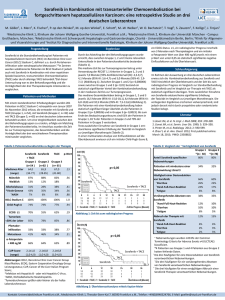
Phase-I-Studie mit Paclitaxel in Kombination mit Bevacizumab und
Werbung

Aus der Medizinischen Klinik III in der Universitätsklinik Marienhospital Herne der Ruhr-Universität Bochum Direktor: Prof. Dr. med. D. Strumberg Phase-I-Studie mit Paclitaxel in Kombination mit Bevacizumab und Sorafenib bei Patienten mit metastasierter oder lokal fortgeschrittener solider Tumorerkrankung Inaugural-Dissertation zur Erlangung des Doktorgrades der Medizin einer Hohen Medizinischen Fakultät der Ruhr-Universität Bochum vorgelegt von Renée Roy aus Bonn 2014 Dekan: Prof. Dr. med. K. Überla Referent: Prof. Dr. med. D. Strumberg Korreferent: Prof. Dr. med. Martin Wegener Tag der mündlichen Prüfung: 21.04.2015 Abstract Roy Renée Phase-I-Studie mit Paclitaxel in Kombination mit Bevacizumab und Sorafenib bei Patienten mit metastasierter oder lokal fortgeschrittener solider Tumorerkrankung. Problem: Die Hemmung der Angiogenese ist ein vielversprechender Therapieansatz bei der Bekämpfung maligner Tumore. Möglichkeiten hierzu sind die Inhibition des Wachstumsfaktors VEGF und seines Rezeptorsystems. Die Effektivität der Therapie lässt sich dadurch erhöhen, dass die Angiogenese an unterschiedlichen Stellen mit potenziell synergistischer Wirkung inhibiert wird. Die vorliegende Arbeit beschreibt eine Phase-IStudie mit dem Chemotherapeutikum Paclitaxel und zwei unterschiedlichen, kombinierten AngiogeneseInhibitoren: dem Tyrosin-Kinase-Inhibitor Sorafenib und dem VEGF-Antikörper Bevacizumab. Methode: Unter ambulanten Bedingungen wurden 400mg Sorafenib (Nexavar®) zweimal täglich zusammen mit 90mg pro m² KOF Paclitaxel (Taxol®) in wöchentlicher Applikationsweise und dosiseskaliert Bevacizumab (Avastin®) alle zwei Wochen verabreicht. Die Bevacizumab-Dosis lag kohortenabhängig bei 0, 1, 2, 4, 5 oder 10mg/kg KG. Ziel ist die Ermittlung der maximal tolerablen Dosis (MTD) und dosislimitierenden Toxizitäten (DLTs) bei Patienten mit fortgeschrittenem Tumorleiden. Die Datenauswertung erfolgte hinsichtlich der Machbarkeit, Sicherheit und Wirksamkeit. Ergebnisse: 28 Patienten mit fortgeschrittener solider Tumorerkrankung wurden zwischen 07/2009 und 03/2012 behandelt; medianes Alter: 61 Jahre; mediane Beobachtungsdauer: 225 Tage; Gesamtzahl zytostatischer Vortherapien: 76 Regime, Median: 2 Regime. In der vorliegenden Studie wurden 71 Zyklen verabreicht, im Median zwei Zyklen je Patient. Bezüglich der Toxizität waren alle Studienteilnehmer auswertbar, 8 Patienten brachen die Therapie wegen schwerer Nebenwirkungen ab. Dosislimitierende Toxizität war das Hand-Fuß-Syndrom (HFS). Diese und andere z. T. schwere Nebenwirkungen führten bei insgesamt 27 Patienten zu einer dauerhaften Dosisreduktion von Sorafenib auf ≤ 50%, eine weitere Reduktion war bei 10 Patienten aus den Kohorten III bis VI notwendig. Häufigste Nebenwirkung war das HFS bei 14 Patienten (bei 8 Patienten Grad 3 und 4), die Fatigue betraf als zweithäufigste Toxizität 11 Patienten (bei 3 Patienten Grad 3 und 4). Unter Grad-3- und Grad-4-Nebenwirkungen allgemein litten 12 bzw. 5 Patienten. Mit einer Toxizitätsrate von 61% war die Therapieverträglichkeit insgesamt schlecht. Eine Assoziation der Nebenwirkungen mit steigender Bevacizumab-Dosis war nicht nachweisbar. Das Ansprechen und die Überlebensrate waren bei 22 Patienten auswertbar. Clinical Benefit hatten 13 Patienten durch Stable Disease, die mediane Überlebenszeit lag bei ca. 38 Wochen und scheint mit der Höhe der Bevacizumab-Dosis und der Differenzierung der Tumorzellen positiv zu korrelieren. Diskussion: Die Medikation ist eine potenziell wirksame Behandlungsalternative bei vorbehandelten Tumorpatienten. Die Ergebnisse zeigen, dass einzelne Patienten von der beschriebenen Medikamentenkombination profitierten. Das hohe Ausmaß der Nebenwirkungsrate schließt jedoch eine weitere Anwendung in dieser Dosierung aus. Vermutlich beruht die hohe Toxizitätsrate auf der Kombination der Medikamente, da die Einzelsubstanzen im Allgemeinen wesentlich besser verträglich sind. Aufgrund der geringen Teilnehmerzahl ist keine ausreichende Beurteilung von Wirksamkeit, Überlebensrate und progressionsfreiem Überleben möglich. Für meine lieben Eltern Inhaltsverzeichnis 1 Einleitung 9 1.1 Historie 9 1.2 Bedeutung der Angiogenese für die Tumorbiologie und Angiogenetischer Switch 9 1.3 VEGF 11 1.4 Bevacizumab (Avastin®) 16 1.5 Sorafenib (Nexavar®) 18 2 Zielsetzung 21 3 Material und Methoden 23 3.1 Teilnehmerauswahl 23 3.2 Diagnostik und Staging 24 3.3 Studiendesign 25 3.4 Studienziele 30 3.5 Sicherheit 31 3.5.1 UAWs / Medikamententoxizitäten und Laborwertveränderungen 31 3.5.2 Schwere unerwünschte Ereignisse 32 3.5.3 Stärkste Nebenwirkung je Patient 32 3.6 Wirksamkeit 4 33 3.6.1 Tumoransprechrate 33 3.6.2 Überlebenszeit 34 3.6.3 Dauer des Tumoransprechens – Progressionsfreies Intervall 35 3.7 Kontrolle des Ansprechens und Fortführung der Therapie 35 3.8 Prüfdauer, Therapieabbruch und Nachsorge 36 3.9 Statistische Methoden 37 Ergebnisse 38 4.1 Patienten 38 4.1.1 Patientenmerkmale 38 4.1.2 Entität, Histologie, Grading 38 4.1.3 Tumorstadien und Metastasierung 40 4.1.4 Vorbehandlung 41 4.2 Therapie 44 1 4.2.1 Anzahl der Zyklen pro Patient 44 4.2.2 Dosisreduktion und Therapiepausen 44 4.2.2.1 Paclitaxel 45 4.2.2.2 Sorafenib 45 4.2.2.3 Bevacizumab 46 4.2.3 Therapiedauer 46 4.2.4 Ursachen für Therapieende 47 4.2.5 Sicherheit 48 4.2.5.1 4.2.5.2 Unerwünschte Arzneimittelnebenwirkungen und Medikamententoxizitäten 48 Laborwertveränderungen 52 4.2.5.2.1 Tumormarker 54 4.2.5.3 Schwere unerwünschte Ereignisse / Stationäre Aufenthalte 56 4.2.5.4 Stärkste Nebenwirkung je Patient 57 4.2.5.4.1 Toxizität in Abhängigkeit von der Dosis des Antikörpers Bevacizumab 58 4.2.6 Wirksamkeit 60 4.2.6.1 Tumoransprechrate 60 4.2.6.2 Überlebenszeit 60 4.2.6.2.1 Überlebenszeit in Abhängigkeit von der Dosis des Antikörpers Bevacizumab 61 4.2.6.2.2 Überlebenszeit in Abhängigkeit vom Tumoransprechen 4.2.6.2.3 Überlebenszeit in Abhängigkeit vom Grading 5 61 62 4.2.7 Anschlusstherapien 62 4.2.8 Todesursachen 62 Diskussion 63 5.1 Kritische Betrachtung des Studienkonzeptes 63 5.2 Kritische Betrachtung der Therapieergebnisse 64 5.2.1 Dosis 64 5.2.2 Unerwünschte Arzneimittelnebenwirkungen / Medikamententoxizitäten 64 5.2.3 Laborwerte 64 2 5.2.4 Individuelle Therapieergebnisse 67 5.2.5 Tumoransprechrate 68 5.2.6 Überlebenszeit 70 5.2.7 Machbarkeit 70 5.3 Ausblick 71 6 Zusammenfassung 73 7 Literaturverzeichnis 75 8 Anhang 82 8.1 Zusätzliche Informationen zu den Medikamenten 8.1.1 Bevacizumab 82 82 8.1.1.1 Chemische Struktur 83 8.1.1.2 Wirkmechanismus 83 8.1.1.3 Pharmakokinetik 84 8.1.1.4 Toxizität 84 8.1.2 Sorafenib 87 8.1.2.1 Chemische Struktur 87 8.1.2.2 Wirkmechanismus 87 8.1.2.3 Pharmakokinetik 88 8.1.2.4 Toxizität 88 8.1.3 Paclitaxel 91 8.1.3.1 Chemische Struktur 91 8.1.3.2 Wirkmechanismus 92 8.1.3.3 Pharmakokinetik 93 8.1.3.4 Toxizität 94 8.1.4 Interaktion 96 Danksagung Lebenslauf 3 Abkürzungsverzeichnis 5 FU / FS 5-Fluoruracil und Folinsäure ALAT Alanin-Aminotransferase ASAT Aspartat-Aminotransferase AZ Allgemeinzustand BWK Brustwirbelkörper CA 15-3 Carbohydrate Antigen 15-3 CA 19-9 Carbohydrate Antigen 19-9 CA 125 Carbohydrate Antigen 125 CEA Carcinoembryonales Antigen CHO chinese hamster ovary CR Complete Response CRC Colorectales Carcinom CRP C-reaktives Protein CT Computertomogramm / -graphie CTCAE Common Terminology Criteria for Adverse Events ECOG Eastern Cooperative Oncology Group EDTA ethylenediamine tetraacetic acid ELISA enzyme-linked immunosorbent assay Gamma-GT Gamma-Glutamyl-Transaminase G-CSF granulocyte-colony stimulating factor GFR glomeruläre Filtrationsrate HER-2 human epidermal growth factor receptor 2 HFS Hand-Fuß-Syndrom 4 i.v. intravenös IFL Irinotecan, 5-Fluoruracil und Leucovorin IgG Immunglobulin Typ G IL-2 Interleukin-2 K. A. keine Angabe KG Körpergewicht KOF Körperoberfläche LDH Lactatdehydrogenase mRNA messenger-RNA MRT Magnetresonanztomogramm / -graphie NA not available NaCl Natriumchlorid NC No Change NCI National Cancer Institute NSCLC non-small cell lung cancer PD Progressive Disease PDGFR-β platelet-derived growth factor receptor typ beta PIGF placenta growth factor PNP Polyneuropathie PR Partial Response PVC Polyvinylchlorid RECIST Response Evaluation Criteria in Solid Tumors SD Stable Disease SUE Schwerwiegendes unerwünschtes Ereignis TKI Tyrosinkinase-Inhibitor 5 TNF-alpha Tumornekrosefaktor-alpha UAW Unerwünschte Arzneimittelnebenwirkung UE Unerwünschtes Ereignis UICC Union Internationale Contre le Cancer VEGF vascular endothelial growth factor VEGFR vascular endothelial growth factor receptor vs. versus Z. n. Zustand nach 6 Abbildungsverzeichnis Abbildung 1: Häufigkeitsverteilung der Tumore (n = 29) 39 Abbildung 2: Häufigkeitsverteilung der Organmetastasierung 41 Abbildung 3: Zusammenfassung der Vortherapien bei jedem Patient (n = 28) 42 Abbildung 4: Anzahl der Patienten mit abgeschlossenen Therapiezyklen 44 Abbildung 5: Therapiedauer pro Patient (n = 28) 47 Abbildung 6: Häufigkeitsverteilung der Toxizitäten unter Therapie (n = 28) 51 Abbildung 7: Anteil der Nebenwirkungen Grad 3 und 4 an den Gesamttoxizitäten (n = 28) 51 Abbildung 8: Häufigkeiten von Laborwerttoxizitäten am Gesamtkollektiv und prozentualer Anteil der Grad 3 und 4 Toxizitäten an der Gesamthäufigkeit 54 Abbildung 9: Tumormarkerentwicklung unter Therapie bei Patient A 55 Abbildung 10: Entwicklung dreier Tumormarker unter Therapie bei Patient B 55 Abbildung 11: Entwicklung des Tumormarkers CA 19-9 unter Therapie bei Patient B 56 Abbildung 12: Stärkste Nebenwirkung je Patient (100% = 28 Patienten) 58 Abbildung 13: Häufigkeit von Toxizitäten Grad 3 und 4 als stärkste Nebenwirkung je Patient in Abhängigkeit von der Bevacizumab-Dosis (n = 28) 59 Abbildung A1: Strukturformel Sorafenib 87 Abbildung A2: Strukturformel Paclitaxel 92 7 Tabellenverzeichnis Tabelle 1: Therapeutische Angriffspunkte im Rahmen der Tumorangiogenese 13 Tabelle 2: Ein- und Ausschlusskriterien der Studienpopulation 24 Tabelle 3: Studiendesign beispielhaft für die ersten beiden Therapiezyklen 27 Tabelle 4: Wöchentliches Therapieschema in der Ambulanz ohne morgendliche Sorafenib-Gabe Tabelle 5: 28 Geplantes und schließlich durchgeführtes Dosiseskalationsdesign (individuelle Dosisreduktionen vorbehalten) 29 Tabelle 6: Endpunkte der vorliegenden Studie 30 Tabelle 7: Remissionskriterien messbarer Tumorparameter nach RECIST, Version 1.1 33 Tabelle 8: Allgemeine Stadieneinteilung bei Tumoren 39 Tabelle 9: Zusammenfassung der Patientencharakteristika 43 Tabelle 10: Dosisreduktion der Medikamente in den unterschiedlichen Kohorten 46 Tabelle 11: Ursachen für eine Beendigung der Therapie (n = 28) 48 Tabelle 12: Häufigkeitsverteilung der Toxizitäten unter Therapie (n = 28) 50 Tabelle 13: Laborwerttoxizitäten unter Therapie nach CTCAE 53 Tabelle 14: Tumoransprechverhalten (n = 22) 60 Tabelle A1: Tumorstadien. Einteilung nach UICC 82 Tabelle A2: Auszug der Common Toxicity Criteria (CTCAE) des National Cancer Institute, Version 3.0 Tabelle A3: Häufigkeit der Nebenwirkungen 8 82 89 1 1.1 Einleitung Historie In den letzten Jahren hat die Bedeutung von Tumorerkrankungen in der Medizin erheblich zugenommen, sie sind derzeit die zweithäufigste Todesursache. Wegen der wachsenden Zahl älter werdender Patienten wird - Schätzungen des Robert-Koch-Instituts zufolge - die Zahl neuer Krebsfälle bis zum Jahr 2020 bei Männern um mindestens 50%, bei Frauen um mindestens 25% steigen. Schon seit Jahrzehnten ist bekannt, dass zwischen Angiogenese und Tumoren ein Zusammenhang besteht und die morphologischen Veränderungen von Tumorgefäßen ein Malignitätskriterium darstellen. 1968 wurde die Hypothese aufgestellt, dass Tumore eine diffusionsfähige angiogenetische Substanz produzieren. Diese Annahme wurde 1971 durch Folkman erhärtet, der sagte, dass Tumorwachstum und Metastasierung allein von der Angiogenese abhängig sind [21]. Demnach sollte durch Blockieren des Gefäßwachstums das Tumorwachstum gebremst werden können. Diese These stieß eine intensive Forschung bezüglich der Suche nach pro- und antiangiogenetischen Substanzen sowie die Suche nach therapeutischen Ansätzen und neuen diagnostischen Methoden an. 1976 zeigte Gullino, dass Zellen in präkanzerösem Gewebe auf dem Weg zur Malignität angiogenetische Kapazität erwerben [11, 25]. 1.2 Bedeutung der Angiogenese für die Tumorbiologie und Angiogenetischer Switch Maßgeblich für das einwandfreie Funktionieren physiologischer Prozesse wie Wachstum, embryonale Entwicklung und Wundheilung ist die Angiogenese. Sie ist aber auch an pathologischen Vorgängen (Entzündung, Diabetes mellitus, Makuladegeneration) beteiligt. Bei akuten Entzündungen trägt die Angiogenese zur Wiederherstellung der Gewebsintegrität bei, während sie bei chronischen Entzündungen, Makuladegeneration, Psoriasis, Lungenfibrose und Tumoren die Krankheit unterhält [15]. Im Gegensatz zur Vaskulogenese, d.h., der Neubildung von Blutgefäßen aus noch undifferenzierten Vorläuferzellen, betrifft die Angioge9 nese das Wachstum bereits bestehender Gefäße durch Migration und Proliferation differenzierter Endothelzellen [11, 35]. Pro- und antiangiogenetische Substanzen werden physiologisch von Endothelzellen, Stromazellen, Blut und extrazellulärer Matrix ausgeschüttet, aber auch Tumorzellen sind dazu in der Lage. Zu den die Angiogenese stimulierenden Faktoren gehören VEGF (siehe Abschnitt 1.3), Endothelin, das Angiopoetin-System, Matrix-Metalloproteinasen und Ephrine; Inhibitoren sind Angiostatin, Endostatin, Thrombospondin-1 und Integrine [15]. Unter physiologischen Bedingungen verläuft die Angiogenese kontrolliert, d.h., es besteht ein Gleichgewicht zwischen pro- und antiangiogenen Faktoren. Die Blutgefäße befinden sich in einer Art Ruhezustand mit geringer Proliferationsrate [15]. Im Fall einer Fremdzellbildung, z.B. bei einem Tumor, verläuft die Angiogenese jedoch anders [11, 22]. Zunächst wird der Tumorzellklon allein durch Diffusion mit Nährstoffen und Sauerstoff versorgt. Ab einer Größe von ca. 2mm 3 muss der Zellklon für das weitere Wachstum durch Sprossen oder intussuszeptives Gefäßwachstum bereits bestehender Blutgefäße eigene Gefäße ausbilden. Angeregt durch verschiedene Signale wie metabolischen (Hypoxie) oder mechanischen (Tumormasse) Stress, entzündliche Immunantworten, genetische Mutationen oder Zellzerfall [11] wird das Gleichgewicht zwischen Stimulatoren und Inhibitoren der Angiogenese zugunsten der Stimulatoren verschoben. Dieses Umschalten wird als Angiogenetischer Switch bezeichnet. Der Überschuss an Angiogenese-stimulierenden Faktoren führt nun zur Auflösung der extrazellulären Matrix und der Basalmembran von bestehenden Blutgefäßen durch Proteasen (z.B. Metalloproteinasen). Anschließend schilfern sich Endothelzellen von der Gefäßwand ab, proliferieren mit einem bis zu 35-mal höheren Faktor und bilden im Tumor gefäßähnliche Strukturen. Zusätzlich schütten die Endothelzellen verschiedene Wachstumsfaktoren aus, die über Stimulation zur exponentiellen Vermehrung der Tumorzellen beitragen [15]. Allerdings hat das oben beschriebene rasche Wachstum der Gefäße Nachteile: Es führt zu unstrukturierten, dünnwandigen Gefäßen, die aufgrund der Durchlässigkeit durch Blutverlust gekennzeichnet sind. Damit lassen sich typische Tumorsymptome wie die Hämaturie bei Blasenkarzinomen, die erst in der Phase der Tumorprogression auftreten, erklären. Außerdem führen durchlässige Gefäße zu einem Flüssigkeitsverlust ins Interstitium, sodass der interstitielle Druck im Tu10 morgewebe ansteigt, die antitumoralen Medikamente erreichen ihren Zielort oft vermindert oder gar nicht [54]. Protektive Mechanismen, wie sie normale Gefäße während des Wachstums ausbilden, fehlen bei Tumorgefäßen, sodass Tumorzellen nicht vor einer wechselnden Sauerstoffzufuhr geschützt sind oder sich dem metabolischen Bedarf anpassen können [15]. Die Tumorgefäße sind durch erhebliche Schwankungen im Durchmesser und blinde Gefäßabbrüche charakterisiert. Auch die Dichte der Gefäßstruktur kann stark variieren, die höchsten Dichten sind in der Tumorperipherie und der Invasionsfront zu finden [15]. Tumorgefäße bestehen meist nicht homogen aus Endothelzellen, sondern entweder vollständig aus Tumorzellen oder aus Tumor- und Endothelzellen (sogenannte Mosaikgefäße, vasculogenic mimicry). Das Vorkommen von Tumorzellen in Tumorgefäßen hat erhebliche Auswirkungen auf die Prognose über frühzeitige Metastasierung [15]. Aus den Unterschieden zwischen physiologischen und tumorösen Gefäßen geht aber auch hervor, dass das Gefäßsystem von malignem Gewebe ein attraktives Ziel bei der Therapie bösartiger Tumore ist [11, 44]. 1.3 VEGF Heute ist bekannt, dass verschiedene molekulare Komponenten an den Mechanismen des Gefäßwachstums beteiligt sind. Eine maßgebliche Rolle spielen dabei Mitglieder der VEGF- (vascular endothelial growth factor) und AngiopoetinFamilie als wichtigste Regulatoren der Angiogenese [11]. Sie stimulieren das Aussprossen und die Verzweigung neuer Blutgefäße. Die Kenntnis der Tumorangiogenese führte zur Bestimmung der antiangiogenen Faktoren VEGF und seiner Rezeptoren VEGFR-1, -2 und -3 in verschiedenen Tumorgeweben. VEGFR-1 ist vor allem an der Vaskulogenese und der physiologischen Angiogenese beteiligt, während VEGFR-3 vorwiegend für die Lymphangiogenese Bedeutung hat und VEGFR-2 ein wesentlicher Mediator bei der Tumorgefäßbildung ist [58]. Inzwischen sind sechs VEGF-Proteine bekannt: A, B, C, D, E und PIGF (Plazenta-Wachstumsfaktor), das wirksamste Protein ist VEGF-A [15]. 11 Studien haben ergeben, dass VEGF in vielen Tumorgeweben hochreguliert ist und häufig mit einer schlechten Prognose einhergeht [58, 62]. Außerdem wurde festgestellt, dass eine erhöhte VEGF-Konzentration mit der Gesamtüberlebensdauer und der Zeit bis zum Tumorprogress korreliert, sodass die VEGFKonzentration als Prognosefaktor gilt [10, 30] (Beispiele für Studien [15]). Im Unterschied zu Chemotherapeutika greift die antiangiogenetische Therapie nicht an der Tumorzelle selbst, sondern an Endothelzellen oder gefäßstabilisierenden muralen Gefäßzellen an [15]. Bei dieser Therapieform funktioniert das lineare Dosis-Wirkungsprinzip nicht wie bei den herkömmlichen Tumortherapien. Außerdem lassen sich die dosislimitierende Toxizität und die daraus ermittelte maximal tolerable Dosis der Substanzen oft nicht bestimmen [15]. Um das Ansprechen der Therapie trotzdem adäquat überwachen zu können, werden prädiktive Biomarker (Isoformen von VEGF-A und -B sowie PIGF) verwendet (sogenannte Surrogatmarker) [15]. Diese sollen die Funktionalität der Tumorgefäße überwachen und so Rückschlüsse auf die Wirkstoffaktivität an der Zielstruktur ermöglichen. Frühere Studien ergaben signifikante Veränderungen der Wachstumsfaktoren unter der Therapie, insbesondere bei Patienten, deren Erkrankung sich stabilisierte [15]. Je höher die antiangiogenen Substanzen dosiert wurden, desto höhere Konzentrationen an VEGF und bFGF (basic fibroblast growth factor) ließen sich im Plasma messen [16, 17]. Weiteren Studien zufolge nahm auch die Konzentration des Serummarkers VEGFR-2 ab [9, 52]. Einige Studien zeigten einen Zusammenhang zwischen der Dosis eines VEGF-Rezeptor-Tyrosinkinase-Inhibitors (TKI) und dem Serumspiegel des löslichen VEGF2-Rezeptors. Je höher die TKI-Dosis war, desto niedriger waren die VEGFR-2-Spiegel [15]. Im Folgenden werden die therapeutischen Möglichkeiten zur Hemmung der Tumorangiogenese vorgestellt [angelehnt an 15]. 