Teil 11
Werbung

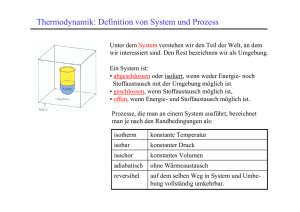
Wärmelehre II Die Wärmelehre (bzw. die Thermodynamik) leidet etwas unter den verschiedensten Begriffen, die in ihr auftauchen. Diese sind soweit noch nicht alle aufgetreten Vorhang auf! Die neu auftretenden Begriffe drücken alle dieselbe Physik aus, nur mit verschiedenen Randbedingungen, z. b. konstantem Druck, konstanter Temperatur. Bevor wir aber die ganze Geschichte nochmals aufrollen, betrachten wir noch einmal die Entropie, wie man sie im Lichte der statistischen Thermodynamik auch verstehen kann. Entropie II Wie groß ist die Wahrscheinlichkeit, dass alle n = 21 Moleküle gleichzeitig wieder in der linken Hälfte sind? Die Wahrscheinlichkeit, dass ein Molekül i sich dort aufhält, ist pi = 1/2. Die Wahrscheinlichkeit, dass neben Molekül i auch Molekül j gerade in der linken Hälfte ist, ist pij = pipj = (1/2)2. Die Wahrscheinlichkeit, dass sich alle Teilchen in der linken Hälfte aufhalten, ist also p = Πnipi = (1/2)n = (1/2)21 ≈ 4.6 · 10−7. Befindet sich im Kasten ein Mol Gas, also N ≈ 6 · 1023 Moleküle, so ist die Wahrscheinlichkeit, dass sich alle N Moleküle in der linken Hälfte 23 aufhalten pN = (1/2)N ≈ 2−6·10 , eine sehr, sehr, sehr, sehr kleine Zahl. . . Repetition Carnot-Prozess Die vier Phasen des Carnot Prozesses sind • isotherme Expansion: Q1 = N kT1 ln VVB A • adiabatische Expansion: Q = 0 • isotherme Kompression: Q2 = N kT2 ln VVC D • adiabatische Kompression: Q = 0 Damit lautet die Summe der aufgenommenen und abgegebenen Wärme Q1 Q2 VB VD + = N k ln + N k ln =0 T1 T2 VA VC weil wir ja auch gesehen haben, dass VC /VD = VB /VA und weil Q = 0 entlang den adiabatischen Ästen. Damit können wir für den gesamten Carnotzyklus schreiben X Qi Ti bzw. als Integral = 0, δQrev = 0, T wo der Index rev andeuten soll, dass die Herleitung nur für reversible Prozesse durchgeführt worden ist. I Thermodynamische Definition der Entropie Alle Zustandsgrößen Z wie Druck, Temperatur, Stoffmenge, Energie etc. in einem reversiblen Prozess (z. B. ein Carnotprozess) sind am Ende des Prozesses wieder gleich, also kann geschrieben werden, I dZ = 0. H Für alle anderen Größen wie Arbeit oder Wärmemenge gilt dies nicht, dW 6= 0. Interessanterweise gilt für die Kombination Q T aber derselbe Sachverhalt wie für Zustandsgrößen, wir haben als eine neue Zustandsvariable gefunden, die Entropie S. δQ oder auch δQ = T dS dS = T irreversibel 1 2 In diesem Zyklus, der aus einem ersten irreversiblen und einem folgenden reversiblen Prozess besteht, muss also gelten I reversibel δQirrev = T Z 1 2 δQirrev + T Z 2 1 δQrev <0 T denn ein irreversibler Prozess hat δQ < 0. Wir können nun die neue Zustandsgröße Entropie in den Zuständen 1 und 2, S1 und S2, einsetzen Z 1 Z 1 2 2 δQirrev + S1 − S2 < 0 T δQirrev < S2 − S1 = ∆S T Ferner nehmen wir an, dass der irreversible Prozess von 1 nach 2 thermisch isoliert stattfinde, also δQirrev = 0 gilt. Dann erhalten wir ∆S = S2 − S1 > 0, ein isoliertes System kann sich