Festkörperphysik
Werbung

Festkörperphysik
Juni 2010
Die Festkörperphysik ist die wohl umfangreichste Einzeldisziplin der gegenwärtigen Physik,
mit Tausenden von aktiven Forschern und einer nahezu unüberschaubaren Zahl von Publikationen. Die stürmische Entwicklung der Festkörperphysik begann in den 1930er Jahren,
also nach der Etablierung der Quantentheorie (obwohl natürlich auch schon früher feste
Stoffe das Interesse der Forscher geweckt haben, insbesondere in der Chemie und Materialkunde). Diese Entwicklung hält bis heute an; in der Tat ist die Festkörperphysik dasjenige
Gebiet der Physik, das am Stärksten unseren Alltag beeinflusst. Man denke an die Erfindung des Transistors, der Magnetspeichertechnik, an hochfeste Werkstoffe, etc. Gemessen
am Umfang des Stoffgebiets kann diese Vorlesung nur einen ganz kleinen Ausschnitt beleuchten.
1
Chemische Bindung in Festkörpern
Was hält die Atome oder Moleküle eines Festkörpers zusammen? Um das zu verstehen,
braucht man auf jeden Fall die Quantentheorie. Man erreicht eine grobe Typisierung der
Festkörper, indem man verschiedene Bindungstypen unterscheidet (siehe Ibach-Lüth, Kap.
1). Das ist praktisch, aber nicht streng durchführbar. Die Metallbindung, die kovalente
Bindung und die Ionenbindung sind streng genommen nur verschiedenen Grenzfälle, bei
denen jeweils an anderer Effekt den Hauptbeitrag zur Bindungsenergie bringt. Aus quantenmechanischer Perspektive werden sie mit den gleichen Methoden behandelt.
Kovalente Bindung: Zwischen Nachbaratomen in einem kovalenten Festkörper besteht
eine chemische Bindung ähnlich zu der Molekülbindung. Es gibt Paare von Elektronen (mit entgegengesetztem Spin), die sich hauptsächlich auf der Verbindungsachse
zwischen zwei Atomen aufhalten.
Beispiele: Diamant, Silizium
Metallbindung: Im Gegensatz zur kovalenten Bindung ist die Wellenfunktion der Valenzelektronen über viele Atome ausgedehnt. Für das Zustandekommen der Bindungsenergie ist zu einem großen Teil die Absenkung der kinetischen Energie der Elektronen
1
verantwortlich, die dann auftritt, wenn die Wellenfunktion räumlich stark ausgedehnt
ist (nachzulesen im Kapitel über Molekülphysik).
Beispiele: Alkalimetalle wie Na und K, Münzmetalle (Cu, Ag, Au). (Die “harten“
Metalle wie Fe weisen zugleich metallische und kovalente Bindungen auf.)
Ionenbindung: Die elektrostatische Anziehung zwischen positiven und negativen Ionen
ist im Wesentlichen für den Zusammenhalt des Kristalls verantwortlich. Zugleich
gibt es kurzreichweitige abstoßende Kräfte quantenmechanischen Ursprungs, die einen
Mindestabstand der Nachbaratome erzwingen.
Beispiel: Kochsalz, MgO (Bei den meisten Oxiden bestehen allerdings Ionenbindung
und kovalente Bindung nebeneinander. Beispiel: Quarz = SiO2 )
van der Waals-Bindung: Der Festkörper besteht aus elektrisch neutralen Atomen oder
Molekülen, die untereinander keine chemische Bindung eingehen können. Trotzdem
gibt es (relative schwache) elektrische Anziehungskräfte: In Folge der “Ortsunschärfe“
des Elektrons in der Quantenmechanik unterliegt die elektronische Ladungsdichte
gewissen Schwankungen. Diese rufen (ähnlich wie bei der elektrostatischen Influenz)
ungleichnamige Schwankungen in Nachbarmolekülen hervor, und daraus resultiert
eine Anziehung.
Beispiele: (Paraffin-)Wachs, polymere Kunststoffe, viele biologische Stoffe
Wasserstoff-Brückenbindung: Sonderform der chemischen Bindung, die nur zwischen
Atomen der Elemente O, N und F auftritt, wenn sich zwischen ihnen ein WasserstoffAtom befinden. Zum Verständnis der Ursachen konsultiere man ein Lehrbuch der
Chemie. Die Stärke der Wasserstoffbrückenbindung kann über einen weiten Bereich
variieren, liegt aber im Allgemeinen zwischen der kovalenten und der van-der-WaalsBindung.
Beispiele: Wasser, Proteine, Bindung zwischen den Strängen des Erbsubstanz-Moleküls
DNA
2
Kristallographie
Einer Beschreibung im Rahmen der Theoretische Physik am besten zugänglich sind kristalline Festkörper, bei denen die atomaren oder molekularen Bausteine periodisch angeordnet
sind. Manche Stoffe kommen bereits in der Natur als Einkristalle vor (Beispiel: Diamant,
Quarz); andere lassen sich in dieser Form züchten. Die Werkstoffe des Alltags sind oft
polykristallin, das heißt sie bestehen aus kleinen, mikrometer- bis millimeter-großen, zueinander unregelmäßig ausgerichteten Kristallen. Festkörper ganz ohne kristalline Ordnung
(z.B. Wachs) bezeichnet man als amorph. Bei manchen amorphen Stoffen, die von den
Physikern als Gläser bezeichnet werden (auch wenn sie nicht transparent sind), handelt es
sich eigentlich um unterkühlte, sehr zähe Flüssigkeiten.