12 Tabelle 1: Therapeutische Angriffspunkte im Rahmen der Tumorangiogenese [angelehnt an 15] Therapieansatz Wirkmechanismus Medikamentenklasse Medikamentenbeispiele Hemmung der an der Hemmung von Antikörper, lösliche Rezeptoren Bevacizumab, IMC-II2Ib, Tumorgefäßbildung Wachstumsfaktoren wie beteiligten Liganden VEGF Hemmung der zuge- Inhibition der a) Rezeptor-Antagonisten b) PTK787/ZK222584, hörigen Rezeptoren Signaltransduktion b) Signaltransduktions-Inhibitoren SU11248, ZV6474, Cediranib Vascular Targeting Zerstörung des Zytoskeletts a) Pharmakotherapie Combrestatin A4, ZD6126, von endothelialem Tubulin b) Ligatur AS1404 Hemmung endothelialer a) TNF-α-Hemmung Adhäsionsmoleküle und junktionale anti-αvβ3, anti-αvβ5, anti-VE- Adhäsionsmoleküle b) Integrin-αvβ3-Hemmung Inhibitoren Cadherin a) Piperidindion (Barbiturat) a) Thalidomid b) Zyklisches Pentapeptid b) Cilengitide Matrix-Metalloproteinase-Inhibitoren Batimastat, Marimastat Von Thrombozyten exprimiertes Thrombospondin-1 Inhibition der Invasion Natürlicher Unklar Angiogenesehemmer VEGF-Trap Protein Selektive Hemmung der Aktivierung der Statine: a) Angiostatin Endothelzellproliferation Matrix-Metalloproteinase a) Plasminogenfragment b) Endostatin und Integrine b) Kollagen-XVIII-Fragment Anmerkung: Unterstrichene Substanzen sind bereits zur Hemmung der Tumorangiogenese zugelassen oder befinden sich in klinischen Phase-III-Studien. 13 Die voraussichtlich wirksamste Strategie zur Hemmung der VEGFvermittelten Tumorangiogenese scheint die Inhibition des Wachstumsfaktors VEGF und seines Rezeptorsystems (VEGFR) zu sein [15]. Diese beiden Mechanismen werden im Folgenden näher erläutert. Der erste Therapieansatz betrifft die Möglichkeit, die Angiogenese von Tumoren zu unterbinden, indem die an der Tumorgefäßneubildung beteiligten Liganden gehemmt werden. VEGF ist für das Überleben der Tumorgefäßzellen essenziell, er schützt sie vor der Apoptose, hält die neu gebildeten Blutgefäße stabil und fördert das Tumorwachstum [15, 54]. Fehlt diese konstante VEGF-Versorgung, sterben die Gefäßendothelzellen ab und die neu gebildeten Mikrogefäße des Tumors zerfallen [15]. Damit wird die Bedeutung der Anti-VEGF-Therapie verständlich: Eine Hemmung des Wachstumsfaktors unterdrückt nicht nur die Proliferation der Gefäßendothelzellen und die Sprossung der Gefäße [2], sondern führt auch zu einer Rückbildung der vorhandenen Tumorgefäßversorgung [15]. Unreife Gefäße werden abgebaut, und der Gefäßaufbau wird strukturiert. Zum einen wird dadurch der Tumor besser durchblutet und so effektiver mit Chemotherapeutika und Sauerstoff versorgt [32], sodass die Ansprechrate auf zytostatische Therapien steigt. Zum anderen führt die verbesserte Gefäßstruktur zu weniger durchlässigen Gefäßen, sodass sich der interstitielle Druck im Tumorgewebe verringert. Dadurch können Zytostatika den Tumor besser durchdringen und effizient wirken [32,54]. Die Normalisierung der Gefäßstruktur und -funktion könnte auch bewirken, dass sich die Wahrscheinlichkeit einer regionalen Lymphknotenmetastasierung verkleinert [15]. Die beschriebenen vaskulären Veränderungen unterstreichen die Bedeutung einer solchen Therapie und die Möglichkeit einer effektiveren Kombination der Anti-VEGF-Therapie mit weiteren antitumoralen Substanzen [15, 67]. Klinische Daten bestätigen die Wirkung der Anti-VEGF-Therapie. Bereits eine Einzelinfusion mit Anti-VEGF beim Menschen zeigte, dass sich die Dichte der Tumormikrogefäße verringerte [67]. Dennoch erfordert die optimale Therapiekontrolle eine kontinuierliche VEGF-Inhibition [15]. Klinische Studien mit dem VEGFA-Antikörper Bevacizumab (Avastin®) ergaben, dass Patienten, die mit dieser hochwirksamen Substanz therapiert wurden, länger überlebten [29, 47, 56] und die Tumorerkrankung später fortschritt als bei der Vergleichsgruppe ohne Bevaci14 zumab. Diese Studien bestätigen die Vermutung, dass die VEGF-Inhibition den Krankheitsverlauf signifikant beeinflussen kann, indem der Wirkstoff das Tumorwachstum anhaltend unterdrückt [46]. Der zweite Therapieansatz gilt nicht den Liganden selbst, sondern den zugehörigen Rezeptoren. Ihre Hemmung unterdrückt auch die Signaltransduktion. Prinzipiell gibt es dafür zwei Möglichkeiten: die Rezeptor-Antagonisten (Antikörper, Peptide, kleine Moleküle) und die Signaltransduktions-Inhibitoren (TyrosinkinaseInhibitoren). Ein Beispiel für die Therapie mit einem Rezeptor-Antagonisten ist die Blockade des VEGF-Rezeptors mit einem Antikörper. Die Firma ImClone entwickelte hierfür den neutralisierenden Anti-KDR-Antikörper IMC-II2Ib, der sich derzeit in der Phase-III-Studie bei Tumorpatienten befindet. Er verhindert die Rezeptoraktivierung, indem er die Bindung des Liganden VEGF an seinen Rezeptor blockiert. Bislang zeigte sich bei guter Therapieverträglichkeit eine vielversprechende Wirksamkeit. Es bleibt allerdings zu klären, ob sich allein mit der Blockade eines einzelnen Angiogenese-relevanten Rezeptors ein therapeutischer Effekt erzielen lässt [15]. Die andere Therapieoption besteht in der Hemmung der Signaltransduktion durch Rezeptor-Tyrosinkinase-Inhibitoren [15]. Das Verständnis dieser Therapie erfordert einige Hintergrundinformationen: Tyrosinkinasen, Enzyme aus der Untergruppe der Proteinkinasen, sind essenziell für die primäre Übermittlung von Signalen aus dem extrazellulären in den intrazellulären Raum. Ihre Aufgabe, die Phosphorylierung von Zielproteinen, ist ein sehr wichtiger Schritt in der Signaltransduktion, denn er steuert Differenzierung, Wachstum, Migration und Apoptosemechanismen der Zellen. Bei vielen malignen Erkrankungen ist das System der Signaltransduktion und der Rezeptor-Tyrosinkinasen im Vergleich zum Ursprungsgewebe verändert. Die Ursachen dafür sind durch Mutationen hervorgerufene Überexpressionen, Genamplifizierungen oder funktionale Veränderungen, die sich schließlich in fehlreguliertem Zellwachstum und Malignomen manifestieren. Inhibitoren der VEGF-Rezeptor-Tyrosinkinasen hemmen die VEGF- induzierte pathologische Angiogenese, indem sie in die zelluläre Signaltransduktion (Phosphorylierung der Zielproteine durch Tyrosinkinasen wird unterbunden) eingreifen. Insbesondere der primäre Endothelzell-mitogene VEGF-Rezeptor 2 15 (KDR) spielt dabei eine große Rolle, denn seine Aktivierung ist der erste intrazelluläre Schritt zur Angiogenese [29]. Zwar ist die orale Bioverfügbarkeit der niedermolekularen KDR-Tyrosinkinase-Inhibitoren im Gegensatz zu anderen antiangiogenen Therapiemöglichkeiten von Vorteil, aber eine gezielte Therapie mit diesen Substanzen ist nur bedingt umzusetzen. Die Ursache dafür ist die Ähnlichkeit der aktiven Zentren von Tyrosinkinase-Enzymen in biologischen Systemen, daher ist die Selektivität der Inhibitoren unzureichend und ihre Aktivität nicht auf das Angriffsziel beschränkt. Dadurch könnten Wirkung und Nebenwirkungen verstärkt werden. Selektive Inhibitoren befinden sich zurzeit noch in der Entwicklung [15]. Breitspektrum-Tyrosinkinase-Inhibitoren greifen hingegen an vielen verschiedenen Tyrosinkinasen an. Hierdurch wird die antitumoröse Wirkung summiert, es bleibt jedoch unklar, welche der gehemmten Kinasen für den antiangiogenen Therapieerfolg entscheidend ist [15]. Zusammenfassend lässt sich sagen, dass die Entwicklung von antiangiogenetischen Substanzen als Monotherapie oder in Kombination mit klassischen Chemotherapien Erfolg versprechend ist. 1.4 Bevacizumab (Avastin®) Die erste Erfolg versprechende Substanz nach jahrelanger Forschung an Hemmstoffen der Angiogenese war Bevacizumab (Avastin®) – ein monoklonaler Antikörper gegen den an der Tumorangiogenese beteiligten Wachstumsfaktor VEGF-A [15]. Die Entwicklung von Bevacizumab basiert auf dem murinen Antikörper A4.6.1, der bei Tierversuchen in Mäusen entdeckt wurde [15]. Dieser Antikörper erkennt eine spezifische Aminosäuresequenz des humanen VEGF-Moleküls und geht mit ihr eine hochaffine Bindung ein, sodass ein Ankoppeln des Liganden an seinen Rezeptor durch die mit der Bindung einhergehende Strukturveränderung inhibiert wird. Der Rezeptor wird nicht aktiviert, und die Signalübertragung ist unterbrochen [15]. Da eine Therapie mit murinen Wirkstoffen für Menschen schlecht verträglich ist, wurde der Antikörper humanisiert. Eine Synthese ergab den zu 93% humanen Antikörper Bevacizumab, der trotz Anpassung ähnlich starke Bindungseigenschaften zum Wachstumsfaktor VEGF aufweist wie der Stammantikörper A4.6.1 [15]. 16 In klinischen Studien zeigte der humane Antikörper beeindruckende Wirksamkeit bei Patienten mit metastasiertem Kolorektalkarzinom und wurde im Jahr 2005 zur Therapie dieser Erkrankung zusammen mit 5-Fluoruracil zugelassen [15]. Bevacizumab ist nicht nur der erste Wirkstoff, der zur Hemmung der Tumorangiogenese zugelassen wurde, sondern stellt zurzeit auch die erfolgreichste antiangiogene Therapieoption dar [15]. Das Prinzip der Antiangiogenese hat sich bis heute bei fünf Tumorentitäten (Darm-, Brust-, Lungen-, Eierstock- und Nierenzelltumoren) als wirksam erwiesen. Bevacizumab ist bei diesen Tumoren seit 2005 bzw. 2007 als fester Bestandteil der Standardtherapie zunehmend etabliert [14, 15, 53]. Die Zulassung zur Behandlung von Patienten mit fortgeschrittenem Ovarialkarzinom in den FIGO-Stadien IIIB-IV erfolgte Ende 2011 [1, 8], die Therapie mit Bevacizumab bei dieser Tumorentität ist weiterhin Bestandteil aktueller Studien [49]. Im Juni 2013 führten die Ergebnisse der randomisierten, doppelblinden Placebo-kontrollierten AVAglio-Studie in Japan zur Zulassung von Bevacizumab bei an Glioblastom oder Gliom erkrankten Patienten [27]. Zur Zulassung für die Erstlinientherapie des Glioblastoms in der Europäischen Union sowie der Schweiz sind derzeit noch klinische Prüfungen ausstehend [15]. Studien bestätigten die Wirksamkeit einer First-Line-Therapie mit Bevacizumab in Kombination mit einer Chemotherapie. Das Überleben von Patienten mit Darm- und Lungentumoren war bei der Kombinationstherapie signifikant länger, das progressionsfreie Überleben bei Patienten mit Ovarialkarzinom konnte um ein Drittel verlängert [1], bei Patienten mit Mamma- und Nierenzellkarzinomen sogar verdoppelt werden [15]. Auch wenn sich noch weitere Substanzen in der klinischen Entwicklung befinden und ihre Verwendung bei anderen Tumorentitäten geprüft wird [15], hat sich Bevacizumab als Pan-Angiogenese-Inhibitor bewährt. Die Annahme, wonach die Substanz die tumoröse Gefäßneubildung unabhängig von der Tumorentität hemmt, konnte bestätigt werden [15]. Zusätzliche Informationen zu Bevacizumab sind dem Abschnitt 7.1.1 im Anhang zu entnehmen. 17 1.5 Sorafenib (Nexavar®) Sorafenib ist ein potenter Multi-Proteinkinase-Inhibitor mit oraler Bioverfüg- barkeit [15]. Er zeigte präklinisch einen dualen Wirkmechanismus mit antiproliferativem Effekt gegen die Tumorzelle selbst und einer antiangiogenen Komponente [15]. Ursächlich hierfür ist die Inhibition der Rezeptor-Tyrosinkinasen, z.B. VEGFR2 und PDGFR-β, die eine wichtige Rolle für die Angiogenese und die Tumorproliferation spielen [15]. Eine erfolgreiche Antitumortherapie mit Sorafenib geht nicht notwendigerweise mit beiden Wirkmechanismen einher, es ist durchaus möglich, dass nur einer der Mechanismen die Tumorverkleinerung bewirkt [15]. Zunächst nahm man an, dass Sorafenib die Serin/Threonin-Kinase Raf-1 hemmt, da festgestellt wurde, dass diese in zahlreichen Tumorentitäten übermäßig aktiviert ist [15]. Dies hat zur Folge, dass auch der Signalweg bei Tumorpatienten verstärkt aktiviert ist, weil Raf-1 eine wichtige Rolle in der Signaltransduktion spielt. Normal regulierte Signalwege bilden die Basis für die einwandfreie Abwicklung der Angiogenese und der Regulation von Zellüberleben und -proliferation. Im Falle einer Überaktivität wird folglich die Tumorentstehung aufgrund der oben genannten Mechanismen begünstigt [15]. Präklinische Studien lieferten jedoch Erkenntnisse über die Hemmung weiterer Kinasen durch Sorafenib. Hierzu zählen beispielsweise Raf-Isoformen sowie die für die Angiogenese relevanten Tyrosinkinasen VEGF-R2 und PDGFR-β [15]. Zu den Eigenschaften von Sorafenib, die u.a. in Xenograft-Modellen nachgewiesen wurden, gehören der Einfluss auf die Autophosphorylierung verschiedener Signalübertragungswege [15], die Induktion der Apoptose über Raf-Hemmung bei humanen Tumorzellen [66], die dosisabhängige Hemmung des Tumorwachstums (Xenograft-Modell) [15] und die Inhibition der Tumorprogression [15]. Studien zeigten bislang folgende Ergebnisse zur Therapie von Nierenzellund Leberkarzinomen mit Sorafenib: Eine Phase-II-Studie (Randomized Discontinued Trial = RDT-Studie) zur Überprüfung der klinischen Wirksamkeit von Sorafenib ergab, dass das mediane progressionsfreie Überleben unabhängig von der Tumorentität bei der Kohorte mit Sorafenib signifikant länger war als bei der Placebo-Gruppe [50]. Die beobachtete antitumorale Wirkung wurde durch eine Phase-III-Studie (Treatment Approaches in Renal cancer Global Evaluation Trial = TARGETs) bestätigt [19], in der die So18 rafenib-Monotherapie gegenüber der Placebo-Therapie bei fortgeschrittenem Nierenzellkarzinom zu einer signifikanten Verlängerung des medianen progressionsfreien Überlebens führte (19,3 vs. 15,9 Monate). Mit der Sorafenib-Behandlung gelang es, die Stabilisierung der Tumorerkrankung von 55% (Placebo) auf 85% zu steigern, die Erkrankung schritt bei Patienten mit Sorafenib-Behandlung langsamer voran als bei Patienten mit Placebo-Therapie (167 vs. 84 Tage). Besonders bemerkenswert ist der Therapieerfolg beim Nierenzellkarzinom, weil dieser Tumor als außerordentlich resistent gegenüber Zytostatika gilt und die Überlebensrate der Patienten mit fortgeschrittenem Karzinom sehr schlecht ist [4]. Weniger als 20% der Patienten überleben länger als zwei Jahre [4]. Die Erfahrungen und Ergebnisse aus diesen Studien führten 2005/2006 zur Zulassung von Sorafenib. Es wird zur Behandlung des fortgeschrittenen Nierenzellkarzinoms nach erfolgloser Therapie mit Interferon-α oder IL-2 oder bei Nichteignung von Patienten für eine Zytokin-Therapie als sogenannte Second-LineTherapie eingesetzt [15]. Sorafenib wird zudem in der Therapie des hepatozellulären Karzinoms eingesetzt. Grundlage dieser Empfehlung sind folgende Erkenntnisse: Eine Phase-II-Studie mit Sorafenib als Monotherapie bei Patienten mit fortgeschrittenem hepatozellulärem Karzinom ergab neben der größeren Ansprechrate und der verlängerten progressionsfreien Zeit ein besseres Gesamtüberleben in der Sorafenib-Gruppe. Die Gruppe der anschließenden Phase-III-Studie (Sorafenib HCC Assessment Randomized Protocol = SHARP-Studie) umfasste Patienten mit unbehandeltem fortgeschrittenem hepatozellulärem Karzinom. Die Resultate dieser Studie wiesen auf ein signifikant höheres mittleres Gesamtüberleben in der Sorafenib-Gruppe hin [51]. Die Wirksamkeit und Tolerabilität von Sorafenib konnten durch eine weitere Phase-III-Studie (Asien-Pazifik-Studie) bestätigt werden [12, 15]. Aufgrund der Erkenntnisse aus der SHARP-Studie wurde Sorafenib als Therapieoption für das hepatozelluläre Karzinom Ende 2007 zugelassen [15]. Seit November 2013 ist Sorafenib in der Therapie des lokal fortgeschrittenen oder metastasierten, differenzierten Schilddrüsenkarzinoms zugelassen, das kein Ansprechen mehr auf eine Radiojodtherapie zeigt. Die DECISION-Studie untersuchte Patienten mit fortgeschrittenem Schilddrüsenkarzinom, die mit Placebo vs. Sorafenib behandelt wurden. Hierbei zeigte sich, dass das progressions19 freie Überleben unter Sorafenib-Therapie im Vergleich zur Placebo-Behandlung nahezu verdoppelt werden konnte [7]. Die Bedeutung von Sorafenib als Mono- und Kombinationstherapie bei anderen Tumorentitäten wird derzeit intensiv erforscht. Von besonderem Interesse sind hierbei das nichtkleinzellige Bronchialkarzinom und das Mammakarzinom sowie die adjuvante Therapie bei Nierenzell- und hepatozellulärem Karzinom [15]. Zusätzliche Informationen zu Sorafenib sind dem Abschnitt 7.1.2 im Anhang zu entnehmen. 20 2 Zielsetzung Mit den in den letzten 40 Jahren identifizierten molekularen Zielstrukturen ist heute eine gezielte Therapie von Krebserkrankungen möglich, die nicht nur besser verträglich als herkömmliche Chemotherapien, sondern auch langfristig wirksam ist [15]. Dennoch erfordert das geringe Gesamtüberleben von Patienten mit fortgeschrittener Tumorerkrankung die Entwicklung neuer Therapien. Eine vielversprechende effektive Tumortherapie scheint derzeit die Behandlung mit antiangiogenetischen Substanzen zu sein. Um diese Therapie aber möglichst effektiv zu gestalten, ist es sinnvoll, das Tumorwachstum an mehreren Stellen zu inhibieren, weil der Tumor bei seiner Entwicklung eine große Anzahl angiogenetischer Moleküle ausschüttet. Wird beispielsweise nur VEGF gehemmt, kann der Tumor auf andere Verbindungen umstellen [11]. Daher wird für die folgende Studie ein Therapieschema verwendet, das die Angiogenese des Tumors an zwei Punkten stört: zum einen über VEGF durch Bevacizumab, zum anderen über die Tyrosinkinasen durch Sorafenib. Eine dritte Substanz, das Chemotherapeutikum Paclitaxel, kann durch die mit Bevacizumab verbesserte Gefäßstruktur die Tumorzellen besser erreichen. Außerdem werden die Zellen zusätzlich mit Sauerstoff versorgt und sind damit vulnerabler für eine Chemotherapie. Paclitaxel soll die Proliferation der Tumorzellen verhindern und die Therapie der malignen Erkrankung damit noch effizienter gestalten. Die nachstehend beschriebene nicht-randomisierte prospektive Open-labelInterventionsstudie der Phase I mit Dosiseskalation soll primär die Machbarkeit („feasibility“) für mehrfach vorbehandelte Patienten mit metastasierter solider Tumorerkrankung bei weiterhin erträglichem Nebenwirkungsprofil untersuchen. Sekundäre Ziele der Studie sind Sicherheit und Wirksamkeit einer Kombinationstherapie aus einem Chemotherapeutikum (Paclitaxel), einem VEGFAntikörper (Bevacizumab) und einem Tyrosin-Kinase-Inhibitor (Sorafenib). Die Rationale für das verwendete Therapiekonzept basiert auf dem präklinischen Synergismus zwischen Paclitaxel und Sorafenib sowie auf dem Prinzip der optimierten Antiangiogenese. Dabei wird die Gefäßneubildung an zwei Stellen gleichzeitig mit potenziell synergistischer Wirkung inhibiert: zum einen durch das als Antiligand 21 wirkende Bevacizumab, zum anderen durch die Hemmung des VEGF-Rezeptors durch Sorafenib. 22 3 3.1 Material und Methoden Teilnehmerauswahl Zur Anwendung der Studienmedikation erfolgte die Auswahl der Probanden anhand bestimmter Kriterien. Die meisten Teilnehmer waren schon vor Studienbeginn Patienten des Marienhospitals Herne und nahmen an unterschiedlichen, teilweise auch experimentellen Chemotherapien teil. Bei vielen Probanden waren die Erst-Linien-Therapie für die jeweilige Tumorentität und die anschließende Entwicklung von Resistenzen gegenüber dem Regime vorausgegangen. Dies äußerte sich radiologisch anhand von Tumorgrößenzunahme oder Bildung neuer Metastasen, laborchemisch durch ansteigende Tumormarker oder klinisch durch Verschlechterung des Allgemeinzustands mit unterschiedlichen Symptomen. Nach Prüfung der Voraussetzungen zur Teilnahme an der Studie wurden die Patienten aufgeklärt und ihnen Bedenkzeit bis zur schriftlichen Einwilligung eingeräumt. Anschließend wurde der Laborstatus der Probanden aktualisiert, sofern dieser älter als 7 Tage war. Die Patienten kamen nicht nüchtern zur Abnahme von < 20ml Blut (1 bis 2 Serumröhrchen, 1 EDTA-Röhrchen zur Blutbildanalyse), der Allgemeinzustand wurde dokumentiert und die neuerliche Therapie begonnen. 