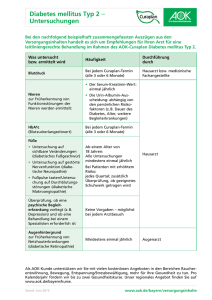
Dissertation D.P. Hoyer
Werbung

Aus dem Zentrum für Innere Medizin der Universität zu Köln Klinik III für Innere Medizin Direktor: Universitätsprofessor Dr. med. E. Erdmann Bedeutung der G-Proteine Gα11 und Gαq für die Entstehung der Myokardhypertrophie bei Typ-1-Diabetes Inaugural-Dissertation zur Erlangung der Doktorwürde der Hohen Medizinischen Fakultät der Universität zu Köln vorgelegt von Dieter Paul Hoyer aus Hannover Promoviert am 13. Januar 2010 Aus dem Zentrum für Innere Medizin der Universität zu Köln Klinik III für Innere Medizin Direktor: Universitätsprofessor Dr. med. E. Erdmann Bedeutung der G-Proteine Gα11 und Gαq für die Entstehung der Myokardhypertrophie bei Typ-1-Diabetes Inaugural-Dissertation zur Erlangung der Doktorwürde der Hohen Medizinischen Fakultät der Universität zu Köln vorgelegt von Dieter Paul Hoyer aus Hannover Promoviert am 13. Januar 2010 Gedruckt mit der Genehmigung der Medizinischen Fakultät der Universität zu Köln, 2010 Dekan: Universitätsprofessor Dr. med. J. Klosterkötter 1. Berichterstatter: Privatdozent Dr. med. H. Reuter 2. Berichterstatter: Universitätsprofessor Dr. med. W. Bloch Erklärung: Ich erkläre hiermit, dass ich die vorliegende Arbeit ohne unzulässige Hilfe Dritter und ohne Benutzung anderer als der angegebenen Hilfsmittel angefertigt habe; die aus fremden Quellen direkt oder indirekt übernommenen Gedanken sind als solche kenntlich gemacht. Bei der Auswahl und Auswertung des Materials sowie bei der Herstellung des Manuskripts habe ich Unterstützungsleistungen von folgenden Personen erhalten: Privatdozent Dr. med. Jochen Müller-Ehmsen, Leiter des Labors für Herzmuskelphysiologie und molekulare Kardiologie, Klinik III für Innere Medizin der Universität zu Köln. Privatdozent Dr. med. Hannes Reuter, wissenschaftlicher Mitarbeiter im Labor für Herzmuskelphysiologie und molekulare Kardiologie, Klinik III für Innere Medizin der Universität zu Köln. Weitere Personen waren an der geistigen Herstellung der vorliegenden Arbeit nicht beteiligt. Insbesondere habe ich nicht die Hilfe eines Promotionsberaters in Anspruch genommen. Dritte haben von mir weder unmittelbar noch mittelbar geldwerte Leistungen für Arbeiten erhalten, die im Zusammenhang mit dem Inhalt der vorgelegten Dissertation stehen. Die Arbeit wurde von mir bisher weder im Inland noch im Ausland in gleicher oder ähnlicher Form einer anderen Prüfungsbehörde vorgelegt und ist auch noch nicht veröffentlicht. Köln, den 22. Februar 2009 Dieter P. Hoyer Die in dieser Arbeit angegebenen Experimente sind nach entsprechender Anleitung durch Herrn Priv.-Doz. Dr. med. Hannes Reuter von mir selbst durchgeführt worden. Danksagung Herrn Professor Dr. med. E. Erdmann danke ich für die großzügige Bereitstellung der Rahmenbedingungen, die diese Arbeit erst ermöglicht haben. Weiterhin gilt mein Dank Herrn Priv.-Doz. Dr. med. Jochen Müller-Ehmsen, Leiter des Labors für Herzmuskelphysiologie und molekulare Kardiologie. Mit freundlicher und konstruktiver Kritik war er stets ein wertvoller Ansprechpartner. Meinen außerordentlichen Dank möchte ich Herrn Priv.-Doz. Dr. med. Hannes Reuter aussprechen. Mit Überlassung dieses Themas ermöglichte er mir, wissenschaftliches Denken und Arbeiten zu erlernen. Durch sachkundige Unterstützung und überaus kompetente, sowie freundliche Betreuung lieferte er mir wichtige Informationen zur Durchführung der Experimente und war ein exzellenter Ansprechpartner im Rahmen wissenschaftlicher Diskussionen. Danken möchte ich Herrn Dr. med. dent. Yüksel Korkmaz, der für die immunhistochemischen Experimente ein wertvoller Mentor war. Den medizinisch-technischen Assistentinnen, Frau Katja Rösler und Frau Esra Koroglu, habe ich für ihre fachliche Unterstützung zu danken. Für Karin und Peter Heike, Felix, Johanna, Cornelia und Katja Inhaltsverzeichnis Verzeichnis der verwendeten Abkürzungen ......................................................... IV 1 Einleitung ........................................................................................................... 1 1.1 Untersuchungen zum Diabetes mellitus und diabetesbedingter Komplikationen am Kleintiermodell nach Streptozotocin-Injektion ................................................... 2 1.2 Bedeutung der G-Proteine Gαq/Gα11 für die Entstehung diabetesbedingter Komplikationen am Herzen ..................................................................................... 3 1.3 Evidenz zur Bedeutung Gαq/Gα11-unabhängiger Mechanismen bei der Entstehung diabetesbedingter Komplikationen am Herzen..................................... 6 1.4 Arbeiten zum Modell der Gαq/Gα11 Doppelknockout Maus.............................. 9 1.5 Proteinkinase C.............................................................................................. 10 1.6 Funktion und Bedeutung von Proteinen der Calcium-induzierten-CalciumFreisetzung und der Ca2+-Homöostase................................................................. 11 1.6.1 Ryanodin-Rezeptor Typ 2 ................................................................... 11 1.6.2 Sorcin .................................................................................................. 12 1.6.3 FKBP12.6............................................................................................ 13 1.6.4 Annexin VII.......................................................................................... 14 1.6.5 Natrium-Calcium-Austauscher ............................................................ 14 1.6.6 Calsequestrin ...................................................................................... 15 1.7 Fragestellungen ............................................................................................. 16 2 Material & Methoden ....................................................................................... 18 2.1 Materialien ..................................................................................................... 18 2.1.1 Versuchstiere ...................................................................................... 18 2.1.2 Verwendete Substanzen und Lösungen ............................................. 18 2.1.2.1 Tierexperimentelle Methoden....................................................... 18 2.1.2.2 DNA-Isolation............................................................................... 18 2.1.2.3 PCR ............................................................................................. 19 2.1.2.4 Herstellung von Homogenaten für Western Blot-Untersuchungen20 2.1.2.5 Bestimmung der Proteinkonzentration ......................................... 20 2.1.2.6 SDS-Gelelektrophorese und Western Blots ................................. 20 2.1.2.7 Quantitative Bestimmung des Ca2+-Verlustes an isolierten Membranpartikeln .......................................................................................... 21 2.1.2.8 Immunhistochemie ....................................................................... 21 2.2 Methoden....................................................................................................... 23 2.2.1 Tierexperimentelle Methoden.............................................................. 23 I Inhaltsverzeichnis 2.2.1.1 Induktion eines Diabetes Mellitus................................................. 23 2.2.1.2 Bestimmung der Blutzuckerwerte und des Körpergewichtes ....... 24 2.2.1.3 Opferung der Tiere und Organentnahme ..................................... 24 2.2.1.4 Echokardiographische Bestimmung funktioneller und morphologischer Veränderungen am Herzen in vivo ..................................... 24 2.2.2 Genotypisierung der Mäuse ................................................................ 26 2.2.2.1 DNA Isolation ............................................................................... 26 2.2.2.2 Polymerase-Kettenreaktion (PCR)............................................... 26 2.2.3 Untersuchungen zur Proteinexpression .............................................. 29 2.2.3.1 Herstellung von Homogenaten für Western Blot-Untersuchungen29 2.2.3.2 Bestimmung der Proteinkonzentration ......................................... 29 2.2.3.3 SDS-Gelelektrophorese ............................................................... 29 2.2.3.4 Western Blot ................................................................................ 30 2.2.4 Untersuchungen zur Funktion ............................................................. 31 2.2.4.1 Herstellung von Homogenaten und Bestimmung der Proteinkonzentration für die quantitative Bestimmung des Ca²+-Verlustes an isolierten Membranvesikeln ........................................................................... 31 2.2.4.2 Quantitative Bestimmung des Ca²+-Verlustes an isolierten Membranvesikeln........................................................................................... 31 3 2.2.5 Immunhistochemie .............................................................................. 32 2.2.6 Statistische Methoden......................................................................... 34 Ergebnisse ....................................................................................................... 35 3.1 Charakterisierung der Versuchstiere.............................................................. 35 3.2 Genotypisierung der Mäuse........................................................................... 40 3.3 Echokardiographische Bestimmung funktioneller und morphologischer Veränderungen am Herzen in vivo........................................................................ 41 3.4 Untersuchungen der Proteinexpression......................................................... 43 3.4.1 Expression des Ryanodinrezeptors Typ 2........................................... 44 3.4.2 Phosporylierung des RYR2 an Serin 2809.......................................... 46 3.4.3 Expression des Sorcin ........................................................................ 48 3.4.4 Expression des FKBP12.6 .................................................................. 50 3.4.5 Expression des Annexin VII ................................................................ 52 3.4.6 Expression des Na+-Ca2+-Austauschers ............................................. 54 3.5 Quantitative Bestimmung des Ca²+-Verlustes an isolierten Membranvesikeln57 II Inhaltsverzeichnis 3.6 Immunhistochemie......................................................................................... 59 3.6.1 Immunhistochemische Ergebnisse zum Einfluss eines Typ1-Diabetes auf die kardiale Expression und Translokation von PKC-Isoformen bei Wildtypmäusen.................................................................................................. 59 3.6.2 Immunhistochemische Ergebnisse zum Einfluss eines Typ1-Diabetes auf die kardiale Expression und Translokation von PKC-Isoformen bei Gα11 Knockoutmäusen............................................................................................... 63 4 Diskussion ....................................................................................................... 71 4.1 Das G-Protein Gα11 ist wesentlich an der Entwicklung einer Hypertrophie des Herzens bei Typ1-Diabetes beteiligt ..................................................................... 71 4.2 Die Signalkaskade der pathophysiologischen Veränderungen bei Typ1Diabetes weisen Hinweise auf eine Beteiligung der Proteinkinas C auf ............... 73 4.3 Die Struktur und die Funktion des Proteinkomplexes der Ca2+-Freisetzung zeigt eine Beeinflussung durch einen Typ1-Diabetes und die G-Proteine Gα11 und Gαq 77 4.4 Klinische Bedeutung der vorliegenden Arbeit ................................................ 80 5 Zusammenfassung.......................................................................................... 81 6 Literaturverzeichnis ........................................................................................ 84 7 Lebenslauf ....................................................................................................... 91 III Verzeichnis der verwendeten Abkürzungen Verzeichnis der verwendeten Abkürzungen ACE Angiotensin-Converting-Enzyme AGEs Advanced Glycation End Products AT1 Angiotensin-II-Rezeptor Typ 1 BSA Bovines Serumalbumin CaMK Calmodulin-abhängige Kinase CICR Calcium-induzierte Calcium-Freisetzung DAG Diacylglycerol DE Densitometrische Einheiten DM Diabetes Mellitus DNA Desoxyribonukleinsäure FKBP12.6 FK506-Binding-Protein 12,6 kDa G-Proteine Guanosintriphosphat-abhängige Proteine HbA1c Hämoglobin A1c IP3 Inositol-1,4,5-triphosphat JNK c-Jun N-terminale Kinase MAPK Mitogen-aktivierte Proteinkinase mRNA messenger-Ribonukleinsäure NCX Natrium-Calcium-Austauscher ND Nicht diabetisierte Tiere NGS Normal Goat Serum PBS Phosphatpuffer-Natriumchlorid PKA Proteinkinase A PKC Proteinkinase C RAAS Renin-Angiotensin-Aldosteron-System RR Ruthenium Rot RT Raumtemperatur RYR1 Ryanodin-Rezeptor-Typ1 RYR2 Ryanodin-Rezeptor-Typ2 SDS Natriumduodecylsulfat SERCA Ca2+-ATPase des Sarkoplasmatischen-Retikulums SR Sarkoplasmatisches Retikulum STZ Streptozotocin Taq-DNA-Polymerase Thermus aquaticus Polymerase IV Einleitung 1 Einleitung Die Zuckerkrankheit, Diabetes mellitus, ist eine Volkskrankheit mit zunehmender Prävalenz in unserer Gesellschaft. Aktuellen Schätzungen zufolge wird sich die heutige Zahl von weltweit 150 Millionen Betroffenen bis zum Jahr 2025 auf 300 Millionen Diabetiker verdoppeln (69). Dies hat weitreichende volkswirtschaftliche Folgen und muss medizinisch als Herausforderung in der Prävention kardiovaskulärer Erkrankungen aufgefasst werden. Unter dem Begriff Diabetes mellitus werden chronische Stoffwechselkrankheiten zusammengefasst, die durch das Leitsymptom der Hyperglykämie charakterisiert sind und zu typischen Komplikationen, dem so genannten diabetischen Spätsyndrom, führen können. Generell können durch einen langjährigen Diabetes mellitus alle Organsysteme geschädigt werden. Als häufige Manifestationen des diabetischen Spätsyndroms ist neben der Vaskulopathie, Nephropathie und Neuropathie die Schädigung des Herzmuskels zu nennen. Viele dieser Komplikationen lassen sich pathophysiologisch als Folge von Gefäßveränderungen, der diabetischen Mikroangiopathie und systemischen Gefäßsklerose, erklären. Eine Besonderheit im klinischen Bild des herzkranken Diabetikers besteht jedoch in der häufigen Vergesellschaftung von kongestiver Herzmuskelinsuffizienz mit fehlender oder nur gering ausgeprägter Koronarsklerose (27, 103). Diabetiker zeigen eine Häufung von Herzinsuffizienz, die im Vergleich zu anderen Patienten auch dann noch gehäuft nachweisbar ist, wenn die Einflüsse von Alter, Blutdruck, Gewicht, Cholesterinwerten und koronarer Herzkrankheit ausgeglichen werden. Bei einer Diabetes-Prävalenz von ca. 6% in der Bevölkerung macht zudem der deutlich erhöhte Anteil an Diabetikern in verschiedenen Herzinsuffizienz-Studien, wie z.B. in der SOLVD-Studie (26%) (119) oder der ATLAS-Studie (19%) (112), die pathophysiologische Bedeutung des Diabetes mellitus für diese Erkrankung deutlich. Diese Beobachtungen unterstützen die Annahme, dass bei Diabetes mellitus direkte zelluläre Veränderungen für die Entwicklung der Herzinsuffizienz von Bedeutung sind und die diabetische Kardiomyopathie eine eigene Entität im Formenkreis der Herzinsuffizienzen darstellt (109). Dabei konnte für beide Formen der Zuckerkrankheit, sowohl dem Insulin-abhängigen Typ 1-Diabetes als auch dem primär nicht Insulin-abhängigen Typ 2-Diabetes die frühzeitige Entwicklung einer diastolischen Dysfunktion und ventrikulären Hypertrophie nachgewiesen werden, der 1 Einleitung im Spätstadium eine systolische Pumpfunktionsstörung folgt (27, 103, 129). Wie im Folgenden näher erläutert wird, kommen ursächlich für die gestörte Kontraktilität des Herzens bei Diabetes mellitus Veränderungen u.a. des Myokardstoffwechsels, der extrazellulären Matrix und der elektromechanischen Kopplung in Betracht. 1.1 Untersuchungen zum Diabetes mellitus und diabetesbedingter Komplikationen am Kleintiermodell nach StreptozotocinInjektion Das Modell des experimentellen Diabetes nach Streptozotocin-Injektion ist eines der am besten untersuchten Tiermodelle zur Identifikation von Veränderungen der Signaltransduktion bei Zuckerkrankheit auf zellulärer Ebene. Streptozotocin (STZ) ist ein Zytotoxin, bestehend aus einem Glukosemolekül mit einer NitrosoharnstoffSeitenkette(38). Dieses Glukosemolekül scheint von Bedeutung für die Anreicherung des STZ in den Inselzellen des Pankreas zu sein, die sodann durch die hochreaktive Nitrosoharnstoff-Seitenkette zerstört werden. Verschiedene Kleintiere wie Ratten und Mäuse zeigen nach Injektion von STZ innerhalb weniger Tage eine ausgeprägte Hyperglykämie (25 bis 30 mmol/l), eine mäßige Hypertriglyzeridämie (200 bis 400 µmol/l) und Ketose bei stark reduzierten Insulinspiegeln (<20 pmol/l) (34, 129). Ähnlich dem Krankheitsbild des Menschen entwickeln diese Tiere zunächst eine diastolische Dysfunktion mit verzögerter isovolumetrischer Relaxation (43, 62). Im Verlauf weniger Wochen entwickelt sich daraus eine manifeste Herzinsuffizienz. Am Rattenmodell des STZ-induzierten Diabetes konnten weder Zeichen des Bluthochdrucks noch der Atherosklerose nachgewiesen werden, so dass die resultierende Kardiomyopathie primär als Folge des Diabetes angesehen werden muss. Es ist wissenschaftlich belegt, dass die gestörte Kontraktilität bei Herzinsuffizienz unterschiedlicher Ätiologie wesentlich durch eine gestörte intrazelluläre Ca2+-Homöostase, strukturelle Veränderungen, u.a. mit Veränderungen der extrazellulären Matrix und einer erhöhten Apoptoserate beeinflusst wird (134). Dies konnte in mehreren Untersuchungen auch am Tiermodell der diabetischen Ratte nach STZ-Injektion nachgewiesen werden. Zu unterschiedlichen Zeitpunkten nach Diabetes-Induktion zeigten sich dabei spezifische Veränderungen von Expression und Funktion Ca2+-regulierender Proteine des Sarkoplasmatischen Retikulums (SR) und der Plasmamembran. So bestand bereits in der frühen Phase (4 - 6 Wochen nach Diabetes-Induktion) eine 2 Einleitung verminderte Ca2+-Aufnahme in das SR durch vermehrte Expression und verminderte Phosphorylierung des Regulatorproteins Phospholamban (137, 151), sowie eine verminderte Expression und Funktion der Ca2+-ATPase des SR (SERCA-2) (138, 151) und des SR Ca2+-Freisetzungskanals (Ryanodin Rezeptor) (8, 151). Diese Veränderungen stellen wahrscheinlich eine wesentliche Ursache für die Abnahme des Ca2+-Gehaltes im SR bei Diabetes mellitus dar (58). Bei Diabetes mellitus kommt es durch die Hyperglykämie und eine vermehrte Stimulation von Angiotensin II-Rezeptoren auch zur Bildung reaktiver Sauerstoffgruppen und einer erhöhten Apoptoserate. Dies wurde von mehreren Arbeitsgruppen u.a. am Myokard der diabetischen Ratte nach STZ-Injektion beschrieben und ist als eine weitere wesentliche Ursache für die Entstehung der kontraktilen Dysfunktion und struktureller Veränderungen bei Diabetes mellitus zu berücksichtigen (37, 66, 102). Zudem beeinflussen Veränderungen der extrazellulären Matrix, insbesondere in der Zusammensetzung der Kollagene, bei Diabetes mellitus wesentlich die mechanischen Eigenschaften des Herzmuskels und sind daher von entscheidender Bedeutung für die Hämodynamik des Herzens (12, 131, 135). Für die Entstehung dieser diabetesbedingten Folgekrankheiten am Herzen könnten sowohl Rezeptor-vermittelte Mechanismen über die G-Proteine Gαq und Gα11, als auch Mechanismen über den veränderten Glukosemetabolismus bei Hyperglykämie verantwortlich sein. Es ist nach wie vor unklar, welchen Stellenwert diese konkurrierenden Mechanismen für die Pathogenese des Diabetes am Herzen haben. Die folgenden Kapitel fassen den aktuellen Stand der Forschung zu diesem Thema zusammen. 1.2 Bedeutung der G-Proteine Gα αq/Gα α11 für die Entstehung diabetesbedingter Komplikationen am Herzen Angiotensin II (Ang II) ist ein inotropes Hormon, das durch Induktion von Hypertrophie und Fibrose eine zentrale Bedeutung für das kardiovaskuläre Remodeling hat, z.B. unter Druckbelastung oder nach Myokardinfarkt (104). Auch bei Patienten mit Diabetes mellitus wurden erhöhte Serumspiegel von Ang II gemessen (56, 136). Auch konnte eine erhöhte Ang II-Rezeptorendichte im Myokard von Diabetikern nachgewiesen werden (105). Zudem zeigte sich u.a. am STZ-Modell der diabetischen Ratte unter Angiotensin-Rezeptorblockade eine signifikante 3 Einleitung Verbesserung sowohl der systolischen als auch der diastolischen Pumpfunktion (78, 98, 102). Am Menschen haben mehrere große klinische Studien darüber hinaus eine signifikante Reduktion kardiovaskulärer Endpunkte, wie Myokardinfarkt, Schlaganfall und kardiovaskulärem Tod, unter Blockade des Renin-Angiotensin Systems (RAS) insbesondere bei Patienten mit Diabetes mellitus zeigen können (48, 57, 73). Ang II aktiviert Rezeptoren an der myokardialen Zelloberfläche, die an die G-Proteine der Gαq-Familie gekoppelt sind. Neben Ang II vermitteln auch andere Hormone, wie z.B. Endothelin und Noradrenalin ihre Wirkung über Gαq. Gαq ist ein übergeordneter Begriff, der die Isoformen Gαq, Gα11, Gα14 und Gα15/16 umfasst. Gαq und Gα11 werden ubiquitär exprimiert, wohingegen Gα14 und Gα15/16 nur in wenigen Geweben, nicht jedoch im Herzen nachgewiesen werden können (4, 142). Die Aktivierung sog. Mitogen-aktivierter Proteinkinasen (MAPKs) und die Stimulation der Phospholipase C zur Synthese von Diacylglycerol (DAG) und Inositol-1,4,5-triphosphat (Ins[1,4,5]P3) sind die zentralen Effektoren der Signalkaskade über Gαq und Gα11 (Abbildung 1;(51)). DAG ist ein direkter Aktivator verschiedener Isoformen der Proteinkinase C (PKC). Ins[1,4,5]P3 vermittelt die Freisetzung von Ca2+ aus intrazellulären Speichern, das wiederum zur Stimulation der Phosphatase Calcineurin und der Calmodulinabhängigen Kinase (CaM-Kinase) benötigt wird. PKC, MAPKs, Calcineurin und CaMKinase stellen nach heutigem Kenntnisstand die wesentlichen Signalwege dar, über die die G-Proteine Gαq und Gα11 zur Induktion von Hypertrophie und Fibrose führen können (51, 126). Abbildung 1 fasst schematisch die Signalwege zusammen, über die o.g. Mediatoren nukleäre Transkriptionsfaktoren aktivieren und das Genexpressionsmuster der Herzmuskelzelle anpassen können. 