Das multifunktionelle Toxin VacA von Helicobacter
Werbung

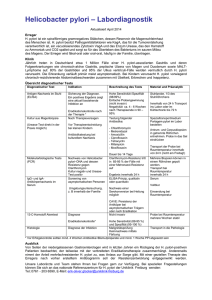
117_163_BIOsp_0209.qxd 152 05.03.2009 15:26 Uhr Seite 152 WISSENSCHAFT · S P E C I A L : P RO T E I N -AU F R E I N I G U NG U N D -A NA LY T I K Bakterielle Immunmodulation Das multifunktionelle Toxin VacA von Helicobacter pylori XAVER SEWALD, RAINER HAAS MAX VON PETTENKOFER-INSTITUT FÜR HYGIENE UND MEDIZINISCHE MIKROBIOLOGIE, LMU MÜNCHEN Die Interaktion des vakuolisierenden Zytotoxins VacA von Helicobacter pylori mit dem Leukozyten-spezifischen β2-Integrin LFA-1 führt zur Hemmung der NFAT-abhängigen Gentranskription humaner T-Zellen. The Helicobacter pylori vacuolating cytotoxin VacA, interacting with the β2-integrin LFA-1, induces down-regulation of NFAT-dependent gene transcription in human T cells. ó Im Jahr 2005 wurden Robin Warren und Barry Marshall für die Entdeckung und Isolierung von Helicobacter pylori aus der Magenmukosa mit dem Nobelpreis für Physiologie und Medizin ausgezeichnet. Weltweit ist etwa jeder zweite Mensch von H. pylori besiedelt. Die Infektion mit H. pylori führt zu einer chronischen Entzündung der Magenschleimhaut (Gastritis), die in 90 Prozent der Fälle symptomlos verläuft. Jedoch entwickeln 15 bis 20 Prozent der infizierten Personen aufgrund dieser chronischen Entzündung Folgeerkrankungen wie Magen- oder Zwölffingerdarmgeschwüre sowie ein bis zwei Prozent ein Magenkarzinom. Spezielle Pathogenitätsfaktoren von H. pylori, wie das mittels eines Typ-IV-Sekretionssystems in eukaryotische Zielzellen übertragene Protein CagA, das Adhäsin BabA sowie das vakuolisierende Zytotoxin VacA, sind dabei signifikant mit der Ausbildung von Magengeschwüren und dem Magenkarzinom assoziiert. Weitere bakterielle Virulenzfaktoren ermöglichen es H. pylori, sowohl den extremen Umweltbedingungen (pH 1–2, Nährstoffknappheit) zu begegnen, als auch dem Immunsystem des Wirts zu entgehen. Die Infektion durch H. pylori induziert eine umfassende Antwort, sowohl des angeborenen als auch des erworbenen Immunsystems. Immunzellen werden dabei von zellulären Zytokinen oder bakteriellen Faktoren angelockt. Die Produktion pro-inflammatorischer Zytokine wie Interleukin(IL)-8, IL1β und Tumornekrosefaktor(TNF)-α führt zum Einwandern von Zellen des angeborenen Immunsystems in die Magenmukosa, darunter Granulozyten, Makrophagen und Mastzellen. Im späteren Verlauf der Infektion werden auch Zellen des adaptiven Immunsystems, wie T-Lymphozyten, zur Infektion rekrutiert. Die starke Immunreaktion kann aber die H. pylori-Infektion nicht beseitigen. Das vakuolisierende Zytotoxin VacA ˚ Abb. 1: Funktionelle Domänen und Prozessierung von VacA. Das reife VacA-Toxin besteht aus p33 und p55. Die Signal-Sequenz (SS, grün) führt zum Sec-abhängigen Transport über die innere Membran (IM) ins Periplasma. Der C-terminal gelegene Autotransporter (braun) katalysiert die Translokation über die äußere Membran (OM). Eine am N-Terminus gelegene hydrophobe Region (orange) aus etwa 30 Aminosäuren vermittelt die Ausbildung der Pore. Die i-Region (rot) ist an der Zelltyp-spezifischen Vakuolisierung beteiligt. Die