23 Tabelle 2: Ein- und Ausschlusskriterien der Studienpopulation Studienpopulation: Patienten mit lokal fortgeschrittener solider Tumorerkrankung der inneren Organe, die sich als nicht resektabel oder metastasiert herausgestellt hatte; palliativer Therapieansatz Einschlusskriterien Ausschlusskriterien - Alter > 18 Jahre - Histologisch gesicherte, solide nahme: Basalzelltumor oder Tumorerkrankung mit mindes- Zervixkarzinom) oder kurative tens einer messbaren Läsion Tumortherapie bereits vor > 5 ECOG 2, sodass ambulante Jahren - - - Betreuung möglich ist - - Schwere Beeinträchtigung von Organen (Leber, Nieren) Z. n. Vortherapien (Chirurgie, - Radiatio, Zytostatika), keine wei- Keine adäquate Knochenmark-Reserve teren wirksamen Standard- chemotherapien möglich, daher Teilnahme an einer anderen Therapiestudie Einschluss in die experimentelle - Studie - Kontraindikationen gegen die Studienmedikamente Adäquate Knochenmark- und Organfunktion - Zweittumorerkrankung (Aus- Keine Einwilligung Effekte der Vortherapie müssen abgeklungen sein, sofern Nachwirkungen zu erwarten sind - 3.2 Schriftliche Einwilligung Diagnostik und Staging Vor Studienbeginn wurde bei allen Patienten ein Staging von Tumor und Me- tastasen mithilfe folgender Diagnostiken durchgeführt: - Anamnese, körperliche Untersuchung, Dokumentation des Allgemeinzustands - Internistisches Standardlabor (großes Blutbild, Elektrolyte, Glukose, Bilirubin gesamt, Harnstoff, Kreatinin, glomeruläre Filtrationsrate, Harnsäure, Ge- 24 samteiweiß, alkalische Phosphatase, Gamma-GT, Transaminasen, LDH, Elektrolyte, Albumin, CRP, spezifische Tumormarker) - MRT - CT Thorax/Abdomen - Sonographie Abdomen - Konventionelles Röntgen des Thorax in zwei Ebenen - Ggf. Skelettszintigraphien Hiermit konnte der Progress der Erkrankung dokumentiert und der Studienein- tritt begründet werden. Der Zeitraum zwischen Staging und Studienbeginn konnte je nach Allgemeinzustand des Patienten zwischen max. 24 Stunden und wenigen Wochen liegen. 3.3 Studiendesign Die Studienmedikation aus 400mg Sorafenib (Nexavar®) zweimal täglich, 90mg pro m² Körperoberfläche Paclitaxel (Taxol®) und dosiseskaliertem Bevacizumab (Avastin®) wurde unter ambulanten Bedingungen verabreicht. Die Probanden nahmen kontinuierlich morgens und abends im häuslichen Umfeld je zwei Tabletten zu 200mg (800mg pro Tag) Sorafenib in 12-stündigem Abstand ein. Beim Auftreten von Toxizitäten konnte die Dosis auf insgesamt 400mg (200mg zweimal täglich), in schweren Fällen auch auf 200mg täglich reduziert werden. Einmal pro Woche, also an Tag 1, 8 und 15 des Therapiezyklus, erschienen die Patienten in der hämato-onkologischen Ambulanz des Marienhospitals Herne. Zunächst wurden sie dem behandelnden Arzt vorgestellt, der neben Anamnese und körperlicher Untersuchung aktuelle Laborwerte bestimmte. Der Allgemeinzustand wurde dokumentiert und Toxizitäten oder Probleme während der letzten Therapiewoche erfragt. Bei regelrechten Resultaten erhielten die Probanden nach geeigneter Prämedikation (Antihistaminikum, Antiemetikum, Kortison) zunächst Paclitaxel als intravenöse Infusion. In der Regel wurde der Portzugang der Patienten genutzt, um die Dosis von ca. 90mg/m2 Körperoberfläche über 90 bis 180 Minuten zu infundieren. Nach kurzer Zwischenspülung mit physiologischer Kochsalzlösung wurde jedoch nur an Tag 1 und 15 des Therapiezyklus mit der Infusion 25 von Bevacizumab fortgefahren. Die Dosis des monoklonalen Antikörpers war von der zugeteilten Kohorte abhängig. Die Probanden wurden in sechs Kohorten mit je vier bis sechs Patienten eingeteilt. Die erste Kohorte mit 6 Teilnehmern erhielt kein Bevacizumab und die zweite mit 5 Patienten 1mg/kg Körpergewicht alle zwei Wochen. Wenn keine Toxizitätsprobleme auftraten, wurde die 14-tägige Dosis des Antikörpers bei den folgenden Kohorten auf 2 (4 Patienten), 4 (5 Patienten), 5 (4 Patienten) bzw. 10mg/kg Körpergewicht (4 Patienten) gesteigert. Die allererste Dosis Bevacizumab erfolgte bei jeder Kohorte im ersten Zyklus nicht an Tag 1, sondern erst an Tag 15, im zweiten Zyklus dann regulär an Tag 1 und 15. Dadurch kann die Verträglichkeit von Sorafenib in Kombination mit Paclitaxel zunächst zwei Wochen lang beobachtet werden. Nach komplikationsfreier Gabe wurden die Patienten 30 Minuten nachbeobachtet, bevor sie in die häusliche Umgebung entlassen wurden. Supportiv erhielten die Teilnehmer fettende, harnstoffhaltige Handcreme zur Vorbeugung des Hand-Fuß-Syndroms unter Sorafenib, nystatinhaltige Mundspüllösung zur Minimierung des Mukositisrisikos und pflegendes Nasenöl gegen trockene Schleimhäute. Die routinemäßige Gabe von Adjuvantien war nicht vorgesehen. Nur bei symptomatischen oder vital gefährdeten Patienten wurden G-CSF sowie Blutzellen (Erythrozyten- und Thrombozytenkonzentrate) appliziert. Sofern keine Nebenwirkungen auftraten, kamen die Patienten einmal pro Woche in die Ambulanz zu Untersuchung, Blutabnahme und Therapie. Bei auftretenden Toxizitäten konnten sie jederzeit den behandelnden Arzt konsultieren. Nach erfolgreichem Abschluss eines Zyklus folgte eine einwöchige Therapiepause, bevor der nächste Kurs begann. Maximal sollten die Patienten 6 Therapiezyklen erhalten, sofern es zu einem Ansprechen oder einer Stabilisierung der Erkrankung kam und die Nebenwirkungen tolerabel waren. In Ausnahmefällen und bei besonders guter Verträglichkeit konnte diese maximale Anzahl überschritten werden. 26 Tabelle 3: Studiendesign beispielhaft für die ersten beiden Therapiezyklen 1. Zyklus Tag Tag Tag Tag Tag Tag 1 2-7 8 9-14 15 16-21 Tag 22-28 Paclitaxel Bevacizumab Sorafenib 2. Zyklus1 Tag Tag Tag Tag Tag Tag Tag 1 2-7 8 9-14 15 16-21 22-28 Paclitaxel Bevacizumab Sorafenib 1 Zyklen 3, 4, 5, 6 nach gleichem Schema 27 Tabelle 4: Wöchentliches Therapieschema in der Ambulanz ohne morgendliche Sorafenib-Gabe Zyklustag Tag 1, 8, 15 Dauer Medikament 30 Minuten 1. Antihistaminikum Dimetinden (Fenistil®) 8mg i.v. Gabe direkt vor 2. Antiallergikum Cimetidin (Tagamet®) 400mg i.v. Chemotherapie 3. Dexamethason (Fortecortin®) 20mg i.v. Tag 90 – 180 Paclitaxel (Taxol®) 90mg/m2 KOF1 i.v. (Portzu- 1, 8, 15 Minuten gang) in 250ml NaCl 0,9% mit PVC-freiem Infusionsbesteck Tag wenige 12, 15 Minuten Tag 90, 60 oder 30 12, 15 Minuten3 Zwischenspülung mit NaCl 0,9% Bevacizumab (Avastin®) 0, 1, 2, 4, 5 oder 10mg/kg Körpergewicht i.v. (Portzugang) in 250ml NaCl 0,9% mit Standardinfusionsbesteck 1 Berechnung der Körperoberfläche (KOF) nach Du Bois & Du Bois [18]: KOF (m2) = (Körpergewicht in kg)0,425 x (Körpergröße in cm)0,725 x 71,84 x 10-4 2 nicht 3 im allerersten Zyklus Die erste Bevacizumab-Infusion läuft über 90 Minuten, bei guter Verträglichkeit kann die Dauer bei folgender Gabe auf 60 Minuten und anschließend auf 30 Minuten reduziert werden. 28 Tabelle 5: Geplantes und schließlich durchgeführtes Dosiseskalationsdesign (individuelle Dosisreduktionen vorbehalten) Anzahl Geplantes Dosiseskalationsdesign Durchgeführtes Studiendesign eingeschlossener Patienten (N = 28) Kohorte Paclitaxel Sorafenib I 90mg/m2 800mg II 90mg/m2 800mg III 90mg/m2 800mg IV 90mg/m2 800mg V 90mg/m2 800mg VI 90mg/m2 800mg 1 Dosisreduktion Bevacizumab 0 mg/kg Körpergewicht 1 mg/kg Körpergewicht 2 mg/kg Körpergewicht 4 mg/kg Körpergewicht 5 mg/kg Körpergewicht 10 mg/kg Körpergewicht Paclitaxel Sorafenib 90mg/m2 400mg1 90mg/m2 400mg1 90mg/m2 400mg1 90mg/m2 400mg1 90mg/m2 400mg1 90mg/m2 400mg1 aufgrund dosislimitierender Toxizitäten (siehe Abschnitt 4.2.2.2) 29 Bevacizumab 0 mg/kg Körpergewicht 1 mg/kg Körpergewicht 2 mg/kg Körpergewicht 4 mg/kg Körpergewicht 5 mg/kg Körpergewicht 10 mg/kg Körpergewicht 6 5 4 5 4 4 3.4 Studienziele Nachfolgend sind die primären und sekundären Studienziele aufgeführt. Tabelle 6: Endpunkte der vorliegenden Studie Endpunkte Primär Machbarkeit der Studie Sicherheit: - Unerwünschte Arzneimittelnebenwirkungen / Medikamententoxizitäten Sekundär - Ausgewählte Laborwertveränderungen - Schwere unerwünschte Ereignisse - Stärkste Nebenwirkung je Patient Wirksamkeit: - Tumoransprechrate (Response Rate) - Überlebenszeit - Progressionsfreies Intervall Die Studie diente auch der Überarbeitung des Therapiekonzeptes und der Medikamentendosierung [57, 63]. Zur routinemäßigen Kontrolle von Sicherheit und Wirksamkeit der Therapie wurde regelmäßig (im Allgemeinen wöchentlich) das genannte breite internistische Standardlabor erstellt. Zusätzlich dienten der wöchentliche Kontakt zu den Probanden und die detaillierte Dokumentation ihrer Beschwerden während des Zyklus zur Überwachung der Nebenwirkungen. Die Dokumentation umfasste neben den Symptomen auch die Dauer der Beschwerden, therapeutische Maßnahmen und die Beurteilung des Zusammenhangs. Zusätzlich mussten Angaben über den Therapieverlauf einschließlich Datum, Dosierungen und eventuellen Abweichungen vom Behandlungsplan erfolgen. 30 3.5 Sicherheit Die Sicherheit der Medikamente stellt einen wesentlichen Aspekt der Stu- dienziele dar. Sie kann mithilfe unerwünschter Arzneimittelnebenwirkungen (UAWs) und Medikamententoxizitäten, Laborwertveränderungen, schwerer unerwünschter Ereignisse (SUEs) und der stärksten Nebenwirkung je Patient bewertet werden. 3.5.1 UAWs / Medikamententoxizitäten und Laborwertveränderungen Die Bewertung von UAWs und Medikamententoxizitäten sowie der Laborwertveränderungen erfolgte mithilfe der Common Toxicity Criteria (CTCAE) des National Cancer Institute, Version 3.0 [48]. Anhand dieser werden akute und subakute Toxizitäten in Schweregrade von 0 bis 4 eingeteilt. Dabei stellt Grad 0 die leichteste und Grad 4 die schwerste Form der Nebenwirkung dar (Details siehe Anhang, Tabelle A2). Zur Auswertung der Studienergebnisse wurden alle Nebenwirkungen während der Therapie berücksichtigt, die im Zusammenhang mit der Chemotherapie entstanden. Obwohl Medikamententoxizitäten durchaus auch erst Wochen nach Therapiebeendigung auftreten können, wurden diese in der Auswertung nicht berücksichtigt, weil ein eindeutiger Zusammenhang mit der Studienmedikation sich nicht in jedem Fall nachweisen lässt. Einige Probanden erhielten nach Abbrechen der Studie eine andere antitumoröse Medikation, sodass nicht mehr eindeutig nachweisbar war, durch welche Medikamente die unerwünschten Wirkungen verursacht wurden. Bei anderen Patienten war eine Nachbeobachtung über beispielsweise 30 Tage gar nicht möglich, weil sie vorher verstarben. Nebenwirkungen, die eindeutig anderer Genese waren, wurden aus der Auswertung ausgeschlossen, unklar dokumentierte Nebenwirkungen blieben ebenfalls unberücksichtigt. Die durch die Paclitaxel-Therapie aufgetretene reversible Alopezie trat bei allen Studienteilnehmern auf und wurde aufgrund rein kosmetischer Probleme nicht in die Auswertung mit einbezogen. Außerdem gab es Patienten, die bereits durch die Vortherapien eine Alopezie erlitten hatten. Bei Schwankungen des Nebenwirkungsgrades nach CTCAE in der ärztlichen Beurteilung wurde stets der höhere Grad herangezogen. Patienten, die 31 während der Studie sowohl an Grad 3 als auch 4 Nebenwirkungen einer Laborwertkategorie (z.B. Leukozyten oder Hämoglobin) litten, wurde in der Auswertung lediglich einmal mit dem schwersten Grad bewertet. 3.5.2 Schwere unerwünschte Ereignisse Zu den unerwünschten Ereignissen zählen jede beobachtete, subjektive oder objektive Befindlichkeitsstörung und jede (klinisch relevante) Laborwertveränderung während der Studienlaufzeit. Das unerwünschte Ereignis muss nicht notwendigerweise im Zusammenhang mit der Medikamenteneinnahme stehen, sondern kann auch eine Unfallfolge sein oder im Anschluss an die Therapie auftreten. Sollte das Medikament Mitverursacher oder Ursache des unerwünschten Ereignisses sein, handelt es sich um eine unerwünschte Arzneimittelnebenwirkung. In der Auswertung der Sicherheit werden diese Ereignisse nicht mit einbezogen, um die Übersichtlichkeit der Nebenwirkungen, die im direkten Zusammenhang mit der Studienmedikation stehen, zu bewahren. Ein schweres unerwünschtes Ereignis führt zu einem stationären Aufenthalt oder zur Verlängerung einer stationären Behandlung. SUEs setzen per definitionem eine vitale Bedrohung, eine dauernde Arbeitsunfähigkeit, eine nachfolgende Behinderung, Missbildung oder einen Zweittumor voraus. Im schlimmsten Fall führen sie zum Tod. Die Sicherheit der Medikamentenkombination wird anhand der Anzahl der SUEs und der Nebenwirkungen Grad 3 und 4 nach CTCAE beurteilt. 3.5.3 Stärkste Nebenwirkung je Patient Eine weitere Möglichkeit, die Toxizität einer Chemotherapie allgemein zu beschreiben, ist die stärkste Nebenwirkung je Patient, d.h., die Nebenwirkung mit dem höchsten Schweregrad während einer zytostatischen Therapie. Daraus lässt sich zusammenfassend die Toxizitätsrate berechnen, sie wird bestimmt durch die Anzahl der Probanden, die vor allem unter Nebenwirkungen Grad 3 und 4 litten. Diese Nebenwirkungen sind von besonderem Interesse, weil sie zu einer akuten 32 Gefährdung der Patienten führen können, ein Beispiel dafür ist die Dehydratation bei Diarrhoen. 3.6 Wirksamkeit Zu den wichtigsten sekundären Endpunkten der Studie zählte die Wirksam- keit der Medikation. Diese kann mithilfe der Tumoransprechrate und der Überlebenszeit bewertet werden. 3.6.1 Tumoransprechrate Die Tumoransprechrate wird mithilfe des unidimensionalen Messverfahrens nach RECIST, Version 1.1 beurteilt (Response Evaluation Criteria in Solid Tumors) [60], das Auskunft über das Ausmaß der radiologischen Tumorantwort unter onkologischer Therapie gibt. Tabelle 7: Remissionskriterien messbarer Tumorparameter nach RECIST, Version1.1 [60] Vollständige Rückbildung jeder klinischen EviComplete Response (CR) denz des Tumors, kein Tumorgewebe mehr nachweisbar Reduktion der Summe der größten Längsdurch- Partial Response (PR) messer der einzelnen beobachteten Läsionen um ≥ 30% Stable Disease (SD) / No Change (NC) < 30%-ige Reduktion und <20%-ige Ausdehnung der Summe der größten Längsdurchmesser der einzelnen beobachteten Läsionen ≥ 20%-ige Zunahme der Summe der größten Progressive Disease (PD) Längsdurchmesser der einzelnen beobachteten Läsionen oder Auftreten einer neuen Läsion Die Bestimmung der messbaren Tumorläsion und die Bewertung des Tumoransprechens gemäß der RECIST-Klassifikation erfolgten durch die behan33 delnden Onkologen / Radiologen und wurden der Aktenlage entnommen. Die Beurteilung der Tumorantwort bezog sich jedoch stets auf das beste Ansprechen während der Therapie. Obwohl die RECIST-Kriterien zur Remission einer Tumorerkrankung vorrangig an dem messbaren Tumordurchmesser festgelegt werden, wurde im Rahmen der Studie eine regelmäßige Kontrolle der spezifischen Tumormarker im Serum ergänzend durchgeführt, um eine vollständige Bewertung der individuellen Tumorantwort unter Therapie zu erhalten. 3.6.2 Überlebenszeit Die Verlängerung der Überlebenszeit der Patienten ist das primäre Ziel einer Chemotherapie und hat neben den RECIST-Kriterien eine wesentliche Bedeutung bei der Bewertung der Effektivität einer Antitumortherapie [60]. Es bleibt zu überprüfen, ob ein auf die Behandlung ansprechendes Tumorverhalten auch mit einer Verlängerung der Überlebenszeit einhergeht. Die Überlebenszeit definiert sich als Zeitspanne zwischen Therapiebeginn bis zum Tod der Probanden oder bis zum letzten Beobachtungszeitpunkt. Bei vielen Patienten konnte mithilfe hausärztlicher Auskunft das genaue Todesdatum bestimmt werden. Patienten, bei denen lediglich der Monat ihres Todes bekannt war, wurden zur Berechnung der Überlebenszeit einbezogen, indem das Todesdatum auf die Mitte des Monats festgelegt wurde. Da nicht alle Patienten im Rahmen der Studie an den Folgen ihrer Erkrankung verstarben, wurde als letzter Beobachtungszeitpunkt der 31.12.2012 definiert. Die noch lebenden Patienten wurden in die Auswertung der Überlebenszeit mit dem Datum des letzten Kontakts zum behandelnden Arzt bis zum 31.12.2012 einbezogen. Die mediane Überlebenszeit (overall survival) wird aus den Überlebenszeiten des Gesamtkollektivs bestimmt und gibt an, zu welchem Zeitpunkt 50% der Patienten noch lebten. Sie kann beim Vergleich verschiedener Therapieformen als wichtiger Parameter herangezogen werden. Bei Zeitangaben werden für ein Jahr 365 Tage und für einen Monat 4,3 Wochen oder 30,4 Tage angenommen. 34 Um die 1-, 2- und 3-Jahresüberlebensraten zu ermitteln, wurden die Überlebenszeiten des untersuchen Patientenkollektivs in Jahresintervalle gruppiert. So kann ein Überblick gegeben werden, wie viel Prozent des Gesamtkollektivs sich nach entsprechender Zeit (z. B. 1 Jahr nach Therapiebeginn) noch unter Beobachtung befanden und somit zu dem Anteil noch lebender Patienten zählte. 3.6.3 Dauer des Tumoransprechens - Progressionsfreies Intervall Um den Erfolg einer zytostatischen Therapie zu messen, dient der Begriff des progressionsfreien Intervalls nach der Union Internationale Contre le Cancer (UICC). Dieses ist definiert als Zeitdauer zwischen Therapiebeginn und Nachweis der Progression oder Todesdatum des Patienten, je nachdem was eher eintrifft. Die Todesursache ist dabei nicht von Bedeutung. Sollte es zum Zeitpunkt des letzten Beobachtungsdatums (31.12.2012) noch zu keiner Progression der Erkrankung gekommen sein, dient das letzte Beobachtungsdatum als Endpunkt. Es werden dabei alle Patienten in die Auswertung einbezogen, die auf die Therapie mit kompletter bzw. partieller Remission oder einer Erkrankungsstabilisierung (Stable Disease) reagierten. 3.7 Kontrolle des Ansprechens und Fortführung der Therapie Die routinemäßige Kontrolle des Therapieerfolgs wurde in der Regel nach dem 2. oder 3. Zyklus mithilfe bildgebender Verfahren durchgeführt. Patienten, die weniger als 2 oder 3 Therapiezyklen absolviert hatten, erhielten ohne dringende Indikation für eine Bildgebung kein neues Staging. Das letzte Staging erfolgte ca. 1-2 Wochen nach der letzten zytostatischen Gabe, wenn ein Tumorprogress vermutet wurde, anderenfalls erst nach drei Wochen. Die spezifischen Tumormarker im Serum wurden in der Regel in 4- bis 10wöchigem Abstand kontrolliert. Mithilfe dieser Daten war es möglich, die Tumorausmaße erneut zu beurteilen und ein Restaging zu erstellen. Unter Therapie wurde der Verlauf der Tumorausdehnung periodisch beobachtet und das weitere Vorgehen abgestimmt. Kam es unter der neuerlichen Behandlung zu einem Erkrankungsprogress, wurde die Therapie abgebrochen und der Patient anderweitig 35 behandelt. Im Fall einer stabilen Erkrankung, einer partiellen Remission oder einer kompletten Remission wurde die Therapie um zwei bis drei Kurse verlängert, bevor ein Restaging erneut über den weiteren Verlauf entschied. 3.8 Prüfdauer, Therapieabbruch und Nachsorge 28 Patienten mit metastasierter Tumorerkrankung und multiplen Vorthera- pien wurden mithilfe des oben beschriebenen Medikamentenschemas zwischen Juli 2009 und März 2012 behandelt. Mangelnde Compliance der Patienten, Auftreten von für Proband und Arzt intolerablen Nebenwirkungen (zzgl. Lebensqualitätseinschränkung, schwerer Allgemeinzustandsverschlechterung) und schwerwiegenden unerwünschten Ereignissen waren Kriterien, die zum Abbruch der Therapie führten. Sofern der Patient die Einwilligung nicht zurückzog oder den Austritt aus der Studie wünschte, traf der behandelnde Arzt die Entscheidung, wann eine Therapie abgebrochen werden musste. Anderenfalls wurde sie bis zum Fortschritt der Erkrankung oder Tod des Patienten fortgeführt. Nach Beendigung oder Abbruch der Therapie wurden alle Teilnehmer bis zu ihrem Tod bzw. bis zum 31.12.2012 als letztes Beobachtungsdatum nachbeobachtet. Die Beobachtungszeit wurde aus der Zeitspanne zwischen Therapiebeginn und letztem Beobachtungszeitpunkt oder Todestag der Probanden ermittelt. Die kürzeste Beobachtungszeit einer Teilnehmerin betrug 13 Tage, am längsten wurde eine Patientin mit Mammakarzinom über 938 Tage nachbeobachtet. Die mediane Beobachtungsdauer (follow-up) betrug 225 Tage. Sowohl die Therapiedauer als auch die Fortsetzung der Kurse (Vorteile versus Toxizitäten) hingen maßgeblich vom Schweregrad der individuell aufgetretenen Nebenwirkungen ab. Nach Abbruch der Studienteilnahme wurden die folgende Tumorbehandlung sowie die geplante Erhaltungstherapie nach erfolgreicher Therapiebeendigung auf die individuelle Situation abgestimmt. 