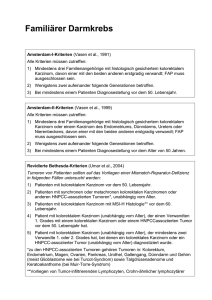
Spezielle Pathologie - broesel
Werbung

Kurs der speziellen Pathologie Aufgaben der Pathologie in der Lehre, Forschung und Krankenversorgung - Pathologie ist die Lehre von den krankhaften Vorgängen im menschlichen Körper - Pathologie ist die Lehre der bei Krankheiten auftretenden morphologisch (Phänotyp) und molekular-pathologisch (Genotyp) fassbaren Organ-, Gewebe- und Zellveränderungen - Pathologie ist ein Fachgebiet der klinischen Medizin (mittelbare Krankenversorgung, Facharztausbildung, Habilitation). Mittelbar heißt, dass man keinen direkten Kontakt zum Patienten hat; das hat Vor- und Nachteile. Der Vorteil ist, dass man das vorliegende Material objektiv und unbeeinflusst beurteilen kann, der Nachteil ist, dass meist einiges an nötigen Informationen fehlt, weshalb man oft nochmal Kontakt mit dem zuständigen Klinker aufnehmen muss und die ganze Krankengeschichte erfragen. - Facharzt / -ärztin für Pathologie (6 Jahre: 5 Jahre Pathologie, 1 Jahr Klinik; Zusatzbezeichnung: Molekulare Pathologie) Aufgaben der Pathologie in der Lehre (neue ÄAO): Allgemeine Pathologie (1. + 2. Semester): Beschreibung der Grundreaktionen und Strukturveränderungen der Organe, Gewebe und Zellen, mit denen der Organismus auf krankmachende Reize antwortet. Spezielle Pathologie (3. + 4. Semester): Darstellung der bei einzelnen Krankheiten auftretenden Zell-, Gewebe und Organveränderungen sowie deren Auswirkungen auf den Gesamtorganismus. Formale Genese: A.Ätiologie: auslösende Ursache B.Pathogenese: Ablauf der Reaktion des Organismus auf die Einwirkung der ätiologischen Noxe C.Klinische Symptomatik, morphologisch fassbare Befunde D.Heilung (wenn man Glück hat) E.Komplikationen (wenn man Pech hat): Defektheilung, chronische Erkrankung, Rezidiv, Tod Aufgaben der Pathologie in der mittelbaren Krankenversorgung (Diagnostik): - Die Pathologie ist integraler Bestandteil der klinischen Medizin. - Die Pathologie ist zuständig für die morphologische (Phänotyp) und molekular-pathologische (Genotyp) Diagnostik von Krankheitsbildern (Biopsie, Zytologie, Autopsie); also wie groß ist ein Tumor, was für ein Tumor ist es, wie aggressiv ist er usw. - Die Pathologie muss dem Kliniker all die Informationen liefern, die eine individuelle, auf den Patienten zugeschnittene Therapie ermöglicht. - Pathologie ist der Weichensteller hinter den Kulissen... Strategien der Diagnostik: Man unterscheidet prinzipiell zwischen der intravitalen Diagnostik und der postmortalen Diagnostik. Intravitale Diagnostik: - Zytopathologie: Mikroskopische Beurteilung weniger Zellverbände oder einzelner Zellen in Ausstrichpräparaten; Bewertungskriterien hauptsächlich zytoplasmatische oder nukleäre Veränderungen. Indikation: prophylaktisches Screening (z.B. Vorsorgeuntersuchungen), minimal invasive Tumordiagnostik, Monitoring (z.B. Tumorerkrankungen) Vorteile: schnell, kostengünstig, hohe diagnostische Aussagefähigkeit, Planbarkeit des weiteren diagnostischen und therapeutischen Vorgehens. Untersuchungsmaterial: Punktionszytologie (Feinnadelpunktion von Gewebe), Exfoliativzytologie (Abstriche von so ziemlich allem was es gibt) - Intraoperative Schnellschnittuntersuchung: Mikroskopische Untersuchungen an entnommenen Gewebeproben während eines operativen Eingriffs. Ziel: Rasche histopathologische Diagnose einer makroskopisch nicht sicher zu beurteilenden Läsion innerhalb von Minuten. Voraussetzung: Enge (auch rämlich) Zusammenarbeit zwischen Operateur und Pathologen, ... Pro Tag werden in Tübingen 20-30 Schnellschnittuntersuchungen angefertigt. - Biopsien Lernziele: - Erklären Sie die formale Pathogenese von Krankheiten. - Wo liegen die Schwerpunkte des Faches Pathologie im Rahmen der Krankenversorgung? - Wo liegen die Indikationtions... bla blub Folie schon wieder weg. Innere Leichenöffnung: Autopsie - Obduktion - Sektion Die drei Begriffe Autopsie - Obduktion - Sektion sind synonym. Lernziele: - äußere Leichenschau: wer / wann / warum - natürlicher Tod: Definition, Konsequenzen - ... Obduktion, Sektion, innere Leichenöffnung: - Verweigerungsrate über 90%, deshalb: - Nennen Sie Gründe aus ärztlicher Sicht für die Durchführung einer klinischen Obduktion; solche Gespräche mit Angehörigen eines kürzlich Verstorbenen sind schwierig. - Welches sind die rechtlichen Voraussetzungen? - Nennen sie Argument, die Sie Angehörigen gegenüber äußern, um die Notwendigkeit der Durchführung einer Obduktion verständlich zu machen. „Rollenspiele“: Situation: Tod nach langer Krankheit Gespräch: Arzt - Angehörige Ziel: Notwendigkeit einer innere Leichenschau Gruppe: 3 Studenten: behandelnder Arzt, 3 Studenten: Angehörige zusätzlich: Was muss ich vor einer Sektion machen? Die Angehörigen wünschen eine Erdbestattung. Ergebnisse: Autopsie kann von Nutzen für nachfolgende Patienten sein, sieht man eher nicht bei einer Erdbestattung. Situation: plötzlicher, unerwarteter, unklarer Tod Gespräch: Arzt - Angehörige Ziel: Notwendigkeit einer inneren Leichenöffnung Gruppe: 3 Studenten: behandelnder Arzt, 3 Studenten: Angehörige zusätzlich: Was muss ich vor einer Sektion machen? Die Angehörigen wünschen eine Feuerbestattung. Ergebnisse: Klärung ob natürlicher oder gewaltsamer Tod, auch versicherungsrechtlich. Notwendig da Feuerbestattung und keine Exhumierung später möglich, Autopsie muss bei unklarer Todesursache auch durchgeführt werden, wenn die Angehörigen nicht zustimmen. Situation: Fehlgeburt / Totgeburt Gespräch: Arzt - Angehörige Ziel: Notwendigkeit einer inneren Leichenöffnung Gruppe: 2 Studenten: behandelnder Arzt, 2 Studenten: Mutter / Vater zusätzlich: Was muss ich vor der Sektion machen? - Und nach der Sektion? Ergebnisse: Nächste Schwangerschaft erfolgreicher durchführen (Ursache der Fehlgeburt?), Qualitätssicherung in der Klinik (Fehler der Neonatologen, Gynäkologen?). Ursachen wie Infektionen, Alkoholkonsum usw. rausfinden. Richtlinien zur Regelung von klinischen Sektionen: Klinische Sektion: Begriff und Aufgaben. 1.Die klinische Sektion ist die letzte ärztliche Handlung im Rahmen der medizinischen Behandlung von Patientinnen und Patienten. Die klinische Sektion ist die ärztliche, fachgerechte Öffnung eines Leichnams, die Entnahme und Untersuchung von Organen und Geweben sowie die äußere Wiederherstellung des Leichnams. In Ausnahmefällen (z.B. unbekannter Primärtumor wird nicht gefunden) werden Organe zurückbehalten, um weitere Untersuchungen durchführen zu können. Die Angehörigen werden in der Regel noch nicht informiert, wenn Organe fehlen. Wenn sie im Vornherein sagen, dass z.B. das Herz nicht entnommen werden darf, richtet man sich aber nach den Wünschen der Angehörigen, auch wenn die Aussagekraft dann eingeschränkt ist. 2.Die klinische Sektion dient der Qualitätssicherung und der Überprüfung ärztlichen Handelns in Hinblick auf Diagnose, Therapie und Todesursache, der Begutachtung, der Epidemiologie, der medizinischen Forschung, der Lehre und Ausbildung der Studentinnen und Studenten sowie der Weiter- und Fortbildung von Ärztinnen und Ärzten. Voraussetzungen: 1.Die Durchführung einer klinischen Sektion setzt voraus: a)die Einwilligung der verstorbenen Person oder ihrer nächsten Angehörigen oder einer von der verstorbenen Person bevollmächtigten Person oder mehrerer solche Personen. b)die vorherige Durchführung der Leichenschau gemäß §22 Bestattungsgesetz Baden-Württemberg: bei Anhaltspunkten für einen nicht natürlichen Tod (§22 Bestattungsgesetz und §9 Abs. 4 Bestattungsverordnung) oder bei ungeklärter Todesart (§9 Abs. 5 Bestattungsverordnung) darf eine klinische Sektion erst nach Freigabe des Leichnams durch die Staatsanwaltschaft erfolgen. ... 4.Bei Meinungsverschiedenheiten über die Durchführung der Sektion zwischen den nächsten Angehörigen und der bevollmächtigten Person darf die Obduktion nicht durchgeführt werden. Eine Leibesfrucht unter 500g ohne Lebenszeichen ist eine Fehlgeburt, eine Leibesfrucht über 500g ohne Lebenszeichen ist eine Totgeburt. Eine Totgeburt ist bestattungspflichtig und muss beim Standesamt beurkundet werden. Fehlgeburten gelten nicht als Tote oder Leichen. Fehlgeburten sind somit (§30 Absatz 2: Bestattungsgesetz) nicht bestattungspflichtig und sich hygienisch einwandfrei und dem sittlichen Empfinden entsprechend zu beseitigen. Leibesfrüchte nach Schwangerschaftsabbrüchen - gleich welchen Geburtsgewichts - sind nicht bestattungspflichtig. Eine Frühgeburt, die Lebenszeichen (schlagendes Herz, pulsierende Nabelschnur) geboten hat, muss bestattet werden. Hier in Tübingen werden aber alle Fehlgeburten, egal wie klein, auf Kosten des Klinikums bestattet. Kardiomyopathien und Myokarditis Kardiomyopathien darf man nicht mit angeborenen Herzfehlern verwechseln. Die korrekte Definition ist: Syn. Myokardiopathie; klin. Bez. für alle Herzmuskelerkrankungen, die nicht durch Koronarsklerose, Erkr. des Perikards, art. od. pulmonale Hypertonie od. angeb. bzw. erworbene Herzfehler bedingt sind. Definition: Kardiomyopathien sind Erkrankungen des Myokards, die klinisch assoziiert sind mit einer kardialen Dysfunktion. Klassifikation: - dilatative Kardiomyopathie: Herzhöhlen erweitert, Wand gleich dick, Koronargefäße normal. - hypertrophe Kardiomyopathie: normaler rechter Ventrikel, linker Ventrikel eingeengt, stark verdickte Wände - restriktive Kardiomyopathie: diastolischer Compliancefehler - arrhythmogene rechtsventrikuläre Kardiomyopathie: überwiegend rechtsventrikulär kombinierter Pumpfehler mit ventrikulären Tachykardien - nichtklassifizierbare Kardiomyopathien: z.B. Fibroelastose, minimal dilatierte Kardiomyopathie, mitochondriale Kardiomyopathie, Karzinoidherz, atriale und rhythmogene Formen. Dilatative Kardiomyopathie: Morphologisch Verlust von Herzmuskelfasern, interstitielle Fibrose, frustrane Hypertrophie der übriggebliebenen Herzmuskelfasern. Wenn mehr als 40% futsch sind, ist es mit dem Leben nicht mehr vereinbar. Eine kausale Therapie ist noch nicht bekannt, man kann nur symptomatisch die übriggebliebenen Herzmuskelfasern pharmakologisch unterstützen. Die Patienten sterben häufig am plötzlichen Herztod durch Rhythmusstörungen. Mögliche Ursachen sind mitochondriale Kardiomyopathien, autosomal dominante Vererbung (Gene auf Ch 14q, 1q, 15q, 11p, 7q), Noonan- und Costello-Syndrom, Lentiginosis. Hypertrophe Kardiomyopathie: Epidemiologie: ♀ > ♂, zu 50% genetisch bedingt Inzidenz: 2,5 Patienten / 100.000 Einwohner / Jahr Prävalenz: 20 Patienten / 100.000 Einwohner Alter: frühe Kindheit bis 7. Lebensjahrzehnt Symptome: ventrikuläre Tachykardien, AV-Überleitungsstörungen, plötzlicher Herztod (Leistungssportler) Diagnostik: invasive Diagnostik nicht obligat, Biopsie möglich (Ausschluss Amyloidose) Eine nicht obstruktive Form kann lebenslang symptomlos bleiben. Obstruktive Formen machen eine funktionelle Stenose des Ausflusstraktes. Morphologisch findet man Texturstörungen der Herzmuskelzellen mit Verlust von Desmin-positiven Myofibrillen. Es gibt auch mitochondrial bedingte hypertrophe Kardiomyopathien, Detektion von Punktmutationen mittels RFLP am mitochondrialen Erbgut. Restriktive Kardiomyopathie: Morphologisch findet sich ventrikuläre Steifheit (z.B. myokardiale Fibrose, Amyloidose) und / oder eine endokardiale Fibro(elasto)se, außerdem vergrößerte Vorhöfe. Folge ist eine Störung der diastolischen Ventrikelfüllung bei normaler systolischer Funktion. Bei der Löffler‘schen Endomyokarditis handelt es sich um die eosinophile Form einer restriktiven Kardiomyopathie; die innere Schicht des Ventrikelmyokards lässt sich eosinophil anfärben. Ein Grund für das Entstehen einer restriktiven Kardiomyopathie kann eine Amyloidose sein, bei der in betroffenen Organen interstitiell Amyloid abgelagert wird, was zu einer Verhärtung und Vergrößerung führt. Arrhthmogene rechtsventrikuläre Kardiomyopathie: Morphologisch: Adipöser rechter Ventrikel, Aneurysmen des rechten Ventrikels, Dilatation des linken Ventrikels. Mögliche Gründe: autosomal dominant vererblich (Kandidatengene sind auf Ch 14, 1, 2, 3), autosomal rezessiv vererblich (Kandidatengene auf Ch 17). Die Klinik ist charakterisiert durch Rhythmusstörungen, plötzlichen Herztod und manchmal Insuffizienz. Die arrhythmogene rechtsventrikuläre Kardiomyopathie ist die häufigste Todesursache beim plötzlichen Tod von Leistungssportlern. Wie es bei denen zu den Fetteinlagerungen in den RV kommt, ist noch ungeklärt. Nichtklassifizierbare Kardiomyopathien: Fallbeispiel endokardiale Fibroelastose: 4 Monate alt gewordenes weibliches Kind, Kammerflimmern, Kammerflattern immer wieder, therapieresistent, bis zum Tod. In der Pathologie war sehr auffallend eine massive Verdickung des Endokards und eine porzellanähnliche Auskleidung der Ventrikel. Die weißen Areale ziehen auch schon in das Myokard ein. In der Histologie sieht man eine massive Endokardfibrose. Spezifische Kardiomyopathien: - ischämische KM - Valvuläre KM - hypertensive KM - entzündliche KM - metabolische KM - Systemerkrankungen - muskuläre Dystrophien - neuromuskuläre Erkrankungen - immunologische und toxische Erkrankungen - peripartale KM Eine toxische Kardiomyopathie entsteht z.B. bei Hochleistungssportlern durch Anabolikaschäden und äußert sich in plötzlich auftretenden Herzrhythmusstörungen. Elektronenmikroskopisch sieht man eine völlig unregelmäßige Anordnung der Querstreifung und Kondensierung von Myofibrillen. Der Schaden heilt problemlos wieder aus, es sei denn, es kommt zur Fibrose. Myokarditis (entzündliche Kardiomyopathien): Definition: Schädigung kardialer Myozyten mit reaktiver entzündlicher Infiltration des Herzmuskels, die klinisch mit einer kardialen Dysfunktion einhergeht. Einteilung in nichtinfektiöse und infektiöse Form. Nichtinfektiöse Myokarditis: Beispiel: Riesenzellmyokarditis Ätiologie: unbekannt Patienten: Jugendliche oder junge Erwachsene, häufig Frauen Klinik: rapide, meist fatal verlaufende Herzinsuffizienz Diagnosesicherung: Endomyokardbiopsie Therapie: hochdosierte Immunsuppression oder Herztransplantation Fallbeispiel: 49 Jahre alt gewordene Patientin; akut einsetzende kardiale Erkrankung mit Dyspnoe, Tachykardien und Vorhoffflimmer mit akuter kardialer Dekompensation; Tod im kardiogenen Schock nach zwei Wochen. Klinische Verdachtsdiagnose war eine dilatative Kardiomyopathie bei Alkoholabusus. Histologisch findet man eine massive Entzündung mit Riesenzellen Beispiel: Rheumatische Myokarditis (Endomyoperikarditis) Ätiologie: Gruppe A-Streptokokken-Infektion mit eitriger Pharyngitis / Tonsillitis; Scharlach Pathogenese: Hypersensitivitätsreaktion gegenüber Zellwandbestandteilen von Gruppe A-Streptokokken; kreuzreagierende Autoantikörper gegen bestimmte typenspezifische M-Proteine und gruppenspezifische Karbohydrate der Gruppe A-Streptokokken mit Glykoproteinen in Herz, Gelenken und neuronalen Strukturen. Krankheitsphasen: exsudative Frühphase / proliferative Phase (rheumatisches Frühinfiltrat) 2-3 Wochen nach Streptokokken-Infekt: degenerative Schädigung des kollagenen Bindegewebes und Veränderungen der interzellulären Grundsubstanz (fibrinoide Nekrose), Umrandung der zentralen fibrinoiden Nekrose durch T-Lymphozyten, Plasmazellen und Histiozyten. Persistenz der rheumatischen Granulome (Aschoff-Geipel-Knötchen): 3-6 Monate Rheumatische Narbe, Endokarditis verrucosa, bei Perikardbeteiligung fibrinöse Perikarditis. Abräumung der Nekrose durch fibroblastenreiches Granulationsgewebe mit Ausbildung kollagener Fasern. Komplikationen: Endomyoperikarditis, Herzrhythmusstörungen, Spätfolgen bei Klappenbefall nach einigen Jahren: Mitral- oder Aortenklappenstenose Mortalität 2% Dann gibts noch einige andere Myokarditiden, z.B. die Überempfindlichkeitsmyokarditis (dosisunabhängige Reaktion auf Medikamente), die hypereosinophile Myokarditis, die granulomatöse Myokarditis (häufig bei systemischer Sarkoidose, Übergang in sekundäre restriktive Kardiomyopathie, Langerhans-Riesenzellen). Infektiöse Myokarditis: Formen: - Virale Myokarditis - Protozoen-Myokarditis - bakterielle und myokotische Myokarditis - Diphterie-Myokarditis Im Prinzip kann sie von fast allem hervorgerufen werden, was da kreucht und fleucht (Viren, Bakterien, Pilze, Parasiten), meist aber Viren. Bakterien eher als Superinfektion, Pilze eher bei Immunsupprimierten. Virale Myokarditis: Klinische Symptomatik: Kein typisches Leitsymptom! Oft blander Verlauf. Die Diagnose lässt sich eindeutig sichern durch eine Herzmuskelbiopsie. Fallbeispiel: 62-jähriger Patient mit unauffälliger Anamnese, Zustand nach Cholezystektomie bei Cholezystolithiasis. 4 Monate später massiv dilatiertes Herz bei akuter viraler Myokarditis. Angeborene Herzfehler (Congenital Heart Diseases = CHD) Herzentwicklung: Septierung der Vorhöfe: - 22. Tag p.c. (post conceptionem): Beginn des Herzschlages - 27.-29. Tag: Beginn der Blutzirkulation - 5. Woche p.c.: Cor commune, äußere Septierung mit Sulcus atrioventricularis und sulcus interventricularis - Dann beginnt die innere Septierung mit Septum primum, das ein Restforamen offen lässt (Foramen primum). - Während das Foramen primum zuwächst, treten Perforationen auf, die man Foramen secundum nennt. - Dann wächst ein Septum secundum entlang des Septum primum, aber nur von einer Seite, und bildet aus dem Foramen primum eine Art Ventil (Valvula foraminis ovalis), das nach links Blut durchlässt. Nach der Geburt strömt Blut in die Lunge, der Lungenkreislauf öffnet sich, das Foramen ovale wird durch den sinkenden Druck im rechten Vorhof verschlossen. - Bei 20% der Menschen kommt es zu einem inkompletten Verschluss, einem funktionell geschlossenen, aber anatomisch offenen Foramen ovale. Bei Druckanstieg rechtsatrial kann das Foramen ovale dann wieder geöffnet werden, was zur paradoxen Embolie führen kann (venöse Embolien gelangen direkt in die Arterien). Bildungs des Ausflusstraktes: - 5. Woche p.c: die Aorta und der Truncus pulmonalis differenzieren sich, der Ductus arteriosus Botalli verbindet aber noch den Truncus pulmonalis mit der Aorta. - 7. Woche p.c.: Das Septum aorticopulmonale bildet sich Bildung der Herzklappen: - Bildung der Segelklappen aus Endokardkissen aus septalen und muralen Kammerwänden: Chordae, Segel und Papillarmuskeln sind Differenzierungsprodukte der ursprünglich spongiös aufgebauten Kammerwand. Das in den Segeln und Chordae tendineae ursprünglich vorhandene Muskelgewebe wird durch straffes Bindegewebe ersetzt. Fetale Zirkulation: Ductus venosus: - Lokalisation: Unter- und Hinterseite der Leber - Trompetenform: engste Stelle am Beginn - Verlauf: Mündung im Bereich des Lebervenenkonfluens - maximaler Flow während der Ventrikelsystole und -diastole - minimaler Flow während der Vorhofkontraktion mit orthograder Strömungsrichtung - Dopplersonsographie: unverzichtbares Kriterium für die Zustandsbeurteilung der Zirkulation des Foeten Ductus arteriosus Botalli: - Prostaglandine bestimmen pränatal die Weite und das Offenbleiben des Ductus arteriosus, deshalb sind Prostaglandin-Antagonisten wie Salicylate, Indometacin, Glukocorticoide in der Schwangerschaft kontraindiziert, sie führen zu Ductusstenose / - verschluss. - Ductusverschluss durch initialen Druckanstieg in A. pulmonalis, Abnahme des Flows in die A. pulmonalis Foramen ovale Sauerstoffgehalt des Blutes: - Voll oxygeniertes Blut fließt durch - venöse Seite der Plazenta - Vena umbilicalis - Ductus venosus Arantii - V. cava inferior = Mischblut - fließt zusammen mit dem rein venösen Blut aus der V. cava superior in den rechten Vorhof - vor allem das Mischblut aus der V. cava inferior fließt dann durch das Foramen ovale in die linke Herzhälfte - von dort aus weiter in den linken Ventrikel, Aorta ascendens und in die obere Körperhälfte / Gehirn - das sauerstoffarme Blut aus der V. cava superior fließt in den rechten Ventrikel und von dort aus über den - Ductus arteriosus Botalli in die Aorta descendens und den unteren Teil des Körpers Postpartale Kreislaufumstellung: - Entfaltung der Lunge nach der Geburt > Abnahme des Lungengefäßwiderstandes und Anstieg des systemischen Gefäßwiderstandes > Zunahme des pulmonal-venösen Rückstromes und Abnahme des Blutzustromes