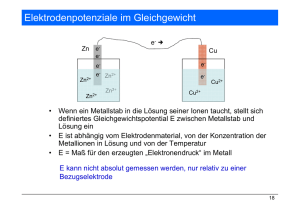
Vorlesung: Allgemeine Chemie für Molekularbiologen
Werbung

Allgemeine Chemie für Molekularbiologen Univ.Prof.Dr Robert Konrat Institut für Theoretische Chemie und Molekulare Strukturbiologie Rennweg 95b, A-1030 Wien Tel.: 01-4277-53002 Email: robert.konrat.univie.ac.at Homepage: http://www.itc.univie.ac.at Allgemeine Chemie für Molekularbiologen Wintersemester 2002/2003 Literatur: Chemie.Das Basiswissen der Chemie (Charles E. Mortimer) (Thieme Verlag, 2001, 7.Auflage) Chemie für Biologen (B.K.Keppler & A.Ding) (Spektrum, Akademischer Verlag, 1997) Was ist Chemie? Chemie:“…….Charakterisierung, Zusammensetzung und Umwandlung von Stoffen“ Gegenstand der Chemie sind die Zusammensetzung und die Struktur von Substanzen sowie die Kräfte, die sie zusammenhalten. Die physikalischen Eigenschaften der Substanzen werden untersucht, denn sie liefern Auskünfte über die Struktur, dienen zu ihrer Identifizierung und Klassifizierung und zeigen Anwendungsmöglichkeiten auf. Chemische Reaktion: …..Stoffumwandlung ( wann, wie schnell, unter welchen Umständen, warum?, Selektivität (gewünschtes Hauptprodukt-unerwünschtes Nebenprodukt) Was ist Chemie? …….Die Chemie hat als Aufgabe und Ziel, das Zusammenwirken der Atome zu verstehen, welches letztendlich das, was wir als Materie, sei sie anorganisch oder biologisch, begreifen, umfasst…… (A Weiss, Vorsitzender der Dt.Bunsen-Gesellschaft) …….Insbesondere die Chemie, aber auch Teile der Biochemie, Astrophysik oder auch Pharmazie (um nur einige zu nennen), ist die Lehre vom Verhalten von Elektronen und Atomkernen ((und ihrer Wechselwirkung miteinander))…… (HW Preuss, Universität Stuttgart) Die Betrachtung der Erscheinungsformen der Materie und ihrer Umwandlungen unter dem Gesichtspunkte der atomaren und molekularen Strukturen, dies ist Chemie. (A von Zelewsky, Universität Freiburg) Aminosäuren Proteine Strukturbiologie Geschichte der Chemie Frühe Anfänge & Antike 2000 v.Chr. Bronzezeit (Kupfer/Zinn) Bronze konnte zum Bau von Werkzeugen und Speerspitzen, Nadeln und Schneidegeräten benutzt werden und löste die Steinwerkzeuge ab. Geschichte der Chemie Frühe Anfänge & Antike 2000 v.Chr. Bronzezeit 1500 v.Chr. Eisenzeit Die Gewinnung von Eisen aus Eisenerz war ein gewaltiger technischer Fortschritt. Durch Erhitzen des Erzes in einem Kohlefeuer wird es reduziert und es entsteht das Metall Eisen Geschichte der Chemie Frühe Anfänge & Antike 2000 v.Chr. Bronzezeit 1500 v.Chr. Eisenzeit 1250 v.Chr. Tapputi-Belatekallin (Parfümherstellerin; „1.Chemikerin“) Tapputi ist die erste schriftlich erwähnte (und namentlich genannte) Chemikerin der Geschichte. Ihr Name findet sich auf einer Keilschrifttafel des alten Mesopotamien, zusammen mit detaillierten Erklärungen der technologischen Aspekte der Parfümherstellung. Geschichte der Chemie Frühe Anfänge & Antike 2000 v.Chr. Bronzezeit 1500 v.Chr. Eisenzeit 1250 v.Chr. Taputti-Belatekallin 6. Jhdt. v.Chr. Thales v. Milet („Naturphilosophie“) Thales ging davon aus, dass hinter aller Vielfalt der Welt eine grundlegende Einheitlichkeit steht (Wasser) Anaximenes (Luft), Heraklit (nichtstofflicher Natur, Feuer), Empedokles (Wasser, Luft, Feuer und Erde), Schüler Pythagoras‘; E. behauptete, nichts in der Welt werde erschaffen oder zerstört – und alle Dinge bestünden aus den 4 Elementen in unterschiedlicher Kombination! Geschichte der Chemie Frühe Anfänge & Antike 2000 v.Chr. Bronzezeit 1500 v.Chr. Eisenzeit 1250 v.Chr. Taputti-Belatekallin 6.Jhdt. v.Chr. Thales v. Milet 5.Jhdt. v.Chr. Leukipp & Demokrit Geht zurück auf die eleatische Philosophie (Parmenides, Zenon), die die veränderliche Welt als bloßen Schein auffaßte. Das Sein/Seiende ist der sinnlichen Erfahrung entrückt und daher nicht-gegenwärtig. Erde:Feststoff; Wasser:Flüssigkeit; Luft:Gas; Feuer:Energie „praktische Klassifizierung der Stoffe“. In der Antike gab es bereits eine chemische Praxis aber keine theoretische Grundlage. Bekannte Elemente waren: z.B. Gold, Silber, Kupfer, Eisen, Blei, Arsen, Quecksilber. Erst Leukipp stellte die Frage nach der Unteilbarkeit der Materie und gelangte zur Vorstellung vom atomos, dem Unteilbaren. Demokrit erweiterte die Theorie seines Lehrers. Es gibt eine unendliche Menge von Atomen, die im Raum in ständiger Bewegung sind, außerdem gibt es unendlich viele Arten von Atomen, alle unterschiedlich in Form und Größe, Gewicht und Temperatur. Sämtliche sichtbaren Veränderungen in der Welt sind Kombinationen und immer neue Kombinationen dieser unwandelbaren Atome. Geschichte der Chemie Frühe Anfänge & Antike 2000 v.Chr. Bronzezeit 1500 v.Chr. Eisenzeit 1250 v.Chr. Taputti-Belatekallin 6.Jhdt. v.Chr. Thales v. Milet 5.Jhdt. v.Chr. Leukipp & Demokrit 5.Jhdt. v.Chr. Sokrates, Platon & Aristoteles Platon: Idealismus (abstraktes Denken, abstrakte Geometrie) Aristoteles: gilt als das erste Universalgenie (Botanik, Geologie, Psychologie und Zoologie, Logik). Äther……flüchtig, ätherisch, luftig, Quintessenz („quinta essentia“, fünftes Element) Geschichte der Chemie Frühe Anfänge & Antike 2000 v.Chr. Bronzezeit 1500 v.Chr. Eisenzeit 1250 v.Chr. Taputti-Belatekallin 6.Jhdt. v.Chr. Thales v. Milet 5.Jhdt. v.Chr. Leukipp & Demokrit 5.Jhdt. v.Chr. Sokrates, Platon & Aristoteles 3.Jhdt. v.Chr. Epikur Epikur‘s Atomismus begrenzt die Zahl der Atomformen; für Größe und Gestalt gelten Ausschlußprinzipien (Materialismus) 1.Jhdt. v.Chr. Lukrez De Rerum Natura Geschichte der Chemie Alchemie („dunkles Zeitalter der Chemie?“) 331 v.Chr. Gründung von Alexandria Die Bibliothek war das Zentrum der antiken Gelehrsamkeit. Hier studierten die wichtigsten Geistesgrößen der hellenistischen Epoche (z.B. Euklid, Archimedes, Aristarch und Hypatia). In Alexandria traf das griechische Denken aber auch auf eine viel ältere Form der Gelehrsamkeit, die „ägyptische Kunst“ oder khemeia (daher unser Wort „Chemie“). Anfangs betraf dieses Wissen vor allem die chemischen Vorgänge beim Einbalsamieren der Toten, ihre Praktiken entwickelten sich aber schon weiter und bezogen auch andere chemische Vorgänge ein, die man im alten Ägypten entdeckt hatte, darunter die Herstellung von Glas, das Färben von Stoffen und insbesondere die Kunst der Metallbearbeitung. In Zusammenhang mit dem ägyptischen Totenkult wurde daraus ein metaphysisches Wissenssystem (Zusammenhang zwischen den 7 Elementen und den damals bekannten sieben Planeten). Man bezeichnete diese okkulten Praktiken auch als ‚hermetische‘ Künste. Ähnlich wie die Astrologen waren die ersten Alchemisten überzeugt, sie hätten mit ihrer Wissenschaft ein Geheimnis des Universums entdeckt (Beziehung zwischen Erde und Menschheit und den sie umgebenden Kosmos). Geschichte der Chemie Alchemie („dunkles Zeitalter der Chemie?“) 331 v.Chr. Gründung von Alexandria 3. Jhdt. v.Chr. Beginn der Alchemie Die Alchemie wurde eigentlich erst geboren, als die philosophische Tradition Griechenlands auf die khemeia traf. Eine der zentralen Bestrebungen war nicht die Suche nach spiritueller Weisheit, sondern die Verwandlung unedler Metalle in Gold (z.T. beruhend auf rätselhaften alten ägyptischen Texten). Es darf jedoch nicht vergessen werden, dass sich ironischerweise dennoch erste Ansätze einer naturwissenschaftlichen Praxis herausbildeten. Geschichte der Chemie Alchemie („dunkles Zeitalter der Chemie?“) 331 v.Chr. Gründung von Alexandria 3. Jhdt. v.Chr. Beginn der Alchemie ~200 v.Chr. Bolos von Mendes (Physica et Mystica) ~300 Zosimos von Panapolis Obwohl letztlich Dokumente der chemischen Scharlatanerie, zeugen diese frühen Schriften der Alchemisten doch von einem gewissen praktischen Wissen über chemische Prozesse. Beispielsweise findet man detaillierte Beschreibungen über Methoden wie Destillation, Filtrieren und Lösen. Ungewollt steht daher die Alchemie nicht nur am Beginn der Chemie sondern auch am Anfang der wissenschaftlichen Pharmazie, der chemische Ansatz zum Heilen. Eine alchemistische Praxis entwickelte sich aber unabhängig von Alexandria auch in anderen Teilen der Welt (Süd-und Mittelamerika, China, Indien). Was waren die Ziele der Alchemie und ihrer Entwicklung? Reichtum, Allheilmittel, Unsterblichkeit u.s.w. Die Alchemie war aber auch das Tätigkeitsgebiet, das uns die Chemie „bescherte“. Geschichte der Chemie Alchemie („dunkles Zeitalter der Chemie?“) 331 v.Chr. Gründung von Alexandria 3. Jhdt. v.Chr. Beginn der Alchemie ~200 v.Chr. Bolos von Mendes (Physica et Mystica) ~300 Zosimos von Panapolis 296 Verbot der Alchemie im Röm.Reich durch Kaiser Diokletian 391 Bibliothek von Alexandria wird geplündert und zerstört (Ende der alexandrinischen Alchemie) 431 Konzil von Ephesos Nestorianismus wird zur Ketzerei erklärt. Nestorianer, die weiterhin Alchemie betreiben, flüchten in den Osten Persiens. 529 Schliessung der Akademie Platons in Athen durch den christlichen Kaiser Justinian (Beginn des dunklen Mittelalters) 670 Belagerung Konstantinopels durch die arabische Flotte. Zerstörung der Flotte durch den Alchemist Callinicus („griechisches Feuer“, destilliertes Rohöl, Petroleum, Kaliumnitrat und gebrannter Kalk) Geschichte der Chemie Alchemie („dunkles Zeitalter der Chemie?“) > 700 Blütezeit der arabischen Naturwissenschaft In ihrem Kern gründet sich die arabische Alchemie auf Die Smaragdtafel, ein Werk des sagenumwobenen Hermes Trismegistos (gr. für „Hermes der Dreimalgroße“). khemeia wird zu al-chemia Djabir ibn Hajjan („Geber“) al-iksir (Elixier). Zuerst ein Stoff (Tinktur), der unedle Metalle in Gold verwandeln kann, später galt das Elixier als Arznei, die viele Krankheiten heilen konnte, dann als Allheilmittel und schließlich als „Lebenselixier“. Das arabische Denken erreichte damit völlig unabhängig das gleiche Stadium, in dem die chinesische Alchemie schon ungefähr 800 Jahre früher angekommen war. Djabir untersuchte z.B. Salmiak (Ammoniumchlorid), Essigsäure und Salpetersäure. Geschichte der Chemie Alchemie („dunkles Zeitalter der Chemie?“) ~900 Blütezeit der arabischen Naturwissenschaft ar-Razi (Rhazes) Er war Oberarzt des größten Spitals in Bagdad und die Medizin verdankt ihm sowohl diagnostische als auch praktische Leistungen. Er erkannte als Erster den Unterschied zwischen Masern und Pocken und beschrieb die Verwendung von Gipsverbänden zur Heilung von Knochenbrüchen. Für die Chemie war sein entscheidender Beitrag die Systematisierung des Fachgebietes. In seiner berühmten Schrift, Das Geheimnis der Geheimnisse, beschreibt Rhazes (1) Apparate und Instrumente, die bis ins 19.Jhdt. den Standard darstellten; (2) chemische Verfahren wie z.B. Destillation, Sublimation und Auflösen fester Stoffe; (3) Klassifizierung aller damals bekannten Chemikalien und Mineralien. Geschichte der Chemie Alchemie („dunkles Zeitalter der Chemie?“) ~900 Blütezeit der arabischen Naturwissenschaft Avicenna (Ibn Sina) Lieferte bedeutende Beiträge zu Medizin, Philosophie, Physik, Politik und Alchemie. Sein wichtigster Beitrag zur Alchemie bestand darin, die Transmutation (Umwandlung unedler Metalle in Gold) in Frage zu stellen. Er formulierte z.B. auch Newton‘s erstes Bewegungsgesetz (das Trägheitsgesetz). Vor allem aber war Avicenna ein überragender Mediziner. Ebenso wie Rhazes hielt er die Medizin für eine Wissenschaft. Avicenna verfasste eine umfassende Aufstellung chemischer Substanzen und ihrer Wirkungen bei der Einnahme als Medikament sowie der Krankheiten, die man mit ihnen heilen konnte. Sein Arzneibuch galt lange als Standardwerk. Geschichte der Chemie Alchemie („dunkles Zeitalter der Chemie?“) 1095 Rückeroberung Toledos durch die Spanier Als das arabische Reich seinen Niedergang erlebte und zerfiel, gingen seine bedeutenden Beiträge zu Naturwissenschaft und Mathematik zu Ende. Die Werke der arabischen Gelehrten gelangten nach und nach in lateinischer Sprache nach Europa (z.T. auch unbekannte Griechische Werke, die nur in arabischer Sprache erhalten geblieben waren). In Europa herrschte zu dieser Zeit eine starre, hierarchische Gesellschaftsordnung, mit der Kirche im Mittelpunkt. Nach ihrer Lehre stand die Erde im Mittelpunkt des Kosmos, und die Antworten auf alle Fragen lagen allein bei Gott. Es war weniger eine wissenschaftsfeindliche als vielmehr eine unwissenschaftliche Epoche. Wissenschaft wurde nur am Rand der von Religiosität durchdrungenen Gesellschaft praktiziert und wahrgenommen. Die Alchemie war einerseits wie geschaffen für die mittelalterliche Denkweise, durch die Verbindung von Metaphysik und Wirklichkeit. Jedoch wollte die Alchemie in frevelhafter Weise die Wirklichkeit auch verändern. Geschichte der Chemie Alchemie („dunkles Zeitalter der Chemie?“) 1095 Rückeroberung Toledos durch die Spanier 1200 Albertus Magnus In Süddeutschland geboren, studierte A.Magnus in Padua und wurde zum ersten großen Alchemisten Europas, und auch heilig gesprochen. A.M. war ein bedeutender Lehrer (Thomas von Aquin war einer seiner Schüler) und ist bis heute der Schutzheilige der Wissenschaftler. Seine chemischen Leistungen waren: höchstwahrscheinlich war er der Erste, der Arsen isolierte. Für A.M. war die Alchemie die einzige wissenschaftliche Tätigkeit, mit deren Hilfe man Erkenntnisse über die Zusammensetzung und den Aufbau der Welt zu finden versuchte. Er war vermutlich der Erste, der das Einzigartige der chemischen Veränderungen erkannte. Geschichte der Chemie Alchemie („dunkles Zeitalter der Chemie?“) 1095 Rückeroberung Toledos durch die Spanier 1200 Albertus Magnus 1214 Roger Bacon Franziskanermönch in Oxford und Paris. Bacons Ideen weisen bemerkenswerte Ähnlichkeiten mit Gedanken auf, die Leonardo da Vinci skizzierte –Dampfschiffe, Autos, U-Boote und sogar Flugapparate. Sein Hauptwerk Opus Majus war lange Zeit verboten und erschien im Jahre 1733! R.B. hat vermutlich in Europa (unabhängig von China) das Schiesspulver entdeckt. Er glaubte an die Transmutation. Geschichte der Chemie Alchemie („dunkles Zeitalter der Chemie?“) 1095 Rückeroberung Toledos durch die Spanier 1200 Albertus Magnus 1214 Roger Bacon 14.Ihdt. Arnaldus von Villanova „Stein der Weisen“ – Es gibt in der Natur eine gewisse Substanz, welche, wenn sie entdeckt und durch die Kunstfertigkeit in ihren vollkommenen Zustand gebracht wird, alle unvollkommenen Körper, welche sie berührt, zur Vollkommenheit verwandelt. Im Laufe der Eroberung Konstantinopels im 4.Kreuzug (1204) gingen unzählige griechische und byzanztinische alchemistische Manuskripte verloren. Dies führte zu einer Neuorientierung und Weiterentwicklung der Alchemie in Europa. Obwohl wissenschaftlich völlig unzulänglich, entwickelten die europäischen Alchemisten die Grundlagen der Chemie im modernen Sinne. Geschichte der Chemie Alchemie („dunkles Zeitalter der Chemie?