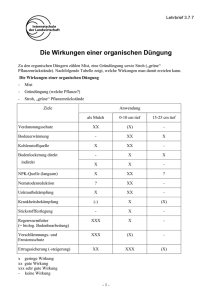
Musterprüfplan - Universität zu Köln
Werbung

Prüfplan-Code xxx Seite 1 von 67 Prüfplan-Template der Universität zu Köln Template Version v7-24-F Hinweise zur Nutzung des Dokuments Diese Vorlage soll die Erstellung von Prüfplänen für Arzneimittelprüfungen der Phase II bis III und anderen interventionellen Studien (z.B. auch Phase I) sowohl im Bereich nichtkommerzieller als auch kommerzieller Studien erleichtern. Die Überschriftentexte berücksichtigen die ICH-Guidelines E3 und E6. Hierbei wurde soweit möglich eine Übertragung von Begrifflichkeiten aus dem Englischen ins Deutsche vorgenommen. Die Überschriftentexte und –nummern sollten nicht verändert werden. Insbesondere sollten keine Überschriften ersten bis dritten Grades hinzugefügt oder entfernt werden. Hiervon ausgenommen sind folgende Anpassungen: Da die Vorlage auf Basis der Regularien, die primär für klinischen Prüfungen von Arzneimitteln gelten, entstanden ist, müssen bei Prüfplänen für die klinische Prüfung von Medizinprodukten Begriffe wie Prüfmedikation etc. auf Medizinprodukte angepasst werden. Dies betrifft z.B. die Kapitelüberschriften 7.2 bis 7.3. Wegen der Vielzahl der hier möglichen, jedoch begrifflich stark unterschiedlichen Vorschriften wurde auf die Erstellung von Vorschlägen verzichtet. Ein eigenes Prüfplantemplate zu Medizinprodukte Studien wird gerade erarbeitet. Weiterhin wurde die Vorlage auf Basis deutscher Regularien erstellt. Bei multinationalen Studien müssen die Verhältnisse für andere Länder gesondert geprüft werden. Erläuterungen sind durch kursive, grüne Schrift hervorgehoben. An einigen Stellen wurden farbig blau gekennzeichnete Beispieltexte bzw. sinnstiftende Platzhalter eingefügt. In beiden Fällen ist die Richtigkeit der getroffenen Aussagen für die aktuelle klinische Prüfung zu verifizieren. Insbesondere gilt dies für die Beispieltexte, die keinesfalls unkritisch übernommen werden sollten. Die Beispieltexte und viele Platzhalter sind formuliert im Hinblick auf die Durchführung einer multizentrischen Studie, bei der die Universität zu Köln Sponsor einer Investigator Initiated Trial (IIT) ist. Viele Aufgaben werden hierbei vom Leiter der klinischen Prüfung und dem Zentrum für klinische Studien Köln übernommen. Sollten Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 2 von 67 Aufgaben von anderen Einrichtungen wahrgenommen werden, so ist dies entsprechend abzubilden. Vor Verwendung dieser Vorlage bitte diesen Text löschen! Für die Richtigkeit der mit diesem Dokument gemachten Vorschläge kann keine Garantie übernommen werden. Jeder Benutzer ist aufgefordert die Richtigkeit anhand der einschlägigen Vorschriften selbst zu prüfen. Hinweise auf Fehler sowie Kommentare bitte an [email protected]. oder eine der unten genannten Autoren-Adressen. Autoren G. Grass, ZKS Köln, Universität zu Köln (Sponsor) Dieses Dokument untersteht der UVM Lizenz für Freie Inhalte (http://www.ifross.de/Lizenzen/LizenzFuerFreieInhalte.html) und darf nur unter Einhaltung dieser Lizenz verwendet werden. Eine Kopie der Lizenz ist bei Weitergabe dieser Vorlage stets beizufügen. Wenn Sie dieses Dokument bei der Erstellung eines Prüfplans oder der Planung einer klinischen Prüfung verwenden, nehmen Sie bitte entsprechenden Hinweis im Prüfplan auf oder stellen Sie ein Zitat in das Literaturverzeichnis. Kontaktadresse der Autoren: [email protected], [email protected], [email protected] Besonderer Dank für wertvolle Hinweise gilt Dr. Claudia Weiß, Prof. Dr. Martin Hellmich. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 3 von 67 Universität zu Köln PRÜFPLAN TITEL DER KLINISCHEN PRÜFUNG Sponsor Leiter der klinischen Prüfung: Firma mit Adresse oder Universität zu Köln Albertus-Magnus-Platz 50923 Köln Name Klinik Uniklinik Köln Kerpener Str. 62 50937 Köln Prüfplan-Code: xxx ISRCTN: xxx Die ISRCTN ist nur anzugeben, wenn diese gemäß Prüfplan-Abschnitt 5.3 beantragt wurde. EudraCT-Nummer: xxx Fassung vom XX.XX.XXXX, Version vX-XX Die Informationen in diesem Prüfplan sind streng vertraulich zu behandeln. Sie dienen nur zur Information des Sponsors, der Prüfer, der Studienmitarbeiter, der Ethikkommission, der Behörden und der Patienten. Dieser Prüfplan darf ohne Zustimmung des Sponsors oder des Leiters der klinischen Prüfung (LKP) nicht an Dritte weitergegeben werden. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 4 von 67 I. Unterschriften Es sollten alle Personen, die maßgeblich an der Erstellung des Prüfplans beteiligt waren unterschreiben. Name (LKP, Leiter der klinischen Prüfung,) Als Bevollmächtigten des Sponsors Unterschrift Datum Universität zu Köln Bei monozentrischen klinischen Prüfungen ist der Hauptverantwortliche für das Prüfzentrum und die gesamte Studie entsprechend dem Arzneimittelgesetz (AMG) als Prüfer zu bezeichnen. In diesem Falle muss hier und überall im Folgenden der „LKP“ / „Leiter der klinischen Prüfung“ durch „Prüfer“ ersetzt werden. Name Medizinischer Ansprechpartner des Sponsors Unterschrift Datum Klinik und Poliklinik für xxx, Uniklinik Köln Ist der medizinische Ansprechpartner des Sponsors gleichzeitig der LKP, dann kann diese zweite Unterschrift entfallen, die Zeile „Medizinischer Ansprechpartner...“ sollte dann oben ergänzt werden. Name Statistiker Unterschrift Datum Institut für Medizinische Statistik, Informatik und Epidemiologie, Universität zu Köln Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 5 von 67 II. Synopsis Sponsor Firma mit Adresse oder Universität zu Köln Albertus-Magnus-Platz 50923 Köln diese vertreten durch: Name (Leiter der klinischen Prüfung, LKP) Klinik und Poliklinik Uniklinik Köln Kerpener Str. 62 50937 Köln Leiter der klinischen Prüfung s. o. Ggf. muss insbesondere bei multinationalen, multizentrischen Prüfungen hier zusätzlich der/die national Bevollmächtigte(n) des Sponsors aufgeführt werden. Titel der klinischen Prüfung: Indikation Phase: Klinische Prüfung der Phase xxx (I bis IV) Handelt es sich um eine klinische Prüfung, die nicht unter die Bestimmungen § 40 ff. AMG fällt, sollten hier entsprechende Angaben gemacht werden, z.B. Anwendungsbeobachtung, nicht-interventionelle Prüfung, klinische Medizinprodukteprüfung etc. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Art der Prüfung, Studiendesign / Methodik: Seite 6 von 67 Mono-/multizentrische, multinationale klinische Prüfung Zwei-/drei-/vierarmig, randomisiert, einfach-/doppelblind, Placebo kontrollierte Parallelgruppen-/ Crossover-Design Patientenanzahl: Je Behandlungsgruppe xxx (insgesamt xxx) Primäres Studienziel Zielgrößen: Primäre Zielgröße: xxx Sekundäre Zielgrößen: xxx Weitere Zielgrößen: Diagnose und Haupt-Einund Ausschlusskriterien: xxx Diagnose: xxx Haupteinschlusskriterien: xxx Hauptausschlusskriterien: xxx Bezeichnung der Prüfmedikation: Prüfpräparat: Dosierung und Applikationsart: Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 7 von 67 Vergleichstherapie, Dosierung und Applikationsart: Dauer der Therapie: Beschreibung der Behandlungsdauer (ggf. unterschiedlich für Prüfpräparat und Vergleichstherapie) und der Nachbeobachtungszeit (Follow-Up), d.h. wie lange dauert die Studie pro Patient insgesamt und wie teilt sich diese Zeit auf. Zeitplan: Einschluss erster Patient III. Quartal 2015 (first patient first visit, FPFV): Einschluss letzter Patient (LPFV): III. Quartal 2017 Letzte Visite des letzten Patienten IV. Quartal 2017 (LPLV): Ende der klinischen Prüfung IV. Quartal 2017 Abschlussbericht/Ergebnisbericht: IV. Quartal 2018 (Vergleiche Kapitel 4.1.1) Statistiker Name Institut für Medizinische Statistik, Informatik und Epidemiolgie Universität zu Köln Kerpener Str. 62 50937 Köln Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Statistische Methoden: Seite 8 von 67 Randomisierung: Blöcke variabler Länge, nach Zentrum und xxx stratifiziert. Die Randomisierung erfolgt zentral. Zwischenauswertung(en): gemäß O’Brien-Fleming-Schranken, evtl. Adaptation des Studiendesigns und Auswertung nach der „inverse-normal method“. Auswertung der primären Zielvariablen: Varianzanalyse (Faktoren: Behandlung und Zentrum; Typ-II-Quadratsummen) GCP-Konformität: Die vorliegende Prüfung wird gemäß der aktuellen Version des Prüfplans, der international anerkannten Leitlinie Good Clinical Practice (ICH-GCP) einschließlich der Archivierung essentieller Dokumente durchgeführt Ggf. Hinweis auf weitere Regularien (AMG, RöV, StrlSchV, u. a.). Finanzierung Prüfplan vX-XX vom XX.XX.XXXX Alle Finanzquellen angeben. Universität zu Köln Prüfplan-Code xxx Seite 9 von 67 III. Inhaltsverzeichnis I. Unterschriften 4 II. Synopsis 5 III. Inhaltsverzeichnis 9 III.a) Verzeichnis der Tabellen 14 III.b) Verzeichnis der Abbildungen 14 IV. Abkürzungsverzeichnis 15 1. Einleitung 16 2. Ziele der klinischen Prüfung 17 3. 2.1. Rationale der klinischen Prüfung 17 2.2. Primäres Ziel 17 2.3. Sekundäre und weitere Ziele 17 Organisationsstruktur 18 3.1. Sponsor 18 3.2. Leiter der klinischen Prüfung 18 3.3. Statistik 18 3.4. Data Monitoring Committee 19 3.5. Weitere Kommitees 20 3.5.1. Steering Committee 20 3.5.2. Advisory Committee 20 3.5.3. Review Board 20 3.6. Prüflabore und sonstige technische Einrichtungen 20 3.7. Zentrale Organisationseinheiten 21 3.8. Prüfer und Prüfzentren 21 Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx 3.8.1. 3.9. 4. Seite 10 von 67 Anforderungen an Prüfer und Prüfzentren 22 Finanzierung 22 Studiendurchführung 4.1. 23 Allgemeines Studiendesign 4.1.1. 23 Zeitplan 23 4.2. Diskussion des Studiendesigns 24 4.3. Auswahl der Studienpopulation 24 4.3.1. Einschlusskriterien 25 4.3.2. Ausschlusskriterien 25 4.4. Nachträglicher Ausschluss von Prüfungsteilnehmern 4.4.1. 27 Verfahren bei vorzeitigem Ende der Behandlung im Rahmen der klinischen Prüfung 4.5. 27 Schließen von Prüfzentren / Vorzeitige Beendigung der klinischen Prüfung 27 4.5.1. Schließen von Prüfzentren 27 4.5.2. Abbruch der gesamten Studie 28 4.6. Behandlungen 28 4.6.1. Angewendete Behandlungen 28 4.6.2. Beschreibung der Prüfmedikation 29 4.6.2.1. Herstellung der Prüfmedikation 29 4.6.2.2. Kennzeichnung der Prüfmedikation 29 4.6.2.3. Lagerung der Prüfmedikation 30 4.6.3. Einhaltung der Therapie / Ausgabe und Rücknahme der Prüfmedikation 30 4.6.4. Methode zur Zuordnung der Patienten zu den Behandlungsgruppen 30 4.6.5. Auswahl der Dosierung der Prüfmedikation 30 Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx 4.6.6. 4.6.7. Seite 11 von 67 Festlegung von Dosierung und Zeitpunkt der Prüfmedikationsgabe für jeden Prüfungsteilnehmer 31 Verblindung 31 4.6.7.1. 4.6.8. Vorangegangene Therapie und Begleittherapie 4.6.8.1. 4.6.9. 4.7. Ausweichtherapie im Notfall Weiterbehandlung nach Ende der klinischen Prüfung Wirksamkeits- und Sicherheitsvariablen 4.7.1. Messung der Wirksamkeits- und Sicherheitsvariablen 31 32 32 32 32 33 4.7.1.1. Primäre Zielgröße 33 4.7.1.2. Sekundäre und weitere Zielgrößen 33 4.7.1.3. Sicherheitsanalyse 33 4.7.1.4. Beschreibung der einzelnen Visiten 33 4.7.2. Angemessenheit der Messverfahren 33 4.7.3. Pharmakokinetik/Bestimmung von Medikamentenspiegeln 34 4.8. Sicherstellung der Datenqualität 34 4.8.1. Monitoring 34 4.8.2. Audits / Inspektionen 36 4.9. 5. Entblindung Dokumentation 36 4.9.1. Datenmanagement 37 4.9.2. Archivierung 38 Ethische und regulatorische Aspekte 39 5.1. Unabhängige Ethikkommissionen 39 5.2. Ethische Durchführung der klinischen Prüfung 39 5.2.1. Berücksichtigte gesetzliche Bestimmungen und Leitlinien Prüfplan vX-XX vom XX.XX.XXXX 39 Universität zu Köln Prüfplan-Code xxx 6. 5.3. Behördenmeldungen, Genehmigungen und Registrierung 40 5.4. Aufklärung und Einwilligung der Prüfungsteilnehmer 41 5.5. Probandenversicherung 41 5.6. Datenschutz 42 Statistische Methoden und Ermittlung der Fallzahl 6.1. 43 Statistischer und analytischer Plan 43 6.1.1. Analysepopulationen 43 6.1.2. Beschreibung des Patientenkollektivs 44 6.1.3. Primäre Zielvariable 44 6.1.4. Sekundäre Zielvariablen 44 6.1.5. Subgruppenanalysen 44 6.1.6. Zwischenauswertung 45 6.2. 7. Seite 12 von 67 Ermittlung der Fallzahl 45 Sicherheit 7.1. 46 Definitionen 46 7.1.1. Unerwünschtes Ereignis 46 7.1.2. Nebenwirkung 47 7.1.3. Schwerwiegendes unerwünschtes Ereignis oder schwerwiegende Nebenwirkung 47 7.1.4. Unerwartete Nebenwirkung 48 7.1.5. Verdachtsfall einer schwerwiegenden unerwarteten Nebenwirkung 48 7.1.6. Andere mögliche prüfungsspezifische Komplikationen und/oder Risiken 48 7.2. Dokumentation und Nachverfolgung von unerwünschten Ereignissen und Nebenwirkungen 7.2.1. Intensität des unerwünschten Ereignisses Prüfplan vX-XX vom XX.XX.XXXX 49 50 Universität zu Köln Prüfplan-Code xxx 7.2.2. 