Vorwort
Werbung

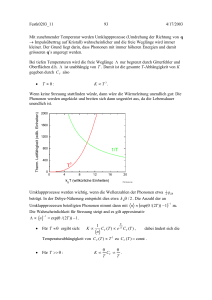
68624702 93 5/15/2016 Mit zunehmender Temperatur werden Umklappprozesse (Umdrehung der Richtung von q Impulsübertrag auf Kristall) wahrscheinlicher und die freie Weglänge wird immer kleiner. Der Grund liegt darin, dass Phononen mit immer höheren Energien und damit grösseren q’s angeregt werden. Bei tiefen Temperaturen wird die freie Weglänge nur begrenzt durch Gitterfehler und Oberflächen d.h. ist unabhängig von T . Damit ist die gesamte T-Abhängigkeit von K gegeben durch CV also K T 3. T 0: Therm. Leitfähigkeit (willk. Einheiten) Wenn keine Streuung stattfinden würde, dann wäre die Wärmeleitung unendlich gut: Die Phononen werden angekickt und breiten sich dann ungestört aus, da die Lebensdauer unendlich ist. 2000 1600 1200 800 1/T 400 T 0 0 4 3 8 12 16 20 kBT (willkürliche Einheiten) File Gauss.opj Umklappprozesse werden wichtig, wenn die Wellenzahlen der Phononen etwa 14 q ZB beträgt. In der Debye-Näherung entspricht dies etwa k B / 2 . Die Anzahl der an Umklappprozessen beteiligten Phononen nimmt dann mit n exp( /( 2T )) 1 zu. Die Wahrscheinlichkeit für Streuung steigt and es gilt approximativ 1 n exp( /( 2T )) 1 . 1 Für T ergibt sich: 1 K CV (T ) e 2T CV (T ) , n dabei ändert sich die Temperaturabhängigkeit von CV (T ) T 3 zu CV (T ) const . Für T : K T CV T . 68624702 94 5/15/2016 nimmt mit T 3 zu, durchläuft ein Maximum und nimmt dann zuerst exponentiell und dann umgekehrt proportional mit der Temperatur ab. Einfache Betrachtungsweise: An der “heissen” Seite werden Phononen mit dem Wärmeinhalt Q CV T ausgesandt und werden dann gestreut. Mit Umklappprozessen besteht dann eine Wahrscheinlichkeit, dass sich das Phonon wieder in Richtung der heissen Seite bewegt, es bringt also keine Wärmeenergie zur “kalten” Seite. In einer Übungsaufgabe wird die freie Weglänge für Diamant ( c 1000 m/s) abgeschätzt und man erhält: T = 100 K (maximale Leitfähigkeit, K 100 W/cm/K, CV 0.1 J/mol/K): l 1 mm T = 300 K: ( K 0.1 W/cm/K, CV 10 J/mol/K): l 10 nm 3.14. Extrem anharmonische Systeme Für kristalline Materialien ist die harmonische Approximation eine gute Näherung um thermische Eigenschaften und Gitterschwingungen von Kristallen zu verstehen. Insbesondere kann zum Beispiel die Temperaturabhängigkeit von CV T 3 erklärt werden. Im Gegensatz dazu beobachtet man in Gläsern bei tiefen Temperaturen CV T (wie Elektronen in Metallen bei T 0 K). CV ist auch viel grösser als in Kristallen. 68624702 95 5/15/2016 Offenbar sind in Gläsern Freiheitsgrade vorhanden, die man nicht harmonischen Oszillatoren zuordnen kann. In der Figur ist eine mögliche Struktur für ein SiO2-Glas aufgezeichnet. Das Fehlen der Periodizität kann zu lokalen Anregungen wie angedeutet führen. Die Positionen x1 und x 2 haben die potentiellen Energien E1 und E2 . Mit zunehmender Temperatur kann das rote Atom immer leichter zwischen x1 und x 2 hin und her springen. Wir haben hier das Modell eines Zweiniveau-Systems. Berechnen wir also im folgenden die spezifische Wärme von N gleichen ZweiniveauSystemen. Die Niveaux sind immer besetzt: N1 N 2 N const . Im thermodynamischen Gleichgewicht ist dann ( E2 E1 ) N1 N N 2 e E1 /(k BT ) E2 /(k BT ) e ( E2 E1 ) /(k BT ) e /(k BT ) . N2 N2 e N N N2 N N 2 /(k BT ) Man erhält damit über 1 . e 1 N2 N2 Die Energie des ganzen Ensembles beträgt 68624702 96 5/15/2016 U N1 E1 N 2 E 2 ( N N 2 ) E1 N 2 E 2 N 2 NE1 N e /( k BT ) 1 const . Damit erhält man für die spezifische Wärme: 2 N e /(k BT ) U CV Nk B . /(k T ) /( k BT ) 1) 2 T V T e B 1 k B T (e Spezifische Wärme (arb. units) 0.5 0.4 C/(NkB) 0.3 U/(N) 0.2 0.1 0.0 0 1 2 3 kBT/ 4 File Gauss.opj k BT : CV 0 , da die Temperatur nicht ausreichend ist, um das obere Niveau anzuregen. k BT : CV 0 , da beide Energieniveaux mit je N / 2 besetzt sind. Eine Temperaturerhöhung ändert die Verteilung nicht mehr. k BT finden die grössten Besetzungszahländerungen statt. CV weisst bei 0.41 einen Buckel auf. Man beobachtet oft eine sogenannte Schottkyanomalie in Kristallen, die Atome oder Ionen enthalten, die mit einer Energieänderung von etwa k B T in einen angeregten Zustand gebracht werden können. Die SchottkyAnomalie überlagert sich dann dem normalen Debye’schen Verlauf. Offensichtlich kann man mit diesem einfachen Modell die spezifische Wärme in Gläsern nicht beschreiben. Im nächsten Schritt beachten wir, dass in einem Glas viele verschiedene Konfigurationen möglich sind und deshalb eine Verteilung von Zweiniveau-Systemen existieren wird. Wir nehmen an, dass diese Systeme mit einer einfachen Verteilungsfunktion n( ) verteilt sind, die gegeben ist durch n( ) 1 / max für 0 max . n( ) ist normiert durch die Bedingung 0 d / max 1 . Damit erhalten wir n( ) N U (T ) N d /(k BT ) e 1 max 0 max d e 0 /( k BT ) 1 N max xmax (k B T ) 2 dx e 0 x x . 1 68624702 97 5/15/2016 Der Integrand fällt mit zunehmendem x so schnell ab, dass wir für unsere Abschätzung x max durch ersetzen können. Das Integral wird dann 0.822 und man erhält: U (T ) 0.822 Nk B2T 2 / max und CV (T ) 1.644 Nk B2T / max T . Es ist klar, dass diese Betrachtungsweise nur für tiefe Temperaturen gilt. Immerhin liefert das Model einen Mechanismus, der zu einer linearen Zunahme von CV bei tiefen Temperaturen führt. Es ist klar, dass zum Beispiel in SiO2 Glas die Atome nicht unabhängig schwingen, sondern miteinander korrelierte Bewegungen ( Phononen) ausführen werden. In den letzten Jahren wurden grosse Fortschritte im Verständnis von Gläsern gemacht, insbesondere mit Hilfe von Neutronenstreuung. 68624702 98 5/15/2016 4. Elektronenzustände in Kristallen Ungefähr ¾ der Elemente liegen in der metallischen Form vor währendem die Mehrheit der Verbindungen nicht-metallisch ist. In diesem Kapitel möchten wir verstehen lernen, warum ein Material metallisch, isolierend, halbleitend oder halbmetallisch sein kann. Es ist klar, dass die Elektronen für das chemische Verhalten von Substanzen verantwortlich sind. Aus diesem Grund müsste man im Prinzip die Schrödingergleichung lösen für rund 1023 Elektronen, die sich im Coulombfeld von ca. 1022 Atomkernen bewegen. Um dem Pauliprinzip zu genügen, müsste man Lösungen finden, die antisymmetrisch sind in Bezug auf die Vertauschung der Koordinaten. Diese Aufgabe ist unlösbar. Wir werden uns im wesentlichen mit der Einelektronnäherung beschäftigen, die trotz ihrer Einfachheit erstaunlich gut funktioniert. 4.1. Klassische Voraussagen Unter der Annahme, dass sich die Elektronen in einem Material wie ein klassisches Gas mit der Geschwindigkeit v (gegeben durch die Maxwell-Boltzmann Verteilung) bewegen, kann man verschiedene Eigenschaften herleiten: spezifische Wärme: Jedes Elektron hat 3 Freiheitsgrade, also wäre CVelekt 32 k B . Die Experimente zeigen, dass die Elektronen nicht wesentlich zur spezifischen Wärme beitragen, ausser in der Nähe von T 0 , wo CVelekt T ist. Elektrische Leitfähigkeit: Ohm’sches Gesetz: j σE ne 2 ne 2 . m m v Für klassisches Gas gilt E v 2 T . Also T 1 / 2 . Beobachtet wird T 1 bei Raumtemperatur. Thermische Leitfähigkeit: K 13 CV v Wie vorhin: v T , also K T 1 / 2 . Beobachtet wird K const . Wiedemann-Franz Gesetz: W und F beobachteten bei Raumtemperatur, dass K / für verschiedene Metalle ähnlich sind. Lorenz zeigte dann (L = Lorenz Zahl): 68624702 99 5/15/2016 2 K k L 3 B . Erstaunlicherweise stimmte diese Zahl sehr gut mit dem Wert T e aus der Theorie der freien Elektronen überein: L 2 kB 2 8 WΩ . 