4 Einleitung Sarkolemm GPCR Gαq/α11 PLC Ins[1,4,5]P3 DAG MAPK Kinasen Ca2+ Calcineurin Calmodulin/CaMK PKC JNK ERK p38 Nukleus T r a n s k r i p t i o n s f a k t o r e n (NFAT, HDAC 4/5/7/9, u.a.) Abbildung 1 Schematische Darstellung wesentlicher Signaltransduktionswege, über die eine Stimulation der G-Proteine Gαq/Gα11 nukleäre Transkriptionsfaktoren aktivieren kann. Es gibt Hinweise, dass diese Signalwege über ein verändertes Genexpressionsmuster die Entstehung von Hypertrophie und Herzinsuffizienz begünstigen können. GPCR: G-Protein gekoppelter Rezeptor, PLC:Phospholipase C, Ins[1,4,5]P3: Inositol-1,4,5triphosphat, DAG: Diacylglycerol, MAPK: Mitogen-aktivierte Proteinkinase, PKC: Proteinkinase C, JNK: c-Jun N-terminale Kinase, ERK: extrazellulär regulierte Kinase, CaMK: Calmodulin-abhängige Kinase. Modifiziert nach Heineke und Molkentin (51). Eine vermehrte Stimulation von Angiotensin II (Typ 1)-Rezeptoren (AT1-Rezeptoren) führt zu einer gesteigerten Expression von Gαq. Entsprechend konnte auch in diabetischen Ratten nach STZ-Injektion eine erhöhte Gαq-Dichte in der myozytären Plasmamembran gezeigt werden (146). Auch transgene Gαq-überexprimierende Mäuse entwickeln den Phänotyp einer kardialen Hypertrophie mit kontraktiler Dysfunktion und gestörten sarkolemmalen und sarkoplasmatischen Ca2+-Strömen (25, 149). Andere Arbeitsgruppen konnten eine gesteigerte Aktivität und Expression der PKC als primären Effektor der Signalkaskade über Gαq und Gα11 in diabetischen Ratten nach STZ-Injektion zeigen (46, 70, 146), die durch Ang II zusätzlich stimuliert werden konnte (80). Diese Ergebnisse unterstützen die viel zitierte Hypothese, dass u.a. Ang II und Endothelin über Gαq und Gα11 einen wesentlichen Einfluss auf die 5 Einleitung Entstehung der diabetischen Kardiomyopathie und anderer diabetischer Komplikationen haben können. Entsprechend sind ACE-Hemmer oder AT1Rezeptorantagonisten heute fester Bestandteil einer leitliniengerechten Therapie zur kardiovaskulären und renalen Protektion bei Patienten mit Diabetes mellitus (106). 1.3 Evidenz zur Bedeutung Gα αq/Gα α11-unabhängiger Mechanismen bei der Entstehung diabetesbedingter Komplikationen am Herzen Neuere Untersuchungen legen die Vermutung nahe, dass der Nutzen einer Behandlung mit ACE-Hemmern nicht allein auf die Blockade des RAS zurückzuführen ist. So konnte unter langfristiger Therapie mit ACE-Hemmern auch in maximaler Dosierung eine Reaktivierung von Ang II im Serum von Patienten mit Herzinsuffizienz nachgewiesen werden (63, 77). Auch ohne Therapie mit ACEHemmern werden bis zu 40% des Ang II unabhängig des Angiotensin converting enzyme (ACE) gebildet und diese alternativen Synthesewege sind bei Diabetes mellitus möglicherweise sogar gesteigert (110). Eine komplette Blockade des RAS scheint somit durch ACE-Hemmer nicht möglich zu sein. Eine Hemmung der Konversion von Ang I zu Ang II durch ACE-Hemmer führt zu einem verminderten Abbau von Bradykinin, einem Kinin, das eine teilweise antagonistische Wirkung zu Ang II vermittelt, u.a. mit verbesserter Koronarperfusion, Regression einer linksventrikulären Hypertrophie und Stimulation der myokardialen Glukoseaufnahme (44, 71, 152). Zudem haben Kinine antifibrotische Eigenschaften. In kultivierten Fibroblasten konnte unter Behandlung mit Bradykinin eine verminderte mRNA Expression von Fibronectin und der Kollagene I und III nachgewiesen werden(68). Entsprechend war die antifibrotische Wirkung einer ACE-Hemmer Therapie in Bradykinin-Rezeptor Knockout Mäusen nach Myokardinfarkt deutlich vermindert (147). Aufgrund dieser Ergebnisse wurde postuliert, dass die kardioprotektive Wirkung der ACE-Hemmer zumindest teilweise auf eine Erhöhung der Kinine zurückzuführen ist (26, 152). Strukturelle und funktionelle Veränderungen im Myokard und der extrazellulären Matrix bei Diabetes mellitus können aber auch direkt Folge der Hyperglykämie sein. Hyperglykämie führt zu einer vermehrten Glukoseoxidation und mitochondrialen Bildung von Superoxid (17, 35, 90). Superoxid wiederum schädigt die nukleäre DNA und aktiviert dadurch das Reparationsenzym Poly-ADP-Ribose-Polymerase (PARP) 6 Einleitung (31). PARP hemmt aber gleichzeitig die Phosphoglycerinaldehyd-Dehydrogenase (GAPDH), ein zentrales Enzym der Glykolyse, so dass Glukose vermehrt über alternative Stoffwechselwege metabolisiert werden kann. Diese alternativen Stoffwechselwege führen zur vermehrten Bildung fortgeschrittener Glykosylierungsendprodukte (Advanced Glycation Endproducts, AGEs), Aktivierung konventioneller Isoformen der PKC und vermehrter Bildung von Sorbitol mit Stimulation des Polyol-Signalweges (31, 90). Tabelle 1 fasst diese alternativen Wege des Glukosestoffwechsels bei Hyperglykämie und ihre Auswirkungen auf die strukturellen und funktionellen Eigenschaften des Herzens zusammen. Diese Zusammenfassung verdeutlicht, dass es anhand der aktuellen Datenlage unklar ist, inwieweit die strukturellen und funktionellen Veränderungen am diabetischen Herzen Folge einer Rezeptor-abhängigen Stimulation der G-Proteine Gαq/Gα11 sind oder Rezeptor-unabhängig über einen veränderten Glukosemetabolismus bei Hyperglykämie vermittelt werden. 7 Einleitung Mediatoren Strukturelle Veränderungen Konsequenz AGE-Bildung (9, Glykosylierung von Verminderte SR Ca2+- 10, 18, 54) Ryanodinrezeptoren Freisetzung und und der SERCA Kontraktilität Glykosylierung von Kollagen Typ III Versteifung der Ventrikel Verminderte ventrikuläre Füllung Vermehrte Apoptoserate Polyol Signalweg Gesteigerter Oxidativer Stress (31, 40, 90) Sorbitol-induzierte Bildung von AGEs Kontraktile Dysfunktion Vermehrte DNA-Strangbrüche Versteifung der Ventrikel Aktivierung der Kardiale Hypertrophie Gestörte Relaxation PKC Verminderte SERCA Aktivität Versteifung der Ventrikel (46, 70, 139) Vermehrte Bildung extrazellulärer Matrix Tabelle 1 Alternative Mechanismen einer Rezeptor-unabhängigen Schädigung des Herzmuskels induziert durch Hyperglykämie AGE: fortgeschrittene Glykosylierungsendprodukte, PKC: Proteinkinase C, SR: sarkoplasmatisches 2+ Retikulum, SERCA: Ca -ATPase des SR 8 Einleitung 1.4 Arbeiten zum Modell der Gα αq/Gα α11 Doppelknockout Maus Das Knockout Mausmodell wurde von der Arbeitsgruppe von Prof. Dr. S. Offermanns, Institut für Pharmakologie der Universität Heidelberg, erstellt und besteht aus einem globalen Gα11 Knockout und einem kardial-spezifischen konditionellen Gαq Knockout auf Basis der Cre/LoxP-Technologie (141). Die Expression der Cre-Rekombinase steht dabei unter Kontrolle des MLC2α-Promotors. Die Variante des konditionellen Gαq Knockout war gewählt worden, da Mäuse mit globalem Knockout beider Gene, Gαq und Gα11, im embryonalen Alter von 11 Tagen mit Zeichen der myokardialen Hypoplasie versterben (92). In einer ersten, hochrangig publizierten Arbeit an diesem Doppelknockout Mausmodell wurde der Einfluss der G-Proteine Gαq und Gα11 auf die Entwicklung der linksventrikulären Hypertrophie unter Druckbelastung durch aortale Konstriktion (aortic banding) untersucht (141). Dabei zeigte sich 14 Tage nach operativer Intervention an den Herzen der Tiere vom Wildtyp erwartungsgemäß eine ausgeprägte Hypertrophie mit Zunahme des Herz-Körpergewichtsquotienten um 58% und einer deutlichen Verschiebung der Druck-Volumen-Beziehung im Sinne einer linksventrikulären Dilatation und Compliancestörung. Unter identischen Bedingungen konnten jedoch an den Herzen der Gαq/Gα11 Doppelknockoutmäuse keine strukturellen oder funktionellen Veränderungen nach Aortenkonstriktion im Vergleich zu shamoperierten Tieren festgestellt werden. Die Unterbrechung der Gαq/Gα11Signaltransduktion durch Doppelknockout hatte somit wahrscheinlich einen protektiven Einfluss auf das kardiale Remodeling unter Druckbelastung. Bei Untersuchungen an genetisch veränderten Tiermodellen muss davon ausgegangen werden, dass in dem sehr komplexen zellulären Stoffwechsel Kompensationsmechanismen auftreten, die auch bei sorgfältiger Untersuchung unerkannt bleiben. Dies stellt generell ein methodisches Problem auch bei Knockout Modellen dar und muss bei der Interpretation der Ergebnisse berücksichtigt werden. 9 Einleitung 1.5 Proteinkinase C Die Mitglieder der Proteinkinase C-Familie spielen bei der hormonellen Signaltransduktion eine wichtige Rolle und sind u.a. in die Regulation von Zellwachstum und Zelldifferenzierung eingeschaltet (23, 74, 91). Derzeitig sind mindestens elf verschiedene Isoformen dieses Enzyms bekannt, die als α-, βI-, βII-, γ-, δ-, ε-, ζ-, η-, θ-, und ι-/λ- Subtypen bezeichnet werden, und differenziert in den unterschiedlichen Geweben exprimiert werden. (24, 32, 93, 96, 97, 99, 100, 117). Auf der Basis ihrer enzymatischen Eigenschaften sind die Säugetier-PKC-Isotypen in kleinere Unterfamilien unterteilt worden. Die am meisten untersuchte Gruppe ist die der konventionellen PKCs (cPKCs), die die Isotypen α, βI, βII und γ umfasst (das PKC β-Gen wird alternativ gespleißt) (24). Sie werden Ca2+-abhängig durch DAG aktiviert. Die neuen PKCs (nPKCs) bestehen aus den Isotypen ε, η, δ, und θ. Sie sind Ca2+-unabhängig, werden aber ebenfalls durch DAG aktiviert (95). Die atypischen PKCs (aPKCs) ι und ζ sind wie die nPKCs Ca2+-unabhängig und zusätzlich auch DAG-unabhängig (94). Die Aktivierung der PKC kann durch Autophosphorylierungen verstärkt werden (111). Man vermutet, dass zwischen der zytosolischen und der membrangebundenen PKC ein Gleichgewicht existiert, welches durch hydrophobe Cofaktoren wie Diacylglycerol in Richtung Membranassoziation verschoben wird und so bei den einzelnen Isoformen zur Translokation führt. Die kardiale Expression umfasst die Isoformen α, βI, βII, δ, ε, λ und ζ (45, 61, 121). Die Isoform θ zeigt neben der deutlichen Expression im Skelettmuskel auch eine schwächere Ausprägung im Herzen (97). Die Funktionen und Substrate der verschiedenen Isoformen der PKC sind letztendlich noch nicht geklärt, jedoch führt eine Aktivierung der kardialen PKC zu Zellwachstum und damit zu kardialer Hypertrophie, zu einer Veränderung der SR-Funktion u.a. über eine verminderte Aktivität der SERCA über Veränderungen am Phospholamban (3, 14), zur Phosphorylierung von Myofilamenten (144) und zur vermehrten Bildung von extrazellulärer Matrix. 10 Einleitung 1.6 Funktion und Bedeutung von Proteinen der Calciuminduzierten-Calcium-Freisetzung und der Ca2+-Homöostase Abbildung 2 Schematische Darstellung des Ryanodinkanals in der Membran des SR und seiner 2+ intrazellulären Interaktion. Nach Ca -Bindung kann Sorcin an den RYR2 binden und diesen 2+ 2+ blockieren. Durch Einflussnahme auf die Ca -Freisetzung kann Sorcin die Ca -Homöostase beeinflussen. Durch Phosphorylierung von Sorcin verliert es seine inhibitorische Eigenschaft auf den 2+ RYR2. Sorcin interagiert mit weiteren Ca -regulierenden Proteinen wie dem NCX, dem L-TypCalciumkanal und Annexin VII. RYR2 = Ryanodin Rezeptor Typ 2; β-AR = beta-Adrenorezeptor; NCX = Natrium-CalciumAustauscher; FKBP12.6 = FK506-Binding-Protein 12,6 kDa 1.6.1 Ryanodin-Rezeptor Typ 2 Nach aktuellem Kenntnisstand bildet die kardiale Isoform des Ryanodin-Rezeptors (RYR2) einen Makromolekularen Komplex, der die Proteinkinase A, FK506-BindingProtein 12,6 (FKBP12.6), ein Ankerprotein mAKAP, die Proteinphosphatase 1 und die Proteinphosphatase 2A beinhaltet (82). Der Ryanodin-Rezeptor Typ 2 selber ist ein in der Membran des SR liegendes Tetramer, deren Untereinheiten jeweils 565 kilo Dalton (kDa) schwer sind, und eine Pore durch die Membran des SR bilden. Durch die Pore setzt der RYR2 das Ca2+ aus dem SR frei, welches für den Kontraktionszyklus gebraucht wird. Die Aktivierung des RYR2 erfolgt über Ca2+, das durch spannungsabhängige Ca2+-Kanäle (Dihydropyridinkanäle) freigesetzt wird (1, 11 Einleitung 33). Dieser, sich in der Theorie selbstverstärkende Mechanismus durch die Ca2+Konzentrationszunahme im Zytoplasma, wird durch bislang unbekannte Faktoren beendet (124). Verschiedene Theorien umfassen u.a. eine Ca2+-abhängige Inaktivierung, eine Inaktivierung durch ein regulierendes Protein wie z.B. Sorcin, eine Wahrscheinlichkeits-abhängige Inaktivierung, oder aber noch gänzlich unbekannte Mechanismen. Ein bestehender Kontakt mehrerer RYR2 untereinander in der Membran des SR konnte inzwischen gezeigt werden und führte zu der Annahme, dass die Kanäle als vereinte Gruppe wirken, und das sogenannte „Coupled Gating“ ausführen (81). Hierbei werden die Kanäle als Einheit gesteuert und agieren gleichzeitig und gleichgerichtet. Durch direkte Phosphorylierungen durch die PKA, die PKC und die Proteinkinase G wird die Öffnungswahrscheinlichkeit des RYR2 erhöht und es kommt zu einer größeren Ca2+-Leckage aus dem SR (128, 130). Unter pathophysiologischen Bedingungen, wie sie bei myokardialer Hypertrophie oder Herzinsuffizienz auftreten, ist das Öffnungsverhalten dieses Kanals verändert mit direktem Einfluss auf die Ca2+Homöostase und Kontraktilität der Herzmuskelzelle (151). 1.6.2 Sorcin Sorcin ist ein 22 kDa schweres Protein der EF-Hand-Familie (150). Es wird in vielen verschiedenen Geweben von Säugetieren, darunter auch der Skelett- und Herzmuskulatur ausgebildet (85). Interessanterweise und auch unerwarteter Weise wurde bei einer experimentellen Überexpression des Sorcins in Fibroblasten eine Expression des RYR2 nachgewiesen (85). In dieser Arbeit konnte auch eine Copräzipitation des RYR2 durch den Sorcinantikörper gezeigt werden. Eine solche physische Verbindung der beiden Proteine legt eine funktionelle Kopplung nahe. Demzufolge ist Sorcin in der Lage eine völlige Blockade des RYR2 zu bewirken (75). Konsequenterweise, nachfolgend durchgeführte Untersuchungen zeigen, dass Sorcin nicht nur die spontane Aktivität des RYR2 (gemessen durch das auftreten von Ca2+-Sparks) hemmt, sondern ebenso die Calcium-induzierte-Calcium-Freisetzung (CICR) hemmen kann (36). Um diese Funktion in vivo auszuführen, muss Sorcin durch eine hohe Ca2+-Konzentration zur Translokation aus dem Zytosol an die Membran befähigt werden. Diese Konzentration von ungefähr 200 mM wird allerdings 12 Einleitung intrazellulär nur an den Dyaden erreicht. Dass Sorcin gerade an diesen durch Immunpräzipitationsexperimente lokalisiert wurde (36) lässt den Schluss zu, dass genau diese Funktion von Sorcin ausgeführt wird. Bis zur vollständigen Hemmung des RYR2 durch Sorcin wird ein Zeitraum von weniger als 20 ms veranschlagt, und somit ist die Möglichkeit gegeben, dass Sorcin zur Beendigung der CICR direkt beiträgt. Durch die Analyse von Modellen einer Sorcinüberexpression herrschen zwei Meinungen über die Funktion von Sorcin in vivo vor. So wird einerseits davon ausgegangen, dass eine Sorcinüberexpression zu hämodynamischen Verschlechterungen führt (84, 115). Andererseits konnte eine kardiale Kontraktionsverbesserung durch eine Sorcinüberexpression nachgewiesen werden (39, 125). 1.6.3 FKBP12.6 FKBP12.6 ist eine Isoform des FKBP12 im Herzmuskel (114). Die Assoziation von FKBP12.6 mit dem RYR2 in der Herzmuskulatur (15, 60, 133) weist darauf hin, dass es als akzessorisches Protein des RYR eine Rolle in der Ca2+-Homöostase spielt. Durch Blockade des FKBP12.6 in Lipidbilayermembranen konnte gezeigt werden, dass es zu einer eingeschränkten Leitfähigkeit einzelner RYR2 kommt, vermutlich durch eine defekte Koordination der vier Untereinheiten des Kanals hervorgerufen (15, 65). Durch Zuführen von FKBP12 unter diesen experimentellen Bedingungen lässt sich die Leitfähigkeit der RYR1 wiederherstellen und es kommt somit zu einer Stabilisierung der Kanäle (15). Aufgrund dieser Erkenntnisse geht man davon aus, dass die Familie der FKBP-Proteine u.a. einer Stabilisierung der RYR-Untereinheiten dienen. Auch mehrere RYR2-Kanäle werden nach der Theorie des „Coupled Gating“ durch FKBP12.6 zu funktionellen Einheiten verbunden (81). Für eine physiologische Bedeutung dieses Mechanismus spricht, dass durch eine Phosphorylierung des RYR2 das FKBP12.6 von den Kanälen dissoziiert und die Öffnungswahrscheinlichkeit der RYR2 erhöht. In vivo Experimente führen diese Hypothese fort. So zeigen Mäuse mit einer FKBP12.6-Defizienz eine erhöhte intrazelluläre Ca2+-Freisetzung, die vorerst mit einer verbesserten Kontraktilität einhergeht, aber bei einer stressinduzierten PKA-Phosphorylierung in einer signifikant häufigeren, tödlichen Arrhythmie mündet, die mit der unkontrollierteren RYR2-Aktion in Verbindung gebracht wird (140). Überexpressionsmodelle des FKBP12.6 in isolierten Kardiomyozyten hingegen führen zu einer Verminderung der 13 Einleitung Ca2+-Sparks der RYR2 und zu einer Stabilisierung der Kanäle im geschlossenen Zustand. Dies mündet in einer Verminderung des Ca2+-Lecks aus dem SR und schließlich zu einer größeren Beladung des SR mit Ca2+, einem höheren Ca2+Transienten und einer verbesserten Kontraktilität (42, 101). 1.6.4 Annexin VII Annexin A7 (Annexin VII) wird in Folge alternativen Spleißens in zwei Transkripten exprimiert. So führt der Einbau eines weiteren Exons zu der größeren Isoform mit 51 kDa gegenüber der kleineren Isoform mit 47 kDa (87). Beide Isoformen sind im Herzen existent (79, 116). Annexin A7 wurde an der Pasmamembran und am TTubulus des Skelettmuskels, nahe sekretorischer Vesikel, in subzellulären Membranstrukturen und auf der Plasmamembran lokalisiert (22, 72). Über eine Ca2+abhängige Bindung an Sorcin ist Annexin A7 wahrscheinlich an der Regulation der elektromechanischen Kopplung und der CICR beteiligt (16, 19, 116) Der Einfluss des Annexins A7 auf die Zellverkürzungs-Frequenz-Beziehung, die in Kardiomyozyten einer Annexin A7-defizienten Mauslinie beschrieben wurde (53), bestätigt die Bedeutung dieses Proteins für die elektromechanische Kopplung. 1.6.5 Natrium-Calcium-Austauscher Das in der Systole in die Zelle einfließende Ca2+muss während der Diastole wieder aus der Zelle ausgeschleust werden, um die Ca2+-Homöostase zu wahren. Der sarkolemmale Natrium-Calcium-Austauscher ist ein passiver Transportmechanismus, der in Abhängigkeit des Membranpotentials und der transmembranären Gradienten von Natrium- und Ca2+-Ionen, diese in beide Richtungen über die Zellmembran transportieren kann. Unter physiologischen Bedingungen führt dies zu einem kurzen systolischen Ca2+-Einstrom gefolgt von einem prominenten Ca2+-Ausstrom in der Diastole. Inwieweit der systolische Ca2+Einstrom unter physiologischen Bedingungen an der CICR beteiligt ist, konnte noch nicht endgültig geklärt werden (108). An einem transgenen Mausmodell mit homozygoter Überexpression des NCX konnte jedoch der Einfluss einer erhöhten Dichte dieses Ionentransporters auf die Ca2+-Freisetzung des SR und die Kinetik des L-Typ Calcium-Kanals eindrucksvoll belegt werden (107). Dies ist wahrscheinlich die direkte Folge verminderter subsarkolemmaler Ca2+-Konzentrationen. Interessanterweise kommt es auch im Überexpressionsmodell des Sorcins zu einer 14 Einleitung Zunahme der NCX-Aktivität, die zu einer verminderten Beladung des SR mit Ca2+ führt (115). 1.6.6 Calsequestrin Calsequestrin ist ein 55 kDa schweres Protein, das im SR der Ca2+-Speicherung dient (64). In der vorliegenden Arbeit wurde es als interner Standard zum Vergleich der aufgetragenen Proteinmengen bei den Immunoblotanalysen verwendet. 15 Einleitung 1.7 Fragestellungen Ziel der vorliegenden Arbeit war es, die Bedeutung der G-Proteine Gαq/Gα11 für die Entwicklung der myokardialen Hypertrophie unter den speziellen Bedingungen des Typ1-Diabetes zu untersuchen. Hierzu wurden funktionelle und biochemische Untersuchungen an kardialem Gewebe von Wildtypmäusen, Gα11 Knockoutmäusen und Gαq/Gα11 Doppelknockoutmäusen durchgeführt. Im einzelnen sollten folgende Fragestellungen in der vorliegenden Arbeit beantwortet werden: 1. Wird die kontraktile Dysfunktion bei Diabetes mellitus primär über eine veränderte Signaltransduktion durch die G-Proteine Gα11 und Gαq vermittelt oder sind Hypertrophie und Herzinsuffizienz bei Typ1-Diabetes im wesentlichen Folge der Hyperglykämie? Ein experimenteller Typ1-Diabetes wurde in Mäusen des Wildtyps sowie in Gα11 Knockoutmäusen und Gαq/Gα11 Doppelknockoutmäusen mittels intraperitonealer Injektion von Streptozotocin induziert. 8 Wochen nach Diabetes-Induktion wurden Herzgewicht und Lungengewicht als Marker für eine Herzhypertrophie und eine Herzinsuffizienz gemessen und die kardiale Funktion dieser Tiere mit Hilfe echokardiographischer Untersuchungen analysiert. 2. Über welche Signalkaskade der G-Proteine Gα11 und Gαq werden die pathophysiologischen Veränderungen (z.B. verändertes Proteinexpressionsmuster) vermittelt? Immunhistochemisch wurden die PKC-Isoformen α, βΙΙ, δ, ε, ζ, θ, sowie die Phosphorylierung der Isoformen δ/θ und ζ/λ untersucht. 3. Welchen Einfluss hat der experimentelle Diabetes mellitus auf den Proteinkomplex und die Funktion des Ca2+-Freisetzungskanals des SR und haben die G-Proteine Gα11 und Gαq eine Bedeutung für diese Veränderungen? 16 Einleitung Die Proteinexpression des RYR2, des Sorcins, des FKBP12.6, des Annexin VII und des NCX wurde gemessen. Durch Bestimmung des quantitativen Verlustes von Ca2+ an isolierten Membranpartikeln wurde die Funktion des Ryanodinkanals in den verschiedenen Versuchsgruppen untersucht. 17 Material & Methoden 2 Material & Methoden 2.1 Materialien 2.1.1 Versuchstiere Die Untersuchungen wurden an acht Wochen alten Gαq/Gα11 Doppelknockout Mäusen, an Gα11 Knockout Mäusen und an Wildtypmäusen des selben Stammes (C57/Bl6) durchgeführt. Die Knockout Mäuse wurden freundlicherweise von Herrn Prof. Dr. S. Offermanns, Direktor des Instituts für Pharmakologie der Universität Heidelberg, zur Verfügung gestellt und in eigener Zucht vermehrt. Die Vorgehensweise zur Generierung der Knockout Mäuse wurde bereits von der Arbeitsgruppe Offermanns ausführlich beschrieben (141). Die Wildtypmäuse wurden bei Charles River Laboratories im Alter von 8 Wochen bestellt. Alle Tiere waren einem künstlichen Tag-Nacht-Rhythmus von 12 Stunden und einer Temperatur von 20-22 °C ausgesetzt. Sie erhielten eine Mäusestandardnahrung und Wasser aus einem Tropfenspender ad libitum. 