Die H-chemische Verschiebung – Bereich & Einflüsse
Werbung

Spektroskopie in der Organischen Chemie
Die 1H-chemische Verschiebung – Bereich & Einflüsse
Der Resonanzbereich der Protonen in organischen Molekülen ist ca. 10 ppm
breit. Nur saure Protonen (z.B. COOH oder SO3H) liegen darüber; Protonensignale von Formylgruppen (z.B. Aldehyde) liegen um δ = 9 bis 11. Auch die
Amidprotonen des Indolrings (z.B. in Tryptophan) sind bei ∼10-11 ppm.
Es gibt eine deutliche Abhängigkeit vom s-Charakter der wasserstofftragenden
Kohlenstoffe, aber keine Korrelation!
sp3 (Alkane):
δ = 0.5 bis 4
2
sp (Alkene/Aromaten):
δ = 4.5 bis 9
sp (Alkine):
δ = ca. 2 bis 3
Innerhalb des sp3-Bereiches werden Protonen durch benachbarte (geminale)
elektronegative oder ungesättigte Substituenten entschirmt:
δ(H-C-F) > δ(H-C-O) > δ(H-C-N) > δ(H-C-C=) > δ(H-C-C-)
1
H-NMR-Spektroskopie
1
Spektroskopie in der Organischen Chemie
Aber auch der Substitutionsgrad spielt eine Rolle:
δ(CHX) > δ(CH2X) > δ(CH3X)
Cyclopropyl-1H-Signale haben ungewöhnlich kleine δ-Werte: -0.5 bis +0.5.
Wie für die C-H Bindungen bewirkt auch bei anderen X-H Bindungen die
Zunahme der Elektronegativität von X die Abnahme der Abschirmung und
entsprechend die Zunahme der chemischen Verschiebung, z.B.
CH3-CH2-OH δ(HO) = 2.56 ppm > CH3-CH2-SH δ(HS) = 1.46 ppm
1
H-NMR-Spektroskopie
2
Spektroskopie in der Organischen Chemie
Bereich der 1H-chemischen Verschiebung (Abb. nach: Friebolin)
1
H-NMR-Spektroskopie
3
Spektroskopie in der Organischen Chemie
Anisotropieeffekte
δ(aromat.-H) > δ(olefin.-H)
Dies ist eine Konsequenz der anisotropen Elektronenverteilung im Molekül und
wird bei Aromaten nach Pople durch den Ringstrom-Effekt erklärt:
Durch das äußere Magnetfeld B0 wird im Aromaten ein Ringstrom induziert, der
seinerseits ein B0 entgegen gerichtetes Magnetfeld erzeugt. Dieses schwächt
B0 oberhalb der Ringebene ab (Abschirmung; kleineres δ), während es B0 in
der Ringebene verstärkt (Entschirmung = geringere Abschirmung; größeres δ).
1
H-NMR-Spektroskopie
4
Spektroskopie in der Organischen Chemie
Beispiel: 1H-NMR-Spektrum von Ethylether
H3C
O-CH2
CH2
O
CH2
CH3
CH3
Man beachte die 1H-chemischen Verschiebungen in Abhängigkeit von der Position des elektronegativen Sauerstoffatoms.
Über den Signalen sind sog. Integrationsstufen aufgezeichnet. Diese stellen ein
relatives Maß für die Zahl der zu den jeweiligen Signalen gehörenden Wasserstoffatome dar.
1
H-NMR-Spektroskopie
5
Spektroskopie in der Organischen Chemie
1
H,1H-Kopplungskonstanten bei skalarer (J-) Kopplung
CH2
CH2
Das 1H-NMR-Spektrum
H3C
O
CH3
von Diethylether (links)
demonstriert, dass das
Spektrum außer der 1HCH3
O-CH2
chemischen Verschiebung und der Integration
noch einen weiteren
Messparameter enthält,
nämlich die Multiplizität
der Signale.
Diese Signalaufspaltungen sind auf skalare
Kopplungen benachbarter Kerne zurückzuführen. Es handelt sich um einen
‚Informationsaustausch’ zwischen Kopplungspartnern über die dazwischen
liegenden Bindungselektronen.
1
H-NMR-Spektroskopie
1
Spektroskopie in der Organischen Chemie
Ein Kern A kann über diesen Mechanismus den Spinzustand seines Nachbarn X
erkennen und reagiert darauf mit seiner Linienposition.
J
magnetische Polarisation der Elektronenhülle ↑Kernmoment
HA bzw. HX
Für jeden der beiden, praktisch gleich populierten Spin- (Energie-)zustände zeigt
A ein einzelnes Signal; es resultiert ein Dublett, dessen Abstand Kopplungskonstante J (in Hz), eine stoffspezifische Konstante, genannt wird. Eine gleich
große Kopplungsaufspaltung tritt auch am Signal des Kerns X auf.