2
Die periodische Anordnung der Bausteine in einem Kristall bezeichnet man als Kristallgitter (siehe Ibach-Lüth, Kap. 2). Man kann das Kristallgitter gedanklich aufbauen durch
“Kopieren und Verschieben“ eines kleinsten Elements, der Einheitszelle des Kristalls. Die
Einheitszelle wird durch die drei Gittervektoren ~a1 , ~a2 , ~a3 aufgespannt. Das Translationsgitter des Kristalls besteht, mathematisch gesprochen, aus der Menge aller Punkte, die
sich durch Linearkombination von ganzzahligen Vielfachen der Gittervektoren darstellen
lassen:
~ = n ~a1 + m ~a1 + o ~a3
R
(1)
Die Wahl der Einheitszelle ist nicht eindeutig. Durch zusätzliche Anforderungen kann man
jedoch eine eindeutige Definition erreichen; man bezeichnet die so erhaltene Einheitszelle
dann als die Wigner-Seitz-Zelle des Kristalls. Die Punkte, die auf der Oberfläche der
Wigner- Seitz-Zelle liegen, sind von (mindestens) zwei Punkten des Translationsgitters
gleich weit entfernt. Die Punkte im Inneren der Wigner-Seitz-Zelle eines Gitterpunkts liegen
näher an diesem Gitterpunkt als an jedem anderen.
Je nach dem Material, das uns interessiert, kann die Einheitszelle mehr als ein Atom enthalten. In der Kristallographie bezeichnet man alle Atome, die in einer Einheitszelle liegen,
als die Basis. (Man beachte, dass dieser Begriff vom Sprachgebrauch in der Mathematik
abweicht.)
Abgesehen von der genauen Ausgestaltung der Basis, für die natürlich unbegrenzt viele
Möglichkeiten bestehen, gibt es nur eine begrenzte Anzahl von Kristallstrukturen. Man
kann sie klassifizieren unter Zuhilfenahme der Konzepte der Gruppentheorie aus der Mathematik. Die Klassifizierung benutzt die Begriffshierarchie Kristallsystem – BravaisGitter – Raumgruppe. Bei den Oberbegriffen Kristallsystem und Bravais-Gitter wird
die Basis nicht betrachtet, sondern nur das Translationsgitter.
Kristallsystem Für das Kristallsystem ist ausschlaggebend, welche Symmetrieeigenschaften (= Verhalten unter Spiegelungen und Drehungen) das Translationsgitter hat. Es
gibt 7 Kristallsysteme; das wichtigste ist das kubische.
Bravais-Gitter Zur Klassifizierung aller Punktmengen, die der Gleichung (1) genügen,
reicht der Begriff des Kristallsystems nicht aus. Zum Beispiel kann man in einem
kubischen Gitter im Zentrum der würfelförmigen Einheitszelle einen Gitterpunkt
hinzufügen. Die Symmetrieeigenschaften bleiben dadurch unverändert, aber man bekommt ein neues Translationsgitter, das wiederum Gl. (1) genügt, mit anderen Gittervektoren.
Die Bravais-Gitter erlauben eine eindeutige Klassifizierung aller Punktmengen, die
durch Gl. (1) erzeugt werden. Entscheidend für die Definition eines Bravais-Gitter
ist: Wenn man auf einem beliebigen Gitterpunkt “steht“ und in eine vorgegebene
Richtung blickt, sieht ein Bravais-Gitter von jedem Gittterpunkt aus gleich aus.
Es gibt insgesamt 14 Bravais-Gitter. Davon gehören 3 zum kubischen Kristallsystem:
3
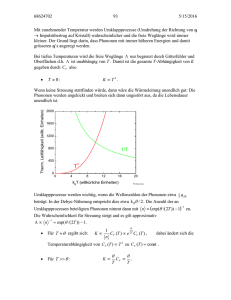
Abbildung 1: zweidimensionales Waben-Gitter und zugehöriges reziprokes Gitter mit erster
Brillouinzone (dünn umrandetes Sechseck)
das einfach-kubische, das kubisch-raumzentrierte (bcc) und das kubisch-flächenzentrierte Bravais-Gitter.
Raumgruppe Zur Bestimmung der Raumgruppe werden die Symmetrieeigenschaften des
Kristallgitters einschließlich der Basis betrachtet. Außer Spiegelungen und Drehungen hat man auch die Gleitspiegelung und die Schraubenachse als Symmetrielemente.
Es gibt 230 verschiedene Raumgruppen.
Aufgabe: Betrachten Sie die Abbildung des Kochsalzkristalls (z.B. in Herrn Mergels Folien). Zu welchem Kristallsystem gehört er? Was ist sein Bravais-Gitter? Wie viele Atome
benötigt man mindestens als Basis? Wenn Sie die Raumgruppe herausfinden wollen, können
Sie auf die Seite http://www.cryst.ehu.es schauen.
Zu jedem Kristallgitter lässt sich das reziproke Gitter konstruieren (Ibach-Lüth, Abschnitt 3.2). Die reziproken Gittervektoren ~b1 , ~b2 und ~b3 , definiert durch
~bi = 2π/Ω (~aj × ~ak ),
{ijk}={123} und zyklische Permutationen der Indices,
(2)
stehen senkrecht auf jeweils zwei der Gittervektoren ~a1 , ~a2 und ~a3 . Das Volumen der Einheitszelle im Ortsraum ist Ω = det(~a1 , ~a2 , ~a3 ). Die Einheitszelle im reziproken Raum, die
z.B. durch die ~bi aufgespannt wird, bezeichnet man als Brillouinzone (Ibach-Lüth, Abschnitt 3.5). Die Wigner-Seitz-Zelle des reziproken Gitters wird als die erste Brillouinzone
bezeichnet.
