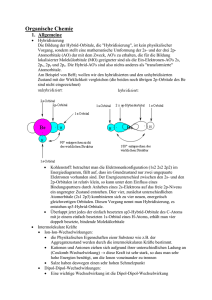
Grundlagen der Organischen Chemie (Reaktionsmechanismen)
Werbung
")
Grundlagen der Organischen Chemie (Reaktionsmechanismen) Themenübersicht A. B. C. D. E. F. G. H. I. Allgemeine Grundlagen Radikalreaktionen Nucleophile Substitutionen Eliminierungen Additionen Aromatische Substitutionen Oxidationen Carbonylreaktionen Umlagerungen Empfehlungen für begleitende Literatur 1. Autorenkollektiv 2. R. Brückner 3. F. A. Carey, R. J. Sundberg 4. J. March 5. K. P. C. Vollhardt, N. E. Schore 6. P. Sykes Organikum 21. Aufl., VCH-Wiley, Weinheim 2001 (ca 108.- DM) Reaktionsmechanismen Spektrum Akademischer Verlag, Heidelberg 1996 (ca. 80.- DM) Organische Chemie VCH-Wiley, Weinheim 1995 (ca 175.- DM) Advanced Organic Chemistry 5. Aufl., Wiley, New York 2001 (ca 170.- DM) Organische Chemie 3. Aufl., VCH-Wiley, Weinheim 2000 (ca 155.- DM) Reaktionsmechanismen der Organischen Chemie 9. überarb. Aufl.,VCH-Wiley, Weinheim Nachdruck 2001 (ca 68.- DM) 1 A. Allgemeine Grundlagen 1. Einteilung Organischer Reaktionen... 1.1. ...nach der strukturellen Änderung a) Substitution: Formal: A-B + C Konvention: A + B-C oder C-A + B Verschiebung eines Elektronenpaars in angegebener Richtung Verschiebung eines Elektrons in angegebener Richtung b) Eliminierung: Formal: X-A-B-Y A=B + X-Y c) Fragmentierung: Formal: A-B-C-D-E A=B + C=D + E A=B + X-Y X-A-B-Y d) Addition: Formal: e) Umlagerungen: Formal: A-B-C B-C-A 2 1.2. ...nach der Art der Reagentien: a) Nucleophile Reagentien: -sind Teilchen (neutrale Atome, Moleküle, Ionen), die über energetisch hochliegende, doppelt besetzte, nicht bindende ("einsame") Elektronenpaare ("lone pairs") oder energetisch hochliegende -Bindungen verfügen. b) Elektrophile Reagentien: -sind Teilchen (neutrale Atome, Nokelüle, Ionen), die energetisch tiefliegende, unbesetzte Molekülorbitale verfügen. c) Oxidationsmittel (Reduktionsmittel) -Oxidation bedeutet Entzug von Elektronen und ist stets mit einer gleichzeitigen Reduktion (Elektronenaufnahme) gekoppelt. Oxidationsmittel sind Elektronenakzeptoren Reduktionsmittel sind Elektronendonoren. 1.3. ...nach der Art der Bindungsspaltung: a) Homolytische Spaltung: Formal: A-B A + B -Bindungen werden gleichsinnig gespalten, wobei aus neutralen Edukten zwei neutrale Radikal-Bruchstücke entstehen. b) Heterolytische Spaltung: Formal: A-B A+ + B- -Bindungen werden ungleichsinnig Bindungselektronen erhält. gespalten, wobei ein Bruchstück beide 3 1.4. ...nach der Selektivität: a) Regioselektivität (auch Chemoselektivität): Bsp.: Regioisomere Frage: Welche von zwei ähnlichen Positionen wird bevorzugt angegriffen? b) Diastereoselektivität: Bsp.: Isoborneol Borneol 89% 11% Diastereomere Frage: Welches von zwei möglichen Diastereomeren wird bevorzugt gebildet? c) Enantioselektivität: Bsp.: Frage: Welches von zwei Enantiomeren wird bevorzugt gebildet? 84% : 16% ~ 68% ee ee = [S]-[R] / [S]+[R] 4 d) Stereospezifität: Eine Reaktion ist stereospezifisch, wenn dabei Konfigurationsisomere A, A' in definierte unterschiedliche Stereoisomere B,B' umgewandelt werden. Bsp.: E-Stilben wird zur anti-Verbindung dibromiert. Diese ist eine achirale meso-Form. Z-Stilben dagegen reagiert mit Brom zum Racemat der chiralen R,R- und S,S-Komponenten. 2. Orbitale: 2.1. Atomorbitale: Nach dem Pauli-Prinzip und der Hund’schen Regel besetzt man Atomorbitale von unten nach oben mit jeweils zwei gepaarten Elektronen. Dabei liegt die Energie der Orbitale in der Reihenfolge (1s)<(2s)<(2p)<(3s)<(3p) etc. Entartete Orbitale werden zunächst einfach besetzt. Die Orbitalenergien entsprechen den Ionisierungsenergien in eV (= zur vollständigen Ablösung aus dem Elektronenverband nötige Energie): Je höher die Elektronegativität des betreffenden Elements, desto niedriger liegen die Orbitalenergien der vergleichbaren Elektronen, denn der Atomkern kann in zunehmendem Maße die Elektronen zu sich hinziehen: 5 Wichtige Elektronegativitäten stehen in jedem Periodensystem: H 2.1 B 2.0 C 2.5 Si 1.8 N 3.0 P 2.1 O 3.5 S 2.5 F Cl Br I 4.0 3.0 2.8 2.4 LCAO Methode: Bei der Annäherung zweier Atomorbitale berühren sich die Elektronenhüllen und ziehen sich je nach Orbitalvorzeichen an oder stoßen sich ab. Dabei entstehen zwei neue Orbitale, eins mit höherer und eins mit niedrigerer Energie, die in der neuen Verbindung wieder nach dem Pauli-Prinzip und der Hund’schen Regel nacheinander aufgefüllt werden. Die Energie der beiden neuen Orbitale kann durch Linearkombination aus den beiden alten Orbitalen berechnet werden. Waren die beiden AO’s einfach besetzt, so wird in der Verbindung nur das bindende, energetisch abgesenkte Orbital besetzt, und Energie wird gewonnen. Waren dagegen beide Orbitale besetzt (Edelgase), so verliert das System Energie, denn die Energieanhebung ist größer als die Absenkung. Das kann man gut am Beispiel der Verbindungen Wasserstoff und Helium sehen: Kompliziertere Systeme funktionieren nach dem gleichen Prinzip. Beim Kohlenstoff gibt es jedoch 3 entartete p-Orbitale. Würden diese zur Bindung benutzt, so könnten Alkane nur 3 Bindungen in einem rechtem Winkel zueinander bilden. Dem weicht der Kohlenstoff durch Hybridisierung aus: Je nach Substituentenzahl mischen sich ein s- und ein p-Orbital zu zwei neuen sp-Hybrid-AO’s und der Kohlenstoff wird zweibindig. Die restlichen p-Orbitale überlappen zu 2 -Bindungen (Alkine). Ein s- und zwei p-Orbitale ergeben drei neue sp2Hybrid-AO’s, die den Kohlenstoff dreibindig machen, bei einer -Bindung (Alkene). Wenn dagegen ein s- mit allen drei p-Orbitalen hybridisiert, entstehen vier neue, gleichwertige sp3Hybrid-AO’s, die den Kohlenstoff vierbindig machen, mit einem Bindungswinkel von ~109°. 6 Die p-Orbitale und ihre Hybride mit s-Orbitalen haben alle zwei Orbitallappen in einer Dimension mit unterschiedlichem Vorzeichen, von denen der rückseitige bei spn-Hybiden jedoch sehr kurz ist. Auch der vorderseitige Orbitallappen wird bei spn-Hybriden mit steigendem s-Charakter immer kürzer. MO-Theorie: Bei der MO-Bildung aus unterschiedlichen Atomorbitalen führt nach der Störungsrechnung die Wechselwirkung zwischen Orbitalen ähnlicher Energie zur größten Energieabsenkung (und Erhöhung). Deshalb bilden solche Orbitale oft die stärksten kovalenten Bindungen. Beispiel: Die C-Cl-Bindung ist allerdings schwächer als die C-F-Bindung, obwohl hier die Energieabsenkung größer ist! Das liegt an dem viel höheren Ausgangsniveau des 3p-Orbitals. So liegt von(C-Cl) > von(C-F) und von(C-Cl) > von(C-F): Die unterschiedliche Stärke der C-X-Bindungen steht also nur in scheinbarem Widerspruch zur unterschiedlich starken Orbitalaufspaltung bei der MO-Bildung, denn trotz größerer Energieabsenkung startet das Cl-Atom von einem viel höheren Niveau als das F-Atom, das auch die größerer Energie-Absenkung nicht einholen kann: Die antibindenden Orbitallappen haben unterschiedliches Vorzeichen. Die bindenden haben gleiches Vorzeichen. Nach der Verschmelzung zieht F aus der Bindung viel Elektronendichte zu sich hinüber, so daß die Orbitallappen am F viel größer werden als die am C. Das ist bei Cl viel weniger der Fall. Dadurch ist die C-Cl-Bindung schwächer polarisiert. 7 Wir werden bei den unterschiedlichen Mechanismen sehr oft die Wechselwirkung zwischen zwei teilweise besetzten Atom- oder Molekülorbitalen behandeln. Dabei handelt es sich meist um das HOMO (Highest occupied MO) des einen Moleküls und das LUMO (Lowest unoccupied MO) des anderen Moleküls, in das die Elektronendichte hineinfließt. Diese HOMO-LUMO-Wechselwirkung beherrscht die attraktive Wechselwirkung zweier Reaktionspartner und erklärt viele experimentelle Befunde hervorragend. Wir werden sie immer wieder verwenden. 3. Stereochemie: Verbindungen mit vier verschiedenen Substituenten an einem tetraedrischen Atom tragen dort ein Chiralitätszentrum oder auch stereogenes Zentrum. Das kann zu Chiralität (Händigkeit, das Auftreten von nicht zur Deckung zu bringendem Bild und Spiegelbild) führen. Bei Zuckern treten mehrere solcher Zentren auf. Trotzdem hat es sich eingebürgert, hier nur die Konfiguration des untersten Chiralitätszentrums zu benennen. Die anderen, relativ dazu auftretenden Konfigurationen bezeichnen die Art des Zuckers. Man zeichnet den Zucker in der Fischerprojektion (Regeln?) und gibt der Verbindung, deren Substituent am untersten Chiralitätszentrum links steht, das Präfix L-, der anderen Form das Präfix D-: Um die absolute Konfiguration jedes Chiralitätszentrums zu benennen, hat sich die CIPNomenklatur eingebürgert (Cahn-Ingold-Prelog): Hier ordnet man die Substituenten am Chiralitätszentrum nach fallender Priorität, bringt den Substituenten niedrigster Priorität nach hinten, und verfolgt den Drehsinn, wenn man vom Substituenten 1 über 2 nach 3 geht. Uhrzeigersinn = R-, dagegen S-: 8 Wenn zwei Verbindungen Enantiomere sind, müssen sich alle Absolutkonfigurationen unterscheiden, und zwar an allen Chiralitätszentren. Solche Verbindungen können nur durch optisch aktive Medien unterschieden werden. Unterscheiden sich dagegen nur einige Absolutkonfigurationen, so handelt es sich bei den beiden Verbindungen um Diastereomere, die physikalisch unterscheidbar sind, z.B. durch Siede- oder Festpunkt, chromatographisch (Polarität) etc. Das macht sich auch im Energiediagramm bemerkbar: Enantiomere haben denselben Energieinhalt, Diastereomere dagegen nicht. Deshalb entstehen in nicht-stereoselektiven Reaktionen immer Racemate. Weitere Arten der Chiralität: a) Axiale Chiralität: Sie tritt vor allem bei Biarylen auf (Atropisomerie) und auch bei Allenen (beachten Sie die senkrecht aufeinander stehenden C=C-Doppelbindungs-MO‘s!) b) Planare Chiralität: Sie tritt vor allem bei Cyclophanen und Ansa-(Henkel-)Verbindungen auf. c) Helicale Chiralität: Sie tritt bei Helices auf; das können künstliche sein (z.B. Helicen), oder natürliche (z.B. DNA, -Helix der Proteine etc.). 9 Doppelbindungen: Hier unterscheidet man zwei Diastereomere, die die Substituenten auf derselben oder auf gegenüberliegenden Seiten der Doppelbindung tragen: Nomenklatur: bei 2 Substituenten nennt man die Diastereomere cis und trans, bei drei Substituenten nach der jeweils höchsten Priorität an jedem Ende der Doppelbindung Z- oder E-: 4. Energieänderungen chemischer Reaktionen. Allgemeines Energieschema: A k B Hier werden zwei Größen sichtbar: a) die Kinetik einer Reaktion gibt ihre Reaktionsgeschwindigkeit wieder und steckt in der Aktivierungsenergie G: 10 v = d[B]/dt = k[A] = -d[A]/dt Sie wird durch die Eyring-Gleichung beschrieben: k = hBT / h e - G/RT = kBT / h e S/RT e -H/RT mit kB = Boltzmann-Konstante; h = Planck'sches Wirkungsquantum. Die Aktivierungsenthalpie spiegelt die Stabilität des ÜZs, die Aktivierungsentropie den Ordnungsgrad des ÜZs wieder. Allgemeiner formuliert es Arrhenius: so erhält man die Gesamtaktivierungsenergie EA (~G) bei einer bestimmten T: k = A e -EA/RT; ln k = lnA - EA / RT Der Graph zeigt den Arrhenius-Plot, also die Auftragung von 1/T gegen lnA. Die Steigung der Geraden liefert EA/R. b) die Thermodynamik einer Reaktion (G°): Die Thermodynamik einer Reaktion gibt die Lage des Gleichgewichtes wieder, wird durch die Gleichgewichtskonstante ausgedrückt und spiegelt sich in der unterschiedlichen thermodynamischen Stabilität der Reaktionsprodukte wieder. Ka = e -G°/RT Thermodynamische und kinetische Produktkontrolle: Ein Edukt oder Zwischenprodukt E geht formal auf zwei Wegen in zwei unterschiedliche Produkte über; der eine Weg hat eine hohe Aktivierungsenergie, führt aber zum thermodynamisch stabilsten Produkt. Der andere hat eine geringe Aktivierungsenergie, führt aber zum weniger stabilen Produkt. P1 k1 E k2 P2 Energiediagramm: 11 Wird das Produktverhältnis durch das Verhältnis der freien Aktivierungsenthalpien G bestimmt, spricht man von kinetischer Kontrolle der Reaktion. Es gewinnt diejenige Reaktion, bei der die Kugel über den niedrigeren Berg rollen muß. Praktisch bedeutet das meistens, daß die Reaktion irreversibel und bei niedriger Temperatur verlaufen muß. Bsp.: Selektivitätbei 25°C in Abhängigkeit von G: G [kcal/mol] [P2] : [P1] 0 1:1 1.4 10 : 1 2.8 100 : 1 4.2 1000 : 1 Wird das Produktverhältnis nur durch das Verhältnis der Produktstabilitäten bestimmt, som spricht man von thermodynamischer Kontrolle. Es gewinnt diejenige Reaktion, bei der das stabilere Endprodukt gebildet wird. Praktisch bedeutet dies, daß man die Reaktionen reversibel und bei hohen Temperaturen führen muß. Bsp: Also gilt: [P2] / [P1] = Ka = e -G° / RT -----------------------------------------Die in der Einführung behandelten grundlegenden Themen Orbitale, Stereochemie und Enegetik werden fast bei jeder Klasse von Reaktionsmechanismen wieder auftauchen; wir werden sie dabei vertiefen und üben. 12 B. Radikalreaktionen 1. Allgemeines: Definition: Radikale sind koordinativ ungesättigte Atome und Moleküle, die über ein ungepaartes Elektron verfügen. Erzeugung von Radikalen: 1.1. Homolytische Bindungsspaltung: A• + B• A-B Eine wichtige Kenngröße ist die Bindungsdissoziationsenergie (BDE) angegeben in kcal/mol. Vorläufer HO-H RO-H H3C-H (H3C)3C-H H2C=CH-CH2-H H3CS-H Bu3Sn-H BDE [kcal/mol] 119 >108 104 92 89 88 74 Vorläufer Et-Br Et-I Me3Pb-Me EtHg-Et Me3CO-OCMe3 Me2C(CN)-N=N-(CN)CMe2 (= AIBN) BDE [kcal/mol] 69 53 49 43 37 32 a) Thermolyse: Dibenzoylperoxid zerfällt in zwei Radikale, die jeweils CO2 abspalten und in das Phenylradikal übergehen. BDE: 30 kcal/mol; t1/2 bei 90°C: 1h. AIBN ('-Azoisobutyronitril) zerfällt in zwei Radikale und Stickstoff. (t1/2 bei 80°C: 1h). b) Photolyse: E = h (Planck'sche Gleichung). Das bedeutet, daß Licht einer bestimmten Wellenlänge eine bestimmte Energie besitzt. Bsp. Bei 300 nm (UV) beträgt die Lichtenergie 96 kcal/mol. Nur absorbierte Strahlung ist aktiv; also benötigt man im Molekül einen Chromophor, der die Lichtenergie aufnimmt. Cl-Cl + h Ph-C(=O)-CH2-Ph + h v 2 Cl• Ph-C(=O) • + •H2C-Ph (Norrish-Typ-1-Spaltung) 13 c) Erzeugung von Radikalen durch Redoxprozesse: Reduktion mit Fe2+ oder Elektronen (SmI2) Analog zu H2O2 können auch t-BuOOH oder Benzoylperoxid so radikalisiert werden. Anodische Oxidation (Kolbe- Reaktion): Carboxylate können anodisch leicht zu Carboxylradikalen oxidiert werden, die anschließend Radikalreaktionen eingehen können. Die Kolbe-Reaktion führt z. B. unter Dimerisierung zu größeren Alkanen. 2. Struktur von Radikalen: Alkylradikale sind im allgemeinen planar, denn die 3 Alkylreste stehen möglichst weit voneinander entfernt. Allerdings ist der Energieaufwand für die Umhybridisieerung von sp2 zu sp3 (Pyramidalisierung) gering (~1-2 kcal/mol). Ausnahme: aus sterischen Gründen sind Radikale in gespannten Ringsystemen pyramidal (d.h. sp3-hybridisiert). Vinylradikale sind meist nicht linear (sp), sondern tatsächlich gewinkelt (sp2), haben aber ebenfalls niedrige Inversionsbarrieren. 14 Ausnahme sind hier sterisch anspruchsvolle trisubstituierte Radikale wie die folgenden: 3. Stabilität von Radikalen: 3.1. Thermodynamische Stabilität: Allgemein hängt die thermodynamische Stabilität von Radikalen von der BDE ihrer Vorläuferalkane R-H ab. Diese korreliert mit dem Substitutionsgrad des entstehenden Radikals: Tertiäre Radikale sind durch multiple Hyperkonjugation stabiler als primäre: (CH3)3C• 92 > (CH3)2CH• 95 > CH3-CH2• 98 > CH3• 105 BDE [kcal/mol] a) So sieht die Hyperkonjugation aus: es ist eine Wechselwirkung zwischen dem einfach besetzten p-Orbital und der übernächsten -Bindung. Energiediagramm: Wichtig: pz- und C-H müssen zur optimalen Überlappung in einer Ebene stehen. Die Wechselwirkung zwischen p- und *-Orbital ist dagegen vernachlässigbar. b) Konjugation mit -Systemen: i) Alkene: Sitzt das einfach besetzte p-Orbital direkt neben einem Alken, so entsteht ein mesomeriestabilisiertes Allylradikal. Die Stabilisierung wird im MO-Bild deutlich, wenn das einfach besetzte p-Orbital (SOMO) mit dem doppelt besetzten -Orbital des Alkens (HOMO) und auch mit dem antibindenden -Orbital des Alkens (LUMO) wechselwirkt. Insgesamt ergeben sich durch die Wechselwirkung von p, und * drei neue Molekülorbitale 1-3. 15 Analog stabilisiert auch ein benachbarter Aromat sehr gut das Radikal (Benzylradikal). ii) Carbonylgruppen: Hier liegt das *-Orbital (LUMO) der Carbonyle in der Nähe des einfach besetzten p-Orbitals (SOMO) und führt zur Stabilisierung. iii) Hyperkonjugation mit den lone pairs eines Donoratoms: Auch hier ist eine Stabilisierung zu beobachten. Das lone pair-HOMO wechselwirkt mit dem einfach besetzten p-Orbital (SOMO). c) Einfluß der Hybridisierung auf die Radikalstabilität: Je höher der Anteil an s-Orbitalcharakter, desto stärker ist eine CH-Bindung. Deshalb ist ein Acetylenradikal am schwersten herzustellen; es tritt bei SR-Reaktionen nicht auf. 3.2. Kinetische Stabilität Die Lebensdauer von Radikalen wird aber auch von der Geschwindigkeit ihrer Rekombination bestimmt; bei sterisch sehr anspruchsvollen Radikalen ist die gegenseitige Annäherung dadurch behindert und das Radikal ist langlebig, und trotzdem sehr reaktiv gegenüber kleineren Molekülen (Persistenz). 16 4. Reaktivität von Radikalen: Radikale rekombinieren im allgemeinen sehr schnell ( k ~1010 s-1). Langsamer ist die Disproportionierung, weil dafür Bindungen gebrochen werden müssen. Cyclisierungsreaktionen verlaufen meist nach 5-exo. Auch intermolekular können Radikale saubere Reaktionen eingehen, z.B. durch Addition an DoBis. Fragmentierungen führen zu kleineren reaktiven Radikalen. Angriff an schwachen Atombindungen schließlich kann zu Atomtransfer-Reaktionen führen. 5. Radikalische Substitution: Der Ersatz eines Substituenten am gesättigten C-Atom durch einen anderen (oft ein Halogen-, aber auch ein Wasserstoffatom) kann im Sinne einer radikalischen Substitution (S R) verlaufen. R-X R-Y a) Zinnhydrid-Reduktion: Halogenide, Selenide, Sulfide und Xanthogenate können milde zu den entsprechenden Kohlenwasserstoffen reduziert werden, wenn das Reagenz Tributylzinnhydrid eingesetzt wird: R-X + Bu3SnH (AIBN) R-H Mit etwa 10% AIBN als Radikalstarter wird zunächst das Tributylzinnhydrid radikalisiert und greift in der Kettenfortpflanzung das Alkylhalogenid an. Das daraus entsehende Radikal abstrahiert nun von weiterem Bu3SnH ein H-Atom und generiert damit weiteres TributylzinnRadikal für die Kette. Abbruchreaktionen beenden wie üblich die Kettenreaktion. 