“) 1095 Rückeroberung Toledos durch die Spanier 1200 Albertus Magnus 1214 Roger Bacon 14.Ihdt. Arnaldus von Villanova 14.Ihdt. Systematische chemische Fortschritte Herstellung von Alkohol (durch A. v. Villanova),Schwefelsäure und Salpetersäure Das Elixier wurde zur zentralen „Substanz“ – Geburtsstunde der Pharmazie (in den Schriften von Johannes von Rupescissa) Geschichte der Chemie Übergangszeit 1493 Theophrastus Bombastus von Hohenheim („Paracelsus“) Iatrochemie (iatros, gr. Arzt), verfolgte das Ziel, die Chemie zum Kernstück der medizinischen Tätigkeit zu machen. Obwohl seine Erkenntnisse eine Mischung aus Altem und Neuem, Kühnem und Verrücktem waren, war er doch von enormer Bedeutung. Vor allem forderte Paracelsus, dass Verbindungen auf genau festgelegte Weise aus reinen Chemikalien herzustellen seien. P. erkannte, dass Zink ein Metall ist. Er beschrieb Kobalt und Wismut (auch wenn er nicht der Entdecker war). Dennoch erkannte er nicht, dass es sich dabei um Elemente handelte. In der Medizin/Pharmazie war P. von noch größerer Bedeutung, zahlreiche Präparate gehen auf ihn zurück: Zinkund Kupfersalze, Blei-und Magnesiumverbindungen, Arsenpräparate gegen Hautleiden und, vor allem, Quecksilber zur Syphilisbehandlung. Obwohl einerseits „modern“, war P dennoch in der mittelalterlichen Denkweise verhaftet: „Signaturenlehre“: z.B. Fliederblätter waren herzförmig, daher eignen sie sich zur Behandlung von Herzkrankheiten, das Schöllkraut mit seinem „gelben Blut“ war die Arznei gegen Gelbsucht. Geschichte der Chemie Der Beginn eines neuen wissenschaftlichen Zeitalters 13. Jhdt. Dietrich von Freiberg (Theodoricus Teutonicus) Dominikanermönch in Freiberg/Sachsen. In seinem Werk De Iride („Über den Regenbogen“) verbinden sich Mathematik und naturwissenschaftliche Beobachtung zu dem ersten bedeutenden Werk über Optik seit Aristoteles (optische Fragen sind auch in späteren Zeiten –bis heute- immer wieder Ausgangspunkt revolutionärer Gedanken). Geschichte der Chemie Der Beginn eines neuen wissenschaftlichen Zeitalters 13. Jhdt. Dietrich von Freiberg (Theodoricus Teutonicus) 1401 Nikolaus von Kues Ein sehr fortschrittlicher Denker, der in seinem späteren Leben Bischof von Brixen wurde. Neben seinen philosophischen Arbeiten machter er Vorschläge zur Kalenderreform, konstruierte konkave Brillengläser, schlug vor, die Pulsfrequenz bei der Diagnose zu berücksichtigen, und zeichnete eine der ersten einigermaßen zuverlässigen Landkarten Europas. Der Höhepunkt seiner praktischen Arbeiten war jedoch ein brillanter Versuch mit der Waage, der für die Chemie entscheidende Konsequenzen haben sollte. Er wog jeden Tag eine Pflanze und beobachtete, dass sich die Pflanze offenbar von Luft ernährte und dass Luft somit ein Gewicht hat. Geschichte der Chemie Der Beginn eines neuen wissenschaftlichen Zeitalters 13. Jhdt. Dietrich von Freiberg (Theodoricus Teutonicus) 1401 Nikolaus von Kues 16.Jhdt. Giordano Bruno (1548, Nola/Neapel; Dominikanermönch) („Personifizierte Widersprüchlichkeit“). Einerseits war er der Prophet der naturwissenschaftlichen Revolution, andererseits war er inspiriert von Mystizismus und Alchemie (Schriften von Hermes Trismegistos). Bedeutsam für die europäische Geistesgeschichte wurde Bruno durch seine Methode des systematischen Denkens, einer Art kreativer Logik, um zu neuen Erkenntnissen zu kommen. Der Ursprung dieser Überzeugung liegt in Nikolaus von Kues‘ Coincidentia Oppositorum, also der Methode, die Dinge unter dem Gesichtspunkt der Vereinigung von Gegensätzen zu betrachten: „Tiefe Magie ist es, das Gegenteil herauszuziehen, nachdem man den Punkt der Vereinigung entdeckt hat“. Geschichte der Chemie Der Beginn eines neuen wissenschaftlichen Zeitalters 13. Jhdt. Dietrich von Freiberg (Theodoricus Teutonicus) 1401 Nikolaus von Kues 16.Jhdt. Giordano Bruno 17.Jhdt. Galileo Galilei, René Descartes, Francis Bacon (1561-2626) B. propagierte (Novum Organum ) ein induktives Vorgehen, um von Beobachtungen zu prinzipielleren und abstrakten Erkenntnissen, zu den ‚Formen‘ der Natur zu gelangen und dadurch Einblick in die Ursachen der Naturprozesse zu gewinnen. Bacon erkannte, dass die Erforschung der Materie (Chemie) im Grundsatz eine praktische Tätigkeit ist und man sich daher auf die Methoden und Entdeckungen der Alchemisten stützen müsste. Dies markiert den Beginn der modernen Chemie! Bacon formulierte auch die praktische Bedeutung der Wissenschaft und deren gesellschaftliche Nützlichkeit, er war auch von der ethischen Vervollkommnung der Gesellschaft durch die Wissenschaft überzeugt. Geschichte der Chemie Moderne Chemie 17.Jhdt. Johan Baptista van Helmot, Franciscus Sylvius, Tachenius Van Helmont verwendete die Waage, um ‚Reaktionen‘ quantitativ zu verfolgen, entdeckte Kohlendioxyd (ohne es so zu nennen) und weitere Gase (leitet sich aus dem gr. Wort für Unordnung, „Chaos“ ab). Dies war ein Zeichen seines Mystizismus‘. Van Helmont erkannte auch eine grundlegende Eigenschaft der Materie: Sie konnte sich zwar während der Experimente verwandeln, aber sie ging niemals verloren (Gesetz der Erhaltung der Masse). Sylvius erkannte, dass viele natürlich vorkommenden Salze durch Reaktionen zwischen Säuren und Basen entstanden waren. Als zusammen gesetzte Stoffe unterschieden sie sich also von Substanzen, die man nicht weiter zerlegen konnte. „Unsterblich“ wurde Sylvius mit seinem Allheilmittel für Nierenerkrankungen: Kornschnaps, der mit Wacholderbeeren aromatisiert war (niederl. Genever –engl. Gin) Durch die Arbeiten von Van Helmont, Sylvius und Tachenius wurde die Chemie zu einer eigenständigen Wissenschaft! Geschichte der Chemie Moderne Chemie 17.Jhdt. Johan Baptista van Helmot, Franciscus Sylvius, Tachenius 1627 Robert Boyle DER Begründer der modernen Chemie! Mit seinem (ebenfalls berühmten) Assistenten Robert Hooke führte Boyle die berühmt gewordenen Gasversuche durch, die heute als BoyleMariotte-Gesetz bezeichnet werden. Er stellte dabei fest, dass Gase Elastizität besitzen und er erkannte, dass, wenn man ein Gas zusammendrücken kann, so muss es aus einzelnen Teilchen bestehen, die sich im leeren Raum bewegen (Allerdings hatten beide einen Vorläufer in der Antike: Heron von Alexandria, 1.Jhdt) In seinem Hauptwerk The Sceptical Chymist lehnte er die aristotelische Theorie der vier Elemente und auch die von Paracelsus erdachte Version mit drei Elementen ab. Stattdessen bezeichnete er in seiner berühmten Definition Elemente als „bestimmte primitive und einfache, oder vollkommen unvermischte Körper; welche nicht aus irgendwelchen anderen Körpern oder auseinander bestehen, welche die Zutaten sind, aus denen sich all jene als vollkommen gemischte Körper bezeichneten unmittelbar zusammensetzen, und in welche sie letztlich zerlegt werden“. Oder: Jede Substanz, die sich nicht mehr in eine einfachere Substanz zerlegen ließ, ist demnach ein Element. Allerdings war auch Boyle ein Anhänger der Alchemie und der Transmutation!!! Geschichte der Chemie Moderne Chemie 17.Jhdt. Johan Baptista van Helmot, Franciscus Sylvius, Tachenius 1627 Robert Boyle 1669 Entdeckung des Phosphors durch Henning Brand 1770 Carl Scheele (Schweden) stellt Chlor her. In weiterer Folge noch: (Molybdän), Mangan, (Wolframsäure), Stickstoff ( zuvor auch durch Daniel Rutherford) und Sauerstoff (wird fälschlicherweise Joseph Priestley zugeschrieben) 1751 Axel Cronstedt erzeugt Nickel und führt zur Analyse das Lötrohr ein (mineralspezifische Färbung; Vater der Mineralogie) 1735 D‘Elhuyar (Fausto und José) Wolfram und Platin Geschichte der Chemie Moderne Chemie 17.Jhdt. Johan Baptista van Helmot, Franciscus Sylvius, Tachenius 1627 Robert Boyle 1669 Entdeckung des Phosphors durch Henning Brand 1770 Carl Scheele 1751 Axel Cronstedt 1735 D‘Elhuyar (Fausto und José) 18.Jhdt. Phlogistontheorie (Metalle=Metallkalk und Phlogiston) Johann Becher & Georg Stahl 18.Jhdt. Henry Cavendish entdeckt Wasserstoff, durch Einwirkung bestimmter Säuren auf Metalle (Er und Priestley zeigen unabhängig voneinander, dass Wasser aus „Luft“ und „brennbarer Luft“ besteht). 18.Jhdt. Joseph Priestley (Pfarrer) Entdeckte Mineralwasser (CO2), Sauerstoff, Stickstoffoxid, Chlorwasserstoff, Ammoniak und Schwefeldioxid. Geschichte der Chemie Moderne Chemie 1743 Antoine de Laurent de Lavoisier Verwendete die Waage als Präzisionsinstrument und entwickelte daraus seine Oxidationstheorie, die beispielsweise die Verkalkung der Metalle und auch die Bildung von Wasser aus „brennbarer Luft“, Wasserstoff, und Sauerstoff, principe oxygène, verständlich machte. Dies war die Grundlage für ein quantitatives Verständnis der chemischen Vorgänge. (Lavoisier räumte allerdings ein, vielleicht niemals herausfinden zu können, „Was“ eigentlich ein Element sei). 1799 Joseph Louis Proust Gesetz der konstanten Proportionen 1803/1807 John Dalton‘s Atomtheorie 19.Jhdt. Jöns Jakob von Berzelius entwickelt die Elektrolyse und entdeckte ausserdem Cer, Selen und Thorium. Berzelius erfand auch die chemische Schreibweise (Symbole), damit hatte die Chemie den gleichen Schritt vollzogen wie die Mathematik beim Wechsel von den römischen zu den arabischen Zahlen. Jetzt hielt die Mathematik Einzug in den innersten Kern der Chemie und versetzte sie in die Lage, ihre Reaktionen genau zu verfolgen. Aber: („Vitalismus“) Geschichte der Chemie Moderne Chemie 19.Jhdt. 19.Jhdt. Friedrich Wöhler synthetisiert Harnstoff, ein organisches anorganischen Ausgangsstoffen. Molekül, Periodensystem der Elemente Dmitrij Iwanowitsch Mendelejew (Vorarbeiten von Johann Döbereiner, Alexandre Béguyer de Chancourtois, John Newlands) aus Emile Definitionen Materie: ausgedehnte Masse; Materie besteht aus Stoffen. Masse: ein Mass für die Menge eines Stoffes und auch ein Mass für die Trägheit (Widerstand gegenüber Bewegung) Gewicht: Anziehungskraft, die die Erde auf einen Körper ausübt (proportional zur Masse des Körpers) Stoffe: bestehen aus einer begrenzten Anzahl einfacher Stoffe, Elemente. „Elemente sind bestimmte primitive und einfache, völlig unvermischte Körper; sie enthalten keine anderen Körper; sie sind die Zutaten, aus denen alle perfekt gemischten Körper zusammengesetzt sind und in welche diese letztlich zerlegt werden“, Robert Boyle (The Sceptical Chymist, 1661). Definitionen Element: ein Stoff, der in keine einfacheren Stoffe zerlegt werden kann Verbindung:entsteht durch das Zusammenfügen von Elementen in definierter Zusammensetzung. Element Symbol Massenanteil (%) Sauerstoff O 49.2 Silicium Si 25.7 Aluminium Al 7.5 Eisen Fe 4.7 Calcium Ca 3.4 Natrium Na 2.6 Kalium K 2.4 Kohlenstoff (C, 0.09); Wasserstoff (H, 0.9); Phosphor (P, 0.1) Definitionen Verbindungen ….bestehen aus Elementen in definierter Zusammensetzung (Gesetz der konstanten Proportionen, Joseph Proust, 1799). Wasser = Wasserstoff:Sauerstoff = 11,19%:88,81% Verbindungen haben andere Eigenschaften als die sie aufbauenden Elemente! Definitionen Stoffe Elemente und Verbindungen sind reine Stoffe. Alle anderen Stoffe sind Gemische. (reine Stoffe in variablem Mengenverhältnis) Heterogenes Gemisch Homogenes Gemisch Unterschiedliche Teile Einheitlich (Lösungen) Sand/Eisenpulver Gold-Silber Legierung Definitionen Phase: abgegrenzte Menge eines einheitlichen oder homogenen Stoffes Phasengrenzfläche: definierte Grenzfläche zwischen den Phasen eines heterogenen Gemisches (z.B. Granit: farblose Quarz-, schwarze Glimmer- und rosafarbene Feldspatkristalle). Materie Phys.Trennung heterogene Gemische homogene Stoffe Phys.Trennung Lösungen (homogenes Gemisch) Reine Stoffe chem.Trennung Verbindung Element Heterogene Gemische fest + fest Gemenge Granit, Sand+Salz Sortieren, Sieben fest + flüssig Suspension Schlamm,Malerfarbe Dekantieren flüssig + flüssig Emulsion Milch fest + gasförmig Aerosol Rauch Filtrieren flüssig + gasförmig Aerosol Nebel, Schaum Sedimentieren Zentrifugieren Atomtheorie POSTULATE: (1) Chemische Elemente bestehen aus kleinsten, nicht weiter zerlegbaren Teilchen, den Atomen. Alle Atome eines Elements sind einander gleich, besitzen also gleiche Masse und gleiche Gestalt. Atome verschiedener Elemente haben verschiedene Eigenschaften. Jedes Element besteht also aus nur einer für das Element typischen Atomsorte. (2) Bei einer chemischen Reaktion werden Atome miteinander verbunden oder voneinander getrennt. Dabei werden nie Atome zerstört oder neu gebildet, und kein Atom eines Elements geht in das eines anderen Elements über. (3) Chemische Verbindungen entstehen durch Reaktion verschiedener Elemente. Eine gegebene Verbindung enthält immer die gleichen Atomsorten, die in einem festen Mengenverhältnis miteinander verknüpft sind. Daltons Theorie ist heute noch gültig, wenn auch das erste Postulat etwas modifiziert wurde. Nach heutiger Kenntnis bestehen die meisten Elemente aus verschiedenen Atomsorten, die sich in ihren Massen unterscheiden (Isotope). Atomtheorie GRUNDGESETZE: (1) Gesetz der Erhaltung der Masse. Während einer chemischen Reaktion bleibt die Gesamtmasse konstant. (Summe der Massen der Ausgangsstoffe/Edukte = Summe der Massen der Produkte) (P2). (2) Gesetz der konstanten Proportion (J.Proust, 1799). In einer Verbindung sind stets die gleichen Elemente im gleichen Massenverhältnis enthalten (P3). (3) Gesetz der multiplen Proportionen. Wenn zwei Elemente A und B mehr als eine Verbindung miteinander eingehen, dann sind die jeweiligen Massenverhältnisse ganzzahlig . (Kohlenmonoxid CO; Kohlendioxid CO 2) Bausteine der Atome Humphry Davy entdeckte 1807-1808 die fünf Elemente Natrium, Kalium, Calcium, Strontium und Barium, als er bestimmte Verbindungen mit elektrischem Strom zersetzte, daher schloß er auf elektrische Anziehungskräfte zwischen den Elementen. Michael Faraday (1832-1833) entdeckte die Gesetze der Elektrolyse und fand einen Zusammenhang zwischen zersetzter Stoffmenge und eingesetzter Strommenge. Darauf basierend schlug George Johnstone Stoney 1874 die Existenz von elektrischen Ladungsträgern vor, die mit Atomen assoziiert sind. 1891 gab er diesen Ladungsträgern den Namen Elektronen. (gr. Elektron=Bernstein, geht zurück auf frühe Versuche von William Gilbert (Ende des 16.Jhdts)). Bausteine der Atome (subatomare Teilchen) Vakuum 1859 Julius Pflücker Kathodenstrahlen Kathode (-) (+) Anode Schnell Teilchen bewegte, Elektronen Emittierte Elektronen sind unabhängig von der Zusammensetzung der Kathode negativ geladene Magnetfeld Elektron N Kathode (-) S Anode (+) Anode (+) (-) Kathode (-) (+) Ablenkung ist abhängig von Ladung q und Masse m (Elektrons) 1897 Joseph Thomson q/m = -1,7588 108 C/g 1909 Messung der Ladung q des Elektrons durch Robert Millikan q = -1,6022 10-19 C Elektrisches Feld Proton Strahlen positiver Ionen (Kanalstrahlen) wurden erstmals 1886 von Eugen Goldstein beobachtet. Die Ablenkung der Kanalstrahlen im elektrischen und im magnetischen Feld wurde von Wilhelm Wien (1898) und J.J.Thomson (1906) untersucht. Sie bestimmten für Wasserstoff Werte für q/m = 9,5791 104 C/g Neutron Da Atome elektrisch neutral sind, muß ein Atom gleich viele Elektronen wie Protonen enthalten. Die tatsächlichen Massen der Atome (ausgenommen Wasserstoff) sind größer als die Summe der Massen der darin enthaltenen Protonen und Elektronen. Daher wurde von Ernest Rutherfold (1920) die Existenz zusätzlicher, ungeladener Teilchen (Neutronen) postuliert. Der experimentelle Nachweis der Neutronen erfolgte durch James Chadwick (1932). Subatomare Teilchen Masse Gramm Atommasseneinheitena Ladungb Elektron 9,1094 10-28 0,00054858 -1 Proton 1,6726 10-24 1,007276 +1 Neutron 1,7649 10-24 1,008665 0 aEine bDie Atommasseneinheit (u) ist 1/12 der Masse des 12C Atoms Einheit der Ladung ist e = 1,602177 10-19 Coulomb Aufbau der Atome Natürliche Radioaktivität Manche Atome bestehen aus instabilen Kombinationen von Elektronen, Protonen und Neutronen und zerfallen unter Abgabe von Strahlung und werden dabei in Atome anderer Elemente umgewandelt. Diese Erscheinung nennt man Radioaktivität und wurde 1896 von Becquerel entdeckt. Rutherford erklärte die Herkunft der Strahlung und man unterscheidet: (α)-, (β)- und (γ)-Strahlung. α-Strahlen bestehen aus Teilchen mit etwa der 4-fachen Masse eines Protons und mit +2 Elementarladungen (d.h. 2 Protonen und 2 Neutronen; Austrittsgeschwindigkeit: 10000-30000 km/s). β-Strahlen bestehen aus Elektronen, die mit etwa 130000 km/s emittiert werden. γ-Strahlen sind Röntgenstrahlen) energiereiche elektromagnetische Strahlen (vergleichbar zu Aufbau der Atome (Rutherford-Atommodell) 1911 Ernest Rutherford Rutherford beschoß 0,004mm dicke Folien aus Aluminium, Gold, Silber oder Kupfer mit α-Strahlen. Während die Mehrzahl der Teilchen die Folie ungehindert durchquerte, wurden einige Teilchen seitwärts abgelenkt und manche in Richtung der Strahlenquelle zurückgeworfen. Atomkern Strahl von α-Teilchen abgelenktes α-Teilchen Aufbau der Atome (Rutherford-Atommodell) Dies führte zu folgenden Annahmen über den Aufbau der Atome: 1. Im Mittelpunkt des Atoms befindet sich ein Atomkern, in dem fast die gesamte Atommasse und die ganze positive Ladung konzentriert ist. Der Zusammenhalt wird durch die starke Kernkraft vermittelt. Sie ist eine der fundamentalen Kräfte der Natur, stärker als die elektrostatische Wechselwirkung und hat eine extrem kurze Reichweite. 