1
H-NMR-Spektroskopie
2
Spektroskopie in der Organischen Chemie
Energieniveauschema (B0 > 0 -> Aufspaltung der magnetischen E-Niveaus α, β):
1-Spinsystem A
E
2-Spinsystem AX
β
E1
A1
E2
α
E = -mhνA
bzw. E in Hz:
E = -mνA
E1 = 1/2 νA
E2 = -1/2 νA
A1: ΔE1-2 = νA
-> 1 Linie bei νA
1
νA
H-NMR-Spektroskopie
E1 = 1/2 νA +1/2 νX
E2 = 1/2 νA -1/2 νX
E3 = -1/2 νA +1/2 νX
E4 = -1/2 νA -1/2 νX
A1: ΔE1-3 = νA usw.
νA
νX
E1 = 1/2 νA +1/2 νX +1/4JAX
E2 = 1/2 νA -1/2 νX -1/4JAX
E3 = -1/2 νA +1/2 νX -1/4JAX
E4 = -1/2 νA -1/2 νX +1/4JAX
A1: ΔE1-3 = νA +1/2 J usw.
J
J
νA
νX
3
Spektroskopie in der Organischen Chemie
Allgemein gilt: Die Multiplizität M eines Signals (Dublett, Triplett, usw.) ergibt
sich aus der Zahl der Kopplungspartner:
Bei n gleichartigen Kopplungspartnern gilt:
M = 2nI+1
I = Spinquantenzahl; hier I = 1/2 , also M = n + 1
Die Multiplizität sowie die relative Intensitätsverteilung innerhalb der Multipletts
lassen sich durch das PASCALsche Dreieck darstellen:
n=0
n=1
n=2
n=3
n=4
n=5
n=6
1
1
1
1
1
1
1
2
3
4
5
6
1
3
6
10
15
1
1
4
10
20
1
5
15
1
6
1
Singulett
Dublett
Triplett
Quartett
Quintett
Sextett
Heptett
Jede Zahl ist immer die Summe der beiden links und rechts darüber stehenden
Zahlen; keine Zahl (außerhalb des Dreiecks) bedeutet 0.
1
H-NMR-Spektroskopie
4
Spektroskopie in der Organischen Chemie
Man kann also aus der Multiplizität eines 1H-Signals der Zahl der jeweiligen
Nachbarkerne errechnen und auf diese Weise ein Netzwerk von 1H,1H-Konnektivitäten im Molekül ermitteln.
Im Falle des Diethylethers findet man für das
linke Signal (OCH2) Multiplizität: Quartett (q), 3 Kopplungspartner, und für das
rechte Signal (CH3) Multiplizität: Triplett (t) 2 Kopplungspartner
Bei nicht gleichartigen Kopplungspartnern:
Hier kann es passieren, dass ein Kern A zwei oder mehr Kopplungspartner M, X
usw. mit unterschiedlichen Kopplungskonstanten J(A,M), J(A,X) usw. hat. Man
interpretiert die Signalaufspaltungen dann nacheinander für jeden Partner, z.B.:
A
J(AX)
J(AM)
1
H-NMR-Spektroskopie
5
Spektroskopie in der Organischen Chemie
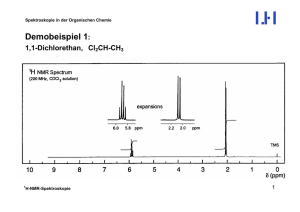
Ein weiteres Beispiel: 1,1-Dichlorethan, Cl2CH-CH3
J
1
H-NMR-Spektroskopie
6
Spektroskopie in der Organischen Chemie
Geminale Kopplungen
Wenn sich die beiden Kopplungspartner (wie Zwillinge; lat.:
gemini) am gleichen Kohlenstoffatom befinden, also nur zwei
H
Bindungen voneinander entfernt sind, nennt man dies eine
C
geminale Kopplung, 2J(1H,1H). In sehr vielen Fällen sind die
beiden Kerne chemisch äquivalent oder enantiotop () und
H
haben deshalb die gleiche chemische Verschiebung; sie
sind isochron. Dann ist das Signal – bei Fehlen weiterer
Kopplungspartner – ein Singulett (A2-System).
Merke, allgemein tritt die skalare Kopplung zwischen magnetisch äquivalenten
Kernen im Spektrum nicht in Erscheinung . (QM-Herleitung – s. Günther)
Isochrone Kerne Ai, die nur eine Spin-Spin-Kopplung zu Nachbarkernen zeigen,
sind magnetisch äquivalent.
