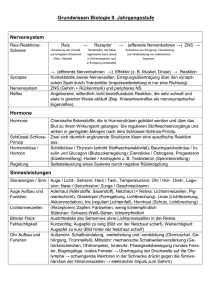
Neues bei paraneoplastischen Syndromen in der Neurologie
Werbung

60 Neues in der Neurologie Neues bei paraneoplastischen Syndromen in der Neurologie New Developments in Paraneoplastic Neurological Diseases Autoren Institute Schlüsselwörter ▶ Paraneoplastische Syndrome ● ▶ Enzephalitis ● ▶ Tumor ● Keywords ▶ paraneoplastic diseases ● ▶ encephalitis ● ▶ tumours ● Bibliografie DOI http://dx.doi.org/ 10.1055/s-0032-1304305 Online-Publikation: 22.2.2012 Akt Neurol 2012; 39: 60–73 © Georg Thieme Verlag KG Stuttgart · New York ISSN 0302-4350 Korrespondenzadresse Dr. Frank Leypoldt Klinik und Poliklinik für Neurologie Universitätsklinik Hamburg-Eppendorf Martinistraße 52 20246 Hamburg [email protected] F. Leypoldt1, K.-P. Wandinger2,3, R. Voltz4 1 Klinik und Poliklinik für Neurologie, Universitätsklinik Hamburg-Eppendorf Institut für Neuroimmunologie und klinische Multiple Sklerose Forschung (INIMS), Universitätsklinik Hamburg-Eppendorf 3 Institut für Experimentelle Immunologie, Euroimmun, Lübeck 4 Klinik für Palliativmedizin, Universität Köln 2 Zusammenfassung Abstract ▼ ▼ In nur wenigen Jahren hat sich das Spektrum und damit das Wissen um paraneoplastische Syndrome in der Neurologie deutlich erweitert. Insbesondere hat die Beschreibung neuer fakultativ paraneoplastischer Subgruppen von Enzephalitiden mit gutem Ansprechen auf immunmodulatorische Therapien das klinische und wissenschaftliche Interesse verstärkt. Das Erkennen dieser Subgruppen mittels neuartiger Autoantikörper hat unmittelbare therapeutische Konsequenzen. Die fundierte Kenntnis paraneoplastischer Syndrome in der Neurologie ist daher äußerst wichtig für den Kliniker. Neue wissenschaftliche Erkenntnisse beleuchten interessante Aspekte der Pathophysiologie paraneoplastischer Prozesse, ihrer möglichen Reversibilität und Initiierung. Neue Registerdaten helfen die Epidemiologie paraneoplastischer neurologischer Syndrome besser einzuschätzen. Der zunehmende Einsatz von „Biologika“ in der Neurologie eröffnet zukünftig neue Therapiechancen. Allerdings führt die mittlerweile entstandene Fülle an neuen Syndromen und Autoantikörpern auch vielfach zu Verwirrung und Unsicherheit. Diese Übersichtsarbeit fasst daher neue und wichtige ältere Aspekte paraneoplastischer neurologischer Syndrome, ihrer Klassifikation, Pathophysiologie und Behandlung zusammen. Recent years have witnessed a growing awareness of new aspects of paraneoplastic processes in neurological diseases. The discovery of new subgroups of paraneoplastic encephalitis syndromes with remarkably good response to immune therapy has spawned renewed clinical and scientific interest. Knowledge of these subgroups and associated autoantibodies is of direct therapeutic consequence and thus of the utmost importance for clinicians. New scientific insights into these syndromes shed light on issues concerning their reversibility, initiation and on paraneoplastic processes in general. Recently published data from a large European registry offer new information on epidemiology. Furthermore, the increasing utility of monoclonal antibodies in neurology is likely to offer new therapeutic avenues for the future. Nevertheless, the abundance of new autoantibodies and syndromes leads to confusion and uncertainty. This review article summarises new and important well-established aspects of paraneoplastic neurological syndromes, their new classification, pathophysiology and treatment. Grundlagen tralen über das periphere Nervensystem, die neuromuskuläre Endplatte bis zum Muskel betroffen sein. Einige klinische Syndrome sind jedoch sehr spezifisch für das Vorliegen einer paraneoplastischen Genese („klassische“ paraneoplastische, ▶ Tab. 1). Sie werden neurologische Syndrome: ● in sichere und mögliche paraneoplastische neu▶ Abb. 1), [1]. rologische Syndrome eingeteilt (● Ein wichtiger Stellenwert kommt dabei unverändert den paraneoplastischen Antikörpern zu. Seit der Erstbeschreibung des Hu-Antikörpers 1985 ▼ Per Definition sind alle nicht-kompressiven, nicht-infiltrativen, nicht-metastatischen, nichtmetabolischen, nicht-infektiösen und nicht-therapieassoziierten Komplikationen eines Tumors paraneoplastische Syndrome. Üblicherweise werden allerdings nur tumorferne, immunvermittelte Erkrankungen des Nervensystems als paraneoplastische neurologische Syndrome bezeichnet. Es kann jede Ebene des Nervensystems, vom zen- Leypoldt F et al. Neues bei paraneoplastischen Syndromen … Akt Neurol 2012; 39: 60–73 Neues in der Neurologie durch Francesc Graus und Jerome B. Posner [2] ist mittlerweile bekannt, dass bei mehr als zwei Dritteln der Patienten mit paraneoplastischen Erkrankungen hochspezifische Antikörper im Serum nachweisbar sind [3]. Historisch bedingt werden die meisten Antikörper nach dem Indexpatienten benannt (AntiHu = Hull, Anti-Yo = Young, Anti-Ri = Richards), allerdings teils auch nach dem Zielantigen (z. B. Anti-CV2/CRMP5). Sie werden eingeteilt in gut-charakterisierte und teil-charakterisierte paraneoplastische Antikörpern (siehe Neues zur Diagnostik und ▶ Tab. 2). Diese Einteilung gilt nicht für Antikörper gegen neuro● nale Oberflächenantigene, die weiter unten vorgestellt werden. Besonders betont werden muss, dass die Klassifikation in gesicherte und mögliche paraneoplastische Syndrome einen Ausschluss relevanter Differenzialdiagnosen fordert. Eine umfassende Differenzialdiagnose sprengt den Umfang dieses Artikels, ▶ Tab. 3. eine kurze Übersicht gibt jedoch ● Neues zur Epidemiologie ▼ Neue epidemiologischen Daten zu paraneoplastischen neurologischen Syndromen wurden kürzlich aus dem europäischen Register von knapp 1 000 Patienten veröffentlicht [3]. Bei der Mehrheit der Patienten findet sich initial ein isoliertes klinisches Tab. 1 „Klassische“ paraneoplastische Syndrome nach [1]. A. Zentrales Nervensystem Enzephalomyelitis Limbische Enzephalitis Subakute Kleinhirndegeneration Opsoklonus-Myoklonus Syndrom B. Peripheres Nervensystem Subakute sensorische Neuronopathie Chronische intestinale Pseudoobstruktion C. Neuromuskuläre Synapse und Muskel Lambert-Eaton-myasthenes Syndrom Dermatomyositis gut charakterisierter AK Mit oder ohne Tumornachweis klassisches Syndrom Ausschluss von Differenzialdiagnosen Teilcharakterisierter AK kein AK Tumornachweis innerhalb von 5 Jahren kein Tumornachweis gesichertes paraneoplastisches Syndrom Mit oder ohne Tumornachweis mögliches paraneoplastisches Syndrom kein AK Tumornachweis und Besserung nach Tumorbehandlung Abb. 1 Diagnosealgorithmus nach [1]. 2004 abhängig vom Vorliegen eines klassischen oder ▶ Tab. 1). nichtklassischen Syndroms (● * Erhöhtes Tumorrisiko: Alter über 40 Jahre, kein Ansprechen auf Immuntherapie. Gut charakteri▶ Tab. 2. sierter/teilcharakterisierter AK siehe ● hohes Tumorrisiko* nicht klassisches Syndrom gut charakterisierter AK ▶ Tab. 4). Bei 10 % der Patienten besteht dagegen Syndrom (90 %; ● von Anfang an eine Mischung mehrerer klinischer Syndrome. Die Häufigsten sind paraneoplastische Kleinhirndegenerationen und sensorische Neuronopathien (jeweils 25 %). Weitere häufige Syndrome sind limbische Enzephalitiden (10 %), Hirnstammenzephalitiden (5 %), generalisierte Enzephalomyelitiden (5 %), autonome Neuropathien (5 %) und Lambert-Eaton-Syndrome (4 %). Die häufigsten paraneoplastischen Antikörper in diesem Register sind Anti-Hu (39 %), Anti-Yo (13 %), anti-CV2/CRMP5 (6 %), Anti-Ri (5 %), Anti-Ma/Ta (4,5 %) sowie Anti-Amphiphysin (3,4 %). Bei 18 % fanden sich keine Antikörper und bei 7 % atypische Antikörper. Die Prävalenz von Autoantikörpern gegen neuronale Oberflächenantigene (s.u.) wurde in dieser Arbeit nicht erfasst. Die neurologischen Symptome zeigen sich bei paraneoplastischen Erkrankungen typischerweise mit subakutem Zeitverlauf und deutlich vor der Tumordiagnose (65 %); zwischen Beginn der neurologischen Symptome und der Tumordiagnose können bis zu 3–4 Jahre liegen. Die Tumordiagnose selbst bei gesicherten paraneoplastischen Syndromen kann im Einzelfall schwierig sein. Die häufigsten assoziierten Tumore sind das kleinzellige Bronchial-Karzinom (SCLC, 38 %), Ovarialtumore (11 %), Mamma-Karzinome (10 %), nicht-kleinzellige Bronchial-Karzinome (8 %), Non-Hodgkin- und Hodgkin-Lymphome, Thymome und Prostata-Karzinome (jeweils 3 %), colorektale, gastro-oesophagale und testikuläre Tumore (jeweils 2 %), Nierenzell-, MerkelZell-Tumore und Neuroblastome (jeweils 1 %) [3]. Das gleichzeitige Auftreten mehrerer paraneoplastischer Erkrankungen (10 %) und mehrerer paraneoplastischer Antikörper ist möglich und kommt vor allem beim kleinzelligen Bronchialkarzinom vor. Paraneoplastische neurologische Erkrankungen sind zwar absolut gesehen selten, treten jedoch bei bestimmten Tumoren gehäuft auf. Beispielsweise tritt ein Lambert-Eaton-MyasthenesSyndrom (LEMS) bei 2–3 % aller Patienten mit kleinzelligem Bronchialkarzinom auf [4]. Tumordiagnose innerhalb von 2 Jahren gesichertes paraneoplastisches Syndrom Ausschluss von Differenzialdiagnosen Teilcharakterisierter AK Tumornachweis innerhalb von 5 Jahren kein Tumornachweis mögliches paraneoplastisches Syndrom Leypoldt F et al. Neues bei paraneoplastischen Syndromen … Akt Neurol 2012; 39: 60–73 61 62 Neues in der Neurologie Tab. 2 Klinisch relevante, paraneoplastisch auftretende Antikörper gemäß. Name (Synonym) Prävalenz* Antigen Klinisches Syndrom häufigste Tumore Gut charakterisierte, paraneoplastische Antikörper (intrazelluläre Antigene) Assoziation mit einem Tumor in > 95 % Lungenkarzinom (85 %), insb. SCLC, Anti-Hu (ANNA-1) 40 % HuD Enzephalomyelitis, limbische Enzephalitis, KleinhirndeNeuroblastom, Prostata-Karzinom, generation, Hirnstammenzephalitis, sensorische NeuMerkel-Zell-Karzinom, weitere ronopathie, sensomotorische Neuropathie, autonome Neuropathie Anti-Yo (PCA-1) 15 % cdr2, cdr62 Paraneoplastische Kleinhirndegeneration, W > M Ovarial-, Mamma-, Uteruskarzinom Anti-CV2 (CRMP5) 5 % CRMP5 Enzephalomyelitis, Polyneuropathie, Optikusneuritis, SCLC, Thymom, weitere limbische Enzephalitis, Kleinhirndegeneration, Chorea Anti-Ma1# 5 % Ma-Proteine Rhombenzephalitis, limbische Enzephalitis, Neuropathie Mammakarzinom, Lungenkarzinom Ma-Proteine Limbische Enzephalitis, Rhombenzephalitis, M > W Keimzelltumor Anti-Ta/Ma2# 5 % Anti-Ri (ANNA-2) 5 % NOVA Opsoklonus-Myoklonus-Syndrom, Rhombenzephalitis, Mammakarzinom, Ovarialkarzinom, Kleinhirndegeneration, Myelitiden SCLC Anti-Amphiphysin 5 % Amphiphysin Stiff-Person-Syndrom; limbische Enzephalitis, RhombenMammakarzinom, SCLC zephalitis, Kleinhirndegeneration, PNP Anti-Recoverin Recoverin Retinopathie Lungenkarzinom Teilcharakterisierte paraneoplastische Antikörper prädiktiver Wert bezüglich Paraneoplasie unklar Anti-Tr(PCA-Tr) 2 % MAZ-Protein