aus Vena umbilicalis > Anlegen des mobilen Septum primum an das Septum secundum > Verschluss des Foramen ovale - Faktoren, die bei normalem Herzen die postnatale Kreislaufumstellung behindern: - Lungenunreife mit Surfactant-Mangel - Neugeboreneninfektion - Verschluss des Ductus arteriosus: O­2-Partialdruck im Blut steigt, dadurch Kontraktion der in der Media liegenden Muskelfasern > Verschluss (nach 10-15 Stunden p.p.). Dann Blutungen und Nekrosen in der Ductuswandung nach 2-3 Wochen, Intimaproliferation und Bindegewebsbildung, Fibrosierung, Umwandlung in das Ligamentum arteriosum. Angeborene Herzfehler: Häufigkeit: - ca. 1% aller Neugeborenen - 20% aller Fehlgeburten haben Herzfehler - 50% aller Fehlgeburten haben Chromosomenanomalien - 10% aller Totgeburten haben Herzfehler Assoziation mit zusätzlichen Fehlbildungen in 25-33% der Fälle Exogene Faktoren: - Röteln in 50% PDA (persistierender Ductus arteriosus) - Diabetes mellitus in 5-8% VSD, TGA (Transposition der großen Arterien) - maternale PKU (Phenylketonurie) - Lupus erythematodes der Mutter - Alkoholabusus in 35% VSD - Antiepileptika - Lithium - monocygote Zwillinge Genetik: - Ursachen: - 8-12% primär genetisch - 2% primär exogen - 90% multifaktoriell - Numerische Chromosomenanomalien: Trisomie 13, 18, 21, Monosomie X - Strukturelle Chromosomenanomalien: Mikrodeletion 22q, Duplikationen - Trisomie 18 in 100% VSD (Ventrikelseptumdefekt) - Trisomie 21 in 40% AVSD (atrioventrikulärer Septumdefekt), ASD II, VSD - Monosomie X in 35% ISTA (Isthmusstenose der Aorta), HLHS (hypoplastisches Linksherzsyndrom) Wiederholungsrisiko: - Beide Eltern haben kein Vitium cordis, 1 Kind mit Vitium cordis: Wiederholungsrisiko 1-4% - Beide Eltern haben kein Vitium cordis, 2 Kinder mit Vitium cordis: Wiederholungsrisiko 12% - Mutter mit Vitium cordis, Risiko 20% Spektrum der häufigsten Herzfehler (%): postnatal pränatal VSD 32 6 PS 9 5 (PS = Pulmonalstenose) ASD 8 0 AVSD Fallot TGA ISTA HLHS 7 7 5 5 4 18 3 2 11 16 Einteilung nach pathophysiologischen Aspekten: - arterio-venöse Shuntvitien: - Vorhofseptumdefekt 6% - Ventrikelseptumdefekt 27% - atrioventrikuläre Septumdefekt 5% - PDA (Persistenz des Ductus arteriosus) 9% - veno-arterielle Shuntvitien - mit vermehrtem Blutfluss in den Lungen: Transposition der großen Arterien (7%), Truncus arteriosus (1%) - mit vermindertem Blutfluss in den Lungen: Fallot‘sche Tetralogie (7%), Trikuspidalatresie (2%) - obstruktive Herzfehler - linksseitige Obstruktion: Aortenstenose, -atresie (5%), hypoplastisches Linksherzsyndrom (3%), ISTA (7%) - rechtsseitige Obstruktion: Pulmonalstenose, -atresie (7%) Vorhofseptumdefekt (ASD): - in 70% mit Chromosomenanomalien wie Trisomie 18 oder Trisomie 21 vergesellschaftet Es gibt vier Formen des ASD: - Ostium primum-Defekt: Septum primum nicht vollständig ausgebildet, Defekt im unteren Anteil des Vorhofseptums - Ostium secundum-Defekt: Septum secundum nicht regelmäßig ausgebildet, Restöffnung im mittleren Teil, ausgeprägter links-rechts-Shunt, Hyperperfusion der Lungengefäße. häufigster ASD - Sinus venosus-Defekt, häufig assoziiert mit Lungenvenenfehlmündung - Sinus coronarius-Defekt, im Bereich der Einmündung des Sinus coronarius Atrioventrikulärer Septumdefekt: = AVSD = Endokardkissendefekt = AV-Kanal - partieller AV-Kanal: Spalt in der Tricuspidalis, Ostium primum-Defekt, Spalt in der Mitralis - totaler AV-Kanal: Spalt auf der kompletten atrioventrikulären Ebene. Im Prinzip ein Ventrikelseptumdefekt kombiniert mit einem Ostium primum-Defekt. - Klinik: nach Wochen Zeichen der Herzinsuffizienz bei großem li-re-Shunt, nach Jahren Shuntumkehr bei pulmonaler Hypertonie (Eisenmenger-Reaktion) - Prognose: unterschiedliche Angaben, abhängig von Fortbestand der Mitral- / Trikuspidalinsuffizienz, ev. späterer Mitralklappenersatz - Kommt oft vor mit Mikrocephalus, Vierfingerfurche, Sandalenfurche, Hypertelorismus, mongoloider Lidachse Ventrikelseptumdefekt: - Klinik: 4-8 Wochen pp Tachydyspnoe, Schwitzen, Dystrophiezeichen, im 1. / 2. Lebensjahr Eisenmengerreaktion: anfängliche Besserung, zunehmende Zyanose - Wenn das Kind im Alter von 3 Monaten einen pulmonale Hypertonie (Aszites, große Leber, Rechtsherzhypertrophei) hat: Echo + Katheter, dann weiteres Prozedere entscheiden, wenn nicht: weiter beobachten - in 8% mit Chromosomenanomalien (Trisomie 13, 18, 21, Di George) - Die muskulären VSD entstehen durch eine gesteigerte myokardiale Apoptose, ev. auch multipel vorkommend („swiss cheese“) Fallot‘sche Tetralogie: - VSD, Pulmonalstenose, reitende Aorta (nach rechts versetzt, reitet auf dem VSD, so dass Blut von beiden Herzkammern hineinfließt), Rechtsherzhypertrophie - Ätiologie: Fehlposition des Conusseptums nach rechts, anterior und oben > Fallot‘sche Monologie könnte man es auch nennen, gibts aber nicht - Klinik: Hämodynamik ist abhängig vom Ausmaß der Pulmonalstenose - Korrektur: Vollkorrektur oder Shuntanlage (bei Verschluss) - in 11% mit Chromosomenanomalien assoziiert (Trisomie 18, Di George) Transposition der großen Arterien: - Entstehung der TGA: Fehlverschmelzung der Conuswülste - Klinik: 1. / 2. Lebenstag: rasch progrediente Zyanose > schnelle Switch-Operation indiziert (dabei auch Heraustrennen der Herzkranzgefäße aus der Lungenschlagader und wieder Annähen an die Aorta) - rettende Notfallmaßnahmen: Herstellen eines Shunts: - Offenhalten des Ductus arteriosus Botalli mit Prostaglandin - Rashkindmanöver: Ballondilatation des Foramen ovale mit Katheter - Prognose: nach Switch: sehr gut; ohne Switch: in wenigen Tagen tot - bei einer TGA kann das Kind sowieso nur überleben, wenn der Ductus arteriosus offen ist und ein VSD oder wenigstens ASD vorliegt Hypoplastisches Linksherzsyndrom (HLHS): Macht 3% aller CHD aus. Entstehung: durch einen verminderten Fluss: Einengung oder Verschluss des Foramen ovale oder Aortenstenose / Aortenatresie. Prinzip: no flow, no growth Deshalb wäre pränatale Therapie indiziert, ist aber noch nicht möglich. Die Embryologie ist noch nicht vollständig bekannt. Man müsste versuchen, das FO oder die Aortenklappe zu sprengen, um einen Fluss sicherzustellen, so dass das linke Herz wachsen kann. Das hypoplastische Linksherzsyndrom oder die ISTA sind relativ typisch für das Turner-Syndrom (X0). Man sieht bei Turner oft: Pterygium colli beidseits, Fußrückenödeme, weiter Mamillenabstand. Beim hypoplastischen Linksherzsyndrom findet man auch relativ häufig eine Endokardfibroelastose (weißer Belag). Auch hinter der atretischen Aortenklappe entwickelt sich die Aorta nicht, sie ist ebenfalls hypoplastisch. Zur operativen Therapie nach Geburt (Norwood-OP) kann man die A. pulmonalis durchtrennen und mit der hypoplastischen Aorta anastomosieren. Es entsteht nur ein Kreislauf mit Mischblut. Die Versorgung der Lungenarterien wird über einen Shunt etwa von der A. brachiocephalica sichergestellt. Die Kinder haben erstaunlicherweise eine gute Lebensqualität, und ohne Operation würden auf jeden Fall alle sehr schnell sterben. Der zweite Schritt ist, dass die V. cava superior an die Pulmonalarterien angeschlossen wird und der rechte Vorhof von der V. cava superior abgetrennt wird. Der dritte Schritt (Norwood III) besteht in der Anastomoselegung zwischen V. cava inferior und Arteria pulmonalis. Folge: Das systemische venöse Blut fließt in die Pulmonalarterie, das pulmonalvenöse Blut geht in den systemischen Ventrikel. Problem ist vor allem die Thrombosierung einer solchen Prothese. Mortalität bei dieser OP ist 10-12% nach 2-3 Jahren. - Varianten: - fehlende atrio-ventrikuläre Konnektion - hypoplast. li. V. mit verschließender Membran im Bereich der Mitralklappe - hypoplast. li. V. mit Aortenatresie - postpartal Perfusion von Aorta und Herzkranzgefäßen nur über den Ductus arteriosus Botalli, wenn man den nicht mit Prostaglandinen offenhält, sterben diese Kinder sehr schnell - Klinik: Innerhalb der ersten Lebenstage Dyspnoe, Hepatomegalie, Zyanose usw. Indikationen für eine fetale Echokardiographie: - Familienanamnese (erhöhtes Wiederholungsrisiko, wenn schon Kind mit Herzfehler auf die Welt kam) - Diabetes mellitus - Infektionen in der Frühschwangerschaft (z.B. Röteln) - anamnestisch bekannte negative Umwelteinflüsse (Medikamentenanamnese, Alkoholabusus) - extrakardiale Fehlbildungen (in einem Drittel der Fälle zusammen mit Herzfehler) - IUWR (intrauterine Wachstumsretardierung) - Nuchal Translucency (= NT, Nackentransparenz) - single-Arterie (nur eine Nabelschnurarterie statt zwei, in 30% mit anderen Fehlbildungen assoziiert) - Hydrops fetalis - Anämie - Mehrlingsschwangerschaft Wenn in der 22./23. SSW ein angeborener Herzfehler diagnostiziert wird, entscheiden sich weit über 50% der Eltern für eine Interruptio. Das hängt natürlich noch von der Schwere des Herzfehlers ab. Lernziele: - Fetale Zirkulation: Diagnostik und Prävention (entsprechende Medikamente nicht einnehmen) - Ursachen der Persistenz der fet. Zirkulation (PFC); persistierende pulmonale Hypertonie des Neugeborenen (PPHN) - Septierungsanomalie: Vitium, Shunt, Folgen, Notfallmaßnahmen - Ductus-abhängige Vitien - CHD + assoziierte Fehlbildungen - Wiederholungsrisiko - Bedeutung der Pränataldiagnostik bezüglich der Prognose der CHD - Ehtik / Problematik der Pränataldiagnostik - Problematik einer objektiven (?) Beratung (Fallbeispiel) Mikroskopieren Präparat 285: Akute Virusmyokarditis Man sieht halt teilweise etwas auseinandergerissene Kardiomyozyten (weils ein Autopsie-Präparat ist), die das gesunde Herzmuskelgewebe darstellen, und dazwischen immer wieder Lücken mit Zellfragmenten und Entzündungszellen (Makrophagen, Granulozyten mit lappigem Kern, Monozyten). In diesem Fall starb die junge Patientin an Parvoviren, den Erregern der Ringelröteln. Präparat 284: Dilatative Kardiomyopathie Etwa 30-40% des Herzmuskelgewebes ist durch hier blau eingefärbtes funktionell minderwertiges Bindegewebe ersetzt; seitlich sieht man das vor allem aus Fett bestehende Epikard (fast nix übrig, Fett rausgelöst) und das fibrotisch verbreiterte Endokard. Schaut man sich die vernarbten Stellen mit dazwischenliegenden Kardiomyozyten genauer an, fällt einem auf, dass diese teilweise atrophisch und sehr dünn, teilweise hypertrophiert sind, mit großem Kaliber und sternförmigem, hyperchromatischem Zellkern. Präparat 113a: Obstruktive hypertrophe Kardiomyopathie Insgesamt zuviele Herzmuskelzellen, keine Nekrosen, unregelmäßige Anordnung von hyperchromatischen Zellen. Das fällt insgesamt als Texturstörung auf. Das Problem an der hypertrophen Kardiomyopathie sind Rhythmusstörungen, wodurch die Patienten normalerweise zuerst auffallen, bis hin zum plötzlichen Herztod. Es ist nicht ganz leicht, bei Sportlern zwischen einer hypertrophen Kardiomyopathie und einem normalen Sportlerherz zu unterscheiden. Beim normalen Sportlerherz liegt nur eine Hypertrophie der Myokardzellen vor, bei der hypertrophen Kardiomyopathie zusätzlich noch die unregelmäßige Anordnung, es sieht einfach komisch aus. Propädeutik, Systematik und Pathologie der Erkrankungen des unteren Atemtrakts Lernziele: Erkrankungen der unteren Atemwege (Trachea, Lunge): Kenntnis der Definitionen, Epidemiologie, Ätiologie, Pathogenese, Klassifikationen, Morphologie, des Verlaufs. III.Obstruktive und restriktive Lungenerkrankungen IV. Pulmonale Infektionen V. Diffuse interstitielle (infiltrative / restriktive) Lungenerkrankungen ... Erkrankungen der Pleura: Kenntnis der Definitionen, Epidemiologie, Ätiologie, Pathogenese, Klassifikationen, Morphologie, des Verlaufs. I. Entzündliche und nicht entzündliche pleurale Effusionen II. Pneumothorax (Ursachen, Folgen) III.Neoplasien der Pleura bla Trachea: - Stenosen - Fremdkörperobturation: - bei Kindern mehr als 50% durch Fremdkörperaspiration - bei Erwachsenen in Folge reduzierter Bewusstseinslage Aspiration von großen Speisebrocken oder sonstigen Zeug - vor allem durch Schlaganfall, Rauschzustände, psychische Erkrankungen - schwerste Ventilationsstörungen - akuter Erstickungstod („Bolustod“) - intratracheale Stenosen (entzündliche Stenosen, Säbelscheidentrachea, Tracheopathia chondropathia plastica): - entzündlich nach Langzeitintubation (iatrogen) - Polypen, Tumoren - überschießende Granulationen nach Tracheotomie - Intubationsgranulome - Verminderung des transversalen trachealen Durchmessers - Säbelscheidentrachea - Verknöcherung der Knorpelspangen nach dorsal - Stenosen durch Wandschwäche - Kompressionsstenose - Tracheitis: - membranöse und pseudomembranöse Infektionen wie etwa Diphterie - ... Lunge: Bedeutung der Lungenpathologie: - alltäglich in klinischer und klinisch-apthologischer Praxis: - primäre respiratorische Infekte - Inhalationspathogene (Zigarettenrauch, Verschmutzung, andere Umweltinhalantien) - chronische Bronchitis - Emphysem - maligne Neoplasien der Lunge: - häufigste tumorbedingte Todesursache bei männlichen und weiblichen Patienten (häufigere Todesursache Mammakarzinom) - Lungenerkrankungen bzw. Veränderungen sind häufigste unmittelbare / terminale todesverursachende Erkrankung: - Lungenödem - Atelektasen - Bronchopneumonien Normale Lunge: - Funktion: Gasaustausch zwischen Atemluft und Blut - Anatomie: - rechts drei Lappen, links zwei Lappen und Lingula - rechts Hauptbronchus vertikaler in Verlängerung der Trachea (darum rechts vermehrt Aspiration) - doppeltes Arteriensystem (bronchial und pulmonal, darum bei Lungenarterienembolie und fehlender Linksherzinsuffizienz kein ischämischer oder hämorrhagischer Infarkt der Lunge) - Gliederung: Hauptbronchus, Bronchi, Bronchioli (kein Knorpel, keine Bronchialdrüsen), Bronchioli terminales (<2mm) - distal der Bronchioli terminales „Acinus“ oder „terminale respriatorische Einheit“ (7mm Durchmesser) - histologisch: mehrreihiges Flimmerepithel, Lamina propria, glatte Muskulatur (Tunica muscularis), gemischte Drüsen, hyaliner Knorpel, um die Bronchien offen zu halten - bis zu den Stimmlippen des Larynx geschichtetes nicht verhornendes Plattenepithel - distal davon Zylinderepithel mit Flimmerbesatz - schleimbildende Epithelien (Becherzellen) - Zusätzlich gibts noch neuroendokrine Zellen, die pathologisch zum kleinzelligen Bronchialkarzinom entarten können. - In der Alveolarwand sind Typ I- und Typ II-Epithelien vorhanden. Bei Beschädigungen regeneriert sich das Gewebe aus den Typ II-Zellen. - Aufbau der Alveolarwand - Alveolarepithel - Alveolarmakrophagen (Teil des MPS, häufig mit phagozytiertem Material beladen) - Alveolarwände sind perforiert (Kohn‘sche Poren, hier Durchtritt von Bakterien, Exsudat usw.) - Surfactant: Auf der Oberfläche des Alveolarepithels (Dipalmitoylphosphatidyl-Cholinphosphatidylglycerol) Kongenitale Anomalien: - Agenesie oder Hypoplasie der Lunge oder von Lungenlappen - tracheale oder bronchiale Anomalien - vaskuläre Anomalien - kongenitale lobäre Überblähung (Emphysem) - kongenitale Zysten (Reste des embryonalen Vordarms) - v.a. bronchiogene Zysten - bis mehr als 5cm durchmessend - den Bronchien benachbart - mit und ohne Verbindung zum Bronchialsystem - schleim- oder luftgefüllt - Komplikationen: eitrige Entzündungen, Lungenabszess - Ruptur in die Lunge, Blutungen, Hämoptysis - Ruptur in die Pleura, Pneumothorax, interstitielles Emphysem - intralobäre und extrapulmonale Sequestration (mit und ohne Verbindung zum normalen Bronchialsystem) - extralobale Sequestration: - überall im Thorax oder Mediastinum - beobachtet bei Kindern als abnorme Raumforderung / Masse - häufig verbunden mit anderen kongenitalen Anomalien - intralobale Sequestration - Atelektasen: - Resorptionsatelektasen: - Folge kompletter Bronchusobstruktion - Verschiebung des Mediastinums zur atelektatischen Lunge hin - häufigste Ursachen: Asthma bronchiale, chronische Bronchitis, Bronchiektase, postoperative Phasen mit Aspiration. - Kompressionsatelektasen: - Verschiebung des Mediastinums von der atelektatischen Lunge weg (Pleuraerguss, Hämatothorax, Pneumothorax, Tumorwachstum) - häufigste Ursachen: Herzversagen, maligne Tumoren - Kontraktionsatelektasen: - fibrotische Veränderungen der Lunge oder Pleura - Kompressions- und Resorptionsatelektasen sind prinzipiell reversibel, Kontraktionsatelektasen nicht! Vaskulär bedingte Lungenerkrankungen (Kreislaufstörungen): - Lungenödem durch: - erhöhter hydrostatischer Druck, z.B. durch Herzinsuffizienz links - erhöhte vaskuläre Permeabilität, z.B. bei Pneumonie - verminderter onkotischer Druck, z.B. bei nephrotischem Syndrom - erhöhter intrapleuraler negativer Druck, etwa bei Atelektasen - verminderte Lymphabfluss, z.B. bei mediastinaler Karzinomatose - Blutstauung (akut und chronisch) - akute Blutstauung: - Ursachen: Herzinfarkt, Herzinsuffizienz als Folge von Linskherzhypertrophie, Schockzustände - Makroskopie: schwere blutreiche Lunge (kongestive Hyperämie) - Folgen: - angioektatische Alveolenkompression - interstitielles Ödem - intraalveoläres Stauungsödem - Übertritt von Erythrozyten in Alveolen - Auftreten von Siderophagen (sogenannten Herzfehlerzellen) - bei Beseitigung der Ursache vollständig reversibel - chronische Blutstauung: - Ursachen: fortbestehende Herzinsuffizienz besonders z.B. bei Stenose der Mitralklappe - Folgen: - - - - chronisches interstitielles Ödem - Aktivierung der Bindegewebszellen im Interstitium - vermehrte Faserbildung (kollagene und elastische Fasern) - Formen: - rote Stauungsinduration - ektatische hyperämische Kapillaren - Stabilisierung der Kapillaren durch interstitielle Fibrose - braune Stauungsinduration - bedingt durch Siderinablagerungen - klinische Folgen: - reduzierter Gasaustausch - Luftnot besonders im Liegen - Funktionsstörung durch Verlängerung der Diffusionsstrecke - Verzögerung des transpulmonalen Blutflusses - reduzierte Lungenvolmina durch Einschränkung der Lungenelastizität - restriktive ventilatorische Insuffizienz - irreversibel! akutes Lungenversagen (ARDS = adult respiratory distress syndrome / diffuse alveolar damage); morphologisch ähnlich dem Atemnotsyndrom des Neugeborenen (IRDS = infant respiratory distress syndrome) Hauptursachen: - Infektion - Sepsis - diffuse pulmonale Infektionen - ... - physikalische Verletzungen - inhalierte Irritantien - chemische Verletzungen - hämatologische Bedingungen - Pankreatitis - Urämie - kardiopulmonärer Bypass Stadien des ARDS: - Normalzustand - Endothelschädigung, Permeabilitätstörung und interstitielles Ödem - Epithelschädigung und beginnendes intraalveoläres Ödem - Ausbildung von hyalinen Membranen - Endothelzellregeneration, interstitielle Fibrose usw. Lungenarterienembolie und Lungeninfarkt Pulmonalarteriensklerose und pulmonale Hypertension - primär / idiopathisch (selten) - sekundär (die Regel) Hämodynamische pulmonale Ödeme / Ödeme durch mikrovaskuläre Schädigungen / akutes Lungenversagen / ARDS / diffuser Alveolarschaden: - Klinischer Verlauf: - initial häufig symptomlos - später Dyspnoe, Tachypnoe, ... - ... Pulmonalarterienembolien, Blutungen und Infarkte: - Verschlüsse von Pulmonalarterien durch Blutkoagel sind immer embolischer Natur - 10% der akuten Todesfälle hospitalisierter Patienten - 10% der Pulmonalarterienembolien verursachen hämorrhagische Lungeninfarkte - Tod durch Blockade des Blutdurchflusses durch die Lunge - Tod durch akutes Cor pulmonale (die nur 4mm dicke rechte Herzkammer kann den akuten Druckanstieg nicht kompensieren, das Herz bleibt stehen) - Komplikation: septischer Infarkt Formen: - fulminante Lungenarterienembolie - Embolien in Segment- und Subsegmentarterien - kleine periphere Lungenarterienembolien Lungenhochdruck und Pulmonalarteriensklerose: - sehr viel seltener als systemischer Bluthochdruck (ca. 1:8) - der pulmonale Hochdruck ist zumeist sekundär - bei Ausschluss anderer Ursachen primärer oder idiopathischer pulmonaler Hochdruck - Ätiologie und Pathogenese: - endotheliale Dysfunktionen - Vasospasmen - verursacht durch Appetitzügler (Aminorex) - Fenfluramin und Phentermin („Anti-Obesitas“-Medikamente) - deshalb meist weibliche Patienten... - Morphologie: - häufig rekanalisierte Thromben - Atherosklerose der Pulmonalarterien - Arteriolosklerose - plexogene pulmonale Arteriopathie - klinischer Verlauf: - am häufigsten