Umweltchemie - Universität Zürich
Werbung

Nebenfach Umweltwissenschaften
Universität Zürich
Prof. Dr. P. Rüedi
Grundlagen der Umweltchemie (PK 2)
(Schwerpunkt Organische Chemie)
Umweltverhalten organischer Verbindungen
A. Allgemeine Einführung
1. Emission-Transmission-Immission
2. Struktur, Reaktivität → Umweltverhalten; Forschungsansätze
B. Bewertung der Umweltrelevanz von Chemikalien
1. Allgemeines
2. Produktion, Mengen, Verfahren; wichtige Xenobiotika
3. Anwendungsmuster
4. Ausbreitung in der Umwelt (Dispersionstendenz)
5. Akkumulation
6. Abbau, Persistenz
C. Umwandlungen organischer Stoffe unter Umweltbedingungen (chemische Reaktionstypen)
1. Allgemeines
2. Abiotische Umwandlungen
2.1. Hydrolyse
2.2. Oxidation
2.3. Reduktion
2.4. Photochemische Reaktionen
3. Biotische Umwandlungen
3.1. Allgemeines
3.2. Oxidation
3.3. Reduktion
3.4. Hydrolyse
3.5. Sekundärreaktionen
3.6. Metabolisierung von Metallen
Literatur: G. Fellenberg, 'Chemie der Umweltbelastung', Teubner Studienbücher Chemie, 1997.
H. Parlar, D. Angerhöfer, 'Chemische Ökotoxikologie', Springer-Verlag, 2. Aufl., 1995
(Quelle der meisten Beilagen).
G. Schwedt, 'Taschenatlas der Umweltchemie', Thieme 1996.
V. Koss, 'Umweltchemie; Eine Einführung für Studium und Praxis', Springer-Verlag, 1997.
17. Nov. 2004/Rü
-1-
Grundlagen der Umweltchemie (PK 2)
Organisch-chemischer Teil
Zusammenfassung der Vorlesung
A. Allgemeine Einführung
A.1. Emission-Transmission-Immission
Zentrales Schema bei der Diskussion von stoffbezogener ("Chemie") Umweltproblematik.
Beilage (S.1a)
A.2. Struktur, Reaktivität, Umweltverhalten; Forschungsansätze;
die Aufgabe der Chemie
Grundfrage:
??
Struktur
Umweltverhalten
"Struktur"
physikalische Eigenschaften
-
chemische Eigenschaften
Reaktivität
Dampfdruck (Sdp., Schmp.)
Löslichkeit (H2O, etc.)
Säure-, bzw. Basenstärke (pKa, pKb)
Verteilungskoeffizienten (z.B. Kow)
- funktionelle Gruppen
- elektronische Substituenten-Einflüsse
- Reaktionsmechanismen
Dispersion in verschiedene Kompartimente
Mobilität, Toxizität
Umwandlungsprodukte
(Abbau)
Chemische Fachkenntnisse sind die Grundlage für das Verständnis des Umweltverhaltens von
Stoffen!
Forschungsansätze
Organische Chemie: enorme Fülle von Stoffen, Varietäten → sehr komplex
Anorganische Chemie: v.a. Mobilisierung von (Schwer)Metallen, 'Speciation'
"Substanz"
Struktur ? Mengen ? qualitativ/quantitativ
Dispersion, Umwandlung
"was" wird "wie" gemessen ?
"wie" ? Produkte ?
Einzelsubstanzen ?
Abnahme Edukt(e) ?
Zunahme Produkt(e)
-2-
Verbindung
Isolierung, Reinheit
(Trenntechniken, z.B.
Chromatographie, etc.)
Umwandlungsprodukte
Metaboliten
oft viel gefährlicher als Edukt!