Aufgabe: Ist das Waben-Gitter ein Bravais-Gitter? Warum nicht? Was ist die Basis dieses
Gitters?
4
Eine Welle ist durch ihren Wellenvektor ~k bestimmt. Wenn man Wellen in einem Kristall
betrachtet, spezialisiert man sich in der Regel auf solche, die bzgl. der Einheitszelle periodischen Randbedingungen genügen. Man kann zeigen: Solche Wellen haben gerade einen
Wellenvektor ~k, der Gl. (3) erfüllt, d.h. der ein Element des reziproken Gitters ist.
2.1
Millersche Indices
Drei Punkte des Translationsgitters, die nicht auf einer Geraden liegen, definieren eine
Ebene, eine sog. Netzebene des Kristalls. Es lässt sich eine Schar von parallelen Netzebenen
definieren, so, dass alle Punkte des Translationsgitters auf einer der Netzebenen liegen. Es
gibt zwei äquivalente Möglichkeiten, eine Netzebenenschar zu spezifizieren:
1. Eine Netzebene schneidet von jeder der drei Koordinatenachsen ein Stück ab, das
ein ganzzahliges Vielfaches (m, n, o-faches) des zugehörigen Gittervektors ist (siehe Ibach-Lüth Abb. 3.5). Durch Erweitern des Tripels (1/m, 1/n, 1/o) mit einem
ganzzahligen Faktor p erhält man ein Tripel (h = p/m, k = p/n, l = p/o) von teilerfremden ganzen Zahlen. (h, k, l) werden als Miller-Indices bezeichnet, und liefern
eine eindeutige Spezifikation der Netzebeneschar. (Anmerkung: Zwischen der so definierten Netzebene und dem Koordinatenursprung liegen p − 1 weitere Ebenen der
gleichen Netzebenenschar.)
~ charakte2. Die Netzebenenschar wird durch ihren (gemeinsamen) Normalenvektor G
risiert. Dieser ist ein ganzzahliges Vielfaches der reziproken Gittervektoren,
~ = h ~b1 + k ~b2 + l ~b3
G
(3)
wobei (h, k, l) die Miller-Indices der Netzebenenschar sind. Der Abstand zweier be~
nachbarter Netzebenen dieser Schar ist d = 2π/|G|.
2.2
Braggsche Deutung der Beugungsbedingung
Konstruktive Interferenz bei der Beugung von Röntgenstrahlung an einem Kristall tritt
genau dann auf, wenn sich der Wellenvektor bei der Beugung genau um einen Vektor des
reziproken Gitters ändert (Ibach-Lüth, Abschnitt 3.3 und 3.4). Das ist gerade dann der Fall,
wenn die Netzebenenschar wie ein Spiegel für die Röntgenstrahlung wirkt (Winkel = −θ
bzw. = θ zwischen der Netzebene und dem einfallenden bzw. reflektierten Strahl), und
der Gangunterschied zweier, an benachbarten Netzebenen reflektierten Strahlen gerade
ein ganzzahliges Vielfaches der Wellenlänge ist. Dies ist die Braggsche Bedingung für
konstruktive Interferenz der gebeugten Wellen:
nλ = 2d sin θ,
n ganzzahlig
5
(4)
Der Experimentator legt durch die Geometrie (relative Ausrichtung des einfallenden Strahls
und des Detektors relativ zum Kristall) fest, an welchen Netzebenen gebeugt wird. Der detektierte Reflex kann eindeutig durch einen Miller-Index bezeichnet werden. Der MillerIndex bezeichnet sowohl den Normalenvektor der Netzebenenschar (im Ortsraum) als
auch den Impulsübertrag des Röntgenquants an das Gitter (in Vielfachen der Vektoren
~bi der Einheitszelle im reziproken Raum). Bei manchen Kristallstrukturen, insbesondere
bei den kubisch-flächenzentrierten und den kubisch-raumzentrierten Kristallen, sind nicht
alle Netzebenenscharen mit einem Beugungsreflex verbunden. Dies liegt daran, dass für
manche Netzebenenscharen, d.h. für manche Kombinationen von Miller-Indices, destruktive Interferenz zwischen den an benachbarten Netzebenen reflektierten Strahlen auftritt.
3
Gitterschwingungen
Bei einer gegebenen Temperatur sind die Atome eines Festkörpers nicht in Ruhe, sondern
führen Schwingungen um ihre Gleichgewichtslage aus. Bei einem Kristall sind die Gleichgewichtslagen identisch mit den Gitterpunkten des Kristallgitters. In einem perfekten Kristall breiten sich die Schwingungen in Form von Wellen aus. Die Dynamik der Atome ist
prinzipiell quantenmechanisch zu behandeln. Im Sinne des Welle-Teilchen-Dualismus der
Quantenmechanik kann man eine Welle auch als eine besondere Form von Teilchen ansehen.
Man sagt, dass den Anregungen des Kristallgitters gewisse Quasi-Teilchen entsprechen, die
Phononen. Schallwellen, die sich in einem Festkörper ausbreiten, sind ein Beispiel für
Phononen. Auch in einem amorphen Festkörper führen die Atome Schwingungen um die
Gleichgewichtslage aus; in diesem Falle werden sich die Schwingungen jedoch nicht immer
als Wellen ausbreiten, sondern können auf bestimmte Atomgruppen oder Raumgebiete
beschränkt bleiben.