2. Elektronen befinden sich ausserhalb des Atomkerns und umkreisen ihn in schneller Bewegung. Die Zahl der Elektronen ist gleich der Zahl der Protonen (Elektroneutralität). Dimensionen (Durchmesser) Atomkern: ~ 1 fm = 10-15 m Atom: ~ 100 – 400 pm = 100 10-12 – 400 10-12 m Aufbau der Atome (Atomsymbole) Ein Atom wird definiert durch: 1. Die Ordnungszahl Z ist gleich der Zahl der positiven Elementarladungen im Atomkern (= Zahl der Protonen). Im neutralen Atom ist die Zahl der Protonen gleich der Zahl der Elektronen. 2. Die Massenzahl A gibt die Gesamtzahl der Nucleonen, d.h. der Protonen und Neutronen zusammen an (und entspricht daher ungefähr der Atommasse in Atommasseneinheiten u, denn Protonen und Neutronen besitzen in etwa die gleiche Masse. 35 17 Cl Atom des Elements Chlor, das Z=17 Protonen und A=35 Nucleonen besitzt, die Zahl der Elektronen ist ebenfalls 17, die Zahl der Neutronen ist A-Z=18. Aufbau der Atome (Ionen) Ein elektrisch geladenes Teilchen, das aus einem oder mehreren Atomen besteht, wird Ion genannt. 32 16 S2- Ion eines Schwefelatoms, das 2 Elektronen aufgenommen hat und Z=16 Protonen und A=32 Nucleonen besitzt, die Zahl der Elektronen ist aber (durch die Elektronenaufnahme!) 18, die Zahl der Neutronen ist A-Z=16. Aufbau der Atome (Isotope) Für die chemischen Eigenschaften eines Atoms ist seine Ordnungszahl entscheidend, nicht jedoch die Masse. Bei einigen Elementen kommen Atome mit unterschiedlichen Massenzahlen (unterschiedliche Anzahl an Neutronen, aber gleiche Ordnungszahl) vor. Atome gleicher Ordnungszahl aber unterschiedlicher Massenzahl nennt man Isotope. 37 17 Cl 35 17 Cl Die chemischen Eigenschaften der Isotope sind ähnlich, Isotope können daher chemisch nicht unterschieden werden. Die meisten natürlichen Elemente bestehen aus Gemischen unterschiedlicher Isotope. Eine Trennung erfolgt aufgrund ihrer unterschiedlichen Masse. Massenspektrometer 1919 Francis W. Aston Die zu untersuchende Substanz wird verdampft, mit einem Elektronenstrahl ionisiert und die entstehenden Molekülionen durch eine angelegte Spannung beschleunigt. Ein Magnetfeld zwingt geladene Teilchen auf eine Kreisbahn, wobei –wie bei Kathodenstrahlen- die Ablenkung vom Verhältnis Ladung zu Masse (q/m) abhängig ist. Nur Ionen mit gleichem q/m-Verhältnis fliegen auf der gleichen Kreisbahn und können durch den Austrittsspalt hindurchtreten. Aufbau der Atome (Atommassen) Einzelne Atome können nicht gewogen werden, jedoch können relative Atommassen bestimmt werden. Dalton wählte Wasserstoff, später wurde Sauerstoff als Bezugselement herangezogen. Heute werden jedoch die relativen Atommassen auf das Kohlenstoffisotop 12 6 C bezogen. Die relativen Atommassen Ar (früher Atomgewichte) sind wichtig für die quantitative Beschreibung chemischer Veränderungen. Die Einheit (u im SIEinheitssystem) ist 1/12 des Kohlenstoffisotops (siehe oben). Anmerkung: Die Atommasse ist nicht einfach die Summe aus Elektronen, Protonen und Neutronen, sondern immer etwas kleiner. Die fehlende Masse (Massendefekt) entspricht der Bindungsenergie des Atomkerns. Diese Energie muss aufgewendet werden, um den Atomkern zu spalten. (Einstein: E=mc2) Aufbau der Atome (mittlere Atommassen) Für Elemente, die als Isotopengemisch in der Natur vorkommen, bestimmen das relative Verhältnis und die jeweiligen relativen Massen die mittlere Atommasse. z.B. Chlor besteht (immer!) zu 75,77% aus 35Cl (Masse: 34,969u) und zu 24,33% aus 37Cl (Masse: 36,966u). Die Atommasse von natürlichem Chlor ergibt sich daher zu: 37 17 Cl 24,33% x 36,966 u = 8,957 u 35 17 Cl 75,77% x 34,969 u = 26,496 u Mittlere Masse = 35,453 u Stöchiometrie Die Stöchiometrie befasst sich mit den Mengenverhältnissen der Elemente in Verbindungen und mit den quantitativen Beziehungen zwischen Elementen und Verbindungen in chemischen Reaktionen („chemisches Rechnen“). (gr. Stoicheion = Element; metron = Messen) Moleküle und Ionen Ein Molekül ist ein Teilchen, das aus zwei oder mehreren Atomen besteht, die fest miteinander verknüpft sind. Sie besitzen definierte chemische und physikalische Eigenschaften. Die Zusammensetzung eines reinen Stoffs wird mit seiner chemischen Formel angegeben, wobei man die folgenden Formelschreibweisen unterscheidet: Molekularformel H2O (wasser), H2 (Wasserstoff), O2 (Sauerstoff), Cl2 (Chlor), S8 (Schwefel), NH3 (Ammoniak) Strukturformel oder Konstitutionsformel H N H Ammoniak H Die relative Molekülmasse Mr (auch in Einheiten von u) ist gleich der Summe der relativen Atommassen aller Atome des Moleküls. Eine empirische Formel gibt das einfachste Zahlenverhältnis an, z.B. Wasserstoffperoxid (H2O2), die empirische Formel ist HO. Moleküle und Ionen Ein Ion ist ein Atom oder Molekül, das eine elektrische Ladung trägt. Man unterscheidet: Ein Kation ist positiv geladen und wird vom Minuspol (Kathode) einer elektrischen Spannung angezogen. Es entsteht durch Elektronenabgabe. Ein Anion ist negativ geladen und wird vom Pluspol (Anode) einer elektrischen Spannung angezogen. Es entsteht durch Elektronenaufnahme. Es gibt sowohl einatomige Ionen (Ca2+) und mehratomige Ionen (auch Molekülionen). NH +4 SO 24 Ammonium-Ion Sulfat-Ion OHHydroxid-Ion Moleküle und Ionen Ionische Verbindungen sind aus Anionen und Kationen aufgebaut und bilden im festen Zustand Kristalle (z.B. NaCl, Kochsalz). Ausschnitt aus einem Natriumchlorid-Kristall Das Mol Die Menge in Gramm eines Elementes, die dem Zahlenwert der relativen Atommasse entspricht, enthält stets die gleiche Zahl von Atomen, NA Atome. NA = Avogadro Zahl, = 6,02214 1023 mol-1 Die Stoffmenge, die aus 6,02214 1023 Teilchen besteht, nennt man ein Mol. Das Mol (eine SI-Basiseinheit) ist als diejenige Stoffmenge definiert, die aus genau so vielen Teilchen besteht, wie Atome in 12g von 126 C enthalten sind. Ein Mol einer molekularen Substanz besteht ebenfalls aus 6,02214.1023 Molekülen und hat die Masse in Gramm, deren Zahlenwert der relativen Molekülmasse entspricht. Die Masse eines Mols nennt man die molare Masse (oder Molmasse). z.B. Die relative Molekülmasse für Wasser beträgt Mr(H2O) = 18,015, d.h. in 18,015 Gramm Wasser sind NA H2O-Moleküle enthalten. (1 H2O-Molekül hat die Masse 18,015 u) Bei ionischen Verbindungen (keine Moleküle vorhanden!) bezieht man sich auf die molare Formelmasse. Das Mol Berechnung der molaren Formelmasse** von BaCl2 1 mol Ba2+ 2 mol Cl- = 137,3 g Barium 2 x 35,45 Cl- 1 mol BaCl2 = 70,9 g Chlor = 208,2 g BaCl2 ** molare (Formel)-Masse des Stoffes; Maßeinheit: Gramm pro Mol (g/mol) Wieviel Gramm sind 0,25 mol Schwefelsäure? M(H2SO4) = 98,08 g/mol m(X) = n(X)M(X) = 0,25 mol x 98,08 g/mol = 24,52 g Zusammensetzung von Verbindungen Wieviel % Eisen sind im Eisen(III)-Oxid enthalten? 1 Mol Fe2O3 enthält 2 mol Fe; 3 Mol O m(Fe)= 2 mol x 55,8 g/mol = 111,6 g m(O) = 3 mol x 16,0 g/mol = 48,0 g m(Fe2O3) = Massenanteil des Fe in Fe2O3 : = m(Fe)/m(Fe2O3) = 0,6993 (69,93 %) 159,6 g Ermittlung chemischer Formeln Bei der chemischen Analyse erhält man deren prozentuale Zusammensetzung (relativen Massenanteile), daraus kann die empirische Formel der Verbindung bestimmt werden: Welches ist die empirische Formel einer Verbindung, die 43,6% P und 56,4% O enthält? 1 Mol Fe2O3 enthält In 100g der Verbindung sind 43,6g P und 56,4g O enthalten. In mol sind das: n(P)= 43,6 g / 30,97 gmol-1 = 1,41 mol n(O)= 56,4 g / 16,00 gmol-1 = 3,53 mol 1,41/1,41 = 1.0 (P) & 3,53/1,41 = 2,50 (O) Empirische Formel (ganzzahliges Verhältnis!!!) : P2O5 Chemische Reaktion Bei der chemischen Reaktion werden Verbindungen ineinander übergeführt. Die Substanzen, die miteinander in Verbindung treten nennt man Reaktanden oder Edukte, die entstehenden Verbindungen heißen Produkte. Der Ablauf der Reaktion wird durch eine chemische Reaktionsgleichung beschrieben, wobei die Edukte auf der linken und die Produkte auf der rechten Seite stehen. 2 H2 + O2 2H2O Diese Gleichung beschreibt, dass aus 2 Molekülen Wasserstoff und 1 Molekül Sauerstoff 2 Moleküle Wasser entstehen. Die Zahlen vor den Molekülsymbolen sind die sogenannten Koeffizienten und bezeichnen die Zahl der beteiligten Moleküle. Um das Gesetz der Erhaltung der Masse zu erfüllen, müssen die Anzahl der Mole jedes Elements auf beiden Seiten übereinstimmen (4 Mol H und 2 Mol O). Chemische Reaktion O H H H H H + O H O O H H Chemische Reaktion 1. Anschreiben der Formeln aller Reaktanden, ein Pfeil und die Formeln aller Produkte. CS2 + Cl2 CCl4 + S2Cl2 Aggregatzustand wird angegeben: (g) gasförmig; (l) flüssig; (s) fest; (aq) in Wasser gelöst 2. Übereinstimmung der Molarität (links und rechts). CS2(l) + 3Cl2(g) CCl4 (l) + S2Cl2 (l) Chemische Reaktion (Verbrennungsprozesse) Bei der Verbrennung einer Substanz an der Luft tritt eine Reaktion mit Sauerstoff ein. Typischerweise entstehen dabei die folgenden Produkte: Kohlenstoff → CO2 (g) Schwefel → SO2 (g) Wasserstoff → H2O (g) oder H2O (l) Bei der Verbrennung von Ethan, C2H6(g), entstehen H2O(g) und CO2 (g), nach der Formel (und unter Berücksichtigung, dass Massenerhaltung, Übereinstimmung der Molaritäten und ganzzahlige Koeffizienten). 2 C2H6(g)+ 7 O2 (g) → 4 CO2 (g) + 6 H2O (g) Chemische Reaktion (Quantifizierung von Umsetzungen) Mithilfe der Reaktion I2O5 (s)+ 5 CO (g) → I2 (s) + 5 CO2 (g) kann die Menge Kohlenmonoxid (CO) in einer Gasprobe bestimmt werden. Wieviel Gramm CO sind vorhanden, wenn 0,192 g Iod (I2) gebildet werden? Molmassen: M(I2) = 254 gmol-1; M(CO) = 28,0 gmol-1 Wieviele Mole I2 werden gebildet? = 0,192 g / 254 gmol-1= 0,756 10-3 mol Nach der Reaktionsgleichung entstehen aus 5 Molen CO 1 Mol I2, daher entstehen 5 x 0,756 10-3 mol = 3,78 10-3 mol CO bzw. 3,78 10-3 mol x 28,0 gmol-1 = 0,106 g CO Chemische Reaktion (Reaktanden im Überschuß) Betrachten wir die Reaktion: 3 Fe + 4 H2O → Fe3O4 + 4 H2 Welcher Reaktand ist ausbeutebegrenzend, bei Einsatz von 4 mol Eisen und 5 mol Wasser? Fe: 4,0 mol / 3 mol = 1,33 > H2O: 5,00 mol / 4 mol = 1,25 Daher ist Wasser begrenzend für die Umsetzung während der Reaktion. Die tatsächliche, maximal umgesetzte Stoffmenge ist abhängig von der Menge an Wasser: Wasserstoff (4H2) = 1,25 x 4 = 5,00 mol. Eisen (obwohl im Überschuß) (3Fe) = 1,25 x 3 = 3,75 mol. Chemische Reaktion (Ausbeute einer chemischen Reaktion) Man unterscheidet eine theoretische (nach der Reaktionsgleichung) und eine tatsächliche Ausbeute. Die prozentuale Ausbeute ist das Verhältnis tatsächliche Ausbeute theoretische Ausbeute X 100 % Chemische Reaktion (Konzentration von Lösungen) Chemische Reaktionen werden (zumeist) in Lösung durchgeführt. Für die quantitativen Berechnungen sind daher Konzentration und Volumina entscheidend. Konzentration c = gelöste Stoffmenge n / Volumen der Lösung V Konzentration wird auch als Molarität bezeichnet. Einheit Mol pro Liter (mol/L) Wieviel Gramm Natriumhydroxid (NaOH) werden benötigt, um 0,45 L einer 0,3 molaren (mol/L) NaOH Lösung herzustellen? Molmasse M(NaOH): 40,0 gmol-1 c(NaOH) = n(NaOH)/0,45 L = 0,3 mol/L n(NaOH) = 0,45 L x 0,3 mol/L = 0,135 mol Menge m(NaOH) = n(NaOH) x M(NaOH) = 0,135 mol x 40,0 gmol-1 = 5,40 g Chemische Reaktion (Konzentration von Lösungen) Herstellung von Verdünnungen: Durch Verdünnung wird das Volumen vergrößert, aber die gelöste Stoffmenge n = c1V1 bleibt erhalten! c2 = n / V2 = c1V1 / V2 Welches Volumen einer Lösung mit c1(HCl) = 12,0 mol/L wird benötigt, um 500 ml einer c 2(HCl) = 3,0 mol/L herzustellen? V1 = c2 V2 / c1 = 3,00 mol/L x 0,5 L / 12,0 mol/L = 0,125 L = 125 mL Energieumsatz bei Chemischen Reaktionen Jede chemische Reaktion oder Stoffumsetzung ist begleitet von einer Energieumsetzung, wobei die freigesetzte oder aufgenommene Energie in verschiedenen Formen in Erscheinung treten kann (Licht, elektrische Energie, mechanische Energie oder Wärme). Die Thermochemie beschäftigt sich mit den ausgetauschten Wärmemengen. Eine Kraft F, die auf einen Körper mit der Masse m wirkt, führt zu einer Bewegungsänderung und Beschleunigung a. F = m.a (Einheit Newton N, 1N = 1 kgm/s2) Die entlang einer Wegstrecke s geleistete Arbeit W ist definiert als W = F.s (Einheit Joule,J, 1J = 1N.m = 1 kgm2/s2) Energieumsatz bei Chemischen Reaktionen Energie ist die Fähigkeit, Arbeit zu leisten. Energiequellen können sein: Bewegung (kinetische E.), Elektrizität (elektrische E.), Chemie (chemische E.) oder Wärme (Wärmeenergie). Energie kann von einer Form in die andere umgewandelt, aber weder erzeugt noch vernichtet werden (erster Hauptsatz der Thermodynamik). z.B. Um einen Körper mit der Masse m auf eine bestimmte Geschwindigkeit zu bringen, ist eine genau definierte Arbeitsleistung notwendig. Die im bewegten Körper steckende kinetische Energie entspricht genau dieser geleisteten Arbeit, daher: Ekin = W = 1/2mv2 geleistete Arbeit W = F.s = m (dv/dt) v.t (a = dv/dt und s ist zeitabhängig v(t).t ) s W= t t ∫ F.ds = m∫(dv(t)/dt) v(t)dt = m/2 ∫ (d/dt) (v ) dt = 1/2mv2 2 0 0 0 Energieumsatz bei Chemischen Reaktionen (Temperatur und Wärme) Zwischen Körpern unterschiedlicher Wärmeenergie kommt es zu einem Energieaustausch, wobei die Temperatur der beiden Körper die Richtung des Energieaustausches festlegt (Wärmeenergie fließt von einem Körper höherer Temperatur zu einem mit geringerer Temperatur). Einheit: Celsius (°C) oder Kelvin (K). °C = K + 273,15. Um einen Körper zu erwärmen muss eine bestimmte Wärmemenge zugeführt werden. Die spezifische Wärme einer Substanz ist die Wärmemenge, die zugeführt werden muss, um 1g der Substanz um 1°C zu erwärmen. Die Kalorie (cal) ist definiert als jene Wärmemenge, die aufgebracht werden muss, um 1g Wasser von 14,5 °C auf 15,5 °C zu erwärmen. Umrechnung in Joule (J): 1 cal = 4,184 J. Energieumsatz bei Chemischen Reaktionen (Wärmekapazität und Kalorimetrie) Die Wärmekapazität C eines Körpers mit der Masse m ist die Wärmemenge, die benötigt wird, um die Temperatur des Körpers um 1°C zu erhöhen, und ist das Produkt aus Masse x spezifischer Wärme. Die erforderliche Wärmemenge Q um einen Körper von der Temperatur T1 auf die Temperatur T2 zu bringen ist Q = C(T2-T1) Spezifische Wärme von Wasser: 4,184 Jg-1K-1. Die Wärmekapazität von 500g Wasser beträgt daher: C = 500 g x 4,184 Jg-1K-1 = 2,092 kJK-1 Die bei einer chemischen Reaktion freigesetzten oder aufgenommenen Wärmemengen werden mithilfe eines Kalorimeters gemessen. Energieumsatz bei Chemischen Reaktionen (Kalorimeter) Ein Bombenkalorimeter wird verwendet, um die bei Verbrennungsprozessen freiwerdende Wärmemenge zu messen. Die chemische Reaktion wird in einer sog. Bombe unter Druck und konstantem Volumen durchgeführt. Zünddraht Rührer Thermometer Isoliertes Gefäß Reaktionsgefäß Wasser Bombe Substanz Aus der Temperaturänderung des Bades nach erfolgter Reaktion kann die umgesetzte Wärmemenge bestimmt werden (Q = CGesamt(T2-T1) ). Energieumsatz bei Chemischen Reaktionen (Reaktionsenergie und Reaktionsenthalpie) Atmosphärendruck p F = A.p Volumensänderung: ∆V = V2 – V1 = A.s Geleistete Arbeit: W = F.s = A.p.s = -∆V .p Volumenarbeit Chemische Reaktion unter Gasentwicklung Das entstehende Gas führt zu einem Druckanstieg und verrichtet daher Arbeit indem es z.B. den Kolben in Bewegung setzt. Die geleistete Arbeit wirkt dem äußeren Luftdruck (p) entgegen. Energieumsatz bei Chemischen Reaktionen (Reaktionsenergie und Reaktionsenthalpie) Chemische Reaktion unter Gasentwicklung Bei Reaktionen in einem geschlossenen Gefäß wird keine mechanische Arbeit geleistet, die gesamte Energie fällt daher als Wärmeenergie an (=Reaktionsenergie). (z.B. Kalorimeter!) Energieumsatz bei Chemischen Reaktionen (Reaktionsenergie und Reaktionsenthalpie) Bei Reaktionen in einem offenen Gefäß wird mechanische Arbeit (und daher Volumensarbeit) geleistet, daher teilt sich die freiwerdende Energie auf Volumenarbeit und einen restlichen Wärmeanteil auf. Diesen Anteil an der gesamten Reaktionsenergie nennt man Reaktionsenthalpie. (W=-∆V.p). Reaktionsenergie Reaktionsenthalpie ∆U = U 2 – U1 ∆H = ∆U + p. ∆V ∆U > 0: Energie wird aufgenommen ∆H > 0: Wärmeenergie aufgenommen (endotherm) wird ∆H < 0: Wärmeenergie abgegeben (exotherm) wird ∆U < 0: Energie wird abgegeben Beispiel Die Verbrennungswärme von CH4(g) wurde bei konstantem Volumen bei 25°C zu ∆U=-885,4 kJ/mol bestimmt. Wie groß ist ∆H? CH4 (g) + 2 O2 (g) → CO2 (g) + 2 H2O (l) ∆U=-885,4 kJ/mol n1 = 3 mol n2 = 1 mol ∆H = ∆U + ∆nRT = -885,4 - 2 x 8,31410-3 x 298,2 = -890,4 kJ/mol Beispiel: Wird bei einer Reaktion 1 mol Gas freigesetzt so entspricht dies bei 25°C und einem Atmosphärendruck von 101 kPa einer Volumensarbeit von 2,5 kJ/mol (p.