(biologische) Aktivierung
Analytik
Identifizierung,
Strukturaufklärung
(Spektroskopie)
Toxizitätstests
Substanzen meist in Spuren
Synthese
CHEMIE
Konsequenz: viele, differenzierte Forschungsaspekte
Aufgaben und Forschungsschwerpunkte in der Umweltchemie
Öko-Diagnostik
Monitoring
Öko-Technologie
Überwachung
Ökotoxikologie
Ökoepidemiologie
Vorkommen und Produktionshöhe
Anwendungsmuster
Ausbreitung in der Umwelt
Aufnahme und Akkumulation in der Umwelt
Toxizität und Ökotoxizität
Persistenz und Abbau
Umwandlungsreaktionen
Prophylaxe
Abfallbehandlung
Therapie
Emissionsvermeidung bei der Produktion
Vermeidung technischer Verunreinigungen
verbesserte Applikationstechniken
Substitution durch umweltkompatible Produkte
verbesserte Abfallbehandlung
Bodenverbesserung durch spezifische Massnahmen
(physikalisch, chemisch, biologisch)
SK 4 im SS: Umweltanalytik, technischer Umweltschutz
-3-
B. Bewertung der Umweltrelevanz von Chemikalien
B.1. Allgemeines
Einbringen von "Materie" in ein System → Veränderung der stofflichen Umweltqualität → Störung
von Gleichgewichten
natürlich/anthropogen
erwünscht/unerwünscht
messbar/nicht messbar → Umweltanalytik
abhängig vom Verhalten der individuellen Verbindungen →bestimmt das Ausmass und die
Dauer von Veränderungen.
Abschätzung und Prognose regionaler oder globaler Veränderungen → Kriterien zur Beurteilung
(vgl. Kap. B.2. – B.6.).
B.2. Produktion, Mengen, Verfahren; Xenobiotika
Basis: Erdöl, Erdgas (fossile Brennstoffe) als
- Brenn- und Treibstoffe
- chemische Grundstoffe (Rohstoffe)
- Petrochemische Industrie: Raffinierien, Aufbereitung von Rohstoffen (Shell, BP, etc.)
- Chemische Industrie: Verarbeitung der Rohstoffe, neue Anwendungsprodukte (Roche, Novartis, etc.)
Probleme: - Veränderung des natürlichen C-Lagers
- gasförmige Emissionen (KW, CO2, NH3 → NOx, H2S → SO2, etc.)
Zahl organischer Verbindungen → "Chemical Abstracts" (seit 1910)
USA 1963: 6 Mio. Substanzen registriert, davon ca. 63'000 im täglichen Gebrauch.
seither: täglich ca. 6'000 neue Registrationen, ca. 1'000 neue synthetische Produkte (Mengen?!).
seit 1967: 16 Mio. Abstracts mit 27 Mio. Substanzen und 3.8 Mio. Reaktionen. Die Datenbank wird
täglich aktualisiert und wächst dabei um 2'500 Abstracts und 11'000 Substanzen.
Aktuell: Nicht nur die Chemie, sondern auch die Biowissenschaften sind vertreten: So stammen
34% der Abstracts aus der Biochemie und bei 26% der Substanzen handelt es sich um
Biosequenzen.
Von den chemischen Substanzen sind heute <100'000 im täglichen Gebrauch
Problematik: - Stoffe, insbesondere Mengen an sich
- Nebenprodukte (unbeabsichtigt), Verunreinigungen (z.B. Lindan, Dioxin)
Generell gilt für "Zahlen":
grösste Vorsicht bei der Interpretation!!
(sind abhängig von der Ermittlungsart
und der dahinter stehenden "Philosophie")
Beilagen (S. 3a – 3e)
- 3d -
Beilage 4.1 zu Kap. B.2.
Grundlagen der Umweltchemie (OC-Teil)
Wichtige Vertreter organischer Xenobiotika
Umweltchemikalien, Xenobiotika
Durch menschliches Zutun (anthropogene Tätigkeit) in relevanten Mengen in die Umwelt emittierte
Stoffe. Störung natürlicher Gleichgewichte und potentielle Gefährdung von Ökosystemen; gelangen
via Nahrungskette in die Stoffkreisläufe. Weitere Klassifizierung aufgrund struktureller Merkmale in
synthetische 'Natur'- (naturidentische oder naturnahe) und eigentliche Fremdstoffe (Xenobiotika).
Letztere sind meist schwer abbaubar, da die entsprechenden Enzyme fehlen.
Kohlenwasserstoffe (KW)*:
aliphatische KW (n- und verzweigte Alkane, -ene, -ine)
(substituierte) Aromaten, v.a. Polyzyklen ('PAH', 'PAK')
(aus der Verbrennung fossiler Brennstoffe, Holz)
z.B.
