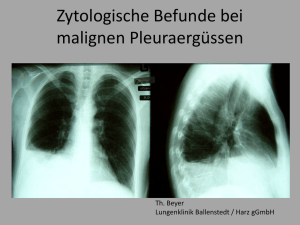
ZIP - FreiDok - Albert-Ludwigs
Werbung

Aus der Neurologischen Universitäts-Klinik der Albert-Ludwigs-Universität Freiburg i. Br. Das demographische, klinische und immunologische Profil von Patienten mit paraneoplastischen neurologischen Syndromen in Assoziation mit gut-charakterisierten antineuronalen Antikörpern - eine Follow-up Studie - INAUGURAL-DISSERTATION zur Erlangung des Medizinischen Doktorgrades der Medizinischen Fakultät der Albert-Ludwigs-Universität Freiburg i. Br. vorgelegt 2010 von Patricia Bischler geboren in Offenburg 1 Dekan: Prof. Dr. Dr. h.c. mult. Hubert Erich Blum 1. Gutachter: Prof. Dr. Sebastian Rauer 2. Gutachter: Fr. Prof. Dr. Almut Zeeck Jahr der Promotion: 2010 2 Inhaltsverzeichnis Liste verwendeter Abkürzungen.......................................................................................... 6 1. Einleitung und Einführung in das Krankheitsbild PNS................................................... 8 1.1 Einleitung und Fragestellung.......................................................................................8 1.2 Einführung in das Krankheitsbild PNS........................................................................9 1.2.1 Krankheitsbild PNS/Definition............................................................................... 9 1.2.2 Epidemiologie......................................................................................................... 10 1.2.3 Immunpathogenese................................................................................................. 11 1.2.4 Spezifische antineuronale paraneoplastische Antikörper....................................... 12 1.2.5 Antikörper-assoziierte Syndrome...................................................................….... 18 1.2.6 Diagnostische Methoden........................................................................................ 31 1.2.7 Therapeutische Optionen........................................................................................ 34 2. Patienten und Methode.................................................................................................... 36 2.1 Patienten...................................................................................................................... 36 2.2 Methoden-Datenbankerstellung................................................................................... 36 2.2.1 Demographische Daten........................................................................................... 36 2.2.2 Paraneoplastische neurologische Syndrome........................................................... 37 2.2.3 Antineuronale Antikörper....................................................................................... 38 2.2.4 Tumor..................................................................................................................... 38 2.2.5 Diagnostik............................................................................................................... 39 2.2.6 Behandlung............................................................................................................. 39 2.2.7 Verlauf/Follow up…………………………………………………….………….. 40 2.3 Statistik…………………………….…..………………………………….……….... 41 3. Ergebnisse........................................................................................................................ 42 3.1 Demographische Daten................................................................................................ 42 3.2 Paraneoplastische neurologische Syndrome................................................................ 43 3.3 Zu Grunde liegende Malignome.................................................................................. 45 3.4 Antineuronale Antikörper............................................................................................ 49 3.4.1 Antikörper und neurologische Syndrome............................................................... 51 3.4.2 Antikörper und Tumorerkrankungen...................................................................... 52 3.5 Ergebnisse der Liquoruntersuchung............................................................................ 54 3.5.1 Liquorergebnisse und neurologische Syndrome..................................................... 54 3.5.2 Liquorergebnisse und Antikörper........................................................................... 56 3 3.6 Weitere Diagnostik...................................................................................................... 57 3.6.1 MRT des Schädels.................................................................................................. 57 3.6.2 MRT der Wirbelsäule............................................................................................. 58 3.6.3 Tumorsuche mittels PET........................................................................................ 58 3.7 Therapie des Malignoms..............................................................................................60 3.8 Behandlung des PNS………………………………………………………………... 62 3.9 Follow up..................................................................................................................... 63 3.9.1 Überlebenszeiten abhängig vom PNS.................................................................... 66 3.9.2 Überlebenszeiten abhängig vom antineuronalen Antikörper................................ 66 3.9.3 Überlebenszeit abhängig vom zu Grunde liegenden Tumor.................................. 67 3.9.4 Überlebenszeit abhängig von der Tumorbehandlung............................................. 67 3.9.5 Überlebenszeit abhängig vom Geschlecht.............................................................. 68 3.9.6 Überlebenszeit abhängig vom Alter bei Diagnosestellung des PNS...................... 68 3.10 Fallvorstellung der langzeitüberlebenden Patienten.................................................. 69 3.10.1 Fall Nr.20.............................................................................................................. 70 3.10.2 Fall Nr.24.............................................................................................................. 71 3.10.3 Fall Nr.25.............................................................................................................. 71 3.10.4 Fall Nr.29.............................................................................................................. 73 3.10.5 Fall Nr.36.............................................................................................................. 73 3.10.6 Fall Nr. 9............................................................................................................... 74 3.10.7 Fall Nr.10.............................................................................................................. 74 3.10.8 Fall Nr.19.............................................................................................................. 75 4. Diskussion........................................................................................................................76 4.1 Studiendesign/Vor- und Nachteile............................................................................... 76 4.2 Demographische Daten................................................................................................ 77 4.3 Paraneoplastische neurologische Syndrome................................................................ 78 4.4 Zu Grunde liegende Malignome.................................................................................. 79 4.5 Antineuronale Antikörper............................................................................................ 80 4.5.1 Häufigkeitsverteilung............................................................................................. 81 4.5.2 Assoziation zu Syndromen..................................................................................... 81 4.5.3 Assoziation zu Tumoren......................................................................................... 82 4.6 Diagnostik.................................................................................................................... 84 4.6.1 Intervall Erstmanifestation des PNS – Tumordiagnose.......................................... 84 4.6.2 Ergebnisse aus der Tumorsuche mittels PET......................................................... 85 4 4.6.3 Ergebnisse der Liquordiagnostik............................................................................ 86 4.6.4 Ergebnisse der Bildgebung mittels MRT............................................................... 87 4.7 Follow up..................................................................................................................... 89 4.7.1 Ergebnisse der Therapie......................................................................................... 89 4.7.2 Überlebenszeitanalyse............................................................................................ 91 a) Überlebenszeit abhängig vom PNS..................................................................... 91 b) Überlebenszeit abhängig vom antineuronalen Antikörper.................................. 92 c) Überlebenszeit abhängig vom assoziierten Karzinom........................................ 92 d) Überlebenszeit abhängig von der Tumorbehandlung.......................................... 94 e) Überlebenszeit abhängig von Geschlecht und Alter bei Diagnosestellung......... 94 4.7.3 Beschreibung der Langzeitüberlebenden................................................................ 95 5. Zusammenfassung........................................................................................................... 98 6. Literaturverzeichnis......................................................................................................... 99 7. Danksagung..................................................................................................................... 107 8. Lebenslauf........................................................................................................................108 9. Anhang............................................................................................................................. 109 5 Liste verwendeter Abkürzungen AK Antikörper ALS amyotrophe Lateralsklerose ANNA antinukleäre neuronale Antikörper anti-MAG anti-Myelin-assoziiertes Glykoprotein ASR Achillessehnenreflex AUP adenocarcinoma of unknown primary CGP chronische gastrointestinale Pseudoobstruktion CIPD chronische inflammatorische demyelinisierende Polyneuropathie CR komplette Remission CRMP collapsin response-mediator protein CT Computertomographie EEG Elektro-Enzephalographie ELISA enzyme-linked immunosorbent assay FDG 18F-Fluordesoxyglucose GBS Guillain-Barré-Syndrom Hu, Ri, Yo, Ma, Tr, CV Initialen der ersten an PNS erkrankten Patienten HWK Halswirbelkörper JÜR Jahres-Überlebens-Rate k.A. keine Angabe kD(a) Kilodalton LEMS Lambert-Eaton-Myasthenie-Syndrom LK Lymphknoten m männlich MG Myasthenia gravis MGUS monoklonale Gammopathie unbestimmter Signifikanz MH Morbus Hodgkin MRT Magnetresonanztomographie MS Multiple Sklerose Ncl. Nucleus NM Neuromyotonie NSCLC nicht-kleinzelliges Bronchialkarzinom 6 OKB oligoklonale Banden OMS Opsoklonus-Myoklonus-Syndrom PCA purkinje-cell-antibody (Purkinjezellen-Antikörper) PEM paraneoplastische Enzephalomyelitis PET Positronen-Emissions-Tomographie PKD paraneoplastische Kleinhirndegeneration PLE paraneoplastische limbische Enzephalitis PLS primäre Lateralsklerose PMA progressive muskuläre Atrophie PNS paraneoplastisches neurologisches Syndrom PR partielle Remission PSR Patellasehnenreflex SCLC kleinzelliges Bronchialkarzinom SPS Stiff-person-Syndrom SSN subakute sensorische Neuropathie u.a. unter anderem V.a. Verdacht auf VGCC spannungsabhängige Kalzium-Kanäle VGKC spannungsabhängige Kalium-Kanäle w weiblich ZNS zentrales Nervensystem 7 1. Einleitung und Einführung in das Krankheitsbild der paraneoplastischen neurologischen Syndrome (PNS) 1.1 Einleitung und Fragestellung Paraneoplastische neurologische Syndrome (PNS) sind seltene tumorassoziierte Erkrankungen. Die Prävalenz wird mit 0,5-3 % der Tumorleiden angegeben (Kaiser, 1999), was sich auch darin widerspiegelt, dass bisher in der Literatur fast ausschließlich klinische Studien an kleineren Fallserien publiziert wurden, meist beschränkt auf eine einzige Tumorentität, einen spezifischen antineuronalen paraneoplastischen Antikörper oder ein bestimmtes paraneoplastisches Syndrom. Außerdem befassen sich nur sehr wenige Arbeiten (Candler et al., 2003) mit dem klinischen Follow up eines Patientenkollektives. Die an der Neurologischen Universitätsklinik Freiburg durchgeführte retrospektive, monozentrische, klinisch und epidemiologisch orientierte Studie untersucht die Daten von 39 Patienten mit der Diagnose eines PNS und dem Nachweis eines oder mehrerer sog. gut charakterisierter antineuronaler Antikörper. Der Erfassungszeitraum erstreckt sich vom 01.08. 1987 bis zum 31.03.2008. Dabei wurden demographische Daten, das klinische Spektrum der PNS, die assoziierten Tumore, der initial bestimmte antineuronale Antikörper, die Befunde aus Liquordiagnostik und Bildgebung, die Therapieformen, das Ansprechen der PNS und der zu Grunde liegenden Tumore auf die Behandlung sowie die Überlebenszeit für jeden einzelnen Patienten zusammengestellt und ausgewertet. So war neben der beschreibenden Darstellung des Kollektives auch eine Betrachtung des Follow up aller Patienten, vor allem jedoch der Anteil der Langzeitüberlebenden, Ziel unserer Untersuchungen. Aus diesem Grund und zur Verlaufsbestimmung der AntikörperKonzentrationen im Vergleich zur Erstuntersuchung bzw. der Suche nach paraneoplastischen Zweitantikörpern wurden die überlebenden Patienten so weit möglich erneut in die Klinik einbestellt, was uns einen genaueren Blick auf die Entwicklung des paraneoplastischen Syndroms und seiner klinischen Auswirkungen ermöglichte. 8 1.2 Einführung in das Thema paraneoplastische neurologische Syndrome 1.2.1 Das Krankheitsbild PNS / Definition Paraneoplastische neurologische Syndrome (PNS) sind seltene tumorassoziierte Erkrankungen des Nervensystems. Man zählt in den Formenkreis der PNS alle diejenigen Komplikationen von Tumorerkrankungen, welche weder durch den Tumor und seine Metastasen noch durch vaskuläre, infektiöse, metabolische oder therapiebedingte Ursachen ausgelöst werden (Voltz, 2002; Darnell, 2003). Vielmehr wird als pathogenetischer Mechanismus eine immunologisch vermittelte Kreuzreaktion zwischen Tumor- und Nervengewebe postuliert (Giometto et al., 1999). Wahrscheinlich führt die ektope Expression neuronaler Antigene durch den Tumor zur Generierung einer Autoimmunantwort mit konsekutiver kreuzreaktiver Nervenzellschädigung (Voltz 2002). Bei etwa 2/3 der Patienten mit paraneoplastischen Erkrankungen lässt sich im Serum eine hochspezifische Antikörperreaktivität nachweisen, welche die paraneoplastische Ätiologie zu fast 100 % beweist (Voltz, 2002) und pathognomonisch ist für ein zum Diagnosezeitpunkt oft noch okkultes Malignom (Kaiser 1999). Der Nachweis eines oder mehrerer dieser zum Teil gut charakterisierten Antikörper im Serum des Patienten ermöglicht es, eine neurologische Symptomatik als paraneoplastisch einzustufen. (Honnorat, 2007). Zudem findet sich häufig eine hochspezifische Assoziation verschiedener Antikörper mit bestimmten Tumortypen, so dass der Nachweis eines definierten Antikörpers die Suche nach einem noch nicht bekannten Tumor erleichtern kann (Voltz, 2002). Die Klinik paraneoplastischer neurologischer Erkrankungen zeichnet sich durch folgende Charakteristika aus (Voltz, 2004): - Es handelt sich um eine seltene Erkrankung, die jedoch bei neurologischen Patienten häufiger ist als angenommen. - Die neurologischen Symptome verlaufen typischerweise subakut und zeigen sich zumeist deutlich vor der Tumordiagnose. Zwischen Beginn der neurologischen Symptomatik und Tumordiagnose können drei bis vier Jahre liegen. 9 - Die Tumordiagnose kann schwierig sein, da das Karzinom oft klein bleibt und nicht metastasiert, was wahrscheinlich Folge der gegen den Tumor gerichteten Immunreaktion ist. - Die neurologische Symptomatik ist im Verlauf oftmals sehr stark ausgeprägt, häufig mit irreversiblen Einschränkungen, so dass vielmals die paraneoplastische Erkrankung und nicht der Tumor zum Tode führt, vor allem wenn das zentrale Nervensystem betroffen ist. - Paraneoplastische Erkrankungen können jedes neurologische System betreffen. Vor allem folgende Syndrome sprechen für eine paraneoplastische Ätiologie: - Depression, Gedächtnisstörung: Limbische Enzephalitis Fieber unklarer Ätiologie: Limbische Enzephalitis Paralytischer Ileus: Autonome Polyneuropahie Schwäche und Mundtrockenheit: Lambert-Eaton-Myasthenie-Syndrom Das gleichzeitige Auftreten mehrerer paraneoplastischer Syndrome ist möglich, sehr häufig ist dies beim kleinzelligen Bronchialkarzinom. Der häufigste einem paraneoplastischen neurologischen Syndrom zugrundeliegende Tumor ist das kleinzellige Bronchialkarzinom (SCLC) (Posner JB, 1995; Candler et al., 2004). Weitere, häufig mit Paraneoplasien assoziierte Tumoren sind unter anderem Mamma- und Ovarialkarzinome, Krebserkrankungen der Prostata oder des Hodens sowie Thymome und lymphoproliferative Erkrankungen (Kaiser 1999). 1.2.2 Epidemiologie Paraneoplastische neurologische Syndrome sind seltene Erkrankungen. Zwar entwickeln etwa 40% der Patienten mit einem kleinzelligen Bronchial-Karzinom im Verlauf ihrer Erkrankung neurologische Symptome wie Schmerzen, Anfälle und Bewusstseinsstörungen, diese sind jedoch meist auf eine Metastasierung oder eine Meningeosis carcinomatosa zurückzuführen (Kaiser, 1999). Die Inzidenz paraneoplastischer Syndrome hingegen wird je nach Studiengröße und Art der untersuchten Karzinome als sehr gering angegeben. R. Kaiser spricht 1999 von einer Häufigkeit von nur etwa 0,5-3% aller Tumorpatienten, Darnell gibt im New England Journal of Medicine 2003 sogar eine Prozentzahl von 0.01% an. Ausnahmen bilden hierbei das 10 Lambert-Eaton-Syndrom, das 3% aller Patienten mit einem kleinzelligen Bronchialkarzinom betrifft (Sculier et al., 1987) und die Myasthenia gravis, die mit etwa 15% der Thymomerkrankungen vergesellschaftet ist (Levy et al., 1998). 1.2.3 Immunpathogenese Seit der Beschreibung von hochspezifischen antineuronalen Antikörpern erscheint zurzeit ein autoimmunpathogenetischer Mechanismus am wahrscheinlichsten. Nach derzeitiger Auffassung führt eine ektope Expression neuronaler Antigene durch den Tumor zur Generierung einer Autoimmunantwort mit konsekutiver kreuzreaktiver Nervenzellschädigung (Voltz 2002). In immunprivilegierten Geweben, z.B. dem ZNS, werden physiologischerweise sogenannte onkoneurale Antigene exprimiert. Kommt es bei der Tumorgenese zu einer pathologischen Expression dieser Antigene im Tumor, entwickelt sich eine Immunreaktion, die sich aufgrund von homologen antigenen Determinanten neben dem Tumor auch gegen Nervengewebe richten kann. Diese Annahme stützt sich unter anderem auf der Tatsache, dass die jeweiligen neuroanatomischen Zielstrukturen der Autoimmunantwort mit den klinisch-neurologischen Symptomen der Patienten korrelieren. Auch wenn bisher eine Vielzahl von verschiedenen antineuronalen Antikörpern beschrieben wurde, scheinen diese selbst wohl eher eine untergeordnete Rolle in der Pathogenese der PNS zu spielen (Voltz 2002). In der Regel führte im Tierversuch weder die Übertragung von Serum noch die direkte Immunisierung mit rekombinanten Antigenen zur Entwicklung einer entsprechenden neurologischen Symptomatik (Tanaka 1995a, Tanaka 1995b). Es gibt inzwischen sogar vermehrt Hinweise darauf, dass primär zytotoxische Zellen für die Schädigung der Neurone verantwortlich sind (Voltz 2002). Histopathologisch werden in den betroffenen Nervenstrukturen entzündliche Infiltrate mit Nachweis von antigenspezifischen zytotoxischen T-Zellen beschrieben (Roberts et al., 2004). Bisher nicht bekannt ist jedoch, ob diese T-Zellen Ausdruck einer sekundären Reaktion sind oder eine primäre Rolle spielen und ob sie mit einem Antigen oder einem Superantigen reagieren. Die T-Zell-Rezeptor-Analyse im Gehirn von anti-Hu-positiven PNS-Patienten zeigte eine oligoklonale Expansion von zytotoxischen Zellen, wahrscheinlich infolge einer primären Reaktion gegen das Antigen Hu (Voltz 2002). Zusätzlich ergaben sich Hinweise auf zytotoxische Hu-spezifische T-Zellen im Blut von anti-Hu-positiven Patienten (Benyahia et 11 al., 1999) und im Blut und Liquor von anti-Yo- positiven Patienten (Albert et al., 1998), so dass die Autoimmunpathogenese der paraneoplastischen Erkrankungen auf dem Boden einer spezifischen T-Zell-Reaktion immer wahrscheinlicher wird. Es besteht die Hypothese, dass THelfer-Zellen die Blut-Hirn-Schranke durchwandern und dort zu einer Aktivierung von BZellen und zytotoxischen T-Zellen führen, wodurch sie den Autoimmunprozess unterhalten (Voltz, 2004). Seit Jahren wird außerdem beobachtet, dass Tumorleiden bei Patienten z.B. mit einem anti-Hu assoziiertem PNS oder einer latenten anti-Hu Immunantwort ohne PNS einen gutartigeren Verlauf nehmen als bei solchen Patienten, die keine Immunantwort gegen das Hu-Antigen aufweisen. So ist im Rahmen der Autoimmunreaktion neben den neurologischen Defiziten ein gewisser tumorsuppressiver Effekt wahrscheinlich (Byrne et al., 1997; Dalmau et a., 1990; Graus et al., 1997). Ein suppressiver Effekt auf die Metastasenausdehnung zeigte sich zum Beispiel auch für die anti-Yo-Autoimmunantwort (Hetzel et al., 1990). Es kann folglich spekuliert werden, ob der ,,biologische Sinn’’ der Autoimmunantwort in der Tumorabwehr durch das Immunsystem liegt. 1.2.4 Spezifische antineuronale paraneoplastische Antikörper Häufig finden sich im Serum und Liquor von Patienten mit paraneoplastischen neurologischen Syndromen antineuronale Antikörper, deren Rolle in der Pathogenese jedoch bis auf wenige Ausnahmen noch nicht vollständig geklärt ist. Nur für Antikörper gegen spannungsabhängige Kalzium-Kanäle beim Lambert-EatonMyasthenie-Syndrom, für anti-Recorvin-Antikörper bei der tumorassoziierten Retinopathie sowie für Antikörper gegen spannungsabhängige Kalium-Kanäle (VGKC) bei der paraneoplastischen Neuromyotonie (Isaacs’ Syndrom) zeigte sich ein direkter ursächlicher Zusammenhang bei der Entstehung der neurologischen Symptomatik (Kaiser 1999). Die antineuronalen Antiköper haben jedoch eine Indikatorfunktion erlangt, da sie zumeist hochspezifisch für ein zugrundeliegendes, jedoch häufig noch unentdecktes Tumorleiden sind (Kaiser 1999). Dadurch besteht die Möglichkeit einer frühen Diagnosestellung mit Einleitung einer entsprechenden Therapie. Außerdem hat die Klassifikation der PNS anhand der Autoantikörper für den Kliniker mittlerweile eine größere Bedeutung erlangt als eine Klassifikation auf der Basis von pathoanatomischen Kriterien. 12 Ob ein Zusammenhang besteht zwischen Änderungen der AK-Konzentration im Verlauf und Entwicklungen von neurologischer Symptomatik oder Tumor sowie Art der Therapie, ist bis jetzt noch umstritten. So konnte eine Untersuchung von 35 Hu-Patienten eine solche Korrelation nicht bestätigen. (Lladó et al., 2004) Erst seit etwa 20 Jahren kennt man spezifische Antikörper als Marker von paraneoplastischen Syndromen, seitdem zeichnet sich dieses Gebiet der Neurologie durch eine ständig wachsende Komplexität aus. Dies hat verschiedene Gründe (Voltz 2002): 1. Es bestehen zwei verschiedene Nomenklaturen, die parallel verwendet werden: Nach Posner werden in der Regel zur Bestimmung der Antikörperreaktivität zwei Nachweismethoden eingesetzt und die ersten beiden Buchstaben der Nachnamen der Indexpatienten verwendet (anti-Hu, Yo, Ma), nach Lennon wird nur eine Methode (Immunhistochemie) eingesetzt und die Reaktivität nach dem Färbeverhalten benannt. Mittlerweile ist man aber auch international immer mehr dazu übergegangen, die Reaktivitäten mit zwei Nachweismethoden zu bestimmen und die Nomenklatur nach Posner zu verwenden. 2. Zu einigen antineuronalen Reaktivitäten gibt es bisher nur Einzelfallbeobachtungen, die klinische Relevanz ist hierbei oft noch unklar. Und auch einige der gut charakterisierten Antikörper sind noch nicht lange beschrieben und die Tests daher erst seit kurzem kommerziell verfügbar. 3. Die Anzahl der bekannten und klinisch relevanten Antikörper nimmt ständig zu und wird somit auch immer unübersichtlicher. 