nur so entwickeln, dass ∆S > 0. Kommt im Zyklus kein irreversibler Vorgang vor, so kann ∆S = 0 sein. Zusammenfassend: • irreversibler Prozess: ∆S > 0 • reversibler Prozess: ∆S = 0 • (unmöglicher Prozess: ∆S < 0) Dies ist der Inhalt des zweiten thermodynamischen Hauptsatzes. Statistische Definition der Entropie Wir unterteilen ein makroskopisches System in viele kleine Untersysteme, welche alle groß genug sind, damit in ihnen eine Mittelbildung noch Sinn macht. Diese Unterteilung geschieht nicht im eigentlichen Raum, sondern in einem Zustandsraum, einem Raum aller möglichen Zustände. Ein Beispiel dafür ist der Phasenraum, der alle möglichen Kombinationen von (~r, ~v ) aufspannt. Er ist also im Allgemeinen sechs-dimensional! Diesen Phasenraum unterteilen wir in lauter kleine Unterräume (∆~x, ∆~ p). Die Anzahl von verschiedenen Zuständen, die den makrospkopischen Gesamtzustand ermöglichen, nennt man statistisches Ensemble. Für jedes Untersystem lässt sich eine Wahrscheinlichkeit angeben, dass es in einem gegebenen Zustand ist, z. B. eine mittlere Energie Ē aufweist, bzw. eine Wahrscheinlichkeit angeben, dass es diese nicht aufweist. Damit lässt sich für jeden Zustand eine Wahrscheinlichkeit angeben, dass er in x Untersystemen auftritt (oder eben auch nicht). Damit ist klar, dass ein makroskopischer Zustand desto wahrscheinlicher wird, je größer die Anzahl statistischer Ensembles wird. (Vgl. das einführende Beispiel mit Gas in den beiden Hälften eines Behälters.) Die Anzahl dieser statistischen Ensembles wird statistische Wahrscheinlichkeit Γ genannt. Γ hat auch etwas mit Ordnung zu tun. Weil die perfekte Anrodnung aller Moleküle in einem Gas nur auf eine Art geschehen kann, ist die Anzahl statistischer Ensembles, die dies ermöglichen, gleich ein, Γ ist also sehr klein verglichen mit einem Γ für einen “normalen”, d. h. ungeordneten Zustand eines Gases, Γ erreicht in diesem Fall sogar sein Minimum. Nun kann man zeigen, dass die Entropie eine Funktion der statistischen Wahrscheinlichkeit ist, S = f (Γ), wo wir f jetzt bestimmen wollen. Wir betrachten wieder unseren Behälter. Slinks = f (Γlinks), Srechts = f (Γrechts), und die Summer der Entropien muss die Entropie des gesamten Systems sein, S = Slinks + Srechts = f (Γ). Wahrscheinlichkeiten sind aber multiplikativ, also muss auch gelten Γ = Γlinks · Γrechts. Diese sog. Funktionalgleichung wird z. B. durch den Logarithmus erfüllt, also definiert man . S = k ln Γ, wo k die Boltzmann-Konstante ist. Zum Vergleich der thermodynamischen und der statistischen Definition überlegen wir uns nochmal, wie groß die Wahrscheinlichkeit und die statistische Wahrscheinlichkeit ist, dass sich ein Teilchen in einem bestimmten Untervolumen V1 = V2/m befindet. Befindet sich genau ein Teilechen in V2, so ist die Wahrscheinlichkeit 1/m. Befinden sich zwei Teilchen in V2, so (1/m)2, allg. ist die Wahrscheinlichkeit p = (1/m)N , wo N die Anzahl Teilchen ist. Die statistische Wahrscheinlichkeit Γ ist gerade