeit reizen
sowie ein Kontakt mit der Haut zu starken Verätzungen
führt.57 Dies hielt Bunsen jedoch nicht davon ab, sein
Produkt genauestens zu analysieren. Dazu gehörte, für
damalige Zeiten durchaus üblich, es zu probieren, d.h. es zu
schmecken!
Abbildung 6.1. Robert Bunsen
(31.3.1811 – 16.8.1899)
So
knoblauchartig
schrieb
schmecke
er,
dass
und
die
sehr
Flüssigkeit
giftig
sei.
Unbeeindruckt forschte Bunsen weiter und synthetisierte
eine Reihe weiterer Derivate, dazu zählte beispielsweise (CH3)2AsCN, dass bei der
Umsetzung mit Hg(CN)2 entsteht.59-61 Bei der Synthese dieser Verbindung ereignete sich auch
ein Laborunfall, bei dem Bunsen durch eine Explosion auf dem rechten Auge teilweise
erblindete und durch die Vergiftung für mehrere Tage in Lebensgefahr schwebte.62 Jedoch
gelang ihm auch in diesem Fall die Synthese des hochgiftigen (CH3)2AsCN. Dieses, so
Bunsen, „bildet wunderschöne, prismatische Kristalle“. Er empfahl, die Aufarbeitung im
Freien vorzunehmen und dabei frische Luft durch ein langes, schnorchelartiges Glasrohr weit
oberhalb der extrem flüchtigen Kristalle einzuatmen. Ein direkter Kontakt sei zu vermeiden,
da in einem geschlossenen Raum bereits 0.065 g zu plötzlicher Taubheit der Hände und Füße
sowie zu einem Schwärzen der Zunge führe und Schwindel bis hin zur Ohnmacht aufträte.
Das handwerkliche Können Bunsens und die guten Arbeitsbedingungen in seinem Labor in
Marburg veranlassten Sir Edward Frankland (18.1.1825 - 9.8.1899) dort in regelmäßigen
Abständen mehrmonatige Forschungsaufenthalte während seiner Doktorarbeit einzulegen.
Hier synthetisierte er auf der Suche nach freien Radikalen unter anderem 1849 das an Luft
entzündliche ZnEt2.63-65 Beim Öffnen des „Autoklaven“ schoss eine Stichflamme hervor, was
6
2. Einleitung
„Because it is there“
auch Frankland nicht davon abhielt auf seinem Forschungsgebiet weiterzuarbeiten. Kurze Zeit
später gelang ihm die Synthese einer Reihe von Metallalkylen, darunter auch HgMe2,66,
67
welches eines der stärksten bekannten Nervengifte ist.
Die ausgewählten Beispiele belegen, dass es Chemiker ganz offensichtlich bereits vor mehr
als 150 Jahren verstanden, mit äußerst empfindlichen und zudem stark toxischen Substanzen
umzugehen. Dabei ergibt sich jedoch die legitime Fragestellung, warum taten sie dies?
Warum forschten sie an Substanzen, die extrem penetrant stinken oder sich an Luft spontan
entzünden? Keinesfalls ist das Forschungsinteresse an Molekülen mit solch extremen
Eigenschaften nur auf Beispiele beschränkt, die sich vor langer Zeit ereigneten. In der
chemischen Literatur finden sich jährlich zahlreiche Artikel, in denen Moleküle zwar nur
flüchtig existieren, ihre kurze Lebensdauer jedoch zu Beifall in der Fachwelt führt. Es finden
sich Moleküle, die hoch toxisch sind oder zu spontanen Explosionen führen. Dazu zählt
beispielsweise das von dem Nobelpreisträger Sir Geoffrey Wilkinson 1976 publizierte
[WMe6], das unvorhersehbar und heftig explodiert, sei es bei Kontakt mit Luft, im Vakuum
oder sogar unter einer „Schutzgasatmosphäre“ wie Argon oder Stickstoff.68 Solche Beispiele
sind selbstverständlich nicht nur auf organometallische oder im Allgemeinen auf
anorganische Substanzen beschränkt. In einem jüngstem Beispiel beschreibt Klaus Banert die
Eigenschaften seines synthetisierten Tetraazidomethans,69 C(N3)4 wie folgt: „Als reine
Substanz ist Tetraazidomethan extrem gefährlich und kann jederzeit – ohne ersichtlichen
Anlass – explodieren. Weniger als ein Tropfen der gaschromatographisch isolierten
Verbindung ist in der Lage, nicht nur die gläserne GC-Falle, sondern auch das Dewar-Gefäß
des Kühlbades komplett zu zerstören. Deshalb sollte die isolierte Substanz nur hinter einer
Schutzscheibe durch Aufdampfen eines Lösemittels, nicht jedoch manuell (mit Pipette oder
Spritze) verdünnt werden. Auch Lösungen von C(N3)4 können bei mechanischer Belastung
(Schraubverschluss) oder nach dem Abdampfen eines leicht flüchtigen Lösemittels, z.B. in
einer Pipette, explodieren.“ Schließlich sei zu beachten, dass es bei den anderen vorgestellten
Aziden ebenfalls zu Explosionen kommen kann. Warum stellen Moleküle mit solch extremen
Eigenschaften lohnenswerte Syntheseziele dar? Größtenteils besitzen sie zunächst keinen
ersichtlichen medizinischen oder technologischen Nutzen, dienen also keinesfalls dazu,
unsere Lebensverhältnisse zu verbessern. Eine Antwort gibt der 1981 mit dem Nobelpreis
geehrte Roald Hoffmann70: „Die Moleküle sind da; sie werden als faszinierend bis bizarr
wahrgenommen, also wollen Chemiker sie herstellen…Die Beweggründe dafür, instabile
Moleküle synthetisieren zu wollen, ist der Wunsch etwas zu tun, was nie zuvor gelang – und
dafür gerühmt zu werden. Ideen und Taten sind unser Aktienkapital…Es liegt in der Natur
7
2. Einleitung
„Because it is there“
des Menschen, dasjenige zu versuchen, das zuvor nie gelungen ist.“ Und schließlich merkt er
an, ist auf der anderen Seite eine Motivation, „die Wissenschaftler stets antreibt, die
Neugierde – ohne einen Gedanken an Belohnung und ohne Streben nach vermeintlichen
Ruhm.“
Diese Neugierde, der Drang zum Erwerb von Wissen, den nur ein grundlagenforschender
Chemiker vergleichsweise „frei“ nachgehen kann, führt letztlich mitunter zur Synthese von
instabilen, anormalen Molekülen. In solchen Verbindungen liegt in einigen Fällen ein Teil der
Atome in ungewöhnlichen Oxidationsstufen vor. Dazu zählt beispielsweise die Reihe der
niedervalenten Gruppe 13 Organlye des Typs EIR (R = sterisch anspruchsvolle und
elektronisch
stabilisierende
Gruppe,
z.B.
Cp*,
C(SiMe3)3,
t
Bu,
2,6-disubstituierte
Arylgruppen).
2.1 Synthese der niedervalenten Gruppe 13 Organyle EIR
Die Synthese von niedervalenten Gruppe 13 Verbindungen mit Hilfe von sterisch
anspruchsvollen und elektronisch stabilisierenden Gruppen R hat ihren Ursprung im Jahr
1957, als E.O. Fischer und H. Meister InCp71 und TlCp72 durch direkte Reaktion der
Halogenide mit Mg(C5H5)2 herstellten. Weitere entsprechende InI-/TlI-Organyle sind leicht
durch Salzmethatesereaktionen von InCl bzw. TlCl mit Alkyltransferreagenzien, wie
Lithiumalkylen, zugänglich.73, 74 Im Jahr 1991 gelang Schnöckel die Synthese von AlCp* aus
AlCl und MgCp*2, der ersten stabilen AlI-Organyl-Verbindung.75 Über die Darstellung der
ersten niedervalenten Gallium-Organyle, darunter GaCp* und verschiedene GaCp*-Derivate,
berichtete 1993 ebenfalls Schnöckel.76 In diesen frühen Arbeiten zeigte sich schnell, dass die
verwendeten 6π-Elektronen-Cyclopentadienid-Anionen (Cp, Cp*, Cp(SiMe3)3, Cp(Benzyl)5)
sich offensichtlich als sterisch anspruchsvolle Elektronendonoren erweisen, die sehr gut zur
Stabilisierung von Metallen in niedrigen Oxidationsstufen geeignet sind. Dies sind jedoch
nicht die einzigen organischen Gruppen, die zur Stabilisierung der niedervalenten Gruppe 13
Spezies dienen können. Die in den Folgejahren von Roesky und Jutzi neu entwickelten
Darstellungsverfahren für AlCp*77 und GaCp*78 durch reduktive Dehalogenierung von
Cp*AlCl2 und Cp*GaI2 in Gegenwart von Kalium lieferten indirekt auch einen Zugang zu
Alkyl-substituierten EIR-Verbindungen (E = Al-In, R = C(SiMe3)3, CH(SiMe3)2, Si(SiMe3)3,
etc.).74,
79-82
Während die oben genannten Gruppe 13 Organyle im Festkörper als Tetramer
bzw. Hexamer vorliegen, synthetisierten Power und Roesky unter Verwendung von sterisch
sehr
anspruchsvollen
organischen
Gruppen,
wie
β-Diketiminaten
(DDP
=
28
Synthese der niedervalenten Gruppe 13 Organyle EIR
2. Einleitung
Diisopropylphenylamino-4-diisopropylphenylimino-2-penten)83-89 oder substituierten Arenen
(Mes* = 2,6-Bis(2,4,6-triisopropylphenyl)phenyl)39, 90-95 die ersten monomeren EIR-Spezies.
Da EIR Verbindungen am sp-hybridisierten Element E zwei unbesetzte p-Orbitale besitzen,
sind sie isolobal zu CO und PR3 (R = Alkyl, Aryl) und können somit auch systematisch als
metalloide, "exotische" Liganden in der Koordinationschemie eingesetzt werden.
π∗
px
py
R
E
O
C
Abbildung 9.1. Isolobalanalogie von EIR und CO
Das Potential als Liganden gegenüber Übergangsmetallen haben die niedrigkoordinierten
Erdmetallverbindungen EIR (E = Al, Ga, In) auch ihrer Stabilität und der damit verbundenen
leichten Handhabbarkeit zu verdanken, da sie im Gegensatz zu den verwandten BorIOrganylen96-101 im freien, ungebundenen Zustand isolierbar sind. Für ein tiefes Verständnis
ihrer Ligandeigenschaften ist es absolut notwendig die Grenzorbitale der EIR Verbindungen
sowie deren Wechselwirkungen mit Übergangsmetallorbitalen entsprechender Symmetrie
genauer zu betrachten.
2.2 Ligandeigenschaften von EIR im freien und gebundenen Zustand
Die niedervalenten Gruppe 13 Organyle EIR besitzen formal ein freies Elektronenpaar in
einem σ-Typ Orbital (HOMO), sowie zwei unbesetzte p-Orbitale (LUMO), die senkrecht zur
E-C Bindungsachse liegen. Daher kann der elektronische Grundzustand als SingulettGrundzustand beschrieben werden, wodurch sich insgesamt die bereits oben erwähnte und in
Abbildung 9.1 gezeigte Isolobalität zu CO ergibt.38, 102, 103 Obwohl die Grenzorbitale von EIR
nicht isolobal zu denen der Singulett-Carbene sind, werden sie häufig in Anlehnung an die
höheren Homologen der 14. Gruppe MR2 (Silylene, Germylene und Stannylene) als
carbenoid bezeichnet, zumal Gemeinsamkeiten in der Chemie beider Spezies vorhanden sind.
Im Jahr 2000 untersuchten Frenking und Mitarbeiter die Bindungsverhältnisse in Komplexen
9
2. Einleitung
Ligandeigenschaften von EIR im freien und gebundenen Zustand
des Typs [LnM-ER] und [M(ER)4] mit verschiedenen organischen Gruppen R (R = Cp, Ph,
N(SiH3)2, Me) und d-Metallzentren M (M = Fe, W, Ni, Pd, Pt) für alle Gruppe 13 Elemente.43,
104
. Abbildung 10.1 zeigt das resultierende Bindungsmodell für die M-EIR Bindung.
R
σ
E
M
π
R
E
M
π'
R
M
E
q(-)
R
E
q(+)
M
Abbildung 10.1. Wechselwirkungen in M-EIR Komplexen
Ein Hauptcharakteristikum der quantenchemischen Analyse ist, dass M-EIR Bindungen stark
sind (z. B. Ni(ECH3)4 : 92 - 36 kcal/mol, Fe(ECH3)5 : 106 - 53 kcal/mol), wobei ihr
Hauptanteil aus Coulomb-Wechselwirkungen resultiert. Aufgrund der sehr unterschiedlichen
Elektronegativitäten der Metallatome M und E ist die M-E-Bindung (Eδ+-Mδ-) vorwiegend
ionisch (kovalente Bindungsordnung < 1). Im Gegensatz zu klassischen Werner-Typ
Komplexen trägt dabei das Gruppe 13 Metallzentrum stets eine positive Partialladung und die
Übergangsmetall-Zentren eine negative Partialladung. Das HOMO des Gruppe 13 Liganden
kann mit dem d(z2)-Orbital des entsprechenden Übergangsmetalls wechselwirken, wodurch
eine σ-Hinbindung resultiert. Der „lone-pair“ Charakter des HOMOs wurde durch Cowley
und Mitarbeiter sowohl rechnerisch als auch experimentell anhand der Synthese der LewisSäure-Base Addukte [Cp*Al→EPhF3] (E = B, Al) bestätigt.103 Aufgrund des steigenden sAnteils sowie der ungünstigeren Hybridisierung wird das freie Elektronenpaar am Gruppe 13
Metallzentrum mit steigender Ordnungszahl zunehmend inert, wodurch die M-E
Bindungsenergie mit steigender Ordnungszahl sinkt. Die freien p-Orbitale stehen prinzipiell
für eine M→EIR π-Rückbindung zur Verfügung. Dabei ist zu beachten, dass die π10
2. Einleitung
Ligandeigenschaften von EIR im freien und gebundenen Zustand
Rückbindungsfähigkeit zum einen aufgrund der geringeren Elektronegativtät der Gruppe 13
Metallatome und zum anderen aufgrund der konkurrierenden π-Donierung der organischen
Substituenten R in die freien p-Orbitale des Gruppe 13 Zentrums limitiert ist. So donieren
beispielsweise die in dieser Arbeit hauptsächlich verwendeten Cp* Substituenten ihrerseits
Elektronen aus ihren C5Me5-π-Orbitalen in beide p-Orbitale des Gruppe 13 Metalls, wodurch
ECp* Liganden bzw. im Allgemeinen EC5R5 Liganden eine deutlich geringere πRückbindungsfähigkeit aufweisen. Allerdings besteht bei Cp-substituierten EIR-Fragmenten
die Möglichkeit, dass sich durch Haptizitätsänderung des C5-Ringes von η5 nach η3 oder η1
die π-Akzeptorfähigkeit erhöht.78 Im Allgemeinen sinkt also innerhalb der Gruppe die πRückbindung M→EIR in der Reihenfolge B > Al > Ga > In >Tl, wobei in Komplexen mit
Alkyl substituierten Gruppe 13 Metallzentren aufgrund der fehlenden π-Donor-Eigenschaften
des organischen Substituenten R starke Rückbindungen vom Übergangsmetall in die leeren pOrbitale des Ligatoratoms vorliegen können.
Seit der ersten Stabilisierung niedervalenter Gruppe 13 Metallzentren durch sterisch
anspruchsvolle und elektronisch stabilisierende Gruppen R, konnte eine Vielzahl von M-EIR
Komplexen hergestellt und anaylsiert werden. Aufgrund der großen Anzahl an Arbeiten in
den letzten 15 Jahren, die sowohl in präparativen als auch in bindungstheoretischen Bereichen
in der EIR-Ligandchemie entstanden sind, beschränkt sich das folgende Kapitel ausschließlich
auf ausgewählte Syntheseprinzipien von M-ECp* Komplexen.
2.3 Synthesen, Strukturen und Reaktivitäten von M-EIR Verbindungen
2.3.1 Substitution labiler Liganden
Aufgrund der Isolobalität von EIR Liganden zu CO, beschäftigten sich anfängliche Arbeiten
zunächst mit dem Reaktionsverhalten von EIR Verbindungen gegenüber ÜbergangsmetallCarbonyl-Systemen und dabei insbesondere gegenüber homoleptischen ÜbergangsmetallCarbonyl-Komplexen. Dabei lag der Fokus zunächst auf der Untersuchung allgemeiner
Eigenschaften dieser neuen Ligandenklasse. So entstehen die Komplexe [Mn2(CO)8{μ2InC(SiMe3)3}2],105 [Co2(CO)6(μ2-ECp*)2] (E = Al,106 Ga78) und [Ni4(μ2-GaCp*)4(CO)6]78
durch Umsetzung der jeweiligen homoleptischen Metallcarbonyle mit EIR. Fortführende
Arbeiten beschäftigten sich mit Ligandensubstitutionen an heteroleptischen CarbonylKomplexen. Als Beispiele dienen hier die Substitutionen von Olefinliganden wie Cycloocten,
Norbornadien,
Cyclooctatetraen
und
Cycloheptatrien,
die
zu
den
Verbindungen
11
2. Einleitung
Substitution labiler Liganden
[Cr(CO)5(ECp*)]
(E
In(C(SiMe3)3)}2]110
=
Al,107
Ga,78,
In108),
[Fe(CO)4(GaCp*)]78
und
[(GaCp*)2Mo(CO)4],109
führen.
Weitere
[Fe2(CO)6{μ2Beispiele
von
Ligandenaustauschreaktionen in heteroleptischen Übergangsmetall-Carbonyl-Komplexen
stellen die Umsetzungen der Acetonitril-haltigen Verbindungen [fac-(RCN)3M(CO)3] (R=Me,
M=Mo; R=Et, M=W) mit GaCp* dar, bei denen die monomeren Komplexe [fac(GaCp*)3M(CO)3] (M=Mo, W) entstehen.111 Der Mo-Komplex [fac-(GaCp*)3Mo(CO)3] ist
hierbei von besonderem Interesse, da er als molekularer Baustein in der Synthese des
dinuklearen Komplexes [Mo2(CO)6{μ2-(GaCp*)}3] aus [fac-(GaCp*)3Mo(CO)3] und [fac(MeCN)3Mo(CO)3] dient.
(RCN)3M(CO)3
GaCp*
GaCp*
M
GaCp*
CO
CO
CO
GaCp*
- 3 RCN
CO
Cp*
Ga
CO
Mo Mo CO
CO
CO
Ga Ga
Cp*
Cp*
(MeCN)3Mo(CO)3
- 3 MeCN
CO
M=Mo, W
Schema 12.1. Synthese von [Mo2(CO)6{μ2-(GaCp*)}3] durch Verwendung des molekularen
Bausteins [fac-(GaCp*)3M(CO)3]
Eine interessante strukturelle Eigenschaft weisen die Additionsprodukte [Cp(CO)2M(μGaCp*)]2 (M=Mo, M=W) auf, welche aus den Umsetzungen von GaCp* mit den Dimeren
Komplexen [CpM(CO)2]2 (M = Mo, W) entstehen.111 Hier tritt infolge der sterischen
Überladung in den Produkten eine Verschiebung der Haptizität von η5 nach η1 der an den GaZentren gebundenen Cp*-Einheiten auf (siehe Schema 12.2).
OC
OC
OC
M
2 GaCp*
M
CO
CO
(M = W)
OC
Ga
M
M
Ga
CO
CO
(M = Mo, W)
Schema 12.2. Sterische Überladung in [Cp(CO)2M(μ-GaCp*)]2 (M=Mo, M=W) führt zu
einer Verschiebung der Haptizität der Cp* Gruppen
12
2. Einleitung
Substitution labiler Liganden
Die gezeigten Beispiele belegen, dass bei der Substitution der starken π-rückbindenden COLiganden durch die starken σ-Donoren ECp* ein intrinsisches Problem auftritt. Wird ein CO
Ligand durch einen ECp* Liganden ersetzt, verstärkt dies die π-Rückbindung des
Übergangsmetallzentrums zu den verbleibenden CO-Gruppen, wodurch ein weiterer
Austausch der übrigen CO-Liganden erschwert bzw. unmöglich wird. Möchte man daher
beispielsweise homoleptische [Mx(ECp*)y] Komplexe synthetisieren, muss man auf den
Einsatz Carbonyl-haltiger Präkursoren verzichten und auf die Substitution labil gebundener
Liganden wie Olefine (COD (COD = Cyclooctadien), NDB (NBD = Norbornadien), C2H4)
zurückgreifen. So lassen sich beispielsweise die COD Liganden in [Ni(COD)2] und
[Pt(COD)2] leicht durch ECp* oder E(C(SiMe3)3) substituieren, was zur Bildung der
tetraedrischen, homoleptischen Komplexe [M(EIR)4] (M = Ni, Pt) führt.38, 109, 112
ER
[M(COD)2]
+ 4 EI R
M
RE
M = Ni, Pt
R = Cp*, C(SiMe3)3
ER
ER
Schema 13.1. Synthese der homoleptischen Komplexe [M(EIR)4] durch Substitution des
labilen Olefinliganden COD
In der Reaktion von [Pd(tmeda)(CH3)2] (tmeda = N,N,N’,N’-tetramethylethylendiamin) mit
ECp* entsteht der analoge homoleptische Pd Komplex [Pd(ECp*)4].113 Bei dieser Reaktion
wird eine weitere Eigenschaft der niedervalenten Gruppe 13 Liganden beobachtet. In der
Synthese findet eine Wanderung der Methylgruppen vom Palladium zum ECp* Liganden
statt. Offensichtlich handelt es sich hier also um eine Redoxreaktion, bei der das
Übergangsmetall Palladium durch den Gruppe 13 Liganden reduziert wird.
Die homoleptischen Komplexe [M(EIR)4] sind aufgrund der stark gebundenen ECp*
Liganden kinetisch inert, so dass weder Substitutionsreaktionen mit E´Cp*, PR3 oder CO
noch ein Koaleszenzverhalten von [M(EIR)4] mit freien EIR im NMR beobachtet wurde. Im
Gegensatz zu den analogen Phosphanverbindungen dienen die Verbindungen [M(GaCp*)4]
(M = Pt, Pd) als molekulare Bausteine zur Synthese von zweikernigen Komplexen des Typs
[MPt(GaCp*)2(μ2-GaCp*)3] (M = Pt, Pd), in denen neben terminalen auch verbrückende
GaCp* Liganden vorhanden sind.113 Diese Verbindungen entstehen bei den Umsetzungen von
[M(GaCp*)4] mit einem molaren Äquivalent [Pt(COD)2] unter anschließender Zugabe von
13
2. Einleitung
Substitution labiler Liganden
GaCp*. Als Pt-Quelle für den zweikernigen Komplex [Pt2(GaCp*)2(μ2-GaCp*)3] dient
ebenfalls [Pt(η2-C2H4)3], welches durch Umsetzung mit überschüssigem GaCp* selektiv zu
[Pt2(GaCp*)2(μ2-GaCp*)3] reagiert. Diese mehrkernigen, homoleptischen Cluster sind die
ersten Beispiele, für die sich weder in der Komplexchemie von Carbonyl- noch von
Phosphanliganden entsprechende Strukturanaloga finden lassen.
xs. GaCp*
Cp*
Ga
GaCp*
M GaCp*
*CpGa
GaCp*
[Pt(COD)2]
Cp*
Ga
GaCp*
*CpGa M
Pt
Ga
Ga Cp*
Cp*
*CpGa M
Pt GaCp*
Ga
Ga Cp*
Cp*
M = Pt, Pd
Schema 14.1. [M(GaCp*)4] als molekularer Bausteine in der Synthese von zweikernigen
Komplexen des Typs [MPt(GaCp*)2(μ2-GaCp*)3]
Die Darstellung eines dreikernigen, gemischtmetallischen Clusters gelang beispielsweise in
der Reaktion von [Pd2(dvds)3] (dvds = 1,3 divinyl-1,1,3,3-tetramethyldisiloxan) mit GaCp* in
Toluol. Bei Raumtemperatur konnte in hohen Ausbeuten der Komplex [Pd3(GaCp*)4(μ²GaCp*)4] isoliert werden, während unter leicht veränderten Reaktionsbedingungen (in Hexan,
bei -30°C) der zweikernige Komplex [Pd2(GaCp*)2(μ2-GaCp*)3] als Hauptprodukt resultiert.
Somit handelt es sich hierbei um ein Beispiel für ein kinetisch kontrolliertes
Clusterwachstum.
Cp*
Ga
Hexan
- 30°C
*CpGa
Pd
Pd
Ga
Cp*
Pd 2 (dvds)3
GaCp*
Ga
Cp*
GaCp*
- 3 dvds
*CpGa
Toluol
RT
Pd
*CpGa
Cp*
Ga
Ga
Cp*
Cp*
Ga
GaCp*
Pd
Pd
Ga
Cp*
GaCp*
Schema 14.2. Synthesen von [Pd2(GaCp*)2(μ2-GaCp*)3] und [Pd3(GaCp*)4(μ²-GaCp*)4]
14
2. Einleitung
Substitution labiler Liganden
Als ein erster Vertreter eines GaCp* Adduktes eines stärker elektrophilen Metallzentrums
konnte die Verbindung [Zn(GaCp*)4][(BArF)2] in der Reaktion von ZnMe2 mit
[H(OEt2)2][BArF] unter anschließender Zugabe von GaCp* synthetisiert werden.114 Der
homoleptische Komplex enthält ein [Zn(GaCp*)4]2+ Kation, welches isostrukturell und
isoelektronisch zu den bereits oben beschriebenen Vertretern der [M(GaCp*)4] (M = Ni, Pd,
Pt) Familie ist. Die Fähigkeit von GaCp* Übergangsmetallkationen in Form von klassischen
Werner-artigen Koordinationsverbindungen zu stabilisieren, wurde jüngst in den Synthesen
der homoleptischen Komplexe [M(GaCp*)4]+ (M = Cu, Ag) und [Rh(GaCp*)5]+ eindrucksvoll
bestätigt.115, 116
GaCp*
[{Rh(COE)2(μ2-(O3SCF3)}2]
GaCp*
*CpGa
Rh
[CF3SO3]-
GaCp*
GaCp*
GaCp*
[Anion]-
GaCp*
[Ag(BPh4)]
GaCp*
M
*CpGa
[Cu(CH3CN)4][BArF]
GaCp*
GaCp*
2+
ZnMe2
2[BArF]-
GaCp*
1. [H(OEt)2][BArF]
2. GaCp*
Zn
*CpGa
GaCp*
GaCp*
Schema 15.1. Synthesen der homoleptischen Kationen [Rh(GaCp*)5]+, [M(GaCp*)4]+ (M =
Cu, Ag) und [Zn(GaCp*)4]2+
2.3.2 Reaktivitäten der homoleptischen Komplexe [Ma(ECp*)b] (M = Pd, Pt)
Bis heute ist wenig über die chemische Reaktivität der M-E Komplexe bekannt, da M-E
Bindungen insbesondere für E = Al, Ga vergleichsweise stark sind und der sterische Anspruch
der EIR Liganden zudem relativ groß ist. So sind beispielsweise die sterisch und elektronisch
gesättigten monomeren Komplexe [M(ECp*)4] kinetisch vollkommen inert.117 Im Gegensatz
15
2. Einleitung
Reaktivitäten der homoleptischen Komplexe [Ma(ECp*)b]
dazu reagieren die mehrkernigen, ungesättigten Komplexe [Ma(ER)b] (b > a > 1) mit einer
Reihe von Liganden (AlCp*, CO, Phosphine, Isonitrile) unter Bildung von zwei- und
dreikernigen Substitutionsprodukten (siehe Schema 16.1).118 In Übereinstimmung mit
quantenchemischen Rechnungen, die belegen, dass eine Substitution von GaCp* oder InCp*
durch AlCp* im Falle der homoleptischen Komplexe thermodynamisch begünstigt ist,43
führen die Umsetzungen von [M2(GaCp*)2(μ2-GaCp*)3] (M = Pd, Pt) mit AlCp* zu der
trimetallischen Verbindung [Pt2(GaCp*)2(μ2-AlCp*)3] sowie zum komplett substituierten
Produkt [Pd2(AlCp*)2(μ2-AlCp*)3].
Cp*
Ga
OC
Cp*
Ga
t
BuN C
Pt
Pt
Ga
Cp*
Pt
Pt
Ga
Cp*
t
CN Bu
CO
Ga
Cp*
Cp*
Al
Ga
Cp*
CNtBu
*CpAl
CO
Cp*
Ga
Ph3P
M
Cp*Al
Pt
Ga
Cp*
PPh3
GaCp*
Pd
Pd
Al
Cp*
M = Pd, Pt
Cp*Al
Cp*
Al
dppe
PR3
Ph2
P
P
Ph2
*CpGa
R3P
Pd
Ga
Cp*
Pt
Pt
Al
Cp*
Cp*
Ga
M
P
Ph2
Al
Cp*
M2(GaCp*)5
Ga
Cp*
Ph2
P
AlCp*
Pd
GaCp*
Al
Cp*
PR3
Ga
Cp*
Schema 16.1. Reaktivität von [M2(GaCp*)2(μ2-GaCp*)3]
Bei der Reaktion von [M2(GaCp*)2(μ2-GaCp*)3] mit PPh3 entstehen unter Erhalt des
Molekülgerüsts die mono- und disubstituierten Komplexe [MPt(GaCp*)(PPh3)(μ2-GaCp*)3]
und [Pd2(PPh3)2(μ2-GaCp*)3]. Aufgrund des in Lösung nachgewiesenen fluktionalen
Verhaltens bei dem sowohl die verbrückenden als auch die terminalen Liganden schnell
austauschen, wird ein dissoziativer Mechanismus der Brückenliganden als wahrscheinlich
angesehen, bei dem ein ungesättigtes Metallzentrum als Intermediat entsteht.
Im Gegensatz zu den Reaktionen der Komplexe [M2(GaCp*)2(μ2-GaCp*)3] mit PR3 (R = Me,
Ph) werden bei den analogen Umsetzungen mit dem chelatisierenden Phosphanligand dppe
16
2. Einleitung
Reaktivitäten der homoleptischen Komplexe [Ma(ECp*)b]
(dppe = bis(diphenylphosphinoethan)) unter Bildung der monomeren Komplexe [M(dppe)2]
(M = Pd, Pt) alle GaCp* Einheiten substituiert.
Die Reaktion von [Pt2(GaCp*)2(μ2-GaCp*)3] mit den starken π-Akzeptoren CNtBu bzw. CO
liefert die zweifach substituierten Produkte [Pt2(CNtBu)2(μ2-GaCp*)3] und [Pt2(CO)2(μ2GaCp*)3], in denen die GaCp* Liganden in verbrückenden Positionen verbleiben.
Während die Molekülstruktur in allen Substitutionsreaktionen der zweikernigen Komplexe
[M2(GaCp*)2(μ2-GaCp*)3] unverändert bleibt, findet bei der Umsetzung von [Pd3(InCp*)8]
mit den Phosphanen PPh3 bzw. dppe (dppe = ddiphenylphosphinoethan) eine Umlagerung des
linearen Pd3 Kerns statt. Dabei entstehen die dreieckigen Pd3 Cluster [Pd3(InCp*)3(PPh3)3]
und
[Pd3(InCp*)3(dppe)2].
In
beiden
Fällen
liegt
eine
trigonale
bipyramidale
Molekülgeometrie vor, in welcher zwei InCp* Liganden oberhalb und unterhalb der Pd3
Dreiecksflächen liegen. Desweiteren werden die Pd Atome abgesättigt durch einen weiteren
verbrückenden InCp* Liganden in der Pd3 Ebene, sowie durch terminal gebundene
Phosphanliganden.
Cp*
In
3 PPh3
- 5 InCp*
PPh3
Pd
Ph3P
Pd
*CpIn
Pd
PPh3
In
Cp*
Pd3(InCp*)8
2 dppe
- 5 InCp*
Ph2
P
Cp*
In
Ph2
P
Pd
Pd
P
Ph2
*CpIn
Pd
P
Ph2
In
Cp*
Schema 17.1. Synthesen von [Pd3(InCp*)3(PPh3)3] und [Pd3(InCp*)3(dppe)2]
2.3.3 Bindungsaktivierungen durch M(ECp*)n
Wie theoretische Rechnungen gezeigt haben, resultiert der größte Anteil der Bindungsenergie
von ECp* an Übergangsmetallzentren aus einer elektrostatischen Wechselwirkung zwischen
dem partiell negativ geladenen Übergangsmetallatom und dem partiell positiv geladenen
17
2. Einleitung
Bindungsaktivierungen durch M(ECp*)n
Gruppe 13 Metall.38, 43, 104, 119-121 Dabei nimmt die Bindungsstärke in der Reihenfolge Al > Ga
> In ab. Die Fähigkeit dieser exotischen Liganden, die Elektronendichte am Metallatom
drastisch zu erhöhen und gleichzeitig sehr elektrophile Zentren zur Koordination von schwach
polaren Substraten zu schaffen, sollte eine hervorragende Basis für Aktivierungsreaktionen
darstellen. Bei der Reaktion von [Ni(COD)2] mit vier Äquivalenten AlCp* in Benzol bildet
sich nicht der erwartete Komplex [Ni(AlCp*)4], sondern es entsteht unter C-H-Aktivierung
eines Benzolmoleküls [Ni(AlCp*)3(H)(Cp*AlPh)].117 Dabei verläuft die Aktivierung des
Benzolmoleküls vermutlich an dem ungesättigten Fragment [Ni(AlCp*)3] über ein
Schlüsselintermediat [(AlCp*)3Ni(H)(C6H5)]. Die Triebkraft der Reaktion ergibt sich aus der
nachfolgenden Wanderung der Phenylgruppe an einen AlCp* Liganden unter Oxidation des
Aluminiums und Ausbildung einer starken Al-C Bindung sowie Koordination des vierten
Äquivalents AlCp*. Das 16e Fragment [Ni(AlCp*)3] lässt sich auch mit anderen geeigneten
Reaktionspartnern wie HSiEt3 abfangen. So entsteht in Gegenwart von HSiEt3 unter Si-H
Aktivierung der Hydrosilyl-Komplex [Ni(AlCp*)3(H)(SiEt3)], in welchem der Hydridligand
in einer terminalen Position vorliegt.
AlCp*
1/4 [{AlCp*}4]
n-hexan
Ni
*CpAl
AlCp*
AlCp*
1/4 [{AlCp*}4]
n-hexan
- HSiEt3,
AlCp*
[Ni(COD)2]
3/4 [{AlCp*}4]
- 2 COD
[Ni(AlCp*)3]
HSiEt3
H Ni
Et3Si
C 6H 6
- HSiEt3
AlCp*
AlCp*
Partielle
Zersetzung
AlCp*
1/4 [{AlCp*}4]
C 6H 6
H Ni
AlCp*
*CpAl
AlCp*
Schema 18.1. C-H und Si-H-Aktivierungen an [Ni(AlCp*)3]
Ein weiteres Beispiel einer C-H-Bindungsaktivierung wurde in der Reaktion von [(η6C6H5CH3)Fe(η4-C4H8)] mit AlCp* beobachtet.122 Bei dieser Reaktion entstehen C-H
18
2. Einleitung
Bindungsaktivierungen durch M(ECp*)n
aktivierte Isomere von [Fe(AlCp*)5], in denen das zentrale Eisenatom verzerrt trigonalbipyramidal umgeben ist. Dabei verknüpfen zwei Methylengruppen aus zwei Cp*-Methyl
Gruppen insgesamt drei AlCp*-Liganden und bilden so das ungewöhnliche tridentate
Chelatsystem
{Cp*Al-CH2(C5Me4)Al-CH2(C5Me4)Al}.
Die
Hydride
nehmen
dabei
verbückende Positionen zwischen den Eisen- und Aluminium-Zentren ein.
Bindungsaktivierungen sind nicht nur auf AlCp* beinhaltende Systeme beschränkt, was die
Umsetzung von [Cp*Rh(CH3)2L] (L = Dimethylsulfoxid, Pyridin) mit GaCp* zeigt.123, 124 Bei
dieser Reaktion entsteht zunächst das isolierbare Intermediat [Cp*Rh(GaCp*)(CH3)2],
welches unter Aktivierung einer C-C-Bindung sowohl in Lösung als auch im Festkörper zur
zwitterionischen Rhodenocenium Spezies [Cp*Rh{(η5-C5Me4)Ga(CH3)3}] weiterreagiert.
Anhand
NMR
spektroskopischer
Kinetikstudien
und
mit
Hilfe
von
detaillierten
Dichtefunktionalrechnungen konnte ein vollständiger Mechanismus vorgestellt werden,
welcher die Aktivierung einer C-C-Bindung an dem Hauptgruppenelement Gallium, assistiert
durch das Übergangsmetallzentrum Rhodium, beinhaltet. Dabei findet im ersten Schritt die
Substitution von Pyridin bzw. dmso im Ausgangskomplex gegen ein Molekül GaCp* unter
Bildung von [Cp*Rh(GaCp*)(CH3)2] statt. Im nachfolgenden Schritt wandern die Rh-CH3
Gruppen
nacheinander
auf
das
Galliumzentrum.
In
dem
dabei
entstehenden
Schlüsselintermediat koordiniert eine Cp*Ga(CH3)2-Einheit als neutrales Dien an ein
Rh(I)Cp* Fragment. Die eigentliche C-C-Aktivierungsreaktion unter Ausbildung des
Endprodukts ist ausgehend von diesem Schlüsselintermediat mit 17.4 kcal/mol energetisch
sehr günstig.
Diese Reaktion ist das erste Beispiel einer C-C-Bindungsaktivierung an einem
Hauptgruppenmetall in Lösung bei milden Bedingungen. Experimentelle und theoretische
Befunde lassen darauf schließen, das die Aktivierung topologisch gesehen klar am
Galliumatom verläuft, der elektronische Beitrag jedoch auf einen kooperativen Effekt
zwischen dem elektronenreichen Rhodiumatom und dem elektrophilen Galliumzentrum
zurückzuführen ist.
19
2. Einleitung
Bindungsaktivierungen durch M(ECp*)n
Rh
Ga
CH3
CH3
Rh
CH3
Ga
CH3
Rh
Rh
Ga(CH3)3
Ga
CH3
CH
3
Schema 20.1. Reaktionsabfolge für die C-C-Bindungsaktivierung in [Cp*Rh(GaCp*)(CH3)2]
2.3.4 Insertionsreaktionen von ECp* in Metall-Halogenid Bindungen
Während C-H-Aktivierungen bisher auf Übergansmetallkomplexe mit AlCp* Liganden
beschränkt sind, gibt es zahlreiche Beispiele für Insertionsreaktionen von ECp* (E = Ga, In)
in Übergangsmetall-Halogenid Bindungen. In Abhängigkeit von der Stöchiometrie sowie von
den Reaktionsbedingungen bilden sich eine Vielfalt von Produkten in den Reaktionen von
ECp* (E = Ga, In) mit den zueinander isolobalen d6-Komplexen [{Cp*RhCl2}2]125,
126
und
[{(p-cymol)RuCl2}2]127. Wird beispielsweise [{RhCp*Cl2}2] mit sechs Äquivalenten ECp* in
Toluol bei Raumtemperatur umgesetzt, führt dies zum monomeren Insertionsprodukt
[RhCp*(ECp*)3Cl2]. Die Reaktion von [{RhCp*Cl2}2] mit nur einem Äquivalent ECp* führt
zu dem Rh(II)-Dimer [{RhCp*Cl}2], welches auch durch Umsetzung von [{RhCp*Cl2}2] mit
Na/Hg oder elementaren Gallium zugänglich ist. Wird [{Cp*RhCl2}2] mit drei Äquivalenten
InCp* umgesetzt, entsteht das Salz [Cp*2Rh][Cp*Rh(InCp*){In2Cl4(μ2-Cp*)}], in welchem
erstmals ein Cp*-Ring in verbrückender Position beobachtet werden konnte. Unter
Verwendung von drei Äquivalenten GaCp* entsteht nicht die analoge Verbindung
[Cp*2Rh][Cp*Rh(GaCp*){Ga2Cl4(μ2-Cp*)}], sondern der strukturell eher klassischen PianoStool-Komplex [Cp*Rh(GaCp*)2(GaCl3)]. In den genannten Beispielen findet eine Reduktion
des Rh(III) Zentrum in [RhCp*Cl2]2 durch ECp* zu einem d8-Rh(I) Fragment statt, welches
dann durch Koordination von ECp* und Bildung eines Lewis-Säure-Base-Addukts stabilisiert
20
2. Einleitung
Insertionsreaktionen von ECp* in Metall-Halogenid Bindungen
wird. Die Umsetzungen des zu [RhCp*Cl2]2 isolobalen [{(p-cymol)RuCl2}2] mit ECp* liefern
ähnliche Insertionsprodukte.
Rh2(GaCp*)2Cl2
Rh2(GaCp*)4(GaCl3)2
Rh2(GaCp*)3
GaCp*
Rh
Rh
Cl Cl
1 ECp*, toluol, 80°C
- ECp*Cl2
-
[(Cp*)2Rh] +
Rh
Cp*In
Cl2In
toluol, 75°C
Cl
Cl
3 InCp*
InCl2
Rh
Cl
Rh
6 ECp*
toluol, RT
Cl
Rh
*CpE
- GaCp*Cl2
ECp*
ECp*Cl2
GaCl3
toluol, RT
3 GaCp*, toluol, 80°C
Rh
Spuren
*CpGa
GaCp*
GaCl3
Schema 21.1. Insertionsreaktionen bei Umsetzung von [{RhCp*Cl2}2] mit ECp*]
Eine weiteres interessantes Produkt bildet sich in der Reaktion von [Ru(PPh3)3Cl2] mit sechs
Äquivalenten GaCp*. Dabei entsteht [Ru(GaCp*)6Cl2] in typischen Ausbeuten von weniger
als 50 %. Unter Verwendung von [Ru(dmso)4Cl2] (dmso = Dimethylsulfoxid) als Quelle für
ein RuCl2 Fragment werden reproduzierbare Ausbeuten von 85% erhalten.128 Das
Rutheniumzentrum in diesem Molekül ist in einer verzerrt oktaedrischen Koordinationssphäre
von sechs GaCp* Liganden umgegeben. Eines der beiden Chlorid-Ionen befindet sich
terminal an einem der GaCp* Liganden, während das zweite Halogenid-Ion zwischen zwei
Ga Zentren verbrückend vorliegt. Drei Cp* Gruppen der GaCp* Liganden befinden sich in
einem η5 Modus, während die verbleibenden drei Einheiten η1 an das Gruppe 13 Metall
binden. Dies ist auf die sterische Überladung in diesem Molekül zurückzuführen.
Interessanterweise befindet sich in der Elementarzelle der Verbindung ein zweites,
kristallographisch unabhängiges Molekül, in welchem vier Cp* Einheiten η1 und nur zwei
21
2. Einleitung
Insertionsreaktionen von ECp* in Metall-Halogenid Bindungen
Cp* Gruppen η5 gebunden sind. Die Flexibilität von koordinierten Cp* Gruppen insbesondere
an Hauptgruppenmetallzentren ist allgemein bekannt.125-127, 129 Die Anwesenheit dieser zwei
unabhängigen Moleküle in einer Elementarzelle belegt, dass die Energiehyperfläche für den
Austausch der Chloride zwischen den Ga Zentren vermutlich extrem flach ist. Dies führt zu
verschiedenen,
energetisch
sehr
ähnlichen
Isomeren,
welche
durch
sehr
kleine
Aktivierungsbarrieren voneinander getrennt sind. Diese Eigenschaft spiegelt sich in der
Lösungsstruktur der Verbindung wider, da ihr NMR Spektrum koaleszierende GaCp* Signale
bis -80 °C enthält. Ein analoges Verhalten wurde bereits zuvor im Falle der
Insertionsprodukte [(p-cymol)Ru(GaCp*)3Cl2] und [RhCp*(GaCp*)3Cl2] beobachtet.125-127
2.3.5 [Ga2Cp*][BArF] als Transferreagenz für „nacktes“ Ga+
Im Jahr 2006 untersuchte Beatrice Buchin im Rahmen ihrer Diplomarbeit Reaktionen von
GaCp* mit [M(NCCH3)6][(BArF)2] (M= V, Cr, Fe, Co, Ni), um auf diesem Weg kationische
Komplexe des Typs [M(ECp*)n]m+ herzustellen.128, 130 Dabei entstand in einigen Fällen, unter
einem redoxneutralen Cp* Transfer vom Ga auf das Übergangsmetall, [Ga2Cp*][BArF] als
Nebenprodukt. Setzt man GaCp* in Fluorbenzol gemäß Schema 23.1 mit einem halben
Äquivalent [H(OEt2)2][BArF] um, kann [Ga2Cp*][BArF] reproduzierbar in 85%iger Ausbeute
dargestellt werden.45 Das 1H-NMR-Spektrum einer Mischung von [Ga2Cp*][BArF] mit einem
molaren Äquivalent GaCp* in Fluorbenzol zeigt bei δ = 1.80 ppm koaleszierende Signale
beider Cp*-Einheiten. Dabei tauschen offensichtlich die Ga+-Ionen schnell zwischen den
GaCp* Einheiten aus, was sowohl auf eine vergleichsweise schwache Ga-Cp*-Bindung als
auch auf eine vollständige Dissoziation von [Ga2Cp*][BArF] in die Ionen schließen lässt. Im
Festkörper hat das Kation eine hochsymmetrische bipyramidale Doppelkegelstruktur. Dabei
befinden sich beide Ga Atome exakt über dem Zentrum des Cp* Ringes, wobei die beiden
Ga-Cp*centr. Abstände mit 2.228 und 2.237 Å deutlich größer sind als in freiem GaCp* (2.081
Å). Das [Ga2Cp*]+ Ion stellt eine stabilisierte Form eines in Lösung bis dahin nicht
bekannten, nahezu „nackten“ Ga+ dar.
22
[Ga2Cp*][BArF] als Transferreagenz für „nacktes“ Ga+
2. Einleitung
Ga
+ [BArF]-
1/2 [H(OEt2)2][BArF]
(C6H5F)
Ga
-
H
Ga
Schema 23.1. Synthese von [Ga2Cp*][BArF]
Die σ-Donor und π-Akzeptoreigenschaften der EIR Liganden hängen stark von der Art des
Substituenten R ab. GaCp* beispielsweise ist ein guter σ-Donor Ligand mit nur schwachen πAkzeptoreigenschaften, da das Cp* Fragment seine π-Elektronen in Richtung der freien pOrbitale des Gallium Zentrums doniert. Diese Konkurrenzsituation ist im Falle von
Alkylsubstituenten nicht vorhanden, so dass beispielsweise GaC(SiMe3)3 der bessere πAkzeptorligand darstellt.38 Daher ergab sich die Fragstellung, welche Ligandeigenschaften ein
terminal koordiniertes, substituentenfreies E+-Kation hätte. Bis zur Synthese von
[Ga2Cp*][BArF] waren nur Übergangsmetallkomplexe vom Typ [(LnM)aE] (a = 2-4) mit
nackten Gruppe 13 Atomen zwischen zwei oder mehr Metallzentren bekannt.30, 131-135 Zudem
handelt es sich hierbei ausschließlich um Carbonylmetallat-Fragmente, in denen das Gruppe
13 Metall E eher in der Oxidationsstufe +III als +I vorliegt. Eine Ausnahme stellen dabei die
„metallophilen“ Bindungen von Tl+ an d8- oder d10-Zentren wie PtII, Pt0 oder AuI in [LnMTl+] dar, die durch Dispersionswechselwirkungen und weitreichende Polarisationseffekte
ohne kovalente Beiträge erklärbar sind.136 Bei der Umsetzung von [Pt(GaCp*)4] mit einer
äquimolaren Menge an [Ga2Cp*][BArF] entsteht in 94% Ausbeute [GaPt(GaCp*)4][BArF].29,
44
Bei der entsprechenden Reaktion von [Pt(GaCp*)4] mit einer äquimolaren Menge der
Bønstedt-Säure [H(OEt2)2][BArF] bildet sich der Komplex [HPt(GaCp*)4][BArF] sowie die
zweikernige Spezies [(Cp*Ga)4Pt(μ-Ga)Pt(GaCp*)3H][(BArF)2], die einen verbrückenden
Ga+-Liganden zwischen zwei Platinzentren enthält. In entsprechender Weise liefert die
Umsetzung von [Pt(PPh3)4] mit dem In+-Transferfreagenz unter Abspaltung eines
Phosphanliganden den Komplex [InPt(PPh3)3][BArF]. In der Festkörperstruktur von
[GaPt(GaCp*)4][BArF] weist das Pt-Zentrum eine leicht verzerrte trigonal-bipyramidale
Koordinationsumgebung auf, in der das nackte Ga+ in axialer Position terminal an das
Übergangsmetall gebunden ist.
23
[Ga2Cp*][BArF] als Transferreagenz für „nacktes“ Ga+
2. Einleitung
GaCp*
Pt
*CpGa
+ [BArF]-
Ga
[Ga2Cp*][BArF]
GaCp*
GaCp*
*CpGa
Pt
GaCp*
GaCp*
GaCp*
[H(OEt2)2][BArF]
+ [BArF]-
H
*CpGa
Pt
GaCp*
GaCp*
GaCp*
*CpGa
+
*CpGa
GaCp*
Pt
Ga
GaCp*
H
Pt
GaCp*
2+
2 [BArF]-
GaCp*
GaCp*
Schema 24.1. Umsetzung von [Pt(GaCp*)4] mit [Ga2Cp*][BArF] und [H(OEt2)2][BArF]
Eine interessante Struktureigenschaft dieses Komplexes ist, dass es sich bei der Pt-Ga+
Bindung mit 2.459(1) Å trotz des geringsten sterischen Anspruches des Ga+-Kations um die
längste Bindung im Komplex handelt. DFT Rechnungen beschreiben die Pt-Ga+
Wechselwirkung als eine schwach polare Donor-Akzeptor Bindung, in der das Ga+ Ion als ein
starker σ- und π-Akzeptor ohne jegliche Donoreigenschaften fungiert. Somit ist das s-artige
freie Elektronenpaar am Ga+ sterisch und chemisch inaktiv, womit solche Systeme durchaus
als Metallanaloga von H+ beschrieben werden können. In neuesten Untersuchungen konnte
eine selektive, oxidative Abspaltung von Cp* unter Bildung von Fulvalen in der Reaktion von
[Pt(GaCp*)4] mit [Cp2Fe][BArF] beobachtet werden, die zum trinuklearen Cluster
[Pt3(Ga)(GaCp*)6][BArF] führt.49 Die Ein-Elektronen-Oxidation von [Pt(GaCp*)4], welche
die zwei leicht oxidierbare Metallzentren Pt(0) und Ga(I) enthält, führt somit eher selektiv zur
Oxidation des anionischen Cp* Liganden anstelle zur Oxidation der Metallzentren. Die
aufgezeigten oxidativen und protolytischen Abspaltungen von Cp* Gruppen koordinierender
GaCp* Liganden sowie der Einsatz von [Ga2Cp*][BArF] als nacktes Ga+ Tranferreagenz,
sollte auf diesem Gebiet schon in naher Zukunft zu neuen Produkten mit ungewöhnlichen
Koordinationsumgebungen führen.
24
[Ga2Cp*][BArF] als Transferreagenz für „nacktes“ Ga+
2. Einleitung
+
[BArF]-
Ga
Ga
F
Pt(GaCp*)4
+ [{FeCp2}{BAr }]
Ga
Ga
Pt
Pt
Ga
Pt
Ga
Ga
Schema 25.1. Oxidative Abspaltung von Cp* führt zu [Pt3(Ga)(GaCp*)6][BArF]
25
3. Ergebnisse
3. Ergebnisse
3.1
Reaktivität von ECp* (E = Al, Ga) gegenüber basischen d8-Metall-
Olefin-Komplexen
Die Chemie der niedervalenten Gruppe 13 Organyle EIR (E = Al, Ga, In; R = Alkyl, Aryl,
Cp*, Amide, β-Diketiminate, Amidinate, Guanidinate) ist ein wachsendes Gebiet in der
modernen anorganischen Chemie. Dabei liegt der Fokus insbesondere auf den
Koordinationseigenschaften
dieser
„exotischen“
30, 31, 33, 34, 37, 84, 85, 112, 137-142
Übergangsmetallzentren.
Liganden
gegenüber
Die einzigartige Bindungssituation in den
resultierenden Metall-Metall-Bindungen wurde in zahlreichen Publikationen beschrieben,
wobei hier hauptsächlich die außergewöhnlichen σ-Donoreigenschaften von EIR sowie der
hohe elektrostatische Charakter der M-E Bindung hervorzuheben ist.42,
104
Ein jüngstes
Highlight stellt der Komplex [η5-{(Me3Si)C5H4}3U−AlCp*] dar, da er die erste ActinidenAluminium Bindung enthält.37 Obwohl die EIR Ligandenklasse formal isolobal zu CO ist,
sind die Koordinationseigenschaften beider Ligandensysteme tatsächlich sehr verschieden. So
unterscheiden sich beispielsweise die Festkörperstrukturen der homoleptischen Cluster
[Ma(ECp*)b] (a = 2 - 6) für die d10 Metalle Ni, Pd und Pt in allen Fällen stark von den
Molekülstrukturen der entsprechenden homoleptischen Carbonyl-Cluster.118 Eine direkte
Substitution von CO durch ECp* ist aufgrund der steigenden π−Rückbindung der
Metallzentren bei Einführung des ECp* Liganden limitiert, wodurch die gewöhnliche
Syntheseroute zu homoleptischen Komplexen des Typs [Ma(ECp*)b] die Substitution von
labileren Olefin-Liganden beinhaltet.117,
118,
122
Nur unter Verwendung der sterisch
anspruchsvollen GaI Liganden Ga(DDP) oder [Ga{[N(Ar)C(H)]2}]− (Ar = C6H3iPr2-2,6)
konnten GaI Übergangsmetallkomplexe isoliert werden, die Olefine als Co-Liganden
beinhalten, wohingegen es im Fall von ECp* stets zu einer vollständigen Substitution kam.143145
Die
sehr
ungewöhnliche
Struktur
des
Ni3
Clusters
[{{μ2-Ga(DDP)}Ni(ethylen)}2Ni(μ2-CH=CH2)(H)], der durch C-H Aktivierung von Ethylen
gebildet wird und einen Vinyl-Liganden in verbrückender Position zu allen drei Nickelzentren
enthält, weist auf die interessante Strukturvielfalt von GaI Clustern in Anwesenheit von
Olefinen hin.143 Daher erschien es vielversprechend als Einstieg in die Dissertation die
Koordinationschemie von ECp* (E = Al, Ga) gegenüber Metall-Olefin-Komplexen zu
untersuchen. Als Ausgangssysteme für die Umsetzungen mit ECp* wurden die d826
Reaktivität von ECp* gegenüber basischen d8-Metall-Olefin-Komplexen
3. Ergebnisse
Übergangsmetallzentren Ru0 und RhI ausgewählt, da sie die Olefin-Liganden aufgrund ihrer
starken Basizität stark an sich binden und somit eine komplette Substitution verhindert
werden sollte. In der Tat reagiert beispielsweise [Ru(η4-butadiene)(PPh3)3] mit GaCp* zum
Substitutionsprodukt
[Ru(η4-butadiene)(PPh3)2(GaCp*)]
(1),
welches
auch
unter
hydrogenolytischen Bedingungen, auch in Anwesenheit von GaCp*, stabil ist. Im Gegensatz
dazu führt die Umsetzung des Bis-Styrol Komplexes [Ru(PPh3)2(styrol)2] mit drei
Äquivalenten
GaCp*
unter
vollständiger
Substitution
der
Olefin-Liganden
zu
[Ru(PPh3)2(GaCp*)3] (2), während die Reaktion von [Ru(η2,η2-COD)(η6-COT)] (COT =
Cyclooctatrien) mit ebenfalls drei Äquivalenten GaCp* unter milden hydrogenolytischen
Bedingungen zu [Ru(η2,η2-COD)(GaCp*)3] (3) führt.
Ru(η4-butadiene)(PPh3)3
GaCp*
Ru
-PPh3
Ph3P
Ph3P
GaCp*
1
PPh3
Ru(η2-styrol)2(PPh3)2
+ 3 GaCp*
- 2 styrol
*CpGa
Ru
GaCp*
GaCp*
PPh3
2
Ru(η2,η2-COD)(η6-COT)
+ GaCp*
3 bar H2, RT, 1h
- C8H16
Ru
*CpGa
*CpGa
GaCp*
3
Schema 27.1. Synthese der Ruthenium Komplexe 1-3
Analog dazu führen die Umsetzungen der RhI Verbindungen [Rh(η2,η2-NBD)(PCy3)2][BArF]
und [Rh(η2,η2-COD)2][BArF] mit den entsprechenden ECp* Liganden (E = Al, Ga) zu den
Komplexen [Rh(η2,η2-NBD)(PCy3)(GaCp*)2][BArF] (4), [Rh(η2,η2-COD)(GaCp*)3][BArF]
(5) und [Rh(η2,η2-COD)(AlCp*)3][BArF] (6).
27
3. Ergebnisse
Reaktivität von ECp* gegenüber basischen d8-Metall-Olefin-Komplexen
[BArF]-
[Rh(η2,η2-NBD)(PCy3)2][BArF]
GaCp*
80 °C, C6H5F
Rh
- PCy3
*CpGa
GaCp*
Cy3P
4
[BArF][Rh(η2,η2-COD)2][BArF]
ECp* (E = Al, Ga)
80 °C, C6H5F
- C8H12
Rh
*CpE
*CpE
ECp*
E = Ga (5)
E = Al (6)
Schema 28.1. Synthese der Rhodium Komplexe 4- 6
Alle neuen Komplexe wurden durch 1H,
13
C und
31
P-NMR Spektroskopie, Elementar- und
Einkristallstrukturanalysen charakterisiert. Die Umsetzungen der Olefin-haltigen d8Metallkomplexe mit GaCp* und AlCp* führen zwar teilweise zu einer Substitution der
Olefin-Liganden durch ECp*, in den meisten Fällen verhindert jedoch die stark basische
Natur der Metallzentren Ru0 und RhI eine vollständige Substitution, wodurch eine Synthese
von ECp* Komplexen mit Olefin-Co-Liganden ermöglicht wird. Die Koordination des
starken σ-Donors GaCp* führt zu einer stärkeren π−Rückbindung vom Metall zum OlefinLiganden, was sich in den C-C Bindungslängen der koordinierten Olefine widerspiegelt. So
wird beispielsweise die Struktur des Butadienkomplex 1 am besten durch ein
Metallacyclopropen und nicht durch einen einfachen π-Komplex beschrieben. Aufgrund
ähnlicher Bindungsenergien von Phosphanen und ECp* an Übergangsmetallen ist eine
vollständige Substitution hier nicht bevorzugt.
3.2 Synthesen und Strukturen elektronenreicher ECp*-RutheniumPolyhydrid-Komplexe und Cluster (E = Al, Ga)
Die Reaktivität von ECp* (E = Al, Ga) gegenüber potentiell reduzierbaren späten
Übergangsmetallkomplexen [LaMXb] (L = neutraler Ligand, X = anionischer Ligand) ist
ein gut untersuchtes Gebiet in der Koordinationschemie niedervalenter Gruppe 13
Organyle EIR (E = Al, Ga; R = anionische Gruppe).33,
114, 128, 130, 146
Dabei dient die
28
3. Ergebnisse
Elektronenreiche ECp*-Ruthenium-Polyhydrid-Komplexe und Cluster
elektropositivere Metallverbindung EIR gewöhnlich als Reduktionsmittel, auf welches die
anionische Gruppe X leicht vom Übergangsmetall übertragen wird. In einigen Fällen sind
Insertionsprodukte des Typs [LaM(ECp*X)b] als Intermediate dieser Redoxreaktionen
isolierbar.
Als
Beispiel
für
ein
solches
Intermediat
dient
der
Komplex
[RhCp*(GaCp*)3Cl2], der sofort bei Raumtemperatur durch Insertion von GaCp* in die
Rh-Cl Bindungen von [RhCp*Cl2]2 entsteht. Die beiden Chloride im Produktkomplex
verbrücken
jeweils
Molekülstruktur
zwei
Galliumzentren,
resultiert.125,
126
Im
was
Gegensatz
in
einer
dazu
stabilen,
reagiert
käfigartigen
der
Komplex
[RhCp*(GaCp*)(CH3)2] unter gleichen Bedingungen deutlich langsamer, wobei im ersten
Schritt ebenfalls eine Insertion von GaCp* in die Rh-CH3 Bindungen stattfindet.123,
124
Das vergleichbar elektrophile Rh-Zentrum dieses Komplexes, welches nicht wie im
vorherigen Fall zusätzlich durch eine π-Donorwirkung der Chloridliganden stabilisiert
wird, folgt jedoch einem unterschiedlichen Reaktionspfad. Dabei entsteht durch C-C
Bindungsaktivierung eines Cp* Ringes das Endprodukt [RhCp*(C5Me4GaMe3)]. In
diesem Zusammenhang erschien es daher sehr interessant, die Reaktivität von ECp*
gegenüber Komplexen mit anderen anionischen Liganden, wie zum Beispiel Hydriden, zu
untersuchen. Es gibt nur zwei in der Literatur bekannte Beispiele, in denen ein ECp*
Ligand
an
einen
Übergangsmetall-Hydrid-Komplex
koordiniert,
nämlich
[Ni(H)(SiEt3)(AlCp*)3]117, der eine terminale Ni-H Bindung enthält, sowie der C-H
aktivierte Komplex [Fe(AlCp*)5]122, welcher Fe-H-Al Brücken aufweist. Wie allgemein
ist also die Beobachtung, dass ECp* nicht in M-H Bindungen insertiert? Könnte die
Verwendung
von
Hydrido-Liganden
einen
Zugang
zu
ECp*
Komplexen
mit
Übergangsmetallen in höheren Oxidationsstufen sein?
Um dieser Fragestellung nachzugehen, wurden Ruthenium-Hydrid-Komplexe mit AlCp*
und GaCp* umgesetzt und die dabei entstandenen Produkte durch 1H,
13
C NMR, IR,
Elementaranalysen und Einkristall-Röntgenstrukturuntersuchungen charaktersiert. Als
Ausgangspunkt
diente
die
Reaktion
des
einkernigen
Ruthenium-Kations
[{Ru(COD)(H)(NH2NMe2)3}]+ mit GaCp*. Nachfolgend wurden Suzuki´s dimerer und
trimerer Polyhydridkomplex, d.h. [{Cp*Ru}2(µ-H)4] und [{Cp*Ru}3(μ-H)3(μ3-H)2], mit
GaCp* und AlCp* umgesetzt. Dabei wurde in keinem Fall eine Insertion in die Ru-HBindung beobachtet, wobei manche Produkte allerdings verbrückende Ru-H-E Einheiten
aufweisen. Der Monohydrid-Komplex [Ru(COD)(H)(NH2NMe2)3]+ reagiert mit GaCp*
zum Substitutionsprodukt [Ru(COD)(H)(GaCp*)3]+ (7), das einen terminal gebundenen
Hydridliganden enthält (siehe Schema 30.1)
29
3. Ergebnisse
Elektronenreiche ECp*-Ruthenium-Polyhydrid-Komplexe und Cluster
-
H
NH2N(CH3)2
Ru
NH2N(CH3)2
BArF
-
H
BArF
GaCp*
+ 3 GaCp*, C6H5F
Ru
- 3 NH2NMe2
GaCp*
NH2N(CH3)2
GaCp*
7
Schema 30.1. Synthese von [Ru(COD)(H)(GaCp*)3][BArF] (7)
Durch Koordination von ECp* an das Dimer [{Cp*Ru}2(µ-H)4] entstehen die
Verbindungen [{Cp*Ru(μ-H)(H)(μ-ECp*)}2] (8, E = Ga; 9, E = Al), welche beide jeweils
zwei terminale und zwei verbrückende Hydride aufweisen (siehe Schema 30.2).
H
H
Ru
Ru
H
Ru
+ 2 ECp*
H
H
E
H
E
Ru
H
H
E = Ga (8), Al (9)
Schema 30.2. Synthesen der Verbindungen 8 und 9
Schließlich entstehen bei den Umsetzungen des trimeren Polyhydrids [{Cp*Ru}3(μH)3(μ3-H)2] mit ECp* Liganden die Additionsprodukte [{Cp*Ru}3(μ-H)5(μ3-ECp*)] (10,
E = Al; 11, E = Ga) (siehe Schema 31.1). Die vorliegenden Ergebnisse verdeutlichen die
prinzipielle Stabilität von ECp*-Übergangsmetallhydrid-Komplexen, da in keinem Fall
eine Insertions- oder Redoxreaktion auftrat. Desweiteren wurde weder eine reduktive
Eliminierung von Cp*H (wie im Falle der Protonierungsreaktion von [Pt(GaCp*)4]44 oder
[(Cp*Ga)4Rh(η1-Cp*GaCH3)]147, siehe Kapitel 3.4) noch ein reduktiver Verlust von H2
beobachtet. Die anionischen Hydridliganden senken offensichtlich die Elektrophilie der
30
3. Ergebnisse
Elektronenreiche ECp*-Ruthenium-Polyhydrid-Komplexe und Cluster
Ru
H
H
H
+ ECp*
H
H
H
Ru
Ru
R
Ru
H
E
H
Ru
H
Ru
H
E = Al (10), Ga (11)
R = η1-Cp*
Schema 31.1. Synthesen der Verbindungen 10 und 11
Übergangsmetallzentren ausreichend, wodurch Cp* Transferreaktionen verhindert werden
(siehe Reaktionen von GaCp* mit kationischen Werner-Komplexen)130. Dies ermöglicht
einen neuen synthetischen Zugang zu Übergangsmetall-ECp* Komplexen in höheren
Oxidationsstufen, welche möglicherweise ein unterschiedliches Reaktionsverhalten
aufweisen als deren nullwertige Analoga. Desweiteren könnten die gemischtmetallischen
Hydrido/Kohlenwasserstoff Komplexe durch reduktive Elimierung einen systematischen
Weg zur Bildung von größeren, gemischtmetallischen Clustern bilden.
3.3
Kontrolliertes
Clusterwachstum
durch
Hydrogenolyse
von
koordinierten GaCp*: Synthese und Struktur des „nackten“ Gallium
Komplexes [Ru2(Ga)(GaCp*)7(H)3] (12).