36 3.9 Statistische Methoden Das Verteilungsmuster vieler Daten der vorliegenden Studie entsprach nicht der Normalverteilung der Grundgesamtheit (unsymmetrische oder schiefe Verteilung). Da Ausreißer den Mittelwert stark beeinflussen, wird der Median aus Messund Erhebungsreihen zur Erfassung statistischer Maße dem Mittelwert vorgezogen, da er von extremen Messwerten kaum beeinflusst wird [37]. Um Streuungsmaße darstellen zu können, wird die Spannweite als Differenz des größten und kleinsten Messwertes innerhalb der Stichprobe genutzt [38]. 37 4 Ergebnisse Im Folgenden werden die Ergebnisse der oben beschriebenen Phase-IStudie vorgestellt. 4.1 Patienten Die nachstehende Probandenauswertung wurde anhand der Aktenlage der Studienteilnehmer vollzogen. Die Angaben beruhen auf der Dokumentation der behandelnden Ärzte. 4.1.1 Patientenmerkmale Auf der Grundlage der in der Einleitung beschriebenen Zusammenhänge wurde die oben beschriebene Phase-I-Studie mit einer Kombinationstherapie aus einem Tyrosinkinase-Inhibitor (Sorafenib), dem VEGF-A-Antikörper (Bevacizumab) und dem Chemotherapeutikum Paclitaxel durchgeführt. Unter den 28 Studienteilnehmer/innen waren 21 Frauen und 7 Männer, ihr Durchschnittsalter bei Therapiebeginn betrug 60 Jahre, die Spannweite lag zwischen 39 und 78 Jahren. Der Median ließ sich bei 61 Jahren ermitteln. In den folgenden Graphiken bezieht sich die Angabe n = x auf die Anzahl der jeweils in die Untersuchung einbezogenen Patienten. 4.1.2 Entität, Histologie, Grading Bei allen Studienteilnehmern wurden histologisch solide, meist weit fortgeschrittene Tumore unterschiedlicher Entität festgestellt. Am häufigsten kam mit 55% das Mammakarzinom vor. 3 Patienten waren an einem Pankreaskarzinom (10%) erkrankt, bei jeweils 2 Patienten wurde ein Urothelkarzinom der Blase, ein Ovarial- bzw. ein Bronchialkarzinom (NSCLC) nachgewiesen (je 7%), je einmal waren Angiosarkom, Prostata-, Cholangio- und Ösophaguskarzinom vertreten (je 3%). Eine Patientin litt sowohl unter einem Ovarial- als auch unter einem beidseiti38 gen Mammakarzinom, deswegen bezieht sich die Anzahl in der folgenden Tabelle nicht auf 28 Patienten, sondern auf n = 29. Im weiteren Verlauf wird das Ovarialkarzinom dieser Patientin nur in die Auswertung der mittleren Erkrankungsdauer und in die Anzahl an Vortherapien einbezogen. Für weitere Ergebnisse ist es nicht von Wichtigkeit, da es sich um eine Nebendiagnose handelt. Abbildung 1: Häufigkeitsverteilung der Tumore (n = 29) Außerdem waren die Tumore vieler Patienten – unabhängig von der Tumorentität – histologisch stark entdifferenziert (Malignitätsgrad mäßig (G2) bis schlecht (G3)). Die Differenzierung konnte bei 26 von 28 Probanden ermittelt werden und verteilte sich auf einen Patienten im G1-Stadium, 15 im G2- und 10 Patienten im G3-Stadium. Tabelle 8: Allgemeine Stadieneinteilung bei Tumoren G1 Niedriger Malignitätsgrad: gute Differenzierung der Tumorzellen G2 Mittlerer Malignitätsgrad: Zwischenform von G1 und G3, in der die Tumorzellen noch mäßig differenziert sind G3 Hoher Malignitätsgrad: schlechte Differenzierung der Tumorzellen G4 Sehr hoher Malignitätsgrad: völlige Entartung der Tumorzellen 39 4.1.3 Tumorstadien und Metastasierung Retrospektiv ließ sich bei mindestens zwei Drittel der Studienteilnehmer bei der Erstdiagnose ein Tumorstadium von III und IV nach der UICC ermitteln (siehe Tabelle A1 im Anhang). Dies entspricht nicht nur einem fortgeschrittenen Erkrankungsstadium mit möglicher Infiltration des Tumors in die Umgebung, sondern geht auch mit einer Lymph- und Fernmetastasierung einher. Eine weitere Differenzierung in die einzelnen Stadien war nicht möglich, da bei vielen Patienten die eindeutige Tumorklassifikation fehlte. Bei 3 Patienten ließ sich das Tumorstadium gänzlich nicht eruieren. Zu Beginn der Studie befanden sich 24 von 28 Patienten (86%) im Tumorstadium IV nach UICC, da Fernmetastasen bekannt waren. Bei lediglich 3 Teilnehmern war der Tumor lymphatisch metastasiert und gehörte damit zum Stadium III. Eine Patientin war metastasenfrei. Wie bereits erwähnt wurde, sind die Stadien III und IV der UICC durch multiple Metastasen gekennzeichnet. Durchschnittlich litt jeder Patient neben dem Primarius unter zwei bis drei verschiedenen Metastasenlokalisationen. Die Tumorabsiedlung in Lymphknoten war mit 71% am häufigsten vertreten (20 von 28 Patienten). Mit 61% folgte der Leberbefall und 14 Teilnehmer litten unter einer ossären Metastasierung (50%). Bei jeweils 6 Patienten wurden Lungenmetastasen bzw. eine Lymphangiosis carcinomatosa diagnostiziert (je 21%) und bei 4 eine Peritonealkarzinose (14%). Muskulatur-, Perikard- und Pleurametastasen waren mit je 7% seltener vertreten, je einmal traten Filiae in Fettgewebe, Knochenmark, Mediastinum, Milz und Zwerchfell auf. 40 Abbildung 2: Häufigkeitsverteilung der Organmetastasierung Anmerkung: n = 78, da 22 Studienteilnehmer mehrere Metastasierungsorte aufwiesen 4.1.4 Vorbehandlung Voraussetzung zur Teilnahme an der Studie mit der Medikamentenkombination Paclitaxel, Sorafenib und Bevacizumab war eine vielfache chemotherapeutische Vorbehandlung der Patienten. Ausgenommen davon waren lediglich zwei Patienten, deren Tumorerkrankung bei Studienbeginn noch nicht zytostatisch vortherapiert war. Ihr Einschluss in die Studie erfolgte als Einzelfallentscheidung bei zu erwartendem individuellen Benefit. Häufig hatten die Teilnehmer neben der zytostatischen Vorbehandlung auch chirurgische und strahlentherapeutische Anwendungen erhalten. Vor Studienbeginn waren 20 von 28 Patienten chirurgisch vorbehandelt (71%), hiervon war bei einer Patientin eine explorative Laparotomie mit dem Ergebnis ‚inoperabel’ durchgeführt worden. Bei 8 Teilnehmern war kein operativer Eingriff erfolgt, überwiegend handelte es sich hierbei um Rezidive einer primär chirurgisch therapierten Erkrankung oder um eine Tumorerkrankung, die für eine 41 operative Sanierung schon zu weit fortgeschritten war. Eine Strahlentherapie war bei ca. zwei Drittel der Studienteilnehmer im Vorfeld bekannt. 10 der 28 Probanden hatten sich keiner Radiatio unterzogen (36%). Ca. 50% der Patienten erhielten sowohl eine Strahlen- als auch eine chirurgische Therapie, 5 wurden nur operiert und drei wurden alleinig bestrahlt. Die meisten Studienteilnehmer waren nicht nur chirurgisch und strahlentherapeutisch, sondern auch polychemotherapeutisch vorbehandelt. Die Anzahl der zytostatischen Vorbehandlungen errechnet sich aus der Addition der einzelnen Chemotherapieregime unabhängig von der Häufigkeit der Einnahmen. Insgesamt gab es 76 Zytostatikatherapien bei 27 Studienpatienten (bei einer Patientin war die Anzahl nicht eruierbar), der Median lag bei 2 Therapielinien. Die individuelle Anzahl variierte jedoch stark zwischen 0 und 13 Therapieregimen. Am häufigsten waren die Patienten mit Gemcitabine, Cyclophosphamid, Paclitaxel, Fluoruracil, Carboplatin oder Capecitabin vorbehandelt. Zusätzlich erhielten 12 von 28 Patienten eine antihormonelle Begleitmedikation. Abbildung 3: Zusammenfassung der Vortherapien bei jedem Patient (n = 28) 42 Tabelle 9: Zusammenfassung der Patientencharakteristika Patientencharakteristika Anzahl der Patienten (n = x) Teilnehmende Patienten - auswertbar bzgl. Toxizität - auswertbar bzgl. Ansprechen / Überleben 28 28 22 Geschlecht Frauen Männer 21 7 Mamma Pankreas Urothel Ovarien TumorlokalisaBronchien tion Prostata Choleangiozellen Ösophagus Angiosarkom 16 3 2 2 2 1 1 1 1 (55%) (10%) (7%) (7%) (7%) (3%) (3%) (3%) (3%) G1 G2 G3 unbekannt 1 15 10 2 (4%) (54%) (36%) (7%) Lymphknoten Leber Knochen Lunge Lymphangiosis carcinomatosa Peritoneum Muskulatur Metastasierung Perikard Pleura Fettgewebe Knochenmark Mediastinum Milz Zwerchfell 20 17 14 6 6 4 2 2 2 1 1 1 1 1 (71%) (61%) (50%) (21%) (21%) (14%) (7%) (7%) (7%) (4%) (4%) (4%) (4%) (4%) 20 18 12 (71%) (64%) (43%) Grading Chirurgie Radiatio antihormonelle Begleitmedikation Vorbehandlung Chemotherapie: Gesamt Median Spannweite 76 2 0-13 43 4.2 Therapie 4.2.1 Anzahl der Zyklen pro Patient Nur drei Studienteilnehmer konnten die geplanten sechs Zyklen abschließen, ein Patient erhielt die Studienmedikation wegen überdurchschnittlich guter Therapieverträglichkeit sogar über acht Zyklen. 21% der Studienteilnehmer konnten schon den ersten Zyklus nicht beenden, ca. 18% schlossen jeweils einen bzw. zwei Zyklen ab, 11% erreichten drei Therapiezyklen, vier Patienten erhielten vier und ein Teilnehmer fünf vollständige Therapiezyklen. Insgesamt wurden 71 vollständige Zyklen verabreicht, der Median lag bei zwei Zyklen pro Patient. Abbildung 4: Anzahl der Patienten mit abgeschlossenen Therapiezyklen 4.2.2 Dosisreduktion und Therapiepausen Bei den meisten Studienteilnehmern war es aus unterschiedlichen Gründen nicht möglich, die Durchführung des Medikamentenschemas einzuhalten. Häufig litten die Patienten unter schweren Nebenwirkungen, sodass die Gabe einer oder mehrerer Wirkstoffe in der Regel für eine Woche ausgesetzt und die Besserung der Symptome abgewartet werden musste. Anschließend erfolgte eine Reevaluation der Beschwerden und gegebenenfalls die Wiederaufnahme der Therapie. 44 4.2.2.1 Paclitaxel Die bei allen Patienten angestrebte Dosierung von 90mg Paclitaxel pro m² Körperoberfläche konnte fast ausnahmslos eingehalten werden. Bei einem Patienten musste laut Dokumentation des behandelnden Arztes die Dosis wegen einer mäßiggradigen Polyneuropathie auf 75% reduziert werden. Die Polyneuropathie unter Paclitaxel war auch mehrfach der Grund für eine Therapiepause. Wegen Nageltoxizität mit Nagelverlust unter Therapie musste das Zytostatikum bei einem anderen Patienten pausiert werden. 4.2.2.2 Sorafenib Bei allen Patienten musste die Sorafenib-Dosierung bereits in der ersten Kohorte wegen der hohen Nebenwirkungsrate um 50% reduziert werden. Auch in der zweiten Kohorte war die ursprünglich geplante Dosis von 800mg Sorafenib nicht tolerabel, sodass auch hier eine Reduktion auf 400mg pro Tag erfolgte. Für die folgenden Kohorten wurde die Anfangsdosis schließlich auf 400mg Sorafenib festgelegt (siehe Abschnitt 3.3, Tabelle 5). Trotzdem war bei vielen Studienteilnehmern aufgrund der schweren Toxizitäten eine weitere Reduktion der Sorafenib-Dosis auf 200mg täglich, in einigen Fällen sogar auf noch weniger (200mg im Abstand von 2 oder 3 Tagen) notwendig. Aus der ärztlichen Dokumentation geht hervor, dass das Hand-Fuß-Syndrom bei mindestens 39% der Patienten der mit Abstand häufigste Grund für die Dosisreduktion des Tyrosinkinase-Inhibitors oder für Therapiepausen war. Die Mukositis war trotz intensiver Mundschleimhautpflege der zweithäufigste Grund, die Therapie mit Sorafenib zu pausieren oder dessen Dosis zu reduzieren. Außerdem sind mehrfach Heiserkeit, depressive Verstimmung und Diarrhoe als Ursache für eine Dosisreduktion bzw. Therapiepause dokumentiert. Weiterhin werden Übelkeit und Erbrechen, Körpergewichtsverlust, Soor, Asthenie und die Entstehung eines Gesäßulkus bzw. Exanthems bei je einem Teilnehmer als Gründe für Therapiepausen oder Dosisreduktion genannt. Im Allgemeinen stellt das Hand-Fuß-Syndrom die dosislimitierende Nebenwirkung von Sorafenib dar. 45 4.2.2.3 Bevacizumab Die häufigste Indikation für eine Therapiepause von Bevacizumab stellten Wundheilungsstörungen bei mehreren Studienteilnehmern dar. Weitere Indikationen waren eine tiefe Venenthrombose und der Wunsch eines Patienten nach Pausieren des Antikörpers. Bei einem Patienten in der Kohorte, die 10mg/kg Körpergewicht Bevacizumab erhielt, war es notwendig, den Antikörper auf die Hälfte der Dosis zu reduzieren. Tabelle 10: Dosisreduktion der Medikamente in den unterschiedlichen Kohorten Kohorte I II III IV V VI (n = 6) (n = 5) (n = 4) (n = 5) (n = 4) (n = 4) Paclitaxel 0 0 0 1 0 0 1 Sorafenib1 6 5 1 3 2 4 21 Bevacizumab2 – 0 0 0 0 1 1 1 Kohorten ∑ I und II Sorafenib-Startdosis: 800mg täglich Kohorten III, IV, V und VI Sorafenib-Startdosis: 400mg täglich 2 Kohorte I: Kein Bevacizumab erhalten 4.2.3 Therapiedauer Die Dauer der Therapie, die sich aus der Zeitspanne zwischen Studienstart und Studienende ergibt, ist unabhängig von der Anzahl der abgeschlossenen Therapiezyklen. Bei einem Teilnehmer wurde die Therapie bereits nach vier Tagen abgebrochen, die längste Therapiedauer betrug 286 Tage. Im Median dauerte die Therapie pro Patient 81 Tage. 46 Abbildung 5: Therapiedauer pro Patient (n = 28) 4.2.4 Ursachen für ein Therapieende Bei insgesamt 25 Patienten musste die Therapie vorzeitig beendet werden. Eine Patientin mit sechs abgeschlossenen Therapiezyklen hatte zwar ebenfalls das offizielle Therapieende erreicht, konnte aber wegen eines schweren HandFuß-Syndroms keine weiteren optionalen Zyklen erhalten und wurde daher in die Kategorie „Therapieende wegen Toxizität“ eingeordnet. Die Medikamententoxizität war insgesamt die häufigste Ursache für das vorzeitige Beenden der Therapie und trat bei weiteren sieben Patienten (insgesamt n = 8) auf. Mehrfach therapielimitierend waren das Hand-Fuß-Syndrom, die Polyneuropathie, Mukositis und Schmerzen, außerdem führten allergische Reaktionen, Heiserkeit, Asthenie und Blutdruckanstieg, Übelkeit und Erbrechen zum Therapieabbruch. Sechs Teilnehmer erkrankten während der Studie an Krankheiten, die eindeutige Kontraindikationen für die weitere Therapie darstellten. Dies waren im Einzelnen: Wundheilungsstörungen von Port- oder Ablatio-Mammae-Wunden (n = 3), Osteonekrose des Kiefers (n = 2) und tiefe Venenthrombose (n = 1). Bei fünf Patienten musste die Therapie vorzeitig beendet werden, weil im Verlauf ein Tumorprogress aufgetreten war, sodass auf weitere Kurse desselben Schemas verzichtet wurde. Eine erhebliche Reduktion des Allgemeinzustandes (AZ) und Erschöpfung führten bei drei Pa47 tienten zu vorzeitigem Therapieende, weitere drei Teilnehmer konnten aufgrund unzureichender Compliance nicht weiterbehandelt werden. Bei ihnen lagen zu lange Therapiepausen zwischen den Zyklen oder sie kamen nicht mehr in die onkologische Ambulanz zur Weiterführung der Therapie. Tabelle 11: Ursachen für eine Beendigung der Therapie (n = 28) Anzahl der Patienten Ursache 8 (29%) Medikamententoxizität 6 (21%) Kontraindikation für weitere Therapie 5 (18%) Tumorprogress 3 (11%) AZ-Reduktion / Erschöpfung 3 (11%) Non Compliance 3 (11%) Therapieende erreicht 4.2.5 Sicherheit Um die Sicherheit der Studienmedikation zu evaluieren, werden im Folgenden die unerwünschten Arzneimittelnebenwirkungen und Medikamententoxizitäten, ausgewählte Laborwertveränderungen, schwere unerwünschte Ereignisse / stationäre Aufenthalte sowie die stärkste Nebenwirkung je Patient näher betrachtet. 4.2.5.1 Unerwünschte Arzneimittelnebenwirkungen und Medikamententoxizitäten Zur Auswertung der unerwünschten Arzneimittelnebenwirkungen und Medikamententoxizitäten ist nicht nur die Gesamthäufigkeit im Patientenkollektiv relevant. Von besonderer Bedeutung ist die Häufigkeit der Nebenwirkungen Grad 3 und 4, weil sie zu einer akuten Gefährdung des Patienten führen können und häufig mit einer starken Einschränkung der Lebensqualität einhergehen. Nachstehend werden die wichtigsten und typischsten Nebenwirkungen der Studienmedikation und ihre Häufigkeit graphisch dargestellt. Die Hämatotoxizität als Nebenwirkung 48 der Studienmedikation wurde nicht in die Graphik aufgenommen, sie wird im Abschnitt „Laborwertveränderungen“ genauer betrachtet. 50% der Studienpatienten entwickelte unter Therapie ein palmoplantares Erythrodysästhesie-Syndrom. Dieses so genannte Hand-Fuß-Syndrom (HFS), eine typische Nebenwirkung des Tyrosinkinase-Inhibitors Sorafenib, ist durch schmerzhafte Erytheme, Schwellungen, Schuppung und Blasenbildung der Haut an Hand- und Fußsohlen gekennzeichnet. Exponierte Bereiche sind vor allem druckbelastete Stellen, zum Beispiel Fersen und Fußballen oder die bevorzugt genutzte Hand. Acht der insgesamt 14 Betroffenen zeigten ein HFS Grad 3 oder 4 nach CTCAE. Abgeschlagenheit, Erschöpfung und Müdigkeit beklagten insgesamt elf Teilnehmer, bei drei von ihnen war ein Grad 3 nach CTCAE dokumentiert. Bei neun der 28 Studienteilnehmer wurde trotz intensiver Pflege der Mundschleimhaut im Laufe der Therapie eine Mukositis diagnostiziert, bei zwei von ihnen hatte sie eine Ausprägung von Grad 3. Die Patienten klagten über Schmerzen im Mund und beim Schlucken. Acht Patienten des Gesamtkollektivs entwickelten Missempfindungen an den Extremitäten im Sinn einer Polyneuropathie (PNP) mit Kribbelparästhesien, Taubheitsgefühlen, Gleichgewichtsstörungen und Gangunsicherheit. Zwei von ihnen litten unter einer PNP Grad 3. Sieben Patienten beklagten eine Heiserkeit, einer von ihnen litt unter Beschwerden Grad 3. Schwere Diarrhoe im Sinne einer Toxizität von Grad 3 konnte bei zwei von sechs an Durchfall erkrankten Patienten beobachtet werden. Jeweils fünf Patienten erkrankten an chemotherapieassoziierter Übelkeit und Erbrechen, ihre Beschwerden waren aber eher diskret. An einem Hautauschlag (RASH) erkrankten ebenfalls fünf Teilnehmer, zwei von ihnen litten unter schweren ekzematösen Veränderungen im Rahmen einer Toxizität Grad 3. Bei zwei Patienten wurde eine ausgeprägte Candida-Infektion des Ösophagus diagnostiziert, sodass eine systemische antimykotische Therapie erforderlich wurde. Beide Patienten beklagten Schluckbeschwerden. Je zweimal waren Schwindel und allergische Reaktionen auf die Studienmedikation beobachtet worden, in drei der vier Fälle war die Symptomatik mode49 rat. Bei einem Patienten mit einer allergischen Reaktion Grad 2 auf Paclitaxel musste die Studie jedoch abgebrochen werden, um den Patienten nicht zu gefährden. Tabelle 12: Häufigkeitsverteilung der Toxizitäten unter Therapie (n = 28) Toxizitäten Grad 1 Grad 2 Grad 3 Grad 4 2 HFS 1 5 6 Hautausschlag 1 2 2 1 1 Allergische Reaktion Soor K. A. Gesamt 14 5 2 2 2 7 Heiserkeit 2 4 1 Diarrhö 2 1 2 Nausea, Emesis 2 3 Mukositis 3 4 2 Fatigue 5 2 3 PNP 2 4 2 Schwindel 1 1 6 5 9 1 8 1 50 11 2 Abbildung 6: Häufigkeitsverteilung der Toxizitäten unter Therapie (n = 28) Abbildung 7: Anteil der Nebenwirkungen Grad 3 und 4 an den Gesamttoxizitäten (n = 28) 51 4.2.5.2 Laborwertveränderungen Leichtgradige Erhöhungen des Kreatininwerts kamen im Patientengut nur selten vor (7%), Erniedrigungen der glomerulären Filtrationsrate waren zwar mit ca. 29% häufiger, gingen aber nicht über Grad 1 und 2 nach CTCAE hinaus. Bei 82% der Patienten wurden Erhöhungen der Aspartat-Aminotransferase (ASAT) beobachtet, bei 17% der Betroffenen handelte es sich um eine Nebenwirkung von Grad 3. Ca. 40% der Studienteilnehmer entwickelten eine Erhöhung der Alanin-Aminotransferase (ALAT), ein Patient war schwerwiegender betroffen (Grad 3). Senkungen des Albumins traten ebenfalls sehr häufig unter Therapie auf, sie betrafen 79% der Patienten, 5% davon schwer (Grad 3). Die häufigste und im Verlauf der Therapie bei allen Patienten zu beobachtende Nebenwirkung war die Anämie. Bei zwei der 28 Betroffenen sank die Hämoglobinkonzentration im Serum auf unter 8g/dl (Grad 3), bei einer Patientin sogar auf 6,4g/dl (Grad 4). Bei ihr war die Gabe von zwei Erythrozytenkonzentraten indiziert, um die klinisch-symptomatische Anämie zu therapieren. Weit gefährlicher und damit bedeutsamer war die Leukozytopenie. Insgesamt 68% der Patienten entwickelten unter Therapie eine Leukozytopenie, davon sank bei einem Patienten die Leukozytenzahlen auf unter 2/nl (Grad 3), drei weitere Patienten litten unter Leukozytopenie Grad 4. Therapeutisch wurde in einigen Fällen mit dem Wachstumsfaktor G-CSF (granulocyte-colony stimulating factor) eingegriffen, um den weiteren und unter Umständen lebensbedrohlichen Abfall der Leukozyten zu verhindern. Eine Patientin entwickelte aufgrund der geringen Leukozytenzahlen ein leukopenisches Fieber, das 25 Tage mit der Gabe von G-CSF stationär behandelt werden musste. Thrombozytopenien entwickelten neun von 28 Patienten, bei einem von ihnen war die Thrombozytenzahl im Serum unter 50/nl gesunken, das entspricht einer Toxizität Grad 3. Die Gabe von Thrombozytenkonzentraten war bei keinem Teilnehmer indiziert. 