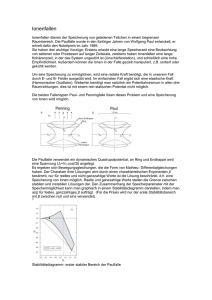
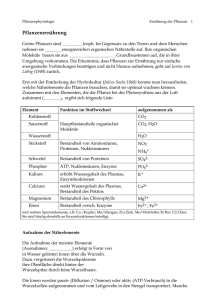
Analytische Chemie III
Werbung

1 AC III – Teil 2 Allmaier Martin Prießner Analytische Chemie III - Teil 2 – Allmaier WS 2010/2011 (VL Nr. 164.253) TUWIS Zugangsschlüssel: AC32010 Massenspektrometrie: Geschichtlich: J. J. Thomson* (Manchester 1856, † Cambridge, 1940) erhielt den Nobelpreis für Physik 1906 für die Erfindung des ersten Massenspektrometer („parabola spectrograph“) der das Verhältnis von Ladung zur Masse (m/z) des Elektrons beschrieb. Zuerst wurde der MS hauptsächlich zur Bestimmung anorganischer Substanzen verwendet. Erst später wurde er für die organische Chemie entdeckt. Der Österreicher K. Biemann half bei der Entwicklung das Massenspektrometer (GC/EI/MS) welcher bei der Marsmission im Jahr 1976 (Viking) eingesetzt wurde, zur Beantwortung der Frage: Gibt es Leben am Mars? (Resultat negativ) Was ist Massenspektrometrie? In einem Massenspektrometer werden Moleküle, die sich in der Gasphase befinden, in „energiereiche“ positive oder negative Ionen übergeführt =>Ionisierung Die gebildeten Ionen können ganz oder teilweise in geladene und ungeladene Bruchstücke zerfallen =>Fragmentierung Die geladenen Teilchen werden durch ein elektrisches Feld beschleunigt und nach ihrer Masse/Ladungszahl im Massenanalysator aufgetrennt und detektiert =>Ionen-Trennung / Massenanalyse Im resultierenden Massenspektrum sind die Massen/Ladungs-zahl-Werte(x-Achse) der Ionen gegen ihre Intensität(y-Achse)dargestellt=>Information über die Probe Chemische Informationen erhältlich mittels MS Informationstypen: 2 AC III – Teil 2 Allmaier Martin Prießner Das Massenspektrum: wird üblicherweise als Strichspektrum dargestellt, wobei die einzelnen Peaks jeweils auf ganzzahligen Massen aufgetragen werden. Den intensivsten Peak bezeichnet man als Basispeak, dessen Höhe wird willkürlich mit 100 angesetzt, alle anderen Peaks werden relativ dazu dargestellt. Dadurch erreicht man eine gut vergleichbare Darstellung der Spektren. Der Basispeak ist nur bei sehr stabilen Verbindungen (wie z. B. polyzyklische Aromaten) gleichzeitig auch der Molekülpeak. Verbindungen die weniger stabil sind, haben oft nur einen kleinen Molekülpeak, bei manchen Substanzklassen kann der Molekülpeak überhaupt fehlen. Da die meisten natürlich vorkommenden Elemente aus einem Isotopengemisch bestehen, findet man neben jedem Fragmentpeak die jeweiligen Isotopenpeaks. Aus dem Intensitätsverhältnis der Isotopenpeaks kann man Rückschlüsse auf die Summenformel einer Verbindung ziehen. (Teach/Me) Der Schemenhafte Aufbau: Auf Grund von überschüssiger Energie kann es zu einer Fragmentierung des Molekülions kommen. Ionen werden beschleunigt und in den Massenanalysator eingebracht. Dort werden sie gemäß Masse zu Ladungszahl getrennt und vom Ionendetektor detektiert. – Signalverarbeitung – Datenausgabe. 1 2 Probe wird über ein Einlasssystem in Ionenquelle transferiert. Dabei gibt es 2 Arten: • Wenn der Analyt in der Gasphase vorhanden ist, muss er nur noch in der Ionenquelle ionisiert werden 1. Kann im Hochvakuum passieren • Wenn flüssig oder fest: kommt es zur Desorption (von 2. Muss im Hochvakuum passieren fest/flüssig in die Gasphase) und Ionisation in einem Schritt Was geschieht mit den Analytmolekülen in einem Elektronenstoßionisations-(EI) massenspektrometer? (EI = Elektroenstoßionisations) 3 AC III – Teil 2 Allmaier Martin Prießner Ionengenerierung mittels Elektronenbeschuss der gasförmiger Moleküle M: + e- →M+•+ 2eM+•→F+ (EE, gerade Elektronenzahl)+ N•(homolytische Spaltung, energetisch bevorzugt) M+•→F+• (OE) + N (gerade Elektronenzahl) (heterolytische Spaltung, bevorzugt bei stabilem Neutralteilchen) Homolytische Spaltung ist energetisch bevorzugt. Detektiert wird nur das geladene Teilchen (Fragmention) Neutralteilchen sind nur indirekt über Massendifferenz messbar. Heterolytische Spaltung findet speziell dann statt, wenn stabile Neutralteilchen (z.B. CO, CO2) abgespalten werden. 3 Methoden der Trennung der Ionen nach Masse/Ladungszahlverhältnis (m/z) basierend auf folgenden physikalischen Prinzipien (J.J. Thomson verwendet) • Ablenkung von Ionen in elektrischen oder magnetischen Feldern (Sektorfeldgeräte) • Filterung von Ionen in elektrischen Wechselfeldern (Quadrupolmassenfilter, Ionenfalle, Ionencyclotron-Resonanz-Analysator) • Trennung aufgrund unterschiedlicher Flugzeiten von Ionen im feldfreien Raum (Flugzeit-Analysatoren) Detektion der getrennten Ionen • Ortsabhängige Detektion o Fotoplatten (J.J. Thomson verwendet. Interpretation schwierig. Konnte trotzdem einige Isotope dadurch nachweisen Ne20, Ne22,) o Array-Detektoren (Jeder Pixel der Fotoplatte wird durch einen eigenen Detektor ersetzt. Effizient aber Teuer => seltener) o Mehrere Detektoren für eine begrenzte Anzahl zu registrierender Ionensorten • Zeitabhängige Detektion o Faraday-Auffänger o Szintillationsdetektor o Sekundärelektronen-vervielfacher (kontinuierlich oder diskontinuierlich) (am häufigsten heute verwendet) an 4 AC III – Teil 2 Allmaier Martin Prießner Datensystem (Aussage: ohne PC geht nichts) • • • Datenerfassung(Sekundärelektronen liefert) Analoge Signal in ein digitale Signale umwandeln Datenmengenreduktion (Daten von Hintergrundionen, Rauschen eleminieren) Datenbearbeitung Normierung auf den Basispeak Subtraktion von BackgroundIonenDateninterpretation (Fragmentierung) Steuerung Instrumentkontrolle Optimierung der Messbedingungen (Computer steuert alles, nur mit PC möglich) Wie bekommt man die prozentuellen Werte? Alle Signalintensitäten werden summiert (von 290 bis 420 = 100%) und daran werden alle Peaks prozentuell verglichen. Z.B. Basispeak = 19% der Gesamtintensität. Dies ist praktisch um Isomere Verbindung zu differenzieren. 3.) EI Massenspektrum von Aceton im Strichspektrumformat Peak mit größter Intensität (=100% bezogen auf relative Intensität) wird „Basispeak“ genannt. Alle anderen Peaks (Fragmentpeaks) werden dann relativ zu dem Basispeak gesehen. Relative Intensität: ist notwendig um Spektren besser vergleichen zu können. Basispeak kann auch ein Fragmention sein, falls dieses die größte Intensität besitzt. Deshalb orientiert sich die relative Intensität in diesem Fall nach dem Fragmention [M-15]+ (Abspaltung einer CH3 Gruppe). Strichspektren sind leicht in Tabellen zu überführen, welche in großen Datenbanken gespeichert werden. 5 AC III – Teil 2 Allmaier Propionaldehyd: besteht aus: • Basispeak Fragmention • 1 Molekülpeak • Kleinen Isotopenpeak 2 • (1.) [M-1]+ Fragmention – entsteht dadurch, dass Verbindung zwischen C-H bricht (labile Bindungen) – führt zur Abgabe eines Radikal Wasserstoffatoms + Fragmentkation • (2.) Spaltung zwischen C-C Bindung bricht. CH3CH2 Radikal kann nicht detektiert werden. OCH Kation mit großer Intensität detektierbar. => C-C Bindung bricht leichter als die C-H Bindung. Durch solche Gesetzmäßigkeiten kann man die Strukturen analysieren. Martin Prießner Molekülion bei 32% Konversionsvorgänge, die in der MS nötig sind Verdampfen durch zuführen thermischer Energie. Manche organische Stoffe sind nicht hitzebeständig. Sie würden pyrolisieren – das bedeutet, dass sie nicht in richtiger Form in der Gasphase vorliegen. Dann muss man eine Desorption durchführen (viel Energie in kurzer Zeit zuführen, damit die Überführung von der Festen in die Gasphase möglich werden kann. Dabei kommt es nicht zur Pyrolyse (Def: thermische Zersetzung von organischen Verbindungen (Wikipedia)). Warum müssen Analyten geladene Teilchen sein? • Der Analyt muss als Ion vorliegen, da er durch die Potentialdifferenz beschleunigt werden muss (anlegen einer Spannung.) • In MS können nur geladene Teilchen detektiert werden • Einige wenige Verbindungen sind von Natur aus geladen, die meisten müssen jedoch ionisiert werden. 1 6 AC III – Teil 2 Allmaier Martin Prießner Distanz zwischen Kollisionen (Mittlere freie Weglänge, MFW) vs. Druck MFW hängt indirekt mit dem Druck zusammen. (siehe Tabelle) Wenn der Druck sinkt steigt die mittlere freie Weglänge. Meistens wird 10^-4 Torr verwendet. Druck bei verschiedenen Ionisations-und D/I-Arten (Ionenquelle) SIMS: Sekondär-IonenMassenspektrometrie Es gibt auch SIMS Verfahren welche auch bei ATM funktionieren. Vakuumanforderungen verschiedenster massenspektrometrischer Analysatoren Warum ist so starkes Vakuum bei ICR/Orbitrap und Magnetfeld notwendig? Ionen durchlaufen eine kreisförmige Bahn (1000x) deshalb legen sie eine lange Strecke zurück – ist nur möglich (ohne mit anderen Teilchen zusammenzustoßen), wenn sehr starkes Vakuum vorherrscht. Warum ist Hochvakuum im Massenanalysator und in Ionendetektor notwendig? Beim Massenanalysator muss das Ion eine gewisse Wegstrecke zurücklegen. Da in der Atmosphäre Teilchen (N2, O2, H2O,…) vorhanden sind, würde es beim durchqueren von den Teilchen stark abgelenkt werden und somit nicht den Detektor erreichen. Molekül könnte auch zerfallen oder reagieren. Im Vakuum kommt es zu keiner Kollision. Warum wird ein Hochvakuum in der MS benötigt? • Ermöglichung der Generierung von freien Ionen und Elektronen in der Gasphase • Verhinderung von Zusammenstößen zwischen den Ionen und von Ionen mit anderen Teilchen (diese Reaktionen würden zu einem komplizierten Massenspektrum führen) 7 AC III – Teil 2 Allmaier • • • Martin Prießner Erhöhung der Empfindlichkeit (Stickstoff-und Sauerstoff-Ionen aus der Luft würden den Detektor übersättigen und dies würde die Gesamtempfindlichkeit des Detektors herabsenken) Erhöhung der Lebensdauer der bei EI-und CI-Messungen eingesetzten Glühkathode Verhinderung der Verschmutzung von Oberflächen durch Kondensation von Probenkomponenten und Schutz der inneren Oberflächen des Gerätes vor Korrosion Anordnung eines MS Pumpsystems 2 Möglichkeiten Vakuum zu erzeugen: 1. Mit einer leistungsfähiger Pumpe 2. Mit zwei Pumpen gestaffelt: 3 Kammern: 1. Normaldruck, 2. Schwaches Vakuum, 3.Hochvakkum. In den Kammern wird zweistufig ein Vakuum erzeugt (günstiger) Pumpsysteme für unterschiedliche Druckbereiche Vakuum kann in drei Druckbereiche unterteilt werden: • Grobvakuum, • Hochvakuum, • ultra Hochvakuum; Für Grobvakuum wird eine „mechanische Rotationspumpe“ verwendet. Für Hochvakuum werden Diffusionspumpen, Turbo(- molekular) Pumpe verwendet. Bis 10^-10 Torr Aufbau eines HV und UHV Vakuumsystems Vorvakuumpumpe generiert zuerst ein Grobvakuum. Danach schaltet man die Hochvakuumpumpe (10^-6 Torr) dazu. Es werden 2 Druckmessgeräte dazwischen geschalten da ein einzelnes Messgerät nicht so niedrige Drücke messen kann. 8 AC III – Teil 2 Allmaier Martin Prießner Prinzip einer Öldrehschieberpumpe für ein Grobvakuum (bis 10^-3Torr) (ist eine Vorvakuumpumpe) Funktion: Für die Erzeugung des Vor- oder Grobvakuums, das man benötigt um die Hochvakuumpumpen betreiben zu können, verwendet man meist eine in Öl gelagerte Drehschieberpumpe (engl. oil sealed pump). Diese besteht aus einem metallischen Hohlzylinder in dem sich ein exzentrisch gelagerter Vollzylinder dreht. Zwei Schieber, die durch eine Feder auseinandergedrückt werden, gleiten entlang der Wand. Dabei schieben sie die am Saugstutzen eintretende Luft vor sich her und geben sie schließlich über das ölüberlagerte Ventil nach außen ab. Das erreichbare Endvakuum beträgt etwa 1 mbar. Normalerweise schaltet man zwei oder drei dieser Pumpen nacheinander (integriert in ein gemeinsames Gehäuse als mehrstufige Pumpe), um ein Endvakuum von 10-2 bis 10-4 mbar zu erreichen. (Teach/Me – Analytische Chemie) Hochvakuumpumpen in der MS Querschnitt durch eine moderne Turbomolekularpumpe Turbomolekularpumpen (oder auch kurz Turbopumpen) sind für den Hochvakuumbereich geeignet und haben einen Arbeitsbereich von 10-1 mbar bis ca. 10-9 mbar. Das Prinzip der Turbomolekularpumpe besteht darin, Gasteilchen durch schnell bewegte, rotierende Oberflächen in die gewünschte Richtung zu bewegen. Wenn die Gasmoleküle mit der Oberfläche des Rotors zusammenstoßen, werden sie beschleunigt und zum Ansaugstutzen der Vorvakuumpumpe befördert. Turbopumpen sind mehrstufig aufgebaut, wobei moderne Designs wie Axialkompressoren arbeiten. (Teach/Me Instrumentelle Analytik) Öldiffusionspumpe Ein Treibmittel (meist Öl oder Quecksilber) wird erhitzt und verdampft. Der Dampf steigt auf und tritt aus den Düsen als Dampfstrahl mit hoher Geschwindigkeit aus und erzeugt einen schirmartigen Treibmittelfluss, der die von oben eindiffundierenden Gasmoleküle mitreißt. Der Dampf kondensiert schließlich an der gekühlten Außenwand. Diffusionspumpen sind mehrstufig gebaut. Das Saugvermögen von Diffusionspumpen ist von der Art des abzupumpenden Gases abhängig. Gase mit niedrigem Molekulargewicht werden schneller abgepumpt als Gase mit hohem Molekulargewicht. Um das Treibmittel nicht in den Rezipienten gelangen zu lassen, wird oft eine Kühlfalle zwischen Pumpe und Rezipienten geschaltet. Bei entsprechendem Aufbau kann ein Enddruck von bis zu 10-7 mbar erreicht werden. 