Viel Arbeit wurde in die Untersuchung der Bindungseigenschaften zwischen monovalenten
Gruppe 13 Organylen EIR (E = Al, Ga, In; R = sperrige Substituenten, z.B. Alkyl, Aryl, Cp*,
β-Diketiminate, Amidinate, Guanidinate) und d/f-Block Übergangsmetallen investiert,
während über ihre Organometallchemie und den damit verbundenen potentiellen
Anwendungen
homoleptischen
dieser
Verbindungsklasse
[Ma(ECp*)b]
deutlich
Verbindungen
weniger
sind
berichtet
aufgrund
wurde.
der
Die
flexiblen
Bindungseigenschaften der Cp* Gruppen am Gruppe 13 Metallatom von besonderem
Interesse.109,
117, 118, 122, 124
Die sanfte chemische Synthese von M/E Hume Rothery Phasen
durch Verwendung von ECp* und ausschließlich Kohlenwasserstoff-Liganden beinhaltenden
Komplexen [MLn] als Präkursoren (n =1, E = Al, Ga; n = 2, E = Zn; M = Cu, Ni)46-48 und die
jüngste Entdeckung von [Mo(ZnMe)9(ZnCp*)3] und [{Mo(CO)4}4(Zn)6(μ-ZnCp*)4], die aus
31
3. Ergebnisse
Synthese und Struktur von [Ru2(Ga)(GaCp*)7(H)3]
den Reaktionen von [(CO)6-nMo(GaCp*)n] mit ZnMe2 hervorgehen (siehe Kapitel 3.6 und
3.7),148, 149 illustrieren einen gezielten molekularen Ansatz zur Darstellung intermetallischer
Nanomaterialien
ausgehend
13/Übergangsmetallchemie.150,
von
151
den
Grundlagen
molekularer
Gruppe
In diesem Zusammenhang ist das Abspalten einer Cp*
Gruppe von koordinierten ECp* Liganden als Initialisierung eines M/E Clusterwachsums ein
wichtiger Aspekt. So führt beispielsweise die selektive Protolyse von koordinierten GaCp* in
[Pt(GaCp*)4]
durch
[H(OEt2)2][BArF]
zum
Gallium
verbrückten
Dimer
[Pt2H(Ga)(GaCp*)7]2+.44 Auch das in Kapitel 3.4 beschriebene [(Cp*Ga)4Rh(GaMe)]+ ist auf
ähnliche Art und Weise aus [(Cp*Ga)4Rh(Ga(Me)(η1-Cp*))] unter Abspaltung von Cp*H
zugänglich.147 Eine selektive, oxidative Abspaltung von Cp* unter Bildung von Fulvalen tritt
in der Reaktion von [Pt(GaCp*)4] mit [Cp2Fe][BArF] auf, was zum trinuklearen Cluster
[Pt3(Ga)(GaCp*)6]+ führt.49 Die sanfte chemische Synthese von M/E Materialien46-48,
152
basiert auf der Co-Hydrogenolyse von [MLn] in Gegenwart von ECp*. Dabei wird die
Anwesenheit von geladenen Spezies ausgeschlossen.46-48 Aus der Tatsache, dass GaCp* inert
gegenüber H2 ist,48 lässt sich schlussfolgern, dass GaCp* haltige Koordinationsverbindungen
als Intermediate in der M/E Materialsynthese vorkommen. Mit dem Ziel frühe Intermediate
solcher
Reaktionen
zu
charakterisieren,
wurde
die
Hydrogenolyse
von
[Ru(η4-COD)(η3-C4H7)2] (η3-C4H7 = 2-methylallyl) in Anwesenheit von GaCp* als ein
Fallbeispiel ausgewählt. Durch Hydrogenolyse von [Ru(η4-COD)(η3-C4H7)2] in Anwesenheit
von GaCp* entsteht [(GaCp*)4(H)Ru(μ-Ga)Ru(H)2(GaCp*)3] (12) in 78 % Ausbeute mit
Cp*H, Cyclooctan und Isobutan als Nebenprodukten (identifiziert durch NMR). Die
Umsetzung von [Ru(η4-COD)(η3-C4H7)2] mit drei molaren Äquivalenten GaCp* in
Abwesenheit
von
H2
führt
selektiv
zu
[Ru(GaCp*)3(TMM)]
(13)
(TMM
=
Trismethylenmethan, η4-C(CH2)3, siehe Kapitel 3.5). Eine entsprechende Hydrogenolyse von
13 führt in Anwesenheit von GaCp* erneut zu 12, Cp*H und Isobutan (siehe Schema 33.1). In
ähnlicher Weise lässt sich [Ru(η2,η2-COD)(η6-COT)] als Rutheniumquelle in der Synthese
von 12 verwenden, wobei zum einen die Intermediate [Ru(GaCp*)(η4-COD)(η4-COT)] und
[Ru(GaCp*)3(η4-COD)] (3) entstehen und zum anderen die Gesamtausbeute von 12
wesentlich geringer ist. Letztlich liefert die Reaktion von 3 mit einer äquimolaren Menge
GaCp* in einer Wasserstoffatmosphäre unter Abspaltung von Cp*H und Cyclooctan erneut
12 (siehe Schema 33.2). Für Röntgenbeugungsuntersuchungen geeignete Einkristalle von 12
entstanden durch langsames mehrtägiges Kühlen einer gesättigten Toluol-Lösung bei -30°C.
32
3. Ergebnisse
Synthese und Struktur von [Ru2(Ga)(GaCp*)7(H)3]
2
+ 8 GaCp*,
3 bar H2, 60 °C, 1h
- Cp*H, - 2 C8H16,
- 4 C4H10
Ru
Cp*Ga
GaCp*
GaCp*
H
Ru
Ru
Ga
Cp*Ga
Cp*Ga
H
GaCp*
GaCp*
-Cp*H, + 2 GaCp*
- C4H10 3 bar H2, 100 °C, 2h
CH2
H2C
+ 6 GaCp*
80 °C, 1h
- C8H12
12
H
CH2
2
Ru
Cp*Ga
GaCp*
Cp*Ga
13
Schema 33.1. Synthesewege der Verbindung 12 mit [Ru(η4-COD)(η3-C4H7)2] als Ru-Quelle
H
Cp*Ga
GaCp*
2
Ru
+ 8 GaCp*
3 bar H2, 100 °C, 2h
- Cp*H, - 4 C8H16
Ru
Cp*Ga
Cp*Ga
12
+ 6 GaCp*
3 bar H2, RT, 1h
2
Ru
- 2 C8H16
GaCp*
+ 4 GaCp*
3 bar H2, RT, 1h
- 2 C8H16
Ga
Cp*Ga
H
+ 2 GaCp*
RT, 30 min
H
Ru
GaCp*
GaCp*
- Cp*H, + 2 GaCp*
- 2 C8H16 3 bar H2, 100 C°, 2h
Ru
2
Cp*Ga
GaCp*
Cp*Ga
3
Schema 33.2. Synthesen der Verbindung 12 mit [Ru(η2,η2-COD)(η6-COT)] als Ru-Quelle
Verbindung 12 kristallisiert in der monoklinen Raumgruppe C2/m. Die Hydridliganden
konnten im Rahmen der Verfeinerung der Festkörperstruktur nicht lokalisiert werden. Aus
den entsprechenden 1H NMR Daten geht hervor, dass in Lösung drei Hydridliganden über
beide Rutheniumzentren verteilt sind. Die Strukturparameter der Ru und Ga Zentren im
33
3. Ergebnisse
Synthese und Struktur von [Ru2(Ga)(GaCp*)7(H)3]
Festkörper, insbesondere die Ga-Ru-Ga Bindungswinkel der Ru-GaCp* Einheiten, deuten
darauf hin, dass jedes Rutheniumatom tatsächlich sechsfach koordiniert ist, was zu den zwei
unterschiedlichen
oktaedrischen
Ru
Fragmenten
[(GaCp*)4(H)Ru(μ-Ga)]
bzw.
[(GaCp*)3(H)2Ru(μ-Ga)] führt.
Abbildung 34.1. Festkörperstruktur von 12. (Thermische Ellipsoide gezeigt mit 50%
Wahrscheinlichkeit, Wasserstoffatome an den Cp* Liganden wurden aus Gründen der
Übersichtlichkeit entfernt). Ausgewählte Bindungslängen (Å) und–winkel (°): Ru(1)-Ga(4)
2.334(1), , Ru(1)-Ga(2) 2.377(1), Ru(1)-Ga(3) 2.383(1), Ru(1)-Ga(1) 2.387(1), Ru(2)-Ga(7) 2.313(1), Ru(2)Ga(1) 2.322(1), Ru(2)-Ga(5) 2.348(1), Ru(2)-Ga(6) 2.440(1) Ga(2)-Cp*Centr. 1.990, Ga(3)-Cp*Centr. 2.011, Ga(4)Cp*Centr. 1.959, Ga(5)-Cp*Centr. 2.029, Ga(6)-Cp*Centr. 2.031, Ga(7)-Cp*Centr. 2.061, Ga(4)-Ru(1)-Ga(4`) 159.0(1),
Ga(4)-Ru(1)-Ga(2) 99.1(1), Ga(4)-Ru(1)-Ga(3) 94.2(1), Ga(2)-Ru(1)-Ga(3) 96.9(1), Ga(4)-Ru(1)-Ga(1) 85.3(1),
Ga(2)-Ru(1)-Ga(1) 86.6(1), Ga(3)-Ru(1)-Ga(1) 176.5(1), Ga(7)-Ru(2)-Ga(1) 145.9(1), Ga(7)-Ru(2)-Ga(5)
99.6(1), Ga(1)-Ru(2)-Ga(5) 94.3(1), Ga(7)-Ru(2)-Ga(6) 33.7(1), Ga(1)-Ru(2)-Ga(6) 114.1(1), Ga(5)-Ru(2)Ga(6) 96.5(1), Ru(2)-Ga(1)-Ru(1) 178.0(1)
Wie aus der Festkörperstruktur ersichtlich, ist scheinbar eine freie Koordinationsstelle am
Ru(1) Zentrum und zwei freie Koordinationsstellen am Ru(2) Zentrum vorhanden. Beide
Rutheniumzentren sind über ein substituentenfreies, „nacktes“ Galliumatom in einer nahezu
perfekten linearen Anordnung (Ru-Ga(1)-Ru 178.0(1)°) miteinander verbunden. An dieser
Stelle sei auf die verwandten elektronenreichen Ru-H Polyhydrid Cluster aus Kapitel 3.2
verwiesen, die AlCp* und GaCp* jedoch kein substituentenfreies Gallium beinhalten. Bei
diesen Verbindungen wurde keine Bildung einer E-H Spezies durch Insertion von ECp* in die
34
3. Ergebnisse
Synthese und Struktur von [Ru2(Ga)(GaCp*)7(H)3]
Ru-H Bindung beobachtet.153 Die Ru-Ga(1) Abstände in 12 betragen 2.322(1) Å (an Ru(2))
und 2.387(1) Å (an Ru(1)), wohingegen die Ru-GaCp* Bindungslängen im Durchschnitt
2.365 Å ergeben (von 2.313(1) Å bis 2.440(1) Å). Diese Daten entsprechen den Ru-Ga
Abständen verwandter Ru-GaCp* Komplexe und Cluster.127,
128, 153, 154
Unter Verwendung
gewöhnlicher Elektronenabzählregeln (mit H Liganden als 2e Donorligand H− und dem
nackten Ga Ligand als 0e Donor Ga+) setzt sich 12 formell zusammen aus einem
ungesättigten neutralen 16e Fragment [(GaCp*)3(H)2RuII] und einem gesättigten 18e
Fragment [(GaCp*)4(H)Ru0(Ga)]. Die lineare Ru-Ga-Ru Anordnung von 12 begründet sich
durch die starken σ− und π-Akzeptoreigenschaften des „nackten“ Ga+, wie DFT Rechnungen
anhand von [(GaCp*)4PtGa]+ und [Pt2H(Ga)(GaCp*)7]2+ belegen.44 Zu beachten ist weiterhin,
dass das Pt Dimer [Pt2H(Ga)(GaCp*)7]2+ isoelektronisch zu 12 ist (mit einer
Gesamtelektronenzahl von 34 Valenzelektronen für die jeweiligen M2Ga8 Kerne).
Das 1H NMR Spektrum von 12 in C6D6 bei Raumtemperatur weist lediglich zwei scharfe
Signale bei δ = 1.91 und -15.51 ppm mit einem Integralverhältnis von ca. 105:3 auf, was
sieben chemisch äquivalenten GaCp* Liganden sowie drei chemisch äquivalenten und
aufgrund der chemischen Verschiebung unzweifelhaft an das Ruthenium gebundenen
Hydridliganden entsprechen. Die NMR Verschiebungen der Hydride und der GaCp* Gruppen
entsprechen den Verschiebungen bereits aus der Literatur bekannten Ru-Komplexe dieser
Liganden.127,
128, 153, 154
In Übereinstimmung mit diesem Spektrum zeigt das
13
C NMR
Spektrum ebenfalls nur zwei Signale bei δ = 117.2 und 10.4 ppm, die den inneren und
äußeren Kohlenstoffatomen der Cp* Gruppen zugeordnet werden können. Der Ursprung der
hochsymmetrischen oder fluktionalen Natur von 12 in Lösung erweist sich als rätselhaft. Eine
chemisch sinnvolle statische Struktur für einen Ru2Ga Kern von solch hoher Symmetrie, der
von sieben GaCp* und drei Hydridliganden umgeben ist, liegt nicht auf der Hand. Auf der
anderen Seite bleibt das
1
H NMR auch bei -100 °C vollkommen unverändert, was
offensichtlich auf einen extrem schnellen fluktionalen Prozess hindeutet, bei dem alle GaCp*
sowie die Hydridliganden über alle drei Metallzentren ihre Positionen tauschen! Wie kann ein
solcher fluktionaler Prozess aussehen? Denkt man zunächst über Dissoziations- und
Assoziationsprozesse nach, müssen reversible Dissoziationen von H2, GaCp* und/oder Cp*H
als mögliche Austauschprozesse der Liganden in Betracht gezogen werden. Jedoch können
alle drei Prozesse aufgrund experimenteller Befunde ausgeschlossen werden. Hochtemperatur
NMR Studien von reinem, isoliertem 12 in Anwesenheit eines Überschusses H2 (3bar),
GaCp* beziehungsweise Cp*H zeigen bis 80 °C keinerlei Koaleszenz mit den freien
Liganden. Desweiteren wird auch kein H/D Austausch in Anwesenheit von D2 (3 bar) oder
35
3. Ergebnisse
Synthese und Struktur von [Ru2(Ga)(GaCp*)7(H)3]
C5Me5D beobachtet, auch nicht bei erhöhten Temperaturen. Die transversale Relaxationszeit
T1 von über 600 ms schließt klar die Anwesenheit eines nicht-klassischen DiwasserstoffLiganden in 12 aus.155, 156 Diese Experimente schließen eindeutig auch intermolekulare Pfade
aus. Ein sinnvoller intramolekularer Prozess konnte ebenfalls nicht mittels DFT Rechnungen
modelliert werden.
Abbildung 36.1. Berechnete Minima und Übergangszustände eines fluktionalen Prozesses
Dabei wurden verschiedene unterschiedliche Reaktionswege berücksichtigt, aber in allen
Fällen war entweder der jeweilige Übergangszustand oder die Thermodynamik der Reaktion
energetisch zu hoch, um einen solch schnellen Prozess erklären zu können (siehe
Experimenteller Teil). Ein solches Beispiel verläuft über das symmetrische Intermediat
[(GaCp)3(H)Ru(μ-GaH)(μ-GaCp)Ru(H)(GaCp)3] (2Cp), das über eine Migration eines
Hydridliganden zum verbrückten Galliumatom und gleichzeitiger Wanderung eines GaCp*
Liganden in eine verbrückende Position zwischen den beiden benachbarten Rutheniumzentren
zugänglich wäre. Die Gesamtenergie dieser Spezies ist der von [(GaCp)4(H)Ru(μGa)Ru(H)2(GaCp)3] (1Cp) sehr ähnlich (-1.6 kcal/mol). Der berechnete Übergangszustand ist
für einen solchen Bewegungsablauf mit +38.2 kcal/mol viel zu hoch, wodurch dieser Prozess
als ein Teil eines schnellen Gleichgewichts ausgeschlossen werden kann (siehe Abbildung
36.1). Ein Vergleich der IR Spektren bei unterschiedlichen Bedingungen (Cyclopentanlösung,
36
3. Ergebnisse
Synthese und Struktur von [Ru2(Ga)(GaCp*)7(H)3]
KBr Pellet, Nujol Dispersion) belegt eindeutig, dass auch in Lösung terminale und nicht
verbrückende Ru-Hydride vorhanden sind, wodurch ein symmetrischer [Ru(μ-H)3Ru]
Komplex als mögliches Intermediat ausgeschlossen werden kann. Dabei ist weiterhin zu
berücksichtigen, dass in der Literatur durchaus Oligohydridkomplexe bekannt sind, in denen
ein schneller Hydridaustausch unterhalb der NMR Zeitskala beobachtet wurde.156
Was ist die Kernaussage in der Synthese von 12, jenseits seiner bemerkenswerten Lösungsund Festkörperstruktur? Erstens wurde die Hydrogenolyse eines GaCp* beinhaltenden
Übergangs-metallkomplexes bisher nicht im Detail untersucht, obwohl kürzlich gezeigt
wurde, dass die Hydrogenolyse von solchen Komplexen unter schärferen Bedingungen zu
M/Ga Legierungs-Nanopartikeln führt (siehe oben). [Ru(η2,η2-COD)(η6-COT)] selbst ist ein
sehr guter Präkursor für sehr kleine und hochreine Ru Nanopartikel.157-159 In der Tat führt
auch die Behandlung von [Ru(η4-COD)(GaCp*)3] mit 3 bar H2 in Mesitylen bei 150 °C im
Verlauf von sieben Tagen zur Bildung von RuGa und RuGa2 Partikeln, während die CoHydrogenolyse von [Ru(η4-COD)(η3-C4H7)2] und GaCp* in einem molaren Verhältnis von
1:2 zu RuGa2 und Ru führt (siehe Experimenteller Teil). Somit kann 12 als ein Intermediat in
einem solchen Legierungsbildungsprozess betrachtet werden, d.h. 12 repräsentiert eine frühe
molekulare Stufe auf dem Weg zu Ru/Ga Nanopartikeln während einer nasschemischen
Synthese. Daher ist es durchaus sehr interessant einen genaueren Blick auf den Mechanismus
der Hydrolyse von koordiniertem GaCp* unter Freisetzung von Cp*H, wie in der Synthese
von 12 beobachtet, zu werfen.
Die Synthese von 12 ausgehend von [Ru(η4-COD)(η3-C4H7)2] führt in einer D2 Atmosphäre
in C6D6 lediglich zu einem 30%igen Einbau der Deuterium-Atome in die Hydrid Positionen,
während es unter Verwendung von [Ru(η2,η2-COD)(η6-COT)] als Startmaterial zu einer
nahezu vollständigen Deuterierung (>90%) kommt. Eine mögliche Erklärung für diesen
Unterschied ist ein schneller H/D Austausch der Deuteride in die Ru(TMM) Gruppe über ein
thermodynamisch stabiles Ru(2-methylallyl) Intermediat. Unter Verwendung von C6H6
gelangt in beiden Fällen signifikant weniger Deuterium ins Produkt, was auf einen H/D
Austausch mit dem Lösemittel in einem frühen Stadium der Reaktion hindeutet, denn 12 ist
kinetisch bemerkenswert inert. Weder ein H/D Austausch mit C6D6, D2 oder Cp*D noch
Ligandenaustauschreaktionen in Anwesenheit von freiem GaCp* oder Cp*H wurden
beobachtet.
Um den Mechanismus der Synthese von 12 zu untersuchen, wurden DFT Rechnungen auf
B3LYP160,
161
/LanL2DZ162 Niveau unter Verwendung von GaCp anstelle von GaCp*
durchgeführt.163 Unter Berücksichtigung, dass Isobutan, Cyclooctan und Cp*H als einzige C37
3. Ergebnisse
Synthese und Struktur von [Ru2(Ga)(GaCp*)7(H)3]
haltigen Nebenprodukte in der Synthese von 12 spektroskopisch bestimmt wurden und in
Anbetracht, dass die Hydrierung von koordinierten Olefinen gewöhnlich ein sehr schneller
Prozess ist, wurde [Ru(GaCp)4(H)2] (IM1) als wahrscheinliches erstes Schlüsselintermediat
vor Abspaltung der CpH Gruppe ausgewählt. Prinzipiell sind zwei unterschiedliche
Reaktionspfade für die CpH Elimierung ausgehend von IM1 denkbar (Schema 38.1).
H
Ga
Weg A
Ga
H
Ru
Ga
H
Ga
IM2
- CpH
Ga
Ga
H
Ru
Ga
H
Ga
Weg B
Ga
Ga
- CpH
Ru
Ga
Ga
H
IM1
IM3
Schema 38.1. Arbeitshypothesen zur Bildung des Schlüsselintermediates in der Synthese von
1Cp
Pfad A repräsentiert einen zweistufigen Mechanismus, der als ersten Schritt einen
Hydridtransfer
vom
Ruthenium-
auf
das
Galliumatom
unter
Bildung
von
[Ru(GaCp)3(H)(GaH(C5H5))] (IM2) beinhaltet. Anschließende CpH Eliminierung am
[GaH(C5H5)] Liganden führt zu einem zweiten Schlüsselintermediat dieser Reaktion, nämlich
[Ru(GaCp)3(H)(Ga)] (IM3). Dieses beinhaltet ein terminal gebundenes Galliumatom. In Pfad
B wird IM3 direkt durch eine konzertierte CpH Eliminierung ausgehend von beiden
Metallzentren in IM1 ohne vorherige Hydridübertragung gebildet. Dimerisierungen von
Intermediaten, die durch GaCp Assoziations-/Dissoziationsgleichgewichte von IM3 und IM1
entstehen, führen schließlich zu [(GaCp)4(H)Ru(μ-Ga)Ru(H)2(GaCp)3] (1Cp) (siehe
Experimenteller Teil).
38
3. Ergebnisse
Synthese und Struktur von [Ru2(Ga)(GaCp*)7(H)3]
Die Hauptkriterien bei der Modellierung der Reaktionsfolge durch DFT Methoden lassen sich
wie folgt zusammenfassen: (a) Die Energie des Übergangszustandes des Hydridtransfers und
der CpH Eliminierung in Pfad A und B sowie (b) die Energiebilanz der GaCp Assoziationsund Dissoziationsprozesse ausgehend von IM1 und IM3. Für Pfad A konnten sowohl das
Intermediat IM2 sowie der entsprechende Übergangszustand der CpH Eliminierung, nämlich
TS1, auf der Energiehyperfläche optimiert werden. IM2 ist um 22.2 kcal/mol energetisch
ungünstiger als IM1, was durch den Verlust der Aromatisierungsenergie des GaCp Ringes
begründbar ist. Der Übergangszustand der CpH Eliminierung TS2 in Pfad A ist mit 51.0
kcal/mol über IM2 (73.2 kcal/mol über IM1) energetisch sehr hoch. Das fünffach
koordinierte [Ru(GaCp)3(H)(Ga)] (IM3) ist dagegen überraschenderweise energetisch sehr
günstig und die Bildung von IM3 ausgehend von IM1 ist nur mit 7.6 kcal/mol endergonisch.
In Pfad B liegt der entsprechende Übergangszustand (TS3) der direkten CpH Eliminierung
von beiden Metallzentren 35.5 kcal/mol über IM1 (siehe Abbildung 40.1). Dabei handelt es
sich um eine bemerkenswert niedrige Aktivierungsbarriere verglichen zu TS2 (Pfad A), aber
dennoch hoch genug, um mit der experimentell beobachteten Tatsache übereinzustimmen,
dass die Synthese von 12 nur unter relativer rauen Reaktionsbedingungen (60 °C, 3 bar H2)
stattfindet. Der Energieaufwand resultiert zum Teil aus dem Wechsel der Haptizität des GaCp
Ringes von η5 in IM2 nach η3 in TS3. Der Hauptprozess in dieser Reaktion ist die Wanderung
des Cp Ringes zum Hydridliganden. Letztlich ist die Assoziation von freiem GaCp an IM3,
die zum oktaedrischen [Ru(GaCp)4(H)(Ga)] führt, um 2.5 kcal/mol exergonisch, während die
Dissoziation von GaCp aus IM1 unter Entstehung von [Ru(GaCp)3(H)2] 17.9 kcal/mol kostet.
Diese Ergebnisse weisen auf die Anwesenheit von parallelen GaCp Assoziations- und
Dissoziationsgleichgewichten in Lösung hin. Die zur Bildung von 1Cp erforderlichen
Intermediate, d. h. [Ru(GaCp)4(H)(Ga)] und [Ru(GaCp)3(H)2], sind energetisch erreichbar.
Bei der abschließenden Dimerisierung dieser beiden Fragmente handelt es sich um einen stark
exergonischen Prozess, bei dem 37.0 kcal/mol frei werden. Leider konnte das vorgeschlagene
Schlüsselintermediat
[Ru(GaCp*)4(H)2]
(IM1,
Schema
38.1)
weder
isoliert
noch
spektroskopisch durch Aufnehmen einer in situ Synthese gemäß Schema 33.1 oder 33.2 in
einem NMR Druckrohr beobachtet werden. Die Synthese des in Kapitel 3.5 berichteten nahe
verwandten Komplexes [Ru(GaCp*)2(PCy3)2(H)2]154 unterstützt jedoch die Annahme eines
IM1 als Intermediat auf dem Weg zum Endprodukt 12.
39
3. Ergebnisse
Synthese und Struktur von [Ru2(Ga)(GaCp*)7(H)3]
Abbildung 40.1. Berechnete Reaktionspfade für die CpH Eliminierung, die zu IM3 führt
Die selektive hydrogenolytische Spaltung eines Cp* Liganden aus einem kooridinierten
GaCp*, die zum neutralen, „nackten“ Gallium verbrückten Komplex [(GaCp*)4(H)Ru(μGa)Ru(H)2(GaCp*)3] (12) führt, deutet auf eine allgemeine Anwendbarkeit dieser Methode
der „Entschützung“ innerhalb der [LaMb(ECp*)c] Verbindungsklasse hin. Dabei sei noch
einmal auf die wichtige Gemeinsamkeit der beschriebenen Hydrogenolyse mit der
protolytischen44,
147
(siehe Kapitel 3.4) und der oxidativen Abspaltung49 von Cp*
hingewiesen. In allen Fällen werden M/Ga Verbindungen oder Cluster höherer Nuklearität
gebildet, die substitutiontenfreie Ga Zentren in verbrückenden Positionen beinhalten, welche
neben M-H Bindungen koexistieren. Die neuen Ergebnisse heben die Eigenschaft von Cp* als
eine entfernbare Schutzgruppe hervor, im Gegensatz zu anderen typischen Substituenten R,
insbesondere der N,N chelatisierenden Bisketiminate und verwandter Spezies, die ebenfalls
40
3. Ergebnisse
Synthese und Struktur von [Ru2(Ga)(GaCp*)7(H)3]
zur Stabilisierung von niedervalenten Gruppe 13 Verbindungen verwendet werden. Ein
tieferes Verständnis dieser Chemie ist ein Schlüssel für die Entwicklung einer sehr
kontrollierten molekularen Bausteinchemie für Hume-Rothery-artige intermetallischeM/E
Cluster und Nanomaterialien.
3.4
Methylgallium als terminaler Ligand in dem all-Gallium koordi-
nierten Rhodium Kation [(Cp*Ga)4Rh(GaCH3)]+ (15)
Carbenoide Gruppe 13 Metallverbindungen EIR (E = Al, Ga, In; R = sterisch anspruchsvoller
Substituent: z.B. Alkyl, Aryl, Cp*, Bisketoiminate, Amidinate, Guanidinate) erwecken als
„exotische“ Liganden gegenüber Übergangsmetallen M großes Interesse, da ihre
Eigenschaften mit denen der verwandten Borylen96-101,
Carbenen (NHCs) verglichen werden können.
34, 84, 85, 137
134, 135
und N-heterozyklischen
Insbesondere Komplexe mit ECp*
Liganden zeigen eine interessante Reaktivität, die durch die schwachen Bindung sowie durch
die Möglichkeit einer haptotropen Verschiebung der Cp* Ringe hervorgerufen wird. Dadurch
verändert sich die Elektrophilie des Gruppe 13 Zentrums E,117, 122, 124 so dass beispielsweise
die selektive Protolyse von koordiniertem GaCp* möglich wird. Die Umsetzung des
Komplexes [Pt(GaCp*)4] mit [H(OEt2)2][BArF] führt dabei über das Intermediat
[HPt(GaCp*)4]+
nach
anschließender
Eliminierung
von
Cp*H
zu
dem
Dimer
[Pt2H(Ga)(GaCp*)7]2+. Mithilfe des Ga+ Transferreagens [Ga2Cp*]+ wurde der Komplex
[GaPt(GaCp*)4]+ erzeugt, in dem nacktes Ga+ als starker σ/π-Akzeptorligand ohne σ-Donor
Eigenschaften bindet.29, 44
In diesem Zusammenhang ist es durchaus denkbar, sonst nur schwer zugängliche GaR
Einheiten durch protolytische Spaltung von Cp*H aus koordinierten Ga(R)Cp* Gruppen zu
erzeugen. Die Auswahl an Substituenten R für monovalente Verbindungen EIR ist aufgrund
der inhärenten Instabilität von EI bezüglich einer Disproportionierung zu E0 und EIII
begrenzt.164 So wurde beispielsweise freies Methylgallium bisher ausschließlich durch
Matrixstudien bei tiefen Temperaturen untersucht.165,
166
Bislang sind nur sehr wenige
Komplexe von EIR Liganden mit kleinen Substituenten R ohne π-Donor/Akzeptor
Eigenschaften bekannt, darunter das Anion [{(CO)4Fe}2GaCH3]- sowie das Dimer
[Cp*IrAlEt]2.167,
168
Dabei ist zu beachten, dass der EIR Ligand in allen Komplexen
verbrückend (dreifach-koordiniert) gebunden ist, was direkte Vergleiche mit terminalen
(zweifach koodinierten) EIR Liganden ausschließt. Analog hierzu wurde erst jüngst über die
Synthese des ersten terminalen Alkylborylen Komplexes [Cp(CO2)MnBtBu] berichtet. Die
41
3. Ergebnisse
Methylgallium als terminaler Ligand
Mn-BIR (R = tBu) Bindung wurde dabei als schwach polar, aber mit signifikanten πRückbindungsanteil beschrieben.100
Die Reaktion von [(COD)(py)Rh(CH3)] (py = Pyridin) mit einem Überschuss GaCp* in
Hexan führt bei Raumtemperatur zu einer Substitution der labilen Liganden Pyridin und 1,5Cyclooctadien sowie zu einer Insertion des Carbenoids GaCp* in die Rh-CH3 Bindung unter
Bildung des ausschließlich Gallium koordinierten Neutralkomplexes [(Cp*Ga)4Rh(η1Cp*GaCH3)] (14) in 89 % Ausbeute (siehe Schema 42.1). Ähnliche Insertionsreaktionen in
Rh-X Bindungen (X= Cl, CH3) wurden bereits zuvor diskutiert.124, 126, 169 Verbindung 14 ist
bei Raumtemperatur in einer Inertgas Atmosphäre (Ar) stabil und löst sich gut in allen
nichtkoordinierenden, unpolaren organischen Lösemitteln wie Hexan, Benzol oder Toluol.
H3C
Ga
[(COD)(py)Rh(CH3)]
+ 5 GaCp*
- COD, Pyridin
*CpGa
Rh
GaCp*
GaCp*
*CpGa
14
Schema 42.1. Synthese von [(Cp*Ga)4Rh(η1-Cp*GaCH3)] (14)
Das 1H NMR Spektrum bei Raumtemperatur in C6D6 zeigt zwei scharfe Singuletts mit einem
Integralverhältnis von ungefähr 75:3, die in d8-Toluol bei -100 °C in drei Signale mit einem
Integralverhältnis von ca. 15:60:3 aufspalten. Diese Beobachtung wird durch einen
fluktionalen Prozess bei Raumtemperatur erklärt, der zum Austausch der CH3 und/oder Cp*
Gruppen zwischen allen Galliumzentren führt. Durch Kühlen einer gesättigten n-Hexan
Lösung von 14 auf -30 °C wurden nach 12 Stunden geeignete Kristalle für
Röntgenbeugungsuntersuchungen erhalten. Verbindung 14 kristallisiert in der tetragonalen
Raumgruppe P4(2)/m. Der Komplex weist im Festkörper eine trigonale-bipyramidale Struktur
auf, was mit den Ergebnissen der Tieftemperatur NMR Studien im Einklang steht (siehe
Abbildung 43.1). Die axialen Positionen werden dabei durch einen GaCp* Liganden und
durch einen η1-Cp*GaMe Liganden besetzt, der aus der Insertionsreaktion resultiert. Dabei
sind die axialen Rh-Ga Bindungslängen mit 2.430(1) Å im Falle des η1-Cp*GaMe sowie mit
2.395(1) Å für GaCp* trotz der unterschiedlichen Koordination überraschenderweise sehr
42
3. Ergebnisse
Methylgallium als terminaler Ligand
ähnlich. Die Zugabe eines Äquivalents an kristallinen [H(OEt2)2][BArF] zu einer FluorbenzolLösung von 14 führt bei -30°C zur Protolyse von 14 unter sofortigen Farbwechsel von Rot zu
Hellgelb (Schema 44.1). Durch langsame Diffusion von n-Hexan in diese Lösung bei 25 °C
konnten in 78% Ausbeute hellgelbe Einkristalle des Salzes [(Cp*Ga)4Rh(GaCH3)][BArF] (15)
isoliert werden. Das 1H NMR Spektrum von isoliertem 15 in Fluorbenzol/C6D6 (10:1) zeigt
bei Raumtemperatur sowohl die Signale des [BArF]- Anions bei δ = 8.27 (ca. 8H) und bei
7.56 ppm (ca. 4H) als auch zwei scharfe Singuletts bei δ = 1.64 und bei 0.54 ppm mit einem
Integralverhältnis von nahezu 60:3, was auf eine fluktionale Struktur in Lösung schließen
lässt.
Abbildung 43.1. Molekülstruktur von [(Cp*Ga)4Rh(η1-Cp*GaCH3)] (14) (Thermische
Ellipsoide gezeigt mit 30% Wahrscheinlichkeit, Wasserstoffatome wurden aus Gründen der
Übersichtlichkeit entfernt). Ausgewählte Bindungslängen (Å) und –winkel (°): Rh(1)-Ga(4)
2.348(1), Rh(1)-Ga(2) 2.357(1), Rh(1)-Ga(3) 2.395(1), Rh(1)-Ga(1) 2.430(1), Ga(1)-C(1) 1.989(12), Ga(1)-C(2)
2.085(18), Ga(4)-Rh(1)-Ga(2) 110.81(2), Ga(4)-Rh(1)-Ga(3) 93.61(3), Ga(2)-Rh(1)-Ga(3) 97.55(5), Ga(4)Rh(1)-Ga(1) 80.19(3), Ga(2)-Rh(1)-Ga(1) 99.95(4), Ga(3)-Rh(1)-Ga(1) 162.49(5), C(1)-Ga(1)-C(2) 109.2(7),
C(1)-Ga(1)-Rh(1) 124.2(4), C(2)-Ga(1)-Rh(1) 124.2(6).
Eine Einkristallstrukturanalyse belegt eine verzerrt trigonale-bipyramidale Struktur für das
Kation [(GaCp*)4Rh(GaCH3)]+, wobei die gewünschte GaCH3 Gruppe eine axiale Position
besetzt (Abbildung 44.1).
43
3. Ergebnisse
Methylgallium als terminaler Ligand
CH3
H 3C
Ga
*CpGa
Rh
[BArF]-
Ga
GaCp*
+ [H(OEt2)2][BArF]
*CpGa
- Cp*H
GaCp*
Rh
GaCp*
GaCp*
*CpGa
Cp*Ga
14
15
Schema 44.1. Synthese von 15 durch Protolyse von 14 in Fluorbenzol.
Abbildung
44.1.
Molekülstruktur
F
[(GaCp*)4Rh(GaCH3)][BAr ]
(15)
des
kationischen
(Thermische
Ellipsoide
Teils
gezeigt
des
mit
Salzes
50%
Wahrscheinlichkeit, Wasserstoffatome wurden aus Gründen der Übersichtlichkeit entfernt).
Ausgewählte Bindungslängen (Å) und –winkel (°): Rh-Ga(1) 2.471(10), Rh-Ga(2). 2.310(7), Rh-Ga(4)
2.282(6), Rh-Ga(5) 2.282(6), Rh-Ga(3) 2.310(7), Ga(1)-C(1) 1.958(11), Ga(1)-Ga(2) 2.766(13), Ga(1)-Ga(5)
2.868(13); C(1)-Ga(1)-Rh 176.09(5), Ga(1)-Rh-Ga(3) 163.58(4), Ga(2)-Rh-Ga(4) 107.91(2), Ga(4)-Rh-Ga(5)
119.59(4).
GaCH3 ist der kleinste mögliche GaR Liganden (mit Ausnahme von R = H), bei dem weder
sterische Wechselwirkungen noch elektronische π Effekte des Substituenten R einen Einfluss
auf die M-Ga Bindung ausüben sollten. Sowohl das bekannte [(Cp*Ga)4PtGa]+ 44 als auch das
neue [(Cp*Ga)4Rh(GaCH3)]+ dienen als isoelektronische (18 Valenzelektronen) und
prinzipiell isostrukturelle Modelle, um M-GaR Bindungsparameter zu vergleichen. Das
44
3. Ergebnisse
Methylgallium als terminaler Ligand
Hauptcharakteristikum der Struktur von [(Cp*Ga)4Rh(GaCH3)]+ liegt zweifelsohne in der
unerwarteten Tatsache, dass die Rh-GaCH3 Bindung mit 2.471(1) Å die mit Abstand längste
Rh-Ga Bindung in dem Komplex ist. Die Rh-Ga(1) Bindung ist um ca. 8 % länger als die
axiale Rh-Ga(3) Bindung und die äquatorialen Rh-Ga-(2,4,5) Bindungen, die sich nur um 1%
voneinander unterscheiden (2.282(1) Å bis 2.310(1) Å). Diese Rh-GaCp* Bindungen sind im
Kation 15 kürzer als in der neutralen Verbindung 14 (2.395(13) Å und 2.347(1) Å).
Die Isolobalität zwischen den Fragmenten L4Rh und CH3 führt zu einer formalen Zerlegung
des Kations [(GaCp*)4Rh(GaCH3)]+ in ein nukleophiles 18 Elektronen Fragment
[(GaCp*)4Rh]- und ein elektrophiles Ion [GaCH3]2+, analog zu der offensichtlichen
Zusammensetzung von [Ga(CH3)2]+ aus einem Ga3+ Ion und zwei Methylanionen. Zu
beachten ist, dass [(Cp*Ga)4Rh(η1-Cp*GaCH3)] (14) als ein Rh-I/GaIII Komplex beschrieben
werden kann, der entsprechend des Syntheseweges durch (formale) oxidative Addition einer
Rh-CH3 Bindung an das Ga(I) Zentrum eines GaCp* Liganden gebildet wird. Die
darauffolgende protolytische Abspaltung von Cp*H, die von 14 zu 15 führt (Schema 44.1),
kann als redoxneutral aufgefasst werden. Unter Vernachlässigung des durchgeführten
Syntheseweges kann das Kation [(Cp*Ga)4Rh(GaCH3)]+ an sich auch als ein RhI/GaI
Komplex
beschrieben
werden,
der
sich
aus
einem
neutralen,
carbenoiden
Zweielektronendonor GaCH3 und dem ungesättigten kationischen 16 Elektronen Fragment
[(GaCp*)4Rh]+ zusammensetzt. In dem zuletzt genannten Fall entsprechen alle GaR Liganden
in 15 einer Ga(I) Spezies.
Welche Betrachtungsweise sollte nun bevorzugt werden? Eine eindeutige Unterscheidung
zwischen beiden Alternativen ist auf der Grundlage von strukturellen oder spektroskopischen
Eigenschaften nicht möglich. Die Ga-CH3 Bindungslängen von 1.978(12) Å für 14 und
1.958(11) Å für 15 sind kleiner als der mit Dichtefunktionalmethoden (DFT) berechnete Wert
von 2.049 Å für freies (monovalentes) GaCH3 in einer Matrix165, 166 und ähneln dem Wert für
die Ga-C Bindungen im trivalenten Ga(CH3)3 (1.967(2) Å).170 Dieser Vergleich mag die
Rh-I/GaIII Sichtweise unterstützen. Um weiteren Aufschluss über die Situation zu erhalten,
wurden wie auch schon zur Bindungssituation im Kation [(Cp*Ga)4PtGa]+44 DFTRechnungen durchgeführt (siehe Abbildung 46.1). Die berechnete Ladung am Galliumatom
der GaCH3 Gruppe (1.06) im Modellkomplex [(CpGa)4Rh(GaCH3)]+ ist höher als an den
GaCp Liganden (0.78/0.79), aber deutlich niedriger als im Kation [Ga(CH3)2]2+ (1.70). Die
berechnete Ladung am Rh beträgt -0.97.
45
3. Ergebnisse
GaMe
GaMe2+
Methylgallium als terminaler Ligand
NBO: q(Ga) = 0.68
GaCp
NBO: q(Ga) = 0.30
Ga NBO Population:
Ga NBO Population:
4s(1.89), 4px(0.40),
4s(1.51), 4px(0.20),
4py(0.01), 4pz(0.01)
4py(0.98), 4pz(0.01)
NBO: q(Ga) = 1.70
15Cp
NBO: q(Ga) = 1.06,
q(Rh) = -0.97, q(Ga-trans)
Ga NBO Population:
= 0.78, q(Ga-cis) = 0.79
4s(1.04), 4px(0.21),
Ga NBO Population:
4py(0.04), 4pz(0.03)
4s(1.12), 4px(0.34),
4py(0.23), 4pz(0.25)
Abbildung 46.1. Berechnete Ladungen am Galliumatom in GaMe, GaCp, GaMe2+ sowie
dem Modellkomplex [(CpGa)4Rh(GaCH3)]+ 15Cp (mit NBO = Natural Bond Orbital)
Die Energy Decomposition Analysis (EDA)171-177 belegt, dass eine Fragmentierung in
[(CpGa)4Rh]+ und GaCH3 (ΔEint = -83.1 kcal/mol) wesentlich weniger Energie erfordert als
46
3. Ergebnisse
Methylgallium als terminaler Ligand
eine homolytische Spaltung (ΔEint = -127.4 kcal/mol), während die benötigte Energie für
einen Zerfall in [(CpGa)4Rh]- und [GaCH3]2+ sehr hoch ist (ΔEint = -457.8 kcal/mol) (siehe
Tabelle 47.1).
Tabelle 47.1. Resultate der EDA auf BP86/TZ2P Niveau. Energiewerte sind in kcal/mol
angegeben.
(GaCp)4Rh(GaMe)+ (in Cs Symmetrie)
ΔEint
ΔEPauli
ΔEelstat[a]
ΔEOrb[a]
ΔEσ[b]
ΔEπ[b]
-127.4
141.7
-117.0 (43.5%)
-152.0 (56.5%)
-122.6 (80.7%)
-29.4 (19.3%)
-83.1
198.0
-172.5 (61.4%)
-108.5 (38.6%)
-95.0 (87.6%)
-13.5 (12.4%)
-457.8
145.8
-264.5 (43.8%)
-339.1 (56.2%)
-277.9 (81.9%)
-61.2 (18.1%)
Fragmente
GaMe+ (d)
(GaCp)4Rh (d)
GaMe (s)
(GaCp)4Rh+ (s)
GaMe2+ (s)
(GaCp)4Rh- (s)
[a]
Die Prozentwerte in Klammern stellen den Anteil aller attraktiven Wecheselwirkungen ΔEelstat+ΔEorb dar.
[b]
Die Prozentwerte in Klammern stellen den Anteil aller Orbitalwechselwirkungen ΔEorb dar.
Zweifellos stützen die berechneten Ladungen und die EDA Resultate nicht ohne Weiteres die
formale Zuordnung einer Oxidationsstufe. An dieser Stelle sei der Leser auf den in diesem
Zusammenhang sehr hilfreichen Artikel von G. Parkin über die Begriffe „Valenz“ und
„Oxidationszahlen“ hingeweisen.178 Zu beachten ist, dass alle Galliumatome in 15 zweifach
koordiniert und trivalent sind, da sie alle drei Valenzelektronen für die Bindung zu deren
Gruppe R und zum Rhodiumzentrum nutzen. Des Weiteren sind alle Galliumatome stärker
positiv geladen als in einer freien GaR Gruppe oder in Neutralkomplexen [M(GaR)n].40, 41, 104,
119, 179
Der stärkere Effekt am Ga(1) gegenüber den anderen Ga Atomen wird durch das
Fehlen der Cp* Gruppe hervorgerufen, die als π-Elektronendonor wirkt. Gerade im Hinblick
auf die Synthese des Kations [(Cp*Ga)5Rh]+ und der konventionellen Sichtweise von [L5Rh]+
Komplexen als RhI-Spezies ist deshalb die Beschreibung von 15 als pseudohomoleptischer
GaI-Komplex zu bevorzugen. Eine ähnliche Diskussion wurde anhand der Komplexe
[(CO)nM(ECp*)] (M = Fe, Cr; E = Al, Ga; n = 4, 5) geführt, bei denen das Gruppe 13
Zentrum basierend auf der Elektronegativität des [(CO)nM] Fragments eher als EIII und
weniger als EI betrachtet wurde.107,
120
Unter Berücksichtigung von Komplexen wie
47
3. Ergebnisse
Methylgallium als terminaler Ligand
[(CO)3M(GaCp*)3]111 (M = Mo, W) erscheint die Beschreibung der gesamten Reihe als EIKomplexe sinnvoller.
Theoretische Studien an Modellverbindungen wie [(Cp*Ga)4PtGa]+44, [(CO)nM(ER)]40, 41, 104,
119, 179
und der homoleptischen Reihe [M(ER)n]104 (Fe, n = 5; Ni, n = 4) belegen, dass die σ-
Donor Stärke von GaCp* (sowie von GaRL2 mit R = CH3 und L = Lewis Base)179 höher ist
als für Ga+ und GaCH3, während sich gleichzeitig die π-Akzeptor Fähigkeit aufgrund der
Überlappung der Cp*-π-Orbitale mit den p Orbitalen des Galliumatoms verringert. Eine
starke σ-Donorwirkung resultiert in einem großen elektrostatischen Beitrag zur gesamten ME Bindungsenergie44,
104
und führt zu kurzen M-E Bindungen, wenn sterische Effekte
vernachlässigbar sind. Die Koordination eines Lewis-basischen Donors L an die GaCH3
Einheit sollte zu einer Verkürzung der entsprechenden M-Ga-Bindungen führen, da das
Ga(CH3)L Fragment ein stärkerer σ-Donor sein sollte als GaCH3.41, 104, 179
Abbildung
48.1.
Molekülstruktur
[(GaCp*)4Rh(Ga(CH3)(py))][BArF]
(16)
des
kationischen
(Thermische
Teils
Ellipsoide
des
gezeigt
Salzes
mit
50%
Wahrscheinlichkeit, Wasserstoffatome wurden aus Gründen der Übersichtlichkeit entfernt).
Ausgewählte Bindungslängen (Å) und –winkel (°): Rh-Ga(1) 2.334(6), Rh-Ga(2). 2.346(6), Rh-Ga(4)
2.333(7), Rh-Ga(5) 2.334(7), Rh-Ga(3) 2.363(7), Ga(1)-C(1) 1.965(5), Ga(1)-N 2.126(4); Ga(1)-Rh-Ga(3)
172.46(3), Ga(4)-Rh-Ga(2) 110.85(3), N-Ga(1)-Rh 117.75(10), C(1)-Ga(1)-Rh 143.69(15).
Die Reaktion von 15 mit einem Äquivalent Pyridin in Et2O bei Raumtemperatur und
anschließender Kristallisation des Produktes durch langsame Diffusion von n-Hexan führt zur
Bildung von großen gelben Einkristallen von [(GaCp*)4Rh(Ga(CH3)(py))][BArF] (16). Eine
48
3. Ergebnisse
Methylgallium als terminaler Ligand
Einkristallstrukturanalyse belegt, dass das Pyridin an der GaCH3 Gruppe koordiniert und
dabei, ähnlich wie in 14, eine Ga(CH3)(py) Einheit entsteht (siehe Abbildung 48.1).
Die Ga-N Bindungslänge von 2.126(4) Å liegt im Rahmen von Gallium-Pyridin Addukten,
während die Ga-CH3 Bindungslänge unverändert bleibt. Wie erwartet führt die Koordination
von Pyridin zu der Verkürzung der Rh-Ga(1) Bindungslänge von 2.471(1) Å in 15 auf
2.334(1) Å in 16, so dass nun alle Rh-Ga Bindungen im Rahmen der Strukturverfeinerung
gleich lang sind! Die Tatsache, dass eine Addition eines neutralen σ-Donors an die GaCH3
Gruppe zu einer Verkürzung der Rh-Ga Bindungslänge führt, zeigt eindeutig, dass die πRückbindung ausgehend vom Rh zum elektrophilen GaCH3 Liganden in 16 schwach ist.
Diese Ergebnisse belegen die Schlussfolgerungen der theoretischen Arbeiten40, 41, 104, 119, 179
darin, dass eine vergleichbar kurze M-ER Bindung nicht einfach einer kovalenten M-E
Mehrfachbindung gleichzusetzen ist.
Der kationische Komplex [(Cp*Ga)4Rh(GaCH3)]+ ist eines von wenigen Beispielen eines
pseudo-homoleptischen Komplexes [MLn] mit einer hohen Koordinationszahl (n > 4) und
Liganden, die über Metallatome an das Zentralmetall binden. Dies ist nicht nur aus
struktureller und bindungstheoretischer Sicht interessant. Die bemerkenswerte selektive
Spaltung eines Cp* Liganden durch Protonierung der Ga(CH3)(Cp*) Gruppe von 14 unter
bevorzugter Freisetzung von Cp*H gegenüber CH4 ist beispiellos und könnte innerhalb dieser
Verbindungsklasse allgemein anwendbar sein. Dies verdeutlicht die einzigartigen
Eigenschaften von Cp* Gruppen im Vergleich zu anderen Substituenten R, die zur
Stabilisierung von niedervalenten Gruppe 13 Verbindungen notwendig sind, insbesondere zu
den N,N chelatisierenden Bisketiminaten und analoger Verbinungen.101 Das fluktionale
Verhalten
an
Hauptgruppenelementen180
koordinierter
Cp*
Einheiten
sowie
die
vergleichsweise schwache Bindung zwischen Cp* und des Gruppe 13 Zentrums ermöglichen
den Einsatz als Schutzgruppen, die unter geeigneten Bedingungen entfernt werden können
(siehe Kapitel 3.3). Daher könnten diese Ergebnisse für die Chemie von Clustern und
Nanopartikeln aus Übergangsmetallen und Gruppe 13 Elementen relevant sein.34
49
Über die Organometallchemie des Ga+ Kations
3. Ergebnisse
3.5
Über die Organometallchemie des Ga+ Kations: Ein- und Ausschalten
des Elektronenpaares des monovalenten Gallium in der Koordinationsumgebung von Ruthenium
Das Konzept der kinetischen Stabilisierung von niedervalenten Gruppe 13 Verbindungen des
Typs EIR durch Verwendung von sterisch anspruchsvollen Substituenten, wie z. B. Cp*,
substituierte Terphenyl- oder Diketiminat-Liganden, führte bislang zu einer umfangreichen
Chemie.84,
85, 99, 109, 112, 137-141, 181, 182
F 147
[(GaCp*)4Rh(GaCH3)][BAr ]
Die in Kapitel 3.4 beschriebene Synthese von
(15) sowie die Darstellung von [Cp*Fe(dppe)(GaI)][BArF]
zeigen, dass sogar sterisch anspruchslose Liganden und schwer zugängliche Spezies wie
GaI183-185 und GaCH3 als terminale Liganden in der Koordinationssphäre eines
Übergangsmetallzentrums abgefangen werden können. Offensichtlich besteht das ultimative
Ziel in dieser Serie in der Chemie von nacktem Ga+.32,
45, 186
. So führt beispielsweise die
Reaktion von [Ga2Cp*][BArF] mit elektronenreichen 18 VE Komplexen [PtL4] (L = PR3,
GaCp*) zu Addukten [L4PtGa]+, welche ein terminal gebundenes Ga+ Ion enthalten.29, 44, 130
DFT Rechnungen beschreiben die Pt-Ga Wechselwirkung als eine schwach polare DonorAkzeptor Bindung, in der das Ga+ Ion als ein starker σ- und π-Akzeptor ohne jegliche
Donoreigenschaften fungiert. Somit ist das s-artige freie Elektronenpaar am Ga+ sterisch und
chemisch inaktiv, wodurch man in dieser Situation die Donoreigenschaften des Ga+ als
„ausgeschaltet“ bezeichnen kann. Eine charakteristische organometallische Chemie des
nackten Kations Ga+ wurde bisher noch nicht untersucht. Das Hauptproblem besteht dabei in
der Redoxlabilität des Ga+, welches sehr leicht durch zahlreiche Reaktionspartner mit
redoxaktiven Übergangsmetallzentren reduziert oder oxidiert werden kann. Nichtsdestotrotz
handelt es sich bei nacktem Ga+ um ein sehr interessantes Synthon für ungewöhnliche
Komplexe des Typs [LnM-GaR’] (M = Übergangsmetall, R’ = anionischer Ligand,
unterschiedlich zu Cp*, etc.), die z. B. durch Insertion von Ga+ in eine M-R’ Bindung oder
durch Addition eines nukleophilen Fragments R’ an einen elektrophilen Komplex [LnMGa]+
zugänglich wären. Obwohl solche Reaktionen für Ga+ bisher unbekannt sind, gibt es
zahlreiche Beispiele von Insertionsreaktionen von neutralen GaIR Spezies, die zumeist zu
Komplexen des Typs [LnM-GaXR] führen.124-128,
147
Bereits bekannte Ausnahmen dieser
allgemeinen Reaktion gehen von Ausgangskomplexen mit zwei (oder mehr) anionischen
Gruppen X aus. Dabei wurde die Oxidation des Galliumliganden unter Bildung von GaIII
Nebenprodukten
RGaX2
und
gleichzeitige
Reduktion
des
jeweiligen
50
Über die Organometallchemie des Ga+ Kations
3. Ergebnisse
Übergangsmetallzentrums beobachtet. Als jüngstes Beispiel dient die Reduktion von SnCl2
durch das Bisketiminat Ga(DDP), welches zu dem ungewöhnlichen Galliumligand
stabilisierten metalloiden Zinn Cluster [Sn17{Ga(DDP)Cl}4] führt.187 Bei den nun folgenden
Reaktionen ging es um die Beantwortung zweier Hauptfragen über die Reaktivität des Ga+
Kations: Erstens, wenn Ga+ sich als ein starkes Elektrophil gegenüber Lewis basischen
Übergangsmetallzentren verhält, wie verhält es sich dann gegenüber nukleophilen πLiganden? Und zweitens, was kann man, unter Berücksichtigung der zahlreichen Beispiele
von Reaktionen mit GaIR gegenüber [LnM-X] Komplexen, über dessen Reaktivität gegenüber
M-X Fragmenten sagen? Als Ausgangspunkt für diese Reaktivitätsstudien von Ga+ gegenüber
organometallischen Substraten diente zum einen der 18 VE Komplex [Ru(GaCp*)3(TMM)]
(13) als ein Beispiel für einen Übergangsmetallkomplex mit einem starken π-Donorliganden
und zum anderen der Ru(II) Komplex [Ru(PCy3)2(GaCp*)2(H)2] (17), der zwei potentiell
reaktive M-H Bindungen enthält.
Synthesen
und
Charakterisierungen
von
[Ru(GaCp*)3(TMM)]
(13)
und
[Ru(PCy3)2(GaCp*)2(H)2] (17).
Die
Reaktion
des
oktaedrischen
RuII
Ausgangskomplexes
[Ru(η4-COD)(η3-
CH2CMeCH2)2]188 mit drei molaren Äquivalenten GaCp* in Toluol bei 80 °C führt innerhalb
einer Stunde in einer Ausbeute von 62% zu [Ru(GaCp*)3(TMM)] (13). Die Bildung des
TMM Liganden durch Übertragung eines Wasserstoffatoms von einer koordinierten 2Methylallyl Gruppe ist hinreichend bekannt, so dass sich in der Literatur zahlreiche Beispiele
finden lassen.189 Die 1H und
13
C Spektren stimmen mit der vorgeschlagenen TMM Struktur
überein und weisen die entsprechenden Signale für den TMM Liganden bei δ = 1.76 ppm
(6H) im 1H NMR Spektrum und bei δ = 84.1 (zentrales C Atom) sowie 24.5 ppm (CH2) im
13
C
NMR
Spektrum
Einkristallstrukturanalyse
auf.
Die
bestimmt
Festkörperstruktur
(siehe
Abbildung
von
13
52.1).
wurde
Die
durch
eine
regenschirmartige
Winkelung des TMM Liganden wird gewöhnlich durch einen sogenannten θ Winkel
quantifiziert, welcher für 13 12.0° beträgt. Dieser Wert stimmt hervorragend mit denen
bekannter TMM Komplexen überein.189,
190
Die Ru-Ga Bindungsabstände betragen im
Durchschnitt 2.348 Å, was vergleichsweise kurz, jedoch noch im Rahmen von bereits
bekannten RuII-GaCp* Komplexen mit symmetrisch am Galliumzentrum koordinierten η5Cp* Liganden ist.191
51
3. Ergebnisse
Über die Organometallchemie des Ga+ Kations
Abbildung 52.1. Molekülstruktur von [Ru(GaCp*)3(TMM)] (13) (Thermische Ellipsoide
gezeigt mit 50% Wahrscheinlichkeit, Wasserstoffatome wurden aus Gründen der
Übersichtlichkeit entfernt). Ausgewählte Bindungslängen (Å) und –winkel (°): Ru(1)-C(2)
2.019(5), Ru(1)-C(3) 2.208(4), Ru(1)-C(1) 2.228(5), Ru(1)-Ga(1) 2.3425(4), Ru(1)-Ga(2) 2.3579(6), Ga(1)Cp*Centr., Ga(2)-Cp*Centr. 1.970, Ga(2)-Cp*Centr. 2.011, C(1)-C(2) 1.444(9), C(2)-C(3), 1.412(6), C(2)-Ru(1)-C(3)
38.70(15), C(2)-Ru(1)-C(1) 39.4(2), Ga(1)-Ru(1)-Ga(2) 95.489(16), Ga(1‘)-Ru(1)-Ga(1) 95.95(2), C(2)-Ru(1)Ga(2) 121.79(19), C(3)-Ru(1)-Ga(1) 159.55(13). C(3‘)-C(2)-C(3) 116.1(6), 115.6(3), C(3)-C(2)-C(1) 115.6(3)
Bei dem zweiten exemplarischen Komplex handelt es sich um den RuII-Hydrid-Komplex
[Ru(PCy3)2(GaCp*)2(H)2] (17), der in hohen Ausbeuten aus der Umsetzung von Chaudret's
Ruthenium Polyhydrid [Ru(PCy3)2(H2)2(H)2]122,
192,
193
(Cy = Cyclohexyl) mit zwei
Äquivalenten GaCp* in Hexan unter Freisetzung von H2 resultiert. Im 1H NMR Spektrum
erscheint das durch eine Kopplung mit den PCy3 Liganden hervorgerufene Triplet für die
Hydride bei δ = -12.74 ppm. Die Verschiebung des Hydridsignals ist dabei in guter
Übereinstimmung mit terminalen Ru-H Komplexen, die in der Literatur berichtet sind.194, 195
Desweiteren liegt die transversale Relaxationszeit T1 über 200 ms, was ein klares Indiz für
eine klassische Dihydrid-Struktur von 17 ist.196,
197
Durch langsames Abkühlen einer
gesättigten Pentanlösung auf -30 °C wurden für Röntgenbeugungsuntersuchungen geeignete
Einkristalle erhalten. Verbindung 17 kristallisiert in der monoklinen Raumgruppe P2(1)/c mit
zwei beinahe identischen Molekülen in der asymmetrischen Einheit. Die Hydridliganden
konnten im Zuge der Verfeinerung der Festkörperstruktur lokalisiert werden. Die
Anwesenheit von terminalen Hydridliganden in der Festkörperstruktur von 17 wurde zudem
durch IR Spektroskopieaufnahmen bestätigt, welche zwei scharfe Absorptionsbanden in der
52
3. Ergebnisse
Über die Organometallchemie des Ga+ Kations
für terminale Ruthenium-Hydridliganden typischen Region zeigt (2026 und 2002 cm-1). Die
Koordinationsumgebung des Metallzentrums wird durch einen verzerrten Oktaeder
beschrieben, der durch zwei Phosphane in trans Position, zwei GaCp* Liganden, die cis
zueinander stehen, sowie zwei zu den GaCp* Einheiten trans liegenden Hydride definiert ist.
Wie bereits in ähnlichen Komplexen beobachtet wurde44, sind die Phosphoratome in Richtung
der Hydridliganden gebogen, was schließlich zu einem P(2)-Ru-P(1) Winkel von 145.54(4)°
führt. Der Winkel zwischen den GaCp* Liganden beträgt 94.67(2)°, während die Ru-Ga
Bindungslängen (Ru(1)-Ga(1) = 2.402(1) Å und Ru(1)-Ga(2) = 2.401(1) Å) im Bereich
anderer Ru-GaCp* Komplexen liegen. Die Cp* Gruppen sind klar η5 an die jeweiligen Ga
Atome gebunden (Ga(1)-Cp*centr. 2.046 Å, Ga(2)-Cp*centr. 2.053 Å) und weisen keine
ungewöhnlichen Merkmale auf.
Abbildung 53.1. Molekülstruktur von [Ru(PCy3)2(GaCp*)2(H)2] (17) (Thermische Ellipsoide
gezeigt mit 50% Wahrscheinlichkeit, Wasserstoffatome mit Ausnahme von H(1) und H(2)
wurden aus Gründen der Übersichtlichkeit entfernt). Ausgewählte Bindungslängen (Å) und –
winkel (°): Ru(1)-P(2) 2.323(1), Ru(1)-P(1) 2.334(1), Ru(1)-Ga(2) 2.401(1), Ru(1)-Ga(1) 2.402(1), P(1)-C(33)
1.859(4), Ga(1)-Cp*centr. 2.046 Å, Ga(2)-Cp*centr. 2.053 Å, P(1)-C(27) 1.872(4), P(1)-C(21) 1.889(4), P(2)-C(51)
1.860(4), P(2)-C(39) 1.864(4), P(2)-C(45) 1.880(4), P(2)-Ru(1)-P(1) 145.54(4), P(2)-Ru(1)-Ga(2) 102.48(4),
P(1)-Ru(1)-Ga(2) 101.25(3), P(2)-Ru(1)-Ga(1) 100.69(3), P(1)-Ru(1)-Ga(1) 101.90(3), Ga(2)-Ru(1)-Ga(1)
94.67(2)
53
Über die Organometallchemie des Ga+ Kations
3. Ergebnisse
Es sollte nicht unerwähnt bleiben, dass die zur Synthese von 17 analoge Reaktion mit AlCp*
anstelle von GaCp* offensichtlich zum entsprechenden Komplex [Ru(PCy3)(AlCp*)2(H)2]
führt, welcher im Vergleich zum Gallium Komplex 17 einen PCy3 Liganden weniger enthält.
1
H,
13
C sowie
31
P NMR Daten des Reaktionsproduktes sind in Übereinstimmung mit der
vorgeschlagenen 16 VE Struktur. Eine Trennung des bei der Reaktion freigewordenen
Phosphans PCy3 sowie eine Isolierung des Aluminiumkomplexes war jedoch nicht möglich,
woraufhin weder eine Elementaranalyse noch eine Röntgenstrukturanalyse des Komplexes
durchgeführt werden konnte.
Die Umsetzung von H+ und Ga+ mit [Ru(GaCp*)3(TMM)] (13).
Da es sich bei dem bis-allyl artigen TMM Liganden um einen starken stabilisierenden, aber
dennoch potentiell reaktiven organometallischen Co-Liganden handelt, erschien 13 als guter
Kandidat, um die Reaktivität des Ga+ Ions zu untersuchen. Da Ga+, ähnlich zu H+, eine starke
Lewis-Säure darstellt,147 wurde zunächst die Protonierung von 13 durch [H(OEt2)2][BArF]
untersucht,
bei
der
in
guten
Ausbeuten
von
ca.
60-70
%
[Ru(GaCp*)4(η3-
(CH2)2C(CH3))][BArF] (18) entsteht (Schema 55.1). Dabei handelt es sich um eine hoch
selektive Reaktion, bei der keine organometallischen Nebenprodukte durch in situ NMR
Spektroskopie nachgewiesen werden konnten. Interessanterweise wurde bei der Reaktion
keine Protolyse der Cp* Gruppe beobachtet, was in starkem Gegensatz zu der in Kapitel 3.4
beschriebenen Reaktion von [Rh(Cp*Ga)4(η1-Cp*GaCH3)] (14) mit [H(OEt2)2][BArF] steht,
bei
der
unter
Freisetzung
von
Cp*H
die
erste
terminale
GaMe
Spezies
[Rh(Cp*Ga)4(GaCH3)][BArF] (15) hergestellt und isoliert werden konnte.198-203.
Dieses Ergebnis zeigt unzweifelhaft, dass die stärker nukleophile Seite in 13 tatsächlich der
TMM Ligand ist. Die Bildung eines η3-π-allylischen Liganden durch Protonierung des η4-πallylischen TMM deutet daraufhin, dass vermutlich nur ein geringer Anteil der πBindungsenergie verloren geht, was sicherlich zur gesamten Triebkraft der Reaktion beiträgt.
Der genaue Ursprung des vierten Äquivalentes GaCp* in 18, welches letztlich zu einem 18
VE Rutheniumzentrum führt, bleibt so weit unklar. Dabei ist eine teilweise Zersetzung von 13
während der Protonierung unter Freisetzung von GaCp* wahrscheinlich. Das 1H NMR
Spektrum enthält die entsprechenden Signale des Methylallyl-Liganden bei δ = 2.52 ppm (d,
syn-H), δ = 2.29 ppm (d, anti-H) und δ = 2.11 ppm (s, CH3) und stimmt somit gut mit der
vorgeschlagenen Struktur von 18 überein. Durch langsame Diffusion von n-Hexan in eine
54
Über die Organometallchemie des Ga+ Kations
3. Ergebnisse
THF-Lösung von 18 entstanden bei Raumtemperatur hellgelbe Einkristalle. Eine
Röntgenstrukturanalyse zeigt eine verzerrte oktaedrische Struktur für das Kation
[Ru(GaCp*)4(η3-(CH2)2C(CH3))]+ (siehe Abbildung 56.1).
H3C
Ru
*CpGa
CH2
*CpGa
CH2
Ru
GaCp*
GaCp*
18
*CpGa
GaCp*
*CpGa
[BArF]-
CH2
+[H(OEt2)2][BArF]
H2C
+
CH2
H2C
H2
C
H2C
13
F
+ [{Ga2Cp*}{BAr }]
*CpGa
*CpGa
GaCp*
GaCp*
Ga
Ru
Ru
Ga
*CpGa
C
H2
2+
2 [BArF]-
GaCp*
CH2
CH2
19
Schema 55.1. Reaktionen von 13 mit [H(OEt2)2][BArF] und [Ga2Cp*][BArF] in Fluorbenzol
Dabei besetzen zwei GaCp* Liganden die axialen Positionen (Ga(2)-Ru(1)-Ga(4) transWinkel von 172.93(2)°), während sich die beiden verbleibenden GaCp* Liganden in der
Äquatorebene trans zu der Methylallyl Gruppe befinden (Ga(1)-Ru(1)-Ga(3) cis-Winkel von
99.09(2)°). Die Ru-Ga Bindungsabstände erstrecken sich über einen Bereich von 2.385(1) Å
für Ru(1)-Ga(2) bis hin zu 2.443(1) Å für Ru(1)-Ga(4) mit einer durchschnittlichen
Bindungslänge von 2.412 Å. Dies entspricht einer Verlängerung der Ru-Ga Bindungslänge
um 2.7 % verglichen zum Startkomplex [Ru(GaCp*)3(TMM)] (13). Die Cp* Gruppen der
niedervalenten Gruppe 13 Liganden kennzeichnen sich eindeutig durch η5 Bindungsmodi mit
Ga-Cp*centr Bindungslängen zwischen 1.960 und 1.995 Å (Durchschnitt 1.977 Å).
Desweiteren fällt auf, dass die an Ga(4) gebundene Cp* Gruppe signifikant in Richtung der
Äquatorebene gekippt ist, was in einem Ru-Ga(4)-Cp*centr. Winkel von 150.56° resultiert. Der
Methylallyl-Ligand ist durch eine typische η3 Koordination charakterisiert und enthält C-C
und Ru-C Bindungsabstände, die vergleichbar zu denen in bereits bekannten RutheniumAllyl-Komplexen sind.147
55
3. Ergebnisse
Über die Organometallchemie des Ga+ Kations
Abbildung 56.1. Molekülstruktur des kationischen Teils von 18 (Thermische Ellipsoide
gezeigt mit 50% Wahrscheinlichkeit, Wasserstoffatome wurden aus Gründen der
Übersichtlichkeit entfernt). Ausgewählte Bindungslängen (Å) und –winkel (°): Ru(1)-C(2)
2.194(4), Ru(1)-C(4) 2.229(5), Ru(1)-C(1) 2.235(4), Ru(1)-Ga(2) 2.3848(6), Ru(1)-Ga(3) 2.4073(6), Ru(1)Ga(1) 2.4113(7), Ru(1)-Ga(4) 2.4433(6), Ga(1)-C(8) 2.314(5), Ga(1)-C(9) 2.327(5), Ga(1)-C(6) 2.328(4), Ga(1)C(7) 2.331(4), Ga(1)-C(5) 2.334(4), Ga(1)-Cp*centr. 1.986 Å, Ga(2)-Cp*centr. 1.960 Å, Ga(3)-Cp*centr. 1.967 Å,
Ga(4)-Cp*centr. 1.995 Å, C(4)-Ru(1)-Ga(2) 99.72(14), C(4)-Ru(1)-Ga(3) 100.06(14), C(1)-Ru(1)-Ga(2)
103.24(13), C(1)-Ru(1)-Ga(1) 93.39(12), C(1)-Ru(1)-Ga(4) 83.82(13) C(1)-Ru(1)-Ga(3) 165.06(13), Ga(2)Ru(1)-Ga(3) 85.16(2), Ga(2)-Ru(1)-Ga(1) 89.44(2), Ga(3)-Ru(1)-Ga(1) 99.09(2), Ga(2)-Ru(1)-Ga(4) 172.93(2),
Ga(3)-Ru(1)-Ga(4) 88.04(2), Ga(1)-Ru(1)-Ga(4) 89.71(2), Ru-Ga(1)-Cp*centr. 169.29, Ru-Ga(2)-Cp*centr. 172.38,
Ru-Ga(3)-Cp*centr.163.15, Ru-Ga(4)-Cp*centr. 150.56
Analog zur Synthese von 18 durch Zugabe von H+, wurde 13 mit einer äquimolaren
Fluorbenzollösung des Ga+ Transferreagenzes [Ga2Cp*][BArF] bei Raumtemperatur
umgesetzt. Anschließende Kristallisation durch langesame Diffusion von n-Hexan in diese
Lösung bei 25°C lieferten die luftempfindlichen, tiefroten Kristalle von [{Ru(GaCp*)3(η3(CH2)2C(CH2(μ-Ga)))}2][(BArF)2] (19) in einer präparativen Ausbeute von ≥ 80% (Schema
55.1). Komplex 19 ist im Festkörper stabil, isomerisiert jedoch in polaren Lösemitteln wie
Fluorbenzol bei höheren Temperaturen (siehe unten). In der Festkörperstruktur von 19 (siehe
Abbildung 57.1) sind beide Rutheniumzentren in einer verzerrt oktaedrischen Umgebung
koordiniert.