2.45 10 3 e K2 Halleffekt: Wenn ein Strom durch einen Leiter fliesst, werden die Elektronen durch das Magnetfeld abgelenkt. Sie erfahren dabei eine Kraft in die y Richtung: e F vH. c Die Ladung e 1.602 10 -19 C ist dabei eine positive Zahl. Es entsteht ein elektrisches Feld 1 . E RH j x mit der Hallkonstanten R H nec z H y v +++++++++++++++++++++ --------------------- I x F E Dabei bedeutet n die Zahl der Elektronen pro Volumeneinheit. Wenn pro Atom ein Elektron zum Ladungstransport beiträgt, entspricht n der Dichte der Atome. Für einige Metalle, wie Na, K, Ag erhält man aus Hallmessungen Werte für n die typisch 10-20% grösser sind. Andererseits ändert zum Beispiel für Al die Hallkonstante sogar das Vorzeichen mit zunehmendem Feld! Be liefert ebenfalls eine negative Dichte. Diese Ergebnisse sind inkompatibel mit dem klassischen Modell. 4.2. Einelektron-Näherung Wir betrachten zuerst ein Atom mit der Kernladung Ze , das von N Elektronen umgeben ist. Die stationären Zustände erhält man dann durch Lösung der Schrödingergleichung N Ze 2 2 e2 (r1 , r2 ,...rN ) j 1 r j 2m i j r j ri E . 68624702 100 5/15/2016 Dieses Problem ist nur analytisch lösbar, wenn man die Wechselwirkung zwischen den Elektronen vernachlässigt. Man spricht dann von unabhängigen Elektronen. Die Lösung ist dann ein Produkt von N Einelektron-Wellenfunktionen j (r j ) die Lösungen von 2 Ze 2 j (r j ) j (r j ) j j (r j ) . 2m rj N Die Lösung des Vielteilchenproblems hat den Energieeigenwert E j und die j 1 Wellenfunktion 1 2 .... N . Es ist klar, dass die Vernachlässigung der Coulombwechselwirkung zwischen den Elektronen sowie des Pauli-Prinzips nicht zulässig ist. Eine wesentlich bessere Beschreibung der Atome erhält man mit Hilfe der Hartree-Fock Näherung. Hartree addierte zur potentiellen Energie der Kerne die potentielle Energie des Elektrons im Ladungsfeld der übrigen Elektronen: e 2 i (ri ) 2 Ze 2 j (r j ) j (r j ) d i j (r j ) j j (r j ) . 2m rj r j ri i j 2 Mit Hilfe eines selbstkonsistenten Verfahrens kann man dann die EinelektronWellenfunktionen optimieren. Die antisymmetrische Produktwellenfunktion erhält man dann durch Bestimmung der Determinanten: 1 (r1 ) 1 (r2 ) ... 1 (rN ) 2 (r1 ) 2 (r2 ) ... 2 (rN ) ... ... ... N (r1 ) 2 (r2 ) ... N (rN ) . Im Prinzip kann man auch in einem Kristall die Hartree-Fock Methode zur Berechnung der Einelektronwellenfunktionen anwenden. Die Wellenfunktionen j wären dann als Kristallorbitale aufzufassen. Auf dieser Ebene muss man aber nicht anfangen. Um dies zu verstehen, betrachten wir die Elektronenkonfiguration 1s 2 2s 2 2 p 6 3s von Na. Wie in der folgenden Figur gezeigt ist, halten sich die Rumpfelektronen 1s 2 2s 2 2 p 6 (Neon) so nahe am Kern auf, dass sie nicht mit den Orbitalen der benachbarten Na Atome überlappen. Deshalb verhalten sie sich im wesentlichen wie atomare Orbitale, die man mit Hartree-Fock Methoden berechnen kann. Die 3s Elektronen hingegen überlappen sehr stark und halten sich bevorzugt zwischen den Atomrümpfen auf. Sie sind nur schwach an die einzelnen Atome gebunden, d.h. sie sind frei (= delokalisiert).