17 b) Barton-McCombie-Deoxygenierung: Alkohole können nach Überführung in Xanthogenate ebenfalls mit Tributylzinnhydrid zu den zugrundeliegenden Kohlenwasserstoffen reduziert werden: Mechanismus: Hier greift das Bu3Sn-Radikal den Schwefel an und führt zu einem O,S-stabilisierten S,OAcetalradikal, welches anschließend fragmentiert und das Alkylradikal freisetzt, welches unter Kettenerhalt Bu3SnH angreift. Als Ersatz für das giftige Zinnhydrid kann auch Tris(trimethylsilyl)silan verwendet werden, welches eine ähnliche BDE besitzt. c) Radikalische Decarboxylierung: Carbonsäuren kann man nach Überführung in Thiohydroxamsäure-Ester (Barton-Ester) ebenfalls mit Bu3SnH zum KW reduzieren. R-COOH R-H 18 Auch hier wird ein mesomeriestabilisiertes S,N-Acetalradikal gebildet. Mechanismus: d) Hunsdiecker-Abbau: Trockene Silbersalze von Carbonsäuren lassen sich in Hypobromite überführen, welche thermisch oder photochemisch zunächst Bromradikale und dann CO2 abspalten, um in die freien Alkylradikale überzugehen, welche weiteres Esterhypobromit angreifen. e) NBS-Bromierung: Alkene werden mit NBS nicht dibromiert, sondern im Sinne einer radikalischen Substitution in Allylstellung monobromiert. Der Mechanismus verläuft ohne Bestrahlung über die Freisetzung von geringen Mengen an Brom aus dem NBS, welches durch AIBN in Radikale zerfällt. Das Bromradikal abstrahiert das allylständige H-Atom, und das entstehende Allylradikal radikalisiert seinerseits weiteres Brom, wobei das Produkt und weiteres Bromradikal gebildet wird, welches die Kette weiterführt. Genauso können Benzylstellungen selektiv angegriffen werden. Dabei gilt die Regel: SSS (Siedehitze, Sonne, Seitenkette) / KKK (Kälte, Katalysator, Kern)! 19 Chlorierungen vcrlaufen viel weniger selektiv als Bromierungen. Am besten geht es mit Sulfurylchlorid, denn hier entsteht kein freies Chlor: Starterradikal + SO2Cl2 SO2Cl• + R-H R• + SO2Cl2 Starter-Cl + SO2Cl• R• + HCl + SO2 R-Cl + SO2Cl• (da capo) f) Chlorierung von Alkanen mit Cl2: H3C-H + Cl2 (h ) H3C-Cl + HCl Mechanismus wie üblich: Die Reaktionsenthalpie einer solchen Reaktion kann leicht aus den Bindungsenthalpien berechnet werden: Aufwand C-H Cl-Cl Summe kcal/mol 104 58 162 Ertrag H-Cl C-Cl Summe kal/mol -103 -84 -187 Netto -25 Die Reaktion ist also mit-25 kcal/mol exotherm. Noch viel reaktiver ist Fluor, während Jod gar keine Wärme mehr freisetzt (endotherm): kcal/mol CH4 + X• • CH3 + X2 Summe • CH3 + HX XCH3 + X• X = F2 X = Cl2 -32 +1 -26 -104 -25 X = Br2 +17 -24 -7 X = I2 +33 -20 +13 Selektivität: Die Selektivität ist meistens der Reaktivität entgegengestzt, d.h. bei milden Reagentien findet man meist die höchste Selektivität. Bsp. 1: R-H + Cl2 + h R-Cl Mit Isopentan ergeben sich drei Angriffsmöglichkeiten des Chlorradikals, an primären, sekundären und tertiären C-Atomen. Die jeweiligen Geschwindigkeitskonstanten liegen wie 20 folgt: kprimär = 1; ksekundär = 4; ktertiär = 7. Das korreliert mit der Stabilität der intermediären Alkylradikale. Bell-Evans-Polanyi haben diesen Zusammenhang als Prinzip festgehalten: EA = f(HR) = a HR + b Anders ausgedrückt: je zunehmend negativer oder Reaktionsenthalpie, desto niedriger die Aktivierungsenthalpie. Bsp. 2: R-H + Br2 + h abnehmend positiver die R-Br Hier ergibt sich für die jeweiligen Geschwindigkeitskonstanten folgendes: kprimär = 1; ksekundär = 80; ktertiär = 1600. Hier ist die H-Abstraktion ein endothermer Prozeß und die Selektivität viel höher! Das liegt daran, daß hier der Übergangszustand spät auf der Reaktionskoordinate kommt. Damit hängt das Hammond-Postulat zusammen: Der ÜZ einer exothermen Reaktion liegt früh auf der Reaktionskoordinate, und ist in Energie und Struktur den Edukten ähnlich. Der Übergangszustand einer endothermen Reaktion hingegen liegt spät auf der Reaktionskoordinate und ist damit in Struktur und Energie den Produkten ähnlich. Das Hammond-Postulat hat Konsequenzen für die Selektivität kinetisch kontrollierter Reaktionen: Wenn also das Verhältnis der Produkte zweier irreversibler Reaktionen ausschließlich durch die jeweiligen Reaktionsgschwindigkeiten bestimmt wird, gewinnt naturgemäß diejenige Reaktion, deren Übergangszustand am niedrigsten liegt. Das Hammond-Postulat läßt nun das Produktverhältnis vorhersagen: Bei einer einstufigen 21 endothermen Reaktion liegt der ÜZ nahe bei den Produkten; es entsteht also rascher das thermodynamisch stabilere Produkt. Ist die Reaktion zweistufig, so liegt der ÜZ des geschwindigkeitsbestimmenden ersten Schritts näher an der reaktiven Zwischenstufe; es gewinnt also die Reaktion mit dem stabileren ZP. a) Einstufige endotherme irreversible Reaktion: b) Zweistufige irreversible Reaktion: Je weniger endotherm eine Reaktion ist, desto weniger stark macht sich diese Selektivität bemerkbar. Allgemein gilt also: Je höher die Reaktivität, desto geringer die Selektivität eines Reagenzes und umgekehrt! Nachtrag zu Regio- und Chemoselektivität: Die Reaktion von NBS mit Toluol liefert nur das Seitenkettenbromierte, nicht das Kernbromierte Produkt. Den bevorzugten Angriff an einer definierten Stelle des Substrats nennt man Regioselektivität: Die Reaktion von NBS mit Propen verläuft nur unter Substitution durch Brom, nicht unter Bromaddition. Umsetzungen, bei denen das Reagenz im Substrat nur eine von mehreren Transformationen bewirkt, nennt man chemoselektiv: 22 Weitere radikalische Substitutionen: g) Autoxidation von aktivierten Alkanen: Cumol kann mit Luftsauerstoff zu Cumolhydroperoxid aufoxidiert werden, welches anschließend zu Phenol und Aceton zerfällt: So ähnlich stellt man sich die Bildung von etherischen Peroxiden vor, die hochexplosiv sind! h) Hydrodeaminierung: Aus primären Aminen kann man über die entsprechenden Isonitrile mit Tributylzinnhydrid die freien KWe machen: Das Tributylzinn-Radikal greift an der N=C-Bindung an und setzt das Alkylradikal frei, welches wie üblich weiteres Bu3SnH angreift und die Kette fortsetzt. Nachtrag: Reaktion von FCKW's mit Ozon - Wie entsteht das Ozonloch? 23 Nachtrag: Polare Einflüsse - nucleophile und elektrophile Radikale Ist die C-H-Bindung von elektronenziehenden oder elektronenschiebenden Substituenten flankiert, so entstehen elektrophile oder nucleophile Radikale, auch wenn sie keine Ladung tragen. Das SOMO wird durch den unterschiedlichen Einfluß der Nachbargruppen entweder angehoben (nucleophile Radikale) oder abgesenkt (elektrophile Radikale). Dadurch reagieren nucleophile Radikale bevorzugt mit elektronenarmen Alkenen, bei denen sie mit dem vergleichsweise tiefliegenden LUMO überlappen können. Umgekehrt reagieren elektrophile Radikale bevorzugt mit elektronenreichen Alkenen, bei denen sie mit dem vergleichsweise hohen HOMO wechselwirken können: So reagieren unsubstituierte Alkylreadikale oder -Alkoxyradikale gern mit Enonen, Acrylaten etc., während Malonesterradikale gern mit Enolethern und Enaminen reagieren: 24 11 Radikalische Additionen 11.1 Cyclisierungen Terminale 1-Bromalkene liefern, wie schon besprochen, mit Tributylzinnhydrid bevorzugt das intramolekulare Cyclisierungsprodukt der 5-exo-Addition (98%) und nur in sehr geringem Maße das Produkt des 6-endo-Angriffs (Baldwin-Regeln: das angreifende Radikal vermag aus stereoelektronischen Gründen mit dem - oder *-MO nur ungenügend zu überlappen, der ÜZ liegt relativ hoch). Daher verläuft der 5-exo Angriff etwa 100-mal schneller als der 6endo-Angriff; die 5-exo-Cyclisierung ist damit die häufigste Reaktion in der Radikalchemie! Beispiele umfassen die intramolekulare Cyclisierung durch den Angriff von in situ erzeugten Arylradikalen (a), Acylradikalen (b) oder auch Alkylradikalen (c): a) b) c) 25 Hier erfolgt zunächst eine Fragmentierung des Barton-Esters, wonach das erzeugte Alkylradikal intramolekular mit der Allylgruppe nach 5-exo cyclisiert. Abschließend wird das stöchiometrisch eingesetzte Reagenz erneut radikalisch angegriffen und so verbleibt das Thiopyridin im Produkt. 3-exo-Cyclisierung: Diese Reaktion ist 10000-mal langsamer als ihre Rückreaktion, bei der natürlich die gesamte Ringspannung frei wird, und findet deshalb gar nicht erst statt. 4-exo-Cyclisierung: Hier ist die Rückreaktion immerhin noch 100-mal schneller und verhindert damit die Bildung eines Vierrings. 6-exo-Cyclisierung: Diese Reaktion geht richtig gut! 26 11.2 Intermolekulare Additionen Unter bestimmten Bedingungen können Radikale auch bevorzugt intermolekular mit Alkenen reagieren, ohne daß dabei Polymerisationen auftreten: Hier liegt das Geheimnis in dem oben diskutierten Wechsel zwischen elektronenarmem angreifenden Radikal und dem bei der Reaktion generierten eletronenreichen Radikalintermediat bzw. umgekehrt. Es gehört also jeweils das Paar von nucleophilem Radikal und elektronenarmer Doppelbindung zusammen sowie das Paar von elektrophilem Radikal und elektronenreichem Alken. Nucleophile Radikale sind Alkyl-, Alkoxy- und Alkylaminosubstituierte C-Radikale. Elektrophile Radikale sind solche mit Elektronenakzeptoren am Radikal-C, wie z. B. Acylradikale. So kann man also mit Tributylzinnhydrid eine gezielte intermolekulare Addition eines Alkylradikals aus einem Alkylhalogenid und einem im Überschuß eingesetzten elektrophilen Alken durchführen. Zur Erzeugung einer niedrigen stationären Zinnhydrid-Konzentration, die wiederum eine niedrige stationäre Radikalkonzentration garantiert, wird eine Spritzenpumpe eingesetzt. Dabei entsteht in guten Ausbeuten das Produkt der gerichteteten C-C-Knüpfung. 27 Interessanterweise bestimmt der Elektronenreichtum aller dabei gebildeten Radikale ihre chemoselektive Reaktion mit genau den richtigen Reagenzien zum richtigen Produkt. Links ist jeweils der hervorragend reagierende, passende Reaktionspartner abgebildet, der die Reaktion vorantreibt, während rechts der unpassende Reaktionspartner steht, der nicht reagiert. Als Beispiel dient die Reaktion von t-Butylbromid mit Acrylnitril oder auch die Reaktion eines Zucker-Selenids mit einem Acrylsäureester. 28 Auch elektronenarme Radikale können so mit nucleophilen Alkenen wie Enolethern intermolekular gerichtet zur Reaktion gebracht werden. 11.3 Radikalische Polymerisationen Wenn das angegriffene Alken im Überschuß vorliegt und das Startradikal nur katalytisch angeboten wird, erfolgt eine Weiterreaktion des imtermediär gebildeten Radikals mit weiterem Alken, vorausgesetzt, die Molekülorbitale liegen günstig. So greift der Initiator (z.B. AIBN oder Peroxid) das Alken in einer intermolekularen Addition an, und das gebildete Intermediärrradikal addiert sofort an ein weiteres Monomer. Wenn keine Übertragungsreagenzien vorhanden sind, läuft die Kette weiter, bis das gesamte Monomer verbraucht ist oder realistischer, eine Dimerisierung oder Disproportionierung stattfindet. 29 Die wichtigsten Polymere im Überblick: Polymer Polyethylen (low density) Monomer Bedingungen Peroxide / O2, hoher Druck, 150-300°C PVC (Polyvinylchlorid) Peroxide Polystyrol Peroxide oder O2 Fluorierte Polymere (Teflon) hoher Druck Polymethacrylate (Plexiglas) AIBN Verwendet man mehrere Monomer-Bausteine, so sind nennt man die zugesetzten Bausteine Comonomere. Sie können die Eigenschaften der Polymere gezielt und stufenlos variieren. Am wichtigsten sind für die Industrie die billigen Comonomere Styrol und Acrylnitril. Im Sinne des oben diskutierten Elektronenbedarfs kann man bei zwei Monomeren, von denen jeweils das eine elektronenreich und das andere elektronenarm ist, eine streng alternierende Copolymerisation erzwingen. Die dabei gebildeten Polymere haben einen sehr regelmäßigen Aufbau und kristalliseren meist gut. Dabei entstehen hervorragende Eigenschaften wie hohe Zugfestigkeit bei geringem Gewicht etc. Solche Polymere bezeichnet man als Hochleistungspolymere. 30 C. Nucleophile Substitutionen 1. Allgemeines: Nu- + R-X Nu-R + X- Nu = Nucleophil R-X = Elektrophil X = Abgangsgruppe (Nucleofug) Nucleophile und Abgangsgruppen können neutral oder geladen sein. Nucleophile: Cl-, Br-, I-, OH-, OR-, SR-, CN-, H2O, ROH, NH3 etc. Abgangsgruppen: Cl-, Br-, I-, OSO3H-, O-SO3R-, OSO2Ph-, OH2, HOR, NR3, N2 etc. Mechanistische Möglichkeiten: a) SN1-Mechanismus: Unimolekulare Reaktion (RG = Reaktionsgeschwindigkeit hängt nur von cSubstrat ab). b) SN2-Mechanismus: Bimolekulare Reaktion (RG hängt von cSubstrat und cNu ab). 2. SN1-Mechanismus: Bindungsbruch vor Bindungsbildung = dissoziativ! A B (Carbeniumion) C Energiediagramm: 31 Erster Schritt ist geschwindigkeitsbestimmend (höchste EA); zweiter Schritt produktbestimmend (das Carbeniumion kann auch umlagern oder H+ eliminieren!). ist SN1 heißt: Geschwindigkeitsgesetz erster Ordnung! -d[R3C-X]/dt = k [R3C-X] Da das Nucleophil Nu- nicht in den geschwindigkeitsbesitimmenden Schritt eingreift, führt eine Erhöhung seiner Konzentration zu keiner Steigerung der RG. 2.1. Wie sehen die Carbeniumionen aus? - planar (außer an Brückenköpfen), d.h. sp2hybridisiertes C-Atom (besonders großer Abstand aller Substituenten): Den Nachweis führten vor allem G. Olah et al. durch NMR-spektroskopische Untersuchungen (Nobelpreis 1994): Angew. Chem. 1973, 85, 183; Angew. Chem. 1995, 107, 1519. 2.2. Was stabilisiert ein Carbeniumion? (das hat auch Einfluß auf die RG) a) Mesomerie mit lone-pairs von benachbarten Heteroatomen: X = OR, NR2 b) Mesomerie mit benachbarten Doppelbindungen = DoBi's: Allylkation, vgl. auch Benzylkation c) Hyperkonjugation: Bindende Wechselwirkung (WW) zwischen leerem p-Orbital und benachbarter -Bindung 32 2.3. Welche Faktoren beeinflussen die SN1-Reaktivität? a) Bell-Evans-Polanyi: Je stabiler das intermediäre Carbeniumion, desto niedriger liegt auch der ÜZ = Übergangszustand des geschwindigkeitsbestimmenden Schritts, desto schneller läuft auch die SN1-Reaktion ab. Tertiäre Substrate reagieren viel schneller als sekundäre, weil die entsprechenden Carbeniumionen durch Hyperkonjugation besser stabilisiert sind. Primäre Substrate reagieren nicht nach SN1. Aber: Tertiäre Carbeniumionen werden andererseits etwas schlechter durch sehr polare Lösungsmittel solvatisiert; das dämpft die Reaktionsbeschleunigung etwas ab. Bsp.: R2CH-Br + Et-OH R3C-Br + Et-OH k = 10-6 s-1 k = 10-2 s-1 R2CH-OEt + HBr R3C-OEt + HBr Eine praktische Anwendung liegt in der Tritylschutzgruppe. Diese kann milde über eine S N1Reaktion eines Alkohols (z.B. ein Nucleosid) mit Tritylchlorid eingeführt werden, und wird ebenfalls über eine SN1-Reaktion milde wieder abgespalten, katalysiert durch schwache Säuren: b) SN1-Reaktion an Brückenköpfen: sind erschwert oder unmöglich, da die planare Geometrie (sp2) der Carbeniumionen nicht realisiert werden kann. krel = 1 10-7 10-13 c) Sterische Hinderung im Substrat: Sperrige Reste im Substrat sind sogar förderlich für die SN1-Reaktion (sterische Beschleunigung), denn im Intermediat führt die Aufweitung der Bindungswinkel von 109° im sp3-Hybrid zu 120° im sp2-Hybrid zu einer Abnahme der störenden WW der Substituenten. 33 Bsp.: krel = 1 < 21 < 580 d) Einfluß des Lösungsmittels (Lömi): Lösungsmittel, die sowohl Kationen (R3C+) als auch Anionen (Y-) gut solvatisieren und damit stabilisieren, beschleunigen die SN1-Reaktion. Dies sind vor allem: - polare, protische Lömi's wie Wasser, Methanol, Ethanol, Carbonsäuren - polare, aprotische Lömi's wie Dimethylformamid (DMF), N-Methylpyrrolidon (NMP), Dimethylsulfoxid (DMSO), Dimethylpropylenharnstoff (DMPU), und das cancerogene Hexamethylphosphorsäuretriamid (HMPT): e) Einfluß der Abgangsgruppe: Je stabiler die Abgangsgruppe, desto leichter tritt SN1-Reaktion ein. Abnehmende Nucleofugie: -N2+ > -OSO2CF3 (Triflat) -OSO2CH3 (Mesylat) -OTos (Tosylat) -I > -Br -OH2+ > Cl -F Dabei korreliert die Abgangsgruppentendenz stark mit dem pKa der korrespondierenden Säuren (H-X): Das kann man sich folgendermaßen verdeutlichen: Bei der Protolysereaktion entstehen aus HX bzw. HY mit etwa demselben Ausgangsniveau die beiden dissoziierten Spezies, die sich in ihrer freien Reaktionsenthalpie G° unterscheiden. Nach dem Massenwirkungsgesetz hängt aber die Gleichgewichtskonstante für diesen Prozess direkt mit e-G°/RT zusammen. Logarithmiert korreliert also der pKa-Wert mit G: Energiediagramm: 34 H2O + HX H3O+ + X- Ka = [H3O+][X-]/[H2O][HX] = e-G°/RT pKa: HI (-10) > HBr (-9) > HCl (-7) HF (+3) H2O (+15.7) NH3 (~35) Also sind Hydroxid OH- und vor allem Amid NH2- sehr schlechte Abgangsgruppen! Wie kann man eine OH-Gruppe verbessern als Abgangsgruppe? Allgemeines Prinzip der elektrophilen Aktivierung durch ein Proton: Nach Protonierung des tertiären Alkohols ist die Abgangsgruppe von OH- zu Wasser OH2 geworden, dessen korrespondierende Säure einen pKa-Wert von ~ -2 aufweist, also schön sauer ist. Lewissäuren können ebenfalls zu einer starken Abgangsgruppentendenz führen; sie wirken besonders gut bei Alkylhalogeniden. So beträgt z.B. der pKa-Wert für HF nur +3.0, der für HFSbF5 jedoch -15! Entsprechend stark wirkt CH3FSbF5 als "magic methyl". f) Konfigurationeller Verlauf der SN1-Reaktion: Optisch aktive Substrate mit einem asymmetrischen C-Atom als Reaktionszentrum racemisieren: über das planare Carbeniumion: Das kann man schön im Energiediagramm zeigen: Aus dem planaren Carbeniumion entstehen über zwei Übergangszustände gleicher Energie die beiden Enantiomere gleicher Energie in gleichem Verhältnis, also als Racemat. Solche ÜZe, die zu Enantiomeren führen, nennt man enantiomorph. Energiediagramm: 35 Das Kohlenstoffatom im Carbeniumion, das drei verschiedene Substituenten trägt, ist dabei prostereogen, d.h. der Angriff eines Reagenzes von einer der beiden Seiten überführt es in ein stereogenes Zentrum. Die Ober- und Unterseite können unterschieden werden; man nennt sie in diesem Fall enantiotop und verwendet für sie die Kürzel Re- und Si-Seite. Zur Benennung: Man bestimmt den Drehsinn nach den CIP-Prioritäten: Aber wenn z. B. in einem unpolareren Medium ein Kontaktionenpaar gebildet wird, erhält man eine partielle Inversion (d.h. partielle SN2-Reaktion). Fazit: SN1-Reaktionen sind vor allem abhängig von: a) der Struktur der Alkylrests; besonders gut reagieren tert. Alkylhalogenide und Arylalkylhalogenide, deutlich schlechter sek. Alkylhalogenide (Grenzfall zur SN2-Reaktion). b) der Art der Abgangsgruppe: Schlechte Abgangsgruppen können in hervorragende überführt werden, wenn Lewissäuren zugegeben werden. Bsp.: F- wird zu SbF6-; NH2 wird durch Diazotierung und Wasserabspaltung zu Diazonium N2+. Insgesamt sind SN1-Reaktionen jedoch seltener und präparativ unbedeutender als SN2Reaktionen; die meisten wichtigen sind Solvolysen. 3. SN2-Mechanismus: Dies ist ein assoziativer Mechanismus, bei dem im ÜZ eine lineare Anordnung von Nucleophil, C-Atom und Abgangsgruppe durchlaufen wird. Die Bindungen Nu-C und C-X sind partiell geknüpft und partiell gebrochen. Diese lineare Anordnung im ÜZ kann man durch Kreuzungsexperimente nachweisen (siehe Brückner): Man braucht dafür ein Molekül, welches theoretisch eine intramolekulare SN2Reaktion eingehen könnte, und ein zweites, welches sowohl im Nucleophil als auch im elektrophilen Substrat modifiziert ist, z. B. durch Deuterium-Labeling. Läßt man nun ein 1:1Gemisch beider Carbanionen in Lösung reagieren, so sollte bei intramolekularem Verlauf der Reaktion nur das vollständig protonierte und das vollständig deuterierte Produkt entstehen. Bei intermolekularem Verlauf würden auch die gekreuzten Produkte entstehen. Genau das findet man hier. Begründung: Beim intramolekularen Angriff würde im ÜZ ein sechsgliedriger Ring entstehen, in dem die teilweise gebildeten bzw. gelösten -Bindungen gewinkelt wären. Dies ist ungünstig. 