Molekularbiologie
Werbung

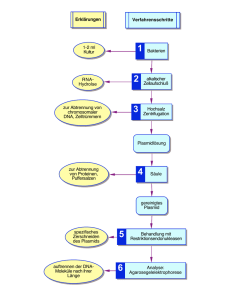
Katja Rey, Christian Menzel, Jan Grolmus 04.12.2002 Biochemisches Praktikum Kurstag: Molekularbiologie Gruppe 8 THEORIE Die Polymerase-Ketten-Reaktion („Polymerase-chain-reation =PCR“) wurde im Jahre 1984 von Kary Mullis entwickelt und ermöglicht in der modernen Genetik die exponentielle Amplifikation (= Vermehrung) von DNA außerhalb der lebenden Zelle. Ablauf der PCR: In einem Reaktionsansatz für eine PCR müssen enthalten sein: Hitzestabile DNA-Polymerase (Taq-Polymerase aus dem Bacillus thermus aquaticus) Doppelsträngige DNA-Matrize Mindestens 2 Primer aus einzelsträngiger DNA Mg2+ Desoxynucleotide Die PCR gliedert sich im einzelnen: 1. Denaturierung der doppelsträngigen DNA bei ca. 94ºC. 2. Anlagerung der Primer bei ca 50-65°C. Die Primer binden an komplementäre DNAAbschnitte und rahmen die zu amplifizierende Sequenz ein. Die geeignete Anlagerungstemperatur ( = annealing temperature) richtet sich nach dem Guanin/Cytosin-Gehalt des Primers. Je höher der G/C-Gehalt, desto höher muss die Temperatur gewählt werden. 3. DNA-Synthese bei ca 72 °C in 5’-3’- Richtung, da bei dieser Temperatur die Polymerase ihre maximale Aktivität aufweist. Bei den ersten beiden Zyklen werden zunächst nur sogenannte „lange Produkte“ hergestellt, da die Polymerase das Ende der zu amplifizierenden Sequenz von alleine nicht erkennen kann. Der Abbruch erfolgt hier durch das erneute Erhitzen des Ansatzes zu Beginn des nächsten Zyklus. Somit treten ab dem dritten Zyklus nur noch Fragmente gewünschter Länge auf. Diesen Zyklus wiederholt man in der Regel bis zu 25 Mal! Die PCR wird vor allem in der Gendiagnostik sowie in der Gerichtsmedizin eingesetzt. 1 Katja Rey, Christian Menzel, Jan Grolmus 04.12.2002 VERSUCHSDURCHFÜHRUNG I Plasmidminipräparation (DNA-Isolierung) Plasmide sind kleine ringförmige DNA-Moleküle in Bakterien mit einer begrenzten Anzahl an Genen (2000-10000 Nukleotide), die oftmals Resistenzfaktoren enthalten. Für diesen Versuch verwenden wir zwei verschiedene Plasmide: Das „normale“ Plasmid enthält das sogenannte PUC18 (β-GalactosidaseGen), was spezifisch einen Zucker-Farbstoff-Komplex spaltet und so den Farbstoff umsetzt. Außerdem enthält es ein Gen für Ampicillin-Resistenz. Das modifiziertes Plasmid enthält eine veränderte PUC18-Sequenz (Es wurden ca. 50 Basenpaare deletiert. Damit wird das Enzym funktionsunfähig und kann den Farbstoff nicht mehr umsetzen.), sowie die Ampicillin-Resistenz. Durchführung: Zentrifugation der Bakterienzellen bei Höchstgeschwindigkeit für drei Minuten in der Mikrozentrifuge. Dabei setzen sich die Bakterien am Boden des Eppendorfgefäßes ab. Der Überstand wird abgeschüttet und der Bodensatz (Bakterien-Pellet) getrocknet. Das Pellet wird mittels eines Respensionspuffers wieder vollständig verflüssigt. Der Resuspensionspuffer enthält vor allem RNAsen, die die Bakterien-RNA zerstören, so dass letztlich nur noch funktionelle DNA in der Suspension vorhanden ist. Die Aufgabe des Resuspensionspuffers besteht in der Destabilisierung der Bakterienzellen. Durch Zugabe eines Lysepuffers werden die Zellen zerstört, so dass die RNAsen nun aktiv werden können und Proteine ausfallen. Der Lysepuffer enthält hauptsächlich SDS und NaOH. Durch das alkalische Milieu entstehen Abstoßungskräfte, die dazu führen, dass die genomische DNA komplett in ihre Einzelstränge zerfällt, während sich die Basen der Plasmide zwar abstoßen, die Einzelringe aber einander angenähert bleiben. Durch Zugabe eines sauren Neutralisationspuffers wird die alkalische Lösung neutralisiert, wobei die Plasmide wieder Doppelstränge bilden. Die genomische DNA sowie die Proteine und die anderen unbrauchbaren Zelltrümmer fallen