Glybera - UniQure
Werbung

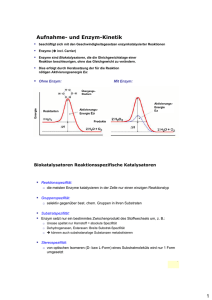
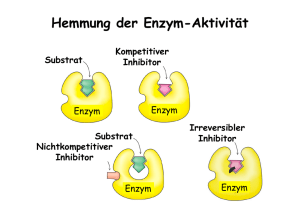
Autumn 2012 Alipogene Tiparvovec (Glybera®) - Gentherapie für LPLD Glybera® (Alipogen Tiparvovec) wurde von der Europäischen Kommission für die Vermarktung in der Europäischen Union zugelassen. Es ist die erste Gentherapie, die in der westlichen Welt zugelassen wurde. Glybera wurde für die Behandlung von Lipoproteinlipase-Defizienz (LPLD) entwickelt, einer sehr seltenen Erbkrankheit, die mit erhöhten Fettwerten im Blut einhergeht. LPLD wird durch Veränderungen des Gens, das für ein Enzym, die sogenannte Lipoproteinlipase (LPL) kodiert, verursacht. Das Enzym LPL spielt eine wichtige Rolle im Zusammenhang mit dem Fett, das wir mit der Nahrung aufnehmen. Funktioniert das Enzym LPL nicht richtig oder ist nicht genug vorhanden, steigen die Blutfettwerte dramatisch an. Glybera führt ein normales, gesundes LPL Gen in den Körper ein, so dass er ein funktionstüchtiges LPL Enzym herstellen kann. Das LPL Gen wird in einen Vektor, der vom adeno-assoziierten Virus (AAV) Serotpyp 1 abgeleitet ist, verpackt, der eine natürliche Propensität zu Muskelzellen hat. Da Muskelzellen normalerweise das wichtigste Gewebe sind, das zur Herstellung von gesunden LPL Enzymen beiträgt, ist dieser spezielle AAV sehr gut für die Korrektur von LPLD geeignet. Glybera wird durch eine einmalige Reihe von kleinen intramuskulären Injektionen in die Beine verabreicht. Glybera Klinische Daten Glybera wurde in drei klinischen Studien in den Niederlanden und in Kanada getestet, an denen insgesamt 27 LPLD Patienten teilnahmen. Eine Folgestudie dauert derzeit noch an. In allen drei klinischen Versuchen wurde Glybera gut vertragen und es wurden keine relevanten Sicherheitssignale beobachtet. Die Daten aus diesen drei klinischen Versuchen weisen darauf hin, dass nach der Therapie die Fettkonzentration im Blut bei beinahe allen Patienten, zwischen 3 und 12 Wochen nach der Injektion von Glybera, reduziert war. Wichtig dabei ist, dass die Verabreichung einer Einzeldosis Glybera zu einer langfristigen Anwesenheit und biologischen Aktivität des Enzyms in dem injizierten Muskel führte. Zudem zeigen die Daten eine klinisch bedeutsame Reduzierung der Häufigkeit von akuter Pankreatitis, der am meisten schädigenden Komplikation von LPLD. Zudem lieferten neue klinische Daten der letzten klinischen Studie in Kanada eine Basis für die Erklärung des Wirkmechanismus. Diese Daten weisen darauf hin, dass eine einzelne Verabreichung von Glybera bei LPLD Patienten zu einer bemerkenswerten, langfristig verbesserten Fähigkeit führt, Chylomikronen aufzuspalten, einen ProteinLipid-Komplex im Blut, der mit der Nahrung aufgenommenes Fett transportiert. LPLD Patienten können Chylomikronen nicht abbauen. Dies führt zu einer Anhäufung dieser Partikel im Blut und kann erhebliche Morbidität und Mortalität verursachen. Insbesondere geht man davon aus, dass eine sehr hohe Konzentration von Chylomikronen, die nach den Mahlzeiten neu produziert werden, der auslösende Faktor für neue Episoden akuter Pankreatitis ist. Durch den Abbau dieser Chylomikronen hilft Glybera, neue Episoden von Pankreatitis zu vermeiden. uniQure B.V. Meibergdreef 61 1105 BA Amsterdam The Netherlands Tel: +31 (0)20 566 7394 [email protected] www.amtbiopharma.com Single administration into leg muscle Über LPLD LPLD betrifft nur etwa einen bis zwei von einer Million Menschen weltweit, obwohl die Erkrankung in bestimmten geographischen Gebieten, wie in Teilen Kanadas, häufiger auftritt. Die Symptome beinhalten unter anderen extreme Bauchschmerzen und, insbesondere, ein erhöhtes Pankreatits-Risiko. LPLD wird durch Veränderungen des Gens, das für ein Enzym, die sogenannte Lipoproteinlipase (LPL), kodiert, verursacht. Das Enzym LPL spielt im Zusammenhang mit dem Fett, das wir mit der Nahrung aufnehmen, eine wichtige Rolle. Funktioniert das Enzym LPL nicht richtig oder ist nicht genug vorhanden, steigen die Blutfettwerte dramatisch an. Da LPLD eine Erbkrankheit ist, ist es in den meisten Fällen die zuverlässigste Diagnosemethode, Gentests an den Personen mit Verdachtssymptomen durchzuführen. Es gibt mehr als 100 verschiedene Veränderungen des LPL Gens, die LPLD hervorrufen können. Um diese Veränderungen festzustellen, wird DNA von einer Blut- oder Speichelprobe isoliert und die gesamte kodierende Region des LPL Gens wird auf Anomalien untersucht. Gentests können Personen mit LPLD detektieren (Personen mit zwei veränderten LPL Genen) und solche, die Träger sind (Personen mit nur einem veränderten LPL Gen). Eltern und Kinder von Menschen mit LPLD sind üblicherweise “Träger” der Krankheit und haben selbst keine LPLD Symptome. Geschwister von Menschen mit LPLD haben eine 25%- Chance, die Krankheit ebenfalls zu haben und eine 50%-Chance, ein Träger zu sein. Sie haben ebenfalls eine 25%-Chance, vollkommen “normale” LPL-Gene zu haben. Da Gentests emotionale, familiäre und soziale Konsequenzen haben können, wird Beratung empfohlen. Die Standard-Krankheitsbekämpfungsstrategie bei LPLD ist es, die Symptome zu reduzieren und das Pankreatits-Risiko zu reduzieren, indem die Chylomikron- und Fettwerte reduziert werden. Dies kann durch eine strenge Beschränkung des Fettgehalts in der Nahrung erreicht werden. Fettsenker, die oft bei anderen Störungen im Zusammenhang mit erhöhten Blutfettwerten verabreicht werden, sind im Allgemeinen im Fall von LPLD nicht wirksam. uniQure B.V. Meibergdreef 61 1105 BA Amsterdam The Netherlands Tel: +31 (0)20 566 7394 [email protected] www.amtbiopharma.com