der Kehrwert dieses Ausdruckes, Γ = mN und gibt an, wieviel Mal wahrscheinlicher es ist, dass sich alle Teilchen in V2 aufhalten (Wahrscheinlichkeit 1) als dass sie sich gerade alle in V2 aufhalten. Wir können auch den natürlichen Logarithmus von Γ bestimmen, µ ¶ µ ¶ V2 nR V2 = · ln ln Γ = N · ln m = N · ln V1 k V1 von daher kommt auch die Definition der Entropie als µ ¶ V2 ∆S = k ln Γ = nR ln V1 Im thermodynamischen Bild hatten wir schon gelernt, dass in einem Prozess, in dem sich ein Gas von einem Volumen V1 auch ein Volumen V2 ausdehnt, sich die Entropie wie folgt ändert. ∆S = S2 − S1 = Z 1 2 δQ = T Z 1 2 dU + Z Z 1 2 pdV T Dabei ist aber dU = Cv dT = 0 weil der Prozess isotherm passiert, ferner ist p = nRT und folglich V ∆S = Z 1 2 µ ¶ nRdV V2 = nR ln , V V1 was dasselbe Resultat liefert, wie im statistischen Fall. Beispiel I: Mischen von Gasen In einem Wärmebad konstanter Temperatur befinde sich ein Behälter mit einer sehr dünnen Trennwand, welche zwei verschiedene Gase der Mengen n1 und n2 in den Volumina V1 und V2 bei identischem Druck trennt. Nun wird die Wand entfernt. Wie ändert sich die Entropie für n1 = n2 und V1 = V2? Nach den vorherigen Überlegungen dehnt sich Gas 1 auf das gesamte Volumen 2 aus, wie auch Gas 2, womit die Entropie für Gas 1 um ∆S1 = n1R ln( V1V+V ) 1 2 ) und somit zunimmt, für Gas 2 gilt ähnlich ∆S2 = n2R ln( V1V+V 2 ∆S = ∆S1 + ∆S2 = 2 n R ln 2 Beispiel II: Schmelzen von Eis Am Rande einer Skipiste schmilzt bei 0◦C ein Kilogramm Eis. Wie groß ist die Entropieänderung? Weil das Schmelzen bei 0◦C geschieht, reicht eine infinitesimale Temperaturänderung, um das Eis zu schmelzen, oder eine infinitesimale Abkühlung um es zum Erstarren zu bringen. Der Prozess ist also reversibel. Damit ∆S = SWasser − SEis = Z 0 Q δQ 1 = T T Z 0 Q Q δQ = T Dabei ist Q natürlich gerade die Schmelzwärme, Q m Lf 1 · 3.338 · 105 kg J = = und damit ∆S = 1222 J/K T T T K kg Intermezzo: Das griechische Alphabet Buchstaben A, α B, β Γ, γ ∆, δ E, ² Z, ζ H, η Θ, θ Name Alpha Beta Gamma Delta Epsilon Zeta Eta Theta Buchstaben I, ι K, κ Λ, λ M, µ N, ν Ξ, ξ O, o Π, π Name Iota Kappa Lambda My Ny Xi Omikron Pi Beispiele: Γυναικων (Frau), Aνδρων (Mann) Buchstaben P, ρ Σ, σ T, τ Υ, υ Φ, φ X, χ Ψ, ψ Ω, ω Name Rho sigma Tau Ypsilon Phi Chi Psi Omega Intermezzo: Warum mal d und mal δ? Die einem Körper zugeführte Wärmemenge δQ trägt zu einer Veränderung der Energie des Körpers bei. Nach dem ersten thermodynamischen Hauptsatz ist die änderung der Energie gleich der zugeführten Wärmemenge und der hineingestecketen Arbeit, dU = δQ + δW = δQ − pdV Die Energie des Körpers bleibt über einen Kreisprozess erhalten, er hängt nur von Anfangs- und Endpunkt eines Prozesses ab. Weil die Wärmemenge und die Arbeit aber durchaus von der Natur des Prozesses abhängig sind, sie unterscheiden sich z. B. in adiabatischen und in isobaren Prozessen, sind sie keine sog. vollständigen Differentiale einer Größe. Die Energie aber schon, sie ist eine