Beobachtung, dass H. pylori-Kulturüberstand die Ausbildung großer, zytoplasmatischer Vakuolen in Epithelzellen induziert, führte 1988 zur Entdeckung des vakuolisierenden Zytotoxins VacA[1]. Es wurde 1992 von Cover und Blaser biochemisch gereinigt und näher charakterisiert[2]. VacA wird als 140 kDa großes Prä-Protoxin produziert, Secabhängig über die innere Membran von H. pylori exportiert und mit einer C-terminal gelegenen Autotransporter-Domäne über die BIOspektrum | 02.09 | 15. Jahrgang 117_163_BIOsp_0209.qxd 05.03.2009 15:26 Uhr Seite 153 153 Epithelzellen Tab. 1: Effekte von VacA auf Epithelzellen. VacA-Effekte Literatur Endosomales System / Sekretion • Hemmung der Carbonat-Sekretion • Hemmung der Säure-Sekretion • Umverteilung des späten endosomalen Kompartiments • Intrazelluläres Wachstum von H. pylori Tuo et al. (2009): J. Infect. Dis., 199. Wang et al. (2008): JBC 283: 26714–26725. Li et al. (2004): Mol. Biol. Cell. 15: 1946–1959. Terebiznik et al. (2006): Infect. Immun. 74: 6599–6614. Programmierter Zelltod (Apoptose) • Hemmung Zell-Zyklus (G1-S) • Aktivierung von Caspase 8 und 9 • Stimulierung pro-apoptotischer Faktoren (Bax/Bak) • Reduktion mitochondriales Membranpotenzial • Ko-Lokalisation von VacA und Mitochondrien • Freisetzung von Cytochrom C Manente et al. (2008): J. Cell. Physiol. 214: 582–587. Yamasaki et al. (2006): JBC 281 11250–11259. Willhite und Blanke (2004): Cell Micro. 6: 143–154. Galmiche et al. (2000): EMBO J. 19, 6361–7630. Willhite et al. (2003): JBC 278: 48204–48209. Zelluläre Signalwege • Aktivierung der MAP-Kinase p38 • Phosphorylierung von Git-1 • Aktivierung PI3K/Akt-Signalweg (Phosphorylierung von PKB, GSK3β) • Kerntranslokation von β-Catenin Hisatsune et al. (2007): Infect. Immun. 75, 4472–4481. Nakayama et al. (2004): JBC 279: 7024-7028. Fujikawa et al. (2003): Nat. Gen. 33: 375–381. Nakayama et al. (2009): JBC 284: 1612–1619. Integrität der Epithelschicht und Zellmembran • Reduktion der transepithelial electric resistance (TER) • Epithel-Durchlässigkeit (Harnstoff und Carbonat) • Ausbildung Anion-spezifischer Kanäle in Plasmamembran (Chlorid, Pyruvat) Papini et al. (1998): J. Clin. Invest. 102: 813–820. Tombola et al. (2001): J. Clin. Invest. 108: 929–937. Guarino et al. (1998): J. Infect. Dis. 178: 1373–1378. Szabo et al. (1999): EMBO J. 18: 5517–5527. Tombola et al. (1999): Bioph. Journ. 76: 1401–1409. äußere Membran sekretiert (Abb. 1). Die Autotransporter-Domäne wird abgespalten, und das Toxin (90 kDa) kann zu hexameren Ringen oligomerisieren. Das reife Toxin besteht aus Domänen der Größe 37 kDa (p33) und 55 kDa (p55), die unterschiedliche Funktionen aufweisen. So ist das p33-Fragment für die Ausbildung einer Membranpore nötig. Essenziell für die Insertion in die Membran ist dabei ein N-terminal gelegener, stark hydrophober Bereich (etwa 30 Aminosäuren) mit drei Wiederholungen eines Gly-XXX-GlyMotivs. Das p55-Fragment ist für die Bindung an die Zelloberfläche sowie die Oligomerisierung durch Interaktion der Untereinheiten verantwortlich. Alle bisher untersuchten H. pylori-Isolate produzieren VacA, wobei deutliche Variationen in der Sequenz