56
3. Ergebnisse
Über die Organometallchemie des Ga+ Kations
Abbildung 57.1. Molekülstruktur des kationischen Teils von 19 (Thermische Ellipsoide
gezeigt mit 50% Wahrscheinlichkeit, Wasserstoffatome wurden aus Gründen der
Übersichtlichkeit entfernt). Ausgewählte Bindungslängen (Å) und –winkel (°): Ru(1)-C(4)
2.227(4), Ru(1)-C(1) 2.238(4), Ru(1)-C(2) 2.238(3), Ru(1)-Ga(1) 2.365(1), Ru(1)-Ga(4) 2.371(1), Ru(1)-Ga(2)
2.377(1), Ru(1)-Ga(3) 2.427(1), Ga(1)-Ga(1‘) 2.584(1), C(1)-C(2) 1.420(5), C(2)-C(4) 1.408(6), C(2)-C(3)
1.498(5), Ga(2)-Cp*centr. 1.916 , Ga(3)-Cp*centr. 1.961 , Ga(4)-Cp*centr. 1.918 , Ru-Ga(2)-Cp*centr. 171.14, RuGa(3)-Cp*centr. 154.26, Ru-Ga(4)-Cp*centr. 176.63, C(4)-Ru(1)-C(1) 65.36(15), C(4)-Ru(1)-C(2) 36.77(14), C(1)Ru(1)-C(2) 36.99(14), C(4)-Ru(1)-Ga(1) 116.86(10), C(1)-Ru(1)-Ga(1) 118.06(10), C(2)-Ru(1)-Ga(1)
105.08(10), C(4)-Ru(1)-Ga(4) 157.97(11), C(1)-Ru(1)-Ga(4) 93.65(10), C(2)-Ru(1)-Ga(4) 126.76(10), Ga(1)Ru(1)-Ga(4) 78.017(17), C(4)-Ru(1)-Ga(2) 94.64(11), C(1)-Ru(1)-Ga(2) 157.94(10), C(2)-Ru(1)-Ga(2)
128.27(10), Ga(1)-Ru(1)-Ga(2) 78.388(17), Ga(4)-Ru(1)-Ga(2) 104.701(19), C(4)-Ru(1)-Ga(3) 86.27(10), C(1)Ru(1)-Ga(3) 85.08(10), C(2)-Ru(1)-Ga(3) 103.01(10), Ga(1)-Ru(1)-Ga(3) 151.918(19), Ga(4)-Ru(1)-Ga(3)
85.248(18), Ga(2)-Ru(1)-Ga(3) 84.360(18)
Dabei koordiniert ein „Gallamethylallyl” Fragment, in welchem ein Proton einer
Methylgruppe des Methylallyl-Liganden durch ein Galliumatom ersetzt ist, in der
Äquatorebene des jeweiligen Rutheniumzentrums trans zu zwei GaCp* Liganden. Das
Galliumatom des Gallamethylallyl-Liganden koordiniert zudem an das benachbarte
Rutheniumzentrum und ist somit Teil von dessen axialer Koordinationsumgebung, welche
durch einen dritten GaCp* Liganden komplettiert wird. Die Ga(1)-C(3) Bindungslänge ist mit
1.959(3) Å nahezu identisch zum Ga-CH3 Bindungsabstand im isoelektronischen und
strukturell verwandten Komplex [Rh(GaCp*)4(GaCH3)][BArF] (15) (Ga-CH3 1.958(11) Å).204
57
3. Ergebnisse
Über die Organometallchemie des Ga+ Kations
Das Hauptcharakteristikum von 19 ist der kurze Ga(1)-Ga(1’) Abstand von 2.584(1) Å.
Vergleichbare Abstände wurden in GaI Clustern gefunden, wie z. B. in [Ga4(SitBu3)4])205
(2.568 Å) oder im GaII-GaII Dimer [Ga2(C6H2iPr3)4]206 (2.515 Å). Wie bereits oben erwähnt
isomerisiert eine Fluorbenzollösung von 19 irreversibel bei 30 minütiger Refluxion, bei der
ein Farbwechsel von rot nach gelb beobachtet wird.
Abbildung 58.1. Molekülstruktur des kationischen Teils von 20 (Thermische Ellipsoide
gezeigt mit 50% Wahrscheinlichkeit, Wasserstoffatome wurden aus Gründen der
Übersichtlichkeit entfernt). Ausgewählte Bindungslängen (Å) und –winkel (°): Ru(1)-C(1) 2.212,
Ru(1)-C(2) 2.244, Ru(1)-C(3) 2.294, Ru(1)-Ga(4) 2.361, Ru(1)-Ga(1) 2.392, Ru(1)-Ga(2) 2.404, Ru(1)-Ga(3)
2.405, Ru(1)-Ga(5) 2.873, Ru(2)-C(35) 2.214, Ru(2)-C(36) 2.240, Ru(2)-C(37) 2.295, Ru(2)-Ga(6) 2.367,
Ru(2)-Ga(5) 2.380, Ru(2)-Ga(8) 2.403, Ru(2)-Ga(7) 2.413, Ru(2)-Ga(1) 2.856, Ga(1)-C(37) 1.906, Ga(1)-Ga(5)
2.540, Ga(5)-C(3) 1.889, C(1)-C(2) 1.421, C(2)-C(3) 1.407, C(2)-C(4) 1.470, C(5)-C(9) 1.411, C(5)-C(6) 1.427,
C(5)-C(10) 1.497, C(1)-Ru(1)-Ga(2) 171.3, C(3)-Ru(1)-Ga(2) 113.8, Ga(4)-Ru(1)-Ga(2) 95.6, Ga(1)-Ru(1)Ga(2) 77.8, C(1)-Ru(1)-Ga(3) 102.7, C(3)-Ru(1)-Ga(4) 149.8, C(1)-Ru(1)-Ga(1) 93.9, C(3)-Ru(1)-Ga(1) 97.5,
Ga(4)-Ru(1)-Ga(1) 82.0, Ga(4)-Ru(1)-Ga(3) 89.9, Ga(1)-Ru(1)-Ga(3) 160.9, Ga(2)-Ru(1)-Ga(3) 85.9, C(1)Ru(1)-Ga(5) 67.6, C(3)-Ru(1)-Ga(5) 41.0, Ga(4)-Ru(1)-Ga(5) 126.9, Ga(1)-Ru(1)-Ga(5) 56.8, Ga(2)-Ru(1)Ga(5) 105.2, Ga(3)-Ru(1)-Ga(5) 138.9, C(3)-C(2)-C(1) 116.3, C(3)-C(2)-C(4) 122.7, C(1)-C(2)-C(4) 120.8
58
Über die Organometallchemie des Ga+ Kations
3. Ergebnisse
Durch langsame Diffusion von n-Hexan in diese Lösung bei 25°C entstanden hellgelbe
Einkristalle des Salzes [{Ru(GaCp*)3(η3-(CH2)(CH(μ-Ga)(CH3))}2][(BArF)2] (20) in einer
Ausbeute von 91 %. Die Molekülstruktur von 20 wurde ebenfalls durch eine
Röntgenstrukturanalyse bestimmt und ist der von 19 sehr ähnlich (siehe Abbildung 58.1). Bei
genauerer Betrachtung von 20 wird deutlich, dass das Galliumatom nicht länger an eine
aliphatische CH2 Gruppe sondern vielmehr an eine Vinylgruppe gebunden ist, d.h. an das
terminale C Atom der Allyl-Einheit. Somit kann die thermische Isomerisierung von 19 → 20
als “Tautomerisierung” eines Allyl-Fragmentes betrachtet werden. Als Resultat dieser
Umlagerung verlängert sich der Ru(1)-Ga(1) Abstand leicht auf 2.392 Å, wohingegen die
entsprechende Ru(1)-Ga(5) (2.873 Å) sowie die Ga(1)-Ga(5) (2.540 Å) Bindung leicht
verkürzt sind. Interessante Charakteristika der beiden Isomere 19 und 20 sind die Winkel
Ru(1)-Ga(1)-C(3`) von 159.0(1)° sowie Ru(1)-Ga(1)-C(37) und Ru(2)-Ga(5)-C(3) von
171.80° bzw. 171.81°. Die Abwinkelung der Ru-Ga-C Einheit in 20 stellt einen signifikanten
Unterschied zur linearen Anordnung in [Rh(GaCp*)4(GaCH3)][BArF] (15) (Rh-Ga-CH3
176.1(5)) dar. Offensichtlich korreliert die Triebkraft der Isomerisierung 19 → 20 mit der
Relaxation des Ru-Ga-C Winkels.
Um einen tieferen Einblick in die Bindungssituation der Isomere 19 und 20 zu erhalten,
wurden DFT Rechnungen für die Modellverbindungen 19M und 20M durchgeführt, in denen
die Cp* Ringe durch Cp Ringe ersetzt worden sind. Dabei lieferten Geometrieoptimierungen
von 19M und 20M unter Verwendung Turbomole (v5.90)206 auf RI-BP86/def2-SVP160, 207, 208
Niveau Bindungslängen und –winkel, welche nahezu identisch zu den experimentell
bestimmten Werten von 19 und 20 sind. Insbesondere der berechnete Ga-Ga Abstand in 19M
(2.580 Å, exp.: 2.584 Å) und 20M (2.577 Å, exp.: 2.540 Å) stimmen sehr gut mit dem
experimentellen Daten überein. Die Rechnungen zeigen, dass Isomer 20M um 5.76 kcal/mol
energetisch günstiger ist als 19M. Dies ist in Übereinstimmung mit dem experimentellen
Befund, dass 19 das kinetische und 20 das thermodynamische Produkt ist. Um die Art und
Weise
der
Ga-Ga
Wechselwirkungen
zu
analysieren,
wurde
209, 210
Bindungssituation in 19M und 20M mit Hilfe der AIM Methode
als
nächstes
die
untersucht. Abbildung
60.1 zeigt das Resultat der Analyse von 19M. Dabei ist zu erkennen, dass es Bindungspfade
und bindungskritische Punkte sowohl für Ga-C und Ga-Ru als auch für Ga-Ga
Wechselwirkungen gibt. Die berechnete Energiedichte von H(rb) = -0.014 Hartree/Å3 am
bindungskritischen Punkt für die Ga-Ga Wechselwirkung deutet auf eine closed-shell
Wechselwirkung mit nur schwachen kovalenten Anteil hin.44 Die berechneten Energiedichten
59
3. Ergebnisse
Über die Organometallchemie des Ga+ Kations
der Ga-Ru Bindung (H(rb) = -0.030 Hartree/Å3) und der Ga-C Bindung (H(rb) = -0.051
Hartree/Å3) belegen, dass die kovalenten Anteile in diesen Bindungen nicht viel größer sind.
Die entsprechenden Resultate für 20M unterscheiden sich nicht grundlegend von denen von
19M, so dass sie an dieser Stelle unberücksichtigt bleiben. Aus den Rechnungen lässt sich
schließen, dass in beiden Verbindungen 19 und 20 attraktive Ga-Ga Wechselwirkungen
vorhanden sind.
Abbildung 60.1. Konturliniendiagramme der Laplace-Verteilungen ∇2ρ(r) von 19M
Dargestellt ist die Laplace-Verteilung der Elektronendichte. Durchgezogene Linien zeigen Bereiche mit
steigender Ladungskonzentration an (∇2ρ(r) < 0), gestrichelte Linien zeigen Bereiche mit fallender
Ladungskonzentration an (∇2ρ(r) > 0). Die durchgezogenen Linien, die Atomkerne miteinander verbinden, sind
Bindungspfade, die durchgezogenen Linien, die Atombereiche voneinander trennen, sind die Schnittestrecken
der Nullflussflächen mit der betrachteten Ebene. Die bindungskritischen Punkte sind die Schnittpunkte der
Bindungspfade mit den Nullflussflächen.
Die Reaktion von Ga+ mit [Ru(PCy3)2(GaCp*)2(H)2] (17).
Die Protonierung des geschlossenschaligen 18VE Komplex 17 durch [H(OEt2)2][BArF] führt
zu einer unselektiven Zersetzung der Ausgangssubstanzen mit kristallinem [HPCy3][BArF] als
einziges isolierbares Zersetzungsprodukt. Die analoge Umsetzung von 17 mit einer
äquimolaren Menge [Ga2Cp*][BArF] führt unter Freisetzung von H2 selektiv zur ionischen
Verbindung [Ru(PCy3)2(GaCp*)2(Ga)][BArF] (21) (siehe Schema 61.1). Das 1H NMR von 21
60
Über die Organometallchemie des Ga+ Kations
3. Ergebnisse
enthält keine Signale in der typischen Region von Ru-H Einheiten, während die typische
Region von Ga-H Spezies durch die breiten PCy3 Signal überdeckt ist. Desweiteren weist das
zugehörige IR Spektrum weder Banden für Ru-H noch für Ga-H Streckschwingungen auf.
Durch langsame Diffusion von n-Hexan in eine Fluorbenzollösung von 21 konnten bei
Raumtemperatur farblose Kristalle von 21 erhalten werden. Das Kation von 21 lässt sich
durch eine verzerrte trigonale-bipyramidale Stuktur mit einem terminalen Ga+ Liganden in
einer äquatorialen Position beschreiben (Abbildung 62.1).
GaCp*
PCy3
H
*CpGa
Ru
*CpGa
H
PCy3
17
+ [Ga2Cp*][BArF]
- H2, GaCp*
[BArF]-
Cy3P
Ru
Ga
Cy3P
GaCp*
21
Schema 61.1. Reaktion von 17 mit [Ga2Cp*][BArF] in Fluorobenzol
Beide GaCp* Liganden besetzen die axialen Positionen in der stark verzerrten trigonalen
bipyramidalen Koordinationsumgebung um das Ru-Zentrum und bilden dabei einen Ga(1)Ru-Ga(2) Winkel von 157.94(3)°. Im Gegensatz zu Verbindung 17 befinden sich die
sperrigen PCy3 Liganden nun in der Äquatorebene, wobei der P(1)-Ru-P(2) Winkel
145.58(4)° beträgt. Die Ru-P Bindungsabstände sind im Vergleich zum Ausgangskomplex 17
leicht verlängert, was möglichweise eine Folge der cis Koordination der Phosphan Liganden
ist (2.329 Å in 2 vs. durchschnittlich 2.372 Å in 6). Die GaCp* Liganden sind signifikant in
Richtung des terminalen Ga+ Liganden gebogen, wobei die daraus resultierenden Ga(3)-RuGa(1) und Ga(3)-Ru-Ga(2) Winkel näher an 80° als an 90° sind. Die Ru-GaCp*
Bindungslängen mitteln sich zu 2.424 Å und sind vergleichbar zu denen in 17 (2.402 Å). Die
Ga-Ga Abstände betragen 2.994(2) Å bzw. 3.015(8) Å. Im Vergleich zu den Ga-Ga
Wechselwirkungen in 19 und 20 sind diese Abstände sehr lang und somit sind schwache
bindende Wechselwirkungen in den Ga+/GaCp* Paaren unwahrscheinlich.
61
3. Ergebnisse
Über die Organometallchemie des Ga+ Kations
Abbildung 62.1. Molekülstruktur des kationischen Teils von 21 (Thermische Ellipsoide
gezeigt mit 50% Wahrscheinlichkeit, Wasserstoffatome wurden aus Gründen der
Übersichtlichkeit entfernt). Ausgewählte Bindungslängen (Å) und –winkel (°): Ru(1)-Ga(3)
2.300(2), Ru(1)-P(1) 2.368(2), Ru(1)-P(2) 2.376(2), Ru(1)-Ga(1) 2.423(1), Ru(1)-Ga(2) 2.424(1), Ga(1)-Ga(3)
2.997(2), Ga(2)-Ga(3) 3.016(1), Ga(1)-Cp*centr. 1.978, Ga(2)-Cp*centr. 1.982, Ga(3)-Ru(1)-P(1) 107.16(6), Ga(3)Ru(1)-P(2) 107.18(6), P(1)-Ru(1)-P(2) 145.65(6), Ga(3)-Ru(1)-Ga(1) 78.68(4), P(1)-Ru(1)-Ga(1) 93.82(4), P(2)Ru(1)-Ga(1) 91.89(5), Ga(3)-Ru(1)-Ga(2) 79.25(5), P(1)-Ru(1)-Ga(2) 93.05(5), P(2)-Ru(1)-Ga(2) 94.20(5),
Ga(1)-Ru(1)-Ga(2) 157.93(4)
Die interessanteste Struktureigenschaft von 21 ist mit 2.300(2) Å sehr kurze Bindungslänge
vom Rutheniumatom zum „nackten“ Ga+ Ion (Ru-Ga(3)), die folglich um mehr als 5 % kürzer
als die Ru-GaCp* Bindungen ist (z. B.. Ru-Ga(1) = 2.421(1) Å). Dabei handelt es sich um
den kürzesten bisher bekannten Ru-Ga Kontakt! Das Kation Fragment in 21 könnte man
formal als ein 16e [Ru(PCy3)2(GaCp*)2] Fragment beschreiben, welches durch die Donierung
des verbliebenen Elektronen Paares des Ga+ elektronisch abgesättigt wird und wodurch
schließlich der formale 18e Komplex [(Ga)Ru(PCy3)2(GaCp*)2]+ gebildet wird. Diese
Interpretation der Situation wäre jedoch falsch. Wie bereits zuvor durch detaillierte
Bindungsanalysen der Platin-Kationen [(Ga)Pt(GaCp)n]+ (n = 3, 4) basierend auf DFT
Rechnungen gezeigt wurde, besitzt der Ga+ Ligand keinerlei Lewis basische Eigenschaften
und fungiert ausschließlich als starker σ und π Akzeptor.44 Für den vorliegenden Fall des
[(Ga)Ru(PCy3)2(GaCp*)2]+ Kations ist dabei insbesondere der formal ungesättigte 16e Platin
62
3. Ergebnisse
Über die Organometallchemie des Ga+ Kations
Komplex [(Ga)Pt(GaCp)3]+ von Interesse. Das Elektronenpaar am Ga+ besitzt hauptsächlich s
Charakter und nimmt nicht an einer koordinativen Bindung teil. Die Energy Decomposition
Analysis (EDA) von [(Ga)Pt(GaCp)n]+ zeigt durch die Zerlegung des ΔEorb Terms in die
Anteile der σ und π Orbitale starke E+-Pt π Wechselwirkungen, die zu einer Ga+-Pt πBindung mit nahezu der gleichen Stärke wie der Ga+-Pt σ-Bindung führen. Dabei ist
außerdem zu beachten, dass das d8 Rutheniumzentrum des [Ru(PCy3)2(GaCp*)2] Fragmentes
vermutlich sogar noch basischer als das Platinzentrum in [Pt(GaCp)3]+ ist. Die Argumentation
der starken σ/π Rückbindung vom basischen Rutheniumatom zum sehr guten σ/π Akzeptor
Liganden Ga+ erklärt damit den sehr kurzen Ru-Ga(3) Bindungsabstand von 2.302(2) Å.
Zusammenfassung
Die Hauptbotschaft der oben gezeigten Ergebnisse kann wie folgt zusammengefasst werden.
Erstens: der Angriff eines nukleophilen Kohlenstoffs an ein elektrophiles Ga+ Ion führt zu
einer RGa Spezies, was zu einem „Einschalten“ des freien Elektronenpaares am GaI führt.
Damit wird das elektrophile Ga+ Ion in ein basisches GaR konvertiert. Daraus resultiert eine
Koordination der monovalenten RGa Einheit an ein benachbartes Rutheniumzentrum und
führt letztlich zu einer Dimerisierung (siehe Schema 55.1, Verbindungen 18 - 20). Zweitens,
und eher unerwartet: sogar koordinierte und somit trivalente RGa Einheiten können via closed
shell Wechselwirkungen schwach aneinander binden. Dieser Effekt erscheint wichtig genug,
dass die Bildung der diskreten dimeren Strukturen 19 und 20 gegenüber der möglicher
Alternativen, wie polymerer Ketten, bevorzugt ist. Letztlich handelt es sich bei dem Komplex
21 um das zweite Beispiel einer Verbindung mit „nacktem“ Ga+, welches terminal an ein
Übergangsmetall koordiniert. Das eher basische Ru0 d8-Metallzentrum sowie die starken σ/πAkzeptor Eigenschaften des Ga+ Liganden führen zu einem extrem kurzen Ru-Ga Abstand.
Dies ist in guter Übereinstimmung mit bereits bekannten Daten des verwandten Platin
Komplexes [(Ga)Pt(GaCp*)4]+.44 Obwohl der Reaktionsmechanismus der zu 21 führenden
Reaktion von [Ru(PCy3)2(GaCp*)2(H)2}] (17) mit dem Ga+ Transferreagenz [Ga2Cp*][BArF]
unbekannt bleibt und keine Intermediate durch in situ NMR Spektroskopie beobachtet
wurden, könnte als logischer erster Schritt dieser Reaktion die Insertion von Ga+ in eine Ru-H
Bindung sein. Dies würde zu einem Intermediat führen, welches zum einen eine saurere Ru-H
und eine basischere Ga-H Einheit enthielte. Eine nachfolgende polare intra- oder
intermolekulare Reaktion würde zu einer H2 Eliminierung und zur Bildung des Endproduktes
führen. An dieser Stelle sollte es nicht unerwähnt bleiben, dass eine an ein Übergangsmetall
63
Über die Organometallchemie des Ga+ Kations
3. Ergebnisse
koordinierte GaH Spezies noch unbekannt bleibt und eine synthetische Herausforderung
darstellt (siehe Kapitel 3.2).192
3.6
Synthese und Struktur von [Mo(GaCp*)6] (22)
Eine Synthesemethode um Übergangsmetall-ECp* Komplexe herzustellen, besteht in der
Substitution labil gebundener Liganden (Nitrile, Olefine u.ä.). So führt beispielsweise die
Substitution der COD Liganden in [M(COD)2] (M = Ni, Pt) durch vier molare Äquivalente
GaCp* zur Bildung der homoleptischen, gemischtmetallischen Verbindungen [M(GaCp*)4].38,
109, 112
Im Zuge dieser Arbeit konnten unter Verwendung von d8-Metall-Olefin-Präkursoren
Übergangsmetall-GaCp* Komplexe isoliert werden, die Olefine als Co-Liganden beinhalten.
Eine komplette Substitution wurde in diesen Fällen aufgrund der starken Basizität der
Übergangsmetalle verhindert (siehe Kapitel 3.1). Desweiteren wurde anhand der
heteroleptischen Präkursoren [Ru(GaCp*)3(TMM)] (13) und [Ru(GaCp*)3(η4-COD)] (3)
gezeigt, dass es möglich ist, verbleibende, koordinierte Olefine unter hydrogenolytischen
Bedingungen abzuspalten. Dies führte bei den obigen Beispielen in Anwesenheit von GaCp*
zum
neutralen,
„nackten“
Gallium
verbrückten
Komplex
[(GaCp*)4(H)Ru(μ-
Ga)Ru(H)2(GaCp*)3] (12) (siehe Kapitel 3.3). Ein weiteres Beispiel, bei dem koordinierte
Olefine ausschließlich unter hydrogenolytischen Bedingungen durch GaCp* Liganden
substituiert werden können, stellt die Reaktion von [Mo(η4-C4H6)3] mit sechs molaren
Äquivalenten GaCp* in Toluol unter einer 3 bar H2 Atmosphäre dar. Dabei entsteht der erste
homoleptische sechsfach koordinierte Übergangsmetall-Gallium-Komplex [Mo(GaCp*)6]
(22) (siehe Schema 64.1).
GaCp*
*CpGa
[Mo(η4-C4H6)3] + 6 GaCp*
Toluol, 110°C, 3 bar H2
GaCp*
Mo
GaCp*
*CpGa
GaCp*
Schema 64.1. Synthese von [Mo(GaCp*)6] (22)
64
3. Ergebnisse
Synthese und Struktur von [Mo(GaCp*)6]
Durch langsames Kühlen einer gesättigten Mesitylenlösung von 22 auf -30 °C wurden nach
12 Stunden für eine Röntgenstrukturanalyse geeignete Einkristalle erhalten. Die wichtigsten
kristallographischen Daten sind in Tabelle 167.1 zusammengfasst und die Molekülstruktur ist
in Abbildung 65.1 ersichtlich.
Abbildung 65.1. Molekülstruktur von [Mo(GaCp*)6] (22) (Thermische Ellipsoide gezeigt mit
50% Wahrscheinlichkeit, Wasserstoffatome wurden aus Gründen der Übersichtlichkeit
entfernt). Ausgewählte Bindungslängen (Å) und –winkel (°): Mo(1)-Ga(4) 2.384(1), Mo(1)-Ga(1)
2.458(1), Mo(1)-Ga(5) 2.474(1), Mo(1)-Ga(3) 2.476(1), Mo(1)-Ga(6) 2.485(1), Mo(1)-Ga(2) 2.493(1), Ga(4)C(31) 2.088(4), Ga(1)-Cp*centr. 2.127, Ga(2)-Cp*centr. 2.091, Ga(3)-Cp*centr. 2.040, Ga(5)-Cp*centr. 2.057, Ga(6)Cp*centr. 2.024, Ga(4)-Mo(1)-Ga(1) 87.92(2), Ga(4)-Mo(1)-Ga(5) 88.77(2), Ga(4)-Mo(1)-Ga(3) 85.12(2), Ga(1)Mo(1)-Ga(5) 175.22(3), Ga(4)-Mo(1)-Ga(6) 174.35(3), Ga(3)-Mo(1)-Ga(2) 176.39(3), Ga(1)-Mo(1)-Ga(3)
90.39(2), Ga(5)-Mo(1)-Ga(3) 92.77(2), Ga(1)-Mo(1)-Ga(6) 91.15(2), Ga(5)-Mo(1)-Ga(6) 92.49(2), C(31)Ga(4)-Mo(1) 168.01(12), Ga(3)-Mo(1)-Ga(6) 89.31(2), Ga(4)-Mo(1)-Ga(2) 91.32(2), Mo-Ga(1)-Cp*centr. 175.21,
Mo-Ga(2)-Cp*centr. 167.42, Mo-Ga(3)-Cp*centr. 170.30, Mo-Ga(5)-Cp*centr. 167.36, Mo-Ga(6)-Cp*centr. 166.04
Das zentrale Molybdänatom ist in einer verzerrt oktaedrischen Anordnung von sechs GaCp*
Liganden umgeben, wobei einer der axialen GaCp* Liganden leicht in die Richtung der
Äquatorebene gebogen ist (Ga(4)-Mo(1)-Ga(3) 85.12(2)°). Die Winkel zwischen den cisLiganden liegen im Bereich von 85.12(2)° und 94.23(2)° (Ga(6)-Mo(1)-Ga(2)), während die
Winkel der trans-Liganden zwischen 174.35(3)° (Ga(4)-Mo(1)-Ga(6) und 176.39(3)° (Ga(3)Mo(1)-Ga(2)) variieren. Die Mo-Ga Abstände liegen zwischen 2.384(1) (Mo-Ga(4)) und
2.493(1) Å (Mo-Ga(2)) und sind somit sogar substantiell kürzer als die Mo-Ga Abstände in
den CO-haltigen Komplexen [fac-(Cp*Ga)3Mo(CO)3]111 (2.523(1) und 2.519(1) Å) und [cisMo(Cp*Ga)2(CO)4]109 (2.554(1) und 2.537(1) Å). Die Cp* Gruppen in 22 sind asymmetrisch
65
3. Ergebnisse
Synthese und Struktur von [Mo(GaCp*)6]
an die Galliumzentren gebunden, wobei die Bindungsmodi klar von η1- (Ga(4)) bis zu einem
nahezu symmetrischen η5-Koordinationsmodus (Ga(1), Ga(2), Ga(3), Ga(5), Ga(6)) variieren.
Dies ist vermutlich eine Konsequenz der sterischen Überfrachtung des Moleküls. Es sollte
zudem nicht unerwähnt bleiben, dass sich die Anzahl der η1-koordinierten Cp* Einheiten je
nach Art des verwendeten Kristallisations-Lösemittels unterscheidet. Die η5-Cp*-Centroid–
Ga Abstände ergeben durchschnittlich 2.069 Å und sind somit nur leicht verkürzt im
Vergleich zum freien Liganden GaCp* (2.081 Å, Gasphase, Monomer). Diese Verkürzung ist
ein indirektes Maß für die Polarität der Mo-Ga Bindung und spiegelt die guten σ-Donor
Eigenschaften von GaCp* Liganden wieder. Das 1H NMR Spektrum von 22 zeigt in C6D6 bei
Raumtemperatur ein scharfes Singulett Signal bei δ = 1.96 ppm, was durch die bereits
bekannte Fluktionalität von Cp* Gruppen an Hauptgruppenmetallen, dabei insbesondere an
Al und Ga, erklärt werden kann.125-129 Das 13C NMR Spektrum zeigt keinerlei ungewöhnliche
Eigenschaften. Die Betrachtung der Strukur von [Mo(GaCp*)6] (22), in der je nach
verwendetem Kristallisations-Lösemittels bis zu drei Cp* Ringe der GaCp* Liganden η1
gebunden
sind,
sowie
die
Synthesen
der
ersten
terminalen
GaMe
Spezies
[(GaCp*)4Rh(GaCH3)][BArF] (15) durch Protolyse eines gebundenen (η1-Cp*GaCH3)
Liganden (siehe Kapitel 3.4) und von Cp*Zn−ZnCp*7, 211, 212 aus ZnCp*2 und ZnEt2, führten
zu der Idee durch Umsetzung von 22 mit ZnMe2 einen weiteren Komplex mit einer oder
mehreren terminalen GaMe Gruppen zu erzeugen. Dabei ist auch zu berücksichtigen, dass
Zink und Gallium Nachbarn im Periodensystem sind und vergleichbare Atomvolumina sowie
Elektronegativitäten (1.7 und 1.8 für Zn bzw. Ga auf der Allred-Rochow Skala) haben. Doch
gemäß Dürrenmatt´s 8. Punkt zu den Physikern („Je planmäßiger die Menschen vorgehen,
desto wirksamer vermag sie der Zufall zu treffen.“) entstand dabei zunächst kein Komplex
mit einer terminalen GaMe Spezies, sondern der Prototyp einer neuen Verbindungsklasse [Mo(ZnCH3)9(ZnCp*)3] (23).
3.7
Zwölf Einelektronenliganden koordinieren ein Metall-zentrum:
Struktur und Bindung von [Mo(ZnCH3)9(ZnCp*)3] (23)
Seit den frühen Tagen von Alfred Werner ist neun die höchste Koordinationszahl für
Metallkomplexe monodentater Liganden.28 Der Begriff „Komplex“ [MLm] bezieht sich dabei
auf ein Molekül mit einem Zentralmetall M, das über Ligatoratome E einen Liganden L durch
66
3. Ergebnisse
Struktur und Bindung von [Mo(ZnCH3)9(ZnCp*)3]
Donor-Akzeptor-Wechselwirkungen bindet, was zu einer Kernstruktur MEn führt.27
Metallatome können auch von einem elektronpräzisen Käfig En umfasst sein. Diese
Verbindungen folgen den Wade-Mingos Regeln und werden typischerweise als endohedrale
Cluster M@En (mit n>9) bezeichnet. Beispiele für die Letzteren sind die kürzlich
synthetisierten Verbindungen M@Pb102- und M@Pb122- (M = Ni, Pd, Pt).213 In diesem Kapitel
geht es um die Synthese eines beispiellosen Moleküls mit einem MoZn12 Kern (Abbildung
69.1), das sich als ein neuartiges Bindeglied zwischen Koordinationsverbindungen und
Cluster-Molekülen anbietet.
Auf
den
ersten
Blick
erinnert
die
ikosaedrische
Struktur
des
Titelmoleküls
[Mo(ZnCH3)9(ZnCp*)3] (23) an die oben zitierten endohedralen Cluster.213 Die tatsächliche
Bindungssituation ist jedoch verblüffenderweise verschieden. Eine quantenchemische
Analyse offenbart eine einzigartige Situation, die am anschaulichsten beschrieben wird als ein
perfekt sd5 hybridisiertes Mo Atom, das sechs Zwei-Elektronen-Drei-Zentren-Bindungen
zwischen Mo und Zn ausbildet. Sechs hochliegende Valenz-MOs, die mit 12 Elektronen
besetzt sind, können klar als Mo-Zn Bindungen identifiziert werden, weitere 6 Elektronen
sind über den Zn Käfig delokalisiert und rufen nur schwache Zn-Zn Wechselwirkungen
hervor (Abbildungen 73.1, 74.1, 76.1). Vor der detailierten Besprechung dieser
Bindungssituation geht es zunächst kurz um die Synthese, sowie die analytischen und
strukturellen Eigenschaften der Titelverbindung .
Durch Umsetzung von [Mo(GaCp*)6] (22) mit einem vierzehnfachen molaren Überschuss an
ZnMe2 in Toluol bei 110 °C über eine Zeitdauer von 2 Stunden kann [Mo(ZnCH3)9(ZnCp*)3]
(23) reproduzierbar in einer Ausbeute von 82% hergestellt werden. Die beiden Ga/Zn
gemischten Verbindungen [Mo(ZnCp*)4(GaMe)4] (24) und [Mo(ZnCp*)4(ZnMe)4(GaMe)2]
(25) sind Intermediate dieser Reaktion und können in quantitativen Ausbeuten mit vier- bzw.
achtfachen molaren Überschüssen an ZnMe2 fast quantitativ isoliert werden (siehe Schema
68.1). Ähnlich wird [Mo(ZnCp*)2(ZnEt)10] (26) aus 22 und einem Überschuß an ZnEt2
gebildet, was auf die prinzipielle Zugänglichkeit einer wirklich homoleptischen Verbindung
[Mo(ZnR)12] unter den richtigen Bedingungen hinweist. Die Reaktionen beinhalten zusätzlich
zur Wanderung von Cp* vom Gallium zum Zink und Me- bzw. Et-Gruppen vom Zink zum
Gallium eine Reduktion von ZnII zu ZnI unter Bildung von Me2GaCp* und GaMe3 als
wichtigste GaIII Nebenprodukte, sowie verschiedener Fulvalenspezies.
67
3. Ergebnisse
Struktur und Bindung von [Mo(ZnCH3)9(ZnCp*)3]
+ 14 ZnEt2
[Mo(GaCp*)6] (22)
+ 14 ZnMe2
[Mo(ZnCp*)2(ZnEt)10] (26)
[Mo(ZnCp*)3(ZnMe)9] (23)
+ 4 ZnMe2
[Mo(GaMe)4(ZnCp*)4] (24)
+ 4 ZnMe2
+ 4 ZnMe2
[Mo(GaMe)2(ZnCp*)4(ZnMe)4] (25)
Schema 68.1. Herstellung der Verbindungen 23 - 26.
Die offensichtlich beteiligten Redoxreaktionen sind in Bezug auf die Vielfalt von
Nebenprodukten nicht sehr selektiv, was auf Radikalmechanismen hindeutet. In Folge werden
die resultierenden monovalenten Zinkbruchstücke ZnMe bzw. ZnEt und ZnCp* abgefangen
und an das Mo0 Zentrum gebunden (Abbildung 69.1). Die treibende Kraft und die hohe
Selektivität der Bildung von 23 aus 22 über die Ga/Zn gemischten Intermediate 24 und 25
hängen sicherlich mit der günstigen Oxidation von GaI zu GaIII und den ziemlich ähnlichen
elektronischen und sterischen Eigenschaften von monovalenten GaR und ZnR Liganden an
einem
Übergangsmetallzentrum
zusammen.
Die
Einkristall-Röntgenstrukturanalyse
(Informationen zu den wichtigsten kristallographischen Daten siehe Tabelle 167.1) von 23
offenbart eine fast vollkommene ikosaedrische Umgebung des Molybdänatoms durch zwölf
ZnR Liganden mit einer Verteilung der neun Methyl und drei η5-Cp* Liganden auf solche Art
und Weise, dass die sterisch anspruchsvollen Cp* Gruppen diejenige Positionen einnehmen,
die so weit wie möglich voneinander entfernt sind, was insgesamt zu einer C3 Symmetrie von
23 führt (Abbildung 69.1). Die Zn-C Abstände der ZnR Einheiten sind innerhalb des
erwarteten Bereichs (siehe Tabelle 70.1). Es sollte hier angemerkt werden, dass eine
Vollelementanalyse von 23 die Anwesenheit von Gallium ausschließt und die MoZn12
Zusammensetzung bestätigt. Die 1H und
13
C NMR Spektren von 23 in Lösung stimmen mit
der Struktur im Festkörper überein und zeigen chemisch gleichwertige Cp* Einheiten sowie
die erwarteten drei Sätze von Methyl-Gruppen. Temperaturabhängige Messungen (25-70 °C)
zeigen eine statische Struktur ohne Austausch der Cp* und Methyl-Positionen. So kann die
Formel von 23 als [Mo(ZnMe)9(ZnCp*)3] beschrieben werden. Die Mo-Zn Abstände sind mit
68
3. Ergebnisse
Struktur und Bindung von [Mo(ZnCH3)9(ZnCp*)3]
2.672(8) - 2.677(13) Å für Mo-ZnCp* und 2.636(9) - 2.648(13) Å für Mo-ZnMe fast gleich
lang (siehe Tabelle 70.1). Diese Werte stimmen mit den Mo-Zn Kontakten von 2.638(3) 2.683(3) Å in der dichtest gepackten intermetallischen Festkörperverbindung MoZn20.44
überein, die auch ikosaedrische MoZn12 Einheiten enthält.214
Abbildung 69.1. Molekülstruktur von [Mo(ZnMe)9(ZnCp*)3] (23) (Thermische Ellipsoide
gezeigt mit 50% Wahrscheinlichkeit, Wasserstoffatome wurden aus Gründen der
Übersichtlichkeit entfernt). Die durchgehenden Linien zwischen den Zinkatomen sind keine
kovalenten Zn-Zn Bindungen, sondern dienen dem Betrachter als Hilfe zur einfachen
Identifizierung der ikosaedrischen Koordination des Mo-Atoms in dieser Perspektive.
Ausgewählte interatomare Abstände (Å) und Winkel (°): Mo-Zn 2.637(1) - 2.677(1) Å (Mittelwert
2.651 Å), Zn-Zn 2.724(1) - 2.853(1) Å (Mittelwert 2.788 Å), Zn-CH3 1.960(10) - 1.968(10) Å (Mittelwert 1.965
Å) and Zn-Cp*(centroid) 1.951 - 1.959 Å (Mittelwert 1.956 Å).
Zwei Beispiele von dinuklearen Mo-Zn Komplexen sind bekannt mit Mo-Zn Abständen von
2.711(1)Å215 und 2.793(3)Å.216 Die Zn-Zn Abstände in 23 stimmen mit 2.724(2) - 2.853(2) Å
mit den jeweiligen Werten in MoZn20.44 überein (2.748(3) - 2.790(3) Å) überein.214 Die Zn-Zn
Einfachbindung im Dimer Cp*Zn-ZnCp* (2.305(3) Å) ist allerdings deutlich kürzer.7
69
3. Ergebnisse
Struktur und Bindung von [Mo(ZnCH3)9(ZnCp*)3]
Tabelle 70.1. Überblick der Bindungslängen in Å ermittelt aus Röntgenstrukturanalysen
M
Metallkern/
M-ZnCp*
Verbindung
M-ZnMe/
M-GaMe
Ga/Zn-Zn
M-ZnEt
MoGa4Zn4 (24)
2.539(1)-2.551(1)
-
2.391(1)-2.405(1)
2.800(1)-3.013(1)
MoGa2Zn8 (25)
2.613(1)-2.633(1)
2.467(1)-2.573(1)
2.467(1)-2.573(1)
2.733(1)-3.035(1)
MoZn12 (23)
2.672(1)-2.677(1)
2.636(1)-2.648(1)
-
2.724(2)-2.853(1)
MoZn12 (27)
2.653(1)-2.657(2)
2.626(1)-2.684(2)
-
2.708(2)-2.833(2)
RuZn10 (28)
2.552(1)-2.575(1)
2.492(1)-2.505(1)
-
2.697(1)-3.010(1)
RuZn8 (29)
2.470(1)-2.488(1)
2.438(1)-2.491(1)
-
2.721(1)-3.145(1)
RhZn9 (30)
2.470(1)-2.502(2)
2.439(2)-2.467(2)
-
2.749(2)-2.986(2)
RhGaZn7 (31)
2.433(1)-2.440(1)
2.383(1)-2.399(1)
2.383(1)-2.399(1)
2.838(1)-3.052(1)
Ni
NiZn8 (32)
2.351(1)-2.370(1)
2.313(1)-2.331(1)
-
2.746(1)-2.917(1)
Pd
PdZn8 (33)
2.447(1)-2.458(1)
2.415(1)-2.422(1)
-
2.822(1)-3.131(1)
Pt
PtZn8 (34)
2.441(1)-2.459(1)
2.402(1)-2.467(1)
-
2.812(2)-3.115(2)
PtZn8 (35)
2.457(1)-2.459(1)
2.425(1)-2.430(0)
-
2.842(1)-3.057(1)
Mo
Ru
Rh
Im Laufe der Jahre befassten sich zahlreiche Arbeiten mit Komplexen des Typs [M(ER)n] (M
= Übergangsmetall; E = Al, Ga, In; R = sterisch anspruchsvoller Substituent), wobei
Metallatome E als Ligatoren für das Zentralatom M dienen und nicht, wie in der
Koordinationschemie üblich, Nichtmetallatome.33 Der neue oktaedrische geschlossenschalige
18 Elektronenkomplex [Mo(GaCp*)6] (22) ist mit den klassischen Metallhexacarbonylen
[M(CO)6] (M = Cr, Mo, W) durch die Isolobalanalogie von CO und der carbenoiden GaI
Spezies GaCp* verwandt. Der MoGa6 Kern von 22 wird durch starke, polare Mo-Ga DonorAkzeptorbindungen zusammengehalten (siehe Kapitel 3.6). Aus diesem Blickwinkel der
Koordinationschemie ist ein Zwei-Elektronen-GaCp* Ligand zu zwei ZnCp* oder ZnMe
Liganden mit jeweils einem Elektron gleichwertig, was auf ein für Mo0 Komplexe erwartetes
18 Elektronensystem für 23 (wie auch für 24, 25 und 26) hinausläuft. Aus dem Blickwinkel
70
3. Ergebnisse
Struktur und Bindung von [Mo(ZnCH3)9(ZnCp*)3]
der klassischen Clusterchemie verlangt die Bindungssituation in einem deltahedralen closoCluster 2n+2 delokalisierte Valenzelekronen, wobei n die Zahl der Ecken des Clusters ist. Für
eine ikosaedrische Struktur sind 26 Valenzelektronen erforderlich, was zusammen mit den
Elektronenpaaren und kovalenten Bindungen der Clusterecken eine Gesamtelektronenanzahl
von 50 ergibt. Dies ist die Situation in typischen endohedralen Zintlclustern wie Pt@Pb122-, in
denen das geschlossenschalige zentrale Pt Atom eher als Templat für den elektronenpräzisen
Pb122- Käfig und nicht als Elektronendonator oder Akzeptor dient.213,
217,
218
Die
Elektronenbilanz für 23 beträgt nur 42. Entsprechend könnte 23 mit hypoelektronischen
Clustern219-221 wie Al13-
222
oder In117-
223
verglichen werden, die eine Gesamtelektronenzahl
von 40, und 16 bzw. 18 Valenzelektronen in Übereinstimmung mit dem Jellium Modell
aufweisen.224 Außerdem können die von Schnöckel et al. entdeckten metalloiden Cluster wie
[Ga19R6]- (R = C(SiMe3)3) zum Vergleich herangezogen werden.19 Dieses spezielle Beispiel
enthält einen Ga13--Kern und sechs flächenverbrückende 2e GaR Liganden mit einer
Gesamtelektronenzahl von 52. Es wird allgemeinhin akzeptiert, dass Struktur und Bindung
solcher metalloiden Cluster nicht einfach über Elektronzähl-Regeln verstanden werden
können, vielmehr muss jeder Fall individuell im Detail studiert werden.225,
226
Eine
-
quantenchemische Analyse der elektronischen Struktur von Al13 zeigt klar, dass die
Bindungssituation in diesem Cluster von der im vorliegenden Molekül 23, wie sie unten
besprochen wird, sehr verschieden ist.227 Zink ist in der Metallclusterchemie ein seltenes
Element, obwohl es viele Legierungen und intermetallische Festkörperverbindungen bildet. In
zinkreichen Übergangsmetall (M) Phasen MaZnb (b > a) sind die Zinkatome zu komplexen
homonuklearen Netzwerken mit hohen Koordinationszahlen verbunden, in denen die
Übergangsmetallatome in einer ikosaedrischen Umgebung von Zinkatomen umfasst
werden.214 Aber interessanterweise hatte diese Situation bis jetzt keinerlei Parallele in der
molekularen Chemie des Zinks. Auch homoleptische Zinkcluster Znn sind unbekannt mit der
Ausnahme einer tetraedrischen Zn4 Einheit, die in Zeolith X stabilisiert wurde.228 Die
geschlossenschalige s2 Elektronkonfiguration zusammen mit der hohen s/p Aufspaltung der
Valenzorbitale von 4 eV benachteiligen stark delokalisierte Zn-Zn Bindungen in nichtausgedehnten vergleichsweise kleinen molekularen Spezies. Der heteroleptische Cluster
[Zn9Bi11]5-
mit
einer
ikosaedrischen
M13
Kernstruktur
Zn(Zn8Bi4)
kann
bemerkenswerterweise als elektronenpräziser Zintl Cluster in Analogie zu Pt@Pb122- gut
beschrieben werden und unterscheidet sich so zweifellos von 23.226 Diese Betrachtungen
werfen die Schlüsselfrage nach der Wichtigkeit der tangentialen Zn-Zn Wechselwirkungen
innerhalb des Zn12 Käfigs im Vergleich zu den radialen Mo-Zn Wechselwirkungen auf.
71
3. Ergebnisse
Struktur und Bindung von [Mo(ZnCH3)9(ZnCp*)3]
Zur Erhellung der Bindungssituation in 23 führte die Arbeitsgruppe um Prof. Gernot Frenking
DFT Rechnungen206, 229 auf RI-BP86/TZVPP Niveau160, 207, 230, 231 durch. Die Optimierung der
Struktur 23 und der Modellverbindungen [Mo(ZnMe)12] (23M) und [Mo(ZnH)12] (23H)
liefern ausgezeichnete Übereinstimmung zwischen berechneten und experimentellen
Parametern für 23 und nur geringe Abweichungen für 23M und 23H (siehe Abbildung 72.1).
Experimentelle Daten
Mo(ZnH)12 (23H), (Ih), i= 0
Mo(ZnMe)12 (23M)
Mo−Zn = 2.651 Å
Mo−Zn = 2.684 Å
Mo−Zn = 2.680-2.682 Å
Zn−Zn = 2.788 Å
Zn−Zn = 2.822 Å
Zn−Zn = 2.818-2.822 Å
Zn−Me = 1.965 Å
Zn−Me = 1.971 Å
Abbildung 72.1. Vergleiche der experimentellen und berechneten [Mo(ZnR)12] Strukturen
Die Ih-Punktgruppe mit identischen Mo-Zn und Zn-Zn Abständen wird für 23H beibehalten.
Dadurch ist es möglich, die elektronische Struktur der Stammverbindung 23H zu analysieren,
um die Bindungssituation in den substituierten Analoga 23H und 23 zu erklären. Verbindung
23H hat 81 doppelt besetzte Valenzorbitale. Die Fragmentorbitalanalyse in ADF160, 207, 230, 231
zeigt, dass nur das HOMO-1 und das HOMO-2 signifikante Beiträge der Molybdän 5s und 4d
Valenzorbitale aufweisen. Die niedrigliegenden Orbitale von 23H sind Kombinationen der
geschlossenschaligen 3d10 Zinkorbitale und der bindenden Zn-H Orbitale. Es gibt 18
Elektronen für Mo-Zn und Zn-Zn Bindungen, wobei 6 Elektronen vom Mo und 12 Elektronen
von den Zn-Atomen kommen. Die Untersuchung der drei höchstliegenden Orbitale von 23H
(siehe Abbildung 73.1 und 74.1) zeigt eine ungewöhnliche Kombination von Metall-Ligand
(Mo-ZnH) und Bindungen innerhalb des Käfigs (Zn-Zn). Das MO-Korrelationsdiagramm für
die Mo-Zn und Zn-Zn Bindungen in 23H in Abbildung 73.1 zeigt, dass das zweithöchste
72
3. Ergebnisse
Struktur und Bindung von [Mo(ZnCH3)9(ZnCp*)3]
Molekülorbital (HOMO-1) ein fünffach entartetes hg-MO ist. Das HOMO-1 wird leicht als
eine Kombination der fünf 4d Valenzorbitale des Mo mit den 4p Valenzorbitalen der Zn
Atome sowie der 1s Funktionen der H Atome erkannt. Ein kleiner Beitrag der 4s(Zn) AOs
polarisiert leicht die 4p(Zn) AOs in Richtung des Mo.
Abbildung 73.1. MO-Korrelationsdiagramm für [Mo(ZnH)12] (23H).
73
3. Ergebnisse
Struktur und Bindung von [Mo(ZnCH3)9(ZnCp*)3]
Abbildung 74.1. HOMO-1 (hg) und HOMO-2 (ag) für [Mo(ZnH)12] (23H).
Die Mullikenanalyse der AO Koeffizienten für die hg MOs ergibt Beiträge von 37.5 % (Mo),
24.9 % (Zn) und 30.6 % (H). Das nächsttieferliegende besetzte ag MO (HOMO-2) ist eine
Kombination der 5s AOs des Mo mit hauptsächlich 4p Orbitalen der Zn Atome sowie 1s
Funktionen der H Atome (Abbildung 74.1). Die Gestalt der Molekülorbitale weist darauf hin,
dass die sechs Valenzelektronen des Mo mit sechs Valenzelektronen der (ZnH)12 Einheit
insgesamt sechs Bindungen ausbilden. Jede ZnH Gruppe trägt dabei ein einzelnes Elektron
bei, wodurch die Frage nach den Wechselwirkungen der verbleibenden sechs Elektronen der
(ZnH)12 Einheit im diamagnetischen (geschlossenschaligen) Molekül aufgeworfen wird.
Die Untersuchung des HOMO von 23H, bei dem es sich um ein dreifach entartetes (t1u)
Orbital handelt, in dem jede Komponente zwei HZn-ZnH Bindungsbeiträge aufweist,
beantwortet die Frage. Die sechs einzelnen Elektronen der ZnH Einheiten, die nicht in Mo-Zn
Bindungen investiert werden, liefern drei Elektronpaare, die über den (ZnH)12 Käfig
delokalisiert sind. Das bedeutet, dass für jedes Zn Atom nur 1/4 Elektronpaar für Zn-ZnBindungen zur Verfügung steht. Das MO Korrelationsdiagramm (Abbildung 73.1 und 74.1),
das berechnete Eigenwerte der Orbitale verwendet, zeigt an, dass die Stabilisierung der
dreifach entarteten t1u Orbitale vom (ZnH)12 durch Wechselwirkung mit den leeren 4p
Funktionen des Mo unwesentlich ist! Die Ergebnisse einer EDA232 stimmen mit diesem
Resultat überein. Die Gesamtorbitalwechselwirkungen zwischen Mo (5s14d5) und (ZnH)12 im
elektronischen Septett-Zustand belaufen sich auf 403.2 kcal/mol, die hauptsächlich aus den hg
Orbitalen (288.0 kcal/mol, 71.5 %) und den ag Orbitalen kommen (96.4 kcal/mol, 23.9 %),
74
3. Ergebnisse
Struktur und Bindung von [Mo(ZnCH3)9(ZnCp*)3]
während die Zn-Zn bindenden t1u Orbitale nur 18.3 kcal/mol (4.5 %) zum Orbitalterm
beitragen. Die Ergebnisse der EDA sind Tabelle 75.1 zu entnehmen.
Tabelle 75.1. EDA Ergebnisse von [Mo(ZnH)12] (23H) auf BP86/TZ2P Niveau. Die
wechselwirkenden Fragmente sind Mo(5s14d5) und (ZnH)12 mit der Elektronenkonfiguration
ag1hg5. Energiewerte sind in kcal/mol angegeben.
ΔEint
ΔEPauli
ΔEelstat[a]
ΔEOrb[a]
ΔEOrb(ag) [b]
ΔEOrb(hg) [b]
ΔEOrb(t1u) [b]
-348.8
594.5
-540.1 (57.2%)
-403.2 (42.8%)
-96.4 (23.9%)
-288.0 (71.5%)
-18.3(4.5%)
[a]
Die Prozentwerte in Klammern stellen den Anteil aller attraktiven Wecheselwirkungen ΔEelstat+ΔEorb dar.
[b]
Die Prozentwerte in Klammern stellen den Anteil aller Orbitalwechselwirkungen ΔEorb dar.
Obwohl die Gesamtelektronenanzahl von 23H auf eine 18-Elektronen-Konfiguration für das
Mo Atom hinweist, zeigt die tatsächliche elektronische Struktur klar, dass sich nur 12
Elektronen in den Valenzorbitalen des Mo befinden.233 Zur Deutung der Wechselwirkung des
Mo mit dem (ZnH)12 Käfig nach dem Valenzbandmodell kann von sechs sd5 Hybridorbitalen
des Mo ausgegangen werden. Intuitiv könnte man vermuten, dass eine sd5 Hybridisierung zu
einer oktaedrischen Symmetrie der sechs Orbitale führt, aber das ist nicht richtig. Bereits vor
langer Zeit konnte gezeigt werden,234-237 dass sd5 Hybridorbitale für Verbindungen MR6 (M =
Übergangsmetall) keine Oh Symmetrie liefern, sondern dass eine niedrigere Symmetrie (C3v
oder C5v) für sechsfach koordinierte Moleküle mit M-R σ-Bindungen bevorzugt wird.
Abbildung 76.1a zeigt die Orientierung der sd5 Hybridorbitale eines Übergangsmetalls und
den entsprechenden Ikosaeder. Es ist offensichtlich, dass die sd5 Hybridorbitale für
Orbitalwechselwirkungen mit zwölf Atomen, die in den Ecken eines Ikosaeders liegen (wie
die Zn Atome in (ZnH)12), ideal geeignet sind. Anders als im Falle von CH4 zum Beispiel, in
dem die vier sp3 Hybridorbitale mit je einem Elektronpaar auftreten, tragen die zwölf Lappen
der sd5 Orbitale des Mo Zentrums in 23H nur jeweils die Hälfte eines Elektronpaares. Mit
75
3. Ergebnisse
Struktur und Bindung von [Mo(ZnCH3)9(ZnCp*)3]
anderen Worten wird der MoZn12 Kern durch sechs Drei-Zentren-Zwei-ElektronenBindungen zusammengehalten, die sich jeweils über die Diagonalen des Ikosaeders
erstrecken.
(a)
(b)
Abbildung 76.1. (a) Schematische Darstellung der sd5 Hybridisierung. (b) Chemische
Bindungen in 23H aus der AIM Analyse. Die roten Punkte stellen bindungskritische Punkte
dar, die schwarzen Linien zeigen die Bindungspfade.
Die elektronische Struktur von 23H wurde auch mittels der AIM-Methode analysiert, die
durch Bader entwickelt wurde.210 Abbildung 76.1b zeigt die chemischen Bindungen in 23H,
wie sie durch AIM erhalten werden. Es gibt zwar Bindungspfade für zwölf Mo-Zn Bindungen
und zwölf Zn-H Bindungen, aber es gibt keine Zn-Zn Bindungspfade! Eine Darstellung der
Laplacefunktion ∇2ρ(r) von 23H in einer Ebene, die durch das Mo Atom und vier ZnH
Liganden aufgespannt wird, ist in Abbildung 77.1 gezeigt. Das Bindungsszenario, wie es
durch die AIM-Analyse erhalten wird, stimmt perfekt mit der sd5 Hybridisierung der
Molybdän-Valenzorbitale (Abbildung 76.1b) überein. Dabei ist zu beachten, dass sich die MO
Analyse (Abbildung 73.1), die schwache Zn-Zn Bindungswechselwirkungen vorschlägt und
die AIM-Analyse nicht widersprechen, sondern einander viel eher ergänzen. Gemäß der MO
76
3. Ergebnisse
Struktur und Bindung von [Mo(ZnCH3)9(ZnCp*)3]
Abbildung 77.1. Konturliniendiagramme der Laplace-Verteilungen ∇2ρ(r) von [Mo(ZnH)12]
(23H) Dargestellt ist die Laplace-Verteilung der Elektronendichte. Durchgezogene Linien zeigen Bereiche mit
steigender Ladungskonzentration an (∇2ρ(r) < 0), gestrichelte Linien zeigen Bereiche mit fallender
Ladungskonzentration an (∇2ρ(r) > 0). Die durchgezogenen Linien, die Atomkerne miteinander verbinden, sind
Bindungspfade, die durchgezogenen Linien, die Atombereiche voneinander trennen, sind die Schnittestrecken
der Nullflussflächen mit der betrachteten Ebene. Die bindungskritischen Punkte sind die Schnittpunkte der
Bindungspfade mit den Nullflussflächen.
Analyse gibt es 6 Elektronpaare in Orbitalen, die entlang den Mo-Zn Achsen liegen, was je
1/2 Elektronpaar für jeden Mo-Zn Kontakt ergibt. Es gibt drei Elektronpaare in Orbitalen, die
Zn-Zn Bindungscharakter haben, d.h. es steht nur 1/10 Elektronpaar für jeden der 30 Zn-ZnKontakte zur Verfügung, was, gemäß der AIM-Analyse, nicht zu einer chemischen Bindung
führt. Eine ähnliche Situation wurde in jüngsten Studien zu Verbindungen mit schwachen
Metall-Metallwechselwirkungen gefunden, die in AIM Berechnungen keinen Bindungspfad
zeigen.238 Entsprechend sind die attraktiven Zn-Zn Wechselwirkungen in 23H nicht stark
genug, um einen leeren (ZnH)12 Käfig zu stabilisieren, der insgesamt nur 36 Elektronen, 14
Elektronen weniger als 50, die "magic number" für stabile, elektronenpräzise ikosaedrische
Cluster. So kann die chemische Bindung in 23H am besten mit der Bindungssituation in
MAu12 (M = Mo, W) verglichen werden, die insgesamt nur 18 Clusterelektronen aufweisen:
Zwölf einzelne Elektronen an den Ligandatomen Au sind für bindende Wechselwirkungen
mit den sechs Valenzelektronen des zentralen Gruppe 6 Atoms verfügbar. Das Molekül
WAu12 (Ih) wurde 2002 durch Pykkö und Runeberg theoretisch vorausgesagt.21 Experimentell
wurde es bereits im selben Jahr durch Wang et al. in der Gasphase nachgewiesen.22 In einer
Studie von Autschbach et al.239 schlugen die Autoren eine geschlossenschalige 18-Elektronen77
3. Ergebnisse
Struktur und Bindung von [Mo(ZnCH3)9(ZnCp*)3]
Bindungssituation für das zentrale Wolfram-Atom mit einer deutlichen Population der p
Valenzorbitale vor. Diese Tatsache ist jedoch für das Molybdän-Atom in 23H anders.
Soll die Modellverbindung 23H und folglich die experimentelle Verbindung 23 als ein
Cluster oder als eine Koordinationsverbindung klassifiziert werden? Eine wesentliche
Charakteristik eines Clusters ist „ein System von Bindungen, die jedes Atom direkt mit seinen
Nachbarn im Polyeder verbinden“.4, 18 Entsprechend sind die wichtigsten Wechselwirkungen
in einem Cluster En diejenigen zwischen den Käfig-Atomen E. Im Gegensatz dazu weist eine
Koordinationsverbindung Donor-Akzeptor Bindungen zwischen dem Zentralmetall und den
Ligandatomen einer Kernstruktur MEn auf. Die Diskussion der elektronischen Struktur deutet
darauf hin, dass 23H weder ein Prototyp eines Clusters noch einer Koordinationsverbindung
ist. Die vorliegenden Daten zeigen, dass es Zn-Zn Bindungswechselwirkungen in 23H gibt,
die aber viel weniger wichtig sind als die Mo-Zn Bindungen. Andererseits sind die Mo-Zn
Bindungen
nicht
typische
Donor-Akzeptorbindungen,
sondern
echte
Elektronenpaarbindungen. Aus den Bindungsanalysen, die unter Verwendung von drei
verschiedenen Methoden, nämlich MO Korrelation, EDA und AIM vorgenommen wurden,
wird der Schluss gezogen, dass 23H am besten als perfekt sd5 hybridisierte
Übergangsmetallverbindung beschrieben wird, in der alle 12 Lappen der Hybridorbitale für
starke kovalente chemische Bindungen verwendet werden. Die achtzehn Valenzelektronen in
23H, die für Mo-Zn und Zn-Zn Bindungswechselwirkungen verfügbar sind, führen zu sechs
2-Elektron-3-Zentrenbindungen zwischen Mo und Zn und drei sehr schwachen delokalisierten
Zn-Zn Bindungen. Diese sind gerade stark genug zur Minimierung von Ligand-Ligand
Abstoßungen und führen so zu der außergewöhnlich hohen Koordinationszahl im Vergleich
zu üblichen Übergangsmetall-komplexen [MLn] monodendater Liganden L.
Zusammenfassend lässt sich sagen, dass die Titelverbindung 23 das erste Molekül mit solch
einer einzigartigen Bindungssituation ist, die in präparativen Mengen synthetisiert und
strukturell durch Einkristall-Röntgenstrukturanalyse charakterisiert worden ist.240 Eine Tür ist
geöffnet worden, die in ein neues Feld metallreicher Moleküle jenseits der Zintl-Grenze
führt,218 welche den Raum zwischen Koordinationsverbindungen und Clustern füllen und zur
weiteren Verknüpfung der Chemie und Physik von Molekülen und Festkörpermaterialien
beitragen werden.
78
3. Ergebnisse
3.8
Synthese und Struktur von [{Mo(CO)4}4(Zn)6(μ-ZnCp*)4]
Molekulare Ausschnitte von Mo/Zn Hume Rothery Phasen: Synthese
und Struktur von [{Mo(CO)4}4(Zn)6(μ-ZnCp*)4] (27)
Zink ist ein wichtiges metallurgisches Element und Bestandteil klassischer Legierungen wie
gewöhnlichem Messing. Mit Übergangsmetallen bildet es eine große Anzahl an binären und
ternären intermetallischen Festkörperverbindungen deren komplizierte Zusammensetzungen
und Strukturen im Konzept der Hume-Rothery Elektronphasen beschrieben werden.241,
242
Beispielsweise sind die Metallatome der Messingphase Cu5Zn8 auf vier verschiedene
kristallographische Positionen verteilt, wobei zwei eine ikosaedrische lokale Umgebung
aufweisen, während die anderen zwei Positionen von elf- bzw. dreizehneckigen Polyedern
niedrigerer Symmetrie umgeben sind.243 Dieser strukturelle Reichtum hatte jedoch - bis jetzt kein Pendant in der molekularen Chemie des Zinks. Es gibt eine mäßige Anzahl von
Molekülen mit Übergangsmetall-Zink Bindungen,244,
245
von denen einige sogar „nackte“
Zinkatome als Strukturelement aufweisen.3, 246-250 Allerdings existieren keine homoleptischen
Zinkcluster, ausgenommen einer tetraedrischen Zn4 Einheit, die in Zeolith X stabilisiert
wurde.228 Die geschlossenschalige s2 Elektronkonfiguration zusammen mit der hohen s/p
Aufspaltung der Valenzorbitale von 4 eV benachteiligen stark delokalisierte Zn-Zn
Bindungen mit Zn in der Oxidationsstufe Null.251 Sogar ligandstabilisierte Zn22+
Verbindungen, formal Zn(I), waren bis zur Entdeckung des stabilen Decamethyldizinkocens,
Cp*Zn−ZnCp* (Cp* = C5Me5),7, 252-255 gefolgt von der Synthese von LZn−ZnL (L = [{(2,6Pri2C6H3)N(Me)C}2CH]), nicht zugänglich.256
Das folgende Kapitel beschäftigt sich mit dem überraschenden Produkt, das bei der Reaktion
des oktaedrischen Mo(0) Komplexes [Mo(CO)4(GaCp*)2] mit vier Moläquivalenten ZnMe2 in
Toluol bei 100 °C entsteht. Die ursprünglich gelbe Reaktionslösung färbte sich innerhalb 1h
dunkelrot. Nach Entfernung aller flüchtigen Stoffe und Kristallisation durch langsame
Diffusion von n-Hexan in eine Fluorbenzollösung des Rohprodukts bei Raumtemperatur
wurden dunkelrote Einkristalle der Cluster-Verbindung [{Mo(CO)4}4(Zn)6(μ-ZnCp*)4] (27)
in reproduzierbaren Ausbeuten von etwa 70 % erhalten.
79
3. Ergebnisse
OC
OC
Synthese und Struktur von [{Mo(CO)4}4(Zn)6(μ-ZnCp*)4]
GaCp*
GaCp*
Mo
CO
CO
(*CpZn)(OC)4Mo
+ ZnMe2
Zn
(*CpZn)(OC)4Mo
Zn
Zn
Zn
Zn
Mo(CO)4(ZnCp*)
Zn
Mo(CO)4(ZnCp*)
27
Schema 80.1. Synthese von Verbindung 27.
Der Komplex 27 ist nur schlecht löslich in üblichen organischen Lösemitteln wie Hexan,
Toluol oder THF und ist bemerkenswert beständig, wenn er unter Edelgasatmosphäre (Ar),
auch ohne Kühlung, gelagert wird. Elementaranalyse sowie AAS schließen das
Vorhandensein von Gallium aus und stimmen ausgezeichnet mit dem aus der
Datenverfeinerung der Röntgenstrukturanalyse abgeleiteten elementaren Aufbau von 27
(siehe Abbildung 81.1) überein. Die Bildung von 27 beinhaltet eine Reduktion von ZnII nach
ZnI und Zn0, unter gleichzeitiger Oxidation von GaCp* zu einer Mischung von Cp*aGaMe3-a
Verbindungen und Bildung von Decamethylfulvalen (durch NMR bestätigt), was insgesamt
auf einen Radikalmechanismus hinweist. Ein sehr ähnlicher Austausch von Ga und Zn sowie
der Kohlenwasserstoff-Liganden mit Bildung der gleichen Nebenprodukte wird beobachtet,
wenn [Mo(GaCp*)6] mit ZnMe2 oder ZnEt2 umgesetzt wird (siehe Kapitel 3.7). Bei diesen
Reaktionen werden von 27 zwar verschiedene, aber doch verwandte Mo/Zn Produkte
erhalten, nämlich die ikosaedrischen [Mo(ZnCp*)3(ZnMe)9] (23) und [Mo(ZnCp*)2(ZnEt)10]
(26). Die molekulare Struktur von 27 im Festkörper ist in Abbildung 81.1 gezeigt.
