V11_Molekuleigen
Werbung

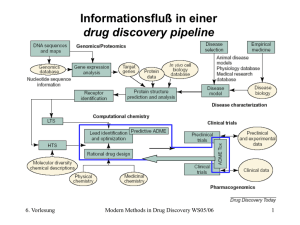
Vorhersage von Moleküleigenschaften (I) Leitmotiv beim rational drug design Prinizipielle Frage ist: Wie hängt der biologische Raum (Aktivität) mit dem chemischen Raum (Struktur) bei Verbindungen zusammen ? Lassen sich aus der Struktur Voraussagen treffen ? QSAR und QSRP 11. Vorlesung Modern Methods in Drug Discovery WS05/06 1 Vorhersage von Moleküleigenschaften (II) Observablen Was sind Moleküleigenschaften ? 11. Vorlesung Molekülmasse MW (aus der Summenformel C12H11N3O2) Schmelzpunkt Siedepunkt Dampfdruck Wasserlöslichkeit Ladung Dipolmoment Unmittelbar aus der Wellenfunktion des Polarisierbarkeit Moleküls Ionisationspotential berechenbar Elektrostatisches Potential Modern Methods in Drug Discovery WS05/06 2 Vorhersage von Moleküleigenschaften (III) Alle Moleküleigenschaften die sich mit physiko-chemischen Methoden messen lassen (sog. Observablen) können umittelbar mit quantenmechanischen Verfahren berechnet werden. Notwendig: Mathematische Beschreibung der Elektronenverteilung z.B. durch die Wellenfunktion des Moleküls 11. Vorlesung Elektronenverteilung Quantenmechanik (QM) Positionen der Kerne Molekülmechanik (MM) Modern Methods in Drug Discovery WS05/06 3 Quantenmechanik (I) Im Rahmen des mathematischen Formalismus sind eine Reihe von Näherungen erfoderlich. Eine der grundlegensten ist die Separierung der Bewegung der Kerne von denen der Elektronen, die sog. Born-Oppenheimer Näherung: Atomkerne sind > 1000 mal so schwer wie Elektronen und folgen den Bewegungen der schnelleren Elektronen Die Wechselwirkung zwischen geladen Teilchen (Elektronen, Kerne) läßt sich durch das Coulombsche Gesetz ausdrücken qi Vij rij qj 11. Vorlesung qi q j rij Modern Methods in Drug Discovery WS05/06 4 Quantenmechanik (II) Die zeitliche Bewegung gemäß der klassischen Mechanik ist entsprechend des 2. Newtonschen Gesetzes gegeben F ma dV 2r m dr t Elektronen sind sehr kleine Partikel (Quanten) die deshalb sowohl Teilchen- als auch Wellencharakteristik aufweisen: 11. Vorlesung Teilchen Welle Galvanische Abscheidung Beugung am Spalt Modern Methods in Drug Discovery WS05/06 5 Schrödinger Gleichung (I) Elektronen können als Wellenfunktion durch die zeitabhängige Schröder Gleichung beschrieben werden H i t Falls der Hamiltonoperator H zeitunabhängig ist, kann die Zeitabhängigkeit der Wellenfunktion als Phasenfaktor ausgeklammert werden, was zur zeitunabhängigen Schrödinger Gleichung führt r, t r e i E t / H r E r 11. Vorlesung Modern Methods in Drug Discovery WS05/06 6 Die Wellenfunktion Die Wellenfunktion ist ein mathematischer Ausdruck der die räumliche Anordnung der (fluktuierenden) Elektronen beschreibt Das Quadrat der Wellenfunktion gibt die Wahrscheinlichkeit P an, das Teilchen (Elektron) an einer bestimmten Stelle im Raum anzutreffen P P ist eine Observable während die Wellenfunktion selbst keine physikalische beobachtbare Größe ist. Integration über den gesammten Raum muß 1 ergeben. d 1 11. Vorlesung Modern Methods in Drug Discovery WS05/06 7 Der Hamiltonoperator Der Hamiltonoperator enthält die kinetische (T) und die potentielle (V) Energie aller betrachteten Partikel i im System 2 2 T Ti i i 1 i 1 2mi N H TV N mit dem Quadrat des Nabla Operators 2 2 2 2 i 2 2 2 