erkennbar sind. Aufgrund verschiedener Sequenz-Polymorphismen lassen sich VacA-Allele unterscheiden, die sich in ihrem Bindungsverhalten an eukaryotische Zellen und ihrer Vakuolisierungsaktivität unterscheiden (Abb. 1). VacA-Polymorphis- BIOspektrum | 02.09 | 15. Jahrgang men treten im Bereich des N-Terminus (s-Region), des mittleren Bereichs (m-Region) sowie in einem intermediären Bereich (i-Region) auf[3]. Polymorphe VacA unterscheiden sich dabei in ihrer Aktivität und dem Spektrum sensitiver Zellen. Epidemiologische Studien ergaben, dass H. pylori mit i1-Allel signifikant mit der Ausbildung von Magenkrebs assoziiert ist. Weitere Aspekte zur Charakterisierung funktioneller VacARegionen ergaben sich aus der Kristallstruktur des p55-VacA-Fragments[4]. Das p55 besteht aus einer lang gestreckten β-helikalen Struktur mehrerer rechtsgedrehter β-Helices, die durch einen Knick von der C-terminalen, globulären Domäne abgesetzt sind. Durch Vergleich oberflächenexponierter Aminosäurereste von 92 VacA-Sequenzen konnten zwei konservierte Bereiche im p55-Fragment bestimmt werden. Vermutet wird in diesen Bereichen eine C-terminal lokalisierte Rezeptor-Bindetasche und ein am N-Terminus gelegener Bereich zur Interaktion mit dem p33Fragment (Abb. 1). VacA als multifunktionales Toxin Nach der Infektion kommt H. pylori zunächst mit Zellen des Magenepithels in Kontakt. Diese Zellschicht besteht hauptsächlich aus Epithelzellen sowie aus Säure- und Magenschleim-produzierenden Zellen. VacA zeigt zahlreiche Effekte auf Epithel- und Endothelzellen, wie die Störung des endosomalen Systems, die Induktion von Apoptose, den Einfluss auf Signaltransduktionswege und die Zerstörung der Membranintegrität (Tab. 1). Der Mechanismus der Vakuolenbildung geht von einer Bindung von VacA an die Zytoplasmamembran und anschließender Aufnahme aus. VacA bildet einen Anionen-spezifischen Kanal, der nach Ansäuerung des Lumens der späten Endosomen zum Einstrom von Chlorid-Ionen (Cl-) führt. Dies hat eine verstärkte Aktivität der vakuolären ATPase (v-ATPase) zur Folge, die vermehrt Protonen ins endosomale Lumen pumpt, um die negative Ladung von Cl- auszugleichen. Durch Einstrom von Wasser kommt es zum osmotischen 117_163_BIOsp_0209.qxd 154 05.03.2009 15:26 Uhr Seite 154 WISSENSCHAFT · S P E C I A L : P RO T E I N -AU F R E I N I G U NG U N D -A NA LY T I K ¯ Abb. 2: VacA-Effekte auf Immunzellen. A, Die Bindung von VacA an den/die Rezeptor/en führt zur Phosphorylierung der MAPK p38 und Erk1/2 sowie zur Aktivierung von Vav-1. B, Aufgenommenes VacA inhibiert die Proliferation von T-Zellen und hemmt die IL-2-Genexpression. C, Die Reifung von H. pylori-beinhaltenden Phagosomen zu Lysosomen wird von aufgenommenen VacA verhindert. D, In B-Zellen verhindert VacA die Antigen-Präsentation durch Störung der Ansäuerung der Vesikel. Vav, GTP-Nukleotid-Austauschfaktor; TACO, tryptophan aspartate-containing coat-Protein; IL-2, Interleukin-2; MIP-1a/b, macrophage inflammatory-Protein-1α/β; NFAT, Nukleärer Faktor aktivierter T-Zellen; Cn, Calcineurin; AP1, aktivierendes Protein 1; NFκB, Nukleärer Faktor kappa B; ER, Endoplasmatisches