7.3. Seite 13 von 67 Zusammenhang des unerwünschten Ereignisses mit dem Prüfpräparat 51 Dokumentation und Meldung von schwerwiegenden unerwünschten Ereignissen, Schwangerschaften/Geburten und Änderungen des Nutzen-Risiko-Verhältnisses53 7.3.1. Meldung des Prüfers an den Sponsor bei schwerwiegenden unerwünschten Ereignissen 8. 54 7.3.2. Meldung des Prüfers an den Sponsor bei Schwangerschaften und Geburten 56 7.3.3. Zweitbegutachtung durch den Sponsor 57 7.3.4. Entblindung bei verblindeten Prüfpräparaten 57 7.3.5. Meldung an die Ethikkommission und die zuständige Bundesoberbehörde 58 7.3.6. Überprüfung und Meldung von Änderungen des Nutzen-Risiko-Verhältnisses59 7.3.7. Information des Data Monitoring Committee 60 7.3.8. Information der Prüfer 60 7.3.9. Information des Zulassungsinhabers 61 7.3.1. Jährlicher Bericht über die Sicherheit der Prüfungsteilnehmer 61 Verwendung der Daten und Publikation 8.1. Berichte 62 62 8.1.1. Zwischenberichte 62 8.1.2. Abschlussbericht 62 8.2. Publikation 63 9. Änderungen des Prüfplans 65 10. Literatur 66 11. Anhänge 67 11.1. Prüfzentren und Prüfer sowie Stellvertreter 67 11.2. Protocol Agreement Form 67 11.3. Steering Committee 67 Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 14 von 67 11.4. Data Monitoring Committee 67 11.5. Advisory Committee 67 11.6. Prüflabore und sonstige technische Einrichtungen 67 11.7. Musteretikett für die Prüfmedikation 67 11.8. Patienteninformation und Einverständniserklärung 67 11.9. Fachinformation 67 11.10. Versicherungsbestätigung 67 11.11. Versicherungsbedingungen 67 III.a) Verzeichnis der Tabellen Tabelle 1: Zeitplan der Studie 23 Tabelle 2: Prüfmedikation 30 Tabelle 3: Untersuchungen im Rahmen der klinischen Prüfung 33 III.b) Abbildung 1: Verzeichnis der Abbildungen Ablaufdiagramm der klinischen Prüfung Prüfplan vX-XX vom XX.XX.XXXX 24 Universität zu Köln Prüfplan-Code xxx Seite 15 von 67 IV. Abkürzungsverzeichnis Abkürzung Bedeutung AE Unerwünschtes Ereignis (Adverse Event) BfArM Bundesinstitut für Arzneimittel und Medizinprodukte (Federal Institute for Drugs and Medical Devices) BOB Bundesoberbehörde (BfArM, PEI) CRF Erhebungsbögen (Case Report Form) DMC Data Monitoring Committee IMP Investigational Medicinal Product LKP Leiter der klinischen Prüfung (Principal Coordinating Investigator) PEI Paul-Ehrlich-Institut SAE Schwerwiegendes unerwünschtes Ereignis (Serious Adverse Event) SUSAR Verdachtsfall einer schwerwiegenden unerwarteten Nebenwirkung (Suspected Unexpected Serious Adverse Reaction) TMF Studienordner des Sponsor (Trial Master File) ISF Prüfarztordner (Investigator Site File) Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 16 von 67 1. Einleitung Die Einleitung sollte möglichst kurz und prägnant gefasst werden. Der Hintergrund der klinischen Prüfung sollte in Bezug auf das Krankheitsbild und auf bereits bekannte Informationen zur Studienintervention geschildert werden. Insbesondere sollten bereits früher durchgeführte Studien, Ergebnisse relevanter klinischer, prä-klinischer und anderer Untersuchungen oder Studien mit vergleichbaren Interventionen Erwähnung finden. Hierbei sollten Referenzen zu relevanten Veröffentlichungen und ggf. anderen Daten zum Hintergrund der klinischen Prüfung gegeben werden. Idealerweise dient ein aktuelles systematisches Review der einschlägigen Untersuchungen als Planungsgrundlage. Die Einleitung sollte stets in Bezug auf das Vorhaben formuliert werden und zur Rationale bzw. zum Ziel der klinischen Prüfung hinführen. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 17 von 67 2. Ziele der klinischen Prüfung 2.1. Rationale der klinischen Prüfung Kurz zusammenfassen, warum die klinische Prüfung durchgeführt wird. xxx 2.2. Primäres Ziel Das primäre Ziel der klinischen Prüfung sollte hier aus klinischer Sicht beschrieben werden (klinische Relevanz, Patienten-Nutzen etc.). Dabei sollte dies die primäre Zielgröße inhaltlich widerspiegeln. Die Eignung der gewählten Größen zur Abbildung des klinischen Ziels sowie deren Messung bzw. die entsprechenden Messverfahren werden im Abschnitt 4.7 beschrieben, deren statistische Auswertung in den Abschnitten 6.1.3 und 6.1.4. xxx 2.3. Sekundäre und weitere Ziele Klinische Beschreibung der sekundären Ziele xxx Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 18 von 67 3. Organisationsstruktur 3.1. Sponsor Sponsor Universität zu Köln diese vertreten durch: Name Klinik und Poliklinik xxx Uniklinik Köln Kerpener Str. 62 50937 Köln 3.2. Leiter der klinischen Prüfung Leiter der klinischen Name Prüfung (LKP): Klinik und Poliklinik Uniklinik Köln Kerpener Str. 62 50937 Köln Im folgenden Text wird der LKP ggf. in seiner jeweiligen Funktion als LKP oder als Bevollmächtigter des Sponsors genannt. 3.3. Statistik Statistiker: xxx Institut für Medizinische Statistik, Informatik und Epidemiolgie Universität zu Köln Kerpener Str. 62 50937 Köln Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 19 von 67 3.4. Data Monitoring Committee Definition und Beschreibung der Aufgaben und Arbeitsweise des Data Monitoring Committees (DMC) (Überwachung des Studienfortschritts, der Sicherheitsdaten, Überwachung von Wirksamkeits-Zielgrößen z.B. im Rahmen der Ergebnisbewertung von Zwischenauswertungen unter Aufrechterhaltung der Verblindung; Beratung des Sponsors/Auftraggebers zur Fortführung, zur Modifikation oder zum Abbruch der klinischen Prüfung im Zusammenhang mit Zwischenauswertungen und/oder Safety Analysen (siehe dazu auch die Guideline EMEA/CHMP/EWP/5872/03 Corr)). Darstellung der Informationswege, ggf. Beachtung der Einbindung des DMC in das Meldeverfahren (Entblindung) zu unerwünschten Ereignissen (unter 7.3.7 zu beschreiben). Hinweis darauf, dass sich das DMC selbst eine Charter (Verfahrensregeln) gibt (siehe auch „The DAMOCLES Study Group. A proposed charter for clinical trial 2005 data monitoring committees: helping them do their job well. Lancet 2005; 365: 711-22"). Ggf. kann hier auch erläutert werden, dass und aus welchen Gründen die Einrichtung eines DMC für nicht erforderlich gehalten wird. Ein Data Monitoring Committee aus unabhängigen Experten wird eingerichtet. Es besteht aus zwei Klinikern und einem Biometriker, die nicht an der Durchführung der Studie beteiligt sind (siehe 11.4). Die Aufgabe des DMC besteht darin, die Sicherheit der Teilnehmer an der klinischen Prüfung zu überwachen, indem durch das DMC die Sicherheit und die Wirksamkeit der Studientherapie beurteilt sowie die Integrität und die Validität der erhobenen Daten und die Durchführung einer klinischen Prüfung fortlaufend überwacht werden. Im Rahmen dieser Prüfungen erstellt das DMC Empfehlungen an den Sponsor hinsichtlich der weiteren Durchführung (z.B. Abbruch oder Modifizierung) der Studie auf Basis der erhobenen Daten. Die für die Aufgaben notwendigen Daten stellt der Sponsor dem DMC nach Maßgabe des DMC zur Verfügung. Dies umfasst insbesondere Listen mit Angaben zu schwerwiegenden unerwünschten Ereignissen und weiteren vom DMC für erforderlich gehaltene Zielgrößen, die mindestens alle 6 Monate sowie zum Zeitpunkt formaler Zwischenauswertungen bereitgestellt werden. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 20 von 67 3.5. Weitere Kommitees Dieses Kapitel und seine Unterkapitel sind optional. 3.5.1. Steering Committee Es kann eine Leitungsgruppe (Steering Committee, SC) bestimmt werden, die die laufende Überwachung der Studie gemeinsam mit dem LKP übernimmt und in allen oder in zu definierenden Fragen z.B. mehrheitlich entscheidet. Die Zusammensetzung des Steering Committees ist in Anhang 11.2 angegeben. 3.5.2. Advisory Committee Häufig wird ein beratendes Gremium zusammengestellt, das insbesondere in die Planung und Bewertung der klinischen Prüfung einbezogen wird. Die Einbindung von Meinungsführern kann das Risiko reduzieren, eine klinische Prüfung durchzuführen, die am Ende nicht auf Akzeptanz trifft. Die Zusammensetzung des Advisory Committees ist in Anhang 11.5 angegeben. 3.5.3. Review Board Häufig wird ein Gremium einbezogen, dass Wirksamkeits- oder Sicherheitsvariablen beurteilt oder entscheidet, ob ein Teilnehmer protokollgerecht behandelt wurde. An dieser Stelle können ggf. auch weitere Gremien genannt werden. Die Zusammensetzung des Review Boards sollte ebenfalls in einem Anhang genannt werden. 3.6. Prüflabore und sonstige technische Einrichtungen An dieser Stelle sollte erwähnt werden, welche Aufgaben durch andere Einrichtungen übernommen werden. Die Bezeichnung und Adresse der Einrichtungen sollte im Anhang 11.6 angegeben werden. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 21 von 67 3.7. Zentrale Organisationseinheiten Hier sollte beschrieben werden, wer die einzelnen Teilaufgaben ggf. übernimmt. Aspekte hierzu unterliegen typischerweise vertraglicher Regelungen (z.B. Sponsorvertrag) und zugehörigen Delegationsplänen. Die einzelnen Ansprechpartner sollten jeweils mit vollständigen Kontaktdaten genannt werden. Im Regelfall sind Änderungen der Kontaktdaten keine bewertungspflichtige Änderung nach § 10 (1) GCP-V. Der federführenden EthikKommission und der zuständigen BOB sollten diese Änderungen unabhängig von der gesetzlichen Pflicht dennoch zur Kenntnis gebracht werden. Projektmanagement: xxx Monitoring: xxx Datenmanagement: xxx SAE-Management: xxx Wissenschaftliche Beratung: Name Zentrum für Klinische Studien Köln (ZKS Köln) Gleueler Str. 269 50935 Köln Tel.: +49 221 478 XXXX Fax: +49 221 478 7983 Email: [email protected] 3.8. Prüfer und Prüfzentren Die klinische Prüfung wird offen mono- / multizentrisch in voraussichtlich xxx Prüfstellen in Deutschland durchgeführt. Falls erforderlich, können weitere qualifizierte Prüfstellen in die Durchführung der klinischen Prüfung einbezogen werden. Eine Liste der Prüfstellen mit Nennung der Prüfer und ihrer Stellvertreter findet sich in Anhang 11.1. Die Informationen zu Prüfstellen, Prüfern, stellvertretenden Prüfern sowie dem weiteren Studienpersonal wird laufend in einer separaten Liste aktualisiert werden. Die endgültige Liste wird dem Abschlussbericht der klinischen Prüfung beigelegt werden. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx 3.8.1. Seite 22 von 67 Anforderungen an Prüfer und Prüfzentren Anforderungen an Prüfzentren und Prüfer beschreiben (z.B. Nachweis regulatorischer Kenntnisse, Erfahrung in der Durchführung klinischer Arzneimittelprüfungen, spezielle Erfahrungen in der Indikation etc.). Die Mindestanforderungen an die Qualifikation für die Aufgaben, die im Rahmen der klinischen Prüfung anfallen, werden durch den Sponsor bzw. durch den LKP als Sponsorbevollmächtigten definiert. Die Nachweise zur Qualifikationen der Mitarbeiter werden sowohl beim Bevollmächtigten des Sponsors (Trial Master File; TMF) als auch an der lokalen Prüfstelle im Investigator Site File (ISF) dokumentiert. Sofern es Prüfer gibt, die nicht Arzt sind, sollte dies entsprechend aufgeführt und begründet werden, warum diese geeignet sind, die Prüfung durchzuführen und welche Aufgaben sie übernehmen und welche nicht (z.B. Aufklärung und Einholen des Einverständnisses). Für die Anforderungen an Prüfer, stellvertretende Prüfer und das weitere Personal der Prüfgruppe muss der Ethikkommission eine Auflistung der Qualifikationsanforderungen vorgelegt werden (http://www.medfak.uni-koeln.de/index.php?id=223). Diese Qualifikationsanforderungen sollen auch hier genannt werden. 3.9. Finanzierung Die klinische Prüfung wird durch Fördermittel der DFG / des BMBF (Aktenzeichen) finanziert. Ein ausführlicher Finanzierungsplan ist nicht Bestandteil des Prüfplans, da dieser separat abgelegt bzw. Gegenstand von Bewilligungen oder Verträgen sowie des Antrags an die Ethik-Kommission ist. Es müssen jedoch alle finanziellen Quellen genannt werden. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 23 von 67 4. Studiendurchführung 4.1. Allgemeines Studiendesign Phase, methodische Studiencharakteristik (z.B. doppelblind, placebokontrolliert, Parallelgruppen / Crossover / Überlegenheit / Nicht-Unterlegenheit etc.), kurze Beschreibung von Verfahren zur Vermeidung bzw. Minimierung von Bias (Randomisierung, Verblindung von xxx). Ein oder zwei Sätze sollten zur Kurzcharakterisierung ausreichen. 