2.1.2 Verwendete Substanzen und Lösungen 2.1.2.1 Tierexperimentelle Methoden Zitronensäure Monohydrat, MG 210,14, Roth, Karlsruhe • Streptozotocin (2-Desoxy-2-(3-methyl-3-nitrosoureido)-D-glucopyranose), MG 265,2, Calbiochem, EMD Biosciences, Inc., San Diego, California, USA • 0,45 µm Filter, Millipore, Billerica, USA • Glukosetestgerät: GlucoMen Glycó, A. Menarini Diagnostics, Florenz, Italien • Teststreifen: GlucoMen Sensor, A. Menarini Diagnostics, Florenz, Italien • EDTA-Blutröhrchen, Sarstedt AG & Co, Nümbrecht • Isofluran, Baxter Deutschland GmbH, Unterschleißheim, Deutschland • Echokardiographiegerät Philips iE33, Schallkopf 11 MHz, Philips, Seoul, Südkorea 2.1.2.2 DNA-Isolation 18 • Phenolchloroform, Roth, Karlsruhe • Ethanol 99,8%, Roth, Karlsruhe • TE-(Tris-EDTA) Puffer: 10 mM Tris HCL, 1 mM EDTA, pH 8,0 Material & Methoden • Lysepuffer: 100 mM Tris pH 8,0, 5 mM EDTA ,1% SDS, 0,2 M NaCl • Proteinase K • Tris Puffer 99.9%, Roth, Karlsruhe • NaCl 99,5%, Roth, Karlsruhe • Natriumduodecylsulfat ultra pure (SDS), Roth, Karlsruhe 2.1.2.3 PCR • Deionisiertes Wasser, Milli Q • MgCl 25 mM, Fermentas, St. Leon-Rot • Taq Puffer, Fermentas, St. Leon-Rot • Taq DNA Polymerase, Roche, Mannheim • dNTP o Desoxyadenosintriphosphat PCR Grade, Roche, Mannheim o Desoxytyrosintriphosphat PCR Grade, Roche, Mannheim o Desoxyguanosintriphosphat PCR Grade, Roche, Mannheim o Desoxycytosintriphosphat PCR Grade, Roche, Mannheim • Primer o Cre1TS (5´-GGACATGTTCAGGGATCGCCAGGCG-´3), Mwg Biotech Ag, Zürich, Schweiz o Cre2TS (5´-GCATAACCAGTGAAACAGCATTGC-´3), Mwg Biotech Ag, Zürich, Schweiz o QIN6C (5´- AGCTTAGTCTGGTGACAGAAGC -´3), Mwg Biotech Ag, Zürich, Schweiz o TVQFL1 (5´- GCATGCGTGTCCTTTATGTGAG -´3), Mwg Biotech Ag, Zürich, Schweiz o TW25 (5´- GCCCCTTGTACAGATGGCAG -´3), Mwg Biotech Ag, Zürich, Schweiz o TW24 (5´- AGCATGCTGTAAGACCGTAG -´3), Mwg Biotech Ag, Zürich, Schweiz • Marker o O´gene ruler 50bp DNA-Ladder, Fermentas, St. Leon-Rot o Middle Range Ladder, Fermentas, St. Leon-Rot • Agarose Gel Loading Dye Solution, Amresco, Solon, Ohio, USA • Universal Agarose, PEQLAB Biotechnologie GmbH, Erlangen 19 Material & Methoden • Ethidiumbromid, Sigma-Aldrich Chemie GmbH, München 2.1.2.4 Herstellung von Homogenaten für Western Blot-Untersuchungen • Präparationspuffer (Zusammensetzung in mmol/l): Saccharose 300, EDTA 10, Phenylmethylsulfonylflourid (PMSF) 1, PIPES 20; pH 7,4, NaH2PO4 50 • Gefrierpuffer (Zusammensetzung in mmol/l): Saccharose 400, HEPES 5, EDTA 10, NaH2PO4 50, Tris/HCL 5; pH 7,2 2.1.2.5 Bestimmung der Proteinkonzentration • Rinderserumalbumin, MG ca. 68000 Dalton, Merck, Darmstadt • Bradford-Reagenz (Bio-Rad Protein-Assay, Dya Reagent Concentrate, BioRad, München) 2.1.2.6 SDS-Gelelektrophorese und Western Blots • 30% Acrylamid/Bis 37, 5:1, Bio-Rad, München • Ammoniumperoxisulfat, MG 228,2, Merck Darmstadt • TEMED, MG 116,21, Roth, Karlsruhe • Natrium-Dodecylsulfat (SDS), MG 288,38, Serva, Heidelberg • PVDF Polyvinylfluorid-Membran, Serva Feinbiochemika, Heidelberg • Polyethylen-Sorbitan Monolaurat (Tween 20) Serva Feinbiochemika, Heidelberg • Bio Magermilch Pulver, Heirler Cenovis GmbH, Radolfzell • Probenpuffer 2× (Zusammensetzung in mmol/l): Tris-HCL 100 (pH 6,8), Glycerin 20%, SDS 4%, Mercaptoethanol 2%, Bromphenolblau 0,2% • Antikörper: o Polyklonaler (Kaninchen) Anti-NCX-Antikörper (IgG), Prof. Dr. K. D. Philipson, University of California, Los Angeles, CA, USA o Monoklonaler (Maus) Anti-Ryanodine-Rezeptor Typ2- Antikörper (IgG1) Affinity Bioreagents Inc., Golden, USA o Monoklonaler (Maus) Anti-Sorcin-Antikörper, Zymed Laboratories Inc., San Francisco, USA o Polyklonaler (Kaninchen) Anti-Calsequestrin-Antikörper, (IgG1), Upstate Biotechnology Inc., Lake Placid, New York, USA 20 Material & Methoden o Polyklonaler (Kaninchen) Anti-FKBP12.6-Antikörper, (IgG1) Affinity Bioreagents Inc., Golden, USA o Anti-Annexin A7-Antikörper (203-217, 45-4), Prof. Dr. A. A. Nögel, Direktorin des Instituts für Biochemie, Universität zu Köln o Polyklonaler (Kaninchen) Anti-phopho-RYR2(Ser-2809)-Antikörper, Badrilla, Leeds, England o Peroxidase-konjugierter Anti-Kaninchen IgG-Antikörper, Sigma Immunochemicals, St. Louis, MO, USA o Peroxidase-konjugierter Anti-Maus IgG-Antikörper, Sigma Immunochemicals, St. Louis, MO, USA • Marker o Page Ruler Prestained Protein Ladder, Fermentas, St. Leon-Rot • Enhanced Chemolumineszenz Kit (ECL), Amersham Life Science, Buckinghamshire, Großbritannien 2.1.2.7 Quantitative Bestimmung des Ca2+-Verlustes an isolierten Membranpartikeln • Imidazole, MG 68,08, Merck, Darmstadt • Kaliumchlorid (KCl) ,MG 74,56, Merck, Darmstadt • Magnesiumchloridhexahydrat, MG 203,3, Merck, Darmstadt • Natriumazid (NaN3), MG 65,01, Merck, Darmstadt • Calciumchlorid (CaCl), MG 147,02, Merck, Darmstadt • EGTA, MG 380,4, Sigma-Aldrich Chemie GmbH, München • Kaliumoxalatmonohydrat, MG 184,24, Fluka Chemie AG, Steinheim • Saccharose, MG 342,3, Roth, Karlsruhe • Kaliumphosphat, MG 136,09, Merck, Darmstadt • Natriumfluorid, MG 41,99, Merck, Darmstadt • Adenosintriphosphat, MG 605,2, Roche, Mannheim • 1,4-Dithiothreit, MG 151,2, Roth, Karlsruhe • Ruthenium Rot, Sigma-Aldrich Chemie GmbH, München • Nitrocellulose Filter 0,45 µm, Millipore, Billerica, USA • 45 Calcium, GE- Healthcare, Buckinghamshire, Großbritannien 2.1.2.8 Immunhistochemie • Phosphatpuffer-Natriumchlorid (PBS): 0.1 M PBS, pH 7.4 21 Material & Methoden • Di-Natriumhydrogenphosphatdihydrat, Merck, Darmstadt • Natriumhydrogenphosphatmonohydrat, Merck, Darmstadt • Natriumchlorid, Merck, Darmstadt • Trispuffer (TB) und Trispuffer-NaCl (TBS): 0.05 M TBS, pH 7.6 • Tris, Roth, Karlsruhe • Tris-HCl Puffer • DAB Lösung (10 mg DAB + 5 µl Nickelsulfat-Lösung (Vector) auf 50 ml 0.05 M Tris/HCl pH 7.6, 6 µl 30% H2O2) Immersionsfixativ-Lösung (4% PFA + 0,1 M PBS): • Paraformaldehyd, Merck, Darmstadt Entwässerungslösungen: • Xylol, Merck, Darmstadt • Ethanol, Merck, Darmstadt Einbettmedien: • Tissue-Tek, O.C.T. Compound, Plano, Tx, USA Eindeckmedien: • Glycerin Gelatine, Merck, Darmstadt • Entellan, Merck, Darmstadt Verwendete Geräte: • Schneidegerät: Kryostat, Jung Frigocut 2800 E, Leica • Messer: Hartmetallmesser C • Objektträger: Shandon adhäsiv Objektträger, Life Sciences International • Kodak Gold 400 (Farbaufnahmen) • Lichtmikroskop: Leitz Orthoplan bzw. Olympus Vanox Lichtmikroskop • Antikörper: o Monoklonaler (Kaninchen) Anti-Proteinkinase C δ-Antikörper, Sigma Immunochemicals, St. Louis, MO, USA o Monoklonaler (Kaninchen) Anti-Proteinkinase C β2-Antikörper, Sigma Immunochemicals, St. Louis, MO, USA o Monoklonaler (Kaninchen) Anti-Phospho-Proteinkinase C δ/θAntikörper, Cell Signaling Technology, Inc., Boston, MA, USA o Monoklonaler (Kaninchen) Anti-Phospho-Proteinkinase C ζ/λAntikörper, Cell Signaling Technology, Inc., Boston, MA, USA 22 Material & Methoden o Monoklonaler (Kaninchen) Anti Proteinkinase C ζ-Antikörper, Cell Signaling Technology, Inc., Boston, MA, USA o Monoklonaler (Kaninchen) Anti Proteinkinase C α-Antikörper, Cell Signaling Technology, Inc., Boston, MA, USA o Monoklonaler (Kaninchen) Anti Proteinkinase C θ-Antikörper, Cell Signaling Technology, Inc., Boston, MA, USA o Monoklonaler (Kaninchen) Anti-Proteinkinase C ε Antikörper, Sigma Immunochemicals, St. Louis, MO, USA 2.2 Methoden 2.2.1 Tierexperimentelle Methoden 2.2.1.1 Induktion eines Diabetes Mellitus Die Induktion des Diabetes Mellitus wurde durch eine einmalige intraperitoneale Injektion des Antibiotikums Streptozotocin (2-Desoxy-2-(3-methyl-3-nitrosoureido)-Dglucopyranose), das einen hochdiabetogenen Effekt durch spezifische β-ZellToxizität durch DNA-Alkylierung und DNA-Strangbrüche zeigt, durchgeführt. Dies hat einen Insulinmangeldiabetes zur Folge, dessen Schweregrad von der Dosis des STZ abhängt. Die gewählte Konzentration betrug 130 mg/kg KG. Dazu wurde die erforderliche Menge in 300 µl Zitratpuffer, 0,1 M, pH 4,5, gelöst, durch einen 0,45 µm Millipore-Filter filtriert und sofort injiziert. Allen Kontrolltieren wurde nur die Trägersubstanz, der Zitratpuffer, injiziert. Als Erfolgskontrolle wurden nach 24 Stunden Blutzuckermessungen durchgeführt. Tiere, die keine erhöhten Blutzuckerspiegel aufwiesen, wurden wiederholt mit STZ behandelt. Als eine erfolgreiche Induktion eines Diabetes Mellitus wurden Blutzuckerwerte von über 300 mg/dl gewertet. Die Tiere waren bei der Induktion des Diabetes acht Wochen alt und wurden für weitere acht Wochen im Tierstall gehalten. Das Tierversuchsprotokoll war vorab von der Bezirksregierung Köln geprüft und genehmigt worden (AZ 50.203.2K47,28/04) 23 Material & Methoden 2.2.1.2 Bestimmung der Blutzuckerwerte und des Körpergewichtes Zur Bestimmung der Blutzuckerwerte wurde eine Schwanzvene der Mäuse punktiert und der gewonnene Tropfen Blut mit Teststreifen (GlucoMen Sensor, A. Menarini Diagnostics) und einem Glucometer (GlucoMen Glycó) analysiert. In der ersten Woche wurden die Werte im zweitägigen Rhythmus und ab der zweiten Woche im wöchentlichen Rhythmus erhoben. Zum selben Zeitpunkt wurde das Gewicht der Mäuse bestimmt. 2.2.1.3 Opferung der Tiere und Organentnahme Im Alter von 16 Wochen wurden die Tiere durch zervikale Dislokation getötet. Die Herzen wurden entnommen, nach dem Wiegen entweder für die spätere Immunoblotanalyse sofort in flüssigem Stickstoff schockgefroren und bei –80°C aufbewahrt, oder für die spätere Immunhistochemie sofort in Immersionsfixativlösung für 24 h bei 4°C fixiert. Das Lungenfeuchtgewicht wurde zur Beurteilung einer kongestiven Herzinsuffizienz bestimmt. Das bei Trennung des Herzens von der Aorta gewonnene Blut wurde mit Hilfe einer Spritze in EDTA-Blutröhrchen überführt. Der HbA1c-Wert wurde aus diesem Blut im Institut für klinische Chemie der Universität zu Köln (Direktor: Prof. Dr. K. Wielckens) bestimmt. Der HbA1c-Wert ist ein Maß für den Anteil an glykosyliertem Hämoglobin im Blut und korreliert mit den durchschnittlichen Blutzuckerwerten der vorangegangenen 2-3 Monaten. Der rechte Tibiaknochen der Mäuse wurde operativ freigelegt und mit Hilfe einer handelsüblichen Schieblehre vermessen. Die anzeigepflichtige Tötung der Versuchstiere war zu Beginn des Projektes seitens der Bezirksregierung Köln bewilligt worden (AZ 50.203.2-K 07/06). 2.2.1.4 Echokardiographische Bestimmung funktioneller und morphologischer Veränderungen am Herzen in vivo Zur Bestimmung funktioneller und morphologischer Veränderungen, die durch den Diabetes über 8 Wochen auftreten können, wurde die Ultraschalluntersuchung der Mäuseherzen in vivo vergleichend sowohl am Tag vor Streptozotocin-Injektion als auch unmittelbar vor Tötung der Tiere durchgeführt. Dabei wurde in der Echokardiographie die Pumpfunktion, die Myokarddicke und die Dimensionen der Herzhöhlen quantifiziert. Für diese Untersuchungen wurden die Tiere narkotisiert, wobei die Dosierung von Isofluran so gering gehalten wurde, dass es zu keiner 24 Material & Methoden Depression der Spontanatmung kam. Die Untersuchungen wurden an einem Echokardiographiegerät (Philips iE33) mit einem hochfrequenten Schallkopf von 11 MHz durchgeführt. Im zweidimensionalen B-mode wurden die parasternale kurze Achse auf midpapillärer Ebene und die parasternale lange Achse aufgezeichnet, mit einer Aufnahmedauer von jeweils mindestens 20 Zyklen. In der parasternalen kurzen Achse wurde das Myokard in sechs Segmente, in der parasternalen langen Achse in sieben Segmente unterteilt (20). Auswertungen erfolgten nur anhand von Aufnahmen, in denen Epikard und Endokard in mindestens fünf Segmenten eindeutig abgegrenzt werden konnte. Im zweidimensionalen Bild wurde auch die Ebene für den eindimensionalen M-Mode auf Höhe der Papillarmuskeln festgelegt. Im M-Mode wurden die linksventrikulären enddiastolischen und endsystolischen Durchmesser der Kammer (LVEDD und LVESD) ausgemessen sowie die Längenverkürzungsfraktion und Wanddicken bestimmt. 25 Material & Methoden 2.2.2 Genotypisierung der Mäuse 2.2.2.1 DNA Isolation Bei den Markierungen der Jungtiere wurde jeweils eine Stanzbiopsie aus dem Ohr gewonnen, die zur Genotypisierung der Tiere verwendet wurde. Das Gewebe wurde über Nacht in einem Lysepuffer mit der Proteinase K lysiert. Hierfür wurden die Proben mit 8000 rpm bei 55°C in einen Thermomixer (Thermomixer 5436, Eppendorf, Hamburg) gegeben. Am nächsten Tag wurde die Proteinase K durch fünfminütiges Erhitzen auf 95°C inaktiviert. Zusammen mit Phenolchloroform wurde für fünf Minuten bei 13000 rpm zentrifugiert (Beckmann Coulter Microfuge R). Die oberste, DNA-haltige der drei Phasen wurde abgetragen und zusammen mit 100% Ethanol zentrifugiert. Nach Entnahme des Ethanols wurde das vorhandene Pellet ein weiteres Mal zentrifugiert, zusammen mit 70% Ethanol. Das Pellet wurde nun in TEPuffer gelöst und bis zur folgenden PCR bei –20°C aufbewahrt. 2.2.2.2 Polymerase-Kettenreaktion (PCR) Für die PCR wurde folgender Reaktionsansatz hergestellt: 1-fach in µl Milli Q (deionisiertes Wasser) 38,5 Taq-Puffer 5 MgCl2 1,5 Primer 1 1 Primer 2 1 dNTP 0,5 Taq-DNA-Polymerase 0,5 Tabelle 2 Reaktionsansatz für die Polymerase-Kettenreaktion Zwei µl der aus dem Ohrgewebe isolierten DNA wurden hinzugegeben, so dass ein Endvolumen von 50µl resultierte. Zur Analyse und dem Nachweis der Gene wurden verschiedene Protokolle verwendet: 26 Material & Methoden Cre PCR Primer: Cre1TS (5´-GGACATGTTCAGGGATCGCCAGGCG-´3) (50 µmol/µl) Cre2TS (5´-GCATAACCAGTGAAACAGCATTGC-´3) (50 µmol/µl) Prg: 94°C 3 min 94°C 20 sek 62°C 20 sek 72°C 1 min 15 sek Wiederholung ab Schritt 2, 29-fach 72°C 10 min Ergebnis: Cre+/- or Cre+/+: Bande bei 270 bp Cre-/-: keine Bande Gα αqflox-PCR Primer: QIN6C (5´- AGCTTAGTCTGGTGACAGAAGC -´3) (50 µmol/µl) TVQFL1 (5´- GCATGCGTGTCCTTTATGTGAG -´3) (50 µmol/µl) Prg: 94°C 3 min 94°C 20 sek 60,5°C 20 sek 72°C 2 min 15 sek Wiederholung ab Schritt 2, 29-fach 72°C 10 min Ergebnis: gαqwt: Bande bei 600 bp gαqflox: Bande bei 700 bp 27 Material & Methoden Gα α11 PCR Wildtyp Allel Primer: TW25 (5´- GCCCCTTGTACAGATGGCAG -´3) (50 µmol/µl) TW24 (5´- AGCATGCTGTAAGACCGTAG -´3) (50 µmol/µl) Prg: 94°C 3 min 94°C 30 sek 64°C 30 sek 72°C 1 min 30 sek Wiederholung ab Schritt 2, 29-fach 72°C 10 min Ergebnis: gα11: Bande bei 820 bp Gα α11 PCR Knockout Allel Primer: 116ExAS (5´-CAGGGGTAGGTGATGATTGTGC-´3) (50 µmol/µl) NeoA (5´-GACTAGTGAGACGTGCTACTTCC-´3) (50 µmol/µl) Prg: 94°C 3 min 94°C 30 sek 57°C 30 sek 72°C 2 min Wiederholung ab Schritt 2, 29-fach 72°C 10 min Ergebnis: gα11KO: Bande bei 450 bp Das PCR-Produkt wurde in einem einprozentigen Agarose-Gel, welches Ethidiumbromid zur Sichtbarmachung enthielt, elektrophoretisch aufgetrennt und das 28 Material & Methoden Ergebnis unter ultraviolettem Licht abgelesen. Ein Marker erlaubte die Größeneinordnung der DNA. 2.2.3 Untersuchungen zur Proteinexpression 2.2.3.1 Herstellung von Homogenaten für Western Blot-Untersuchungen Nach der Entnahme der Herzen aus dem Thorax wurden diese von Geweberestbeständen befreit, in flüssigem Stickstoff schockgefroren und bei – 80°C gelagert. Zur Bestimmung der Proteinexpression wurden zu einem späteren Zeitpunkt Anteile der Herzen in einem Metallgefäß unter ständiger Kühlung in flüssigem Stickstoff mit einem Hammer zerkleinert. Das hierbei entstehende pulvrige Produkt wurde im zehnfachen Verhältnis mit Präparationspuffer versetzt und sofort auf Eis gestellt. Hier wurde die weitere Homogenisierung mit einem Mixer UltraTurrax T8 (Janke & Kunkel KG, IKA-Werke, Staufen) vier mal über jeweils 20 Sekunden durchgeführt. Anschließend wurde Gefrierpuffer in gleichem Volumen dazugegeben. Die Proben wurden nun entweder direkt zur Western Blot Analyse weiterverarbeitet oder zur späteren Verwertung aliquotiert und bei –80°C gelagert. 2.2.3.2 Bestimmung der Proteinkonzentration Um zu gewährleisten, dass für die folgenden Western Blots die Proben gleiche Mengen an Protein enthielten, wurde die Proteinkonzentration nach der Methode von Bradford bestimmt (13). Alle Proben wurden mit Hilfe des Gefrierpuffers auf eine Konzentration von 2000-4000 µg/ml eingestellt. Als Standard diente Rinderserumalbumin durch welches eine lineare Eichgerade erstellt wurde (r > 0,99). 2.2.3.3 SDS-Gelelektrophorese Die SDS-Gelelektrophorese dient der quantitativen Bestimmung einzelner Proteine in einem Homogenat. Durch Anlegen einer Spannung an ein SDS-Polyacrylamidgel werden dabei die Proteine nach ihrem Molekulargewicht aufgetrennt. Zur Vorbereitung der Proben wurden diese mit einem Probenpuffer im Verhältnis 1:2 versetzt. Für die Gelelektrophorese wurden Trenngele von 8-12 % Polyarcylamid und Sammelgele von 4 % Polyacrylamid verwendet. 29 Material & Methoden Nach Herstellung der entsprechenden Polyacrylamidgele und deren Aushärtung, wurden die Proben mit einer Proteinmenge von 50 µg aufgetragen, bei 80 mA für ca. eine Stunde gesammelt und bei 120 mA für fünf bis sechs Stunden getrennt. Neben den Proben wurde ein Marker in den Gelen aufgetrennt, der eine Größenidentifizierung der Proteine zuließ. 2.2.3.4 Western Blot Der SDS-Polyacrylamidgelelektrophorese schloss sich der Transfer der Proteine von den Gelen auf eine Polyvinyldienfluorid-Membran (PVDF-Membran) im NassblotVerfahren an. Es wurde über einen Zeitraum von 16 Stunden mit einem Strom von 64 mA bzw. 100 mA bei 4°C geblottet. Hierbei fand eine Hoefer-Transfer-Einheit (Hoefer, San Francisco, CA, USA) Verwendung. Um einen erfolgreichen Transfer nachzuweisen wurden die Gele mit Coomassie Brilliant Blue eingefärbt. Um unspezifische Bindungsstellen der PVDF-Membran zu blocken, wurden die Membranen nach dem Nassblot mit 5%iger Milch in TBST für 2 Stunden bei Raumtemperatur geschüttelt. Anschließend wurden die Membranen zweimal mit TBST für zehn Minuten gewaschen. Auf einem Schüttler wurde nun wiederum bei 4°C für mindestens 12 Stunden die Inkubation mit dem spezifischen Antikörper, der jeweils gegen das zu untersuchende Protein gerichtet war, vorgenommen. Es folgten sechs jeweils fünfminütige Waschungen mit TBST, eine weitere Blockung mit 5%iger Milch für 20 Minuten und eine zehnminütige Waschung mit TBS. Die Membranen wurden nun mit dem zweiten Antikörper bei Raumtemperatur für zwei Stunden inkubiert. Noch einmal schlossen sich Waschvorgänge an, zwei mal zehn Minuten mit TBST und einmal zehn Minuten mit TBS. In einem weiteren Schritt folgte die Inkubation der Membranen für eine Minute in einer Chemiluminiszenz-Lösung (ECL, Amersham-Buchler, Braunschweig) und dann die Exposition auf einem Röntgenfilm. Nach Entwicklung der Filme ließ sich durch Einscannen und durch eine computerunterstützte densitometrische Analyse (ImageQuant, PDI Imaging System, Krefeld) eine Quantifizierung der Proteinexpression vornehmen. 30 Material & Methoden Antikörper Spezies Molekulargewicht Poly/Monoklonal Verdünnung Ryanodin-Typ-2 Maus 565 kDa Monoklonal 1:2000 NCX Kaninchen 72,120,160 kDa Monoklonal 1:1000 Calsequestrin Kaninchen 55 kDa Polyklonal 1:2000 Sorcin Maus 22 kDa Monoklonal 1:1000 FKBP12.6 Kaninchen 12,6 kDa Polyklonal 1:2000 Phospho-RYR2- Kaninchen 565 kDa Polyklonal 1:2500 Annexin VII Maus 47, 51 kDa Monoklonal 1:1000 Kaninchen IgG Ziege - Monoklonal 1:2000 Maus IgG Schaf - Monoklonal 1:2000 Serine 2809 Tabelle 3 Charakteristik der verwendeten Antikörper für den Western Blot 2.2.4 Untersuchungen zur Funktion 2.2.4.1 Herstellung von Homogenaten und Bestimmung der Proteinkonzentration für die quantitative Bestimmung des Ca²+-Verlustes an isolierten Membranvesikeln Die Herstellung von Homogenaten folgte dem gleichen Prinzip wie die Herstellung der Homogenate für die Western Blots (siehe 2.2.3.1). Es wurde aber statt des Gefrier- und des Präparationspuffers ein Kaliumphosphat-Puffer verwendet. Die Proteinkonzentrationsbestimmung wurde ebenfalls unter Verwendung des KaliumPhosphat-Puffers durchgeführt, wie in 2.2.3.2 beschrieben. 2.2.4.2 Quantitative Bestimmung des Ca²+-Verlustes an isolierten Membranvesikeln Die Bestimmung des Ca²+-Verlustes wurde mit der Millipore-Filtrationstechnik durchgeführt wie bei Luo et al. beschrieben (76). 31 Material & Methoden Es wurde folgender Reaktionsansatz erstellt: Imidazol pH 7.0 40 mM KCl 95 mM NaN3 5 mM MgCl2 5 mM EGTA 0,5 mM K+Oxalat 5mM 2+ Tabelle 4 Reaktionsansatz für die quantitative Bestimmung des Ca -Verlustes an isolierten Membranpartikeln. Die Untersuchungen wurden bei pCa 5,5 durchgeführt. Um die Leckage und damit die Funktion des Ryanodinkanals zu prüfen wurde jede Probe einmal mit, und einmal ohne Ruthenium Rot, welches den Ryanodinkanal blockiert, in einer Konzentration von 1 µM gemessen. Der Reaktionsansatz wurde nach Zugabe der Probe für eine Minute bei 37°C inkubiert. Vor dem Hinzufügen von ATP (Konzentration: 5 mM) wurde eine unspezifische Messung durchgeführt. Das ATP sorgt für die Initiierung der Ca²+-Aufnahme über die SERCA. Messungen wurden bei 1, 2, 4, 6 und 8 Minuten unternommen. Der unspezifische Messwert wurde von den erhobenen Messwerten abgezogen. Die Ca2+-Aufnahme wurde nach der Durchführung eines Curve Fit der 1, 2, 4, 6 und 8 Minutenwerte berechnet. Der Ca2+-Verlust des Ryanodinkanals konnte berechnet werden, indem man die Werte der Ca2+-Aufnahme ohne Ruthenium Rot von den Werten der Ca2+-Aufnahme mit Ruthenium Rot subtrahierte. 2.2.5 Immunhistochemie Nach Opferung der Mäuse durch zervikale Dislokation wurde das Herzgewicht bestimmt. Möglichst schnell wurden die Herzen anschließend in Immersionsfixativlösung für 24 h bei 4°C fixiert. Nach dem Waschen in 0,1 M PBS, pH 7,4, 24 h, bei 4°C, wurden die Proben mit 20% Saccharoselösung in 0,1 M PBS, pH 7,4, 48 h, bei 4°C, kryoprotektiert. Die Proben wurden dann einzeln in Tissue-Tek eingebettet und sofort in flüssigem Stickstoff tiefgefroren. Die tiefgefrorenen Blöcke wurden bei -80°C gelagert. 32 Material & Methoden Es wurden Schnitte von 7 µm Dicke mit dem Kryostat angefertigt und auf Objektträgern aufgefangen. Die Schnitte wurden bis zur Durchführung der immunhistochemischen Untersuchungen bei –20°C aufbewahrt. Für die immunhistochemische Inkubation wurde die Avidin-Biotin-PeroxidaseKomplex-Methode (Elite ABC Kit, Vector Labor, Peterborough, UK) angewendet, die folgende Schritte umfasste: 1. 2x15 Min. bei RT waschen in 0,05 M TBS pH 7,6 2. 20 Min. inkubieren bei RT in 0,3% H2O2 verdünnt in 0,05 M TBS zur Blockierung der endogenen Peroxidase. 3. 30 Min. inkubieren bei RT mit 10% normal goat serum (NGS) + 0,05 M TBS pH 7,6 + 1% bovine serum albumine (BSA). 4. 24 Std. Inkubation bei 4 °C mit primärem Antikörper, verdünnt in 1% NGS + 0,2% Triton X-100 + 0,05 M TBS pH 7,6. 5. 2x15 Min. bei RT waschen in 0,05 M TBS, pH 7,6. 6. 1 Std. Inkubation bei RT mit sekundärem Antikörper, verdünnt in 1% NGS + 0,05 M TBS pH 7,6. 7. 2x15 Min. bei RT waschen in 0,05 M TBS pH 7,6. 8. 1 Std. Inkubation bei RT mit dem ABC-Komplex, Vectastain Elite ABC Reagenz (1:100; Vector), verdünnt in 0,05 M TBS pH 7,6. 9. 2x15 Min. bei RT waschen in 0,05 M TBS pH 7,6. 10. 10 Min. (die Zeit wurde für jeden Antikörper gleich festgelegt) bei RT mit der DAB-Lösung inkubieren. 11. 2x15 Min. bei RT waschen in 0,05 M TBS pH 7,6. 