Retikulum; MHCII, Haupthistokompatibilitätskomplex II; P, Phosphat. Anschwellen der Membranvesikel. VacA stört somit die Organisation des intrazellulären Vesikeltransports, was als mögliche Ursache für die reduzierte Sekretion von Säure bzw. Carbonat angesehen wird. Neben dem Einfluss auf das endosomale Kompartiment kann VacA durch Freisetzung von CytochromC Apoptose in Epithelzellen induzieren. Dabei spielen eine Reduktion des mitochondrialen Membranpotenzials sowie die Aktivierung pro-apoptotischer Faktoren wie Bax/Bak und der Caspasen 8 und 9 eine Rolle (Tab. 1). Insgesamt ist die Bedeutung der verschiedenen VacA-Effekte auf Epithelzellen für die Infektion des Magens durch H. pylori in vivo jedoch noch unzureichend verstanden. Aufgrund der vielen unterschiedlichen Effekte wurde VacA als multifunktionales Toxin bezeichnet. VacA als Immunotoxin Die Besonderheit der H. pylori-Infektion liegt in der chronischen, meist lebenslangen Entzündung. Bei der Kontrolle einer H. pyloriInfektion spielen neben Makrophagen und Antigen-präsentierenden Zellen (B-Lymphozyten, Dendritische Zellen), CD4+-T-Lymphozyten eine zentrale Rolle. Die Effekte von VacA auf verschiedene Typen von Immunzellen sind in Abbildung 2 zusammengefasst. Wie für Epithelzellen gezeigt, führt VacA auch bei T-Lymphozyten zur Aktivierung der Mitogen-aktivierten Proteinkinase (MAPK) p38 ¯ Abb. 3: Spezies-spezifische Wirkung von VacA auf T-Zellen. A, In humanen T-Zellen bindet VacA vermutlich über Glykolipide an die Zelloberfläche. Der Leukozyten-spezifische Rezeptor CD18, eine Untereinheit des β2-Integrins hLFA-1, vermittelt die funktionelle Aufnahme. VacA führt zur Ausbildung saurer Vakuolen und zur Hemmung der Kerntranslokation des Transkriptionsfaktors NFAT. Dies hemmt die IL-2-Expression nach T-Zell-Aktivierung. B, In murinen T-Zellen kann VacA nicht binden und aufgenommen werden. Die Zellen weisen keine Hemmung der IL-2-Expression und Proliferation auf. C, Expression von humanem CD18 in murinen T-Zellen führt zur Ausbildung saurer Vakuolen. VacA bindet Spezies-spezifisch an humanes CD18. Eine Hemmung der IL-2Expression findet nicht statt[6]. hLFA-1, humanes LFA-1; mLFA-1, murines LFA-1; VacA p33, rot; p55, blau; IL-2, Interleukin-2; NFAT, Nukleärer Faktor aktivierter T-Zellen. BIOspektrum | 02.09 | 15. Jahrgang 117_163_BIOsp_0209.qxd 05.03.2009 15:26 Uhr Seite 155 155 und Erk1/2 sowie des GTP-Nukleotid-Austauschfaktors (GEF) Vav-1 (Abb. 2A). Diese Aktivierung ist unabhängig von der Aufnahme des Toxins und wird vermutlich über die Bindung von VacA an Oberflächenrezeptoren vermittelt. Dies könnte durch die beschriebenen Rezeptoren Sphingomyelin und/oder LFA-1 erfolgen[5, 6]. Von Makrophagen phagozytierte H. pylori können in einem intrazellulären Kompartiment überdauern, da ein Abtöten in Lysosomen durch VacA verhindert wird (Abb. 2C). Weiter wurde gezeigt, dass VacA die proteolytische Prozessierung von Antigenen und somit die Antigenpräsentation zur Aktivierung von T- und B-Lymphozyten stört[7], was auf Veränderungen des endosomalen Transports zurückgeführt wird (Abb. 2D). Als ein direkter Effekt von VacA auf