bei Frauen im Alter zwischen 20 und 40 Jahren - erste Symptome sind Dyspnoe, Müdigkeit und pektanginöse Beschwerden - später respiratorische Insuffizienz, Zyanose, Rechtsherzhypertrophie und Tod durch dekompensiertes Cor pulmonale - häufig kompliziert durch Thrombembolien und Pneumonien - ca. 80% der Patienten versterben 2-5 Jahre nach Ausbruch der Erkrankung - einzelne Patienten profitieren von kontinuierlicher vasodilatatorischer Therapie und antithrombotischer medikation - Cor pulmonale (Rechtsherzhypertrophie, die jederzeit zum Tod führen kann) Obstruktive und restriktive Lungenerkrankungen 1.obstruktive Lungenerkrankungen: - erhöhter Widerstand auf jedem Niveau in den Luftwegen durch partielle oder komplette Obstruktion: Emphysem, chronische Bronchitis, Bronchektasen, Asthma bronchiale > Reduktion des exspiratorischen Sekundenvolumens. - chronisch obstruktive Lungenerkrankungen (air way disease, COPD, COLD): - Emphyseme (Aufblähungen der Lunge mit möglicherweise blasigem Umbau ganzer Lungenläppchen im subpleuralen Bereich) Emphysemarten: - zentriazinär / zentrilobulär (95% aller Emphyseme, typisch für Raucher, chronische Bronchitis und Pneumokoniosen) - panazinäres / panlobuläres Emphysem (Alpha-1-Antitrypsinmangel) - paraseptales / distal-azinäres Emphysem (der proximale Anteil des Azinus ist normal, der distale dilatiert), entsteht häufig in der Nachbarschaft von Fibrose, Vernarbungen oder Atelektasen - irreguläres Emphysem (Narbenemphysem) Inzidenz: Autoptisch bei 50% der Verstorbenen (panazinär und zentriazinär) - in 6,5% todesursächlich bei diesen Patienten - klare positive Korrelation zwischen Zigarettenkonsum sowie Schwere des Emphysems - Pathogenese: Destruktion der Alveolarwände durch den Protease-Antiproteinase-Mechanismus - Morphologie / klinischer Verlauf: - chronischer Husten häufig erstes Symptom - Pfeifen des Atemgeräusches (häufig Verwechslung mit Asthma bronchiale) - verlässliches Zeichen ist die Reduktion des respiratorischen Sekundenvolumens - Todesursachen: - respiratorische Azidose, Koma und Tod - Rechtsherzversagen - Lungenkollaps infolge Pneumothorax (Emphysem-Bullae subpleural platzen) andere Emphysemtypen: - kompensatorisches Emphysem (z.B. Restlunge nach Lobektomie) - seniles Emphysem (sog. Alterslunge) - obstruktive Überblähung / Überinflation (z.B. Bronchusverlegung durch Fremdkörper) - bullöses Emphysem (durch Konfluenz vor allem peripherer Emphysemblasen) - interstitielles Emphysem (z.B. nach Überdruckbeatmung) - chronische Bronchitis: - klinisch definiert (chronischer Husten mit Sputumproduktion in mehr als 3 Monaten in den letzten 2 aufeinander folgenden Jahren) - einfache Bronchitis (keine Obstruktion) - chronische asthmoide Bronchitis (hyperreaktive Luftwege) - obstruktive chronische Bronhcitis (gewöhnlich mit Emphysem verbunden, vor allem bei „heavy smokers“) - Pathogenese: chronische Irritation, mikrobiologische Infekte: - Hypersekretion - Becherzellhyperplasie bis in die Bronchiolen - Bronchiolitis und „small air ways disease“ 2.restriktive Lungenerkrankungen: - reduzierte Totalkapazität der Lungen - exspiratorisches Sekundenvolumen normal oder lediglich proportional reduziert - Hauptursachen: Brustwandstörungen bei normaler Lunge (neuromuskuläre Erkrankungen), hochgradige Obesitas, Pleuralerkrankungen, Kyphoskoliose, interstitielle Lungenerkrankungen 3.interstitielle und infiltrative Lungenerkrankungen - akut (klassisches Beispiel ARDS) - chronisch (Staublungenerkrankungen, Pneumokoliosen, chronisch-infiltrative Lungenerkrankungen) In der Klinik verwendete Abkürzungen für chronisch bronchopulmonale Erkrankungen: - chronic obstructive lung disease (COLD) - chronisches unspezifisches respiratorisches Syndrom (CURS) - chronic non-specific lung disease (CNSLD) - chronic airway obstruction (CAO) - chronic obstructive airway disease (COAD) - chronic obstrutive pulmonary disease (COPD) Asthma bronchiale: - Definition: Asthma bronchiale ist eine chronische rezidivierend entzündliche Erkrankung, welche durch hyperreaktive Bronchien charakterisiert ist. Sie führt zu episodischen Bronchospasmen und ... Treffen Allergene nach einer einmal erfolgten Sensibilisierung auf die schon konditionierten Mastzellen und Eosinophilen Granulozyten, werden massiv Zytokine und Mediatoren ausgeschüttet, die zu Bronchospasmen und einer Destruktion des Flimmerepithels führen, außerdem zu einer starken Schleimproduktion der Becherzellen. - extrinsisches Asthma induziert durch Typ i Hypersensibilitätsreaktion, induziert durch Exposition gegenüber einem extrinsischen Antigen - atopisches / allergisches Asthma - allergische bronchopulmonale Aspergillose - intrinsisches Asthma: - verursacht durch nicht-immunologische Mechanismen - Einnahme von ASS - pulmonale Infekte (vornehmlich viral) - Erkältungskrankheiten - inhalierte Irritantien - berufsbedingtes Asthma - Stress und körperliche Anstrengung - Pathogenese: - atopisches / allergisches Asthma bronchiale: - induziert durch Umweltirritation (Staub, Pollen, Tierhaare, Nahrungsmittel usw.) - häufig positive Familienanamnese - nicht-atopisches Asthma: - Folie weg :( - Medikamenten-induziertes Asthma bronchiale: - Folie weg :( - Umwelt- bzw. berufsbedingtes Asthma: - Folie weg :( - Morphologie: - Basalmembranverdickung - Ödem und Entzündung mit Eosinophilie (5-50% der inflammatorischen Zellen) - Hyperplasie der submukösen Drüsen - Hypertrophie der Bronchusmuskulatur (Folge rezidivierender prolongierter Bronchuskonstriktionen) - Klinik: - selten Spontanheilung - Asthmaanfälle (bis Stunden) - Status asthmaticus (Tage; evtl. schwere Zyanose und Tod) - progressive Überblähung, Emphysembildung - Zeichen bakterieller Infekte, chronische persistierende Bronchitis - Bronchiektasen, Pneumonie, gelegentlich Cor pulmonale und Tod im Herz- / Rechtsherzversagen Maligne Tumoren Epithel: ... ist praktisch die Matrix, auf der sich epitheliale Tumoren entwickeln: - Hyperplasie - Metaplasie (es bildet sich Plattenepithel, Übergangsepithel z.B. aus Zylinderepithel im Respirationstrakt, so kann es sogar in der Lunge verhornende Plattenepithelkarzinome geben) - Dysplasie (Grading, Präneoplasie) - CIS (carcinoma in situ): wird nach 10-15 Jahren zu 68% zum Carcinom Mit immunhistochemischer Färbung kann man in histologischen Präparaten etwa untersuchen, in welchen Schichten das Epithel proliferiert. In allen Schichten proliferierendes Epithel ist typisch für maligne Entartungen. Lungentumoren: Epitheliale Lungentumoren - Klassifikation: 1.1 Benigne - Papillom - Adenom - andere 1.2 Präinvasive Läsionen - Dysplasie - CIS - atypische adenomatöse Hyperplasie - diffuse idiopathische neuroendokrine HP 1.3 Maligne: - Plattenepithel-Ca (35-60%) Es können sich oft multiple Metastasen in der Haut entwickeln. - kleinzelliges Ca (20-25%) Hochmaligne neuroendokrine Karzinome; können z.B. Bombesin oder Serotonin sekretieren. Makroskopisch eine sehr dichte, fleischartige Masse, zellreicher Tumor. Ähnelt eher einem Sarkom als einem Karzinom. Histologisch sieht man bei Formalinfixierung fast kein Zytoplasma mehr, nur Zellkerne. Lässt sich gut mit Chromogranin anfärben (Diagnostik). Wenn man mit Proliferationsmarkern anfärbt, sieht man sehr viele Zellen, die aus der G0-Phase herausgetreten sind, auch wenn man sonst recht wenige Mitosen finden wird. - Adeno-Ca (6-32%) - großzelliges Ca (10-15%) Kann man noch unterscheiden in anaplastische und neuroendokrine großzellige Ca. - Carcinom-Sarkom - Pulmoblastom Lungen-Karzinome: - - - - - - - - - - - 13% aller Malignome Altersgipfel 7. Lebensjahrzehnt 50.000 per anno Neuerkrankungen in Deutschland 95% Raucher Raucher: 30-60fach höheres Risiko (3000-6000%...) 70% zentral (hiliär), 20-30% peripher 2-3% pneumonisch, das heißt diffuses Wachstum (besonders tückisch, gebärdet sich wie eine Pneumonie) zentrale Tumor-Vernarbung (Narben-Ca!) Pancoast-Typ (wenn etwa Lungentumoren übergreifen auf die Clavicularregion manchmal pseudomesotheliomatöses Wachstum lockere Korrelation Typ-Wachstumsform Rundherd-Histologie: - maligne: 49% primäre Ca. 38% andere Tumoren 2% Metastasen 9% - benigne 51% Tumor 14% nicht-tumorös 37% Tumoren / Neoplasien der Lunge: - 90-95% der Lungentumoren sind bronchiogene Karzinome - 5% sind bronchiale Karzinoide und 2-5% mesenchymale und andere Neoplasien Bronchiogene Karzinome: - in der sogenannten ersten Welt wichtigster maligner Tumor - ca. 30% aller tumorbedingten Todesfälle - mehr als 7% aller Todesfälle (sowohl männlich als auch weiblich) Ätiologie und Pathogenese: - Tabakrauchen!!! - Asbestexposition! Vornehmlich im Zusammenhang mit Tabakrauchen (ohne Tabakrauch-Exposition 5fach gesteigertes Bronchialkarzinomrisiko, mit Tabakrauchen 50-90fach gesteigertes Bronchialkarzinomrisiko) - industrielle Risiken - Strahlung - Uranexposition - Nickel, Chromat, Kohlenstaub, Senfgas, Arsen, Beryllium, Eisenoxyde, Drucker, Arbeiter in Goldminen, Lösungsmittel - Luftverschmutzung - Radonexposition in geschlossenen Räumen Haupttypen des Lungenkarzinoms und klinische Besonderheiten: Herkömmlicherweise wird zwischen 4 Haupttypen des Lungenkarzinoms unterschieden, chen. Plattenepithelkarzinom Adenokarzinom Großzelliges Karzinom Assoziation mit 96% 75% 93% Nikotinabusus Anteil weiblicher 18% 36% 21% Patienten Hilusnahe Lokali- ~80% ~25% ~25% sation Metastasierung spät, vorwiegend lym- früh, lymphogen früh und hämatogen phogen Molekulargenetik: die insgesamt 95% ausmaKleinzelliges Karzinom 96% 19% ~85% ... - - - - - bei klinischer Manifestation 10-20 genetische Mutationen dominante Onkogene C-myc in kleinzelligen Karzinomen und k-ras in Adenokarzinomen Inaktivierung von p53, Retinoblastomgen und anderen Genen auf dem kurzen Arm des Chromosoms 3 gelegentlich familiäre Häufung Vernarbung (sog. Narbenkarzinom) Stadieneinteilung der kleinzelligen Karzinome: - Limited Disease (LD) Der Tumor ist auf einen Hemithorax begrenzt - Extensive Disease (ED) Der Tumor dehnt sich über beide Thoraxhälften aus. R-Klassifikation: Beschreibt, ob die Tumoren bei chirurgischer Therapie mikroskopisch residual (R1) oder makroskopisch residual (R2) entfernt wurden; R0 = kein Residualtumor nachweisbar. Metastasierung: - lymphogen - hämatogen - kavitär (Symptom: hämorrhagischer Erguss) - kanalikulär - Impfmetastasen Endokrine Tumoren: - ADH, macht Hyponatriämie wegen überschüssiger ADH-Sekretion - ACTH, führt zu Cushing-Syndrom - Parathormon - ... Metastasen in der Lunge: - häufigster Tumor in der Lunge - 30% aller Malignome in der Lunge - besonders Hoden-, Knochentumoren und Melanome - zwei Drittel der Metastasen in der Lunge im ersten bis zweiten Jahr nach OP des extrapulmonalen Tumors - Metastasierung meist nach peripher, oft multipel vorhanden Pleura: - primäre intrapleurale bakterielle Infektionen - primäre Neoplasmen der Pleura - pleurale Effusionen - Ein Pleuraerguss entsteht unter 5 Bedingungen: 1.gesteigerter hydrostatischer Druck, z.B. bei kongestivem Herzversagen 2.gesteigerte vaskuläre Permeabilität, z.B. bei Pneumonien 3.verminderte onkotischer Druck, z.B. bei nephrotischen Syndromen 4.reduzierter intrapleuraler negativer Druck 5.Lymphangiosis carcinomatosa Pleuratumor: - solitärer fibröser Tumor (sog. pleurales Fibrom) - malignes Mesotheliom - es entsteht in viszeraler und parietaler Pleura - in bis zu 90% der Fälle Asbestexposition nachweisbar - zwischen Asbestexposition und Tumormanifestation liegen 25 bis 45 Jahre Fallbesprechung Sektion Sektionen, auch bei nicht unklarer Todesursache, sind auch heute noch zur Qualitätssicherung wichtig. Fallbesprechung: - - - - - - - - - - - - - - - - Patientin, 56 Jahre alt 12. Juni (samstags) mittags von Notarzt in Status asthmaticus eingeliefert in der Aufnahme massive Dyspnoe, 83% Sauerstoffsättigung, laut Ehemann den ganzen Morgen Atemnot anamnestisch Nikotinabusus von 30-40 Packyears bis vor ein paar Jahren regelmäßiger Alkoholabusus stark adipös, etwa 100kg unter 1,70m COPD (chronisch obstruktive Lungenerkrankung), wurde mit Steroiden und Theophyllin therapiert Pulse an Extremitäten schlecht palpabel, Unterschenkelödeme, Ulzera dann tachykard, starkes Schwitzen im Labor LDH über 7000 Hepatitisserologie negativ, CMV-Diagnostik negativ am 13. Juni stellt der Oberarzt fest, dass die Lebervenen perfundierbar sind und das angenommene Budd-Chiari-Syndrom ausgeschlossen ist (wieder runter von Leber-TX-Liste) Blut-pH wird azidotisch (7,31), Urinausscheidung nimmt ab, Leberwerte sind fallend, die Temperatur steigt über 39°C trotz Augmentan nachts um 1 Uhr am 14. Juni bekommt die Patienten einen Katheter in die V. subclavia, um 6 Uhr muss reanimiert werden, weil die Patienten eine Pulsfrequenz von 190 hatte bei einem mittleren arteriellen Druck von 30. wahrscheinlich lag eine Rechtsherzinsuffizienz vor mit sekundärer Leberschädigung und Nierenversagen Der Ehemann sprach sich gegen eine lebensverlängernde Therapie aus, am 15. Juni stirbt die Patientin im Kreis ihrer Familie. Weil die Lunge bei einer COPD nicht mehr richtig belüftet wird, sinkt auch der Blutdurchfluss (Euler-LiljestrandReflex), das führt zur pulmonalen Hypertonie mit Rechtsherzbelastung, im Lauf der Jahre damit zu einer Rechtsherzinsuffizienz. Die COPD entstand wohl wegen Nikotinabusus (z.B. über 40 Jahre lang jeden Tag eine Packung = 40 Packyears), wodurch das Flimmerepithel zu Grunde geht (vor allem die Zilien zuerst). Dadurch kann der produzierte Schleim nicht mehr nach außen transportiert werden, die Lunge kann nicht mehr gut belüftet werden, der Sauerstoffaustausch ist gestört. Außerdem bietet sich damit ein guter Nährboden für Bakterien = Entzündung = Schwellung = weitere Einengung der Atemwege, Verlegung weiterer Bronchiolen. Unter COPD können sich auch Emphyseme bilden, wobei Alveolarsepten immer dünner werden, reißen können, die Kapillarfläche nimmt ab, der Druck im Lungenkreislauf nimmt weiter zu, die Rechtsherzbelastung steigt noch weiter. Die Patientin kam primär wegen Status asthmaticus in die Klinik; das ist einfach ein akuter Asthmaanfall. Darunter versteht man das anfallsweise Auftreten und schwere Atmen durch hyperreaktive Entzündungsprozesse in den Bronchiolen / Alveolen. Auf die eigentlich Ursache kam man dann mit einer Echokardiographie, in der nachgewiesen wurde, dass der rechte Ventrikel einfach nicht mehr genug Blutdurchfluss schafft. Auch der linke Ventrikel war in seiner Pumpfunktion diffus eingeschränkt (statt 50% Kontraktilität wie bei gesunden Menschen nur 20%). Ein Aspekt, der für die Kliniker sehr überraschend war: Die Fulminanz des Leberzerfalls und des Verfalls der Funktion des rechten Ventrikels. Sie hatte auch ein erhöhtes CRP und wahrscheinlich Infektionsprozesse laufen; die akute Störung könnte auch durch eine Myokarditis hervorgerufen worden sein. Pathologie: - Lunge: li 650g, re 700g (etwa Normalgewicht). Hauptbronchien erweitert, „Muskelrippen“ in den Bronchien = Hypertrophie der bronchialen Muskeln im Rahmen der COPD, keine ausgeprägte Entzündung. Veränderungen im Bereich der A. pulmonalis (Arteriosklerose), was in der A. pulmonalis absolut untypisch ist, also pulmonale Hypertonie. Histologisch zerstörte Alveolarsepten, also Lungenemphysem. In den Pulmonalarterien sieht man Thromben, die allerdings entweder frisch sind, oder Leichengerinnsel, oder bei denen der Umbau nicht so vonstatten gehen konnte wie normal (kann an der Steroidtherapie liegen). - Herz: Gewicht 550g (normal wäre etwa 320g), 500g wäre das kritische Herzgewicht (Perfusion vom Myokard eingeschränkt, Kapillarversorgung reicht nicht mehr aus für die hypertrophierten Muskelfasern); Wandstärke rechter Ventrikel: 5mm (normal 2-3mm). Linker Ventrikel Wandstärke 16mm (normal 12mm). Man sieht einen „romanischen“ Bogen, keinen gotischen Spitzbogen im linken Ventrikel, also auch Hypertrophie und Dilatation. Keine Narben, also kein Z.n. Herzinfarkt, auch keine frischen Herde. Mitralklappe unauffällig, stark prominente Trabekelmuskulatur. Myokard histologisch im Großen und Ganzen unauffällig. - Leber: Man sieht makroskopisch ein Muster aus gelb und rot = „Muskatnussleber“ = schwere subakute Blutstauung als Zeichen der Rechtsherzinsuffizinz. Das Parenchym ist an vielen Stellen nekrotisch und von Erythrozyten durchsetzt. - Milz: 90g Gewicht, Reduktion der weißen Pulpa. Zusammenfassung: - langjährige COPD als Grunderkrankung - Exazerbation der Grunderkrankung möglicherweise durch eine akute Infektion - Patientin verstarb an einer subakuten Rechtsherzinsuffizienz, entstanden auf dem Boden der langjährigen COPD Mikroskopieren Präparat 239: Lunge, „normale“ Histomorphologie und plattenepitheliale Metaplasie Eigentlich sieht man nur das, was schon in der Überschrift steht. Das Präparat gibt es in meiner Sammlung auch gar nicht, deshalb hab ich nur die Powerpoint-Folien zum Anguggen. Präparat 285: Lunge, Plattenepithelkarzinom G2 Altersgipfel für Bronchialkarzinome 7. Lebensjahrzehnt. Man verfolgt im Präparat das Zylinderepithel und sieht, dass es an einer Stelle plötzlich sehr breit wird. Geht man in den Tumor rein, sieht man eine ausgeprägt Kernpleiomorphie. Präparat 285 war zwar schon die akute Virusmyokarditis, ... naja. Jedenfalls... wenn man ein bisschen rumsucht, sieht man auch Hornschichten, ist also ein verhornendes PEK. Präparat 286: Lunge, Adenokarzinom G2 286 ist zwar gleichzeitig auch Koronarsklerose, aber die scheinen hier gerne doppelt zu belegen, dass man es in der Klausur nicht zu einfach hat oder warum auch immer. In niedriger Vergrößerung sucht man erstmal den Tumor und sieht, dass er sehr unruhig aussieht, also unregelmäßig und mit verschiedenen Strukturen. Geht man näher ran, erkennt man eine Anisochromasie, das Chromatin ist also in den Kernen unregelmäßig verteilt, es gibt dunkle und helle Areale. Man erkennt verteilt über den Tumor ein paar drüsige Strukturen, das Karzinom ist also noch nicht sehr entdifferenziert. Wenn man es mit einem fortgeschritteneren Adenokarzinom zu tun hat, wächst es oft so massiv wie ein PEK und man muss Immunhistochemie zur Klassifizierung einsetzen. Präparat 154: Lunge, kleinzelliges (neuroendokrines) Karzinom Kleinzellige Karzinome sind hochmaligne Tumoren aus anaplastischen zytoplasmaarmen Zellen mit Resten einer endokrinen Differenzierung. Sie haben ein hohes Wachstumstempo bei einer frühen lymphogenen und hämatogenen Streuung. Das Wachstumsmuster ist typisch: Man sieht Pseudorosetten, Tubuli, Rippen als Anklang an die endokrine Herkunft. Die Zellgrenzen sind schlecht erkennbar, die Zellkerne sind dicht, homogen und heterochromatisch. Es gibt zwei Versionen von 154er-Präparaten: In einer ist der Tumor fast über das ganze Präparat, in der anderen sieht man zwei kleine Stellen mit Tumor. Niere Epitheliale Nierentumoren des Erwachsenen: Nierenparenchym: - Nierenzell-Adenome - Nierenzell-Karzinom Nierenbeckenzeug ... Pathogenese: - Ausgang vom Epithel der Tubuli oder Sammelrohre - Adenom-Karzinom-Sequenz, die Abgrenzung in der Zytologie und vom Wachstumsmuster Adenom-Karzinom ist hier sehr schwer Nomenklatur Adenom vs Karzinom: - G1 <0,5cm = Nierenzell-Adenom - G1 0,5-2cm = Nierenzell-Karzinom mit geringer maligner Potenz - G1 >2cm = Nierenzell-Karzinom - G2/3 jede Größe = Nierenzell-Karzinom - klarzellig-solid jede Größe = Nierenzell-Karzinom Auch die sogenannten Nierenzell-Adenome können schon metastasieren. Nierenzell-Adenome: - papilläres / tubulopapilläres Adenom Papillen sind in diesem Fall schmale, bindegewebige Septen, die von Tumorzellen überzogen werden. Insgesamt kann das Adenom auch rund sein, Hauptsache im Inneren finden sich lauter kleine Papillen. Nicht verwechseln mit dem papillären Schilddrüsenkarzinom... dort finden sich „Milchglaskerne“, die weiß imponieren. Oft auch mit Siderose (Eisenablagerung) und Schaumzellen in den Septen. - onkozytäres Adenom = Onkozytom - makroskopisch kleine, meistens direkt unter der Kapsel liegende Erhebungen, die eher grau imponieren - oft Zufallsbefunde in einem Nierenpräparat Nierenzellkarzinom: Synonym (sollte nicht verwendet werden): - Grawitz-Tumor - Hypernephroides Karzinom - Hypernephrom Kommt daher, dass er makroskopisch aussieht wie ein Nebennierenrinden-Tumor (gelbe Farbe, klarzellig), deshalb dachte man, dass die NNR „Keim“ des Tumors sei (andere Fehlbezeichnung: Lipom, wegen der gelben Farbe). Epidemiologie: - 6.