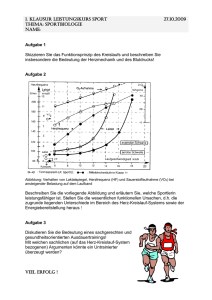
Dokument_43.
Werbung

Konditionelle Gendeletion des Ryanodinrezeptors RyR2 im Herzen der Maus Der Naturwissenschaftlichen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg zur Erlangung des Doktorgrades Dr. rer. nat. vorgelegt von Anita Milbradt aus Temeschburg Als Dissertation genehmigt von der Naturwissenschaftlichen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg Tag der mündlichen Prüfung: 11. November 2011 Vorsitzender der Promotionskommission: Prof. Dr. Rainer Fink Erstberichterstatter: Prof. Dr. Andreas Ludwig Zweitberichterstatter: Prof. Dr. Johann Helmut Brandstätter Inhaltsverzeichnis I Inhaltsverzeichnis Inhaltsverzeichnis I Zusammenfassung IV Summary V 1. Einleitung 1 1.1 Das Herz 1 1.2 Die elektromechanische Kopplung des Herzens 1 1.3 Der Ryanodinrezeptor 3 1.4 Die Rolle von RyR2 im Reizleitungssystem 4 1.5 Konditionelle Knockout-Tiere: das Cre/loxP-System 6 1.6 Induzierbare konditionelle RyR2-Knockoutmäuse 7 1.7 Ziel dieser Arbeit 8 2. Material und Methoden 9 2.1 DNA-Techniken 9 2.1.1 Isolierung genomischer DNA zur Genotypisierung 9 2.1.2 Isolierung genomischer DNA aus Mausherz 9 2.1.3 Polymerasekettenreaktion (PCR) 10 2.1.4 Agarose-Gelelektrophorese 11 2.2 RNA-Techniken 12 2.2.1 Isolierung von Gesamt-RNA 12 2.2.2 Semiquantitative RT-PCR aus Gewebe 12 2.2.3 Semiquantitative RT-PCR aus isolierten Sinusknotenzellen 13 2.3 Protein-Techniken 14 2.3.1 Isolierung und Präparation von Proteinen 14 2.3.2 Proteinbestimmung mit BCA 14 2.3.3 SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE) 14 2.3.4 Western-Blot Analyse 16 2.4 Histologische Färbungen 17 2.4.1 Herstellung von Gefrierschnitten 17 2.4.2 Herstellung von Paraffinschnitten 18 2.4.3 HE-Färbung 18 2.4.4 Sirius-Red-Färbung 19 2.4.5 Lektin-Färbung 19 2.5 Isolierung von Sinusknotenzellen 20 2.6 Messung der Ca2+-Konzentration in Sinusknotenzellen 21 Inhaltsverzeichnis II 2.7 Gewinnung von Kardiomyozyten durch Langendorff-Perfusion 21 2.8 Telemetrische Elektrokardiogramm Ableitungen 23 2.9 Blutdruckmessungen mit Schwanzmanschette (Tail Cuff) 25 2.10 Wiegen und Ausmessen der Herzkompartimente 26 2.11 Haltung und Zucht von Mäusen 26 2.11.1 Haltung 26 2.11.2 Zucht von konditionellen RyR2-Knockout-Mäusen 26 2.11.3 RyR3-Knockout-Mäuse 28 2.11.4 Tamoxifen-Injektion 28 2.12 Statistische Analyse 28 3. Ergebnisse 29 3.1 Charakterisierung einer herzspezifischen RyR2-KO-Mauslinie (RyR2MerCreMer) 29 3.1.1 Überlebensrate der RyR2MerCreMer-KO-Mäuse 29 3.1.2 Analyse der RyR2 Expression im Herzen nach Deletion 30 3.1.2.1 Analyse der RNA-Expression der drei RyR-Isoformen 30 3.1.2.2 Analyse der RyR2-Proteinexpression 31 3.1.3 Untersuchung des Herzgewichts 32 3.1.4 Analyse der Herzfrequenz und der EKG-Parameter 34 3.1.5 Analyse der strukturellen Veränderungen des Herzens 36 3.1.6 Untersuchung der Morphologie und Größe der Kardiomyozyten 37 3.2 Charakterisierung einer reizleitungs-spezifischen RyR2-KO-Mauslinie (RyR2KiT) 39 3.2.1 Analyse der Expression von RyR2 in RyR2KiT-Mäusen 39 3.2.1.1 Analyse der RNA-Expression der drei RyR-Isoformen 39 3.2.1.2 Analyse der RyR2 Proteinexpression 41 3.2.2 Untersuchung der strukturellen Veränderungen der Herzen 42 3.2.3 Analyse der Herzfrequenz und der EKG-Parameter 43 3.2.4 Analyse der Herzfrequenz-Modulation durch verschiedene Pharmaka 46 3.2.5 Analyse der Herzfrequenzvariabilität 47 3.2.6 Auswirkung der RyR2-Deletion auf den Blutdruck 49 3.2.7 Phosphorylierungstatus von Phospholamban 49 2+ 3.2.8 Ermittlung des basalen intrazellulären Ca -Gehalts in Sinusknotenzellen 50 3.3 RyR2-Rekombination ohne Induktion 52 3.3.1 RyR2-Rekombination ohne Induktion in der RyR2MerCreMer-Mauslinie 52 3.3.2 RyR2-Rekombination ohne Induktion in der RyR2KiT-Mauslinie 54 3.3.3 RyR2-Rekombination ohne Induktion in der ubiquitär aktiven Mauslinie 3.4 RyR2CAGG-Cre 56 Analyse der RyR3-Expression im Herzen 56 Inhaltsverzeichnis III 3.4.1 Nachweis der RyR3-Proteinexpression im Herzen 57 3.4.2 Nachweis der RyR3-Expression in isolierten Sinusknotenzellen 57 3.5 EKG-Untersuchungen an RyR3KO-Mäusen 58 3.5.1 Analyse der Herzfrequenz und der Aktivität der RyR3KO-Mäuse 58 3.5.2 Untersuchung der Herzfrequenz-Modulation in RyR3KO-Mäusen 59 3.5.3 Analyse der Herzfrequenzvariabilität 61 4. Diskussion 63 4.1 Charakterisierung der RyR2MerCreMer-KO-Mäuse 63 4.2 Charakterisierung der RyR2KiT-KO-Mäuse 66 4.3 Rekombination des gefloxten RyR2-Locus ohne Induktion 70 4.4 Die Rolle von RyR3 im Herzen 71 4.5 Ausblick 73 5. Anhang 74 5.1 Verwendete Primer 74 5.2 Verwendete Antikörper 75 5.3 Verwendete transgene Mauslinien 76 5.4 Abkürzungsverzeichnis 77 6. Abbildungsverzeichnis 81 7. Literaturverzeichnis 84 8. Eigene Veröffentlichungen 90 9. Lebenslauf 91 10. Danksagung 92 Zusammenfassung IV Zusammenfassung Der intrazelluläre Ca2+-Kanal RyR2 steuert die Ca2+-Freisetzung aus dem SR und spielt eine große Rolle bei der Kontraktion des Herzens. Neben seiner Rolle bei der Kontraktion soll er auch eine wichtige Funktion bei der Generierung des Schrittmacherpotentials im Reizbildungs- und leitungssystem haben. Da die globale Deletion von RyR2 embryonal letal ist, konnte bis jetzt seine genaue Funktion im Herzen anhand eines Mausmodells nicht untersucht werden. Das Ziel der vorliegenden Arbeit war es die Funktion von RyR2 sowohl im ganzen Herzen als auch selektiv nur im Reizbildungs- und leitungssystem zu charakterisieren. Dazu wurden zwei konditionelle KO-Mauslinien verwendet, bei denen der Knockout im adulten Tier durch Tamoxifen induzierbar war. Zusätzlich sollte die Expression von RyR3 im Herzen auf Protein-Ebene bestätigt werden und seine Funktion mittels EKGTelemetrie untersucht werden. Zur Analyse von Mäusen mit einer Deletion von RyR2 im ganzen Herzen wurde die RyR2MerCreMer-Mauslinie verwendet. Es zeigte sich, dass RyR2MerCreMer-KO-Tiere sehr schnell nach der Tamoxifen-Behandlung eine Herzinsuffizienz entwickelten und starben. Dabei zeigten die Herzen typische morphologische Merkmale, wie eine Dilatation der Vorhöfe oder Bildung von Fibrose. Außerdem waren auf zellulärer Ebene ebenfalls Unterschiede aufgetreten, so dass die Kardiomyozyten vergrößert und unförmig wirkten. Beim selektiven RyR2-KO im Reizbildungs- und leitungssystem (RyR2KiT-KO) waren die Tiere lebensfähig und zeigten keine morphologischen Unterschiede zu den Kontrollen. Telemetrische EKG-Messungen zeigten jedoch, dass die Deletion von RyR2 zu einer Bradykardie führte. Zusätzlich konnte gezeigt werden, dass eine Herzfrequenz-Modulation durch die Gabe von Pharmaka in den RyR2KiT-KO-Tieren zwar noch möglich war, allerdings erreichten die KO-Tiere nicht die gleichen maximalen Frequenzen wie die Kontrollen. Außerdem wiesen die RyR2KiT-KO-Tiere eine geringere Herzfrequenzvariabilität auf. Messungen der intrazellulären basalen Ca2+-Konzentration in isolierten Sinusknotenzellen zeigten, dass die RyR2KiT-KO-Tiere einen niedrigeren basalen Ca2+-Gehalt hatten als die Kontrollen. Somit konnte gezeigt werden, dass RyR2 sowohl bei der Kontraktion des Herzens, als auch bei der Generierung des Schrittmacherpotentials eine wichtige Rolle spielt. Neben RyR2 wurde auch die Rolle von RyR3 im Herzen untersucht. Es konnte zum ersten Mal auf Protein-Ebene die Expression von RyR3 im Sinusknoten und in den Vorhöfen nachgewiesen werden und somit die bereits publizierten RNA-Daten bestätigt werden. EKGUntersuchungen an globalen RyR3KO-Tieren zeigten allerdings keinen Unterschied in der Herzfrequenz oder Herzfrequenzvariabilität der KO-Tiere im Vergleich zu Wildtyp-Mäusen. Summary V Summary The intracellular calcium release channel RyR2 governs the release of Ca2+ from the sarcoplasmic reticulum which initiates muscle contraction. In addition, RyR2 is proposed to play a crucial role in controlling sinoatrial node (SAN) pacemaking. Since global deletion of RyR2 in mouse models is embryonically lethal, the role of RyR2 in the adult mouse heart, especially in the SAN, has been unknown. Objectives of this thesis were to study the role of RyR2 in the whole heart and only in the pacemaker conduction system using two different tamoxifen inducible mouse lines. In addition, expression of isoform RyR3 in the heart was to be confirmed and its role analysed by telemetric electrocardiogram recordings. The RyR2MerCreMer-mouse line was used to study the role of the channel in the whole heart. Mice with cardiomyocyte specific knockout of RyR2 died within two weeks after knockout induction and showed significant atrial dilation and fibrosis. Moreover, the degenerated morphology and increased size of isolated cardiomyocytes indicated alterations induced by the knockout also on the cellular level. In contrast, mice with deletion only in the pacemaker conduction system (RyR2KiT-KO) were viable and had no morphological changes of the heart or SAN. Telemetric electrocardiogram recordings revealed that RyR2 deletion results in bradycardia. Heart rate modulation with pharmacological substances was still possible in RyR2KiT-Cre KO mice. However, heart rates never reached similar values as the controls. In addition RyR2KiT-KO mice had reduced heart rate variability. Analysis of the intracellular Ca2+ levels demonstrated reduced basal Ca2+ levels measured in isolated RyR2KO SAN cells. These results strongly suggest that sarcoplasmic reticulum Ca 2+ release by RyR2 plays a dominant role in contraction of the heart as well as in sinoatrial pacemaking. In addition, it could be demonstrated that the protein of the isoform RyR3 is also expressed in the SAN and atria, thus confirming published RNA-data on the subject. Electrocardiogram recordings of RyR3 knockout mice, however showed no differences in the heart rate or heart rate variability compared to wild type mice. Einleitung 1 1. Einleitung 1.1 Das Herz Die Aufgabe des Herzens ist es, regelmäßig und koordiniert Blut in die Organe zu pumpen und so jede Zelle des Körpers mit Sauerstoff und Nährstoffen zu versorgen. Es besteht aus zwei Vorhöfen (rechtes und linkes Atrium) und zwei Herzkammern (rechter und linker Ventrikel), durch welche ständig Blut in das Kreislaufsystem gepumpt wird. Dies wird durch einen fortwährenden Ablauf von Kontraktion (Systole) und Relaxation (Diastole) des Herzmuskels gewährleistet. Jeder Herzschlag wird durch eine elektrische Erregung im Sinusknoten (SAN, sinoatrial node) ausgelöst. Dieser arbeitet autonom und wird auch als Schrittmacher des Herzens bezeichnet. Durch die spontane Depolarisation im Sinusknoten wird ein Aktionspotential (AP) generiert und das Signal über den Atrioventrikularknoten (AV-Knoten), das His-Bündel und die Purkinje-Fasern an das Myokard weitergegeben, woraus die Kontraktion des Herzens resultiert. Die kontraktile Funktion der Herzmuskelzellen wird in erster Linie durch die intrazelluläre Regulation von Calcium-Ionen (Ca2+) gesteuert. Eine Vielzahl membranständiger und cytosolischer Proteine ist daran beteiligt, die CalciumHomöostase aufrecht zu erhalten. 1.2 Die elektromechanische Kopplung des Herzens Der auslösende elektrische Reiz, der zur Kontraktion im Herzen führt, ist das Aktionspotential. Die elektromechanische Kopplung beschreibt den Prozess zwischen dem AP einer Herzmuskelzelle und der darauf folgenden Kontraktion. Um eine normale Herzfunktion zu 2+ gewährleisten, ist eine exakte Kontrolle der intrazellulären 2+ Ca -Konzentration [Ca ]i notwendig. Sobald ein Aktionspotential ausgelöst wird, erhöht sich kurzzeitig die Permeabilität der Zellmembran für Natriumionen (Na+). Durch den folgenden Na+-Einstrom ins Cytosol wird die Zellmembran, die den Intrazellulärraum von der extrazellulären Matrix abgrenzt und an einigen Stellen tubuläre Einstülpungen (T-Tubuli) aufweist, depolarisiert. Im Bereich der T-Tubuli sind spannungsabhängige L-Typ Ca2+-Kanäle lokalisiert, die sich bei der Depolarisation der Zellmambran öffnen. Ca2+ fließt als Einwärtsstrom (ICa) in das Cytosol der Zelle und trägt zur Plateauphase des Aktionspotentials der Herzmuskelzellen bei (Fabiato 1983, Bers 2002, Kushnir und Marks 2010). Durch den Anstieg der Ca2+-Konzentration im Cytosol werden die Ryanodinrezeptoren (RyR), welche die Membran des sarkoplasmatischen Retikulums (SR) durchspannen, aktiviert und Ca2+ strömt aus dem SR ebenfalls in das Cytosol. Dies wird als Ca2+-induzierte Ca2+-Freisetzung (calcium induced calcium release (CICR)) bezeichnet (Fabiato und Fabiato, Einleitung 2 1975; Sham et al, 1995). Das SR fungiert dabei als Ca2+-Speicher der Zelle. Die erhöhte cytoplasmatische Ca2+-Konzentration wiederum bewirkt, dass die Aktin-Myosin-Interaktion in den Myofibrillen aktiviert wird. Dabei wird Ca2+ durch die calciumbindende Untereinheit des Troponins, das Troponin C, gebunden und es kommt zu einer Konformationsänderung des Troponinkomplexes, wodurch die Wechselwirkung zwischen Tropomyosin und Aktin beeinflusst wird. In Folge dessen kann Myosin an Aktin binden und löst so die Kontraktion des Herzens aus. Damit das Myokard nach der Kontraktion wieder relaxieren kann, muss die Ca2+-Konzentration im Cytosol wieder erniedrigt werden. Dabei ist der Rücktransport von cytosolischem Ca2+ in das SR von größter Bedeutung. Die SR-Ca2+-ATPase (SERCA) stellt dabei den Hauptmechanismus zur Entfernung des Ca2+ aus dem Cytosol dar. Dies erfolgt unter Verbrauch von einem Molekül ATP (Adenosintriphosphat) pro Transport von zwei Ca2+. Die Aktivität der SERCA wird durch das Protein Phospholamban (PLB) kontrolliert (Tada et al, 1974). In seiner unphosphorylierten Form bindet PLB an die SERCA und inhibiert bei niedriger cytoplasmatischer Ca2+-Konzentration die SERCA-Aktivität. Kommt es zu einer Phosphorylierung von PLB durch die Protein Kinase A (PKA) oder Calmodulinkinase II (CaMKII), löst sich das PLB von der SERCA, die Hemmung der SERCA wird aufgehoben und Ca2+ kann zurück in das SR gepumpt werden. Zusätzlich zur SERCA kommt der Na+/Ca2+-Austauscher (NCX) in der Plasmamembran zum Einsatz. Dieser pumpt ein Ca 2+ im Austausch zu drei Na+ aus dem Cytosol heraus. Der zusätzliche Ca2+-Ausstrom durch die Ca2+-ATPase in der Zellmembran und die Aufnahme von Ca2+ über Uniporter in die Mitochondrien, spielen beim Ca2+-Transport aus dem Cytosol nur eine untergeordnete Rolle (Bers, 2002). Wichtig bei diesem System ist, dass die Menge an Ca 2+, welche in die Zelle eintritt, unter normalen Bedingungen genau dieselbe ist, die durch den NCX wieder herausgepumpt wird. Eine schematische Darstellung der intrazellulären Ca2+-Bewegungen im Myokard im Verlauf eines Herzzyklus ist in Abbildung 1.1 gezeigt. Einleitung 3 Abbildung 1.1: Schematische Darstellung der elektromechanischen Kopplung im Herzen. Die roten Pfeile stellen den Ca2+-Einstrom und die Ca2+-induzierte Ca2+-Freisetzung während der Systole dar. Die grünen Pfeile stellen den Rücktransport des Ca2+ ins Sarkoplasmatische Retikulum (SR) und den Ca2+-Ausstrom durch den NCX während der Relaxation dar. RyR, Ryanodin-Rezeptor; NCX, Na+-Ca2+-Austauscher; ATP, Ca2+-ATPase; PLB, Phospholamban. Im Kasten ist die Beziehung von Aktionspotential (AP, schwarz) zu intrazellulärer Ca2+-Konzentration ([Ca]I, blau), und zur Kontraktion (rot) verdeutlicht (modifiziert nach Bers, 2002). 1.3 Der Ryanodinrezeptor Ryanodinrezeptoren (RyR) sind intrazelluläre Ca2+-Freisetzungskanäle, die die Membran des endoplasmatischen Retikulums (ER) oder des SR durchspannen und für den Ca2+- Ausstrom aus dem ER/SR in das Cytosol verantwortlich sind (Bers, 2002). Sie existieren als Homotetramere, wobei jede Untereinheit aus etwa 5000 Aminosäuren mit einer molekularen Masse von ca. 565 kDa besteht (Fill und Copello, 2002). Damit gehören sie zu den größten bisher identifizierten Ionenkanälen. RyR-Monomere bestehen zu 4/5 aus einer großen cytoplasmatischen Domäne, die Bindungsstellen für unterschiedliche Modulatoren von RyR beinhaltet und zu 1/5 aus einer Transmembrandomäne. Der Name der Rezeptoren wurde von dem pflanzlichen Alkaloid Ryanodin abgeleitet, welches von RyR mit hoher Affinität gebunden wird und bei einer Konzentration von über 10 µM zu einer Inhibition der Ca2+Freisetzung führt (Li et al, 1997; Rubenstein und Lipsius, 1989). Bisher sind drei RyRIsoformen bekannt: RyR1, RyR2 und RyR3. Die drei RyR-Isoformen besitzen untereinander eine Homologie von ca. 65 %. RyR1 wird hauptsächlich in der Skelettmuskulatur exprimiert, wurde aber auch in geringen Mengen in anderen Geweben nachgewiesen (Takeshima et al, Einleitung 4 1989). RyR2 wird als die kardiale Form angesehen, kommt aber auch in glatten Muskel- und Gehirnzellen vor (Otsu et al, 1990; Tunwell et al, 1996). RyR3 wird hauptsächlich im Gehirn exprimiert (Sorrentino und Volpe, 1993; Takeshima et al, 1998). Zudem konnte, bis jetzt allerdings nur auf RNA-Ebene, nachgewiesen werden, dass neben RyR2 ebenfalls RyR3 im Herzen vorkommt. Im Gegensatz zu RyR2, der im ganzen Herzen exprimiert wird, konnte RyR3 allerdings nur im Sinusknoten und in den Vorhöfen nahgewiesen werden (Tellez et al, 2006). Welche wichtige Rolle RyR hat, konnte durch folgende Beispiele gezeigt werden: Mäuse, bei denen RyR1 deletiert ist, sterben perinatal an Ateminsuffizienz, welche durch Versagen der Skelettmuskulatur bedingt ist (Takeshima et al, 1994). Ein Fehlen des Gens für den RyR2 führte zur embryonalen Letalität (Takeshima et al, 1998). Im Gegensatz dazu sind RyR3-Knockout-Mäuse lebensfähig und zeigen keine veränderten Muskelfunktionen (Takeshima et al, 1996; Bertocchini et al, 1997). Fehlerhafte Regulation von RyR2 und die damit verbundene Störung in der Ca2+-Homöostase wird als Ursache für Herzinsuffizienz, Hypertrophie und Arrhythmien angesehen (Kushnir et al, 2010; Betzenhauser und Marks, 2010). 1.4 Die Rolle von RyR2 im Reizleitungssystem Die kardiale Isoform RyR2 soll neben ihrer Rolle bei der Kontraktion des Herzmuskels, auch eine wichtige Rolle bei der Generierung des Schrittmacherpotentials im Sinusknoten spielen (Maltsev et al, 2006; Lakatta et al, 2010). Wie bereits erwähnt sind Sinusknotenzellen spontan aktiv und jeder Herzschlag beginnt mit einem Aktionspotential im Sinusknoten. Das Aktionspotential einer Sinusknotenzelle zeichnet sich durch eine langsame, spontane Depolarisationsphase und ein relativ positives Ruhemembranpotential aus (Abb. 1.2). Im Vergleich zu Myokardzellen fehlt dem Sinusknotenaktionspotential eine ausgeprägte Plateauphase. Die Frequenz, mit der das Herz schlägt wird über die Zeitspanne zwischen zwei Aktionspotentialen definiert. Verkürzt sich diese, so erhöht sich die Frequenz und das Herz schlägt schneller. Die Länge dieser Zeitspanne ist dabei insbesondere abhängig von der diastolischen Depolarisation (DD). Eine Modulation der Steigung der DD z.B. mittels Pharmaka oder sportlicher Betätigung, moduliert die Herzfrequenz (Abb.1.2). Einleitung 5 Abbildung 1.2: Schematische Darstellung eines Aktionspotentials und die dazu beitragenden Ionenströme und Ca2+-Freisetzungen in einer Sinusknotenzelle. Während der diastolischen Depolarisation führen die Inaktivierung des Kaliumstroms (IK) und die Aktivierung des HCN-Stroms (If) zur Depolarisation der Zelle. Der anschließende Ca2+-Einstrom über L- und T-Typ-Ca2+*-Kanäle (ICa,L und ICa,T), sowie die Ca2+-Freisetzung über die RyR sind für die Aufstrichphase verantwortlich. Durch Rücktransport des Ca2+ in das SR und durch Aktivierung der Kaliumkanäle wird die Zelle im Anschluss repolarisiert (modifiziert nach Maltsev und Lakatta, 2007). Welche Komponenten Einfluss auf die Regulation der DD haben, konnte bis jetzt noch nicht eindeutig geklärt werden. Einerseits wird dem If-Strom, der durch die spannungsabhängigen HCN-Kanäle (Hyperpolarization-activated and cyclic nucleotide gated cation channels) (Brown et al, 1979; Ludwig et al, 1998) generiert wird, eine tragende Rolle bei der DD zugeschrieben, weshalb er als so genannter Schrittmacherstrom bezeichnet wird. Andererseits soll dabei auch die intrazelluläre Ca2+-Freisetzung über den RyR2 von großer Bedeutung sein (Lakatta und DiFrancesco, 2009). Zur Funktion von RyR2 im Sinusknoten gibt es zwei Theorien: Erstens konnte gezeigt werden, dass im SAN von Kaninchen mehr zyklisches Adenosinmonophosphat (cAMP) gebildet wird als in Kardiomyozyten (Vinogradova et al, 2006). Dies hat zur Folge, dass durch den erhöhten cAMP-Gehalt die Phosphorylierung verschiedener Ca2+-regulierender Proteine gesteigert wird. So werden sowohl die membranständigen Ca2+-Kanäle, RyR2, als auch der NCX stärker aktiviert. Dadurch gelangt zum einen mehr Ca2+ durch die Ca2+-Kanäle in das Zellinnere, zum anderen strömt durch die stärkere Aktivierung von RyR2 vermehrt Ca2+ aus dem SR in das Cytosol. Damit die Zelle wieder relaxieren kann, muss somit der NCX ebenfalls verstärkt zum Einsatz kommen. Dieser pumpt wie bereits in Abschnitt 1.2 beschrieben ein Ca 2+ aus der Zelle heraus, im Austausch jedoch drei Na+ wieder in die Zelle herein. Durch die verstärkte Aktivität des NCX wird daher ein Netto-Einwärtsstrom erzeugt welcher die DD beeinflussen soll (Yang et al, 2002; Bogdanov et al., 2001 und 2006). Als Zweites konnte gezeigt werden, dass im Sinusknoten von Kaninchen die Adenylatcyclase-Isoformen (AC) fünf und sechs, Einleitung 6 sowie die Isoformen eins und acht exprimiert werden, welche die Synthese von cAMP katalysieren können. Im Gegensatz zu AC5 und AC6 können AC1 und AC8 allerdings direkt durch Ca2+ aktiviert werden (Younes et al, 2008). Da der intrazelluläre Ca2+-Gehalt einer Zelle hauptsächlich vom Ca2+-Ausstrom aus dem SR durch den RyR2 abhängt, soll RyR2 somit auch direkt den cAMP-Spiegel beeinflussen können. cAMP reguliert zum einen den IfStrom, der durch HCN-Kanäle generiert wird, zum anderen beeinflusst cAMP über die PKA die Phosphorylierung verschiedenster Proteine, die am Calcium-Signalweg beteiligt sind, sowie die Regulation weiterer Ionenkanäle (Ludwig et al, 1998; Lakatta und DiFrancesco, 2009). Eine schematische Darstellung der Signalwege, die bei der Generierung des Schrittmacherpotentials beteiligt sein sollen, ist in Abb. 1.3 dargestellt. Abbildung 1.3: Schematische Darstellung der basalen Schrittmacherregulation. Der cAMP-Gehalt einer Sinusknotenzellen wird durch intrazelluläre Ca2+-Signalwege (rot) und verschiedene Ionenkanäle beeinflusst. Im Sinusknoten sollen im Vergleich zum restlichen Herzen zusätzlich auch die Adenylatcylasen AC1 und AC8 exprimiert werden (modifiziert nach Vinogradova und Lakatta, 2009). 