4. Die Sensitivität der klinisch relevanten Antikörper liegt bei ungefähr 50-60%, so dass auch ohne Nachweis einer Antikörperreaktivität die Diagnose eines paraneoplastischen Syndroms gestellt werden kann. Die Spezifität hingegen liegt bei fast 100%. Somit ist bei Nachweis eines spezifischen Antikörpers von einem Tumorgeschehen auszugehen. Die folgende Liste (Tabelle 1; Voltz, 2002) gibt einen kurzen Überblick über die klinisch relevanten Antikörper-Reaktivitäten als Marker einer paraneoplastischen Ätiologie: 13 Tabelle 1 Überblick über die klinisch relevanten Antikörper-Reaktivitäten Name alternative Antigen Neurologische Syndrome Bezeichnung 170 kDa Neuropathie, Kleinhirndegeneration, ANNA-3 PLE Amphiphysin Stiff-man-Syndrom; verschiedene Anti-Amphiphysin anti-CRMP5 CRMP5 PLE, CGP, sensorische Neuropathie, Anti-CV2 Kleinhirndegeneration, LEMS ANNA-1 Hu Proteine Sensorische Neuropathie, PLE Anti-Hu Kleinhirndegeneration, CGP Ma-Proteine Hirnstammenzephalitis, PLE Anti-Ma1 37 kDa Kleinhirndegeneration Ma/Ta Ma Proteine Hirnstammenzephalitis, PLE, Anti-Ma2 40 kDa Kleinhirndegeneration Recoverin Retinopathie Anti-Recoverin ANNA-2 NOVA Zerebelläre Degeneration, OMS, Anti-Ri Hirnstammenzephalitis Titin Muskelfilament Myasthenia gravis Anti-Titin PCA-Tr Unbekannt Kleinhirndegeneration Anti-Tr PCA-1 cdr2, cdr62 Kleinhirndegeneration, Neuropathie Anti-Yo 280 kDa Enzephalitis, LEMS, Neuropathie PCA-2 Abkürzungen: ANNA: CRMP: Hu, Ri, Yo, Ma, Tr, CV: PCA: PLE: CGP: OMS: LEMS: antinukleäre neuronale Antikörper collapsin response-mediator protein Initialen der ersten Patienten, bei denen diese Syndrome beschrieben wurden purkinje-cell-antibody (Purkinjezellen-Antikörper) paraneoplstische limbische Enzephalitis chronische gastrointestinale Pseudoobstruktion Opsoklonus-Myoklonus-Syndrom Lambert-Eaton-Myasthenie-Syndrom Aufgrund der oftmals unspezifischen neurologischen Symptomatik und bei noch nicht erbrachtem Tumornachweis kann die Diagnosesicherung eines paraneoplastischen Syndroms jedoch nach wie vor Probleme bereiten. Von einer europäischen Konsensusgruppe wurden deshalb kürzlich Diagnosekriterien vorgeschlagen, welche zwischen einem „definitiven“ und einem „möglichen“ PNS unterscheiden. Ein wichtiger Aspekt dieser Kriterien ist, ob bei den zu untersuchenden Patienten sogenannte „gut charakterisierte antineuronale Antikörper“ vorliegen (Graus et al., 2004). So sollte bei fehlendem Nachweis eines Tumors nur bei Nachweis eines dieser Antikörper von einem definitiven paraneoplastischen Syndrom ausgegangen werden. Die Klassifikation beruht auf verschiedenen Gesichtspunkten: 1. Dem Vorliegen charakteristischer Muster in der Immunhistochemie und dem Einsatz eines Western-Blot mit rekombinanten Proteinen zur Charakterisierung der Spezifität 2. Der Häufigkeit der Assoziation mit verschiedenen Tumoren 3. Der Beschreibung von klassischen PNS, die mit diesen Antikörpern assoziiert sind 4. Der eindeutigen Identifikation in verschiedenen Studien 14 5. Der Häufigkeit der Antikörper bei Patienten ohne Tumornachweis Zu diesen gut charakterisierten Antikörper-Reaktivitäten zählt man neben anti-Hu als der häufigsten auch anti-Ri, -Yo, -CV2, -Amphiphysin und -Ma2. Tabelle 2 (Graus, Delattre, Antoine, 2004) zeigt eine genauere Darstellung dieser gut charakterisierten antineuronalen Antikörper. Tabelle 2 Überblick über die 6 „gut charakterisierten“ paraneoplastischen antineuronalen Antikörper Antikörper Anzahl beschriebener Fälle > 600 Neurologisches Syndrom häufig assoziierte Karzinome (Ca) Antigeneigenschaften Subakute sensible Polyneuropathie Subakute zerebelläre Degeneration Limbische Enzephalitis Hirnstammenzephalitis Enzephalomyelitis Autonome Neuropathie Kleinzelliges Bronchial-Ca neuronen-spezifisches RNABindungsprotein Yo (PCA-1)* > 200 Subakute zerebelläre Degeneration Polyneuropathie Ovarial-Ca Mamma-Ca. 62 kDa = DNA-Bindungsprotein CV2 (CRMP-5) > 100 Kleinzelliges Bronchial-Ca Thymom soll bei der Entwicklung des Nervensystems eine Rolle spielen Ma-2 (Ma/Ta) ca. 70 Subakute zerebelläre Degeneration Limbische Enzephalitis Enzephalomyelitis Sensible Polyneuropathie Sensomotorische Neuropathie Autonome Neuropathie Chorea Hirnstammenzephalitis Limbische Enzephalitis Subakute zerebelläre Degeneration Seminom Bronchial-Ca soll bei der Steuerung der Apoptose eine Rolle spielen Ri (ANNA-2)* > 60 Amphiphysin ca. 20 Subakute zerebelläre Degeneration Hirnstammenzephalitis Opsoklonus-Myoklonus-Syndrom Stiff-Person-Syndrom u.a. Mamma-Ca > kleinzelliges RNA-Bindungsprotein, Bronchial-Ca Bedeutung für die Reifung und Differenzierung von Neuronen Mamma-Ca, kleinzelliges Bindung an Dynamin: Bronchial-Ca, Endozytose von synaptischen Vesikeln Hu (ANNA-1)* * diese Bezeichnungen sind nicht als synonym zu verstehen, da ihnen verschiedene Nachweismethoden zu Grunde liegen Abkürzungen: ANNA: APCA: CRMP: Hu, Ri, Yo, Ma, CV: PCA: antinukleäre neuronale Antikörper anti-purkinje-cell-antibody (Anti-Purkinjezellen-Antikörper) collapsin response-mediator protein Initialen der ersten Patienten, bei denen diese Syndrome beschrieben wurden purkinje-cell-antibody (Purkinjezellen-Antikörper) Da diese sechs gut charakterisierten antineuronalen Antikörper die Grundlage der vorliegenden Studie bildeten, werden im Folgenden Häufigkeit, zugrundeliegende Tumoren sowie die assoziierten paraneoplastischen neurologischen Syndrome genauer beschrieben: 15 Hu-Antikörper Benannt nach dem Indexpatienten ‚Hull’ (Graus et al, 1985) sind diese Antikörper die erste beschriebene und gleichzeitig häufigste Antikörper-Reaktivität. Mittlerweile wurden in der Literatur über 600 Fälle beschrieben. Der häufigste assoziierte Tumor ist mit etwa 70% das kleinzellige Bronchialkarzinom, weitere Tumoren sind Neuroblastome, Prostatakarzinome und andere neuroendokrine Tumoren (Kaiser, 1999). Trotz der Tatsache, dass kleinzellige Bronchialkarzinome bei Männern häufiger auftreten, findet sich eine Prädominanz von Hu-Antikörpern bei Frauen (Rosenblum, 1993). Initialsymptom bei 2/3 der Patienten und häufigstes paraneoplastisches Syndrom ist die subakute sensible Neuropathie, aber wie Tabelle 2 zu entnehmen ist, spielen auch verschiedene Enzephalitiden eine Rolle. So finden sich neben Affektionen von Hirnstamm und Kleinhirn auch limbische Enzephalitiden (Gultekin et al., 2000). Die Hu-Proteine sind neuroonkonale Proteine und somit konstitutiv in zentralen und peripheren Neuronen exprimiert. Außerdem finden sich die spezifischen Antigene ektop in assozierten Tumorgeweben (Voltz, 2002). Die Immunfluoreszenz zeigt, dass Hu-Antikörper mit den Kernen neuronaler Zellen sowohl des zentralen als auch des peripheren Nervensystems reagieren (Graus et al., 1985). Während alle kleinzelligen Bronchialkarzinome Hu-Proteine exprimieren, zeigten sich bei ca. 16% der neurologisch unauffälligen Patienten niedrige Titer von Hu-Antikörpern. Hier imponierte eine positive Assoziation zwischen der Prävalenz von Hu-Antikörpern und dem Ansprechen auf die Tumorbehandlung sowie dem Gesamtüberleben der Patienten (Graus et al., 1997). Yo-Antikörper Die am zweithäufigsten vorkommende Antikörperreaktivität wurde nach der Indexpatientin ‚Young’ benannt (Grenlee J. 1983). Seither wurden mehr als 200 Fälle mit dieser AntikörperReaktivität beschrieben. Typischerweise finden sich Yo-Antikörper bei Frauen mit Ovarial- oder Mammakarzinomen, selten wurde auch über männliche Patienten berichtet, dann aber assoziiert mit anderen Tumoren, z.B. Adenokarzinomen des Ösophagus (Sutton et al., 2001). Das typische neurologische Syndrom bei Yo-positiven Patientinnen ist die paraneoplastische Kleinhirndegeneration, deren Verlauf initial subakut ist mit Erreichen eines stabil schlechten Niveaus nach Wochen bis Monaten (Voltz, 2002). In der Immunfluoreszenz binden YoAntikörper an zytoplasmatische Strukturen von Purkinjezellen, in der Elektronenmikroskopie 16 erkennt man eine Bindung an Ribosomen, granuläres endoplasmatisches Retikulum und an die trans-Seite von Vesikeln des Golgiapparates (Rodriguez et al., 1988). Im Gegensatz zu den Hu-Antikörpern zeigte sich, dass der Nachweis von Yo-Antikörpern nicht mit einer verbesserten Tumorprognose assoziiert ist (Rojas et al., 2000). Letztendlich versterben die meisten Patienten an den Folgen der schweren neurologischen Grunderkrankung (Terence et al., 1995). Aufgrund der hohen Wahrscheinlichkeit für das Vorliegen eines gynäkologischen Tumors empfiehlt Posner bei gesichertem Nachweis des Antikörpers trotz negativer Tumorsuche eine gynäkologische Totaloperation durchzuführen (Hysterektomie, Salpingo-Ovarektomie). Ri-Antikörper Diese Antikörperreaktivität wurde erstmals 1988 beschrieben und trägt seit 1991 die Initialen der Indexpatientin ‚Richards’ (Luque et al., 1991). Ri-Antikörper sind wie auch Yo-Antikörper ein Hinweis auf das Vorliegen eines gynäkologischen Tumors, in vielen Fällen findet sich hierbei ein Mammakarzinom (Voltz, 2002). Andere zu Grunde liegende Tumoren können ein Bronchialkarzinom (z.B. Stourac et al. 2001), ein Schilddrüsenkarzinom oder ein Morbus Hodgkin sein (Kaiser; 1999). Die häufigsten assoziierten neurologischen Syndrome sind die subakute zerebelläre Degeneration, die Hirnstammenzephalitis und das Opsoklonus-Myoklonus-Syndrom. Im Gegensatz zu Hu-Antikörpern ist die Reaktivität der Ri-Antikörper auf die Kerne von Neuronen des zentralen Nervensystems beschränkt (Voltz, 2002). Ma1 bzw. Ma2-Antikörper [Ma2=Ta] Diese noch nicht sehr lange bekannten Antikörper sind nach der Indexpatientin ‚Margret’ benannt und richten sich gegen verschiedene Antigene (Ma1:37 kDa; Ma2:40 kDa) aus der Familie der Ma-Proteine (Voltz, 2004). Aufgrund einer sehr fleckförmigen Verteilung dieser Antigene sind die Antikörper immunhistochemisch relativ schwer zu bestimmen. Sicherer ist hier die serologische Diagnosestellung mittels Western-Blot mit rekombinanten Ma-Proteinen (Voltz 2002), wobei allerdings die Kreuzreaktivität von Ma1 mit Ma2/Ta zu beachten ist. Eine alleinige Reaktivität gegen Ma2 weist mit relativ hoher Spezifität auf Keimzelltumoren des Hodens hin, während Ma1-Antikörper unter anderem bei Bronchial-, Mamma-, Kolonund Parotistumoren nachzuweisen sind (Voltz, 2004; Kaiser 1999) 17 Sowohl Ma1- als auch Ma2-Antikörper finden sich bei Enzephalitiden im Bereich des Kleinhirns und Hirnstamms, kommen aber auch bei Patienten mit Affektion des limbischen Systems vor (Dalmau, 1999; Rosenfeld et al., 2001; Voltz et al., 1999). Amphiphysin-Antikörper Zu Beginn wurde diese Antikörper-Reaktivität, die mit einem Synapsen-Protein von 128 kDa reagiert, in Zusammenhang mit einem Mammakarzinom beschrieben und führte dort zu einem Stiff-Person-Syndrom (Folli et al., 1993). Inzwischen weiß man jedoch, dass noch weitere Tumoren wie ein Bronchialkarzinom oder ein Ovarialkarzinom der paraneoplastischen Symptomatik zu Grunde liegen können (Antoine et al., 1999; Saiz, 1999). Als häufigste assoziierte neurologische Syndrome stellten sich Enzephalitiden des Kleinhirns, Hirnstamms oder limbischen Systems heraus, aber auch sensible Polyneuropathien wurden beschrieben (Antoine et al., 1999). CV2-Antikörper Diese Reaktivität, die erstmals von einer französischen Arbeitsgruppe beschrieben wurde, erkennt zum einen ein oligodendrozytäres Protein, welches am axonalen Tranport beteiligt ist (Honnorat et al., 1996), zum anderen reagiert es direkt mit Neuronen des peripheren Nervensystems (Antoine, 2001). Die neurologische Symptomatik ist vielfältig und reicht von Enzephalitiden (limbisch, Kleinhirn, Chorea, diffus) über das Lambert-Eaton-Syndrom und Optikusneuritiden bis hin zu peripheren sensiblen Polyneuropathien (Kaiser, 1999). Als zu Grunde liegende Karzinome wurden Lungentumoren, Uterussarkome oder Thymome beschrieben (Honnorat et al., 1996; Vernino, 2002; Yu et al., 2001). 1.2.5 Antikörper-assoziierte Syndrome So vielfältig wie die verschiedenen Antikörper sind auch die damit assoziierten Symptome. Es hat sich gezeigt, dass paraneoplastische neurologische Syndrome jede Ebene des Nervensystems betreffen können. 18 Doch gerade diese Symptome - und nicht der Tumor - sind es, die den Patienten zum Arzt führen. Die Diagnosestellung des zu Grunde liegenden Karzinoms erfolgt in den meisten Fällen erst später (Honnorat et al., 2007). Ein subakuter Verlauf der Erkrankung ist typisch und die neurologische Symptomatik oftmals sehr stark ausgeprägt und irreversibel (Robert et. al., 2003). Zudem ist das gleichzeitige Auftreten mehrerer Syndrome möglich (Voltz, 2002). Obwohl eine Großzahl der Patienten letztendlich an den Folgen des zu Grunde liegenden Tumors verstirbt, kann auch die neurologische Symptomatik so stark ausgeprägt sein, dass sie mit dem Leben nicht vereinbar ist (Robert et al., 2003). Im Folgenden werden die wichtigsten paraneoplastischen neurologischen Syndrome nach deren Lokalisationen aufgelistet und kurz dargestellt. Dabei ist es wichtig, zwischen den sogenannten „klassischen“ und „nicht-klassischen“ PNS zu unterscheiden. Graus führt in einer Arbeit von 2004 folgende Kriterien zur Unterscheidung auf: - die häufige Assoziation mit einer Tumorerkrankung - unabhängig vom Antikörper-Status ist eine intensive Tumorsuche zwingend erforderlich - jedes klassische Syndrom betreffend die Erfüllung bestimmter, von früheren Arbeitsgruppen beschriebenen Diagnosekriterien, hier am Beispiel folgender häufig beobachteter PNS: Enzephalomyelitis: klinische oder pathologische Dysfunktion verschiedener Anteile des Nervensystems/Affektion verschiedener Anteile des ZNS mit zusätzlicher Beteiligung von Hinterwurzelganglien oder Plexus myentericus. Limbische Enzephalitis: subakutes Einsetzen von Krämpfen, Störungen im Kurzzeitgedächtnis, Verwirrung und psychiatrischer Symptomatik innerhalb von Tagen bis zu 12 Wochen, zusätzlich pathologische Befunde im Bereich des limbischen Systems in neuropathologischen oder neuroradiologischen Untersuchungen. Subakute Kleinhirndegeneration: eine sich innerhalb von weniger als 12 Wochen entwickelnde, schwere zerebelläre Symptomatik ohne Korrelat in Form einer Kleinhirnatrophie oder einer anderen nicht altersentsprechenden Veränderung im MRT. Sensorische Neuropathie: subakutes, asymmetrisches Einsetzen mit Erreichen eines Rankin scale von höchstens drei vor Ablauf von 12 Wochen, Anzeichen von Sensibilitätsstörungen und meist Schmerzen, Beteiligung der oberen Extremitäten, Verlust der Propriozeption in den 19 betroffenen Arealen und Anzeichen einer Schädigung sensibler Neurone in elektrophysiologischen Untersuchungen. Tabelle 3 zeigt eine Übersicht über die klassischen paraneoplastischen Syndrome, die möglichen Antikörper-Reaktivitäten und zu Grunde liegenden Tumoren. In der nachfolgenden Auflistung sind die klassischen PNS durch Unterstreichen kenntlich gemacht, die Erläuterung im Anschluss berücksichtigt vor allem diese klassischen Syndrome. Tabelle 3 Übersicht über die klassischen neurologischen Syndrome, Antikörper und die Wahrscheinlichkeit einer paraneoplastischen Ätiologie (Voltz, 2002) Syndrom Lambert-Eaton-Syndrom Subakute Kleinhirndegeneration Klinische Wahrscheinlichkeit einer paraneoplastischen Ätiologie 60% 50% Dermatomyositis Opsoklonus/Myoklonus (Kind) Opsoklonus/Myoklonus (Erwachsener) 30% 50% Subakute sensible Neuropathie 20% Limbische Enzephalopathie 20% Enzephalomyelitis 10% 20% Sinnvolle AKBestimmungen WahrscheinPrimäre Tumorsuche lichkeit bei positivem AKErgebnis anti-VGCC SCLC anti-Hu >95% SCLC, Prostata anti-PCA-2 >95% SCLC anti-CRMP5/-CV2 >95% SCLC, Thymom ANNA-3 >95% SCLC anti-Yo >95% Ovar, Mamma anti-Amphiphysin >95% SCLC anti-Ta/-Ma2 >95% Hoden anti-Ma2 >95% verschiedene anti-Ri >95% Mamma anti-Tr >95% M. Hodgkin n.v. Ovar, Lunge,Pankreas anti-Hu >95% Neuroblastom anti.Ri ant-Hu anti-Ma anti-Ta/-Ma2 anti-Hu anti-Ampiphysin ANNA-3 anti-CRMP5/-CV2 anti-Hu anti-Ta/-Ma2 ANNA-3 anti-CRMP5/-CV2 anti-Amphiphysin anti-Hu 20 >95% >95% >95% >95% >95% >95% >95% >95% >95% >95% >95% >95% >95% >95% Mamma, Ovar SCLC, Prostata verschiedene Hoden SCLC, Prostata SCLC SCLC SCLC, Thymom SCLC, Prostata Hoden SCLC SCLC,Thymom SCLC SCLC I.) 1) Syndrome des zentralen Nervensystems: Limbische Enzephalitis 2) Hirnstamm-Enzephalitis 3) Paraneoplastische subakute Kleinhirndegeneration 4) Opsoklonus-Myoklonus-Syndrom 5) Retinopathie 6) Stiff-person-Syndrom 7) Paraneoplastische Enzephalomyelitis II.) Syndrome des peripheren Nervensystems: 8) Subakute sensorische Neuropathie 9) Subakute/chronische sensomotorische Neuropathien 10) Autonome Neuropathie/Chronische gastrointestinale Pseudoobstruktion 11) Motoneuronerkrankungen 12) Neuromyotonie III.) Syndrome der neuromuskulären Synapse und des Muskels 13) Lambert-Eaton-Myasthenie-Syndrom 14) Myasthenia gravis 15) Dermatomyositis I.) Syndrome des zentralen Nervensystems 1) Limbische Enzephalitis (PLE) Die limbische Enzephalitis zeigt sich in Störungen des Affekts (Depressionen, Ängstlichkeit, Persönlichkeitsveränderungen) und der Gedächtnisfunktion (Kurzzeitgedächtnis, retrograde Amnesie). In etwa 50% der Fälle kommt es zudem zu komplex-fokalen und sekundär- 21 generalisierten epileptischen Anfällen (Voltz, 2002). Bei einem Teil der Patienten treten Verwirrtheitszustände, Halluzinationen und paranoide Denkstörungen auf. Typisch ist ein subakuter Verlauf, der sich zu Beginn variabel, im Verlauf jedoch meist progredient zeigt mit Demenz und Störungen des Bewusstseins (Kaiser, 1999). Die limbische Enzephalitis kann isoliert oder zusammen mit einer Rhombenzephalitis oder einer sensiblen Polyneuropathie (s.u.) auftreten (Kaiser, 1999). Nach einer Studie von Gultekin von 2000 sind zur Diagnose einer paraneoplastischen limbischen Enzephalitis folgende Kriterien erforderlich: Typische klinische Symptome Weniger als vier Jahre bis zur Tumordiagnose Ausschluss von Differentialdiagnosen Ein pathologisches Ergebnis von Lumbalpunktion, MRT oder EEG Bei etwa 50% der Patienten findet sich – oftmals erst nach Beginn der Symptomatik- ein kleinzelliger Tumor der Lunge, in 20% ein Hodenkarzinom und in 8% ein Mammakarzinom (Gultekin et al., 2000). Als Antikörperreaktivitäten wurden bisher neben Hu-Antikörpern auch Ma-, CV2-, Amphiphysin-, Tr- sowie VGKC-Antikörper beschrieben (Dalmau et al., 1998; Deodhare, 1996; Voltz et al., 1998; Pozo-Rosich et al., 2003). Letztere Antikörper sind jedoch nicht zwingend pathognomonisch für ein zu Grunde liegendes Malignom (Tan et al., 2008). 2) Hirnstammenzephalitis Manifestiert sich eine Enzephalitis primär im Hirnstamm, kann es zu Symptomen wie Augenbewegungsstörungen (Nystagmus, Blickparesen, Opsoklonus), Sensibilitätsstörungen im Bereich des N. trigeminus, bulbären Symptomen wie Dysarthrie und Dysphagie sowie zu zentralen Paresen oder sogar zu bedrohlichen Atemdepressionen kommen (Ball et al., 1994; Lucchinetti, 1998; Kaiser, 1999). Eine Hirnstammenzephalitis tritt häufig zusammen mit anderen paraneoplastischen Syndromen und seltener als isoliertes Syndrom auf. Zu den assoziierten Antikörpern gehören Hu-, Ma- und Ri-Antikörper (Voltz, 2002). Unter den assoziierten Krebserkrankungen finden sich neben kleinzelligen Bronchialkarzinomen als häufigsten Tumoren auch Mammakarzinome und andere gynäkologische Tumoren sowie Hodenkarzinome beim Mann (PNS Euronet[1]). 22 3) Subakute Kleinhirndegeneration (PKD) Typisch für dieses häufige Syndrom ist die rasche Progredienz innerhalb von Tagen bis Wochen mit Erreichen eines stabil schlechten Niveaus. Neben einer meist erheblichen zerebellären Ataxie finden sich als Anzeichen der Schädigung weiterer zerebellärer Strukturen Schwindel, Doppelbilder, Nystagmus (horizontal, vertikal, rotatorisch), Dysphagie und Dysarthrie sowie Pyramidenbahnzeichen, Polyneuropathien, Rigor, Tremor und Myoklonien (Peterson, 1992; Rosenblum 1993). Es kann zudem vorkommen, dass eine PKD mit einem Lambert-Eaton-Syndrom vergesellschaftet ist, was vor allem in Assoziation mit einem kleinzelligen Bronchialkarzinomen beschrieben wird (Mason et al., 1997). Im Frühstadium der Erkrankung kann eine Kernspintomographie unauffällig sein, der Nachweis einer zerebellären Atrophie gelingt oft erst Monate nach Einsetzen der Symptomatik (Benzing, 1998). Bei der PKD kommen – je nach zugrundeliegendem Tumor- verschiedene AntikörperReaktivitäten vor: Yo-Antikörper finden sich vorwiegend bei Frauen mit einem Mamma- oder Ovarialkarzinom (Peterson, 1992). Tr-Antikörper wurden bei Männern mit Morbus Hodgkin und PKD beschrieben (Bernal et al., 2003). Hier ist auch das Manifestationsalter der neurologischen Symptomatik mit durchschnittlich 30 Jahren deutlich geringer als bei Patientinnen mit PKD in Assoziation mit einem gynäkologischen Tumor (Kaiser, 1999). Charakteristisch für eine PKD bei einem kleinzelligen Bronchialkarzinom sind sowohl Huals auch CV2-Antikörper (Mason et al., 1997; Honnorat, 1996). Auch Ma2- und VGCCAntikörper finden sich gehäuft bei Patienten mit Lungenkarzinomen und PKD (Honnorat, 2007). Ri-Antikörper können unter anderem auf ein Mamma- oder ein Bronchialkarzinom hinweisen (Kaiser, 1999). Eine Antikörperbestimmung kann aufgrund dieser Assoziationen folglich hilfreich sein bei der Suche nach dem zu Grunde liegendem Tumor. 4) Opsoklonus-Myoklonus-Syndrom (OMS) Ein Opsoklonus ist definiert als schnelle, konjugierte Augenbewegungen unterschiedlicher Frequenz und Amplitude in alle Richtungen. Zusätzlich zu diesem Symptom finden sich beim OMS meist Myoklonien von Rumpf und Extremitäten sowie eine Ataxie. Wahrscheinlich 23 liegt dieser Symptomatik eine Störung im Nucleus fastigii des Kleinhirns zugrunde (Voltz, 2002). Das Opsoklonus-Myoklonus-Syndrom wurde bei drei verschiedenen klinischen Gruppen beobachtet (Honnorat, 2007): - Pädiatrische Patienten mit einem Neuroblastom - Erwachsene Patientinnen mit Nachweis von Ri-Antikörpern, wobei meist ein Mammakarzinom zu Grunde liegt - Erwachsene Patienten ohne Nachweis eines Antikörpers, überwiegend assoziiert mit einem kleinzelligen Bronchialkarzinom Ebenfalls beobachtet wurde dieses klassische Syndrom bei Patienten mit Nachweis eines Huoder Yo-Antikörpers (Dalmau et al., 1995; Hers et al., 1994). Der Beginn der Symptomatik ist zumeist akut, der bildmorphologische Nachweis einer Affektion von Kleinhirn oder Hirnstamm (Neuronendegeneration in der Olive) gelingt auch bei intensiver Suche in vielen Fällen nicht (Kaiser, 1999). Eine Rückbildungstendenz des Opsoklonus-Myoklonus-Syndroms bei Kindern zeigt sich häufig nach Entfernung des Tumors mit anschließender Chemotherapie (Hammer, 1995). Bei Erwachsenen kann es -neben einer Spontanremission- unter Tumortherapie und Behandlung des neurologischen Syndroms (s.u.) sowohl zu einer Besserung der klinischen Symptomatik als auch zu einer Progredienz kommen (Dropcho, 1993; Anderson et al., 1988). 5) Retinopathie Die paraneoplastische Retinopathie ist zum einem mit verschiedenen Tumoren wie SCLC, Ovarial-, Mammakarzinom und endokrinen Tumoren vergesellschaftet (Cancer associated retinopathy, CAR), zum anderen kann sie mit einem Melanom (Melanoma associated retinopathy, MAR) assoziiert sein (Voltz, 2002). Während visuelle Störungen bei einer MAR oft erst Jahre nach der Tumordiagnose auftreten, zeigen sich bei einer CAR Symptome wie erhöhte Lichtempfindlichkeit, Farbsehstörungen, Visusminderung (Zäpfchen) oder Verlängerung der Dunkeladaptation und Ringskotome (Stäbchen) bis zu zwei Jahre vor der Tumordiagnose (Jacobson, 1990). Unter den vielen bisher beschriebenen Antikörpern, die sich gegen verschiedene Retinaproteine richten, ist der erste und am häufigsten mit CAR assoziierte das AntiRecorvin, das an einem 23 kDa-Photorezeptorprotein angreift (Thirkill et al., 1989). 24 Die paraneoplastische Retinopathie ist relativ selten, der Verlauf zeigt sich über Monate hinweg progredient und führt am Ende in den meisten Fällen zur schmerzlosen Erblindung (Kaiser, 1999). Besserungen werden unter einer Therapie mit Prednisolon beschrieben (Jacobson, 1990; Rizzo, 1992). 6) Stiff-person-Syndrom (SPS) Dieses relativ seltene Syndrom zeigt sich durch Steifigkeit vor allem der Rücken- und der proximalen Extremitätenmusklatur, außerdem imponieren sehr schmerzhafte Spasmen getriggert durch sensible und emotionale Reize. Diese können so heftig sein, dass Gelenkdeformierungen und Kontrakturen bis hin zu Muskelrissen und Knochenbrüchen möglich sind (Layzer, 1988). Der Verlauf der Erkrankung ist zumeist fluktuierend mit sukzessiver Progredienz und vor allem in Assoziation mit einem SCLC zeigen sich nicht selten zusätzlich Anzeichen einer Enzephalomyelitis oder einer sensiblen Polyneuropathie (Kaiser, 1999). Bei nahezu 70% der Patienten, vor allem in Assoziation mit einem Diabetes mellitus und als Marker eines idiopathischen SPS, sind im Serum Antikörper gegen die GlutamatDehydrogenase (GAD) nachweisbar (Honnorat, 2007; Voltz, 2002). Dagegen werden als Auslöser eines paraneoplastischen SPS und dann als Marker für ein zu Grunde liegendes Mammakarzinom Amphiphysin-Antikörper beschrieben (Folli et al., 1993). Ebenfalls beobachtet wurde das Stiff-person-Syndrom in Assoziation mit einem Lungen- oder Kolonkarzinom sowie mit einem Morbus Hodgkin und malignen Thymomen (Honnorat, 2007). 7) Paraneoplastische Enzephalomyelitis (PEM) Charakteristisch für eine Enzephalomyelitis ist die Tatsache, dass meist gleichzeitig verschiedene Strukturen wie Hippokampus, Hirnstamm, Rückenmark oder auch Hinterwurzeln betroffen sein können (Graus, 2004), was eine Vielfalt an möglichen Symptomen abhängig von der Lokalisation der Affektion nach sich zieht. Häufig finden sich Anzeichen einer sensorischen Neuropathie, limbischen Enzephalitis, Myelitis, chronischen gastrointestinalen Pseudoobstruktion oder von anderen autonomen Dysfunktionen (30%, Orthostase, Pupillenanomalien, Impotenz, Mundtrockenheit, u.a.) (Honnorat, 2007). Manifestationen einer PEM können außerdem 25 zerebelläre Symptome oder eine Hirnstammenzephalitis sein, welche sich durch Schwindel, Nystagmus, Ataxie, Diplopie, Dysarthrie und Dysphagie äußert. Als typischer assoziierter Tumor gilt das kleinzellige Bronchialkarzinom, der häufigste Antikörper ist anti-Hu (Graus et al., 2001), aber auch anti-CV2, -Amphiphysin oder –Ri können vorkommen (Graus, Dalmau, 2007). II.) Syndrome des Peripheren Nervensystems 8) Subakute sensorische Neuropathie Diese erstmals 1948 von Denny Brown beschriebene klassische paraneoplastische Polyneuropathie zeichnet sich durch einen subakuten Verlauf aus mit zu Beginn meist asymmetrisch verteilten schmerzhaften Par- und Dysästhesien vor allem im Bereich der Extremitäten. Im Verlauf kommt es meist zum symmetrischen Verlust aller Reflexe und sensiblen Qualitäten. Der Verlust der Propriozeption mit der damit verbundenen sensiblen Ataxie trifft die Patienten dabei besonders schwer. Während bei einer paraneoplastischen Erkrankung alle sensiblen Qualitäten verloren gehen, zeichnet sich eine differentialdiagnostisch ebenfalls zu erwägende iatrogene Neuropathie unter Chemotherapie, z.B. die cis-Platin induzierte Polyneuropathie, durch den Erhalt von Schmerz- und Temperaturempfindung aus (Rosenblum, 1993). Der häufigste assoziierte Antikörper ist anti-Hu, wobei es große Unterschiede in Zeitverlauf und Verteilung der Symptomatik gibt (Voltz, 2002). Beschrieben wurden auch Fälle mit dem Nachweis von CV2- oder Amphiphysin-Antikörpern (Honnorat, 1996; Antoine et al., 1999). Häufigster mit einer paraneoplastischen sensiblen Neuropathie assoziierter Tumor ist mit Sicherheit das kleinzellige Bronchialkarzinom (Dalmau et al., 1992). Die auch als DennyBrown-Syndrom bezeichnete Krankheit tritt jedoch nur selten isoliert in Erscheinung, viel häufiger finden sich zusätzliche Anzeichen einer Enzephalomyelitis, Zerebellitis oder autonomen Neuropathie, seltener auch eines Lambert-Eaton-Syndroms, welche die Patienten zusätzlich einschränken (Dalmau, 1992; Lucchinetti, 1998). 26 9) Subakute/chronische sensomotorische Neuropathie Zeigen sich neben Symptomen einer sensiblen Polyneuropathie zusätzlich Anzeichen für den Verlust motorischer Neurone, handelt es sich um eine sensomotorische Polyneuropathie. Diese kann jedoch verschiedene Gründe haben, unter anderem auf einem infiltrativen Tumorwachstum, Medikamentennebenwirkungen oder einer Kachexie beruhen, so dass man auch hier erst nach Ausschluss anderer Ursachen oder nach sicherem Nachweis einer Antikörper-Reaktivität von einer paraneoplastischen Genese ausgehen kann (Voltz, 2002). Antoine et al. zeigten in einer Arbeit von 1999, dass nur bei etwa 1/3 der Patienten mit paraneoplastischer sensomotorischer Neuropathie Antikörper wie anti-Hu oder anti-CV2 im Serum nachweisbar waren. 15-30% der Hu-Patienten mit einer peripheren Neuropathie zeigen Anzeichen sowohl einer sensorischen als auch einer motorischen Komponente, wobei eine asymmetrische und multifokale Manifestation leicht als Mononeuritis multiplex oder Polyradikulopathie, ein akuter und schwerer Verlauf alle Extremitäten betreffend leicht als Guillain-Barré-Syndrom fehlgedeutet werden kann (PNS Euronet[2]). Die sensomotorische Neuropathie in Assoziation mit anti-CV2 betrifft überwiegend die unteren Extremitäten, Schmerzen werden in diesem Fall seltener beobachtet als bei Hu-Antikörpern (PNS Euronet[2]). 10) Autonome Neuropathie/ Chronische gastrointestinale Pseudoobstruktion Ebenfalls Symptom einer paraneoplastischen Erkrankung können Störungen im autonomen Nervensystem sein. Diese sind z.T. schwerwiegend, wie z.B. ein plötzlicher Atem- oder Herzstillstand (kardiale Arrhythmie) und können so auch zum Tode führen (Kaiser, 1999). Häufiger sind jedoch klinische Zeichen wie orthostatische Probleme, Pupillotonie, Hyperhidrosis, Impotenz, Harnretention und gastrointestinale Beschwerden wie Diarrhöe oder Obstipation (Dalmau, 1990; Fagius, 1983; Graus et al., 1987; Honnorat, 2007). Letztere auch als chronische gastrointestinale Pseudoobstruktion (CGP) bezeichnete autonome Störung gehört zu den klassischen PNS und findet sich nach Honnorat und Antoine (2007) insbesondere in Verbindung mit einem kleinzelligen Bronchialkarzinom und dem Nachweis von Hu- oder CV2-Antikörpern. Symptome sind in erster Linie eine chronische schwere Obstipation aufgrund der Zerstörung enterischer Nerven sowie Gewichtsverlust, aber auch Dysphagie, Übelkeit, Erbrechen und Störungen der Motilität von Ösophagus und Magen können auftreten. 27 11) Motoneuronerkrankungen Generell äußern sich Motoneuronerkrankungen durch progrediente isolierte zentrale und periphere Paresen ohne relevante sensible Defizite aufgrund einer isolierten Degeneration des ersten und/oder zweiten Motoneurons. Es handelt sich um ein nicht-klassisches PNS und es ist umstritten, ob verschiedene isolierte Erkrankungen wie eine amyotrophe Lateralsklerose (ALS), eine primäre Lateralsklerose (PLS) oder eine progressive muskuläre Atrophie (PMA) bei Koinzidenz mit einem Karzinom als paraneoplastisch einzuordnen sind (PNS Euronet[3]). So zeigt etwa eine aktuelle Studie von 2006 keine Assoziation der gut-etablierten antineuronalen Antikörper mit einer ALS (Stich et al., 2007). Motorische Syndrome sind zwar häufig, treten jedoch meist zusammen mit anderen Beschwerden auf. So findet sich zum Beispiel eine Affektion von erstem und zweitem Motoneuron häufig im Rahmen einer PEM vorwiegend bei Hu-Patienten (PNS Euronet [3]). Forsyth et al. unterscheiden in einer Veröffentlichung von 1997 drei verschiedene Patientengruppen mit möglichen paraneoplastischen Erkrankungen der Motoneurone: Patienten mit entsprechender Symptomatik und Nachweis von HuAntikörpern. Hier gilt die paraneoplastische Ursache als bewiesen. Frauen mit Mammakarzinom und Beteiligung des 1. Motoneurons, mit Symptomen ähnlich einer primären Lateralsklerose (keine assoziierten AK beschrieben). Patienten mit einer Tumorerkrankung und einer ALS-ähnlichen Symptomatik. Hier ist die Assoziation jedoch eher zufällig und die Tumortherapie beeinflusst nicht den Verlauf der neurologischen Erkrankung. 