12. Schnitte auf beschichtete Objektträger übertragen. 13. Schnitte an der Luft trocknen. 14. Schnitte in aufsteigender Ethanolreihe dehydrieren: 30%, 50%, 70%, 90%, 95%, 100% Ethanol je 5 Min. 15. Schnitte in Xylene klären. 16. Schnitte in Entellan eindecken. Um eine unspezifische Bindung der Antikörper auszuschließen, wurden Schnitte angefertigt, bei denen jeweils nicht mit dem primären oder nicht mit dem sekundären Antikörper inkubiert wurde, sowie ein Schnitt, der mit keinem Antikörper inkubiert 33 Material & Methoden wurde. Die Befunde wurden im Lichtmikroskop in Übersichts- und Detailaufnahmen (400-fache Vergrößerung) ausgewertet und mikrophotographisch dokumentiert. 2.2.6 Statistische Methoden Zur Auswertung der Versuche wurde der arithmetische Mittelwert und der dazugehörige Standardfehler des Mittelwertes (SEM) für die jeweiligen Proben errechnet. Der Student t-Test für verbundene oder unverbundene Stichproben diente zur Ermittlung der statistischen Signifikanz. Als statistisch signifikant wurde ein pWert < 0,05 betrachtet. 34 Ergebnisse 3 Ergebnisse 3.1 Charakterisierung der Versuchstiere Die Versuchstiere wurden im Alter von 16 Wochen durch zervikale Dislokation getötet. Hierbei wurden der Blutzuckerwert sowie der HbA1c–Wert zur Überprüfung der Stoffwechsellage der Tiere ermittelt. Das Herzfeuchtgewicht diente als Marker einer Herzhypertrophie und das Lungenfeuchtgewicht als Marker einer Herzinsuffizienz. Um diese Größen zu normieren wurde die Länge des Tibiaknochens bestimmt. Tabelle 5 fasst die erhobenen Daten zum Zeitpunkt des Todes der Tiere zusammen. Wildtyp Wildtyp Gα11 Gα11 Gα11/Gαq Diabetes Kontrolle Knockout Knockout Doppelknockout Doppelknockout Diabetes Kontrolle Diabetes Kontrolle n = 13 n = 13 n=4 n=4 n = 14 n = 10 Gα11/Gαq Gewicht (g) 20,5 ± 2,6 ∗ 31,6 ± 2,5 25,2 ± 3,3 ∗ 32,0 ± 5,1 30,5 ± 0,2 31,5 ± 6,3 Blutzucker (mg/dl) 491 ± 91 ∗ 432 ± 107 ∗ 109 ± 9 453 ± 120 ∗ 111 ± 3 Herz/Tibia (g/cm) 0,07 ± 0,0 ∗ 0,05 ± 0,0 0,09 ± 0,01 0,09 ± 0,01 0,13 ± 0,0 ∗ 0,08 ± 0,0 Lunge/Tibia (g/cm) 0,09 ± 0,0 0,09 ± 0,01 0,11 ± 0,01 0,10 ± 0,01 0,11 ± 0,0 ∗ 0,08 ± 0,0 103 ± 12 ∗ p < 0,05 vs. Kontrolle Tabelle 5 Allgemeine Daten der verschiedenen Tiergruppen. Die Werte sind als Mittelwerte ± SEM zum Opferungszeitpunkt der Tiere angegeben. Das Körpergewicht der diabeteskranken Tiere ist sowohl bei den Wildtypmäusen, als auch bei den Gα11 Knockoutmäusen gegenüber der Kontrollgruppe zum Opferungszeitpunkt signifikant vermindert. Der Verlauf des Körpergewichtes über den 8-wöchigen Versuchszeitraum ist in Abbildung 4 für ein Tier nach Induktion eines Diabetes mellitus und in Abbildung 3 für ein Kontrolltier exemplarisch dargestellt. Es zeigt sich eine Gewichtsreduktion des Körpergewichtes bei dem diabetischen Tier gegenüber einer leichten 35 Ergebnisse Gewichtszunahme des Kontrolltieres. Dies ist ein Hinweis auf die typische katabole Stoffwechsellage bei Typ 1 Diabetes Mellitus. Alle Tiere zeigten zwei Tage nach einer STZ-Injektion Blutzuckerwerte von mindestens 200 mg/dl. Im Verlauf von fünf Tagen wiesen alle diese Tiere Blutzuckerwerte von 300 mg/dl und mehr auf. Der typische Verlauf der Blutzuckerwerte ist ebenfalls exemplarisch in Abbildung 4 für ein Tier, dem ein Diabetes Mellitus induziert wurde, dargestellt. Abbildung 3 zeigt die entsprechenden Messungen einer Kontrollmaus. Bei der Opferung der Mäuse wurde das Herzfeuchtgewicht und das Lungenfeuchtgewicht ermittelt und auf die Länge der Tibia des jeweiligen Tieres normiert. Die Ergebnisse hierzu sind in Abbildung 5 und 6 dargestellt. Es zeigt sich, dass diabeteskranke Wildtypmäuse im Vergleich zu Kontrollmäusen ein erhöhtes Herzgewicht aufweisen. Dies ist ein deutlicher Hinweis auf eine Myokardhypertrophie in dieser Gruppe. Das Lungenfeuchtgewicht wies bei diesen Versuchsgruppen keinen signifikanten Unterschied auf. Dieser Befund wurde bei den diabeteskranken Gα11 Knockoutmäusen nicht beobachtet. In dieser Gruppe hatten sowohl die diabeteskranken Tiere, als auch die Kontrolltiere äquivalente Messwerte. Bei den diabetischen Gα11/Gαq Doppelknockoutmäusen wiederum ergab sich ein signifikant höheres Herzgewicht als auch ein signifikant erhöhtes Lungengewicht gegenüber den Doppelknockoutmäusen ohne Diabetes Mellitus. Zusammengefasst ergibt sich aus der Quantifizierung von Herz- und Lungengewicht bei Wildtypmäusen der Hinweis auf eine deutliche Myokardhypertrophie bei den diabeteskranken Tieren. Gα11/Gαq Doppelknockoutmäuse entwickeln nach Induktion eines Diabetes eine deutliche Myokardhypertrophie und weisen sogar Zeichen der kongestiven Herzinsuffizienz auf. Nur Mäuse mit isolierter Deletion des Gα11-Proteins entwickeln 8 Wochen nach Induktion eines Diabetes Mellitus keinen kardialen Phänotyp. Zur Kontrolle der Blutzuckerstoffwechsellage bei Mäusen mit und ohne Diabetes mellitus wurde in beiden Gruppen der HbA1c-Wert bestimmt. Abbildung 7 zeigt den mittleren HbA1c-Wert beider Gruppen, der 8 Wochen nach Induktion eines Diabetes einen Anstieg von 240 % aufwies. 36 Ergebnisse 600 30 500 28 400 26 300 24 200 100 22 0 20 1 7 14 21 35 42 49 Körpergewicht (g) Blutzucker (mg/dl) Kontrolle 56 Tag Blutzucker (mg/dl) Körpergewicht (g) Abbildung 3 Repräsentativer Verlauf des Blutzuckers und des Körpergewichts eines Kontrolltieres. Der Verlauf von Blutzucker und Körpergewicht ist repräsentativ sowohl für Tiere des Wildtyps, als auch für Gα11- und Gα11/Gαq Doppelknockoutmäuse. STZ-induzierter Diabetes Mellitus 600 30 28 400 26 300 24 200 Körpergewicht (g) Blutzucker (mg/dl) 500 22 100 0 20 1 7 14 21 28 35 42 49 56 Tag Blutzucker (mg/dl) Körpergewicht (g) Abbildung 4 Repräsentativer Verlauf des Blutzuckers und des Körpergewichts eines Tieres nach Induktion eines Diabetes Mellitus durch intraperitoneale Injektion von Streptozotocin (STZ) an Tag 1. Der Verlauf von Blutzucker und Körpergewicht ist repräsentativ sowohl für Tiere des Wildtyps, als auch für Gα11- und Gα11/Gαq Doppelknockoutmäuse. 37 Ergebnisse H erz gew icht *** Herzgewicht / Tibialänge g/cm 0,14 0,12 0,1 * 0,08 0,06 0,04 0,02 0 Ga11/Gaq-KO Gα11/Gαq KO Kontr. Kontr.nn==44 Ga11/Gaq-KO Gα11/Gαq KODM DM Ga11-KO Gα11 KOKontr. Kontr. nn == 44 nn==13 13 Ga11-KO DM Gα11 KO DM nn==13 13 Wildty p Kontr. Wildtyp Kontr. n n= =1010 Wildty p DM Wildtyp DM n n= =1414 Abbildung 5: Darstellung des Mittelwertes des Herzgewichtes ± SEM der Tiere normiert auf die Tibialänge zum Zeitpunkt der Opferung. Die Gα11/Gαq Doppelknockoutmäuse zeigten eine hochsignifikante Zunahme des Herzgewichtes nach Induktion eines DM. Die Wildtypmäuse zeigten eine signifikante Zunahme des Herzgewichtes nach Induktion eines DM. Die Gα11 Knockoutmäuse zeigten auch nach 8 Wochen Diabetes keine Veränderungen des Herzgewichtes. ∗ p < 0,05 vs. Kontrolle; ∗∗∗ p < 0,001 vs. Kontrolle; KO = Knockout; DM = Diabetes Mellitus; Kontr. = Kontrolle Lungengew icht Lungengewicht /Tibialänge g/cm 0,14 ** 0,12 0,1 0,08 0,06 0,04 0,02 0 Ga11/Gaq-KO Gα 11/Gαq KO Kontr.nn==44 Kontr. Ga11/Gaq-KO DM Gα 11/Gαq KO DM nn==44 Ga11-KO Kontr. Gα 11 KO Kontr. nn==13 13 GαGa11-KO 11 KO DMDM nn==13 13 Wildty pKontr. Kontr. Wildtyp n n= =1010 Wildty pDM DM Wildtyp n =n 14 = 14 Abbildung 6: Darstellung des Mittelwertes des Lungengewichtes ± SEM der Tiere normiert auf die Tibialänge zum Zeitpunkt der Opferung. Nur bei den Gα11/Gαq Doppelknockoutmäusen zeigte sich eine signifikante Zunahme des Lungengewichtes nach 8 Wochen Diabetes. Die anderen Versuchsgruppen hatten jeweils äquivalente Werte zu den Kontrollen. ∗∗ p < 0,01 vs. Kontrolle; KO = Knockout; DM = Diabetes Mellitus; Kontr. = Kontrolle 38 Ergebnisse Wildtypmäuse *** 12 10 HbA1c [%] 8 6 4 2 0 Kontrolle n = 9 Diabetes Mellitus n = 7 Abbildung 7: Anteil an glykosyliertem Hämoglobin (HbA1c) im Blut von Mäusen 8 Wochen nach Induktion eines Diabetes Mellitus und nicht diabeteskranker Kontrolltiere. Durch Induktion eines Diabetes Mellitus kommt es zu einer signifikanten Zunahme des HbA1c um 240 % relativ gegenüber der Kontrolle. ∗∗∗ p < 0,001 vs. Kontrolle 39 Ergebnisse 3.2 Genotypisierung der Mäuse Um die in eigener Zucht vermehrten Mäuse zu genotypisieren, wurden rein qualitative PCR-Untersuchungen durchgeführt. Aus dem Ohrgewebe, welches bei der Markierung der Mäuse gewonnen wurde, wurde die DNA isoliert und für die PCR verwendet. Vier Gene der Mäuse wurden untersucht. Das Cre-Gen, als Nachweis eines konditionellen Gα11/Gαq-Doppelknockouts. Das Gαq-flox-Gen als Nachweis der spezifischen Schnittstellen für das Cre-Enzym, gleichzeitig diente das Gαq-flox-Gen als Nachweis einer gelungenen DNA-Isolation, da dieses Gen bei allen Tieren exprimiert wurde. Das Gα11-Wildtyp-Gen, welches nur bei Wildtypmäusen dargestellt werden kann, um die Gα11-Defizienz zu überprüfen. Das Gα11-Knockout-Gen, das nur bei Gα11 Knockoutmäusen nachgewiesen werden kann. Abbildung 8 zeigt beispielhaft die Fotographie eines Agarosegels, in welchem die PCR-Produkte verschiedener Mäuse aufgetragen wurden. Bei ca. 280 bp kommt das PCR-Produkt des Cre-Gens als Bande zur Darstellung. M - + + + - + - + - - LK PK 300 bp 200 bp M = Marker LK = Leerkontrolle PK = Positivkontrolle + = Cre positiv - = Cre negativ Abbildung 8 Repräsentative Fotographie einer das Cre-Gen nachweisenden PCR. 40 Ergebnisse 3.3 Echokardiographische Bestimmung funktioneller und morphologischer Veränderungen am Herzen in vivo Die Ultraschalluntersuchung der Mäuseherzen in vivo wurde vergleichend sowohl am Tag vor Streptozotocin-Injektion als auch unmittelbar vor Tötung der Tiere durchgeführt, um morphologische und funktionelle Veränderungen, die durch den Diabetes über 8 Wochen auftreten können, zu untersuchen. Die Myokarddicke und die Dimensionen der Herzhöhlen wurden quantifiziert. Im zweidimensionalen B-mode wurden die parasternale kurze Achse auf midpapillärer Ebene und die parasternale lange Achse aufgezeichnet. Im zweidimensionalen Bild wurde auch die Ebene für den eindimensionalen M-Mode auf Höhe der Papillarmuskeln festgelegt. Im M-Mode wurden die linksventrikulären enddiastolischen und endsystolischen Durchmesser der Kammer (LVEDD und LVESD) ausgemessen sowie die Längenverkürzungsfraktion und Wanddicken bestimmt. Eine Übersicht über die Ergebnisse liefert Tabelle 6 und Abbildung 9. Systolisch Diastolisch Gα11 Knockout Gα11 Knockout Wildtyp Kontrolle Wildtyp Diabetes Kontrolle Diabetes Septum (cm) 0.155 ± 0.007 0.178 ± 0.006 0.165 ± 0.005 0.142 ± 0.004 ** LVESD (cm) 0.182 ± 0.009 0.133 ± 0.016 0.178 ± 0.011 0.170 ± 0.013 LVPW (cm) 0.140 ± 0.004 0.169 ± 0.007 ** 0.123 ± 0.006 0.136 ± 0.003 Septum (cm) 0.072 ± 0.004 0.102 ± 0.004 *** 0.083 ± 0.003 0.087 ± 0.005 LVEDD (cm) 0.381 ± 0.009 0.315 ± 0.017* 0.383 ± 0.012 0.351 ± 0.012 LVPW (cm) 0.080 ± 0.004 0.117 ± 0.005 *** 0.083 ± 0.003 0.085 ± 0.008 FS % 0.523 ±0.019 0.585 ± 0.031 0.536 ± 0.026 0.519 ± 0.028 ∗ p < 0,05 vs. Control; ∗∗ p < 0,01 vs. Control, ∗∗∗ p < 0,001 vs. Control Tabelle 6 Echokardiographische Daten (Mittelwert ± SEM) nach 8 Wochen eines experimentellen Diabetes bei Wildtypmäusen und Gα11 Knockoutmäusen im Vergleich zu nicht diabeteskranken Kontrolltieren. Die Wildtypmäuse zeigten eine signifikante Zunahme der diastolischen und systolischen LVPW und der diastolischen Septumdicke. Der LVEDD war vermindert. Diese Alterationen konnten nicht in der Gα11 Knockoutgruppe nach 8 Wochen Diabetes beobachtet werden. Hier zeigte sich eine signifikante Abnahme der systolischen Septumdicke. LVESD = linksventrikulärer endsystolischer Durchmesser LVEDD = linksventrikulärer enddiastolischer Durchmesser LVPW = Dicke der linksventrikulären Hinterwand FS = Längenverkürzungsfraktion 41 Ergebnisse Es zeigte sich nach 8 Wochen Diabetes eine signifikante Zunahme sowohl der systolischen als auch der diastolischen Dicke der linksventrikulären Hinterwand (LVPW) bei den Wildtypmäusen. Diese Tiere zeigten ebenfalls einen verminderten linksventrikulären enddiastolischen Durchmesser im Vergleich zu den nicht diabeteskranken Kontrolltieren. Diese Veränderungen, die als Hinweis auf eine Herzhypertrophie interpretiert werden können, zeigten sich nicht bei den Gα11 Knockoutmäusen nach 8 Wochen Diabetes im Vergleich zu den nicht diabeteskranken Kontrolltieren. Bei den Gα11 Knockoutmäusen ließ sich nach 8 Wochen eines experimentellen Diabetes lediglich eine Verminderung der systolischen Septumdicke nachweisen. Die Längenverkürzungsfraktion blieb in allen untersuchten Tiergruppen unverändert, was für eine unveränderte kardiale Funktion spricht. Wildtyp Gα11 -Knockout Kontrolle Diabetes Abbildung 9 Exemplarische Darstellung des linken Ventrikels im Querschnitt in den untersuchten Tiergruppen. Es zeigt sich bei den Wildtypmäusen eine signifikante Zunahme der Dicke der Ventrikelwand (Pfeile) nach 8 Wochen Diabetes im Vergleich zu nicht diabeteskranken Kontrolltieren. Dieses konnte bei den Gα11 Knockouttieren nicht beobachtet werden. 42 Ergebnisse 3.4 Untersuchungen der Proteinexpression Bei Typ 1-Diabetes kann die Proteinexpression der Herzmuskelzelle direkt durch die Hyperglykämie als auch durch die Hypoinsulinämie moduliert werden (89, 123). Gleichzeitig kann aber auch die gesteigerte neurohumorale Aktivität bei Diabetes mellitus die Genexpression des Herzens wesentlich beeinflussen. Insbesondere die G-Proteine Gα11 und Gαq, die bei Diabetes in ihrer Expression und Aktivität gesteigert sind, haben einen wesentlichen Einfluss auf die Stimulation zahlreicher Transkriptionsfaktoren. Über diese Signalkaskade können Veränderungen in der Expression Ca2+-regulierender Proteine des SR vermittelt werden, die am Modell der STZ-induzierten Ratte von mehreren Arbeitsgruppen beschrieben wurden (8, 137, 151). Diese Veränderungen kommen ursächlich für die gestörte diastolische und systolische Pumpfunktion des diabetischen Herzens in Betracht. Um den Einfluss einer diabetischen Stoffwechsellage auf die Proteinexpression zu untersuchen, wurden die Herzen von diabetischen Wildtyptieren analysiert. Außerdem konnte durch den Vergleich mit Herzen des Gα11/Gαq Knockoutmodells die Bedeutung der G-Proteine Gα11 und Gαq auf die Proteinexpression in einem diabetischen Zustand untersucht werden. Besonderes Augenmerk lag hierbei auf dem Proteinkomplex des SR-Calcium-Freisetzungskanals. Nachfolgend werden im einzelnen die Ergebnisse zur Expression des Ryanodinrezeptors Typ 2, des Sorcins, des FKBP12.6, des Annexin VII und des Na+/Ca2+-Austauschers dargestellt. Die Western Blots wurden sämtlich mit Calsequestrin als interner Standard durchgeführt und alle dargestellten Ergebnisse auf Calsequestrin normiert abgebildet. 43 Ergebnisse 3.4.1 Expression des Ryanodinrezeptors Typ 2 Der Ryanodinrezeptor, der im Herzen als Isoform 2 exprimiert wird, spielt die zentrale Rolle in der Vermittlung der Calcium-induzierten-Calcium-Freisetzung (CICR) aus dem Sarkoplasmatischen Retikulum. Der Einfluss eines Diabetes mellitus auf dieses 565 kDa schwere Protein wurde durch die vergleichende Messung der Proteinexpression von diabeteskranken Wildtypmäusen mit Kontrollwildtypmäusen untersucht (RYR2 [densitometrische Einheiten] ND: 4,02 ± 0,18 ; DM: 4,22 ± 0,36; p= 0,6 ) (Abb. 10). Der Einfluss der GProteine Gα11 und Gαq unter diabetischen Bedingungen wurde durch vergleichende Messungen der Proteinexpression von Gα11 Knockoutmäusen (RYR2 [densitometrische Einheiten] ND: 5,58 ± 1,03; DM: 7,02 ± 0,8; p= 0,28) (Abb. 11) und Gα11/ Gαq Doppelknockoutmäusen untersucht (RYR2 [densitometrische Einheiten] ND: 1,17 ± 0,04; DM: 1,22 ± 0,02; p= 0,25) (Abb. 12). Die Proteinexpression war in allen drei Gruppen unverändert. RYR2 / Calsequestrin (Densitometrische Einheiten) Wildtyp Ryanodin-Expression 1,2 1 0,8 0,6 0,4 0,2 0 Kontrolle n = 10 Diabetes mellitus n = 8 565 kDa RYR2 55 kDa Calsequestrin DM K DM K DM K DM K DM K DM K DM K DM K K K Abbildung 10 Relative Expression des RYR 2 bei Wildtypmäusen 8 Wochen nach Induktion eines Diabetes mellitus und nicht diabeteskranken Kontrollen. 8 Wochen nach der Induktion eines Diabetes mellitus zeigte sich keine Veränderung der kardialen Ryanodin-Expression von Wildtypmäusen im Vergleich mit nicht diabeteskranken Kontrollen. DM = Diabetes Mellitus; K = Kontrolle 44 Ergebnisse RYR2 / Calsequestrin (Densitometrische Einheiten) Gα α 11 Knockout Ryanodin-Expression 1,6 1,4 1,2 1 0,8 0,6 0,4 0,2 0 Kontrolle n = 7 Diabetes mellitus n = 8 565 kDa RYR2 55 kDa K DM K DM K DM K DM K DM K DM K DM DM Calsequestrin Abbildung 11 Relative Expression des RYR 2 bei Gα11 Knockoutmäusen 8 Wochen nach Induktion eines Diabetes mellitus und nicht diabeteskranken Kontrollen. 8 Wochen nach der Induktion eines Diabetes mellitus zeigte sich keine signifikante Veränderung der kardialen Ryanodin-Expression von Gα11 Knockoutmäusen im Vergleich mit nicht diabeteskranken Kontrollen. DM = Diabetes Mellitus; K = Kontrolle RYR2 / Calsequestrin (Densitometrische Einheiten) Gα α 11/Gα α q Knockout Ryanodin-Expression 1,2 1 0,8 0,6 0,4 0,2 0 Kontrolle n = 4 Diabetes mellitus n = 4 RYR2 Calsequestrin 565 kDa 55 kDa DM K DM K DM K DM K DM Abbildung 12 Relative Expression des RYR 2 bei Gα11/Gαq Doppelknockoutmäusen 8 Wochen nach Induktion eines Diabetes mellitus und nicht diabeteskranken Kontrollen. 8 Wochen nach der Induktion eines Diabetes mellitus zeigte sich keine signifikante Veränderung der kardialen Ryanodin-Expression von Gα11/Gαq Doppelknockoutmäusen im Vergleich mit nicht diabeteskranken Kontrollen. DM = Diabetes Mellitus; K = Kontrolle 45 Ergebnisse 3.4.2 Phosporylierung des RYR2 an Serin 2809 Die Phosphorylierungsstelle Serin 2809 wird als wichtiger Regulationsfaktor des RYR2 bewertet. Die Phosphorylierung wird durch die Proteinkinase A und die Calmodulin-abhängige Kinase vorgenommen (55, 83, 145). Außerdem wird eine schwächere Phosphorylierung durch weitere Enzyme wie der Proteinkinase C und der Proteinkinase G vorgenommen (128, 130). Durch diesen Mechanismus der Phosphorylierung wird die Öffnungswahrscheinlichkeit des RYR2 erhöht und es kommt zu einer größeren Ca2+-Leckage aus dem SR. Die Untersuchungen durch einen spezifischen Antikörper, der nur am phosphorylierten Serin 2809 bindet, konnten keine Unterschiede zwischen diabetischen Tieren und Kontrolltieren, weder in Wildtypen (Serin 2809 [densitometrische Einheiten] ND: 4,25 ± 0,84; DM:4,41 ± 0,70; p= 0,88) (Abb.13), noch in den beiden Knockoutmodellen (Serin 2809 [densitometrische Einheiten] ND: 1,04 ± 0,11; DM: 1,13 ± 0,09; p= 0,56) (Abb. 14) (Serin 2809 [densitometrische Einheiten] ND: 0,90 ± 0,03; DM: 0,93 ± 0,02; p= 0,37) (Abb. 15) nachweisen. Die Ergebnisse wurden jeweils sowohl auf die Expression des RYR2 insgesamt, als auch auf die Dichte des Calsequestrin als interner Standard bezogen ausgewertet. Phosphoserin 2809 / Calsequestrin (Densitometrische Einheiten) Wildtyp RYR2-Phosphorylierung 1,4 1,2 1 0,8 0,6 0,4 0,2 0 Kontrolle n = 10 Diabetes mellitus n = 8 565 kDa Serin 2809 55 kDa Calsequestrin DM K DM K DM K DM K DM K DM K DM K DM K K K Abbildung 13 Relative Phosphorylierung des RYR2 an Serin 2809 bei Wildtypmäusen 8 Wochen nach Induktion eines Diabetes mellitus und nicht diabeteskranken Kontrollen. 8 Wochen nach der Induktion eines Diabetes mellitus zeigte sich keine signifikante Veränderung der kardialen Phosphorylierung des RYR2 an Serin 2809 von Wildtypmäusen im Vergleich mit nicht diabeteskranken Kontrollen. DM = Diabetes Mellitus; K = Kontrolle 46 Ergebnisse Phosphoserin 2809 / Calsequestrin (Densitometrische Einheiten) Gα α11 Knockout RYR2-Phosphorylierung 1,4 1,2 1 0,8 0,6 0,4 0,2 0 Kontrolle n = 7 Diabetes mellitus n = 8 Phosphoserin 565 kDa Calsequestrin 55 kDa K DM K DM K DM K DM K DM K DM K DM DM Abbildung 14 Relative Phosphorylierung des RYR2 an Serin 2809 bei Gα11 Knockoutmäusen 8 Wochen nach Induktion eines Diabetes mellitus und nicht diabeteskranken Kontrollen. 8 Wochen nach der Induktion eines Diabetes mellitus zeigte sich keine signifikante Veränderung der kardialen Phosphorylierung des RYR2 an Serin 2809 von Gα11 Knockoutmäusen im Vergleich mit nicht diabeteskranken Kontrollen. DM = Diabetes Mellitus; K = Kontrolle Phosphoserin 2809 / Calsequestrin (Densitometrische Einheiten) Gα α11/Gα αq Knockout RYR2-Phosphorylierung 1,2 1 0,8 0,6 0,4 0,2 0 Kontrolle n = 4 Diabetes mellitus n = 4 565 kDa Phosphoserin 55 kDa Calsequestrin DM K DM K DM K DM K Abbildung 15 Relative Phosphorylierung des RYR2 an Serin 2809 bei Gα11/Gαq Doppelknockoutmäusen 8 Wochen nach Induktion eines Diabetes mellitus und nicht diabeteskranken Kontrollen. 8 Wochen nach der Induktion eines Diabetes mellitus zeigte sich keine signifikante Veränderung der kardialen Phosphorylierung des RYR2 an Serin 2809 von Gα11/Gαq Doppelknockoutmäusen im Vergleich mit nicht diabeteskranken Kontrollen. DM = Diabetes Mellitus; K = Kontrolle 47 Ergebnisse 3.4.3 Expression des Sorcin Sorcin ist ein 22 kDa schweres Protein. Während die Funktion noch nicht vollständig aufgeklärt ist, ist die Assoziation des Proteins mit dem RYR2 und seine Ca2+abhängige Translokation unumstritten. Es wurde die Expression dieses potentiellen Modulators des RYR2 unter diabetischen Bedingungen bei Wildtypmäusen, sowie bei Gα11 Knockoutmäusen und bei Gα11/Gαq Doppelknockoutmäusen untersucht und mit den jeweiligen Kontrollmäusen verglichen. Interessanterweise zeigte sich eine unveränderte Expression bei den Wildtypmäusen (Sorcin [densitometrische Einheiten] ND: 1,33 ± 0,08; DM: 2,18 ± 0,82; p= 0,21) (Abb. 16) und bei den Gα11 Knockoutmäusen (Sorcin [densitometrische Einheiten] ND: 0,4 ± 0,01; DM: 0,4 ± 0,03; p= 0,98) (Abb. 17), hingegen bei den Gα11/Gαq Doppelknockoutmäusen eine starke Expressionszunahme (Sorcin [densitometrische Einheiten] ND: 0,26 ± 0,11; DM: 0,77 ± 0,08; p= 0,01) nach 8 Wochen Diabetes (Abb.18). Sorcin / Calsequestrin (Densitometrische Einheiten) Wildtyp Sorcin-Expression 2,5 2 1,5 1 0,5 0 Kontrolle n = 10 Diabetes mellitus n = 6 Sorcin Calsequestrin 22 kDa 55 kDa DM K DM K DM K DM K DM K DM K K K K K Abbildung 16 Relative Expression des Sorcins bei Wildtypmäusen 8 Wochen nach Induktion eines Diabetes mellitus und nicht diabeteskranken Kontrollen. 8 Wochen nach der Induktion eines Diabetes mellitus zeigte sich keine signifikante Veränderung der kardialen Sorcin-Expression von Wildtypmäusen im Vergleich mit nicht diabeteskranken Kontrollen. DM = Diabetes Mellitus; K = Kontrolle 48 Ergebnisse Sorcin / Calsequestrin (Densitometrische Einheiten) Gα α11 Knockout Sorcin-Expression 1,2 1 0,8 0,6 0,4 0,2 0 Kontrolle n = 7 Diabetes mellitus n = 8 22 kDa Sorcin Calsequestrin 55 kDa K DM K DM K DM K DM K DM K DM K DM DM Abbildung 17 Relative Expression des Sorcins bei Gα11 Knockoutmäusen 8 Wochen nach Induktion eines Diabetes mellitus und nicht diabeteskranken Kontrollen. 