1.5 Konditionelle Knockout-Tiere: das Cre/loxP-System Die Möglichkeit, das Mausgenom durch zufällige Integration von Transgenen oder gezielt über homologe Rekombination in embryonalen Stammzellen zu verändern, hat entscheidend dazu beigetragen, die Funktion von Genen besser zu verstehen. Da diese Techniken eine permanente genetische Modifikation in der Keimbahn der Maus erzeugen, führt das konventionelle Ausschalten eines Gens (globaler Knockout) in allen Zellen des Tieres während der prä- und postnatalen Entwicklung zum Verlust der Genfunktion. Ein solcher Einleitung 7 Gen-Knockout kann zu embryonalem Tod oder Tieren mit schweren Defekten und stark eingeschränkter Lebensfähigkeit führen. Eine Alternative zu totalen Knockout-Tieren bietet das Cre/loxP-System. Die Cre-Rekombinase (cyclization recombination) ist ein Protein des Bakteriophagen P1 und katalysiert die Rekombination zwischen zwei loxP-Erkennungssequenzen („locus of crossing over of P1“), ohne dass die Rekombinase hierzu einen Cofaktor benötigt. Abhängig von der Orientierung der loxP-Sequenzen untereinander und dem Ort ihrer Integration kann man durch Cre-vermittelte Rekombination eine Inversion, Exzision/Integration oder Translokation von DNA erzeugen. Um Veränderungen des Genoms in vivo durchzuführen, wird das Herausschneiden von loxP-flankierten („gefloxten“) DNA-Segmenten verwendet (Hoess and Abremski, 1990). In Abbildung 1.4 ist die Exzision mit Hilfe des Cre/loxP-Rekombinasesystems schematisch dargestellt. Abbildung 1.4: Schema des Cre/loxPRekombinationssystems. Cre verursacht die Rekombination zwischen zwei loxP Erkennungssequenzen (Dreiecke). Die loxP Sequenz (unten) besteht aus zwei 13 bp Palindromen (waagrechte Pfeile), die eine asymmetrische 8 bp „core“ Region flankieren. Die Phosphodiesterbindungen, die Cre spaltet, sind durch senkrechte Pfeile gekennzeichnet. Die Rekombination zweier gleichgerichteter loxP Sequenzen in derselben DNA führt zur Exzision des flankierten Segments und es bleibt lediglich eine loxP Sequenz übrig. Um eine gewebespezifische Deletion zu erreichen, muss die Cre-Rekombinase unter einem gewebsspezifischen Promoter exprimiert werden. Dadurch wird die Rekombinase ebenfalls nur in einem bestimmten Gewebe exprimiert und deletiert nur dort die gefloxte DNA-Region (Metzger und Feil, 1999). 1.6 Induzierbare konditionelle RyR2-Knockoutmäuse In dieser Arbeit wurden zwei unterschiedliche konditionelle Mauslinien generiert und charakterisiert, in denen zum einen RyR2 im ganzen Herzen, zum anderen RyR2 spezifisch nur im Reizleitungssystem deletiert wurde. Mit Hilfe der konditionellen Methode war es möglich, eine Gendeletion im adulten Tier hervorzurufen. Bei der Deletion im ganzen Herzen wurde ein Cre-Fusionsprotein verwendet, das zwischen zwei mutierten Östrogenrezeptorbindungsdomänen (MerCreMer) liegt. Die Transkription dieses Transgens steht unter der Kontrolle des herzspezifischen α-Myosin schweren Kette (αMHC) Promotors und wird somit nur im Herzen exprimiert (Sohal et al, 2001). Zum anderen wurde als CreProtein das HCN4KiT-Cre, das mit einer modifizierten Ligandenbindungsdomäne Einleitung 8 (Punktmutationen: G400V/M543A/L544A) des Östrogenrezeptors fusioniert ist, verwendet (Hoesl et al, 2008). Diese Cre-Rekombinase steht unter der Kontrolle des endogenen HCN4Promotors. Da HCN4 im Herzen ein auf das Reizleitungssystem beschränktes Expressionsmuster besitzt, findet der Knockout nur im Reizleitungssystem statt. Durch die Modifikationen besitzt endogenes Östrogen bei beiden Cre-Fusionsproteinen keine Affinität mehr zu den Ligandenbindungsdomänen, die Affinität des SERMs Tamoxifen ist jedoch hoch (Feil et al, 1997; Kühbandner et al, 2000). Erst nach Applikation und Bindung von Tamoxifen an die Ligandenbindungsdomäne des Fusionsproteins wird der Proteinkomplex aktiviert, das Fusionsprotein kann in den Zellkern gelangen und vermittelt die Rekombination an den loxPSequenzen. Mit Hilfe der beiden unterschiedlichen Mauslinien RyR2MerCreMer und RyR2KiT können adulte herz- bzw. reizleitungsspezifische RyR2-KO-Mäuse generiert und auf funktionaler und molekularer Ebene charakterisiert werden. 1.7 Ziel dieser Arbeit Ryanodinrezeptoren (RyR) spielen eine wichtige Rolle bei der Funktion des Herzens. Dabei sollen sie sowohl bei der Kontraktion des Herzmuskels als auch bei der Generierung des Schrittmacherpotentials wichtig sein, ihre genaue Funktion ist allerdings noch unklar. Welche spezifische Rolle der Ryanodinrezeptor RyR2 sowohl bei der Kontraktion des Herzens als auch bei der Generierung eines Schrittmacherpotentials hat, sollte im Rahmen dieser Arbeit untersucht werden. Da die globale Deletion von RyR2 embryonal letal ist (Takeshima et al, 1998), sollten in dieser Arbeit zwei konditionelle Mauslinien generiert werden, bei denen die Deletion von RyR2 erst im adulten Tier erfolgt. Der herzspezifische sowie der reizleitungsspezifische RyR2-Knockout sollte mit Hilfe molekularbiologischer Methoden bestätigt werden. Die Auswirkungen der RyR2-Deletion sollten auf morphologischer als auch funktioneller Ebene bestimmt werden. Zudem sollte die Rolle einer weiteren RyR Isoform RyR3 bestimmt werden, über deren Funktion im Herzen bisher wenig bekannt war. Material und Methoden 2. 9 Material und Methoden Sämtliche gentechnische Versuche nach S1 wurden von der Regierung von Mittelfranken nach Aktenzeichen 50-8791.2.38/39 genehmigt. 2.1 DNA-Techniken 2.1.1 Isolierung genomischer DNA zur Genotypisierung Zur Genotypisierung wurde das Mausgewebe (Zehen- oder Schwanzbiopsie) über Nacht bei 55 °C in 100 µl Lysepuffer verdaut (Schüttler: Thermomixer Compact, Eppendorf). Am nächsten Tag wurde das Lysat zur Inaktivierung der Proteinase K bei 95 °C für 15 min gekocht. Die isolierte DNA konnte bei 4 °C gelagert werden. Lysepuffer: Proteinase K (20 mg/ml, Roth) 2 µl 10x PCR Puffer 10 µl H2O 88 µl 10x PCR Puffer pH 9,0: Tris/HCL 1,58 g (100 mM) MgCl2 0,304 g (15 mM) KCl 3,73 g (500 mM) H2O ad 100 ml 2.1.2 Isolierung genomischer DNA aus Mausherz Gesamt DNA aus den unterschiedlichen Kompartimenten des Herzens (rechter und linker Ventrikel, rechter und linker Vorhof und Sinusknotengewebe) wurde mit Hilfe des DNeasy Tissue Kits (Qiagen) nach Herstellerangaben isoliert. Die eluierte DNA wurde bei 4 °C gelagert. Material und Methoden 10 2.1.3 Polymerasekettenreaktion (PCR) Die Polymerasekettenreaktion (PCR) ist eine in vitro Technik zur Amplifizierung einer spezifischen Polynukleotidsequenz (Mullis et al, 1986). Sie erfolgt in drei Schritten, deren Abfolge temperaturgesteuert ist: Im ersten Schritt wird die doppelsträngige Nukleinsäuresequenz durch Hitzeeinwirkung (90-94 °C) aufgeschmolzen (= Denaturierung). Beim anschließenden Absinken der Temperatur hybridisieren zwei Oligonukleotid-Primer an komplementäre Sequenzen der Ziel-Sequenz (= Primer-Anlagerung). Nach dem Anlagerungsschritt werden die hybridisierten Primer an ihrem 3`-Ende durch die hitzestabile DNA-Polymerase aus dem Bakterium Thermophilus aquaticus (Taq-Polymerase) verlängert (= Elongation). Die beiden entstandenen Nukleotid-Stränge bilden die Matrize für den nächsten Zyklus. Bei einer Zyklenzahl von 30-50 kommt es auf diese Weise zu einer exponentiellen Vervielfältigung der Sequenz. Die PCR-Produkte wurden mittels Gelelektrophorese aufgetrennt und mittels Ethidiumbromid unter UV-Licht sichtbar gemacht. PCR-Ansatz: Primer 1 (10 pmol/µl) 1,25 µl Primer 2 (10 pmol/µl) 1,25 µl Primer 3 (10 pmol/µl) (abhängig vom Genotyp) 1,25 µl 10x PCR Puffer 2,5 µl dNTPs (1,25 mM) 4,0 µl H2O 12,4 µl Taq DNA-Polymerase (5 U/µl ) (selbst hergestellt) DNA (~100 ng/µl) 0,35 µl 2,0 µl PCR-Programm für Genotypisierung: 95 °C 5 min 95 °C 30 s 65 °C -1 °C / Zyklus 30 s 72 °C 30 s 10x Material und Methoden 11 95 °C 30 s 55 °C 30 s 72 °C 30 s 72 °C 5 min 28x Die Reaktionen wurden in einer PCR-Maschine (Mastercycler, Eppendorf) durchgeführt. Ein Aliquot der PCR-Reaktion (10 µl) wurde auf ein ethidiumbromidhaltiges 2 % Agarosegel aufgetragen und elektrophoretisch aufgetrennt. Das Gel wurde unter UV-Licht dokumentiert. PCR-Primer: Die in der vorliegenden Arbeit verwendeten Primer wurden bei MWG-Biotech, Ebersberg, synthetisiert. Die Sequenzen sind im Anhang (siehe Abschnitt 5.1) aufgeführt. Die lyophilisierte Primer-DNA wurde in soviel H2Odest. gelöst, dass die Konzentration 100 pmol/µl betrug. Davon wurden Arbeitslösungen von 10 pmol/µl hergestellt. 2.1.4 Agarose-Gelelektrophorese Die DNA-Proben wurden mit 6x Probenpuffer versetzt und zur Analyse in 1 % oder 2 % TBEgepufferten Agarose-Gelen elektrophoretisch aufgetrennt. Für die Größenbestimmung der DNA wurde ein DNA-Längenstandard (Fermentas) verwendet. Die aufgetrennte DNA konnte durch den interkalierenden Fluoreszenzfarbstoff Ethidiumbromid (Roth) unter UV-Licht als Bande sichtbar gemacht werden. Zur Dokumentation wurde das Agarose-Gel auf einem UV-Transilluminator aufgenommen. 10x TBE-Elektrophorese-Puffer pH 8,0: Tris (Roth) 108 g (890 mM) Borsäure (Roth) 55 g (890 mM) EDTA (Roth) 7.4 g (20 M) H2O ad 1000 ml Ficoll Typ 400 (AppliChem) 18 g (18 %) EDTA (Roth) 24 ml (0,12 mM) 10x TBE 60 ml Bromphenolblau (Sigma) 3 ml (aus 50 mg/ml Lösung) Xylencyanol FF (Sigma) 3 ml (aus 50 mg/ml Lösung) 6x Probenpuffer: Material und Methoden 2.2 12 RNA-Techniken 2.2.1 Isolierung von Gesamt-RNA Zur RNA-Isolierung aus Gewebe wurde das Herz in eiskaltem PBS in rechten und linken Ventrikel, rechter und linker Vorhof und Sinusknoten geteilt. Die Isolierung von RNA wurde mit dem RNeasy Micro Kit (Qiagen) für Gewebe laut Herstellerangaben durchgeführt. Zum Schluss wurde die RNA in 30 μl RNase-freiem Wasser aufgenommen. 2.2.2 Semiquantitative RT-PCR aus Gewebe Da RNA nicht als Template in einer PCR fungieren kann, muss sie erst in ihre komplementäre DNA (cDNA) umgeschrieben werden. Dies geschieht mit Hilfe der Reversen Transkriptase, einer RNA-abhängigen DNA-Polymerase. Die reverse Transkriptions-PCR (RT-PCR) wurde mit dem „OneStep RT-PCR Kit“ (Qiagen) nach Herstellerangaben durchgeführt. Nach der PCR-Reaktion wurden die Proben auf einem Agarosegel aufgetrennt und visualisiert. Komponenten: Endkonzentration: 5x OneStep RT-PCR Puffer 1x dNTPs 400 μM dNTP Primer 1 0,6 μM Primer 2 0,6 μM OneStep RT-PCR Enzym Mix RNase Inhibitor 5-10 U/Reaktion Template RNA 1 pg bis 2 μg H2O, RNase frei ad 25 μl Material und Methoden 13 RT-PCR-Programm: 50 °C 30 min 95 °C 15 min 94 °C 30 s 65 °C -1 °C / Zyklus 30 s 72 °C 30 s 94 °C 30 s 55 °C 30 s 72 °C 30 s 72 °C 10min 10x 28x 2.2.3 Semiquantitative RT-PCR aus isolierten Sinusknotenzellen Zum Nachweis von RNA aus einzelnen isolierten Sinusknotenzellen wurden die Zellen, wie in Abschnitt 2.5 beschrieben, isoliert. Danach wurden die Zellen behutsam mechanisch vereinzelt, auf Poly-L-Lysin beschichtete Deckgläschen ausgesät und mit einer Pipette einzeln aufgesaugt. Anschließend wurden die Zellen in ein Eppendorfgefäß mit 4 µl des 5x Puffers („OneStep RT-PCR Kit“ (Qiagen)) und 16 µl H2O gegeben und in flüssigem Stickstoff schockgefroren. Die PCR wurde mit dem „OneStep RT-PCR Kit“ (Qiagen) nach Herstellerangaben durchgeführt. RT-PCR-Programm: 50 °C 30 min 50 °C 30 min 95 °C 15 min 95 °C 30 s 65 °C -1 °C / Zyklus 30 s 72 °C 1 min 95 °C 30 s 55 °C 30 s 72 °C 1 min 72 °C 5 min 10x 28x Material und Methoden 2.3 14 Protein-Techniken 2.3.1 Isolierung und Präparation von Proteinen Für die Proteinisolation aus Herzgewebe wurden jeweils kleine Stücke des rechten und linken Ventrikels, sowie des rechten und linken Vorhofs verwendet. Für Protein aus Sinusknotengewebe wurde ein Gewebestück (~1 mm2) vom Übergang der Vena cava superior in den rechten Vorhof unter einem Stereomikroskop (Zeiss) exzidiert. Die isolierten Gewebestückchen wurden in Lysepuffer (2 % SDS / 50 mM Tris, pH 7,5 + 1x Protease Inhibitor Cocktail (Roche)) gegeben und für 2 min in einem Gewebehomogenisator homogenisiert. Anschließend wurde das Lysat 10 min bei 95 °C erhitzt und bei 13000 rpm für 5 min zentrifugiert. Der Überstand, in dem sich die Proteine befanden, wurde aliquotiert und bei -80 °C gelagert. 2.3.2 Proteinbestimmung mit BCA Um die Proteinkonzentration bestimmen zu können, wurde der BCA-Test (BCA Protein Assay Kit; Pierce) durchgeführt. Dieser Proteinbestimmung liegt zu Grunde, dass Cu2+-Ionen in alkalischer Umgebung von Proteinen zu Cu+-Ionen reduziert werden. Durch die Zugabe von BCA (2,2-Bis-(chinolin-4-) carbonsäure) entsteht ein BCA-Cu+-Komplex, der sich violett färbt und mit Hilfe von BSA-Standardproben (Albumin Standard, Pierce) quantifiziert werden kann. Die Extinktion dieses Komplexes kann bei 550 nm bis 630 nm gemessen werden. Da die Intensität der Färbung von Reaktionszeit und Temperatur abhängt, muss bei jeder Bestimmung eine Eichkurve mit Lösungen bekannter BSA-Konzentration angefertigt werden. Das instabile BCA Reagenz wurde jeweils kurz vor Gebrauch aus dem Reagenz A und B (BCA Protein Assay Kit, Pierce) im Verhältnis 50 zu 1 angesetzt, mit der verdünnten Proteinlösung (1:50 bis 1:500) gemischt und für 60 min bei 60 °C inkubiert. Die Farbreaktion wurde über einen ELISA-Reader (Dynatech Laboratories) ausgewertet. Anhand der Werte der Standards konnte die einzusetzende Proteinkonzentration berechnet werden. 2.3.3 SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE) Die Auftrennung von Proteinen nach ihrem Molekulargewicht erfolgte durch eine SDSPolyacrylamid-Gelelektrophorese nach Laemmli (Laemmli, 1970). Bei dieser Methode wird das Detergens SDS zugegeben, welches die natürliche Ladung der Proteine überdeckt und somit die Proteine nur aufgrund ihres Molekulargewichts aufgetrennt werden. Es wurde eine Gel Apparatur der Firma Biorad (Mini Protean II Cell) verwendet. Zur Herstellung der SDSPolyacrylamidgele wurde das Trenngel bis ca. 2 cm unter den oberen Rand gegossen und Material und Methoden 15 mit Ethanol überschichtet. Nach der Polymerisation wurde das Ethanol abgehoben, das Sammelgel darauf gegossen und der Kamm eingesetzt. Benutzt wurde ein 4 %-iges Trenngel für den Nachweis von RyR2 und RyR3 sowie ein 15 %-iges Trenngel für den Nachweis von Phospholamban. Die vorbereiteten Proteinlysate (mit 6x Probenpuffer versehen und 5 min bei 95 °C inkubiert) sowie ein Proteinstandard (Precision Plus Protein Standard, Biorad) wurden auf das Gel aufgetragen. Die Proteinlysate wurden bei 130 V elektrophoretisch getrennt. 4x Trenngelpuffer (pH 8,8): 4x Sammelgelpuffer (pH 6,8): Tris/Base 18,2 g (1,5 M) Tris/Base 3,02 g (0,5 M) SDS 0,4 g (0,4 %) SDS 0,2 g (0,2 %) H2O ad 100 ml H2O ad 50 ml 15 % bzw. 4 % Trenngel: Sammelgel: Acrylamid 7,5 bzw. 2,0 ml Acrylamid 4x Trenngelpuffer 3,75 ml 4x Sammelgelpuffer 1,25 ml H2O 3,75 bzw. 9,25 ml H2O 3,05 ml 20 % APS (Roth) 25 μl 20 % APS (Roth) 12,5 μl TEMED (Roth) 10 μl TEMED (Roth) 5 μl 6x Laemmli-Puffer: 0,65 ml 10x SDS-Laufpuffer pH 8,3: 4x Tris/SDS, pH 6,8 7 ml Tris/Base (Roth) 30,2 g (250 mM) Glycerin (VWR) 3 ml (30 %) Glycin (Roth) 144 g (1,92 M) DTT (Roth) 0,93 g (0,6 M) SDS (AppliChem) 10 g (1 %) Bromphenolblau (Sigma) 1,2 mg (0,012 %) H2O ad 1000 ml H2O ad 10 ml Acrylamid: 30 % Acrylamid / 0,8 % Bisacrylamid (Rotiphorese Gel 30, Roth) Material und Methoden 16 2.3.4 Western-Blot Analyse Der Protein-Transfer wurde im Semidry-Elektroblot (V20 SDB, Biometra) durchgeführt. Dazu wurden drei Filterpapiere (G002, Whatman) in Anodenpuffer I (AP I), zwei in Anodenpuffer II (AP II) und fünf in Kathodenpuffer (KP) präinkubiert. Die Membran wurde zuerst in Methanol, dann in Anodenpuffer II getunkt. Auf die Anode wurden zuerst die Filterpapiere aus dem Anodenpuffer I und II gelegt. Darauf wurden die Membran (Immobilon-P, Millipore) und das Gel gelegt. Zum Schluss folgten die im Kathodenpuffer präinkubierten Filterpapiere (Abb. 2.1). Abbildung 2.1: Aufbau der Blotting Apparatur. Luftblasen zwischen den einzelnen Schichten wurden sorgfältig entfernt. Das Elektroblotting erfolgte bei 32 mA (0,76 mA pro 1 cm2 Membran) für 1,5 h. Anodenpuffer I, pH 10,4: Anodenpuffer II, pH 10,4: Tris/Base 36,3 g (300 mM) Tris/Base 3,03 g (25 mM) Methanol (Roth) 200 ml Methanol (Roth) 200 ml H2O ad 1000 ml H2O ad 1000 ml Kathodenpuffer, pH 7,6: TBST, pH 8,2: γ-Aminohexansäure (Sigma) 5,2 g (40 mM) 10x TBS 100 ml Tris/Base 3,03 g (25 mM) Tween 20 (VWR) 1 ml Methanol (Roth) 200 ml H2O ad 900 ml H2O ad 1000 ml Material und Methoden 17 10x TBS, pH 8,2: Tris/Base 12,1 g (10 mM) NaCl 87,6 g (150 mM) H2O ad 1000 ml Immundetektion: Nach dem Blotten wurde die Membran 2 h bei Raumtemperatur in Blockinglösung (3 % BSA (Bovines Serum Albumin) in 1x TBST) geschwenkt. Die Membran wurde anschließend mit 1x TBST gewaschen und mit einem spezifischen primären Antikörper (Antikörper und Verdünnungen siehe Anhang 5.2) über Nacht bei 4 °C inkubiert. Am nächsten Tag wurde nach drei zehnminütigen Waschschritten mit 1x TBST die Membran mit dem jeweiligen Meerettich-Peroxidase-gekoppelten, sekundären Antikörper für 1 h bei RT inkubiert. Nach drei Waschschritten (jeweils 10 min in 1x TBST) wurden die nachzuweisenden Proteine mit Hilfe einer Chemilumineszenz Reaktion der Meerrettich-Peroxidase detektiert. Dazu wurden die beiden Lösungen des Western Lightning Chemiluminsecence Reagenz (PerkinElmer) 1:1 gemischt und für 1 min mit der Membran inkubiert. Anschließend wurde ein Film (Hyperfilm ECL, Amersham) auf der Membran exponiert und in einer Entwicklermaschine (Agfa) entwickelt. 2.4 Histologische Färbungen Für morphologische Untersuchungen von Gewebeschnitten wurden im Rahmen dieser Arbeit sowohl Gefrierschnitte als auch Paraffinschnitte angefertigt. Anschließend wurden drei unterschiedliche Färbungen verwendet. 