4.1.1. Zeitplan Kurze, übersichtliche Beschreibung des Zeitplans der gesamten Studie: Kurze Darstellung von Abfolge und Dauer der Intervention, ggf. einzelner Prüfungsphasen, sowie Nachbeobachtung (follow-up), Visitenplan (Zeitpunkt und Angabe des tolerierbaren Zeitfensters) und Beschreibung von Labor- und ggf. weiterer Diagnostik, es empfiehlt sich die tabellarische und graphische Darstellung. Tabelle 1: Zeitplan der Studie Einschluss erster Patient III. Quartal 2015 (first patient first visit, FPFV): Einschluss letzter Patient (LPFV): III. Quartal 2017 Letzte Visite des letzten Patienten (LPLV): IV. Quartal 2017 Ende der klinischen Prüfung IV. Quartal 2017 Abschlussbericht/Ergebnisbericht: IV. Quartal 2018 (vgl. II. Synopsis) Ende der klinischen Prüfung Als Ende der Prüfung wird der Zeitpunkt von 30 Tagen nach der letzten Visite des letzten Patienten (LPLV) definiert, da zu diesem Zeitpunkt die reguläre Nachbeobachtungszeit eventueller SAEs innerhalb der klinischen Prüfung endet. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 24 von 67 An dieser Stelle ist das Ende der klinischen Prüfung zu definieren, das die Meldepflicht gegenüber den Behörden terminiert. In der Regel wird als Prüfungsende die letzte Visite des letzten Patienten (LPLV) angenommen, ggf. verlängert um die Zeit, innerhalb derer AEs/SAEs nachverfolgt werden (siehe Kapitel 7.2). Abbildung 1: Ablaufdiagramm der klinischen Prüfung 4.2. Diskussion des Studiendesigns Hilfreich bei der Diskussion ist es zu argumentieren, welche Maßnahmen zur Vermeidung von Bias getroffen worden sind (Verblindung, Randomisierung, etc.) und warum diese als notwendig erachtet werden. Die gewählte Fallzahl (Verweis auf statistische Fallzahlplanung) und der Vergleich (Parallelgruppen, Crossover) sollten begründet werden. Auch die Wahl der Kontrollgruppe sollte begründet werden. Hierbei ist ggf. die ethische Vertretbarkeit der Placebo-Kontrolle zu begründen bzw. darzustellen, dass bzw. ob die verwendete Kontrolle Standard der regulären Heilfürsorge ist. 4.3. Auswahl der Studienpopulation Die Studienpopulation sollte mit klinischen Worten umschrieben werden. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 25 von 67 Es muss ggf. begründet werden, warum z.B. Kinder oder Nicht-Einwilligungsfähige in die Prüfung einbezogen werden und wie deren besondere Schutzwürdigkeit berücksichtigt wird. Begründung der Geschlechterverteilung Die erwartete Geschlechterverteilung benennen und begründen, warum diese Verteilung in der Gruppe der betroffenen Personen zur Feststellung möglicher geschlechtsspezifischer Unterschiede bei der Wirksamkeit oder Unbedenklichkeit des geprüften Arzneimittels geeignet erscheint. Ggf. auch auf das Kapitel 6.1.5 verweisen. 4.3.1. Einschlusskriterien Studienindikation Generell gilt, dass Ein- wie auch Ausschlusskriterien möglichst eindeutig und mit wenig Interpretationsspielraum beschrieben werden sollten. Auch ggf. Messgrößen oder Diagnostik beschreiben, anhand derer dieses Kriterium geprüft werden kann (z.B. Befund gesichert anhand einer Koronarangiographie, die in den Akten vorliegen muss, nicht älter als xxx / Entscheidung durch Einschätzung des Prüfers / eines ärztlichen Mitglieds der Prüfgruppe im Rahmen der Anamnese). Es ist zu berücksichtigen, dass die Festlegung von Ein- und Ausschlusskriterien ggf. zu studienbedingten z.B. diagnostischen Maßnahmen führt, die in der Beschreibung der Visiten berücksichtigt werden müssen und erst nach Einwilligung durchgeführt werden dürfen. Alter größer oder gleich 18 Jahre, Geschäftsfähigkeit, Vorliegen einer schriftlichen Einwilligungserklärung des Probanden/Patienten Falls gesunde Probanden eingeschlossen werden, muss definiert werden, wer als „gesund“ gilt und wie der Status festgestellt wird. 4.3.2. Ausschlusskriterien Teilnahme an anderen interventionellen Prüfungen Kontraindikationen gemäß Fachinformation oder Investigator’s Brochure Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 26 von 67 Arzneimittel, mit denen Wechselwirkungen zu erwarten sind Erkrankungen / Befunde, die z.B. die Zielgröße maßgeblich verändern und damit den zu untersuchenden Therapieeffekt verwischen oder ggf. verhindern könnten Schwangerschaft oder Stillzeit Fehlende sichere Empfängnisverhütungsmaßnahmen (je nach Prüfpräparat gilt das auch für Partner/Partnerinnen von Prüfungsteilnehmern). Als sichere Schwangerschaftsverhütungsmaßnahmen gelten Verfahren mit einem PearlIndex kleiner 1 %: o Orale hormonelle Kontrazeption („Pille“), (falls keine Beeinträchtigung der Wirksamkeit im Rahmen der Studie angenommen wird, z.B. kann bei Prüfpräparaten, die zu Erbrechen und Durchfall führen, nicht mehr von ausreichender Sicherheit ausgegangen werden) o Dermale hormonelle Kontrazeption, o Vaginale hormonelle Kontrazeption (NuvaRing®) o Kontrazeptionspflaster, o Langzeit wirksame, injizierbare Kontrazeptiva, o Progesteron abgebendes Implantat (Implanon®), o Tubenligatur (weibliche Sterilisation), o Hormon abgebendes Intrauterinpessar („Hormonspirale“), o Doppelte Barrieremethoden Als nicht zuverlässig gelten daher beispielsweise: Kondom plus Spermizid, einfache Barriere Methoden (Scheidenpessar, Kondom, weibliches Kondom), Kupferspirale, Rhythmusmethoden, Basaltemperaturmethode, Coitus interruptus. Die Regelungen in Bezug auf Empfängnisverhütung leiten sich von Guideline ICH E8 Chapter 3.2.2.1 Selection of Subjects in Verbindung mit ICH M3 Note 4 ab. Personen, die in einem Abhängigkeits- / Arbeitsverhältnis zum Sponsor oder Prüfer stehen Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 27 von 67 Unterbringung in einer Anstalt aufgrund gerichtlicher oder behördlicher Anordnung. 4.4. Nachträglicher Ausschluss von Prüfungsteilnehmern (A) Ausschluss von der Therapie (B) Ausschluss von den Untersuchungen (C) Ausschluss von der Dokumentation Hier sollte festgelegt werden: a) wann und wie Prüfungsteilnehmer von der klinischen Prüfung/Behandlung mit dem/den Prüfpräparat(en) auszuschließen sind, b) welche Daten ausgeschlossener Prüfungsteilnehmer zu welchen Zeitpunkten zu erheben sind, c) ob und wie Prüfungsteilnehmer zu ersetzen sind. CAVE: Der nachträgliche Ausschluss von Prüfungsteilnehmern ist – so irgend möglich – zu vermeiden, um eine aussagekräftige Analyse gemäß des Intention-to-Treat-Prinzips zu ermöglichen. Ein Ausschluss von der Therapie bedeutet z.B. nicht notwendigerweise ein Ausschluss von den weiteren Untersuchungen oder der weiteren Dokumentation. 4.4.1. Verfahren bei vorzeitigem Ende der Behandlung im Rahmen der klinischen Prüfung Dokumentation der entsprechenden Daten mit zugehöriger Begründung, weitere Behandlung, Nachbeobachtung des Patienten etc. 4.5. Schließen von Prüfzentren / Vorzeitige Beendigung der klinischen Prüfung 4.5.1. Schließen von Prüfzentren Hier sind z.B. Kriterien, die zur Erwägung des Schließens führen, Eskalationskette etc. zu benennen. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx 4.5.2. Seite 28 von 67 Abbruch der gesamten Studie Der Sponsor ist berechtigt, die Prüfung aufgrund relevanter medizinischer oder ethischer Bedenken oder mangelnder Durchführbarkeit der Studie vorzeitig zu beenden. In einem solchen Fall werden die Gründe für die vorzeitige Beendigung der Prüfung detailliert dokumentiert. Patientinnen/Patienten, die zum Zeitpunkt des Abbruchs der Prüfung noch in Behandlung stehen, sollen sich einer Abschlussuntersuchung unterziehen, die entsprechend dokumentiert wird. Bestehen bei einem Prüfer ethische Bedenken bezüglich der Weiterführung der Prüfung, so muss dies unverzüglich dem LKP angezeigt werden. Die vorzeitige Beendigung der klinischen Prüfung wird in Erwägung gezogen, wenn sich das Nutzen-Risiko-Verhältnis für die Patienten deutlich verändert, die Anwendung der Prüfmedikation nicht länger vertretbar ist, der Sponsor aus Sicherheitsgründen (z.B. auf Anraten des DMC) einen Abbruch der klinischen Prüfung für notwendig erachtet, der frühzeitige Nachweis einer Überlegenheit bzw. Unterlegenheit einer Behandlungsgruppe durch eine Zwischenauswertung oder andere Forschungsergebnisse erbracht wird, die klinische Prüfung sich als nicht durchführbar erweist Über den Abbruch der Prüfung entscheidet der Sponsor im Benehmen mit dem LKP, DMC / SC / Advisory Board / Statistiker. 4.6. Behandlungen 4.6.1. Angewendete Behandlungen Eine genaue, nachvollziehbare Beschreibung der Behandlungen, die in jeder Behandlungsgruppe der klinischen Prüfung durchgeführt werden, ist erforderlich. Dies schließt die gesamte Prüfungsdauer sowie die Applikationsform, Dauer und Dosierung ein. Dabei ist auch abzugrenzen, wie lange die Studienbehandlung fortgeführt wird und wie lange ggf. die Nachbeobachtungszeit pro Patient dauert. Hierbei ist klarzustellen, welche Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 29 von 67 Behandlungsmaßnahmen von der klinischen Praxis / vom klinischen Standard bzw. Leitlinien abweichen. 4.6.2. Beschreibung der Prüfmedikation Beschreibung der Prüfmedikation (Zusammensetzung, Konzentration. Herkunft des Placebos bzw. der Vergleichsmedikation. Insbesondere bei lang dauernden klinischen Prüfungen Angaben zur Haltbarkeit und Verfahren bei abgelaufenem Haltbarkeitsdatum). Hierbei ist zu beachten, dass als Prüfmedikation auch eventuelle Vergleichsmedikation zählt. Handelsname: Nur angeben, wenn ein bestimmtes Handelspräparat verwendet werden soll Wirkstoff: ATC-Code: Darreichungsform: Dosis: Hersteller: Kann entfallen, wenn Handelsware von frei wählbaren Herstellern verwendet werden soll und kein Herstellungsschritt erfolgt. Bereits zugelassen für folgende Indikation: Falls die Prüfmedikation bereits für die Behandlung am Menschen zugelassen ist, sollten hier Indikation und ggf. Kontraindikationen zusammenfassend dargestellt werden. 4.6.2.1. Herstellung der Prüfmedikation Sind Herstellungsprozesse im Sinne des § 4 (14) AMG erforderlich (auch z.B. Verpackung, Umverpackung oder Kennzeichnung), so benötigt man dafür eine Herstellungserlaubnis für Prüfpräparate, d.h. es muss eine entsprechende Einrichtung eingebunden werden. 4.6.2.2. Kennzeichnung der Prüfmedikation Hier sollte beschrieben werden, wer für die Kennzeichnung verantwortlich ist. Ferner sollte der geplante Text eines Musteretiketts oder ein Musterbegleitschreiben in der Anlage 11.7 beigefügt werden. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 30 von 67 Die Kennzeichnung der Prüfmedikation erfolgt entsprechend § 5 GCP-V. 4.6.2.3. Lagerung der Prüfmedikation Sind besondere Lagerungsbedingungen zu berücksichtigen, so sollten diese hier beschrieben werden (Kühlkette und deren Einhaltung etc.), ebenso z.B. die Maßnahmen, die zur Kontrolle ergriffen werden müssen (z.B. Temperaturaufzeichnungen o.ä.). 4.6.3. Einhaltung der Therapie / Ausgabe und Rücknahme der Prüfmedikation Hier sollten Strategien benannt werden, die zur Verbesserung der Therapie-Adhärenz sowie zu deren Monitoring eingesetzt werden. Beschreibung von Dokumentation und Verfahren zur Ausgabe und Rücknahme der Prüfmedikation beim einzelnen Patienten sowie beim Prüfzentrum. Hierbei ist auch zu beschreiben, was mit zurückgenommener oder nicht mehr benötigter Medikation geschieht. Beschreibung der Verantwortlichkeit für die Drug Accountability (Prüfmedikation / Vergleichsmedikation / Placebo). Darüber hinaus sollte ein Verfahren beschrieben werden, das eine unverzügliche Rücknahme der Prüfpräparate ermöglicht, falls dies erforderlich wird. 4.6.4. Methode zur Zuordnung der Patienten zu den Behandlungsgruppen Hier ist ggf. das Randomisierungsverfahren praktisch zu beschreiben (wer teilt wann und wie welche Behandlungen zu; Zuteilungsverhältnis; Geheimhaltung; Struktur der Patientenidentifikation (PatID). Ferner sollte ggf. die Stratifizierung begründet werden. Die angegebenen Details dürfen die Vorhersagbarkeit der Zuteilung nicht ermöglichen (z.B. sollte die Blocklänge nicht angegeben werden). 4.6.5. Tabelle 2: Auswahl der Dosierung der Prüfmedikation Prüfmedikation morgens Prüfplan vX-XX vom XX.XX.XXXX mittags abends zur Nacht Universität zu Köln Prüfplan-Code xxx Seite 31 von 67 Tag 1 Tag 2 Tag 3 Die Dosierung und auch die Dauer der Therapie muss detailliert begründet werden. 4.6.6. Festlegung von Dosierung und Zeitpunkt der Prüfmedikationsgabe für jeden Prüfungsteilnehmer Hier ist zu erläutern wie ggf. eine Dosierungsanpassung beim einzelnen Prüfungsteilnehmer erfolgt. Hier sollten auch Kriterien zur Modifikation/Beendigung der zugeteilten Behandlung genannt werden (z.B. Veränderung der Dosierung infolge von unerwünschten Nebenwirkungen, Wunsch des Patienten oder Verbesserung/Verschlechterung der Erkrankung). Ggf. muss bezüglich der Beendigung auch Rückbezug auf den Abschnitt 4.4 genommen werden. 