8 Wochen nach der Induktion eines Diabetes mellitus zeigte sich keine signifikante Veränderung der kardialen Sorcin-Expression von Gα11 Knockoutmäusen im Vergleich mit nicht diabeteskranken Kontrollen. DM = Diabetes Mellitus; K = Kontrolle Sorcin / Calsequestrin (Densitometrische Einheiten) Ga11/Gaq Knockout Sorcin-Expression ** 3,5 3 2,5 2 1,5 1 0,5 0 Kontrolle n = 4 Diabetes mellitus n = 4 Sorcin 22 kDa 55 kDa Calsequestrin DM K DM K DM K DM K DM Abbildung 18 Relative Expression des Sorcins bei Gα11/Gαq Doppelknockoutmäusen 8 Wochen nach Induktion eines Diabetes mellitus und nicht diabeteskranken Kontrollen. 8 Wochen nach der Induktion eines Diabetes mellitus zeigte sich eine signifikante, kardiale Expressionszunahme des Sorcins bei Gα11/Gαq Doppelknockoutmäusen im Vergleich mit nicht diabeteskranken Kontrollen. ∗∗ p < 0,01 vs. Kontrolle DM = Diabetes Mellitus; K = Kontrolle 49 Ergebnisse 3.4.4 Expression des FKBP12.6 Das FK506-binding-Protein, FKBP12.6, ist mit dem RYR2 assoziiert. Es scheint, bei Verbindung mit dem RYR2, diesen in einem geschlossenen Zustand zu halten. Seine Rolle als potentieller Antagonist zum Sorcin wird diskutiert. Auch dieses 12,6 kDa schwere Protein wurde in allen drei Gruppen untersucht. Nach Induktion eines Diabetes mellitus wird FKBP12.6 in Wildtypmäusen signifikant vermindert exprimiert (FKBP12.6 [densitometrische Einheiten] ND: 3,79 ± 0,43; DM:1,37 ± 0,34; p= 0,002 ) (Abb. 19). Bei Gα11 Knockoutmäusen konnte diese Alteration nicht nachgewiesen werden (FKBP12.6 [densitometrische Einheiten] ND: 1,15 ± 0,2; DM:0,84 ± 0,03; p= 0,09) (Abb. 20). Gα11/Gαq Doppelknockoutmäuse zeigten diesen Unterschied ebenfalls nicht (FKBP12.6 [densitometrische Einheiten] ND: 0,18 ± 0,02; DM:0,19 ± 0,03; p= 0,63) (Abb. 21). FKBP12.6 / Calsequestrin (Densitometrische Einheiten) Wildtyp FKBP12.6-Expression 1,2 1 0,8 0,6 * 0,4 0,2 0 Kontrolle n = 10 Diabetes mellitus n = 6 12 kDa FKBP12.6 55 kDa Calsequestrin DM DM DM DM DM DM K K K K K K K K K K Abbildung 19 Relative Expression des FKBP12.6 bei Wildtypmäusen 8 Wochen nach Induktion eines Diabetes mellitus und nicht diabeteskranken Kontrollen. 8 Wochen nach der Induktion eines Diabetes mellitus zeigte sich eine signifikante, kardiale Expressionsreduktion des FKBP12.6 bei Wildtypmäusen im Vergleich zu nicht diabeteskranken Kontrollen. ∗ p < 0,05 vs. Kontrolle; DM = Diabetes Mellitus; K = Kontrolle 50 Ergebnisse FKBP12.6 / Calsequestrin (Densitometrische Einheiten) Gα α11 Knockout FKBP12.6-Expression 1,4 1,2 1 0,8 0,6 0,4 0,2 0 Kontrolle n = 7 Diabetes mellitus n = 8 12 kDa FKBP12.6 55 kDa Calsequestrin K K K K K K K DM DM DM DM DM DM DM DM Abbildung 20 Relative Expression des FKBP12.6 bei Gα11 Knockoutmäusen 8 Wochen nach Induktion eines Diabetes mellitus und nicht diabeteskranken Kontrollen. 8 Wochen nach der Induktion eines Diabetes mellitus zeigte sich keine signifikante Veränderung der kardialen FKBP12.6-Expression von Gα11 Knockoutmäusen im Vergleich mit nicht diabeteskranken Kontrollen. DM = Diabetes Mellitus; K = Kontrolle FKBP12.6 / Calsequestrin (Densitometrische Einheiten) Gα α 11/Gα α q Knockout FKBP12.6-Expression 1,4 1,2 1 0,8 0,6 0,4 0,2 0 Kontrolle n = 4 Diabetes mellitus n = 4 12 kDa FKBP12.6 Calsequestrin 55 kDa DM K DM K DM K DM K DM Abbildung 21 Relative Expression des FKBP12.6 bei Gα11/Gαq Doppelknockoutmäusen 8 Wochen nach Induktion eines Diabetes mellitus und nicht diabeteskranken Kontrollen. 8 Wochen nach der Induktion eines Diabetes mellitus zeigte sich keine signifikante Veränderung der kardialen FKBP12.6-Expression von Gα11/Gαq Doppelknockoutmäusen im Vergleich mit nicht diabeteskranken Kontrollen. DM = Diabetes Mellitus; K = Kontrolle 51 Ergebnisse 3.4.5 Expression des Annexin VII Die schon beschriebene Lokalisation des Annexins A7 sowie die Ca2+konzentrationsabhängigen Bindung an Sorcin (16) macht eine Beteiligung des Proteins an der CICR wahrscheinlich. Der Einfluss des Annexins A7 auf die Zellverkürzungs-Frequenz-Beziehung, die in Kardiomyozyten einer Annexin A7defizienten Mauslinie beschrieben wurde (53), zeigt ebenfalls die Bedeutung für die elektromechanische Kopplung. Annexin VII wird durch alternatives Spleißen bedingt in zwei Isoformen, 47 kDa und 51 kDa, ausgeprägt. Die Untersuchungen der drei Gruppen konnte zeigen, dass Annexin VII nach Induktion eines Diabetes mellitus in den Herzen der Wildtypmäuse vermindert ausgeprägt vorlag (Annexin VII [densitometrische Einheiten] ND: 1,09 ± 0,04; DM: 0,58 ± 0,06; p= 0,00 ) (Abb.22). Diese Veränderung wurde nicht in den Herzen von Gα11 Knockoutmäusen beobachtet (Annexin VII [densitometrische Einheiten] ND: 6,19 ± 1,77 ; DM: 5,36 ± 1,37; p= 0,71) (Abb. 23). In den Herzen der Gα11/Gαq Doppelknockoutmäusen konnte Annexin VII verstärkt in der 47 kDa Isoform nachgewiesen werden (Annexin VII [densitometrische Einheiten] ND: 0,28 ± 0,06; DM: 0,54 ± 0,03; p= 0,008) (Abb. 24), die 51 kDa Isoform hingegen zeigte keine Veränderungen (Annexin VII [densitometrische Einheiten] ND: 0,72 ± 0,09; DM: 0,76 ± 0,05; p= 0,46) (nicht gesondert dargestellt). Annexin VII / Calsequestrin (Densitometrische Einheiten) Wildtyp AnnexinVII-Expression 47 kDa 1,2 1 0,8 *** 0,6 0,4 0,2 0 Kontrolle n = 10 Diabetes mellitus n = 6 50 kDa Annexin VII 55 kDa Calsequestrin DM DM DM DM DM DM K K K K K K K K K K Abbildung 22 Relative Expression des Annexin VII bei Wildtypmäusen 8 Wochen nach Induktion eines Diabetes mellitus und nicht diabeteskranken Kontrollen. 8 Wochen nach der Induktion eines Diabetes mellitus zeigte sich eine signifikante, kardiale Expressionsreduktion des Annexin VII bei Wildtypmäusen im Vergleich zu nicht diabeteskranken Kontrollen. ∗∗∗ p < 0,001 vs. Kontrolle; DM = Diabetes Mellitus; K = Kontrolle 52 Ergebnisse Annexin VII / Calsequestrin (Densitometrische Einheiten) Gα α11 Knockout AnnexinVII Expression, 47 kDa 1,4 1,2 1 0,8 0,6 0,4 0,2 0 Kontrolle n = 7 Diabetes mellitus n = 8 50 kDa Annexin VII 55 kDa Calsequestrin K DM K DM K DM K DM K DM K DM K DM DM Abbildung 23 Relative Expression des Annexin VII bei Gα11 Knockoutmäusen 8 Wochen nach Induktion eines Diabetes mellitus und nicht diabeteskranken Kontrollen. 8 Wochen nach der Induktion eines Diabetes mellitus zeigte sich keine signifikante Veränderung der kardialen Annexin VII-Expression von Gα11 Knockoutmäusen im Vergleich mit nicht diabeteskranken Kontrollen. DM = Diabetes Mellitus; K = Kontrolle Annexin VII / Calsequestrin (Densitometrische Einheiten) Gα α11/Gα αq Knockout AnnexinVII-Expression, 47 kDa ** 2,5 2 1,5 1 0,5 0 Kontrolle n = 4 Diabetes mellitus n = 4 50 kDa Annexin VII 55 kDa Calsequestrin DM K DM K DM K DM K DM Abbildung 24 Relative Expression des Annexin VII bei Gα11/Gαq Doppelknockoutmäusen 8 Wochen nach Induktion eines Diabetes mellitus und nicht diabeteskranken Kontrollen. 8 Wochen nach der Induktion eines Diabetes mellitus zeigte sich eine signifikante, kardiale Expressionszunahme des AnnexinVII bei Gα11/Gαq Doppelknockoutmäusen im Vergleich mit nicht diabeteskranken Kontrollen. Das Säulendiagramm zeigt die quantitative Analyse der Annexin VII-Isoform bei 47 kDa. ∗∗ p < 0,01 vs. Kontrolle; DM = Diabetes Mellitus; K = Kontrolle 53 Ergebnisse 3.4.6 Expression des Na+-Ca2+-Austauschers Der Na+-Ca2+-Austauscher (NCX) dient der Na+-gradientenabhängigen Elimination von Ca2+ aus der Zelle. Der NCX setzt sich im Western Blot aus drei Banden zusammen (70 kDa, 120 kDa, 160 kDa). Die densitometrische Auswertung wurde mit der 120 kDa-Bande vorgenommen, da diese den funktionellen, adulten NCX im Herzen repräsentiert. Es zeigte sich eine deutliche Expressionsminderung in den Herzen der diabeteskranken Wildtypmäuse gegenüber den Kontrollwildtypmäusen (NCX 120 [densitometrische Einheiten] ND: 1,04 ± 0,03 ;DM: 0,81 ± 0,09; p= 0,02) (Abb.25). Dies wurde bei den Gα11 Knockoutmäusen nicht beobachtet (NCX 120 [densitometrische Einheiten] ND: 0,68 ± 0,12 ;DM: 0,62 ± 0,12; p= 0,71) (Abb. 26). Auch bei den Gα11/Gαq Doppelknockoutmäusen wurde dieser Unterschied in der Expression nicht nachgewiesen (NCX 120 [densitometrische Einheiten] ND: 0,8 ± 0,07; DM:0,82 ± 0,09; p= 0,83) (Abb. 27). NCX / Calsequestrin (Densitometrische Einheiten) Wildtyp NCX-Expression 1,2 * 1 0,8 0,6 0,4 0,2 0 Kontrolle n = 10 Diabetes mellitus n = 6 120 kDa NCX 55 kDa Calsequestrin DM DM DM DM DM DM K K K K K K K K K K Abbildung 25 Relative Expression des NCX bei Wildtypmäusen 8 Wochen nach Induktion eines Diabetes mellitus und nicht diabeteskranken Kontrollen. 8 Wochen nach der Induktion eines Diabetes mellitus zeigte sich eine signifikante, kardiale Expressionsreduktion des NCX bei Wildtypmäusen im Vergleich zu nicht diabeteskranken Kontrollen. ∗ p < 0,05 vs. Kontrolle; DM = Diabetes Mellitus; K = Kontrolle 54 Ergebnisse NCX / Calsequestrin (Densitometrische Einheiten) Gα α11 Knockout NCX-Expression 1,4 1,2 1 0,8 0,6 0,4 0,2 0 Kontrolle n = 7 Diabetes mellitus n = 8 120 kDa NCX 55 kDa Calsequestrin K K K K K K K DM DM DM DM DM DM DM DM Abbildung 26 Relative Expression des NCX bei Gα11 Knockoutmäusen 8 Wochen nach Induktion eines Diabetes mellitus und nicht diabeteskranken Kontrollen. 8 Wochen nach der Induktion eines Diabetes mellitus zeigte sich keine signifikante Veränderung der kardialen NCX-Expression von Gα11 Knockoutmäusen im Vergleich mit nicht diabeteskranken Kontrollen. DM = Diabetes Mellitus; K = Kontrolle NCX / Calsequestrin (Densitometrische Einheiten) Gα 11/Gα q Knockout NCX-Expression 1,4 1,2 1 0,8 0,6 0,4 0,2 0 Kontrolle = 4 Diabetes mellitus n = 4 NCX 120 kDa Calsequestrin 55 kDa DM K DM K DM K DM K DM Abbildung 27 Relative Expression des NCX bei Gα11/Gαq Doppelknockoutmäusen 8 Wochen nach Induktion eines Diabetes mellitus und nicht diabeteskranken Kontrollen. 8 Wochen nach der Induktion eines Diabetes mellitus zeigte sich keine signifikante Veränderung der kardialen NCX-Expression von Gα11/Gαq Doppelknockoutmäusen im Vergleich mit nicht diabeteskranken Kontrollen. DM = Diabetes Mellitus; K = Kontrolle 55 Ergebnisse Wildtyp Gα11 Knockout Gα11/Gαq Knockout RYR 2 ⇒ ⇒ ⇒ Phosphorylierung des ⇒ ⇒ ⇒ Sorcin ⇒ ⇒ ⇑ FKBP 12.6 ⇓ ⇒ ⇒ Annexin VII ⇓ ⇒ ⇑ NCX ⇓ ⇒ ⇒ RYR2 an Serin 2809 Tabelle 7 Übersicht über die vergleichende Analyse der Expression kardialer Proteine im Western Blot-Verfahren. Dargestellt ist die signifikante, kardiale Proteinexpressionsveränderungen der diabetischen Versuchsgruppen im Vergleich zu den Kontrollgruppen. 56 Ergebnisse 3.5 Quantitative Bestimmung des Ca²+-Verlustes an isolierten Membranvesikeln Die Funktion des RYR2 ist im Zusammenhang mit der Entstehung des kardialen Phänotyps bei Diabetes Mellitus als wichtiger pathogenetischer Faktor zu deuten (68). Durch eine gestörte Funktion dieses Kanals, der für die Freisetzung des Ca2+ aus dem SR im Kontraktionszyklus zuständig ist, kann es zu einer gestörten Ca2+Homöostase kommen und damit zu einem veränderten, pathologischen Kontraktionsverhalten (118). Um die Funktion des RYR2 zu untersuchen wurde an isolierten Membranvesikeln die Fähigkeit zur Ca2+-Aufnahme sowohl in An- als auch in Abwesenheit von Ruthenium Rot, einem selektiven Hemmstoff des RYR2, durch radioaktive Ca2+-Aufnahme-Versuche gemessen. Die Differenz der Aufnahme des Ca2+ in die Membranvesikel bei An- und Abwesenheit des Ruthenium Rots stellt den Ca2+-Verlust über den RYR2 dar. Dieser Verlust kann unter pathologischen Bedingungen gesteigert sein und damit zu einer veränderten Ca2+-Homöostase und pathologischem Kontraktionsveralten der Kardiomyozyten führen. Die Berechnung des Ca2+-Verlustes bei den Wildtypmäusen (Abb. 28) konnte eine signifikante Zunahme des Ca2+-Verlustes auf 300% über den RYR2 nach 8wöchigem Diabetes mellitus nachweisen (Ca2+-Verlust [nmol/mg] ND: 15,53 ± 1,98; DM: 45,72 ± 6,7; p= 0,02). Bei den Gα11 Knockoutmäusen konnte diese Veränderung des Ca2+Verlustes nicht nachgewiesen werden (Ca2+-Verlust [nmol/mg] ND: 38,71 ± 4,83; DM: 48,44 ± 6,94; p= 0,27). Die diabeteskranken Tiere hatten äquivalente Werte zu den Kontrolltieren (Abb. 29). Ein ähnliches Bild wurde in der Versuchsgruppe der Gα11/Gαq Doppelknockoutmäusen beobachtet. Auch hier bestand kein signifikanter Unterschied zwischen dem Ca2+-Verlust der diabeteskranken Tiere und dem Ca2+Verlust der Kontrolltiere (Ca2+-Verlust [nmol/mg] ND: 87,56 ± 24,85; DM: 77,18 ± 37,21; p= 0,82) (Abb. 30). 57 Ergebnisse Wildtyp Kontrolle Kontrolle Wildtyp 0 n=4 Wildtyp DM Wildtyp DM n=6 relativer Ca²+ Verlust 0,5 1 1,5 DM = Diabetes Mellitus ∗ p < 0,05 vs. Kontrolle 2 2,5 3 3,5 * 4 Gα11 KO Kontrolle Ga11 Kontrolle 0 n=6 Gα DM 11 KO DM Ga11 n=4 0,2 relativer Ca²+ Verlust 2+ Abbildung 28 Darstellung des Mittelwertes des Ca Verlustes ± SEM bei Kontrollwildtypmäusen, sowie 8 Wochen nach Induktion eines Diabetes mellitus bei Wildtypmäusen aus isolierten Membranvesikeln des Herzmuskelgewebes. Es kommt zu einer signifikanten 2+ Vergrößerung des Ca -Verlustes über den RYR2 nach der Induktion des Diabetes mellitus. 0,4 0,6 2+ Abbildung 29 Darstellung des Mittelwertes des Ca Verlustes ± SEM bei Gα11 Knockoutmäusen, sowie bei Gα11 Knockoutmäusen 8 Wochen nach Induktion eines Diabetes mellitus aus isolierten Membranvesikeln des Herzmuskelgewebes. Es kommt nicht zu einer 2+ Veränderung des Ca -Verlustes nach der Induktion des Diabetes mellitus. DM = Diabetes Mellitus 0,8 KO = Knockout 1 1,2 1,4 1,6 Gα11/GαqKontrolle KO Kontrolle Gα 11/Gαq KO Ga11/q Ga11/q DMDM 0 relativer Ca²+ Verlust 0,2 0,4 0,6 n=4 2+ Abbildung 30 Darstellung des Mittelwertes des Ca Verlustes (nmol/mg Protein) ± SEM bei Gα11/Gαq Knockoutmäusen, sowie bei Gα11/Gαq Knockoutmäusen 8 Wochen nach Induktion eines Diabetes mellitus aus isolierten Membranvesikeln des Herzmuskelgewebes. Es kommt nicht zu einer 2+ Veränderung des Ca -Verlustes nach der Induktion des Diabetes mellitus. DM = Diabetes Mellitus 0,8 1 1,2 1,4 58 n=4 KO = Knockout Ergebnisse 3.6 Immunhistochemie Bei Diabetes mellitus ist die Aktivität der PKC gesteigert (46, 70, 146). Dies ist wahrscheinlich von wesentlicher Bedeutung für die Entstehung der Hypertrophie, sowie für Veränderungen im Kontraktionsverhalten und der intrazellulären Ca2+Regulation des diabetischen Herzens (29). Die Aktivierung der PKC kann sowohl Rezeptor-vermittelt durch Stimulation mit Angiotensin II, Endothelin-1, u.a. erfolgen (29, 127), Rezeptor-unabhängig direkte Folge der Hyperglykämie sein (30, 90) oder additiv durch beide Faktoren begünstigt werden (80). Verschiedene Stimuli unterscheiden sich dabei durch die Aktivierung spezifischer Isoformen der PKC (29, 30, 90). Nach Aktivierung wird in Abhängigkeit der Isoform und des Stimulus eine Translokation der Kinase aus dem Zytosol an die Plasmamembran, das Zytoskelett oder in den Zellkern beobachtet, die dort ihre spezifische Wirkung entfaltet. Um die verschiedenen Isoformen der PKC in ihrem Expressionsmuster, sowie ihre intrazelluläre Verteilung im Herzen unter diabetischen Bedingungen zu untersuchen, wurden immunhistochemische Färbungen angewandt. Außerdem wurde die Bedeutung des G-Proteins Gα11 auf die Expression und Translokation der PKC anhand des Gα11 Knockout-Mausmodells sowohl unter diabetischen als auch unter Kontrollbedingungen analysiert. 3.6.1 Immunhistochemische Ergebnisse zum Einfluss eines Typ1-Diabetes auf die kardiale Expression und Translokation von PKC-Isoformen bei Wildtypmäusen Die Expression der PKC α war im Herzen von Wildtypmäusen gleichmäßig über das Myokard verteilt. Durch die Induktion eines Diabetes mellitus wurde die Expression der PKC α in den Kardiomyozyten signifikant gesteigert (PKC α [densitometrische Einheiten] ND: 16,14 ± 3,33; DM: 29,12 ± 1,62; p= 0,006). Die intrazelluläre Verteilung hingegen war nicht wesentlich verändert. Die Expression der PKC βII war im Herzen deutlich messbar, ohne jedoch durch einen Diabetes mellitus beeinflusst zu werden (PKC βII [densitometrische Einheiten] ND: 8,65 ± 0,76; DM: 9,67 ± 2,46; p= 0,7). Die PKC δ zeigte tendenziell eine stärkere Ausprägung unter diabetischen Bedingungen in den Herzen von Wildtypmäusen, doch war dieser Unterschied nicht signifikant (PKC δ [densitometrische Einheiten] ND: 25,3 ± 1,6; DM: 31,18 ± 5,32; p= 0,31). Die PKC ε wurde im Herzen stark exprimiert. Einen Einfluss des Diabetes mellitus auf die Ausprägung konnte man 59 Ergebnisse nicht erkennen (PKC ε [densitometrische Einheiten] ND: 24,36 ± 2,19; DM: 18,2 ± 2,5; p= 0,1). Allerdings ist anzumerken, dass sich die intrazelluläre Verteilung deutlich verschoben hatte, so dass von einer Translokation ausgegangen werden muss, die abhängig vom Diabetes mellitus zu sein scheint. Die Lokalisation der PKC ε zeigte am immunhistochemischen Präparat nach Induktion eines Diabetes mellitus eine stärkere Membranbindung (Abb. 31). Die PKC θ zeigte eine nur mäßige Ausprägung im Herzen. Die Expression war offensichtlich unabhängig von der Induktion eines Diabetes (PKC θ [densitometrische Einheiten] ND: 9,44 ± 0,42; DM: 8,53 ± 1,67; p= 0,61). Die PKC ζ zeigte in den Herzen von Wildtypmäusen nur eine geringe Expression, die unter diabetischen Bedingungen deutlich und signifikant zunahm (PKC ζ [densitometrische Einheiten] ND: 32,16 ± 4,65; DM: 48,8 ± 3,98; p= 0,02). Auch bei dieser Isoform der PKC zeigte sich das schon bei der PKC ε beschriebene Phänomen der Translokation. Die PKC kann u.a. durch Phosphorylierung aktiviert werden. Die Phosphorylierung der PKC ζ und λ war unter Kontrollbedingungen kaum zu beobachten. Ein experimenteller Diabetes mellitus führte zu einer starken Phosphorylierung dieser Isoformen im Herzen (Phospho-PKC ζ/λ [densitometrische Einheiten] ND: 14,5 ± 1,95; DM: 29,23 ± 3,84; p= 0,006). Die Isoform ζ zeigte somit nach Induktion eines Diabetes mellitus sowohl eine vermehrte Expression, als auch eine vermehrte Phosphorylierung. Um zu analysieren, ob die Phosphorylierung unabhängig von der Expressionszunahme vermehrt auftrat, wurden die jeweiligen Ergebnisse im Verhältnis zueinander betrachtet. Es zeigte sich, dass die Zunahme der Phosphorylierung mit der Expressionszunahme in gleichem Maße einherging. Das Verhältnis der phosphorylierten PKC ζ−Form zur nicht-phosphorylierten PKC ζ-Form blieb also konstant (PKC ζ/Phospho-PKC ζ/λ [densitometrische Einheiten] ND: 2,44 ± 0,52; DM: 1,73 ± 0,11; p= 0,18) es kam also zu keiner zusätzlichen Aktivierung dieser Isoform durch die Induktion des Typ1Diabetes. Die Phosphorylierung der Isoformen δ und θ war sowohl bei Kontrolltieren als auch diabetischen Tieren nur in geringem Ausmaß zu beobachten (Phospho-PKC δ/θ [densitometrische Einheiten] ND: 14,90 ± 1,42; DM: 13,56 ± 2,14; p= 0,61) (Abb. 32). 60 Ergebnisse Densitometrische Einheiten 60 * 50 40 ** 30 20 10 0 PKC PKCαa Kontrolle Kontrolle n=6 PKCaαDM PKC DM n=6 PKC α PKC b βΙΙII PKC Kontrolle Kontrolle n=6 PKC PKC b IIβΙΙDM PKC βΙΙ DM n=6 PKCeε PKC Kontrolle Kontrolle n=6 PKC ε PKC e DM DM n=6 PKC ε PKC PKCζz Kontrolle Kontrolle n=6 PKCz ζDM PKC DM n=6 PKC ζ Kontrolle DM Abbildung 31 Immunhistochemische Färbungen der PKC Isoformen im Herzen bei Wildtypmäusen unter Kontrollbedingungen, sowie nach Induktion eines Typ 1-Diabetes. Die Intensität der Färbung spiegelt die Stärke der Expression wider. 400-fache Vergrößerung. Die PKC Isoform α und ζ zeigten eine signifikante, kardiale Expressionzunahme nach 8 Wochen DM im Vergleich zu der Kontrollgruppe. Die PKC Isoformen βII und ε hingegen zeigten keine veränderte Expression. ∗ p < 0,05 vs. Kontrolle; ∗∗ p < 0,01 vs. Kontrolle; DM = Diabetes mellitus 61 Ergebnisse Densitometrische Einheiten 40 35 ** 30 25 20 15 10 5 0 Phospho-ζ/λ Phospho-PKC Phospho- ζ/λ Phospho-PKC Phospho- δ/θ Phospho-PKC Phospho- δ/θ PKC Phospho-PKC PKCdδ PKC-z/l PKC-z/l PKC-d/q PKC-d/q Kontrolle Kontrolle DM DM Kontrolle DM DM n=6 Kontrolle n=6 PhosphoPKC-ζ/λ n=6 Kontrolle n=6 PhosphoPKC-δ/θ n=6 PKC d DM PKC δ DM n=6 PKC δ PKC PKCqθ Kontrolle Kontrolle n=6 PKC q DM PKC θ DM n=6 PKC θ Kontrolle DM Abbildung 32 Immunhistochemische Färbungen der PKC Isoformen im Herzen bei Wildtypmäusen unter Kontrollbedingungen, sowie nach Induktion eines Typ 1-Diabetes. Die Intensität der Färbung spiegelt die Stärke der Expression bzw. der Phosphorylierung wider. 400-fache Vergrößerung. Die Phosphorylierung der PKC Isoformen ζ/λ zeigten eine starke kardiale Zunahme nach 8 Wochen DM im Vergleich mit der Kontrollgruppe. Die Phosphorylierung der Isoformen δ/θ veränderte sicht nicht. Ebenso veränderte sich die Expression der PKC Isoformen δ und θ nach 8 Wochen DM nicht. ∗∗ p < 0,01 vs. Kontrolle; DM = Diabetes mellitus 62 Ergebnisse 3.6.2 Immunhistochemische Ergebnisse zum Einfluss eines Typ1-Diabetes auf die kardiale Expression und Translokation von PKC-Isoformen bei Gα α11 Knockoutmäusen Im Gegensatz zu Herzmuskelgeweben von Wildtypmäusen wurde die PKC α in Herzen von Gα11 Knockoutmäusen nur sehr schwach exprimiert. Auch nach Induktion eines Diabetes mellitus änderte sich dies nicht (PKC α [densitometrische Einheiten] ND: 6,99 ± 1,14; DM: 8,16 ± 1,13; p= 0,48). Dies deutet auf eine Gα11abhängige Stimulation der PKC α hin. Die PKC βII zeigte im Herzen der Gα11 Knockoutmäuse das gleiche Verhalten wie die PKC α (PKC βII [densitometrische Einheiten] ND: 7,09 ± 1,17; DM: 7,38 ± 0,68; p= 0,83). PKC βII ist sowohl in Wildtyp als auch in Knockout-Herzen gering exprimiert und scheint demnach durch das Knockout nicht beeinflusst zu werden. Im Vergleich zu Herzmuskelgewebe von Mäusen des Wildtyps wurde die PKC δ in Knockoutmäusen deutlich schwächer exprimiert. Nach Induktion eines Diabetes kam es hier zu einer signifikanten Zunahme der PKC δ−Dichte, die jedoch selbst unter diesen Bedingungen nur das Expressionsniveau der Wildtypmäuse erreichte. Dieses Ergebnis deutet daraufhin, dass die basale Expression der PKC δ Gα11-abhängig reguliert wird. Die Zunahme der PKC δ−Dichte bei Typ-1 Diabetes scheint jedoch unabhängig von der Signalkaskade über Gα11 zu erfolgen. Die PKC ε wurde in den Herzen der Gα11 Knockoutmäuse stark ausgebildet. Eine Tendenz der vermehrten Expression durch den Diabetes mellitus ließ sich erkennen, allerdings war dieser Unterschied nicht signifikant (PKC ε [densitometrische Einheiten] ND: 21,5 ± 1,84; DM: 28,87 ± 3,18; p= 0,06). Zu erwähnen ist auch bei den Gα11 Knockoutmäusen eine Translokation der PKC ε an die Plasmamembran, wie sie auch im Herzen von Wildtypmäusen zu beobachten war. Die PKC θ zeigte genauso wie bei den Wildtypmäusen nur ein niedriges Expressionsniveau, das auch nicht durch den Diabetes mellitus beeinflusst wurde (PKC θ [densitometrische Einheiten] ND: 10,29 ± 1,25; DM: 8,95 ± 1,07; p= 0,43). Die PKC ζ wurde in ihrem hohen Expressionsniveau nicht durch einen Diabetes mellitus beeinflusst (PKC ζ [densitometrische Einheiten] ND: 27,04 ± 2,25; DM: 21,38 ± 1,31; p= 0,055). Allerdings wiesen die diabeteskranken Knockouttiere im Gegensatz zu der erhöhten Expression bei diabeteskranken Mäusen des Wildtyps sogar die Tendenz zu einer schwächeren PKC ζ Ausbildung auf. Die Phosphorylierung der PKC ζ und λ bzw. der PKC δ und θ war bei den Gα11 63 Ergebnisse Knockoutmäuse lediglich auf einem niedrigen Niveau nachzuweisen, ohne eine Änderung bei Diabetes mellitus erkennen zu lassen (Phospho-PKC ζ/λ [densitometrische Einheiten] ND: 12,60 ± 1,09; DM: 14,60 ± 1,6; p= 0,32) (PhosphoPKC δ/θ [densitometrische Einheiten] ND: 9,15 ± 0,80; DM: 9,84 ± 1,93; p= 0,73). Augenfällig ist hierbei, dass es zu einer Expressionszunahme der PKC δ bei Diabetes mellitus kam, ohne dass jedoch eine vermehrte Phosphorylierung der PKC δ auftrat. Also nahm die Gesamtmenge des Proteins zu, der relative Anteil der phosphorylierten, und damit wahrscheinlich aktivierten Form des Proteins, nahm hingegen ab (Abb. 33 und Abb. 34). 