9 AC III – Teil 2 Allmaier Martin Prießner Allgemeine Kriterien zur Beurteilung von Massenspektrometern • • • • • • • • Auflösung (Ist ausschlaggebend dafür, wie gut man Ionen bei unterschiedlicher Masse zu Ladungszahl trennen kann. R = m/∆m ( R … Auflösung)) Transmission (gibt an, wie viele Ionen zum Detektor kommen (in Prozent)) Massengenauigkeit (setzt sich aus 2 Dinge zusammen: Massenrichtigkeit (wie richtig der gefunden Wert zum theoretischen Wert passt) Massenpräzision (wie präzise die Messung ist)) Massenbereich Empfindlichkeit (LOD) (: Limit of Detection (wie empfindlich kann ich messen?) Linearität (LOQ): Limit of Quantification (in welchem Bereich ist der Response direkt proportional zur Menge des Analyten) Scangeschwindigkeit Tandem MS bzw. Mehrstufen MS Fähigkeit Einige wichtige MS relevante Termini • Molekülion • Protoniertes (De) Molekülion • Quasimolekülion • Vorläuferion • Fragmention • • • • • Molekülmasse: Nominelle Masse (C 12) Durchschnittliche Masse (C 12.011) Monoisotope Masse (C 12.0000) Molekülion ([M]+): M: + e- → M+• + 2eEI: Durch WW der Stoßelektronen mit dem Probenmolekül M entsteht ein Molekülion M+• (Radikalkation), dass die gleiche Molekularmasse wie das Molekül hat. Protoniertes Molekülion ([M+H]+): CI: Durch Kollision der Probenmoleküle mit dem durch einen Elektronenstrahl ionisierten Reaktandgas erfolgt eine Anlagerung eines Proton an das Molekülion. Quasi-Molekülion: CI: Es können nicht nur Protonen angelagert werden, sondern auch andere Kationen, wie etwa Na+, die immer vorhanden sind. [M-H]+: CI: Es können auch Protonen vom neutralen Analyt-Molekül auf das kationische Reaktandgas-Molekül übertragen werden. Am Beispiel Methan: CH5+ + M [M-H]+ + CH4 + H2 Fragmention: Wenn die zugeführte Energie die zur Ionisation des Moleküls notwendige Energie übersteigt, zerfällt das gebildete Radikalkation durch den Überschuss in Fragmentionen. 10 AC III – Teil 2 Allmaier Martin Prießner M+• → F+ + N• Radikalkation zerfällt durch homolytische Bindungsspaltung in ein detektierbares Kation und ein radikalisches Neutralteilchen. M+•→F+• + N Radikalkation spaltet eine kleines, stabiles Neutralteilchen durch heterolytische Bindungsspaltung ab, zurück bleibt wieder ein detektierbares Radikalkation. Vor der Bindungsspaltung oder Abspaltung eines Neutralteilchens kann es auch innerhalb des Molkülions zu einer Umlagerung kommen, falls der dafür benötigte Energiebedarf geringer als für die Fragmentierung selbst ist. [M+nH]n+: mehrfach protonierte Molekülionen entstehen vor allem bei der Elektronensprayionisation großer Moleküle, die mehrere positive Ladungen an ihrer Oberfläche stabilisieren können. m/z: Masse-zu-Ladung-Verhältnis, wird in einem Massenspektrum auf der x-Achse gegen die Intensität aufgetragen. Basispeak: Das intensivste Signal im Spektrum, dem willkürlich eine Intensität von 100% zugeordnet wird. Relative Intensität: die Intensitäten der Peaks werden relativ zu dem willkürlich auf 100% gesetzten Basispeak aufgetragen. Monoisotopes MG: wird durch Addition der exakten Atommasse der am häufigsten vorkommenden Isotope berechnet. Durchschnittliches MG: wird durch Addition der durchschnittlichen Atommasse aller Isotope berechnet. Nominelles MG: wird durch Addition der auf ganzzahlige Werte gerundeten Atommasse der häufigsten Isotope berechnet. Durchschnittliche und monoisotope m/z Werte 5 x Alanin: 1. mit berechnetem 1 Monoisotopen Werte. Durchschnittliche und exakte Masse passen mit dem Basispeak überein. 50 x Alanin: 2. breite Isotopenverteilung. Maximum der „Isotopeneinhüllenden“ ist nahe 2 der durchschnittlichen Molekülmasse. Je größer die Molekülmasse desto besser passt die durchschnittliche Masse für die Berechnung (Monoisotope 3 11 AC III – Teil 2 Allmaier Martin Prießner Masse ist dann nicht sehr hilfreich) (Grafik 3: Je größer der M/Z Wert desto großer wird die Diskrepanz. (3-4 Dalton) –bis MM 1800 kann mit monoisotopen Massen gerechnet werden. Einlasssysteme • für Gase/Flüssigkeiten zur direkten Ionisation (z.B. via GC) • für Flüssigkeiten/Feststoffe zur Desorption und Ionisation (in einem Schritt) • als Lösung via HPLC oder CE(Kapillarelektrophorese) (z.B. ESI) (Elektrosprayionisation) • als Feststoff oder in schwer verdampfbare Lösung via Probenträger (z.B. MALDI) Einlasssysteme: a) Direkter Einlass – für feste und flüssige Proben Die Probe wird mit Hilfe einer speziellen beheiz- und kühlbaren Schubstange in die Ionenquelle eingebracht. Die Probe wird zuerst in eine Probenschleuse vor der Ionenquelle eingeführt, die mittels Vorvakuumpumpe evakuiert ist. Erst danach wird der Verschlussdeckel zum Hochvakuumsystem der Ionenquelle geöffnet und der Träger eingeschoben. Aufgrund des geringen Drucks gehen leichter flüchtige Proben von selbst in die Gasphase über, oder der Träger wird elektrisch aufgeheizt bis die Probe verdampft. (Für sehr leicht flüchtige Substanzen, die schon im Vorvakuum in die Gasphase wechseln würden, wird die Schubstande gekühlt) Die Heizung wird über ein spezielles Temperaturprogramm gesteuert: i) kontinuierliche Aufheizung = linearer Temperaturgradient (alle Komponenten einer unbekannten Probe, d.h. mit unbekanntem Siedepunkt sollen in die Gasphase übergehen) ii) diskontinuierliche Aufheizung = Stufentemperaturgradienten (langsames Verdampfen einer Komponente deren Siedepunkt bekannt ist, d.h. die Temperatur bleibt über längere Zeit konstant auf der Siedetemperatur bevor zur nächsten Stufe der Komponente mit dem höheren Siedepunkt aufgeheizt wird) 12 AC III – Teil 2 Allmaier Martin Prießner b) Indirekter Einlass (flüssige und gasförmige Proben) An der Ionenquelle befindet sich ein heizbares Vorratsgefäß mit einem Volumen von etwa 1 cm3. Dieses Gefäß wird durch Öffnen von Ventil 1 mit Hilfe einer Vorvakuumpumpe evakuiert. Nach dem Schließen des Ventils wird die flüssige oder gasförmige Probe durch ein Septum mit einer Spritze eingebracht. Die Temperatur des Vorratsgefäßes wird im Bereich von 20°C bis 250°C so eingestellt, dass der Dampfdruck der Substanz etwa 0,1 Pa beträgt. Dann wird die Probe durch Öffnen des Ventils 2 in den Hochvakuumbereich der Ionenquelle überführt. Der Substanzstrom bleibt über längere Zeit (einige Stunden) konstant, wenn das Vorratsgefäß genügend groß ist. Nach Beendigung der Messung wird das Ventil 2 geschlossen und das Vorratsgefäß durch Öffnen von Ventil 1 erneut unter Vorvakuum gesetzt. Dabei werden die restlichen Probenmoleküle abgepumpt. 2 Beispiele indirekter Probeneinlass (links, Erdgas und rechts, leicht flüchtige Flüssigkeit) Tetrachlorethylen DCI Emitterschubstange (direct chemical ionisation) An der Spitze der Schubstange befindet sich ein beheizbarer Draht. Auf diesem wird die gelöste Probe draufgegeben und in die Ionenquelle eingebracht dort aufgeheizt, wobei es zur Desorption kommt (Überführung in die Gasphase) (Problem: Pyrolyse). Anwendungsmöglichkeiten: Nicht viel (eines z.B. C60 Fullerene oder Fulleren mit Molekülfulleren Kation (ein Na befindet sich im Zentrum des Fullerens). Es ist leicht bestimmbar ob Na vorliegt – da sich die Masse von 720 (+ 23) auf 743 vergrößert. 13 AC III – Teil 2 Allmaier Martin Prießner Wie viel Probenmaterial wird durchschnittlich für massenspektrometrische Analysen benötigt? Indirekter Probeneinlass benötigt viel Probe! Generierung von Ionen für die massenspektrometrische Analyse • Gasphasenionisationstechniken „harte Technik • EI, CI, FI und API(FI – Feldionisation, API - „atmosphere pressure ionisation“, CI – Chemische Ionisation) • Desorption/Ionisationstechniken „weiche Technik“ • SIMS (Sekundärionenmassenspektrometrie)-Techniken • Spray-Techniken Weiche und Harte Ionenquellen: Ionenquellen werden oft als Hart oder weich kategorisiert. Hauptquelle des harten Typs ist der Elektronenstoß. Harte Quellen geben hohe Energien an die gebildeten Ionen ab und versetzen diese in hochangeregte Schwingungs- und Rotationszustünde. Die Relaxation dieser Ionen wird von einem beträchtlichen Anteil an Fragmentierung begleitet, aus dem sich komplexe Massenspektren ergeben. Im Gegensatz dazu erzeugen weiche Quellen, etwa chemische Ionisierung oder Desorption, relativ geringe angeregte Ionen. Daher tritt auch nur geringe Fragmentierung auf, und die Spektren sind einfach. Beide Spektrentypen sind brauchbar. Das einfache Spektrum weicher Quellen erlaubt die schnelle Bestimmung der Molekularmasse des Analyte. Das komplexere Spektralmuster harter Quellen erlaubt dagegen oft die zweifelsfreie Identifizierung eines Analyten. (Skoog Leary – Instrumentelle Analytik S. 473) Gasphasenionisationstechniken • Elektronenstoßionisation (EI) • Chemische Ionisation (CI) (relevant) • Feldionisation (FI) • Atmospheric pressure ionization (API) (Nicht so relevant) Gasphasenionisations- und Desorptionstechniken unterscheiden sich wesentlich in ihren Prinzipien: In der Gasphasenionisation, welche auch als „harte Technik“ bekannt ist, werden aus neutralen Molekülen Elektronen herausgeschlagen, was zu Ionisierung und anschließenden Fragmentierung führt. Die Ionen und Fragmentionen können dann in einem Analysator detektiert werden. Gasphasenionisationsquellen werden oft als hart oder weich kategorisiert. 14 AC III – Teil 2 Allmaier Martin Prießner Hauptquelle des harten Typs ist die Elektronenstoßionisation. Harte Quellen geben hohe Energien an die gebildeten Ionen ab und versetzen diese in hochangeregte Schwingungs- und Rotationszustände. Die Relaxation dieser Ionen wird von einem beträchtlichen Anteil an Fragmentierung begleitet, aus dem sich komplexe Massenspektren ergeben. Das komplexere Spektralmuster harter Quellen erlaubt oft die zweifelsfreie Identifizierung eines Analyten. Im Gegensatz dazu erzeugen weiche Quellen, etwa chemische Ionisierung, relativ geringe angeregte Ionen. Daher tritt auch nur geringe Fragmentierung auf, und die Spektren sind einfach. Beide Spektrentypen sind brauchbar. Das einfache Spektrum weicher Quellen erlaubt die schnelle Bestimmung der Molekularmasse des Analyten. Aufbau eines MS. Desorption/Ionisationstechniken, auch bekannt als „weiche Technik“, teilt sich in zwei Gebiete: SIMS-Technik SIMS hat sich sowohl zur Bestimmung der atomaren als auch molekularen Zusammensetzung fester Oberflächen bewährt. Die Technik beruht darauf, dass einer Oberfläche einer Probe mit einem 5-20 keV Ionenstrahl (z.B. aus Cs+, Ar+, diese werden in einer Ionenkanone gebildet) beschossen wird. Durch den Aufprall dieser positiven Primärionen wird die Atomlage an der Oberfläche der Probe abgetragen, wobei sich auch Sekundärionen entstehen die zur Analyse in das Spektrometer überführt werden. Der Energietransfer erfolgt in sehr kurzer Zeit (Picosekunden). Vorteil gegenüber thermischer Energie: Thermische Energie hat eine sehr lange Übertragungszeiten. Bei dieser Technik kommt es dadurch zu keinen kovalenten Bindungsbrüchen z.B. durch Laserstrahl. Spraybasierende Desorption/Ionisationstechniken Kapillare mit Probelösung Elektrisch: Zwischen Kapillare und einer Gegenelektrode wird Spannung anlegen (einige kV). Durch diese Spannung entstehen Aerosoltröpfchen, die die Analytmoleküle enthalten. Pneumatisch: Gas wird mit hoher 15 AC III – Teil 2 Allmaier Martin Prießner Geschwindigkeit koaxial oder ortogonal vorbeigeleitet, dabei werden auch Aeroloströpfchen gebildet (nm bis Mikrometer) SIMS basierende Desorption/Ionisationstechniken Die drei roten sind wichtig. Spraybasierende Desorption/Ionisationstechniken Gasphasenionisationstechniken • Elektronenstoßionisation (EI) (electron impact) • • • Chemische Ionisation (CI) Feldionisation (FI) Atmospheric pressure ionization (API) Elektronenstoßionisation Elektronenstoßionisation (engl. electron impact, EI) ist die älteste und am meisten verwendete Ionisierungstechnik. Dabei wird ein Elektronenstrahl durch die (im Hochvakuum) verdampfte Probe geschickt (Druck von 10-2 bis 10-5 mbar). Bei der Kollision der Elektronen mit den Probenmolekülen werden aus den neutralen Molekülen Elektronen herausgeschlagen und führen zu einer Ionisierung der Moleküle (Radikalkationen mit ungerader Elektronenzahl). Die ionisierten können je nach Stabilität der Moleküle und abhängig von der Energie der Elektronen weiter zerfallen und Fragmentionen bilden. 