Verbindung 27 kristallisiert in der tetragonalen Raumgruppe P-4. Weitere relevante
kristallographische Daten sind in Tabelle 168.1 zusammengestellt. Die asymmetrische Einheit
enthält zwei kristallographisch unabhängige Moleküle mit fast identischen strukturellen
Eigenschaften. Der Cluster kann als Supertetraeder beschrieben werden, das durch vier
[Mo(CO)4] Einheiten definiert wird (siehe Abbildung 82.1) und Mo-Mo Abstände zwischen
5.223 und 5.285 Å sowie perfekte Tetraederwinkel von 60.0° und 109.5° (im Durchschnitt)
aufweist. In den Kanten des Mo4 Tetraeders befinden sich Zinkatome, die jeweils zwei
Molybdänatome fast linear und symmetrisch verbrücken. Diese Situation führt zu einem
verzerrten Zn6 Oktaeder, wenn alle Zn-Zn Kontakte hervorgehoben werden. Die Verzerrung
vom idealen Oktaeder ist eine Konsequenz von vier zusätzlichen ZnCp* Liganden, die vier
80
3. Ergebnisse
Synthese und Struktur von [{Mo(CO)4}4(Zn)6(μ-ZnCp*)4]
der zwölf Mo-Zn Bindungen verbrücken, welche jeweils als "halbe Supertetraederkanten“
(Mo4) angesehen werden können. Die Zn-Zn Abstände im Zn6 Oktaeder reichen von 2.625(1)
Å bis 3.026(1) Å mit einem durchschnittlichen Zn-Zn Abstand von 2.871 Å. Die Zn-Zn
Abstände zwischen den verbrückenden ZnCp* Liganden und den nur an Metallatome
koordinierenden „nackten“ Zinkatomen betragen 2.586(1) Å und 2.626(1) Å und sind also mit
den kurzen Abständen im Zn6 Oktaeder vergleichbar. Insgesamt sind die Zn-Zn Abstände nur
wenig länger als die jeweiligen Zn-Zn Kontakte in der zinkreichen, dichtest gepackten
intermetallischen Festkörperverbindung MoZn20.44, die eine ikosaedrische MoZn12 Einheit mit
Zn-Zn Abständen von 2.748 - 2.790 Å aufweist.214, 257
Abbildung 81.1. Molekulare Struktur von [{Mo(CO)4}4(Zn)6(μ-ZnCp*)4] (27) wie sie durch
Röntgenstrukturanalyse bestimmt wurde (thermische Ellipsoide für 50% Wahrscheinlichkeit,
die Wasserstoffatome sind aus Gründen der Übersichtlichkeit nicht gezeigt). Ausgewählte
interatomare Abstände (Å) und Winkel (°): Mo-Zn 2.637(1) - 2.677(1) Å (Mittelwert 2.651 Å), Zn-Zn
2.724(1) - 2.853(1) Å (Mittelwert 2.788 Å), Zn-CH3 1.960(10) - 1.968(10) Å (Mittelwert 1.965 Å) und ZnCp*(centroid) 1.951 - 1.959 Å (Mittelwert 1.956 Å).
81
3. Ergebnisse
Synthese und Struktur von [{Mo(CO)4}4(Zn)6(μ-ZnCp*)4]
Abbildung 82.1. Der Mo4 Tetraeder (links) und der Zn6 Oktaeder (rechts) in der molekularen
Struktur von [{Mo(CO)4}4(Zn)6(μ-ZnCp*)4] (27) wie sie durch Einkristallröntgenstrukturanalyse bestimmt wurde.
Im Vergleich dazu ist der Zn-Zn Abstand im Dimer Cp*Zn−ZnCp* (2.305 Å) deutlich kürzer,
was zweifellos auf das Vorhandensein einer starken lokalisierten 2e2c Zn-Zn σ-Bindung
zwischen den monovalenten, als Zn(I) beschreibbaren, Cp*Zn Einheiten zurückzuführen ist.7,
212, 253, 254
Übergangsmetallkomplexe, die zwei oder mehr Zinkatome in unmittelbarer
Nachbarschaft zueinander aufweisen zeigen normalerweise keine bedeutenden Zn-Zn
Interaktionen. Ein repräsentatives Beispiel für diese Verbindungsklasse ist [(NiCp)2(ZnCp)4],
in dem Zn-Zn Abstände von 2.854 und 2.873 Å als nicht bindend besprochen wurden.258, 259
Bemerkenswerterweise sind die Mo-Zn Bindungsabstände mit 2.664(1) - 2.681(1) Å für MoZnCp* (Durchschnitt 2.673 Å) fast identisch mit denen für Mo-Zn6 (2.617(1) - 2.716(1) Å;
Durchschnitt 2.641 Å). Diese Werte sind nah an den Mo-Zn Abständen von MoZn20.44 (2.638
- 2.683 Å).214 Es gibt nur wenige Beispiele für molekulare Mo-Zn Komplexe und deren MoZn Abstände liegen zwischen 2.538(1) - 2.793(3) Å.215,
260
Die Mo-C-O Winkel der vier
Mo(CO)4 Fragmente sind alle im Bereich von 176.1(4)° bis 178.1(4)°, was sicherlich
ausschließt, daß Mo-Zn Bindungen mit CO Liganden verbrückt sind. Dies stimmt mit dem
Infrarotspektrum von 27 überein, das vier scharfe Absorptionsbanden bei 2001, 1970, 1931
und 1908 cm-1 aufweist, was dem typischen Bereich für terminale CO Liganden entspricht.
Offensichtlich gibt es strukturelle Analogien bezüglich der Koordinationsumgebung der
Metallatome von 27 mit der Situation in Mo/Zn Hume Rothery Phasen. Die Zinkatome in
MoZn20.44 besetzen 16 verschiedene Positionen mit Koordinationszahlen von 10 bis 16.214 Die
Mo-Atome werden in nur zwei Positionen gefunden, wobei die stärker besetzte eine
82
3. Ergebnisse
Synthese und Struktur von [{Mo(CO)4}4(Zn)6(μ-ZnCp*)4]
ikosaedrische
MoZn12
Einheit
darstellt.
Dieses
Arrangement
wird
auch
in
[Mo(ZnCp*)3(ZnMe)9] (23) angetroffen (siehe Kapitel 3.7). Stellen Sie sich ein
dreidimensionales Polymer vor, das aus (leicht verzerrten) Mo4Zn6 Supertetraedern gebildet
wird (Abbildung 83.1, links). Solch eine Anordnung enthält die Mo-Atome in einer
ikosaedrischen Zn12 Umgebung (Abbildung 83.1, Mitte) und entsprechend sitzen die
Zinkatome
in
einer
zweifach
verkappten
pentagonal-prismatischen
Zn10Mo2
Koordinationsumgebung (Abbildung 83.1, rechts). Beides sind auch die dominierenden
Motive in der Festkörperstruktur von MoZn20.44.
Abbildung 83.1. Ausschnitt aus einer (imaginären) Mo/Zn Legierugsstruktur, die aus
flächenverknüpften Mo4Zn6 super-Tetraedern (links) aufgebaut ist. Die ikosaedrische Zn12
Umgebung für die Mo Atome (Mitte) und die zweifach verkappte pentagonal-prismatischen
Zn10Mo2 Umgebung (rechts) für die Zn Atome.
Es scheint, dass die stark gebundenen CO Liganden an den Mo(CO)4 Einheiten um den Zn6
Kern ein weiteres Clusterwachstum verhindern und so 27 als einen speziellen, molekularen
„Ausschnitt“ einer polymeren intermetallischen Mo/Zn Verbindung stabilisieren.
Was gibt es über die Elektronenzahl des Molybdäns in Verbindung 27 zu sagen? ZnCp* und
CO sind Ein- bzw. Zwei-Elektronendonorliganden und jedes der „nackten“ Zinkatome trägt
zwei 4s Elektronen für kovalente Bindungen bei, was insgesamt zu 48e führt, d. h. jedes d6
Mo0 Atom erhält 12e von den Liganden. Die Molybdänzentren erfüllen also perfekt die 18
Valenzelektronenregel. Auf Basis dieser naiven ersten Interpretation ist es sinnvoll
anzunehmen, daß die als d10-Zn(0) formulierten Zn-Zentren nur über schwache „closed-shell“
Wechselwirkungen miteinander interagieren. Die Frage allerdings, ob es nicht doch
erhebliche kovalente Zn-Zn Bindungen257 innerhalb des Zn6 Oktaeders gibt, die entweder
83
3. Ergebnisse
Synthese und Struktur von [{Mo(CO)4}4(Zn)6(μ-ZnCp*)4]
verschieden oder vergleichbar mit denen im ikosaedrischen [Mo(ZnCp*)3(ZnMe)9] (23) sind,
kann nicht ohne ausführliche theoretische Rechnungen beantwortet werden. Verbindung 27 ist
präzendenzlos als erster Übergangsmetallcluster der viele Zinkatome in unmittelbarer
Nachbarschaft zueinander als Kern enthält, die offensichtlich koordinieren und miteinander
wechselwirken. Somit stellen 23 und 27 molekulare Modelle dar, Ausschnitte einer
intermetallischen
Hume-Rothery-Verbindung.
Betrachtet
man
die
beiden
neuen
ligandenstabilisierten Mo/Zn reichen Moleküle 23 und 27, so zeigt sich das hohe Potential der
Molekülfamilie [LaMb(GaCp*)c], die als Ausgangsmaterialien von hohem synthetischen
Nutzen sind. Sie dienen als Synthone für andere zu 23 und 27 ähnliche Verbindungen, die die
molekulare
Koordinations-
und
Clusterchemie
mit
intermetallischer
Festkörper-
Materialchemie auf eine neue Art verbinden.
3.9
Die
neue
Stoffklasse
der
hochkoordinierten
Zink-reichen
Übergangsmetallkomplexe
Die Untersuchung der Chemie von Komplexen der niedervalenten Gruppe 13 Verbindungen
[Mx(ER)y] führte zur Synthese der bereits in Kapitel 3.7 genannten Schlüsselverbindung
[Mo(ZnMe)9(ZnCp*)3] (23) sowie zu dem strukturverwandten, heteroleptischen Komplex
[{Mo(CO)4}4(Zn)6(μ-ZnCp*)4] (27) (siehe Kapitel 3.8). Für einen synthetischen Zugang ist
die Verwendung der Molekülfamilie [LaMb(ECp*)c] als Ausgangsmaterialien zwingend
notwendig, da die kombinierten Eigenschaften der Liganden ECp* (starke Donoren,
intrinsische Reduktionsmittel, labil gebundener Cp*-Rest) zum Tragen kommen. Die
Triebkraft in den hochselektiven Synthesen von 23 und 27 ist zweifelsohne die günstige
Oxidation von Ga(I) zu Ga(III) kombiniert mit den sehr ähnlichen elektronischen und
sterischen Eigenschaften der GaR und ZnR Liganden. Da nun zahlreiche homoleptische
einkernige Startkomplexe [M(ER)n] sowie heteroleptische Analoga [LmM(ER)n] verfügbar
waren, ergab sich im Folgenden die Fragestellung, ob das Synthesekonzept auf andere
homologe bzw. analoge Fälle ausdehnbar sei und sich somit eine neue Stoffklasse von
hochkoordinierten
Übergangsmetallkomplexen
mit
ungewöhnlichen
ZnIR
Liganden
erschließen ließe.
84
Synthesen und Strukturen der M-ZnR Komplexe der d6-Metalle
3. Ergebnisse
3.9.1 Synthesen und Strukturen der M-ZnR Komplexe der d6-Metalle
Wie bereits in Kapitel 3.7 berichtet wurde, entsteht bei der Reaktion von [Mo(GaCp*)6] (22)
mit einem vierzehnfachen molaren Überschuss an ZnMe2 in Toluol bei 110 °C über einen
Zeitraum von 2 Stunden der pseudo-homoleptische Komplex [Mo(ZnMe)9(ZnCp*)3] (23),
welcher in Form von großen, gelben, prismatischen Einkristallen in sehr guter Ausbeute bei 30 °C auskristallisiert. Wird die eingesetzte Menge an ZnMe2 verringert, führt dies zur
Bildung von zwei verschiedenen, isolierbaren Intermediaten, die sich im Gallium/Zink
Verhältnis in ihrer Ligandensphäre unterscheiden. So bildet sich bei Umsetzung von
[Mo(GaCp*)6] (22) mit vier Äquivalenten ZnMe2 in heißem Toluol der Komplex
[Mo(ZnCp*)4(GaMe)4] (24), während bei der Reaktion von 22 mit sechs molaren
Äquivalenten ZnMe2 das achtfach koordinierte Produkt [Mo(ZnCp*)4(ZnMe)4(GaMe)2] (25)
entsteht (siehe Schema 68.1). Beide Produkte, 24 und 25, bilden sich dabei in nahezu
quantitativen Ausbeuten. Bei der Analyse der neuen Verbindungen 24 und 25, sowie der
verwandter Produkte (siehe unten), ist zu berücksichtigen, dass die Natur des jeweiligen
Metalls in der Ligandensphäre (Zn vs. Ga) nicht allein durch eine Einkristallstrukturanalyse
bestimmt werden kann, da es sich bei den Elementen um Nachbarn im Periodensystem
handelt
und
sie
somit
ähnliche
Elektronendichten
aufweisen.
Die
Elementzusammensetzungen von 24 und 25 wurden durch Element-Vollanalysen sowie durch
AAS Analysen bestimmt und stimmen gut mit den vorgeschlagenen Zusammensetzungen
überein. Das 1H NMR Spektrum von 24 in C6D6 enthält zwei scharfe Singulet-Signale bei δ =
2.07 und 0.39 ppm, womit die beiden möglichen Isomere [Mo(ZnCp*)4(GaMe)4] und
[Mo(GaCp*)4(ZnMe)4] denkbar sind. DFT Berechnungen zeigen, dass das GaMe enthaltene
Isomer [Mo(ZnCp*)4(GaMe)4] um 20 kcal/mol stabiler ist als der analoge ZnMe Komplex.
Daher beschränken sich alle weiteren Diskussionen von gemischtmetallischen Ga/Zn
Komplexen auf die Annahme von GaMe Spezies anstelle von ZnMe Liganden. Durch Kühlen
einer gesättigten Hexanlösung von 24 für 12 Stunden bei -30 °C konnten Einkristalle isoliert
werden, die für röntgenkristallographische Untersuchungen geeignet waren. Wichtige
kristallographische
Daten
sind
in
Tabelle
167.1
zusammengefasst,
während
die
Molekülstruktur in Abbildung 86.1 ersichtlich ist. Komplex 24 kristallisiert in der monoklinen
Raumgruppe P2(1)/c. In der Festkörperstruktur von 24 ist das Molybdänatom von vier GaMe
und vier ZnCp* Liganden in der Art umgeben, dass die sterisch anspruchsvollen Cp*
Gruppen möglichst weit voneinander entfernt sind. Diese Anordnung führt zu einer leicht
85
3. Ergebnisse
Synthesen und Strukturen der M-ZnR Komplexe der d6-Metalle
verzerrten trigonalen dodekaedrischen Struktur mit überlagerten Zn4 und Ga4 Tetraedern (acht
Ecken, zwölf Dreiecksflächen, siehe Abbildung 86.1 und Abbildung 100.1) mit Ga2Zn2
Torsionswinkeln von 29.24° für Ga(1)-Zn(2)-Ga(2)-Zn(3) und 30.60° für Ga(3)-Zn(1)-Ga(4)Zn(4). Die Mo-ZnCp* Abstände liegen im Bereich von 2.539(1)-2.551(1) Å und sind somit
im Vergleich zu denen in [Mo(ZnCp*)3(ZnMe)9] (23) (2.672(8)-2.677(13) Å) oder
[{Mo(CO)4}4(Zn)6(μ-ZnCp*)4] (27) (2.664(1)-2.681(1) Å) deutlich verkürzt, was vermutlich
auf die niedrigere Koordinationszahl des Molybdänatoms zurückzuführen ist. In den wenigen
bekannten Beispielen von Mo-Zn Komplexen liegen die Mo-Zn Bindungsabstände mit
2.538(1)-2.793(3) Å in einem vergleichbaren Bereich.215, 216, 260-264 Die Mo-GaMe Abstände
betragen 2.391(1) - 2.405(1) Å mit einem Durchschnittswert von 2.398 Å, wohingegen die
Ga-Zn Abstände im Ga4Zn4 Metallkern zwischen 2.800(1) und 3.013(1) Å liegen (siehe
Tabelle 70.1).
Abbildung 86.1. Molekulare Struktur von [Mo(ZnCp*)4(GaMe)4] (24) wie sie durch
Röntgenstrukturanalyse bestimmt wurde (thermische Ellipsoide für 50% Wahrscheinlichkeit,
die Wasserstoffatome sind aus Gründen der Übersichtlichkeit nicht gezeigt). Ausgewählte
interatomare Abstände (Å) und Winkel (°): Mo-Zn 2.539(1) - 2.551(1) Å (Mittelwert 2.549 Å), Ga/ZnZn 2.800(1) - 3.013(1) Å (Mittelwert 2.916 Å), Mo-GaCH3 2.391(1) - 2.405(1) Å (Mittelwert 2.398 Å), Ga-CH3
1.919(6) - 1.968(5) Å (Mittelwert 1.948 Å) und Zn-Cp*(centroid) 1.950 - 1.988 Å (Mittelwert 1.973 Å).
Unter Verwendung von acht molaren Äquivalenten von ZnMe2 anstelle von vier bzw.
vierzehn molaren Äquivalenten kann ein zweites Intermediat in der Reaktion von 22 mit
86
3. Ergebnisse
Synthesen und Strukturen der M-ZnR Komplexe der d6-Metalle
ZnMe2 in hohen Ausbeuten isoliert werden. Dabei handelt es sich um den zehnfach
koordinierten Komplex [Mo(ZnCp*)4(ZnMe)4(GaMe)2] (25), welcher zusätzlich aus der
Umsetzung von 24 mit zwei molaren Äquivalenten ZnMe2 zugänglich ist (siehe Schema
68.1). Durch Kühlen einer gesättigten Toluollösung von 25 bei -30 °C für 12 Stunden konnten
Einkristalle isoliert werden, die für röntgenkristallographische Untersuchungen geeignet
waren. Wichtige kristallographische Daten sind in Tabelle 167.1 zusammengefasst, während
die die Molekülstruktur in Abbildung 87.1 ersichtlich ist. Komplex 24 kristallisiert in der
monoklinen Raumgruppe P2(1)/n.
Abbildung 87.1. Molekulare Struktur von [Mo(ZnCp*)4(ZnMe)4(GaMe)2] (25) wie sie durch
Röntgenstrukturanalyse bestimmt wurde (thermische Ellipsoide für 50% Wahrscheinlichkeit,
die Wasserstoffatome sind aus Gründen der Übersichtlichkeit nicht gezeigt). Ausgewählte
interatomare Abstände (Å) und Winkel (°): Mo-Ga/Zn 2.467(1) - 2.633(1) Å (Mittelwert 2.569 Å), ZnGa/Zn 2.733(1) - 3.035(1) Å (Mittelwert 2.844 Å), Ga/Zn-CH3 1.946(5) - 1.961(5) Å (Mittelwert 1.953 Å) und
Zn-Cp*(centroid) 1.953 - 1.987 Å (Mittelwert 1.967 Å).
Die Röntgenstrukturanalyse von 25 zeigt, dass die zehn Liganden einen Zn8Ga2 Käfig um das
Molybdänzentrum bilden, der am geeignetsten durch ein verzerrtes, zweifach verkapptes
Antiprisma beschrieben werden kann. Das Antiprisma wird charakterisiert durch
Torsionswinkel für Zn(1)-Zn(3)-Zn(2)-Zn(4) von 0.05° und 11.35° für Zn(5)-Ga(1)-Zn(6)Zn(8) sowie einen Zn(6)-Zn(2)-Zn(3) Winkel von 55.01(2)°. Die verkappenden ZnCp* und
GaMe Liganden befinden sich in leicht zuneinander gebogenen trans Positionen mit einem
87
3. Ergebnisse
Synthesen und Strukturen der M-ZnR Komplexe der d6-Metalle
resultierenden Ga(2)-Mo(1)-Zn(7) Winkel von 161.73(3)°. Die Mo-ZnCp* sowie die MoGaMe Bindungslängen sind in 25 aufgrund der höheren Koordinationszahl des
Molybdänzentrums im Vergleich zu 24 signifkikant länger (siehe Tabelle 70.1), während die
Metall-Metall Abstände im Ga2Zn8 Käfig bemerkenswerterweise nahezu unverändert bleiben
(2.733(1) - 3.035(1) Å). Das 1H NMR Spektrum von 25 zeigt in C6D6 bei Raumtemperatur
drei Signale bei δ = 2.14 (30H), 2.07 (30H) und 1.91 ppm (15H), die den Cp* Gruppen
zugeordnet werden können. Desweiteren enthält das Spektrum ein Signal für beide GaMe
Einheiten bei δ = 0.56 ppm (6H) sowie drei Signale für die vier ZnMe Gruppen bei δ = 0.14
(3H), 0.02 (6H) und -0.01 ppm (3H). Es sollte nicht unerwähnt bleiben, dass das Spektrum bei
70 °C ein fluktionales Verhalten des Moleküls aufweist, welches zu einem Signal für die Cp*
Gruppen bei δ = 2.09 ppm sowie zu einem Signal für die ZnMe Einheiten bei δ = -0.03 ppm
führt, während das Signal für die GaMe Liganden unverändert bleibt. Aufgrund der
schlechten Löslichkeit von Verbindung 25 in nahezu allen organischen Lösemitteln konnte
kein 13C Spektrum aufgenommen werden.
Der Vergleich der Festkörperstrukuren des Eduktes 22, des Intermediats 25 und des
Endprodukts 23 zeigt, dass sich in der Reaktion von 22 mit ZnMe2 mit steigender
Koordinationszahl des Molybdänzentrums das Raumangebot in der Ligandensphäre verringert
und dadurch ein sukzessiver Austausch der sterisch anspruchsvollen MCp* Einheiten gegen
vergleichsweise kleine MMe Spezies stattfindet (M = Zn, Ga). Dies führte zu der Idee,
homoleptische Komplexe des Typs [Mo(ZnR)12] durch Umsetzung von 22 mit Dialkyl-ZinkSpezies ZnR2, welche sterisch anspruchsvolle Gruppen R beinhalten, zu erzeugen. In der Tat
entsteht bei der Reaktion von 22 mit einem vierzehnfachen molaren Überschuss an ZnEt2 der
ikosaedrische Komplex [Mo(ZnCp*)2(ZnEt)10] (26) (siehe Schema 68.1, Abbildung 89.1), in
welchem nur zwei Cp* Gruppen vorhanden sind. Offensichtlich führt die Verwendung von
sterisch anspruchsvolleren ZnEt Gruppen zu weniger ZnCp* Gruppen im Endprodukt,
wodurch ein prinzipieller Zugang zu homoleptischen Komplexen [Mo(ZnR)12] durch
Auswahl eines geeigneten Substituenten R als durchaus möglich erscheint. Die weiteren
strukturellen Eigenschaften von 26 sind nahezu identisch zu denen in 23 (siehe Tabelle 70.1)
und somit wird an dieser Stelle auf eine ausführliche Strukturdiskussion verzichtet.
Das 1H NMR Spektrum in C6D6 zeigt bei Raumtemperatur die Signale verschiedener Isomere,
die aufgrund der Kompliziertheit des Spektrums nicht weiter zugeordnet bzw. identifiziert
werden können.
88
3. Ergebnisse
Synthesen und Strukturen der M-ZnR Komplexe der d6-Metalle
Abbildung 89.1. Molekulare Struktur von [Mo(ZnCp*)2(ZnEt)10] (26) wie sie durch
Röntgenstrukturanalyse bestimmt wurde (thermische Ellipsoide für 50% Wahrscheinlichkeit,
die Wasserstoffatome sind aus Gründen der Übersichtlichkeit nicht gezeigt). Ausgewählte
interatomare Abstände (Å) und Winkel (°): Mo-Zn 2.626(1) - 2.684(2) Å (Mittelwert 2.651 Å), Zn-Zn
2.708(2) - 2.833(2) Å (Mittelwert 2.787 Å), Zn-C2H5 1.95(2) - 2.17(3) Å (Mittelwert 2.021 Å) und ZnCp*(centroid) 1.935 - 1.942 Å (Mittelwert 1.939 Å).
3.9.2 Synthesen und Strukturen der M-ZnR Komplexe der d8-Metalle
Analog zu den Synthesen der pseudohomoleptischen Komplexe 23 und 26 führt die Reaktion
von [(GaCp*)4(H)Ru(μ-Ga)Ru(H)2(GaCp*)3] (12) mit 22 molaren Äquivalenten an ZnMe2 in
Toluol bei 80 °C in einer Ausbeute von 72% zum verwandten, ausschließlich Zinkkoordinierten Komplex [Ru(ZnCp*)4(ZnMe)6] (28) (siehe Schema 90.1). Eine alternative
Darstellung von 28 besteht in der Verwendung von [Ru(GaCp*)6(Cl)2] als Rutheniumquelle,
wobei sich 28 nach dieser Methode in wesentlich geringeren Ausbeuten isolieren lässt (siehe
Experimenteller Teil).
89
Synthesen und Strukturen der M-ZnR Komplexe der d8-Metalle
3. Ergebnisse
[Ru 2(Ga)(GaCp*)7 (H)3]
+22 ZnMe 2
2 [Ru(ZnCp*)4 (ZnMe)6] (28)
+ 16 ZnMe 2
+ 2 ZnMe 2
2 [Ru(ZnCp*) 4(ZnMe)4(H)2] (29)
Schema 90.1. Synthesen der hochkoordinierten Komplexe [Ru(ZnCp*)4(ZnMe)6] (28) und
[Ru(ZnCp*)4(ZnMe)4(H)2] (29)
Die Festkörperstruktur von 28 konnte durch eine Einkristallstrukturanalyse bestimmt werden.
Eine POVRAY Darstellung von 28 ist in Abbildung 90.1 gezeigt und wichtige
kristallographische Daten sind in Tabelle 168.1 aufgeführt.
Abbildung 90.1. Molekulare Struktur von [Ru(ZnCp*)4(ZnMe)6] (28) wie sie durch
Röntgenstrukturanalyse bestimmt wurde (thermische Ellipsoide für 50% Wahrscheinlichkeit,
die Wasserstoffatome sind aus Gründen der Übersichtlichkeit nicht gezeigt). Ausgewählte
interatomare Abstände (Å) und Winkel (°):Ru-Zn 2.492(1) - 2.575(1) Å (Mittelwert 2.521 Å), Zn-Zn
2.697(1) - 3.010(1) Å (Mittelwert 2.802 Å), Zn-CH3 1.931(7) - 1.973(8) Å (Mittelwert 1.950 Å) und ZnCp*(centroid) 1.945 - 2.032 Å (Mittelwert 1.973 Å).
Die Einkristall-Röntgenstrukturanalyse von 28 offenbart eine verzerrte, zweifach-verkappte,
quadratisch antiprismatische Koordinationsumgebung des Rutheniumzentrums durch zehn
ZnR Liganden. Das quadratische Antiprisma wird definiert durch Zn4 Torsionswinkel von
90
Synthesen und Strukturen der M-ZnR Komplexe der d8-Metalle
3. Ergebnisse
0.06° für Zn(1)-Zn(2)-Zn(3)-Zn(4) und 9.12° für Zn(7)-Zn(5)-Zn(10)-Zn(8) sowie durch
einen Zn(1)-Zn(2)-Zn(5) Winkel von 59.67(2)°. Die verkappenden Liganden ZnCp* und
ZnMe sind in trans Positionen leicht zueinander gebogen, was in einem Zn(9)-Ru(1)-Zn(6)
Winkel von 160.15(2)° resultiert. Bei einem Vergleich der Ru-Zn Abstände in Komplex 28
fällt auf, dass im Falle der Cp* substituierten ZnIR Liganden der Abstand leicht vergrößert ist
als bei den entsprechenden ZnMe Spezies (2.552(1)-2.575(1) Å für ZnCp* vs. 2.492(1)2.505(1) Å für ZnMe, siehe Tabelle 70.1). Insgesamt sind die Ru-Zn Abstände jedoch
deutlich kürzer als die Ru-ZnEt Bindungsabstände in [{(η5-C5Me5)Ru}3(μ3-ZnEt)(μ-H)3(μ3H)] (2.675(1) und 2.656(1) Å) und [{(η5-C5Me5)Ru}3(μ3-ZnEt)2(μ-H)3] (2.641(1) Å).244 Die
Zn-C Abstände der ZnR Einheiten sind innerhalb des erwarteten Bereichs (siehe Tabelle
70.1). Die 1H und
13
C NMR Spektren in Lösung stimmen mit der Struktur im Festkörper
überein und zeigen chemisch gleichwertige Cp* und Methyl Einheiten bei δ = 2.07 (60H,
ZnCp*) und -0.19 ppm (18H, ZnMe) im 1H NMR Spektrum sowie bei δ = 110.6 (C5Me5),
14.6 (CH3) und 10.6 ppm (C5Me5) im 13C NMR Spektrum.
Wird in der Umsetzung von 12 mit ZnMe2 die Menge des eingesetzten ZnMe2 auf einen 16fachen molaren Überschuss reduziert, kann das Intermediat [Ru(ZnCp*)4(ZnMe)4(H)2] (29) in
guten Ausbeuten isoliert werden (siehe Schema 90.1). Das 1H NMR Spektrum von Komplex
29 enthält bei Raumtemperatur in C6D6 drei scharfe Singulett Signale bei δ = 2.00, -0.11 und
-15.89 ppm in einem Integralverhältnis von 60:12:2, welche den vier ZnCp*, vier ZnMe
sowie den beiden Hydridliganden zugeordnet werden können. Die chemische Verschiebung
der Hydridliganden ist charakteristisch für terminal gebundene Hydridliganden.156,
265
Die
Linienbreite und die chemische Verschiebungen der Signale zeigt keine Veränderung beim
Herunterkühlen einer d8-Toluollösung auf -100 °C, was auf eine symmetrische
Lösungsstruktur von 29 hindeutet. Desweiteren schließt die transversale Relaxationszeit T1
von über 300 ms die Anwesenheit eines nicht-klassischen Diwasserstoff-Liganden in 29 klar
aus.155, 156 Die Anwesenheit von terminalen Ru-H Einheiten in der Festkörperstruktur von 29
wurde zudem durch IR Spektroskopieaufnahmen bestätigt, welche zwei scharfe
Absorptionsbanden in der für terminale Ruthenium-Hydridliganden typischen Region (1923
und 1901 cm-1) zeigt.
Durch langsames Abkühlen einer gesättigten Toluollösung auf -30 °C wurden für
Röntgenbeugungsuntersuchungen
geeignete
Einkristalle
erhalten.
Wichtige
kristallographische Daten sind in Tabelle 168.1 zusammgefasst und eine Darstellung der
Molekülstruktur von 29 ist in Abbildung 92.1 ersichtlich. Komplex 30 kristallisiert in der
91
3. Ergebnisse
Synthesen und Strukturen der M-ZnR Komplexe der d8-Metalle
monoklinen Raumgruppe P2(1)/n. Die Koordinationsumgebung des Zentralmetalls wird, wie
bereits im achtfach koordinierten Komplex [Mo(ZnCp*)4(GaMe)4] (24), durch einen
verzerrten, trigonalen Dodekaeder beschrieben, in welchem sich ZnMe und ZnCp* Einheiten
aufgrund des hohen sterischen Anspruchs der Cp* Gruppen auf den Eckpositionen
alternierend abwechseln. Die für einen Dodekaeder charakteristischen Torsionswinkel
betragen für Zn(1)-Zn(7)-Zn(2)-Zn(8) 39.30° und für Zn(3)-Zn(5)-Zn(4)-Zn(6) 29.59°.
Aufgrund der niedrigeren Koordinationszahl des Rutheniumzentrums in 29 im Vergleich zu
28 erhöht sich das Platzangebot in der Ligandensphäre, wodurch sich die Zn-Zn Abstände im
Zn8 Käfig in 29 im Vergleich zu dem Zn10 Käfig in 28 verlängern, während sich gleichzeitig
die Ru-Zn Abstände deutlich verkürzen (siehe Tabelle 70.1).
Abbildung 92.1. Molekulare Struktur von [Ru(ZnCp*)4(ZnMe)4(H)2] (29) wie sie durch
Röntgenstrukturanalyse bestimmt wurde (thermische Ellipsoide für 50% Wahrscheinlichkeit,
die Wasserstoffatome sind aus Gründen der Übersichtlichkeit nicht gezeigt). Ausgewählte
interatomare Abstände (Å) und Winkel (°): Ru-Zn 2.438(1) - 2.491(1) Å (Mittelwert 2.470 Å), Zn-Zn
2.721(1) - 3.145(1) Å (Mittelwert 2.857 Å), Zn-CH3 1.951(6) - 1.967(6) Å (Mittelwert 1.957 Å) und ZnCp*(centroid) 1.917 - 1.945 Å (Mittelwert 1.937 Å).
Desweiteren ist es durchaus bemerkenswert, dass sich das zehnfach koordinierte Produkt 28
bei der Reaktion von 29 mit weiteren ZnMe2 bildet. Daraus kann gefolgert werden, dass in der
Umsetzung von 12 mit ZnMe2 zuerst die GaCp* Liganden in 12 mit ZnMe2 reagieren und
dabei intermediär der heteroleptische Komplex 29 entsteht. Die anschließende Reaktion
92
3. Ergebnisse
Synthesen und Strukturen der M-ZnR Komplexe der d8-Metalle
zwischen den Ru-H Spezies und ZnMe2, welche schließlich zum Endprodukt 28 führt, ist eine
bereits aus der Literatur bekannte Methode Übergangsmetall-Zink-Bindungen zu knüpfen.193,
244
3.9.3 Synthesen und Strukturen der M-ZnR Komplexe der d9-Metalle
Die Reaktion des ausschließlich Ga-Liganden beinhaltenden Komplexes [(Cp*Ga)4Rh(η1Cp*GaCH3)] (14) mit einem siebenfachen molaren Überschuss an ZnMe2 führt in Toluol bei
110 °C über einen Zeitraum von einer Stunde in einer Ausbeute von 80 % zu dem neunfach
koordinierten Komplex [Rh(ZnCp*)3(ZnMe)6] (30). Das Ga/Zn gemischtmetallische
Intermediat [Rh(GaMe)(ZnCp*)4(ZnMe)3] (31) kann in dieser Reaktion unter Verwendung
von sieben anstelle von elf molaren Äquivalenten an ZnMe2 in nahezu quantitativen
Ausbeuten isoliert werden (siehe Schema 93.1).
[(Cp*Ga)4Rh(η 1-Cp*GaCH 3)]
+11 ZnMe 2
[Rh(ZnCp*)3(ZnMe)6 ] (30)
+ 7 ZnMe 2
+ 2 ZnMe 2
[Rh(GaMe)(ZnCp*)4(ZnMe)3 ] (31)
Schema 93.1. Darstellung der hochkoordinierten Komplexe [Rh(ZnCp*)3(ZnMe)6] (30) und
[Rh(GaMe)(ZnCp*)4(ZnMe)3] (31)
Durch langsames Kühlen einer gesättigten Toluollösung auf -30 °C wurden für
röntgenkristallographische Untersuchungen geeignete Einkristalle von 30 erhalten. Wichtige
kristallographische Daten sind in Tabelle 169.1 zusammengefasst und eine Darstellung der
Molekülstruktur ist in Abbildung 94.1 ersichtlich. Die Einkristallstrukturanalyse von 30
offenbart eine nahezu vollkommene einfach verkappte quadratisch antiprismatische
Koordinationsumgebung des Rhodiumatoms durch neun ZnR Liganden mit einer Verteilung
von sechs Methylgruppen und der Cp* Substituenten in der Art und Weise, dass die η593
3. Ergebnisse
Synthesen und Strukturen der M-ZnR Komplexe der d9-Metalle
koordinierten, sterisch anspruchsvollen Cp* Gruppen sich in denjenigen Positionen befinden,
die möglichst weit voneinander entfernt sind. Diese Anordnung führt insgesamt zu einer C3
Symmetrie von 30 (siehe auch Abbildung 100.1). Das quadratische Antiprisma wird durch
perfekte Torsionswinkel von nahezu 0° für die jeweiligen Zn Ebenen und einem Zn1-Zn1’Zn4 Winkel von 56.73° definiert.
Abbildung 94.1. Molekulare Struktur von [Rh(ZnCp*)3(ZnMe)6] (30) wie sie durch
Röntgenstrukturanalyse bestimmt wurde (thermische Ellipsoide für 50% Wahrscheinlichkeit,
die Wasserstoffatome sind aus Gründen der Übersichtlichkeit nicht gezeigt). Ausgewählte
interatomare Abstände (Å) und Winkel (°): Rh-Zn 2.439(2) - 2.502(2) Å (Mittelwert 2.459 Å), Zn-Zn
2.749(2) - 2.986(2) Å (Mittelwert 2.844 Å), Zn-CH3 1.952(14) - 2.020(14) Å (Mittelwert 1.979 Å) und ZnCp*(centroid) 1.911-1.965 Å (Mittelwert 1.928 Å).
Der verkappende ZnCp* Ligand befindet sich direkt oberhalb des Zentrums der Zn1-Zn1’Zn2-Zn2’ Ebene mit einem resultierenden Rh-(Zn1-Zn1’-Zn2-Zn2’)centroid-Zn5 Winkel von
174.40°. Die Rh-Zn Abstände sind mit 2.470(1) - 2.502(2) Å für Rh-ZnCp* und 2.439(2) 2.467(2) Å für Rh-ZnMe fast gleich lang (siehe Tabelle 70.1). Bisher sind zwei Beispiele mit
Rh-ZnR Einheiten bekannt, in denen allerdings verbrückende Rh-Zn-Rh Bindungen auftreten.
Die Bindungsabstände in den genannten Beispielen betragen 2.558(1) und 2.513(1) Å in
[{({Pri2P(CH2)3PPri2)Rh}2(μ-H)2(μ-ZnCH2Ph)2]266 sowie 2.453(1) - 2.6115(8) Å in
[{(dippp)Rh}2(μ-H)2(μ-ZnC5H5)2]267 und sind somit vergleichbar zu denen in 30. Die
interatomaren Zn-Zn Abstände im Zn9 Käfig betragen 2.749(2) - 2.986(2) Å. Die 1H und 13C
94
3. Ergebnisse
Synthesen und Strukturen der M-ZnR Komplexe der d9-Metalle
NMR Spektren des Produktes sind in Übereinstimmung mit der Festkörperstruktur und zeigen
neben den chemisch äquivalenten Cp* Gruppen ein Signal für die ebenfalls chemisch
äquivalenten Methylgruppen. Temperaturabhängige Messungen (25-70 °C) zeigen eine
statische Struktur ohne Austausch der Cp* und Methyl-Positionen.
Die
Molekülstruktur
des
Intermediats
31
konnte
ebenfalls
durch
eine
Einkristallröntgenstrukturanalyse bestätigt werden.
Abbildung 95.1. Molekulare Struktur von [Rh(GaMe)(ZnCp*)4(ZnMe)3] (31) wie sie durch
Röntgenstrukturanalyse bestimmt wurde (thermische Ellipsoide für 50% Wahrscheinlichkeit,
die Wasserstoffatome sind aus Gründen der Übersichtlichkeit nicht gezeigt). Ausgewählte
interatomare Abstände (Å) und Winkel (°): Rh-Zn/Ga 2.383(1) - 2.440(1) Å (Mittelwert 2.415 Å), ZnGa/Zn 2.838(1) - 3.052(1) Å (Mittelwert 2.899 Å), Ga/Zn-CH3 1.953(4) - 1.957(4) Å (Mittelwert 1.955 Å) und
Zn-Cp*(centroid) 1.919 - 1.942 Å (Mittelwert 1.927 Å).
Eine exakte Zuordnung des GaMe Liganden in der Festkörperstruktur ist aus den vorhanden
Datensätzen leider nicht möglich. Eine AAS Analyse stimmt jedoch gut mit der
vorgeschlagenen Elementzusammensetzung überein. In dem achtfach koordinierten Komplex
31 ist das Zentralmetall Rh von seinen acht Liganden in einer verzerrten, trigonal
dodekaedrischen Koordinationssphäre umgeben (siehe Abbildung 95.1), womit Verbindung
31 isostrukturell zum Molybdän-Komplex 24 ist. Die Torsionswinkel betragen 19.62° für
Zn(1)-Zn(5)-Ga(1)-Zn(7) und 20.63° für Zn(4)-Zn(2)-Zn(6)-Zn(3), während der Zn(3)-Zn(7)95
3. Ergebnisse
Synthesen und Strukturen der M-ZnR Komplexe der d9-Metalle
Ga(1) Winkel 63.80(2)° beträgt. Wie bereits anhand der gemischmetallischen Mo-Ga/Zn
Komplexe 24, 25 und 23 sowie der Ru-Ga/Zn Verbindungen 28 und 29 gezeigt wurde, führt
auch im Falle der entsprechenden Rh-Ga/Zn Verbindungen 30 und 31 eine niedrigere
Koordinationszahl des Übergangsmetalls zu einer Verkürzung der Metall-Ligand-Bindungen,
wohingegen sich die Ligand-Ligand Abstände im Käfig aufgrund des erhöhten Raumangebots
vergrößern. So betragen die Rh-Zn Abstände 2.433(1) - 2.440(1) Å für Rh-ZnCp* und
2.394(1) - 2.399(1) Å für Rh-ZnMe, während die Ga/Zn-Zn Kontakte 2.838(1) - 3.052(1) Å
voneinander entfernt sind (siehe Tabelle 70.1). Im 1H NMR Spektrum können die Signalsätze
zweier Isomere im Verhältnis 3:1 identifiziert werden (siehe Experimenteller Teil).
3.9.4 Synthesen und Strukturen der M-ZnR Komplexe der d10 Metalle
Durch Umsetzungen der homoleptischen d10-Komplexe [M(GaCp*)4] (M = Ni, Pd, Pt) mit
jeweils zehn molaren Äquivalenten an ZnMe2 entstehen in Toluol bei 80°C die
entsprechenden pseudo-homoleptischen ZnIR-haltigen Komplexe [M(ZnCp*)4(ZnMe)4] (M =
Ni (32), Pd (33), Pt (34)) (siehe Schema 96.1) reproduzierbar in hohen Ausbeuten.
[M(GaCp*)4]
+10 ZnMe2
[M(ZnCp*)4(ZnMe)4] (M = Ni (32), Pd (33), Pt (34))
Schema 96.1. Synthese der pseudo-homoleptischen Komplexe [M(ZnCp*)4(ZnMe)4] (M = Ni
(32), Pd (33), Pt (34)
Da die neuen Verbindungen 32, 33 und 34 isostrukturell zueinander sind, wird an dieser Stelle
nur [Pd(ZnCp*)4(ZnMe)4] (33) als ein repräsentatives Beispiel diskutiert. Durch Kühlen einer
gesättigten Toluollösung auf -30 °C konnten für eine Röntgenstrukturanalyse geeignete
Einkristalle von 33 isoliert werden. Wichtige kristallographische Daten sind in Tabelle 169.1
zusammengefasst und die Molekülstruktur ist in Abbildung 97.1 gezeigt.
96
Synthesen und Strukturen der M-ZnR Komplexe der d10-Metalle
3. Ergebnisse
Abbildung 97.1. Molekulare Struktur von [Pd(ZnCp*)4(ZnMe)4] (33) wie sie durch
Röntgenstrukturanalyse bestimmt wurde (thermische Ellipsoide für 50% Wahrscheinlichkeit,
die Wasserstoffatome sind aus Gründen der Übersichtlichkeit nicht gezeigt). Ausgewählte
interatomare Abstände (Å) und Winkel (°): Pd-Zn 2.415(1) - 2.458(1) Å (Mittelwert 2.435 Å), Zn-Zn
2.822(1) - 3.131(1) Å (Mittelwert 2.897 Å), Zn-CH3 1.950(5) - 1.967(5) Å (Mittelwert 1.959 Å) und ZnCp*(centroid) 1.920 - 1.946 Å (Mittelwert 1.934 Å).
Verbindung
33
kristallisiert
in
der
monoklinen
Raumgruppe
P2(1)/n.
In
der
Festkörperstruktur von 33 ist das Zentralatom Palladium von seinen acht Liganden in
Übereinstimmung mit den isostrukturellen achtfach koordinierten Komplexen 24 und 31 in
einer leicht verzerrten, trigonalen dodekaedrischen Struktur mit überlagerten (ZnCp*)4 und
(ZnMe)4 Tetraedern umgeben. Die Zn4 Torsionswinkel betragen 21.41° für Zn(5)-Zn(3)Zn(7)-Zn(1) und 20.15° für Zn(4)-Zn(6)-Zn(2)-Zn(8), während der Zn(1)-Zn(5)-Zn(8) Winkel
56.96° beträgt. Die Pd-Zn Bindungslängen sind mit 2.447(1) - 2.458(1) Å für Pd-ZnCp* und
mit 2.415(1) - 2.422(1) Å für Pd-ZnMe fast gleich lang, aber insgesamt jedoch deutlich kürzer
als die Pd-Zn Abstände in einer ZnPd Legierung (2.65 A°).268 Bis zur Abgabe dieser Arbeit
gab es lediglich ein weiteres Beispiel einer molekularen, kovalenten Pd-Zn Bindung, nämlich
in (FPNP)Pd–Zn–Pd(PNPF), in welchem die Pd-Zn Abstände mit 2.379(1) Å und 2.372(10) Å
nur wenig kürzer sind als in 33.3 Die Zn-Zn Abstände innerhalb des Zn8 Käfigs betragen
2.822(1) - 3.131(1) Å und sind somit vergleichbar zu den bereits beschriebenen interatomaren
Abständen in der Ligandenhülle der achtfach koordinierten Komplexe 24 und 31 (siehe
Tabelle 70.1) Das 1H NMR Spektrum von 33 enthält bei Raumtemperatur in C6D6,
entsprechend der Symmetrie der Festkörperstruktur, zwei Signale bei δ = 2.08 (ZnCp*) und
97
3. Ergebnisse
Synthesen und Strukturen der M-ZnR Komplexe der d10-Metalle
0.04 ppm (ZnMe) in dem erwarteten Integralverhältnis von 60:12. Das
13
C NMR Spektrum
enthält keinerlei ungewöhnliche Merkmale.
An dieser Stelle sollte es nicht unerwähnt bleiben, dass die Reaktion von [Pt(GaCp*)4] mit
ZnEt2 anstelle von ZnMe2 zur Bildung der analogen, isostrukturellen Verbindung
[Pt(ZnCp*)4(ZnEt)4] (35) führt (siehe Abbildung 98.1). Aufgrund nahezu identischer
Strukturparameter wird auf eine detaillierte Besprechung der Molekülstruktur verzichtet und
auf Tabelle 70.1 verwiesen.
Abbildung 98.1. Molekulare Struktur von [Pt(ZnCp*)4(ZnEt)4] (35) wie sie durch
Röntgenstrukturanalyse bestimmt wurde (thermische Ellipsoide für 50% Wahrscheinlichkeit,
die Wasserstoffatome sind aus Gründen der Übersichtlichkeit nicht gezeigt). Ausgewählte
interatomare Abstände (Å) und Winkel (°): Pt-Zn 2.425(1) - 2.459(1) Å (Mittelwert 2.443 Å), Zn-Zn
2.842(1) - 3.057(1) Å (Mittelwert 2.938 Å), Zn-C2H5 1.949(3) - 1.959(3) Å (Mittelwert 1.954 Å) und ZnCp*(centroid) 1.930-1.931 Å (Mittelwert 1.931 Å).
3.9.5 Zusammenfassende Betrachtung zu den Synthesen und Strukturen der
Stoffklasse der hochkoordinierten Zink-reichen Übergangsmetallkomplexe
Seit der Begründung der Koordinationschemie durch Alfred Werner im Jahre 189328 ist der
Stand der Forschung und damit auch des Lehrbuchwissens, dass Koordinationszahlen (KZ) n
98
3. Ergebnisse
Synthesen und Strukturen der M-ZnR Komplexe - Zusammenfassung
≥ 10 in diskreten Molekülen bzw. Komplexen [MLn] für einzähnige Liganden L unbekannt
sind. Höhere Koordination ist strikt auf Komplexe mit mehrzähnigen Liganden beschränkt
bzw. auf dicht oder dichtest gepackte Festkörperverbindungen. Die höchste Koordinationszahl
in Polyhydridkomplexen ist bekanntermaßen 9 in [ReH9]2-.269 Die jüngst in einer
Wasserstoffmatrix bei 4 K beobachtete Spezies WH12 ist jedoch eigentlich als WH4(H2)4 zu
verstehen.270,
271
Ausgehend von homo- oder heteroleptischen Komplexen des Typs
[LyM(GaCp*)x] wurde eine Familie von neuartigen, hochkoordinierten einkernigen
Verbindungen der allgemeinen Formel [M(M’R1)a(ER2)b] (M’ = Zn) entdeckt, deren Prototyp
das zwölfach koordinierte [Mo(ZnMe)9(ZnCp*)3] (23) ist. Für einen synthetischen Zugang ist
die Verwendung der Molekülfamilie [LaMb(ECp*)c] als Ausgangsmaterialien zwingend
notwendig, da hier die kombinierten Eigenschaften der Liganden ECp* (starke Donoren,
intrinsische Reduktionsmittel, labil gebundener Cp*-Rest) zum Tragen kommen. Die
Reaktionen beinhalten zusätzlich zur Wanderung von Cp* vom Gallium zum Zink und Mebzw. Et-Gruppen vom Zink zum Gallium eine Reduktion von ZnII zu ZnI unter Bildung von
Me2GaCp* und GaMe3 als wichtigste GaIII Nebenprodukte, sowie verschiedener
Fulvalenspezies. Die Triebkraft in den hochselektiven Synthesen ist zweifelsohne die günstige
Oxidation von Ga(I) zu Ga(III) kombiniert mit den sehr ähnlichen elektronischen und
sterischen Eigenschaften der GaR und ZnR Liganden. Die Gesamtzahl an ZnR Liganden in
den resultierenden Übergangsmetallkomplexen ist ausschließlich von der Zahl der
Galliumliganden im Präkursor sowie von der in der Reaktion eingesetzten Menge an ZnMe2
abhängig. Aus dem Blickwinkel der Koordinationschemie ist ein Zwei-Elektronen GaCp*
Ligand zu zwei ZnCp* oder ZnMe Liganden mit jeweils einem Elektron gleichwertig, so dass
es bei den Umsetzungen von Komplexen des Typs [M(GaCp*)x] mit einem Überschuss an
ZnR2 > 2x stets zur Bildung der pseudo-homoleptischen, 18e Komplexe [M(ZnCp*)a(ZnR)b]
kommt. Werden in den Reaktionen geringere Mengen an ZnMe2 eingesetzt, entstehen dabei,
elektronisch ebenfalls abgesättigte, gemischtmetallische Ga/Zn Intermediate, wie die
Isolierung von [Mo(ZnCp*)4(GaMe)4] (24), [Mo(ZnCp*)4(ZnMe)4(GaMe)2] (25) und
[Rh(GaMe)(ZnCp*)4(ZnMe)3] (31) zeigt. Die Existenz der Intermediate, die reproduzierbar in
hohen Ausbeuten hergestellt werden konnten, verdeutlicht eine unterschiedliche Reaktivität
der GaCp* Liganden. Desweiteren belegt die Synthese von [Ru(ZnCp*)4(ZnMe)4(H)2] (29),
dass in der Reaktion von [(GaCp*)4(H)Ru(μ-Ga)Ru(H)2(GaCp*)3] (12) zunächst die GaCp*Liganden und erst darauf folgend die Hydride mit ZnMe2 reagieren.
99
3. Ergebnisse
Synthesen und Strukturen der M-ZnR Komplexe - Zusammenfassung
Tabelle 100.1. Elektronenanzahl in den synthetisierten metallreichen Molekülen
Verbindung
Metallkern
Elektronenanzahl
e(M)+e(GaR)+e(ZnR)
[Mo(GaMe)4(ZnCp*)4] (24)
[Mo(GaMe)2(ZnCp*)4(ZnMe)4] (25)
[Mo(ZnCp*)3(ZnMe)9] (23)
[Mo(ZnCp*)2(ZnEt)10] (26)
[Ru(ZnCp*)4(ZnMe)6] (28)
[Ru(ZnCp*)4(ZnMe)4(H)2] (29)
[Rh(ZnCp*)3(ZnMe)6] (30)
[Rh(GaMe)(ZnCp*)4(ZnMe)3] (31)
[Ni(ZnCp*)4(ZnMe)4] (32)
[Pd(ZnCp*)4(ZnMe)4] (33)
[Pt(ZnCp*)4(ZnMe)4] (34)
[Pt(ZnCp*)4(ZnEt)4] (35)
MoGa4Zn4
MoGa2Zn8
MoZn12
MoZn12
RuZn10
RuZn8
RhZn9
RhGaZn7
NiZn8
PdZn8
PtZn8
PtZn8
6 + 8 + 4 = 18
6 + 4 + 8 = 18
6 + 12 = 18
6 + 12 = 18
6 + 8+ 4 = 18
8 + 10 = 18
9 + 9 = 18
9 + 2 + 7 = 18
10 + 8 = 18
10 + 8 = 18
10 + 8 = 18
10 + 8 = 18
Da es sich bei den Metallkäfigen in den Festkörperstrukturen aller bisher erhaltenen
Komplexe um platonische Körper handelt (siehe Abbildung 100.1) und die Produkte formal
die 18 Elektronenregel erfüllen (siehe Tabelle 100.1), befolgen die neuen Moleküle
offensichtlich die VSEPR Regeln.
Abbildung 100.1. Platonische Körper der pseudo-homoleptischen ausschließlich Zink
koordinierten 4d Übergangsmetallkomplexe wie sie durch Röntgenstrukturanalyse bestimmt
wurden: Ikosaeder (gelb, z.B. in [Mo(ZnCp*)3(ZnMe)9] (23)), ein zweifach verkapptes
quadratisches Antiprisma (rot, z.B. in [Ru(ZnCp*)4(ZnMe)6] (28)), ein einfach verkapptes
quadratisches Antiprisma (blau, z.B. in [Rh(ZnCp*)3(ZnMe)6] (31)) und ein trigonales
Dodekaeder (grün, z.B. in [Pd(ZnCp*)4(ZnMe)4] (33))
100
3. Ergebnisse
Synthesen und Strukturen der M-ZnR Komplexe - Zusammenfassung
Obwohl alle Moleküle formal die Edelgasregel befolgen, belegen quantenchemische
Untersuchungen anhand der Schlüsselverbindung 23, dass die Bindungssituation deutlich
komplexer ist. Bei dem ikosaedrische Komplex 23 handelt es sich um ein perfekt sd5
hybridisiertes Metallzentrum (Mo), wobei alle 12 Orbitallappen für die Bindung der zwölf
„Liganden“ (ZnR) benutzt werden, indem 12 Elektronen auf sechs 2e-3z Bindungen verteilt
sind, die in Richtung der Raumdiagonalen durch die Ikosaederstruktur des für Moleküle
präzedenzlosen MoZn12 Kerns ausgebildet werden. Die restlichen 6 Elektronen rufen nur sehr
schwache Zn-Zn Bindungen hervor und sind ohne Mo-Zn Beitrag über den (ZnR)12 „Käfig“
delokalisiert. Aufgrund der außergewöhnlichen Bindungssituation in 23 befindet sich das
Molekül auf der Grenzfläche zwischen einer Koordinationsverbindung und einem Cluster. In
Zusammenarbeit mit Prof. Gernot Frenking und Mitarbeitern wurde die Bindungssituation der
weiteren pseudo-homoleptischen Komplexe analysiert. Die daraus resultierenden Ergebnisse
werden im folgenden Kapitel kurz vorgestellt.
3.9.6 Über die Bindungssituationen in den pseudohomoleptischen d8-d10 MetallZinkkomplexen
In
Kapitel
3.7
wurden
die
Bindungsverhältnisse
im
Metallkern
in
Komplex
[Mo(ZnCp*)3(ZnMe)9] (23) anhand der Modellverbindung [Mo(ZnH)12] (23H) beschrieben.
Unter
Verwendung
von
AIM
und
MO
Korrelationsdiagrammen
konnte
in
der
Bindungsanalyse für 23H gezeigt werden, dass die achtzehn Valenzelektronen, welche für
Mo-Zn und Zn-Zn Wechselwirkungen zur Verfügung stehen, zu sechs 2e3z Mo-Zn
Bindungen sowie zu drei schwachen, delokalisierten Zn-Zn Bindungen führen. Die AIM
Analyse für 23H zeigt interessanterweise zwölf Bindungspfade für die Mo-ZnH Kontakte,
jedoch keine Bindungspfade für Zn-Zn Wechselwirkungen. Die schwachen Zn-Zn Bindungen
dienen hauptsächlich dazu die Ligand-Ligand Abstoßungen zu minimieren, was schließlich zu
der für Metallkomplexe mit monodentaten Liganden L beispiellos hohen Koordinationszahl in
23 führt. Weiterhin wurde der Schluss gezogen, dass die Schlüsselverbindung 23 am besten
als perfekt sd5 hybridisierte Übergangsmetallverbindung beschrieben wird, in der alle 12
Lappen der Hybridorbitale, die in die Ecken des Ikosaeders zeigen, für starke kovalente
chemische
Bindungen
verwendet
werden.
Aufgrund
dieser
ungewöhnlichen
Bindungssituation, die weder auf einen prototypischen Cluster noch auf eine gewöhnliche
101
Bindungssituationen in den d8-d10 Metall-Zinkkomplexen
3. Ergebnisse
Koordinationsverbindung schließen lässt, ergab sich im Folgenden die Fragestellung, ob die
Bindungssituation in den MZnn Käfigen mit n = 8, 9, 10 in ähnlicher Art und Weise wie für
23H interpretiert werden kann.
Tabelle 102.1. Vergleich der experimentellen Bindungslängen mit den berechneten Daten,
wobei in den Rechnungen Cp* gegen Cp substituiert wurde (berechnet in kursiver Schrift).
M
Metallkern
M-ZnCp*
M-ZnMe
M-GaMe
Ga/Zn-Zn
MoZn12 (23)
2.672(1)-2.677(1)
2.666-2.667
2.636(1)-2.648(1)
2.673-2.678
-
2.724(2)-2.853(1)
2.796-2.832
MoGa4Zn4 (24)
2.539(1)-2.551(1)
2.588-2.590
-
2.391(1)-2.405(1)
2.426-2.427
2.800(1)-3.013(1)
2.880-2.932
MoGa2Zn8 (25)
2.613(1)-2.633(1)
2.622-2.646
2.467(1)-2.573(1)
2.628-2.647
2.521(1)-2.572(1)
2.450-2.457
2.733(1)-3.035(1)
2.729-3.053
MoZn12 (26)
2.653(1)-2.657(2)
2.626(1)-2.684(2)
-
2.708(2)-2.833(2)
RuZn10 (28)
2.552(1)-2.575(1)
2.554-2.564
2.492(1)-2.505(1)
2.524-2.544
-
2.697(1)-3.010(1)
2.723-3.036
RuZn8 (29)
2.470(1)-2.488(1)
2.438(1)-2.491(1)
-
2.721(1)-3.145(1)
RhZn9 (30)
2.470(1)-2.502(2)
2.506-2.508
2.439(2)-2.467(2)
2.488-2.490
-
2.749(2)-2.986(2)
2.850-2.995
RhGaZn7 (31)
2.433(1)-2.440(1)
2.476-2.493
2.394(1)-2.399(1)
2.468
2.383(1)
2.309
2.838(1)-3.052(1)
2.850-2.942
Ni
NiZn8 (32)
2.351(1)-2.370(1)
2.380
2.313(1)-2.331(1)
2.357
-
2.746(1)-2.917(1)
2.772-2.836
Pd
PdZn8 (33)
2.447(1)-2.458(1)
2.492-2.493
2.415(1)-2.422(1)
2.458-2.459
-
2.822(1)-3.131(1)
2.866-2.953
PtZn8 (34)
2.441(1)-2.459(1)
2.506-2.508
2.402(1)-2.467(1)
2.476-2.477
-
2.812(2)-3.115(2)
2.888-2.956
PtZn8 (35)
2.457(1)-2.459(1)
2.425(1)-2.430(0)
-
2.842(1)-3.057(1)
Mo
Ru
Rh
Pt
Für eine Analyse der Bindungssituation in den hochkoordinierten Verbindungen 28, 30 sowie
32-34,
welche
Koordinationszahlen
zwischen
acht
und
zehn
besitzen,
wurden
quantenchemische DFT Berechnungen auf RI-BP86/def2-TZVPP und auf BP86/TZ2P+
Niveau durchgeführt. Eine Berechnung der verwandten Verbindungen 26, 29 und 35 fand
nicht statt, da sie für eine Interpretation der Bindungssituation unwichtig sind. Im ersten
Schritt wurden zunächst die Cp* Gruppen gegen Cp Gruppen substituiert. Ein Vergleich der
102
Bindungssituationen in den d8-d10 Metall-Zinkkomplexen
3. Ergebnisse
berechneten Bindungslängen der MZnn Kerne mit den experimentell gemessenen Daten zeigt
nur äußerst geringe Unterschiede (siehe Tabelle 102.1).
Als nächstes wurden die Geometrien der repräsentativen Modellverbindungen 28H, 20H
32H-34H optimiert, in welchen die Cp Liganden gegen Wasserstoffatome ersetzt wurden.
Tabelle 103.1 verdeutlicht, dass die Bindungslängen in den Metallkernen der Wasserstoff
substituierten System sehr ähnlich zu den Werten der berechneten Cp enthaltenden
Verbindungen sind.
Tabelle 103.1. Berechnete Bindungslängen in den Modellverbindungen
Verbindung
Mo(ZnH)12
23H RI-BP86/def2-TZVPP
Mo(GaH)4(ZnH)4 24H
Mo(GaH)2(ZnH)8 25H
Ru(ZnH)10
28H
Rh(ZnH)9
30H
Rh(GaH)(ZnH)7
31H
Ni(ZnH)8
32H
Pd(ZnH)8
33H
Pt(ZnH)8
34H
(Ih)
RI-BP86/def2-TZVPP
(D2)
RI-BP86/def2-TZVPP
(Cs)
RI-BP86/def2-TZVPP
(D4d)
RI-BP86/def2-TZVPP
(D3h)
RI-BP86/def2-TZVPP
(Cs)
RI-BP86/def2-TZVPP
(D4d)
RI-BP86/def2-TZVPP
(D4d)
RI-BP86/def2-TZVPP
(D4d)
M-Zn
M-Ga
Zn-Zn
Ga-Ga
2.684
-
2.822
-
2.586
2.417
3.143
3.279
2.6272.651
2.541;
2.584
2.497;
2.516
2.4812.486
2.434;
2.440
2.374
-
2.482
-
2.496
-
2.7633.124
2.740;
2.798
2.853;
3.044
2.8353.142
2.821;
2.993
2.936;
3.149
2.951;
3.172
2.308
-
Ga-Zn
2.9283.116
2.8243.000
-
-
-
-
-
2.989;
3.244
-
-
-
-
-
-
Somit ist es durchaus gerechtfertigt, die Bindungssituation in den Modellverbindungen,
welche Hydridliganden beinhalten, zu analysieren, um die Natur der Metall-Ligand
Bindungen in den realen Komplexen, welche organische Gruppen besitzen, zu erklären. Der
Fokus der vorliegenden Bindungsanalyse liegt auf dem Vergleich der Metall-Ligand
Wechselwirkungen in den zwölfach koordinierten, ikosaedrischen, Strukturen mit denjenigen
pseudo-homoleptischen Komplexen, welche eine Koordinationszahl von acht, neun und zehn
besitzen. Zu diesem Zweck wurden die elektronischen Strukturen der isoelektronischen
Modellverbindungen [Mo(ZnH)12] (23H), [Ru(ZnH)10] (28H), [Rh(ZnH)9] (30H) und
[Pd(ZnH)8] (33H) analysiert und miteinander verglichen (siehe Abbildung 104.1). Dabei ist
zu berücksichtigen, dass die Verbindungen 28H und 33H eine D4d Symmetrie besitzen,
103
Bindungssituationen in den d8-d10 Metall-Zinkkomplexen
3. Ergebnisse
während 30H eine D3h Symmetrie aufweist. Diese hohen Symmetrien der homoleptischen
Verbindungen ermöglicht es, die Metall-Ligand Orbitalwechselwirkungen im Detail zu
untersuchen
23H (Ih)
28H (D4d)
30H (D3h)
33H (D4d)
Abbildung 104.1. Geometrieoptimierte Minima der Modellverbindungen [M(ZnH)n]:
[Mo(ZnH)12] (Ih) (23H), [Ru(ZnH)10] (D4d) (28H), [Rh(ZnH)9] (D3h) (30H) und [Pd(ZnH)8]
(D4d) (33H).
Die Gleichgewichtsstruktur von [Ru(ZnH)10] (28H) besitzt D4d Symmetrie, was zu einer
Aufhebung der Entartung derjenigen MO´s führt, die p und d Valenzorbitale des Ru
beinhalten. Die Molekülorbitale, welche p(Ru) Beiträge beinhalten, haben b2(pz) oder e1(px,
py) Symmetrie, während die MO´s mit d(Ru) Funktionen e2(dxy, dx2-y2), e3(dxz, dyz) oder
a1(dz2) Symmetrie besitzen. Desweiteren ist zu berücksichtigen, dass die a1 Orbitale auch
einen Beitrag der s(Ru) Atomorbitale besitzen könnten, da letztere ebenfalls a1 symmetrisch
sind. Aus diesem Grund können die EDA Ergebnisse nicht zwischen diesen Orbitalen
unterscheiden. Die wichtigsten besetzten Valenzorbitale von 28H, welche Beiträge aus den
Ruthenium Valenzorbitalen besitzen, sind in Abbildung 105.1 gezeigt. Eine Betrachtung der
besetzten
MO´s
zeigt,
dass
sich
die
Bindungssituationen
für
die
Ru-(ZnH)10
Wechselwirkungen in 28H und die Mo-(ZnH)12 Wechselwirkungen in 23H ähneln. Die MO´s
20e1(HOMO) und 20b2(HOMO-1) von 28H sind hauptsächlich Orbitale des Käfigs mit nur
sehr kleinen Beiträgen der p(Ru) Atomorbitale. Sie ähneln dabei dem 12t1u (HOMO) von 23H
(siehe Abbildung 73.1 und 74.1). Die Orbitale 19e3 (HOMO-2), 18e2 (HOMO-3) und 21a1
(HOMO-4) von 28H besitzen signifikante Koeffizienten aus den d(Ru) Valenzorbitalen und
entsprechen dabei dem 12hg (HOMO-1) Orbital von 23H. Die nächst tiefer liegenden
104
3. Ergebnisse
Bindungssituationen in den d8-d10 Metall-Zinkkomplexen
Valenzorbitale von 28H, welche einen großen Anteil aus den d(Ru) AOs haben, sind 20a1,
18e3, 17e2 und 19a1, wobei die a1 Orbitale starke Koeffizienten aus den s(Ru) und dz2(Ru)
AOs besitzen. Bei den darunter liegenden 19e1 und 18b2 Molekülorbitalen handelt es sich
hauptsächlich um Käfigorbitale mit einem vernachlässigbar kleinen Koeffizienten am
Ruthenium. Desweiteren enthält Abbildung 105.1 Orbitale, welche einen großen Anteil aus
den s(Ru) und d(Ru) Valenz-Atomorbitalen besitzen. Dabei handelt es sich um die Orbitale
18a1, 10e2, 12e3, 15a1 und 14a1. Die verbleibenden besetzten Valenzorbitale, welche a1, e2
oder e3 Symmetrie aufweisen, besitzen leichte Koeffizienten an den Ru AOs.
Abbildung 105.1. Ausgewählte besetzte Valenzorbitale von [Ru(ZnH)10] (28H)
105
3. Ergebnisse
Bindungssituationen in den d8-d10 Metall-Zinkkomplexen
Die EDA von 28H ist schwieriger zu interpretieren als die EDA von 23H, da die Auswahl der
elektronischen Zustände der Fragmente für die Ru-(ZnH)10 Wechselwirkungen nicht eindeutig
zu treffen ist. Daher wurden EDA Berechnungen für unterschiedliche Ladungen und
elektronischen Zustände für die jeweiligen Fragmente durchgeführt. Tabelle 107.1 enthält die
numerischen Ergebnisse für die Wechselwirkungen zwischen Ru2+ (s1d5) und (ZnH)102- in
Septettzuständen. Die EDA Ergebnisse für Ru(ZnH)10 (28H) lassen sich mit den Ergebnissen
der Modellverbindung Mo(ZnH)12 (23H) (Tabelle 75.1 und 107.1) vergleichen, da in beiden
Modellen die wechselwirkenden Fragmente elektronische Septettzustände aufweisen. Die
wichtigsten kovalenten Beiträge stammen dabei aus den Valenz d-Orbitale des Rutheniums.
