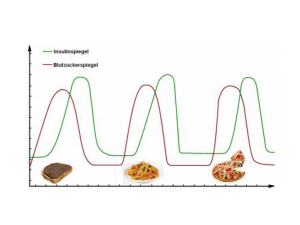
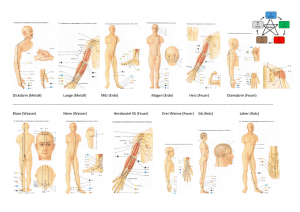
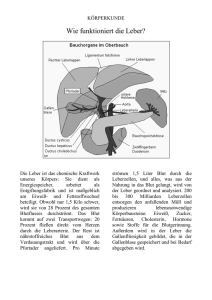
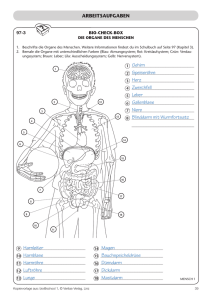
Pathophysiologie für Ernährungswissenschaftler
Werbung

Skript zur Vorlesung Pathophysiologie für Ernährungswissenschaftler Universitätsklinikum Gießen Vorwort Sehr geehrte Studierende der Ernährungswissenschaften, anbei finden Sie die letzte Version des Skriptes "Pathophysiologie für Ernährungswissenschaftler" noch im alten Format. Möglicherweise ergeben sich durch die Neustrukturierung des Studiengangs Änderungen, die dann von den neuen (und alten) Dozenten vorgenommen werden können. Da die Inhalte prüfungsrelevant sind, ist es günstig, dass sie von den gegenwärtigen Prüfern akzeptiert sind. Damit ist dieses Skript bis auf weiteres die Grundlage insbesondere der schriftlichen Prüfungen im Fach Pathophysiologie. Skripte sind natürlich nie perfekt und sollten auch mit der Zeit an neuere Entwicklungen angepasst werden. Wenn Sie Vorschläge und Verbesserungs-wünsche haben wenden Sie sich an Prof. Dr. Dr. N. Katz Direktor Institut für Klinische Chemie und Pathobiochemie Gaffkystr. 11c oder Prof. Dr. Thomas Linn Med. Klinik und Poliklinik 3 Rodthohl 6 Gießen, den 18.02.2003 Seite 1 VORWORT 1 1. DER LIPIDSTOFFWECHSEL 7 1.1 Aufnahme, Synthese, Funktionen, Ausscheidung 8 1.2 Der Transport der Lipide 9 1.3 Abnormale Lipoproteine 11 1.4 Elektrophorese 12 1.5 Die Frederickson-Einteilung der Hyperlipoproteinämien 12 1.6 Folgen der HLP • Lipidablagerungen im Gewebe • Mikrozirkulationsstörungen 14 14 14 1.7 Therapie bei hohem LDL-/IDL-Spiegel 14 1.8 Diättherapie bei HLP 14 1.9 Dyslipoproteinämie 15 1.10 Nierensteine (Harnsteine) & Gicht 15 1.11 Nierensteine • Symptome der Harnleitersteine und Nierenkolik • Arten der Nierensteine • Therapie der Nephrolithiasis 16 17 17 18 1.12 Hyperurikämie und Gicht • Pathogenese • Entzündungs (reaktion) • Primäre Hyperurikämie • Sekundäre Hyperurikämie (5%) • Hyperurikämie • Renale Harnsäureausscheidung: • Klinik und Therapie der Hyperurikämie 19 20 20 21 21 21 21 22 1.13 Arteriosklerose 23 1.14 WHO-Definition 23 1.15 Risikofaktoren der Atherosklerose • Reversible Faktoren: • Teilweise reversible Faktoren: • Nichtreversible Faktoren: • Andere mögliche Faktoren: 24 24 24 24 24 1.16 Regulation des Cholesterinstoffwechsels 25 1.17 Pathomechanismus 26 1.18 SMC (Smooth Muscle Cell) 28 Seite 2 1.19 Krankheitsbilder der Atherosklerose (eingeteilt nach Arealen) • AS der Koronararterien = Koronare Herzkrankheit (KHK) • AS der Gehirn-versorgenden Arterien = Cerebralsklerose • AS der Becken- und Beinarterien = Arterielle Verschlußkrankheit (AVK) 28 28 29 29 1.19 Therapie der Atherosklerose • Beseitigung der Risikofaktoren • Behandlung der Erkrankung 29 29 29 2. KOHLENHYDRATSTOFFWECHSEL UND DIABETES MELLITUS 32 2.1 Kohlenhydratstoffwechsel • Insulin • Insulinwirkung I • Insulinwirkung II • Synthese • Sekretionssteuerung • Formen des Insulins 32 32 33 34 34 35 35 2.2 Diabetes mellitus 36 2.3 Typen des Diabetes mellitus • WHO: Klassifikation Diabetes (1985) • Typ 1 Diabetes • Pathogenese des Typ 1 Diabetes • Typ 2 Diabetes • Pathogenese Typ 2a • Pathogenese Typ 2b 37 37 38 39 40 40 40 2.4 Diagnose des Diabetes mellitus • Harnglucosebestimmung • HbA1c 46 46 47 2.5 Diabetische Spätschäden • Makroangiopathie • Mikroangiopathie • Neuropathie 48 48 48 50 2.6 Koma bei Diabetes 51 2.7 Therapieformen • Diät • Unterstützung von Muskelarbeit, Ausdauersport • Tabletten • Insulin 52 52 52 53 53 2.8 Der diabetische Fuß 53 3 PROTEIN- UND AMINOSÄURESTOFFWECHSEL 54 3.1 Protein-Umsatz 54 3.2 Eiweiß-/N-Bilanz 55 3.3 Eiweiß-Restriktion 55 3.4 Verteilung der Körperproteine 56 Seite 3 3.5 Funktionen der Proteine 56 3.6 Störungen des Proteinstoffwechsels • A: Protein-Mangel • B: Störung der Serumprotein-Zusammensetzung • C: Genetische Defekte im Abbau der AS 57 57 57 61 4. VERDAUUNG 64 4.1 Definitionen • Normaler Stuhlgang • Obstipation • Diarrhoe 64 64 64 64 4.2 Ursachen der Obstipation • Funktionell • Organisch 65 65 65 4.3 Therapie der Obstipation 66 4.4 Diarrhoe • Maldigestion • Malabsorption • Magenteilresektion 67 68 68 71 4.5 Chronische entzündliche Darmerkrankungen • Einheimische Sprue • Morbus Crohn • Colitis Ulcerosa 71 71 71 72 5. LEBER UND ALKOHOL 75 5.1 Funktionen der Leber • Regulation des Energiestoffwechsels • Baustoffwechsel • Entgiftung 76 76 76 76 5.2 Leberversagen • Glucose-Homoiostase • Aminosäure-Homoiostase und Degradation • Lipidstoffwechsel • Die Cholesterinsynthese und -ausscheidung • Die Gallensäurensynthese und -ausscheidung • Hormonsynthese • Entgiftung • Leberversagen 76 77 77 78 78 78 79 79 79 5.3 Erkrankungen der Leber • Leberzirrhose = chronische Leberinsuffizienz • Folgen • KH-Stoffwechsel • AS-Stoffwechsel und Harnstoffsynthese • Synthese und Sekretion von Proteinen • Entgiftung und Abbau 79 79 81 83 84 85 86 5.4 Störungen des Leberstoffwechsels durch Alkohol • Erhöhung des NADH/NAD+-Quotienten 86 87 Seite 4 • Bildung von Acetaldehyd 88 6. DIE NIERE 89 6.1 Funktionen der Niere • Ausscheidung • Regulation • Hormonsynthese 89 89 89 89 6.2 Die Harnbildung 90 6.3 Die Clearance 92 6.4 Blutdruckregelung durch die Niere 94 6.5 Chronische Niereninsuffizienz (CNI) • Ursachen • Symptome • Ernährungsrelevante Probleme • Stadien der CNI • Therapie der CNI 95 95 95 96 96 97 6.6 Akutes Nierenversagen (ANV) • Ursachen • Verlauf • Therapie 98 98 98 98 6.7 Nephrotisches Syndrom 98 7. OSTEOPOROSE 100 7.1 Vitamin D • Funktionen 100 100 7.2 Hormone des Ca-Stoffwechsels 100 7.3 Osteoporose • Stadien • Therapie 101 101 101 8. PARENTERALE ERNÄHRUNG UND SONDENKOST 103 8.1 Parenterale Ernährung 103 8.2 Inhaltsstoffe der Nährlösungen 103 8.3 Probleme bei der parenteralen Ernährung 104 8.4 Sondenkost 105 8.5 Probleme bei Sondenkost 107 8.6 Vitamine 107 8.7 Spurenelemente 108 Seite 5 9. SCHILDDRÜSE 110 9.1 Anatomie 110 9.2 Biosynthese der Hormone • Regulation 110 110 9.3 T3 und T4 111 9.4 Diagnostik • TRH-Test • Immunometrischer Assay • TBG-Plasmakonzentration 112 112 113 113 9.5 Erkrankungen • Euthyreote Schilddrüsenvergrößerung • Hyperthyreose - Morbus Basedow • Hypothyreose 114 114 115 117 10. EßSTÖRUNGEN 119 10.1 Adipositas 119 10.2 Magersucht • Anorexia Nervosa • Bulimia nervosa 119 119 120 Seite 6 1. Der Lipidstoffwechsel Seite 7 1.1 Aufnahme, Synthese, Funktionen, Ausscheidung Die Lipide der menschlichen Nahrung lassen sich in drei Gruppen einteilen: 1. Triglyceride 2. Phosphatide und Sphingolipide 3. Steroide Die Zufuhr von Lipiden liegt bei einem Energieanteil von durchschnittlich 40-50%, das entspricht ca.150g/d . Die DGE empfiehlt, maximal 30 Energieprozente in Form von Lipiden aufzunehmen. Mit dieser überhöhten Lipidversorgung korreliert eine übermäßige Zufuhr von Cholesterin (~560 mg/d). Cholesterin wird sowohl aus der Nahrung (tierische Lebensmittel) resorbiert als auch endogen synthetisiert. Praktisch alle Körperzellen sind zur Cholesterinsynthese befähigt. Der Hauptanteil der Neusynthese von ca.1g/d findet im Darm (ca.400 mg) und in der Leber (ca.600 mg) statt. Cholesterin hat im Organismus wichtige Aufgaben zu erfüllen: • Es ist Bestandteil der Zellmembran und reguliert deren Stabilität und Fluidität. • In der Nebenniere und den Keimdrüsen werden aus Cholesterin die Steroidhormone synthetisiert. • Es dient als Vitamin D-Vorstufe. • In der Leber wird es zu den primären Gallensäuren umgebaut. • Es ist Bestandteil der Plasma-Lipoproteine. Die Ausscheidung erfolgt über • den Darm als neutrale Steroide oder Gallensäuren • den Harn als Abbauprodukte der Steroidhormone, • die Haut als Cholesterin und Squalen, v.a. über die Talgdrüsen. Normalerweise stehen Aufnahme und endogene Produktion mit der Ausscheidung im Gleichgewicht. Verschiebt sich das Gleichgewicht in Richtung Aufnahme und Produktion, so kommt es zur Hypercholesterinämie. Seite 8 1.2 Der Transport der Lipide Triglyceride, Phospholipide und Cholesterin (frei und verestert) werden im Blut als Bestandteil von Lipoproteinen transportiert. Diese enthalten immer alle 4 Lipidklassen (allerdings in unterschiedlichem Ausmaß) und einen Proteinanteil, das Apoprotein. Die Triglyceride und Cholesterinester befinden sich im Kern der kugelförmigen Teilchen, die anderen Substanzen sind amphipolar und machen die Hülle aus. Sie orientieren sich mit ihren hydrophoben Polen nach innen, mit den hydrophilen (Hydroxylgruppen) nach außen. Die Lipoproteine werden nach ihrer Zusammensetzung in verschiedene Klassen eingeteilt und haben im Stoffwechsel unterschiedliche Aufgaben. Triglyceridreiche Lipoproteine besitzen den größten Lipidkern und den geringsten Proteinanteil. Dies verleiht ihnen das geringste spezifische Gewicht und den größten Partikelradius. Ihre Akkumulation führt zu einer Trübung des Serums. Seite 9 Die Chylomikronen sind die größten Lipoproteine. Sie entstehen in den Mucosazellen des Darms und dienen dem Transport der resorbierten Nahrungslipide, v.a. der Triglyceride, die auch ihren größten Anteil ausmachen. Sie versorgen aus dem Darm das Energiebedürfnis der Zellen und die Speicher. Die VLDL (Very Low Density Lipoproteins) werden von der Leber gebildet und dienen der Umverteilung. Sie transportieren v.a. in der Leber synthetisierte Triglyceride, aber auch Cholesterin und Cholesterinester von der Leber in andere Organe. Die Lipoproteinlipase an der Oberfläche der Endothelzellen baut die Triglyceride der VLDL und Chylomikronen zu Glycerin und freien Fettsäuren ab. Durch diesen Vorgang werden die Chylomikronen in sog. Remnants, die VLDL in IDL (Intermediate Density Lipoproteins) und weiter in LDL (Low Density Lipoproteins) umgewandelt. Diese Lipoproteine sind mit Cholesterinestern angereichert. Die Remnants werden von der Leberzelle über spezifische Rezeptoren aufgenommen und in den Lysosomen abgebaut. Die LDL werden praktisch in alle extrahepatischen Zellen aufgenommen und beliefern sie mit Cholesterin. LDL wird zu 80% über den LDL-Rezeptor in die Zelle aufgenommen. Das Apoprotein wird abgespalten, Cholesterinester werden durch saure Lipase hydrolysiert und das freie Cholesterin ins Cytoplasma abgegeben. Freies Cholesterin hemmt nun in der Zelle die Synthese der HMG-CoA-Reduktase (HMG-CoA = 3-Hydroxy-3-methyl-glutaryl-CoA), die den limitierenden Schritt der endogenen Cholesterinsynthese katalysiert, und aktiviert die Acyl-CoA-Cholesterinacyl-Transferase (ACAT), die das Cholesterin mit freien Fettsäuren verestert. Auch die Synthese neuer LDL-Rezeptoren und die Funktion der bereits vorhandenen wird durch freies Cholesterin gehemmt, so daß die Überladung der Zelle mit Cholesterin verhindert wird. 20% der Aufnahme in die Zelle erfolgt über den Scavenger-Pathway und ist von der Zelle nicht kontrollierbar. Die einströmende Menge ist allein von der Plasma-Konzentration abhängig. Auch fehlt hier die negative Rückkopplung zur Rezeptorsynthese und zur Synthese der HMG-CoA-Reduktase. Über diesen Scavenger-Pathway werden beim Gesunden bevorzugt glycosiliertes und oxidiertes LDL aufgenommen. Er spielt vor allem beim genetischen Defekt des LDLRezeptors eine große Rolle. Als Folge des Defekts steigt die LDL-Plasmakonzentration an und der Scavenger-Pathway wird vermehrt genutzt. Die Zellen mit einer hohen Aufnahmekapazität über Scavenger (wie Makrophagen) nehmen zu viel LDL auf, werden zu Schaumzellen und können sogar platzen. Die HDL (High Density Lipoproteins) sind besonders reich an Apoproteinen. Sie werden direkt von den Mukosazellen des Darms und von den Leberzellen gebildet und modifiziert durch Kontkt mit Chylomikronen und VLDL. Sie transportieren Cholesterin aus den Zellen zur Leber, wo es über die Galle ausgeschieden wird. Seite 10 1.3 Abnormale Lipoproteine Lp(a) ist das atherogenste Lipoprotein. Es stammt aus der Leber und ist eine Untervariante des LDL. 1-5% aller Menschen haben eine genetisch bedingte vermehrte Konzentration dieses Lipoproteins. Unter den Betroffenen treten Herzinfarkte häufig schon zwischen dem 30. und 50. Lebensjahr auf. Die Lp(a) können allenfalls gering therapiert werden. Das IDL heißt auch flotierendes Lipoprotein und kommt normalerweise im Organismus nur in sehr kleinen Mengen vor. Es entsteht als Zwischenstufe bei der Umwandlung von VLDL in LDL. 50% des entstandenen IDL werden direkt über spezifische Rezeptoren in die Leber aufgenommen und dort metabolisiert. LPX tritt mit hoher diagnostischer Spezifität bei Stau des Gallenabflusses (Cholestase) auf. Es entsteht im Blut bei hohen Konzentrationen an Gallensäuren, die durch die Cholestase auftreten. Seite 11 1.4 Elektrophorese Man kann die einzelnen Lipoproteine aufgrund ihres Verhaltens mittels Elektrophorese trennen. Es ergibt sich ein typisches Muster, wobei sich die HDL in der aFraktion, die VLDL in der prä-ßFraktion und die LDL in der ßFraktion finden . Die Chylomikronen wandern nicht im elektrischen Feld. 1.5 Die Frederickson-Einteilung der Hyperlipoproteinämien Sind eine oder mehrere Lipoproteinfraktionen im Serum erhöht, so spricht man von einer Hyperlipoproteinämie (HLP). Man unterscheidet zunächst zwei Formen: Bei der primären HLP liegt eine genetische Ursache vor, die sekundäre HLP ist die Folge einer anderen Grunderkrankung, z.B. Übergewicht, Diabetes mellitus, Pankreatitis, Adipositas, Alkoholismus, Hyperurikämie, Leber-und Nierenerkrankungen, Hypothyreose etc. Seite 12 Nach Art des hauptsächlich vermehrten Lipoproteins unterscheidet man nach Frederickson und Levy die Typen IV. Die Typen IIa, IIb und IV machen ca. 95% aller HLP aus. Die primäre HLP Typ I stellt einen Defekt der Lipoproteinlipase dar, so daß der Abbau der Chylomikronen verlangsamt ist. Die Fließeigenschaft des Blutes ist beeinträchtigt, das Plasma ist milchig-trüb und rahmt leicht auf. Das Atheroskleroserisiko ist gering. Es kommt zu Durchblutungsstörungen in Darm und Pankreas, was zu Koliken und Pankreatitis führen kann. Nach fettreicher Nahrung ist der Gehalt der Chylomikronen im Serum besonders stark erhöht, es handelt sich also um eine lipidinduzierte HLP. Bei den primären HLP IIa und IIb liegt ein LDL-Rezeptordefekt vor, so daß die LDL nur über den Scavenger-Pathway in die Zellen aufgenommen werden. Beim Typ IIa sind die Triglyceridkonzentrationen normal, das Cholesterin stark erhöht, das Serum ist klar. Es kommt zu Cholesterin-Ablagerungen in den Geweben, Xanthelasmen, Xanthomen der Achillessehne etc. Beim Typ IIb findet ein Rückstau bis zu den VLDL statt, so daß auch die Triglyceride erhöht sind. Das Plasma ist klar bis trüb. Beide Typen sind mit einem hohen Atheroskleroserisiko verknüpft. Beim Typ III wird ein abnormes Apoprotein E gebildet. Apoprotein E ist für die schnelle Elimination der IDL aus der Blutbahn wichtig, so daß hier vor allem die IDL erhöht sind. Es bilden sich Handlinien- und Druckstellen-Xanthome. Auch dabei ist das Atheroskleroserisiko sehr hoch. Beim Typ IV ist eine Anreicherung von VLDL festzustellen, das vermehrt gebildet wird. (Z.B. bei Diabetes: Lipolyse wird nicht genügend gehemmt, da zu wenig Insulin vorhanden ist.) Dieser Typ tritt v.a. nach KH-reicher Nahrung auf. Deshalb handelt es sich hierbei um eine KH-induzierte HLP. Das Plasma ist trüb und milchig. Das Atheroskleroserisiko ist deutlich erhöht. Typ V stellt gewissermaßen eine Kombination der Typen I und IV dar, ist sowohl lipid-als auch KH-induziert. VLDL und Chylomikronen sind erhöht, das Plasma ist trüb und milchig und rahmt wegen seines Gehaltes an Chylomikronen auf. Es besteht kein erhöhtes Atheroskleroserisiko. Seite 13 Übersicht über die Typen der HLP nach Frederickson 1.6 Folgen der HLP Lipidablagerungen im Gewebe intra-/extrazellulär A: Haut: Xanthome (intraz.) Sehnen: Xanthome (intraz.) Cornea: Arcus lipoides (extraz.) B: Gefäßintima: Atherom (intraz., bei Zellzerfall auch extraz.) Mikrozirkulationsstörungen A: B: Abdominalkoliken Pankreatitis 1.7 Therapie bei hohem LDL-/IDL-Spiegel 1. Beseitigung der Grunderkrankung bei sekundärer HLP, zusätzlich Diät und Medikamente (s.u.) 2. Bei primärer HLP: Senkung des Cholesterinspiegels direkt -cholesterinarme Diät (wenig tierische Produkte) -Cholesterin-Resorption senken +faserreiche Kost (so wird die Ausscheidung generell erhöht, damit auch die des Cholesterins) +Cholestyramin, Cholestipol etc ( Anionen-Tauscher, binden Gallensalze im Darm und verstärken so die Cholesterinausscheidung) +Sitosterin bildet mit Cholesterin nicht-resorbierbare Komplexe - Hemmung der Hormon-sensitiven Lipase durch Nicotinsäure - Beschleunigung des VLDL- und LDL-Metabolismus: Fibrate - Hemmung der HMG-CoA-Reduktase: Mevinolin (lebertoxische Nebenwirkungen) - LDL-Apherese (Blutreinigungsverfahren) 1.8 Diättherapie bei HLP Typ I Die Senkung der Chylomikronenbildung ist das erste Prinzip. Die Zufuhr von langkettigen Fettsäuren muß auf ca. 25g/d reduziert werden, die entstehende Energielücke kann durch Zusatz von MCT-Fetten geschlossen werden (mittelkettige Fettsäuren werden nicht in Chylomikronen transportiert). Bei abdominellen Beschwerden sollte auf eine geeignete Lebensmittelauswahl geachtet werden. Typ IIa Die Zufuhr von Nahrungscholesterin sollte eingeschränkt werden. Eine Einschränkung der Fettzufuhr und eine Modifizierung in Richtung v.a. der Seite 14 Monoensäuren ist sinnvoll. Eine erhöhte Zufuhr von Ballaststoffen in einem praktikablen Rahmen hat direkt keinen Einfluß. Typ IIb/III Die Vorschriften zur Ernährung bei Typ IIa und IV sind zu beachten. Typ IV Das Körpergewicht muß normalisiert werden, die Zufuhr von leichtresorbierbaren KH und Alkohol sind einzuschränken. Typ V Sowohl die Energie-, als auch die Fett- und KH-Zufuhr muß eingeschränkt werden. 1.9 Dyslipoproteinämie Ein erhöhtes Risiko für coronare Herzerkrankungen kann allerdings auch vorliegen, wenn keine HLP vorliegt, aber die Zusammensetzung der Lipoproteine verändert ist (Dyslipoproteinämie). Gefährlich ist hierbei ein vermindertes HDL- bei erhöhtem LDLCholesterin. 