36 Allgemeines Energiediagramm der SN2-Reaktion: SN2 heißt Geschwindigkeitsgesetz zweiter Ordnung: -d[R3C-X]/dt = k [Nu-] [R3C-X] 3.1. Substituenteneinflüsse: Der ÜZ trägt eine negative Ladung, deshalb stabilisieren alle Substituenten, die eine negative Ladung stabilisieren (Elektronenakzeptoren), auch den ÜZ und führen so zu einer niedrigeren EA und damit einer schnelleren Reaktion. Besonders gut sind z.B. Allyl- oder Benzylhalogenide, Halogenmethylether oder auch Halogenacetonitrile. Bsp.: krel = 1 krel = 35000 37 Die Reaktion von p-Nitrobenzylchlorid mit KI ist in Aceton ~10mal so schnell wie die von Benzylchlorid: 3.2. Einfluss des Nucleophils: Wegen des Zeitgesetzes zweiter Ordnung hat jetzt das Nucleopil einen großen Einfluß auf die RG. Energiediagramm: Wichtig: Ein gutes Nucleophil hat ein hoch liegendes Highest Occupied Molecular Orbital (HOMO), d.h. eine niedrige Elektronegativität! Bsp.: innerhalb einer Periode: R2N- > RO- > F-; RS- > Clinnerhalb einer Gruppe: I- > Br- > Cl- > FDie Nucleophilie von harten Nucleophilen wird zusätzlich verstärkt, wenn das Nachbaratom zum Nucleophil ebenfalls ein Elektronendonator ist. Bsp.: HOO- > OH- ; H2N-NH2 > H-NH2. Anders ausgedrückt: Ein Nucleophil reagiert eher nach einem SN2-Mechanismus, wenn es weich gemäß HSAB ist. Das HSAB-Prinzip (Pearson) sagt folgendes: Harte Basen (Nucleophile) reagieren am liebsten mit harten Säuren (Elektrophilen), weiche Basen (Nucleophile) dagegen lieber mit weichen Säuren (Elektrophilen). Auch hier gilt wieder: Harte Basen (Nucleophile) haben ein tiefliegendes HOMO, d.h. eine hohe Ionisierungsenergie, damit verbunden eine hohe Ladungsdichte und eine geringe Polarisierbarkeit. Bsp.: F-, RO-, RCO2-. Harte Säuren (Elektrophile) haben ein hochliegendes LUMO, damit verbunden eine niedrige Elektronegativität und eine geringe Polarisierbarkeit. Bsp.: H+, Li+, BF3, AlCl3. Weiche Basen (Nucleophile) dagegen haben ein hochliegendes HOMO, d.h. eine niedrige Ionisierungsenergie, damit verbunden eine hohe Polarisierbarkeit und eine niedrige EN des Zentralatoms. Bsp.: R2S, RSH, RS-, CN-, R3P. Weiche Säuren (Elektrophile) dagegen haben ein tiefliegendes LUMO und sind polarisierbar. Bsp.: Cu+, Ag+, Hg+, Pd2+ etc. Nur beim selben Zentralatom gehen Nucleophilie und Basizität einher. Bsp.: RO- > ArO- > R-CO2- >> ROH > RSO3Anwendungen: Reaktionen von ambidenten Nucleophilen: Das kann man ausnutzen, indem man die Reaktion von ambidenten Nucleophilen steuert. Solche Teilchen haben meistens ein hartes und weiches Ende und gehorchen daher dem HSAB-Prinzip. Ein gutes Beispiel ist das Enolation: 38 Die Base (Nucleophil) Enolat reagiert mit dem weichen C- und greift am weichen Ethyliodid an (C-Alkylierung von Enolaten); mit dem harten O- greift sie dagegen am harten Ethylsulfat an (O-Alkylierung von Enolaten). Das Enolation ist ein Spezialfall des Allylanions. Die MO's des Allylsystems sollte man kennen. Sie werden im Allylkation, -radikal oder anion nacheinander mit Elektronen aufgefüllt. Beim Enolat zeigt sich nun im 1-MO, daß die höchste Elektronendichte am elektronegativen O sitzt; dies zeigt das harte Nucleophil an. Im HOMO dagegen ist der Orbitalkoeffizient und damit der Orbitallappen am Enolat-C am größten, und zeigt das beste Nucleophil an: Ein weiteres Beispiel für ambidentes Verhalten ist das Nitritanion: Das Nitritanion greift als harte Base (Nucleophil) am harten Carbeniumion an und führt so zum Alkylnitril; als weiche Base greift es mit dem N- am weichen Alkylhaloganid an und führt so zum Nitroalkan. 3.3. Konfigurationeller Verlauf der SN2-Reaktion: Bei einer streng nach SN2 verlaufenden Reaktion an optisch reinen Verbindungen mit asymmetrischem C-Atom am Reaktionszentrum findet immer vollständige Inversion an diesem Zentrum statt (Walden-Umkehr der Konfiguration). Das nennt man stereospezifisch! 39 Wie läßt sich der Rückseitenangriff des Nucleophils erklären? Bei der günstigen HOMOLUMO-WW findet eine gute bindende Überlappung des lone pairs am Nucleophil mit dem leeren *-Orbital der C-X-Bindung nur dann statt, wenn sich das Nucleophil linear von hinten annähert (back side attack). Dazu kommt die abstoßende WW mit dem *-Orbital bei einem Frontalangriff (front side attack)und die elektrostatische Anziehung durch das positiv polarisierte C-Atom. 3.4. Sterische Hinderung: Die SN2-Reaktion ist natürlich sehr von der sterischen Zugänglichkeit des Reaktionszentrums für den Angriff des Nucleophils abhängig: Deshalb reagieren primäre Alkylhalogenide immer viel schneller als sekundäre, die wieder als tertiäre, und bei diesen wiederum besonders schwerfällig die hochsubstituierten Neopentylhalogenide: krel = 1 0.03 10-3 10-5 10-7 (Das Nucleophil muß an den störenden CH-Bindungen in -Stellung vorbei) 3.5. Lösungsmitteleffekte: 40 Dipolar aprotische Lösungsmittel eignen sich am besten für die SN2-Reaktion. Dies sind z.B. DMF, DMSO, HMPT (s.o.). Der Grund: Diese Lösungsmittel haben stark polarisierte C=O, S=O oder P=O-Bindungen, die Kationen hervorragend solvatisieren (gute Löslichkeit anorganischer Metallsalze). Die positive Partialladung sitzt aber relativ unzugänglich im Inneren der Lömi-Moleküle, so daß die Anionen kaum solvatisiert werden. Man spricht auch von "nackten" Anionen, die natürlich dadurch sehr nucleophil sind. Das gleiche kann man mit Cosolventien wie Kronenethern und Kryptanden erreichen. So werden harte Nucleophile verbessert: Man bekommt eine neue Reihung der Nucleophilie in solchen dipolar aprotischen Lösungsmitteln: CN- > AcO- > N3- > F- > Cl- > Br- > I- > SCNNormale, also apolare Lösungsmittel wie Aceton, Ether, Benzol und halogenierte Kohlenwasserstoffe dagegen verbessern die Nucleophilie weicher Nucleophile: HS- > I- > CN- > R2Se > Br- > N3- > R2S > H3N > Cl- > FWie kann man unlösliche geladene Nucleophile in organische Lösungsmittel bringen? Man benutzt dafür die Phasentransferkatalyse. Tetraalkylammoniumkationen transportieren auch sehr polare Nucleophile wie Carboxylate in die organische Phase, wo sie mit ihrem organischen unpolaren Substrat reagieren können. Anschließend nehmen sie das Nucleofug als Gegenion wieder zurück mit in die wäßrige Phase und stehen für einen neuen Transportzyklus zur Verfügung: 3.6. Nachbargruppeneffekte: Wenn sich nucleophile Gruppen des Substrats in der Nachbarschaft zum Reaktionszentrum befinden, können sie sich an der Reaktion beteiligen. Dies ist entropisch günstig und führt meist zu zweistufigen Mechanismen mit geringerer EA: Bsp.: krel = 1 0.001 Mechanismus: 41 Das intermediär gebildete Episulfoniumion ist sehr reaktiv und wird selbst durch das schlechte Nucleophil Wasser leicht geöffnet. Bei optisch reinen Substraten findet man hier durch doppelte Inversion eine Nettoretention der Konfiguration! Fazit: SN2-Reaktionen hängen vor allem von der Struktur des Alkylrests im Substrat ab: Besonders gut gehen prim. Alkyl-, Allyl- und Benzylreste, schlechter sek. Alkylreste. Die präparativ wichtigsten Abgangsgruppen sind: Iodid und Sulfonate. Dipolar aprotische Lösungsmittel begünstigen harte Nucleophile. Die Nucleophilie wird dabei bestimmt durch a) die Ladung und b) die HOMO-Energie. 4. Beispiele zu nucleophilen Substitutionsreaktionen 4.1. Halodehydroxylierung (I-, Br- und Cl- als Nucleophile) Allgemein: R-OH + X- R-X + OH- Dabei ist OH- natürlich eine sehr schlechte Abgangsgruppe und muß erst in eine gute umgewandelt werden: a) Umsetzung mit HX: R-OH + HBr R-Br + H2O Nun ist H2O eine relativ gute Abgangsgruppe (s.o.) und die Reaktion verläuft je nach Substituent R nach SN1 (sekundär, tertiär) oder SN2 (primär). Analog verläuft die Reaktion mit H-I. H-Cl dagegen ist weniger reaktiv (schlechteres Nucleophil) und nur tertiäre oder benzylische Alkohole reagieren. b) Umsetzung mit anorganischen Säurehalogeniden: R-OH + PX3 R-X + HOPX2 PX3 = Phosphortrihalogenide mit X = Cl, Br, I. Die Gruppe -OPX2 ist eine sehr gute Abgangsgruppe und wird vom dritten X- nucleophil angegiffen. PI3 und PBr3 werden oft in situ hergestellt aus Prot und X2: ------------ 42 Thionylchlorid kann mit oder ohne Hilfsbase verwendet werden. Mit Pyridin als Hilfsbase wird das Proton des Alkohols abgefangen und das freie Chloridion greift unter Inversion nucleophil den Chlorsulfinsäureester von hinten an. Ohne Pyridin wird HCl frei und der Chlorsufinsäureester durchläuft eine (innere) SNi-Reaktion, die aus sterischen Gründen unter Retention verläuft. Appel-Reaktion: Eine milde moderne Methode verwendet die Reagentienkombination Triphenylphosphin/Tetrahalogenomethan: R-OH + CX4 / PPh3 (CH2Cl2) R-X + HCX3 + O=PPh3 Mechanismus: (Angew. Chem. 1975, 87, 863) Hier wird das Br-Atom nucleophil angegriffen, die Abgangsgruppe TrihalogenomethylCarbanion deprotoniert sofort den Alkohol, welcher das Halogenid am Halogenotriphenylphosphoniumion substituiert. Das freie Halogenid seinerseits wift die nun geschaffene gute Abgangsgruppe Triphenylphosphinoxid heraus. Das geht mit Br- und I-. 4.2. Nucleophile Substitutionen an Alkylhalogeniden, -sulfaten und -sulfonaten a) Finkelstein-Reaktion: R-Cl + NaI R-I + NaCl Dies ist natürlich eine Gleichgewichtsreaktion. In Aceton ist jedoch NaCl im Gegensatz zu NaI unlöslich; dadurch wird das Gleichgewicht auf die Produktseite verschoben. Diese Reaktion wird oft in situ angewendet, um die wenig reaktiven Chloride in die viel reaktiveren Iodide zu überführen. b) Reaktionen mit Sulfonsäure-Abgangsgruppen - Herstellung der Edukte: R-OH + Cl-SO2-R' R-O-SO2-R' (+ Nu-) R-Nu + -O-SO2-R' Herstellung der Sulfonate: Tosylate: Tos-Cl/py; Mesylate: Mes-Cl/CH2Cl2/NEt3; Triflate: (Tfa)2O/py c) Sulfate (reaktiver als Sulfonate): Wichtige Reagentien sind z.B. das klassische Dimethylsulfat (cancerogen) und das starke Methylierungsreagenz "magic methyl" Fluorsulfonsäuremethylester. In Marburg sollte aber das Meerwein-Reagenz (Trimethyloxonium-tetrafluoroborat) nicht vergessen werden. Hier ist die Methylgruppe extrem hart und O-alkyliert z.B. selektiv Enolate oder Amide etc.: 43 4.3. Hydrodehalogenierung: R-CH2-I + LiAlH4 (THF) R-CH2-H + LiAlH3I Mechanismus: LAH kann viermal ein echtes Hydridion zur Verfügung stellen, welches als Nucleophil unter SN2 substituiert. Ersatz und milder: Tributylzinnhydrid (später). 4.4. Ethersynthese (Alkoholate als Nucleophile): R-OH + R'-X R-OH + NaH R-O-R' (Williamson'sche Ethersynthese) R-ONa (+ R'-X) R-O-R' + NaX Ebenso geht die Reaktion mit Phenolen zu aromatischen Ethern. So kann man unsymmetrische Ether darstellen. Phenole reagieren wegen ihres niedrigeren pKa-Wertes von ~9.2 mit milderen Basen (z.B. KOH) als Alkohole mit einem pKa von 15-17 (z.B. NaH). Ether werden häufig als Schutzgruppe für Alkohole verwendet. Besonders beliebt sind die Benzyl- und die Tritylschutzgruppe, weil sie milde abspaltbar sind (Benzyl: hydrogenolytisch, d.h. Pd-C/ H2; Trityl: milde Säure, RT): Ähnlich kann man mit unreaktiven Carboxylaten Ester herstellen. Entweder man führt die Reaktion mit Alkylhalogeniden in dipolar aprotischen Lösungsmitteln durch (DMSO etc.), oder man benutzt das hochreaktive Diazomethan, welches erst die Carbonsäure deprotoniert und anschließend als Diazoniumion vom Carboxylat angegriffen wird. Dabei entsteht sofort der Methylester und N2. 44 4.5. Synthese von Aminen (N-Nucleophile): R-X + NH3 R-NH2 R2NH R4N+X- R3 N Diese SN-Reaktion ist nicht selektiv, denn sekundäre Amine sind aufgrund des +I-Effekts ihrer Alkylgruppen reaktiver als primäre Amine usw. Deshalb geht die Reaktion oft durch bis zum quartären Ammnoiumsalz (erschöpfende Methylierung; früher oft gefolgt von HofmannEliminierung zur Strukturaufklärung von Alkaloiden). a) Primäre und sekundäre Amine - Gabriel-Synthese: Phthalimid ist sauer aufgrund der doppelten Carbonylanziehung, sein Kaliumsalz greift als Nucleophil Alkylhalogenide direkt nach SN2 an. Anschließende saure Aufarbeitung oder Hydrazinolyse in Alkoholen führt zum schwerlöslichen Phthalylhydrazid und dem freien Amin. b) Primäre Amine über die Azidsynthese: R-X + NaN3 R-N3 + NaX (+ LAH oder Pd-C/H2 R-NH2) Die hochreaktiven Azide (explosiv!) werden meist nicht isoliert, sondern sofort weiterverarbeitet. c) Sekundäre Amine über die Sulfonamid-Synthese: Ar-SO2-NHR + KOH oder NaH ArSO2NRR' + H3O+ Ar-SO2NR-M+ (+ R'-X) H2NRR'+ Sulfonamide sind sehr sauer durch die stark elektronenziehende Sulfongruppe und können bereits mit KOH deprotoniert werden. Monoalkylierung und anschließende saure Hydrolyse führen zu den freien sekundären Ammoniumhydrohalogeniden. 4.6. Reaktionen mit C-Nucleophilen: a) Kolbe Nitrilsynthese: R-X + NaCN R-CN + NaX (+ HCl R-COOH) Das ganze geht gut in DMSO (s.o.). Die Nitrile sind Vorläufer für Carbonsäuren. b) Alkylierung CH-acider Verbindungen: allgemein: 45 Malonester: pKa~11 Malondinitril: pKa~11 Cyanessigester: pKa ~11 Hier bietet sich als Base NaOMe (pKa~16) an. ---------------Ketone: pKa~20 - stärkere Base: LDA (36) Dithiane: pKa~30 - stärkere Base: BuLi (45) Acetylene: pKa~25: stärkere Base: BuLi (45). c) Alkylierung mit Cupraten: Unsymmetrische Alkane kann man am besten aus Alkylhalogeniden und den weichen Cupraten herstellen (Corey-House-Reaktion). Nebenreaktionen wie Eliminierungen und Transmetallierungen werden so vermieden: Darstellung des Cuprats: Vorteile: breit anwendbar, hohe Toleranz von funktionellen Gruppen, sehr viel bessere Ausbeuten als mit RLi oder RMgX. Inversion der Konfiguration am sek. R-Br und R-Cl. Chemoselektivität: bevorzugter Angriff am reaktiveren Halogenid: 4.7. Reaktionen mit P-Nucleophilen: a) Wittig-Vorläufer: Phosphoniumsalze Ph3P + R-CH2-X Ph3P+-CH2-R X- Daraus entstehen mit starken Basen die Ylide für die Wittig-Olefinierung. b) Wittig-Horner-Vorläufer: Arbuzow-Reaktion 46 (RO)3P + R-CH2-X (RO)2P(=O)-CH2-R + R-X via: Phosphonsäureester entstehen über den nucleophilen Angriff von Trialkylphosphiten an Alkylhalogeniden, gefolgt von einem zweiten nucleophilen Angriff des Bromids am Phosphoniumsalz. 4.8. Reaktionen mit S-Nucleophilen: a) Thioether wie Williamson: R-X + HS-R' + Base R-S-R' + HBase+X- Achtung: Thiole sind viel saurer als Alkohole (pKa~10.5). b) Thiole aus Alkylhalogeniden über S-Alkylthiouroniumsalze: R-X + H2N-C=S-NH2 R-SH + H2N-C=O-NH2 + HX Mechanismus: Das nucleophile S-Atom greift am Alkylhalogenid an, das S-Alkylthiouroniumsalz wird von Natronlauge angegriffen und wirft das Thiolat heraus, welches bei der Aufarbeitung protoniert wird. Gute Abgangsgruppe: Harnstoff. 5. Nucleophile Substitution an substituierten Silanen: Allgemein: R'-OH + Cl-SiR3 + NEt3 R'-O-SiR3 + HNEt3+Cl- Silylether sind wichtige Schutzgruppen in der Synthese. Mechanismus-Vorschlag: Hier greift zuerst das Nucleophil an und generiert eine pentakoordinierte Si-Spezies. Das geht wegen der zusätzlichen d-Orbitale am Si. Mit der Base wird im zweiten Schritt HX eliminiert. Das ganze ist also keine SN2-Reaktion, sondern ein Additions-Eliminierungs-Mechanismus. 47 Wie spaltet man Silylether? mit H3O+ oder mit F--Anionen. Merke: Fluor hat eine hohe Assoziation zu Si: Bindungsenergie Si-F: 136 kcal/mol. Praktisch z.B. mit TBAF oder HF/py: R'-O-SiR3 + Bu4N+F- R'-OH + FSiR3 So werden oft am Ende von Synthesen die Silylschutzgruppen milde entfernt. Wie spaltet man Ether allgemein? mit HI: Cyclische Ether: HI oder TMSI Aromatische Methylether: BBr3 mit Dimethylsulfid 48 D. Eliminierungen 1. Einteilung: a) 1,1-Eliminierung (-Eliminierung): Zwei unterschiedliche Gruppen werden von demselben C-Atom abgespalten. Dabei entsteht ein Carben. b) 1,2-Eliminierung (-Eliminierung, häufig): Zwei unterschiedliche Gruppen werden von benachbarten (vicinalen) C-Atomen abgespalten. Dabei entsteht ein Olefin. c) 1,3-Eliminierung (-Eliminierung): Zwei unterschiedliche Gruppen werden von 1,3-ständigen C-Atomen abgespalten. Es entsteht ein Cyclopropan. 2. Ionische -Eliminierungen: Sehr oft wird H-X von benachbarten C-Atomen abgespalten. Das geschieht mechanistisch auf dreierlei Weise: E1, E1cb, E-Eliminierungen. 2.1. E1-Eliminierung: Im geschwindigkeitsbestimmenden ersten Schritt der Reaktion wird genau wie bei der S N1Reaktion die C-X-Bindung gespalten. Dies geschieht im Gegensatz zu den anderen Eliminierungsreaktionen vor der Abspaltung des Protons. Der zweite Schritt entscheidet nun über den weiteren Verlauf: wird aus dem Carbeniumion ein Proton abgespalten (E1) oder ein wartendes Nucleophil addiert (SN1)? Hier kann man vor allem über die Temperatur steuern: hohe Temperaturen begünstigen immer die Eliminierungsreaktion! Grund: Gibbs-Helmholtz: G = H -TS . Bei E1-Reaktionen entstehen aus dem Carbeniumion zwei Teilchen (S>0), bei SN1-Reaktionen entsteht dagegen aus Carbeniumion und Nucleophil nur ein Produktteilchen (S<0). 49 Bsp.: tBu-Cl + 80% wäßriges Ethanol, T = 78°C T 25°C 50°C 65°C SN1 83% 76% 64% tBu-OH + Isobuten E1 17% 24% 36% Mechanismus: Kinetik: Eine E1-Reaktion hat ihren Namen von der Kinetik erster Ordnung. Die Reaktionsgeschwindigkeit der E1-Reaktion hängt also nur von der Konzentration des Substrats ab, nicht von der der Base! Regioselektivität: Bei Eliminierungsreaktionen unter E1-Bedingungen wird bevorzugt das höher substituierte Alken gebildet (Saytzew-Regel). Der Grund: Höher substituierte Alkene sind durch den +I-Effekt der Alkylgruppen thermodynamisch stabiler. Hier haben wir aber nur scheinbar ein gutes Beispiel für die thermodynamische Kontrolle. In Wirklichkeit ist die Reaktion kinetisch kontrolliert. Dafür greift hier das Evans-Bell-Polanyi-Konzept: Die Reaktion mit der niedrigsten Produktenergie hat auch den niedrigsten ÜZ. Energiediagramm: Der erste ÜZ ist geschwindigkeitsbestimmend, der zweite ÜZ dagegen produktbestimmend. Bsp.: 50 Hier entstehen Saytzew- und Hofmann-Produkt (das mit der niedriger substituierten DoBi) im Verhältnis 99:1. Das entspricht einem G von 2.8 kcal/mol. Der +I-Effekt der Alkylgruppen wird durch eine Wechselwirkung des leeren *-MO's der DoBi mit der übernächsten C-H oder C-C--Bindung erklärt. Elektronendichte fließt also aus der -Bindung ins *-Orbital: Lösungsmittel: Genau wie die SN1-Reaktion verläuft auch die E1-Reaktion besonders gut in polaren protischen Lösungsmitteln (sowohl Carbeniumion als auch die Abgangsgruppe werden dabei gut solvatisiert). Modernes Anwendungsbeispiel aus der Schutzgruppentechnik: Mit wäßrigen Säuren kann man milde t-Butylether, -ester oder -carbamate spalten. Es entsteht jeweils das tButylcarbeniumion, welches ein Proton eliminiert ud zu Isobuten wird. 2.2. E1cb-Mechanismus: Hier wird zuerst die CH-Bindung gespalten, erst danach die C-X-Bindung. Das kann zwei Gründe haben: erstens kann die CH-Bindung geschwächt sein, wie es bei CH-aciden Verbindungen der Fall ist; zweitens kann die C-X-Bindung besonders stark sein, wie es bei schlechten Abgangsgruppen X der Fall ist. Dieser Mechanismus ist jedoch eher selten. Ein Beispiel ist die Reaktion mit R 1 = Nitro = guter -Akzeptor: 51 Nitroalkane haben einen pKa-Wert von ~10, dadurch wird der erste Deprotonierungsschritt schnell, denn er führt zum mesomeriestabilisierten Nitronat-Anion. Erst im zweiten Schritt wird die Gesamtreaktionsgeschwindigkeit bestimmt, denn OH- ist eine schlechte Abgangsgruppe. Dadurch erklärt sich das Kürzel: Die Reaktionsgeschwindigkeit hängt nur von der Konzentration dieser Zwischenstufe ab, nämlich der korrespondierenden Base zum Edukt. Andere wichtige Beispiele sind die Hofmann-Eliminierung, bei der die Abspaltung des Trialkylammoniumions wegen der starken C-N-Bindung langsam verläuft, und die Aldolkondensation, bei der die Carbonylgruppe für CH-Acidität sorgt und gleichzeitig eine schlechte Abgangsgruppe (das Hydroxidanion) vorliegt. Ähnlich verläuft die KnoevenagelKondensation. Modernes Anwendungsbeispiel: Abspaltung der Fmoc-Schutzgruppe in der Peptidchemie. Nach Deprotonierung mit Morpholin entsteht das mesomeriestabilisierte Fluorenylanion (korrespondierende Base), welches leicht CO2 und das Peptid abspaltet: 2.3. E2-Mechanismus: Hier werden C-X und C-H-Bindung gleichzeitig (konzertiert) gespalten. Dadurch entsteht eine Kinetik zweiter Ordnung wie bei der SN2-Reaktion: Die Reaktionsgeschwindigkeit hängt von der Konzentration beider Reaktionspartner ab, nämlich sowohl der Base als auch des Substrats: v = k [Edukt] [Base] Das dazugehörige Energiediagramm ähnelt sehr dem der SN2-Reaktion: 52 Welche Faktoren bestimmen hier das Verhältnis von Hofmann und Saytzew-Produkt? Wichtig sind Acidität und sterische Hinderung des abzuspaltenden H-Atoms. Außerdem spielt die Güte der Abgangsgruppe X und natürlich die Stärke und Raumerfüllung der Base eine entscheidende Rolle – viele Faktoren! Abgangsgruppeneffekt: Abgangsgruppe X F Cl Br I Hofmann-Anteil 70 % 33 % 28 % 19 % Saytzew-Anteil 30 % 67 % 72 % 81 % Je schlechter die Abgangsgruppe (z.B. Fluorid), desto mehr ist die C-H-Bindung im ÜZ bereits gebrochen. Primäre Alkylgruppen haben aber acidere Protonen als sekundäre, denn das primäre Carbanion ist weniger durch Hyperkonjugation benachbarter Alkylreste destabilisiert. Also gewinnt hier das Hofmann-Produkt. Auch die Trialkylammonium- und die Dialkylsulfonium-Gruppe sind schlechte Abgangsgruppen. Im Extremfall verläuft die Reaktion nicht mehr nach E2, sondern nach E1cb. Iodid ist dagegen eine sehr gute Abgangsgruppe; im ÜZ ist deshalb sowohl die CH-als auch die C-X-Bindung bereits teilweise gebrochen und der ÜZ weist dadurch partiellen Alkencharakter auf (MO-Bild: die sp3-Hybridatomorbitale gehen kontinuierlich in p-Orbitale über, die bei anti-Stellung von H und X bereits zum entstehenden -MO überlappen können product development control). Dadurch wird das stabilere Alken gebildet (Saytzew-Produkt). 