So belaufen sich die Gesamtorbitalwechselwirkungen zwischen Ru2+ (s1d5) und (ZnH)102- im
elektronischen Septett-Zustand auf 886.3 kcal/mol, wobei diese hauptsächlich aus den
(e2)(dx2-y2,dxy) (291.7 kcal/mol, 32.9 %) und (e3)(dxz,dyz) Atomorbitalen (280.9 kcal/mol, 31.7
%) stammen. Zudem wird ersichtlich, dass das dz2 Atomorbital von ähnlicher Wichtigkeit ist
wie die anderen d-AO´s des Rutheniums (212.2 kcal/mol, 24.0 %). Somit stammen die
Gesamtorbitalwechselwirkungen des Ru in 28H aus dessen d-Orbitalen, welche insgesamt
80.8% des ΔEOrb Terms ausmachen. Dem entgegen stehen die Beiträge des s(Ru) AO und der
p(Ru) AO´s, die mit 8.8 % und 4.1 % wesentlich kleiner sind.
Diese Resultate beziehen sich auf die Auswahl von geladenen Fragmenten. Tabelle 107.1
enthält auch EDA Ergebnisse für die Wechselwirkung zwischen den Fragmenten der
neutralen Spezies Ru(s0d8) und (ZnH)10, welche entweder im elektronischen Triplett- oder im
Singulettzustand vorliegen können. Dabei muss berücksichtigt werden, dass sich bei der
Wechselwirkung von geladenen bzw. ungeladenen Fragmenten die elektrostatischen Anteile
an den Bindungswechselwirkungen verschieben. Während zwei Drittel der attraktiven
Wechselwirkungen zwischen Ru2+ und (ZnH)102- aus ΔEOrb und ca. ein Drittel aus ΔEelstat.
stammen, führt eine Wechselwirkung zwischen neutralen Fragmenten zu einer Umkehr der
attraktiven Beiträge, so dass nun ΔEelstat einen wesentlich größeren Anteil als ΔEOrb. hat.
Dennoch liefern die EDA Ergebnisse für neutrale Fragmente ein sehr ähnliches Bild ab wie
die EDA Ergebnisse der geladenen Spezies. Die wichtigsten Orbitalwechselwirkungen
zwischen dem Metall und dem Käfig stammt aus den d-Orbitalen, welche insgesamt knapp 80
% des ΔEOrb. Terms ausmachen. Dabei wird weiterhin deutlich, dass die Wechselwirkungen
zwischen den Triplett-Fragmenten den absolut kleinsten Wert für ΔEOrb. ergeben. Damit lässt
sich die Bindungssituation in [Ru(ZnH)10] (28H) als Wechselwirkung zwischen den neutralen
Triplett-Fragmenten Ru(s0d8) und (ZnH)10 beschreiben, in welcher neben den kovalenten
106
Bindungssituationen in den d8-d10 Metall-Zinkkomplexen
3. Ergebnisse
Wechselwirkungen
der
vier
ungepaarten
Elektronen
auch
Donor-Akzeptor-
Wechselwirkungen zwischen den doppelt besetzten und den leeren Orbitalen des Ru und des
(ZnH)10 Käfigs vorliegen. Die Annahme von neutralen Spezies wird durch die berechneten
Partialladungen der Atome in den Modellverbindungen 23H-25H, 28H, 30H-34H bestätigt.
Wie Tabelle 108.1 verdeutlicht, tragen dabei die Übergangsmetalle eine kleine negative
Ladung zwischen -0.1e und -0.2e, wobei hier die Modellverbindung [Pd(ZnH)8] (33H) die
einzige Ausnahme darstellt (q(Pd) = +0.096e).
Tabelle 107.1. Ergebnisse der EDA Analysen von [Ru(ZnH)10] (28H) auf BP86/TZ2P+
Niveau mit unterschiedlichen elektronischen Zuständen der interagierenden Fragmente.
Energien in kcal/mol.
Ru2+ (s1 d5) +
(ZnH)102- (Septett)
Ru(s0 d8)[a] +
(ZnH)10 (Triplett)
Ru(s0 d8) [b] +
(ZnH)10 (Singulett)
ΔEint
-956.2
-342.3
-392.6
ΔEPauli
448.1
511.3
477.4
ΔEelstat[c]
-518.0 (36.9%)
-542.0 (63.5%)
-531.1 (61.1%)
ΔEOrb[c]
-886.3 (63.1%)
-311.5 (36.5%)
-338.9 (39.0%)
ΔEOrb (a1)(s,dz2) [d]
-212.2 (24.0%)
-78.6 (25.2%)
-94.1 (27.8%)
ΔEOrb (a2) [d]
-0.3 (<0.1%)
-0.0 (0.0%)
-0.0 (0.0%)
ΔEOrb (b1) [d]
-0.5 (0.1%)
-0.0 (0.0%)
-0.0 (0.0%)
ΔEOrb (b2)(pz) [d]
-35.2 (4.0%)
-4.2 (1.4%)
-5.2 (1.5%)
ΔEOrb (e1)(px,py) [d]
-65.4 (7.4%)
-7.0 (2.2%)
-7.7 (2.3%)
-291.7 (32.9%)
-126.0 (40.5%)
-115.4 (34.1%)
-280.9 (31.7%)
-95.6 (30.7%)
-116.5 (34.4%)
ΔEOrb (e2)(dx2-y2,dxy) [d]
ΔEOrb (e3)(dxz,dyz)
[d]
[a]
In Ru(s0d8) (Triplett) sind die dxz und dyz (e3) AO´s einfach besetzt.
[b]
In Ru(s0d8) (Singulett) ist das dz2 AO unbesetzt.
[c]
Die Prozentwerte in Klammern stellen den Anteil aller attraktiven Wecheselwirkungen ΔEelstat+ΔEorb dar.
[d]
Die Prozentwerte in Klammern stellen den Anteil aller Orbitalwechselwirkungen ΔEorb dar.
107
Bindungssituationen in den d8-d10 Metall-Zinkkomplexen
3. Ergebnisse
Tabelle 108.1. Die berechneten atomaren Partialladungen der Modellverbindungen
Verbindung
M
Ga
Zn
H(Ga)
H(Zn)
[Mo(ZnH)12] (23H)
-0.228
-
0.159
-
-0.140
[Mo(GaH)2(ZnH)8] (24H)
-0.112
0.160;0.170
0.162 - 0.172
-0.108;-0.106
-0.166 - -0.170
[Mo(GaH)4(ZnH)4] (25H)
-0.177
0.178
0.162
-0.117
-0.178
[Ru(ZnH)10] (28H)
-0.146
-
0.176
[Rh(ZnH)9] (30H)
-0.172
-
0.180;0.184
-
-0.163;-0.164
[Rh(GaH)(ZnH)7] (31H)
-0.196
0.221
0.178 - 0.183
-0.111
-0.170
[Ni(ZnH)8] (32H)
-0.121
-
0.178
-
-0.163
[Pd(ZnH)8] (33H)
0.096
-
0.154
-
-0.166
[Pt(ZnH)8] (34H)
-0.117
-
0.179
-
-0.165
-0.157;-0.163
Ein verblüffendes Ergebnis bei der Anaylse der Bindungssituation in 23H lieferten die AIM
Rechnungen (siehe Kapitel 3.7). Wie Abbildung 109.1 verdeutlicht sind nur Bindungspfade
für die Mo-Zn und die Zn-H Wechselwirkungen vorhanden, es liegen jedoch keine
entsprechenden Zn-Zn Bindungspfade vor. Abbildung 109.1b enthält die LaplaceVerteilungen, die Bindungspfade, die bindungskritischen Punkte sowie die Nullflussflächen in
der Molekülebene von 28H. Wie in 23H gibt es Bindungen zwischen dem
Übergangsmetallatom und jedem Zinkatom sowie entsprechende Pfade in den Zn-H
Fragmenten, jedoch keine Bindungspfade zwischen den Zinkatomen innerhalb des (ZnH)10
Käfigs. Dabei ist auch zu beachten, dass die Zn-Zn Abstände in 28H (2.740 Å und 2.798 Å)
sogar kürzer sind als in 23H (2.822 Å; Tabelle 103.1). Die AIM Analyse unterstützt somit die
oben dargelegte Bindungssituation, in der sowohl kovalente Mehrzentrenbindungen als auch
Donor-Akzeptor-Bindungen vorliegen. Die Zn-Zn Wechselwirkungen in 28H sind somit wie
in 23H nur sehr schwach.
108
3. Ergebnisse
Bindungssituationen in den d8-d10 Metall-Zinkkomplexen
(a) [Mo(ZnH)12] (23H)
(b) [Ru(ZnH)10] (28H)
(c) [Pd(ZnH)8] (28H)
(d) [Rh(ZnH)9] (30H)
Abbildung 109.1. Konturliniendiagramme der Laplace-Verteilungen ∇2ρ(r) der Komplexe (a)
[Mo(ZnH)12] (23H), (b) [Ru(ZnH)10] (28H), (c) [Pd(ZnH)8] (33H), (d) [Rh(ZnH)9] (30H).
Dargestellt ist die Laplace-Verteilung der Elektronendichte. Durchgezogene Linien zeigen Bereiche mit
steigender Ladungskonzentration an (∇2ρ(r) < 0), gestrichelte Linien zeigen Bereiche mit fallender
Ladungskonzentration an (∇2ρ(r) > 0). Die durchgezogenen Linien, die Atomkerne miteinander verbinden, sind
Bindungspfade, die durchgezogenen Linien, die Atombereiche voneinander trennen, sind die Schnittestrecken
der Nullflussflächen mit der betrachteten Ebene. Die bindungskritischen Punkte sind die Schnittpunkte der
Bindungspfade mit den Nullflussflächen.
109
3. Ergebnisse
Bindungssituationen in den d8-d10 Metall-Zinkkomplexen
Die achtfach koordinierte Modellverbindung [Pd(ZnH)8] (33H) besitzt ebenfalls D4d
Symmetrie (siehe Abbildung 104.1). Die Zn-Zn Abstände der direkt benachbarten Zinkatome
in 33H sind deutlich länger als in 23H und 28H. Es ist somit nicht verwunderlich, dass es
ebenfalls keine Bindungspfade im (ZnH)8 gibt. Es sind ausschließlich jeweils acht
Bindungspfade zwischen den Pd und Zn Atomen sowie zwischen den Zn und H Atomen
vorhanden.
Eine Betrachtung des Molekülorbitalschemas von 33H legt eine ähnliche Bindungssituation
wie in 23H und 28H nahe. Die wichtigsten besetzten Orbitale von 33H, die einen Anteil aus
den Palladium Atomorbitalen haben, sind in Abbildung 111.1 gezeigt. Die 13b2(HOMO) und
17e1(HOMO-1) Orbitale sind hauptsächlich Käfigorbitale und weisen lediglich einen kleinen
Beitrag aus den p(Pd) Atomorbitalen auf. Sie ähneln dabei dem 12t1u Orbital (HOMO) von
23H (siehe Abbildung 73.1). Unterhalb des HOMO und des HOMO-1 befinden sich zehn
Orbitale, welche große Koeffizienten aus den s(Pd) und d(Pd) Valenz-Atomorbitalen besitzen.
Sie können in Gruppen von jeweils fünf Orbitalen ähnlicher Energie zusammengefasst
werden. Dabei handelt es sich um die 14a1 (HOMO-2), 16e2 (HOMO-3), und 16e3(HOMO-4)
Orbitale (Gruppe 1) sowie um die 13a1, 15e2, und 15e3 Orbitale (Gruppe 2). Bei den nächst
tiefer liegenden Molekülorbitalen, welche einen Beitrag aus den p(Pd) Valenz-Atomorbitalen
beinhalten, handelt es sich um die 12b2 und 16e1 Käfigorbitale mit vernachlässigbaren
Koeffizienten am Pd. Darunter befinden sich die 12a1 und 9a1 MO´s, welche große
Koeffizienten am s(Pd) Valenz-Atomorbital beinhalten. Zwischen den beiden zuletzt
genannten Orbitalen befindet sich ein weiterer Satz von fünf energetisch sehr nahe liegenden
MO´s (10a1, 10e2, und 10e3), welche große Koeffizienten aus den d(Pd) AO´s aufweisen und
somit zu der Gesamtorbitalwechselwirkung in 33H beitragen.
110
3. Ergebnisse
Bindungssituationen in den d8-d10 Metall-Zinkkomplexen
Abbildung 111.1. Ausgewählte besetzte Valenzorbitale des Moleküls [Pd(ZnH)8] (33H).
Die Ergebnisse der EDA Berechnungen für 33H sind in Tabelle 112.1 zusammengefasst. Im
Gegensatz zu der EDA Interpretation von 28H ist hier die Auswahl der elektronischen
Zustände der wechselwirkenden Fragmente leichter zu treffen, da es sich bei dem
111
Bindungssituationen in den d8-d10 Metall-Zinkkomplexen
3. Ergebnisse
Grundzustand von Palladium um eine geschlossen schalige s0d10 Konfiguration handelt,
welche für Donor-Akzeptor Wechselwirkungen mit einem (ZnH)8 Käfig im elektronischen
Singulett-Zustand sehr gut geeignet ist. Die EDA Berechnungen zeigen, dass die Pd-(ZnH)8
Bindung hauptsächlich aus einer elektrostatischen Wechselwirkung (ΔEint = 77.5 %)
resultiert, während nur 22.5 % der attraktiven Wechselwirkung aus dem ΔEOrb. Term
stammen. Mit 71.9 % kommt der größte Beitrag in der Orbitalwechselwirkung aus der
Donierung der e2(dxy, dx2 −y2) und e3(dxz, dyz) Orbitale des Palladiums in die leeren Orbitale
des (ZnH)8 Käfigs. Die Orbitalwechselwirkung der a1(s, dz2) Orbitale betragen nur 16.0 %, da
eine Donierung der besetzten Käfigorbitale in die s(Pd) AO offensichtlich nur geringfügig zur
Stabilität des Moleküls beiträgt. Tabelle 112.1 enthält außerdem die EDA Ergebnisse der
anderen homoleptischen [M(ZnH)8] Komplexe der Gruppe 10 Übergangsmetalle, wobei die
berechneten Werte denen von 33H sehr ähnlich sind.
Tabelle 112.1. EDA Ergebnisse auf BP86/TZ2P Niveau für die Komplexe [M(ZnH)8] (M =
Ni, Pd, Pt) mit den Fragmenten M (s0d10) und (ZnH)10 im elektronischen Singulett-Zustand.
Energien in kcal/mol.
Ni
Pd
Pt
∆Eint
-246.3
-201.2
-279.0
∆EPauli
208.1
402.4
486.0
-310.8 (68.4%)
-467.7 (77.5%)
-583.4 (76.3%)
-143.6 (31.6%)
-135.8 (22.5%)
-181.6 (23.7%)
-23.4 (16.3%)
-21.7 (16.0%)
-43.8 (24.1%)
0.0 (0.0%)
0.0 (0.0%)
0.0 (0.0%)
0.0 (0.0%)
0.0 (0.0%)
0.0 (0.0%)
-5.9 (4.1%)
-5.4 (4.0%)
-8.2 (4.5%)
-12.0 (8.3%)
-11.0 (8.1%)
-16.7 (9.2%)
-46.5 (32.4%)
-43.6 (32.1%)
-51.2 (28.2%)
-55.8 (38.9%)
-54.1 (39.8%)
-61.7 (34.0%)
∆Eelstat
∆Eorb
[a]
[a]
∆E(a1) (s, dz2)
∆E(a2)
[b]
[b]
∆E(b1)[b]
∆E(b2) (pz)
[b]
∆E(e1) (px, py)
[b]
∆E(e2) (dxy, dx2 −y2)
∆E(e3) (dxz, dyz)
[b]
[b]
[a]
Die Prozentwerte in Klammern stellen den Anteil aller attraktiven Wecheselwirkungen ΔEelstat+ΔEorb dar.
[b]
Die Prozentwerte in Klammern stellen den Anteil aller Orbitalwechselwirkungen ΔEorb dar.
112
3. Ergebnisse
Bindungssituationen in den d8-d10 Metall-Zinkkomplexen
Abbildung 113.1. Ausgewählte besetzte Valenzorbitale des Moleküls [Rh(ZnH)9] (30H).
113
3. Ergebnisse
Bindungssituationen in den d8-d10 Metall-Zinkkomplexen
Die neunfach koordinierte Verbindung [Rh(ZnH)9] (30H) besitzt eine D3h Symmetrie (siehe
Abbildung 104.1). Die Zn-Zn Abstände der direkt benachbarten Zinkatome (2.853 Å und
3.044 Å) sind in einem ähnlichen Bereich wie in 33H (siehe Tabelle 103.1). Abbildung
109.1d verdeutlicht, dass die AIM Analyse analog zu der Bindungssituation in 23H, 28H und
33H keine Zn-Zn Bindungspfade, sondern ausschließlich Rh-Zn und Zn-H Bindungen
aufweist. Obwohl 30H eine niedrigere Symmetrie als die anderen drei Modellverbindungen
besitzt, ist es möglich die Beiträge aus den verschiedenen AO´s des Zentralmetalls Rh an den
einzelnen MO´s zu unterscheiden. Abbildung 113.1 enthält die wichtigsten besetzten MO´s,
welche einen Beitrag am Rhodium enthalten. Das 29e’(HOMO) von 30H hat einen hohe
Koeffizienten aus den px(Rh) und py(Rh) AO´s mit kleinen Anteilen aus den dxy(Rh) und dx2
−y2(Rh)
Orbitalen. Das 16a2” (HOMO-1) besitzt nur einen kleinen Beitrag aus dem pz(Rh) AO.
Die nahezu entarteten Orbitale sind vergleichbar zu dem 12t1u (HOMO) von [Ru(ZnH)12]
(28H) (siehe Abbildung 105.1). Darunter befindet sich das 22a1’(HOMO-2) Orbital, welches
nur kleine Beiträge aus dem s(Rh) und dem dz2(Rh) AOs (Abbildung 113.1) aufweist. Bei den
nächst tiefer liegenden Molekülorbitalen handelt es sich um das 28e’(HOMO-3),
21a’(HOMO-4) sowie dem 20e”(HOMO-5). Sie können leicht als Orbitale mit starken
Beiträgen aus den d(Rh) AO´s identifiziert werden und entsprechen dabei in umgekehrter
Reihenfolge den 12hg (HOMO-1) und 9ag (HOMO-2) von 30H. Abbilung 113.1 enthält
zudem zwei weitere Orbitalsätze, welche aus je fünf MOs mit signifikanten Koeffizienten am
d(Rh) AO´s bestehen (20a1’, 27e’, 19e” und 15a1’, 18e’, 12e”). Desweiteren gibt es zwei
tiefer liegende Orbitalsätze mit nahezu entarteten Orbitalen (16a2”, 27e’ und 11a2”, 19e’) mit
kleinen Koeffizienten aus dem p(Rh) AO´s. Die beiden letzten MO´s mit Beiträgen aus den
s(Rh) Valenz-Atomorbitalen sind die 19a1’ und 14a1’ Orbitale.
Die EDA Ergebnisse für 30H sind in Tabelle 115.1 zusammengefasst. Die Rechnungen
beziehen sich dabei auf die geschlossenschaligen, geladenen Fragmente Rh- (s0d10) und
(ZnH)9+ in elektronischen Singulett-Zuständen sowie auf die offenschaligen, neutralen
Fragmente Rh (s0d9) und (ZnH)9 im elektronischen Dublett-Zustand. Beide EDA Rechnungen
zeigen, dass die elektrostatischen Wechselwirkung stärker sind als die attraktiven
(kovalenten) Orbitalwechselwirkungen. Da der Orbitalterm ΔEOrb. für die geladenen
Fragmente wesentlich kleiner ist (-172.8 kcal/mol vs. -241.0 kcal/mol), liegt die Vermutung
nahe, dass es sich bei der Bindungssituation in 30H um eine Rh--(ZnH)9+ Wechselwirkung
handelt. Die EDA Ergebnisse belegen, dass der größte Beitrag zum ΔEOrb. Term aus dem
∆E(e1“) (dxz, dyz) Orbitalen stammt, wohingegen der ∆E(a2“) (pz) Beitrag vernachlässigbar ist.
114
Bindungssituationen in den d8-d10 Metall-Zinkkomplexen
3. Ergebnisse
Somit stammt der große Wert für ∆E(e1‘) (px, py, dxy, dx2-y2) hauptsächlich aus den d(Rh)
Atomorbitalen, während die großen Beträge von ∆E(a1‘) (s, dz2) aus dem dz2(Rh) AO
stammen.
Somit
lässt
sich
schlussfolgern,
dass
es
sich
bei
den
wichtigsten
Orbitalwechselwirkungen in 30H um eine Donierung der d(Rh) Orbitale in die leeren Orbitale
des (ZnH)9 Käfigs handelt.
Tabelle 115.1. EDA Ergebnisse auf BP86/TZ2P+ Niveau für den Komplex [Rh(ZnH)9]
(30H) mit D3h Symmetrie BP86/TZ2P+ unter Berücksichtigung unterschiedlicher
elektronischer Zustände der Fragmente. Energien in kcal/mol.
Rh- (s0 d10) + (ZnH)9+
(Singulett)
Rh (s0 d9) + (ZnH)9
(Doublett)
∆Eint
-457.4
-294.5
∆EPauli
598.4
465.1
∆Eelstat [a]
-883.0 (83.6%)
-518.5 (68.3%)
∆Eorb [a]
-172.8 (16.4%)
-241.0 (31.7%)
∆E(a1‘) (s, dz2) [b]
-28.8 (16.7%)
-62.0 (25.7%)
-0.2 (0.1%)
-0.3 (0.1%)
-65.9 (38.2%)
-85.5 (35.5%)
-0.1 (0.0%)
-0.0 (0.0%)
1.0 (-0.6%)
-5.2 (2.2%)
-78.8 (45.6%)
-88.1 (36.6%)
∆E(a2‘) [b]
∆E(e1‘) (px, py, dxy, dx2-y2) [b]
∆E(a1“)
“
[b]
∆E(a2 ) (pz)
[b]
∆E(e1“) (dxz, dyz)
[b]
[a]
Die Prozentwerte in Klammern stellen den Anteil aller attraktiven Wecheselwirkungen ΔEelstat+ΔEorb dar.
[b]
Die Prozentwerte in Klammern stellen den Anteil aller Orbitalwechselwirkungen ΔEorb dar.
3.9.7 Synthesen und Strukturen von heteroleptischen Komplexen mit ZnR Liganden
Ausgehend von homoleptischen Übergangsmetallkomplexen des Typs [M(GaCp*)x]
entstehen bei den Umsetzungen mit einem hohen Überschuss an ZnMe2 einkernige, pseudohomoleptische Verbindungen der allgemeinen Formel [M(M’R1)a(ER2)b] (M’ = Zn). In
Kapitel 3.8 wird die Synthese der Cluster-Verbindung [{Mo(CO)4}4(Zn)6(μ-ZnCp*)4] (27)
115
3. Ergebnisse
Synthesen und Strukturen von heteroleptischen M-ZnR Komplexen
beschrieben, welche aus der Umsetzung des heteroleptischen Mo(0) Komplexes
[Mo(CO)4(GaCp*)2] mit vier Moläquivalenten ZnMe2 entsteht. In der Festkörperstruktur von
27 bilden die vier Molybdänatome einen Mo4 Tetraeder, auf dessen Kanten sich sechs
substituentenfreie, „nackte“ Zinkatome befinden. Diese Anordnung führt zu einem verzerrten
Zn6 Oktaeder. In dem vorliegenden Fall scheint es, dass die stark gebundenen CO-Liganden
an den {Mo(CO)4}-Einheiten um den Zn6-Kern ein weiteres Clusterwachstum verhindern und
so 27 als einen molekularen „Ausschnitt“ einer polymeren intermetallischen Mo/ZnVerbindung stabilisieren. Im Zuge dieser Untersuchungen, ergab sich die Fragestelllung,
inwieweit sich das Synthesekonzept zur Erzeugung von Übergangsmetall-ZnR-Komplexen
auf heteroleptische Verbindungen erweitern lässt.
Zu diesem Zweck wurde [fac-Mo(CO)3(GaCp*)3] mit einem achtfachen Überschuss an
ZnMe2 in Toluol bei 100 °C umgesetzt. Im Gegensatz zur Synthese von 27 verfärbte sich die
gelbe Reaktionslösung während der Reaktion nicht. Nach Entfernung aller flüchtigen Stoffe
und Kristallisation durch langsames Kühlen einer gesättigten n-Hexanlösung auf -30 °C
wurden
gelbe
Einkristalle
der
neunfach
koordinierten
Komplexverbindung
[Mo(CO)3(ZnCp*)3(ZnMe)3] (36) in reproduzierbaren Ausbeuten von etwa 80% erhalten. Der
Komplex 36 ist in allen gängigen organischen Lösungsmitteln wie Hexan, Toluol oder THF
löslich und ist bemerkenswert beständig, wenn er unter Argonatmosphäre, auch ohne
Kühlung, gelagert wird. Eine Elementaranalyse sowie AAS schließen das Vorhandensein von
Gallium aus und stimmen ausgezeichnet mit dem Aufbau aus der Röntgenstrukturlösung
überein.
[fac-Mo(CO)3(GaCp*)3]
+8 ZnMe2
[Mo(CO)3(ZnCp*)3(ZnMe)3] (36)
Schema 116.1. Synthese des heteroleptischen Komplexes [Mo(CO)3(ZnCp*)3(ZnMe)3] (36)
In analoger Weise zu den Synthesen der pseudo-homoleptischen Verbindungen 23, 26, 28, 30
und 33-35, die ebenfalls unter Verwendung eines großen Überschusses an ZnMe2 hergestellt
werden, findet auch hier eine vollständige Substitution der GaCp* Liganden im Edukt durch
die doppelte Anzahl an ZnR Spezies statt. Die molekulare Struktur von 36 im Festkörper ist in
Abbildung 117.1 gezeigt. Verbindung 36 kristallisiert in der triklinen Raumgruppe P-1.
Weitere relevante kristallographische Daten sind in Tabelle 170.1 zusammengestellt.
116
3. Ergebnisse
Synthesen und Strukturen von heteroleptischen M-ZnR Komplexen
Abbildung 117.1. Molekulare Struktur von [Mo(CO)3(ZnCp*)3(ZnMe)3] (36) wie sie durch
Röntgenstrukturanalyse bestimmt wurde (thermische Ellipsoide für 50% Wahrscheinlichkeit,
die Wasserstoffatome sind aus Gründen der Übersichtlichkeit nicht gezeigt). Ausgewählte
interatomare Abstände (Å) und Winkel (°): Mo-C(3) 1.993(4), Mo-C(2) 2.005(4), Mo-C(1) 2.013(4),
Mo-Zn(1) 2.617(1), Mo-Zn(5) 2.624(1), Mo-Zn(3) 2.625(1), Mo-Zn(2) 2.628(1), Mo-Zn(6) 2.628(1), Mo-Zn(4)
2.628(1), Zn(2)-C(4) 1.952(4), Zn(3)-C(5) 1.956(4), Zn(5)-C(6) 1.949(4), Zn-Cp*(centroid) 1.899-1.984 Å
(Mittelwert 1.933 Å), O(1)-C(1)-Mo 176.7(3), O(2)-C(2)-Mo(1) 174.4(3), O(3)-C(3)-Mo(1) 177.9(3)
Die Festkörperstruktur von 36 belegt, dass in dem Komplex das Zentralmetall Mo von drei
CO Liganden, drei ZnCp* sowie von drei ZnMe Liganden umgeben ist. Dabei ist in dem
Molekül zunächst kein Koordinationspolyeder zu erkennen, wie es beispielsweise in den
pseudo-homoleptischen oder gemischtmetallischen Komplexen der Fall war (siehe Kapitel
3.9-3.9.5). Bei einer genaueren Betrachtung der Molekülstruktur von 36 fällt allerdings auf,
dass sich die CO Liganden auf Deckung oberhalb der ZnMe Spezies befinden. Desweiteren
ordnen sich die sterisch anspruchsvollen ZnCp* Liganden in der Art und Weise in der Ebene
zwischen den drei CO und drei ZnMe Liganden an, dass daraus insgesamt eine C3 Symmetrie
des Moleküls resultiert (siehe Abbildung 117.1).. Es fällt auf, dass die Anordnung der CO
Liganden im Vergleich zum Edukt [fac-Mo(CO)3(GaCp*)3] erhalten bleibt, so dass sie sich
im Produkt ebenfalls in cis Positionen zueinander befinden. Die Mo-Zn Abstände sind mit
2.617(1) - 2.628(1) Å für Mo-ZnCp* und mit 2.625(1) - 2.628(6) Å für Mo-ZnMe nahezu
identisch. Damit sind sie im Vergleich zu den Mo-Zn Abständen im verwandten Cluster 27
117
3. Ergebnisse
Synthesen und Strukturen von heteroleptischen M-ZnR Komplexen
leicht verkürzt (2.641 Å für Mo-Zn6 bzw. 2.664 und 2.681 für Mo-ZnCp*), liegen jedoch im
Rahmen der wenigen, bereits bekannten Beispiele für molekulare Mo-Zn Komplexe (Mo-Zn
2.538(1) - 2.793(3) Å).215, 260 Die RZn-ZnR Abstände in 36 betragen im Durchschnitt 2.745 Å
und stimmen mit den jeweiligen Werten der in Kapitel 3.9 vorgestellten Mo-Zn Komplexen
überein (siehe Tabelle 70.1). Die Cp* Gruppen der ZnCp* Liganden kennzeichnen sich
eindeutig durch η5 Bindungsmodi mit Zn-Cp*centr. Bindungslängen zwischen 1.899 und 1.984
Å (Durchschnitt 1.933 Å). Desweiteren fällt auf, dass alle drei Cp* Gruppen signifikant
gekippt sind, was in Mo-Zn-Cp*centr. Winkeln von 157.23° bis 166.28° resultiert. Die Mo-C-O
Winkel der drei Mo-CO Fragmente sind alle im Bereich von 174.4(1)° bis 177.9(1)°, wodurch
sicherlich ausgeschlossen werden kann, dass Mo-Zn Bindungen mit CO Liganden verbrückt
sind. Dies stimmt mit dem Infrarotspektrum von 36 überein, das drei Absorptionsbanden bei
1967, 1923, und 1915 cm-1 aufweist, was dem typischen Bereich für terminale CO Liganden
entspricht. Die 1H und
Festkörper
überein
13
C NMR Spektren von 36 in Lösung stimmen mit der Struktur im
und
zeigen
chemisch
gleichwertige
Cp*
und
Methyl.
Temperaturabhängige Messungen (25-70 °C) zeigen eine statische Struktur ohne Austausch
der Cp* und Methyl-Positionen.
Ist diese Reaktion nur eine Ausnahme, die auf heteroleptische Edukte mit CO als CoLiganden beschränkt ist oder ist das Synthesekonzept allgemein auf GaCp* beinhaltende
Verbindungen anwendbar? Im Rahmen dieser Fragestellung wurde der zu [facMo(CO)3(GaCp*)3] isoleketronische Komplex [Ru(GaCp*)3(TMM)] (13) ebenfalls mit acht
Äquivalenten ZnMe2 umgesetzt und für eine Stunde in Toluol bei 100 °C gerührt. Nach
Aufarbeitung der Reaktionsmischung wurden durch mehrstündiges Kühlen einer gesättigten
n-Hexan Lösung für röntgenkristallographische Untersuchungen geeignete Einkristalle
erhalten. Die Röntgenstrukturlösung zeigt, dass es sich bei dem gemessenen Kristall um den
Komplex [Ru(TMM)3(ZnCp*)3(ZnMe)3] (37) handelt, in welchem offensichtlich der TMM
Ligand erhalten bleibt. Eine Elementaranalyse sowie AAS Untersuchungen schließen das
Vorhandensein von Gallium aus. In 37 wird das Ruthenium-Zentrum asymmetrisch von drei
ZnMe, drei ZnCp* sowie dem TMM Liganden koordiniert. Die regenschirmartige Winkelung
des TMM Liganden, die durch einen Winkel θ quantifiziert wird, ist mit 11.2° nahezu
identisch zu der in 13. Dieser Wert stimmt desweiteren hervorragend mit denen bekannter
TMM Komplexen überein.189, 190 Die Ru-C Bindungslängen in 37 (Ru-C(1)) 2.073(4) Å, RuC(2-4) 2.265(4)-2.285(4) Å) sind ebenfalls fast unverändert verglichen zu denen in 13 (Ru118
3. Ergebnisse
Synthesen und Strukturen von heteroleptischen M-ZnR Komplexen
C(1)) 2.019(5) Å, Ru-C(2-4) 2.208(4)- 2.228(5) Å). Die Ru-Zn Bindungsabstände betragen
im Durchschnitt 2.477 Å, wobei die Ru-ZnMe (durchschnittlich 2.453 Å) gegenüber den RuZnCp* (durchschnittlich 2.500 Å) Abständen leicht verkürzt sind. Diese Werte stimmen sehr
gut mit den entsprechenden Daten der bereits bekannten Ru-Zn Komplexe überein (siehe
Kapitel 3.9.2, Tabelle 70.1). Die Cp* Gruppen der ZnCp* Liganden sind eindeutig η5 an die
Zn Atome gebunden, wobei die Bindungslängen zwischen 1.955 und 1.997 Å (Durchschnitt
1.978 Å) betragen.
Abbildung 119.1. Molekulare Struktur von [Ru(TMM)3(ZnCp*)3(ZnMe)3] (37) wie sie durch
Röntgenstrukturanalyse bestimmt wurde (thermische Ellipsoide für 50% Wahrscheinlichkeit,
die Wasserstoffatome sind aus Gründen der Übersichtlichkeit nicht gezeigt). Ausgewählte
interatomare Abstände (Å) und Winkel (°): Ru-C(1) 2.073(4), Ru-C(4) 2.265(4), Ru-C(2) 2.269(4), RuC(3) 2.285(4), Ru-Zn(1) 2.448(1), Ru-Zn(2) 2.449(1), Ru-Zn(3) 2.462(1), Ru-Zn(5) 2.485(1), Ru-Zn(4)
2.498(1), Ru-Zn(6) 2.517(1), Zn(1)-C(5) 1.955(4), Zn(2)-C(6) 1.952(4), Zn(3)-C(7) 1.963(4), Zn-Cp*(centroid)
1.982-1.955 Å (Mittelwert 1.973 Å), C(2)-C(1)-C(3) 115.8(4), C(2)-C(1)-C(4) 116.5(4), C(3)-C(1)-C(4)
116.5(4), C(1)-Ru-Zn(1) 103.66(11), C(4)-Ru-Zn(1) 131.79(12), C(2)-Ru-Zn(1) 68.84(11), C(4)-Ru-Zn(2)
131.31(12), C(2)-Ru-Zn(2) 105.70(11), C(3)-Ru-Zn(2) 68.67(11), Zn(1)-Ru-Zn(3) 124.345(19)
Das 1H NMR Spektrum der isolierten Kristalle enthält in C6D6 bei Raumtemperatur drei
Signale bei δ = 2.039, 1.292 und -0.112 ppm im Integralverhältnis von 45:6:9, die unter
Annahme einer C3 symmetrischen Lösungsstruktur von 37 chemisch äquivalenten ZnCp* und
ZnMe Gruppen sowie dem TMM Liganden zugeordnet werden können. Desweiteren ist im 1H
119
3. Ergebnisse
Synthesen und Strukturen von heteroleptischen M-ZnR Komplexen
NMR Spektrum ein weiterer Signalsatz mit drei Signalen bei δ = 2.021, 0.971 und -0.085 ppm
im Integralverhältnis von 30:6:12 vorhanden. Somit ist bei der Reaktion von 13 mit ZnMe2
ein zweites Isomer entstanden, welches aufgrund der chemisch äquivalenten Cp* und
Methylgruppen im NMR Spektrum in Lösung symmetrisch ist und der Summenformel
[Ru(TMM)(ZnCp*)2(ZnMe)4] (38) folgt. Werden die TMM Signale beider Isomere integriert
und miteinander verglichen, ergibt sich ein Isomerenverhältnis von 60:40 bezogen auf das
Hauptisomer 38. Diese Annahmen sind in Übereinstimmung mit den erhaltenen Werten aus
der Elementaranalyse und AAS (siehe Experimenteller Teil). Eine röntgenkristallographische
Untersuchung von 38 steht noch aus.
40 % [Ru(TMM)3(ZnCp*)3(ZnMe)3] (37)
[Ru(GaCp*)3(TMM)] (13)
+8 ZnMe2
+
60 % [Ru(TMM)3(ZnCp*)2(ZnMe)4] (38)
Schema 120.1. Synthese der heteroleptischen Komplexe [Ru(TMM)3(ZnCp*)3(ZnMe)3] (37)
und [Ru(TMM)3(ZnCp*)2(ZnMe)4] (38)
Welche Schlussfolgerungen lassen sich aus den Synthesen von 36-38 ziehen? Zunächst
einmal ist festzustellen, dass es bei den Umsetzungen von [fac-Mo(CO)3(GaCp*)3] und
[Ru(GaCp*)3(TMM)] mit Überschüssen an ZnMe2 zu einer vollständigen Substitution der
GaCp* Liganden durch die doppelte Anzahl an ZnR Liganden kommt, wie es bereits zuvor in
den Synthesen der pseudo-homoleptsichen Komplexe 23, 26, 28, 30 sowie 33-35 beobachtet
wurde. Somit handelt es sich bei den neuen Verbindungen 36-38 um isoelektronische und
quasi-isostrukturelle 18 Elektronen Komplexe. In den Strukturen ist ein d6-Metallzentrum
durch sechs ZnR Liganden sowie durch stark gebundene Co-Liganden umgeben.
Offensichtlich ist das Synthesekonzept sehr allgemein und somit auch auf heteroleptische
Eduktkomplexe des Typs [LaMb(GaCp*)c] anwendbar. Einzige, jedoch zwingende
Voraussetzung ist, dass die Ausgangsmaterialien GaCp* enthalten, da nur dann die
erforderlichen Redoxprozesse ablaufen können (siehe Kapitel 3.7). Aufgrund der Fülle an
bereits verfügbaren Startverbindungen, die eine Vielzahl von verschiedenen Co-Liganden
beinhalten, ist in Zukunft eine Ausweitung auf ein großes Spektrum unterscherschiedlicher
Komplexe mit ZnR Liganden zu erwarten.
120
4. Zusammenfassung
4. Zusammenfassung
Die vorliegende Arbeit befasst sich mit der Synthese und Charakterisierung von
Übergangsmetallkomplexen, welche niedervalente Aluminium- und Galliumorganyle
beinhalten.
Die
Koordinationschemie
dieser
exotischen
Ligandensysteme
„ist
ein
hochaktuelles Gebiet der modernen Hauptgruppenchemie, das in den letzten Jahren
beträchtliche Fortschritte verzeichnete.“29-34 Jüngste Glanzlichter in diesem Bereich stellen
die Synthesen der ersten Nd-Ga,35 U-Ga36 oder U-Al37 Bindungen dar. Während über die
Struktur- und Bindungseigenschaften der Komplexe [LaMb(ER)c] im Laufe der letzten 15
Jahre viele Arbeiten entstanden sind, ist bisher sehr viel weniger über ihre
Organometallchemie und den damit verbundenen potentiellen Anwendungen berichtet
worden. Der Schwerpunkt dieser Arbeit lag daher nicht nur auf der Erschließung neuer
Synthesewege für homo- und heteroleptische ECp* Komplexe (E = Al, Ga), sondern vielmehr
auf der Folgechemie der sich daraus ergebenen Produkte. Im besonderen Fokus lagen dabei
die einzigartigen Eigenschaften der Cp* Schutzgruppen, die zur Stabilisierung von
niedervalenten Gruppe 13 Verbindungen notwendig sind und die sich unter geeigneten
Bedingungen entfernen ließen. Im Zuge dieser Studien gelang es, eine neue Stoffklasse von
hochkoordinierten Übergangsmetallkomplexen mit ungewöhnlichen ZnIR Liganden zu
erschließen, die die molekulare Koordinations- und Clusterchemie mit der intermetallischen
Festkörper-Materialchemie auf eine neue Art verbindet.
4.1
Reaktivität von ECp* (E = Al, Ga) gegenüber Metall-Olefin-
Komplexen
Bislang gelang es nur unter Verwendung von sterisch anspruchsvollen GaI Liganden
Ga(DDP) oder [Ga{[N(Ar)C(H)]2}]− (Ar = C6H3iPr2-2,6) Übergangsmetallkomplexe zu
isolieren, die Olefine als Co-Liganden beinhalten, wohingegen es im Fall von ECp* stets zu
einer vollständigen Substitution kam.143-145 In der vorliegenden Arbeit wurde nun gezeigt,
dass die Reaktionen von Olefinen enthaltenden, basischen d8-Übergangsmetallkomplexen mit
GaCp* und AlCp* zwar teilweise zu einer Substitution der Olefin-Liganden durch ECp*
führen, in den meisten Fällen wird jedoch aufgrund der starken Basizität der Metallzentren
121
4. Zusammenfassung
Ru0 und RhI eine vollständige Substitution verhindert. So reagiert beispielsweise [Ru(η4butadiene)(PPh3)3] mit GaCp* zum Substitutionsprodukt [Ru(η4-butadiene)(PPh3)2(GaCp*)]
(1), welches auch unter hydrogenolytischen Bedingungen, auch in Anwesenheit von GaCp*,
stabil ist. Im Gegensatz dazu führt die Umsetzung des Bis-Styrol Komplexes
[Ru(PPh3)2(styrol)2] mit drei Äquivalenten GaCp* unter vollständiger Substitution der OlefinLiganden zu [Ru(PPh3)2(GaCp*)3] (2), während die Reaktionen der RhI Verbindungen
[Rh(η2,η2-NBD)(PCy3)2][BArF] und [Rh(η2,η2-COD)2][BArF] mit den entsprechenden ECp*
Liganden (E = Al, Ga) zu den Komplexen [Rh(η2,η2-NBD)(PCy3)(GaCp*)2][BArF] (4),
[Rh(η2,η2-COD)(GaCp*)3][BArF] (5) und [Rh(η2,η2-COD)(AlCp*)3][BArF] (6) führen.
Aufgrund der Koordination des starken σ-Donors ECp* wird die π−Rückbindung vom
Übergangsmetall zum Olefin-Liganden verstärkt, was sich u. a. in den C-C Bindungslängen
der koordinierten Olefine widerspiegelt. So lässt sich beispielsweise die Festkörperstruktur
des Butadienkomplexes 1 am geeignetsten durch ein Metallacyclopropen und nicht durch
einen einfachen π-Komplex beschreiben.
Desweiteren gelang es verbleibende Olefine unter hydrogenolytischen Bedingungen
abzuspalten. So entstand beispielsweise bei der Reaktion von [Mo(η4-C4H6)3] mit sechs
molaren Äquivalenten GaCp* in Toluol unter einer 3 bar H2 Atmosphäre der homoleptische,
sechsfach koordinierte Übergangsmetall-Gallium-Komplex [Mo(GaCp*)6] (22).
4.2
Synthesen und Strukturen elektronenreicher ECp*-Ruthenium-
Polyhydrid-Komplexe und Cluster (E = Al, Ga)
Während das Reaktionsverhalten von ECp* (E = Al, Ga) gegenüber potentiell
reduzierbaren späten Übergangsmetallkomplexen [LaMXb] (L = neutraler Ligand, X =
Halogenid,
Methyl)
ein
gut
untersuchtes
Gebiet
in
der
Koordinationschemie
I
niedervalenter Gruppe 13 Organyle E R (E = Al, Ga; R = organische Gruppe) ist,33, 114, 128,
130, 146
war bisher wenig über deren Reaktivität gegenüber Komplexen mit anderen
anionischen Liganden, wie zum Beispiel Hydriden, bekannt. Als Startmaterialien für diese
Untersuchungen wurden die Ruthenium-Komplexe [Ru(COD)(H)(NH2NMe2)3][BArF],
[Ru(PCy3)2(H2)2(H)2], [{Cp*Ru}2(µ-H)4] und [{Cp*Ru}3(μ-H)3(μ3-H)2] ausgewählt. In
den Umsetzungen konnten zwei unterschiedliche Reaktionstypen festgestellt werden. Die
einkernigen Komplexe [Ru(COD)(H)(NH2NMe2)3][BArF] und [Ru(PCy3)2(H2)2(H)2]
122
4. Zusammenfassung
reagieren unter Substitution der labilen Hydrazin bzw. H2 Liganden zu den monomeren
Komplexen [Ru(COD)(H)(GaCp*)3][BArF] (7) und [Ru(PCy3)2(GaCp*)2(H)2] (17),
während die mehrkernigen Moleküle [{Cp*Ru}2(µ-H)4] und [{Cp*Ru}3(μ-H)3(μ3-H)2]
unter Addition von ECp* zu den elektronenreichen Polyhydrid Clustern [{Cp*Ru(μH)(H)(μ-ECp*)}2] (8, E = Ga; 9, E = Al) und [{Cp*Ru}3(μ-H)5(μ3-ECp*)] (10, E = Al;
11, E = Ga) reagieren. In den untersuchten Fälle wurden weder Redoxreaktionen noch
Insertionen in die Ru-H Bindungen beobachtet, wobei manche Produkte allerdings
verbrückende Ru-H-E Einheiten aufweisen. Die Stabilität der entstehenden Moleküle wird
auch dadurch verdeutlicht, dass weder eine reduktive Eliminierung von Cp*H noch ein
reduktiver
Verlust
von
H2
stattfindet.
Offensichtlich
senken
die
anionischen
Hydridliganden die Elektrophilie der Übergangsmetallzentren ausreichend, um Cp*
Transferreaktionen zu verhindern (siehe Reaktionen von GaCp* mit kationischen WernerKomplexen)130. Die gezeigten Ergebnisse offenbaren einen neuen synthetischen Zugang
zu Übergangsmetall-ECp* Komplexen in höheren Oxidationsstufen, deren Folgechemie
von Interesse gegenwärtiger Untersuchungen ist.
4.3
Cp* als entfernbare Schutzgruppe
4.3.1 Kontrolliertes Clusterwachstum durch Hydrogenolyse von koordinierten GaCp*
Bei der sanften chemischen Synthese von M/E Hume Rothery Phasen46-48, 152 basierend auf
der
Co-Hydrogenolyse
von
ECp*
und
ausschließlich
Kohlenwasserstoff-Liganden
beinhaltenden Komplexen [MLn] als Präkursoren (n =1, E = Al, Ga; n = 2, E = Zn; M = Cu,
Ni) handelt es sich um einen gezielten molekularen Ansatz zur Darstellung intermetallischer
Nanomaterialien
ausgehend
13/Übergangsmetallchemie.150,
151
von
den
Grundlagen
molekularer
Gruppe
Im Zuge dieser Arbeit wurde eine Hydrogenolyse eines
GaCp* beinhaltenden Übergangsmetallkomplexes im Detail untersucht. So entsteht bei der
Hydrogenolyse
von
[Ru(η4-COD)(η3-C4H7)2]
in
Anwesenheit
von
GaCp*
[(GaCp*)4(H)Ru(μ-Ga)Ru(H)2(GaCp*)3] (12) mit Cp*H, Cyclooctan und Isobutan als
Nebenprodukten.
In
ähnlicher
Weise
lässt
sich
[Ru(η2,η2-COD)(η6-COT)]
als
Rutheniumquelle in der Synthese von 12 verwenden, wobei unter leicht veränderten
Reaktionsbedingungen
die
Intermediate
[Ru(GaCp*)(η4-COD)(η4-COT)]
und
123
4. Zusammenfassung
[Ru(GaCp*)3(η4-COD)] (3) entstehen. Eine spektroskopische und strukturelle Untersuchung
von 12 offenbart zwei oktaedrisch koordinierte Ruthenium-Zentren, welche durch ein
„nacktes“, substituentenfreies Galliumatom in einer nahezu linearen Anordnung miteinander
verbunden sind. Dabei koordinieren insgesamt sieben GaCp* und drei Hydridliganden in der
Art und Weise an zwei Rutheniumzentren, dass dabei die beiden unterschiedlichen Ru
Fragmente [(GaCp*)4(H)Ru(μ-Ga)] bzw. [(GaCp*)3(H)2Ru(μ-Ga)] entstehen.
2
+ 8 GaCp*,
3 bar H2, 60 °C, 1h
- Cp*H, - 2 C8H16,
- 4 C4H10
Ru
Cp*Ga
GaCp*
GaCp*
H
Ru
Ru
Ga
Cp*Ga
Cp*Ga
H
+ 6 GaCp*
80 °C, 1h
- C8H12
GaCp*
GaCp*
-Cp*H, + 2 GaCp*
- C4H10 3 bar H2, 100 °C, 2h
CH2
H2 C
12
H
CH2
2
Ru
Cp*Ga
GaCp*
Cp*Ga
13
H
Cp*Ga
GaCp*
2
Ru
+ 8 GaCp*
3 bar H2, 100 °C, 2h
Ru
Cp*Ga
- Cp*H, - 4 C8H16
Cp*Ga
12
+ 6 GaCp*
3 bar H2, RT, 1h
2
Ru
- 2 C8H16
GaCp*
+ 4 GaCp*
3 bar H2, RT, 1h
- 2 C8H16
Ga
Cp*Ga
H
+ 2 GaCp*
RT, 30 min
H
Ru
GaCp*
GaCp*
- Cp*H, + 2 GaCp*
- 2 C8H16 3 bar H2, 100 C°, 2h
Ru
2
Cp*Ga
GaCp*
Cp*Ga
3
Schema 124.1. Clusterwachstum durch Hydrogenolyse von koordinierten Liganden
124
4. Zusammenfassung
1
H NMR spektroskopische Untersuchungen belegen, dass auch bei -100 °C ein extrem
schneller fluktionaler Prozess in Lösung stattfindet, bei dem alle GaCp* Liganden sowie die
Hydridliganden über alle drei Metallzentren ihre Positionen tauschen. Eine CoHydrogenolyse von [Ru(η4-COD)(η3-C4H7)2] und GaCp* in einem molaren Verhältnis von
1:2 führt zu RuGa2 und Ru Partikeln, womit 12 als ein Intermediat in einem solchen
Legierungsbildungsprozess aufgefasst werden kann. Somit repräsentiert 12 eine frühe
molekulare Stufe auf dem Weg zu Ru/Ga Nanopartikeln während einer nasschemischen
Synthese.
Desweiteren wurde der Mechanismus der Hydrogenolyse von koordinierten GaCp* unter
Freisetzung von Cp*H, wie in der Synthese von 12 beobachtet, im Detail durch
Dichtefunktionalmethoden
untersucht.
Dabei
konnte
ein
sinnvoller
Mechanismus
vorgeschlagen werden, der ausgehend von dem Schlüsselintermediat [Ru(GaCp)4(H)2] (IM1)
über
eine
konzertierte
CpH
Eliminierung
und
anschließende
Assoziations-
/Dissoziationsgleichgewichte schließlich zu der Modellverbindung [(GaCp)4(H)Ru(μGa)Ru(H)2(GaCp)3] (1Cp) führt.
4.3.2 Methylgallium als terminaler Ligand in dem all-Gallium koordinierten Rhodium
Kation [(Cp*Ga)4Rh(GaCH3)]+ (15) durch protolytische Spaltung von Cp*H
Ausgehend von den jüngsten Ergebnissen zu [Ga2Cp*][BArF]45 und [Pt(GaCp*)4Ga][BArF] 32,
45, 186
konnte ein terminal koordiniertes Methylgallium erzeugt und kristallographisch
charakterisiert werden. Der kationische Komplex [(Cp*Ga)4Rh(GaCH3)]+ (15) ist eines von
wenigen Beispielen eines pseudo-homoleptischen Komplexes [MLn] mit einer hohen
Koordinationszahl (n > 4) und Liganden, die über Metallatome an das Zentralmetall binden.
Die Röntgenstrukturanalysen der Verbindung [Rh(GaCp*)4(GaMe)][BArF] (15) und ihres
Pyridinaddukts [(GaCp*)4Rh(Ga(CH3)(py))][BArF] (16) belegen die kontraintuitive Tatsache,
dass Methylgallium, als kleinster Vertreter der Alkylgallium(I) Familie, ein guter σ-Donor,
jedoch ein schlechter π-Rückbinder ist. Dies ist nicht nur aus struktureller und
bindungstheoretischer Sicht interessant. Die bemerkenswerte selektive Spaltung eines Cp*
Liganden durch Protonierung einer Ga(CH3)(Cp*) Gruppe in [(Cp*Ga)4Rh(η1-Cp*GaCH3)]
(14) unter bevorzugter Freisetzung von Cp*H gegenüber CH4 und der daraus resultierenden
125
4. Zusammenfassung
Bildung von 15 ermöglicht einen Zugang zu sterisch anspruchslosen Alkylgallium(I)
Verbindungen, die in freier Form instabil und nicht isolierbar sind.
CH3
H3C
Ga
[(COD)(py)Rh(CH3)]
+ 5 GaCp*
- COD, Pyridin
*CpGa
Rh
[BArF]-
Ga
GaCp*
GaCp*
+ [H(OEt2)2][BArF]
- Cp*H
*CpGa
*CpGa
Rh
GaCp*
GaCp*
Cp*Ga
14
15
Schema 126.1. Terminales Methylgallium durch Protolyse eines koordinierten (η1Cp*GaCH3) Liganden
Die neuen Ergebnisse heben die einzigartigen Eigenschaften von Cp* Gruppen im Vergleich
zu anderen Substituenten R hervor, die zur Stabilisierung von niedervalenten Gruppe 13
Verbindungen notwendig sind, insbesondere zu den N,N chelatisierenden Bisketiminaten und
analoger
Verbinungen.101
Das
fluktionale
Verhalten
an
Hauptgruppenelementen180
koordinierter Cp* Einheiten sowie die vergleichsweise schwache Bindung zwischen Cp* und
des Gruppe 13 Zentrums ermöglichen den Einsatz als Schutzgruppen, die unter geeigneten
Bedingungen entfernt werden können. Die gezeigten Ergebnisse deuten auf eine allgemeine
Anwendbarkeit
dieser
Methode
der „Entschützung“ innerhalb der [LaMb(ECp*)c]
Verbindungsklasse hin. Somit ist ein tieferes Verständnis dieser Chemie ein Schlüssel für die
Entwicklung einer sehr kontrollierten molekularen Bausteinchemie für Hume Rothery artige
intermetallische M/E Cluster und Nanomaterialien.
4.4
Über die Organometallchemie des Ga+ Kations
Eine charakteristische organometallische Chemie des nackten Kations Ga+ wurde bisher noch
nicht untersucht. Das Hauptproblem besteht dabei in der Redoxlabilität des Ga+, welches sehr
leicht durch zahlreiche Reaktionspartner mit redoxaktiven Übergangsmetallzentren reduziert
oder oxidiert werden kann. Nichtsdestotrotz handelt es sich bei nacktem Ga+ um ein sehr
126
4. Zusammenfassung
interessantes Synthon für ungewöhnliche Komplexe des Typs [LnM-GaR’] (M =
Übergangsmetall, R’ = anionischer Ligand, unterschiedlich zu Cp*, etc.), die z. B. durch
Insertion von Ga+ in eine M-R’ Bindung oder durch Addition eines nukleophilen Fragments
R’ an einen elektrophilen Komplex [LnMGa]+ zugänglich wären. Tatsächlich führt die
Reaktion des Ga+ Transferreagenz [Ga2Cp*][BArF] mit [Ru(GaCp*)3(TMM)] (13) zu einem
Angriff eines nukleophilen Kohlenstoffs an ein elektrophiles Ga+ Ion und zur Ausbildung
einer RGa Spezies, was zu einem „Einschalten“ des freien Elektronenpaares am GaI führt.
Damit wird das elektrophile Ga+ Ion in ein basisches GaR konvertiert. Dies führt zu einer
Koordination der monovalenten RGa Einheit an ein benachbartes Rutheniumzentrum und
schließlich zu der Bildung des Dimers [{Ru(GaCp*)3(η3-(CH2)2C(CH2(μ-Ga)))}2][(BArF)2]
(19). Komplex 19 ist im Festkörper stabil, isomerisiert jedoch in polaren Lösemitteln wie
Fluorbenzol
bei
höheren
Temperaturen
[{Ru(GaCp*)3(η3-(CH2)(CH(μ-
zu
Ga)(CH3))}2][(BArF)2] (20). Dabei ist das Galliumatom nicht länger an eine aliphatische CH2
Gruppe sondern vielmehr an eine Vinylgruppe gebunden, wodurch die thermische
Isomerisierung von 19 → 20 als “Tautomerisierung” eines Allyl-Fragmentes betrachtet
werden kann.
H2C
CH2
H2C
H2C
CH2
+ [Ga2Cp*][BArF]
Ru
*CpGa
*CpGa
GaCp*
GaCp*
GaCp*
Ga
Ru
Ru
*CpGa
*CpGa
H2
C
Ga
*CpGa
2+
2 [BArF]-
GaCp*
CH2
C
H2
CH2
19
13
GaCp*
PCy3
H
*CpGa
Ru
*CpGa
H
PCy3
17
+ [Ga2Cp*][BArF]
- H2, GaCp*
[BArF]-
Cy3P
Ru
Ga
Cy3P
GaCp*
21
Schema 127.1. Oben: Das Ga+ Transferreagenz [Ga2Cp*][BArF] als Synthon für
ungewöhnliche Komplexe des Typs [LnM-GaR’] Unten: Synthese eines 16 Elektronen
Komplexes mit terminalen, „nacktem“ Ga+ via reduktiver H2 Eliminierung
127
4. Zusammenfassung
Die RGa Einheiten in 19 und 20 binden via closed shell Wechselwirkungen schwach
aneinander, wobei dieser Effekt groß genug erscheint, um die Bildung der diskreten dimeren
Strukturen 19 und 20 gegenüber der möglicher Alternativen, wie polymerer Ketten, zu
begünstigen.
Desweiteren
entsteht
bei
der
Reaktion
von
[Ru(PCy3)2(GaCp*)2(H)2]
(17)
mit
[Ga2Cp*][BArF] unter reduktiver Eliminierung von H2 selektiv die ionische Verbindung
[Ru(PCy3)2(GaCp*)2(Ga)][BArF] (21). Dabei handelt es sich bei dem Komplex 21 um das
zweite Beispiel einer Verbindung mit “nacktem” Ga+, welches terminal an ein
Übergangsmetall koordiniert. Das eher basische Ru0 d8-Metallzentrum sowie die starken σ/πAkzeptor Eigenschaften des Ga+ Liganden führen in Übereinstimmung mit bereits bekannten
Daten des verwandten Platin Komplexes [(Ga)Pt(GaCp*)4][BArF]44 zu dem kürzesten bisher
bekannten Ru-Ga Kontakt.
4.5
Die
neue
Stoffklasse
der
hochkoordinierten
Zink-reichen
Übergangsmetallkomplexe
Ausgehend von homo- oder heteroleptischen Komplexen des Typs [LyM(GaCp*)x] wurde
eine Stoffklasse von hochkoordinierten einkernigen Verbindungen der allgemeinen Formel
[M(M’R1)a(ER2)b] (M’ = Zn) erschlossen, deren Prototyp das zwölfach koordinierte
[Mo(ZnMe)9(ZnCp*)3] (23) ist. Bei diesen Verbindungen handelt es sich nicht um eine
Laborkuriosität, welche nur in der Gasphase oder in einer Tieftemperaturmatrix isoliert
werden können. Sie können in hohen Ausbeuten in einem präparativen Maßstab isoliert
werden, welche tiefergehende Untersuchungen ihrer Folgechemie oder ihren physikalischen
Eigenschaften ermöglicht. Für einen synthetischen Zugang ist die Verwendung der
Molekülfamilie [LaMb(ECp*)c] als Ausgangsmaterialien zwingend notwendig, da hier die
kombinierten
Eigenschaften
der
Liganden
ECp*
(starke
Donoren,
intrinsische
Reduktionsmittel, labil gebundener Cp*-Rest) zum Tragen kommen. Die Reaktionen
beinhalten zusätzlich zur Wanderung von Cp* vom Gallium zum Zink und Me- bzw. EtGruppen vom Zink zum Gallium eine Reduktion von ZnII zu ZnI unter Bildung von
Me2GaCp* und GaMe3 als wichtigste GaIII Nebenprodukte, sowie verschiedener
Fulvalenspezies.
Die
Vielfalt
der
Nebenprodukte
deutet
dabei
auf
beteiligte
128
4. Zusammenfassung
Radikalmechanismen hin. Die entstehenden monovalenten Zinkfragmente ZnMe bzw. ZnEt
und ZnCp* werden abgefangen und an das jeweilige Übergangsmetallzentrum gebunden. Die
Triebkraft sowie die hohe Selektivität der Bildung der entsprechenden Moleküle hängen
sicherlich mit der günstigen Oxidation von GaI zu GaIII und den sehr ähnlichen elektronischen
und sterischen Eigenschaften von monovalenten GaR und ZnR Liganden an einem
Übergangsmetallzentrum zusammen. Desweiteren ist die Gesamtzahl an ZnR Liganden in den
resultierenden Übergangsmetallkomplexen ausschließlich von der Zahl der Galliumliganden
im Präkursor sowie von der in der Reaktion eingesetzten Menge an ZnMe2 abhängig. Aus
dem Blickwinkel der Koordinationschemie ist ein Zwei-Elektronen-GaCp* Ligand zu zwei
ZnCp* oder ZnMe Liganden mit jeweils einem Elektron gleichwertig, so dass es bei den
Umsetzungen von Komplexen des Typs [M(GaCp*)x] mit einem Überschuss an ZnR2 > 2x
stets zur Bildung der pseudo-homoleptischen 18e Komplexe [M(ZnCp*)a(ZnR)b] kommt.
Abbildung 129.1. Festkörperstrukturen der pseudo-homoleptischen Übergangsmetall-ZnR
Komplexe wie sie durch Röntgenstrukturanalyse bestimmt wurde (thermische Ellipsoide für
50% Wahrscheinlichkeit, die Wasserstoffatome sind aus Gründen der Übersichtlichkeit nicht
gezeigt).
129
4. Zusammenfassung
Unter Verwendung von geringeren Mengen an ZnMe2 entstehen dabei gemischtmetallische
Ga/Zn Intermediate, deren Existenz eine unterschiedliche Reaktivität der GaCp* Liganden
verdeutlicht. Desweiteren belegt die Synthese von [Ru(ZnCp*)4(ZnMe)4(H)2] (29), dass in der
Reaktion von [(GaCp*)4(H)Ru(μ-Ga)Ru(H)2(GaCp*)3] (12) zunächst die GaCp*-Liganden
und erst darauf folgend die Hydride mit ZnMe2 reagieren.
Abbildung 130.1. Festkörperstrukturen der gemischtmetallischen Ga/Zn Intermediate wie sie
durch
Röntgenstrukturanalyse
bestimmt
wurde
(thermische
Ellipsoide
für
50%
Wahrscheinlichkeit, die Wasserstoffatome sind aus Gründen der Übersichtlichkeit nicht
gezeigt).
Es ist beachtenswert, dass die Moleküle die VSEPR Regeln befolgen, so dass es sich bei den
Metallkäfigen in den Festkörperstrukturen um platonische Körper handelt. Desweiteren
offenbaren
die
Koordinationsumgebungen
der
Metallatome
in
den
Komplexen
[Mo(ZnCp*)3(ZnMe)9] (23) und [{Mo(CO)4}4(Zn)6(μ-ZnCp*)4] (27) strukturelle Analogien
bezüglich der Situation in Mo/Zn Hume Rothery Phasen. So enthält die experimentell
ermittelte Struktur der MoZn20.44 Phase ikosaedrische MoZn12 Einheiten, welche auch in
[Mo(ZnCp*)3(ZnMe)9] (23) vorhanden sind. Eine gedankliche Flächenverknüpfung des in
[{Mo(CO)4}4(Zn)6(μ-ZnCp*)4] (27) angetroffenen Mo4Zn6 Supertetraeders führt zu einer
zweifach verkappten pentagonal-prismatischen Zn10Mo2 Koordinationsumgebung, welche
auch ein dominierendes Motiv in der Festkörperstruktur von MoZn20.44 ist (siehe Abbildung
131.1).
130
4. Zusammenfassung
Abbildung 131.1. Oben: Der Mo4 Tetraeder (links) und der Zn6 Oktaeder (rechts) in der
molekularen
Struktur
von
[{Mo(CO)4}4(Zn)6(μ-ZnCp*)4]
(27)
wie
sie
durch
Einkristallröntgenstrukturanalyse bestimmt wurde. Unten: Ausschnitt aus einer (imaginären)
Mo/Zn Legierugsstruktur, die aus flächenverknüpften Mo4Zn6 super-Tetraedern (links)
aufgebaut ist. Die ikosaedrische Zn12 Umgebung für die Mo Atome (Mitte) und die zweifach
verkappte pentagonal-prismatischen Zn10Mo2 Umgebung (rechts) für die Zn Atome.
Obwohl alle Moleküle formal die Edelgasregel befolgen, belegen quantenchemische
Untersuchungen, dass die Bindungssituationen deutlich komplexer sind. Aus den
Bindungsanalysen, die unter Verwendung von drei verschiedenen Methoden, nämlich MO
Korrelation, EDA und AIM vorgenommen wurden, wird der Schluss gezogen, dass
beispielsweise die ikosaedrischen Schlüsselverbindung 23 am besten als eine perfekt sd5
hybridisierte Übergangsmetallverbindung beschrieben wird, in der alle 12 Lappen der
Hybridorbitale für starke kovalente chemische Bindungen verwendet werden. Die achtzehn
Valenzelektronen in 23, die für Mo-Zn und Zn-Zn Bindungswechselwirkungen verfügbar
sind, führen zu sechs 2-Elektron-3-Zentrenbindungen zwischen Mo und Zn und drei sehr
schwachen delokalisierten Zn-Zn Bindungen. Diese sind gerade stark genug zur Minimierung
von Ligand-Ligand Abstoßungen und führen so zu der hohen Koordinationszahl im Vergleich
zu üblichen Übergangsmetallkomplexen [MLn] monodendater Liganden L. Die Diskussion
131
4. Zusammenfassung
der elektronischen Struktur belegt, dass 23 weder ein Prototyp eines Clusters noch einer
Koordinationsverbindung ist.
Die strukturellen und elektronischen Eigenschaften der neuen Molekülfamiile zeigen, dass ein
neues Feld metallreicher Moleküle jenseits der Zintl-Grenze betreten worden ist,218 welche
den Raum zwischen Koordinationsverbindungen und Clustern füllen und zur weiteren
Verknüpfung der Chemie und Physik von Molekülen und Festkörpermaterialien beitragen
werden.