12) Neuromyotonie (Isaac`s Syndrom) (NM) Symptome einer Neuromyotonie sind unter anderem Schwäche und Steifigkeit der Muskulatur sowie Zuckungen und Krämpfe insbesondere im Bereich der distalen Extremitäten (Kaiser, 1999). Zusätzlich zeigen sich in Folge der verzögerten Relaxation der Muskulatur häufig Haltungsanomalien. Diese Beschwerden, die nach mehrmaliger Kontraktion wieder abnehmen, persistieren unter Narkose, im Schlaf und bei Blockade peripherer Neurone (Newsom-Davis, 1993), was vor allem der Unterscheidung zu einem Stiff-person-Syndrom dienen kann. 28 Bei etwa 50% der Patienten mit einer Neuromyotonie lassen sich im Serum Antikörper gegen spannungsabhängige Kaliumkanäle (VGKC) nachweisen (Shillito, 1995), die jedoch keine Marker einer paraneoplastischen Ätiologie sind, da sie nicht zwingend tumorassoziiert sind. Das Isaac-Syndrom kann auftreten in Assoziation mit einem SCLC, einem Thymom oder einem M. Hodgkin (Voltz, 2002). III.) Syndrome der neuromuskulären Synapse und des Muskels 13) Lambert-Eaton-Myasthenie-Syndrom (LEMS) Leitsymptom des LEMS ist eine Schwäche vor allem der proximalen Muskulatur, wobei häufiger die unteren Extremitäten betroffen sind. Die Patienten beklagen unter anderem eine rasche Ermüdbarkeit beim Gehen und Treppensteigen, nicht selten auch Schmerzen bei vermehrter Belastung der Muskulatur. Zusätzlich kommt es bei etwa 80% der Patienten zu Störungen der autonomen Regulation wie Mundtrockenheit, Impotenz, Harnverhalt, Obstipation oder orthostatischen Kreislaufproblemen. Eine zuverlässige diagnostische Methode und vor allem hilfreich bei der Abgrenzung zu einer Myasthenia gravis ist die Elektrophysiologie. Bei niederfrequenter Nervenstimulation (3 Hz) zeigt sich bei beiden Erkrankungen eine Abnahme von Amplitude und Dauer des Muskelaktionspotentials (Dekrement), bei höherfrequenter Stimulation über 20 Hz jedoch kommt es beim LEMS zu einer vorübergehenden Zunahme der Amplitude auf über 100% (Inkrement), bevor sich auch hier eine posttetanische Erschöpfung einstellt (O’Neill, 1988; Newsom-Davis, 2004). Bei 50-70% der Patienten mit einem Lambert-Eaton-Syndrom findet sich ein assoziierter Tumor, mit bis zu 90% ist das kleinzellige Bronchialkarzinom am häufigsten (O’Neill, 1988). Seltener diagnostiziert werden Lymphome oder andere Tumoren. Ursache der Muskelschwäche beim LEMS ist eine verminderte Freisetzung von Acetylcholin an der präsynaptischen Membran der neuromuskulären Synapse. In bis zu 90% der Patienten finden sich -auch wenn kein Tumor nachweisbar ist- Antikörper gegen spannungsabhängige Kalziumkanäle (anti-VGCC) (Kaiser, 1999). Diese AK können jedoch nicht als serologische 29 Marker für die paraneoplastische Ätiologie eines LEMS dienen, da sie sowohl bei der paraneoplastischen als auch bei der nicht paraneoplastischen Form auftreten. 14) Myasthenia gravis (MG) Auch bei der Myasthenia gravis (MG) ist eines der Leitsymptome die rasche Ermüdbarkeit der proximalen Muskulatur, häufiger als beim LEMS finden sich hier jedoch Symptome wie Doppelbilder, Ptosis, Dysphagie, Dysphonie und Dysarthrie. Während das LEMS eher bei Männern zu beobachten ist, ist die Myasthenie bei Frauen bis zu zweimal häufiger (Kaiser, 1999). Wie bereits beschrieben findet sich bei der MG in der elektrophysiologischen Diagnostik kein Inkrement des Muskelaktionspotentials bei Nervenstimulation über 20 Hz. Ursache der Muskelschwäche ist hier die verminderte Aufnahme von Acetylcholin im Bereich der postsynaptischen Membran, bei nahezu allen Patienten mit der generalisierten Form der Erkrankung lassen sich im Serum Antikörper gegen Acetylcholin-Rezeptoren nachweisen (Kaiser, 1999). Etwa 15% der MG-Erkrankungen sind mit einem Thymom assoziiert (Willcox et al., 1987), und bei ca. 50% der Patienten mit einem Thymom finden sich Symptome einer Myasthenie. Eine Tumorentfernung kann zu einer Heilung beitragen, hierbei haben sich Titin-Antikörper als hilfreich erwiesen, da sie eine hohe Aussagekraft im Bezug auf das Vorliegen eines Thymoms haben (Skeie et al., 1997; Voltz et al., 1997). So kann neben der Bildgebung auch der Nachweis dieses Antikörpers bei der Diagnosefindung hilfreich sein. 15) Dermatomyositis Die motorische Komponente dieses klassischen Syndroms manifestiert sich durch eine meist symmetrische, die proximalen Muskeln betreffende und über Wochen bis Monate nur langsam fortschreitende Myopathie (PNS Euronet [4]). Bei etwa einem Drittel der Patienten können zusätzlich Paresen der distalen Muskulatur auftreten. Neben Schluckstörungen und einer Schwäche der Atemmuskulatur kann es zur Beeinträchtigung der Gesichts- und Augenmuskulatur sowie Muskelschmerzen kommen, letztere werden bei Kindern wesentlich häufiger beschrieben (F. Blaes et al., 2005). Zusätzlich zu diesen Symptomen treten Hautveränderungen wie ein Schmetterlingserythem, Teleangiektasien im Bereich der Nägel, erythematöse und squamöse Veränderungen über den 30 Fingerendgelenken und umschriebenen Hautatrophien im Bereich der Fingergelenke auf (F. Blaes et al.,2005). Eine paraneoplastische Ursache der Erkrankung wird in bis zu 30% der Fälle vermutet (Hill et al, 2001), assoziierte Tumoren sind dann Ovarial-. Bronchial-, Pankreas-, Magen- oder kolorektale Karzinome und Non-Hodgkin-Lymphome. Zwar konnten bei der Dermatomyositis bisher auch einige Antikörper nachgewiesen werden, diese waren jedoch alle nicht mit einem Tumor vergesellschaftet. 1.2.6 Diagnostische Methoden Neben den klinischen Charakteristika einer paraneoplastischen neurologischen Erkrankung dient in erster Linie der Nachweis eines gut charakterisierten antineuronalen Antikörpers als richtungsweisend im Bezug auf die Diagnose eines PNS. Diese Antikörper sind so spezifisch, dass bei deren Nachweis mit an Sicherheit grenzender Wahrscheinlichkeit von einer paraneoplastischen Erkrankung auszugehen ist (Graus et al., 2004). In diesem Fall sind paraneoplastische neurologische Syndrome nicht mehr wie früher alleine eine Ausschlussdiagnose. In einem europäischen Konsensuspaper wurden die Kriterien für die Unterscheidung zwischen einem möglichen und einem definitiven paraneoplastischen Syndrom diskutiert (Graus et al., 2004). Folgende für die Diagnosestellung relevanten Fragen wurden kürzlich vom europäischen PNS-Netzwerk zu paraneoplastischen neurologischen Erkrankungen (PNS Euronetwork) festgelegt (Quelle: Voltz, 2004) o Liegen andere Differentialdiagnosen vor (Tabelle 4)? o Ist das neurologische Syndrom "klassisch" paraneoplastisch, d. h. relativ häufig mit einem Tumor assoziiert? o Liegen gut charakterisierte antineuronale Antikörper vor, d. h. von mindestens zwei Arbeitsgruppen bestätigte und an einer ausreichenden Anzahl von Patienten und Kontrollen getestete Antikörperreaktivitäten (Tabelle 2)? o Ist ein für dieses neurologische Syndrom typischer Tumor diagnostiziert? o Wann im Verhältnis zum Beginn des neurologischen Syndroms wurde der Tumor diagnostiziert? o Bessert sich das neurologische Syndrom unter Tumortherapie? 31 Tabelle 4 Überblick über mögliche Differentialdiagnosen paraneoplastischer neurologischer Syndrome Paraneoplastisches Syndrom Limbische Enzephalitis Rhombenzephalitis Opsoklonus-/Myoklonus-Syndrom Enzephalomyelitis Subakute Kleinhirndegeneration Retinopathie Subakute sensible Neuropathie Autonome Neuropathie Motoneuronerkrankungen Neuromyotonie Stiff-person-Syndrom Lambert-Eaton-Myasthenie-Syndrom Myasthenia gravis Differentialdiagnosen infektiös, v. a. Herpes-Enzephalitis; autoimmunologisch, z. B. anti-VGKC positiv; Hirntumor; Demenz infektiöse Enzephalitis, z. B. Listerien, Toxoplasmose; basale Meningitis; Miller-FisherSyndrom; Myasthenia gravis, Hirntumor Opsoklonus: viral, medikamentös Myoklonien: physiologisch, hereditär, sporadisch, Epilepsie-Syndrome, Enzephalopathien verschiedener Ätiologie, degenerative Erkrankungen (spinozerebelläre Degenerationen u. a.) Infektiös; parainfektiös; MS genetisch; infektiös (Epstein-Barr-Virus); äthyltoxisch vaskulär; Neuritis N. optici z.B. bei Multipler Sklerose CIDP; MGUS; anti-MAG-Neuropathie Diabetes mellitus; GBS; CIDP multifokale motorische Neuropathie; ALS Crampi; Myotone Dystrophien Myotone Dystrophien Myasthenia gravis autoimmun; medikamenteninduziert, kongenital, LEMS (Klassische paraneoplastische neurologische Syndrome sind durch Unterstreichen kenntlich gemacht) Unter Berücksichtigung der genannten Kriterien wurde im Konsensuspaper von Graus et al., 2004 ein diagnostischer Algorithmus vorschlagen, anhand dessen die diagnostische Sicherheit bestimmt werden kann (Abbildung 1). Zur Testung der antineuronalen Antikörper gibt es folgende Empfehlungen (Voltz, 2002): - Das Serum sollte sowohl mithilfe der Immunhistochemie als auch eines Western Blot auf Antikörper-Reaktivitäten untersucht werden. - Es ist immer ein Bestätigungstest durchzuführen, der in der jeweils anderen Nachweismethode besteht; für anti-Hu kann statt eines Western Blot ein ELISA angewandt werden. 32 Abbildung 1 Algorithmus zur Diagnostik paraneoplastischer neurologischer Syndrome (Voltz 2004) Wie aus dem Flussdiagramm ersichtlich, spielt neben dem Nachweis eines Antikörpers ebenso die Tumordiagnose eine große Rolle. Nach Honnorat (2007) geht in etwa 80% der Fälle die neurologische Symptomatik einer Tumordiagnose voraus, die meisten Tumoren werden innerhalb von vier bis sechs Monaten diagnostiziert. Sowohl bei Nachweis eines antineuronalen Antikörpers als auch bei einer klassischen neurologischen Symptomatik ohne Nachweis eines Antikörpers sollte mittels konventioneller Bildgebung (Sonographie, Röntgen, CT, Szintigraphie) und Bestimmung von Tumormarkern nach einer Tumorentität gesucht werden (Voltz, 2002). Eine mögliche Antikörper-Reaktivität kann aufgrund der gehäuften Assoziation mit bestimmten Tumoren zusätzlich zur Diagnosefindung beitragen. Da gerade im Zuge der paraneoplastischen Erkrankung infolge einer möglicherweise tumorsuppressiven Wirkung der Tumor oftmals klein und lokalisiert bleibt (ChartrandLefebvre et al., 1998) hat sich bei positivem Antikörpernachweis bzw. bei klassischem PNS und negativer Routinediagnostik neben einem invasiven diagnostischen Vorgehen das Ganzkörper-FDG-PET als hilfreich erwiesen. Jedoch lässt sich auch bei negativem Ergebnis ein zu Grunde liegender Tumor nicht mit 100%iger Sicherheit ausschließen (Voltz, 2002). Wie der Algorithmus deutlich macht, kann auch bei fehlendem Antikörpernachweis bei Bestätigung eines bestimmten Tumors in Kombination mit einer klassischen neurologischen Symptomatik von einer paraneoplastischen Ätiologie ausgegangen werden (siehe Abb.1). 33 Dies ist insofern wichtig, als dass bei fast 50% der Patienten mit einem PNS kein gut charakterisierter Antikörper nachgewiesen werden kann (Graus et al., 2004). Generell wird eine paraneoplastische Ätiologie wahrscheinlicher, falls die Symptome innerhalb von vier Jahren vor oder nach einer Tumordiagnose auftreten, der Liquor Entzündungszeichen (Zellzahlerhöhung, Nachweis einer intrathekalen Immunglobulinsynthese, Schrankenstörung) aufweist, Differentialdiagnosen ausgeschlossen wurden und sich die neurologischen Symptome im Zuge einer Tumortherapie verbessern (Voltz, 2002). Ebenfalls eine Rolle bei der Diagnostik einer paraneoplastischen Erkrankung spielt die Bildgebung von Schädel und Rückenmark. Dalmau et al. (2002) beschreiben die Ergebnisse von CT und MRT des Schädels bei 71 Hu-Patienten, dabei zeigten -vor allem im MRTdiejenigen eine bildmorphologische pathologische Veränderung, die an einer limbischen Enzephalitis litten. Zu ähnlichen Ergebnissen kommt auch Candler in einer aktuelleren Arbeit von 2004. Hier konnte bei einem Patienten mit zerebellärer Symptomatik im MRT zusätzlich der Nachweis einer Kleinhirnatrophie erbracht werden, welche jedoch mit einer generalisierten Hirnatrophie einherging. So wird vor allem bei klinischem Verdacht auf eine limbische Enzephalitis die zerebrale Bildgebung empfohlen, wobei das MRT eine größere Sensitivität aufweist (Voltz, 2002). Neben der Suche nach diesen pathologischen Veränderungen kann die Bildgebung auch dem Ausschluss von Differentialdiagnosen wie Metastasen u.a. dienen (Voltz 2002). Ein normales MRT schließt eine paraneoplastische Erkrankung nicht aus, es empfiehlt sich also immer eine zusätzliche Liquordiagnostik und ein Antikörperscreening aus dem Serum. Der Nachweis struktureller Veränderungen peripherer neurologischer paraneoplastischer Erkrankungen kann mit elektrophysiologischen Methoden wie Elektromyographie und Neurographie geführt werden. 1.2.7 Therapeutische Optionen [nach Leitlinien der DGN; http://www.dgn.org/images/stories/dgn/leitlinien/LL2008/ll08kap_085.pdf] Die Behandlung von Patienten mit paraneoplastischen neurologischen Syndromen ist oftmals schwierig und sollte rasch erfolgen. Ebenfalls beachtet werden sollte die Tatsache, dass jedes Syndrom und jede Antikörperreaktivität einen anderen klinischen Verlauf mit sich bringen kann, dass ein natürlicher Verlauf fluktuieren kann und auch eine spontane Verbesserung der neurologischen Symptomatik möglich ist. Im Zuge der Immunreaktion gegen das Karzinom 34 sind sogar spontane Tumorregressionen beschrieben worden. So sollte jeder Patient individuell behandelt werden. Die Therapie besteht grundsätzlich aus zwei großen Säulen: zum einen der Behandlung des zu Grunde liegenden Tumors, zum anderen der Immunmodulation: Die Tumorbehandlung spielt nach wie vor die Hauptrolle im Therapiekonzept. Die besten Aussichten auf eine Stabilisierung oder gar Besserung der neurologischen Symptomatik hat der Versuch einer erfolgreichen Tumorelimination (Keime-Guibert et al., 2000), am besten in enger Zusammenarbeit mit dem Onkologen. Dabei ist von entscheidender Rolle, dass das Karzinom schnellstmöglich diagnostiziert und einer entsprechenden Therapie zugeführt wird. Denn nur bei einer Tumorentfernung und der damit verbundenen Elimination der durch diesen exprimierten neuronalen Antigene handelt es sich um eine kausale Therapie mit Aussicht auf eine erfolgreiche Beseitigung des antigenen Stimulus. Die Tatsache, dass es sich bei paraneoplastischen Erkrankungen aller Wahrscheinlichkeit nach um einen Autoimmunprozess handelt, eröffnet dem Kliniker eine zweite Therapieoption in Form einer Immunmodulation/Immunsuppression. Diese kann durch Steroide, intravenöse Immunglobuline, Cyclophosphamid und/oder eine Plasmapherese erfolgen. In Ermangelung guter evidenzbasierter Daten wird in den DGN-Leitlinien folgender Therapievorschlag gemacht: Man beginnt mit einem Zyklus Steroidtherapie (5 500mg Methylprednisolon i.v.). Nach einer Woche kann bei ausbleibendem Erfolg die Gabe von intravenösen Immunglobulinen angeschlossen werden (2g/kg Körpergewicht verteilt über 5 Tage). Nachdem man wiederum eine Woche abgewartet hat, kann bei weiterhin mangelndem Ansprechen eine Plasmapherese oder eine Behandlung mit Cyclophosphamid angeschlossen werden. Je eher die Therapie begonnen wird, desto besser sind die Aussichten auf Erfolg. Leider kommt es schon sehr früh zur meist irreversiblen Zerstörung neuronaler Strukturen, was den mangelnder Effekt der Immuntherapie bei der überwiegenden Zahl der Patienten erklärt (Keime-Guibert et al., 2000). Vor allem bei Syndromen des zentralen Nervensystems zeigt sich selten eine Besserung, bei folgenden Syndromen des peripheren Nervensystems ist eine Immuntherapie aufgrund bisheriger therapeutischer Erfolge jedoch definitiv indiziert (Voltz, 2002): Sensomotorische Neuropathie, Lambert-Eaton-Syndrom, Myasthenia gravis, Dermato-/Polymyositis, Neuromyotonie und Stiff-person-Syndrom. Im Rahmen einer Immunmodulation gilt bereits eine Stabilisierung der Symptomatik als Erfolg, zeigt sich keine Wirkung, ist von einer immunologischen Dauerbehandlung abzuraten. 35 2. Patienten und Methode 2.1 Patienten Die nachfolgende Darstellung stützt sich auf eine retrospektive, klinisch und epidemiologisch orientierte, monozentrische Studie, in der die Daten von Patienten mit paraneoplastischen neurologischen Syndromen untersucht wurden. Ausgehend von Krankenakten vom 01.08.1987 bis zum 31.03.2008 am Neurozentrum der Universitätsklinik Freiburg wurde ein Kollektiv von Patienten mit paraneoplastischen neurologischen Syndromen und zugleich positiven Befunden der sog. gut etablierten antineuronalen Antikörper (anti-Hu, Ri, Yo, CV2/CRMP5, Amphiphysin und Ma2) im Serum zusammengestellt. Die Basisdaten wurden hierbei aus archivierten Patientenunterlagen bzw. Arztbriefen der Neurologischen Universitätsklinik Freiburg entnommen, die Verlaufsdaten wurden anhand ärztlicher Verlaufsberichte nach mündlicher sowie schriftlicher Einverständniserklärung durch Patienten/Angehörige ergänzt. Die Studie wurde von der Ethikkommission der Universität Freiburg genehmigt (19.12.2007; Antrags-Nr.: 367/07). 2.2 Methoden-Datenbankerstellung Die Datenbank basiert auf dem Programm Microsoft Excel, mit dem anhand einer DatenbankMaske folgende Parameter erfasst wurden: 2.2.1 Demographische Daten Geschlecht, Alter bei Erstmanifestation des PNS, Alter bei Diagnosestellung des PNS, Alter bei Diagnosestellung des Malignoms, Manifestation des PNS vor oder nach Tumordiagnose sowie das Intervall zwischen PNS Erstmanifestation und Tumordiagnose wurden untersucht. 36 2.2.2 Paraneoplastische neurologische Syndrome Neben der Unterscheidung zwischen peripherer und zentraler Manifestation wurden anhand der zu Grunde liegenden Krankenakten/Arztbriefe bzw. der klinischen Untersuchung der überlebenden Patienten folgende paraneoplastische neurologische Syndrome erfasst (klassische Syndrome sind unterstrichen): Zentrale Manifestation Hirnstammenzephalitis Paraneoplastische Enzephalomyelitis/ Myelitis (PEM) Limbische Enzephalitis Subakute Kleinhirndegeneration (PCD) Opsoklonus-Myoklonus-Syndrom Rhombenzephalitis Optikusneuritis Subakute Retinadegeneration, Retinopathie Nekrotisierende Myelopathie Motoneuron-Erkrankung Stiff-person-Syndrom Periphere Manifestation Sensomotorische Neuropathie Subakute sensorische Neuropathie (Denny-Brown-Syndrom) Autonome Neuropathie/Chronische gastrointestinale Pseudoobstruktion Mononeuritis multiplex Akute/chronische Polyradikulitis (GBS, CIDP) Neuromyotonie (Isaacs-Syndrom) Dermatomyositis/Polymyositis Nekrotisierende Myopathie Lambert-Eaton-Myasthenie-Syndrom 37 2.2.3 Antineuronale Antikörper Die Patienten wurden im Rahmen der Routinediagnostik mittels Immunfluoreszenz, Immunoblot oder ELISA auf das Vorliegen eines oder mehrerer der gut etablierten antineuronalen Antikörper in Serum untersucht: - anti-HuD - anti-Yo - anti-Ri - anti-CV2/CRMP5 - anti-Ma2 - anti-Amphiphysin Erfasst wurde die den Unterlagen zu entnehmende Anzahl und Entität der antineuronalen Antikörper. Bei einem Teil der überlebenden Patienten konnte zusätzlich nach Einverständniserklärung eine Verlaufsuntersuchung der antineuronalen Antikörper im Serum nach erneuter Blutentnahme erfolgen. 2.2.4 Tumor Es wurde nach folgenden Tumorerkrankungen eingeteilt: - Kleinzelliges Bronchialkarzinom - Nicht-kleinzelliges Bronchialkarzinom - Mammakarzinom - Ovarialkarzinom - Endometriumkarzinom - Karzinoid/neuroendokriner Tumor - Prostatakarzinom - Seminom - Melanom - Hodgkin-/Non-Hodgkin-Lymphom - Metastase eines Adenokarzinoms bei unklarem Primärtumor - Tumorverdächtige Herde im PET - Keine Neoplasie detektiert - Keine Angaben 38 Metastasen bei ungeklärtem Primärtumor oder ein verdächtiger Befund in der PET wurden ebenfalls in die Beschreibung mit aufgenommen. Konnte keine Neoplasie gefunden werden, wurde dies vermerkt. 2.2.5 Diagnostik In die diagnostische Beschreibung mit aufgenommen wurden Ergebnisse der Bildgebung aus - Schädel-MRT - Wirbelsäulen-MRT - PET. Dabei wurde jede relevante pathologische Veränderung aus allen durchgeführten Untersuchungen festgehalten. Bei der Schädel-MRT erfolgte eine Differenzierung zwischen hyper- bzw. hypointensen Läsionen und einer Atrophie sowohl des Groß- als auch des Kleinhirns. Bei der MRT der Wirbelsäule wurden eventuelle hyper- bzw. hypointense Läsionen erfasst. Im Falle einer positiven Bildgebung in der PET wurde außerdem erfasst, ob diese Veränderung mit der Routinebildgebung korrelierte oder alleinig die PET tumorverdächtige Ergebnisse erbrachte. Zusätzlich zur Bildgebung erfolgte die Erfassung des jeweils erstmalig erhobenen Liquorbefundes. Dabei wurden folgende Parameter untersucht: - Zellzahl pro µl - Gesamteiweiß (mg/l) - IgG-Index - Vorliegen oligoklonaler Banden Im Rahmen der Analyse der oligoklonalen Banden wurde differenziert zwischen isoliertem Auftreten im Liquor und positivem Befund sowohl im Liquor als auch im Serum. 2.2.6 Behandlung Die Behandlung eines PNS besteht aus zwei Komponenten: der Tumortherapie und der Immuntherapie/-modulation. Vor diesem Hintergrund wurden jeweils folgende TherapieParameter erfasst: 39 Tumorbehandlung - Resektion - Chemotherapie - Radiatio - keine PNS-Behandlung - Immunsuppression - Steroide - Plasmapherese - Immunglobuline - keine 2.2.7 Verlauf/Follow up Betrachtet wurde in erster Linie der Krankheitsverlauf in Bezug auf die zu Grunde liegende Therapie. Dabei wurde unterschieden zwischen dem Einfluss der Tumortherapie und dem der Immuntherapie, da sich in den bisherigen Studien in erster Linie die Behandlung des Karzinoms als erfolgversprechend erwiesen hat. Die dabei unterschiedenen Parameter waren - kein Ansprechen - partielles/ transientes Ansprechen - Stabilisierung - Verbesserung Zusätzlich erfasst wurden Daten wie die Dauer des Follow up seit der Diagnosestellung des PNS, die Überlebenszeit nach der Erstmanifestation von neurologischen Symptomen und die Überlebenszeit nach der Diagnosestellung des PNS. Bei den verstorbenen Patienten wurde untersucht, ob der Tumor oder die neurologische Erkrankung zum Tode führten und welches Ergebnis eine eventuell durchgeführte Autopsie erbracht hatte. Die überlebenden Patienten wurden telefonisch kontaktiert und nach mündlicher und schriftlicher Einverständniserklärung so weit möglich zu einer Nachuntersuchung in die Neurologische Universitätsklinik einbestellt. Hierbei wurde neben der Erhebung eines aktuellen neurologischen Status eine Serumprobe zur Bestimmung des derzeitigen Antikörperstatus entnommen. 40 War ein ambulanter Termin in der Klinik nicht möglich, wurde dem Patienten die Serumprobe vor Ort durch den Hausarzt entnommen. 2.3 Statistik Angesichts der relativ geringen Fallzahl von 39 Patienten beschränkte sich die Studie überwiegend auf eine beschreibende Darstellung der Ergebnisse. Ferner führten wir eine Kaplan-Meier-Analyse zur Überlebenszeit unserer Patienten ab dem Zeitpunkt der Erstmanifestation der neurologischen Symptomatik durch. 41 3. Ergebnisse Der Ergebnisteil paraneoplastischen umfasst die neurologischen genaue Darstellung Syndromen, von zu Grunde demographischen liegenden Daten, Malignomen, antineuronalen Antikörpern, Resultaten aus Liquoruntersuchung und weiterer Diagnostik wie MRT und PET, Therapie des Malignoms und Behandlung des PNS sowie der Beschreibung des Follow up der Patienten. Eine Übersichtstabelle im Anhang bietet einen Überblick über die wichtigsten Ergebnisse (Tabelle 21 [Quelldaten]). 3.1 Demographische Daten Insgesamt umfasst die Studie klinische Daten von 39 Patienten mit einem paraneoplastischen neurologischen Syndrom bei Nachweis eines oder mehrerer der gut charakterisierten antineuronalen Antikörper. Das Alter lag zum Ende des Follow up zwischen 29 und 84 Jahren. Von den anfänglich 43 Patienten mussten im Verlauf vier Patienten aus folgenden Gründen aus der Statistik ausgeschlossen werden (siehe Übersichtstabelle im Anhang): In zwei Fällen waren die zur Verfügung stehenden Unterlagen zur Aufnahme in die Studie nicht ausreichend (Nr.1; Nr.42). In zwei Fällen konnte neben einer mangelhaften Datenlage die neurologische Symptomatik nicht eindeutig einem bestimmten paraneoplastischen neurologischen Syndrom zugeordnet werden (Nr.2; Nr.43). Ferner ergaben sich bei Patient Nr.43 nach Auswertung der Daten Zweifel an der paraneoplastischen Genese der Erkrankung. Von den verbleibenden 39 Patienten waren 21 Fälle (53,8%) weiblichen und 18 Fälle (46,2%) männlichen Geschlechts. Das Durchschnittsalter aller Patienten zum retrospektiv erfassten Zeitpunkt der Erstmanifestation des PNS lag bei 60,7 Jahren, der jüngste Patient war hierbei 17,8 Jahre, die älteste Patientin 74,6 Jahre alt. Im Mittel waren die Frauen zum Zeitpunkt der Erstmanifestation mit 60,5 Jahren etwas jünger als die Männer mit 60,9 Jahren. 42 Das Durchschnittsalter der Patienten zum Zeitpunkt der Diagnosestellung der paraneoplastischen neurologischen Erkrankung lag bei 61,7 Jahren, der jüngste Patient war hierbei 20,3 Jahre, die älteste Patientin 75,3 Jahre alt. Auch hier lag das Alter der Frauen mit im Mittel 61,2 Jahren und das der Männer mit 62,3 Jahren nicht weit voneinander entfernt. Zum Zeitpunkt der Diagnosestellung waren nur fünf der 39 Patienten jünger als 50 Jahre, in neun Fällen lag das Alter über 70 Jahren. 3.2 Paraneoplastische neurologische Syndrome Die Differenzierung einer isoliert peripheren, einer isoliert zentralen Manifestation oder einer Beteiligung des zentralen und zugleich peripheren Nervensystems ergab folgende Verteilung: Bei 17 (43,6%) der 39 Patienten war ausschließlich das zentrale Nervensystem betroffen, bei 12 (30,8%) Patienten fand sich eine Affektion des peripheren Nervensystems und bei 10 (25,6%) der 39 Patienten waren sowohl das zentrale als auch das periphere Nervensystem betroffen. Eine sensomotorische Neuropathie fand sich bei acht der 12 Patienten mit isolierter Beteiligung des peripheren Nervensystems, drei weitere Patienten litten unter einer subakuten sensorischen Neuropathie (Denny-Brown-Syndrom). Der letzte Patient aus dieser Gruppe zeigte sowohl Anzeichen einer chronischen Polyradikulitis als auch einer autonomen Neuropathie, welche sich in erster Linie in einer Störung der Magen-Darm-Peristaltik (Chronische gastrointestinale Pseudoobstruktion) äußerte. Sechs der Patienten, bei denen ausschließlich das zentrale Nervensystem betroffen war, hatten eine subakute Kleinhirndegeneration, drei eine Hirnstammenzephalitis, drei eine paraneoplastische Enzephalomyelitis und drei eine limbische Enzephalitis. Bei einer Patientin fanden sich Symptome einer subakuten Retinadegeneration bzw. Retinopathie und ein Patient war nur schwer einem der klassischen bzw. nicht-klassischen Syndrome zuzuordnen. Seine Symptomatik lässt sich am ehesten als choreatisches Syndrom beschreiben. Die Kombination aus einer subakuten Kleinhirndegeneration und einer sensomotorischen Neuropathie zeigten drei Patienten, ebenfalls drei Patienten litten unter einer subakuten Kleinhirndegeneration und einer sensorischen Neuropathie. Eine sensomotorische Neuropathie fand sich bei drei weiteren der Patienten mit Affektion beider Systeme, davon jedoch bei einem in Kombination mit einer Hirnstammenzephalitis, bei einem mit einer limbischen Enzephalitis und beim dritten zusammen mit einer paraneoplastischen 43 Enzephalomyelitis. Die Kombination aus subakuter sensorischer Neuropathie und Hirnstammenzephalitis fand sich bei einem der Patienten. Die anderen paraneoplastischen neurologischen Syndrome, die in der Kriterienliste im Methodenteil aufgeführt sind, konnten bei keinem der Patienten gefunden werden. Somit wird deutlich, dass die sensomotorische Neuropathie mit 14 Fällen (35,9%) das am stärksten vertretene Syndrom ist, gefolgt von der subakuten Kleinhirndegeneration mit 12 Fällen (30,8%), sechs davon in isolierter Form. Auch die subakute rein sensorische Neuropathie war mit insgesamt sieben Fällen häufig vertreten, wobei in drei Fällen eine zusätzliche subakute Kleinhirndegeneration, in einem Fall eine zusätzliche Hirnstammenzephalitis hinzukam. Eine deutliche Geschlechtspräferenz bei einem der neurologischen Syndrome konnte nicht beobachtet werden. Folglich fanden sich bei den 39 Patienten insgesamt 50 verschiedene neurologische Syndrome, entweder isoliert oder in Kombination. Dabei handelte es sich in 28 Fällen um ein klassisches PNS. Die Tabellen 5 und 6 bieten einen Überblick über oben beschriebene Verteilung der PNS bzw. die verschiedenen Kombinationen bei Manifestation mehrerer Syndrome. Tabelle 5 Häufigkeitsverteilung der PNS Syndrom Häufigkeit Prozent Sensomotorische Neuropathie 14 28 Sensorische Neuropathie 7 14 Autonome Neuropathie 1 2 Chronische Polyradikulitis 1 2 Subakute Kleinhirndegeneration 12 24 Hirnstammenzephalitis 5 10 Enzephalomyelitis 4 8 Limbische Enzephalitis 4 8 Subakute Retinadegeneration 1 2 ‚‚Choreatisches Syndrom’’ 1 2 Peripher Zentral 44 Tabelle 6 Kombinationen bei Manifestation mehrerer Syndrome PatientenNr. Geschlecht Antikörper 1. Syndrom 2. Syndrom 10 w Hu Kleinhirndegeneration Sensorische Neuropathie 12 m Hu Kleinhirndegeneration Sensorische Neuropathie 13 m Amphiphysin Hirnstammenzephalitis Sensorische Neuropathie 14 m Hu Chronische Polyradikulitis Autonome Neuropathie 16 m Hu Kleinhirndegeneration Sensomotorische Neuropathie 18 w Hu Kleinhirndegeneration Sensorische Neuropathie 19 m Hu Limbische Enzephalitis Sensomotorische Neuropathie 22 m Hu Enzephalomyelitis Sensomotorische Neuropathie 25 w CV2 Kleinhirndegeneration Sensomotorische Neuropathie 30 w Hu Hirnstammenzephalitis Sensomotorische Neuropathie 39 m Hu Kleinhirndegeneration Sensomotorische Neuropathie 3.3 Zugrundeliegende Malignome Insgesamt wurde im Beobachtungszeitraum bei 27 der 39 Patienten (69,2%) ein definitiver Primärtumor diagnostiziert, in zwei weiteren Fällen fanden sich histologische Hinweise auf lokale Lymphknoten-Metastasen eines SCLC (5,1%). Bei zehn Patienten (25,7%) ergaben sich in der Routinediagnostik keine sicher wegweisenden Befunde. Bei jeweils acht Patientinnen und acht Patienten wurde ein kleinzelliges Bronchialkarzinom diagnostiziert, drei Männer und drei Frauen waren an einem nicht-kleinzelligen