Bei der Berechnung der Phononen gibt es Analogien zur Behandlung der Molekülschwingungen: Auch für Phononen benutzt man die Born-Oppenheimer-Näherung. Sie ist nicht
streng gültig; wenn man die Zusatzterme nicht mehr vernachlässigt, erhält man zusätzliche Terme, die in diesem Kontext als Elektron-Phonon-Kopplung bezeichnet werden.
Elektronen und Phononen können Energie miteinander austauschen. Bei Raumtemperatur
ist der elektrische Widerstand eines Metalls in den meisten Fällen durch die ElektronPhonon-Kopplung dominiert: Die von der angelegten elektrischen Spannung beschleunigten Elektronen streuen an den Phononen, wobei man sich die Phononen als Quasiteilchen
vorstellen kann, mit denen die Elektronen kollidieren.
Die Potentialenergiefläche, die sich im Rahmen der Born-Oppenheimer-Näherung ergibt,
wird im Sinne einer Taylor-Entwicklung um die Gleichgewichtslage (das Potentialminimum)
entwickelt (Ibach-Lüth, Abschn. 4.1). Dabei geht man meistens bis zur zweiten Ordnung,
d.h. bis zum quadratischen Term in der Auslenkung un des Atoms. Für manche physikalischen Effekte sind aber auch die höheren Terme der Entwicklung wichtig, z.B. für die
Wärmeausdehnung der Festkörper sowie für ihre Wärmeleitfähigkeit.
6
3.1
Dispersionsrelation für Phononen
Für jedes Wellenphänomen gibt es einen naturgesetzlichen Zusammenhang zwischen der
Frequenz der Welle und dem Wellenvektor. Dieser Zusammenhang ω(~k) wird als Disperisonsrelation bezeichnet. Als vereinfachtes Modell für das dreidimensionale Kristallgitter
wird oft eine eindimensionale Kette betrachtet. Die Kette besteht aus Massen m, die durch
Federn mit der Federkonstante D verbunden sind. Wir betrachten Auslenkungen der Massen in Richtung des Wellenvektors ~k entlang der Kette, also sog. longitudinale Wellen.
Die Federn sollen dem Hookschen Gesetz genügen; mit anderen Worten: Wir betrachten
longitudinale Phononen in harmonischer Näherung.
Die gekoppelten Bewegungsgleichungen für die Massen lauten
mün − D (un+1 − un ) − (un − un−1 ) = 0
(5)
Man erhält ein geschlossenes Gleichungssystem, indem man N Massen mit periodischen
Randbedingungen betrachtet (d.h. eine geschlossene Kette). Mit dem Ansatz
un (t) = Ceikna e−iωt
(6)
erhält man Lösungen der Bewegungsgleichung in Form von ebenen Wellen, falls
ω2 = 2
D
D
(1 − cos ka) = 4 sin2 (ka/2)
m
m
(7)
Dies ist die Dispersionrelation. Wie bei allen Wellenphänomenen, so gilt auch hier, dass
sich ein Wellenpaket mit der Gruppengeschwindigkeit
vg =
dω
dk
(8)
ausbreitet. Bei einer Schallwelle handelt es sich physikalisch um komprimierte bzw. gedehnte Bereiche des Gitters, die sich durch den Kristall bewegen, wobei diese Bereiche sehr viel
größer als die atomaren Abstände sind. Eine solche Kompression oder Dehnung, also eine
lokale Volumenänderung, entspricht immer einer longitudinalen Welle. Der Wellenvektor
einer Schallwelle ist betragsmäßig sehr viel kleiner als die Abmessungen der Brillouinzone.
Folglich erhält man die Schallgeschwindigkeit als
vSchall = lim
|k|→0
dω
dk
(9)
Etwas komplizierter sind die Verhältnisse, wenn man eine Kette betrachtet, die abwechselnd
aus zwei verschiedenen Massen M und m besteht (Ibach-Lüth, Abschn. 4.3). In diesem
Fall kann man die Bewegungsgleichungen nur“ bis auf ein System von zwei gekoppelten
”
Gleichungen reduzieren.
−M ω 2 + 2D −2D cos(ka/2)
C1
=0
(10)
−2D cos(ka/2) −mω 2 + 2D
C2
7
Dies kann auch als ein Eigenwertproblem für den Eigenwert ω aufgefasst werden. Es gibt
zwei qualitativ verschiedene Lösungen, die zu zwei Zweigen der Disperionsrelation führen.
Der akustische Zweig verhält sich qualitativ ähnlich wie die Kette aus gleichen Massen.
Der optische Zweig hat höhere Frequenzen und eine geringere Dispersion (Das bedeutet:
Die Frequenz des optischen Zweiges hängt weniger vom Wellenvektor ab.) Die charakteristischen Merkmale der beiden Zweige sind: Im akustischen Zweig bewegen sich bei
betragsmäßig kleinem ~k zwei benachbarte Massen gleichphasig, im optischen Zweig gegenphasig. 1 Am Rande der ersten Brillouinzone ist hingegen das Bewegungsmuster im
akustischen und im optischen Zweig ähnlich. In einem Kristall mit zweiatomiger Basis,
aber gleichen Massen kann sogar der Fall auftreten, dass sich akustischer Zweig und optischer Zweig am Rand der ersten Brillouinzone berühren; bei verschiedenen Massen bleiben
optischer und akustischer Zweig aber stets durch eine Energielücke getrennt.