1.10 Nierensteine (Harnsteine) & Gicht Substanzen, die schlecht löslich sind und im Urin hoch konzentriert werden, bilden Kristalle, wenn ihre kritische Übersättigungsgrenze überschritten wird. Aggregation solcher Kristalle führt zu Harnsteinen, die u.U. den Harnabfluß gefährden. Sie bestehen meist aus Calciumoxalat, seltener aus Harnsäure oder Cystin. Der Harn enthält Substanzen, die die Löslichkeit und die kritische Übersättigungsgrenze erhöhen sowie die Kristallaggregation hemmen Seite 15 1.11 Nierensteine Die Nierensteine bilden sich oft in den Nierenkelchen oder im Nierenbecken. Ausgußsteine füllen das gesamte Nierenbecken aus. Da der Harn nicht mehr abfließen kann, kommt es zu einem Rückstau bis ins Nierenparenchym. Harn ist ein guter Nährboden für Bakterien. Sie werden mit dem Blut antransportiert oder es kann eine ascendierende Infektion von der Harnröhre in die Niere entstehen (Schmierinfektion). Gerade, wenn der Harnfluß fehlt, bieten sich für Bakterien gute Möglichkeiten, in der Harnröhre aufzusteigen. Die Steine werden durch den Harn transportiert. Gelegentlich können sie aufgrund ihrer Größe den Ureter nicht passieren (Harnleiterstein). Sie können aber auch bis in die Blase Seite 16 transportiert oder mit dem Urin ausgeschieden werden. Man kann sie entfernen, indem man sie herauszieht oder durch Ultraschall zertrümmert. Ein charakteristisches Symptom ist die Steinkolik. Eine Kolik ist durch einen Schmerz charakterisiert, der langsam ansteigt, eine Spitze erreicht und dann wieder abfällt. Diese Schmerzen werden dadurch hervorgerufen, daß ein muskuläres Hohlorgan (hier: die Blase, der Ureter) einen Fremdkörper vertreiben will. In diesem Fall wird in der glatten Muskulatur ein Tonus aufgebaut, der dann die Schmerzen verursacht. Symptome der Harnleitersteine und Nierenkolik • • • • Schmerzen (Kolik) Blut im Urin Aufstau (Hydronephrose) Infektion (Pyelonephritis) Arten der Nierensteine Die Steine haben unterschiedliche Zusammensetzung. Häufigster Bestandteil ist Calcium (80%). Calcium-Oxalat: Calcium-Phosphat: Harnsäuresteine: Struvitsteine: Cystin-Steine: 60% 20% 5-10% 10-15% ca.1% Obwohl die Cystinsteine die seltenste Form der Nierensteine darstellen, kennt man ihre Ursachen mit am Besten. Sie entstehen bei Cystinurie, einem genetischen Defekt eines intestinalen und nephralen Transportsystems für dibasische Aminosäuren. Davon sind also Cystin, Ornithin, Lysin und Arginin (einfach zu merken:COLA) betroffen. Bei diesen Menschen wird Cystin nicht aus dem Darm aufgenommen, sondern aus Methionin synthetisiert. Da die Ausscheidung gehemmt ist, bilden sich Ablagerungen in Form der Cystinsteine. Die Cystinurie stellt einen angeborenen Defekt dar, deshalb tritt diese Form der Nierensteine bereits bei jungen Menschen auf; das Durchschnittsalter beträgt 28 Jahre. Bei den anderen Arten der Nierensteine sind die betroffenen Menschen meist bereits älter als 50 Jahre. Calcium-Steine treten am häufigsten auf. Ursache ist meist eine Hyperkalzurie durch endokrine Störungen oder gesteigerte Freisetzung aus Knochen. Ihre Bildung wird nicht wesentlich durch die Calcium-Zufuhr mit der Nahrung beeinflußt, dennoch gibt es diätetische Empfehlungen. Hierbei gelten für fast alle Steinarten gleiche Verhaltensmaßnahmen. Vor allem die Zufuhr an tierischem Protein und Natrium sollte reduziert werden. Seite 17 Therapie der Nephrolithiasis akut • Spasmolytika (BuscopanR, BaralginR) • Schlingenextraktion • Operation • Lithotrypsie (Ultraschall-Zerstörung) chronisch (Prophylaxe) • Flüssigkeitszufuhr (es sollten 2 l Urin pro Tag produziert werden) • Reduktion von Natrium, Oxalsäure (Schokolade, Spinat, Rhabarber, schwarzer Tee) • Verminderung von tierischem Eiweiß • Alkalisierung des Urin-pH (Citronensäure- oder Bicarbonat-Salze, verbessert die Löslichkeit aller Steine außer der Infektsteine) • Calciumhaltige Steine: Thiazide (vermehrte Ca-Rückresorption), Allopurinol (Xanthinoxidasehemmer) • Infekt (Struvit-)steine: Antibiotika, Harnansäuerung auf pH<6 (L-Methionin), ggf. Operation • Harnsäuresteine: purinarme Kost, Allopurinol, Alkalisierung auf pH 6.2-6.8 (Uralyt-UR) • Cystinsteine: Alkalisierung auf pH>7.4, massive Flüsssigkeitszufuhr (4 l Urin pro Tag), evtl. D-Penicillamin (NW!) Seite 18 1.12 Hyperurikämie und Gicht Aus purinreicher Kost entsteht im Stoffwechsel über die Xanthinoxidase die Harnsäure. Ist ein erhöhter Harnsäurespiegel im Blut feststellbar, spricht man von einer Hyperurikämie. Seite 19 Das Auftreten von Gicht in einer Population ist umso häufiger, je höher die Serumharnsäurekonzentration ist. Einfluß haben aber auch andere Faktoren wie Alter, Gewicht, Cholesterinspiegel und Alkoholkonsum. Deshalb kann Gicht auch ohne Hyperurikämie entstehen, bzw. eine Hyperurikämie ohne Gicht verlaufen. Es sind drei Stadien unterscheidbar: 1) Hyperurikämie ohne Symptome 2) akuter Gichtanfall 3) chronische Gicht Pathogenese Im akuten Gichtanfall weist das betroffene Gelenk die typischen Entzündungszeichen (Erwärmung, Rötung, Schmerz) auf. Der Anfall verläuft in einer Kettenreaktion. Zunächst kommt es zu einem Ausfall von Harnsäurekristallen in den Gelenkhäuten, die wie ein Filter wirken. Durch niedrige Temperaturen, niedrigen pH-Wert (6.4) und schlechte Durchblutung wird dieser Vorgang begünstigt. Diese Einflußfaktoren bedingen sich auch gegenseitig: wenn die Durchblutung schlecht ist, können die Schlacken nicht abtransportiert werden, was eine Senkung des pH-Wertes zur Folge hat; auch Entzündungen bewirken einen Abfall des pHWertes. Deshalb tritt Gicht meist zuerst an den Akren = Enden des Körpers auf, z.B. an der großen Zehe. Um die Harnsäurekristalle wieder aus den Gelenkhäuten zu entfernen, wandern Leukocyten ein und phagocytieren sie. Doch oft werden die Membranen der entstehenden Phagolysosomen von den Kristallen zerrissen und lysosomale Enzyme können austreten. So kommt es im umgebenden Gewebe zu Schädigungen und Entzündungen. Auch die von den Phagocyten freigesetzten Leukotriene und Prostaglandine fördern Entzündungsreaktionen. Die dadurch bedingte Erniedrigung des pH-Wertes führt zu einer verstärkten Ausfällung von Harnsäure. Dies kann durch eine Verminderung der Purinzufuhr und Alkoholkarenz unterbrochen werden. Die Harnsäure wandert auch in die Niere, wo sich Nierensteine bilden und es zu einem Ausfall im Parenchym kommt. Das Nierenbecken ist im Vergleich zur Rinde schlecht durchblutet. Außerdem passiert die Harnsäure ständig den Tubulusapparat, so daß in dieser Region viel davon zurück ins Parenchym diffundiert. Dadurch steigt hier die HarnsäureKonzentration, womit die Bedingungen für eine Entzündung gegeben sind (Pyelonephritis = Nierenbeckenentzündung). Chronische Gicht endet meist mit einer Niereninsuffizienz. Entzündungs (reaktion) Wichtiger Mechanismus des Körpers zur Beseitigung von Gewebsschäden und deren Ursachen. Ursachen: Trauma (Operation, Verletzung) Mikroorganismen (Bakterien, Viren, Pilze, Protozoen) Immunreaktionen (Allergie, Autoimmunität) Stoffwechselendprodukte (Harnsäure) Symptome: Rubor Rötung Calor Überwärmung Tumor Schwellung Dolor Schmerzen Functio laesa gestörte Funktion Mediatoren: Leukocyten: Granulocyten (neutrophil, eosinophil, basophil) Monocyten Lymphocyten Eicosanoide (Prostaglandine, Leukotriene), Tumor- Nekrose-Faktor (TNF), Interleukine, Immunglobuline, Complementsystem Seite 20 Primäre Hyperurikämie verminderte renale Ausscheidung (95-98%) gesteigerte Synthese (1-2%), z.B. Lesch-Nyhan-Syndrom Begleiterkrankungen: Hypertonie (40-80%), Adipositas (70%), Diabetes mellitus (10-25%), Fettleber (60-90%), Hyperlipoproteinämie (40-100%) Sekundäre Hyperurikämie (5%) vermehrte Produktion: Polyzythämie, CML(chronisch-myelotische Leukämie), OMF (Osteomyelofibrose) Hämolytische Anämie Zytostase/Radiatio mal. Tumoren Zuckeraustauschstoffe (Fructose, Sorbit, Xylit) Fasten/Nulldiät (+Azidose) verminderte Ausscheidung: chronische Niereninsuffizienz Medikamente (z.B. Diuretika. Salizylate) Vergiftungen (Blei, Cadmium, Beryllium) Hyperparathyreoidismus Alkohol, Laktatazidose dekompensierte Herzinsuffizienz Das Lesch-Nyhan-Syndrom stellt einen Mangel des Enzyms HGPT dar. Dadurch kommt es zu verminderten intrazellulären Konzentrationen von AMP und GMP. Außerdem entfällt die Hemmung der Adenosin-Phospho-Ribosyl-transferase (APRT). Es kommt zu einer etwa 20fach erhöhten de novo-Purinsynthese. Diese Krankheit wird x-chromosomal rezessiv vererbt. Hyperurikämie Harnsäure i. S. Normalwerte empfohlene obere Grenzwerte Männer 3,6-8,2 mg% 7,0 mg% Frauen 2,3-6,1 mg% 6,0 mg% (ein Serumwert von 6,4 mg% entspricht dem Löslichkeitsprodukt von Natriumurat in Plasmawasser bei 37°C, d.h. Gichtanfälle unterhalb dieses Wertes unwahrscheinlich = sinnvoller Grenzwert) Harnsäure i.S. (mg%) 6,0-6,9 Gichtarthritis: Prävalenz 1,8% Inzidenz (%/Jahr) 0,1% Nierensteinanamnese 7,0-7,9 >9,0 11,8% 0,5% 12,7% 36% 4,9% 40% Renale Harnsäureausscheidung: Harnsäure-Clearance: Glomerulum: Proximaler Tubulus: Distaler Tubulus: Beim Gichtkranken: 8-10 ml/min (6-12% der glomerulär filtrierten Harnsäuremenge erscheinen im Urin). vollständige Filtration nahezu vollständige Rückresorption Sekretion und teilweise Rückresorption Störung der tubulären Sekretion In 95% der Fälle handelt es sich um primäre Gicht, d.h. einer chronisch verlaufenden erblichen Störung des Purinstoffwechsels. Sie ist charakterisiert durch eine Erhöhung des Uratgesamtpools. Normalerweise beträgt er ~1g, bei Gichtkranken kann er auf bis zu 30g ansteigen. Das Transportsystem, das hier eine Rolle spielt, ist weitgehend unbekannt. Seite 21 Sekundäre Gicht ist nicht direkt genetisch bedingt. Sie tritt z.B. im Zusammenhang mit Zuständen auf, in denen ein verstärkter Abbau von Zellen stattfindet (z.B.Tumor), weil dann auch kernhaltiges Material anfällt, bei dessen Abbau Purine frei werden. Ursache kann auch eine verminderte Harnsäureausscheidung durch chronische Nierenerkrankungen oder bestimmte Arzneimittel sein. Klinik und Therapie der Hyperurikämie Seite 22 1.13 Arteriosklerose 1.14 WHO-Definition Arteriosklerose/Atherosklerose ist eine variable Kombination von Veränderungen der Intima, bestehend aus herdförmiger Ansammlung von Fettsubstanzen, komplexen Kohlenhydraten, Blut und Blutbestandteilen, Bindegewebe und Calciumablagerungen, verbunden mit Veränderungen der Arterienmedia. Lokalisation Intima der Arterien, kurz hinter Verzweigungen (Turbulenzen!) Art der Veränderung herdförmige Verdickung (Plaque) Bestandteile Fette, Wasser (Ödem) Zellen (Monocyten, Thrombocyten, glatte Muskelzellen, Endothelzellen) Bindegewebsgrundsubstanz, Blut, Calcium Betroffen größere bis mittlere Arterien: Aorta und periphere Arterien der unteren Extremitäten: Arterielle Verschlußkrankheit Arteria carotis zum ZNS: Zerebralsklerose Coronarien (Herzkranzgefäße): Coronarsklerose Seite 23 1.15 Risikofaktoren der Atherosklerose Die Risikofaktoren für Atherosklerose lassen sich nach zwei Schemen einteilen: a) nach der Ordnung: Risikofaktoren erster Ordnung sind bedeutsamer als die der 2. Ordnung b) nach Reversibilität Reversible Faktoren: • Zigarettenrauchen (1.Ordnung) • Arterielle Hypertonie (1.Ordnung) • Adipositas • Teilweise reversible Faktoren: • Hyperlipoproteinämie, quantitativ (1.Ordnung) • Dyslipoproteinämie, qualitativ (1.Ordnung) • Diabetes mellitus • Nichtreversible Faktoren: • Alter (bei Männern ab dem 20. Lj.) • Männliches Geschlecht • Genetische Faktoren • Andere mögliche Faktoren: • Physische Inaktivität • Stress (evtl. über blutdrucksteigernde Wirkung), Persönlichkeitstyp Seite 24 1.16 Regulation des Cholesterinstoffwechsels Die Aufnahme von Cholesterin über den LDL- Rezeptor • hemmt die Ausbildung neuer LDL-Rezeptoren • aktiviert die Cholesterin-Acyl-Transferase • hemmt die Cholesterin-Neubildung Bei Aufnahme über den Scavenger-Way ist eine Überladung der Zelle möglich, in der Zelle bilden sich Fettröpfchen. Löst man zur Untersuchung das Fett heraus, bleiben bienenwabenartige Löcher zurück: daher die Bezeichnung “Schaumzelle”. (Siehe auch Kapitel “Lipidstoffwechsel”.) Ist LDL oxidiert, acetyliert oder Neuraminsäure abgespalten, so bezeichnet man es als modifiziertes LDL. Dieses kann nicht an Apo B binden und wird daher nur über den Scavenger Rezeptor aufgenommen, ist also besonders atherogen. Seite 25 1.17 Pathomechanismus Hier spielen 4 verschiedene Zellarten eine Rolle: 1. Endothelzellen 2. Thrombocyten 3. Monocyt des Blutes, der zum Makrophagen des Gewebes wird 4. Muskelzellen (normale glatte oder transformierte) Durch Hypertonie, O2-Mangel, mechanische Reize etc. kann es zu Läsionen des Endothels kommen. • Über diese Lücke wandern Monocyten in die Intima ein und bilden Mediatoren (Leukotrien = LT B4, phänotyp-verändernden Faktor PMF und makrophage-derived growth factor MDGF). Damit stimulieren sie sich selbst zur Umwandlung zu Makrophagen. • Der von den Makrophagen abgegebene PMF wirkt auf die glatte Muskelzelle (1). Sie verliert ihre Kontraktilität und synthetisiert Kollagen, was zu einer Bindegewebsvermehrung führt. Die Elastica Interna wird durchbrochen, die Muskelzellen wachsen aus der Media heraus in die Intima, unterstützt durch PDGF (platelet-derived growth factor, 2) und MDGF (3). Seite 26 • Auch in die Gefäßwand eingewanderte Leukocyten stimulieren über Wachstumsfaktoren am Ort der Schädigung vermehrt auftretende Bindegewebszellen, die in gesteigertem Maß Grundsubstanz synthetisieren. • Aufgrund der Läsion wird die Permeabilität für Makromoleküle gesteigert. LDL wandern ein und können von Makrophagen, die viele Scavenger-Rezeptoren haben, aufgenommen werden. Sie entwickeln sich so zu Schaumzellen. Da die LDL in der Intima auch schnell modifiziert werden, wird dieser Vorgang noch beschleunigt. Die Akkumulation von Lipiden führt schließlich zum sog. atherosklerotischen Plaque, in dem sich die Schaumzellen anreichern (“fatty streaks”). • Calcium wird verstärkt eingelagert, da aus LDL vermehrt Fettsäuren freigesetzt werden. Diese binden das Ca2+ und Wasser (“Verkalkung”). • Um den Endotheldefekt zu verschließen, lagern sich Thrombocyten an, die sich selbst über Thromboxan (TX A2) aktivieren. Es kann passieren, daß sie sich überstimulieren und so das Gefäß verstopfen. So ein Thrombus ist der Hauptausgangspunkt für einen akuten Myokardinfarkt. Die Endothelzellen bilden Prostaglandin (PG I2). Dies wirkt anti-aggregatorisch und antikoagulatorisch und verhindert normalerweise eine zu heftige Anlagerung der Thrombocyten. • Die Verdickung der Arterienwand sowie die Bildung von Thromben können das Gefäßlumen völlig verschließen. Thromben, die sich von größeren Gefäßen ablösen, verursachen den Verschluß kleinerer Gefäße in der Peripherie. Seite 27 1.18 SMC (Smooth Muscle Cell) 1.19 Krankheitsbilder der Atherosklerose (eingeteilt nach Arealen) AS der Koronararterien = Koronare Herzkrankheit (KHK) a) Angina pectoris (intermittierender Blutmangel mit Schmerz): hier ist noch keine Nekrose vorhanden; auf Verabreichung von Nitropräparaten, die NO (den wichtigsten gefäßerweiternden Faktor endogener Art) freisetzen, läßt der Schmerz nach b) Herzinfarkt (Myokardnekrose durch Arterienverschluß): Beseitigung des Schmerzes durch Medikamente kaum möglich, nicht reversibel • Vorderwandinfarkt (Ramus interventrikularis anterior der linken Koronararterie) • Seitenwandinfarkt (Ramus circumflexus der linken Koronararterie) • Hinterwandinfarkt (rechte Koronararterie) c) Herzinsuffizienz (sukzessiver Untergang kleiner Myokardareale und deren Ersatz durch Bindegewebe): kontraktiles Gewebe fehlt Seite 28 AS der Gehirn-versorgenden Arterien = Cerebralsklerose a) Transitorische ischämische1 Attacken (TIA; Dauer ≤ 24h): Patient ist bewußtlos, kann sich an nichts mehr erinnern, evtl. Lähmungserscheinungen etc. b) Prolongierter reversibler ischämischer Insult (PRIND; Dauer 1 Tag bis 1 Woche) c) Progredienter Hirninsult (seltenere Form des Schlaganfalls, Blutung ins Gehirn, gleiche Symptome) AS der Becken- und Beinarterien = Arterielle Verschlußkrankheit (AVK) (Stadium I: ohne Syptome) a) Claudicatio intermittens (Stadium II): Schmerz in den Beinen tritt nach Belastung auf, verschwindet bei Erholung wieder, je nach O2-VersorgungÆSchaufensterguckerKrankheit b) Ruheschmerzen (Stadium III): Sauerstoffversorgung reicht selbst in Ruhe nicht mehr aus c) Nekrosen/ Gangrän (Stadium IV): das Bein stirbt bereits ab Je nach betroffenen Arterien unterscheidet man den Becken-, Oberschenkel- und Unterschenkeltyp. 1.19 Therapie der Atherosklerose Beseitigung der Risikofaktoren Hypercholesterinämie • Gewichtsnormalisierung • Cholesterin runter, P/S-Quotient hoch • EPA (zwei Fischmahlzeiten pro Woche) Hypertonie • Gewichtsnormalisierung • Kochsalzrestriktion • Medikamente: ß-Blocker, Diuretika, Ca2+-Antagonisten, ACEAngiotensin Converting Enzyme, s. Kapitel „Niere“) Inhibitoren (ACE = Zigarettenrauchen einstellen, insbesondere bei Einnahme hormoneller Antikonzeptiva Diabetes mellitus • Gewichtsnormalisierung • strenge Einstellung (HbA1c), nicht bei alten Patienten Behandlung der Erkrankung • Training (besonders bei AVK; Kollateralen-Bildung!) • Durchblutungsfördernde Medikamente (besonders bei KHK, weniger bei AVK, sinnlos bei CS) • Antikoagulantien, Fibrinolyse (besonders bei Verschlüssen) • Perkutane transluminale (coronare) Angioplastie [PT(C)A; besonders bei KHK und AVK] • Operation: Bypass, Endarteriektomie Bypass am Herz: ACVB = aorto-coronarer Venenbypass LIMA = left internal mammarial artery 1 durch mangelnde Blutversorgung hervorgerufen Seite 29 Die antiaggregatorische Wirkung der Eicosapentaensäure beruht auf der Bildung der Serie 3 Prostaglandine. TX A3 hat keine Wirkung, PG I3 wirkt wie PG I2 (vgl. “Pathomechanismus”). Seite 30 Seefische enthalten also viel Lipide, davon sind ca.10% Eicosapentaensäure. Das ist ein viel höherer Prozentsatz als bei allen anderen Lipiden pflanzlicher oder tierischer Herkunft. Seite 31 2. Kohlenhydratstoffwechsel und Diabetes mellitus 2.1 Kohlenhydratstoffwechsel Kohlenhydrate sind der größte mit der Nahrung zugeführte Energielieferant. Für sämtliche Stoffwechselvorgänge dient Glucose als wichtigster Energieträger. Der Blutglucosespiegel beträgt normalerweise 70-120 mg% (das entspricht 3,9-6,7 mmol/l). Die meisten Organe können neben Glucose auch Ketonkörper und freie Fettsäuren als Energielieferanten nutzen. Erythrocyten und Nierenmark sind jedoch absolut glucoseabhängig. Sie bauen Glucose anaerob zu Lactat ab, das dann in der Leber zur Gluconeogenese genutzt wird. Auch das ZNS ist unter normalen Bedingungen auf die kontinuierliche Zufuhr von Glucose angewiesen. Der Glucoseverbrauch des menschlichen Gehirns beträgt etwa 144 g/d. Bei einem plötzlichen Abfall des Blutglucosespiegels kommt es deshalb zur Hypoglycämie. Unter längerfristigen Hungerbedingungen kann das Gehirn seinen Energiebedarf jedoch zum Teil durch Verstoffwechselung von Ketonkörpern decken. Der Kohlenhydratstoffwechsel wird über Hormone gesteuert. Eine senkende Wirkung auf die Blutglucosekonzentration kommt dabei dem Insulin zu, da dieses u.a. die Aufnahme durch die Zelle steigert. Glucagon, STH (Somatotropes Hormon) und Glucocorticoide fördern hingegen die Glucosefreisetzung und bewirken so eine Erhöhung des Blutzuckerspiegels. Insulin und Glucagon werden im Pankreas gebildet, Insulin in den B-Zellen (ca.80% des Inselorgans), Glucagon in den A-Zellen. Auch Somatostatin, das regulierend auf die Bildung von STH in der Adenohypophyse wirkt, wird in der Bauchspeicheldrüse produziert. Die Glucocorticoide werden in der Nebennierenrinde gebildet. Insulin Insulin beeinflußt sowohl den Kohlenhydrat- als auch den Eiweiß- und Lipidstoffwechsel. Es besteht aus 2 Peptidketten, die über 2 Disulfidbrücken miteinander verbunden sind. Die AKette besteht aus 21 Aminosäuren, die B-Kette aus 30. Aufbau des menschlichen Insulins Zur Therapie wird heute überwiegend Humaninsulin verwendet, früher benutzte man auch Rinder- und Schweineinsulin. Diese unterscheiden sich nur gering vom Humaninsulin, und ihre Funktion ist weitgehen identisch. Es gibt zwei Sorten, das U40 und U100 Insulin. U100 bedeutet, daß sich 100 Einheiten Insulin in einem ml Lösung befinden. Dieser Typ wird häufiger verwendet. Seite 32 Insulinwirkung I Seite 33 Insulinwirkung II Glycogensynthese Fett-Synthese Hemmung des Eiweiß-Abbaus Glucosetransport Aminosäuretransport Lipolyse Leber + + + ø ø ø Muskel + ø + + + - Fettgewebe + + + + + - Synthese Insulin wird auf zwei Weisen hergestellt: a) gentechnisch durch Colibakterien b) chemisch, indem man Insulin aus Rinder- bzw. Schweineorganen isoliert und die abweichenden Aminosäuren austauscht. In den B-Zellen der Bauchspeicheldrüse wird zunächst das einkettige Präproinsulin hergestellt. Es setzt sich aus Präpeptid, B-Kette, C-Peptid und A-Kette zusammen. Das Präpeptid dient als Signalpeptid. Nach der Biosynthese des Präproinsulins an den Ribosomen des rauhen ER ist es für die Einschleusung der Peptidkette in das Lumen des ER verantwortlich. Dabei wird es selber abgespalten und bleibt im Cytosol zurück. Durch Abspaltung des C-Peptids entsteht Insulin. Dementsprechend wird das C-Peptid äquimolar zu Insulin abgegeben und wird zur Messung der endogenen Insulin-Sekretion verwendet. Das Insulin wird in Granula gespeichert. Man kann zwei Phasen der Insulinsekretion unterscheiden, wenn man das Pankreas isoliert und mit Glucoselösung (300mg/dl) perfundiert. In der frühen Phase kommt es zu einer schnellen, plötzlichen Abgabe ins Blut. Dabei werden sämtliche Speichergranula entleert. Es folgt ein Abfall für ca.5 Minuten, dann setzt die Spätphase ein, in der frisch produziertes Insulin ausgeschüttet wird. Diese Phase hält etwa 20 Minuten an (bei ständigem weiteren Glucosereiz), dann ist eine Erschöpfung der Synthese festzustellen. Seite 34 Sekretionssteuerung Stimulierend wirken • Zucker (Glucose, Mannose) • Hormone (STH, ACTH, Glucagon) • Medikamente (Sulfonylharnstoffe) • Aminosäuren (Arginin) • Enterohormone (GIP, VIP, GLP-1) Hemmend wirken • 2-Desoxyglucose • Medikamente (Diazoxid) • Hormone (Adrenalin, Insulin) Glucose stellt den wichtigsten Stimulator der Sekretion dar. Antagonisten des Insulins sind Somatostatin, Catecholamine und Glucagon. Durch ihre antagonistische Wirkung treiben sie indirekt den Blutzucker in die Höhe und wirken dementsprechend wieder stimulierend auf die Insulinausschüttung, so daß sich ein Gleichgewicht einstellen kann. 1. Insulin bildet mit den hochspezifischen, membranständigen Rezeptoren einen InsulinRezeptor-Komplex. Es aktiviert dabei als first messenger einen Überträger, der die Aufnahme von z.B.Glucose in die Zelle und die dort stattfindende Umwandlung in die speicherfähige Form fördert. 2. Glucagon fungiert ebenfalls als first messenger. Wie Insulin beeinflußt es den Zellstoffwechsel über einen Überträger. Es stimuliert die Adenylatcyclase, die für die Bildung von cAMP aus ATP verantwortlich ist. Bei Bildung von Insulin-Rezeptor-Komplexen werden in einigen Geweben (Leber, Fettgewebe) die durch Glucagon stimulierten Überträger durch einen dritten Überträger gehemmt. Dadurch wird auch die Bildung des für die Gluconeogenese wichtigen cAMP gehemmt. 3. Unter Einfluß von Insulin wird über einen nicht näher bekannten second messenger außerdem die Phosphodiesterase (PDE) aktiviert, die cAMP zu AMP abbaut. Formen des Insulins Es gibt 2 Formen des Insulins: Einmal das schnell wirkende Normalinsulin und das Verzögerungsinsulin, das Protamin oder Zinkkomplexe enthält. Es gibt auch chemisch Seite 35 veränderte Insulinanaloge, die bereits nach 5 Minuten oder über 24 Stunden wirksam werden. Bei der konventionellen Insulintherapie wird zweimal täglich eine Gabe von Normal- und Verzögerungsinsulin gespritzt. Bei einer intensivierten Therapie wird zu jeder Mahlzeit Normal- und über Nacht reines Verzögerungsinsulin gespritzt. 