132
4. Zusammenfassung
Tabelle 133.1. Überblick über die neu synthetisierten und charakterisierten Verbindungen
#
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
Kapitel / Seite
Verbindung
Exp. Daten
4
3.1 / 27
141
[Ru(η -but)(PPh3)2(GaCp*)]
3.1 / 27
[Ru(PPh3)2(GaCp*)3]
141
4
3.1 / 27
141
[Ru(η -COD)(GaCp*)3]
2
2
F
3.1
/
27
142
[Rh(η , η -NBD)(PCy3)(GaCp*)2][BAr ]
2
2
F
3.1 / 27
142
[Rh(η , η -COD)(GaCp*)3][BAr ]
2
2
F
3.1 / 27
143
[Rh(η , η -COD)(AlCp*)3][(BAr ]
F
3.2 / 29
[(COD)Ru(H)(GaCp*)3][BAr ]
143
3.2
/
30
[{Cp*Ru(µ-GaCp*)}2(µ-H)4]
143
3.2 / 30
[{Cp*Ru(µ-AlCp*)}2(µ-H)4]
144
3
3
3.2 / 30
[{Cp*Ru(µ-H)}3(µ -AlCp*)(µ -H)2]
144
3
3
3.2 / 30
[{Cp*Ru(µ-H)}3(µ -GaCp*)(µ -H)2]
144
3.3
/
32
145
[(GaCp*)4(H)Ru(μ-Ga)Ru(H)2(GaCp*)3
3.3 / 32; 3.5 / 51
[Ru(GaCp*)3(TMM)]
154
1
3.4 / 42
[(Cp*Ga)4Rh(η -Cp*GaCH3)]
154
F
3.4 / 43
[(Cp*Ga)4Rh(GaCH3)][BAr ]
154
F
3.4
/
48
[(Cp*Ga)4Rh(Ga(CH3)(py))][BAr ]
155
3.5 / 52
[Ru(PCy3)2(GaCp*)2(H)2]
155
3
F
3.5 / 54
156
[Ru(GaCp*)4(η -(CH2)2C(CH3))][BAr ]
3
F
3.5 / 56
156
[Ru(GaCp*)3(η -(CH2)2C(CH2(μ-Ga)))2][(BAr )2]
3
F
3.5
/
58
156
[Ru(GaCp*)3(η -(CH2)(CH(μ-Ga)(CH3))}2][(BAr )2]
F
3.5 / 61
[Ru(PCy3)2(GaCp*)2(Ga)][BAr ]
157
3.6 / 64
[Mo(GaCp*)6]
157
3.7 / 67
[Mo(ZnCp*)3(ZnMe)9]
158
3.7
/
67;
3.9.1
/
85
[Mo(GaMe)4(ZnCp*)4]
158
3.7 / 67; 3.9.1 / 85
[Mo(GaMe)2(ZnCp*)4(ZnMe)4]
158
3.7 / 67; 3.9.1 / 88
159
[Mo(ZnCp*)2(ZnEt10)]
3.8 / 80
159
[{Mo(CO)4}4(Zn)6(μ-ZnCp*)4]
3.9.2
/
89;
3.9.6
/
106
[Ru(ZnCp*)4(ZnMe)6]
160
3.9.2 / 91
[Ru(ZnCp*)4(ZnMe)4(H)2]
160
3.9.3 / 93; 3.9.6 / 114
[Rh(ZnCp*)3(ZnMe)6]
160
3.9.3 / 93
[Rh(GaMe)(ZnCp*)4(ZnMe)3]
161
3.9.4
/
94
[Ni(ZnCp*)4(ZnMe)4]
161
3.9.4 / 94; 3.9.6 / 110
[Pd(ZnCp*)4(ZnMe)4]
161
3.9.4 / 94
[Pt(ZnCp*)4(ZnMe)4]
162
3.9.4 / 98
[Pt(ZnCp*)4(ZnEt)4]
162
3.9.7
/
116
[Mo (ZnCp*)3(ZnMe)3(CO)3]
162
3.9.7 / 118
[Ru(ZnCp*)3(ZnMe)3(TMM)]
162
133
5. Ausblick
5. Ausblick
Die durch diese Arbeit eingeführte Stoffklasse der hochkoordinierten Zink-reichen
Übergangsmetallkomplexe ist zum gegenwärtigen Zeitpunkt beispiellos. Daher ist es zunächst
nicht absehbar, in welche Richtungen sich die Chemie dieser Molekülfamilie entwickeln wird.
Am naheliegendsten erscheint eine Erweiterung dieser Stoffklasse auf Übergangsmetalle der
Gruppen 3-5. Da für einen synthetischen Zugang die Verwendung der Molekülfamilie
[LaMb(ECp*)c] als Ausgangsmaterialien zwingend notwendig ist, müssen zuvor die
entsprechenden [M(ECp*)n] Komplexe hergestellt werden. Gelingt beispielsweise die
Synthese des hypothetischen, elektronisch gesättigeten Komplexes [Zr(GaCp*)7], würde einer
die Bildung eines vierzehnfach koordinierten (formal) 18-Elektronenkomplexes [Zr(ZnR)14]
nichts mehr entgegen stehen. Entsprechendes gilt für die Übertragung auf die schweren fElemente wie Th und U, zumal jüngst die erste U-AlCp* Bindung beschrieben wurde.37
Wird [Mo(GaCp*)6] (22) mit einem Überschuss an ZnEt2 umgesetzt, bildet sich der
ikosaedrische
Komplex
[Mo(ZnCp*)2(ZnEt)10]
(26),
welcher
im
Vergleich
zu
[Mo(ZnCp*)3(ZnMe)9] (23) eine ZnCp* Einheit weniger enthält. Dies weist auf die
prinzipielle Zugänglichkeit einer wirklich homoleptischen Verbindung [Mo(ZnR)12] unter den
richtigen Bedingungen hin. Daher erscheint es sinnvoll das Synthesekonzept auf andere Reste
R auszudehen (z. B. C6H5, iPr, usw.).
Eine äußerst interessante Fragestellung ist, inwieweit sich das vorliegende Synthesekonzept
auf andere homologe bzw. isolobale Fälle erweitern lässt. Erste erfolgsversprechende
Hinweise liefern die Synthesen und strukturelle Charakterisierungen von hochkoordinerten
Übergangsmetallkomplexen, welche mit CdR Liganden beinhalten. So entsteht bei der
Umsetzung von [Pt(GaCp*)4] mit dem CdMe2 analog zur Synthese von [Pt(ZnCp*)4(ZnMe)4]
Abbildung 134.1. Hochkoordinierte Übergangsmetallkomplexe mit CdR Liganden
134
5. Ausblick
(34)
das
Cd-Homologe
[Pd(ZnCp*)4(ZnMe)4]
[Pt(CdCp*)4(CdMe)4],
(33)
mit
CdMe2
während
der
bei
der
Reaktion
gemischtmetallische
von
Komplex
[Pd(ZnCp*)4(CdMe)4] entsteht. Als letztes Beispiel dient der ikosaedrische Komplex
[Mo(ZnCp*)3(CdMe)9], der sich bei der Umsetzung von [Mo(GaMe)4(ZnCp*)4] (24) mit
CdMe2 bildet.
Im Zuge dieser Expansion lautet eine weitere Schlüsselfrage, ob es möglich ist, das
Synthesekonzept auf isolobale, nicht homologe Fragmente auszudehnen. Erste theoretische
Untersuchungen belegen, dass es sich bei [Mo(MgH)12] wie auch bei [Mo(ZnH)12] (23H) um
ein Minimum handelt. Eine Synthese der zu 23 analogen Mg-Verbindung stellt sicherlich eine
interessante Herausforderung dar, wobei die erforderliche Startverbindung MgMe2 unter
Schlenkbedingungen jedoch leicht zugänglich ist.
Einen weiteren Beleg des hohen Synthesepotentials dieser neuen Stoffklasse liefert die
Reaktion
von
24
mit
ClAuPPh3,
bei
der
der
ikosaedrische
Komplex
[Mo{Au(PPh3)}8(AuCl)2(EMe)2] (E = Ga oder Zn) entsteht. Dabei ist zu beachten, dass es
sich bei den koordinierenden AuL Fragmenten ebenfalls um Synthesebausteine handelt, die
isolobal zu den ZnR Spezies sind. Gelingt in folgenden Experimenten die Synthese des
hypothetischen Komplexes [W(GaCp*)6], könnte ausgehend von einem Wolfram analogen
Komplex
[W(ZnMe)9(ZnCp*)3]
das
ligandgeschütze
Derivat
[W(AuL)12]
der
Gasphasenspezies WAu1222 zugänglich sein. Offensichtlich erscheint eine Ausweitung des
Syntheseprinzips auf eine Verwendung von Übergangsmetallfragmenten, welche isolobal zu
den ZnR Liganden sind, als durchaus möglich.
Abbildung 135.1 Molekülstrukur von [Mo{Au(PPh3)}8(AuCl)2(EMe)2] (E = Ga oder Zn)
135
5. Ausblick
Ein spannendes Forschungsgebiet ist sicherlich eng mit einer ausgedehnten Untersuchung der
Folgechemie dieser neuen Stoffklasse verbunden. Dabei ergeben sich eine Vielzahl von
möglichen Fragestellungen. Lassen sich beispielsweise die Methyl- bzw. Cp* Reste der ZnRFragmente
substituieren?
Wie
reagiert
beispielsweise
der
ikosaedrische
Komplex
[Mo(ZnMe)9(ZnCp*)3] (23) mit stöchiometrischen Mengen Iod oder mit anderen HalogenidTransferreagenzien? Ist dabei ein Alkyl/Halogen-Austausch möglich? Ein Meilenstein in
diesem Bereich wäre sicherlich die Synthese eines homoleptischen Komplexes des Typs
[M(ZnH)x]. Als mögliche Ausgangsmaterialen könnten dabei die Reagenzien L2ZnH2 oder
[Cp*ZnH]2 dienen.
Ein Blick auf die elektronische Struktur von 23 verrät, dass das HOMO hinsichtlich Mo-Zn
nichtbindend ist und die über den Zn12-Käfig delokalisierten vier Elektronen nur zu sehr
schwachen Zn-Zn Bindungen führen. Daraus ergibt sich die Fragestellung, wie sich
Redoxchemie der Stoffklasse im Allgemeinen und von 23 im Speziellen verhält. Eine
Oxidation sollte aufgrund der oben beschriebenen elekronischen Situation prinzipiell zu
keiner merklichen Beeinträchtigung der Stabilität führen und somit sollte auch die Oxidation
des Moleküls ohne Zerfall der Gesamtstruktur möglich sein.
Hochinteressant sind sicherlich auch die Umsetzungen der pseudo-homoleptischen Komplexe
23, 26, 28, 30 und 32-35 mit Dicarbonsäuren. Resultieren hier bei der Auswahl einer
geeigneten Stöchiometrie ähnliche Netzwerkstrukturen, wie sie im Falle der porösen
metallorganischen Gerüstpolymere (MOF´s) vorhanden sind? Eine sekundäre Baueinheit des
Typs MoZn12 oder PtZn8 würde gewiss zu kompizierteren Festkörperstrukturen sowie zu
veränderten elektronischen und physikalischen Eigenschaften der entstehenden Netzwerke
führen.
136
6. Experimentelles
6. Experimentelles
1.
Allgemeine Arbeitstechniken
Alle Umsetzungen mit metallorganischen Verbindungen wurden, falls nicht anders vermerkt
unter rigorosem Ausschluss von Luft- und Feuchtigkeit in ausgeheizten Glasapparaturen
(Schlenk-, Vakuum-Linie- und Glove-Box-Techniken) durchgeführt. Als Inertgas wurde
Argon (nachgereinigt über Cu-Katalysator und über Molekularsieb 4 Å getrocknet; O2- bzw.
H2O Gehalt < 1ppm) verwendet. Tieftemperaturreaktionen wurden im Aceton/Trockeneisbad
bei der angegebenen Temperatur durchgeführt. Die Trocknung und Destillation aller
Verbindungen erfolgte im Hochvakuum (Drehschieberpumpe der Fa. Edwards (p < 2ڄ10-2
mbar).
Lösemittel
Alle verwendeten Lösemittel (Hexan, Toluol, Et2O, THF) wurden mit Hilfe des MBraun SPS
– Solvent Purification-Systems getrocknet. Bei diesem System werden Lösemittel der Qualität
p.a. (>99% Reinheit) mit Hilfe eines Inertgases (Argon) mit 300 – 500 mbar Überdruck aus
den Vorratsgefäßen durch eine Reihenschaltung von zwei Filterkolonnen gedrückt. Die,
abhängig von dem verwendeten Lösemittel, mit Aluminiumoxid (Toluol, THF),
Molekularsieb oder Kupferkatalysator (Hexan, Pentan) gefüllten Kolonnen trocknen und
befreien die Lösemittel von Restsauerstoff. Der Trocknungsgrad aller Lösemittel wurde durch
Titration nach Karl-Fischer überprüft. Nur Lösemittel mit einem Wassergehalt von < 5 ppm
kamen zum Einsatz.
2.
Routineanalysemethoden
Kernresonanzspektroskopie (NMR)
Die Proben zur Aufnahme von Kernresonanzspektren wurden in hochreinen, deuterierten
Lösemitteln der Firma Merck (Uvasol®) und der Firma Deutero gelöst. Die Lösemittel
137
6. Experimentelles
wurden zuvor mit Molekularsieb 4 Å getrocknet und mit Argon gesättigt. Zur Aufnahme der
Spektren wurde ein Bruker Advance 250 MHz NMR-Spektrometer („Bruker Analytik“,
Rheinstetten) verwendet. Die Angabe der chemischen Verschiebung δ erfolgt in ppm relativ
zur eingestrahlten Frequenz, wobei bei 1H- und
13
C-NMR Spektren die Restsignale der
deuterierten Lösemittel als interne Standards verwendet wurden (1H, 250.1 MHz;
13
C, 62.9
13
MHz; C6D6 bei 7.15 ppm und 128.02 ppm). Alle C-NMR-Spektren sind, sofern nicht anders
angegeben, protonenbreitbandentkoppelt. Die chemische Verschiebung δ der
31
P-NMR
Spektren beziehen sich auf einen externen Standard (85 % Orthophosphorsäure). Für die
Angabe der Signalmultiplizitäten werden folgende Abkürzungen verwendet: s = Singulett, d =
Dublett, t = Triplett, q = Quartett, m = Multiplett und br = breites Signal. Die Werte der
Kopplungskonstanten werden in Hz angegeben und sind als Beträge zu verstehen. Die
Vorzeichen der Kopplungskonstanten wurden nicht bestimmt.
Elementaranalyse (EA):
Elementaranalysen aller Verbindungen wurden in der Service-Center-Abteilung der RuhrUniversität Bochum an einem C, H, N-Analyseautomat vom Typ „CHNSO Vario EL 1998“
der Firma „Elementar“ (Hanau) sowie in der AG Elemantaranalyse der Universität
Duisburg/Essen an einem C, H, N-Analyseautomat vom Typ EA 1110 CHNSO der Firma
„Carlo Erba Instruments“ durchgeführt.
Einkristall-Röntgenkristallstrukturanalyse (RSA):
Die Einkristall-Röntgenstrukturanalysen wurden unter Verwendung von Mo-Kα-Strahlung (λ
= 0.7107 Å, Graphit Monochromator) an einem Oxford Xcalibur 2 CCD/PD Diffraktometer
durchgeführt. Die Strukturlösung und -verfeinerung erfolgte unter Verwendung der
Programme SHELXS-97 und SHELXL-97.272, 273 Alle Verfeinerungen wurden mit Hilfe der
Full-Matrix-Least-Squares-Methode gegen F2 durchgeführt, alle nicht-Wasserstoff-Atome
wurden
unter
Verwendung
von
anisotropen
thermischen
Parametern
verfeinert.
Wasserstoffatome wurden in berechneten Lagen isotrop verfeinert. Die finalen Strukturwerte
138
6. Experimentelles
R1, wR2 und der Goodness-of-fit-Faktor (GOF) wurden mit Hilfe der folgenden Formeln
berechnet:
R1 = Σ( Fbeob. − Fber . ) / Σ Fbeob.
1/ 2
2
2
2
2
2 1/ 2
wR2 = [Σ w ( Fbeob
. − Fber . ) / Σ w ( Fbeob . ) ]
2
2
2
1/ 2
GOF = [Σ w ( Fbeob
. − Fber . ) /( N beob. − N ber )]
3.
Zur
Ausgangsverbindungen
Darstellung
der
metallorganischen
Ausgangsprodukte
wurden
handelsübliche
Basischemikalien verwendet, die, wenn nicht anders angegeben, ohne weitere Reinigung
eingesetzt wurden. Kommerziell nicht erhältliche Laborpräparate sind nachfolgend aufgelistet
und wurden gemäß der Literatur synthetisiert.
AlCp*274, 275
[Ru(η4-COD)(η3-C4H7)2]188
GaCp*78, 276
[Ru(PCy3)2(H2)2(H)2}]265, 286
[Ga2Cp*][BArF]45
[Ru(η2,η2-COD)(η6-COT)]287
[Cp*H]277
[Ru(η4-COD)(η4-COT)(GaCp*)]122
[H(OEt2)2][BArF]278
[Ru(GaCp*)6(Cl)2]128
[cis-Mo(GaCp*)2(CO)4]109
[(COD)(py)Rh(CH3)]288
[Mo(η4-C4H6)3]279
[Rh(COD)2][BArF]289
[fac-Mo(GaCp*)3(CO)3]111
[Rh(PCy3)2(η2,η2NBD)][BArF]290, 291
[Ru(η4-C4H6)(PPh3)3]280, 281
[Ni(GaCp*)4]109
[{Cp*Ru}2(µ-H)4]282, 283
[Pd(GaCp*)4]113
[Ru(COD)(H)(NH2NMe2)3][BArF]284
[Pt(GaCp*)4]113
[Ru(styr)2(PPh3)2]285
139
6. Experimentelles
4.
Arbeitsvorschriften und analytische Daten
Arbeitsvorschriften und analytische Daten neuer Verbindungen
[Ru(η4-but)(PPh3)2(GaCp*)] (1). Eine Lösung von [Ru(η4-but)(PPh3)3] (0.200 g, 0.212
mmol) in Toluol (6 mL) wurde mit GaCp* (0.052 g, 0.254 mmol) versetzt und für 1 h bei 80
°C gerührt. Anschließend wurden alle flüchtigen Bestandteile im Vakuum entfernt, der
Rückstand mit wenig Hexan (3 x 2 mL) gewaschen und im Vakuum getrocknet. Der
verbliebene weiße Feststoff wurde in Toluol gelöst durch Abkühlen auf -30°C kristallisiert.
Ausbeute: 0.137 g (73 %). Elementaranalyse: ber. für C50H51GaP2Ru: C, 67.88; H, 5.81. gef.:
C, 66.98; H, 5.04. 1H NMR δH(C6D6), 7.85-6.95 (m, 30H, PPh3), 4.98 (br, 2H, syn 1,4-CH2),
1.81 (s, 15H, GaCp*), 1.51 (br, 2H, CH), -0.66 (br, 2H, anti 1,4-CH2).13C{1H} NMR
δC{H}(C6D6), 143.9 (d, J = 30.5 Hz), 134.0 (d, J = 11.5 Hz), 127.7 (d, J = 27.4 Hz), 114.0
(C5Me5), 75.1 (s, CH=CH2), 36.5 (s, CH=CH2) 10.1 (C5Me5). 31P{1H} NMR δP(C6D6) = 58.9
(PPh3).
[Ru(PPh3)2(GaCp*)3] (2). Eine Lösung von frisch hergestelltem [Ru(styr)2(PPh3)2] (0.300 g,
0.360 mmol) in Toluol (5 mL) wurde bei -30 °C mit GaCp* (0.236 g, 1.151 mmol) versetzt.
Nach langsamen Erwärmen auf Raumtemperatur wurde die Reaktionslösung für weitere 30
min bei 60 °C. Anschließend wurden alle flüchtigen Bestandteile im Vakuum entfernt, der
Rückstand mit wenig Hexan (3 x 2 mL) gewaschen und im Vakuum getrocknet. Das rote
Rohprodukt wurde in heißem Hexan gelöst und durch Abkühlen auf -30 °C kristallisiert.
Ausbeute: 0.300 g (67 %) Elementaranalyse: ber. für C66H75Ga3P2Ru: C, 63.90; H, 6.09. gef.:
C, 63.11; H, 5.70. 1H NMR δH(C6D6) 8.06-7.00 (m, 30H, PPh3), 1.67 (s, 45H, GaCp*).
13
C{1H} NMR δC{H}(C6D6), = 139.8 (d, J = 29.4 Hz), 135.0 (d, J = 10.7 Hz), 129.7 (d, J = 2.4
Hz), 114.5 (C5Me5), 11.2 (C5Me5). 31P{1H} NMR δP(d8-thf) = 63.6 (s, PPh3).
[Ru(η4-COD)(GaCp*)3] (3). Eine Lösung von [Ru(η4-COD)(η6-COT)] (0.500 g; 1.585
mmol) in Hexan (12 ml) wurde in einer Fisher-Porter-Bottle mit GaCp* (1.040 g; 5.073
mmol) versetzt und anschließend unter H2-Atmosphäre (3 bar Arbeitsdruck) für 1 h bei
Raumtemperatur gerührt. Dabei bildete sich eine orange-rote Lösung, sowie ein orangefarbener Niederschlag. Die Suspension wurde in ein Schlenkrohr überführt, das Lösemittel im
140
6. Experimentelles
Arbeitsvorschriften und analytische Daten
Vakuum entfernt, der Rückstand mit wenig kaltem Hexan gewaschen und letztlich im
Vakuum getrocknet. Ausbeute 0.927 g (71 %). Elementaranalyse: ber. für C38H57Ga3Ru: C,
55.38; H, 6.97. gef.: C, 55.52; H, 7.62. 1H NMR δH(C6D6) 3.90 (s, 4H, COD), 2.17 (s, 8H,
COD), 1.96 (s, 45H, GaCp*).
13
C{1H} NMR δC{H}(C6D6) 113.5 (C5Me5), 45.4 (CH, COD),
34.9 (s, CH2, COD), 10.3 ppm (C5Me5).
[Rh(η2, η2-NBD)(PCy3)(GaCp*)2][BArF] (4). Zu einer Lösung von [Rh(PCy3)2(η2,η2-
NBD)][BArF] (0.280 g, 0.173 mmol) in Fluorobenzol (6 mL) wurde GaCp* (0.078 g, 0.380
mmol) zugegeben. Die Reaktionsmischung wurde für 1 h bei 80 °C gerührt. Nach Entfernung
des Lösemittels im Vakuum wurde der Rückstand mit Hexan gewaschen (3x4 mL). Das
Produkt wurde durch Umkristallisation des Rohprodukts durch langsame Diffusion von
Hexan in eine Fluorbenzollösung gereinigt. Ausbeute: 0.239 g (79 %). Elementaranalyse: ber.
für C77H83BF24Ga2PRu: C, 52.89; H, 4.78 gef.: C, 51.98; H, 4.19. 1H NMR δH(d8-thf) 7.79 (s,
8 H, BArF), 7.58 (s, 4H, BArF), 3.64 (br s, 4H, NBD), 3.35 (br s, 2H, NBD), 2.10 (s, 30H,
GaCp*), 1.63 (s, 33H, PCy3).13C{1H} NMR δC{H}(d8-thf) 163.0 (q, J=49.7 Hz, [BArF]), 135.8
([BArF]), 130.2 (q, J=31.7 Hz, [BArF]), 125.7 (q, J=272.2 Hz, [BArF]), 118.4 ([BArF]), 115.9
(C5Me5), 62.1 (s), 48.0 (s), 36.7 (t, J=6.0 Hz), 32.7 (br), 27.7 (q, J=59.8 Hz), 10.4 (C5Me5).
31
P{1H} NMR δP(d8-thf) = 62.0 (d, J=124.1 Hz, PCy3).
[Rh(η2, η2-COD)(GaCp*)3][BArF] (5). Eine Lösung von [Rh(η4-COD)2][BArF] (0.300 g,
0.254 mmol) in Fluorbenzol (5 mL) wurde bei Raumtemperatur mit GaCp* versetzt (0.172 g,
0.839 mmol) und zunächst für 1 h gerührt. Anschließend wurde das Lösemittel im Vakuum
auf ca. 2 mL reduziert und das Produkt durch Zugabe von n-Hexan ausgefällt. Das Lösemittel
wurde abfiltriert und der orangene Rückstand mit Hexan gewaschen (2 x 2 mL) und
anschließend im Vakuum getrocknet. Das Produkt wurde durch Umkristallisation des
Rohprodukts durch langsame Diffusion von Hexan in eine Fluorbenzollösung gereinigt.
Ausbeute: 0.378 g (88 %). Elementaranalyse: ber. für C70H69BF24Ga3Rh: C, 49.77; H, 4.12.
gef.: C, 50.12; H, 4.41. 1H NMR δH(d8-thf) 7.79 (s, 8 H, BArF), 7.58 (s, 4H, BArF), 4.52 (s,
4H, COD), 2.14 (s, 8H, COD), 2.06 (s, 45H, GaCp*). 13C{1H} NMR δC{H}(d8-thf) 162.8 (q,
J=49.6 Hz, [BArF]), 135.6 ([BArF]), 130.0 (q, J=31.7 Hz, [BArF]), 125.5 (q, J=272.3 Hz,
[BArF]), 118.2 ([BArF]) 115.7 (C5Me5), 66.1 (d, J=8.7 Hz, CH, COD), 33.9 (s, CH2, COD),
10.0 ppm (C5Me5).
141
6. Experimentelles
Arbeitsvorschriften und analytische Daten
[Rh(η2, η2-COD)(AlCp*)3][BArF] (6). Eine Suspension von [Rh(η4-COD)2][BArF] (0.300 g,
0.254 mmol) und AlCp* (0.123 g, 0.761 mmol) in Fluorbenzol (5 mL) wurde für 1 h bei 70
°C gerührt. Nach Abkühlen der Reaktionsmischung auf Raumtemperatur wurde das
Lösemittel im Vakuum auf ca. 2 mL reduziert und das Produkt durch Zugabe von n-Hexan
ausgefällt. Das Lösemittel wurde abfiltriert und der orangene Rückstand mit Hexan
gewaschen (2 x 2 mL) und anschließend im Vakuum getrocknet. Das Produkt wurde durch
Umkristallisation des Rohprodukts durch langsame Diffusion von Hexan in eine
Fluorbenzollösung gereinigt. Ausbeute 0.248 g (63 %). Elementaranalyse: ber. für: C, 53.86;
H, 4.46. gef.: C, 54.66; H, 4.48. 1H NMR δH(CD2Cl2) 7.75 (s, 8 H, BArF), 7.58 (s, 4H, BArF),
4.17 (s, 4H, COD), 2.15 (s, 8H, COD), 2.02 (s, 45H, GaCp*). 13C{1H} NMR δC{H}(CD2Cl2)
162.3 (q, J=49.7 Hz, [BArF]), 135.4 ([BArF]), 129.4 (q, J=31.4 Hz, [BArF]), 125.2 (q, J=272.4
Hz, [BArF]), 118.0 ([BArF]) 116.0 (C5Me5), 63.1 (d, J=6.8 Hz, CH, COD), 34.6 (s, CH2,
COD), 10.6 ppm (C5Me5).
[(COD)Ru(H)(GaCp*)3][BArF] (7). Eine Lösung von [Ru(η4-COD)(H)(NH2NMe2)3][BArF]
(0.300 g, 0.239 mmol) in Fluorbenzol (6 mL) wurde mit GaCp* (0.162 g, 0.789 mmol)
versetzt und für 2 h bei 60 °C gerührt. Das Lösemittel wurde im Vakuum auf ca. 2 mL
reduziert und das Produkt durch Zugabe von n-Hexan ausgefällt. Das Lösemittel wurde
abfiltriert und der farblose Rückstand mit Hexan gewaschen (2 x 2 mL) und anschließend im
Vakuum getrocknet. Das Produkt wurde durch Umkristallisation des Rohprodukts durch
langsame Diffusion von Hexan in eine Fluorbenzollösung gereinigt. Ausbeute: 0.384 g (86 %)
farblose kristalline Nadeln. Elementaranalyse: ber. für C70H70BF24RuGa3: C, 49.80; H, 4.18.
gef.: C, 50.02; H, 4.23. 1H NMR δH(d8-thf) 7.79 (s, 8 H, BArF), 7.58 (s, 4H, BArF), 3.69 (s,
4H, COD), 2.14 (s, 15H, GaCp*), 2.11 (s, 8H, COD), 2.03 (s, 30H, GaCp*), -7.43 (s, 1H).
13
C{1H} NMR δC{H}(C6D6) 162.8 (q, J=49.6 Hz, [BArF]), 135.6 ([BArF]), 130.1 (q, J=31.5
Hz, [BArF]), 125.6 (q, J=272.3 Hz, [BArF]), 118.2 ([BArF]) 115.8 (eq.-C5Me5), 115.7 (ax.C5Me5), , 10.1 (ax.-C5Me5), 10.0 ppm (eq.-C5Me5). IR ν = 2005 cm-1 (vs, H)
[{Cp*Ru(µ-GaCp*)}2(µ-H)4] (8). Eine Lösung von [{Cp*Ru}2(µ-H)4] (0.300 g, 0.629
mmol) in 6 mL Toluol wurde mit GaCp* (0.284 g, 1.386 mmol) bei Raumtemperatur
versetzt. Die Reaktionsmischung wurde langsam auf 60 °C erwärmt und für 30 min
gerührt. Nach Abkühlen der dunkelroten Lösung auf Raumtemperatur wurden alle
142
6. Experimentelles
Arbeitsvorschriften und analytische Daten
flüchtigen Bestandteile im Vakuum entfernt und der dunkelrote Rückstand mit wenig
kaltem n-Hexan gewaschen. Der dunkelrote Feststoff wurde in Toluol gelöst und das
Produkt durch Abkühlen auf -30°C kristallisiert. Ausbeute: 0.401 g rote Kristalle (72 %).
Elementaranalyse: ber. für C40H64Ru2Ga2: C, 54.19; H, 7.28. gef.: C, 53.64; H, 6.98. 1H
NMR δH(C6D6) 2.07 (s, 30H, RuCp*), 1.99 (s, 30H, GaCp*), -14.42 (s, 4H).
13
C{1H}
NMR δC{H}(C6D6) 121.7 (C5Me5, Cp*Ga), 92.0 (C5Me5, Cp*Ru), 13.9 (C5Me5, Cp*Ru),
13.1 (C5Me5, Cp*Ga). IR ν = 1823, 1598 cm-1 (vs, H)
[{Cp*Ru(µ-AlCp*)}2(µ-H)4] (9). Eine Supension von [{Cp*Ru}2(µ-H)4] (0.300 g, 0.629
mmol) und AlCp* (0.204 g, 1.258 mmol) in Toluol (10 mL) wurde für 45 min refluxiert.
Die Lösung wurde abfiltriert und alle flüchtigen Bestandteile im Vakuum entfernt. Der
dunkelrote Rückstand wurde mit wenig kaltem n-Hexan gewaschen. Anschließend wurde
der Feststoff wieder in Toluol gelöst und das Produkt durch Abkühlen auf -30 °C
kristallisiert. Ausbeute: 0.358 g rote Kristalle (71 %). Elementaranalyse: ber. für
C40H64Ru2Al2: C, 59.98; H, 8.05. gef.: C, 59.14; H, 8.78. 1H NMR δH(C6D6) 2.09 (s, 30H,
RuCp*), 2.02 (s, 30H, AlCp*), -16.30 (s, 4H).
13
C{1H} NMR δC{H}(C6D6) 117.2 (C5Me5,
Cp*Al), 91.0(C5Me5, Cp*Ru), 14.0 (C5Me5, Cp*Ru), 12.3 (C5Me5, Cp*Al). IR ν = 1820
cm-1 (vs, H)
[{Cp*Ru(µ-H)}3(µ3-AlCp*)(µ3-H)2] (10). Eine Suspension bestehend aus [{Cp*Ru(µ-
H)}3(µ3-H)2] (0.300 g, 0.629 mmol) und AlCp* (0.102 g, 0.629 mmol) in 6 mL Toluol
suspendiert wurde für 1 h refluxiert. Die dunkelrote Lösung wurde auf Raumtemperatur
abgekühlt, wobei sich ein dunkelroter Niederschlag bildet. Die überstehende Lösung
wurde abkanuliert, der Rückstand mit kaltem Hexan gewaschen und im Vakuum
getrocknet. Ausbeute 0.333 g dunkelrote Kristalle (83 %). Elementaranalyse: ber. für
C40H65Ru3Al: C, 54.83; H, 7.48. gef.: C, 55.32; H, 8.23. 1H NMR δH(C6D6) 1.96 (s, 15H,
AlCp*), 1.92 (s, 45H, RuCp*), -13.61 (s, 4H).
13
C{1H} NMR δC{H}(C6D6) 120.4 (ring
atoms, Cp*Al), 86.5 (ing atoms Cp*Ru), 13.2 (CH3, Cp*Ru), 13.0 (CH3, Cp*Al). IR ν =
2051, 1948, 1747 cm-1 (vs, H)
[{Cp*Ru(µ-H)}3(µ3-GaCp*)(µ3-H)2] (11). Eine Lösung von [{Cp*Ru(µ-H)}3(µ3-H)2]
(0.300 g, 0.420 mmol) in 6 mL Toluol wurde bei Raumtemperatur mit GaCp* (0.112 g,
143
6. Experimentelles
Arbeitsvorschriften und analytische Daten
0.546 mmol) versetzt und anschließend bei 60 °C für 1 h gerührt. Alle flüchtigen
Bestandteile wurden im Vakuum entfernt. Das Produkt wurde mit kaltem Hexan
gewaschen, in Toluol gelöst und durch langsames Abkühlen auf -30°C kristallisiert.
Ausbeute: 0.312 g dunkelrote Kristalle (81 %). Elementaranalyse: ber. für C40H65Ru3Ga:
C, 52.28; H, 7.13. gef: C, 51.36; H, 7.97. 1H NMR δH(d8-thf) 1.90 (s, 45H, RuCp*), 1.70
(s, 15H, GaCp*), -12.62 (s, 5H).
13
C{1H} NMR δC{H}( d8-thf) 122.6 (ring atoms, Cp*Ga),
87.8 (ring atoms Cp*Ru), 13.5 (CH3, Cp*Ru), 13.3 (CH3, Cp*Ga). IR ν = 1944, 1893,
1552 cm-1 (vs, H)
[(GaCp*)4(H)Ru(μ-Ga)Ru(H)2(GaCp*)3 (12).
1)
Eine Lösung von [Ru(η4-COD)(η3-C4H7)2] (0.300 g; 0.939 mmol) in Hexan (8 ml)
wurde in einer Fisher-Porter-Bottle mit 4.2 molaren Äquivalenten GaCp* (0.808 g; 3.94
mmol) versetzt und anschließend unter H2-Atmosphäre (3 bar Arbeitsdruck) für 1 h bei 60 °C
gerührt. Nach Abkühlen auf Raumtemperatur wurde die nun dunkelrote Lösung in ein
Schlenkrohr überführt, das Lösemittel im Vakuum entfernt, der Rückstand mit wenig kaltem
Hexan gewaschen und letztlich im Vakuum getrocknet. Der gelbe Feststoff wurde in heißem
Hexan gelöst und das Produkt durch Abkühlen auf -30°C kristallisiert. Ausbeute: 0.625 g
gelbe Kristalle (78 %). Elementaranalyse: ber. für C70H108Ga8Ru2: C, 49.18; H, 6.37, Ga
32.69, Ru 11.85. gef.: C, 48.43; H, 6.72, Ga 32.01, Ru 10.57. 1H NMR δH(C6D6) 1.91 (s,
GaCp*), -15.51 (s, Ru-H).
13
C{1H} NMR δC{H}(C6D6) 117.2 (C5Me5), 10.4 (C5Me5). IR
(nujol): ν = 1818 und 1808 cm-1 (vs, Ru-H).
2)
Eine Lösung von [Ru(η2,η2-COD)(η6-COT)] (0.300 g; 0.95 mmol) in Toluol (12 ml)
wurde in einer Fisher-Porter-Bottle mit GaCp* (0.800 g; 3.90 mmol) versetzt und
anschließend unter H2-Atmosphäre (3 bar Arbeitsdruck) für 2 h bei 100 °C gerührt. Nach
Abkühlen auf Raumtemperatur wurde die nun dunkelrote Lösung in ein Schlenkrohr
überführt, das Lösemittel im Vakuum entfernt, der Rückstand mit wenig kaltem Hexan
gewaschen und letztlich im Vakuum getrocknet. Der gelbe Feststoff wurde in heißem Hexan
gelöst und das Produkt durch Abkühlen auf -30°C kristallisiert. Ausbeute: 0.398 g gelbe
Kristalle (49 %).
144
6. Experimentelles
Arbeitsvorschriften und analytische Daten
Abbildung 145.1. Pulverdiffraktogramm einer Mischung aus RuGa2 und RuGa Partikeln,
welche aus der Umsetzung von [Ru(η4-COD)(GaCp*)3] (3) mit 3 bar H2 in Mesitylene bei
150 °C nach 7 Tagen und anschließender Calcinierung bei 1000 °C für 10 Stunden
resultieren. Es sollte nicht unerwähnt bleiben, dass ein aufgenommenes Pulverdiffraktogramm
vor dem Calcinieren nur sehr breite Reflexe enthielt, was vermutlich auf eine kleine
Partikelgröße zurückgeführt werden kann.
Abbildung 145.2. Pulverdiffraktogramm einer Mischung aus RuGa2 und Ru Partikeln,
welche durch Co-Hydrogenolyse von [Ru(η4-COD)(η3-C4H7)2] und GaCp* in einem molaren
Verhältnis von 1:2 (3 bar H2, Mesitylene, 150 °C, 7 Tage) nach anschließender Calcinierung
(10 h, 1000 °C) erhalten wurden. Es sollte nicht unerwähnt bleiben, dass ein aufgenommenes
Pulverdiffraktogramm vor dem Calcinieren nur sehr breite Reflexe enthielt, was vermutlich
auf eine kleine Partikelgröße zurückgeführt werden kann.
145
6. Experimentelles
Arbeitsvorschriften und analytische Daten
EDX Analysen zeigen, dass sich die Elementzusammensetzungen beider Proben, d.h. vor und
nach Calcinierung, unter Berücksichtigung der allgemeinen Genauigkeit einer EDX Analyse
nur leicht voneinander unterscheiden und einer Gesamtzusammensetzung von Ru1Ga2
entsprechen.
Spectrum processing :
Peak possibly omitted : 1.743 keV
Processing option : All elements analyzed (Normalised)
Number of iterations = 2
Spectrum processing :
Peak possibly omitted : 1.745 keV
Processing option : All elements analyzed (Normalised)
Number of iterations = 2
Element
Gewichtsprozent Atomprozent
Ga K
Ru L
55.53
44.47
Gesamt
100.00
Element
Gewichtsprozent Atomprozent
Ga K
Ru L
62.29
37.71
Gesamt
100.00
64.42
35.58
70.54
29.46
Abbildung 146.1. EDX Analysen der uncalcinierten (oben) und der calcinierten Probe
(unten), welche durch Co-Hydrogenolyse von [Ru(η4-COD)(η3-C4H7)2] und GaCp* in einem
molaren Verhältnis von 1:2 erhalten wurden (3 bar H2, Mesitylen, 150 °C, 7 Tage).
146
6. Experimentelles
Arbeitsvorschriften und analytische Daten
Computational Details sowie zusätzliche Resultate zu den Rechnungen zu Komplex 12
Alle DFT-Berechnungen wurden mit dem Programmpaket Gaussian 03 durchgeführt. Bei
allen
Geometrieoptimierungen
160, 161
Hybridfunktional B3LYP
wurden
ohne
sowie
bei
allen
Frequenzberechnungen
162
und der Basissatz Lanl2DZ
Symmetrieeinschränkungen
optimiert.
wurde
das
verwendet. Alle Strukturen
Die
Berechnungen
der
Übergangszustände wurden durch QST2 oder QST3 Ansätze durchgeführt.
Abbildung 147.1. Berechnete parallele GaCp Assoziations-/Dissoziations-Gleichgewichte in
Lösung, die schließlich zu 1Cp führen
147
6. Experimentelles
Arbeitsvorschriften und analytische Daten
Abbildung 148.1. Berechnete parallele GaCp Assoziations-/Dissoziations-Gleichgewichte in
Lösung, die schließlich zu 1Cp führen
Abbildung 148.2. Berechnete Minima für einen fluktionalen Prozesses
148
6. Experimentelles
Arbeitsvorschriften und analytische Daten
Abbildung 149.1. Berechnete Minima für einen fluktionalen Prozesses
Berechnete Geometrien aller Minima und Übergangszustände
CpH
C
C
C
C
C
H
H
H
H
H
H
1.2264
0.3661
-0.9503
-1.0501
0.2058
1.8899
1.8805
0.7636
-1.8020
-1.9840
0.4646
-0.0827
1.1673
0.8050
-0.6703
-1.2059
-0.1266
-0.1257
2.1765
1.4780
-1.2235
-2.2593
0.0003
0.0000
-0.0012
0.0008
-0.0019
-0.8777
0.8861
-0.0003
-0.0004
0.0047
-0.0009
GaCp
Ga
C
C
C
H
C
H
C
H
H
H
-1.1722 -0.0000 -0.0000
1.0339 -0.8457 -0.8800
1.0341 0.5756 -1.0762
1.0341 -1.0982 0.5323
1.0638 -1.5958 -1.6600
1.0339 1.2015 0.2149
1.0641 1.0859 -2.0306
1.0341 0.1669 1.2090
1.0639 -2.0720 1.0045
1.0632 2.2668 0.4050
1.0639 0.3153 2.2810
1Cp
Ru
H
Ga
Ga
Ga
Ga
Ru
H
H
Ga
Ga
Ga
Ga
C
C
C
H
C
2.2989 -0.1222 -0.1314
2.1127 -0.2662 -1.7582
-0.1388 -0.0457 -0.0915
1.9869 -2.4584 -0.3398
4.6937 -0.2695 -0.4059
2.3058 0.0831 2.2716
-2.4655 -0.2352 0.1044
-1.9063 -1.7807 0.0807
-1.9044 -0.3074 1.6480
2.2102 2.1552 -0.7658
-4.3973 -1.2857 1.0411
-2.9895 -0.4017 -2.2360
-3.0794 2.0562 0.4823
2.9671 -0.5915 4.4982
3.1714 0.8286 4.4033
1.5560 -0.8435 4.3918
3.7379 -1.3359 4.6501
1.8855 1.4499 4.2385
H
C
H
H
H
C
C
C
H
C
H
C
H
H
H
C
C
C
H
C
H
C
H
H
H
C
C
C
H
C
H
C
H
H
H
C
C
C
H
C
H
C
H
H
H
C
C
C
4.1224
0.8888
1.0763
1.6995
-0.1781
3.2690
2.3065
2.5528
4.3406
0.9972
2.5264
1.1517
2.9928
0.0605
0.3506
6.6762
6.5837
6.9397
6.5886
6.7894
6.4094
7.0097
7.0819
6.8029
7.2152
0.4014
1.3914
1.0548
-0.6483
2.6593
1.2135
2.4487
0.5763
3.5973
3.2043
-2.0587
-2.6521
-3.1008
-1.0079
-4.0648
-2.1321
-4.3416
-2.9775
-4.7952
-5.3176
-6.2467
-5.2938
-6.8654
1.3405
0.4168
-1.8123
2.5126
0.5594
4.1470
3.7122
4.5347
4.1957
3.8352
3.3785
4.3426
4.9181
3.5934
4.5467
-1.6335
-0.6200
-0.9859
-2.7006
0.6540
-0.7883
0.4278
-1.4790
1.6142
1.1878
-4.2084
-4.2163
-4.5399
-3.9744
-4.5567
-4.0193
-4.7577
-4.6159
-4.6702
-5.0406
-0.3733
-1.6593
0.6105
-0.1813
-1.4707
-2.6088
-0.0658
1.6763
-2.2521
0.3970
-2.9515
-3.3402
-1.7302
4.4708
4.2327
4.4375
4.1530
4.1223
-1.5729
-2.5510
-0.3887
-1.7142
-1.9682
-3.5565
-0.6336
0.5226
-2.4528
0.0634
-0.9284
-1.9410
0.3259
-1.0872
-1.3121
-2.9959
0.0889
1.2790
-1.8114
0.8317
-0.7715
-1.8134
0.4633
-0.8889
-1.2210
-2.8623
0.1883
1.4307
-1.7482
0.9094
-4.5623
-4.3348
-4.5203
-4.7389
-4.1542
-4.3229
-4.2692
-4.6651
-3.9878
-4.1993
0.9654
1.9678
1.3907
H
C
H
C
H
H
H
C
C
C
H
C
H
C
H
H
H
-6.4663
-5.3261
-4.6813
-6.2982
-7.6278
-4.7404
-6.5622
-4.7134
-3.6616
-4.1644
-5.7428
-2.4644
-3.7600
-2.7761
-4.7072
-1.5035
-2.0926
-3.4938
-2.3543
-4.2326
-1.3601
-1.1826
-2.3733
-0.4895
3.7905
3.8250
4.2140
3.5102
4.2715
3.5817
4.5116
4.3042
4.4200
4.8745
0.0546
3.0143
1.9533
2.6542
0.8513
3.9244
3.2408
1.1000
2.0775
-0.1562
1.2827
1.4228
3.1277
0.0429
-1.0884
1.8996
-0.7144
2Cp
Ru 1.4919 -0.1987 -0.0038
H 1.1952 -0.5069 -1.5821
Ga -0.1642 -1.8315 0.8333
Ga -0.0658 1.6470 -0.3476
Ru -1.7463 -0.0862 0.1255
H -1.5071 0.3004 1.6971
C 2.0888 4.4737 -0.9407
C 1.0819 3.9406 -1.7486
C 1.5671 4.6482 0.3977
H 3.0877 4.7492 -1.2628
C -0.1061 3.7034 -0.9217
H 1.1443 3.7414 -2.8135
C 0.2374 4.2312 0.4173
H 2.1226 5.0450 1.2407
H -1.1065 3.8160 -1.3396
H -0.4400 4.2814 1.2636
H -0.2024 -3.2587 1.5791
Ga -3.3283 -1.6810 0.8892
Ga -1.8655 -0.6932 -2.2168
Ga -3.3223 1.6787 0.1847
Ga 3.1045 1.2252 -0.9445
C -5.4532 2.5069 0.9469
C -4.4422 3.3873 1.4685
C -5.4721 2.6524 -0.4825
H -6.1054 1.8710 1.5314
C -3.8414 4.0762 0.3604
H -4.1990 3.5274 2.5138
C -4.4767 3.6209 -0.8427
149
6. Experimentelles
H -6.1330 2.1346 -1.1655
H -3.0596 4.8219 0.4235
H -4.2540 3.9588 -1.8467
C -3.9328 -3.8543 1.7971
C -4.2990 -2.8681 2.7744
C -4.7406 -3.6422 0.6283
H -3.1817 -4.6228 1.9218
C -5.3371 -2.0492 2.2122
H -3.8777 -2.7696 3.7662
C -5.6102 -2.5270 0.8856
H -4.7148 -4.2324 -0.2784
H -5.8372 -1.2279 2.7089
H -6.3528 -2.1297 0.2059
C -1.3752 -0.1730 -4.5468
C -0.8626 -1.4782 -4.2256
C -2.8082 -0.2402 -4.5182
H -0.7811 0.6977 -4.7932
C -1.9866 -2.3471 -3.9956
H 0.1813 -1.7617 -4.1954
C -3.1850 -1.5782 -4.1780
H -3.4856 0.5800 -4.7188
H -1.9347 -3.4017 -3.7569
H -4.1974 -1.9480 -4.0783
C 5.9545 0.3606 -1.7574
C 5.0808 0.8966 -2.7269
C 6.0480 1.2810 -0.6635
H 6.4620 -0.5948 -1.8212
C 4.5748 2.1647 -2.2343
H 4.8135 0.4450 -3.6751
C 5.2323 2.4024 -0.9426
H 6.6369 1.1393 0.2351
H 4.1142 2.9319 -2.8464
H 5.1538 3.3085 -0.3542
Ga 2.8553 -2.1190 -0.1811
Ga 1.9896 0.4085 2.2836
C 1.9855 2.0112 4.0742
C 1.1526 0.9342 4.5318
C 3.3297 1.5107 3.9536
H 1.6651 3.0257 3.8771
C 1.9776 -0.2277 4.6932
H 0.0889 0.9919 4.7237
C 3.3211 0.1260 4.3373
H 4.1981 2.0857 3.6593
H 1.6445 -1.2013 5.0287
H 4.1820 -0.5294 4.3666
C 2.9850 -4.6514 0.0085
C 3.2171 -4.2667 -1.3509
C 4.0050 -4.0607 0.8213
H 2.1667 -5.2627 0.3653
C 4.3879 -3.4328 -1.3869
H 2.6212 -4.5562 -2.2069
C 4.8790 -3.3053 -0.0355
H 4.1060 -4.1683 1.8934
H 4.8459 -3.0116 -2.2721
H 5.7704 -2.7727 0.2692
IM 1
Ru 0.0002
H -0.0007
H 0.0005
Ga -2.2727
Ga 0.0001
Ga 0.0009
Ga 2.2729
C 1.1592
C 0.0008
C 0.7106
H 2.1896
C -1.1635
H 0.0046
C -0.7246
H 1.3440
H -2.1915
H -1.3643
C -4.4004
0.0001 -0.5246
1.0810 -1.7527
-1.0811 -1.7526
-0.0013 -1.1421
1.8485 1.0327
-1.8478 1.0333
0.0006 -1.1428
3.7956 1.9823
4.2859 1.2900
3.0056 3.0942
4.0018 1.7227
3.7988 1.9745
4.9251 0.4164
3.0076 3.0894
2.5090 3.8179
4.0077 1.7078
2.5125 3.8086
1.1595 -1.4622
Arbeitsvorschriften und analytische Daten
C
C
H
C
H
C
H
H
H
C
C
C
H
C
H
C
H
H
H
C
C
C
H
C
H
C
H
H
H
-3.8848
-4.7174
-4.5352
-3.8842
-3.5778
-4.3994
-5.1231
-3.5766
-4.5332
-1.1582
0.0086
-0.7235
-2.1853
1.1644
0.0156
0.7118
-1.3657
2.1958
1.3426
3.8864
3.8868
4.4009
3.5798
4.4015
3.5806
4.7181
4.5345
4.5358
5.1229
0.7168
-0.0009
2.1892
-0.7232
1.3513
-1.1631
0.0002
-1.3596
-2.1922
-3.8000
-4.2849
-3.0073
-4.0114
-3.7916
-4.9244
-3.0021
-2.5134
-3.9955
-2.5037
-0.7204
0.7197
-1.1603
-1.3568
1.1623
1.3542
0.0018
-2.1894
2.1919
0.0029
-2.7293
-0.6810
-1.1580
-2.7274
-3.5503
-1.4592
0.3223
-3.5467
-1.1522
1.9740
1.2921
3.0894
1.7053
1.9866
0.4189
3.0972
3.8071
1.7291
3.8219
-2.7255
-2.7271
-1.4569
-3.5451
-1.4596
-3.5483
-0.6783
-1.1499
-1.1551
0.3253
IM 2
Ru 0.2445 -0.1897 -0.4926
H -0.1213 -0.2294 -2.0888
H 3.5693 1.4149 -1.7659
Ga -1.6735 -1.6540 -0.3568
Ga -0.8076 1.8761 -0.7220
Ga 0.6064 0.0108 1.9044
Ga 2.3100 0.6253 -1.2443
C -0.7375 4.1484 -1.4249
C -1.7869 3.4854 -2.1551
C -1.1242 4.1975 -0.0415
H 0.1688 4.5606 -1.8485
C -2.8201 3.1313 -1.2190
H -1.8087 3.3147 -3.2234
C -2.4094 3.5710 0.0836
H -0.5509 4.6428 0.7610
H -3.7493 2.6316 -1.4594
H -2.9726 3.4535 1.0001
C -4.0793 -1.8644 -0.6148
C -3.4678 -2.7446 -1.5724
C -3.7860 -2.3664 0.6993
H -4.6799 -0.9940 -0.8450
C -2.7970 -3.7883 -0.8482
H -3.5222 -2.6508 -2.6492
C -2.9952 -3.5542 0.5536
H -4.1191 -1.9340 1.6340
H -2.2512 -4.6145 -1.2852
H -2.6209 -4.1729 1.3588
C 0.2241 -0.7104 4.2110
C 1.6486 -0.6946 4.0373
C -0.2542 0.6380 4.0782
H -0.3817 -1.5820 4.4225
C 2.0531 0.6617 3.8009
H 2.3044 -1.5540 4.0906
C 0.8792 1.4866 3.8259
H -1.2814 0.9624 4.1833
H 3.0676 1.0045 3.6442
H 0.8545 2.5616 3.7022
C 4.6632 -1.6927 -1.2524
C 4.1548 -1.7499 -2.5978
C 3.5826 -1.5908 -0.3646
H 5.7108 -1.7097 -0.9742
C 2.7549 -1.6848 -2.5564
H 4.7586 -1.8160 -3.4958
C 2.3400 -1.5720 -1.1510
H 3.6273 -1.5796 0.7182
H 2.0712 -1.7493 -3.3945
H 1.4143 -2.0573 -0.7839
IM 3
C 4.7253 -0.5800 0.1295
C 4.5207 -0.3495 -1.2706
C 4.0512 -1.5723 -1.8604
C 3.9681 -2.5632 -0.8223
C 4.3826 -1.9481 0.4104
Ga 2.3303 -0.9139 -0.2792
Ru 0.0205 -0.4602 0.0691
Ga -2.2690 -1.0026 -0.2808
Ga -0.0409 1.9627 -0.0300
Ga 0.0214 -0.5932 2.3611
C -3.8254 -2.5020 -1.3091
C -4.1002 -2.4781 0.1026
C -4.5914 -1.1668 0.4352
C -4.6191 -0.3853 -0.7669
C -4.1449 -1.2063 -1.8439
C 0.3684 3.9606 -1.3949
C 1.0752 4.1389 -0.1579
C 0.1061 4.2228 0.8987
C -1.2000 4.0988 0.3135
C -1.0376 3.9369 -1.1031
H 0.0521 -2.0788 -0.2388
H 0.3209 4.3744 1.9486
H -2.1411 4.1383 0.8465
H 2.1486 4.2158 -0.0435
H 3.8153 -1.7264 -2.9052
H 3.6669 -3.5948 -0.9488
H 4.6910 0.5824 -1.7936
H -1.8347 3.8304 -1.8275
H 0.8150 3.8774 -2.3772
H -3.9865 -3.3085 0.7870
H -4.8986 -0.8354 1.4184
H -3.4616 -3.3517 -1.8717
H -4.9401 0.6449 -0.8476
H -4.0527 -0.9078 -2.8800
H 4.4511 -2.4376 1.3730
H 5.0820 0.1452 0.8492
IM 4
u -0.0007 -0.5174 -0.4982
H -0.0010 -2.1563 -0.4981
H -0.0005 -0.6439 1.0794
Ga 0.0013 1.9102 -0.2018
Ga -2.2935 -1.0121 -0.1256
C 1.1629 3.9080 0.6325
C -0.0058 3.7016 1.4431
C 0.7295 4.2444 -0.6928
H 2.1894 3.8416 0.9692
H -0.0144 3.4606 2.4983
H 1.3724 4.4666 -1.5347
C -3.9012 -2.7450 0.2141
C -4.4965 -1.8945 -0.7793
C -3.7842 -1.9880 1.4326
H -3.6102 -3.7782 0.0780
C -4.7518 -0.6173 -0.1773
H -4.7208 -2.1735 -1.8005
C -4.3123 -0.6714 1.1865
H -3.4007 -2.3552 2.3754
H -5.1951 0.2391 -0.6684
H -4.3798 0.1293 1.9111
Ga 2.2917 -1.0146 -0.1262
C -1.1609 3.9155 0.6151
C -0.7054 4.2490 -0.7035
H -2.1928 3.8556 0.9363
H -1.3342 4.4754 -1.5550
C 3.8972 -2.7491 0.2140
C 4.4938 -1.8990 -0.7790
C 3.7808 -1.9924 1.4326
150
6. Experimentelles
H 3.6052 -3.7820 0.0775
C 4.7504 -0.6223 -0.1766
H 4.7182 -2.1781 -1.8002
C 4.3104 -0.6763 1.1870
H 3.3964 -2.3593 2.3752
H 5.1948 0.2337 -0.6673
H 4.3785 0.1242 1.9119
IM 5
Ru -0.0275 -0.1488 -0.4374
H -0.0296 0.5416 -1.9337
Ga -0.0217 -1.2151 1.7278
Ga 2.2797 -0.1476 -0.9642
Ga 0.0681 2.1181 0.5090
Ga -0.1099 -2.3432 -1.5632
C 1.1677 -2.6035 3.3737
C 0.0135 -3.3634 2.9906
C 0.7160 -1.3982 4.0125
H 2.1995 -2.8932 3.2227
H 0.0217 -4.3228 2.4898
H 1.3450 -0.6281 4.4399
C 3.9293 0.0459 -2.6965
C 4.0836 -1.3090 -2.2516
C 4.2611 0.9157 -1.5960
H 3.6388 0.3632 -3.6895
C 4.5063 -1.2849 -0.8830
H 3.9031 -2.1953 -2.8458
C 4.6194 0.0852 -0.4726
H 4.2908 1.9970 -1.6246
H 4.7052 -2.1505 -0.2646
H 4.9431 0.4376 0.4982
C -0.9840 4.3093 1.1991
C -0.1736 4.5706 0.0438
C -0.1129 3.9423 2.2780
H -2.0615 4.4024 1.2543
H -0.5309 4.8950 -0.9256
H -0.4183 3.7027 3.2889
Ga -2.3459 -0.0074 -0.8960
C -1.1537 -2.6316 3.3886
C -0.7229 -1.4153 4.0215
H -2.1801 -2.9466 3.2518
H -1.3647 -0.6613 4.4582
C -4.0246 0.2928 -2.5877
C -4.2411 -1.0578 -2.1567
C -4.2815 1.1636 -1.4680
H -3.7406 0.6081 -3.5832
C -4.6287 -1.0308 -0.7780
H -4.1230 -1.9440 -2.7665
C -4.6568 0.3372 -0.3466
H -4.2541 2.2452 -1.4825
H -4.8589 -1.8933 -0.1661
H -4.9373 0.6925 0.6364
C 1.1984 4.3655 0.4106
C 1.2350 3.9771 1.7912
H 2.0563 4.5094 -0.2343
H 2.1260 3.7695 2.3706
Arbeitsvorschriften und analytische Daten
C -4.5261
C -2.9334
H -5.1207
C -3.2371
H -5.4749
C -2.2510
H -2.4685
H -3.0434
H -1.1843
C -4.2856
C -3.3671
C -3.5199
H -5.3652
C -2.0326
H -3.6334
C -2.1280
H -3.9218
H -1.1175
H -1.2956
C -0.7914
C -1.5895
C -1.6342
H 0.2578
C -2.9254
H -1.2425
C -2.9501
H -1.3243
H -3.7586
H -3.8097
C 1.8464
C 2.6242
C 2.7026
H 0.7981
C 3.9637
H 2.2693
C 4.0119
H 2.4167
H 4.7961
H 4.8868
C 5.6023
C 4.5572
C 6.0587
H 5.9863
C 4.3697
H 4.0243
C 5.2985
H 6.8410
H 3.6682
H 5.4115
C 3.4348
C 2.1481
C 3.2430
H 4.3836
C 1.1647
H 1.9575
C 1.8418
H 4.0219
H 0.1015
H 1.3821
IM 7
0.0604
-1.6301
-2.1036
0.6721
0.5787
-0.3728
-2.6072
1.7325
-0.2387
3.9448
3.6129
4.3412
3.9139
3.7995
3.2906
4.2493
4.6591
3.6501
4.4967
-4.3049
-4.1671
-4.8298
-4.0593
-4.6024
-3.8144
-5.0139
-5.0517
-4.6348
-5.4010
0.1438
-1.0486
1.2711
0.1848
-0.6585
-2.0645
0.7766
2.3128
-1.3272
1.3786
-2.7219
-3.3216
-1.5507
-3.0982
-2.5167
-4.2329
-1.4227
-0.8808
-2.7141
-0.6451
3.8385
3.7606
4.3530
3.5780
4.2286
3.4328
4.5936
4.5371
4.3071
4.9940
-3.9107
-3.9808
-3.7537
-4.0714
-3.8599
-4.1124
-3.9911
-4.1668
-4.2359
1.6778
2.7329
0.5296
1.7431
2.2341
3.7308
0.8696
-0.4234
2.7917
0.2247
1.3856
2.5747
0.3457
1.2895
2.2658
3.5368
0.8878
-0.6669
2.9552
0.3568
4.4539
4.2762
4.2218
4.7217
3.9356
4.3944
3.9017
4.2911
3.7581
3.6923
-1.1601
-1.9436
-1.8509
-0.2208
-3.1205
-1.7047
-3.0603
-1.5181
-3.9208
-3.8047
-2.1245
-2.7598
-0.7983
-2.5756
-1.8244
-3.7736
-0.6135
-0.0695
-2.0138
0.2815
IM 6
Ru -2.8535 -0.3314 0.5213
H -2.6453 -0.3092 2.1534
Ga -0.5338 -0.1012 0.2053
Ga -2.4362 -2.6345 0.9824
Ga -3.2169 -0.4093 -1.8922
Ru 1.7834 -0.1431 -0.2117
H 1.3828 -1.7091 0.0776
H 1.1144 -0.4745 -1.6730
Ga -2.9769 2.0054 0.9963
Ga 3.6547 -1.2081 -1.2182
Ga 2.5403 0.0487 2.0671
Ga 2.1509 2.1206 -0.9436
C -4.3381 -1.3629 -3.8549
C
C
C
C
C
Ga
Ru
Ga
Ga
Ru
Ga
Ga
C
C
6.2555 -1.7143 -0.0345
5.6903 -2.0333 1.2449
4.7961 -3.1472 1.0702
4.8126 -3.5119 -0.3209
5.7136 -2.6252 -1.0006
3.7905 -1.3220 -0.1067
1.8646 -0.0088 -0.5455
2.4865 1.8878 0.8616
0.6832 -1.0360 1.2310
-1.3505 0.0337 0.1340
-3.1839 1.3600 -0.5625
-0.0721 1.0341 -1.6248
0.6522 1.7404 -3.4650
1.8949 0.9495 -3.2250
C 2.8728 1.8216 -2.7135
C 2.3297 3.1658 -2.6909
C 1.0236 3.1351 -3.1689
Ga -2.3868 -0.6945 2.1360
Ga -1.8104 -1.9622 -1.1691
C -2.6528 -0.4726 4.5056
C -2.4126 -1.8635 4.2318
C -3.5294 -2.3656 3.4798
C -4.4623 -1.2889 3.2956
C -3.9217 -0.1194 3.9277
C -5.4120 2.1963 -0.2088
C -4.5317 3.3342 -0.2403
C -4.0060 3.4488 -1.5721
C -4.5564 2.3847 -2.3608
C -5.4240 1.6099 -1.5211
C -1.0608 -3.8167 -2.4997
C -1.9796 -3.0873 -3.3303
C -3.2888 -3.2037 -2.7528
C -3.1824 -3.9999 -1.5664
C -1.8063 -4.3795 -1.4065
C 2.3454 4.2232 1.5672
C 1.8204 3.4264 2.6406
C 2.8984 2.6475 3.1818
C 4.0883 2.9639 2.4457
C 3.7483 3.9362 1.4475
H 1.4454 -1.3588 -1.3703
H -0.9470 1.3270 1.0406
H 3.8879 1.5452 -2.4564
H 2.0439 -0.0837 -3.5126
H 2.8574 4.0441 -2.3367
H -0.0353 1.4888 -4.2719
H 0.3606 3.9861 -3.2821
H 0.8522 -1.9716 2.5347
H -5.9876 1.8608 0.6441
H -4.3253 4.0023 0.5857
H -6.0014 0.7463 -1.8250
H -3.3245 4.2139 -1.9203
H -4.3588 2.2024 -3.4091
H -1.5455 -2.4313 4.5414
H -2.0053 0.1854 5.0702
H -3.6535 -3.3824 3.1307
H -4.3911 0.8545 3.9760
H -5.4102 -1.3514 2.7768
H -1.7301 -2.5642 -4.2445
H -0.0001 -3.9375 -2.6768
H -4.1986 -2.7720 -3.1497
H -1.4038 -5.0035 -0.6190
H -3.9979 -4.2773 -0.9110
H 1.7863 4.9306 0.9685
H 0.7949 3.4242 2.9873
H 4.4321 4.3892 0.7409
H 2.8285 1.9541 4.0102
H 5.0730 2.5493 2.6200
H 4.2362 -3.6425 1.8528
H 4.2573 -4.3245 -0.7711
H 5.9123 -1.5398 2.1821
H 5.9469 -2.6450 -2.0573
H 6.9693 -0.9257 -0.2349
IM 8
Ru
H
Ga
Ru
H
H
Ga
Ga
Ga
Ga
Ga
Ga
C
C
-1.6744 -0.2781 -0.2356
-1.3996 -0.0314 -1.8278
0.0006 -1.9083 -0.5581
1.6730 -0.3499 0.0269
0.0008 0.4452 0.0722
1.3750 -0.9777 1.5067
2.2478 1.5392 1.3743
2.1133 0.5851 -2.1834
3.5867 -1.7440 0.1659
-2.0833 -0.6467 2.1398
-3.5917 -1.4000 -1.0641
-2.2586 2.0366 -0.3613
3.9621 2.7988 2.6701
3.3277 3.7565 1.8124
151
6. Experimentelles
C
H
C
H
C
H
H
H
C
C
C
H
C
H
C
H
H
H
C
C
C
H
C
H
C
H
H
H
C
C
C
H
C
H
C
H
H
H
C
C
C
H
C
H
C
H
H
H
C
C
C
H
C
H
C
H
H
H
2.9728
5.0074
1.9439
3.8090
1.7217
3.1406
1.2014
0.7863
4.6405
5.5346
4.5356
4.1419
5.9846
5.8291
5.3666
3.9480
6.6795
5.5165
1.3380
1.8535
2.4369
0.3065
3.2691
1.2789
3.6287
2.3775
3.9480
4.6268
-1.2967
-1.8094
-2.3993
-0.2643
-3.2274
-1.2316
-3.5906
-2.3423
-3.9053
-4.5904
-2.2691
-3.6407
-1.5489
-1.8562
-3.7683
-4.4397
-2.4779
-0.4973
-4.6807
-2.2473
-4.5749
-5.2223
-4.9416
-3.9447
-5.9872
-5.1537
-5.8155
-4.6347
-6.5908
-6.2715
2.2859
2.5192
3.8370
4.3240
2.9269
1.5597
4.4870
2.7776
-3.8989
-2.8811
-3.7672
-4.6424
-2.1203
-2.7245
-2.6675
-4.3961
-1.2910
-2.3259
1.1655
2.3730
0.2589
0.9787
2.2109
3.2580
0.9053
-0.7314
2.9502
0.4868
-1.3544
-0.0209
-2.2173
-1.6564
-0.0623
0.8551
-1.4183
-3.2858
0.7797
-1.7785
4.0201
3.7878
4.3531
3.9741
3.9794
3.5319
4.3289
4.6027
3.8849
4.5478
-3.2270
-3.2247
-2.0150
-4.0133
-2.0159
-4.0050
-1.2689
-1.7298
-1.7202
-0.3148
3.5749
2.6479
2.1817
1.0264
3.2729
4.3598
1.7371
3.7965
-0.3077
-0.7837
1.1184
-0.9158
0.3486
-1.8132
1.5248
1.7746
0.3228
2.5406
-4.4175
-3.8341
-4.5999
-4.6872
-3.6569
-3.5923
-4.1292
-5.0328
-3.2516
-4.1424
4.3311
4.4845
4.0118
4.4515
4.2609
4.7498
3.9682
3.8481
4.3178
3.7620
-1.7539
-1.3958
-0.5552
-2.7534
0.0206
-2.0795
0.5404
-0.4957
0.5952
1.5752
-2.2718
-0.9886
-2.9550
-2.6662
-0.8795
-0.2417
-2.0922
-3.9529
-0.0312
-2.3226
TS1
C -0.7690 -3.0248 3.1586
C -1.2186 -1.8093 3.7726
C -0.0625 -1.0404 4.1358
C 1.1049 -1.7848 3.7475
C 0.6661 -3.0118 3.1436
Ga -0.0275 -1.1517 1.6940
Ru -0.0547 -0.0248 -0.4601
Ga 0.0936 -1.8861 -1.9383
C -2.9035 -1.1639 -1.5447
C -2.7615 -2.5869 -1.1863
C -2.2002 -3.2561 -2.2675
C -2.1107 -2.3100 -3.4016
C -2.6291 -1.0862 -2.9894
Ga 2.2696 0.5147 -0.5276
Arbeitsvorschriften und analytische Daten
Ga -1.3339 1.8888 0.1501
C -3.5330 3.0683 0.2766
C -2.6848 3.7505 -0.6591
C -1.5569 4.2722 0.0630
C -1.7128 3.9096 1.4477
C -2.9348 3.1677 1.5756
C 3.9275 2.0747 -1.3700
C 4.3537 0.7700 -1.7955
C 4.7147 0.0186 -0.6268
C 4.5115 0.8554 0.5198
C 4.0255 2.1264 0.0627
H -0.0412 0.8561 -1.8447
H -1.7862 -0.5470 -0.9717
H 4.4146 0.4247 -2.8196
H 3.6135 2.8837 -2.0170
H 5.0862 -0.9980 -0.6152
H 3.7930 2.9811 0.6850
H 4.7028 0.5816 1.5494
H -0.7518 4.8631 -0.3540
H -2.8692 3.8685 -1.7190
H -1.0404 4.1756 2.2530
H -4.4682 2.5760 0.0430
H -3.3363 2.7594 2.4941
H -0.0667 -0.0810 4.6370
H -2.2493 -1.5259 3.9421
H 2.1327 -1.4870 3.9100
H -1.4007 -3.8205 2.7853
H 1.3060 -3.7966 2.7611
H -3.6477 -0.5294 -1.0689
H -2.6901 -0.1809 -3.5809
H -2.9514 -2.9970 -0.2019
H -1.8005 -2.5741 -4.4063
H -1.9584 -4.3116 -2.3200
TS2
Ru -0.2172 0.0668 -0.5218
H 0.1726 0.7131 -1.9759
H -2.5263 -1.3145 -2.3147
Ga 0.3553 2.2517 0.2921
Ga 1.9515 -0.7674 -0.7419
Ga -0.6835 -0.9751 1.6277
Ga -0.9704 -1.8055 -1.9322
C 3.3842 -2.4297 -1.7306
C 3.8067 -1.1247 -2.1618
C 3.5904 -2.5180 -0.3125
H 2.9848 -3.2091 -2.3658
C 4.2816 -0.4086 -1.0058
H 3.7970 -0.7555 -3.1788
C 4.1465 -1.2719 0.1362
H 3.3733 -3.3778 0.3074
H 4.6918 0.5925 -1.0038
H 4.4262 -1.0283 1.1526
C 1.9807 3.9565 0.9487
C 1.3749 4.3830 -0.2825
C 0.9636 3.9474 1.9636
H 3.0254 3.7132 1.0929
C -0.0158 4.6346 -0.0287
H 1.8804 4.5102 -1.2310
C -0.2696 4.3654 1.3594
H 1.1064 3.6893 3.0049
H -0.7427 4.9797 -0.7523
H -1.2217 4.4730 1.8624
C -1.7991 -1.0571 3.7988
C -2.1887 -2.2514 3.1027
C -0.3828 -1.1191 4.0277
H -2.4601 -0.2592 4.1113
C -1.0157 -3.0524 2.9025
H -3.1943 -2.5094 2.7970
C 0.1023 -2.3529 3.4705
H 0.2105 -0.3804 4.5508
H -0.9820 -4.0195 2.4178
H 1.1248 -2.7063 3.5040
C -4.9716 0.0770 -1.5096
C -4.4911 0.7956 -2.6924
C
H
C
H
C
H
H
H
-3.8897
-6.0128
-3.1143
-5.1244
-2.6469
-3.9080
-2.4513
-1.9262
-0.2831
-0.1400
0.8759
1.1886
0.1313
-0.8111
1.3550
0.7577
-0.7314
-1.2948
-2.6407
-3.4807
-1.4372
0.2151
-3.3512
-0.7834
TS(1T-2T) / TS(2T-1T)
Ru -2.0434 0.0256 -0.4651
H -1.9859 0.0647 -2.1037
Ga 0.4401 0.0748 -0.7562
Ru 2.6067 0.0406 0.3186
H 1.6605 0.2457 1.6361
H 1.1954 0.1480 -2.2258
Ga 2.7691 2.3523 0.8323
Ga 4.2886 -0.2962 -1.5030
Ga 2.4118 -2.1007 1.2825
Ga -1.8023 -2.2499 -1.0391
Ga -1.8487 0.0190 1.9421
Ga -1.9296 2.3327 -0.9325
Ga -4.4451 -0.0501 -0.5450
C -2.5044 -3.9403 -2.5906
C -1.1074 -3.6953 -2.8235
C -2.6424 -4.5071 -1.2767
H -3.3076 -3.7492 -3.2899
C -0.3828 -4.1090 -1.6549
H -0.6756 -3.2780 -3.7235
C -1.3312 -4.6086 -0.6974
H -3.5687 -4.8182 -0.8115
H 0.6892 -4.0514 -1.5222
H -1.0995 -5.0144 0.2784
C -6.5956 1.0609 -1.0416
C -6.4689 -0.0341 -1.9607
C -6.6987 0.5189 0.2841
H -6.6305 2.1105 -1.3035
C -6.4946 -1.2532 -1.2037
H -6.3866 0.0448 -3.0372
C -6.6363 -0.9118 0.1840
H -6.8238 1.0880 1.1963
H -6.4410 -2.2551 -1.6100
H -6.7059 -1.6105 1.0078
C -2.9832 4.1863 -2.0497
C -2.7002 4.6284 -0.7120
C -1.7308 3.9254 -2.7054
H -3.9649 4.0838 -2.4929
C -1.2744 4.6414 -0.5410
H -3.4323 4.9132 0.0322
C -0.6744 4.2084 -1.7735
H -1.6036 3.5893 -3.7259
H -0.7470 4.9413 0.3545
H 0.3849 4.1149 -1.9727
C 3.1166 3.8324 2.6930
C 4.2674 4.0541 1.8626
C 1.9567 4.2811 1.9699
H 3.1227 3.4225 3.6947
C 3.8240 4.6438 0.6349
H 5.2911 3.8196 2.1240
C 2.4000 4.7851 0.6955
H 0.9401 4.2831 2.3417
H 4.4543 4.9271 -0.1980
H 1.7723 5.2235 -0.0691
C 3.3631 -3.5409 3.0328
C 1.9864 -3.2440 3.3199
C 3.4054 -4.3604 1.8573
H 4.2158 -3.2114 3.6120
C 1.1791 -3.8802 2.3112
H 1.6231 -2.6740 4.1652
C 2.0622 -4.5723 1.4120
H 4.2997 -4.7464 1.3858
H 0.0980 -3.8716 2.2626
H 1.7623 -5.1633 0.5564
C -0.6504 0.1515 3.9845
152
6. Experimentelles
C
C
H
C
H
C
H
H
-1.6317
-1.3507
0.4217
-2.9272
-1.4237
-2.7540
-0.8955
-3.8706
1.2015
-1.1050
0.2834
0.5929
2.2637
-0.8269
-2.0868
1.1186
4.0560
4.0388
3.9219
4.1587
4.0642
4.1482
4.0335
4.2319
Arbeitsvorschriften und analytische Daten
H
C
C
C
H
C
H
C
-3.5435
5.2466
4.8831
6.3253
4.7978
5.7440
4.1163
6.6310
-1.5647
-1.6675
-0.3931
-1.4551
-2.6204
0.6029
-0.2200
-0.0536
4.2112
-3.2884
-3.8477
-2.3673
-3.5371
-3.2720
-4.5916
-2.3560
H
H
H
6.8277 -2.2203 -1.7895
5.7319 1.6609 -3.5005
7.4034 0.4236 -1.7666
[Ru(GaCp*)3(TMM)] (13). Eine Lösung von [Ru(η4-COD)(η3-CH2CMeCH2)2] (0.300 g,
0.939 mmol) in Toluol (6 mL) wurde bei Raumtemperatur mit GaCp* (0.616 g, 3.005 mmol)
versetzt und anschließend für 1 h bei 80 °C refluxiert. Das Lösemittel wurde im Vakuum
entfernt und der rote Rückstand mit wenig kaltem n-Hexan gewaschen. Der verbliebene gelbe
Feststoff wurde in n-Hexan gelöst und langsam auf -30 °C gekühlt, wobei 13 in Form von
hellgelben Prismen auskristallisiert. Ausbeute: 0.448 g gelbe Kristalle (62 %).