53 Einfluß der Base: Eine sperrige Base attackiert natürlich bevorzugt das leichter zugängliche terminale Proton und begünstigt damit die Bildung des Hofmann-Produkts: Base K-OEt K-OtBu K-O-CH(C2H5)3 Hofmann-Anteil 38 % 73 % 89 % Saytzew-Anteil 62 % 27 % 11 % Sperrige Basen verhindern aber auch wirksam die unerwünschte Nebenreaktion über SN2, wenn eigentlich nur eine Deprotonierung erwünscht ist. Wichtige Beispiele: Hünigbase (Ethylsiisopropylamin) KOtBu DBN (Diazabicyclononen) DBU (Diazabicycloundecen) 2.4. Stereochemischer Verlauf der E2-Eliminierung: Es gibt grundsätzlich zwei konzertierte Eliminierungsgeometrien: a) anti-Eliminierung (erfolgt aus einer gestaffelten Konformation): b) syn-Eliminierung (erfolgt aus einer ungünstigen ekliptischen Konformation): zu a) Acyclische Substrate gehen meist die anti-Eliminierung ein: 54 Im obigen Beispiel liefert die Meso-Verbindung das E-Olefin, die R,R-Verbindung dagegen das Z-Olefin. Das bedeutet, die anti-Eliminierung ist streng stereospezifisch. Cyclohexanderivate können nur dann anti-eliminieren, wenn die Abgangsgruppe eine transdiaxiale Anordnung zur gespaltenen C-H-Bindung einnimmt (Barton-Regel): Obwohl das Gleichgewicht nur zu 1/5000 auf der rechten Seite liegt, verläuft die Reaktion zu 100% aus dieser Konformation und liefert das stereochemisch eindeutige Produkt der antiEliminierung. b) Cyclische Derivate gehen eine syn-Eliminierung ein, wenn die anti-Eliminierung aus sterischen Gründen unterdrückt wird: Am starren Norbornangerüst ist kein Umklappen möglich; folgerichtig wird hier die syn-E2Eliminierung von DX zu 94 % gegenüber der nicht korrekten anti-E2-Eliminierung von HX zu 6 % bevorzugt. 2.5. -Eliminierungen von Het1/Het2: Diese Reaktionen werden oft auch als Fragmentierungen bezeichnet: Mg + Br-CH2-CH2-Br [Br-Mg-CH2-CH2-Br] MgBr2 + H2C=CH2 Mg + Br-CH2-CH2-X [Br-Mg-CH2-CH2-X] MgBrX + H2C=CH2 Die intermediär gebildeten -heterosubstituiertem gesättigten Organometallverbindungen sind natürlich instabil: sie enthalten ja eine Base und eine Abgangsgruppe im selben Molekül. Metalle sind zum Beispiel: Li+, ZnX+ etc. So kann man auch Ketene generieren: 55 R-CHBr-C(=O)Br + Zn R-CH=C=O + ZnBr2 2.6. Thermisch induzierte -Eliminierungen: Hier fungiert die Abgangsgruppe als interne Base. Deshalb läuft die Reaktion rein thermisch ab, ohne äußere Baseneinwirkung. Die Reaktion geht über einen 5- oder 6-gliedrigen ÜZ und verläuft im Sinne einer syn-Eliminierung. a) Esterpyrolyse: Diese Reaktion erfordert hohe Temperaturen von 400-500°C und ist von daher synthetisch schlecht einsetzbar. b) Tschugajew-Reaktion: Die Pyrolyse eines Xanthogenats verläuft wesentlich milder bei 150-200 °C. Das Xanthogenat entsteht aus der Reaktion eines Alkoholats mit CS2 und anschließender Alkylierung mit MeI. c) Decarboxylierung von -Ketoestern (Fragmentierung): Diese Reaktion verläuft ähnlich wie die Esterpyrolyse. Es handelt sich um eine Retro-EnReaktion, bei der zunächst die Enolform gebildet wird, welche anschließend tautomerisiert. 56 d) Cope-Eliminierung: Eine weitere syn-Eliminierung ist die Cope-Eliminierung von Aminoxiden, welche durch Oxidation von Aminen mit H2O2 (oft auch mcpba) zugänglich sind. Bei 120°C greift das Aminoxid als innere Base das -ständige H an und eliminiert anschließend sich selbst. Über eine 5-gliedrigen ÜZ entsteht hier stereospezifisch das Z-Olefin. e) Selenoxid-Pyrolyse: Doppelbindungen kann man auch über 2,3-sigmatrope Umlagerungen aus Selenoxiden herstellen. Auch hier fungiert das Selenoxid als innere Base und greift das -ständige Proton an und eliminiert anschließend sich selbst. Großer Vorteil: Diese Reaktion läuft bereits bei RT ab. Es entsteht das Alken und Phenylseleninsäure. f) Nachtrag: Zur milden Dehydratisierung von Alkoholen verwendet man Burgers Reagenz: Hier wird in einer cyclischen syn-Eliminierung die neu ins Molekül eingeführte Abgangsgruppe durch intramolekularen Angriff des Imidanions eliminiert. g) Nachtrag: Ebenfalls über syn-Eliminierungen verläuft die Peterson Olefinierung. Hier kann man mit Säure oder Base selektiv die Stereochemie der Eliminierung vorbestimmen: 57 E. Additionen 3. Allgemeines Eine unsymmetrische Verbindung X+-Y- wird an eine Doppelbindung C=C addiert. A) Elektrophile Addition: Zuerst greift X+ an, dann erst Y-. Die Reaktion verläuft über folgende Zwischenstufen: oder mit R3/R4 = Donorsubstituenten Onium-Ion B) Nucleophile Addition: Zuerst greift Y- an, dann erst X+. Die Reaktion verläuft über folgende Zwischenstufen: mit R1/R2 = Akzeptorsubstituenten C) Radikalische Addition: Zuerst greift X oder Y an, dann erst Y oder X. Die Reaktion verläuft über folgende Zwischenstufen: D) [4+2], [2+2]-Cycloadditionen: Diese Reaktionen verlaufen konzertiert, ohne Zwischenstufen (no mechanism reactions!): 58 4. Thermodynamische Betrachtungen: Man kann die Reaktionsenthalpie berechnen, wenn man Aufwand und Ertrag voneinander subtrahiert: -Bindung -Bindung -Bindung Aufwand -Bindung Ertrag Bsp.: Addition von HCl an Ethen: C-C = +64 kcal/mol Ertrag: C-Cl = -81 kcal/mol H-Cl = +103 kcal/mol C-H = -99 kcal/mol ______________________________________________________________________ Bilanz: -13 kcal/mol (=exotherm, günstig!) Aufwand: Ebenso kann man z.B. die Additionen von Halogenen an Ethen berechnen und findet: Br2 + C=C I2 + C=C H2O + C=C Br-C-C-Br I-C-C-I HO-C-C-H (-26 kcal/mol) (- 4 kcal/mol) (-10 kcal/mol) Alle Reaktionen verlaufen exotherm, aber die Iodaddition ist viel ungünstiger als die Bromaddition. Hier sind aber noch keine Entropieglieder drin, also auch keine TBerücksichtigung! (Gibbs-Helmholtz !) 5. Elektrophile Additionen an C=C-Doppelbindungen: 3.1. Allgemeiner Mechanismus: -Komplex, überbrücktes nichtklassisches Kation anti-Addition 59 Der -Komplex, also das nichtklassische verbrückte Kation, steht im Gleichgewicht mit dem klassischen Carbeniumion. Wenn dieses gebildet wird, ist der Angriff des Nucleophils von beiden Seiten möglich, und es gibt keine stereochemische Kontrolle des konfigurationellen Verlaufs der Reaktion. 3.2. Säurekatalysierte Hydatisierung von Alkenen: Diese Reaktion ist reversibel; tiefe Temperaturen begünstigen dabei die Addition; bei hohen Temperaturen kann allerdings die Reaktion auch in die Eliminierung hineinlaufen; dies ist ein Entropieeffekt! Denn bei der Addition entsteht aus dem Carbeniumion und dem Nucleophil ein neues Teilchen, während bei der Eliminierung aus dem Carbeniumion zwei neue Teilchen entstehen; hier ist die Entropieänderung also positiv. Bei T-Erhöhung fällt damit das günstige Entropieglied der Gibbs-Helmholtz-Gleichung -TS immer mehr ins Gewicht, und lenkt die Reaktion in Richtung Eliminierung. -Komplex, überbrücktes nichtklassisches Kation Carbeniumion Das planare Carbeniumion (s.o.) kann natürlich von oben und von unten angegriffen werden, und führt so zu einem racemischen Gemisch, falls nicht noch ein weiteres Stereozentrum gebildet wird (Diastereomere). Regioselektivität bei der elektrophilen Addition: Isobuten z.B. liefert bei der Wasseraddition (Hydratisierung) ausschließlich t-Butanol und nicht Isobutanol. Das liegt an der Stabilisierung der positiven Ladung im intermediären Carbeniumion. Regel von Markownikow: Die Protonierung der Doppelbindung führt stets zum stabileren, höhersubstituierten Carbeniumion. Da die elektrophile Addition an Doppelbindungen aber kinetisch kontrolliert ist, muß man sich die Kontrolle des geschwindigkeitsbestimmenden Schritts ansehen: In diesem Fall ist das die Protonierung der DoBi zum Carbeniumion. Die Reaktion mit dem niedrigeren ÜZ gewinnt. Welche ist das? Hammond-Postulat: Späte ÜZe sind produktähnlich, frühe eduktähnlich. In unserem Fall ist also der ÜZ1, der zum dem primären Carbeniumion führt, ungünstiger, als der ÜZ2, der zum tertiären Carbenium führt. Die Differenz der Aktivierungsenthalpien G≠ 60 beträgt ~28 kcal/mol. Dieser Unterschied reicht aus, um die Reaktion ganz regioselektiv zum t-Butanol zu lenken. Beispiele: a) Enolether liefern bei der sauren Hydratisierung Aldehyde, weil das Carbeniumion neben der Methoxygruppe mesomeriestabilisiert ist; die Hydrolyse zum Halbacetal liefert indirekt den Aldehyd: b) Tetrahydropyrane liefern bei der sauren Alkoholyse regioselektiv die 2-THP-Ether. THP ist eine gute Acetal-Schutzgruppe für Alkohole, die man sauer milde wieder abspalten kann. Auch hier wird das Carbeniumion über die Alkoxygruppe mesomeriestabilisiert. c) Herstellung von t-Butylestern (Schutzgruppen für Carbonsäuren, Peptidchemie!): Hier ist das Nucleophil Nu-H die Carbonsäure selbst (RCOO-H). Sie greift das protonierte Alken an und verliert anschließend selbst ihr Proton. 61 d) Addition von HBr an Doppelbindungen: Hier ist das Nucleophil Nu-H das H-Br, wobei strenge Markownikow-Kontrolle beobachtet wird, denn das sekundäre Carbeniumion ist stets stabiler als das primäre. 5.3. Diastereoselektive trans-Additionen: Allgemein orientiert sich das Molekül X+-Y- mit der positiven Partialladung zur elektronenreichen DoBi hin. Nach der Ausbildung des Onium-Ions wird dieses von der Rückseite her angegriffen; dabei kann jedes der beiden ursprünglichen DoBi-C-Atome angegriffen werden. Als Elektrophile kommen u.a. Cl+, Br+, I+ und Hg(OAc)+ in Frage. a) Reaktion von Alkenen mit Brom in apolaren Lösungsmitteln: Bei achiralen Resten R1 und R2 sind die beiden Seiten der Doppelbindung enantiotop; d.h., ein Angriff auf der Ober- oder Unterseite würde zu enantiomeren Zwischenprodukten führen. Durch Angriff des Br+-Kations entsteht ein Epibromoniumkation, welches 3 Zentren mit zwei Elektronen verbindet (nichtklassisches überbrücktes Kation; in der Mesomeriedarstellung sieht man, daß die positive Ladung auf allen 3 Atomen des Dreirings liegen kann). Damit ist die freie Drehbarkeit um die C-C-Bindung stark eingeshcränkt, und der Angriff des Bromidanions findet im Sinne einer SN2-Reaktionvon der Rückseite her statt (insgesamt antiAddition). Bei unterschiedlichen Resten R1 und R2 führt der Angriff an den C-Atomen a oder b zu enantiomeren Produkten. 62 Die Bromaddition an Alkene läuft also stereospezifisch ab. Das bedeutet, daß ein stereochemisch einheitliches Edukt (z.B. cis-Alken) ein stereochemisch einheitliches Produkt (also ein definiertes trans-Addukt) ergibt. Ein Spezialfall tritt bei gleichen Resten R1 und R2 auf: Im unteren Beispiel ist gezeigt, wie das symmetrische cis-Buten ein Enantiomerenpaar im Verhältnis 1:1 liefert (C2-Symmetrie), das trans Alken dagegen zur achiralen meso-Form führt, die spiegelsymmetrisch ist (CS). cis-Buten: trans-Buten: b) Additionen von Brom an Alkene in protischen Lösungsmitteln: 1. Halogenether: In Methanol als überschüssigem Lösungsmittel hat das entstehende Bromoniumion keine Chance, mit weiterem Bromid zu reagieren,weil der riesige Überschuß Methanol trotz seiner schwächeren Nucleophilie gewinnt. Über den Angriff an C-Atom a oder b entstehen wieder enantiomere -Halogenether. Ein Produkt hat nun die R,R- das andere die S,S-Konfiguration an den beiden neuen Stereozentren. 2. Halogenhydrine: NBS zerfällt in protischen und polaren Lösungsmitteln in das Bromkation und das mesomeriestabilisierte Succinimidanion. Das entstehende Bromoniumkation kann nun 63 aufgrund der sterisch abgeschirmten Neopentylstellung nur auf der anderen Seite angegriffen werden, und zwar wieder SN2-artig von der Rückseite, so daß in eindeutiger Weise das transBromhydrin entsteht, natürlich als Racemat. Das Succimidanion deprotoniert dabei hilfreich das Wasser, welches als Hydroxidion im zweiten Schritt angreift. 3. Halogenlactonisierung: Das Iodoniumkation wird intramolekular vom Carboxylat angegriffen (entropisch günstig). 4. Solvomercurierung von Olefinen: Die DoBi greift das Hg2+-Kation nucleophil an und wirft ein Acetation heraus. Dabei entsteht ein Oxymercuriniumkation, welches wieder eine offene und eine überbrückte Grenzstruktur besitzt. Wasserangriff öffnet SN2-artig dieses Kation von unten und führt zum Hg-haltigen Zwischenprodukt, welches normalerweise mit NaBH4 reduziert wird. Bei unterschiedlich substituierten Doppelbindungen erfolgt die Addition nach Markownikow: Man fand heraus, daß die C-Hg-Bindung zum höher substituierten C-Atom länger ist als zum weniger substituierten C-Atom, was einen größeren Beitrag des klassischen Carbeniums am höher substituierten C-Atom anzeigt. Diese Bindung ist deshalb schwächer und wird bevorzugt von hinten angegriffen. 64 5.4. Diastereoselektive cis-Additionen a) Hydroborierung von Alkenen BH3 ist ein starkes Elektrophil wegen seines Elektronensextetts am Bor (Elektronenmangelverbindung). Es stabilisiert sich daher über 2-Elektronen-3Zentrenbindungen zum dimeren Diboran. In etherischen Lömi's wird es aber komplexiert und deaggregiert: Nur monomere Borane sind zur cis-Addition befähigt! Mechanismus: Nach einer cis-Addition an die DoBi kann die BH2-Gruppe noch zwei weitere Alkene anlagern, so daß zum Schluß ein Trialkylboran entsteht. Bei sterisch gehinderten Alkenen werden oft nur Mono- oder Dialkylborane gebildet, welche selbst als Hydroborierungsmittel dienen können. 2,3-Dimethylbuten führt so zum Thexylboran, 1,4-Cyclooctadien zum 9-Bicycloboranonan. Regioselektivität bei der Hydroborierung: Generell wird Boran am weniger substituierten Ende der DoBi angelagert (AntiMarkownikow): Das hat erstens sterische Gründe: Die BH2-Gruppe würde bei der sehr engen seitlichen Annäherung des Reagenzes an das Alken mit dem Rest R ins Gedränge kommen. Diesen Beitrag kann man quantifizieren, indem man sterisch unterschiedlich anspruchsvolle 65 Hydroborierungsreagenzien miteinander vergleicht. So führt Boran zu 80%, 9-BBN dagegen zu über 99% zum Anti-Markownikow-Produkt. Das hat aber auch elektronische Gründe: Obwohl man von einer nahezu gleichzeitigen konzertierten Addition spricht, induziert natürlich die Annäherung des Borans an die Doppelbindung im anzugreifenden Atom eine entgegengesetzte Partialladung. Die ist gegenüber dem elektrophilen Bor natürlich negativ, gegenüber dem negativierten B-Hdagegen positiv. Diese positive Partialladung ist aber wieder am höher substituierten C-Atom besser stabilisiert, so daß auch hier wieder die positive Partialladung am höher substituierten C besser stabilisiert wird. Transformation von Alkylboranen: Die wichtigste Umsetzung ist die basische oxidative Aufarbeitung zu Alkoholen. Insgesamt führt die Reaktion eines Trialkylborans mit alkalischem Wasserstoffperoxid zum Alkohol: Mechanismus: Das Hydroperoxid-Anion greift das leere p-Orbital des Borans an. Das dabei entstehende Anion geht eine der Wagner-Meerwein-Umlagerung ähnliche Reaktion ein, bei der der Rest R anionotrop auf den Peroxy-Sauerstoff wandert ( dieser ist positiviert) und OH- abspaltet. 66 Wir haben also jetzt zwei Methoden kennengelernt, mit denen wir terminale Alkene regioselektiv zu primären oder sekundären Alkoholen hydratisieren können: b) Heterogen katalysierte Hydrierung: Die cis-Addition von Wasserstoff an Alkene läuft ohne Katalysator nicht ab! Üblicherweise benutzt man dafür einen heterogenen (unlöslichen) Katalysator, nämlich Pd auf Aktivkohle, feinverteilt (Pd-C). Man nimmt an, daß das Pd oder Pt den Wasserstoff homolytisch spaltet und im Innern des Metalls atomar speichert. Mit Deuterium erhält man beispielsweise eine stereospezifische cis-Addition an cis-Buten: Das wird dadurch erklärt, daß das Alken auf der Katalysatorobefläche andocken muß, und während dieses Andockens zweimal hintereinander von derselben Seite angegriffen wird. c) cis-Hydroxylierung von Alkenen: Man nimmt am besten Osmiumtetroxid, eine leichtflüchtige, kristalline, hochgiftige Substanz. Diese kann katalytisch oder stöchiometrisch eingesetzt werden. In beiden Fällen entsteht zunächst im Sinne einer Cycloaddition ein Osmatester. Dieser kann nun in Anwesenheit von N-Methylmorpholin-N-oxid hydrolytisch aufgearbeitet werden, wobei das NMO das Osmium zurück zum OsO4 oxidiert, oder aber reduktiv, wobei H2S die Protonen liefert, und das reduzierte Osmiumdioxid gebildet wird: Analog kann auch Permanganat verwendet werden; dieses Reagenz führt aber oft zu Nebenreaktionen. 