4.6.7. Verblindung Beschreibung der Verblindung (wer wird verblindet und wie – Teilnehmer, Behandler, Bewerter, Auswerter), falls möglich und sinnvoll, ggf. auch Begründung, warum z.B. eine doppelte Verblindung nicht möglich ist und wie man trotzdem Bias minimiert (etwa Verblindung von Patient und Nachuntersucher), Verfahren, Sicherstellung der Aufrechterhaltung (Regelung des Zugangs zu vertraulichen Unterlagen wie der Randomisierungsliste, Beschreibung der praktischen Umsetzung (wer macht was)) und Erfolgskontrolle der Verblindung bzw. Verweis auf entsprechende Manuals, die Details regeln. 4.6.7.1. Entblindung Beschreibung des Entblindungsverfahrens auf Seiten des Prüfzentrums im Notfall (z.B. Notfallumschläge, andere Entblindungsmaßnahmen) unter Berücksichtigung der größtmöglichen Aufrechterhaltung der Verblindung, ggf. Rücksprache mit dem Sponsor, Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 32 von 67 Meldeverpflichtungen bei Entblindung. Ggf. sind mit der Entblindung verbundene Meldeprozesse zu beschreiben (vgl. auch 7.3.4). 4.6.8. Vorangegangene Therapie und Begleittherapie Beschreibung, welche vorangegangene Therapie und welche Begleittherapie zulässig sind. 4.6.8.1. Ausweichtherapie im Notfall Beschreibung der Ausweichtherapie. 4.6.9. Weiterbehandlung nach Ende der klinischen Prüfung Hier ist zu beschreiben, wie die Prüfungsteilnehmer nach Ende der klinischen Prüfung weiterbehandelt werden. Hierbei ist auch zu bedenken, dass Prüfmedikamente ggf. ausgeschlichen oder möglicherweise nicht ohne weiteres abgesetzt werden können. 4.7. Wirksamkeits- und Sicherheitsvariablen In dem vorliegenden Kapitel soll beschrieben werden, wie die klinischen Ziele im Sinne von messbaren Größen operationalisiert werden. Methoden und Zeitpunkte für die Bewertung, Aufzeichnung und Auswertung sollen beschrieben werden einschließlich der Begründung, warum diese Größen geeignet sind, die zu untersuchenden Ziele zu erfassen (cave: konsistente Darstellung mit der Synopse und Statistik beachten). Gemäß „Chan A et al. RESEARCH METHODS AND REPORTING SPIRIT 2013 explanation and elaboration : guidance for protocols of clinical trials. 2013;1–42“ werden Ergebnisvariablen durch vier Aspekte bestimmt, (1) die spezifische Messgröße (z.B. systolischer Blutdruck), (2) die Analyse-Metrik (z.B. Veränderung vom Startwert, Schlusswert, Zeit bis zum Ereignis), (3) die Methode der Aggregation (z.B. Median, Proportion) und (4) den Zeitpunkt des Ergebnisses. Die klinische Relevanz der gewählten Ergebnisvariablen zur Wirksamkeit und Sicherheit sollte erklärt werden. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx 4.7.1. 4.7.1.1. Seite 33 von 67 Messung der Wirksamkeits- und Sicherheitsvariablen Primäre Zielgröße Hier ist zu beschreiben, welches die primäre Zielgröße ist und wie sie gemessen wird (s.o.). 4.7.1.2. Sekundäre und weitere Zielgrößen Hier ist zu beschreiben, welches die sekundären Zielgrößen sind und wie sie gemessen werden (s.o.). 4.7.1.3. Sicherheitsanalyse Hier ist zu beschreiben, welche Größen der Sicherheitsanalyse zugrunde gelegt und wie sie gemessen werden (s.o.). 4.7.1.4. Beschreibung der einzelnen Visiten Die Visiten werden zu folgenden Zeitpunkten durchgeführt (s. Tabelle 3). Ggf. weitere Beschreibung der Visiten einfügen Tabelle 3: Untersuchungen im Rahmen der klinischen Prüfung Visite Zeitpunkt 0 1 2 Tag 0 Tag 7 Tag 14 (+/- 3) (+/-3) X X Aufklärung und Einverständnis X Untersuchung 1 X Untersuchung 2 X 3 4 5 6 X X X X X X Dauer der klinischen Prüfung beim individuellen Patienten Die Dauer der klinischen Prüfung beim individuellen Patienten sollte kurz angegeben werden. 4.7.2. Angemessenheit der Messverfahren Diskussion und Bewertung der Messverfahren der Wirksamkeits- und Sicherheitsvariablen. Empfehlung: die fachgebietsspezifischen Leitlinien und Guidelines (z.B. des Committee for Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 34 von 67 Medicinal Products for Human Use / CHMP und/oder der Core Outcome Sets der COMETinitiative (http://www.comet-initiative.org) sollten möglichst berücksichtigt werden. 4.7.3. Pharmakokinetik/Bestimmung von Medikamentenspiegeln Sofern pharmakokinetische Untersuchungen oder andere Bestimmungen von Medikamentenspiegeln vorgesehen sind, sind diese hier zu beschreiben. Anderenfalls kann dieses Kapitel einschließlich der Überschrift entfallen. 4.8. Sicherstellung der Datenqualität 4.8.1. Monitoring Zur Qualitätssicherung der Studie wird ein Monitoring in den Prüfzentren durchgeführt. Ziel des Monitorings ist die Überprüfung der Sicherstellung und des Schutzes der Rechte und Sicherheit der Prüfungsteilnehmer, der Validität, Nachprüfbarkeit und Vollständigkeit der Studiendaten sowie der Übereinstimmung der Studiendurchführung mit dem Prüfplan, GCP und den geltenden gesetzlichen Bestimmungen. Falls für die Studie ein risikoadaptiertes Monitoring, z.B. in Anlehnung / nach ADAMON (http://ctj.sagepub.com/content/6/6/585.abstract) geplant ist, sollte dies entsprechend unter Angabe der Risikoklassifizierung beschrieben werden, darüber hinaus ggf. ein Hinweis auf Maßnahmen der zentralen Qualitätssicherung. Das Monitoring erfolgt risikoadaptiert und wird flankiert durch Maßnahmen der zentralen Qualitätssicherung entsprechend des ADAMON-Konzeptes, wie z.B. zeitnahe Mahnung ausstehender Dokumentation und Vergleich der Dokumentations-Qualität zwischen den Zentren. Der genaue Umfang und die Art des Monitorings werden in einem gesonderten MonitoringManual beschrieben. Hier sind jedoch die Grundzüge wiederzugeben: Mindestumfang der Monitoringaktivitäten, Maßnahmen zur zentralen Qualitätssicherung sowie Informationswege. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 35 von 67 Alle Prüfer erklären sich damit einverstanden, dass der Monitor in regelmäßigen Abständen das Prüfzentrum besucht und dabei seitens des Prüfzentrums in geeigneter Weise unterstützt wird. Dies wird in den Prüfzentrumsverträgen vereinbart. Es wird eine entsprechende Passage in die Einwilligungserklärung (siehe Kap. 5.4) aufgenommen, die dem Monitor das Recht gewährt, unter Berücksichtigung des Datenschutzgesetzes die Dokumentationsbögen (CRF) mit den Originalunterlagen zu vergleichen (Krankenblätter, EKG, Laborausdrucke etc.). Die Prüfer ermöglichen dem Monitor für das prüfungsbezogene Monitoring direkten Zugang zu allen erforderlichen Unterlagen. Sinn und Zweck dieser Besuche sind insbesondere: die Prüfung der Einwilligungserklärungen, die Prüfung der Patientensicherheit (Auftreten und Dokumentation / Meldung von AEs bzw. SAEs), die Überprüfung der CRFs auf Genauigkeit und Vollständigkeit, die Validierung der CRFs gegenüber den Originaldaten (Source Data Verification, SDV)), die Überprüfung der Handhabung der Prüfmedikation. die Evaluation des Prüfungsfortgangs, die Kontrolle der Compliance mit dem Prüfplan, die GCP-konforme Durchführung der Studie am Prüfzentrum Besprechung mit dem Prüfer über die Durchführung der Studie und ggf. festgestellte Mängel Über jeden Besuch wird ein Monitorbericht erstellt, der den Fortschritt der klinischen Prüfung dokumentiert und über alle aufgetretenen Schwierigkeiten (z.B. Verweigerung der Einsichtnahme) unterrichtet. Der Prüfer wird zur Behebung eventueller Mängel die vom Monitor vorgeschlagenen Maßnahmen angemessen berücksichtigen. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx 4.8.2. Seite 36 von 67 Audits / Inspektionen Der Sponsor hat das Recht, im Rahmen der Qualitätssicherung Audits am Prüfzentrum und anderen ggf. an der Studie beteiligten Einrichtungen durchzuführen. Ziel von Audits ist die Kontrolle der Validität, Nachprüfbarkeit und Vollständigkeit der Daten und der Glaubwürdigkeit der klinischen Prüfung sowie die Prüfung der Sicherstellung der Patientenrechte und Gewährleistung der Patientensicherheit. Der Sponsor kann hierzu Personen beauftragen, die ansonsten nicht in die klinische Prüfung eingebunden sind (Auditoren). Diesen wird gestattet, in sämtliche prüfungsbezogene Unterlagen Einsicht zu nehmen (insbesondere: Prüfplan, Erhebungsbögen, Patientenakten, Dokumentation der Prüfmedikation, prüfungsbezogene Korrespondenz). Der Sponsor und alle teilnehmenden Prüfzentren verpflichten sich, Auditoren und Inspektionen von zuständigen Behörden zu unterstützen und in diesem Zusammenhang den beauftragten Personen Zugang zu den Originaldokumenten zu gewähren. Alle Audits durchführenden Personen verpflichten sich, personenbezogene Daten sowie weitere Daten vertraulich zu behandeln. 4.9. Dokumentation Alle prüfungsrelevanten Daten werden von einem zuständigen Mitglied der Prüfgruppe zeitnah in den bereitgestellten (ggf. elektronischen) Dokumentationsbögen erfasst. Die Erhebungsbögen werden vom Prüfer bzw. autorisierten, ärztlichen Mitglied der Prüfgruppe persönlich unterzeichnet. Geben Sie hier ggf. auch Ausfüll- und Korrekturhinweise (z.B. vollständiges Ausfüllen, Korrektur muss den ursprünglichen Eintrag lesbar lassen und abgezeichnet werden; siehe auch ICH-GCP E6). Hier werden die Quelldaten für die Studie festgelegt. Dies umfasst auch die Definition, welche Daten ggf. ausschließlich in den Prüfbögen (CRF) erfasst werden. Dies bedeutet, dass ggf. Teile des CRF als Quelldaten (Source Data) deklariert werden. In der Regel wird dies beispielsweise für vom Patienten selbst ausgefüllte Bögen oder Patienten-Tagebücher zutreffen. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx 4.9.1. Seite 37 von 67 Datenmanagement Die folgenden Angaben sind in der Regel zutreffend, wenn das Datenmanagement im ZKS Köln durchgeführt wird. Sowohl die IT-Infrastruktur als auch das Personal für das Datenmanagement wird vom ZKS Köln gestellt. Die Studiendatenbank wird vor Dateneingabe vom ZKS Köln gemäß eigener Standard Operating Procedures entwickelt und validiert. Das Datenmanagementsystem basiert auf einer kommerziellen Studiensoftware und speichert die Daten in einer Datenbank. Alle Änderungen, die an den Daten vorgenommen werden, werden in einem Audittrail gespeichert. Die Studiensoftware verfügt über ein studienspezifisch anpassbares Benutzerund Rollenkonzept. Die Datenbank ist in ein allgemeines IT-Infrastruktur- und Sicherheitskonzept mit Firewall und Backupsystem eingebunden. Die Daten werden täglich gesichert. Nach der Vervollständigung und Bereinigung der Daten wird die Datenbank geschlossen und die Daten für die statistische Analyse exportiert. Im ZKS Köln werden alle Dokumentationsbögen registriert und auf Vollständigkeit geprüft. Die Daten werden von unabhängigen Dateneingabekräften doppelt in eine validierte Studiendatenbank eingegeben und abgeglichen. Zusätzlich werden Plausibilitäts-Checks in der Datenbank durchgeführt. Unstimmigkeiten und Unplausibilitäten werden vom Datenmanager schriftlich mit dem Zentrum abgeklärt. Diese Rückfragen (Queries) sind vom Zentrum zeitnah zu beantworten. Details werden im Datenmanagement-Manual geregelt. Alternativ für RDE-Studien lautet der letzte Absatz: Die Daten werden in den Zentren online über das Internet erfasst. Während der Eingabe erfolgen Plausibilitätsprüfungen, so dass viele Unstimmigkeiten sofort geklärt werden. Ein Datenmanager des ZKS Köln führt später weitere Prüfungen auf Vollständigkeit und Plausibilität durch und klärt Unstimmigkeiten mit dem Zentrum auf elektronischem Wege über die Studiensoftware. Diese elektronischen Rückfragen (Queries) sind vom Zentrum zeitnah zu beantworten. Details werden im Datenmanagement-Manual geregelt. Beschreiben Sie hier auch Notfall-Prozesse für den Fall eines Defekts des elektronischen Datenerfassungs-Systems Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 38 von 67 Im Fall eines Defekts des elektronischen Datenerfassungs-Systems werden Ausdrucke der Erfassungsmasken zur Verfügung gestellt, sodass bei einem Systemausfall die Daten auf Papier zwischen dokumentiert werden können. 4.9.2. Archivierung Alle Dokumentationsbögen, Einverständniserklärungen sowie weitere wichtige Prüfungsunterlagen werden gemäß § 13 (10) GCP-V mindestens 10 Jahre aufbewahrt. Die Prüfstelle wird alle wesentlichen Unterlagen der klinischen Prüfung nach der Beendigung oder dem Abbruch der Prüfung mindestens 10 Jahre aufbewahren, es sei denn, es gelten aus anderen Vorschriften längere Aufbewahrungspflichten. Für klinische Prüfungen von Arzneimitteln, deren Ergebnisse in einem Zulassungsantrag verwendet werden gelten weiterführende Archivierungspflichten. Für klinische Prüfungen, in denen zum Zwecke der medizinischen Forschung Röntgenstrahlung, radioaktive Stoffe oder ionisierende Strahlung am Menschen eingesetzt wird, gelten die entsprechenden Archivierungszeiten, die in der Röntgenverordnung (RöV) bzw. der Strahlenschutzverordnung (StrlSchV) festgelegt sind. U.a. sind nähere Hinweise zur Archivierung auf der Homepage des Site-ManagementSystems des ZKS Köln (https://clinicalsite.org/) unter Dokumente/Externes Archiv der klinikinternen SOP „Archivierung von papierbasierten Studienunterlagen in einem externen Archiv (Fa. Hasenkamp)“ Vx-xx-F zu entnehmen. Zu beachten ist, dass eine eventuelle Digitalisierung behördensicher erfolgen muss und ggf. aufgrund gesetzlicher oder anderer Vorschriften die Originale erhalten bleiben müssen. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 39 von 67 5. Ethische und regulatorische Aspekte 5.1. Unabhängige Ethikkommissionen Die klinische Prüfung wird erst nach Vorliegen einer zustimmenden Bewertung der zuständigen Ethikkommission begonnen. In jedem weiteren Prüfzentrum wird die klinische Prüfung erst durchgeführt, nachdem die zuständige beteiligte Ethikkommission die Eignung der Prüfstelle und die Qualifikation des Prüfers und seines Stellvertreters festgestellt hat. 5.2. Ethische Durchführung der klinischen Prüfung Der vorliegende Prüfplan sowie ggf. nachfolgende Änderungen des Prüfplans wurden bzw. werden in Übereinstimmung mit der Deklaration von Helsinki in der Fassung von 1996 verfasst. Bei Studien gemäß AMG muss auf die Deklaration von Helsinki in der Fassung vom Oktober 1996 (48th General Assembly, Somerset West, Republic of South Africa, October 1996) verwiesen werden. Ggf. sind hier kritische ethische Aspekte der klinischen Prüfung nochmals aufzugreifen. 5.2.1. Berücksichtigte gesetzliche Bestimmungen und Leitlinien Die vorliegende klinische Prüfung wird in Übereinstimmung mit den veröffentlichten Grundsätzen der Good Clinical Practice (ICH-GCP)-Leitlinie und den zutreffenden gesetzlichen Bestimmungen (insbesondere dem Arzneimittelgesetz und der GCPVerordnung) durchgeführt. Diese Grundsätze betreffen unter anderem EthikkommissionsVorgänge, Patientenaufklärung und Einwilligungserklärung, Befolgen des Protokolls, administrative Dokumente, Dokumentation der Prüfmedikation, Datenerhebung, Patientenakte (Quelldokumente), Erfassung und Meldung unerwünschter Ereignisse (AE), Vorbereitung von Inspektionen und Audits sowie Aufbewahrung von Unterlagen. Alle Prüfer und weiteres unmittelbar mit der Prüfung befasstes Personal werden darüber informiert, dass Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 40 von 67 in- und ausländische Überwachungs-, die zuständigen Bundesbehörden sowie vom Sponsor der klinischen Prüfung autorisiertes Personal Studiendokumente und Patientenakten jederzeit einzusehen berechtigt sind. 5.3. Behördenmeldungen, Genehmigungen und Registrierung Vor Beginn der klinischen Prüfung werden die entsprechenden Unterlagen der zuständigen Bundesoberbehörde (Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) bzw. PaulEhrlich-Institut (PEI) zur Genehmigung vorgelegt. Außerdem erfolgt eine Anzeige bei den zuständigen Behörden in den Ländern, in denen sich ein Prüfzentrum befindet. Bitte beachten, dass ggf. weitere Genehmigungen/Meldungen z.B. an die Bundesopiumstelle, das Bundesamt für Strahlenschutz, Landesumweltamt etc. erforderlich sein können. Vor Beginn der klinischen Prüfung wird die Registrierung der klinischen Prüfung bei einem von der WHO anerkannten Studienregister veranlasst (http://www.who.int/ictrp/en/). Die Meldung der Studie in einem öffentlich zugänglichen Register und die Angabe der ISRCTN-Nummer oder einer entsprechenden Registrierungsnummer ist bei klinischen Prüfungen mit Arzneimitteln, die mit Erwachsenen in den Phasen II bis IV und mit Kindern und Jugendlichen der Phasen I bis IV durchgeführt werden, optional, da diese Studien an EudraCT gemeldet werden und seit dem 22.03.2011 über das EU Clinical Trials Register (EU-CTR: www.clinicaltrialsregister.eu) öffentlich zugänglich sind. EU-CTR umfasst den WHO-Datensatz und ist von der ICMJE als öffentlich zugängliches Register anerkannt. Das Deutsche Register Klinische Studien (DRKS: www.drks.de) übernimmt auf Antrag die Daten aus dem EU Clinical Trials Register (und anderen Registern) in die DRKS-Datenbank, inkl. einer deutschen Übersetzung, und macht sie damit für Interessenten in Deutschland einfacher verfügbar. Es reicht dazu eine E-Mail an das DRKS ([email protected]) mit Angabe der EudraCT-Nummer bzw. der jeweiligen Registrierungsnummer. Aus dem Betreff sollte hervorgehen, dass es sich um den Datenimport aus einem anderen Register handelt. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 41 von 67 Im Falle einer von der Universität zu Köln gesponserten Studie muss der Prüfplan-Code bei der Qualitätssicherungseinheit des Sponsors beantragt werden. 5.4. Aufklärung und Einwilligung der Prüfungsteilnehmer Ein Patient kann nur in die klinische Prüfung aufgenommen werden, wenn er die Einwilligung hierzu erteilt hat, nachdem er durch einen Prüfer / ärztliches Mitglied der Prüfgruppe mündlich und schriftlich über Wesen, Bedeutung und Tragweite der klinischen Prüfung in angemessener und verständlicher Weise aufgeklärt worden ist. Er muss mit der Einwilligung zugleich erklärt haben, dass er mit der im Rahmen der klinischen Prüfung erfolgenden Aufzeichnung von Daten und ihrer Überprüfung durch vom Sponsor beauftragte Personen (z.B. Monitor, Auditor) sowie durch die zuständige Überwachungs- oder Bundesbehörde einverstanden ist. Der Patient wird über den potentiellen Nutzen und die Nebenwirkungen von Prüfmedikation und Placebo sowie über die Notwendigkeit und Bedeutung einer placebokontrollierten klinischen Prüfung unterrichtet. Es muss ihm klar sein, dass er seine Einwilligung jederzeit und ohne Angabe von Gründen zurückziehen kann, ohne dass ihm hieraus Nachteile erwachsen. Das Original der schriftlichen Einwilligung wird im Studienordner des Prüfzentrums verwahrt. Dem Patienten wird eine Kopie der schriftlichen Patientenaufklärung und Einwilligungserklärung ausgehändigt. Zudem wird eine Kopie in der Patientenakte abgelegt. Patienteninformation und Einwilligungserklärung sind als Anlage (11.6) beigefügt. Patienteninformation und Einwilligungserklärung, alle weiteren Unterlagen, die Teilnehmer erhalten sowie evtl. Rekrutierungsanzeigen werden vor Verwendung der zuständigen Ethikkommission zur zustimmenden Bewertung vorgelegt. Im Rahmen des Monitorings wird überprüft, ob die jeweils aktuelle Einwilligung vor Beginn der klinischen Prüfung vom betroffenen Patienten eigenhändig datiert und unterzeichnet wurde. 5.5. Probandenversicherung Eine Probandenversicherung wird für alle eingeschlossenen Patienten nach § 40 AMG im Rahmen des Gruppenversicherungsvertrages der Uniklinik Köln mit der xxx Versicherung Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 42 von 67 AG abgeschlossen. Sitz, Police-Nr., Telefon- und Faxnummer der Versicherungsgesellschaft werden in die Patienteninformation aufgenommen (siehe dort). Ggf. werden zusätzlich eine Wege-Unfall-Versicherung (bei studienbedingten Wegen z.B. zu Nachsorgeuntersuchungen) und/oder eine Versicherung nach StrlSchV/RöV erforderlich. 5.6. Datenschutz Die Bestimmungen der Datenschutzgesetze werden beachtet. Es wird sichergestellt, dass alle Untersuchungsmaterialen und -daten entsprechend der Datenschutzbestimmungen vor wissenschaftlichen Verwertungen adäquat pseudonymisiert werden. Die Prüfungsteilnehmer werden über die Weitergabe ihrer pseudonymisierten Daten im Rahmen der Dokumentations- und Mitteilungspflichten nach § 12 und § 13 GCP-V an die dort genannten Empfänger aufgeklärt. Personen, die der Weitergabe nicht zustimmen, werden nicht in die klinische Prüfung eingeschlossen. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 43 von 67 6. Statistische Methoden und Ermittlung der Fallzahl 6.1. Statistischer und analytischer Plan Ausführliche Informationen zur Planung, Durchführung und Ergebnisdarstellung der statistischen Analyse sind den Dokumenten ICH E9 und E3 zu entnehmen. Die wesentlichen, verwendeten statistischen Verfahren sind durch Literaturhinweise zu spezifizieren. 6.1.1. Analysepopulationen Alle Analysen werden an drei Populationen durchgeführt: Der primäre Auswertungsdatensatz ist die Intention-to-Treat-Population (ITT; Full Analysis Set, FAS). Dieser Datensatz umfasst alle Patienten, die in die klinische Prüfung aufgenommen und randomisiert wurden. Die Auswertung erfolgt strikt gemäß der Zuteilung nach Randomisierung. Es können ggf. Ausnahmen definiert werden, z.B. Patienten, die die Studienbehandlung trotz Randomisierung nie erhalten haben oder Verletzung von wesentlichen Ein- oder Ausschlusskriterien (s. ICH E9). Die eingeschränkte oder fehlende Auswertbarkeit von Studienteilnehmern („attrition“, „drop-out“, „lost-to-follow-up“) sollte definiert und/oder die Behandlung der entsprechend fehlenden Zielvariablen in der statistischen Analyse festgelegt werden. Der sekundäre Auswertungsdatensatz ist die Per-Protocol-Population (Per-Protocol Set, PPS). Dieser Datensatz umfasst alle Patienten, die dem Protokoll entsprechend behandelt wurden. Sofern Studienteilnehmer trotz Abweichungen von der Behandlung gemäß Protokoll dem PPS zugeordnet werden sollen, so sind hierfür begründet die Kriterien auszuführen. Die ITT-Analyse ist Standard bei konfirmatorischen Studien zur Überlegenheit, bei NichtUnterlegenheitsstudien sind ITT- und PPS-Datensatz gleichwertig. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 44 von 67 Der tertiäre Auswertungsdatensatz (Safety-Population) umfasst alle Patienten, die die Prüfmedikation bzw. Studienbehandlung bekommen haben. Diese werden dann ausgewertet wie behandelt. Es sollte hier beschrieben werden, wie die Zuordnung der Patienten zu diesen Populationen erfolgen soll: Welche Arten von möglichen Prüfplanverletzungen gibt es und wie sind diese zu werten. Häufig ist es sinnvoll, ein Gremium zu benennen (z.B. LKP und ein oder zwei weitere Experten), das die Verdachtsfälle von Prüfplanverletzungen im Konsensverfahren möglichst verblindet begutachtet, die nicht mittels eines Algorithmus anhand der Daten eindeutig zu klären sind. Details, die während der Prüfplan-Erstellung nicht klar sind, können und müssen später etwa im statistischen Analyseplan festgeschrieben werden. 6.1.2. Beschreibung des Patientenkollektivs Es ist darzustellen, anhand welcher Parameter das Patientenkollektiv beschrieben wird; z.B. Alter, Geschlecht, Größe, Gewicht, krankheitscharakterisierende Größen. 6.1.3. Primäre Zielvariable Beschreibung der Hypothesen, Test- und Schätzverfahren (adjustiert / nicht-adjustiert), ggf. weitergehender Verweis auf den statistischen Analyseplan. Strategie zur Behandlung fehlender Werte (einfache/mehrfache Imputation, best/worst case scenario), Umgang mit kleinen Strata; Sensitivitätsanalysen. 6.1.4. Sekundäre Zielvariablen 6.1.5. Subgruppenanalysen s.o. Im Prüfplan dargestellte Subgruppenanalysen (Hypothesen angeben) sind regelhaft verlässlicher als reine Post-hoc-Analysen. Eine Subgruppenanalyse nach Geschlecht sollte stets durchgeführt werden (vgl. § 42 (1) Nr. 2 AMG, § 7 (2) Nr. 12 GCP-Verordnung). Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx 6.1.6. Seite 45 von 67 Zwischenauswertung Soll eine „geplante“ Zwischenauswertung durchgeführt werden? Beschreibung der statistischen Methodik (ggf. Verweis auf 6.2, insbesondere wenn eine adaptives Design gewählt wurde). 6.2. Ermittlung der Fallzahl Statistische Begründung der Fallzahl in Abhängigkeit vom gewählten Studiendesign. Die zugrunde gelegten Annahmen (z.B. Effektgröße, Anteil und Einfluss eingeschränkt- oder nicht-auswertbarer Studienteilnehmer („attrition“, „drop-out“, „lost-to-follow-up“)) sollten beschrieben und begründet werden, ebenso die statistischen Verfahren zur Ermittlung der Fallzahl bzw. Power der klinischen Prüfung (Testverfahren, Fehlerarten, verwendete Software). Die Fallzahl sollte auch klinisch begründet werden (z.B. anhand des klinischrelevanten Unterschieds). Neben der geplanten Anzahl der Prüfungsteilnehmer - pro Gruppe und insgesamt - sollten auch Annahmen zur Anzahl im Prüfzentrum (im Durchschnitt und Range) dargestellt werden. Hier sind ggf. Strata, Zwischenauswertungen und SubgruppenAnalysen in die Begründung einzubeziehen. Hier auch begründen, warum die gewählte Geschlechterverteilung angemessen erscheint, in der Gruppe der betroffenen Personen den Nachweis möglicher geschlechtsspezifischer Unterschiede bei der Wirksamkeit oder Unbedenklichkeit des geprüften Arzneimittels zu erbringen (vgl. § 42 (1) Nr. 2 AMG, § 7 (2) Nr. 12 GCP-V). Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 46 von 67 7. Sicherheit 7.1. Definitionen 7.1.1. Unerwünschtes Ereignis Unerwünschtes Ereignis (Adverse Event, AE) ist jedes nachteilige medizinische Vorkommnis, das einer betroffenen Person widerfährt, der ein Prüfpräparat verabreicht wurde, und das nicht notwendigerweise in ursächlichem Zusammenhang mit dieser Behandlung steht. In der Regel unterliegen alle AEs (vgl. § 3 (6) GCP-V) der Dokumentationspflicht (vgl. § 12 (4) GCP-V). Als Ausnahme können an dieser Stelle, mit entsprechender Begründung, Ereignisse definiert werden, die im Rahmen der klinischen Prüfung nicht der Dokumentationspflicht im CRF unterliegen bzw. nicht als AEs zu verstehen sind. Als Beispiel können dies Laborabweichungen sein, die im Rahmen der Grunderkrankung (z.B. in der Onkologie) erwartet werden. Diese müssten dann z.B. erst ab einer zu definierenden Grenze (zB. ab einem bestimmten CTCAE Grad) als AEs dokumentiert werden. Eine Ausnahme dieser AEs von der Dokumentationspflicht im CRF entbindet den Prüfer nicht von seinen berufsrechtlichen Dokumentationspflichten (§ 10 Musterberufsordnung für die in Deutschland tätigen Ärztinnen und Ärzte). Begleiterkrankungen Die Verschlechterung einer vorbestehenden Erkrankung wird als unerwünschtes Ereignis definiert. Als unerwünschtes Ereignis gilt jedoch nicht eine vorbestehende Erkrankung, die bereits vor Einschluss in die klinische Prüfung zur Planung einer Behandlungsmaßnahme, z.B. eines stationären Aufenthalts, geführt hat. Schwangerschaft Das Eintreten einer Schwangerschaft gilt im Rahmen dieser klinischen Prüfung aus Gründen der Arzneimittelsicherheit als unerwünschtes Ereignis. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 47 von 67 Die Weiterbehandlung und ggf. das Absetzen des Prüfpräparats sowie die Nachbeobachtung sollte in Absprache zwischen dem Prüfer, Sponsor, ggf. auch dem Hersteller erfolgen. Sollte es aufgrund der pharmakologischen Eigenschaften des Prüfpräparates notwendig sein, auch Schwangerschaften bei Partnerinnen männlicher Prüfungsteilnehmer zu erfassen, sollte dies hier festgelegt werden. Dies ist z.B. dann der Fall, wenn das Prüfpräparat in relevanter Konzentration im Sperma ausgeschieden wird oder erbgutschädigende Wirkungen bekannt sind. 7.1.2. Nebenwirkung Nebenwirkung (Adverse Drug Reaction, ADR) ist jede nachteilige und unbeabsichtigte Reaktion auf ein Prüfpräparat, unabhängig von dessen Dosierung. Hierbei ist unter Reaktion ein Ereignis gemeint, das in einem mindestens möglichen Zusammenhang mit der Gabe des Prüfpräparates steht. (vgl. § 3 (7) GCP-V, § 4 (13) AMG) 7.1.3. Schwerwiegendes unerwünschtes Ereignis oder schwerwiegende Nebenwirkung Schwerwiegendes unerwünschtes Ereignis (Serious Adverse Event, SAE) oder schwerwiegende Nebenwirkung (Serious Adverse Drug Reaction, SADR) ist jedes unerwünschte Ereignis oder jede Nebenwirkung, die 1. zum Tode geführt hat, 2. lebensbedrohend ist, 3. eine stationäre Behandlung oder deren Verlängerung erforderlich macht, 4. zu bleibender oder schwerwiegender Behinderung oder Invalidität führt, 5. eine kongenitale Anomalie oder einen Geburtsfehler zur Folge hat, 6. oder jedes andere Ereignis, das von medizinischer Bedeutung ist. 1.-5.: § 3(8) GCP-V; 6. ICH E2A Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 48 von 67 Als lebensbedrohlich im obigen Zusammenhang werden Ereignisse betrachtet, bei denen die Gefahr zu sterben zum Zeitpunkt des Ereignisses bestand. Als Krankenhausaufnahme wird jeder stationäre Aufenthalt eines Prüfungsteilnehmers angesehen, der mindestens eine Nacht (0 – 6 Uhr) umfasst hat. Bereits vor der ersten Gabe des Prüfpräparates geplante Krankenhausaufnahmen gelten nicht als schwerwiegende unerwünschte Ereignisse, erfordern aber eine entsprechende Dokumentation in Patientenakte und CRF (siehe 7.1.1). 7.1.4. Unerwartete Nebenwirkung Unerwartete Nebenwirkung ist eine Nebenwirkung, die nach Art oder Schweregrad nicht mit der vorliegenden Information über das Prüfpräparat übereinstimmt (§ 3 (9) GCP-V). Die erwarteten Nebenwirkungen sind in dem entsprechenden Referenzdokument aufgeführt. Referenzdokument zum Prüfpräparat: Dokument benennen (Fachinformation oder Investigator’s Brochure) in der jeweils aktuellen Version. Referenzdokument zum Vergleichspräparat: Dokument benennen 7.1.5. Verdachtsfall einer schwerwiegenden unerwarteten Nebenwirkung Verdachtsfall einer schwerwiegenden unerwarteten Nebenwirkung (Suspected Unexpected Adverse Reaction, SUSAR) ist ein unerwünschtes Ereignis, das nach Art oder Schweregrad nicht mit der vorliegenden Information über das Prüfpräparat übereinstimmt, als schwerwiegend beurteilt wird und bei dem ein Zusammenhang mit der Prüfmedikation wenigstens als möglich eingeschätzt wird. §4 (13) AMG 7.1.6. Andere mögliche prüfungsspezifische Komplikationen und/oder Risiken Dieses Kapitel ist optional. Erwähnt werden können z.B. Überdosierung oder Intoxikationen der Studienmedikation, substanzspezifische schwere Nebenwirkungen Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 49 von 67 7.2. Dokumentation und Nachverfolgung von unerwünschten Ereignissen und Nebenwirkungen Der Sponsor trägt dafür Sorge, dass alle Personen, die an der Behandlung der Prüfungsteilnehmer beteiligt sind, adäquat über die Verantwortlichkeiten bei Auftreten unerwünschter Ereignisse informiert sind. Bei jeder Visite werden die Patienten befragt, ob unerwünschte oder schwerwiegende unerwünschte Ereignisse aufgetreten sind. Unerwünschte Ereignisse werden sowohl in der Patientenakte als auch in den Erhebungsbögen dokumentiert. Zum Umgang mit schwerwiegenden unerwünschten Ereignissen sei auf Abschnitt 7.3 verwiesen. Zur Sicherheitsanalyse sei auf Abschnitt 4.7.1.3 verwiesen. Ggf. ist es sinnvoll, unerwünschte Ereignisse ab der Abgabe der Einwilligungserklärung zu dokumentieren. Dies gilt z.B. für klinische Prüfungen, bei denen vor Gabe des Prüfpräparats prüfungsspezifische Maßnahmen erfolgen. Daher sollte hier definiert werden, ab welchem Zeitpunkt die Dokumentation / das Meldeverfahren beginnen soll. Ebenso sollte hier das Ende der Meldeverpflichtung beschrieben werden, und zwar für die Nachverfolgung bereits eingetretener Ereignisse ebenso wie für die Meldeverpflichtung für nach dem Studienende eingetretene Ereignisse (z.B. für beide 4 Wochen nach der letzten Studienvisite (LPLV), siehe auch 7.2.1;). Alle unerwünschten Ereignisse eines Prüfungsteilnehmers, die zwischen der Abgabe der Einwilligungserklärung / der ersten Einnahme des Prüfpräparates / der ersten prüfungsspezifischen Maßnahme / andere Definition und 30 Tage / anderer Zeitraum (abhängig von der Pharmakokinetik des Prüfpräparates) nach der letzten Visite / andere Definition des Prüfungsteilnehmers auftreten, werden dokumentiert und innerhalb des obengenannten Zeitraums nachverfolgt. Grundsätzlich besteht die Dokumentationspflicht während der gesamten Laufzeit der klinischen Prüfung, nicht nur während der Therapiephase zuzüglich einer Karenzzeit. SAEs sind ausnahmslos während der gesamten Dauer der klinischen Prüfung zu erfassen. Es können lediglich in begründeten Fällen Ausnahmen von der unverzüglichen Meldepflicht definiert werden. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 50 von 67 Alle unerwünschten Ereignisse innerhalb des oben definierten Beobachtungszeitraumes werden im CRF einschließlich der unten aufgelisteten Parameter dokumentiert. Ausgenommen hiervon sind die AEs, die unter Punkt 7.1.1 explizit genannt worden sind. Unerwünschte Ereignisse werden im CRF einschließlich folgender Parameter erfasst: Beginn und Ende, Intensität, Zusammenhang mit dem Prüfpräparat, schwerwiegend oder nicht-schwerwiegend, Unterbrechung oder Absetzen der Prüfpräparatgabe bzw. andere ergriffene Maßnahmen. Bei Auftreten eines unerwünschten Ereignisses muss der betreffende Prüfungsteilnehmer, unabhängig vom Kausalzusammenhang zwischen AE und Prüfpräparat, innerhalb des oben definierten Zeitraums so lange beobachtet werden, bis die Symptome abgeklungen sind, pathologische Laborwerte auf die Ausgangswerte zurückgegangen sind, sich eine plausible Erklärung für das unerwünschte Ereignis ergeben hat, bis zum Tode des Prüfungsteilnehmers oder bis die klinische Prüfung einschließlich des obengenannten Zeitraums bei dem betroffenen Patienten beendet ist. Erkrankungen, die vor Verabreichung des Prüfpräparates bestanden, werden nicht als AE sondern als Begleiterkrankung dokumentiert. Jede neue Erkrankung oder jede Verschlechterung einer bereits bestehenden Erkrankung wird im Verlauf der klinischen Prüfung als AE dokumentiert. Wenn ein unerwünschtes Ereignis als schwerwiegend eingeschätzt wird, wird das Ereignis zusätzlich zur AE-Dokumentation auf einem gesonderten SAE-Bogen dokumentiert. SAEs unterliegen einer gesetzlich vorgegebenen Meldepflicht (Verfahren dazu: siehe 7.3) 7.2.1. Intensität des unerwünschten Ereignisses Ggf. ist hier auf etablierte Codierungen wie beispielsweise die Common Terminology Criteria for Adverse Events (CTCAE) (ehemals Common Toxicity Criteria, CTC) zu verweisen. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 51 von 67 Es ist zu beachten, dass die Begriffe, die hier auftauchen, später genauso im CRF Verwendung finden müssen. Kategorien zur Bewertung von Schweregrad und Kausalität (siehe auch 7.2.2), sollten also so bezeichnet werden, wie sie auf den Meldebögen erscheinen sollen. CAVE: Die Intensität („severe/schwer“) darf nicht verwechselt werden mit dem Kriterium „schwerwiegend“ (seriousness, siehe 7.1.3), das zur Abgrenzung AE versus SAE geprüft wird! Der Prüfer (bzw. das ärztliche Mitglied der Prüfgruppe) wird die aufgetretenen unerwünschten Ereignisse hinsichtlich ihrer Intensität wie folgt einstufen: Leicht: Klinisches Symptom oder Zeichen, das gut toleriert wird. Mittel: Klinisches Symptom oder Zeichen, das ausreichend ist, die normale Aktivität zu beeinträchtigen. Schwer: Klinisches Symptom oder Zeichen, das zu einer starken Beeinträchtigung oder zur Arbeitsunfähigkeit oder der Unfähigkeit, alltägliche Verrichtungen durchzuführen führt. Der Prüfer (bzw. das ärztliche Mitglied der Prüfgruppe) wird die aufgetretenen unerwünschten Ereignisse hinsichtlich ihrer Intensität gemäß den NCI CTC Kriterien, Version 4.0, wie folgt einstufen: Grade 1 = mild Grade 2 = moderate Grade 3 = severe Grade 4 = life-threatening or disabling Grade 5 = death related to AE 7.2.2. Zusammenhang des unerwünschten Ereignisses mit dem Prüfpräparat Jedes unerwünschte Ereignis wird vom Prüfer (bzw. dem ärztlichen Mitglied der Prüfgruppe) beurteilt, ob ein Zusammenhang mit dem Prüfpräparat vermutet werden kann oder nicht. Dabei müssen die Art und das Muster der Reaktion, der zeitliche Zusammenhang zur Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 52 von 67 Verabreichung des Prüfpräparates, der klinische Status des Patienten, die Begleitmedikation und andere relevante klinische Parameter in Betracht gezogen werden. Wenn das Ereignis auf Grund mangelnder Effektivität oder auf Basis der Grunderkrankung eingetreten ist, wird es als nicht zusammenhängend beurteilt. Bei doppelblinden Studien ist zu beachten, dass die Kausalitätsbewertung seitens des Prüfers (bzw. des ärztlichen Mitglieds der Prüfgruppe) sowohl unter der Annahme erfolgt, dass es sich um das Verum handelt, als auch unter der Annahme, dass es sich um das Plazebo handelt (wenn nicht aus medizinischen Gründen eine sofortige Notfall-Entblindung erfolgen muss). Für die Kausalitätsbeurteilung des unerwünschten Ereignisses mit dem Prüfpräparat gelten folgende Begriffsbestimmungen (zur Erfassung im CRF siehe auch 7.2.1) (WHO Causality Assessment of Suspected Adverse Reactions): Ggf. kann auch ein anderes Klassifikationsschema verwendet werden. Das vorliegende Schema ist jedoch für die von der Universität zu Köln initiierten Studien vorgesehen. Sicher: Ein Ereignis, das einem nachvollziehbaren zeitlichen Ablauf nach der Anwendung des Prüfpräparates folgt oder bei der die Arzneimittelkonzentration in Körpergewebe oder -flüssigkeit gemessen wurde, einem bekannten oder erwarteten Antwortmuster auf das Prüfpräparat folgt und nach Absetzen oder Dosisreduktion verschwindet und bei erneuter Exposition wieder auftritt. Wahrscheinlich: Ein Ereignis, das einem nachvollziehbaren zeitlichen Ablauf nach der Anwendung des Prüfpräparates folgt, einem bekannten oder erwarteten Antwortmuster auf das verdächtigte Prüfpräparat folgt und nach Absetzen oder Dosisreduktion verschwindet und nicht durch die bekannten Merkmale des klinischen Zustandes des Probanden/Patienten erklärt werden kann. Möglich: Ein Ereignis, das einem nachvollziehbaren zeitlichen Ablauf nach der Anwendung des Prüfpräparates folgt, einem bekannten oder erwarteten Antwortmuster auf das verdächtigte Prüfpräparat folgt, das aber leicht auch durch eine Reihe anderer Faktoren hervorgerufen worden sein könnte. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 53 von 67 Unwahrscheinlich: Ein Ereignis, bei dem ausreichend Informationen vorliegen für die Annahme, dass kein Zusammenhang mit dem Prüfpräparat besteht. Nicht beurteilt: Ein Ereignis, das als unerwünschtes Ereignis gemeldet wurde, bei dem eine Beurteilung des Zusammenhangs zum Zeitpunkt der Meldung nicht erfolgt ist, weil weitere Daten notwendig sind oder zurzeit erhoben werden. Nicht beurteilbar: Eine Einschätzung des Zusammenhangs ist nicht möglich. Ein Verdachtsfall einer Nebenwirkung besteht, wenn der Kausalzusammenhang wenigstens als „möglich“ oder als „nicht beurteilbar“ bzw. „nicht beurteilt“ eingeschätzt wird. Ereignisse, die bezüglich eines kausalen Zusammenhangs als „unwahrscheinlich“ klassifiziert werden, gelten nicht als Verdachtsfälle einer Nebenwirkung. 7.3. Dokumentation und Meldung von schwerwiegenden unerwünschten Ereignissen, Schwangerschaften/Geburten und Änderungen des Nutzen-Risiko-Verhältnisses Details zu studienspezifischen Prozessen auf Seiten des Sponsors, wie z.B. Informationsund Kommunikationswege, Schnittstellen und Verweise auf Verantwortlichkeiten (Ansprechpartner, Einbindung DMC etc.) werden sinnvollerweise in einem studienspezifischen Manual geregelt. Auf ein solches sollte dann hier verwiesen werden. Darüber hinaus bietet sich die Darstellung in Form einer Graphik (flow chart) der Meldewege und –fristen an. Wird das SAE-Management von einer anderen Stelle durchgeführt, so ist der Text im gesamten Kapitel 7.3. entsprechend anzupassen. Im Folgenden werden die für die Studie relevanten Eckpunkte und Prozesse zur Dokumentation und Meldung von schwerwiegenden unerwünschten Ereignissen, Schwangerschaften/Geburten und Änderungen des Nutzen-Risiko-Verhältnisses definiert. Weitere Details zu studienspezifischen Prozessen auf Seiten des Sponsors, wie z.B. Informations- und Kommunikationswege, Schnittstellen und Verweise auf Verantwortlichkeiten (Ansprechpartner, Einbindung DMC etc.) sind in einem studienspezifischen Manual geregelt. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx 7.3.1. Seite 54 von 67 Meldung des Prüfers an den Sponsor bei schwerwiegenden unerwünschten Ereignissen Die Meldung eines schwerwiegenden unerwünschten Ereignisses (SAE) erfolgt auf separaten Meldebögen. Diese Meldebögen sind in Englisch auszufüllen. Mindestinformationen für eine SAE-Meldung: Gültige EUDRACT-Nummer Studiennummer des Sponsors Identifizierbarer Patienten/Probanden Identifizierbarer Berichterstatter Mutmaßliches schwerwiegendes unerwünschtes Ereignis Verdächtiges Prüfpräparat Bewertung des Kausalzusammenhang Unabhängig vom vermuteten Kausalzusammenhang muss jedes schwerwiegende unerwünschte Ereignis im Verlauf der klinischen Prüfung im entsprechenden CRF-Teil dokumentiert und umgehend, spätestens innerhalb von 24 Stunden nach Bekanntwerden auf einem SAE-Bogen an den Sponsor gemeldet werden. Bei einem bereits gemeldeten SAE ist jede Änderung u.a. das Auftreten einer Komplikation, eine Verschlechterung oder die Beendigung des SAEs als Follow-Up des ursprünglichen SAEs umgehend, spätestens 24 Stunden nach Bekanntwerden an den Sponsor zu melden. Auf dem Bogen wird dokumentiert, dass es sich um ein Follow-Up handelt, unter Angabe der ursprünglichen SAE-Nummer. Ein SAE, das als unzusammenhängend zu einem bereits gemeldeten SAE eingeschätzt wird, wird als neues SAE dokumentiert und gemeldet. Beim Wiederauftreten eines bereits gemeldeten Ereignisses ist im Einzelfall aufgrund der medizinischen Hintergründe zu entscheiden, ob es sich um eine Follow-Up Meldung des ursprünglichen SAE oder ein neues SAE handelt. Ein schwerwiegendes unerwünschtes Ereignis besteht fort, bis das Ereignis nicht mehr als schwerwiegend einzustufen ist, bis zum Tode des Prüfungsteilnehmers oder bis die klinische Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 55 von 67 Prüfung einschließlich des obengenannten Zeitraums bei dem betroffenen Patienten beendet ist. Alle SAE- und Follow-Up-Berichte sind umgehend, spätestens innerhalb von 24 Stunden nach Bekanntwerden, an die folgende Adresse zu melden: Zuständige Stelle: xxx Fax: xxx Falls SAE- und andere Berichte parallel an andere Stellen, z.B. an die das Prüfmedikament stellende Pharmafirma, gemeldet werden müssen, muss der folgende Abschnitt entsprechend angepasst werden (ggf. ergänzt um Angaben, innerhalb welcher Zeit die Meldung erfolgen muss). Parallel erfolgt eine Meldung an die folgende Adresse: Zuständige Stelle: xxx Fax: xxx Das Original des Meldebogens und die Faxbestätigung müssen bei dem CRF des Patienten / im Investigator Site File (ISF) abgelegt werden. Von der unverzüglichen Meldung als SAE sind die folgenden Ereignisse ausgenommen: XXX Es können Ereignisse von der unverzüglichen Meldepflicht ausgenommen werden (vgl. § 12 (4) GCP-V), z.B. die Hospitalisierung zur Durchführung einer Chemotherapie im Rahmen der Grunderkrankung bei onkologischen klinischen Prüfungen oder Nebenwirkungen einer nicht studienbedingt, jedoch bei jedem Patienten applizierten Therapie (z.B. BasisChemotherapie). Für diese sind entsprechende Referenzdokumente anzugeben. Umgekehrt kann es sinnvoll sein, es zur Pflicht zu machen, hier aufzuführende Ereignisse zeitnah zu melden, die für besonders sicherheitsrelevant gehalten werden, für sich genommen jedoch noch nicht die Kriterien eines SAE erfüllen würden (z.B. Leberwertveränderungen, wenn Lebertoxizitäten erwartet werden. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx 7.3.2. Seite 56 von 67 Meldung des Prüfers an den Sponsor bei Schwangerschaften und Geburten Die Meldung von Schwangerschaften und Geburten erfolgt auf jeweils separaten Meldebögen. Diese Meldebögen sind in Englisch auszufüllen. Jede Schwangerschaft im Verlauf der klinischen Prüfung muss dokumentiert und umgehend, spätestens innerhalb von 24 Stunden nach Bekanntwerden an den Sponsor gemeldet werden. Falls für das Prüfpräparat nicht ausgeschlossen werden kann, dass über einen männlichen Prüfungsteilnehmer auch Schwangerschaften der Partnerinnen der männlichen Prüfungsteilnehmer beeinflusst werden, muss hier ergänzt werden: Dies betrifft auch Schwangerschaften von Partnerinnen männlicher Prüfungsteilnehmer, die im Verlauf der Studienbehandlung des männlichen Prüfungsteilnehmers auftreten. Nach der Entbindung muss der Prüfer (bzw. das ärztliche Mitglied der Prüfgruppe) auf einem separaten Berichtsbogen den Verlauf der Geburt und die Gesundheit des Kindes dokumentieren und umgehend, spätestens innerhalb von 24 Stunden nach Kenntnis über die Geburt des Kindes, an den Sponsor melden. Ergibt die Bewertung des Schwangerschaftsausgangs, dass es sich um ein schwerwiegendes Ereignis (SAE) der Mutter oder des Kindes handelt oder gegebenenfalls sogar um den Verdachtsfall einer unerwarteten schwerwiegenden Nebenwirkung (SUSAR) handelt, muss dieser Fall entsprechend den gesetzlichen Vorgaben gemeldet werden (siehe 0). Der Dokumentationszeitraum endet 4 Wochen nach der Geburt des Kindes. Die Dokumentationsdauer sollte wenigstens den Zeitraum bis zur U2 umfassen. Der Zeitraum der Nachbeobachtung und die Häufigkeit der Dokumentation während dieses Zeitraums muss abhängig vom Risikoprofil des Prüfpräparates festgelegt werden. Für die Nachverfolgung des Ausgangs der Schwangerschaft wird eine gesonderte Einverständniserklärung nach erneuter Aufklärung der Schwangeren eingeholt. Für die Nachverfolgung des Kindes nach der Geburt wird eine gesonderte Einwilligung beider Erziehungsberechtigten nach erneuter Aufklärung eingeholt. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 57 von 67 Für die Nachverfolgung der Schwangerschaft bis zur Geburt reicht die Einverständniserklärung der Schwangeren aus. Alle Schwangerschafts- und Geburtsberichte inkl. Follow-Ups sind umgehend, spätestens innerhalb von 24 Stunden nach Bekanntwerden, an die folgende Adresse zu melden: Zuständige Stelle: xxx Fax: xxx Falls Schwangerschafts- und Geburtsberichte parallel an andere Stellen, z.B. an die das Prüfmedikament stellende Pharmafirma, gemeldet werden müssen, muss der folgende Abschnitt entsprechend angepasst werden (ggf. ergänzt um Angaben, innerhalb welcher Zeit die Meldung erfolgen muss). Parallel erfolgt eine Meldung an die folgende Adresse: Zuständige Stelle: xxx Fax: xxx Das Original des Meldebogens und die Faxbestätigung müssen bei dem CRF des Patienten / im Investigator Site File (ISF) abgelegt werden. 7.3.3. Zweitbegutachtung durch den Sponsor Alle SAE-Mitteilungen (schwerwiegende unerwünschte Ereignisse) werden vom Sponsor/LKP hinsichtlich der Kriterien schwerwiegend (siehe 7.1.3), kausal (siehe 7.2.2) und unerwartet (siehe 7.1.4) unabhängig von der Einschätzung des Prüfers / des ärztlichen Mitglieds der Prüfgruppe begutachtet. Bei verblindeten Prüfpräparaten sollte die Bewertung für den Sponsor soweit möglich nicht von einem Prüfer übernommen werden. 7.3.4. Entblindung bei verblindeten Prüfpräparaten In doppelblinden klinischen Prüfungen gemäß AMG muss die Entblindung vorgenommen werden, sofern Prüfer / das beurteilende ärztliche Mitglied der Prüfgruppe oder Sponsor das Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 58 von 67 Ereignis als schwerwiegend erachten und ein Kausalzusammenhang möglich ist sowie der Sponsor das Ereignis als unerwartet einschätzt (siehe 7.3.4). Bei Verdachtsfällen von schwerwiegenden unerwarteten Nebenwirkungen (SUSARs) erfolgt die Meldung an die Ethikkommission und die zuständige Bundesoberbehörde sowie an XXX (DMC, Pharmafirma) unter Offenlegung der Gruppenzugehörigkeit des Patienten, in unverblindeter Form. Ggf. soll hier und im Folgenden die entsprechend zuständige Stelle eintragen werden, die für die Entblindung zuständig ist. Die Verfahrensweise der Kausalitätsbewertung und Entblindung bzw. Zweitbegutachtung ist entweder an dieser Stelle im Prüfplan oder in anderen Dokumenten der klinischen Prüfung (z.B. Manuals, ggf. darauf verweisen) zu beschreiben. 7.3.5. Meldung an die Ethikkommission und die zuständige Bundesoberbehörde Während der klinischen Prüfung wird jeder bekannt gewordene Verdachtsfall einer schwerwiegenden unerwarteten Nebenwirkung (SUSAR) vom Sponsor/LKP der zuständigen Bundesoberbehörde sowie der Ethikkommission angezeigt. Bei multinationalen klinischen Prüfungen sind überdies die Meldepflichten in den anderen Staaten zu beachten (Behörden, Ethikkommissionen und eventuell weitere Einrichtungen). Diese sind frühzeitig zu ermitteln, um das Meldewesen vor Beginn der klinischen Prüfung entsprechend organisieren zu können. Tödliche oder lebensbedrohliche SUSARs Die zuständige Bundesoberbehörde und die zuständige Ethikkommission werden vom Sponsor/LKP über alle tödlichen oder lebensbedrohlichen SUSARs informiert. Dies erfolgt umgehend, spätestens jedoch innerhalb von 7 Kalendertagen nach Bekanntwerden der Minimalkriterien für die umgehende Meldung beim Sponsor/LKP. In allen Fällen wird weitere relevante Information gesucht und der zuständigen Bundesoberbehörde sowie der Ethikkommission innerhalb von höchstens acht weiteren Tagen übermittelt. Im Falle des Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 59 von 67 Todes einer betroffenen Person erfolgt darüber hinaus eine Information an die beteiligte Ethik-Kommission, in deren Zuständigkeitsbereich der Todesfall aufgetreten ist. Nicht-tödliche und nicht-lebensbedrohliche SUSARs Die zuständige Bundesoberbehörde und die zuständige Ethikkommission werden vom Sponsor/LKP über alle anderen SUSARs informiert, und zwar umgehend, spätestens jedoch innerhalb von 15 Kalendertagen nach Bekanntwerden der Minimalkriterien für die umgehende Meldung beim Sponsor/LKP. Weitere relevante nachfolgende Information wird so bald als möglich übermittelt. Im Falle unvollständiger Information zum Zeitpunkt der Meldung wird beim Meldenden oder anderen verfügbaren Quellen ergänzende Information, die zur adäquaten Analyse notwendig ist, nachgefragt. 