Pyren
Benzo[a]pyren
(stark kanzerogen)
Benzo[e]pyren
Coronen
Halogenierte KW*: R-F, R-Cl, R-Br, z.B. Fluor-Chlor-KW ('FCKW', Kühlmittel ('Freone'), Treibgase)
'PER' (Lösungs- und chem. Reinigungsmittel
Aromaten, z.B. 1,4-Dichlorbenzol (Mottenschutz, WC-Reiniger)
Cln
Polychlorierte Biphenyle*:
('PCPs')
Clm
Wärmeleiter (Transformatorenöle)
Hydraulik- und Getriebeöle
Isomerengemisch (209)
OH
Polychlorierte Phenole*:
z.B.
Cl
Cl
Cl
Cl
(pKa = 4,8 )
Verrottungshemmer (Holzschutz)
Cl
Dibenzofurane*:
Xm
Xn
O
Dibenzodioxine*:
Xn
2
3
O
O
X = H, Cl
Isomerengemisch (135)
Reaktionsnebenprodukte,
Kehrichtverbrennung
X = H, Cl
Isomerengemisch (75)
Reaktionsnebenprodukte,
Kehrichtverbrennung
Xm
8
7
z.B. 2,3,7,8-TCDD
(«Dioxin»)
- 3e -
COOR
Phthalate*:
Beilage 4.2 zu Kap. B.2.
Weichmacher (in Kunststoffen)
Lösemittel, Schmieröle, Trägersubstanzen
COOR'
R = oder ≠ R', n- oder verzweigte Alkane
Detergentien:
(Tenside, oberflächenaktive Stoffe)
SO3
anionische:
C10 - C18
SO3
R
z.B. lineare Alkylsulfonsäuren ('LAS')
H 3C
kationische:
CH3
R1, R2 = C16 - C18
N
R1
R2
O
neutrale:
(C 8-C 10)
O
OH
n = 4-20
Komplexbildner:
HOOCH 2C
CH 2COOH
CH 2COOH
HOOCH2C
N-CH2-CH2-N
HOOCH 2C
N
CH 2COOH
CH2COOH
'EDTA'
'NTA'
Pestizide : Herbizide, Insektizide, Fungizide
Strukturell sehr verschiedenartige Verbindungen
(ca. 55%) (ca. 37%)
(ca. 8%)
v.a Organo(thio)phosphate, Carbamate,
Halogenide,Heterocyclen, etc.
Nahrungsmittel-Zusätze:
Stabilisatoren, Emulgatoren, Konservierungsmittel,
Antioxidantien, Farbstoffe, Duftstoffe, etc.
Strukturell verschiedenartige, meist aromatische Verbindungen
(verursachen in vielen Fällen Allergien!)
* = 'Ubiquisten', aufgrund hoher Mobilität nahezu überall vorhanden
-4-
B.3. Anwendungsmuster
Quantitative Erfassung der Einsatzbereiche von Einzelchemikalien im Endverbrauch.
Relativ wenig Chemikalien sind gut untersucht.... v.a. chlorierte KW ( → 80-90% für Reinigung).
Beilage (S. 4a)
B.4. Ausbreitung in der Umwelt (Dispersionstendenz)
Transmission ohne Stoffumwandundlungen
Verlassen des eigentlichen Applikationsbereiches
Transport zwischen Umweltkompartimenten: Luft-Wasser-Boden
Luft
trockene Deposition
Volailität
Adsorption
Wasser
Boden
allg. physikalisch sehr komplexe Gleichgewichte (Diffusion, Transportphänomene)
entscheidend: physikalische Konstanten (vgl. Kap. A.2.)
z.B.
OH
OH
O
H 2O
pKa≈10
H2O-Löslichkeit!
Cl
OH
Cl
Cl
NO2
O2N
Cl
Cl
pKa=4,8
NO2
pKa≈1 (!)
pKa = f (elektronische Substituenten-Effekte) → Art und Lage der Substituenten
e--Donatoren → pKa steigt (pKb sinkt) → weniger sauer (basischer)
e--Akzeptoren → pKa sinkt (pKb steigt) → stärker sauer
(vgl. die pKa-Werte von Halogen-substituierten Essigsäuren (Repetitoriumsaufgabe!); s. Kap. C.2.1. ).
