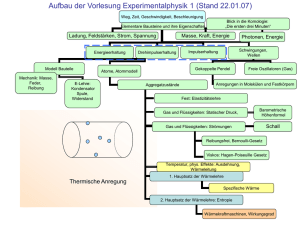
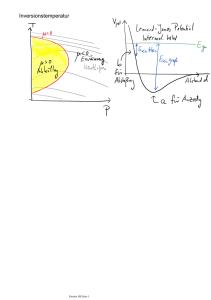
Vorlesung (pdf, aktualisiert 14-02-17)
Werbung
")
Physikalische Chemie I: Thermodynamik, Thermochemie Michael Bredol Fachhochschule Münster – Fachbereich Chemieingenieurwesen Bredol (FH-MS) PC-I 1 / 186 Lehrbücher P.W.Atkins, Physikalische Chemie, VCH G. Wedler, Lehrbuch der Physikalischen Chemie, VCH Engel / Reid, Physikalische Chemie, Pearson Czeslik / Seemann / Winter, Basiswissen Physikalische Chemie, Teubner Nickel, Lehrbuch der Thermodynamik, PhysChem Verlag Daubenfeld / Zenker, Reiseführer Physikalische Chemie, Springer Themen: Energie, Temperatur, Weg- und Zustandsfunktionen, Hauptsätze, Innere Energie, Reversible / irreversible Prozesse, Ideale und Reale Gase, Entropie, Freie Enthalpie und Energie, Gleichgewichte Bredol (FH-MS) PC-I 2 / 186 Workload Leistungspunkte im Modul: 6 −→ 180 Arbeitsstunden insgesamt Kontaktzeit Vorlesung / Übungen: 60 hrs Vorbereitung der Übungen, lesen, Arbeit mit den Vorlesungsunterlagen: 60 hrs Kontaktzeit Praktikum: 30 hrs Praktikumsbericht: 10 hrs Vorbereitung Prüfung: 20 hrs Bredol (FH-MS) PC-I 3 / 186 1. Rückblick Begriffe Zentrale physikalische Begriffe in der Thermodynamik: Innere Energie (U): Fähigkeit eines Systems, Arbeit zu verrichten Arbeit (W ): Energieumsatz bei der Überwindung einer Gegenkraft längs eines Weges ~ ) : elementare physikalische Wechselwirkungen Kräfte (F Energieformen: Potenzielle Energie, kinetische Energie, Wärmeübergang, chemische Arbeit, elektrische Arbeit, ..... Bredol (FH-MS) PC-I 4 / 186 1. Rückblick Begriffe Wärme (-übergang) (Q): beruht auf ungeordneter molekularer Bewegung Temperatur (T ): Maß für Intensität der ungeordneten Bewegung Aus dieser Betrachtung folgt, dass es einen absoluten Nullpunkt der Temperatur geben muss. Die darauf fußende Temperatur-Einheit ist das Kelvin (K). Energie, Arbeit und Kraft sind sowohl auf der mikroskopischen als auch der makroskopischen Skala definiert Wärme ist ein Sonderfall: als statistisches Phänomen mikroskopisch interpretierbar, aber nur makroskopisch sinnvoll Bredol (FH-MS) PC-I 5 / 186 1. Rückblick Begriffe Auf mikroskopischer Ebene: Wärme ist die Art der Energieübertragung, die mit ungeordneter Bewegung von Molekülen verbunden ist Arbeit ist die Art der Energieübertragung, die mit koordinierter Bewegung von Molekülen verbunden ist Unterschiedliche Formen der Arbeit (z.B. mechanische, elektrische) können prinizipiell verlustfrei ineinander umgewandelt werden Arbeit kann stets quantitativ in Wärme umgewandelt werden (Beispiel: elektrische Arbeit zur Beheizung) Bredol (FH-MS) PC-I 6 / 186 1. Rückblick Prozesse Wärme kann in zyklischen Prozessen nicht vollständig in Arbeit umgewandelt werden (Beispiel: Wärmekraftwerk) Gibt ein System Wärme ab, wird ihr Betrag mit negativem Vorzeichen angeschrieben (das System “verliert Energie”): exothermer Prozess Nimmt ein System Wärme auf, wird ihr Betrag mit positivem Vorzeichen angeschrieben (das System “gewinnt Energie”): endothermer Prozess Ein System kann daran gehindert werden, Wärme auszutauschen (durch Isolierung, durch sehr schnellen Prozessablauf, in einem abgeschlossenen System): adiabatischer Prozess Bredol (FH-MS) PC-I 7 / 186 1. Rückblick Weg- und Zustandsfunktion Arbeit und Wärme sind Weg- oder Prozessfunktionen; ihre Beträge hängen von der Art der Prozessführung (dem Weg, der Vorgeschichte) ab Um einen Zustand unabhängig von Weg und Vorgeschichte zu charakterisieren, benutzt die Thermodynamik Zustandsfunktionen. Eine zentrale Zustandsfunktion ist die Innere Energie U eines Systems, die man als seine Gesamtenergie ansehen kann Die Innere Energie als Zustandsfunktion ist nur von den Zustandsvariablen abhängig. Wenn die Zustandsvariablen eines Systems sich nicht mehr mit der Zeit ändern, ist ein stationärer Zustand erreicht. Bredol (FH-MS) PC-I 8 / 186 1. Rückblick Gleichgewicht Wenn der stationäre Zustand stabil ist (d.h. kein anderer stationärer Zustand nach Störung des Systems eingenommen werden kann), spricht man von einem Gleichgewicht. Thermisches Gleichgewicht: wenn zwei Systeme die gleiche Temperatur aufweisen, findet kein Wärmeübertrag zwischen ihnen statt Nullter Hauptsatz der Thermodynamik: Wenn System A im thermischen Gleichgewicht mit System B ist und desgleichen System B mit System C, dann sind auch die Systeme A und C miteinander im thermischen Gleichgewicht. Bredol (FH-MS) PC-I 9 / 186 2. Der Erste Hauptsatz Energieerhaltung Der Erste Hauptsatz der Thermodynamik ist axiomatischer Natur und nur durch die widerspruchsfreie Beobachtung gedeckt Eine der möglichen Formulierungen erinnert stark an den Energieerhaltungssatz der Physik: Die Innere Energie U eines abgeschlossenen Systems ist konstant Lassen wir dagegen Wärmeaustausch und Arbeit durch das System zu (geschlossenes System), sind Änderungen von U erlaubt Bredol (FH-MS) PC-I 10 / 186 2. Der Erste Hauptsatz Energieerhaltung Änderungen der Zustandsfunktion U sind gleichberechtigt durch die Prozessfunktionen Wärme Q und Arbeit W zu realisieren: ∆U = Q + W (1) Diese Form des Ersten Hauptsatzes verknüpft also Prozess- und Zustandsfunktionen Die Formulierung ist gleichbedeutend damit, dass kein Perpetuum Mobile konstruiert werden kann, also keine Maschine, die ohne Energiequelle Arbeit verrichtet. Bredol (FH-MS) PC-I 11 / 186 2. Der Erste Hauptsatz Differenzielle Schreibweise Wenn nur infinitesimal kleine Änderungen betrachtet werden sollen: dU = δQ + δW (2) Der Operator d vor U beschreibt das Differenzial einer Zustandsfunktion (vollständiges Differenzial), der Operator δ vor Q und W die Differenziale einer Wegfunktion (unvollständige Differenziale, die Wegangabe fehlt!) Statt δ sind auch andere Symbole gebräuchlich, etwa ð, d̄ oder d Um den Ersten Hauptsatz auf konkrete Prozesse anwenden zu können, müssen Q und W aus diesen Prozessen ableitbar sein. Bredol (FH-MS) PC-I 12 / 186 2. Der Erste Hauptsatz Arbeit Arbeit, die von einem oder an einem System geleistet wird, kann unterschiedliche Formen annehmen: elektrische Arbeit, Volumenarbeit, Oberflächenarbeit ... Zunächst wird ausschließlich die Volumenarbeit betrachtet Darunter ist z.B. die Arbeit zu verstehen, die ein System bei seiner Expansion gegen einen äußeren Druck leistet: besonders wichtig bei Beteiligung von Gasen Um die Volumenarbeit näher beschreiben zu können, wird zunächst ein einfaches Arbeitsmedium für die Expansion oder Kompression definiert: das Ideale Gas Bredol (FH-MS) PC-I 2. Der Erste Hauptsatz 13 / 186 2.1. Das Ideale Gas Zustandsgleichung Beobachtung: Mit fallendem Druck und steigender Temperatur verlieren alle Gase ihre Individualität Alle Zustandsvariablen werden in einer einfachen Zustandsgleichung miteinander verknüpft: PV = nRT (3) R: allgemeine Gaskonstante (8.314 J K −1 mol −1 ), P: Gasdruck, V : Gasvolumen, n: Stoffmenge des Gases, T : Temperatur des Gases Bredol (FH-MS) PC-I 14 / 186 2. Der Erste Hauptsatz 2.2. Volumenarbeit Reversible Prozesse Ein reversibler Prozess kann jederzeit durch eine Gegenkraft umgekehrt werden, er verläuft zu jedem Zeitpunkt durch Gleichgewichtszustände Ein irreversibler Prozess führt durch Nicht–Gleichgewichtszustände zu nicht umkehrbaren Veränderungen Reversible Expansion (oder Kompression) eines Gases erfordert demnach eine Gegenkraft, mit deren Hilfe die Expansion (oder Kompression) jederzeit wieder rückgängig gemacht werden kann Im Gedankenexperiment ist das gegeben, wenn ein in einem Zylinder befindliches Gas durch sehr langsames Verschieben eines Kolbens gegen eine Gegenkraft expandiert oder komprimiert wird Bredol (FH-MS) PC-I 2. Der Erste Hauptsatz 15 / 186 2.2. Volumenarbeit Reversible Expansion Geleistete Arbeit kann am Kolben abgenommen werden; gleichzeitig kann die Gegenkraft am Kolben jederzeit den Prozess umkehren Irreversible Expansion liegt z.B. vor, wenn das Gas unter seinem eigenen Druck frei in das Vakuum ausströmt: es wird keine Arbeit geleistet, der Prozess ist nicht durch eine Gegenkraft umkehrbar Bredol (FH-MS) PC-I 16 / 186 2. Der Erste Hauptsatz 2.2. Volumenarbeit Reversible Expansion Für reversible Expansion läßt sich die geleistete Volumenarbeit aus den mechanischen Gesetzen ableiten: dW = −F dz (4) Die infinitesimal geleistete Arbeit ist gleich dem Produkt aus der infinitesimalen Wegstrecke und der überwundenen Gegenkraft (negatives Vorzeichen, die Innere Energie sinkt, da Arbeit geleistet wird, um die Gegenkraft zu überwinden) Wenn die Gegenkraft F durch einen äußeren Druck bereitgestellt wird [Druck (P) = Kraft(F ) pro Fläche (A)], dann gilt dW = −Pex Adz (5) PC-I 17 / 186 Bredol (FH-MS) 2. Der Erste Hauptsatz 2.2. Volumenarbeit Reversible Expansion Adz wiederum entspricht genau der Volumenänderung dV , so dass schließlich folgt: dW = −Pex dV (6) Bei reversibler Expansion / Kompression muss der äußere Gegendruck stets gleich dem Innendruck des Systems sein, so dass im reversiblen Fall auch gilt: dWrev = −PdV (7) Freie Expansion in das Vakuum bedeutet, dass keine Gegenkraft (Gegendruck) auf das System wirkt: Pex = 0 und somit auch dW = 0 zu jedem Zeitpunkt Expansion gegen einen konstanten äußeren Druck ist ebenfalls irreversibel, da keine ausbalancierte Gegenkraft auf das System wirkt Bredol (FH-MS) PC-I 18 / 186 2. Der Erste Hauptsatz 2.2. Volumenarbeit Reversible Expansion Reversible, isotherme Volumenarbeit mit einem Idealen Gas kann berechnet werden, da die Zustandsgleichung des Systems (Verknüpfung von n, P, V , T ) bekannt ist: nRT V (8) dV V2 = −nRT ln V V1 (9) PV = nRT W = −nRT P= ZV2 V1 Ist das Endvolumen größer als das Anfangsvolumen (Expansion), dann ist W < 0: das System leistet Arbeit an seiner Umgebung Entsprechend wird bei isothermer, reversibler Kompression Arbeit am System geleistet, W > 0 Bredol (FH-MS) PC-I 2. Der Erste Hauptsatz 19 / 186 2.2. Volumenarbeit P/V-Diagramm Reversible Arbeit im P/V-Diagramm: Integral / Fläche unter der Isothermen zwischen den beiden Endvolumina Wird nicht reversibel gearbeitet, sondern gegen einen konstanten Druck Pex (z.B. Atmosphäre): irreversible Volumenarbeit ist Pex ∆V P Reversible Volumenarbeit (grün) Irreversible Volumenarbeit bei konstantem Druck (Rechteck) Pex 0 0 V1 V2 V Bredol (FH-MS) PC-I 20 / 186 2. Der Erste Hauptsatz 2.2. Volumenarbeit Maximale Arbeit Die reversible Volumenarbeit liefert stets den maximal möglichen Betrag an Volumenarbeit Das gilt auch für alle anderen Medien und für alle Arten von Arbeit: reversible Prozessführung liefert stets den größtmöglichen Ertrag an Arbeit Technische Prozesse zur Erzeugung von Arbeit (Kraftwerke, Akkumulatoren, elektrische Batterien...) versuchen daher möglichst nahe an Gleichgewichtszuständen zu arbeiten (reversible Prozessführung) Bredol (FH-MS) PC-I 2. Der Erste Hauptsatz 21 / 186 2.3. Reale Gase Abweichung vom Idealverhalten Ausgangspunkt Ideales Gas: es gibt keine Wechselwirkungen (Kräfte) zwischen den Teilchen; die Innere Energie ist ausschließlich von der Temperatur abhängig; die Teilchen besitzen scheinbar keine Ausdehnung Diese Voraussetzungen sind bei hohem Druck und kleinen Temperaturen nicht mehr erfüllt; es werden neue Zustandsgleichungen benötigt Beobachtungen: Gase kühlen sich bei Entspannung ab oder heizen sich auf, ohne dass die Enthalpie verändert wird Gase können auch kondensieren: Anziehungskräfte! Gase können nicht beliebig weit komprimiert werden: Abstoßung! Bredol (FH-MS) PC-I 22 / 186 2. Der Erste Hauptsatz 2.3. Reale Gase Abweichung vom Idealverhalten Empirischer Ansatz zur Korrektur: Abweichungen werden durch (meist druckabhängige) Korrekturkoeffizienten in der Zustandsgleichung beschrieben, z.B. durch Reihenentwicklung (Z -Faktor, Virialgleichung....) Mikroskopischer Ansatz zur Korrektur: Abweichungen werden durch Beschreibung der wirksamen Kräfte zwischen den Teilchen erfasst, daraus wird eine neue Zustandsgleichung gewonnen (z.B. van-der-Waals–Gleichung). Bredol (FH-MS) PC-I 2. Der Erste Hauptsatz 23 / 186 2.3. Reale Gase Kräfte Ursprung der wirksamen Kräfte: Abstoßung durch Volumenausschluss (“harte oder weiche Kugeln”) Anziehung durch London–van-der-Waals’sche Dispersionskräfte Clusterbildung (chemische Kräfte) Coulomb–Kräfte (dipolar und multipolar) Empirische Beschreibung mit Kompressionsfaktor Z : Z ≡ PVm RT (10) Im Idealen Gas ist Z stets gleich Eins