64 Ergebnisse Densitometrische Einheiten 35 30 25 20 15 10 5 0 PKC PKCαa Kontrolle Kontrolle n=6 PKCaαDM PKC DM n=6 PKC α PKC βΙΙ PKC b II Kontrolle Kontrolle n=6 PKCb βΙΙ PKC II DM DM n=6 PKC βΙΙ PKC PKCε e Kontrolle Kontrolle n=6 PKC ε PKC e DM DM n=6 PKC ε PKC z ζ PKC Kontrolle Kontrolle PKC ζ PKC z DM n=6 DM n=6 PKC ζ Kontrolle DM Abbildung 33 Immunhistochemische Färbungen der PKC Isoformen im Herzen bei Gα11 Knockoutmäusen unter Kontrollbedingungen, sowie 8 Wochen nach Induktion eines Typ 1-Diabetes. Die Intensität der Färbung spiegelt die Stärke der Expression wider. 400-fache Vergrößerung. Bei Gα11 Knockoutmäusen konnte keine Änderung der Expression der PKC Isoformen α, βII, ε und ζ nach 8 Wochen Diabetes mellitus beobachtet werden. DM = Diabetes mellitus 65 Ergebnisse ** Densitometrische Einheiten 30 25 20 15 10 5 0 Phospho-PKC ζ/λ Phospho-PKC PKC δd PhosphoPhospho-ζ/λ Phospho-PKC Phospho- δ/θ Phospho-PKC Phospho- δ/θ PKC Kontrolle DM DM Kontrolle DM DM Kontrolle PKC-z/l PKC-z/l PKC-d/q PKC-d/q Kontrolle n=6 Kontrolle n=6 PhosphoPKC-ζ/λ n=6 Kontrolle n=6 PhosphoPKC-δ/θ n=6 PKCd δDM PKC DM n=6 PKC δ PKC θq PKC Kontrolle Kontrolle n=6 PKC PKC q θDM DM n=6 PKC θ Kontrolle DM Abbildung 34 Immunhistochemische Färbungen der PKC Isoformen im Herzen bei Gα11 Knockoutmäusen unter Kontrollbedingungen, sowie 8 Wochen nach Induktion eines Typ 1-Diabetes. Die Intensität der Färbung spiegelt die Stärke der Expression bzw. der Phosphorylierung wider. 400-fache Vergrößerung. Bei den Gα11 Knockoutmäusen nahm die Expression der PKC Isoform δ nach 8 Wochen DM signifikant zu. Die PKC Isoform θ zeigte keine signifikante Veränderung. Die Phosphorylierung der PKC Isoformen ζ/λ und δ/θ zeigte nach der Diabetesinduktion keine Veränderungen. ∗∗ p < 0,01 vs. Kontrolle; DM = Diabetes mellitus 66 Ergebnisse Vergleicht man die beiden Versuchsgruppen miteinander, so fällt auf, dass die veränderte und jeweils verstärkte Expression der PKC α und ζ sowie die erhöhte Phosphorylierung der Isoformen ζ/λ bei den Wildtypmäusen nach Induktion eines Typ1-Diabetes bei den Gα11 Knockoutmäusen unter den selben experimentellen Bedingungen nicht hervortreten. Dies deutet darauf hin, dass bei den Wildtypmäusen diese Veränderungen in Abhängigkeit vom G-Protein Gα11 entstehen und dementsprechend bei den Knockoutmäusen nicht auftreten können. Der direkte Vergleich der Gewebeschnitte von Mäusen des Wildtyps und von Gα11 Knockoutmäusen unter Kontrollbedingungen zeigte eine verminderte Dichte der PKC α (PKC α [densitometrische Einheiten] WT: 16,14 ± 3,33; KO: 6,98 ± 1,14; p= 0,04) und δ (PKC δ [densitometrische Einheiten] WT: 31,18 ± 5,32; KO: 17,02 ± 1,22; p= 0,03) in den Knockouttieren. Dabei war die PKC δ bei Gα11 Knockoutmäusen sowohl in Bezug auf die Expression des Proteins insgesamt, als auch in ihrer phosphorylierten Form in Herzmuskelgewebe vermindert (Phospho-PKC δ/θ [densitometrische Einheiten] WT: 14,91 ± 1,42; KO: 9,15 ± 0,8; p= 0,005). Nach Induktion eines Typ1-Diabetes wird die Proteinexpression auf das Niveau von Kontrolltieren des Wildtyps angehoben. Die absolute Menge an phosphorylierter PKC δ bleibt durch die Hyperglykämie bei Gα11 Knockoutmäusen jedoch unbeeinflusst. Es ist somit zu vermuten, dass in Abwesenheit von Gα11 auch bei vermehrter Expression von PKC δ nach Induktion eines Typ1-Diabetes eine Phosphorylierung und damit Aktivierung dieses second messengers ausbleibt (Abb. 35 und Abb. 36). 67 Ergebnisse Densitometrische Einheiten 40 35 30 25 20 15 * 10 5 0 PKC PKCαaWildtyp Wildtyp n=6 PKC PKCαaKO KO n=6 PKC α PKCbβΙΙ PKCb βΙΙ PKC ε Wildtyp PKCeεKO KO PKC Wildtyp PKC II WWildtyp ildtyp PKC II KOKO PKC eW ildtyp PKC PKCzζW ildtyp n=6 n=6 PKC βΙΙ n=6 n=6 PKC ε n=6 PKCz KO ζ KO PKC n=6 PKC ζ Wildtyp Knockout Abbildung 35 Immunhistochemische Färbungen der PKC Isoformen im Herzen unter Kontrollbedingungen bei Wildtypmäusen und Gα11 Knockoutmäusen. Die Intensität der Färbung spiegelt die Stärke der Expression wider. 400-fache Vergrößerung. Die PKC Isoform α zeigte eine signifikante, kardiale Expressionsreduktion bei Gα11 Knockoutmäusen im Vergleich zu Wildtypmäusen. Die PKC Isoformen βII, ε und ζ waren nicht signifikant verändert bei Gα11 Knockoutmäusen im Vergleich zu Wildtypmäusen. ∗ p < 0,05 vs. Kontrolle; KO = Knockout 68 Densitometrische Einheiten Ergebnisse 40 35 30 25 ** 20 * 15 10 5 0 Phospho- Phospho- Phospho- Phospho- PKC d PKC d KO PKC δ n=6 Wildtyp nKO =6 n=6 Wildtyp nKO =6 n=6 KO n=6 Phospho-PKC ζ/λ Phospho-PKC ζ/λ Phospho-PKC δ/θ Phospho-PKC δ/θ PKC δ PKC-z/l PKC-z/l PKC-d/q PKC-d/q Wildtyp Wildtyp KO Wildtyp KO Wildtyp PhosphoPKC-ζ/λ PhosphoPKC-δ/θ PKC δ PKC q PKC θ Wildtyp Wildtyp PKC q KO PKC θ n=6 KO n=6 PKC θ Wildtyp Knockout Abbildung 36 Immunhistochemische Färbungen der PKC Isoformen im Herzen unter Kontrollbedingungen bei Wildtypmäusen und Gα11 Knockoutmäusen. Die Intensität der Färbung spiegelt die Stärke der Expression bzw. der Phosphorylierung wider. 400-fache Vergrößerung. PKC δ war sowohl in Bezug auf die Expression des Proteins insgesamt als auch in ihrer phosphorylierten Form in Herzen von Gα11 Knockoutmäusen signifikant vermindert. Bei den Gα11 Knockoutmäusen war die Phosphorylierung der PKC Isoformen ζ/λ und die Expression der PKC Isoform θ gegenüber den Wildtypmäusen hingegen unverändert. ∗ p < 0,05 vs. Kontrolle; ∗∗ p < 0,01 vs. Kontrolle; KO = Knockout 69 Ergebnisse DM Wildtyp DM Gα11 Knockout PKC α ⇑ ⇒ PKC βΙΙ ⇒ ⇒ PKC δ ⇒ ⇑ PKC ε ⇒ ⇒ PKC θ ⇒ ⇒ PKC ζ ⇑ ⇒ Phospho-PKC-ζ/λ ⇑ ⇒ Phospho-PKC-δ/θ ⇒ ⇒ Tabelle 8 Signifikante Expressionsveränderung der PKC Isoformen im Myokard von Wildtypmäusen und von Gα11 Knockoutmäusen im Vergleich zu den jeweiligen Kontrollgruppen. Gα11 Knockout PKC α ⇓ PKC βΙΙ ⇒ PKC δ ⇓ PKC ε ⇒ PKC θ ⇒ PKC ζ ⇒ Phospho-PKC-ζ/λ ⇒ Phospho-PKC-δ/θ ⇓ Tabelle 9 Signifikante Expressionsveränderung der PKC Isoformen im Myokard von Gα11 Knockoutmäusen im Vergleich zu Mäusen des Wildtyps jeweils unter Kontrollbedingungen. 70 Diskussion 4 Diskussion Ziel der vorliegenden Arbeit war es, die Bedeutung der G-Proteine Gα11 und Gαq für die Entwicklung der myokardialen Hypertrophie unter den speziellen Bedingungen des Typ1-Diabetes zu untersuchen. Hierzu wurden funktionelle und biochemische Untersuchungen an kardialem Gewebe von Wildtypmäusen, Gα11 Knockoutmäusen und Gα11/Gαq Doppelknockoutmäusen durchgeführt. 4.1 Das G-Protein Gα α11 ist wesentlich an der Entwicklung einer Hypertrophie des Herzens bei Typ1-Diabetes beteiligt Die Hypertrophie und diastolische Dysfunktion des Herzmuskels gehören zu den kardialen Manifestationsformen, die frühzeitig bei Diabetes mellitus zu beobachten sind. In der vorliegenden Arbeit konnte durch intraperitoneale Injektion von Streptozotocin in Mäusen eine stabile Hyperglykämie erzeugt werden, die über den Verlauf von 8 Wochen zur Entwicklung einer ausgeprägten myokardialen Hypertrophie führte. Dies konnte sowohl in vivo mittels echokardiographischer Bestimmung der Septumdicke, der Wanddicken und der linksventrikulären Diameter als auch durch direkte Messung des Herzgewichtes nachgewiesen werden. Zeichen der Herzinsuffizienz, wie sie unter ähnlichen experimentellen Bedingungen am Modell der Ratte beschrieben wurden, konnten in der aktuellen Untersuchung nicht nachgewiesen werden. Dies könnte auf die verschiedenen Spezies oder die unterschiedliche Dosierung und Applikation des Streptozotocin zurückzuführen sein. Der Pathomechanismus, der der Entstehung einer Myokardhypertrophie bei Diabetes mellitus zugrunde liegt, ist noch nicht bekannt. Zahlreiche Untersuchungen belegen, dass bereits die alleinige Hyperglykämie durch Stimulation des Polyol-Pathways, der PKC als auch der Bildung von AGEs eine zentrale Rolle bei der Entwicklung diabetesbedingter Folgekrankheiten am Herzen spielt (9, 90, 139). Auch der direkte Einfluss einer Hyperglykämie auf die kardiovaskuläre Genexpression konnte gezeigt werden (123). Demgegenüber spricht ein hohes Maß an Evidenz für die Bedeutung neurohumoraler Mechanismen, wie z.B. einer vermehrten Stimulation und Expression des AT1-Rezeptors, bei der Entstehung der Hypertrophie am diabetischen Herzen (105) oder erhöhter Endothelin-1-Serumspiegel bei Patienten 71 Diskussion mit Diabetes mellitus (86). Angiotensin- und Endothelin-Rezeptoren gehören zur Gruppe der G-Protein-gekoppelten Rezeptoren, die ihr intrazelluläres Signal über die G-Proteine Gα11 und Gαq vermitteln. Die vorliegenden Ergebnisse unterstreichen die Bedeutung der neurohumoralen Aktivierung für die Hypertrophieentwicklung in der frühen Phase eines Typ1-Diabetes, da unter identischen experimentellen Bedingungen für das Gα11 Knockout Mausmodell im Gegensatz zum Wildtyp kein kardialer Phänotyp nachgewiesen werden konnte. Gα11 wird in allen Organsystemen zusammen mit Gαq co-exprimiert und reguliert wie Gαq die funktionelle Kopplung G-Protein-gekoppelter Rezeptoren mit der Phospholipase C. Da die gezielte Deletion von Gα11 im Genom der Maus keinen Phänotyp zeigte, wurde vermutet, dass in Anbetracht ihrer sehr ähnlichen Rezeptoren- und Effektoren-Spezifität Gαq die gestörte Signaltransduktion über Gα11 vollständig kompensieren kann (11, 52, 92, 143). Die vorliegenden Ergebnisse zeigen eindrucksvoll, dass eine entsprechende Kompensation unter den pathophysiologischen Bedingungen eines Typ1-Diabetes jedoch nicht stattfindet. Die alleinige Deletion von Gα11 zeigt sowohl im Phänotyp, in der Aktivierung spezifischer PKC-Isoformen, als auch in der Proteinexpression eindeutige Unterschiede im Vergleich zu Kontrolltieren des Wildtyps, die zumindest in Bezug auf den Phänotyp kardioprotektiv sind. Mäuse mit kombiniertem Knockout von Gα11 und Gαq hingegen zeigten in der vorliegenden Arbeit 8 Wochen nach Induktion eines Diabetes einen Anstieg von Herz- und Lungengewicht als Hinweis auf die Entwicklung einer kongestiven Herzinsuffizienz. Dies deutet darauf hin, dass zumindest eine partielle Aktivität der G-Proteine Gα11 und Gαq von Bedeutung für den Erhalt der Pumpfunktion des Herzens in der frühen Phase des Diabetes mellitus ist. Interessanterweise zeigte eine frühere Arbeit am selben Mausmodell, dass die kombinierte Deletion von Gα11 und Gαq nach operativer Aortenkonstriktion über den Beobachtungszeitraum von zwei Wochen der Entwicklung einer Hypertrophie entgegenwirken und im Gegensatz zu Kontrolltieren des Wildtyps den kardialen Phänotyp weitgehend erhalten konnte eine alleinige Deletion von Gα11 dazu aber nicht ausreichte (141). 72 Diskussion 4.2 Die Signalkaskade der pathophysiologischen Veränderungen bei Typ1-Diabetes weisen Hinweise auf eine Beteiligung der Proteinkinas C auf Die G-Proteine Gα11 und Gαq vermitteln intrazellulär diverse Signalkaskaden über die eine Beeinflussung der Genexpression möglich erscheint. Zu den relevanten Mechanismen, durch die die G-Proteine Gα11 und Gαq zur Induktion von Hypertrophie und Fibrose führen können (51, 126) zählen die Aktivierung der PKC, der MAPKs, des Calcineurin und der CaM-Kinase. Die Auswirkungen eines Diabetes mellitus auf intrazelluläre Signalkaskaden sind Gegenstand der aktuellen Forschung und noch nicht vollständig aufgeklärt. In zahlreichen Arbeiten konnte die PKC als bedeutungsvoller second messenger im Zusammenhang mit der Entstehung kardialer Folgekrankheiten bei Diabetes mellitus dokumentiert werden (46, 70, 146) und es konnte interessanterweise auch ein kardioprotektiver Effekt bei Diabetes mellitus durch eine unspezifische Blockade der PKC erreicht werden (148). In der vorliegenden Arbeit war das Expressionsmuster der PKC und ihrer Isoformen in Abhängigkeit eines Typ1-Diabetes und des G-Proteins Gα11 Gegenstand der Analyse. Bei Mäusen des Wildtyps wurde nach Induktion eines Diabetes mellitus über 8 Wochen eine Expressionszunahme der PKC Isoformen α und ζ beobachtet. Die Zunahme der Phosphorylierung der PKC ζ/λ erfolgte in der gleichen Relation wie die Expressionszunahme, sodass von keiner zusätzlichen Aktivierung dieser Isoform ausgegangen werden kann. Die spezifischen Funktionen der einzelnen Isoformen der PKC sind noch Gegenstand aktueller Forschung, dabei werden die Isoformen α und ζ mit einer Stimulation der Wachstumsfaktoren für Bindegewebe und einem damit verbundenen Remodelling und schließlich einer Fibrose assoziiert (50). Für die PKC α ist ebenso eine pathologische Bedeutung bei Diabetes mellitus auf das Gefäßsystem durch Erhöhung der endothelialen Permeabilität nachgewiesen (46, 67). Auch eine PKC α−vermittelte Reaktion der Kardiomyozyten auf die Hyperglykämie wurde beschrieben (41) die genaue zelluläre Funktion ist jedoch noch nicht endgültig geklärt. Die übrigen in dieser Arbeit untersuchten Isoformen βII, δ, ε und θ sowie die Phosphorylierung und damit Aktivierung der Isoformen δ/θ zeigten keine Veränderung durch die Induktion eines Typ 1-Diabetes. Assoziiert man den kardialen 73 Diskussion Phänotyp der Wildtypmäuse mit der Veränderung der Signalkaskade der G-Proteine Gα11 und Gαq, so scheint ein Zusammenhang zwischen einer gesteigerten Expression der PKC α und der PKC ζ und einem Remodelling, das zur Hypertrophie führt, möglich. Die vorliegenden Daten stehen im Einklang mit Ergebnissen verschiedener Publikationen, die eine Steigerung der Expression der PKC α (67), sowie eine unveränderte Expression der Isoformen βII und δ (61, 67) am diabeteskranken Herzen zeigen konnten. Neben diesen liegen allerdings auch divergente Berichte vor, wie eine verminderte Expression der PKC ε und PKC βII (113) am Herzen bei Typ1-Diabetes. Sicherlich müssen unterschiedliche Faktoren wie u.a. die untersuchte Spezies in diesem Zusammenhang berücksichtigt werden. In der vorliegenden Arbeit wurde neben der Analyse der PKC Isoformen des Wildtyps das Expressionsmuster auch bei Gα11-Deletion untersucht. Hierbei kam es nicht zu einer Zunahme der Expression der PKC Isoformen α und ζ nach Induktion eines Typ1-Diabetes. Die Regulation dieser Isoformen der PKC ist also unter diabetischen Bedingungen beim Wildtyp abhängig vom G-Protein Gα11. Lediglich die PKC δ zeigte nach Induktion des Typ1-Diabetes eine Expressionszunahme bei Gα11-Deletion, die jedoch dem Expressionsniveau des Wildtyps entsprach. Damit liegt also bei Gα11Deletion eine verminderte Expression der PKC δ vor, die durch den Typ1-Diabetes auf das Kontrollniveau zurückgeführt wurde. Die PKC δ kann also eine Expressionszunahme bei Hyperglykämie trotz Deletion des Gα11 erfahren, die im Wildtyp nicht zu beobachten war. In diesem Zusammenhang ist wichtig zu erwähnen, dass ein Anstieg der phosphorylierten Form der PKC δ/θ bei Gα11-Deletion ausblieb. Dies bedeutet, dass der relative Anteil der phosphoryliert vorliegenden und damit aktivierten Form der PKC δ abnahm. Die Funktion der PKC δ schließt den Einfluss auf Dihydropiridinkanäle ein und hat damit vielleicht auch Auswirkung auf die Ca2+Homöostase (41). Es wird von der Zunahme des Ca2+-Einstroms über Dihydropiridinkanäle ausgegangen (120) was eine Zunahme der Ca2+-Beladung der Zelle zur Folge haben kann. Ebenso ist eine Assoziation der PKC δ mit einer kardialen Hypertrophie gezeigt worden (122). Dies könnte einen Erklärungsansatz für Beobachtungen in der vorliegenden Arbeit darstellen. Somit besteht die Möglichkeit, dass der kardiale Phänotyp nach Induktion eines Typ1-Diabetes bei Gα11-Deletion, der im Gegensatz zum Phänotyp beim Wildtyp keine Hypertrophie aufwies, im 74 Diskussion Zusammenhang mit dem Vorliegen einer geringeren phosphorylierten bzw. aktivierten PKC δ steht. Das hier unterschiedliche Expressionsmuster der beiden Versuchsgruppen könnte eine Erklärung für den unterschiedlichen Phänotyp nach Induktion eines Typ1Diabetes darstellen: Die bekannte Wachstumsstimulierende und Fibrosestimulierende Wirkung der PKC (51) könnte durch die verminderte Expression bei den Gα11 Knockoutmäusen eingeschränkt sein und dies würde konsekutiv nicht zu einer Hypertrophie des Herzens führen. Vergleicht man die kardialen Expressionsmuster der PKC-Isoformen bei nicht diabeteskranken Mäusen des Wildtyps und nicht diabeteskranken Gα11 Knockoutmäusen, kann für die untersuchten Isoformen die Bedeutung für die Expression und Phosphorylierung und damit Aktivierung des G-Proteins Gα11 unter Basalbedingungen beurteilt werden. Die Gα11 Knockoutmäuse zeigten eine verminderte Expression der Isoformen α und δ, sowie eine Verminderung der Phosphorylierung der Isoformen δ/θ. Dies weist auf eine Abhängigkeit der Regulation der genannten Isoformen der PKC unter Basalbedingungen vom G-Protein Gα11 hin. Bedeutsam ist der Nachweis einer kongruenten Verminderung sowohl der Expression als auch der Phosphorylierung der PKC δ. Hier kann sich also schon unter Kontrollbedingungen ein niederes Expressionsniveau dieser Wachstums- und Fibrosefördernden Isoformen α und δ bei Gα11-Deletion abzeichnen, dass nach Induktion des Typ1-Diabetes sich nicht verändert (PKC α) oder sich allenfalls auf das Kontrollniveau des Wildtyps angleicht ohne eine weitere Phosphorylierung bzw. Aktivierung aufzuzeigen (PKC δ). Die kardioprotektive Wirkung einer Gα11-Deletion bei Typ1-Diabetes, die sich in einem unveränderten kardialen Phänotyp im Gegensatz zur Hypertrophie des Herzens beim Wildtyp äußert, könnte teilweise auf diese Beobachtungen zurückzuführen sein. In der vorliegenden Arbeit ließen sich für zwei Isoformen der PKC Veränderungen in der intrazellulären bzw. interzellulären Verteilung nachweisen. Die PKC ε zeigte nach Induktion des Diabetes sowohl bei den Wildtypmäusen, als auch bei Gα11 Knockouttieren eine intrazellulären Translokation. Durch Analyse der immunhistochemischen Ergebnisse erschien eine Translokation an die interzellulären Kontaktzonen der Zellen als wahrscheinlich als Hinweis auf eine Rolle der PKC ε in der interzellulären Signalvermittlung. Die Bedeutung der PKC ε wird bis jetzt in einer 75 Diskussion Kardioprotektion gesehen, die über verschiedene Signalkaskaden wie u.a. der Aktivierung von anti-Apoptose Transkriptionsfaktoren wie NF-κB vermittelt werden soll (2). Allerdings ist ebenfalls eine exzessive Phosphorylierung des Connexin-43, einem Molekül der interzellulären Kontaktzonen der Kardiomyozyten, beschrieben worden, die bei Diabetes mellitus durch die PKC vermittelt werden soll und zu Dysfunktionen der interzellulären Kommunikation führen könnte (59). Die spezielle Rolle der PKC ε konnte in diesem Zusammenhang ebenfalls nachgewiesen werden (28) und zeigt die Bedeutung der Ergebnisse in der vorliegenden Arbeit. Dieser Mechanismus der pathophysiologisch zur Manifestation des Diabetes mellitus am Herzen beitragen soll (59) tritt sowohl im Wildtyp, als auch nach Gα11-Deletion auf. Der kardiale Phänotyp der Gα11 Knockouttiere bei Typ1-Diabetes ist also unabhängig vom vorliegen dieses Mechanismus, welcher möglicherweise über andere Signaltransduktionen kompensiert wird. Auch für die PKC ζ konnten Veränderungen der intrazellulären Verteilung im Wildtyp nach Induktion eines Typ1-Diabetes nachgewiesen werden. Die PKC ζ konnte vermehrt perinukleär nachgewiesen werden und macht eine Einflussnahme dieser Isoform auf die Genexpression der Zelle möglich. Die Gα11-Deletion verhinderte die Ausprägung dieses Bildes. Dieser Mechanismus scheint also über das G-Protein Gα11 vermittelt zu werden und muss in seiner Bedeutung noch weiter untersucht werden. Die in der vorliegenden Arbeit beobachteten Veränderung in der Expression, Phosphorylierung und Translokation der PKC bei Typ1-Diabetes machen die Bedeutung dieser Enzymfamilie als wesentlichen Signalweg für die Entstehung der Hypertrophie als frühe kardiale Manifestationsform bei Typ1-Diabetes deutlich und zeigen die zentrale Bedeutung des G-Proteins Gα11 für die Aktivierung der PKC und konsekutiv die Induktion der Hypertrophie. Das komplexe Zusammenspiel auch mit anderen Signalkaskaden der G-Proteine Gα11 und Gαq z.B. über MAPKs, Calcineurin und die CAMkinase, ist in diesem Zusammenhang zu berücksichtigen. Dies war jedoch nicht Gegenstand der vorliegenden Arbeit. 