xi yi zi N N V Vij i 1 j i mit Vij qi q j rij Als Folge der Born-Oppenheimer Näherung kann auch der Hamilton Operator in Kern- und elektronischen Anteil separiert werden 11. Vorlesung Modern Methods in Drug Discovery WS05/06 8 Die Wellenfunktion (II) Jeglicher mathematischer Ausdrück für die Wellenfunktion muß bestimmte Kriterien erfüllen, um die physikalische Natur der Elektronen korrekt wiedergeben zu können. Zur Vereinfachung nimmt man die Wellenfunktion aller Elektronen im Molekül als Produkt von Einelektronenfunktionen an, die jeweils ein Elektron beschreiben 1 2 ... N Diese müßen folgende Kriterien erfüllen: • Elektronen sind nicht voneinander zu unterscheiden • Sie stoßen sich gegenseitig ab • Es gilt das Pauliprinzip (zwei Elektronen mit unterschiedlichem Spin können sich einen Zustand (Orbital) teilen) 11. Vorlesung Modern Methods in Drug Discovery WS05/06 9 Schrödinger Gleichung (II) Gemäß der Schrödingergleichung muß es verschiedene Energieniveaus für die Elektronen im Molekül geben. Diese Energien erhalten wir durch Integration und umformen zu 2 H d E d H d E d 2 Die zu erhaltende Energien sind allerdings abhängig von der Güte der verwendeten Wellenfunktion und deshalb immer größer oder gleich der tatsächlichen Energie. Im einfachsten Fall wählen wir zur Beschreibung der Einelektonenfunktionen 1s-Orbitale als Basissatz 11. Vorlesung Modern Methods in Drug Discovery WS05/06 10 Molekül Orbital Theorie (I) Molekülorbitale lassen sich als Linearkombination von Atomorbitalen (LCAO-Ansatz) oder anderen Basisfunktionen darstellen cA A cB B ... c N N z.B. für H2 cA1s A cB1s B 1sA 1sB K allgemeiner Ausdruck für ein MO i c i 1 mit dem Atomorbital 11. Vorlesung Modern Methods in Drug Discovery WS05/06 11 Molekül Orbital Theorie (II) Benutzt man den LCAO Ansatz für die Wellenfunktion so erhält man für H2 2 d cA A cB B cA A cB B d c 2A A2 c 2B B2 2c A c B A B d c 2A A2 d c 2B B2 d 2c A c B A Bd =1 =1 Überlappungsintegral S Wegen der Normierung der Wellenfunktion über den gesammten Raum 2 2 2 d c c A B 2c A c BS 11. Vorlesung Modern Methods in Drug Discovery WS05/06 12 Molekül Orbital Theorie (III) Allgemeine Form in Matrix-schreibweise: H ES 0 Die Lösungen der Säkulargleichungen für E ergeben die Energien der bindenden und antibindenden MOs E 0 Der hauptsächliche numerische Aufwand besteht in der Suche nach geeigneten Koeffizienten (cA, cB, ...) die vernünftige Orbital Energien ergeben. Variationsprinzip Hartree-Fock-Gleichungen Self Consistent Field (SCF) Verfahren 11. Vorlesung Modern Methods in Drug Discovery WS05/06 13 Hückel Theorie (I) (1931) Limitiert auf planare, konjugierte -Systeme, Orbitale werden vernachlässigt. Ursprüngliches Ziel war die Deutung der nicht-additiven Eigenschaften von aromatischen Verbindungen (z.B. Benzol gegenüber “Cyclohexatrien”) Die -Orbitale werden als Linearkombination aus Atomorbitalen (pz-Orbitale) erhalten (LCAO). Die Elektronen bewegen sich in einem Feld, das von den Elektronen und den Atomkernen erzeugt wird 11. Vorlesung Modern Methods in Drug Discovery WS05/06 14 Hückel Theorie (II) Beispiel Ethen H2C=CH2 E pz pz 11. Vorlesung Modern Methods in Drug Discovery WS05/06 15 Hückel Theorie (III) Innerhalb der Hückeltheorie enthält die Fockmatrix genau soviele Spalten und