16 AC III – Teil 2 Allmaier Martin Prießner Verwendet man eine genau definierte Elektronenenergie (typischerweise 70 eV), so ist die Art und Häufigkeit der Fragmentionen sehr gut reproduzierbar. Senkt man die Elektronenenergie, so werden zunehmend weniger Fragmentionen gebildet, bis nur mehr Molekülionen auftreten. Unterschreitet man die Ionisierungsenergie des Moleküls (ca. 10 eV) unterbleibt die Ionisierung. EI ist deshalb die am meisten genutzte Ionisierungstechnik, weil eine sehr große Zahl organischer Verbindungen durch EI ionisiert werden können. Als Grenze für die Einsetzbarkeit der EI gilt, dass die betreffende Substanz zumindest einen Dampfdruck von 10-6 Torr aufweisen sollte. Um diesen Dampfdruck zu erreichen, kann die Probe auch aufgeheizt werden (bis ca. 300 Grad Celsius). Allerdings können thermisch labile Substanzen dabei eventuell zerfallen. Die Zahl der gebildeten Molekülionen im Vergleich zu den Fragmentionen, kann durch Änderung der Elektronenenergie verändert werden. Dieser Effekt kann auch ausgenützt werden um Molekülionen zu erkennen. Nach der Ionisierung werden die gebildeten Ionen durch eine Extraktionselektrode mit einer Spannung von wenigen Volt aus der Ionisationskammer abgesaugt und beschleunigt. Der Strahl wird mit elektrischen Blenden fokussiert, so dass die Dichte des Strahls möglichst gleichmäßig ist. Damit bekommen die Ionen eine konstante kinetische Energie, mit der sie in den Analysator eintreten. Die auftretende Unschärfe der kinetischen Energie ist entscheidend für die mögliche Auflösung des Instruments. (Teach/Me) Elektronstoßionisation von organischen Molekülen M: →M+•+ e M+•→F1++ N1• M+•→F2+•+ N2 Organische gasförmige Molekül interagiert mit dem Elektronenstrahl. Dadurch entsteht ein Radikalkation. Dieses Radikalkation kann kovalente Bindungen im Molekül brechen – generieren Fragmente (auch neutrale Fragmente, welche nicht detektiert werden können. Z.B. H2O CO, CO2) Oder kleine Moleküle die aus großem Molekül herausbrechen können. 17 AC III – Teil 2 Allmaier Martin Prießner EI Fragmentierung organischer Moleküle 1. Es kann zu Bindungsspaltungen kommen. Alles was + geladen ist (Fragmentionen) kann detektiert werden. 2. Zeigt wenn es Neutralteilchen verliert 3. Oder es kommt zu Umlagerungen innerhalb des Moleküls. Das passiert in der Gasphase - die schlussendlich zu Fragmentionen führen. Fragmentierung läuft nach Gesetzmäßigkeiten ab. Damit kann man Fragmentierungen gut erklären (im Fall organischer Verbindungen) Typische EI Ionisationseffizienz als Funktion der Elektronenenergie Energie des Elektronenstrahls wird gegen die Ionisationseffizient aufgetragen (100% = alle Analytmoleküle werden in Ionen überführt) In der Realität sind es 1-3% . Der Rest ist neutral und stehen nicht zur Analyse zu Verfügung. Ab 10 eV kommt es zu einer kontinuierlichen Zunahme (der erste Wert bei dem Ionen gebildet) bei 50 eV max. – danach nimmt der Wert wieder ab (weil so viel überschüssige Energie führt zu Spontanfragmentierungen – deshalb nimmt die Ionisationseffizienz ab. 70 eV - optimale Ausbeute. (ein Standard für den Elektronenstrahl ) Positive EI Massenspektren von 2-Aminobenzoesäure mit unterschiedlichen Elektronenstrahlenergien Mithilfe der EI können eine Vielzahl organischer Moleküle ionisiert werden, vorausgesetzt sie haben mindestens einen Dampfdruck von 10-6 Torr, d.h. sie liegen im Hochvakuum in der Gasphase vor. Das kann auch mithilfe von Erwärmung geschehen, was jedoch wiederum thermisch instabile Proben ausschließt. 18 AC III – Teil 2 Allmaier Martin Prießner Das Spektrum ändert sich entsprechend der Spannung. Wird die Energie des Ionenstrahls erhöht, kommt es zu stärkeren Fragmentierungen (ein Fragmention wird zum Basispeak) und zu einer höheren Intensität der Peaks. Die Standard-Spannung von 70eV liefert eine gute Ausbeute und Intensität, außerdem wurde durch die Vereinheitlichung der Spektren der Aufbau einer großen Datenbank mit Vergleichsmöglichkeiten möglich. Positives EI Massenspektrum eines sek. Alkohols mit praktisch keine Molekülion Bsp.: Sekundärer Alkohol Molekülion besitzt in diesem Diagramm eine relative Intensität von 0,3%. Das ist zu gering um das Gewicht bestimmen zu können. Das bedeutet, dass in diesem Fall die Anregungsenergie zu groß gewählt wurde und sich zu viele Fragmentionen gebildet haben. Massenspektren von Dipeptidderivat bei unterschiedlichen Ionenquellentemperaturen Temperatur des Ionenquellenblocks beeinflusst ebenfalls die Anzahl der gebildeten Fragmente. Beheizt wird um Ablagerungen zu verhindern. Zweites Spektrum ist nicht so aussagekräftig, weil der Molekülwert zu niedrig ist. Auch der Druck in der Ionenquelle ein wichtiger Faktor (gutes Vakuum) 19 AC III – Teil 2 Allmaier Martin Prießner Vor-und Nachteile der Elektronenstoßionisation Vorteile o Subpicomol-Empfindlichkeit(pico = 10^-12) o Riesige Datenbank ( (bei 70 eV) über 300000 Massenspektren) o Fragmentierung (Fingerabdruck eines Moleküls – bei Identifizierung sehr hilfreich) o Strukturinformation aus Fragmentierung Nachteile o Eingeschränkter Molekulargewichts-Bereich (das mögliche Molekülgewicht ist von der Flüchtigkeit abhängig) o Thermische Zersetzung während der Verdampfung o Zu starke Fragmentierung, kein Molekülion (dann erhält man keine Information über Molekulargewicht– Ausgangspunkt einer Strukturaufklärung.) Elektronenstoßquellen sind einfach in der Anwendung, liefern hohe Ionenströme und ermöglichen daher auch hohe Empfindlichkeiten. Die umfangreiche Fragmentierung und die daraus folgende hohe Signalzahl ist ebenfalls ein Vorteil, weil sie die zweifelsfeie Identifizierung des Analyten ermöglicht. Diese Fragmentierung kann jedoch auch ein Nachteil sein, wenn das Signal des Molekülions Verschwindet, und die Molekularmasse des Analyten deshalb nicht zu bestimmen ist. Eine weitere Einschränkung für den Einsatz von Elektronenstoßquellen ist der Umstand, dass die Probe verflüchtigt werden muss, was zu thermischen Abbau einiger Analyten noch vor der Ionisierung führen kann. Die thermische Zersetzung kann bisweilen dadurch minimiert werden, dass die Verdampfung von einer geheizten Sonde aus stattfindet, die nahe am Eintrittsspalt des Spektometers angebracht ist. Durch den niedrigen Durch im Quellbereich erfolgt auch die Verflüchtigung bei tieferer Temperatur. Zudem steht weniger Zeit für eine thermische Zersetzung zur Verfügung. Wie bereits zuvor angemerkt, lassen sich Elektronenstoßquellen nur bei Analyten mit Molmassen kleiner als 10^3 Dalton verwenden. (Skoog Leary: S.477) Chemische Ionisation (CI) Die chemische Ionisation findet durch Kollision der gasförmigen Atome einer Probe mit Ionen statt, die durch Elektronenbeschuss eines im Überschuss vorliegenden Reaktantgases erzeugt wurden. (Skoog Leary S. 478) (weitere Erklärung:->) Die chemische Ionisation (CI) erzeugt ionisierte Probenmoleküle nicht durch direkte Ionisation (wie bei der Elektronenstoßionisation), sondern durch Transfer von Protonen von einem Reaktandgas auf die Probe (oder durch Entzug eines Protons durch das Reaktantgas). Als Reaktantgase werden meist Methan, Isobutan und Ammoniak eingesetzt. Die entstehenden Probenmoleküle sind 20 AC III – Teil 2 Allmaier Martin Prießner Pseudomolekülionen [MH]+ mit gerader Elektronenzahl. Ein wichtiger Unterschied zwischen diesen Pseudomolekülionen und den M+ Ionen, die bei EI entstehen, besteht darin, dass bei CI die gebildeten Ionen eine geringere Überschussenergie haben und dadurch weniger stark fragmentieren. Bei der chemischen Ionisation entstehen auch negative Ionen, die man durch Umpolen der Spannungen in der Ionenquelle detektieren kann. Charakteristika der chemischen Ionisation: • einfache Spektren mit geringer Fragmentierung • hohe Empfindlichkeit • geringe Reproduzierbarkeit (dadurch keine Bibliothekssuche möglich) (Teach/Me) Chemische Ionisation von organischen Molekülen CH4 + e → CH4+• + 2e CH4+• + CH4 → CH5+ + CH3• CH5++ M → MH+ + CH4 CH5++ M → [M-H]++ CH4 + H2 C2H5++ M → [M-H]++ C2H6 Nebenreaktionen Chemische Ionisation von organischen Molekülen mit komplexer Adduktbildung CH4 +•→CH3++ H• CH3++ CH4 →C2H5++ H2 C2H5++ M →MH++ C2H4 C2H5++ M →MC2H5+ Massenspektren der Aminosäure Prolin (oben, EI; unten, CI) 1 2 Prolinmethabolismus: Untersuchung bei gesunden und kranken Menschen. Es wurde qualitativ und quantitativ das Plasma untersucht. 1. Aminosäure wurde als Reinsubstanz eingebracht. Es konnte jedoch kein Molekülion generiert werden (bei EI zu starke Fragmentierung) 2. Bei CI Verwendung (mit Methan) sehr intensives Molekülion generierbar. Molekülion um eins größer. Außerdem bilden sich 2 Fragmentionen. 21 AC III – Teil 2 Allmaier Martin Prießner Typisches CI Massenspektrum mit Methan An diesem Beispiel ist auch ersichtlich, dass ebenfalls Moleküle mit einem größeren M/Z Wert (als das Molekülion) generiert werden können. Erklärung: Durch eine Nebenreaktion lagert sich eine Verbindung an das neutrale M an (M+29). Durch die Massendifferenz der 2 Peaks kann man einfach dieses zusätzliche Ion bestimmen werden In Abhängigkeit vom verwendeten Reaktandgas erhält man ebenfalls unterschiedliche Spektren: Resultate klar zu sehen. Reaktantgas Methan und Isobutan. 1. Basispeak Fragmention. 2. Basispeak das Molekülions (hat eine erhöhte Signalintensität) Methan: • gut geeignet für die meisten organischen Verbindungen • stärkere Fragmentierung der Probenmoleküle • erzeugt [MH]+, [MCH3]+ Addukt-Ionen • die Addukt-Ionen sind nicht bei allen Substanzen gut ausgeprägt Isobutan: • erzeugt [MH]+ und [MC4H9]+ Addukt-Ionen • eher geringere Fragmentierung • die Addukt-Ionen sind nicht deutlich besser ausgeprägt als bei Methan • nicht so universell einsetzbar wie Methan (Teach/Me) 22 AC III – Teil 2 Allmaier Martin Prießner Bei diesem Beispiel wurde Butylmethylacrylat in gesättigten KW auf verschiedene Arten untersucht. Dieser Stoff ist für die Polymerherstellung sehr wichtig. Fragestellung: Wie viel freies BM ist noch vorhanden? 1. Vielzahl von anderen Signalen stören bei der Bestimmung der Retentionszeit. Außerdem ist die Intensität sehr niedrig und dadurch auch schwer bestimmbar. 2. Bessere Signalintensität als bei 1 3. Alle Verunreinigungen werden nur schlecht ionisiert – gut für die Quantifizierung. Durch Wechsel des Reaktantgases kann man die Selektivität beeinflussen. SIMS basierende Desorption/Ionisationstechniken Fast Atom Bombardment (FAB) und Liquid Sekundärionenmassenspektrometrie (LSIMS) Vakuum Matrixunterstützte Laserdesorption/ionization (MALDI) Atmospheric Pressure Matrixunterstützte Laserdesorption/ionization (AP MALDI) Desorption Electrospray Ionization (DESI) In den 70er Jahre entwickelte sich die SIMS Technik. Dabei wird die Probe auf der Oberfläche mit einem hochenergentischen Primärionenstrahl beschossen. Nachteil: viel Probe wird zerstört. Lösung: Man könnte eine Matrix verwenden um den Analyten zu puffern. Erste Technik die sich somit entwickelte war FAB (Primärionenstrahl mit Xenonteilchen). (LSIMS mit Cesium als Primärionenstrahl) 3. Maldi mit Flugzeitmassenspektroskopie. Konnte sehr große Moleküle untersuchen. 4. AP Maldi werden mit IonenfallenMS oder auch bei FlugzeitMS verwendet. 