Lungentumor erkrankt und drei Patientinnen litten an einem Mammakarzinom. Bei einer Patientin fand sich ein Ovarialkarzinom und beim letzten Patienten ein Morbus Hodgkin. In vier Fällen wurde die Diagnose bei schon hochgradigem Verdacht in der Routinebildgebung durch eine PET bestätigt. Folglich wurde keine der Tumorerkrankungen in der Gruppe der Patienten mit gesichertem Primärtumor ausschließlich anhand einer PETUntersuchung diagnostiziert. Das Durchschnittsalter dieser 27 Patienten betrug zum Zeitpunkt der Tumordiagnose 60,3 Jahre, der jüngste Patient (Nr.29, Morbus Hodgkin) war 20,3 Jahre, die älteste Patientin (Nr.40, NSCLC) 75,3 Jahre alt. 45 Bei Patient Nr.23 und Patientin Nr.25 zeigten Routinediagnostik und Histologie statt eines Primärtumors nur Metastasen eines kleinzelligen Bronchialkarzinoms, in einem Fall bestätigt durch eine auffällige PET-Untersuchung. Tabelle 7 fasst die Häufigkeitsverteilung der diagnostizierten Tumorerkrankungen nochmals zusammen. Tabelle 7 Häufigkeitsverteilung der Tumorerkrankungen Tumor Häufigkeit Prozent SCLC 16 55,2 NSCLC 6 20,7 Mammakarzinom 3 10,4 Ovarialkarzinom 1 3,4 Morbus Hodgkin 1 3,4 LK-Metastasen eines SCLC 2 6,9 SCLC: Small cellular lung cancer NSCLC: Non small cellular lung cancer Bei fünf Patientinnen und fünf Patienten blieb die Suche nach einem Primärtumor mittels konventioneller Bildgebung laut den uns zur Verfügung stehenden Unterlagen nicht eindeutig oder erfolglos. Von diesen zehn Patienten ohne wegweisenden Befund wurden fünf mittels PET auf ein lokales Tumorgeschehen untersucht. In zwei Fällen blieb diese Untersuchung ohne Ergebnis (Nr.6, CV2; Nr.27, Hu), bei einer Patientin fanden sich bei negativer Routinebildgebung Signalanreicherungen im Bereich von Thoraxwand und Pleura (Nr.32, Yo), bei einer weiteren Yo-Patientin ergab sich der Verdacht auf paraaortale Metastasen eines Adenokarzinoms a.e. im Sinne eines Ovarial- oder Blasenkarzinoms (Nr.26). In diesem Fall fanden sich auch in der Routinebildgebung Hinweise auf einen lokalen Tumorprozess. Der fünfte Patient zeigte Signalanreicherungen sowohl im Bereich des Ileozoekalpols als auch mediastinaler und hilärer Lymphknoten (Nr.9, Hu, Amphiphysin), die Routinebildgebung war hier ohne Befund geblieben. Und zuletzt ergaben sich bei einem der Patienten mit in vivo fehlendem Tumornachweis in der Autopsie tumorverdächtige mediastinale Lymphknoten, deren histologisches Ergebnis jedoch nicht eruiert werden konnte. Zusammenfassend fanden sich unter Aufwendung aller zur Verfügung stehender Untersuchungsmethoden wie Routinebildgebung, PET und Autopsie im Studienkollektiv ein 46 gesichertes Malignom oder ein höchstgradiger Tumorverdacht bei 33/39 Patienten (84,6%). Lediglich bei sechs Patienten (15,4%) konnte keinerlei Hinweis auf das assoziierte Karzinom gefunden werden. Bei allen 27 Patienten mit definitivem Tumor ließ sich der Ausdehnungsgrad des Karzinoms zum Zeitpunkt der Diagnosestellung ausmachen. Bei zehn Patienten war der Tumor lokal begrenzt (fünf SCLC, vier NSCLC, ein Mammakarzinom), bei 13 Patienten war der Tumor bereits in einem lokal fortgeschrittenen Stadium, bzw. fanden sich lokale LymphknotenMetastasen (neun SCLC, zwei NSCLC, ein Mammakarzinom und ein Morbus Hodgkin) und Fernmetastasen konnten in vier Fällen nachgewiesen werden, davon zwei SCLC, ein Mammakarzinom und ein Ovarialkarzinom. Bei zwei weiteren Patienten fanden sich zum Zeitpunkt der Tumordiagnose nur positive lokale Lymphknoten. Das Ergebnis der histologischen Untersuchung deutete jedoch wie bereits erwähnt in beiden Fällen auf ein zugrundeliegendes SCLC hin, so dass in beiden Fällen von einem regional fortgeschrittenen Tumorstadium mit lokalen LymphknotenMetastasen ausgegangen werden kann. Bei nur vier Patienten manifestierte sich die neurologische Symptomatik nach Diagnosestellung des Tumors (Choreatisches Syndrom [SCLC + Hu]; Hirnstammenzephalitis [Mamma-Ca + Ri]; limbische Enzephalitis [NSCLC + Ri]; Kleinhirndegeneration [Ovarial-Ca + Yo]). Das durchschnittliche Intervall zwischen Tumordiagnose und Erstmanifestation des PNS betrug dabei im Mittel 67 Monate (7-120 Monate). Bei 35 Patienten (89,7%) war zum Zeitpunkt der neurologischen Erstsymptomatik noch kein Tumor bekannt. In dieser Gruppe fand sich nur bei 25 Patienten im Verlauf ein definitiver Primärtumor (bei zwei Personen wurden lokale Lymphknoten-Metastasen eines SCLC beschrieben). Das Intervall zwischen Erstmanifestation des PNS und Tumordiagnose betrug bei diesen 25 Patienten im Mittel 17,5 Monate (0,5-133 Monate). Tabelle 8.1 bietet einen Überblick über die einzelnen Tumorerkrankungen, Tabelle 8.2 einen Überblick über die Patienten ohne Tumordiagnose und den jeweils assoziierten PNS. 47 Tabelle 8.1 Überblick über die Patienten mit Tumordiagnose und assoziierten PNS Nr. Geschlecht 3 w Hu Sensomotorische Neuropathie SCLC 4 m Hu Sensomotorische Neuropathie NSCLC 4 5 w Mamma-Ca 12 8 w Ri,Hu,Am Enzephalomyelitis phiphysin Hu Limbische Enzephalitis SCLC 1,5 10 w Hu SCLC 133 11 m Ri Subakute Kleinhirndegeneration Subakute sensorische Neuropathie Limbische Enzephalitis 13 m Amphiphysin SCLC 10,5 14 m Hu SCLC 14 15 w Hu Hirnstammenzephalitis Subakute sensorische Neuropathie Chronische Polyradikulitis Autonome Neuropathie Sensomotorische Neuropathie SCLC 3 16 m Hu NSCLC 10 17 w Hu Subakute Kleinhirndegeneration Sensomotorische Neuropathie Sensomotorische Neuropathie SCLC 2,5 18 w Hu SCLC 4 19 m Hu SCLC 53,5 20 m SCLC 43,5 21 w CV-2 Hu Yo Subakute Kleinhirndegeneration Subakute sensorische Neuropathie Limbische Enzephalitis Sensomotorische Neuropathie Enzephalomyelitis Subakute Kleinhirndegeneration Ovarial-Ca 29 m Hu Subakute Kleinhirndegeneration M. Hodgkin 30,5 30 w Hu NSCLC 33,5 31 m Hu Hirnstammenzephalitis Sensomotorische Neuropathie Sensomotorische Neuropathie 33 w Ri Hirnstammenzephalitis Mamma-Ca 34 m Hu Choreatisches Syndrom SCLC 35 w CV-2 Subakute Kleinhirndegeneration NSCLC 9 36 w Hu SCLC 3 37 w Hu SCLC 0,5 38 w Hu Subakute sensorische Neuropathie Subakute sensorische Neuropathie Subakute Kleinhirndegeneration Mamma-Ca 2 39 m Hu SCLC 8 40 w Hu Subakute Kleinhirndegeneration Sensomotorische Neuropathie Subakute Retinadegeneration NSCLC 8 41 m Hu Limbische Enzephalitis SCLC 1 23 m Hu Sensomotorische Neuropathie 9 25 w CV-2 Subakute Kleinhirndegeneration Sensomotorische Neuropathie LK-Met. eines SCLC LK-Met. eines SCLC SCLC: AK PNS Tumor 10,5 NSCLC SCLC Small cellular lung cancer; NSCLC: Non-small cellular lung cancer 48 Intervall PNSIntervall ErstmanifestaTumortion – Tumor- diagnose - PNSdiagnose Erstmanifesta(Monate) tion (Monate) 118 23 2 120 7 27,8 Tabelle 8.2 Überblick über die Patienten ohne definitive Tumordiagnose Nr. Geschlecht AK PNS Tumorverdächtige Veränderungen 6 7 9 w m m Subakute sensorische Neuropathie Sensomotorische Neuropathie Hirnstammenzephalitis 12 m CV2 Hu Hu, Amphiphysin Hu 22 m Hu 24 w Hu Subakute Kleinhirndegeneration Subakute sensorische Neuropathie Enzephalomyelitis Sensomotorische Neuropathie Sensomotorische Neuropathie ----PET pos. im Bereich des Ileozoekalpol sowie mediastinaler und hilärer Lymphknoten --- 26 w Yo Subakute Kleinhirndegeneration 27 28 32 m w w Hu Hu Yo CV2 Hirnstammenzephalitis Enzephalomyelitis Subakute Kleinhirndegeneration AUP: auffällige Autopsie mediastinaler LK --V.a. paraaortale Metastasen eines Ovarialoder Blasenkarzinoms/AUP (PET) ----PET pos. im Bereich Thoraxwand und Pleura adenocarcinoma of unknown primary 3.4 Antineuronale Antikörper Bei 35 der 39 Patienten konnte im Verlauf der Erkrankung ein einzelner Antikörper im Serum nachgewiesen werden, drei Patienten zeigten zwei Antikörperreaktivitäten und in einem Fall fanden sich Hinweise auf drei verschiedene Antikörper. Der Nachweis erfolgte in der Regel mittels eines Screening-Verfahrens Immunfluoreszenz) und einem (Western Bestätigungstest Blot mit (Western Cerebellum-Extrakt Blot oder ELISA oder mit rekombinanten Antigenen). Hu-Antikörper ließen sich in insgesamt 30 Fällen eruieren, davon zweimal in Kombination mit einem weiteren Antikörper (CV2; Amphiphysin) und einmal zusammen mit zwei anderen Reaktivitäten (Ri, Amphiphysin). Nur bei einem dieser drei Patienten (Nr.9) war Hu jedoch der „Hauptantikörper“ mit der höchsten Reaktivität in den Untersuchungen (siehe Tabelle 10). Ri-Antikörper fanden sich bei drei Patienten, zweimal isoliert und einmal zusammen mit anti-Hu und –Amphiphysin. Bei letzterer Patientin wurde anti-Ri mit der stärksten Reaktivität als Hauptantikörper angegeben. Yo-Antikörper traten in drei Fällen auf, zweimal alleine und einmal in Kombination mit anti-CV2, wobei dieser Antikörper im Vergleich zu anti-Yo nur eine schwache Reaktivität zeigte. 49 CV2-Antikörper fanden sich bei insgesamt 5 Patienten, davon zweimal zusammen mit jeweils einem weiteren Antikörper (Hu und Yo). Bei Patientin Nr.32 war anti-CV2 neben anti-Yo jedoch nur schwach reaktiv. Anti-Amphiphysin war in drei Fällen nachweisbar, dabei einmal isoliert, einmal in Kombination mit anti-Hu und bei einem weiteren Patienten mit Hu- und RiAntikörpern. In beiden Fällen war die Reaktivität von anti-Amphiphysin geringer als die der anderen Antikörper. Tabelle 9 veranschaulicht die Häufigkeiten der einzelnen Antikörpern, Tabelle 10 nochmals genauer die Verteilung unter Beachtung von Mehrfachauftreten bei einem Patienten. Tabelle 9 Häufigkeiten der einzelnen Antikörper Antikörper Hu Ri Yo CV2 Amphiphysin Häufigkeit 30 3 3 5 3 Prozent 68 7 7 11 7 14:16 2:1 3:0 4:1 1:2 Geschlecht (w:m) Tabelle 10 Isolierte Antikörper und Kombinationen Patienten-Nr. Antikörper Häufigkeit Geschlecht (w:m) 1 Antikörper Hu 27 13:14 Ri 2 1:1 Yo 2 2:0 CV2 3 3:0 Amphiphysin 1 0:1 2 Antikörper 20 CV2 +Hu 1 0:1 9 Hu+Amphiphysin 1 0:1 32 Yo+CV2 1 1:0 1 1:0 3 Antikörper 5 Ri+Hu+Amphiphysin 50 3.4.1 Antikörper und neurologische Syndrome Hu-Antikörper fanden sich bei 39 paraneoplastischen Syndromen, davon in 13 Fällen bei einer sensomotorischen Neuropathie, in fünf bei einer sensorischen Neuropathie und jeweils einmal bei einer autonomen Neuropathie und einer chronischen Polyradikulitis. Bei zentralen Syndromen ließ sich der Antikörper siebenmal bei einer Kleinhirndegeneration, viermal bei einer Enzephalomyelitis, jeweils dreimal bei einer Kleinhirndegeneration und limbischen Enzephalitis und jeweils einmal bei einer Retinadegeneration und einem choreatischen Syndrom nachweisen. Ri-Antikörper fanden sich jeweils einmal bei einer Hirnstammenzephalitis, einer Enzephalomyelitis und einer limbischen Enzephalitis. Yo-Antikörper ließen sich in drei Fällen mit einer subakuten Kleinhirndegenration nachweisen. CV2-Antikörper konnten jeweils einmal bei einer sensomotorischen und subakut sensorischen Neuropathie, dreimal bei einer Kleinhirndegeneration und einmal bei einer Enzephalomyelitis nachgewiesen werden. Amphiphysin-Antikörper fanden sich einmal bei einem Patienten mit einer sensorischen Neuropathie, zweimal bei einer Hirnstammenzephalitis und einmal bei einer Enzephalomyelitis. Tabelle 11 Vorkommen der verschiedenen Antikörper bei den einzelnen Syndromen (berücksichtigt auch Zweitantikörper und das Zusammentreffen mehrerer Syndrome bei einem Patienten) Hu Ri Yo CV2 Amphiphysin Peripher Sensomotorische Neuropathie 13 1 Sensorische Neuropathie 5 1 Autonome Neuropathie 1 Chronische Polyradikulitis 1 1 Zentral Subakute Kleinhirndegeneration 7 Hirnstammenzephalitis 3 1 Enzephalomyelitis 4 1 Limbische Enzephalitis 3 1 Subakute Retinadegeneration 1 Choreatisches Syndrom 1 Vorkommen des AK insgesamt 39 3 3 51 3 2 3 1 1 6 4 Wie ersichtlich, führt die Tabelle alle aufgetretenen Verbindungen von Antikörpern und Syndromen auf, berücksichtigt folglich auch jene Patienten mit mehreren Antikörperreaktivitäten und/oder verschiedenen paraneoplastischen Syndromen (siehe Tabelle 6), so dass die Zahl der Antikörper in Verbindung mit einem bestimmten Syndrom die Anzahl der Patienten übersteigt. Tabelle 12 führt nochmals die Fälle von Antikörper-Kombinationen bei einem Patienten in Zusammenhang mit paraneoplastischen Syndrom und Tumor auf. Interessanterweise war gerade bei diesen Patienten ausschließlich das zentrale Nervensystem betroffen. Tabelle 12 Mehrfachantikörper (mit Haupt- und Nebenantikörper) und damit verbundene PNS/Tumoren Patienten-Nr. Geschlecht AK PNS Tumor 5 w 9 m 20 m 32 w Ri Hu Amphiphysin Hu Amphiphysin Enzephalomyelitis Mamma-Ca Hirnstammenzephalitis CV2 Hu Yo CV2 Enzephalomyelitis PET pos. im Bereich Ileozoekalpol sowie mediastinaler und hilärer Lymphknoten SCLC Kleinhirndegeneration PET pos. im Bereich Thoraxwand und Pleura 3.4.2 Antikörper und Tumorerkrankungen Hu-Antikörper fanden sich in 15 Fällen bei einem kleinzelligen Bronchialkarzinom, viermal bei einem nicht-kleinzelligen Bronchialkarzinom, zweimal bei einem Mammakarzinom und jeweils einmal bei Morbus Hodgkin und Metastasen eines SCLC. In sieben Fällen gelang kein sicherer Nachweis eines Tumors. Hierbei ergab sich jedoch bei einem Patienten in der PET und bei einem weiteren in der Autopsie ein dringender Tumorverdacht. Ri-Antikörper konnten einmal bei einem NSCLC und zweimal bei einem Mammakarzinom nachgewiesen werden Yo-Antikörper fanden sich nur einmal zusammen mit einem Ovarialkarzinom, bei den beiden anderen Patientinnen blieb die Suche nach einem Primärtumor ohne Erfolg. Es zeigten sich jedoch mittels PET zumindest Hinweise auf eine Tumorerkrankung, in einem Fall bei zusätzlicher positiver Routinediagnostik (siehe Tabelle 16). 52 CV2-Antikörper konnten jeweils einmal bei einem SCLC, einem NSCLC und Metastasen eines SCLC nachgewiesen werden. Auch hier fand sich bei zwei Patienten kein Primärtumor, bei einer Patientin jedoch eine auffällige PET im Bereich Thoraxwand/Pleura. Amphiphysin-Antikörper traten einmal zusammen mit einem kleinzelligen Bronchialkarzinom und einmal mit einem Mammakarzinom auf. Bei einem Patienten (mit zus. Hu-AK) blieb die Suche nach einem Karzinom erfolglos. Die PETUntersuchung zeigte hier jedoch Signalanreicherungen am Ileozoekalpol und im Bereich mediastinaler sowie hilärer Lymphknoten. Tabelle 13 Antikörper und Assoziation mit verschiedenen Tumorerkrankungen (unter Berücksichtigung von AKKombinationen bei vier Patienten) Hu SCLC NSCLC Mamma -Ca 15 4 2 1 2 Ri Yo Ovarial Morbus -Ca Hodgkin 1 Metast. eines SCLC 1 1 Amphiphysin 1 2 Kein Tumornachweis 5 Total 30 3 1 CV2 Tumorverdächtige Befunde bei unkl. Primarius 2 1 1 1 1 1 3 1 5 3 Das Intervall zwischen Erstmanifestation des PNS und Tumordiagnose war mit durchschnittlich 26,8 Monaten bei anti-CV2-Nachweis größer als bei anti-Hu (18,5 Monate), anti-Ri (12 Monate) und anti-Amphiphysin (10,5 Monate). Im Fall der Patientin mit anti-Yo und Ovarialkarzinom ging die Tumordiagnose dem Krankheitsbeginn hingegen um 23 Monate voraus. 53 3.5 Ergebnisse der Liquoruntersuchung Untersucht wurden Zellzahl (pro µl), Gesamteiweiß (mg/l) und als Hinweis auf eine intrathekale Immunglobulinsynthese das Vorliegen oligoklonaler Banden, der IgG-Index, und Anzeichen einer lokalen IgG, -A und/oder IgM-Synthese mittels des Reiber-Schema. In insgesamt 36 Fällen waren Ergebnisse aus einer Liquoruntersuchung vorliegend: Bei 15 Patienten (41,7%) war die Zellzahl erhöht (>5/mm³), der Mittelwert lag bei 22,5/mm³ Zellen (6-64/ mm³). Ein Gesamteiweiß von mehr als 450 mg/l fand sich in 21 Fällen (58,3%) mit im Mittel 850,9 mg/l (473-2060 mg/l). In neun von 36 Liquorbefunden war der IgG-Index nicht bestimmt worden bzw. war nicht zu eruieren, bei einem Patienten (3,7%) lag dieser unter 0,4, bei 17 Patienten (63%) zwischen 0,4 und 0,8 und in neun Fällen (33,3%) bei 0,8 oder darüber. Anhand der Aktenlage war der Befund der oligoklonalen Banden in 34 von 36 Fällen (94,4%) verfügbar. Hierbei wiesen 19 Patienten (55,9%) Banden isoliert im Liquor auf, zwei Patienten (5,9%, Hu und Yo) zeigten sowohl im Liquor als auch im Serum oligoklonale Banden (Nr.21, Yo [8 Banden im Liquor, 6 identische Banden in Serum und Liquor], Nr.31, Hu [Banden im Serum > Banden im Liquor]), während in den übrigen 13 Fällen (38,2%) keine oligoklonalen Banden nachweisbar waren. Bei neun Patienten fanden sich zusätzlich zu oligoklonalen Banden im Reiber-Schema quantitative Hinweise auf eine lokale Immunglobulinsynthese. Hierbei lag in allen Fällen eine autochthone IgG-Synthese mit im Mittel 48% und in zwei Fällen zusätzlich eine lokale IgASynthese mit durchschnittlich 25,5% vor. 3.5.1 Liquorergebnisse und neurologische Syndrome Bei den 15 Patienten mit Erhöhung der Zellzahl handelte es sich ausschließlich um Patienten mit Affektion des ZNS, in acht Fällen isoliert, in sieben Fällen mit Beteiligung sowohl zentraler als auch peripherer Strukturen. Bei Betrachtung der mit dieser Erhöhung verbundenen neurologischen Syndrome ergab sich weder eine spezielle Häufung eines bestimmten PNS noch andere Auffälligkeiten. Auch in Bezug auf die Erhöhung des Gesamteiweiß zeigte sich eine Prädominanz bei Syndromen mit Beteiligung des ZNS, ein Wert von über 450 mg/l fand sich hierbei bei 15 Patienten (71,4%). In sechs weiteren Fällen (28,6) war jedoch ausschließlich das periphere 54 Nervensystem betroffen (dreimal sensomotorische Neuropathie, dreimal sensorische Neuropathie). Bei den 21 Patienten mit Nachweis von oligoklonalen Banden im Liquor war in 20 Fällen (58,8%) von einer intrathekalen Immunglobulin-Synthese auszugehen. Da sich bei Patient Nr.31 (anti-Hu, sensomotorische Neuropathie) ein nahezu identisches Muster der Banden in Liquor und Serum mit mehr Banden im Serum fand, lag hier keine intrathekale Synthese vor. Bei 16 der 20 Patienten (80%) mit sicherer intrathekaler Antikörperproduktion konnte eine Involvierung des zentralen Nervensystems beobachtet werden, elfmal in isolierter Form und fünfmal in Kombination mit Syndromen des peripheren Nervensystems. Bei Patientin Nr.21 mit Yo-Antikörpern und einer Kleinhirndegeneration fanden sich zusätzlich oligoklonale Banden im Serum, jedoch in geringerer Anzahl als im Liquor. Eine isolierte periphere Symptomatik fand sich bei vier Patienten (20%). Waren keine oligoklonalen Banden im Liquor detektierbar bzw. zeigten sich identische Banden in Liquor und Serum, so war die neurologische Symptomatik in sechs von 14 Fällen (42,9%) rein auf das periphere Nervensystem beschränkt, bei drei Patienten (21,4%) fand sich zusätzlich eine zentrale Symptomatik und bei den restlichen fünf Patienten (35,7%) war ausschließlich das zentrale Nervensystem betroffen. Zusammenfassend fanden sich Anzeichen einer intrathekalen Immunglobulinsynthese in Fällen mit ausschließlicher Affektion des ZNS bei 68,8% (11/16), bei zusätzlicher Beteiligung des peripheren Nervensystems bei 62,5% (5/8) und bei ausschließlicher peripherer Symptomatik bei 40% (4/10). Wie zu erwarten, handelte es sich bei den neun Syndromen mit begleitender starker Erhöhung des IgG-Index auf 0,8 oder höher ausschließlich um Fälle mit Beteiligung des ZNS, hierbei war bei sechs Patienten (66,7%) isoliert das ZNS betroffen, bei drei Patienten (33,3%) zusätzlich das periphere Nervensystem. War ein Reiber-Schema verfügbar, zeigte dieses immer Hinweise auf eine intrathekale IgG-Synthese, in zwei Fällen (subakute Retinadegeneration, Kleinhirndegeneration + sensomotorische Neuropathie) auch Anzeichen für eine lokale IgA-Synthese. Bei dem einzigen Patienten mit einem IgG-G-Index von unter 0,4 (Patient Nr.9, Hirnstammenzephalitis) fanden sich weder oligoklonale Banden noch war das Reiber-Schema auffällig. Tabelle 14 bietet einen Überblick über alle Patienten mit Nachweis von oligoklonalen Banden in Liquor und/oder Serum. 55 Tabelle 14 PNS und Antikörper bei Nachweis von oligoklonalen Banden Nr. OKB IgGIndex lokale IgSynthese (%) IgA - AK PNS 3 Liquor + Serum - 0,61 IgG 0 zentral 5 + - 1,12 35,5 0 6 + - 0,66 0 - Ri,Hu,Am Enzephalomyelitis phiphysin CV2 8 15 + + - 0,45 0 - Hu Hu Limbische Enzephalitis 16 + - - - - Hu Subakute Kleinhirndegeneration 19 + - 0,8 - - Hu Limbische Enzephalitis 20 21 22 + + + + - 1,6 3,74 - 52,2 80 - 0 0 - CV2, Hu Yo Hu Enzephalomyelitis Subakute Kleinhirndegeneration Enzephalomyelitis 23 + - - - - Hu 25 + - 2,3 68 31 CV2 Subakute Kleinhirndegeneration 26 27 30 + + + - 0,7 0,47 1,4 0 0 47 0 0 Yo Hu Hu Subakute Kleinhirndegeneration Hirnstammenzephalitis Hirnstammenzephalitis 31 + + 0,58 0 - Hu 32 33 35 40 + + + + - 0,81 1,0 0,63 0,87 68 30 0 30 0 0 20 Yo, CV2 Ri CV2 Hu Subakute Kleinhirndegeneration Hirnstammenzephalitis Subakute Kleinhirndegeneration Subakute Retinadegeneration 41 + - 0,7 21 0 Hu Limbische Enzephalitis Hu peripher Sensomotorische Neuropathie Subakute sensorische Neuropathie Sensomotorische Neuropathie Sensomotorische Neuropathie Sensomotorische Neuropathie Sensomotorische Neuropathie Sensomotorische Neuropathie Sensomotorische Neuropathie Sensomotorische Neuropathie Sensomotorische Neuropathie 3.5.2 Liquorergebnisse und Antikörper Hu-Antikörper fanden sich zusammen mit verschiedensten Liquorbefunden, von den 21 Patienten mit positiven oligoklonalen Banden konnten in 14 Fällen (66,6%) Hu-Antikörper nachgewiesen werden (siehe Tabelle 14). Besonders auffällig war außerdem die Tatsache, dass es sich bis auf eine Ausnahme (Nr.11, Anti-Ri) bei den Fällen ohne Nachweis von oligoklonalen Banden um Hu-Patienten handelte. Von diesen zwölf Patienten litten vier (33,3%) an einer isolierten zentralen Symptomatik, fünf (41,7%) an einem Syndrom des peripheren Nervensystems und in drei (25%) Fällen waren sowohl zentrales als auch peripheres Nervensystem betroffen. Angesichts der im Vergleich zu anti-Hu geringen Prävalenz der anderen Antikörper lassen sich an dieser Stelle im Bezug auf die Liquorergebnisse keine verwertbaren Aussagen treffen. 56 3.6 Weitere Diagnostik 3.6.1 MRT des Schädels In 31 Fällen lagen uns Ergebnisse einer MR-Bildgebung des Schädels vor. Davon fanden sich bei 20 Patienten pathologische Befunde. Bei acht der elf Patienten mit hyper- bzw. hypointensen Läsionen handelte es sich in erster Linie um eher unspezifische zerebrale Veränderungen, nur bei drei Patienten konnte eine Assoziation zum neurologischen Syndrom hergestellt werden: Patientin Nr.8 (Hu, limbische Enzephalitis) zeigte eine deutliche Hyperintensität im Bereich des medialen Temporallappens links und der Inselrinde links, Patientin Nr.28 (Hu, schwere Enzephalomyelitis) wies vor allem subkortikal bis kortikal gelegene, disseminierte Hyperintensitäten auf und Patient Nr.34 (Hu, Choreatisches Syndrom) zeigte eine symmetrische Hyperintensität im Bereich des Nucleus caudatus als Zeichen einer möglichen Stammganglienaffektion. Eine Atrophie zerebraler Strukturen fand sich bei insgesamt acht Patienten. Sechs Patienten zeigten eine zerebelläre Atrophie, in fünf Fällen bei assoziierter subakuter Kleinhirndegeneration. Eine Kleinhirnatrophie zeigte außerdem Patientin Nr.30 (Hu, Hirnstammenzephalitis und sensomotorische Neuropathie). Des Weiteren ließ sich eine geringe fronto-temporale Atrophie bei Patient Nr.11 (Ri) mit einer limbischen Enzephalitis nachweisen. Auch Patientin Nr.6 (CV2) zeigte eine temporale Atrophie, jedoch hier ohne klare klinisch-bildmorphologische Korrelation in Bezug auf ihre subakute sensorische Neuropathie. Und zuletzt fand sich bei Patient Nr.41 (Hu) eine fraglich verstrichene Rinden-Mark-Grenze im Bereich des linken Temporalpols, was sich in Einklang mit der klinischen Symptomatik einer limbischen Enzephalitis bringen lässt. Zur Veranschaulichung dieser Befunde dient Tabelle 15, die einen Überblick über die verschiedenen pathologischen Veränderungen im MRT-Schädel mit Korrelation zum neurologischen Befund bietet. 57 Tabelle 15 Pathologische Veränderungen im MRT-Schädel mit Korrelation zum neurologischen Befund Patienten- Geschlecht AK PNS MRT-Befund Nr. Atrophie 10 w Hu 25 w CV2 35 29 30 w m w CV2 Hu Hu 39 m Hu 11 6 m w Ri CV2 Subakute Kleinhirndegeneration Sensorische Neuropathie Subakute Kleinhirndegeneration Sensomotorische Neuropathie Subakute Kleinhirndegeneration Subakute Kleinhirndegeneration Hirnstammenzephalitis Sensomotorische Neuropathie Subakute Kleinhirndegeneration Sensomotorische Neuropathie Limbische Enzephalitis Sensorische Neuropathie 8 w Hu Limbische Enzephalitis 28 w Hu Enzephalomyelitis 34 m Hu Choreatisches Syndrom 41 m Hu Limbische Enzephalitis Kleinhirnatrophie Kleinhirnatrophie Kleinhirnatrophie Atrophie des Kleinhirnwurmes mäßiggradige zerebelläre Atrophie betonte Furchung des Kleinhirnes geringe fronto-temporale Atrophie temporal betonte Atrophie Hypo-/Hyperintense Läsionen deutliche Hyperintensität im medialen Temporallappen links und Inselrinde links multiple im Marklager/subkortikal bis kortikal liegende Hyperintensitäten symm. Hyperintensität im Bereich des Ncl. caudatus Andere fraglich verstrichene Rinden-MarkGrenze linker Temporalpol 3.6.2 MRT der Wirbelsäule In 17 Fällen lagen Befunde einer Untersuchung der Wirbelsäule vor, die jedoch bis auf drei Ausnahmen unauffällig waren. Hierbei waren bei zwei von drei Patienten die bildmorphologischen Befunde vereinbar mit dem klinischen paraneoplastischen Syndrom. Patient Nr.20 (Hu+CV2; Enzephalomyelitis) hatte ein fragliches hyperintenses Myelopathiesignal in Höhe BWK 2-4, bei Patient Nr.23 (Hu, sensomotorische Neuropathie) fand man diskogene und spondylogene Raumforderungen im Bereich der mittleren HWS und bei Patientin Nr.33 (Ri, Hirnstammenzephalitis) zeigte sich ein fragliches Myelopathiesignal in Höhe HWK 3/4. 3.6.3 Tumorsuche mittels PET Die Resultate aus den PET-Untersuchungen wurden bereits unter 3.3 (zugrundeliegende Malignome) genauer dargestellt. 58 Von den zehn vorliegenden Untersuchungsergebnissen fanden sich in acht Fällen pathologische Befunde, wobei bei zwei dieser Patienten (Nr.9, Anreicherung am Ileozoekalpol und mediast./hilären LK; Nr.32, Anreicherung im Bereich Thoraxwand/Pleura) die initiale Routinediagnostik zur Tumorsuche ohne Befund geblieben war. Tabelle 16 dient zur Veranschaulichung der uns vorliegenden Untersuchungsergebnisse. Tabelle 16 Ergebnisse der PET-Untersuchungen Nr. Geschlecht AK PNS Tumor 6 w CV2 Subakut sensorische Neuropathie 9 m Hu, Amphiphysin Hirnstammenzephalitis 13 m 25 w Amphiphysin CV2 26 w Yo 27 m Hu 30 w Hu 31 32 m w Hu Yo, CV2 35 w CV2 Hirnstammenzephalitis Sensorische Neuropathie Subakute Kleinhirndegeneration LK-Metastasen Sensomotorische Neuropathie eines SCLC Subakute Kleinhirndegeneration Metastasen eines Adeno-Ca. bei unkl. Primarius (pos. Routine) Hirnstammenzephalitis Kein Tumorhinweis Hirnstammenzephalitis NSCLC Sensomotorische Neuropathie Sensomotorische Neuropathie SCLC Subakute Kleinhirndegeneration Anreicherung im Bereich Thoraxwand/Pleura Subakute Kleinhirndegeneration NSCLC 59 Kein Tumorhinweis Anreicherung am Ileozoekalpol und mediast./hilären LK SCLC PET-Befund positiv positiv bei negativer Routinediagnostik - - + + + - + - + - - - + - + + + + - 3.7 Therapie des Malignoms In 29 Fällen (davon 28 Patienten mit gesichertem Karzinom bzw. Metastasen eines SCLC) kam eine Tumortherapie in Form von Resektion, Chemotherapie oder Bestrahlung zum Einsatz. Patientin Nr.26 (Metastasen eines Adenokarzinoms bei unklarem Primärtumor) wurde bei bestehendem Verdacht auf ein Ovarialkarzinom mit Carboplatin/Taxol behandelt. Zehn Patienten erhielten keine Tumortherapie. Bei vierzehn Patienten setzte man auf eine Monotherapie, zwölf Patienten wurden mit zwei Methoden therapiert und in drei Fällen kamen gegen den Tumor OP, Chemotherapie und Radiatio zum Einsatz. Elf Patienten wurden operiert (3 SCLC, 4 NSCLC, 3 Mamma-Ca, 1 Ovarial-Ca), 22 Patienten erhielten eine Chemotherapie (14 SCLC, 1 NSCLC, 2 Mamma-Ca, 1 Ovarial-Ca, 1 Morbus Hodgkin, 2 bei Metastasen eines SCLC, 1 bei Metastasen eines Adeno-Ca bei unklarem Primärtumor [Nr.26]), und in drei Fällen setzte man zusätzlich zu Operation und Chemotherapie eine Bestrahlung ein (1 SCLC, 2 Mamma-Ca). Acht der zehn zum Zeitpunkt des Studienabschlusses noch lebenden Patienten hatten im Verlauf der Erkrankung eine Tumortherapie erhalten. Hierbei konnte in sechs Fällen eine komplette, bei einer Patientin eine partielle Remission beobachtet werden. Von den 29 Patienten, die eine Tumortherapie erhalten hatten, waren bei Ende des Follow up 20 Patienten verstorben. Doch auch hier war in zwei Fällen eine komplette Remission des Karzinoms (Nr.18+21), in einem Fall eine komplette Remission mit darauffolgendem Rezidiv (Nr. 39) beschrieben worden. In drei Fällen (Nr.7, 16 und 27) konnte nicht eruiert werden, ob die Patienten zum Zeitpunkt des Studienendes noch lebten. In einem Fall (Nr.16) war der zu Grunde liegende Tumor behandelt worden und den Arztbriefen war eine komplette Remission des Karzinoms zu entnehmen. Tabelle 17 bietet einen Überblick über diejenigen Patienten, bei denen ein Ansprechen des Karzinoms auf die Tumortherapie zu beobachten war und zeigt ferner, ob sich im Zuge der Tumorregression eine Besserung der neurologischen Symptomatik einstellte. 