67 d) Epoxidierung von Alkenen: Organische Persäuren enthalten einen positivierten Sauerstoff. Sie reagieren mit elektronenreichen Alkenen, indem sie in einer konzertierten Reaktion eben diesen Sauerstoff+ übertragen und dabei in normale Carbonsäuren übergehen. Wieder findet eine syn-Addition statt, wobei ein sogenannter eingeschnürter ÜZ durchlaufen wird. Da in diesem ÜZ die Persäure eine negative Partialladung bekommt, stabilisieren Elektronenkzeptoren an ihr den ÜZ und beschleunigen damit die Reaktion. Das Alken wird durch Elektronendonoren aktiviert, denn am Peroxysauerstoff befindet sich auch im ÜZ eine positive Partialladung. Also: Trifluoressigsäure ist besser als Essigsäure. Auf der anderen Seite reagieren die höher substituierten Alkene, die sonst schwer zur Reaktion zu bringen sind, mit den Persäuren am liebsten: 6. Cycloadditionen: 4.1. Diels-Alder-Reaktion = [4+2]Cycloaddition: Dien + Dienophil Cyclohexen Die Diels-Alder-Reaktion ist ein Beispiel für pericyclische Reaktionen. Sie zeichnen sich durch einen cyclischen ÜZ aus, bei dem ein aromatischer oder antiaromatischer ÜZ durchlaufen wird (Woodward-Hoffmann-Regeln). Bei aromatischen ÜZen haben wir 4n+2 Elektronen, bei den antiaromatischen 4n -Elektronen. Das Energiediagramm dieser konzertierten Reaktion zeigt keine Zwischenstufe. Die Reaktionspartner lagern sich flach übereinander an (suprafacial), so daß die oberen und unteren Orbitallappen der -MO's miteinander wechselwirken. 68 Grenzorbital-WWen: Nach der Störungstheorie üben Orbitale ähnlicher Energie eine große stabilisierende und destabilisierende WW aufeinander aus, wenn die entsprechenden Moleküle sich annähern. Das ist aber nur dann günstig, wenn nur eines der beiden Orbitale besetzt ist, weil dann im ÜZ gerade der stabilere Zustand besetzt wird, was einen Energiegewinn bringt. Dies ist bei der WW zwischen HOMO und LUMO der Fall. Die Vorzeichen der äußeren Enden müssen dabei für eine erlaubte Reaktion im Sinne der Woodward-Hoffmann-Regeln miteinander übereinstimmen (bindende Überlappung). Das gilt bei der Diels-Alder-Reaktion für beide Fälle. Meistens jedoch werden elektronenreiche Diene mit hochliegenden HOMO's und elektronenarme Dienophile mit niedrigen LUMO's verwendet. Es gibt aber auch die D.-A.-Reaktion mit inversem Elektronenbedarf. Die D.-A.-Reaktion ist wegen der suprafacialen Anordnung im ÜZ stereospezifisch: Aus cisDienophil entsteht ein cis-Produkt, aus trans-Dienophil ein trans-Produkt. Zusätzlich findet man oft eine bevorzugte Orientierung des Dienophils mit seiner elektronenziehenden Gruppe unterhalb (endo) des Dien--Systems. Dies wird mit einer zusätzlichen bindenden Sekundär-Wechselwirkung der beiden Molekülorbitale erklärt, die bei der exo-Annäherung unmöglich ist: 69 4.2. Cyclopropanierung = [2+1]-Cycloaddition: Carbene haben ein sp2-konfiguriertes C-Atom und zwei Liganden. Das freie Elektronenpaar kann entweder gepaart im sp2-Hybridatomorbital sitzen (Singlett-Carben) oder ungepaart je eines im sp2, das andere im p-Orbital (Triplett-Carben). Für pericyclische Reaktionen eignet sich nur das Singlett-Carben. Im ÜZ lagert sich das Carben seitlich an die DoBi an (Raumschiff-Enterprise), denn nur so kann eine bindende Überlappung der Orbitale gewährleistet werden. Dichlorcarben z.B. kann mit seinem LUMO (das leere p-Orbital) am HOMO (der -Bindung des Alkens) angreifen. Herstellung von CCl2: HCCl3 + RO- ROH + CCl3- CCl2 4.3. Ozonolyse: Ozon ist ein fabloses, giftiges Gas; in organischen Lösungsmitteln sieht es blau aus. Es ist ein starkes Elektrophil und Oxidationsmittel. O3 ist eine 1,3-dipolare Verbindung. Damit kann es sehr leicht 1,3-dipolare Cycloadditionen eingehen: Schon bei tiefen Temperaturen bildet sich so ein Primärozonid, der aber instabil ist und durch eine Cycloreversion einen Aldehyd und eine Peroxospezies erzeugt, die erneut cyclisieren und ein Sekundärozonid erzeugen. Diese kann z.B. reduktiv aufgearbeitet werden und führt so zu Aldehyden. 70 Die reduktive Aufarbeitung z.B. mit Dimethylsulfid entzieht dem Sekundärozonid den positivierten Sauerstoff und liefert DMSO als stabiles Endprodukt: 5. Radikalische Additionen: 5.1. Intramolekulare Cycloaddition: Terminale -Bromalkene können mit Tributylzinnhydrid und AIBN regioselektiv zum 5exo-Produkt cyclisiert werden. das 6-endo-Produkt entsteht praktisch überhaupt nicht. Wie geht das? Das Tributylzinn-Radikal spaltet die C-Br-Bindung, und das entstehende C-Radikal greift intramolekular seine terminale DoBi an. Hier ist aus stereoelektronischen Gründen die 5-exoCyclisierung der 6-endo-Cyclisierung fast immer überlegen. (Baldwin-Regeln) Die entstehenden primären Alkylradikale greifen noch nicht umgesetztes Tributylzinnhydrid an und entreißen ihm ein H-Radikal; dabei entsteht neues Tributylzinn-Radikal für die Kettenreaktion. Ein Anwendungsbeispiel in Kombination mit der Oxymercurierung: 71 Hier greift Hg-Acetat selektiv die elektronenreiche DoBi im Enolether an; das Mercuriniumion wird vom Lömi Methanol geöffnet und führt zum dargestellten Zwischenprodukt. Dessen Reaktion mit NaBH4 führt nun aber nicht zum Alkan, sondern zum Cycloalkan. Der Grund: Die Reduktion des Organo-Hg-Acetats führt über ein Hg-Hydrid unter Erwärmen zum Alkylradikal, welches anschließend intramolekular die elektronenarme DoBi angreift und cyclisiert. Normalerweise entreißt es weiterem Organo-Hg-Hydrid das Wasserstoffatom und geht in ein Alkan über, während elementares Hg gebildet wird und ein weiteres Alkylradikal die Kette fortsetzt. 5.2. Intermolekulare Reaktionen: Allgemeines: Ein Radikal soll an eine terminale Doppelbindung anlagern und ein neues Radikal bilden: Hier können zwei Fälle auftreten: Entweder man hat eine elektronenarme DoBi mit einem tiefliegenden LUMO, welches mit dem SOMO eines nucleophilen Radikals reagiert (R= Donoren!). Oder man hat eine elektronenreiche DoBi mit einem hochliegenden HOMO, welches mit dem SOMO des elektrophilen Radikals reagiert (R = Akzeptoren!). 72 Beide Fälle werden beobachtet: a) Das t-Butylradikal ist nucleophil und greift daher gern die elektronenarme DoBi des Acrylnitrils an: b) Das Malonester-Radikal ist elektrophil und greift daher gerne die elektronenreiche DoBi des Enolethers an: Beide Reaktionen sind präparativ wichtig und werden oft durchgeführt. 73 F. Aromatische Substitutionen 1. Allgemeine Betrachtungen zu Aromaten: Definition (Hückel): Ein Aromat ist ein konjugiertes cyclisches -Elektronensystem mit 4n+2 -Elektronen. Prototyp Benzol: Hinweis auf die Delokalisation der -Elektronen: Die ortho-Disubstitutionsprodukte sind ununterscheidbar. Weitere rätselhafte Eigenschaften: Benzol reagiert nicht wie andere Alkene mit Bromwasser oder KMnO4; in Kristallstrukturen findet man gleiche Bindungslängen von 140 pm; das liegt genau zwischen der Länge einer C-C-Einfachbindung mit 154 pm und der einer C=C-Doppelbindung mit 133 pm. Offensichtlich trägt diese Bindungssituation zur Stabilität von Benzol bei. Wieviel stabiler ist Benzol als das theoretische Cyclohexatrien? Das kann man anhand von Hydrierwärmen herausfinden: Hydrierwärmen: Cyclohexen hat mit 28.6 kcal/mol Hydrierwärme weit mehr als ein Drittel der Hydrierwärme von Benzol (-49.8.kcal). Die Differenz von (3 x 28.6 =) 85.8 kcal –49.8 kcal = 36.0 kcal/mol entspricht der Resonanzenergie aus der Mesomeriestabilisierung. Diese besondere Stabilität von Aromaten ist der Grund dafür, daß bei den meisten Aromatenreaktionen das aromatische System erhalten bleibt oder am Schluß wiederhergestellt wird. Bsp.: Additionsreaktionen zerstören den Aromaten und sind äußerst selten. Die Verbindung X-Y führt viel leichter eine aromatische Substitution durch, wobei als Nebenprodukt H-Y gebildet wird. 74 MO-Betrachtung des Benzols (nach Frost und Mussulin): Bei vollständiger sp2-Konfiguration aller 6 C-Atome überlappen die übrigbleibenden 6 pOrbitale miteinander und bilden ein 6-Elektronensystem. Das beschreibt man mit 6 MO's 1-6. Sie entstehen, indem man 0,1,2 und 3 Knotenebenen durch das -System legt. Bei 1 und 2 Knotenebenen gibt es je zwei Möglichkeiten: die Knotenebene kann durch die C-Atome schneiden oder durch die C-C-Bindungen. Deswegen sind also 2 und 3 wie auch 4 und 5 entartet. Nach der Hund'schen Regel und dem Pauli-Prinzip füllt man nun von unten nach oben auf, und findet, daß die untersten 3 MO's doppelt besetzt werden können. Diese 3 bilden aber gerade die Gruppe der bindenden MO's, denn alle benachbarten p-Orbitale haben gleiches Vorzeichen. Nur die unbesetzten MO's 4-6 sind antibindend: Man kann zeigen, daß man allgemein die Aromaten als Polygone symmetrisch auf ihre Spitze stellen kann; die Abfolge der C-Atome ergibt die Energielage der MO's. Dabei liefern Cyclobutadien oder auch Cyclooctatetraen mit 4n -Elektronen Diradikale mit nichtbindendem Charakter. Interessanterweise sind solche Systeme so instabil, daß sie der Antiaromatizität ausweichen, z.B. durch Verzerrung des Cyclobutadiens zum Rechteck oder durch Faltung des vorher planaren Cyclooctatetraens. Nur Systeme mit 4n+2 -Elektronen sind planar, denn so entsteht maximale Überlappung und damit maximale Aromatizität. 75 Das hat Konsequenzen für das NMR: Ein äußeres angelegtes Magnetfeld H0 induziert nämlich im cyclischen -Elektronensystem des Aromaten einen Ringstrom. Dieser wieder erzeugt ein Gegenfeld, welches im Innern des Benzols dem äußeren Magnetfeld entgegenwirkt (Abschirmung = Hochfeldverschiebung von Protonensignalen, z.B. beim [18]Annulen sichtbar), in der Peripherie jedoch das äußere Magnetfeld verstärkt (Entschirmung = Tieffeldshift der aromatischen Protonen gegenüber olefinischen Protonen von 5-7 ppm!). Fassen wir noch einmal die Kriterien für Aromatizität zusammen: 1. Stabilisierung aus der Delokalisation von -Elektronen 2. Bindungslängenausgleich 3. Ringstrom 4. 4n+2 -Elektronen 5. Planare Geometrie Beispiele für Aromaten mit steigender Zahl von 4n+2 -Elektronen: 76 Neben Benzol gibt es noch eine große Anzahl von Heteroaromaten, die vor allem in vielen Naturstoffen und Medikamenten eine große Rolle spielen: Pyridin Thiophen Furan Pyrrol Achtung! Das freie Elektronenpaar am Heteroatom kann zum aromatischen System gehören (z.B. Pyrrol) und sitzt dann in einem pz-Orbital, oder es kann als lone pair in einem sp2Orbital vorliegen und wirkt dann basisch (Pyridin). 2. Elektrophile aromatische Substitution (SEAr): Besonders elektronenreiche Aromaten mit einer hohen Elektronendichte im -System reagieren leicht mit Elektrophilen nach folgendem allgemeinen Mechanismus: Energiediagramm: In einem schnellen vorgelagerten Gleichgewichtsschritt bildet sich der sogenannte Komplex, eine lose vor allem auf elektrostatischer Anziehung beruhende Anordnung des Elektrophils oberhalb des aromatischen -Systems. Im langsamen, geschwindigkeitsbestimmenden Schritt öffnet sich das aromatische System und bindet das Elektrophil kovalent (-Komplex). Das dabei entstehende Pentadienylkation ist immer noch resonanzstabilisiert, hat aber eine große Tendenz zur Rearomatisierung. Diese gelingt anschließend schnell, indem ein Proton eliminiert wird. 2.1. Substituierte Aromaten – Regioselektivität der Zweitsubstitution a) Elektronische Faktoren: Die Ladungsverteilung im -Komplex kann durch Mesomeriestrukturen qualitativ abgeschätzt werden und auch quantenmechanisch 77 berechnet werden. Sie konzentriert die positive Ladung vorwiegend auf die p- und oPositionen. Mesomere Grenzstrukturen: Ladungsverteilung im -Komplex: Das hat zur Folge, daß das Elektrophil in Donorsubstituierten (aktivierten) Aromaten bevorzugt in der p- und o-Position eintritt: Im Gegensatz dazu destabilisiert ein Akzeptorsubstituierter (desaktivierter) Aromat diese Positionen, und lenkt die Zweitsubstitution in die m-Stellung: Allgemein: Donorsubstituenten = OH, OR, NR2, NH2, SR, Ph, Alkyl Akzeptorsubstituenten = NR3+, C(=O)-R, C(=O)-X, CN, SO3H, NO2 Chamäleonsubstituenten: Diese haben einen +I und einen -I-Effekt (Bsp.: Chlormethyl) bzw. einen +M- und einen -I-Effekt (Bsp. Chlor-, Brom-). Hier überwiegt eher der starke induktive Effekt. Und wenn es mehr als einen Substituenten gibt? Dann können die beiden kooperativ (verstärkend) oder kompetitiv wirken (wer ist stärker?). Bsp.1 - Kooperative Wirkung (+Iund -M-Substituent in para-Stellung): Beide lenken ortho zum +I-Substituenten: 78 Bsp.2 - Kompetitive Wirkung (+I- und +M-Substituent in para-Stellung): Meist ist der mesomere Effekt stärker und setzt sich durch: b) Sterische Faktoren: Oft ist die o-Substitution durch die sterische Hinderung zwischen Substituent und der eintretenden Gruppe in o-Stellung stark benachteiligt. Das entsprechende C-Atom ist im -Komplex zudem sp3-hybridisiert. So läßt sich Toluol z. B. praktisch nur in p-Stellung acetylieren. c) Statistische Faktoren: Man muß natürlich zusätzlich berücksichtigen, daß für eine Zweitsubstitution nur eine p-Position, aber jeweils zwei o- und m-Positionen zur Verfügung stehen. a-c) Beispiel: Die Reaktion eines alkylsubstituierten Benzols mit Brom in Trifluoressigsäure läuft bei Raumtemperatur (RT) ab. In der Tabelle wird deutlich, wie bei höherem sterischen Anspruch des Erstsubstituenten der Anteil an p-Produkt zunimmt, obwohl dieses statistisch benachteiligt ist. 79 R= Me Et iPr tBu o-Produkt (%) 18 13 8 0 m-Produkt (%) 0 0 0 0 p-Produkt (%) 82 87 92 100 2.2. Wichtige elektrophile aromatische Substitutionen: a) Ar-Hal-Bindungsknüpfung (Halogenierung): Donorsubstituierte Aromaten (z.B. Anilin, Phenol, Acetanilid) reagieren bereits ohne Katalysatoren mit Chlor oder Brom: So kann man alle drei aktivierten Positionen substituieren. Bei nichtaktivierten Aromaten benötigt man einen Katalysator, der das Brommolekül polarisiert und Br+ erzeugt: Br2 + FeBr3 Br+---Br---FeBr3- So gelingt z. B. bei p-Nitrotoluol die regioselektive Monobromierung ortho zur Methylgruppe: Dabei kann hier mit einem anderen Reagenz auch die Methylgruppe bromiert werden. Dies funktioniert allerdings nach dem Mechanismus einer radikalischen Substitution und kann deshalb durch die Wahl der Reaktionsbedingungen gesteuert werden. Man sagt, es gilt die KKK/SSS-Regel: „Kälte und Katalysator führen zur Kernbromierung, Siedehitze und Sonne dagegen zur Seitenkettenbromierung.“ b) Ar-SO3H-Bindungsknüpfung (Sulfonierung): Mit konzentrierter (98%) oder sogar rauchender Schwefelsäure (SO3 in H2SO4, Oleum) kann das SO3H+-Kation elektrophil in Aromaten eingeführt werden: 80 Bei Naphthalin entsteht dabei zunächst das kinetisch kontrollierte Produkt, welches sich im Laufe der Zeit aber in das thermodynamisch stabilere umwandelt. Das ist nur durch die Reversibilität der Reaktion möglich; alle Schritte sind Gleichgewichtsreaktionen. Das kinetisch bevorzugte -Produkt weist im -Komplex 2 Grenzstrukturen mit intaktem Aromaten auf. (Nach dem Hammond-Postulat ist ein später Übergangszustand produktähnlich, so daß hier die Stabilisierung der Zwischenstufe auf den späten ÜZ übertragen werden kann). Das thermodynamische -Produkt ist dagegen weniger sterischer Spannung zwischen den beiden benachbarten -Positionen ausgesetzt. c) Ar-NO2-Bindungsknüpfung (Nitrierung): Nitroaromaten kann man mit mehreren Reagentien herstellen - sie alle liefern ein NO2+Kation: HO-NO2, O2N-O-NO2, NO2+BF4-. Nitriersäure ist besonders reaktiv: HNO3 + H2SO4 NO2+ + H3O+ + HSO4- Zur Vermeidung von Nebenreaktionen sollte man das Nitrierreagenz der Reaktivität des Aromaten anpassen. Einige Beispiele: Veratrol wird von wäßriger Salpetersäure nur einmal nitriert, dann ist der Aromat zu sehr desaktiviert: Toluol liefert mit Nitriersäure ein Gemisch von o- und p-Produkt im Verhältnis 2:1 (= Statistik). 81 Nitroniumtetrafluoroborat schafft es, Dinitrobenzol zum Trinitroderivat in 61% Ausbeute binnen 3 h umzusetzen, Nitriersäure liefert dagegen selbst nach 5 Tagen bei 110°C nur 45 %. d) Ar-Alkyl-Bindungsknüpfung (Friedel-Crafts-Alkylierung): Zur Einführung von Alkylsubstituenten müssen wir möglichst freie Carbeniumionen generieren. Wie gelingt das? - mit Alkylhalogeniden oder –sulfonaten und Lewissäuren - mit Alkoholen und Brønsted-Säuren - mit Olefinen und Brønsted-Säuren R-Cl + AlCl3 R-OH + H-X R+---Cl---AlCl3+ R---O(H)---H---X R-CH=CH2 + H+ R+ AlCl4R+ + H2O + X- R-CH+-CH3 Probleme: - Nach der Einführung der ersten Alkylgruppe wird der Aromat zusätzlich aktiviert, dadurch entsteht die Gefahr der Polyalkylierung. - Die Carbenium-Zwischenstufen können umlagern. Anwendungsbeispiele: Biphenyl wird selektiv p-tbutyliert; die sterisch anspruchsvolle o-Position wird nicht angegriffen. Nach Wasserabspaltung wird das tertiäre Carbeniumion entropisch günstig intramolekular angegriffen, und zwar p zum aktivierenden Methoxy-Substituent. 82 e) Ar-Acyl-Bindungsknüpfung (Friedel-Crafts-Acylierung): Säurechloride können noch leichter durch Lewissäuren aktiviert werden als Alkylhalogenide. AlCl3 zieht das Chloridanion so stark zum lewissauren Al-Atom, daß man fast von freien Acylium-Kationen sprechen kann: Auch Anhydride kann man so aktivieren: f) Ar-N=N-Bindungsknüpfung (Azokupplung): Diazoniumsalze sind schwache Elektrophile; deshalb reagieren sie nur mit reaktiven Aromaten wie Phenolaten und sekundären oder tertiären aromatischen Aminen. Dabei wird eine Azoverbindung gebildet. Die Diazoniumsalze stellt man klassisch über die Umsetzung von aromatischen Aminen mit salpetriger Säure in HCl her: Hier ist die Synthese des Azo-Farbstoffs Orange 1. Die Sulfonatfunktion gewährleistet ausreichende Wasserlöslichkeit, das Naphtholatanion die ausreichende Aktivierung des zu substituierenden Aromaten: Das delokalisierte -System dieses Farbstoffs (Farbträger = Chromophor) reicht von der elektronenschiebenden Hydroxygruppe (Farbverstärker = Auxochrom) zur elektronenziehenden Sulfonsäurefunktion. 83 g) gemischte Beispiele – Namensreaktionen: i) Formylierungen: Ar-H Ar-CHO Verschiedene Wege führen zu Formylkation-Äquivalenten; z.B. Gattermann-Koch: HCl + C=O + CuCl + AlCl3 Gattermann: HCN + HCl + AlCl3 (ZnCl2) „H-CO+“ H-C(=NH)-Cl (auch mit RCN) Vilsmeier: DMF und POCl3 reagieren miteinander unter Generierung eines ChloriminiumIons (Vilsmeier-Reagenz), welches nach Substitution zur Formylgruppe hydrolysiert wird. So kann z.B. Methoxyphenol p-formyliert werden (warum nicht p-zur Methoxygruppe?): Bischler-Napieralski: Säureamide werden mit POCl3 ebenfalls zu Chloriminiumsalzen umgesetzt und können einen bereits vorhandenen Benzolrest intramolekular angreifen; so entstehen Isochinoline (Alkaloide). ii) Chlormethylierung: Formaldehyd und HCl bzw. eine Lewissäure führen nach erfolgter Hydroxyalkylierung direkt weiter zu Benzylchloriden (Eintopfreaktion): 84 h) Ipso-Substitution: Wenn ein tertiärer Alkylrest oder noch besser ein Trialkylsilylrest am Aromaten sitzt, lenkt dieser den Angriff des Elektophils oft in eben diese (ipso)-Position, denn die in Nachbarschaft entstehende positive Ladung (eigentlich ein Pentadienylkation) wird durch Hyperkonjugation stabilisiert (beim Silicium durch das große -Bindungs-MO noch besser: >20 kcal/mol; Effekt des Si). So kann man z.B. m-TMS-Anisol selektiv in der eigentlich ungünstigen m-Stellung FriedelCrafts-acylieren: Die ipso-Substitution ist nicht nur an tButylresten, sondern auch an Sulfonsäuren möglich. In der Calixarenchemie macht man sich diese Reaktion bei der selektiven Dealkylierung der Calixaren-Grundkörper zunutze. Anschließend kann durch vielfältige SEAr-Reaktionen funktionalisiert werden: 3. Nucleophile aromatische Substitution über Meisenheimer Komplexe: Wegen des meist elektronenreichen Charakters von Aromaten sind nucleophile Reaktionen eher selten. Trotzdem gibt es mindestens drei verschiedene Wege, wie ein Nucleophil an 85 Benzolderivaten angreifen kann. Hier ist der direkte Angriff (Additions-EliminierungsMechanismus): Hierfür müssen zwei Voraussetzungen erfüllt sein: Erstens muß ein stark elektronenziehender –I-Substituent den Angriff des Nucleophils im ersten geschwindigkeitsbestimmenden Schritt erleichtern (am besten ist F); zweitens muß die negative Ladung des entstehenden Meisenheimer Komplexes durch –M-Substituenten (am besten Nitro) stabilisiert sein: Dafür ist es natürlich am besten, wenn der/die –M-Substituenten in o-/p-Position zur Abgangsgruppe stehen. Ein ganz wichtiges Anwendungsbeispiel ist das Sanger-Reagenz in der Peptidanalytik: 2,4Dinitrofluorbenzol reagiert nämlich mit der N-terminalen Aminogruppe eines Peptids unter Bildung des Meisenheimer Salzes und anschließender Abspaltung des Fluorid-Anions, so daß ein N-terminales (fluoreszierendes) 2,4-Dinitrophenylanilin entsteht. 