-5-
B.5. Akkumulation
Teilaspekt der Dispersion aus der Sicht des Organismus: Unerwünschte Anreicherung von Xenobiotika.
Der meistbenutzte Parameter für Umwelt-Diskussionen:
Kow-Wert: n-Octanol als Modell für die biologische Membran. Je höher der Kow desto grösseres
umweltgefährdendes Potential.
z.B. Phthalate Kow ≈ 30 - 80'000 ( Pow = 1,5 - 4,9);
PCB's
≈ 100'000 (Pow = 5)
Beilagen (S. 5a – 5c)
B.6. Abbau, Persistenz
Aufenthaltszeit einer Chemikalie in einem definierten Umweltbereich. Abhängig von der
Geschwindigkeit des Abbaus → Stoffumwandlung. Hohe Persistenz bedeutet meist
Akkumulation. Es gibt kein absolutes Mass für Persistenz, nur relative, z.B. t1/2.
Erwünschte Persistenz: bis optimale Wirkung erreicht (z.B. Dünger, Pestizide).
Unerwünschte Persistenz: Stabilität von Verbindungen → Nebenwirkungen, etc.
"Abbau": vollständige Eliminierung aus dem Umweltbereich.
Mineralisierung:
vollständiger Abbau zu CO2, H2O und Zuwachs an Biomasse der beteiligten
Organismen (biotische Umwandlung, vgl. Kap. C.2.), eher selten.
meistens: indirekt via Metaboliten (niedermolekulare Verbindungen, die in die Stoffkreisläufe
eingehen. Die Bildung von CO2 und H2O ist eine Zeitfrage! d.h. jeder organische Stoff
ist vollständig abbaubar.
Abschätzung der (möglichen) Geschwindigkeit: → "Chemie"; Kenntnis funktioneller Gruppen,
Reaktivität und Reaktionsmechanismen.
Wichtigste Abbaureaktion: Oxidation (s. Kap. C.1.3., C.2.2.).
resistent
Halogen-KW
(allg. C-Hal)
C
H
C
C
leichter abbaubar (reaktiver)
ungesättigte KW
C OH
O
O
C
C
C
C
C
C (Verzweigungen)
C
SH
C
S
C
O
C
H
S
S
C
allg. reaktive funktionelle Gruppen
mit N,S,P,O
Beilage (S. 5d)
OH
-6-
C. Stoffumwandlungen unter Umweltbedingungen
C.1. Allgemeines
vgl. Abbau, Metaboliten (Umwandlungsprodukte) → → "Chemie": Verständnis
Stoffkenntnisse
Reaktionsmechanismen
Das reaktive Verhalten einer chemischen Verbindung in der Umwelt ist sehr komplex und
entsprechend schwierig zu untersuchen. Beilage (S.6a)
2 grundlegende Wege:
abiotisch
biotisch
in Kompartimenten Luft, Wasser Boden
→ Temperatur, Licht
in Organismen
→ Enzyme (spezifisch)
C.2. Abiotische Umwandlungen
C.2.1. Hydrolyse
Bindungsspaltung mit "Wasser": H2O als Nucleophil (säure-, oder basenkatalysiert)
meist
C
X
X ≠ C,H
X = O, N, S, Hal (elektronegative Elemente)
Säuren, Alkohole, Amine
CO2 + .........
CO2 + .........
je nach Rest
H3PO4 + .......
Ester, Amide
Carbamate
Carbonate
Phosphate
Abhängig von den Bedingungen:
- sauer →
Metaboliten
- basisch → "Verseifung" (irreversibel, MWG!)
"Hydrolyse" (Gleichgewicht)
Mechanistisch: Addition-Eliminierung via sp2 → sp3 (tetragonales Zwischenprodukt) → sp2
v Hydr. = f (pH, T, e--Donor, e--Akzeptor)
+σ, +π
-σ,-π
je höher, desto rascher
langsamer
Elektrophilie am angegriffenen Zentrum: erniedrigt
rascher
erhöht
Substituenteneffekte!