60 Tabelle 17 Einfluss auf das PNS bei Ansprechen des Tumors auf eine Therapie Nr. 4 Geschlecht m 5 w 20 m 25 AK Hu PNS Sensomotorische Neuropathie Enzephalomyelitis Tumor NSCLC Ri,Hu, Amphiphysin CV2 Enzephalomyelitis Hu Mamma-Ca w CV2 29 m Hu Kleinhirndegeneration Sensomotorische Neuropathie Kleinhirndegeneration LKMetastasen eines SCLC M. Hodgkin 36 w Hu Sensorische Neuropathie SCLC 37 w Hu Sensorische Neuropathie SCLC 18 w Hu SCLC 21 w Yo Kleinhirndegeneration Sensorische Neuropathie Kleinhirndegeneration 39 m Hu SCLC 16 m Hu Kleinhirndegeneration Sensomotorische Neuropathie Kleinhirndegeneration Sensomotorische Neuropathie SCLC Ovarial-Ca NSCLC Tumortherapie Status Resektion Radiatio Resektion Chemotherapie Radiatio Resektion Chemotherapie Radiatio Chemotherapie Radiatio CR neurologische Symptomatik Transientes Ansprechen CR Stabilisierung CR Stabilisierung CR Transientes Ansprechen Chemotherapie Radiatio Resektion Chemotherapie Chemotherapie Radiatio Chemotherapie Radiatio Resektion Chemotherapie Resektion Chemotherapie CR Stabilisierung CR Stabilisierung PR Kein Ansprechen CR Kein Ansprechen CR Kein Ansprechen Resektion CR CR mit Transientes Ansprechen Rezidiv Kein Ansprechen CR: Complete remission; PR: Partial remission Zusätzlich erhielten viele dieser Patienten eine Therapie ihrer neurologischen Symptomatik in Form von Immunsuppression, Steroiden, Plasmapherese und/oder Immunglobulinen. Abschnitt 3.8 beleuchtet genauer, welcher Form diese Therapie war und in welchen Fällen sich eine Verbesserung der neurologischen Symptomatik einstellte. Die Unterscheidung, ob dieses Ansprechen primär der Therapie des Tumors oder aber auch der Modulation des Autoimmunprozesses zuzuschreiben ist, war jedoch nicht immer uneingeschränkt möglich. 61 3.8 Behandlung des PNS In 21 Fällen kam eine immunmodulatorische Therapie zum Einsatz. Bei zehn Patienten wurde die neurologische Symptomatik mittels Monotherapie, bei sechs Patienten mit zwei verschiedenen Ansätzen und in fünf Fällen mit drei verschiedenen Methoden behandelt. Zum Einsatz immunsuppressiver Medikamente kam es bei zehn Patienten, 19-mal wurde mit Steroiden behandelt. Der Versuch einer Behandlung mittels Plasmapherese wurde in nur zwei Fällen gemacht und auch dann immer in Kombination mit Immunsuppression und Steroiden. Immunglobuline kamen bei acht Patienten zum Einsatz und wurden ausnahmslos ergänzend zu Immunsuppression und/oder Steroiden eingesetzt. Leider konnte bei keinem der 39 Patienten eine deutliche und dauerhafte Verbesserung der neurologischen Symptomatik erzielt werden, sei es durch eine Tumortherapie oder durch die Immunmodulation. Es konnte allenfalls eine Stabilisierung auf hohem Niveau beobachtet werden. Tabelle 18 beleuchtet all jene Patienten, bei denen ein Ansprechen im Rahmen der Therapie erkennbar war und welcher Form diese Behandlung war. In den meisten Fällen war vor allem dann eine positive Veränderung zu verzeichnen, wenn das Karzinom (erfolgreich) behandelt wurde, aber auch im Zuge einer alleinigen immunmodulatorischen Therapie zeigte sich bei einer Patientin, bei der die Tumorsuche erfolglos geblieben war (Nr.24), eine Stabilisierung der neurologischen Symptomatik. Bei insgesamt 17 Patienten war der Einfluss einer Therapie (Tumor- und/oder PNSBehandlung) auf das neurologische Syndrom zu erkennen, davon zeigten elf ein partielles/transientes Ansprechen und sechs Patienten eine Stabilisierung auf unterschiedlichem Niveau. Tabelle 18 bietet einen Überblick über diese Fälle und macht den Einfluss von Tumortherapie und Immunmodulation deutlich. 62 Tabelle 18 Fälle mit Ansprechen der neurologischen Symptomatik in Verbindung mit der Behandlung Nr. 3 GeAK schlecht w Hu 4 m Hu 5 w 15 w Ri,Hu,Amphiphysin Hu 17 w Hu 20 m CV2 PNS Tumortherapie + Sensomotorische Neuropathie Sensomotorische Neuropathie Enzephalomyelitis + + Sensomotorische Neuropathie Sensomotorische Neuropathie + Enzephalomyelitis Sensomotorische Neuropathie Sensomotorische Neuropathie Immunmodulation Immunsuppression Steroide Steroide Immunglobuline Steroide Ansprechen PNS Transientes Ansprechen Transientes Ansprechen Stabilisierung Transientes Ansprechen Transientes Ansprechen + Steroide Immunglobuline Immunsuppression Steroide Plasmapherese keine + Steroide Transientes Ansprechen Stabilisierung + Stabilisierung Hu 23 m Hu 24 w Hu 25 w CV2 29 31 m m Hu Hu 32 w 33 w Yo CV2 Ri 34 m 35 36 39 w w m kein Tumor + Immunsuppression Steroide Plasmapherese Immunsuppression Steroide Immunglobuline keine keine Kleinhirndegeneration Sensomotorische Neuropathie Kleinhirndegeneration Sensomotorische Neuropathie Kleinhirndegeneration kein Tumor Hirnstammenzephalitis + Hu Choreatisches Syndrom + Immunsuppression Steroide Immunglobuline Steroide CV2 Hu Hu Kleinhirndegeneration Sensorische Neuropathie Kleinhirndegeneration Sensomotorische Neuropathie + + + Steroide keine keine + + Steroide Transientes Ansprechen Stabilisierung Transientes Ansprechen Transientes Ansprechen Transientes Ansprechen Transientes Ansprechen Stabilisierung Stabilisierung Transientes Ansprechen 3.9 Follow up Von allen 39 Patienten konnten Follow up-Daten gesammelt und ausgewertet werden. Die Gesamtdauer des Follow up ab der Erstmanifestation des PNS betrug im Mittel 42,5 Monate (1,8-194,4 Monate). Das Follow up der überlebenden Patienten lag im Durchschnitt bei 83,4 Monaten (12,8-194,4 Monate), das der Verstorbenen bei 25 Monaten (1,8-133,2 Monate). 26 Patienten waren zum Zeitpunkt des Studienendes verstorben, zehn Patienten am Leben und in drei Fällen konnte nicht eruiert werden, ob und wie lange die Patienten überlebt hatten. 63 Bei den 26 verstorbenen Patienten lag die Todesursache in drei Fällen bei den Folgen der neurologischen Erkrankung (Nr.17: Hu+sensomotorische Neuropathie; Nr.21: Yo+subakute Kleinhirndegeneration; Nr.22:Hu+Enzephalomyelitis/sensomotorische Neuropathie), in elf Fällen war der Tumor selbst nicht mit dem Leben vereinbar, wobei es sich hierbei ausschließlich um Bronchialkarzinome handelte. In zwölf Fällen konnte die Todesursache nicht eruiert werden. In nur zwei Fällen (Nr.22 + 41) wurde eine Autopsie durchgeführt, wobei nur bei einem Patienten der Zugang zum Autopsiebericht möglich war. Beim Hu-Patienten Nr.22 mit einer Enzephalomyelitis und einer sensomotorischen Neuropathie konnte durch die Autopsie ein mit malignen Zellen infiltrierter mediastinaler Lymphknoten nachgewiesen werden, ein Hinweis auf einen Primärtumor ergab sich jedoch nicht, nachdem die Suche schon zu Lebzeiten ohne Erfolg geblieben war. Der Patient verstarb infolge seiner neurologischen Erkrankung. Tabelle 19 veranschaulicht die Zusammenhänge zwischen Todesursache, Antikörper, Karzinom und paraneoplastischem Syndrom. Tabelle 19 Todesursache (soweit bekannt) in Assoziation mit Antikörper, PNS und Tumor Nr. Geschlecht AK PNS Tumor Todesursache 3 8 w w Hu Hu Sensomotorische Neuropathie Limbische Enzephalitis SCLC SCLC Malignom Malignom 10 w Hu SCLC Malignom 14 m Hu SCLC Malignom 15 w Hu Subakute Kleinhirndegeneration Subakute sensorische Neuropathie Chronische Polyradikulitis Autonome Neuropathie Sensomotorische Neuropathie SCLC Malignom 17 w Hu Sensomotorische Neuropathie SCLC neurologisch 18 w Hu SCLC Malignom 21 w Yo Subakute Kleinhirndegeneration Subakute sensorische Neuropathie Subakute Kleinhirndegeneration Ovarial-Ca neurologisch 22 m Hu kein Tumor neurologisch 23 m Hu Enzephalomyelitis Sensomotorische Neuropathie Sensomotorische Neuropathie Malignom 30 w Hu LK-Metastasen eines SCLC NSCLC 34 m Hu SCLC Malignom 39 m Hu SCLC Malignom 41 m Hu SCLC Malignom Hirnstammenzephalitis Sensomotorische Neuropathie Choreatisches Syndrom Subakute Kleinhirndegeneration Sensomotorische Neuropathie Limbische Enzephalitis 64 Malignom Die Überlebenszeit nach Erstmanifestation des PNS war wie bereits erwähnt bei 36 Patienten bekannt und betrug bei Ende des Follow up durchschnittlich 46,5 Monate (1,8-196,8 Monate). Die Überlebenszeit der noch Lebenden lag dabei im Mittel bei 85,2 Monaten (5,5-196,8), die der Verstorbenen bei 31,6 Monaten (1,8-133,2 Monate). Kein Ansprechen des neurologischen Syndroms auf eine Therapie zeigte sich in 22 Fällen, bei elf Patienten stellte sich eine vorübergehende Besserung ein und bei sechs Patienten stabilisierte sich die Symptomatik auf unterschiedlichem Niveau (s. Tabelle 18). Die nachfolgenden Ausführungen umfassen eine Überlebenszeit-Analyse der 36 Patienten, bei denen Tod oder Überleben bei Studienabschluss bekannt war, abhängig von PNS, AK, zu Grunde liegendem Tumor, Geschlecht und Alter zum Zeitpunkt der Erstmanifestation. Abbildung 2 zeigt als Überblick eine Kaplan-Meier-Kurve dieser 36 Patienten, welche auf die Überlebenszeit ab dem Zeitpunkt der Erstmanifestation der neurologischen Symptomatik Bezug nimmt. Abbildung 2 Kaplan-Meier-Analyse 65 3.9.1 Überlebenszeiten abhängig vom PNS Die mittlere Überlebenszeit der zwölf Patienten mit einer (isolierten oder kombinierten) sensomotorischen Neuropathie betrug 52 Monate (4,3-196,8 Monate), neun Patienten waren bei Studienabschluss verstorben. Die mittlere Überlebenszeit der sieben Patienten mit einer subakuten sensorischen Neuropathie betrug 50,6 Monate (5,5-133,2 Monate), vier Patienten waren bei Studienabschluss verstorben, wobei es sich hierbei ausnahmslos um diejenigen Fälle handelte, bei denen zusätzlich eine zentrale Symptomatik vorherrschend war (Hirnstammenzephalitis, Kleinhirndegeneration). Die Überlebenszeit des Patienten mit einer chronischen Polyradikulitis in Kombination mit einer autonomen Neuropathie (Nr.14) betrug 14,4 Monate, bei Abschluss der Studie war er bereits verstorben. Die mittlere Überlebenszeit der elf Patienten mit einer (isolierten oder kombinierten) subakuten Kleinhirndegeneration betrug 50,9 Monate (4,8-143,4 Monate), acht Patienten waren bei Studienabschluss verstorben. Die mittlere Überlebenszeit der vier Patienten mit einer (isolierten oder kombinierten) Hirnstammenzephalitis betrug 54 Monate (30-92,4 Monate), alle vier Patienten waren bei Studienabschluss verstorben. Die mittlere Überlebenszeit der vier Patienten mit einer (isolierten oder kombinierten) Enzephalomyelitis betrug 51,3 Monate (4,3-157,8 Monate), zwei Patienten waren bei Studienabschluss verstorben. Die mittlere Überlebenszeit der vier Patienten mit einer (isolierten oder kombinierten) limbischen Enzephalitis betrug 28 Monate (1,8-66 Monate), alle vier Patienten waren bei Studienabschluss verstorben. Die Überlebenszeit der Patientin mit einer Retinadegeneration (Nr.40) betrug 12 Monate, die des Patienten mit einem ‚‚choreatischem Syndrom’’ (Nr.34) 21,6 Monate. Beide waren bei Abschluss der Studie verstorben. 3.9.2 Überlebenszeiten abhängig vom antineuronalen Antikörper Die nachfolgende Darstellung berücksichtigt den jeweiligen Hauptantikörper der Patienten. 66 Die mittlere Überlebenszeit der 25 Patienten mit Hu-Antikörpern betrug 45,9 Monate (1,8-196,8 Monate), 20 Patienten waren bei Studienabschluss verstorben. Die mittlere Überlebenszeit der drei Patientinnen mit Yo-Antikörpern betrug 9,4 Monate (4,8-15 Monate), zwei Patientinnen waren bei Studienabschluss verstorben. Die mittlere Überlebenszeit der drei Patienten mit Ri-Antikörpern betrug 46,6 Monate (39,6-60 Monate), zwei Patienten waren bei Studienabschluss verstorben. Die mittlere Überlebenszeit der vier Patienten mit CV2-Antikörpern betrug 81,7 Monate (12,8-157,8 Monate), eine Patientin war bei Studienabschluss verstorben. Die Überlebenszeit des Patienten mit Amphiphysin-Antikörpern betrug 30 Monate, er war bei Studienabschluss verstorben. 3.9.3 Überlebenszeit abhängig vom zu Grunde liegenden Tumor Die mittlere Überlebenszeit der 18 Patienten mit einem SCLC oder Metastasen eines SCLC betrug 49,8 Monate (1,8-157,8 Monate), 14 Patienten waren bei Studienabschluss verstorben. Die mittlere Überlebenszeit der fünf Patienten mit einem NSCLC betrug 28 Monate (16,4-39,6 Monate), vier Patienten waren bei Studienabschluss verstorben. Die mittlere Überlebenszeit der drei Patientinnen mit einem Mammakarzinom betrug 46,2 Monate (38,4-60 Monate), zwei Patientinnen waren bei Studienabschluss verstorben. Die Überlebenszeit der Patientin mit einem Ovarialkarzinom betrug 4,8 Monate, sie war bei Studienabschluss verstorben. Die Überlebenszeit des Patienten mit einem Morbus Hodgkin betrug 143,4 Monate, er war bei Studienabschluss am Leben. Die mittlere Überlebenszeit der acht Patienten ohne Tumordiagnose betrug 43,8 Monate (3-196,8 Monate), fünf Patienten waren bei Studienabschluss verstorben. 3.9.4 Überlebenszeit abhängig von der Tumorbehandlung Die mittlere Überlebenszeit der acht Patienten, die keine Tumortherapie erhalten hatten, betrug 42,2 Monate (1,8-196,8 Monate), sechs Patienten waren bei Studienabschluss verstorben. 67 Die mittlere Überlebenszeit der zehn Patienten, deren Tumor operiert wurde, betrug 57 Monate (4,8-146 Monate), sechs Patienten waren bei Studienabschluss verstorben. Die mittlere Überlebenszeit der 22 Patienten, die eine Chemotherapie erhielten, betrug 53,7 Monate (4,8-157,8 Monate), 15 Patienten waren bei Studienabschluss verstorben. Die mittlere Überlebenszeit der 13 Patienten, deren Tumor mit einer Bestrahlung behandelt wurde, betrug 57 Monate (5,5-157,8 Monate), acht Patienten waren bei Studienabschluss verstorben. Erhielten die Patienten nur eine Form der Tumorbehandlung, betrug die mittlere Überlebenszeit 33,6 Monate (4,8-133,2 Monate), bei zwei Therapieformern lag sie bei 53,5 Monaten (4,8-143,4 Monate) und setzte man alle drei Methoden ein, betrug die durchschnittliche Überlebenszeit 86 Monate (40,1-157,8 Monate). 3.9.5 Überlebenszeit abhängig vom Geschlecht Bei den 21 Frauen betrug die mittlere Überlebenszeit 47,5 Monate (3-196,8 Monate), vierzehn Patientinnen waren bei Studienabschluss verstorben. Bei den 15 Männern betrug die mittlere Überlebenszeit 45,1 Monate (1,8-157,8 Monate), zwölf Patienten waren bei Studienabschluss verstorben. 3.9.6 Überlebenszeit abhängig vom Alter bei Diagnosestellung des PNS Fünf Patienten waren zum Zeitpunkt der Diagnosestellung des PNS unter 50 Jahre alt, bei einem Durchschnittsalter von 39,5 betrug die mittlere Überlebenszeit 105,4 Monate. Zwei Patienten waren bei Studienabschluss verstorben. 31 Patienten waren zum Zeitpunkt der Diagnosestellung des PNS über 50 Jahre alt, bei einem Durchschnittsalter von 65,3 betrug die mittlere Überlebenszeit 37 Monate. 24 Patienten waren bei Studienabschluss verstorben. a. Ein Patient war zum Zeitpunkt der Diagnosestellung des PNS unter 30 Jahre alt (Nr.29 [Hu/Kleinhirndegeneration/M. Hodgkin); 20,3 Jahre). Seine Überlebenszeit betrug bei Studienabschluss 143,4 Monate, zu diesem Zeitpunkt war er noch am Leben. b. Zwei Patientinnen waren zum Zeitpunkt der Diagnosestellung des PNS zwischen 30 und 40 Jahre alt (Nr.3; Nr.24), das Durchschnittsalter lag bei 39,2 Jahren. Die mittlere 68 Überlebenszeit betrug 108,6 Monate, eine Patientin war bei Studienabschluss verstorben. c. Zwei Patienten waren zum Zeitpunkt der Diagnosestellung des PNS zwischen 40 und 50 Jahre alt (Nr.18; Nr.20), das Durchschnittsalter lag bei 49,6 Jahren. Die mittlere Überlebenszeit betrug 83,1 Monate, ein Patient war bei Studienabschluss verstorben. d. Sechs Patienten waren zum Zeitpunkt der Diagnosestellung des PNS zwischen 50 und 60 Jahre alt, das Durchschnittsalter lag bei 55,5 Jahren. Die mittlere Überlebenszeit betrug 36,7 Monate, vier Patienten waren bei Studienabschluss verstorben. e. Siebzehn Patienten waren zum Zeitpunkt der Diagnosestellung des PNS zwischen 60 und 70 Jahre alt, das Durchschnittsalter lag bei 65,2 Jahren. Die mittlere Überlebenszeit betrug 34 Monate, vierzehn Patienten waren bei Studienabschluss verstorben. f. Acht Patienten waren zum Zeitpunkt der Diagnosestellung des PNS über 70 Jahre alt, das Durchschnittsalter lag bei 72,4 Jahren. Die mittlere Überlebenszeit betrug 47,8 Monate, sechs Patienten waren bei Studienabschluss verstorben. 3.10 Fallvorstellung der langzeitüberlebenden Patienten Acht der 39 Patienten überlebten die paraneoplastische neurologische Erkrankung mit oder ohne Diagnose eines Primärtumors länger als fünf Jahre ab dem Zeitpunkt der Diagnosestellung des PNS. Davon waren bei Beendigung des Follow up noch fünf am Leben. In vier Fällen (Nr.20, 24, 25 und 36) konnten wir im Rahmen einer Nachuntersuchung eine erneute Serumprobe gewinnen. Hierbei fanden sich bei allen vier Patienten unverändert die gleichen Antikörper im Serum. Die durchschnittliche Dauer zwischen erstmaligem Antikörpernachweis und aktueller Kontrolle lag bei 123 Monaten [35-171 Monate]. Dieser teils sehr lange Zeitraum spricht folglich für eine langjährige Persistenz antineuronaler Antikörper auch bei klinischer Stabilisierung der Symptomatik bzw. bei Patienten im Stadium der Komplettremission ihres Karzinoms. Bei Patientin Nr.24 konnten bei noch immer ausstehender Tumordiagnose und stabiler neurologischer Symptomatik nach 171 Monaten erneut Hu-Antikörper nachgewiesen werden. Tabelle 20 zeigt eine Übersicht, im Anschluss findet sich eine kurze Einzelfallbeschreibung der acht langzeitüberlebenden Patienten. 69 Tabelle 20 Übersicht über die Langzeitüberlebenden Nr Geschlecht Geburtsdatum 9 m 04.12.43 10 w 13.05.30 19 m 12.10.28 20 m 27.01.49 24 25 w w 21.04.55 11.09.27 29 36 m w 21.07.78 23.02.24 AK PNS Hu, Hirnstammenzephalitis Amphi physin Hu Kleinhirndegeneration Sensorische Neuropathie Hu Limbische Enzephalitis Sensomotorische Neuropathie CV2 Enzephalomyelitis Hu Hu Sensomotorische Neuropathie CV2 Kleinhirndegeneration Sensomotorische Neuropathie Hu Kleinhirndegeneration Hu Sensorische Neuropathie Tumor Status Überlebenszeit seit Erstmanifestation (Monate) kein Tumor verstor ben 92,4 SCLC verstor ben verstor ben CR 133,2 CR 196,8 117,9 CR CR 143,4 146 SCLC SCLC kein Tumor LK-Metastasen eines SCLC M. Hodgkin SCLC 66 157,8 CR: Complete remission 3.10.1 Fall Nr. 20 (CV2+Hu; Enzephalomyelitis; SCLC) Erste Symptome traten bei diesem Patienten 12/1994 im Alter von 46 Jahren auf. Sie zeigten sich in Form einer subakut aufgetretenen Gangunsicherheit mit Progredienz im folgenden Jahr, hinzukommend nun transiente Doppelbilder, eine spastische Paraparese und eine deutliche Pallhypästhesie. Zu diesem Zeitpunkt wurde angesichts einer intrathekalen IgGSynthese bei unauffälligem Schädel-MR die Verdachtsdiagnose einer Enzephalomyelitis disseminata mit spinalem Verlauf gestellt. 09/98 wurde bei dem Raucher nach siebenmonatiger entsprechender Anamnese die Diagnose eines kleinzelligen Bronchialkarzinoms rechts hilär im Stadium der „limited disease“ gestellt, die Gangunsicherheit war bis zu diesem Zeitpunkt unverändert geblieben. In einer retrospektiven Untersuchung einer Serumprobe von 03/1996 und einer aktuellen Probe ließen sich schließlich 10/98 HuD-Antikörper nachweisen. Zusätzlich fanden sich in beiden Proben damals noch nicht identifizierte CV2-Antikörper. Die neurologische Symptomatik war zu diesem Zeitpunkt geprägt durch unscharf begrenzte Hypästhesien der unteren Extremitäten, einer Gangataxie und Fallneigung sowie positiven Babinski-Reflexen vor allem rechts bei normalem Befund der oberen Extremitäten und der Hirnnerven, so dass sie am ehesten dem klinischen Bild einer paraneoplastischen (Enzephalo)Myelitis entsprach. Der Tumor wurde in Folge mit vier Zyklen einer Chemotherapie nach dem VIP-E-Protokoll (Epirubicin, Etoposid, Cisplatin und Ifosfamid), einer Pneumonektomie rechts sowie einer 70 Bestrahlung von Mediastinum und Neurokranium therapiert, eine immunmodulatorische Therapie erhielt der Patient nicht. Seither befindet sich der Patient im Stadium der kompletten Remission, die neurologische Symptomatik hat sich im Wesentlichen stabilisiert, was sich auch in unserer Kontrolluntersuchung 02/2008 bestätigte. Die spastische Paraparese mit spastisch-ataktischem Gangbild sowie Pallhypästhesie zeigten sich nahezu unverändert. Eine erneute Serumkontrolle ergab eine stärkere CV2-Bande in Kombination mit einer schwachen HuD-Bande. 3.10.2 Fall Nr. 24 (Hu; sensomotorische Neuropathie; kein Tumor) Die paraneoplastische Erkrankung äußerte sich bei dieser Patientin erstmals 11/91 im Alter von 36,6 Jahren in Form einer Gangunsicherheit, Schmerzen und Verspannungsgefühlen im Bereich der rechten Schulter und des Rückens, einem Taubheitsgefühl des Daumens und kleinen Fingers rechts sowie der Beine. Die Liquordiagnostik zeigte eine Eiweißerhöhung und den Nachweis oligoklonaler Banden. Im Laufe des Jahres 1992 nahmen Dysästhesien und Unsicherheit in Armen und Beinen zu, hinzu kam eine Kraftminderung der rechten Hand ohne Atrophie. Eine Cortisontherapie Anfang des Jahres zeigte nur vorübergehenden Einfluss. 11/94 wurden mittels Western-Blot Hu-Antikörper in Serum und Liquor nachgewiesen. Eine intensive Tumorsuche blieb jedoch ohne Erfolg. In den Jahren bis 1998 machte man wiederholte Therapieversuche mit SteroidHochdosis-Behandlungen, einer Plasmapherese, und drei Zyklen Immunadsorption in Kombination mit Cyclophosphamid-Gaben. Es zeigte sich keine Besserung der Symptomatik, eine wiederholte Tumorsuche blieb ohne Erfolg. Seit 1998 beschränkte sich die Behandlung der Patientin auf regelmäßige physiotherapeutische Sitzungen und halbjährliche Onko-PET-Untersuchungen. In unserer Untersuchung 02/2008 fanden sich strumpf- und handschuhförmige Hypästhesien und Hypalgesien der Extremitäten sowie eine Pallanästhesie und Herabsetzung des Lagesinns, außerdem ataktische Extremitäten mit ataktischem Gangbild. Die motorische Komponente war zu jenem Zeitpunkt jedoch unauffällig, so dass bei der Patientin eine Ataxie bedingt durch den Verlust sensibler Neurone deutlich im Vordergrund stand. In der Serumkontrolle ließen sich erneut Hu-Antikörper nachweisen. 3.10.3 Fall Nr. 25 (CV2; PKD+sensomotorische Neuropathie; SCLC-Metastasen) 07/98 im Alter von 70,8 Jahren zeigten sich bei dieser Patientin erstmals Symptome einer neurologischen Erkrankung in Form einer Hypästhesie und Hyperalgesie im Bereich der 71 rechten Oberschenkelaußenseite und Leiste. Außerdem beschrieb die Patientin diffus gürtelförmig ausstrahlende Missempfindungen in Höhe Th11/12 und im Bereich des linken Gesäßes. Die Gehfähigkeit war deutlich eingeschränkt, es fanden sich Paresen der Hüftbeuger bds. und eine Schwäche der Oberschenkelabduktion rechts. ASR links und PSR bds. waren nicht auslösbar. Bis 01/2000 zeigte sich die neurologische Symptomatik deutlich progredient. In der Untersuchung fanden sich alle Anzeichen einer sensomotorischen Polyneuropathie mit einer einseitigen Betonung der Symptomatik in Form von Atrophien, Paresen, fehlenden Muskeleigenreflexen und Sensibilitätsstörungen vor allem der oberen Extremität rechts. 02/2000 therapierte man in rascher Folge mit Hochdosis-Steroiden, i.v.-Immunglobulinen sowie einer Cyclophosphamid-Puls-Therapie, was in Kombination mit Rehabilitations– maßnahmen zu einer Stabilisierung und vorübergehenden Besserung der Symptomatik führte. Der Liquor mit einem IgG-Index von 2,3 und dem Nachweis oligoklonaler Banden war hinweisend auf einen entzündlichen Prozess im ZNS. Die Wiederholung der Therapie mit Cyclophosphamid und Immunglobulinen zeigte sich zusammen mit einer intensiven Rehabilitation auch weiterhin als hilfreich. Im Rahmen einer deutlichen Verschlechterung der neurologischen Symptomatik Ende 2000 fanden sich schließlich 11/2000 Lymphknotenmetastasen eines kleinzelligen Karzinoms mediastinal und rechts hilär bei V.a. ein okkultes primäres Bronchialkarzinom. Zu diesem Zeitpunkt ließen sich im Liquor noch keine spezifische AK nachweisen, neben der intrathekalen IgG-Synthese war nun zusätzlich eine IgA-Synthese nachweisbar. Unter einer bis 05/01 durchgeführten kombinierten Bestrahlungs- und Chemotherapie war der Tumor rückläufig, die neurologische Symptomatik zeigte sich jedoch weiter progredient. 03/2005 konnten schließlich CV2-AK in Serum nachgewiesen werden, die Patientin hatte bis zu diesem Zeitpunkt zusätzlich zerebelläre Symptome in Form einer ausgeprägten Ataxie entwickelt. Unsere Untersuchung am 02/2008 bestätigte den progredienten Verlauf der Erkrankung mit ausgeprägten Symptomen einer sensomotorischen Neuropathie und Kleinhirndegeneration trotz erfolgreicher Behandlung der Metastasen bei weiterhin mangelndem Nachweis eines Primärtumors. Anhand einer Serumkontrolle konnten erneut CV2-Antikörper nachgewiesen werden. 72 3.10.4 Fall Nr. 29 (Hu; PKD; Morbus Hodgkin) Die zerebelläre Symptomatik äußerte sich bei diesem Patienten erstmals 07/96 im Alter von 17,8 Jahren in Form einer subakut aufgetretenen Gangunsicherheit, Doppelbildern und Nystagmus, hinzukommend ein starker Tremor vor allem im Stamm- und Kopfbereich. Ein Schädel-MRT von 04/98 zeigte eine deutliche Atrophie des Kleinhirns vor allem im Bereich des Wurmes. 11/98 wurde die Diagnose eines nodulär sklerosierenden Hodgkin-Lymphoms gestellt, es fanden sich multiple Lymphome mediastinal, supraklavikulär, axillär und iliakal bds. sowie im Abdomen, die Suche nach antineuronalen Antikörpern blieb jedoch ohne Erfolg. Ab Anfang 12/98 behandelte man den Patienten mit acht Zyklen einer Chemotherapie nach dem BEACOPP-Schema, es folgten Nachbestrahlungen 04-07/99. In den Kontrolluntersuchungen zeigte sich eine komplette Remission. Die neurologische Symptomatik hatte sich zu diesem Zeitpunkt weitgehend stabilisiert, eine deutliche Verbesserung war jedoch nicht zu verzeichnen. Wann im Verlauf die antineuronalen HuAntikörper erstmals detektiert wurden, war nicht eruierbar. Seit sechs Jahren meidet der Patient jeglichen Arztkontakt, laut Angehörigen sei sein Zustand jedoch stabil und kein Rezidiv des Tumors aufgetreten. Auch eine Nachuntersuchung von unserer Seite lehnte der Patient ab, so dass weder ein aktueller neurologischer Status erhoben noch eine Serumkontrolle durchgeführt werden konnte. 3.10.5 Fall Nr. 36 (Hu, sensorische Neuropathie; SCLC) Bei dieser Patientin zeigten sich 02/96 im Alter von 72 Jahren erstmals Symptome in Form einer rasch progredienten Gangunsicherheit, einer Hypästhesie beider Beine sowie nach proximal aufsteigenden, strumpf- und handschuhförmigen Kribbelparästhesien und Hyperalgesien. Außerdem fanden sich in der Untersuchung Anzeichen einer diskreten beinbetonten Spastik. Bereits zwei Monate später konnten im Rahmen der Diagnostik ein kleinzelliges Bronchialkarzinom im Stadium „limited disease“ und Hu-Antikörper in Serum und Liquor nachgewiesen werden. 6/96 wurde eine Tumorresektion durchgeführt, an die sich mehrere Zyklen einer adjuvanten Chemotherapie mit Epirubicin und Cyclophosphamid anschlossen. Im Rahmen dieser Maßnahmen kam es zu einer Stabilisierung der Symptomatik. Eine immunmodulatorische Therapie wurde nicht durchgeführt. Einer Nachuntersuchung stimmte die Patientin nicht zu, jedoch konnte sie uns mitteilen, dass es zu keinem Tumorrezidiv gekommen sei und sich auch die neurologische Symptomatik 73 weitestgehend stabilisiert habe. Die Kontrolle einer vom Hausarzt vor Ort abgenommenen Blutprobe ergab den erneuten Nachweis von Hu-Antikörpern. 3.10.6 Fall Nr. 9 (Hu+Amphiphysin; Hirnstammenzephalitis; kein Tumor) Erste Symptome äußerten sich in Form von Kribbelparästhesien im Bereich der Extremitäten verbunden mit einem leichten Taubheitsgefühl 05/96 im Alter von 52,4 Jahren. Diese überwiegend sensible Symptomatik zeigte sich im weiteren Verlauf progredient, bis 1998 kam es außerdem zu Paresen und vor allem im Beinbereich zu Faszikulationen der Muskulatur. Bis 1999 traten zu nun deutlichen Atrophien an Beinen und Händen eine Gangund Standataxie sowie Dysmetrie und Dysarthrie hinzu. Zwischenzeitlich stellte man die Verdachtsdiagnose einer ALS bzw. einer funikulären Myelose. Doch auch unter Vitamin B12-Substitution war die Symptomatik progredient, die spastisch-ataktische Gangstörung führte Ende 2002 zur Rollstuhlpflichtigkeit. Außerdem fanden sich neuropsychologische Defizite in Form von Wesensveränderungen, Verwirrtheit und Konzentrationsstörungen. Im MRT zeigten sich Korrelate für eine ausgedehnte Leukenzephalopathie. Am 05.05.2003 konnte man schließlich Hu-Antikörper im Liquor nachweisen. In einer daraufhin durchgeführten PET-Untersuchung zeigten sich Mehranreicherungen im Bereich des Ileozoekalpols sowie der mediastinalen und hilären Lymphknoten, die Suche nach einem definitiven Primärtumor blieb erfolglos. Im Laufe des Jahres 2003 kam es zu einer raschen Verschlechterung der neurologischen Symptomatik, eine Therapie in Form einer Immunmodulation wurde nicht eingesetzt. Der Patient verstarb 01/2004 an den Folgen der schwerwiegenden neurologischen Symptomatik. 