-häufigster maligner Tumor bei Männern - 12.-häufigster maligner Tumor bei Frauen - 3% aller malignen Tumoren - Inzidenz 10:100.000 Einwohner - Männer : Frauen 2-3:1 - 80% aller NZK zwischen dem 49. und 69. Lebensjahr Ätiologie: - unbekannt - Risikofaktoren (aber nur minimal): - Cadmiumexposition - Arsenexposition - Nitroso- und Arylamidverbindungen - Zigarettenrauchen (40% der Fälle beim Mann) - Adipositas (Risiko 2-fach höher) - erworbene Nierenzysten bei Dialysepatienten Genetisch: - papilläres NZK: Trisomie 7, 17 - hereditäres papilläres NCC: multiple bilaterale papilläre NZK - klarzelliges NZK bei Hippel-Lindau-Syndrom - klarzelliges NZK bei 3p-Deletion Klinische Symptome: - Klassische Trias (60%): Hämaturie, Schmerzen, Flankenmasse - Systemische Symptome (40%): Gewichtsverlust (Kachexie), Fieber - Paraneoplastische Syndrome: Hyperkalziämie, Erythrocytose, Hypertonie, Gynäkomastie - CUP-Syndrom: (carcinoma of unknown primary) Primärtumor erst sekundär über Metastase entdeckt - BSG-Erhöhung - Leberfunktionsstörung Deswegen meist bei Primärdiagnose schon sehr groß (unspezifische, unauffällige Symptome). Nicht selten Zufallsdiagnose in einer Sonographie. Bricht ein NZK in die V. renalis ein, spricht man von einer Venangiosis carcinomatosa. Gewebeversprengungen (Choristie): Synonym: - Heterotopie - Ektopie - Dystopie Beispiele: - Glia - Ektopia cordis - Mamma (akzessorisch) - NN (Ni, Le, Samenstr.), Mg, Pa - Endometrium Besonders gefährlich in Lymphknoten. Histologische Typen des NZK: - klarzelliges Karzinom (73%) - papilläres (chromophiles) Karzinom (12%) - basophiler Typ (Subtyp 1) - eosinophiler Typ (Subtyp 2) - chromophobes Karzinom (5%) (haloartige perinukleäre Aufhellungen, sonst eher eosinophiles Plasma. Liegt an kolloidal gelösten Eisenpartikeln) - sarkomatoides (spindelzelliges) Karzinom - Sammelrohrkarzinom (Ductus-Bellini-Karzinom) (1%) - unklassifizierbares Karzinom Das Grading findet nach der Differenzierung statt; bei G1 sind die Kerne noch klein und wenig variabel, bei G2 sieht man häufiger Nucleoli in den Zellkernen, bei G3 sind die Kerne unregelmäßig groß und zeigen prominente Nucleoli. Typisch für Nierenzellkarzinome ist, dass sie sehr gut vaskularisiert sind und nicht selten Einblutungen haben. Bei papillären Karzinomen muss man aufpassen, dass man eingewanderte Makrophagen / Schaumzellen nicht für klarzellige Karzinomzellen hält, da sie auch recht hell sind. Ductus Bellini-Karzinom: Histogenese: ausgehend von den Sammelrohrepithelien, distale Tubulussegmente Häufigkeit: selten Morphologische Merkmale: - Lokalisation: nahe dem Nierenbecken / -mark - unscharfe Tumorgrenze - kettenartige Tumornester - variable Histologie (papillär / kleinzystisch / kompakt / kettenartig / tubulär / desmoplastisch) - reichliches Stroma, faserreich, wirkt narbig, derb Klinische Merkmale: - sehr schlechte Prognose Sarkomatoides / spindelzellartiges NZK: Per definitionem immer G3, also schlecht differenziert. Die Tumorzellen sehen halt länglich / spindelig aus, also eher mesenchymal, es ist aber noch ein Karzinom! Man kann sie mittels Immunhistochemie anfärben, sie produzieren Keratin, bei weiterer Entdifferenzierung zunehmend ersetzt durch Vimentin als Intermediärfilament-Komponente. Metastasierung: - Lokal: Kapseldurchbruch, Nierenbeckeneinbruch - lymphogene Metastasierung hämatogene Metastasierung - - - - - - - - Lunge 55% Lymphknoten 34% Leber 33% Knochen 32% kontralaterale Niere 11% Gehirn 6% Milz 5% Haut 3% Knochenmetastasen sind besonders unangenehm und schmerzhaft; sie können auch zu pathologischen Frakturen führen. Knochenmetastasen können auch viele Jahre nach erfolgreicher kompletter Resektion des Primärtumors noch auftreten. Je nach Lokalisation lassen sich diese Metastasen auch extrem schlecht sanieren, z.B. ins Os sakrum... Prognose des Nierenzellkarzinoms: Dabei gibt es zwei Komplexe, die entscheidend sind: - pathologisch-anatomische Faktoren (Ausbreitung, Metastasen, Grading, ...) - Stadium („staging“) - histologischer Tumortyp - Differenzierungsgrad = Grading (insbesondere Kernmorphologie) - Architektur / Wachstumsmuster - Angiosis carcinomatosa - biomolekulare Faktoren: - proliferative Aktivität (Ki67-MiB1) - DNA-Gehalt (Ploidie) - Tumorsuppressorgene (p53) - Wachstumsfaktoren (EGF-R, c-kit, bcl-2) - Tumorangiogenese Bei Stadium IV (Fernmetastasen) ist die 5-Jahres-Überlebensrate <5%, bei Stadium I (kleiner Tumor innerhalb der Niere) liegt sie bei etwa 80%. Die biomolekularen Faktoren sind zwar gut bestimmbar und hören sich toll an, sind aber bisher prognostisch / therapeutisch im Prinzip ohne Bedeutung. Differenzierung Zyste / NZK: Da sind die Urologen oft am Verzweifeln, denn Zysten sind verdammt häufig. Zysten entstehen entweder als Aufweitungen von Tubuli bei Abflussbehinderungen oder als genetisch determinierte Zysten und sind in der Regel völlig harmlos. Es gibt dummerweise auch Nierenzellkarzinome, die in Zysten entstehen (Klarzellen kleiden Zyste aus oder liegen in der Wandung), und darüber hinaus auch primär zystische Nierenzellkarzinome, die Zysten bilden. - primäres multilokuläres zystisches klarzelliges NZK (<1% der NZK) - pseudozystische Umwandlung eines NZK (Einblutung, Nekrose = Regression) Onkozytäres Adenom = Onkozytom: Makroskopie: - solitärer dunkelbrauner, fixiert hellbrauner Tumor, zentrale sternförmige Narbe, keine Nekrosen, keine Kapsel Mikroskopie: - solide-trabekulär tubulär wachsend, große Zellen mit grob-granulärem eosinophilem (mitochondrienreich) Zytoplasma, z.T. Kernpleomorphie, selten Mitosen - Immunhistochemie: Keratin +, Vimentin Dieser Tumor ist gutartig, deshalb ist es ganz nützlich, ihn richtig zu diagnostizieren, dann muss man dem Patient nämlich nicht die komplette Niere resezieren. Schwierig ist die Abgrenzung gegenüber dem papillären NZK, da das Zytoplasma ähnlich aussieht, besonders ähnlich wie das chromophobe NZK. Deshalb hier ne Tabelle: Vimentin Keratin CK7 Mitosen Polymorphie Papillen Zellrasse Karzinom Onkozytom +/+ + +/+ sehr selten + (+) + - (nie!) mehrere möglich uniform Metastasen in der Niere: Karzinommetastasen im Obduktionsgut bei 4,6 - 7,8% der Fälle. Primärtumor in: - Mamma 24,5% - Lunge 24,0% - Darm 10,5% - andere Niere 7,6% - Magen 6,6% Nicht-epitheliale Tumoren: Lymphome und myeloproliferate Erkrankungen gehen häufig mit einer Infiltration der Nieren (>2/3 der Fälle) einher. Bisweilen extreme Vergrößerung der Nieren. Mesenchymale Nierentumoren: Angiomyolipom = benigner Tumor - Makroskopie: - lobulierte Tumoren variabler Zusammensetzung, gelb = fettreich, graubraun = muskulär. Nekrosen, Blutungen - meist solitär, bis 20cm groß, wenn multipel häufig assoziiert mit tuberöser Sklerose - LK-, Leber- und Lungenmanifestation möglich - Mikroskopie: - dickwandige Blutgefäße, faszikulär angeordnete glatte Muskelfasern, die typischerweise manschettenförmig um diese dicken Blutgefäße liegen, reife Fettzellen. Pleomorphien und Mitosen können vorkommen - IH: HMB45 + Andere benigne mesenchymale Tumoren: - Leiomyom (zumeist in Kapsel = „Kapsulom“) - Lipom - renomedullärer Interstitialzelltumor = Markfibrom (von Prostaglandin sezernierenden Interstitialzellen ausgehend) - Hämangiom - juxtaglomerulärer Tumor Maligne mesenchymale Tumoren (selten): - Leiomyosarkom - Angiosarkom - Liposarkom (häufigster in der Niere) - Rhabdomyosarkom - Fibrosarkom Tumorartige Veränderungen: Inflammatorischer myofibroblastischer Pseudotumor: - tumorartige Konfiguration bestehend aus Myofibroblasten (IH: SM-Aktin +), Plasmazellen, Lymphozyten und kollagenen Fasern Xanthogranulomatöse Pyelonephritis (XGP): - tumorartiger inflammatorischer Prozess bestehend aus Xanthomzellen = großen Histiozyten mit schaumigem Zytoplasma, Riesenzellen, Nekrosen, Abszessen, Plasmazellen und Lymphozyten Harnblase Urotheliale Hyperplasien: Vor allem bei chronischer Urozystitis (bei Frauen häufiger) kommt es zu Hyperplasien, bei denen der grundsätzliche Aufbau der Harnblasenwand aber noch erhalten ist (intakte Umbrella-Zellschicht). Bei einer akuten Entzündung sieht man ein Infiltrat mit vor allem neutrophilen Granulozyten, bei einer chronischen Entzündung Lymphozyten und Plasmazellen, bei Granulation noch Kapillarproliferate, bei einer fibrosierenden Entzündung noch Fibroblasten / Fibrozyten. Urothelmetaplasien: - Plattenepithelmetaplasie - intestinale Metaplasie (Zylinderepithel mit Becherzellen wie im Colon und Rektum) - nephrogene Metaplasie (nephrogenes Adenom, nur in den ableitenden Harnwegen vorkommend) Eine solche Metaplasie, besonders die Plattenepithelmetaplasie, kann dem Pathologen wie eine Neoplasie vorkommen. Man erkennt, dass es keine Haut ist, weil keine Hautanhangsgebilde vorhanden sind (Talgdrüsen, Haarfollikel usw.). Die Plattenepithelhyperplasie ist besonders bei älteren Frauen im Trigonumbereich häufig. Die intestinale Metaplasie kann so weit gehen, dass die von Brunn‘schen Zellnester mit einbezogen und komplett mit intestinalem Epithel ausgekleidet werden, so dass sie wie Krypten im Dickdarm aussehen (Zystitis glandularis). Benigne epitheliale Tumoren: - urotheliales Papillom - plattenepitheliales Papillom - villöses Adenom (aus Drüsenepithel; bei Drüsenepithel ist papillär = villös) Beim urothelialen Papillom sieht man filigrane Strukturen, die in die Harnblase hineinragen; der Aufbau des Urothels ist normal. Beim plattenepithelialen Papillom sieht man ab und zu Koilozyten bei einer HPV-Infektion. Intraepitheliale Neoplasie des Urothels: Der Übergang von Normal über Dysplasie zu CiS ist wie immer fließend. Der Begriff Dysplasie wird zunehmend ersetzt durch „intraepitheliale Neoplasie“. Die Zellen werden zunehmend rund statt vertikal länglich, die Umbrellazellschicht geht verloren, man sieht zunehmend Mitosen, die Kern-Plasma-Relation wird höher. Grundregeln zur Diagnostik: - flache, oft multifokale Läsionen - immer gering differenziert (G3) Die Diagnose „CiS des Urothels“ ist, bei Bestätigung, schon ausreichend für die Indikation der Zystektomie. Maligne epitheliale Tumoren: - Urotheliales Karzinom: - papilläres urotheliales Karzinom (wächst vorwiegend in die Harnblase hinein) - solides urotheliales Karzinom (geschlossene Tumorzellaggregate ohne Drüsen, wachsen invasiver) - Varianten urothelialer Karzinome Epidemiologie: Männer 5. Stelle / Frauen 11. Stelle Zum Zeitpunkt der Diagnosestellung: - 70% „oberflächlich“ - 30% muskelinvasiv - 15% metastasiert Jegliche Form der Hämaturie muss abgeklärt werden durch eine Zystoskopie. Varianten des Urothelkarzinoms: - Urothelkarzionom mit squamöser Differenzierung (Metaplasie), auch wenn 95% wie ein Plattenepithelkarzinom - Urothelkarzinom mit glandulärer Differenzierung (Metaplasie) - Sarkomatoides (spindelzelliges) Karzinom Sonstige (nicht urotheliale) Karzinome: - reines Plattenepithelkarzinom - reines Adenokarzinom - Urachuskarzinom - Klarzellkarzinom - kleinzelliges Karzinom - undifferenziertes Karzinom Ätiologie: - Zigarettenrauchen ist für ca. 50% der Harnblasenkarzinome ursächlich - Pfeife- und Zigarettenrauchen nur geringe Risikoerhöhung - 4-Aminobiphenyl - ... Histologische Klassifikation von Tumoren der Harnblase: - mesenchymale Tumoren: - Rhabdomyosarkom (häufig im Kleinkindesalter) - Leiomyosarkom (eher bei Älteren) - Liposarkom (auch eher bei Älteren) sehr selten Sonstige seltene Tumoren: - Paragangliom - Lymphome - Karzinosarkom - malignes Melanom Und metastatische Tumoren und sekundäre Tumorinfiltrate. Einteilung der Lymphome: Diagnose Low-grade Lymphome des Malt Diffuses großzelliges B-Zell-Lymphom CD 45 + - CD 20 + + CD 5 - CD 10 + Kappa/Lambda + + Tumorartige Läsionen der Harnblase: - polypoide Zystitis (massive Entzündung mit ödematöser Infiltration des Urothels, Papillen) - follikuläre Zystitis (Hyperplasie des MALT-Gewebes) - Malakoplakie - Amyloidose - Myofibroblastäre Proliferationen - myofibroblastärer Tumor (echter Tumor) - postoperativer Spindelzellknoten - Müllerianose (Endometriose) Malakoplakie, Diagnosekriterien: - oft multiple gelb-braune polypöse weiche Läsionen, <2cm Durchmesser - monotone zytoplasmareiche Infiltrate mit PAS-positiven Makrophagen, deren Zytoplasma Michaelis-Gutmann-Körperchen enthält (verkalkte, nicht vollständig abgebaute Bakterienanteile, vor allem von Kolibakterien) - im Rahmen eines Schleimhautimmundefekts - Symptome: gelegentliche Hämaturie, Harndrang, Dysurie oder Pollakisurie, Flankenschmerzen Müllerianose der Harnblase: - meist Frauen zwischen 20-50 Jahren - oft intramuskuläre Lokalisation - - Durchmesser ca. 1-4 cm Drei Müller-Epithel-Typen: - endometrial = Endometriose - endozervikal = Endozervikose - endosalpingial = Endosalpingiose Lernziele: - epitheliale Abnormalitäten der Harnblase - benigne und maligne epitheliale Tumoren der Harnblase - sonstige Tumoren der Harnblase - tumorartige Veränderungen der Harnblase Prostata In der normalen Prostata sind 30-50 tubuloalveoläre Drüsen in ein Stroma aus Bindegewebe (Fibroblasten) und glatter Muskulatur eingebettet, in der Mitte der Prostata findet man natürlich Urothel um den Harnleiter. Das sekretorische Epithel ist zylindrisch, zur Basalmembran hin begrenzt von einer Basalzellschicht, die sehr flach ist und auch Lücken aufweist. Die Drüsenausführungsgänge werden mündungsnahe auch von Urothel ausgekleidet. Oft kommt es zu gutartigen Prostatahyperplasien, fast bei jedem Mann ab einem bestimmten Alter. Dabei kommt es zu Einfaltungen des Epithels in das Lumen. Weit über 90% der Neoplasien der Prostata sind Adenokarzinome, es gibt aber logischerweise auch primäre Urothelkarzinome der Prostata. Tumorartige Veränderungen der Prostata: Benigne Prostatahyperplasie (BPH), Synonym: Adenomyomatose, noduläre Hyperplasie Morphologie: - Hyperplastische Drüsen mit vergrößerten, z.T. zystisch aufgeweiteten Azini und papillären Epitheleinfältelungen mit Basalzellen und sekretorischen Zellen - Hyperplasie des fibromuskulären Stromas mit diffuser fibromuskulärer Stromahyperplasie und nodulärere fibröser / muskulärer / fibromuskulärer Stromahyperplasie Es können sowohl die zwei Seitenlappen als auch der Mittellappen hyperplasieren. Manchmal kommt es bei einer starken Hyperplasie zur sogenannten Balkenharnblase, wenn die Prostata stark in den Harnblasenboden drückt. Diagnostizieren lässt sich sowas über Ultraschall oder eine digitale rektale Untersuchung. Auch eine BPH ist knotig, aber nicht so hart und außen höckerig wie ein Prostatakarzinom. Durch die BPH kommt es zu einem erhöhten Widerstand im Harnleiter, wogegen die Harnblase dann ankämpfen muss (Hyperplasie), später kommt es zu objektiven Beschwerden wie Harnverhalt, Schwächung des Harnstrahls, Restharnbildung usw. Bedingt durch die Volumenzunahme in dem hyperplastischen Organ können Areale auftreten, die nicht mehr ideal durchblutet werden, es entstehen Infarktareale mit Einblutungen. Die Restharnbildung durch die Aufstauung kann auch zu einer Aufweitung der Uretheren (Megauretheren) bis hin zu einer Aufstauung des Nierenbeckens (Hydronephrose) führen. Komplikationen sind natürlich Entzündungen bis hin zur Pyelonephritis. Auch ohne Entzündung kann die Niere bei einem chronischen Aufstau atrophisch werden, die Niere fibrosiert und kann kaum noch eine Funktion erfüllen. Benigner epithelialer Tumor der Prostata: Zuerst einmal: Benigne Tumoren der Prostata sind absolute Raritäten. Papilläres Adenom: - Vorkommen: junge Männer - Lokalisation: prostatische Urethra - Morphologie: papillärer Tumor aus benignen papillären Prostatazellen Prostatakarzinom: Epidemiologie: - nach dem 70. Lebensjahr der häufigste bösartige Tumor beim Mann - häufiger bei Dunkel- als bei Hellhäutigen (bei weißer Bevölkerung des Westens 20-45 / 100.000, bei schwarzer Bevölkerung der USA 100 / 100.000) - Mortalität steigt linear mit dem Alter Kategorien des Prostatakarzinoms: - klinisches Karzinom: Diagnosestellung primär klinisch, Bestätigung durch histologische Untersuchung - okkultes Karzinom: Tumor manifestiert sich durch Metastasen - latentes Karzinom: entdeckt durch Autopsie - inzidentes Karzinom: zufällig entdeckt, z.B. im Resektat bei BPH (bei TUR = transurethraler Resektion) Die Diagnose lässt sich auch durch den PSA-Nachweis stellen (prostata-spezifisches Antigen), das allerdings auch bei anderen entzündlichen Prozessen (Prostatitis) erhöht sein kann, auch bei einer massiven BPH ist es deutlich erhöht. Das PSA sollte unter 2,5 ng/ml liegen, maximal 4 ng/ml. Das Prostatakarzinom ist im Tastbefund sehr derb und höckerig (Vergleich: etwa wie ein Fingerknöchel) und enthält makroskopisch graue Bereiche, das liegt an der desmoplastischen Stromareaktion und der vermehrten Bildung von kollagenem Bindegewebe v.a. um die versorgenden neugebildeten Blutgefäße. Karzinome entstehen vermehrt peripher in den Seitenlappen oder im Hinterlappen, nur 10% in der vorderen Kommissur. Vorläuferläsion des Prostatakarzinoms: Prostatische intraepitheliale Neoplasie (PIN) = Carcinoma in situ Morphologie: - proliferierende atypische Zellen, begrenzt auf Drüsen - erhaltene Basalzellschicht (CK 5/6 pos.) (← Wichtig!) - nukleäre Anaplasie (Größe, Form) - Makronukleolen (so große wie beim Prostatakarzinom weist kaum eine andere Neoplasie auf) Adenokarzinom der Prostata: = invasiver Tumor der sekretorischen Zellen (keine Basalzellen) Die allermeisten Prostatakarzinome sind natürlich Adenokarzinome, viel mehr Gewebe, das entarten kann, hats da ja nicht. Sie wachsen entweder tubulär, kribriform (siebartig), papillär oder solide. Morphologie: - nukleäre Anaplasie - Kerne oft groß und variabel - Nukleoli oft groß (Makronukleolen!), variabel, doppelt - Cave: manche Karzinome ohne Anaplasie - Drüsenarchitektur - variierende Größe und Form mit gewinkelter Kontur: tubulär, kribriform, papillär, fusioniert, wodurch Löcher entstehen, das Ganze sieht dann etwa wie ein Schweizer Käse aus. - variable Dichte - pathologisches Sekret in Lumina (eher fleckig, eher basophil = dunkel) - Stromainvasion, Tumorausbreitung - unregelmäßige Verteilung (aufgehobene lobuläre Architektur) - perineurales Wachstum, für CA beweisend wenn zirkulär um den Nerv - extraprostatische Invasion (Fettbindegewebe ohne glatte Muskulatur) - Infiltration der Samenblasen - Subtypen nach Wachstumsmuster: - tubulär (acinär): = G1 wenn uniform (Gleason 1 oder 2) - papillär: = G2 (Gleason 2 oder 3) - kribriform: große, tubuläre Drüsen mit Brückenbildungen der Tumorzellen = G2 (Gleason 3) - fusionierte Drüsen: dichtgepackte Gruppen von Tubuli ohne trennendes Stroma = G3 (Gleason 4) - solide / trabekulär: = G3 (Gleason 5) Varianten: - muzinöses Adenokarzinom, enthält reichlich extrazellulären Schleim - spindelzelliges Karzinom, enthält Spindelzellen, ähnelt einem Sarkom, fokal Areale mit Drüsen - Prostatakarzinom mit ektoper plazentarer Glykoproteinbildung, enthält bizarre, z.T. mehrkernige Zellen (wird nicht gefragt) Grading: - Grading nach Gleason, basierend auf Wachstumsmuster - 1 bis 5, differenzierter als andere Klassifizierungen, Drüsenmuster auch relevant für Prognose bei Prostata-Ca - setzt sich zusammen aus dem Punktwert des am häufigsten vorkommenden Wachstumsmusters plus des am zweithäufigsten vorkommenden Wachstumsmusters, also höchster Wert mit „nur solide“ ist 5+5=10. Niedrigster Wert ist 2. - Grading nach WHO, basierend auf nukleärer Anaplasie und glandulärer Differenzierung - G1 = hoch (gut) differenziert (Gleason 1-4) - G2 = mäßig differenziert (Gleason 5+6) - G3 = gering (schlecht) differenziert (ab Gleason 7) Immunhistochemie des Prostatakarzinoms: - PSA, SPP (prostataspezifisches Antigen, saure Prostataphosphatase): positiv (normal: positiv), damit kann man differenzieren, ob es überhaupt ein Prostatakarzinom oder die Metastase eines anderen Adenokarzinoms ist. Auch Metastasen z.B. in Knochen kann man damit untersuchen und beweisen, dass ein Prostata-Ca vorliegt. - CK 5/6: negativ (normal: positiv in den Basalzellen) Sonstige Karzinome der Prostata: - Urothelzell-Karzinom (Transitionalzell-Karzinom) - Ausgang von prostatischer Urethra mit Einwachsen in Prostatagänge > Stromainvasion der Prostata - Ausgang Harnblase oder prostatische Urethra mit direkter Stromainvasion der Prostata - Plattenepithelkarzinom - reine Form sehr selten - eher plattenepitheliale Metaplasie (Differenzierung) eines Adenokarzinoms als Therapiefolge - Basalzellkarzinom - Karzinom der prostatischen Basalzellen - sehr selten, gering differenziert (anaplastische Zellen) - kleinzelliges Karzinom - kleinzelliges neuroendokrines Karzinom (identische Morphologie wie kleinzelliges Bronchialkarzinom) - undifferenziertes Karzinom - vollständig entdifferenziert, keine Eingliederung in eine Gruppe möglich Mesenchymale Tumoren der Prostata: - benigne: fraglich, ob echte Fibrome oder Leiomyome existieren > eher fibröse / leiomyomatöse Hyperplasien - maligne (Sarkome), sehr selten - Rhabdomyosarkom - Leiomyosarkom - Stromasarkom: spindelzelliges Sarkom ohne myogene Differenzierung Tumorartige Veränderungen der Prostata: Atrophie der Prostata: - Morphologie: - kleine Drüsen, lobulär gegliedert um zentralen Gang - kleine Zytoplasmakerne oder so oder Zellen oder so Sonderformen der Hyperplasie: - Basalzellhyperplasie (oft verwechselt mit einer Drüsenatrophie) - atypische Hyperplasie: nukleäre Veränderungen noch nicht ausreichend für prostatische intraepitheliale Hyperplasie - mikroglanduläre Hyperplasie (= atypische adenomatöse Hyperplasie AAH) - atypical small acinar proliferation (ASAP) Prostatitis: - akute (eitrige) Prostatitis - Infiltrate aus neutrophilen Granulozyten - chronische Prostatitis - Infiltrate aus Lymphozyten, Plasmazellen, Makrophagen - unspezifische granulomatöse Prostatitis - keine spezifische Ätiologie - Infiltrat aus Lymphozyten, Plasmazellen, Makrophagen, Epitheloidzellen, Riesenzellen um destruierten Gang / Acinus - spezifische granulomatöse Prostatitis - spezifische Ätiologie, meist säurefeste Stäbchen (Calmette-Guérin-Bazillus-Therapie) Verursachen auch eine PSA-Erhöhung. Hoden Aufbau: Die meisten Neoplasien und auch die mit dem höchsten malignen Potential entstehen in den Hodenkanälchen: Keimzelltumoren. Von außen nach innen findet man in den Kanälchen Fußzellen (Sertolizellen) und Spermatogonien auf der Basalmembran, dann Spermatozyten, Spermatiden und am Lumen reife Spermien. Zwischen den Kanälchen liegen Interstitialzellen des Rete testis, die eher flach sind; dort finden sich auch die Leydig-Zellen. Bedingt durch die unterschiedlichen Reifestadien wirkt das Zellbild in einem normalen Hoden etwas unruhig. Das Testosteron wird von den Leydig-Zellen gebildet. Leydig-Zell-Tumoren sind in aller Regel gutartig. Testikuläre intraepitheliale Neoplasie (TIN): = Vorstufe invasiver Tumoren - Aneuploidie - Seminoma in situ - embryonales Carcinoma in situ - 50% nach 5 Jahren → invasiv - kein β-HCG (human chorionic gonadotropine) oder AFP (Alpha-Feto-Protein) - PLAP-Färbung (placenta-like alcalic phosphatase) wird eingesetzt, um die atypischen Keimzellen anzufärben, was die Diagnostik extrem erleichtert. WHO-Klassifikation der Hodentumoren - Keimzelltumoren: A. Tumoren mit einheitlichem histologischem Bau 1.Seminom 2.spermatozytisches Seminom (S + STRZ; anapl. S) 3.embryonales Karzinom 4.Dottersacktumor (Yolk-Sac-Tumor) 5.Polyembryom 6.Choriokarzinom 7.Teratom a) reif b) unreif c) mit maligner Transformation B. Tumoren mit histologisch unterschiedlicher Bauart 1.embryonales Karzinom und Teratom (Teratokarzinom) 2.Choriokarzinom kombiniert mit anderen Keimzelltumoren 3.andere Kombinationstumoren Keimzelltumoren stellen 95% der Tumoren des Hodens, Sertolizelltumoren 2%, Leydigzelltumoren 3%. Von den Keimzelltumoren stellen Seminome 45%, bei denen nur ein Zelltyp vorkommt. Keimzellschichten bilden die embryonalen Karzinome (40%), das sehr unreif und sehr maligne ist. Folgt dabei eine Differenzierung in Richtung somatische Strukturen, resultieren Teratome, bei Differenzierung in richtung extraembryonal entstehen z.B. Dottersacktumoren oder Choriokarzinome. Seminom: Die Zellen liegen in geschlossenen, soliden, breiten Verbänden, eher rundliche Zellen mit hellem Zytoplasma, haben Ähnlichkeit mit Spermatogonien. Man sieht um die Gefäßwandungen viele kleine Zellen, die sich dort ansammeln: Lymphozyten. Typisch für das Seminom sind diese Ansammlungen von Lymphozyten und Plasmazellen im Stroma (lymphozytäre Stromareaktion, im Gegensatz zur häufigeren desmoplastischen Stromareaktion wie bei Mammakarzinom, Prostatakarzinom etc.). Metastasen setzen sich vor allem in die Lungen ab, typischerweise aber vorher noch eine Metastasierung in die regionären Lymphknoten (parailiakal, paraaortal). Embryonales Karzinom: Manche der Tumorzellen weisen hyaline Globuli auf. Das Wachstumsmuster ist variabel, meistens dichtgepackt solide, manchmal auch tubulär. Der Tumor ist sehr aggressiv, man sieht viele Mitosen. Große Tumorzellen, starke Pleomorphie, Nekrosen. Dottersacktumor: Enthält zystische Strukturen, wirkt makroskopisch „eklig-glibberig“. Man findet Schiller-Duval-Körperchen: Außen Tumorzellen, dann lockeres, helles Stroma, in der Mitte ein Blutgefäß. Dottersacktumoren exprimieren AFP in recht hohen Mengen. Polyembryom: Kann man als Variante des Dottersacktumors auffassen. Es bilden sich Embryoidkörperchen, die wie Keimanlagen aussehen (Keimblattschichtungen) Choriokarzinom: Vermutlich die höchst malignen Tumoren, die beim Menschen vorkommen. Makroskopisch meist schon ausgedehnte Einblutungen und Nekrosen. Sie enthalten mehrkernige Riesenzellen (wie Synzytiotrophoblasten der Plazentazotten) und kleinere, rundliche Zellen (als Pendant zum Zytotrophoblast). Man sieht plumpe, zottenartige Formationen mit Riesenzellen außen und kleineren, dichtliegenden Tumorzellen innen. Immer gut vaskularisiert. Typischerweise eine Expression von β-HCG. Die Serumwerte sind dann exorbitant hoch. Teratom: Schwer zu verstehender Tumor. Der Name kommt aus dem griechischen und bedeutet „Wundergeschwulst“. Es sind Keimzelltumoren, die eine somatische Liniendifferenzierung zeigen. In einem reifen Teratom findet man differenzierte, reife Gewebskomponenten wie in einem adulten Organismus. Es kann eigentlich alles vorkommen. Per definitionem muss ein reifes Teratom Vorkommen von zwei verschiedenen Keimblattepithelien haben. Beim unreifen Teratom hat man Komponenten, die nicht in einem adulten Organismus vorkommen, z.B. Gewebe, das fetal aussieht (z.B. Gliazellverbände auf fetaler Stufe). Das Gewebe sieht nicht maligne aus, wie bei unreifen, undifferenzierten Tumoren. Bei Teratomen mit maligner Transformation liegt reifes Gewebe vor, das entartet ist, wie z.B. reife Drüsen mit Adenokarzinom. Grundsätzlich ist nach der Pubertät jedes Teratom ein maligner Tumor, auch ein reifes Teratom. Vor der Pubertät verhalten sich reife Teratome gutartig. Das reife Teratom der Ovarien ist ein sehr häufiger Tumor und verhält sich gutartig. Mikroskopieren Präparat 155: Nierenzellkarzinom Dass es Niere ist, erkennt man schnell an den Glomerula. Die häufigsten Tumoren der Niere (maligne wie benigne) leiten sich von epithelialen Strukturen ab, und das sind in der Niere nunmal Tubulusepithelien. Maligne + epithelial = Karzinom. Hier dominieren Zellen mit kaum angefärbtem Zytoplasma, also ein klarzelliges Nierenkarzinom. Man findet aber auch Zellen mit einem rötlich angefärbten Zytoplasma, was als eosinophile Variante bezeichnet wird; diese sind recht scharf von den klarzelligen Anteilen getrennt. Die Tumoren exprimieren Vimentin (typisch für mesenchymale Tumoren) und Keratin (für epitheliale Tumoren typisch). Diese Koexpression machen nur ganz wenige Karzinome, darunter eben Nierenkarzinome. Präparat 155 kommt sehr wahrscheinlich in der Prüfung dran, da es relativ leicht ist. Präparat 241: Peniskarzinom An einer Stelle geht das normale geringgradig verhornende Plattenepithel deutlich in ein invasiv wachsendes, verhornendes Karzinomgewebe über. Verhornung ist hier ein Zeichen für noch gute Differenzierung, da das Ausgangsgewebe ja auch Keratin bildet. An der „Invasionsfront“ des Karzinom sind die entarteten Zellen noch in einzelnen Strängen angeordnet, dort kann man gut beobachten, wie sie das normale Gewebe infiltrieren. Auch dieses Präparat kommt wahrscheinlich in der Prüfung dran. Präparat 245: Prostatahyperplasie Hier erzählt PD Dr. Wehrmann zuerst noch einmal allgemein etwas über die Prostata und Prostatakarzinome, weil am Mittwoch so viele gefehlt haben... Die häufigste Erkrankung bei Männern über 70 Jahren ist die benigne Prostatahyperplasie, die zur Ausbildung eines prominenten Mittellappens führt, der dann in die Harnblase hineinragt; die ableitenden Harnwege werden zusammengedrückt und der Harnfluss behindert. Histologisch sieht man knotige Veränderungen und aufgeweitete Drüsen, die Epitheleinfältelungen (papilläre Einfältelungen) aufweisen. Man findet auch Drüsen mit abgeflachten Epithelien, in denen sich retiniertes und eingedicktes Prostatasekret befindet. Die häufigsten Karzinome der Prostata (über 90%) sind Adenokarzinome; diese werden nach der Morphologie der Drüsen, also dem Wachstumsmuster unterteilt. Es gibt 5 Typen: - tubulär (runde Drüsen, klein/groß) - kribriform (siebförmig, Zellbrücken durch die Drüsen, sieht aus wie Schweizer Käse) - papillär (bindegewebige Grundstöcke, auf denen die Tumorzellen liegen, fingerartig) - fusioniert (so dicht, dass dazwischen kein Bindegewebe mehr liegt, reine Adenokarzinomzellen mit Drüsenbildung) - solide (es werden gar keine Drüsen mehr gebildet, die Zellen liegen nur noch in Strängen oder Haufen) Eine gute Prognose haben tubuläre, eine schlechte haben fusionierte und solide Tumoren. Adenokarzinome der Prostata haben typischerweise ganz große Nukleolen (Makronukleolen, könnte in einer Prüfungsfrage enthalten sein). Präparat 243: Prostatakarzinom Mit kribriformen und fusionierten Arealen, sollte man erkennen, auch wenn das Präparat schwach gefärbt ist. Man findet fast nie nur eine Art von Wachstum, deshalb berechnet man den Gleason-Score (dieser legt die 5 Wachstumsmuster zu Grunde) durch Addition der Punktezahlen der beiden häufigsten Wachstumsmuster. 10 wäre das am schlechtesten differenzierte, 2 das am besten differenzierte Karzinom. Präparat 194: Seminom Die häufigsten Tumoren im Hoden sind maligne Keimzelltumoren. Da gibt es wieder verschiedene Subtypen: - Seminom (häufigster maligner Keimzelltumor) - embryonales Karzinom - Dottersacktumor - Polyembryom (sehr selten) - Choriokarzinom (aggressivste Variante) - Teratom (= „Wundergeschwulst“) a) reif, enthält z.B. Knorpel, Knochen, Zähne, Dickdarmschleimhaut, normale Haut mit Haaren usw. b) unreif, enthält unreifes Gewebe, wie es so in einem normalen Organismus nicht zu finden ist c) mit maligner Transformation Für das Seminom sind die vielen kleinen Zellen in den unterteilenden Bindegewebssepten typisch, wobei es sich um Lymphozyten handelt (man geht von einer lymphozytären Reaktion aus) Präparat 238: reifes Teratom des Hodens Man findet Talgdrüsen und reifes Schilddrüsengewebe, dazu Plattenepithel, das in mehrreihiges Flimmerepithel übergeht und außerdem noch mukoalveoläre Drüsen, alles im Hoden... Hämatopathologie Leukämien sind die häufigsten Krebserkrankungen bei Kindern, deshalb ebenfalls sehr wichtig (hier kommt die typische Kuchenstatistik, die bei Beginn jeder Vorlesung zeigt, wie wichtig das Thema ist). In den lymphatischen Organen findet die Ontogenese = Entwicklung und Reifung lymphatischer Zellen statt. Lymphatisches Gewebe findet sich an vielen Stellen. Primäre lymphatische Organe: - Knochenmark - Thymus Sekundäre: - Lymphknoten - Milz - MALTs (mukosa-assoziiertes, lymphatisches Gewebe) im Darm, lymphatisches Gewebe im Rachenring usw. In der 10. Schwangerschaftswoche bilden sich schon mesenchymale Knötchen, in denen Myelopoiese stattfindet, ab der 14. bilden sich T-Zell-Areale. In der 18. SSW kann man schon follikuläre dendritische Zellen nachweisen. Organisiertes lymphoides Gewebe ist für adaptive Immunantworten verantwortlich: 1. MALT - Kontrolle der mukosalen Grenzen 2. Lymphknoten - Kontrolle der Haut und intestinaler Flüssigkeiten 3. Milz - Kontrolle des Blutes. Die Milz wird am Mittwoch besprochen, heute Lymphknoten. Lymphknoten: Am Pol ziehen Blutgefäße raus und rein, der LK wird von einer Kapsel umgeben, durch die Vas afferentia (zuführende Lymphbahnen) ziehen. Unter der Kapsel findet sich der Randsinus, darunter die Rinde, in der sich Lymphfollikel befinden, und zwar Primär- und Sekundärfollikel mit B-Lymphozyten. Unscharf von der Rinde abgegrenzt findet sich der Paracortex, in dem sich T-Lymphozyten aufhalten, darunter noch ist das Mark, von Lymphsinus durchsetzt, die von Retikulumzellen begrenzt werden. Antikörperbildung: Reife B-Zellen mit Antigenen gelangen in einen Primärfollikel, wo sie eine Zentroblasten-Proliferation bewirken, der Follikel wird zum Sekundärfollikel. Aus diesen differenzieren sich Zentrozyten, die zu Plasmazellen und Memory-B-Zellen differenzieren. Die Lymphozyten, die apoptotisch werden, werden ständig von „Sternhimmel“-Makrophagen phagozytiert, die so wegen ihres hellen Zytoplasmas heißen. In den Follikeln finden sich ständig viele Mitosen, vor allem von Zentroblasten. Die Zentrozyten fallen meistens der Apoptose anheim, was übrig bleibt, sollte für körpereigene Zellen nicht mehr gefährlich sein und differenziert dann weiter zu Plasmazellen und Memoryzellen. Eine follikuläre lymphatische Hyperplasie spricht für eine rheumatische Erkrankung wie etwa rheumatoide Arthritis. Bei der angiofollikulären Lymphadenopathie ist das Keimzentrum fast atroph, während sich fast zwiebelschalenartig darum herum eine Hyperplasie mit vielen Mitosen bildet, die Lymphknoten schwellen (Castleman-Tumor, geht von Plasmazellen aus). Diese Erkrankung ist gutartig, selten und selbstlimitierend, aber man sollte es mal gehört haben. Sinushyperplasie / Sinuskatarrh: Ansammlung von Makrophagen in den Sinus, unspezifische Reaktion mit Makrophagenvermehrung im Abflussgebiet entzündlicher und neoplastischer Prozesse. Sinusinfiltrate: Die Lymphknoten lassen kaum noch eine Kapsel erkennen und sind sehr aufgelockert. In den Lymphknotensinus finden sich reichlich breit zytoplasmatische Histiozyten, die in der PAS-Reaktion granuläres PAS-positives Material enthalten (PAS färbt Glykogen und Schleim an, damit kriegt man viele Bakterien durch das glykogenreiche Zytoplasma). Wenn man zusätzlich noch eine Zottenatrophie mit Bakterieninfiltraten findet, ist die zu Grunde liegende Krankheit sehr wahrscheinlich ein Morbus Whipple. Sinusinfiltrate können auch auf ein großzellig-anaplastisches Lymphom zurückgehen oder ein paar andere Erkrankungen wie Sinushistiozytose. Granulomatöse Entzündung: In den Granulomen müssen sich Riesenzellen oder Epithelzellen (Epitheloidzellgranulom) finden. Zu Grunde liegende Erkrankungen können M. Crohn, Sarkoidose (M. Boeck), Tuberkulose oder Wegener-Granulomatose sein. Die Sarkoidose etwa befällt typischerweise bei jungen Frauen (20-30 Jahre alt) die intrathorakalen Lymphknoten, häufig in der Lunge. Behandelt man sie nicht frühzeitig, kommt es zur irreversiblen Lungenfibrose. Kleinherdige Epitheloidzellreaktion: Kann zusammen mit einer sog. „unreifen Sinushistiozytose“ (wobei es sich in Wirklichkeit um monozytäre B-Zellen handelt) auftreten, das dahinterliegende Krankheitsbild ist Toxoplasmose, die sog. Piringer-Adenitis. Entzündungen mit Nekrosen: Käsige Nekrosen treten bei der Tuberkulose auf. Wenn retikulozytär abszedierende Nekrosen mit vielen Plasmazellen auftreten, handelt es sich wahrscheinlich um die „Katzenkratzkrankheit“, nicht allzu selten. Systematik der malignen Lymphome: Bei den neoplastischen Erkrankungen des hämatopoetischen und lymphatischen Systems werden vier Entitäten unterschieden: - myeloische Neoplasien - lymphatische Neoplasien - Mastzellerkrankungen - histiozytäre Neoplasien Die Grundentitäten der malignen Lymphome sind: Non-Hodgkin-Lymphome (ca. 85%) Hodgkin-Lymphome (ca. 15%) B-Zell- und T-Zell-Lymphome werden unterteilt in: - Vorläufer-Neoplasien (B/T), zu denen lymphoblastische Neoplasien, lymphoblastische Leukämien / Lymphome, pro B-ALL, cALL, pre-BALL, pro-TALL, TALL und lymphoblastische Lymphome gehören - reife (periphere) Neoplasien (B- / T- / NK-Zellreihe) werden gemäß der klinischen Manifestationsform unterteilt in: - vorwiegend disseminiert / leukämisch - primär extranodal (intestinal, kutan) - vorwiegend nodal umfasst etwa 30 Entitäten Diagnostik von Lymphomen: - Morphologie: Wachstumsmuster, Befallsmuster, Hodgkin- vs Non-Hodgkin-Lymphom, Subklassifizierung - Immunhistochemie: B-Zell- vs T-Zell-Lymphome - molekulare Zusatzuntersuchungen - klinisches Erscheinungsbild Ein follikuläres Lymphom kann man von einer follikulären Hyperplasie gut unterscheiden, wenn man sich die Follikel genau anschaut. Beim Lymphom findet man keine scharfe Follikelgrenze mehr, keine Sternhimmelmakrophagen und keine dunkle Hälfte mit Zentroblasten und eine helle Hälfte mit Zentrozyten. Ein solches Lymphom entsteht, indem die B-Zellen, die nicht hochaffin reagieren, keine Apoptose mehr machen und gefressen werden können. Eine maligne Aberration im Genom kann dazu führen, dass BCL-2 überexprimiert wird, ein Anti-Apoptoseprotein, so dass keine Apoptosen mehr stattfinden, wodurch der Tumor zwar langsam, aber stetig wächst. Das follikuläre Lymphom ist mit 22% aller NHL das häufigste indolente Lymphom. Plasmozytom: Häufigster Knochentumor des Menschen, geht vom Knochenmark aus. Dieser Tumor tritt meist zwischen 60-70 Jahren auf und ist nicht allzu schlimm, muss auch meist nicht therapiert werden; die Menschen sterben daran auch normalerweise nicht. Er kann zwar zu Wirbelkörperbrüchen und sowas führen, den Betroffenen geht es auch schlecht, aber man stirbt nicht daran wie an den meisten anderen Lymphomen. Zum Nachweis kann man eine Leichtketten-Restriktionsanalyse auf Ebene der Proteinexpression machen (leichte Ketten der Immunglobuline), dann sieht man, dass der Tumor monoklonal ist, also nur eine Sorte der leichten Ketten produziert. Lymphozytisches Infiltrat im Magen: Im Magen kommt physiologischerweise kein lymphozytäres Gewebe vor; trifft man es doch an und wächst es auch noch in die Drüsen ein, handelt es sich um ein MALT-Lymphom. Diese Tumoren kann man mit einer Antibiose (!) behandeln, da sie im Magen meist durch Helicobacter pylori hervorgerufen werden. Das Lymphom geht dann tatsächlich zurück. Die T-Zellen im Infiltrat sind spezifisch auf H. pylori, die dann die B-Zell-Proliferation induzieren. Häufigkeit der nodalen und extranodalen Lymphome: 60% der NHL sind nodal, 40% sind extranodal. ... Mantel-Zell-Lymphom: Gehört zu den „bösen“, gefährlichen Lymphomen, obwohl es nicht sehr schnell proliferiert und kleinzellig ist. Es führt zur lymphomatösen Polypose, das auf den Kliniker wie ein Karzinom wirkt. Man findet neoplastische Zellen um ein reaktives Keimzentrum herum. In der Immunhistochemie kann man Cyclin D1 nachweisen, das dort überexprimiert wird (ein nukleäres Enzym). Es bewirkt, dass diese Zellen sich nicht mehr in die Ruhephase begeben können, so dass der Tumor ununterbrochen proliferiert, ohne dass er deswegen großzellig sein müsste. Richter-Syndrom / Richter-Transformation: Dabei kann es bei einer niedrig malignen Grunderkrankung, etwa einer B-Zell chronisch lymphatischen Leukämie niedriger Malignität, zu einer Transformation zu einer hoch malignen, stark invasiv wachsenden Zellart kommen, die dann großzellig, mit großen Nukleolen und nekrotisierend wächst. Nur die Hälfte solcher Fälle entsteht aus der vorbestehenden BCLL, bei der anderen Hälfte handelt es sich um ein denovo high-grade Lymphom (Zweitneoplasie). Eine BCLL prädisponiert nämlich auch zu Zweitneoplasien, das Risiko auf irgendeine andere Krebserkrankung ist bei solchen Patienten etwa um das Zehnfache erhöht. Die Hälfte der BCLLs geht aus unmutierten B-Zellen, die andere aus mutierten B-Zellen hervor. Damit ist gemeint, dass es sich entweder um naive B-Zellen handelt, die noch nicht im Keimzentrum war und ihr Genom mutiert hat, um viele verschiedene Antikörperketten hervorzubringen, oder um quasi-reife B-Zellen, die schon im Keimzentrum waren. Die „reife“ BCLL hat eine wesentlich bessere Prognose, das mediane Überleben liegt bei 7 Jahren, die unreife wächst aggressiver und hat ein medianes Überleben von nur 3 Jahren. Burkitt-Tumor: Das Burkitt-Lymphom tritt meist endemisch in Afrika auf, bei Kindern und Jugendlichen, und beginnt im Gesicht oder am Hals. Dieser Tumor wächst über das Wochenende ins Gehirn rein und der Patient ist tot, sehr gefährlich. Der endemische Burkitt-Tumor ist EBV-assoziiert (Epstein-Barr-Virus), er kann allerdings auch bei uns auftreten ist dort nicht EBV-assoziiert. Lässt sich sehr gut behandeln, obwohl er so schnell wächst. Man muss nur rechtzeitig mit der Therapie anfangen, bevor es zu spät ist. Molekulargenetik der Lymphome: Beim follikulären Lymphom liegt eine Genaberration auf Chromosom 14 vor, bzw. ein crossing over von Chromosom 14 und 18. In dem betroffenen Teilstück auf 14 werden die Immunglobuline, besonders die schweren Ketten, repräsentiert. Es gibt konstante Regionen, Verbindungsregionen und variable Regionen. In der „joining region“ liegt ein Enhancer, ein Verstärker, der die Expression der nachgeschalteten Gene erst bewerkstelligt, in diesem Fall eine extreme Überexpression der nachgeschalteten Gene für die B-Zell-Ontogenese einleitet. Im Chromosom 18 liegt das Gen für das Antiapoptose-Protein BCL-2. Was jetzt kommt, ist klar: Durch ein crossing over von Chromosom 14 und 18 kommt das BCL-2 in direkte Nachbarschaft des Enhancers, wodurch es überexprimiert wird und die Zellen unfähig zur Apoptose werden > Tumorzellen. Deshalb kann man durch eine positive Reaktion auf BCL-2 in der Immunhistologie follikuläre Lymphome erkennen. Zytopenien: Werden nach der WHO-Klassifikation von 2001 oder nach der FAP-Klassifikation eingeordnet. - RA refraktäre Anämie - RARS RA mit Ringsideroblasten - RAEB RA mit Blastenexzess - ... Eisenmangelanämie: Pathogenese: - Fe-Mangel - Hb-Bildung reduziert - weniger Hb pro Erythrozyt - Gewebe-Hypoxämie - Epo erhöht - Thrombozytose - Therapie: Fe-Substitution Dabei sind die Erys hypochrom und mikrozytär. Perniziöse Anämie: Komplexes Krankheitsbild; wird auch megaloblastäre Anämie genannt, weil Megaloblasten vorliegen. Es handelt sich meist um eine Vitaminmangelanämie. Zytosen: 1. Stammzell-Erkrankungen neoplastisch: CML (chronisch myeloische Leukämie), überschwemmt das gesamte Knochenmark Chronisch idiopathische Myelofibrose: Dazu brauchen wir erstmal die normale Knochenmark-Zytologie, um das Krankheitsbild zu verstehen. 