Elementaranalyse: ber. für C34H51Ga3Ru: C, 53.03; H, 6.68. gef.: C, 52.14; H, 6.54. 1H NMR
δH(C6D6) 1.89 (s, 45H, GaCp*), 1.76 (s, 6H, CH2). 13C{1H} NMR δC{H}(C6D6) 113.5 (C5Me5),
84.1(Czentral, TMM), 24.5 (CH2, TMM), 10.2 (C5Me5).
[(Cp*Ga)4Rh(η1-Cp*GaCH3)] (14). [(η4-COD)(py)Rh(CH3)] (0.300 g, 0.98 mmol) wurde in
n-Hexan (5 mL) gelöst und mit 6 mol Äquivalenten GaCp* (1.209 g, 5.90 mmol) versetzt.
Dabei trat ein sofortiger Farbwechsel von Orange nach Rot ein. Die Reaktionsmischung
wurde 30 min bei Raumtemperatur gerührt. Anschließend wurde das Lösemittel im Vakuum
entfernt und der Rückstand mit Isopentan extrahiert. Das Lösemittelvolumen wurde auf ca. 3
mL eingeengt. Die Lösung wurde für 16 h bei -40 °C gekühlt, woraufhin ein roter
mikrokristalliner Niederschlag entstand. Durch Rekristallisation in n-Hexan wurden geeignete
Kristalle für eine Einkristallstrukturanalyse erhalten. Ausbeute: 0.896 g (89 %).
Elementaranalyse: ber. für C51H78Ga5Rh: C, 53.61; H, 6.88. gef.: C, 53.29; H, 6.70. 1H NMR
(C6D6, 25°C): δ = 1.92 (75H, C5Me5), 0.34 (3H, GaMe) 1H NMR (Toluol-d8, -100°C): δ =
2.21 (15H, η1-C5Me5) 1.93 (60H, C5Me5), -0.05 (3H, GaMe).
13
C NMR (C6D6, 25°C): δ =
113.83 (C5Me5), 11.75 (C5Me5), 10.48 (GaMe).
[(Cp*Ga)4Rh(GaCH3)][BArF] (15). Zu einer Lösung von 14 (0.500 g, 0.44 mmol) in
Fluorbenzol (4 mL) wurde unter starken Rühren bei -30°C eine Lösung von [H(OEt2)2][BArF]
(0.443 g, 0.44 mmol) in Fluorbenzol (4 mL) hinzukanulliert. Nach langsamen Erwärmen auf
Raumtemperatur wurde die Reaktionsmischung für weitere 30 min gerührt. Das Lösemittel
153
6. Experimentelles
Arbeitsvorschriften und analytische Daten
wurde im Vakuum entfernt und der feste Rückstand mit wenig Hexan gewaschen (3 x 3 mL)
und anschließend im Vakuum getrocknet. Einkristalle wurden durch langsame Diffusion von
n-Hexan in eine Flurbenzollösung erhalten. Ausbeute: 0.638 g (78 %). Elementaranalyse: ber.
für C73H75BF24Ga5Rh: C, 46.87; H, 4.04. gef.: C, 46.37; H, 3.65. 1H NMR (Fluorbenzol/C6D6,
25°C): δ = 8.27 (8H, [BArF]), 7.56 (4H, [BArF]), 1.64 (60H, C5Me5), 0.54 (3H, GaMe)
F
13
C
F
NMR (Et2O/C6D6, 25°C): δ = 162.5 (q, J = 49.8 [BAr ]), 135.4 ([BAr ]), 129.8 (q, J = 31.6
Hz [BArF]), 125.2 (q, J = 272.2 Hz [BArF]), 117.8 ([BArF]), 114.7 (C5Me5), 15.4 (C5Me5), 9.8
(GaMe).
[(Cp*Ga)4Rh(Ga(CH3)(py))][BArF] (16). Zu einer Lösung von 15 (0.200 g, 0.11 mmol) in
Fluorbenzol (4 mL) wurde bei -30 °C Pyridin (10 μL, 0.128 mmol) hinzugegeben. Nachdem
die Lösung bei Raumtemperatur für 30 min gerührt wurde, wurden alle flüchtigen Substanzen
im Vakuum entfernt. Der hellorange Niederschlag wurde in Fluorbenzol (ca. 4 mL) gelöst
und das Produkt durch langsame Diffusion von n-Hexan in diese Lösung auskristallisiert.
Ausbeute: 0.175 g (84 %). Elementaranalyse: ber. für C73H75BF24Ga5Rh: C, 47.83; H, 3.96, N
0.72. gef.: C, 46.99; H, 4.04, N 0.98. 1H NMR (thf-d5, 25°C): δ = 8.80 (3H, Pyridin), 8.19
(2H, Pyridin), 7.80 (8H, [BArF]), 7.58 (4H, [BArF]), 1.86 (60H, C5Me5), 0.70 (3H, GaMe) 13C
NMR (thf-d5, 25°C): δ = 163.0 (q, J = 49.8 [BArF]), 149.3 (C5H5N), 135.8 ([BArF]), 130.2 (q,
J = 28.7 Hz [BArF]), 127.1 (C5H5N), 125.7 (q, J = 272.2 Hz [BArF]), 118.4 ([BArF]), 115.9
(C5Me5), 115.4 (C5H5N), 10.6 (C5Me5), 10.3 (GaMe).
[Ru(PCy3)2(GaCp*)2(H)2] (17). Eine Lösung von [Ru(PCy3)2(H2)2(H)2] (0.300 g, 0.449
mmol) in Hexan (6 mL) wurde bei Raumtemperatur langsam mit GaCp* (0.203 g, 0.988
mmol) versetzt. Die Reaktionsmischung wurde für weitere 30 min bei 60 °C gerührt, wobei
ein farbloser Niederschlag ausfiel. Nach Abkühlen der dunkelroten Lösung auf
Raumtemperatur wurden alle flüchtigen Bestandteile im Vakuum entfernt und der farblose
Rückstand mit wenig kaltem n-Hexan gewaschen. Das Produkt wurde in Pentan gelöst und
durch langsames Abkühlen auf -30°C kristallisiert. Ausbeute: 0.405 g (84 %) farblose
Kristalle. Elementaranalyse: ber. für C55H96Ga2P2Ru: C, 62.33; H, 9.13. gef.: C, 62.02; H,
8.74. 1H NMR δH(C6D6) 2.07 (s, 30H, GaCp*), 1.63 (s, 66H, PCy3), -12.74 (triplet, 2J(P-H) =
26.31 Hz, 2H)
13
C{1H} NMR δC{H}(C6D6) 113.7 (C5Me5), 39.3 (PCy3), 32.5 (PCy3), 31.0
(PCy3), 28.4 (PCy3), 27.6 (PCy3), 10.8 (C5Me5). 31P{1H} NMR δP(C6D6, 298 K, 101.25 MHz)
154
6. Experimentelles
Arbeitsvorschriften und analytische Daten
= 87.04 (s) 13C{1H} NMR δC{H}(C6D6) 113.5 (C5Me5), 42.6 (br, PCy3), 39.4 (br, PCy3), 32.4
(br, PCy3), 31.2 (br, PCy3), 29.4 (br, PCy3), 28.5 (br, PCy3), 27.6 (s, PCy3), 27.3 (s, PCy3),
27.1 (s, PCy3), 26.9 (s, PCy3), 26.5 (s, PCy3), 10.8 (C5Me5). ν = 2026 und 2002 cm-1 (vs, RuH)
[Ru(GaCp*)4(η3-(CH2)2C(CH3))][BArF] (18). Zu einer Mischung von frisch hergestelltem
[Ru(GaCp*)3(TMM)] (x) (0.300 g, 0.390 mmol) und [H(OEt2)2][BArF] (0.394 g, 0.390 mmol)
wurde
Fluorbenzol
bei
-30
°C
hinzugegeben.
Nach
langsamen
Erwärmen
auf
Raumtemperatur wurde die Reaktionsmischung für weitere 2h gerührt. Anschließend wurden
alle flüchtigen Komponenten im Vakuum entfernt und der hellorangene Rückstand mit Hexan
gewaschen (3 x 5 mL). Das Rohprodukt wurde durch langsame Diffusion von Hexan in eine
Fluorbenzollösung umkristallisiert. Ausbeute: 0.423 g orangene Kristalle (59 %).
Elementaranalyse: ber. für C76H79BF24Ga4Ru: C, 49.63; H, 4.33. gef.: C, 50.39 H, 3.98. 1H
NMR δH(d8-thf) 7.80 (s, 8 H, BArF), 7.58 (s, 4H, BArF), 2.52 (t, 3J(H-H) = 1.22 Hz, 2H, synCH2), 2.29 (t, 2H, 3J(H-H) = 1.22 Hz, anti-CH2), 2.11 (s, 3H, CH3), 2.01 (s, 60H, GaCp*),
13
C{1H} NMR δC{H}(d8-thf) 162.8 (q, J=49.8 Hz, [BArF]), 135.6 ([BArF]), 130.0 (q, J=34.4
Hz, [BArF]), 125.5 (q, J=272.2 Hz, [BArF]), 118.2 ([BArF]) 116.5 (C5Me5), 92.9 (C,
Methylallyl) 68.2 (CH2, Methylallyl), 19.5 (CH3, Metyhlallyl), 10.9 (C5Me5).
[{Ru(GaCp*)3(η3-(CH2)2C(CH2(μ-Ga)))}2][(BArF)2] (19). Zu einer Mischung von frisch
hergestelltem [Ru(GaCp*)3(TMM)] (13) (0.300 g, 0.390 mmol) und [Ga2Cp*][BArF] (0.443
g, 0.390 mmol) wurde Fluorbenzol bei Raumtemperatur hinzugegeben und für 30 min
gerührt. Anschließend wurden alle flüchtigen Komponenten im Vakuum entfernt und der
hellorangene Rückstand mit Hexan gewaschen (3 x 5 mL). Das Rohprodukt wurde durch
langsame Diffusion von Hexan in eine Fluorbenzollösung umkristallisiert. Ausbeute 0.531 g
dunkelrote Kristalle (80 %). Elementaranalyse: ber. für C76H79BF24Ga4Ru: C, 46.55; H, 3.73.
gef.: C, 46.31 H, 3.34. Das 1H NMR Spektrum von 19 in einer CD2Cl2 oder FluorbenzolLösung ist aufgrund wahrscheinlicher fluktionaler Prozesses unerwartet kompliziert. Dennoch
können die Signale des Hauptproduktes zu der entsprechenden Festkörperstruktur von 19
zugeordnet werden. Tieftemperaturspektren sowie ein
13
C Spektrum konnten aufgrund der
schlechten Löslichkeit der Verbindung nicht aufgenommen werden. 1H NMR δH(CD2Cl2)
155
6. Experimentelles
Arbeitsvorschriften und analytische Daten
7.73 (s, 8 H, BArF), 7.56 (s, 4H, BArF), 2.44 (br, 4H), 2.28 (br, 4H), 2.25 (4H), 2.04 (s, 30H),
1.94 (60H).
[{Ru(GaCp*)3(η3-(CH2)(CH(μ-Ga)(CH3))}2][(BArF)2] (20). Eine Lösung von frisch
hergestelltem 19 (0.300 g, 0.088 mmol) in Fluorbenzol (5 mL) wurde für 1 h refluxiert.
Anschießend wurden alle flüchtigen Bestandteile im Vakuum entfernt und der gelbe
Rückstand mit Hexan gewaschen (3 x 5mL). Das Rohprodukt wurde in Fluorbenzol gelöst
und durch langsame Diffusion von n-Hexan in diese Lösung kristallisiert. Ausbeute: 0.273 g
gelbe Kristalle (91 %). Elementaranalyse: ber. für C76H79BF24Ga4Ru: C, 46.55; H, 3.73. gef.:
C, 46.51 H, 3.52. 1H NMR δH(CD2Cl2) 7.73 (s, 16 H, BArF), 7.56 (s, 8H, BArF), 5.16 (d, 3J(HH)
3
= 3.04 Hz, ca. 2H, syn-CH2), 2.45 ppm (s, 6H, CH3), 1.98 (s, 90H, GaCp*), 1.68 (t, 2H,
J(H-H) = 3.22 Hz, anti-CH2), -2.09 (d, 3J(H-H) = 3.41 Hz, 2H).
13
C{1H} NMR δC{H}(CD2Cl2)
164.6 (q, J=49.8 Hz, [BArF]), 137.7 ([BArF]), 131.7 (q, J=31.5 Hz, [BArF]), 127.5 (q, J=272.4
Hz, [BArF]), 120.3 ([BArF]) 118.8 (br, C5Me5), 106.7 (Gallallyl), 104.8 (Gallallyl), 37.8
(Gallallyl), 23.4 (CH3, Gallallyl), 13.4 (C5Me5).
[Ru(PCy3)2(GaCp*)2(Ga)][BArF] (21). Zu einer Lösung von [Ru(PCy3)2(GaCp*)2(H)2}]
(17) (0.300 g, 0.283 mmol) in 4 mL Fluorbenzol wurde eine Lösung von [Ga2Cp*][BArF]
(0.322 g, 0.283 mmol) in Fluorbenzol (3 mL) bei Raumtemperatur hinzukanulliert, wobei eine
sofortige Gasentwicklung sowie ein Farbumschlag von gelb nach rot einsetzte. Die
Reaktionsmischung wurde für eine weitere Stunde bei Raumtemperatur gerührt. Das
Lösemittel wurde auf ca. 2 mL eingeengt und das Produkt duch Hinzugabe von n-Hexan
ausgefällt. Die überstehende Lösung wurde durch Filtration entfernt, der Rückstand mit
Hexan gewaschen (2 x 2 mL) und anschließend im Vakuum getrocknet. Das Rohprodukt
wurde durch langsame Diffusion von Hexan in eine Fluorbenzollösung umkristallisiert,
woraufhin nadelförmige Kristalle entstanden. Ausbeute: 0.440 g (78 %). Elementaranalyse:
ber. für C87H106BF24Ga3P2Ru: C, 52.49; H, 5.37. gef: C, 52.00; H, 6.08. 1H NMR
δH(C6H5F/C6D6, 10:1) 8.32 (s, 8 H, BArF), 7.67 (s, 4H, BArF), 1.97 (s, 30H, GaCp*), 1.36 (s,
66H, PCy3).
31
P NMR δP(C6H5F/C6D6, 10:1) = 66.9 (s, PCy3).
13
C{1H} NMR δC{H}(CD2Cl2
10:1) 162.2 (q, J=49.2 Hz, [BArF]), 135.2 ([BArF]), 130.4 (q, J=34.1 Hz, [BArF]), 125.9 (q,
J=272.2 Hz, [BArF]), 118.3 ([BArF]) 119.5 (C5Me5), 36.8 (s, PCy3), 33.9 (s, PCy3), 31.8 (s,
156
6. Experimentelles
Arbeitsvorschriften und analytische Daten
PCy3), 31.4 (s, PCy3), 30.6 (s, PCy3), 30.5 (s, PCy3), 29.3 (s, PCy3), 28.6 (s, PCy3), 28.4 (s,
PCy3), 27.1 (s, PCy3), 13.2 (C5Me5).
[Mo(GaCp*)6] (22). Frisch hergestelltes [Mo(η4-C4H6)3] (0.300 g, 1.162 mmol) wurde in
einer Fischer-Porter Druckflasche in Toluol (12 mL) gelöst. Nach der Zugabe von GaCp*
(1.666 g, 8.129 mmol) wurde die Reaktionsmischung mit 3 bar H2 versetzt. Die orangefarbene
Lösung wird auf 100 °C erwärmt, wobei sich ein roter mikrokristalliner Niederschlag bildete.
Nachdem die Mischung für weitere 16 h bei 80 °C gerührt worden war, wurde sie in ein
Schlenkrohr überführt. Die roten Kristalle wurden durch Kannulation isoliert, mit wenig nHexan gewaschen und im Vakuum getrocknet. Umkristallisation aus Mesitylen lieferte
wohlgeformte, rote, nadelförmige Einkristalle. Ausbeute: 0.785 g (51 %). Elementaranalyse
ber. für C60H90Ga6Mo: C, 54.36; H, 6.84; gef.: C, 53.86; H, 6.24.1H NMR (C6D6, 25°C): δ =
1.96 (90H, C5Me5); 13C NMR (C6D6, 25°C): δ = 117.11 (C5Me5), 11.91 (C5Me5);
[Mo(ZnCp*)3(ZnMe)9] (23). 22 (0.300 g, 0.226 mmol) wurde in Toluol gelöst (5 mL) und
bei Raumtemperatur mit 1.58 ml einer 2M ZnMe2 Lösung in Toluol versetzt (14 eq, 3.168
mmol). Die Mischung wurde auf 100 °C für 2h erwärmt, wobei sich eine gelbe Lösung
bildete. Nach dem Abkühlen auf Raumtemperatur wurde die Lösung filtriert und das
Lösemittel im Vakuum entfernt. Das Rohprodukt wurde aus Hexan umkristallisiert, wobei das
Produkt durch langsames Kühlen auf -30°C kristallisierte. Ausbeute: 0.263 g (82 %). 1H
NMR (C6D6, 25°C): δ = 2.14 (45H, C5Me5), 0.24 (9H, Me), 0.19 (9H, ZnMe), 0.03 (9H, Me);
13
C NMR (C6D6, 25°C): δ =113.71 (C5Me5), 23.82 (Me), 23.37 (Me), 22.19 (Me), 11.38
(C5Me5). Elementaranalyse: ber. für C39H72Zn12Mo: C, 32.95; H, 5.10; Zn, 55.20; Mo, 6.75;
gef.: C, 32.66; H, 5.64, Zn, 55.01; Mo, 6.44; es wurde kein Gallium detektiert.
[Mo(GaMe)4(ZnCp*)4] (24). Die Herstellung verlief analog zu 23 (s.o.) unter Verwendung
von 4 molaren Äquivalenten ZnMe2 anstelle von 14. Ausbeute: 0.237 g (85 %).
Elementaranalyse ber. für C44H72Ga4Zn4Mo: C, 42.71; H, 5.86; Ga, 22.54; Zn, 21.14; Mo,
7.75; gef.: C, 42.56; H, 5.82; Ga, 22.36; Zn, 21.25; Mo, 7.871H NMR (C6D6, 25°C): δ = 2.07
(60H, C5Me5), 0.39 (12H, Me);
13
C NMR (C6D6, 25°C): δ = 109.75 (C5Me5), 34.10 (Me),
11.06 (C5Me5);.
157
6. Experimentelles
Arbeitsvorschriften und analytische Daten
[Mo(GaMe)2(ZnCp*)4(ZnMe)4] (25). Eine frisch hergestellte Probe von 22 (0.300 g, 0.226
mmol) wurde in Benzol (5 mL) bei Raumtemperatur mit 0.67 ml einer 2M ZnMe2 Lösung in
Toluol (8 eq, 1.356 mmol) versetzt. Die Reaktionsmischung wurde für 30 min auf Rückfluss
erhitzt, wobei sich ein gelber, mikrokristalliner Niederschlag bildete. Der Niederschlag wurde
mittels Kannulation von der Lösung befreit, zweimal mit wenig Hexan gewaschen und im
Vakuum getrocknet. Das Produkt wurde aus heißem Benzol durch langsames Kühlen auf
Raumtemperatur kristallisiert. Ausbeute: 0.261 g (83 %). Elementaranalyse ber. für
C46H78Ga2Zn8Mo: C, 39.76; H, 5.66, Zn, 37.64; gef.: C, 39.99; H, 5.94, Zn 37.11. 1H NMR
(C6D6, 25°C): δ = 2.15 (30H, C5Me5), 2.07 (15H, C5Me5) 1.91 (15H, C5Me5), 0.57 (6H, Me),
0.14 (3H, Me), 0.02 (6H, Me), -0.14 (3H, Me).
[Mo(ZnCp*)2(ZnEt10)] (26). Eine Suspension von 22 (0.300 g, 0.226 mmol) in Toluol (5
mL) wurde bei Raumtemperatur mit 3.16 ml einer 1M ZnEt2 Lösung in Hexan (14eq, 3.168
mmol) versetzt. Die Reaktionsmischung wurde bei 80°C für 30 min gerührt, wobei sich eine
gelbe Lösung bildete. Nach Abkühken auf Raumtemperatur wurde die Lösung filtriert, das
Lösemittel im Vakuum entfernt und der gelbe Rückstand im Vakuum getrocknet. Das
Rohprodukt wurde in Hexan gelöst und langsam auf -30°C abgekühlt. Ausbeute: 0.263 g (82
%). Elementaranalyse ber. für C40H80Zn12Mo: C, 33.32; H, 5.59; Zn, 54.43; gef.: C, 33.11; H,
5.24, Zn, 54.28; Das 1H NMR Spektrum von 26 in C6D6 enthält sowohl bei Raumtemperatur
als auch bei 80 °C Signale verschiedener Isomere, die aufgrund der Kompliziertheit des
Spektrums nicht zugeordnet werden konnten. 1H NMR (C6D6, 25°C): δ = 2.21 (s, 45H,
C5Me5), 2.19 (s, 45H, C5Me5), 2.15 (s, 45H, C5Me5), 2.12 (s, 45H, C5Me5), 1.65 (m), 1.52
(m), 1.27 (m), 1.03 (m), 0.89 (m), 0.72 (m), 0.51 (m); 13C NMR (C6D6, 25°C): δ =114.30 (s),
114.08 (s), 113.91 (s), 111.98 (s), 41.06 (s), 38.43 (s), 38.35 (s), 37.50 (s), 36.49 (s), 36.38 (s),
34.97 (s), 34.60 (s), 34.33 (s), 34.11 (s), 31.95 (s), 29.55 (s), 29.41 (s), 27.22 (s), 25.62 (s),
23.04 (s), 22.78 (s), 20.83 (s), 18.89 (s), 14.52 (s), 14.34 (s), 11.64 (s), 11.40 (s), 11.12 (s),
10.89 (s), 10.85 (s), 10.54 (s), 10.43 (s), 10.40 (s), 9.58 (s)
[{Mo(CO)4}4(Zn)6(μ-ZnCp*)4] (27). Eine Lösung von [Mo(CO)4(GaCp*)2] (0.500 g, 0.809
mmol) in 4 mL Toluol wurde mit 1.70 ml einer 2M ZnMe2 Lösung in Toluol bei
Raumtemperatur versetzt (4.2 äq., 3.4 mmol). Die Reaktionsmischung wurde auf 100 °C für 1
h erwärmt, wobei sich eine dunkelrote Lösung bildete. Nach dem Abkühlen auf
Raumtemperatur wurde die Lösung filtriert und das Lösemittel im Vakuum entfernt. Das
158
6. Experimentelles
Arbeitsvorschriften und analytische Daten
Rohprodukt wurde zweimal mit wenig kaltem n-Hexan gewaschen (2 x 3 mL) und in
Fluorbenzol gelöst. Umkristallisieren durch langsame Diffusion von n-Hexan in diese
Fluorbenzollösung
lieferte
wohlgeformte
Nadeln.
Ausbeute:
0.283
g
(69
%).
Elementaranalyse ber. für C56H60O16Mo4Zn10: C, 33.19; H, 2.98; Zn, 32.26. Gefunden: C,
32.89; H, 3.11; Zn, 33.11. 1H NMR δH(C6D6) 2.10 (s, ZnCp*,
13
C{1H} NMR δC{H}(C6D6)
111.2 (C5Me5), , 10.0 (C5Me5). ν = 2001, 1970, 1931 und 1908 cm-1 (vs, CO).
[Ru(ZnCp*)4(ZnMe)6] (28).
1)
Eine frisch hergestellte Probe von 12 (0.300 g, 0.175 mmol) wurde in Toluol (5 mL)
bei Raumtemperatur mit 1.93 ml einer 2M ZnMe2 Lösung in Toluol (22 äq, 3.861 mmol)
versetzt. Die Reaktionsmischung wurde für 45 min auf Rückfluss erhitzt, wobei sich ein
gelber, mikrokristalliner Niederschlag bildete. Der Niederschlag wurde mittels Kannulation
von der Lösung befreit, zweimal mit wenig Hexan gewaschen und im Vakuum getrocknet.
Das Produkt wurde aus heißem Toluol durch langsames Kühlen auf 30 °C kristallisiert.
Ausbeute: 0.349 g (72 %). Elementaranalyse ber. für C46H78Zn10Ru: C, 39.86; H, 5.67, Zn,
47.18; gef.: C, 39.47; H, 5.61, Zn 47.67. 1H NMR (C6D6, 25°C): δ = 2.07 (s, 60H, C5Me5), 0.19 (s, 3H, ZnMe); 13C NMR (C6D6, 25°C): δ = 110.56 (s, C5Me5), 14.56 (s, ZnMe), 10.64
(s, C5Me5).
2)
Eine frisch hergestellte Probe von [Ru(GaCp*)6(Cl)2] (0.250 g, 0.178 mmol) wurde in
6 mL Toluol gelöst und anschließend bei Raumtemperatur mit 1.78 ml einer 2M ZnMe2
Lösung in Toluol (20 äq, 3.567 mmol) versetzt. Die Reaktionsmischung wurde für 45 min auf
Rückfluss erhitzt, wobei sich ein gelber, mikrokristalliner Niederschlag bildet. Der
Niederschlag wurde mittels Kannulation von der Lösung befreit, zweimal mit wenig Hexan
gewaschen und im Vakuum getrocknet. Das Produkt wurde aus heißem Toluol durch
langsames Kühlen auf 30 °C kristallisiert. Ausbeute:0.204 g (59 %).
[Ru(ZnCp*)4(ZnMe)4(H)2] (29). Die Herstellung verlief analog zu 28 (s.o., Syntheseweg 1)
unter Verwendung von 16 molaren Äquivalenten ZnMe2 anstelle von 22. Ausbeute: 0.367 g
(84 %). Elementaranalyse ber. für C44H74Zn8Ru: C, 43.06; H, 6.08; Zn, 42.63; gef.: C, 43.41;
H, 6.11, Zn, 42.391H NMR (C6D6, 25°C): δ = 2.00 (s, 60H, C5Me5), -0.11 (s, 12H, ZnMe), 15.89 (s, 2H, Ru-H); 13C NMR (C6D6, 25°C): δ =110.80 (s, C5Me5), 13.41 (s, ZnMe), 10.88
(s, C5Me5). IR ν = 1923, 1901 cm-1 (vs, H)
159
6. Experimentelles
Arbeitsvorschriften und analytische Daten
[Rh(ZnCp*)3(ZnMe)6] (30). Eine frisch hergestellte Probe von 14 (0.300 g, 0.253 mmol)
wurde in 6 mL Toluol gelöst und mit 1.45 ml (11 mol äq.) einer 2M ZnMe2 Lösung in Toluol
versetzt. Die Reaktionsmischung wurde für 1 h bei 80 °C gerührt. Nach dem Abkühlen auf
Raumtemperatur wurde die Lösung filtriert und das Lösemittel im Vakuum entfernt. Das
Rohprodukt wurde aus Hexan umkristallisiert, wobei das Produkt durch langsames Kühlen
auf -30°C kristallisierte. Ausbeute: 0.250 g gelbe Kristalle (80 %). Elementaranalyse ber. für
C36H63Zn9Rh: C, 36.42; H, 5.35; Zn, 49.57; gef.: C, 36.53; H, 5.48, Zn, 37.48 1H NMR (C6D6,
25°C): δ = 2.07 (s, 45H, C5Me5), -0.04 (s, 18H, ZnMe); 13C NMR (C6D6, 25°C): δ =111.29 (s,
C5Me5), 23.82 (d, ZnMe, 2J(Rh-C) = 5.01 Hz,), 10.78 (s, C5Me5).
[Rh(GaMe)(ZnCp*)4(ZnMe)3] (31). Die Herstellung verlief analog zu 30 (s.o.) unter
Verwendung von 7 molaren Äquivalenten ZnMe2 anstelle von 11. Ausbeute 0.223 g gelbe
Kristalle (74 %). Elementaranalyse ber. für C44H72GaZn7Rh: C, 42.92; H, 5.98; Zn, 37.17;
gef.: C, 42.17; H, 5.48, Zn, 37.48 In Lösung liegen zwei Isomere vor, deren Signale sich im
1
H NMR Spektrum wie folgt unterscheiden lassen. Isomer 1 (Hauptisomer): 1H NMR (C6D6,
25°C): δ = 2.15 (s, 15H, C5Me5), 2.14 (s, 15H, C5Me5), 2.06 (s, 30H, C5Me5), 0.42 (s, 3H,
GaMe), -0.11 (s, 9H, ZnMe). Isomer 2: 1H NMR (C6D6, 25°C): δ = 2.06 (s, 60H, C5Me5), 0.06 (s, 3H, GaMe), -0.17 (s, 9H, ZnMe). Aufgrund des komplizierten 13C Spektrums können
die Signale den einzelnen Isomeren nicht zugeordnet werden.
13
C NMR (C6D6, 25°C): δ
=110.17 (s, C5Me5), 110.11 (s, C5Me5), 32.08 (d, Ga/ZnMe, 2J(Rh-C) = 7.62 Hz), 31.98 (d,
Ga/ZnMe, 2J(Rh-C) = 4.32 Hz), 17.84 (d, Ga/ZnMe, 2J(Rh-C) = 4.59 Hz), 17.37 (d,
Ga/ZnMe, 2J(Rh-C) = 5.77 Hz), 11.09 (s, C5Me5), 10.99 (s, C5Me5), 10.94 (C5Me5)
[Ni(ZnCp*)4(ZnMe)4] (32): Eine Lösung von [Ni(GaCp*)4] (0.511 g, 0.582 mmol) in Toluol
(8 mL) wurde bei Raumtemperatur mit 2.47 ml einer 2M ZnMe2 Lösung versetzt (10 mol äq.,
4.944 mmol). Die Reaktionsmischung wurde bei 80 °C für 1h gerührt, wobei sich eine gelbe
Lösung und wenig schwarzer Niederschlag bildete. Nach Abkühlen auf Raumtemperatur
wurde die Lösung filtriert, das Lösemittel im Vakuum entfernt und der gelbe Rückstand im
Vakuum getrocknet. Das Rohprodukt wurde in wenig Toluol gelöst und langsam auf -30°C
abgekühlt. Ausbeute: 0.434 g (63 %). Elementaranalyse: ber. für C44H72Zn8Ni: C, 44.68; H,
6.14; Zn, 44.23; gef.: C, 44.13; H, 6.19, Zn, 44.19 1H NMR (C6D6, 25°C): δ = 2.07 (s, 60H,
C5Me5), 0.00 (s, 12H, ZnMe);
13
C NMR (C6D6, 25°C): δ =110.91 (s, C5Me5), 13.21 (s,
ZnMe), 10.96 (s, C5Me5).
160
6. Experimentelles
Arbeitsvorschriften und analytische Daten
[Pd(ZnCp*)4(ZnMe)4] (33). Die Herstellung verlief analog zu 32 unter Verwendung von
[Pd(GaCp*)4]
anstelle
von
[Ni(GaCp*)4].
Ausbeute:
79
%
farbloser
Kristalle.
Elementaranalyse: ber. für C44H72PdZn8: C, 42.94; H, 5.90; Zn, 42.51; gef.: C, 43.28; H, 5.81,
Zn, 42.11 1H NMR (C6D6, 25°C): δ = 2.08 (60H, C5Me5), -0.04 (12H, ZnMe);
13
C NMR
(C6D6, 25°C): δ =110.26 (C5Me5), 15.71 (ZnMe), 10.86 (C5Me5).
[Pt(ZnCp*)4(ZnMe)4] (34). Die Herstellung verlief analog zu 32 unter Verwendung von
[Pt(GaCp*)4]
anstelle
von
[Ni(GaCp*)4].
Ausbeute:
79
%
farbloser
Kristalle.
Elementaranalyse: ber. für C44H72Zn8Pt: C, 40.06; H, 5.50; Zn, 39.65; gef.: C, 40.89; H, 5.71,
Zn, 39.00 1H NMR (C6D6, 25°C): δ = 2.06 (s, 60H, C5Me5), 0.18 (s, 12H, ZnMe); 13C NMR
(C6D6, 25°C): δ =109.92 (s, C5Me5), 22.03 (tr, ZnMe, 2J(Pt-C) = 74.06 Hz), 10.82 (s, C5Me5).
[Pt(ZnCp*)4(ZnEt)4] (35). Die Herstellung verlief analog zu 34 unter Verwendung von
ZnEt2 anstelle von ZnMe2. Ausbeute: 85 % gelber Kristalle. Elementaranalyse: ber. für
C48H80Zn8Pt: C, 41.92; H, 5.86; Zn, 38.04; gef.: C, 41.44; H, 5.39, Zn, 38.62 1H NMR (C6D6,
25°C): δ = 2.10 (s, 60H, C5Me5), 1.57 (m, 6H, Zn-CH2CH3), 1.46 (m, 6H, Zn-CH2CH3), 0.41
(m, 8H, Zn-CH2CH3). Das
13
C NMR Spektrum zeigt neben den Signalen der inneren Cp*-
Ringatone drei Sätze von Doublets, was auf die Existenz zweier Isomere hindeutet. 13C NMR
(C6D6, 25°C): δ =110.34 (s, C5Me5), 33.95 (s), 32.83 (s), 11.12 (s), 10.78 (s), 10.24 (s), 9.33
(s).
[Mo(CO)3(ZnCp*)3(ZnMe)3] (36). Eine Lösung von [fac-Mo(CO)3(GaCp*)3] (0.300 g,
0.377 mmol) in 6 mL Toluol wurde mit 1.51 ml einer 2M ZnMe2 Lösung in Toluol bei
Raumtemperatur versetzt (8 Äq., 3.0 mmol). Die Reaktionsmischung wurde auf 100 °C für 1
h erwärmt. Nach dem Abkühlen auf Raumtemperatur wurde die Lösung filtriert und das
Lösemittel im Vakuum entfernt. Das Rohprodukt wurde zweimal mit wenig kaltem n-Hexan
gewaschen (2 x 3 mL), in wenig n-Hexan gelöst und langsam auf -30°C abgekühlt. Ausbeute:
Ausbeute: 0.313 g (81 %). Elementaranalyse ber. für C36H54O3MoZn6: C, 42.26; H, 5.32; Zn,
38.35. Gefunden: C, 42.34; H, 5.11; Zn, 38.00. 1H NMR δH(C6D6) 2.08 (s, ZnCp*, 45H), 0.02 (s, ZnMe, 9H).
13
C{1H} NMR δC{H}(C6D6) 214.6 (CO), 110.7 (s, C5Me5), 17.8 (s,
ZnMe), 10.2 (s, C5Me5). ν = 1967, 1923 und 1915 cm-1 (vs, CO)
161
6. Experimentelles
Arbeitsvorschriften und analytische Daten
[Ru(TMM)3(ZnCp*)3(ZnMe)3] (37), [Ru(TMM)3(ZnCp*)2(ZnMe)4] (38). Eine Lösung
von 13 (0.500 g, 0.649 mmol) in Toluol (6 mL) wurde bei Raumtemperatur mit 2.60 mL (8
Äq., 5.195 mmol) einer 2M Toluollösung von ZnMe2 versetzt und anschließend für 1 h bei
100 °C gerührt. Das Lösemittel wurde im Vakuum entfernt und der hellgelbe Rückstand mit
wenig kaltem n-Hexan gewaschen. Der verbliebene gelbe Feststoff wurde in n-Hexan gelöst
und langsam auf -30 °C gekühlt, wobei ein Isomerengemisch von 37 und 38 in Form von
hellgelben Prismen auskristallisiert. Gesamtausbeute: 0.460 g gelbe Kristalle (davon ca. 60 %
38, bestimmt durch NMR). Elementaranalyse: ber. für 37: C37H60Zn6Ru: C, 44.52; H, 6.06;
Zn, 39.30. ber. für 38: C28H48Zn6Ru: C, 38.30; H, 5.51; Zn, 44.68. ber. für eine Mischung aus
60% 38 und 40% 37: C, 40.76; H, 5.73; Zn, 42.53. gef.: C, 40.54; H, 5.61; Zn, 42.09. 1H
NMR δH(C6D6) von 37: 2.04 (s, 45H, ZnCp*), 1.30 (s, 6H, CH2), -0.11 (s, 9H, ZnMe). 1H
NMR δH(C6D6) von 38: 2.02 (s, 30H, ZnCp*), 0.97 (s, 6H, CH2), -0.09 (s, 9H, ZnMe).
13
C{1H} NMR δC{H}(C6D6) beider Isomere: 111.1 (C5Me5), 110.6 (C5Me5), 91.2 (Czentral,
TMM), 89.5 (Czentral, TMM), 29.6 (CH2, TMM), 29.1 (CH2, TMM), 11.2 (C5Me5), 10.8
(ZnMe), 10.6 (C5Me5), 10.4 (ZnMe).
162
7. Ergänzende kristallographische Daten
7. Ergänzende kristallographische Daten
Tabelle 163.1. Ergänzende kristallographische Daten zu Verbindungen 1, 3-6
Verbindung
1
3
4
5
6
Empirische Formel
C50H51GaP2Ru
C38H57Ga3Ru
C77H83BF24Ga2PRh
C70H69BF24Ga3Rh
C70H69Al3BF24Rh
Molmasse [g/mol]
884.64
824.07
1748.56
1689.13
1560.91
Temperatur [K]
100(2)
113(2)
100(2)
106(2)
105(2)
Wellenlänge Mo-Kα [Å]
0.71073
0.71073
0.71073
0.71073
0.71073
Kristallgröße [mm]
0.15 × 0.1 × 0.1
0.25 × 0.15 × 0.1
0.2 × 0.15 × 0.1
0.2 × 0.15 × 0.1
0.3 × 0.2 × 0.15
Kristallsystem
Triklin
Triklin
Monoklin
Orthorhombisch
Orthorhombisch
Raumgruppe
P-1
P-1
P2(1)/n
Pna2(1)
Pna2(1)
a (Å)
11.345(3)
10.693(4)
15.8148(4)
24.941(7)
25.022(6)
b (Å)
13.493(3)
11.111(4)
25.0768(9)
19.361(6)
19.373(5)
c (Å)
14.575(4)
17.202(6)
19.0581(8)
14.669(5)
14.526(5)
α(°)
89.35(2)
81.52(3)
90
90
90
β (°)
68.43(2)
76.88(3)
92.723(3)
90
90
γ (°)
83.562(19)
67.23(3)
90
90
90
Zellvolumen [Å3]
2060.7(9)
1831.3(11)
7549.6(5)
7083(4)
7042(3)
Z
2
2
4
4
4
Dichte ρcalc. [g cm-3]
1.426
1.494
1.538
1.584
1.472
1.132
2.612
1.048
1.461
0.384
F (000)
912
844
3552
3392
3176
2θ Messbereich [°]
3.04 - 27.56
3.39 - 36.86
3.31 - 27.68
3.01 - 25.05
3.01 - 25.06
-14≤h≤14,
-17≤h≤15,
-20≤h≤20,
-20≤h≤29,
-28≤h≤17,
-17≤ k ≤17,
-13≤ k ≤16,
-32≤k≤15,
-22≤k≤22,
-15≤k≤23,
-16≤l≤18
-27≤l≤24
-21≤l≤24
-17≤l≤15
-17≤l≤13
20754
24884
45596
19982
16068
9394
13183
17010
10037
10527
[Rint = 0.0687]
[Rint = 0.0393]
[Rint = 0.1255]
[Rint = 0.0521]
[Rint = 0.0367]
Full-matrix least-
Full-matrix least-
Full-matrix least-
Full-matrix least-
Full-matrix least-
squares on F2
squares on F2
squares on F2
squares on F2
squares on F2
Daten/Restraints/Parameter
9394/0/487
13183/0/379
17010/12/1039
10037/1/892
10527/1/892
Absorptionskorrektur
Empirisch
Empirisch
Empirisch
Empirisch
Empirisch
0.751
0.855
0.669
0.783
0.819
R1 = 0.0448,
R1 = 0.0438,
R1 = 0.0443,
R1 = 0.0425,
R1 = 0.0435,
wR2 = 0.0528
wR2 = 0.0854
wR2 = 0.0481
wR2 = 0.0633
wR2 = 0.0699
R1 = 0.1236,
R1 = 0.0921,
R1 = 0.1272,
R1 = 0.0712,
R1 = 0.0718,
wR2 = 0.0690
wR2 = 0.0975
wR2 = 0.0565
wR2 = 0.0693
wR2 = 0.0758
0.436 und -0.610
1.497 und -0.858
0.710 und -0.949
0.897 und -0.571
1.001 und -0.410
Absorptionskoeffizient
μ [mm-1]
hkl Messbereich
Anzahl der Reflexe
unabhängige Reflexe
Verfeinerungsmethode
2
Goodness-of-fit on F
(GOF)
Finale R Werte [I>2σ(I)]
R Werte (alle Daten)
Restelektronendichte [e/Å3]
163
7. Ergänzende kristallographische Daten
Tabelle 164.1. Ergänzende kristallographische Daten zu Verbindungen 7-11
Verbindung
7
8
9
10
11
Empirische Formel
C70H70BF24Ga3Ru
C40H64Ga2Ru2
C80H128Al4Ru4
C40H65AlRu3
C40H65GaRu3
Molmasse [g/mol]
1688.30
886.49
1602.02
876.11
913.81
Temperatur [K]
113(2)
105(2)
113(2)
100(2)
103(2)
Wellenlänge Mo-Kα [Å])
0.71073
0.71073
0.71073
0.71073
0.71073
Kristallgröße [mm]
0.25 × 0.15 × 0.1
0.4 × 0.2 × 0.1
0.25 × 0.2 × 0.15
0.3 × 0.2 × 0.15
0.2 × 0.1 × 0.1
Kristallsystem
Monoklin
Monoklin
Triklin
Triklin
Triklin
Raumgruppe
P2(1)/c
C2/c
P-1
P-1
P-1
a (Å)
12.3213(4)
22.547(4)
11.1012(7)
11.5481(4)
10.9515(14)
b (Å)
15.2459(5)
10.278(2)
16.2246(9)
18.8345(5)
18.382(3)
c (Å)
38.0827(18)
17.1391(15)
22.2073(12)
19.5558(8)
19.955(3)
α (°)
90
90
86.070(4)
112.383(4)
87.387(11)
β (°)
96.276(3)
96.283(12)
78.525(5)
91.855(3)
88.635(11)
γ (°)
90
90
85.984(5)
92.893(3)
89.897(11)
Zellvolumen [Å3]
7110.9(5)
3947.9(11)
3904.1(4)
3921.8(2)
4011.9(9)
4
4
2
4
4
1.577
1.491
1.363
1.484
1.513
1.436
2.126
0.844
1.188
1.796
F (000)
3392
1800
1672
1800
1852
2θ Messbereich [°]
2.86 - 27.56
3.31 - 25.10
2.59 - 25.06
2.70 - 27.58
2.80 - 25.05
-14≤h≤16,
-26≤h≤26,
-13≤h≤13,
-15≤h≤15,
-11≤h≤13,
-19≤k≤19,
-12≤ k ≤11,
-19≤k≤19,
-24≤k≤24,
-21≤k≤21,
-49≤l≤49
-20≤l≤15
-26≤l≤26
-25≤l≤25
-21≤l≤23
69067
15648
63685
75251
32896
16319
3501
13767
17983
14154
[Rint = 0.0828]
[Rint = 0.0490]
[Rint = 0.0971]
[Rint = 0.0584]
[Rint = 0.0710]
Full-matrix least-
Full-matrix least-
Full-matrix least-
Full-matrix least-
Full-matrix least-
squares on F2
squares on F2
squares on F2
squares on F2
squares on F2
Daten/Restraints/Parameter
16319/0/924
3501/48/298
13767/50/513
17983/18/878
14154/36/793
Absorptionskorrektur
Empirisch
Empirisch
Empirisch
Empirisch
Empirisch
1.120
1.045
0.894
0.882
0.904
R1 = 0.0933,
R1 = 0.0451,
R1 = 0.0760,
R1 = 0.0293,
R1 = 0.0575,
wR2 = 0.1532
wR2 = 0.0958
wR2 = 0.1893
wR2 = 0.0609
wR2 = 0.0856
R1 = 0.1432,
R1 = 0.0661,
R1 = 0.1590,
R1 = 0.0579,
R1 = 0.1362,
wR2 = 0.1629
wR2 = 0.1054
wR2 = 0.2243
wR2 = 0.0640
wR2 = 0.1052
1.304 und -1.458
1.458 und -0.832
1.357 und -0.606
1.918 und -0.606
2.250 und -1.498
Z
Dichte ρcalc. [g cm ]
-3
Absorptionskoeffizient
μ [mm-1]
hkl Messbereich
Anzahl der Reflexe
unabhängige Reflexe
Verfeinerungsmethode
2
Goodness-of-fit on F
(GOF)
Finale R Werte [I>2σ(I)]
R Werte (alle Daten)
Restelektronendichte [e/Å3]
164
7. Ergänzende kristallographische Daten
Tabelle 165.1. Ergänzende kristallographische Daten zu Verbindungen 12-16
Verbindung
12
13
14
15
16
Empirische Formel
C70H105Ga8Ru2
C34H30Ga3Ru
C51H78Ga5Rh
C73H75BF24GaRh
C78H80BF24Ga5NRh
Molmasse [g/mol]
1706.44
748.81
1142.64
1870.65
1949.75
Temperatur [K]
105(2)
113(2)
113(2)
100(2)
103(2)
Wellenlänge Mo-Kα [Å]
0.71073
0.71073
0.71073
0.71073
0.71073
Kristallgröße [mm]
0.2 × 0.1 × 0.1
0.25 × 0.2 × 0.15
0.25 × 0.2 × 0.15
0.15 × 0.1 × 0.1
0.15 × 0.15 × 0.1
Kristallsystem
Monoklin
Tetragonal
Tetragonal
Monoklin
Monoklin
Raumgruppe
C2/m
I4/m
P-4
P2/n
P2(1)/c
a (Å)
25.774(3)
19.8445(6)
18.956(2)
15.8518(10)
20.9830(16)
b (Å)
16.266(4)
19.8445(6)
18.956(2)
12.8256(8)
20.522(2)
c (Å)
22.000(5)
18.8042(7)
16.057(2)
19.4342(6)
20.064(2)
α (°)
90
90
90
90
90
β (°)
108.555(14)
90
90
98.661(4)
107.195(11)
γ (°)
90
90
90
90
90
Zellvolumen [Å3]
8744(3)
7405.2(4)
5769.8(11)
3906.1(4)
8254.0(14)
4
8
4
2
4
1.296
1.343
1.315
1.590
1.569
2.788
2.577
2.608
2.003
1.900
F (000)
3444
2968
2336
1868
3904
2θ Messbereich [°]
2.94 - 27.58
2.54 - 27.58
3.22 - 25.08
2.81 - 27.60
2.74 - 27.63
-33≤h≤33,
-25≤h≤25,
-22≤h≤22,
-20≤h≤18,
-26≤h≤27,
-11k≤21,
-22≤k≤25,
-17≤k≤22,
-16≤k≤16,
-26≤k≤16,
-28≤l≤28
-24≤l≤24
-19≤l≤19
-18≤l≤25
-26≤l≤26
44051
37838
40625
39237
70713
10408
4402
5306
9036
19130
[Rint = 0.0675]
[Rint = 0.0438]
[Rint = 0.0457]
[Rint = 0.0651]
[Rint = 0.0740]
Full-matrix least-
Full-matrix least-
Full-matrix least-
Full-matrix least-
Full-matrix least-
squares on F2
squares on F2
squares on F2
squares on F2
squares on F2
Daten/Restraints/Parameter
10408/24/451
4402/0/200
5306/0/361
9036/24/569
19130/12/1042
Absorptionskorrektur
Empirisch
Empirisch
Empirisch
Empirisch
Empirisch
1.002
0.983
1.065
0.874
0.889
R1 = 0.0730,
R1 = 0.0334,
R1 = 0.0635,
R1 = 0.0536,
R1 = 0.0416,
wR2 = 0.1920
wR2 = 0.0936
wR2 = 0.1775
wR2 = 0.1186
wR2 = 0.0811
R1 = 0.1003,
R1 = 0.0505,
R1 = 0.0823,
R1 = 0.1261,
R1 = 0.1233,
wR2 = 0.2144
wR2 = 0.0980
wR2 = 0.1904
wR2 = 0.1348
wR2 = 0.1048
3.807 und -1.584
1.710 und -0.459
2.314 und -1.137
1.058 und -1.931
1.045 und -0.678
Z
Dichte ρcalc. [g cm ]
-3
Absorptionskoeffizient
μ [mm-1]
hkl Messbereich
Anzahl der Reflexe
unabhängige Reflexe
Verfeinerungsmethode
2
Goodness-of-fit on F
(GOF)
Finale R Werte [I>2σ(I)]
R Werte (alle Daten)
Restelektronendichte [e/Å3]
165
7. Ergänzende kristallographische Daten
Tabelle 166.1. Ergänzende kristallographische Daten zu Verbindungen 17-20
Verbindung
17
18
19
20
Empirische Formel
C117H208P4Ga4Ru2
C76H79BF24Ga4Ru
C144H136B2F50Ga8Ru2
C147H137B2F50.50Ga8Ru2
Molmasse [g/mol]
2219.73
1839.15
3598.05
3644.59
Temperatur [K]
113(2)
113(2)
100(2)
103(2)
Wellenlänge Mo-Kα [Å]
0.71073
0.71073
0.71073
0.71073
Kristallgröße [mm]
0.25 × 0.15 × 0.1
0.25 × 0.2 × 0.15
0.3 × 0.2 × 0.15
0.2 × 0.1 × 0.1
Kristallsystem
Monoklin
Orthorhombisch
Monoklin
Triklin
Raumgruppe
P2(1)/c
Pbca
P2(1)/c
P-1
a (Å)
20.9566(18)
25.546(3)
15.3517(4)
14.1090(6)
b (Å)
24.925(3)
21.2722(8)
18.2016(6)
14.7960(7)
c (Å)
23.0088(19)
29.176(2)
27.2028(10)
36.0416(18)
α (°)
90
90
90
86.712(4)
β (°)
92.018(7)
90
102.118(3)
80.878(4)
γ (°)
90
90
90
88.267(4)
Zellvolumen [Å3]
12011.1(19)
15855(2)
7431.8(4)
7414.8(6)
4
8
2
2
1.228
1.541
1.608
1.632
1.225
1.625
1.733
1.739
F (000)
4728
7392
3592
3639
2θ Messbereich [°]
2.68 - 27.70
2.67 - 27.59
2.81 - 27.66
2.74 - 25.05
-27≤h≤23,
-33≤h≤32,
-20≤h≤20,
-16≤h≤11,
-32≤ k ≤32,
-27≤k≤27,
-23≤k≤23,
-17≤k≤16,
-29≤l≤29
-38≤l≤37
-35≤l≤35
-42≤l≤42
95267
290868
74060
30540
27422
18306
17247
24536
[Rint = 0.1130]
[Rint = 0.1514]
[Rint = 0.0435]
[Rint = 0.1137]
Full-matrix least-
Full-matrix least-
Full-matrix least-
Full-matrix least-
squares on F2
squares on F2
squares on F2
squares on F2
Daten/Restraints/Parameter
27422/0/1160
18306/0/955
17247/0/988
24536/0/1896
Absorptionskorrektur
Empirisch
Empirisch
Empirisch
Empirisch
0.659
0.824
1.309
0.861
R1 = 0.0446,
R1 = 0.0483,
R1 = 0.0449,
R1 = 0.0701,
wR2 = 0.0729
wR2 = 0.0927
wR2 = 0.1069
wR2 = 0.1403
Z
Dichte ρcalc. [g cm ]
-3
Absorptionskoeffizient
μ [mm-1]
hkl Messbereich
Anzahl der Reflexe
unabhängige Reflexe
Verfeinerungsmethode
2
Goodness-of-fit on F
(GOF)
Finale R Werte [I>2σ(I)]
R Werte (alle Daten)
Restelektronendichte [e/Å3]
R1 = 0.1516,
R1 = 0.1247,
R1 = 0.0849,
R1 = 0.1494,
wR2 = 0.0828
wR2 = 0.1050
wR2 = 0.1197
wR2 = 0.1548
0.933 und -0.734
0.959 und -0.541
1.345 und -0.667
0.968 und -0.975
166
7. Ergänzende kristallographische Daten
Tabelle 167.1. Ergänzende kristallographische Daten zu Verbindungen 21-25
Verbindung
21
22
23
24
25
Empirische Formel
C100H118BF26Ga3P2Ru
C87H126Ga6Mo
C39H72MoZn12
C44H72Ga4MoZn4
C52H84Ga2MoZn8
Molmasse [g/mol]
2196.92
1686.14
1421.35
1237.32
1467.53
Temperatur [K]
113(2)
113(2)
113(2)
113(2)
113(2)
Wellenlänge Mo-Kα [Å]
0.71073
0.71073
0.71073
0.71073
0.71073
Kristallgröße [mm]
0.4 × 0.2 × 0.1
0.25 × 0.2 × 0.15
0.2 × 0.1 × 0.1
0.25 × 0.2 × 0.1
0.35 × 0.25 × 0.15
Kristallsystem
Monoklin
Triklin
Orthorhombisch
Monoklin
Monoklin
Raumgruppe
P2(1)/c
P-1
Pnma
P2(1)/n
P2(1)/n
a (Å)
22.929(5)
12.2106(9)
25.0480(7)
12.7822(16)
17.3360(8)
b (Å)
17.448(3)
14.7410(12)
14.7952(4)
23.5489(15)
17.4952(12)
c (Å)
25.47(3)
21.1396(17)
15.1100(4)
18.963(4)
18.8614(12)
α (°)
90
79.893(7)
90
90
90
β (°)
101.84(4)
87.157(7)
90
94.042(13)
94.730(4)
γ (°)
90
77.266(7)
90
90
90
Zellvolumen [Å3]
9972(10)
3653.6(5)
5599.6(3)
5693.9(13)
5701.1(6)
4
2
4
4
4
1.463
1.533
1.686
1.443
1.710
1.076
2.391
5.273
3.751
4.471
F (000)
4496
1752
2832
2488
2960
2θ Messbereich [°]
2.69 - 27.62
2.61- 27.53
2.79 - 27.57
2.54 - 27.64
2.80 - 27.61
-18≤h≤29,
-15≤h≤15,
-29≤h≤32,
-16≤h≤16,
-16≤h≤22,
-22≤ k ≤22,
-19≤k≤19,
-19≤k≤15,
-30≤ k ≤19,
-22≤k≤22,
-33≤l≤33
-27≤l≤27
-19≤l≤19
-24≤l≤24
-24≤l≤24
Z
Dichte ρcalc. [g cm ]
-3
Absorptionskoeffizient
μ [mm-1]
hkl Messbereich
Anzahl der Reflexe
90864
69567
55527
57463
57679
22956
16576
6692
13180
13174
[Rint = 0.0823]
[Rint = 0.0984]
[Rint = 0.0642]
[Rint = 0.1798]
[Rint = 0.1138]
Full-matrix least-
Full-matrix least-
Full-matrix least-
Full-matrix least-
Full-matrix least-
squares on F2
squares on F2
squares on F2
squares on F2
squares on F2
Daten/Restraints/Parameter
22956/0/1198
16576/0/766
6692/0/250
13180/0/478
13174/0/568
Absorptionskorrektur
Empirisch
Empirisch
Empirisch
Empirisch
Empirisch
0.806
0.756
1.098
0.686
0.644
R1 = 0.0410,
R1 = 0.0429,
R1 = 0.0399,
R1 = 0.0473,
R1 = 0.0353,
wR2 = 0.0838
wR2 = 0.0733
wR2 = 0.1148
wR2 = 0.1019
wR2 = 0.0553
unabhängige Reflexe
Verfeinerungsmethode
Goodness-of-fit on F2
(GOF)
Finale R Werte [I>2σ(I)]
R Werte (alle Daten)
R1 = 0.1074,
R1 = 0.1231,
R1 = 0.0839,
R1 = 0.1678,
R1 = 0.1131,
wR2 = 0.0927
wR2 = 0.0828
wR2 = 0.1417
wR2 = 0.1159
wR2 = 0.0608
1.149 und -0.972
2.616 und -0.788
1.813 und -0.824
0.682 und -0.668
Restelektronendichte [e/Å3] 1.434 und -1.157
167
7. Ergänzende kristallographische Daten
Tabelle 168.1. Ergänzende kristallographische Daten zu Verbindungen 26-29
Verbindung
26
27
28
29
Empirische Formel
C40H30MoZn12
C56H60Mo4O16Zn10
C52H84RuZn10
C44H72RuZn8
Molmasse [g/mol]
1391.02
2026.50
1463.96
1225.05
Temperatur [K]
113(2)
105(2)
113(2)
105(2)
Wellenlänge Mo-Kα [Å]
0.71073
0.71073
0.71073
0.71073
Kristallgröße [mm]
0.35 × 0.25 × 0.1
0.2 × 0.15 × 0.1
0.25 × 0.25 × 0.15
0.2 × 0.2 × 0.1
Kristallsystem
Monoklin
Tetragonal
Monoklin
Monoklin
Raumgruppe
P2(1)/c
P-4
P2(1)/n
P2(1)/n
a (Å)
12.1242(8)
18.04440(10)
17.2946(11)
12.5205(4)
b (Å)
21.3988(11)
18.04440(10)
17.390(2)
23.2905(7)
c (Å)
23.125(3)
10.0674(2)
18.725(2)
22.5218(8)
α (°)
90
90
90
90
β (°)
90
90
94.516(7)
123.522(2)
γ (°)
90
90
90
90
Zellvolumen [Å3]
5999.7(9)
3277.95(7)
5614.4(10)
5475.2(3)
4
2
4
4
1.540
2.053
1.732
1.486
4.920
4.381
4.481
3.731
F (000)
2688
1984
2960
2480
2θ Messbereich [°]
2.69 - 27.63
3.19 - 27.60
2.64 - 27.74
2.79 - 27.63
Z
Dichte ρcalc. [g cm ]
-3
Absorptionskoeffizient
μ [mm-1]
-15≤h≤11,
-23≤h≤23,
-22≤h≤22,
-15≤h≤16,
hkl Messbereich
-27≤ k ≤27,
-23k≤16,
-22≤k≤17,
-30≤ k ≤21,
-30≤l≤30
-13≤l≤13
-24≤l≤23
-29≤l≤29
Anzahl der Reflexe
60546
26852
57251
55315
13810
16576
13102
12631
[Rint = 0.0681]
[Rint = 0.0532]
[Rint = 0.1274]
[Rint = 0.1074]
Full-matrix least-
Full-matrix least-
Full-matrix least-
Full-matrix least-
squares on F2
squares on F2
squares on F2
squares on F2
Daten/Restraints/Parameter
13810/0/473
7596/0/389
13102/0/303
12631/0/502
Absorptionskorrektur
Empirisch
Empirisch
Empirisch
Empirisch
0.853
0.768
0.742
0.884
R1 = 0.0689,
R1 = 0.0277,
R1 = 0.0430,
R1 = 0.0502,
wR2 = 0.1818
wR2 = 0.0353
wR2 = 0.0718
wR2 = 0.1158
unabhängige Reflexe
Verfeinerungsmethode
2
Goodness-of-fit on F
(GOF)
Finale R Werte [I>2σ(I)]
R Werte (alle Daten)
Restelektronendichte [e/Å3]
R1 = 0.1433,
R1 = 0.0440,
R1 = 0.1541,
R1 = 0.0925,
wR2 = 0.2063
wR2 = 0.0369
wR2 = 0.0925
wR2 = 0.1245
2.427 und -1.844
0.647 und -0.514
1.194 und -0.914
2.698 und -1.494
168
7. Ergänzende kristallographische Daten
Tabelle 169.1. Ergänzende kristallographische Daten zu Verbindungen 30-33
Verbindung
30
31
32
33
Empirische Formel
C36RhZn9
C44H72GaRhZn7
C109H166Ni2Zn16
C44H72PdZn8
Molmasse [g/mol]
1123.60
1231.24
2639.76
1230.38
Temperatur [K]
113(2)
113(2)
105(2)
113(2)
Wellenlänge Mo-Kα [Å]
0.71073
0.71073
0.71073
0.71073
Kristallgröße [mm]
0.3 × 0.25 × 0.15
0.2 × 0.15 × 0.1
0.15 × 0.15 × 0.1
0.25 × 0.15 × 0.1
Kristallsystem
Orthorhombisch
Monoklin
Monoklin
Monoklin
Raumgruppe
Pmc2(1)
P2(1)/c
C2/c
P2(1)/n
a (Å)
19.5840(7)
16.9525(7)
29.9453(13)
12.6106(3)
b (Å)
12.5029(5)
16.5830(4)
20.8547(6)
23.3762(9)
c (Å)
17.3314(6)
23.4379(9)
22.1767(7)
18.9914(7)
α (°)
90
90
90
90
β (°)
90
110.783(4)
126.644(3)
93.411(3)
γ (°)
90
90
90
90
Zellvolumen [Å3]
4243.7(3)
6160.2(4)
11112.2(7)
5588.5(3)
4
4
4
4
1.759
1.328
1.578
1.462
5.386
3.387
3.753
3.707
F (000)
2124
2488
5424
2488
2θ Messbereich [°]
3.12 - 27.65
2.85 - 27.60
2.59 - 27.71
2.77 - 27.56
-25≤h≤25,
-19≤h≤22,
-38≤h≤24,
-12≤h≤16,
-16≤k≤16,
-17≤ k ≤21,
-27k≤27,
-30≤k≤28,
-16≤l≤22
-30≤l≤30
-28≤l≤28
-24≤l≤24
43385
61573
40710
55911
8613
14058
12834
12787
[Rint = 0.1004]
[Rint = 0.0530]
[Rint = 0.0724]
[Rint = 0.0999]
Full-matrix least-
Full-matrix least-
Full-matrix least-
Full-matrix least-
squares on F2
squares on F2
squares on F2
squares on F2
Daten/Restraints/Parameter 8613/13/442
14058/0/478
12834/0/584
12787/0/478
Absorptionskorrektur
Empirisch
Empirisch
Empirisch
Empirisch
0.782
0.793
0.715
0.886
R1 = 0.0401,
R1 = 0.0347,
R1 = 0.0355,
R1 = 0.0426,
wR2 = 0.0625
wR2 = 0.0713
wR2 = 0.0546
wR2 = 0.0976
R1 = 0.0902,
R1 = 0.0695,
R1 = 0.0887,
R1 = 0.0790,
wR2 = 0.0700
wR2 = 0.0752
wR2 = 0.0600
wR2 = 0.1037
1.774 und -1.287
1.971 und -0.608
0.620 und -0.462
1.886 und -0.987
Z
Dichte ρcalc. [g cm ]
-3
Absorptionskoeffizient
μ [mm-1]
hkl Messbereich
Anzahl der Reflexe
unabhängige Reflexe
Verfeinerungsmethode
2
Goodness-of-fit on F
(GOF)
Finale R Werte [I>2σ(I)]
R Werte (alle Daten)
Restelektronendichte
[e/Å3]
169
7. Ergänzende kristallographische Daten
Tabelle 170.1. Ergänzende kristallographische Daten zu Verbindungen 34-37
Verbindung
34
35
Empirische Formel
C44H72PtZn8
C52H90PtZn8
C36H54MoO3Zn6
C37H60RuZn6
Molmasse [g/mol]
1319.07
1449.29
1022.95
998.14
Temperatur [K]
105(2)
113(2)
113(2)
113(2)
Wellenlänge Mo-Kα [Å]
0.71073
0.71073
0.71073
0.71073
Kristallgröße [mm]
0.40 × 0.35 × 0.2
0.25 × 0.15 × 0.15
0.1 × 0.1 × 0.1
0.2 × 0.15 × 0.1
Kristallsystem
Monoklin
Orthorhombisch
Triklin
Monoklin
Raumgruppe
P2(1)/n
Pbcn
P-1
P2(1)/n
a (Å)
12.5896(12)
17.2256(3)
12.2736(17)
9.4592(3)
b (Å)
23.1888(8)
17.7766(3)
12.7396(10)
17.2181(7)
c (Å)
19.066(4)
18.5676(3)
16.037(3)
24.8610(10)
α (°)
90
90
92.183(15)
90
β (°)
93.262(11)
90
110.003(17)
99.901(3)
γ (°)
90
90
118.080(13)
90
Zellvolumen [Å3]
5557.0(12)
5685.63(17)
2018.1(5)
3988.8(3)
4
4
2
4
1.577
1.693
1.683
1.662
5.919
5.795
3.836
3.937
F (000)
2616
2912
1032
2024
2θ Messbereich [°]
2.58 - 27.65
2.80 - 27.61
2.92 - 27.58
3.19 - 27.57
-16≤h≤15,
-19≤h≤22,
-13≤h≤15,
-12≤h≤12,
-21≤ k ≤30,
-23≤k≤23,
-16≤ k ≤11,
-22≤ k ≤22,
-24≤l≤24
-24≤l≤23
-20≤l≤15
-27≤l≤32
56261
55459
13875
39989
12875
6591
9054
9222
[Rint = 0.0459]
[Rint = 0.0383]
[Rint = 0.0249]
[R(int) = 0.0754]
Full-matrix least-
Full-matrix least-
Full-matrix least-
Full-matrix least-
squares on F2
squares on F2
squares on F2
squares on F2
Daten/Restraints/Parameter 12875/0/478
6591/0/281
9054/54/483
9222/24/488
Absorptionskorrektur
Empirisch
Empirisch
Empirisch
Empirisch
1.074
0.949
0.912
0.833
R1 = 0.0556,
R1 = 0.0234,
R1 = 0.0301,
R1 = 0.0327,
wR2 = 0.1578
wR2 = 0.0532
wR2 = 0.0630
wR2 = 0.0665
R1 = 0.0902,
R1 = 0.0435,
R1 = 0.0564,
R1 = 0.0695,
wR2 = 0.1729
wR2 = 0.0570
wR2 = 0.0702
wR2 = 0.0708
7.395 und -4.148
0.973 und -0.717
0.748 und -0.592
1.276 und -0.538
Z
Dichte ρcalc. [g cm ]
-3
Absorptionskoeffizient
μ [mm-1]
hkl Messbereich
Anzahl der Reflexe
unabhängige Reflexe
Verfeinerungsmethode
36
37
2
Goodness-of-fit on F
(GOF)
Finale R Werte [I>2σ(I)]
R Werte (alle Daten)
Restelektronendichte
[e/Å3]
170
8. Literaturverzeichnis
8. Literaturverzeichnis
[1]
S. Shaik, J. Comp. Chem. 2007, 28, 51-61.
[2]
G. N. Lewis, J. Am. Chem. Soc. 1916, 38, 762-785.
[3]
C. M. Fafard, C.-H. Chen, B. M. Foxman, O. V. Ozerov, Chem. Comm. 2007, 4465-4467.
[4]
F. A. Cotton, G. Wilkinson, C. A. Murillo, M. Bochmann, Advanced Inorganic Chemistry 2005, 6.
Aufl., Wiley, New York.