Kleinhirndegeneration Hodgkinlymphom, NHL Anti-Zic4 Zic1-4 Kleinhirndegeneration SCLC Anti-SOX-1 (AGNA) SOX-1 Sensitivität 67 %, Spezifität 95 % bezüglich vorliegen eines SCLC, Bronchial-Karzinoid SCLC bei nachgewiesenem LEMS PCA-2 280 kD Enzephalitis, Lambert-Eaton myasthenes Syndrom, PNP SCLC ANNA-3 170 kD Neuropathie, Kleinhirndegeneration, limb. Enzephalitis SCLC Anti-ganglionäre AchR-AK ganglionäre Autonome Neuropathie (Sensitivität 50 %, Spezifität bei SCLC, Thymom, Lymphom, Blasen-, AchR (α3) Titer über 1 nmol/l > 90 %), nur 30 % paraneoplastisch Mamma-, Prostata-, Rektumkarzinom Fakultativ paraneoplastische Antikörper (Antikörper die mit oder ohne Tumor auftreten, neuronale Oberflächenantigene) Anti-NMDA-R NMDA-R NR1a Limbische Enzephalitis, W»M, Gedächtnisstörungen, 20-50 % Ovarial-Teratom > männl. Bewegungsstörung, Katatonie, psychiatrisch Keimzelltumor Anti-VGKC-Komplex LGI1, CASPR2 Limbische Enzephalitis, M > W, Hyponatriämie, SCLC, Thymom Neuromyotonie, Morvan Syndrom Anti-AMPA-R GluR1 Limbische Enzephalitis, W > M, Atypische Psychose, SCLC, Mammakarzinom, Thymom GluR2 häufige Rezidive Anti-GABAB-R GABAB Limbische Enzephalitis, häufig Anfälle. Koinzident oft SCLC, Thymom, neuroendokrine Tumore GAD-AK *Gemäß Prävalenz [3]. Prävalenz bezogen auf Anti-Ma und Anti-Ma2/Ta zusammen; W: Frauen; M: Männer; PNP: Polyneuropathie. # Neues zu paraneoplastischen Enzephalitiden ▼ Bei Enzephalitiden spielt mittlerweile die Autoantikörperdiagnostik eine besonders wichtige Rolle. In den letzten Jahren wurden paraneoplastische Syndrome mit Autoantikörpern gerichtet gegen neue Antigene beschrieben [5]. Im Unterschied zu den oben genannten „klassischen“ paraneoplastischen Antikörpern kommen sie auch bei nicht-paraneoplastischen Syndromen vor und richten sich gegen Zielantigene auf der Oberfläche neuronaler Zellen, meist gegen Kanal- oder Rezeptorproteine. Sie sind in der Literatur unter dem Namen ihres Zielantigens bekannt geworden: Glutamatrezeptoren: N-Methyl-D-Aspartat (NMDA)Rezeptor Antikörper und α-Amino-3-hydroxy-5-methyl-4isoxazol-Propionsäure (AMPA)-Rezeptor Antikörper, spannungsabhängige Kalium-Kanal (VGKC)-Komplex Antikörper z. B. Lgi1, Caspr2, γ-Amino-Butyrat(b) (GABA(b)) Rezeptor Antikörper. ▶ Tab. 2 wider. Eine Übersicht gibt ● Insgesamt etabliert sich in der Literatur zunehmend die Unterscheidung in klassische paraneoplastische neurologische Syndrome mit Antikörpern gegen intrazelluläre Antigene (Hu, CV2/ CRMP5, Yo, Ri etc.) und in fakultativ paraneoplastische neurologische Syndrome mit Antikörpern gegen Oberflächenantigene (engl.: neuronal surface antigen antibodies) [5]. Letztere Erkrankungen werden überbegrifflich auch als synaptische Autoimmunität (eng.: synaptic autoimmunity) bezeichnet. Im Folgenden werden beide Gruppen daher getrennt vorgestellt. Die Enzephalitiden mit ausschließlich Antikörpern gegen das intrazellulär lokalisierte Enzym Glutamatdecarboxylase (GAD) kommen vermutlich nicht paraneoplastisch vor, sie werden in dieser Übersichtsarbeit nicht berücksichtigt. Trotz dieser neuen Antikörper findet sich allerdings weiterhin bei 5–10 % autoimmuner limbischer Enzephalitiden kein derzeit nachweisbarer antineuronaler Antikörper [6]. Dieser Prozentsatz wird sich in Zukunft mit der Entdeckung neuer antikörperdefinierter Subgruppen wahrscheinlich weiter verringern. Enzephalitiden mit Antikörpern gegen intrazelluläre Antigene (klassische paraneoplastische Antikörper) ▼ Werden bei Patienten mit einem passenden klinischen Bild gut charakterisierte paraneoplastische AK nachgewiesen, sind dies am häufigsten Hu-Antikörper (50 % [7]). Bei diesen Patienten ist die Enzephalitis oft nur initial auf limbische Areale begrenzt und entwickelt sich häufig im Verlauf zu einer generalisierten Enzephalomyelitis. Ähnliches gilt für CV2/CRMP5-Antikörper, die oft auch zusammen mit Anti-Hu auftreten können. Hierbei sind assoziierte Optikusneuritiden und Chorea beschrieben [8–10]. Wichtig sind des weiteren Anti-Ma/Ta in bis zu 30 % der Fälle meist bei jungen Männern (Anti-Ta/Ma2) oder ohne Alters- und Leypoldt F et al. Neues bei paraneoplastischen Syndromen … Akt Neurol 2012; 39: 60–73 Neues in der Neurologie Tab. 3 Differenzialdiagnosen paraneoplastischer Syndrome. Klinisches Syndrom Differenzialdiagnosen Limbische Enzephalitis (LE) Infektiöse Enzephalitis, v. a. HSV, VZV, HHV6 bei Immunsuppression, Lues, CJD*, HIV/PML, WNV. Lupus erythematodes; Sjögren-Syndrom; Hashimoto-Enzephalopathie; Gliom; Wernicke Korsakoff Syndrom Infektiöse Enzephalitis, v. a. Listerien, Toxoplasmose; basale Meningitis TBC; M. Whipple Miller-Fisher/Bickerstaff; Myasthenia gravis, M. Behçet, Gliom Opsoklonus: u. a. HIV, Hepatitis C, WNV, M. Whipple, Borreliose, postinfektiös (Mycoplasmen, Salmonellen, Streptokokken), medikamentös (v. a. Lithium und Tricyclica) Myoklonien: physiologisch, hereditär, sporadisch, Epilepsie-Syndrome, Enzephalopathien, CJD*, neurodegenerative Erkrankungen, Glutensensitive Enteropathie, Lance-Adams-Syndrom post anoxisch Infektiös; parainfektiös; MS; NMO; spinale Durafistel, Syringomyelie, Tethered cord Syndrom, Spinalis-anteriorSyndrom Alkohol, Medikamentös (Lithium, Antikonvulsiva, 5-FU, Cytosin Arabinosid), Vit. B1, B12, Folsäure, Vit. E, Antikörper-assoziiert (GAD; anti-Ca#, Homer-3, mGluR1), Infektiös (VZV, EBV, Lues, M. Whipple), CJD*, meningeale Siderose, MSA-C, hereditär Vaskulär; Neuritis N. optici, LHON, Medikamentös, Tabak-Alkohol-Amblyopie Diabetes mellitus, CIDP; MGUS; Anti-MAG-Neuropathie; Sjögren-Syndrom, Zöliakie, Cryoglobulinämie, Medikamentös/Toxisch (Cisplatin, Amiodaron), Hypervitaminosis B6; Vit. B12 Mangel, HIV Diabetes mellitus; GBS; CIDP, Sjögren-Syndrom, Porphyrie, MSA-A, Mitochondriopathie Multifokale motorische Neuropathie; ALS; Bulbospinale Muskelatrophie Kennedy; Primäre Lateralsklerose Krampi; Krampus-Faszikulationssyndrom; Radiogene Plexusaffektion; Myotonie; Neuromyotonie und ZNS (Morvan Syndrom) Myotonie, Neuropathien (Areflexie), PERM Myasthenia gravis, Episodische Ataxie Typ 2, Botulismus Myopathie, z. B. durch Steroide, IBM, immunmediierte nekrotisierende Myopathien Rhombenzephalitis Opsoklonus-/Myoklonus-Syndrom Myelitis Subakute Kleinhirndegeneration (PKD) Retino-/Optikopathie Subakute sensorische Neuronopathie Autonom. Neuropath Motoneuron-Erkrank Neuromyotonie Stiff-person-Syndrom Lambert-Eaton-Syndrom Poly-/Dermatomyositis 5-FU: 5-Fluoruracil; ALS: Amyotrophe Lateralsklerose; CIDP: chronisch inflammatorische demyelinisierende Polyneuropathie; CJD: Creutzfeld-Jakob-Disease; EBV: Ebstein-Barr Virus; GBS: Guillain-Barre-Syndrom; HIV: Humanes Immundefizienzsyndrom Virus; IBM: Einschlusskörpermyositis; JC/HIV: Progressive multifokale Leukenzephalopathie bei HIV; LEMS: Lambert-Eaton-Myasthenie-Syndrom; LHON: Lebersche Hereditäre Optikusneuropathie; MAG: Myelin assoziiertes Glycoprotein; MGUS: Monoklonale Gammopathie unklarer Signifikanz ; MSA: Multisystematrophie; NMO: Neuromyelitis optica; PERM: Progressive Encephalomyelitis mit Rigidität und Myoklonien; TBC: Tuberkulose; VZV: Varizella zoster Virus; WNV: West Nile Virus; *Bei PKD Protein 14-3-3 in 14 % Falsch-Positiv; # 1. Jarius S et al.: A new Purkinje cell antibody (anti-Ca) associated with subacute cerebellar ataxia: immunological characterization. J Neuroinflammation 2010, 7:21 Geschlechterpräferenz (Anti-Ma). Bei letzteren treten zu den Symptomen der PLE gehäuft Hirnstammsymptomen hinzu [11, 12]. Selten finden sich auch Amphiphysin-AK, oft ebenfalls mit weiteren paraneoplastischen Antikörpern [13]. Die häufigsten, assoziierten Tumoren sind Lungen-Karzinome (meist kleinzellige Bronchialkarzinome = SCLC), gedacht werden muss jedoch insbesondere bei jungen Männern auch an HodenKarzinome (Ma2) und bei Frauen an Mamma-Karzinome. Auch neuroektodermale Hauttumoren (Merkel-Zell-Karzinome) sind beschrieben. Eine paraneoplastische Enzephalitis kann sich auch primär und isoliert im Hirnstamm (Rhombenzephalitis) manifestieren (5,6 % aller paraneoplastischer Syndrome [3]) und führt dann zu Blickparesen, Doppelbildern, Dysarthrie oder gar lebensbedrohlichen Bildern mit Atemdepression [14, 15]. Meist tritt eine Hirnstammenzephalitis jedoch als Teil einer generalisierten Enzephalomyelitis auf. Assoziierte AK können sein: AntiHu, Anti-Yo, Anti-Ma, Anti-CV2/CRMP5 und Anti-Ri. Enzephalitiden mit Antikörpern gegen neuronale Oberflächenantigene (synaptische Autoimmunität) – NMDA-Rezeptor Antikörper Enzephalitis ▼ Das klinische Spektrum der Enzephalitiden mit Autoantikörpern gegen neuronale Oberflächenantigene ist weit gefächert, gemeinsam ist ihnen jedoch das gute Ansprechen auf Immunsuppression und das Auftreten als idiopathische und paraneoplastische Variante [5]. Die NMDA-Rezeptor Antikörper assoziierte Enzephalitis ist häufig, 1 % der Patienten einer neurologischen, deutschen Intensivstation [16] und 4 % aller Enzephaliti- den in einer multizentrischen englischen Studie [17] waren betroffen. Diese Enzephalitiden mit IgG-Antikörpern gerichtet gegen den N-Methyl-D-Aspartat (NMDA)-Rezeptor (NR1 Untereinheit) zeigen häufig einen prototypischen Verlauf, dessen Kenntnis daher wichtig ist. Die Erkrankung betrifft insbesondere Frauen (85 %), davon 33 % jünger als 18 Jahre und Kinder ( < 14 Jahre) in 20 % [18]; Männer (15 %) und ältere Patienten beider Geschlechter können jedoch ebenso erkranken [18, 19]. Oft tritt ein Prodrom ähnlich einer Virusinfektion auf. Tage später folgen Ängstlichkeit, Stimmungsschwankungen sowie weitere psychiatrische Symptome: Psychosen, affektive Störungen, Zwangshandlungen/-gedanken etc. Meist innerhalb der ersten beiden Wochen werden diese gefolgt von Gedächtnisstörungen, epileptischen Anfällen, dissoziativen Bewusstseinsminderungen, Bewegungsstörungen (rhythmische orofaziolinguale und appendikuläre Bewegungen) [20], autonome Instabilität und häufig beatmungspflichtigen Hypoventilation. NMDA-Rezeptor Antikörper Enzephalitiden werden häufig als virale Enzephalitis, malignes neuroleptisches Syndrom oder katatoner Stupor oder Enzephalitis lethargica von Economo [21] fehldiagnostiziert. Die Häufigkeit einer paraneoplastischen Variante ist altersabhängig; ca. 60 % der Patientinnen älter als 18 Jahre, 30 % der Patientinnen jünger als 18 Jahre und nur 15 % der Patientinnen bis zum 14 Lebensjahr haben uni- oder bilaterale ovariale Teratome [18]. Nur 5 % der Männer haben meist gonadale Tumore/Teratome, selten kann ein Hodgkin-Lymphom zugrunde liegen [18, 22]. Die klinische Präsentation zwischen idiopathischen und paraneoplastischen Varianten unterscheidet sich nicht, jedoch kommt es bei idiopathischen Patienten häufiger zu Rezidiven (25 % [18, 23]). Leypoldt F et al. Neues bei paraneoplastischen Syndromen … Akt