Bredol (FH-MS) PC-I 24 / 186 2. Der Erste Hauptsatz 2.3. Reale Gase Kräfte Für die die meisten realen Gase bei mittleren Temperaturen gilt: Z < 1 bei niedrigem Druck (Anziehung überwiegt, das benötigte molare Volumen kleiner als im Idealfall) Z > 1 bei hohem Druck (Abstoßung überwiegt, molares Volumen größer als im Idealfall) Wichtige Ausnahmen: Wasserstoff, Helium Bei sehr kleinen Temperaturen (wenige K) sind quantenmechanische Effekte zu beachten (Suprafluidität) Bredol (FH-MS) PC-I 2. Der Erste Hauptsatz 25 / 186 2.3. Reale Gase Kompressionsfaktor Schematisch: 2 T=273K 1.8 1.6 1.4 Z 1.2 1 0.8 0.6 Ideales Gas CH4 H2 NH3 0.4 0.2 0 200 400 600 800 1000 P/bar Bredol (FH-MS) PC-I 26 / 186 2. Der Erste Hauptsatz 2.4. Virialgleichung Definition der Virialgleichung Das reale Verhalten kann empirisch durch Virialgleichungen dargestellt werden, meistens als Funktion des Drucks: Z = 1 + BP + CP 2 + . . . (11) B heißt “zweiter Virialkoeffizient”, C “dritter Virialkoefizient” usw. Meist (nicht zu hoher Druck!) wird nur B(T ) verwendet. Beispiele: H2 Ar N2 CO2 B/Pa−1 (T=273K) 6.04 ∗ 10−9 −9.56 ∗ 10−9 −4.63 ∗ 10−9 −6.59 ∗ 10−8 Bredol (FH-MS) B/Pa−1 (T=600K) 7.94 ∗ 10−9 2.39 ∗ 10−9 9.56 ∗ 10−9 −2.49 ∗ 10−9 PC-I 2. Der Erste Hauptsatz 27 / 186 2.4. Virialgleichung Realverhalten −→ Idealverhalten für P −→ 0 und T −→ ∞ nähern sich alle Gase idealem Verhalten an, es gilt dann stets dZ /dP = 0, da stets Z = 1 Z =1 (singulär, d.h. in einem einzelnen Punkt auf der Druckachse), wenn PVm = RT . Aber dZ /dP 6= 0 ! Virialgleichung: dZ /dP = B + 2CP + . . . T = TB : bei der Boyle–Temperatur TB ist in einem großen Druckbereich dZ /dP = 0; das reale Gas verhält sich weitgehend wie ein ideales Gas, da Anziehung und Abstoßung sich aufheben In der einfachen Virialgleichung ist bei der Boyle–Temperatur B(T ) = 0 Beispiel: TB (Luft)=346.8 K Luft in der Nähe von Raumtemperatur läßt sich daher in sehr guter Näherung mit der Zustandsgleichung idealer Gase beschreiben! Bredol (FH-MS) PC-I 28 / 186 2. Der Erste Hauptsatz 2.4. Virialgleichung Kompressionsfaktor Kompressionsfaktor bei verschiedenen Temperaturen, schematisch: Z Singularität 1 Ideales Gas T<TB T=TB T>TB 0 P Bredol (FH-MS) PC-I 2. Der Erste Hauptsatz 29 / 186 2.5. van-der-Waals–Gleichung Abstoßung und Anziehung Die van-der-Waals’sche Zustandsgleichung berücksichtigt mikroskopisch wirksame Kräfte und korrigiert damit die Zustandsgleichung des Idealen Gases: Abstoßende Kräfte werden durch ein unzugängliches (Eigen–) Volumen harter Kugeln dargestellt (Parameter b, Covolumen) Anziehende Kräfte werden durch einen nach innen gerichteten, mit der Dichte zunehmenden “Binnendruck” a/Vm2 (Parameter a) dargestellt Bredol (FH-MS) a P+ 2 Vm (Vm − b) = RT PC-I (12) 30 / 186 2. Der Erste Hauptsatz 2.5. van-der-Waals–Gleichung Gleichgewicht Im P/V –Diagramm ist die van-der-Waals–Gleichung ein Polynom dritten Grades: T3 T2 T1 P T3 > T2 > T1 • • GG (l) GG (g) Vm Bredol (FH-MS) PC-I 2. Der Erste Hauptsatz 31 / 186 2.5. van-der-Waals–Gleichung Gleichgewicht Bei niedrigen Temperaturen wird die P/V –Kurve mehrdeutig: es gibt Extremwerte und einen Wendepunkt Interpretation: Gleichgewicht g/l, Ersatz des mehrdeutigen Teils der P/V -Kurve durch eine Gerade, die ein Integral mit der Kurve von Null einschließt (keine Arbeit im GG, reversible Umwandlung!) Die Temperatur, bei der Extremwerte und Wendepunkt in einen Sattelpunkt zusammenfallen, heißt kritische Temperatur TK , die zugehörigen Werte für P und Vm heißen PK und Vm,K Bei Temperaturen oberhalb von TK ist keine Verflüssigung möglich, unterhalb von TK können Flüssigkeit und Gas im Gleichgewicht koexistieren (Siedegleichgewicht) Bredol (FH-MS) PC-I 32 / 186 2. Der Erste Hauptsatz 2.5. van-der-Waals–Gleichung Beispiel CO2 a = 0.3688 m6 Pa mol−2 , b = 4.267 ∗ 10−5 m3 mol−1 200 permanentes Gas 150 P / bar 850K 700K 100 600K 500K Flüssigkeit 400K 350K 290K 250K Gas 50 Zweiphasengebiet (l/g) 0 0.05 0.1 0.15 0.2 0.25 0.3 3 Vm / (m /mol) Bredol (FH-MS) 0.35 0.4 0.45 0.5 PC-I 2. Der Erste Hauptsatz 33 / 186 2.5. van-der-Waals–Gleichung Theorem der übereinstimmenden Zustände Die kritische Isotherme besitzt einen Sattelpunkt: erste und zweite Ableitung sind gleichzeitig Null Daraus lassen sich die kritischen Daten als Funktion der v.d.W.–Parameter bestimmen: PK = a 27b 2 Vm,K = 3b TK = 8a 27bR (13) Durch Bezug der Zustandsvariablen auf die jeweilige kritische Größe gewinnt man die reduzierte v.d.Waals–Gleichung: Tred = T /TK Pred = P/PK Pred = Bredol (FH-MS) Vm,red = Vm /Vm,K 3 8Tred − 2 3Vm,red − 1 Vm,red PC-I (14) (15) 34 / 186 2. Der Erste Hauptsatz 2.5. van-der-Waals–Gleichung van-der-Waals–Gleichung und Virialentwicklung Weitere, verfeinerte und erweiterte Zustandsgleichungen sind vorgeschlagen worden, sie werden in diversen technischen Anwendungen genutzt Die v.d.W.–Gleichung entspricht einer Virialentwicklung in P bis zum quadratischen Term (in den Koeffizienten wird teils das ideale Gasgesetz benutzt): aP ab ab a 2 P + PV = RT + bP − + P = RT + b − P2 2 2 RT RT (RT ) (RT ) (16) Der quadratische Term wird häufig vernachlässigt. Näherungsweise läßt sich so die Boyle–Temperatur eines v.d.W.-Gases angeben (der lineare Koeffizient muss Null werden): TB = Bredol (FH-MS) a Rb PC-I 2. Der Erste Hauptsatz (17) 35 / 186 2.6. Wärmekapazität Wärmekapazität Das Ergebnis aus der Diskussion der Volumenarbeit eingesetzt in den Ersten Hauptsatz: dU = δQ + δWe − PdV (18) We fasst alle Arten von Nicht-Volumenarbeit zusammen Wenn das betrachtete System keine We verrichten kann und zusätzlich dV = 0 gilt (isochore Prozesse), dann reduziert sich die Gleichung zu Bredol (FH-MS) dU = δQ(V = const, keinWe ) (19) dU = dQV (20) PC-I 36 / 186 2. Der Erste Hauptsatz 2.6. Wärmekapazität Wärmekapazität Wenn demnach in einem System konstanten Volumens die aufgenommene oder abgegebene Wärmemenge gemessen wird, dann bestimmt man direkt die Änderung der Inneren Energie des Systems In der Praxis geschehen solche Messungen z.B. in einem Bombenkalorimeter. Die Bombe ist ein Stahlreaktor, der konstantes Volumen garantiert. Sie befindet sich in einem äußeren Wasserbad, dessen Temperaturgang zur Bestimmung des Wärmeumsatzes ausgenutzt wird Die Eichung solcher Kalorimeter kann zweckmäßig durch elektrische Heizung oder chemische Eichprozesse erfolgen Bredol (FH-MS) PC-I 2. Der Erste Hauptsatz 37 / 186 2.6. Wärmekapazität Wärmekapazität Zufuhr von Wärme bei konstantem Volumen erhöht die Innere Energie und die Temperatur eines Systems. Wie stark die Innere Energie mit der Temperatur zunimmt, ist eine spezifische System- (und Stoff-) Eigenschaft Sie wird charakterisiert durch die Wärmekapazität CV des Systems (genauer: Wärmekapazität bei konstantem Volumen): ∂U (21) CV = ∂T V Die Definition enthält die partielle Ableitung der Inneren Energie nach der Temperatur bei konstantem Volumen Bredol (FH-MS) PC-I 38 / 186 2. Der Erste Hauptsatz 2.6. Wärmekapazität Wärmekapazität Bei bekannter Wärmekapazität des Systems erhält man die Änderung der Inneren Energie des Systems bei Temperaturänderung: dU = CV dT (22) Bei konstantem Volumen ist die Änderung der Inneren Energie äquivalent zum Wärmeaustausch, so dass auch gilt: dQV = CV dT ∂Q CV = ∂T V (23) (24) Aus dieser Schreibweise resultiert die Bezeichnung Wärmekapazität. Bezieht man sich dabei auf ein Mol eines Stoffes, erhält man die molare Wärmekapazität CV ,m = CV /n Bredol (FH-MS) PC-I 2. Der Erste Hauptsatz 39 / 186 2.6. Wärmekapazität Temperaturabhängigkeit der Wärmekapazität Im Allgemeinen sind Wärmekapazitäten temperaturabhängig, so dass zur Ermittlung von ∆U entsprechend integriert werden muss: ∆U = ZT2 CV dT (25) T1 Wärmekapazitäten (reiner Stoffe) findet man daher tabellarisch häufig in Form empirischer Reihenentwicklungen nach der Temperatur Bredol (FH-MS) PC-I 40 / 186 2. Der Erste Hauptsatz 2.7. Vollständige Differenziale Formaler Ansatz Die Wärmekapazität bei konstantem Volumen als partielle Ableitung der Inneren Energie ist einer der Koeffizienten des vollständigen (auch total oder exakt genannt) Differenzials von U bezüglich der Zustandsvariablen V und T : ∂U ∂U dU = dT + dV (26) ∂T V ∂V T Vollständige Differenziale einer Funktion zweier Variablen beschreiben die Tangentialebene in einem Punkt der durch die Funktion aufgespannten Fläche Bredol (FH-MS) PC-I 2. Der Erste Hauptsatz 41 / 186 2.7. Vollständige Differenziale Beispiel: ideales Gas Druck als Funktion von T und Vm : P = RT /Vm , also ∂P RT ∂P R =− 2 = ∂T Vm Vm ∂Vm T Vm dP = RT R dT − 2 dVm Vm Vm (27) (28) Tangentialebene an (P0 , T0 , Vm,0 ): P − P0 = Bredol (FH-MS) R RT (T − T0 ) − 2 (Vm − Vm,0 ) Vm,0 Vm,0 PC-I (29) 42 / 186 2. Der Erste Hauptsatz 2.7. Vollständige Differenziale Beispiel: ideales Gas Mit den Koordinaten T0 = 650 K und Vm,0 = 11 l/mol: 15 P/bar 10 5 0 −5 900 5 10 T0 Vm,0 700 600 15 Vm / (l/mol) 800 500 20 400 25 Bredol (FH-MS) T/K 300 30 200 PC-I 2. Der Erste Hauptsatz 43 / 186 2.7. Vollständige Differenziale Schwarz’scher Satz Für ein vollständiges Differenzial (Zustandsfunktionen!) gilt der Satz von Schwarz: Die zweiten gemischten partiellen Ableitungen in einem vollständigen Differenzial sind gleich Formuliert für U: ∂2U ∂ ∂U ∂ ∂U ∂2U = = = ∂V ∂T V T ∂T ∂V T V ∂V ∂T ∂T ∂V (30) Ob ein Differenzial vollständig (total, exakt) ist, erkennt man also daran, ob der Schwarz’sche Satz erfüllt ist. Ist er nicht erfüllt, liegt ein unvollständiges Differenzial (Prozessfunktionen, Wegfunktionen!) vor. Bredol (FH-MS) PC-I 44 / 186 2. Der Erste Hauptsatz 2.7. Vollständige Differenziale Kurvenintegrale Für vollständige Differenziale gilt auch, dass das Kurvenintegral wegunabhängig nur von Anfangs- und Endpunkt der Kurve bestimmt ist. Für geschlossene Kurven (Anfangs- gleich Endzustand) ist das Kurvenintegral einer Zustandsfuktion Null: E=A I dU = 0 (31) A Für Prozessfunktionen gilt jeweils das Gegenteil: es werden unvollständige Differenziale gebildet, das Kurvenintegral ist wegabhängig, die Integration über eine geschlossene Kurve ist ungleich null Bredol (FH-MS) PC-I 2. Der Erste Hauptsatz 45 / 186 2.7. Vollständige Differenziale Ersatzprozesse Ein Prozess 1 −→ 3 mit einem idealen Gas kann durch die Abfolge eines isochoren Schritts 1 −→ 2 und eines isothermen Schritts 2 −→ 3 ersetzt werden: isotherm isochor Prozess P 2 1 3 Vm Es werden nun die Differenziale von Q und U verglichen Bredol (FH-MS) PC-I 46 / 186 2. Der Erste Hauptsatz 2.7. Vollständige Differenziale Ersatzprozesse Im isochoren Schritt sind sowohl die zuzuführende Wärmemenge als auch die Änderung der Inneren Energie allein durch die Wärmekapazität bestimmt: dU = dQV = CV ,mdT (32) Im isothermen Schritt muss die geleistete Arbeit durch eine gleich große Wärmezufuhr kompensiert werden, da im idealen Gas U nur von der Temperatur und nicht vom Volumen abhängt: dU = δQ + δW = 0 −→ δQ = PdVm = Bredol (FH-MS) RT dVm Vm PC-I 2. Der Erste Hauptsatz (33) 47 / 186 2.7. Vollständige Differenziale Ersatzprozesse Die Differenziale für U und Q werden nun als vollständiges Differenzial oder aus den beiden Teilschritten zusammengesetzt: dU = CV ,m dT + 0dVm δQ = δQ1−→2 + δQ2−→3 = CV ,m dT + (34) RT dVm Vm (35) Zur Prüfung mit dem Schwarz’schen Satz werden die zweiten gemischten partiellen Ableitungen untersucht: CV ,m ist nicht volumenabhängig, die Ableitung nach Vm ist gleich null: U bildet ein vollständiges Differenzial RT /Vm ist abgeleitet nach T gleich R/Vm : Q bildet ein unvollständiges Differenzial Bredol (FH-MS) PC-I 48 / 186 2. Der Erste Hauptsatz 2.8. Enthalpie H Definition In der chemischen Praxis ist das Arbeiten bei konstantem (Atmosphären-) Druck weit verbreitet. Es wird daher untersucht (ausgehend vom Ersten Hauptsatz), wie sich U bei einem Prozess unter konstantem (äußeren) Druck P unter Berücksichtigung der Volumenarbeit verändert: dU = δQ − PdV ZU2 U1 = ZQP δQ − P 0 (36) ZV2 dV (37) V1 Die Angabe der Integrationsgrenze QP am Integral über dem unvollständigen Differenzial δQ gibt explizit den eingeschlagenen Weg an: konstanter Druck. Bredol (FH-MS) PC-I 2. Der Erste Hauptsatz 49 / 186 2.8. Enthalpie H Definition Ausführung der Integration liefert: U2 − U1 = QP − P(V2 − V1 ) (38) (U2 + PV2 ) − (U1 + PV1 ) = QP (39) oder umgestellt: Der Wärmeumsatz bei konstantem Druck QP entspricht daher der Differenz einer Größe U + PV . Daraus folgt die Definition