Musterlösung zu ‚Das Werner-Syndrom
Werbung

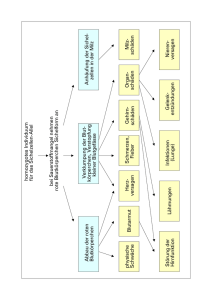
Musterlösung zu ‚Das Werner-Syndrom’ Übungsaufgaben für das Abitur 6 Der Prüfling 1. ... analysiert den Erbgang und ... Analysefragen: a) Kommt das Merkmal (hier: Werner-Syndrom/ WS) in jeder Generation vor? Nein → Hinweis auf rezessive Vererbung b) Wie ist das Verhältnis von Merkmalsträgern (MT) zu Nicht-Merkmalsträgern (NMT)? 4 MT : 27 NMT → Hinweis auf rezessive Vererbung c) Wie ist das Verhältnis von männlichen MT zu weiblichen MT? 2 : 2 → Hinweis auf autosomale Vererbung ... entwickelt eine Hypothese zum Vererbungsmodus ... Hypothese: Das WS wird autosomal-rezessiv vererbt. ... überprüft Hypothese an besonders geeigneten Stellen ... a) Das WS wird autosomal-rezessiv vererbt. Allel für gesund: A Allel für WS: a Alle MT haben den Genotyp aa. Da alle Eltern von MT gesund sind, müssen sie heterozygot sein. Das gehäufte Auftreten von MT in der 5. Generation ist auf Verwandtenehen zurückzuführen. → Hypothese ist mit dem Stammbaum vereinbar. H. ist richtig. b) Das WS wird autosomal-dominant vererbt. Allel für gesund: a Allel für WS: A Alle MT müssen den Genotyp AA oder Aa haben. Zumindest ein Elter muss im Genotyp A haben und selber MT sein. Stimmt mit den Angaben in Stammbaum nicht überein. → Hypothese ist falsch. c) Das WS wird gonosomal-dominant vererbt. Allel für WS: A Allel für gesund: a Symbol für das y-Chromosom: < Alle männlichen MT haben den Genotyp A<. Der Genotyp der Mutter eines männlichen MT muss AA oder Aa sein. Mutter muss krank sein. Stimmt mit den Angaben im Stammbaum nicht überein. → Hypothese ist falsch. d) Das WS wird gonosomal-rezessiv vererbt. Allel für WS: a Allel für gesund: A Symbol für das y-Chromosom: < Ein gesunder NMT kann keine MT-Tochter haben. Bei einem rezessiven Erbgang muss diese homozygot rezessiv sein, d. h. ein rezessives Allel vom Vater bekommen. Da dieser nur ein Allel für dieses Merkmal hat, müsste er ebenfalls MT sein. In der vierten Generation haben zwei männliche NMT MT-Töchter. → Hypothese ist falsch. Zusammenfassung: Das WS wird autosomal-rezessiv vererbt. Dieses Ergebnis wird gestützt durch die Aussage in Material A. bei dem Werner-Syndrom handele es sich um ein in Mitteleuropa sehr seltenes Syndrom (Häufigkeit 1 : 1.000.000). In dem dargestellten Familienstammbaum taucht die Krankheit erst nach mehrmaliger Verwandtenehe auf, ebenfalls ein Hinweis auf rezessive Vererbung. 2. ... beschreibt den Aufbau eines Eukaryoten-Gens und ... Ein Eukaryoten-Gen besteht aus den Promotor fernen Kontrollelementen, dem Enhancer, den Promotor nahen Kontrollelementen, der Promotorregion mit der TATA-Box und dem Startpunkt für die Transkription, dem Gen im engeren Sinne und der Terminatorregion. Alle Teile, außer dem Gen im engeren Sinne, haben regulatorische Funktion. Das Gen im engeren Sinne umfasst die für Genprodukte codierenden Abschnitte, die Exons, und die die Exons unterbrechenden nicht codierenden Abschnitte, die Introns. Abb. 1: Eukaroyoten-Gen Quelle: Campbell, Biologie ... leitet aus Material B Besonderheiten des WNR-Gens ab. Abbildung 2 zeigt eine schematische Darstellung des WNR-Gens und eine Auswahl möglicher Mutationen. Der gesamte Abschnitt umfasst 4299 Basenpaare. Farbig markiert sind fünf Gene, dargestellt ohne die im ersten Teil der Aufgabe beschriebenen Feinstrukturen. Vor, nach und zwischen den Genen liegen sechs weiß dargestellte, nicht codierende Zwischen-Genabschnitte (ZGA). Die ersten beiden Gene (schwarz/ violett) codieren für 3’-5’-Exonuclease und Helicase. Für die Gene 3 bis 5 liegen keine Angaben zu den Genprodukten vor. Angegeben sind 12 mögliche Mutationen, von denen 8 Substitutionen und 4 Deletionen sind. Diese Mutationen liegen vor im ZGA 1, im Gen für 3’-5’-Exonuclease, im ZGA 2, im Gen für Helicase, im ZGA 3 sowie im Gen ‚gelb’. Sie können einzeln oder kombiniert auftreten. Auffällig ist, dass unterschiedliche Mutationen in verschiedenen, hinter einander liegenden Genen bzw. ZGAs das WS verursachen können. Es gibt offensichtlich mehrere molekulare Ursachen für das WS. 3. ... stellt die Vorgänge