Neurol 2012; 39: 60–73 63 64 Neues in der Neurologie Tab. 4 Klinisch-neurologische Syndrome mit den jeweils sinnvollen Antikörper-Tests und häufigsten Tumoren. Syndrom und Häufigkeit* Sinnvolle Antikörper (S = Serum, L = Liquor) Paraneoplastische Genese in % Häufige assoziierte Tumore Subakute sensorische Neuronopathie 25 % Intrazelluläre Antigene (S): Hu, Amphiphysin, CRMP5/CV2, Ma/Ta Subakute Kleinhirndegeneration 25 % Intrazelluläre Antigene (S): Hu, Yo, CRMP5/CV2, Ma/ Ta, Ri, Tr, Amphiphysin, PCA-2, ANNA-3, Zic4 Oberflächen-Antigene (S): VGCC Intrazelluläre Antigene (S): Hu, Amphiphysin, CRMP5/CV2, Ma/Ta, SOX1 Intrazelluläre Antigene (S): Hu, CRMP5/CV2, Amphiphysin, Ma/Ta Sensomotorische Neuro-/radikulopathie 16 % Enzephalomyelitis und Hirnstammenzephalitis 11 % Limbische Enzephalitis 10 % Autonome Neuropathie 5 % Lambert-Eaton-Myasthenes-Syndrom 5 % Opsoklonus/Myoklonus (Erwachse) 2 % Intrazelluläre Antigene (S): Hu, Ma/Ta, CRMP5/-CV2, Ri, Amphiphysin Oberflächen-Antigene (S+L): NMDA-R, VGKC#, GABA-R, AMPA-R Intrazelluläre Antigene (S): Hu, Peripherin Oberflächen-Antigene (S): Ganglionäre AchR Intrazelluläre Antigene (S): SOX-1 Oberflächen-Antigene (S): VGCC Intrazelluläre Antigene (S): Ri, Hu, Ma/Ta Oberflächen-Antigene (S+L): NMDA-R Opsoklonus/Myoklonus (Kind) Intrazelluläre Antigene (S): vereinzelt Hu Dermatomyositis 2 % Intrazelluläre, nicht-paraneoplastische Antigene (S): Jo1, Mi2, SRP Retinopathie 1 % Intrazelluläre Antigene (S): Hu, CRMP5/CV2, Recoverin Intrazelluläre Antigene (S): Amphiphysin Intrazelluläre, nicht-paraneoplastische AK (S + L): GAD Intrazelluläre Antigene (S): Titin Oberflächen-Antigene (S): AchR, MUSK Stiff-person-Syndrom 1 % Myasthenia gravis In 20 % SCLC, Thymom, Lymphom, Mammakarzinom oder Keimzelltumor In 50 % SCLC, Mammakarzinom, Keimzelltumor (Hoden/ Ovar), Thymom oder Lymphom In 10 % SCLC, Thymom oder Lymphom In 10 % SCLC, Thymom, Mammakarzinom, Keimzelltumor (Hoden/Ovar), Merkelzellkarzinome (Haut) oder Lymphom In 20 % SCLC, Keimzelltumor (Hoden/Ovar), Thymom, Mammaka., Lymphom, Prostatakarzinom In 2–30 % SCLC, Thymom, Lymphom, Blasen-, Mamma-, Prostata- oder Rektumkarzinom In 60 % SCLC In 20 % Mammakarzinom, SCLC oder Keimzelltumor (Hoden/Ovar) In 50 % Neuroblastome In 30 % Ovarial-, Lungen-, Pankreaska. oder andere Tumore In 5 % SCLC, Thymom oder Melanom In 5 % Mammakarzinom, SCLC In 15 % Thymom *Häufigkeit bezogen auf alle paraneoplastischen Erkrankungen [3], klinische Syndrome kommen in 10 % der paraneoplastischen Fälle kombiniert vor, daher Gesamtprävalenz > 100 %. # Antikörper gegen VGKC-Komplex (u. a. LGI1, CASPR2, Contactin-2). Diagnostisch zeigen ca. 50 % MRT Auffälligkeiten und nur 25 % im T2/FLAIR gewichtetem cMRT Signalanhebungen im mesiotemporalen Kortex [18, 23]. Auch kontrastmittelaufnehmende Herde kortikal, meningeal oder in den Basalganglien sowie seltener auch spinal können auftreten [18, 24]. Der Liquor ist zu 90 % auffällig, in der Frühphase jedoch nur bei 70 %. Bei 90 % zeigt sich eine milde, lymphozytäre Pleozytose, in 30 % der Fälle Schrankenstörungen und 9–40 % oligoklonale Banden. Die Pleozytose ist häufig nur in der Frühphase nachweisbar, im Verlauf kommt es oft zum Auftreten von oligoklonalen Banden. Das EEG ist bei 80–90 % mit häufig diffuser Delta-Theta-Aktivität pathologisch verändert [18, 23]. Es findet sich bei schweren Fällen ein typischer frontookzipitaler Gradient des Glucosemetabolismus, der sich im FDG-PET als frontotemporaler Hypermetabolismus und okzipitaler Hypometabolismus darstellt [25]. In der Mehrzahl der Fälle kommt es zu einer weitgehenden Rückbildung der Symptomatik, allerdings bleiben bei ca. einem Viertel anhaltende schwere Defizite oder es kommt zu einem letalen Ausgang meist durch intensivmedizinische Komplikationen oder die autonome Dysfunktion (4 %) [18]. Enzephalitiden mit Antikörpern gegen andere neuronale Oberflächenantigene (synaptische Autoimmunität) – AMPA, GABA(b), VGKC-Komplex- Antikörper Enzephalitis ▼ Enzephalitiden mit AMPA-, GABA(b)-, VGKC-Komplex (Lgi1, ▶ Tab. 2) sind klinisch nur bedingt vonCASPR2)-Antikörpern (● einander zu unterscheiden. Allen gemeinsam ist der limbische Schwerpunkt mit psychiatrischen, mnestischen und epileptischen Symptomen, das Ansprechen auf Immunsuppression sowie idiopathische und paraneoplastische Fälle [5]. Letztlich unterscheidet erst der Antikörperbefund sicher zwischen diesen Enzephalitiden. Einige Aspekte helfen jedoch bei der Unterscheidung und sind eher typisch für eine der Unterformen. Bei AMPARezeptor Antikörper assoziierte Enzephalitiden finden sich eher Lungentumore und Thymome als zugrundeliegende Tumore [26, 27]. Lgi1-Antikörper Enzephalitiden gehen oft mit einer Hyponatriämie einher, betreffen eher ältere Männer, haben geringere Liquorauffälligkeiten und sind nur selten paraneoplastischer Genese [28]. CASPR2 Antikörper assoziierte Syndrome zeigen oft eine Neuromyotonie zusammen mit einer Enzephali- Leypoldt F et al. Neues bei paraneoplastischen Syndromen … Akt Neurol 2012; 39: 60–73 Neues in der Neurologie tis (Morvan Syndrom) [29, 30]. GABA(b) Rezeptor Enzephalitiden gehen immer mit epileptischen Anfällen oder sogar einem Status epilepticus einher, treten bei älteren Patienten mit Lungentumoren oder jüngeren Patienten idiopathisch auf [31, 32]. Enzephalitiden bei VGKC-Komplex Antikörper Nach neuen Erkenntnissen sind bei der limbischen Enzephalitis mit Antikörpern gegen spannungsabhängige Kaliumkanäle (voltage gated potassium channel; VGKC) die Antikörper gegen VGKC-assoziierte Proteine und nicht gegen die nur kleine extrazelluläre Domäne des Kanals selbst gerichtet. Daher werden diese Antikörper auch VGKC-Komplex-Antikörper oder nach dem Zielantigen Lgi1 [28] oder Caspr2 [29, 30] benannt. Es sind überwiegend ältere Männer betroffen (m:w 3:1; Mittel 60. LJ). Klinisch zeigen sich häufig epileptische Anfälle (90 %), Gedächtnisstörungen ( > 90 %) sowie eine Hyponatriämie (40–60 %). Die epileptischen Anfälle treten teils mit der ungewöhnlicher Semiologie einer faziobrachialen, paroxysmalen Dystonie auf, dies kann der eigentlichen Enzephalitis vorausgehen [33]. Seltener treten Myoklonien (40 %) und vegetative Zeichen (10 %) auf [28, 30, 34]. Im meist pathologischen EEG (70 %) zeigen sich epilepsietypische Potenziale (30 %) und auch Allgemeinveränderungen oder Herdbefunde (30 %). Im Liquor findet sich in einem kleineren Prozentsatz als bei den anderen Enzephalitiden eine Auffälligkeit. Nur bei 40 % der Patienten werden Schrankenstörungen, seltener Pleozytosen und fast nie oligoklonale Banden nachgewiesen [35]. Hilfreich sind cMRT Untersuchungen, die bei ca. 70–80 % eine T2-Hyperintensität der medialen Temporallappen aufweisen. Nur in der Minderzahl der Fälle liegt eine paraneoplastische Ursache vor (5–10 %) [28, 30]. Es handelt sich dabei um unterschiedliche Tumoren wie Lungen, Schilddrüsen, Mamma oder Nierenzellkarzinome aber auch ovariale Teratome oder Thymome sind beschrieben worden [28, 35]. Die Prognose nach erfolgter Immuntherapie ist gut (bis zu 80 % gute Erholung), jedoch sind auch schwere Residuen und letale Verläufe beschrieben. Enzephalitiden bei AMPA-Rezeptor Antikörpern Bei Patienten mit Enzephalitiden, die mit Antikörpern gegen die Glutamatrezeptoruntereinheiten 1 und 2 (GluR1/2) des α-Amino3-hydroxy-5-methyl-4-isoxazol-Propionsäure (AMPA)-Rezeptors assoziiert sind, handelt es sich in der Regel um Frauen (90 %) im mittleren Alter (38–87, median 60 Jahre), die eine akute limbische Dysfunktion entwickeln [26, 27, 36]. Diese Enzephalitiden treten meist paraneoplastisch auf (80 %), zugrunde liegend können dann ein Bronchial-Karzinome, Mamma-Karzinome oder Thymome sein. Im Liquor findet sich meist eine lymphozytäre milde Pleozytose (80 %), oligoklonale Banden sind selten. Bis zu 75 % der Patientinnen zeigen typische mesiotemporale MRTAuffälligkeiten und im EEG fanden sich bei 75 % Auffälligkeiten. Die neurologischen Symptome sprechen gut auf eine Tumorbehandlung und Immunsuppression an, zeigen aber eine Tendenz zu Rezidiven (60 %), welches wiederum gut auf Immuntherapie anspricht. Schübe sind dabei nicht sicher mit einem Tumorrezidiv assoziiert [26]. Enzephalitiden mit GABA(b) Rezeptor Antikörpern Schließlich können bei Enzephalitiden teils auch Antikörpern gerichtet gegen GABA(b)-Rezeptoren nachgewiesen werden. Es handelt sich scheinbar um die größte Subgruppe paraneoplastischer bisher seronegativer limbischer Enzephalitiden [32]. Diese zeigen insbesondere das Bild einer limbischen Dysfunktion (Verwirrtheit und Gedächtnisstörungen) und früh im Verlauf auftretenden Krampfanfällen [31]. Männer und Frauen scheinen ähnlich häufig betroffen zu sein mit einem mittleren Erkrankungsalter von 60 Jahren. Im Liquor fand sich bei 70 % eine lymphozytäre Liquorpleozytose, bei 30 % oligoklonale Banden und ebenfalls bei 30 % eine Schrankenstörung. Auch hier fanden sich bei 70 % der Patienten typische, mesiotemporale MRT Auffälligkeiten, allerdings war das initiale MRT oft negativ. Das EEG zeigte häufig (75 %) Temporallappenanfälle, temporale epileptische Erregbarkeitssteigerungen oder temporale Verlangsamungen. Bei der Hälfte der Patienten konnte im Verlauf ein kleinzelliges Bronchialkarzinom oder ein neuroendokriner Tumor der Lunge gefunden werden, die andere Hälfte der Patienten war jünger und ohne zugrundeliegenden Tumor. Die Hälfte der Patienten mit GABA(b)-Rezeptor Antikörpern hatten zusätzliche Autoantikörper im Serum (TPO, GAD65, SOX1 oder N-Typ spannungsabhängige Kalziumkanäle). Neues zur paraneoplastischen Kleinhirndegeneration ▼ Bei der paraneoplastischen Kleinhirndegeneration (PKD) handelt es sich um das häufigste paraneoplastische, neurologische Syndrom mit Antikörpern gegen intrazelluläre Antigene (24 % [3]). Meist entsteht subakut über Tage bis wenige Wochen ein schwerstes zerebelläres Syndrom mit ausgeprägter Beteiligung der Arme, Verlust des Gehvermögens, Dysarthrie, sakkadierter Blickfolge und Nystagmus (meist Downbeat-Nystagmus). Gelegentlich besteht eine heterogene Prodomalsymptomatik ähnlich einem grippalen Infekt. Oft stabilisiert sich die Erkrankung nach mehreren Monaten auf einem sehr schlechten, zerebellären Funktionsniveau (90 % Rollstuhlabhängig). Das Fehlen einer Atrophie im MRT ist in frühen Stadien die Regel und gut mit einer PKD vereinbar. Späte Bildgebung kann dann eine Kleinhirnatrophie darstellen. In Einzelfällen kann ein FDG-Hirnstoffwechsel-PET bereits früh zerebelläre Hypermetabolismen in frühen Stadien einer PKD nachweisen [37]. Ein klassisches Überlappungssyndrom besteht zwischen einer PKD und einem LEMS bei Patienten mit einem SCLC [38]. Eine neue Beobachtung ist, dass in einer großen Fallserie im Gegensatz zu früheren Fallserien [39, 40], bei bis zu 23 % der Patientinnen initial eine extrazerebelläre Symptomatik (Hirnstamm, Hirnnerven) besteht und bei 10 % sogar Neuropathien parallel oder zeitlich vorausgehend nachgewiesen werden können [41]. Allerdings bestätigt auch diese Fallserie, dass die funktionelle Erholung bei PKD unabhängig vom assoziierten Antikörper auch bei Tumorbehandlung und/oder Immunsuppression gering ist. Neues zum Opsoklonus-Myoklonus-Syndrom ▼ Das seltene paraneoplastische Opsoklonus-Myoklonus-Syndrom (POMS) (2,3 %) wird durch chaotische, konjugierte, hochamplitudige, unwillkürliche Sakkaden und meist generalisierte Myoklonien charakterisiert (eng. Dancing eyes – Dancing feet). Die Erkrankung entsteht u. a. durch eine Funktionsstörung im Nucleus fastigii des Kleinhirns. Es kommt in 20 % der Fälle paraneoplastisch vor, die übrigen sind meist postinfektiös. Für die paraneoplastische Variante sprechen ein Alter über 50 Jahren und das Vorliegen einer Enzephalopathie. Beim Erwachsenen finden sich assoziierte Tumore meist im Bereich der Lunge oder der Mamma [42]. Bei Kindern sind in 50 % Neuroblastome ur- Leypoldt F et al. Neues bei paraneoplastischen Syndromen … Akt Neurol 2012; 39: 60–73 65 66 Neues in der Neurologie sächlich. Anti-Hu, Anti-Ri oder Anti-Ma2 sowie viele weitere, nicht charakterisierte AK können assoziiert sein [42, 43]. Neu ist, dass in zwei Einzelfallberichten ein Opsoklonus-Myoklonus Syndrom bei nachgewiesenen NMDA-Rezeptor Antikörpern beschrieben wurde [44, 45]. kann mit einem Karzinom (KAR) oder einem Melanom (Melanom assoziierte Retinopathie, MAR) assoziiert sein. Neu und bemerkenswert, allerdings bisher nur kasuistisch beschrieben, ist das paraneoplastische Syndrom einer essentiellen Hypernatriämie, welches mit Antikörpern gegen spezielle Natriumkanäle in den circumventrikulären Organen und einem Ganglioneurom mit ektoper Expression dieser Kanäle einherging [52]. Neues zum Stiff-Person-Syndrom ▼ Progredienter axialer und appendikulärer Rigor mit lumbaler Hyperlordose und paroxysmale, schmerzhafte einschießende Spasmen der Extremitäten getriggert durch Willkürbewegungen, Aufregung oder taktile/auditorische Stimuli werden als Stiff-Person-Syndrom (SPS) bezeichnet. Das Syndrom kommt idiopathisch und paraneoplastisch vor. Frauen sind häufiger als Männer betroffen (7:3) und es existieren verschiedene Varianten, sogenannte SPS-Plus Syndrome z. B. Stiff-Limb-Syndrom und PERM (Progrediente Enzephalomyelitis mit Rigor und Myoklonien). Antikörper gegen Glutamatdecarboxylase (GAD) und nach neueren Erkenntnissen auch Glycin Rezeptoren sind serologische Marker eines idiopathischen SMS (50–90 %) [46, 47]. In seiner paraneoplastischen Form (5 % der SPS, 0,6 % der paraneoplastischen Syndrome) können die Patienten Antikörper gegen Amphiphysin (mit oder ohne Anti-GAD) zeigen, dies ist dann ein Marker für ein zugrunde liegendes Mamma-Karzinom, seltener ein SCLC oder ein Thymom [13]. Es existieren jedoch weiterhin keine verlässlichen Marker für eine Unterscheidung zwischen idiopathisch und paraneoplastisch. Seltenere paraneoplastische Syndrome des zentralen Nervensystems ▼ Sehr selten – nur in 0,3 % aller paraneoplastischen neurologischen Syndrome – manifestiert sich die paraneoplastische Symptomatik als isolierte Myelitis. Bei paraneoplastischen Myelitiden ist dann meist die graue Substanz unter Beteiligung der Vorderhörner langstreckig betroffen. Bei dieser nekrotisierenden Myelopathie gibt es keine spezifischen assoziierten Antikörper und keine streng assoziierten Tumoren. Sie kommen auch idiopathisch mit besserer Prognose vor [48]. Kasuistisch wurden neuerdings auch NMO-AK positive Myelitiden in Zusammenhang mit Tumoren nachgewiesen. Obwohl selten, handelt es sich damit bei der Neuromyelitis optica mit Aquaporin4-Antikörpern um eine fakultativ paraneoplastische Erkrankung [49]. Extrapyramidal-motorische Syndrome sind ebenfalls nur selten Ausdruck eines paraneoplastischen Syndroms (1,5 %) mit Antikörpern gegen intrazelluläre Antigene [3]. Jedoch finden sich diese gehäuft bei idiopathischen oder paraneoplastischen Syndromen mit Antikörpern gegen Oberflächenantigene (s. o.). Eine generalisierte Chorea, fokale und segmentale Dystonien und Hemiballismus können die Erstmanifestation einer paraneoplastischen Enzephalitis sein. Bei Nachweis von CV2/CRMP5-AK zeigt sich in bis zu 70 % der Fälle initial eine meist asymmetrische oder einseitige Chorea, im Verlauf entwickelt sich bei 90 % der Patienten jedoch eine Beteiligung weiterer Systeme [50]. Auch subakut progrediente progressive supranukleäre Paralyse-ähnliche Erkrankungsbilder sind paraneoplastisch bei Lymphomen beschrieben (Anti-Ma/Ma2 [51]). Während etwa jeder zehnte Tumorpatient über Visusstörungen klagt, ist die paraneoplastische Retinopathie relativ selten. Sie Neues zu paraneoplastischen Neuropathien ▼ Bei über 40 % der gesicherten paraneoplastischen Syndrome tritt eine Affektion des peripheren Nervensystems auf [3]. Grundsätzlich können paraneoplastische Syndrome am peripheren Nervensystem sehr heterogen sein und jedes Syndrom verursachen. Insbesondere sind proximale und distale Leitungsverzögerungen ähnlich einer akuten oder chronisch inflammatorischen demyelinisierenden Polyradikulopathie bei NonHodgkin Lymphomen [3, 53, 54] oder auch Mononeuritis multiplex-Syndrome beschrieben [55, 56]. Die sensible Neuronopathie (SN, „Hinterstrang-Ganglionitis“) stellt allerdings die häufigste paraneoplastische Manifestation am peripheren Nervensystem dar (25 % [3]). Hierbei kommt es zu asymmetrischen, meist schmerzhaften Hyp- und Parästhesien der Arme, Beine, des Rumpfes und des Gesichtes mit schwerer Lageempfindungsstörung, einer sensiblen Ataxie und pseudochoreoathetotischen Bewegungen der ausgestreckten Arme und Beine. Es wurden kürzlich diagnostische Kriterien publiziert [57]. Sie ist meist mit Anti-Hu, Anti-CV2/CRMP5 oder Anti-Amphiphysin assoziiert [58, 59]. Ebenfalls hochverdächtig auf eine paraneoplastische Genese ist eine ausgeprägte autonome Neuropathie (AN, 5,2 % der paraneoplastischen Syndrome [3]) isoliert oder zusammen mit einer SN. Diese kann zu chronischer, intestinaler Pseudoobstruktion, orthostatischen Dysregulation oder kardialen Arhythmien, Mundtrockenheit, erektilen Dysfunktion oder Dyshidrose [60] führen. Meist findet sich auch hier ein Anti-Hu-AK seltener ein Anti-CV2/CRMP5 oder Anti-Amphiphysin-AK. Mittlerweile ist gezeigt, dass bei einem kleinen Teil dieser Patienten mit einer Pandysautonomie hoch-titrige Antikörper gegen ganglionäre nikotinerge Acetylcholin-Rezeptoren (α3-AchR) nachgewiesen werden können. Sie treten paraneoplastisch und idiopathisch auf [61]. Ein weiterer neuer Autoantikörper assoziiert mit diesem Syndrom ist gegen Peripherin gerichtet, bei diesen Patienten finden sich oft begleitende Endokrinopathien [62]. Häufig kommen AN auch zusammen mit einem LEMS vor. Assoziierte Tumoren sind die üblicherweise mit Anti-Hu und CV2/CRMP5 auftretenden Tumortypen. AN haben eine schlechte Prognose [3]. Isolierte, chronisch progrediente Motoneuronsyndrome sind in den allermeisten Fällen neurodegenerativer Genese [63]. Insbesondere sollte an eine paraneoplastische Genese gedacht werden bei (1) rasch progredientem Verlauf mit (2) einem isolierten zweiten Motoneuron Syndrom und (3) sensibler Beteiligung sowie (4) systemischen oder enzephalopathischen Symptomen [64]. Ursächlich sind dann vor allem Non-Hodgkin-Lymphome [65]. In diesem Fall werden im Verlauf jedoch meist auch andere Gebiete des ZNS und PNS betroffen und der Liquor zeigt häufig entzündliche Veränderungen. Spezifische Antikörper sind nicht bekannt, gelegentlich findet sich ein Anti-Hu AK. Bei der Neuromyotonie (NM, Isaac-Syndrom) bestehen zu unterschiedlichen Graden Muskelkrämpfe, Muskelzuckungen/-wogen, Steifigkeit und Schwäche der Extremitäten und des Rump- Leypoldt F et al. Neues bei paraneoplastischen Syndromen … Akt Neurol 2012; 39: 60–73 Neues in der Neurologie fes mit oder ohne autonome Dysfunktion. Elektrophysiologisch werden repetitive Entladungen von Motoneuronen mit hoher Intraburst-Frequenz nachgewiesen. Neben hereditären Formen sind die meisten Erkrankungen autoimmuner Ursache. 60 % haben Autoantikörper gegen den VGKC-Komplex (insbesondere CASPR2) und 25 % der antikörperpositiven Patienten eine paraneoplastische Ursache, meist Thymome, seltener Bronchial-Karzinome oder Lymphome. Zwar sind diese Antikörper SerumMarker einer NM, jedoch nicht für deren paraneoplastische Ätiologie [29, 66, 67]. Hinweisend auf eine paraneoplastische Variante einer NM sind höheres Alter, autonome Störungen und sensible Beteiligung. Das Zusammentreffen mit zentralnervösen Syndromen und autonomen Beschwerden wird als Morvan-Syndrom bezeichnet [66]. Bei einem Lambert-Eaton-Myasthenie-Syndrom (LEMS) ist der präsynaptische Teil der neuromuskulären Endplatte betroffen. Meist kommt es zu langsam progredienten Hüft- und Schultergürtelschwächen mit Verminderung der Muskeleigenreflexe. Die externen Augenmuskeln sind nur gering und nicht bereits initial betroffen. Oft kommt es zu autonomen Störungen. Etwa 3 % aller Patienten mit einem SCLC haben klinisch und elektrophysiologisch ein LEMS [68] und 60 % der Patienten mit einem LEMS haben einen assoziierten Tumor, meist ein SCLC oder Lymphom. Neues zur Pathophysiologie ▼ Paraneoplastische, neurologische Erkrankungen gestatten einen faszinierenden Einblick in die natürliche immunvermittelte Tumorabwehr des Menschen. Es kommt zu einer „ektopen“ Expression von Antigenen im Tumorgewebe, die ansonsten nur durch Nervenzellen exprimiert werden. Dies sind die sogenannten onkoneuralen Antigene. Im Rahmen einer „guten“ Immunreaktion gegen den Tumor entsteht dann eine „falsche“ Autoimmunantwort gegen das Nervensystem. Dass eine Immunreaktion gegen Tumoren nicht selten und auch pathologisch relevant ist, zeigen die hohen Inzidenzen von Anti-Hu Antikörpern bei Patienten mit kleinzelligen Bronchialkarzinomen ohne neurologische Symptomatik (bis zu 29 % [69]) und der Überlebensvorteil von Patienten mit diesen Antikörpern [70, 71]. Warum jedoch einzelne Patienten mit einem SCLC ein manifestes paraneoplastisches Syndrom mit hochtitrigen Anti-Hu Antikörpern entwickelt, während andere nur niedrige oder gar keine Anti-Hu Antikörper ohne neurologische Symptomatik entwickeln bleibt derzeit nur ungenügend erklärt. Interessant sind neueste Arbeiten, die zeigen, dass kleinzellige Bronchialkarzinome der Ratte – genau wie beim Menschen – konstitutiv HuD-Antigene exprimieren und sich spontan Anti-Hu Antikörper bei 16 % der Tiere bilden. Allerdings kommt es auch hier nicht zu neurologischen Symptomen [72]. Eine mögliche Erklärung könnte analog zu anderen Autoimmunerkrankungen in