76 Diskussion 4.3 Die Struktur und die Funktion des Proteinkomplexes der Ca2+Freisetzung zeigt eine Beeinflussung durch einen Typ1Diabetes und die G-Proteine Gα α11 und Gα αq Die Manifestationsform des Diabetes mellitus am Herzen zeigt sich früh als Hypertrophie, die im Verlauf der Erkrankung in eine Herzinsuffizienz übergehen kann. Die gestörte Kontraktilität bei Herzinsuffizienz kann erwiesenermaßen auf eine gestörte intrazelluläre Ca2+-Homöostase, sowie auf strukturelle Veränderungen, u.a. mit Veränderungen der extrazellulären Matrix und einer erhöhten Apoptoserate (134) zurückgeführt werden. Auch im Tiermodell der diabetischen Ratte nach Injektion von Streptozotocin konnten diese Veränderungen nachgewiesen werden. Die gestörte Ca2+-Homöostase zeigte sich hierbei auch in spezifischen Veränderungen der Expression und Funktion Ca2+-regulierender Proteine des SR wie der SERCA und ihrem Regulatorprotein Phospholamban (137, 138, 151) und des SR-CalciumFreisetzungskanals (RYR2) (8, 151). Diese Veränderungen stellen wahrscheinlich eine wesentliche Ursache für die Abnahme des Ca2+-Gehaltes im SR bei Diabetes mellitus dar (58, 118). Die RYR2 bilden nach aktuellem Erkenntnisstand makromolekulare Komplexe, die u.a. FKBP12.6 enthalten und durch Proteine wie Sorcin reguliert werden (36, 82). Der Komplex des Ca2+-Freisetzungskanals wurde in der vorliegenden Arbeit unter dem Einfluss eines Typ1-Diabetes durch Analyse der Proteinexpression mit Hilfe des WesternBlot-Verfahrens und durch Analyse der Funktion durch Messung des Ca2+-Verlustes aus isolierten Membranvesikeln untersucht. Das Proteinexpressionsmuster wies nach 8 Wochen Diabetes mellitus Veränderungen auf. So wurde das regulierende Protein FKBP12.6 sowie das Annexin VII vermindert im Western Blot-Verfahren nachgewiesen. FKBP12.6 stabilisiert die Aktion mehrerer RYR2, so dass diese in einer funktionellen Einheit wirken (81). Dieser Mechanismus soll zu einer Verminderung des Ca2+-Lecks aus dem SR und schließlich zu einer größeren Beladung des SR mit Ca2+, einem höheren Ca2+-Transienten und einer verbesserten Kontraktilität führen (42, 101). Der Nachweis der verminderten Expression des FKBP12.6 steht demnach im Einklang mit der ebenfalls nachgewiesenen veränderten Funktion des Ca2+Freisetzungskanals, die sich in einer größeren Ca2+-Leckage des RYR2 äußerte. Dies könnte direkt eine Verschlechterung der Kontraktilität bewirken (42, 101). Interessanterweise dissoziiert das FKBP12.6 durch Phosphorylierung des RYR2 von 77 Diskussion diesem (140). Eine Veränderung der Phosphorylierung des RYR2 an Serin 2809 konnte in dieser Arbeit jedoch exemplarisch nicht nachgewiesen werden. Annexin VII hingegen scheint in der Lage zu sein, Sorcin zu binden und damit die Interaktion mit dem RYR2 zu ermöglichen (53). Sorcin ist ein Inhibitor des RYR2, der die CICR beenden kann. Eine verminderte Expression von Annexin VII, die eine verschlechterte Bindung von Sorcin an den RYR2 zur Folge hätte (53), könnte demnach auch zu einer größeren Leckage von Ca2+ aus dem RYR2 führen, wie sie in der vorliegenden Arbeit beobachtet werden konnte. Hinzuzufügen bleibt, dass Sorcin selbst unverändert exprimiert wurde. Die Veränderungen des Ca2+Freisetzungskomplexes nach 8 Wochen Typ1-Diabetes haben somit auch einen Einfluss auf die Ca2+-Homöostase. Dies könnte ursächlich zum beobachteten Phänotyp der Herzhypertrophie bei Wildtypmäusen beitragen. Der Phänotyp bei Gα11 Knockoutmäusen zeigte nach 8 Wochen Typ1-Diabetes keine Veränderungen im Vergleich mit nicht diabeteskranken Gα11 Knockoutmäusen. Auch bei diesen Tieren wurde die Funktion und die Expression des Ca2+-Freisetzungs-komplexes analysiert. Die Ergebnisse konnten weder eine Veränderung der Funktion bzw. eine erhöhte Ca2+-Leckage noch eine Veränderung der Proteinexpression von RYR2, Sorcin, Annexin VII oder FKBP12.6 nachweisen. Die Deletion des Gα11 scheint also einen günstigen Einfluss auf die Proteinexpression und Funktion dieser Proteine bei Typ1Diabetes zu haben. Der Phänotyp bei Gα11/ Gαq Doppelknockoutmäusen zeigte diesen protektiven Einfluss nach 8 Wochen Diabetes mellitus nicht, sondern stellte sich mit Zeichen der Herzinsuffizienz dar. Die kardiale Expression und Funktion des Ca2+-Freisetzungskanals von Gα11/ Gαq Doppelknockoutmäusen nach 8 Wochen Typ1-Diabetes wurde ebenfalls analysiert. Auch hier konnte ein spezifisches Expressionsmuster nachgewiesen werden. Sorcin und Annexin VII zeigten eine signifikante Steigerung in ihrer Expression. Da Annexin VII die physiologische Wirkung von Sorcin am RYR2 möglich machen soll (53), ist es vielleicht möglich eine kongruente Expressionssteigerung beider Proteine mit einer stärkeren Wirkung des Sorcins zu assoziieren. Sorcin führt zu einer Inhibition des RYR2 und möglicherweise zur Beendigung der CICR (36). Eine pathologische Steigerung der Funktion dieses Proteins könnte demnach zu einer übermäßigen Hemmung des RYR2 führen. Allerdings konnte keine Veränderung der Ca2+-Leckage aus isolierten Membranvesikeln bei Mäusen dieses Genotyps über den RYR2 nach 8 Wochen Diabetes mellitus nachgewiesen werden. Möglich erscheint als Erklärung, dass die 78 Diskussion starke Expression beider Proteine einen Kompensationsmechanismus darstellen, der zu einem Erhalt der Funktion des RYR2 bei Diabetes mellitus führt. Im Zusammenhang mit einem Diabetes mellitus kann die Sorcinüberexpression sogar zu einer Wiederherstellung normaler Kontraktionsverhältnisse führen (125). Allerdings herrschen interessanterweise zwei gegensätzliche Meinungen über die Wirkung einer Sorcinüberexpression vor. So wird einerseits eine hämodynamische Verschlechterung mit der Sorcinüberexpression (84, 115), andererseits eine Verbesserung der Kontraktilität (39, 125) mit der Sorcinüberexpression assoziiert. Die vorliegenden Daten stehen in Einklang mit der aktuellen Literatur. Shao und Mitarbeiter zeigten bereits eine verminderte Funktion bei unveränderter Expression des RYR2 nach Induktion eines Typ-1-Diabetes mellitus (118). Diese unveränderte Expression des Kanals ist allerdings von anderen Arbeitsgruppen nicht (132) oder zeitabhängig (5, 6, 8, 21, 88, 151) nachgewiesen worden. Bei Diabetes mellitus können auch morphologische Veränderungen an Proteinen, wie z.B. die Bildung von Disulfidbrücken und Advanced Glycation End Products (AGEs) auftreten (7, 9), die mit funktionellen Störungen des RYR2 einhergehen (6). Eine mögliche Ursache für die unterschiedlichen Ergebnisse könnte die Wahl der Spezies sein. So ist die Proteinexpressionsverminderung des RYR2 in der Regel in Rattenherzen nachgewiesen worden. In der vorliegenden Arbeit wurde jedoch Mausgewebe analysiert. Ebenfalls ein divergierender Faktor in den verschiedenen Arbeiten ist die gewählte Dosis und Applikationsform des STZ. Mit einer einmaligen intraperitonealen Gabe von 130 mg/kg KG STZ wurde in der vorliegenden Arbeit eine niedrige Dosis gewählt, die allerdings zu äquivalenten Blutzuckerwerten führte, wie in den angeführten Arbeiten, die in der Regel höhere Dosen verwandten und diese intravenös applizierten. Zusammenfassend bestehen deutliche Unterschiede im Proteinexpressionsmuster des Ca2+-Freisetzungskanals der drei unterschiedlichen Versuchsgruppen nach Induktion eines Diabetes mellitus. Diabetesbedingt kann es bei Mäusen des Wildtyps zur Verminderung von Annexin VII und FKBP12.6 und damit zu einer erhöhten Ca2+Leckage des SR kommen. Diese Veränderungen konnten in der Gruppe der Gα11 Knockoutmäuse nicht beobachtet werden. In dieser Versuchsgruppe waren keine diabetesabhängigen Veränderungen in der Expression der untersuchten Proteine zu erkennen. Die Gruppe der Gα11/Gαq Doppelknockoutmäuse zeigte keine Abschwächung der Proteinexpression, sondern zusätzlich eine Zunahme der 79 Diskussion Expression von Sorcin und Annexin VII, die für die beobachteten Zeichen der Kongestion bei diesen Tieren von Bedeutung sein könnten. 4.4 Klinische Bedeutung der vorliegenden Arbeit Die in der vorliegenden Arbeit erarbeitete kardioprotektive Wirkung des G-Proteins Gα11 mag einen Hinweis über die Pathogenese einer Hypertrophie durch eben dieses Proteins im Rahmen eines Diabetes mellitus geben, die sich im vorliegenden Modell auf Ebene des Phänotyp, auf Ebene der intrazellulären Signalvermittlung über die Proteinkinase C und auf Ebene der Proteinexpression zeigte. Die G-Proteine Gα11 und Gαq vermitteln das Signal diverser Hormone. Neben Endothelin und Noradrenalin wird auch das intrazelluläre Signal von Angiotensin II über den AT1Rezeptor und damit über die G-Proteine Gα11 und Gαq vermittelt. Das Knockout des AT1-Rezeptors, welches zu einer Hemmung des RAAS führt, kann allerdings die Entstehung einer Hypertrophie im Tiermodell nicht verhindern (47, 49) im Gegensatz zu der hier beobachteten kardioprotektiven Wirkung einer Gα11-Deletion. Das auffällige Benefit der diabetischen Population durch die Gabe von ACE-Hemmern bzw. AT1-Rezeptor-Antagonisten, also einer Hemmung des RAAS, in großen klinischen Studien (48, 57, 73) könnte also im Zusammenhang mit der hier nachgewiesenen kardioprotektive Wirkung einer Gα11-Deletion stehen. 80 Zusammenfassung 5 Zusammenfassung Hintergrund: Der Prävalenz des Diabetes mellitus wird für die nächsten Jahre eine deutliche Zunahme vorausgesagt. Zu den multiplen Manifestationen dieser Erkrankung gehört auch eine kardiale Kontraktionsstörung, die sich unabhängig von einer koronaren Makroangiopathie entwickelt. Bei dieser Form der Herzinsuffizienz, auch als diabetische Kardiomyopathie bezeichnet, zeigt sich früh eine Myokardhypertrophie mit isolierter Relaxationsstörung, aus der sich mit zunehmender Manifestationsdauer das Vollbild einer kongestiven Herzmuskelinsuffizienz entwickelt. Die klinische Beobachtung einer kardioprotektiven Wirkung von ACEHemmern und AT1-Rezeptorantagonisten bei Patienten mit Diabetes mellitus legt die Vermutung nahe, dass neurohumorale Mechanismen bei der Entstehung einer solchen diabetischen Kardiomyopathie von zentraler Bedeutung sind. Gleichzeitig gibt es zahlreiche Hinweise, dass die strukturellen und funktionellen Veränderungen im Myokard bei Diabetes mellitus auch direkt Folge der Hyperglykämie sein können. Ziel der vorliegenden Arbeit war es daher, die Signalwege zu identifizieren, über die eine Hypertrophie und Herzmuskelinsuffizienz bei Diabetes mellitus primär vermittelt werden. Methoden: Der Angiotensin II-Typ 1-Rezeptor (AT1-Rezeptor) gehört zur Familie der G-Protein gekoppelten Rezeptoren und vermittelt sein Signal primär über die GProteine Gαq und Gα11. Die vorliegenden Untersuchungen wurden daher an Knockoutmäusen mit spezifischer Deletion des Gα11, einem Mausmodell mit kombinierter Deletion der G-Proteine Gαq und Gα11, sowie an Wildtyp Mäusen durchgeführt. In diesen Tieren wurde ein experimenteller Typ-1-Diabetes durch intraperitoneale Injektion von Streptozotocin induziert. 8 Wochen nach Induktion eines Diabetes wurden Herz- und Lungengewicht als Marker für Myokardhypertrophie und Herzinsuffizienz gemessen, sowie echokardiographische Untersuchungen zur Analyse der kardialen Funktion durchgeführt. Die Isoformen α, βΙΙ, δ, ε, ζ, θ, sowie die Phosphorylierung der Isoformen δ/θ und ζ/λ der Proteinkinase C wurden immunhistochemisch in ihrem Expressionsmuster in den verschiedenen Versuchsgruppen untersucht. Zur Bestimmung funktioneller Veränderungen wurde der Einfluss des experimentellen Diabetes auf den intrazellulären Calciumstoffwechsel exemplarisch am Calcium-Freisetzungskanal des sarkoplasmatischen Retikulums analysiert. Hierzu wurde die Proteinexpression von Ryanodin (Isoform 2; RYR2), Sorcin, FKBP12.6, Annexin VII bestimmt, die Phosphorylierung von RYR2 an Serin 2809 81 Zusammenfassung analysiert, und funktionell der Verlust von Calcium an isolierten Membranvesikeln in einem Versuchsansatz mit radioaktiv markiertem Calcium in Gegenwart und in Abwesenheit von Ruthenium Rot quantitativ in den verschiedenen Untersuchungsgruppen verglichen. Ergebnisse: 8 Wochen nach Induktion eines Typ-1-Diabetes konnte bei Wildtypmäusen erwartungsgemäß eine konzentrische Myokardhypertrophie mit erhaltener Kontraktilität nachgewiesen werden. Gα11 Knockoutmäuse hingegen zeigten unter identischen experimentellen Bedingungen keinen kardialen Phänotyp. Im Gegensatz hierzu entwickelten Gα11/Gαq Doppelknockouttiere Zeichen einer kongestiven Herzmuskelinsuffizienz. Unter den untersuchten Isoformen der Proteinkinase C kam es bei Mäusen des Wildtyps nach Induktion eines Diabetes mellitus zu einer Expressionszunahme der Isoformen α und ζ. Zudem konnte eine spezifische Translokation der Isoformen ε und ζ nachgewiesen werden. Gα11 Knockoutmäuse wiederum zeigten keine Veränderung in der Expression der Proteinkinase C. Lediglich die Translokation der Isoform ε zeigte in diesen Tieren bei Diabetes ein Muster, vergleichbar mit dem der Wildtyp Mäuse. Der Proteinkomplex des Calciumfreisetzungskanals zeigte nach 8-wöchigem Diabetes bei den Wildtypmäusen im Vergleich zu den nicht diabeteskranken Kontrolltieren eine signifikant verminderte Expression von FKBP12.6 und Annexin VII. Zudem zeigte sich funktionell ein vermehrter Verlust von 45Calcium an isolierten Membranvesikeln. Entsprechende funktionelle und molekulare Veränderungen konnten bei den Gα11 Knockoutmäusen nicht nachgewiesen werden. Gα11/ Gαq Doppelknockoutmäuse wiederum zeigten ein verändertes Proteinexpressionsmuster nach 8-wöchigem Diabetes. Hier kam es jedoch im Gegensatz zu den Wildtyp Mäusen zu einer vermehrten Expression von Sorcin und Annexin VII. Hinweise auf eine veränderte Funktion des Calciumfreisetzungskanals ergaben sich bei Gα11/ Gαq Doppelknockoutmäusen nicht. Schlussfolgerungen: Das G-Protein Gα11 ist wesentlich an der Entwicklung einer Hypertrophie des Herzens bei Typ1-Diabetes beteiligt, da eine Deletion dieses Gens das Auftreten von diabetesbedingten makroskopischen und molekularen, strukturellen und funktionellen Veränderungen im Modell verhindern kann. Eine partielle Aktivität der G-Proteine Gα11 und Gαq ist von Bedeutung für den Erhalt der Pumpfunktion des Herzens in der frühen Phase des Diabetes mellitus, da die 82 Zusammenfassung Deletion beider Gene zu einem Phänotyp mit Zeichen der kongestiven Herzmuskelinsuffizienz führte. 83 Literaturverzeichnis 6 Literaturverzeichnis 1. Adachi-Akahane S, Cleemann L, and Morad M. Cross-signaling between L-type Ca2+ channels and ryanodine receptors in rat ventricular myocytes. J Gen Physiol 108: 435-454, 1996. 2. Akita Y. Protein kinase C-epsilon (PKC-epsilon): its unique structure and function. J Biochem 132: 847-852, 2002. 3. Allen BG and Katz S. Phosphorylation of cardiac junctional and free sarcoplasmic reticulum by PKC alpha, PKC beta, PKA and the Ca2+/calmodulin-dependent protein kinase. Mol Cell Biochem 155: 91-103, 1996. 4. Amatruda TT, 3rd, Steele DA, Slepak VZ, and Simon MI. G alpha 16, a G protein alpha subunit specifically expressed in hematopoietic cells. In: Proc Natl Acad Sci U S A, 1991, p. 5587-5591. 5. Belke DD and Dillmann WH. Altered cardiac calcium handling in diabetes. Curr Hypertens Rep 6: 424-429, 2004. 6. Bidasee KR, Dincer UD, and Besch HR, Jr. Ryanodine receptor dysfunction in hearts of streptozotocin-induced diabetic rats. Mol Pharmacol 60: 1356-1364, 2001. 7. Bidasee KR, Nallani K, Besch HR, Jr., and Dincer UD. Streptozotocin-induced diabetes increases disulfide bond formation on cardiac ryanodine receptor (RyR2). J Pharmacol Exp Ther 305: 989998, 2003. 8. Bidasee KR, Nallani K, Henry B, Dincer UD, and Besch HR, Jr. Chronic diabetes alters function and expression of ryanodine receptor calcium-release channels in rat hearts. Mol Cell Biochem 249: 113-123, 2003. 9. Bidasee KR, Nallani K, Yu Y, Cocklin RR, Zhang Y, Wang M, Dincer UD, and Besch HR, Jr. Chronic diabetes increases advanced glycation end products on cardiac ryanodine receptors/calcium-release channels. Diabetes 52: 1825-1836, 2003. 10. Bidasee KR, Zhang Y, Shao CH, Wang M, Patel KP, Dincer UD, and Besch HR, Jr. Diabetes increases formation of advanced glycation end products on Sarco(endo)plasmic reticulum Ca2+ATPase. In: Diabetes, 2004, p. 463-473. 11. Blank JL, Ross AH, and Exton JH. Purification and characterization of two G-proteins that activate the beta 1 isozyme of phosphoinositide-specific phospholipase C. Identification as members of the Gq class. J Biol Chem 266: 18206-18216, 1991. 12. Bollano E, Omerovic E, Svensson H, Waagstein F, and Fu M. Cardiac remodeling rather than disturbed myocardial energy metabolism is associated with cardiac dysfunction in diabetic rats. In: Int J Cardiol, 2006. 13. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248-254, 1976. 14. Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, Kimball TF, Lorenz JN, Nairn AC, Liggett SB, Bodi I, Wang S, Schwartz A, Lakatta EG, DePaoli-Roach AA, Robbins J, Hewett TE, Bibb JA, Westfall MV, Kranias EG, and Molkentin JD. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat Med 10: 248-254, 2004. 15. Brillantes AB, Ondrias K, Scott A, Kobrinsky E, Ondriasova E, Moschella MC, Jayaraman T, Landers M, Ehrlich BE, and Marks AR. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell 77: 513-523, 1994. 16. Brownawell AM and Creutz CE. Calcium-dependent binding of sorcin to the N-terminal domain of synexin (annexin VII). J Biol Chem 272: 22182-22190, 1997. 17. Cai L and Kang YJ. Oxidative stress and diabetic cardiomyopathy: a brief review. In: Cardiovasc Toxicol, 2001, p. 181-193. 18. Candido R, Forbes JM, Thomas MC, Thallas V, Dean RG, Burns WC, Tikellis C, Ritchie RH, Twigg SM, Cooper ME, and Burrell LM. A breaker of advanced glycation end products attenuates diabetes-induced myocardial structural changes. In: Circ Res, 2003, p. 785-792. 19. Caohuy H, Srivastava M, and Pollard HB. Membrane fusion protein synexin (annexin VII) as a Ca2+/GTP sensor in exocytotic secretion. Proc Natl Acad Sci U S A 93: 10797-10802, 1996. 20. Cerqueira MD, Weissman NJ, Dilsizian V, Jacobs AK, Kaul S, Laskey WK, Pennell DJ, Rumberger JA, Ryan T, and Verani MS. Standardized myocardial segmentation and nomenclature for tomographic imaging of the heart: a statement for healthcare professionals from the Cardiac Imaging Committee of the Council on Clinical Cardiology of the American Heart Association. J Nucl Cardiol 9: 240-245, 2002. 21. Choi KM, Zhong Y, Hoit BD, Grupp IL, Hahn H, Dilly KW, Guatimosim S, Lederer WJ, and Matlib MA. Defective intracellular Ca(2+) signaling contributes to cardiomyopathy in Type 1 diabetic rats. Am J Physiol Heart Circ Physiol 283: H1398-1408, 2002. 84 Literaturverzeichnis 22. Clemen CS, Hofmann A, Zamparelli C, and Noegel AA. Expression and localisation of annexin VII (synexin) isoforms in differentiating myoblasts. J Muscle Res Cell Motil 20: 669-679, 1999. 23. Clemens MJ, Trayner I, and Menaya J. The role of protein kinase C isoenzymes in the regulation of cell proliferation and differentiation. J Cell Sci 103 ( Pt 4): 881-887, 1992. 24. Coussens L, Rhee L, Parker PJ, and Ullrich A. Alternative splicing increases the diversity of the human protein kinase C family. DNA 6: 389-394, 1987. 25. D'Angelo DD, Sakata Y, Lorenz JN, Boivin GP, Walsh RA, Liggett SB, and Dorn GW, 2nd. Transgenic Galphaq overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci U S A 94: 8121-8126, 1997. 26. Dell'Italia LJ and Oparil S. Bradykinin in the heart: friend or foe? In: Circulation, 1999, p. 23052307. 27. Dhalla NS, Liu X, Panagia V, and Takeda N. Subcellular remodeling and heart dysfunction in chronic diabetes. In: Cardiovasc Res, 1998, p. 239-247. 28. Doble BW, Ping P, and Kardami E. The epsilon subtype of protein kinase C is required for cardiomyocyte connexin-43 phosphorylation. Circ Res 86: 293-301, 2000. 29. Dorn GW, 2nd and Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest 115: 527-537, 2005. 30. Du J, Cai S, Suzuki H, Akhand AA, Ma X, Takagi Y, Miyata T, Nakashima I, and Nagase F. Involvement of MEKK1/ERK/P21Waf1/Cip1 signal transduction pathway in inhibition of IGF-Imediated cell growth response by methylglyoxal. J Cell Biochem 88: 1235-1246, 2003. 31. Du X, Matsumura T, Edelstein D, Rossetti L, Zsengeller Z, Szabo C, and Brownlee M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. In: J Clin Invest, 2003, p. 1049-1057. 32. Dzau VJ. Tissue renin-angiotensin system in myocardial hypertrophy and failure. Arch Intern Med 153: 937-942, 1993. 33. Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am J Physiol 245: C1-14, 1983. 34. Fang ZY, Prins JB, and Marwick TH. Diabetic cardiomyopathy: evidence, mechanisms, and therapeutic implications. In: Endocr Rev, 2004, p. 543-567. 35. Farhangkhoee H, Khan ZA, Mukherjee S, Cukiernik M, Barbin YP, Karmazyn M, and Chakrabarti S. Heme oxygenase in diabetes-induced oxidative stress in the heart. In: J Mol Cell Cardiol, 2003, p. 1439-1448. 36. Farrell EF, Antaramian A, Rueda A, Gomez AM, and Valdivia HH. Sorcin inhibits calcium release and modulates excitation-contraction coupling in the heart. J Biol Chem 278: 34660-34666, 2003. 37. Fiordaliso F, Li B, Latini R, Sonnenblick EH, Anversa P, Leri A, and Kajstura J. Myocyte death in streptozotocin-induced diabetes in rats is angiotensin II- dependent. In: Lab Invest, 2000, p. 513527. 38. Fischer LJ and Rickert DE. Pancreatic islet-cell toxicity. In: CRC Crit Rev Toxicol, 1975, p. 231263. 39. Frank KF, Bolck B, Ding Z, Krause D, Hattebuhr N, Malik A, Brixius K, Hajjar RJ, Schrader J, and Schwinger RH. Overexpression of sorcin enhances cardiac contractility in vivo and in vitro. J Mol Cell Cardiol 38: 607-615, 2005. 40. Galvez AS, Ulloa JA, Chiong M, Criollo A, Eisner V, Barros LF, and Lavandero S. Aldose reductase induced by hyperosmotic stress mediates cardiomyocyte apoptosis: differential effects of sorbitol and mannitol. In: J Biol Chem, 2003, p. 38484-38494. 41. Giles TD, Ouyang J, Kerut EK, Given MB, Allen GE, McIlwain EF, and Greenberg SS. Changes in protein kinase C in early cardiomyopathy and in gracilis muscle in the BB/Wor diabetic rat. Am J Physiol 274: H295-307, 1998. 42. Gomez AM, Schuster I, Fauconnier J, Prestle J, Hasenfuss G, and Richard S. FKBP12.6 overexpression decreases Ca2+ spark amplitude but enhances [Ca2+]i transient in rat cardiac myocytes. Am J Physiol Heart Circ Physiol 287: H1987-1993, 2004. 43. Grimm D, Jabusch HC, Kossmehl P, Huber M, Fredersdorf S, Griese DP, Kramer BK, and Kromer EP. Experimental diabetes and left ventricular hypertrophy: effects of beta-receptor blockade. In: Cardiovasc Pathol, 2002, p. 229-237. 44. Groves P, Kurz S, Just H, and Drexler H. Role of endogenous bradykinin in human coronary vasomotor control. In: Circulation, 1995, p. 3424-3430. 45. Gu X and Bishop SP. Increased protein kinase C and isozyme redistribution in pressure-overload cardiac hypertrophy in the rat. Circ Res 75: 926-931, 1994. 46. Guo M, Wu MH, Korompai F, and Yuan SY. Upregulation of PKC genes and isozymes in cardiovascular tissues during early stages of experimental diabetes. Physiol Genomics 12: 139146, 2003. 85 Literaturverzeichnis 47. Hamawaki M, Coffman TM, Lashus A, Koide M, Zile MR, Oliverio MI, DeFreyte G, Cooper Gt, and Carabello BA. Pressure-overload hypertrophy is unabated in mice devoid of AT1A receptors. Am J Physiol 274: H868-873, 1998. 48. Hansson L, Lindholm LH, Niskanen L, Lanke J, Hedner T, Niklason A, Luomanmaki K, Dahlof B, de Faire U, Morlin C, Karlberg BE, Wester PO, and Bjorck JE. Effect of angiotensin-convertingenzyme inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: the Captopril Prevention Project (CAPPP) randomised trial. In: Lancet, 1999, p. 611616. 