Zeilen, wie Atome in Molekül vorhanden sind. Alle Diagonalelemente entsprechen einem Atom i und werden auf den Wert gesetzt. Nichtdiagonalelemente sind nur dann nicht Null, wenn zwischen den Atomen i und j eine Bindung existiert. Dieser Resonanzparameter wird auf den Wert (<0) gesetzt. Werte für kann man aus UV/VIS-Spektren erhalten ( -4.62 eV) Beispiel Butadien: 1 2 3 4 2 1 11. Vorlesung 4 3 1 0 0 2 0 3 0 4 0 0 Modern Methods in Drug Discovery WS05/06 16 Hückel Theorie (IV) Für ein cyclisches -System, wie etwa Benzol ergeben sich die Orbitalenergien und Orbitalkoeffizienten zu 2k ; mit k 0,1,..., N 1 N 2 1k 1 ci exp N N i 2 cos E 0 Daraus ergibt sich auch die Hückelregel, die besagt, daß ein System mit [4n+2] -Elektronen aromatisch ist 11. Vorlesung Modern Methods in Drug Discovery WS05/06 17 Hückel Theorie (V) • Anwendungen der Hückelmethode zur Vorhersage und Interpretation von UV/VIS-Spektren • Unterschiedliche Parameter für unterschiedliche Atome (C,N,O) ermöglichen die Anwendung der Hückeltheorie auf weitere Moleküle • Experimentell können Orbitalenergien direkt durch Photoelektronenspektroskopie (PES) bestimmt werden, und damit auch (Ionisationspotential) und 11. Vorlesung Modern Methods in Drug Discovery WS05/06 18 Hartree-Fock basierte Methoden H = E Born-Oppenheimer Näherung Ein-Determinanten Ansatz ZDO-Nährung Valenzelektronen Parameter Hartree-Fock-Gleichungen RHF Semiempirische Methoden mit minimalem Basissatz Optimierte Basissätze all electron Ab initio Methoden mit endlichem Basissatz Multi-Determinanten Ansätze Valenzelektronen UHF ECP spin (,) space Semiempirische CI Verfahren 11. Vorlesung CI MCSCF Modern Methods in Drug Discovery WS05/06 CASSCF 19 Semiempirische Methoden (I) Das Problem von ab initio Rechnungen ist die N4 Abhängigkeit von der Anzahl der Zwei-Elektronen Integrale. Diese sind durch die Anzahl der Basisfunktionen und die Wechselwirkungen zwischen Elektronen auf verschiedenen Atomen bedingt. In semiempirischen Verfahren wird der numerische Aufwand durch Annahmen und Näherungen stark reduziert: 1. Nur Valenzelektronen werden betrachtet, Rumpfelektronen werden mittels der effektiven Kernladung beschrieben die ein effektives Potential bilden (frozen core) 2. Minimaler Basissatz wird verwendet (ein s und drei p-Orbitale pro Atom), aber dafür exakte STOs die orthogonal zueinander sind. 3. Zero Differential Overlap (ZDO) Näherung 11. Vorlesung Modern Methods in Drug Discovery WS05/06 20 Semiempirische Methoden (II) Seit 1965 wurde eine Reihe von semiempirischen Verfahren vorgestellt die zum Teil noch heute für die Vorhersage und Simulation von elektromagnetischen Spektren von Bedeutung sind: CNDO/S, INDO/S, ZINDO Als besonders erfolgreich zur Berechnung von Moleküleigenschaften haben sich seit ihrer Einführung MNDO (Modified Neglect of Diatomic Overlap) Thiel et al. 1975, AM1 (Austin Model 1) Dewar et al. 1985 und PM3 (Parameterized Method 3) J.P.P. Stewart 1989 herausgestellt. Das liegt u.a. auch an ihrer weitläufigen Verbreitung durch das Programmpaket MOPAC und dessen spätere Konkurrenten. Alle drei basieren auf derselben Näherungsstufe NDDO und unterscheiden sich vor allem in der jeweiligen Parametrisierung der einzelnen Atome. 