-7-
Quantitativer Zusammenhang: Hammett-Beziehung (s. Lehrbücher); Säurestärken (pKa) von
substituierten Benzoesäuren und Hydrolysegeschwindigkeiten ihrer Ester.
Massgebende Parameter:
- Art (Struktur) des Substitutenten (Donor, Akzeptor)
- Ort der Substitution (ortho/para: grosser Einfluss auf Elektronendichte im aromatischen π-System; meta: weniger)
Spaltung von C-Halogen
Nucleophile Substitition (sp3 → sp3, SN1, SN2; mechanistisch keine eigentliche Hydrolyse)
C
H 2O
X
C
OH
+
HX
Beispiel: C4H9Cl (5 Isomere)
CH3CH2CH2CH2
Cl
CH3CH2CHCH3
CH3
CH3
t1/2
CHCH2
≈ 1 Jahr
Cl
Cl
(2 Enantiomere)
CH3
CH3 C
CH3
≈ 1 Monat
Grund: SN2 → SN1 Solvolyse; Stabilität der kationischen Zwischenprodukte;
tert. >> sek. > prim. ("Markovnikov-Regel")
Beilage (S. 7a)
≈ 30 sec
Cl
-8-
C.2.2. Oxidation
.
Reaktion mit "Sauerstoff". Es existieren verschiedene Spezies: 3O2, 1O2, atomarer O, OH , O3.
Nur bedingt getrennte Betrachtung zulässig, da durch verschiedene Prozesse die Spezies z.T. in
Gleichgewichten vorliegen (vgl. PC-Teil) → Transmission!
Wichtig: Molekularer O2 ist im Grundzustand ein Biradikal (Triplett, d.h. 2 ungepaarte e- in
einem antibindenden π-Orbital) → paramagnetisch.
AO
AO
MO
σ*2p
π*2p
2px2
2py
π*2p
∆E
2pz
2pz
π2p
2py
2. angeregter Zustand
τGas ≈ 10 sec, τLsg ≈ 1 nsec
2px2
≈ 160 kJ
(760 nm)
π2p
1. angeregter Zustand: Singulett-O2
τGas ≈ 45 min, τLsg ≈ 1 msec
σ2p
O
2s2
σ*2s2
2s2
π*
Grundzustand: Triplett-O2
τ≈∞
O
σ*1s2
(1O2)
≈ 92 kJ
(1270 nm)
σ2s2
1s2
O
O
(3O2)
1s2
σ1s2
Quantenmechanik:
Reaktionen von verschiedenen Spinzuständen (Tripletts mit Singuletts) sind
verboten. Die allermeisten Moleküle liegen im stabilen Grundzustand als
Singulett (gepaarte e-) vor. Falls O2 im Grundzustand als Singulett vorliegen
würde → Reaktion ("Verbrennen").
Unterschiedliche Reaktionsweise und -Produkte der verschiedenen Sauerstoff-Spezies:
Grundzustand:
Autoxidation
Singulett :
(Cyclo)Addition
Atomarer O:
Epoxide (Oxirane)
OH. :
H-Abstraktion, Addition, Anlagerung an Olefine
Ozon:
C-C-Spaltung von Olefinen → Ketone, Aldehyde → Säuren
Beilage (S. 8a)
-9-
– Molekularer Sauerstoff (Grundzustand, Triplett-O2 (3O2))
Relativ langsame Reaktion (τ ≈∞, 21%) → Autoxidation, meist (Schwer)metallkatalysiert.
R H + O2
R O-OH
R
+
OOH
"Kette"
"Abbruch"
(Radikalkettenreaktion)
M+I
M+II
R O + M+II + OH
+ M+I + H
R O-O
Typische Redox-Reaktion; v.a. bedeutend bei Lipiden:
R CH2-CH=CH
Zellmembran
Membranschädigungen
Lipidperoxidation
Alterungsprozesse
R CH-CH=CH
R CH-CH=CH
O
(Allylstellung,
stabilisiertes Radikal)
O
R'-H
R CH-CH=CH
R CH-CH=CH
OH
O
+
R'
OH
Verschiebung von Doppelbindungen
H
H
H
H
vgl. Arachidonsäure-Metabolismus → Schmerzempfindung, Entzündungen, etc.