52 Tabelle 13: Laborwerttoxizitäten unter Therapie nach CTCAE [48] Laborwert Referenz Einheit Kreatinin 0,5 - 1,1 GFR Grad 2 mg/dl 1 1 2 (7%) 90 – 140 ml/min/1,73m² 7 1 8 (29%) OT / ASAT 5 – 35 U/l 16 3 4 23 (82%) 4 (17%) PT / ALAT 0 – 45 U/l 8 2 1 11 (39%) 1 (9%) Albumin 3,5 – 5 g/dl 6 15 1 22 (79%) 1 (5%) Hämoglobin 14 – 18 g/dl 18 7 2 1 28 (100%) 3 (11%) Leukozyten 4 – 10 /nl 5 10 1 3 19 (68%) 4 (21%) /nl 8 1 9 (32%) 1 (11%) 11 (39%) 2 (18%) 15 (54%) 4 (27%) Thrombozyten 140 – 440 Grad 3 Neutrophile 1,8 – 7 /nl 1 8 2 Lymphozyten 1–4 /nl 3 8 3 53 Grad 4 1 Gesamt Anteil der Grad Grad 1 3 + 4 Betroffenen Abbildung 8: Häufigkeiten von Laborwerttoxizitäten am Gesamtkollektiv und prozentualer Anteil der Grad 3 + 4 Toxizitäten an der Gesamthäufigkeit 4.2.5.2.1 Tumormarker Im Rahmen der Studie wurden zur vollständigen Bewertung der individuellen Tumorantwort unter Therapie die spezifischen Tumormarker im Serum bei 25 von 28 Studienteilnehmern bestimmt. Da in dieser Studie aber auch der individuelle Therapieerfolg ausschlaggebend war, werden im Folgenden die Ergebnisse der Tumormarkerentwicklung unter Therapie bei zwei Patienten vorgestellt. Die Diagramme zeigen, dass die Marker in diesen beiden individuellen Fällen unter Studienmedikation (Bereich rot markiert) signifikant sanken, was auf ein Ansprechen des Tumors auf die Therapie schließen lässt. 54 Abbildung 9: Tumormarkerentwicklung unter Therapie bei Patient A Abbildung 10: Entwicklung dreier Tumormarker unter Therapie bei Patient B 55 Abbildung 11: Entwicklung des Tumormarkers CA 19-9 unter Therapie bei Patient B 4.2.5.3 Schwere unerwünschte Ereignisse / Stationäre Aufenthalte Bei den meisten Patienten konnte die Therapie unter ambulanten Bedingungen problemlos verabreicht werden. Bei ca. der Hälfte des Gesamtkollektivs (13 von 28, 46%) kam es jedoch während der Einnahme der Studienmedikation zu Ereignissen, die eine stationäre Behandlung erforderten. Folgende Ursachen führten zu insgesamt 21 stationären Aufenthalten: Starke Nebenwirkungen unter Studienmedikation (n = 4) o Mukositis o Hand-Fuß-Syndrom o Gangstörungen Allgemeinzustandsverschlechterung (n = 4) in Kombination mit o Dyspnoe o Schmerzen o Appetitlosigkeit und Schwäche o Schwindel, Übelkeit und Diarrhoe 56 Portinfekt (n = 3) Stationäre Fortführung der Chemotherapie (n = 3) Frakturereignis (n = 2) o vermutlich osteoporotische BWK-12-Fraktur o pathologische Humerus-Fraktur Transfusionspflichtige Epistaxis (n = 1) Zustand nach Kollapsereignis (n = 1) Pneumothorax nach Pleurapunktion (n = 1) Port-Operation mit anschließendem Pneumothorax und Portspitzenthrombose (n = 1) Tiefe Beinvenenthrombose (n = 1) 4.2.5.4 Stärkste Nebenwirkung je Patient In die Auswertung der stärksten Nebenwirkung je Patient wurden nicht nur die oben genannten unerwünschten Arzneimittelnebenwirkungen und Medikamententoxizitäten einbezogen, sofern sie vom behandelnden Arzt in Grade entsprechend der National Cancer Institutes (CTCAE) eingeteilt worden waren, sondern auch ausgewählte Laborparameter (siehe Abschnitt 4.2.5.2). Sehr diskrete Beschwerden (Grad 1 nach CTCAE) konnten bei drei Patienten festgestellt werden (11%), es bedurfte hierbei aber keiner weiteren Therapie. Symptome von Grad 2 nach CTCAE, die in der Regel ebenfalls noch als geringgradig zu werten sind, betrafen acht Studienteilnehmer (29%). Somit zeigte sich bei insgesamt elf der 28 Patienten (39%) eine sehr gute Therapieverträglichkeit. Grad-3Nebenwirkungen und damit deutliche Beschwerden zeigten zwölf der 28 Teilnehmer (43%). Fünf Patienten (18%) litten während der Therapie unter Toxizitäten Grad 4. 57 Abbildung 12: Stärkste Nebenwirkung je Patient (100% = 28 Patienten) Anmerkung: Aufgrund von Rundungen beträgt die Summe der Einzelwerte 101% Die Toxizitätsrate wurde aus der Anzahl der Patienten mit Nebenwirkungen Grad 3 und 4 nach CTCAE in Relation zum Gesamtkollektiv ermittelt. 17 von 28 Teilnehmern litten unter dritt- und viertgradigen Nebenwirkungen, sodass sich eine Toxizitätsrate von 61% ergab. Todesfälle, die in einen kausalen Zusammenhang mit der Therapie gebracht werden konnten, wurden nicht beobachtet. 4.2.5.4.1 Toxizität in Abhängigkeit von der Dosis des Antikörpers Bevacizumab Von besonderer Bedeutung ist die Frage, ob die Toxizität der zytostatischen Therapie von der steigenden Dosis des Antikörpers Bevacizumab abhängt. In Abbildung 13 ist der Prozentsatz der Studienteilnehmer in den verschiedenen Kohorten dargestellt, die als stärkste Nebenwirkung je Patient mindestens eine Grad-3oder sogar eine Grad-4-Toxizität nach CTCAE erreichten. Dabei ergibt sich, dass 67% der Patienten der ersten Kohorte ohne Antikörpertherapie unter Nebenwir58 kungen von Grad 3 oder 4 als schwerste Toxizität pro Patient litten. In der 2. Kohorte, die 1mg/kg KG Bevacizumab erhielt, wurde bei allen Teilnehmern als schwerste Nebenwirkung pro Patient ein Grad 3 oder 4 diagnostiziert. Die Abbildung zeigt, dass trotz steigender Bevacizumab-Dosis in der Studienmedikation die Patienten der folgenden Kohorten weniger schwere Nebenwirkungen entwickelten. 50 bis 60% der Teilnehmer aus den Kohorten 3 bis 6 zeigten als stärkste Nebenwirkung je Patient höchstens ein Grad 2 nach CTCAE. Tendenziell konnte demnach bisher nicht nachgewiesen werden, dass die aufgetretenen Toxizitäten mit der Antikörperdosis assoziiert sind. Abbildung 13: Häufigkeit von Toxizitäten Grad 3 und 4 als stärkste Nebenwirkung je Patient in Abhängigkeit von der Bevacizumab-Dosis (n = 28) 59 4.2.6 Wirksamkeit In die Auswertung der Tumoransprechrate und der Überlebenszeit konnten lediglich die Teilnehmer einbezogen werden, die mindestens einen Therapiezyklus abgeschlossen hatten; dies war bei 22 der 28 Patienten der Fall. 4.2.6.1 Tumoransprechrate Bei 13 der 22 Patienten konnte mithilfe der Studienmedikation eine Stabilisierung der Tumorerkrankung erreicht werden (SD). Bei sieben Teilnehmern kam es zu einem Progress der Erkrankung (PD), sodass eine sofortige Änderung des Therapieplans erforderlich war. Bei zwei Patienten war die Wirksamkeit trotz ausreichender Anzahl an abgeschlossenen Therapiezyklen nicht zu bewerten (NA), weil während des Studienverlaufs keine ausreichende Diagnostik stattgefunden hatte. Zu kompletten oder partiellen Remissionen kam es im Rahmen der Studie nicht. Tabelle 14: Tumoransprechverhalten (n = 22) Tumoransprechen 4.2.6.2 Anzahl der Patienten SD 13 (46%) PD 7 (25%) NA 2 (7%) Überlebenszeit Die Überlebenszeit der diesbezüglich auswertbaren Studienteilnehmer (n = 22) erstreckte sich zwischen 104 und 938 Tagen. Die mediane Überlebenszeit betrug 266 Tage, somit lebten ca. 37 Wochen nach Studienende noch 50% des untersuchten Kollektivs. 60 4.2.6.2.1 Überlebenszeit in Abhängigkeit von der Dosis des Antikörpers Bevacizumab Von besonderem Interesse ist die Überlebenszeit der Patienten in Abhängigkeit von der Dosis an Bevacizumab. 0 mg/kg Körpergewicht Bevacizumab (n = 6) 217 Tage 1 mg/kg Körpergewicht Bevacizumab (n = 5) 208 Tage 2 mg/kg Körpergewicht Bevacizumab (n = 2) 291 Tage 4 mg/kg Körpergewicht Bevacizumab (n =3) 280 Tage 5 mg/kg Körpergewicht Bevacizumab (n = 3) 395 Tage 10 mg/kg Körpergewicht Bevacizumab (n = 3) 574 Tage Auch wenn sich aufgrund der geringen Patientenzahl keine Signifikanz berechnen lässt, zeigt sich die Tendenz, dass die mediane Überlebenszeit mit der Dosierung von Bevacizumab steigt. Patienten, die die Höchstmenge des Antikörpers im Rahmen der Studie erhielten, lebten länger als Patienten, die kein oder nur wenig Bevacizumab erhielten. 4.2.6.2.2 Überlebenszeit in Abhängigkeit vom Tumoransprechen Im Hinblick auf den Zusammenhang zwischen der Überlebensrate und dem Tumoransprechen ergeben sich folgende mediane Überlebenszeiten: - SD (n = 13) 234 Tage - PD (n = 7) 363 Tage - NA (n = 2) 221 Tage Auch hier ist aufgrund der geringen Patientenanzahl die Berechnung einer Signifikanz nicht möglich, aber die Aufstellung lässt erkennen, dass die mediane Überlebenszeit offenbar unabhängig vom Tumoransprechen ist: Patienten, die eine Progression ihrer Erkrankung während der Therapie erfuhren, hatten trotzdem eine längere mediane Überlebenszeit als Patienten, die auf die Studienmedikation mit einer Stabilisierung reagierten. 61 4.2.6.2.3 Überlebenszeit in Abhängigkeit vom Grading Betrachtet man die Überlebenszeit in Abhängigkeit vom Grading der Tumorerkrankung, so ergeben sich folgende mediane Überlebenszeiten: - hoch differenzierte G1-Tumore (n = 1) 572 Tage - niedrig differenzierte G2-Tumore (n = 11) 363 Tage - entdifferenzierte G3-Tumore (n = 8) 243 Tage - keine Angabe (n = 2) 162 Tage Auch hier ist die Berechnung der Signifikanz aufgrund der geringen Teilnehmerzahl nicht möglich. Es zeigt sich aber die Tendenz, dass Patienten mit hoch differenzierten Tumoren eine höhere mediane Überlebenszeit haben als Patienten mit entdifferenzierten Tumoren. 4.2.7 Anschlusstherapien Im Anschluss an die abgeschlossene oder abgebrochene Therapie mit Paclitaxel, Sorafenib und Bevacizumab erhielten die Patienten je nach individueller Situation folgende weitere Behandlungen: Chemotherapie (n = 14) Radiatio und antihormonelle Therapie (n = 2) Tumorresektion und Radiatio (n = 1) Keine Therapie (n = 8) Keine Angaben zur weiteren Therapie (n = 3) 4.2.8 Todesursachen Von den in der Auswertung berücksichtigten 22 Patienten waren 16 Patienten zum Zeitpunkt des Studienendes am 31.12.2012 verstorben. Dabei war bei nahezu allen Patienten von einer Todesursache im Rahmen des Tumorprogresses auszugehen. 62 5 Diskussion Die vorliegende Studie untersucht neben dem primären Ziel (Machbarkeit) auch sekundäre Ziele (Sicherheit und Wirksamkeit) der zytostatischen Kombinationstherapie aus Paclitaxel, Sorafenib und Bevacizumab bei 28 Patienten mit metastasierter oder lokal fortgeschrittener solider Tumorerkrankung. Neben der kritischen Betrachtung des Studiendesigns und der Studienergebnisse werden in diesem Kapitel Bezüge zur aktuellen Literatur hergestellt und die Relevanz der vorliegenden Medikamentenkombination für zukünftige zytostatische Therapieplanungen diskutiert. 5.1 Kritische Betrachtung des Studienkonzeptes Die heterogene Verteilung der Studienpopulation und die geringe Anzahl an Studienteilnehmern (28 Patienten) entspricht charakteristischerweise einer PhaseI-Studie. In der Geschlechterverteilung bildeten sich zwei ungleich große Gruppen mit 7 Männern und 21 Frauen, das Alter variierte stark zwischen 39 und 78 Jahren. Außerdem wurden Patienten mit insgesamt 9 verschiedenen Tumorentitäten in die Studie aufgenommen, um unter anderem auch die panangiogenetische Inhibition durch Bevacizumab nachzuweisen. Auch hier waren die Gruppen sehr unterschiedlich groß und ließen daher einen Vergleich der Ergebnisse untereinander nicht zu. Gleiches gilt für die uneinheitlichen Voraussetzungen bei Grading, Tumorstadien nach UICC und Vorbehandlung der Patienten. Bezüglich des primären Therapieziels – der Machbarkeit der Studie – hat die Heterogenität keine Auswirkung. Eine regelmäßige und langfristige Therapieapplikation wurde durch die geringe Lebenserwartung und den zeitweise schlechten Allgemeinzustand der schwerkranken komorbiden und mehrfach vortherapierten Patienten beeinträchtigt. Außerdem kann nicht überprüft werden, inwieweit die Einnahme von Sorafenib im häuslichen Umfeld nach Schema erfolgte. 63 5.2 Kritische Betrachtung der Therapieergebnisse 5.2.1 Dosis Vier offene Dosisfindungsstudien an Patienten mit fortgeschrittener solider Tumorerkrankung ermittelten eine optimale Sorafenib-Dosis von 400mg Wirkstoff zweimal täglich als Monotherapie. Häufigste wirkstoffabhängige Toxizitäten waren Fatigue (35%), Diarrhö (35%) und Hand-Fuß-Syndrom (35%). Diesen Studien zufolge ist bei dem geringen Nebenwirkungsprofil von Sorafenib als Monotherapie eine kontinuierliche Behandlung ohne Therapiepausen möglich. Dosislimitierende Nebenwirkungen traten erst bei Gaben von über 600mg Sorafenib zweimal täglich auf [65]. Während die Dosis von Sorafenib in Kombination mit Bevacizumab und Paclitaxel in unserer Studie bei nahezu allen Patienten aufgrund von Toxizitäten reduziert werden musste, konnten Paclitaxel und Bevacizumab in der vorgegebenen Dosierung meist problemlos verabreicht werden. Für zukünftige Untersuchungen kann die Dosierung von Paclitaxel und Bevacizumab wie in der vorliegenden Studie beibehalten werden, Sorafenib sollte jedoch in Kombination mit diesen beiden Wirkstoffen in weiteren Studien auf 200mg täglich reduziert werden. 5.2.2 Unerwünschte Arzneimittelnebenwirkungen / Medikamententoxizitäten Eine Kombination der drei Wirkstoffe Paclitaxel, Sorafenib und Bevacizumab, wie sie in der vorliegenden Studie untersucht wurde, ist mit zahlreichen und häufig schwerwiegenden Nebenwirkungen verbunden. Auch wenn die therapieassoziierten Toxizitäten aufgrund der Medikamentenkombination im Nachhinein nicht eindeutig den einzelnen Wirkstoffen zugeordnet werden können, zeigen die Abbildungen 7 und 8 im Ergebnisteil zahlreiche Grad 3 und 4 Toxizitäten nach CTCAE. 5.2.3 Laborwerte Laborwertveränderungen unter Chemotherapie sind häufig und im Ausmaß variabel. Ein bedeutender Faktor für die Wirksamkeit zytostatischer Therapie ist ihre Auswirkung auf sich schnell teilende Zellen. Die heutigen Forschungsergeb64 nisse ermöglichen bislang nicht, die Wirkung von Zytostatika allein auf Tumorzellen zu beschränken. Sie behindern daher nicht nur das Wachstum der malignen Zellen, sondern schädigen auch körpereigene Zellen. Davon sind am stärksten die sich schnell teilenden Blut- und Schleimhautzellen betroffen. Dies erklärt die hohe Anzahl an Patienten, die unter Therapie an einer Anämie litten. Weitaus gefährdeter waren jeweils vier Patienten aus der vorliegenden Studie, die unter Leukozytopenien und Lymphozytopenien Grad 3 und 4 mit entsprechend hohem Risiko für fieberhafte Infekte litten. In einer multizentrischen randomisierten „Discontinuation“-Phase-II-Studie (RDT-Studie) wurde die klinische Wirksamkeit von Sorafenib als Monotherapie bei unterschiedlichen Tumorentitäten untersucht [31]. Das Hauptaugenmerk in dieser Studie lag auf Patienten mit Nierenzellkarzinom. Zwar war in der vorliegenden Phase-I-Studie diese Tumorentität nicht vertreten, interessant ist aber der Vergleich der aufgetretenen Nebenwirkungen in beiden Studien. In der RDT-Studie war eine gute Therapieverträglichkeit von Sorafenib bei einer Dosierung von 400mg zweimal täglich dokumentiert, aufgetretene Nebenwirkungen waren Fatigue (73%), Hautausschläge (66%), Hand-Fuß-Syndrom (62%), Schmerzen (58%) und Diarrhö (58%). Die häufigste Toxizität Grad 3 oder 4 war Bluthochdruck. Vor allem das Hand-Fuß-Syndrom (50%) und die Fatigue (39%) waren auch in der vorliegenden Studie mit der Kombinationstherapie aus Sorafenib, Bevacizumab und Paclitaxel überaus häufig vertreten. Hautausschläge, Diarrhö und Bluthochdruck wurden seltener beobachtet. Die häufigste Nebenwirkung vom Grad 3 oder 4 nach CTCAE war in der vorliegenden Phase-I-Studie das Hand-Fuß-Syndrom. Eine weitere Studie mit einer wöchentlichen Bevacizumab- (2mg/kg KG) und Paclitaxel- (80mg/m²) / Carboplatin-Gabe mit vs. ohne Sorafenib (200mg täglich) bei multipel vorbehandelten Patienten mit metastasiertem Zervixkarzinom [34] kann zum Vergleich von beobachteten Toxizitäten unter einer Kombinationstherapie herangezogen werden. In dieser Studie wurden hämatologische Nebenwirkungen, die den Grad 1 nach CTCAE überschritten, nicht beobachtet. Bluthochdruck und Nasenbluten, die auf die Wirkung des Bevacizumab zurückzuführen sind, wurden regelmäßig dokumentiert. Die Hälfte der mit Sorafenib therapierten Patienten entwickelte ein Hand-Fuß-Syndrom, allerdings war eine Behandlung der Hauterscheinungen nicht erforderlich. Diese Untersuchung zeigte, dass die Medika65 mentenkombination bei Patienten mit stark vortherapiertem Zervixkarzinom wirksam und die Nebenwirkungen tolerabel waren. Anders hingegen in der vorliegenden Phase-I-Studie: Anämien, Leuko- und Lymphozytopenien vom Grad 3 und 4 entwickelten jeweils 3 bis 4 Studienteilnehmer. Bluthochdruck und Nasenbluten wurden auch hier beobachtet. Ein Patient aus der ersten Kohorte (ohne Gabe von Bevacizumab) litt unter einer hämoglobinrelevanten Epistaxis, die mit zwei Erythrozytenkonzentraten therapiert werden musste. Vergleichbar ist die hohe Rate an Hand-Fuß-Syndromen in beiden Studien, sie betrug in der vorliegenden Phase-I-Studie 50%, führte jedoch mehrfach zu Pausen, Abbrüchen oder Reduktion der Therapie. Das Syndrom musste in vielen Fällen langwierig behandelt werden. In einer doppelblinden randomisierten Phase-IIb-Studie (NU07B1-Studie) wurden Patienten mit HER2-negativem Mammakarzinom mit Paclitaxel (90mg/m² wöchentlich) mit vs. ohne Sorafenib (400mg zweimal täglich) therapiert [23]. Resultat der NU07B1-Studie war, dass die Kombination aus Paclitaxel mit Sorafenib zu einer verbesserten Krankheitskontrolle führte. Die Inzidenz von Grad 3 und 4 Nebenwirkungen war in beiden Gruppen (Sorafenib plus Paclitaxel vs. Placebo plus Paclitaxel) im Allgemeinen vergleichbar. Einzelne Nebenwirkungen von Grad 3 oder 4 traten jedoch bei der Kombination von Paclitaxel mit Sorafenib häufiger auf als unter alleiniger Paclitaxel-Therapie. Das Hand-Fuß-Syndrom (HFS) war mit 55% nicht nur die häufigste Nebenwirkung unter Therapie (vs. 7% in der PlaceboGruppe), sondern auch die häufigste Toxizität Grad 3 und 4 nach CTCAE (31% vs. 3%). Weitere schwere Nebenwirkungen waren Neutrozytopenien, Anämien und Asthenie. Im Verlauf dieser Studie musste die Sorafenib-Dosis bei ca. 50% der Patienten reduziert werden. Mithilfe dieser Dosisreduktion, durch Einlegen von Therapiepausen und unterstützende Maßnahmen waren die Nebenwirkungen zu kontrollieren. Bei Patienten, die die Kombinationstherapie erhielten, musste zur Beherrschung der Therapietoxizitäten häufiger die Medikamentendosis reduziert werden als in der Placebo-kontrollierten Gruppe. Auch in der vorliegenden Studie konnten bestätigt werden, dass die Toxizitätsrate unter Therapie mit Sorafenib steigt. Das HFS stellte mit 50% die häufigste Nebenwirkung unter Therapie dar, ca. 29% der Studienteilnehmer entwickelten ein 66 HFS von Grad 3 oder 4. Auch wenn das HFS ein reversibles Ereignis ist und sich nicht direkt auf das Überleben auswirkt, schränkt es doch die Lebensqualität der Patienten stark ein. Außerdem führen häufige Dosisreduktionen zu einer verminderten Dosisdichte und somit zu eingeschränkter Wirksamkeit. Die hohe Rate an Hand-Fuß-Syndromen in der vorliegenden Studie ist vergleichbar mit derjenigen aus der NU07B1-Studie. Diese Toxizität gilt als klinisch signifikant, weil sie nicht nur bei vielen Patienten zu Dosisreduktionen und Behandlungsunterbrechungen führte, sondern sogar mehrfach Therapieabbrüche notwendig machte. Weitere häufige schwerwiegende Toxizitäten waren in der vorliegenden Studie – ähnlich wie in der NU07B1-Studie – neben der Anämie die Lympho-, Leuko-, Neutro- und Thrombozytopenie. Asthenie trat im Gegensatz zur NU07B1-Studie weniger häufig auf, dafür war die Fatigue eine häufig beobachtete Nebenwirkung. Die in dieser Arbeit diskutierte Studie zeigt auch, dass die ursprünglich geplante Sorafenib-Dosis nicht einzuhalten ist. Auch hier musste bei nahezu allen Patienten die Dosis im Studienverlauf aufgrund von Nebenwirkungen reduziert werden. Dies und die Therapiepausen führten aber in den meisten Fällen ebenso wie in der NU07B1-Studie zur Besserung der Symptomatik und tolerablen Nebenwirkungen. 22% der Teilnehmer aus der NU07B1-Studie konnten die Therapie aufgrund schwerer Toxizitäten nicht beenden. In der vorliegenden Studie mit Bevacizumab, Paclitaxel und Sorafenib konnten vergleichbare Zahlen erfasst werden. 