Sind die beiden Wasserstoffkerne dagegen diastereotop, sind sie anisochron,
bilden ein AX- bzw. AB-Spinsystem bzw. -Teilsysteme, und die Kopplung ist ermittelbar.
1
H-NMR-Spektroskopie
1
Spektroskopie in der Organischen Chemie
Beispiele für geminale Kopplungskonstanten:
Einfluss des s-Charakters (s. Günther):
H
H
H
H
C
H2C C
C
H
-12.4 Hz
H
H
H
+2.5 Hz
-4.5 Hz
Einfluss der Substitution (s. Günther):
H
HO
H
C
H
-10.8 Hz
H
O
Cl
H
C C
C
H
+5.5 Hz
H
H
-1.3 Hz
z.B. CH4 – J nicht aus Spektrum -> Abschätzung z.B. basierend auf CD2H2
1
H-NMR-Spektroskopie
2
Spektroskopie in der Organischen Chemie
Vicinale Kopplungen
H
C
H
C
1
C
ϕ
H
H-NMR-Spektroskopie
H
Wenn sich die beiden Kopplungspartner an benachbarten Kohlenstoffatomen (lat.: vicinus, der Nachbar)
befinden, also drei Bindungen voneinander entfernt
sind, nennt man dies eine vicinale Kopplung,
3 1
J( H,1H)-Werte sind immer positiv.
Die Besonderheit der vicinalen Kopplung ist, dass sie
eine starke Abhängigkeit vom Torsionswinkel ϕ zwischen den beiden C-H-Bindungen besitzt. Diese wird
durch die sog. KARPLUS-Beziehung beschrieben
(nächste Seite). In den Abbildungen links und unten ist
der Torsionswinkel ca. 600; die beiden Wasserstoffatome stehen gauche zueinander.
3
Spektroskopie in der Organischen Chemie
KARPLUS-Beziehung
3J(1H,1H)
in Hz
ϕ
C
H
H
C
Karplus-Conroy-Kurve
allgemein:
3
J = A + Bcos ϕ + Ccos2ϕ
A = 4.5, B = -0.5, C = 4.5
ϕ
3
J(1H,1H) = 8.5 cos2 ϕ - 0.28
bei
00 ≤ ϕ ≤ 900
3
bei
900 ≤ ϕ ≤ 1800
J(1H,1H) = 9.5 cos2 ϕ - 0.28
Merke: Die KARPLUS-Beziehung liefert für eine experimentell ermittelte Kopplung
keinen exakten Torsionswinkel, sondern immer nur Winkelbereiche!
Wenn nicht eindeutig -> Zusatzinformationen notwendig.
1
H-NMR-Spektroskopie
4
Spektroskopie in der Organischen Chemie
Die KARPLUS-Abhängigkeit gilt auch für olefinische Wasserstoffatome:
Z
H
H
C C
H
E
H
z.B.:
C C
H
7-12 Hz
H
Ph
C C
Ph
14-19 Hz
H
C C
COOH H
12.3 Hz
COOH
15.8 Hz
Merke: Die 3J(1H,1H)-Kopplungskonstante ist ein hervorragender Parameter zur
Unterscheidung von cis- (ϕ = 0°) & trans- (ϕ = 180°) Olefinen (bzw. Z & E).
Es gibt aber auch hier Abhängigkeiten vom s-Charakter und von Substitution:
C
C
H
H
11.6 Hz
1
H-NMR-Spektroskopie
H
C
C
C
C
H
2.8 Hz
H
H
5.1 Hz
C
C
8.8 Hz
H
H
H
H
7.5 Hz
5
Spektroskopie in der Organischen Chemie
C
C
H
H
7-8.5 Hz
C
O
C
C
H
O
H
1-3 Hz
C
H
H
5-8 Hz
H
H
C
H
H
2-5 Hz
H
H
H
2-5 Hz
H
9-12 Hz
H
H
H
C
C
C
H
Bei frei drehbaren Teilstrukturen (Kohlenwasserstoff-Ketten) beobachtet man
3 1
J( H,1H) = 7 bis 8 Hz als Durchschnittswert.
1
H-NMR-Spektroskopie
6
Spektroskopie in der Organischen Chemie
Es gibt aber auch eine Abhängigkeit von benachbarten elektronegativen Substituenten:
H
H
H
Bei den Zuckern sind die 3J(1H,1H)Kopplungskonstanten wegen der
Existenz der Sauerstoffatome eher
am unteren Ende des Erwartungsbereiches.