für die Enthalpie H: H = U + PV (40) und damit ∆H = QP Bredol (FH-MS) PC-I (41) 50 / 186 2. Der Erste Hauptsatz 2.8. Enthalpie H Definition In Worten: Die Enthalpieänderung eines Systems ist gleich der zugeführten oder abgeführten Wärmemenge bei konstantem Druck, wenn das System ausschließlich Volumenarbeit verrichtet Da die Enthalpie aus den Zustandsfunktionen und Zustandsvariablen U, P und V zusammengesetzt ist, ist sie selbst auch eine Zustandsfunktion Die differenzielle Schreibweise für H läßt sich aus der Differenzierung der Definitionsgleichung und Einsetzen des Ersten Hauptsatzes (auschließlich Volumenarbeit) gewinnen: Bredol (FH-MS) PC-I 2. Der Erste Hauptsatz 51 / 186 2.8. Enthalpie H Wärmekapazität CP dH = dU + PdV + V dP (42) dH = δQ − PdV + PdV + V dP (43) dH = δQ + V dP (44) Daraus folgt unter konstantem Druck sofort dH = δQP Es ist zweckmäßig, H als Zustandsfunktion von T und P: ∂H ∂H dT + dP dH = ∂T P ∂P T Der erste Koeffizient definiert die (isobare) Wärmekapazität CP : ∂H CP = ∂T P Bredol (FH-MS) PC-I (45) (46) (47) 52 / 186 2. Der Erste Hauptsatz 2.8. Enthalpie H Wärmekapazität CP Es folgen analog zur Inneren Energie U die Beziehungen dH = CP dT (wenn P = const) (48) dQP = CP dT (wenn P = const) (49) ∆H = ZT2 (wenn P = const) CP dT (50) T1 dH = CP dT + ∂H ∂P dP (51) T Die Enthalpie H verhält sich bezüglich der Inneren Energie U komplementär, wenn Volumen und Druck vertauscht werden Bredol (FH-MS) PC-I 2. Der Erste Hauptsatz 53 / 186 2.8. Enthalpie H Beziehung zwischen H und U In einem idealen Gas läßt sich durch Einsetzen der Zustandsgleichung ein einfacher Zusammenhang zwischen H und U herstellen: H = U + PV = U + nRT (52) dH = dU + RndT + RT dn (53) dH = dU + RT dn (54) (wenn T = const) Sind bei einem Prozess (ideale) Gase beteiligt, dann lassen sich mit dieser Beziehung Änderungen von Enthalpie H in solche der Inneren Energie U näherungsweise umrechnen und umgekehrt Sind ausschließlich kondensierte Phasen an einem Prozess beteiligt, dann sind die Volumenänderungen gewöhnlich klein, genau wie der Unterschied zwischen ∆H und ∆U Bredol (FH-MS) PC-I 54 / 186 2. Der Erste Hauptsatz 2.8. Enthalpie H Freiheitsgrade und Wärmekapazität CV ist die Änderung der Inneren Energie U mit der Temperatur T Aus der kinetischen Gastheorie bzw. der statistischen Thermodynamik läßt sich das Ergebnis entnehmen, dass pro (energetischem) Freiheitsgrad eines Moleküls CV,m gerade R/(2NA ) beträgt In einatomigen idealen Gasen liegen in der Nähe der Raumtemperatur nur die drei Tranlationsfreiheitsgrade vor (keine Schwingungen möglich, da thermische Energie zu gering), so dass die molare Wärmekapazität 3R/2 beträgt In zweiatomigen Gasen sind zusätzlich zwei Rotationsfreiheitsgrade verfügbar; die molare Wärmekapazität CV,m beträgt in diesem Fall daher 5/2R Für Gase mehratomiger Moleküle steigt dieser Wert auf über 3R Bredol (FH-MS) PC-I 2. Der Erste Hauptsatz 55 / 186 2.8. Enthalpie H CP und CV Die Differenz zwischen CP und CV läßt sich für Ideale Gase aus den Definitionen ermitteln: ∂U ∂U ∂V ∂U ∂H − = +P − (55) CP − CV = ∂T P ∂T V ∂T P ∂T P ∂T V Das vollständige Differenzial von U bezüglich T und V läßt sich geschickt umstellen: ∂U ∂U dU = dT + dV ∂T V ∂V T Bredol (FH-MS) ∂U ∂T = P ∂U ∂T + V PC-I ∂U ∂V T ∂V ∂T (56) (57) P 56 / 186 2. Der Erste Hauptsatz 2.8. Enthalpie H CP und CV Das Ergebnis aus Gl.57 wird in Gl.55 eingesetzt und liefert ∂U ∂V ∂V + CP − CV = P ∂T P ∂V T ∂T P ∂U ∂V P+ = ∂T P ∂V T (58) Im Idealen Gas fällt der zweite Summand in der eckigen Klammer weg, und der Differenzialquotient vor der Klammer läßt sich aus der Zustandsgleichung berechnen: nRT nR ∂V V = −→ = −→ P ∂T P P CP − CV = nR CP,m − CV ,m = R Bredol (FH-MS) PC-I 2. Der Erste Hauptsatz (59) (60) 57 / 186 2.9. Adiabatische Zustandsgleichung Adiabatische Prozesse In adiabatischen Prozessen ist δQ = 0, daher gilt in einem reversiblen adiabatischen Prozess (Erster Hauptsatz): dU = δW = −PdV (61) Für dU läßt sich das vollständige Differenzial unter Nutzung von CV einsetzen: ∂U dV = −PdV (62) CV dT + ∂V T In einem ideales Gas verschwindet der zweite Summand auf der linken Seite, und man erhält: CV dT = −PdV = − Bredol (FH-MS) PC-I nRT dV V (63) 58 / 186 2. Der Erste Hauptsatz 2.9. Adiabatische Zustandsgleichung Adiabatische Prozesse Variablentrennung und Integration: nR T2 V2 V1 CV dT = − dV −→ CV ln = −nR ln = nR ln T V T1 V1 V2 (64) In Exponentialform: T2 T1 CV = V1 V2 nR (65) und umgestellt mit molarer Wärmekapazität als adiabatische Zustandsgleichung für ideale Gase: C V2 T2 V ,m /R C = V1 T1 V ,m Bredol (FH-MS) /R = const PC-I 2. Der Erste Hauptsatz (66) 59 / 186 2.9. Adiabatische Zustandsgleichung Adiabatische Prozesse Diese Gleichung ist bereits eine der Formen der adiabatischen Zustandsgleichung für ideale Gase. Zur weiteren Vereinfachung wird noch ein Koeffizient γ eingeführt: γ≡ im idealen Gas : CP CV CP,m − CV ,m R = =γ−1 CV ,m CV ,m (67) (68) Damit wird aus Gl.66: VT CV ,m/R = Bredol (FH-MS) V T γ−1 PC-I = const (69) 60 / 186 2. Der Erste Hauptsatz 2.9. Adiabatische Zustandsgleichung Poisson’sche Zustandsgleichung Einsetzen der Zustandsgleichung des Idealen Gases für T liefert die Poisson’sche Zustandsgleichung für adiabatische Prozesse in idealen Gasen: PV 1/(γ−1) CV ,m /R = const VT =V nR −→ V γ P = const (70) Im P/V –Diagramm verlaufen Adiabaten stets steiler als Isothermen (γ > 1) Adiabatische Prozesse: Wärmeaustausch ist unterbunden (thermische Isolation) oder auf Grund der Geschwindigkeit des Prozesses unmöglich (z.B. Ausbreitung von Schallwellen in Luft) Bredol (FH-MS) PC-I 2. Der Erste Hauptsatz 61 / 186 2.9. Adiabatische Zustandsgleichung Koeffizienten Für technische Zwecke existieren noch einige Koeffizienten zur Beschreibung von Ausdehnung und Kompressibilität: 1 ∂V Isobare thermische Ausdehnung α= V ∂T P Für ein Ideales Gas ist α = 1/T 1 ∂V κT = − Isotherme Kompressibilität V ∂P T (71) (72) Für ein Ideales Gas ist κT = 1/P Bredol (FH-MS) PC-I 62 / 186 2. Der Erste Hauptsatz 2.9. Adiabatische Zustandsgleichung Koeffizienten Auch für eine adiabatische Zustandsänderung läßt sich eine Kompressibilität angeben: ∂ ln V 1 ∂V =− Adiabatische Kompressibilität κad = − V ∂P ad ∂P ad (73) Aus der adiabatischen Zustandsgleichung 70 folgt, dass γ ln V + ln P = const −→ κad = 1 γP (74) Für ein Ideales Gas unterscheiden sich die adiabatische und die isotherme Kompressibilität daher um den Faktor 1/γ. Bredol (FH-MS) PC-I 2. Der Erste Hauptsatz 63 / 186 2.10. Joule-Thomson–Prozess Drosselentspannung Joule-Thomson–Prozess: Expansion durch ein Drosselventil: ein Gas wird von höherem Druck P1 durch ein den Rückschlag verhinderndes Drosselventil auf einen kleineren Druck P2 entspannt Das System sei thermisch isoliert: irreversible adiabatische Expansion Bredol (FH-MS) PC-I 64 / 186 2. Der Erste Hauptsatz 2.10. Joule-Thomson–Prozess Drosselentspannung Die auf der Hochdruck– und Niederdruckseite umgesetzte Arbeit wird getrennt angeschrieben Am System geleistete Arbeit (Kompression auf Volumen null): W1 = −P1 ∆V = −P1 (0 − V1 ) = P1 V1 (75) Vom System geleistete Arbeit (Expansion auf das Endvolumen): W2 = −P2 ∆V = −P2 (V2 − 0) = −P2 V2 (76) Da Wärmefluss unterbunden ist (adiabatischer Prozess, δQ = 0), muss für die Änderung der Inneren Energie durch den Prozess gelten: ∆U = W1 + W2 = P1 V1 − P2 V2 (77) oder nach Umstellung: U2 + P2 V2 = U1 + P1 V1 −→ H2 = H1 Bredol (FH-MS) PC-I 2. Der Erste Hauptsatz (78) 65 / 186 2.10. Joule-Thomson–Prozess Joule-Thomson-Koeffizient Es handelt sich demnach um einen isenthalpischen Prozess: die Enthalpie des Systems ändert sich nicht Daher läßt sich aus dem vollständigen Differenzial der Enthalpie direkt angeben, wie sich die Temperatur durch den Prozess ändert; das Ergebnis heißt Joule-Thomson–Koeffizient: ∂H dP −→ dH = CP dT + ∂P T Bredol (FH-MS) ∂T ∂P H 1 =− CP PC-I ∂H ∂P (79) T 66 / 186 2. Der Erste Hauptsatz 2.10. Joule-Thomson–Prozess Joule-Thomson-Koeffizient In idealen Gasen ist (∂H/∂P)T gleich Null: die Enthalpie ist wie die Innere Energie nur von der Temperatur abhängig, nicht vom Druck oder dem Volumen Entsprechend verändert sich die Temperatur eines idealen Gases nicht, wenn es einem Joule-Thomson–Prozess unterworfen wird In realen Gasen treten allerdings Wechselwirkungen zwischen den Gasteilchen auf; sie sind in den meisten Fällen insgesamt anziehend Diese Anziehungskraft muss überwunden werden; die dafür nötige Energie kann nur der Inneren Energie des Systems entnommen werden; es kühlt sich daher durch die Expansion ab Bredol (FH-MS) PC-I 2. Der Erste Hauptsatz 67 / 186 2.10. Joule-Thomson–Prozess Joule-Thomson-Koeffizient In einigen wenigen Gasen überwiegen die Abstoßungskräfte zwischen den Teilchen; bei der Expansion wird Energie aus der gespeicherten Abstoßungsenergie frei, das System erwärmt sich Die wirksamen Kräfte sind sehr klein, auch der Joule-Thomson–Effekt ist gewöhnlich klein. Typische Werte bei Zimmertemperatur: N2 : 0.25 K/bar He: -0.062 K/bar Stickstoff bei Raumtemperatur ist ein Vertreter der realen Gase mit überwiegend anziehenden Kräften zwischen den Teilchen: der Joule-Thomson–Koeffizient ist positiv, das Gas kühlt sich bei Drosselentspannung ab Bredol (FH-MS) PC-I 68 / 186 2. Der Erste Hauptsatz 2.10. Joule-Thomson–Prozess Einfluss der Temperatur Helium weist bei Raumtemperatur überwiegend abstoßende Kräfte auf; daher erwärmt sich das Gas bei der Drosselentspannung; ein anderer wichtiger Vertreter dieser Gruppe ist Wasserstoff Ob in einem Gas überwiegend Abstoßung oder Anziehung vorliegt, wird u.a. durch die Temperatur eingestellt Alle Gase besitzen daher eine Inversionstemperatur Ti , an der sich anstoßende und abziehende Wechselwirkungen gerade aufheben Oberhalb von Ti überwiegen abstoßende Kräfte, unterhalb von Ti dominieren anziehende Kräfte Bredol (FH-MS) PC-I 2. Der Erste Hauptsatz 69 / 186 2.10. Joule-Thomson–Prozess Einfluss der Temperatur Typische Werte für Ti : He: 30 K N2 : 617 K H2 : 202 K Ar: 740 K N2 und Ar lassen sich daher durch Drosselentspannung abkühlen und verflüssigen H2 und He müssen zunächst vorgekühlt werden, um unterhalb der Inversionstemperatur operieren zu können Abkühlung durch Drosselentspannung ist Grundlage des Linde-Verfahrens zur Luftverflüssigung. Dabei wird das zu verflüssigende Gas im Kreis geführt und mehrfach durch ein Drosselventil entspannt, bis dass Kondensation eintritt Bredol (FH-MS) PC-I 70 / 186 3. Thermochemische Größen 3.1. Grundlagen Standardzustände Absolute Werte für die Enthalpie sind ebenso wie solche für die Innere Energie nicht bestimmbar Um einen festen Bezugspunkt zu erzeugen, werden Standardzustände für alle Stoffe definiert Standardzustände können bei beliebiger Temperatur angegeben werden; will man auch die Temperatur festlegen, dann wird die Verwendung von T=298 K als Normtemperatur empfohlen Alle (auch nur theoretisch denkbaren) Prozesse besitzen eine Standardprozessenthalpie für die Überführung der Edukte im jeweiligen Standardzustand in Produkte im jeweiligen Standardzustand Bredol (FH-MS) PC-I 3. Thermochemische Größen 71 / 186 3.1. Grundlagen Zustandsfunktionen Aus der Eigenschaft der Enthalpie, eine wegunabhängige Zustandsfunktion zu sein, läßt sich eine wichtige Schlussfolgerung ziehen: Für die (Standard)-Prozessenthalpien ist es gleichgültig, auf welchem Weg die Edukte in die Produkte überführt werden Insbesondere gilt daher, dass Prozesse zusammengesetzt werden dürfen Daher muss z.B. gelten: ∆sub H = ∆sm H + ∆V H (80) Weiterhin muss auch gelten, dass bei Prozessumkehr sich nur das Vorzeichen umkehrt, nicht aber der Betrag Bredol (FH-MS) PC-I 72 / 186 3. Thermochemische Größen 3.1. Grundlagen Zustandsfunktionen Daher z.B. : ∆V H = −∆Kondensation H (81) Zu den physikalischen Zustandsänderungen gehören auch Lösungsvorgänge: eine Substanz wird aus dem reinen in den gelösten Zustand überführt Unter der Lösungsenthalpie bei unendlicher Verdünnung versteht man die Überführung in eine so weit verdünnte Lösung, dass alle Wechselwirkungen zwischen den gelösten Teilchen vernachlässigbar klein werden. Symbol: ∅ Beispiel: für HCl(g) −−→ HCl(aq) ist ∆L H Raumtemperatur Bredol (FH-MS) ∅ ∆L H = -75.14 kJ/mol bei PC-I 3. Thermochemische Größen 73 / 186 3.1. Grundlagen Zustandsfunktionen Entsprechend lassen sich weitere Zustandsänderungen physikalischer