[5]
F. A. Cotton, C. A. Murillo, R. A. Walton, Multiple Bonds Between Metal Atoms 2005, 3. Aufl.,
Springer, New York.
[6]
Z. Zhu, R. C. Fischer, J. C. Fettinger, E. Rivard, M. Brynda, P. P. Power, J. Am. Chem. Soc. 2006, 128,
15068-15069.
[7]
I. Resa, E. Carmona, E. Gutierrez-Puebla, A. Monge, Science 2004, 305, 1136-1138.
[8]
Z. Zhu, M. Brynda, R. J. Wright, R. C. Fischer, W. A. Merrill, E. Rivard, R. Wolf, J. C. Fettinger, M.
M. Olmstead, P. P. Power, J. Am. Chem. Soc. 2007, 129, 10847-10857.
[9]
S. P. Green, C. Jones, A. Stasch, Science 2007, 318, 1754-1757.
[10]
F. A. Cotton, N. F. Curtis, C. B. Harris, B. F. G. Johnson, S. J. Lippard, J. T. Mague, W. R. Robinson, J.
S. Wood, Science 1964, 145, 1305-1307.
[11]
F. A. Cotton, Inorg. Chem. 1965, 4, 334-336.
[12]
F. A. Cotton, J. Chem. Ed. 1983, 60, 713-720.
[13]
T. Nguyen, A. D. Sutton, M. Brynda, J. C. Fettinger, G. J. Long, P. P. Power, Science 2005, 310, 844847.
[14]
G. Frenking, Science 2005, 310, 796-797.
[15]
M. Brynda, L. Gagliardi, P.-O. Widmark, P. P. Power, B. O. Roos, Angew. Chem. Int. Ed. 2006, 45,
3804-3807.
[16]
C. R. Landis, F. Weinhold, J. Am. Chem. Soc. 2006, 128, 7335-7345.
[17]
U. Radius, F. Breher, Angew. Chem. Int. Ed. 2006, 45, 3006-3010.
[18]
F. A. Cotton, Quart. Rev., Chem. Soc. 1966, 20, 389-401.
[19]
A. Schnepf, G. Stoesser, H. Schnöckel, J. Am. Chem. Soc. 2000, 122, 9178-9181.
[20]
A. Purath, R. Koppe, H. Schnockel, Angew. Chem. Int. Ed. 1999, 38, 2926-2928.
[21]
P. Pyykko, N. Runeberg, Angew. Chem. Int. Ed. 2002, 41, 2174-2176.
[22]
X. Li, B. Kiran, J. Li, H.-J. Zhai, L.-S. Wang, Angew. Chem. Int. Ed. 2002, 41, 4786-4789.
[23]
G. Schmid, Chem. Rev. 1992, 92, 1709-1727.
[24]
N. T. Tran, D. R. Powell, L. F. Dahl, Angew. Chem. Int. Ed. 2000, 39, 4121-4125.
[25]
H. Schnöckel, E. Weckert, A. Ecker, Nature 1997, 387, 379-381.
[26]
A. Schnepf, H. Schnöckel, Angew. Chem. Int. Ed. 2001, 40, 712-715.
[27]
J. R. Gispert, Coordination Chemistry 2008, Wiley-VCH, 1. Aufl., Weinheim.
[28]
A. Werner, Z. Anorg. Chem. 1893, 3, 267-330.
[29]
S. Aldridge, Angew. Chem. Int. Ed. 2006, 45, 8097-8099.
[30]
R. A. Fischer, J. Weiss, Angew. Chem. Int. Ed. 1999, 38, 2831-2850.
171
8. Literaturverzeichnis
[31]
G. Linti, H. Schnöckel, Coord. Chem. Rev. 2000, 206-207, 285-319.
[32]
A. H. Cowley, Chem. Comm. 2004, 2369-2375.
[33]
C. Gemel, T. Steinke, M. Cokoja, A. Kempter, R. A. Fischer, Euro. J. Inorg. Chem. 2004, 4161-4176.
[34]
R. J. Baker, C. Jones, Coord. Chem. Rev. 2005, 249, 1857-1869.
[35]
P. L. Arnold, S. T. Liddle, J. McMaster, C. Jones, D. P. Mills, J. Am. Chem. Soc. 2007, 129, 5360-5361.
[36]
S. T. Liddle, J. McMaster, D. P. Mills, A. J. Blake, C. Jones, W. D. Woodul, Angew. Chem. Int. Ed.
2009, 121, 1097 - 1100.
[37]
S. G. Minasian, J. L. Krinsky, V. A. Williams, J. Arnold, J. Am. Chem. Soc. 2008, 130, 10086-10087.
[38]
W. Uhl, M. Benter, S. Melle, W. Saak, G. Frenking, J. Uddin, Organometallics 1999, 18, 3778-3780.
[39]
J. Su, X.-W. Li, R. C. Crittendon, C. F. Campana, G. H. Robinson, Organometallics 1997, 16, 45114513.
[40]
F. A. Cotton, X. Feng, Organometallics 1998, 17, 128-130.
[41]
C. L. B. Macdonald, A. H. Cowley, J. Am. Chem. Soc. 1999, 121, 12113-12126.
[42]
G. Frenking, N. Froehlich, Chem. Rev. 2000, 100, 717-774.
[43]
J. Uddin, G. Frenking, J. Am. Chem. Soc. 2001, 123, 1683-1693.
[44]
B. Buchin, C. Gemel, T. Cadenbach, I. Fernandez, G. Frenking, R. A. Fischer, Angew. Chem. Int. Ed.
2006, 45, 5207-5210.
[45]
B. Buchin, C. Gemel, T. Cadenbach, R. Schmid, R. A. Fischer, Angew. Chem. Int. Ed. 2006, 45, 10741076.
[46]
M. Cokoja, H. Parala, M.-K. Schroeter, A. Birkner, M. W. E. Van den Berg, W. Gruenert, R. A.
Fischer, Chem. Mater. 2006, 18, 1634-1642.
[47]
M. Cokoja, H. Parala, M. K. Schroeter, A. Birkner, M. W. E. van den Berg, K. V. Klementiev, W.
Gruenert, R. A. Fischer, J. Mater. Chem. 2006, 16, 2420-2428.
[48]
M. Cokoja, B. R. Jagirdar, H. Parala, A. Birkner, R. A. Fischer, Eur. J. Inorg. Chem. 2008, 3330-3339.
[49]
M. Halbherr, T. Bollermann, C. Gemel, R. A. Fischer, Chem. Comm. 2009, submitted.
[50]
D. Seyferth, Organometallics 2001, 20, 1488-1498.
[51]
L. Cadet de Gassicourt, Memoires de Mathématique et de Physique. Presentés à l'Acadédemie Royale
des Sciences par diverse Savans et lûs dans ses Assemblées 1760, 623.
[52]
J. J. Berzelius, Jahresber. 1841, 20, 526.
[53]
L. B. Guyton de Morveau, H. Maret, J. F. Durande, Élemens de Chymie théoretique et pratique 1778,
Dijon, Vol. III, 39.
[54]
A. v. Baeyer, Ostwald's Klassiker der exakten Wissenschaften 1891, Wilhelm Engelmann, Leipzig.
[55]
R. W. Bunsen, Poggendorf´s Ann. 1837, 40, 219.
[56]
R. W. Bunsen, Poggendorf´s Ann. 1837, 42, 145.
[57]
R. W. Bunsen, Ann. 1837, 24, 271.
[58]
R. W. Bunsen, Ann. 1839, 31, 175.
[59]
R. W. Bunsen, Ann. 1841, 39, 1.
[60]
R. W. Bunsen, Ann. 1842, 42, 14.
[61]
R. W. Bunsen, Ann. 1843, 46, 1.
[62]
H. Roscoe, J. Chem. Soc. 1900, 77, 513-554.
[63]
E. Frankland, Ann. 1849, 17, 171.
172
8. Literaturverzeichnis
[64]
E. Frankland, J. Chem. Soc. 1850, 2, 263.
[65]
D. Seyferth, Organometallics 2001, 20, 2940-2955.
[66]
E. Frankland, B. F. Duppa, Ann. 1864, 130, 104.
[67]
E. Frankland, J. Chem. Soc. 1864, 17, 29.
[68]
A. L. Galyer, G. Wilkinson, Dalton Trans. 1976, 2235-2238.
[69]
K. Banert, Y.-H. Joo, T. Rueffer, B. Wlafort, H. Lang, Angew. Chem. Int. Ed. 2007, 46, 1168-1171.
[70]
R. Hoffmann, H. Hopf, Angew. Chem. Int. Ed. 2008, 47, 4474-4481.
[71]
E. O. Fischer, H. P. Hofmann, Angew. Chem. 1957, 69, 639-640.
[72]
H. Meister, Angewandte Chemie 1957, 69, 533-534.
[73]
O. T. Beachley, Jr., R. Blom, M. R. Churchill, K. Faegri, Jr., J. C. Fettinger, J. C. Pazik, L. Victoriano,
Organometallics 1989, 8, 346-356.
[74]
W. Uhl, R. Graupner, M. Layh, U. Schuetz, J. Organomet. Chem. 1995, 493, C1-5.
[75]
C. Dohmeier, C. Robl, M. Tacke, H. Schnöckel, Angew. Chem. 1991, 103, 594-595
[76]
D. Loos, H. Schnöckel, J. Organomet. Chem. 1993, 463, 37-40.
[77]
S. Schulz, H. W. Roesky, H. J. Koch, G. M. Sheldrick, D. Stalke, A. Kuhn, Angew. Chem. 1993, 105,
1828-1830.
[78]
P. Jutzi, B. Neumann, G. Reumann, H.-G. Stammler, Organometallics 1998, 17, 1305-1314.
[79]
C. Schnitter, H. W. Roesky, C. Ropken, R. Herbst-Irmer, H.-G. Schmidt, M. Moltemeyer, Angew.
Chem. Int. Ed. 1998, 37, 1952-1955.
[80]
W. Uhl, A. Jantschak, J. Organomet. Chem. 1998, 555, 263-269.
[81]
W. Uhl, W. Hiller, M. Layh, W. Schwarz, Angew. Chem. 1992, 104, 1378-1380
[82]
G. Linti, J. Organomet. Chem. 1996, 520, 107-113.
[83]
C. Cui, H. W. Roesky, H.-G. Schmidt, M. Noltemeyer, H. Hao, F. Cimpoesu, Angew. Chem. Int. Ed.
2000, 39, 4274-4276.
[84]
N. J. Hardman, B. E. Eichler, P. P. Power, Chem. Comm. 2000, 1991-1992.
[85]
N. J. Hardman, R. J. Wright, A. D. Phillips, P. P. Power, J. Am. Chem. Soc. 2003, 125, 2667-2679.
[86]
N. J. Hardman, P. P. Power, Inorg. Chem. 2001, 40, 2474-2475.
[87]
N. J. Hardman, C. Cui, H. W. Roesky, W. H. Fink, P. P. Power, Angew. Chem. Int. Ed. 2001, 40, 21722174.
[88]
N. J. Hardman, P. P. Power, Chem. Comm. 2001, 1184-1185.
[89]
M. Stender, B. E. Eichler, N. J. Hardman, P. P. Power, J. Prust, M. Noltemeyer, H. W. Roesky, Inorg.
Chem. 2001, 40, 2794-2799.
[90]
J. Su, X.-W. Li, R. C. Crittendon, G. H. Robinson, J. Am. Chem. Soc. 1997, 119, 5471-5472.
[91]
X.-W. Li, W. T. Pennington, G. H. Robinson, J. Am. Chem. Soc. 1995, 117, 7578-7579.
[92]
B. Twamley, P. P. Power, Angew. Chem. Int. Ed. 2000, 39, 3500-3503.
[93]
M. Niemeyer, P. P. Power, Angew. Chem. Int. Ed. 1998, 37, 1277-1279.
[94]
S. T. Haubrich, P. P. Power, J. Am. Chem. Soc. 1998, 120, 2202-2203.
[95]
T. Yamaguchi, K. Ueno, H. Ogino, Organometallics 2001, 20, 501-507.
[96]
H. Braunschweig, Angew. Chem. Int. Ed. 1998, 37, 1786-1801.
[97]
H. Braunschweig, M. Colling, Coord. Chem. Rev. 2001, 223, 1-51.
[98]
H. Braunschweig, M. Colling, Eur. J. Inorg. Chem. 2003, 393-403.
173
8. Literaturverzeichnis
[99]
H. Braunschweig, C. Kollann, D. Rais, Angew. Chem. Int. Ed. 2006, 45, 5254-5274.
[100]
H. Braunschweig, M. Burzler, T. Kupfer, K. Radacki, F. Seeler, Angew. Chem. Int. Ed. 2007, 46, 77857787.
[101]
H. Braunschweig, K. Radacki, K. Uttinger, Angew. Chem. Int. Ed. 2007, 46, 3979-3982.
[102]
A. J. Lupinetti, G. Frenking, S. H. Strauss, Angew. Chem. Int. Ed. 1998, 37, 2113-2116.
[103]
J. D. Gorden, C. L. B. Macdonald, A. H. Cowley, Chem. Comm. 2001, 75-76.
[104]
J. Uddin, C. Boehme, G. Frenking, Organometallics 2000, 19, 571-582.
[105]
W. Uhl, S. U. Keimling, W. Hiller, M. Neumayer, Chem. Ber. 1995, 128, 1137-1139.
[106]
C. Üffing, A. Ecker, R. Köppe, H. Schnöckel, Organometallics 1998, 17, 2373-2375.
[107]
Q. Yu, A. Purath, A. Donchev, H. Schnöckel, J. Organom. Chem. 1999, 584, 94-97.
[108]
P. Jutzi, B. Neumann, G. Reumann, L. O. Schebaum, H.-G. Stammler, Organometallics 1999, 18, 25502552.
[109]
P. Jutzi, B. Neumann, L. O. Schebaum, A. Stammler, H.-G. Stammler, Organometallics 1999, 18, 44624464.
[110]
W. Uhl, M. Pohlmann, Organometallics 1997, 16, 2478-2480.
[111]
M. Cokoja, T. Steinke, C. Gemel, T. Welzel, M. Winter, K. Merz, R. A. Fischer, J. Organomet. Chem.
2003, 684, 277-286.
[112]
W. Uhl, M. Pohlmann, R. Wartchow, Angew. Chem. Int. Ed. 1998, 37, 961-963.
[113]
C. Gemel, T. Steinke, D. Weiss, M. Cokoja, M. Winter, R. A. Fischer, Organometallics 2003, 22, 27052710.
[114]
A. Kempter, C. Gemel, T. Cadenbach, R. A. Fischer, Inorg. Chem. 2007, 46, 9481-9487.
[115]
T. Bollermann, A. Puls, C. Gemel, T. Cadenbach, A. Fischer Roland, Dalton Trans. 2009, 1372 - 1377
[116]
T. Bollermann, Dissertation, Ruhr-Universität-Bochum, unveröffentlicht.
[117]
T. Steinke, C. Gemel, M. Cokoja, M. Winter, R. A. Fischer, Angew. Chem. Int. Ed. 2004, 43, 22992302.
[118]
T. Steinke, C. Gemel, M. Winter, R. A. Fischer, Chem. Eur. J. 2005, 11, 1636-1646.
[119]
C. Boehme, G. Frenking, Chem. Eur. J. 1999, 5, 2184-2190.
[120]
J. Weiss, D. Stetzkamp, B. Nuber, R. A. Fischer, C. Boehme, G. Frenking, Angew. Chem. Int. Ed. 1997,
36, 70-72.
[121]
W. Uhl, S. Melle, G. Frenking, M. Hartmann, Inorg. Chem. 2001, 40, 750-755.
[122]
T. Steinke, M. Cokoja, C. Gemel, A. Kempter, A. Krapp, G. Frenking, U. Zenneck, R. A. Fischer,
Angew. Chem. Int. Ed. 2005, 44, 2943-2946.
[123]
T. Cadenbach, C. Gemel, R. Schmid, S. Block, R. A. Fischer, Dalton Trans. 2004, 3171-3172.
[124]
T. Cadenbach, C. Gemel, R. Schmid, R. A. Fischer, J. Am. Chem. Soc. 2005, 127, 17068-17078.
[125]
T. Steinke, C. Gemel, M. Cokoja, M. Winter, R. A. Fischer, Chem. Comm. 2003, 1066-1067.
[126]
T. Steinke, C. Gemel, M. Cokoja, M. Winter, R. A. Fischer, Dalton Trans. 2005, 55-62.
[127]
M. Cokoja, C. Gemel, T. Steinke, F. Schroeder, R. A. Fischer, Dalton Trans. 2005, 44-54.
[128]
B. Buchin, C. Gemel, A. Kempter, T. Cadenbach, R. A. Fischer, Inorg. Chim. Acta 2006, 359, 48334839.
[129]
P. Jutzi, B. Neumann, L. O. Schebaum, A. Stammler, H.-G. Stammler, Organometallics 2000, 19, 14451447.
174
8. Literaturverzeichnis
[130]
B. Buchin, Diplomarbeit 2005, Ruhr-Universität-Bochum.
[131]
N. A. Compton, R. J. Errington, N. C. Norman, Adv. Organomet. Chem. 1990, 31, 91-182.
[132]
K. Ueno, T. Watanabe, H. Tobita, H. Ogino, Organometallics 2003, 22, 4375-4377.
[133]
N. R. Bunn, S. Aldridge, D. L. Kays, N. D. Coombs, A. Rossin, D. J. Willock, J. K. Day, C. Jones, L.-l.
Ooi, Organometallics 2005, 24, 5891-5900.
[134]
H. Braunschweig, G. R. Whittell, Chem. Eur. J. 2005, 11, 6128-6133.
[135]
H. Braunschweig, K. Radacki, D. Rais, F. Seeler, Angew. Chem. Int. Ed. 2006, 45, 1066-1069.
[136]
J. R. Stork, M. M. Olmstead, A. L. Balch, J. Am. Chem. Soc. 2005, 127, 6512-6513.
[137]
C. Jones, P. C. Junk, J. A. Platts, A. Stasch, J. Am. Chem. Soc. 2006, 128, 2206-2207.
[138]
R. J. Baker, R. D. Farley, C. Jones, M. Kloth, D. M. Murphy, Dalton Trans. 2002, 3844-3850.
[139]
M. C. Kuchta, J. B. Bonanno, G. Parkin, J. Am. Chem. Soc. 1996, 118, 10914-10915.
[140]
E. S. Schmidt, A. Jockisch, H. Schmidbaur, J. Am. Chem. Soc. 1999, 121, 9758-9759.
[141]
C. Dohmeier, D. Loos, H. Schnöckel, Angew. Chem. Int. Ed. 1996, 35, 129-149.
[142]
A. Seifert, G. Linti, Inorg. Chem. 2008, 47, 11398-11404.
[143]
A. Kempter, C. Gemel, T. Cadenbach, R. A. Fischer, Organometallics 2007, 26, 4257-4264.
[144]
C. Jones, D. P. Mills, R. P. Rose, A. Stasch, Dalton Trans. 2008, 4395-4408.
[145]
A. Kempter, C. Gemel, R. A. Fischer, Chem. Eur. J. 2007, 13, 2990-3000.
[146]
A. Kempter, C. Gemel, R. A. Fischer, Inorg. Chem. 2005, 44, 163-165.
[147]
T. Cadenbach, C. Gemel, D. Zacher, R. A. Fischer, Angew. Chem. Int. Ed. 2008, 47, 3438-3441.
[148]
T. Cadenbach, T. Bollermann, C. Gemel, I. Fernandez, M. von Hopffgarten, G. Frenking, A. Fischer
Roland, Angew. Chem. Int. Ed. 2008, 47, 9150-9154.
[149]
T. Cadenbach, C. Gemel, A. Fischer Roland, Angew. Chem. Int. Ed. 2008, 47, 9146-9149.
[150]
R. A. Fischer, J. Behm, T. Priermeier, W. Scherer, Angew. Chem. Int. Ed. 1993, 32, 746-748.
[151]
R. A. Fischer, W. Scherer, M. Kleine, Angew. Chem. Int. Ed. 1993, 32, 748-750.
[152]
M. Cokoja, H. Parala, A. Birkner, O. Shekhah, M. W. E. van den Berg, R. A. Fischer, Chem. Mater.
2007, 19, 5721-5733.
[153]
T. Cadenbach, T. Bollermann, C. Gemel, R. A. Fischer, Dalton Trans. 2009, 322 - 329.
[154]
T. Cadenbach, C. Gemel, T. Bollermann, I. Fernandez, G. Frenking, A. Fischer Roland, Chem. Eur. J.
2008, 14, 10789-10796.
[155]
D. G. Hamilton, R. H. Crabtree, J. Am. Chem. Soc. 1988, 110, 4126-4133.
[156]
S. Sabo-Etienne, B. Chaudret, Mod. Coord. Chem. 2002, 45-58.
[157]
C. Pan, K. Pelzer, K. Philippot, B. Chaudret, F. Dassenoy, P. Lecante, M.-J. Casanove, J. Am. Chem.
Soc. 2001, 123, 7584-7593.
[158]
J. Garcia-Anton, M. R. Axet, S. Jansat, K. Philippot, B. Chaudret, T. Pery, G. Buntkowsky, H.-H.
Limbach, Angew. Chem. Int. Ed. 2008, 47, 2074-2078.
[159]
F. Schroder, D. Esken, M. Cokoja, W. E. van den Berg Maurits, I. Lebedev Oleg, G. Van Tendeloo, B.
Walaszek, G. Buntkowsky, H.-H. Limbach, B. Chaudret, R. A. Fischer, J. Am. Chem. Soc. 2008, 130,
6119-6130.
[160]
A. D. Becke, Phys. Rev. A 1988, 38, 3098-3100.
[161]
R. G. Parr, W. Yang, C. Lee, Phys. Rev. B 1988, 37, 785.
[162]
T. H. Dunning, P. J. Hay, Theoretical Chemistry, Vol. 3, Plenum, New York 1976.
175
8. Literaturverzeichnis
[163]
Alle Energien werden als relative freie Energie ΔGo angegeben.
[164]
A. Schnepf, H. Schnöckel, Angew. Chem. Int. Ed. 2002, 41, 3532-3552.
[165]
H.-J. Himmel, A. J. Downs, T. M. Greene, L. Andrews, Organometallics 2000, 19, 1060-1070.
[166]
H.-J. Himmel, A. J. Downs, T. M. Greene, L. Andrews, Chem. Comm. 1999, 2243-2244.
[167]
R. A. Fischer, M. M. Schulte, E. Herdtweck, M. R. Mattner, Inorg. Chem. 1997, 36, 2010-2017.
[168]
J. T. Golden, T. H. Peterson, P. L. Holland, R. G. Bergman, R. A. Andersen, J. Am. Chem. Soc. 1998,
120, 223-224.
[169]
A. Kempter, C. Gemel, N. J. Hardman, R. A. Fischer, Inorg. Chem. 2006, 45, 3133-3138.
[170]
B. Beagley, D. G. Schmidling, I. A. Steer, J. Mol. Struc. 1974, 21, 437-444.
[171]
J. Krijn, E. J. Baerends, Fit Functions in the HFS-Method, Internal Report, Vrije Universiteit
Amsterdam, Niederlande, 1984, Alle Berechnungen wurden unter Verwendug des Programmpakets
ADF ausgeführt. .
[172]
F. M. Bickelhaupt, E. J. Baerends, Rev. in Comp. Chem. 2000, 15, 1-86.
[173]
G. Te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. Van Gisbergen, J. G.
Snijders, T. Ziegler, J. Comp. Chem. 2001, 22, 931-967.
[174]
F. L. Hirshfeld, Theoret. Chim. Acta 1977, 44, 129-138.
[175]
T. Ziegler, A. Rauk, Theoret. Chim. Acta 1977, 46, 1-10.
[176]
K. Morokuma, J. Chem. Phys. 1971, 55, 1236-1244.
[177]
Die EDA teilt die Bindungsenergie zwischen Fragmenten ΔEint auf drei Beiträge auf: die PauliAbstoßung ΔEPauli, die quasi-klassische elektrostatische Wechselwirkung ΔEelstat und den kovalenten
(Orbital-)- Beitrag ΔEorb. Dieser letzte Term setzt sich zusammen aus den Beiträgen ΔEσ und ΔEp für
Orbitale mit σ- bzw. π-Symmetrie.
[178]
G. Parkin, J. Chem. Ed. 2006, 83, 791-799.
[179]
R. A. Fischer, M. M. Schulte, J. Weiss, L. Zsolnai, A. Jacobi, G. Huttner, G. Frenking, C. Boehme, S. F.
Vyboishchikov, J. Am. Chem. Soc. 1998, 120, 1237-1248.
[180]
P. Jutzi, G. Reumann, Dalton Trans. 2000, 2237-2244.
[181]
R. J. Baker, C. Jones, M. Kloth, J. A. Platts, Angew. Chem. Int. Ed. 2003, 42, 2660-2663.
[182]
R. J. Baker, C. Jones, J. A. Platts, J. Am. Chem. Soc. 2003, 125, 10534-10535.
[183]
H.-J. Himmel, G. Linti, Angew. Chem. Int. Ed. 2008, 47, 6326-6328.
[184]
N. D. Coombs, W. Clegg, A. L. Thompson, D. J. Willock, S. Aldridge, J. Am. Chem. Soc. 2008, 130,
5449-5451.
[185]
N. D. Coombs, D. Vidovic, J. K. Day, A. L. Thompson, D. D. Le Pevelen, A. Stasch, W. Clegg, L.
Russo, L. Male, M. B. Hursthouse, D. J. Willock, S. Aldridge, J. Am. Chem. Soc. 2008, 130, 1611116124.
[186]
I. Fernandez, E. Cerpa, G. Merino, G. Frenking, Organometallics 2008, 27, 1106-1111.
[187]
G. Prabusankar, A. Kempter, C. Gemel, M.-K. Schroeter, R. A. Fischer, Angew. Chem. Int. Ed. 2008,
47, 7234-7237.
[188]
K. S. MacFarlane, S. J. Rettig, Z. Liu, B. R. James, J. Organomet. Chem. 1998, 557, 213-219.
[189]
M. D. Jones, R. D. W. Kemmitt, A. W. G. Platt, Dalton Trans. 1986, 1411-1418.
[190]
K. McNeill, R. A. Andersen, R. G. Bergman, J. Am. Chem. Soc. 1997, 119, 11244-11254.
176
8. Literaturverzeichnis
[191]
A. F. Borowski, S. Sabo-Etienne, M. L. Christ, B. Donnadieu, B. Chaudret, Organometallics 1996, 15,
1427-1434.
[192]
T. Cadenbach, T. Bollermann, C. Gemel, R. A. Fischer, Dalton Trans. 2009, 322 - 329.
[193]
M. Ohashi, K. Matsubara, T. Iizuka, H. Suzuki, Angew. Chem. Int. Ed. 2003, 42, 937-940.
[194]
R. H. Crabtree, D. G. Hamilton, J. Am. Chem. Soc. 1986, 108, 3124-3125.
[195]
G. J. Kubas, Acc. Chem. Res. 1988, 21, 120-128.
[196]
M. Grellier, L. Vendier, B. Chaudret, A. Albinati, S. Rizzato, S. Mason, S. Sabo-Etienne, J. Am. Chem.
Soc. 2005, 127, 17592-17593.
[197]
J. Eckert, C. M. Jensen, T. F. Koetzle, T. L. Husebo, J. Nicol, P. Wu, J. Am. Chem. Soc. 1995, 117,
7271-7272.
[198]
T. Braun, G. Muench, B. Windmueller, O. Gevert, M. Laubender, H. Werner, Chem. Eur. J. 2003, 9,
2516-2530.
[199]
V. Cadierno, P. Crochet, J. Diez, S. E. Garcia-Garrido, J. Gimeno, S. Garcia-Granda, Organometallics
2003, 22, 5226-5234.
[200]
T. Kondo, H. Ono, N. Satake, T.-a. Mitsudo, Y. Watanabe, Organometallics 1995, 14, 1945-1953.
[201]
M. D. Mbaye, B. Demerseman, J.-L. Renaud, L. Toupet, C. Bruneau, Angew. Chem. Int. Ed. 2003, 42,
5066-5068.
[202]
H. Sasabe, S. Nakanishi, T. Takata, Inorg. Chem. 2003, 6, 1140-1143.
[203]
P. Xue, S. Bi, H. H. Y. Sung, I. D. Williams, Z. Lin, G. Jia, Organometallics 2004, 23, 4735-4743.
[204]
N. Wiberg, K. Amelunxen, H. W. Lerner, H. Noth, W. Ponikwar, H. Schwenk, J. Organomet. Chem.
1999, 574, 246-251.
[205]
X. He, R. A. Bartlett, M. M. Olmstead, K. Ruhlandt-Senge, B. E. Sturgeon, P. P. Power, Angew. Chem.
Int. Ed. 1993, 32, 717-719.
[206]
R. Ahlrichs, M. Baer, M. Haeser, H. Horn, C. Koelmel, Chem. Phys. Lett. 1989, 162, 165-169.
[207]
J. P. Perdew, Phys. Rev. B 1986, 33, 8822.
[208]
F. Weigend, R. Ahlrichs, Phys. Chem. Chem. Phys. 2005, 7, 3297.
[209]
D. Cremer, E. Kraka, Angew. Chem. 1984, 96, 612-614.
[210]
R. F. W. Bader, Atoms in Molecules. A Quantum Theory 1990, Oxford University Press, Oxford.
[211]
D. del Rio, A. Galindo, I. Resa, E. Carmona, Angew. Chem. Int. Ed. 2005, 44, 1244-1247.
[212]
A. Grirrane, I. Resa, A. Rodriguez, E. Carmona, E. Alvarez, E. Gutierrez-Puebla, A. Monge, A.
Galindo, D. Del Rio, R. A. Andersen, J. Am. Chem. Soc. 2007, 129, 693-703.
[213]
E. N. Esenturk, J. Fettinger, Y.-F. Lam, B. Eichhorn, Angew. Chem. Int. Ed. 2004, 43, 2132-2134.
[214]
T. Nasch, W. Jeitschko, J. Solid State Chem. 1999, 143, 95-103.
[215]
D. E. Crotty, E. R. Corey, T. J. Anderson, M. D. Glick, J. P. Oliver, Inorg. Chem. 1977, 16, 920-924.
[216]
D. E. Crotty, T. J. Anderson, M. D. Glick, J. P. Oliver, Inorg. Chem. 1977, 16, 2346-2350.
[217]
A. Spiekermann, S. D. Hoffmann, T. F. Faessler, Angew. Chem. Int. Ed. 2006, 45, 3459-3462.
[218]
T. F. Fässler, S. D. Hoffmann, Angew. Chem. Int. Ed. 2004, 43, 6242-6248.
[219]
Der Ausdruck “hypoelektronisch” meint im Allgemeinen eine elektronendefizitäre Bindungssituation in
Clustern und intermetallischen Festkörperverbindungen.
[220]
R. B. King, I. Silaghi-Dumitrescu, M. M. Uta, J. Chem. Theory and Comp. 2008, 4, 209-215.
[221]
J. D. Corbett, Q. S. Lin, Inorg. Chem. 2003, 42, 8762-8767.
177
8. Literaturverzeichnis
[222]
H.-P. Chen, R. S. Berry, R. L. Whetten, Phys. Rev. B 1991, 10647-10653.
[223]
S. C. Sevov, J. D. Corbett, Inorg. Chem. 1991, 4875-4877.
[224]
M. Brack, Rev. Mod. Phys. 1993, 677-732.
[225]
J. R. Hartig, A. Stößer, P. Hauser, H. Schnöckel, Angew. Chem. Int. Ed. 2007, 46, 1658-1662.
[226]
J. M. Goicoechea, S. C. Sevov, Angew. Chem. Int. Ed. 2006, 45, 5147-5150.
[227]
Die AIM Analyse von Al13- liefert Al-Al Bindungspfade im Al12 Käfig. Die EDA Resulate legen nahe,
dass die Bindungssituation am besten als endohedraler Komplex aufzufassen ist, in dem sich ein Al
Kation in einem Al122- Käfig befindet. M. v. Hopffgarten, G. Frenking, to be published.
[228]
S. Zhen, K. Seff, J. Phys. Chem. B 1999, 103, 6493-6497.
[229]
Die Geometrieoptimierung und Energieberechnungen wurden mit dem Programmpaket TURBOMOLE
(12)
Version
15.18
durchgeführt,
das
bei
Cosmologic
erhältlich
ist:
http://www.cosmologic.de/QuantumChemistry/main_turbomole.html.
[230]
F. Weigend, R. Ahlrichs, Phys. Chem. Chem. Phys. 2005, 7, 3297-3305.
[231]
K. Eichkorn, O. Treutler, H. Ohm, M. Häser, R. Ahlrichs, Chem. Phys. Lett. 1995, 242, 652-660.
[232]
T. Ziegler, A. Rauk, Inorg. Chem. 1979, 18, 1558-1565.
[233]
P. Pyykkö, J. Organomet. Chem. 2006, 691, 4336-4340 „…the preferred closed-shell electron
structures, such as those in typical high-symmetry 4318-electron systems, are driven both by the
bonding contributions to the centre and by the kinetic-energy (nodal-structure) terms in the ligand
subsystem, Ln. The latter imposes a filling order s < p < d, even for the MLn complex. Then the 4318e
principle can be right for the right reasons even without any np contributions at the central atom.“
[234]
A. C. Tang, H. K. Lu, J. Chin. Chem. Soc. 1950, 17, 251.
[235]
A. C. Tang, J. Chin. Chem. Soc. 1951, 18, 15.
[236]
C.-G. Zhan, F. Liu, Z.-M. Hu, Int. J. Quantum Chem. 1987, 32, 13.
[237]
T. K. Firman, C. R. Landis, J. Am. Chem. Soc. 1998, 120, 12650-12656.
[238]
J. Reinhold, O. Kluge, C. Mealli, Inorg. Chem. 2007, 47, 7142-7147.
[239]
J. Autschbach, B. A. Hess, M. P. Johansson, J. Neugebauer, M. Patzschke, P. Pyykkö, M. Reiher, D.
Sundholm, Phys. Chem. Chem. Phys. 2004, 6, 11-22.
[240]
Verbindungen MH12 (M = Cr, Mo, W) besitzen auch zwölf Einelektronenliganden um ein
Metallzentrum, aber Bindungsanalysen zeigen, dass es sich dort um vier klassische M-H und vier
nichtklassische M-(H2) Bindungen handelt: L. Gagliardi, P. Pyykkö, J. Am. Chem. Soc. 2004, 2126,
15014. Die Wolframverbindung [WH12] konnte kürzlich in FTIR-Studien durch Kondensation von
Wolframatomen in einer Wasserstoffmatrix bei 15014K nachgewisen werden: X. Wang, L. Andrews, I.
Infante, L. Gagliardi, J. Am. Chem. Soc. 12008, 15130, 11972-11978.
[241]
H. Jones, The Theory of Brillouin Zones and Electron States in Crystals 1962, (Amsterdam: North
Holland).
[242]
N. F. Mott, H. Jones, The Theory of the Properties of Metals and Alloys 1936, (London: Oxford
University Press).
[243]
J. K. Brandon, R. Y. Brizard, P. C. Chieh, R. K. McMillan, W. B. Pearson, Acta Cryst. 1974, 30, Pt. 6,
1412-1417.
[244]
M. Ohashi, K. Matsubara, H. Suzuki, Organometallics 2007, 26, 2330-2339.
178
8. Literaturverzeichnis
[245]
Y. Wang, B. Quillian, C. S. Wannere, P. Wei, P. v. R. Schleyer, G. H. Robinson, Organometallics 2007,
26, 3054-3056.
[246]
J. M. Burlitch, J. Am. Chem. Soc. 1969, 91, 4562-4563.
[247]
J. M. Burlitch, A. Ferrari, Inorg. Chem. 1970, 9, 563-569.
[248]
T. Heumann, H. W. Schleicher, H. Venker, Z. fuer Metallkunde 1969, 60, 438-441.
[249]
M. J. Mays, A. T. T. Hsieh, J. Chem. Soc. A 1971, 2648-2653.
[250]
P. H. M. Budzelaar, H. J. Alberts-Jansen, J. Boersma, G. J. M. Van der Kerk, Polyhedron 1982, 1, 563566.
[251]
C. E. Moore, Atomic Energy Levels, National Bureau of Standards, Washington DC, 1971.
[252]
A. Grirrane, I. Resa, A. Rodriguez, E. Carmona, E. Alvarez, E. Gutierrez-Puebla, A. Monge, A.
Galindo, D. del Rio, A. Andersen Richard, J. Am. Chem. Soc. 2007, 129, 693-703.
[253]
A. Grirrane, I. Resa, A. Rodriguez, E. Carmona, Coord. Chem. Rev. 2008, 252, 1532-1539.
[254]
A. Galindo, E. Carmona, Angew. Chem. Int. Ed. 2008, 47, 6526-6536.
[255]
R. Fernández, A. Grirrane, I. Resa, A. Rodriguez, E. Carmona, E. Alvarez, E. Gutierrez-Puebla, A.
Monge, J. M. López del Amo, H.-H. Limbach, A. Lledós, F. Maseras, D. Del Rio, Eur. J. Chem. 2009,
15, 924-935.
[256]
Y. Wang, B. Quillian, P. Wei, H. Wang, X.-J. Yang, Y. Xie, R. B. King, P. V. R. Schleyer, H. F.
Schaefer, III, G. H. Robinson, J. Am. Chem. Soc. 2005, 127, 11944-11945.
[257]
Zusätzlich ist anzumerken, dass der Atomradius von Zn 1.33 Å beträgt. Beobachtete Zn-Zn Abstände
von ca. 32.62 Å (das entspricht dem kürzesten Abstand im Zn36 Oktaeder) und Abstände von 32.5932.63 Å der Cp*Zn Einheiten zu den "nackten" Zn Atomen sind also im Bereich bindender Abstände.
[258]
P. H. M. Budzelaar, J. Boersma, G. J. M. Van der Kerk, A. L. Spek, A. J. M. Duisenberg,
Organometallics 1985, 4, 680-683.
[259]
P. H. M. Budzelaar, J. Boersma, G. J. M. Van der Kerk, Angew. Chem. 1983, 95, 335-336.
[260]
J. S. Denis, W. Butler, M. D. Glick, J. P. Oliver, J. Am. Chem. Soc. 1974, 96, 5427-5436.
[261]
S. Alvarez, M. Ferrer, R. Reina, O. Rossell, M. Seco, X. Solans, J. Organomet. Chem. 1989, 377, 291303.
[262]
J. M. Burlitch, S. E. Hayes, G. E. Whitwell, II, Organometallics 1982, 1, 1074-1083.
[263]
D. E. Crotty, J. P. Oliver, Inorg. Chem. 1977, 16, 2501-2506.
[264]
P. H. M. Budzelaar, J. Boersma, G. J. M. Van der Kerk, J. Organomet. Chem. 1980, 202, C71-C72.
[265]
S. Sabo-Etienne, B. Chaudret, Coord. Chem. Rev. 1998, 178-180, 381-407.
[266]
M. D. Fryzuk, D. H. McConville, S. J. Rettig, Organometallics 1990, 9, 1359-1360.
[267]
M. D. Fryzuk, D. H. McConville, S. J. Rettig, Organometallics 1993, 12, 2152-2161.
[268]
J. A. Rodriguez, M. Kuhn, J. Phys. Chem. 1996, 100, 381.
[269]
W. Bronger, L. á Brassard, P. Müller, Z. Anorg. Allg. Chemie 1999, 625, 1143-1146.
[270]
L. Gagliardi, P. Pyykkö, J. Am. Chem. Soc. 2004, 126, 15014.
[271]
X. Wang, L. Andrews, I. Infante, L. Gagliardi, J. Am. Chem. Soc. 2008, 130, 1972.
[272]
G. M. Sheldrick, SHELXL-97, Program for Crystal Structure Refinement, Universität Göttingen 1997.
[273]
G. M. Sheldrick, SHELXS-97, Program for the Solution of Crystal Structures, Universitaet Goettingen
1997.
179
8. Literaturverzeichnis
[274]
S. Schulz, H. W. Roesky, H. J. Koch, G. M. Sheldrick, D. Stalke, A. Kuhn, Angew. Chem. Int. Ed.
1993, 32, 1729-1731.
[275]
M. Schormann, K. S. Klimek, H. Hatop, S. P. Varkey, H. W. Roesky, C. Lehmann, C. Roepken, R.
Herbst-Irmer, M. Noltemeyer, J. Solid State Chem. 2001, 162, 225-236.
[276]
P. Jutzi, L. O. Schebaum, J. Organomet. Chem. 2002, 654, 176-179.
[277]
F. X. Kohl, P. Jutzi, J. Organomet. Chem. 1983, 243, 119-121.
[278]
M. Brookhart, B. Grant, A. F. Volpe, Jr., Organometallics 1992, 11, 3920-3922.
[279]
W. Gausing, G. Wilke, Angew. Chem. 1981, 93, 201-202.
[280]
D. J. Cole-Hamilton, G. Wilkinson, Nouv. J. Chim. 1977, 1, 141-155.
[281]
D. J. Cole-Hamilton, G. Wilkinson, Chem. Comm. 1977, 59-60.
[282]
H. Suzuki, H. Omori, D. H. Lee, Y. Yoshida, M. Fukushima, M. Tanaka, Y. Moro-oka,
Organometallics 1994, 13, 1129-1146.
[283]
H. Suzuki, H. Omori, D. H. Lee, Y. Yoshida, Y. Morooka, Organometallics 1988, 7, 2243-2245.
[284]
T. V. Ashworth, E. Singleton, J. J. Hough, Dalton Trans. 1977, 1809-1815.
[285]
B. N. Chaudret, D. J. Cole-Hamilton, G. Wilkinson, Dalton Trans. 1978, 1739-1745.
[286]
B. Chaudret, R. Poilblanc, Organometallics 1985, 4, 1722-1726.
[287]
P. Pertici, G. Vitulli, M. Paci, L. Porri, Dalton Trans. 1980, 1961-1964.
[288]
M. A. Kulzick, R. T. Price, R. A. Andersen, E. L. Muetterties, J. Organomet. Chem. 1987, 333, 105118.
[289]
B. Guzel, M. A. Omary, J. P. Fackler, A. Akgerman, Inorg. Chim. Acta 2001, 325, 45-50.
[290]
M. J. Ingleson, S. K. Brayshaw, M. F. Mahon, G. D. Ruggiero, A. S. Weller, Inorg. Chem. 2005, 44,
3162-3171.
[291]
S. K. Brayshaw, M. J. Ingleson, J. C. Green, J. S. McIndoe, P. R. Raithby, G. Kociok-Koehn, A. S.
Weller, J. Am. Chem. Soc. 2006, 128, 6247-6263.
180
9. Anhang
9. Anhang
9.1
Posterbeiträge und Vorträge
•
Cadenbach, T.; Bollermann, T.; Gemel, C.; Fischer, R.A.; Fernandez, I.;
Hopffgarten, M. v.; Frenking, G., Colloquium of the Analytical & Inorganic
Chemistry, Ruhr-University Bochum, Germany „Linkages Between Coordination
Compounds and Metal Clusters“, 12. Dez. 2008, (Vortrag)
•
Cadenbach, T.; Bollermann, T.; Gemel, C.; Fischer, R.A.; Fernandez, I.;
Hopffgarten, M. v.; Frenking, G., Zing Conference, Organometallics at the centre, ,
Cancun, Mexico „A New Class Of Mixed Metal Cluster Molecules“, 13-16 März
2008 (Vortrag)
•
Cadenbach, T.; Bollermann, T.; Gemel, C.; Fischer, R.A.; Fernandez, I.;
Hopffgarten, M. v.; Frenking, G., Zing Conference, Organometallics at the centre,
Cancun, Mexico „A Linkage Between Coordination Compounds and Metal Clusters:
Synthesis of an Endohedral MoZn12 Cage Molecule“ 13-16 März 2008 (Poster)
•
Cadenbach, T.; Bollermann, T.; Gemel, C.; Fischer, R.A.; Fernandez, I.;
Hopffgarten, M. v.; Frenking, G., UNAM University, Mexico City, Mexico, „A
Linkage Between Coordination Compounds and Metal Clusters: Synthesis of an
Endohedral MoZn12 Cage Molecule“ 4. März 2008 (Vortrag)
•
Cadenbach, T.; Gemel, C.; Schmid, R.; Fischer, R.A., 1st Section Days, Ruhr-
University Research School, Bochum, „A New Class Of Mixed Metal Cluster
Molecules“, 5-7. Nov. 2007 (Poster)
•
Cadenbach, T.; Gemel, C.; Schmid, R.; Fischer, R.A., 9th FIGIPAS Meeting in
Inorganic Chemistry, Vienna, Austria, „(H)(GaCp*)4Ru(Ga)Ru(GaCp*)3(H)2: From
Bimetallic Clusters To Alloy Nanoparticles“ 4.-7. Juli 2007 (Poster)
181
9. Anhang
•
Cadenbach, T.; Gemel, C.; Schmid, R.; Fischer, R.A., FECHEM, Conference on
Organometallic Chemistry, Budapest, Hungary, „Mechanistic Insights into an
Unprecedented C-C Bond Activation on a Rh/Ga Bimetallic Complex“,3.-8. Sept.
2005 (Poster)
9.2
Publikationsliste
Erstauthor:
•
Cadenbach, T.; Gemel, C.; Schmid, R.; Halbherr, M.; Yusenko, K.; Cokoja, M.;
Fischer, R. A., „Substituent-free Ga by Hydrogenolysis of Coordinated GaCp*:
Synthesis and Structure of Highly Fluxional [Ru2(Ga)(GaCp*)7(H)3].”, Angew.
Chem. Int. Ed. 2009, DOI: 10.1002/anie.200805605, in press. (Dieser Artikel wurde
für ein Cover ausgewählt)
•
Cadenbach, T.; Bollermann, T.; Gemel, C.; Fischer, R. A. „ Syntheses and Crystal
Structures of Ruthenium and Rhodium Olefin Complexes Containing GaCp*”,
Inorganic Chemistry 2009, ic-2008-02283r.R1, in press.
•
Cadenbach, T.; Bollermann, T.; Gemel, C.; Fischer, R. A., „Synthesis and structure
of electron rich ruthenium polyhydride complexes and clusters“, Dalton Trans.
2009, (2), 322-329.
•
Cadenbach, T.; Bollermann, T.; Gemel, C.; Fernandez, I.; von Hopffgarten, M.;
Grenking, G.; Fischer, R. A. „Twelve one-electron ligands coordinating one metal
center: structure and bonding of [Mo(ZnCH3)9(ZnCp*)3]”, Angew. Chem. Int. Ed.
2008, 47(47), 9150-9154. (Dieser Artikel wurde für ein Cover ausgewählt und ge-
highlighted in S. K. Ritter, C & EN News 2008, 86 (44) sowie D.L. Kays, S.
Aldridge Angew. Chem. Int. Ed. 2009, DOI: 10.1002/anie.200900491
•
Cadenbach, T.; Gemel, C.; Fischer, R. A. “Molecular cut-outs of Mo/Zn hume-
rothery phases: synthesis and structure of [{Mo(CO)4}4(Zn)6(μ-ZnCp*)4]”, Angew.
Chem. Int. Ed. 2008, 47(47), 9146-9149. (als Hot Paper ausgewählt) sowie
highlighted in D.L. Kays, S. Aldridge Angew. Chem. Int. Ed. 2009, DOI:
10.1002/anie.200900491
182
9. Anhang
•
Cadenbach, T.; Gemel, C.; Bollermann, T.; Fernandez, I.; Frenking, G.; Fischer, R.
A., „Organometallic chemistry of Ga+: formation of an unusual gallium dimer in the
coordination sphere of ruthenium.“, Chemistry – A European Journal 2008, 14(34),
10789-10796.
•
Cadenbach, T.; Gemel, C.; Zacher, D.; Fischer R., A., „Methylgallium as a terminal
ligand in [(Cp*Ga)4Rh(GaCH3)]+“, Angew. Chem. Int. Ed. 2008, 47(18), 3438-3441.
•
Cadenbach, T.; Gemel, C.; Schmid, R.; Fischer, R. A., „Mechanistic Insights into
an Unprecedented C-C Bond Activation on a Rh/Ga Bimetallic Complex: A
Combined Experimental/Computational Approach“, J. Am. Chem. Soc. 2005,
127(48), 17068-17078.
•
Cadenbach, T.; Gemel, C.; Schmid, R.; Block, S.; Fischer, R. A., „The reaction of
RhCp*(CH3)2(L) (L = pyridine, dmso) with GaCp* and AlCp*: A new type of
carbon-carbon bond activation reaction“, Dalton Trans. 2004, (20), 3171-3172.
Co-Author:
•
Bollermann, T.; Puls, A.; Gemel, C.; Cadenbach, T.; Fischer, R. A., „Reactions of
cationic transition metal acetonitrile complexes [M(CH3CN)n]m+ with GaCp*: novel
gallium complexes of iron, cobalt, copper and silver“, Dalton Trans. 2009, 322 - 329
•
Zhang, W.-H.; Zhang, X.; Hua, Z.; Harish, P.; Schroeder, F.; Hermes, S.;
Cadenbach, T.; Shi, J.; Fischer, R. A., „Synthesis, Bifunctionalization, and
Application of Isocyanurate-Based Periodic Mesoporous Organosilicas“, Chem. of
Mat. 2007, 19(10), 2663-2670.
•
Kempter, A.; Gemel, C.; Cadenbach, T.; Fischer, R. A., „Nickel Olefin Complexes
Supported by GaI(DDP)“, Organometallics 2007, 26(17), 4257-4264.
•
Kempter, A.; Gemel, C.; Cadenbach, T.; Fischer, R. A., „Synthesis and Structure of
New Compounds with Zn-Ga Bonds: Insertion of the Gallium(I) Bisimidinate
183
9. Anhang
Ga(DDP) into Zn-X (X = CH3, Cl) and the Homoleptic Complex Cation
[Zn(GaCp)4]2+“, Inorg. Chem. 2007, 46(22), 9481-9487.
•
Buchin, B.; Gemel, C.; Kempter, A.; Cadenbach, T.; Fischer, R. A., „Organo group
13 complexes of transition metals. Part XLV. Reaction of iron and ruthenium
halogenide complexes with GaCp* and AlCp*: Insertion, Cp* transfer reactions and
orthometallation“, Inorg. Chim. Acta 2006, 359, (15), 4833-4839.
•
Buchin, B.; Gemel, C.; Cadenbach, T.; Fernandez, I.; Frenking, G.; Fischer, R. A.,
„Organic group 13 complexes of d-block elements, XLIV. "Naked" Ga+ and In+ as
pure acceptor ligands: structure and bonding of [GaPt(GaCp*)4][BArF]“, Angew.
Chem. Int. Ed. 2006, 45, (31), 5207-5210. (Dieser Artikel wurde ge-highlighted in S.
Aldridge, Angew. Chem. Int. Ed. Engl. 2006, 45, 8097-8099)
•
Buchin, B.; Gemel, C.; Cadenbach, T.; Schmid, R.; Fischer, R. A., „The
[Ga2(C5Me5)]+ ion: bipyramidal double-cone structure and weakly coordinated,
monovalent Ga+“, Angew. Chem. Int. Ed. 2006, 45, (7), 1074-1076.
•
Buchin, B.; Gemel, C.; Cadenbach, T.; Fischer, R. A., „Organo Group 13
Complexes of Transition Metals. XL. Coordination Chemistry of Ga(C5Me4Ph):
Novel Homoleptic d10 Cluster Complexes of Palladium“, Inorg. Chem. 2006, 45, (4),
1789-1794.
•
Buchin, B.; Steinke, T.; Gemel, C.; Cadenbach, T.; Fischer, R. A., „Organo Group
13 complexes of transition metals. XXXVIII: Synthesis and characterization of the
novel AlI compound Al(C5Me4Ph): comparison of the coordination chemistry of
Al(C5Me5) and Al(C5Me4Ph) at d10 metal centers“, Z. Anorg. Allgem. Chem. 2005,
631, (13-14), 2756-2762.
•
Mukinovic, M.; Brenner, G.; Khanderi, J.; Spoellmann, S.; Fischer, R. A.;
Tafipolsky, M.; Cadenbach, T.; Schmid, R., „A multiscale simulation approach for
the MOCVD of GaN using a single-molecule precursor in a vertical stagnation flow
reactor“, Chem. Vap. Dep. 2005, 11, (6-7), 306-316.
184
9. Anhang
Manuskripte in Vorbereitung
•
Cadenbach, T.; Bollermann, T.; Gemel, C.; Tombul, M.; Fernandez, I.; von
Hopffgarten, M.; Grenking, G.; Fischer, R. A. „Structures and Bonding of Highly
Coordinated Transition Metal Molecules Containing Zn(I) Ligands”, J. Am. Chem.
Soc. 2009, in Vorbereitung.
•
Bollermann, T.; Cadenbach, T.; Gemel, C.; Fernandez, I.; von Hopffgarten, M.;
Grenking, G.; Fischer, R. A. „Structures and Bonding of Highly Coordinated
Transition Metal Molecules Containing Cd(I) and Au(I) Ligands”, J. Am. Chem.
Soc. 2009, in Vorbereitung.
185
9. Anhang
9.3 Lebenslauf
Anschrift:
Name:
Thomas Cadenbach
Adresse:
Kronprinzenstrasse 44
44135 Dortmund
Telefon:
0231 524155
Mobiltelefon:
0170 2965279
Email-Adresse:
[email protected]
Persönliche Daten:
Geburtsdatum:
25.11.1978
Geburtsort:
Dortmund
Staatsangehörigkeit:
deutsch
Familienstand:
ledig
Schulausbildung:
1985-1989
Lessing-Grundschule, Dortmund
1989-1993
Helmholtz-Gymnasium, Dortmund
1993-1996
Gymnasium Johanneum, Lüneburg
1996-1999
Stadtgymnasium, Dortmund
19.05.1999
Allgemeine Hochschulreife, Note: 2.1
186
9. Anhang
Zivildienst:
02.08.1999-30.06.2000
Caritasverband Dortmund, ganztägige Betreuung
von geistig- und körperbehinderten Kindern
Hochschulausbildung:
01.10.2000
Beginn des Diplomstudiengangs Chemie an der
Ruhr- Universität Bochum
02.10.2002
Diplom-Vorprüfung, Note: sehr gut
31.03.2005
Diplom-Hauptprüfung, Note: mit Auszeichnung
Thema der Diplomarbeit: „Eine ungewöhnliche CC Bindungsaktivierung an einem Rh-Ga-Komplex
– Experimentelle und Theoretische Studien“
Seit 01.10.2005
Promotion,
Thema
der
Promotionsarbeit:
„Zwischen
Koordinationsverbindungen
und
Clustern – Prinzipien und Konzepte zur Synthese
hochkoordinierter, metallreicher Moleküle.“
Berufserfahrung:
01.06.1999-31.07.1999
Zeitarbeiter bei Randstad Zeitarbeit
01.07.2000-31.08.2000
Hilfsarbeiter bei Caspar Hessel Betonwerke
GmbH & Co. KG
03.02.2003-30.06.2004
Studentische Hilfskraft
15.08.2004-30.06.2005
Studentische Hilfskraft
01.07.2005-31.10.2005
Wissenschaftlicher Mitarbeiter
02.04.2007-31.08.2007
Wissenschaftliche Hilfskraft
Seit 15.10.2007
Wissenschaftlicher Mitarbeiter
187
9. Anhang
Stipendien, Auszeichnungen, Preise:
01.07.2005
Studienabschlussstipendium der Ruth und Gerth
Massenberg Stiftung für den besten Notendurchschnitt des Jahrgangs
21.06.2005
Preise an Studierende der Ruhr Universität
Bochum für die beste Diplomarbeit des Jahres im
Fach Chemie
01.10.2005-30.09.2007
Chemiefonds Stipendium für Doktoranden der
Stiftung Stipendien-Fonds des Verbandes der
Chemischen-Industrie
seit 01.02.2007
Mitglied der Ruhr-University Research School
188
Übersicht über die synthetisierten Verbindungen
#
Verbindung
Kapitel / Seite
Exp. Daten
4
1
[Ru(η -but)(PPh3)2(GaCp*)]
3.1 / 27
141
2
[Ru(PPh3)2(GaCp*)3]
3.1 / 27
141
3
[Ru(η4-COD)(GaCp*)3]
3.1 / 27
141
4
[Rh(η2, η2-NBD)(PCy3)(GaCp*)2][BArF]
5
6
3.1 / 27
142
2
2
F
3.1 / 27
142
2
2
F
3.1 / 27
143
[Rh(η , η -COD)(GaCp*)3][BAr ]
[Rh(η , η -COD)(AlCp*)3][(BAr ]
F
7
[(COD)Ru(H)(GaCp*)3][BAr ]
3.2 / 29
143
8
[{Cp*Ru(µ-GaCp*)}2(µ-H)4]
3.2 / 30
143
9
[{Cp*Ru(µ-AlCp*)}2(µ-H)4]
3.2 / 30
144
3
3
3.2 / 30
144
3
3
11 [{Cp*Ru(µ-H)}3(µ -GaCp*)(µ -H)2]
3.2 / 30
144
12 [(GaCp*)4(H)Ru(μ-Ga)Ru(H)2(GaCp*)3
3.3 / 32
145
3.3 / 32; 3.5 / 51
154
3.4 / 42
154
15 [(Cp*Ga)4Rh(GaCH3)][BAr ]
3.4 / 43
154
16 [(Cp*Ga)4Rh(Ga(CH3)(py))][BArF]
3.4 / 48
155
3.5 / 52
155
18 [Ru(GaCp*)4(η -(CH2)2C(CH3))][BAr ]
3.5 / 54
156
19 [Ru(GaCp*)3(η3-(CH2)2C(CH2(μ-Ga)))2][(BArF)2]
3.5 / 56
156
20 [Ru(GaCp*)3(η3-(CH2)(CH(μ-Ga)(CH3))}2][(BArF)2]
3.5 / 58
156
21 [Ru(PCy3)2(GaCp*)2(Ga)][BAr ]
3.5 / 61
157
22 [Mo(GaCp*)6]
3.6 / 64
157
23 [Mo(ZnCp*)3(ZnMe)9]
3.7 / 67
158
24 [Mo(GaMe)4(ZnCp*)4]
3.7 / 67; 3.9.1 / 85
158
25 [Mo(GaMe)2(ZnCp*)4(ZnMe)4]
3.7 / 67; 3.9.1 / 85
158
26 [Mo(ZnCp*)2(ZnEt10)]
3.7 / 67; 3.9.1 / 88
159
3.8 / 80
159
3.9.2 / 89; 3.9.6 / 106
160
3.9.2 / 91
160
3.9.3 / 93; 3.9.6 / 114
160
31 [Rh(GaMe)(ZnCp*)4(ZnMe)3]
3.9.3 / 93
161
32 [Ni(ZnCp*)4(ZnMe)4]
3.9.4 / 94
161
33 [Pd(ZnCp*)4(ZnMe)4]
3.9.4 / 94; 3.9.6 / 110
161
34 [Pt(ZnCp*)4(ZnMe)4]
3.9.4 / 94
162
35 [Pt(ZnCp*)4(ZnEt)4]
3.9.4 / 98
162
36 [Mo (ZnCp*)3(ZnMe)3(CO)3]
3.9.7 / 116
162
37 [Ru(ZnCp*)3(ZnMe)3(TMM)]
3.9.7 / 118
162
10 [{Cp*Ru(µ-H)}3(µ -AlCp*)(µ -H)2]
13 [Ru(GaCp*)3(TMM)]
1
14 [(Cp*Ga)4Rh(η -Cp*GaCH3)]
F
17 [Ru(PCy3)2(GaCp*)2(H)2]
3
F
F
27 [{Mo(CO)4}4(Zn)6(μ-ZnCp*)4]
28 [Ru(ZnCp*)4(ZnMe)6]
29 [Ru(ZnCp*)4(ZnMe)4(H)2]
30 [Rh(ZnCp*)3(ZnMe)6]
Übersicht über die synthetisierten Verbindungen
# Verbindung
4
Kapitel / Seite
Exp. Daten
1
[Ru(η -but)(PPh3)2(GaCp*)]
3.1 / 27
141
2
[Ru(PPh3)2(GaCp*)3]
3.1 / 27
141
3
[Ru(η4-COD)(GaCp*)3]
3.1 / 27
141
4
5
6
2
2
F
3.1 / 27
142
2
2
F
3.1 / 27
142
2
2
F
3.1 / 27
143
[Rh(η , η -NBD)(PCy3)(GaCp*)2][BAr ]
[Rh(η , η -COD)(GaCp*)3][BAr ]
[Rh(η , η -COD)(AlCp*)3][(BAr ]
F
7
[(COD)Ru(H)(GaCp*)3][BAr ]
3.2 / 29
143
8
[{Cp*Ru(µ-GaCp*)}2(µ-H)4]
3.2 / 30
143
9
[{Cp*Ru(µ-AlCp*)}2(µ-H)4]
3.2 / 30
144
3
3
3.2 / 30
144
3
3
11 [{Cp*Ru(µ-H)}3(µ -GaCp*)(µ -H)2]
3.2 / 30
144
12 [(GaCp*)4(H)Ru(μ-Ga)Ru(H)2(GaCp*)3
3.3 / 32
145
3.3 / 32; 3.5 / 51
154
3.4 / 42
154
3.4 / 43
154
3.4 / 48
155
3.5 / 52
155
3.5 / 54
156
3.5 / 56
156
3.5 / 58
156
21 [Ru(PCy3)2(GaCp*)2(Ga)][BAr ]
3.5 / 61
157
22 [Mo(GaCp*)6]
3.6 / 64
157
23 [Mo(ZnCp*)3(ZnMe)9]
3.7 / 67
158
24 [Mo(GaMe)4(ZnCp*)4]
3.7 / 67; 3.9.1 / 85
158
25 [Mo(GaMe)2(ZnCp*)4(ZnMe)4]
3.7 / 67; 3.9.1 / 85
158
26 [Mo(ZnCp*)2(ZnEt10)]
3.7 / 67; 3.9.1 / 88
159
3.8 / 80
159
3.9.2 / 89; 3.9.6 / 106
160
3.9.2 / 91
160
3.9.3 / 93; 3.9.6 / 114
160
31 [Rh(GaMe)(ZnCp*)4(ZnMe)3]
3.9.3 / 93
161
32 [Ni(ZnCp*)4(ZnMe)4]
3.9.4 / 94
161
33 [Pd(ZnCp*)4(ZnMe)4]
3.9.4 / 94; 3.9.6 / 110
161
34 [Pt(ZnCp*)4(ZnMe)4]
3.9.4 / 94
162
35 [Pt(ZnCp*)4(ZnEt)4]
3.9.4 / 98
162
36 [Mo (ZnCp*)3(ZnMe)3(CO)3]
3.9.7 / 116
162
37 [Ru(ZnCp*)3(ZnMe)3(TMM)]
3.9.7 / 118
162
10 [{Cp*Ru(µ-H)}3(µ -AlCp*)(µ -H)2]
13 [Ru(GaCp*)3(TMM)]
1
14 [(Cp*Ga)4Rh(η -Cp*GaCH3)]
F
15 [(Cp*Ga)4Rh(GaCH3)][BAr ]
F
16 [(Cp*Ga)4Rh(Ga(CH3)(py))][BAr ]
17 [Ru(PCy3)2(GaCp*)2(H)2]
3
F
18 [Ru(GaCp*)4(η -(CH2)2C(CH3))][BAr ]
19 [Ru(GaCp*)3(η3-(CH2)2C(CH2(μ-Ga)))2][(BArF)2]
3
F
20 [Ru(GaCp*)3(η -(CH2)(CH(μ-Ga)(CH3))}2][(BAr )2]
F
27 [{Mo(CO)4}4(Zn)6(μ-ZnCp*)4]
28 [Ru(ZnCp*)4(ZnMe)6]
29 [Ru(ZnCp*)4(ZnMe)4(H)2]
30 [Rh(ZnCp*)3(ZnMe)6]