bei der Replikation dar und ... Die Darstellung der Vorgänge bei der Replikation kann auf unterschiedlicher Ebene erfolgen. elektronenmikroskopische Ebene: Untersucht man die Replikation von Prokaryoten mithilfe des EM, erkennt man die ringförmige, abschnittweise replizierte DNA sowie die Replikationsgabeln. Experimentelle Ebene (Meselson-Stahl-Experiment): Die für die Untersuchung verwendeten Bakterien lässt man für eine gewisse Zeit in einem Medium 15 wachsen, welches als einzige Stickstoffquelle Ammoniumchlorid mit dem schweren N-Isotop enthält. Danach werden die Bakterien in ein Medium mit normalem Stickstoff überführt. Mithilfe einer 15 14 Dichtegradientenzentrifugation lässt sich die ‚schwere’ N-haltige DNA von der leichteren N-haltigen DNA unterscheiden. Zentrifugiert man die so behandelten Bakterien, so sammelt sich ihre DNA bandenförmig in dem Bereich des Dichtegradienten, der ihrer eigenen Dichte entspricht. Beobachtungen nach Probenentnahme aus 15N-haltigen Medium einer Zellteilung zwei Zellteilungen drei Zellteilungen eine Bande unten eine Bande in der Mitte eine Bande in der Mitte, eine Bande oben eine Bande in der Mitte, eine dicke Bande oben Erklärung: DNA wird semi-konservativ repliziert. Der DNA-Doppelstrang wird phasenweise in zwei Einzelstränge getrennt. An jeden Einzelstrang lagern sich nach dem Prinzip der komplementären Basenpaarung komplementäre Nucleotide an und ergänzen den Einzelstrang zum Doppelstrang. Molekulare Ebene: Problem bei der Replikation auf molekularer Ebene: DNA-Polymerase arbeitet nur in 5’-3’-Richtung. Die beiden Einzelstränge der DNA verlaufen antiparallel. Die beiden Einzelstränge müssen folglich unterschiedlich repliziert werden. - Helicase entwindet den DNA-Doppelstrang in zwei Einzelstränge - Einzelstrangbindende Proteine stabilisieren die Einzelstränge und verhindern die Ausbildung von Wasserstoffbrücken zwischen den komplementären Nucleotiden der Einzelstränge. Vorgänge am Leitstrang: - Synthese eines Primers an den Anfang des Leitstranges - Anlagerung komplementärer Nucleotide an den Leitstrang - Kontinuierliche Synthese des Leitstranges in 5’-3’-Richtung durch die DNA-Polymerase Vorgänge am Folgestrang: - Synthese kurzer RNA-Primer durch Primase - Anlagerung komplementärer Nucleotide an den Folgestrang Verlängerung der Primer zu Okazaki-Fragmenten durch DNA-Polymerase in 5’-3’-Richtung Ersetzen der RNA-Nucleotide der Primer durch DNA-Nucleotide durch DNA-Polymerase Verbindung der Okazaki-Fragmente mit dem wachsenden Strang durch Ligase ... veranschaulicht die Replikation mit Skizzen. beschriftete Skizzen zu den beobachtbaren Vorgängen auf den unterschiedlichen Ebenen, siehe BioBuch Größe: je Skizze mindestens ½ Seite 4. ... erläutert die Auswirkungen der angegebenen Mutationen auf struktureller Ebene sowie für die Funktion der Genprodukte. Substitution: Austausch von Basen, Punktmutation Bei Punktmutationen muss unterschieden werden zwischen Mutationen mit Wirkung und stummen Mutationen. Ob eine Mutation stumm ist oder nicht, kann auf mehreren Ebenen untersucht werden: - auf struktureller Ebene (genetischer Code) - auf der Ebene der Funktion der Genprodukte d. h. der Bildung der Tertiärstruktur eines Polypeptids/ Proteins. Klärung der Auswirkung von Mutationen auf struktureller Ebene (genetischer Code) Der genetische Code ist degeneriert. Es ist deshalb von Bedeutung, an welcher Stelle eines DNATripletts die Mutation erfolgt. Beispiel: CG* codiert für Arginin, * steht für beliebige Base. Austausch der dritten Base → keine Änderung der codierten AS → stumme Mutation Austausch der zweiten Base z. B. in CU* → Änderung der codierten Base in Leucin → eventuell Mutation mit Wirkung Austausch der ersten Base z. B. in GG* → Änderung der codierten Base in Glycin → eventuell Mutation mit Wirkung Klärung der Auswirkung von Mutationen auf der Ebene der Bildung der Tertiärstruktur eines Polypeptids/ Proteins Nicht jede AS in der Primärstruktur eines Polypeptids/ Proteins ist über die Reaktion ihrer Restgruppe an der Bildung der Tertiärstruktur beteiligt. Proteine wirken aber i. d. R. über ihre 