11. Vorlesung Modern Methods in Drug Discovery WS05/06 21 AM1 (Austin Model 1) Dewar, Stewart et al. J.Am.Chem.Soc. 107 (1985) 3902 Vorteile gegenüber MDNO: + bessere Molekülgeometrien v.a. bei hypervalenten Elementen (P, S) + H-Brücken (aber mit der Tendenz zur Gabelung) + Aktivierungsenergien bei chemischen Reaktionen Schwächen von AM1 (und allen Verfahren auf NDDO Basis): - hypervalente Elementen allgemein, da keine d-Orbitale - Verbindungen mit freien Elektronenpaaren - NO2-Verbindungen 11. Vorlesung Modern Methods in Drug Discovery WS05/06 22 PM3 (Parameterized Method 3) J.J.P. Stewart J.Comput.Chem. 10 (1989) 209 Im Gegensatz zu den bisherigen Methoden erfolgte die Parametrisierung stärker durch statistisches Kalkül Vorteile gegenüber AM1: + bessere Molekülgeometrien bei C, H, P und S + NO2-Verbindungen besser Nachteile gegenüber AM1: - Alle übrigen N-haltigen Verbindungen schlechter - höhere Atomladungen führen zu polarerem Charakter der Moleküle - Nicht alle parametrisierten Elemente (Mg, Al) liefern zuverläßige Ergebnisse für alle Substanzklassen 11. Vorlesung Modern Methods in Drug Discovery WS05/06 23 Moleküleigenschaften aus semiempirischen QM-Rechnungen (I) Im Gegensatz zu ab initio Rechnungen wurden die semiemprischen Methoden MNDO, AM1 und PM3 an experimentellen Daten kalibriert: • Bildungswärmen • Molekülgeometrien (Bindungslängen, Winkel) • Dipolmomente • Ionisationspotentiale Dadurch sind die Ergebnisse von semiempirischen Methoden bei diesen Eigenschaften oft denen von ab initio Rechnungen auf niedrigem Niveau (mit vergleichbatem Rechenaufwand) überlegen. 11. Vorlesung Modern Methods in Drug Discovery WS05/06 24 Bildungswärmen Berechnung der Bildungswärmen von Verbindungen bei 25° C H f o ( Molekül ) E elec ( Molekül ) Atome E elec ( Atome) Atome H fo ( Atome) Atomisierungs Bildungswärmen energien der Elemente Experimentell bekannt Berechnet werden muss also die elektronische Energie O H O Atomisierung H 11. Vorlesung H Eelec(Molekül) H Modern Methods in Drug Discovery WS05/06 O H H 25 Vergleich der Methoden Berechnung der Bildungswärmen von Verbindungen bei 25° C Durchschnittlicher absoluter Fehler (in kcal/mol) Anzahl Verbindungen Methode (C, H, N, O, sowie) MNDO AM1 PM3 Al (29) 22.1 10.5 16.4 4.9 Si (84) 12.0 8.5 6.0 6.3 P (43) 38.7 14.5 17.1 7.6 S (99) 48.4 10.3 7.5 5.6 Cl (85) 39.4 29.1 10.4 3.9 Br (51) 16.2 15.2 8.1 3.4 I (42) 25.4 21.7 13.4 4.0 Zn (18) 21.0 16.9 14.7 4.9 Hg (37) 13.7 9.0 7.7 2.2 Mg (48) 9.3 15.4 12.0 9.3 11. Vorlesung Modern Methods in Drug Discovery WS05/06 MNDO/d 26 Neue Semiempirische Methoden seit 1995 MNDO/d Thiel & Voityuk J.Phys.Chem. 100 (1996) 616 Erweitert die MNDO-Methode um d-Obitale und ist “kompatibel” mit den bereits für MNDO parametrisierten Atomen PM3(tm), PM5 d-Orbitale für Elemente der Übergangsreihen (Transition Metals) SAM1 Semi ab initio Method 1 Bestimmte Integrale werden konsequent berechnet, deshalb auch für Übergangsmetalle (Cu, Fe) anwendbar AM1* Winget, Horn et al J.Mol.Model. 