2.2 Diabetes mellitus Der Diabetes mellitus ist eine der häufigsten und bedeutendsten Stoffwechselstörungen und beruht auf einem Insulinmangel. Dabei unterscheidet man zwischen absolutem und relativem Insulinmangel. Ein absoluter Insulinmangel liegt lediglich dann vor, wenn die Bauchspeicheldrüse nicht mehr in der Lage ist, Insulin zu produzieren (1). Ein relativer Insulinmangel ist zunächst dann gegeben, wenn die Insulinsynthese zu gering (1) oder die Sekretion gestört ist (2). Dies kann u.a. durch einen Defekt der (noch hypothetischen) Glucose-Rezeptoren der B-Zellen hervorgerufen werden. Relativer Insulinmangel tritt auch dann auf, wenn die Zahl der Insulinrezeptoren am Erfolgsorgan vermindert ist oder ein Rezeptorendefekt besteht (3). Außerdem kann es im Körper zu einem vermehrten Abbau des Insulins kommen (4) oder die Wirkung durch Antikörper oder Antagonisten vermindert werden. Man versucht, Diabetes bereits in einer Frühphase zu erfassen. Man kann Menschen bestimmen, die genetisch bedingt ein hohes Risiko haben, Diabetes zu bekommen. Ist der Vater beispielsweise ein Typ 1 Diabetiker, so besteht bei dem Kind eine Wahrscheinlichkeit von 6%, Diabetes Typ 1 zu bekommen. Ist der Diabetes des Vaters allerdings bereits in jungen Jahren aufgetreten, erhöht sich die Wahrscheinlichkeit auf 8%. Zeigt das Kind eine positive Reaktion auf ICA (Islet-Cell-Antibodies), so ist sein Risiko auf 30% erhöht. Seite 36 Man kann auch den intravenösen Glucosetoleranztest durchführen. Dabei werden 0.5g Glucose pro kg Körpergewicht injiziert, dann die Insulinsekretion gemessen. Dabei ist nur die erste Phase bedeutsam. Fällt sie vollständig aus, oder liegt sie unter der 10. Perzentile der Normalbevölkerung, so besteht für die betreffende Person eine Wahrscheinlichkeit von 80%, in den nächsten 5 Jahren Typ 1 zu bekommen, da die B-Zellen kurz vor der Zerstörung stehen. In diesem Fall kann man beispielsweise Nicotinamid anwenden; es wirkt als Radikalfänger und schont so die B-Zellen. 2.3 Typen des Diabetes mellitus WHO: Klassifikation Diabetes (1985) 1. 1.1 1.2 1.2.1 1.2.2 1.3 1.4 2. 3. Manifester DM IDDM (Typ 1, Insulin-Dependent DM, insulinpflichtig) NIDDM (Typ 2, Non-Insulin-Dependent DM, nicht insulinpflichtig) ohne Adipositas mit Adipositas MRDM (Malnutrition Related DM) weitere Formen (z.B. chronische Pankreatitis) IGT (pathologische Glucosetoleranz) GDM (Gestationsdiabetes, Schwangerschaftsdiabetes) Seite 37 Inselzellantikörper, zelluläre Immunphänomene Die Linie stellt den zunehmenden Verlust an Inselzellmasse dar. Die B-Zellen werden durch eine unbekannte Initialläsion (z.B. Virus) geschädigt. Eine Autoimmunerkrankung entsteht allerdings erst, wenn die Umweltbedingungen und die genetischen Voraussetzungen gegeben sind. Zu einer klinischen Manifestation kommt es erst, wenn >90% der Inselzellen zerstört sind; ab hier wird Insulin gegeben. Dies führt zu einer individuellen Erholung (Remission), die ab und zu ausreichen kann, um den Stoffwechsel für einige Zeit aufrecht zu erhalten, so daß keine Insulingaben nötig sind. Typ 1 Diabetes (10-15% der DM-Fälle in USA/Europa) Typ 1 ist der typische Insulinmangeldiabetes; früher sprach man von juvenilem Diabetes, da er bereits im Jugendalter (bis ca.25 Jahre) auftritt. Er entsteht in den meisten Fällen durch Autoimmunprozesse. Dabei sind sowohl humorale (Antikörper) als auch zelluläre (zytotoxische Lymphocyten, Makrophagen) Faktoren beteiligt. Ursache sind wahrscheinlich Slow Virus Infekte, bei denen die für die B-Zellen toxischen Lymphocyten nicht ausgeschaltet wurden. Diese Art von Diabetes tritt auch als polyendokrines Syndrom auf, d.h. auch Nebenniere und Schilddrüse werden geschädigt. Seite 38 Pathogenese des Typ 1 Diabetes Die MHC ( Major Histocompatibility Complex) sind Moleküle auf der Membran von Leukocyten, die zwischen körpereigen und -fremd unterscheiden. Davon gibt es 3 Klassen, die alle auf dem Chromosom 6 lokalisiert sind. Bestimmte MHC-Gene sind mit einer erhöhten Empfänglichkeit für das Auftreten eines Typ 1 Diabetes assoziiert. Dies betrifft besonders die Gene der Klasse II. Es besteht eine bevorzugte Verbindung des Diabetesrisikos mit den Allelen DR3 und/oder DR4. Die Assoziation von DR4 mit dem Allel DQB1 und DQA1 weist eine besonders enge Beziehung zum Typ 1 Diabetes auf. Bei der durch das Gen DQB1 codierten DQ-ß-Kette ist das in Position 57 befindliche Aspartat gegen neutrale Aminosäuren ausgetauscht, in der DQ- -Kette befindet sich in Position 52 Arginin. Entscheidend hierbei ist also die Konformationsänderung des von den DQ-a- und DQ-ß-Ketten gebildeten Klasse-II-MHCProteins. Beim Typ 1 handelt es sich um eine Autoimmunerkrankung. Die im Blut auftretenden Inselzellantikörper sind ein guter Marker für die Autoimmunität, aber die eigentlich schädigenden Zellen sind Makrophagen und T-Lymphocyten. Zuerst tauchen in den Langerhansschen Inseln Makrophagen auf, vermutlich vermittelt durch vermehrte Expression von MHC Klasse II-Molekülen auf den Endothelzellen und ICAM-1 (=Adhäsionsprotein, wird bei allen Entzündungsvorgängen eingesetzt). Diese prozessieren phagocytierte B-Zell-Eiweiße (putatives Antigen), präsentieren sie auf ihrer Oberfläche den T-Helferzellen und aktivieren sie so. Die T-Helferzellen ihrerseits geben die Information weiter an cytotoxische T-Zellen, die die endokrinen B-Zellen nun angreifen. Außerdem aktivieren die T-Helferzellen die B-Lymphocyten, die Inselzellantikörper bilden (nicht zelltoxisch). Es gibt zwei Formen der T-Helferzellen (Th’s): Einerseits die Th1 (diese Zellen könnte man als bad guys bezeichnen); sie produzieren IFN (Interferon g) und IL2 (Interleukin 2), die die cytotoxischen T-Zellen stimulieren und so letztendlich zur Zerstörung der insulinproduzierenden B-Zellen führen.IFN ist zusammen mit IL1 direkt zytotoxisch für die B-Zellen. IFN und IL2 hemmen außerdem die Th2 (die die good guys darstellen). Sie produzieren IL4 und IL10, die die Autoimmunprozesse und die cytotoxischen Zellen hemmen. Seite 39 Typ 2 Diabetes Die Vorstufe des Typ 2 ist das metabolische Syndrom, das gemeinsame Auftreten von • Hypertonie • Diabetes • Übergewicht • Hyperlipidämie Die gemeinsame Ursache ist eine Insulinresistenz. Insulin beeinflußt die Na/K-Pumpe in der Niere,und bewirkt so eine vermehrte Natrium-Rückresorption, die das Blutvolumen und damit den Blutdruck erhöht. Da Insulin ein anaboles Hormon darstellt, führt ein hoher InsulinSpiegel direkt zu Übergewicht und Hyperlipidämie. Typ 2-Diabetes ist also im wesentlichen genetisch bedingt. Hier finden wir zwei Erscheinungsformen: Der Typ 2a tritt vorwiegend bei Normal- und Untergewichtigen auf. Es kommt dabei zu einer Sekretionsstarre. 80% der Typ 2-Patienten leiden jedoch unter dem Typ 2b. Die Hauptursache für diesen Diabetes ist eine Adipositas (Fettsucht). Pathogenese Typ 2a (ca.15% der DM-Fälle in USA/Europa) Hier ist der grundlegende Mechanismus eine gestörte Insulinsekretion, d.h. die B-Zellen sind noch vorhanden. Glucose, bestimmte Aminosäuren und gastrointestinale Hormone stimulieren das Pankreas. Der Typ 2a stellt eine Blindheit der B-Zellen gegenüber Glucose dar, d.h. sie reagieren nur noch auf andere Stimuli. Dadurch wird weniger Insulin und sehr viel Glucagon gebildet (da Insulin kaum noch seinen hemmenden Einfluß auf die Glucagon-Bildung ausüben kann). Die Langerhansschen Inseln haben eine spezifische Art der Durchblutung: Das Blut tritt von außen ein, wandert auf dem schnellsten Wege zum Kern und zweigt sich von hier nach außen auf. Da die A-Zellen am äußeren Rand der Inseln liegen und die B-Zellen innen, werden die B-Zellen also zuerst mit Blut versorgt. Dabei nimmt das Blut Insulin auf und gelangt im nächsten Schritt zu den A-Zellen. Hier regelt der Insulin-Gehalt des Blutes die Glucagon-Sekretion. Durch die hohe Glucagon-Sekretion wird die Glucose-Produktion der Leber angekurbelt, doch die peripheren Gewebe werden trotzdem nicht ausreichend versorgt, da Insulin für die Aufnahme in die Zelle fehlt. Möglicherweise handelt es sich beim Typ 2a zusätzlich auch um eine Störung im ZNS. Das Inselorgan steht unter Kontrolle des N.Vagus. Dieser könnte die Hyperglucagonämie verstärken. Pathogenese Typ 2b (ca.50% der DM-Fälle in USA/Europa) Beim Typ 2b handelt es sich um eine Insulinresistenz. Deshalb verwendet man hierbei die Hyperinsulinämie als Marker. Der Insulinrezeptor besteht aus 2a- und 2ß- Untereinheiten; die ß-Einheiten sind in der Membran verankert, die a-Einheiten ihnen über Disulfidbrücken angeschlossen. Seite 40 Die intrazellulären Anteile haben Tyrosinkinaseaktivität, d.h. sie können Phosphat von ATP auf die OH-Gruppe von Tyrosinresten in Proteinen übertragen. Dies ist nötig, damit Insulin seine Wirkung ausüben kann. Die Phosphorylierung setzt Mediatoren (Serin-ThreoninKinasen) in Gang, die für die Bildung von Glucose-Kanälen und deren Translokation verantwortlich sind. Bei den Postrezeptorvorgängen können die verschiedensten Störungen auftreten, die dann den Typ 2b ausmachen. Seite 41 Bei Hyperglycämie kann es zu einer Herabsetzung der Insulinrezeptoren kommen. Das ist darauf zurückzuführen, daß zunächst aufgrund eines hohen Blutglucosespiegels vermehrt Insulin produziert wird. Dadurch entsteht ein Mißverhältnis zwischen Insulin und den Rezeptoren. Da die Zelle nur begrenzt Glucose aufnehmen und umsetzen kann, wird die Zahl der Insulinrezeptoren herabgesetzt. Diesen Vorgang bezeichnet man als DownRegulation. Sie führt zunächst zu einer weiteren Vermehrung der Insulinproduktion, bis das Pankreas mit der Insulinproduktion überfordert ist und diese herabsetzt. Der Gestationsdiabetes stellt meist eine frühe Form des Typ 2 dar. Während der Schwangerschaft kommt es zu einer vermehrten Produktion von Hormonen, die die Insulinwirkung hemmen (HPL, Östrogene, Progesteron, Glucocorticoide...). Charakteristisch beim Gestationsdiabetes ist, daß die Symptome nach der Entbindung zunächst einmal verschwinden. Das Risiko der Mutter, später Diabetes zu bekommen, ist aber deutlich erhöht. Bei Kaukasiern ist das Risiko gering; innerhalb der nächsten 5 Jahre nach der Entbindung tritt er nur bei 10% erneut auf; in Asien ist er kaum verbreitet. Die gestörte Glucosetoleranz besteht bei 3-5% der Bevölkerung der westlichen Industrieländer und ist mit einem erhöhten Diabetes-Risiko assoziiert. Sie besteht, wenn 2 Stunden nach einer oralen Belastung mit 75g Glucose ein Blutzuckerspiegel von 140-200 mg/dl festzustellen ist. Wird in diesem Fall das Verhalten nicht geändert, so besteht eine Wahrscheinlichkeit von 30%, Typ 2 zu bekommen. Dieses Risiko steigt allerdings bei Vorliegen anderer Risikofaktoren. Für den MRDM sind verschiedene Nahrungstoxine (z.B.die Nitrosamine im Maniok), die die B-Zellen angreifen, sowie Eiweißmangel verantwortlich. Die chronische Pankreatitis wird z.B. durch zu hohen Alkoholkonsum hervorgerufen. Dabei wird zuerst das exokrine, später auch das endokrine Pankreas geschädigt. Seite 42 Normalzustand Typ-2a-Diabetes Seite 43 Substratfluß Im nüchternen Zustand werden 70% der Glucose von nicht-insulinabhängigen Geweben aufgenommen. Die Abbildungen 1-4 zeigen den Substratfluß während der Glucoseabsorption, Postabsorption und bei postabsorptiver Bewegung (S.25,Skript). Die nach der Nahrungsaufnahme absorbierte Glucose wird teilweise direkt als Energielieferant genutzt, zum Teil jedoch in Glycogen umgewandelt. Während die im ZNS direkt zugeführte Glucose unter aeroben Bedingungen abgebaut wird, erfolgt der Abbau in den Erythrocyten anaerob. Dabei entsteht Lactat, das über den Blutkreislauf in die Leber gelangt und dort zur Gluconeogenese genutzt werden kann. In der Leber und in der Muskulatur wird die Glucose in die speicherfähige Form Glycogen überführt. Glycogen ist im Gegensatz zu Glucose nicht osmotisch aktiv und entzieht dem Körper somit kein Wasser. Die überschüssige Glucose wird in Triglyceride umgebaut und im Fettgewebe gespeichert. Abb.2 zeigt den Substratfluß bei kurzzeitigem Hungerzustand, wie er z.B. über Nacht eintritt. In einer solchen Phase werden Gehirn und Erythrocyten von der Leber mit Glucose (aus der Gluconeogenese) versorgt. Als Ausgangssubstrat dienen dabei das in der Leber gespeicherte Glycogen, Aminosäuren, Glycerol und Lactat. Das Muskelgewebe setzt in dieser Zeit Aminosäuren frei. Der Energiebedarf von Leber und Muskulatur wird durch Fettsäuren gedeckt, die aus den Triglyceriden des Fettgewebes stammen. Bei der Verbrennung der Fettsäuren entsteht, wie beim aeroben Glucoseabbau, CO2. Seite 44 In Abb.3 ist der Substratfluß nach längerfristiger Nahrungskarenz dargestellt. Da nach längerem Hungern die Glycogenspeicher der Leber aufgebraucht sind (nach etwa 1-2 Tagen), kann nur noch über die Verstoffwechselung von Aminosäuren, Glycerol und Lactat Glucose synthetisiert werden. Zusätzlich kommt es zur Glucosefreisetzung durch die Niere. Die gebildete Glucose reicht jedoch nicht zur vollständigen Energieversorgung von Erythrocyten und ZNS (vollständig glucoseabhängig) aus. Das Gehirn stellt sich unter diesen Bedingungen teilweise auf die Verstoffwechselung von Ketonkörpern ein. Außerdem wird die Seite 45 Glucose im Gehirn vermehrt anaerob abgebaut, was zur Freisetzung von Lactat führt und der Leber als Substrat zur Gluconeogenese dient. Bei ausreichender Triglyceridspeicherung kann der Mensch bis zu 50 Tage ohne Nahrungszufuhr überleben. Kommt es während des kurzzeitigen Hungerzustandes zu körperlicher Aktivität (Abb.4), setzt verstärkt die Verstoffwechselung von Glycogen, Aminosäuren, Glycerol und Lactat ein. In diesem Fall wird auch das Muskelglycogen freigesetzt und vermehrt Fettsäuren im Muskel umgesetzt. Muskelglycogen wird nur für den Eigenbedarf des Muskels zur Verfügung gestellt. Die Glucosezufuhr für Erythrocyten und ZNS sowie der Fettsäureabbau in der Leber entsprechen den Stofwechselvorgängen des normalen kurzfristigen Hungerzustandes. Im Muskel kommt es in erster Linie zum aeroben Abbau der Energieträger. Teilweise erfolgt der Abbau der Glucose jedoch anaerob, was wiederum zur Bildung von Lactat führt. Glucose ist der einzige Brennstoff, der von der Skelettmuskulatur unter anaeroben Bedingungen genutzt werden kann. 2.4 Diagnose des Diabetes mellitus Harnglucosebestimmung Kommt es im Serum zu Glucosewerten von 150-180 mg/dl (9,4-10,0 mmol/l) wird die Nierenschwelle überschritten. Es kommt dann zur Ausscheidung von Glucose über den Harn (Glucosurie). Die Glucosurie tritt dann ein, wenn das Ultrafiltrat des Glomerulum mehr Glucose enthält als im Tubulussystem rückresorbiert werden kann (max. Rückresorption = 350 mg Glucose/min.). Die Harnzuckerbestimmung wird nur angewandt, um erhöhte Zuckerwerte festzustellen. Erhöhte Harnglucosekonzentrationen können auch Folge eines renalen Diabetes sein. Darunter versteht man eine Störung der Tubulusrückresorption, die meist mit anderen Störungen einhergeht. Seite 46 HbA1c Die Abb.22 zeigt die Varianten des humanen Hämoglobins und die prozentualen Anteile der Varianten beim Gesunden. Diese Abbildung stellt die Biosyntheseschritte des HbA1c dar. Die Bindung ist irreversibel, sobald die Aldiminform in die Ketoaminform übergegangen ist (AMADORI- Umlagerung). Da die Bildung des HbA1c mit der Blutglucosekonzentration korreliert, ist sie ein genauerer Parameter zur Diagnose der diabetischen Stoffwechsellage als die Blutglucosekonzentration und das Auftreten von Glucose im Harn. Diese beiden geben lediglich Auskunft darüber, ob eine erhöhte Belastung in den letzten Stunden vorlag oder nicht. Sie vermitteln ungenaue Anhaltsdaten. Die Bestimmung der HbA1c-Werte erlaubt eine retrospektive Beurteilung der diabetischen Stoffwechsellage während der letzten 4-6 Wochen. Abbildung: Hb-Auftrennung mittels HPLC HbA1c <7% gut oder < Mittelwert + 2 SD (Standardabw.) 7-8% akzeptabel < Mittelwert + 4 SD >8% inakzeptabel > Mittelwert + 4 SD (für TypII-Diabetes nach Gries, 1985) mittlerer Blutzucker = 33,3 x HbA1c - 86 (lineare Regression nach Nathan, 1984) Seite 47 2.5 Diabetische Spätschäden Weniger als 1% der Diabetiker sterben an der primären Stoffwechselstörung und durch Insulinmangel verursachte Folgesymptome. Den Krankheitsverlauf bestimmen vor allem vaskuläre (d.h.die Blutgefäße betreffende) Komplikationen. Ihr Auftreten und der Komplikationsgrad sind u.a. von der Dauer des Diabetes und der Stoffwechsellage des Diabetikers abhängig. Selbst bei guter Einstellung lassen sich Angiopathien (arteriosklerotische Gefäßveränderungen) nicht völlig vermeiden. Betreffen sie die größeren Gefäße, spricht man von einer Makroangiopathie. Bei der Mikroangiopahtie treten die Schäden v.a. in den Endabschnitten, also den kleineren Gefäßen, auf. Makroangiopathie Die Makroangiopathie entspricht dem morphologischen und klinischen Krankheitsbild des Nichtdiabetikers. Sie tritt beim Diabetiker jedoch häufiger und zumeist früher auf. Die sekundäre Hyperlipoproteinamie ist beim unbehandelten Diabetes mellitus durch 2 Faktoren bedingt: 1. Die Aktivität der Fettgewebs-Lipoprotein-Lipase ist erniedrigt. Dieses Enzym wirkt v.a. in den Kapillarendothelzellen und an der Plasmamembran der extrahepatischen Gewebe. Es katalysiert die Spaltung von Triglyceriden. Die Erniedrigung der Aktivität wird vermutlich durch den Insulinmangel ausgelöst, da sie sich bei Gabe von Sulfonylharnstoff oder Insulin normalisiert. 2. Aufgrund des herrschenden Energiemangels kommt es beim Diabetes mellitus zur exzessiven Fettgewebsmobilisation, so daß die Leber ständig unter einem erhöhten Fettsäureangebot steht. Dies begünstigt die Triglyceridsynthese und Abgabe von VLDL in das Blut. Mikroangiopathie Die Mikroangiopathie ist gekennzeichnet durch eine Verdickung v.a. der Basalmembran und der Intima. Die Verdickung verlängert die Diffusionsstrecke zwischen Erythrocyten und Körperzellen, so daß der O2-Transport erschwert ist. Sie manifestiert sich überwiegend an der Netzhaut, der Niere und peripheren Nerven. (Komplikationen treten nicht in den Seite 48 Geweben mit Insulin-abhängigem Glucose-Transport auf.) Häufige Folge nach langjährigem Verlauf ist die diabetische Nephropathie, die durch eine Albuminurie gekennzeichnet ist. Parallel dazu entwickelt sich meist eine Retinopathie. Dabei werden folgende Stadien durchlaufen: 1. Venenerweiterung im Hintergrund des Auges, auch Blutungen 2. Harte Exudate = Narben entstehen 3. Proliferative Retinopathie = Neubildung von Gefäßen, um die minderdurchbluteten Gebiete zu versorgen. Die neuen Gefäße sind meist instabil und reißen leicht. 4. Glaskörperblutung, Blindheit Der Mechanismus der Entstehung der Mikroangiopathie ist noch nicht vollständig geklärt. Eine zenrale Bedeutung kommt dabei einer gesteigerten nichtenzymatischen Glycosylierung von Proteinen zu. Dabei reagiert der reduzierende Zucker Glucose mit freien Aminogruppen (z.B. des Lysins). Die entstehenden AGE’s (Advanced Glycosylation End Products) schädigen den Körper auf vielfältige Weise. V.a. intrazellulär treten Querverbindungen von Matrixproteinen auf, was Funktionsänderungen zur Folge hat. Typ IV Kollagen und Laminin beispielsweise können nicht weiter abgebaut werden und akkumulieren im Körper. • Glycosyliertes Hämoglobin zeigt eine erhöhte 02-Bindungs- und verminderte O2Dissoziationsfähigkeit; dadurch sind bei Diabetikern Heilungsvorgänge verlangsamt • Glycosylierte LDL werden schlechter abgebaut als normale • Glykierung von Albumin erhöht seine Durchtrittsrate durch die Glomerulummembran • Glykierung von Faktor VIII erhöht die Plättchenaggregationsneigung • Bei Glykierung von Leuko- und Lymphocytenmembranen werden Immunreaktionen gestört, Glykierung von Immunglobulinen vermindert deren Bindungsfähigkeit • Glucose wird durch Aldosereduktase in Sorbitol umgewandelt. So kommt es zu einer Anhäufung von Sorbit in Zellen, deren Membran für Glucose frei durchlässig ist. Die Seite 49 erhöhte Konzentration führt über seinen osmotischen Effekt zu Verquellungen (resultiert z.B. in einem diabetischen Katarakt) • In Glomerulus, Retina und Nerv ist die Sorbitolakkumulation mit einer Abnahme des zellulären Myoinositols assoziiert. Myoinositol reguliert die Na/K-ATPase, die wiederum das Zellmembranpotential aufrechterhält (kann zu eingeschränkter Nervenleitfähigkeit führen) • Kollagen-gebundene AGE’s inaktivieren Stickoxid, das einen entspannenden Einfluß auf glatte Muskelzellen hat und daher blutdrucksenkend wirkt Neuropathie Die häufigste Komplikation ist die diabetische Neuropathie; sie tritt bei 60-90% der Diabetiker auf. Es gibt 3 Formen , wovon 2 auf eine Mikroangiopathie zurückzuführen sind. 1) Symmetrische sensible Neuropathie: Hierbei tritt in Ruhe ein vermindertes Schmerz-, Temperatur- und Berührungsempfinden auf. Es kommt zu “Ameisenlaufen” an den Beinen, meist den Unterschenkeln. 