29% der Studienteilnehmer brachen die Therapie aufgrund von Medikamententoxizitäten ab. 5.2.4 Individuelle Therapieergebnisse Ziel jeder Studie mit zytostatischen Wirkstoffen ist neben einem signifikanten statistischen Ergebnis vor allem der individuelle Therapieerfolg. In der vorliegenden Studie konnte mithilfe der neuerlichen Therapie bei einigen Patienten zumindest vorübergehend eine deutliche Befundbesserung erzielt werden. Ein Patient mit hepatisch metastasiertem Urothelkarzinom des Nierenbeckens klagte zu Beginn der Studie über Leberkapselschmerz. Unter Therapie verschwand die 67 Schmerzsymptomatik gänzlich und die erhöhten Kreatininwerte normalisierten sich. Der Patient berichtete über einen deutlich verbesserten Allgemeinzustand. Auch ein Patient mit Prostatakarzinom berichtete über steigende Belastbarkeit, psychische und physische Befundbesserung. Bei einer Patientin mit Mammakarzinom war eine rasch rückläufige Schmerzsymptomatik im rechten Oberbauch bei hepatischen Filiae dokumentiert. Bei einer weiteren, ebenfalls an Brustkrebs erkrankten Teilnehmerin berichtete der behandelnde Arzt von klinisch deutlichen Hinweisen auf eine Tumorregression. Auch die Tumormarker der Patientin waren leicht rückläufig. Die Patientin mit Ovarial- und beidseitigen Mammakarzinom erklärte sogar, dass sie sich zum Zeitpunkt der Therapie besser fühlte als vor Therapiebeginn. Diese und weitere Beispiele zeigen, dass das neuerliche Therapiekonzept zwar aufgrund der geringen Patientenzahl nicht unbedingt statistisch signifikante Ergebnisse lieferte, jedoch individuell zu Befund- und Allgemeinzustandsverbesserung führte. Auch wenn sich die Studienmedikation möglicherweise nicht auf das progressionsfreie Überleben ausgewirkt hat, ist für Patienten die Lebensqualität der verbleibenden Zeit der entscheidende Faktor. Um die nachhaltig verbesserte Lebensqualität sicher zu stellen, mussten allerdings die unerwünschten Arzneimittelnebenwirkungen der Wirkstoffe eng kontrolliert werden. 5.2.5 Tumoransprechrate Trotz einer erkennbaren positiven Tendenz der Ergebnisse zur Wirksamkeit der Therapie lassen die Struktur einer Phase-I-Studie und die geringe Teilnehmerzahl keine ausreichende Beurteilung der Wirksamkeit zu. Die Ergebnisse der vorliegenden Studie zeigen, dass 46% der bezüglich des Ansprechens auswertbaren Patienten zumindest zeitweise eine Stabilisierung der Tumorerkrankung erreichten (Clinical Benefit). Unter den oben genannten Gesichtspunkten hat demnach ca. die Hälfte der Patienten auf die Therapie angesprochen und nur ein Viertel mit einer Progression unter der zytostatischen Therapie reagiert. In Anbetracht der Situation, dass die Patienten unter stark vorbehandelten und weit fortgeschrittenen Tumorerkrankungen mit geringer Lebenserwartung litten, ist dieses Ergebnis der Studie zufriedenstellend. 68 Die Tumoransprechrate scheint allerdings nicht von der verabreichten Bevacizumab-Dosis abhängig zu sein. Es zeigt sich weder die Tendenz, dass Patienten mit hoher Antikörperdosis vermehrt auf die Therapie ansprechen, noch dass sie mit einem Tumorprogress reagieren. Die Annahme, dass Bevacizumab die tumoröse Gefäßneubildung unabhängig von der Tumorentität hemmt, konnte auch in dieser Studie bestätigt werden: Unter den Patienten, die auf die Studienmedikation mit einer Stabilisierung ihrer Tumorerkrankung ansprachen, waren verschiedenste Tumorentitäten vertreten: Urothelkarzinom des Nierenbeckens, Angiosarkom, Pankreas-, Prostata-, Ösophagus- und Mammakarzinom. Eine Phase-I-Studie untersuchte Patienten mit fortgeschrittenen soliden Tumorerkrankungen unter Therapie mit Telatinib in Kombination mit Bevacizumab [39]. Diese Studie, die ebenfalls die Wirksamkeit von Bevacizumab in Kombination mit einem Tyrosin-Kinase-Inhibitor prüfte, ergab ein Clinical Benefit von 46%. Hier erreichten also 46% der Patienten unter Therapie mindestens eine Stabilisierung ihrer Tumorerkrankung. Ein vergleichbares Ergebnis lieferte die vorliegende Phase-I-Studie, in der Bevacizumab in Kombination mit Paclitaxel und einem TyrosinKinase-Inhibitor getestet wurde. Auch hier hatten 46% der Teilnehmer ein Clinical Benefit. Ein vergleichbares Clinical Benefit von 53% erreichte eine Phase-I-Studie, in der Bevacizumab (5mg/kg KG alle 14 Tage) und Sorafenib (bis zu 200mg zweimal täglich) bei Patienten mit fortgeschrittener solider Tumorerkrankung verabreicht wurden [40]. Die doppelblinde randomisierte Phase-IIb-Studie (NU07B1), die eine Therapie bei Patienten mit HER2-negativem Mammakarzinom mit Paclitaxel (90mg/m² wöchentlich) mit vs. ohne Sorafenib (400mg zweimal täglich) untersuchte [23], zeigte eine höhere objektive Tumoransprechrate in der Patientengruppe, die Sorafenib und Paclitaxel erhielten. In der vorliegenden Phase-I-Studie wurden alle Patienten mit Sorafenib und Paclitaxel therapiert, allein die Bevacizumab-Dosis variierte. Aufgrund der Ergebnisse der NU07B1-Studie ist davon auszugehen, dass die objektive Tumoransprechrate in der vorliegenden Phase-I-Studie durch die Kombination des Chemotherapeutikums Paclitaxel mit dem Tyrosin-KinaseInhibitor Sorafenib ebenfalls gestiegen ist. 69 5.2.6 Überlebenszeit Die mediane Überlebenszeit von ca. 38 Wochen (entspricht ca. 8,8 Monaten) bei Patienten, die mit Sorafenib, Bevacizumab und Paclitaxel therapiert worden sind, ist vielversprechend und mit Ergebnissen anderer Studien vergleichbar. So hat eine aktuelle Studie aus dem Jahr 2013 ergeben, dass die Behandlung von Patienten mit metastasiertem Mammakarzinom mit Paclitaxel (90mg/m² an Tag 1, 8 und 15) und Bevacizumab (10mg/kg KG an Tag 1 und 15) als ErstlinienTherapie zu einer medianen Überlebenszeit von 12,3 Monaten führte [42]. Auch die Spannweite der Überlebenszeit in dieser Studie von 2013 (4,6 bis 44,8 Monate) ist vergleichbar mit den Ergebnissen der vorliegenden Phase-I-Studie, in der die Medikation mit Sorafenib erweitert wurde (3,5 bis 31,2 Monate) [42]. Die Ergebnisse bezüglich der Überlebenszeit in Abhängigkeit vom Grading zeigen, dass Patienten mit hochdifferenzierten Tumoren eine höhere mediane Überlebenszeit haben als Patienten, deren Tumorzellen entdifferenziert sind. Dieses Resultat ist erwartet, denn Tumore, die dem Ursprungsgewebe ähnlich sind, sind weniger maligne als Tumore, die mit dem Ursprungsgewebe kaum noch Übereinstimmungen aufweisen. Allerdings muss man bei der Auswertung auch beachten, dass die Gruppe „G1“ mit nur einem Patienten keine ausreichende Anzahl an Patienten darstellt, um einen validen Vergleich der Gruppen untereinander zu ermöglichen. Jedoch zeigt auch die Gegenüberstellung der Gruppen „G2“ und „G3“ mit wesentlich mehr Patienten, dass die mediane Überlebenszeit mit der Differenzierung des Tumors steigt. Die doppelblinde randomisierte NU07B1-Studie untersuchte Paclitaxel (90mg/m² wöchentlich) mit vs. ohne Sorafenib (400mg zweimal täglich) bei Patienten mit HER2-negativem Mammakarzinom [23]. Diese Phase-IIb-Studie zeigte keinen signifikanten Unterschied im Gesamtüberleben bei Patienten aus der Paclitaxel-Gruppe gegenüber denen, die zusätzlich Sorafenib erhielten. 5.2.7 Machbarkeit Primäres Ziel der oben beschriebenen Phase-I-Studie ist die Beurteilung der klinischen Machbarkeit. Zusammenfassend kann man sagen, dass es sich bei der Kombination aus Paclitaxel, Sorafenib und Bevacizumab um ein potenziell 70 wirksames Therapiekonzept handelt, welches aber in weiteren klinischen Studien untersucht werden muss. Während Paclitaxel und Bevacizumab fast ausnahmslos in der vorgegebenen Dosierung verabreicht werden konnten, musste die Sorafenib-Dosis wiederholt wegen Toxizität reduziert werden. So war zum Beispiel das Hand-FußSyndrom unter Therapie die am häufigsten zu beobachtende Nebenwirkung und dosislimitierende Toxizität in der vorliegenden Studie. Die Kombination der drei Wirkstoffe führte zu einer hohen Nebenwirkungsrate, die in diesem Ausmaß für weitere Untersuchungen nicht zu tolerieren ist. Die hohe Toxizitätsrate von 61% und die Feststellung, dass 29% der Teilnehmer aufgrund von Nebenwirkungen die Studie abbrachen, erfordern bei zukünftigen Studien mit dieser Kombination eine Dosismodifikation. Um die Wirksamkeit des Therapieschemas und das Überleben der Patienten, die mit diesem Therapiekonzeptes behandelt werden, mit anderen ähnlichen Behandlungsschemata vergleichen zu können, müssen die Daten an einem größeren und enger definierten Patientenkollektiv im Rahmen einer Phase-II-Studie erhoben werden. Ursprünglich war bereits zu Beginn der oben beschriebenen Untersuchung der Übergang in eine randomisierte Phase-II-Studie geplant. Nach Feststellung der hohen Therapietoxizität konnte eine anschließende Untersuchung mit den gleichen Wirkstoffen in gleicher Dosierung nicht durchgeführt werden. 5.3 Ausblick Aus dem Verständnis der Tumorangiogenese und der Möglichkeit, diese zu unterbinden, entwickelte sich die Idee einer vielversprechenden Behandlungsalternative bei malignen Tumorerkrankungen. Die Erforschung antiangiogenetischer Wirkstoffe in der Präklinik zeigte erfolgversprechende Ergebnisse [15], die sich in klinischen Studien nur teilweise bestätigen ließen [45]. Die Hemmung von VEGF und seinem Rezeptorsystem etablierte sich vor allem bei malignen Tumoren mit palliativem Therapiekonzept [20, 61]. Über die Dauer einer antiangiogenetischen Therapie gibt es aktuell unterschiedliche Ansichten, Studienergebnissen zufolge ist eine Behandlung bis zum Progress der Tumorerkrankung zu empfehlen. Inwiefern jedoch eine Therapie 71 darüber hinaus – bei Tumorprogress – sinnvoll ist, ist aktuell noch unklar. Möglicherweise führt das plötzliche Absetzen der Angiogenesehemmer aber zu einer überschießenden Gefäßneubildung des Tumorgewebes mit Krankheitsprogress [15]. Zur Abschätzung der biologischen Aktivität können Surrogatmarker dienen, die allerdings bisher nur in Phase-I-Studien eingesetzt worden sind [15]. Zukünftig sollen jedoch auch Phase-III-Studien mit löslichen Markern durchgeführt werden [15]. Die Bestimmung beispielsweise der VEGF-Konzentration im Serum ist in Studien mit antiangiogenetischen Wirkstoffen mittlerweile etabliert. Für die ursprünglich geplante Phase II der oben beschriebenen Studie wäre von besonderem Interesse, inwiefern die VEGF-Konzentration von der verabreichten Bevacizumab-Dosis abhängig ist. Es wird angenommen, dass Patienten mit Tumorerkrankungen im Frühstadium weniger proangiogene Faktoren exprimieren und somit stärker VEGF-abhängig sind. Bevacizumab könnte bei Patienten im Frühstadium der Erkrankung sehr viel effektiver sein [15]. Patienten mit mehrfach chemotherapeutisch vorbehandelter schwerer refraktärer Tumorerkrankung sind möglicherweise durch die Vielzahl an exprimierten proangiogenen Faktoren weniger VEGF-abhängig. Die Konzentration von VEGF im Serum ist dadurch unter Umständen verfälscht [15]. Eine weitere Möglichkeit, die Wirkung der antiangiogenetischen Substanzen an den Tumorgefäßen in der ursprünglich geplanten Phase-II-Studie zu verifizieren, bietet die dynamische Kontrastmittel-Perfusionsbildgebung (DCE-MRI). Erste klinische Daten zeigen, dass hiermit das Ansprechen der Erkrankung vor allem individuell prognostiziert werden kann [15]. 72 6 Zusammenfassung Die vorliegende Studie untersucht die Machbarkeit, Sicherheit und Wirksamkeit einer Kombinationstherapie mit einem Chemotherapeutikum (Paclitaxel) zusammen mit einem VEGF-Antikörper (Bevacizumab) und einem Tyrosin-KinaseInhibitor (Sorafenib) bei vorbehandelten Patienten mit metastasierter solider Tumorerkrankung. Das verwendete Therapiekonzept hemmt die Angiogenese an zwei Stellen gleichzeitig mit potenziell synergistischer Wirkung. Insgesamt 28 Patienten (21 Frauen, 7 Männer) mit fortgeschrittener solider Tumorerkrankung wurden im Zeitraum Juli 2009 bis März 2012 mit der oben genannten Wirkstoffkombination behandelt. Das mediane Alter zu Therapiebeginn betrug 61 Jahre (Spanne: 39-78 Jahre), die mediane Beobachtungsdauer 225 Tage. Im Median hatte jeder Patient zwei zytostatische Vortherapien erhalten (Spanne: 0-13 Chemotherapien). Paclitaxel und Bevacizumab konnten dabei fast ausnahmslos in der vorgegebenen Dosierung verabreicht werden. Hingegen musste die Sorafenib-Dosis bei 21 von 28 Patienten aufgrund der hohen Nebenwirkungsrate reduziert werden. Die häufigsten Therapieabbrüche waren auf Medikamentennebenwirkungen zurückzuführen, von besonderer Bedeutung war hier das Hand-Fuß-Syndrom unter Sorafenib. Bezüglich der Toxizität waren alle 28 Studienteilnehmer auswertbar. Es ergab sich, dass ca. 43% der Patienten unter Grad 3 Nebenwirkungen litten und ca. 18% schwere Nebenwirkungen vom Grad 4 aufwiesen. Die Therapieverträglichkeit war mit einer Gesamttoxizitätsrate von 61% vergleichsweise schlecht. Eine Assoziation der Nebenwirkungen mit steigender Bevacizumab-Dosis konnte nicht nachgewiesen werden. Dosislimitierende Toxizität von Sorafenib war das Hand-FußSyndrom. Bezüglich Ansprechen und Überlebensrate waren 22 von 28 Patienten auswertbar. Es ergab sich ein Clinical Benefit von 46% und eine mediane Überlebenszeit von ca. 38 Wochen. Tendenziell scheint die mediane Überlebenszeit von der Bevacizumab-Dosis und der Differenzierung der Tumorzellen abhängig zu sein. 73 Insgesamt ist von einer potenziell wirksamen Behandlungsalternative auszugehen, deren assoziierte Nebenwirkungen in dieser Dosierung nicht tolerabel sind. Individuelle Ergebnisse zeigen aber, dass einzelne Patienten von dieser Therapie profitierten. Die Struktur der vorliegenden Phase-I-Studie lässt eine abschließende Bewertung des Therapiekonzeptes offen. Für die Behandlung von Patienten mit maligner Tumorerkrankung ist auch zukünftig ein Therapieansatz mit antiangiogenetischen Wirkstoffen sinnvoll. Ein vielversprechender Ansatz sind Studien mit Konzentrationsbestimmungen von VEGF im Serum unter antiangiogenetischer Therapie. 74 7 [1] Literaturverzeichnis Aghajanian, C., Blank, S. V., Goff, B. A., Judson, P. L., Teneriello, M. G, Husain, A. et al. (2012). OCEANS: a randomized, double-blind, placebocontrolled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J Clin Oncol 30 (17), 2039-2045 [2] Baluk, P., Hashizume, H., McDonald, D. M. (2005). Cellular abnormalities of blood vessels as targets in cancer. Curr Opin Genet Dev 15 (1), 102-111 [3] Bartsch, V. (2004). Das Taxol®-Buch. 2. Auflage. Georg Thieme Verlag KG, Stuttgart & New York [4] Bayer (Schweiz) AG (Hrsg.). (Januar 2013). Fachinformation Sorafenib [5] Berry, S. R., Van Cutsem, E., Kretzschmar, A., Michael, M., Rivera, F., DiBartolomeo, M. et al. (2008). Final efficacy results for bevacizumab plus standard first-line chemotherapies in patients with metastatic colorectal cancer: First BEAT. J Clin Oncol (ASCO Meeting Abstracts) 26 (15 Suppl), abstr 4025 [6] Bristol-Myers Squibb (Hrsg.). (November 2013). Fachinformation Paclitaxel [7] Brose, M. S., Nutting, C. M., Jarzab, B., Elisei, R., Siena, S., Bastholt, L. et al. (2014). Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet Oncol, Epub ahead of print. [8] Burger, R. A., Brady, M. F., Bookmann, M. A., Fleming, G. F., Monk, B. J., Huang, H. et al. (2011). Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med 365 (26), 2473-2483 [9] Burstein, H. J., Elias, A. D., Rugo, H. S., Cobleigh, M. A., Wolff, A. C., Eisenberg, P. D. et al. (2008). Phase II study of sunitinib malate, an oral multitargeted tyrosine kinase inhibitor, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol 26 (11), 1810-1816 75 [10] Carmeliet, P. (2003). Angiogenesis in health and disease. Nat Med 9 (6), 653-660 [11] Carmeliet, P., Jain, R. K. (2000). Angiogenesis in cancer and other diseases. Nature 407 (6801), 249-257 [12] Cheng, A. L., Kang, Y. K., Chen, Z., Tsao, C. J., Qin, S., Kim, J. S. et al. (2009). Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol 10 (1), 25-34 [13] Choy, H., Roderiguez, F. F., Koester, S., Hilsenbeck, S., von Hoff, D. D. (1993). Investigation of taxol as a potential radiation sensitizer. Cancer 71 (11), 3774-3778 [14] Dank, M., Budi, L., Piko, B., Mangel, L., Erfan, J., Cseh, J. (2014). Firstline bevacizumab-paclitaxel in 220 patients with metastatic breast cancer: results from the AVAREG study. Anticancer Res 34 (3), 1275-1280 [15] Drevs, J. (2010). Tumorangiogenese – Grundlagen und therapeutische Ansätze. 3. Auflage. UNI-MED-Verlag AG, Bremen [16] Drevs, J., Schmidt-Gersbach, C., Mross, K., Henry, A., Dugan, M., Laurent, D. et al. (2001). VEGF and sTIE-2 are useful blood markers for hypoxia under antiangiogenic treatment with a specific VEGF receptor tyrosine kinase inhibitor (PTK787/ZK222584) in cancer patients. Clinical Cancer Research, abstr 291 [17] Drevs, J., Schmidt-Gersbach, C., Mross, K., Henry, A., Dugan, M., Lee, L. et al. (2002). Biomarkers (VEGF, bFGF) for assessing the biological activity of PTK787/ZK222584 (PTK/ZK), a vascular endothelial growth factor (VEGF) receptor inhibitor, in tumours known to overexpress VEGF. Eur J Cancer 38 (Suppl 7), 78 [18] DuBois, D., DuBois E. F. (1916). A formula to estimate the approximate surface area if height and weight be known. Arch Int Med 1916, 17, 863871 [19] Escudier, B., Eisen, T., Stadler, W. M., Szczylik, C., Oudard, S., Staehler, M. (2009). Sorafenib for treatment of renal cell carcinoma: final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J Clin Oncol 27 (20), 3312-3318 76 [20] Ferrara, N., Hillan, K. J., Gerber, H. P., Novotny, W. (2004). Discovery and development of Bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov 3 (5), 391-400 [21] Folkman, J. (1971). Tumor angiogenesis: therapeutic implications. N Engl J Med 285 (21), 1182-1186 [22] Folkman, J. (1972). Anti-angiogenesis: new concept for therapy of solid tumors. Ann Surg 175 (3), 409-416 [23] Gradishar, W. J., Kaklamani, V., Sahoo, T. P., Lokanatha, D., Raina, V., Bondarde, S. et al. (2013). A double-blind, randomised, placebocontrolled, phase 2b study evaluating sorafenib in combination with paclitaxel as a first-line therapy in patients with HER2-negative advanced breast cancer. Eur J Cancer 49 (2), 312-322 [24] Grothey, A., Sugrue, M. M., Purdie, D. M., Dong, W., Sargent, D., Hedrick, E., Kozloff, M. (2008). Bevacizumab beyond first progression is associated with prolonged overall survival in metastatic colorectal cancer: results from a large observational cohort study (BRiTE). J Clin Oncol 26 (33), 5326-5334 [25] Gullino, P. M., Gimbrone, M. A. Jr. (1976). Angiogenic capacity of preneoplastic lesions of the murine mammary gland as a marker of neoplastic transformation. Cancer Res 36 (7 PT 2), 2611-2620 [26] Hartenstein, R. Platin-Verbindungen. In: Huhn, D., Herrmann, R. (Hrsg.). (1995). Medikamentöse Therapie maligner Erkrankungen. 3. Auflage. Gustav-Fischer-Verlag, Stuttgart, 54-60 [27] Henriksson, R., Bottomley, A., Mason, W., Saran, F., Wick, W., Nishikawa, R. et al. (2013). Progression-free survival and health-related quality of life in AVAglio, a phase III study of bevacizumab, temozolomide, and radiotherapy in newly diagnosed glioblastoma. J Clin Oncol 31 (20), 2635-2636 [28] Herrmann, R. Taxoide. In: Huhn, D., Herrmann, R. (Hrsg.). (1995). Medikamentöse Therapie maligner Erkrankungen., 3. Auflage. GustavFischer-Verlag, Stuttgart, 62-64 [29] Hurwitz, H., Fehrenbacher, L., Novotyn, W., Cartwright, T., Hainsworth, J., Heim, W. et al. (2004). Bevacizumab plus irinotecan, fluorouracil, and 77 leucovorin for metastatic colorectal cancer. N Engl J Med 350 (23), 2335-2342 [30] Iruela-Arispe, M. L., Dvorak, H. F. (1997). Angiogenesis: a dynamic balance of stimulators and inhibitors. Thromb Haemost 78 (1), 672-677 [31] Jain, L., Venitz, J., Figg, W. D. (2006). Randomized discontinuation trial of sorafenib (BAY 43-9006). Cancer Biol Ther 5 (10), 1270-1272 [32] Jain, R. K. (2001). Normalizing tumor vasculature with antiangiogenic therapy: a new paradigm for combination therapy. Nat Med 7 (9), 987989 [33] Karow, T., Lang-Roth, R. (2009). Allgemeine und Spezielle Pharmakologie und Toxikologie. 