9 (2003) 408. d-Orbitale für Elemente ab der 3. Periode (P,S, Cl) 11. Vorlesung Modern Methods in Drug Discovery WS05/06 27 Elektronische Moleküleigenschaften (I) Neben der Struktur von Molekülen lassen sich auch alle elektronischen Eigenschaften berechnen. Viele davon ergeben sich als Antwort des Moleküls auf eine äußere Störung: Entfernen eines Elektrons Ionisationspotential Allgemein läßt sich eine Störung durch ein äußeres Feld in einer Taylor Reihe entwickeln. Im Falle eines äußeren elektrischen Feldes F erhält man ein induziertes Dipolmoment ind: ind o F 12 F 2 ... o permanentes Dipolmoment des Moleküls Polarisierbarkeit (erste) Hyperpolarisierbarkeit 11. Vorlesung Modern Methods in Drug Discovery WS05/06 28 Elektronische Moleküleigenschaften (II) Auswahl von Eigenschaften die sich aus den n-ten Ableitungen der Energie nach äußeren Feldern berechnen lassen Elektr. Magn. K.Spin Koord. Eigenschaft 0 1 0 0 0 2 3 0 1 1 0 0 0 0 1 0 0 0 0 0 0 1 0 1 11. Vorlesung 0 0 0 1 0 0 0 0 0 0 2 1 0 0 0 0 1 0 0 2 1 0 0 0 Energie Elektrisches Dipolmoment Magentisches Dipolmoment Hyperfeinkopplungskonstanten Gradient der Energie (Optimierung) Elektrische Polarisierbarkeit (erste) Hyperpolarisierbarkeit harmonische Schwingungen (IR) IR Absorptionsintensitäten Circularer Dichroismus (CD) Kernspin-Kopplung (J) Kernmagnetische Abschirmung Modern Methods in Drug Discovery WS05/06 29 Molekulares Elektrostatisches Potential (I) Durch die Kerne Z und Elektronen i eines Moleküls entsteht eine Ladungsverteilung im Raum. An jedem beliebigen Punkt r kann man das dadurch enstehende Potential V(r) bestimmen: VESP r Kerne A ri 2 ZA dri r RA r ri Während der Kernanteil lediglich die Ladungen der Kerne enthält, ist für den elektronischen Teil eine Wellenfunktion notwendig. Zur Erinnerung: In Kraftfeldern benutzt man Atomladungen (auf den Atomen) die die elektrischen Multipole wiedergeben 11. Vorlesung Modern Methods in Drug Discovery WS05/06 30 Molekulares Elektrostatisches Potential (II) Zur Bestimmung des MEP an einem Punkt r ersetzt man in der Praxis die Integration durch eine Summation über hinreichend kleine Volumenelemente. Zur Visualisierung gibt man das MEP beispielsweise auf der van der Waals Oberfläche an Eine weitere Möglichkeit ist die Darstellung von Oberflächen mit jeweils gleichem Potential (Isocontur) Aus: A. Leach, Molecular Modelling, 2nd ed. 11. Vorlesung Modern Methods in Drug Discovery WS05/06 31 Molekulares Elektrostatisches Potential (III) Die Kenntnis dieser Oberflächenladungen ermöglich ihrerseits die Bestimmung von Atomladungen (z.B. für Kraftfelder) ESP derived atomic charges Diese müssen wiederum die elektrischen Multipole wiedergeben (iteratives Verfahren) Literatur: Cox & Williams J.Comput.Chem. 2 (1981) 304 Bieneman & Wiberg J.Comput.Chem. 11 (1990) 361 CHELPG Verfahren Singh & Kollman J.Comput.Chem. 5 (1984) 129 RESP Verfahren Ladungen für AMBER Kraftfeld 11. Vorlesung Modern Methods in Drug Discovery WS05/06 32 Quantenmechanische Deskriptoren (Auswahl) Atomladungen (partial atomic charges) Keine Observablen ! Mulliken Populationsanalyse Electrostatic potential (ESP) derived charges E Dipolmoment LUMO Polarisierbarkeit HOMO HOMO / LUMO (eV) der Grenzorbitale WienerJEnergien (Pfad Nummer) Covalent hydrogen bond acidity/basicity Donor Differenz der HOMO/LUMO Energien zu Wasser H-Brücken Eigenschaften Akzeptor Lit: M. Karelson et al. Chem.Rev. 96 (1996) 1027 11. Vorlesung Modern Methods in Drug Discovery WS05/06 33 Moleküleigenschaften aus semiempirischen Methoden (II) Welche Methode für welchen Zweck ? Nur strukturelle Eigenschaften (Molekülgeometrie): PM3 v.a. bei NO2-Verbindungen Elektronische Eigenschaften: MNDO bei halogenhaltigen Verbindungen (Cl, Br, I) AM1 bei hypervalenten Elementen, H-Brücken Die gleichen Deskriptoren aus unterschiedlichen Methoden sind nicht direkt vergleichbar ! 