Wenn man von der eindimensionalen Kette zum realistischen dreidimensionalen Gitter
übergeht, muss man beachten, dass die Auslenkungen der Atome nun auch senkrecht auf
dem Wellenvektor ~k stehen können. Solche Moden bezeichnet man als transversal; es
gibt zwei solche Moden. Die transversalen akustischen Moden bewirken keine lokale Volumenänderung, und haben normalerweise eine niedrigere Frequenz als die longitudinal
akustische Mode. In Kristallen mit einer Basis aus nb Atomen gibt es zusätzlich 3(nb − 1)
optische Moden, d.h. auch bei den optischen Moden gibt es eine longitudinale und zwei
transversale. Charakteristisch für die optischen Moden ist ferner, dass ihre Frequenz auch
bei |~k| = 0, also im Zentrum der Brillouinzone, endlich bleibt, während die Frequenzen der
akustischen Zweige dort gegen Null streben. Die saubere Trennung zwischen transversalen
und longitudinalen Moden ist im dreidimensionalen Kristall nur entlang der Richtungen
hoher Gittersymmetrie möglich; für beliebige Richtungen von ~k bekommt man Moden, die
gemischten transversalen und longitudinalen Charakter haben können.
Eigentlich hätten wir zur Berechnung der Gitterschwingungen die Atomkerne quantenmechanisch behandeln müssen. Die obigen Dispersionsrelationen bleiben aber auch in der
quantenmechanischen Behandlung gültig. Als Folge der Quantentheorie ergibt sich, dass
die Energie eines bestimmten Phonons proportional zu seiner Frequenz ist, ähnlich wie
auch die Energie des Lichtquants propotional zur Frequenz des Lichts ist. Die Energie in
einer bestimmten Schwingungsmode, charakterisiert durch den Wellenvektor ~k und die Frequenz ω, muss immer ein ganzzahliges Vielfaches des Energiequants ~ω(~k) sein, zuzüglich
der Nullpunktsenergie des harmonischen Oszillators.
1
Letzteres gibt dem optischen Zweig seinen Namen: In Ionenkristallen können bestimmte Gitterschwingungen, bei denen sich positive und negative Ionen gegenphasig bewegen, mit Hilfe von elektromagnetischer
Strahlung, also optisch, angeregt werden. Die Bezeichnung optisches Phononen“ hat sich allgemein ein”
gebürgert, auch wenn eine Anregung mit Hilfe von Licht nicht möglich ist.
8
3.2
Thermodynamik der Gitterschwingungen
Die Wärmeenergie äußert sich bei Kristallen als eine ungeordnete Bewegung der Atome
um ihre Gleichgewichtslage. Dies kann man sich so vorstellen, dass eine zufällige Anzahl
verschiedener Phononen angeregt ist. Durch die Zufälligkeit unterscheidet sich die Wärmebewegung von der Anregung einer einzigen Phonon-Mode, wie sie beispielsweise in einer
Schallwelle vorliegt. Die innere Energie eines Kristalls ist
X
1
~
U=
~ω(k) n~k +
,
(11)
2
~k
wobei n~k die Zahl von angeregten Phononen mit Wellenvektor ~k ist. Diese ist eine nicht determinierte ganze Zahl, die im thermodynamischen Gleichgewicht eine Funktion der Temperatur ist. Die Wahrscheinlichkeit, ein Phonon der Energie ~ω(~k) anzuregen, folgt der
Boltzmann-Verteilung (siehe Notizen zur Thermodynamik, 2.2),
∞
X
1
~ω(n + 1/2)
~ω(n + 1/2)
Pn = exp −
mit
Z=
exp −
(12)
Z
kB T
kB T
n=0
Im thermischen Gleichgewicht können sich in einer Schwingungsmode jede beliebige Anzahl
nk = 1, · · · ∞ an Quanten befinden. Anders gesagt: Jeder Phonon-Zustand kann beliebig
oft besetzt sein. Immer wenn (nicht wechselwirkende) quantenmechanische Anregungen
sich in dieser Weise verhalten, resultiert daraus die Bose-Einstein-Verteilung. Anders
ausgedrückt: Die Phononen gehören zu einer bestimmten Familie von (Quasi-)Teilchen,
den Bosonen, welche die Eigenschaft haben, dass sich beliebig viele davon im gleichen
quantenmechanischen Zustand befinden können. Man erhält die mittlere Besetzung hni
im thermodynamischen Gleichgewicht, indem man eine geometrische Reihe aufsummiert
(Ibach-Lüth, Abschn. 5.2).
1
hni = ~ω/(k T )
(13)
B
e
−1
Mit diesen Kenntnissen kann man die innere Energie und die spezifische Wärme eines
Kristalls berechnen (Ibach-Lüth, Abschn. 5.3). Bei großen Temperaturen ist die mittlere Besetzungswahrscheinlichkeit proportional zur Temperatur (Taylorentwicklung von hni
bzgl. (kB T )−1 ). Die spezifische Wärme ist dann unabhängig von der Temperatur und
strebt gegen den Grenzwert 3nB N kB /V , wobei N die Zahl der Einheitszellen ist, aus
der das Kristallvolumen V aufgebaut ist, und nB die Anzahl der Atome in der Basis.
Diesen Hochtemperatur-Grenzwert bekommt man auch einfach, indem man das Äquipartitionsprinzip anwendet (siehe Notizen zur Thermodynamik, Abschnitt 2.3). Bei niedrigen
Temperaturen ist das klassische Äquipartitionsprinzip jedoch nicht anwendbar, weil dann
die mittlere Besetzungszahl hni klein (von der Größenordnung Eins) ist, und sich so die
quantenmechanischen Eigenschaften der Materie bemerkbar machen. Im Grenzfall T → 0
findet man, dass die spezifische Wärme gemäß einem T 3 -Gesetz anwächst.