∆V). (Molvolumen!) Energieumsatz bei Chemischen Reaktionen (Satz von Hess) 1840 formulierte Germain H. Hess (aufgrund experimenteller Befunde) das Gesetz der konstanten Wärmesummen. Nach dem Satz von Hess ist die Reaktionsenthalpie einer Reaktion unabhängig von Zwischenstufen und daher eine Konstante. C (Graphit) + O2 (g) → CO2 (g) ∆H = -393,5 kJ/mol Der Prozeß kann auch in 2 Stufen ablaufen: C (Graphit) + ½ O2 (g) → CO (g) ∆H = -110,5 kJ/mol → CO2 (g) ∆H = -283,0 kJ/mol CO (g) + ½ O2 (g) C (Graphit) + O2 (g) → CO2 (g) ∆H = -393,5 kJ/mol Energieumsatz bei Chemischen Reaktionen (Satz von Hess) Der Satz von Hess kann aufgrund der Additivität der Reaktionsenthalpien dazu verwendet werden, um Reaktionsenthalpien zu bestimmen, die nicht direkt zugänglich sind. C (Graphit) + O2 (g) → CO2 (g) ∆H = - 393,5 kJ/mol 2 H2 (g) + O2 (g) → 2 H2O (l) ∆H = - 571,8 kJ/mol CO2 (g) + 2 H2O (l) → CH4 (g) + 2O2 (g) ∆H = + 890,4 kJ/mol C (Graphit) + 2 H2 (g) → CH4 (g) ∆H = - 74,9 kJ/mol Energieumsatz bei Chemischen Reaktionen (Bildungsenthalpien) Reaktionsenthalpien können anhand tabellierter Bildungsenthalpien, StandardBildungsenthalpien (∆Hf0), berechnet werden. (Standardbedingungen: Normaldruck: 101,325 kPa; Standard-Temperatur: 25°C). Verbindung Al2O3 (s) ∆Hf0 kJ.mol-1 Verbindung ∆Hf0 kJ.mol-1 -1669,8 HJ (g) +25,9 CH4 (g) -74,85 HBr (g) -36,2 C2H2 (g) +226,7 HCl (g) -92,3 CO2 (g) -393,5 HF (g) -269 Fe2O3 (s) -822,2 NaCl (s) -411,0 Energieumsatz bei Chemischen Reaktionen (Bindungsenergien) Die Atome in Molekülen sind durch chemische Bindungen verbunden. Die Energie, die notwendig ist, um eine chemische Bindung in einem zweiatomigen Molekül aufzubrechen, wird Dissoziationsenergie genannt. H-H (g) → 2 H (g) ∆H = + 435 kJ/mol Cl-Cl (g) → 2 Cl (g) ∆H = + 243 kJ/mol H-Cl (g) → H(g) + Cl (g) ∆H = + 431 kJ/mol H2 (g) + Cl2 (g) → 2 HCl (g) ∆H = - 184 kJ/mol exotherme Reaktion (∆H = 435 + 243 – 2x431 = -184 kJ/mol) Energieumsatz bei Chemischen Reaktionen (Bildungsenthalpien) Mittlere Bindungsenergien von Molekülen in der Gasphase. Bindung Bindungsenergie kJ.mol -1 C-C 347 C=C 619 C-H 414 C-F 485 O-H 463 Grundlagen der Thermodynamik (Die Zustände der Gase) Der Zustand eines Gases ist definiert durch: Volumen V (der Raum, den das Gas einnimmt) Stoffmenge n (Molzahl) Druck p (SI-Einheit: Pascal, Pa=1Nm-2; 1 bar=105 Pa, 1 atm = 101,325 kPa = 760 Torr) Temperatur T wobei aber nicht alle Parameter unabhängig voneinander sind, sondern über eine Zustandsgleichung zusammenhängen. Gasgesetze Gay-Lussac: Die Volumina von Gasen, die bei chemischen Reaktionen umgesetzt werden, stehen in einem ganzzahligen Verhältnis zueinander (konst. Temperatur und konst.Druck). z.B. 1 Vol H2 + 1 Vol Cl2 → 2 Vol HCl Avogadro: Gleiche Volumina beliebiger Gase enthalten bei gleicher Temperatur und gleichem Druck die gleiche Anzahl von Molekülen. (V=Konstante x n) (Diese Beziehung zwischen Koeffizienten und Stoffvolumina gilt aber nur für Gase! Boyle-Mariotte: Das Volumen eines Gases ist umgekehrt proportional zum Druck. (p.V=Konstante) Charles: Das Volumen eines Gases hängt linear von der Temperatur ab. (V=Konstante x T) Zustandsgleichung des idealen Gases Die Gesetze von Boyle-Mariotte, Charles und Avogadro können zusammengefasst werden zu: p V = Konstante n T Der für alle Gase konstante Faktor wird als Gaskonstante R bezeichnet pV=nRT R = 8,31451 J K-1mol-1 (=1,98722 cal K-1mol-1 ). (bei konstantem Volumen und Teilchenzahl): p1/T1 = p2/T2 Das molare Volumen Vm bei Standardbedingungen (25°C, 1 bar) beträgt Vm = 22,414 L mol-1 Dalton Gesetz und Partialdruck Der Druck einer Mischung idealer Gase ist gleich der Summe der Drücke, die die Einzelkomponenten ausüben, wenn sie das Volumen der Mischung jeweils alleine ausfüllen. Dieser Einzeldruck wird Partialdruck genannt (Ideales Gas!). p = pA + pB ……….Σ pj pj = nj RT/ V Ein Gefäß des Volumens V=10L enthält bei 298K 1,00 mol N2 und 3,00 mol H2. Wie groß ist der Gesamtdruck (wenn sich beide Gase ideal verhalten)? RT/ V = 2,48x105 Pa mol-1 p(N2) = 0,248 Mpa p(H2) = 0,744 Mpa Gesamtdruck (p(N2) + p(H2) ) : = 0,992 MPa Die kinetische Gastheorie Quantifizierung der Eigenschaften eines idealen Gases. Annahme: (1) Das Gas besteht aus Molekülen der Masse m und des Durchmessers d in kontinuierlicher, zufälliger Bewegung. (2) Die Größe der Moleküle ist vernachlässigbar klein gegenüber ihrer mittleren freien Weglänge (zurückgelegte Strecke zwischen zwei Zusammenstößen) (3) Es gibt keine Wechselwirkungen zwischen verschiedenen Gasmolekülen ausser elastischen Stößen. Die Ableitung der kinetischen Gastheorie beruht auf der Änderung des Impulses der Gasteilchen (=MassexGeschwindigkeit). Je größer der Impuls, umso größer ist auch die Stoßkraft beim Aufprall auf die Gefäßwand. 1. Berechnung der Impulsänderung; 2. Gesamtzahl der Stöße/Fläche/Zeit; 3.Impulsänderung =Kraft/Fläche Die kinetische Gastheorie vx∆t Vor dem Aufprall mvx erreicht die Wand -mvx Nach dem Aufprall Koordinate x Impulsänderung 2mvx Avx∆t erreicht die Wand nicht Koordinate x Teilchen = Avx∆tnNA/ V 50% 50% Fläche A Die kinetische Gastheorie Gesamtimpulsänderung= n NA A vx ∆t 2mvx 2V 2 Gesamtimpulsänderung= n M A vx ∆t V Geschwindigkeit der Gesamtimpulsänderung = Kraft (F=m.a) Druck= Kraft/Fläche Geschwindigkeit der Gesamtimpulsänderung 2 = n M A vx V Die kinetische Gastheorie Gesamtimpulsänderung= n NA A vx ∆t 2mvx 2V 2 Gesamtimpulsänderung= n M A vx ∆t V Geschwindigkeit der Gesamtimpulsänderung = Kraft (F=m.a) Druck= Kraft/Fläche Druck = n M vx2 < vx2 > = 1/3 c2 V p V = 1/3 n M c2 Die kinetische Gastheorie p V = 1/3 n M c2 pV= nRT c = (3RT/M) 1/2 z.B.: CO2 bei 298K: M(CO2) = 44,01 g mol-1: c = 411 m s -1 Vergleich: Schallwellen in Luft (Höhe des Meeresspiegels): 340 ms-1 Die kinetische Gastheorie Zusammenfassung: Ein typisches Gas (N2 oder H2) bei 1 bar und 25°C kann als eine Menge von Molekülen betrachtet werden, die sich mit einer mittleren Geschwindigkeit von 350 ms-1 fortbewegen. Jedes Molekül trifft etwa jede Nanosekunde einmal auf ein anderes, und dazwischen legt es eine Wegstrecke (~70nm) von etwa 103 Moleküldurchmessern zurück. Maxwell-Boltzmann-Geschwindigkeitsverteilung Die kinetische Gastheorie beschreibt die quadratisch gemittelte Geschwindigkeit der Moleküle. In Wirklichkeit sind die Geschwindigkeiten (bedingt durch die zufälligen Zusammenstöße) über einen weiten Bereich verteilt. Die Geschwindigkeitsverteilung wurde von James Clerk Maxwell abgeleitet: f(s) = 4π (M/2πRT)3/2 s2 exp(-Ms2/2RT) Die mittlere Geschwindigkeit ist daher: ∞ c =∫ s f(s) ds; -∞ z.B.: N2 (25°C) = 475 ms -1 Maxwell-Boltzmann-Geschwindigkeitsverteilung f(s) = 4π (M/2πRT)3/2 s2 exp(-Ms2/2RT) Der flüssige Zustand Gase → Flüssigkeiten → Festkörper Molekulare Beweglichkeit nimmt ab Ordnung nimmt zu Die Moleküle in einer Flüssigkeit bewegen sich langsam und werden daher durch zwischenmolekulare Kräfte innerhalb eines bestimmten Volumens zusammengehalten. Dennoch existiert keine fixierte räumliche Verteilung der Moleküle. Eine Flüssigkeit (1) beansprucht ein gewisses Volumen, (2) behält die Form aber nicht bei; (3) eine Druckänderung hat nur geringen Einfluß auf das Volumen; (4) Temperaturerhöhung bewirkt eine leichte Zunahme des Volumens. (5) Die Diffusionsgeschwindigkeit von Flüssigkeiten ist erheblich langsamer als bei Gasen (freie Weglänge kurz). Der flüssige Zustand Viskosität („Fließwiderstand“) Die Viskosität ist eine Folge der zwischenmolekularen Anziehungs-Kräfte (und ist daher ein Maß für die Stärke dieser Kräfte!). Diese Kohäsionskräfte nehmen mit zunehmender Teilchengeschwindigkeit ab. Temperaturerhöhung führt daher zu einer Abnahme der Viskosität, Druckerhöhung bewirkt eine Zunahme der Viskosität. Oberflächenspannung Moleküle an der Flüssigkeitsoberfläche erfahren eine einseitige Anziehung in das Innere der Flüssigkeit, während Moleküle im Inneren eine gleichmäßige Anziehung in alle Richtungen zeigen. Flüssigkeiten tendieren daher dazu, die Oberfläche zu minimieren (kugelförmiger Flüssigkeitstropfen!). Sie nimmt mit steigender Temperatur ab, da die schnelle Molekülbewegung der zwischenmolekularen Anziehung entgegenwirkt. Der flüssige Zustand Verdampfung Anzahl der Moleküle Die kinetische Energie der Moleküle in einer Flüssigkeit folgt auch (wie Gase) einer Maxwell-Boltzmann-Verteilung. Es gibt daher zu jedem Zeitpunkt Teilchen mit höherer und niedriger Energie (und einen permanenten Energie-Austausch). Moleküle, deren Energie hoch genug ist, werden die flüssige Phase verlassen und in die Gasphase übertreten. Dieser Verlust an energiereichen Molekülen führt zu einer Reduktion der kinetischen Energie der Flüssigkeit (Temperaturabsenkung; Abkühlung der Körpertemperatur durch „Schwitzen“), der nur durch Wärmezufuhr aus der Umgebung ausgeglichen werden kann. Mit steigender Temperatur nimmt die Verdampfungsgeschwindigkeit zu, da die Anzahl der Teilchen, die die erforderliche Energie zum Übertritt in die Gasphase besitzen, zunimmt. Energie Der flüssige Zustand Verdampfung Dieser Prozess setzt sich fort, bis die ganze Flüssigkeit verdampft ist. Die zuzuführende Energiemenge, die ein Mol einer Flüssigkeit bei gegebener Temperatur verdampfen läßt, wird als molare Verdampfungsenthalpie ∆HV dieser Flüssigkeit bezeichnet. H2O (l) → H2O (g) ∆HV (25°C) = +43,8 kJ/mol Der flüssige Zustand Dampfdruck Während der Verdampfung treten Moleküle in die Gasphase über. In einem geschlossenen Gefäß bleiben die verdampfenden Moleküle allerdings in der Nähe der Flüssigkeit und können, aufgrund ihrer ungeordneten Bewegung, wiederum in die Flüssigkeitsphase zurückkehren. H2O (l) H2O (g) Die Zahl der Moleküle, die pro Zeiteinheit aus der Gasphase in die Flüssigkeit zurückkehren, hängt von ihrer Konzentration in der Gasphase ab. Zu Beginn des Verdampfungsprozesses sind wenige Moleküle in der Gasphase und nur wenige kehren daher zurück. Nach einiger Zeit wird ein Gleichgewichtszustand erreicht (Verdampfungs- und Kondensationsgeschwindigkeit sind gleich groß). In diesem Zustand bleibt die Zahl der Moleküle in der Gasphase konstant, weil gleich viele Teilchen verdampfen und wiederum in die Flüssigkeit zurückkehren. Die in der Gasphase befindlichen Teilchen üben einen Druck aus („Dampfdruck“). Der flüssige Zustand Dampfdruck Dampfdruck (logP) / (mbar) Dampfdruck nimmt mit der Temperatur zu (Gasgesetze!). Der Dampfdruck hängt von der Stärke der zwischenmolekularen Wechselwirkungen ab (Diethylether < Wasser). Diethylether Wasser Temperatur Der flüssige Zustand Siedepunkt (Sdp.) ist definiert als die Temperatur, bei welcher der Dampfdruck einer Flüssigkeit dem äußeren Atmosphärendruck entspricht. Der Siedepunkt ist daher abhängig vom äußeren Druck (und keine Konstante!). Die Temperatur einer siedenden Flüssigkeit bleibt ebenso konstant. z.B. Sdp.(Wasser): 280m Seehöhe: 99°C; 3000m: 90,2°C; 4807 (Mont Blanc): 84°C. Diese Tatsache wird z.B. bei der Vakuumdestillation verwendet, oder auch beim Entwässern von Nahrungsmitteln. Der flüssige Zustand Verdampfungsenthalpie Beziehen sich auf den normalen Siedepunkt. mol. Verdampfungsenthalpie ∆HV /kJ mol-1 Flüssigkeit Siedepunkt Wasser 100,0 40,7 Ethanol 78,3 38,6 Chloroform 61,3 29,4 Diethylether 34,6 26,0 Die freigesetzte Energie bei der Kondensation eines Mols Dampf zu einer Flüssigkeit nennt man molare Kondensationsenthalpie und hat den gleichen Betrag wie die molare Verdampfungsenthalpie, allerdings umgekehrtes Vorzeichen. Der flüssige Zustand Die Clausius-Clapeyron-Gleichung beschreibt den Zusammenhang zwischen Dampfdruck einer Flüssigkeit und der Temperatur. (Näherung: molare Verdampfungsenthalpie ∆HV ist über den betrachteten Temperaturbereich konstant) log P = - ∆HV /(2,303xRT) + C C…..substanzspezifische Größe log (P2/P1) = [∆HV/(2,303xR)] [(T2-T1)/ T2T1] Anwendungsbeispiele: Berechnung der Verdampfungsenthalpie. Der feste Zustand Gefrierpunkt ist definiert als die Temperatur, bei der Flüssigkeit und Festkörper (bei Normaldruck, 101,3 kPa) miteinander im Gleichgewicht sind. dem äußeren Atmosphärendruck entspricht. Die entzogene Wärmemenge (1 Mol Substanz) ist die molare Kristallisationsenthalpie. Während des Gefrierens bleibt die Temperatur konstant (siedende Flüssigkeit). Es existieren aber auch unterkühlte Flüssigkeiten (Schmelzen). In diesem Fall erfolgt die Kristallisation durch “Animpfen“(Kristall der Verbindung, Staubteilchen oder Glasabrieb). Amorphe Feststoffe (Gläser): bestehen aus komplizierten Molekülen oder Ionen, die sich nicht oder nur schlecht ordnen (z.B. Glas, Teer oder viele Kunststoffe). Sie existieren daher als Flüssigkeiten mit extrem hoher Viskosität, besitzen keinen definierten Schmelzpunkt, d.h. die Viskosität nimmt allmählich ab. Der feste Zustand Schmelzpunkt (Smp.) Durch Erwärmen schmilzt eine kristalline Substanz bei der gleichen Temperatur, bei der sie gefriert. Die Wärmemenge, die dazu notwendig ist, ist die molare Schmelzenthalpie (oder molare Schmelzwärme); gleicher Betrag aber umgekehrtes Vorzeichen wie die molare Kristallisationsenthalpie. molare Schmelzenthalpie /kJ mol-1 Feststoff Schmelzpunkt Wasser 0,0 6,02 Ethanol -117,2 4,60 Chloroform -63,5 9,20 Diethylether -116,3 7,26 (klein im Vergleich zu den Verdampfungsenthalpien. Volumensänderung und daher keine Volumensarbeit!) Grund: geringe Phasendiagramme (Druck-Temperatur-Diagramme) beschreiben die Zustände gasförmig, flüssig und fest einer Substanz. Existenz-Bedingungen der Sublimation: Festkörper → Dampf (benötigte Wärmemenge: molare Sublimationsenthalpie). Schmelzpunktskurve K Kritischer Pkt. B Metastabiler Druck Flüssigkeit Feststoff U Zustand G S T Dampfdruckkurve Gas von Wasser A