4. Nucleophile aromatische Mechanismus): Substitution über Arine (Eliminierungs-Additions- Stehen in einem Aromaten H und X (= gute Abgangsgruppe, z.B. Bromid) direkt nebeneinander, so kann eine starke Base zunächst deprotonieren und HX abspalten. Nucleophile Addition des Nucleophils als H-B an diese Dreifachbindung ergibt direkt den substituierten Aromaten, oft als Gemisch der beiden Regioisomeren: 86 Anwendungsbeispiele: Chlorbenzol kann bei hoher Temperatur sogar von der relativ schwachen Base NaOH dehydrohalogeniert werden. Die überschüssigen Hydroxidionen greifen anschließend sofort das Arin an und führen zu Phenolen (das ist präparativ natürlich selten erwünscht). Auch intramolekular käßt sich die Reaktion führen: Das erste Äquivalent BuLi deprotoniert den Halgenaromaten,das zweite das aromatische Amin und nun kann das Imidanion intramolekular an die Dreifachbindung addieren: 87 G. Oxidationen 1. Allgemeine Betrachtungen: Oxidation bedeutet Elektronenentzug und ist stets mit einer Reduktion (Aufnahmen von Elektronen) gekoppelt. Das Oxidationsmittel wird selbst reduziert. Bsp.: R3N + Fe3+ R3N• + + Fe2+ Bei organischen Reaktionen gibt es eine formale Ableitung von Oxidationszahlen, die etwas anders funktioniert als in der Anorganik (Organikum S. 382): Regel: In jeder kovalenten Bindung rechnet man stets dem elektronegativeren Bindungspartner sämtliche Elektronen des Bindungselektronenpaars zu. Dann addiert man die Elektronenzahlen für jedes Atom und zieht diese Zahl von der Zahl der Valenzelektronen ab; so erhält man die positive oder negative Oxidationsstufe. Bei gleichen Bindungspartnern erhält jedes Atom die Hälfte aller Bindungselektronen. Bsp.: Methan: C bekommt alle Bindungselektronen = 8. Es hat 4 Valenzelektronen, minus 8 = Ox.stufe –4! In Ethan bekommt jedes C-Atom zwei der vier Bindungselektronen. In Ameisensäure behält das C-Atom nur die zwei Bindungselektronen aus der C-H-Bindung. So kann man in Reaktionen sofort Oxidationen und Reduktionen an der Zu- oder Abnahme der Oxidationszahlen feststellen: a) b) c) 2. Oxidationen an nichtfunktionalisierten Kohlenstoffatomen: Dies ist ein sehr anspruchsvolles, aktuelles Kapitel der organischen Synthesechemie. Erst im letzten Jahr wurden erste, vielversprechende Lösungen vorgestellt. Die Oxidierbarkeit der 88 Alkylgruppe wird allerdings wesentlich erleichtert, wenn in Nachbarschaft eine Doppelbindung oder ein Aromat stehen. So ist die Methylgruppe in Butan sehr schwierig selektiv zu oxidieren, dagegen im Acetophenon (CH-acide), Propen (Allylstellung) oder Toluol (Benzylstellung) recht einfach zu funktionalisieren. Beispiele folgen: a) Oxidation von p-Chlortoluol mit Salpetersäure zum Benzoesäurederivat. b) Oxidation von m-Methylpyridin mit Kaliumpermanganat zur Nicotinsäure. c) Oxidation von p-Butyltoluol mit Kaliumpermanganat zur Terephthalsäure. Hier wird gleichzeitig die C-C-Bindung gespalten (später Kap.4). d) Oxidation von 2-Alkenen mit Selendioxid zu Allylalkoholen. Der Mechanismus verläuft über eine sogenannte En-Reaktion (vgl. Diels-Alder, siehe Additionen) des Allylteils an die Se=ODoppelbindung, gefolgt von einer 2,3-sigmatropen Umlagerung (pericyclische Reaktion, siehe später unter dem Thema Umlagerungen). Der gebildetete Seleninsäureester wird hydrolytisch gespalten: 89 e) Oxidation von Alkylketonen mit Selendioxid zu 1,2-Diketonen. Diese Reaktion verläuft über den nucleophilen Angriff des Enols am SeO2, der gebildete Selenigsäure-Enolester geht wieder eine 2,3-sigmatrope Umlagerung ein und zerfällt schließlich in einer Fragmentierungsreaktion unter Bildung von elementarem Selen zum 1,2-Diketon. 3. Oxidation der C=C-Doppelbindung: Hier gehören wichtige Additionsreaktionen hin, wie z. B.: a) die Epoxidierung mit Persäuren b) die Ozonolyse mit Ozon c) die cis-Hydroxylierung mit Permanganat oder milder mit Osmiumtetroxid Alle diese Reaktionen finden sich schon beim Thema Additionsreaktionen. 4. Oxidation von Alkoholen zu Aldehyden, Ketonen oder carbonsäuren: a) Oxidation mit Chrom-Reagenzien: -Chromtrioxid CrO3 (Oxstufe +VI) Im sauren oxidieren CrO3-haltige Reagenzien sekundäre Alkohole glatt zu Ketonen. Hierfür wurden einige mildere Reagenzien entwickelt, die aber alle auf Cr+VI beruhen, z.B. H 2CrO4 / H2SO4 aq. = Jones Reagenz. Aldehyde werden von diesem starken Oxidationsmittel aber sofort weiteroxidiert zur Carbonsäure. 90 Der Mechanismus ist noch nicht vollständig geklärt; er beruht wahrscheinlich auf der intermediären Bildung eines Chromatesters, der anschließend fragmentiert und den Aldehyd und Cr-IV-Säure liefert. -modifizierte CrVI-Reagenzien: Mildere Varianten haben elektronenschiebende Substituenten am Chrom bzw. vermeiden den stark sauren Charakter des Oxidationsmittels: -Collins-Reagenz: CrO3 • py -Pyridiniumchlorochromat (PCC): -Pyridiniumdichromat (PDC): ist etwas milder als PCC und nicht sauer! Anwendungsbeispiele: In Methylenchlorid kann ein primärer Alkohol milde zum Aldehyd oxidiert werden, ohne dabei einen Enolether anzugreifen: In DMF dagegen geht die Reaktion durch bis zur Carbonsäure: 91 b) Swern-Oxidation: Sehr universell einsetzbar und milde ist die Swern-Oxidation, heute eines der besten Standardverfahren zur selektiven Oxidation von primären Alkoholen zu Aldehyden. Man benötigt die Reagenzienkombination: Oxalylchlorid (hochgiftig!), DMSO und Triethylamin in Methylenchlorid, und arbeitet meist bei –60°C. Der Mechanismus ist interessant und beginnt mit der Bildung der eigentlichen reaktiven Spezies aus Oxalylchlorid und DMSO, die gleichzeitig CO und CO2 eliminieren; das dabei gebildete Dimethylchlorsulfoniumkation wird SN-artig vom primären Alkohol angegriffen. Anschließend deprotoniert die milde Base Triethylamin am aciden C-H neben dem positiv geladenen S-Atom und generiert ein Schwefelylid. Dieses eliminiert als innere Base Dimethylsulfid als stabiles Nebenprodukt und hinterläßt meist in hoher Reinheit den Aldehyd. Da alle Nebenprodukte flüchtig sind, kann durch einfaches Eindampfen der Lösung aufgearbeitet werden. Anstelle von Oxalylchlorid wird oft auch Trifluoressigsäureanhydrid verwendet. Auch hier greift DMSO im ersten Schritt nucleophil an und generiert ein Dimethylsulfoniumion mit guter Abgangsgruppe: c) Oxidation mit TEMPO: Auch mit persistenten Radikalen kann man Alkohole zu Carbonylverbindungen oxidieren. Im ersten Schritt entreißt dabei hypochlorige Säure dem Aminoxid-Sauerstoff sein freies Elektron. Damit entsteht ein Dialkylnitrosylkation, welches nucleophil vom Alkohol angegriffen und zum Aminoxid-Zwitterion geöffnet wird. Hier wird 92 ähnlich wie bei der Cope-Eliminierung durch die innere Base Nitroxid eine synEliminierungsreaktion ausgelöst, die direkt zur Carbonylverbindung führt. Das dabei entstehende Hydroxylamin wird anschließend von hypochloriger Säure direkt zum Dialkylnitrosylkation reoxidiert, so daß das teure TEMPO-Edukt nur katalytisch eingestzt werden muß: d) Oxidation mit Perruthenaten: Nach dem Chemiker S. Ley wird die milde Oxidation von primären Alkoholen zu Aldehyden mit der Kombination aus katalytischen Mengen an Tetraalkylammonium-Perruthenaten und stöchiometrischen Mengen an NMethylmorpholin-N-oxid genannt: Bu4N+ RuVIIO4- (TBAP, Tetrabutylammoniumperruthenat) Pr4N+ RuVIIO4- (TPAP) NMO = Dabei wird ein Perruthenat-Ester gebildet, der ähnlich wie der Chromatester oxidativ fragmentiert: NMO oxidiert anschließend das katalytisch eingesetzte Ruthenium zurück zur Oxstufe +VII. e) Dess-Martin-Periodinan: Eine weitere sehr milde und effiziente Methode zur selektiven Oxidation primärer Alkohole zu Aldehyden macht von hypervalenten Iodverbindungen Gebrauch. Dabei wird SN-artig wieder ein Periodinan-Ester gebildet, welcher anschließend vom Nucleofug Acetat als milder Base angegriffen wird und eine reduzierte Iod-I-Spezies eliminiert: 93 f) Oxidation allylischer und benzylischer Alkohole: MnO2 mit der Oxstufe +IV eignet sich hervorragend zur Oxidation von allylischen oder benzylischen Alkoholen zu den entsprechenden Carbonylverbindungen. Die Reaktion verläuft im neutralen und bei RT und liefert aus primären Allylalkoholen -ungesättigte Aldehyde: 4. Oxidative Spaltun von C-C-Einfachbindungen (Glycolspaltung): Glycole (1,2-Diole) sind leicht über Epoxide herstellbar und können mit Natriumperiodat oder Bleitetraacetat oxidativ zu Carbonylverbindungen gespalten werden: Reagenzien: a) NaIO4, H2O, Methanol; b) Pb(OAc)4, CH2Cl2. Nach der Lemieux-RudloffVariante kann dabei das teure Periodat von stöchiometrischen Mengen an KMnO4 reoxidiert werden. Der Mechanismus verläuft, wie so oft, zunächst über die Bildung eines cyclischen Esters, der anschließend im Sinne einer [4+2]-Cycloreversion auseinanderfällt und direkt die Carbonylverbindung liefert, die bei cyclischen Diolen auch eine -Struktur haben kann: 94 H. Carbonyle 5. Allgemeine Betrachtungen: Die Carbonylgruppe von Reaktionsmöglichkeiten: Carbonylverbindungen verleiht ihnen mindestens zwei a) als Lewissäure: Nucleophile greifen am elektrophilen C+ an - nucleophile Additionen: b) als Brönsted-Säure: Basen deprotonieren das acide C-H in -Stellung - Enolatchemie: Beide Atome der Carbonylgruppe sind sp2-hybridisiert, d.h. auch die lone pairs des Sauerstoffs. Nucleophile greifen mit ihrem HOMO (z.B. ein freies sp3-Hybrid-AO) das LUMO der Carbonylgruppe an (natürlich das *-MO). Dabei fliegen sie von hinten im schrägen 105°-Winkel an (Bürgi-Dunitz-Trajektorie). Die Carbonylgruppe ist wegen ihrer Planarität viel reaktiver als eine CH2-X-Gruppe in SN2-Reaktionen! Im Energiediagramm wechselwirkt also das relativ niedrig sitzende HOMO des Nucleophils mit dem relativ hoch sitzenden LUMO der Carbonylgruppe. Ergo: Elektronenziehende Substituenten an der Carbonylgruppe erhöhen deren Elektrophilie; besonders schiebende Substituenten im Nucleophil erhöhen dagegen dessen HOMO-Energie und damit ebenfalls dessen Reaktivität. 95 2. Reaktivität von Carbonylverbindungen gegenüber Nucleophilen: a) sterische Einflüsse: Große -Substituenten erschweren besonders für sperrige Nucleophile den Anflug in der Bürgi-Dunitz-Trajektorie: kleines Nucleophil CN-: krel. = 1 0.67 0.03 großes Nucleophil BH4-: krel. = 1 0.006 0.0002 b) elektronische Substituenteneinflüsse: Induktiver Effekt Die Elektrophilie des Carbonyl-C-Atoms wird durch Substituenten mit Akzeptoreigenschaften erhöht (-I-Effekt). Das kann man z.B. an der Esterhydrolyse sehen: Substrat krel. = 1 >61 16000 Substituenten mit-Donoreigenschaften erniedrigen dagegen die Carbonyl-Elektrophilie das kann man z.B. am Übergang von Formaldehyd über Acetaldehyd zu Aceton sehen: Die Einführung von elektronenschiebenden +I-Substituenten (Methylgruppen) erniedrigt die Elektrophilie deutlich. Ergo: Ketone sind generell reaktiver als Aldehyde! Mesomerer Effekt -Donorsubstituenten (+M) in Carbonsäurederivaten erniedrigen ihre Reaktivitt gegenüber Nucleophilen - das kann man z.B. bei Estern und Amiden sehen: 96 Der einfach gebundene Ester-Sauerstoff schiebt Elektronen in die C=O-Doppelbindung; dadurch entsteht eine partielle DB, die für ihre Rotation ~12 kcal/mol benötigt. Analog wirken die Heteroatome mit freiem Elektronenpaaren in Carbonsäuren und Carboxylaten (-OH, -O-), Thioestern (-SR), Amiden (-NR2), Hydroxamsäuren (NH-OH), Hydraziden (NH-NH2) etc. In Abhängigkeit von der Stärke des +M-Effekts ergibt sich folgende Reihenfolge der Reaktivität: Carboxylate sind am unreaktivsten, Säurechloride am reaktivsten. c) Addition oder Substitution? i) Ketone und Aldehyde addieren gerne Nucleophile: Dabei kann das Nucleophil neutral oder anionisch geladen sein. ii) Ketene und Heterocumulene addieren ebenfalls gerne Nucleophile: 97 iii) Carbonsäure- und Kohlensäurederivate reagieren lieber unter Substitution: 3. Wie verläuft hier der Mechanismus? Es ist ganz anders als bei den SN-Reaktionen an gesättigten C-Atomen: Hier wird erst das Nucleophil addiert, dann die Abgangsgruppe eliminiert (Additions-Eliminierungs-Mechanismus). Nur in einigen Fällen kann die dabei durchlaufene tetraedrische Zwischenstufe isoliert werden (Boche et al.: N,N,N - S,N,N etc.): a) Reaktionen im protischen, nicht-sauren Milieu: Es entsteht durch Nucleophil-Addition eine tetraedrische Zwischenstufe, die je nach pH-Wert entweder schnell protoniert wird und anschließend X- und H+ verliert oder im basischen direkt X- eliminiert. b) Reaktionen über stabile Tetraeder-Zwischenstufen c) H+-katalysierte Reaktionen an C=O-Kohlenstoff: Nach Protonenaktivierung greift das Nucleophil am Carbonyl-C an, eliminiert H-X und verliert im letzten Schritt sein Nu-H-Proton. Jetzt zu praktischen Beispielen: 98 4. Hydrate, Acetale, Imine, Enamine: a) Bildung von Hydraten: In einem reversiblen Prozess entsteht das Hydrat; die Lage des Gleichgewichts hängt von der thermodynamischen Stabilität von Edukt und Produkt ab (thermodynamische Kontrolle). Nur bei reaktiven Carbonylverbindungen mit -I- bzw. -M-Substituenten liegt das Gleichgewicht auf der Produktseite. So liegt Aceton nur zu <0.1% als Hydrat vor, Chloral dagegen zu 100%. % Hydrat im Gleichgewicht: <0.1 58 Auch das Aminosäure-Nachweisreagenz Glyoxalsäureester. 100 Ninhydrin ist 100 (Chloral) ein Hydrat, genauso wie b) Bildung von Acetalen: (Acetale von Ketonen würden früher als Ketale bezeichnet). Die Acetalisierung ist ein sauer katalysierter Prozess; d.h., Acetale sind unter basischen Bedingungen stabil und werden deshalb oft als Schutzgruppen für die C=O-Doppelbindung verwendet. Der Mechanismus der Acetalisierung verläuft nach Protonenaktivierung über die Anlagerung eines Äquivalents an Alkohol zum protonierten Halbacetal und über dessen Protonierung und weitere AlkoholAnlagerung zum Vollacetal. Dies ist dann nur noch sauer spaltbar: 99 Bsp.: Cyclohexanon kann sehr leicht am Wasserabscheider mit Ethylenglycol acetalisiert werden; dabei werden katalytische Mengen an p-Toluolsulfonsäure gebraucht. Das Cyclohexanon-Ethylenglycol-Diacetal entsteht in >95% Ausbeute als Spiroverbindung. Verwendung von Orthoestern: Im sauren acetalisiert der Orthoester die meisten Carbonylverbindungen glatt und in hohen Ausbeuten. Das dabei gebildete Wasser reagiert mit weiterem Orthoester zu Alkohol und Ameisensäure und wird so effizient weggefangen. Dadurch verschiebt sich das Gleichgewicht auf die Produktseite. Normalerweise ist die Stufe des Halbacetals instabil; Ausnahme: bei intramolekularer Halbacetalbildung werden 5- und 6-gliedrige Ringe ohne große Ringspannung aufgebaut; sie sind über den sogenannten anomeren Effekt stabilisiert (später mehr bei der Zuckerchemie). Ein gutes Beispiel ist die Glucose. Sie liegt zu >99% in der Halbacetalform vor. Die dadurch entstehenden Epimere (ein unterschiedliches Stereozentrum) nennt man - und -Form. 100 Bildung von Thioacetalen: Ganz analog zu den normalen Acetalen bilden sich auch Thioacetale, z.B. über eine Lewissäure-Aktivierung. Sie werden als Schutzgruppen für Ketone verwendet und dienen anschließend zur C-C-Knüpfung unter Umpolung zu Acylanionen-Äquivalenten: Die Umpolung geschieht am Carbonyl C-H, welches zunächst natürlich stark positiv polarisiert ist. Im Thioketal dagegen ist es CH-acide und kann zum Carbanion deprotoniert werden (Corey-Seebach-Methode). Die abschließende Hydrolyse der Thioketale ist der unangenehmste Schritt der ganzen Sequenz und benötigt Hg2+-Salze: Exkurs: Acetale als Schutzgruppen und anomerer Effekt: Man verwendet gerne Aldehyde oder Ketone zur Diolschützung, die gezielt 6- bzw. 5-Ringe bilden. Das kann über die thermodynamische Kontrolle zum stabilsten Endprodukt erklärt werden: Aldehyde bilden gerne den stabilsten 6-Ring und legen den Alkyl- oder Arylrest in die equatoriale Position. Bei ketonen würde der zweite Alkylrest axial sitzen und damit ungünstige 1,3-diaxiale Wechselwirkungen eingehen, deswegen weicht das System auf den 5Ring aus: 101 Anomerer Effekt: In acetalischen Strukturen mit zwei Heteroatomen am selben C-Atom sind bestimmte räumliche Anordnungen besonders günstig. Das betrifft besonders die -Anomere bei Zuckern, die aufgrund der axialen Stellung des Substituenten eigentlich fast gar nicht auftreten sollten. Der sogenannte anomere Effekt ist ein stereoelektronisches Phänomen und wird meistens über eine günstige n-*-Überlappung erklärt. Das nichtbindende freie Elektronenpaar am Ring-Sauerstoff überlappt mit dem rückseitigen Orbitallappen im *Orbital der C-X-Bindung. Das bedeutet, daß gute +M-Substituenten mit nucleophilen und deswegen angehobenen HOMO's am besten mit guten -I-Substituenten und deswegen abgesenkten *-LUMO's wechselwirken und die stärksten anomeren Effekte ergeben: Bei Glucose findet sich daher im Mutarotations-Gleichgewicht neben Spuren der offenkettigen Aldehydform 63% -Form und immerhin 37% -Form! c) Bildung von Iminen: Primäre Amin reagieren mit Carbonylverbindungen über die Stufe des Halbaminals (besser: N,O-Acetals) zu Iminen (bei aromatischen Carbonylen: Schiff'’sche Basen): Bsp: 102 In der thermodynamisch kontrollierten Reaktion entsteht im Gleichgewicht natürlich das sterisch günstigere E-Isomer. d) Bildung von Enaminen: Bei der Reaktion von sekundären Aminen mit Carbonylverbindungen fehlt in der Zwischenstufe ein Proton am Stickstoff zur Eliminierung, und dafür wird das Proton am -CAtom abgespalten. Es entsteht ebenfalls im Gleichgewicht das thermodynamisch stabilere Enamin (regioselektiv): Bsp.: Die Reaktion von Cyclohexanon mit Morpholin gelingt unter Protonenkatalyse am Wasserabscheider in guter Ausbeute: e) Reaktionen mit anderen N-Nucleophilen: In analoger Form kann man mit Carbonylverbindungen eine ganze Reihe von N-Nucleophilen umsetzen. Bei substituierten Hydroxylaminen muß das Stickstoffatom die OH-Gruppe nucleophil eliminieren. Das gebildete Hydroxidanion deprotoniert anschließend die saure OHGruppe am Iminiumion und generiert ein Nitron (1,3-Dipol). 