23 AC III – Teil 2 Allmaier Martin Prießner Fast Atom Bombardment (In der Vorlesung wurde nicht so genau darauf eingegangen) (FAB) ist eine Technik um schwerflüchtige und/oder thermisch labile Substanzen (z.B. Peptide) ionisieren zu können. FAB gehörte zu den historisch gesehen ersten Ionisationsmethoden, die für Substanzen verwendet werden konnte, bei denen Elektronenstoßionisation oder chemische Ionisation nicht funktionieren. Am besten funktioniert FAB bei polaren Substanzen mit höherem Molekulargewicht, wie z.B. Peptiden, ionischen Organometallverbindungen, oder Biomolekülen. Generell gehört FAB zu den besten Ionisierungsmethoden für ionische Teilchen. Das Prinzip von FAB beruht auf dem Beschuss der Probe mit schnellen, elektrisch neutralen Atomen (z.B. Argon oder Xenon). Dazu wird die Probe in einem Tropfen Glyzerin aufgelöst. Durch den Beschuss mit dem Neutralteilchenstrahl werden Probenmoleküle aus der Matrix geschlagen und ionisiert. Die Vorgänge bei der Ionisation sind ziemlich komplex und nur teilweise bekannt. Neben den unvermeidlichen Fragmenten der Matrixmoleküle, bilden sich typischerweise meist Pseudomolekülionen [MH]+ und eher weniger Fragmente aus. Bei Anwesenheit von Salzen können sich auch die entsprechenden schwereren Adduktionen ausbilden (z.B. [MNa]+). Außerdem beobachtet man sehr oft Clusterbildungen, wo mehrere Moleküle zusammen "kleben" und mit einer gemeinsamen Ladung bei der entsprechenden höheren Masse detektiert werden. Ein wichtiger Aspekt bei der FAB-Ionisation ist die Auswahl einer geeigneten Matrix (meist Glyzerin, Thioglyzerin, oder 3-Nitrobenzyl-Alkohol). Die Matrix sollte die Probe auflösen und die Ionisierung der Probenmoleküle unterstützen, aber keine chemische Reaktion mit der Probe eingehen, und (was sehr wichtig ist) eine geringe Flüchtigkeit aufweisen, da die Matrix sonst im Hochvakuum in Bruchteilen einer Sekunde verdampfen würde. Manchmal wird zur besseren Ionisation auch Trifluoressigsäure zur Matrix hinzugefügt. (Teach/Me) MALDI (Matrix-assisted laser desorption/ionization): Die Probenmoleküle werden mittels Laserbeschuss aus einer niedermolekularen Matrix heraus unzerstört verdampft. Dazu wird die Probenlösung mit einer konzentrierten Matrixlösung gemischt (eventuell noch Zusatz von Salzen zur Verbesserung der Ionisation) und auf einen Probenträger aufgebracht. Die Matrix muss eine Substanze mit einem Chromophor, der die Laserenergie absorbiert, enthalten. Die durch die Matrix absorbierte Laserenergie bildet ein Plasma, in dem die Probe relativ unfragmentiert ionisiert wird. Das Signal-Rausch-Verhältnis kann durch mehrmaliges Beschießen der Probe mit einem Laserpuls verbessert werden. Es werden Laser im UV- (Anregung des aromatischen π-ElektronenSystems) und im IR-Bereich (Anregung der OH-Bindungen von flüssigen oder gefrorenen Matrices) eingesetzt. Da die Ionisation schlagartig und gepulst erfolgt, muss mit ebenfalls gepulst arbeitenden Analysatoren (z.B. TOF) gearbeitet werden. Es muss außerdem eine Mindestenergie (Schwellwert) eingestrahlt werden, sonst komm es zu keiner Ionisierung. Danach bleibt die Intensität der Molekülionen relativ konstant. 24 AC III – Teil 2 Allmaier Martin Prießner Anwendung: hochmolekulare Verbindungen: synthetische und biologische Polymere Oberflächen, Proteine und Peptide Hochvakuum MALDI Konzept • Matrix absorbiert stark bei der Wellenlänge des Lasers • Die von der Matrix in extrem kurzer Zeit absorbierte Energie initiert den Phasenübergang(= Desorption) und die Ionisation (Energie wird in Pikosekunden absorbiert. Wenn Energie in so kurzer Zeit aufgenommen wird, entsteht kaum thermische Belastung für die Analytmoleküle) • Intakte Analytmoleküle werden durch die Matrixstrahlexpansion in die Gasphase übergeführt (gewisses Volumen der Matrix gelangen ebenfalls in die Gasphase. Auf diesem Weg können auch sehr große Moleküle in die Gasphase überführt werden.) • Verschiedenste parallel ablaufende Mechanismen (e-Transfer, Protonentransfer, Adduktionen-Anlagerung, ...) führen zu intakten Molekülionen, die dann in den Analysator hinein beschleunigt werden Einteilung der MALDI Technik in Abhängigkeit vom Ionenquellendruck • AP (Atmosphärendruck) MALDI: D/I bei Atmosphären-Normaldruck (unterstützt durch Stickstoffgegenstrom) • Mitteldruck MALDI: D/I bei 1 –100 mTorr • Vakuum MALDI(„KLASSISCHE FORM“): D/I im Hochvakuum (≈10^-6Torr) 1. Stickstoffgegenstrom wird verwendet da die Probe wegen zu hohen Temperaturen zerstört werden kann MALDI Volumenpräparationstechnik („Dried droplet“) Die massenspektrometrische Probenvorbereitung dient der Optimierung einer Probe für die Analyse in einem Massenspektrometer. Diese haben, je nach Ionisationsverfahren, unterschiedliche Anforderungen an Volumen, Konzentration und Zusammensetzung der Analyt-Lösung. (Wikipedia) Es soll so sein das Matrix und Analyt homogen kristallisieren. Wenn inhomogen kristallisiert gehen viele Schüsse ins Leere. 25 AC III – Teil 2 Allmaier Martin Prießner Molekulardynamische Simulation des Desorptionsvorganges in der MALDI mit 40 Jm-2 Es werden Cluster von Ionen raus geschossen. Wichtig Oberfläche sollte flach sein Sonst stimmt die Flugzeit nicht mehr überein weil sie unterschiedlich starten Positive und negative Molekülionen, die den Analyten (M) enthalten MALDI ist an verschiedene Laser gekoppelt. Wichtig dabei ist das Zusammenspiel: Wellenlänge/Laser. Dabei soll die Probe ein Absorptionsmaximum besitzen die der Frequenz des Lasers entspricht. Großes Protein: ähnliche Informationen bei beiden. Man kann nicht sagen, welches Spektrum besser ist weil eine andere Skalierung verwendet wurde. Vorteil: beim IR kann man Probe in Wasser einfrieren somit Wasser als Matrix verwenden. 26 AC III – Teil 2 Allmaier Martin Prießner Anforderungen an die Matrixlösungen • • • • • Lösungsmittelqualität (z.B. Wasser, EtOH, ACN) Matrixkonzentrationen: mg/ml bis zu gesättigten Lösungen. Molar Ratio Analyt: Matrix 1:100 -1:100 000 Ansäuern der Probe mit TFA, FA oder HCl im positiven Ionenmodus Lagerung der Matrixlösungen (kann begrenzt gelagert werden: max 1 Woche) Kristallformen zweier MALDI Matrices Manche Verbindungen kristallisieren nicht mit glatten Oberflächen sondern mit länglichen Kristallen. Dann bekommt man kaum eine brauchbare Auflösung. Negatives MALDI Massenspektrum eines Oligonukleotides p(dT)18mit der Matrix HPA Negatives MALDI Massenspektrum eines Oligonukleotides mit der Matrix norHarman 27 AC III – Teil 2 Allmaier Martin Prießner 3-D Struktur von humanem Hämoglobin, einem nichtkovalenten Heterokomplex Positives MALDI Massenspektrum von verdünntem humanem Vollblut (1/500) MALDI Massenspektrum eines Pflanzenöls (Gemisch) mit einer binären flüssigen Matrix Bsp.: mittels MALDI kann man neutrale Verbindungen messen. (Pflanzenöle neutral) durch Zugabe von Na Salz werden sie detektierbar. MALDI ist auch für kleine Moleküle geeignet Positives MALDI Massenspektrum eines technischen Polymers (Poly(methylmethacrylat)) mit der Matrix 3Indolylacrylsäure Technisches Polymer ist ein Gemisch dessen Spektrum eine Gaußverteilung hat und bei welchem der Abstand zwischen den Peaks immer gleich ist) 28 AC III – Teil 2 Allmaier Martin Prießner MALDI Massenspektren von Vitamin C mit der Matrix 2,5-DHB (oben) und dem binären, flüssigen Matrixsystem Co Nanopulver/Glycerin (unten) 2. Ascorbinsäure Vitamin C Beim ersten Diagramm ist bei 2 Peaks die Zuordnung des Fragmention nicht eindeutig zuordenbar. Um nun die richtige Antwort zu bekommen kann man eine andere Technik zum Messen verwenden oder eine andere Matrix. MALDI Massenspektrum des größten, je erfolgreich analysierten monodispersen Moleküls (Ig M, 355 nm, Matrix: Sinapinsäure Target: Stahl) Mit MALDI kann man auch sehr große Moleküle analysieren. 1 Megadalton. Auflösung sehr schlecht. Vor-und Nachteile von MALDI 29 AC III – Teil 2 Allmaier Martin Prießner Häufig gebildete Molekülionen in Vakuum MALDI Einfache Protonierung [MH]+ Statt Protonen können sich Kationen anlagern Entfernung eines Protons [M -H]- Einfache und mehrfache Kationisierung(Cat = Na, K, Ag, Cs, etc.) [M -(n-1)H + nCat]+ Desorption Electrospray Ionization (DESI) DESI (Desorption Electrospray Ionisation): Durch ESI wird ein Strahl an Nano-Teilchen erzeugt, der in einem gewissen Winkel auf die Probe aufschlägt und diese abträgt (Desorption, aber sehr schmutzig). Die gelösten AnalytMoleküle gelangen in einem gewissen Winkel in das MS und können analysiert werden. Englische Erklärung: Desorption electrospray ionization (DESI) is a desorption ionization (DI) method by nature, and, like matrix-assisted laser desorption/ionization(MALDI), it is used for the analysis of material present on a surface. DESI includes features reminiscent of electrospray ionization (ESI) in respect to both its instrumental and mechanistic aspects. However, the analyte in the DESI experiment is not in solution as in ESI. Instead, a microelectrospray ion source is used to produce charged droplets, ionic clusters, and/or gas-phase ions (depending on chosen experimental conditions), and these are directed at the sample surface. The sample ispresent in the ambient environment. An electrical potential of several kilovolts (kV) is applied to the spray solution, and pneumatic nebulization is used to assist in desolvation. Ionization of molecules present on the sample surface occurs upon the impact of the ESIoriginated, charged particles with the surface. Surfaces include deposited samples on sample holder targets as well as surfaces of natural objects such as biologicaltissues or minerals. 30 AC III – Teil 2 Allmaier Martin Prießner Wichtige Parameter bei DESI • • • • • • • Elektrosprayspannung Cluster-Lösungsmitteltyp und -Fluss Abstand Spraynadelspitze und Targetoberfläche (Punkt/Fläche des Primärteilcheneinschläge) (mm-Bereich) Abstand Targetoberfläche (Punkt/Fläche der D/I) und MS Eingang (mm-Bereich) α-und β-Winkeln des Primär-und Sekundärstrahls Temperatur am Eingang des Massenspektrometers (wichtig hält der Analyt die Temperatur aus Typ des Oberflächenmaterials Spraybasierende Desorption/Ionisationstechniken Thermospray (TS) Particle Beam (PB) Atmospheric pressure chemical ionization (APCI) Elektrosprayionisation (ESI) Nanoelektrosprayionisation (nESI) Atmospheric pressure photoionization (APPI) Bei Atmosphärendruck (braucht nicht in Hochvakuum generieren. Geht bei kleinen und hochmolekularen Verbindungen ESI - Elektrosprayionisation Schema des angenommenen elektrisch getriebenen Sprayvorganges im positiven Ionenmodus Bei der Elektrosprayionisation wird eine Analytlösung durch eine Metallkapillare geleitet, an deren Spitze eine Spannung angelegt ist. Durch die Spannung kommt es zur Bildung eines elektrischen Feldes zwischen der Kapillare und einer Gegenelektrode. Das elektrische Feld durchdringt die Analytlösung und in ihr befindliche Ionen bewegen sich elektrophoretisch auf die Gegenelektrode zu. Dabei bildet sich an der Spitze der Kapillare ein Überschuss gleichartig geladener Ionen, die sich gegenseitig abstoßen und über die Bildung eines Taylor-Kegels (Taylor-Cone) als feines Aerosol (etwa 10 µm Tropfengröße) aus der Kapillare austreten. (stabil von 2,9 bis 3,6 kV, abhängig von den Eigenschaften der Probe). Dadurch lassen sich auch sehr große Molekülionen gut detektieren, da das m/z-Verhältnis in einen Bereich verschoben wird, der sich auch mit gängigen Analysatoren erfassen lässt. Ein neutrales Trägergas wie Stickstoff wird häufig benutzt um die Vernebelung der Lösung und die Verdampfung des Lösungsmittels zu unterstützen. Aufgrund der Verdampfung des Lösungsmittels verkleinert sich die Tropfengröße, während die Dichte des elektrischen Feldes auf der TropfenOberfläche zunimmt. Wenn der Radius der Tropfen kleiner als das sogenannte Rayleigh- 31 AC III – Teil 2 Allmaier Martin Prießner Limit wird, zerfallen die Tropfen wegen der Abstoßung von gleichartigen Ladungen (Coulomb-Explosionen) in kleinere Tröpfchen. Für die Bildung freier Ionen in der Gasphase existieren mehrere Modellvorstellungen. (Wikipedia) Flussraten: ESI: 200-1000 μL/min μESI: 0,2-20 μL/min nESI: 3-20 nL/min Anwendung als Desorptions/Ionisations-Methode: Kopplung mit LC. 2 Konzepte: wie Ionen in Gasphase übergehen. 