11. Vorlesung Modern Methods in Drug Discovery WS05/06 34 Beispiele für dir Vorhersage von Moleküleigenschaften Deskriptoren aus semiempirischen Methoden und allgemein gebräuchliche Variablen Klassifikationsverfahren mittels Entscheidungsbäumen Vorteil: Oft mehr exper. Daten verfügbar als bei quantitativen QSAR Meßgrößen • in vitro Mutagenizität von MX-Verbindungen • CNS Permeabilität von Substanzen • QT-Intervall Verlängerung (hERG-Kanal Inhibitoren) 11. Vorlesung Modern Methods in Drug Discovery WS05/06 35 Quantum QSAR Generierung der molekularen Eigenschaften für die QSARGleichung aus quantenmechanischen Daten. Bsp: Mutagenizität von MX-Verbindungen Cl2CH Cl R2 H R1 H O ln TA100 [ revertants/nmol ] O HO 10 O R3 O 8 6 O O 4 2 0 -2 ln(TA100) = -13.57 E(LUMO) –12.98 ; r = 0.82 -4 -6 -8 -0.3 -0.5 -0.7 -0.9 -1.1 -1.3 -1.5 E(LUMO) AM1 calculation [ eV ] Lit.: K. Tuppurainen et al. Mutat. Res. 247 (1991) 97. 11. Vorlesung Modern Methods in Drug Discovery WS05/06 36 CNS Permeability 95% CNS– 91% CNS+ 82% hlsurf 99% 72% vxbal 99% qsum+ qsumo ar5 96% 96% 11. Vorlesung 89% qsum+ qsum+ 100% 100% 100% 100% 88% qsum– 100% pcgc 100% qsum– mpolar 79% 100% 89% 77% cooh 83% dipdens 89% qsum– 80% hbdon size & shape 99% qsum+ 86% dipm electrostatic H-bonds mde34 kap3a 92% 100% 100% 100% 100% 100% 100% 100% mde13 Modern Methods in Drug Discovery WS05/06 100% 94% 37 Decision tree for QT-prolonging drugs 89% size & shape 88% electrostatic 75% 73% H-bonds hacsurf 86% 71% 99% QT+ QT– t1e 88% Level of accuracy in % 11. Vorlesung MR mde23 100% 100% 100% logP 100% 100% qsumn 100% 100% MR 100% 92% 83% MR 100% t2e 99% 93% MR 100% 96% mpolar sgeca 95% 87% dipdens 82% 89% 96% 89% SMARTS 90% 100% chbba hlsurf hy mghbd logP MR Modern Methods in Drug Discovery WS05/06 qsumn 96% 100% 100% 100% 100% 38 Common structural features of QT-prolonging drugs F NH2 Cl F O H N Cl N O HO O N N H OH N O N O N N N HO N O N Astemizole N O N N O F H Sertindole F Terfenadine Cisapride H Grepafloxacin Derived common substructure expressed as SMARTS string 11. Vorlesung Modern Methods in Drug Discovery WS05/06 39 Moleküleigenschaften aus Kraftfeldern Prinzipbedingt weisen Kraftfelder eine noch stärkere Abhängigkeit von der Parametrisierung auf. Dadurch lassen sich eigentlich nur Voraussagen über die Struktur ( Docking) und, in begrenztem Umfang, über Schwingungsspektren (IR) machen Aufgrund des geringen Rechenaufwandes sind Kraftfelder aber sehr gut geeignet, um Konformationssuchen durchzuführen. 4D-QSAR (verschiedene gedockte Konformationen, z.B. in Cytochrom P450) 11. Vorlesung Modern Methods in Drug Discovery WS05/06 40 Moleküleigenschaften aus Moleküldynamik Simulationen Bindungsaffinitäten, genauer gesagt freie Bindungsenergien G von Liganden an Enzyme aus Free Energy Perturbation Rechnungen Vorteil: relativ genaue Vorhersagen Nachteil: sehr rechenzeitaufwenig, daher nur wenige Liganden durchführbar Lit.: A.R. Leach Molecular Modelling, Longman. 11. Vorlesung Modern Methods in Drug Discovery WS05/06 41