49. Harada K, Komuro I, Shiojima I, Hayashi D, Kudoh S, Mizuno T, Kijima K, Matsubara H, Sugaya T, Murakami K, and Yazaki Y. Pressure overload induces cardiac hypertrophy in angiotensin II type 1A receptor knockout mice. Circulation 97: 1952-1959, 1998. 50. He Z, Way KJ, Arikawa E, Chou E, Opland DM, Clermont A, Isshiki K, Ma RC, Scott JA, Schoen FJ, Feener EP, and King GL. Differential regulation of angiotensin II-induced expression of connective tissue growth factor by protein kinase C isoforms in the myocardium. J Biol Chem 280: 15719-15726, 2005. 51. Heineke J and Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. In: Nat Rev Mol Cell Biol, 2006, p. 589-600. 52. Hepler JR, Kozasa T, Smrcka AV, Simon MI, Rhee SG, Sternweis PC, and Gilman AG. Purification from Sf9 cells and characterization of recombinant Gq alpha and G11 alpha. Activation of purified phospholipase C isozymes by G alpha subunits. J Biol Chem 268: 14367-14375, 1993. 53. Herr C, Smyth N, Ullrich S, Yun F, Sasse P, Hescheler J, Fleischmann B, Lasek K, Brixius K, Schwinger RH, Fassler R, Schroder R, and Noegel AA. Loss of annexin A7 leads to alterations in frequency-induced shortening of isolated murine cardiomyocytes. Mol Cell Biol 21: 4119-4128, 2001. 54. Herrmann KL, McCulloch AD, and Omens JH. Glycated collagen cross-linking alters cardiac mechanics in volume-overload hypertrophy. In: Am J Physiol Heart Circ Physiol, 2003, p. H12771284. 55. Hohenegger M and Suko J. Phosphorylation of the purified cardiac ryanodine receptor by exogenous and endogenous protein kinases. Biochem J 296 ( Pt 2): 303-308, 1993. 56. Hollenberg NK, Stevanovic R, Agarwal A, Lansang MC, Price DA, Laffel LM, Williams GH, and Fisher ND. Plasma aldosterone concentration in the patient with diabetes mellitus. In: Kidney Int, 2004, p. 1435-1439. 57. HOPE Investigators. Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy. In: Lancet, 2000, p. 253-259. 58. Horackova M and Murphy MG. Effects of chronic diabetes mellitus on the electrical and contractile activities, 45Ca2+ transport, fatty acid profiles and ultrastructure of isolated rat ventricular myocytes. In: Pflugers Arch, 1988, p. 564-572. 59. Inoguchi T, Yu HY, Imamura M, Kakimoto M, Kuroki T, Maruyama T, and Nawata H. Altered gap junction activity in cardiovascular tissues of diabetes. Med Electron Microsc 34: 86-91, 2001. 60. Jayaraman T, Brillantes AM, Timerman AP, Fleischer S, Erdjument-Bromage H, Tempst P, and Marks AR. FK506 binding protein associated with the calcium release channel (ryanodine receptor). J Biol Chem 267: 9474-9477, 1992. 61. Jiang J, Yuen V, Xiang H, and McNeill JH. Improvement in cardiac function of diabetic rats by bosentan is not associated with changes in the activation of PKC isoforms. Mol Cell Biochem 282: 177-185, 2006. 62. Joffe, II, Travers KE, Perreault-Micale CL, Hampton T, Katz SE, Morgan JP, and Douglas PS. Abnormal cardiac function in the streptozotocin-induced non-insulin-dependent diabetic rat: noninvasive assessment with doppler echocardiography and contribution of the nitric oxide pathway. In: J Am Coll Cardiol, 1999, p. 2111-2119. 63. Jorde UP, Vittorio T, Katz SD, Colombo PC, Latif F, and Le Jemtel TH. Elevated plasma aldosterone levels despite complete inhibition of the vascular angiotensin-converting enzyme in chronic heart failure. In: Circulation, 2002, p. 1055-1057. 64. Jorgensen AO, Broderick R, Somlyo AP, and Somlyo AV. Two structurally distinct calcium storage sites in rat cardiac sarcoplasmic reticulum: an electron microprobe analysis study. Circ Res 63: 1060-1069, 1988. 65. Kaftan E, Marks AR, and Ehrlich BE. Effects of rapamycin on ryanodine receptor/Ca(2+)-release channels from cardiac muscle. Circ Res 78: 990-997, 1996. 66. Kajstura J, Fiordaliso F, Andreoli AM, Li B, Chimenti S, Medow MS, Limana F, Nadal-Ginard B, Leri A, and Anversa P. IGF-1 overexpression inhibits the development of diabetic cardiomyopathy and angiotensin II-mediated oxidative stress. In: Diabetes, 2001, p. 1414-1424. 86 Literaturverzeichnis 67. Kang N, Alexander G, Park JK, Maasch C, Buchwalow I, Luft FC, and Haller H. Differential expression of protein kinase C isoforms in streptozotocin-induced diabetic rats. Kidney Int 56: 1737-1750, 1999. 68. Kim NN, Villegas S, Summerour SR, and Villarreal FJ. Regulation of cardiac fibroblast extracellular matrix production by bradykinin and nitric oxide. In: J Mol Cell Cardiol, 1999, p. 457466. 69. King H, Aubert RE, and Herman WH. Global burden of diabetes, 1995-2025: prevalence, numerical estimates, and projections. Diabetes Care 21: 1414-1431, 1998. 70. Koya D and King GL. Protein kinase C activation and the development of diabetic complications. Diabetes 47: 859-866, 1998. 71. Kudoh A and Matsuki A. Effects of angiotensin-converting enzyme inhibitors on glucose uptake. In: Hypertension, 2000, p. 239-244. 72. Kuijpers GA, Lee G, and Pollard HB. Immunolocalization of synexin (annexin VII) in adrenal chromaffin granules and chromaffin cells: evidence for a dynamic role in the secretory process. Cell Tissue Res 269: 323-330, 1992. 73. Lindholm LH, Ibsen H, Dahlof B, Devereux RB, Beevers G, de Faire U, Fyhrquist F, Julius S, Kjeldsen SE, Kristiansson K, Lederballe-Pedersen O, Nieminen MS, Omvik P, Oparil S, Wedel H, Aurup P, Edelman J, and Snapinn S. Cardiovascular morbidity and mortality in patients with diabetes in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet 359: 1004-1010, 2002. 74. Livneh E and Fishman DD. Linking protein kinase C to cell-cycle control. Eur J Biochem 248: 1-9, 1997. 75. Lokuta AJ, Meyers MB, Sander PR, Fishman GI, and Valdivia HH. Modulation of cardiac ryanodine receptors by sorcin. J Biol Chem 272: 25333-25338, 1997. 76. Luo W, Grupp IL, Harrer J, Ponniah S, Grupp G, Duffy JJ, Doetschman T, and Kranias EG. Targeted ablation of the phospholamban gene is associated with markedly enhanced myocardial contractility and loss of beta-agonist stimulation. Circ Res 75: 401-409, 1994. 77. MacFadyen RJ, Lee AF, Morton JJ, Pringle SD, and Struthers AD. How often are angiotensin II and aldosterone concentrations raised during chronic ACE inhibitor treatment in cardiac failure? In: Heart, 1999, p. 57-61. 78. Machackova J, Liu X, Lukas A, and Dhalla NS. Renin-angiotensin blockade attenuates cardiac myofibrillar remodelling in chronic diabetes. In: Mol Cell Biochem, 2004, p. 271-278. 79. Magendzo K, Shirvan A, Cultraro C, Srivastava M, Pollard HB, and Burns AL. Alternative splicing of human synexin mRNA in brain, cardiac, and skeletal muscle alters the unique N-terminal domain. J Biol Chem 266: 3228-3232, 1991. 80. Malhotra A, Kang BP, Cheung S, Opawumi D, and Meggs LG. Angiotensin II promotes glucoseinduced activation of cardiac protein kinase C isozymes and phosphorylation of troponin I. Diabetes 50: 1918-1926, 2001. 81. Marx SO, Gaburjakova J, Gaburjakova M, Henrikson C, Ondrias K, and Marks AR. Coupled gating between cardiac calcium release channels (ryanodine receptors). Circ Res 88: 1151-1158, 2001. 82. Marx SO and Marks AR. Regulation of the ryanodine receptor in heart failure. Basic Res Cardiol 97 Suppl 1: I49-51, 2002. 83. Meissner G. Ryanodine receptor/Ca2+ release channels and their regulation by endogenous effectors. Annu Rev Physiol 56: 485-508, 1994. 84. Meyers MB, Fischer A, Sun YJ, Lopes CM, Rohacs T, Nakamura TY, Zhou YY, Lee PC, Altschuld RA, McCune SA, Coetzee WA, and Fishman GI. Sorcin regulates excitation-contraction coupling in the heart. J Biol Chem 278: 28865-28871, 2003. 85. Meyers MB, Pickel VM, Sheu SS, Sharma VK, Scotto KW, and Fishman GI. Association of sorcin with the cardiac ryanodine receptor. J Biol Chem 270: 26411-26418, 1995. 86. Migdalis IN, Kalogeropoulou K, Karmaniolas KD, Varvarigos N, Mortzos G, and Cordopatis P. Plasma levels of endothelin and early carotid atherosclerosis in diabetic patients. Res Commun Mol Pathol Pharmacol 108: 15-25, 2000. 87. Moss SE and Crumpton MJ. Alternative splicing gives rise to two forms of the p68 Ca2(+)-binding protein. FEBS Lett 261: 299-302, 1990. 88. Netticadan T, Temsah RM, Kent A, Elimban V, and Dhalla NS. Depressed levels of Ca2+-cycling proteins may underlie sarcoplasmic reticulum dysfunction in the diabetic heart. Diabetes 50: 21332138, 2001. 89. Nickenig G, Roling J, Strehlow K, Schnabel P, and Bohm M. Insulin induces upregulation of vascular AT1 receptor gene expression by posttranscriptional mechanisms. Circulation 98: 24532460, 1998. 87 Literaturverzeichnis 90. Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, and Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. In: Nature, 2000, p. 787-790. 91. Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science 258: 607-614, 1992. 92. Offermanns S, Zhao LP, Gohla A, Sarosi I, Simon MI, and Wilkie TM. Embryonic cardiomyocyte hypoplasia and craniofacial defects in G alpha q/G alpha 11-mutant mice. Embo J 17: 4304-4312, 1998. 93. Ono Y, Fujii T, Ogita K, Kikkawa U, Igarashi K, and Nishizuka Y. Identification of three additional members of rat protein kinase C family: delta-, epsilon- and zeta-subspecies. FEBS Lett 226: 125128, 1987. 94. Ono Y, Fujii T, Ogita K, Kikkawa U, Igarashi K, and Nishizuka Y. Protein kinase C zeta subspecies from rat brain: its structure, expression, and properties. Proc Natl Acad Sci U S A 86: 3099-3103, 1989. 95. Ono Y, Fujii T, Ogita K, Kikkawa U, Igarashi K, and Nishizuka Y. The structure, expression, and properties of additional members of the protein kinase C family. J Biol Chem 263: 6927-6932, 1988. 96. Osada S, Mizuno K, Saido TC, Akita Y, Suzuki K, Kuroki T, and Ohno S. A phorbol ester receptor/protein kinase, nPKC eta, a new member of the protein kinase C family predominantly expressed in lung and skin. J Biol Chem 265: 22434-22440, 1990. 97. Osada S, Mizuno K, Saido TC, Suzuki K, Kuroki T, and Ohno S. A new member of the protein kinase C family, nPKC theta, predominantly expressed in skeletal muscle. Mol Cell Biol 12: 39303938, 1992. 98. Özdemir S, Ugur M, Gurdal H, and Turan B. Treatment with AT(1) receptor blocker restores diabetes-induced alterations in intracellular Ca(2+) transients and contractile function of rat myocardium. In: Arch Biochem Biophys, 2005, p. 166-174. 99. Parker PJ. Protein kinase C phosphorylation: an introduction. Methods Mol Biol 233: 159-162, 2003. 100.Parker PJ, Coussens L, Totty N, Rhee L, Young S, Chen E, Stabel S, Waterfield MD, and Ullrich A. The complete primary structure of protein kinase C--the major phorbol ester receptor. Science 233: 853-859, 1986. 101.Prestle J, Janssen PM, Janssen AP, Zeitz O, Lehnart SE, Bruce L, Smith GL, and Hasenfuss G. Overexpression of FK506-binding protein FKBP12.6 in cardiomyocytes reduces ryanodine receptor-mediated Ca(2+) leak from the sarcoplasmic reticulum and increases contractility. Circ Res 88: 188-194, 2001. 102.Privratsky JR, Wold LE, Sowers JR, Quinn MT, and Ren J. AT1 blockade prevents glucoseinduced cardiac dysfunction in ventricular myocytes: role of the AT1 receptor and NADPH oxidase. In: Hypertension, 2003, p. 206-212. 103.Regan TJ, Ahmed S, Haider B, Moschos C, and Weisse A. Diabetic cardiomyopathy: experimental and clinical observations. In: N J Med, 1994, p. 776-778. 104.Remme WJ. Pharmacological modulation of cardiovascular remodeling: a guide to heart failure therapy. In: Cardiovasc Drugs Ther, 2003, p. 349-360. 105.Reuter H, Adam C, Gronke S, Zobel C, Frank KF, Muller-Ehmsen J, Brabender J, and Schwinger RH. The increased angiotensin II (type 1) receptor density in myocardium of type 2 diabetic patients is prevented by blockade of the renin-angiotensin system. Diabetologia 49: 3067-3074, 2006. 106.Reuter H, Grönke S, and Schwinger RH. Behandlung der Hypertonie bei Diabetes mellitus - was ist evidenzbasiert? In: Diabetes, Stoffwechsel und Herz, 2006, p. 23-42. 107.Reuter H, Han T, Motter C, Philipson KD, and Goldhaber JI. Mice overexpressing the cardiac sodium-calcium exchanger: defects in excitation-contraction coupling. J Physiol 554: 779-789, 2004. 108.Reuter H, Pott C, Goldhaber JI, Henderson SA, Philipson KD, and Schwinger RH. Na(+)--Ca2+ exchange in the regulation of cardiac excitation-contraction coupling. Cardiovasc Res 67: 198207, 2005. 109.Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O'Connell J, Olsen E, Thiene G, Goodwin J, Gyarfas I, Martin I, and Nordet P. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. In: Circulation, 1996, p. 841-842. 110.Rincon-Choles H, Kasinath BS, Gorin Y, and Abboud HE. Angiotensin II and growth factors in the pathogenesis of diabetic nephropathy. In: Kidney Int Suppl, 2002, p. 8-11. 88 Literaturverzeichnis 111.Rybin VO, Guo J, Gertsberg Z, Elouardighi H, and Steinberg SF. Protein kinase Cepsilon (PKCepsilon) and Src control PKCdelta activation loop phosphorylation in cardiomyocytes. J Biol Chem 282: 23631-23638, 2007. 112.Ryden L, Armstrong PW, Cleland JG, Horowitz JD, Massie BM, Packer M, and Poole-Wilson PA. Efficacy and safety of high-dose lisinopril in chronic heart failure patients at high cardiovascular risk, including those with diabetes mellitus. Results from the ATLAS trial. Eur Heart J 21: 19671978, 2000. 113.Schaffer SW, Ballard C, and Mozaffari MS. Is there a link between impaired glucose metabolism and protein kinase C activity in the diabetic heart? Mol Cell Biochem 176: 219-225, 1997. 114.Schreiber SL. Chemistry and biology of the immunophilins and their immunosuppressive ligands. Science 251: 283-287, 1991. 115.Seidler T, Miller SL, Loughrey CM, Kania A, Burow A, Kettlewell S, Teucher N, Wagner S, Kogler H, Meyers MB, Hasenfuss G, and Smith GL. Effects of adenovirus-mediated sorcin overexpression on excitation-contraction coupling in isolated rabbit cardiomyocytes. Circ Res 93: 132-139, 2003. 116.Selbert S, Fischer P, Pongratz D, Stewart M, and Noegel AA. Expression and localization of annexin VII (synexin) in muscle cells. J Cell Sci 108 ( Pt 1): 85-95, 1995. 117.Selbie LA, Schmitz-Peiffer C, Sheng Y, and Biden TJ. Molecular cloning and characterization of PKC iota, an atypical isoform of protein kinase C derived from insulin-secreting cells. J Biol Chem 268: 24296-24302, 1993. 118.Shao CH, Rozanski GJ, Patel KP, and Bidasee KR. Dyssynchronous (non-uniform) Ca2+ release in myocytes from streptozotocin-induced diabetic rats. J Mol Cell Cardiol 42: 234-246, 2007. 119.Shindler DM, Kostis JB, Yusuf S, Quinones MA, Pitt B, Stewart D, Pinkett T, Ghali JK, and Wilson AC. Diabetes mellitus, a predictor of morbidity and mortality in the Studies of Left Ventricular Dysfunction (SOLVD) Trials and Registry. Am J Cardiol 77: 1017-1020, 1996. 120.Singer-Lahat D, Gershon E, Lotan I, Hullin R, Biel M, Flockerzi V, Hofmann F, and Dascal N. Modulation of cardiac Ca2+ channels in Xenopus oocytes by protein kinase C. FEBS Lett 306: 113-118, 1992. 121.Sodha NR, Clements RT, Bianchi C, and Sellke FW. Cardiopulmonary bypass with cardioplegic arrest activates protein kinase C in the human myocardium. J Am Coll Surg 206: 33-41, 2008. 122.Steinberg SF, Goldberg M, and Rybin VO. Protein kinase C isoform diversity in the heart. J Mol Cell Cardiol 27: 141-153, 1995. 123.Stenina OI. Regulation of vascular genes by glucose. Curr Pharm Des 11: 2367-2381, 2005. 124. Stern MD and Cheng H. Putting out the fire: what terminates calcium-induced calcium release in cardiac muscle? Cell Calcium 35: 591-601, 2004. 125.Suarez J, Belke DD, Gloss B, Dieterle T, McDonough PM, Kim YK, Brunton LL, and Dillmann WH. In vivo adenoviral transfer of sorcin reverses cardiac contractile abnormalities of diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol 286: H68-75, 2004. 126.Sugden PH. Signaling in myocardial hypertrophy: life after calcineurin? In: Circ Res, 1999, p. 633646. 127.Sugden PH and Clerk A. Endothelin signalling in the cardiac myocyte and its pathophysiological relevance. Curr Vasc Pharmacol 3: 343-351, 2005. 128.Suko J, Maurer-Fogy I, Plank B, Bertel O, Wyskovsky W, Hohenegger M, and Hellmann G. Phosphorylation of serine 2843 in ryanodine receptor-calcium release channel of skeletal muscle by cAMP-, cGMP- and CaM-dependent protein kinase. Biochim Biophys Acta 1175: 193-206, 1993. 129.Tahiliani AG and McNeill JH. Diabetes-induced abnormalities in the myocardium. In: Life Sci, 1986, p. 959-974. 130.Takasago T, Imagawa T, Furukawa K, Ogurusu T, and Shigekawa M. Regulation of the cardiac ryanodine receptor by protein kinase-dependent phosphorylation. J Biochem 109: 163-170, 1991. 131.Tayebjee MH, Lip GY, and MacFadyen RJ. What role do extracellular matrix changes contribute to the cardiovascular disease burden of diabetes mellitus? In: Diabet Med, 2005, p. 1628-1635. 132.Teshima Y, Takahashi N, Saikawa T, Hara M, Yasunaga S, Hidaka S, and Sakata T. Diminished expression of sarcoplasmic reticulum Ca(2+)-ATPase and ryanodine sensitive Ca(2+)Channel mRNA in streptozotocin-induced diabetic rat heart. J Mol Cell Cardiol 32: 655-664, 2000. 133.Timerman AP, Ogunbumni E, Freund E, Wiederrecht G, Marks AR, and Fleischer S. The calcium release channel of sarcoplasmic reticulum is modulated by FK-506-binding protein. Dissociation and reconstitution of FKBP-12 to the calcium release channel of skeletal muscle sarcoplasmic reticulum. J Biol Chem 268: 22992-22999, 1993. 134.Towbin JA and Bowles NE. The failing heart. In: Nature, 2002, p. 227-233. 135.Tschöpe C, Walther T, Königer J, Spillmann F, Westermann D, Escher F, Pauschinger M, Pesquero JB, Bader M, Schultheiss HP, and Noutsias M. Prevention of cardiac fibrosis and left 89 Literaturverzeichnis ventricular dysfunction in diabetic cardiomyopathy in rats by transgenic expression of the human tissue kallikrein gene. In: Faseb J, 2004, p. 828-835. 136.Van Dyk DJ, Erman A, Erman T, Chen-Gal B, Sulkes J, and Boner G. Increased serum angiotensin converting enzyme activity in type I insulin-dependent diabetes mellitus: its relation to metabolic control and diabetic complications. In: Eur J Clin Invest, 1994, p. 463-467. 137.Vasanji Z, Dhalla NS, and Netticadan T. Increased inhibition of SERCA2 by phospholamban in the type I diabetic heart. In: Mol Cell Biochem, 2004, p. 245-249. 138.Vetter R, Rehfeld U, Reissfelder C, Weiss W, Wagner KD, Gunther J, Hammes A, Tschöpe C, Dillmann W, and Paul M. Transgenic overexpression of the sarcoplasmic reticulum Ca2+ATPase improves reticular Ca2+ handling in normal and diabetic rat hearts. In: Faseb J, 2002, p. 16571659. 139.Wakasaki H, Koya D, Schoen FJ, Jirousek MR, Ways DK, Hoit BD, Walsh RA, and King GL. Targeted overexpression of protein kinase C beta2 isoform in myocardium causes cardiomyopathy. Proc Natl Acad Sci U S A 94: 9320-9325, 1997. 140.Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, Sun J, Guatimosim S, Song LS, Rosemblit N, D'Armiento JM, Napolitano C, Memmi M, Priori SG, Lederer WJ, and Marks AR. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell 113: 829-840, 2003. 141.Wettschureck N, Rutten H, Zywietz A, Gehring D, Wilkie TM, Chen J, Chien KR, and Offermanns S. Absence of pressure overload induced myocardial hypertrophy after conditional inactivation of Galphaq/Galpha11 in cardiomyocytes. Nat Med 7: 1236-1240, 2001. 142.Wilkie TM, Scherle PA, Strathmann MP, Slepak VZ, and Simon MI. Characterization of G-protein alpha subunits in the Gq class: expression in murine tissues and in stromal and hematopoietic cell lines. In: Proc Natl Acad Sci U S A, 1991, p. 10049-10053. 143.Wu D, Katz A, Lee CH, and Simon MI. Activation of phospholipase C by alpha 1-adrenergic receptors is mediated by the alpha subunits of Gq family. J Biol Chem 267: 25798-25802, 1992. 144.Wu SC and Solaro RJ. Protein kinase C zeta. A novel regulator of both phosphorylation and dephosphorylation of cardiac sarcomeric proteins. J Biol Chem 282: 30691-30698, 2007. 145.Xiao B, Jiang MT, Zhao M, Yang D, Sutherland C, Lai FA, Walsh MP, Warltier DC, Cheng H, and Chen SR. Characterization of a novel PKA phosphorylation site, serine-2030, reveals no PKA hyperphosphorylation of the cardiac ryanodine receptor in canine heart failure. Circ Res 96: 847855, 2005. 146.Yang JM, Cho CH, Kong KA, Jang IS, Kim HW, and Juhnn YS. Increased expression of Galphaq protein in the heart of streptozotocin-induced diabetic rats. Exp Mol Med 31: 179-184, 1999. 147.Yang XP, Liu YH, Mehta D, Cavasin MA, Shesely E, Xu J, Liu F, and Carretero OA. Diminished cardioprotective response to inhibition of angiotensin-converting enzyme and angiotensin II type 1 receptor in B(2) kinin receptor gene knockout mice. In: Circ Res, 2001, p. 1072-1079. 148.Yaras N, Bilginoglu A, Vassort G, and Turan B. Restoration of diabetes-induced abnormal local Ca2+ release in cardiomyocytes by angiotensin II receptor blockade. Am J Physiol Heart Circ Physiol 292: H912-920, 2007. 149.Yatani A, Frank K, Sako H, Kranias EG, and Dorn GW, 2nd. Cardiac-specific overexpression of Galphaq alters excitation-contraction coupling in isolated cardiac myocytes. J Mol Cell Cardiol 31: 1327-1336, 1999. 150.Zamparelli C, Ilari A, Verzili D, Vecchini P, and Chiancone E. Calcium- and pH-linked oligomerization of sorcin causing translocation from cytosol to membranes. FEBS Lett 409: 1-6, 1997. 151.Zhong Y, Ahmed S, Grupp IL, and Matlib MA. Altered SR protein expression associated with contractile dysfunction in diabetic rat hearts. Am J Physiol Heart Circ Physiol 281: H1137-1147, 2001. 152.Zuraw B. Bradykinin in protection against left-ventricular hypertrophy. In: Lancet, 2001, p. 11161118. 90 Lebenslauf 7 Lebenslauf Name: Dieter Paul Hoyer Geburtsdatum: 22.08.1983 Geburtsort: Hannover Staatsangehörigkeit: Deutsch Eltern: Univ.-Prof. Dr. med. Peter F. Hoyer Dr. rer. nat. Karin Hoyer Schulische Bildung: 1989-1993 Grundschule Groß-BuchholzerKirchweg, Hannover 1993-1995 Orientierungsstufe Rehmer Feld, Hannover 1995-1998 Gymnasium Sophienschule, Hannover 1998-2002 Gymnasium Goetheschule, Essen Juni 2002 Abitur Zivildienst: Juli 2002 - März 2003, Zentrallabor, Universitätsklinikum Essen Studium: April 2003 Studium der Humanmedizin an der Universität zu Köln April 2005 Ärztliche Vorprüfung Juni 2005 Beginn einer experimentellen Doktorarbeit im Labor für Herzmuskelphysiologie und molekulare Kardiologie unter Herrn PD Dr. med. H. Reuter an der Klinik III für Innere Medizin der Universität zu Köln (Direktor: Univ.-Prof. Dr. med. E. Erdmann) Februar 2008 - Januar 2009 Praktisches Jahr Staatsexamen im Juni 2009 91