Natur durch die Angabe der Enthalpieänderung charakterisieren Zu den insbesondere in der Chemie wichtigen Typen gehören die Ionisierungsenthalpie und die Elektronenanlagerungsenthalpie, z.B. Na(g) −−→ Na + (g) + e – Cl(g) + e – −−→ Cl – (g) Ein Index wie ”I” gibt an, dass es sich um die Entfernung oder Anlagerung des “ersten” Elektrons handelt: erste, zweite usw. Ionisierungsenthalpie(z.B. 496 kJ/mol und 4563 kJ/mol für Na) Im Fall der Elektronenanlagerungsenthalpie ∆EA H kann diese sowohl negativ sein (elektronegative Elemente) als auch positiv ∅ Bredol (FH-MS) PC-I 74 / 186 3. Thermochemische Größen 3.2. Chemische Reaktionen Enthalpieänderungen ∅ Standardreaktionsenthalpie ∆R H für chemische Reaktionen: Wenn 1 mol Formelumsatz der Edukte im Standardzustand unter Standarddruck in 1 mol Formelumsatz der Produkte im Standardzustand unter Standarddruck überführt wird, dann ist die zugehörige Enthalpieänderung die molare Standardreaktionsenthalpie Wichtig: ∆R H bezieht sich immer auf getrennte Produkte und Edukte: Mischeffekte werden nicht berücksichtigt ∅ Bredol (FH-MS) PC-I 3. Thermochemische Größen 75 / 186 3.2. Chemische Reaktionen Enthalpieänderungen Bestimmte chemische Reaktionen erhalten einen eigenen Namen: oder ∆f H ∅ Standard - Hydrierungsenthalpie ∆Hydrier H ∅ ∅ Standard - Verbrennungsenthalpie ∆C H ∅ Standard - Bildungsenthalpie ∆B H Diese Werte liegen oft tabelliert vor Vereinbarungsgemäß ist der stöchiometrische Koeffizient der zu verbrennenden oder zu hydrierenden Edukte gleich -1 Die Temperatur wird als Index an H angegeben (z.B. ∆B H298 ) ∅ Bredol (FH-MS) PC-I 76 / 186 3. Thermochemische Größen 3.3. Hess’scher Satz Kombinationen von Reaktionen H ist eine Zustandsfunktion; für Enthalpieänderungen durch chemische Reaktionen folgt daraus sofort der Hess’sche Satz: Die Standardenthalpie einer Reaktion ist gleich der Summe der Standardenthalpien einer Folge von Reaktionen, in die die betreffende Reaktion formal zerlegt werden kann Beispiel für die indirekte Bestimmung einer Standardenthalpie: Bredol (FH-MS) PC-I 3. Thermochemische Größen 77 / 186 3.3. Hess’scher Satz Kreisprozesse Um Enthalpieänderungen bei Prozessen zu bestimmen, die nicht direkt messbar sind, werden entweder Ersatzprozesse oder zyklische Prozesse definiert, deren einzige Unbekannte die gesuchte Größe ist Kreisprozess: ein System wird durch eine Folge von Prozessen / Reaktionen in sich selbst überführt; Anfangs- und Endzustand sind gleich Da es sich bei H um eine Zustandsfunktion handelt, muss die Summe aller Änderungen von Standardenthalpien in Kreisprozessen Null sein Beispiel: Bestimmung der Hydrierungsenthalpie von flüssigem Benzol aus den (leicht messbaren) Verbrennungsenthalpien von Benzol, Cyclohexan und Wasserstoff Bredol (FH-MS) PC-I 78 / 186 3. Thermochemische Größen 3.3. Hess’scher Satz Kreisprozesse C6 H6 + 15 2 O2 −−→ 6 CO2 + 3 H2 O −3268 kJ/mol 6 CO2 + 6 H2 O −−→ C6 H12 + 9 O2 3 H2 + 23 O2 −−→ 3 H2 O 3902 kJ/mol −3 ∗ 286 kJ/mol C6 H12 −−→ C6 H6 + 3 H2 −∆Hydrier H (C6 H6 ) ————————————————————————— Ø −−→ Ø 0 ∅ ∆Hydrier H (C6 H6 ) = −224 kJ/mol ∅ −→ Bredol (FH-MS) PC-I 3. Thermochemische Größen 79 / 186 3.4. Kirchhoff’scher Satz Bildungsenthalpien Definition der Bildungsenthalpie als einer speziellen Reaktionsenthalpie: Die Standardbildungsenthalpie ∆B H oder ∆f H eines Stoffes ist die Standardreaktionsenthalpie seiner Bildung aus den Elementen in ihrem jeweiligen Referenzzustand ∅ ∅ Der Referenzzustand der Elemente ist die stabilste Form bei der gegebenen Prozesstemperatur und dem Standarddruck von 1 bar; werden die Elemente nicht im Referenzzustand eingesetzt, ist die entsprechende Umwandlungsenthalpie zu berücksichtigen. Bredol (FH-MS) PC-I 80 / 186 3. Thermochemische Größen 3.4. Kirchhoff’scher Satz Bildungsenthalpien Aus meist bei T = 298 K tabellierten Bildungsenthalpien lassen sich dem Hess’schen Satz zufolge Standardreaktionsenthalpien berechnen: N X νi ∆ B H i (82) ∆R H = ∅ ∅ i=1 Standardreaktionsenthalpien bei anderen Temperaturen können aus der Temperaturabhängigkeit der Enthalpie berechnet werden Benötigt wird dazu das vollständige Differenzial der Enthalpie nit der Formulierung bei konstantem Druck: dH = CP dT + ∂H ∂P dP −→ H(T2 ) − H(T1 ) = T ZT2 CP dT (83) T1 Bredol (FH-MS) PC-I 3. Thermochemische Größen 81 / 186 3.4. Kirchhoff’scher Satz Kirchhoff’scher Satz Solche Beziehungen lassen sich für alle Reaktionsteilnehmer aufstellen. Nach Zusammenfassung: ∆R H(T2 ) = ∆R H(T1 ) + ZT2 ∆R CP dT (84) T1 ∆R C P = N X νi CP,m,i (85) i=1 Aus diesem Grund finden sich in thermochemischen Tabellen Wärmekapazitäten stets zusammen mit Standardenthalpien (meist bei 298 K) Bredol (FH-MS) PC-I 82 / 186 3. Thermochemische Größen 3.4. Kirchhoff’scher Satz Beispiel: Wasser Häufig kann ∆R CP als temperaturunabhängig betrachtet und das Integral damit stark vereinfacht werden Beispiel : Verdampfungsenthalpie von Wasser 298 K: ∆V H = 44.02 kJ/mol ∅ Um die Verdampfungsenthalpie am Siedepunkt zu berechnen, sind entsprechend der “Reaktion“ H2 O(l) −−→ H2 O(g) die Wärmekapazitäten von flüssigem und gasförmigem Wasser nötig: CP (Dampf ) = 35.5 J K −1 mol −1 , CP (Fl üssigkeit) = 75.3 J K −1 mol −1 −→ ∆R CP = −39.8J K −1 mol −1 Damit: Verdampfungsenthalpie am Siedepunkt (373 K) 41.04 kJ/mol (tatsächlich: 40.656 kJ/mol) Die verbleibende Diskrepanz beruht vor allem auf der Temperaturabhängigkeit der Wärmekapazitäten selbst Bredol (FH-MS) PC-I 3. Thermochemische Größen 83 / 186 3.5. Born-Haber-Kreisprozess Gitterenthalpie Die Gitterenthalpie von Ionenkristallen ist ein Beispiel für eine Größe, die definitiv nicht direkt bestimmt werden kann und nur durch einen Kreisprozess zugänglich ist Definition: MX(s) −−→ M + (g) + X – (g) Für das Beispiel NaCl wird ausgehend von festem NaCl zunächst die Bildungsreaktion umgekehrt, dann die Elemente in gasförmige Ionen überführt, und diese schließlich wieder zum NaCl vereint: NaCl(s) −−→ Na(s) + 21 Cl2 (g) −∆B H = 411.2 kJ/mol Na(s) −−→ Na(g) ∆sub H = 107.3 kJ/mol + Na(g) −−→ Na (g) + e – (g) ∆ion H = 498.3 kJ/mol 1 −−→ Cl(g) 0.5∆diss H = 121.7 kJ/mol 2 Cl2 (g) Cl(g) + e – (g) −−→ Cl – (g) ∆EA H = −351.2 kJ/mol – + Na (g) + Cl (g) −−→ NaCl(s) −∆Gitter H =? P 0 −−→ 0 0 ∅ ∅ ∅ ∅ ∅ ∅ Bredol (FH-MS) PC-I 84 / 186 3. Thermochemische Größen 3.5. Born-Haber-Kreisprozess Solvatationsenthalpie Alle Enthalpien bis auf die Gitterenthalpie sind aus unabhängigen Quellen bekannt. Zustandsfunktionen ändern sich in Kreisprozessen nicht, daher für NaCl: ∆Gitter H = 787.3 kJ/mol ∅ Für Gitter aus höher geladenen Ionen liegen die Werte deutlich höher, z.B. für MgO: ∆Gitter H = 3850 kJ/mol ∅ Analog kann man in ähnlich gelagerten Fällen vorgehen, z.B. für die Standardsolvatationenthalpie von ionischen Verbindungen: ∅ ∅ ∅ ∅ Bredol (FH-MS) −∆B H = 407.3 kJ/mol ∆sub H = 107.3 kJ/mol ∆ion H = 498.3 kJ/mol 0.5∆diss H = 121.7 kJ/mol ∆EA H = −351.2 kJ/mol ∆Solv H =? 0 ∅ −−→ Na(s) + 12 Cl2 (g) −−→ Na(g) −−→ Na + (g) + e – (g) −−→ Cl(g) −−→ Cl – (g) −−→ NaCl(aq) −−→ 0 ∅ NaCl(aq) Na(s) Na(g) 1 2 Cl2 (g) Cl(g) + e – (g) Na + (g) + Cl – (g) P 0 PC-I 3. Thermochemische Größen 85 / 186 3.5. Born-Haber-Kreisprozess Solvatationsenthalpie Standardsolvatationsenthalpie von NaCl ist daher -783.4 kJ/mol In diesen Kreisprozessen sind keine Wechselwirkungen zwischen den Ionen in Lösung berücksichtigt; so ermittelte Werte gelten daher nur für unendlich stark verdünnte Lösungen Die Standardbildungsenthalpien gelöster Stoffe sind eindeutig definiert; für Lösungen vollständig dissoziierter Stoffe wären jedoch Standardbildungsenthalpien einzelner Ionen sinnvoll, um komplexe Salzgemische einfach beschreiben zu können Zu diesem Zweck wird die Standardbildungsenthalpie einer gelösten ionischen Verbindung auf die entstehenden Ionen aufgeteilt Dazu ist jedoch ein neuer Bezugspunkt notwendig, und man definiert: Bredol (FH-MS) ∅ ∆B H + H (aq) ≡ 0 PC-I (86) 86 / 186 3. Thermochemische Größen 3.5. Born-Haber-Kreisprozess Solvatationsenthalpie Von diesem definierten Bezugspunkt aus könnnen die Standardbildungsenthalpien aller Ionen angegeben werden So wird ∆B H (Cl – ) aus der Standardbildungsenthalpie von HCl(aq) ermittelt, die von Na + dann aus der Standardbildungsenthalpie von NaCl(aq) usw. ∅ Die ermittelten Werte verteilen sich über einen großen Bereich, mit positiven und negativen Vorzeichen Bredol (FH-MS) PC-I 4. Der Zweite Hauptsatz 87 / 186 4.1. Definitionen Hintergrund Der Erste Hauptsatz mit seinen Konsequenzen gestattet es, mit den Wegfunktionen Wärme und Arbeit umzugehen Bei Anwendung auf physikalische Zustandänderungen oder chemische Prozesse werden Aussagen über den damit verbundenen Wärmeumsatz möglich Der Erste Hauptsatz ermöglicht es aber noch nicht, auch die Triebkräfte für einen Prozess anzugeben Exotherme und endotherme Prozesse können nämlich gleichermaßen spontan ablaufen oder aber auch nicht; es muss folglich eine weitere Größe geben, die die Prozesse antreibt Bredol (FH-MS) PC-I 88 / 186 4. Der Zweite Hauptsatz 4.1. Definitionen Hintergrund Offene Fragen, für deren Beantwortung die Entropie erforderlich ist: Warum fließt Wärme niemals freiwillig vom kälteren zum wärmeren Bereich? Warum neigt Diffusion in den meisten Fällen dazu, Konzentrationsunterschiede einzuebnen statt aufzusteilen? Warum strömt ein ideales Gas freiwillig und irreversibel in das Vakuum aus, obwohl die Innere Energie sich dabei nicht ändert? Offenbar reicht die Untersuchung der Energie (oder auch Enthalpie) nicht aus, um die Triebkräfte dieser und ähnlicher Prozesse zu analysieren Bredol (FH-MS) PC-I 4. Der Zweite Hauptsatz 89 / 186 4.1. Definitionen Eigenschaften Um das erforderliche Maß für die Freiwilligkeit eines Prozesses zu erhalten, definieren wir eine neue Funktion, die Entropie S Ihre wichtigste Eigenschaft wird durch den Zweiten Hauptsatz der Thermodynamik vorgegeben: Bei einer freiwilligen Zustandsänderung nimmt die Entropie eines abgeschlossenen Systems zu: ∆Sgesamt > 0 Irreversible Prozesse laufen stets freiwillig ab, daher muss gelten: Irreversible Prozesse produzieren Entropie Reversible Prozesse produzieren keine Entropie Bredol (FH-MS) PC-I 90 / 186 4. Der Zweite Hauptsatz 4.1. Definitionen Eigenschaften Für reversible Prozesse gilt, dass sie zu jedem Zeitpunkt durch Gegenkräfte ausbalanciert sind; von diesem Gleichgewicht entfernt sich das System nicht freiwillig Infinitesimal kleine Schritte auf einem reversiblen Weg lassen entsprechend die Entropie nicht zunehmen Deutung der Entropie mit den Methoden der Statistischen Thermodynamik: S = K ln W (87) W ist dabei die Thermodynamische Wahrscheinlichkeit eines Zustandes, k eine Naturkonstante: R = kNA Bredol (FH-MS) PC-I 4. Der Zweite Hauptsatz 91 / 186 4.1. Definitionen Konfigurationen Freiwilligkeit bedeutet in einem abgeschlossenen System daher: es wird ein insgesamt wahrscheinlicherer Zustand eingenommen Wahrscheinlichkeit ist eng mit dem Begriff der Ordnung verknüpft: vollständige Ordnung ist unwahrscheinlich Ungeordnete Zustände sind wahrscheinlicher, da es dafür mehrere Konfigurationen gibt W ist in diesem Zusammenhang die Anzahl der möglichen Konfigurationen Der Zusammenhang zwischen Entropie und Ordnungszustand legt nahe, dass die Entropie nicht direkt mit der Arbeit (geordnete Teilchenbewegung), sehr wohl aber mit der Wärme (ungeordnete Teilchenbewegung) verknüpft ist Bredol (FH-MS) PC-I 92 / 186 4. Der Zweite Hauptsatz 4.1. Definitionen Entropie und Wärme Wärmezufuhr intensiviert die ungeordnete Bewegung innerhalb eines Systems, daher ist anzunehmen, dass Entropieänderungen proportional zu zu- oder abgeführten Wärmemengen sind Zur quantitativen Beschreibung muss zwischen dem betrachteten System und dem darin ablaufenden Prozess und seiner Umgebung (die Größen dort sind mit einem Apostroph markiert) unterschieden werden Die Summe aus System und Umgebung ist stets ein abgeschlossenes System: Uges ist konstant, Sges muss stets steigen oder gleich bleiben Bredol (FH-MS) PC-I 4. Der Zweite Hauptsatz 93 / 186 4.1. Definitionen Entropie und Wärme Wenn die Umgebung sehr groß ist, dann wird in die (isotherme) Umgebung übergehende Wärme dort proportional die Entropie erhöhen: Bredol (FH-MS) dS ′ ∝ δQ ′ (88) δQ ′ = −δQ (89) PC-I 94 / 186 4. Der Zweite Hauptsatz 4.1. Definitionen Entropie und Wärme Aus der Beobachtung, dass