103 Reaktionen mit Dicarbonylverbindungen: Wichtige Methoden zur Synthese von Stickstoffheterocyclen beruhen auf nucleophilen Additionen von Aminen an Dicarbonyle: Bsp. 1: Chinoxaline entstehen durch doppelten Angriff von o-Phenylendiaminen an 1,2Diketone: Bsp. 2: 3,5-Dialkylpyrazole entstehen durch doppelten Angriff von Hydrazin an 1,3-Diketone: Bsp. 3: 3,5-Dialkylisoxazole entstehen durch doppelten Anmgriff von Hydroxylaminen an 1,3-Diketone: Bsp. 4: Pyrrole entstehen durch doppelten Angriff von Ammoniak an 1,4-Diketone: (Lit.: T. L. Gilchrist, Heterocyclenchemie, VCH, Weinheim 1995) 5. Derivate von Carbonsäuren Reaktivitätsreihenfolge (zur Erinnerung): 104 a) Herstellung von Carbonsäurechloriden: Carbonsäuren kann man direkt in ihre Säurechloride überführen. Dazu verwendet man üblicherweise Thionylchlorid SOCl2 oder das reaktivere, aber auch giftigere Oxalylchlorid (COCl)2. Der basenkatalysierte und basenfreie Mechanismus wurde schon behandelt. b) Herstellung von Carbonsäureestern: i) Säurekatalysierte Veresterung von Carbonsäuren mit Alkoholen: Mechanismus (AAc2 = Säurekatalysierte Acylknüpfung mit Reaktionsgeschwindigkeitsgesetz zweiter Ordnung): Da alle Schritte dieser Reaktion reversibel sind, herrscht thermodynamische Kontrolle. Das Reaktionsprodukt wird also meist unter Verwendung eines Alkohol-Überschusses als Lösungsmittel gewonnen. Bsp.: Auch azeotrop kann man übrigens das Reaktionswasser entfernen, wenn man als Solvens z.B. Chloroform nimmt. ii) aus Säurechloriden bzw. Anhydriden: Das Amin ist in diesem Fall sterisch gehindert, kann also nicht als Acylierungskatalysator wirken, sondern entfernt lediglich das Proton aus der tetraedrischen Zwischenstufe: 105 iii) aus Säurechloriden mit Pyridinderivaten: Jetzt ist das Amin nucleophil und bereitet die gute Abgangsgruppe Pyridin vor, die anschließend erneut nucleophil am Säurechlorid angreift. Noch besser fungiert das hochreaktive p-Dimethylaminopyridin (DMAP). Bsp.: iv) Veresterung mit Diazomethan: Diazomethan deprotoniert im ersten Schtritt die Carbonsäure; anschließend greift das Carboxylatanion nucleophil am Diazonium-C-Atom an und eliminiert die gute Abgangsgruppe Stickstoff: Achtung: Diazomethan ist hochgiftig und sehr explosiv!!! v) mit DCC (Dicyclohexylcarbodiimid): DCC wird auch von der wenig nucleophilen Carbonsäure angegriffen und bereitet seinerseits im Zwischenprodukt die gute Abgangsgruppe Dicyclohexylharnstoff vor, die beim 106 anschließenden Angriff des Alkohols freigesetzt wird. DCU ist schwerlöslich und fällt aus. DCC ist aber noch wichtiger bei der Amidsynthese (s.u.) vi) Veresterung nach Mukaiyama: 2-Chloro-N-methylpyridiniumsalze werden ebenfalls von Carbonsäuren nucleophil angegriffen und eliminieren das Chlorid. Damit bereiten sie die gute Abgangsgruppe NMethylpyridon vor, die anschließend beim Angriff des Alkohols freigesetzt wird: vii) Veresterung nach Yamaguchi: Besonders gut für Lactone ist das hochreaktive Säurechlorid der 2,4,6-Trichlorbenzoesäure, denn es bildet mit der Carbonsäure leicht ein gemischtes Anhydrid, welches am Aromaten so sperrig ist, daß es automatisch vom Alkohol am richtigen Carbonyl-C-Atom angegriffen wird und das stabile Trichlorbenzoation eliminiert: c) Herstellung von Carbonsäureamiden: i) aus Carbonsäurechloriden: 107 ii) aus Carbonsäureestern: Hier ist ein Überschuß Amin auch nicht nötig, weil die Rückreaktion unwahrscheinlich ist, denn das Amid ist viel weniger reaktiv als ein Ester. Bsp.: Pentafluorphenylester werden gerne zur Amidknüpfung verwendet (Peptidsynthese): iii) Peptidsynthese mit DCC: Wenn eine Aminosäure N-geschützt ist, kann selektiv an der Carbonsäuregruppe verlängert werden. Dafür benutzt man gerne Kupplungsreagentien wie DCC, die die Carbonsäuregruppe aktivieren, damit im zweiten Schritt ein Aminosäureester nucleophil angreifen kann und die Peptidbindung knüpft: Achtung: In der reaktiven Zwischenstufe besteht die Gefahr der Racemisierung, besonders, wenn mit der Hilfsbase ein Oxazolinon gebildet wird! 108 6. Hydrolyse von Carbonsäurederivaten: a) Verseifung: Die basenkatalysierte Verseifung produziert Carboxylate (Seifen) über den BAc2-Mechanismus (Basenkatalysierte Acylspaltung mit Reaktionsgeschwindigkeitsgesetz zweiter Ordnung): b) Saure Hydrolyse: Diese Reaktion ist einfach die Umkehr der sauer katalysierten Veresterung (AAc2). c) Esterspaltung sterisch anspruchsvoller Ester (AAl1): t-Butylester werden meist mit Trifluoressigsäure milde gespalten. Dabei greift das Proton den basischsten Sauerstoff in der Carbonylgruppe an, und die Elektronen klappen in seine Richtung, so daß das t-Butylkation übrigbleibt, welches entweder ein Proton eliminiert oder Wasser addiert. 7. Nucleophile Addition metallorganischer Verbindungen an die Carbonylgruppe: a) Herstellung metallorganischer Verbindungen (Mg / Li): i) Alkylmagnesium-Verbindungen (Grignards): 109 Über einen radikalischen Mechanismus greift das Mg-Atom in zwei ElektronentransferSchritten am Alkylhalogenid an. dabei wird intermediär ein Alkylradikal gebildet, wlches auch spontan dimerisiert (Nebenprodukte). Die Reaktivität der Alkylhalogenide steigt vom Chlorid zum Iodid an (nur mit sehr reaktivem Mg können Alkylfluoride Grignardreagentien bilden). ii) Organolithium-Verbindungen: Sie können entweder durch direkte Metallierung oder auch durch den Brom-LithiumAustausch gebildet werden. Im letzten Fall geht die Reaktion über At-Komplexe. b) Umsetzung mit Ketonen und Aldehyden: Aldehyde bilden mit Metallorganylen sekundäre Alkohole, Ketone tertiäre Alkohole: c) Umsetzung mit Säurederivaten: i) mit Carbonsäureestern: Angriff des Metallorganyls an einem Ester führt zu einer tetraedrischen Zwischenstufe, die Alkoholat eliminiert und zum Keton wird. Dieses ist aber noch reaktiver als der Ester und addiert sofort ein weiteres Äquivalent Metallorganyl. Dabei entsteht ein tertiärer Alkohol: 110 Bsp.: Das Lacton wird geöffnet und als Keton-Zwischenprodukt erneut methyliert: ii) mit Amiden: Monoalkylamide haben ein acides H und werden deprotoniert. Das Amidenolat ist aber zu basisch und kann nicht von weiterem Metallorganyl angegeriffen werden: Anders bei Dialkylamiden oder noch besser beim Weinreb-Amid: Hier ist das Amid aus Hydroxylamin gebaut und bietet mit dem Alkoxyrest eine weitere Koordinationsstelle für das Metallkation, nachdem das Metallorganyl einmal angegriffen hat. Über ein stabiles tetraedrisches Zwischenprodukt entsteht bei der Hydrolyse ein Keton! Bsp.: Heute weiß man allerdings, daß auch die tetraedrischen Zwischenstufen anderer Dialkylamide stabil sind; es scheint also doch eine Stabilisierung des Übergangszustands zu sein, die die Weinrebamide so reaktiv und sauber in ihrer Monoalkylierung macht. iii) mit Carbonsäuren: Mit Grignardreagentien findet lediglich Deprotonierung statt; mit OrganolithiumVerbindungen erhält man jedoch Ketone: Dabei entsteht hier ein stabiles doppelt deprotoniertes Hydrat, welches gegen einen weiteren nuceophilen Angriff immun ist. Saure Aufarbeitung liefert das Keton. 111 Bsp.: iv) mit CO2 (Carboxylierung): CO2 ist ebenfalls ein starkes Elektrophil und liefert bei der Reaktion mit Carbanionen sofort Carboxylate: v) mit Nitrilen: Genauso können Nitrile mit Carbanionen reagieren. Es entsteht zunächst ein metalliertes Imin, welches mit Säure zum Keton hydrolysiert: Bsp.: 8. Enolate von Carbonylverbindungen als Nucleophile: a) Allgemeines: Je nach Substituent X spricht man von unterschiedlichen Enolaten, wenn CH-acide Carbonylverbindungen deprotoniert werden: 112 X -H -Alkyl / -Aryl -O-Alkyl / -O-Aryl -NR1R2 Name Aldehyd-Enolat Keton-Enolat Ester-Enolat Amid-Enolat Sperrige Basen sind für die Enolatbildung ideal, denn so wird der direkte Angriff des Metallorganyls auf die C=O-Doppelbindung verhindert. Beispiele sind: KOtBu NaH, KH Natriumamid, LDA Alkyllithium (n-BuLi, t-BuLi) Wichtig: Die Basenstärke wird der Acidität des Protons an der CH-aciden Verbindung angepaßt! Der pKa der Base muß mindestens 1-2 Größenordnungen über dem der CH-aciden Verbindung liegen. Eine Alternative zur vollständigen Deprotonierung sind irreversible Reaktionen, bei denen gasförmige Produkte entstehen, z.B. die Deprotonierung mit Hydriden oder Amide. Bsp.: R-H + NaH R-Na + H2 ↑ R-H + Li-NH2 R-Li + NH3 ↑ b) Ambidente Enolate: Molekülorbitale: Das Enolation ist dem Allylanion vergleichbar. Während aber im Allylanion eine symmetrische Verteilung der Elektonendichte vorliegt, polarisiert das Sauerstoffatom die MO's im Enolation stark. Das hat zur Folge, daß aufgrund der hohen Elektronegativität von O im ψ1 der Koeffizient am O sehr groß, der an C-2 aber sehr klein ist. Im nächsthöheren ψ2 (dem HOMO) dagegen ist es umgekehrt, wenn auch weniger dramatisch: hier trägt der Sauerstoff den kleineren Koeffizienten als das C-2: 113 Das aber hat Folgen: Harte Elektrophile, die eine ionische Wechselwirkung suchen, finden in der Summe über die besetzten MO's ψ1 und ψ2 die höchste Elektronendichte am Sauerstoff und greifen auch dort an (Bsp.: Protonen, Carbeniumionen). Weiche Elektrophile (weiche Säuren) mit einem tiefliegenden LUMO können am besten mit dem C-2 im Enolat-HOMO überlappen und greifen zur Knüpfung einer kovalenten Bindung bevorzugt dort an (Bsp.: Alkylhalogenide, Aldehyde, Ketone etc.) Das bedeutet C- vs. O-Alkylierung! Die Enolat-Grenzformel ist generell besser, denn die negative Ladung ist am elektronegativeren Sauerstoff viel besser stabilisiert. Die Metall-C--Bindung ist entsprechend instabiler als die Metall-O--Bindung. Regel 1: Je freier das Enolation ist (z. B. durch eine bessere Solvatation des Gegenkations), desto mehr O-Alkylierungsprodukt beobachtet man: HMPT solvatisiert in diesem Fall das Kaliumion viel besser als THF. Regel 2: Je weicher das Elektrophil ist (HSAB-Prinzip), desto mehr C-Alkylierung beobachtet man: 114 X= Cl Br I 46:54 33:67 <1:99 Ein weiches Elektrophil hat ein niedriges LUMO, macht also mit Carbanionen bindende Wechselwirkungen mit hohem kovalenten Charakter. Die Weichheit nimmt bei den Halogenen von Cl nach I zu. Insgesamt sind harte Abgangsgruppen die Sulfate und Tosylate, weiche vor allem Iodide. Zunehmende Weichheit Auch Trialkylsilylchloride sind hart (später mehr). Regel 3: Bei intramolekularen Reaktionen bestimmen stereoelektronische Faktoren die Angriffsrichtung. Nach J. E. Baldwin (Chem. Commun. 1977, 233) findet die C-2Alkylierung eines Enolats orthogonal zur Substituentenebene statt, indem das Elektrophil am doppelt gefüllten pz-Orbital angreift. Harte Elektrophile dagegen greifen den Sauerstoff an seinen beiden sp2-lone pairs an, d.h. in der Substituentenebene! Bei Cyclisierungen ist bei weichen Abgangsgruppen grundsätzlich die C-Alkylierung bevorzugt. Kann aber das Nucleophil (das Enolation) das Alkylhalogenid nicht genau von hinten angreifen (SN2), so weicht die Reaktion auf die O-Alkylierung aus, die ja in der Ebene stattfindet. Bsp.1: Bsp.2: Regioselektivität der Enolatbildung: Thermodynamische vs. kinetische Kontrolle 115 Bei unsymmetricshen Ketonen u.ä. hat man grundsätzlich die Möglichkeit, zwei regioisomere Enolate zu generieren, die sich vor allem im Substitutionsgrad der Doppelbindung unterscheiden. Diese Enolatbildung läßt sich steuern. Wenn man das stabilere der beiden haben will (das ist aufgrund der Saytzew-Regel das höhersubstituierte), muß man thermodynamische Kontrolle ausüben. Das heißt also Gleichgewichtsbedingungen schaffen, z.B. durch hohe Temperaturen, protische Lösungsmittel und lange Reaktionszeiten. Will man das niedriger substituierte Enolat erzeugen, so muß man kinetische Kontrolle ausüben, das heißt also eine irreversible Reaktionsführung durch niedrige Temperaturen, aprotische Lösungsmittel, langsames Zutropfen des Ketons zum LDA. Hier entsteht bevorzugt das Produkt mit der geringsten Aktivierungsenergie, also das im Angriff sterisch weniger gehinderte. Problem: Lösung: c) Alkylierungen von -Dicarbonylverbindungen: c.1. Malonsäureester-Synthese: Malonsäureester haben ein sehr acides Proton, das schon mit Alkoholaten vollständig entfernt werden kann. Das Ester-Enolat greift Alkylhalogenide an und wird in -Stellung alkyliert: 116 Achtung: Das Produkt ist immer noch CH-acide und kann mit dem Esterenolat equilibrieren. Das nun entstehende alkylierte Esterenolat ist allerdings weniger reaktiv als das ursprüngliche Esterenolat und reagiert normalerweise nicht unter Zweitalkylierung, außer bei sehr reaktiven Elektrophilen: Anschließend kann man durch saure Verseifung beide Estergruppen spalten; die dabei entstehende -Ketosäure decarboxyliert leicht im Sinne einer Retro-En-Reaktion und liefert eine verlängerte Essigsäure: c.2. Acetessigester-Synthese (analog): Genauso kann man auch Acetessigester alkylieren und nach Verseifung des Esters einer Ketonspaltung unter Decarboxylierung zuführen: Der Vorteil dieser Varianten der Ketonalkylierung ist die regioselektive Deprotonierung; die ist nämlich bei Methylethylketon äußerst schwierig, bei Acetessigester dagegen ein Kinderspiel: 117 c.3. Aldoladdition: Allgemeines: Aldehyd- und Keton-Enolate reagieren mit weiterer Carbonylkomponente unter Aldolbildung, der sich oft eine Kondensation anschließt: Aldolisierung: Darunter versteht man die basen- oder säurekatalysierte Selbstkondensation von Aldehyden und Ketonen im Sinne der Aldolkondensation. Falls aber Enolat- und Carbonylkomponente aus verschiedenen Vorläufern stammen, hat man eine gekreuzte Aldolreaktion; die ist schwieriger zu realisieren. Problem: Chemoselektive Deprotonierung der gewünschten Verbindung. Wie schafft man das? 1. Es gibt nur eine acide Stelle. Wenn nun die Carbonylgruppe der Enolatkomponente auch noch sterisch abgeschirmt wird, so daß sie weniger elektrophil reagiert, fungiert nur die andere Komponente als Elektrophil: 2. Das Enolat wird zuerst unter kinetischen Bedingungen erzeugt (irreversible Reaktion bei tiefen Temperaturen in aprotischen Lösungsmitteln). Dann wird bei tiefen Temperaturen das Elektrophil zugegeben und reagiert direkt mit der Enolatkomponente ab. 3. Reaktion über Silylenolether (Mukaiyama-Variante): Das harte Elektrophil TMS-Cl wird vom Enolat unter O-Alkylierung angegriffen und macht den Silylenolether. Dieser wird mit der Carbonylkomponente und equimolaren Mengen der Lewissäure TiCl4 umgesetzt. Dabei aktiviert die Lewissäure einerseits die Carbonylgruppe und spaltet mit dem Chloridion andererseits gleichzeitig milde den Silylenolether. Das freigesetzte Enolat greift in situ an und macht die gerichtete Aldolreaktion. 118 c.4. Mannich-Reaktion: Die Mannich-Reaktion funktioniert analog der säurekatalysierten Aldol-Reaktion, nur daß hier das Elektrophil ein Iminiumsalz ist: Das Enol greift also bei leicht saurem pH das Iminiumsalz aus Formaldehyd und einem sekundären Amin an; dabei entsteht eine Mannich-Base, d.h. ein -Aminoketon. Bsp.: Im letzten Schritt wird das Hydrochlorid der Mannich-Base mit Kalilauge deprotoniert zur freien Mannich-Base. c.5. Reaktionen über Aza-Enolate: Imine können mit starken Basen deprotoniert werden. Die dabei gebildeten Aza-Enolate sind hochreaktiv und greifen alle möglichen Elektrophile an. Besonders wichtig sind metallierte Hydrazone, denn über diesen Weg hat Enders seine gesamte SAMP/RAMP-Chemie aufgebaut (chirale Hydrazone zur enantioselektiven Alkylierung von Carbonylderivaten): 119 c.6. Reaktionen zwischen Ketonen und Säurederivaten: In Anwesenheit eines Esters ist die chemoselektive Deprotonierung eines Ketons leicht möglich (pKa: Keton < pKa: Ester): Das nach Deprotonierung des Endprodukts entstehende delokalisierte Cabanion ist stabil und muß zur Freisetzung des Produkts mit äquimolaren Menge Säure umgesetzt werden. Je nach Substituent liegt die resultierende -Dicarbonylverbindung auch als Enol-Tautomer vor. i) Falls aber der Ester durch eine -Ketogruppe zusätzlich aktiviert ist (wie im -Ketoester), fungiert er natürlich selbst als Enolatkomponente. Eine solche Reaktion einer 1,3Dicarbonylverbindung mit einem Keton oder Aldehyd nennt man KnoevenagelKondensation: Bsp.: Dies ist also eine Erweiterung der Acetessigester-Chemie. ii) Darzens‘ Glycidester-Synthese: -Chlorester haben eine erhöhte CH-Acidität und lassen sich leichter deprotonieren. Die Reaktion mit einem Epoxid führt zunächst über dessen nucleophile Ringöffnung. Das gebildete Alkoholatanion greift seinerseits nucleophil am Chlorid an und wirft es SN2-artig hinaus (NGE): 120 iii) Reformatzky-Reaktion: -Bromester reagieren mit elementarem Zink zu einer Grignardanalogen Organozinkverbindung, welche nicht sich selbst, sondern nur reaktivere Ketone und Aldehyde angreift: iv) Dieselbe Reaktionsfolge kann man auch durch Erzeugung des Esterenolats bei tiefen Temperaturen erzwingen. Anschließend erfolgt bei –78°C die Zugabe des Aldehyds oder Ketons: c.7. Reaktionen zwischen Carbonsäurederivaten: i) Claisen-Kondensation: Ester können mit starken Basen deprotoniert werden und mit sich selbst reagieren; zum Schluß wird angesäuert: Diese Reaktion kann entropisch günstiger auch intramolekular gefahren werden; dann heißt sie Dieckmann-Kondensation: 121 c.8. Michael-Addition: -ungesättigte Carbonylverbindungen wie Enale und Enone können prinzipiell in Art einer 1,2-Addition an die Carbonylgruppe oder aber auch nach Art einer 1,4-Addition an den Kohlenstoff angegriffen werden. Im letzten Fall wirkt sich die positive Teilladung aus, die durch das Vinylogie-Prinzip (Mesomerie) in Stellung anliegt. Meist kann man die 1,2Addition mit reaktiveren Nucleophilen kinetisch kontrolliert erzwingen, während die 1,4Addition unter thermodynamischer Kontrolle abläuft. Bsp.: -78°C: +25°C: 63% 0% : : 7% 85% Bei thermodynamischer Produktkontrolle, also bei entsprechend hohen Temperaturen, erhält man in derselben Reaktion vorwiegend das Produkt der 1,4-Addition. Allgemein gilt: Weiche CH-acide Verbindungen reagieren bevorzugt mit -ungesättigten Aldehyden, Ketonen und Nitrilen (HSAB-Prinzip): 122 Exkurs Cuprat-Addition: Weiche Metallorganyle sind solche mit hohen kovalenten Bindungsanteilen. Hier haben sich besonders die Cuprate bewährt. Während Grignardverbindungen mit Enonen unter 1,2-Addition reagieren, erhält man bei der Umsetzung mit Cupraten meist in hohen Ausbeuten das Produkt der 1,4-(Michael)-Addition. Der Mechanismus verläuft nach der Aktivierung der Carbonylfunktion über die Lewis-Säure Lithiumkation zunächst unter Metall-Addition an der Doppelbindung; danach erfolgt eine reduktive Eliminierung der Organokupfer-Spezies unter Freisetzung des alkylierten Enolats, welches abschließend protoniert wird: Cuprate sind also weiche Reagentien und bevorzugen die Michael-Addition; Grignards und Alkyllithiumverbindungen sind dagegen harte Basen und bevorzugen die 1,2-Addition. Eine wichtige Michael-Addition ist die Umsetzung von Enonen mit Enolaten. Wenn hier im ersten Schritt Methylvinylketon (MVK, Vorsicht: polymerisiert leicht!) verwendet wird, kann nach Protonierung und erneuter Deprotonierung im selben Reaktionsgefäß eine Aldolkondensation angeschlossen werden, die zur Ringanellierung führt. Diese als RobinsonAnellierung bekanntgewordene Reaktion wird auch heute noch gern für den Aufbau des ABRingsystems von Steroiden genutzt: 123 i) Enamine in der Michael-Addition: Auch reaktive Enamine können mit reaktiven Michael-Akzeptoren unter 1,4-Addition reagieren; so werden z. B. -Ketonitrile zugänglich: c.9. Umsetzung von Ketonen und Aldehyden mit Phosphor- und Schwefel-Yliden: Hier sind wichtige Olefinierungs und Dreiring-Synthesen versteckt! Phosphor-Ylide macht man meist durch Reaktion von Phosphinen mit Alkylhalogeniden und anschließender Deprotonierung der Phosphoniumsalze: 124 Analog funktioniert die Herstellung von Schwefel-Yliden durch die Alkylierung von Thioethern und anschließende Deprotonierung mit starken Basen: i) Wittig-Reakion: Phosphor-Ylide können mit Ketonen und Aldehyden unter Bildung eines Betain-Addukts reagieren. Dieses cyclisiert zum nachgewiesenen Oxaphosphetan, welches anschließend im Sinne einer Cycloreversion zum Alken und Phosphinoxid-Nebenprodukt geöffnet wird: Bei substituierten Phosphor-Yliden können E-und Z-Alkene gebildet werden. Diese Reaktion kann man in gewissen Grenzen durch die elektronische Natur der Substituenten steuern: a) nicht stabilisierte Ylide: sind solche mit einfachen Alkylsubstituenten, die das Carbanion nicht stabilisieren. Hier wird meist das Z-Produkt gebildet. Das kann über die kinetische Steuerung erklärt werden, denn hier ist auch der erste Schritt der Addition an die Carbonylverbindung schnell; es wird der günstigste Übergangszustand durchlaufen. 