den MHC Haplotypen liegen. Bei Patienten mit einem paraneoplastischen Syndrom mit AntiHu Antikörpern und SCLC findet sich gehäuft HLA-DQ2 und HLA-DR3 [73, 74]. Vermutlich sind die paraneoplastischen Antikörper bei paraneoplastischen neurologischen Syndromen mit Autoantikörpern gerichtet gegen intrazelluläre Antigene nur ein (für die Diagnose hilfreiches) „Epiphänomen“ und zeigen die letztlich primär zytotoxische, zellvermittelte lymphozytäre Immunreaktion gegen den Tumor (und damit auch gegen das Nervensystem) an. Hierfür spricht, dass die meisten paraneoplastischen Antigene intra- zellulär und damit außerhalb der Reichweite von Antikörpern liegen. Passend dazu konnte bisher nur für Anti-Recoverin und Anti-Amphiphysin eine direkte pathogene Rolle in vitro und in vivo gezeigt werden. Ob die anderen paraneoplastischen Antikörper gegen intrazelluläre Antigene (Anti-Hu, -Yo, -CV2/ CRMP5, -Ma2 usw.) tatsächlich auch pathogen sind, bleibt weiterhin umstritten, jedoch sprechen die meisten Befunde dagegen [75–77]. Alle Studien fanden tatsächlich hohe AntikörperTiter im Serum, jedoch keine klinische oder subklinische Enzephalitis [78, 79]. Im Gegensatz dazu gibt es inzwischen mehrere Hinweise darauf, dass es zytotoxische T-Zellen sind, die primär für die Schädigung der Neurone verantwortlich sind [80–82]. Ein Transfer von TZellen, die spezifisch für onkoneurale Proteine sind, führt tatsächlich im Tiermodell zu einer Enzephalitis [83]. Tierexperimentell sind autoimmune zytotoxische T-Zellen gegen das HuD Antigen im T-Zell-Repertoire gesunder Mäuse vorhanden aber üblicherweise über Toleranzmechanismen supprimiert [84]. Diese Toleranz bricht im Rahmen einer Tumorantwort gegen HuD positive Tumore vermutlich weg. Beim Menschen finden sich bei Patienten mit paraneoplastischen Syndromen quantitative und funktionelle Veränderungen an T-Zell Subtypen [85], sowie typische und atypische zytotoxische T-Zellen, die gegen HuD Antigene gerichtet sind [86]. Parallel sind regulatorische T-Zellen supprimiert [87]. Letztlich steht der endgültige, wenn auch schwer zu führende – Beweis für eine primär T-Zell vermittelte Immunreaktion als zugrunde liegendes Agens von paraneoplastischen neurologischen Syndromen mit Antikörpern gegen intrazelluläre Antigene derzeit noch aus, jedoch mehren sich die Hinweise. Pathophysiologie der Enzephalitiden mit neuronalen Oberflächenantigen-Antikörpern ▼ Anders stellt sich die Situation bei paraneoplastischen Syndromen mit Autoantikörpern gegen neuronale Oberflächenantigene dar. Viele Hinweise unterstützen die pathophysiologische Bedeutung der Autoantikörper bei dieser Erkrankung. Reversible Rezeptor-Internalisierung und damit Inaktivierung als Reaktion auf Antikörper-vermittelte Vernetzung der NMDA-Rezeptoren wurde kürzlich als Erkrankungsmechanismus identifiziert [88]. Passenderweise zeigen die wenigen vorhandenen autoptischen Untersuchungen keinen neuronalen Untergang, KomplementAblagerung oder entzündliche Infiltrate [88, 89]. Dies spricht gegen eine zellulär vermittelte Immunreaktion und stellt aufgrund des Fehlens struktureller, neuronaler Schädigung vermutlich die Basis des guten Ansprechens auf eine Immunsuppression dar. Dass eine intrathekale Synthese der Antikörper eine größere Rolle spielt als die systemische Produktion ist hochwahrscheinlich. So kann bei den meisten Patienten sowohl eine erhöhte intrathekale Gesamt-IgG als auch NMDA-Rezeptor-AK-spezifische IgG-Konzentration nachgewiesen werden. Auch der häufige Nachweis oligoklonaler Banden und die kürzliche, bioptische Demonstration vieler meningealer und perivaskulärer Plasmazellen beim Menschen weist in diese Richtung [23, 90]. Bei paraneoplastischer Genese ist der Auslöser vermutlich ähnlich wie oben geschildert die ektope Expression von eigentlich neuronal restringierten Rezeptoren auf der Oberfläche von Tumorzellen [31, 32, 89, 91]. Das auslösende Agens im Falle der idiopathischen Fälle ist unbekannt. Allerdings beschreiben viele Leypoldt F et al. Neues bei paraneoplastischen Syndromen … Akt Neurol 2012; 39: 60–73 67 68 Neues in der Neurologie Patienten eine prodromale Erkrankung mehrere Tage vor Ausbruch der Enzephalitis. Diese möglicherweise virale Infektion könnte eine Rolle bei der Entstehung der autoreaktiven B-Zellen spielen [23]. Auch Fälle einer post-vakzinösen NMDA-Rezeptor Enzephalitis sind beschrieben [18, 92]. Die endgültige Entschlüsselung des Entstehungsmechanismus der idiopathischen Variante dieser Erkrankung steht jedoch noch aus. Neues zur Diagnostik ▼ Aufgrund der Vielfalt der bei paraneoplastischen Syndromen betroffenen Systeme richtet sich die Diagnostik naturgemäß nach dem klinischen Syndrom. Notwendig ist immer eine umfassen▶ Tab. 3). Bei paraneoplastischen Enzede Differenzialdiagnose (● phalomyelitiden existieren zumeist keine spezifischen Auffälligkeiten in der Bildgebung. Zum Ausschluss von Differenzialdiagnosen sind allerdings insbesondere MRT Darstellung des Gehirns und Rückenmarks wichtig. Ein unauffälliges cMRT schließt ein paraneoplastisches Syndrom nicht aus, bei einzelnen Syndromen (z. B. Opsoklonus-Myoklonus Syndrom oder Kleinhirndegeneration) ist das MRT initial immer unauffällig und ein auffälliger MRT-Befund spricht eher gegen die Diagnose. Die Liquoruntersuchung ist sensitiver, jedoch ebenso unspezifisch. Hier helfen neuere Daten, die Häufigkeit und Art von Liquorauffälligkeiten bei paraneoplastischen Syndromen besser einzuschätzen [93]. Der Liquor ist meist entzündlich verändert, häufig bestehen geringe lymphozytäre Pleozytosen und/oder oligoklonale Banden, gelegentlich auch nur Schrankenstörungen. Bei 90 % der paraneoplastischen Erkrankungen ist zumindest einer dieser 3 Parameter auffällig. Die lymphozytäre Pleozytose nimmt mit zunehmender Dauer der Erkrankung ab (50 % während der ersten 3 Monate, 30 % danach), während die Inzidenz der Schrankenstörung zunimmt. Bei 7 % ist der Liquor allerdings komplett unauffällig [93]. Die weiteren Untersuchungen hängen vom betroffenen System ab. Das EEG ist bei limbischen Enzephalitiden und Enzephalomyelitiden meist auffällig, die visuell evozierten Potenziale bei Optikusneuritiden, bei peripheren Syndromen ist eine Elektroneuro- und myografie wichtig. Bei Retinopathien können ein Elektroretinogramm und eine optische Kohärenztomografie (OCT) helfen. Eine Schlüsselstellung bei der Differenzialdiagnostik und mittlerweile auch Differenzialtherapie von paraneoplastischen Syndromen und Enzephalitiden nimmt die Autoantikörperdiagnostik ein. Bei 80 % der Patienten mit gesichertem PNS lassen sich hochspezifische Antikörper (AK) nachweisen [3]. In den meisten Fällen genügt die Einsendung von Serum, bei limbischen Enzephalitiden und bei Stiff-Person-Syndrom sollte ein Liquor und Serum-Pärchen eingesandt werden. Der Versand von Liquor und abzentrifugiertem Serum kann bei Raumtemperatur, mehrtägige Lagerung bei 4 °C erfolgen. Die Bestimmung der paraneoplastischen Antikörper soll in einem in der Diagnostik erfahrenen Labor erfolgen und auf zwei unabhängigen Labormethoden (Immunoblot bzw. Line-Assay und Immunhistochemie für paraneoplastische Antikörper, Immunfluoreszenz/-histochemie und zell-basierte Assays für Oberflächen-AK) beruhen. Aufgrund symptomatischer Überlappungen der verschiedenen Krankheitsentitäten ist es sinnvoll, alle diese Parameter bei jedem Patienten parallel zu bestimmen (Autoantikörper-Profile) [94]. Während der Nachweis eines gut-charakterisierten Antikörpers gegen ein intrazelluläres Antigen immer eine zugrundeliegende Paraneoplasie anzeigt, ist dieser Beweis für teil-charakterisierte Antikörper nicht endgültig erbracht. In den letzten Jahren ist allerdings insbesondere der Nachweis von Anti-Tr oder seltener ▶ Tab. 2) bei subakuten Kleinhirndegeneratioauch Anti-Zic4 (● nen als deutlicher Hinweis auf einen zugrundeliegenden Tumor (Anti-Tr Non-Hodgkin Lymphom, Anti-Zic4 SCLC) wahrgenommen worden [65]. Auch der Nachweis von Anti-SOX-1 wird mittlerweile als starker Indikator eines zugrundeliegenden Tumor, meist eines SCLC, angesehen [95, 96], allerdings ist damit kein typischen klinisches Syndrom assoziiert. Bei fakultativ paraneoplastischen Erkrankung (LEMS, autonome Neuropathien usw.) kann dieser Antikörper hilfreich sein. Im positiven Fall ist ein paraneoplastisches LEMS durch ein SCLC sehr wahrscheinlich. Ein unauffälliger SOX-1 Antikörper schließt allerdings eine paraneoplastische Genese nicht aus. Unbedingt beachtet werden muss, dass paraneoplastische onkoneurale Antikörper auch bei Patienten mit Tumoren ohne PNS – meist in niedrigerer Konzentration – nachgewiesen werden können, beispielsweise bei 29 % der Patienten mit einem kleinzelligen Bronchialkarzinom [69]. In diesem Fall liegt kein paraneoplastisches Syndrom, sondern lediglich ein immunologisches Epiphänomen unklarer Signifikanz vor. Der Tumorsuche kommt bei gesicherten oder möglichen paraneoplastischen Syndromen oder fakultative paraneoplastischen Syndromen ein hoher Stellenwert zu. Hier sollte ein abgestuftes ▶ Tab. 5). In Abhängigkeit der Vorgehen durchgeführt werden (● aufgrund des klinischen Syndroms und insbesondere des nachgewiesenen Autoantikörpers vermuteten Tumorlokalisation sollte die Wahl der geeigneten Bildgebung erfolgen. Auch Risikofaktoren wie Rauchen, Familienanamnese sollten in diese Überlegungen mit einfließen. Bei unauffälliger Primärdiagnostik schließt sich dann die Sekundärdiagnostik an. Da die Immunantwort gegen den Tumor offensichtlich biologisch sehr effektiv ist, können die Tumore für lange Zeit histologisch klein und damit bildmorphologisch nur schwer zu diagnostizieren bleiben. In einem direkten Vergleich bei der Tumordiagnose von Antikörperpositiven Patienten war das FDG-PET signifikant dem CT überlegen, hat aber auch seine Grenzen. Die FDG-PET oder FDG-PET/CT bietet bei unauffälliger Primärdiagnostik eine zusätzliche Sensitivität (20–40 %) bei hoher Spezifität (85 %) [97], dies gilt nicht für Mamma-Karzinome, nicht-metastasierte Hauttumore oder differenzierte Teratome. Bei letzteren Tumoren kommen andere ▶ Tab. 5). Auch bei AK-negativen PatienVerfahren zum Einsatz (● ten mit wahrscheinlichen, paraneoplastischen Syndromen ist die Verwendung eines Ganzkörper-FDG-PET als hochsensitive Suchmethode für okkulte Tumore eine wertvolle Hilfe in der Differenzialdiagnose [97]. Eine rasche bioptische Sicherung ist in der Situation eines suspekten FDG-PET Befundes unbedingt notwendig. Erfahrungsgemäß können Biopsien von FDG-anreichernden, suspekten Läsionen oft nur floride Entzündung ohne Malignitätszeichen zeigen. Im entsprechenden Kontext eines gesicherten paraneoplastischen neurologischen Syndroms sollte dies nicht davon abhalten, eine erneute bioptische Sicherung