2) Neuropathie des autonomen Nervensystems: Diese Neuropathie ist weniger häufig und kann in vielen unterschiedlichen Formen auftreten. • Herz-Kreislauf-Symptome, die Herzfrequenz ist variabilitätsvermindert, es kann zu Narkosezwischenfällen kommen • Gastrointestinale autonome Neuropathie: Atonie der glatten Muskulatur des Darmbereichs, postprandiale Hyperglycämie, Verstopfung • Urogenitale Neuropathie: führt bei Männern zu Impotenz, bei Frauen zur Überlaufblase (die Muskeln sind unterversorgt und daher zu schwach, Urinretention ist begünstigend für Infektionen) • Autonome Neuropathie der Sudomotorik: Sudomotorneurone innervieren die Schweißdrüsen; verstärkte Schweißsekretion in der oberen Körperhälfte, in der unteren dagegen eine verminderte; Pupilleninnervation beeinträchtigt, daher schlechte Einstellung auf unterschiedliche Lichtverhältnisse 3) Diabetische Mononeuropathie: nur ein Nerv ist betroffen, meist ein Gesichtsnerv; es handelt sich um eine Makroangiopathie des versorgenden Gefäßes Seite 50 2.6 Koma bei Diabetes Beim Diabetes mellitus kann es verursacht durch Unter- oder Überzuckerung zu Stoffwechselentgleisungen kommen. Eine Unterzuckerung führt zum hypoglycämischen Schock (Coma hypoglycämicum), eine Überzuckerung zum Coma diabeticum. Eine Hypoglycämie kann durch Steigerung der Insulindosis ohne Anpassung der Kohlenhydratzufuhr, durch Reduzierung der Kohlenhydratzufuhr ohne Anpassung der Insulindosis oder durch gesteigerte körperliche Aktivität ohne vorherige Anpassung der Kohlenhydrataufnahme ausgelöst werden. Schockgefahr besteht, wenn die Blutglucosekonzentration unter 60 mg% (=3,3 mmol) sinkt. Der Glucosemangel wirkt sich insbesondere auf das Nervensystem aus. Kommt es zum Auftreten von Voranzeichen eines Schocks (Herzklopfen, Reizbarkeit..), kann durch Einnahme von Zucker oder Traubenzucker vorgebeugt werden. Befindet sich der Patient bereits im Koma, so muß die Kohlenhydratzufuhr intravenös erfolgen. Beim Coma diabeticum unterscheidet man zwischen • ketoazidotischem • nicht azidotischem (hyperosmolarem) • lactazidotischem Koma. Das ketoazidotische Koma basiert auf einem absoluten Insulinmangel und tritt deshalb fast nur bei Typ 1 Diabetikern auf. Es kommt zur verstärkten Lipolyse und somit zur Ketogenese. Bei normaler Stoffwechsellage würden die entstehenden Ketonkörper dem Fettsäureabbau zugeführt. Jetzt wird aber Acetacetat in die Blutgefäße abgegeben und führt zur metabolischen Azidose. Der Körper wirkt dieser Azidose auf zweierlei Weise entgegen: 1. Acetacetat wird zu Aceton und CO2 abgebaut. CO2 wird vermehrt abgeatmet (Hyperventilation) 2. Durch Abgabe von Na+- und K+-Ionen wird die Ionenbilanz ausgeglichen. Es kommt zur Diurese, was einen Wasser- und Ionenverlust bedeutet. Das nicht azidotische, hyperosmolare Koma tritt vorwiegend beim Typ 2 auf. Es ist charakterisiert durch stark erhöhte Blutglucosewerte (>600mg/dl). Dieser Anstieg bewirkt eine Glucosurie, da die Nierenschwelle überschritten wird; gleichzeitig wird vermehrt Wasser ausgeschieden. Zusätzlich kommt es durch den erhöhten osmotischen Druck zur Dehydratation der Zellen, was wiederum zur Hypervolämie (Plasmaexpansion) führt. Hypervolämie hemmt die Aldosteronausschüttung. Da Aldosteron die Na-Rückresorption in Seite 51 der Niere induziert, kommt es unter Hemmung dieses Hormons zu einem Natriumverlust. Glucosurie und NaCl-Verlust wirken sich wiederum osmotisch auf den Wasserhaushalt aus; dem Körper wird durch Polyurie Wasser entzogen; der Hypervolämie folgt eine Hypovolämie. Bei fortschreitender Zelldehydratation kommt es zum Koma. Ursache des laktazidotischen Koma ist fast immer eine Zirkulationsstörung im Bereich der peripheren Gewebe und wird vorwiegend durch Kreislaufinsuffizienz ausgelöst. Es wird außerdem durch die Behandlung mit Biguaniden begünstigt, die Zusammenhänge sind jedoch nicht geklärt. Beim laktazidotischen Koma sinkt der Blut-pH unter 7,25 durch Erhöhung des Laktat-Spiegels, ohne daß die Glucosekonzentration erhöht ist. Die Gefahr eines Coma Diabeticum ist bei Streßzuständen, wie starke körperliche Anstrengung, Infektionskrankheiten, operativen Eingriffen etc besonders groß. Da die unmittelbare Folge des Koma die Dehydratation ist, besteht die Behandlung in Wasser- und Elektrolytzufuhr, sowie in der Gabe von Insulin zur Förderung des Glucoseabbaus. Außerdem führt man Bicarbonate zur Normalisierung des pH-Wertes zu. 2.7 Therapieformen Diät • KH-Anteil >50%, möglichst langsam resorbierbar • der Patient muß lernen, die Broteinheiten und seinen Insulinbedarf pro BE zu schätzen (1BE = 12g KH) Unterstützung von Muskelarbeit, Ausdauersport (führt zu Up-Regulation; allein dadurch kann der Insulinbedarf bis zu 80% absinken!) Seite 52 Tabletten • Sulfonylharnstoffe zur Förderung der Insulinsekretion (Typ 2a) • Biguanide zur Steigerung der nicht Insulin-abhängigen KH-Aufnahme (Typ 2b) in Muskel und Fettgewebe • Acarbose zur Verzögerung der enteralen KH-Aufnahme = Hemmer der a-Glucosidase Insulin 2.8 Der diabetische Fuß Seite 53 3 Protein- und Aminosäurestoffwechsel Mit der Nahrung aufgenommene Proteine werden in Aminosäuren gespalten, die durch die Mucosazellen ins Pfortaderblut absorbiert werden und so in alle Zellen gelangen, hauptsächlich in Leber und Niere. Sie sind die Hauptorgane des Proteinstoffwechsels. Die metabolischen Reaktionen der AS können grob in drei Kategorien eingeteilt werden: 1. Freie AS aus dem Pool werden zur Bildung von Körpereiweiß verwandt, wobei gleichzeitig Körpereiweiß wieder zu AS abgebaut wird. 2. Weitere freie AS aus dem Pool werden oxidativ desaminiert oder transaminiert und so in a-Ketosäuren überführt. Durch oxidative Decarboxylierug entstehen daraus FettsäureCoA-Thioester, die entweder zur Energiegewinnung zu CO2 und H2O oxidiert oder in Ketonkörper, Fettsäuren oder Glucose umgewandelt werden. Die NH2-Gruppe bzw. das NH3 (freigesetzt durch die oxidative Desaminierung) muß entgiftet werden (neurotoxisch bei erhöhter Konzentration), indem es als Harnstoff (in der Leber synthetisiert aus NH3 und CO2) über den Urin ausgeschieden wird. (Dies geschieht mit 90% des NH3. Die restlichen 10% gelangen direkt als NH4+ in den Urin.) 3. Einige AS werden für die Synthese anderer stickstoffhaltiger Verbindungen, wie Purinbasen, Kreatin, Adrenalin u.a. verwendet, die letztlich auch ausgeschieden werden. Zusätzlich werden nicht essentielle AS mit Hilfe der Aminogruppen anderer AS synthetisiert. 3.1 Protein-Umsatz Mit gemischter Nahrung werden in Deutschland ca.100 g Nahrungsprotein (=16 g N) aufgenommen (die WHO-Empfehlung liegt bei 0,75 g/kg KG), was 125 g AS entspricht. Die Gewichtszunahme resultiert aus der H2O-Anlagerung bei der Hydrolyse im Darm. Sie gelangen in den AS-Pool, in dem sich auch die freigesetzten AS aus dem Abbau des Körpereiweißes befinden. Pro Tag werden ca. 400 g Körpereiweiß abgebaut. Im Idealfall findet genausoviel Neusynthese aus dem AS-Pool wie Abbau statt; dann befindet sich der Körper im Stickstoff-Gleichgewicht. Die restlichen 125 g AS werden umgebaut in Seite 54 Glucose (glucoplastische AS) oder Ketonkörper (ketoplastische AS) oder werden abgebaut (s.o.). Der Ammoniak wird hauptsächlich als Harnstoff entgiftet; knapp 10% des NH3 werden direkt über die Niere als NH4+ eliminiert. 3.2 Eiweiß-/N-Bilanz NB = Normalbilanz bei einer bei uns üblichen Zufuhr EM = endogenes Minimum BM = Bilanzminimum Der Stickstoff der Nahrung ist fast vollständig Proteinstickstoff. Nur zu einem geringen Teil stammt er aus Nucleinsäuren, Aminozuckern etc. Deshalb wird zur Beurteilung des Proteinstoffwechsels anstelle der kaum meßbaren Proteinbilanz die befriedigend meßbare N-Bilanz herangezogen. Da ca.1/6 des Proteins aus N aufgebaut ist, entsprechen 100 g Protein 16 g N. Durch die Stickstoffbilanz kann ermittelt werden, inwieweit der Proteinbedarf des Körpers gedeckt ist: aufgenommene Menge - ausgeschiedene Menge im Kot = N-Bilanz Die N-Ausscheidung mit dem Fäces beträgt beim gesunden Erwachsenen maximal 2g/24 h. Sie ist vergleichsweise gering, ist meßtechnich schwer zu bestimmen und wird daher vernachlässigt. Ist die Zufuhr an Eiweiß bzw.N größer als der Abbau von Eiweiß bzw. die Ausscheidung von N, so bedeutet dies Anabolismus, also Wachstum. Eiweiß wird im Körper neu gebildet. Dieser Zustand liegt z.B. bei Kindern vor, im Erwachsenenalter kaum (nur in Genesungsphasen). Ist die Zufuhr geringer als der Abbau, liegt ein Katabolismus vor, der längerfristig zur Kachexie (Auszehrung) führt. 2-3 kg Protein kann ohne Gefahr bei Hunger abgebaut werden, darüber ist es gefährlich. Im Bereich oberhalb von 4g entsprechen sich Zufuhr und Ausscheidung. Bei dem Bilanzminimum von 4g (24g Protein) ist die Bilanz minimal noch erhalten. Bei einer geringeren Zufuhr übersteigen Abbau und Ausscheidung die Zufuhr: die Zufuhr kann auf 0 absinken, die Ausscheidung aber nicht; sie bleibt ungefähr bei 3g N/24 h. 3.3 Eiweiß-Restriktion Eine gezielte Reduktion der Eiweißzufuhr ist nötig, wenn AS-Stoffwechsel oder Harnstoffsynthese gestört sind oder die Ausscheidung harnpflichtiger Substanzen vermindert ist. Ein Anstieg des Ammoniak-Spiegels sollte verhindert werden, da er toxisch wirkt. Die Eiweiß-Restriktion kann das Fortschreiten des Krankheitsprozesses erheblich verzögern. Sie wird angewandt bei 1. Niereninsuffizienz 2. Störung der Harnstoffsynthese a) erworben: z.B. durch Leberzirrhose b) genetisch: geringer als 1:10.000 3. Störung des AS-Stoffwechsels: erblich, relativ selten (ca. 1:5 000 - 1:10 000) Seite 55 Bei der eiweißarmen Diät sind drei Punkte zu beachten: 1. Das Bilanz-Minimum darf nicht unterschritten werden 2. Der Bedarf an essentiellen AS muß trotzdem optimal gedeckt sein 3. Eventuell zusätzliche Zufuhr von a-Ketosäuren (da sie Ammoniak aufnehmen können, so daß die Entgiftungsmechanismen entlastet werden) 3.4 Verteilung der Körperproteine intrazellulär interstitiell intravasal Urin % Gesamtprotein 94 2 4 - %H2O 66 27 7 - Prot.Konz (g/dl) 15-25 0,2-2 6-8 <10mg/dl 3.5 Funktionen der Proteine 1. als Struktur 2. zur Bewegung 3. Katalyse (als Enzyme) zellulär Tubulin, Aktin u. a. -> Cytoskelettbildung Aktin, Myosin -> an Muskelkontraktionen beteiligt (in allen Zellen Bewegungen, z.B.Cytoplasmaströmungen möglich, da Aktin und Myosin überall vorhanden sind) a) strukturgebunden (d.h. an Membranen verankert, z.B. bei der Atmungskette) b) löslich (-> größerer Anteil der Enzyme, z.B. bei Hydrolyse, AS-Abbau) 4. Transport Membranenzyme mit Transportfunktion Hämoglobin 5. als Puffer Hämoglobin (puffert in Erythrocyten die bei der COBindung freiwerdenden HIonen ab) - 6. Infektabwehr extrazellulär Kollagen, Elastin, Laminin -> liegen in der Bindegewebsgrundsubstanz zur Strukturerhaltung - a) strukturgebunden (d.h. an Lipoproteine assoziiert/LCAT) b) löslich (-> Sekretenzyme zur Blutgerinnung, in der Leber gebildet -> Überlauf: Zellen gehen zugrunde, geben Enzyme ins Plasma ab, z.B. CK, GOT, GPT, Amylase) Albumin (transportiert FS, Schilddrüsenhormone...) Transferrin (Eisen) Transcortin (Steroidhormone der NNR) Haptoglobin Albumin (bindet und neuteralisiert Protonen) spezifisch: Immunglobuline unspezifisch: Komplementfaktoren (gelöste Stoffe Akut-Phase-Proteine: a-1Antitrypsin, Haptoglobin, Coeruloplasmin, C-reaktives Protein Seite 56 7. Signalvermittler Hormon-Rezeptoren Coeruloplasmin, C-reaktives Protein Peptidhormone (z.B. Insulin, wirkt aber nur, wenn zellulär andere Proteine als Rezeptoren vorhanden sind) 3.6 Störungen des Proteinstoffwechsels A: Protein-Mangel (quantitativ) Ursachen: 1. verminderte Zufuhr 2. gestiegene Verluste a) Niere: bei Proteinurie können bis zu 40g ausgeschieden werden b) verminderte Resorption bei Darmerkrankungen c) Wunden geben ein sehr proteinreiches Sekret ab 3. gestiegener Abbau (extreme Stoffwechselsteigerung: T3, T4, Katecholamine) Folgen: 1) kurzfristig: endogener Proteinabbau sinkt auf 25 g/Tag 2) langfristig:Verlust von > 1-2 kg von 6-12 kg Gesamtkörpereiweiß führt zu folgenden Krankheitssymptomen: 2.1) beim TRANSPORT: Wasserretention in Gewebe = Ödeme in Körperhöhlen = Ascites 2.2) STRUKTUR, BEWEGUNG, KATALYSE Leber, Niere, Muskelabbau (führt zu Herzversagen) 2.3) ABWEHR Infektionsrisiko steigt B: Störung der Serumprotein-Zusammensetzung (qualitativ/quantitativ) Seite 57 Abbildung A Abbildung A zeigt die normale Konstellation der Serumeiweiße. Albumin ist ein monoklonales Protein und zeigt deshalb eine einheitliche Zacke. In allen anderen Fraktionen sind mehrere Proteinarten enthalten. Abbildung B zeigt die Veränderungen des Serumeiweißbildes bei Erkrankungen. Bei einer akuten Entzündung sind die a1 und a2 Fraktion erhöht; bei einer chronischen Entzündung fallen sie wieder ab, aber durch die produzierten Immunglobuline steigt die g-Fraktion. Bei einer Zirrhose ist die Albuminsynthese der Leber und damit auch die Konzentration im Serum verringert. Auch hier steigen die Globuline (Antikörper gegen Hepatitis-Virus). Das nephrotische Syndrom ist durch einen starken Abfall des Serumalbumins gekennzeichnet. Da das Filterorgan Niere gestört ist, werden auch Proteine mit dem Urin ausgeschieden. Albumin ist davon wegen seiner geringen Größe als erstes betroffen. a2Makroglobulin dagegen ist groß und kann die Niere nicht passieren, was zu einem Rückstau führt. Bei der monoklonalen Gammopathie zeigt sich eine spezifische Erhöhung der Ig’s. Es handelt sich hierbei um einen Tumor, bei dem eine einzelne Zelle entartet und sich vermehrt. Jede der Tochterzellen bildet das gleiche Ig monoklonals Immunglobulin). Seite 58 n). Abbildung B Seite 59 Bei a1-Antitrypsin-Mangel werden die Elastasen des Gewebes nicht genug blockiert. Dabei wird z.B. das elastische Gerüst der Lunge geschädigt, ein passives Zusammenziehen funktioniert nicht mehr. Morbus Wilson: Durch verminderte Cu-Ausscheidung in die Galle und den niedrigen Coeruloplasminspiegel kommt es zu Cu-Ablagerung in Leber und Hirn, was zu Leberzirrhose und Intelligenz-Defekten führt. Seite 60 C: Genetische Defekte im Abbau der AS 1.Phenylketonurie (Brenztraubensäureschwachsinn, PKU) Die Phenylketonurie tritt mit einer Häufigkeit von 1: 10.000 auf und ist damit die häufigste AS-Stoffwechselstörung. Sie wird rezessiv vererbt. Es handelt sich hierbei um einen Defekt der Metabolisierung des Phenylalanins. Die häufigste Störung ist die der PhenylalaninHydroxylase (1) in der Leber. Phenylalanin wird also nicht in Tyrosin umgewandelt und stattdessen von der Transaminase, die normalerweise Tyrosin umsetzt, als Substrat verwendet. Dabei entstehen Phenyl- und Hydroxyphenylacetat. Diese beiden Metaboliten wirken im Laufe von Monaten hirntoxisch. Ihr vermehrtes Auftreten im Urin hat der Krankheit ihren Namen gegeben. Durch diesen Defekt wird Tyrosin, woraus normalerweise Dopachinon und Katecholamine hergestellt werden, zur essentiellen AS. Dopachinon ist eine Vorstufe des Melanins, so daß die betroffenen Kinder eine verminderte Pigmentierung zeigen. Durch die fehlenden Neurotransmitter lassen die erlernten Eigenschaften des Säuglings nach (die Kinder sind “blond, blöd und blauäugig”); ab ca. einem Jahr zeigt er Krämpfe und stirbt ohne Behandlung im Alter von 5-6 Jahren. Eine frühzeitige Diagnose bei Neugeborenen ist wichtig, um sofort die Erährung umzustellen und die erst später in Erscheinung tretenden schweren Schäden zu vermeiden. Deshalb führt man den mikrobiologischen Hemmtest nach Guthrie durch . Hierbei handelt es sich um eine Bakterienkultur (Bacillus subtilis) auf einer Pappkarte, die durch Thienylalanin gehemmt wird. Durch Zugabe von phenylalaninreichem Blut beginnt sie zu wachsen. Je stärker die Präzipitationssichel ausgeprägt ist, desto höher ist die Konzentration des Phenylalanins im Blut. Der Test kann erst nach dem 5. Lebenstag durchgeführt werden, da der Fötus intrauterin von der Mutter versorgt wird und sie somit auch das beim Fötus anfallende Phenylalanin mit abbaut. Erst postnatal kann es daher akkumulieren. Ist der Guthrie-Hemmtest positiv, so muß nicht eine PKU vorliegen. Es kann sich auch um eine transiente PKU bzw. eine Hyperphenylalaninämie ohne PKU handeln, die auf eine zu hohe Zufuhr in den ersten Tagen zurückzuführen ist. Die Therapie besteht in einer Kost, in der alle essentiellen AS in ausreichender Menge enthalten sind, der Gehalt an Phenylalanin aber dem individuellen Bedarf des Kindes angepaßt ist. Es gibt spezielle Lebensmittel, aus denen Phenylalanin durch Adsorption an Kohle vollständig entfernt wurde und denen Fette, KH und Mineralstoffe zugesetzt wurden. 2. Ahornsirupkrankheit (Verzweigtketten-Ketonurie) Die verzweigtkettigen AS Valin, Leucin und Isoleucin werden im Gegensatz zu den übrigen essentiellen AS vorwiegend in peripheren Organen (Skelettmuskulatur, Niere...) abgebaut. Seite 61 Der erste Abbauschritt besteht in einer Transaminierung zu den entsprechenden Ketosäuren. Bei der Ahornsirupkrankheit liegt ein Defekt der dehydrierenden Decarboxylierung aller drei Ketosäuren vor (Schritt 2). Die Aminosäuren selbst bzw. ihre Metaboliten werden vermehrt mit dem Harn ausgeschieden und verleihen ihm einen Geruch von Ahornsirup (ähnlich Maggi). Außerdem tritt in Plasma und Urin die ungewöhnliche AS Alloisoleucin auf. Eine frühe Diagnose und entsprechende Diät sind auch hier ausgesprochen wichtig. Ohne Therapie kommt es zu schweren zentralnervösen Schädigungen, die bereits in den ersten Lebenswochen zum Tod führen können. Seite 62 Abbauwege der verzweigtkettigen Aminosäuren Seite 63 4. Verdauung Die Bilanz wird ermittelt durch eine Magensonde und 24-h-Sammelurin. Es ist wichtig, sie bei parenteraler Ernährung zu beachten. Bei Darmoperationen, Anus praeter etc muß eine andere Flüssigkeitsbilanz zugrunde gelegt werden. Je nach fehlendem oder erkranktem Organ muß berechnet werden, wieviel Flüssigkeit mehr oder weniger zugeführt werden sollte. 4.1 Definitionen Normaler Stuhlgang Frequenz: Gewicht: Konsistenz: 3 x täglich bis 3 x wöchentlich 100-200 g/d breiig, geformt Obstipation Frequenz: < 3 x wöchentlich Gewicht: < 50 g/d Der Begriff Obstipation umfaßt folgende auf den Stuhlgang bezogene Beschwerden: - zu selten, zu hart, zu wenig und zu unregelmäßig Diarrhoe Frequenz: Gewicht: Konsistenz: akut: chronisch: >3x täglich > 250 g/d durch erhöhten Flüssigkeitsanteil ungeformt, flüssig (> 80% Wasser) < 2 Wochen (z.B. bei Cholera, Salmonellen...) > 2 Wochen (bei Darmerkrankungen) Seite 64 4.2 Ursachen der Obstipation Funktionell 1. Bewegungsmangel: Bewegung aktiviert den Sympathicus, wodurch vorerst die Darmmotorik gehemmt wird; gegenreflektorisch wird aber der Parasympathikus aktiviert, der einen fördernden Einfluß auf die Darmmotorik ausübt 2. Ballaststoffmangel: Wasser kann nicht gebunden werden und führt zu erhöhtem intraluminalem Druck. Unter ballaststoffreicher Nahrung hingegen kommt es zu einem hohen Wasserbindungsvermögen. Dieses hat einen voluminösen Stuhl sowie einen geringeren intraluminalen Druck zur Folge. Je größer das Stuhlvolumen, desto geringer der Druck in der Darmwand. 3. Psychogen: chronisch unwillkürliche Unterdrückung des Stuhlreflexes, z.B. durch Streß. Wird der Defäkationsreiz öfter unterdrückt, so wird dieser Reflex abgeschwächt. Dies hat zur Folge, daß der Darminhalt über längere Zeit nicht entleert wird; es kommt zu Wasserverlust und Obstipation. 4. Colon irritabile: hierbei treten Diarrhoe und Obstipation im Wechsel auf, zusammen mit Schmerzen im Bauchbereich. Meist führt man eine Sonographie durch, wobei man die Fläche des Magens durch Ultraschall bestimmt und daraus das Volumen schätzt. Man verabreicht einen Trunk, durch den man dann messen kann, wie schnell sich der Magen ausdehnt bzw. wieder zusammenzieht, was die Passagezeit bedingt. Beim Colon irritabile ist die Propulsion stark verzögert. Organisch Darmerkrankungen 1. Kolonkarzinom (Dickdarmkrebs) Das Karzinom geht von der Schleimhaut aus und wächst nach innen, was schließlich zu einem Verschluß des Darmlumens führt. Es treten Diarrhoe und Obstipation im Wechsel auf, Teerstühle, Blutabgang mit dem Stuhl, Schwächegefühl und Blutarmut. 