125 Bsp.: b) stabilisierte Ylide: sind solche mit elektronenziehenden Substituenten, besonders gut sind –M-Substituenten. Hier wird meist das E-Produkt gebildet. Dies wird im allgemeinen über die thermodynamische Kontrolle erklärt: Es entsteht im langsamsten ersten Schritt, der in diesem Fall reversibel ist (Förderung durch hohe Temperatur, Salzusatz sowie protische Lösugsmittel) das stabilste Betain, in dem natürlich die Substituenten möglichst weit voneinander entfernt stehen: Bsp.: ii) Horner-Wadsworth-Emmons-Variante: Wenn aber stabilisierte Ylide mit Ketonen umgesetzt werden, findet man oft keine vollständige Reaktion. Man benötigt in diesem Fall etwas reaktiveres als ein Ylid, in dem ja immer auch die neutrale Grenzformel einen Beitrag hat; am besten wäre etwas permanent anionisches. Das haben Horner et al. mit Phosphonatanionen gefunden: 126 Zunächst wird diesmal ein Trialkylphosphit mit einem Alkylhalogenid umgesetzt, und das Phosphoniumsalz wird anschließend direkt vom Bromidanion dealkyliert. Das entstehende Phosphonat ist CH-acide und kann milde deprotoniert werden. Dieses Carbanion aber ist immer noch reaktiv genug, um Ketone nucleophil anzugreifen. Erneut wird das stabilere Alkoxid gebildet, welches im folgenden stereospezifisch über das Oxaphosphetananion (hier ein Phosphoran mit 5-bindigem P) zum Alken zerfällt. Das andere Nebenprodukt ist hier ein wasserlösliches Phosphatanion, welches leicht durch Ausschütteln abgetrennt werden kann. Über diese Variante erhält man also vorwiegend E-Produkte. iii) Schwefel-Ylide: Schwefel-Ylide geben mit Ketonen Epoxide, nachdem der erzeugte Alkoholat-Sauerstoff intramolekular Dialkylsulfid herausgeworfen hat: d) Reduktion von Carbonylverbindungen 1. Reduktion von Aldehyden und Ketonen: Eine wichtige Reaktion von Carbonylverbindungen ist natürlich auch die Rückreaktion zu Alkoholen oder gar Alkanen im Sinne der Oxidationsumkehr, d.h. also die Entfernung von Sauerstoff (Deoxygenierung) oder die Anlagerung von Wasserstoff (Hydrierung). 127 i) Reduktion zu Alkoholen: Diese Transformation kann durch Hydridlieferanten leicht erreicht werden. Allerdings sind diese oft sehr reaktiv und können auch zu Nebenreaktionen führen. Weniger reaktiv als Lithiumaluminiumhydrid (in THF, Ether) ist z.B. das gerne verwendete Natriumborhydrid (in THF oder auch Ethanol). Mechanismus: Auch hier ist wieder zunächst die Komplexierung der Carbonylfunktion durch das Metallkation wichtig (so ist z.B. LiBH4 ein stärkeres Reduktionsmittel als NaBH4. In Ethanol kann auch das protische Solvens diese Aufgabe übernehmen). Dann überträgt das komplexe Hydrid ein echtes Hydridanion und lagert sich anschließend an das gebildete Alkoholat an. Da es immer noch negativ geladen ist, kann es erneut ein Hydridion übertragen, bis alle verbraucht sind. Während der Reaktion treten also mehrere unterschiedliche Reduktionsmittel als Zwischenstufen auf: Genauso verläuft die Reaktion mit LAH. Chemoselektivität: Ein Aldehyd ist für einen nucleophilen Angriff reaktiver als ein Keton, wird also von milden Hydridüberträgern bevorzugt angegriffen (kinetische Kontrolle): ii) Deoxygenierung: ii.1) Clemmensen-Reduktion: Mit Zn und Salzsäure kann man Ketone wirksam komplett zu den entsprechenden Alkanen reduzieren. Der Mechanismus ist nicht genau bekannt. Auch 128 Zinkamalgam kann Steroidsynthesen. verwendet werden (milder). Ein Anwendungsgebiet sind ii.2) Wolff-Kishner-Reduktion: Das gleiche Resultat kann man auch basisch erzielen: Hier braucht man zunächst das Hydrazon, welches anschließend mit Base (festem KOH) zersetzt wird. Huang-Minlon-Variante: Mit KOtBu in DMSO läßt sich diese Reaktion schon bei niedrigeren Temperaturen durchführen. Natürlich wird dabei die gute Abgangsgruppe Stickstoff freigesetzt: ii.3) über S,S-Acetale: Thioketale können nicht nur zu Ketonen hydrolysiert werden, sonder auch Nickel-katalysiert zu Alkanen hydriert werden. 2. Reduktion von Iminen: Aus Carbonylverbindungen kann man leicht Imine herstellen; deren Reduktion führt im Sinne einer reduktiven Aminierung der Carbonylverbindung über deren Iminiumsalz zu gesättigten Aminen: 129 Das Problem bei der Herstellung sekundärer Amine ist die Überalkylierung bei der HofmannAlkylierung primärer Amine. Durch die Reduktion des entsprechenden Imins mit Natriumcyanoborhydrid gelingt die selektive Monoalkylierung. 3. Reduktion von Carbonsäureestern: Carbonsäurederivate können ebenfalls zu Alkoholen reduziert werden; spannender und schwieriger ist aber die partielle Reduktion zu Aldehyden. Dies gelingt zum Beispiel bei tiefen Temperaturen mit DIBAH (Diisobutylaluminiumhydrid). Es hydroaluminiert die C=O-Doppelbindung (vgl. der Hydroborierung) und generiert ein metalliertes Acetal, welches nicht weiter reduziert wird, sondern mit milder Säure zum Aldehyd hydrolysiert wird. Im ersten Schritt wird dabei die Aluminium-Lewissäure an die Carbonylgruppe angelagert. Anschließend wird das Hydrid übertragen: Bsp.: DIBAH eignet sich hervorragend zur Reduktion von Lactonen zu Lactolen: 130 Mit LAH (in THF) geht die Reaktion dagegen durch bis zum Alkohol: Hier wird nach dem ersten Angriff Aluminiumalkoholat eliminiert und ein Aldehyd gebildet, welcher reaktiver ist als der Ester und direkt weiterreduziert wird. (Auch DIBAH reagiert bei RT in CH2Cl2 meistens durch bis zum Alkohol). 4. Reduktion von Carbonsäuren: Die meistverwendeten Reagentien sind LAH in Ether oder THF und BH3. Der Mechanismus verläuft analog dem der Ester-Reduktion. 5. Reduktion von Amiden: Weinreb-Amide können leicht zu Aldehyden reduziert werden, genau wie sie auch leicht zu Ketonen monoalkyliert werden. Dafür kann man sogar LAH verwenden, oder aber das mildere DIBAH. Wieder ist die landläufige Erklärung die Bildung einer stabilisierten Zwischenstufe mit einem am Methoxy-Sauerstoff koordinierten Aluminiumkation: a) Normale sekundäre Amide lassen sich ebenfalls mit LAH reduzieren. Allerdings entsteht hierbei ein primäres Amin, denn hier wird kein Amidanion, sondern andersherum lieber ein Aluminiumalkoholat eliminiert. Das gebildete Iminiumkation lagert nun leicht ein zweites Hydridanion an: 131 b) Primäre Amide tun das gleiche: Die tetraedrische zusätzlich am N deprotonierte Zwischenstufe eliminiert wieder das Aluminiumalkoholat, geht dabei in ein Imin über, welches seinerseits zum Amidanion reduziert wird. 7. Reduktion von Nitrilen: Nitrile können ebenfalls durch LAH zu primären Aminen reduziert werden. Achtung: Die zwischenzeitlich entstehenden Imine können selber von Aminen angegriffen werden. Dabei entstehen sekundäre Amin-Nebenprodukte. 8. Reduktionen unter C-C-Knüpfung: a) Acyloin-Kondensation: Eine wichtige Methode zur Herstellung mittlerer (gespannter) und größerer Ringe ist die Acyloinkondensation. Letzlich wird hier eine Verknüpfung zweier Estergruppen über die Reduktion durch metallisches Natrium vorgenommen. Mechanismus: Das durch ET entstandene metallierte Ketylradikal dimerisiert und eliminiert zweimal Natriumalkoholat. Das Diketon wird erneut reduktiv angegriffen und bildet ein doppelt metalliertes Endiol, dessen Protonierung zum -Hydroxyketon führt. 132 Bsp.: Dies ist die Rühlmann-Variante, bei der das hochreaktive, zu Nebenreaktionen neigende (welche?) Endiolat mit Trimethylsilylchlorid abgefangen wird. Anschließende milde Aufarbeitung ergibt das Acyloin (=-Hydroxyketon). b) McMurry-Kupplung (reduktive C=C-Kupplung): Welche niedervalenten Titan-Derivate nimmt man? Meist wird eine Mischung aus TiCl4 und TiCl3 mit Reduktionsmitteln wie LAH oder Mg, Zn oder K, Na umgesetzt. Der Mechanismus ist nicht ganz geklärt. Vorschlag: Auf der Titanoberfläche findet ein ET statt und erzeugt aus zwei Carbonylen zwei Alkoholatradikale, welche dimerisieren. Anschließend passiert das gleiche noch einmal und ein Alken entsteht, wobei die beiden negativ geladenen Sauerstoffatome auf der Titanoberfläche Titanoxid bilden. 133 Bsp.: Exkurs: Reduktionen mit Alkalimetallen in NH3 (fl) oder EtNH2 / THF: Zunächst entsteht aus Na und Ammoniak eine tiefblaue Lösung von solvatisierten Elektronen. Diese greifen reduktiv alles an, was Beine hat. i) Reduktion von Enonen Reduktionsmittel! zu Ketonen: Solvatisierte Elektronen sind weiche ii) Reduktion von 1,3-Dienen zu 2-Alkenen: Der Angriff am Diensystem erzeugt ein Allylradikalanion, welches mit Protonen gequencht wird, erneut reduziert und ein zweites Mal gequencht wird. Dabei entsteht ein Gemisch aus cis- und trans-Alken: iii) Birch-Reduktion: Eine sehr wichtige Reaktion reduziert Aromaten zu 1,4-Dienen (Dihydrobenzolen). Genau wie bei den Dienen verläuft diese Reaktion unter Generierung von Allylradikalanionen, welche anschließend regioselektiv zu 1,4-Dienen protoniert, erneut reduziert und erneut protoniert werden. Donorsubstituierte Aromaten lenken die Reduktion in o-Stellung (OR, NR2, Alkyl): 134 Mechanismus: Nach der ersten ET-Reaktion greift ein Proton aus dem Solvens (NH3 oder Spuren ROH) an, das entstehende Pentadienylradikal wird erneut reduziert (ET) und das Pentadienylanion erneut protoniert. Die negative Ladung sitzt dabei immer neben dem elektronenschiebenden Substituenten. Akzeptorsubstituierte Aromaten lenken entsprechend anders (COR, SO2R, CN): 135 I. Umlagerungen 1. Sextett-Umlagerungen: Allgemeines: Der einfachste Fall einer Sextett-Umlagerung ist die anionotrope 1,2Wanderung einer Gruppe mit freiem Elektronenpaar zu einem ElektronenpaarmangelZentrum (Sextett): -Y = Atom mit EP-Lücke (C, N, O, also Kation, Carben, Nitren): Carbeniumion Carben Nitren -RN = (als Anion) wandernde Gruppe -CA = Ausgangspunkt der Wanderung = Atom, an dem die EP-Lücke besser stabilisiert ist als an Y. Stereochemie: Bei einer 1,2-Wanderung wandert RN unter Retention der Konfiguration im wandernden Rest (das wird durch die Woodward-Hoffmann-Regeln festgelegt): Aber: Die Wanderung verläuft am empfangenden Nachbaratom häufig unter Inversion (SN2): 2. Wagner-Meerwein-Umlagerung (1,2-Umlagerung von H oder Alkylrest): Bei der Wagner-Meerwein-Umlagerung (WMU) wandert ein Rest R zum benachbarten Carbeniumion. Das dabei entstehende neue Carbeniumion stabilisiert sich oft durch unterschiedliche Folgereaktionen, z. B. durch Protonenabspaltung: 136 Bsp.: Die Triebkraft der Umlagerung ist meist die Bildung des stabileren Carbeniumions. Je stabiler der wandernde Substituent (d.h., je leichter der Substituent ein EP zur Verfügung stellen kann oder je nucleophiler er ist), desto leichter geht er die Umlagerung ein. Die Reihenfolge der Wanderungstendenz sieht also so aus: H< prim. Alkyl < sek. Alkyl < tert-Alkyl < Vinyl < Aryl (dabei ist Ar-NO2 < Ar-OMe) Bsp.: Ein primäres Carbeniumion lagert sich gerne zu einem sekundären um. Mechanismus: Zur günstigen bindenden Orbitalüberlappung muß die wandernde Gruppe mit ihrem Bindungselektronenpaar coplanar zum leeren p-Orbital des Carbeniumions stehen! Beispiel: 137 Noch vor der eigentlichen Friedel-Crafts-Alkylierung lagert das mit der Lewissäure entstehende primäre Propylcarbeniumion um in das sekundäre Isopropylcarbeniumion. Dessen Angriff am Aromaten führt zur bevorzugten Bildung von Isopropylbenzol. 3. Pinakol-Pinakolon-Umlagerung: Glykole bilden mit Säure Carbeniumionen, die einen 1,2-Alkylshift eingehen, so daß die positive Ladung von einem OH-Substituenten mesomeriestabilisiert werden kann. Abschließende Deprotonierung führt an der Stelle zum Keton. Bsp.: Auch der Aromat kann anionotrop wandern; das stellt man sich über das Homobenzylkation vor, dessen mesomere Grenzstrukturen auch eine Spiroverbindung mit Pentadienylkation im Sechsring enthalten: 4. Umlagerungen von Epoxiden: Nach Lewissäure-Angriff am Epoxid-Sauerstoff öffnet der Dreiring und liefert ein Carbeniumion, welches sich über die Bildung einer Carbonylgruppe und gleichzeitigen 1,2Alkylshift stabilisiert (Semipinakol-Umlagerung): 138 Bsp.: Hier wird die positive Ladung natürlich in Benzylstellung am besten stabilisiert; nach Dissoziation der Lewissäure wandert hier das Proton und liefert einen -Ketoester. 5. Tiffeneau-Demianov-Umlagerung: 1,2-Aminoalkohole können nach Diazotierung Stickstoff verlieren, wenn gleichzeitig unter Deprotonierung des Alkohols ein Keton entsteht und der O-ständige Alkylrest anionotrop zum Carbeniumion 1,2-wandert: Bsp.: So macht man aus Ketonen ihre Homologen: aus dem ursprünglichen Keton generiert man nämlich den 1,2-Aminoalkohol durch HCN-Addition und anschließende LAH-Reduktion. Bei cyclischen Ketonen erhält man das ringerweiterte Keton. 6. Diazoketone (Wolff-Umlagerung): Aus Säurechloriden erhält man durch Substitution mit Diazomethan Diazoketone, welche leicht Stickstoff verlieren (Licht- oder Ag-I-katalysiert). Dabei entsteht formal ein Acylcarben, welches unter 1,2-Wanderung des Alkylrests zum elektrophilen Carben139 Kohlenstoff ein Keten bildet, welches anschließend mit Alkoholen zum Ester abgefangen werden kann. Diese Umlagerung verläuft allerdings nach neueren Erkenntnissen konzertiert, vermeidet also die Bildung des hochreaktiven Carbens. Immerhin wird dabei formal das Elektronensextett am Carben in das günstigere Oktett am Keten überführt. Insgesamt ergibt sich ausgehend vom Säurechlorid eine Homologisierung des Carbonsäurederivats, auch genannt Arndt-Eistert-Synthese. So kann man z. B. aus der natürlichen -Form -Aminosäuren machen. 7. Umlagerungen unter Wanderung zum N-Atom: a) Hofmann-Umlagerung: Carbonsäureamide können mit Hypobromit zu -Halogenamiden umgesetzt werden, welche leicht im Sinne einer -Eliminierung HBr abspalten. Dabei wird formal ein Acylnitren gebildet, welches die gleiche Umlagerung eingeht wie ein Acylcarben: der Alkylrest wandert anionotrop zum elektrophilen Nitren-Stickstoff und liefert dabei ein Isocyanat, welches anschließend mit Wasser zur Carbaminsäure abgefangen wird. Diese verliert spontan CO2 und bildet das Amin-Endprodukt. Heute weiß man, daß auch diese Reaktion konzertiert abläuft und die Bildung des hochreaktiven freien Acylnitrens vermeidet. 140 b) Curtius-Umlagerung: Genauso kann man auch von Carbonsäureaziden ausgehen, welche aus Carbonsäurechloriden und Natriumazid entstehen. Nach Stickstoff-Abspaltung entsteht auch hier wieder formal das Acylnitren und lagert um wie gehabt: Der Curtius-Abbau hat den Vorteil, daß man kein Wasser im Reaktionsgefäß hat, die gebildeten Isocyanate sind also stabil und können z.B. mit Diolen zur Polyaddition herangezogen werden. i) Lossen-Abbau: Auch über acylgeschützte Hydroxamsäuren kann man durch 1,1Eliminierung Acylnitrene generieren: c) Beckmann-Umlagerung: Oxime können nach Tosylierung eine gute Abgangsgruppe verlieren; die dabei gebildeten Nitrencarbeniumionen lagern spontan um und bilden Nitriliumkationen, welche wiederum Wasser anlagern und Carbonsäureamide bilden. Bsp.: So wird -Caprolactam für die Nylon-6-Synthese gemacht. 141 8. Umlagerungen unter Wanderung zum O-Atom: a) Hydroperoxid-Umlagerung: Radikalisch gebildete Alkylhydroperoxide werden am endständigen OH protoniert, verlieren Wasser und lagern den am besten wandernden Alkyl- oder Arylrest um. Das bei der Arylwanderung gebildete Arylethercarbeniumion lagert Wasser an und zerfällt zu Keton und Phenol („Hock’sche Phenolsynthese“). b) Baeyer-Villiger-Oxidation: Die nucleophile Addition von Persäuren an Ketone führt zum Peroxyhalbacetal, welches unter O,O-Spaltung formal zum O-Insertionsprodukt in den Keton-Grundkörper weiterreagiert. Dies geschieht unter 1,2-Wanderung des Alkylrests vom Kohlenstoff zum elektrophilen Peroxy-Sauerstoff. Dabei wandert immer die nucleophilste Gruppe mit der höchsten Wanderungstendenz: c) Oxidation von Alkylboranen: Die klassische Aufarbeitung von Alkylboranen mit alkalischer H2O2-Lösung erfolgt ebenfalls über die 1,2-Wanderung eines Alkylrests vom Bor zum Sauerstoff; dabei wird die energiereiche O,O-Bindung gespalten. Wichtig: Diese Wanderung erfolgt unter Retention der Konfiguration in der wandernden Gruppe! 142 9. Sigmatrope Umlagerungen: Sigmatrope Umlagerungen bilden eine große Gruppe von pericyclischen, konzertierten Reaktionen, bei denen 4n+2 -Elektronen thermisch erlaubt cyclisch wandern. a) [3,3]-sigmatrope Umlagerungen: Wenn die Gruppe X = CR2, NR oder O, handelt es sich um die Cope, Aza-Cope- oder Claisen-Umlagerung. Diese Reaktionen sind alle reversibel und unterliegen deshalb der thermodynamischen Produktkontrolle. i) Cope-Umlagerung: Im unten abgebildeten Doppelbindung die treibende Kraft. Beispiel ist die konjugierte neue Oxy-Cope: Noch leichter geht die Cope-Umlagerung mit OH-Gruppen in 1-Stellung, am besten nach Deprotonierung zum Alkoholat: Das dabei gebildete Enol tautomerisiert anschließend irreversibel zum Aldehyd. ii) Claisen-Umlagerung: Hier ist die Ausbildung der C=O-Doppelbindung die treibende Kraft. 143 Bsp.1.: Bsp. 2: Fischer’sche Indolsynthese: Hier lagert auch ein Aza-Claisen-System 3,3-sigmatrop um; allerdings ist die treibende Kraft hier nicht die Ausbildung des Imins, sondern die durch Tautomerisierung zum Enamin bewirkte Rearomatisierung. Das Anilin greift anschließend das zweite Imin an und liefert nach Ammoniak-Abspaltung Indol. b) [2,3]-sigmatrope Umlagerungen: i) Wittig-Umlagerung: Mäßig acide Allylalkylether lagern nach Deprotonierung im Alkylteil zu Alkoholaten um, deren pKa-Wert viel niedriger liegt (~17) als der des Alkylethers (>30). Die Triebkraft ist also die Bildung des stabileren Anions: Die Reaktion ist ein schönes Beispiel für einen kationotrop wandernden Rest, und verläuft über einen 5-gliedrigen Übergangszustand, aus dem heraus auch die hohe Stereoselektivität erklärt werden kann: 144