anzustreben. Bei fehlendem Tumornachweis sollte basierend auf den europäischen Leitlinien [98] initial nach 3–6 Monaten für mindestens 4 ▶ Tab. 5) Jahre eine Tumorsuche mit den geeigneten Verfahren (● durchgeführt werden. Diese Empfehlungen basieren auf Expertenmeinungen. Eine Studie zeigt, dass beim LEMS eine 2-jährige Nachbeobachtungszeit ausreicht [99]. Wie lange und in welchem Intervall eine Nachbeobachtung Teratom-negativer Patientinnen mit einer NMDA Rezeptor Antikörper Enzephalitis Leypoldt F et al. Neues bei paraneoplastischen Syndromen … Akt Neurol 2012; 39: 60–73 Neues in der Neurologie Tab. 5 Abgestufte Tumordiagnostik in Abhängigkeit von der vermuteten Lokalisation für ausgewählte Tumore. Sensitivität in Klammern soweit bekannt. Verweis auf existierende bei der AWMF registrierte S2- und S3-Leitlinien. EB-US: Endobronchialer Ultraschall. Tumor Diagnostik Siehe auch separate Leitlinien – AWMF Registernummer Primäre Sekundäre Tertiäre CT-Thorax (80–85 %) MRT Thorax CT-Thorax (75–90 %), MRT Thorax Mammografie (80 %), Ultraschall FDG-PET oder FDPPET/CT Bronchoskopie/EB-US und ggfs. Feinnadelpunktion Ggfs. Mediastinoskopie CT-Becken/Abdomen Ovarial-Teratom Transvag. Ultraschall (69–90 %) + CA-125 Transvag. Ultraschall (69–90 %) Testis-Tumore Ultraschall (72 %) + β-HCG, AFP CT-Becken/Abdomen (76 %), MRT-Abdomen Lymphome CT-Thorax/Abdomen Ultraschall dermatologische Untersuchung Ggfs. Biopsie FDG-PET oder FDGPET/CT Bronchial-Karzinom Thymom Mamma-Karzinom Ovarial-Karzinom Hauttumore (MerkelZell-Karzinom) FDG-PET oder FDPPET/CT Mamma-MRT MRT (93–98 %) erfolgen sollte ist unklar, wir untersuchen alle 6 Monate für 3 Jahre. Neues zur Behandlung ▼ Sicherlich trägt die Beschreibung therapie-responsiver Subgruppen der paraneoplastischen Enzephalitiden zu einem gewissen Umdenken bei. Therapeutischer Nihilismus aufgrund „schlechter Prognose“ ist bei paraneoplastischen neurologischen Syndromen nicht grundsätzlich gerechtfertigt. Einzelne klinische Syndrome sind besser reversibel als allgemein angenommen (z. B. Enzephalitiden). Die suffiziente Tumorbehandlung ist mit einer Besserung oder Stabilisierung der Erkrankung selbst bei Anti-Hu assoziierten Enzephalomyelitiden korreliert [59]. Zwar sind einzelne Syndrome funktionell nur selten reversibel (z. B. Kleinhirndegeneration bei Anti-Yo in nur 15 % der Fälle) [41], allerdings kann auch hier nicht vorhergesagt werden, welcher Patient doch anspricht. Die Therapie paraneoplastischer Syndrome teilt sich nach wie vor ein in (1) die Tumorbehandlung (2) Immunsuppression bei fehlendem Tumornachweis und (3) symptomatische Therapien. Tumortherapie Die therapeutische Wirksamkeit einer Tumorbehandlung lässt sich pathogenetisch mit einer Entfernung der onkoneuralen Antigenquelle erklären. Die Evidenz der klinischen Wirksamkeit einer suffizienten Tumorbehandlung leitet sich für die klassischen paraneoplastischen neurologischen Syndrome mit Antikörpern gegen intrazelluläre Antigene aus großen retrospektiven Fallserien (u. a. [59]) oder großen Registerdaten [100] ab. Eine Tumorbehandlung ist mit einer Odds Ratio von 4,56 (95 % Konfidenzintervall 1,62–12,86) mit einer Stabilisierung oder Besserung einer paraneoplastischen Enzephalomyelitis mit anti-Hu korreliert [59]. Bei den Enzephalitiden mit Oberflächenantigen-Antikörpern ist für die NMDA-Rezeptor Antikörper in einer sehr großen Fallserie gezeigt, dass die rasche Resektion eines FDG-PET oder FDG-PET/CT CT-Thorax (extrapelvische Teratome) ggf. FDG-PET oder FDG-PET/CT (maligne Teratome) S3 - Prävention, Diagnostik, Therapie des Lungenkar-zinoms 020 - 007 S3 - Diagnostik, Therapie des Mammakarzinoms der Frau - 032045OL S2K - Diagnostik und Therapie maligner Ovarialtumoren - 032-035 Teratoms (innerhalb von 4 Monaten) mit verminderten Rezidiven und einem günstigeren Verlauf einhergeht [18, 23]. Die Tumortherapie sollte eng mit den Onkologen abgestimmt werden, derzeit gibt es keine abweichenden Empfehlungen in der Tumorbehandlung onkologischer Patienten mit oder ohne paraneoplastische Syndrome. Welcher Art die Tumorbehandlung (Operation, Chemotherapie, Radiatio) ist, hängt primär vom Tumortyp und nicht vom paraneoplastischen Syndrom ab. Immuntherapie Bei paraneoplastischen Syndromen mit Antikörpern gegen intrazelluläre Antigene ist die Wirksamkeit einer Immunsuppression nicht evidenzbasiert ist, aber insbesondere bei fehlendem Nachweis eines Tumors empfohlen. Ob eine Immunsuppression das Tumorwachstum beschleunigt, ist nicht abschließend erforscht. Hierüber sollte eine Aufklärung erfolgen. Bezüglich der Art der Immunmodulation gibt es keine Daten aus kontrollierten Studien, sie beruhen auf retrospektiven Fallserien [59, 101] und kleinen unkontrollierten Studien [102, 103]. Beim LEMS kann eine Behandlung mit ivIG versucht werden [104]. Bei der idiopathischen Form des Stiff-Person-Syndroms kann eine Behandlung mit ivIG wirksam sein, dies ist für die paraneoplastische Form nicht bewiesen [105]. Letztlich sind aus pathophysiologischen Überlegungen monoklonale Antikörper bei paraneoplastischen Syndromen sinnvolle Therapiealternativen. Etabliert hat sich hier insbesondere Rituximab bei kindlichen Opsoklonus-Myoklonus-Syndromen ohne Neuroblastom und bei NMDA-Rezeptor Antikörper Enzephalitiden. Zu anderen therapeutischen Antikörpern sind mit Ausnahme von Alemtuzumab bei einer Karzinom-assoziierten Retinopathie [106] jedoch bisher keine Fälle bekannt. Immuntherapien und Dosierungen sowie Dosisintervalle sind ▶ Tab. 6 zusammengefasst. Sensorische Neuronopathien, in ● sensomotorische Neuropathien und autonome Neuropathien sowie zentralnervöse PNS werden pragmatisch mit Steroidstößen (5 × 1 g Methylprednisolon i. v.) ggf. mit oraler Fortsetzung (1mg/kg KG Prednisolonäquivalent) und individuellem Aus- Leypoldt F et al. Neues bei paraneoplastischen Syndromen … Akt Neurol 2012; 39: 60–73 69 70 Neues in der Neurologie Tab. 6 Übersicht, Dosierungen und Intervalle immunsuppressiver Substanzen und Verfahren. Substanz/Verfahren Dosierungen Laboruntersuchungen, Vorsorge Wichtige unerwünschte Wirkungen Methylprednisolon [101] 1 000–2 000 mg/d oder 20–40 mg/kg KG/d i. v. für 3–5 Tage Protonenpumpenhemmer (Magenschutz); ggfs.Thromboseprophylaxe Prednisolon (z. B. Decortin H®) initial 1 mg/kg/d; langsame, individuelle Dosisreduktion bei Ansprechen. Längerfristige Behandlung mit 0,1 mg/ kg KG/d Vitamin D, Kalzium; jährliche Knochendichte, ggfs. Bisphosphonate; Protonenpumpenhemmer intravenöse Immunglobuline [103, 108] 2 g/kg Induktion 3–5 Tage, Erhaltung 0,5–1 g/kg 1–3 Tage alle 4–5 Wochen; keine bekannten, präparatespezifischen Unterschiede 5 Zyklen Plasmaseparation gegen Albumin oder Frischplasma vor Erstgabe Ausschluss IgA Mangel Schlafstörungen, Stimmungswandel, Sehstörungen, Dyspepsie, Bluthochdruck,Blutzuckerentgleisung Kurzfristig: siehe Methylprednisolon Längerfristig: Magenulzeration, Osteoporose, Myopathie, Flüssigkeitsretention, Gewichtszunahme, Diabetes mellitus, Bluthochdruck, Infektionsrisiko, Katharakt, Hautveränderungen allergische Reaktionen, Kopfschmerzen, sterile Meningitis, vorübergehende Nierenfunktionsstörung Monitoring, CRP, Leukozyten, Fibrinogen, kein Fieber, Compliance Katheteranlage, Blutung, Sepsis, Änderung von Antikonvulsivaspiegeln Rituximab 375 mg/m2KO/Monat für 4 Monate [102] oder 1 000 mg Rituximab d1 und d15 [109] Wiederholung individuell. Nach 12 Monaten oder bei Nachweis steigender CD19-Zellen im Blut max. Kumulativdosis 30 g ( − 50 g); 750–1 000 mg/m2 i. v. alle 4 Wochen. Dosissteigerung bis Leukozytennadir < 4/nl; Reduktion um 125 mg/m2 bei Leukos < 3/nl, Aussetzen bei Leukos < 1,5/μl. Reduktion 30 % bei Krea-Cl < 30 ml/min oder Alter > 65J. Behandlungsdauer: bis zur Remission, > 6 Monate Monitorüberwachung, Vorbehandlung: 1 000 mg Paracetamol, 2 mg Clemastin, 50 mg Ranitidin, 100 g Prednisolon. CD19, CD20 Bestimmung Exantheme, Bronchospasmus, Fieber (50 %), Schüttelfrost, Rigor, Übelkeit Kopfschmerz und Blutdruckabfall (10 %) Nadir an Tag 12–16. Blutbild, Leberfunktion, Entzündungswert, Urinanalyse (wöchentlich Monat 1, danach monatlich); engmaschiger bei GFR 10–30 ml/min Übelkeit, Erbrechen, Diarrhoe, Fatigue, Zystitis, Hämaturie, Knochenmarksuppression, Urothel-Karzinom Plasmapherese oder Protein A Immunadsorption [107] Rituximab [102] Cyclophosphamid [101, 106] schleichschema behandelt. Nach 2 Wochen ohne Besserung oder Stabilisierung eines zuvor progredienten Syndroms ist bei zentralnervösen Syndromen eine Behandlung mit Cyclophosphamid üblich. Meist erfolgt eine intravenöse Stoßtherapie mit 750– 1 000 mg/m2 Körperoberfläche in Einzelfällen bis 2 000 mg/m2 alle 3–4 Wochen [107]. Bei isolierten peripher-neurologischen Manifestationen können therapeutisch ivIG (2 g/kg KG verteilt über 5 Tage) oder eine Plasmapherese/Immunadsorption eingesetzt warden [108]. In Einzelfällen wurden ivIG auch mit Erfolg bei Hirnstamm- und Kleinhirnmanifestationen angewandt [109]. Zu monoklonalen Antikörpertherapien existieren einzelne Kasuistiken und eine unkontrollierte Studie. Als Alternative zu Cyclophosphamid kann Rituximab entweder 375 mg/m2KOF monatlich über 4 Monate [102] oder 1 000 mg absolut an Tag 1 und Tag 15 analog zu dem Vorgehen bei Neuromyelitis optica [110] erwogen werden, ein Vorteil eines dieser Regime ist nicht belegt. Obwohl in diesem Kontext nicht evidenzbasiert, sollte bei längerfristiger Immunsuppression zur Vermeidung von steroidalen Nebenwirkungen zusätzlich zu oder anstatt Steroiden mit Steroid-sparenden Agenzien behandelt werden (MTX, Azathioprin, Mycophenolat Mofetil, Cyclosporin A). Im Rahmen einer systemischen Tumorbehandlung ( = Chemotherapie) ist eine zusätzliche Immunsuppression mit weiteren Agenzien aus pathophysiologischen Überlegungen meist nicht sinnvoll. Bei isolierter Radiatio oder Operation von Tumoren kann eine adjuvante Immunsuppression sinnvoll sein. Die Enzephalitiden mit Antikörpern gegen Oberflächenantigene sprechen zwar besser auf eine Immuntherapie an, es gibt jedoch auch hier bisher keine kontrollierten randomisierten prospektiven Therapiestudien sondern lediglich Fallberichte und -serien. Aus der größten vorhandenen Fallserie der NMDA Rezeptor Antikörper assoziierten Enzephalitiden kann abgeleitet werden, dass eine frühe Therapie mit Steroiden, humanen Immunglobulinen oder Plasmapherese sinnvoll ist. Der frühe Beginn einer immunsuppressiven Therapie ( < 40 Tage) kann den Verlauf günstig beeinflussen und den Aufenthalt auf der Intensivstation deutlich reduzieren [19]. Bei Patienten, die nicht frühzeitig – also innerhalb der ersten 8 Wochen – behandelt wurden oder keinen nachweisbaren Tumor haben, wird eine Immunsuppression mit Cyclophosphamid oder dem monoklonalen CD20 Antikörper Rituximab empfohlen [18]. Insbesondere die gute Verträglichkeit und die lange Wirksamkeit sowie das Fehlen einer Gonadentoxizität bei jungen Frauen sprechen für letztere Substanz. Nach eigener Erfahrung ist die Wirksamkeit sehr gut. Wenig ist allerdings über die Langzeittherapie bekannt. Meist muss nach einer Resektion eines Teratoms bei einer paraneoplastischen NMDA-Rezeptor Antikörper Enzephalitis nicht dauerhaft immunsuppressiv behandelt werden. Bei Patientinnen ohne nachweisbares Teratom wird nach einer intravenösen Cyclophosphamidtherapie oft eine orale Immunsuppression mit Azathioprin, MTX oder Mycophenolat Mofetil über 1–2 Jahre durchgeführt. Rituximab kann individuell nach 3–12 Monaten erneut appliziert werden, oft wird der erneute Anstieg von CD19 positiven Zellen im Blut als Surrogatparameter für eine sich abschwächende Wirkung des Rituximab interpretiert. Leypoldt F et al. Neues bei paraneoplastischen Syndromen … Akt Neurol 2012; 39: 60–73 Neues in der Neurologie Literatur Key Messages 1. Bei paraneoplastischen Syndromen kommt der Autoantikörperdiagnostik herausragende Bedeutung zu. 2. Bei Enzephalitiden wird zwischen Autoantikörpern gegen intrazelluläre Antigene (Hu, Yo, CV2/CRMP5, Ri, usw.) und Oberflächenantigene (NMDA-Rezeptor, AMPA-Rezeptor, usw.) unterschieden. 