2. Divertikulitis Es handelt sich hierbei um säckchenförmige Ausstülpungen der Colonwand, die vorwiegend im Bereich des Colon sigmoideum zu finden sind. Die Ursache der Divertikulitis ist Ballaststoffmangel. Er führt zu einem hohen intrakolischen Druck, der bewirkt, daß sich die Dickdarmschleimhaut zwischen den Gefäßen im weichen Bindegewebe nach außen drückt. In diesen Ausstülpungen kann sich Stuhl ansammeln. Der Divertikelhals, der im Bereich der Muskelschicht liegt, wird durch die Muskulatur eingeengt. Da die Divertikel selbst keine Muskulatur besitzen, ist eine selbständige Entleerung nicht möglich. Durch die lange Verweildauer des Darminhalts in den Divertikeln besteht die Gefahr einer Entzündung. Kann der Körper sie nicht begrenzen, ist eine Operation nötig. Im schlimmsten Fall kommt es zur Perforation der Divertikel, was zu einer Peritonitis (Bauchfellentzündung) und lokaler Abszeßbildung führt. Die Divertikulitis ist mit Schmerzen und Fieber verbunden. Therapie Erkennen der Divertikel (durch Röntgenuntersuchung, Endoskopie..), Erhöhung der zugeführten Ballaststoffe, Regulierung der Stuhlfrequenz... 3. Ileus (Darmverschluß): a) mechanischer Ileus: Verlegung des Darmlumens (z.B. durch Tumoren, Fremdkörper...) b) paralytischer Ileus: Lähmung, die Darmperistaltik ist aufgehoben (z.B.durch Sepsis) Seite 65 medikamentös Viele Arzneimittel wirken direkt oder durch ihre Nebenwirkungen auf den Darm • Opiate • Antazida: säurehemmend; sie enthalten beispielsweise AlHCO3, das bei zu geringer Flüssigkeitszufuhr ausfallen kann • Diuretika: wassertreibend, so daß weniger Flüssigkeit im Darm vorhanden ist • Psychopharmaka: starke vegetative Nebenwirkungen, behindern die Darmmotorik • Eisen-Präparate: bilden Ausfällungen, wenn zu wenig Flüssigkeit zugeführt wird • Laxantien: stuhltreibend; schädigen das autonome Nervensystem und wirken in der Phase des Absetzens verstopfend endokrin/metabolisch (immer mit anderen charakteristischen Symptomen vergesellschaftet) • Hypothyreose: Grundumsatzverminderung, Myopathie (Muskelschwäche), beides führt zu einer verminderten Propulsionskraft im Darm • Hypercalcämie • Schwangerschaft: hier sind die Ursachen v.a. mechanisch, da der Druck im Bauch steigt • Amyloidose (Zellschäden durch Ablagerung von ß2-Mikroglobulin in Leber, Gefäßen etc.) • Porphyrie (angeborene oder erworbene Störung des Bilirubinstoffwechsels) neurologisch • ZNS-Erkrankungen (Meningitis, Atherosklerose) • Rückenmarkserkrankungen: Aktivität des autonomen Nervensystems in Darm ist unterbrochen, verminderte Sendefrequenz vom Rückenmark; der Eigenimpulsgeber ist langsamer als das Rückenmark, v.a. der Dickdarm ist betroffen. 4.3 Therapie der Obstipation organische Ursachen: funktionelle Ursachen: Therapie des Grundleidens Aufklärung (Reflex einüben) körperliche Bewegung (Sport) Ballaststoffe Verbot von Laxantien Seite 66 Circulus vitiosus bei Obstipation 4.4 Diarrhoe Ergänzung zu Tabelle 6-14. Laktulose, Mannit, Sorbit, Citrat, Antazida wirken wasserziehend. Gastrinom sitzt in den endokrinen Zellen des Magens und Dünndarms, produziert Hormone, die die Sekretion von Wasser anregen. Karzinoide können beispielsweise Hormone produzieren, die zu einer gesteigerten Aktivität des Nervensystems und einer Hypermotilität führen. Parasiten können ähnliche Stoffe absondern Toxine steigern die transzelluläre Sekretion von Cl- Seite 67 VIPom Leckflux Lymphom Tumor der Pankreasinselzellen, der VIP (vasoactive intestinal peptide) produziert; dies führt im Endeffekt zu einer Cl--Sekretion und so zur Diarrhoe. Schleimhautdefekte, Wasser aus dem Interstitium hat freien Zugang zum Darmlumen Wucherung des Lymphgewebes Maldigestion Störungen des enteralen enzymatischen Abbaus von Kohlenhydraten, Proteinen und Lipiden z.B. bei Pankreasinsuffizienz oder Erkrankung der Galle ableitenden Wege; es findet kein ausreichender Aufschluß der Nahrung statt, so daß die Dünndarmschleimhaut ihre Arbeit nicht verrichten kann. Malabsorption Mangelhafte epitheliale Resorption von Nährstoffen, Ionen und/oder Vitaminen aus dem Darm, entweder durch Fehlen einzelner epithelialer Transportmechanismen oder durch Verlust resorptiver Epithelfläche. Seite 68 Je nach Ort der Erkrankung treten spezifische Mängel auf. Seite 69 Biliäre Insuffizienz: Fettstühle, oft als Diarrhoe interpretiert. Seite 70 Magenteilresektion Bei der Resektion nach Billroth I werden das Antrum und Teile des Korpus entfernt und eine physiologische Passage mit verringertem Volumen hergestellt. Nach Billroth II wird keine physiologische Passage erzeugt, sondern ein Ende endet blind. Hier kommt es oft zu einer Stumpfgastritis. Gallensäuren werden durch die bakterielle Überwucherung in großer Menge dekonjugiert und so inaktiviert. Sie können auch laxierend wirken. Durch fehlendes Reservoir des Magens ist die Verweildauer verkürzt. Es kommt zu einer raschen Entleerung ins Jejunum, wodurch dieses plötzlich überdehnt wird und infolge der Hyperosmolarität des Speisebreis dem Plasma größere Flüssigkeitsmengen entzogen werden (Dumping Syndrom). Die Kohlenhydrataufnahmezeit ist geringer; bei gleichbleibender Insulin- (VIP-, GIP-) Sekretion kommt es gehäuft zu Hypoglycämien. 4.5 Chronische entzündliche Darmerkrankungen Einheimische Sprue (auch als Zöliakie bezeichnet; da dieser Begriff aber auch auf andere Krankheiten angewandt wird, sollte man ihn vermeiden. Besser: Glutenenteropathie) Bei der Sprue handelt es sich um eine Glutenenteropathie. Es liegt nicht, wie früher vermutet, eine Enzymdefekt vor, sondern eine Unverträglichkeitsreaktion gegen das Getreideproteingemisch Gluten. Es werden Antikörper gegen Glutenabbauprodukte gebildet, die eine Entzündung in der Dünndarmschleimhaut hervorrufen. Wie bei den meisten Entzündungen, wird dadurch die resorptive Fähigkeit der Dünndarmzellen zerstört. Symptome sind Diarrhoe, Gewichtsabnahme, abdominelle Beschwerden und Blähsucht. Außerdem treten eine Reihe von Mangelerscheinungen als Folge der Malabsorption auf. Morbus Crohn Morbus Crohn (granulomatöse Colitis2, Ileitis regionalis, Colitis Crohn) ist eine chronische Entzündung einzelner Segmente des Dünn- und/oder Dickdarms. Jeder Darmabschnitt kann betroffen sein, zu 90% ist es jedoch das distale Ileum.Zwischen Dünn- und Dickdarm besteht ein Ventilmechanismus; hier tritt Morbus Crohn am häufigsten auf.Durch die Entzündung wird das Ventil zerstört, so daß der Bolus vom Dickdarm zurück in den Dünndarm fließen kann. Alle Schichten der Darmwand, evtl. auch Mesenterium und Lymphgewebe sind betroffen. Es bilden sich Geschwüre und Fisteln (unphysiologische Verbindungen zwischen Organen, können auch durch die Haut brechen), die tief in die Submucosa und Muskulatur eindringen. Wenn auch das Bauchfell vom Entzündungsprozeß betroffen ist, kommt es zu schlimmen Schmerzen. Nach dem Entzündungsschub beruhigt sich das System wieder, bei Regeneration der Entzündung bildet sich eine Narbe durch eingesetztes Bindegewebe. Dieses Bindegewebe 2 Die granulomatöse Colitis ist der M.Crohn des Dickdarms. Die Bildung von Granulomen ist eine spezifische Abwehrform des Immunsystems, die auch bei mikrobiell hervorgerufenen Colitiden auftritt. Seite 71 hat konstringierende Eigenschaften, so daß sich der Darm irreversibel verkürzt. Durch diesen Narbenzug oder Schwellungen kommt es zu Stenosen (Verengungen des Darmlumens). Symptome: abdominelle Beschwerden, Diarrhoe, Gewichtsabnahme, Fieberschübe. Auch hier findet man oft Zeichen einer Unterversorgung mit essentiellen Nährstoffen als Folge einer unzureichenden Zufuhr (Appetitlosigkeit, Erbrechen, Angst vor dem Essen...) aber auch gestörter Resorption. Morbus Crohn tritt im Alter von 15-25 Jahren auf und kommt bei Männern und Frauen gleich häufig vor. Die Ursachen sind noch unbekannt. Colitis Ulcerosa Die Colitis Ulcerosa (chronische Entzündung der Colonschleimhaut) kann sowohl das gesamte als auch nur bestimmte Abschnitte des Organs befallen, wobei das Rectum immer mit angegriffen ist. Die Ursache der Colitis Ulcerosa ist unbekannt. Man nimmt an, daß Autoimmunmechanismen und Streß die Erkrankung auslösen. Die Krankheit tritt in jedem Lebensalter auf. Am häufigsten ist der Beginn zwischen dem 20. und 40. Lebensjahr. Das Verhältnis Männer : Frauen beträgt 1.3 : 1. Das Hauptsymptom sind unter krampfartigen Schmerzen abgesetzte, blutschleimige Stühle. Hierdurch bedingte Blut-, Mineralstoff- und Wasserverluste führen zu Anämie, Hypoproteinämie und Störungen des Elektrolythaushalts. Bei Colitis Ulcerosa kommt es zu Entzündungen von Mucosa und Submucosa mit Geschwürbildung. Seite 72 bei C: + A B C (bei C: +) Seite 73 Unterschiede zwischen Colitis Ulcerosa und Morbus Crohn Beginn Rektale Blutung Massive Blutung Häufigste Lokalisation segmentale Lokalisation Stenosebildung Innere Fisteln Anorektale Fisteln Perianale Abszesse als Erstsymptom Tox. Colondilatation Karzinomrisiko Colitis Ulcerosa allmählich/akut regelmäßig 3% Rektum mit kont. Ausbr. nach prox. sehr ungewöhnlich selten ungewöhnlich relativ selten 3-4% sehr ungewöhnlich 2-10% erhöht Morbus Crohn allmählich relativ selten ungewöhnlich Ileokolitis, segmentale Kolitis häufig sehr häufig häufig sehr häufig 20-25% 25% < 1% gering Colondilatation: toxische Erweiterung des Dickdarms durch Versagen der neuromuskulären Einheit. Die Toxine legen Nerven zeitweise lahm, so daß die glatten Muskelzellen keinen Tonus mehr haben. Bei Morbus Crohn geschieht das selten, da es sich hier meist um eine kleinere Entzündungsfläche handelt. Die Stärke des Schubs korreliert negativ mit dem Körpergewicht; je stärker der Schub, umso mehr Blutverlust tritt auf. Seite 74 5. Leber und Alkohol Symptome bei Leberschädigung Die Leber ist das zentrale Organ des Intermediärstoffwechsels. Sie synthetisiert AS, Gallensalze, Cholesterin, Phospholipide und Proteine, baut AS ab und speichert Nährstoffe (Glycogen, Vitamin A). Außerdem hat sie eine wichtige Funktion bei der Entgiftung und Ausscheidung körpereigener und -fremder Substanzen. Durch die Produktion der Gallenflüssigkeit ist sie die größte exokrine Drüse des Körpers. Seite 75 Bei Leberschädigung treten folgende Symptome auf (vgl. vorhergehende Seite): - der Patient wird psychisch auffällig: desorientiert, schwer ansprechbar, Muskelzuckungen: hepatische Encephalopathie - Hautveränderungen: gelb (Bilirubin wird in der Haut abgelagert) - kleine Gefäßerweiterungen: spider nevi = Lebersternchen - verminderte Körperbehaarung: Gynäkomastie (Brustwachstum mit Feminisierung) - Erweiterung kleinster Gefäße in der Hand und Zunge: nicht isoliert sichtbar, aber Handflächen und Zunge sind knallrot - Blutergüsse: Blutungen in der Haut, die nicht schnell resorbiert werden - Vermehrung des Bauchumfangs (Ascites) Die genauen Ursachen für diese Symptome werden im Folgenden besprochen. 5.1 Funktionen der Leber Regulation des Energiestoffwechsels 1. Glucose-Homoiostase: Glycogen-Synthese (≤150g, 10% der Leber, 25g/h) Glycogenolyse (≤20g/h) Gluconeogenese (≤5g/h) Steuerung: Ins, Ggn 2. AS-Homoiostase und Degradation NH3-Entgiftung (ca 15g/dÆ30g Hst/d) HCO3-- Elimination: (pH-Regulation) 3. Lipidstoffwechsel: GlcÆFS ASÆFS, KK FSÆKK FSÆTGÆVLDL Baustoffwechsel 1. Proteinsynthese und -sekretion: 40% der synthetisierten Proteine: Plasmaprotein = Albumin 10-20 g/d; HWZ: 14 d Fibrinogen 1-2 g/d; HWZ: 4 d Gerinnungsfaktoren II, V, VII, X; HWZ: h-d Transportproteine: Thyroxin-bindendes Globulin, Coeruloplasmin, Transferrin... Cholinesterase (CHE) LCAT 2. Cholesterin-Synthese /-Ausscheidung Gallensäuren-Synthese/-Ausscheidung 3. Hormonsynthese z.B. Somatomedin Entgiftung Oxidation,Reduktion, Hydroxylierung: Steroidhormone, Fremdstoffe Glucuronidierung: Bilirubin.. Sulfatierung: Steroidhormone Degradation: Hormone (Peptidhormon, T3, T4...), Bakterientoxine Hst-Synthese: s.o. 5.2 Leberversagen Akut: Chronisch: selten, z.B. Vergiftung (CCl4, Knollenblätterpilz) Sepsis Akute schwerste Hepatitis häufig Ausfall von >2/3 Lebergewebe bei Leberzirrhose: nach Hepatitis Seite 76 autoimmunologisch: Speichererkrankungen: bei chron. Alkoholabusus bei chron. Gallestau Primär biliäre Zirrhose (P.b.C.) Fe2+, Cu2+ Glucose-Homoiostase Das vom Verdauungstrakt zurückströmende venöse Blut sammelt sich in der Pfortader, die es zur Leber führt. Nach Aufteilung in ein Kapillarsystem verläßt das Blut die Leber wieder über die Lebervene. Diese mündet in die untere Hohlvene. Mit dem Pfortaderblut werden sämtliche resorbierten Nährstoffe (abgesehen von den wenigen, die auf dem Lymphweg transportiert werden) und auch die im Darm produzierten und resorbierten Toxine zur Leber transportiert. Die so zur Leber gelangenden Nährstoffe werden zum Teil schon beim ersten Durchgang durch das Organ eliminiert und in den Stoffwechsel eingeschleust. Das gleiche gilt für die insbesondere beim bakteriellen Eiweißabbau im Darm anfallenden Toxine. Bei der Verdauung von Polysacchariden entsteht vor allem Glucose, die vom Darmepithel resorbiert wird und zur Leber gelangt, die sie unter ATP-Verbrauch zu Glucose-6-Phosphat phosphoryliert. Dieses kann in Glucose-1-Phosphat umgewandelt und zur GlycogenSynthese verwendet werden oder zu Acetyl-CoA verstoffwechselt und in Form von VLDL exportiert werden. Glycogen als hydrophile Substanz kann nur zusammen mit Wasser in der Zelle gelagert werden und enthält nur 1-2 kcal/g. Fett ist hydrophob und daher besser als Speichersubstanz geeignet. Die Fähigkeit der Leber und der Muskulatur, Glycogen zu speichern, ist im Vergleich zur Energiespeicherung im Fettgewebe relativ gering. In Extremfällen kann 10% der Leber aus Glycogen bestehen. Entsprechend dieser geringen Speichermenge sind die Glycogenspeicher bei Nahrungskarenz schnell aufgebraucht; das Muskelglycogen wird vorwiegend bei körperlicher Aktivität mobilisiert. Beim ersten Durchgang durch die Leber werden dem Pfortaderblut nur ca.10% der Glucose entzogen, aber ein großer Teil wird aus den peripheren Geweben wieder in Form von Lactat, Pyruvat und Alanin an die Leber zurückgeliefert und dort wieder in Glucose oder Glycogen umgewandelt (Gluconeogenese bzw. Glucogenese). Die Synthesegeschwindigkeit bei der Gluconeogenese beträgt 5g/h. Das entspricht der Menge, die die absolut glucoseabhängigen Organe benötigen. Welcher Weg eingeschlagen wird, ist hormonell gesteuert. Nach Aufnahme einer KH-reichen Mahlzeit sind Glucose- und Insulinkonzentration im Pfortaderblut erhöht, die Glucagonkonzentration gegenüber dem Zustand vor der Mahlzeit erniedrigt. Insulin erhöht die Speicherung in Form von Glycogen, wobei gleichzeitig die Gluconeogenese gehemmt wird. Nach kurzfristigem Hungerzustand (z.B. morgens vor dem Frühstück) sind die Konzentrationen von Glucose und Insulin im Pfortaderblut stark abgefallen, während die Glucagonkonzentration angestiegen ist. In diesem Fall gibt die Leber vermehrt Glucose ab (erhöhter Glycogenabbau, vermehrte Gluconeogenese, verminderter Glucoseabbau). Hinter der Leber liegt also eine komplett andere Blutzusammensetzung vor als in der Pfortader. Die Leber scheidet endogene Metaboliten/ Abbauprodukte in den Darm aus. Aminosäure-Homoiostase und Degradation (s. auch Kapitel “Protein- und AS-Stoffwechsel”) Die Nahrungsproteine werden im Darm zu freien AS gespalten, in dieser Form zur Leber transportiert und dort entweder verstoffwechselt oder weiter im Körper verteilt. (Aromatische AS werden in der Leber abgebaut, verzweigtkettige überwiegend in der Muskulatur.) Seite 77 Eine Eiweißspeicherung in der Leber erfolgt nicht. Liegen aber Proteinmangelbedingungen vor, kommt es, bevor der Eiweißgehalt anderer Organe oder des Plasmas abnimmt, schnell zu einer Mobilisation von Proteinen in der Leber. Sind die Gewebeproteine in normaler Menge vorhanden, werden zusätzlich aufgenommene AS für die Gewinnung metabolischer Energie verwendet oder in Fett bzw. Glycogen umgebaut und in dieser Form gespeichert. Die Aminogruppen, die bei diesem Prozeß frei werden, werden letztlich zu Ammoniumionen umgewandelt, die über den Harnstoffzyklus in Harnstoff eingebaut und über den Urin ausgeschieden werden. Lipidstoffwechsel (s. auch Kapitel “Lipidstoffwechsel”) Die Aufgabe der Leber im Fettstoffwechsel besteht darin, Fettsäuren aus dem Plasma aufzunehmen, zu reverestern und in Form von Triglyceriden, an Lipoproteine gebunden, wieder ans Blut abzugeben, Fettsäuren zu metabolisieren und zu synthetisieren. Ausgangssubstanz ist dabei das beim Abbau von KH und AS anfallende Acetyl-CoA. Eine hohe Kohlenhydratzufuhr steigert, eine hohe Fettzufuhr senkt die Neubildungsrate. In der Leber können Acetyl-CoA-Einheiten auch zu Acetoacetyl-CoA kondensiert und zu Ketonkörpern umgewandelt werden. Diese dienen anderen Geweben, im Hunger vor allem dem Gehirn, als Energiequelle. Des weiteren wird in der Leber das Enzym LCAT (Lecithin-Cholesterin-Acyl-Transferase) synthetisiert. Es katalysiert die Übertragung eines Fettsäureesters aus der ß-Position des Lecithins auf Cholesterin unter Bildung von Cholesterinestern. Es ist auch für die Veresterung von Cholesterin in den Plasmalipoproteinen verantwortlich. Bei schweren Leberschäden mit eingeschränkter Proteinsynthese ist die Aktivität der LCAT im Blut erniedrigt; es kommt so zu einem Abfall der Cholesterinester im Blutserum (Estersturz). Die Cholesterinsynthese und -ausscheidung (s. auch Kapitel “Lipidstoffwechsel”) Die Leber ist auch im Hinblick auf das Cholesterin das zentrale Stoffwechselorgan. Sie ist der Hauptort der Synthese, das Zentrum für den Abbau und die Ausscheidung mit der Galle. Leber und Dünndarm geben HDL an die Blutbahn ab, die in den verschiedenen Geweben des Körpers Cholesterin aufnehmen und zur Leber transportieren. Hier dient Cholesterin als Ausgangssubstanz für die Gallensäurensynthese. Das Cholesterin zirkuliert im Organismus zwischen Darm und Leber über den sog. enterohepatischen Kreislauf. Täglich werden etwa 2g Cholesterin mit der Galle aus der Leber in den Darm sezerniert. Nur 1/4 davon wird mit dem Fäzes ausgeschieden; der Rest wird reabsorbiert und gelangt über das Pfortadersystem oder durch die Lymphe, an Chylomikronen gebunden, zurück zur Leber. Die Gallensäurensynthese und -ausscheidung Die Gallenflüssigkeit wird von den Leberzellen sezerniert, in der Gallenblase zwischengelagert und bei Bedarf ins Duodenum abgegeben. Pro Tag werden vom Erwachsenen maximal 1-1,2 l Gallenflüssigkeit produziert. Neben Wasser und Elektrolyten enthält die Lebergalle hauptsächlich Gallensalze, Cholesterin, Phospholipide, Steroide sowie Ausscheidungsprodukte wie Bilirubin und viele Fremdstoffe. Sie ist isoton und hat eine ähnliche Elektrolytzusammensetzung wie das Plasma. Ihr pH-Wert ist neutral bis leicht alkalisch. Gallensäuren werden in der Leber aus Cholesterin gebildet, mit Taurin und Glycin konjugiert und in die Galle sezerniert. Durch die Konjugation sind die Moleküle auf ihrer AS-Seite stark polar, was die Wasserlöslichkeit fördert, während das Steroidgerüst lipophil ist. So können sie als Detergentien für Lipide dienen. Die in der Leber gebildeten primären Gallensäuren Cholsäure und Chenodesoxycholsäure werden im Dünn- und Dickdarm teilweise zu sekundären Gallensäuren umgewandelt (Desoxycholsäure, Lithocholsäure...). Ca. 90% der Seite 78 Gallensäuren werden im distalen Ileum fast vollständig resorbiert und im Pfortaderblut gebunden an HDL und Albumin zur Leber zurücktransportiert. Dort werden sie von Hepatocyten aufgenommen und erneut in die Galle abgegeben. Bei einem Ausfall des Ileums, dem alleinigen Resorptionsort der Gallensäuren, kommt es zu einer Unterbrechung des enterohepatischen Kreislaufs. Die einsetzende unphysiologisch hohe Gallensalzausscheidung führt zu einer Poolverringerung und einer Beeinträchtigung der Fettverdauung und -resorption. Das Bilirubin stammt hauptsächlich aus dem Hämoglobinabbau in den Zellen des retikuloendothelialen Systems von Milz, Leber und Knochenmark. Der Hämring des Hämoglobins wird abgespalten und geöffnet, wodurch Biliverdin entsteht, das anschließend in Bilirubin umgewandelt wird. Es wird an Albumin gebunden in die Leber transportiert, dort zu wasserlöslichen Mono- und Diglucuroniden konjugiert und über die Galle ausgeschieden. Hormonsynthese Das Wachstumshormon Somatotropin steuert des Skelettwachstum und andere Stoffwechselprozesse. Seine Wirkungen löst es z.T. über die Bildung wachstumsfördernder Faktoren in der Leber, der Somatomedine, aus. Das wichtigste Somatomedin ist Somatomedin C. Es stimuliert in allen Körperzellen die Zellteilung über eine Vermehrung der Proteinsynthese. Außerdem hemmt Somatomedin C die Somatotropin-Freisetzung im Hypophysenvorderlappen. Entgiftung Viele Stoffe müssen erst im Entgiftungsstoffwechsel von Leber und Niere metabolisch aufgearbeitet werden, ehe sie ausgeschieden werden können. Wasserunlösliche Substanzen werden in eine wasserlösliche Form überführt, was ihre Ausscheidung über Galle und Urin ermöglicht. Dies geschieht über Oxidation, Reduktion und Hydroxylierung, Koppelung an Glucuronsäure, Sulfat und Gluthathion. Außerdem findet in der Leber der Abbau von Alkohol statt (s.u.). Leberversagen • akut: Die akute Hepatitis umfaßt alle hepatozellulären Erkrankungen, die mit einer akuten Entzündung der Leber einhergehen. Auslöser für eine Hepatitis können