2.4.1 Herstellung von Gefrierschnitten Das isolierte Mausherz wurde zunächst in Tyrode III (siehe Abschnitt 2.5) und anschließend in Sucrose-Lösung (20 % in Tyrode III) gewaschen. Um Risse im Gewebe zu vermeiden wurde das Herz direkt in TissueTek-Einbettmedium (Jung) eingebettet und bei -20 °C eingefroren. Die Schnitte wurden im Kryostat (HM560, Microm) bei einer Blocktemperatur von -20 °C angefertigt. Dabei lag die Schnittdicke bei 14 μm. Danach wurden die Schnitte auf Polylysin-beschichtete Objekträger (VWR) aufgezogen, getrocknet und feuchtigkeitsgeschützt mit Hilfe von Trockenperlen (Silica Gel Orange (Roth) eingefroren. Material und Methoden 18 2.4.2 Herstellung von Paraffinschnitten Das Mausherz wurde isoliert, in PBS gewaschen und über Nacht in 4 % Paraformaldehydlösung (PFA, Roth) fixiert. Anschließend wurde das Gewebe über folgende aufsteigende Ethanolreihe dehydriert: Ethanolreihe: 70 %, 90 %, 2x 100 % je 60 min bei 4 °C 1:1 Ethanol/Toluol 30 min bei RT 100 % Toluol 30 min bei RT Paraffin I, II, III je 4 bis 16 h bei 60 °C Danach wurde das Gewebe in Paraffin eingebettet. Mit einem Rotationsmicrotom (HM335E, Microm) wurden 10 μm dicke Schnitte angefertigt, die im Wasserbad bei 42 °C gestreckt und auf Polylysin-beschichtete Objektträger (VWR) gegeben wurden. Die Schnitte wurden anschließend für 3h bei 60 °C auf die Objektträger fixiert. Unter Lichtausschluss konnten die Schnitte einige Monate aufbewahrt werden. 2.4.3 HE-Färbung Bei der Hämatoxylin/Eosin (HE) Färbung werden die Kerne blauviolett und das Zytoplasma rosa gefärbt. Hämatoxylin-Lösung nach Harris (Sigma): Hämatoxylin 7,5 g/l Eosin-Lösung: Eosin Y 6,0 g Phloxin B 1,5 g 70 % Ethanol ad 250 ml Die Färbung wurde nach folgendem Protokoll durchgeführt: Durchführung: 15 sek. Hämatoxylinlsg. 2x2 min in H2O 5 sek. in 0,1 % Amoniaklösung (frisch) 2x2 min in H2O für 15 sek. in Eosinlösung Material und Methoden 19 8x in 95 % Ethanol tauchen 8x in 100 % Ethanol tauchen 2 min in 100 % Ethanol 2x2 min in Xylol Nach der Färbung wurden die Gewebeschnitte mit DePeX-Harz (Serva) und Deckgläschen bedeckt. 2.4.4 Sirius-Red-Färbung Bei dieser Färbung stellen sich Kollagenfasern rot dar, während Muskelfasern und Zytoplasma gelb erscheinen. Die Schnitte wurden 60 min mit Sirius Red gefärbt, kurz mit 1 % HCl gespült und anschließend mittels einer aufsteigenden Alkoholreihe (70 %-iges Ethanol, 80 %-iges Ethanol, 96 %-iges Ethanol, 100 %-iger Ethanol, Xylol) entwässert. Nach der Färbung wurden die Schnitte mit DePeX-Harz (Serva) und Deckgläschen bedeckt. 2.4.5 Lektin-Färbung Lektine sind Proteine, die spezifisch mit Zuckerresten, z.B. von Glykoproteinen der Zellmembran interagieren. Bei dieser Färbung wurden FITC-konjugierte Lektine verwendet, die es möglich machten den Zellumriss und somit die Zellgröße zu veranschaulichen. Die Paraffin-Schnitte wurden in Xylol entparaffiniert, dann einer absteigenden Alkoholreihe (100 %-igen Ethanol, 90 %-igen Ethanol, 70 %-igen Ethanol) unterworfen und mit H2O dest. gespült. Im Anschluss daran wurden die Schnitte für 2 h bei RT und unter Lichtausschluss mit 0,1 mg/ml Weizen-Lektin (gelöst in PBS; Sigma Aldrich, FITC-konjugiert) inkubiert, dreimal 5 min in PBS gewaschen und anschließend mit fluoreszierendem Einbettmedium (Vectashield, Vectorlabs) eingedeckelt. Die fluorochromen Konjugate wurden mit einem konfokalen Lasermikroskop (Zeiss) bei einer Wellenlänge von 488 nm angeregt und aufgenommen. 10x PBS, pH 7,4: NaCl (Roth) 80 g KCl (Roth) 2,0 g Na2HPO4 (Roth) 6,2 g KH2PO4 (Roth) 2,0 g H2O ad 1000 ml Material und Methoden 2.5 20 Isolierung von Sinusknotenzellen Der Sinusknoten wurde wie in Abschnitt 2.3.1 beschrieben isoliert, anschließend in mehrere kleine Stücke geschnitten und in Tyrode-Low-Lösung für 5 min bei 35 °C inkubiert. Danach wurden 1,75 mg/ml Collagenase B, 0,4 mg/ml Elastase (Roche) und 1 mg/ml BSA in die Tyrode-Low-Lösung gegeben und für 25 min bei 35 °C verdaut. Nach dem Verdau wurden die Gewebestücke in mehreren Wasch-/ Zentrifugenschritten von den Enzymen gereinigt und in KB-Lösung für 2 h bei 4 °C aufbewahrt. Tyrode-Low, pH 6,9: Tyrode III, pH 7,4: NaCl 0,82 g (140 mM) NaCl 4,1 g (140 mM) KCl 0,04 g (5,4 mM) KCl 0,2 g (5,4 mM) CaCl2 0,02 g (0,2 mM) CaCl2 0,13 g (1,8 mM) MgCl2 0,05 g (0,5 mM) MgCl2 0,1 g (1 mM) KH2PO4 0.08 g (1,2 mM) HEPES 0,6 g (5 mM) Taurin 0,63 g (50 mM) D-Glucose 0,54 g (5,5 mM) HEPES 0,12 g (5 mM) H2O ad 500 ml D-Glucose 0,11 g (5,5 mM) H2O ad 100 ml KB (mod. Kraftbrühe) pH 7,4: KCl 0,19 g (25mM) L-Glutamat 1,18 g (80 mM) Taurin 0,25 g (20 mM) KH2PO4 0,14 g (10 mM) MgCl2 0,06 g (3 mM) D-Glucose 0,20 g (10 mM) HEPES 0,24 g (10 mM) EGTA 0,02 g (0,5 mM) H2O ad 100 ml Material und Methoden 2.6 21 Messung der Ca2+-Konzentration in Sinusknotenzellen Die Untersuchung der intrazellulären Ca2+-Konzentration wurde in Zusammenarbeit mit Frau Dr. Eberhardt aus der Arbeitsgruppe von Prof. Dr. Peter Reeh vom Institut für Physiologie der FAU durchgeführt. Die Untersuchung erfolgte über eine Fluoreszenzmessung. Dazu wurde der Farbstoff Fura-2 Acetoxymethylester (AM) verwendet. Dieser bildet einen ChelatKomplex mit den Ca2+-Ionen der Zelle und kann nach Anregung mit einer definierten Wellenlänge detektiert werden. Für die Messungen wurden die isolierten Sinusknotenzellen mit einer Pipette behutsam mechanisch vereinzelt und auf Poly-L-Lysin beschichtete Deckgläschen ausgesät. Anschließend wurden sie für 45 min in Dunkelheit mit 5 µM Fura2AM und 0.02 % Pluronic (Invitrogen) versetzt, um eine Aufspaltung der Esterbindung des Fura-2AM durch endogene Esterasen und die Freisetzung von Fura zu erlauben. Die Deckgläschen wurden auf ein Olympus IX71 Mikroskop montiert und konstant bei 37 °C mit Tyrode III Lösung umspült. Fura-2 wurde bei 340 und 380 nm mit einem Polychrom V Monochromator angeregt und Aufnahmen für 4 ms, bei 20 Hz mit einer 12bit CCD-Kamera gemacht. Für die Aufnahme und Analyse der Daten wurde die TILLvisION 4.0.1.3 Software (Till Photonics) verwendet. Spontane Transienten der Sinusknotenzellen wurden für 2 min aufgenommen. Für die Auswertung der Ca2+-Konzentration wurde die zelluläre Autofluoreszenz subtrahiert und der Quotient der beiden Anregungswellenlängen gebildet. Aus dem ermittelten Quotient F340/F380 (ratio F340/F380) lässt sich die zelluläre Ca 2+Konzentration abschätzen. Als repräsentativ für den basalen cytosolischen Ca2+-Gehalt wurden die untersten zehn Prozent des gemessenen Quotienten von F340/F380 berechnet. Um auszuschießen, dass der gemessene Ca2+-Gehalt von der Schlagfrequenz der einzelnen Sinusknotenzellen abhängt, wurden die Zellen, zur Erniedrigung der Schlagfrequenz, mit 1 mM Zatebradin (gelöst in Tyrode) über ein Perfusionssystem umspült und der Ca2+-Gehalt gemessen. 2.7 Gewinnung von Kardiomyozyten durch Langendorff-Perfusion Die Isolation adulter Kardiomyozyten wurde mittels retrograder Perfusion nach Langendorff durchgeführt. Das Mausherz wurde isoliert und eine abgerundete 20G-Nadel (BD Microlance, 0,9x40 mm, BD) in den Aortenbogen eingeführt, ohne die Aortenklappe zu durchstechen. Über eine Kanüle wurde das Herz für acht Minuten mit Perfusionpuffer perfundiert (Flussrate konstant 3 ml/min, 37 °C). Danach erfolgte eine ebenfalls achtminütige Perfusion mit enzymhaltigem Verdaupuffer. Nach diesen beiden Perfusionsschritten wurde das Herz von der Kanüle entfernt und durch vorsichtige Pipettierschritte vereinzelt und anschließend filtriert. Es folgte ein Zentrifugationsschritt (1 min bei 180 G) bevor die Material und Methoden 22 Myozyten in Stop-Puffer I und anschließend in Stop-Puffer II überführt wurden. Am Ende wurden die Zellen für 2 h in KB-Lösung (siehe Abschnitt 2.5) im Kühlschrank gelagert. Um die Größe und Morphologie der Zellen genau zu ermitteln, wurde die Kardiomyozyten- Lösung in eine 12-well-Platte pipettiert und unter dem Mikroskop (Zeiss Axiovert 10) bei einer 400-fachen Vergrößerung ausgemessen. Die Prozedurschritte und Lösungsprotokolle wurden in Anlehnung an Protokolle der AfCS (Alliance for Cellular Signaling) durchgeführt. Perfusionspuffer (Stock), pH 7,46: NaCl (Merck) 6,60 g (113 mM) KCl (Roth) 0,35 g (4,7 mM) KH2PO4 (Normapor) 0,08 g (0,6 mM) Na2HPO4 (Roth) 0,11 g (0,6 mM) MgSO4 (B. Schmidt) 0,30 g (1,2 mM) NaHCO3 (Roth) 1,01 g (12 mM) HEPES (Roth) 10,00 ml (10 mM) Taurin (Fluka AG) 3,75 g (30 mM) H2O ad 1000 ml 1x Perfusionspuffer: Perfusionspuffer (Stock) 245,0 ml Glukose (Fluka AG) 3,75 g (5,5 mM) BDM (Roth) 5,0 ml (10 mM) H2O ad 250 ml Verdaupuffer: Collagenase B (Roche) 0,25 mg/ml CaCl2 (Merck) (Stock 100 mM) 3,12 µl (12,5 µM) 1x Perfusionspuffer ad 25 ml Stop-Puffer 1: BSA (Roth) 10,0 mg CaCl2 (Merck) (Stock 10 mM) 6,25 μl (12,5 μM) 1x Perfusionspuffer ad 5 ml Material und Methoden 23 Stop-Puffer 2: BSA (Roth) 10,0 mg CaCl2 (Merck) (Stock 10 mM) 12,5 μl (12,5 μM) 1x Perfusionspuffer 2.8 ad 10 ml Telemetrische Elektrokardiogramm Ableitungen Mit Hilfe des Elektrokardiogramms (EKG) kann die elektrische Aktivität des Herzens aufgezeichnet werden. In dieser Arbeit wurden telemetrische EKG-Messungen durchgeführt. Den Mäusen wurde im Alter von 2-3 Monaten ein Radiotransmitter (TA10EA-F20, Data Sciences International, Abb. 2.2.B) intraperitonal unter Isoflurannarkose implantiert. Die ableitenden Elektroden wurden subkutan im Bereich der rechten Schulter und auf dem linken Bauchmuskel unterhalb des Rippenbogens angebracht (Abb. 2.2). Bei der telemetrischen EKG-Messung wird das EKG-Signal vom Transmitter an eine Empfängerplatte gesendet und an einen Computer weitergeleitet. Anschließend wurden die Daten mit der Dataquest A.R.T. Software ausgewertet. Abbildung 2.2: Telemetrische EKG-Ableitung. (A) Röntgenbild einer Maus mit implantiertem Transmitter (modifiziert nach Kramer et al, 2003). (B) Aufnahme eines EKG-Transmitters Material und Methoden 24 Drei Wochen nach der Operation wurde mit den EKG-Messungen begonnen. Bei den RyR2MerCreMer-Mäusen wurde dabei mit einer Messreihe von sieben Tagen begonnen. Im Anschluss daran wurde an fünf aufeinander folgenden Tagen zur Induktion der Rekombination je 2 mg Tamoxifen/100 µl Miglyol intraperitoneal injiziert. Die Messungen wurden bis zum Tod der Knockout-Tiere weitergeführt, wobei alle 10 Minuten ein 20 s langes EKG-Signal aufgenommen wurde. Für die Bestimmung der EKG-Parameter wurden mehrere repräsentative 20 s lange EKG-Segmente ausgewertet (Abb. 2.1). Die erste Messreihe bei den RyR2KiT-Tieren begann ebenfalls mit einer Dauermessung von sieben Tagen, wobei alle 10 Minuten ein 20 s langes EKG-Segment aufgenommen wurde. Anschließend wurde mit der Applikation von Pharmaka begonnen. Hierbei wurde jeweils 60 min vor der Pharmaka-Injektion, während der gesamten Wirkdauer und bis mindestens 60 min nach Abklingen der Wirkung jede Minute ein 20 s langer EKG Abschnitt aufgezeichnet. Zwischen den Injektionen der einzelnen Substanzen wurde jeweils ein Tag Pause eingelegt. Nach der abgeschlossenen Messreihe wurde den Tieren Tamoxifen an fünf aufeinander folgenden Tagen injiziert. Vier Wochen nach der ersten Tamoxifen-Injektion wurden die Dauermessung und die Pharmaka-Messreihe wiederholt. Pharmaka Konzentrationen: NaCl (Vehikel) 0,9 % Isoprenalin 0,5 mg/kg Carbachol 0,5 mg/kg CCPA 0,3 mg/kg Atropin 1 mg/kg Zatebradin 2,0 mg/kg Die EKG-Messungen an RyR3KO-Tieren starteten ebenfalls mit einer Dauermessung von sieben Tagen, gefolgt von der Pharmaka-Aplikation. Die verwendeten Pharmaka und Konzentrationen waren dieselben wie bei der RyR2KiT-Mauslinie. Für die Bestimmung der EKG-Parameter wurden mehrere repräsentative 20 s lange EKGSegmente ausgewertet. Dazu wurden die einzelnen Parameter P, PQ, QRS, QT, RR und TP ermittelt (Abb. 2.3). Material und Methoden 25 Abbildung 2.3: Definition der EKG-Wellen und -Intervalle. Die Erregung des Herzens beginnt im Sinusknoten. Aufgrund der wenigen Zellen des Sinusknotens und der damit insgesamt geringen Potentialänderung bezogen auf das gesamte Herz ist keine Abweichung von der isoelektrischen Linie im EKG bei der Sinusknoten-Erregung zu detektieren. Erst wenn das rechte Atrium die Erregungswelle weiterleitet, wird dies als P-Welle sichtbar. Die atrioventrikuläre Überleitungszeit und die anhaltende Erregung der Vorhöfe werden durch die PQ-Dauer angezeigt. Der QRS-Komplex spiegelt die Erregungsausbreitung über die Ventrikel wider. Die Erregungsrückbildung der Ventrikel wird durch die T-Welle angezeigt. Da das QT-Intervall frequenzabhängig ist, kann zum besseren Vergleich ein korrigiertes QT-Intervall (QTc) berechnet werden. Das frequenzunabhängige QTc-Intervall ergibt sich aus der Gleichung QT (Mitchell et al, RR / 100 1998). Die Herzfrequenz (Schläge pro Minute, bpm) ergibt sich aus dem zeitlichen Abstand von R- zu R-Zacke. Das TP-Intervall spiegelt die Dauer vom Ende eines EKG-Komplexes bis zum Beginn der folgenden Erregung wider. Die Standardabweichung der R-R Dauer (RRSD) wurde als Maß für die Herzfrequenzvariabilität verwendet. Neben dem EKG kann mit Hilfe des implantierten Transmitters auch die Aktivität der Tiere gemessen werden. Dabei wird die horizontale Bewegung des implantierten Transmitters über die Empfängerplatte erfasst und ausgewertet. 2.9 Blutdruckmessungen mit Schwanzmanschette (Tail Cuff) Unter der Blutdruckmessung mittels Schwanzmanschette versteht man eine nicht-invasive Methode der Blutdruckerfassung bei Kleintieren. Die Mäuse befanden sich während der gesamten Messphase in einer auf 37 °C erwärmten Wärmeröhre. Gemessen wurde anhand einer Blutdruckmanschette am Schwanz der Tiere (Blutdruckmessgerät und Software der Material und Methoden 26 Firma „Softron Corporation“). Beim automatischen Aufpumpen der Manschette steigt die Druckkurve an und im Verlauf des abfallenden Manschettendrucks öffnen sich die Schwanzgefäße und ein Puls sowie der Blutdruck sind messbar. Die Tiere wurden zunächst über mehrere Tage an die Messungen gewöhnt, bis die Messwerte stabil waren. Ausgewertet wurde der systolische Blutdruck. 2.10 Wiegen und Ausmessen der Herzkompartimente Die Tiere wurde gewogen, anschließend getötet und das Herz in warme Tyrode III (37 °C; s. Punkt 2.5) gegeben. In der warmen Tyrode konnte das Herz weiter schlagen und wurde von geronnenem Blut befreit. Anschließend wurden die Herzen nach vorsichtigem Ausdrücken der Tyrode-Lösung in einer Petrischale gewogen und ausgemessen. Danach wurde das Herz in die unterschiedlichen Kompartimente (rechter Vorhof, linker Vorhof, rechter Ventrikel, linker Ventikel und Septum) geteilt und das Gewicht und die Dicke der einzelnen Kompartimente ermittelt. Zusätzlich wurde das Herzgewicht (mg) durch das Körpergewicht (g) der jeweiligen Maus geteilt und somit das Verhältnis zwischen Herz- und Körpergewicht ermittelt (Heart-Bodyweight-Ratio, HBR, (mg/g)). 2.11 Haltung und Zucht von Mäusen 2.11.1 Haltung Die Mäuse wurden im Tierstall des Instituts für Experimentelle und Klinische Pharmakologie und Toxikologie der Universität Erlangen gehalten und gezüchtet (12 Stunden Tag/NachtRhythmus, 23 °C). Eine Genehmigung für sämtliche Tierversuche der Regierung von Mittelfranken lag nach Aktenzeichen: 54-2531.31-8/07 vor. Die Tiere wurden ad libitum mit einer Zuchtdiät für Ratten und Mäuse (ssniff) gefüttert und mit Trinkwasser versorgt. Die Verpaarung erfolgte ab einem Alter von sechs Wochen. Die Genotypisierung der neugeborenen Tiere erfolgte im Alter von 10 Tagen mittels PCR. Im Alter von 3-4 Wochen wurden die Jungen abgesetzt, mit Ohrmarken versehen und nach Geschlechtern getrennt gehalten. 2.11.2 Zucht von konditionellen RyR2-Knockout-Mäusen Zur Zucht RyR2 L2/L2 von gewebespezifischen ,KiT) wurden gefloxte RyR2 L2/L2 Knockout-Mäusen (RyR2L2/L2,MerCreMer bzw. -Mäuse (Takeshima et al, 1998) mit den Cre- Recombinase exprimierenden Tieren MerCreMer (Sohal et al, 2001) oder HCN4KiT-Cre Material und Methoden 27 (nachfolgend als KiT bezeichnet) (Hoesl et al, 2008) verpaart. Die daraus entstandenen RyR2L2/+,MerCreMer bzw. -KiT-Tiere wurden wiederum mit gefloxten RyR2L2/L2-Mäusen gekreuzt. Durch die gewebespezifische Expression der tamoxifen-induzierbaren CreRekombinasen MerCreMer im ganzen Herzen und KiT im Reizleitungssystem konnte der RyR2-Knockout zeitlich kontrolliert und gewebespezifisch erreicht werden (Abb. 2.4). Zusätzlich wurde eine Verkreuzung von RyR2L2/L2-Mäusen mit Mäusen, die das ubiquitäre CAGG-Cre exprimieren durchgeführt. Abbildung 2.4: Verpaarungsschema zur Generierung von konditionellen RyR2-KO-Mäusen. Gefloxte RyR2-Mäuse wurden mit MerCreMer- oder KiT-Mäusen verpaart. Die RyR2L2/+,MerCreMer bzw. RyR2L2/+,KiT x RyR2L2/L2 Verpaarung ergibt 25 % RyR2L2/L2,MerCreMer- bzw. RyR2L2/L2,KiTMäuse. Mit Hilfe von Tamoxifen kann die Deletion zu einem beliebigen Zeitpunkt induziert werden. Material und Methoden 28 2.11.3 RyR3-Knockout-Mäuse Zur Untersuchung des RyR3 im Herzen wurden konventionelle RyR3-Knockout-Mäuse verwendet, bei denen das Exon 6 des RyR3 deletiert war (Bertocchini et al, 1997). Die Mäuse wurden von Maik Gollasch (Max-Delbrück-Center für molekulare Medizin, Berlin) bezogen. 2.11.4 Tamoxifen-Injektion Durch die Injektion von Tamoxifen (Sigma) wurde die Cre-vermittelte Rekombination aktiviert. Tamoxifen (2 mg/100 µl Miglyol; immer frisch angesetzt) wurde gevortext und in ein Ultraschallbad gegeben bis sich alle Kristalle vollständig gelöst hatten. Die Injektion erfolgte intraperitoneal an fünf aufeinander folgenden Tagen im Alter von sechs bis acht Wochen. Die Experimente mit RyR2KiT-KO-Tieren starteten im Normalfall vier Wochen nach der ersten Tamoxifen Injektion. Für Untersuchungen an RyR2MerCreMer-KO-Mäusen wurden die Tiere kurz vor ihrem zu erwartenden Tod getötet. 2.12 Statistische Analyse Die statistischen Auswertungen erfolgten mit Origin6.1 (Origin Lab Corporation). Die ermittelten Werte sind als Mittelwert ± Standardabweichung angegeben. Die statistische Signifikanz wurde mittels Student`s t-Test berechnet, wobei Unterschiede mit einem pWert kleiner 0,05 als signifikant angesehen wurden. Ergebnisse 29 3. Ergebnisse 3.1 Charakterisierung einer herzspezifischen RyR2-KO-Mauslinie (RyR2MerCreMer) Um die genaue Funktion von RyR2 im Herzen von adulten Tieren untersuchen zu können, wurden konditionale RyR2-Knockout-Mäuse mit Hilfe des Cre/loxP-Systems generiert. Für die herzspezifische Deletion von RyR2 wurde eine Mauslinie verwendet, bei der das Exon 1 von RyR2 von loxP-Sequenzen flankiert ist (Takeshima et al, 1998). Durch Verpaarung dieses Stammes mit Mäusen, die das MerCreMer-Fusionsprotein unter der Kontrolle des Promotors für die herzspezifische α-Myosin schwere Kette (αMHC) exprimierten (Sohal et al, 2001), wurde die Sequenz innerhalb der loxP-Schnittstellen durch die Cre-Rekombinase spezifisch nur im Herzen exzidiert. Die dadurch entstandenen homozygoten Mäuse (RyR2L2/L2,MerCreMer) sowie deren heterozygoten Geschwistertiere (RyR2L2/+,MerCreMer) wurden im Alter von 6 bis 8 Wochen an fünf aufeinander folgenden Tagen mit je 2 mg Tamoxifen intraperitoneal behandelt. Der dadurch hervorgerufene RyR2-Knockout der RyR2L2/L2,MerCreMer-Mäuse (nachfolgend als RyR2MerCreMer-KO bezeichnet) und deren Kontrollen (RyR2L2/+,MerCreMer) wurden anschließend auf funktioneller, histologischer und molekularer Ebene charakterisiert. 3.1.1 Überlebensrate der RyR2MerCreMer-KO-Mäuse Um die Auswirkung der RyR2-Deletion im Herzen von RyR2MerCreMer-KO-Tieren zu untersuchen, wurden sowohl die RyR2L2/L2,MerCreMer-Mäuse als auch deren Kontrollen (RyR2L2/+,MerCreMer) ab Beginn der Tamoxifen-Behandlung beobachtet. Dabei zeigte sich, dass die Tamoxifen-Injektion keinen Einfluss auf die Überlebensrate der Kontrolltiere (n=8) hatte. Allerdings führte die RyR2-Deletion bereits innerhalb von zwei Wochen zum Tod aller KO-Tiere (n=8) (durchschnittliche Überlebensdauer 12,7±2,8 Tage). In Abbildung 3.1 ist die Überlebensrate der RyR2MerCreMer-Tiere in Prozent angegeben. Ergebnisse 30 Abbildung 3.1. Überlebensrate der RyR2MerCreMer-KO-Mäuse. Von Tag 0 bis Tag 5 fanden die Tamoxifen-Injektionen statt. Alle KO-Tiere (n=8) starben innerhalb von etwa 2 Wochen nach Injektionsbeginn. Das Überleben der Kontrollen (n=8) war nicht beeinflusst. 3.1.2 Analyse der RyR2 Expression im Herzen nach Deletion Für die weiteren molekularbiologischen und histologischen Versuche wurden die RyR2MerCreMer-KO-Tiere kurz vor ihrem zu erwartenden spontanen Tod getötet und das Herz exzidiert. 