9
4
Elektronische Struktur von Festkörpern
Die Eigenschaften eines Festkörpers, wie z.B. seine Härte, elektrische Leitfähigkeit oder
optische Eigenschaften sind letztlich alle auf die elektronische Struktur zurückzuführen.
Darum nimmt die Berechnung der elektronischen Struktur eine zentrale Stellung in der
Festkörperphysik ein. Wenn wir einmal die Born-Oppenheimer-Näherung als gültig betrachten, müssen wir “nur noch“ die Schrödinger-Gleichung für alle Elektronen lösen. Die
Elektronen “spüren“ dabei das elektrostatische Potential der Atomkerne, die als fest an
ihren Gitterpositionen angenommen werden, sowie die Coulomb-Abstoßung untereinander.
Dieses mathematische Problem ist allerdings viel zu kompliziert, um es lösen zu können.
Häufig betrachtet man die Elektronen im Festkörper in der Näherung unabhängiger, nicht
wechselwirkender Teilchen (Ibach-Lüth, Kapitel 6). Es gibt zwei verschiedene Argumentationslinien: Zum Einen interessiert man sich oft nur für den Grundzustand der Elektronen;
und man kann zeigen, dass man dann die Elektron-Elektron-Wechselwirkung wie ein effektives Potential behandeln kann (Dichtefunktionaltheorie). Bei anderen Fragestellungen,
z.B. die Leitfähigkeit betreffend, kann man die Wechselwirkung der Elektronen untereinander zwar nicht vernachlässigen, aber man kann wieder bestimmte quantenmechanische
Quasiteilchen einführen, in deren Konstruktion wesentliche Aspekte der Wechselwirkung
bereits schon verarbeitet sind. Diese Quasiteilchen haben dann evtl. von den freien Elektronen abweichende Eigenschaften (z.B. eine andere Masse m∗ 6= me ), können aber in erster
Näherung wiederum als nicht wechselwirkend angesehen werden. In beiden Argumentationslinien gelangt man zu einem quantenmechanischen Einteilchen-Bild, mit dem man dann
mathematisch etwas leichter weiterarbeiten kann. Dennoch muss man immer im Gedächtnis behalten, dass die Vorstellung unabhängiger Elektronen im Festkörper eine (ziemlich
drastische) Näherungsannahme ist. Bestimmte physikalische Phänomene, wie z.B. den Magnetismus oder die Supraleitung, kann man nur verstehen, wenn man über dieses (allzu)
einfache Bild hinausgeht.
Im Einteilchenbild der Elektronen kann man zwischen Rumpfelektronen und Valenzelektronen unterscheiden. Die Ersteren sind so stark an ein bestimmtes Atom gebunden (sie
befinden sich in einem energetisch tiefliegenden atomaren Energieniveau), dass man sie
einfach einem bestimmten Atom zuordnen kann. Die Wellenfunktion der Valenzelektronen
ist dagegen über viele Atome (in Metallen über den ganzen Kristall) ausgedehnt. In der
Regel werden die Elektronen in der äußersten Schale der (freien) Atome beim Übergang
zum Festkörper als Valenzelektronen behandelt. Die Rumpfelektronen bewirken, dass das
elektrostatische Potential des Kerns abgeschirmt wird bis auf einen Rest, der viel flacher
(weniger räumlich veränderlich) ist, als das Potential der Atomkerne alleine.
In einem perfekten Kristall bewegen sich die Valenzelektronen in einem räumlich periodischen Potential. Die Schrödinger-Gleichung für ein Teilchen in einem periodischen Potential besitzt nur Lösungen für bestimmte Energie-Eigenwerte. Für andere Werte der Energie
hat die Schrödinger-Gleichung keine (periodischen) Lösungen. Wenn man die elektronische
Struktur im Einteilchenbild diskutiert, spricht man deshalb oft von Energiebändern: Ein
10
Band ist ein bestimmtes Intervall auf der Energieachse, in dem Lösungen existieren. Bänder
können überlappen, oder durch eine Energielücke getrennt sein.
4.1
Metalle mit nahezu unabhängigen Valenzelektronen
Im Folgenden wollen wir einfache Metalle (z.B. Alkali- und Münzmetalle) etwas genauer
betrachten. Diese haben nur ein Valenzelektron pro Atom. Das Potential, dass die Valenzelektronen spüren, ist so flach, dass man es als räumlich konstant ansehen kann. Die Lösung
der Schrödinger-Gleichung in einem konstanten Potential sind ebene Wellen.
Frage: Wenn wir den Festkörper als unendlich ausgedehnt2 betrachten, wie viele solche
Lösungen gibt es dann bezogen auf ein bestimmtes Kristallvolumen?