Wärme immer vom warmen zum kalten Körper fließt, schließen wir, dass bei hoher Temperatur Wärmezufuhr die bestehende Ordnung in geringerem Maße erhöht als bei niedrigerer Temperatur Die inverse Temperatur dient daher als integrierender Faktor zur Definition der Entropie: δQ ′ dS = T ′ (90) Einheit der Entropie ist demnach J/K Die Gleichung ist allgemein gültig, solange sich die Umgebung im inneren Gleichgewicht befindet; der Prozess im System selbst darf reversibel oder irreversibel sein Bredol (FH-MS) PC-I 4. Der Zweite Hauptsatz 95 / 186 4.1. Definitionen Entropie und Wärme Beispiel: der betrachtete Prozess sei eine exotherme chemische Reaktion. Unter konstantem Druck und im thermischen Gleichgewicht mit der Umgebung wird dadurch die Entropie der Umgebung erhöht: ∆ H R ∆S ′ = − (91) T Ein endothermer Prozess würde entsprechend die Entropie der Umgebung verringern Die Entropieänderung des Systems selbst läßt sich berechnen, da S eine Zustandsfunktion und es daher gleichgültig ist, ob der Prozess irreversibel oder reversibel geführt wird Für den reversiblen Fall gilt: Bredol (FH-MS) PC-I 96 / 186 4. Der Zweite Hauptsatz 4.1. Definitionen Entropie und Wärme dS ′ = − dQrev T (92) Wenn auch der Wärmeaustausch reversibel erfolgt (T = T ′ ), gilt dS ′ = −dS (93) und damit für das System selbst der Zweite Hauptsatz der Thermodynamik: dS = Bredol (FH-MS) dQrev T (94) PC-I 4. Der Zweite Hauptsatz 97 / 186 4.2. Der Carnot-Prozess Carnot-Zyklus Um zu zeigen, dass S eine Zustandsfunktion ist, wird ein spezieller Kreisprozess herangezogen, der Carnot-Prozess, hier für ein einatomiges ideales Gas mit T1 = 1000 K und T2 = 630 K: 8000 P/Pa 6000 •A Adiabatische Kompression Isotherme Expansion (T1) 4000 2000 D• Adiabatische Expansion •B •C Isotherme Kompression (T2) 0 2 4 6 8 10 3 Vm/m Bredol (FH-MS) PC-I 98 / 186 4. Der Zweite Hauptsatz 4.2. Der Carnot-Prozess Carnot-Zyklus Enthalten sind zwei adiabatische und zwei isotherme Schritte; der Prozess ist der Prototyp für alle zyklisch arbeitenden Wärme-Kraft-Maschinen In den adiabatischen Schritten wird keine Wärme ausgetauscht: Q = 0 In der Expansion A −−→ B wird dem System die Wärme Q1 zugeführt, bei der Kompression C −−→ D vom System die Wärme Q2 abgeführt Wenn alle Prozessschritte reversibel ablaufen: ∆S = V V Q1 Q2 + = R ln B + R ln D T1 T2 VA VC Bredol (FH-MS) (95) PC-I 4. Der Zweite Hauptsatz 99 / 186 4.2. Der Carnot-Prozess Entropiebilanz Die Verhältnisse der Volumina lassen sich aus der Zustandsgleichung der Adiabaten durch Division ermitteln: C /R VA T1 V C /R C /R VB T1 V = VD T2 V C /R = VC T2 V VA V = D VB VC und damit: I dQrev = ∆S = 0 T (96) (97) (98) Es läßt sich zeigen, dass jeder denkbare Kreisprozess sich aus Carnot-Prozessen zusammensetzen läßt; bei infinitesimal kleinen Carnot-Prozessen ist dieser Ansatz exakt. Daher ist die Entropieänderung in allen denkbaren Kreisprozesen gleich Null: die Entropie ist eine Zustandsfunktion Bredol (FH-MS) PC-I 100 / 186 4. Der Zweite Hauptsatz 4.3. Wirkungsgrade Arbeit im Carnot-Zyklus Aus einem Carnot-Kreisprozess läßt sich Arbeit gewinnen: −W = −(WAB + WBC + WCD + WDA ) (99) Für ideale Gase ist in adiabatischen Prozessen W = ∆U = CV dT , also nur von den beiden Temperaturniveaus abhängig: WBC + WDA = 0 Die verbleibenden Beiträge der Arbeit aus den isothermen Schritten lassen sich leicht berechnen: VB V + RT2 ln D VA VC −W = RT1 ln (100) Mit den bereits bestimmten Verhältnissen der Volumina (Gl.97) wird daraus V −W = R ln B (T1 − T2 ) (101) VA Bredol (FH-MS) PC-I 4. Der Zweite Hauptsatz 101 / 186 4.3. Wirkungsgrade Arbeit im Carnot-Zyklus Um diese Arbeit gewinen zu können, muss dem System die Wärme Q1 zugeführt und die Wärme Q2 abgeführt werden Daher wird der Wirkungsgrad η des Prozesses definiert als η= −W Q1 (102) Mit Q1 = −W1 = RT1 ln(VB /VA ) folgt daraus: η= T1 − T2 T2 =1− T1 T1 (103) Der Wirkungsgrad eines Carnot-Prozesses ist demnach nur von dem Verhältnis der beteiligten Temperaturniveaus abhängig! Bredol (FH-MS) PC-I 102 / 186 4. Der Zweite Hauptsatz 4.3. Wirkungsgrade Temperatur und Wirkungsgrad Aus den Überlegungen folgt eine alternative Formulierung des Zweiten Hauptsatzes: Alle reversiblen Carnot-Prozesse, die zwischen den selben Ausgangs- und Endtemperaturen ablaufen, haben den selben Wirkungsgrad Die vom Carnot-Prozess geleistete Arbeit entspricht der im P/V-Diagramm eingeschlossenen Fläche ABCD Der Carnot’sche Kreisprozess läßt sich sehr einfach auch in einem sogenannten T/S- (Wärme-)diagramm darstellen: Bredol (FH-MS) PC-I 4. Der Zweite Hauptsatz 103 / 186 4.3. Wirkungsgrade Wärmediagramm A B D C T T1 T2 E 0 0 F S1 S2 S Für ein ideales Gas erscheinen (reversible) Adiabaten als Linien konstanter Entropie, während Isothermen Linien konstanter Temperatur sind Bredol (FH-MS) PC-I 104 / 186 4. Der Zweite Hauptsatz 4.3. Wirkungsgrade Wärmediagramm Aus dem Wärmediagramm lassen sich die Wärmemengen ablesen, die aus dem wärmeren Reservoir aufgenommen (Rechteck ABEF) und die an das kältere Reservoir abgegeben werden (Rechteck DCEF) Die Differenz (Rechteck ABCD) ist gleich der abgegebenen Arbeit In der eingezeichneten Richtung ist der Carnot-Prozess eine Wärmekraftmaschine: aus einem Wärmefluss zwischen warmem und kaltem Reservoir wird Arbeit gewonnen In der umgekehrten Richtung wird durch aufgewandte Arbeit Wärme von einem kälteren Niveau auf ein wärmeres Niveau gebracht Wenn dieser Prozess dazu dient, das kältere Temperaturniveau weiter abzukühlen, spricht man von einer Kältemaschine; wird dagegen das wärmere Niveau weiter aufgeheizt, spricht man von einer Wärmepumpe Bredol (FH-MS) PC-I 4. Der Zweite Hauptsatz 105 / 186 4.4. Zeitrichtung Deutung der Entropie Der Begriff “Entropie” stammt aus dem griechischen Wort für “Umwandlung” (τρoπή): nach Rudolf Clausius wird stets Wärme bei hoher Temperatur in solche bei niedriger Temperatur “verwandelt” Das chinesische Schriftzeichen für die Entropie (PinYin–Umschrift: shang) ist moderner, da es das Verhältnis von H und T und den Hinweis auf Wärme enthält: Links: Feuer (Radikal) Bredol (FH-MS) Rechts: Quotient (Phonetik) PC-I 106 / 186 4. Der Zweite Hauptsatz 4.4. Zeitrichtung Deutung der Entropie Carnot selbst war nicht auf der Höhe der Zeit und glaubte an die Kalorik (Lehre von der Wärme als gewichtslosem Wärmestoff Trotzdem zog Carnot die richtigen Schlüsse. Seine Argumente sind jedoch “über weite Strecken unverständlich, und wo sie verständlich sind, sind sie falsch”, wie in späteren Lehrbüchern festgehalten wurde (Müller, Grundzüge der Thermodynamik, Springer 1994) Die Entropieungleichung (2. Hauptsatz) impliziert im Gegensatz zum Rest der klassischen Physik eine Vorzugsrichtung der Zeit und sagt für das Universum den “Wärmetod” voraus; daher war die Entropie häufig auch Gegenstand philosophischer Spekulationen Heute wird sie vor allem als Informationsmaß gedeutet, u.a. auch in der theoretischen Informatik (1 bit=k ˆ ln 2) Bredol (FH-MS) PC-I 4. Der Zweite Hauptsatz 107 / 186 4.4. Zeitrichtung Degradation und Pessimismus Pessimisten formulieren die Hauptsätze auf ihre eigene Weise: (Müller, Grundzüge der Thermodynamik, Springer 1994) 1st law : You can’t win 2nd law : You shouldn’t even try Oder als zwingende kulturelle Degradation: Shakespeare / Hamlet: to be or not to be ? Friedrich Nietzsche: to be is to do Jean Paul Sartre: to do is to be Frank Sinatra: do be do be do be do Bredol (FH-MS) PC-I 108 / 186 4. Der Zweite Hauptsatz 4.5. Irreversible Prozesse Wärmetransport Systeme und ihre Umgebung insgesamt unterscheiden eindeutig zwischen irreversiblen und reversiblen Prozessen: Irreversibel : dS + dS ′ > 0 oder dS > −dS ′ (104) Reversibel : dS + dS ′ = 0 oder dS = −dS ′ (105) Ein typischer irreversibler Prozess ist der Wärmeübergang von einem wärmeren auf einen kälteren Körper. Mit der Definition der Entropie kann man anschreiben, wie die (positive) Wärmemenge δQ übergeht: δQ 1 δQ 1 dS = = − − δQ (106) Tkalt Twarm Tkalt Twarm Für diesen Prozess ist daher stets dS > 0, er ist irreversibel und läuft spontan ab Bredol (FH-MS) PC-I 4. Der Zweite Hauptsatz 109 / 186 4.5. Irreversible Prozesse Expansion eines Idealen Gases Wenn ein ideales Gas reversibel und isotherm von einem Anfangsvolumen VA auf das Endvolumen VE expandiert wird, ist dabei die Arbeit −nRT ln(VE /VA ) zu leisten Im Idealen Gas ist die Innere Energie ausschließlich temperaturabhängig, daher im isothermen Fall: Qrev = −Wrev Demzufolge gilt dann 1 ∆S = T ZE dQrev = V Qrev = nR ln E T VA (107) A Da die Entropie eine Zustandsfunktion ist, muss das hergeleitete Ergebnis aber auch für irreversible Expansion gelten! Bredol (FH-MS) PC-I 110 / 186 4. Der Zweite Hauptsatz 4.5. Irreversible Prozesse Irreversible Expansion eines Idealen Gases Im reversiblen Fall ist die Entropiezunahme des Systems genau mit einer Entropieabnahme der Umgebung ausbalanciert, da der Umgebung Wärme entnommen wird und als Volumenarbeit wieder abgegeben wird: ∆S = −∆S ′ (108) Bei isothermer Expansion in das Vakuum wird keine Arbeit geleistet (Pex = 0) und folglich auch keine Wärme ausgetauscht. Die Entropieänderung des Systems ist dann gleich der Gesamt–Entropieänderung dieses irreversiblen Prozesses: ∆Sgesamt = nR ln VE VA (109) Da die Entropie eines Systems eine Zustandsfunktion ist, gibt diese Gleichung die Entropieänderung eines idealen Gases bei jeder Expansion an Bredol (FH-MS) PC-I 4. Der Zweite Hauptsatz 111 / 186 4.6. Entropieänderungen bei Phasenübergängen Übergangsentropie Phasenübergänge wie Schmelzen, Sublimieren und Verdampfen gehen offensichtlich mit der Veränderung des Ordnungszustandes des Systems einher Im Gleichgewicht zwischen zwei Phasen erfolgt jede Systemänderung reversibel. Bei konstantem Druck gilt dann für die Entropieänderung bei der Phasentransformation: ∆trans S = Qtrans,T ∆trans H = Ttrans Ttrans (110) Schmelzen, Verdampfen und Sublimieren sind endotherme Vorgänge; folglich ist die Transformationsentropie dieser Vorgänge in Übereinstimmung mit der Intuition stets positiv Bredol (FH-MS) PC-I 112 / 186 4. Der Zweite Hauptsatz 4.6. Entropieänderungen bei Phasenübergängen Ähnlichkeiten Dabei kommen in den entsprechenden Zahlenwerten auch die relativen Ähnlichkeiten der Phasen zum Ausdruck; daher sind z.B. Verdampfungsentropien stets größer als Schmelzentropien Beispiel Wasser : Schmelzentropie bei 273 K ist 22.0 J/(mol K), Verdampfungsentropie bei 373 K ist 109 J/(mol K) Für eine große Zahl von Flüssigkeiten liegen die Standardverdampfungsentropien am jeweiligen Siedepunkt sehr nahe beieinander, meist etwa zwischen 85 und 88 J/(mol K) Darin kommt die allgemeine Ähnlichkeit der Abnahme des Ordnungszustandes durch Verdampfung zum Ausdruck Bredol (FH-MS) PC-I 4. Der Zweite Hauptsatz 113 / 186 4.6. Entropieänderungen bei Phasenübergängen Ähnlichkeiten Dieser empirische Zusammenhang ist als Pictet-Trouton’sche Regel bekant und lautet ∆V S ∼ = 10 R oder ∆V H ∼ = 10 RTS (111) Assoziierende Flssigkeiten wie Wasser oder niedere Alkohole fallen nicht unter diese Regel; die Verdampfungsentropie ist hier größer, da die Assoziate den Ordnungsgrad der Flüssigkeit erhöhen Für Wasser ist ∆V H/RTS etwa 13 Die resultierende große Verdampfungsenthalpie für Wasser ist Konsequenz der Assoziatbildung und gewissermaßen unsere Lebensgrundlage Bredol (FH-MS) PC-I 114 / 186 4. Der Zweite Hauptsatz 4.7. Die Temperaturabhängigkeit der Entropie Grundlage Der Zusammenhang zwischen Entropie und Temperatur ist unmittelbar durch die thermodynamische Definition der Entropie gegeben, in integrierter Form: ZE dQrev S(TE ) = S(TA ) + (112) T A Wenn die Erwärmung unter konstantem Druck stattfindet, gilt dQP = CP dT (113) S(TE ) = S(TA ) + ZTE CP dT T (114) TA Bredol (FH-MS) PC-I 4. Der Zweite Hauptsatz 115 / 186 4.7. Die Temperaturabhängigkeit der Entropie Integration Alternativ gilt bei konstantem Volumen während der Erwärmung: S(TE ) = S(TA ) + ZTE CV dT T (115) TA Damit ist die Bestimmung der Entropie bei beliebiger Temperatur auf die (temperaturabhängige) Bestimmung der Wärmekapazität zurückgeführt Wenn die Wärmekapazitäten im interessierenden (kleinen) Temperaturintervall selbst nicht von der Temperatur abhängen, läßt sich einfach integrieren: S(TE ) = S(TA ) + CP ln Bredol (FH-MS) PC-I TE TA (116) 116 / 186 4. Der Zweite Hauptsatz 4.7. Die Temperaturabhängigkeit der Entropie