H
9 - 12 Hz
2 - 5 Hz
aber:
HOH2C
HO
HO
H
HOH2C
O
H
HO
HO
H
O
HO OH
HO H
α: 3.5 Hz
β: 7.7 Hz
OH
D-Glucose
1
H-NMR-Spektroskopie
7
Spektroskopie in der Organischen Chemie
Fernkopplungen (Long-Range-Kopplungen)
Sind die beiden Kopplungspartner mehr als drei Bindungen voneinander entfernt,
spricht man von Fernkopplungen (long-range-Kopplungen).
n 1
J( H,1H) mit n > 3
Meist sind diese Kopplungen sehr klein (< 0.5 Hz), unter bestimmten Umständen
können sie jedoch Werte annehmen, die im Spektrum erkennbare Signalaufspaltungen verursachen.
Unter folgenden strukturellen Voraussetzungen können long-range-Kopplungen
beobachtet werden:
(a) W-Kopplung (4J): Die Wasserstoff- und die
dazwischen liegenden Kohlenstoffatome bilden
C
H
H
eine Anordnung wie der Buchstabe W.
C
C
Achtung: Das ganze Strukturelement muss weit1-4 Hz
gehend koplanar sein. Geringe Abweichungen
werden aber toleriert.
1
H-NMR-Spektroskopie
8
Spektroskopie in der Organischen Chemie
(b) Allyl-Kopplung (4J):
(c) Homoallyl-Kopplung (5J):
H
H
-3 bis +2 Hz
-3,5 bis +2.5 Hz H
C
C C
H
1
H-NMR-Spektroskopie
0 bis 2.5 Hz
H
H
1 bis 2 Hz
C
C C
H
H
C
H
C C C C
1 bis 3 Hz
H
H
0 bis 1 Hz
9
Spektroskopie in der Organischen Chemie
13
C-NMR-Spektroskopie
Der 13C-Kern ist in seinen wichtigsten Kerneigenschaften dem 1H ähnlich. Er ist
ebenso ein Spin-1/2-Kern, weist also im äußeren Magnetfeld B0 nur zwei
Energiezustände mit geringem Populationsunterschied auf. Er hat kein
Quadrupolmoment, liefert also hochaufgelöste Signale.
Im Vergleich zur 1H-NMR-Spektroskopie gibt es aber zwei wesentliche Unterschiede:
γ (1H )
Das magnetogyrische Verhältnis ist viel kleiner:
≈ 4. Dadurch ist bei
13
γ ( C)
gleicher Magnetfeldstärke die Resonanzfrequenz entsprechend kleiner; z.B:
400 MHz 1H, aber 100.6 MHz 13C.
Wegen einer ungefähren Abhängigkeit von
gere Messempfindlichkeit.
13
C-NMR-Spektroskopie
2
B03, resultiert eine deutlich gerin-
1
Spektroskopie in der Organischen Chemie
Gravierender jedoch ist die Tatsache, dass die natürliche Häufigkeit des Isotops 13C nur ca. 1.1% ist. (Das Hauptisotop 12C mit 98.9% Häufigkeit hat eine
Spinquantenzahl I = 0; es ist also NMR-inaktiv.)
Zusammen mit der geringeren Empfindlichkeit ergibt sich unter sonst gleichen
Messbedingungen (gleiche Substanzkonzentration, gleiches Spektrometer) für
den Vergleich der Sensitivitäten S:
S(1H)/S(13C) ≈ 5700
Es kommt noch eine weitere Erschwernis hinzu: 13C-Signale sind wegen der
zahlreichen und zum Teil großen 13C,1H-Kopplungen (siehe später) oft stark
aufgespalten; das heißt, die ohnehin relativ geringe Signalintensität verteilt sich
auf mehrere, womöglich durch Fernkopplung sogar noch verbreiterte Teilsignale.
Dies sind die Gründe, warum die 13C-NMR-Spektroskopie erst in den 1970er
Jahren, also mehr als ein Jahrzehnt später als die 1H-NMR-Spektroskopie eine
Routinemethode wurde.
Anfangs standen deshalb auch messtechnische Verbesserungen des SignalRausch-(S/N)-Verhältnisses im Vordergrund.
13
C-NMR-Spektroskopie
2
Spektroskopie in der Organischen Chemie
Viele 13C NMR Spektren werden mit {1H}-Breitband (BB)-Entkopplung
aufgenommen. Hierbei wird während der 13C-NMR-Messung (Acquisition) ein
Entkopplerfeld auf den gesamten Protonenbereich gelegt, das so intensiv ist,
dass es alle Protonen gleichzeitig sättigen und damit entkoppeln kann.