3.1.2.1 Analyse der RNA-Expression der drei RyR-Isoformen Die Deletion von RyR2 wurde durch RT-PCR Experimente überprüft (Abb. 3.2). Dazu wurde Herzgewebe von RyR2MerCreMer-KO-Tieren präpariert, in die einzelnen Kompartimente getrennt und daraus RNA isoliert. Bei diesem Versuchsansatz wurden zusätzlich zur Expression von RyR2 die anderen beiden RyR-Isoformen untersucht (RyR1 und RyR3). Bei der Analyse von RyR2-mRNA konnte gezeigt werden, dass die Deletion von RyR2 sowohl in den Ventrikeln als auch im linken Vorhof und im Sinusknoten erfolgreich war. Dagegen ist eine schwache Bande im rechten Vorhof nachweisbar, was dadurch erklärt werden kann, dass die MerCreMer-Recombinase dort in geringer Konzentration und/oder als Mosaik exprimiert ist. Die RyR1-mRNA konnte im ganzen Herzen nicht detektiert werden. Bei RyR3 dagegen konnte gezeigt werden, dass im SAN- und Vorhof-Bereich neben RyR2 auch RyR3 exprimiert wird. Im Vergleich zu Kontrollen (3.2.1.1) wiesen die KO-Mäuse keinen Unterschied in der Expression der Isoformen RyR1 und RyR3 auf. Ergebnisse 31 Abbildung 3.2: RT-PCR zum Nachweis der RyR-Isoformen im Herzen der RyR2MerCreMer-KOMäuse. Verwendete Primerkombinationen und resultierende Bandengröße: RyR1 (for/rev) 719 bp, RyR2 (G/H) 300 bp, RyR3 (Mf/Mr) 380 bp, Oben: Kein Nachweis von RyR1-mRNA. Mitte: Keine RyR2Banden sichtbar, außer einer schwachen Bande im rechten Vorhof. Unten: RyR3 kommt im Sinusknoten und den Vorhöfen vor. 3.1.2.2 Analyse der RyR2-Proteinexpression Als Nachweis, ob im Herzgewebe der RyR2MerCreMer-KO-Tiere tatsächlich auch eine Deletion des RyR2-Proteins stattgefunden hat, wurde eine Western-Blot Analyse durchgeführt. Dazu wurde Herzgewebe von KO-Tieren und von heterozygoten Kontrollen verwendet. Die Western-Blot Analyse (Abb. 3.3) zeigte deutliche Unterschiede in der Proteinexpression zwischen den beiden Genotypen. Im Vergleich zu den Kontrollen lässt sich das RyR2-Protein in den Ventrikeln der KO-Mäuse nicht mehr detektieren. Dagegen kann eine RyR2-Bande in den Vorhöfen noch nachgewiesen werden. Demgegenüber konnte bei den Kontrollen RyR2-Protein sowohl in den Vorhöfen als auch im Ventrikel weiterhin detektiert werden. Abbildung 3.3: Expression von RyR2-Protein im Herzen der RyR2MerCreMer-KO-Mäuse. Im Western-Blot von Herzlysaten der Kontrolltiere wurde mittels RyR-Antikörper das 565 kDa große RyR2-Protein nachgewiesen. In den Ventrikeln der KO-Tiere wurde kein RyR2-Protein mehr detektiert. In den Vorhöfen wurde RyR2 in geringerer Expression weiterhin nachgewiesen. Tubulin diente als Ladekontrolle. Ergebnisse 32 3.1.3 Untersuchung des Herzgewichts Als nächstes wurde die Morphologie der Herzen von RyR2MerCreMer-KO-Tiere im Vergleich zu Kontrollen untersucht. Dazu wurden die Tiere zuerst gewogen, anschließend getötet, die Herzen entnommen und diese ebenfalls gewogen, in die einzelnen Kompartimente geteilt und ausgemessen. Die Herzen der KO-Tiere waren vergrößert, wobei vor allem die Vorhöfe stark vergrößert waren. Insbesondere der linke Vorhof wirkte sehr stark dilatiert und war mit Thromben gefüllt (Abb. 3.4). Die Veränderungen in der Morphologie der KO-Herzen lassen darauf schließen, dass die KO-Tiere an den Folgen einer Herzinsuffizienz starben. Abbildung 3.4: Repräsentative Aufnahmen der RyR2MerCreMer-KO-Herzen. (A) Kontrollherz. (B) RyR2MerCreMer-KO-Herz. Das KO-Herz weist dilatierte und mit Thromben gefüllte Vorhöfe auf. Das Myokard der KO-Tiere erscheint nur durch fotografier-bedingte Lichtreflexe heller als das Myokard der Kontrollen, die augenscheinliche Durchblutung war nicht verändert. r Vo, rechter Vorhof; l Vo, linker Vorhof. Zum besseren Vergleich wurde das Herzgewicht auf das Körpergewicht der jeweiligen Maus normalisiert. Das Verhältnis von Herzgewicht zu Körpergewicht (Heart – Bodyweight – Ratio, HBR) war um mehr als das 3-fache erhöht (Ctr: 5,1 ± 0,6 mg/g; n=23; KO: 16,1 ± 2,3 mg/g; n=12; p< 0,001). Besonders auffällig waren die Größe und das Gewicht des linken Vorhofs der KO-Tiere erhöht (Abb.3.5 und Tabelle 1). Aber auch der rechte Vorhof und der Ventrikel waren im Vergleich zu den Kontrollherzen signifikant vergrößert. Ergebnisse 33 Abbildung 3.5: Herz-Gewicht und HBR der RyR2MerCreMer-KO-Tiere. Das Gewicht der Ventrikel und Vorhöfe sowie der HBR-Wert der KO-Tiere waren signifikant vergrößert im Vergleich zu den Kontrollen. **, p < 0.01; ***, p < 0.001. Das Körpergewicht der Kontrolltiere (25,3±4,9 g) war zudem signifikant höher als das Körpergewicht der KO-Mäuse (20,1±1,9 g). Dieser Unterschied kann darauf zurückgeführt werden, dass die KO-Tiere zum Zeitpunkt der Herzentnahme durch die Herzinsuffizienz bereits körperlich beeinträchtigt waren und dadurch innerhalb weniger Tage mehrere Gramm an Gewicht verloren hatten. Ctr (n=23) KO (n=12) pWert Herzlänge (mm) Herzdurchmesser (mm) Länge l Vorhof (mm) Länge r Vorhof (mm) linke Ventrikelwand (mm) rechte Ventrikelwand (mm) Septumdicke (mm) 7.6±0.9 5.1±0.4 2.4±0.3 3.0±0.5 1.9±0.4 0.6±0.1 1.7±0.2 8.5±0.7 5.8±0.5 6.8±1.1 4.2±0.8 2.1±0.4 0.7±0.1 1.8±0.4 < 0.001 < 0.001 < 0.001 < 0.001 > 0.1 < 0.05 > 0.1 Tabelle 1: Biometrische Daten der Ventrikel und Vorhöfe. Die Länge und Breite der Herzen, sowie die Dicke der einzelnen Kompartimente war verändert in RyR2MerCreMer-KO-Tieren. Sowohl die Länge als auch der Durchmesser der KO-Herzen waren etwas vergrößert. Beim Vermessen der Ventrikelwände und des Septums konnte gezeigt werden, dass zwar die linke Ventrikelwand und das Septum der KO-Tiere tendenziell dicker waren, der Unterschied war jedoch nicht signifikant. Lediglich die Dicke der rechten Ventrikelwand war wenig vergrößert (Tabelle 1). Ergebnisse 34 3.1.4 Analyse der Herzfrequenz und der EKG-Parameter Mittels EKG-Telemetrie wurde die Herzfrequenz und die lokomotorische Aktivität der Tiere untersucht. Dazu wurden Kontrollen und KO-Tiere mit Telemetrie-Transmittern versehen und das EKG kontinuierlich ab Beginn der Tamoxifen-Injektion aufgezeichnet (Abb. 3.6). Abbildung 3.6: Telemetrische EKG-Ableitungen an wachen Tieren während der letzten sieben Tage vor dem Tod der KO-Tiere. Absinken der Herzfrequenz (oben) und der Aktivität (unten) der RyR2MerCreMer-KO-Tiere im Vergleich zu den Kontrollen. Die KO-Tiere zeigen keinen Tag-Nacht-Rhythmus mehr. Dabei ist zu erkennen, dass die Herzfrequenz der KO-Tiere bis zu deren Tod kontinuierlich abnimmt. Daneben nahm auch die lokomotorische Aktivität der KO-Mäuse immer weiter ab, was als ein deutliches Zeichen für einen stark eingeschränkten Allgemeinzustand gewertet werden kann. Während die Kontrolltiere einen normalen Tag-Nacht-Rhythmus, mit höheren Herzfrequenzen nachts und niedrigeren Herzfrequenzen tagsüber aufwiesen, konnte bei den KO-Tieren kein Tag-Nacht-Rhythmus mehr festgestellt werden. Des Weiteren wurden die EKG-Parameter anhand mehrerer 20 s langer EKG-Abschnitte ausgewertet (Abb 3.7). Bei den KO-Mäusen wurden für die Auswertung EKG-Abschnitte aus Messungen zum Zeitpunkt sieben Tage, sowie zwei Tage vor dem Tod der Tiere verwendet. Die P-Dauer, RR- ,PQ-, QRS-, QT- und TP-Intervalle wurden ermittelt und das herzfrequenz-korrigierte QTc-Intervall wurde berechnet. Bereits eine Woche vor dem Tod der KO-Tiere konnte eine Sinusbradykardie festgestellt werden, zwei Tage vor dem Tod war die Herzfrequenz noch weiter gesunken. Dies lässt sich anhand des RR-Intervalls zeigen, welches eine Woche vor Ergebnisse 35 dem Tod der KO-Tiere um 24 ms, zwei Tage vor dem Tod bereits um 182 ms verlängert war. Dies entspricht einer Abnahme der Herzfrequenz um 68 bpm sieben Tage vor dem Tod der KO-Tiere und einer Abnahme um 253 bpm, zwei Tage vor deren Tod. Neben dem RRIntervall waren die QT- und TP-Zeit der KO-Mäuse signifikant verlangsamt. Abbildung 3.7: EKG-Parameter der RyR2MerCreMer-KO-Mäuse. Es wurden EKG-Abschnitte zu dem Zeitpunkt sieben Tage bzw. zwei Tage vor dem Tod der KO-Tiere ausgewertet. (A) Mittelwerte ± Standardabweichung von P-Dauer, PQ, QRS, QT, TP und QTc (korrigiertes QT-Intervall). (B) Das RR-Intervall bezeichnet die Zeit zwischen den beiden R-R-Zacken. Die Dauer der einzelnen Parameter in A und B ist in ms angegeben. *, p < 0.05; ***, p < 0.001. In Abbildung 3.8 sind EKG-Beispiele aus Messungen zwei Tage vor dem Tod der KO-Mäuse im Vergleich zu deren Kontrollen dargestellt. Während das EKG der Kontrolle normale EKGParameter aufweist, ist das EKG der KO-Maus sichtbar verändert. Besonders auffällig sind dabei neben der Bradykardie sowohl verlängerte und morphologisch veränderte T-Wellen, als auch die P-Wellen, die nach unten zeigen. In den KO-Tieren treten also massive Störungen der Erregungsbildung- und Rückbildung auf. Ergebnisse 36 Abbildung 3.8: Beispiele telemetrischer EKG-Ableitungen. (A) EKG-Spur einer Kontroll-Maus. (B) EKG-Spur einer RyR2MerCreMer-KO-Maus. Die P-Welle und das Ende der T-Welle sind durch Pfeile markiert. 3.1.5 Analyse der strukturellen Veränderungen des Herzens Um genauere Aufschlüsse über die Auswirkung der RyR2-Deletion auf das Herz der KOTiere zu erhalten, wurden die Herzen eingebettet, geschnitten und histologisch mittels HEund Sirius-Red-Färbung analysiert. Bei der HE-Färbung (Abb. 3.9A) konnten deutliche morphologische Veränderungen festgestellt werden. Vor allem im stark vergrößerten linken Vorhof konnten große organisierte Thromben nachgewiesen werden. Der rechte Vorhof war zwar nicht so stark vergrößert wie der linke, jedoch waren auch hier Thromben zu erkennen. Das Auftreten von Kollagen und Fibrose im Myokard wurde anhand der Sirius-Red-Färbung untersucht. Durch diese Färbung wird Kollagen rot und das Zytoplasma der Herzmuskelzellen gelb angefärbt. Kollagene gehören zu den Strukturproteinen und kommen vor allem im Bindegewebe sowie in der extrazellulären Matrix des Herzens vor und können als Fibrosemarker verwendet werden. Die Abbildungen 3.9B und 3.9C zeigen repräsentative Aufnahmen der Herzen nach einer Sirius-Red-Färbung. Bei den Kontroll-Herzen wurden keine vermehrten Kollageneinlagerungen in den zellulären Zwischenräumen nachgewiesen. Wie zu erwarten, war jedoch das Bindegewebe z.B. um Blutgefäße rot angefärbt. Bei den KO-Tieren dagegen konnte pathologische Fibrosierung in den Ventrikeln detektiert werden, die das gesamte Myokard durchzog. Ergebnisse 37 Abbildung 3.9: Atriale Thrombose und vermehrte Fibrose im Ventrikel der RyRMerCreMer-KOMäuse. (A) HE-Färbung an Kryoschnitten von KO- und Kontroll-Herzen. (B) Sirius-Red-Färbung an Kryoschnitten von KO- und Kontroll-Herzen. (C) Vergrößerung aus linker Ventrikelwand von (B). Ao, Aorta; r Vo, rechter Vorhof; l Vo, linker Vorhof; r Ven, rechter Ventrikel; l Ven, linker Ventrikel. 3.1.6 Untersuchung der Morphologie und Größe der Kardiomyozyten Um die Veränderungen auf zellulärer Ebene zu untersuchen, wurde die Morphologie und Größe von über Langendorff-Perfusion frisch isolierten Kardiomyozyten untersucht. Während die Kardiomyozyten der Kontrolltiere (n=5 Mäuse) unter einem Phasenmikroskop typische Strukturen wie eine Querstreifung, aufwiesen, waren die Myozyten der KO-Tiere (n=6 Mäuse) pathologisch verändert. Die KO-Kardiomyozyten wirkten ödematös und mit Vakuolen gefüllt. Zudem konnte keine Querstreifung mehr erkannt werden (Abb. 3.10 A). Ergebnisse 38 Abbildung 3.10 Analyse der isolierten Kardiomyozyten. (A) Abbildung einer Kontroll- und einer KO-Kardiomyozyte. (B) Tabelle mit der gemessenen Breite und Länge (in µm) der Kontrollen (n=350) und KO-Kardiomyozyten (n=420). Maßstabsbalken, 25 μm.*, p < 0.05; ***, p < 0.001. Das Vermessen der Zellbreite und Zelllänge isolierter Myozyten bestätigte eine leichte Größenzunahme der KO-Myozyten (Abb. 3.10B). Die Zellen waren signifikant länger und breiter im Vergleich zu den Kontroll-Myozyten. Desweiteren wurde die Morphologie der Kardiomyozyten anhand von Paraffin-Schnitten durch eine Lektin-Färbung untersucht, wodurch die Zellmembran der ventrikulären Kardiomyozyten genauer dargestellt werden konnte (Abb. 3.11). Auch hierbei konnte deutliche Veränderungen der KO-Kardiomyozyten festgestellt werden. Die Zellen der KO-Präparate waren stark vergrößert und unförmig. Abbildung 3.11: Lektin–Färbung von Paraffinschnitten durch das ventrikuläre Myokard zur Darstellung der Zellmembran. Links: Kontroll-Kardiomyozyten. Rechts: KO-Kardiomyozyten. Die Zellembran ist grün dargestellt. Maßstabsbalken, 50 μm. Ergebnisse 3.2 39 Charakterisierung einer reizleitungs-spezifischen RyR2-KOMauslinie (RyR2KiT) RyR2 soll neben seiner Funktion im Myokard auch eine wichtige Rolle bei der Generierung des Schrittmacherpotentials im Reizleitungssystem spielen (Maltsev et al, 2006; Lakatta et al, 2010). Um die Funktion von RyR2 im Reizleitungssystem besser untersuchen zu können, wurde neben der RyR2MerCreMer-Mauslinie eine zweite Mauslinie generiert. Dazu wurde mit Hilfe eines konditionellen Knockout-Systems die Deletion von RyR2 selektiv nur im Reizbildungs- und Reizleitungssystem hervorgerufen. Zum einen wurde die gefloxte RyR2Mauslinie verwendet (Takeshima et al, 1998), zum anderen die HCN4KiT-Cre Mauslinie (Hoesl et al, 2008). Bei der HCN4KiT-Cre Mauslinie wird nach Induktion mit Tamoxifen nur in Zellen des Sinus- und AV-Knotens ein RyR2-Knockout (RyR2KiT-KO) erzeugt. Für die Zielgendeletion wurden homozygote Mäuse (RyR2L2/L2,KiT) generiert und nach Induktion mit Tamoxifen untersucht. Als Kontrollen dienten die heterozygoten Geschwister-Tiere (RyR2L2/+,KiT), die ebenfalls mit Tamoxifen behandelt wurden. Im Gegensatz zu den herzspezifischen RyR2MerCreMer-KO-Mäusen sind die RyR2KiT-KOTiere nach der Tamoxifen-Behandlung lebensfähig und zeigen keine phänotypischen Auffälligkeiten. 3.2.1 Analyse der Expression von RyR2 in RyR2KiT-Mäusen 3.2.1.1 Analyse der RNA-Expression der drei RyR-Isoformen Um die Deletion von RyR2 im Reizleitungssystem auf RNA-Ebene nachzuweisen, wurde eine RT-PCR mit RNA aus verschiedenen Bereichen des Herzens durchgeführt. Zusätzlich wurde untersucht, ob in RyR2KiT-KO-Tieren eine kompensatorische Hochregulation der anderen RyR-Isoformen RyR1 und RyR3 stattgefunden hatte. Ergebnisse 40 Abbildung 3.12: RT-PCR zum Nachweis der RyR-Isoformen im Sinusknotengewebe der RyR2KiT-KO-Tiere. Keine Änderung der RyR1 und RyR3 RNA-Expression in RyR2KiT-KO-Tieren im Vergleich zu den Kontrollen. Die RyR2 Deletion in den KO-Tieren konnte auf RNA-Ebene nicht nachgewiesen werden. Wie schon in Abschnitt 3.1.2.1 gezeigt, wird RyR1 im Herzen nicht exprimiert, RyR3 kommt dagegen im Sinusknoten und den Vorhöfen vor. Wie in Abb. 3.12 zu sehen ist, konnte auch nach Induktion des RyR2-Knockouts keine RyR1-RNA im Sinusknoten der KO-Mäuse detektiert werden. RyR3-RNA konnte sowohl im Sinusknotengewebe der KO-Mäuse als auch in der Kontrolle nachgewiesen werden, war in den KO-Tieren jedoch nicht stärker oder schwächer exprimiert als in den Kontrollen. Die Deletion von RyR2 konnte mit RT-PCR nicht bestätigt werden, da RyR2-RNA im Sinusknoten beider Gruppen detektiert werden konnte. Das lässt sich dadurch erklären, dass sich in isolierten Sinusknoten, aus denen die RNA gewonnen wurde, viele Vorhofmyozyten befanden, in denen RyR2 weiterhin exprimiert wird. Diese geringe mRNA-Menge reicht aus, um unter den angewanden RT-PCR-Bedingungen ein Signal zu generieren. Auf Protein-Ebene (siehe folgender Abschnitt 3.2.2.2) war die Deletion allerdings eindeutig nachweisbar. Da RyR1 wie bereits beschrieben im Herzen nicht vorkommt (Tellez et al, 2006), wurde für die Proben aus dem Vorhof und Ventrikel nur die RNA-Expression von RyR2 und RyR3 untersucht (Abb. 3.13). In den Vorhöfen und in den Ventrikeln fand wie erwartet keine RyR2-Deletion statt, so dass die RyR2-RNA in den KOTieren weiterhin detektiert werden konnte. Die Isoform RyR3 konnte wie bereits beschrieben, zwar in den Vorhöfen, jedoch nicht in den Ventrikeln detektiert werden. Ergebnisse 41 Abbildung 3.13: RT-PCR zum Nachweis der RyR-Isoformen im Herzgewebe der RyR2KiT-KOTiere. Oben: RyR2-RNA war bei den KO-Tieren und den Kontrollen in allen Kompartimenten zu detektieren. Unten: RyR3-RNA konnte nur in den Vorhöfen beider Gruppen nachgewiesen werden. 3.2.1.2 Analyse der RyR2 Proteinexpression Die Deletion von RyR2 sollte bei der RyR2KiT-KO-Maus nur im Reizleitungssystem, nicht jedoch im restlichen Herzen stattfinden. Zum Nachweis, dass der RyR2-Knockout erfolgreich war, wurde eine Western-Blot Analyse durchgeführt. Zusätzlich zum Sinusknoten wurde auch Gewebe aus dem rechten und linken Vorhof der Mausherzen untersucht. Im Gegensatz zur RNA-Detektion mittels RT-PCR war auf Protein-Ebene keine Bande im Gewebe des Sinusknotens der KO-Mäuse mehr zu erkennen (Abb. 3.14). Im Vergleich dazu konnte im Sinusknoten der Kontrollen eine starke RyR2-Bande detektiert werden. RyR2 konnte unverändert in den Vorhöfen der Kontrollen als auch in denen der KO-Tiere nachgewiesen werden. Somit war RyR2 wie erwartet im Sinusknoten deletiert, die RyR2 Expression in den Vorhöfen der KO-Tiere war jedoch nicht beeinflusst. Abbildung 3.14: RyR2-Proteinexpression im Herzen der RyR2KiT-KO-Mäuse. Nur im SAN der KO-Mäuse war das RyR2-Protein deletiert. In den Vorhöfen der KO-Tiere konnte das 565 kDa große RyR2-Protein weiterhin detektiert werden. Tubulin diente als Ladekontrolle. Ergebnisse 42 3.2.2 Untersuchung der strukturellen Veränderungen der Herzen Um strukturelle Veränderungen in den Herzen zu untersuchen, wurden Kryoschnitte von RyR2KiT-KO- und Kontroll-Herzen mit HE gefärbt. Nach lichtmikroskopischer Betrachtung konnten keine Veränderungen der Herzmuskulatur oder des Sinusknotengewebes nach RyR2-Deletion festgestellt werden (Abb. 3.15 oben). Pathologische Prozesse am Herzen gehen häufig mit einem Umbau der Herzmuskulatur einher, wobei Herzmuskelgewebe durch Bindegewebe ersetzt wird. Durch eine Sirius-Red-Färbung werden solche kollagenen Fasern durch eine Rotfärbung dargestellt und grenzen sich daher deutlich vom Zytoplasma der Kardiomyozyten ab. Außer den physiologisch vorhandenen Bindegewebsstrukturen, wie z.B. an den Gefäßen, war bei den KO-Herzen kein vermehrtes Bindegewebe und somit keine Fibrosierung zu beobachten (Abb. 3.15 unten). Abbildung 3.15: Morphologie der RyR2KiT-KO-Herzen. (A) HE-Färbung an Kryoschnitten von Kontroll- und KO-Herzen. (B) Sirius-Red-Färbung an Kryoschnitten von Kontroll- und KO-Herzen. Es konnten keine Unterschiede in der Morphologie oder dem Fibrosierungsgrad festgestellt werden. Ao, Aorta; r Vo, rechter Vorhof; l Vo, linker Vorhof. Ergebnisse 43 3.2.3 Analyse der Herzfrequenz und der EKG-Parameter Mittels telemetrischer EKG-Transmitter wurden die Herzfrequenz und die lokomotorische Aktivität der RyR2KiT-KO-Mäuse im Vergleich zu den heterozygoten Kontrollen untersucht. Da die RyR2KiT-Mauslinie generiert wurde, um spezifisch im Reizleitungssystem den RyR2 zu deletieren, wurde untersucht ob die Rhythmus-Generierung und Reizweiterleitung im Herzen durch den Knockout beeinflusst wurden. Dazu wurden RyR2L2/L2,KiT- und RyR2L2/+,KiT-Mäuse mit EKG-Telemetrie-Transmittern versehen. Zur Bestimmung der basalen Herzfrequenz wurde ihr EKG kontinuierlich eine Woche aufgezeichnet (Abb. 3.16). Anschließend wurden den Mäusen verschiedene Pharmaka verabreicht, die die Herzfrequenz modulieren sollten. Um den Knockout zu generieren, wurde den Tieren Tamoxifen injiziert und alle Messungen wurden vier Wochen nach den Tamoxifen-Injektionen wiederholt. Die EKG-Messungen vor der Tamoxifenbehandlung werden in Abschnitt 3.3.2 näher behandelt. Abbildung 3.16: Telemetrische EKG-Ableitungen an wachen RyR2KiT-KO- und Kontrolltieren. Oben: Die mittlere Herzfrequenz der KO-Tiere ist deutlich niedriger als die Herzfrequenz der Kontrollen. Unten: Die Aktivität der KO-Tiere im Vergleich zu den Kontrollen ist dagegen nicht verändert. Es gab keinen Unterschied im Tag-Nacht-Rhythmus. Die Analyse der Herzfrequenz und der lokomotorische Aktivität der Tiere vier Wochen nach der Tamoxifen-Behandlung zeigte, dass RyR2 defiziente Mäuse einen normalen Sinusrhythmus und keine Anzeichen für Arrhythmien hatten. Allerdings ist zu erkennen, dass die mittlere Herzfrequenz der KO-Tiere deutlich niedriger war als die der Kontrollen (Abb. Ergebnisse 44 3.16). Die KO-Tiere wiesen somit eine Bradykardie auf. Es konnte kein Unterschied bei der lokomotorischen Aktivität der beiden Gruppen festgestellt werden. Somit lässt sich die Bradykardie nicht auf eine geringere Aktivität der KO-Tiere im Vergleich zu den Kontrollen zurückführen. Die mittlere Herzfrequenz über eine Woche betrug bei den KO-Mäusen 330 bpm, bei den Kontrollen 456 bpm (p<0,05). Obwohl die KO-Mäuse eine deutliche