Integration bzw. bei konstantem Volumen S(TE ) = S(TA ) + CV ln TE TA (117) S(T = 0) ist zunächst unbekannt Für T > 0 läßt sich die Entropie eines Stoffes nun entwickeln, wenn alle Phasenübergänge zwischen 0 K und der interessierenden Temperatur berücksichtigt werden Wenn keine Pasenübergänge im festen Zustand auftauchen (einfachster Fall), gilt dann für ein Gas: Bredol (FH-MS) PC-I 4. Der Zweite Hauptsatz 117 / 186 4.7. Die Temperaturabhängigkeit der Entropie Integration S(T ) = S(0)+ TSm Z 0 ∆ H CP (s)dT + Sm + T TSm ZTS TSm CP (l)dT ∆ H + V + T TS ZT CP (g)dT T (118) TS Für eine Flüssigkeit wird entsprechend die Entwicklung nach dem Schmelzübergang abgebrochen, für Festkörper vor dem Schmelzübergang, aber nach etwaigen Phasenumwandlungen des Festkörpers Auftragungen von Messdaten zur einfachen graphischen Integration erfolgen entsprechend entweder in der Form CP /T über T oder in der Form CP über ln T Bredol (FH-MS) PC-I 118 / 186 4. Der Zweite Hauptsatz 4.7. Die Temperaturabhängigkeit der Entropie Tiefe Temperaturen Bei sehr tiefen Temperaturen gilt ein (auch theoretisch begründetes) Gesetz, wonach CP proportional ist zur dritten Potenz der Temperatur (Debye’sches T 3 –Gesetz): CP = aT 3 T < 10K : (119) Diesen Zusammenhang kann man häufig heranziehen, um Messwerte bei tiefer Temperatur zu noch tieferen Werten hin zu extrapolieren Mit kombinierten Daten aus Messung und Extrapolation lassen sich (bis auf den noch unbekannten Wert bei T = 0) Standardentropien für alle Substanzen ermitteln Bredol (FH-MS) PC-I 4. Der Zweite Hauptsatz 119 / 186 4.7. Die Temperaturabhängigkeit der Entropie Beiträge zur Standardentropie Beispiel Stickstoff im Referenzzustand bei Normtemperatur 298 K: Sm /(J K−1 mol−1 ) 1.92 25.25 6.43 23.88 11.42 11.41 72.13 39.20 191.04 ∅ Debye’sches Gesetz (0–10K) Integration 10K – 35.61K Phasenübergang fest/fest bei 35.61K Integration 35.61K –63.14K Schmelzübergang (63.14K) Integration 63.14K – 77.32K Verdampfungsübergang (77.32K) CP konstant bis 298K (ideales Gas) Summe Demnach ist also Sm (298K) = Sm (0) + 191.04 J K−1 mol−1 ∅ Bredol (FH-MS) PC-I 120 / 186 5. Der Dritte Hauptsatz 5.1. Annäherung an den Nullpunkt der Temperatur Qualitative Ableitung Der Wert der Entropie bei T = 0K läßt sich qualitativ ermitteln: Am absouten Nullpunkt ist sämtliche Wärmebewegung vollständig zum Stillstand gekommen; alle Teilchen (in einem Kristall) sind daher regelmäßig in einem starren Gitter angeordnet Da es in einem Kristall so nur eine mögliche Anordnung der Teilchen gibt, ist die thermodynamische Wahrscheinlichkeit nach Boltzmann gleich eins und somit die Entropie gleich Null Diese Überlegung vernachlässigt jedoch quantenmechanische Effekte, wie sie z.B. in der Nullpunktsenergie des harmonischen Oszillators zum Ausdruck kommen Bredol (FH-MS) PC-I 5. Der Dritte Hauptsatz 121 / 186 5.1. Annäherung an den Nullpunkt der Temperatur Qualitative Ableitung Die statistische Überlegung zur Entropie am absoluten Nullpunkt ist historisch etwas anders entstanden; Ausgangspunkt war das rein empirische Nernst’sche Wärmetheorem, das Aussagen über Entropiedifferenzen macht: Bei Annäherung an den absoluten Nullpunkt der Temperatur nähern sich auch alle Entropiedifferenzen physikalischer oder chemischer Stoffumwandlungen dem Wert Null an In Theorie und Experiment strebt auch die Wärmekapazität fester Stoffe dem Wert Null bei Annäherung an den absoluten Nullpunkt zu (siehe z.B. Gl.119). Bredol (FH-MS) PC-I 122 / 186 5. Der Dritte Hauptsatz 5.1. Annäherung an den Nullpunkt der Temperatur Qualitative Ableitung Am absoluten Nullpunkt können im Gleichgewicht aus den Elementen nur ideal kristalline Verbindungen entstehen Da die Entropiedifferenzen aller Prozesse gegen Null streben, müssen somit auch alle ideal kristallinen Verbindungen die Entropie der konstituierenden Elemente besitzen Wenn man annimmt, dass die gemeinsame Entropie der Elemente und der ideal kristallinen Verbindungen bei T=0 exakt gleich dem Wert Null ist, ergibt sich an keiner Stelle ein Widerspruch zu messbarem Verhalten Bredol (FH-MS) PC-I 5. Der Dritte Hauptsatz 123 / 186 5.2. Entropie bei T=0 Hauptsatz Diese Zusammenhänge werden im Dritten Hauptsatz der Thermodynamik zusammengefasst: Wenn man die Entropie jedes Elements in seinem stabilen Zustand bei T=0 gleich Null setzt, besitzt jeder reine Stoff eine positive Entropie, die bei T=0 den Wert Null erreichen kann, wenn es sich um ideal kristalline Verbindungen handelt Der Dritte Hauptsatz besagt demnach, dass wir im Gegensatz zu U und H im Fall der Entropie absolute Werte angeben können Bredol (FH-MS) PC-I 124 / 186 5. Der Dritte Hauptsatz 5.2. Entropie bei T=0 Hauptsatz Entropien, die sich auf den so definierten Nullpunkt beziehen, werden exakt als Entropien nach dem Dritten Hauptsatz (third law entropies) bezeichnet Befindet sich der betreffende Stoff i in seinem Standardzustand und bei der Temperatur T , spricht man von der Standardentropie Si (T ) nach dem Dritten Hauptsatz ∅ Die Elemente besitzen daher ebenfalls eine Standardentropie Ausnahme: für einzelne Ionen lassen sich die Überlegungen des Dritten Hauptsatzes nicht sinnvoll anwenden Bredol (FH-MS) PC-I 5. Der Dritte Hauptsatz 125 / 186 5.2. Entropie bei T=0 Ionen Definition: Die Standardentropie des in Wasser gelösten Wasserstoffions ist bei allen Temperaturen gleich Null Da Ionen in der Chemie nur gepaart (insgesamt elektroneutral) auftreten, wird der durch die obige Definition erzeugte unbekannte Fehler stets ausgeglichen Im Gegensatz zu allen anderen Stoffen können daher einzelne Ionen auch negative Standardentropien aufweisen, um den obigen Fehler zu kompensieren Damit ergeben sich stets eindeutige Reaktionsentropien, auch bei Beteiligung von Ionen: N X ∆R = (120) νi S i ∅ ∅ i=1 Bredol (FH-MS) PC-I 126 / 186 6. Freie Enthalpie G und Freie Energie A 6.1. Hintergrund Ableitung Die Gesamtentropieänderung in einem abgeschlossenen System erlaubt die Entscheidung darüber, in welcher Richtung ein Prozess im abgeschlossenen System spontan und irreversibel abläuft Die Beschreibung der Eigenschaften der Umgebung des eigentlichen Systems ist jedoch sehr unhandlich; daher wird eine thermodynamische Funktion gesucht, die allein aus Informationen des Systems heraus vorhersagt, in welcher Richtung Prozesse ablaufen Dazu wird der Zweite Hauptsatz in Form der Clausius’schen Ungleichung für ein System analysiert, das infolge einer Zustandsänderung Wärme mit der Umgebung austauscht: dSges = dS + dS ′ = dS − Bredol (FH-MS) δQ >0 T PC-I 6. Freie Enthalpie G und Freie Energie A (121) 127 / 186 6.1. Hintergrund Ableitung Die Ungleichung kann für zwei Spezialfälle ausgewertet werden, wenn keine anderen als Volumenarbeiten auftreten. Bei konstantem Druck gilt: dS − dH >0 T (122) Bei konstantem Volumen dagegen gilt: dS − dU >0 T (123) In beiden Ungleichungen tauchen nur noch Zustandsfunktionen des Systems auf! Bredol (FH-MS) PC-I 128 / 186 6. Freie Enthalpie G und Freie Energie A 6.1. Hintergrund Freiwillige Prozesse Aus den beiden Ungleichungen lassen sich Kriterien für freiwillige Prozesse im System ablesen: Bei konstantem Druck: Bei konstanter Entropie muss die Enthalpie abnehmen, bei konstanter Enthalpie muss die Entropie zunehmen Bei konstantem Volumen: Bei konstanter Entropie muss die Innere Energie abnehmen, bei konstanter Innerer Energie muss die Entropie zunehmen Mit diesen Aussagen liegen Kriterien für freiwillige Prozesse vor, die Veränderungen der Entropie im System mit solchen der Enthalpie bzw. der Inneren Energie im System verknüpfen! Bredol (FH-MS) PC-I 6. Freie Enthalpie G und Freie Energie A 129 / 186 6.1. Hintergrund Definitionen Nach nochmaliger Umformung lassen sich vor diesem Hintergrund neue, zusammenfassende Zustandsfunktionen definieren Bei konstantem Druck: T dS > dH (124) T dS > dU (125) Bei konstantem Volumen: Deshalb G = H − TS Freie Enthalpie (Gibbs − Energie) (126) A = U − TS Freie Energie (Helmholtz − Energie) (127) Für A findet man gelegentlich auch das Symbol F Bredol (FH-MS) PC-I 130 / 186 6. Freie Enthalpie G und Freie Energie A 6.1. Hintergrund Folgerungen Für isotherme Prozesse folgt aus den Definitionen dG = dH − T dS dA = dU − T dS (128) Setzt man diese Gleichungen nun in die Ungleichungen für freiwillige Prozesse (Gl.124 und Gl.125) im System ein, dann folgt bei konstantem Druck (und konstanter Temperatur): dG 6 0 (129) sowie bei konstantem Volumen (und konstanter Temperatur): dA 6 0 (130) Diese Ungleichungen vereinigen die Systemeigenschaften in einer Zustandsfunktion und geben die Richtung freiwillig ablaufender isothermer Prozesse an Bredol (FH-MS) PC-I 6. Freie Enthalpie G und Freie Energie A 131 / 186 6.1. Hintergrund Gleichgewicht Die Ergebnisse lassen sich auch zur Angabe von Gleichgewichtsbedingungen nutzen: Bei konstantem Druck und konstanter Temperatur ist ein System dann im chemischen Gleichgewicht, wenn G ein Minimum erreicht (dG = 0) Bei konstantem Volumen und konstanter Temperatur ist ein System dann im chemischen Gleichgewicht, wenn A ein Minimum erreicht (dA = 0) Die meisten chemischen Prozesse verlaufen isobar und isotherm, daher spielt G in der Chemie eine zentrale Rolle Bredol (FH-MS) PC-I 132 / 186 6. Freie Enthalpie G und Freie Energie A 6.1. Hintergrund Endotherm vs. exotherm Nun läßt sich auch begründen, warum auch endotherme Prozesse freiwillig und spontan ablaufen können: Endotherme Prozesse führen zwar zu einer Entropieabnahme in der Umgebung; wird diese aber durch die Entropiezunahme im System überkompensiert, verläuft der Prozess dennoch freiwillig Andererseits wird auch klar, wie in einem geschlossenen System der Ordnungsgrad erhöht werden kann: Nimmt im System durch den Prozess die Entropie ab, dann muss ein freiwillig ablaufender Prozess exotherm sein, da nur so die Entropie der Umgebung erhöht wird und die Abnahme im System überkompensiert werden kann Bredol (FH-MS) PC-I 6. Freie Enthalpie G und Freie Energie A 133 / 186 6.2. Freiwilligkeit Prozessrichtung Generelle Regeln: Prozesse, die die Entropie des Systems erhöhen und exotherm verlaufen, verlaufen immer freiwillig Prozesse, die die Entropie des Systems erniedrigen und endotherm verlaufen, können niemals freiwillig verlaufen Zustände niedriger Enthalpie oder niedriger Innerer Energie sind demnach nicht generell begünstigt – entscheidend ist die Erhöhung der Gesamtentropie Erlaubte (“freiwillige”) Prozesse können allerdings mit unmessbar kleiner Geschwindigkeit ablaufen; die Ausgangszustände nennt man dann metastabil oder kinetisch stabil Bredol (FH-MS) PC-I 134 / 186 6. Freie Enthalpie G und Freie Energie A 6.2. Freiwilligkeit Maximale Arbeit Mit Hilfe der Größe A lassen sich auch Aussagen machen über die maximal mögliche Arbeit, die ein Prozess leisten kann Dazu wird Gl.121 in den Ersten Hauptsatz der Thermodynamik eingesetzt: δQ = dU − δW T dS >δQ −→ T dS > dU − δW (131) (132) oder auch δW > dU − T dS bzw. Bredol (FH-MS) δW > dA bei T = const PC-I 6. Freie Enthalpie G und Freie Energie A (133) 135 / 186 6.2. Freiwilligkeit Maximale Arbeit Die größtmögliche (reversible!) Arbeit, die ein System leisten kann, ist daher der (vom Betrag her) größtmögliche negative Wert von δW . Daher folgt: dWmax = dU − T dS (134) Bei konstanter Termperatur ergibt die Integration: Wmax = ∆A (135) Die Änderung der Freien Energie A durch einen isothermen Prozess gibt an, wieviel Arbeit der Prozess maximal leisten kann Bredol (FH-MS) PC-I 136 / 186 6. Freie Enthalpie G und Freie Energie A 6.2. Freiwilligkeit Maximale Nicht-Volumen–Arbeit Eine ähnliche Überlegung läßt sich für die Freie Enthalpie anstellen, ausgehend von der Enthalpie: dH = dU + d(PV ) = δQ + δW + d(PV ) (136) Die Arbeitsbeiträge lassen sich aufteilen in Volumenarbeit −PdV und Nicht-Volumenarbeit We : dH = δQ + δWe − PdV + d(PV ) = δQ + δWe + V dP (137) In reversiblen (isothermen) Prozessen läßt sich δQ durch T dS ersetzen (Zweiter Hauptsatz): dH = T dS + δWe + V dP (138) dG = dH − T dS = δWe + V dP (139) und damit Bredol (FH-MS) PC-I 6. Freie Enthalpie G und Freie Energie A 137 / 186 6.2. Freiwilligkeit Maximale Nicht-Volumen–Arbeit Daraus folgt, dass für einen (isothermen und isobaren) Prozess dG die maximal mögliche Nicht-Volumenarbeit dWe,max angibt (maximal, weil reversible Prozessführung vorausgesetzt wurde): We,max = ∆G (140) Die Nicht-Volumenarbeit kann z.B. elektrische Arbeit sein (Elektrochemie, Brennstoffzellen, ...)