aus (milchige Trübung). Durch Zentrifugation setzten sich die ausgefallenen „Abfälle“ als Bodensatz ab, während sich die Plasmide im Überstand befinden. Der Überstand wird in eine QIAprep spin – Säule überführt, wobei die Plasmide an der Säule binden. Es folgt ein Reinigungsschritt mit einem Waschpuffer. Durch einen Eluationspuffer werden die Plasmide aus der Säule gewaschen (Stammlösung der Plasmide) und mit Aqua dest. auf 1:200 verdünnt. Diese Lösung stellt die Matrize für die PCR dar. 2 Katja Rey, Christian Menzel, Jan Grolmus 04.12.2002 II. PCR Der Reaktion werden wie zuvor beschrieben verschiedene Primer („Primermix“) zugesetzt, die an komplementären Stellen im Bereich des Gens der β- Galaktosidase ansetzen: Der Primer SE (sense) bindet „upstream“ am 5’-Ende der DNA –Sequenz. Der Primer AS 1 (antisense 1) bindet im Bereich der Deletion im PUC18. Der Primer AS 2 (antinsense 2) bindet „downstream“ am 3’-Ende der DNA-Sequenz. Im modifierten Plasmid kann sich der Primer AS 1 also nicht anlagern , da die Ansatzstelle genau im Bereich der Deletion liegt. Somit erwarten wir für die noch kommende Gelelektrophorese folgendes Bandenschema: Normales Plamsid: Zwei unterschiedlich lange Banden! Beim normalen Plasmid kann der Primer SE, AS 1 und AS 2 ansetzen, so dass zwei unterschiedlich lange Fragmente entstehen, die entsprechend ihrer Größe in der Elektrophorese wandern. Modifiziertes Plasmid: Eine lange Bande! Da lediglich die Primer SE1 und AS 2 ansetzen könne, werden sich zwei Fragmente gleicher Länge bilden, die in der Elektrophoerese auch gleich weit laufen werden Statt der separaten Zugabe der einzelnen PCR-Komponenten wurde bei der Durchführung ein sogenannter „Mastermix“ verwendet, der alle notwenigen Komponenten in geeignetem Maße enthält. Nun erfolgt die PCR in einem Zeitraum von 75 Minuten, wobei ca. 25 Zyklen durchlaufen werden. III. Restriktionsverdau Neben der PCR haben wir zur Plasmidanalyse noch einen Restriktionsverdau mit zwei verschiedenen Enzymen durchgeführt: Bam HI: Dieses Enzym besitzt nur eine Schnittstelle auf dem Plasmid und zwar genau im Bereich der Deletion im PUC18. RsaI: dieses Enzym besitzt drei Schnittstellen auf dem Plasmid, wobei die eine ebenfalls im Bereich der Deletion des PUC18 liegt. Da im modifizierten Plasmid eine Schnittstelle genau im Bereich der Deletion liegt, fehlt ihm somit eine Schnittstelle für beide Enzyme. Damit können wir für die Banden in der noch folgenden Gelelektrophorese folgendes Schema erwarten: Rsa1 BamHI Normales Plasmid 3 Banden 1 Bande Unmodifiziertes Plasmid 2 Banden 2 Banden Für den Restriktionsverdau wurden folgende Ansätze hergestellt: A) Stammlösung der Plasmide + Puffer Y + Rsa1 B) Stammlösung der Plasmide + Puffer 2 + BamHI Die Ansätze wurden bei 37°C im Heizblock zwei Stunden lang inkubiert. Anschließend wurde vom Assistenten ein 0,8%-iges Agarose-Gel angefertigt, auf dem dem die folgende Gelelekrophorese stattfindet. 3 Katja Rey, Christian Menzel, Jan Grolmus 04.12.2002 IV. Laden des Gels Das PCR-Produkt und die beiden Restriktionsansätze wurden jeweils mit einem Ladepuffer versetzt und für ca. eine Stunde bei 90V in der Gelelektrophoerese entwickelt. V. Transformation Als Transformation bezeichnet man die Übertragung fremder DNA mittels Vektoren (z.B. Plasmide) in andere Organismen. Für diesem Versuch wurden erneut die oben genannten Plasmide verwendet: „Nomales“ Plasmid enthält das sogenannte PUC18 (β-GalactosidaseGen), was spezifisch einen Zucker-Farbstoff-Komplex spaltet uns so den Farbstoff umsetzt. Außerdem enthält es ein Gen für AmpicillinResistenz Modifiziertes Plasmid enthält die veränderte PUC18-Sequenz (d.h. es wurden ca. 50 Basenpaare deletiert. Damit wird das Enzym funktionsunfähig und kann den Farbstoff nicht mehr umsetzen), sowie die Ampicillin-Resistenz. Durchführung: 1. Die kompetenten Bakterien Zellen wurden auf Eis aufgetaut. 