Bradykardie zeigten, blieb der Tag-Nacht-Rhythmus mit höheren Herzfrequenzen nachts (aktive Phase) und niedrigeren Herzfrequenzen tagsüber weiterhin erhalten. Dies lässt darauf schließen, dass die Möglichkeit einer Modulation der Herzfrequenz nicht verloren gegangen war. Der signifikante Unterschied in der Herzfrequenz konnte ebenfalls beobachtet werden, wenn man die Mittelwerte der Herzfrequenzen nachts und während des Tages miteinander verglich (Abb. 3.17A). Dabei wurden für die Tagphase Messungen von 7 Uhr bis 19 Uhr verwendet, für die Nachtphase Werte von 19 Uhr bis 7 Uhr. Überraschenderweise gab es einen signifikanten Unterschied bei der Untersuchung der Herzfrequenzvariabilität zwischen den beiden Gruppen. Die Herzfrequenz der KO-Mäuse wies dabei eine geringere Variabilität im Vergleich zu den Kontrollen auf. Dieser Unterschied ist in Abb. 3.17B anhand der Standardabweichung der RR-Intervalle von Messungen während der Tag- bzw. Nacht-Phase dargestellt. Abbildung 3.17 Mittlere Herzfrequenz und Herzfrequenzvariabilität der RYR2KiT-KO-Tiere. (A) Mittlere Herzfrequenzen nachts und tagsüber. (B) Darstellung der Herzfrequenzvariabilität anhand der Standardabweichung der RR-Intervalle. Die KO-Tiere (n=7) haben eine niedrigere Herzfrequenzvariabilität als die Kontrollen (n=7). *, p < 0.05. Um herauszufinden, welcher EKG-Abschnitt die Bradykardie der RyR2KiT-KO-Tiere verursacht, wurden die einzelnen EKG-Parameter anhand 20 s langer EKG-Abschnitte aller Mäuse ausgewertet und die PQ-, QRS-, QT-, TP und RR-Intervalle ermittelt (Abb. 3.18). Ergebnisse 45 Abbildung 3.18: EKG-Parameter von RyR2KiT-KO- und Kontrolltieren. Die Mittelwerte ± Standardabweichung von P-Dauer, PQ, QRS, QT, TP und RR sind angegeben. Die signifikante Verlängerung des RR-Intervalls (und damit der Herzfrequenz) ist auf eine Verlängerung der PQ- und TP- Intervalle zurückzuführen. *, p < 0.05; **, p < 0.01. Bei ansonsten normalem EKG (Abb. 3.19) konnte eine signifikant verlängerte PQ-Dauer um 6.4 ms und vor allem ein um 28.1 ms stark verlängertes TP-Intervall der KO-Tiere im Vergleich zu den Kontrollen festgestellt werden. Die TP-Zeit spiegelt den Abstand zwischen dem Ende einer T-Welle bis zur darauf folgenden P-Welle wider, also die Zeit, in der der Sinusknoten aktiv ist. Insgesamt ergibt sich daraus die beobachtete Bradykardie mit einer Verlängerung des RR-Intervalls der KO-Tiere um 37 ms im Vergleich zu den Kontrollen. Die übrigen EKG Parameter waren nicht verändert. Daraus lässt sich schließen, dass die Deletion von RyR2 starke Auswirkungen auf die Generierung des Aktionspotentials im Sinusknoten hat. Abbildung 3.19: Beispiele telemetrischer EKG-Ableitungen. EKG-Spur einer Kontroll-Maus (links) und einer RyR2KiT-KO-Maus (rechts). Pfeile markieren die PWelle und das Ende der T-Welle. Ergebnisse 46 3.2.4 Analyse der Herzfrequenz-Modulation durch verschiedene Pharmaka Um zu überprüfen, ob eine Herzfrequenz-Modulation noch möglich ist, wurde den Mäusen verschiedene Pharmaka appliziert. Eine Herzfrequenz-Steigerung wurde zuerst mit dem βAdrenorezeptor-Agonisten Isoprenalin (0,5 mg/kg) untersucht (Abb. 3.20, 3.21). Sowohl die Kontrolltiere als auch die RyR2KiT-KO-Tiere konnten nach Injektion von Isoprenalin ihre Herzfrequenz steigern, die Frequenzen der KO-Tiere waren jedoch stets niedriger als bei den Kontrolltieren. Während die Kontrollen eine Herzfrequenz von bis zu 700 bpm erreichten, überstieg die Herzrate bei den KO-Tieren nie 500 bpm. Dabei ist jedoch zu beachten, dass die Basalfrequenz der KO-Tiere, wie bereits in Abschnitt 3.2.4 beschrieben, von vornherein niedriger war, als die Frequenz der Kontrollen. Abbildung 3.20: Herzfrequenz-Steigerung mittels Isoprenalin. Dargestellt sind die mittleren Herzfrequenzen der RyR2KiT-KO-Mäuse (n=7) und der Kontrollen (n=7) eine Stunde vor und drei Stunden nach Injektion von 0,5 mg/kg Isoprenalin. Zum Zeitpunkt t=0 wurde Isoprenalin injiziert. Die KO-Tiere konnten ebenso wie die Kontrollen ihre Herzfrequenz nach Isoprenalin-Injektion steigern, erreichten aber nicht die gleichen hohen Frequenzen wie die Kontrollen. Neben Isoprenalin wurden auch der muscarinische Acetylcholin-Rezeptor Antagonist Atropin (1 mg/kg), der If–Blocker Zatebradin (2 mg/kg), der A1-Adenosin-Rezeptor Agonist CCPA (0,3 mg/kg) und der muscarinische M2- Rezeptor Agonist Carbachol (0,5 mg/kg) verabreicht (Abb. 3.21A). Mit Atropin konnte die Herzfrequenz ähnlich wie mit Isoprenalin gesteigert werden. Allerdings war die Steigerung mit Atropin weniger effizient als mit Isoprenalin, so dass lediglich Werte von 600 bpm bei den Kontrolltieren erreicht werden konnten. Auch mit Atropin konnte bei den KO-Tieren aber keine vergleichbar hohen Herzfrequenzen wie bei den Kontrollen erzielt werden. Mit den getesteten Pharmaka Zatebradin, CCPA und Carbachol kann die Herzfrequenz gesenkt werden. Dabei fiel auf, dass die KO-Tiere niedrigere Herzfrequenzen erreichten als die Kontrollen. Somit war zwar mit allen verabreichten Pharmaka eine Modulation der Herzfrequenz bei den KO-Tieren erzielt Ergebnisse 47 worden, allerdings konnten nicht dieselben absoluten Frequenzen wie bei den Kontrollen erreicht werden. Um zu überprüfen, ob die Unterschiede darauf zurückzuführen sind, dass die KO-Tiere von vornherein eine niedrigere Herzfrequenz haben, wurde ermittelt, ob der Prozentsatz, mit dem die Herzfrequenz gesteigert oder gesenkt wurde, ebenfalls unterschiedlich ist. Hierbei konnte kein signifikanter Unterschied bei der Berechnung der relativen Veränderung festgestellt werden (Abb. 3.21B). Die Herzfrequenz-Steigerung oder Senkung hielt bei allen Pharmaka bei beiden Gruppen gleich lange an. Die Herzfrequenz der KO-Tiere konnte auch mit höheren Dosierungen, wie z.B. 1 mg/kg Isoprenalin (Ctr: 689±29 bpm; RyR2KiT-KO: 461±18 bpm) nicht weiter gesteigert werden. Dies lässt darauf schließen, dass zwar die Deletion von RyR2 im Reizleitungssystem zu einer Sinusbradykardie führt, die Signalwege jedoch, die zu einer Modulation der Herzfrequenz benötigt werden, nicht beeinträchtigt sind. Abbildung. 3.21: Herzfrequenz-Modulation in RYR2KiT-KO-Mäusen. (A) Mittlere Herzfrequenz im Vergleich Ctr (n=7) und RyR2KiT-KO (n=7). Die basale Herzfrequenz und die Herzfrequenz nach der Gabe von 0,5 mg/kg Isoprenalin (Iso), 1 mg/kg Atropin (Atro), 2 mg/kg Zatebradin (Zate), 0,3 mg/kg CCPA oder 0,5 mg/kg Carbachol (Carb) waren zwischen den beiden Genotypen signifikant verändert. (B) Prozentuale Änderung der Herzfrequenz. Kein Unterschied dagegen bestand bei der relativen Herzfrequenz-Änderung in Prozent. Dargestellt sind die Mittelwerte ± Standardabweichung der ersten 30 min nach Pharmaka-Injektion. n.s., nicht signifikant; *, p < 0,05. 3.2.5 Analyse der Herzfrequenzvariabilität Die Injektion von Carbachol bewirkt, wie oben bereits beschrieben, dass die Herzfrequenz gesenkt wird. Carbachol ist ein Strukturanalogon von Acetylcholin und ein Agonist am muscarinischen M2-Rezeptor. Wenn M2-Rezeptoren mit Carbachol interagieren, wird der cAMP-Spiegel in einer Myokardzelle gesenkt, was letztendlich in einer HerzfrequenzSenkung resultiert (Abb. 3.22A). Ergebnisse 48 Abbildung 3.22: Änderung der Herzfrequenzvariabilität bei RyR2KiT-KO-Tieren. (A) Mittlere Herzfrequenzen der KO-Tiere und Kontrollen vor und nach Carbachol-Injektion (0,5 mg/kg). Zum Zeitpunkt t=0 wurde Carbachol injiziert. Die Striche markieren den Zeitraum, der für die Auswertung von (B) verwendet wurde. (B) Darstellung der Dauer der RR-Intervalle und Standardabweichung der RR-Dauer (Kasten). RyR2KiT-KO-Mäuse haben eine geringere Herzfrequenz-Variabilität als die Kontroll-Mäuse. **, p < 0,01. Überraschenderweise fiel beim Betrachten der einzelnen EKG-Komplexe nach Injektion von Carbachol auf, dass es Unterschiede in der Herzfrequenzvariabilität zwischen den Kontrollen und den KO-Tieren gibt. Besonders auffällig waren diese Unterschiede etwa 15 min nach der Injektion von Carbachol, wenn die Herzfrequenz wieder begann auf das Basalniveau anzusteigen. Während die Kontrolltiere in dieser Phase Schwierigkeiten hatten streng rhythmische Schläge zu generieren, wirkte der Herzschlag der KO-Mäuse gleichmäßiger. Dies konnte anhand der Dauer des RR-Intervals bestimmt werden (Abb. 3.22B), die Auswertung bei den KO-Tiere weniger „verrauscht“ wirkte. Die Herzfrequenzvariabilität kann über die Standardabweichung der RR-Werte (RR-SD) definiert werden. Während die Kontrolltiere einen mittleren RR-SD-Wert von 54 hatten, wiesen die KO-Tiere lediglich einen mittleren RR-SD-Wert von 25 auf (Abb. 3.22 Kasten). Beispiele telemetrischer EKGAbleitungen nach Abklingen der Carbachol-Wirkung sind in Abbildung 3.23 dargestellt. Während das EKG der KO-Maus gleichmäßig wirkt, ist das EKG der Kontrolle dysrhythmisch mit teils kurzen Sinuspausen und schnelleren bzw. langsameren Herzfrequenzen. Ergebnisse 49 Abbildung 3.23: Beispiele telemetrischer EKG-Ableitungen nach Abklingen der CarbacholWirkung. (A) EKG-Spur einer Kontroll-Maus. (B) EKG-Spur einer RyR2KiT-KO-Maus mit weniger Herzfrequenzvariabilität. Die EKG-Beispiele wurden ca. 15 Minuten nach der Carbachol-Injektion (0,5 mg/kg) aufgezeichnet. 3.2.6 Auswirkung der RyR2-Deletion auf den Blutdruck Um festzustellen, ob die aufgetretene Bradykardie der RyR2KiT-KO-Tiere auch einen Einfluss auf den systolischen Blutdruck hat, wurde eine nicht-invasive Blutdruckbestimmungen mittels einer Blutdruckmanschette am Schwanz wacher Tiere durchgeführt. Dabei konnten keine signifikanten Unterschiede bei den systolischen Blutdruckwerten zwischen RyR2KiT-KO- und Kontrolltieren festgestellt werden (Abb. 3.24). Abbildung 3.24: Basale Blutdruckmessungen an RyR2KiT-KO-Tieren. Dargestellt sind systolische Blutdruckwerte am wachen Tier, gemessen Blutdruckmanschette am Schwanz. n.s., nicht signifikant. mittels einer 3.2.7 Phosphorylierungstatus von Phospholamban Viele funktionell wichtige Proteine im Herzen werden durch cAMP-vermittelte Phosphorylierung reguliert. Eines dieser Proteine ist Phospholamban (PLB), wobei der Anteil an phosphoryliertem PLB (P-PLB) als indirekter Nachweis für funktionelles cAMP verwendet Ergebnisse 50 werden kann. Um zu überprüfen, ob die Deletion von RyR2 im Reizleitungssystem einen Einfluss auf die Phosphorylierung von PLB hat, wurde eine Western-Blot Analyse durchgeführt (Abb. 3.25). Während der Proteinlevel von PLB sowohl im Sinusknoten als auch im Vorhof bei beiden Gruppen nicht unterschiedlich war, war die Proteinexpression des P-PLB im Sinusknoten von KO-Mäusen im Vergleich zu den Kontrollen deutlich reduziert (n= 3 Westernblots). Bei den Vorhöfen beider Gruppen gab es dagegen keinen Unterschied. Man kann daraus schließen, dass durch die Deletion von RyR2 die cAMP-abhängige Phosphorylierung selektiv im Sinusknoten der KO-Tiere erniedrigt ist. Abbildung 3.25: Western-Blot zur Ermittlung der Expression von Phospho-Phospholamban und Phospholamban. Die Expression von P-PLB ist im Sinusknoten der RyR2KiT-KO-Tiere erniedrigt. Die PLB Expression ist in allen Proben gleich stark. Tubulin diente als Ladekontrolle. Detektion mittels ECL-System. 3.2.8 Ermittlung des basalen intrazellulären Ca2+-Gehalts in Sinusknotenzellen Es konnte nachgewiesen werden, dass im Sinusknoten von Kaninchen die Adenylatcyclasen AC5, AC6, AC1 und AC8 exprimiert werden. AC5 und AC6 werden durch Ca2+ gehemmt, AC1 und AC8 dagegen sind direkt durch Ca2+ aktivierbar (Younes et al, 2008). Da die intrazelluläre Ca2+-Konzentration einer Zelle hauptsächlich von der Ca2+-Freisetzung aus dem SR durch den RyR abhängt, sollte somit RyR2 direkt den cAMP-Spiegel beeinflussen können (Lakatta et al, 2010). Um zu untersuchen, ob die Deletion von RyR2 Auswirkungen auf den Ca2+-Gehalt der Sinusknotenzellen hat, wurden Sinusknotenzellen isoliert, mit dem Fluoreszenzfarbstoff Fura-2AM beladen und der basale Ca2+-Gehalt Ergebnisse 51 gemessen. In Abb. 3.26A ist ein Beispiel einer Messung einer KO- sowie einer KontrollSinusknotenzelle dargestellt, bei der ein deutlicher Unterschied im basalen Ca 2+-Gehalt zu erkennen ist. Dieser Unterschied zwischen den beiden Gruppen war nicht in allen Messungen eindeutig nachzuweisen. So ist z.B. in Abb. 3.26B ein Beispiel einer Ca 2+Messung dargestellt, bei der der basale Ca2+-Gehalt sowie die Frequenz der KO-Zelle höher erscheint als bei der Kontroll-Sinusknotenzelle. Aufgrund der Trägheit des verwendeten Detektionssystems konnte keine sichere Aussage über die Amplitude der Ca 2+-Transienten gemacht werden. Abbildung 3.26: Beispiel von zwei verschiedenen Ca2+-Messungen an Sinusknotenzellen. (A) Die basale Ca2+-Konzentration der RyR2KiT-KO (rot) ist eindeutig niedriger als die der Kontrolle (schwarz). (B) Kein eindeutiger Unterschied im basalen Ca2+-Gehalt der RyR2KiT-KO im Vergleich zu der Kontrolle. Allerdings war der Mittelwert der gemessenen basalen Ca2+-Konzentration der KOSinusknotenzellen (n=14) signifikant erniedrigt gegenüber den Kontroll-Zellen (n=12). Dagegen konnte bei Ca2+-Messungen von Vorhofzellen (Ctr: n=16; KO: n=11) kein Unterschied zwischen KO- und Kontrolltieren festgestellt werden (Abb. 3.27A). Ein weiterer Unterschied konnte bei der Schlagfrequenz der einzelnen Sinusknotenzellen gemessen werden und zwar war die Frequenz der KO-Zellen meist niedriger als die der Kontrollen. Um auszuschließen, dass alleine eine niedrigere Schlagfrequenz für den niedrigeren basalen Ca2+-Gehalt verantwortlich war, wurde die Schlagfrequenz der Kontroll-Sinusknotenzellen mit Hilfe des If-Blockers Zatebradin (1 mM) in der externen Lösung gesenkt. So konnte mittels Zatebradin die Schlagfrequenz der Kontroll-Sinusknotenzellen von 150 bpm auf 79 bpm gesenkt werden und entsprach in etwa der Schlagfrequenz der KO-Sinusknotenzellen (82 bpm). Nach Senkung der Schlagfrequenz mit Zatebradin gab es zwar tatsächlich einen basal niedrigeren Ca2+-Gehalt in den Kontroll-Zellen, jedoch war der Unterschied nicht signifikant (Abb. 3.27B). Diese Ergebnisse geben einen ersten Hinweis darauf, dass die KO- Ergebnisse 52 Sinusknotenzellen auch unabhängig von ihrer Schlagfrequenz einen tendenziell niedrigeren Ca2+-Gehalt als die Kontroll-Sinusknotenzellen hatten. Abbildung 3.27: Mittelwerte der basalen Ca2+-Konzentration von Sinusknoten- und Vorhofzellen. (A) Basale Ca2+-Konzentration der Sinusknoten- und Vorhofzellen von RyR2KiT-KO-Mäusen und Kontrollen. (B) Kontroll-Sinusknoten-Zellen (n=12) ohne Zugabe von Zatebradin und KontrollSinusknoten-Zellen (n=8) nach Zugabe von 1 mM Zatebradin in die externe Lösung. Dargestellt sind die Mittelwerte ± Standardfehler. n.s., nicht signifikant; **, p<0,01. 3.3 RyR2-Rekombination ohne Induktion 3.3.1 RyR2-Rekombination ohne Induktion in der RyR2MerCreMer-Mauslinie Die Verwendung des Cre/loxP-Systems zur Steuerung der Rekombination ist nicht nur von der Gewebespezifität, sondern zusätzlich von der Stringenz der Kontrollierbarkeit der Rekombinaseaktivität abhängig. Es besteht die Gefahr, dass es auch ohne eine direkte Aktivierung der Cre-Rekombinase durch Tamoxifen möglicherweise zu einer unkontrollierten Rekombination kommt. Tatsächlich entwickelten die homozygoten RyR2MerCreMer-Tiere (RyR2L2/L2,MerCreMer) auch ohne Injektion von Tamoxifen eine Kardiomyopathie und starben im Alter von etwa drei Monaten. Dabei zeigten die Herzen die gleichen Anzeichen für Fibrose und atriale Dilatation wie die KO-Herzen, denen Tamoxifen injiziert wurde. Um die Rekombination auf genomischer Ebene zu untersuchen, wurde DNA aus unterschiedlichen Kompartimenten des Herzens isoliert und mit Hilfe einer PCR amplifiziert. Als Kontrolle diente DNA aus Muskel- und Lebergewebe. Dabei konnten in einer Reaktion RyR2L1-, RyR2L2- und Wildtyp-Allele amplifiziert werden (Abb. 3.28). Mit L2 wird das Allel mit Ergebnisse 53 dem „gefloxten" RyR2-Exon bezeichnet. L1 dagegen steht für das deletierte Allel mit einer verbliebenen RyR2 L2/L2 loxP-Stelle. Die DNA-Untersuchungen zeigten, dass sowohl bei ,MerCreMer-Tieren (Abb. 3.28B), als auch bei den heterozygoten Kontrollen, RyR2L2/+,MerCreMer (Abb. 3.28A), eine geringe Rekombination im rechten und linken Ventrikel detektiert werden konnte, ohne dass vorher Tamoxifen gespritzt wurde. Nach einer einmaligen Injektion und verstärkt nach mehrmaligem Injizieren von Tamoxifen, konnten dann im ganzen Herzen Banden für die erfolgreiche Deletion (L1) nachgewiesen werden. In der Leber und im Muskelgewebe dagegen konnte, wie zu erwarten, nur Banden für das gefloxte RyR2-Gen (L2) amplifiziert werden. Dadurch konnte gezeigt werden, dass auch vor der Behandlung mit Tamoxifen bereits eine geringe Deletion im Ventrikel stattgefunden hatte, d.h. eine Aktivität der Cre-Rekombinase ohne Tamoxifen-vermittelte Induktion stattgefunden hatte. Abbildung 3.28: PCR mit genomischer DNA aus RyR2MerCreMer-KO-Herzen. (A) PCR mit DNA aus Herzgewebe einer RyR2L2/+,MerCreMer-Maus (B) PCR mit DNA aus Gewebe einer RyR2L2/L2,MerCreMer-Maus. Es wurden die Allele RyR2L1 (220 bp), RyR2L2 (340 bp) und RyR2WT (300 bp) nachgewiesen. Somit fand auch ohne Induktion durch Tamoxifen eine Rekombination statt. Ergebnisse 54 3.3.2 RyR2-Rekombination ohne Induktion in der RyR2KiT-Mauslinie Wie bereits in Abschnitt 3.2.3 erwähnt wurde bei EKG-Messungen der RyR2KiT-Linie bereits vor der Behandlung mit Tamoxifen eine EKG-Dauermessung durchgeführt. Dabei fiel auf, dass die basale Herzfrequenz der homozygoten RyR2KiT-Tiere (RyR2L2/L2,KiT) bereits vor der gezielten Deletion von RyR2 im Reizleitungssystem etwas erniedrigt war im Vergleich zu den Kontrollen (Abb. 3.29). Demgegenüber war die basale Herzfrequenz der Kontrolltiere (RyR2L2/+,KiT) nach Tamoxifen-Gabe im Vergleich zu den Messungen vor der TamoxifenBehandlung nicht wesentlich verändert. Ein Grund dafür könnte sein, dass wie bereits für die RyR2MerCreMer-Mauslinie gezeigt, vor der Tamoxifen-Behandlung eine Rekombination des gefloxten RyR2-Locus stattgefunden hatte. Es muss jedoch klar festgehalten werden, dass die Herzfrequenz der RyR2KiT-KO-Tiere nach Injektion von Tamoxifen deutlich weiter erniedrigt. Abbildung 3.29: Telemetrische EKG-Ableitungen an wachen RyR2KiT-Mäusen. Dargestellt ist die Herzfrequenz der RyR2L2/L2,KiT-Tiere (n=7) und Kontrollen (n=7) vor und nach Tamoxifen-Behandlung, gemessen während einer EKG-Dauermessung über sieben Tage. Auch ohne Tamoxifen-Injektion (rosa) ist die Herzfrequenz der RyR2L2/L2,KiT-Tiere niedriger, als die der Kontrollen (schwarz ohne Tamoxifen, grau mit Tamoxifen). Zur besseren Darstellung wurden die Daten durch Mitteln von 50 benachbarten Punkten geglättet. Tam, Tamoxifen. Um nachzuprüfen, ob die erniedrigte Herzfrequenz tatsächlich, wie vermutet, durch eine Rekombination ausgelöst wurde, wurde DNA aus Sinusknoten-Gewebe von RyR2L2/L2,KiTTieren ohne Tamoxifen-Injektion isoliert und eine PCR durchgeführt (Abb. 3.30A). Es konnte in allen drei isolierten Sinusknotenproben neben der L2-Bande auch eine schwache L1- Ergebnisse 55 Bande nachgewiesen werden. Somit fand tatsächlich auch ohne Tamoxifen eine Rekombination im Sinusknoten statt. Um die Rekombination genauer zu untersuchen, wurde, wie für die RyR2MerCreMer-Mauslinie, DNA zu unterschiedlichen Zeitpunkten während dem Zeitraum der Tamoxifen-Behandlung bzw. sechs Wochen nach Beginn der Tamoxifen-Injektion, isoliert und mit Hilfe einer PCR amplifiziert. Die DNA-Untersuchungen zeigten, dass bei RyR2L2/L2,KiT-Tieren nach einmaligem Spritzen eine deutliche Rekombination im Sinusknoten zu erkennen ist. Zudem konnte eine Woche nach Beginn der Tamoxifen-Behandlung sowie sechs Wochen nach der Behandlung neben dem Sinusknoten auch im rechten Vorhof eine sehr schwache L1-Bande nachgewiesen werden (Abb. 3.30B). Somit könnte sich der Knockout nicht wie beabsichtigt nur auf das Reizleitungssystem, sondern in einem geringen Ausmaß auch auf den rechten Vorhof erstrecken. Andererseits ist nicht auszuschließen, dass bei der Isolation des rechten Vorhofs zusätzlich auch Sinusknotenzellen mit isoliert wurden. Abbildung 3.30: PCR mit genomischer DNA aus Herzgewebe von RyR2L2/L2,KiT-Mäusen. (A) PCR mit DNA aus Sinusknoten-Gewebe von drei verschiedenen RyR2L2/L2,KiT-Mäusen. (B) PCR mit DNA aus Herzgewebe einer RyR2L2/L2,KiT-Maus. Es wurde DNA aus den angegebenen Geweben isoliert und mit einer PCR die Allele RyR2L1 (220 bp) und RyR2L2 (340 bp) nachgewiesen. Es fand auch ohne Tamoxifen eine geringe Rekombination im Sinusknoten statt. Ergebnisse 56 3.3.3 RyR2-Rekombination ohne Induktion in der ubiquitär aktiven Mauslinie RyR2CAGG-Cre Um zu überprüfen, ob die Rekombination auf ein ungewöhnlich leicht rekombinierbares gefloxtes RyR2-Allel zurückzuführen ist, wurde die gefloxte RyR2-Mauslinie mit einer dritten Cre-Linie, der CAGGCre-ERTM-Linie verpaart. Das CAGG-Cre (Hayashi und McMahon, 2002) ist ubiquitär aktiv und kann ebenfalls durch Tamoxifen erst im adulten Tier aktiviert werden. Unerwarteterweise Nachkommen gezüchtet konnten werden. jedoch Dagegen keine konnten homozygoten bei der RyR2L2/L2,CAGG- Genotypisierung der Nachkommen RyR2L1/+,CAGG-Tiere, bei denen also bereits ein Allel von RyR2 deletiert war, nachgewiesen werden. Daraus lässt sich schließen, dass bereits während der Embryonalentwicklung, ohne Induktion durch Tamoxifen, eine so starke Rekombination des RyR2-Gens stattgefunden hatte, dass RyR2 deletiert wurde und zur embryonalen Letalität geführt hat. Zur genauen Untersuchung wurde eine DNA-Analyse der Herzen der RyR2L1/+,CAGG-Mäuse durchgeführt. Im Herzgewebe konnten wie erwartet L1-Banden, jedoch keine Banden für L2 nachgewiesen werden. Somit konnte bei allen drei untersuchten Cre-Linien (MerCreMer, HCN4KiT-Cre und CAGG-Cre) unerwarteterweise eine Rekombination des RyR2-Locus auch ohne Tamoxifen-Behandlung nachgewiesen werden. Abbildung 3.31: PCR mit DNA aus Ventrikelgewebe zweier RyR2L1/+,CAGG-Mäuse. Es konnten in beiden Mäusen Banden für wt und RyR2L1 nachgewiesen werden, nicht jedoch für das gefloxte RyR2L2. 