– Singulett-Sauerstoff (angeregter Zustand (1O2))
Gebildet durch sog. Sensibilisierung
3O
2
Sens.
1O
2
Sens.: photochemischer Katalysator, Energie-Überträger (strukturell sehr verschiedenartig, auch
heterogen)
Wichtig bei Umweltreaktionen → Transmission
[1O2]
in Atmosphäre
≈ 10-5 ppm
in Gewässern
≈ 10-12 ppm
(vgl. Konz. Masse!)
- 10 -
Strukturell:
1O hat gepaarte Aussenelektronen → verhält sich wie ein hochreaktives "Olefin".
2
1O hat ein rel. tief liegendes leeres π*-Orbital, d.h. leichte e--Aufnahme → starkes
2
Elektrophil.
Reaktivität:
- Addition an Olefine (v.a. bei allylischen H)
H
O
O
H
O
O
- Cycloaddition (Diels-Alder Dienophil)
O
O
O
O
→ Abbau von natürlichen und anthropogenen Dienen (z.B. Lipide, etc.)
Beilage (S. 8a)
Die Reaktion ist sterisch und elektronisch anspruchsvoll → kein Umsatz mit hochsubstituierten
und/oder perhalogenierten Olefinen (z.B. PER)!
– Atomarer Sauerstoff
v.a. in Atmosphäre
UV
O2
in Bodennähe
2 O
vgl. PC-Teil
λ<430nm
NO2
NO + O
Verantwortlich für die Bildung von Ozon.
Reaktivität: Addition an Olefine; ähnlich wie 3O2,1O2; es resultieren aber andere Reaktionsprodukte → Epoxide (Oxirane).
H
H
H
O
O
H
O
H
H
Beilage (S.8a)
– Hydroxyl-Radikale (OH.)
v. a. in Troposphäre, τ ≈ 1 sec
NOH. ≈ konstant ≈ 106/cm3
Grund: - stat. Konz. H2O
- Photodissoziation von O3
- Löschung von 1O2
(Z
l S
i
i PC T il!)
- 11 -
Reaktivität:
- H-Abstraktion bei Alkanen,
Alkoholen, Aminen, Halogen-KW
HO
+
- Anlagerung an Doppelbindungen
HO
+
R-H
H2O
+
R
OH
HO
- Addition an Aromaten
+
H
Hydroxylierung
OH
Beilage (S. 11a)
- Ozon (O3)
Bildung, etc. s. PC-Teil
Reaktivität: C-C-Spaltung von Doppelbindungen → Reaktion mit Spuren-Olefinen (→ PAN).
Beilage (S. 11a)
O
O
O
O3 ist ein Dipolarophil.
+
R
O
O
R = H → Aldehyde → Säuren
R ≠ H → Ketone
O O
O3
R
R
O
R
Reaktionsmechanismus (s. Allg. Chemie B, 2.Teil):
O3
C C
R
R
R C C R
O
O
C
O
R
' Primärozonid'
(1,3-dipolarophile Addition)
O
Aldehyde (R = H),
Ketone (R ≠ H)
red.
O O
C
ox.
R
Carbonsäuren (R = H),
Ketone (R ≠ H)
'Molozonid', ein Peroxi-acetal
(nach Umlagerung)
Allgemeines für Reaktionen mit Sauerstoff-Spezies:
Die Reaktionen verlaufen sehr ähnlich, führen aber zu unterschiedlichen Produkten in
unterschiedlichen Mengen. Erst eine Produkteanalyse zeigt, welche Spezies reagiert hat!
OOH
Ox
+
+
OOH
3O
2
1
:
O2 :
OOH
5%
55 %
40 %
90 %
-
10 %
- 12 -
C.2.3. Reduktion
Allgemein: immer REDOX-Reaktionen! Reduktion aus der Sicht des Substrats.
Reduktionen sind bei Xenobiotika nicht häufig und weniger wichtig als die Oxidation.
Reduktionen laufen in anaerobem Milieu ab, meist in Sedimenten.
Wichtigstes Redoxsystem Fe2+ → Fe3+ (komplexiert in Porphyrinen, Proteinen, etc.)