T-Lymphozyten ist die Hemmung der Proliferation von T-Zellen zu nennen, wobei der Mechanismus nicht im Detail geklärt ist[8]. VacA hemmt die Expression des Zytokins IL-2, das normalerweise nach Aktivierung des T-ZellRezeptors(TCR)/Korezeptors exprimiert wird. Die Transkription von IL-2 hängt von der Aktivierung der Ca2+-abhängigen Phosphatase Calcineurin ab, die zur Dephosphorylierung des Transkriptionsfaktors NFAT führt. VacA verhindert die Dephosphorylierung und somit die Translokation von NFAT in den Nukleus. Genexpressionsstudien ergaben, dass VacA die Aktivierung von Calcineurin, ähnlich der Wirkung des immunsuppressiven Moleküls FK506 inhibiert[8]. Neben der Expression von IL-2 ist auch die Produktion weiterer Zytokine wie MIP-1α und MIP-1β betroffen (Abb. 2B). Spezies-spezifische Interaktion von VacA mit T-Lymphozyten Die Interaktion mit der Zellmembran ist der erste wichtige Schritt für ein Toxin. Häufig sind an der Aufnahme eines Toxins spezifische zelluläre Interaktionspartner beteiligt, die als Rezeptor und/oder Korezeptor bezeichnet werden. Durch die Bindung an einen spezifischen Rezeptor kommt es meistens zur rezeptorvermittelten Aufnahme des Toxins in das endosomale System. So dienen beispielsweise für das Botulinum-NeurotoxinA Ganglioside als niedrigaffine Rezeptoren zur Zellbindung und das synaptic vesicle protein als Proteinrezeptor zur Aufnahme in Neuronen. Für VacA wurde in vitro die Interaktion mit Glykosphingolipiden beschrieben. Außerdem konnte mit Sphingomyelin ein Korezeptor auf Endothelzellen identifiziert werden[5]. SphinBIOspektrum | 02.09 | 15. Jahrgang gomyelin könnte auch ein möglicher Korezeptor auf Leukozyten darstellen, der die initiale Bindung von VacA an die Zelloberfläche vermittelt. Die funktionelle Aufnahme von VacA in T-Lymphozyten erfolgt jedoch über CD18, der β-Untereinheit Leukozyten-spezifischer Integrine[6]. Das auf Immunzellen dominierende Integrin LFA-1, bestehend aus den Untereinheiten β2 (CD18) und αL (CD11a), stellt somit einen zentralen Rezeptor für VacA auf verschiedenen Immunzellen dar. Wie im Modell (Abb. 3) dargestellt, kommt es zur initialen Interaktion von VacA mit einem Korezeptor (Glykolipid) auf Lymphozyten. Die Bindung führt zu strukturellen Umlagerungen und zur Übertragung auf den Leukozytenspezifischen VacA-Rezeptor CD18. Das Integrin LFA-1 vermittelt die funktionelle Aufnahme von VacA, was die Ausbildung saurer Vakuolen und die Hemmung der ZytokinExpression auslöst (Abb. 3A). In murinen T-Zellen führt VacA nicht zur Ausbildung saurer Vakuolen und zur Hemmung der IL-2-Expression. Diese Spezies-Spezifität von VacA wird durch die Beobachtung gestützt, dass ausschließlich H. pylori ein vacA-Gen besitzt, nicht jedoch andere Helicobacter-Spezies, wie Helicobacter felis, der im Magen von Katzen und Hunden vorkommt. VacA bindet weder an die Zelloberfläche von primären murinen T-Zellen, noch wird es aufgenommen (Abb. 3B). Eine mögliche Ursache hierfür ist die fehlende Bindung an den spezifischen Rezeptor oder Korezeptor. Werden die humanen β2-Integrine (hLFA-1, hMac1) in Maus-T-Zellen exprimiert, kann man eine verstärkte