1.Megakaryozyt (lockeres Chromatin, weites Zytoplasma, riesige Zelle) 2.Erythrone (Inseln von erythropoetische Zellen zusammen mit Ammenzellen, die das Eisen abgeben) - basophile Proerythroblasten (riesige Zelle, großer Kern mit ausgeprägtem Nucleolus, dunkel basophiles Plasma) - basophile Erythroblasten - polychromatischer Erythroblast (kleine Zelle, großer runter Kern, bläuliches Zytoplasma) - orthochromatischer Erythroblast (sieht schon fast aus wie fertige Erys, hat aber noch einen Kern) - cup-shaped Erythrozyt 3.Granulopoese - Neutrophiler - Stabkerniger (stark eingekerbter Kern) - Metamyelozyt - Myelozyt - Promyelozyt: 4a Eosinophiler (zweifach gelappter Kern, auch wenn man das eosinophile Plasma selten erkennt) 4b Lymphozyt (Kern so groß wie die Zelle, fast kein Zytoplasma) 4c Plasmazelle (stark und unregelmäßig angefärbt, exzentrisch gelagerter Kern) Bild bei der chronisch idiopathischen Myelofibrose: Man sieht abnorme, große Megakaryozyten, die geclustert liegen. Man beobachtet außerdem eine Fettzelldepletion wie bei der CML, das Knochenmark wird rot statt gelb. Die Megakaryozyten haben eine vergrößerte Kern-Plasma-Relation, der Kern ist hyperchromatisch. Zellen werden in die Megakaryozyten eingestülpt, was ein Zeichen des vermehrten Umsatzes ist. Man sieht außerdem viele eosinophile Granulozyten. Deshalb nennt man diese Erkrankung auch megakaryozyäre Myelose, die anderen Stammzellreihen sind aber auch betroffen. Man erkennt in einer Silberfärbung eine Markfibrose, lauter kleine Fäden, die ein Netz bilden, das immer dichter wird. Die Megakaryozyten induzieren eine Fibrose im Knochenmark, wodurch es bald keinen Platz mehr für die Hämatopoese gibt; sie wird dann ausgelagert. Im Spätstadium sieht man im Knochenmark nur noch eine Osteosklerose und kann gar keine hämatopoetischen Zellen mehr unterscheiden. Durch die Auslagerung der Hämatopoese kommt es zur Splenomegalie, in der dann die extramedulläre Blutbildung stattfindet. Man findet eine Linksverschiebung, ein leukoplastisches Blutbild. Fallbesprechung Sektion Die Grunderkrankung war ein Peutz-Jeghers-Syndrom (erbliche Tumorerkrankung, die man an Hyperpigmentationen im Mundbereich und an den Handflächen erkennen kann), auf dessen Grundlage sich ein Pankreastumor mit Magen- wandinfiltration (angeblich; in der Pathologie aber nicht nachweisbar) bildete, auf Grund des erhöhten Risikos für alle malignen Entartungen. Sie bekam jahrelang Marcumar wegen chronischem Vorhofflimmern. Durch die paraneoplastischen Veränderungen bildeten sich bei ihr Thrombosen an vielen Stellen, in beiden V. femorales bis hoch in die V. cava inferior und außerdem in der Lungenarterie. Es kam durch eine zusätzlich vorhandene Linksherzinsuffizienz zu einem hämorrhagischen Infarkt des Lungengewebes und einer Einblutung in die Pleurahöhle rechts, ein blutiger Pleuraerguss von etwa 1,5l fand sich. Im Darm fanden sich viele Polypen (wegen Peutz-Jeghers-Syndrom), die aber nicht maligne entartet waren. Der rechte Ventrikel war hypertrophiert, die Lungenarterie wies atherosklerotische Veränderungen und Plaques auf, also lag schon länger eine pulmonale Hypertonie vor. An beiden Beinen waren starke Ödeme und eine trockene Gangrän an den Füßen, die sich durch Mikrothromben erklären lässt. Der Pankreaskopf war knotig verändert, der Tumor erstreckte sich bis zum Ligamentum hepatoduodenale, in dem die ganzen Lymphknoten Metastasen aufwiesen. Es finden sich weiterhin intravitale Thromben im Herz und in den Vv. jugulares. Es fanden sich Zeichen für eine rezidivierende Lungenarterienembolie, einige Thromben waren schon bindegewebig organisiert. Der Gallengang war abschnittsweise komplett von dem Tumor infiltriert und ulzeriert. Der linke Ventrikel ist hypertrophiert und dilatiert, die Herzspitze ein romanischer, kein gotischer Bogen mehr. In der Niere finden sich Infarkte und frische Embolien in den Arterien; eine Ursache für Thromben in den Arterien könnte ein persistierendes Foramen ovale sein, hier liegt es aber eher an der allgemeinen Gerinnungsstörung durch die Paraneoplasien. Die Häufigkeit für Peutz-Jeghers-Syndrom ist 1:120.000 Geburten, es ist autosomal dominant erblich. Als weiter Komplikationen zu den oben genannten können Invaginationen des Darms auftreten, die mit kolikartigen Bauchschmerzen einhergehen und zum Ileus führen können. Es gehört zur Gruppe der Phakomatosen: Oberbegriff für neurokutane Syndrome mit ektodermalen bzw. mesenchymalen Tumoren sowie kongenitalen Gefäßveränderungen an Haut, Augen u. ZNS. Mikroskopieren Präparat 302: Follikuläres Lymphom Man sieht unscharfe Follikelgrenzen und insgesamt eine recht zerstörte Lymphknotenstruktur. Man sieht in den neoplastischen Follikeln (die man vielleicht mit Primärfollikeln verwechseln könnte, aber dazu sind es zu viele) keine Sternhimmelmakrophagen und kein Keimzentrum. Differentialdiagnose wäre die follikuläre lymphatische Hyperplasie bei rheumatoiden Erkrankungen, aber da ist die LK-Struktur erhalten. Präparat 301: Hodgkin-Lymphom Bei Hodgkin-Lymphomen müssen Hodgkin-Zellen und/oder Reed-Sternberg-Zellen (multinukleär) vorhanden sein. Der Lymphknoten ist komplett von einer Fibrose umgeben. Die Hodgkin-Zellen erkennt man daran, dass sie verhältnismäßig riesig sind und einen großen Kern mit stark prominentem Nucleolus haben. Weiterhin sieht man in dem Lymphknoten viele eosinophile Granulozyten (rote Areale). Präparat 17126/97: reaktives Knochenmark In unsere Billigpräparaten sollte man wenigstens eine Steigerung der Erythropoese sehen oder so, mehr kann man net sehen. Dazu kommt es durch eine Aktivierung des Knochenmarks durch erhöhte Erythropoietin-Ausschüttung. Präparat K 7953/98: autoimmunhämolytische Anämie Hat anscheinend keiner, das Präparat. Sieht aber schön bunt aus auf den Folien. Präparat 12674/00: megakaryozytäre Myelose Man sieht halt sehr viele Megakaryozyten, die in Gruppen gelagert sind. Dazwischen erkennt man noch viele Fettzellen, das Stadium der Erkrankung ist anscheinend noch nicht fortgeschritten. Es handelt sich im Prinzip um eine 3-LinienErkrankung, d.h. auch die Granulopoese und die Erythropoese ist gesteigert, aber man diagnostiziert sie anhand der Megakaryozyten; ganz typisch sind Megakaryozyten mit Wolkenkernen. Die Erkrankung wird in die zelluläre und die fibrotische Phase unterschieden. Das Präparat hier ist noch nicht einmal in der zellulären Phase, sondern befindet sich noch in einer Frühphase. Präparat 17442/00: Milz bei CIM (chronisch idiopathischer Myelofibrose) Die Milz hat normalerweise rote und weiße Pulpa, eine Kapsel und Gefäße. Hier sieht man aber eine Milz mit mehreren Infarkten. In den Hohlräumen (= Sinusoiden) der roten Pulpa finden sich Megakaryozyten und Vorläuferzellen. Gynäkopathologie: Schwerpunkt: Ovar, Uterus/Zervix, Mamma Grundkenntnisse der Anatomie und Physiologie Ätiologie und Pathogenese der wichtigstesn Tumoren Malignome, relative Häufigkeiten: - 1. Stelle: Mamma-Ca - 4. Stelle: Corpus-Ca - 5. Stelle: Ovarial-Ca - 8. Stelle: Zervix-Ca (weltweit dritte (?) Stelle, aber bei uns bzw. den Industrienationen relativ weit zurückgedrängt worden, weil man in der Lage ist, die Vorstufen gut zu diagnostizieren und zu behandeln) Embryologie: Befruchtung: → Festlegung d. gonadalen Geschlechts Embryonalentwicklung: → bis zur 7. Woche identisch (Indifferenzstadium) Ausbildung primärer Geschlechtsmerkmale: Y-Chromosom → primäre Keimstränge + Mark → Hoden kein Y-Chromosom → sekundäre Keimstränge + Mark → Ovar (zwei X-Chromosomen notwendig!) Die Keimdrüsen, sowohl Hoden als auch Ovar, bestehen feingeweblich aus drei Anteilen: - Coelomepithel - Mesenchym - Urgeschlechtszellen Das erklärt die Vielfalt der Ovarial- und Hodentumoren! In der 5. Embryonalwoche verdickt sich das Coelomepithel (=Peritonealepithel) und wird zum sogenannten „Keimepithel“. Dann kommt es lateral zu einer Einstülpung des Coelomepithels zu Müller‘schen Gängen. Anfang der 6. Woche kommt es zu einem Einwandern der Urgeschlechtszellen aus dem Dottersack in das Keimdrüsenblastem, dann proliferiert das Keim- oder Coelomepithel und Mesenchym (die Genitalleiste vergrößert sich). Das alles führt zur Ausbildung der primären Keimstränge. Die verhalten sich dann je nach Geschlecht unterschiedlich. Wenn ein Y-Chromosom vorhanden ist, läuft die Entwicklung Richtung Hoden: - prim. Keimstränge → Samenstränge (Tub. seminiferi, Tub. recti, Rete testis) - Entwicklung Tunica albuginea (frühes diagnostisches Merkmal in der Sonographie) - Urnieregang (=Wolff‘scher Gang) (Dd. efferentes, D. epididymidis, D. deferens) - Mark → Lydig‘sche Zwischenzellen - Rückbildung Müller‘scher Gang Falls kein Y-Chromosom vorhanden ist, läuft die Entwicklung Richtung Ovar: - Degeneration der primären Keimstränge - Ausbildung sekundärer Keimstränge (Follikelepithel) - sekundäre Keimstränge zerfallen in Eiballen (Oogonie + Follikelepithel) - Mark → ovarielles Stroma - Rückbildung Urnierengang (Wolff‘scher Gang) - Müller‘sche Gänge proliferieren (es entstehen daraus beide Tuben, Uterus, Zervix, proximale Vagina) weiblich Oogonie Follikelepithel Ovarielles Stroma embryonal Urgeschlechtszellen „Keimepithel“ Mesenchym d. Genitalleiste männlich Spermatogonie Sertoli-Stützzellen, Samenstränge Leydig‘sche Zwischenzellen --- Urnierengänge (Wolff-Gänge) Tuben, Uterus, Zervix, prox. Vagina Müller‘sche Gänge Ductus epididymidis, Ductus deferens --- Aufbau des Ovars: - Keimepithel (=Müller‘sches Epithel) - Tunica albuginea (rudimentär) - Kortex (Follikel + Stroma) - Mark (Bindegewebe, Gefäße, Nerven) Follikelreifung: - Primärfollikel (Oogonie + Follikelepithel, einreihig) - Sekundärfollikel (Oogonie + Follikelepithel, mehrreihig) - Tertiärfollikel (Graafscher F.) (Oogonie + Stratum granulosum + Cumulus oophorus + Antrum folliculare + Theka folliculi) Gelbkörperbildung: - Ovulation (Oogonie + Follikelepithelmantel) - Corpus luteum (nach 3-4 Tagen) (Follikelepithel → Graunulosa-Luteinzellen, Thekazellen → Theka-Luteinzellen) - Corpus luteum graviditatis (3-4 Monate) - Corpus luteum cyclicum (10-12 Tage) Aufgaben der Ovarien: - reproduktive Funktionen - endokrine Funktionen → Östrogen (vorwiegend in der 1. Zyklushälfte) → Progesteron (vorwiegend in der 2. Zyklushälfte) Ovar Ovarialzysten: DD: Malignom! a) Zysten des Follikels: - Follikelzysten - Granulosa-Luteinzysten - Theka-Luteinzysten - Corpus-luteum-Zysten - Corpus-albicans-Zysten b) Zysten des Müller-Epithels: - Inklusionszysten (man nimmt an, dass aus diesen epitheliale Ovarialtumore entstehen, die häufigsten) - Endometriosezysten Komplikationen: - Größenzunahme - Ruptur (kann durch Reizung des Peritoneums extrem schmerzhaft sein und peritonitische Symptome verursachen) - Torsion (hämorrhagische Infarzierung) - Hormonproduktion (Zyklus- oder Blutungsstörungen) Ovarialtumoren Epithelial: - 60-70% insgesamt - Anteil an den malignen Tumoren des Ovars: >90% - 60.-70. LJ Keimzell: - 10-20% insgesamt - Anteil an den malignen Tumoren des Ovars: <5% - 0-25. LJ Keimstrang-Stroma: - 5-10% insgesamt - Anteil an den malignen Tumoren des Ovars: <5% - jedes Lebensalter Metastasen: - 5-10% insgesamt - Anteil an den malignen Tumoren des Ovars: 5% - variables Lebensalter 1. Epitheliale Tumoren: (ausgehend vom Müller‘schen Oberflächenepithel, man geht davon aus, dass sie aus Inklusionszysten entstehen) - seröser Tumor (mit Abstand der häufigste) - muzinöser Tumor - endometrioider Tumor - hellzelliger Tumor - Brenner-Tumor (solide Tumoren, die urothelial wirken) Stadien: a Normales Müller-Oberflächenepithel. Kleine Inklusionszysten des Ovars stellen die Ausgangszellen der epithelialen Ovarialtumoren dar. b Zystadenom. Mit einem einschichtigen Epithel der zystischen Drüse. Können sehr groß werden, manche 20cm. c Borderline-Tumor. Mit mehrschichtigem Epithel und unregelmäßigen Brückenbildungen. Praktisch ein CiS. Basalmembran intakt. d Adenokarzinom. Mit infiltrativem Wachstum und Einbruch in die Serosa. Benigne: - seröses Zystadenom: 15% aller Ovarialtumoren, 60% aller benignen = häufigster Ovarialtumor - serös-papilläres Zystadenom: Sieht makroskopisch maligne aus wegen den Proliferaten auf oder in den Zysten. - mzinöses Zystadenom: Zweihäufigster benigner Ovarialtumor. Einreihiges schleimproduzierendes Epithel. Können gigantische Größen annehmen, ohne zu entarten. - Brenner-Tumor: 2% der epithelialen Tumoren, sind zu 99% benigne - 10-20% bilateral - Makroskopie: meist klein, fibromartig, gelegentlich zystig - Histologie: Inseln von übergangsepithelähnlichen Zellen in overial-stroma-ähnlichem BGW - Merke: Extrem selten Niedrig maligne = Borderline: - niedrig maligner muzinöser Ovarialtumor. In der immunhistologischen Anfärbung mit p53 (Tumorsuppresorprotein) sind manche Zellen stark angefärbt = überexprimiert = genetisch instabil. Außerdem sieht man kleine Abschnürungen. Maligne: - serös (50% insgesamt, in der Hälfte aller Fälle bilateral) - muzinös (5%, 20% bilateral) - endometrioid (10%, 25% bilateral) - hellzellig (5%, meist unilateral) - maligner Brenner-Tumor (Rarität) - Karzinosarkom - Merke: Bei Diagnose in 80% aller Fälle schon größer als 15cm im Durchmesser! pT1a = Oberfläche intakt, glatt und spiegelnd, einzelne Teile der Kapsel fibrosiert pT1c = Tumor auf Oberfläche, Kapsel durchbrochen 2. Keimstrang-Stroma-Tumoren: - Fibrom - Granulosa- / Thekazelltumoren - Sertoli- / Leydigzell-Tumoren (Androblastom, können durch Differenzierungsstörungen im Ovar auftreten) - Merke: Zwei Drittel sind hormonproduzierend! Granulosa- / Thekazell-Tumoren: - alle Altersgruppen - 80% Östrogenbildung - 90% im Stadium I entdeckt - Dignität nicht vorhersehbar - Klinik: Pubertas praecox, Zyklusstörungen, Postmenopausenblutungen - Makroskopie: ähnelt Nebennierenrinde, häufig Blutungen, Nekrosen Sertoli- / Leydigzell-Tumor (Androblastom): - meist 20.-30. LJ - Androgenproduktion - 20% maligne - vermännlichtes Erscheinungsbild („durfte nicht mehr auf die Damentoilette“) 3. Keimzell-Tumoren: - Teratom (mit Abstand am häufigsten) - Dysgerminom - Chorionepitheliom - endometrialer Sinustumor - insgesamt 10-20% der Ovarialtumoren, meist im Kindes- oder Jugendalter, verschwindend geringer maligner Anteil Teratom: - 20-25% aller Ovarialtumoren und damit die zweithäufigste Gruppe (Kindesalter, Adoleszenz, Schwangerschaft bis 50%!) - 99% benigne - 10% bilateral - Reife Teratome können jedes Körpergewebe bilden. Oft findet man Haut und Hautanhangsgebilde (Haare, Talgdrüsen), neuronale Anteile wie Kleinhirn, oder alle Bindegewebe wie Fettgewebe, Knorpel, Knochen. Dysgerminom: - „Seminom des Hodens“ - „entartete primordiale Eizelle“ - maligne: 2-3% - sensibel für Strahlen- und Chemotherapie Chorionepitheliom / -karzinom: - Tumormarker: ß-HCG! - hochmaligne - strahlen- und chemoresistent Endometrialer Sinustumor (syn. Dottersacktumor): - ... Zervix Das Zervixkarzinom war in Deutschland nicht primär, wie heute, das achthäufigste Karzinom, sondern vor einigen Jahrzehnten noch an dritter Stelle und ist es in vielen Ländern noch heute. Anatomie des Zervix: Der Anteil des Zervix, der in die Vagina hineinragt, ist die Portio. Man findet dort ektozervikal Plattenepithel, endozervikal Drüsenepithel. Der Übergang ist abrupt und unruhig, deswegen ziemlich vulnerabel. Plattenepithel-Ca. Am häufigsten ist das Plattenepithelkarzinom der Zervix, das meist nichtverhornend ist, da es auch von nichtverhornendem Plattenepithel ausgeht. Es macht wohl über 90% der Zervixkarzinome aus. Wenn es schlecht differenziert ist (G3), sind die Zellen spindelförmig, und man hat bisweilen Probleme, es von einem mesenchymalen Tumor zu unterscheiden. Adeno-Ca. Selten, geht von den Drüsen des Endozervix aus FIGO-Stadium Häufigkeit Ib / T1b 45% (34-70%) II / T2 29% (16-41%) III / T3 IV 22% (10-40%) Häufige Begleitkomplikationen - Therapie 5-Jahres-Überleben (%) 80-90% Operation (Strahlentherapie) (eventuell Harnwegs- Operation und / oder 60-75% komplikationen) Strahlentherapie ... Strahlentherapie 30-40% symptomat. Maßnah- 0-15% men (evtl. Strahlentherapie) Epidemiologie: - Neuerkrankungen 15 / 100.000 Frauen / Jahr, das sind hier etwa 6000 / Jahr - 2000 Todesfälle / Jahr - 40% Stadium I diagnostiziert - Neuerkrankungen Präkanzerosen insgesamt: 25.000 / Jahr - weltweit Rang zwei bei Frauen, besonders in Südamerika und Afrika Besonderheiten: - bekannte Risikofaktoren (HPV) → Cx-Ca (RR: 70) (zum Vergleich: Nikotin → Bronchial-Ca RR: 39) - eigentlich gilt: Ohne HPV kein Cervixkarzinom, allerdings gibt es viele verschiedene HPV-Typen, die ein unterschiedliches Ca-Risiko beinhalten. Es sind 120 HPV-Genotypen bekannt, ca. 35 genitale HPVs, ca. 12-14 high-risk HPVs - Die Gene E6 und E7 im HPV sind die Onkogene. E6 aut p53 ab (Tumorsuppressorgen), E7 pRb. Das führt zu einem Verlust der Zellzykluskontrolle, überschießender Proliferation des Plattenepithels und natürlich einem gesteigerten Risiko der malignen Entartung. Im Zuge der Integration der zyklischen Virus-DNA in das humane Genom muss sie an einer Stelle aufbrechen, um sich linear integrieren zu können. Man vermutet, dass es durch einen Aufbruch im Bereich der E1- oder E2-Gene zu Neoplasien kommt, da die Produkte dieser Gene E6 und E7 kontrollieren. Damit kommt es endgültig zu einem Verlust der Kontrolle über den Zellzyklus. - bekannte Krebsvorstufen (CIN = cervikale intraepitheliale Neoplasien) - exponierte Lokalisation (Portio), deshalb Vorsorge durch nichtinvasive Methode (zytologischer Abstrich) → nicht-invasive (zytologische) Früherkennung Risikofaktoren Virus: - HPV-Typ (high risk, z.B. -16, -18) - HPV-Persistenz, z.B. für ein Jahr: 100-fach höheres Risiko für CIN. Bei zweimaligem Nachweis einer high-risk-HPVInfektion kommt es innerhalb von 10 Jahren bei 50% zum CIN. - „viral load“, Anzahl der Viruskopien pro Zelle - Mehrfachinfektion - Onkogenaktivität (E6 / E7) Präkanzerosen: Man geht ganz mechanistisch davon aus, dass sich das Plattenepithel mit der Zeit immer stärker entdifferenziert, wobei es zu einer höheren Kern-Plasma-Relation kommt, außerdem wirken die Zellen unreif. Man unterscheidet je nachdem, in welche Epithelschicht die Dysplasien reichen; sie beginnen unten und breiten sich von dort über das mittlere ins obere Epitheldrittel aus, entsprechend spricht man von CINs Grad 1 bis 3. Sobald die Zellen die Basalmembran durchbrochen haben und in das Stroma infiltrieren, handelt es sich nicht mehr um ein Carcinoma in situ, sondern ein Karzinom, die Prognose wird automatisch schlechter, da im Stroma Lymph- und Blutgefäße sind, über die das Karzinom fernmetastasieren kann. Auch wenn die Dysplasie noch nicht in die oberen Schichten des Epithels vorgedrungen ist, finden sich dort oft veränderte Zellen, sogenannte Koilozyten, die pathognomonisch sind und auf eine Infektion mit HPV hinweisen. Sie haben hyperchromatische, vergrößerte Zellkerne und eine perinukleäre Plasmaaufhellung. Früherkennung: Im zytologischen Abstrich kann man die oberste Zellschicht des Epithels beurteilen. Die Unterteilung ist Pap I für unauffällig, Pap II für unauffällig / Entzündung o.