Dadurch kollabieren die durch die 13C,1H-Kopplungen aufgespaltenen 13CMultipletts zu schmalen Singuletts. Gleichzeitig wird auch noch ein zweiter
begrüßenswerter Effekt erzielt, die Erhöhung der Signalintensität durch den
Kern-Overhauser-Effekt (siehe IM3). Er kann wegen
ηmax = 1/2· γ(S)/γ(I) und γ(S)[1H]/γ(I)[13C] ≈ 4
ηmax ≈ 2
das 13C-NMR-Signal auf das Dreifache (1+ηmax) erhöhen; allerdings nur bei
wasserstofftragenden Kohlenstoffatomen. Dann ist die räumliche Entfernung
zwischen dem 13C-Kern und den NOE-erzeugenden Protonen sehr kurz, was
die zentrale Voraussetzung für eine effektive NOE-Wechselwirkung ist. Quartäre
Kohlenstoffatome erfahren nur geringe NOE-Effekte und haben meist viel kleinere Signale.
Merke: Integrale von 13C-NMR-Signalen sollten deshalb nicht zur Bestimmung
der Zahl der jeweils zugehörigen C-Atome verwendet werden.
13
C-NMR-Spektroskopie
3
Spektroskopie in der Organischen Chemie
13
C-NMR-Spektren von 2-Butanol
{1H}-BB-entkoppelt (unten) und 1H-gekoppelt (oben)
OH
H3C
13
C-NMR-Spektroskopie
CH
CH2
CH3
4
Spektroskopie in der Organischen Chemie
„Off-Resonance“-entkoppelt (unten) und DEPT135 (oben)
13
C-NMR-Spektroskopie
5
Spektroskopie in der Organischen Chemie
{1H}-BB-entkoppelte 13C-NMR-Spektren weisen also im Vergleich zur Messung
1
H-gekoppelter Spektren ein deutlich verbessertes S/N-Verhältnis und eine größere Übersichtlichkeit auf.
Allerdings geht bei dieser Technik die gesamte Information über die Zahl der
am jeweiligen Kohlenstoffatom befindlichen Wasserstoffe, die sich im 1H-gekoppelten Spektrum in den Signalmultiplizitäten ausdrückt, verloren. Aus diesem
Grund hat sich schon früh die routinemäßige Messung von Begleitexperimenten, den sog. „Off-Resonance“-Spektren, durchgesetzt. Hierbei wird die 1HEntkoppler-Frequenz nicht wie bei BB-Messungen auf den 1H-Resonanzbereich
gesetzt, sondern daneben („off resonance“). Dadurch werden die Protonen nur
teilweise entkoppelt; sie werden im Vergleich zu den 1H-gekoppelten Signalen
schmaler (geringere Gefahr der Überlappung), behalten aber eine Restkopplung, sodass die Multiplizität noch erkennbar ist.
Seit den 1980er Jahren hat sich jedoch eine fortschrittlichere Technik, DEPT,
durchgesetzt, bei der die Information über die Zahl der anhängenden Wasserstoffe nicht durch (manchmal nur schwer erkennbare) Restaufspaltungen, sondern durch Signalphasen von Singuletts erhältlich ist.
13
C-NMR-Spektroskopie
6
Spektroskopie in der Organischen Chemie
Wichtig: in DEPT-Spektren gibt es keine Signale von quartären Kohlenstoffatomen (siehe CDCl3-Signal)!
Es gibt eine zwei gängige DEPT-Varianten: DEPT90, das nur Signale von CHAtomen abbildet, und DEPT135, bei dem CH und CH3 positive und CH2
negative Signale gibt. Zusammen mit dem BB-entkoppelten 13C-NMRSpektrum lassen sich die 13C-Signale auch komplexer Verbindungen eindeutig
zuordnen:
DEPT90
z.B.: 13C 1D NMR Spektren
Oleanolsäurederivat
(nur Aliphatenteil)
DEPT135
COOCH3
BB
13
C-NMR-Spektroskopie
AcO
nur in BB
7
Spektroskopie in der Organischen Chemie
Die 13C-chemische Verschiebung
Im Vergleich zur 1H-chemischen Verschiebung ist der δ(13C)-Bereich sehr viel
größer. Er erstreckt sich über ca. 250 ppm und zeigt eine ausgeprägte Differenzierung für Alkan- (δ = 0 - 80) und Alken-/Aromaten-Kohlenstoffatome (δ =
100 - 160). Es sei erwähnt, dass es hier keine wesentlichen Unterschiede
zwischen Alkenen und Aromaten gibt, weil der aus der 1H-NMR bekannte
Anisotropieeffekt bei Aromaten (Ringstrom) hier aufgrund des viel größeren Resonanzbereichs nicht mehr identifizierbar ist. Die chemische Verschiebung der
Alkan- und Alken-/Aromatenkohlenstoffe ist zudem von der Existenz
benachbarter Heteroatome abhängig.