3. Anti-Tr, Anti-SOX1, Anti-Zic4, PCA-2, ANNA-3 oder ganglionäre ACh-Rezeptor Antikörper gelten als teilcharakterisierte paraneoplastische Antikörper, sollten jedoch trotzdem als starker Hinweis auf ein paraneoplastisches Syndrom gewertet werden. 4. Die Tumorsuche sollte nach vermuteter Lokalisation ge▶ Tab. 5). staffelt erfolgen (● 5. Ein Tumorscreening sollte alle 3–6 Monate für mindestens 4 Jahre erfolgen. Bei LEMS sind 2 Jahre ausreichend. 6. Die Tumortherapie (systemische Tumortherapie, Radiatio, Operation) ist Therapie der ersten Wahl bei paraneoplastischen Syndromen. Die Art der Tumortherapie richtet sich nach dem Tumor und nicht nach dem paraneoplastischen Syndrom. 7. Immunmodulatorische Therapie paraneoplastischer Syndrome ist üblich obwohl nicht evidenzgesichert. Sie sollte als cyclophosphamidbasiertes Regime durchgeführt werden. Eine Immunsuppression zusätzlich zu einer systemischen Tumortherapie sollte nicht durchgeführt werden. 8. Enzephalitiden mit Antikörpern gegen Oberflächenantige▶ Tab. 2) sollten frühzeitig immunsuppressiv (Steroine (● de, Plasmapherese, ivIG) behandelt werden. Bei fehlendem Ansprechen und fehlendem Tumornachweis wird eine Eskalation mit Cyclophosphamid oder Rituximab empfohlen, bei Tumornachweis eine rasche Tumorentfernung. Zur Person Dr. Frank Leypoldt ist Facharzt für Neurologie und seit 2004 in der Klinik für Neurologie in der Universitätsklinik Hamburg-Eppendorf tätig. Sein klinisches Interesse gilt insbesondere Enzephalitiden, paraneoplastischen Syndromen, rheumatologischen Erkrankungen und Immunneuropathien, seit 2005 betreut er die neuroimmunologische Sprechstunde. Besonderen Wert legt er auf die interdisziplinäre Zusammenarbeit mit Rheumatologen, Dermatologen und anderen „autoimmunen“ Disziplinen. Wissenschaftlich beschäftigt er sich neben Enzephalitiden auch mit sekundären Immunmechanismen im Schlaganfall. Interessenkonflikt ▼ Frank Leypoldt gibt keine Interessenkonflikte an. Klaus-Peter Wandinger besitzt Aktien der Firma Euroimmun AG. Raymond Voltz gibt keine Interessenkonflikte an. 1 Graus F, Delattre JY, Antoine JC et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatr 2004; 75: 1135–1140 2 Graus F, Cordon-Cardo C, Posner J. Neuronal antinuclear antibody in sensory neuronopathy from lung cancer. Neurology 1985; 35: 538– 543 3 Giometto B, Grisold W, Vitaliani R et al. Paraneoplastic neurologic syndrome in the PNS Euronetwork database: a European study from 20 centers. Arch Neurol 2010; 67: 330–335 4 van Oosterhout AG, van de Pol M, Velde ten GP et al. Neurologic disorders in 203 consecutive patients with small cell lung cancer. Results of a longitudinal study. Cancer 1996; 77: 1434–1441 5 Rosenfeld MR, Dalmau J. Anti-NMDA-Receptor Encephalitis and Other Synaptic Autoimmune Disorders. Curr Treat Options Neurol 2011 6 Bataller L, Kleopa KA, Wu GF et al. Autoimmune limbic encephalitis in 39 patients: immunophenotypes and outcomes. J Neurol Neurosurg Psychiatr 2007; 78: 381–385 7 Gultekin SH, Rosenfeld MR, Voltz R et al. Paraneoplastic limbic encephalitis: neurological symptoms, immunological findings and tumour association in 50 patients. Brain 2000; 123 (Pt 7): 1481–1494 8 Cross SA, Salomao DR, Parisi JE et al. Paraneoplastic autoimmune optic neuritis with retinitis defined by CRMP-5-IgG. Ann Neurol 2003; 54: 38–50 9 Honnorat J, Antoine J, Derrington E et al. Antibodies to a subpopulation of glial cells and a 66 kDa developmental protein in patients with paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry 1996; 61: 270–278 10 Kinirons P, Fulton A, Keoghan M et al. Paraneoplastic limbic encephalitis (PLE) and chorea associated with CRMP-5 neuronal antibody. Neurology 2003; 61: 1623–1624 11 Dalmau J, Graus F, Villarejo A et al. Clinical analysis of anti-Ma2associated encephalitis. Brain 2004; 127: 1831–1844 12 Hoffmann LA, Jarius S, Pellkofer HL et al. Anti-Ma and anti-Ta associated paraneoplastic neurological syndromes: 22 newly diagnosed patients and review of previous cases. J Neurol Neurosurg Psychiatr 2008; 79: 767–773 13 Pittock SJ, Lucchinetti CF, Parisi JE et al. Amphiphysin autoimmunity: paraneoplastic accompaniments. Ann Neurol 2005; 58: 96–107 14 Saiz A, Bruna J, Stourac P et al. Anti-Hu-associated brainstem encephalitis. J Neurol Neurosurg Psychiatr 2009; 80: 404–407 15 Lucchinetti C, Kimmel D, Lennon V. Paraneoplastic and oncologic profiles of patients seropositive for type 1 antineuronal nuclear autoantibodies. Neurology 1998; 50: 652–657 16 Prüss H, Dalmau J, Harms L et al. Retrospective analysis of NMDA receptor antibodies in encephalitis of unknown origin. Neurology 2010; 75: 1735–1739 17 Granerod J, Ambrose HE, Davies NW et al. Causes of encephalitis and differences in their clinical presentations in England: a multicentre, population-based prospective study. Lancet Infect Dis 2010; 10: 835–844 18 Dalmau J, Lancaster E, Martinez-Hernandez E et al. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol 2011; 10: 63–74 19 Irani SR, Bera K, Waters P et al. N-methyl-D-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain 2010; 133: 1655–1667 20 Kleinig TJ, Thompson PD, Matar W et al. The distinctive movement disorder of ovarian teratoma-associated encephalitis. Mov Disord 2008; 23: 1256–1261 21 Dale RC, Irani SR, Brilot F et al. N-methyl-D-aspartate receptor antibodies in pediatric dyskinetic encephalitis lethargica. Ann Neurol 2009; 66: 704–709 22 Zandi MS, Irani SR, Follows G et al. Limbic encephalitis associated with antibodies to the NMDA receptor in Hodgkin lymphoma. Neurology 2009; 73: 2039–2040 23 Dalmau J, Gleichman AJ, Hughes EG et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol 2008; 7: 1091–1098 24 Kruer MC, Koch TK, Bourdette DN et al. NMDA receptor encephalitis mimicking seronegative neuromyelitis optica. Neurology 2010; 74: 1473–1475 25 Leypoldt F, Buchert R, Kleiter I et al. Anti-NMDA receptor encephalitis exhibits frontotemporo-occipital gradient of glucose metabolism. JNNP submitted Leypoldt F et al. Neues bei paraneoplastischen Syndromen … Akt Neurol 2012; 39: 60–73 71 72 Neues in der Neurologie 26 Lai M, Hughes EG, Peng X et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol 2009; 65: 424–434 27 Bataller L, Galiano R, García-Escrig M et al. Reversible paraneoplastic limbic encephalitis associated with antibodies to the AMPA receptor. Neurology 2010; 74: 265–267 28 Lai M, Huijbers MGM, Lancaster E et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol 2010; 9: 776–785 29 Lancaster E, Huijbers MGM, Bar V et al. Investigations of caspr2, an autoantigen of encephalitis and neuromyotonia. Ann Neurol 2011; 69: 303–311 30 Irani SR, Alexander S, Waters P et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis. Morvan’s syndrome and acquired neuromyotonia Brain 2010 31 Lancaster E, Lai M, Peng X et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol 2009 32 Boronat A, Sabater L, Saiz A et al. GABAB receptor antibodies in limbic encephalitis and anti-GAD-associated neurologic disorders. Neurology 2011; 76: 795–800 33 Irani SR, Michell AW, Lang B et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 2011; 69: 892–900 34 Vincent A, Buckley C, Schott JM et al. Potassium channel antibodyassociated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain 2004; 127: 701–712 35 Jarius S, Hoffmann L, Clover L et al. CSF findings in patients with voltage gated potassium channel antibody associated limbic encephalitis. J Neurol Sci 2008; 268: 74–77 36 Graus F, Boronat A, Xifró X et al. The expanding clinical profile of anti-AMPA receptor encephalitis. Neurology 2010; 74: 857–859 37 Choi K-D, Kim JS, Park S-H et al. Cerebellar hypermetabolism in paraneoplastic cerebellar degeneration. J Neurol Neurosurg Psychiatr 2006; 77: 525–528 38 Mason WP, Graus F, Lang B et al. Small-cell lung cancer, paraneoplastic cerebellar degeneration and the Lambert-Eaton myasthenic syndrome. Brain 1997; 120 (Pt 8): 1279–1300 39 Peterson K, Rosenblum M, Kotanides H et al. Paraneoplastic cerebellar degeneration. I. A clinical analysis of 55 anti-Yo antibody-positive patients. Neurology 1992; 42: 1931–1937 40 Rojas-Marcos I, Rousseau A, Keime-Guibert F et al. Spectrum of paraneoplastic neurologic disorders in women with breast and gynecologic cancer. Medicine (Baltimore) 2003; 82: 216–223 41 McKeon A, Tracy JA, Pittock SJ et al. Purkinje cell cytoplasmic autoantibody type 1 accompaniments: the cerebellum and beyond. Arch Neurol 2011; 68: 1282–1289 42 Bataller L, Graus F, Saiz A et al. Clinical outcome in adult onset idiopathic or paraneoplastic opsoclonus-myoclonus. Brain 2001; 124: 437–443 43 Bataller L, Rosenfeld MR, Graus F et al. Autoantigen diversity in the opsoclonus-myoclonus syndrome. Ann Neurol 2003; 53: 347–353 44 Kurian M, Lalive PH, Dalmau J. Opsoclonus-myoclonus syndrome in anti-N-methyl-D-aspartate receptor encephalitis. Arch Neurol 2010; 67: 118–121 45 Smith JH, Dhamija R, Moseley BD et al. N-methyl-D-aspartate receptor autoimmune encephalitis presenting with opsoclonus-myoclonus: treatment response to plasmapheresis. Arch Neurol 2011; 68: 1069–1072 46 Mas N, Saiz A, Leite MI et al. Antiglycine-receptor encephalomyelitis with rigidity. J Neurol Neurosurg Psychiatr 2010 47 Turner MR, Irani SR, Leite MI et al. Progressive encephalomyelitis with rigidity and myoclonus: glycine and NMDA receptor antibodies. Neurology 2011; 77: 439–443 48 Katz JD, Ropper