17. Auflage. Thomas Karow Verlag, Pulheim, 911 [34] Kikuchi, Y., Takano, M., Goto, T., Kouta, H., Kikuchi, R., Kudoh, K. et al. (2011). Effects of weekly bevacizumab and paclitaxel/carboplatin with or without sorafenib on heavily pretreated patients with recurrent or persistent cervical cancer. J Clin Oncol (ASCO Meeting Abstract) 29 (Suppl), abstr 5058 [35] Kim, K. J., Li, B., Winer, J., Armanini, M., Gillett, N., Phillips, H. S., Ferrara, N. (1993). Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 362 (6423), 841-844 [36] Kohler, D. R., Goldspiel, B. R. (1994). Paclitaxel (Taxol). Pharmacotherapy 14 (1), 3-34 [37] Lange, S., Bender, R. (2001). Median oder Mittelwert?. Dtsch Med Wochenschr 132, e1-e2 [38] Lange, S., Bender, R. (2001). Variabilitätsmaße. Dtsch Med Wochenschr 126, T29-T30 [39] Langenberg, M. H., Witteveen, P. O., Roodhart, J., Lolkema, M. P., Verheul, H. M., Mergui-Roelvink, M. et al. (2011). Phase I evaluation of telatinib, a VEGF receptor tyrosine kinase inhibitor, in combination with bevacizumab in subjects with advanced solid tumors. Ann Oncol 22 (11), 2508-2515 [40] Lee, J. M., Sarosy, G. A., Annunziata, C. M., Azad, N., Minasian, L., Kotz, H. et al. (2010). Combination therapy: intermittent sorafenib with 78 bevacizumab yields activity and decreased toxicity. Br J Cancer 102 (3), 495-499 [41] Liebmann, J., Cook, J. A., Fisher, J., Teague, D., Mitchell, J. B. (1994). In vitro studies of Taxol as a radiation sensitizer in human tumor cells. J Natl Cancer Inst 86 (6), 441-446 [42] Livi, L., Bonomo, P., Meattini, I., Simontacchi, G., Greto, D., Desideri, I. et al. (2013). Paclitaxel and bevacizumab in first-line treatment for HER2-negative metastatic breast cancer: single-center experience. Med Oncol 30 (1), 434 [43] Lüllmann, H., Mohr, K., Hein, L. (2006). Pharmakologie und Toxikologie. 16. Auflage. George Thieme Verlag, Stuttgart, 426 [44] Marmé, D. (1996). Tumor angiogenesis: the pivotal role of vascular endothelial growth factor. World J Urol 14 (3), 166-174 [45] Marmé, D. (2003). The impact of antiangiogenic agents on cancer therapy. J Cancer Res Clin Oncol 129 (11), 607-620 [46] Mass, R. D., Sarkar, S., Holden, S. N., Hurwitz, H. (2005). Clincial benefit from bevacizumab (BV) in responding and non-responding (NR) patients (pts) with metastatic colorectal cancer (mCRC). J Clin Oncol (ASCO Meeting Abstracts) 23 (16 Suppl), abstr 3514, 249s [47] Miller, K. D., Wang, M., Gralow, J., Dickler, M., Cobleigh, M., Perez, E. A. et al. (2007). Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med 357 (26), 2666-2676 [48] National Cancer Institute (NCI). (1993). Investigator’s Handbook. A manual for participants in clinical trials of investigational agents. National Cancer Institute, Bethesda, Maryland (USA) [49] Pujade-Lauraine, E., Hilpert, F., Weber, B., Reuss, A., Poveda, A., Kristensen, G. et al. (2014). Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer. The AURELIA Open-Label randomized Phase III Trial. J Clin Oncol, Epub ahead of print. [50] Ratain, M. J., Eisen, T., Stadler, W. M., Flaherty, K. T., Kaye, S. B., Rosner, G. L. et al. (2006). Phase II placebo-controlled randomized discontinuation trial of sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol 24 (16), 2505-2512 79 [51] Rimassa, L., Sanotoro, A. (2009). Sorafenib therapy in advanced hepatocellular carcinoma: the SHARP trial. Expert Rev Anticancer Ther 9 (6), 739-745 [52] Rini, B. I., Michaelson, M. D., Rosenberg, J. E., Bukowski, R. M., Sosman, J. A., Stadler, W. M. et al. (2008). Antitumor activity and biomarker analysis of sunitinib in patients with bevacizumab-refractory metastatic renal cell carcinoma. J Clin Oncol 26 (22), 3743-3748 [53] Ritzwoller, D. P., Carroll, N. M., Delate, T., Hornbrook, M. C., Kushi, L., Bowles, E. J. et al. (2014). Comparative effectiviness of adjuvantive Bevacizumab for advanced lung cancer: The cancer research network experience. J Thorac Oncol 9 (5), 692-701 [54] Roche Pharma (Schweiz) AG (Hrsg.). (September 2013). Fachinformation Bevacizumab [55] Rowinsky, E. K., Eisenhauer, E. A., Chaudhry, V., Arbuck, S. G., Donehower, R. C. (1993). Clinical toxicities encountered with paclitaxel (Taxol). Semin Oncol 20 (4 Suppl 3), 1-15 [56] Sandler, A., Gray, R., Perry, M. C., Brahmer, J., Schiller, J. H., Dowlati, A. et al. (2006). Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med 355 (24), 2542-2550 [57] Schuhmacher, M., Schulgen, G. (2008). Methodik klinischer Studien. Methodische Grundlagen der Planung, Durchführung und Auswertung. 3. Auflage. Springer-Verlag Berlin, Heidelberg, 16-17 [58] Takahashi, Y., Kitadai, Y., Bucana, C. D., Cleary, K. R., Ellis, L. M. (1995). Expression of vascular endothelial growth factor and its receptors, KDR, correlates with vascularity, metastasis, and proliferation of human colon cancer. Cancer Res 55 (18), 3964-3968 [59] ten Bokkel Huinink, W. W., Eisenhauer, E., Swenerton, K. (1993). Preliminary evaluation of a multicenter, randomized comparative study of TAXOL (paclitaxel) dose and infusion length in platinum-treated ovarian cancer. Canadian-European Taxol Cooperative Trial Group. Cancer Treat Rev 19 (Suppl C), 79-86 [60] Therasse, P., Arbuck, S. G. , Eisenhauer, E. A., Wanders, J., Kaplan, R. S., Rubinstein, L. et al. (2000). New Guidelines to Evaluate the Re80 sponse to Treatment in Solid Tumors (RECIST Guidelines). J Natl Cancer Inst 92 (3), 205-216 [61] Thomas, A. L., Morgan, B., Drevs, J., Unger, C., Wiedenmann, B., Vanhoefer, U. et al. (2003). Vascular endothelial growth factor receptor tyrosine kinase inhibitors: PTK787/ZK 222584. Semin Oncol 30 (3 Suppl 6), 32-38 [62] Thomas, K. A. (1996). Vascular endothelial growth factor, a potent and selective angiogenic agent. J Biol Chem 271 (2), 603-606 [63] Trampisch, H. J., Windeler, J. (2000). Medizinische Statistik. 2. Auflage. Springer-Verlag Berlin, Heidelberg, New York, 45 [64] Van Cutsem, E., Rivera, F., Berry, S., Kretzschmar, A., Michael, M., DiBartolomeo, M. et al. (2009). Safety and efficacy of first-line bevacizumab with FOLFOX, XELOX, FOLFIRI and fluoropyrimidines in metastatic colorectal cancer: the BEAT study. Ann Oncol 20 (11), 1842-1847 [65] Wilhelm, S., Carter, C., Lynch, M., Lowinger, T., Dumas, J., Smith, R. A. et al. (2006). Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov 5 (10), 835-844 [66] Wilhelm, S. M., Carter, C., Tang, L., Wilkie, D., McNabola, A., Rong, H. et al. (2004). BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 64 (19), 7099-7109 [67] Willet, C. G., Boucher Y., di Tomaso, E., Duda, D. G., Munn, L. L., Tong, R. T. et al. (2004). Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med 10 (2), 145-147 [68] Wittekind, C., Meyer, H.-J. (2010). TNM Klassifikation maligner Tumoren. 7. Auflage. Wiley-VCH Verlag, Weinheim 81 8 Anhang Tabelle A1: Tumorstadien. Einteilung nach UICC [68] Dukes Stadium 0 T N M Tis N0 M0 Stadium I A T1, T2 N0 M0 Stadium II B T3, T4 N0 M0 Stadium III C Jedes T N1, N2 M0 Stadium IV (D) Jedes T Jedes N M1 Tabelle A2: Auszug der Common Toxicity Criteria (CTCAE) des National Cancer Institute, Version 3.0 [48] Grad I Grad II Grad III Grad IV Kreatinin [mg/dl] > 1,1 – 1,65 > 1,65 – 3,3 > 3,3 – 6,6 GFR [ml/min/1,73m²] < 67,5 – 45 < 45 – 22,5 < 22,5 ASAT [U/l] > 35 – 87,5 > 87,5 – 175 >175 – 700 > 700 ALAT [U/l] > 45 – 112,5 > 112,5 – 225 > 225 – 900 > 900 Albumin [g/dl] < 3,5 – 3 <3–2 <2 Hämoglobin [g/dl] < 14 – 10 < 10 – 8 < 8 – 6,5 < 6,5 Leukozyten [x 109 /L] <4–3 <3–2 <2–1 <1 Thrombozyten [x 109 /L] < 140 – 75 < 75 – 50 < 50 – 25 < 25 < 1,8 – 1,5 < 1,5 – 1 < 1 – 0,5 < 0,5 < 1 – 0,8 < 0,8 – 0,5 < 0,5 – 0,2 < 0,2 Neutrophile Granulozyten [x 109 /L] Lymphozyten [x 109 /L] 8.1 > 6,6 Zusätzliche Informationen zu den Medikamenten 8.1.1 Bevacizumab Bevacizumab ist ein antineoplastisch wirksamer monoklonaler Antikörper, der zielgerichtet gegen bestimmte Zellen oder Strukturen wirkt, während die Che82 motherapie meist unspezifisch alle wachsenden Zellen stört [54]. Gentechnisch lässt sich die Substanz mittels (Mikro-)Organismen herstellen (Rekombination) und aus Ovarialzellen des chinesischen Hamsters, den so genannten CHO-Zellen, gewinnen [54]. Da der Antikörper eine ausgeprägte antitumorale Aktivität gegen menschliche Krebsarten aufweist, wird die antiangiogenetische Wirkung von Bevacizumab genutzt, um Tumore von Kolon, Pankreas, Mamma, Ovarien und Prostata sowie nicht kleinzellige Bronchial- und fortgeschrittene Nierenzellkarzinome zu behandeln [54]. Bevacizumab wird unter dem Handelsnamen Avastin® von der Firma Roche Pharma AG (Schweiz) vertrieben. 8.1.1.1 Chemische Struktur Bevacizumab ist ein humanisierter monoklonaler Antiköper (monoclonal antibody = mab) mit einer Molmasse von 149.000 Dalton [54]. Die intravenöse Applikation erfolgt in Verdünnung mit 0,9%iger Kochsalzlösung [54]. 8.1.1.2 Wirkmechanismus Bevacizumab ist eine antiangiogen wirkende Substanz, die über Inhibition des Wachstumsfaktors VEGF-A in die Tumorgefäßneubildung eingreift [54]. Diese Wirkung beruht darauf, dass der Antikörper an den Gefäßwachstumsfaktor VEGFA (vascular endothelial growth factor) bindet und dieser deshalb nicht mehr an seine zugehörigen Rezeptoren VEGFR-1 und VEGFR-2 auf der Endothelzelloberfläche binden kann. Da die biologische Aktivität von VEGF durch dieses Ereignis neutralisiert wird und VEGF der entscheidende Faktor der Vaskulogenese und Angiogenese ist, wird das Tumorgefäßsystem normalisiert und die Vaskularisierung reduziert. Die weniger effektive Versorgung der Tumorzellen mit Sauerstoff und Nährstoffen führt zu einer Wachstumshemmung des Tumors. Die mikrovaskuläre Permeabilität und somit der Druck im Tumorgewebe sinken, sodass Chemotherapeutika das Zielgewebe besser erreichen können. Die Tumorprogression metastasierter Erkrankungen wird gehemmt [54]. 83 8.1.1.3 Pharmakokinetik Folgende pharmakokinetische Daten für Bevacizumab entstammen mehreren klinischen Studien mit Patienten mit soliden Tumoren, denen Bevacizumab intravenös appliziert wurde [54]. Die Studien zeigten, dass der Antikörper in einem Dosisbereich von 1 bis 10mg/kg eine lineare Pharmakokinetik aufweist. Auffällig war, dass das Stoffwechselprofil des Antikörpers dem eines nativen, nicht VEGFbindenden IgG-Moleküls entspricht. Im Einzelnen bedeutet das, dass weder der Metabolismus noch die Eliminierung von Bevacizumab primär über Leber und Nieren, sondern – ähnlich wie endogene IgGs – hauptsächlich proteolytisch im gesamten Körper einschließlich der Endothelzellen erfolgt [54]. Die Pharmakokinetik war vom Alter der Probanden unabhängig, weder bei Kindern noch bei älteren Patienten konnten Unterschiede im Verteilungsvolumen und in der Clearance von Bevacizumab festgestellt werden. Studien zu pharmakokinetischen Auswirkungen bei Leber- und Niereninsuffizienz liegen nicht vor [54]. Eine Dosisanpassung bei älteren Patienten ist nicht notwendig. [54]. 8.1.1.4 Toxizität Bisherige klinische Untersuchungen seit der Markteinführung 2005 zeigten, dass Bevacizumab bei einer empfohlenen Dosis (abhängig von der Tumorentität 5-10mg/kg Körpergewicht einmalig alle zwei Wochen oder 15mg/kg Körpergewicht einmalig alle drei Wochen [54]) als intravenöse Infusion ein überschaubares Nebenwirkungsprofil hat [15]. Die Therapie wurde sowohl bei einmaliger als auch bei wiederholter Gabe des Angiogenese-Hemmers von der Mehrzahl der Patienten äußerst gut vertragen [15]. Dies ist ein Vorteil gegenüber der klassischen Chemotherapie und anderen monoklonalen Antikörpern sowie niedermolekularen Verbindungen. Zudem macht das gut dokumentierte Wirksamkeits- und Sicherheitsprofil, das auch bei älteren Patienten durch zwei große Phase-IV-Beobachtungsstudien (BRiTE, First BEAT) bestätigt wurde [5, 24, 64], Bevacizumab zu einem bevorzugten Pharmakon in der Tumortherapie [54]. Nachfolgend sind die häufigsten und schwerwiegendsten Toxizitäten unter Bevacizumab-Therapie aufgelistet [24, 54]. 84 - Wundheilungsstörungen: Die Bevacizumab-Therapie sollte vor Operationen abgesetzt und frühestens 28 Tage nach größeren Operationen oder besser erst nach völliger Abheilung der Wunde wieder aufgenommen werden, da anderenfalls die Inzidenz für postoperative Blutungen und Wundheilungsstörungen mit 10-20% erhöht ist. - Erkrankungen des Gastrointestinaltrakts: Magen-Darm-Perforationen (1-2%), Fisteln (bis zu 2%) meist innerhalb der ersten sechs Behandlungsmonate, außerdem Ileus, Übelkeit, Erbrechen, Obstipation, Diarrhö, Stomatitis, Rektalblutung - Erkrankungen des Nervensystems: periphere sensorische Neuropathie, Apoplex, Synkope, Somnolenz, Kopfschmerz, Dysgeusie, reversibles posteriores Leukoenzephalopathie-Syndrom - Gefäßerkrankungen: o Blutungen der Schleimhaut (20-40%), der Vagina und des Zahnfleisches sind typisch. Bei Blutungen Grad 3-5 (0,4-5%) und intrakraniellen Blutungen sollte Bevacizumab dauerhaft abgesetzt werden. o Hypertonie ist vermutlich dosisabhängig und mit bis zu 34% häufig. Hypertonien Grad 3 und 4 kommen bei 3-17,9% und hypertensive Krisen bei bis zu 1% der Patienten vor. o Erkrankungen der Arterien: Arterielle Thromboembolie (3,8%), zerebrale Insulte einschließlich transitorischer ischämischer Attacken (2,3%) und Myokardinfarkte (1,4%) o Erkrankungen der Venen: Venöse Thromboembolien (tiefe Beinvenenthrombosen, Lungenembolie und Thrombophlebitis) in 2,8-17,3% der Fälle - Erkrankungen der Atemwege, des Brustraums und Mediastinums: Lungeneinblutung, Bluthusten, Dyspnoe, Hypoxie, Epistaxis, Rhinitis. - Herzerkrankungen: Kongestive Herzinsuffizienz bei bis zu 3,5% der Fälle, vor allem bei metastasiertem Mammakarzinom, supraventrikuläre Tachykardie - Erkrankungen des Blutes und Lymphsystems und Infektionen: schwere febrile Neutropenien teils mit Infektion (z. B. Sepsis, Abszess), Anämien, Leukopenien, Thrombozytopenien 85 Unerwünschte Arzneimittelwirkungen unter Bevacizumab-Gabe sind im Allgemeinen von geringem bis mäßigem Schweregrad und können klinisch mithilfe von Standard- oder Akutbehandlung gut kontrolliert werden [24]. Therapeutisch wird eine Bevacizumab-Pause und bei sehr schwerwiegenden Nebenwirkungen ein dauerhaftes Absetzen empfohlen, von einer alleinigen Dosisreduktion des Antikörpers ist abzusehen. Wenn die Therapie für den Probanden gut verträglich ist, wird empfohlen, diese bis zur Tumorprogression fortzuführen, auch wenn begleitende Chemotherapien aufgrund von Nebenwirkungen vorzeitig abgesetzt werden. Die genannten Nebenwirkungen wurden überwiegend bei Patienten beobachtet, die im Rahmen der Studien Bevacizumab in Kombination mit einer Chemotherapie erhielten. Daher überschneiden sich einige Nebenwirkungen mit denen, die häufig unter Chemotherapie auftreten, z. B. palmoplantares ErythrodysästhesieSyndrom unter Capecitabin, periphere sensorische Neuropathie unter Paclitaxel oder Oxaliplatin. Eine Symptomverschlimmerung durch Bevacizumab ist aber nicht auszuschließen [54]. Zu den schwerwiegendsten in Studien beobachteten Nebenwirkungen unter Bevacizumab-Therapie zählten die Magen-Darm-Perforation, hämorrhagische Ereignisse (tumorassoziierte Blutungen, Lungeneinblutungen und Bluthusten bei NSCLC [24]) und arterielle Thromboembolien [24, 54]. Die häufigsten beobachteten unerwünschten Arzneimittelwirkungen bei einer Bevacizumab-Behandlung waren Hypertonie, Fatigue oder Asthenie, Diarrhö und Bauchschmerzen [24, 54]. Sie traten in über 10% der Fälle auf. Laborwertveränderungen wie eine reduzierte Anzahl neutrophiler Granulozyten und Leukozyten und das Auftreten von Protein im Urin können auf die Bevacizumab-Therapie zurückgeführt werden. Weitere Laborauffälligkeiten 3. und 4. Grades, die im Unterschied zur Kontrollgruppe (ohne Bevacizumab) bei mindestens 2% der Patienten unter Bevacizumab-Therapie auftraten, waren Hyperglykämie, erniedrigter Hämoglobinwert, Hypokaliämie, Hyponatriämie, erniedrigte Leukozytenanzahl und ein erhöhter INR-Wert [54]. 86 8.1.2 Sorafenib Sorafenib ist ein potenter Protein-Kinase-Inhibitor mit multimodalem Therapieansatz, der neben dem antiproliferativen Effekt auch eine antiangiogene Komponente besitzt [15]. Die Substanz wurde gemeinsam von der Bayer HealthCare AG und Onyx Pharmaceuticals entwickelt [15] und im Juli 2006 zur Therapie des fortgeschrittenen Nierenzellkarzinoms und ein Jahr später zur Behandlung des Leberzellkarzioms zugelassen [4]. Seit November 2013 ist Sorafenib zugelassen in der Therapie des lokal fortgeschrittenen oder metastasierten, differenzierten Schilddrüsenkarzinoms, das kein Ansprechen mehr auf eine Radiojodtherapie zeigt [7]. Sorafenib wird unter dem Handelsnamen Nexavar® von der Bayer HealthCare AG vertrieben [4]. 8.1.2.1 Chemische Struktur Sorafenib ist ein synthetischer Wirkstoff aus der Gruppe der Diarylharnstoffe in der Darreichungsform einer Tablette [15]. Aus seiner Summenformel C21H16ClF3N4O3 errechnet sich ein Molekulargewicht von 464,83g/mol. Abbildung A1: Strukturformel Sorafenib 8.1.2.2 Wirkmechanismus Sorafenib ist ein Multikinase-Inhibitor, der durch Hemmung mehrerer Enzyme mit Kinase-Aktivität auf molekularer Ebene die Signalwirkung von Wachstumsfaktoren ausschaltet und so in die Zellteilung antiproliferativ eingreift [4]. Außerdem ist seine antiangiogenetische Wirkung beschrieben, die therapeutisch genutzt wird, um den Tumor von seinen versorgenden Blutgefäßen abzuschneiden [4]. Dadurch wird der Tumor in seinem Wachstum begrenzt [4]. 87 8.1.2.3 Pharmakokinetik Der Metabolismus von Sorafenib erfolgt primär in der Leber [4] durch oxidativen Abbau über CYP3A4- und UGT1A1/UGT1A9-vermittelte Glukuronidierung [4, 15]. Die anschließende Ausscheidung erfolgt zu 77% über den Stuhl, während 19% der Dosis als Glukuronide im Urin ausgeschieden werden [4, 15]. 51% der Gesamtdosis von Sorafenib findet sich unverändert in den Fäzes wieder, nicht aber im Urin. Diese Tatsache weist darauf hin, dass die Ausscheidung von unverändertem Sorafenib über die Galle zur Elimination beitragen kann. Bisherige Analysen deuten darauf hin, dass die Pharmakokinetik von Sorafenib weder vom Alter der Patienten (bis 65 Jahre) noch vom Geschlecht oder dem Körpergewicht abhängt [4]. Eine Dosisanpassung ist auch bei älteren Patienten (über 65 Jahre) nicht erforderlich [4], jedoch gibt es für diese Altersgruppe bisher nur wenige Erfahrungen. Da in einigen Fällen über Nierenversagen berichtet wurde, ist Vorsicht geboten [4]. Nieren- oder Leberfunktionsstörungen erfordern ebenfalls keine Dosisanpassung, Daten zu schwerer Leberfunktionsstörung liegen nicht vor [4]. 8.1.2.4 Toxizität Verschiedene Studien der Phasen I-III testeten die klinische Wirksamkeit von Sorafenib an Patienten mit unterschiedlichen soliden und fortgeschrittenen Tumorentitäten [15]. In vier offenen Dosisfindungsstudien konnte eine kontinuierliche Sorafenib-Dosis von 400mg beim Erwachsenen zweimal täglich empfohlen werden, wobei eine Tablette 200mg des Wirkstoffs enthält [4, 65]. Dosislimitierende Nebenwirkungen wurden bei Dosierungen >600mg zweimal täglich beobachtet [65]. Bei der empfohlenen Tagesgesamtdosis von 800mg [4] oder weniger wies Sorafenib ein vertretbares Nebenwirkungsprofil auf [4]. Die gute Verträglichkeit und das gut beherrschbare Sicherheitsprofil der Substanz, vor allem in Hinblick auf die Schwere der Erkrankung [4], ermöglicht eine kontinuierliche SorafenibTherapie ohne Pausen [15, 65]. Nachfolgend sind einige typische Nebenwirkungen unter SorafenibTherapie aufgelistet, zur Bedeutung der Häufigkeit von Nebenwirkungen siehe Tabelle A3. 