→ Grenze zwischen abiotischen und biotischen Reaktionen.
Reduktive Prozesse sind noch relativ wenig untersucht.
Typische Beispiele:
NO2
C
Base, Nucleophil → Weiterreaktion
polymerisiert leicht → Bindung an Huminstoffe
NH2
Hal
C
Dehalogenierung
wichtig bei Insektiziden (DDT, Aldrin, etc.)
H
Cl
Cl
Cl
Cl
Cl
Cl
Lindan
Die Synthese des Insektizids Lindan aus Cl2/hν ergibt ein Gemisch von allen möglichen
Stereoisomeren; das wirksame Agens (aaaeee) wird nur zu ca. 15% erhalten. Eingesetzt wird
das Rohgemisch.
Cl
Cl
Cl
Cl
Cl
Cl
- 13 -
C.2.4. Photochemie
Allgemein:
thermische Reaktion
photochemische Reaktion
"Wärme", ∆T
Grundzustand (HOMO)
"Licht", hν
angeregter Zustand (LUMO)
Produkte aus thermischen und photochemischen Prozessen verschieden. Thermisch "verbotene"
Reaktionen sind meist photochemisch "erlaubt" (und umgekehrt).
Beilage (das 1. Diagramm soll nur die grosse Komplexizität der Reaktionswege aufzeigen!)
Photochemische Raktionen sind typische Transmissionsreaktionen:
- Photooxidation (via O2!)
- Photolyse/Photodissoziation (Bindungsspaltung mit hν) → Dealkylierung, Dehalogenierung
- viele Reaktionen mit Schwermetallen
z.B. Methylierung von Quecksilber:
Hg2+ + CH3COO
hν
hν
+ CO2
[HgCH3]
(CH3)2Hg
hν
C2H6
+
{Hg0}
geht ähnlich auch mit Pb, As, Se, Sn, etc. (vgl. Kap, C.3.6.)
Abbau von Atrazin:
Cl
N
N
H
hν
N
N
OH
OH
N
H
H2O
N
N
H
hν
N
N
N
H
hν
Dealkylierungen
N
H2N
N
N
NH2
Nur wenige Abbauwege wurden bisher photochemisch untersucht.
Wichtiger umweltrelevanter Reaktionstyp:
Photomineralisierung → Totalabbau von Xenobiotika in adsorbiertem Zustand.
Adsorption wirkt als Sensibilisator: Verschiebung des UV/VIS-Spektrums der adsorbierten
Verbindungen nach längeren Wellen → weniger Energie zur Anregung erforderlich.
Beilage (S. 13a)
- 15 -
Auch bei allen biotischen Prozessen gilt: unterschiedliche Reaktionsbedingungen geben
verschiedene Metaboliten!
z.B.
O2
CH3
COOH
rasch
anaerob
langsam
ungiftig, H2O-löslich
toxische Produkte: Chinone, Phenole, etc.
Cl
Cl
O2
Cl
Cl
Mikroorg.
CO2 +
HCl
(Totalabbau)
"PER"
Cl
H
Cl
Cl
"TRI"
O
Cl
Oxidase
Leber
H
biotisch
Cl
Cl
unsymmetr. Epoxid
stark genotoxisch!
Interkalation mit DNA, RNA
mutagen, kanzerogen
(vgl. auch Vinylchlorid!)
H
O
Cl
Cl C CHO
Cl
Ox
Cl3C-COOH
thermisch
Cl
H C CHO
Cl
Ox
Cl2HC-COOH
H
Cl
H
C.3.3. Reduktion
Reduktive Prozesse wenig untersucht. Enzyme sind Reduktasen (NAD(P)-abhängig).
Hydrogenasen - Dehydrogenasen → REDOX.