Temperatur (°C) Dampfdruckkurve von Eis Wasser Phasendiagramme (Druck-Temperatur-Diagramme) beschreiben die Zustände gasförmig, flüssig und fest einer Substanz. B Existenz-Bedingungen K 22,1 MPa 101,3kPa 0,61 kPa Druck Flüssigkeit Feststoff U G S T Gas A 0,0025 100 Temperatur (°C) 0,01 374 Wasser der Phasendiagramme für Kohlendioxid Anwendungen: überkritisches CO2 als mobile Phase in der überkritischen Flüssigkeitschromatographie (Trenn- und/oder Extraktionsverfahren). Anomalie des Wassers (TB) beschreibt ein Absinken des Smp. bei steigendem Druck (ähnliches Verhalten zeigt Gallium und Bismut). Wasser dehnt sich beim Gefrieren aus. (Lockere Packung der Wassermoleküle im Eis, Wasserstoffbrücken; Gletscher, Schneeball). (Molvolumen:18,0 cm3 → 19,63 cm3). Gegenteil CO2: B K Druck Flüssigkeit Feststoff U G S T Gas A Temperatur (°C) Absinken des Smp. steigendem Druck Kristalline Feststoffe Kristalle sind aus Atomen, Ionen oder Molekülen aufgebaut. Die Kristalle werden anhand der Bausteine und der zusammenhaltenden Kräfte unterteilt: Kristalline Feststoffe Ionenkristalle Positive und negative Ionen werden durch elektrostatische Wechselwirkungen zusammengehalten (starke Wechselwirkungen, hohe Smp., spröde, elektrisch leitend nur im geschmolzenen oder gelösten Zustand).) Kristalline Feststoffe Ionenkristalle Molekülkristalle Aus Molekülen aufgebaut und durch schwache (London-Kräfte, DipolDipol,Wasserstoff-Brücken) zusammengehalten (weich, niedrige Smp., nicht leitend) Kristalline Feststoffe Ionenkristalle Molekülkristalle Gerüststrukturen Atome werden durch ein Netzwerk von chemischen Bindungen zusammengehalten (z.B. Diamant). (hart, hohe Smp., schlecht oder nicht leitend). Kristalline Feststoffe Ionenkristalle Molekülkristalle Gerüststrukturen Schichtstrukturen Die chemische Vernetzung ist auf 2 Dimensionen beschränkt (Kohlenstoff im Graphit). Diese Verbindungen haben hohe Schmelzpunkte, können entweder weich (Schichten können gegenseitig verschoben werden) oder hart sein (Glimmer, elektrisch geladene Schichten!). Kristalline Feststoffe Ionenkristalle Molekülkristalle Gerüststrukturen Schichtstrukturen Kettenstrukturen Die chemische Vernetzung erfolgt eindimensional, wobei die parallel gebündelten Ketten entweder durch sog. London-Kräfte oder durch Gegenionen zusammengehalten werden können (oft faserig und hohe Schmelzpunkte). Kristalline Feststoffe Ionenkristalle Molekülkristalle Gerüststrukturen Schichtstrukturen Kettenstrukturen Metallische Kristalle Besondere Form der Bindung (metallische Bindung) zwischen den Metallatomen (Elektronen sind Teil einer ‚Elektronenwolke‘ und daher im Kristall frei beweglich). Die positiven Metallatome nehmen feste Plätze im Kristall ein (hohe Smp., hohe Dichten, gut deformierbar, hämmern, schmieden, etc. gute elektrische Leiter!). Kristallgitter In einem Kristall sind Teilchen in symmetrischer und geordneter Weise in einem sich wiederholenden, dreidimensionalen Muster angeordnet (Kristallstruktur). Die Symmetrie des Kristalls wird mithilfe des Kristallgitters beschrieben. Ein Gitter ist eine 3D-Anordnung von Punkten, die untereinander völlig equivalent sind. Kristallgitter In einem Kristall sind Teilchen in symmetrischer und geordneter Weise in einem sich wiederholenden, dreidimensionalen Muster angeordnet (Kristallstruktur). Die Symmetrie des Kristalls wird mithilfe des Kristallgitters beschrieben. Ein Gitter ist eine 3D-Anordnung von Punkten, die untereinander völlig equivalent sind. Anhand ihrer Symmetrie werden Kristallgitter in Kristallsysteme eingeteilt. Kubisch-primitives Gitter (a=b=c; α=β=γ=90°) Elementarzelle Kristallsysteme Kubisch (a = b = c; α = β = γ = 90°) Tetragonal (a = b ≠ c; α = β = γ = 90°) Hexagonal,trigonal (a = b ≠ c; α = β = γ = 120°) (ortho-)rhombisch (a ≠ b ≠ c; α = β = γ = 90°) Monoklin (a ≠ b ≠ c; α = γ = 90°, β ≠ 90°) Triklin (a ≠ b ≠ c; α ≠ β ≠ γ ≠ 90°) Lösungen Lösungen sind homogene Gemische (Lösungsmittel u. gelöste Stoffe). Löslichkeit = maximale Stoffmenge, die bei gegebener Temperatur noch lösbar ist. Konzentration = gelöste Stoffmenge (verdünnte/konzentrierte Lösungen). Bei höherer Stoffmenge stellt sich ein Gleichgewicht ein zw. Gelöstem Anteil und Rest (fest, flüssig oder gasförmig). Die Konzentration in der Lösung ist konstant = gesättigte Lösung; geringer Stoffmenge: ungesättigte Lösung; übersättigte Lösung (metastabil). Lösungen + - + - + - + - + - + - + - + Wasser (H2O) Ionen sind hydratisiert Auflösen - + - + + - Lösungen Hydratationsenthalpie Energie für den (hypothetischen) Prozeß, in welchem Ionen aus der Gasphase in gelöste, hydratisierte Ionen übergeführt werden. Sie ist abhängig von der Stärke der Wechselwirkungen zwischen Ionen und den Wassermolekülen. (allg. Solvatationsenthalpie) K+ (g) + Cl- (g) → K+ (aq) + Cl- (aq) ∆H = -684,1 kJ/mol Kristallhydrate/Kristallwasser Beim Eindampfen wässriger Lösungen werden kristalline Substanzen erhalten, in denen assoziierte Wassermoleküle vorliegen, das eingebaute Wasser wird Kristallwasser genannt. (z.B. BaCl2 . 2H2O, d.h. pro Molekül Bariumchlorid werden im Kristall 2 Moleküle Wasser eingebaut). Lösungen Lösungsenthalpie Energie, die beim Auflösen einer Substanz in einem Lösungsmittel entweder freigesetzt oder aufgenommen wird (konst. Druck). So wie die Hydratationsenthalpie ist auch die Lösungsenthalpie abhängig von der Konzentration der erhaltenen Lösung. 2 Beiträge: (1): Energie, die notwendig ist um die Kristallstruktur aufzubrechen und gasförmige Ionen zu bilden KCl (s) → K+ (g) + Cl- (g) ∆H = 701,2 kJ/mol (2): freigesetzte Hydratationsenthalpie Wasserstoffbrücken) (Summe aller Prozesse, K+ (g) + Cl- (g) → K+ (aq) + Cl- (aq) ∆H = -684,1 kJ/mol Gesamtenergie: ∆H = +17,1 kJ/mol Lösungsenthalpien von Gasen sind exotherm. z.B. Lösungen Löslichkeit ist abhängig von Druck und Temperatur Endotherme Lösungsvorgänge: Temperaturerhöhung führt zu einer Erhöhung der Löslichkeit Exotherme Lösungsvorgänge: Temperaturerhöhung führt zu einer Erniedrigung der Löslichkeit (Gase: bei höherer Temperatur schlechter löslich). Druckerhöhung spielt vor allem bei Gasen eine Rolle: Henry-Dalton-Gesetz Konzentration ( c) = Konstante (K) x Partialdruck (p) Konzentration von Lösungen 1. Massenanteil (Massenprozente) Massenanteil des Stoffes / Gesamtmasse der Lösung 10% NaCl = 10g + 90g Wasser = 100g Lösung 2. Stoffmengenanteil (Molenbruch) Stoffmenge des gelösten Stoffes / Gesamte Stoffmenge aller Stoffe der Lösung 36,5 g HCl + 36,0 g H2O; entspricht 1 mol HCl und 2 mol H2O: Stoffmengenanteil von HCl (0,333) und H2O (0,667). 3. Stoffmengenkonzentration: (Molarität, molare Konzentration) Stoffmenge / Volumen der Lösung c = n/V. 4. Molalität: Stoffmenge / kg Lösung 5a. Massenkonzentration β= Masse / Volumen der Lösung 5b. Volumenkonzentration δ= Volumenanteil der Substanz / Volumen der Lösung 5c. Volumenanteil ϕ (bei Gasen gleich δ) Dampfdruck von Lösungen Raoult-Gesetz Gesamtdampfdruck der Lösung = Summe aller Partialdampfdrücke p = p(A) + p(B) p(A) = x(A) x p0(A) x(A) Stoffmengenanteil von A p(B) = x(B) x p0(B) x(B) Stoffmengenanteil von B p = x(A) x p0(A) + x(B) x p0(B) In einer idealen Lösung: A-B = A-A = B-B p0(A) Druck p(Gesamt) p(A) p0(B) p(B) Stoffmengenanteil x(A) Reale Lösungen Positive Abweichungen Negative Abweichungen (A-B < A-A + B-B) (A-B > A-A + B-B) p(Gesamt) p0(B) p0(A) Druck Druck p0(A) p(A) p(B) p(Gesamt) p(A) p0(B) p(B) Stoffmengenanteil x(A) Stoffmengenanteil x(A) Dampfdruckdiagramme Flüssigkeit Druck a b Dampf Stoffmengenanteil x(A) im Dampf XA YA Stoffmengenanteil x(A) 50 10 p0(A)/p0(B) = 1 Stoffmengenanteil x(A) in der Flüssigkeit Siedediagramme Dampf Temperatur 1 2 3 4 Flüssigkeit B Fraktionierte Destillation (Zahl der theoretischen Böden: Effizienz der Destillationsanlage) Rektifikation A Azeotrope Azeotrop Temperatur Dampfzusammensetzung Siedepunkt der Flüssigkeit Stoffmengenanteil x(A) Azeotrop: Zusammensetzung der beiden Phasen ändert sich nicht! Siedepunktsmaximum: (fl.Phase durch A-B Wechselwirkungen stabilisiert; Chloroform/Aceton; Salpetersäure/Wasser) Siedepunktsminimum: (fl.Phase durch A-B Wechselwirkungen destabilisiert; Dioxan/Wasser, Ethanol/Wasser [4%Wasser;96% Ethanol]) Dampfdruck von Lösungen Dampfdruck einer verdünnten Lösung (gelöst ist ein nicht-flüchtiger Stoff, p(B)=0); [ x(A) + x(B) = 1, Stoffmengenanteil ] p = p(A) + p(B) p = x(A) x p 0(A) p = (1 - x(B) ) x p0(A) p = p 0(A) - x(B) x p0(A) Dampfdruckerniedrigung der Lösung führt zu einer Siedepunktserhöhung bzw. Gefrierpunktserniedrigung. Das Ausmaß der Änderung ist abhängig vom Lösungsmittel und der Konzentration des gelösten Stoffes, nicht aber von der Natur des gelösten Stoffes. Dampfdruck von Lösungen Molmassenbestimmung (mittels Siedepunktserhöhung –Ebullioskopie- und Gefrierpunktserniedrigung Kryoskopie) ∆TS = ES x b ∆TG = EG x b ES molale Siedepunktserhöhung, EG molale Gefrierpunktserniedrigung, b molale Konzentration des gelösten Stoffes Eigenschaften von Lösungen Osmose (Eigenschaft, die von der Konzentration des gelösten Stoffes abhängig ist)) Niveaudifferenz („osmotischer Druck“) reines Lösungsmittel Lösung semipermeable Membran (durchlässig für Lösungsmittel nicht aber für gelösten Stoff) Eigenschaften von Lösungen Osmose Niveaudifferenz („osmotischer Druck“) Mehr H2O / Volumen Druckanstieg! H2O H2O Eigenschaften von Lösungen Osmotischer Druck (Jacobus van‘t Hoff) Zustandsgleichung des idealen Gases Niveaudifferenz („osmotischer Druck“, π) πV = n R T π=cRT Beispiel: Die wässrige Lösung von 30,0g eines Proteins in 1,0 L hat bei 25°C einen osmotischen Druck von 1,69 kPa. Welche Molmasse hat das Protein? n = πV/RT = 6,82 10-4 mol ; Masse = 30,0g /6,8210-4 mol = 4,40 104 g/mol (44000 Dalton (Da); 44 kDa) Reaktionskinetik Lehre von der Geschwindigkeit chemischer Reaktionen. Reaktionsmechanismus = Details der chemischen Umsetzungen (gebrochene und gebildete Bindungen) Reaktionsgeschwindigkeit = ∆c/∆t A2 (g) + X2 (g) → 2 AX (g) Reaktionsgeschwindigkeit v für die Reaktion wird ausgedrückt durch v(AX) = ∆c(AX)/∆t (Intervall, Differenz → Differential; Geschwindigkeiten ändern sich!) v(AX) = dc(AX)/dt v(A2) = v(X2)= -dc(A2)/dt = -dc(X2)/dt Reaktionsgeschwindigkeit: (mol L-1 s-1) Reaktionskinetik c Mol L-1 Geschwindigkeit = Steigung der Kurve (Tangente) v(A2) = -dc(A2)/dt c(AX) ∆c(A2)/∆t1) ∆c(A2)/∆t2) ∆c(A2)/∆t3) ∆c(A2) Zeit ∆t ∆c(A2)/∆t1) >∆c(A2)/∆t2) > ∆c(A2)/∆t3) c(A2) oder c(X2) v(A2) = -∆c(A2)/∆t Reaktionskinetik Konzentrationsabhängigkeit der Reaktionsgeschwindigkeit wird durch ein Geschwindigkeitsgesetz beschrieben (Beziehung zw. Konzentrationen der Reaktanden und Geschwindigkeit) 2 N2O5 (g) → 4 NO2 (g) + O2 (g) v(NO2) = k x c(N2O5) Proportionalitätskonstante k = Geschwindigkeitskonstante; substanzspezifisch und temperatur-(druck-) abhängig. k ist Reaktionskinetik Reaktionsordnung: Summe der Exponenten der Konzentrationsparameter im Geschwindigkeitsgesetz. Das Geschwindigkeitsgesetz kann NICHT! Aus der Reaktionsgleichung abgeleitet werden, sondern muss experimentell bestimmt werden. NO2 (g) + 2 HCl (g) → NO (g) + H 2O (g) + Cl2 (g) v(NO) = k x c(NO2) x c(HCl) (2.Ordnung) 2NO (g) + 2 H2 (g) → N2 (g) + 2 H2O (g) v(N2) = k x c2(NO) x c(H2) (3.Ordnung) CH3CHO (g) → CH4 (g) + CO (g) v(CH3CHO) = k x c3/2(CH3CHO) (3/2.Ordnung) 0.Ordnung: konzentrationsunabhängig! Reaktionskinetik Geschwindigkeitsgesetz beschreibt die Abhängigkeit der Reaktionsgeschwindigkeit von der Zeit Reaktion 1.Ordnung: v(A) = -dc(A)/dt = k c(A) dc(A)/c(A) = -k dt Integration der Gleichung liefert c0(A) c(A) = c0(A) e c(A) ln c(A) = -k t + ln c0(A) ln c(A) ln c0(A) ln [c(A)/c0(A) ] = -k t Halbwertszeit: t 1/2=ln2/k = 0,693/k c0(A)/2 Zeit -kt t1/2 Zeit Reaktionskinetik v(A) = -dc(A)/dt = k c2(A) Reaktion 2.Ordnung: Integration der Gleichung liefert 1/c(A) 1/c(A) = k t + 1/c0(A) Halbwertszeit: t 1/2=1 / (k c0(A) ) 1/c0(A) Zeit Reaktionskinetik v(A) = -dc(A)/dt = k Reaktion 0.Ordnung: Integration der Gleichung liefert c(A) = -k t + c0(A) c0(A) c(A) Halbwertszeit: t 1/2 = c 0(A) / (2k) Zeit Reaktionskinetik Charakteristische Beziehungen für ausgewählte Reaktionstypen: Ordnung v-Gesetz Zeitabhängigkeit Linearisierung t 1/2 c(A) = - k t + c0(A) c(A) vs t c0(A)/2k ln2/k 0. v=k 1. v = kc(A) ln(c0(A)/c(A)) = k t ln c(A) vs t 2. v = kc2(A) 1/c(A) = k t + 1/c0(A) 1/c(A) vs t 1/(kc0(A)) Reaktionsmechanismus Einstufige Reaktionen: CH3–Br + OH- → CH3–OH + Br- Mehrstufige Reaktionen: NO (g) + F 2 (g) → ONF (g) + F (g) ON (g) + F (g) → ONF (g) 2 NO (g) + F2 (g) → 2 ONF (g) Bei mehrstufigen Reaktionen treten Zwischenprodukte oder Zwischenstufen auf. Kollisionstheorie A2 (g) + X2 (g) → 2 AX (g) Effektive Kollision: d.h. Kinetische Energie der Teilchen (Temperatur!) muss ausreichend sein, um Bindungsbruch zu ermöglichen und die relative Orientierung der aufprallenden Teilchen. Übergangszustand A2 (g) + X2 (g) → 2 AX (g) Die Reaktion verläuft über einen aktivierten Komplex „A2X2“ A X A X A2 (g) + X2 (g) → 2 AX (g) „A2X2“ Der Übergangszustand („A2X2“) besitzt eine relativ hohe Energie. Die Bindungen A-A und X-X sind geschwächt und die Bindung A-X ist bereits partiell gebildet. Reaktionsdiagramm A2 (g) + X2 (g) [ A2X2 ] → 2 AX (g) Potentielle Energie [ A2X2 ] Energie des Übergangszustands Ea,h Ea,r A2 + X2 ∆U = Ea,h - Ea,h 2 AX Reaktionskoordinate Ea,h Aktivierungsenergie Die kinetische Gastheorie Quantifizierung der Eigenschaften eines idealen Gases. Annahme: (1) Das Gas besteht aus Molekülen der Masse m und des Durchmessers d in kontinuierlicher, zufälliger Bewegung. (2) Die Größe der Moleküle ist vernachlässigbar klein gegenüber ihrer mittleren freien Weglänge (zurückgelegte Strecke zwischen zwei Zusammenstößen) (3) Es gibt keine Wechselwirkungen zwischen verschiedenen Gasmolekülen ausser elastischen Stößen. Die Ableitung der kinetischen Gastheorie beruht auf der Änderung des Impulses der Gasteilchen (=MassexGeschwindigkeit). Je größer der Impuls, umso größer ist auch die Stoßkraft beim Aufprall auf die Gefäßwand. 