Vakuolisierung dieser „humanisierten“ murinen T-Zellen beobachten. Eine Hemmung der IL-2-Expression durch VacA ist jedoch weiterhin nicht möglich (Abb. 3C). Als Ursache hierfür sind Variationen in der Zielstruktur (Signalwege, Calcineurin usw.) in Betracht zu ziehen, sodass VacA trotz funktioneller Aufnahme die Expression von IL-2 nicht hemmen kann. Zusätzlich könnte das Fehlen des VacA-spezifischen Korezeptors auf murinen T-Zellen zu einer uneffizienten Aufnahme führen. Danksagung Unsere Arbeiten wurden durch die Deutsche Forschungsgemeinschaft unterstützt. ó Literatur [1] Leunk, R. D., Johnson, P. T., David, B. C., Kraft, W. G., Morgan, D. R. (1988): Cytotoxic activity in broth-culture filtrates of Campylobacter pylori. J. Med. Microbiol. 26: 93–99. [2] Cover, T. L., Blaser, M. J. (1992): Purification and characterization of the vacuolating toxin from Helicobacter pylori. J. Biol. Chem. 267: 10570–10575. [3] Rhead, J. L., Letley, D. P., Mohammadi, M., Hussein, N., Mohagheghi, M. A., Eshagh, H. M., Atherton, J. C. (2007): A new Helicobacter pylori vacuolating cytotoxin determinant, the intermediate region, is associated with gastric cancer. Gastroenterology 133: 926–936. [4] Gangwer, K. A., Mushrush, D. J., Stauff, D. L., Spiller, B., McClain, M. S., Cover, T. L., Lacy, D. B. (2007): Crystal structure of the Helicobacter pylori vacuolating toxin p55 domain. Proc. Natl. Acad. Sci. USA 104: 16293–16298. [5] Gupta, V. R., Patel, H. K., Kostolansky, S. S., Ballivian, R. A., Eichberg, J., Blanke, S. R. (2008): Sphingomyelin functions as a novel receptor for Helicobacter pylori VacA. PLoS. Pathog. 4: e1000073. [6] Sewald, X., Gebert-Vogl, B., Prassl, S., Barwig, I., Weiss, E., Fabbri, M., Osicka, R., Schiemann, M., Busch, D. H., Semmrich, M., Holzmann, B., Sebo, P., Haas, R. (2008): Integrin subunit CD18 Is the T-lymphocyte receptor for the Helicobacter pylori vacuolating cytotoxin. Cell Host Microbe 3: 20–29. [7] Molinari, M., Salio, M., Galli, C., Norais, N., Rappuoli, R., Lanzavecchia, A. Montecucco, C. (1998): Selective inhibition of Ii-dependent antigen presentation by Helicobacter pylori toxin VacA. J. Exp. Med. 187: 135–140. [8] Gebert, B., Fischer, W., Weiss, E., Hoffmann, R., Haas, R. (2003): Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science 301: 1099–1102. Korrespondenzadresse: Dr. Xaver Sewald LMU München Lehrstuhl Bakteriologie Max von Pettenkofer-Institut Pettenkoferstraße 9A D-80336 München Tel.: 089-5160-5420 [email protected] AUTOREN Rainer Haas Xaver Sewald 1977–1984 Studium der Biologie. 1987 Promotion an der Universität Tübingen. 1992– 1993 Auslandsaufenthalt in Lausanne, Schweiz. 1996 Habilitation im Fach Mikrobiologie. Seit 1997 Professor für Medizinische Mikrobiologie an der LMU München. Schwerpunkte: Pathogenitätsmechanismen in Helicobacter pylori. 1998–2003 Studium der Biologie an der LMU München. 2003 Diplomarbeit am Institut für Genetik und Mikrobiologie, LMU München. 2004–2008 Promotion am Max von Pettenkofer-Institut, LMU München in der AG von Prof. Dr. Haas. Ab April 2009 Postdoc in der AG von Prof. Dr. Mothes, Yale University, New Haven, USA.