3.4 Analyse der RyR3-Expression im Herzen Neben dem RyR2 wird im Sinusknoten und in den Vorhöfen auch der RyR3 exprimiert. Bis jetzt konnte dies jedoch nur auf RNA-Ebene (Tellez et al, 2006) nachgewiesen werden. Wie in Abschnitt 3.13 konnten diese Ergebnisse auch in der vorliegenden Arbeit bei RNA- Ergebnisse 57 Analysen bestätigt werden. Um zu überprüfen, welche Rolle der RyR3 im Herzen spielt, sollte einerseits seine Expression genauer untersucht und andererseits eine RyR3-KO-Maus analysiert werden. 3.4.1 Nachweis der RyR3-Proteinexpression im Herzen Da Untersuchungen auf Transkriptebene nur bedingt Rückschlüsse auf das Proteinniveau geben, wurde die Expression von RyR3 im Herzen auch auf Proteinebene analysiert. Dazu wurden Proteinextrakte aus verschiedenen Kompartimenten des Herzens hergestellt und eine Western-Blot Analyse durchgeführt. Dabei konnte gezeigt werden, dass tatsächlich neben RyR2- auch RyR3-Protein im Herzen der Mäuse exprimiert wird. Im Gegensatz zu RyR2, der im ganzen Herzen exprimiert wird, konnte RyR3 jedoch selektiv im Sinusknoten und in den Vorhöfen detektiert werden. Lebergewebe diente als Negativkontrolle, in der weder RyR2 noch RyR3 exprimiert werden (Abb. 3.32). Abbildung 3.32: RyR3-Proteinexpression im Herzen. Nachweis von RyR3-Protein (565 kDA) im SAN und in den Vorhöfen. Im Ventrikel konnte RyR3 nicht detektiert werden. r und l bezeichnen den rechten bzw. linken Vorhof oder Ventrikel. 3.4.2 Nachweis der RyR3-Expression in isolierten Sinusknotenzellen In isoliertem Sinusknoten-Gewebe können sich neben Sinusknotenzellen auch noch andere Zellarten wie Vorhofmyozyten oder Bindegewebszellen befinden. Um zu untersuchen, ob RyR3 tatsächlich in einzelnen Sinusknotenzellen exprimiert wird, wurde das Verfahren der Einzelzell-RT-PCR durchgeführt. Dazu wurden einzelne Sinusknotenzellen isoliert, eingesammelt und eine RT-PCR durchgeführt. Dabei dienten Primer, die den Sinusknoten spezifischen-Ionenkanal HCN4 erkennen, als Marker für Sinusknotenzellen. Anhand der RTPCR konnte gezeigt werden, dass einzelne Sinusknotenzellen tastsächlich neben dem Sinusknotenmarker HCN4 auch RyR3 exprimieren (Abb. 3.33). Ergebnisse 58 Abbildung 3.33: RyR3-mRNA Expression in isolierten Sinusknotenzellen. Abgebildet sind die RT-PCR-Ansätze von zwei isolierten Sinusknotenzellen mit spezifischen Primern für RyR3 (Mf/Mr) und HCN4 (rtE/rtF). RyR3 wird in Sinuskontenzellen exprimiert. 3.5 EKG-Untersuchungen an RyR3KO-Mäusen Im Gegensatz zum RyR2-total-Knockout sind RyR3KO-Mäuse lebensfähig und zeigen keine veränderte Muskelfunktion (Takeshima et al, 1996; Bertocchini et al, 1997). Da gezeigt werden konnte, dass RyR3 im Sinusknoten und Vorhof exprimiert wird (s. Punkt 3.4), wurde nun untersucht, ob die Deletion von RyR3 einen Einfluss auf die Herzfunktion hat. Dazu wurden mittels telemetrischer EKG-Ableitung die Herzfrequenz und die Aktivität von globalen RyR3KO-Mäusen (Bertocchini et al, 1997) im Vergleich zu Wildtyp-Mäusen untersucht. 3.5.1 Analyse der Herzfrequenz und der Aktivität der RyR3KO-Mäuse Das EKG der RyR3KO-Mäuse und der Wildtyp-Tiere wurde eine Woche lang kontinuierlich aufgezeichnet. Anschließend wurde die Applikation verschiedener Pharmaka durchgeführt. Es konnte kein Unterschied in der Herzfrequenz oder der Aktivität der RyR3KO-Tiere im Vergleich mit den Kontrollen festgestellt werden (Abb. 3.34). Die basale Herzfrequenz lag bei Wildtyp-Mäusen bei 567±58 bpm und bei RyR3KO-Mäusen bei 537±56 bpm. Außerdem zeigten die RyR3KO-Mäuse keine Veränderungen im Tag-Nacht-Rhythmus. Ergebnisse 59 Abbildung 3.34: Herzfrequenz und Aktivität der RyR3KO- und Wildtyp-Mäuse. Kein Unterschied in der mittleren Herzfrequenz und der Aktivität der RyR3KO-Mäuse (rot) zu den Kontrollen (schwarz). Des Weiteren wurden EKG-Parameter ausgewertet (Tab. 3). Sowohl die P-Dauer als auch die PQ-, QRS- und QT- und TP-Intervalle wurden ermittelt. Das Herzfrequenz-korrigierte QTc-Intervall wurde berechnet. Auch hier wurden keine signifikanten Unterschiede festgestellt. Daraus lässt sich schließen, dass RyR3 keinen Einfluss auf die HerzfrequenzRegulation oder Reizweiterleitung hat. P-Dauer PQ QRS QT TP WT (n=4) 11,4±2,5 24,7±1,9 6,6±0,3 41,1±5,1 27,6±5,3 RyR3KO (n=6) 11,8±1,1 27,7±1,0 7,5±0,7 37,8±3,7 31,8±6,4 Tabelle 2: EKG-Parameter von RyR3KO- und Wildtyp-Mäusen. Die Werte sind als Mittelwerte ± Standardabweichung in ms angegeben. P-Dauer, PQ, QRS und QT bezeichnen die dazu gehörigen EKG-Intervalle. 3.5.2 Untersuchung der Herzfrequenz-Modulation in RyR3KO-Mäusen Die Herzfrequenz-Steigerung wurde als erstes mit dem β-Agonisten Isoprenalin untersucht (Abb. 3.35). Es konnten sowohl die Wildtyp-Mäuse als auch die RyR3KO-Tiere die Ergebnisse 60 Herzfrequenz auf ungefähr 700 bpm steigern. Folglich war die Herzfrequenz-Steigerung durch den β-Agonisten in diesen Tieren nicht beeinflusst. Die Herzfrequenz-Senkung wird am Beispiel von Carbachol gezeigt. Es trat kein signifikanter Unterschied in der Herzfrequenz-Senkung im Vergleich zwischen RyR3KO-Tieren (182,8±27,2 bpm) und Wildtyp-Mäusen (174,2±19,3 bpm) auf. Abbildung 3.35: Herzfrequenz-Modulation in den RyR3KO-Tieren. (A) Herzfrequenz-Steigerung mittels Isoprenalin (0,5 mg/kg) (B) Herzfrequenzsenkung mit Carbachol (0,5 mg/kg). Isoprenalin und Carbachol wurden jeweils zum Zeitpunkt t=0 injiziert. RyR3KO-Mäuse (rot, n=6) konnten ebenso wie die Wildtyp-Tiere (schwarz, n=4) ihre Herzfrequenz nach IsoprenalinInjektion steigern bzw. nach Carbachol-Injektion senken. Neben Isoprenalin und Carbachol wurde zusätzlich auch die Wirkung der Pharmaka CCPA und Atropin getestet. Es konnten jedoch auch bei diesen Substanzen kein Unterschied zwischen den RyR3KO-Mäusen und den Wildtyp-Tieren gemessen werden. Durch den A1Adenosin Rezeptor Agonisten CCPA (0,3 mg/kg) wurde in beiden Gruppen eine Senkung der Herzfrequenz auf circa 140 bpm erreicht, durch den Acetylcholin-Receptor Antagonist Atropin (1 mg/kg) ließ sich die Herzfrequenz jeweils auf ca. 710 bpm steigern (Abb. 3.36). Es waren somit keine signifikanten Unterschiede zwischen den Mauslinien vorhanden. Ergebnisse 61 Abbildung 3.36: Herzfrequenz-Modulation durch verschiedene Pharmaka in RyR3KO-Tieren. Mittlere Herzfrequenz im Vergleich zwischen Wildtyp (schwarz, n=4) und RyR3KO-Tieren (rot, n=6). Die basale Herzfrequenz und die Herzfrequenz nach der Gabe von 0,5 mg/kg Isoprenalin (Iso), 1 mg/kg Atropin (Atro), 2 mg/kg Zatebradin (Zate), 0,3 mg/kg CCPA oder 0,5 mg/kg Carbachol (Carb), waren zwischen den beiden Genotypen nicht signifikant verändert. Dargestellt sind die Mittelwerte ± Standardabweichung der ersten 30 min nach der Pharmaka-Injektion. n.s., nicht signifikant. 3.5.3 Analyse der Herzfrequenzvariabilität Als nächstes wurde untersucht, ob es bei den RyR3KO-Mäusen ähnlich wie bei den RyR2KiT-KO-Tieren zu Unterschieden in der Herzfrequenzvariabilität kommt. Hierzu wurden die EKG-Ableitungen nach der Injektion von Carbachol, wenn die Herzfrequenz wieder begann auf die Basalwerte anzusteigen, untersucht. Die Dauer des RR-Intervale wurde bestimmt und die Herzfrequenzvariabilität als Standardabweichung der RR-Werte (RR-SD) dargestellt. Es konnte kein Unterschied in der Herzfrequenzvariabilität zwischen den RyR3KO-Tieren und den Wildtyp-Mäusen nachgewiesen werden. Beide Gruppen hatten Schwierigkeiten in dieser Phase streng rhythmische Herzschläge zu generieren (Abb. 3.37). Somit konnte gezeigt werden, dass RyR3 alleine keinen Einfluss auf die Herzfrequenz oder Rhythmogenese hat. Weitere Experimente sind nötig um die genaue Rolle von RyR3 im Herzen bestimm bestimmen zu können. Ergebnisse 62 Abbildung 3.37: Darstellung der Herzfrequenzvariabilität. (A) Dauer der RR-Intervalle nach Abklingen der Carbachol-Wirkung. (B) Standardabweichung der RRDauer. Bei RyR3KO-Mäusen war keine veränderte Herzfrequenzvariabilität nachzuweisen. n.s., nicht signifikant. Diskussion 4. 63 Diskussion Der Ryanodinrezeptor RyR2 ist ein intrazellulärer Ca2+-Kanal, der die Ca2+-Freisetzung aus dem SR steuert und eine große Rolle bei der Kontraktion des Herzens spielt (Bers, 2002). Da die globale RyR2-Deletion in Mäusen embryonal letal ist (Takeshima et al, 1998), konnte bis jetzt die genaue Funktion von RyR2 im Herzen anhand eines Mausmodells nicht untersucht werden. Neben seiner Rolle bei der Kontraktion wird darüber diskutiert, ob RyR2 auch eine wichtige Funktion bei der Generierung des Schrittmacherpotentials im Sinusknoten hat (Maltsev et al, 2006; Lakatta et al, 2010). Daher war es das Ziel dieser Arbeit, die Funktion von RyR2 sowohl im ganzen Herzen als auch selektiv nur im Erregungsbildungs- und leitungssystem mit Hilfe zweier RyR2-KO-Modelle zu charakterisieren. Dazu wurden zwei konditionelle Mauslinien verwendet, bei denen der Knockout im adulten Tier durch Tamoxifen induzierbar war. Mit Hilfe dieser beiden Mauslinien konnte die RyR2-Deletion sowohl zeitlich als auch gewebespezifisch kontrolliert werden. Zusätzlich sollte die Expression einer weiteren Ryanodinrezeptor-Isoform RyR3 im Herzen auf Protein-Ebene analysiert und seine Funktion mittels EKG-Telemetrie untersucht werden. 4.1 Charakterisierung der RyR2MerCreMer-KO-Mäuse Die erfolgreiche Deletion in diesem Mausmordell konnte dadurch gezeigt werden, dass sowohl die mRNA- als auch die Protein-Expression von RyR2 in den Ventrikeln der KO-Tiere stark erniedrigt war. Allerdings war RyR2-mRNA und RyR2-Protein in den Vorhöfen noch nachweisbar. Dies kann dadurch erklärt werden, dass die MerCreMer-Recombinase in den Vorhöfen in geringer Konzentration und/oder als Mosaik exprimiert ist. mRNA- Untersuchungen der beiden Isoformen RyR1 und RyR3 in RyR2MerCreMer-KO-Mäusen zeigten keine kompensatorische Hochregulierung der beiden Isoformen nach Deletion von RyR2. Da es sich bei der durchgeführten RT-PCR jedoch um eine semiquantitative Methode handelte, konnte die Expression der einzelnen Isoformen nicht genau quantifiziert werden. Dies könnte mit Hilfe einer quantitativen Real-Time-RT-PCR durchgeführt werden. Ventrikuläre RyR2-Deletion führt zu Herzinsuffizienz Die globale Deletion von RyR2 ist vermutlich embryonal letal aufgrund der wichtigen Rolle der intrazellulären Ca2+-Homöostase für die Kontraktion des Herzens (Takeshima et al, Diskussion 64 1998). Auch die in dieser Arbeit durchgeführte konditionelle Deletion von RyR2 im Ventrikel führte zum Tod der KO-Mäuse. Überraschend war, dass bereits innerhalb von zwei Wochen nach Beginn der Tamoxifen-Injektion eine so starke Beeinträchtigung der Herzfunktion bei den RyR2MerCreMer-KO-Tieren vorlag, dass die Tiere starben. Die KO-Tiere zeigten Zeichen einer Herzinsuffizienz mit leicht vergrößerten Ventrikeln und massiv dilatierten und mit Blut und Thromben gefüllten Vorhöfen. Sowohl das Herzgewicht als auch das Herz/Körpergewichtverhältnis der KO-Tiere waren signifikant erhöht. Der Grund dafür war vor allem der sehr schwere linke Vorhof. Durch die Deletion ist die Ca 2+-Homöostase beeinträchtigt, was in einer Störung des Kontraktionsvermögens resultiert. Anhand der Sirius-Red-Färbung konnte gezeigt werden, dass durch die Deletion von RyR2 im Herzen der KO-Mäuse eine Fibrosierung im Myokard entstanden war. Das Vorhandensein einer Fibrose ist ein deutlicher Hinweis darauf, dass im Herzen Umbauprozesse ablaufen. Dabei werden die Proteine der extrazellulären Matrix, deren Hauptbestandteil Kollagene darstellen, im Herzen stark überexprimiert (Burlew und Weber, 2002). Es kommt dadurch zu einer vermehrten Versteifung und auch zu einer Massenzunahme des Ventrikels. Dies bedingt eine unzureichende Füllung des Herzens während der Diastole (= diastolische Dysfunktion) und eine verminderte Auswurfleistung (= systolische Dysfunktion) (Janicki and Brower, 2002). Durch den Rückstau kommt es zur Dilatation der Vorhöfe und zur Bildung von Thromben, was ausgeprägt in RyR2MerCreMer-KO-Mäusen zu beobachten war. Außerdem waren Unterschiede auf zellulärer Ebene zu erkennen. So konnte bei der Lektin-Färbung und der Untersuchung isolierter Kardiomyozyten gezeigt werden, dass die Zellen der RyR2MerCreMer-KO-Tiere vergrößert und deformiert waren. Dies ist ein weiteres Anzeichen für die beobachtete Kardiomyopathie der Mutanten. Aufgrund der höheren Belastung der Kardiomyozyten während der Kardiomyopthie kann es zu einer vermehrten Apoptose der Zellen kommen. Durch die Apoptose wiederum könnte die Reizleitung im Herz gestört werden, wodurch die funktionellen Eigenschaften gestört sind (Duszanska et al, 2008). Ebenfalls führt die Beschädigung der Kardiomyozyten zu einem Verlust an gap junctions, was in einer systolischen Dysfunktion resultiert. Hinzu kommt die Tatsache, dass sich durch die Fibrose der elektrische Widerstand erhöht, so dass die elektrophysiologische Kommunikation der Kardiomyozyten untereinander gestört wird (Lee et al, 1998; Bradford et al, 2007). Die morphologischen Veränderungen lassen somit auf eine Störung in der elektromechanischen Kopplung nach Verlust von RyR2 schließen. Diskussion 65 Therapeutische Beeinflussung der Umbauprozesse im Myokard Aufgrund der schnellen Entwicklung einer Kardiomyopathie stellt die RyR2MerCreMer-KOMauslinie ein geeignetes Modell dar, Pharmaka zu untersuchen, die sich positiv auf die aufgetretenen Umbauprozesse im Myokard auswirken könnten (Jones et al, 2004; Hoshikawa et al, 2011). Dazu wurde in unserer Arbeitsgruppe bereits in ersten Versuchen RyR2MerCreMerL2/L2-Tieren ab dem Alter von vier Wochen der ACE-Hemmer Captopril (0,25 mg/ml) zusammen mit dem Betablocker Metoprolol (0,5 mg/ml) oral im Trinkwasser verabreicht. Im Alter von zwei Monaten wurde mit der Tamoxifen-Behandlung begonnen und das Überleben der KO-Tiere dokumentiert. Es konnte jedoch bisher kein Unterschied bei der Überlebensrate der RyR2MerCreMer-KO-Tiere, die mit den Pharmaka behandelt wurden, im Vergleich zu KO-Tieren, die Captopril und Metoprolol nicht erhalten hatten, nachgewiesen werden. Ein Grund könnte darin liegen, dass die Umbauprozesse, welche durch das Fehlen von RyR2 aufgetretenen sind, zu schnell abliefen, so dass die KO-Tiere bereits kurze Zeit nach der Tamoxifen-Behandlung starben. Außerdem fanden, wie in Abschnitt 3.3 beschrieben, auch ohne Tamoxifen-Injektion Rekombinationsereignisse statt. Dadurch könnte die Herzfunktion auch schon vor dem Verabreichen der Pharmaka stark beeinträchtigt gewesen sein. Dagegen konnten mit Candesartan, einem AT1-RezeptorAntagonisten, bereits erste Unterschiede in der Überlebensdauer der KO-Tiere festgestellt werden (R. Hummel, unveröffentlichte Daten). Dabei wurden RyR2L2L2,MerCreMer-Tiere ohne Injektion von Tamoxifen verwendet. Die Tiere entwickelten auch ohne Tamoxifen eine Herzinsuffizienz, im Gegensatz zu den behandelten Kardiomyopathie jedoch deutlich langsamer. Den RyR2 L2/L2 Tieren entwickelt sich die ,MerCreMer-Tieren wurde dazu ab einem Alter von 6-8 Wochen täglich 2,5 mg/kg Candesartan oral verabreicht. Es zeigte sich, dass durch die Gabe von Candesartan die Überlebensdauer der Tiere um 17 Tage verlängert werden konnte. In weiteren Versuchen wird sich zeigen, wie ausgeprägt die Umbauprozesse im Myokard der Mutanten durch die Pharmaka-Wirkung beeinflusst werden. Beeinträchtigung der Herzfunktion Da vermutet wird, dass RyR2 auch wichtig bei der Generierung des Schrittmacherpotentials ist, wurde versucht, auch diese Rolle mit Hilfe der RyR2MerCreMer-KO-Tiere zu untersuchen. Dazu wurden EKG-Analysen an RyR2MerCreMer-KO-Mäusen durchgeführt. Dabei zeigten sich bereits wenige Tage nach Beginn der Tamoxifen-Injektion Unterschiede in den EKG-Ableitungen der KO-Tiere im Vergleich zu den Kontrollen. Die KO-Mäuse entwickelten eine Bradykardie, wobei die Herzfrequenz immer weiter absank. Untersuchungen der einzelnen EKG-Abschnitte zeigten, dass neben der RR-Zeit vor allem Diskussion 66 das TP-Intervall stark verlängert und die Morphologie der T-Welle verändert war. Das TPIntervall spiegelt den Zeitraum vom Ende eines EKG-Komplexes bis zum Beginn des folgenden EKG-Abschnitts wider. Die Verlängerung der TP-Zeit könnte durch eine fehlgesteuerte Aktionspotential-Generierung im Sinusknoten erklärt werden. Aufgrund der Morphologie-Änderung der T-Welle scheint auch die Erregungsrückbildung, welche durch die T-Welle angezeigt wird, beeinträchtigt. Auffällig war auch, dass besonders häufig negative PWellen auftraten. Die Vorhöfe werden in diesem Fall nicht vom Sinusknoten aus depolarisiert, sondern retrograd über den AV-Knoten. Dies deutet ebenfalls auf eine Dysfunktion des Sinusknotens hin. Neben der immer langsamer werdenden Herzfrequenz war bei den KO-Tieren bereits kurze Zeit nach der Tamoxifen-Injektion keine circadiane Rhythmik mehr zu erkennen. Dies lässt darauf schließen, dass bei den KO-Tieren keine Modulation der Herzfrequenz mehr möglich ist. Die Veränderungen im EKG lassen somit auf eine Störung der Rhythmogenese nach Verlust von RyR2 schließen. Jedoch ist nicht auszuschließen, dass die aufgetretene Störung in der Rhythmogenese auch ein sekundärer Effekt der Herzinsuffizienz ist. 4.2 Charakterisierung der RyR2KiT-KO-Mäuse Der Mechanismus, der der spontanen Aktivität der Sinusknotenzellen zugrunde liegt, beruht auf einem Zusammenspiel von Ionenkanälen, intrazellulärer Ca 2+-Freisetzung und Ionentransportern. Die Rolle von RyR2 wird dabei in der Literatur kontrovers diskutiert (Maltsev und Lakatta, 2008; Mangoni und Nargeot, 2008; Lakatta und DiFrancesco, 2009). Es wird jedoch vermutet, dass die intrazelluläre Ca2+-Freisetzung über den RyR2 eine wichtige Rolle bei der Generierung des Schrittmacherpotentials spielt (Vinogradova et al, 2006). Da nicht genau geklärt werden konnte, ob die Störung in der Rhythmogenese bei der RyR2MerCreMer-KO-Mauslinie ein sekundärer Effekt der Herzinsuffizienz ist oder RyR2 tatsächlich auch eine große Rolle bei der Generierung des Schrittmacherpotentials spielt, wurde in dieser Arbeit neben den RyR2MerCreMer-KO-Mäusen, auch die RyR2KiTMauslinie untersucht. Bei diesen Mäusen wird RyR2 nach Injektion von Tamoxifen spezifisch nur in Zellen des Reizbildungs- und leitungssystems ausgeschaltet. Im Gegensatz zu den RyR2MerCreMer-KO-Mäusen waren die RyR2KiT-KO-Tiere lebensfähig und zeigten keine offensichtlichen phänotypischen Auffälligkeiten. Es konnten ebenfalls keine Anzeichen für Veränderungen in der Morphologie der Herzen oder des Sinusknotens zwischen den beiden Gruppen bei histologischen Färbungen (HE- und Sirius-Red-Färbung) festgestellt werden. Die erfolgreiche Deletion von RyR2 konnte anhand einer Western-Blot Analyse gezeigt werden, wobei das RyR2-Protein wie