! Die Änderung der Freien Enthalpie durch einen isothermen und isobaren Prozess gibt an, wieviel Nicht-Volumenarbeit der Prozess maximal leisten kann Bredol (FH-MS) PC-I 138 / 186 6. Freie Enthalpie G und Freie Energie A 6.3. Fundamentalgleichungen Verbindung von Erstem und Zweitem Hauptsatz Um Ersten und Zweiten Hauptsatz gemeinsam nutzen zu können, wird auf die Innere Energie zurückgegriffen; für einen reversiblen Prozess lassen sich Arbeit und Wärme darin geeignet ersetzen: dU = δQ + δW reversible Prozesse −→ dU = T dS − PdV (141) Diese Gleichung ist als Fundamentalgleichung der Thermodynamik bekannt (hier nur Volumenarbeit berücksichtigt) U ist eine Zustandsfunktion, daher muss die Fundamentalgleichung für alle (nicht nur reversible) Prozesse gelten, solange ausschließlich Volumenarbeit auftritt Die Gleichung heißt Fundamentalgleichung, da sie dU nun ausschließlich mit Zustandsfunktionen darstellt Bredol (FH-MS) PC-I 6. Freie Enthalpie G und Freie Energie A 139 / 186 6.3. Fundamentalgleichungen Natürliche Variablen Es wird deutlich, dass die Innere Energie zwar als Funktion beliebiger Variablen entwickelt werden kann (die Kombination aus T und V wurde bereits benutzt, um Wärmekapazitäten zu definieren), Entropie S und Volumen V jedoch die “natürliche” Wahl darstellen: ∂U ∂U dU = dS + dV ∂S V ∂V S Koeffizientenvergleich mit Gl.141: ∂U =T ∂S V ∂U ∂V = −P (142) S Der erste Koeffizient läßt sich heranziehen, um die Temperatur rein thermodynamisch zu definieren Bredol (FH-MS) PC-I 140 / 186 6. Freie Enthalpie G und Freie Energie A 6.3. Fundamentalgleichungen Fundamentalgleichung für H Ausgehend von der Fundamentalgleichung für U läßt sich eine ähnliche Beziehung auch für die Enthalpie ableiten, wenn keine andere Arbeit als Volumenarbeit verrichtet wird: dH = d(U + PV ) = dU + PdV + V dP = T dS − PdV + PdV + V dP = T dS + V dP (143) Entsprechend sind die natürlichen Variablen, in denen die Enthalpie dargestellt wird, Entropie S und Druck P Bredol (FH-MS) PC-I 6. Freie Enthalpie G und Freie Energie A 141 / 186 6.3. Fundamentalgleichungen Natürliche Variablen für H Die Bedeutung der Koeffizienten des vollständigen Differenzials von H wird wieder durch Vergleich mit der Fundamentalgleichung für H entnommen: ∂H ∂H dH = dS + dP ∂S P ∂P S Koeffizientenvergleich mit Gl.143: ∂H =T ∂S P ∂H ∂P =V (144) S Der erste Koeffizient läßt sich wieder heranziehen, um die Temperatur rein thermodynamisch zu definieren Bredol (FH-MS) PC-I 142 / 186 6. Freie Enthalpie G und Freie Energie A 6.3. Fundamentalgleichungen Freie Energie A In analoger Weise erhält man für die Freie Energie A: dA = d(U − TS) = T dS − PdV − T dS − SdT = −PdV − SdT (145) (146) Natürliche Variablen der Freien Energie sind somit Temperatur und Volumen. Bedeutung der Koeffizienten wieder durch Vergleich mit dem vollständigen Differenzial: ∂A ∂A dA = dV + dT ∂V T ∂T V ∂A ∂V = −P T Bredol (FH-MS) ∂A ∂T = −S V PC-I 6. Freie Enthalpie G und Freie Energie A (147) 143 / 186 6.3. Fundamentalgleichungen Freie Enthalpie G Schließlich läßt sich auch das vollständige Differenzial der Freien Enthalpie G entsprechend formulieren: dG = dH − T dS − SdT = −SdT + V dP (148) sowie die entsprechenden Koeffizienten: ∂G ∂G dG = dT + dP ∂T P ∂P T ∂G ∂T = −S P ∂G ∂P =V (149) T Die “natürlichen” Variablen der Freien Enthalpie sind demnach Druck und Temperatur, also die beiden wichtigsten Zustandsvariablen in der Chemie! Bredol (FH-MS) PC-I 144 / 186 6. Freie Enthalpie G und Freie Energie A 6.3. Fundamentalgleichungen Freie Enthalpie G Die Temperaturabhängigkeit der Freien Enthalpie ist durch die (negative) Entropie gegeben. Da die Entropie eines Systems eine positive Größe ist, muss die Freie Enthalpie eines Systems mit der Temperatur stets kleiner werden Für Gase mit ihrer hohen Entropie ist das offenbar ausgeprägter als für Flüssigkeiten, für Flüssigkeiten wiederum ausgeprägter als für Festkörper Die Druckabhängigkeit der Freien Enthalpie eines Systems ist durch das Volumen gegeben. Da Volumina stets positiv sind, muss die Freie Enthalpie eines Systems mit dem Druck stets zunehmen Wiederum gilt, dass Gase ein höheres Volumen aufweisen als die kondensierten Phasen, so dass die Druckabhängigkeit in Gasen sehr ausgeprägt sein wird Bredol (FH-MS) PC-I 6. Freie Enthalpie G und Freie Energie A 145 / 186 6.3. Fundamentalgleichungen G/T In vielen Fällen ist es bequemer, nicht für G, sondern für G/T die Temperatur-Abhängigkeit zu betrachten: ∂G G−H G = H − TS −→ = −S = (150) ∂T P T oder umgestellt ∂G ∂T − P G H =− T T (151) Mit der Produktregel erhält man: Bredol (FH-MS) PC-I 146 / 186 6. Freie Enthalpie G und Freie Energie A 6.3. Fundamentalgleichungen G/T ∂ ∂T G T P 1 ∂G ∂ 1 = +G T ∂T P ∂T T P 1 ∂G G = − 2 T ∂T P T ∂G G 1 − = T ∂T P T (152) (153) (154) und schließlich durch Einsetzen des Zwischenergebnisses aus Gl.151 die Gibbs-Helmholtz–Gleichung: ∂ H G (155) =− 2 ∂T T T P Die T -Abhängigkeit von G läßt sich also sowohl durch S als auch durch H beschreiben! Bredol (FH-MS) PC-I 6. Freie Enthalpie G und Freie Energie A 147 / 186 6.3. Fundamentalgleichungen G/T Die Ergebnisse lassen sich nicht nur für Absolutwerte von Enthalpie und Freier Enthalpie, sondern auch für Zustandsänderungen formulieren: ∂ ∆G ∂ G2 − G1 = (156) ∂T T ∂T T P P G2 ∂ G1 ∂ − (157) = ∂T T ∂T T P P H2 H1 =− 2 + 2 (158) T T ∆H =− 2 (159) T Solche Zusammenhänge werden nützlich sein, wenn Gleichgewichte zu beschreiben sind, da ln K = −∆R G /RT ∅ Bredol (FH-MS) PC-I 148 / 186 6. Freie Enthalpie G und Freie Energie A 6.3. Fundamentalgleichungen A/T Für die Freie Energie A läßt sich eine entsprechende Beziehung herleiten: ∂ A U (160) =− 2 ∂T T T V Die Temperaturabhängigkeit von G und A ist in der Praxis sehr wichtig, da sie experimentellen Zugriff auf die Entropie oder Enthalpie ermöglicht Die Druckabhängigkeit ist in der Praxis von geringerer Bedeutung, da sie generell eher kleiner ist bzw. bei isobaren Prozessen auch ganz entfällt Die bedeutendsten Beiträge sind bei Beteiligung von Gasen zu erwarten Bredol (FH-MS) PC-I 6. Freie Enthalpie G und Freie Energie A 149 / 186 6.3. Fundamentalgleichungen Druckabhängigkeit von G in Gasen Für einen isothermen Prozess mit einem idealen Gas ist anzusetzen: G(P2 ) = G(P1 ) + ZP2 V dP (161) nRT dP P (162) P1 = G(P1 ) + ZP2 P1 = G(P1 ) + nRT ln P2 P1 (163) Dieser Zusammenhang wird die Grundlage der Thermodynamik der Mischphasen und chemischen Gleichgewichte darstellen! Bredol (FH-MS) PC-I 150 / 186 6. Freie Enthalpie G und Freie Energie A 6.3. Fundamentalgleichungen Druckabhängigkeit von G in kondensierter Phase In kondensierten Phasen kann das entsprechende Integral ebenfalls näherungsweise ausgewertet werden, da Flüssigkeiten und Festkörper nur wenig kompressibel sind (V ist annähernd druckunabhängig): G(P2 ) = G(P1 ) + ZP2 V dP ≃ G(P1 ) + V (P2 − P1 ) (164) P1 Anwendung auf Anfangs- und Endzustand für eine Zustandsänderung mit kondensierten Phasen liefert: ∆G(P2 ) = ∆G(P1 ) + ∆V (P2 − P1 ) Bredol (FH-MS) PC-I 6. Freie Enthalpie G und Freie Energie A (165) 151 / 186 6.4. Maxwell’sche Gleichungen Herleitung Aus den vier Fundamentalgleichungen der Thermodynamik für U, H, G und A lassen sich weitere nützliche Beziehungen durch Anwendung des Schwarz’schen Satzes herleiten. Beispiel U: ∂U ∂U dS + dV = T dS − PdV dU = ∂S V ∂V S ∂ ∂V ∂U ∂S ∂ = ∂S Bredol (FH-MS) V S ∂T ∂V =− S ∂U ∂V ∂P ∂S (166) S V (167) V PC-I 152 / 186 6. Freie Enthalpie G und Freie Energie A 6.4. Maxwell’sche Gleichungen Herleitung Ähnliche Beziehungen gewinnt man aus H, A und G: ∂V ∂T = ∂P S ∂S P ∂S ∂V ∂S ∂P = T ∂P ∂T =− T (168) (169) V ∂V ∂T (170) P Diese vier Gleichungen zusammen heißen Maxwell’sche Gleichungen. Damit können auch Koeffizienten bestimmt werden, deren Bedeutung bisher nicht klar war, z.B. die Volumenabhängigkeit von U bei konstanter Temperatur: Bredol (FH-MS) PC-I 6. Freie Enthalpie G und Freie Energie A 153 / 186 6.4. Maxwell’sche Gleichungen Folgerungen dU = CV dT + ∂U ∂V dV (171) T Umstellung des vollständigen Differenzials für U in den natürlichen Variablen: ∂U ∂U dS + dV dU = ∂S V ∂V S dU −→ = dV Bredol (FH-MS) ∂U ∂S PC-I V dS + dV ∂U ∂V (172) S 154 / 186 6. Freie Enthalpie G und Freie Energie A 6.4. Maxwell’sche Gleichungen Folgerungen Einschränkung auf isotherme Prozesse: ∂U ∂S ∂U ∂U = + T = const −→ ∂V T ∂S V ∂V T ∂V S =T ∂S ∂V −P (173) (174) T Hier kann man nun eine der Maxwell’schen Gleichungen nutzen, um den unhandlichen Koeffizienten mit der Entropie in einen bequemeren mit Druck und Temperatur umzuwandeln: ∂P ∂U =T −P (175) ∂V T ∂T V Bredol (FH-MS) PC-I 6. Freie Enthalpie G und Freie Energie A 155 / 186 6.5. Thermodynamische Zustandsgleichungen Allgemeine Zustandsgleichungen Gl.175 und verwandte Gleichungen heißen Thermodynamische Zustandsgleichungen, weil sie allgemeingültig eine thermodynamische Größe (hier: U) durch die bequemen Zustandsvariablen P,T und V beschreiben. Im Übrigen ermöglicht der Zusammenhang nun die Interpretation des vollständigen Differenzials von U auch in den (handlicheren) Variablen T und V : ∂P dU = CV dT + T − P dV (176) ∂T V Eine entsprechende Beziehung kann auch für das vollständige Differenzial der Enthalpie abgeleitet werden: Bredol (FH-MS) PC-I 156 / 186 6. Freie Enthalpie G und Freie Energie A 6.5. Thermodynamische Zustandsgleichungen Enthalpie dH = −→ ∂H ∂P = T ∂H ∂S ∂H ∂S = −T dS + P P ∂V ∂T ∂S ∂P ∂H ∂P S + ∂H ∂P T dP (177) S +V P Daraus schließlich das vollständige Differenzial von H in den “bequemen” Variablen T und P: ∂V dH = CP dT + −T + V dP ∂T P Bredol (FH-MS) PC-I 6. Freie Enthalpie G und Freie Energie A (178) (179) 157 / 186 6.5. Thermodynamische Zustandsgleichungen Reale Systeme Die gefundenen Koeffizienten für die Volumenabhängigkeit von U sowie die Druckabhängigkeit von H werden immer dann wichtig, wenn die Eigenschaften realer Systeme zu beschreiben sind Liegen empirische oder theoretische Zustandsgleichungen für die realen Systeme vor, lassen sich die Koeffizienten daraus direkt berechnen Beispiel: Kohlendioxid, beschrieben mit der (ausmultiplizierten und in den Koeffizienten vereinfachten) v.d.W.–Gleichung (a = 0.386 m6 Pa/mol 2 , b = 4.267 ∗ 10−5 m3 /mol): an abn2 aP abP 2 PV PV ∼ − bP + − − bP + − 2 2 = RT = n V n RT V2 R T Bredol (FH-MS) PC-I (180) 158 / 186 6. Freie Enthalpie G und Freie Energie A 6.5. Thermodynamische Zustandsgleichungen Reale Systeme nRT an nabP (181) + nb − + 2 2 P RT R T Der für das vollständige Differenzial von H in P und T erforderliche Koeffizient läßt sich daraus nun berechnen: ∂V nR an 2nabP = + − (182) ∂T P P RT 2 R 2T 3 −→ V = Damit sowie mit Gl.179 und Gl.181 wird nun das vollständige Differenzial konkret beschrieben: 2an 3nabP + 2 2 dP dH = CP dT + nb − RT R T Bredol (FH-MS) PC-I 6. Freie Enthalpie G und Freie Energie A (183) 159 / 186 6.5. Thermodynamische Zustandsgleichungen Reale Systeme Mit den v.d.W.