Die Antwort darauf gibt die elektronische Zustandsdichte. Wir brauchen eine Methode,
um die Ebene-Welle-Lösungen abzuzählen (siehe Ibach-Lüth, Abschn. 6.1). Da der Kristall
periodisch ist, fordert man üblicherweise, dass die Wellen periodischen Randbedingungen
auf der Einheitszelle genügen3 . Betrachten wir der Einfachheit halber eine würfelförmige
Einheitszelle mit dem Volumen Ω = a3 . Die Wellenfunktion exp(ikx x) ist periodisch auf
dem Intervall [0, a], wenn
kx = nx
2π
a
mit
nx
ganze Zahl;
(14)
und Gleiches gilt für ky und kz mit unabhängigen ganzen Zahlen ny und nz . Die Wellenvektoren liegen also auf einem diskreten Gitter; wir können sie in der Reihenfolge aufsteigender
Energie abzählen. Die Zustandsdichte D(E) ist so definiert, dass wir die Abzählung einfach
durch eine Energie-Integration bewerkstelligen können:
Z
Z
X
3
d r dE D(E) :=
g
(15)
Kristall
kx ,ky ,kz
Dabei ist V = N Ω das Volumen des ganzen Kristalls, bestehend aus N Elementarzellen,
und die Konstante g ist ein Entartungsfaktor, der berücksichtigt, dass es in der Quantentheorie entartete Zustände geben kann. Wenn wir konstantes Potential v0 annehmen, lautet
die Energie-Impuls-Beziehung einfach
ε~k =
~2 (kx2 + ky2 + kz2 )
+ v0
2m∗
2
(16)
Wir wollen uns mathematische und physikalische Komplikationen durch die Existenz von Oberflächen
ersparen. Die Oberflächenphysik ist ein eigenes Wissensgebiet, dass sich mit den interessanten Phänomenen
an Oberflächen beschäftigt, die über die Volumeneigenschaften hinausgehen.
3
Man kann aber auch andere Randbedingungen fordern; am Ergebnis ändert das nichts.
11
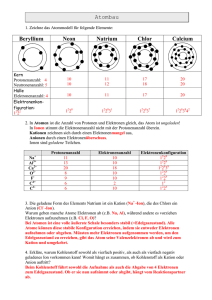
Abbildung 2: Die Summation über die ~k-Vektoren (Gitterpunkte, Kästchengröße (∆k)2 )
wird mittels der Zustandsdichte in eine eindimensionale Integration über die Energie (Kreise mit zunehmendem Radius) überführt. Anschaulich steht die Zustandsdichte D(ε) für die
Dichte der Gitterpunkte, die zwischen den (infinitesimal benachbarten) Kreisen liegen.
Damit können wir die Summe über den ~k-Raum zunächst in eine Integration über die erste
Brillouin-Zone umwandeln (im Sinne der Diskretisierung des Integrals mit einer Schrittweite ∆k = 2π(N Ω)−1/3 = 2π/(N 1/3 a) ), und anschließend in ein Integral über die Energie,
Z
Z
Z
X
d3 k
gV
dε −1 2m∗ ( − v0 )
g ↔ gN Ω
↔ 2 dε
= V dE D(E) .
(17)
(2π)3
2π
d|k|
~2
kx ,ky ,kz
Daraus ergibt sich schließlich
g
D(E) = 2
4π
2m∗
~2
3/2
E 1/2
(18)
Die Integrationsvariable E wurde dabei mit ε − v0 identifiziert; d.h. im Folgenden beziehen
sich alle Energien auf v0 . Physikalisch heißt das, wir wählen die tiefste Stelle des Energiebandes als Nullpunkt für die Energieskala. Die Zustandsdichte wächst also im Falle eines
dreidimensionalen kristallinen Materials mit unabhängigen Elektronen mit der Wurzel der
Energie an. Die Elektronen (sowohl freie Elektronen als auch die davon abgeleiteten Quasiteilchen) gehören zur Familie der Fermionen; daher unterliegen sie dem Pauli-Prinzip
(siehe Quantentheorie-Skript von Herrn Prof. Guhr): Jeder Quantenzustand kann nur einmal besetzt werden. Wir haben bisher noch nicht über den Spin gesprochen: Da es zwei
Spin-Zustände gibt, kann jeder Ebene-Wellen-Zustand genau zweimal besetzt werden. Deshalb müssen wir in Gl. (18) g = 2 setzen.
12
Im thermischen Gleichgewicht muss man die Valenzelektronen mit Hilfe der BoltzmannVerteilung behandeln. Für die mittlere Besetzungszahl hne i erhält man auf Grund des
Pauli-Prinzips die Fermi-Dirac-Verteilung f (ε, T )
hne i(εk ) =
1
X
n=0
n Pn =
e−(εk −µ)/(kB T )
1
=
= f (εk , T )
1 + e−(εk −µ)/(kB T )
e(εk −µ)/(kB T ) + 1
(19)
Hierbei wurde noch ein chemisches Potential µ berücksichtigt, das der Tatsache Rechnung
trägt, dass die Elektronen nicht frei, sondern in einem Potential gebunden sind4 . Wenn man
sich nur für Energie-Eigenwerte εk interessiert, die deutlich oberhalb oder unterhalb von µ
liegen, d.h. die mehr als kB T von µ weg sind, dann ist f (εk , T ) in guter Näherung entweder
Null oder Eins. Bei sehr kleiner Temperatur, T → 0, geht die Fermi-Dirac-Verteilung in eine
Stufenfunktion über, mit einem Sprung von Eins auf Null bei µ. Physikalisch ausgedrückt
heißt das: Für T = 0 sind die energetisch am tiefsten liegenden Zustände besetzt, von unten
aufsteigend bis zu einem höchsten Zustand, so dass alle Ne Teilchen “untergebracht“ sind.
Die Energie des höchsten Zustandes, der noch besetzt ist, wird als die Fermi-Energie EF
bezeichnet. In Formeln ausgedrückt lautet dies
Z ∞
Ne =
dE D(E)f (E, T ) ,
(20)
−∞
wobei der Parameter µ so zu wählen ist, dass obige Gleichung erfüllt wird. Dazu muss µ
von der temperatur abhängen, wenn auch meistens nur sehr schwach. Bei T = 0 gilt
Z ∞
Z EF
Ne =
dE D(E)f (E, 0) =
dE D(E) .