1. Charge residue model: gut geeignet um mehrfachgeladenen und große Ionen zu generieren. Die schematische Darstellung zeigt Aerosoltröpfchen (an der Oberfläche positive Ladungen) gelöst sind 3 Analytmoleküle (weiß). Es verdampft LM Durchmesser und der Tropfen wird kleiner, jedoch die Anzahl der Ladungen bleibt gleich. Ladungen kommen so knapp aneinander, dass es zu einer Coulombsche Explosion kommt. (Aerosoltröpfchen zerfallen in kleinere Aerosoltröpfchen). Am Ende hat man ein Analytmolekül (ohne LM aber 5 fach protoniert (5+)) (neutrales Molekül + 5 Protonen) /+3/+1. (Bild abgezählt) Modell gilt ab Molekulargewicht von 1000. Das Charged Residue Modell geht davon aus, dass letztlich winzige Tropfen von etwa 1 nm Durchmesser übrigbleiben, die nur ein ionisiertes Analytmolekül enthalten. (Wikipedia) 2. Konzept: „Ion evaporation model“: Passiert darauf, dass an der Oberfläche der Tröpfchen Ladung in Form von Protonen sind. Oder die Analytmoleküle sind von sich aus selbst geladen. An der Oberfläche des geladenes Analytmolekül lagert sich z.B. H2O dipolartig an. Auf der Oberfläche wirkt ein elektrisches Feld, welches angelegt wird. Dabei kann es passieren, dass ein geladenes Teilchen von der 32 AC III – Teil 2 Allmaier Martin Prießner Flüssigkeitsoberfläche in die Gasphase übertritt. Somit wird ein einfach geladene Ionen in der Gasphase generiert. MG <1000 Beim „Ion Evaporation Model“ wird angenommen, dass bereits aus größeren geladenen Tropfen freie Ionen in die Gasphase emittiert werden. Die erzeugten Ionen werden durch die Potentialdifferenz zwischen Spraykapillare und Orifice in das MS gelenkt. Gebildete Molekülionen in ESI (und nESI) Einfache und mehrfache Protonierung Einfache und mehrfache Deprotonierung Adduktbildung mit Kationen (Kationen = Li, Na, K, etc.) (sind unerwünscht) Ladungstransfer (wie bei EI) Adduktbildung mit LM (z.B. Wasser) oder organische Moleküle (OM) [MH]+ or [M + nH]n+ [M -H]-or [M -nH]n[M + nC]n+ [M -(n-1)H + nC]+ [M]+ [M + H + nH2O]+ [M + H + nOM]+ Y= Relative Intensität X= Masse zu Ladungszahl. Gemisch aus 2 Proteinen liegt vor Sichtbar: Serie von Mehrfachprotonierten Molekülionen. 8 fach bis 16fach (13 = Basispeak) Das zweite Protein erkennt man bei genauerer Betrachtung. (4 fach und 3 fach protoniertes Molekülion) Zuordnung der Protonierungszahlen funktioniert nur, wenn man weiß was drinnen ist. Wenn man das weiß dekonvoliert man das Spektrum. Man wandelt Ionen die mehrfachgeladen sind in MG um. (unteres Diagramm: Y = Molekulargewicht X = Intensität) in diesem Diagramm sind 2 33 AC III – Teil 2 Allmaier Martin Prießner Proteine mit unterschiedlichem MG sichtbar. Der Algoritmus ist auf einfachen Vorgang zurückzuführen. Berechnete Molekulargewichte aus einem ESI Massenspektrum Beispiel von oben ausgeführt. (Spalte 4 von oben: MG = 16949,9 Wenn man von allen Paaren die MG berechnet und den Durchschnitt berechnet erhält man ein sehr genaues MG des tatsächlichen Moleküls. MG unterscheiden sich minimal (Mittelwert ist gute Näherung zum wirklichen MG) Aber großer aufwand (mehr als MALDI) Bsp. eines ESI MS negativ (DNS) sind Polyanionen. (mehrfach negativ geladen) sind gut geeignet um in ESI analysiert zu werden. Im Diagramm sieht man ein 6,7 -11 fach deprotoniertes Molekülion. „Wenn Struktur passt kann man in negativen Modus gute Resultate erzielen“ Bsp. Proteinkomplex. 3D Struktur. Ausschnitt des MS im Bereich M/Z 13000 – 18000. (Verteilung von mehrfachgeladenen Ionen) Komplex kann 70 Protonen an der Oberfläche stabilisieren. Aus System kann man mit ESI das MG berechnen (MG ist 1,02 Megadalton.) Diese Methode somit kann auch sehr große Moleküle analysieren. 34 AC III – Teil 2 Allmaier Martin Prießner Vor-und Nachteile von ESI Vorteile • Subpicomol-Empfindlichkeit • Molekülion fast immer gebildet (wenn Proben in Lösung sind gelingt es fast immer Molekülion zu bildet) • Geringe Fragmentierung (sehen ausschließlich Molekül) • Molekulargewichts-Bereich bis 100 kDa (Kilodalton) Nachteile • Für apolare (wenn z.B. keine Amino-, Carboxy-gruppierungen aufweisen dann funktioniert die Ionisation nur sehr schlecht) und niedermolekulare (MG: 100 - 300) Verbindungen nicht optimal • Kaum Fragmentierung (können keinen Fingerprint herstellen) • Zeitlimitierung für MS/MS (wenn bestimmtes Volumen versprüht ist gibt es keine Möglichkeit noch andere Analysen durchzuführen) • Keine Wiederholbarkeit • Probenflussbegrenzung Vor-und Nachteile von Nano ESI Vorteile • Femtomol-Empfindlichkeit (um 3 Zehnerpotenzen bessere Empfindlichkeit) • Molekülion fast immer gebildet • Geringe Fragmentierung • Molekulargewichts-Bereich bis 100 kDa • Keine Zeitlimitierung für MS/MS (wegen der geringen Flussraten) • CZE (Kapillarzonenelekrophorese) und Nano HPLC koppelbar Nachteile • Für apolare und niedermolekulare Verbindungen nicht optimal • Kaum Fragmentierung • Keine Wiederholbarkeit • Probenflußbegrenzung • Manueller Vorgang (Herstellung der Spraykapillare / das Befüllen sehr aufwendig)(Ausnahme nESI Chip, CZE und Nano HPLC (mit Roboter – sehr teuer ~300000€)) Probeneinbringung in die Spraynadel via • Kolbenspritze (einfachste und robuste) • Fließinjektion (eine Pumpe die Permanent LM in die Spraykapillare hineinpumpt. – kontinuierliche Bildung eines Taylorkonus. In LM Strom injiziert man die Analytprobe) • Trennmethoden (z.B. HPLC, CE, ITP) 35 AC III – Teil 2 Allmaier • Martin Prießner Pipette/Injektionsspritze in nESI Spraynadel und dann Kapillarkräfte/Spannung/Druck (in der Nano ESI zu transportieren) Voraussetzung Probelösung muss partikelfrei und komplett gelöst sein, sonst wird die Spraynadel verstopft Typen von massenspektrometrischen Analysatoren • Flugzeit (Time-of-flight, TOF) • Quadrupol (Q) • Ionenfalle oder Paulfalle (Ion trap, IT) • Sektor(Magnet)feld (B) (vor 20 Jahren / heute nur noch in der Dioxinanalytik und Dopinganalyse verwendet) • Fouriertransformation-Ionencyclotron-resonanz (FT ICR) (komplexe Analysatoren nicht häufig verwendet) • Orbitrap (ist erst im Kommen) Auflösungsdefinitionen bei Massenspektrometrischen Analysatoren Beispiel: Auflösung (resolution) allgemein: ܴ = : beschreibt das Vermögen eine ∆ MS, Ionen mit unterschiedlichem m/zVerhältnis zu trennen. Der Wert von ∆݉ kann auf zwei unterschiedliche Arten bestimmt werden. Bei Angabe einer Auflösung ist daher wichtig, auch anzugeben, auf welches m/z sich der Wert bezieht und mit welcher Methode ∆݉ ermittelt wurde. R10% Tal: Bei dieser Methode betrachtet man 2 Peaks, die sich so überlappen, dass das Tal zwischen den beiden Peaks 10% der Intensität des kleineren Peaks aufweist. ∆݉ wird dann als ݉ଶ − ݉ଵ ermittelt, wobei ݉ଶ > ݉ଵ ist. R50% Tal: Wie R10% Tal nur muss das Tal 50% der Intensität ausmachen. Diese Methode wird besonders bei niederauflösenden Massenanalysatoren verwendet. RFWHM: FWHM (Full Width at Half Maximum) ist die Halbwertsbreite des Peaks, d.h. die Breite des Peaks bei 50% der Gesamthöhe. ∆݉ ist eben diese Halbwertsbreite. 36 AC III – Teil 2 Allmaier Martin Prießner Prinzip eines linearen Flugzeitanalysators (TOF) Ein Flugzeitmassenspektrometer (engl. time of flight - TOF) basiert auf der Tatsache, dass Teilchen gleicher kinetischer Energie mit unterschiedlicher Geschwindigkeit fliegen, wenn sie unterschiedliche Masse aufweisen. Beschleunigt man Ionen in einem elektrischen Feld mit der Spannung V so haben die Ionen beim Verlassen des Feldes die kinetische Energie q.V (q ist die Ladung eines Ions), die gleich ½.m.v2 sein muss: Wenn nun die Ionen unterschiedliche Masse aufweisen, müssen Sie entsprechend unterschiedliche Geschwindigkeiten haben, da ja alle anderen Größen in der Gleichung konstant sind. Damit lässt sich aus der Flugzeit der Ionen über eine bestimmte Strecke die Masse dieser Ionen bestimmen. Prinzip eines klassischen Reflektors (Ionenspiegel, RTOF) Trennung aufgrund unterschiedlicher Flugzeit im feldfreien Raum Flugzeitanalysatoren (TOF=time of flight): Die in der Ionenquelle (bevorzugter Einsatz von MALDI, da auch das TOF-Gerät im gepulsten Modus arbeitet) erzeugten Protonen werden durch einen elektrisches Impuls mit 103 bis 104 Volt (und gleicher Frequenz wie der Ionisationsimpuls) beschleunigt und treten in das feldfreie Driftrohr (1m langes Stahlrohr mit Hochvakuum) ein. Weil alle Ionen beim Eintritt in das Rohr die gleiche kinetische Energie besitzen, variieren ihre Geschwindigkeiten umgekehrt proportional zu ihrer Masse, d.h. die leichteren Ionen erreichen den Detektor vor den schwereren Ionen. Die Flugzeiten liegen im Mikrosekunden-Bereich, deshalb muss die digitale Datenaufzeichnung sehr schnell erfolgen. Durch Verlängerung der Flugzeit kann man auch eine bessere Auftrennung der Ionen erreichen. Um dies ohne Verlängerung des Driftrohres zu realisieren, werden Reflektoren (=Ionenspiegel) eingesetzt. 37 AC III – Teil 2 Allmaier Martin Prießner Am Ende des Rohres wird ein elektrisches Feld angelegt, das der Beschleunigungsrichtung entgegengesetzt ist und so die Ionen zuerst abbremst und dann in die entgegengesetzt Richtung beschleunigt. Die Ionen besitzen nicht die exakt gleiche kinetische Energie, das kann aber im Reflektor ausgeglichen werden, wodurch eine bessere Energieschärfe erreicht wird.) Einsatz von Flugzeitanalysatoren • Gasphasenionisationstechniken: EI und CI (wenn in wenigen Sekunden gebraucht wird (schnelle Analysen) • Desorption/Ionisationstechnik: LDI, MALDI, SIMS, ESI und nESI • Kombination mit (z.B. ultraschnellen) Trenntechniken: GC, CE, nano und μHPLC • Imaging/Profiling Massenspektrometrie (auf atomarer und molekularer Ebene) (Ein molekulare oder atomares Bild einer Oberfläche zu generieren) Prinzip eines Quadrupolanalysators/filters Ein weiteres Prinzip zur Auftrennung von Massen besteht - neben der Trennung im Magnetfeld und der Trennung über die Flugzeit - in der Trennung in hochfrequenten elektrischen Feldern. Die Felder regen die Ionen zu oszillierenden Flugbahnen an, die nur für einen bestimmten Massenbereich stabil sind und nur diesen Ionen erlaubt, das Massenfilter zu passieren. Quadrupolmassenspektrometer sind der häufigste Massenspektrometer-Typ, da die Geräte kompakt und kostengünstig gebaut werden können. Quadrupole sind außerdem schnell genug, um mit der Gaschromatographie gekoppelt zu werden. Ein Quadrupol-MS besteht im Wesentlichen aus vier hyperbolischen Metallstäben, die paarweise als Elektroden dienen. An die jeweils genüberliegenden Stäbe wird eine positive bzw. negative Gleichspannung angelegt, die mit einer Wechselspannung überlagert ist. Hierdurch ergibt sich im Inneren dieser vier Stäbe nur für ein bestimmtes Masse/LadungsVerhältnis eine stabile Flugbahn, alle anderen Ionen werden ausgeblendet (sie prallen auf 38 AC III – Teil 2 Allmaier Martin Prießner die Stäbe und werden entladen). Durch Variation der angelegten Spannungen kann man den gesamten Massenbereich scannen. (Teach/Me) Modulation wird erreicht durch Kombination aus Gleichspannung und RF Spannung (= radiofrequente Wechselspannung). Einsatz von Quadrupolanalysatoren • Gasphasenionisationstechniken: EI und CI • Desorption/Ionisationstechnik: SIMS, ESI und APCI (MALDI) • Kombination mit Trenntechniken: GC und HPLC wegen hoher Scanrate • Isotopendetektion (nur ein Isotop wird durchgelassen) • Gasanalysen Das Prinzip der Quadrupolionenfalle 3D Quadrupolionenfalle: „Iontrap“ / „Paulfalle“ Das Prinzip einer Ionenfalle (engl. ion trap) beruht darauf, Ionen in einem Quadrupolfeld "gefangen" zu halten. (Lissajou Figuren Im schwarzen Bild) Je nach Art der einwirkenden Felder kann man entweder nur Ionen einer bestimmten Masse gefangen halten, oder aber sämtliche Ionen in der Falle vorrätig halten und durch geeignete Veränderung der Felder Ionen mit einer bestimmten Masse dazu zu bringen, den Iontrap zu verlassen. Dadurch ist es möglich, gezielt den Vorrat der Ionen Massen aufgetrennt zu scannen. Die Ionenfalle besteht aus drei Elektroden, einer Ringelektrode und zwei hyperbolischen Deckkappen (Einlass/Auslasselektrode). Die Deckkappen haben jeweils ein Loch für den Eintritt und den Austritt der Ionen. (Teach/Me) Vorteile des Ion-Traps liegen in der Möglichkeit Mehrfach-Stoßexperimente durchzuführen und im besseren Signal-Rauschverhältnis der Messung. Eine Verbesserung dieses Analysators wird durch das Einleiten von Helium Gas in die Falle erreicht. Reicht nämlich das Feld im Inneren der Ionenfalle nicht aus um Ionen mit hoher 39 AC III – Teil 2 Allmaier Martin Prießner kinetischer Energie in eine Kreisbahn zu lenken, wird durch die bremsenden Stoßprozesse mit den Helium Atomen eine Verdichtung der Ionen im Zentrum der Falle herbeigeführt (Ionenkühlung). Vorteile der Ionenfalle liegen in der Möglichkeit Mehrfach-Stoßexperimente durchzuführen und im besseren Signal-Rauschverhältnis der Messung Eine weitere Änderungen: 1. Quadropolfilter wurden vorgeschalten die in der Lage waren einen Ionenstrahl mit großer Parallelität zu generieren. Wie trennt man die so gefangenen Ionen wieder auf? – Das Quadrupolfeld wird kontinuierlich erhöhen, wodurch die Ionen anfangen an zu oszillieren (zuerst die kleinen Ionen dann die Größeren). Abhängig von der Masse erreichen die Ionen durch die Bewegung die Öffnung bei der kein Feld mehr herrscht und gelangen schlussendlich zum Detektor. Einsatz von Quadrupolionenfallenanalysatoren • Gasphasenionisationstechniken: EI(Elektorenstoßionisation) und CI (chemische Ionisation.) • Desorption/Ionisationstechnik: SIMS, ESI, APCI, DESI und AP-MALDI • Kombination mit Trenntechniken: GC, CE und HPLC wegen hoher Scanrate (schnelle Trennrate) • MSn in der Zeitdomäne mit einem Analysator (Mehrstufen MS, „n“ kann von 2-5 sein) Charakteristika der besprochenen massenspektrometrischen Analysatoren. 