AH. Progressive necrotic myelopathy: clinical course in 9 patients. Arch Neurol 2000; 57: 355–361 49 Pittock SJ, Lennon VA. Aquaporin-4 autoantibodies in a paraneoplastic context. Arch Neurol 2008; 65: 629–632 50 Panzer J, Dalmau J. Movement disorders in paraneoplastic and autoimmune disease. Curr Opin Neurol 2011; 24: 346–353 51 Grant R, Graus F. Paraneoplastic movement disorders. Mov Disord 2009; 24: 1715–1724 52 Hiyama TY, Matsuda S, Fujikawa A et al. Autoimmunity to the sodium-level sensor in the brain causes essential hypernatremia. Neuron 2010; 66: 508–522 53 Camdessanché J-P, Antoine J-C, Honnorat J et al. Paraneoplastic peripheral neuropathy associated with anti-Hu antibodies. A clinical and electrophysiological study of 20 patients. Brain 2002; 125: 166–175 54 Vallat JM, De Mascarel HA, Bordessoule D et al. Non-Hodgkin malignant lymphomas and peripheral neuropathies – 13 cases. Brain 1995; 118 (Pt 5): 1233–1245 55 Oh SJ. Paraneoplastic vasculitis of the peripheral nervous system. Neurol Clin 1997; 15: 849–863 56 Leypoldt F, Friese MA, Böhm J et al. Multiple enlarged nerves on neurosonography: An unusual paraneoplastic case. Muscle Nerve 2011; 43: 756–757 57 Camdessanché J-P, Jousserand G, Ferraud K et al. The pattern and diagnostic criteria of sensory neuronopathy: a case-control study. Brain 2009; 132: 1723–1733 58 Antoine JC, Honnorat J, Camdessanché JP et al. Paraneoplastic antiCV2 antibodies react with peripheral nerve and are associated with a mixed axonal and demyelinating peripheral neuropathy. Ann Neurol 2001; 49: 214–221 59 Graus F, Keime-Guibert F, Reñe R et al. Anti-Hu-associated paraneoplastic encephalomyelitis: analysis of 200 patients. Brain 2001; 124: 1138–1148 60 Lee HR, Lennon VA, Camilleri M et al. Paraneoplastic gastrointestinal motor dysfunction: clinical and laboratory characteristics. Am J Gastroenterol 2001; 96: 373–379 61 McKeon A, Lennon VA, Lachance DH et al. Ganglionic acetylcholine receptor autoantibody: oncological, neurological, and serological accompaniments. Arch Neurol 2009; 66: 735–741 62 Chamberlain JL, Pittock SJ, Oprescu A-M et al. Peripherin-IgG association with neurologic and endocrine autoimmunity. Journal of autoimmunity 2010 63 Stich O, Kleer B, Rauer S. Absence of paraneoplastic antineuronal antibodies in sera of 145 patients with motor neuron disease. J Neurol Neurosurg Psychiatr 2007; 78: 883–885 64 Younger DS. Motor neuron disease and malignancy. Muscle Nerve 2000; 23: 658–660 65 Briani C, Vitaliani R, Grisold W et al. Spectrum of paraneoplastic disease associated with lymphoma. Neurology 2011; 76: 705–710 66 Hart IK, Maddison P, Newsom-Davis J et al. Phenotypic variants of autoimmune peripheral nerve hyperexcitability. Brain 2002; 125: 1887–1895 67 Loukaides P, Schiza N, Pettingill P et al. Morvan’s syndrome associated with antibodies to multiple components of the voltage-gated potassium channel complex. J Neurol Sci 2011 68 Payne M, Bradbury P, Lang B et al. Prospective Study into the Incidence of Lambert Eaton Myasthenic Syndrome in Small Cell Lung Cancer. J Thorac Oncol 2009; 5: 34–38 69 Monstad SE, Knudsen A, Salvesen HB et al. Onconeural antibodies in sera from patients with various types of tumours. Cancer Immunol Immunother 2009; 58: 1795–1800 70 Graus F, Dalmou J, Rene R et al. Anti-Hu antibodies in patients with small-cell lung cancer: association with complete response to therapy and improved survival. J Clin Oncol 1997; 15: 2866–2872 71 Honnorat J, Cartalat-Carel S, Ricard D et al. Onco-neural antibodies and tumour type determine survival and neurological symptoms in paraneoplastic neurological syndromes with Hu or CV2/CRMP5 antibodies. J Neurol Neurosurg Psychiatr 2009; 80: 412–416 72 Kazarian M, Calbo J, Proost N et al. Immune response in lung cancer mouse model mimics human anti-Hu reactivity. J Neuroimmunol 2009; 217: 38–45 73 de Graaf MT, de Beukelaar JWK, Haasnoot GW et al. HLA-DQ2(+) individuals are susceptible to Hu-Ab associated paraneoplastic neurological syndromes. J Neuroimmunol 2010 74 Dalmau J, Graus F, Cheung N et al. Major histocompatibility proteins, anti-Hu antibodies, and paraneoplastic encephalomyelitis in neuroblastoma and small cell lung cancer. Cancer 1995; 75: 99–109 75 Greenlee J, Parks T, Jaeckle K. Type IIa (“anti-Hu”) antineuronal antibodies produce destruction of rat cerebellar granule neurons in vitro. Neurology 1993; 43: 2049–2054 76 Hormigo A, Lieberman F. Nuclear localization of anti-Hu antibody is not associated with in vitro cytotoxicity. J Neuroimmunol 1994; 55: 205–212 77 Verschuuren JJ, Dalmau J, Hoard R et al. Paraneoplastic anti-Hu serum: studies on human tumor cell lines. J Neuroimmunol 1997; 79: 202–210 78 Sillevis Smitt P, Manley G, Posner J. Immunization with the paraneoplastic encephalomyelitis antigen HuD does not cause neurologic disease in mice. Neurology 1995; 45: 1873–1878 Leypoldt F et al. Neues bei paraneoplastischen Syndromen … Akt Neurol 2012; 39: 60–73 Neues in der Neurologie 79 Tanaka M, Tanaka K, Onodera O et al. Trial to establish an animal model of paraneoplastic cerebellar degeneration with anti-Yo antibody. 1. Mouse strains bearing different MHC molecules produce antibodies on immunization with recombinant Yo protein, but do not cause Purkinje cell loss. Clin Neurol Neurosurg 1995; 97: 95–100 80 Voltz R, Dalmau J, Posner JB et al. T-cell receptor analysis in anti-Hu associated paraneoplastic encephalomyelitis. Neurology 1998; 51: 1146–1150 81 Albert ML, Darnell JC, Bender A et al. Tumor-specific killer cells in paraneoplastic cerebellar degeneration. Nat Med 1998; 4: 1321– 1324 82 Benyahia B, Liblau R, Merle-Béral H et al. Cell-mediated autoimmunity in paraneoplastic neurological syndromes with anti-Hu antibodies. Ann Neurol 1999; 45: 162–167 83 Pellkofer H, Schubart AS, Höftberger R et al. Modelling paraneoplastic CNS disease: T-cells specific for the onconeuronal antigen PNMA1 mediate autoimmune encephalomyelitis in the rat. Brain 2004; 127: 1822–1830 84 DeLuca I, Blachère NE, Santomasso B et al. Tolerance to the neuronspecific paraneoplastic HuD antigen. PLoS ONE 2009; 4: e5739 85 de Beukelaar JW, Verjans GM, van Norden Y et al. No evidence for circulating HuD-specific CD8+ T cells in patients with paraneoplastic neurological syndromes and Hu antibodies. Cancer Immunol Immunother 2007; 56: 1501–1506 86 Roberts WK, Deluca IJ, Thomas A et al. Patients with lung cancer and paraneoplastic Hu syndrome harbor HuD-specific type 2 CD8+ T cells. J Clin Invest 2009; 119: 2042–2051 87 Tani T, Tanaka K, Idezuka J et al. Regulatory T cells in paraneoplastic neurological syndromes. J Neuroimmunol 2008; 196: 166–169 88 Hughes EG, Peng X, Gleichman AJ et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci 2010; 30: 5866–5875 89 Tüzün E, Zhou L, Baehring JM et al. Evidence for antibody-mediated pathogenesis in anti-NMDAR encephalitis associated with ovarian teratoma. Acta Neuropathol 2009; 118: 737–743 90 Martinez-Hernandez E, Horvath J, Shiloh-Malawsky Y et al. Analysis of complement and plasma cells in the brain of patients with antiNMDAR encephalitis. Neurology 2011; 77: 589–593 91 de Lange R, Dimoudis N, Weidle UH. Identification of genes associated with enhanced metastasis of a large cell lung carcinoma cell line. Anticancer Res 2003; 23: 187–194 92 Hofmann C, Baur M-O, Schroten H. Anti-NMDA receptor encephalitis after TdaP-IPV booster vaccination: cause or coincidence? J Neurol 2011; 258: 500–501 93 Psimaras D, Carpentier A, Rossi C. CSF study in paraneoplastic syndromes. J Neurol Neurosurg Psychiatr 2009 94 Wandinger KPL, Klingbeil C, Gneiss C et al. New serological markers for the differential diagnosis of autoimmune limbic encephalitis. J Lab Med 2011; 35: 329–342 95 Sabater L, Titulaer M, Saiz A et al. SOX1 antibodies are markers of paraneoplastic Lambert-Eaton myasthenic syndrome. Neurology 2008; 70: 924–928 96 Titulaer MJ, Klooster R, Potman M et al. SOX antibodies in small-cell lung cancer and Lambert-Eaton myasthenic syndrome: frequency and relation with survival. J Clin Oncol 2009; 27: 4260–4267 97 Hadjivassiliou M, Alder SJ, Van Beek EJR et al. PET scan in clinically suspected paraneoplastic neurological syndromes: a 6-year prospective study in a regional neuroscience unit. Acta Neurologica Scandinavica 2009; 119: 186–193 98 Titulaer MJ, Soffietti R, Dalmau J et al. Screening for tumours in paraneoplastic syndromes: report of an EFNS task force. Eur J Neurol 2011; 18: 19–e3 99 Titulaer MJ, Wirtz PW, Willems LNA et al. Screening for small-cell lung cancer: a follow-up study of patients with Lambert-Eaton myasthenic syndrome. J Clin Oncol 2008; 26: 4276–4281 100 Candler P, Hart P, Barnett M et al. A follow up study of patients with paraneoplastic neurological disease in the United Kingdom. J Neurol Neurosurg Psychiatry 2004; 75: 1411–1415 101 Sillevis Smitt P, Grefkens J, De Leeuw B et al. Survival and outcome in 73 anti-Hu positive patients with paraneoplastic encephalomyelitis/sensory neuronopathy. J Neurol 2002; 249: 745–753 102 Keime-Guibert F, Graus F, Fleury A et al. Treatment of paraneoplastic neurological syndromes with antineuronal antibodies (Anti-Hu, anti-Yo) with a combination of immunoglobulins, cyclophosphamide, and methylprednisolone. J Neurol Neurosurg Psychiatr 2000; 68: 479–482 103 Shamsili S, de Beukelaar J, Gratama JW et al. An uncontrolled trial of rituximab for antibody associated paraneoplastic neurological syndromes. J Neurol 2006; 253: 16–20 104 Keogh M, Sedehizadeh S, Maddison P. Treatment for Lambert-Eaton myasthenic syndrome. Cochrane Database Syst Rev 2011; 2: CD003279 105 Dalakas MC, Koffman B, Fujii M et al. A controlled study of intravenous immunoglobulin combined with prednisone in the treatment of IBM. Neurology 2001; 56: 323–327 106 Espandar L, O’Brien S, Thirkill C et al. Successful treatment of cancerassociated retinopathy with alemtuzumab. J Neurooncol 2007; 83: 295–302 107 Stark E, Wurster U, Patzold U et al. Immunological and clinical response to immunosuppressive treatment in paraneoplastic cerebellar degeneration. Arch Neurol 1995; 52: 814–818 108 Cher L, Hochberg F, Teruya J et al. Therapy for paraneoplastic neurologic syndromes in six patients with protein A column immunoadsorption. Cancer 1995; 75: 1678–1683 109 Uchuya M, Graus F, Vega F et al. Intravenous immunoglobulin treatment in paraneoplastic neurological syndromes with antineuronal autoantibodies. J Neurol Neurosurg Psychiatry 1996; 60: 388–392 110 Cree BAC, Lamb S, Morgan K et al. An open label study of the effects of rituximab in neuromyelitis optica. Neurology 2005; 64: 1270–1272 Leypoldt F et al. Neues bei paraneoplastischen Syndromen … Akt Neurol 2012; 39: 60–73 73