-Parametern für Kohlendioxid ergibt sich so z.B. bei 350 K und 1 bar: dHm = CP,m dT − 2.22 ∗ 10−4 m3 dP (184) Der ermittelte Zahlenwert zeigt, dass auch in realen Gasen die Druckabhängigkeit der Enthalpie nur gering ist und daher bei nicht zu großen Druckintervallen vernachlässigt werden darf Auch die Differenz von CP und CV läßt sich nun allgemein angeben. Aus dem Ersten Hauptsatz kombiniert mit dem vollständigen Differenzial für U: ∂U ∂U δQ = dV + dT + PdV (185) ∂V T ∂T V Bredol (FH-MS) PC-I 160 / 186 6. Freie Enthalpie G und Freie Energie A 6.5. Thermodynamische Zustandsgleichungen CP − CV Umstellung und Beschränkung auf konstanten Druck liefert: ∂Q ∂U ∂U ∂V ∂V = + +P ∂T P ∂V T ∂T P ∂T V ∂T P CP = ∂U ∂T + V ∂V ∂T P ∂U ∂V +P T (186) (187) Hier kann nun die thermodynamische Zustandsgleichung (Gl.175) im Klammerausdruck eingesetzt werden und man erhält ∂P ∂V T (188) CP − CV = ∂T P ∂T V Bredol (FH-MS) PC-I 6. Freie Enthalpie G und Freie Energie A 161 / 186 6.5. Thermodynamische Zustandsgleichungen CP − CV Der erste Koeffizient ist der isobare Expansionskoeffizient α (Gl.71) multipliziert mit dem Volumen, der zweite heißt isochorer Spannungskoeffizient β: ∂P CP − CV = V αT β (189) β= ∂T V In diese (thermodynamisch streng gültige!) Gleichung können nun (reale, messbare) Stoffeigenschaften für kondensierte Phasen eingesetzt werden Der Spannungskoeffizient kann alternativ auch durch α und die Kompressibilität (Gl.72) dargestellt werden: α β= κ Bredol (FH-MS) α2 CP − CV = VT κ PC-I (190) 162 / 186 6. Freie Enthalpie G und Freie Energie A 6.5. Thermodynamische Zustandsgleichungen Joule-Thomson–Koeffizient Der Joule-Thomson–Koeffizient (Gl.79) kann ebenfalls direkt aus der entsprechenden Zustandsgleichung bestimt werden: ∂H T ∂V ∂P T ∂T P − V ∂T = (191) =− ∂P H CP CP Bei Kenntnis empirischer oder theoretischer Zustandsgleichungen für das betreffende Gas kann aus dem thermischen Expansionskoeffizienten die Inversionstemperatur entnommen werden Für CO2 bei 350 K und 1 bar (CP = 37.11 J K−1 mol−1 ) folgt beispielsweise: ∂T = 5.98 ∗ 10−6 K /Pa ∂P H Bredol (FH-MS) (192) PC-I 6. Freie Enthalpie G und Freie Energie A 163 / 186 6.5. Thermodynamische Zustandsgleichungen CP (P) und CV (V ) Durch die Maxwell’schen Gleichungen erhält man auch die Druckabhängigkeit von CP sowie die Volumenabhängigkeit von CV : ∂ ∂ ∂(G + TS) ∂H ∂CP = = (193) ∂P T ∂T ∂T T P ∂T ∂P T P ∂ ∂V = +T = V −T ∂T ∂T P P T T P (194) 2 2 ∂V ∂ V ∂T ∂V ∂ V = = −T (195) − −T ∂T P ∂T ∂T P ∂T 2 P ∂T 2 P ∂ ∂T ∂G ∂P ∂S ∂P Für ein ideales Gas ergibt sich null, ansonsten werden empirische oder theoretische Zustandsgleichungen eingesetzt Bredol (FH-MS) PC-I 164 / 186 6. Freie Enthalpie G und Freie Energie A 6.5. Thermodynamische Zustandsgleichungen CP (P) und CV (V ) Völlig analog ergibt sich auch: 2 ∂ P ∂CV =T ∂V T ∂T 2 V (196) Alle diese Gleichungen sind thermodynamisch streng gültig; erst nach Einsetzen von spezifischen Stoffeigenschaften bzw. Zustandsgleichungen erfolgt die Einschränkung auf spezifische Systeme Bredol (FH-MS) PC-I 165 / 186 7. Zusammenfassung Thermodynamik Thermodynamische Potenziale U, H, G, A heißen thermodynamische Potenziale, da für sie folgender Satz gilt: Ist ein thermodynamisches Potenzial eines Systems als Funktion aller seiner natürlichen Variablen bekannt, so ist alles über das System bekannt. Die bisher abgeleiteten Grundgleichungen lassen sich auch im Huckenheim–Merkschema darstellen; darin sind alle thermodynamischen Potenziale von ihren natürlichen Variablen “eingerahmt”: Bredol (FH-MS) PC-I 166 / 186 7. Zusammenfassung Thermodynamik Merkschema S H P – U G V A T + Schon unter Varus hatten alle progressiven Germanen Taschenrechner Die Koeffizienten der vollständigen Differenziale der Potenziale erhält man durch Aufstellung entlang einer Seite des Schemas und Ablesung auf der Diagonalen, die von der bezogenen Variablen ausgeht; dabei sind die Vorzeichen zu beachten Die vier Maxwell–Gleichungen der Zustandsvariablen sind ebenfalls abzulesen, wenn zwei Dreiecke mit den Variablen gegenläufig so verschränkt werden, dass ein thermodmisches Potential eingeschlossen wird Bredol (FH-MS) PC-I 167 / 186 7. Zusammenfassung Thermodynamik Alle Ergebnisse der Kombination von Erstem und Zweitem Hauptsatz: dU = T dS − PdV dA = −SdT − PdV ∂U = −P ∂V S ∂H =V ∂P S ∂A = −S ∂T V ∂H =T ∂S P Bredol (FH-MS) PC-I dH = T dS + V dP (197) dG = −SdT + V dP (198) =T (199) = −S (200) = −P (201) =V (202) ∂U ∂S V ∂G ∂T P ∂A ∂V T ∂G ∂P T 168 / 186 7. Zusammenfassung Thermodynamik sowie die Maxwell–Gleichungen und die thermodynamischen Zustandsgleichungen: ∂P ∂T ∂V ∂T =− = ∂V S ∂S V ∂P S ∂S P ∂S ∂S ∂P ∂V = =− ∂V T ∂T V ∂P T ∂T P ∂U ∂P =T −P ∂V T ∂T V ∂H ∂V = −T +V ∂P T ∂T P Bredol (FH-MS) PC-I 8. Phasengleichgewichte in Reinstoffen (203) (204) (205) (206) 169 / 186 8.1. Gleichgewichtsbedingungen Definitionen Eine thermodynamische Phase bezeichnet einen Teil eines Systems, der bis in molekulare Bereiche hinein physikalisch homogen aufgebaut ist Eine Phase kann mehrere räumlich getrennte Gebiete umfassen (Emulsionen, Ausscheidungen in Metallen usw.); der Phasenbegriff wird im kolloidalen Bereich gelegentlich unscharf Stehen in einem Reinstoff Phasen miteinander im Gleichgewicht, dann sind in allen beteiligten Phasen P, T und Gm gleich (hier formuliert für zwei Phasen α und β): mechanisches Gleichgewicht : Pα = Pβ (207) thermisches Gleichgewicht : Tα = Tβ (208) chemisches Gleichgewicht : β α Gm = Gm (209) Bredol (FH-MS) PC-I 170 / 186 8. Phasengleichgewichte in Reinstoffen 8.1. Gleichgewichtsbedingungen Verschobene Gleichgewichte Bei (lansamer, reversibler) Verschiebung auf einen neuen Gleichgewichts–Zustand unter ständigem Erhalt des Gleichgewichts müssen dann auch Änderungen der Freien Enthalpie in den beteiligten Phasen gleich sein: β α dGm = dGm (210) Unter Nutzung der Fundamentalgleichung erhält man daraus: β α −Sm dT + Vmα dP = −Sm dT + Vmβ dP oder nach Sortierung β β α α Vm − Vm dP = Sm − Sm dT Bredol (FH-MS) (211) (212) PC-I 8. Phasengleichgewichte in Reinstoffen 171 / 186 8.1. Gleichgewichtsbedingungen Gleichung von Clausius und Clapeyron Nach Umstellung erhält man daraus die Clausius–Clapeyron’sche Gleichung: α − Sβ Sm ∆Sm dP m = (213) = β α dT ∆V m Vm − Vm Diese Gleichung beschreibt die Druck/Temperatur-Abhängigkeiten von Phasenübergängen jeder erdenklichen Art! Zur Anwendung auf spezielle Phasenübergänge müssen die Randbedingungen berücksichtigt werden und dann die Integration durchgführt werden Bredol (FH-MS) PC-I 172 / 186 8. Phasengleichgewichte in Reinstoffen 8.2. Siedegleichgewicht Reversible Verdampfung Wenn die Verdampfung am Siedepunkt reversibel und isobar verläuft, dann gilt: ∆ H ∆V S = V (214) T Wenn α die gasförmige und β die flüssige Phase bezeichnet, dann folgt daraus: ∆ H dP = V (215) β dT α T V −V m m Das Volumen der flüssigen Phase kann im Siedevorgang vernachlässigt werden, wenn man weit vom kritischen Punkt entfernt ist; das Volumen der gasförmigen Phase wird durch das ideale Gasgesetz abgeschätzt: P∆V H dP = dT RT 2 Bredol (FH-MS) PC-I 8. Phasengleichgewichte in Reinstoffen (216) 173 / 186 8.2. Siedegleichgewicht Dampfdruckkurve Nun läßt sich nach Variablentrennung unter der Voraussetzung integrieren, dass ∆V H temperaturunabhängig ist: ∆V H 1 1 P2 = − ln P1 R T1 T2 (217) Logarithmische Auftragungen des Dampfdruckes über einer 1/T -Achse liefern daher im Allgemeinen eine Gerade, aus deren Steigung die Verdampfungsenthalpie entnommen werden kann Wird der Dampfdruck auf den Siedepunkt TS bei Standarddruck (1bar) bezogen, dann ergibt sich die folgende Form: Bredol (FH-MS) PC-I 174 / 186 8. Phasengleichgewichte in Reinstoffen 8.2. Siedegleichgewicht Dampfdruckkurve P ∆ H ln = V bar R ln(P/bar) = − 1 1 − TS T (218) ∆ S(TS ) ∆V H + V RT R (219) In parametrisierter Form ist diese Gleichung als August’sche Gleichung bekannt: ln(P/bar) = − A +B T /K (220) Beispiel: Wasser. Die Standardverdampfungsenthalpie beträgt bei 273 K etwa 44 kJ/mol, bei 373 K etwa 40 kJ/mol. Bredol (FH-MS) PC-I 8. Phasengleichgewichte in Reinstoffen 175 / 186 8.2. Siedegleichgewicht Dampfdruckkurve Logarithmische Auftragung des Dampfdruckes über 1/T : 200 175 150 125 100 T (oC) 75 50 25 0 3 2 1 ln(P/bar) 0 −1 −2 −3 −4 −5 0.0022 0.0024 0.0026 0.0028 0.003 0.0032 0.0034 0.0036 K/T Bredol (FH-MS) PC-I 176 / 186 8. Phasengleichgewichte in Reinstoffen 8.2. Siedegleichgewicht Dampfdruckkurve Wie bereits auf anderem Wege in der Allgemeinen Chemie gezeigt, ist die tatsächliche lineare Abhängigkeit erheblich steiler: 0 25 50 75 T (oC) 100 125 150 175 200 18 16 14 P/bar 12 10 8 6 4 2 0 300 350 400 450 T/K Bredol (FH-MS) PC-I 8. Phasengleichgewichte in Reinstoffen 177 / 186 8.2. Siedegleichgewicht Antoine–Gleichung Da ∆V H in der Realität merklich temperaturabhängig ist, sind für größere T –Intervalle empirische Gleichungen in Gebrauch Die am weitesten verbreitete Form ist die Antoine–Gleichung, deren Koeffizienten häufig in Tabellenwerken auftauchen: log(P/bar) = − A +B C + T /K (221) Allerdings ist damit keine linearisierte Auftragung mehr möglich Bredol (FH-MS) PC-I 178 / 186 8. Phasengleichgewichte in Reinstoffen 8.3. Schmelzgleichgewicht Randbedingungen Für den Übergang flüssig/fest sind die Verhältnisse etwas unübersichtlicher (variierende Schmelzvolumina, Schmelzanomalien) Unter der Annahme, dass Schmelzenthalpie und Schmelzvolumen temperaturunabhängig sind, erhält man aus Gl.213: ZP2 dP = ∆sm H ∆sm V ZT2 dT T (222) ∆sm H T2 ln ∆sm V T1 (223) P1 T1 P2 = P1 + Schmelzkurven verlaufen daher ganz anders als Siedekurven! Bredol (FH-MS) PC-I 8. Phasengleichgewichte in Reinstoffen 179 / 186 8.4. Sublimationsgleichgewicht Gemeinsamkeiten Da Enthalpien Zustandfunktionen sind, sind Sublimationsenthalpien die Summe aus Schmelz– und Verdampfungsenthalpie Sublimationskurven verlaufen daher bezüglich der Temperatur stets steiler als die zugehrigen Siedekurven, aber mit gleicher mathematischer Form In Pasendiagrammen von Reinstoffen (übliche Auftragung: Druck über Temperatur, oft mit logarithmischer Druckachse) tauchen alle Kurventypen nebeneinander auf, zeigen aber jeweils die erarbeiteten charakteristischen Verläufe Bredol (FH-MS) PC-I 180 / 186 8. Phasengleichgewichte in Reinstoffen 8.5. Phasendiagramme CO2 Ein sehr einfaches Beispiel ist Kohlendioxid (leicht schematisiert): o T/ C −78.5 −56.6 31.1 1000 100 kritischer Punkt P/bar flüssig (l) fest (s) 10 Tripelpunkt gas (g) 1 Standard−Sublimationspunkt 0.1 150 200 Bredol (FH-MS) 250 T/K 300 PC-I 8. Phasengleichgewichte in Reinstoffen 350 181 / 186 8.5. Phasendiagramme Reinstoffe Reinstoff-Phasendiagramme sind ausgezeichnet durch (mindestens) einen Tripelpunkt (drei Phasen stehen im Gleichgewicht) sowie einen kritischen Punkt des Siedegleichgewichtes Am kritischen Punkt verschwinden die Unterschiede zwischen Gas und Flüssigkeit: die Standardverdampfungsenthalpie geht gegen Null, die Dichteunterschiede verschwinden, es findet keine Phasentrennung mehr statt Oberhalb der kritischen Temperatur spricht man von einem permanenten Gas – Verflüssigung ist nicht mehr möglich (z.B. Luft) Wichtige Anwendung: in der Nähe des kritischen Punktes lassen sich Gase als überkritische Lösungsmittel einsetzen. Technisch: Kohlendioxid, Lebensmitteltechnik (Hopfen, Coffein u.a.). Bredol (FH-MS) PC-I 182 / 186 8. Phasengleichgewichte in Reinstoffen 8.5. Phasendiagramme Beispiel CO2 Verhalten eines Fluids um den kritischen Punkt herum in einem transparenten Behälter: www.youtube.com/watch?v=-gCTKteN5Y4 Einsatz von überkritischem CO2 zur schonenden Naturstoffextraktion: Quelle: Spektrum der Wissenschaft, Mai 1999 Bredol (FH-MS) PC-I 8. Phasengleichgewichte in Reinstoffen 183 / 186 8.5. Phasendiagramme Wasser Im Gleichgewicht können auch mehrere feste Phasen (Modifikationen) existieren. Dabei entstehen weitere Gleichgewichtslinien und Tripelpunkte (Beispiel: Kohlenstoff) Besonders wichtig: das Phasendiagramm von Wasser mit mehreren festen Modifikationen Das Schmelzvolumen in der Nähe des Normaldruckes ist negativ (Anomalie des Wassers), “Eis schwimmt auf Wasser”. Hydrothermalsynthese: Wasser als Lösungsmittel unter Druck Bredol (FH-MS) PC-I 184 / 186 8. Phasengleichgewichte in Reinstoffen 8.5. Phasendiagramme H2 O Phasendiagramm leicht schematisiert: o T/ C Eis VI 10000 0.01 100.0 373.9 Eis V Eis III Eis II 1000 kritischer Punkt 100 flüssig (l) P/bar 10 1 Standard−Schmelzpunkt Eis I 0.1 gas (g) 0.01 Tripelpunkt 0.001 0.0001 200 300 Bredol (FH-MS) 400 500 T/K PC-I 600 700 185 / 186 Inhaltsverzeichnis 1. Rückblick 2. Der Erste Hauptsatz 2.1. Das Ideale Gas 2.2. Volumenarbeit 2.3. Reale Gase 2.4. Virialgleichung 2.5. van-der-Waals–Gleichung 2.6. Wärmekapazität 2.7. Vollständige Differenziale 2.8. Enthalpie H 2.9. Adiabatische Zustandsgleichung 2.10. Joule-Thomson–Prozess 3. Thermochemische Größen 3.1. Grundlagen 3.2. Chemische Reaktionen 3.3. Hess’scher Satz 3.4. Kirchhoff’scher Satz 3.5. Born-Haber-Kreisprozess 5. 6. 7. 8. 4. Der Zweite Hauptsatz 4.1. Definitionen 4.2. Der Carnot-Prozess Bredol (FH-MS) PC-I 4.3. Wirkungsgrade 4.4. Zeitrichtung 4.5. Irreversible Prozesse 4.6. Entropieänderungen bei Phasenübergängen 4.7. Die Temperaturabhängigkeit der Entropie Der Dritte Hauptsatz 5.1. Annäherung an den Nullpunkt der Temperatur 5.2. Entropie bei T=0 Freie Enthalpie G und Freie Energie A 6.1. Hintergrund 6.2. Freiwilligkeit 6.3. Fundamentalgleichungen 6.4. Maxwell’sche Gleichungen 6.5. Thermodynamische Zustandsgleichungen Zusammenfassung Thermodynamik Phasengleichgewichte in Reinstoffen 8.1. Gleichgewichtsbedingungen 8.2. Siedegleichgewicht 8.3. Schmelzgleichgewicht 8.4. Sublimationsgleichgewicht 8.5. Phasendiagramme 186 / 186