40 AC III – Teil 2 Allmaier Martin Prießner Ablenkung der Ionenstrahlen im magnetischen oder elektrischen Feld Sektorfeldgeräte: Bei SektorAnalysatoren wird ein Permanentoder Elektromagnet verwendet um den Strahl der Ionenquelle auf einen Kreisbogen von 180°, 90° oder 60° zu führen. Das passiert in einem gebogenen Metallrohr, das unter Hochvakuum steht. Ionen mit unterschiedlichem m/z- Verhältnis fliegen im Magnetfeld auf Kreisbahnen mit unterschiedlichen Radien, welche neben der Masse auch noch von der magnetischen Feldstärke und von der Beschleunigungsspannung abhängen. Variiert man diese nun kann man genau einen Typ von Ionen mit einem bestimmten Masse/Ladungsverhältnis auf den Ionendetektor bringen. Beziehung zwischen Ionentransmission und Auflösung R Beziehung zwischen Ionentransmission und Auflösung R Es gibt eine strenge Beziehung zwischen Ionentransmission und Auflösung (wie viele Ionen schick ich rein / wie viele kann ich detektieren). Bild: Wenn man die Auflösung erhöht wird die Transmission schlechter und umgekehrt. Bei einer Auflösung von z.B. 1000 hat man eine Transmissionsrate von 35 %. Viele Ionen gehen verloren. Konsequenz: Empfindlichkeit geht ebenfalls verloren. Man kann die Auflösung nicht verbessern ohne auch Verluste bei Empfindlichkeit hinzunehmen. Man kann versuchen ein Mittelmaß zu finden. Bei hoher Auflösung wird das Isotopenmuster deutlich dargestellt. Aus dem Muster kann man auf eventuell vorhandenen Heteroatome (Cl, Br,…) schließen, als auch auf den ungefähren C-Gehalt (M+1 Peak relativ zu M Peak in Prozent umrechnen und durch 1,1 dividieren) Bei schlechterer Auflösung überlagern sich die Peaks und die eigentlichen Isotopenpeaks werden von einer Umhüllenden umgeben, es geht Information verloren. 41 AC III – Teil 2 Allmaier Martin Prießner Eines der Hauptprobleme in der Spurenanalytik ist die mangelnde Selektivität bei Proben mit einer komplexen Matrix, wie sie bei vielen Umweltproben oder bei biologischen oder medizinischen Proben normalerweise vorliegen. Um die Selektivität drastisch zu erhöhen, kann man zwei Massenspektrometer hintereinander schalten (TandemMS, oder MS/MS). Dabei wählt das erste Spektrometer Ionen einer bestimmten Masse aus, die dann im zweiten Spektrometer zu weiterem Zerfall angeregt werden. (Teach/Me) Tandem-MS = MS-MS-Kopplung: Um die Selektivität bei Proben mit komplexer Matrix drastisch zu erhöhen, kann man zwei Massenspektrometer hintereinander schalten. Dabei können im ersten Spektrometer Ionen einer bestimmten Masse ausgewählt werden, die dann im zweiten Spektrometer zu weiterem Zerfall angeregt werden. Es können somit jeweils ein Massenspektrum pro Molekülion aus dem ersten MS erhalten werden. Das erste MS übernimmt sozusagen die Funktion des GC. Die zu bestimmende Probe wird im ersten Massenspektrometer mit Hilfe weicher Ionisation überwiegend in Molekülionen und protonierte Molekülionen überführt. Dabei entsteht eine sehr große Anzahl verschiedener Ionen, von denen jene Masse im ersten Massenspektrometer (manchmal auch Massenfilter genannt) ausgewählt wird, die die substanzspezifischen Ionen enthält. Die ausgewählten Ionen werden dann in eine feldfreie Stoßkammer (=Ionisationskammer des zweiten MS) geleitet, wo sie mit einem neutralen Gas (Helium, Argon) zusammenstoßen und weiter zerfallen. Die Spektren dieser Zerfallsprodukte werden im nachgeschalteten zweiten Spektrometer aufgezeichnet. Man erzeugt also ein vollständiges Spektrum der vom ersten Massenfilter ausgewählten Ionen. Dieses Spektrum wird auch CID-Spektrum (engl. collision induced dissociation) genannt und ist für die betreffenden Ionen genauso charakteristisch wie ein normales Massenspektrum für neutrale Moleküle. MS/MS lässt sich kostengünstig in so genannten "Triple-stage Quadrupolgeräten" verwirklichen. Dabei sind drei Quadrupol-Massenfilter unmittelbar hintereinander geschaltet. Das mittlere wird als Stoßzelle benützt. Unterschiedliche Möglichkeiten der Kopplung: Tochterionen MS-MS: Das erste MS ist fix auf eine Masse eingestellt und filtert das zu dieser Masse gehörende Mutterion heraus, das zweite MS scannt die Tochterionen, d.h. die in der Stoßkammer erhaltenen Fragmente. Mutterionen MS-MS: Das erste MS scannt einen Spektralbereich ab, während das zweite MS fix auf die Masse eines der Tochterionen eingestellt ist. Dadurch werden alle für ein bestimmtes Tochterion in Frage kommenden Mutterionen registriert. Nahe verwandte 42 AC III – Teil 2 Allmaier Martin Prießner Verbindungen ergeben normalerweise mehrere Tochterionen des gleichen Typs, daher eignet sich dieses Verfahren zur Feststellung der Identität und Konzentration einer Klasse eng verwandter Verbindungen. Typen von Tandem- bzw. Mehrstufenmassenspekrometrie. Am einfachsten wenn Tandem MS in der Raumdomäne. Das bedeutet es wird im ersten MS ein Vorläuferion selektioniert. Dieses gelangt in die Kollusionszelle in der die weitere Fragmentierung passiert und zum Schluss werden die Fragmente in das zweite MS gebracht. Dies ist auch in der Zeitdomäne durchführbar und kann mit der Quadropolionenfalle = Pausfalle durchgeführt werden. Dabei wird im ersten Zeitbereich ein gewisses Ion herausgefiltert. Dann im zweiten Zeitbereich kommt es zur Kollision mit Gasmoleküle (Siehe Funktionsweise Paulfalle) Dann erflogt die Energieanregung in der Reaktionszone, was zur Fragmentierung führt. Am Ende werden die einzelnen Fragmentionen aus der Ionenfalle zum Detektor gebracht. Wenn Ionen mit Neutralteilchen zusammenstoßen, wird ein Teil der kinetischen Energie der Ionen in innere Energie umgeformt und führt (falls die zugeführte Energie ausreichend ist) zum Zerfall dieser Ionen. Man spricht bei diesem Vorgang von Stoßaktivierung (engl. collisional activation) wenn die zugeführte Energie zu gering für einen Zerfall der Ionen war, oder von stoßinduzierten Zerfällen (engl. CID - collision-induced dissociation, oder CAD - collisionally activated dissociation), wenn die Ionen durch den Stoß zerfallen. (Teach/Me) 43 AC III – Teil 2 Allmaier Martin Prießner 2 Möglichkeiten 1. Hochenergie CID: Ionen die hineingeschossen werden kollidieren mit den Gasemolekülen (He, N2, Ar,) Die Energie die dabei entsteht, wird dazu verwendet das Vorläuferion zu Fragmentieren. 2. LE CID: bei dieser Methode ist die Energie, die die Ionen mitbringen, geringer. Das Gasmolekül interagiert mit dem Ion und bildet einen Komplex in der Gasphase, welcher zu rotieren beginnt. Anschließend kommt es zu einer Schwingungsanregung –welche zum Bruch von labilen kovalenten Bindungen und somit zur Fragmentation führt. Energie kann auch durch Laserpulse oder Elektronenstrahl zugeführt werden. Aber CID hat den Vorteil, dass man die Fragmentierung steuern kann. Z.B. Durch Variation der Gase, wie viel Gas, wie lange die Ionen in Reaktionszone verweilen. Usw. Warum Tandemmassenspektrometrie bzw. Mehrstufenmassenspektrometrie? • Empfindlichkeit/Selektivität • Strukturinformation bei gleichzeitig hoher Empfindlichkeit • Gemischanalyse ohne Vortrennung Beziehung zwischen Signalintensität Rauschen und Zahl der Analysatorstufen Aus diesem Diagramm ist ersichtlich, dass das Rauschen und das Signal mit der Anzahl der Analysatorstufen Exponentiell abfällt. (Rauschen wesentlich schneller). Die uns interessierende Kurve ist das Signal/Rauschverhältnis welches exponentiell steigt. Das bedeutet, dass die Zahl der MS Stufen die Qualität der Analysen verbessert. Beispiel: Kombination von B-, E-, RTOF-, und QAnalysatoren für Tandem- und Mehrstufenmassenspektrometrie (Hybridinstrumente) Hybridinstrumente: dabei werden mehrere Analysatortypen kombiniert. 44 AC III – Teil 2 Allmaier Martin Prießner Fragmentionennomenklatur für Peptide nach Fohlman-Roepstorff-Biemann (http://www.diss.fu-berlin.de/diss/servlets/MCRFileNodeServlet/FUDISS_derivate_000000000171/1_kap1.pdf;jsessionid=65061830780377D1D905411B1E541373?hosts=) Als C-Terminus oder Carboxy-Terminus wird jenes Ende eines Proteins oder Polypeptids bezeichnet, welches eine Aminosäure mit einer freien Carboxygruppe (COOH) besitzt. Als N-Terminus oder Amino-Terminus, wird jenes Ende eines Proteins oder Polypeptids bezeichnet, welches eine Aminosäure mit einer freien Aminogruppe (NH2) besitzt. (Wikipedia) Mehrstufenmassenspektrometer (QIT) für LE CID Experimente in der Zeitdomäne Mehrstufen MS: Eine QP Ionenfalle wird verwendet um ein CID Experimente in der Zeitdomäne zu machen. Bringen Ion ein, selektionieren das Vorläuferion und injizieren einen Puls von He-, Ar-, Atomen in das Zentrum der Zelle und erzeugen eine RF Spannung. Dadurch kommt es zur Fragmentierung und die gebildeten Fragmente werden dann aufgetrennt und analysiert. Ablauf eines LE CIDMS/MS bzw. MSn Experiments in einer QIT Selektionieren des Vorläuferions erfolgt dadurch, dass Teilchen mit nicht passendem M/Z Wert aus der Ionenfalle geworfen werden. Danach kommt der Gaspuls und eine Modifikation der Anregungsfrequenz (CID basierend) Dadurch sind die Ionen in der Ionenfalle gut verteilt. Anschließend versuchen wir eine Fragmentionenkühlung Durchzuführen (alle Fragmentionen werden im Zentrum der Falle gesammelt) und ein Fragmention nach dem anderen werden zum Detektor geschickt. 