3-D-Struktur (siehe Schlüssel-Schloss-Prinzip). Mutationen in Tripletts, die für Cystein codieren, verursachen überdurchschnittlich häufig Veränderungen in der 3-D-Struktur eines Polypeptids/ Proteins, da Cystein an der Bildung der Disulfidbrücken beteiligt ist. Mutationen in Tripletts, die für Tryptophan codieren, verursachen selten Veränderungen in der 3-D-Struktur eines Polypeptids/ Proteins, da die Restgruppe von Tryptophan kaum mit Restgruppen anderer AS reagiert. Mutationen, die die 3-D-Struktur eines Genprodukts verändern, führen entweder zu einer Funktionsminderung bzw. zu einem Funktionsverlust (loss of function) oder zu einer Funktionssteigerung (gain of function). Deletion: Verlust von Basen (einzelnen bzw. lichtmikroskopisch sichtbar fehlenden DNA-Abschnitten)/ Rastermutation Der Verlust von einer oder mehreren Basen (vor allem wenn nicht teilbar durch drei) führt zu einer Verschiebung des Leserasters der Tripletts und damit zu einer stark veränderten Primärstruktur des Genprodukts. Eine stark veränderte Primärstruktur hat eine Veränderung der Tertiärstruktur des Genprodukts zur Folge und damit i. d. R. eine Funktionsminderung bzw. einen Funktionsverlust. 5. .. bewertet den hohen zeitlichen und finanziellen Aufwand für die Erforschung seltener Erbkrankheiten. In der Medizinethik haben sich vier Prinzipien bei Bewertungen etabliert. Diese vier Prinzipien sind bei der gegebenen Aufgabe anzuwenden. 1.) Eindeutige Mehrung des Nutzens für die Erkrankten: Die Erforschung des WS bietet die Möglichkeit, eindeutige Zusammenhänge zwischen den Symptomen und ihrer genetischen Grundlage herzustellen, also die Ursache der Symptome zu ermitteln und zu verstehen. Das Verständnis der Ursachen ermöglicht in vielen Fällen, Medikamente, Prophylaxemaßnahmen u. a. zu entwickeln. Dies mehrt den Nutzen für die Betroffenen. 2.) Vermeidung von physischen und psychischen Schäden bei allen Risikopersonen: Zur Vermeidung derartiger Schäden erfolgt die Erforschung der Ursachen sowie die Erprobung von Medikamenten und Maßnahmen an Modellorganismen wie Mäusen. Menschen sind erst in einem späten Stadium von den Forschungsmaßnahmen betroffen. 3.) Respekt für die Entscheidungsautonomie der von der Maßnahmen direkt oder indirekt Betroffenen: Das Recht auf Entscheidungsautonomie bezieht sich nicht auf die Grundlagenforschung, sondern auf den Anwendungsbereich. Es muss jedem selbst überlassen bleiben, ob er Maßnahmen oder Medikamente anwenden möchte oder nicht. 4.) Berücksichtigung aller Bürgerinnen und Bürger im Hinblick auf ihre gerechte Teilhabe an den insgesamt verfügbaren medizinischen Ressourcen: Alle BürgerInnen haben das Recht auf medizinische Versorgung. Dieses Recht ist unbestritten. Das Problem bei diesem Prinzip liegt in den Begriffen ‚gerechte Teilhabe’ und ‚insgesamt verfügbare Ressource’. Diese Begriffe sind zu präzisieren, da sie völlig unterschiedlich interpretiert werden können. So kann man unter ‚gerechter Teilhabe’ verstehen, entsprechend des Anteils an der Gesamtbevölkerung oder entsprechend des gleichen Rechts auf medizinische Versorgung. Im ersten Fall wären Personen mit seltenen Erbkrankheiten in einer ungünstigen Position, weil die Erforschung dieser Krankheit unverhältnismäßig viel Geld und Forschungszeit benötigt von den ‚insgesamt verfügbaren medizinischen Ressourcen’. Im zweiten Fall spielt die Häufigkeit der Erkrankung keine Rolle, da der Focus nicht auf die Krankheit gerichtet, alle Erkrankungen werden gleich gewichtet, sondern auf den Erkrankten. Vor dem Gesetz sind alle gleich. Zu beachten ist bei der Bewertung, dass die Symptome des WS denen typischer altersbedingter Krankheiten entsprechen. Es gibt sehr viel mehr Personen mit dem dargestellten Krankheitsbild als nur durch das WS bedingt. Ein weiter gehendes Verständnis von Alterungsprozessen und ihren Ursachen könnte u. U. die Kosten für diesen medizinischen Bereich deutlich mindern und Personen ein längeres Erwerbsleben ermöglichen und damit die ‚insgesamt verfügbare medizinische Ressource’ in Form von Krankenkassenbeiträgen erhöhen. Autoren: N. Stukenbröker, A.-L. Bergmann LK Bio 13