88 Tabelle A3: Häufigkeit der Nebenwirkungen [4] Sehr häufig ≥ 1/10 - Häufig Gelegentlich Selten ≥ 1/100 ≥ 1/1000 ≥ 1/10.000 < 1/10 <1/100 < 1/1000 Sehr selten <1/10.000 Erkrankungen des Gastrointestinaltrakts: Durchfall, Übelkeit und Erbrechen werden sehr häufig [4], Verdauungsstörungen, Obstipation, Dyspepsie, Schluckbeschwerden, entzündeter oder trockener Mund, Stomatitis, Glossodynie, Appetitlosigkeit und Gewichtsverlust häufig beobachtet [4]. Ein weiteres unerwünschtes Ereignis ist die gastrointestinale Perforation mit einer Häufigkeit von weniger als 1% [4]. - Erkrankungen der Haut: Sehr häufig werden unter Therapie Alopezie, Pruritus, Hauterytheme, palmoplantares Erythrodysästhesie-Syndrom und Hautausschlag beobachtet. Bei den beiden letzteren handelt es sich üblicherweise um Nebenwirkungen Grad 1 und 2 nach der CTCAE. Sie treten im Allgemeinen während der ersten 6 Wochen der Behandlung auf und sprechen auf symptomatisch topische Therapie gut an [4]. Anderenfalls kann die Sorafenib-Dosis reduziert oder die Gabe pausiert werden [4]. Weniger häufig sind Akne, entzündete, trockene oder abschuppende Haut und exfoliative Dermatitis [4]. - Erkrankungen des Blutes und Lymphsystems und Infektionen: Lymphopenien unter Sorafenib-Gabe sind sehr häufig [4]. Von grippeähnliche Erkrankungen, Fieber und Asthenie wird häufig berichtet [4]. - Gefäßerkrankungen: o Blutungen einschließlich des Gehirns, Magens, Darms oder der Atemwege [4] sind sehr häufig. Wenn die Hämorrhagie behandlungspflichtig wird, sollte die Therapie pausiert werden [4]. o Hypertonie: Leichte bis mäßige Hypertonie tritt bevorzugt in der frühen Phase der Behandlung auf [4]. Diese sehr häufige Toxizität erfordert regelmäßige Blutdruckkontrollen, lässt sich aber mithilfe antihypertensiver Standardtherapien gut behandeln [4, 15]. Gelegentlich auftretende hypertensive Krisen oder schwere andauernde Hypertonien, die therapeu- 89 tisch nicht beherrschbar sind, erfordern eine dauerhafte Unterbrechung der Sorafenib-Gabe [4]. - Erkrankungen des Nervensystems: Sehr häufig berichten Patienten über Schmerzen in Mund, Abdomen, Kopf, Knochen oder Tumor und Müdigkeit [4]. Häufig klagen Probanden über Depressionen, erektile Dysfunktion und periphere sensorische Neuropathie [4]. - Herzerkrankungen: Gelegentlich werden unter Therapie myokardiale Ischämien, Myokardinfarkte und Herzinsuffizienz beobachtet [4]. Weiterhin wird häufig über Heiserkeit, Nierenversagen, Tinnitus, Arthralgie und Myalgie [4] berichtet. Bisher gibt es keinerlei Erkenntnisse, ob und wie sich Sorafenib auf die Wundheilung auswirkt. Trotzdem wird vorsorglich empfohlen, das Pharmakon vor größeren chirurgischen Eingriffen abzusetzen. Wann mit der Behandlung fortgefahren werden kann, ist bisher nicht genau bekannt [4]. Die meisten Nebenwirkungen unter Sorafenib-Therapie sind mithilfe symptomatischer Behandlung zu kontrollieren. Wenn die Toxizitäten jedoch nicht mit entsprechender Therapie beherrschbar sind, muss die Sorafenib-Gabe, vor allem bei schweren Nebenwirkungen mit potenzieller Lebensgefahr, pausiert oder dauerhaft beendet werden. Alternativ kann bei schweren Nebenwirkungen die Dosis auf zwei Tabletten à 200mg einmal täglich reduziert werden [4]. Solange die Nebenwirkungen vertretbar sind und der Patient von der Behandlung profitiert, wird die Sorafenib-Gabe fortgesetzt [4]. Zu den häufigsten Nebenwirkungen unter Sorafenib-Therapie zählen mit mehr als 10% gastrointestinale Ereignisse (Diarrhö (35%), Übelkeit, Erbrechen), Hautreaktionen (Hautausschlag, Pruritus, Hauterythem, Alopezie, palmoplantares Erythrodysästhesie-Syndrom (35%)), Fatigue (35%), Hypertonie, Hämorrhagie und Schmerzen [4]. Auch bei sehr hohen Dosen wurde hauptsächlich über Durchfall und dermatologische Ereignisse geklagt. Häufigste Nebenwirkung in der SHARP-Studie waren Diarrhö, Gewichtsverlust, palmoplantares Erythro- dysästhesie-Syndrom und Hypophosphatämie [15, 65]. Die erwähnten Laborwertveränderungen zählen zu den schwerwiegenden Toxizitäten unter Sorafenib-Therapie. Sehr häufig werden Anstiege 3. und 4. Grades der Lipase und Amylase im Serum sowie Hypophosphatämien (35-45% unter So90 rafenib-Therapie vs. 11-12% in der Placebo-Gruppe) beobachtet [4]. Häufig kommt es zu einem vorübergehenden Anstieg der Transaminasen, zu Lymphozytopenie (13% vs. 7%), Leukopenie, Neutropenie (5% vs. 2%), Anämie (2% vs. 4%) und Thrombozytopenie (1% vs. 0%) [4]. 8.1.3 Paclitaxel Das antineoplastisch wirksame Chemotherapeutikum Paclitaxel gehört zur Gruppe der Taxane, eine neuere Klasse von Pflanzenalkaloiden. Die Pflanzenextrakte wurden ursprünglich aus der Rinde der Pazifischen Eibe (Taxus brevifolia) isoliert, entsprechend prekär war die Produktion. Die heute im Handel erhältlichen Präparate sind jedoch semisynthetisch und werden auf der Grundlage von Pflanzenteilen (Zweige und Nadeln der europäischen Eibe Taxus baccata) erzeugt [28, 33, 36]. Paclitaxel wird von der Firma Bristol-Myers Squibb GmbH unter dem Handelsnamen Taxol® verkauft und ist für die Therapie des Ovarial- und Mammakarzinoms, des NSCLC und des AIDS-assoziierten Kaposi-Sarkoms zugelassen [6, 33]. 8.1.3.1 Chemische Struktur Paclitaxel ist ein Diterpen-Pseudoalkaloid mit komplexer Ringstruktur. Da die Substanz wasserunlöslich ist, erfolgt die intravenöse Applikation in Ethanol und polyoxyethyliertem Rizinusöl (Cremophor) [26] und anschließender Verdünnung mit 0,9%iger Natriumchlorid-Lösung und/oder 5%iger Glucose-Lösung [6]. Aus der Summenformel C47H51NO14 errechnet sich ein Molekulargewicht von 853,9g/mol [3]. 91 Abbildung A2: Strukturformel Paclitaxel 8.1.3.2 Wirkmechanismus Wie alle Chemotherapeutika greift auch Paclitaxel in den Vermehrungszyklus von Körperzellen ein. Die Substanz gehört mit ihren antimikrotubulären Eigenschaften zur Gruppe der Taxane [6]. Mikrotubuli im Zellinneren spielen eine wichtige Rolle während der Mitose, sie dienen der Ausbildung der mitotischen Zellspindel und sind der spezifische Angriffspunkt von Paclitaxel. In Anwesenheit dieses Zytostatikums wird der Aufbau der Mikrotubuli durch Zusammenlagerung von Tubulindimeren stimuliert [3, 6]. Allerdings bindet Paclitaxel reversibel an die β-Tubulinuntereinheit bereits aufgebauter Mikrotubuli und verändert so die Tubulusstruktur [43]. Die Depolymerisation der Mikrotubuli wird gehemmt [6], sodass das dynamische Gleichgewicht zwischen Auf- und Abbau der intrazellulären Mikrotubuli gestört ist. Die Mikrotubuli werden stabilisiert [6, 43] und ihr Abbau behindert. Durch die abnorme Stabilisierung und die Akkumulation atypischer [6, 43] funktionsgestörter Mikrotubuli kann das mikrotubuläre Netzwerk während der vitalen Interphase der Mitose nicht dynamisch reorganisiert werden [6], d.h., die korrekte Ausbildung des Spindelapparats wird verhindert [33]. Die Mikrotubuli organisieren sich während des Zellzyklus zu multiplen funktionslosen Mikrotubulibündeln und -sternen, den so genannten Astern [3, 6]. Die korrekte Anordnung der Chromosomen wird verhindert und die Zellteilung kann nicht ordnungsgemäß beendet werden. Sie wird in der späten G2- oder M-Phase irreversibel blockiert, sodass viele Zellen in dieser Teilungsphase verbleiben. Häufig resultiert der Eintritt in die 92 Apoptose. Bei anderen Zellen erfolgt der Wiedereintritt in die G1-Phase, wobei jedoch funktionslose Zellen mit vielen oder zu kleinen Zellkernen entstehen [3]. Aufgrund dieses Wirkungsprinzips werden Taxane auch als Spindelgifte bezeichnet. Paclitaxel wird neben der Eigenschaft, den programmierten Zelltod zu fördern, auch noch eine strahlensensibilisierende Wirkung zugesprochen [3, 13, 41]. Ursächlich hierfür ist die höchste Strahlensensibilität der Zellen in der G2- oder MPhase der Mitose, deren Anzahl Paclitaxel durch Hemmung der Ausbildung des Spindelapparats erheblich erhöht. 8.1.3.3 Pharmakokinetik Die Umverteilung von Paclitaxel aus dem zentralen in das periphere Kompartiment, also von intravasal nach extravasal, erfolgt kurz nach intravenöser Gabe. Paclitaxel gelangt in die meisten Gewebe, die genaue Verteilung im menschlichen Organismus ist bisher aber noch nicht vollständig aufgeklärt [6]. In-vitroStudien zeigten mit 89-98% eine auffallend hohe Proteinbindung von Paclitaxel an menschliches Serumeiweiß [6]. In der Leber wird Paclitaxel vorrangig über Cytochrom-P450-Isoenzyme zu aktiven Hydroxyverbindungen verstoffwechselt [6]. Studien ergaben, dass die renale Ausscheidung der Substanz nur in geringem Umfang zur systemischen Clearance beiträgt, da im Harn lediglich 1,3-12,6% der verabreichten Dosis von Paclitaxel in unveränderter Form vorzufinden sind. Vermutlich sind der Metabolismus über die Leber und die anschließende Elimination über die Galle die Hauptmechanismen der Verstoffwechselung von Paclitaxel [6]. Auswirkungen von Nieren- oder Leberinsuffizienz wurden bislang nicht genauer untersucht [6]. Dennoch wird von einer Paclitaxel-Gabe bei schwerer Leberinsuffizienz abgeraten, weil Toxizitäten, vor allem Myelosuppressionen Grad 3 und 4, gehäuft auftreten [6]. Obwohl bisher keine Veränderung im Metabolismus oder der Elimination festgestellt werden konnte, wenn Paclitaxel gleichzeitig mit anderen Arzneimitteln verabreicht wurde, die am Stoffwechselweg beteiligte Enzyme inhibieren oder induzieren, ist bei gleichzeitiger Anwendung Vorsicht geboten [6]. Weiterhin konnte eine Akkumulation von Paclitaxel bei wiederholten Therapiezyklen 93 nicht beobachtet werden [6], auch sind eindeutige Zusammenhänge zwischen Pharmakokinetik und Wirkung nicht nachgewiesen. 8.1.3.4 Toxizität Klinische Studien mit Paclitaxel als Monotherapie bei soliden Tumoren werden seit der Markteinführung 1994 zur Ermittlung der Toxizitäten durchgeführt [6]. Dabei ergab sich, dass das Nebenwirkungsspektrum bei der empfohlenen Dosis von 175mg Paclitaxel pro m² Körperoberfläche als 3-stündige Infusion im Abstand von drei Wochen [6] relativ breit ist. Trotzdem wird die Behandlung von den meisten Patienten gut vertragen, auch von umfangreich vorbehandelten Probanden oder solchen mit schlechtem Allgemeinzustand [55]. Bei keiner Toxizität konnte eindeutig nachgewiesen werden, dass sie vom Alter der Patienten abhängig war [6]. Die Prozentzahlen der nachfolgend aufgeführten häufigen und schwerwiegenden Nebenwirkungen unter Paclitaxel-Therapie gelten für das NSCLC, das Ovarial- und Mammakarzinom [6]. Auf die prozentualen Abweichungen beim AIDS-assoziierten Kaposi-Syndrom wird nicht weiter eingegangen [6]. - Erkrankungen des Blutes und Lymphsystems: Knochenmarksuppression mit Anämie (64%), schwere Anämie mit Hämoglobinwerten <8,05g/dl bzw. <5mmol/l (6%), schwere Neutropenie (<500 Zellen/mm³) ohne fiebrige Episode (28%), schwere Neutropenie mit Dauer ≥7 Tage (1%), Thrombozytopenie (11%), Thrombozytenzahlen unter 50.000 Zellen/mm³ (3%), Blutungen, febrile Neutropenie, akute myeloische Leukämie und myelodysplastisches Syndrom [6]. - Erkrankungen des Immunsystems: schwere Überempfindlichkeitsreaktionen bei 1% der Probanden. Diese sind assoziiert mit Dyspnoe, Hypo- oder Hypertonie, Angioödem, generalisierter Urtikaria, Schwitzen, Schüttelfrost, Schmerzen, Tachykardie und potenziell letal. Leichte allergische Reaktionen mit Flush und Hautauschlag beobachtet man bei 34% der Patienten [6]. Mithilfe der Prämedikation von Glukokortikoiden, H1- und H2-Rezeptorantagonisten kann die Häufigkeit erheblich reduziert werden. Selten werden anaphylaktische Reaktionen und anaphylatischer Schock beobachtet [6]. 94 - Erkrankungen des Nervensystems: Neurotoxizität (hauptsächlich Polyneuropathie (66%)), aber auch motorische Neuropathie, autonome Neuropathie, Grand-Mal-Anfälle, Konvulsionen, Enzephalopathie, Schwindel, Ataxie und Kopfschmerzen [6] - Herzerkrankungen: Arrhythmien (Bradykardien, atrioventrikuläre Leitungsstörungen) in 2% der Fälle, aber für gewöhnlich asymptomatisch [55], Myokardinfarkt, Synkope, Kardiomyopathie, Tachykardien, Vorhofflimmern [6] - Erkrankungen des Gastrointestinaltrakts: Nebenwirkungen wie Diarrhö (28%), Nausea / Emesis (43%), Übelkeit und Mukositis (18%) [6] sind zwar sehr häufig, aber üblicherweise geringen Ausmaßes (Grad 1 bis 2), seltener sind Ileus, Darmperforation, ischämische Kolitis und Pankreatitis [6]. In Einzelfällen wurden Thrombose im Mesenterium, pseudomembranöse und neutropenische Kolitiden, Aszites, Ösophagitis und Obstipation festgestellt [6]. - Leber und Gallenerkrankungen: Hepatische Nekrose und hepatische Enzephalopathie mit möglichem letalem Ausgang sind sehr selten [6]. - Erkrankungen der Atemwege, des Brustraums und Mediastinums: In seltenen Fällen Ateminsuffizienz, Lungenembolie, Lungenfibrose, interstitielle Pneumonie bei Kombination mit Bestrahlung der Lunge, Dyspnoe, Pleuraerguss und Husten [6] - Erkrankungen von Skelettmuskulatur und Knochen: Arthralgien und Myalgien gehäuft bei höheren Dosierungen (60%), davon 13% mit schwerer Ausprägung [6] - Infektionen: Infektionen vor allem der Harnwege und des oberen Respirationstraktes sind sehr häufig, Fälle mit septischem Schock und letalem Ausgang sind dokumentiert; selten entwickeln Patienten eine Sepsis, Peritonitis oder eine Pneumonie [6]. - Gefäßerkrankungen: Hypotonie, Hypertonie, Thrombose, Thrombophlebitis und Schock [6] - Erkrankungen der Haut: Die reversible Alopezie ist bei fast allen Patienten zu beobachten [6]. Unerwünschte Arzneimittelnebenwirkungen unter Paclitaxel erfordern häufig therapeutische Interventionen. Sofern Toxizitäten nicht mit entsprechender Be95 handlung reguliert werden können, muss die Therapie mit Paclitaxel, vor allem bei schweren Nebenwirkungen mit potenzieller Lebensgefahr, dauerhaft beendet werden. Alternativ kann bei weniger schweren Ereignissen zunächst eine Dosisreduktion in den folgenden Behandlungskursen um 20% versucht werden [6]. Die häufigste schwerwiegende Nebenwirkung unter Paclitaxel-Therapie ist die Myelosuppression [6]. Sie stellt meist den dosislimitierenden Faktor der Behandlung dar [6]. Nach einer Studie ist die myelosuppressive Wirkung von Paclitaxel (135 oder 175mg/m²) bei 3-stündiger Infusion geringer als bei 24-stündiger Infusion [3, 59]. Wöchentliche Blutbildkontrollen sind zu empfehlen, um lebensbedrohliche Komplikationen zu vermeiden. Bei schwerwiegenden Granulozytopenien sollte eine Behandlung mit Wachstumsfaktoren erfolgen [6]. Obwohl periphere Neuropathien zu den häufigsten Nebenwirkungen zählen, verursachen sie nur selten schwere Symptome. Sie können bereits während des ersten Behandlungskurses auftreten und mit der Häufigkeit der Paclitaxel-Gaben zu kumulativen Effekten führen [55]. Weiterhin wurde in Studien die Dosisabhängigkeit der Neurotoxizität festgestellt. Die Ergebnisse der Studie zeigen, dass bei höherer Paclitaxel-Dosis 85% der Patienten Neurotoxizitäten entwickelten (15% davon schwer), während bei geringer Dosis nur 25% erkrankten (3% schwer) [6]. Therapieabbrüche aufgrund von Neuropathien sind eher selten, aber in schweren Fällen wird zu einer Dosisreduktion von 20% in allen folgenden Behandlungskursen geraten [6]. Üblicherweise bessern sich die Empfindungsstörungen innerhalb einiger Monate nach Absetzen der Substanz, bestenfalls verschwinden sie vollständig [6]. Die genannten Laborwertveränderungen betreffen vor allem das blutbildende System, aber auch eine starke Erhöhung von ASAT und alkalischer Phosphatase, starke Bilirubinwerterhöhung sowie Anstiege des Kreatinin-Spiegels werden beschrieben [6]. 8.1.4 Interaktion Bei der folgenden Besprechung der Wechselwirkungen sind ausschließlich solche berücksichtigt, die für die vorliegende Studie relevant sind. 96 Zur Überprüfung klinisch relevanter pharmakokinetischer Wechselwirkungen zwischen Bevacizumab und einer gleichzeitigen Chemotherapie wurde mithilfe von Studien ermittelt, dass die Verfügbarkeit von Bevacizumab während einer Chemotherapie nicht beeinflusst wird. Bezüglich der Clearance dieses Antikörpers zeigten sich keine Unterschiede bei den Patienten mit zusätzlicher Zytostatikatherapie zu solchen, die mit Bevacizumab als Monotherapie behandelt wurden. Die Wirkung aller gleichzeitig angewendeten Chemotherapien auf die BevacizumabClearance wird als klinisch nicht signifikant angesehen [54]. Reversible unerwünschte Nebenwirkungen traten in einer Kombinationsbehandlung von Bevacizumab mit dem Rezeptor-Tyrosinkinase-Inhibitor Sunitinibmaleat auf. Dazu zählen neben Anämie und Thrombozytopenie auch mikroangiopathische hämolytische Anämien und Hypertonie mit hypertensiven Krisen, erhöhte Kreatininwerte und neurologische Symptome [54]. Ersten Ergebnissen aus Phase-I-Studien zufolge kommt es bei einer Kombination von Sorafenib mit Standardchemotherapeutika wie Oxaliplatin und Gemcitabin nicht zu einer gegenseitigen pharmakokinetischen Beeinflussung. Vorsicht ist allerdings dann geboten, wenn Sorafenib mit Arzneimitteln kombiniert wird, die vorwiegend über den UGT1A1- oder UGT1A9-Stoffwechselweg metabolisiert werden, z. B. Irinotecan, Estradiol, Docetaxel oder Paclitaxel [4, 15]. Interaktionen zwischen diesen Medikamenten und Sorafenib sind bekannt und führen zu verstärkten Nebenwirkungen [15]. Bisher ist unklar, ob Paclitaxel die Plasmakonzentration von Sorafenib erhöht [4]. Die gleichzeitige Anwendung von Arzneimitteln gegen Magenübersäuerung kann die Plasmakonzentration von Sorafenib verringern, außerdem können auch Medikamente, die den Metabolismus von Sorafenib verstärken oder herabsetzen, dessen Konzentration im Plasma beeinflussen. Hierzu gehören unter anderem Rifampicin, Hypericumperforatum, Phenytoin, Carbamazepin, Phenobarbital und Dexamethason [4, 15]. Paclitaxel interagiert ebenfalls mit verschieden Zytostatika, z. B. Cisplatin, die aber in der vorliegenden Studienmedikation nicht verwendet wurden. Die Recherche bisheriger Untersuchungen ergab keine Daten zur Interaktion der drei Studienmedikamente untereinander. 97 Danksagung Die Untersuchungen für diese Arbeit wurden in der Klinik für Onkologie und Hämatologie des Marienhospitals in Herne (Direktor: Prof. Dr. med. D. Strumberg) – Universitätsklinikum der Ruhr-Universität Bochum – durchgeführt. Den Mitarbeitern dieser Abteilung danke ich für die Einarbeitung und Unterstützung sowie die Bereitstellung der Patientenakten. Meinem Doktorvater Herrn Prof. Dr. med. D. Strumberg danke ich für die Bereitstellung des Promotionsthemas, die damit verbundene Möglichkeit, in der Klinik für Onkologie und Hämatologie zu promovieren, sowie die gute fachliche Betreuung im Verlauf dieser Arbeit. Mein besonderer Dank gilt Frau Dr. B. Schultheis für die Mühe, die Zeit und Unterstützung beim Verfassen dieser Dissertation. Des Weiteren möchte ich allen Patienten danken, die trotz ihrer schweren Erkrankung den Mut hatten, an dieser Studie teilzunehmen. Ohne ihre Hilfe wäre diese Studie nicht möglich gewesen. Von Herzen danke ich meinen lieben Eltern für die geistige und finanzielle Unterstützung während meines Studiums und bei dieser Arbeit und besonders meiner Mutter für die Durchsicht des Manuskripts. Lebenslauf Persönliche Daten: Name: Renée Roy Geburtsdatum, -ort: 09.09.1988, Bonn Staatsangehörigkeit: deutsch Familienstand: ledig Schulausbildung: 1994 – 1998 Brüder-Grimm-Grundschule in Langenfeld 1998 – 2006 Konrad-Adenauer-Gymnasium in Langenfeld Abschluss: Abitur Hochschulausbildung: 10/2006 – 10/2012 Studium der Humanmedizin an der Ruhr-Universität Bochum 2006 – 2008 Vorklinik Herbst 2008 Erster Abschnitt der Ärztlichen Prüfung 2008– 2012 Klinik 08/2011 – 07/2012 Praktisches Jahr 1. Tertial Innere Medizin (Marienhospital Herne) 2. Tertial Chirurgie (Marienhospital Herne) 3. Tertial Geriatrie (Marienhospital Herne) Herbst 2012 Zweiter Abschnitt der Ärztlichen Prüfung und Approbation Facharztausbildung: seit 03/13 Assistenzärztin für Innere Medizin im Marienhospital Herne, Klinik für Altersmedizin und Frührehabilitation, Universitätsklinikum der RuhrUniversität Bochum