3.10.7 Fall Nr. 10 (Hu; PKD+sensorische Neuropathie; SCLC) Bereits 01/86 im Alter von 55,1 Jahren zeigten sich bei dieser Patientin zerebelläre Symptome in Form einer zunehmenden Gangunsicherheit/Ataxie und Dysmetrie. Die Bildgebung zeigte eine diskrete Kleinhirnatrophie, nebenbefundlich fand sich eine zervikale Spinalstenose auf Höhe HWK 3/4 bis 6/7. Eine genauere Tumorsuche wurde nicht durchgeführt. Die Datenlage bis 1996 war leider gering, eine wahrscheinlich einmalig durchgeführte Steroidtherapie zeigte keine Wirkung. Im Jahre 1996 kam es zu einer deutlichen Verschlechterung der zerebellären Symptomatik, zusätzlich fanden sich Symptome einer sensiblen Polyneuropathie. Eine aufgrund von Dyspnoe und thorakalen Schmerzen durchgeführte Tumorsuche ergab 07/96 die Diagnose eines den linken Stammbronchus komprimierenden kleinzelligen Bronchialkarzinomes. Eine kurzfristig eingeleitete Chemotherapie nach dem EPICO74 Protokoll konnte nicht verhindern, dass die Patientin aufgrund der respiratorischen Situation und eines septischen Krankheitsbildes am 30.07.1996 an einem Herz-Kreislaufversagen bei ausgeprägter Tumorkachexie verstarb. 3.10.8 Fall Nr. 19 (Hu; PLE+sensomotorische Neuropathie, SCLC) Ein epileptischer Grand mal-Anfall 09/87 im Alter von 58,9 Jahren war bei diesem Patienten das erste Anzeichen seiner neurologischen Erkrankung. In Folge kam es zu einem zweiten Grand mal-Anfall und zu wiederholten, fast täglich auftretenden fokal-motorischen Anfällen. Zusätzlich zu einer vorwiegend motorischen Aphasie und Störungen des Kurzzeitgedächtnisses entwickelten sich psychotischen Symptome wie Unruhe, Gespanntheit und Angstzustände. Unter Phenytoin und Carbamazepin blieb ein erneuter großer Anfall aus, die fokalen Anfälle persistierten. Im Jahre 1990 entwickelten sich zudem sensible Defizite in Form von Hypästhesien aller vier Extremitäten und Paresen vor allem des linken Armes. 02/92 konnte schließlich ein kleinzelliges Bronchialkarzinom diagnostiziert werden, das im Anschluss mit vier Zyklen einer Chemotherapie therapiert wurde. Ein deutliches Ansprechen sowohl des Tumors als auch der neurologischen Erkrankung auf diese Therapie zeigte sich jedoch nicht. Der Zeitpunkt des Nachweises der Hu-Antikörper konnte nicht eruiert werden. Der Patient verstarb am 20.03.93 nach einer Überlebenszeit von 66 Monaten, die genaue Todesursache blieb unklar. 75 4. Diskussion 4.1 Studiendesign/Vor- und Nachteile In der vorliegenden an der Neurologischen Universitätsklinik Freiburg durchgeführten retrospektiven, monozentrischen, klinisch und epidemiologisch orientierten Studie wurden die Daten von 39 Patienten mit der Diagnose eines PNS und dem Nachweis eines oder mehrerer der sog. gut charakterisierten antineuronalen Antikörpern untersucht. Dabei wurden demographische Daten, das klinische Spektrum der paraneoplastischen neurologischen Syndrome, die assoziierten Tumoren, initial bestimmte antineuronale Antikörper, die Befunde aus Liquordiagnostik und Bildgebung, die Therapieformen, das Ansprechen der PNS und der zu Grunde liegenden Tumoren auf die Behandlung sowie die Überlebenszeit für jeden einzelnen Patienten zusammengestellt und ausgewertet. Paraneoplastische neurologische Syndrome (PNS) sind seltene tumorassoziierte Erkrankungen. Die Prävalenz wird mit nur 0,5-3% der Tumorleiden angegeben (Kaiser, 1999), was sich auch darin widerspiegelt, dass bisher in der Literatur fast ausschließlich klinische Studien an kleineren Fallserien publiziert wurden, meist beschränkt auf eine einzige Tumorentität, einen spezifischen antineuronalen paraneoplastischen Antikörper oder ein bestimmtes paraneoplastisches Syndrom. Außerdem befassen sich nur sehr wenige Arbeiten (Candler et al., 2003, Sillevis Smitt et al., 2002) mit dem klinischen Follow up eines Patientenkollektives. Unser Fokus lag auf der retrospektiven Untersuchung eines Patientenkollektives mit Nachweis eines oder mehrerer der sechs gut charakterisierten antineuronalen Antikörper (antiHu, -Yo, -Ri, -Ma2, -CV2, -Amphiphysin) und der Beschreibung der assoziierten gut etablierten PNS. Die Studie betrachtet ferner das Follow up der Patienten und geht hierbei insbesondere auf die Beschreibung der acht Langzeitüberlebenden ein (siehe 3.10). Die vorliegende Arbeit basiert auf der Analyse eines Schwerpunktlabors, es handelt sich folglich nicht um eine epidemiologische Studie im klassischen Sinne. Da ein Großteil der Daten retrospektiv erhoben wurde, besteht durchaus die Möglichkeit, dass verschiedene Patientendaten unvollständig oder nicht zuverlässig sind. Diese Fehleranfälligkeit und die Tatsache, dass unsere Studie nur zur Generierung bzw. Stärkung von Hypothesen, nicht jedoch zur Beweisfindung genügt, müssen im Zusammenhang mit dieser Arbeit 76 berücksichtigt werden. Nicht untersucht wurden Patienten mit Nachweis von nicht gut charakterisierten antineuronalen Antikörpern. Die niedrige Fallzahl in unserer Studie ist mit Sicherheit auch auf die geringe Prävalenz paraneoplastischer neurologischer Syndrome zurückzuführen, was sich in bereits publizierten Fallserien mit vergleichbaren Zahlen wiederspiegelt. Diese Tatsache geht mit einer eingeschränkten Möglichkeit der statistischen Auswertung einher, was zu einem mehr beschreibenden Charakter der Untersuchungsergebnisse führte. Ferner beschränkten wir uns aufgrund der teils unvollständigen Datenlage auf die unter Abschnitt 2, Patienten und Methoden, aufgeführten Kriterien. 4.2 Demographische Daten Die Analyse ergab mit einem Verhältnis von etwa 1,2:1 eine leichte Prädominanz der Erkrankung bei Frauen, was auch schon in einer Follow up-Studie von Candler et al. (2004) beobachtet wurde, wohingegen dort mit einem Verhältnis von 2,3:1 ein wesentlich deutlicheres Ergebnis beschrieben wird. Betrachtet man die Geschlechterverteilung im Zusammenhang mit dem jeweiligen Antikörper, dann decken sich die Ergebnisse mit vielen Studien: Mit einem Verhältnis Frauen/Männer von 3:0 Patienten bei anti-Yo und 2:1 bei antiRi wird die Assoziation dieser Syndrome mit gynäkologischen Tumorerkrankungen deutlich (Voltz, 2002; Honnorat, 2007; Graus, 2004). Auch bei anti-CV2 (4:1) bestätigte sich das Überwiegen dieses Syndroms beim weiblichen Geschlecht (Voltz, 2002). Bei anti-Hu zeigte sich mit einem Verhältnis von 14:16 (w:m) Patienten die bereits mehrfach beschriebene Prädominanz bei Männern (Graus et al., 2001, Camdessanché et al., 2002). Im Gegensatz dazu wurde jedoch auch schon ein Überwiegen dieses Antikörpers beim weiblichen Geschlecht beschrieben (Dalmau et al., 1992; Rosenblum, 1993; Voltz, 2002). Mit einem durchschnittlichen Alter von 61,7 Jahren zum Zeitpunkt der Diagnosestellung und nur fünf Patienten mit einem Alter von unter 50 Jahren weichen unsere Ergebnisse kaum von Resultaten anderer Studien ab, bei denen ein ähnliches mittleres Erkrankungsalter beschrieben wird (Stourac et al., 2001; Candler et al., 2004). Einzig der Fall eines jungen Mannes mit Nachweis von Anti-Hu bei Morbus Hodgkin und Kleinhirndegeneration, der zum Zeitpunkt der Erstmanifestation erst 17,8 Jahre alt war, ist hier gesondert zu erwähnen. Fälle mit ähnlich jungen Patienten mit paraneoplastischen 77 neurologischen Syndromen bei Morbus Hodgkin sind bereits bekannt und werden in anderen, älteren Studien beschrieben (Horwich et al., 1966; Trotter et al., 1976; Greenberg, 1984). Der Nachweis von anti-Hu bei Morbus Hodgkin hingegen ist ungewöhnlich, eine detaillierte Beschreibung ähnlicher Fälle konnte in der Literatur nicht gefunden werden, jedoch findet diese Assoziation in einer Arbeit von Senties-Madrid (2001) kurz Erwähnung. In der Regel lassen sich bei mit dieser Krebserkrankung assoziierten PNS Tr-Antikörper nachweisen. Diese nach ihrem Erstbeschreiber Trotter benannte Reaktivität ist ein Marker für einen M. Hodgkin bei meist männlichen Patienten, die an einer Kleinhirndegeneration leiden (Voltz, 2002). 4.3 Paraneoplastische neurologische Syndrome Als häufigstes Syndrom des ZNS fand sich in 24% der Fälle eine subakute Kleinhirndegeneration, was sich mit früheren Beobachtungen deckt (Honnorat, 2007; de Beukelaar, 2006). Doch auch die Hirnstammenzephalitis mit 10% und die Enzephalomyelitis sowie die limbische Enzephalitis mit jeweils 8% waren in unserem Kollektiv gehäuft vorzufinden. Als seltenere Syndrome fanden sich bei einer Patientin eine subakute Retinadegeneration (2%) und bei einem Patienten ein choreatisches Syndrom. Letztere Symptomatik zeigt sich eher selten in Assoziation mit einem Tumor, wurde jedoch bereits in einer Fallserie von Tremont-Lukas et al. (2002) beschrieben. Wie auch in unserer Studie waren dort ausschließlich kleinzellige Bronchialkarzinome assoziiert, als Antikörper wird in zwei der vier berichteten Fälle anti-Hu angegeben, was sich mit der Beobachtung bei unserem Patienten deckt. Interessant ist die Verteilung der peripheren Syndrome: Entgegen der Erwartungen zeigte sich bei uns nicht die subakute sensorische Neuropathie als häufigstes Syndrom (14%), sondern die sensomotorische Neuropathie mit 28% der Patienten. In einer Analyse von 200 HuPatienten wurde bei nur 4,5% der Patienten eine isolierte sensomotorische Neuropathie (Graus et al., 2001) beschrieben, Candler et al. (2004) geben gar einen Prozentsatz von 1,6% an. Im Gegensatz dazu liegt der Anteil bei Lucchinetti et al. (1998) mit 22-41% wesentlich höher. Wie bekannt ist, findet sich beim Denny-Brown-Syndrom elektrophysiologisch häufig eine zusätzliche, klinisch nicht manifeste motorische Beteiligung (Camdessanche et al., 2002), doch zeigten alle unsere Patienten in der PNS-Gruppe „sensomotorische Neuropathie“ deutliche klinische Defizite im motorischen Bereich. So ist das Ergebnis unserer Studie nicht 78 etwa nur auf neurophysiologische Beobachtungen, sondern auf eine tatsächliche klinische Manifestation der motorischen Komponente zurückzuführen. Bei einem weiteren Patienten (Nr.11; Hu + SCLC) fand sich eine chronische Polyradikulitis in Kombination mit einer autonomen Neuropathie. Mit nur einem Fall von autonomer Neuropathie bei anti-Hu, was einem Anteil von 2,6% entspricht, fanden wir dieses klassische Syndrom eher selten. Smitt et al. beschreiben in einer Arbeit von 2002 zum Beispiel einen Prozentsatz von 23% mit autonomen Dysfunktionen. Isoliert traten sensorische bzw. sensomotorische Neuropathien bei 52% der Patienten auf, bei 48% fand sich eine zusätzliche zentrale Affektion (60% zerebelläre Degeneration, 20% Hirnstammenzephalitis, 10% Enzephalomyelitis und 10% limbische Enzephalitis). Dieses multifokale Auftreten ist nicht ungewöhnlich. Es findet sich vor allem bei Hu-Antikörpern, da Hu-Proteine konstitutiv in Neuronen des zentralen und peripheren Nervensystem exprimiert werden, und wurde bereits vielfach beschrieben (Kaiser, 1999; Graus et al., 2001, Sillevis Smitt et al., 2002). Die Untersuchung der neurologischen Symptomatik unserer Patienten macht deutlich, dass sich im Zusammenhang mit dem Nachweis von antineuronalen Antikörpern neben den klassischen paraneoplastischen Syndromen nicht selten auch eine atypische, nicht-klassische Symptomatik zeigen kann. So sollte man immer auch bei einer untypischen neurologischen Symptomatik unklarer Genese an ein paraneoplastisches neurologisches Syndrom denken und eine Tumorsuche bzw. den Nachweis antineuronaler Antikörper anstreben. 4.4 Zugrundeliegende Malignome Bei 10 von 39 Patienten (25,6%) konnte kein definitiver Primärtumor detektiert werden, wobei das Geschlechterverhältnis mit jeweils fünf Frauen und Männern ausgewogen war. Es fanden sich jedoch bei vier weiteren Patienten entweder in PET oder Autopsie Hinweise auf ein lokales Tumorgeschehen. So blieben letztlich sechs Patienten (15,4%) ohne jeglichen Tumorhinweis. Dieser Prozentsatz liegt vergleichend in anderen Studien zwischen 2% bei anti-Hu und –Yo (Graus et al., 2001; Shams’ili et al., 2003; Rojas-Marcos et al., 2003), 3% bei anti-Ri (Dropcho 1993; Pittock et al., 2003) und 5% bei anti-Amphiphysin (Folli et al., 1993; Prego et al., 2002). Eine neurologische Symptomatik unbekannter Genese gilt jedoch auch im Falle des mangelnden Tumorhinweises bei Nachweis eines klassischen antineuronalen Antikörpers als definitives PNS (Graus, 2004). Dies erklärt die 79 pathognomonische Bedeutung klassischer Antikörper und verdeutlicht die Wichtigkeit einer entsprechenden Diagnostik. Mit einem Anteil von fast 83% bestätigten sich Bronchialkarzinome als häufigste Tumorentität (Posner 1995). Wie zu erwarten konnte in dieser Gruppe bei 18 von 24 Patienten (75%) als häufigster Tumor ein SCLC nachgewiesen werden, in sechs weiteren Fällen (25%) fand sich ein nicht-kleinzelliger Bronchialtumor. Mit einem Mammakarzinom bei drei Patientinnen (10,4%), einem Ovarialkarzinom bei einer Patientin (3,4%) und einem Fall von Morbus Hodgkin (3,4%) fand sich in unserer Arbeit keine Tumorentität, die bisher noch nicht im Zusammenhang mit einem paraneoplastischen neurologischen Syndrom beschrieben wurde. Erwartungsgemäß handelte es sich in überwiegender Anzahl um Bronchialkarzinome und gynäkologische Tumoren (Posner, 1995; Voltz, 2002). Bei nur vier der 29 Patienten mit einer Tumordiagnose fanden sich zum Diagnosezeitpunkt Fernmetastasen, in zehn Fällen war der Tumor vielmehr klein und/oder lokal begrenzt (davon fünf SCLC). Diese Beobachtung einer lokalen Beschränkung des Karzinoms (wohlmöglich als Folge des immunologischen Abwehrmechanismus im Rahmen der Erkrankung) wird auch in anderen Veröffentlichungen beschrieben (Graus et al., 1997; Smitt et al., 2002; Pittock et al., 2005). Der „biologische Sinn“ der paraneoplastischen Autoimmunantwort könnte folglich in der Tumorabwehr durch das Immunsystem liegen. 4.5 Antineuronale Antikörper Trotz der teilweisen Überlappungen ist bei klinischem Verdacht auf ein PNS die Bestimmung einer möglichen Antikörperreaktivität unerlässlich. Die gut charakterisierten antineuronalen Antikörper sind pathognomonisch für ein zu Grunde liegendes Karzinom, beweisend für das Vorliegen eines PNS und mit Einschränkungen assoziiert mit bestimmten PNS (Graus et al., 2006). Aufgrund der bisherigen Beobachtungen im Bezug auf die Häufung verschiedener Antikörper bei bestimmten Tumorerkrankungen kann deren Entität bei der Diagnostik des zu Grunde liegenden Karzinoms hilfreich sein. Eher unspezifisch sind die Antikörper hingegen im Bezug auf die assoziierten Syndrome, einzig anti-Yo steht in eindeutigem Zusammenhang mit einer subakuten Kleinhirndegeneration (Terence et al., 1995). 80 4.5.1 Häufigkeitsverteilung Hu-Antikörper fanden sich bei insgesamt 30 Patienten (68%) und waren somit erwartungsgemäß auch in unserer Arbeit die häufigste Reaktivität (Voltz, 2002). Seltener waren CV2-Antikörper mit fünf Fällen (11%) und anti-Yo, -Ri und Amphiphysin, die bei jeweils drei Patienten (7%) nachgewiesen wurden. Bei Candler et al. (2003) fanden sich zum Vergleich anti-Hu bei 64%, anti-Yo bei 24% und anti-Amphiphysin bei 2,5% der Patienten. Eine Studie von Stourac et al. (2001) fand folgende Häufigkeiten vor: anti-Hu in 50%, antiYo in 40%, anti-Ri in 20% und anti-CV-2 in 10% der Fälle. Bei vier unserer Patienten ließen sich mehrere Antikörper nachweisen. An Kombinationen fanden sich CV2+Hu (SCLC), Yo+CV2 (kein Tumor), Hu+Amphiphysin (kein gesicherter Tumor) und Ri+Hu+Amphiphysin (Mamma-Ca). In einer Arbeit von 2004 beschreiben Pittock et al. die bei der Untersuchung von 553 Patientenseren gefundenen Antikörperkombinationen. Während sich in dieser Untersuchung bei 31% der Patienten mehrere Antikörper fanden, war bei uns in 10,3% der Fälle eine Koexistenz nachweisbar. Bis auf die Ausnahme der Kombination von anti-Yo und anti-CV2 (Nr.32) konnten in jener Studie alle auch bei uns vorgefundenen Koexistenzen nachgewiesen werden. Diese Kombination findet hingegen in einer älteren Arbeit aus dem Jahre 2000 kurz Erwähnung (Rogemond et al., 2000). Folglich trat bei keinem unserer Patienten eine Antikörperkombination auf, die nicht schon zuvor beschrieben wurde. Alle vier Patienten litten an einer Erkrankungen des zentralen Nervensystems (siehe Tabelle 12). Ursächlich für die Koprävalenz mehrerer antineuronaler Antikörper bei ein und demselben Patienten ist am ehesten die Expression multipler onkoneuraler Antigene durch den Tumor mit konsekutiver Bildung verschiedener Antikörper durch das Immunsystem (Smitt et al., 2002). 4.5.2 Assoziation zu Syndromen Anti-Hu positive Patienten litten in der Mehrzahl (51%) unter einer peripheren Neuropathie (sensomotorisch (13), sensorisch (5), autonom (1), Polyradikulopathie (1)), dem wahrscheinlich am häufigsten assoziierten paraneoplastischen Syndrom (Younger, 1994; Voltz, 2002). Auch die Fälle von Kleinhirndegeneration (7), Enzephalomyelitis (4), Hirnstammenzephalitis (3) und limbischer Enzephalitis (3) gelten als häufige Erkrankungen in Verbindung mit diesem Antikörper (Gultekin et al., 2000; Graus, 2001). 81 Zu den eher seltenen Syndromen zählen die subakute Retinadegeneration (Nr. 40, NSCLC) und das choreatische Syndrom, von uns jeweils einmal beobachtet. Letzteres findet bereits unter 4.3 Erwähnung. Eine Retinopathie kann gehäuft in Assoziation mit Bronchialkarzinomen, meist SCLC auftreten (Chan et al., 2003). Fälle von Retinopathien bei anti-Hu-Nachweis ließen sich in der Literatur nicht finden, häufiger beobachtet werden in diesem Zusammenhang anti-CV2 (Ko et al., 2008). Da Hu-Proteine jedoch konstitutiv in peripheren und zentralen Neuronen exprimiert sind, scheint auch eine Assoziation mit einer Retinopathie naheliegend und möglich (Voltz, 2002). Yo-Antikörper konnten ausschließlich bei drei Patientinnen mit subakuter Kleinhirndegeneration gefunden werden, was diesen Antikörper als spezifischen Marker einer paraneoplastischen zerebellären Degeneration bestätigt (Terence et al., 1995; Voltz, 2002; Pittock et al., 2004). Ri-Antikörper, deren Reaktivität auf Kerne von Neuronen des ZNS beschränkt ist (Voltz, 2002), fanden sich erwartungsgemäß jeweils einmal bei Hirnstammenzephalitis, limbischer Enzephalitis und Enzephalomyelitis. Ein Opsoklonus-Myoklonus-Syndrom, bei dem anti-Ri erstmals identifiziert und als Marker einer gynäkologischen Erkrankung beschrieben wurde, konnte bei keiner Patientin gefunden werden (Voltz, 2002; Honnorat, 2007). CV2-Antikörper, die bei einer Vielzahl von PNS, u.a. bei Enzephalomyelitiden, Kleinhirndegeneration, limbischer Enzephalitis, Chorea und peripheren Neuropathien nachgewiesen werden (Yu et al., 2001; Dalmau, 2008), traten auch bei uns in keiner bisher unbekannten Konstellation auf (sensomotorische Neuropathie (1), sensorische Neuropathie (1), Kleinhirndegeneration (3), Enzephalomyelitis (1)). Die mit Amphiphysin-Antikörpern assoziierten Fälle von Hirnstammenzephalitis (2), Enzephalomyelitis (1) und sensorischer Neuropathie decken sich ebenfalls mit verschiedenen Angaben aus der Literatur (Antoine et al., 1999; Pittock et al., 2005). 4.5.3 Assoziation zu Tumoren In Assoziation mit anti-Hu konnte als häufigster Tumor das Bronchialkarzinom gefunden werden (67%), des Weiteren litten zwei Patientinnen an einem Mammakarzinom und ein Patient an Morbus Hodgkin. Bei insgesamt sieben Patienten mit Nachweis dieses Antikörpers konnte kein definitiver Tumor gefunden werden, jedoch waren auch hier in zwei Fällen die Untersuchungsbefunde hinweisend auf ein Bronchialkarzinom (siehe Tabelle 8.2). 82 Lungentumoren, insbesondere das SCLC, sind als häufigste Tumorentität bei anti-HuNachweis bekannt (Voltz, 2002; Graus et al., 2001; Sillevis Smitt et al., 2002). Ein Auftreten bei Mammakarzinomen und auch bei Morbus Hodgkin findet in der Literatur kurz Erwähnung (Candler et al., 2004; Graus, 2001 [Hu bei Mamma-Ca]; Senties-Madrid, 2001 [Hu bei MH]). Wie jedoch bereits unter 4.2 erläutert, zeigt sich ein Morbus Hodgkin in erster Linie in Assoziation mit Tr-Antikörpern, Angaben zum Auftreten in Verbindung mit anti-Hu werden in der Literatur kaum gemacht. Ri- und Yo-Antikörper waren erwartungsgemäß bei Frauen mit Verdacht auf bzw. bestätigtem gynäkologischem Tumor vorzufinden (Luque et al., 1991; Peterson et al., 1992; Rojas et al., 2000). Jedoch gelang der Nachweis von Ri-Antikörpern in einem Fall auch bei einem männlichen Patienten (Nr.11, limbische Enzephalitis, NSCLC), wobei die Tumordiagnose der neurologischen Erstmanifestation um 118 Monate vorauseilte. Im Primärtumor dieses Patienten, welcher mittels Oberlappenresektion behandelt wurde, konnte retrospektiv eine Ri-Expression nachgewiesen werden, was im Fall eines Rezidives, das knapp zehn Jahre später im Rahmen der neu aufgetretenen neurologischen Symptomatik diagnostiziert wurde, nicht gelang (Harloff, Hummel, Rauer, 2005). Bronchialkarzinome in Assoziation mit anti-Ri werden u.a. bei Stourac et al., 2001 und bei Graus et al., 2004 beschrieben. Bei einer Patientin (Nr.32) mit Yo-Antikörpern war die PET-Untersuchung hinweisend auf ein Bronchialkarzinom, eine definitive Diagnose konnte jedoch nicht eruiert werden. Jedoch fanden sich bei ihr zusätzlich CV2-Antikörper, die häufig mit einem Lungentumor assoziiert sind (Rogemond, 2000). So wurden in der unserer Studie auch in drei weiteren Fällen mit anti-CV2-Nachweis Bronchialkarzinome diagnostiziert (SCLC (2), NSCLC (1)). Amphiphysin-Antikörper konnten jeweils einmal bei SCLC und bei Mammakarzinom gefunden werden, den neben dem Ovarialkarzinom am häufigsten mit diesem Antikörper assoziierten Tumoren (Folli et al., 1993; Antoine et al., 1999; Saiz, 1999). Die in bisherigen Arbeiten bereits mehrfach beschriebenen und auch bei uns beobachteten Assoziationen zwischen den verschiedenen gut-charakterisierten Antikörpern und bestimmten Tumoren macht nochmals die pathognomonische Bedeutung der verschiedenen Reaktivitäten und deren Relevanz im Bezug auf die Diagnostik der Krebserkrankung deutlich. Letztendlich konnte in nur 15,4% unseres Kollektives keinerlei Tumorhinweis gefunden werden, in den anderen Fällen zeigten sich in überwiegender Anzahl bereits beschriebene Assoziationen. 83 4.6 Diagnostik Keine klinische Symptomatik alleine ist beweisend für eine paraneoplastische Ätiologie der neurologischen Erkrankung, obwohl vor allem die klassischen paraneoplastischen neurologischen Syndrome relativ häufig in Assoziation mit bestimmten Tumoren vorzufinden sind. So ist es von großer Wichtigkeit, eine Untersuchung auf paraneoplastische antineuronale Antikörper zu veranlassen, da diese einerseits pathognomonisch für ein zu Grunde liegendes Karzinom sind und zum anderen einen wertvollen Hinweis auf dessen Entität bieten können. Neben einer Computer- und/oder Kernspintomographie ist in der Bildgebung im Rahmen der Tumorsuche auch eine Untersuchung mittels PET hilfreich (siehe 4.6.2). Bei der genaueren Diagnostik bzw. Klassifizierung des neurologischen Syndroms können neben der Art des assoziierten Antikörpers auch die Untersuchung des Liquors (4.6.3) auf entzündliche Veränderungen als Anzeichen einer immunvermittelten Reaktion sowie Ergebnisse aus der Bildgebung mittels MRT (4.6.4) hilfreich sein. 4.6.1 Intervall Erstmanifestation des PNS - Tumordiagnose Das Einsetzen der neurologischen Symptomatik vor einer Tumordiagnose konnte bei 35 der 39 Patienten (89,7%) beobachtet werden, was im Vergleich zu anderen Studien ein relativ hoher Anteil zu sein scheint (Candler et al., 2004 [69%]); Graus et al., 2001 [71,5%]; Sillevis Smitt et al., 2002 [70%]). Im Zuge der Tumorsuche konnte bei 25 Patienten ein assoziiertes Karzinom diagnostiziert werden. Die Latenz bis zur Diagnose lag im Mittel bei 17,5 Monaten (0,5-133 Monate) und somit über dem Durchschnitt anderer Arbeiten (Graus et al., 2001 [6,5 Monate]; Sillevis Smitt et al., 2002 [3,5 Monate]). Mit 26,8 Monaten bei anti-CV2 und 18,5 Monaten bei anti-Hu war die Latenz bis zur Tumordiagnose bei diesen Patienten besonders lange. Doch gerade diese Zeitspanne sollte möglichst kurz sein, da sich in den meisten Fällen nur eine effektive Tumortherapie als erfolgversprechend im Bezug auf eine Verbesserung der neurologischen Symptomatik gezeigt hat (Voltz, 2002). Die frühzeitige Suche nach spezifischen antineuronalen Antikörpern bei neurologischen Erkrankungen ungeklärter Ätiologie kann bei der Karzinomdiagnose hilfreich sein, auch weil verschiedene bereits bekannte Korrelationen auf die Tumorentität hinweisen können, z.B. anti-Hu bei SCLC und PEM/SM (Sillevis Smitt et al., 2002) oder anti-Yo/-Ri bei gynäkologischen Tumoren (RojasMarcos et al., 2003). 84 Bei nur vier Patienten setzte die neurologische Symptomatik nach der Tumordiagnose ein. Eine Yo-Patientin litt an einem Ovarialkarzinom, dass 23 Monate zuvor diagnostiziert worden war. Anscheinend kann bei Yo-Patientinnen die Tumordiagnose der neurologischen Erstsymptomatik häufiger vorauseilen. So ist in einer Arbeit von Shams’ili et al. (2003) von einem Prozentsatz von 67% zu lesen. Und auch bei anti-Ri-Patienten finden sich nicht selten entsprechende Verhältnisse (Pittock et al., 2003). Bei zwei unserer drei Patienten mit RiAntikörpern (Mamma-Ca [120 Monate]; NSCLC [188 Monate]) war das Intervall Tumordiagnose-neurologische Erstsymptomatik mit durchschnittlich 154 Monaten besonders lange. Eine Ursache dieser langen Latenz könnte hierbei die Tatsache spielen, dass zwar ein Tumor diagnostiziert wird, der neuronale Verlust im Rahmen der begleitenden Immunreaktion jedoch (noch) nicht hinreichend ist für die klinische Manifestation einer neurologischen Symptomatik. Dies spielt vor allem dann eine Rolle, wenn die Tumormasse und folglich auch die Antigenlast im Rahmen einer effektiven Tumortherapie auf ein Minimum reduziert wurden. Eine weitere These wäre eine zwischenzeitliche Tumorzellmutation in Primärtumor oder Rezidiv, die zur erstmaligen Expression onkoneuraler Antigene mit konsekutivem Nervenzellverlust führt. So wurde zum Beispiel Patient Nr.11 erst im Rahmen eines Rezidivtumors neurologisch auffällig (Harloff, Hummel, Rauer, 2005). Der Beweis dieser These ließe sich erbringen, wenn sich bei Untersuchung des Primärtumorgewebes keine Expression onkoneuraler Antigene nachweisen lässt, sich diese Antigene jedoch bei einem nach Behandlung verbleibenden Tumorrest oder einem Rezidiv finden lassen. Die damit verbundene späte Immunreaktion könnte die teils lange Latenz zwischen Tumordiagnose und Erstmanifestation der neurologischen Symptomatik erklären. 4.6.2 Ergebnisse aus der Tumorsuche mittels PET Falls bei einem Patienten mit klinischerseits hochgradigem Verdacht auf ein PNS und/oder Nachweis eines gut charakterisierten antineuronalen Antikörpers die Tumorsuche mittels konventioneller Diagnosemethoden erfolglos bleibt, ist eine PET indiziert (S. Younes-Mhenni et al., 2004). Die Studie von Younes-Mhenni zeigte hierbei im Bezug auf die Tumordetektion eine Sensitivität der PET von über 83% und somit einer verbesserte Diagnosestellung des zu Grunde liegenden Karzinoms. In unserer Studie lagen in zehn Fällen (26%) die Untersuchungsergebnisse einer PET vor, wobei sich in acht Fällen pathologische Befunde vorfanden (Tabelle 16). In sechs Fällen 85 diente diese Bildgebung der Bestätigung einer bereits auffälligen Routinediagnostik, wobei sich bei einer Patientin trotz bestätigtem Verdacht auf paraaortale Metastasen weiterhin kein Primärtumor nachweisen ließ (Nr.26; Yo). Aufgrund der paraaortal verlaufenden Lymphabflusswege des Retroperitoneums sowie unter Berücksichtigung der Spezifität von anti-Yo ist bei dieser Patientin mit hoher Wahrscheinlichkeit von einem latenten Ovarialkarzinom auszugehen. In Fällen wie diesem, sprich beim Nachweis von YoAntikörpern bei Patientinnen ohne definitiven Primärtumor ist als Therapieansatz eine prophylaktische Salpingo-Oophor-Hysterektomie in Erwägung zu ziehen. Bei zwei weiteren Patienten (Nr.9; Nr.32) war die vorhergehende Diagnostik unauffällig geblieben. Hier fanden sich zwar in der PET auffällige Befunde, die Diagnosestellung eines definitiven Primärtumors jedoch war in beiden Fällen nicht möglich (Nr. 32 PET pos. im Bereich von Thoraxwand und Pleura [Nr.32, Yo+CV2]; PET pos. im Bereich des Ileozoekalpols/ mediastinaler und hilärer Lymphknoten [Nr. 9, Hu+Amphiphysin]). Obwohl demnach bei einigen Patienten die Tumormasse im Rahmen der paraneoplastischen Erkrankung mit ihrer postulierten Anti-Tumor-Aktivität relativ gering geblieben und somit schwer zu detektieren war, ergaben sich in einigen Fällen mithilfe einer PET erste Hinweise auf ein Malignom. Somit ist gerade bei diesen Patienten eine engmaschige klinische und bildgebende Verlaufskontrolle höchst indiziert. 