sog. Zustandsgröße. Zustandsgrößen werden als vollst. Differential geschrieben, Nicht-Zustandsgrößen eben nicht. Das vollst. Differential einer Funktion f (x1, x2, x3, . . .) ist ∂f ∂f ∂f df = dx1 + dx2 + dx2 + . . . ∂x1 ∂x2 ∂x2 Offensichtlich muss für ein vollständiges Differential gelten I df (~x) = 0 über jede geschlossene Kurve, denn dann sind ja Anfangs- und Endpunkt identisch. Die Notation δQ und δW unterscheidet diese Größen von den Zustandsgrößen, damit man nicht in Versuchung gerät, sie als Zustandsgrößen aufzufassen. Arbeit, innere Energie und Wärmemenge: δW = dU − δQ Schwierigkeiten im Verständnis der Thermodynamik erklären sich hauptsächlich dadurch, dass der Energiebegriff hier nicht mehr so einfach ist, wie in der Mechanik. Während das Anheben eines Steins in der Mechanik einfach zu einer Erhöhung der potentiellen Energie desselben führt, die erst noch ausschließlich von Anfangs- und Endzustand abhängen. Leider ist ein ähnlicher Sachverhalt in der Thermodynamik deutlich komplizierter, weil die Energie eines Gases je nach Prozess verschieden in geleistete oder aufgenommene Arbeit und aufgenommene oder abgegebene Wärme unterteilt werden kann. Man stelle sich einen Carnotprozess mit einem Stein vor! Die totale Energieänderung ist natürlich gleich Null, die abgegebene oder aufgenommene Wärme oder Arbeit muss aber nicht zwingend verschwinden. Bleibt bei einem Prozess das Volumen erhalten, so ist die vom Körper aufgenommene Wärme gerade gleich der Änderung seiner Energie. Die innere Energie (isochore Prozesse) Findet in einem Prozess keine Volumenveränderung (isochorer Prozess) statt, so ist die geleistete Arbeit gleich Null, folglich dU = δQ. Dies ist aber gerade dU = CV dT , also erhalten wir die bekannte Beziehung µ ¶ ∂U CV = . ∂T V Ferner gilt mit dU = δQ − P dV und δQ ≤ T dS für konstantes Volumen (dV = 0) dU ≤ T dS, für adiabatische Prozesse ist sogar dU ≤ 0 (denn dS = 0). Im thermodynamischen Gleichgewicht eines Systems mit konstanter Entropie und Volumen ist also die innere Energie minimal. Umgekehrt kommt jede Veränderung des Systems bei konstanter innerer Energie und Volumen erst zum Stillstand, wenn die Entropie maximal wird (dS ≥ 0). Die Enthalpie (isobare Prozesse) Bleibt bei einem Prozess nicht das Volume konstant, sondern der Druck, so kann die Wärmemenge als Differential δQ = d(U + pV ) = dH einer Größe H = U + pV geschrieben werden, die Enthalpie genannt wird. Also ist die Änderung der Enthalpie (H) bei Prozessen bei konstantem Druck gleich der Wärmemenge, die durch den Körper aufgenommen wird, denn dH = dU + pdV + V dp = dU + pdV = δQ und folglich (aus den gleichen Überlegungen wie für die innere Energie) µ ¶ ∂H Cp = ∂T p Vergleich innere Energie und Enthalpie Für ein System, dem eine bestimmte Wärmemenge δQ zugeführt wird, gilt: • bei konstantem Volumen dU = δQ − pdV = δQ also dU = δQ • bei konstantem Druck dH = dU + pdV = δQ − pdV + pdV = δQ also dH = δQ Die Schmelzwärme bei konstantem Druck heißt deshalb zum Beispiel