45 AC III – Teil 2 Allmaier Martin Prießner Typen von Detektoren in der MS Die Detektion der Fragmentionen in einem für Massenspektrometer erfolgt meist über die Umwandlung der Ionen in Elektronen und anschließender Vervielfachung und Zählung der Elektronen. Alternativ dazu (allerdings mit geringerer Empfindlichkeit) können die Fragmentionen aber auch direkt gemessen werden, z.B. mit Faraday-Bechern oder Photoplatten. Die folgende Tabelle gibt einen Überblick zu den wichtigsten Detektoren. Channeltron Ein Channeltron ist eine horn-förmige Dynoden-Struktur die im Inneren mit einer Elektronen-emittierenden Schicht überzogen ist. Durch einen Lawineneffekt werden Sekundärelektronen vervielfacht. Sekundärelektronenvervielfacher (SEV) SEVs (engl. electron multiplier tube, EMT) sind ähnlich konstruiert wie Photomultiplier. Sie bestehen aus einer Kaskade von Dynoden, die durch einen Lawineneffekt der Sekundärelektronen die eintreffende Ladung der Ionen verstärken. Faraday-Becher Der Faraday-Becher wird zum direkten Auffangen der Ionen verwendet. Ein angeschlossenes Elektrometer misst damit direkt die Ladung der einfliegenden Ionen. Mit dem FaradayBecher können keine Einzelimpulse registriert werden, da er um einige Größenordnungen unempfindlicher als z.B. ein SEV ist. Daly-Detektor Der Daly-Detektor besteht aus einem Metallknopf der durch Einschlag von Ionen Sekundärelektronen emittiert. Die emittierten Elektronen werden in einem Szintillator durch einen Photomultiplier gezählt. Der Vorteil dieser Anordnung besteht im Schutz des Photomultipliers vor dem Geschehen im Massenspektrometers. Dadurch kann - bei gleicher Empfindlichkeit - eine Lebensdauer des Photomultipliers von 5 Jahren erreicht werden (Channeltrons müssen je nach Betrieb etwa alle 6 Monate getauscht werden). Channelplate Ein Channelplate besteht aus einer Matrix aus Glaskapillaren (ca. 20 μm Innendurchmesser), die innen mit einer Elektronenemittierenden Schicht beschichtet sind. An die Kapillaren wird eine hohe Spannung angelegt, sodass die Sekundärelektronen beschleunigt und vervielfacht werden. • • • • Photographische Platten und Faraday-Zylinder (sehr alt/ wird nicht mehr verwendet) Sekundärelektronenvervielfacher und MCP(häufig in MS) Dalydetektor (Photomultiplier mit Konversionsdynode) (selten verwendet) Arraydetektoren 46 AC III – Teil 2 Allmaier Martin Prießner Prinzip eines Sekundärelektronenvervielfacher (SEV) mit diskreten Dynoden und einer kontinuierlichen Dynode Sekundärelektronenvervielfacher (SEV) engl. electron multiplier tube (EMT) funktionieren nach folgendem Prinzip: das zu detektierende Ion wird zuerst auf eine spezielle Elektrode gelenkt (die. sog. Konversionsdynode) und schlägt dort beim Aufprall ein oder mehrere Elektronen heraus. Diese Elektronen werden nun durch die nächste Elektrode (Dynode) beschleunigt und prallen dort auf. Beim Aufschlag schlägt jedes Elektron wieder jeweils 1 bis 3 Elektronen heraus, die von der nächsten Dynode wiederum beschleunigt werden und dort aufprallen. Dieser Prozess wird über 15 bis 20 Dynoden fortgesetzt, wodurch sich eine lawinenartige Vermehrung der Elektronen ergibt. Dadurch erzeugt nach 20 Stoßvorgängen (bei 20 Dynoden), ein primäres Teilchen (das zu detektierende Ion) ca. 106 Elektronen am Ausgang des SEVs. Die Verstärkung des SEVs lässt sich durch Änderung der Spannungen an den Dynoden leicht einstellen. (Teach/Me) Variante 2: Channeltrons (in der Vorlesung wird es kontinuierliche Dynode genannt) sind vom Prinzip her vergleichbar mit Channelplates, allerdings ist die aktive Schicht nicht in Kapillaren angeordnet sind sondern in Form eines gebogenen Trichters. Das Ion wird an einer Konversionsdynode in Sekundärelektronen umgewandelt, die dann in den Trichter hinein beschleunigt werden (Foto). Ein Channeltron eignet sich sehr gut zur Detektion einzelner Ionen. Nachteil: Sekundärelektronenvervielfacher hält nur 1 Jahr weil die Schicht abnutzt. Micro channel plate (MCP) Detektor Zwischen beiden metallisierten Plattenseiten liegt eine Beschleunigungsspannung an, die Platte selbst besteht aus einem Halbleiter und ist ähnlich wie ein Sieb durchlöchert beziehungsweise durchzogen von mikroskopisch feinen Kanälen, die typischerweise einen Lochabstand von ca. 10 µm und einen Durchmesser von ca. 6-8 µm besitzen. Die Platte hat eine Dicke von wenigen Zehntel Millimetern und die Kanäle 47 AC III – Teil 2 Allmaier Martin Prießner sind um ca. 10° gegen die Plattenachse verkippt, so dass die einfallenden Elektronen mit Sicherheit mehrmals die Kanalwand treffen. Sie werden dann von einer zwischen den Platten längs der Kanäle anliegenden elektrischen Spannung beschleunigt und vervielfachen sich bei jedem Wandstoß, jeder einzelne Kanal verhält sich somit wie ein mikroskopischer Elektronenvervielfacher, wie er beispielsweise im Fotovervielfacher eingesetzt wird. An der Austrittsseite hat sich die Zahl der Elektronen durch Vielfachstöße mit der Kanalwand um das ca. 1000-fache erhöht. Durch eine Nachbeschleunigungsstrecke werden die verstärkten (=vervielfachten) Elektronen auf den eigentlichen Detektor gelenkt. (Wikipedia) (gleich Erklärung Wie VO nur bessere Formulierung) Charakteristika von MS Detektoren Signalverarbeitung der Detektorsignale Heute werden nicht mehr Analogdaten aufgezeichnet. Analog Digitalkonversion. Um Strichspektrum generieren zu können. 2.2. Zerfallsmechanismen und Grundregeln der EI-Fragmentierung zur Strukturaufklärung organischer Moleküle Generelles Protokoll zur Interpretation von Massenspektren 1. Identifikation des Molekülions (Auswahl der Technik und Reinsubstanz wichtig!(falls es ein Gemisch ist, muss man eine Vortrennung durchführen) 2. Bestimme die Elementarzusammensetzung (ein MS Analysator muss eine gute Auflösung und Genauigkeit haben, um eine Elementarzusammensetzung zu liefern. (geht nicht mit Quadrupolanalysator.) 3. Bestimme den Basispeak (wichtige Info weil sie zum Fingerprint gehört) 48 AC III – Teil 2 Allmaier Martin Prießner 4. Suche nach charakterist eristischen Isotopenmustern (Molekülion und d Fr Fragmentionen) (Chlor Brom (79:81 = 1:1) 1:1 Blei (Chlor 35:37 = 3:1) Silber (107:109 = 1 1:1) das bedeutet, (z.B. Cl) wenn man im A Abstand von 2 Masseeinheiten 2 Peaks im Ver erhältnis 3:1 vorliegen hat, ist dass ein Indikator für Chlor? 5. Identifiziere Neutralteilc teilchenverlust vom Molekülion (die vom Molek olekülion abgespalten werden.. FFalls eine Abspaltung von CO oder CO2 irgendw endwo im ursprünglichen Molekül ekül diese funktionellen Gruppen vorliegen.) 6. Identifiziere charakteris teristische Niedermassen-Ionen 7. Identifiziere charakteris teristische m/z Werte (z.B. 14 (CH2), 26 (C2H2), 2), 28 2 (CO, N2, C2H4), 29 CHO, ...) Man an kontrolliert, ob gewisse Massendifferenzen zen vorliegen. z.B. wenn wir Abstände von 14 Dalton finden weist das auf CH2 hin. (Iso Isobare Masse: das bedeutet das unterschie schiedliche Ionen zwar nominell dieselbe Masse sse haben, aber eine andere Zusammensetzu etzung besitzen( z.B. m/z 28)) Wenn man die Ma Masse ganz genau bestimmt (auf 4 Nachko chkommastellen), dann kann man diese erst genauer gen unterscheiden. senspektrum mit Referenzspektren (Im Fall vom om EI werden die 8. Vergleiche das Massens Spektren mit Datenbank banken verglichen) 9. Verknüpfe alle anderen ren Probeninformation mit den massenspektrom trometrischen Daten und prüfe die Konsisten stenz Zusammensetzung eines organ rganischen Moleküls mit einem Molekülion zwischen zwis m/z 180 und 180,2 (folgende Elemente nte sind s möglich C, H, N, O) X – Achse ein n sehr seh kleiner Massenbereich ich angegeben. a In diesem kleinen en M Massenfenster kann man eine ne ggroße Zahl von verschiedenen en Verbindungen V hineinprojizieren ieren. So kann, aus der auf die 4.. Ko Kommastelle bestimmten Masse, Mas das Molekül genau zugeordn rdnet werden. Man braucht eine seh ehr große Messgenauigkeit. (je mehr Elemente Elem vorkommen dürfen umso mehr Möglic öglichkeiten ergeben sich). Bis zu 500-600 Dalton kan kann man dieses Verfahren verwenden. Im größ größeren Maß ist es nicht mehr möglich weil sonst nst d die Zahl der möglichen Verbindungen immer mer größer wird. Theoretisches Isotopenmuster ster ffür C1, C100, C300, C400, C500, Wenn man das Isotopenmus muster von Kohlenstoff ansieht, dann n be besteht er aus C12 und C13 in einem Verhältnis ltnis 99:1. (kann gut im MS unterschieden werden en B Bild 1) Wenn man nun 100 C Atome tome betrachtet erhält man das zweite Isotopenmu nmuster. Der erste Strich symbolisiert 100 C12 12 A Atome, der zweite 99 C12 und 1 C13… usw. 49 AC III – Teil 2 Allmaier Martin Prießner Man soll nach charakteristischem Isotopenmuster suchen. Isotopenmuster von reinen Chlor (35:37 – 3:1) Wenn eine Verbindung 2 Cl Atome enthält bekommt man die 2. Darstellung: 1. Peak besteht aus 2 Chlor (35:37 = 3:1) Natürliche Isotopenmusterkombinationen Hier sind die Isotopenmuster der einzelnen Cl/Br Verbindungen aufgezeichnet. Bei der Mischung aus Cl und Br wird das Isotopenmuster sehr komplex, aber weiterhin charakteristisch 1. Peak nur aus CL35 2. Peak aus einem Cl37 und einem Cl 35 3. Peak nur aus Cl37 Abhängig davon bei welchem m/z Wert dieses Muster kommt, kann man auf die Verbindung schließen. Brom (79:81 = 1:1) Chlor (35:37 = 3:1) Hier sind die Isotopenmuster der einzelnen Cl/Br Verbindungen aufgezeichnet. Bei der Mischung aus Cl und Br wird das Isotopenmuster sehr komplex, aber weiterhin charakteristisch 50 AC III – Teil 2 Allmaier Martin Prießner Isotopenmuster von Chlor: Berechnen Sie das Isotopenmuster (m/z-Werte und die dazugehörigen resultierenden Intensitäten) von Cl3, von Cl2, von BrCl bzw. von Br2 (79Br : 81 Br = 1:1; 35Cl : 37Cl = 3:1). Für Verbindungen mit nur einer Atomsorte kann die Formel ሺܽ + ܾሻ angewendet werden. Für ܽ und ܾ werden die Häufigkeitsanteile eingesetzt. Cl2: a = 3, b = 1 a2 : 2ab : b2 Isotopenmuster: 9:6:1 Cl3 a3 : 3a2b : 3ab2 : b3 Isotopenmuster: 27:27:9:1 also ca. 3:3:1 Br2: a = b = 1 a2 : 2ab : b2 Isotopenmuster: 1:2:1 Bei verschiedenen Atomsorten verwendet man: ሺܽ + ܾሻ ሺܿ + ݀ሻ BrCl: a = b = 1, c = 3, d = 1 ac : bc + ad : bd Isotopenmuster: 3:4:1 Alternativ kann man auch alle möglichen Kombinationen aufschreiben und deren Häufigkeit bestimmen. 51 AC III – Teil 2 Allmaier Martin Prießner Charakteristisches Isotopenmuster in einem EI Massenspektrum eines organischen Moleküls Basispeak Beispiel eines organischen Moleküls: Komplexes Isotopenmuster. Genau die M/Z Werte sind gegeben. Somit kann man die Elementarzusammensetzung der einzelnen Isotopen berechnet. Die ist nützlich, weil es sich bei dieser Verbindung um eine metallorganische Verbindung handelt, bei welche 3 Metallatome in dem organischen Molekülverband eingebettet sind. Wenn man das Isotopenmuster der Metalle hineinlegt ist klar, dass dieses Isotopenmuster zu einer bestimmten Elementarzusammensetzung passt. Man kann auch bestimmte Isotopenmuster in Fragmentionen suchen. Jedoch wenn im Molekülion das Muster nicht vorhanden ist, dann wird es im Fragmention ebenfalls nicht der Fall sein. Aus dem Molekülradikalkation wird ein [M-X]+ wobei X die Abspaltung des Neutralteilchen ist. Wenn wir jedoch stattdessen M+1 finden, dann deutet das darauf hin, dass folgende Komponenten vorliegen können (siehe Tabelle) So kann man bei jeder Massenzahl in der Tabelle nachsehen, um welche Verbindung es sich handel könnte. 52 AC III – Teil 2 Allmaier Martin Prießner Nicht plausible Verluste vom Molekülion • • • • [M-7]+ [M-3]+ [M-6]+ ... meist von Verunreinigungen oder Rauschen (in Wirklichkeit handelt es sich oft um 2 Molekülionen Verluste zwischen 3 -14Da und 21 -26Da sind selten plausibel „Stickstoffregel“ -wichtige Regel für die Strukturaufklärung Elemente O, C, S: gerade Wertigkeit, gerades Atomgewicht (wenn man ganzzahliges Atomgewicht betrachtet) Elemente H, F, Cl, Br, I: ungerade Wertigkeit, ungerades Atomgewicht -Verbindungen, die nur diese Elemente enthalten (z.B. O2, H2 oder Cl2), haben immer eine geradzahlige Molekülmasse Ausnahme: N ungerade Wertigkeit, (obwohl) gerades Atomgewicht -Verbindungen mit einer ungeraden Anzahl von N-Atomen haben stets eine ungeradzahlige Molekülmasse Die sogenannte „Stickstoffregel“ (gültig für EI) Da in organischen Verbindungen N das einzige Element mit ungerader Bindigkeit und gerader Massenzahl ist, gilt die Stickstoffregel: Eine ungerade Massenzahl des Molekülions weist auf eine ungerade Zahl von N-Atomen im Molekül hin, bei einer geraden Massenzahl enthält das Molekül entweder gar keinen Stickstoff oder eine gerade Zahl von N-Atomen. Die McLafferty Umlagerung: Die Mc-Lafferty Umlagerung ist eine H-Umlagerung, die bei vielen Carbonyl-Verbindungen auftritt. Dabei wandert das γ-Wasserstoffatom auf den Sauerstoff der Carbonylverbindung 53 AC III – Teil 2 Allmaier Martin Prießner und gleichzeitig wird die Bindung zwischen α und β-Atom gespalten. Das Radikalkation zerfällt in ein Neutralteilchen (meist ein Alken) und in ein Radikalkation. Allgemein sind für eine McLafferty-Umlagerung eine Doppelbindung und ein H am γ-C notwendig, die noch dazu einen 6-gliedrigen Übergangszustand eingehen können. Hier ein Beispiel zur McLafferty Umlagerung: Gegeben ist ein Butylalkohol. Bei m/z 74 befindet sich das Molekülion, welches jedoch nicht den Basispeak darstellt. Man sieht dass CH3 bei m/z von 15 am Ende dieser Struktur abgespalten wird. Das ist so nieder, dass das Signal nur mit großer Vorsicht zu betrachten ist. Eine weitere Abspaltung ist die einer Propylgruppierung bei m/z 43 (relativ intensives Signal). Diese Gruppe ist ein Kation und der andere Teil ist bei m/z 31 sichtbar, welcher den Basispeak dieser Verbindung darstellt. Ebenfalls sichtbar ist ein Peak bei m/z 41, 42 und 56. 