Auch für die Carbonylgruppen gibt es einen separaten Bereich, der je nach Oxidationsstufe des Carbonyl-C´s sogar noch weiter unterteilt ist. Dadurch wird es
sehr einfach, Kohlensäure- und Carbonsäurederivate (δ = 150-180) von Aldehyden (δ = 180-200) und Ketonen (δ = 190-220) zu unterscheiden.
Noch größere δ-Werte haben nur Thioketone (δ = 250-270) und Carbeniumionen (δ = 200-400).
13
C-NMR-Spektroskopie
1
Spektroskopie in der Organischen Chemie
Man beachte, dass alle genannten Bereiche nur als typische Bereiche anzusehen sind. Es gibt für alle Substanzklassen Molelülstrukturen, deren Resonanzen Ausreißer nach oben und unten sein können!
13
C-NMR-Spektroskopie
2
Spektroskopie in der Organischen Chemie
Ein ungewöhnliches Verhalten zeigen die α-Effekte der schweren Halogene
(„Schweratom-Effekt“), wie hier am Beispiel
der Halogenmethane (CH4-nXn) gezeigt.
δ(13C)
Während bei den Chloriden mit zunehmender
Substitution die jeweiligen Inkremente additiv
bleiben (ca. +25 ppm), gilt dies für X = Br nur
bis CH2Br2 (ca. + 10 ppm); danach werden die
Inkremente negativ. Bei den Iodiden sind sie
von Anfang an negativ und nehmen in ihren
Absolutwerten sogar stark zu. Das Maximum
ist Br4 mit δ = -292, ein solitärer Wert weit ab
vom üblichen Resonanzbereich.
Hierfür werden relativistische Effekte verantwortlich gemacht.
n
13
C-NMR-Spektroskopie
3
Spektroskopie in der Organischen Chemie
Wegen des großen Resonanzbereichs ist die Signalzuordnung ein wesentliches
Problem, ohne dessen Lösung Strukturbestimmungen oft fragwürdig bleiben. Es
ist daher sehr wichtig, empirisch ermittelte Strukturabhängigkeiten und Inkrementenregeln zu kennen, die oft spezifisch für bestimmte Substanzklassen sind.
Im folgenden werden einige dieser Regeln vorgestellt ( NMR-1 in IM-2).
Man beachte, dass in den letzten zwei Jahrzehnten ein- und zweidimensionale
Multipuls-NMR-Experimente entwickelt wurden, die empirische Zuordnungen
teilweise oder sogar ganz entbehrlich machen. Sie beruhen auf Spin-Spin-Korrelationen durch die zwischen den Kernen liegenden Bindungen (skalare Kopplung) oder durch den Raum (dipolare Kopplung, NOE) und erlauben dadurch,
ein dreidimensionales Netzwerk der 1H- und 13C-Kerne aufzustellen. Zu den
wichtigsten dieser Methoden gehören COSY, HETCOR, HMQC, HMBC, NOESY
und einige andere ( NMR in IM-3).
13
C-NMR-Spektroskopie
4
Spektroskopie in der Organischen Chemie
Beispiele:
13
C-NMR-Spektrum von Cyanessigsäureethylester. Ordne die Signale zu.
s-Singulett, t-Terzett, q-Quartett
13
C-NMR-Spektroskopie
5
Spektroskopie in der Organischen Chemie
13
C-NMR-Spektren von ortho-Bromphenol. -> Substitutionsmuster
13
C-NMR-Spektroskopie
6
Spektroskopie in der Organischen Chemie
Aufgabe:
Entscheide anhand der
beiden 13C-NMR-Spektren,
um welche der alternativen
Strukturen es sich jeweils
handelt.
-> chem. äquiv.
Lösung:
Abzählen der Peaks gibt
(a) Cycloheptatrienderivat,
rechts
(b) Cyclohexandienderivat,
links
13
C-NMR-Spektroskopie
7
Spektroskopie in der Organischen Chemie
NMR-Lösungsmittel
Für die NMR-Spektroskopie in der OC werden i.a. deuterierte Lösungsmittel
verwendet. Die Substitution der leichten durch die schweren Wasserstoffatome
hat zwei Vorteile:
- Deuterium als Spin-1-Kern hat ebenfalls ein magnetisches Moment, aber
seine Resonanzfrequenz ist sehr weit von der der Protonen entfernt. Es
kann deshalb dazu benutzt werden, das Verhältnis von Magnetfeld und Radiofrequenz konstant zu halten (Lock), ein Routineverfahren zur elektronischen Stabilisierung der Messmethode.