erwartet lediglich im Sinusknoten der KO-Mäuse Diskussion 67 deletiert war, dagegen im Sinusknoten der Kontrollen sowie in den Vorhöfen beider Gruppen weiterhin exprimiert wurde. Dagegen wurde bei RT-PCR-Untersuchungen weiterhin RyR2RNA im Sinusknoten der RyR2KiT-KO-Tiere nachgewiesen. Dies könnte damit erklärt werden, dass in isoliertem Sinusknotengewebe immer noch verhältnismäßig viele Vorhofmyozyten zu finden sind, in denen RyR2-mRNA weiterhin exprimiert wird, da in diesen Zellen keine KiT-Cre vermittelte Rekombination stattfindet. Zudem ist die Methode der RTPCR sehr sensitiv, so dass bereits geringste Mengen an mRNA detektiert werden können. Wie oben dargestellt ist auf der Proteinebene aber kein RyR2 mehr nachweisbar. RNAUntersuchungen der Isoformen RyR1 und RyR3 zeigten keine Unterschiede zwischen den Kontrollen und den KO-Tieren. Es gab somit keine kompensatorische Hochregulation dieser Isoformen in den KO-Tieren. RyR2-Deletion im Sinusknoten führt zu Bradykardie Mittels EKG-Telemetrie konnte gezeigt werden, dass die Deletion von RyR2 im Sinus- (und AV-) Knoten zu einer deutlichen Bradykardie führte. Dieses Ergebnis ist mit den Untersuchungen von Li et al, (1997) in Einklang zu bringen, die zeigen konnten, dass bei Zugabe von Ryanodin, mit dem man RyR2 blockieren kann, die Schlagfrequenz von kultivierten Kaninchen-Sinusknotenzellen erniedrigt wird. Die Ursache für die Bradykardie könnte möglicherweise in einer Verzögerung der Impulsleitung liegen. Die in dieser Arbeit durchgeführten EKG-Messungen zeigten tatsächlich eine verlängerte PQ-Zeit bei den RyR2KiT-KO-Mäusen. Die PQ-Zeit ist das Intervall vom Beginn der Vorhoferregung bis zum Beginn der Kammererregung, sie gibt Auskunft über die sogenannte Überleitungszeit. Es zeigte sich jedoch, dass die Bradykardie hauptsächlich durch eine Verlängerung der TPDauer zustande kam. Es war also nicht nur die Erregungsüberleitung von den Vorhöfen auf die Ventrikel, sondern insbesondere die Zeit zwischen dem Ende eines EKG-Komplexes bis zum Beginn der nächsten Erregung (TP-Zeit) verlangsamt. Dies lässt darauf schließen, dass die Funktion des Sinusknotens gestört ist. So kann es z.B. sein, dass das Zusammenspiel der einzelnen Sinusknotenzellen aufgrund der Deletion nicht mehr richtig funktioniert und es somit längert dauert, bis ein adäquates, koordiniertes Aktionspotential generiert wird. Ein weiterer Grund für die verlängerte TP-Zeit könnte sein, dass zwar ein Signal im Sinusknoten generiert wird, das Signal jedoch nicht aus dem Sinusknoten-Bereich weitergeleitet wird. Obwohl die KO-Mäuse eine Bradykardie aufwiesen, blieb der Tag-Nacht-Rhythmus weiterhin erhalten. Dies ist ein Anzeichen dafür, dass die Möglichkeit einer Modulation der Herzfrequenz nicht verloren gegangen war. Somit konnte eindeutig gezeigt werden, dass der RyR2 eine wichtige Rolle bei der Generierung des Schrittmacherpotentials spielt. Diskussion 68 Herzfrequenz-Modulation Die Herzfrequenz kann mit Hilfe verschiedener Neurotransmitter und Pharmaka moduliert werden. So kann z.B. durch den β-Adrenorezeptor-Agonisten Isoprenalin oder den muscarinischen Acetylcholin-Rezeptor-Antagonisten Atropin die Herzfrequenz gesteigert werden. Eine Herzfrequenz-Modulation war in den RyR2KiT-KO-Mäusen jedoch nur eingeschränkt möglich. Zwar ließ sich die Herzfrequenz der KO-Tiere genauso wie die der Kontrollen durch Isoprenalin oder Atropin steigern, es konnten jedoch nicht die gleichen maximalen Frequenzen erreicht werden. Zudem konnte die Herzfrequenz durch höhere Pharmaka-Konzentrationen nicht weiter gesteigert werden. Dies stimmt nur zum Teil mit den Studien von Vinogradova et al (2002) überein. Diese zeigten zwar ebenfalls, dass der RyR2 eine wichtige Rolle bei der Modulation des Herzrhythmus spielt, allerdings beschrieben sie, dass die Ca2+-Freisetzung aus dem SR über den RyR2 hauptsächlich für den positiv chronotrophen Effekt des β-adrenergen Signalwegs verantwortlich sei. Dazu schalteten sie den RyR mit Hilfe von Ryanodin (3 µmol/L) während β-adrenerger Stimulation funktionell aus, so dass kein Ca2+ mehr aus dem SR freigesetzt wurde. Dagegen konnte in der vorliegenden Arbeit gezeigt werden, dass eine Steigerung der Herzfrequenz durch Isoprenalin nach Deletion von RyR2 immer noch möglich war. Ein ähnliches Ergebnis konnte auch mit Pharmaka erzielt werden, die die Herzfrequenz senken. In diesem Fall, erreichten die KO-Tiere niedrigere Herzfrequenzen als die Kontrollen. Jedoch war aufgrund der Bradykardie die Herzfrequenz der KO-Tiere bereits vor der Pharmaka-Applikation im Vergleich zu den Kontrollen erniedrigt. Um diesen Effekt zu berücksichtigen, wurde berechnet, um welchen Prozentsatz sich die Herzfrequenz durch die Pharmaka-Injektion verändert hatte. Dabei konnte gezeigt werden, dass der relative Anstieg oder Abfall der Herzfrequenzen zwischen den Kontrollen und den KO-Mäusen nach Injektion der verschiedenen Pharmaka nicht unterschiedlich war. Somit sind die beobachteten Unterschiede in den Herzfrequenzen auf die Bradykardie der KO-Mäuse zurückzuführen. Dies spricht stark dafür, dass die Herzfrequenz-Stimulation nicht alleine von RyR2 abhängt, sondern, dass dabei auch andere Mechanismen oder Ionenkanäle eine Rolle spielen. So könnten z.B. die HCN-Kanäle, die T- und L-Typ Ca2+-Kanäle, der NCX1 oder sogar RyR3 der ebenfalls im Sinusknoten exprimiert wird, wichtig für die Herzfrequenz-Regulierung sein (Mangoni und Nargeot, 2008). RyR2 dagegen scheint eher für die Generierung der basalen Herzfrequenz als für deren Modulation verantwortlich zu sein. Verringerte Herzfrequenzvariabilität bei RyR2KiT-KO-Mäusen Diskussion 69 Die Herzfrequenz passt sich beständig momentanen Erfordernissen an. So hat z.B. körperliche Beanspruchung in der Regel eine Erhöhung der Herzfrequenz zur Folge, die bei Entlastung und Entspannung normalerweise wieder sinkt. Dabei zeigt sich eine höhere Anpassungsfähigkeit an Belastungen in einer größeren Variabilität der Herzfrequenz. Überraschenderweise wiesen die RyR2KiT-KO-Mäuse eine geringere Herzfrequenzvariabilität auf als die Kontrollen. Dies zeigte sich besonders deutlich, wenn die Herzfrequenz nach Abklingen der Wirkung von Carbachol wieder begann auf das Basalniveau anzusteigen. Während die Herzschläge der Kontroll-Tiere in dieser Phase ungleichmäßig erschienen, war die Herzfrequenz der KO-Mäuse rhythmischer. Im Gegensatz zu den Ergebnissen dieser Arbeit werden Mutationen im RyR2-Gen oft mit Arrhythmien, wie z.B. der catecholaminergen polymorphen ventrikulären Tachykardie (CPVT), in Verbindung gebracht (Priori et al, 2001). Der Unterschied könnte daran liegen, dass in der vorliegenden Arbeit eine Deletion von RyR2 gezielt im Reizleitungssystem stattfand, während bei den anderen Studien das RyR2-Gen im ganzen Herzen mutiert vorliegt. Außerdem konnte bei Studien von Watanabe et al (2009) gezeigt werden, dass die pharmakologische Hemmung von RyR2 bei Patienten, die an CPVT erkrankt waren, einen antiarrhythmischen Effekt hat. Dies könnte ebenfalls darauf hindeuten, dass eine Deletion von RyR2 eine antiarrhythmische Wirkung hat. Eine erhöhte Herzfrequenzvariabilität spiegelt ein funktionierendes autonomes Nervensystem wider (Gehrmann et al, 2000). Eine niedrigere Herzfrequenzvariabilität wie bei den RyR2KiT-KO-Tieren dagegen wird oft mit Krankheiten wie Herzinsuffizienz in Verbindung gebracht (Rich et al, 1988; Binkley et al, 1991). Die Höhe der Ruheherzfrequenz scheint jedoch ein entscheidender Faktor im Hinblick auf die Lebenserwartung zu sein, so dass möglicherweise eine anhaltend niedrigere Herzfrequenz eine erhöhte Lebenserwartung zur Folge hätte. So wurde in größeren Studien gezeigt, dass eine erhöhte Herzfrequenz ein Risikofaktor für die kardiovaskulär bedingte Mortalität ist (Kannel et al, 1987; Benetos et al, 1999). Mit Hilfe der RyR2KiT-KO-Mäuse könnte untersucht werden, ob tatsächlich ein Zusammenhang zwischen einer niedrigeren Herzfrequenz und einer erhöhten Lebenserwartung besteht. Beeinflussung des intrazellulären Ca2+-Gehalts und cAMP durch RyR2 Es wird vermutet, dass RyR2 durch die Kontrolle des intrazellulären Ca2+ einen Einfluss auf den cAMP-Gehalt und damit auch auf die Schrittmacheraktivität im Sinusknoten hat. Interessanterweise wurde bereits gezeigt, dass im Sinusknoten von Kaninchen mehr cAMP gebildet wird als in Kardiomyozyten (Vinogradova et al, 2006). Dies wiederum hätte zur Folge, dass im Sinusknoten durch den erhöhten cAMP-Gehalt essenzielle Regulatorproteine Diskussion 70 stärker phosphoryliert werden könnten als im restlichen Herzen. Eines dieser Proteine ist Phospholamban (PLB). Somit kann der Phosphorylierungslevel von PLB als indirekter Nachweis für den funktionellen cAMP-Gehalt verwendet werden. In dieser Arbeit konnte gezeigt werden, das P-PLB im Sinusknoten von RyR2KiT-KO-Tieren im Vergleich zu Kontrollen deutlich reduziert war. Dies spricht dafür, dass der cAMP-Gehalt im Sinusknoten der KO-Tiere ebenfalls reduziert ist. Der RyR2 scheint somit tatsächlich einen direkten Einfluss auf den cAMP-Gehalt zu haben. Zusätzlich konnten Younes et al (2008) zeigen, dass im Sinusknoten von Kaninchen, im Gegensatz zum Ventrikel, neben den Adenylatcyclasen AC5 und AC6 auch AC1 und AC8 exprimiert werden. AC5 und AC6 werden durch Ca2+ gehemmt, AC1 und AC8 dagegen direkt durch Ca2+ aktiviert. Um zu untersuchen, ob in den Sinusknotenzellen der KO-Tiere der basale Ca2+-Gehalt verändert ist und somit einen Einfluss auf die Aktivität der unterschiedlichen AC-Isoformen hat, wurden fluorometrische Ca2+-Messungen durchgeführt. Es konnte gezeigt werden, dass in Sinusknoten-Zellen von RyR2KiT-KO-Mäusen der basale Ca2+-Gehalt niedriger war als in den Kontrollzellen. Jedoch war der Unterschied nicht immer eindeutig. So gab es z.B. auch Messungen, bei denen der basale Ca2+-Gehalt sowie die Frequenz der KO-Zellen höher erschien, als bei den Kontroll-Sinusknotenzellen. Der niedrigere intrazellulärer Ca2+-Spiegel in den KO-Mäusen könnte allerdings einen Hinweis darauf geben, dass die AC1 und AC8 weniger aktiviert werden, was somit zu einem geringeren cAMP-Gehalt in den KO-Zellen führen könnte. Der erniedrigte cAMP-Gehalt könnte schließlich zu einer geringeren Aktivierung von If und dadurch zu einer Verlangsamung der diastolischen Depolarisationsrate des SAN führen. Dies wiederum könnte eine Erklärung für die Bradykardie der RyR2KiT-KO-Mäuse darstellen. Allerdings konnten bis jetzt nur in Sinusknotenzellen von Kaninchen die AC1 und AC8 nachgewiesen werden, somit ist noch nicht geklärt, ob im Sinusknoten der Maus die beiden Isoformen ebenfalls exprimiert werden. Außerdem werden im Sinusknoten der Maus auch die Isoformen AC5 und AC6 exprimiert, die ebenfalls durch Ca2+ beeinflusst werden können und so ebenfalls den cAMP-Spiegel steuern können. 4.3 Rekombination des gefloxten RyR2-Locus ohne Induktion Erstaunlicherweise starben RyR2L2/L2,MerCreMer-Tiere auch ohne Injektion von Tamoxifen im Alter von etwa drei Monaten. Dabei zeigten sie die gleichen Anzeichen für eine Kardiomyopathie und Herzinsuffizienz wie die KO-Tiere, denen Tamoxifen injiziert wurde. Auch die RyR2L2/L2,KiT-Tiere wiesen vor Tamoxifen bereits eine leichte Bradykardie auf. Ein Diskussion 71 Grund dafür könnte sein, dass bereits ohne Tamoxifen eine Rekombination des gefloxten RyR2-Allels stattgefunden hatte. Tatsächlich konnte dies auf DNA-Ebene sowohl für die RyR2MerCreMer- als auch für die RyR2KiT-Mauslinie bestätigt werden. Zusätzlich wurde ein induzierbarer ubiquitärer RyR2-KO mit Hilfe der CAGG-Cre-Mauslinie erzeugt. Auch in dieser Line fand eine Rekombination ohne Induktion mit Tamoxifen statt. Im Gegensatz zu den beiden anderen Mauslinien war die Rekombination jedoch bereits im Embryonalstadium so ausgeprägt, dass keine homozygoten RyR2L2/L2,CAGG-Nachkommen gezüchtet werden konnten. Dies deutet in allen drei RyR2-KO-Linien darauf hin, dass die verschiedenen CreRekombinasen “leaky” sind, also eine Rekombinaseaktivität auch ohne Tamoxifenvermittelte Induktion stattgefunden hatte. Eine mögliche Erklärung für diese unerwartete Beobachtung verschiedener bietet Gene. die Der Tatsache unterschiedlicher RyR2-Locus weist Chromatinstrukturen vermutlich eine leicht innerhalb zugängliche Chromatinstruktur auf, wodurch bereits durch eine sehr geringe Cre-Aktivität eine Rekombination hervorgerufen werden kann. Dass die Rekombination ohne Tamoxifen auf den außergewöhnlich leicht zugänglichen RyR2-Locus zurückzuführen ist, konnte auch dadurch bestätigt werden, dass bei weiteren Versuchen in unserer Arbeitsgruppe, z.B. bei der Deletion des Ca2+-Kanals Cav1.2 mit Hilfe der MerCreMer-Mauslinie, keine Rekombination ohne Tamoxifen beobachtet wurde. Auch bei der Verwendung der KiT-CreMaus zur Rekombination verschiedener anderer Loci konnte dieses Phänomen nicht beobachtet werden (Hoesl et al, 2008; Hermann et al, 2011). Da die RyR2 L2/L2,MerCreMerTiere auch vor einer Behandlung mit Tamoxifen eine Kardiomyopathie entwickelten, war es schwierig die zeitliche Entwicklung der Umbauprozesse im Herzen nach Tamoxifen genau zu bestimmen. Außerdem ist es sehr wahrscheinlich, dass auch bei der Verpaarung der RyR2L2/L2-Tiere mit anderen Cre-Linien zur Untersuchung von RyR2 z.B. im glatten Muskel oder im Gehirn schon vor einer Tamoxifen-Injektion eine Rekombinaton stattfinden wird. Diese Ergebnisse weisen darauf hin, dass generell bei der Verwendung konditioneller Mausmutanten genau überprüft werden muss, wann und inwieweit eine Rekombination stattgefunden hat, um Ergebnisse richtig interpretieren zu können. 4.4 Die Rolle von RyR3 im Herzen Bereits in früheren Studien konnte auf RNA-Ebene nachgewiesen werden, dass neben RyR2 auch RyR3 im Sinusknoten und in den Vorhöfen exprimiert wird (Mangoni und Nargeot, 2008; Tellez et al, 2006). Es konnte zudem gezeigt werden, dass der mRNA-Gehalt an RyR3 im zentralen Sinusknotengewebe sogar höher ist als der mRNA-Gehalt von RyR2. In der vorliegenden Arbeit konnte RyR3 zum ersten Mal auch auf Protein-Ebene im Sinusknoten- Diskussion 72 und Vorhof-Gewebe, nicht jedoch im Ventrikel, nachgewiesen werden. Zusätzlich konnte RyR3 auch in einzelnen isolierten Sinusknotenzellen detektiert werden. In isoliertem Sinusknotengewebe können sich immer noch Vorhofmyozyten befinden, daher war es wichtig zu zeigen, dass RyR3 auch in einzelnen Sinusknotenzellen vorkommt. Welche Rolle RyR3 in der Funktion des Herzens hat, wurde in ersten Versuchen anhand von EKG-Messungen von RyR3KO-Tieren untersucht. Es konnten sowohl in der Herzfrequenz als auch in der Aktivität der RyR3KO-Mäuse kein Unterschied zu den Kontrollen festgestellt werden. Auch nach Applikation verschiedener Pharmaka zeigten sich keine Unterschiede in der Herzfrequenz-Modulation. Im Gegensatz zu den RyR2KiT-KO-Tieren konnten auch keine Unterschiede in der Herzfrequenzvariabilität detektiert werden. Diese Ergebnisse stimmen zum Teil mit einem Bericht von Kihwan et al (1997) überein, der ebenfalls keine Unterschiede in der Herzfrequenz von RyR3KO-Tieren mit Wildtyp-Mäusen zeigen konnten. Dagegen wurde im Gegensatz zu dieser Arbeit eine verringerte Herzfrequenzvariabilität bei RyR3KO-Tieren im Vergleich zu den Kontrollen festgestellt. Der Unterschied könnte darin liegen, dass in der vorliegenden Arbeit eine RyR3KO-Mauslinie verwendet wurde bei der das Exon 6 und somit das Carboxy-Ende des Proteins deletiert wurde (Bertocchini et al, 1997). Im Gegensatz dazu wurde bei der RyR3KO-Mauslinie, die bei der Arbeit von Kihwan et al (1997) verwendet wurde, das Exon 1 deletiert (Takeshima et al, 1996). Daraus lässt sich schließen, dass RyR3 alleine keinen Einfluss auf die Herzfrequenz-Regulation oder Reizweiterleitung hat. Seine Rolle im Herzen bleibt jedoch weiterhin unklar. Es könnte sein, dass RyR3 und RyR2 sich gegenseitig beeinflussen und somit die Analyse einer Mauslinie, bei der sowohl der RyR2 als auch der RyR3 gleichzeitig deletiert sind, weitere Aufschlüsse über die Funktion von RyR3 im Herzen liefern könnte. Diskussion 4.5 73 Ausblick Die RyR2MerCreMer-Mauslinie kann in Zukunft für die Untersuchung von Pharmaka verwendet werden, die positiv auf die Umbauprozesse im Myokard wirken, wie z.B. ATIIAntagonisten oder ACE-Hemmer. Dabei kann die RyR2MerCreMer-Mauslinie (aufgrund der nachgewiesenen Deletion von RyR2 auch ohne Tamoxifen) ein geeignetes Modell darstellen diese Pharmaka in einer sich langsam entwickelnden Herzinsuffizienz zu untersuchen. Um die Rolle von RyR2 im Reizleitungssystem genauer zu untersuchen, könnten zusätzlich spontane Aktionspotentiale von einzelnen RyR2KiT-KO- und Kontroll-Sinusknotenzellen aufgezeichnet und analysiert werden. Zusätzlich könnte versucht werden, den cAMP-Gehalt im Sinusknoten der RyR2KiT-KO-Tiere genauer zu bestimmen. Weitere Studien sind außerdem nötig, um für die Rolle von RyR3 während der autonomen Herzaktion ein größeres Verständnis zu bekommen. Die funktionelle Analyse eines DoppelKnockouts, bei dem sowohl RyR3 als auch RyR2 im Reizleitungssystem deletiert ist, könnte hierbei wichtige Hinweise liefern. Dazu sollten Doppel-KO-Mäuse auf funktioneller, histologischer und molekularer Ebene charakterisiert werden. Anhang 74 5. Anhang 5.1 Verwendete Primer Die Primer der folgenden Tabellen wurden von der Firma MWG Biotech bezogen. Die Arbeitslösungen der Primer waren auf 10 pmol/µl verdünnt. „f“ steht für forward und „r“ für reverse. Primer für Genotypisierung: Primerbezeichnung Sequenzen 5´- GAC CCG AGT AGC AGC CGT TGCAG GGA GGA CTG RyR 143 RyR 142 RyR JS4r RyR3 M1f RyR3 M2r GCC ATA ACT AT - 3` 5´- CCG CAG CCG GTT GTC ACT CTCAC CCA AAA GGA GGA CAG TAG AGT - 3´ 5´- ATT GGG ACC ACG CCA GTG AACTG CCC TCA TCC AGT CGC TAG AC - 3´ 5´- CAC ATC CCA ATC TCC TTT ACT CC GTA CGC ATC GTG AAC CTC ATT G - 3 5´- GCT TAT TCT GCC CTA ATG CCA C TTT CGG CAG TTA AAG TTG ATG - 3´ RyR3 pGK_JS1 5´- CGT GCT ACT TCC ATT TGT CAC GTC - 3´ Cre 800 5´- GCT GCC ACG ACC AAG TGA CAG CAA TG - 3´ Cre 1200 5´- GTA GTT ATT CGG ATC ATC AGC TAC AC - 3´ Gabra1F 5´- AAC ACA CAC TGG AGG ACT GGC TAG G - 3´ Gabra1R 5´- CAA TGG TAG GCT CAC TCT GGG AGA TGA TA - 3´ Tabelle 3: Verwendete Primer für Genotypisierung. Anhang 75 Primer für RT-PCR: Primerbezeichnung Sequenzen RyR1 for 5´- TGA ATA CAA CGC CTG CTC TG - 3` RyR1 rev 5´- ATG TGT GGG ACT TGG ACC AG - 3´ RyR2 G 5´- GCG AAG GCG AGG ATG AGA TC - 3´ RyR2 H 5´- CGT AGC AAT ATG GCG TGT CC - 3 RyR3 Mf 5´- CAA CAA CAA CCT GGG CAT TGA T - 3´ RyR3 Mr 5´- GAA TGA AGA CCT CGG CCA CCA - 3´ HCN4 rt E 5´- ACA AAG TTG GGA TCT GCG TT - 3´ HCN4 rt F 5´- GAC AGC GCA TCC ATG ACT AC - 3´ Tabelle 4: Verwendete Primer für RT-PCR. 5.2 Verwendete Antikörper Antikörper gegen Spezies Bezeichnung/Verdünnung Herkunft RyR * Maus MA3-916 / 1:500 Thermo Scientific α-Tubulin Kaninchen 11H10 / 1:1000 PLB Maus MA3-922 / 1:200 Thermo Scientific p-PLB Kaninchen 07-052 / 1:200 Upstate RyR3 Kaninchen 1:1000 Kaninchen-IgG Meerrettichperoxidase Ziege goat anti-rabbit IgG-HRP/ 1:50.000 New England Biolabs Giannini et al, 1995 Dianova Anhang 76 Maus-IgG Meerrettichperoxidase Ziege goat anti-mouse IgG-HRP/ 1:50.000 Dianova Tabelle 5: Verwendete Antikörper. * Der RyR-Antikörper wird von Thermo Scientific so beschrieben, dass er RyR2 sowie schwach RyR1 im Gewebe von Amphibien, Hunden, Hühnern, Fischen, Meerschweinchen und Ratten erkennen soll. Somit sollte er im Herzen der Maus spezifisch RyR2 detektieren. 