2. Anschließend wurde das Plasmidgemisch zupipettiert und erneut in Eis inkubiert. 3. Durch Hitzeschock im Heizblock sollen die Plasmide in die Bakterien aufgenommen werden. 4. Anschließen mussten sie nochmals für kurze Zeit im Eisbad inkubiert werde. 5. Ausplattieren auf den Kulturplatten. Da die Kulturlplatten neben dem Zuckerfarbstoff-Komplex auch das Antibiotikum Ampicillin enthalten, können nur diejenigen Bakterien sich zu Kolonien entwickeln, die zuvor eines der beiden Plasmide aufgenommen haben. Diejenigen Bakterien, die das normale Plasmid aufgenommen haben, haben zusätzlich noch die Eigenschaft den Zucker-Farbstoffkomplex zu spalten und sind somit als blaue Kolonie zu erkennen. Die anderen Bakterien haben diese Eigenschaft nicht und sind auf der Kulturplatte als einfache weiße Kolonien zu sehen. 4 Katja Rey, Christian Menzel, Jan Grolmus 04.12.2002 VERSUCHSAUSWERTUNG Auswertung der PCR: Die Gelelektrophorese zeigt, dass eine deutliche Bande vorhanden ist. In diesem Fall bedeutet dies, dass es sich um das modifizierte Plasmid handeln muss. Da eine Deletion vorhanden ist, können lediglich Fragmente zwischen SE1 und AS2 amplifiziert werden. Beim unmodifizierten Plasmid kann der Primer AS1 nur an der nicht deletierten Stelle ansetzen, und so das Plasmid in zwei Fragmente schneiden, die in der Gelelektrophoerese als zwei Banden sichtbar werden. Aus dem Auswertungsdiagramm lässt sich schließen, dass die Fragmente ungefähr eine Länge von 0,6 Kb haben. Auswertung Restriktionsverdau A: Da wir das modifizierte Plasmid als Versuchsplasmid bekommen haben, sollten beim Restriktionsansatz A mit dem Enzym RSA1 zwei Banden sichtbar sein. Auf dem Auswertungsdiagramm sind zwei Banden sichtbar. Die untere Bande zeigt eine intensivere Absorption als die obere. Die Länge der beiden Fragmente betragen ungefähr 2 und 3 Kb. B: Der Verdau mit dem Enzym BamHI erbrachte zwei Banden, eine etwas intensiver obere Bande und eine weniger intensive untere Bande. Hier ist kein Schnitt erfolgt, so dass die zwei Banden aus den zwei unterschiedlichen Formen des intakten Plasmids („Cyclisch und Supercoiled“) resultieren .Die „supercoiled“-Form wird wegen ihrer kleineren Größe weiterlaufen ist und liegt daher häufiger vor, ist also höher konzentriert. Die Lange der Fragmente betragen ebenfalls 2 und 4 Kilobasen. Auswertung der Transformation Beim Auszählen der Kolonien auf der Kulturplatte fanden wir eine blaue und vier weiße Kolonien vor. Die blaue Kolonie geht auf ein Bakterium zurück, dass ein normales Plasmid ( ohne Deletion) aufgenommen haben muss und daher in Lage ist, den auf der Kulturplatte befindlichen ZuckerFarbstoff-Komplex zu spalten und so den Farbstoff umzusetzen. Die weißen Kolonien haben folglich ein Plasmid mit Deletion aufgenommen. Berechnung der Transformationseffizienz: Es wird angenommen, dass 15ng Plasmide eingesetzt wurden. Wie viele Kolonien würden sich dann in 1 μg DANN befinden? 1 μg entsprechen 1000ng. 5 Katja Rey, Christian Menzel, Jan Grolmus 04.12.2002 Für die blaue Kolonie gilt: In 15 ng fanden wir eine blaue Kolonie Folglich fänden wir in 1000ng ca. 67 Kolonien Ebenso verhält es sich mit den weißen Kolonien: In 15ng fanden wir vier weiße Kolonien. In 1000ng würden wir also ca. 268 weiße Kolonien auffinden. Aus dem Verhältnis von 67 blauen Kolonien zur Summe aller Kolonien (335) ergibt sich: 20% unmodifizierte Plasmide im Reaktionsansatz. und damit 80% modifizierte Plasmide im Reaktionsansatz. Schaubild der Gelelektrophorese: Unsere Versuchsansätze sind die vorletzten in der ersten Reihe (markiert)! 6