(21)
−∞
−∞
Bei kleinen Temperaturen, und bei dreidimensionalen Metallen praktisch immer, kann man
das chemische Potential µ in sehr guter Näherung mit der Fermi-Energie identifizieren. Man
sollte aber immer daran denken, dass diese beiden Konzept verschiedenen physikalische
Bedeutung haben.
Das Charakteristische an einem Metall ist, dass die Fermi-Energie einen Wert annimmt, bei
dem die Zustandsdichte nicht Null ist. D.h. es gibt Zustände sowohl direkt über wie auch
direkt unter der Fermi-Energie. In der Sprache der Energiebänder bedeutet das: In einem
Metall kreuzt (mindestens) ein Energieband die Fermi-Energie. Charakteristisch für einen
Halbleiter oder Isolator ist die Existenz einer Energielücke: Hier liegt das chemische Potential gerade so, dass ein Band ganz gefüllt ist, und dieses Band ist vom nächsten Band durch
die Energielücke getrennt. In Halbleitern ist die dagegen die Energielücke nicht besonders
gross, so dass es möglich ist, dass einige Elektronen durch eine thermische Anregung (bei
Raumtemperatur) über die Lücke in das ansonsten unbesetzte Band angehoben werden
4
In thermodynamischer Sichtweise ist in einem homogenen System (unendlich ausgedehnter Festkörper)
das chemische Potential µ gleich dem Gibbs-Potential (der freien Enthalpie) pro Teilchen, G/Ne .
13
können. Bei Isolatoren ist das nicht (bzw. nur bei sehr hohen Temperaturen) möglich. Dies
erklärt die stark verschiedenen elektrischen Eigenschaften von Metallen, Halbleitern und
Isolatoren: In der quantenmechanischen Betrachtungsweise tragen nämlich nur die Elektronen in der Umgebung der Fermi-Energie zum Stromfluß bei. Bei Metallen sind solche
Elektronen immer vorhanden, bei Halbleitern nur nach thermischer Anregung (oder, alternativ, nach einer Dotierung mit Fremdatomen), und bei einem Isolator praktisch gar
nicht.
4.2
Thermodynamik eines Metalls
Bei der spezifischen Wärme eines Metalls macht es sich bemerkbar, dass es dort viele
Möglichkeiten gibt, die Elektronen in angeregte Zustände zu überführen, jedenfalls deutlich mehr als bei Halbleitern oder Isolatoren, wo die Energielücke die Wahrscheinlichkeit
von thermischen Anregungen klein hält. Der Beitrag der Elektronen kommt zum Beitrag
der Gitterschwingungen hinzu, der in allen Materialklassen vorhanden und oberhalb der
Raumtemperatur relativ ähnlich ist (nämlich bei den meisten Materialien 3nB (N/V )kB ,
grob gesprochen).
Die innere Energie der Valenzelektronen ist gegeben durch
Z ∞
U=
dE D(E)f (E, T )E ,
(22)
0
wobei der tiefste Punkt des Valenzbandes als Energienullpunkt gewählt ist.
Aufgabe: Drücken Sie die innere Energie bei T = 0 durch die Fermi-Energie aus!
Für die spezifische Wärme erhält man damit
Z ∞
∂f (E, T )
cV =
dE D(E)
E
∂T
0
(23)
Bei der Berechnung der spezifischen Wärme (siehe Ibach-Lüth, Abschn. 6.4) kann man
ausnutzen, dass die Fermi-Energie in einem Metall, bezogen auf das Valenzbandminimum,
sehr viel größer ist als die thermischen Energien, für die man sich interessiert5 , EF kB T .
Zur Durchführung einer Taylorentwicklung in dem kleinen Parameter x = (E − EF )/(kB T )
schreibt man zunächst
Z ∞
∂f (E, T )
cV =
dE D(E)
(E − EF )
(24)
∂T
0
(was erlaubt ist, da das Hinzufügen des konstanten Faktors EF den Wert des Integrals
nicht ändert), und erhält dann für niedrige Temperaturen das Ergebnis
cV ≈
5
π2
2
D(EF )kB
T
3
Die Größenordnung von EF /kB ist ∼ 104 · · · 105 .
14
(25)
Bei tiefen Temperaturen erwartet man also einen in T linearen Beitrag der Elektronen
zur spezifischen Wärme. Da der Beitrag der Gitterschwingungen bei tiefen Temperaturen
mit T 3 zunimmt, gibt es einen Temperaturbereich, bei dem der elektronische Beitrag zu
cV merklich ist. cV /T geht im Limes T → 0 gegen eine Konstante γ, in deren Wert sich
Besonderheiten der elektronischen Struktur widerspiegeln. Bemerkenswert ist, dass in γ
über die Zustandsdichte D(EF ), Gl. (18), die Masse m∗ der Quasiteilchen eingeht. Das bedeutet, man kann durch eine Messung von γ feststellen, ob die Quasiteilchen eine von der
freien Elektronenmasse abweichende Masse haben, und kann damit etwas über quantenmechanische Vielteilcheneffekte lernen. In der Tat beobachtet man bei manchen Materialien
eine sehr hohe effektive Masse der Quasiteilchen an der Fermi-Energie (“Schwere-FermionSysteme“).
Literatur
[1] H. Ibach und H. Lüth, Festkörperphysik – Einführung in die Grundlagen, Springer,
Heidelberg, 6. Auglage, 2002.
[2] N. W. Ashcroft und N. D. Mermin, Festkörperphysik , Oldenbourg, München, 3. Auflage, 2007.
15