oft auch Schmelzenthalpie. Ist der Prozess adiabatisch (δQ = 0), so wird H minimal. Die freie Energie (T und V konstant) Nun halten wir neben dem Volumen (→ innere Energie erhalten) auch noch das Volumen konstant. Z 1 2 δQ ≤ S2 − S1 wegen T = const. also Q = T Z 2 δQ ≤ T (S2 − S1) 1 wo das Gleichheitszeichen für reversible Prozesse gilt. In δW = dU − δQ δW ≥ U2 − U1 − T (S2 − S1) die Arbeit, die nötig ist, um ein System vom Zustand 1 in den Zustand 2 zu bringen. Wir können nun eine weitere Zustandsgröße F definieren, die freie Energie . F = U − T S, δW ≥ F2 − F1 = ∆F, die angibt • wie viel Arbeit ein isothermes System nach außen abgeben kann (nämlich eben gerade ∆F ) • wie viel Arbeit in das System hineingesteckt worden ist (nämlich eben gerade ∆F ) Weil F offenbar die verfügbare Energie ist, eben die “freie” Energie, heißt F eben freie Energie. Ist ∆V = 0, so gilt wegen 0 = δW ≥ dF , dass die freie Energie abnimmt (irreversible Prozesse) oder erhalten bleibt (reversible Prozesse). Wegen T S = U − F nimmt die Entropie zu. Damit herrscht im thermodynamischen Gleichgewicht ein Zustand minimaler freier Energie. Die freie Enthalpie (T und p konstant) Prozesse, in denen die Temperatur und der Druck konstant bleiben, sind in der Natur sehr häufig. In Analogie zu Definition der Enthalpie H = U + pV und der Einführung der freien Energie F = U −T S führen wir nun die sog. freie Enthalpie G = U − T S + pV = F + pV = H − T S ein. Bei konstanter Temperatur und Druck gilt: dG = dU − T dS + pdV = dF + pdV Bei konstantem Druck ist die verrichtete Arbeit W = −p (V2 − V1), wir können dies wegen der konstanten Temperatur auch schreiben als pV2 − pV1 ≤ F1 − F2, bzw. (F2 + pV2) − (F1 + pV1) ≤ 0 weshalb G = F + pV in einem isothermen und isobaren Prozess minimal ist, denn G2 ≤ G1 . Chemische Reaktionen In chemischen Reaktionen muss die Stoffmenge nicht unbedingt erhalten bleiben, 2H2 + O2 → 2H2O ist ein Beispiel in dem n1 = 2 plus n2 = 1 Mol zu n3 = 2 Mol werden. Es ist klar, dass in einem solchen Gas die bisher beschriebenen Gesetze etwas modifiziert werden müssen. Betrachten wir z. B. die innere Energie, die nun von drei Zustandsgrößen abhängt, der Entropie S, dem Volumen V und der Stoffmenge n. Das Differential von U lautet nun µ ¶ ∂U dU = T dS − P dV + dn ∂n S,V Die Klammer wird of abgekürzt als . µ= µ ∂U ∂n ¶ S,V und heisst chemisches Potential. Besteht das Gas aus mehreren Stoffen i, so hat jeder Stoff eine bestimmte Stoffmenge und folglich dU = T dS − pdV + X µidni i Offensichtlich kann nun auch in einem abgeschlossenen System (δQ = 0) die Entropie sich ändern, wenn eine Reaktion die Mischverhältnisse der verschiedenen Stoffe verändert. Thermodynamische Potentiale Die Größen S, U , H, F und G heißen thermodynamische Potentiale weil sie es erlauben, formal ähnliche Beziehungen aufzustellen, wie ein Potential dies in der Mechanik erlaubt. Sie beschreiben zwar alle dieselbe Physik (die Thermodynamik) aber eben für verschieden ablaufende