1. Berechnung der Impulsänderung; 2. Gesamtzahl der Stöße/Fläche/Zeit; 3.Impulsänderung =Kraft/Fläche Die kinetische Gastheorie vx∆t Vor dem Aufprall mvx erreicht die Wand -mvx Nach dem Aufprall Koordinate x Impulsänderung 2mvx Avx∆t erreicht die Wand nicht Koordinate x Teilchen = Avx∆tnNA/ V 50% 50% Fläche A Die kinetische Gastheorie Gesamtimpulsänderung= n NA A vx ∆t 2mvx 2V 2 Gesamtimpulsänderung= n M A vx ∆t V Geschwindigkeit der Gesamtimpulsänderung = Kraft (F=m.a) Druck= Kraft/Fläche Geschwindigkeit der Gesamtimpulsänderung 2 = n M A vx V Die kinetische Gastheorie Gesamtimpulsänderung= n NA A vx ∆t 2mvx 2V 2 Gesamtimpulsänderung= n M A vx ∆t V Geschwindigkeit der Gesamtimpulsänderung = Kraft (F=m.a) Druck= Kraft/Fläche Druck = n M vx2 < vx2 > = 1/3 c2 V p V = 1/3 n M c2 Die kinetische Gastheorie p V = 1/3 n M c2 pV= nRT c = (3RT/M) 1/2 z.B.: CO2 bei 298K: M(CO2) = 44,01 g mol-1: c = 411 m s -1 Vergleich: Schallwellen in Luft (Höhe des Meeresspiegels): 340 ms-1 Die kinetische Gastheorie Zusammenfassung: Ein typisches Gas (N2 oder H2) bei 1 bar und 25°C kann als eine Menge von Molekülen betrachtet werden, die sich mit einer mittleren Geschwindigkeit von 350 ms-1 fortbewegen. Jedes Molekül trifft etwa jede Nanosekunde einmal auf ein anderes, und dazwischen legt es eine Wegstrecke (~70nm) von etwa 103 Moleküldurchmessern zurück. Maxwell-Boltzmann-Geschwindigkeitsverteilung Die kinetische Gastheorie beschreibt die quadratisch gemittelte Geschwindigkeit der Moleküle. In Wirklichkeit sind die Geschwindigkeiten (bedingt durch die zufälligen Zusammenstöße) über einen weiten Bereich verteilt. Die Geschwindigkeitsverteilung wurde von James Clerk Maxwell abgeleitet: f(s) = 4π (M/2πRT)3/2 s2 exp(-Ms2/2RT) Die mittlere Geschwindigkeit ist daher: ∞ c =∫ s f(s) ds; -∞ z.B.: N2 (25°C) = 475 ms -1 Maxwell-Boltzmann-Geschwindigkeitsverteilung f(s) = 4π (M/2πRT)3/2 s2 exp(-Ms2/2RT) Grundlagen der Thermodynamik Die Thermodynamik ist die Lehre der Energieänderungen im Verlaufe von physikalischen und chemischen Vorgängen. System (Reaktionsgefäß, Maschine, elektrochemische Zelle, biologische Zelle) Umgebung durch eine Grenzfläche vom System getrennt. Umgebung Umgebung Umgebung Stoff Energie System Offenes Energie System Geschlossen System Abgeschlossen Grundlagen der Thermodynamik Umgebung Energie System Geschlossen In einem geschlossenen System ist die Änderung der inneren Energie U gleich der Energie, die in Form von Wärme (q) oder Arbeit (w) durch die Wände des Systems übertragen wird. ∆U = q + w Die innere Energie U hängt vom Zustand des Systems ab, nicht jedoch davon, wie er erreicht wurde. Sie ist daher eine Zustandsfunktion. Arbeit, Wärme und Energie Energie ist die Fähigkeit, Arbeit zu leisten. Wenn sich die Energie eines Systems als Folge einer Temperaturdifferenz zur Umgebung ändert, sagt man: Energie wurde in Form von Wärme übertragen. Energieerhöhung wird also durch eine Temperaturdifferenz und die dadurch verursachte Wärmeübertragung hervorgerufen. Aus molekularer Sicht (kinetische Gastheorie!) ist Wärme diejenige Art der Energieübertragung, die durch zufällige Bewegung der Moleküle in der Umgebung vermittelt wird (thermische Bewegung). Aus molekularer Sicht ist die Arbeit, diejenige Art der Energieübertragung, die durch koordinierte (oder geordnete) Teilchenbewegung vermittelt wird. (Gewicht wird angehoben oder abgesenkt, elektrischer Strom). System Arbeit (geordneter Energietransfer) System Wärme (ungeordneter Energietransfer) Grundlagen der Thermodynamik Erster Hauptsatz der Thermodynamik: Energie kann von einer Form in eine andere umgewandelt werden, sie kann aber weder erzeugt noch vernichtet werden. Die innere Energie (U) eines abgeschlossenen Systems ist konstant. Die innere Energie setzt sich zusammen aus: Anziehungs- und Abstoßungskräfte zwischen Atomen, Molekülen, Ionen und subatomaren Teilchen, kinetische Energie. U ist nicht messbar; die Thermodynamik befasst sich mit Änderungen von U! H = U + p.V Grundlagen der Thermodynamik Die Entropie: Die zentrale Größe der Thermodynamik ist die Wärme q, allerdings ist die Wärme keine Zustandsfunktion, sie ist daher wegabhängig. Übertragung von Energie durch Arbeit erfolgt durch geordnete Arbeit und ist daher mit KEINER Entropieänderung verbunden. Wird jedoch dem System Wärme zugeführt, so wird die thermische Bewegung der Moleküle angeregt (ungeordnet, kinetische Gastheorie), daher ist dS ∝ dq. Wärme fließt freiwillig von einem warmen Körper zu einem kalten; eine bestimmte Wärmemenge, die bei höherer Temperatur gespeichert ist, bedeutet also eine kleinere Entropie. Daraus folgt: Wenn eine Energie dq‘ einem Körper bei niedriger Temperatur zugeführt wird, ist die damit verbundene Entropieänderung größer, als wenn diese Energie dem Körper bei höherer Temperatur zugeführt wird. dS = dq‘/T (Carnot Kreisprozess!, dS ist eine Zustandsfunktion). z.B. System im Gleichgewicht: Exotherme Reaktion; abgegebene Wärmemenge q‘=-∆H, daher Entropieänderung in der Umgebung: ∆S‘=-∆H/T) Grundlagen der Thermodynamik Die Entropie: Die Entropie ist ein Maß für die Wärme (die zwischen System und Umgebung ausgetauscht wird), aber auch für den Ordnungszustand W. S = klnW (Ludwig Boltzmann) Gleichverteilung Grundlagen der Thermodynamik Die Entropie: Die Kenntnis der statistischen Definition der Entropie erlaubt es, den Ordnungszustand W zu ermitteln. HCl (T=0): CO (T=0): S=0 (W=1, lnW=0) W=2x2x…=2N S = kln2N = Nkln2 = 5,76 JK -1 Grundlagen der Thermodynamik Zweiter Hauptsatz der Thermodynamik: Wärme kann nicht von selbst vom kälteren zum heißeren Körper fließen (Claudius) Es ist unmöglich, eine periodisch arbeitende Maschine zu bauen, die nichts anderes tut, als einem Wärmereservoir Wärme zu entziehen und dabei Arbeit zu leisten (Planck, Kelvin) (Perpetuum mobile II.Art). Bei einer spontanen Zustandsänderung vergrößert sich die Entropie. Freiwillig stellt sich somit immer nur ein Zustand mit geringerer Ordnung ein. Der 2.Hauptsatz definiert, ob ein Prozess abläuft oder nicht (im Gegensatz zum 1.Hauptsatz) Grundlagen der Thermodynamik Die freie Enthalpie ∆SUmg ∆SSys Energie ∆SGesamt = ∆S Umg+ ∆S Sys ; ∆SUmg= - ∆H/T (∆H =Reaktionsenthalpie) ∆SGesamt = ∆S - ∆H/T (∆S = ∆SSys); T∆SGesamt = T∆S - ∆H ∆G = - T∆SGesamt Freie Reaktionsenthalpie: ∆G = ∆H - T∆S Freie Enthalpie: G = H –TS (Zustandsgröße) Grundlagen der Thermodynamik Die freie Energie Für Reaktionen bei konstantem Volumen gilt: Freie Reaktionsenergie: ∆F = ∆U - T∆S Freie Enthalpie: F = U –TS (Zustandsgröße) Grundlagen der Thermodynamik Prozess erlaubt oder nicht?: ∆G < 0, Reaktion läuft freiwillig ab. ∆G = 0, System ist im Gleichgewicht. ∆G > 0, Reaktion läuft nicht freiwillig ab (wohl aber in umgekehrter Richtung). Die freie Reaktionsenthalpie berücksichtigt 2 Faktoren, die für die Freiwilligkeit einer Reaktion entscheidend sind: (a) Energieminimum (∆H <0) (b) Maximum an Unordnung (∆S >0) Grundlagen der Thermodynamik Thermodynamische Reaktionsdaten (kJ/mol): Reaktion ∆H T∆S ∆G H2(g) + Br2(g) → 2HBr(g) -72,5 34,0 -106,5 2H2(g) + O2(g) → 2H2O(l) -571,7 -97,3 -474,4 2Ag2O(s) → 4 Ag(s) + O2(g) +61,2 +39,5 +21,7 Die freie Standard-Reaktionsenthalpie, ∆G0, ist die freie Reaktionsenthalpie bei 25°C und 101,3 kPa. Grundlagen der Thermodynamik Absolute Entropien: Die Entropie einer perfekten kristallinen Substanz verschwindet beim absoluten Nullpunkt (3.Hauptsatz der Thermodynamik; Walter Nernst) und nimmt mit der Temperatur zu. Absolute Werte können aus der Wärmekapazität berechnet werden (d∆H/dT = Cp). Absolute Standard-Entropie S0: Entropie bei 25°C und 101,3kPa. Standard-Reaktionsentropie ∆S0 = Summe der absoluten Entropien der Produkte minus der Summe der absoluten Entropien der Edukte. Absolute Entropie eines Elements ist NICHT gleich Null. [Absolute Entropien: ~ 20 – 250 J/(mol K) ] Entropieänderung bei der Bildung einer Verbindung aus den Elementen ist NICHT gleich der absoluten Entropie dieser Verbindung! z.B. Hg (l) + 1/2O2 (g) → HgO (s) ∆H0 = -90,7 kJ/mol; ∆G0 = ?; ∆S0 = ?; ∆S0 = S0(HgO) – S0(Hg) – 1/2 S0(O2) = -107,9 J/(mol K) ∆G0 = ∆H0 – T ∆S0 = -58,5 kJ/mol Grundlagen der Thermodynamik Das chemische Potential: Da die freie Enthalpie eine zentrale Rolle insbesondere für physikalische und chemische Vorgänge (Reaktionen) spielt, hat man der molaren Größe einen besonderen Namen gegeben: das chemische Potential ∆µ = ∆G / n Für Gleichgewichte ist ∆µ immer Null. Das chemische Potential ist vor allem für Betrachtungen der freien Enthalpie in Stoffgemischen wichtig. Säure-Base-Gleichgewichte Das Ionenprodukt des Wassers. pH-Wert Wasser zeigt eine Eigendissoziation. 2 H2O c (H3O+) c(OH -) =K H3O+ + OH- K Gleichgewichtskonstante c2(H2O) Das Gleichgewicht liegt auf der linken Seite. In reinem Wasser und in verdünnten Lösungen wird die Konzentration der Wasser-Moleküle als konstant angenommen (und in die Gleichgewichtskonstante integriert). c(H+).c(OH-) = K.c2(H2O) = KW Ionenprodukt: KW = 1,0 10-14 mol2/L2 (25°C) Säure-Base-Gleichgewichte In reinem Wasser: c(H+) = c(OH-); c2(H+) = 1,0 10-14 mol2/L2 (25°C) Daher: c(H+) = 1,0 10-7 mol/L Wird eine Säure gelöst erhöht sich die H+-Ionenkonzentration. H+–Konzentration ist daher größer als 1,0 10-7 mol/L (im basischen Bereich ist es umgekehrt). Der pH-Wert ist definiert als pH = -log c(H+) pOH = -log c(OH -) pH 0 7 14 c(H+) 1,0 100 1,0 10-7 1,0 10-14 c(OH-) 1,0 10-14 1,0 10-7 1,0 100 sauer neutral basisch Säure-Base-Gleichgewichte Beispiel: Wie groß sind c(H+), c(OH-), pH und pOH für Salzsäure mit 0,02 mol/L HCl? c(H+) = 2,0 10-2 mol/L; pH = -log(2,0 10-2) = -log 2,0 – log 10-2 = -0,30 + 2 = 1,7 c(OH-) = KW/c(H+) = 5,0 10-13 mol/L; pOH = 14 – pH = 12,3 HCL = Starker Elektrolyt Starke Elektrolyte sind in Lösung vollständig dissoziiert. Beispiel: Eine Lösung von 0,01 mol/L CaCl2 enthält 0,01 mol/L Ca2+ und 0,02 mol/L Cl- Ionen. Säure-Base-Gleichgewichte Schwache Elektrolyte sind unvollständig dissoziiert. Beispiel: Essigsäure (CH3COOH) H3O+ + CH3COO- CH3COOH + H 2O c (H3O+) c(CH3COO-) c(H2O) c(CH3COOH) =K K Gleichgewichtskonstante Es gilt: c(H2O) = konst. Und c(H+) = c(H3O+). c (H+) c(CH3COO-) = K c(H2O) c(CH3COOH) = KS KS Dissoziationskonstante Säurekonstante CH3COOH H+ + CH3COO- Säure-Base-Gleichgewichte Essigsäure pKS = -log KS = 4,74 CH3COOH H+ + CH3COO- D.h bei einer Gesamtkonzentration von 0,1 mol/L (bei 25°C) ist Essigsäure zu 1,34% dissoziiert. Säure-Base-Gleichgewichte Dissoziationsgrad (α) Der Dissoziationsgrad eines schwachen Elektrolyten (ionenbildender Stoff) ist der Bruchteil der gesamten Stoffmenge (c 0), der in Ionen dissoziiert ist (in % angegeben). c(CH3COO-) c0(CH3COOH) =α α Dissoziationsgrad undissoziiert: c(CH3COOH) = c0 - x mol/L dissoziiert: c(CH3COO-) = x mol/L c(H+) c(CH3COO-) x2 = c(CH3COOH) c0 - x = KS x2 + K Sx – KSc0 = 0 x = c(H+) = 1/2KS + √(1/4KS2 + KSc0) = 0 Bei schwachen Säuren (kl. K S): x = c(H+) = √KSc0 pH ≈ ½( pKS – log c0) Säure-Base-Gleichgewichte Ionenkonzentration und Dissoziationsgrad von Essigsäure bei 25°C c0 mol/L C(H+)=c(CH3COO-) mol/L pH Dissoziationsgrad α 1,00 0,00426 2,37 0,426 0,100 0,00134 2,87 1,34 0,0100 0,000418 3,83 4,18 0,00100 0,000126 3,90 12,6 Säure-Base-Gleichgewichte Basenkonstante In wäßrigen Lösungen von schwachen Basen stellt sich ein Gleichgewicht ein, an dem OH--Ionen beteiligt sind. NH4+ + OH- NH3 + H 2O c (NH4+) c(OH -) c(NH3) c(H2O) =K K Gleichgewichtskonstante c(H2O) = konst. c (NH4+) c(OH -) = K c(H2O) c(NH3) = KB KB Basenkonstante Bei schwachen Basen (kl. KB): x = c(OH-) = √KBc 0 pOH ≈ ½( pKB – log c0) Säure-Base-Gleichgewichte Säure-Base-Konjugation NH3 + H 2O Base NH4+ + OHKonjugierte Säure (Kation einer schwachen Base) Konjugierte Base (Anion einer schwachen Säure) Säure CH3COOH + H 2O H3O+ + CH3COO- Säure-Base-Gleichgewichte Säure-Base-Konjugation Zwischen der Basenkonstante einer Base und der Säuredissoziationskonstante ihrer konjugierten Säure besteht ein einfacher Zusammenhang. AH H+ + A- c(H+) c(A-) c(AH KS KB A- + H 2O AH + OH- c(AH) c(OH -) = K B c(A-) = KS c(H+) c(A-) = c(AH c(AH) c(OH -) c(A-) = KW = 10-14 pKS + pKB = pKW = 14 Säure-Base-Gleichgewichte Zusammenhang zwischen Säure-Base-Stärke Je größer KS für eine Säure ist, desto kleiner ist KB für die konjugierte Base (und umgekehrt) pKS + pKB = pKW = 14 Starke Säure ↔ Schwache Säure ↔ Schwache Base Starke Base Säure-Base-Gleichgewichte Beispiel: Welchen pH-Wert hat eine Lösung von 0,1 mol Natriumacetat pro Liter? (Acetat-Ion ist eine schwache Base) CH3COO- + H 2O OH- + CH3COOH pKS (CH3COOH) = 4,74. pKB = 14 – pKS = 9,26. pOH ~ ½ (pKB – log c0) = 5,13 pH = 14 – pOH ~ 8,87 Säure-Base-Gleichgewichte Beispiele: Säure pKS Flußsäure HF Salpetrige Säure HNO2 Blausäure HCN 3,2 H + + FH+ + NO2- 3,4 9,4 H + + CN- Basen Methylamin CH3NH2 + H 2O Ammoniak NH3 + H 2O Hydrazin N2H4 + H 2O CH3NH3+ + OHNH4+ + OHN2H5+ + OH- 3,3 4,7 6,0 Säure-Base-Gleichgewichte Indikatoren: Indikatoren sind (organische) Farbstoffe, deren Farbe pH-abhängig ist. Universalindikator = Mischung bestimmter Indikatoren, die je nach pH-Wert einen bestimmten Farbton besitzt. H+ + Ind- HInd c(H+) c(Ind-) = KS = 10-7 mol/L c(HInd) c(Ind -) c(HInd) pH = 5 c(Ind -) c(HInd) = ROT 10-7 c(H+) = 10-7 c(H+) pH = 8 pH = 7 = 10-2 PURPUR c(Ind -) c(HInd) = BLAU 10-7 c(H+) = 10 Säure-Base-Gleichgewichte Pufferlösungen: Pufferlösungen bewirken, dass der pH-Wert einer Lösung konstant bleibt. (z.B.: Lösung von CO2 in Wasser, CO2 + H2O ↔ H2CO3. Eine Pufferlösung ist eine Lösung, die eine relativ hohe Konzentration einer schwachen Säure UND ihrer konjugierten Base enthält. CH3COOH H+ + CH3COO- c(H+) c(CH3COO-) x mol/L = c(H+) c(CH3COOH) x mol/L = KS = 1,8 10-5 mol/L pH = pKS = 4,74 Für eine Lösung, die eine schwache Säure und ihre konjugierte Base im Stoffmengenverhältnis 1:1 enthält, gilt: pH = pKS Säure-Base-Gleichgewichte Pufferkapazität: 1,0 mol Essigsäure und 1,0 mol Natriumacetat (pH = 4,742). Es wird HCl zugegeben, wie ändert sich der pH? c(H+) c(CH3COO-) = KS c(CH3COOH) c(H+) = KS c(CH3COOH) c(CH3COO-) pH = pKS - log c(CH3COOH) c(CH3COO-) Zusatz von 0,01 mol/L HCl: pH = pKS – log(1,01/0,99) = 4,733. (0,01 mol/L : pH = 2!) Zusatz von 0,1 mol/L HCl: pH = pKS – log(1,10/0,90) = 4,655. (0,1 mol/L : pH = 1!) Zusatz von 0,01 mol/L NaOH: pH = pKS – log(0,99/1,01) = 4,751. (0,01 mol/L : pH = 12!!!) Säure-Base-Gleichgewichte Henderson-Hasselbach-Gleichung: pH = pKS - log c(HA) c(A-) Aus Cyansäure (HOCN) und Kaliumcyanat (KOCN) soll eine Pufferlösung mit pH=3,5 hergestellt werden. pKS=3,92. pH = pKS - log c(HOCN) c(NCO-) log [c(HOCN)/c(NCO-)] = pKS – pH = 3,92 – 3,50 = 0,42 c(HOCN) = 2,63 c(NCO-) Säure-Base-Gleichgewichte Mehrprotonige Säuren: enthalten mehr als ein dissoziierbares Wasserstoffatom (H2SO4, H3PO4), d.h. setzen mehr als ein Wasserstoffatom pro Molekül frei. H3PO4 H+ + H 2PO4- c (H+) c(H2PO4-) c(H3PO4) = KS1 H2PO4- H+ + HPO42- c (H+) c(HPO42-) c(H2PO4-) = KS2 HPO42- H+ + PO43- c (H+) c(PO43-) c(H2PO42-) = KS3 = 7,5 10-3 mol/L = 6,2 10-8 mol/L = 1,0 10-12 mol/L Säure-Base-Gleichgewichte Phosphorsäure: 0,1 mol/L Phosphorsäure; wie groß sind die Konzentrationen der einzelnen Ionen? H3PO4 0,1 - x H+ + H 2PO4x x mol/L x = c(H+) = 1/2KS + √(1/4KS2 + KSc0) = 0 x = 2,4 10-2 mol/L c(H3PO4) = 7,6 10-2 mol/L H2PO4- H+ + HPO42- c(HPO42-) = 6,2 10-8 mol/L HPO42- H+ + PO43- c(PO43-) = 2,6 10-18 mol/L Säure-Base-Gleichgewichte Kohlensäure: Lösungen von Kohlendioxid und Wasser reagieren sauer. CO2 + H 2O c (H+) c(HCO3-) c(CO2) HCO3c (H+) c(CO32-) c(HCO3-) H+ + HCO3- = KS1 = 4,2 10-7 mol/L H+ + CO32= KS2 = 4,8 10-11 mol/L Säure-Base-Gleichgewichte Salze schwacher Säuren und Basen: Lösungen von Salzen schwacher Säuren reagieren basisch. Anionen schwacher Säuren reagieren basisch. Kationen schwacher Basen reagieren sauer. z.B. CH3COO- + H 2O OH- + CH3COOH Hydroxid-Ionen werden gebildet! NH3 + H 2O NH4+ + OHHydroxid-Ionen werden verbraucht! NH4+ NH3 + H + Eine 0,30 mol/L NH4Cl-Lösung hat einen pH-Wert von? pKS(NH4+) = 14 – pKB(NH3) = 14 – 4,7 = 9,3 pH = ½ ( pKS – log 0,30) = 4,9 Salze starker Säuren oder Basen reagieren nicht und verändern daher den pH-Wert nicht! Säure-Base-Gleichgewichte Säure-Base-Titration: Neutralisation der H+ (oder OH-) Ionen durch zugegebene OH- (oder H+) Ionen. Schrittweise Neutralisation bis zum Äquivalenzpunkt (Konzentration an H+ gleich OH-), beim Äquivalenzpunkt (Na+=Cl-) ist die (H+,OH-) Ionenkonzentration durch die Eigendissoziation (Ionenprodukt) des Wassers bestimmt. z.B. HCl + NaOH 2 H2O H3O+ + OH- durch schrittweises Zugeben von NaOH werden H+-Ionen teilweise neutralisiert und verdünnt! Basischer Bereich pH = 7 pH Äquivalenzpunkt (Na+=Cl-) („Wendepunkt“) Volumen NaOH Teilweise Verdünnung Säure-Base-Gleichgewichte Säure-Base-Titration (schwache Säure und starke Base): z.B. Essigsäure und NaOH. 0,1 mol/L Essigsäure, pH = 2,87 pH = ½ (pKS – log c0) Beim Äquivalenzpunkt: Gesamtvolumen: doppelt so groß; CH3COO-: 0,05 mol/L pOH = ½ (pKB – log 0,05) = 5,28 ; pH = 15 – pOH = 8,72 Äquivalenzpunkt (nur Acetat-Ionen!) („Wendepunkt“) pH = 8,72 pH Bereich geringer! Volumen NaOH Pufferlösung (Henderson-Hasselbach-Gl.) Säure-Base-Gleichgewichte Säure-Base-Titration (schwache Säure und schwache Base): z.B. Essigsäure und Ammoniak. Kein steiler Anstieg! pH pH = 7,0 Volumen NH3 Säure-Base-Gleichgewichte Starke Säure & Starke Base Detektion durch Indikatoren Schwache Säure & Starke Base pH Volumen Base Gut detektierbar: Schwache Säure & Schwache Base Starke Säure/Starke Base & Schwache Säure/Starke Base & Starke Säure/Schwache Base Schlecht detektierbar: Schwache Säure/Schwache Base Löslichkeit Löslichkeitsprodukt: Manche Substanzen sind in Wasser nur geringfügig löslich (teilweise Dissoziation in Ionen). Aus diesem Grund existiert ein Gleichgewicht zwischen gesättigter Lösung und ungelöstem Feststoff. z.B. Silberchlorid (AgCl). AgCl c (Ag+) c(Cl-) c(AgCl Ag+ (aq) + Cl- (aq) =K c (Ag+)c(Cl-) = Kc(AgCl) = L L Löslichkeitsprodukt Bei 25°C lösen sich 0,00188g AgCl in 1L Wasser. Wie groß ist L? n(AgCl) = m(AgCl)/M(AgCl) = 0,00188g / 143 gmol-1 = 1,31 10-5 mol c(Ag+) = c(Cl-) = 1,31 10-5 mol/L; L = 1,7 10-5 mol2/L2 Löslichkeit Löslichkeitsprodukt: Die tatsächliche Löslichkeit eines Salzes wird zusätzlich durch Salzeffekte (Zusatz eines weiteren Elektrolyten, Abschirmung der Ionen, Aktivitätsverringerung) oder durch Reaktion eines gebildeten Ions beeinflusst. BaCO3 (s) CO32- (aq) + H 2O Ba2+ (aq) + CO32- (aq) HCO3- (aq) + OH- (aq) Die Konzentration an CO32- wird durch die Reaktion mit Wasser verringert. Löslichkeit Fällungsreaktionen: Durch Zugabe des entsprechenden Gegenions kann ein Ion ‚ausgefällt‘ werden. Z.B. z.B. 10 mL einer 0,01 mol/L AgNO3-Lösung werden mit 10 mL einer 0,0001 mol/L NaClLösung versetzt. L(AgCl) = 1,7 10-10 mol2/L2. Gesamtvolumen: 20 mL c(Ag+)c(Cl-) = 5,0 10-3 . 