- Das Lösungsmittel hat bei 100%iger Substitution kein eigenes Signal in der
1
H-NMR. Bei der Verwendung der normalen, nichtdeuterierten Lösungsmittel wäre bei den üblichen Substratkonzentrationen das Lösungsmittelsignal
das bei weitem größte, was zu unerwünschten Signalüberlagerungen, aber
auch zu Problemen bei der Darstellung kleinerer Signale nach der FourierTransformierung des FIDs führen kann.
Schreibweisen am Beispiel des Aceton: (CD3)2CO, Aceton-d6 oder [D6]-Aceton.
NMR-Spektroskopie
1
Spektroskopie in der Organischen Chemie
Dennoch haben die üblicherweise verwendeten deuterierten Lösungsmittel ein
– wenn auch kleines – 1H-Signal, weil der Deuterierungsgrad der üblichen kommerziellen Lösungsmittel nicht 100%, sondern nur 99% bis 99,9% ist.
Einige 1H-chemische Verschiebungen häufig benutzter Lösungsmittel (bei RT):
CHCl3
δ = 7.24
Methanol-d3 (CHD2OD)
δ = 3.35
Aceton-d5
δ = 2.04
Benzol-d5
δ = 7.27
CHDCl2
δ = 5.32
CHD2CN
δ = 1.93
HDO
δ = 4.65
Merke: Die Lösungsmittelmoleküle, deren 1H-Signale man beobachtet, besitzen einen 1H-Kern und eine um 1 verminderte Zahl von Deuteriumatomen, also
z.B. Aceton-d5.
NMR-Spektroskopie
2
Spektroskopie in der Organischen Chemie
Die 1H-Signale der deuterierten Lösungsmittel sind jedoch keine Singuletts,
wenn die Gesamtzahl der Wasserstoffatome im Molekül meist größer als 1 ist,
also auch noch Deuteriumatome (2H) vorhanden sind.
Beispiel CHDCl2: Deuterium hat die Spinquantenzahl 1. Das bedeutet, dass es
für einen Deuteriumkern drei ähnlich stark populierte Energieniveaus gibt:
m = -1, 0 und +1.
Das Signal eines 1H-Kern, der mit einem 2H koppelt, besteht also aus drei äquidistanten, praktisch gleich intensiven Linien (1:1:1-Triplett):
2J (2H,1H)
ν(1H)
2
H,1H-Kopplungskonstanten sind ungefähr um den Faktor 1/6.5 kleiner als die
entsprechenden 1H,1H-Kopplungskonstanten, weil γ(1H)/ γ(2H) ≈ 6.5.
NMR-Spektroskopie
3
Spektroskopie in der Organischen Chemie
Bei mehr als einem Deuterium-Kopplungspartner wird das 1H-Signal mit jedem
neuen Deuterium-Kopplungspartner in ein 1:1:1-Triplett aufgespalten.
Beispiel: Methanol-d, CHD2-OD
ν(1H)
Es entsteht ein 1:2:3:2:1-Quintett.
NMR-Spektroskopie
4
Spektroskopie in der Organischen Chemie
Die Multiplizitäten und die relativen Peakintensitäten können bei Kopplung mit
Spin-1-Kernen aus einer dem PASCALschen Dreieck ähnlichen Darstellung abgeleitet werden:
n=0
n=1
n=2
n=3
n=4
n=5
n=6
1
1
1 4
1 5 15
6 21 50
1
3
10
30
90
1
2
6
16
45
126
1
1
3
7
19
51
141
1
2
6
16
45
126
1
3
10
30
90
1
4 1
15 5 1
50 21 6
1
Jede Zahl ist immer die Summe der unmittelbar über ihr sowie der beiden links
und rechts davon stehenden Zahlen; keine Zahl (außerhalb des Dreiecks) bedeutet 0.
NMR-Spektroskopie
5
Spektroskopie in der Organischen Chemie
Schwieriger ist es bei Lösungsmitteln, die chemisch unterschiedliche Deuteriumpositionen enthalten. Beispiel: Deuterobenzol, C6HD5
H
D
D
D
D
D
NMR-Spektroskopie
Das Proton (oben) koppelt mit zwei ortho-,
zwei meta- und einem para-ständigen Deuteriumkern. Da die ortho- und die meta-2H-Kerne
paarweise auch noch magnetisch nichtäquivalent () sind, resultiert ein außerordentlich
komplexes 1H-Signal das aus vielen Einzellinien besteht (siehe links simuliertes Signal).