Prozesse. Aus ihnen können alle anderen Zustandsgrößen hergeleitet werden, ein Bsp.: dU T µ ∂U ∂S ¶ µ ¶ µ ¶ ∂U ∂U dS + dV + dn = ∂V ∂n V,n S,n S,V µ ¶ µ ¶ µ ¶ ∂U ∂U ∂U = ; p=− ; µ= ∂S V,n ∂V S,n ∂n S,V Wir können das Differential von U , dU auch nach dS auflösen und finden dS 1 T "µ ¶ #−1 " µ ∂U ∂U · dU − = ∂S V,n ∂V µ ¶ µ ¶ ∂S ∂S p = ; = ; ∂U V,n T ∂V U,n ¶ µ ¶ ∂U dV − dn ∂n S,n S,V µ ¶ ∂S µ =− T ∂n U,V # Ähnlich können alle Zustandsvariablen auch in Abhängigkeit von der Enthalpie geschrieben werden, µ ¶ µ ¶ µ ¶ ∂H ∂H ∂H dS + dp + dn dH = ∂S V,n ∂V S,n ∂n S,V µ ¶ µ ¶ µ ¶ ∂H ∂H ∂H T = ; V = ; µ= , ∂S p,n ∂p S,n ∂n S,p und dasselbe für die freie Energie dF S µ ¶ µ ¶ µ ¶ µ ¶ ∂F ∂F ∂F = dT + dV + dn ∂T V,n ∂V S,n ∂n S,V µ ¶ µ ¶ µ ¶ ∂F ∂F ∂F = − ; p=− ; µ= ∂T V,n ∂V T,n ∂n T,V und natürlich auch für die freie Enthalpie ¶ µ ¶ ∂G ∂G ∂G dT + dp + dn ∂T p,n ∂p T,n ∂n T,p µ ¶ µ ¶ µ ¶ ∂G ∂G ∂G = − ; V = ; µ= ∂T p,n ∂p T,n ∂n T,p dG = S µ Beispiel: Die Strahlung eines schwarzen Körpers Wir fassen die Strahlung als Gas auf, als Gas von Photonen. Denn auch Photonen üben einen Druck aus, das Gas verfügt über eine innere Energie, etc. Strahlung verhält sich oft wie ein Gas! Auch Photonen haben einen Impuls p~, treffen sie auf eine Wand und werden dort reflektiert, so ändert sich ihr Impuls auch um 2px. Dies entspricht einem Kraftstoß F dt. Die Zeit dt ist gerade die Zeit, die das Photon braucht, um zur Wand zu kommen. Folglich ist die Anzahl Stöße von Photonen mit der Wand gleich der Anzahl Photonen, die die Wand innerhalb von dt erreichen können, nämlich nAvxdt, wo n die Teilchenzahldichte bedeutet. Mit P = F/A erhalten wir also den Druck auf eine Wand in x-Richtung Px = 2npxvx. Den Druck an jeder Stelle finden wir wieder durch Mittelung aller Impulse und Geschwindigkeiten, P V = N h~ p · ~v i/3 Nun stellt sich die frage, was denn für Photonen p~ · ~v bedeutet? Impuls und Geschwindigkeit zeigen in dieselbe Richtung, die Geschwindigkeit ist natürlich c und folglich (wie für alle relativistischen Teilehcne ohne Ruhemasse) E = pc. Also ist der Druck gerade gleich einem Drittel der inneren Energie des Photonengases! U PV = 3 Wir können nun mit Hilfe des ersten Hauptsatzes und unter Anwendung der freien Energie zeigen, dass die Energiedichte u = U/V proportional zur vierten Potenz der Temperatur ist, u(T ) ∝ T 4. Denn µ ∂U ∂V ¶ = u(T ) T aus dem erstan Hauptsatz haben wir dU = T dS − pdV und folglich µ ∂U ∂V ¶ = u(T ) = T T µ ∂S ∂V ¶ − p. T Jetzt haben wir gesehen, dass die freie Energie dF = −SdT − pdV ein vollst. Differential ist. Für vollst. Differentiale gilt df ∂A ∂y = ∂f ∂f dx + dy ∂x ∂y ∂f 2 ∂f 2 = A(x, y)dx + B(x, y)dy und wegen = gilt ∂x∂y ∂y∂x µ ¶ µ ¶ ∂B ∂S ∂p = und somit = ∂x ∂V T ∂T V Dies setzen wir oben ein, µ ∂U ∂V ¶ = u(T ) = T T µ ∂p ∂T ¶ − p. V Wie wir aber schon herausgefunden haben, ist der Druck der Wärmestrahlung gegeben durch p = u/3, womit wir erhalten, dass T du u u = − 3 dT 3 du dT = 4 u T ln u = 4 ln T + const. u = const.T 4