5,0 10-5 = 2,5 10-7 mol2/L2 > L („Fällung von AgCl“) Ionenzusätze: Gleichionige Zusätze verschieben das Gleichgewicht nach links („Fällung“) AgCl Ag+ (aq) + Cl- (aq) Löslichkeitsprodukt: Solange das System im Gleichgewicht ist, ist L unabhängig vom Reaktionsmechanismus! Komplexgleichgewichte Komplexe: Verbindungen, die aus Liganden, die an ein Zentralatom koordiniert sind. Komplexe können geladen (Summe der Ladungen des Zentralatoms und der Liganden) bzw. neutral sein. Hydratisierte Metallkationen sind als Komplexverbindungen zu betrachten. Fe2+ (aq) + 6 CN- (aq) [Fe(CN)6]4- Nettoladung: 2(+) - 6(-) = 4(-) [Ag(NH3)2]+ [Cu(NH3)4]2+ [Fe(CN)6]3- [Zn(OH)4]2- [CdCl4]2- [Cu(OH2)4]2+ Ligandenaustausch: In wäßriger Lösung kann es zu einem Austausch der Liganden kommen. z.B. [Cu(OH2)6]2+ + 4 NH3 Cu2+ + 4 NH3 [Cu(OH2)2 (NH3)4]2+ + 4 H 2O [Cu(NH3)4]2+ (Fe3+) Komplexgleichgewichte Dissoziationskonstante: Komplexverbindungen zerfallen stufenweise. Die Gleichgewichtskonstante für die Bruttoreaktion wird Komplexzerfalls- oder dissoziationskonstante genannt. [Ag(NH3)2]+ [Ag(NH3)]+ + NH3 c([Ag(NH3)]+) c(NH3) = KD1 = 1,4 10-4 mol/L c([Ag(NH3)2]+) [Ag(NH3)]+ c(Ag+) c(NH3) c([Ag(NH3)]+) Ag+ + NH3 = KD2 = 4,3 10-4 mol/L (Komplex-) Dissoziationskonstante: K D = K D1KD2 = 6,0 10-8 mol 2/L2 (Komplex-) Bildungskonstante: K K = 1/KD = 1,6 107 mol -2/L-2 Komplexgleichgewichte Beispiele: Schwerlösliche Verbindungen können durch Komplexbildung in Lösung gebracht werden. Löslichkeit von AgCl in Ammoniak-Lösung, c(NH3) = 0,10 mol/L? Ag+ (aq) + Cl- (aq) AgCl (s) Ag+ (aq) + 2 NH3 (aq) c (Ag+)c(Cl-) = L [Ag(NH3)2]+ (aq) c([Ag(NH3)2]+) c(Ag+)c2(NH3) Gesamtreaktion! AgCl (s) + 2 NH3 (aq) [Ag(NH3)2]+ (aq) + Cl- (aq) c([Ag(NH3)2]+)c(Cl-) c2(NH3) = LKK = KK Komplexgleichgewichte [Ag(NH3)2]+ (aq) + Cl- (aq) AgCl (s) + 2 NH3 (aq) 1. 2. c([Ag(NH3)2]+) = c(Cl-) c(NH3) = 0,10 mol/L – 2 c(Cl-) c2(Cl-) [0,10 – 2c(Cl-)]2 = LKK = 1,67 107 . 1,7 10-10 c(Cl-) = 4,8 10-3 mol/L c(Cl-) = 1,3 10-5 mol/L (reines Wasser) Elektrochemie Redoxvorgänge Seit Boyle bildete das Phänomen der Verbrennung eines der wichtigsten Probleme der Chemie. Lavoisier erkannte, dass bei einer Verbrennung Sauerstoff verbraucht wird. Er führte daher den Begriff Oxidation ein und übertrug den Namen auf alle Vorgänge, bei denen sich eine Substanz mit Sauerstoff verbindet. Festlegung der Oxidationszahlen (1) Elementatome haben die Oxidationsstufe 0 (2) In einfachen Ionen entspricht die Ladung der Oxidationsstufe (3) Wasserstoff hat im allg. die Oxidationsstufe 0 (Metallhydride) (4) Sauerstoff (5) Die Summe der Oxidationsstufen einer neutralen Verbindung ist Null (Ausnahme: komplexe Ionen) Säure-Base-Gleichgewichte In reinem Wasser: c(H+) = c(OH-); c2(H+) = 1,0 10-14 mol2/L2 (25°C) Daher: c(H+) = 1,0 10-7 mol/L Wird eine Säure gelöst erhöht sich die H+-Ionenkonzentration. H+–Konzentration ist daher größer als 1,0 10-7 mol/L (im basischen Bereich ist es umgekehrt). Der pH-Wert ist definiert als pH = -log c(H+) pOH = -log c(OH -) pH 0 7 14 c(H+) 1,0 100 1,0 10-7 1,0 10-14 c(OH-) 1,0 10-14 1,0 10-7 1,0 100 sauer neutral basisch Säure-Base-Gleichgewichte Beispiel: Wie groß sind c(H+), c(OH-), pH und pOH für Salzsäure mit 0,02 mol/L HCl? c(H+) = 2,0 10-2 mol/L; pH = -log(2,0 10-2) = -log 2,0 – log 10-2 = -0,30 + 2 = 1,7 c(OH-) = KW/c(H+) = 5,0 10-13 mol/L; pOH = 14 – pH = 12,3 HCL = Starker Elektrolyt Starke Elektrolyte sind in Lösung vollständig dissoziiert. Beispiel: Eine Lösung von 0,01 mol/L CaCl2 enthält 0,01 mol/L Ca2+ und 0,02 mol/L Cl- Ionen. Säure-Base-Gleichgewichte Schwache Elektrolyte sind unvollständig dissoziiert. Beispiel: Essigsäure (CH3COOH) H3O+ + CH3COO- CH3COOH + H 2O c (H3O+) c(CH3COO-) c(H2O) c(CH3COOH) =K K Gleichgewichtskonstante Es gilt: c(H2O) = konst. Und c(H+) = c(H3O+). c (H+) c(CH3COO-) = K c(H2O) c(CH3COOH) = KS KS Dissoziationskonstante Säurekonstante CH3COOH H+ + CH3COO- Säure-Base-Gleichgewichte Essigsäure pKS = -log KS = 4,74 CH3COOH H+ + CH3COO- D.h bei einer Gesamtkonzentration von 0,1 mol/L (bei 25°C) ist Essigsäure zu 1,34% dissoziiert. Säure-Base-Gleichgewichte Dissoziationsgrad (α) Der Dissoziationsgrad eines schwachen Elektrolyten (ionenbildender Stoff) ist der Bruchteil der gesamten Stoffmenge (c 0), der in Ionen dissoziiert ist (in % angegeben). c(CH3COO-) c0(CH3COOH) =α α Dissoziationsgrad undissoziiert: c(CH3COOH) = c0 - x mol/L dissoziiert: c(CH3COO-) = x mol/L c(H+) c(CH3COO-) x2 = c(CH3COOH) c0 - x = KS x2 + K Sx – KSc0 = 0 x = c(H+) = 1/2KS + √(1/4KS2 + KSc0) = 0 Bei schwachen Säuren (kl. K S): x = c(H+) = √KSc0 pH ≈ ½( pKS – log c0) Säure-Base-Gleichgewichte Ionenkonzentration und Dissoziationsgrad von Essigsäure bei 25°C c0 mol/L C(H+)=c(CH3COO-) mol/L pH Dissoziationsgrad α 1,00 0,00426 2,37 0,426 0,100 0,00134 2,87 1,34 0,0100 0,000418 3,83 4,18 0,00100 0,000126 3,90 12,6 Säure-Base-Gleichgewichte Basenkonstante In wäßrigen Lösungen von schwachen Basen stellt sich ein Gleichgewicht ein, an dem OH--Ionen beteiligt sind. NH4+ + OH- NH3 + H 2O c (NH4+) c(OH -) c(NH3) c(H2O) =K K Gleichgewichtskonstante c(H2O) = konst. c (NH4+) c(OH -) = K c(H2O) c(NH3) = KB KB Basenkonstante Bei schwachen Basen (kl. KB): x = c(OH-) = √KBc 0 pOH ≈ ½( pKB – log c0) Säure-Base-Gleichgewichte Säure-Base-Konjugation NH3 + H 2O Base NH4+ + OHKonjugierte Säure (Kation einer schwachen Base) Konjugierte Base (Anion einer schwachen Säure) Säure CH3COOH + H 2O H3O+ + CH3COO- Säure-Base-Gleichgewichte Säure-Base-Konjugation Zwischen der Basenkonstante einer Base und der Säuredissoziationskonstante ihrer konjugierten Säure besteht ein einfacher Zusammenhang. AH H+ + A- c(H+) c(A-) c(AH KS KB A- + H 2O AH + OH- c(AH) c(OH -) = K B c(A-) = KS c(H+) c(A-) = c(AH c(AH) c(OH -) c(A-) = KW = 10-14 pKS + pKB = pKW = 14 Säure-Base-Gleichgewichte Zusammenhang zwischen Säure-Base-Stärke Je größer KS für eine Säure ist, desto kleiner ist KB für die konjugierte Base (und umgekehrt) pKS + pKB = pKW = 14 Starke Säure ↔ Schwache Säure ↔ Schwache Base Starke Base Säure-Base-Gleichgewichte Beispiel: Welchen pH-Wert hat eine Lösung von 0,1 mol Natriumacetat pro Liter? (Acetat-Ion ist eine schwache Base) CH3COO- + H 2O OH- + CH3COOH pKS (CH3COOH) = 4,74. pKB = 14 – pKS = 9,26. pOH ~ ½ (pKB – log c0) = 5,13 pH = 14 – pOH ~ 8,87 Säure-Base-Gleichgewichte Beispiele: Säure pKS Flußsäure HF Salpetrige Säure HNO2 Blausäure HCN 3,2 H + + FH+ + NO2- 3,4 9,4 H + + CN- Basen Methylamin CH3NH2 + H 2O Ammoniak NH3 + H 2O Hydrazin N2H4 + H 2O CH3NH3+ + OHNH4+ + OHN2H5+ + OH- 3,3 4,7 6,0 Säure-Base-Gleichgewichte Indikatoren: Indikatoren sind (organische) Farbstoffe, deren Farbe pH-abhängig ist. Universalindikator = Mischung bestimmter Indikatoren, die je nach pH-Wert einen bestimmten Farbton besitzt. H+ + Ind- HInd c(H+) c(Ind-) = KS = 10-7 mol/L c(HInd) c(Ind -) c(HInd) pH = 5 c(Ind -) c(HInd) = ROT 10-7 c(H+) = 10-7 c(H+) pH = 8 pH = 7 = 10-2 PURPUR c(Ind -) c(HInd) = BLAU 10-7 c(H+) = 10 Säure-Base-Gleichgewichte Pufferlösungen: Pufferlösungen bewirken, dass der pH-Wert einer Lösung konstant bleibt. (z.B.: Lösung von CO2 in Wasser, CO2 + H2O ↔ H2CO3. Eine Pufferlösung ist eine Lösung, die eine relativ hohe Konzentration einer schwachen Säure UND ihrer konjugierten Base enthält. CH3COOH H+ + CH3COO- c(H+) c(CH3COO-) x mol/L = c(H+) c(CH3COOH) x mol/L = KS = 1,8 10-5 mol/L pH = pKS = 4,74 Für eine Lösung, die eine schwache Säure und ihre konjugierte Base im Stoffmengenverhältnis 1:1 enthält, gilt: pH = pKS Säure-Base-Gleichgewichte Pufferkapazität: 1,0 mol Essigsäure und 1,0 mol Natriumacetat (pH = 4,742). Es wird HCl zugegeben, wie ändert sich der pH? c(H+) c(CH3COO-) = KS c(CH3COOH) c(H+) = KS c(CH3COOH) c(CH3COO-) pH = pKS - log c(CH3COOH) c(CH3COO-) Zusatz von 0,01 mol/L HCl: pH = pKS – log(1,01/0,99) = 4,733. (0,01 mol/L : pH = 2!) Zusatz von 0,1 mol/L HCl: pH = pKS – log(1,10/0,90) = 4,655. (0,1 mol/L : pH = 1!) Zusatz von 0,01 mol/L NaOH: pH = pKS – log(0,99/1,01) = 4,751. (0,01 mol/L : pH = 12!!!) Säure-Base-Gleichgewichte Henderson-Hasselbach-Gleichung: pH = pKS - log c(HA) c(A-) Aus Cyansäure (HOCN) und Kaliumcyanat (KOCN) soll eine Pufferlösung mit pH=3,5 hergestellt werden. pKS=3,92. pH = pKS - log c(HOCN) c(NCO-) log [c(HOCN)/c(NCO-)] = pKS – pH = 3,92 – 3,50 = 0,42 c(HOCN) = 2,63 c(NCO-) Säure-Base-Gleichgewichte Mehrprotonige Säuren: enthalten mehr als ein dissoziierbares Wasserstoffatom (H2SO4, H3PO4), d.h. setzen mehr als ein Wasserstoffatom pro Molekül frei. H3PO4 H+ + H 2PO4- c (H+) c(H2PO4-) c(H3PO4) = KS1 H2PO4- H+ + HPO42- c (H+) c(HPO42-) c(H2PO4-) = KS2 HPO42- H+ + PO43- c (H+) c(PO43-) c(H2PO42-) = KS3 = 7,5 10-3 mol/L = 6,2 10-8 mol/L = 1,0 10-12 mol/L Säure-Base-Gleichgewichte Phosphorsäure: 0,1 mol/L Phosphorsäure; wie groß sind die Konzentrationen der einzelnen Ionen? H3PO4 0,1 - x H+ + H 2PO4x x mol/L x = c(H+) = 1/2KS + √(1/4KS2 + KSc0) = 0 x = 2,4 10-2 mol/L c(H3PO4) = 7,6 10-2 mol/L H2PO4- H+ + HPO42- c(HPO42-) = 6,2 10-8 mol/L HPO42- H+ + PO43- c(PO43-) = 2,6 10-18 mol/L Säure-Base-Gleichgewichte Kohlensäure: Lösungen von Kohlendioxid und Wasser reagieren sauer. CO2 + H 2O c (H+) c(HCO3-) c(CO2) HCO3c (H+) c(CO32-) c(HCO3-) H+ + HCO3- = KS1 = 4,2 10-7 mol/L H+ + CO32= KS2 = 4,8 10-11 mol/L Säure-Base-Gleichgewichte Salze schwacher Säuren und Basen: Lösungen von Salzen schwacher Säuren reagieren basisch. Anionen schwacher Säuren reagieren basisch. Kationen schwacher Basen reagieren sauer. z.B. CH3COO- + H 2O OH- + CH3COOH Hydroxid-Ionen werden gebildet! NH3 + H 2O NH4+ + OHHydroxid-Ionen werden verbraucht! NH4+ NH3 + H + Eine 0,30 mol/L NH4Cl-Lösung hat einen pH-Wert von? pKS(NH4+) = 14 – pKB(NH3) = 14 – 4,7 = 9,3 pH = ½ ( pKS – log 0,30) = 4,9 Salze starker Säuren oder Basen reagieren nicht und verändern daher den pH-Wert nicht! Säure-Base-Gleichgewichte Säure-Base-Titration: Neutralisation der H+ (oder OH-) Ionen durch zugegebene OH- (oder H+) Ionen. Schrittweise Neutralisation bis zum Äquivalenzpunkt (Konzentration an H+ gleich OH-), beim Äquivalenzpunkt (Na+=Cl-) ist die (H+,OH-) Ionenkonzentration durch die Eigendissoziation (Ionenprodukt) des Wassers bestimmt. z.B. HCl + NaOH 2 H2O H3O+ + OH- durch schrittweises Zugeben von NaOH werden H+-Ionen teilweise neutralisiert und verdünnt! Basischer Bereich pH = 7 pH Äquivalenzpunkt (Na+=Cl-) („Wendepunkt“) Volumen NaOH Teilweise Verdünnung Säure-Base-Gleichgewichte Säure-Base-Titration (schwache Säure und starke Base): z.B. Essigsäure und NaOH. 0,1 mol/L Essigsäure, pH = 2,87 pH = ½ (pKS – log c0) Beim Äquivalenzpunkt: Gesamtvolumen: doppelt so groß; CH3COO-: 0,05 mol/L pOH = ½ (pKB – log 0,05) = 5,28 ; pH = 15 – pOH = 8,72 Äquivalenzpunkt (nur Acetat-Ionen!) („Wendepunkt“) pH = 8,72 pH Bereich geringer! Volumen NaOH Pufferlösung (Henderson-Hasselbach-Gl.) Säure-Base-Gleichgewichte Säure-Base-Titration (schwache Säure und schwache Base): z.B. Essigsäure und Ammoniak. Kein steiler Anstieg! pH pH = 7,0 Volumen NH3 Säure-Base-Gleichgewichte Starke Säure & Starke Base Detektion durch Indikatoren Schwache Säure & Starke Base pH Volumen Base Gut detektierbar: Schwache Säure & Schwache Base Starke Säure/Starke Base & Schwache Säure/Starke Base & Starke Säure/Schwache Base Schlecht detektierbar: Schwache Säure/Schwache Base Löslichkeit Löslichkeitsprodukt: Manche Substanzen sind in Wasser nur geringfügig löslich (teilweise Dissoziation in Ionen). Aus diesem Grund existiert ein Gleichgewicht zwischen gesättigter Lösung und ungelöstem Feststoff. z.B. Silberchlorid (AgCl). AgCl c (Ag+) c(Cl-) c(AgCl Ag+ (aq) + Cl- (aq) =K c (Ag+)c(Cl-) = Kc(AgCl) = L L Löslichkeitsprodukt Bei 25°C lösen sich 0,00188g AgCl in 1L Wasser. Wie groß ist L? n(AgCl) = m(AgCl)/M(AgCl) = 0,00188g / 143 gmol-1 = 1,31 10-5 mol c(Ag+) = c(Cl-) = 1,31 10-5 mol/L; L = 1,7 10-5 mol2/L2 Löslichkeit Löslichkeitsprodukt: Die tatsächliche Löslichkeit eines Salzes wird zusätzlich durch Salzeffekte (Zusatz eines weiteren Elektrolyten, Abschirmung der Ionen, Aktivitätsverringerung) oder durch Reaktion eines gebildeten Ions beeinflusst. BaCO3 (s) CO32- (aq) + H 2O Ba2+ (aq) + CO32- (aq) HCO3- (aq) + OH- (aq) Die Konzentration an CO32- wird durch die Reaktion mit Wasser verringert. Löslichkeit Fällungsreaktionen: Durch Zugabe des entsprechenden Gegenions kann ein Ion ‚ausgefällt‘ werden. Z.B. z.B. 10 mL einer 0,01 mol/L AgNO3-Lösung werden mit 10 mL einer 0,0001 mol/L NaClLösung versetzt. L(AgCl) = 1,7 10-10 mol2/L2. Gesamtvolumen: 20 mL c(Ag+)c(Cl-) = 5,0 10-3 . 5,0 10-5 = 2,5 10-7 mol2/L2 > L („Fällung von AgCl“) Ionenzusätze: Gleichionige Zusätze verschieben das Gleichgewicht nach links („Fällung“) AgCl Ag+ (aq) + Cl- (aq) Löslichkeitsprodukt: Solange das System im Gleichgewicht ist, ist L unabhängig vom Reaktionsmechanismus! Komplexgleichgewichte Komplexe: Verbindungen, die aus Liganden, die an ein Zentralatom koordiniert sind. Komplexe können geladen (Summe der Ladungen des Zentralatoms und der Liganden) bzw. neutral sein. Hydratisierte Metallkationen sind als Komplexverbindungen zu betrachten. Fe2+ (aq) + 6 CN- (aq) [Fe(CN)6]4- Nettoladung: 2(+) - 6(-) = 4(-) [Ag(NH3)2]+ [Cu(NH3)4]2+ [Fe(CN)6]3- [Zn(OH)4]2- [CdCl4]2- [Cu(OH2)4]2+ Ligandenaustausch: In wäßriger Lösung kann es zu einem Austausch der Liganden kommen. z.B. [Cu(OH2)6]2+ + 4 NH3 Cu2+ + 4 NH3 [Cu(OH2)2 (NH3)4]2+ + 4 H 2O [Cu(NH3)4]2+ (Fe3+) Komplexgleichgewichte Dissoziationskonstante: Komplexverbindungen zerfallen stufenweise. Die Gleichgewichtskonstante für die Bruttoreaktion wird Komplexzerfalls- oder dissoziationskonstante genannt. [Ag(NH3)2]+ [Ag(NH3)]+ + NH3 c([Ag(NH3)]+) c(NH3) = KD1 = 1,4 10-4 mol/L c([Ag(NH3)2]+) [Ag(NH3)]+ c(Ag+) c(NH3) c([Ag(NH3)]+) Ag+ + NH3 = KD2 = 4,3 10-4 mol/L (Komplex-) Dissoziationskonstante: K D = K D1KD2 = 6,0 10-8 mol 2/L2 (Komplex-) Bildungskonstante: K K = 1/KD = 1,6 107 mol -2/L-2 Komplexgleichgewichte Beispiele: Schwerlösliche Verbindungen können durch Komplexbildung in Lösung gebracht werden. Löslichkeit von AgCl in Ammoniak-Lösung, c(NH3) = 0,10 mol/L? Ag+ (aq) + Cl- (aq) AgCl (s) Ag+ (aq) + 2 NH3 (aq) c (Ag+)c(Cl-) = L [Ag(NH3)2]+ (aq) c([Ag(NH3)2]+) c(Ag+)c2(NH3) Gesamtreaktion! AgCl (s) + 2 NH3 (aq) [Ag(NH3)2]+ (aq) + Cl- (aq) c([Ag(NH3)2]+)c(Cl-) c2(NH3) = LKK = KK Komplexgleichgewichte [Ag(NH3)2]+ (aq) + Cl- (aq) AgCl (s) + 2 NH3 (aq) 1. 