5.3 Verwendete transgene Mauslinien Mauslinie Promotor Eigenschaften Referenz Takeshima et al, 1998 RyR2 L2/L2 RyR3 Das vierte Exon des (erhalten von K. Chien, RyR2-Gens ist von loxP- Cardiovascular Research Sequenzen flankiert Center, Boston) Das sechste Exon des -/- RyR3-Gens ist deletiert Bertocchini et al, 1997 (erhalten von M. Gollasch, MDC, Berlin) Hoesl et al, 2008 HCN4KiT-Cre HCN4 Exprimiert Cre-ERT2 im (Generiert von AG Reizleitungssystem Ludwig) Sohal et al, 2001 MerCreMer α- MHC Exprimiert MerCreMer im ganzen Herzen Enhancer und Exprimiert Cre-ERT2 Huhn-Actin ubiquitär Promotor/Enhancer Tabelle 6: Verwendete Mauslinien. Cardiovascular Research Center, Boston) Hayashi und McMahon, Cytomegalovirus CAGG-Cre (erhalten von K. Chien, 2002 (bezogen von Charles River) Anhang 5.4 77 Abkürzungsverzeichnis: Abkürzung Erklärung °C Grad Celsius Abb. Abbildung AC Adenylatcyclase AP Aktionspotential AP I Anodenpuffer 1 AP II Anodenpuffer 2 APS Ammoniumperoxidsulfat ATP Adenosintriphosphat Atro Atropin AV-Knoten Atrioventrikularknoten BCA Bicinchoninsäure BDM Diacetylmonoxim bp Basenpaar bpm beats per minute BSA Bovines Serum Albumin cAMP zyklisches Adenosin-3', 5'-Monophosphat Carb Carbachol CCPA 2-Chloro-N6-Cyclo-pentyl-Adenosin cDNA Komplementäre Desoxyribonukleinsäure Cre Cre-Rekombinase CreERT2 Tamoxifen-induzierbare Cre-Rekombinase C-Terminus Carboxy-Terminus Ctr Kontrolle dATP Desoxyadenosintriphosphat dCTP Desoxycytosintriphosphat dest destilliert dGTP Desoxyguanosintriphosphat DMSO Dimethylsulfoxid DNA Desoxyribonukleinsäure DNAse Desoxyribonuclease dNTP Desoxynukleosidtriphosphat DTT Dithiothreitol dTTP Desoxythymidintriphosphat Anhang 78 ECL enhanced chemiluminescence EDTA Natrium-Ethylendiamin-N,N,N´,N´-Tetraacetat EKG Elektrokardiogramm T2 ER Fura 2 AM Modifizierte Ligandenbindungsdomäne des Östrogenrezeptors Acetoxymethylester des Fluoreszenzfarbstoffes Fura 2 g Gramm GABRA GABAA1-Rezeptor GAPDH Glycerinaldehyd-3-phosphat-Dehydrogenase gefloxt loxP flankiert h Stunde HBR Heart Bodyweight Ratio HCN Hyperpolarisations-aktivierter durch zyklische Nukleotide modulierter Kationenkanal HE Hämatoxylin / Eosin HRP Meerrettichperoxidase i.p. Intraperitoneal If "Funny" Strom der HCN-Kanäle IgG Immunglobulin G Iso Isoprenalin kb Kilobasen KB Kraftbrühe kDa Kilodalton kg Kilogramm KiT Knock-in induzierbar durch Tamoxifen KO Knockout KP Kathodenpuffer loxP Locus of crossover (x) of Phage P LSM Laser Scan Mikroskopie M Mol mA Milliampere Mer mutierter Östrogenrezeptor mg Milligramm MHC myosin heavy chain min Minute ml Milliliter Anhang 79 mm Millimeter mM Millimol mRNA Messenger RNA n.s. Nicht signifikant PAGE Polyacrylamid-Gelelektrophorese PBS Phosphat-gepufferte Kochsalzlösung PCR Polymerasekettenreaktion PFA Paraformaldehyd PLB Phospholamban P-PLB Phospho-Phospholamban PVDF Polyvinylidendifluorid RNA Ribonukleinsäure RNAse Ribonuklease rpm Umdrehungen pro Minute RT Raumtemperatur RT-PCR Reverse Transkriptions-PCR RyR Ryanodinrezeptor s Sekunde SAN Sinusknoten SD Standardabweichung SDS Natriumdodecylsulfat SERCA sarcoendoplasmatische Retikulum-CalciumATPase SERMs Selective Etrogen Receptor Modulators TAM Tamoxifen TBE Tris-Borat-EDTA-Puffer TBST Tris-Borat-Puffer mit Tween TE Tris-EDTA TEMED N,N,N´,N´,Tetramethylethyldiamin tg Transgen Tris Tris-(hydroxymethyl)-methylamin Tub Tubulin U Einheit ÜN über Nacht UV Ultraviolett Anhang 80 V Volt / Ventrikel WT Wildtyp Zate Zatebradin μg Microgramm μl Microliter μm Micrometer μM Micromolar μm2 Quadratmicrometer Tabelle 7: Verwendete Abkürzungen. Abbildungsverzeichnis 6. 81 Abbildungsverzeichnis Einleitung: Abbildung 1.1: Schematische Darstellung der elektromechanischen Kopplung im Herzen. Abbildung 1.2: Schematische Darstellung beitragenden Ionenströme eines Aktionspotentials und Ca2+-Freisetzungen und die dazu in einer Sinusknotenzelle. Abbildung 1.3: Schematische Darstellung der basalen Schrittmacherregulation. Abbildung 1.4: Schema des Cre/loxP-Rekombinationssystems. Material und Methoden: Abbildung 2.1: Aufbau der Blotting Apparatur. Abbildung 2.2: Telemetrische EKG-Ableitung. Abbildung 2.3: Definition der EKG-Wellen und -Intervalle. Abbildung 2.4: Verpaarungsschema zur Generierung von konditionellen RyR2-KOMäusen. Ergebnisse: Abbildung 3.1: Überlebensrate der RyR2MerCreMer-KO-Mäuse. Abbildung 3.2: RT-PCR zum Nachweis der RyR-Isoformen im Herzen der RyR2MerCreMer-KO-Mäuse. Abbildung 3.3: Expression von RyR2-Protein im Herzen der RyR2MerCreMer-KO-Mäuse. Abbildung 3.4: Repräsentative Aufnahmen der RyR2MerCreMer-KO-Herzen. Abbildung 3.5: Herz-Gewicht und HBR der RyR2MerCreMer-KO-Tiere. Abbildung 3.6: Telemetrische EKG-Ableitungen an wachen Tieren während der letzten sieben Tage vor dem Tod der KO-Tiere. Abbildung 3.7: EKG-Parameter der RyR2MerCreMer-KO-Mäuse. Abbildung 3.8: Beispiele telemetrischer EKG-Ableitungen. Abbildung 3.9: Atriale Thrombose und vermehrte Fibrose im Ventrikel der RyRMerCreMer-KO-Mäuse. Abbildung 3.10: Analyse der isolierten Kardiomyozyten. Abbildung 3.11: Lektin–Färbung von Paraffinschnitten durch das ventrikuläre Myokard zur Darstellung der Zellmembran. Abbildung 3.12: RT-PCR zum Nachweis der RyR-Isoformen im Sinusknotengewebe der RyR2KiT-KO-Tiere. Abbildungsverzeichnis Abbildung 3.13: 82 RT-PCR zum Nachweis der RyR-Isoformen im Herzgewebe der RyR2KiTKO-Tiere. Abbildung 3.14: RyR2-Proteinexpression im Herzen der RyR2KiT-KO-Mäuse. Abbildung 3.15: Morphologie der RyR2KiT-KO-Herzen. Abbildung 3.16: Telemetrische EKG-Ableitungen an wachen RyR2KiT-KO- und Kontrolltieren. Abbildung 3.17: Mittlere Herzfrequenz und Herzfrequenzvariabilität der RYR2KiT-KOTiere. Abbildung 3.18: EKG-Parameter von RyR2KiT-KO- und Kontrolltieren. Abbildung 3.19: Beispiele telemetrischer EKG-Ableitungen. Abbildung 3.20: Herzfrequenz-Steigerung mittels Isoprenalin. Abbildung 3.21: Herzfrequenz-Modulation in RYR2KiT-KO-Mäusen. Abbildung 3.22: Änderung der Herzfrequenzvariabilität bei RyR2KiT-KO-Tieren. Abbildung 3.23: Beispiele telemetrischer EKG-Ableitungen nach Abklingen der CarbacholWirkung. Abbildung 3.24: Basale Blutdruckmessungen an RyR2KiT-KO-Tieren. Abbildung 3.25: Western-Blot zur Ermittlung der Expression von Phospho-Phospholamban und Phospholamban. Abbildung 3.26: Beispiel von zwei verschiedenen Ca2+-Messungen an Sinusknotenzellen. Abbildung 3.27: Mittelwerte der basalen Ca2+-Konzentration von Sinusknoten- und Vorhofzellen. Abbildung 3.28: PCR mit genomischer DNA aus RyR2MerCreMer-KO-Herzen. Abbildung 3.29: Telemetrische EKG-Ableitungen an wachen RyR2KiT-Mäusen. Abbildung 3.30: PCR mit genomischer DNA aus Herzgewebe von RyR2L2/L2,KiT-Mäusen. Abbildung 3.31: PCR mit DNA aus Ventrikelgewebe zweier RyR2L1/+,CAGG-Mäuse. Abbildung 3.32: RyR3-Proteinexpression im Herzen. Abbildung 3.33: RyR3-mRNA Expression in isolierten Sinisknotenzellen. Abbildung 3.34: Herzfrequenz und Aktivität der RyR3KO- und Wildtyp-Mäuse. Abbildung 3.35: Herzfrequenz-Modulation in den RYR3KO-Tieren. Abbildung 3.36: Herzfrequenz-Modulation durch verschiedene Pharmaka in RyR3KOTieren. Abbildung 3.37: Darstellung der Herzfrequenzvariabilität. Tabellen: Tabelle 1: Biometrische Daten der Ventrikel und Vorhöfe. Tabelle 2: EKG-Parameter von RyR3KO- und Wildtyp-Mäusen. Tabelle 3: Verwendete Primer für Genotypisierung. Tabelle 4: Verwendete Primer für RT-PCR. Abbildungsverzeichnis Tabelle 5: Verwendete Antikörper. Tabelle 6: Verwendete Mauslinien. Tabelle 7: Verwendete Abkürzungen. 83 Literaturverzeichnis 7. 84 Literaturverzeichnis Benetos A, Rudnichi A, Thomas F, Safar M, Guize L. (1999) Influence of heart rate on mortality in a French population: role of age, gender, and blood pressure. Hypertension. 33(1):44-52. Bertocchini F, Ovitt CE, Conti A, Barone V, Schöler HR, Bottinelli R, Reggiani C, Sorrentino (1997) Requirement for the ryanodine receptor type 3 for efficient contraction in neonatal skeletal muscles. EMBO J. 16(23):6956-6963. Bers, DM (2002) Cardiac excitation–contraction coupling. Nature 415:198–205. Betzenhauser MJ, Marks AR (2010) Ryanodine receptor channelopathies. Pflugers Arch. 460(2):467-480. Binkley PF, Nunziata E, Haas GJ, Nelson SD, Cody RJ (1991) Parasympathetic withdrawal is an integral component of autonomic imbalance in congestive heart failure: demonstration in human subjects and verification in a paced canine model of ventricular failure. J Am Coll Cardiol. 18(2):464-472. Bogdanov KY, Vinogradova TM, Lakatta EG. (2001) Sinoatrial nodal cell ryanodine receptor and Na(+)-Ca(2+) exchanger: molecular partners in pacemaker regulation. Circ Res. 88: 1254–1258. Bogdanov KY, Maltsev VA, Vinogradova TM, Lyashkov AE, Spurgeon HA, Stern MD, Lakatta EG. (2006) Membrane potential fluctuations resulting from submembrane Ca2+ releases in rabbit sinoatrial nodal cells impart an exponential phase to the late diastolic depolarization that controls their chronotropic state. Circ Res. 99(9):979-987. Brown HF, Difrancesco D, Noble SJ (1979) How does adrenaline accelerate the heart? Nature 280:235-236. Burlew BS, Weber KT (2002) Cardiac fibrosis as a cause of diastolic dysfunction. Herz 27: 92-98. Fabiato A (1983) Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol. 245(1):C1-14. Literaturverzeichnis 85 Fabiato A, Fabiato F (1975) Contractions induced by a calcium-triggered release of calcium from the sarcoplasmic reticulum of single skinned cardiac cells. J Physiol Lond. 249(3):469495. Feil R, Wagner J, Metzger D, Chambon P (1997) Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem Biophys ResCommun. 237(3): 752-757. Fill M, Copello JA (2002) Ryanodine receptor calcium release channels. Physiol Rev. 82(4):893-922. Herrmann S, Fabritz L, Layh B, Kirchhof P, Ludwig A (2011) Insights into sick sinus syndrome from an inducible mouse model. Cardiovasc. Res. 90(1):38-48. Hoess RH, Abremski K. (1985) Mechanism of strand cleavage and exchange in the Cre-lox site-specific recombination system. J Mol Biol. 181(3):351-362. Hoesl E, Stieber J, Herrmann S, Feil S, Tybl E, Hofmann F, Feil R, Ludwig A. (2008 ) Tamoxifen-inducible gene deletion in the cardiac conduction system. J. Mol Cell Cardiol. 45(1):62-69. Hoshikawa E, Matsumura Y, Kubo T, Okawa M, Yamasaki N, Kitaoka H, Furuno T, Takata J, Doi YL. (2011) Effect of Left Ventricular Reverse Remodeling on Long-Term Prognosis After Therapy With Angiotensin-Converting Enzyme Inhibitors or Angiotensin II Receptor Blockers and β Blockers in Patients With Idiopathic Dilated Cardiomyopathy. Am J Cardiol. 107(7):1065-1070. Janicki JS, Brower GI (2002) The role of myocardial fibrillar collagen in ventricular remodeling and function. Journal of Cardiac Failure 8:319-325. Jones ES, Black MJ, Widdop RE (2004) Angiotensin AT2 receptor contributes to cardiovascular remodelling of aged rats during chronic AT1 receptor blockade. J Mol Cell Cardiol. 37(5):1023-1030. Kannel WB, Kannel C, Paffenbarger RS Jr, Cupples LA. (1987) Heart rate and cardiovascular mortality: the Framingham Study. Am Heart J. 113(6):1489-1494. Literaturverzeichnis 86 Kihwan Ju, Kuwaki T, Takeshima H, Iino M, Kumada M (1997) Power spectral analysis of heart rate variability in ryanodine receptor type 3 knockout mice. Biomedical Engineering Conference P. 7 - 10 Kramer K, Kinter LB (2003) Evaluation and applications of radiotelemetry in small laboratory animals. Physiological Genomics 13:197-205. Kushnir A, Betzenhauser MJ, Marks AR (2010) Ryanodine receptor studies using genetically engineered mice. FEBS Lett. 584(10):1956-1965. Kühbandner S, Brummer S, Metzger D, Chambon P, Hofmann F, Feil R (2000) Temporally controlled somatic mutagenesis in smooth muscle. Genesis 28(1):15-22. Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227(5259):680-685. Lakatta EG, DiFrancesco D (2009) What keeps us ticking: a funny current, a calcium clock, or both? J Mol Cell Cardiol. 47(2):157-170. Lakatta EG, Maltsev VA, Vinogradova TM (2010) A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart's pacemaker. Circ Res. 106(4):659-673. Li J, Qu J, Nathan RD. (1997) Ionic basis of ryanodine’s negative chronotropic effect on pacemaker cells isolated from the sinoatrial node. Am J Physiol. 273: H2481–H2489. Ludwig A, Zong X, Jeglitsch M, Hofmann F, Biel M (1998) A family of hyperpolarizationactivated mammalian cation channels. Nature 393:587–591. Maltsev VA, Lakatta EG (2008) Dynamic interactions of an intracellular Ca2+ clock and membrane ion channel clock underlie robust initiation and regulation of cardiac pacemaker function. Cardiovasc Res. 77(2):274-284. Maltsev VA, Vinogradova TM, Lakatta EG (2006) The emergence of a general theory of the initiation and strength of the heartbeat. J Pharmacol Sci. 100(5):338-369. Literaturverzeichnis 87 Mangoni ME, Nargeot J (2008) Genesis and regulation of the heart automaticity. Physiol Rev. 88(3):919–982. Metzger D, Feil R (1999) Engineering the mouse genome by site-specific recombination. Current opinions in Biotechnology 10:470-476. Mitchell GF, Jeron A, Koren G. (1998) Measurement of heart rate and Q-T interval in the conscious mouse. Am J Physiol. 274:H747-H751. Mullis K, Faloona F, Scharf S, Saiki R, Horn G, Erlich H (1986) Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harb Symp Quant Biol. 1:263-273. O’Connell TD, Ni Y, Lin KM, Han H, Yan Z. (2003) Isolation and culture of adult mouse cardiac myocytes for signaling studies. AFCS Research Reports 1:1–9. Otsu K, Willard H, Khanna V, Zorato F, Green N (1990) Molecular cloning of cDNA encoding the Ca2+ release channel (ryanodine receptor) of rabbit cardiac muscle sarcoplasmic reticulum. J Biol Chem. 265:13472–13483. Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V, Danieli GA (2001) Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 103:196–200. Rich MW, Saini JS, Kleiger RE, Carney RM, teVelde, Freedland KE. (1988) Correlation of HRV with clinical and angiographic variables and late mortality after myocardial infarction. Am J Cardiol. 62:714–717. Rubenstein DS, Lipsius SL (1989) Mechanisms of automaticity in subsidiary pacemakers from cat right atrium. Circ Res. 64:648-657. Sham JSK, Cleemann L, Morad M (1995) Functional coupling of Ca2+ channels and ryanodine receptors in cardiac myocytes. Proc Natl Acad Sci U.S.A. 92:121-125. Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, Penninger JM, Molkentin JD (2001) Temporally regulated and tissuespecific gene manipulations in the adult and embryonic heart using a tamoxifen inducible Cre protein. Circ Res. 89:20-25. Literaturverzeichnis 88 Sorrentino V, Volpe P (1993) Ryanodine receptors: how many, where and why? Trends Pharmacol.Sci. 14:98-103. Tada M, Kirchberger MA, Repke DI, Katz AM (1974) The stimulation of calcium transport in cardiac sarcoplasmic reticulum by adenosine 3':5'-monophosphate-dependent protein kinase. J. Biol. Chem. 249:6174-6180. Takeshima H, Ikemoto T, Nishi M, Nishiyama N, Shimuta M, Sugitani Y, Kuno J, Saito I, Saito H, Endo M, Iino M, Noda T. (1996) Generation and characterization of mutant mice lacking ryanodine receptor type 3. J Biol Chem. 271(33):19649-19652. Takeshima, H., Iino, M., Takekura, H., Nishi, M., Kuno, J., Minowa, O., Takano, H. and Noda, T (1994) Excitation–contraction uncoupling and muscular degeneration in mice lacking functional skeletal muscle ryanodine-receptor gene. Nature 369:556–559. Takeshima H, Komazaki S, Hirose K, Nishi M, Noda T, Iino M (1998) Embryonic lethality and abnormal cardiac myocytes in mice lacking ryanodine receptor type 2. Embo J. 17:33093316. Takeshima H, Nishimura S, Matsumoto T, Ishida H, Kangawa K, Minamino N, Matsuo H, Ueda M, Hanaoka M, Hirose T, Numa S (1989) Primary structure and expression from complementary DNA of skeletal muscle ryanodine receptor. Nature 339: 439-445. Tellez JO, Dobrzynski H, Greener ID, Graham GM, Laing E, Honjo H, Hubbard SJ, Boyett MR, Billeter R (2006) Differential expression of ion channel transcripts in atrial muscle and sinoatrial node in rabbit. Circ Res. 99(12):1384-1393. Tunwell E, Wickenden C, Bertrand B, Shevchenko V, Walsh M, Allen P (1996) The human cardiac muscle ryanodine receptor-calcium release channel: identification, primary structure and topological analysis. Biochem J. 318:477–487. Vinogradova TM, Bogdanov KY, Lakatta EG (2002) beta-Adrenergic stimulation modulates ryanodine receptor Ca2+ release during diastolic depolarization to accelerate pacemaker activity in rabbit sinoatrial nodal cells. Circ Res. 90: 73–79. Literaturverzeichnis 89 Vinogradova TM, Lakatta EG (2009) Regulation of basal and reserve cardiac pacemaker function by interactions of cAMP-mediated PKA-dependent Ca2+ cycling with surface membrane channels. J Mol Cell Cardiol. 47(4):456-74. Vinogradova TM, Lyashkov AE, Zhu W, Ruknudin AM, Sirenko S, Yang D, Deo S, Barlow M, Johnson S, Caffrey JL, Zhou YY, Xiao RP, Cheng H, Stern MD, Maltsev VA, Lakatta EG (2006) High basal protein kinase A-dependent phosphorylation drives rhythmic internal Ca2+ store oscillations and spontaneous beating of cardiac pacemaker cells. Circ Res. 98:505– 514. Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE, Duff HJ, Roden DM, Wilde AA, Knollmann BC (2009) Flecainide prevents catecholaminergic polymorphic ventricular tachycardia in mice and humans. Nat Med. 15(4):380-383. Yang HT, Tweedie D, Wang S, Guia A, Vinogradova T, Bogdanov K, Allen PD, Stern MD, Lakatta EG, Boheler KR (2002) The ryanodine receptor modulates the spontaneous beating rate of cardiomyocytes during development. Proc Natl Acad Sci USA. 99(14):9225-9230. Younes A, Lyashkov A, Graham D, Sheydina A, Volkova M, Mitsak M, Vinogradova T, Lukyanenko Y, Li Y, Ruknudin A, Boheler K, van Eyk J, Lakatta EG (2008) Ca2+-stimulated basal adenylyl cyclase activity localization in membrane lipid microdomains of cardiac sinoatrial nodal pacemaker cells. J Biol Chem. 283:14461–14469. Eigene Veröffentlichungen 8. 90 Eigene Veröffentlichungen Milbradt A, Eberhardt M, Teutsch C, Chien K, Reeh P, Ludwig A, Stieber J. Ryanodine receptor 2 deletion in the adult mouse heart results in bradycardia and heart failure. EMBO J. (eingereicht) Milbradt A, Ludwig A, Stieber J (2010) Deletion of RyR2 in the SAN/AV-node results in moderate bradycardia without arrhythmias. Naunyn-Schmiedeberg´s Arch Pharmacol 381:233 Hösl E, Kilian A, Ludwig A, Stieber J (2008) Conditional deletion of potential pacemaker channels in the mouse heart via the Cre/loxP system using a SAN/AV node specific Cre mouse. Naunyn-Schmiedeberg´s Arch Pharmacol 377:260 Böhm C, Hayer S, Kilian A, Zaiss MM, Finger S, Hess A, Engelke K, Kollias G, Krönke G, Zwerina J, Schett G, David JP. (2009) The alpha-isoform of p38 MAPK specifically regulates arthritic bone loss. J Immunol. 183(9):5938-5947 Polzer K, Soleiman A, Baum W, Axmann R, Distler J, Redlich J, Kilian A, Krönke G, Schett G, Zwerina J (2008) Selective p38MAPK isoform expression and activation in ANCAassociated crescentic glomerulonephritis: Role of p38MAPK α. Ann Rheum Dis. 67(5):602608 Lebenslauf 91 9. Lebenslauf Anita Milbradt geb. Kilian Geburtsdatum, -ort 10. Januar 1981, Temeschburg Nationalität deutsch Familienstand verheiratet Ausbildung 1987-1990 Grundschule Temeschburg, Rumänien 1990-1992 Grundschule Guldeinstraße, München 1992-2001 Wittelsbacher Gymnasium, München Erhalt der Allgemeinen Hochschulreife 09/2001-09/2003 Friedrich-Alexander-Universität (FAU), Erlangen Grundstudium (Biologie, Diplom) 10/2003-09/2006 Friedrich-Alexander-Universiät (FAU), Erlangen Hauptstudium (Biologie, Diplom) 04/2006 Friedrich-Alexander-Universiät (FAU), Erlangen Diplomprüfung im Nebenfächern Hauptfach Biotechnik, Biochemie und Immunologie den und Entwicklungsbiologie 08/2006-06/2007 Medizinische Klinik III, Erlangen Abschluss des Studiums zur Diplom-Biologin mit Anfertigung der Diplomarbeit „Die Rolle der p38MAPK-Isoformen in der Osteoklastogenese“ Seit 11/2007 Institut für Pharmakologie und Toxikologie, Erlangen Promotion mit dem Titel: “Konditionelle Gendeletion des Ryanodinrezeptors RyR2 im Herzen der Maus“ Danksagung 92 Danksagung Herrn Prof. Dr. Andreas Ludwig danke ich für sein großes Interesse an meiner Arbeit, seine freundliche Unterstützung sowie seine hervorragende Betreuung. Frau PD Dr. Juliane Stieber danke ich für ihre Geduld, die großartige Betreuung und, dass sie stets ein offenes Ohr für alles (auch Privates) hatte. Vielen Dank für Alles, Juliane. Herrn Prof. Dr. Brandstätter danke ich für seine Bereitschaft das Zweitgutachten zu erstellen. Frau Dr. Mirjam Eberhardt danke ich herzlich für die Hilfe bei den Ca2+-Messungen. Mein besonderer Dank gilt vor allem Susanne, Sabine, Claudia, Andrea und Hugo für all die lustigen Stunden im und außerhalb des Labors. Allen derzeitigen und ehemaligen Mitarbeitern der AG Ludwig danke ich für das angenehme Arbeitsklima, für wissenschaftliche sowie auch für weniger wissenschaftliche Diskussionen und für gesellige Aktivitäten abseits des Laboralltags. Mein besonderer Dank gilt meiner Familie und meinen Freunden die immer zu mir gehalten haben, danke für alles. Jens, danke, dass du immer für mich da bist.