Die D,H-Kopplungskonstanten sind aber recht
klein, nämlich nur ca. 15% (1/6.5) der entsprechenden J(1H,1H)-Werte. Deshalb beobachtet
man i. a. ein deutlich verbreitertes Singulett
(die Einhüllende).
6
Spektroskopie in der Organischen Chemie
Lösungsmittelsignale in der 13C-NMR
Für die Aufspaltungen der 13C-NMR-Signale von deuterierten Lösungsmitteln
gilt Analoges wie bei Protonen, weil beide Kerne einen Kernspin von 1/2 haben.
Allerdings bekommt man für nicht oder nur teilweise deuterierte Spezies separate Signale (Isotopenverschiebung) mit der jeweiligen Deuterium-Aufspaltung.
Angesichts der üblicherweise sehr hohen Deuterierungsgrade kommerzieller
Lösungsmittel spielt dies aber kaum eine Rolle.
Das 13C-Signal des gebräuchlichsten Lösungsmittels CDCl3 (Deuterochloroform) ist ein Triplett bei δ = 77.0:
1J (13C,2H)
ν(13C)
Auch hier gilt: 1J(13C,2H) ∼ 1J(13C,1H)/6.5 (∼210.6 Hz/ 6.5 )
NMR-Spektroskopie
7
Spektroskopie in der Organischen Chemie
13
C-chemische Verschiebungen, Multiplizitäten und 1J(13C,2H)-Werte (in Hz)
einiger deuterierter Lösungsmittel:
CDCl3
CD2Cl2
CD3OD
CD3-CO-CD3
C6D6
CD3-CO-CD3
CD3CN
Pyridin-d5
NMR-Spektroskopie
77.0
53.5
49.3
29.3
206.3
128.0
39.7
1.3
117.7
123.5
135.5
149.5
ppm
t
quint
sept
sept
m
t
sept
sept
m
t
t
t
32
27
21
20
24
21
21
25
24
27
Hz
8
Spektroskopie in der Organischen Chemie
Beispiele für Lösungsmittelsignale (Aceton) mit unterschiedlichem Deuterierungsgrad:
*: C1HD2-CO-CD3
o: C1H2D-CO-CD3
NMR-Spektroskopie
*: CD3-CO-CD3
o: C1HD2-CO-CD3
9
Zusammenfassung1: Magnetisierungstransfer bzw.
Korrelation von einem Kern mit benachbarten Kernen in
einem Protein
• Skalare bzw. J-Kopplung - via Bindungselektronen ->
Bestimmung der chemischen Verschiebungen
einzelener Kernen und welche Kerne über chemische
Bindungen verknüpft sind
• Dipolare Kopplung - durch den Raum zwischen
Kernen, die sich räumlich nahe sind. Z.B. NOE-Effekt
zur Gewinnung struktureller Informationen in Form
von interatomaren Distanzen (-> NOESY)
R
N
C!
C
HN
H!
O
Zusammenfassung 2:
Strukturinformationen aus NMR-Daten
Distanzen
(NOEs)
Dihedrale
Winkel
(3J)
Wasserstoffbrücken
(3J, HEX)
Orientierung von
Bindungsvektoren
(RDCs)
Ionisierungskonstante
-COOH
-COO- + H+
Dynamik
Diffusionskonstanten
RDCs
Dissoziationskonstanten
Anhang:
Auf den folgenden Seiten sind einige ergänzende
Informationen
Atomkerne können eine magnetisches Moment haben
Resonanz- bzw.
Larmorfrequenz
Magnetfeld B0
Für Kerne mit Spin 1/2,
z.B. 1H, 15N, 13C, 31P
Aus H. Kessler !Skriptum zur Vorlesung OCIV", TU München
Aufbau eines Kernspinresonanzspektrometers
Temperierbarer
Probenkopf
Für Kerne mit Spin 1/2,
z.B. 1H, 15N, 13C, 31P
Computer zur
Steuerung und
Datensicherung
Supraleitender
Magnet
Elektronik
(Temperaturkontrolle, Freqzenzsynthesizer, AD Konverter, Verstärker)
Aus H. Kessler !Skriptum zur Vorlesung OCIV", TU München
Isotopenmarkierung
Zu gering -> Ausweg: Isotopenmarkierung
Zum Beispiel durch Produktion des gewünschten
Proteins in gentechnisch veränderten Bakterienzellen
(Escherichia coli)
-> einzige Stickstoffquelle 15N-Ammoniumchlorid
-> einzige Kohlenstoffquelle 13C-Glucose
Erzeugung detektierbarer Magnetisierung
1D 1H Spektrum
eines Proteins