4.6.3 Ergebnisse der Liquordiagnostik Zusätzlich zur Bildgebung kann auch die Untersuchung des Liquors hilfreich sein im Bezug auf die Diagnosestellung eines PNS. Zum einen dient diese dem Ausschluss anderer Ursachen der neurologischen Symptomatik wie Infektionen mit verschiedenen Erregern oder malignen Infiltrationen, zum anderen lassen sich eventuell Anzeichen einer allgemeinen entzündlichen Veränderung finden, die in diesem Fall meist in leicht erhöhten Werten bei Zellzahl (3040/mm³) und Eiweiß (50-100 mg/dl) sowie im Nachweis von oligoklonalen Banden und einem erhöhten IgG-Index bestehen (Darnell, 2003; Stich et al., 2003). So fand sich in unserer Studie bei 41,7% der Patienten eine auf durchschnittlich 22,5/mm³ erhöhte Zellzahl, bei 58,3% ein auf durchschnittlich 851 mg/dl erhöhtes Gesamteiweiß und bei 33,3% ein IgG-Index von 0,8 oder darüber. Bei fast 62% ließen sich oligoklonale Banden im Liquor nachweisen, davon bei zwei Patienten (Nr.31, Hu; Nr.21; Yo) zusätzlich oligoklonale Banden im Serum. Die Analyse des Reiber-Schemas ergab bei neun Patienten 86 quantitative Hinweise auf eine autochthone IgG-Synthese, zwei dieser Patienten zeigten eine zusätzliche IgA-Synthese (Tabelle 14). Alle drei Patientinnen mit Yo-Antikörpern und Nachweis von oligoklonalen Banden litten an einer Kleinhirndegeneration. Diese Konstellation scheint häufig zu sein, wurde sie doch bereits in einer Studie von Stourac et al. (2001) in drei Fällen vorgefunden. Ob ein Zusammenhang besteht und ob Patienten mit Nachweis von oligoklonalen Banden generell unter schwereren neurologischen Symptomen leiden, wie in besagter Studie beobachtet, ließ sich von unserer Seite nicht bestätigen. Unsere Analyse ergab, dass sich pathologische Veränderungen im Liquorbefund erwartungsgemäß in erster Linie auf Patienten mit Affektion des ZNS konzentrieren (Dalmau et al., 1992; Pittock et al., 2003; Dalmau, 2008), doch auch bei Syndromen des peripheren Nervensystems lassen sich bisweilen pathologische Veränderungen finden (Sillevis Smitt et al., 2002; Vedeler et al., 2006). So auch bei sechs unserer Patienten, bei denen das Liquoreiweiß auf über 450 mg/dl erhöht war (sensomotorische Neuropathie (3), sensorische Neuropathie (3)). In einer Studie von Stich et al. (2007), in der mittels ELISA Liquorproben auf eine spezifische Synthese der gut charakterisierten Antikörper untersucht wurden, konnte bei 88% der Patienten mit überwiegender Affektion des ZNS eine Erhöhung des spezifischen Antikörperindex nachgewiesen werden. Im Gegensatz dazu fand sich bei ausschließlicher peripherer Beteiligung keine signifikante Erhöhung des Index, was die Theorie eines Autoimmunprozesses auf dem Boden einer spezifischen Antikörperproduktion durch eine intrathekale B-Zell-Population bei PNS unterstützt (Stich et al., 2007). Im Vergleich dazu lag bei uns die Prävalenz einer intrathekalen Gesamt-IgG-Synthese in Form des Nachweises oligoklonaler Banden im Liquor bei Syndromen mit Beteiligung des zentralen Nervensystems bei 66,7% (16/24), hierbei im Falle einer ausschließlichen Affektion des ZNS bei 68,8%, bei zusätzlicher peripherer Symptomatik bei 62,5%. War ausschließlich das periphere Nervensystem betroffen, konnten in unserer Studie immerhin noch bei 50% (5/10) der Patienten oligoklonale Banden nachgewiesen werden, während in der Untersuchung aus dem Jahr 2007 keine Erhöhung des spezifischen Antikörperindex bei einer ausschließlich peripheren Symptomatik gefunden werden konnte. 87 4.6.4 Ergebnisse der Bildgebung mittels MRT Die Ergebnisse der Untersuchung der Wirbelsäule bei 17 Patienten blieben bis auf drei Ausnahmen ohne pathologischen Befund. Die Veränderungen in diesen drei Fällen waren diskret bzw. fraglich, bei Patient Nr.20 (Hu+CV2; Enzephalomyelitis) mit einem fraglichen hyperintensen Myelopathiesignal in Höhe BWK 2-4 und bei Patientin Nr.33 (Ri; Hirnstammenzephalitis) mit einem fraglichen Myelopathiesignal in Höhe HWK 3/4 sind die Veränderungen durchaus mit der Klinik des PNS vereinbar. Die Bildgebung des Schädels hingegen führte zu spezifischeren Ergebnissen. Tabelle 15 gibt einen Überblick über die auffälligen Befunde bei 39% der Patienten, bei denen ein MRT des Schädels durchgeführt worden war. Bei insgesamt fünf Patienten mit einer subakuten zerebellären Degeneration fanden sich Anzeichen einer Kleinhirnatrophie. Diese Patienten hatten entweder anti-Hu (3 Fälle) oder anti-CV2 (2 Fälle) im Serum. Bei allen drei Patientinnen mit anti-Yo und zerebellärer Degeneration jedoch zeigten sich bildmorphologisch keine pathologischen Befunde. Im Rahmen einer Kleinhirndegeneration kann das MRT zu Beginn noch normal sein, im Verlauf lassen sich jedoch oftmals Veränderungen wie eine Atrophie und/oder vermehrte Kontrastmittelaufnahme in der Bildgebung beobachten (Graus, 2007; Dalmau, 2008). Unsere Beobachtungen decken sich hierbei mit den Ergebnissen anderer Studien, in denen ebenfalls über oben beschriebene Veränderungen im Rahmen einer Kleinhirndegeneration berichtet wird (Rojas-Marcos et al., 2003; Candler et al., 2004). Weiterhin zeigten drei Patienten mit limbischer Enzephalitis pathologische Veränderungen (fronto-temporale Atrophie [Ri]; hyperintense Läsionen im medialen Temporallappen links [Hu]; verstrichene Rinden-Mark-Grenze im Temporallappen links [Hu]). So unterstreichen unsere Ergebnisse im Einklang mit Beobachtungen aus anderen Studien (Gultekin et al., 2000; Candler et al., 2004) die Tatsache, dass sich bei einer limbischen Enzephalitis in bis zu 80% der Fälle eine Atrophie oder hyperintense Signale im Bereich der Temporallappen finden lassen. Auch die Betonung einer Hirnhemisphäre ist nichts Ungewöhnliches und wird häufiger beschrieben (Vedeler et al., 2006; Tüzün, 2007). Diffuse Veränderungen wie bei einem Fall von Enzephalomyelitis (multiple Läsionen in Marklager und Kortex [Hu]) konnten auch Candler et al. in ihrer Arbeit bei einem Patienten beobachten. In einem weiteren Fall mit choreatischer Symptomatik (Hu, SCLC) zeigte das MRT hyperintense Läsionen im Bereich des Ncl. caudatus beidseits. Dies ist nicht die erste Beschreibung von pathologischen Befunden in diesem Zusammenhang, schon bei Heckmann 88 et al. (1997) wird über eine Atrophie des Ncl. caudatus bei anti-Hu berichtet. Auch bei Croteau et al. (2001) fanden sich hyperintense Signale im Bereich des Neostriatums, hier in Assoziation mit einem 78-kD-Antigen. In beiden Fällen war wie bei unserem Patienten ein SCLC nachgewiesen worden. Trotz dieser Beobachtungen ist die konventionelle Bildgebung der Neuraxis bei paraneoplastischen neurologischen Syndromen leider in vielen Fällen nicht wegweisend. Dies liegt vor allem daran, dass Veränderungen erst dann sichtbar werden, wenn bereits ein größerer Anteil an Neuronen untergegangen ist. Mit Ausnahme der limbischen Enzephalitis ist in den meisten Fällen die klinische Symptomatik aufgrund der meist subakuten Manifestation sensitiver als die Bildgebung. 4.7 Follow up Die vorliegende Studie ist eine der wenigen, die sich mit dem klinischen Follow up eines größeren Patientenkollektives mit einem Spektrum an verschiedenen Syndromen und Antikörpern beschäftigt, die bisherigen Arbeiten waren in erster Linie Einzelfallberichte oder konzentrierten sich auf einen bestimmten Antikörper bzw. ein einziges paraneoplastisches neurologisches Syndrom. 4.7.1 Ergebnisse der Therapie 29 Patienten erhielten eine Therapie des zu Grunde liegenden Tumors, bei 21 Patienten kam eines immunmodulatorische Therapie zum Einsatz, ein Einfluss der Behandlung auf die neurologische Symptomatik zeigte sich bei insgesamt 17 Patienten. Es war jedoch nicht immer uneingeschränkt möglich, zwischen dem Effekt von Tumorbehandlung und immunmodulatorischer Therapie zu unterscheiden. Alle Patienten, bei denen eine vorübergehende Besserung bzw. eine längerfristige Stabilisierung der Symptomatik zu beobachten war, hatten in irgend einer Form eine Tumortherapie mit oder ohne kombinierter immunmodulatorischer Behandlung erhalten, was bisherige Beschreibungen in anderen Studien unterstreicht (Candler et al., 2004; Shams'ili et al., 2003; Gultekin et al., 2000). Aus Tabelle 20 wird zudem deutlich, dass sich alle noch nicht verstorbenen Langzeitüberlebenden, bei denen ein Tumor gefunden und behandelt worden war, im Stadium der Komplettremission befinden. Nur bei zwei Patienten, bei denen jedoch 89 kein definitiver Tumor gefunden wurde (Nr.24+32; Tabelle 18), konnte der Einfluss einer alleinigen Immuntherapie auf die neurologische Symptomatik beobachtet werden, was jedoch auch an anderer Stelle in der Literatur beschrieben wurde (Oh et al., 1997). So scheint bei ausstehender Tumordiagnose eine alleinige immunmodulatorische Therapie ratsam (Sillevis Smitt et al., 2002). Generell stellt sich hier die Frage, ob bei paraneoplastischen neurologischen Syndromen der fehlende Hinweis auf einen Tumor mit einer verlängerten Überlebenszeit einhergeht. In unserer Studie konnte in sechs Fällen kein definitiver Primärtumor gefunden werden. Hierbei war in vier Fällen die Überlebenszeit bekannt: zwei Patienten (50%) waren bei Studienende verstorben, die mittlere Überlebenszeit lag bei 10,5 Monaten [3; 18], zwei Patienten (50%) waren am Leben, die mittlere Überlebenszeit lag bei 104,8 Monaten [12,8; 196,8]. Die Gesamtüberlebenszeit dieser vier Patienten lag folglich bei im Mittel 57,7 Monaten. Vergleichend dazu fand sich bei 32 von 33 Patienten mit auffälliger PET/Autopsie bzw. definitivem Tumornachweis eine durchschnittliche Überlebenszeit von nur 45,1 Monaten [1,8-157,8]. In einen Fall war die Überlebenszeit nicht bekannt. 24 von 32 Patienten (75%) waren bei Studienende verstorben, die mittlere Überlebenszeit lag bei 33,4 Monaten [1,8133,2], acht von 32 Patienten (25%) waren noch am Leben, die mittlere Überlebenszeit lag bei 80,25 Monaten [5,5-157,8]. Unsere Fallzahl ist zwar sehr gering, doch die hier aufgeführten Zahlen enthalten zumindest einen Hinweis darauf, dass ein fehlender Tumorhinweis ein prognostisch günstiger Faktor hinsichtlich der Überlebenszeit sein könnte. Wichtig ist neben einer frühen Diagnosestellung eines Karzinoms bzw. des PNS weiterhin eine rasche Behandlung von sowohl dem Tumor als auch der neurologischer Symptomatik. Neben einer erfolgreichen Tumorelimination als Kausaltherapie kann sich hierbei in Einzelfällen zusätzlich eine frühe Therapie mittels Immunmodulation als hilfreich erweisen, wahrscheinlich weil auf diese Weise die irreversible Zerstörung neuronaler Strukturen in einem frühen Stadium verhindert wird. Leider fand sich in unserem Kollektiv kein Fall einer wirklichen, dauerhaften Verbesserung der neurologischen Symptomatik, allenfalls eine Stabilisierung auf unterschiedlichem Niveau, was sich jedoch in der Literatur in den meisten Fällen ähnlich darstellt (Darnell et al., 2003; Candler et al., 2004; Sillevis Smitt et al., 2002). Die Prognose einer paraneoplastischen neurologischen Erkrankung ist oftmals schlecht und hängt im Wesentlichen von der frühen Diagnose und Therapie des assoziierten Karzinoms ab, bevor es zu irreversiblen neuronalen Schäden kommen kann. 90 4.7.2 Überlebenszeitanalyse Wir betrachteten die Überlebenszeiten abhängig von PNS, antineuronalem Antikörper, assoziiertem Karzinom, Tumorbehandlung, Geschlecht und Alter bei Diagnosestellung des paraneoplastischen Syndroms. Ferner erstellten wir eine Kaplan-Meier-Kurve zur Überlebenszeit aller Patienten, welche im Ergebnisteil unter 3.9 zu finden ist (Abbildung 2). a) Überlebenszeit abhängig vom PNS Bei Vorliegen einer sensomotorischen oder einer subakut sensorischen Neuropathie lag die mittlere Überlebenszeit bei 52 bzw. 51 Monaten. Candler et al. fanden bei Patienten mit SSN eine längere Überlebenszeit von durchschnittlich 64 Monaten. Die Betrachtung der Patienten mit einer zusätzlichen zentralen Symptomatik zeigte bei uns mit einer durchschnittlichen Überlebenszeit von 49,4 Monaten eine vergleichbare Prognose. Syndrome, die das zentrale Nervensystem betreffen, sprechen auf verschiedene Therapieansätze oftmals eher schlecht an (Darnell et al., 2003). Dies kann daran liegen, dass eingesetzte Immunmodulatoren bzw. Immunsupressiva aufgrund einer schlechten Liquorgängigkeit keine ausreichenden Konzentrationen an Neuronen des ZNS erreichen. Vor allem jedoch wird in diesem Zusammenhang ein zum Zeitpunkt des Therapiebeginns bereits fortgeschrittener, irreversibler Neuronenuntergang eine Rolle spielen (Keime-Guibert et al., 2000). Sowohl bei der limbischen Enzephalitis (Dalmau et al., 2008: kein Benefit von Tumor- oder Immuntherapie bei anti-Hu, -CV2 und –Amphiphysin) als auch bei der subakuten Kleinhirndegeneration ist eine immunmodulatorische Therapie häufig frustran. Letztere zeigt sich vor allem in Verbindung mit Yo-AK vielfach als therapierefraktär (Peterson et al., 1992; Posner et al., 1995; Cao et al., 1999; Rojas-Marcos et al., 2000). Die mittlere Überlebenszeit lag bei unseren Patienten mit Kleinhirndegeneration bei etwa 51 Monaten (Candler et al., 2001: 42 Monate), die Patienten mit PLE lebten durchschnittlich nur 28 Monate und waren zum Zeitpunkt des Studienendes alle verstorben. Graus (2007) beschreibt zwar ein Ansprechen der limbischen Enzephalitis auf eine Therapie von Tumor und PNS in bis zu 30% der Fälle, was bei uns jedoch nicht beobachtet werden konnte. Die Patienten mit einer Enzephalomyelitis lebten im Mittel 51 Monate, was deutlich länger ist als Angaben aus der Literatur (Candler et al., 2001: 30 Monate; de Beukelaar et al., ca. 12 91 Monate), diejenigen mit einer Hirnstammenzephalitis 54 Monate, wobei alle vier Patienten bei Studienabschluss verstorben waren. b) Überlebenszeit abhängig vom antineuronalen Antikörper Der Vergleich mit Angaben aus der Literatur war nur bedingt möglich, da hier in den meisten Fällen die Arbeiten auf ein bestimmtes Syndrom beschränkt sind, nicht jedoch Überlebenszeiten in Bezug auf verschiedene Antikörper und bei Vorliegen aller möglichen Syndrome untersucht wurden, wie das in unserer Studie der Fall war. Die längste Überlebenszeit hatten bei uns Patienten mit CV2-AK mit durchschnittlich 82 Monaten, die kürzeste diejenigen mit Yo-Antikörpern mit im Mittel nur 9,4 Monaten. So beschreibt Cartalat-Carel et al. 2003 bei Patienten mit SCLC und anti-CV2 eine 2,5-mal längere Überlebenszeit als beim Nachweis von anti-Hu. Und die schlechte Prognose beim Vorliegen von Yo-Antikörpern zeigte sich auch in einer Untersuchung von Shams’ili et al. (2003), der die Überlebenszeiten von Patienten mit PCD und Vorliegen von Hu-, Ri- und YoAntikörpern untersuchte. Ob bei Auftreten von anti-Yo und PCD die Prognose für Patientinnen mit Mammakarzinomen besser ist als bei Assoziation mit anderen gynäkologischen Tumoren (de Beukelaar et al., 2006), konnten wir nicht bestätigen. Die Überlebenszeiten von Patienten mit anti-Hu (46 Monate) und anti-Ri (47 Monate) lagen in unserer Studie nicht sehr weit auseinander. Shams’ili gibt bei Vorliegen einer Kleinhirndegeneration für anti-Hu durchschnittlich nur sieben Monate, für anti-Ri jedoch 69 Monate Überlebenszeit an, wobei dieses Ergebnis den Angaben zufolge jedoch nicht signifikant sei (Shams’ili et al., 2003). In einer weiteren Arbeit von Pittock et al. (2003) findet sich eine mittlere Überlebenszeit bei Patienten mit Ri-Antikörpern von nur 22 Monaten. Unser Patient mit Nachweis von anti-Amphiphysin überlebte 30 Monate lang, was somit kaum von einer Angabe aus der Literatur mit einer durchschnittlichen Überlebensdauer von 33 Monaten abweicht (Pittock et al., 2005). c) Überlebenszeit abhängig vom assoziierten Karzinom Eine wichtige Frage ist neben der Analyse der Überlebenszeiten im Vergleich zu anderen Angaben aus der Literatur die Frage, ob sich die Prognosen von Patienten mit 92 Tumorerkrankungen, jedoch ohne den Nachweis von assoziierten paraneoplastischen neurologischen Syndromen von denen bei begleitender neurologischer Symptomatik unterscheiden, ob das Vorliegen von antineuronalen Antikörpern folglich einen Effekt auf das Langzeitüberleben von Patienten hat (Darnell et al., 2003). Die Patienten ohne definitive Tumordiagnose überlebten im Mittel 44 Monate. Bei vier dieser Patienten fanden sich jedoch Hinweise auf ein lokales Tumorgeschehen. Hier lag die mittlere Überlebenszeit bei nur 30 Monaten. Mit durchschnittlich 57,7 Monaten überlebten vier der sechs Patienten ohne jeglichen Tumorhinweis länger, in zwei Fällen war die Überlebenszeit nicht bekannt. Candler et al. fanden im Vergleich bei anti-Hu, dem auch bei uns am häufigsten nachgewiesenen Antikörper, bei mangelndem Tumornachweis eine Überlebenszeit von nur 16 Monaten. Sillevis-Smitt (2002) beschreibt hingegen für Hu-Patienten ohne Karzinomdiagnose eine signifikant bessere Prognose als bei Nachweis des assoziierten Tumors. Angaben zu genaueren Überlebenszeiten für die einzelnen Krebserkrankungen waren in der Literatur schwer zu finden, so dass sich folgende Beschreibung auf die wenigen in der Fachliteratur angegebenen Prognosen für verschiedene Karzinome ohne begleitendes PNS beschränken muss. So wird für das Bronchialkarzinom eine durchschnittliche 5-JÜR von etwa 10% angegeben (Herold, 2007; Pschyrembel, 2002), wobei die Prognose bei Vorliegen eines SCLC noch schlechter ist, da hier in 70% der Fälle der Tumor bei Diagnose bereits inoperabel ist. Beim SCLC im Stadium limited disease können mittels aggressiver Therapie bestehend aus Bestrahlung und Chemotherapie 5-10% definite Heilungen beobachtet werden. Drei unserer 18 Patienten, also etwa 17% mit Nachweis eines SCLC, überlebten länger als fünf Jahre, ob man deshalb jedoch von einer besseren Prognose bei gleichzeitigem Vorliegen eines PNS sprechen kann, ist fraglich. Die mittlere Überlebenszeit betrug hierbei etwa 50 Monate. NSCLC und Ovarialkarzinom waren mit einer hohen Sterblichkeit verbunden, kein Patient lebte länger als fünf Monate. Da bei Morbus Hodgkin in über 70% mit einer langfristigen Remission gerechnet werden kann, ist auch die lange Überlebenszeit unseres Patienten von 143 Monaten nichts Außergewöhnliches. Und zuletzt fanden wir bei Patientinnen mit Mammakarzinomen eine mittlere Überlebenszeit von nur 46 Monaten, was kürzer ist als Angaben aus der Literatur mit einer bis zu 80%igen 5JÜR. Wir können nach Auswertung der Daten folglich nicht mit Sicherheit sagen, ob das Vorliegen eines paraneoplastischen neurologischen Syndroms bei einer Tumorerkrankung mit 93 einer verbesserten Prognose einhergeht. Dies liegt jedoch auch an der in unserer Studie begrenzten Anzahl an Patienten und an der Tatsache, dass oftmals auch die neurologische Symptomatik zum Tode führen kann. d) Überlebenszeit abhängig von der Tumorbehandlung Jene Patienten, bei denen im Rahmen der Erkrankung keinerlei Tumortherapie zum Einsatz kam, überlebten im Mittel 42 Monate. Acht der zehn zum Zeitpunkt des Studienabschlusses noch lebenden Patienten hatten im Verlauf der Erkrankung eine Tumortherapie erhalten, wobei in sechs Fällen eine komplette Remission beobachtet werden konnte. Erhielten die Patienten nur eine Form der Tumorbehandlung, betrug die mittlere Überlebenszeit 33,6 Monate, bei zwei Therapieformen lag sie bei 53,5 Monaten und setzte man alle drei Methoden ein, betrug die durchschnittliche Überlebenszeit 86 Monate. Diese Zahlen machen deutlich, was bereits in der Literatur vielfach beschrieben wurde, und zwar im Zusammenhang mit jedem Antikörper und den meisten neurologischen Syndromen: Patienten, die eine Tumortherapie erhalten, leben signifikant länger und eine Verbesserung der Symptomatik ist vor allem bei Erreichen einer Komplettremission zu beobachten (Shams’ili et al., 2003, Keime-Guibert et al., 1999). Graus (2007) beschreibt den positiven Effekt einer Tumortherapie vor allem bei Opsoklonus, PEM und Lambert-Eaton-Syndrom, eine limbische Enzephalitis hingegen sei im Rahmen einer Therapie selten mit einer Verbesserung verbunden (Dalmau et al., 2008). In vielen Fällen, so auch in unserer Studie, ist jedoch selbst bei einer konsequenten Tumortherapie höchstens mit einer Stabilisierung, weniger mit einer Verbesserung der neurologischen Symptomatik zu rechnen. e) Überlebenszeit abhängig von Geschlecht und Alter bei Diagnosestellung Bei Betrachtung der Überlebenszeit abhängig vom Geschlecht fanden sich mit 48 Monaten bei den weiblichen und 45 Monaten bei den männlichen Patienten vergleichbare Zahlen. Von den Frauen waren soweit bekannt bei Studienende nur 67% verstorben, von den Männern hingegen 80%. Bekanntermaßen ist das weibliche Geschlecht jedoch generell mit einer längeren Überlebenszeit verbunden, es bleibt also unklar, ob die beobachtete Differenz zum Teil nicht auch mit diesem Effekt zu erklären ist. Candler fand zum Beispiel 43% der Männer, 94 jedoch nur 38% der Frauen zum Zeitpunkt des Studienendes in einem stabilen neurologischen Zustand vor, und auch in anderen Arbeiten wurden verschiedene Ergebnisse beschrieben. Wie zu erwarten zeigte sich, dass die Überlebenszeiten mit steigendem Alter bei Erstsymptomatik verkürzt waren, wobei man jedoch das bei höherem Alter ohnehin kürzere Überleben nicht außer Acht lassen darf. So beschreibt auch Shams’ili bei Patienten über 60 Jahren ein schlechteres Outcome und eine kürzere Überlebenszeit als bei jüngeren Patienten (Shams’ili et al., 2003). Unsere Untersuchungen unterstreichen diese Aussage: Waren die Patienten zum Diagnosezeitpunkt unter 50 Jahre alt (durchschnittlich 40 Jahre), lebten sie im Mittel 105 Monate, waren sie älter (durchschnittlich 65 Jahre), betrug die mittlere Überlebenszeit hingegen nur 37 Monate. 4.7.3 Beschreibung der Langzeitüberlebenden Bei den acht Patienten, die seit Beginn der neurologischen Symptomatik länger als fünf Jahre überlebt hatten, wurden als Hauptantikörper ausschließlich anti-Hu oder –CV2 gefunden, wobei ein Patient an einem Morbus Hodgkin erkrankt war, fünf Patienten ein SCLC vorwiesen und in zwei Fällen kein Tumor nachweisbar war. Zwar fanden wir für Patienten mit CV2-Antikörpern wie bereits unter 4.7.2 b beschrieben eine etwas längere Überlebenszeit, ein um den Faktor 2,5 verlängertes Überleben für Patienten mit CV2 und SCLC im Vergleich zu Hu und SCLC, wie in einer Arbeit von Cartalat-Carel et al. (2004) beschrieben, konnten wir hingegen nicht finden. Zum Zeitpunkt des Studienendes waren drei der Erkrankten aus der Gruppe der Langzeitüberlebenden verstorben, fünf Patienten noch am Leben. Patientin Nr.24 (Hu+sensomotorische Neuropathie), bei der bisher kein Tumor diagnostiziert werden konnte und deren Symptomatik bei der Abschlussuntersuchung keine Progredienz zeigte, hatte mit fast 200 Monaten die längste Überlebenszeit. So konnte auch in einer Untersuchung von 73 anti-Hu-Patienten mit PEM bzw. sensorischer Neuropathie eine signifikant bessere Prognose und längere Überlebenszeit bei Patienten ohne Karzinomnachweis gefunden werden (SillevisSmitt et al., 2002). Der Einsatz einer intensiven immunmodulatorischen Therapie bei mangelndem Tumornachweis (Sillevis-Smitt et al., 2002) zeigte bei dieser Patientin allenfalls vorübergehenden Einfluss auf die neurologische Symptomatik. Diese Beobachtung einer wenig erfolgreichen Immuntherapie bei PNS wurde in anderen Arbeiten bereits mehrfach beschreiben (Voltz, 2002; Graus et al., 2007; Keime-Guibert et al., 2000). 95 Patient Nr.9 (Hu+Hirnstammenzephalitis) war ebenfalls ohne Tumordiagnose geblieben, überlebte jedoch nur 92 Monate und war bei Ende des Follow up bereits verstorben. Die Patienten Nr.10 und 19 litten beide an einem SCLC bei Nachweis von Hu-Antikörpern, waren jedoch bei Studienende bereits verstorben. Hier hatte sich kein Ansprechen des Karzinoms auf den Einsatz der gegen den Tumor gerichteten Therapie gezeigt, ganz im Gegensatz zu den vier Patienten, die bei Ende des Follow up noch am Leben waren und die sich zu diesem Zeitpunkt im Stadium der Komplettremission befanden (durchschnittliche Überlebenszeit: 141 Monate). Bei den Patienten Nr.20, 29 und 36 fand sich im Rahmen der Tumortherapie eine Stabilisierung der neurologischen Symptomatik, es war in keinem Fall eine immunmodulatorische Therapie zum Einsatz gekommen. So zeigt sich auch hier die Bedeutung einer frühen und adäquaten Tumortherapie zur Verbesserung der Prognose und Stabilisierung, in Einzelfällen eventuell sogar Verbesserung der neurologischen Erkrankung (Keime-Guibert et al., 1999; Shams’ili et al., 2003; Graus et al., 2007). Bei Patientin Nr.25 (CV2+Kleinhirndegeneration/sensomotorische Neuropathie) war die neurologische Symptomatik bei Ende des Follow up trotz Erreichen des Status der Komplettremission progredient, hier hatte sich jedoch im Rahmen einer zuvor eingesetzten aggressiven Immuntherapie mit Hochdosis-Steroiden, Immunglobulinen sowie Cyclophosphamid eine zeitweilige Verbesserung der neurologischen Symptomatik gezeigt. Dieses entgegen der sonst überwiegend negativen Beschreibungen registrierte Ansprechen auf einen solchen Therapieansatz wird jedoch selten beobachtet und ist in manchen Fällen wohl zusätzlich auf begleitende Faktoren wie Physiotherapie etc. zurückzuführen. Voltz empfiehlt den möglichst frühzeitigen Einsatz von intravenösen Immunglobulinen (Voltz et al, 2006). Auch andere immunmodulatorische Therapieansätze sollten ohne Verzögerung eingesetzt werden, um die irreversible Schädigung neuronaler Strukturen zu verringern. Doch auch dann ist in den meisten Fällen nur mit einer Stabilisierung, nicht jedoch mit einer Verbesserung der Symptomatik zu rechnen (Keime-Guibert et al., 2000). Die immunmodulatorische Therapie wird auch weiterhin nur Therapieansatz zweiter Wahl bleiben, im Vordergrund muss eine möglichst frühzeitige und effektive Tumorbekämpfung als kausaler Therapieansatz einer paraneoplastischen neurologischen Erkrankung stehen. Da häufig auch die Krebserkrankung zum Tode führt, spielt eine frühzeitige Tumordiagnose und –therapie umso mehr eine wichtige Rolle. Unsere Arbeit verdeutlicht letztlich die Wichtigkeit einer frühen und adäquaten Therapie vor allem der zu Grunde liegenden Tumorerkrankung. Basis einer effektiven Therapie ist es 96 jedoch, eine paraneoplastische neurologische Erkrankung – auch im Falle einer wie hier mehrfach beobachteten nicht-klassischen Symptomatik - auch rechtzeitig zu diagnostizieren. Jede subakut einsetzende und progrediente ungeklärte neurologische Symptomatik des zentralen und/oder peripheren Nervensystems sollte den Kliniker zumindest an ein PNS denken lassen und eine weiterführende Diagnostik vor allem mittels Antikörpernachweis und Tumorsuche bis hin zum Einsatz einer PET nach sich ziehen. Anhand der verschiedenen pathognomonischen Antikörper sind wir im Besitz eines hervorragenden diagnostischen Mittels, das neben einer erfolgreichen Tumorsuche zu einer frühzeitigen Diagnose eines paraneoplastischen neurologischen Syndroms führen und so zumindest bei einigen Patienten zu einer Verbesserung der Prognose bezüglich Lebensqualität und Überlebensdauer beitragen kann. 97 5. Zusammenfassung Paraneoplastische neurologische Syndrome (PNS) sind seltene tumorassoziierte Erkrankungen, welche auf einer immunologischen Kreuzreaktion zwischen Antigenen des Tumors und Nervengewebe beruhen. Unsere retrospektive, klinisch und epidemiologisch orientierte, monozentrische Studie umfasste demographische Daten, das klinische Spektrum der PNS, die assoziierten Tumoren, initial bestimmte antineuronale Antikörper, Befunde aus Liquordiagnostik und Bildgebung, Therapieformen, Ansprechen der PNS und der zu Grunde liegenden Karzinome auf die Behandlung sowie die Überlebenszeit von 39 Patienten mit dem Nachweis eines oder mehrerer der sog. gut etablierten antineuronalen Antikörper. Im Rahmen des Follow up untersuchten wir Serumproben überlebender Patienten auf aktuelle AKKonzentrationen bzw. Auftreten eines paraneoplastischen Zweitantikörpers. Neben den klassischen PNS fanden wir mit Polyradikulitis, Hirnstammenzephalitis, Retinadegeneration, Chorea und vor allem sensomotorischer Neuropathie in 44% der Fälle ein nicht-klassisches PNS vor. In überwiegender Anzahl (68%) waren Hu-AK anzutreffen, als häufigstes mit PNS assoziiertes Karzinom bestätigte sich das Bronchialkarzinom (83%), meist ein SCLC. Bereits bekannte Assoziationen fanden sich auch in unserer Studie, u.a. anti-Hu bei Bronchialkarzinomen oder anti-Yo bei Frauen mit Kleinhirndegeneration. Die neurologische Symptomatik manifestierte sich in 89,7% der Fälle vor einer Karzinomdiagnose. In nur 15,4% fand sich keinerlei Tumorhinweis, wobei sich im Rahmen der Diagnostik eine PET in einigen Fällen als hilfreich erwiesen hatte. Mittels MRT waren pathologische Veränderungen vor allem bei Kleinhirndegeneration sowie limbischer Enzephalitis zu beobachten. Eine verbesserte Prognose im Vergleich zu Patienten mit entsprechenden Tumoren, jedoch ohne PNS konnte nicht bestätigt werden. Die schlechteste Prognose zeigte sich bei Erkrankungen des ZNS, v.a. bei limbischer Enzephalitis. Die längste Überlebenszeit fand sich bei Nachweis von CV2- (82 Monate), die kürzeste bei Yo-Antikörpern (9,4 Monate). Der Benefit einer immunmodulierenden Therapie war gering, eine längerfristige Stabilisierung zeigte sich meist nur im Rahmen einer (aggressiven) Tumortherapie, es wurde jedoch kein einziger Fall einer dauerhaften Verbesserung der neurologischen Symptomatik beobachtet. So sollte man, auch bei einer nicht-klassischen Symptomatik, zumindest an ein PNS denken und mit Hilfe der pathognomonischen Antikörper eine rasche Tumordiagnose anstreben, denn in der überwiegenden Anzahl der Fälle stellt nur die Behandlung des assoziierten Karzinoms als auslösendem Faktor eine auf Dauer erfolgversprechende Therapie dar. 98 6. Literaturverzeichnis 1. Albert ML, Darnell JC, Bender A et al. (1998) Tumor-specific killer cells in paraneoplastic cerebellar degeneration. Nat Med 4:1321–1324. 