Ä., Pap IIID für geringe / mäßige Dysplasie (kann nur sehr schwer unterschieden werden in der Zytologie), Pap IVa für schwere Dysplasie, Pap V bei manifestem Karzinom, fast ausschließlich atypische Zellen im Abstrichmaterial, hyperchromatische Kerne, Mitosen, bizarre Zellen (Pap kommt von George Papanicolao, 1883-1962). Die frühen Dysplasien erkennt man nur deshalb, weil die Kohäsivität der dysplastischen Epithelzellen untereinander abnimmt und man deshalb nicht nur, wie bei gesundem Gewebe, die oberste Zellschicht, sondern auch die tieferen, dysplastischen, mit erwischt. Bei einer schweren Dysplasie spricht man dann in der Zytologie von Pap IVa, in der Histologie von CIN III, was auch einem Carcinoma in situ entspricht; die Polarität ist nicht mehr gegeben. Pap IIID entspricht z.B. CIN I mit Koilozyten, das sind pathognomonische Veränderungen der Epithelien durch die Virusinfektion (HPV). Spätestens im Stadium CIN III wird therapiert, da hier eine Präkanzerose vorliegt. <holger schreibt> Plattenepithel-CA, schwere Dysplasie: - mikroinvasives Wachstum - breitflächig invasiv Therapie ist immer die Hysterektomie mit Lymphknotenentfernung! Chirurgisch: Konisation: Konisches Gewebsstück aus Portio wird Pathologen gegeben und histologisch untersucht (ist Invasion vorhanden?) </holger schreibt> Wenn keine Invasion vorhanden ist und die Absetzungsgrenzen frei sind, ist damit die Therapie beendet. Wenn nicht, folgt die Hysterektomie, es sei denn, die Patientin ist jung und hat noch einen Kinderwunsch. Das ist allerdings natürlich riskant. Die Sensitivität der Zytologie ist ziemlich niedrig, die Spezifität aber hoch. Der HPV-Test dagegen hat eine hohe Sensitivität (Kranke als krank erkennen), aber eine niedrige Spezifität (Gesunde als gesund erkennen). Problem an der HPVTestung sind die 3-fach höheren Kosten gegenüber der Zytologie, die mögliche Stigmatisierung (kanzerogener Virus, der sexuell übertragen werden kann). Warum untersucht man nicht immer zytologisch und mit HPV-Test? - Kosten - Stigmatisierung, die ein positiver Befund mit sich bringt (sexuell übertragbarer Virus, der kanzerogen ist) - Ø Therapie der Infektion HPV-Vakzine: Gegenstand intensiver Forschung und erster klinischer Studien: - Prophylaktische Impfung mit VLPs („virus-like particles“, L1/L2) - Impfung von Mädchen und Jungen (vor erstem Viruskontakt) - signifikanter Effekt erst nach Jahrzehnten zu erwarten - vielversprechende erste Studienergebnisse - Therapeutische Impfung mit Vakzinen gegen Onkoprotein E6 / E7 - Impfung nach Viruskontakt (post expositionem) - Therapie von CIN und Zervixkarzinom - bisher keine zufriedenstellenden Ergebnisse HPV, Epidemiologie: - HPV-Prävalenz bei normaler Zytologie: <25 J. ~40% 25-29 J. ~22% >45 J. ~3% - HPV-Prävalenz nimmt mit steigender Anzahl der Sexualpartner zu - HPV-Infektion, „life time risk“: 85-90% - HPV-Infektionen latent (8-15 Monate): 60-80%, verschwindet spontan wieder - persistierende high risk HPV-Infektion (>12 Monate) → 100-fach höheres Risiko für CIN! - persistierende HPV-Infektion → 20% CIN - multiple HPV-Typen: 20-45% - CIN → CxCa ~10 Jahre (Zeitspanne zur Früherkennung ist lang) Das HPV-Virus ist an ein ausreifendes Plattenepithel gebunden, auf anderen Epitheltypen kann es sich gar nicht manifestieren. Deshalb ist es im Labor auch schwer zu untersuchen, man bekommt noch kein richtig ausreifendes polares Plattenepithel in vitro hin, das der HPV-Virus bräuchte. Vermeidung: Ziel ist es, die Inzidenz auf 0 zu drücken. Dazu gibt es zwei Möglichkeiten: a)Beseitigung der Risikofaktoren / Ursache (HPV) → Vakzinierung b)Erkennung und Behandlung der Vorstufen (CIN) → Früherkennung (Zytologie! +/- HPV?) Erfolge der Früherkennung: Früherkennung seit Finnland: 1965 England 1987 USA: 1970er Jahre Rückgang der Todesfälle um 60% 60% 40% Natürlicher Verlauf der Vorstufen des Cervix-Cas (Plattenepithel-Ca): Regression (%) Persistenz (%) Progression in CIS, (%) Progression in Ca, (%) CIN I 57 32 11 1 CIN II 44 35 22 5 CIN III 32 <56 12 Vor allem bei jungen Frauen mit vorhandenem Kinderwunsch eher abwarten und keine Konisation durchführen. Uterus Menstruationszyklus: Unter ansteigendem Östrogenspiegel hat man eine Proliferation des Endometriums, die Zellen teilen sich, das auskleidende Epithel ist nicht mehr nur einreihig, sondern zwei- oder mehrreihig. Dann steigt der Progesteronspiegel zusammen mit der basalen Körpertemperatur, und es kommt zur Auflösung der Schleimhaut, das Endometrium zerfällt, es kommt zu Blutungen, daran schließt sich wieder die Regeneration der Schleimhautoberfläche an. Man hat eine Proliferationsphase, eine frühe und eine späte Sekretionsphase (sägezahnartiges Muster in den Drüsenschläuchen), auf die die Abbruchblutung folgt. Bei Einnistung einer Eizelle in der Sekretionsphase wandelt sich die Stroma in die Dezidua um, die Stromazellen werden größer, haben mehr Zytoplasma und wirken flächig, eigentlich fast wie Tumorzellen. In der frühen Sekretionsphase findet man subnukleäre Vakuolen in den Drüsenzellen, so dass die Zellkerne nicht mehr direkt basal auf der Basalmembran liegen, sondern etwa in der Mitte der Zelle. In der späten Sekretionsphase liegen die Zellkerne wieder basal und die Vakuolen sind apikal. Uterine Blutung, Ursachen: - physiologisch - hormonelle Imbalance (gestörter enzymatischer Steroidabbau) - - - - - - - - - - Schleimhaut-Hyperplasien Uterus-Tumoren Endometriose hormonproduzierender Ovarial-Tumor (Granulosa-, Thekazell-Tumor) Schwangerschaft Plazenta-Tumor Verletzungen Infektion (Tbc, Syphilis) hämatologische Systemerkrankungen, z.B. mit Störungen des Gerinnungssystems psychogen Diagnostik: Durch Abschabung, Endometriumbiopsie mit der Strichkürette. Man schabt zuerst im Bereich des Cervix, erst danach und getrennt im Bereich des Endometriums aus, um die Lokalisation eines eventuellen Befundes besser eingrenzen zu können. Je nach Zyklusphase bekommt man mehr oder weniger Material, das der Pathologe dann untersuchen kann, um ein Karzinom auszuschließen oder zu diagnostizieren. Wichtig für die Beurteilung ist, dass man das Endometrium in seiner ganzen Dicke erfasst, d.h. im Präparat sollte auch immer etwas Myometrium sein. Tumoren des Uterus: Tumoren des Endometriums: 1.Epitheliale Tumoren a)gutartig: Polypen, oft zystisch b)bösartig: Endometriumkarzinom (endometrioide Ca 80-90%, adenokankroides Ca ca. 10%, adenosquamöses Ca ca. 5%, mukoepidermoides Ca 5-7%, serös-papilläres Ca 1-4%, klarzelliges Ca 2-5%, muzinöses Ca 1%) 2.Stromatumoren a)gutartig: Stromaproliferate b)bösartig: Stromasarkome (≥ 10 Mitosen / 10 HPF = high power field, Gesichtsfelder bei max. Vergrößerung) 3.Karzinosarkom (maligner Müller-Mischtumor) Tumoren des Myometriums: a)gutartig: Leiomyom (sehr häufig) b)bösartig: Leiomyosarkom (≥ 10 Mitosen / 10 HPF) c) ferner: Adenomatoidtumor (benigne?) Metastasen (i.d.R. anderer Adeno-Cas) Infiltration durch Cervix-Ca Myome: Myome an sich haben keine Entartungstendenz, können aber durch ihre Größenprogredienz Probleme machen und auch zu Durchblutungsstörungen führen. Vor einer geplanten Schwangerschaft sollte man auf jeden Fall sonographisch abklären, ob Myome vorhanden sind, da sie während der Schwangerschaft oder bei der Geburt Probleme machen können. Außerdem sollte eine Dysplasie, wenn sie gefunden wird, auf jeden Fall vor der Schwangerschaft behandelt werden, da Dysplasien und Tumoren während einer Schwangerschaft geradezu aufblühen. Ein Leiomyom von einem Leiomyosarkom zu unterscheiden, ist richtig schwer; man muss die Mitosen zählen (beim Leiomyom, auch bei der zellreichen Variante, wird man kaum eine finden) oder Proliferationsmarker einsetzen. Korpuskarzinom, Epidemiologie: - häufigstes Genitalkarzinom - Neuerkrankungen: 25 / 100.000 Frauen / Jahr, in Deutschland 10.000 Frauen / Jahr - Altersgipfel 65-70 Jahre (15% prämenopausal, 3% < 40 Jahre) - 75% im Stadium I diagnostiziert Korpuskarzinom, Risikofaktoren: - frühe Menarche - späte Menopause - - - - - - Nulliparität Adipositas Diabetes mellitus Hypertonie Langzeitgabe von Östrogenen ohne Gestagen Tamoxifentherapie (Antiöstrogen), Tamoxifen wird in der adjuvanten Therapie des hormonrezeptorpositiven MammaCa eingesetzt. Tamoxifen hat offensichtlich noch eine restintrinsische Aktivität am Corpusendometrium. Korpuskarzinom, Globale Inzidenz: Um so höher der Lebensstandard, um so höher die Inzidenz; in den Ländern der „1. Welt“ am häufigsten. Der „westliche Lebensstil“ begünstigt das Auftreten von Corpus-Cas. Korpuskarzinom, Symptome: - postmenopausale Blutung - ... Korpuskarzinom, Diagnostik: - gynäkologische Untersuchung - transvaginaler Ultraschall - Hysteroskopie - fraktionierte Abrasio - Röntgen-Thorax wegen Metastasen... - ggf. internistische Abklärung - Zystoskopie - Rektoskopie Korpuskarzinom, Präkanzerosen: Paar Stufen zum Karzinom: - einfache Proliferation - einfache glandulär-zystische Hyperplasie (sieht aus wie ein Schweizer Käse-Muster) - komplexe adenomatöse Hyperplasie (ohne Atypie), Dichte der Drüsen und Größe der Drüsenzellen nimmt weiter zu - (komplexe) atypische adenomatöse Hyperplasie, obligate Präkanzerose, Drüsen liegen dicht an dicht, man erkennt kein Stroma mehr dazwischen, konfluierende Drüsen, zahlreiche Mitosen. Therapie der Wahl: Hysterektomie. Korpuskarzinom, Histologie: - endometrioides Adeno-Ca - adenosquamöses Ca - Karzinosarkom, in der Desmin-Färbung sieht man den mesenchymalen Anteil Korpuskarzinom, Therapie: Stadium Ia, b Hysterektomie, Adnexektomie bds. Stadium Ic, IIIc Hysterektomie, Adnexektomie bds. (LK-Entfernung pelvin, paraaortal) Stadium II Radikale Hysterektomie, Adnexektomie bds. (LK-Entfernung pelvin, paraaortal) ... Die meisten Corpus-Cas werden im Stadium I diagnostiziert. Bei Stadium Ib ist weniger als die Hälfte des Myometriums infiltriert, bei Ic ist auch die äußere Hälfte, die sehr viel blutgefäßreicher ist, infiltriert, das Metastasenrisiko steigt stark. Mamma Die weibliche Brust besteht im Prinzip aus modifizierten Schweißdrüsen. Ihr Wachstum wird beeinflusst durch weibliche Hormone (Östrogen, Progesteron, Prolactin, Oxytocin) Sie besteht aus Drüsenparenchym, fibrösem Stroma und Fettgewebe. Ein Lobus ist eine funktionelle Einheit aus Ausführungsgang, Gangsystem und Lobuli. Im Drüsenparenchym finden sich 15-25 Lobi, d.h. auch 15-25 separate Ausführungsgänge (Porus excretorius) in der Mamille. Jeder Lobulus besteht wiederum aus 10-100 Azini und einem Gangsystem mit terminalem Ductulus; hier entstehen die meisten Mammakarzinome. Der Lobulus wird auch terminale ductulo-lobuläre Einheit = TDLE genannt. Ein Azinus wird gebildet aus apokrinem Epithel, Myoepithel und Basalmembran. Beim invasiven Karzinom gehen die Myoepithelzellen verloren. Pathologie der Mamma: - angeborene Störungen (z.B. Polythelie, Poly- / Amastie, Mikro- / Makromastie, Fehlbildungen d. Mamille / Areola) - fibrös-zystische Masthopathie - benigne proliferative Mammaläsionen (ductale Hyperplase, radiäre Narbe, Adenose / Skleradenose) - Mastitis - ... Fibrös-zystische Mammopathie: Definition: Der Begriff umfasst fibröse und zystische Umbauprozesse der Mamma verschiedenen Ausmaßes. a)Zystenbildung: Vergröberung der Lobuli und Azini (wie wenn man einen Ballon aufbläst), zahlreiche Zysten von mikroskopisch bis zu 1cm Größe, Verkalkungen in den Zysten b)Fibrose des spezialisierten Stromas c)apokrine Metaplasie des Drüsenepithels: Kleine, monomorphe Kerne, stark eosinophiles Plasma, mikropapilläre Fältelungen Ursache: hormonelle Fehlsteuerung der TDLE (terminale duktulo-lobuläre Einheit) Komplikationen: - Schmerzen - Ruptur - Verkalkung - „Knotenbildung“ Klinisch-pathologische Korrelation: - Normvariante? Weil so häufig - kein gesteigertes Krebsrisiko! Benigne proliferative Mammaläsionen: Als da wären: - duktale Hyperplasie - radiäre Narbe (im Bereich der Lobuli und Azini) - Adenose / Skleradenose Duktale Hyperplasie („Epitheliose“): - makroskopisch uncharakteristisch - histologisch: papilläre, kribriforme, solide Proliferate - „buntes Zellbild“ (basaloide und luminale Epithelien), aber ungefährlich - Ursache: hormonelle Stimulation - Häufigkeitsgipfel: Adoleszenz Radiäre Narbe: - Definition: Umschriebene narbige oder strahlige Läsion, die mammografisch und im Schnellschnitt mit einem invasiven Karzinom verwechselt werden kann. - In das Bindegewebe sind Drüsen eingebettet, die durch den Vernarbungsprozess deformiert werden. Das kann sehr schwer von einem Karzinom zu trennen sein, z.B. im Schnellschnitt. Deshalb versucht man allgemein suspekte Mammabefunde präoperativ mit Stanzbiopsien abzuklären, die Dignität zu sichern. - sternförmige Anordnung der Drüsenproliferate - mit / ohne epitheliale Proliferate - zentrale Stromafibrose und Elastose Adenose / Skleradenose: - Definition: Epithelial-myoepitheliale Proliferation der TDLE mit erhaltener (Adenose) oder aufgehobener (Sklerade- - - - - - nose) Läppchenstruktur. deutlich vergrößerte Läppchen (Lobuli) evtl. zystische Ektasie der Azini unscharfe Begrenzung der Läppchen durch Epithel-Myoepithel-Proliferate in das Stroma evtl. psammomatöse Verkalkungen kann auch schwer von einem Karzinom zu unterscheiden sein → im Zweifelsfall Immunhistologie Komplikationen bei den obigen Veränderungen: - knotige Konsistenzzunahme des Drüsenkörpers - Verkalkung → Ausschluss von Malignität durch (Mikro-) Biopsie Klinisch-pathologische Korrelation: - gering gesteigertes Krebsrisiko (RR: 1,6 - 4,5)! - fibrös-zystische Mastopathie und proliferative Mammaläsionen treten sehr oft gemeinsam auf Benigne Tumoren der Mamma: - (z.B. tubuläre, duktale, pleomorphe) Adenome - fibroepitheliale Tumoren! (vor allem Fibroadenom) - papilläre intraduktale Läsionen - mesenchymale Tumoren (Lipom, Hämangiom, etc.) - adenomyoepitheliale Tumoren (sehr selten) Fibroadenom: - häufigster benigner Tumor der Mamma - Häufigkeitsgipfel < 30. Lebensjahr - rund, scharf begrenzt, verschieblich - makroskopisch: grauweiß-streifig bis glasig-granulär - histologisch: perikanalikulärer und intrakanalikulärer Typ (dann scheint das Bindegewebe umgeben zu sein von komprimierten Drüsengängen - evtl. epitheliale Proliferate - evtl. Verkalkung - verlängerte Ausführungsgänge, die durch das proliferierende Bindegewebe „hirschgeweihartig“ deformiert werden Phylloides-Tumor: - Synonym: „Cystosarcoma phylloides“ - selten! - oft relativ groß (> 10cm), höhere Proliferationsrate als Fibroadenom - weich, wird später entdeckt - Rezidivneigung - ca. 15% metastasieren - Makro: grauweiß-gelappt - Histo: „fibrosarkom“-artiger Tumor mit zystischen Drüsen („leaf-like“-Aspekt, blattartige Struktur) - besteht aus Bindegewebe, das von ein- oder zweireihigem Epithel überdeckt wird - meist relativ gutartig, bei „sarkomatösem“ Stroma aber aggressiver und selten Metastasen - Risiko auf malignen Tumor dieses Typs nimmt mit Größe zu Intraduktales Papillom: - papilläre epitheliale / myoepitheliale Proliferate mit fibrovaskulärem Stroma in zystisch erweiterten Drüsen - (solitäres) zentrales Papillom („Milchgangspapillom“) → häufig Mamillensekretion, evtl. blutig → Galaktografie: Gangabbruch - (multiple) periphere Papillome → tastbare Knoten und/oder mammografische Verdichtungen - geringes Krebsrisiko, aber: Abgrenzung papilläres DCIS (duktales Carcinoma in situ)! Der Marker SM-Aktin (smooth muscle actine) fehlt beim DCIS. Die einzige Klausurfrage aus dieser Woche kommt zu einem benignen Tumor der Mamma. Mammakarzinome: - häufigste maligne Neoplasie der Frau (ca. 25%) → jede 8. Frau erkrankt, 50% versterben - häufigste Todesursache bei Frauen zw. 35 und 55 Jahren - heterogene Tumorerkrankung - Entstehung über präinvasive Stadien (Ca in situ) → durch Mammografie-Screening: 15-30% aller Cas werden entdeckt (man sieht nicht die Cis, nur die Mikroverkalkungen) - Ätiologie: Risikofaktoren sind Strahlenexposition, Alkohol, „westliche Diät“, Übergewicht, große Körpergröße, frühe Menarche, Nulliparität, späte Menopause, orale Kontrazeptiva, regulärer Menstruationszyklus (?), benigne Brustveränderungen, familiäre Belastung Beziehung zw. prolif. Mammaläsionen und Mamma-Ca: - gleichzeitiges Vorkommen - „höhergradige“ Epithelproliferate bei Mamma-Ca - retrospektiv Diagnose von florider DH (ductaler Hyperplasie?) und ADH - parallele Inzidenzraten bei verschiedenen Populationen - (partiell) gleichartige molekulare Veränderungen - höhere Ca-Inzidenz bei konservativer Therapie Empfehlung „College of American Pathologists“: - keine oder geringe Hyperplasie: Ca-Risiko normal - einfache duct./lob. Hyperplasie: 1,5-2x - atypische duct./lob. Hyperplasie (kein Zytokeratin 5/6 → SM-Aktin-negativ): 5x - DCIS / CLIS: 8-10x DCIS ist das ductale Carcinoma in situ (95%) → ductale Differenzierung, kohäsiv wachsende Tumorzellen CLIS das Carcinoma lobulare in situ (5%) → lobuläre Differenzierung, Verlust des Zellzusammenhangs Invasive Karzinome: Symptome: - „suspekter Mammabefund“ → tastbarer Knoten → mammografische Verdichtungen / Verkalkungen - abnorme Mamillensekretion - Hautinfiltration / -ulzeration, Mamillenretraktion - Fixierung des Tumors an der Thoraxwand - inflammatorisches Karzinom (klinische Diagnose) - Metastasen: lymphogen / hämatogen Histologische Subtypen: - invasives lobuläres Ca., ca. 15% - invasives duktales Ca. ca. 85% → invasiv ductal NOS (not otherwise specified, 80%) → tubulär → papillär → muzinös → medullär Auf der Basis eines intraduktalen papillären Carcinoma in situ entwickelt sich z.B. normalerweise eine papilläres Karzinom. Die CIS wachsen, obwohl noch nicht invasiv, manchmal in der gesamten Mamma entlang der Ausführungsgänge, wodurch eine brusterhaltende Therapie erschwert wird. Medulläres Karzinom: - synzytiales Wachstumsmuster (>75%) - keine glandulären Strukturen - lympho- / plasmazelluläres Infiltrat, das Immunsystem kämpft anscheinend dagegen, deshalb ist die Prognose etwas besser - nukleäre Pleomorphie (G 2-3) - glatte Begrenzung Inflammatorisches Karzinom (= klinische Diagnose) - Lymphangiosis carcinomatosa liegt oft zu Grunde (Orangenhaut) Bei der TNM-Klassifikation gibt es die Spezialität, dass es zwischen pN0 und pN1 noch „pN0 (ITC)“ gibt, bei der isolierte Tumorzellen / -cluster <0,2mm in den Lymphknoten gefunden wurde; dabei ist die klinische Relevanz und der Einfluss auf die Prognose noch nicht klar. Mammakarzinom-Therapie: - komplette chirurgische Exzision - evtl. neoadjuvante (Chemo-)Therapie - BET (+ SN; +/- ALNE) - Mastektomie / Ablatio (+ SN; +/- ALNE) - adjuvante Therapie - Radiatio - Chemotherapie - anti-hormonelle Therapie (Tamoxifen) - Antikörpertherapie (Herceptin) Probleme der täglichen Routine bei der Mammadiagnostik: Probleme sind im Allgemeinen: - Mikrokalk - der suspekte Herdbefund - der intraoperative Schnellschnitt - Invasion ja / nein? Ist ein duktales CIS wirklich noch ein CIS oder schon mikroinvasiv? - Resektion in sano? Besonders ein Problem bei den in situ-Karzinomen, die sich entlang der Gänge ausbreiten - Bestimmung von Prognoseparametern - seltene / atypische Tumoren Die häufigsten invasiven Tumoren der Mamma sind das invasiv duktale und das invasiv lobuläre Adenokarzinom. Mikrokalk: Verkalkungen stehen nicht immer für Malignität, auch wenn in der Regel ein Malignom hinter einer Verkalkung steckt. Kalk kann auch von normalem Epithel eingelagert werden oder z.B. bei apokrinen Metaplasien mit Verkalkungen. Auch regressiv veränderte Fibroadenome, die fast nur noch aus hyalinem Bindegewebe bestehen, verkalken oft. Beim duktalen CIS kommt es zunächst zu zentralen Nekrosen in den befallenen Ausführungsgängen, die dann sekundär verkalken. Bei Vakuumbiopsien, die bis zu zweieinhalb Zentimeter durchmessende Gewebszylinder liefern, wird in der Pathologie noch eine „Blockradiografie“ durchgeführt, wobei die Gewebeprobe nochmals geröntgt und auf nicht angeschnittenen Mikrokalk untersucht wird. Die ersten radiologisch fassbaren Veränderungen sind z.B. beim einem ductalen CiS, bei dem Nekrosen auftreten mit kleinen dystrophischen Verkalkungsherden. Der suspekte Herdbefund: Umschriebene, eher glattrandige Befunde in der Mammografie sind eher benigne, hochgradig suspekt sind dagegen unscharf begrenzte, strahlenförmig in das umgebende Gewebe ausgreifende Verdichtungen. Dann werden in der Regel Mikrobiopsien entnommen, die in der Pathologie fixiert werden, das Gewebswasser wird entzogen, dann werden sie in Paraffin eingegossen (das Wasser im Gewebe wird durch Paraffin ersetzt). Dann werden Schnitte angefertigt, gefärbt und untersucht. Das Ganze dauert etwa einen Tag. Zur Not können Präparate auch nach 3h schon durch das ganze Procedere geschleust werden, dann ist die Qualität aber etwas schlechter. Es werden 5-10 Mikrobiopsien pro Befund entnommen. Meistens werden auch kleine 5mm-Befunde sehr gut getroffen mit der Stanze, manchmal gehts aber auch daneben; der Pathologe kann nur diagnostizieren, was ihm auch an Material vorliegt. So kann manchmal ein CIS in der Stanzbiopsie vorliegen, während die Diagnose nach der Operation dann invasiv duktales Karzinom lautet („sampling error“). Der intraoperative Schnellschnitt: - Kann benutzt werden, um eine Tumordiagnose zu stellen. - Dient dazu, die Resektionsränder zu beurteilen. - Sentinel-Lymphknoten werden damit untersucht, um eine komplette Ausräumung der Lymphknoten zu entscheiden. Die Hälfte wird formalinfixiert und für die endgültige Beurteilung aufbewahrt, die andere Hälfte wird in ein Gel eingegossen und auf eine Metallunterlage gesetzt, die dann in eine Tiefgefrierkammer kommt. Sobald der Lymphknoten eingefroren ist, werden dünne Schnitte angefertigt und auf einen Glasobjektträger aufgebracht. Problematisch wird die Sache bei Mikrometastasen, die man vielleicht in der gewählten Schnittebene überhaupt nicht angeschnitten hat. Dann werden weitere Schnitte angefertigt und mit Keratinanfärbung untersucht. Findet man Mikrometastasen, ist es noch strittig, was gemacht werden sollte, aber zur Zeit wird sicherheitshalber dann nachoperiert und die Axilla ausgeräumt. - Ergebnis nach ca. 15 Minuten. - gut beurteilt werden können die Resektionsränder, dagegen kann das Grading des Tumors nur schlecht beurteilt werden, da die Qualität des tiefgefrorenen Schnellschnittpräparats einiges schlechter ist als das eines formalinfixierten, in Paraffin eingegossenen Präparats. Invasion? SM-Actin markiert vor allem Myoepithelien, aber auch schwächer das sekretorische Drüsenepithel. In normalen Ausführungs- / Drüsengängen markiert es halt die äußere Myoepithelzellschicht und das sekretorische Drüsenepithel, sonst nichts. Man kann in dieser Anfärbung erkennen, ob es sich um einen in situ-Prozess oder einen invasiven handelt. Wenn die äußere Hülle von Myoepithel und Drüsenepithel intakt ist, handelt es sich nicht um einen invasiven Prozess. In der ck 5/6 (Cytokeratin)-Färbung werden alle epithelialen Zellen mit Zytokeratinskelett angefärbt, also vor allem die apokrinen Drüsenepithelzellen. Wenn alle Zellen einer Hyperplasie in der ck 5/6-Färbung positiv sind, liegt eine einfache duktale Hyperplasie vor, wenn nicht, also wenn sie die Expression verloren haben, ist es eine atypische duktale Hyperplasie. Mann kann auch Antikörper gegen den Östrogenrezeptor zur Anfärbung benutzen, um sekretorisches Drüsenepithel anzufärben, und danach SM-Actin, um zu schauen, ob alle Drüsenepithelien von Myoepithel umgeben sind. Ist dies nicht der Fall, handelt es sich schonmal um einen malignen Prozess. Resektion in sano? Die Ränder des Operationsresektats werden mit Farbstoff angefärbt und für die räumliche Orientierung mit drei Fadenmarkierungen versehen. Die Ränder werden mikroskopisch auf Tumorfreiheit untersucht, wobei alle Karzinomzellen einen gewissen Abstand vom Resektionsrand haben müssen, sonst muss nachoperiert werden. Bestimmung von Prognoseparametern: Am HE-Präparat wird immer das TNM-Stadium bestimmt, die Grundlage jeder weiteren Therapie und der Prognose. Dazu kommt die immunhistologische Bestimmung der Hormonrezeptoren, in erster Linie der Östrogen- und Progesteronrezeptoren. Exprimiert der Tumor den Östrogenrezeptor, besteht in der Adjuvanz die Möglichkeit der hormonellen Therapie (Tamoxifen). Man bestimmt den Prozentsatz der ER-positiven Zellen und die Stärke der Anfärbung (0 = nicht angefärbt, 3 = stark angefärbt). Dann wird noch auf Her 2 untersucht, ein Wachstumsfaktorrezeptor auf der Zelloberfläche. Jedenfalls kann die entsprechende Genamplifikation, die der Vermehrung der Wachstumsfaktorrezeptoren zu Grunde liegt, auch in der FISH (floureszenz in situ-Hybridisierung) nachgewiesen werden. Die entsprechenden Patientinnen, bei denen eine hochgradige Genamplifikation des Her 2-Gens bzw. Überamplifikation von Her 2 vorliegt, können dann auch mit einem Antikörper (Herceptin) gegen diesen Rezeptor behandelt werden, wodurch die Zellproliferation möglicherweise bereits disseminierter Tumorzellen gehemmt wird. Sehr teuer, aber sehr effektiv. Seltene / atypische Tumoren: - Metastasen von malignen Melanomen - Infiltration der Mamma mit malignen B-Zell-Lymphomen (Antikörperfärbung gegen B-Lymphozyten, CD20) Molekulare Pathologie: - Zervixkarzinom → HPV-assoziierte Karzinogenese - - - Corpuskarzinom → „mismatch repair“-Gendefekte Ovarialkarzinom → BRCA1 / BRCA2 Mammakarzinom → BRCA1 / BRCA2 Molekulare Mechanismen: - Corpus-Ca - Ovarial-Ca - Mamma-Ca → 90% sporadisch, 5-10% familiär Fallbesprechung Sektion 80-jähriger Mann, der Zimmermann war und am 6. Juli gestorben ist. Bekannt war COPD und Asbestose, Vorhofflimmern bei absoluter Arrythmie, Herzinsuffizienz, Glaukom, Prostatahyperplasie, Z.n. Prostataresektion und Harnröhrenstriktur. Er bekam dementsprechend Medikamente gegen Herzinsuffizienz, Marcumar usw. Er wurde am 6.6. ins Paul-Lechler-Krankenhaus wegen Atembeschwerden eingewiesen. Er hatte außerdem Exsikkose, Harnverhalt und noch ein paar Sachen, eine Pneumonie wurde diagnostiziert. Er wurde nach ein paar Tagen entlassen. Am 1.7. wurde er wieder eingewiesen wegen Hämatinerbrechen: Abdomen war gebläht, aber weich, kein Tumor, kein Teerstuhl. Im Röntgen Abdomen eine massiv geblähte Magenblase, im Röntgen Thorax eine beginnende Stauung. Dann wurde eine Gastroskopie durchgeführt mit den üblichen Medikamenten (Dormicum), er benötigte schon anfangs 4l Sauerstoff, Refluxösophagitis wurde festgestellt, makroskopisch eine Pangastritis, im Magen selbst war kein Blut. Sie verlief allerdings recht dramatisch, nach etwa einer Viertelstunde rutschte der Patient mit einer HF von 123 und einer Sauerstoffsättigung von 70% in die Hypoxie und war etwa eine Stunde später tot, Reanimation war erfolglos. Makroskopisch sieht man ein Hautemphysem am Hals links und thorakal die Stauung. In der Sektion fand sich makroskopisch an der Lunge nichts auffälliges außer einer Pneumonie (Gewebe brüchig, viel Flüssigkeit) und ein paar weiße Flecken, die entweder von einer Tbc oder der Asbestose kamen. Die Lunge war halt insgesamt ödematös verändert, was an der Herzinsuffizienz lag (Linksherzinsuffizienz). Die Pulmonalarterie war sklerotisch. Das Herz war vergrößert und dilatiert (465g), die Kammerwand war dicker (links 15mm), das Trabekelwerk abgeflacht, romanischer statt gotischer Bogen an der Herzspitze, Thromben waren keine zu finden, die Aorta war dilatiert, die Carotis war unauffällig, und Infarktzeichen konnte man auch keine feststellen. Die Niere war mit kleinen Zysten durchsetzt, die Rinden-Mark-Grenze war undeutlich und man sah Einziehungen an der Oberfläche; der Urether war unauffällig. Pankreas und Nebenniere waren unauffällig. An der Leber fanden sich Schnürfurchen (durch hypertrophiertes Zwerchfell, das die Leber eindrückt) und gelbe Punkte (Cholestase). Ösophagus und Magen waren unauffällig bis auf ein leicht abgeflachtes Schleimhautrelief. Der Inhalt im Dünndarm war blutig und der Dünndarm gedehnt, das Colon weit und das Relief abgeflacht. Histologisch sieht man im Herz keine frischen Infarkte, aber hypertrophe Muskelfasern; der Befund spricht für eine chronische ischämische Schädigung. In der Lunge werden histologisch Zeichen der Linksherzinsuffizienz gefunden (Aufspleißung des Bindegewebes um die Gefäße), und zwar der akuten und der chronischen. Es finden sich Hämosiderophagen in der Lunge als Zeichen der chronischen Stauung. Interessant und auffällig war der Dünndarm. Die gesamte Dünndarmwand war gerötet (segmentale Veränderung, die große Teile betroffen hat). Die Schleimhaut zeigt in den betroffenen Gebieten schon makroskopisch einzelne kleine Defekte. Histologisch sieht man hyperämische Kapillaren und Blutungen in das Gewebe und letzendlich ins Darmlumen. Die Blutungsquelle waren also große Teile des Dünndarms, besonders das Jejunum. Obduktionsbefund: Grundleiden: - COPD - Pleuraplaques (Asbest? ließ sich histologisch nicht nachweisen) - arterielle Hypertonie Folgeleiden: - rezidivierte Pneumonien - globale Herzhypertrophie / -insuffizienz → Rhythmusstörungen → non-okklusive ischämische Enteropathie Todesursache: Dekompensiertes Cor pulmonale bzw. dekompensierte globale Herzinsuffizienz. Cor pulmonale bedeutet, dass das Herz hypertrophiert ist, weil der Widerstand in den Lungengefäßen über lange Zeit erhöht war. Mikroskopieren Präparat 216: CIN III / Carcinoma in situ der Portio Man bemerkt, wenn man am Epithel außen entlangfährt, einen abrupten Übergang von normalem, mehrschichtigem unverhorntem Plattenepithel zu schwer dysplastischem Plattenepithel. Wichtig bei solchen Funden: Die Basalmembran ansehen, die muss intakt sein; wenn sie es hier nicht wäre, hätte man kein CIS, sondern schon ein invasives Karzinom. Präparat 145: Plattenepithelkarzinom der Cervix uteri Hier sieht man im Prinzip dasselbe, nur dass der Tumor flächenhaft invasiv wächst und „Finger“ ins tieferliegende Gewebe aussendet; die Basalmembran ist durchbrochen. Außerdem sieht man in dem Präparat noch, dass Lymphgefäße infiltriert und erweitert sind, wahrscheinlich sind also schon Lymphknotenmetastasen vorhanden. HPV ist der wesentliche ätiologische Faktor beim Zervixkarzinom; eine Erkennung der Krebsfrühstufen ist möglich, und die Behandlung ebenfalls. Präparat 125: Endometrium, Proliferationsphase Man sieht Drüsenlumina mit regelmäßigem, einreihigem Epithel, eingelagert in den Großteil des endometrialen Stromas. Präparat 150B: Adenokarzinom des Corpus uteri Man erkennt nichts mehr von dem geordneten Bild das vorherigen Präparats; man erkennt noch Reste von Stroma (allerdings Myometrium-Stroma, d.h. der Tumor infiltriert bereits das Myometrium). Das Epithel bildet papilläre Auswüchse in die unregelmäßigen Drüsenlumina. Solche Tumoren entstehen auf Östrogeneinfluss hin, z.B. auf Therapie mit Östrogenen oder auch mit Tamoxifen (AntiÖstrogen). Präparat 139: Phylloides-Tumor der Mamma Typisch ist die blattartige Struktur mit Fältelungen. Das Präparat wird nur kurz angesprochen. Präparat 146: DCIS + invasives duktales Mamma-Ca Intraduktal sieht man den Tumor, der die Ausführungsgänge „tapetenartig“ ausfüllt. In der Mitte bilden sich häufig Nekrosen; diese nennt man Komedonekrosen, weil man sie aus den Gängen auspressen kann... Wenn man eine Weile sucht, sieht man Ausführungsgänge, bei denen die Basalmembran durch das atypische Tumorepithel durchbrochen wird, das wären dann mikroinvasive Regionen des Tumors. Wenn man ein bisschen rumsucht, sieht man Regionen, in denen die Bereiche mit den atypischen, großkernigen Epithelzellen nicht mehr schön rund wie Ausführungsgänge aussehen, sondern im gesamten Stroma verstreut sind; das ist der invasive Anteil des Tumors. Magentumoren ... Magenkarzinom: Etwa 40% der Magenkarzinome sind vom diffusen, 60% vom intestinalen Typ. Beim diffusen Typ muss der Chirurg einen größeren Sicherheitsabstand beim Exzidieren halten. Die Magenkarzinome vom diffusen Typ wachsen drüsig und sind häufig Siegelringkarzinome (Siegelringzellen erkennt man am charakteristischen Zellkern, sieht halt aus wie ein Siegelring). Magenfrühkarzinom: - Karzinom beschränkt auf Mukosa (Magenfrühkarzinom vom Mukosa-Typ) - Karzinom beschränkt auf Mukosa und Submukosa (Magenkarzinom vom Submukosa-Typ) - Günstige Prognose: 5-Jahres Überleben 95% bzw. 90% Ein guter Grund für regelmäßige Gastroskopie-Vorsorgeuntersuchungen. Mesenchymale Tumoren des Magens: gutartig Gastrointestinaler Stromatumor (GIST) Fibrom Leiomyom Lipom Hämangiom Neurinom (Schwannom) / Neurofibrom bösartig maligner GIST Fibrosarkom Leiomyosarkom Liposarkom Hämangiosarkom malignes Neurinom / Neurofibrosarkom maligne Lymphome Fehlbildungen / Anomalien des Dünndarms - - - - - - - Gewebsheterotopien (z.B. Magenschleimhaut in einem Meckel-Divertikel o.Ä.) Malrotation (unwichtig) Divertikel konnatale und erworbene Dünndarmstenosen Dünndarmatresie membranöse Stenosen (unwichtig) Meckel-Divertikel: - Rest des Ductus omphaloentericus - ca. 60cm oral der Bauhinschen Klappe - bei Appendektomie: ca. 4% - heterotope Magenschleimhaut (häufig) - heterotopes Pankreasgewebe (selten) - gefährlich nur, wenn es einen Ulkus entwickelt und dann okkulte Blutungen verursacht Kreislaufstörungen: Hämorrhagischer Dünndarminfarkt: - Thrombose oder Thrombembolie einer Mesenterialarterie - Thrombose einer Mesenterialvene - inkarzerierte Hernie Ein hämorrhagischer Infarkt kann nur bei einem Verschluss von Gefäßen in Organen mit doppelter Blutversorgung oder ausgedehntem Kollateralkreislauf auftreten. Ein hämorrhagischer Infarkt ist im Prinzip eine Nekrose, die eingeblutet ist. Entzündungen des Dünndarms: Es werden nur zwei besprochen: - Lambliasis (Diardiasis) - Morbus Whipple Gibt aber noch viel mehr: - Duodenitis - Salmonellosen - Cholera - Enteritis necrotisans - hämorrhagisch-nekrotisierende Säuglingskolitis - ... Lambliasis (Giardiasis): - Lamblia intestinalis ist ein Protozoon aus der Gruppe der Flagellaten - Trinkwasser, Salat, Gemüse, fäkal-oral - vor allem Kinder betroffen - Diarrhoen, Malabsorption, krampfartige Schmerzen Die Viecher schauen einen unter dem Mikroskop richtig an, sieht aus wie ein Gesicht. Morbus Whipple: - bakteriell verursachte Krankheit, die sich konstant am Dünndarm manifestiert, aber auch alle anderen Organe betreffen kann - Erreger: Whipple-Bakterium = Tropheryma whippelii (grampositiv, gehört zu den Aktinomyzeten), lässt sich noch nicht anzüchten, Nachweis nur über PCR - Diagnose am besten durch Dünndarmbiopsie mit pathognomonischen „SPC-Zellen“ (sickle-form particles containing cells) - mittleres Lebensalter (am häufigsten 40-50 Jahre) - Männer : Frauen wie 5-8 : 1 - Hauptsymptom: chronisch-rezidivierende Diarrhoen, Malabsorption, Gewichtsverlust, häufig auch Gelenkschmerzen (!), Hypotonie, Lymphadenopathie, neurologische Symptomatik (!), Hautpigmentierungen, periphere Ödeme usw. Epitheliale Tumoren des Dünndarms: gutartig bösartig Adenome neuroendokrine Tumore („Karzinoide) und nochwas Die neuroendokrinen Tumoren kommen auch im Bronchialsystem vor. - 2% aller malignen Tumore - gutartige Tumore auch möglich - langsam wachsende Karzinome - oder: hochmaligne (niedrigdifferenziertes neuroendokrines Karzinom, z.B. kleinzelliges Bronchialkarzinom) - die meisten Karzinoide treten auf im Appendix (34%) und im Dünndarm, und da vor allem im terminalen Ileum (49%) Krankheiten des Dickdarms Kreislaufstörungen: - hämorrhagischer Infarkt - ischämische Colitis Fehlbildungen des Dickdarms: - Atresien - Stenosen Dickdarmdivertikel: - multiple Dickdarmdivertikel = Divertikulose - Entzündung der Divertikel = Divertikulitis - Divertikelkomplikationen! Die Divertikel gehen durch die Muskularis hindurch; die Divertikelwand besteht nur noch aus Mukosa, Submukosa und Serosa. Sie bilden sich rein mechanisch durch schweren, harten Stuhlgang (wenn man ne Woche lang keine Ballaststoffe zu sich nimmt). Es kann zu Blutungen, Stenosen, Abszessen, Perforationen und Fisteln kommen als Divertikelkomplikation. Entzündliche Dickdarmerkrankungen: - Kolitiden komplexer Ätiologie und Pathogenese: - antibiotikainduzierte pseudomembranöse Enterokolitis - urämische Enterokolitis - idiopathische chronisch-entzündliche Darmerkrankungen: - Colitis ulcerosa - Morbus Crohn Pseudomembranöse Kolitis: - Antibiotika (Clindamycin, Lincomycin, Chloramphenicol u.a.) unterdrücken und zerstören die normale Darmflora - überschießendes Wachstum von Clostridien, Enterobacter, Proteus u.a. - Clostridium difficile: Enterotoxine! - profuse wässrig-blutige Diarrhoen Colitis ulcerosa: - chronisch rezidivierende Entzündung der Mukosa und Submukosa des Dickdarms (mukosale Colitis) - Inzidenz: 3-9 / 100.000 / Jahr - Prävalenz: 40-90 Fälle / 100.000 - 20. - 40. Lebensjahr - Frauen : Männer wie 1,5 : 1 Morbus Crohn: - chronisch rezidivierende Entzündung des gesamten Gastrointestinaltraktes (vor allem des terminalen Ileums, „Ileitis terminalis“) - segmentale und transmurale Entzündung, d.h. alle Wandschichten werden betroffen, nicht nur die Mukosa und Submukosa, sondern auch die Muscularis mucosae und die Serosa. - Inzidenz: 2-4 / 100.000 / Jahr - Prävalenz: 20-40 / 100.000 - Jugendliche, junge Erwachsene - Pflastersteinrelief der Schleimhaut - Ätiologie unklar - systemische Manifestationen - schubweiser Verlauf - fissurale Ulzerationen - Epitheloidzellgranulome Epitheliale Tumoren des Dickdarms: Gutartig: Adenom Bösartig: Folie weg Familiäre adenomatöse Poliposis coli mit tausenden von Adenomen nebeneinander... diese Patienten müssen rechtzeitig colektomiert werden, sonst entwickeln sich daraus lauter Adenokarzinome. Lynch-Syndrom („HNPCC = hereditary non-polyposis colorectal cancer-syndrome“): - autosomal-dominant (2p) - Kolonkarzinom vor dem 45. Lebensjahr - Zökum und Colon ascendens - „flat adenoma-syndrome“ - familiär: Endometrium- und / oder Ovarialkarzinome - Lynch I: rechtsseitig lokalisierte Kolonkarzinome - Lynch II: Kolonkarzinome + andere maligne Tumore (Endometrium, Ovar, Pankreas)- Zervixkarzinom → HPV-assoziierte Karzinogenese - Corpuskarzinom → „mismatch repair“-Gendefekte - Ovarialkarzinom → BRCA1 / BRCA2 - Mammakarzinom → BRCA1 / BRCA2 Molekulare Mechanismen: - Corpus-Ca - Ovarial-Ca - Mamma-Ca → 90% sporadisch, 5-10% familiär HNPCC (Lynch-) Syndrom: hereditary nonpolyposis colorectal cancer - autosomal dominante Erkrankung - erhöhtes Risiko für Kolorektale Karzinome, Endometrium-Ca u.a. - Auftreten oft schon in frühem Alter - Ursache: Mutationen in Mismatch-Repair-Genen (MMR) wie hMLH-1, hMSH-2, hMSH-6 - - - - - - - Folge: Replikationsfehler (RER = replication errors) Diagnose: in ca. 90% Mikrosatelliten-Instabilität (MSI) Anamnese → positive Eigen- / Familienanamnese humangenetische Beratung MSI-Analyse + IH MMR-Gen-Expression → MSI high (≥ 2/5 Loci) Mutationsanalyse MMR-Gene → Nachweis von MMR-Mutationen intensiviertes Krebsvorsorge-Screening