Viren, Bakterien, Alkohol, Arzneimittel (Halotan), oder Gifte (CCl4 = Tetrachlorkohlenstoff, z.B. in Fleckenwasser, a-Amanithin: Gift der Knollenblätterpilze) etc. sein • chronisch: häufig kommt es bei Leberzirrhose zum Ausfall von mehr als 2/3 des Lebergewebes, z.B. nach chronischer Hepatitis. Als chronische Hepatitis bezeichnet man eine langsam fortschreitende Entzündung der Leber, die mit einem ständigen Untergang von Leberzellen einhergeht und deren Ursache unbekannt ist. Die Symptome sind unspezifisch. Im Serum kommt es insbesondere zu einer g-Globulinvermehrung und einer Erhöhung der GOT- und GPT-Aktivität als Hinweis auf einen Untergang von Leberzellen. Zirrhose: chronische Entzündung, narbige Veränderung 5.3 Erkrankungen der Leber Leberzirrhose = chronische Leberinsuffizienz Die Leber ist aus Leberläppchen von 1-2 mm Durchmesser aufgebaut, die auf Querschnitten durch das Lebergewebe annähernd sechseckig erscheinen. Die menschliche Leber setzt sich aus 50 000 bis 100 000 solcher Läppchen zusammen, die durch schwach ausgebildete Bindegewebszüge voneinander getrennt sind. An den Stellen, an denen mehrere Läppchen Seite 79 mit ihren Kanten zusammenstoßen, verdichtet sich das Bindegewebe zu kleinen Dreiecken und bildet die sog. periportalen Felder. In diesen verlaufen die Äste der Pfortader, der Leberarterie und der intrahepatischen Gallengänge. Im Läppchenzentrum liegt die Vena centralis; auf dem Weg vom portalen Dreieck zu der zentralen Vene passiert das Blut über ein radiäres Kapillarnetz die Leberzellen Alkoholbedingt oder posthepatitisch kommt es zu Einzelzellnekrosen (Schädigung bzw. Untergang der Leberzelle). Diese lösen durch Chemotaxis eine Einwanderung von Granulocyten, Lymphocyten und Makrophagen aus. Die von den Makrophagen und Lymphocyten abgegebenen Lymphokine stimulieren die Bildung von Myofibroblasten und die Proliferation eingewanderter Fibroblasten. Diese beiden Zelltypen synthetisieren vermehrt Kollagen (v.a. Typ I, III, IV und V). Die vermehrte Bildung von Bindegewebe (Fibrose) schnürt die Leberläppchen ein; da sie weiter in die Richtung regenerieren, in die sie weggedrängt werden, wird ihre Struktur nachhaltig geschädigt. Durch die entstehende Narbe fließt kein Blut. An den Stellen, die für das Blut noch durchlässig sind, wird die Diffusionsstrecke zur Zentralvene immer länger, was zu einem weiteren Absterben von Lebergewebe führt. Die Leber vernarbt durch und durch und schrumpft schließlich bis auf 1/5 ihrer ursprünglichen Größe zusammen. Seite 80 Folgen 1. Parenchymuntergang und -verlust Æ Funktionsverminderung 2. Bindegewebseinschnürung : führt zu a)Minderperfusion Æ O2 Ø Æ weitere Nekrosen b)portaler Hypertension Æ Umgehungskreislauf c)Gallestau Æ perikapilläre Entzündung Æ Bindegewebsnarbe↑ Durch den behinderten Blutfluß steigt der Druck in der Pfortader, so daß es zu erhöhter Diffusion in den Kapillaren des Bauchraumes kommt. Blutwasser wird in die freie Bauchhöhle abgedrückt und ein Wasserbauch entsteht (Ascites). Die durch Parenchymschädigung hervorgerufene verminderte Albuminsynthese der Leber hat den gleichen Effekt. Der kolloidosmotische Druck des Plasmas sinkt und damit auch die Reabsorption von Wasser aus dem Extravasalraum. Seite 81 Seite 82 KH-Stoffwechsel Hepatogener Diabetes Da die Glucosehomöostase des Körpers vorwiegend durch die Leber reguliert wird, tritt bei Patienten mit chronischer Lebererkrankung trotz erhöhter Seruminsulinkonzentration oft eine gestörte Glucosetoleranz oder manifester Diabetes mellitus auf. Charakteristisch ist, daß Insulingaben zu keinem drastischen Abfall des Blutglucosespiegels führen, weil das Zielorgan Leber nicht auf Insulin mit normaler Glycogensynthese reagiert. Da der Patient keine Speicher anlegen kann, kommt es zu einer postprandialen Hyper- und Nüchternhypoglycämie. Seite 83 Die bei Lebererkrankungen gestörte Gluconeogenese verstärkt eine Hypoglycämie und führt außerdem zu einem erhöhten Lactatspiegel. Da Galactose nur in der Leber in Glucose umgewandelt werden kann, wird der GlucoseAnstieg im Serum nach einer Galactose-Gabe als Parameter für die Leberfunktion genutzt (40 g Galactose Æ <30 mg Glucose/dl Serum nach 90 Minuten). Die Umsetzung der Galactose ist allerdings interindividuell sehr unterschiedlich und daher kein guter Parameter für die Leberfunktion insgesamt. Veränderungen des Zustands eines Patienten lassen sich aber gut ablesen. AS-Stoffwechsel und Harnstoffsynthese Störungen des AS-Stoffwechsels und der NH3-Entgiftung führen typischerweise zur hepatischen Encephalopathie. a) Störung des Abbaus aromatischer AS Aromatische AS wie Phe, Tyr und Trp werden bevorzugt in der Leber, verzweigte AS wie Leu, Ile und Val bevorzugt im Muskel abgebaut. Bei Leberschädigung steigen Phe, Tyr und Trp im Serum an. Ile, Leu und Val sind normal bis erniedrigt. Dadurch wird über das Transportsystem im ZNS vermehrt Phe und Tyr aufgenommen. Phe hemmt die Umwandlung von Tyr zu Noradrenalin. Stattdessen wird aus Tyr der pathologische Neurotransmitter Octopamin gebildet (s.Abb.1). b) Trp wirkt in pathologisch hohen Konzentrationen somnolent (daher wird es auch als mildes Schlafmittel benutzt). c) Durch Störung der Harnstoffsynthese steigt NH4+ an. Bei durchschnittlicher Zufuhr von 100 g Nahrungsprotein/d wird 16 g NH3/d gebildet, das hauptsächlich durch Harnstoffsynthese entgiftet wird. 100 g Protein Æ 16 g NH3 Æ 1 g NH4+ im Urin... Æ 30 g Harnstoff aus Leber über Niere In der Frühphase einer Leberzirrhose wird der Verlust von Lebergewebe durch Steigerung der Aktivität des verbliebenen Parenchyms kompensiert. Ammoniak im Blut steigt erst noch oraler Belastung mit 5 g NH4+Ac an. Vorsicht: Bei Leberzirrhose kann darauf eine hepatische Encephalopathie ausgelöst werden! Im Spätstadium ist dann Ammoniak auch ohne Belastung erhöht.. Dies hat dann zur Folge: 1) Hemmung des GABA(g - Aminobuttersäure)-abhängigen Chloridtransports an synaptischen Membranen (s.Abb.2). 2) Intrazelluläre Anreicherung von NH4+ durch höhere H+-Konzentration in der Zelle. NH3 tritt durch die zellmembran hindurch, NH4+ jedoch nicht. Dadurch kommt es zu vermehrter Bildung von Glutamin und a-Ketoglutaramid, die die Neurotransmission vor allem über GABA hemmen. Abb. 2 Abb. 1 Seite 84 Die Toxizität von Ammoniak ist zunächst durch den Verlust an Lebergewebe bedingt; sie wird jedoch verstärkt durch: • Alkalose: Im Blut liegt bevorzugt NH3 vor, das in die Zellen eindringt und wegen des dort niedrigeren pH zu NH4+ umgewandelt wird. Da NH4+nicht durch Membranen diffundieren kann, wird es intrazellulär angereichert. Die Alkalose beruht auf einer verminderten HCO3--Elimination in der Harnstoffsynthese. Weiterhin beruht sie auf: • Hyperaldosteronismus: Aldosteron wird vermindert in der Leber abgebaut. Durch erhöhtes Aldosteron wird im Blut Na+ ansteigen und K+ abfallen. Damit K+ nicht lebenskritisch abfällt, wird es aus den Zellen ins Blutplasma nachgeliefert. Im Tausch wird H+ in die Zelle transportiert und bindet dort NH3 als NH4+. • Gesteigerte Glutaminase-Aktivität: K-Mangel aktiviert die Glutaminase der Niere, die dann vermehrt NH4+ aus Glutamin freisetzt. • Perihepatische Umgehungskreisläufe leiten das sich vor der Leber stauende Pfortaderblut an der Leber vorbei. Der Stau ist durch Verminderung des intrahepatischen Blutflusses aufgrund bindegewebigen Umbaus bedingt. Umgehungskreisläufe (Shunts) senken zwar den Pfortaderdruck, leiten aber das Blut ohne hepatische Entgiftung in die Hohlvene. Der wichtigste Umgehungskreislauf führt von der Pfortader über die Magenvene und die Oesophagusvenen in die Hohlvene. Die Venen erweitern sich zu Oesophagusvarizen, die rupturieren können, was lebensgefährliche Blutungen zur Folge hat. • Vermehrte Ammoniakbildung im Darm: Blut aus Oesophagusvarizenblutung ist schwer verdaulich und gelangt nur teilweise verdaut ins Colon. Dort bauen Bakterien das Protein unter Freisetzung von NH3 ab. Dies verstärkt (typischerweise wenige Tage nach der Blutung) die hepatische Encephalopathie. Weitere weniger wichtige und gefährliche Umgehungskreisläufe verwenden a) periumbilikale Venen, die normalerweise nach der Geburt nicht mehr benötigt werden. erkennbar sind diese an Venenerweiterungen auf der Bauchdecke (Caput medusae). b) perianale Venen, die zu Hämorrhoiden erweitert werden. Die hepatische Encephalopathie äußert sich in folgenden Symptomen: Stadium 1: Hypomimie, Persönlichkeitsveränderungen, Desorientiertheit, Schreib- und Sprachstörungen Stadium 2: Schlagender Tremor = Flapping Tremor Stadium 3: Unruhe, Aggressivität Stadium 4: Koma hepaticum mit Foetor hepaticus (fauliger Geruch der Atemluft von zerfallendem Lebergewebe) Synthese und Sekretion von Proteinen Bei akutem Leberversagen fallen nach wenigen Stunden die in der Leber gebildeten Gerinnungsfaktoren ab, da ihre Halbwertszeit kurz ist. Albumin und Cholinesterase bleiben wegen ihrer Halbwertszeit von 12-14 Tagen noch mehrere Tage im Normbereich. Bei chronischem Leberversagen wird in der Frühphase der Ausfall von Lebergewebe durch das übrige Gewebe kompensiert; in der Spätphase kommt es zum Abfall von Albumin im Blutserum mit der Folge verminderter Wasserbindung im Gefäßsystem und Ödem- bzw. Ascitesbildung. Weiterhin fallen Gerinnungsfaktoren ab, was zur Blutungsneigung führt (u.a. Oesophagusvarizen). Seite 85 Ascites wird bedingt durch 1. Portale Hypertension 2. Albuminmangel 3. Hyperaldosteronismus durch verminderten Aldosteronabbau in der Leber, was zu Na- und H2O-Retention führt. Durch Rückstau von Blut in die Milz kommt es zu Splenomegalie (Vergrößerung der Milz) und beschleunigtem Abbau von Blutzellen (Erythrocyten und Thrombocyten) in der Milz. Entgiftung und Abbau a) gestörte Harnstoffsynthese s.o. b) verminderte Bilirubin-Glucuronidierung und -Ausscheidung mit Folge von Gelbsucht (Ikterus) durch Bilirubin-Ablagerung u.a. in der Haut und den Skleren des Auges. c) verminderter Steroidhormonabbau: der Östrogenspiegel erhöht sich, was zu einer Feminisierung (Brust, Behaarung, Testesatrophie) führt. Der erhöhte Aldosteronspiegel hat o.g. Folgen. d) nur noch unzureichender Fremdstoffmetabolismus, verminderter Medikamentenabbau. 5.4 Störungen des Leberstoffwechsels durch Alkohol Alkohol wird zu mehr als 90% in der Leber abgebaut, wobei als Abbauwege • die Alkoholdehydrogenase (ADH), • das mikrosomale ethanoloxidierende System (MEOS) und • die Katalase zur Verfügung stehen. Normalerweise erfolgt der Alkoholabbau fast ausschließlich über die ADH. Erst bei chronischem Alkoholkonsum spielt der Abbau über das MEOS eine wesentliche Rolle. Seite 86 Frauen sollten nicht mehr als 30 g, Männer nicht mehr als 60 g Alkohol/d zu sich nehmen (0,5 l Bier = 20 g Alkohol), da sonst die Gefahr einer chronischen Leberschädigung drastisch zunimmt. 80% des Alkoholabbaus laufen über die ADH. Hiervon liegen drei Isoenzyme vor (ADH1-3), wobei das ADH3 besonders aktiv ist. Es kommt bevorzugt bei Japanern vor, bei denen die rasche Bildung von Acetaldehyd zu Flush und Übelkeit führen kann. Intoxikationen können über verschiedene Mechanismen erfolgen: Erhöhung des NADH/NAD+-Quotienten Alkoholabbau über die ADH liefert NADH, welches in der Atmungskette unter Bildung von ATP oxidiert werden kann. Übersteigt die NADH-Bildung den zellulären ATP-Bedarf, so kommt es zu einer Erhöhung des NADH/NAD+-Quotienten, wodurch andere NADHabhängige Reaktionen beeinflußt werden: a) Pyruvat wird vermindert oxidiert und somit vermehrt Laktat gebildet, was in einer LacAcidose resultieren kann. Die gesteigerte Ketonkörpersynthese aus Acetyl-CoA führt über die Bildung von ßHydroxybutyrat zu einer Ketoacidose. Lactat und ß-Hydroxybutyrat vermindern kompetitiv die Harnsäuresekretion im Nierentubulus, wodurch es zum Gichtanfall kommen kann. Lactat hemmt außerdem die Prolinoxidase, so daß die Fibrose verstärkt wird. b) NADH hemmt die Glycolyse auf der Ebene der GADPH. Seite 87 c) NADH hemmt die Gluconeogenese aus Lactat, da Lactat nicht zu Pyruvat umgesetzt wird. So kommt es zu einer Hypoglycämie. Bildung von Acetaldehyd Der entstehende Acetaldehyd • führt zu Gefäßerweiterung (Flush, Säufernase) • deinduziert die mitochondriale Aldehyddehydrogenase und hemmt dadurch den eigenen Abbau • zerstört die Mitochondrien (Ort des Acetaldehydabbaus!) und Mikrotubuli • bildet Gluthathion, das als GSH nicht mehr für die Reduktion und Entgiftung zur Verfügung steht: die Radikalprotektion ist vermindert Die vermehrte Bildung von Acetyl-CoA führt zu einer gesteigerten FS-Synthese, während gleichzeitig der FS-Abbau durch ß-Oxidation infolge des erhöhten NADH/NAD+-Quotienten gehemmt ist. Aufgrund der erhöhten NADH-Konzentration wird vermehrt L-Glycerin-3Phosphat aus Dihydroxyacetonphosphat bereitgestellt, so daß auch die Triglyceridsynthese gesteigert ist. Wegen der Schädigung des mikrotubulären Zytoskeletts ist die Sekretion in Form von Lipoproteinen jedoch gestört, so daß diese in den Leberparenchymzellen akkumulieren. Es bildet sich eine Fettleber. Seite 88 6. Die Niere Die Niere ist ein paarig angelegtes, bohnenförmiges Organ. Sie wird versorgt von einem großen arteriellen Gefäß, entsorgt von Urether und Nierenvene. Sie ist eins der bestdurchblutetsten Organe: Sie erhält 20% dessen, was das Herz pro Minute ausstößt. Durchblutung: 1 l/min Primärharn: 120 l/d Endharn: 1,2 l/d Jede Niere hat in ihrer konkaven Seite eine Vertiefung (Hilus), durch den die Nierenarterie, die Nierenvene, Lymphgefäße und Nerven ein- bzw. austreten. Hier ist auch die Austrittsstelle des Urethers aus dem Nierenbecken, über den der gesammelte Urin der Harnblase zugeleitet wird. Die Niere ist unterteilt in Rinde, Mark und Becken und von einer derben bindegewebigen Kapsel überzogen. In der Rinde befinden sich die Nephrone, in denen die Harnbildung stattfindet. Ein Nephron besteht aus dem runden Nierenkörperchen (Filtration des Primärharns), in das ein Knäuel von Blutkapillaren, der Glomerulus, eingestülpt ist und einem dort entspringenden Tubulus. Jede Niere besitzt mehr als 1 Million solcher Nephrone. Das Mark ist durch Säulen der Rindensubstanz in pyramidenförmige Lappen unterteilt, deren Spitzen zum Zentrum hin konvergieren. Diese Spitzen (Nierenpapillen) sind von schlauchförmigen Nierenkelchen überzogen. Sie fangen den fertigen Harn auf und leiten ihn in das Nierenbecken. Von der Harnblase wird der Urin über die Harnröhre abgeleitet. Harnleiter, Blase und Harnröhre verändern den Harn nicht mehr. 6.1 Funktionen der Niere Ausscheidung Stoffwechselendprodukte Körperfremde Substanzen (z.B. Medikamente) Regulation Elektrolyt-/ Wasserhaushalt/Blutdruck Säure-/Base-Haushalt Hormonsynthese 1,25- Dihydroxycholecalciferol Renin Erythropoietin Die Aufgabe der Niere besteht in erster Linie in der Konstanthaltung der Zusammensetzung der extrazellulären Flüssigkeit. Sie dient der Ausscheidung wasserlöslicher, nicht proteingebundener Substanzen. Harnstoff, Harnsäure und Kreatinin stellen die harnpflichtigen Stoffwechselendprodukte dar. (Kreatinin wird in dem Umfang ausgeschieden, wie Muskulatur im Körper vorhanden ist, da Kreatinphosphat als kurzfristiger Energiespeicher dient. Es reicht z.B. aus, um den Energiebedarf für einen 100m-Lauf zu decken; bei einem 200m-Lauf setzt dagegen Lactatbildung ein.) Körperfremde Substanzen führen oft zu einer Verfärbung des Harns (rote Bete) oder zu Geruchsveränderungen (Antibiotika). Die Regulation des Säure-Basen-Gleichgewichts erfolgt über die Ausscheidung von Protonen bzw. Bicarbonat. Allerdings ist die Niere für die Säureregulation nur sekundär wichtig; die größere Bedeutung hat hier die Lunge. Seite 89 Die Niere stellt durch die Hormonproduktion auch ein endokrines Organ dar. 1,25-Dihxydroxycholecalciferol führt zu vermehrter Ca- Rückresorption in der Niere, erhöhter Resorption im Darm und Knochenabbau. Ein Mangel führt zu Rachitis. Renin wirkt blutdrucksteigernd und damit einer Hypotonie entgegen. Erythropoietin beschleunigt den Reifungsprozeß der Erythrocytenvorstufen im Knochenmark, so daß eine Niereninsuffizienz mit einer Anämie einhergeht. 6.2 Die Harnbildung Die Harnbildung beginnt im Glomerulus (1). Hier wird durch den Blutdruck in den Kapillaren aus dem durchfließenden Blutplasma der Primärharn abgepreßt. Die Glomeruluskapillaren sind von einem Kelchsystem umgeben, der Bowman’schen Kapsel. Zwischen Kapillarlumen und dem Innenraum der Bowman’schen Kapsel liegt eine Trennschicht aus Kapillarendothel, Basalmembran und Epithel der Bowman’schen Kapsel, die die Filtrationsbarriere darstellt. Die Durchlässigkeit ist durch Porengröße und Wandladung bestimmt. Frei filtriert werden nur Stoffe, deren Molekülradius kleiner als 1,6-1,8 nm ist. Makromoleküle und Blutzellen werden also zurückgehalten. Bei Molekülen mit einem Radius von <4,4 nm ist die Filtraion ladungsabhängig, was auf die fixen negativen Wandladungen des Filters zurückzuführen ist. Die Plasmaproteine haben eine Größe von Seite 90 60000-1 Million Dalton, der Primärharn ist also extrem eiweißarm. Andere Proteine ( bis ca 60000 MG) werden teilweise wieder rückresorbiert. Elektrolytkonzentration im Serum Gesunder Elektrolyt mmol/l Natrium 135,0-155,0 Kalium 3,5-5,5 Kalzium 2,0-2,75 Chlorid 97,0-108,0 Phosphat 0,8-1,5 mg% 310,0-357,0 17,0-22,0 8,0-12,0 355,0-380 2,5-4,2 Der Primärharn entspricht in seiner Zusammensetzung weitgehend dem Plasmawasser. Entlang des Tubulusrohrs (2) werden nun noch sowohl Substanzen in den Harn hineinsezerniert als auch rückresorbiert. Der proximale Tubulus ist mit einem dichten Bürstensaum besetzt, so daß hier große Salzund Wassermengen resorbiert werden können. Die Tubuluszellen haben besonders viele Mitochondrien, die das für die Na-K-Pumpe benötigte ATP produzieren. Das Ultrafiltrat enthält ca 145 mmol/l Natriumionen und als begleitende Anionen v.a. Chlorid und Bicarbonat. Dazu kommen etwa 5 mmol/l Glucose und AS, sowie K+, Ca2+ usw. Die Resorption dieser Stoffe erfolgt gekoppelt an die des Na+ aktiv über eine Reihe von Carriern. Der Na+-Einstrom in die Zelle ist dabei passiv. Über einen Na-H-Austausch-Carrier wird für jedes resorbierte Na+- ein H+-Ion aktiv sezerniert. Dies wird fast ausschließlich dazu verwendet, filtriertes Bicarbonat zu resorbieren. Da bei diesen Transportprozessen v.a. positive Ladungen in die Zelle geschleust werden, wird nun ein Teil des filtrierten Chlorids passiv nachgezogen. Durch die Resorption all dieser gelösten Stoffe kommt es zu einem osmotischen Wasserstrom, der wiederum andere gelöste Stoffe passiv mitreißen kann (Solvent drag). AS und Glucose werden relativ vollständig rückresorbiert (sofern der Glucose-Spiegel nicht übernormal erhöht ist). Harnsäure und Harnstoff werden passiv vollständig rückresorbiert und erst in einem zweiten Schritt wieder sezerniert. Im proximalen Tubulus sind Carrier vorhanden, die organische Säuren und Basen aktiv ins Tubuluslumen sezernieren. Diese Mechanismen sind sehr effektiv, so daß eine rasche Ausscheidung von Gift- und Abfallstoffen möglich ist. Hier erfolgt auch die Ausscheidung der Harnsäure. Da für Harnsäure, Milchsäure und Ketonkörper die gleichen Transportmechanismen gelten, kann Gicht auftreten, wenn zu viel Milchsäure und Ketonkörper im Blut vorliegen. Die anschließende Henle-Schleife, die von der Rinde ins Mark und wieder zurück zieht, dient der Konzentrierung des Harns. Vor allem im absteigenden Schleifenschenkel zieht das umgebende hypertone Milieu osmotisch Wasser. Das Blut hat also bei Verlassen der Niere nur sehr wenig an Volumen verloren. Die Harnkonzentrierung findet im Gegenstromprinzip statt. Im dicken aufsteigenden Teil der Schleife, der wasserundurchlässig ist, werden ca. 30% des filtrierten NaCl resorbiert, und zwar mit Hilfe eines aktiven Kotransportsystems. Dabei werden ein Natriumion, ein Kaliumion und zwei Chloridionen gemeinsam in die Zelle geschleust. In diesem dicken, aufsteigenden Schleifenschenkel wird auch bevorzugt Magnesium resorbiert. Im distalen Konvolut, dem Verbindungsstück und Sammelrohr (3) wird die Wasser- und Na+Rückresorption durch Hormone und andere Signale an den Bedarf angepaßt. Große Bedeutung kommt hier dem Aldosteron-Angiotensin-Renin-System (s.u.) zu. Je nach Zustand des Organismus (Alkalose/Acidose) werden H+ sezerniert oder resorbiert. Hier Seite 91 kommt es auch zu einer Abgabe von Kalium in den Harn, was einen lebenswichtigen Vorgang darstellt. Transportprozesse im Nephron Lokalisation proximaler Tubulus Henlesche Schleife distaler Tubulus und Sammelrohr Aktive Resorption Sekretion Aminosäuren, (p-AminoProteine, Hippursäure, Glucose, Na+, K+, Penicillin) 2+ 2+ Ca , Mg , HCO3-, Phosphat, Sulfat Passive Resorption Sekretion Harnstoff, Cl , NH3 H2O H2O Na+, K+, Ca2+, Cl- H+ Harnstoff, Na+, H2O Harnstoff, Na+, Cl+ H , NH3 6.3 Die Clearance Die renale Clearance ist ein