41 ist so zu erklären, dass aus diesem Fragment auch 2 Protonen abgespalten werden, weil eine Doppelbindung gebildet wird. 42 und 56 kann durch diese McLafferty Umlagerung erklärt werden. Wie bereits beschrieben ist die Bildung eines 6er Rings möglich und das Wasserstoffatom wandert was schlussendlich zu einem Bindungsbruch führt, bei welchem 2 Fragmente entstehen. (m/z 42 und m/z 56) somit kann man alle Fragmentionen in diesem Beispiel erklären. Beispiel einer Gesetzmäßigkeit: Retro-Diels-Alder Reaktion in der Massenspektrometrie Voraussetzungen:6-gliedriges und cyclisches System in dem Doppelbindungen vorliegen. Retro Diels Alder Reaktion. Grundkonzept: Es ist ein Molekülradikalkation vorliegend und BAD mit einer Doppelbindung. Bei dieser Reaktion kommt es zum Bindungsbruch (an den eingezeichneten Stellen) Das führt zu einem Neutralteilchen, welches jedoch nicht detektiert werden kann und zu einem Fragmention mit 2 Doppelbindung. Das kann auch bei Verbindungen die verschiedenen Substituenten besitzen passieren. Das ist ebenfalls eine Gesetzmäßigkeit bei der Rückschlüsse auf die Substituenten gemacht werden können Die möglichen Abspaltungen von neutralen Fragmentteilchen sind in Listen festgehalten. Wenn eine gewisse Massendifferenz beobachtet wird kann anhand dieser Tabellen auf das jeweilige Molekül geschlossen werden. 54 AC III – Teil 2 Allmaier Martin Prießner Basierend auf den Gesetzmäßigkeiten wird ein Fragmentierungsbaum aufgebaut Wenn alle Fragmente analysiert worden sind, kann man einen Fragmentierungsbaum aufstellen. Dieser Baum starte mit dem Molekülion gefolgt von den Abspaltungen von z.B. Fragmentionen (A+, B+, C+). Die können dann weiter fragmentieren und zu weitere Fragmente führen. Wenn man einen solchen Baum erst mal erstellt hat, kann man viele Strukturen eindeutig zuordnen und eine unbekannte Verbindung komplett in der Struktur aufklären. Oft ist es jedoch nur möglich Teile der Verbindung zu analysieren. Dann muss mit anderen Methoden weiterprobiert werden. Beispiel eines EI Massenspektrums von Benzoesäuremethylester In diesem Massenspektrum sieht man ein Molekülion und als Basispeak ein Fragmention. Charakteristisch ist die COH3 Abspaltung. Wenn, wie in diesem Fall eine solche Massendifferenz vorliegt, kann man davon ausgehen, dass es sich auch um eine Verbindung handelt die mit einem Ester verknüpft ist. Ein weiteres Fragment ist die CO Abspaltung, die sich auf die Carbonylgruppe in der Verbindung bezieht. Das letzte Fragment ist die Abspaltung des (und zusätzliche Aufspaltung) des aromatischen Ringsystems mit C2H2. Mit diesen Informationen kann man sagen, dass es sich bei der gesuchte Verbindung um eine Carbonsäure + einen Ethylester handelt und m/z 77 ist charakteristisch für ein aromatisches Ringsystem. Weitere Informationen können NMR oder IR Spektroskopie liefern. Diese Techniken zusammen erlauben eine komplette Strukturaufklärung von kleineren Molekülen 55 AC III – Teil 2 Allmaier Martin Prießner 2.3. On-line Kopplungstechniken: GC/MS und HPLC/MS Chromatographische und elektrophoretische on-line Kopplungstechniken mit Massenspektrometern Zwei Methoden sind bei den On-line Kopplungen besonders wichtig: Gaschromatographie mit Massenspektroskopie und die HPLC mit MS (In der Klammer stehen die Möglichkeiten der Kopplung) (APCI = Atmospheric Pressure Chemical Ionisation, APPI = Atmospheric Pressure Photon Ionisation, SEC = Size-Exclusion Chromatography, GPC = Gelpermeationschromatographie= SEC, CEC = Kapillarelektrochromatographie, TLC =Dünnschichtchromatographie,) Trennung von organischen Molekülen 20% aller organischen Verbindungen kann man unzerstört aus der flüssigen/festen Phase in die Gasphase überführen und mit GC analysieren. Die restlichen 80% können nur in flüssiger Form analysiert werden, weil sie nicht in die Gasphase überführt werden können. direkte Kopplung mit GC – Gasphasenionisation (EI, CI) direkte Kopplung mit LC – ESI; LC: HPLC, CZE, IC Kopplungstechniken: GC-EI-MS: Bei der Kopplung von GC mit einem MS, dient der vorgeschaltete Gaschromatograph zu Auftrennung des Substanzgemisches und das Massenspektrometer zu Identifizierung und Quantifizierung der einzelnen Komponenten. Kapillarsäulen: Die Fließgeschwindigkeit der Eluenten ist in der Regel gering genug um den Säulenausgang des GC direkt über eine geheizte Transferleitung mit der Ionisierungskammer des MS zu verbinden. Das leichte Trägergas kann dort abgesaugt werden. Gepackte Säulen: Da gepackte Säulen einen höheren Gasdruck benötigen ist die austretende Gasmenge zu groß um direkt in die Ionenquelle geleitet zu werden. Es muss ein Separator vorgeschaltet werden um einen Großteil des Trägergases von dem Analyten abzutrennen. Für jeden Peak des GC und somit für jede Substanz kann ein Spektrum erstellt werden, das im Rechner gespeichert und später verarbeitet wird. GC-EI-MS-Geräte werden vor allem zur Identifizierung komplexerer Verbindungen in komplizierten Matrices verwendet, die in natürlichen oder biologischen Systemen 56 AC III – Teil 2 Allmaier Martin Prießner vorkommen. Der Einsatz erfolgt in der Lebensmitteltechnologie, Umweltforschung und medizinischen Diagnosen. Bei genügend schneller Scanrate können mehrere Scans pro eluierter Substanz durchgeführt werden. Die Intensitäten der m/z Peaks steigen dabei stetig und fallen dann wieder ab. Durch eine gewisse Verzögerung verzerren sich die GC-Peaks nach rechts: LC-ESI-MS: 80% der organischen Verbindungen können nicht zerstörungsfrei in die Gasphase gebracht werden und müssen daher mittel HPLS getrennt werden. Weitere Trenntechniken in flüssiger Phase sind die Kapillar-Zonen-Elektrophorese und die Ionenchromatographie. Die gelösten AnalytMolekülen eigenen sich zur Ionisierung mittels ESI. Nach Anpassen der Flussrate können unterschiedlich dicke Kapillaren verwendet werden. Die Ionisierung kann zusätzlich durch eine unterstützende Flüssigkeit (sheath liquid) oder Gas (nebulizing gas) gefördert werden. Zusätzlich kann die Ionisierung durch Ultraschall verbessert werden. Diese Kopplungstechniken erhöhen den Informationsgehalt über die Analyten. Ein MS liefert klarerweise mehr Informationen als ein simpler UV-Detektor. Noch mehr Möglichkeiten ergeben sich mit Mehrstufen-MS. Kopplungsarten Offene Kopplung („open-split“) GC/MS (EI oder CI) Diese Technik ist geeignet um einen Gaschromatographen mit EI oder CI zu koppeln. Durch das Ende der GC Säule werden die eluierenden Peaks herantransportiert. Dieses Rohr kommt in eine etwas größere Kapillare. In der Mitte trifft sie sich mit der sogenannten „Transferkapillare“, welche direkt in den Massenspektrometer führt (wenn die Tranferkapillare kleiner im ID als die GC Kapillare ist, kommt es zu große Transferverlusten) Zuerst herrscht Atmosphärendruck und nach dieser 2. Kapillare herrscht Hochvakuum. Problem: über die Transferkapillare wird kontinuierlich Luft angesaugt. Somit gelangt auch O2 in die EI Ionenquelle, welche ionisiert wird und ein starkes Rauschen liefert. Um das zu beheben führt man eine He – Spülung ein die damit den Sauerstoff verdrängt. He hat ein so geringes Ionisierungspotential, dass es bei der Messung zu keiner Störung kommt. Vorteil dieser Technik: robust, man kann schnell bei bedarf die GC Säule wechseln. 57 AC III – Teil 2 Allmaier Martin Prießner Direkte Kopplung für Kapillar GC/MS (EI oder CI) Um 100% Transfereffizienz zu erreichen (was bei der offenen Kopplung nicht möglich ist) gibt es die direkte Kopplung. Dabei kann man ausschließlich GC mit EI oder CI koppeln. In der Abbildung sieht man eine GC Säule (die 10, 50, 100 lang sein kann) mit einer beheizten Transferleitung (Grund: sonst würden sich die Analytmoleküle ablagern) durch die die GC Säule in die Ionenquelle des Ms gelangt. Somit gelangen die eluierenden Teilchen auf direkten Weg und ohne Verluste in den Analysator. Diese Methode ist nur deshalb möglich, weil das Material der GC-Säule so inert ist, dass es nicht in der Ionenquelle abgetragen werden kann. Scanrate: Massenspektren über einen GC Peak Hier sieht man einen chromatographischen (GC) Peak, der eine gewisse Peakbreite besitzt. Beim linken Bild gibt man dem massenspektrischen Analysator lange Zeit um ein Spektrum zu generieren. Beim rechten Bild wird ein sehr viel kürzerer Scan gemacht und man erhält ein anderes Massenspektrum. Man sieht, dass bei beiden Spektren je 2 Peaks vorhanden sind, welche jedoch abhängig von der Scanzeit unterschiedliche Intensitäten besitzen. Dabei passt das Spektrum welches in kürzerer Zeit aufgenommen wurde besser mit dem tatsächlichem (wurde mit Direkteinlass generiert) zusammen. Das bedeutet, dass abhängig davon, wie lange man während eines Peak misst verändert sich das jeweilige erhaltende Spektrum. In den rechten Spektren wird dieser Sachverhalt noch mal genauer verdeutlicht. Beim ersten Bild ist eingezeichnet, wann die darunterliegenden Spektren generiert worden sind. Dabei ist genau ersichtlich, dass abhängig davon, wann im eluierenden Peak das Spektrum erstellt worden ist, sich die Intensitäten stark unterscheiden. 58 AC III – Teil 2 Allmaier Martin Prießner Um ein aussagekräftiges Spektrum zu erhalten, addiert man in der Praxis alle erhaltenen Spektren und erhält ein MS, welches repräsentativ für den ganzen GC Peak ist. Ein solches Spektrum hat eine große Ähnlichkeit mit dem Spektrum, welches durch Direkteinlass generiert wurde. 247: Molekülion 73, 147: Fragmentionen 73: Basispeak Einfluss der Scanrate auf einen chromato-graphischen oder elektrophoretischen Peak Ein weiterer wichtiger Punkt ist die Scangeschwindigkeit. Besonders wichtig wird diese, wenn es zu keiner guten und eindeutigen Trennung zweier Substanzen kommt (was in der Realität oft der Fall ist). Bei sehr langsamer Scanrate, bekommt man eine Peakform, die kein eindeutiges Anzeichen dafür gibt, dass es sich dabei um 2 Komponenten handelt (rechts bei sehr hohen Scangeschwindigkeit jedoch schon. Je mehr Spektren aufgenommen werden, umso leichter kann man die ursprüngliche Peakform rekonstruieren. Auch die Empfindlichkeit ist mit der Scangeschwindigkeit gekoppelt. Wenn langsam gescannt wird, bekommt man eine bessere Empfindlichkeit als mit schnellen Scans. Die Entscheidung über die Scangeschwindigkeit ist von der Situation abhängig (z.B. vom Signal/Rausch Verhältnis.) TIC (total ion current) Chromatogramm und “selected ion” Chromatogramme eines GC/EI/MS Analyse Das TIC Chromatogramm repräsentiert die Summe aller Intensitäten, die während eines Durchlaufes aufgenommen werden. (Wikipedia eng.) Im Gegensatz dazu werden beim SI (selected ion) nur ein bestimmter m/z-Wert dargestellt. . 59 AC III – Teil 2 Allmaier Martin Prießner Mit GC/MS kann man auch leicht Vergleiche von Massenspektren mit Datenbanken durchführen. Bei einem guten GC erhält man als eluierende Komponenten die Reinstoffe sind. Häufig sind die Reinkomponenten zur Vergleichsanalyse nicht zugänglich. Jedoch sind diese oft schon in einer Datenbank gespeichert. Wenn man also eine unbekannte Verbindung analysiert, kann man Abgleiche mit Datenbanken machen. Dann bekommt man eine Liste von Vorschlägen, die eine gewisse Ähnlichkeit mit dem erhaltenen MS haben. Die einzelnen Vorschläge werden mit einer gewissen Wahrscheinlichkeit angegeben, wie sehr es zu dem erhaltenen Spektrum passt. (max. ist 1000 = 100% gleich) Für welchen Vorschlag man sich dann entscheidet, hängt auch davon ab, ob es überhaupt in meiner Komponente möglich ist, so ein Produkt zu enthalten. Um einen Vergleich mit Datenbanken machen zu können, muss mit der Standardenergie von 79 eV untersucht werden. Anhang: Vergleich der Massenspektren von 1-Decanol: a) Chemische Ionisation mit Isobutan als Reaktantgas b) 70 eV-Elektronenstoß-Ionisation 60 AC III – Teil 2 Allmaier Martin Prießner