2. Alexander KR, Barnes CS, Fishman GA, Pokorny J, Smith VC (2004) Contrast-processing deficits in melanoma-associated retinopathy. Invest Ophthalmol Vis Sci 45:305–310. 3. Anderson NE, Budde-Steffen C, Rosenblum MK, Graus F, Ford D, Synek BJ, Posner JB (3-1988) Opsoclonus, myoclonus, ataxia, and encephalopathy in adults with cancer: a distinct paraneoplastic syndrome. Medicine 67:100-109. 4. Antoine JC, Absi L, Honnorat J, Boulesteix JM, de Brouker T, Vial C, Butler M, De Camilli P, Michel D (1999) Antiamphiphysin antibodies are associated with various paraneoplastic neurological syndromes and tumors. Arch Neurol 56:172–177. 5. Antoine JC, Mosnier JF, Absi L et al. (1999) Carcinoma associated paraneoplastic peripheral neuropathies in patients with and without anti-onconeural antibodies. J Neurol Neurosurg Psychiatry 67:7–14. 6. Antoine JC, Honnorat J, Camdessanché JP, Magistris M, Absi L, Mosnier JF, Petiot P, Kopp N, Michel D (2001) Paraneoplastic anti-CV2 antibodies react with peripheral nerve and are associated with a mixed axonal and demyelinating peripheral neuropathy. Ann Neurol. 49(2):21421. 7. Ball JA,Warner T, Reid P et al. (1994) Central alveolar hypoventilation associated with paraneoplastic brain-stem encephalitis and anti-Hu antibodies. J Neurol 241:561–566. 8. Benyahia B, Liblau R, Merle-Beral H et al. (1999) Cell-mediated autoimmunity in paraneoplastic neurological syndromes with anti-Hu antibodies. Ann Neurol 45:162–167. 9. Benzing T,Rump LC,Kaiser R,Peter HH (1998) Paraneoplastic cerebellar degeneration in Hodgkin’s disease. Dtsch Med Wochenschr 123: 493-496. 10. Bernal F, Shams'ili S, Rojas I, Sanchez-Valle R, Saiz A, Dalmau J, Honnorat J, Sillevis Smitt P, Graus F (2003) Anti-Tr antibodies as markers of paraneoplastic cerebellar degeneration and Hodgkin's disease. Neurology 60:230–234. 11. Blaes F., Stoll G., Sommer N., Voltz R. (2005) Paraneoplastische neuromuskuläre Erkrankungen. Nervenheilkunde Heft 5:351-450. 12. Byrne T, Mason WP, Posner JB, Dalmau J (1997) Spontaneous neurological improvement in antiHu associated encephalomyelitis. J.Neurol.Neurosurg.Psychiatry 62:276-8. 13. Camdessanche JP, Antoine JC, Honnorat J et al. (2002) Paraneoplastic peripheral neuropathy associated with anti-Hu antibodies. A clinical and electrophysiological study of 20 patients. Brain 125:166–175. 14. Candler PM, Hart PE, Barnett M, Weil R and Rees JH (2004) A follow up study of patients with paraneoplastic neurological disease in the United Kingdom. J. Neurol. Neurosurg. Psychiatry 75;1411-1415. 99 15. Cao Yangming, M.D., Abbas Jalal, M.D., Wu Xiaoling, M.D, Dooley Joseph, M.D., van Amburg Albert L., M.D. (1999) Anti-Yo Positive Paraneoplastic Cerebellar Degeneration associated with Ovarian Carcinoma: Case Report and Review of the Literature. Gynecologic Oncology 75:178– 183. 16. Cartalat-Carel S, Camdessanche JP, Ricard D, et al. (2004) The paraneoplastic neurological syndrome associated with anti-CV2 (anti-CRMP5) antibodies is different from that of anti-Hu antibodies. Neurology 62(Suppl 5):A476-A477. 17. Chan Jane W., MD (2003) Paraneoplastic Retinopathies and Optic Neuropathies. Survey of ophthalmology. Volume 48 Number 1. 18. Chartrand-Lefebvre C, Howarth N, Grenier P, Keime F, Orcel B, Beigelman C (1998) Association of small cell lung cancer and the anti-Hu paraneoplastic syndrome: radiological and CT findings. AJR Am J Roentgenol. 170:1513–1517. 19. Croteau, D., Owainati, A., Dalmau, J., and Rogers, L.R. (2001) Response to cancer therapy in a patient with a paraneoplastic choreiform disorder. Neurology 57:719-722. 20. Dalmau J, Furneaux HM, Gralla RJ, Kris MG, Posner JB (1990) Detection of the anti-Hu antibody in the serum of patients with small cell lung cancer--a quantitative western blot analysis. Annals of Neurology 27:544-52. 21. Dalmau J, Graus F, Cheung NK, Rosenblum MK,Ho A (1995) Major histocompatibility proteins, anti-Hu antibodies, and paraneoplastic encephalomyelitis in neuroblastoma and small cell lung cancer. Cancer 75:99-109. 22. Dalmau J, Graus F,Rosenblum MK,Posner JB (1992) Anti-Hu–associated paraneoplastic encephalomyelitis/ sensory neuronopathy. A clinical study of 71 patients. Medicine (Baltimore) 71:59–72. 23. Dalmau J, Gultekin SH,Voltz R et al. (1999) Ma1, a novel neuronal and testis specific protein, is recognized by the serum of patients with paraneoplastic neurologic disorders. Brain 122:27-29. 24. Dalmau Josep, Myrna R Rosenfeld (2008) Paraneoplastic syndromes of the CNS. Lancet Neurol 7: 327–40 25. Darnell RB, Posner JB Paraneoplastic syndromes involving the nervous system (2003) N Engl J Med. 349(16):1543-54. 26. de Beukelaar Janet W., Sillevis Smitt Peter A. (2006) Managing Paraneoplastic Neurological Disorders. The Oncologist 11:292–305. 27. Deodhare S, O’Connor P, Ghazarian D, Bilbao JM (1996) Paraneoplastic limbic encephalitis in Hodgkin’s disease. Can J Neurol Sci 23:138-140. 28. Dropcho EJ, Kline L,Riser J (1993) Antineuronal (anti-Ri) antibodies in a patient with steroidresponsive opsoclonusmyoclonus. Neurology 43:207-211. 29. Fagius J,Westerberg E, Olsson Y (1983) Acute pandysautonomia and severe sensory deficit with poor recovery: a clinical, neurophysiological and pathological case study. J Neurol Neuros Psychiatry 46:725-733. 30. Folli F, Solimena M, Cofiell R, Austoni M, Tallini G, Fassetta G, Bates D, Cartlidge N, Bottazzo GF, Piccolo G, et al. (1993) Autoantibodies to a 128-kd synaptic protein in three women with the stiff-man syndrome and breast cancer. N Engl J Med 328:546–551. 100 31. Forsyth PA,Dalmau J, Graus F et al. (1997) Motor neuron syndromes in cancer patients. Ann Neurol 41: 722–730. 32. Giometto B, Taraloto B, Graus F (1999): Autoimmunity in paraneoplastic neurological syndromes [Review]. Brain Pathology 9(2):261-73. 33. Graus F, Dalmou J, Rene R, Tora M, Malats N, Verschuuren JJ, Cardenal F, Vinolas N, Garcia del Muro J, Vadell C, Mason WP, Rosell R, Posner JB, Real FX (1997) Anti-Hu antibodies in patients with small-cell lung cancer: association with complete response to therapy and improved survival. J Clin Oncol 15(8):2866-72. 34. Graus F, Delattre JY, Antoine JC, et al. (2004) Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry 75(8):1135-40. 35. Graus F, Elkon K, Lioberes P,Risbalta T,Torres A,Ussetti P,Valls J, Obach J,Agusti-Vidal A (1987) Neuronal antinuclear antibody (anti-Hu) in paraneoplastic encephalomyelitis simulating acute polyneuritis. Acta Neurol Scand 75:249-252. 36. Graus F, Keime-Guibert F, Rene R, et al. (2001) Anti-Hu-associated paraneoplastic encephalomyelitis: analysis of 200 patients. Brain 124:1138–1148. 37. Graus F,Cordon-Cardo C,Posner JB (1985) Neuronal antinuclear antibody in sensory neuronopathy from lung cancer. Neurology 35:538–543. 38. Greenberg HS (1984) Paraneoplastic cerebellar degeneration: a clinical and CT study. J Neuooncol 2:377-382. 39. Greenlee J, Brashear R (1983) Antibodies to cerebellar Purkinje cells in patients with paraneoplastic cerebellar degeneration and ovarian carcinoma. Ann Neurol 14:609-613. 40. Gultekin SH, Rosenfeld MR, Voltz R et al. (2000) Paraneoplastic limbic encephalitis: tumor association, neurological symptoms, and immunological findings in 50 patients. Brain 123:1481– 1494. 41. Hammer M,Larsen M, Stack C (1995) Outcome of children with opsoclonus-myoclonus regardless of etiology. Pediatr Neurol 13: 21-24. 42. Harloff A, Hummel S, Kleinschmidt M, Rauer S (2005) Anti-Ri antibodies and limbic encephalitis in a patient with carcinoid tumour of the lung. J Neurol. 252(11):1404-5. 43. Heckmann, J.G., Lang, C.J., Druschky, A., Claus, D., Bartels, O., and Neundorfer, B. (1997) Chorea resulting from paraneoplastic encephalitis. Mov. Disord. 12:464-466. 44. Herold Gerd und Mitarbeiter, Innere Medizin, 2007 45. Hersh B, Dalmau J,Dangond F, Gultekin S, Geller E,Wen PY (1994) Paraneoplastic opsoclonusmyoclonus associated with anti- Hu antibody. Neurology 44:1754-1755. 46. Hetzel DJ, Stanhope CR, O'Neill BP, Lennon VA (1990) Gynecologic cancer in patients with subacute cerebellar degeneration predicted by anti-Purkinje cell antibodies and limited in metastatic volume. Mayo Clin.Proc. 65:1558-63. 47. Hill CL, Zhang Y, Sigurgeirsson B, Pukkala E, Mellemkjaer L, Airio A, Evans S, Felson D (2001) Frequency of specific cancer types in dermatomyositis and polymyositis: a population-based study. Lancet 357:96–100. 48. Honnorat J, Antoine JC (2007) Paraneoplastic neurological syndromes. Orphanet J Rare Dis 2: 22. Published online 2007 May 4. doi: 10.1186/1750-1172-2-22. 101 49. Honnorat J,Antoine JC,Derrington E,Aguera M, Belin MF (1996) Antibodies to a subpopulation of glial cells and a 66 kDa developmental protein in patients with paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry 61:270-278. 50. Horwich RL, Buxton PH, Ryan GMS (1966) Cerebellar degeneration in Hodgkin’s disease. J Neurol Neurosurg Psychiatry 29:45-51. 51. Ivo W. Tremont-Lukats, Gregory N. Fuller, Teresa Ribalta, Pierre Giglio, and Morris D. Groves (2002) Paraneoplastic chorea: Case study with autopsy confirmation. Neuro-Oncology 4:192–195. 52. Jacobson DM,Thirkill CE,Tipping SJ (1990) A clinical triad to diagnose paraneoplastic retinopathy. Ann Neurol 28:162-167. 53. Kaiser R. (1999) Paraneoplastic neurologic syndromes. Diagnostic and pathogenetic significance of autoantibodies. Nervenarzt 70(8):688-701. 54. Keime-Guibert F, Graus F, Broet P, Rene P, Molinuevo JL, Ascaso C, Delattre JY (1999) Clinical outcome of patients with anti-Hu-associated encephalomyelitis after treatment of the tumor. Neurology 53(8):1719-23. 55. Keime-Guibert F, Graus F, Fleury A, Rene R, Honnorat J, Broet P, Delattre JY (2000) Treatment of paraneoplastic neurological syndromes with antineuronal antibodies (anti-Hu, anti-Yo) with a combination of immunoglobulins, cyclophosphamide, and methylprednisolone. J Neurol Neurosurg Psychiatry 68:479–482. 56. Ko Melissa W., MD, Josep Dalmau, MD, PhD, and Steven L. Galetta, MD (2008) NeuroOphthalmologic Manifestations of Paraneoplastic Syndromes. J Neuro-Ophthalmol 28:58–68. 57. Layzer R (1988) Stiff-man syndrome – an autoimmune disease? N Engl J Med 318:1060-1062. 58. Levy Y, Afek A, Sherer Y, et al. (1998) Malignant thymoma associated with autoimmune diseases: a retrospective study and review of the literature. Semin Arthritis Rheum 28:73-79. 59. Lisak RP, Mitchell M, Zweiman B, Asbury AK (1977) Guillain Barre syndrome and Hodgkin's disease; three cases with immunological studies. Ann Neurol 1:72-8. 60. Lladó, MD; P. Mannucci, MD; A.F. Carpentier, MD; S. Paris, BSc; Y. Blanco, MD; A. Saiz, MD; J.Y. Delattre, MD; and F. Graus, MD (2004) Value of Hu antibody determinations in the follow-up of paraneoplastic neurologic syndromes. Neurology 63:1947–1949. 61. Lucchinetti CF, Kimmel DW, Lennon VA (1998) Paraneoplastic and oncologic profiles of patients seropositive for type 1 antineuronal nuclear autoantibodies.Neurology 50:652–657. 62. Luque FA, Furneaux HM, Ferziger R, Rosenblum MK, Wray SH, Schold S Jr, et al. (1991) AntiRi: an antibody associated withparaneoplastic opsoclonus and breast cancer. Ann Neurol 29: 241±51. 63. Luque FA, Furneaux HM, Ferziger R, Rosenblum MK,Wray SH, Schold SC Jr, Glantz MJ, Jaeckle KA,Biran H, Lesser M (1991) Anti-Ri: an antibody associated with paraneoplastic opsoclonus and breast cancer. Ann Neurol 29:241-251. 64. Mason WP, Graus F, Lang B et al. (1997) Small-cell lung cancer, paraneoplastic cerebellar degeneration and the Lambert-Eaton myasthenic syndrome. Brain 120:1279–1300. 65. Newsom-Davis J,Mills KR (1993) Immunological associations of acquired neuromyotonia (Isaacs’ syndrome). Report of five cases and literature review. Brain 116:453-469. 66. Newsom-Davis J. Lambert-Eaton myasthenic syndrome. Rev Neurol (Paris). 2004;160:177–180. 102 67. Oh, Shin J., Dropcho, Edward J. and Claussen, Gwen C (1997) Anti-Hu-associated paraneoplastic sensory neuronopathy responding to early aggressive immunotherapie: Report of two cases and review of literature Department of Neurology, The University of Alabama. Muscle Nerve 20: 1576–1582. 68. O'Neill JH, Murray NM, Newsom-Davis J (1988) The Lambert-Eaton myasthenic syndrome. A review of 50 cases. Brain 111:577–596. 69. Peterson K, Rosenblum MK,Kotanides H,Posner J (1992) Paraneoplastic cerebellar degeneration. I. A clinical analysis of 55 anti-Yo antibody- positive patients. Neurology 42: 1931- 1937. 70. Pittock Sean J. Pittock, MD, Claudia F. Lucchinetti, MD, and Vanda A. Lennon, MD (2003) AntiNeuronal Nuclear Autoantibody Type 2: Paraneoplastic Accompaniments. Ann Neurol 53:580– 587. 71. Pittock SJ, Lucchinetti CF, Parisi JE, Benarroch EE, Mokri B, Stephan CL, Kim KK, Kilimann MW, Lennon VA (2005) Amphiphysin autoimmunity: paraneoplastic accompaniments. Ann Neurol. 58(1):96-107. 72. Pittock SJ, Kryzer TJ, Lennon VA (2004) Paraneoplastic antibodies coexist and predict cancer, not neurological syndrome. Ann Neurol. 56(5):715-9. 73. PNS Euronet[1], http://www.pnseuronet.org/professionals/be.html; Brainstem encephalitis 74. PNS Euronet[2], http://www.pnseuronet.org/professionals/mneuropathy.htm; Paraneoplastic sensory-motor neuropathies 75. PNS Euronet[3], http://www.pnseuronet.org/professionals/mnd.html; Motor neuron diseases 76. PNS Euronet[4], http://www.pnseuronet.org/professionals/dermatomyositis.htm; Dermatomyositis 77. Posner JB (1995) Paraneoplastic syndromes. In: Posner JB, ed. Neurologic complications of cancer. Philadelphia: F. A. Davis Company 353±85. 78. Pozo-Rosich P et al. (2003) Voltage-gated potassium channel antibodies in limbic encephalitis. Ann Neurol 54(4):530-3. 79. Prego L, Previtali SC, Nemni R, et al (2002) Autoantibodies to amphiphysin I and amphiphysin II in a patient with sensory-motor neuropathy. Eur. Neurol 47:196–200. 80. Pschyrembel, Klinisches Wörterbuch 259. Auflage, de Gruyter 2002 81. Rees JH, Hain SF, Johnson MR, Hughes RA, Costa DC, Ell PJ, Keir G, Rudge P (2001) The role of [18 F]fluoro-2-deoxyglucose-PET scanning in the diagnosis of paraneoplastic neurological disorders. Brain 124:2223–2231. doi: 10.1093/brain/124.11.2223. 82. Rizzo J, Gittinger J (1992) Selective immunohistochemical staining in the paraneoplastic retinopathy syndrome. Ophthalmology 99: 1286-1295. 83. Roberts WK, Darnell RB (2004) Neuroimmunology of the paraneoplastic neurological degenerations. Curr Opin Immunol. 16(5):616-22. 84. Rodriguez M,Truh L, O’Neill,BP, Lennon V (1988) Autoimmune paraneoplastic cerebellar degeneration: ultrastructural localization of antibody binding sites in Purkinje cells. Neurology 38:1380-1386. 85. Rogemond Véronique,1 and Honnorat Jérôme (2000) Anti-CV2 Autoantibodies and Paraneoplastic Neurological Syndromes. Clinical Reviews in Allergy and Immunology, Volume 19 103 86. Rojas I, Graus F, Keime-Guibert F et al. (2000) Long-term clinical outcome of paraneoplastic cerebellar degeneration and anti-Yo antibodies. Neurology 55:713–715. 87. Rojas-Marcos I, Rousseau A, Keime-Guibert F, et al. Spectrum of paraneoplastic neurological disorders in women with breast and gynaecological cancer. Medicine 2003;82:216–33. 88. Rosenblum MK (1993) Paraneoplasia and autoimmunologic injury of the nervous system: the antiHu syndrome. Brain Pathol 3:199-212. 89. Rosenfeld MR,Eichen JG,Wade DF et al. (2001) Molecular and clinical diversity in paraneoplas-tic immunity to Ma proteins. Ann Neurol 50:339–348. 90. Saiz A, Dalmau J, Butler MH et al. (1999) Anti-amphiphysin I antibodies in patients with paraneoplastic neurological disorders associated with small cell lung carcinoma. J Neurol Neurosurg Psychiatry 66:214–7. 91. Sculier J-P, Feld R., Evans WK, et al. (1987) Neurologic disorders in patients with small cell lung cancer. Cancer 60:2275-2283. 92. Senties-Madrid Horacio MD and Vega-Boada Felipe MD (2001) Paraneoplastic Syndromes Associated with Anti-Hu Antibodies. IMAJ 2001;3:94-103. 93. Shams'ili Setareh, Joost Grefkens, Bertie de Leeuw, Martin van den Bent, Herbert Hooijkaas, Bronno van der Holt, Charles Vecht and Peter Sillevis Smitt (2003) Paraneoplastic cerebellar degeneration associated with antineuronal antibodies: analysis of 50 patients. Brain 126, 1409±1418. 94. Shillito P, Molenaar PC,Vincent A, Leys K, Zheng W, van den Berg RJ, Plomp JJ, van Kempen GT, Chauplannaz G,Wintzen AR (1995) Acquired neuromyotonia: evidence for autoantibodies directed against K+ channels of peripheral nerves. Ann Neurol 38:714-722. 95. Sillevis Smitt Peter, Grefkens Joost, de Leeuw Bertie, van den Bent Martin, Putten van, Hooijkaas Herbert, Vecht Charles (2002) Survival and outcome in 73 anti-Hu positive patients with paraneoplastic encephalomyelitis/sensory neuronopathy. J Neurol 249:745–753. 96. Skeie GO, Freiburg A,Kolmerer B, Labeit S, Aarli JA, Appiah-Boadu S, Gilhus NE (1997) Titin transcripts in thymomas. J Autoimmun 10:551-557. 97. Stich O, Graus F, Rasiah C, Rauer S (2003) Qualitative evidence of anti- Yo-specific intrathecal antibody synthesis in patients with paraneoplastic cerebellar degeneration. J Neuroimmunol 141:165-169. 98. Stich Oliver, Jarius Sven, Kleer Barbara, Rasiah Christiane, Voltz Raymond, Rauer Sebastian (2007) Specific antibody index in cerebrospinal fluid from patients with central and peripheral paraneoplastic neurological syndromes. Journal of Neuroimmunology, Volume 183, Issues 12, February 2007, Pages 220-224 99. Stourac P, Kadanka Z, Palyza V (2001) Paraneoplastic neurological syndromes-patients' cohort profile in the Czech Republic. Acta Neurol Scand 104: 72-77. 100. Sutton IJ, Fursdon Davis CJ, Esiri MM et al. (2001) Anti-Yo antibodies and cerebellar degeneration in a man with adenocarcinoma of the esophagus. Ann Neurol 49:253–257. 101. Tan et al. (2008) Clinical spectrum of voltage-gated potassium channel autoimmunity. Neurology 70(20):1883-90. 104 102. Tanaka K, Tanaka M, Igarashi S, Onodera O, Miyatake T, Tsuji S (1995) Trial to establish an animal model of paraneoplastic cerebellar degeneation with anti-Yo antibody. 2. Passive transfer of murine mononuclear cells acitvated with recombinant Yo protein to paraneoplastic cerebellar degeneration lymphocytes in severe combined immunodeficiency mice. Clin Neurol Neurosurg 1995a. 97(1):101-5. 103. Tanaka M, Tanaka K, Onodera O, Tsuji S (1995) Trial to establish an animal model of paraneoplastic cerebellar degeneration with anti-Yo antibody. 1. Mouse strains bearing different MHC molecules produce antibodies on immunization with recominant Yo protein, but do not cause Purkinje cell loss. Clin Neurol Neurosurg 1995b. 97(1):95-100. 104. Terence J. O'Brien, Bill Pasaliaris, Anthony D'Apice, Edward Byrne (1995) Anti-Yo positive paraneoplastic cerebellar degeneration: a report of three cases and review of the literature. Journal of Clinical Neuroscience 2 (4):316-320. 105. Thirkill CE, FitzGerald P, Sergott RC et al. (1989) Cancer-associated retinopathy (CAR syndrome) with antibodies reacting with retinal, optic-nerve, and cancer cells. N Engl J med 321:1589–1594. 106. TrotterRL, Hendlin BA, Osterland CK (1976) Cerebellar degeneration with Hodgkin’s disease: an immuological study. Arch Neurol 33:660-661. 107. Tüzün Erdem and Dalmau Josep (2007) Limbic Encephalitis and Variants: Classification, Diagnosis and Treatment. The Neurologist 13:261–271. 108. Vedeler C. A., J. C. Antoine, B. Giometto, F. Graus, W. Grisold, I. K. Hart, J. Honnorat, P. A. E. Sillevis Smitt, J. J. G. M. Verschuuren and R. Voltz (2006) Management of paraneoplastic neurological syndromes: report of an EFNS Task Force. European Journal of Neurology 13:682– 690. 109. Vernino S,Tuite P,Adler CH et al. (2002) Paraneoplastic chorea associated with CRMP-5 neuronal antibody and lung carcinoma. Ann Neurol 51:625–630. 110. Voltz R. (2006) Intravenous immunoglobulin therapy in paraneoplastic neurological syndromes. J Neurol (Suppl 5):V/33–V/38. 111. Voltz R, Gultekin S,Posner J, Rosenfeld M,Dalmau J (1998) Sera of patients with testicular cancer and paraneoplastic limbic/brainstem encephalitis harbor an antineuronal antibody (anti-Ta) that recognizes a novel neuronal protein. J Neurol 245:379. 112. Voltz R, Gultekin SH,Rosenfeld MR et al. (1999) A serologic marker of paraneoplastic limbic and brain-stem encephalitis in patients with testicular cancer.N Engl Jmed 340:1788–1795. 113. Voltz RD (2002) Paraneoplastic neurological syndromes: an update on diagnosis, pathogenesis, and therapy. Lancet Neurology 1:294-305. 114. Voltz RD (2004) Paraneoplastic neurological autoimmune diseases. Dtsch. Med. Wochenschr. 129: 1743-1746. 115. Voltz RD, Albrich WC, Nagele A et al. (1997) Paraneoplastic myasthenia gravis: detection of antiMGT30 (titin) antibodies predicts thymic epithelial tumor. Neurology 49:1454–1457. 116. Willcox N, Schluep M,Ritter M, Schurman H,Newsom- Davis J, Christensson B (1987) Myasthenic and nonmyasthenic thymoma.An expansion of a minor cortical epithelial cell subset? Am J Pathol 127:447-460. 105 117. Younes-Mhenni S., M. F. Janier, L. Cinotti, J. C. Antoine, F. Tronc, V. Cottin, P. J. Ternamian, P. Trouillas and J. Honnorat (2004) FDG-PET improves tumour detection in patients with paraneoplastic neurological syndromes. Brain 127, 2331–2338. 118. Younger DS J, Inhirami G, Sherman WH, Hays AP (1994) Anti-Hu associated peripheral nerve and muscle microvasculitis. Neurology 44:181±3. 119. Yu Z, Kryzer TJ, Griesmann GE et al. (2001) CRMP-5 neuronal autoantibody: marker of lung cancer and thymoma-related autoimmunity. Ann Neurol 49:146–154. 106 7. Danksagung Ein herzlicher Dank gilt meinem Doktorvater Herrn Prof. Sebastian Rauer für die Überlassung des Themas und die Möglichkeit, meine Doktorarbeit in seiner Abteilung zu erstellen. Ganz besonders bedanken möchte ich mich an dieser Stelle bei meinem Betreuer Dr. Oliver Stich. Nur mit seiner freundlichen und hilfsbereiten Unterstützung war es mir möglich, diese Dissertation durchzuführen. Seine scheinbar unerschöpfliche Geduld, seine stets konstruktive Kritik und seine Ratschläge waren für mich eine große Hilfe, vor allem in Situationen, in denen ich mit meinem Latein am Ende war. Bei Fr. Prof. Almut Zeeck bedanke ich mich für die Übernahme der Zweitkorrektur meiner Dissertation. Ein Dank gilt auch Prof. Schulte-Mönting für die freundliche Beratung und Unterstützung in statistischen Fragen. Und zuletzt möchte ich natürlich all jenen danken, die mir während der Arbeit an dieser Dissertation auf irgendeine Art beratend oder unterstützend zur Seite standen, insbesondere jedoch meinen Eltern und Geschwistern, ohne deren Verständnis und Unterstützung ich dieses Ziel wohl nicht erreicht hätte. Patricia Bischler; Offenburg, Mai 2010 107 Die Seite 108 (Lebenslauf) enthält persönliche Daten. Sie ist deshalb nicht Bestandteil der Online-Veröffentlichung. 108 9. Anhang - Tabelle 21 [Quelldaten] 109 m m w m m w w w m w m w m w w m w w w w m 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 m w 18 43 w 17 m m 16 42 w 15 w m 14 m m 13 41 m 12 40 m 11 7 w m 6 10 w 5 w w 4 m m 3 9 w 2 8 w m 1 Geschlecht Nr. +1,5 SCLC Limbische Enzephalitis [Hu?] Hu Hu Hu Hu Hu Hu Hu CV-2 Hu Ri Limbische Enzephalitis Sensomotorische Neuropathie Autonome Neuropathie unklare psychiatrische Symptomatik Subakute Kleinhirndegeneration Subakute Kleinhirndegeneration, Sensomotorische Neuropathie Subakute Retimadegeneration Subakute sensorische Neuropathie Subakute sensorische Neuropathie Subakute Kleinhirndegeneration Choreatisches Syndrom Hirnstammenzephalitis Subakute Kleinhirndegeneration Yo, CV-2 Hu Hu Subakute Kleinhirndegeneration Hirnstammenzephalitis, Sensomotorische Neuropathie Sensomotorische Neuropathie Enzephalomyelitis Hirnstammenzephalitis Sensomotorische Neuropathie Subakute Kleinhirndegeneration, Sensomotorische Neuropathie Subakute Kleinhirndegeneration Subakute Kleinhirndegeneration Enzephalomyelitis, Sensomotorische Neuropathie Sensomotorische Neuropathie Hirnstammenzephalitis Subakute Kleinhirndegeneration, Subakute sensorische Neuropathie Limbische Enzephalitis Subakute Kleinhirndegeneration, Subakute sensorische Neuropathie Hirnstammenzephalitis Subakute sensorische Neuropathie Chronische Polyradikulitis Autonome Neuropathie Sensomotorische Neuropathie Subakute Kleinhirndegeneration, Sensomotorische Neuropathie Sensomotorische Neuropathie Subakute Kleinhirndegeneration, Subakute sensorische Neuropathie Limbische Enzephalitis, Sensomotorische Neuropathie Enzephalomyelitis ------+30,5 PET+ [V.a. Metastasen eines Ovarial- oder Blasen-Ca] kein Tumorhinweis kein Tumorhinweis M. Hodgkin -4 --- kein Tumorhinweis NSCLC +1 +8 +8 +2 +0,5 +3 +9 -7 SCLC NSCLC SCLC Mamma-Ca SCLC SCLC NSCLC SCLC -120 --- PET+ im Bereich Thoraxwand und Pleura Mamma-Ca +2 SCLC +33,5 +27,8 NSCLC --- LK-Metastasen eines SCLC +9 --- -23 +43,5 +53,5 +4 +2,5 +10 +3 +14 kein Tumorhinweis auffällige Autopsie mediastinaler LK LK-Metastasen eines SCLC Ovarial-Ca SCLC SCLC SCLC SCLC NSCLC SCLC SCLC +10,5 --- kein Tumorhinweis SCLC -118 +133 NSCLC SCLC --- --- kein Tumorhinweis Sensomotorische Neuropathie PET + [Ileozoekalpol/hiläre und medistinale LK] --- kein Tumorhinweis Subakute sensorische Neuropathie Hu Hu Hu Yo CV-2 Hu Hu Hu Yo CV-2, Hu Hu Hu Hu Hu Hu Hu Amphiphysin Hu Ri Hu Hu Amhiphysin Hu Hu CV-2 +4 +12 Enzephalomyelitis Ri, Hu, Amphiphysin NSCLC +10,5 --- SCLC unbekannt Autopsie: V.a. primäres Lymphom Intervall PNS-ManifestationTumordiagnose (Monate) Ovarial-Ca Tumor Mamma-Ca Sensomotorische Neuropathie Subakute Kleinhirndegeneration Anosmie, Pupillotonie, Potenzminderung, Sinusbradykardie Sensomotorische Neuropathie PNS Hu Hu [Hu?] [Yo?] AK --- --betonte Furchung des Kleinhirns --temporal li. verstrichene Rinden-Mark-Grenze --- --- --- --Signal+ [Höhe BWK 3-4] Signal+ [symmetr. im Bereich Ncl. caudatus] Kleinhirnatrophie --Signal+ [Marklager/ sub kortikal bis kortikal] Atrophie des Kleinhirnwurmes mäßiggradige Kleinhirnatrophie --- --- Kleinhirnatrophie --- --- --- --Signal+ [Höhe BWK 2-4] --- --- --- --- --- --- --- Kleinhirnatrophie geringe frontotemporale Atrophie --- --temporal betonte Atrophie --Signal+ [temporal und Inselrinde links] --- --- --- ----- MRT-Schädel/WS --- 57 370 320 2060 49 1900 509 292 685 419 49 630 516 207 313 559 1024 664 32 56 1044 520 839 61 1800 1352 157 79 --- 657 125 480 --- 473 572 46 500 512 --- 573 --- 1044 Liquoreiweiß (mg/l) --- k.A. 0,7 0,87 k.A. k.A. 0,52 0,47 0,63 k.A. 1 0,81 0,58 1,4 0,53 0,47 0.47 0,7 2,3 0,48 k.A. k.A. 3,74 1,6 0,8 k.A. 0,63 k.A. 0,45 --- 0,51 k.A. 0,47 --- 0,38 k.A. 0,49 0,66 1,12 --- 0,61 --- 1,05 Ig-GIndex --- - + + - k.A. - - + - + + +[L+S] + - - + + + - + + +[L+S] + + - - + + --- k.A. - - --- - + - + + --- + --- +[L+S] OKB unbek. neurol. onkol. unbek. kein Ansprechen partielles/transientes Ansprechen kein Ansprechen kein Ansprechen onkol. kein Ansprechen kein Ansprechen unbekannt unklar unbek. 12 unbek. unbekannt 33,6 onkol. partielles/transientes Ansprechen 0,5 unbekannt 1,8 38,4 5,5 unbek. kein Ansprechen 146 lebend kein Ansprechen 38,4 21,6 lebend unbek. 59,9 8,4 6 33,6 143,3 3 unbekannt 15 117,9 196,8 31,2 4,3 4,8 157,8 66 8,4 57,7 unbekannt 39,6 14,4 30 18 39,6 133,2 92,4 4,8 unbekannt 12,8 40,1 16,4 20,4 8 unbekannt Überlebenszeit seit Erst manifestation (Mon.) Stabilisierung Stabilisierung onkol. unbek. unbek. unbek. onkol. kein Ansprechen partielles/transientes Ansprechen partielles/transientes Ansprechen partielles/transientes Ansprechen partielles/transientes Ansprechen lebend unbek. unbek. lebend lebend Stabilisierung kein Ansprechen kein Ansprechen kein Ansprechen partielles/transientes Ansprechen Stabilisierung onkol. partielles/transientes Ansprechen lebend neurol. kein Ansprechen neurol. onkol. partielles/transientes Ansprechen kein Ansprechen onkol. kein Ansprechen lebend unbek. kein Ansprechen Stabilisierung unbek. unbek. kein Ansprechen kein Ansprechen onkol. kein Ansprechen kein Ansprechen unbek. onkol. kein Ansprechen unbek. lebend lebend lebend onkol. unbek. Todesursache unbek. kein Ansprechen kein Ansprechen Stabilisierung partielles/transientes Ansprechen partielles/transientes Ansprechen unbekannt unbekannt Ansprechen des PNS auf Therapie PNS u/o Tumor Tabelle 21 Quelldaten