Maß für die Elimination eines Stoffes aus dem Blutplasma bei der Nierenpassage, stellt also ein Maß für die Klärfunktion der Niere dar. Der ClearanceWert gibt den Teil des renalen Plasmaflusses an, der pro Minute von dem betreffenden Stoff völlig befreit wird. Oft verwendet man für diese Messung die Bearbeitung/Ausscheidung des Kreatinins, da es ausschließlich glomerulär filtriert wird und tubulär praktisch nicht mehr verändert. Creatinin-Clearance (ml/min) = CreaUrin (mg%) • VolUrin (ml / d) CreaSerum (mg%) •1440(min/ d) Schätzformel: Creatinin-Clearance = KG(kg) • (140 - Alter) 72 • CreaSerum (mg%) Der Normalwert der Clearance beträgt 120 ml/min. Sinkt er auf 60 ml/min, so ist die Niere zu 50% geschädigt; bei einer Schädigung von 75% sinkt er auf 30 ml/min. Seite 92 Substanz Harnstoff/Kreatinin Glucose/Harnsäure Insulin Myoglobin Hämoglobin Serumalbumin Molare Masse 60 180 5500 17000 68000 69000 Molekülradius (nm) 0,16 0,36 1,46 1,95 3,25 3,55 Konzentration Filtrat /Plasma 1,0 1,0 0,98 0,75 0,03 0,01 Wird ein Stoff bei einem einzigen Nierendurchgang vollständig eliminiert, entspricht der Clearance-Wert dem renalen Plasmafluß. Um die Durchblutung der Niere abschätzen zu können, verwendet man die PAH-Clearance (PAH = Paraaminohippursäure). Der Normwert für die PAH-Clearance liegt bei 650 ml/min. Seite 93 6.4 Blutdruckregelung durch die Niere Auf den Gefäßschlingen des Glomerulums sitzt der juxtaglomeruläre Apparat. Bei Abfall des Blutdrucks im Vas afferens bilden diese Zellgruppen Renin. Renin spaltet von dem aus der Leber stammenden Angiotensinogen das Angiotensin I ab. Dies wird durch eine zweite Protease, das ständig vorhandene Converting Enzyme, erneut Seite 94 gespalten, so daß das Achterpeptid Angiotensin II entsteht. Dieses Hormon hat verschiedene Wirkungsmechanismen: • es gehört zu den wirksamsten gefäßverengenden Substanzen und bewirkt die allgemeine Vasokonstriktion mit Ausnahme der Herzkranzgefäße • es führt zu einer Aldosteronfreisetzung in der NNR ‡ Rückresorption von Na, damit osmotische Wasserretention • über Gefäßkonstriktion Verringerung der glomerulären Filtrationsrate • mehr Durst und Salzappetit 6.5 Chronische Niereninsuffizienz (CNI) Die meisten Nierenerkrankungen können, unabhängig von ihrer Ursache, in einer chronischen Niereninsuffizienz enden. Primär glomeruläre Erkrankungen sind die häufigste Ursache. Bei der CNI handelt es sich um eine irreversible, langsam fortschreitende Einschränkung der Nierenfunktion durch Ausfall funktionstüchtiger Nephrone. Durch den Parenchymverlust entwickelt sich schließlich das Bild der Schrumpfniere. Ohne weitere Einwirkung der primären schädigenden Ursache kann es zum Stillstand des Krankheitsprozesses kommen (stationäre Nierenfunktionseinschränkung) oder durch funktionelle Überlastung der Restnephrone zu einer Progression. Ursachen • • • • • • Glomerulonephritis/-sklerose Hypertonie/Nephrosklerose Diabetes mellitus Zystennieren Pyelonephritis, Interstitielle Nephritis Andere Ursachen oder unbekannt 26% 17% 15% 9% 8% 25% Am häufigsten treten Veränderungen im Bereich des Glomerulums auf. Die Glomerulonephritis kann durch immunpathogenetische Mechanismen entstehen. Beispielsweise kann es 12-14 Tage nach einer Rachenentzündung zum Auftreten von Blut im Harn kommen. Dabei wurden Antigene gegen die die Entzündung auslösenden Streptokokken gebildet, die aber ebenfalls gegen Strukturen der Glomeruli reagieren. Hypertonie führt über atherosklerotische Veränderung der Gefäße in der Bowman’schen Kapsel zu einer Einschränkung der Nierenfunktion. Bei der interstitiellen Nephritis handelt es sich um eine Nierenentzündung, bei der primär das interstitielle Gewebe betroffen ist, später aber auch Glomeruli, Tubuli und Gefäße geschädigt werden können. Symptome • • • • • Retention harnpflichtiger Substanzen Hypervolämie/Hypovolämie Metabolische Acidose Hypocalcämie/Hyperphosphatämie Hyperparathyreoidismus/Osteomalazie • • • • • • Hypertonie Akzelerierte Atherosklerose Neuromuskuläre Störungen (zentral, peripher) Gastrointestinale Störungen (z.B.Gastroenteritis) Hauterscheinungen (z.B. Juckreiz) Hämatologische Veränderungen (Anämie, Leukopenie, Blutungsneigung) Seite 95 Bei CNI treten typische Veränderungen der Körperflüssigkeiten auf. Übersteigt die Natriumaufnahme die Ausscheidungskapazität, kommt es zur Wasserretention und damit zur Hypertonie; auch Hyperkaliämie tritt häufiger auf, allerdings erst im fortgeschrittenen Stadium (da der größte Teil tubulär sezerniert wird). Um die Hypocalcämie zu kompensieren, wird der Knochen abgebaut, wodurch Knochenerweichung auftritt. Bei einer Filtrationsrate <15% der Norm tritt eine Intoxikation (Urämie) auf. Es kommt zu Urinablagerung in der Haut, so daß diese eine grün-braune Färbung annimmt und die Patienten urämisch riechen. Ernährungsrelevante Probleme 1. Natrium-/Wasser-Haushalt 2. Kalium-Haushalt 3. Calcium-/Phosphat-Haushalt 4. Kalorienzufuhr 5. Eiweißzufuhr Ödeme, Hypertonie Hyperkaliämie Osteopathie Extraossäre Verkalkungen Katabolismus Harnstoff, Progression Stadien der CNI Es besteht zwar keine strenge Korrelation zwischen dem Ausmaß der Nierenschädigung und den Symptomen, aber es lassen sich trotzdem gewisse Stadien angeben. Kreatinin stellt einen guten Indikator für eine späte, nicht für eine frühe Funktionsänderung dar. Erst wenn >50% der Nephrone geschädigt sind, treten Symptome auf. Die Menge des Seite 96 Plasmas, die von Kreatinin befreit werden kann, sinkt immer mehr. Es kommt zu einem Rückstau und somit zu einem Anstieg der Konzentration im Serum. Therapie der CNI - Kompensierte Retention/Präurämie: Diät, Medikamente - Präurämie/Urämie: Dialyse, Nierentransplantation Diät: Eiweißeinschränkung bis ca. 25-45g, sehr hochwertige Kombinationen wichtig Biologische Wertigkeit von Proteinen und Proteingemischen: • Vollei/Kartoffel 137 Seite 97 • • • • • Vollei/Milch Vollei/Weizen Bohnen und Mais Vollei Kartoffel 122 118 101 100 90-100 Bei der Dialyse erfolgt ein Entzug niedermolekularer harnpflichtiger Substanzen aus dem Blut. In der Dialyseflüssigkeit müssen also Ionen, AS und Glucose in der gleichen Konzentration wie im Plasma vorhanden sein. Es gibt im wesentlichen zwei Formen der Dialyse: • Hämodialyse • Peritonealdialyse: Dialyseflüssigkeit wird in die Bauchhöhle eingeleitet, die Dialyse findet über das Bauchfell statt (nicht so effektiv) Die Durchführung der Dialyse ist wöchentlich 1-3 mal nötig, wobei das Befinden mit der Konzentration an Abfallstoffen im Blut stark schwankt. 6.6 Akutes Nierenversagen (ANV) Ursachen • Schock (hypovolämisch, hämorrhagisch, septisch, kardiogen) 50% • Toxisch (Antibiotika, Kontrastmittel) 25% • Akute schwere Nierenerkrankung • Akute Urin-Abflußstörung • Hämolyse, Rhabdomyolyse, Paraproteine 20% (Hämoglobin ist ein Tetramer und wird normalerweise nicht filtriert. Bei einem Transfusionszwischenfall beispielsweise kann es aber in die Untereinheiten zerfallen, so filtriert werden und das Glomerulum verstopfen.) Verlauf 1. Anurie/Oligurie (<100/<400 ml Urin/Tag) Dauer: 2-3 Wochen 2. Polyurie (>2000 ml Urin/Tag) Dauer: 1-3 Wochen 3. Normurie mit langsamer Normalisierung der harnpflichtigen Substanzen Therapie • Grundkrankheit • Anurie/Oligurie • Polyurie Medikamente Dialyse parenterale Ernährung (35g AS, davon 60% essentiell) ausreichende Flüssigkeitszufuhr 6.7 Nephrotisches Syndrom Beim nephrotischen Syndrom wird die selektive Permeabilität des Glomerulums verändert. Durch die Erkrankung wird die negative Wandladung abgeschafft und die Anzahl der Poren vermindert. Die großen Proteine werden nun nicht mehr abgestoßen und besetzen sozusagen die Poren, wodurch vermehrt große und weniger kleine Moleküle filtriert werden. So kommt es zu einer Proteinurie. Seite 98 Glomeruläre Schädigung Eiweißverlust (Albumin ‡ Proteinurie) Eiweißmangel (‡ Hypoproteinämie) Eiweißsynthesesteigerung (‡ Hyperlipoproteinämie) verminderter onkotischer Druck/ sekundärer Hyperaldosteronismus (‡Ödeme) Seite 99 7. Osteoporose 7.1 Vitamin D Ergocalciferol = Vitamin D2, Cholecalciferol = Vitamin D3 Vitamin D ist zusammen mit dem Parathormon der wichtigste Regulator für die Kalziumhomöostase. Unter Einwirkung von UV-Strahlung kann es in der Haut aus dem Provitamin 7Dehydrocholesterin gebildet werden. Ca 50% des benötigten Vitamin D wird so im Körper hergestellt, der Rest muß über die Nahrung zugeführt werden. Das intestinal resorbierte und in der Haut synthetisierte Vitamin D3 gelangt in die Leber und wird dort zu 25-Hydroxycholecalciferol hydroxyliert. Das nicht umgewandelte Vitamin D3 wird in der Muskulatur und im Fettgewebe gespeichert. 25-OH-D3 ist die wichtigste Transportform des Vitamin D. In der Niere wird diese Verbindung nochmals hydroxyliert zu 1,25Dihydroxycholecalciferol. Dieser Metabolit ist die eigentliche Wirkform. Durch bestimmte Regulationsmechanismen wird die Syntheserate dem Bedarf angepaßt. Parathormon beispielsweise fördert die Umwandlung von 25-Hydroxycholecalciferol in 1,25Dihydroxycholecalciferol. Es wird in das Blut sezerniert und gelangt auf diese Weise zu allen Erfolgsorganen. Der Calcium-Bedarf liegt bei 800 mg/d, durchschnittlich aufgenommen werden 500-700 mg. In der Schwangerschaft ist der Bedarf auf 1000-1500 mg erhöht, ebenso bei Personen mit Osteoporose. Vitamin D-Mangel verursacht bei Säuglingen und Kleinkindern Rachitis, bei Erwachsenen Osteomalazie. Funktionen • • • • • antirachitisch (Ca-P-Stoffwechsel gestört, Entkalkung der Knochen) fördert den Ca- Einbau in die organische Matrix des Knochens steigert die Ca-Aufnahme im Darm erhöht die Ca-Rückresorption in der Niere erhöht den Ca-Spiegel im Blut (bei Überdosierung: Ca-Abbau auch im Knochen) 7.2 Hormone des Ca-Stoffwechsels Hormon PTH (aus Nebenschilddrüse) Vitamin D Calcitonin (C-Zellen, Schilddrüse) Effekt (Blut) Ca ≠ , Ph Ø Ca ≠ , Ph ≠ Ca Ø , Ph (+-) Antag. des PTH Kontrolle durch Ca im Blut Ø Ph Ø , PTH ≠ Ca ≠ Der wichtigste Unterschied zwischen PTH und Vitamin D besteht darin, daß Vitamin D in physiologischen Konzentrationen die intestinale Kalziumaufnahme erhöht und PTH nicht. Als wichtigste und stärkste Kontrollparameter bei der Bildung der Hormone wirken der Kalziumund Phosphat-Spiegel im Blut. Seite 100 7.3 Osteoporose Osteoporose ist die häufigste Skeletterkrankung des Menschen. Sie kann durch Kalziummangel oder Immobilitätbegünstigt werden. Bei Osteoporose handelt es sich aber nicht um Vitamin D-Mangel! Zur Behandlung der Osteoporose werden sowohl Calcitonin als auch Vitamin D als Medikamente eingesetzt. Der Knochen stellt kein statisches Gewebe dar, sondern wird ständig ab- und umgebaut (Bone Remodeling). Osteoklasten bauen die verknöcherte Matrix ab, während Osteoblasten neue, Ca-arme Matrix bilden, in die dann unter Vitamin D-Einfluß Kalziumsalze eingelagert werden. Die Aktivität dieser Knochenzellen und damit die Skelettmasse ist allerdings abhängig vom Lebensalter und den Sexualhormonen. Die maximale Knochenmasse ist zwischen dem 25. und 30. Lebensjahr erreicht und nimmt anschließend etwa um 0,5-1% pro Jahr ab. Frauen sind hierbei noch zusätzlich durch die Menopause belastet, da dann die Östrogene als Schutzfaktor für den Knochen entfallen. Die Frakturschwelle wird mit ca 80 Jahren erreicht. Osteoporose unterteilt man in primäre und sekundäre Formen. Bei der primären unterscheidet man zwei Typen: Osteoporose Typ I ist die postmenopausale Osteoporose, die bei über 50jährigen Frauen auftritt. Hierbei steigt die Verlustrate des Knochengewebes rasch an und betrifft v.a. die Trabekel, also den inneren Teil des Knochens. Die Dichte des Knochens ist deshalb deutlich vermindert (WHO: >2 Standardabweichungen unter der Altersnorm). Es kommt in erster Linie zu Wirbelfrakturen und Zahnverlust. Die senile Form (Osteoporose Typ II) betrifft überwiegend Frauen ab dem 70. Lebensjahr. Hier schreiten die Verluste langsamer voran und beziehen sich v.a. auf die Corticalis. Die Dichte des Knochens ist niedrig normal. Brüche treten im Bereich der Wirbelsäule und Hüfte auf. Sekundäre Osteoporose tritt z.B. bei Hyperparathyreoidismus auf. Der erhöhte Spiegel an Parathormon beschleunigt den Knochenumsatz, wobei der Abbau den Anbau überwiegt. Auch durch eine Cortisonbehandlung kann es zur Osteoporose kommen. Cortison wirkt antagonistisch zum Vitamin D, es hemmt die Kalziumaufnahme im Darm und stört den Anbau des Knochens. Stadien Es werden fünf Stadien unterschieden: 1. Beschwerdefreiheit, Knochendichteverminderung 2. Schmerzen, normale Mobilität 3. Schmerzen mit Mobilitätseinschränkung 4. Schmerzen, Immobilität nach Knochenbruch 5. allg. Immobilität, Schmerzen, Knochenbrüche, manifeste neurolog. Probleme (KnochendeformationÆ Druck auf periphere NervenÆ Lähmung) Therapie • Kalziumaufnahme erhöhen auf 1500mg (Æ Milch: 300 mg in 250 ml, Joghurt: 180 mg in 150 ml) • draußen aufhalten, Sport • gegen Schmerzen: physikalische Methoden (meist gehen Muskelschmerzen, Rheuma, Bandscheibenvorfälle mit Osteoporose einher Æ Fango, Massage, Reizstrom...) Seite 101 • Calcitoninpräparate, um die Heilung osteoporotischer Brüche zu beschleunigen • in Europa Einsatz von Natriumfluorid: in bestimmten Konzentrationen und über einen bestimmten Zeitraum hinweg stimulierende Wirkung auf Osteoblasten • Bisphosphonate zur Hemmung der Osteolyse, knochenbaufördernd Beispiel: 40jährige Frau im ersten Stadium Als erstes sollte man erfragen, ob die betroffene Frau sich in der Menopause befindet oder die Östrogene aus anderen Gründen erniedrigt sind. Eine frühzeitige Östrogensubstitution kann den raschen Verlust an Knochenmasse vermindern, da die Aktivierung der Osteoklasten vermindert wird. Da diese Präparate aber Nebenwirkungen haben, sollte man prüfen, in welchen Fällen sie wirklich angebracht sind. Auch die anderen Geschlechtshormone haben Einfluß auf den Zustand des Knochens. Bei Männern wird der Abbau überwiegend durch Testosteronmangel hervorgerufen. Entscheidend sind dabei die Wechselwirkungen der einzelnen Hormone. Seite 102 8. Parenterale Ernährung und Sondenkost 8.1 Parenterale Ernährung Als parenterale Ernährung bezeichnet man die Zufuhr von Nährstoffen unter Umgehung des Verdauungstraktes direkt in die Blutbahn. Dies ist erforderlich bei Erkrankungen der Abdominalorgane, bei postoperativen Zuständen, in denen der Gastrointestinaltrakt vorübergehend außer Funktion gesetzt werden muß oder bei erheblichen Störungen der Verdauung oder Resorption. Bei dieser Art der Ernährung wird auch die Leber als Regulationsorgan umgangen. Deshalb muß die zugeführte Nährlösung optimal zusammengesetzt sein; sie muß den Bedarf an Energie, essentiellen Nährstoffen, Vitaminen, Mineralstoffen, Spurenelementen und AS decken. 8.2 Inhaltsstoffe der Nährlösungen Kohlenhydrate Die Kohlenhydrate haben eine große Bedeutung, da sie schnell verwertbare Energieträger darstellen. Dafür stehen Glucose, Fructose und die Zuckeralkohole Xylit und Sorbit zur Verfügung. Eine Deckung des Energiebedarfs allein mit diesen Kohlenhydraten ist allerdings nicht möglich. Fette Über die Lipidemulsion kann man ausreichend Energie zuführen. 20%ige Fettemulsionen werden noch toleriert, so daß hier das Problem der zu hohen Flüssigkeitszufuhr umgangen werden kann. Es werden Triglyceride, meist Soja-Lipide verwendet. Aminosäuren In der Aminosäurelösung müssen alle essentiellen AS in ausreichender Menge vorhanden sein (40-50% der AS-Lösung), daneben auch nichtessentielle als zusätzliche Stickstoffquelle. Kinder benötigen Histidin als essentielle AS, Frühgeburten Cystein. Dabei ist zu beachten, daß unter den Bedingungen der parenteralen Ernährung nur vier der nichtessentiellen AS ausreichend synthetisiert werden, nämlich Asparaginsäure, Glutaminsäure, Serin und Glycin. Je nach Vorerkrankung können spezielle AS-Gemische verwendet werden; Leber- und Nierenkranke erhalten beispielsweise einen erhöhten Anteil verzweigtkettiger AS. Seite 103 Energiebilanzen Wichtig für die exakte Dosierung ist die Erfassung des Ernährungszustands des Patienten (da eventuell Mängel ausgeglichen werden müssen) und seines Energiebedarfs. Der Basalbedarf entspricht dem Energiebedarf eines gesunden Menschen. Durch Verletzungen, Krankheiten und Streß steigt der Kalorienbedarf erheblich an. Erwachsene benötigen etwa 20-45 ml Wasser pro kg Körpergewicht, Säuglinge 130-180 ml/kg. Diese Menge sollte durch die Nährlösung nicht wesentlich überschritten werden. Normale Lösung bei Stoffwechselgesunden - 50% Glucose (bei Kindern: 20%) - 10% AS - 10% / 20% Lipidemulsion in der Regel: 500ml G 50% 500ml AS10% 400ml LE 20% 1400 ml 950 kcal 205 kcal 745 kcal 1900 kcal = 50 en% = 11 en% = 39 en% 8.3 Probleme bei der parenteralen Ernährung Ein 70 kg schwerer Mensch mit Sepsis hätte einen Bedarf von 3500 kcal pro Tag, was eine “Energielücke” von 1600 kcal bedeutet. Dieser wird durch eine erhöhte Menge an Nährlösung gedeckt. Die Konzentration läßt sich nicht erhöhen, da es schon bei den oben angegebenen Konzentrationen zu osmotisch bedingten Venenreizungen kommen kann. Die hohe Volumenzufuhr muß durch Medikamente kompensiert werden. Kohlenhydrate Bei mehrwöchiger Gabe von >400g Glucose / Tag kann sich eine Fettleber entwickeln. Durch die hohe Zufuhr ist die Glycogenspeicherkapazität bald erschöpft; die Leber wandelt die Glucose dann auch in Fettsäuren und Triglyceride um. Seite 104 Die hohe Glucosekonzentration im Blut (Hyperglycämie) kann zu einem Diabetes führen. Es muß exogen Insulin zugeführt werden. Bei Verwendung von Zuckeraustauschstoffen kann es zum Abfall des zellulären ATP, zur Hyperurikämie und Hyperlaktatämie kommen. Aminosäuren Hierbei ist die hohe Volumenzufuhr das größte Problem. Um 100g AS zuzuführen, benötigt man einen Liter Nährlösung. Fette Durch Gabe der Lipidemulsion wird eine Hypertriglyceridämie hervorgerufen. Man sollte nach dem ersten Tag mit parenteraler Fettzufuhr das Plasma untersuchen. Ist es milchig, spricht dies für eine nicht ausreichende Fettelimination. Die Fettdosis muß dementsprechend reduziert werden. Emulsionen mit MCT-Fetten werden schneller aus dem Blut eliminiert. Heute wird parenterale Ernährung zunehmend mit Sondenkost kombiniert, um diese Probleme zu umgehen. 8.4 Sondenkost Sondenkost wird bei intakter Funktion der Verdauungs- und Resorptionsorgane eingesetzt, wenn eine ausreichende perorale Nährstoffaufnahme nicht möglich ist. Die Nahrung wird in flüssiger Form über eine durch Nase, Rachen und Speiseröhre in den Magen bzw. Dünndarm eingeführte Sonde verabreicht. In seltenen Fällen werden Sonden auch durch die Bauchdecke direkt in den Magen gelegt. Seite 105 Die Zusammensetzung der Sondenkost richtet sich nach den Grundsätzen einer optimalen Ernährung. Liegen Verdauungs- und Stoffwechselerkrankungen vor oder andere Abweichungen des Bedarfs von der Norm, so wird die Zusmmensetzung der Kost darauf abgestimmt. Ernährt wird entweder mit sondengängig gemachter Normalkost oder einer industriell hergestellten bilanzierten Diät. Selbst hergestellte Sondenkost hat sich in der Klinik nicht durchgesetzt, weil hohe Personalkosten anfallen und die Gefahr einer bakteriellen Kontamination besteht. Bei der nährstoffdefinierten Diät werden hochmolekulare Nahrungsmittel angeboten. Diese Ernährungsform setzt einen funktionierenden Magen-Darm-Trakt voraus. Die chemisch definierte Diät wird bei Vorliegen einer Malabsorption angewandt; die Nahrungsmittel liegen hier niedermolekular vor. Sondenkost (Inhalt pro 2000 ml) Peptide (g, en%) Fett (g, en%) Fresubin (nährstoffdefiniert, hochmolekular) flüssig 76 (15) 68 (30) instant 74 (14) 16 (7) (2100ml) plus 76 (15) 68 (30) 750 MCT 112,5 (20) 90 (35) (1500ml) DFN (1600ml) 89,6 (20) 75,2 (37) KH (g, en%) En.-Dichte (kcal/ml) Osmolarität (mosmol/l) 276 (55) 414 (79) 1 1 350 300 276 (55) 255 (45) 1 1,5 250 (12g BS) 300 192 (43) 1,12 290 1 1 400 300 1,3 450 Survimed (chemisch definiert, niedermolekular) OPD 90 (18) 52 (22) 300 (60) instant 74 (14) 23 (10) 408 (76) (2100ml) renal 31 (6) 23 (10) 414 (84) (1500ml) Seite 106 8.5 Probleme bei Sondenkost Generell ist der Sondenkost der Vorzug vor der parenteralen Ernährung zu geben, da sie physiologischer, sicherer und einfacherer ist. Das größte Problem hierbei ist die hohe osmotische Wirksamkeit: Die Sondenkost bindet Wasser im Darm, wodurch eine osmotische Diarrhoe auftreten kann. Dies kann man durch eine langsame Anpassung des Patienten an die Sondenkost verringern. Bei zu hoher Osmolarität der Nahrung kann auch das Dumping Syndrom auftreten. Spurenelemente und Vitamine werden sowohl den Nährlösungen als auch der Sondenkost in Form von Konzentraten zugesetzt. 