H
H
CONH2
Aktives Zentrum:
mechanistisch: Hydrid-Übertragung
N
Rib-P-P-Rib-Adenin
wichtigste Reduktionen (vgl. auch C.2.3.):
NO2
C
Hal
(Die Nitrogruppe ist eine typisch
anthropogene funktionelle Gruppe)
NH2
C
H
Beilage (S. 15a)
- 16 -
C.3.4. Hydrolyse
Enzyme sind Hydrolasen (Esterasen, Amidasen, Proteasen). Für Xenobiotika nicht selektiv; stark
abhängig von der Struktur der abzubauenden Verbindungen:
z.B. Hydrolyse von Phthalaten:
O
O
O
( )n
Hydrolase
( )n
Leber
COOH
+ 2
COOH
( )n
O
O
O
O
O
Beilage (S.15a)
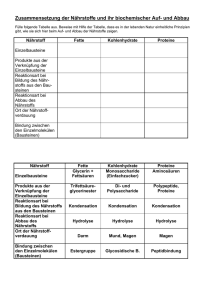
C.3.5. Sekundärreaktionen
Reaktion von Xenobiotika und deren Metaboliten mit Makromolekülen, sog. Konjugation
(in einigen Fällen können Xenobiotika auch untereinander reagieren; Frage der Verdünnung)
Biotransformation in H2O-lösliche Derivate → Ausscheidung (Eliminierung, "Entgiftung")
Beilage (S. 15a)
Diskussion einiger komplexerer Beispiele - Abbau von Ethen-bis-thiocarbamat
- Umwandlung von DDT
- Abbau von Parathion
- Abbau von aromatischen KW
- Abbau von polycyclischen Aromaten
- Abbau von 2,4-D
Beilagen (S. 16a – 16c)
OH
- 17 -
Fallstudie: Chlorphenoxyessigsäuren
X
OCH2COOH
Cl
Cl
X = H: "2,4-D"
X = Cl: "2,4,5-T"
Verbindungen sind Wachstumshormone → Einsatz als potente Herbizide (Pflanze wächst sich
"zu Tode"); rasche Wirkung, problemloser Abbau (τ1/2 = 1-2 Monate)
X
OCH2COOH
Cl
X
biotisch
oder hν
Cl
OH
COOH
+
Cl
COOH
Cl
(Oxalsäure)
rascher mikrobieller Abbau
(s. Beilage)
somit eigentlich "ideales" Herbizid
Problematik: Synthese!
X
Cl
Cl
Cl
X
NaOH
OH
Cl
Cl
X
O
Cl
Cl
Cl-CH2-COOH
2,4-D
2,4,5-T
erwünschte Reaktion
unerwünschte Nebenreaktion:
X
O
X
O
Cl
Cl
Cl
Cl
O
X
2
Dioxine
R = Cl: 2,3,7,8-Tetrachlorodibenzodioxin
(TCDD, "Dioxin")
Bildungsmengen abhängig von der Reaktionsführung (Druck, Temperatur):
T < 500° → 5000 ppm TCDD, T > 800° → 40 ppm
weitere Nebenprodukte:
Cl
Cl
Cl
O
Cl
O
Polychlor-dibenzofurane
Polychlor-diphenylether
- 19 -
Toxische Wirkungen (Auswahl):
Hg:
MeHgCl aus Abwässern angereichert in Thunfisch → ZNS-Schäden ("Minimata-Krankheit",
Japan)
Cd:
Anreicherung in Reis (0,3 ppm) nach Bewässerung aus Bergwerk-Abraumhalden →
Zerstörung des Knochenmarks, rasche Entkalkung, Knochenschrumpfung, sehr
schmerzhaft ("Itai-Itai-Krankheit", Japan)
Pb:
Antiklopfmittel, Wasserleitungen, Farben → ersetzt Calcium in der Knochensubstanz
(Erweichung)
Sn:
Farbanstriche (z.B. Schutz bei Schiffen) → ZNS-Schäden, Fische, Molluskizide Wirkung,
Fertilität (!)
Kurse über Umwelt-Toxikologie (PK3, SK4).
Zusammenfassung
Dispersion, Akkumulation
Reaktionsverhalten
Wirkung
stark abhängig von den physikalischen Eigenschaften
(Kow, Sdp., pKa, etc.); diese folgen aus der Struktur
extrem abhängig von der Struktur und den Reaktionsbedingungen
→ Chemie (elektronische, sterische Effekte)
abhängig von chemischen und physikalischen Eigenschaften einer Verbindung,
nie voraussehbar! (Zelle, Organismus, Individuum? Population, Ökosystem?)