7.3.6. Überprüfung und Meldung von Änderungen des Nutzen-RisikoVerhältnisses Der Sponsor/LKP unterrichtet unverzüglich, spätestens aber innerhalb von 15 Tagen nach Bekanntwerden, die zuständige Bundesoberbehörde, die zuständige Ethik-Kommission und die zuständigen Behörden anderer Mitgliedstaaten der Europäischen Union und anderer Vertragsstaaten des Abkommens über den Europäischen Wirtschaftsraum, in deren Hoheitsgebiet die klinische Prüfung durchgeführt wird, über jeden Sachverhalt, der eine erneute Überprüfung der Nutzen-Risiko-Bewertung des Prüfpräparates erfordert. Hierzu gehören insbesondere: Einzelfallberichte von erwarteten schwerwiegenden Nebenwirkungen mit einem unerwarteten Ausgang, eine Erhöhung der Häufigkeit erwarteter schwerwiegender Nebenwirkungen, die als klinisch relevant bewertet wird, Verdachtsfälle schwerwiegender unerwarteter Nebenwirkungen, die sich ereigneten, nachdem die betroffene Person die klinische Prüfung bereits beendet hat, Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 60 von 67 Ereignisse im Zusammenhang mit der Studiendurchführung oder der Entwicklung des Prüfpräparates, die möglicherweise die Sicherheit der betroffenen Personen beeinträchtigen können. 7.3.7. Information des Data Monitoring Committee Das DMC wird vom Sponsor/LKP über alle sicherheitsrelevanten Ereignisse informiert (siehe auch Kapitel 3.4). Wie das DMC ggf. eingebunden wird und was im Einzelnen zu berichten ist, muss festgelegt werden (z.B. alle SAEs, bestimmte SAEs, SUSARs und alle weiteren zu definierenden Umstände). Details sollten z.B. in Manualen für SAE-Management und DMC geregelt sein, auf die dann hier verwiesen wird (siehe auch 3.4). 7.3.8. Information der Prüfer Der Sponsor/LKP informiert alle Prüfzentren über alle SUSARs einschließlich relevanter weiterer Information innerhalb der für die zuständige Bundesoberbehörde geltenden Fristen. Der Prüfer des jeweiligen Zentrums ist verantwortlich für die Weiterleitung der Information innerhalb der Prüfgruppe des jeweiligen Zentrums. Falls neue Information bekannt wird, die von der den Prüfzentren gegebenen wissenschaftlichen Information abweicht, oder Informationen über SUSAR-Meldungen aus anderen Studien unter derselben Sponsorschaft zu dem hier verwendeten Prüfpräparat bekannt werden, informiert der Sponsor/LKP alle Prüfzentren hierüber. Darüber hinaus informiert an der Universität zu Köln der Sponsor/LKP im Rahmen der Verpflichtung des Sponsors nach §13 (2) und (3) GCP-V jeden Leiter einer klinischen Prüfung unter universitärer Sponsorfunktion, der denselben Wirkstoff als Prüfpräparat verwendet, über jedes ihm bekannt gewordene SUSAR (http://www.medfak.unikoeln.de/index.php?id=730&L=0). Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx 7.3.9. Seite 61 von 67 Information des Zulassungsinhabers Das Kapitel ist optional. Falls vertraglich mit dem Zulassungsinhaber eine Meldung von SUSARs vereinbart worden ist, kann der folgenden Satz eingefügt werden: Darüber hinaus informiert der Sponsor/LKP den Zulassungsinhaber über alle SUSARs einschließlich der Information über die Meldung an die zuständige Bundesoberbehörde und Ethikkommission entsprechend den vertraglichen Vereinbarungen. Ggf. darüber hinaus gehende Meldeverpflichtungen sollten vertraglich geregelt und hier entsprechend dargestellt werden. 7.3.1. Jährlicher Bericht über die Sicherheit der Prüfungsteilnehmer Der Sponsor/LKP legt einmal jährlich oder auf Verlangen der zuständigen Bundesoberbehörde und den zuständigen Behörden anderer Mitgliedstaaten der Europäischen Union und anderer Vertragsstaaten des Abkommens über den Europäischen Wirtschaftsraum, in deren Hoheitsgebiet die klinische Prüfung durchgeführt wird, einen Bericht zur Sicherheit der Prüfungsteilnehmer vor, der alle verfügbaren relevanten Information zur Sicherheit der Patienten während des Berichtszeitraums berücksichtigt. Der Bericht wird auch der federführenden Ethikkommission vorgelegt. Der jährliche Sicherheitsbericht wird entsprechend der Vorgaben der ICH Guideline E2F „Development Safety Update Report – DSUR“ und gemäß § 13 (6) GCP-V erstellt. Der Beginn für den ersten Berichtszeitraum ist das Genehmigungsdatum der ersten klinischen Prüfung des selben Sponsors zu einem Prüfarzneimittels durch eine zuständige Oberbehörde eines Landes, in dem die klinische Prüfung durchgeführt wird. Der Berichtszeitraum darf 1 Jahr nicht überschreiten. Alle weiteren Berichtzeiträume beginnen am Data Lock Point des Folgejahres. Der Jahressicherheitsbericht wird beim BfArM und der zuständigen Ethikkommission vorgelegt. Alle Berichte werden parallel an jeden Leiter einer klinischen Prüfung unter derselben Sponsorschaft, der denselben Wirkstoff als Prüfpräparat verwendet, versendet. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 62 von 67 8. Verwendung der Daten und Publikation 8.1. Berichte 8.1.1. Zwischenberichte Zum Jährlichen Bericht über die Sicherheit der Prüfungsteilnehmer siehe 7.3.1. Ggf. Bericht an den finanziellen Förderer über den Stand der Rekrutierung, Berichte zu Zwischenauswertungen, Berichte für das DMC etc. erläutern. 8.1.2. Abschlussbericht Die zuständige Behörde und die Ethikkommission werden gemäß § 13 (8) GCP-Verordnung innerhalb von 90 Tagen über die Beendigung der klinischen Prüfung informiert. Innerhalb einen Jahres nach Abschluss der klinischen Prüfung wird der Abschlussbericht unter Berücksichtigung der ICH Guideline E3 erstellt. Der zuständigen Bundesoberbehörde und der Ethikkommission wird gemäß § 13 (9) GCP-V innerhalb der gleichen Frist die Zusammenfassung des Abschlussberichtes über die klinische Prüfung übermittelt, der alle wesentlichen Ergebnisse der Prüfung abdeckt. Ferner wird der Bundesoberbehörde der Bericht zur Veröffentlichung nach § 42b (2) AMG innerhalb eines Jahres nach Ende der klinischen Prüfung übermittelt. Bei der Erstellung des Abschlussberichts soll die ICH Guideline E3 berücksichtigt werden. Der Abschlussbericht und der zur Veröffentlichung nach § 67a (2) AMG vorgesehene Bericht können unterschiedliche Formate aufweisen. Bei klinischen Prüfungen mit zugelassenen Prüfpräparaten werden der zuständigen Bundesoberbehörde und der Ethikkommission innerhalb eines Jahres nach Beendigung der klinischen Prüfung (Definition siehe Kapitel 4.1.1) die Ergebnisse der klinischen Prüfung entsprechend den Vorgaben des § 42b (1) AMG zur Verfügung gestellt (d.h. über das Webportal „PharmNet.Bund“ (http://www.pharmnet-bund.de/dynamic/de/ergebnisberichte/ index.htm öffentlich zugänglich gemacht). Dieser Ergebnisbericht enthält alle wesentlichen Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 63 von 67 Ereignisse der Prüfung sowie die Namen aller beteiligten Prüfer und deren Einrichtungen. In der Regel ist vom Sponsor keine zusätzliche Einreichung eines Berichts nach § 13 (9) GCPV erforderlich, wenn er seine Verpflichtungen aus § 42b (2) AMG vollständig erfüllt, es sei denn die Bundesoberbehörde fordert weitergehende Informationen an. Bei noch nicht zugelassenen Prüfpräparaten ist der Bericht zur Veröffentlichung nach § 67a (2) AMG innerhalb von 6 Monaten nach Erteilung oder Änderung der Zulassung vorzulegen, soweit es sich um konfirmatorische klinische Prüfungen handelt (§42b (1) AMG), 8.2. Publikation Es ist vorgesehen, die Ergebnisse der klinischen Prüfung zu gegebener Zeit und nach gegenseitiger Abstimmung mit dem LKP in einer wissenschaftlichen Fachzeitschrift und/oder bei deutschen und internationalen Kongressen vorzustellen. Grundsätzlich ist einer Gesamtpublikation der klinischen Prüfung Vorzug zu geben. Die „Uniform requirements for manuscripts submitted to biomedical journals (International Committee of Medical Journal Editors" (ICMJE) [JAMA 1997;277:927-34]) werden berücksichtigt. Auch eine Registrierung der klinischen Prüfung in einem öffentlichen Register entsprechend der Empfehlungen des ICMJE ist vorgesehen (siehe auch 5.3). Für alle Veröffentlichungen gilt, dass der Datenschutz sowohl für alle Daten von betroffenen Personen als auch für die Daten der teilnehmenden Prüfer und andere Mitglieder der Prüfgruppe gewahrt bleibt. Die Erfolgsraten bzw. Einzelergebnisse der teilnehmenden Prüfzentren sind nur dem Sponsor bekannt. Ggf. können hier Personen oder Einrichtungen genannt werden, die im Auftrage des Sponsors die Daten erhalten und daher über diese Sachverhalte Kenntnis haben. Die Veröffentlichung oder ein Vortrag der Ergebnisse aus dieser klinischen Prüfung, einschließlich einer Veröffentlichung oder eines Vortrags eines einzelnen Prüfzentrums, bedürfen der vorherigen Kenntnisnahme und einer vorgehenden Kommentierung und Genehmigung durch den Sponsor. Der Prüfer erklärt mit Unterzeichnung des Prüfstellenvertrags stellvertretend für alle Mitglieder der Prüfgruppe des Prüfzentrums sein Einverständnis, dass die Ergebnisse dieser Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 64 von 67 klinischen Prüfung bei nationalen und internationalen Zulassungs- und Überwachungsbehörden, der Bundesärztekammer, der Kassenärztlichen Bundesvereinigung und den Krankenkassen vorgelegt werden können. Gleichzeitig erklärt sich der Prüfer damit einverstanden, dass in diesem Zusammenhang sein Name, seine Anschrift, seine Qualifikationsmerkmale und der Umfang seiner Beteiligung an der klinischen Prüfung bekannt gegeben werden. Falls das ZKS Köln / IMSIE in die Studie eingebunden ist, muss folgender Satz eingefügt werden: Bei Veröffentlichungen wird die Unterstützung durch das ZKS Köln / IMSIE angegeben. Soweit zutreffend werden die Mitarbeiter des ZKS Köln / IMSIE als Mitautoren aufgeführt und das BMBF-Förderkürzel des ZKS Köln (01KN1106) anzugeben. Falls die Mitarbeiter nicht Mitautoren des Artikels sind, werden sie namentlich in der Danksagung genannt. Von allen Projektveröffentlichungen ist dem ZKS Köln eine Kopie zur Verfügung zu stellen. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 65 von 67 9. Änderungen des Prüfplans Zum Zweck der Sicherstellung weitgehend vergleichbarer Bedingungen in allen Prüfzentren sowie im Interesse einer einwandfreien Datenauswertung ist eine Änderung der vereinbarten und im Prüfplan niedergelegten Prüfungsbedingungen nicht vorgesehen. In Ausnahmefällen sind jedoch Änderungen der Prüfungsbedingungen möglich. Diese erfolgen nur nach gegenseitiger Abstimmung zwischen dem Sponsor, dem LKP und dem Biometriker sowie allen Unterzeichnenden (Autoren) dieses Prüfplans. Jede Änderung der im Prüfplan vorgesehenen Studienprozeduren muss schriftlich unter Angabe der jeweiligen Gründe erfolgen und von allen Autoren dieses Prüfplans unterschrieben werden (Amendment). Nach §§ 10 (1) und 4 GCP-V genehmigungspflichtige nachträgliche Änderungen werden der Ethikkommission und der Bundesoberbehörde zur Genehmigung vorgelegt und erst nach deren Genehmigung umgesetzt. Hiervon unbenommen sind Änderungen, die zur Abwendung unmittelbarer Gefahr notwendig sind. Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 66 von 67 10. Literatur 1. The European Agency for the Evaluation of Medicinal Product. Note for Guidance on Good Clinical Practice (CPMP/ICH/135/95). 2. The European Agency for the Evaluation of Medicinal Product. Note for Guidance Structure and Content of Clinical Study Reports (CPMP/ICH/137/95). 3. National Cancer Institute. Protocol Templates, Applications and Guidelines http://ctep.cancer.gov/guidelines/templates.html. 4. EMEA-Guideline On Data Monitoring Committees: EMEA/CHMP/EWP/5872/03 Corr 5. The DAMOCLES Study Group. A proposed charter for clinical trial 2005 data monitoring committees: helping them do their job well. Lancet 2005; 365: 711-22 6. Clinical trial registration: a statement from the International Committee of Medical Journal Editors. Accessed at http://www.icmje.org/clin_trial.pdf on 22 May 2007. 7. WHO. Causality Assessment of Suspected Adverse Reactions. http://www.whoumc.org/DynPage.aspx?id=22682 8. Chan A et al. RESEARCH METHODS AND REPORTING SPIRIT 2013 explanation and elaboration : guidance for protocols of clinical trials. 2013;1-42 Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln Prüfplan-Code xxx Seite 67 von 67 11. Anhänge 11.1. Prüfzentren und Prüfer sowie Stellvertreter 11.2. Protocol Agreement Form 11.3. Steering Committee 11.4. Data Monitoring Committee 11.5. Advisory Committee 11.6. Prüflabore und sonstige technische Einrichtungen 11.7. Musteretikett für die Prüfmedikation 11.8. Patienteninformation und Einverständniserklärung 11.9. Fachinformation 11.10. Versicherungsbestätigung 11.11. Versicherungsbedingungen Prüfplan vX-XX vom XX.XX.XXXX Universität zu Köln