2. c([Ag(NH3)2]+) = c(Cl-) c(NH3) = 0,10 mol/L – 2 c(Cl-) c2(Cl-) [0,10 – 2c(Cl-)]2 = LKK = 1,67 107 . 1,7 10-10 c(Cl-) = 4,8 10-3 mol/L c(Cl-) = 1,3 10-5 mol/L (reines Wasser) Elektrochemie Redoxvorgänge Seit Boyle bildete das Phänomen der Verbrennung eines der wichtigsten Probleme der Chemie. Lavoisier erkannte, dass bei einer Verbrennung Sauerstoff verbraucht wird. Er führte daher den Begriff Oxidation ein und übertrug den Namen auf alle Vorgänge, bei denen sich eine Substanz mit Sauerstoff verbindet. Der Ausdruck Reduktion, mit dem man ursprünglich nur die Zurückführung eines Metalloxids auf das entsprechende Metall (d.h. Metallgewinnung aus dem Oxid) bezeichnet hatte, wurde allmählich für jede Abspaltung von Sauerstoff aus einer Verbindung verwendet und erlangte damit die gegenteilige Bedeutung des Begriffes Oxidation. Jedoch gibt es viele Reaktionen (auch eigentliche Verbrennungen, Na in Chloratmosphäre), die sich äußerlich nicht von Verbrennungen unterscheiden, daher wurden die Begriffe Reduktion und Oxidation erweitert. Elektrochemie Redoxvorgänge Das gemeinsame Merkmal all dieser Vorgänge, ist die Tatsache, dass die Metallatome ihre Außenelektronen abgeben und dadurch zu positiven Ionen werden. Diese freigesetzten Elektronen werden von den Nicht-Metall-Atomen aufgenommen. z.B. Na + Na Mg + Na+ O O + Na+ Mg2+ + O2- O2- Nicht immer tritt eine vollständige Abgabe von Elektronen auf. Ganz allgemein ist daher eine Oxidation ein Vorgang, bei dem einem Teilchen Elektronen (oder Elektronendichte) entzogen werden. Elektrochemie OXIDATION = Elektronenabgabe REDUKTION = Elektronenaufnahme Allerdings kann ein Teilchen nur Elektronen abgeben, wenn diese gleichzeitig von anderen Teilchen aufgenommen werden. Oxidation und Reduktion verlaufen daher stets gekoppelt. Diese Reaktionen nennt man daher Redoxreaktionen (Redoxvorgänge): Eine Redoxreaktion ist eine Elektronenübertragung Elektrochemie Beispiele für Redoxreaktionen: Verbrennung von Metallen (oder Wasserstoff) in Sauerstoff oder auch Chlor; Bildung von Metallsulfiden aus Metall und Schwefel. Reduktion von Metalloxiden mit unedlen Metallen, Wasserstoff oder Kohle. Cu O + H2 → Cu + H2O Al2O3 + 6 Na → Al + 3 Na2O Thermolyse von HgO 2 HgO → Hg + O2 Verdrängungsreaktionen Fe + Cu2+ → Fe2+ + Cu 2 I- + Br2 → I2 + 2 Br Photochemische Zersetzung der Silberhalogenide AgCl, AgBr und AgI färben sich bei Einwirkung von Licht allmählich dunkel und schließlich schwarz, wobei die Reaktionsgeschwindigkeit von AgCl zu AgI zunimmt. Durch die bereitgestellte Lichtenergie wird ein Elektron auf ein Ag+-Ion übertragen. Elektrochemie Vergleich zwischen Säure-Base-Reaktionen und Redoxreaktionen: Ebenso wie die Protolyse verlaufen auch die meisten Redoxvorgänge reversibel (umkehrbar). Oxidationsmittel/Reduktionsmittel = Base/konjugierte Säure Redoxpaar: Säure/Base-Paar: Red Ox + n e - AH H+ + A- Na Na+ + e- HCl H+ + Cl- 2 Cl- Cl 2 + 2 e - NH4+ H+ + NH3 Elektrochemie Wie bei einer Säure-Base Reaktion sind auch bei einer Redoxreaktion zwei Redoxpaare beteiligt. Redoxpaar: Ox(2) + Red(1) HA + B Red(2) + Ox(1) A- + HB+ Säure/Base-Paar: Elektrochemie Grundbegriffe: Elektrischer Strom ist ein Fluß von elektrischer Ladung. (z.B. Elektronen in einem Metalldraht). Elektrische Spannung (Potential) induziert einen Stromfluß und bewirkt die Bewegung der Elektronen z.B. in einem Draht. Einheit: Volt (V). Elektrische Ladung („Ladungsmenge, Elektrizitätsmenge“); Einheit: Coulomb (C). Stromstärke = Stromfluß/Zeit; Einheit: Ampére (A). 1 Ampére entspricht einem Stromfluß von 1 Coulomb pro Sekunde. V2 V1 Potential (V) Um elektrische Ladungen zu bewegen, ist Energie erforderlich. Um eine Ladung von 1C gegen ein Potential von 1 Volt zu bewegen, muß eine Energie von 1 Joule aufgebracht werden (1J = 1V.C). Elektrochemie Grundbegriffe: Elektrischer Widerstand ist der Widerstand, den ein Stoff (z.B. Metall) einem Stromfluß entgegensetzt. Je größer der Widerstand, umso höher das Potential, um eine bestimmte Stromstärke aufrechtzuerhalten. U = R.I (U: Potential; R: Widerstand; I: Stromstärke). Einheit: Ohm (Ω) 1 Ω = 1 V /A. Die Leitfähigkeit ist der Kehrwert des Widerstands, Einheit: Siemens (S). Elektrochemie Elektrolytische Leitung In Elektrolytlösungen erfolgt der elektrische Stromfluß durch (wandernde) Ionen. Die elektrolytische Leitung findet daher in Elektrolytlösungen oder in geschmolzenen Salzen statt. Elektrode e- Gleichstromquelle eElektrode (-) (+) Elektrolyt Cl- Na+ Na+ (-) Cl- Kathode Na+ + e- → Na (+) Anode 2 Cl- → Cl2 + 2 e- Elektrochemie Elektrolyse einer Natriumchlorid-Schmelze Temperatur: ~600°C e- Gleichstromquelle (-) Flüssiges Natrium e(+) Cl- Na+ (geringere Dichte) Na+ (-) Cl- Kathode Na+ + e- → Na (+) Anode 2 Cl- → Cl2 + 2 e- Chlorgas Elektrochemie Die Redoxreaktion kann in zwei Halbreaktionen zerlegt werden. Reduktion des Na+: Na+ + e- → Na Oxidation des Cl-: 2 Cl- → Cl2 + 2 e- Elektrolytische Zelle: Eine Reaktion, die freiwillig nicht ablaufen würde, wird mithilfe einer äußeren Stromquellen erzwungen (Energie). Galvanische Zelle: erzeugt einen Strom mittels einer freiwillig ablaufenden chemischen Reaktion. In beiden Fällen spricht man von elektrochemischen Zellen. Elektrochemie In der elektrochemischen Zelle verläuft der Reduktions- und Oxidationsschritt räumlich voneinander getrennt. Jede Reaktion findet in einem eigenen Elektrodenraum statt. Während der Reaktion fließen die Elektronen, die in der Halbreaktion Red1 →Ox1 + efreigesetzt werden, durch den äußeren Stromkreis und treten durch die andere Elektrode wiederum in die Zelle ein. Dort werden sie zur Reduktion des Oxidationsmittels verwendet. Ox2 + e- → Red2 Anodenreaktion (Oxidation): Kathodenreaktion (Reduktion): Red1 →Ox1 + eOx2 + e- → Red2 Reduktion des Na+: Na+ + e- → Na Oxidation des Cl-: 2 Cl- → Cl2 + 2 e- Elektrochemie Elektrodentypen Eine Metall/Metallionen-Elektrode besteht aus einem Metall in Kontakt mit einer Lösung eines seiner Salze (z.B. Cu im Kontakt mit Cu2+-Ionen). Die Bezeichnung dafür ist M | M+(aq). Gaselektrode besteht aus einem Inertmetall (zumeist Platin, Pt), das als Quelle oder Senke für Elektronen fungiert, und Gas im Gleichgewicht mit einer Lösung seiner Ionen (z.B. H2 und H+). Das Metall wirkt nur als Katalysator, nimmt aber nicht direkt daran teil. Ein Beispiel hierfür ist die Wasserstoffelektrode: gasförmiger Wasserstoff wird in eine Lösung eingeleitet, die Wasserstoff-Ionen enthält, das Redoxpaar ist H+/H2. Die symbolische Schreibweise für diese Elektrode lautet Pt | H2(g) | H+ (aq). Elektrochemie Elektrolyse In einer wäßrigen Lösung von Natriumsulfat wandern Na+-Ionen zur Kathode und SO42--Ionen zur Anode. Allerdings sind in der Lösung auch H3O+ und OHvorhanden, die leichter entladen werden können als die Ionen aus dem Natriumsulfat. Kathoden-Prozess: 2H2O 2 H+ + 2 e- Anoden-Prozess: 2 H+ + 2 OH - 2H2O H2 (g) 2 OH- Gesamtreaktion: H2O H2 (g) + ½ O2 (g) 2 H+ + 2 OH ½ O2 (g) + H 2O + 2 e - Elektrochemie Elektrolyse wäßriger Salzlösung 1. Na2SO4: H2O → H2 (g) + ½ O2 (g) 2. NaCl: 2H2O + 2 Na+ + 2 Cl- → H2 (g) + Cl2 (g) + 2 Na+ + 2 OH - 3. CuSO4: H2O + Cu2+ → ½ O2 (g) + Cu (s) + 2 H + 4. CuCl2: Cu2+ + 2 Cl- → Cu (s) + Cl2 (g) Alternativreaktionen an der Anode 2 SO42- → S2O82- + 2 e H2O → ½ O2 (g) + 2 H + + 2 e Cu (s) → Cu2+ e- Gleichstromquelle (-) e(+) vorher Kupfer (Cu) Kupfer (Cu) SO42Cu2+ Cu2+ Verwendung: Elektrolytische Raffination von Rohkupfer (95-%ig); „Versilbern“ Elektrochemie Stöchiometrie Michael Faraday Quantitativer Zusammenhang zwischen chemischen Umsetzungen und umgesetzten elektrischen Ladungsmengen. Z.B. bei der Elektrolyse von 1 mol NaCl (zu Na und Cl2) werden 1 mol Elektronen benötigt. Die Ladungsmenge (in Coulomb, C) wird als Faraday-Konstante (F) bezeichnet. F = 96485 C/mol = 6,022 1023 mol-1 . 1,6022 10-19 C (Avogadro-Zahl x Elementarladung) Faraday-Gesetz m= Elektrizitätsmenge L: L = I (Stromstärke) . Zeit (Sekunden) M L z F m = abgeschiedene Menge M = Molmasse z = umgesetzte Elektronen F = Faraday-Konstante L = Elektrizitätsmenge Elektrochemie Beispiel: Wieviel Kupfer scheidet sich ab, wenn ein Strom von 0,75 A 10 Minuten lang durch eine wäßrige Kupfersulfat-Lösung geleitet wird? Elektrizitätsmenge: 450 C z = 2 Elektronen; M = 63,5 g/mol m(Cu) = 63,5 . 450 / (2 96485) = 0,148 g Elektrochemische Zellen Salzbrücke Metallischer Leiter Elektrodenräume Elektrolyt Elektrochemische Zellen e- eGleichstromquelle (-) (+) Ag+ e- (-) (+) Cu2+ NO3- SO42- Kathode Anode Ag+ + e- → Ag 2H2O → O2 (g) + 4 H+ + 4 e- Kathode Cu2+ + 2e- → Cu Silber-Coulombmeter Anode 2H2O → O2 (g) + 4 H+ + 4 e- Silber-Coulombmeter Beispiel: Wieviel Kupfer scheidet sich bei der Elektrolyse von Kupfersulfat ab, wenn gleichzeitig 1,00 Ag abgeschieden wird? Faraday-Gesetz: m = (M/z) (L/F); L = 894,5C für 1,00g Ag. m(Cu) = 63,5 . 894,5 / (2 96485) = 0,294 g Wie lange dauert der Vorgang bei einer Stromstärke von 1,6 A? L = I.t: t = 894,5 C/ (1,60 A) = 559,1 s Galvanische Zellen e(-) (+) Voltmeter Daniell-Element Kathode/Pluspol: Reduktion Zn2+ SO42- Cu2+ SO42- Anode/Minuspol: Oxidation Anode Zn → Zn2+ + 2e- Kathode Cu2+ + 2e- → Cu Trennwand (Durchtritt von Ionen) Zellspannung oder Elektromotorische Kraft (EMK) Elektrolyse ↔ Galvanische Zelle Elektrolyse erfolgt unter Aufwendung elektrischer Energie (Elektronen werden von der Anode zur Kathode gepumpt). Die zur Elektrolyse erforderliche Spannung muß größer als die entgegengerichtete Zellspannung sein. Ist die angelegte Spannung kleiner arbeitet die elektrochemische Zelle als galvanische Zelle. (-) (+) H2 (-) Cl2 H+ Cl- (+) H2 Cl2 H+ Cl- Elektrolytische Zelle Galvanische Zelle 2 HCl → H2 + Cl2 H2 + Cl2 → 2 HCl Zellspannung oder Elektromotorische Kraft (EMK) Elektrolyse ↔ Galvanische Zelle Ist die angelegte Spannung kleiner arbeitet die elektrochemische Zelle als galvanische Zelle. Die freigesetzte elektrische Energie (an den Elektroden auftretende Zellspannung oder Elektromotorische Kraft ) entspricht der freien Reaktionsenthalpie der chemischen Reaktion. Möglichkeit zur direkten experimentellen Bestimmung der freien Reaktionsenthalpie, über die geleistete Arbeit). Merke: ∆G = ∆H – T∆S ; ∆G …… maximale Arbeit, die das System leisten kann. Elektrolyse 2 HCl H2 + Cl2 Galvanische Zelle Chemisches Gleichgewicht liegt vor, wenn keinerlei chemische Reaktion vor sich geht, wenn also die angelegte Spannung gleich der Zellspannung ist. Wird bei konstantem Druck und konstanter Temperatur bsp. Die Konzentration eines Stoffes verändert, so kommt das chemische System aus dem Gleichgewicht und es erfolgt eine Reaktion in die eine oder andere Richtung. Die Reaktion läuft solange bis das Gleichgewicht erreicht ist. Zur Definition des chem. Gleichgewichtes kommt jetzt auch die elektrische Energie hinzu! Elektrochemische Gleichgewichtsbedingung ∆G < 0: chemische Energie wird in elektrische Energie übergeführt und in der Batterie gesopeichert ( galvanische Zelle). ∆G > 0: der Batterie wird elektrische Energie entnommen (Elektrolyse). ∆G = 0: Gleichgewicht, freie Reaktionsenthalpie ist gleich der elektrischen Energieänderung der Batterie, ∆Eel. Elektrochemische Gleichgewichtsbedingung: ∆G + ∆Eel = 0 Elektrochemische Gleichgewichtsbedingung Werden bei einer Reaktion je molarem Formelumsatz NA Elektronen ausgetauscht, so beträgt die vom System (reversibel) geleistete Arbeit ∆Eel = NAe∆E da die elektrische Arbeit bzw. Energie durch Spannung mal Ladung (1J = 1VC) definiert ist (∆E=Zellspannung=Potentialdifferenz). Da F= NAe ∆G + F ∆E = 0 oder ∆G = - F ∆E oder bei nNA Elektronen je Formelumsatz ∆G + nF ∆E = 0 ∆E = -∆G / nF ∆E elektromotorische Kraft (EMK) oder Zellspannung Elektrochemische Gleichgewichtsbedingung Reaktionen in der elektrochemischen Zelle werden von der Batteriespannung und der elektromotorischen Kraft bestimmt. ∆G + nF ∆E < 0 Galvanische Zelle ∆G + nF ∆E > 0 Elektrolytische Zelle Daher: Messung der freien Reaktionsenthalpie aus Spannungsmessungen!!!! Messung der Elektromotorischen Kraft (EMK) Für die Messung der elektromotorischen Kraft ist es erforderlich, dass sie stromlos durchgeführt wird. Bei Stromentnahme aus der Zelle (endlicher Widerstand), laufen Reaktionen ab und das System gerät aus dem Gleichgewicht. Zur Messung der EMK verwendet man daher Potentiometer. Batterie Schiebewiderstand Amperemeter elektrochemische Zelle Messung der Elektromotorischen Kraft (EMK) Problem: Zahlreiche Zellen arbeiten in der Realität nicht immer reversibel (d.h. keine Stromlosigkeit wenn Gegenspannung = Zellspannung). Diese Erscheinung wird als Polarisation (oder Überspannung) bezeichnet. Zumeist ist die Ursache eine kinetische Hemmung eines heterogenen Elektrodenteilschrittes und abhängig von der Stromdichte und der Beschaffenheit der Elektrodenoberfläche. Die Polarisation entspricht einer irreversibel aufgewendeten Energie und kann bei der Umkehr, dem galvanischen Prozess, nicht zurückgewonnen werden. Elektrodenpotentiale Oxidation an der Anode: Zink-Ionen gehen in Lösung und Elektronen verbleiben im Zinkstab, dadurch entsteht ein elektrisches Potential, das der weiteren Auflösung von Zink-Ionen entgegenwirkt. - Zn2+ + + + + + Anode Zn → Zn2+ + 2e- Das entstehende Potential ist abhängig von: 1. Konzentration an Zink 2. Temperatur Elektrodenpotentiale Die entstehenden Elektronen werden abgeführt. (Stromfluss, „Elektronendruck“). Wird an das System eine weitere Halbzelle angeschlossen, so werden in Abhängigkeit vom 2.System mehr oder weniger Elektronen abgegeben, d.h. das resultierende Potential ist abhängig vom „Gegendruck“. Das absolute Potential kann nicht gemessen werden, nur das relative Potential zu einer Referenzelektrode. e- - Zn2+ + + + + + Anode Zn → Zn2+ + 2e- Referenzelektrode! Norm-Wasserstoff-Elektrode Als Referenzelektrode dient die Norm-Wasserstoff-Elektrode. Sie besteht aus Wasserstoffgas, das bei einem Druck von 101,3 kPa eine Platinelektrode umspült, die ihrerseits in eine Säure-Lösung mit einer H+(aq)-Ionenaktivität von a(H+)=1 eingetaucht ist. e- e- H2 Cu Salzbrücke (K+ Cl-) Cu2+ H+ Pt Anode H2 → 2H+ + 2e- Kathode + 2e- → Cu Cu2+ Pt|H 2|H+ || Cu2+|Cu Norm-Wasserstoff-Elektrode Für die Norm-Wasserstoff-Elektrode wurde willkürlich das Elektrodenpotential E0 = 0,00 Volt festgelegt. Die EMK einer Standard-Elektrode gegen die Norm-WasserstoffElektrode wird Normalpotential genannt, und mit dem Symbol E0 bezeichnet. z.B. besitzt die Cu2+|Cu-Elektrode das Normpotential E0 = +,0.34 V. Das Vorzeichen zeigt an, dass die Kupfer-Elektrode der Pluspol (Kathode) im Vergleich zur Wasserstoff-Elektrode ist. Ein positives Vorzeichen bezieht sich somit auf eine erlaubte Reduktion, im Vergleich zur Norm-Wasserstoff-Elektrode. Die E0–Werte sind in der sog. elektrochemischen Spannungsreihe tabelliert. Normalpotentiale können auch durch Messung gegen Elektroden bekannter Metalle gemessen werden. z.B. Ni|Ni2+ || Cu2+|Cu = 0,59V E0(Cu2+|Cu) - E0(Ni2+|Ni ) = 0,59V E0(Ni2+|Ni ) = -0,59V + 0,34 V = -0,25V