8.6 Vitamine Reservekapazität des Menschen für einige Vitamine: Vitamin B1 4-10 Tage Vitamin B2 2-6 Wochen Niacin 2-6 Wochen Vitamin B6 2-6 Wochen Vitamin C 4-8 Wochen Folsäure 3-4 Monate Vitamin E 1 Jahr Vitamin A 1-2 Jahre Vitamin B12 3-5 Jahre Vitamin K Vitamin K ist ein Überbegriff für drei Substanzen: Phyllochinon (K1), Menachinon (K2) und Menadion (K3). Da Vitamin K ein fettlösliches Vitamin darstellt, ist zur Resorption das Beisein von Fett und die normale Gallensekretion wichtig. Die Gerinnungsfaktoren VII, IX, X und Prothrombin können in der Leber nur in Gegenwart von Vitamin K hergestellt werden. Somit kommt es bei einem Mangel an Vitamin K zu einer erhöhten Blutungsneigung. Vitamin K ist aber ebenso nötig bei der Synthese der Proteine C und S, die die Gerinnung hemmen. Der tägliche Bedarf ist sehr gering, er beträgt nur 0,03-0,15 mg/kg KG. Das von den Darmbakterien hergestellte Vitamin K trägt zur Bedarfsdeckung bei, so daß ein Mangel selten auftritt. Er kann in der Schwangerschaft aber zu Mißbildungen führen. Ursachen für einen Mangel • chronische Darmerkrankungen (intakter Dünndarm ist für die Resorption wichtig) • Neugeborene (besitzen wenig Vitamin K und davon abhängige Gerinnungsfaktoren) • Zufuhr von Vitamin K-Antagonisten (Markumar, Sintrom zur Verhinderung der Blutgerinnselbildung als Prophylaxe und Therapie bei Thrombosen, Embolien, Herzinfarkt etc.) • Antibiotika (Zerstörung der Darmflora und damit der endogenen Vitamin K-Produktion) Vitamin B12 = Cobalamin Cobalamin ist ein wasserlösliches Vitamin, das hauptsächlich in tierischen Lebensmitteln vorkommt. Der Bedarf liegt bei etwa 1mg/Tag. Das mit der Nahrung aufgenommene Vitamin B12 (Extrinsic Factor) verbindet sich mit dem von der Magenschleimhaut gebildeten Intrinsic Factor zu einem Komplex, der dann im terminalen Ileum in das Blut aufgenommen wird. Seite 107 Vitamin B12 ist an der Nukleinsäuresynthese beteiligt und damit an der Neubildung von Zellkernen. Außerdem ist es ein wichtiger Cofaktor für viele enzymatische Reaktionen, z.B. die Übertragung von Methylgruppen bei der Methioninsynthese. Der Mangelzustand kann eintreten durch • operative Entfernung des Magens bzw. des terminalen Ileums • schwere Ileumerkrankungen • Fischbandwurm • streng vegetarische Kost (B12-Spiegel im Plasma meist niedrig normal, echter Mangel nur selten) Bei einem Mangel kommt es zu einer Verringerung der Zellteilung im Knochenmark und damit zur perniziösen Anämie. Die Erythrocyten sind groß und hyperchrom. Als weiteres Symptom tritt funikuläre Myelose auf (Funiculi = Stränge des Rückenmarks). 8.7 Spurenelemente Eisen Verteilung im Körper Gesamtbestand Hämeisen Speichereisen Blut: Transferrin Gewebe: Ferritin Funktionseisen 3,5 g 2,1 g 1,0 g (2,5 mg) 0,4 g Funktionseisen umfaßt das Eisen in Myoglobin, Cytochromen, Katalasen, Peroxidasen etc. Aus der Nahrung werden täglich etwa 2,5 mg Fe resorbiert (möglich ist eine Resorption bis zu 3mg). Der Eisen-Speicher im Körper beträgt 1g, so daß eine vollständige Füllung 400 Tage in Anspruch nehmen würde. Den größten Speicher stellt das Ferritin dar, v.a. in den retikuloendothelialen Zellen der Leber. Dieses Eisen wird bei Bedarf zuerst abgebaut. Der tägliche Umsatz liegt bei etwa 25 mg. Fe2+ wird oral besser resorbiert, da es auch bei pH7 resorptionsfähig ist. Fe3+ dagegen wird nur in Magennähe im sauren Milieu resorbiert. Pflanzliche Produkte enthalten vorwiegend Fe3+, so daß man bei vegetarischer Ernährung auf eine ausreichende Vitamin C-Zufuhr achten sollte. Dies reduziert Fe3+ zu Fe2+ und bildet mit Eisen gut lösliche Komplexe. Ursachen des Mangels • Blutverlust (2 ml Blut = 1 mg Eisen) oder Blutspende • GIT-Blutungen (M.Crohn, Colitis Ulcerosa, Divertikel...) • Schwangerschaft (500 mg) • vegetarische Ernährung Prälatenter Eisenmangel (= Speichereisenmangel): Speichereisen < 200 mg Latenter Eisenmangel (= Transporteisenmangel): Serumeisen < 60 mg/100ml Manifester Eisenmangel: Hb < 12 g/l Seite 108 Symptome • Mundwinkelrhagaden (Schrunden) • Störungen des Haar- und Nagelwachstums • Atrophie von Haut und Schleimhäuten • Müdigkeit • Atemnot • Schwindel • Kopfschmerzen • selten Schluckstörungen • Erythrocyten hypochrom und klein Zink täglicher Bedarf: 2,2 mg (WHO) Resorption: 7% empfohlene Aufnahme: 15 mg/d Ursachen von Zinkmangel • Darmerkrankungen (M.Crohn, Colitis Ulcerosa, Sprue) • Parenterale Ernährung, Sondenkost • (vegetarische Ernährung, Weizenkleie) Symptome • Akrodermatitis • gestörte Wundheilung • Haarausfall • Minderwuchs Zink ist Bestandteil vieler Metalloenzyme (DNA-, RNA-Polymerase, Carboanhydrase, alkalische Phosphatase, Phospholipasen...) und dient der Enzymaktivierung. So hat es eine große Bedeutung für den Eiweiß-, Fett- und Kohlenhydratstoffwechsel und den Säure-BaseHaushalt. Seite 109 9. Schilddrüse 9.1 Anatomie Die Schilddrüse befindet sich vor der Luftröhre und umschließt diese mit zwei Seitenlappen dicht unterhalb des Schildknorpels. Sie liegt in der Nähe der Stimmritze, so daß Aphonie nach einer Operation möglich ist. Durch gefäßhaltige Bindegewebsscheidewände (Septen) wird die Schilddrüse in verschieden große Läppchenbezirke gegliedert. Die funktionelle Einheit des Drüsengewebes ist der Follikel. Von diesen rund angeordneten Zellanhäufungen sind etwa eine Million vorhanden. In ihrem Inneren befindet sich eine homogene Masse, das Kolloid. Dessen Hauptbestandteil ist Thyreoglobulin, das Speichereiweiß für die Schilddrüsenhormone. Die eigentlichen hormonbildenden Drüsenzellen umgeben dieses Kolloid als einschichtiges Epithel. 9.2 Biosynthese der Hormone Jodidionen werden in die Follikelzelle aus dem Blut durch aktiven Transport gegen ein Konzentrationsgefälle aufgenommen (Jodination). Dieser Transport kann durch andere Anionen (Perchlorate, Thiocyanat) blockiert werden. Durch eine Jodperoxidase entsteht elementares Jod, das in die Tyrosinreste von Thyreoglobulin eingeführt wird (Jodisation). Jetzt werden entweder zwei Moleküle Dijodtyrosin zu Thyroxin (T4) oder ein monojodinierter mit einem dijodierten Tyrosylrest zu Trijodthyronin (T3) kondensiert. Thyreoglobulin, das T3- und T4-Reste sowie Mono- und Dijodtyrosylreste enthält, wird im Kolloid gespeichert. Werden Hormone benötigt, werden sie daraus wieder freigesetzt, indem lysosomale Proteasen das Thyreoglobulin abbauen. Regulation Neben der Regelung über das Hypothalamus-Hypophysen-System mit den Hormonen TRH (Thyreotropin-Releasing-Hormon, Thyreoliberin) und TSH (thyreoideastimulierendes Hormon) wird die Aktivität der Schilddrüsenhormone auch durch andere periphere Mechanismen bestimmt. Hierzu gehören die Balance des Jodhaushalts über das Kolloid und Seite 110 die Jodzufuhr in der Nahrung sowie die Umwandlung von T4 in der Peripherie zu T3 oder zu rT3. 9.3 T3 und T4 Von den Schilddrüsenfollikeln werden zwei stoffwechselsteuernde Hormone abgegeben, Thyroxin (T4) und Trijodthyronin (T3). In den parafollikulären C-Zellen wird Calcitonin produziert. T4 ist das Hauptprodukt der Schilddrüse und wird nur in dieser synthetisiert. Es ist biologisch nicht sehr aktiv, wird zu 99% an Plasmaproteine gebunden und zu den Zielzellen transportiert. Als Transportproteine dienen thyroxinbindendes Globulin (TBG, 60%), thyroxinbindendes Präalbumin (TBPA, 30%) und Albumin (10%). Die biologisch wirksame Form, das T3, entsteht zum größten Teil in seinen Zielzellen durch Dejodierung. Nach Bindung an einen intranukleären T3-Rezeptor wirkt es direkt auf die Transkription ein. Neben dem normalen T3 wird noch ein biologisch inaktives, das sog. reverse T3 (rT3) gebildet. Diese Form steigt bei schweren Erkrankungen und Erschöpfungszuständen stark an. Die Schilddrüsenhormone sind entscheidend an normalen Entwicklungsprozessen wie Differenzierung und Wachstum beteiligt. Beim Erwachsenen greifen sie stabilisierend in alle metabolischen Prozesse ein und haben daher Rezeptoren in nahezu allen Zellen des Organismus. Seite 111 Die Funktion des T3 besteht v.a. in der Beschleunigung oxidativer Stoffwechselprozesse in den meisten Zellen. Darüber hinaus stimuliert es die RNA-Synthese und damit die Proteinbildung. Bei Kindern fördern die Schilddrüsenhormone auch das körperliche Wachstum und die Gehirnentwicklung. T3 und T4 können nur in ihrer freien Form wirken. Sie unterliegen keinen zirkadianen Rhythmen. 9.4 Diagnostik TRH-Test TRH ist ein sehr einfach aufgebautes Peptidhormon. Es besteht nur aus drei AS (Glu-His-Pro). Deshalb ist es leicht zu synthetisieren und wird diagnostisch eingesetzt bei dem sog. TRH-Test. Dabei werden 200 mg TRH nasal appliziert und nach 30 Minuten Blut entnommen. Beim Gesunden steigt der TSH-Spiegel an (vgl. Abb. Regelkreis). Bei Hyperthyreose (Überfunktion) dagegen steigt die TSH-Konzentration nicht an. Hier liegt eine negative Rückkopplung vor: die Hemmung durch die erhöhten Mengen an T3 und T4 kann durch das applizierte TRH nicht aufgehoben werden. Für eine Hypothyreose ist ein hoher TSH-Spiegel mit überschießender Stimulierbarkeit durch TRH charakteristisch. Seite 112 Immunometrischer Assay Früher war es nur möglich, die Gesamthormonmenge nachzuweisen, nicht aber ausschließlich das freie Hormon. Heute ist eine Bestimmung des freien Hormons über den immunometrischen Assay möglich. Dabei werden zwei monoklonale Antikörper verwendet, die an unterschiedlichen Stellen des Hormons binden. Nur wenn Bindungsstellen für beide Antikörper vorhanden sind, liegt das richtige Hormon vor und wird auch gezählt. So wird weder rT3 noch das proteingebundene Hormon gemessen. TBG-Plasmakonzentration Auch die Bestimmung des TBG im Plasma hat diagnostischen Wert. Bei Abweichungen vom Normwert lassen sich Rückschlüsse auf eventuelle Erkrankungen ziehen Seite 113 9.5 Erkrankungen Die Schilddrüsenerkrankungen werden in drei Hauptgruppen eingeteilt: a) Schilddrüsenvergrößerung, Struma (mit oder ohne Funktionsstörung) b) Schilddrüsenunterfunktion (mit latenter oder manifester Hypothyreose) c) Schilddrüsenüberfunktion (mit latenter oder manifester Hyperthyreose) Die Begriffe Hyperthyreose und Hypothyreose bezeichnen lediglich ein pathologisch erhöhtes bzw. erniedrigtes Angebot der Schilddrüsenhormone an die Körperzellen. Allerdings wird weder etwas über die Ursachen noch über die Symptome ausgesagt. Eine Hypothyreose kann z.B. auch entstehen, ohne daß in der Schilddrüse pathologische Vorgänge stattfinden (s.u.). Euthyreote Schilddrüsenvergrößerung Die euthyreote Schilddrüsenvergrößerung entsteht selten aufgrund genetischer Defekte (Jodaufnahme-, Peroxidase-, Kondensationsdefekte). Häufiger ist Jodmangel die Ursache. Die DGE empfiehlt eine Zufuhr von 200 mg/d. In der BRD liegt die durchschnittliche Aufnahme allerdings nur bei 80 mg/d absolut. Die Messung erfolgt über den Gehalt im 24 h-Sammelurin. Um Sammelfehlern vorzubeugen, bezieht man den Jodgehalt auf die Menge an ausgeschiedenem Kreatinin. Der Normalwert liegt hier bei etwa 50 mg/g Kreatinin. Ursachen für die verminderte Jodaufnahme sind v.a. zu geringe Gehalte in den Lebensmitteln und im Trinkwasser (v.a. in küstenfernen Regionen). Ab einem Gehalt von <100 mg Jod tritt endemisch Struma auf. Bei ausschließlicher Verwendung von jodiertem Speisesalz könnte der Bedarf allerdings gedeckt werden. Allein schon der Einsatz im Haushalt würde wahrscheinlich ausreichen (1 g jodiertes Salz = 20 mg Jod). Als Regelhormon bei allen Synthese- und Sekretionsprozessen der Schilddrüse dient TSH aus dem Hypophysenvorderlappen. Jodmangel wirkt als unabhängiger Wachstumsfaktor auf die Follikel. Generell finden zwei Formen des Wachstums an den Follikelzellen statt: 1) Hyperplasie: mehr Zellen entstehen 2) Hypertrophie: die Zellen werden relativ größer TSH und Jodmangel wirken sich auf diese zwei Arten des Wachstums unterschiedlich stark aus. Durch den unspezifischen Reiz des Jodmangels kommt es also vorwiegend zur Hyperplasie, wobei alle Zellen proliferieren. Im Hinblick auf die Stimulierbarkeit durch TSH verhält sich der Zellverband allerdings heterogen. So kann es passieren, daß im Jodmangel nur Zellen entstehen, die sehr TSH-empfindlich sind oder Follikel, die gar nicht auf TSH reagieren. Solche Zellen, die nicht durch TSH stimuliert werden können und somit nicht dem körpereigenen Regulationssystem Seite 114 unterliegen, bezeichnet man als autonom. Jodmangel ist der wichtigste Faktor bei der Entstehung der Autonomie. Sind mehr als 5g autonome Bezirke vorhanden, liegt eine manifeste Überfunktion vor. Die WHO unterscheidet drei Stadien der Struma nach der Größe: Stadium 0: Keine Struma Stadium 1: Struma tastbar oder per Ultraschall festzustellen, aber nur sichtbar bei zurückgeneigtem Kopf Stadium 2: bei normaler Kopfhaltung deutlich sichtbar Stadium 3: mechanische Komplikationen (Behinderung bei Schluckvorgang oder Atmung; Blutstau durch Wachstum in den Brustkorb, wo große Gefäße abgedrückt werden) 1988 wurde an einigen tausend Menschen in Deutschland eine Ultraschalluntersuchung durchgeführt und dabei eine andere Definition festgelegt. Danach lag eine Struma bei mehr als der dreifachen Standardabweichung über dem Mittelwert vor. Frauen Männer 13jährige 6jährige 18 ml 25 ml 8 ml 4 ml Nach beiden Einteilungen liegt bei ca 30% der Bevölkerung eine Struma vor! Hyperthyreose - Morbus Basedow Die Hyperthyreose (Thyreotoxikose) wird durch ein überhöhtes Angebot an Schilddrüsenhormonen hervorgerufen. Dadurch sind der Basisstoffwechsel, die Körpertemperatur, das Herzzeitvolumen und die Herzfrequenz erhöht, die Erregbarkeit gesteigert. Es treten v.a. vegetative Symptome auf: die Betroffenen sind meist hektisch, nervös und dürr und essen, ohne zuzunehmen. Sie leiden unter Herzklopfen, Schlaflosigkeit, Haarausfall und schwitzen leicht. Typisch ist ein feinschlägiger Tremor. Mögliche Ursachen einer Hyperthyreose sind: • eine Autoimmunerkrankung (M.Basedow) • eine autonome Struma • eine Überdosierung mit Schilddrüsenhormonen • eine unkontrollierte TSH-Sekretion durch die Hypophyse • Metastasen Seite 115 Morbus Basedow Morbus Basedow ist eine Autoimmunerkrankung. Die Schilddrüse wird durch Antikörper, die gegen den TSH-Rezeptor gerichtet sind, unkontrolliert stimuliert. Hierdurch kommt es zu einer Mehrproduktion und Sekretion von T3 und T4. Wodurch die Störung zustande kommt, ist unzureichend geklärt. Es treten drei Symptome auf: - Struma - TSH-Rezeptor-Antikörper im Plasma (bei ca 80% der Betroffenen) - Exophthalmus (vorstehende Augen) Die Basedow-Orbitopathie ist eine eigenständige Autoimmunerkrankung, die aber sehr häufig gemeinsam mit M.Basedow auftritt. Zeitlich kann die Orbitopathie vor oder nach Beginn der Hyperthyreose oder erst viel später auftreten. Bei voller Ausprägung entwickelt sich ein Exophthalmus. Der Hauptgrund hierfür ist eine Muskelschwellung aufgrund der durch die Antikörper hervorgerufenen Entzündung. Die entzündliche Bindegewebsvermehrung innerhalb und außerhalb der Augenmuskeln führt zu einer weiteren Einengung der Augenhöhle. Man unterscheidet sechs Stadien: Stadium I: leichte Retraktion des Oberlids Stadium II: Lidschwellung, Photophobie, vermehrte Tränensekretion Stadium III: Vorstehen des Augapfels (Zehntelmm); Entzündung wird nicht empfunden, keine Symptome! Stadium IV: erste Komplikationen beim Sehen (Augenmuskelblockade, Doppeltsehen) Seite 116 Stadium V: Stadium VI: kleine Defekte in der Hornhaut (Austrocknen, da der Lidschluß nicht mehr richtig funktioniert) Sehausfälle, Schädigung des Sehnervs durch steigenden Druck in der Höhle Autonome Struma Die autonome Struma entwickelt sich meist nach dem oben genannten Pathomechanismus aus dem Jodmangel. Dabei unterscheidet man je nach Art der autonomen Bezirke drei Ausprägungen: - umschriebener Bezirk: autonomes Adenom - mehrere Knoten: multifokales Adenom - nicht abgrenzbar: disseminierte Autonomie (nur 10%) Die autonomen Bezirke raffen die vorhandenen Jodidionen an sich und produzieren unkontrolliert Hormone, wodurch der Plasmaspiegel an T3 und T4 hoch ist. Die TSHabhängigen Bezirke werden aufgrund dieser hohen Konzentrationen immer stärker herunterreguliert. Therapiemöglichkeiten - Operation (v.a. angewandt bei einzelnem Knoten) - Tabletten: Thyreostatika (bei disseminierter Autonomie) Peroxidasehemmer (Thiamazol) Jod-Aufnahmehemmer (Perchlorat) - radioaktives Jod (zerstrahlt in genügend großer Menge die Schilddrüsenzellen; da die autonomen Bezirke fast alles Jod aufnehmen, werden auch nur sie beeinträchtigt. Diese Methode wird auch bei Schildrüsentumoren angewandt, solange die Zellen noch genügend differenziert sind.) Hypothyreose Hypothyreose umfaßt alle Zustände, die durch ein vermindertes Angebot an Schilddrüsenhormonen hervorgerufen werden. primär: Endorgan (Schilddrüse) ist gestört sekundär: Störung in Hirnanhangdrüse (Hypophyse) tertiär: Störung im Hypothalamus Um neonatale Hypothyreosen festzustellen, wird ein Neugeborenen-Screening durchgeführt. Am fünften Tag nach der Geburt wird der Gehalt des Blutes an TSH festgestellt. Bei Vorliegen einer Hypothyreose ist er erhöht. Wenn dies der Fall ist, wird ein genauer Hormontest durchgeführt. Noch vor der 12. Lebenswoche muß mit T4-Gaben begonnen werden, da sonst die Hirnentwicklung gefährdet ist. Ohne frühzeitige Behandlung treten auch noch weitere Seite 117 Defekte auf wie Skelettveränderungen und eine Veränderung des neuromuskulären Kontaktes, so daß die allgemeine Entwicklung zurückbleibt. Es kommt zum Kretinismus. Die häufigste Ursache einer Hypothyreose im Erwachsenenalter ist der Verlust an Schilddrüsengewebe nach einer Struma-Operation oder nach Radiojod-Behandlung. Symptome • Herabsetzung des Grundumsatzes • Gewichtszunahme • Blässe, trockene Haut • verminderte geistige Beweglichkeit • Kälteempfindlichkeit Das Vollbild der Hypothyreose wird wegen der dabei zu beobachtenden eigentümlichen Verdickung und Schwellung der Haut als Myxödem bezeichnet. Seite 118 10. Eßstörungen 10.1 Adipositas Die Zahl der Fettsüchtigen hat während der letzten Jahrzehnte in den hochzivilisierten Industrieländern ständig zugenommen und liegt zur Zeit (je nach Statistik und Definition) bei 30-50% der Gesamtbevölkerung. Die Adipositas entsteht durch eine positive Energiebilanz, d.h. es wird mehr Energie aufgenommen als verbraucht. Der Grund dafür ist die zunehmende Verringerung des Energieverbrauchs, bedingt durch immer geringer werdende körperliche Arbeit bei weitgehend konstanter Energiezufuhr. Die überschüssige Energie wird in Form von Fett gespeichert. Die Neigung, Fett zu speichern, ist allerdings individuell unterschiedlich. Die Adipositas wird in eine endogene und eine exogene Form unterteilt. Die endogene Form ist selten. Sie beruht z.B. auf einer Hypothyreose. Auch eine Störung im Sättigungszentrum kann die Ursache sein, wodurch die Regulation der Nahrungsaufnahme beeinträchtigt ist. Für endogene Faktoren spricht auch die familiäre Häufung. Eine über dem Bedarf liegende Energiezufuhr ist nur bei der exogenen Form ersichtlich. Hierbei sind psychische Störungen und Abweichungen vom normalen Eßverhalten beteiligt (z.B. durch seelische Belastungen ausgelöste gesteigerte Nahrungsaufnahme). Oft spielen auch falsche erzieherische Maßnahmen eine Rolle. Es zeigt sich eine erhöhte Außenreizabhängigkeit, d.h. die Nahrungsaufnahme findet nicht mehr aufgrund von Hungergefühl statt sondern Appetit entsteht durch Signale der Umwelt. 10.2 Magersucht Für die Magersucht ist eine negative Energiebilanz und die damit einhergehende Reduktion des Körpergewichts verantwortlich. Es kommt zu einer verringerten Nahrungszufuhr durch - reduziertes Hungergefühl (Schädigung des Hungerzentrums = zerebrale Magersucht) - Nahrungsmangel - Malassimilationssyndrom - erhöhten Energieverbrauch (z.B. durch Schilddrüsenüberfunktion) Anorexia Nervosa Die Anorexia nervosa entsteht aus einer psychischen Fehlhaltung. Sie tritt fast ausschließlich bei Frauen, vorzugsweise während der Pubertät auf. Dabei kommt es zu einer hochgradigen Abneigung gegen die Nahrung, die sogar zum Erbrechen nach dem Essen führen kann. Hier zeigen sich fließende Übergänge zum Krankheitsbild der Bulimie. Als Ursache wird eine Trotz- und Protestreaktion angenommen. Merkmale • Verlust von mindestens 25% des ursprünglichen Gewichts • intensive Angst, dick zu werden • sekundäre Amenorrhoe • gesteigerte körperliche Energieentfaltung • depressive Verstimmungen • Körperschemastörungen (auch bei Untergewicht sieht man sich noch als zu dick an) Seite 119 Bulimia nervosa Auch die Patienten mit Bulimie haben Angst vor zu hohem Körpergewicht. Bei dieser Krankheit kommt es zu Phasen exzessiver Nahrungsaufnahme (innerhalb von 1-2 Stunden bis zu 20000 Kalorien). Die dann drohende Gewichtszunahme wird durch selbst ausgelöstes Erbrechen bzw. Einnahme von Laxantien verhindert. Folgen der Bulimie und Anorexie • Schädigung des Ösophagus • Störungen des Elektrolyt-, Säure-/Basen- und Wasserhaushalts • Karies • Überempfindlichkeit der Zähne Letzte Bearbeitung ©AOS 19.02.2003 13:26:27 Uhr Seite 120